Изобретение относится к гликоконъюгатам 20(S)-камптотецина, в которых элемент 3-О-метилированной β-L-фукозы связан с гидроксильной группой производного камптотецина в положении 20 посредством модифицированного тиомочевиной пептидного мостика. Также изобретение относится к способу получения соединений данного изобретения и их применению в качестве лекарственных средств при лечении раковых заболеваний.
20(S)-камптотецином является пентациклический алкалоид, который был выделен в 1966 году Wall et al. (J.Am.Chem.Soc. 88, 3888 (1966)). Он обладает высоким противоопухолевым активным действием, что продемонстрировано в многочисленных опытах in vitro и in vivo. Однако попытки реализации этого действия при клинических исследованиях потерпели неудачу из-за возникших проблем токсичности и растворимости.
Через расщепление кольца Е-лактона и образование соли натрия было получено соединение, растворимое в воде, которое находилось, в зависимости от баланса уровня рН, в форме замкнутого кольца. Но клинические испытания также не привели к успеху.
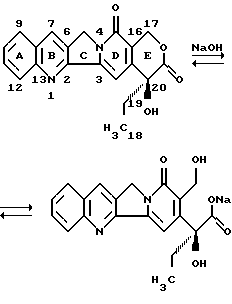
20 лет спустя было найдено, что биологическая активность в ферментных ингибиторах топоизомераз I является обратимой. После этого исследовательские изыскания возобновились, чтобы получить совместимые и in vivo активные производные камптотецина.
Соли производных камптотецина, модифицированные кольцами А и В, а также производные 20-O-ацила с ионизированными группами, которые используют для улучшения растворимости в воде, описаны (Vishnuvajjala et al. патент США US 4943579). Последние пролекарственные концепции позднее были перенесены на модифицированные производные камптотецина (Wani et al. международная заявка, WO 9602546). Но описанные пролекарства 20-O-ацила in vivo имеют очень короткий период полураспада и очень быстро расщепляются до основы.
В международной заявке WO 9631532 Al описана модифицированная сахаром цитостатика, при которой связывание различных цитотоксичных или цитостатичных активных соединений с, например, региоселективно модифицированными углеводными элементами посредством определенного мостика приводит к улучшению опухолевой селективности. Из описанных в международной заявке WO 9631532 Al комбинаций углевода, мостика и активного вещества было неожиданно обнаружено, что связывание модифицированных элементов β-L-фукозы в положении 3 с гидроксильной группой в положении 20 20(S)-камптотецина посредством модифицированного тиомочевиной пептидного мостика, состоящего из полностью стереометрической неполярной и основной аминокислоты, приводит к получению наиболее предпочтительных конъюгатов со следующими свойствами:
- эфирное связывание носителя радикала с гидроксильной группой в положении 20 стабилизирует действие важного лактонового кольца в частях камптотецина;
- специальная конформация дипептидного мостика показывает, что конъюгаты в экстраклеточной среде и в крови в сравнении с подобными, описанными в международной заявке WO 9631532 конъюгатами с другими мостиками, обладают четко улучшенной стабильностью. Конъюгаты данного изобретения являются более стабильными, чем описанные в патенте США US 4943579 пролекарства 20-O-ацила камптотецина;
- конъюгаты данного изобретения лучше растворяются в воде, чем подобные конъюгаты из международной заявки WO 963I532;
- конъюгаты данного изобретения in vivo показывают высокую активность в отношении опухолевых клеток и опухолевого ксенотрансплантата;
- конъюгаты данного изобретения in vivo при внутривенном введении демонстрируют эксклеточную терапевтическую эффективность при больших дозах в отношении различных опухолей;
- конъюгаты данного изобретения в отношении основного токсофора демонстрируют высокую совместимость и опухолевую селективность, особенно при токсичности костного мозга.
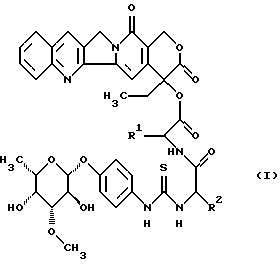
Изобретение относится к соединениям общей формулы (I)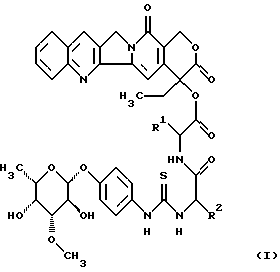
в которой R1 является полностью стереометрической неполярной боковой цепью аминокислоты и
R2 является основной боковой цепью аминокислоты, а также к их солям, стереоизомерам и смесям стереоизомеров.
Предпочтительными являются соединения общей формулы (I), в которой R1 является разветвленным алкильным радикалом, который имеет до 4 атомов углерода и R2 является радикалом формулы -(CH2)n-R3, причем R3 является -NH2,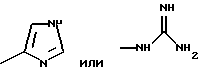
n равно 1-4.
Наиболее предпочтительными являются соединения общей формулы (I), в которой R1 является разветвленным алкильным радикалом формулы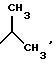
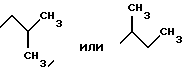
R2 является остатком формулы
-(CH2)2-NH2, -(CH2)3-NH2, -(CH2)4-NH2,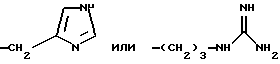
Элемент камптотецина может быть представлен в конфигурации 20(R) или 20(S) или в виде смеси этих обеих стереоизомерных форм. Предпочтительной является конфигурация 20(S).
Аминокислоты могут быть представлены в конфигурации L или D или также в виде смеси форм L и D.
Понятие "аминокислоты" обозначает широко распространенные в природе α-аминокислоты, но включает также и их гомологи, изомеры и производные. Примерами изомеров могут являться энантиомеры. Производными могут быть, например, аминокислоты, имеющие защитные группы.
Под аминокислотами с "полностью стереометрическими" боковыми цепями понимают такие аминокислоты, чье разветвление боковых цепей находится в положении β или γ; например, валин и изолеуцин или леуцин.
Примерами аминокислот с неполярными боковыми цепями являются: аланин, валин, леуцин, изолеуцин, пролин, триптофан, фенилаланин, метионин. Примерами аминокислот с основными боковыми цепями являются: лизин, аргинин, гистидин, орнитин, диаминомасляная кислота.
Соединения данного изобретения представлены предпочтительно в виде их солей, например, солей органических или неорганических кислот.
К кислотам, которые могут быть добавлены, принадлежат предпочтительно галогеноводородные кислоты, например, соляная кислота или бромистоводородная кислота, предпочтительно, соляная кислота; кроме того, фосфорная, азотная и серная кислоты, моно и бифункциональные карбоновые кислоты и гидроксикарбоновые кислоты, например, уксусная кислота, трифторуксусная кислота, малеиновая кислота, малоновая кислота, щавелевая кислота, глюконовая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, салициловая кислота, сорбиновая кислота и молочная кислота; а также сульфоновые кислоты, например, п-толуолсульфоновая кислота, 1,5-нафталиндисульфоновая кислота или камфарсульфоновая кислота.
Согласно данному изобретению гликоконъюгаты могут быть получены, например, путем связывания 20(S)-камптотецина с активированными компонентами карбоксила, части которых могут быть представлены защищенными аминокислотами, пептидами или пептидами, модифицированными углеводами.
Предпочтительно, при последовательном проведении синтеза гликоконъюгатов по обычным методикам, вначале проводят ацилирование 20(S)-камптотецина N-защищенным активированным карбоксильным элементом неполярной, полностью стереометрической аминокислоты в подходящем растворителе, в данном случае, в присутствии основания. Затем по известным методикам выборочно отщепляют аминозащитную группу. Затем связывают, при необходимости, с подходящим защищенным элементом основной аминокислоты и в конце деблокируют функциональную группу α-аминокислоты при сохранении защитной группы в боковых цепях. На заключительной стадии проводят связывание с углеводным радикалом путем превращения п-аминофенил-3-О-метил-β-L-фукопиранозида в соответствующий изотиоцианат и затем связывают с деблокированной α-аминогруппой пептидилкамптотецина. В данном случае оставшиеся в боковых цепях защитные группы отщепляют и свободную аминогруппу превращают в подходящую соль аммония.
Таким образом, изобретение относится к способу получения гликоконъюгатов общей формулы (I)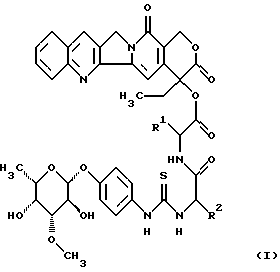
в которой R1 является полностью стереометрической неполярной боковой цепью аминокислоты и R2 является основной цепью аминокислоты
или их солей, отличающийся тем, что изотиоцианат формулы (II)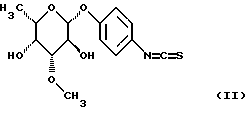
взаимодействует с имеющим в боковой цепи защитную группу пептидилкамптотецином формулы (III)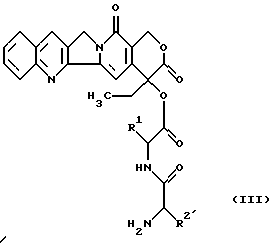
в которой Rl имеет вышеуказанное значение и R2 имеет значение вышеуказанного радикала R2, который кроме этого может находиться в основной группе обычной в химии пептидов защитной группы до получения гликоконъюгата формулы (IV)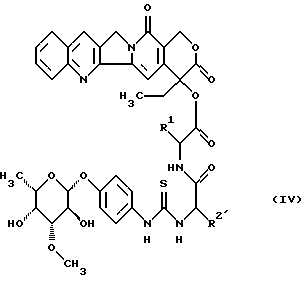
в которой R1 и R2 имеют вышеуказанные значения,
в данном случае, находящуюся в боковых цепях аминозащитную группу отщепляют по обычным методикам, и полученное соединение превращают в предпочтительную соль.
Допустимой является смена стадий реакций для получения целевого соединения. Таким образом, согласно одному также предпочтительному варианту п-изотиоцианатофенил -3-О-метил-β-L-фукозид вначале связывают с подходящей защищенной конечной основной аминокислотой, и затем этот элемент превращают со свободной аминогруппой конъюгата аминокислоты из 20(S)-камптотецина и неполярной, полностью стереометрической аминокислоты. В данном случае, находящиеся в боковых цепях защитные группы отщепляют и свободную аминогруппу превращают в подходящую соль аммония.
Таким образом, далее изобретение относится к альтернативному способу получения соединений общей формулы (I) или их солей, отличающемуся тем, что изотиоцианат формулы (II)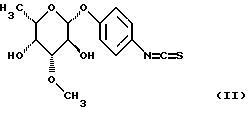
взаимодействует с подходящей защищенной конечной основной аминокислотой формулы (V)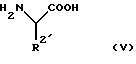
в которой R2 является основной боковой цепью аминокислоты, основные группы которой могут быть защищены, до получения конъюгата аминокислоты формулы (VI)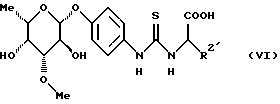
в которой R2 имеет вышеуказанное значение,
и затем этот конъюгат превращают с конъюгатами аминокислоты формулы (VII)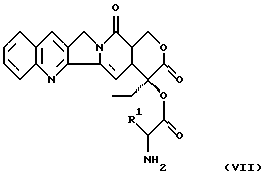
в которой R1 имеет вышеуказанное значение, защитную группу боковых цепей отщепляют и соединение превращают в подходящую соль.
В частности, после присоединения первой аминокислоты к камптотецину можно получить смеси диастереомеров. Чистые диастереомеры соединений данного изобретения получают, например, по вышеуказанному способу, по которому после присоединения первого элемента аминокислоты к камптотецину и последующего отщепления защитной группы диастереомеры разделяют подходящим способом. Из промежуточной смеси чистых диастереомеров по вышеуказанному способу можно получить целевую смесь чистых диастереомеров.
Также возможно разделение смеси диастереомеров целевого соединения обычным способом на отдельные диастереомеры.
Проведение реакций возможно при разных отношениях давления и температуры, например, давление 0,5-2 бар, соответственно температура от -30oС до +100oС, в подходящих растворителях, например, диметилформамиде, тетрагидрофуране, дихлорметане, хлороформе, низших спиртах, ацетонитриле, диоксане, воде или в смесях названных растворителей. Как правило, реакции предпочтительно проводят в диметилформамиде, дихлорметане или тетрагидрофуране/дихлорметане при комнатной температуре и нормальном давлении.
Для активирования карбоксильных групп используют известные в химии пептидов соединяющие реагенты, как, например, в Jakubke/Jeschkeit: аминокислоты, пептиды, протеины; описаны в Verlag Chemie 1982 или Tetrahedr. Lett. 34, 6705 (1993). Предпочтительными являются, например, ангидриды N-карбоновой кислоты, хлорангидриды или смешанные ангидриды.
В дальнейшем подходящим для активирования карбоксильных групп является образование аддуктов карбодиимидов, например, N,N'-диэтилкарбодиимид, N, N'-диизопропилкарбодиимид, N, N'-дициклогексилкарбодиимид, N-(3-диметиламинопропил)-N'-этил-карбодиимид-гидрохлорид, N-циклогексил-N'-(2-морфолиноэтил)-карбодиимид-мето-п-толуолсульфонат, или такие соединения карбонила, как, например, карбонилдиимидазол, или 1,2-оксазольные соединения, например, 2-этил-5-фенил-1,2-оксазол-3-сульфат или 2-трет-бутил-5-метил-изоксазолил-перхлорат, или ациламиновые соединения, например, 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин, или ангидрид пропанфосфоновой кислоты, или изобутиловый эфир хлормуравьиной кислоты, или бензотриазолилокси-трис-(диметиламино)-фосфоний-гексафторофосфат, 1-гидроксибензотриазоловый эфир или N-гидроксисукцинимидовый эфир. В дальнейшем, возможно использование компонентов аминокислот в виде чистых ангидридов.
В качестве оснований возможно использование, например, триэтиламина, этилдиизопропиламина, пиридина, N,N-диметиламинопиридина или других.
В качестве защитных групп для третичного функционирования аминокислот возможно использование известных в химии пептидов защитных групп, например, уретаны, алкилы, ацилы, сложные эфиры или амиды.
В рамках данного изобретения аминозащитными группами являются используемые обычно в химии пептидов аминозащитные группы.
К ним предпочтительно принадлежат: бензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, 3,5-диметоксибензилоксикарбонил, 2,4-диметоксибензилоксикарбонил, 4-метоксибензилоксикарбонил, 4-нитробензилоксикарбонил, 2- нитробензилоксикарбонил, 2-нитро-4,5-диметоксибензилоксикарбонил, метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил (БОК), аллилоксикарбонил, винилоксикарбонил, 3,4,5-триметоксибензилоксикарбонил, фталоил, 2,2,2-трихлорэтоксикарбонил, 2,2,2-трихлор-трет-бутоксикарбонил, метилоксикарбонил, 4-нитрофеноксикарбонил, фторенил-9-метоксикарбонил (ФМОК), формил, ацетил, пропионил, пивалоил, 2-хлорацетил, 2-бромацетил, 2,2,2-трифторацетил, 2,2,2-трихлорацетил, бензоил, бензил, 4-хлорбензоил, 4-бромбензоил, 4-нитробензоил, фталимид, изовалероил или бензилоксиметилен, 4-нитробензил, 2,4-динитробензил, 4-нитрофенил или 2-нитрофенилсульфенил. Наиболее предпочтительными являются группа (ФМОК) и (БОК).
Предпочтительными защитными группами карбоксила являются алкильные эфиры, имеющие 1-4 атомов углерода, с прямой или разветвленной цепью.
Отщепление защитной группы на соответствующей стадии реакции проводят, например, путем обработки кислотой или основанием гидрогенолитическим или другим способом.
Гликоконъюгаты данного изобретения показывают как in vitro, так и in vivo неожиданно сильный противоопухолевый эффект в отношении различных опухолей, в частности, при опухолях легких, молочных желез, поджелудочной железы и толстой кишки, связанный с высокой селективностью в отношении доброкачественных клеток.
Также возможно использование гликоконъюгатов данного изобретения при лечении раковых заболеваний как у людей, так и у животных.
Данное изобретение относится к фармацевтическим препаративным формам, которые содержат нетоксичные инертные фармацевтически приемлемые носители одного или нескольких соединений данного изобретения или которые состоят из одного или нескольких активных веществ данного изобретения, а также к способу получения этих препаративных форм.
Активные вещества могут быть представлены в одном или нескольких вышеуказанных носителях, а также в микрокапсулах.
Терапевтически активные соединения должны присутствовать в вышеуказанных фармацевтических препаративных формах в концентрации от приблизительно 0,1 до 99,5, предпочтительно, от приблизительно 0,5 до 95 мас.% общей смеси.
Вышеуказанные фармацевтические препаративные формы могут содержать кроме соединений данного изобретения также и другие активные вещества.
В общем, как при лечении людей, так и животных, предпочтительным для достижения эффективного результата является использование активного вещества данного изобретения в общей массе от приблизительно 0,5 до приблизительно 500, предпочтительно 5-100 мг/кг веса тела каждые 24 часа, в данном случае, в виде нескольких единичных дозированных форм. Единичная дозированная форма содержит вышеуказанные активные вещества данного изобретения предпочтительно в количестве от приблизительно 1 до приблизительно 80, наиболее предпочтительно 3-30 мг/кг веса тела.
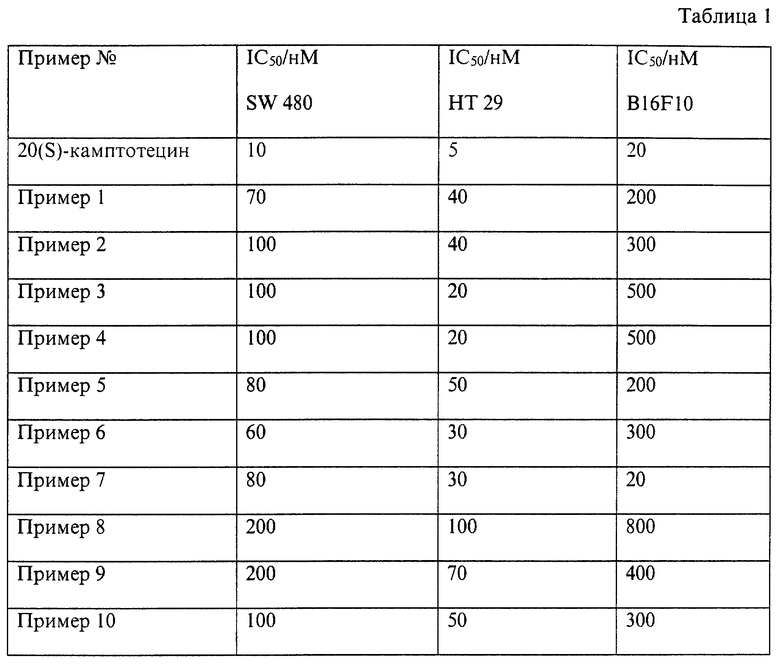
Биологические испытания.
1. Тест ингибитора роста для определения цитотоксичных свойств:
Соединения человеческих клеток толстой кишки SW 480 и НТ 29 (АТСС-Nr.CCL 228 и НВТ 38), а также соединения клеток меланомы мыши B16F10 (CRL 6475) помещают в Roux-ванночки в среду RPMI 1640 с примесью 10% FCS. Затем трипсинируют и помещают в RPMI плюс 10% FCS с числом клеток 50000 клеток/мл для SW 480 и НТ 29 и 20000 клеток для B16F10. 100 мкл клеточной суспензии/волну помещают в микроволновую печь 96 и инкубируют в течение 1 дня при температуре 37oС в инкубаторе с CO2. Затем 100 мкл среды RPMI и 1 мкл DMSO добавляют к тестируемой субстанции. Спустя 6 дней проверяют рост. Для этого на каждую микроволну добавляют 25 мкл раствора МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолинбромид) с исходной концентрацией 5 мг/мл воды, инкубируют в течение 5 часов при температуре 37oС в инкубаторе с СО2. Затем среду отфильтровывают и добавляют 100 мкл i-пропанол/волну, подвергают вибрации со 100 мкл воды в течение 30 минут и измеряют экстинкцию при 595 нм при помощи Multiplate Reader (Bio..)3550-UV.
Цитотоксичный эффект приведен в таблице 1 как показатель IC50 для клеток SW 480, НТ 29 и B16F10.
2. Гематопоэтическая активность гликоконъюгатов в сравнении с основным активным веществом:
Материал и методики:
Из бедра мыши получают клетки костного мозга. Клетки 105 инкубируют в среде McCoy 5A (0,3% агар) вместе с рекомбинационными составляющими GM-CSF (Genzyme: образование колоний штамм клеток) и субстанцией (10-4-100 мкг/мл) при температуре 37oС и 7% CO2. Спустя 7 дней считают колонии (<50 клеток) и кластер (17-50 клеток).
Результат:
Как представлено в таблице 2, исследуемые гликоконъюгаты показывают в отношении основного активного вещества характерно уменьшающуюся ингибицию пролиферации штамма клеток костного мозга.
3. Ингибирование роста опухолей у голых мышей (in vivo)
Материал:
Для всех экспериментов (in vivo) при исследовании ингибирования роста опухолей используют голых мышей с врожденным отсутствием вилочковой железы (NMRI nu/nu-штамм). Отобранную многоклеточную опухоль легкого LXFL 529 развивают путем последовательного введения в мышей. Человеческую опухоль распределяют изоферментативными и иммуногистохимическими методиками.
Экспериментальный синтез:
Опухоль имплантируют в мышей (в возрасте 6-8 недель) с двух сторон. Лечение начинают в зависимости от времени дублирования, как только опухоли достигнут диаметра 5-7 мм. Часть мышей подвергают рандомизации, а часть мышей оставляют в качестве контрольных (5 мышей на группу из 8-10 обработанных). Отдельные опухоли контрольной группы мышей растут все прогрессивнее.
Величину опухолей измеряют в двойном измерении при помощи штангенциркуля. Объем опухолей, который хорошо коррелируется с клеточным числом, используют затем для всех анализов. Объемы вычисляют по формуле "длина х ширина х ширина /2" ({ахb2}/2, а или b обозначают два прямоугольно расположенных диаметра).
Показатели относительного объема опухолей вычисляют для каждой отдельной опухоли путем деления величины опухоли ко дню Х на величину опухоли дня 0 (с момента рандомизации). Затем средние показатели относительного объема опухолей используют для дальнейшего анализа.
Ингибирование объема опухолей (относительный объем опухолей тестируемой группы/контрольной группе х100, Т/К в %) является конечной измеряемой величиной (см. табл. 3).
Лечение:
Введение соединений проводят внутривенно, например, на день 0, 1 и 2 после рандомизации, причем общую дозировку дня разделяют больше 2 раз.
Результат:
Терапевтическая эффективность гликоконъюгатов данного изобретения из примеров 1 и 2 представлена на примере ксенографта многоклеточной человеческой опухоли легких LXFL 529. Терапию проводят при максимально допустимой дозе, а при половине максимально допустимой дозы до отчетливой опухолевой ремиссии. В отношении других опухолей также демонстрируется эксклеточная активность.
Продолжительная комплексная ремиссия соединения из примера 1 при дозе от 16 до 8 мг/кг и зависимость действия от дозы в дальнейшем эксперименте с планируемым количеством дней терапии 1-3 продемонстрированы на чертеже.
4. Гидролитическая стабильность:
Соединения данного изобретения из примеров 1, 2, 8 и 9 растворяют в воде и после 24-часового отстаивания при комнатной температуре с помощью жидкостной хроматографии высокого разрешения (ЖХВР) показывают четкое ниже 1% выделение камптотецина от поверхностного процента.
При растворении 10 мкM соединений из примеров 1 и 2 в среде RPMI плюс 10% FCS и 30%-ной человеческой цельной крови в буферном растворе PBS после 24-часового отстаивания происходит выделение камптотецина ниже 1%.
Методика:
ЖХВР System Hewlett Packard HP 1050
Колонка: нуклеозил 120-5 С 18 250 мм х 4 мм (Macherey & Nagel; Germany) Элюент: А: 0,01 м КН2РO4 в воде (Н2=милли-пора)
В: 80% ацетонитрил/20% элюент А
Поток: 1,2 мл
Градиент: to: 20% B - t40: 100% B
t45 : 100% B - t47 : 20% В
Определение: 240 нм или 370 нм
5. Стабилизация лактона:
Гликоконъюгаты данного изобретения из примера 1, 2, 8 и 9 растворяют в 80% воды и 20% ацетонитрила и соединяют с 2 эквивалентами раствора едкого натра с рН 9. После отстаивания в течение 1 часа при комнатной температуре происходит расщепление лактонового кольца ниже 5% (определение по вышеуказанной методике).
Примеры
Углеродный аддукт:
I) п-аминофенил-3-О-метил L-фукопиранозид:
L-фукопиранозид: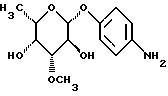
I.a) п-нитрофенил-3-О-метил-β-L-фукопиранознд:
6 г (21 ммоль) п-нитрофенил-β-L-фукопиранозида в 300 мл абсолютного метанола смешивают с 7,84 г (31,5 ммоль) дибутилцинноксида и нагревают в течение 2 часов в присутствии флегмы. Затем концентрируют, остаток сушат и помещают в 300 мл диметилформамида, добавляют 15,7 мл метилиодида и смесь перемешивают в течение 40 часов при температуре 70oС. Растворитель удаляют в вакууме и остаток помещают в 300 мл дихлорметана. Суспензию фильтруют, оставшийся раствор снова концентрируют и подвергают флэш-хроматографии (дихлорметан/метанол 99/1). После концентрации получают 3,82 г (61%) целевого продукта.
1.b) п-аминофенил-3-О-метил-β-L-фукопиранозид:
3,81 г (12,73 ммоль) п-нитрофенил-β-L-фукопиранозида растворяют в метаноле, добавляют диоксид платины и гидрируют в атмосфере водорода при немного повышенном давлении. После фильтрации катализатора и осаждения смеси эфиром получают 3 г (88%) целевого продукта. (Тонкослойная хроматография: дихлорметан/метанол 9/1, Rf=0,53)
Аддукты пептидил-камптотецина:
II) 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил]-L-лизил-L/D-лeyцил}-камптотецин, трифторацетат:
II) L 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил]-L-лизил-L-лeyцил}-камптотецин, трифторацетат:
II) D 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил]-L-лизил-D-лeyцил}-камптотецин, трифторацетат:
II.а) 20(S)-20-O-[N-(трет-бутоксикарбонил)-L/D-леуцил]-камптотецин:
Суспензию 10 г (28,7 ммоль) 20(S)-камптотецина в 250 мл абсолютного диметилформамида добавляют при перемешивании к 11,1 г (43 ммоль) ангидрида N-(трет-бутоксикарбонил)-леуцин-N-карбоновой кислоты, а также 1 г 4-(N,N-диметиламино)-пиридина. Смесь обрабатывают в ультразвуковой бане в течение 16 часов при комнатной температуре, снова добавляют 3,7 г ангидрида N-(трет-бутоксикарбонил)-леуцин-N-карбоновой кислоты и снова обрабатывают в течение 2 часов при комнатной температуре. Затем оставшийся нерастворенным камптотецин отделяют и фильтрат концентрируют в вакууме. Остаток очищают путем флэш-хроматографии (петролейный эфир/этилацетат 1/1>1/4) и получают 13,55 г (84%) целевого соединения IIа). (Тонкослойная хроматография: ацетонитрил Rf= 0,47)
II.b) 20(S)-20-O-L/D-леуцил-камптотецин, трифторацетат:
II.b) L 20(S)-20-O-L-леуцил-камптотецин, трифторацетат:
II.b) D 20(S)-20-O-D-леуцил-камптотецин, трифторацетат:
Раствор соединения II. а) (13,55 г, 24,1 ммоль) в смеси из 100 мл дихлорметана и 40 мл безводной трифторуксусной кислоты перемешивают в течение 30 минут при комнатной температуре. После концентрации в вакууме полученный продукт осаждают диэтиловым эфиром и тщательно промывают диэтиловым эфиром. Из тонкослойной хроматограммы определяют двойное пятно с показателем Rf от 0,4 и 0,32. (Выход: 9,5 г (68%)).
Через многократное осаждение диэтиловым эфиром из дихлорметана/метанола возможно разделение смеси на два отдельных компонента II.b) L и II.b) D. Оба компонента представлены в виде диастереомеров, которые дают различные спектры 1H-NMR:
полярный диастереомер:
400-MHz-1H-NMR(CD2Cl2/CD3OD 1:1): δ
sС-Н (кольцо В) 8,63 ппм,
sС-Н (кольцо D) 7,4 ппм,
неполярный диастереомер:
400-MHz-1H-NMR(CD2Cl2/CD3OD 1:1):δ
sС-Н (кольцо В) 8,60 ппм,
sC-H (кольцо D) 7,32 ппм.
На дальнейших стадиях возможно использование как смеси этих двух форм, так и очищенных диастереомеров в чистом виде.
II.с) 20(S)-20-O-[Nα-(трет-бутоксикарбонил)-Nε-(фторенил-9-метоксикарбонил)-L-лизил-L/D-леуцил]-камптотецин:
II. c) L 20(S)-20-O-[Nα-(тpeт-бyтoкcикapбoнил)-Nε-(фтopeнил-9-метоксикарбонил)-L-лизил-L-леуцил]-камптотецин:
II.c) D 20(S)-20-O-[Nα-(тpeт-бyтoкcикapбoнил)-Nε-(фтopeнил-9-метоксикарбонил)-L-лизил-D-леуцил]-камптотецин:
25,6 г (54,6 ммоль) Nα-(трет-бутоксикарбонил)-Nε-(фторенил-9-метоксикарбонил)-L-лизина и 11,1 г гидрата 1-гидрокси-1Н-бензотриазола растворяют в 500 мл диметилформамида. Добавляют 12,6 г (1,2 экв) гидрохлорида N-этил-N'-(диметиламинопропил)-карбодиимида и перемешивают в течение 1 часа при комнатной температуре. Затем добавляют соединение II.b (26,2 г, 0,83 экв) и 7,83 мл (1 экв) этил-диизопропиламина, и смесь перемешивают в течение 16 часов при комнатной температуре. Концентрируют в вакууме и очищают флэш-хроматографией (петролейный эфир/этилацетат 1:2>этилацетат). Получают 43,5 г (87%) целевого соединения (тонкослойная хроматография: Rf=0,44).
Так же как и в дальнейших примерах возможно аналогичное превращение смеси с каждой формой очищенных диастереомеров в чистом виде в II.b).
II) 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил] -L-лизил-L/D-лeyцил} -камптотецин, трифторацетат:
II) L 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил] -L-лизил-L-лeyцил}-камптотецин, трифторацетат:
II) D 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил] -L-лизил-D-лeyцил}-камптотецин, трифторацетат:
Соединение II. с) (43,3 г, 47,5 ммоль) помещают в 150 мл дихлорметана и добавляют 50 мл безводной трифторуксусной кислоты. Полученный раствор перемешивают в течение 1 часа при комнатной температуре. Концентрируют в вакууме и продукт осаждают диэтиловым эфиром. Получают 39,4 г (90%) целевого соединения (тонкослойная хроматография: ацетонитрил/вода 10:1 Rf=0,35).
III) 20(S)-20-O-(L-гистидил-L/D-валил)-камптотецин, трифторацетат:
III.a) 20(S)-20-O-[N-(трет-бутоксикарбонил)-L/D-валил)-камптотецин:
Суспензию 10 г (28,7 ммоль) 20(S)-камптотецина в 500 мл абсолютного диметилформамида перемешивают с 21,5 г (3 экв) ангидрида N-(трет-бутоксикарбонил)-валин-N-карбоновой кислоты, а также 1 г 4-(N,N-диметиламино)-пиридина. В течение 16 часов обрабатывают в ультразвуковой бане при комнатной температуре, отделяют остаточный нерастворенный камптотецин и фильтрат концентрируют в вакууме. Остаток очищают флэш-хроматографией (петролейный эфир/этилацетат 1:3>этилацетат). Получают 11,65 г (73%) целевого соединения (тонкослойная хроматография: ацетонитрил Rf=0,44). Подобную реакцию проводят в дихлорметане вместо диметилформамида в присутствии флегмы, что способствует образованию диастереомера элемента валина конфигурации L.
III.b) 20(S)-20-O-L/D-валил-камптотецин, трифторацетат:
III.b) L 20(S)-20-O-L-валил-камптотецин, трифторацетат:
III.b) D 20(S)-20-O-D-валил-камптотецин, трифторацетат:
Раствор соединения III. а) (11,65 г, 21 ммоль) в смеси из 100 мл дихлорметана и 100 мл безводной трифторуксусной кислоты перемешивают в течение 1 часа при комнатной температуре. После концентрации в вакууме осаждают диэтиловым эфиром из дихлорметана/метанола и изолируют в виде смеси диастереомеров. Выход: 10,9 г (92%). (Тонкослойная хроматография: ацетонитрил/вода 20: 1 Rf=0,31 и 0,39). На этой стадии возможен более высокий уровень очистки неполярного диастереомера элемента валина конфигурации L через кристаллизацию из метанола:
8 г полученного продукта с концентрированным неполярным диастереомером растворяют в 80 мл метанола, охлаждают при температуре 0oС и помещают в 10 мл диэтилового эфира. Затем смесь осаждают 60 мл диэтилового эфира, выпавший продукт фильтруют и сушат. Получают 4,6 г (58%) чистого неполярного диастереомера элемента валина конфигурации L. После осаждения маточного раствора диэтиловым эфиром получают 730 мг (9%) фракции продукта с отношением к диастереомеру 1:18.
В дальнейших реакциях возможно использование как смеси диастереомеров, так и очищенного неполярного или полярного диастереомера. Превращения проводят аналогично.
III. c) 20(S)-20-O-[N-(трет-бутоксикарбонил)-L-гистидил-L/D-валил] -камптотецин:
III. c) L 20(S)-20-O-[N-(трет-бутоксикарбонил)-L-гистидил-L-валил] -камптотецин:
III. c) D 20(S)-20-O-[N-(трет-бутоксикарбонил)-L-гистидил-D-валил] -камптотецин:
5,95 г (23,3 ммоль)N-(трет-бутоксикарбонил)-L-гистидина и 4,73 г гидрата 1-гидрокси-1Н-бензотриазола растворяют в 200 мл диметилформамида. Добавляют 5,38 г гидрохлорида N-этил-N'-(диметиламинопропил)-карбодиимида и перемешивают в течение 1 часа при комнатной температуре. Затем добавляют соединение III.b (10,9 г, 19,44 ммоль) и 6,7 мл этилдиизопропиламина и смесь перемешивают в течение 16 часов при комнатной температуре. После концентрации в вакууме и очистке флэш-хроматографией (ацетонитрил/вода 50:1>20:1) получают целевое соединение (тонкослойная хроматография: ацетонитрил/вода 10:1 Rf= 0,42), которое немедленно подвергают дальнейшему превращению.
III) 20(S)-20-O-(L-гистидил-L/D-валил)-камптотецин, бис-фторацетат:
III) L 20(S)-20-O-(L-гистидил-L-валил)-камптотецин, бис-фторацетат:
III) D 20(S)-20-O-(L-гистидил-D-валил)-камптотецин, бис-фторацетат:
Соединение III. c) помещают в 100 мл дихлорметана и соединяют с 50 мл безводной трифторуксусной кислоты. Полученный раствор перемешивают в течение 30 минут при комнатной температуре. После концентрации в вакууме полученный продукт осаждают диэтиловым эфиром. Получают 13,05 г (83%) целевого продукта (тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:1:2 Rf=0,2).
IV) 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил]-D-лизил-L/D-лeyцил}-камптотецин, трифторацетат:
IV) L 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил]-D-лизил-L-лeyцил} -камптотецин, трифторацетат:
IV) D 20(S)-20-O-{Nε-[фтоpeнил-9-метоксикарбонил]-D-лизил-D-леуцил} -камптотецин, трифторацетат:
Синтез этого соединения проводят полностью аналогично соединению диастереомеров II. На стадии II.с) используют соответствующий изомер D Nα-(трет-бутоксикарбонил)-Nε-(фторенил-9-метоксикарбонил)-L-лизина (тонкослойная хроматография: ацетонитрил/вода 10:1 Rf=0,4).
V) 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил] -L-opнитил-L/D-лeyцил}-камптотецин, трифторацетат:
V) L 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил]-L-opнитил-L-лeyцил} -камптотецин, трифторацетат:
V) D 20(S)-20-O-{Nε-[фтopeнил-9-мeтoкcикapбoнил]-L-opнитил-D-лeyцил}-камптотецин, трифторацетат:
Синтез соединения проводят полностью аналогично соответствующему соединению лизина II. На стадии II.с) используют Nα-(трет-бутоксикарбонил)-Nε-(фторенил-9-метоксикарбонил)-L-орнитин-Nα-(трет-бутоксикарбонил)-Nε-(фторенил-9-метоксикарбонил)-L-лизина. Тонкослойная хроматография: ацетонитрил/вода 20:1 Rf=0,125.
VI) 20(S)-20-O-{L-аргинил-L/D-леуцил}-камптотецин, три-трифторацетат:
VI) L 20(S)-20-O-{L-аргинил-L-леуцил}-камптотецин, три-трифторацетат:
VI) D 20(S)-20-O-{L-аргинил-D-леуцил}-камптотецин, три-трифторацетат:
VI. a) 20(S)-20-O-(три-трет-бутоксикарбонил-L-аргинил-L/D-леуцил}-камптотецин
VI. a) L 20(S)-20-O-(три-трет-бутоксикарбонил-L-аргинил-L-леуцил}-камптотецин
VI. a) D 20(S)-20-O-(три-трет-бутоксикарбонил-L-аргинил-D-леуцил}-камптотецин
200 мг (0,42 ммоль) три-трет-бутоксикарбонил-L-аргинина и 80 мг гидрата 1-гидрокси-1Н-бензотриазола растворяют в 20 мл диметилформамида. Добавляют 97 мг гидрохлорида N-этил-N'-(диметиламинопропил)-карбодиимида и перемешивают в течение 1 часа при комнатной температуре. Затем добавляют соединение II. b) (200 мг, 0,35 ммоль) и 217 мкл этилдиизопропиламина и смесь перемешивают в течение 3 часов при комнатной температуре. После концентрации и обработки водой получают целевое соединение (тонкослойная хроматография: ацетонитрил/вода 5:1:0,2 Rf=0,77), которое немедленно подвергают дальнейшему превращению.
VI) 20(S)-20-O-{L-аргинил-L/D)-леуцил}-камптотецин, три-трифторацетат:
VI) L 20(S)-20-O-{L-аргинил-L-леуцил}-камптотецин, три-трифторацетат:
VI) D 20(S)-20-O-{L-аргинил-D-леуцил}-камптотецин, три-трифторацетат:
0,35 ммоль соединения VI. а) перемешивают в 10 мл дихлорметана в 5 мл безводной трифторуксусной кислоты в течение 2 часов при комнатной температуре. Затем смесь концентрируют и дважды осаждают диэтиловым эфиром из дихлорметана/метанола. Получают 280 мг (82%) целевого соединения (тонкослойная хроматография: ацетонитрил/вода 10:3:1,5 Rf=0,42).
VII) L 20(S)-20-O-{Nε-[фторенил-9-метоксикарбонил)-L-лизил-L-валил}-камптотецин, трифторацетат:
VIIa) L 20(S)-20-O-[Nα-(тpeт-бyтoкcикapбoнил)-Nε-/ (фтopeнил-9-мeтоксикарбонил)-L-лизил-L-валил]-камптотецин:
10,02 г (21,4 ммоль) N-(трет-бутоксикарбонил)-N-(фторенил-9-метоксикарбонил)-L-лизина и 4,4 г (32,1 ммоль) гидрата 1-гидрокси-1Н-бензотриазола растворяют в 400 мл диметилформамида. Добавляют 4,92 г (1,2 экв) гидрохлорида N-этил-N'-(диметиламинопропил)-карбодиимида и перемешивают в течение 30 минут при температуре 0oС. Затем соединяют с 10 г (17,8 ммоль) неполярного диастереомера III. b) L соединения III.b) и 9,2 мл (3 экв) этилдиизопропиламина и смесь перемешивают в течение 16 часов при комнатной температуре. Концентрируют в вакууме, остаток перемешивают с 500 мл воды в течение 30 минут и отфильтровывают. Продукт помещают в 400 мл дихлорметана, остаточную воду удаляют, раствор концентрируют до 200 мл и осаждают 800 мл диэтилового эфира. Отфильтровывают и остаток промывают диэтиловым эфиром. Получают 14,712 г целевого соединения (92%), тонкослойная хроматография: ацетонитрил Rf=0,6.
VII) L 20(S)-20-O-{ N-[фторенил-9-метоксикарбонил] -L-лизил-L-валил}-камптотецин, трифторацетат:
Соединение VII.a) L (14,65 г, 16,3 ммоль) помещают в 300 мл дихлорметана и соединяют с 50 мл трифторуксусной кислоты при температуре 0oС. Полученный раствор перемешивают в течение 2 часов охлажденным. Концентрируют в вакууме, полученный продукт осаждают диэтиловым эфиром. Получают 13,8 г (93%) целевого соединения, тонкослойная хроматография: ацетонит-рил/вода 10:1 Rf=0,35.
Пример 1
20(S)-20-O-{Nα-[О-(3-O-метил-β-L-фукопиранозил)-4-гидрокси-фениламинокарбонил]-L-лизил-L/D)-леуцил}-камптотецин, гидрохлорид: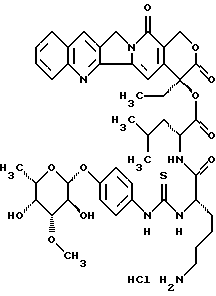
1. a) 20(S)-20-O-{Nα-[O-(3-O-метил -β-L-фукопиранозил)-4-гидроксифениламинотиокарбонил] -Nε-[фторенил-9-метоксикарбонил] -L-лизил-L/D-леуцил} -камптотецин:
Раствор из 9,82 г (36,5 ммоль) п-аминофенил-3-О-метил -β-L-фукопиранозид (пример 1) в 500 мл диоксан/вода 1:1 перемешивают с 3,94 мл тиофосгена (1,4 экв). Через 10 минут добавляют 4 экв этилдиизопропиламина, концентрируют в вакууме и остаток сушат в вакууме масляного пресса в течение 1 часа. Полученный изотиоцианат растворяют в 500 мл абсолютного диметилформамида и добавляют 30,4 г (32,8 ммоль) соединения II, а также 22,6 мл этилдиизопропиламина. Смесь перемешивают в течение 16 часов при комнатной температуре, затем концентрируют в вакууме и перемешивают с водой. Остаток очищают флэш-хроматографией (ацетонитрил>ацетонитрил/вода 30:1). Этот продукт осаждают диэтиловым эфиром из дихлорметана/метанола и промывают диэтиловым эфиром. Получают 28,7 г (78%) целевого соединения (тонкослойная хроматография: ацетонитрил/вода 10:1 Rf=0,44).
l. b) 20(S)-20-O-{Nα-[O-(3-O-метил -β-L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-L-лизил-L/D)-леуцил}-камптотецин:
28,6 г (25,5 ммоль) конъюгата I.a) помещают в 200 мл диметилформамида с 5 мл пиперидина, перемешивают в течение 2 часов при комнатной температуре, смесь концентрируют и остаток дважды дигерируют дихлорметаном, затем добавляют диэтиловый эфир. Смесь помещают в диметилформамид/дихлорметан и осаждают эфиром, этот процесс повторяют дважды. Продукт фильтруют и промывают эфиром. Выход: 19,5 г (85%), тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:1:0,2 Rf=0,3.
1) 20(S)-20-O-{ -{Nα--[O-(3-O-метил -β-L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-L-лизил-L/D)-леуцил}-камптотецин, гидрохлорид:
10 г (11,1 ммоль) соединения из примера l.b) помещают в 100 мл воды, добавляют 11,1 мл (1 экв) раствора lN HCl и лиофилизируют. Затем продукт несколько раз осаждают диэтиловым эфиром из дихлорметана/метанола. Выход: 9,15 г (88%), тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:1:0,2 Rf =0,3.
Пример 2
20(S)-20-O-{N-[O-(3-O-метил -β-L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-L-гистидил-L/D-валил}-камптотецин, гидрохлорид: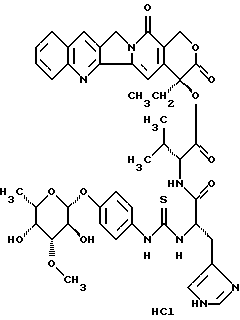
Раствор из 7,14 г (26,5 ммоль) п-аминофенил-3-О-метил- -β-L-фукопиранозида (углеводный аддукт I) в 300 мл диоксан/вода 1:1 перемешивают с 2,86 мл тиофосгена (1,4 экв). Через 10 минут добавляют 4 экв этилдиизопропиламина, смесь концентрируют в вакууме и остаток сушат в вакууме масляного пресса в течение 1 часа. Полученный изотиоцианат растворяют в 500 мл абсолютного диметилформамида и соединяют с 17,45 г (22 ммоль) соединения III, а также 22,7 мл этилдиизопропиламина. Смесь перемешивают в течение 16 часов при комнатной температуре, концентрируют в вакууме и остаток растворяют в воде. Смесь доводят до уровня рН 7,8 с 1N водного раствора едкого натра и фильтруют. Остаток сушат в высоком вакууме, дважды осаждают диэтиловым эфиром из дихлорметана/метанола и промывают диэтиловым эфиром. Получают 17,97 г (91%) целевого соединения, которое затем превращают с 1 экв О, 1N водного раствора соляной кислоты в гидрохлорид (тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:1:0,2 Rf=0,3), (FAB-MS: m/z=896(M+H+)).
Пример 3
20(S)-20-O/-{Nα-[O-(3-O-метил -β-L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-D-лизил-D-леуцил}-камптотецин, гидрохлорид: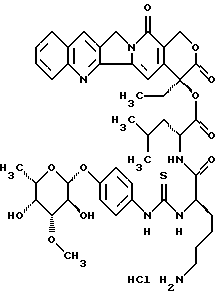
Синтез проводят аналогично примеру 1, при этом получают аддукты 1 и IV. )D, причем со стадии получения 20-O-леуцил-камптотецина II.b) используют полярный диастереомер II. b)D. Тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:1:0,2 Rf=0,31, FAB-MS: m/z=901=M+H+.
Пример 4
20(S)-20-O -{Nα-[O-(3-O-метил -β-L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-L-орнитил-D-леуцил}-камптотецин, гидрохлорид: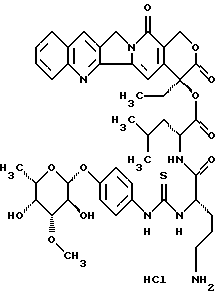
Синтез проводят полностью аналогично примеру 1, при этом получают аддукты I и V), причем со стадии получения 20-O-леуцил-камптотецина II.b) используют полярный диастереомер II. b)D с D конфигурацией леуцина. Тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:l:0,2 Rf= 0,25.
Пример 5
20(S)-20-O -{Nα-[O-(3-О-метил/-β-L-фукопиранозил)-4-гидрокси-фениламино-карбонил]-L-аргинил-D-леуцил}-камптотецин: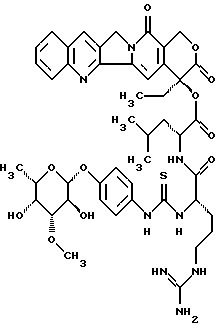
Раствор из 73 мг (0,27 ммоль) р-аминофенил-3-О-метил -β-L-фукопиранозида (аддукт I) в 20 мл диоксан/вода 1:1 перемешивают с 30 мкл тиофосгена (1,4 экв). Через 10 минут добавляют 4 экв этилдиизопропиламина, концентрируют в вакууме и остаток сушат в вакууме масляного пресса в течение 1 часа. Полученный изотиоцианат растворяют в 20 мл абсолютного диметилформамида и перемешивают с 175 мг (0,18 ммоль) соединения VI)D, а также 620 мкл этилдиизопропиламина. Для синтеза соединения VI)D со стадии получения 20-О-леуцил-камптотецина II.b) используют полярный диастереомер II.b)D. Смесь перемешивают в течение 16 часов при комнатной температуре, концентрируют в вакууме и перемешивают с дихлорметаном. Остаток осаждают диэтиловым эфиром из дихлорметана/метанола. Затем лиофилизируют из диоксана/воды и вновь кристаллизуют диэтиловым эфиром из дихлорметана/метанола. Получают 154 мг (90%) целевого соединения, тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:1:0,2 Rf=0,39, FAB-MS: m/z=929=M+H+.
Пример 6
20(S)-20-O -{Nα-[O-(3-O-мeтил -β-L-фyкoпиpaнoзил)-4-гидpoкcи-фeнилaминoтиокарбонил]-L-лизил-D-леуцил}-камптотецин, гидрохлорид: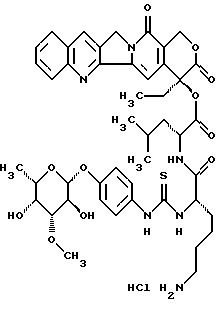
Соединение получают аналогично примеру 1, причем на стадии получения 20-O-леуцил-камптотецина II. b) используют полярный диастереомер II.b)D с D конфигурацией леуцина.
Возможно альтернативное разделение соединения из примера 1 на отдельные изомерные формы, например, с помощью препаративной ЖХВР.
Условия:
разделительная колонка: Macherey & Nagel 250х21 мм нуклеозил 100-7 С18 АВ,
растворитель А: Н2O+0,1 М КН2РO4.
Растворитель В: ацетонитрил/вода 4:1+0,02 M KH2PO4,
поток: 10 мл/мин,
инъекционный объем: 1500 мкл,
градиент: 0-30% В - 4-30; 20-90; 22-90; 24-30,
длина волны: 215 нм.
После разделения препаративной ЖХВР соответствующие фракции лиофилизируют и остаток несколько раз осаждают диэтиловым эфиром из дихлорметана/метанола. Затем регулируют уровень рН 8-9, смесь фильтруют и остаток превращают с 0,1 N соляной кислоты в гидрохлорид.
Изомеры в примерах 6 и 7 в спектре 1H-NMR показывают различные химические перемещения, в частности, для обоих синглетов ароматических атомов Н в B или D кольце камптотецина.
Диастереомер с леуцином D:
400-MHz-1H-NMR(CD2Cl2/CD3OD 1:1): δ
sС-Н (кольцо В) 8,52 ппм,
sС-Н (кольцо D) 7,42 ппм.
FAB-MS: m/z=901=M+H+.
Пример 7
20(S)-20-O-{Nα-[O-(3-O-метил L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-L-лизил-L-леуцил}-камптотецин, гидрохлорид:
L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-L-лизил-L-леуцил}-камптотецин, гидрохлорид: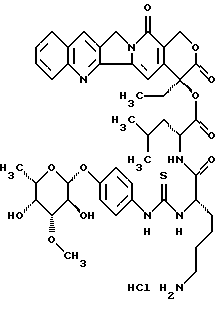
Соединение получают аналогично примеру 1, причем на стадии получения 20-O-леуцил-камптотецина II.b) используют неполярный диастереомер II.b)L с L конфигурацией леуцина.
Возможно альтернативное разделение соединения из примера 1 на отдельные изомерные формы, например, с помощью препаративной ЖХВР.
Условия: аналогичны примеру 6.
После разделения препаративной ЖХВР соответствующие фракции лиофилизируют и остаток несколько раз осаждают диэтиловым эфиром из дихлорметана/метанола. Затем регулируют уровень рН 8-9, смесь фильтруют и остаток превращают с 0,1 N соляной кислоты в гидрохлорид.
Изомеры в примерах 6 и 7 в спектре 1H-NMR показывают различные химические перемещения, в частности, для обоих синглетов ароматических атомов Н в B или в D кольце камптотецина.
Диастереомер с леуцином L:
400-MHz-1H-NMR(CD2Cl2/CD3OD 1:1): δ
sС-Н (кольцо В) 8,6 ппм,
sС-Н (кольцо D) 7,35ппм.
FAB-MS:m/z=901=M+H+.
Пример 8
20(S)-20-O-{Nα-[O-(3-O-мeтил-β-L-фyкoпиpaнoзил)-4-гидpoкcи-фeнилaминo-тиокарбонил]-L-лизил-L-валил}-камптотецин, гидрохлорид: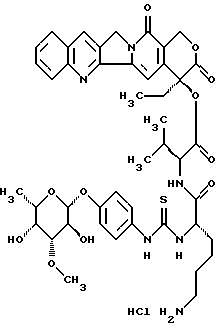
Соединение получают аналогично примеру 1, используя неполярное соединение VII)L.
Отношение диастереомеров можно определить путем аналитической ЖХВР. В данном случае, кристаллизацией из метанола возможно дальнейшее очищение диастереомера с L конфигурацией валина (>20:1).
Условия ЖХВР:
разделительная колонка: Macherey&Nagel 250х4 мм нуклеозил 100-7 С 18 АВ,
растворитель А: Н2О+0,1 М КН2РO4
растворитель В: ацетонитрил/вода 4:1+0,02 М КН2РO4,
поток: 1 мл/мин,
инъекционный объем: 15 мкл.
Чистый диастереомер в спектре 1H-NMR показывает только сигнальный осадок, в частности, для обоих синглетов ароматических атомов водорода в B или в D кольце камптотецина.
400-MHz-1H-NMR(CD2Cl2/CD3OD 1:1): δ
sС-Н (кольцо В) 8,55 ппм,
sС-Н (кольцо D) 7,35 ппм,
FAB-MS: m/z=887=M+H+.
Также возможен альтернативный предшествующий способ при синтезе соединения из примера 8. При этом углеводный элемент из примера 1 вначале связывают с находящимся в боковых цепях защищенным производным лизина.
8а) Nα-(O-(3-O-метил-β-L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-Nε-[фторенил-9-метоксикарбонил]-L-лизин
Раствор из 6,8 г (25,3 ммоль) п-аминофенил-3-О-метил-β-L-фукопиранозида (пример I) в 600 мл диоксан/вода 1:1 перемешивают с 2,72 мл тиофосгена (1,4 экв). Через 10 минут добавляют 26 мл этилдиизопропиламина, перемешивают в течение 5 минут при комнатной температуре и концентрируют в вакууме до объема 260 мл. Добавляют 800 мл дихлорметана и разделяют фазы. Органическую фазу дважды промывают водой, сушат над сульфатом натрия и концентрируют. Остаток перемешивают с 200 мл метил-трет-бутилового эфира и 200 мл петролейного эфира и фильтруют. Получают 7,26 г (92%) изотиоцианата.
2,92 г (9,4 ммоль) полученного изотиоцианата растворяют в 200 мл диоксан/вода 1:1 и добавляют 3,11 г (0,9 экв) Nε-[фтopeнил-9-мeтoкcикapбoнил]-лизина, а также 3,2 мл этилдиизопропиламина. Смесь перемешивают в течение 16 часов при комнатной температуре, концентрируют в вакууме и остаток растворяют в воде. рН регулируют с 1 N НСl до 2, продукт осаждают и через тридцать минут фильтруют. Остаток суспендируют в дихлорметане и растворитель дважды удаляют. После многократного промывания простым и петролейным эфиром получают 5,3 г (92%) целевого продукта (тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:1:0,2 Rf=0,69).
8b) 20(S)-20-O-{Nα-[O-(3-O-метил-β-L-фукопиранозил)-4-гидрокси- фениламино-тиокарбонил] -Nε-/ фторенил-9-метоксикарбонил] -L-лизил-L-валил} -камптотецин
1,2 г (1,76 ммоль) Nα-[О-(3-O-метил-β-L-фукопиранозил)-4-гидроксифениламино-тиокарбонил]-Nε-[фторенил-9-метоксикарбонил]-L-лизина (пример 8а) и 675 мг (1,2 ммоль) соединения III.b)L растворяют в 50 мл диметилформамида, смесь охлаждают до 0oС и затем соединяют с 346 мг (1,8 ммоль) гидрохлорида N-этил-N'-(диметиламинопропил)-карбодиимида. Смесь перемешивают в течение ночи, повышая температуру до комнатной. Растворитель удаляют в вакууме и остаток перемешивают с водой. Смесь очищают флэш-хроматографией над силикагелем, причем вначале в качестве растворителя используют ацетонитрил, а затем ацетонитрил/вода 50: 1. Затем соответствующие фракции концентрируют и получают 1,06 г (76%) целевого продукта (тонкослойная хроматография: ацетонитрил/вода 20:1 Rf=0,34).
Пример 9
20(S)-20-O-{Nα-[O-(3-O-мeтил-β-L-фyкoпиpaнoзил)-4-гидpoкcи-фeнилaминo-тиокарбонил]-L-гистидил-L-валил}-камптотецин, гидрохлорид: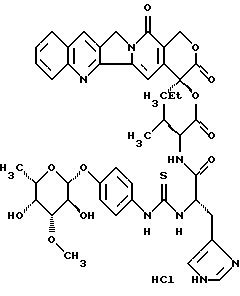
Соединение получают аналогично примеру 2 из неполярного диастереомера 20-O-валил-камптотецина, трифторацетата III.b)L.
Отношение диастереомеров можно определить путем аналитической ЖХВР. В данном случае, кристаллизацией из метанола возможно дальнейшее очищение диастереомера валина L конфигурации (>20:1).
Условия ЖХВР:
разделительная колонка: Macherey & Nagel 250х4 мм нуклеозил 100-7 С 18 АВ,
растворитель А: Н2O+0,1 М КН2РO4,
растворитель В: ацетонитрил/вода 4:1+0,02 М КН2РO4,
поток: 1 мл/мин,
инъекционный объем: 15 мкл.
Чистый диастереомер в спектре 1H-NMR показывает только сигнальный осадок, в частности, для обоих синглетов ароматических атомов H в В или в D кольце камптотецина.
400-MHz-1H-NMR(CD2Cl2/CD3OD 1:1): δ
sС-Н (кольцо В) 8,58 ppm (наложение ароматического СН гистидина),
sС-Н (кольцо D) 7,35 ppm.
Тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5: 1:0,2, Rf=0,36, FAB-MS: m/z=896=M+H+.
Пример 10
20(S)-20-O-{Nα-[O-(3-O-метил-β-L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил]-L-аргинил-L-леуцил}-камптотецин,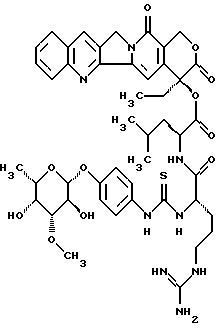
Продукт получают аналогично примеру 5. Причем на стадии получения 20-О-леуцил-камптотецина II. b) используют неполярный диастереомер II.b)L. Тонкослойная хроматография: ацетонитрил/вода/ледяная уксусная кислота 5:1: 0,2, Rf= 0,4, FAB-MS: m/z=929=M+H+. С помощью приведенной в примерах 8 и 9 препаративной ЖХВР можно разделить диастереомеры из примеров 5 и 10.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАЩИЩЕННЫЕ ПРОИЗВОДНЫЕ ВАЗОПРЕССИНА | 1997 |
|
RU2123498C1 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2096415C1 |
| ЛИПОТРИПЕПТИДЫ НА ОСНОВЕ ДИЭФИРОВ L-ГЛУТАМИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2575851C1 |
| ЛИПОТЕТРАПЕПТИДЫ НА ОСНОВЕ ДИЭФИРОВ L-ГЛУТАМИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2533554C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛОВОГО ЭФИРА О-ТРЕТ-БУТИЛ-N-(N-ТРЕТ-БУТОКСИКАРБОНИЛ-L-ЛИЗИЛ)-L-ТРЕОНИНА | 2010 |
|
RU2436794C1 |
| АМИНОАЦИЛЬНЫЕ ПРОИЗВОДНЫЕ ПРОЛЕКАРСТВА И ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ТРОМБОЭМБОЛИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2435768C2 |
| АГЛЮКОДАЛЬБАГЕПТИДЫ И/ИЛИ ИХ СОЛИ С КИСЛОТАМИ ИЛИ ОСНОВАНИЯМИ, ИЛИ ИХ ВНУТРЕННИЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2126419C1 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОМПЛЕКСЫ С ОСТАТКАМИ САХАРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2280644C2 |
| СРЕДСТВО ДЛЯ ИНАКТИВАЦИИ ВИРУСОВ, ОБЛАДАЮЩЕЕ ОДНОВРЕМЕННОЙ РИБОНУКЛЕАЗНОЙ, МЕМБРАНОЛИТИЧЕСКОЙ И ПРОТИВОВИРУСНОЙ АКТИВНОСТЯМИ | 2008 |
|
RU2399388C2 |
| АМИНОАЦИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ПРОЛЕКАРСТВ И ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ТРОМБОЭМБОЛИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2456283C9 |


Изобретение относится к группе новых соединений камтотецингликоконъюгатам общей формулы I, где R1 - полностью стереометрическая неполярная боковая цепь аминокислоты, представленная алкильным радикалом, который имеет до 4 атомов углерода; R2 - основная боковая цепь аминокислоты, являющейся радикалом формулы -(CH2)n-R3, причем R3 = NH2,
и n равно 1-4, а также их соли, стереоизомеры и смеси стереоизомеров; двум способам получения этих соединений, а также лекарственному средству, обладающему способностью ингибировать рост объема опухолей или замедлять их рост. 4 с. и 3 з.п. ф-лы, 3 табл., 1 ил.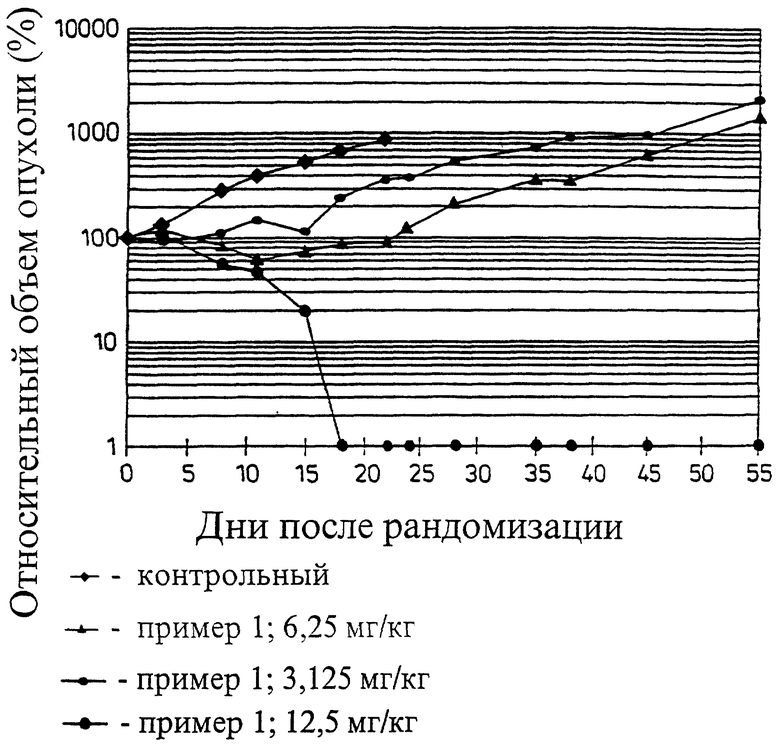
в которой R1 является полностью стереометрической неполярной боковой цепью аминокислоты, представленной алкильным радикалом, который имеет до 4 атомов углерода;
R2 является основной боковой цепью аминокислоты, являющейся радикалом формулы -(СН2)n-R3, причем R3 является -NН2,
n равно 1-4,
а также их соли, стереоизомеры и смеси стереоизомеров.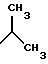
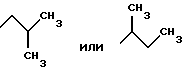
R2 является радикалом формулы -(CH2)2-NH2, -(CH2)3-NH2, -(CH2)4-NH2, 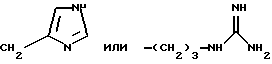
и их соли, стереоизомеры и смеси стереоизомеров. L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил] -L-лизил-L-валил} -камптотецином формулы
L-фукопиранозил)-4-гидрокси-фениламино-тиокарбонил] -L-лизил-L-валил} -камптотецином формулы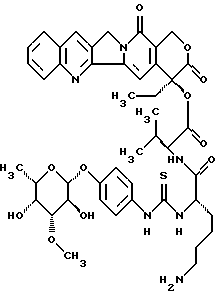
и его солями.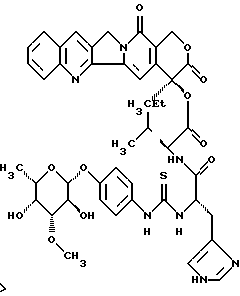
и его солями.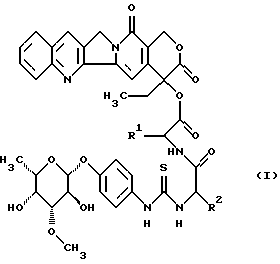
в которой R1 является полностью стереометрической неполярной боковой цепью аминокислоты, представленной алкильным радикалом, который имеет до 4 атомов углерода;
R2' является основной боковой цепью аминокислоты, представленной радикалом формулы -(CH2)n-R3, причем R3 является -NH2,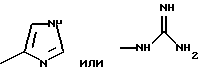
n равно 1-4,
или их солей, отличающийся тем, что изотиоцианат формулы (II)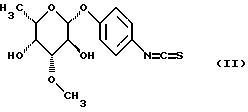
взаимодействует с имеющим в боковой цепи защитную группу пептидилкамптотецином формулы (III)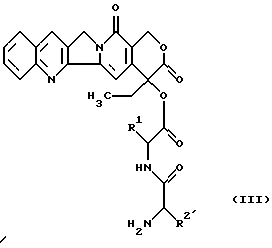
в которой R1 имеет вышеуказанное значение;
R2 имеет значение вышеуказанного основного радикала R2, который кроме этого может находиться в основной группе обычной в химии пептидов защитной группы,
до получения гликоконъюгата формулы (IV)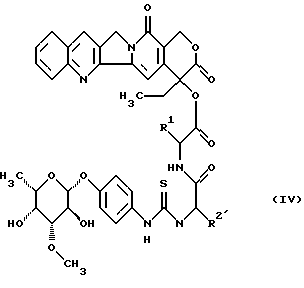
в которой R1 и R2' имеют вышеуказанные значения,
при необходимости находящуюся в боковых цепях аминозащитную группу отщепляют по обычным методикам и полученное соединение превращают в предпочтительную соль.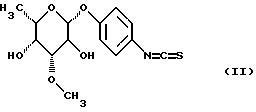
взаимодействует с конечной основной аминокислотой формулы (V), которая может иметь подходящую защитную группу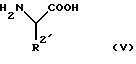
в которой R2' является основной боковой цепью аминокислоты, основные группы которой могут быть защищены,
до получения конъюгата аминокислоты формулы (VI)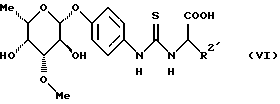
в которой R2' имеет вышеуказанное значение,
и затем этот конъюгат превращают с конъюгатами аминокислоты формулы (VII)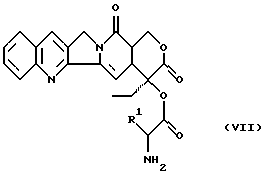
в которой R1 имеет вышеуказанное в п. 1 значение,
при необходимости группу боковых цепей отщепляют и при необходимости соединение превращают в подходящую соль.
Приоритет по пунктам:
14.05.1997 - по пп. 1-5 и 7;
14.01.1998 - по п. 6.
| SU 727647 А, 18.04.1980 | |||
| RU 95112573 А1, 20.02.1997 | |||
| Центробежная форсунка | 1972 |
|
SU501250A1 |
| ЕР 0757049 А, 05.02.1997 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |

