Область техники
Настоящее изобретение применимо в области медицины. Точнее говоря, производные дигидропиразолопиримидинона по изобретению применимы в области лечения различных злокачественных опухолей в качестве ингибитора киназы, особенно в качестве ингибитора Weel-киназы.
Уровень техники
Клетки обладают контролирующим механизмом, заключающимся в том, что в случае, когда повреждена их ДНК, клетки временно останавливают клеточный цикл и устраняют повреждение ДНК (Cell Proliferation, Vol. 33, pp. 261-274). Примерно в половине случаев злокачественных опухолей человека ген-супрессор злокачественной опухоли p53 подвергся мутации или утрачен, и клетки вследствие этого потеряли их G1 контролирующую функцию. Несмотря на это, такие клетки злокачественной опухоли все еще сохраняют их оставшуюся G2 контролирующую функцию, которая рассматривается в качестве единственного фактора понижения чувствительности клеток к противоопухолевым агентам, активным в отношении ДНК, и к облучениям.
Weel-киназа представляет собой тирозиновую киназу, которая участвует в G2 контроле клеточного цикла. Weel фосфорилирует Cdc2(Cdk1) тирозин 15, который участвует в продвижении к M стадии от G2 стадии в клеточном цикле, вследствие этого инактивируя Cdc2 и временно останавливая клеточный цикл на G2 стадии (The EMBO Journal, Vol. 12, pp. 75-85). Таким образом, для клеток злокачественной опухоли, утративших p53 функцию как таковую, полагают, что G2 контролирующая функция, осуществляемая Weel, важна при устранении поврежденной ДНК, для того чтобы избежать гибели клетки. До настоящего времени сообщалось, что снижение экспрессии Weel посредством воздействия на РНК или ингибирование Weel соединениями может повысить чувствительность клеток злокачественной опухоли к адриамицину, рентгеновскому облучению и гамма-облучению (Cancer Biology & Therapy, Vol. 3, pp. 305-313; Cancer Research, Vol. 61, pp. 8211-8217). Исходя из вышесказанного, полагают, что ингибитор Weel может ингибировать G2 контролирующую функцию утративших p53 клеток злокачественной опухоли, вследствие этого увеличивая чувствительность клеток к противоопухолевым агентам, активным в отношении ДНК, и к облучениям.
В качестве низкомолекулярного ингибитора Weel-киназы известны, например, соединения, описанные в заявке США 2005/0250836, WO 2003/091255, Cancer Research, Vol. 61, pp. 8211-8217, или в Bioorg. & Med. Chem. Lett., Vol. 15, pp. 1931-1935. Однако соединения, описанные в этих ссылках, достаточно сильно отличаются от соединений по изобретению с точки зрения их структур.
С другой стороны, WO 2004/056786, WO 2005/021532 или WO 2006/091737 раскрывают различные соединения, такие как дигидропиразолопиридины, которые довольно похожи на соединения по изобретению с точки зрения основных конструкций их структуры. Однако эти ссылки также не раскрывают конкретно и не предлагают какое-либо ингибирующее воздействие на Weel-киназу этих соединений, а также и не раскрывают соединений по изобретению.
Сущность изобретения
Целью изобретения является получение нового агента против злокачественной опухоли, обладающего ингибирующим воздействием на киназу, в частности ингибирующим воздействием на Weel-киназу.
В качестве результата кропотливых исследований авторы настоящего изобретения обнаружили, что соединения общей формулы (I) обладают превосходным ингибирующим воздействием на киназу, в особенности превосходным ингибирующим воздействием на Weel-киназу, и выполнили настоящее изобретение:
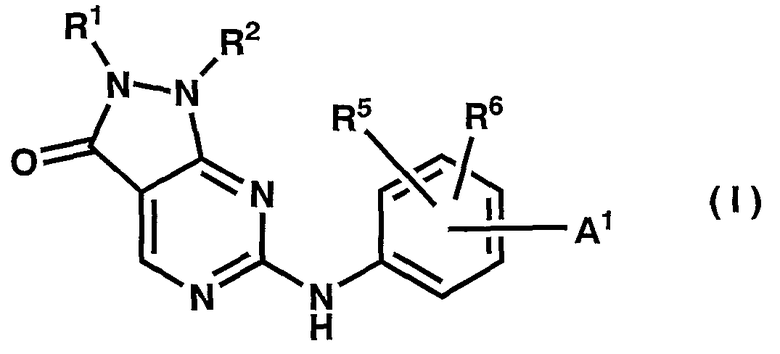
где
A1 выбирают из следующей формулы (aa1):

R1c представляет собой атом водорода, низшую алкенильную группу или группу -Q3-A3(R1d)R1e;
A3 представляет собой атом азота или представляет собой метин или 1-винил-2-илиденовую группу, необязательно замещенную гидроксильной группой, низшей алкильной группой или гидроксилсодержащей низшей алкильной группой;
Q3 представляет собой одинарную связь или низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, карбонильной группой, сульфинильной группой или сульфонильной группой, и/или замещены атомом галогена, цианогруппой, гидроксильной группой или низшей алкильной группой;
R1d и R1e представляют собой независимо атом водорода, атом галогена, цианогруппу, гидроксильную группу, низшую алкильную группу или гидроксилсодержащую низшую алкильную группу, или вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, сульфинильной группой, сульфонильной группой, карбонильной группой, виниленовой группой или группой -N(R1f)-, и/или замещены гидроксильной группой или низшей алкильной группой;
R1f представляет собой атом водорода, низшую алкильную группу, галогенсодержащую низшую алкильную группу, низшую алкенильную группу или низшую алканоильную группу;
R1 представляет собой низшую алкенильную группу или низшую алкинильную группу;
R2 представляет собой фенильную, пиридильную или тиенильную группу, которая может содержать группу -Q4-A4(R1g)R1h ;
A4 представляет собой атом азота или метиновую группу, необязательно замещенную атомом галогена, гидроксильной группой, низшей алкильной группой или гидрокси-низшей алкильной группой;
Q4 представляет собой одинарную связь или низшую алкиленовую группу, в которой одна или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода или карбонильной группой, и/или замещены низшей алкильной группой;
R1g и R1h представляют собой независимо атом водорода, атом галогена, цианогруппу, гидроксильную группу, низшую алкильную группу, содержащую низшую алкоксигруппу низшую алкильную группу, низшую алканоильную группу, низшую алкоксикарбонильную группу или низшую алкилсульфонильную группу, или вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, сульфинильной группой, сульфонильной группой, карбонильной группой или группой -N(R1i)-, и/или замещены атомом галогена или низшей алкильной группой;
R1i представляет собой атом водорода, низшую алкильную группу или галогенсодержащую низшую алкильную группу;
R5 и R6 представляют собой независимо атом водорода, низшую алкильную группу или гидроксилсодержащую низшую алкильную группу.
Соединения (I) по изобретению обладают ингибирующим воздействием на киназу, в особенности ингибирующим воздействием на Well-киназу, и вследствие этого применимы в качестве лекарственных средств при различных злокачественных опухолях, таких как рак головного мозга, шейноцеребральный рак, рак пищевода, рак щитовидной железы, мелкоклеточный рак, немелкоклеточный рак, рак молочной железы, рак легкого, рак желудка, рак желчного пузыря/желчного протока, рак печени, рак поджелудочной железы, рак толстой кишки, рак прямой кишки, рак яичников, хориокарцинома, рак матки, рак шейки матки, рак почечной лоханки/мочеточника, рак мочевого пузыря, рак предстательной железы, рак полового члена, рак яичка, фетальный рак, рак Вильмса, рак кожи, злокачественная меланома, нейробластома, остеосаркома, опухоль Юинга, саркома мягких тканей, острый лейкоз, хронический лимфолейкоз, хронический миелоцитарный лейкоз, лимфома Ходжкина.
В частности, соединения (I) по изобретению применимы в качестве лекарственных средств, например, при раке молочной железы, раке легкого, раке поджелудочной железы, раке толстой кишки, раке яичников, остром лейкозе, хроническом лимфолейкозе, хроническом миелоцитарном лейкозе, лимфоме Ходжкина.
Изобретение относится к соединениям формулы (I) и их солям, а также к способам их получения и их применения.
Значения терминов, использованных в настоящем описании, описаны ниже, и изобретение описано более подробно ниже.
"Атом галогена" обозначает атом фтора, атом хлора, атом брома и атом иода.
"Низшая алкильная группа" обозначает прямую или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода, включая, например, метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, пентильную группу, изопентильную группу, гексильную группу, изогексильную группу.
"Галогенсодержащая низшая алкильная группа" обозначает вышеупомянутую низшую алкильную группу, в которой какое-либо замещаемое положение замещено одним, или двумя, или несколькими, предпочтительно от 1 до 3, одинаковыми или различными, вышеупомянутыми атомами галогена, включая, например, фторметильную группу, дифторметильную группу, трифторметильную группу, 2-фторэтильную группу, 1,2-дифторэтильную группу, хлорметильную группу, 2-хлорэтильную группу, 1,2-дихлорэтильную группу, бромметильную группу, иодметильную группу.
"Гидроксилсодержащая низшая алкильная группа" обозначает вышеупомянутую низшую алкильную группу, в которой какое-либо замещаемое положение замещено одной, или двумя, или несколькими, предпочтительно 1 или 2, гидроксильными группами, включая, например, гидроксиметильную группу, 2-гидроксиэтильную группу, 1-гидрокси-1-метилэтильную группу, 1,2-дигидроксиэтильную группу, 3-гидроксипропильную группу.
"Низшая алкоксильная группа" обозначает прямую или разветвленную алкоксильную группу, имеющую от 1 до 6 атомов углерода, включая, например, метоксильную группу, этоксильную группу, пропоксильную группу, изопропоксильную группу, бутоксильную группу, втор-бутоксильную группу, изобутоксильную группу, трет-бутоксильную группу, пентилоксильную группу, изопентилоксильную группу, гексилоксильную группу, изогексилоксильную группу.
"Низшая алканоильная группа" обозначает алканоильную группу, имеющую вышеупомянутую низшую алкильную группу, или такую, которая представляет собой алканоильную группу, имеющую от 2 до 7 атомов углерода, включая, например, ацетильную группу, пропионильную группу, бутирильную группу, изобутирильную группу, валерильную группу, изовалерильную группу, пивалоильную группу.
"Низшая алкиленовая группа" обозначает прямую или разветвленную алкиленовую группу, имеющую от 1 дo 6 атомов углерода, включая, например, метиленовую группу, этиленовую группу, триметиленовую группу, тетраметиленовую группу, пентаметиленовую группу, гексаметиленовую группу.
"Низшая алкенильная группа" обозначает прямую или разветвленную алкенильную группу, имеющую от 2 до 6 атомов углерода, включая, например, винильную группу, 1-пропенильную группу, аллильную группу, изопропенильную группу, 3-бутенильную группу, 2-бутенильную группу, 1-бутенильную группу, 1-метил-2- пропенильную группу, 1-метил-1-пропенильную группу, 1-этил-1-этенильную группу, 2-метил-2-пропенильную группу, 2-метил-1-пропенильную группу, 3-метил-2-бутенильную группу, 4-пентенильную группу.
"Низшая алкинильная группа" обозначает прямую или разветвленную алкинильную группу, имеющую от 2 до 6 атомов углерода, включая, например, этинильную группу, 1-пропинильную группу, 2-пропинильную группу, 3-бутинильную группу, 2-бутинильную группу, 1-бутинильную группу, 1-метил-2-пропинильную группу, 1-этил-2-пропинильную группу, 1-метил-2-бутинильную группу, 4-пентинильную группу.
"Содержащая низшую алкоксильную низшая алкильная группа" обозначает вышеупомянутую низшую алкинильную группу, в которой какое-либо из замещаемых положений замещено одной, или двумя, или более, предпочтительно 1 или 2, одинаковыми или различными, вышеупомянутыми низшими алкоксильными группами, включая, например, метоксиметильную группу, этоксиметильную группу, 2-метоксиэтильную группу, 2-этоксиэтильную группу, 1-метокси-1-метилэтильную группу, 1,2-диметоксиэтильную группу, 3-метоксипропильную группу.
"Низшая алкоксикарбонильная группа" обозначает алкоксикарбонильную группу, имеющую вышеупомянутую низшую алкоксильную группу, или такую, которая представляет собой алкоксикарбонильную группу, имеющую от 2 до 7 атомов углерода, включая, например, метоксикарбонильную группу, этоксикарбонильную группу, пропоксикарбонильную группу, изопропоксикарбонильную группу, бутоксикарбонильную группу, изобутоксикарбонильную группу, трет-бутоксикарбонильную группу, пентилоксикарбонильную группу.
"Низшая алкилсульфонильная группа" обозначает прямую или разветвленную алкилсульфонильную группу, имеющую от 1 до 6 атомов углерода, включая, например, метилсульфонильную группу, этилсульфонильную группу, пропилсульфонильную группу, изопропилсульфонильную группу, бутилсульфонильную группу, втор-бутилсульфонильную группу, изобутилсульфонильную группу, трет-бутилсульфонильную группу, пентилсульфонильную группу, изопентилсульфонильную группу, гексилсульфонильную группу, изогексилсульфонильную группу.
"Соли" соединений по изобретению обозначают обычные фармацевтически приемлемые соли. Например, в случае, когда соединения имеют карбоксильную группу, гидроксильную группу или кислотную гетероциклическую группу, такую как тетразолильная группа, они могут образовывать основно-аддитивные соли по карбоксильной группе, гидроксильной группе или кислотной гетероциклической группе; или в случае, когда соединения имеют аминогруппу или основную гетероциклическую группу, они могут образовывать кислотно-аддитивные соли по аминогруппе или основной гетероциклической группе.
Основно-аддитивные соли включают, например, соли щелочного металла, такие как соли натрия, соли калия; соли щелочноземельного металла, такие как соли кальция, соли магния; соли аммония; и соли органического амина, такие как соли триметиламина, соли триэтиламина, соли дициклогексиламина, соли этаноламина, соли диэтаноламина, соли триэтаноламина, соли прокаина, соли N,N'-дибензилэтилендиамина.
Кислотно-аддитивные соли включают, например, соли неорганической кислоты, такие как гидрохлориды, сульфаты, нитраты, фосфаты, перхлораты; соли органической кислоты, такие как малеаты, фумараты, тартраты, цитраты, аскорбаты, трифторацетаты; и сульфонаты, такие как метансульфонаты, изетионаты, бензолсульфонаты, п-толуолсульфонаты.
"Сложные эфиры" соединений по изобретению обозначают обычные фармацевтически приемлемые сложные эфиры по карбоксильной группе, если таковая имеется, соединений. Они включают, например, сложные эфиры с низшей алкильной группой, такой как метильная группа, этильная группа, пропильная группа, изопропильная группа, бутильная группа, втор-бутильная группа, трет-бутильная группа, пентильная группа, изопентильная группа, неопентильная группа, циклопропильная группа, циклобутильная группа, циклопентильная группа; сложные эфиры с аралкильной группой, такой как бензильная группа, фенетильная группа; сложные эфиры с низшей алкенильной группой, такой как аллильная группа, 2-бутенильная группа; сложные эфиры с содержащей низшую алкоксильную группу низшей алкильной группой, такой как метоксиметильная группа, 2-метоксиэтильная группа, 2-этоксиэтильная группа; сложные эфиры с содержащей низшую алканоилоксигруппу низшей алкильной группой, такой как ацетоксиметильная группа, пивалоилоксиметильная группа, 1-пивалоилоксиэтильная группа; сложные эфиры с содержащей низшую алкоксикарбонильную группу низшей алкильной группой, такой как метоксикарбонилметильная группа, изопропоксикарбонилметильная группа; сложные эфиры с содержащей карбоксигруппу низшей алкильной группой, такой как карбоксиметильная группа; сложные эфиры с содержащей низшую алкоксикарбонилоксигруппу низшей алкильной группой, такой как 1-(этоксикарбонилокси)этильная группа, 1-(циклогексилоксикарбонилокси)этильная группа; сложные эфиры с содержащей карбамоилоксигруппу низшей алкильной группой, такой как карбамоилоксиметильная группа; сложные эфиры с фталидильной группой; сложные эфиры с (5-замещенной-2-оксо-1,3-диоксол-4-ил)метильной группой, такой как (5-метил-2-оксо-1,3-диоксол-4-ил)метильная группа.
Для того чтобы проиллюстрировать соединения по изобретению более конкретно, предпочтительные примеры с символами, использованными в формуле (I), и другие описаны ниже более подробно.
R1 представляет собой низшую алкенильную группу или низшую алкинильную группу.
"Низшая алкенильная группа" для R1 представляет собой, например, предпочтительно, аллильную группу, 2-метил-2-пропенильную группу, 3-метил-2-бутенильную группу, особенно предпочтительно, аллильную группу.
"Низшая алкинильная группа" для R1 представляет собой, например, предпочтительно, 2-пропинильную группу.
В предпочтительных примерах осуществления изобретения R1 представляют собой, например, низшую алкенильную группу, необязательно замещенную атомом галогена, более конкретно, аллильную группу, 2-метил-2-пропенильную группу, 3-метил-2-бутенильную группу; более предпочтительно, аллильную группу.
В других предпочтительных примерах осуществления изобретения R1 представляют собой, например, низшую алкинильную группу, необязательно замещенную атомом галогена, более конкретно, 2-пропинильную группу.
В особенности низшая алкенильная группа, такая как аллильная группа и др., является предпочтительной для R1.
R1 представляет собой, например, предпочтительно, аллильную группу, 2-метил-2-пропенильную группу, 3-метил-2-бутенильную группу, 2-пропинильную группу, более предпочтительно, аллильную группу.
R2 представляет собой фенильную, пиридильную или тиенильную группу, которая может иметь группу -Q4-A4(R1g)R1h.
"Фенильная, пиридильная или тиенильная группа, которая может иметь группу -Q4-A4(R1g)R1h" для R2 обозначает вышеупомянутую незамещенную фенильную, пиридильную или тиенильную группу, или вышеупомянутую фенильную, пиридильную или тиенильную группу, замещенную группой -Q4-A4(R1g)R1h. Группа может быть одинаковой или различной, в количестве одной, или двух, или несколько, предпочтительно 1 или 2 группы -Q4-A4(R1g)R1h в каком-либо замещаемом положении в ней.
В группе -Q4-A4(R1g)R1h заместитель может быть такой, что A4 представляет собой атом азота или представляет собой метиновую группу, необязательно замещенную атомом галогена, гидроксильной группой, низшей алкильной группой или гидроксилсодержащей низшей алкильной группой; Q4 представляет собой одинарную связь или низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода или карбонильной группой, и/или замещены низшей алкильной группой; R1g и R1h представляют собой независимо атом водорода, атом галогена, цианогруппу, гидроксильную группу, низшую алкильную группу, содержащую низшую алкоксигруппу низшую алкильную группу, низшую алканоильную группу, низшую алкоксикарбонильную группу или низшую алкилсульфонильную группу, или вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, сульфинильной группой, сульфонильной группой, карбонильной группой или группой -N(R1i)-, и/или замещены атомом галогена или низшей алкильной группой.
"Метиновая группа, необязательно замещенная атомом галогена, гидроксильной группой, низшей алкильной группой или гидроксилсодержащей низшей алкильной группой" для A4 обозначает незамещенную метиновую группу или метиновую группу, имеющую заместитель, выбранный из группы, состоящей из атома галогена, гидроксильной группы, низшей алкильной группы и гидроксилсодержащей низшей алкильной группы.
Одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу для Q4, могут быть независимо заменены атомом кислорода или карбонильной группой, и/или замещены низшей алкильной группой.
Низшая алкиленовая группа, которую R1g и R1h вместе образуют, представляет собой, например, предпочтительно, этиленовую группу, триметиленовую группу, тетраметиленовую группу, пентаметиленовую группу. Когда "A4", с которой они связаны, представляет собой атом азота, тогда они образуют совместно с атомом азота 1-азиридинильную группу, 1-азетидинильную группу, 1-пирролидинильную группу, пиперидиновую группу; когда "A4" представляет собой метиновую группу, они образуют совместно с метиновой группой циклопропильную группу, циклобутильную группу, циклопентильную группу, циклогексильную группу. В основном, более предпочтительными являются 1-пирролидинильная группа, пиперидиновая группа, циклобутильная группа, циклогексильная группа.
Одна, или две, или несколько метиленовых групп, образующих вышеупомянутую низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, сульфинильной группой или сульфонильной группой, карбонильной группой или группой -N(R1i)-, и/или замещены атомом галогена или низшей алкильной группой. Примеры замененных или замещенных групп представляют собой, предпочтительно, выбранные из следующей формулы (bb3'):

R1i в группе -N(R1i)- представляет собой атом водорода, низшую алкильную группу или галогенсодержащую низшую алкильную группу.
Низшая алкильная группа для R1i представляет собой, например, предпочтительно, метильную группу, этильную группу.
Галогенсодержащая низшая алкильная группа для R1i представляет собой, например, предпочтительно, фторметильную группу, дифторметильную группу.
Предпочтительные примеры осуществления изобретения для группы -Q4-A4(R1g)R1h представляют собой, например, как следующие ниже:
(i) A4 представляет собой атом азота, Q4 представляет собой одинарную связь или метиленовую группу, необязательно замещенную низшей алкильной группой, и R1g и R1h представляют собой независимо атом водорода, низшую алкильную группу, низшую алканоильную группу, низшую алкоксикарбонильную группу или низшую алкилсульфонильную группу;
(ii) A4 представляет собой атом азота или представляет собой метиновую группу, необязательно замещенную атомом галогена, гидроксильной группой, низшей алкильной группой или гидроксилсодержащей низшей алкильной группой, Q4 представляет собой карбонильную группу, и R1g и R1h представляют собой независимо атом водорода или низшую алкильную группу;
(iii) A4 представляет собой метиновую группу, необязательно замещенную атомом галогена, гидроксильной группой, низшей алкильной группой или гидроксилсодержащей низшей алкильной группой, Q4 представляет собой одинарную связь или метиленовую группу, необязательно замененную атомом кислорода, и R1g и R1h представляют собой независимо атом водорода, атом галогена, гидроксильную группу, низшую алкильную группу или низшую алкоксикарбонильную группу;
(iv) A4 представляет собой метиновую группу, необязательно замещенную атомом галогена, гидроксильной группой, низшей алкильной группой или гидроксилсодержащей низшей алкильной группой, Q4 представляет собой одинарную связь, и R1g и R1h вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода или группой -N(R1i)-; или
(v) A4 представляет собой атом азота, Q4 представляет собой одинарную связь, и R1g и R1h вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены карбонильной группой или группой -N(R1i); более предпочтительный вышеуказанный (iii).
Более конкретно, группа -Q4-A4(R1g)R1h представляет собой, например, предпочтительно, аминогруппу, диметиламинометильную группу, метилсульфониламиногруппу, N-метил-N- метилсульфониламинометильную группу, карбамоильную группу, диметилкарбамоильную группу, метильную группу, 1-фтор-1-метилэтильную группу, гидроксиметильную группу, 1-гидрокси-1-метилэтильную группу, 2-гидрокси-1,1-диметилэтильную группу, 2-гидрокси-2-метилпропильную группу, 2-гидрокси-1,1- диметилпропильную группу, метоксигруппу, 2-гидроксиэтоксигруппу, 1-гидроксициклобутильную группу, 2-оксо-1-пирролидинильную группу или 3-метил-2-оксоимидазолидин-1-ильную группу, даже более предпочтительна 1-гидрокси-1-метилэтильная группа и др.
Предпочтительные примеры осуществления изобретения для R2 представляют собой, например, фенил или пиридил, более предпочтительна пиридильная группа, имеющая группу -Q4-A4(R1g)R1h.
Конкретнее, таким образом, фенильная, пиридильная или тиенильная группа, которая может иметь группу -Q4-A4(R1g)R1h для R2, представляет собой, например, предпочтительно, фенильную группу, 3-диметиламинометилфенильную группу, 3-диметилкарбамоилфенильную группу, 3-(1-гидрокси-1-метилэтил)фенильную группу, 3-тиенильную группу, 2-пиридильную группу, 6-амино-2-пиридильную группу, 6-(N-метил-N-метилсульфониламинометил)-2-пиридильную группу, 6-метил-2-пиридильную группу, 6-(1-гидрокси-1-метилэтил)-2-пиридильную группу, 6-(2-гидрокси-1,1-диметилэтил)-2-пиридильную группу, 6-(2-гидрокси-2-метилпропил)-2-пиридильную группу, 6-(2-гидрокси-1,1-диметилпропил)-2-пиридильную группу, 6-(2-гидроксиэтокси)-2-пиридильную группу, 6-(1-гидроксициклобутил)-2-пиридильную группу, 6-(2-оксо-1-пирролидинил)-2-пиридильную группу или 6-(3-метил-2-оксоимидазолидин-1-ил)-2-пиридильную группу, более предпочтительна 6-(1-гидрокси-1-метилэтил)-2-пиридильная группа и др.
Предпочтительные примеры осуществления изобретения для R1 и R2 в формуле (I) представляют собой, например, R1 представляет собой низшую алкенильную или низшую алкинильную, более предпочтительно, низшую алкенильную группу, и R2 представляет собой фенильную или пиридильную, более предпочтительно, пиридильную группу, имеющую группу -Q4-A4(R1g)R1h.
A1 в формуле (I) выбирают из следующей формулы (aa1):
R1с в группе -N(R1c)- представляет собой атом водорода, низшую алкенильную группу или группу -Q3-A3(R1d)R1e.
Низшая алкенильная группа для R1с представляет собой, например, предпочтительно, винильную группу, аллильную группу.
В группе -Q3-A3(R1d)R1e для R1c A3 представляет собой атом азота или представляет собой метин или 1-винил-2-илиденовую группу, необязательно замещенную гидроксильной группой, низшей алкильной группой или гидроксилсодержащей низшей алкильной группой; Q3 представляет собой одинарную связь или низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, карбонильной группой, сульфинильной группой или сульфонильной группой, и/или замещены атомом галогена, цианогруппой, гидроксильной группой или низшей алкильной группой; R1d и R1e представляют собой независимо атом водорода атом галогена, цианогруппу, гидроксильную группу, низшую алкильную группу или гидроксилсодержащую низшую алкильную группу, или вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, сульфинильной группой, сульфонильной группой, карбонильной группой, виниленовой группой или группой -N(R1f)-, и/или замещены гидроксильной группой или низшей алкильной группой.
"Метиновая или 1-винил-2-илиденовая группа, необязательно замещенная гидроксильной группой, низшей алкильной группой или гидроксилсодержащей низшей алкильной группой" для A3 обозначает незамещенные метиновую или 1-винил-2-илиденовую группу, или метиновую или 1-винил-2-илиденовую группу, имеющую заместитель, выбранный из группы, состоящей из гидроксильной группы, низшей алкильной группы и гидроксилсодержащей низшей алкильной группы.
Заместитель представляет собой, предпочтительно, гидроксильную группу, низшую алкильную группу, такую как метильная группа и этильная группа.
Низшая алкиленовая группа для Q3 представляет собой, например, предпочтительно, метиленовую группу, этиленовую группу, триметиленовую группу.
Одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу для Q3, могут быть независимо заменены атомом кислорода, атомом серы, карбонильной группой, сульфинильной группой или сульфонильной группой, и/или замещены атомом галогена, цианогруппой, гидроксильной группой или низшей алкильной группой.
Атом галогена для R1d или R1e представляет собой, например, предпочтительно, атом фтора, атом хлора.
Низший алкил для R1d или R1e представляет собой, например, предпочтительно, метильную группу, этильную группу.
Гидроксилсодержащая низшая алкильная группа для R1d или R1e представляет собой, например, предпочтительно, гидроксиметильную группу, 2-гидроксиэтильную группу.
Низшая алкиленовая группа, которую R1d и R1e вместе образуют, представляет собой, например, предпочтительно, этиленовую группу, триметиленовую группу, тетраметиленовую группу. Когда "A3", с которой они связаны, представляет собой атом азота, тогда они образуют совместно с атомом азота 1-азиридинильную группу, 1-азетидинильную группу, 1-пирролидинильную группу; когда "A3" представляет собой метиновую группу, они образуют совместно с метиновой группой циклопропильную группу, циклобутильную группу, циклопентильную группу; когда "A3" представляет собой 1-винил-2-илиденовую группу, тогда они образуют совместно с 1-винил-2-илиденовой группой 1-циклобутенильную группу, 1-циклопентенильную группу, 1-циклогексенильную группу. В основном, более предпочтительными являются циклопропильная группа, циклобутильная группа, циклопентильная группа.
Одна, или две, или несколько метиленовых групп, образующих вышеупомянутую низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, сульфинильной группой или сульфонильной группой, карбонильной группой, виниленовой группой или группой -N(R1f)-, и/или замещены гидроксильной группой или низшей алкильной группой.
R1f в группе -N(R1f)- представляет собой атом водорода, низшую алкильную группу, галогенсодержащую низшую алкильную группу, низшую алкенильную группу или низшую алканоильную группу.
Предпочтительные примеры осуществления изобретения для группы -Q3-A3(R1d)R1e представляют собой, например, такие как нижеследующие:
(i) A3 представляет собой метиновую группу, необязательно замещенную гидроксильной группой или низшей алкильной группой, Q3 представляет собой одинарную связь, и R1d и R1e представляют собой независимо атом водорода или низшую алкильную группу; или
(ii) A3 представляет собой метиновую группу, необязательно замещенную гидроксильной группой или низшей алкильной группой, Q3 представляет собой низшую алкиленовую группу, в которой одна или две метиленовых группы, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, карбонильной группой или сульфонильной группой, и/или замещены гидроксильной группой, и R1d и R1e представляют собой независимо атом водорода, атом галогена, цианогруппу или низшую алкильную группу; более предпочтительно вышеуказанное (i).
Более конкретно, группа -Q3-A3(R1d)R1e представляет собой, например, предпочтительно, метильную группу, этильную группу, пропильную группу, изопропильную группу, трет-бутильную группу, гидроксиметильную группу, 1-гидрокси-1-метилэтильную группу, 2-гидроксиэтильную группу, 2-метоксиэтильную группу, 2-этоксиэтильную группу, 2-гидрокси-2-метилпропильную группу, 3-фтор-2-гидроксипропильную группу, ацетильную группу, пропионильную группу, 2-метоксиацетильную группу, трет-бутоксикарбонильную группу, метилсульфонильную группу, 2-(метилсульфонил)этильную группу; более предпочтительно, метильную группу, этильную группу, трет-бутильную группу, 2-гидроксиэтильную группу, 2-метоксиэтильную группу, ацетильтную группу; более предпочтительно, метильную группу.
R1с представляет собой, предпочтительно, атом водорода или группу -Q3-A3(R1d)R1e, более предпочтительно, группу -Q3-A3(R1d)R1e.
Предпочтительные примеры осуществления изобретения для A1 представляют собой, например, 1-пиперазинильную группу, 4-метил-1-пиперазинильную группу, 4-этил-1-пиперазинильную группу, 4-изопропил-1-пиперазинильную группу, 4-трет-бутил-1-пиперазинильную группу, 4-циклопропил-1-пиперазинильную группу, 4-циклобутил-1-пиперазинильную группу, 4-циклопропилметил-1-пиперазинильную группу, 4-(2-гидроксиэтил)-1-пиперазинильную группу, 4-(2-метоксиэтил)-1-пиперазинильную группу, 4-(2-метоксиацетил)-1-пиперазинильную группу, 4-ацетил-1-пиперазинильную группу, 4-метилсульфонил-1-пиперазинильную группу, 4-метил-3-оксо-1-пиперазинильную группу, 4-пиперидильную группу, 1-метил-4-пиперидильную группу, 1-(2-гидроксиэтил)-4-пиперидильную группу, 4-гидрокси-1-метил-4-пиперидильную группу, более предпочтительно, 4-метил-1-пиперазинильную группу, 4-этил-1-пиперазинильную группу, 4-(2-гидроксиэтил)-1-пиперазинильную группу, 4-ацетил-1-пиперазинильную группу, 1-метил-4-пиперидильную группу.
R5 и R6 представляют собой независимо атом водорода, низшую алкильную группу или гидроксилсодержащую низшую алкильную группу.
Предпочтительные примеры осуществления изобретения для R5 и R6 представляют собой, например, такие, когда они оба представляют собой атомы водорода, или когда какой-либо из них представляет собой атом водорода и другой представляет собой низшую алкильную группу, такую как метильная группа и этильная группа, или гидроксилсодержащую низшую алкильную группу, такую как гидроксиметильная группа и 2-гидроксиэтильная группа.
В соединении формулы (I) R5, R6 и A1 может быть в каком-либо замещаемом положении соседней фенильной группой.
Группа формулы
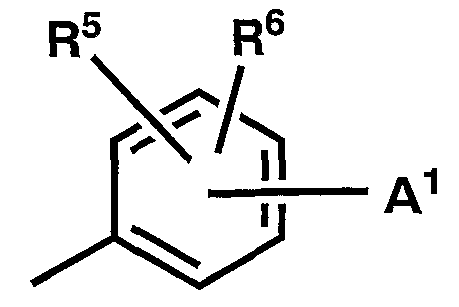
представляет собой, предпочтительно, 4-(4-метил-1-пиперазинил)фенильную группу, 3-метил-4-(4-метил-1- пиперазинил)фенильную группу, 3-гидроксиметил-4-(4-метил-1-пиперазинил)фенильную группу, 4-(4-этил-1-пиперазинил)фенильную группу, 4-(4-(2-гидроксиэтил)-1-пиперазинил)фенильную группу, 4-(4-ацетил-1-пиперазинил)фенильную группу, 4-(1-метил-4-пиперидил)фенильную группу.
Термин "какое-либо замещаемое положение" обозначает положения, имеющие замещаемые водород(ы) при атоме(ах) углерода, азота, кислорода и/или серы, где замещение водорода химически допустимо и результаты замещения представляют собой стабильное соединение.
В соединениях по изобретению замена метиленовой группы (групп), образующих низшую алкиленовую группу, различными радикалами, такими как кислород, сера, сульфинил, сульфонил, карбонил, винилен и замещенный или незамещенный имин, допустима в случае, когда замена химически допустима и результаты замены представляют собой стабильное соединение.
В зависимости от типа заместителя и от его солевой формы соединения по изобретению могут быть в форме стереоизомеров и таутомеров, таких как оптические изомеры, диастереомеры, геометрические изомеры; и соединения по изобретению включают все эти стереоизомеры и таутомеры и их смеси.
Изобретение включает различные кристаллы, аморфные формы, соли, гидраты и сольваты соединений по изобретению. Кроме того, пролекарства соединений по изобретению находятся в рамках объема изобретения.
Примеры соединений формулы (I) и их солей представляют собой, например, соединений и их соли, описанные в примерах; и более предпочтительными являются нижеследующие соединения:
3-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил)-N,N-диметилбензамид,
2-аллил-6-{[3-(гидроксиметил)-4-(4-метилпиперазин-1-ил)фенил]амино}-1-(3-тиенил)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-1-[3-(1-гидрокси-1-метилэтил)фенил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-1-[3-(диметиламинометил)фенил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-6-{[3-гидроксиметил-4-(4-метилпиперазин-1-ил)фенил]амино}-1-пиридин-2-ил-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-1-(6-аминопиридин-2-ил)-6-[{4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-6-{[4-(4-этилпиперазин-1-ил)фенил]амино}-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
6-{[4-(4-ацетилпиперазин-1-ил)фенил]амино}-2-аллил-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-6-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]фенил}амино)-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-1-[6-(2-гидрокси-2-метилпропил)пиридин-2-ил]-6-{[4-(1-метилпиперидин-4-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1-[6-(2-оксопирролидин-1-ил)пиридин-2-ил]-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он,
N-{[6-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил)пиридин-2-ил]метил}-N-метилметансульфонамид,
1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-2-(2-пропинил)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он и
2-аллил-1-[6-(3-метил-2-оксоимидазолидин-1-ил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он.
Способы получения соединений по изобретению описаны ниже.
Соединения (I) по изобретению могут быть получены, например, в соответствии со способами получения, упомянутыми ниже, или в соответствии со способами, указанными в примерах и в примерах производственного получения. Однако способы получения для соединений (I) по изобретению не должны ограничиваться этими примерами реакций.
Способ получения 1
Соединение общей формулы (II)

где L1 представляет собой уходящую группу; R1p представляет собой низшую алкенильную группу или низшую алкинильную группу; R2p представляет собой фенильную, пиридильную или тиенильную группу, которая может содержать группу -Q4p-A4p(R1gp)R1hp; Q4p представляет собой одинарную связь или низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода или необязательно защищенной карбонильной группой, и/или замещены низшей алкильной группой; R1gр и R1hp представляют собой независимо атом водорода, атом галогена, цианогруппу, необязательно защищенную гидроксильную группу, низшую алкильную группу, содержащую низшую алкоксигруппу низшую алкильную группу, низшую алканоильную группу, низшую алкоксикарбонильную группу или низшую алкилсульфонильную группу, или вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, сульфинильной группой, сульфонильной группой, необязательно защищенной карбонильной группой или группой -N(R1ip)-, и/или замещены атомом галогена или низшей алкильной группой,
реагирует с соединением общей формулы (III) или его солью
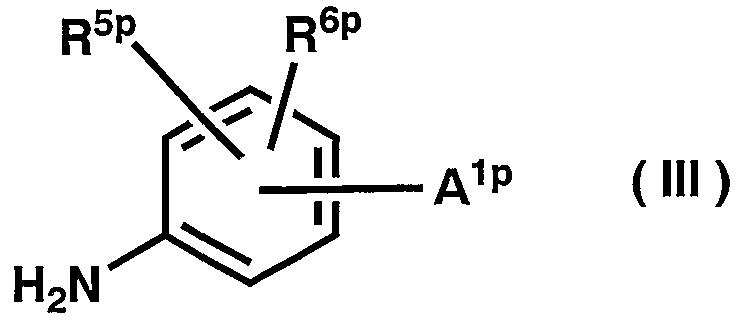
где A1p выбирают из следующей формулы (aap1):

R1сp представляет собой атом водорода, низшую алкенильную группу или группу -Q3p-A3p(R1dp)R1ep; А3p представляет собой атом азота или представляет собой метиновую группу или 1-винил-2-илиденовую группу, которая может быть замещена необязательно защищенной гидроксильной группой, низшей алкильной группой или содержащей необязательно защищенную гидроксигруппу низшей алкильной группой; Q3p представляет собой одинарную связь или низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, необязательно защищенной карбонильной группой, сульфинильной группой или сульфонильной группой, и/или замещены атомом галогена, цианогруппой, необязательно защищенной гидроксильной группой или низшей алкильной группой; R1dp и R1ep представляют собой независимо атом галогена, цианогруппу, необязательно защищенную гидроксильную группу, низшую алкильную группу или содержащую необязательно защищенную гидроксигруппу низшую алкильную группу, или вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, образующих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода, атомом серы, сульфинильной группой, сульфонильной группой, необязательно защищенной карбонильной группой, виниленовой группой или группой -N(R1fp)-, и/или замещены необязательно защищенной гидроксильной группой или низшей алкильной группой; R1fp представляет собой имино-защитную группу, атом водорода, низшую алкильную группу, галогенсодержащую низшую алкильную группу, низшую алкенильную группу или низшую алканоильную группу; R5p и R6ρ представляют собой независимо атом водорода, низшую алкильную группу или содержащую необязательно защищенную гидроксигруппу низшую алкильную группу, приводя к образованию соединения общей формулы (IV)

где A1p, R1p, R2p, R5p и R6p имеют те же значения, как указано выше, и, необязательно, защитная группа удалена из нее с получением соединения общей формулы (I)
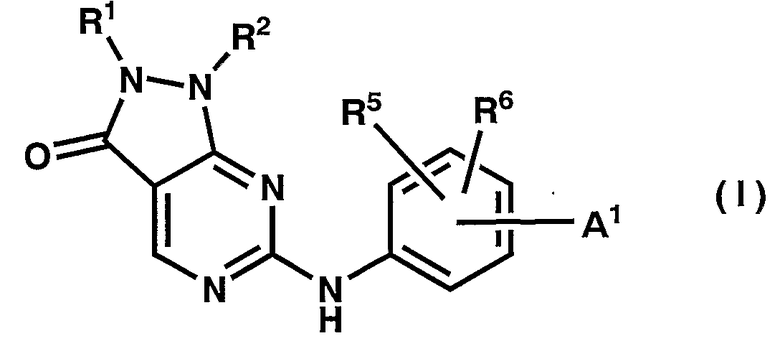
где A1, R1, R2, R5 и R6 имеют те же значения, как указано выше.
Уходящая группа L1 включает, например, атом галогена, такой как атом хлора, атом брома, атом иода; органическую сульфонильную группу, такую как метилсульфинильную группу, метилсульфонильную группу, этилсульфонильную группу, фенилсульфонильную группу; и органическую сульфонилоксигруппу, такую как метилсульфонилоксигруппу, трифторметилсульфонилоксигруппу, п-толилсульфонилоксигруппу; предпочтительно, атом хлора, метилсульфинильную группу, метилсульфонильную группу.
Способ получения представляет собой общий способ для получения соединений формулы (I).
В вышеуказанной реакции, когда реагенты имеют аминогруппу, иминогруппу, гидроксильную группу, карбоксильную группу, карбонильную группу или им подобные, которые не участвуют в реакции, тогда аминогруппа, иминогруппа, гидроксильная группа, карбоксильная группа и карбонильная группа могут быть подходящим образом защищены амино- или имино-защитной группой, гидроксил-защитной группой, карбоксил-защитной группой или карбонил-защитной группой, и после этого реагенты могут реагировать, и после реакции защитные группы могут быть удалены.
Конкретно не определенная "амино- или имино-защитная группа" может быть какой-либо группой, имеющей ее функцию. Например, она включает аралкильную группу, такую как бензильная группа, п-метоксибензильная группа, 3,4-диметоксибензильная группа, o-нитробензильная группа, п-нитробензильная группа, бензгидрильная группа, тритильная группа; низшую алканоильную группу, такую как формильная группа, ацетильная группа, пропионильная группа, бутирильная группа, пивалоильная группа; бензоильную группу; арилалканоильную группу, такую как фенилацетильная группа, феноксиацетильная группа; низшую алкоксикарбонильную группу, такую как метоксикарбонильная группа, этоксикарбонильная группа, пропилоксикарбонильная группа, трет-бутоксикарбонильная группа; аралкилоксикарбонильную группу, такую как бензилоксикарбонильная группа, п-нитробензилоксикарбонильная группа, фенетилоксикарбонильная группа; низшую алкилсилильную группу, такую как триметилсилильная группа, трет-бутилдиметилсилильная группа; тетрагидропиранильную группу; триметилсилилэтоксиметильную группу; низшую алкилсульфонильную группу, такую как метилсульфонильная группа, этилсульфонильная группа; арилсульфонильную группу, такую как бензолсульфонильная группа, толуолсульфонильная группа; и особенно предпочтительно, ацетильную группу, бензоильную группу, трет-бутоксикарбонильную группу, триметилсилилэтоксиметильную группу, метилсульфонильную группу.
Конкретно не определенная "гидроксил-защитная группа" может быть какой-либо группой, имеющей ее функцию. Например, она включает низшую алкильную группу, такую как метильная группа, этильная группа, пропильная группа, изопропильная группа, трет-бутильная группа; низшую алкилсилильную группу, такую как триметилсилильная группа, трет-бутилдиметилсилильная группа; низшую алкоксиметильную группу, такую как метоксиметильная группа, 2-метоксиэтоксиметильная группа; тетрагидропиранильную группу; триметилсилилэтоксиметильную группу; аралкильную группу, такую как бензильная группа, п-метоксибензильная группа, 2,3-диметоксибензильная группа, o-нитробензильная группа, п-нитробензильная группа, тритильная группа; ацильную группу, такую как формильная группа, ацетильная группа; и особенно предпочтительно, метильную группу, метоксиметильную группу, тетрагидропиранильную группу, тритильную группу, триметилсилилэтоксиметильную группу, трет-бутилдиметилсилильную группу, ацетильную группу.
Конкретно не определенная "карбоксил-защитная группа" может быть какой-либо группой, имеющей ее функцию. Например, она включает низшую алкильную группу, такую как метильная группа, этильная группа, пропильная группа, изопропильная группа, трет-бутильная группа; галогенсодержащую низшую алкильную группу, такую как 2,2,2-трихлорэтильная группа; низшую алкенильную группу, такую как аллильная группа; аралкильную группу, такую как бензильная группа, п-метоксибензильная группа, п-нитробензильная группа, бензгидрильная группа, тритильная группа; и особенно предпочтительно, метильную группу, этильную группу, трет-бутильную группу, аллильную группу, бензильную группу, п-метоксибензильную группу, бензгидрильную группу.
Конкретно не определенная "карбонил-защитная группа" может быть какой-либо группой, имеющей ее функцию. Например, она включает ацетали и кетали, такие как этиленкеталь, триметиленкеталь, диметиленкеталь.
Для реакции соединения формулы (II) и соединения формулы (III), как правило, эквимолярное или избыточное молярное количество, предпочтительно, от эквимолярного количества до 1,5 молей соединения (III) используется по отношению к одному молю соединения (II).
Реакция осуществляется обычно в инертном растворителе. Инертный растворитель представляет собой, например, предпочтительно, толуол, бензол, хлористый метилен, хлороформ, тетрагидрофуран, диоксан, диметилформамид, N-метилпирролидон, диметилсульфоксид и смешанные из них растворители.
Предпочтительно, реакция осуществляется в присутствии основания. Основание включает, например, органические основания, такие как триэтиламин, диизопропилэтиламин, пиридин, 4- диметиламинопиридин; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия.
Количество используемого основания может быть обычно от эквимолярного количества до избыточного молярного количества, предпочтительно, от 1 до 3 молей по отношению к одному молю соединения формулы (II).
Температура реакции может быть, как правило, от 0°C до 200°C, предпочтительно, от 20°C до 150°C.
Время реакции может быть, как правило, от 5 минут до 7 дней, предпочтительно, от 30 минут до 24 часов.
После реакции система может быть обработана обычным методом с получением неочищенного продукта соединения формулы (IV). Полученное таким образом соединение формулы (IV) очищают обычным методом, или не очищают, необязательно подвергают удалению защитных групп с аминогруппы, гидроксильной группы, карбоксильной группы и карбонильной группы, на которых они есть, необязательно, хотя удобно совмещают при этом получение соединения формулы (I).
Способ удаления защитной группы различается в зависимости от типа защитной группы и от стабильности намеченного соединения (I). Например, снятие защиты может быть достигнуто в соответствии со способами, описанными в ссылках [см. Protective Groups in Organic Synthesis, 3rd. Ed., by T. W. Greene, John Wiley & Sons (1999)], или в соответствии со способами, подобными этим. Например, в этом документе реально применим способ сольволиза с кислотой или основанием, который включает в себя обработку защищенного соединения кислотой в количестве от 0,01 молей до значительного избыточного количества, предпочтительно, трифторуксусной кислотой, муравьиной кислотой или хлористоводородной кислотой, или обработку основанием в количестве от эквимолярного до значительного избыточного количества, предпочтительно, гидроксидом калия или гидроксидом кальция; и способ химического восстановления комплексом гидрида металла или каталитического восстановления с катализатором палладий на угле или с катализатором никель Ренея.
Соединения формулы (I) могут быть легко выделены и очищены каким-либо обычным способом выделения. Примеры способа представляют собой, например, экстракцию растворителем, перекристаллизацию, колоночную хроматографию, препаративную тонкослойную хроматографию.
Соединения могут быть превращены в их фармацевтически приемлемые соли или эфиры обычным методом; и, наоборот, их соли или эфиры также могут быть превращены в свободные соединения обычным методом.
"Соли" соединения формулы (III) подразумевают обычные соли, используемые в области органической химии. Например, когда соединение имеет аминогруппу или основную гетероциклическую аминогруппу, тогда его соли представляют собой кислотно-аддитивные соли по аминогруппе или по основной гетероциклической группе.
Кислотно-аддитивные соли включают, например, соли неорганической кислоты, такие как гидрохлориды, сульфаты, нитраты, фосфаты, перхлораты, соли органической кислоты, такие как малеаты, фумараты, тартраты, цитраты, аскорбаты, трифторацетаты; сульфонаты, такие как метансульфонаты, изетионаты, бензолсульфонаты, п-толуолсульфонаты.
Соединения формул (II) и (III) могут быть коммерчески доступными или могут быть получены в соответствии со способами, описанными в ссылках [см. WO 2006/004040, WO 2003/037872; Journal of Medicinal Chemistry, Vol. 48, pp. 2371-2387; Bioorg. & Med. Chem. Lett., Vol. 14, pp. 5793-5797; Journal of the Chemical Society, Perkin Transaction II, Vol. 3, p. 843], или в соответствии со способами, подобными этим, или в соответствии со способами, описанными ниже, или в соответствии со способами, описанными в примерах и примерах производственного получения, необязательно, хотя, возможно, объединяемыми.
Способ получения A

где Et представляет собой этильную группу; L2 представляет собой уходящую группу; Me представляет собой метильную группу; R1p и R2p имеют те же значения, как указано выше.
Способ получения A представляет собой способ для получения соединения формулы (II), в котором уходящая группа для L1 представляет собой метилсульфинильную группу, или способ для получения соединения формулы (II-1).
В соответствии с этим способом получения соединение формулы (II-1) может быть получено при реакции соединения формулы (1) и гидразинового производного формулы (2) в присутствии основания, с образованием соединения формулы (3), и после этого введением группы R1p в соединение (3), с образованием соединения (5), и в конечном итоге окислением метилтиогруппы в соединении (5) до метилсульфинильной группы.
На стадии реагирования соединения формулы (1) и гидразинового производного формулы (2) в присутствии основания с образованием соединения формулы (3), как правило, от 0,5 молей до избыточного молярного количества, предпочтительно, от эквимолярного количества до 3,0 молей гидразинового производного (2) применяют по отношении к одному молю соединения (1).
Обычно реакцию осуществляют в инертном растворителе. Инертный растворитель представляет собой, например, предпочтительно, хлористый метилен, хлороформ, тетрагидрофуран, диэтиловый эфир, бензол, толуол, диметилформамид или смешанные растворители из них.
Предпочтительно, реакцию осуществляют в присутствии основания. Основание включает, например, органические основания, такие как триэтиламин, диизопропилэтиламин, пиридин, 4-диметиламинопиридин; неорганические основания, такие как гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, гидрокарбонат натрия.
Обычно количество используемого основания составляет, предпочтительно, от эквимолярного количества до избыточного молярного количества по отношению к одному молю соединения (1). Когда основание представляет собой жидкость, тогда основание может также служить в качестве растворителя.
Температура реакции может быть, как правило, от -78°C до 100°C, предпочтительно, от 20°C до 80°C.
Время реакции может быть, как правило, от 5 минут до 7 дней, предпочтительно, от 30 минут до 24 часов.
На стадии реагирования соединения (3) и соединения (4) с образованием соединения (5), обычно, от 0,5 молей до избыточного молярного количества, предпочтительно, от 2,0 молей до 5,0 молей соединения (4) применяют по отношению к одному молю соединения (3).
Уходящая группа для L2 представляет собой, предпочтительно, атом галогена, такой как атом хлора, атом брома, атом иода.
Обычно реакция может осуществляться в инертном растворителе, таком как тетрагидрофуран, бензол, толуол, ацетонитрил, диметилформамид, в присутствии основания, такого как гидрид натрия, амид натрия, алкоксид натрия, или в растворителе, таком как метанол, этанол, ацетонитрил, в присутствии основания, такого как гидроксид натрия, гидроксид калия, карбонат калия.
Обычно температура реакции составляет, предпочтительно, от 0°C до температуры кипения растворителя, используемого в реакции; и, обычно, время реакции составляет, предпочтительно, от 1 часа до 48 часов.
Для стадии окисления метилтиогруппы в соединении (5) для получения соединения (II-1), применимым является способ окисления метилтиогруппы в метилсульфинильную группу или метилсульфонильную группу, как таковой хорошо известный в области органической химии. Обычно, например, в инертном растворителе, таком как бензол, толуол, хлористый метилен, хлороформ, тетрагидрофуран, ацетонитрил или диметилформамид, от 0,5 молей до избыточного молярного количества, предпочтительно, от эквимолярного количества до 1,5 молей окисляющего агента, такого как метахлорпербензойная кислота или оксон, могут использоваться по отношению к одному молю соединения (5) для окисления.
Температура реакции составляет, обычно, предпочтительно, от 0°C до точки кипения растворителя, используемого в реакции, и, обычно, время реакции составляет, предпочтительно, от 30 минут до 8 часов.
Соединения формул (1) и (2) могут быть коммерчески доступными или могут быть получены в соответствии с известными способами, или в соответствии со способами, описанными в примерах, или в соответствии со способами, им подобными, необязательно, хотя, возможно, объединяемыми.
Способ получения B

где M представляет собой обычно встречающийся в органических соединениях атом металла; RP представляет собой атом водорода или имино-защитную группу; Et, M, Me, R1p и R2p имеют те же значения, как указано выше.
Имино-защитная группа Rр представляет собой, например, предпочтительно, бензильную группу, пара-метоксибензильную группу, трет-бутоксикарбонильную группу, бензилоксикарбонильную группу.
Способ получения B представляет собой способ для получения соединения формулы (II-1).
В соответствии с этим способом получения соединение формулы (II-1) может быть получено при реакции соединения формулы (1) и гидразинового производного формулы (8) в присутствии основания, затем гидролиза полученного в результате соединения и циклизации его с образованием соединения формулы (9), и после этого при реакции соединения (9) с металлоорганическим соединением формулы (10) в присутствии катализатора для введения, таким образом, группы R2p в него, с образованием соединения (5), и, в конечном итоге, окислением метилтиогруппы в соединении (5) до метилсульфонильной группы.
На стадии реакции соединения формулы (1) и гидразинового производного формулы (8) в присутствии основания, как правило, используемое количество гидразинового производного (8) может быть от 0,5 молей до избыточного молярного количества, предпочтительно, от эквимолярного количества до 1,5 молей по отношению к одному молю соединения (1).
Реакция обычно может осуществляться в присутствии органического основания, такого как триэтиламин, диизопропилэтиламин, пиридин, 4-диметиламинопиридин, или неорганического основания, такого как гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, гидрокарбонат натрия, в инертном растворителе, таком как хлористый метилен, хлороформ, тетрагидрофуран, диэтиловый эфир, бензол, толуол, диметилформамид или из них смешанные растворители.
Обычно количество используемого основания составляет, предпочтительно, от эквимолярного количества до избыточного молярного количества по отношению к одному молю соединения (1). Когда основание представляет собой жидкость, тогда основание может также служить в качестве растворителя.
Температура реакции может быть, как правило, от -78°C до 200°C, предпочтительно, от 20°C до 100°C.
Время реакции может быть, как правило, от 5 минут до 7 дней, предпочтительно, от 8 часов до 24 часов.
Для стадии гидролиза соединения, полученного в предшествующей реакции, применимым является способ гидролиза карбоксилатов, сам по себе известный в области органической химии. Обычно в растворителе, таком как метанол, этанол, тетрагидрофуран, диоксан, вода или в смешанном из них растворителе, соединение может быть обработано кислотой, такой как хлористоводородная кислота или серная кислота, или основанием, таким как гидроксид натрия, гидроксид калия или гидроксид кальция.
Обычно температура реакции составляет, предпочтительно, от 50°C до температуры кипения растворителя, используемого в реакции; и обычно время реакции составляет, предпочтительно, от 1 часа до 48 часов.
После гидролиза полученное в результате соединение циклизуется с получением соединения (9). Для этого реакционная жидкая среда может быть подкислена после гидролиза, при этом собственно циклизация может проходить. В случае, когда циклизация не происходит, тогда гидролизованное соединение может быть нагрето с обратным холодильником в присутствии уксусного ангидрида, или гидролизованное соединение может быть обработано тионилхлоридом для достижения предполагаемой циклизации соединения.
При циклизации с уксусным ангидридом количество используемого уксусного ангидрида составляет, предпочтительно, избыточное молярное количество, и время реакции обычно составляет, предпочтительно, от 1 часа до 48 часов.
В случае, когда гидролизованное соединение обрабатывается тионилхлоридом, количество используемого тионилхлорида составляет, предпочтительно, избыточное молярное количество, и время реакции обычно составляет, предпочтительно, от 1 часа до 48 часов.
Стадия реакции соединения (9) с металлоорганическим соединением формулы (10) в присутствии катализатора для введения, таким образом, группы R2p в него, с образованием соединения (5), может осуществляться с использованием галоидного соединения, имеющего группу R2p, вместо металлоорганического соединения формулы (10). Когда такое галоидное соединение используется, тогда в качестве катализатора предпочтительным является комплекс иодид меди(I)-диамин.
Стадия окисления метилтиогруппы в соединении (5) с получением соединения (II-1) может быть осуществлена таким же методом, как и стадия окисления метилтиогруппы в соединении (5) с получением соединения (II-1) в способе получения A.
Соединение формулы (8) может быть коммерчески доступным или может быть получено в соответствии с известными способами, или в соответствии со способами, описанными в примерах, или в соответствии со способами им подобными, необязательно, хотя, возможно, объединяемыми.
Примеры фармацевтического теста для соединений по изобретению показаны ниже.
Фармацевтический тест 1 (ингибирующее воздействие на Weel-киназу)
(1) Очистка Weel-киназы:
кДНК Weel-киназы с глутатион-S-трансферазой (GST), присоединенной по ее амино-концу, была введена в бакуловирусный экспрессионный вектор, чтобы сконструировать рекомбинантный бакуловирус, которым были инфицированы клетки насекомых клеточной линии Sf9 для достижения высокого уровня экспрессии в них. Инфицированные клетки извлекали и солюбилизировали, и затем белок GST-меченой Weel-киназы адсорбировали на глутатионовую колонку и элюировали с колонки глутатионом, и активную фракцию обессоливали на обессоливающей колонке, получая очищенный фермент.
(2) Определение активности Weel-киназы:
При определении активности Weel-киназы в качестве субстрата использовали синтетический пептид Поли(Lys,Tyr)гидробромид (Lys:Tyr (4:1)), приобретенный от фирмы Sigma.
Количество реакционной смеси составляло 21,1 мкл; и состав реакционного буфера был следующий: 50 мM Трис-HCl буфера (pH 7,4)/10 мM хлорида магния/1 мM дитиотреитола. Очищенную Weel-киназу, 2,5 мкг субстратного пептида, 10 мкM немеченого аденозинтрифосфата (ATФ) и 1 мкКюри [γ-33Р]-меченого ATФ (2500 Кюри/ммоль или более) добавляли к нему и инкубировали при 30°C в течение 30 минут. Далее 10 мкл 350 мM фосфатного буфера добавляли к реакционной смеси для остановки реакции. Субстратный пептид адсорбировали на P81 бумажный фильтр 96-луночного планшета, затем промывали несколько раз 130 мM фосфатным буфером и его радиоактивность подсчитывали с помощью жидкостного сцинтилляционного счетчика. [γ-33Р]-меченый ATФ приобретался от фирмы Amersham Bioscience.
Для добавления тестового соединения в реакционную систему соединение разбавляли диметилсульфоксидом (ДМСО) для приготовления серии разбавлений. 1,1 мкл раствора каждого разбавления добавляли к реакционной системе. В качестве контроля 1,1 мкл ДМСО добавляли к реакционной системе.
Как видно из таблицы 1, соединения по изобретению показывают превосходную Weel-ингибирующую активность.
Фармацевтический тест 2 (ингибирующее воздействие на рост опухоли)
Клетки рака человека толстой кишки WiDr (полученные от ATCC) были имплантированы подкожно на спине F344/N Jcl-rnu бестимусных крыс. Спустя восемь дней после имплантации им внутривенно вводили гемцитабин (50 мг/кг, Gemzar injection, Eli-Lilly); и спустя 24 часа тестовое соединение растворяли в растворителе (5% глюкоза) и вводили им посредством непрерывной внутривенной инъекции в течение 8 часов. Объем опухоли (0,5 × (наибольший диаметр) × (наименьший диаметр)2) определяли на 0, 3, 6, 10 и 13 дни. День 0 обозначает день, в который вводили гемцитабин. Относительный объем опухоли представляет собой относительную величину, которая рассчитана на основании того, что объем опухоли равен 1 в 0 день. Процент роста опухоли (%T/C) определяли в соответствии с следующей формулой:
Когда изменение объема опухоли, считая от 0 дня в группе, которая подвергалась введению тестового соединения, составляло более чем 0 (>0):
% T/C = [(изменение объема опухоли в случае тестовых соединений на 3, 6, 10, 13 день)/(изменение объема опухоли в контроле на 3, 6, 10, 13 день)] × 100.
Когда изменение объема опухоли считая от 0 дня в группе, которая подвергалась введению тестового соединения, составляло менее чем 0 (<0):
%T/C = [(изменение объема опухоли в случае тестовых соединений на 3, 6, 10, 13 день)/(изменение объема опухоли в случае тестовых соединений в 0 день)] × 100.
Данные по ингибирующему воздействию на рост опухоли показаны в таблице 2.
Введение гемцитабина понижало процент роста опухоли, но когда гемцитабин комбинировали с соединением по изобретению, тогда процент роста опухоли еще дополнительно снижался. А именно в группе, где химическая доза была высокой, животные показывали инволюцию опухоли.
Как отмечено выше, соединение по изобретению в комбинации с другим противораковым агентом увеличивает воздействие другого противоракового агента.
Фармацевтический тест 3 (способ определения эффективности лекарства с клетками (сенсибилизирующее воздействие рентгеновского облучения)
a) Реагенты:
Зародышевую бычью сыворотку (FBS) получали от Morgate; RPMI 1640 среда и 0,25% трипсин EDTA были от Invitrogen; набор реактивов для циклического теста плюс ДНК был от Becton Dickinson и полиамидный сетчатый фильтр был от Millipore.
b) Клетки:
Клетки немелкоклеточного рака легких человека (NCI-H1299) получали от ATCC.
c) Способ определения воздействия:
NCI-H1299 клетки суспендировали в RPMI 1640 среде с 10% FBS-добавкой и клеточную суспензию наносили на 6-луночный Nunclondelta-процессированный пластмассовый планшет от Nunc в количестве 100000 клеток/2 мл/на лунку и инкубировали в течение ночи в атмосфере 5% CO2 - 95% воздух при 37°C. Используя Softex's M-150WE, клетки подвергали рентгеновскому облучению 5000 рентген и затем дополнительно инкубировали в атмосфере 5% CO2 - 95% воздух при 37°C в течение 16 часов. Тестируемое соединение поэтапно разбавляли ДМСО и наносили в количестве 2 мл на планшет с посеянными клетками, подвергшимися рентгеновскому облучению. Инкубировали в атмосфере 5% CO2 - 95% воздух при 37°C в течение 8 часов и затем клеточную культуру частично отбирали. 0,25% Трипсин добавляли к клеткам, оставшимся в лунке, в количестве 600 мкл, и оставляли в статическом состоянии при комнатной температуре для приготовления разбавленной клеточной суспензии. Разбавленную клеточную суспензию и предварительно отобранную клеточную культуру смешивали для каждого образца, затем центрифугировали и супернатант удаляли. Отбор образцов таким образом был выполнен. Образец суспендировали в буфере (1 мл) набора реактивов для циклического теста плюс ДНК, замораживали и хранили при -80°C. Сохраняемый образец размораживали в день тестирования, центрифугировали и супернатант удаляли и осадок суспендировали в растворе А (250 мкл) для циклического теста, оставляли в статических условиях при комнатной температуре на 10 минут, и затем туда же добавляли раствор B (150 мкл) и еще оставляли в статических условиях при комнатной температуре на 10 минут. Далее раствор C (150 мкл) добавляли к анализируемому образцу, оставляли в статических условиях при 4°C на 10 минут и затем фильтровали через полиамидный сетчатый фильтр, таким образом, осуществляя окрашивание ДНК. Используя Becton Dickinson's FACS Calibur, количественно определяли количество ДНК в каждой клетке в соответствии с FACS процессом и определяли относительное число клеток с осуществленной фрагментацией ДНК.
Как показано в таблице 3, соединение по изобретению имеет превосходное индуктирующее воздействие на фрагментацию ДНК клеток, полученных из раковых клеток человека (NCI-H1299).
Как отмечено выше, соединение по изобретению в комбинации с рентгеновским облучением усиливает воздействие рентгеновского облучения.
Соединения формулы (I) могут быть введены перорально или парентерально, и после создания препаратов, подходящих для введения такими способами, соединения могут использоваться как фармацевтические композиции и противораковые агенты.
Термин "злокачественная опухоль", как на него ссылаются в настоящем описании, включает многообразные саркому и карциному и включает солидный рак и гематопоэтический рак. Солидный рак, как на него ссылаются в данном документе, включает, например, опухоль головного мозга, шейноцеребральный рак, рак пищевода, рак щитовидной железы, мелкоклеточный рак, немелкоклеточный рак, рак молочной железы, рак легкого, рак желудка, рак желчного пузыря/желчного протока, рак печени, рак поджелудочной железы, рак толстой кишки, рак прямой кишки, рак яичников, хориокарциному, рак матки, рак шейки матки, рак почечной лоханки/мочеточника, рак мочевого пузыря, рак предстательной железы, рак полового члена, рак яичка, фетальный рак, опухоль Вильямса, рак кожи, злокачественную меланому, нейробластому, остеосаркому, опухоль Юинга, саркому мягких тканей. С другой стороны, гематопоэтический рак включает, например, острый лейкоз, хронический лимфолейкоз, хронический миелоцитарный лейкоз, истинную полицетемию, злокачественную лимфому, множественную миелому, лимфому Ходжкина, неходжкинскую лимфому.
Термин "лечение злокачественной опухоли", как на него ссылаются в данном описании, обозначает, что противораковый агент вводят пациенту со злокачественной опухолью для того, чтобы ингибировать рост раковых клеток у пациента. Предпочтительно, когда лечение приводит в результате к регрессии роста злокачественной опухоли или когда лечение приводит к уменьшению размера обнаруживаемой злокачественной опухоли. Более предпочтительно, когда лечение приводит в результате к полному исчезновению злокачественной опухоли.
Соединения по изобретению, как ожидается, должны быть эффективны особенно в случае солидного рака человека. Солидный рак человека включает, например, рак головного мозга, шейноцеребральный рак, рак пищевода, рак щитовидной железы, мелкоклеточный рак, немелкоклеточный рак, рак молочной железы, рак легкого, рак желудка, рак желчного пузыря/желчного протока, рак печени, рак поджелудочной железы, рак толстой кишки, рак прямой кишки, рак яичников, хориокарциному, рак матки, рак шейки матки, рак почечной лоханки/мочеточника, рак мочевого пузыря, рак предстательной железы, рак полового члена, рак яичка, фетальный рак, опухоль Вильямса, рак кожи, злокачественную меланому, нейробластому, остеосаркому, опухоль Юинга, саркому мягких тканей, острый лейкоз, хронический лимфолейкоз, хронический миелоцитарный лейкоз, лимфому Ходжкина.
Фармацевтическая композиция и противораковый агент изобретения могут содержать фармацевтически приемлемый носитель или разбавитель. В данном документе "фармацевтически приемлемый носитель или разбавитель" относятся к вспомогательным веществам [например, жиры, пчелиный воск, полутвердые и жидкие полиолы, природные или гидрогенезированные масла и др.]; воде (например, дистиллированная вода, особым образом дистиллированная вода для инъекции и др.), физиологическому солевому раствору, спирту (например, этанол), глицерину, полиолам, водному раствору глюкозы, манниту, растительным маслам и др.); добавкам [например, разжижающий агент, дезинтегрирующий агент, связующее вещество, скользящее вещество, увлажняющий агент, стабилизатор, эмульгатор, диспергирующее вещество, консервирующее вещество, подсластитель, окрашивающее вещество, пряный агент или ароматизатор, концентрирующий агент, разбавитель, буферное вещество, растворяющий или солюбилизирующий агент, химикат для достижения сохранности, соль для модификации осмотического давления, покрывающий агент или антиоксидант] и им подобные.
В отношении каждого получения фармацевтической композиции и противоракового агента изобретения различные формы лекарственного препарата могут быть выбраны, и примеры их включают пероральные лекарственные препараты, такие как таблетки, капсулы, порошки, гранулы или жидкости; или стерилизованные жидкие парентеральные лекарственные препараты, такие как растворы или суспензии; суппозитории, мази и им подобные.
Твердые лекарственные препараты могут быть получены в формах таблетки, капсулы, гранулы и порошка без каких-либо дополнительных вспомогательных веществ, или получены с использованием подходящих наполнителей (вспомогательных веществ). Примеры таких наполнителей (вспомогательных веществ) могут включать сахариды, такие как лактоза или глюкоза; крахмал кукурузы, пшеницы или риса; жирные кислоты, такие как стеариновая кислота; неорганические соли, такие как метасиликаталюминат магния или безводный фосфат кальция; синтетические полимеры, такие как поливинилпирролидон или полиалкиленгликоль; спирты, такие как стеариловый спирт или бензиловый спирт; синтетические производные целлюлозы, такие как метилцеллюлоза, карбоксиметилцеллюлоза, этилцеллюлоза или гидроксипропилметилцеллюлоза; и другие обычно используемые вспомогательные вещества, такие как желатин, тальк, растительное масло и аравийская камедь.
Эти твердые лекарственные препараты, такие как таблетки, капсулы, гранулы и порошки, могут, как правило, содержать, например, от 0,1 до 100% масс., и предпочтительно, от 5 до 98% масс. соединения вышеприведенной формулы (I) в качестве активного ингредиента, исходя из общей массы лекарственного препарата.
Жидкие лекарственные препараты получают в формах суспензии, сиропа, инъекции и капельной внутривенной инфузии (внутривенного раствора), используя подходящие вспомогательные вещества, которые обычно используются в жидких лекарственных препаратах, такие как вода, спирт или полученное из растений масло, такое как соевое масло, арахисовое масло и кунжутное масло.
А именно когда лекарственный препарат вводится парентерально в форме внутримышечной инъекции, внутривенной инъекции или подкожной инъекции, подходящий растворитель или разбавитель может быть представлен дистиллированной водой для инъекции, водным раствором гидрохлорида лидокаина (для внутримышечной инъекции), физиологическим раствором, водным раствором глюкозы, этанолом, полиэтиленгликолем, пропиленгликолем, раствором для внутривенной инъекции (например, водный раствор лимонной кислоты, цитрата натрия или им подобных), или раствором электролита (для внутривенной капельной инфузии и внутривенной инъекции), или смешанным из них раствором.
Такая инъекция может быть в форме предварительно приготовленного раствора, или в форме порошка в чистом виде, или порошка, соединенного с подходящим наполнителем (вспомогательным веществом), который растворяют в момент использования. Инъекционный раствор может содержать, например, от 0,1 до 10% масс. активного ингредиента, исходя из общей массы лекарственного препарата.
Жидкие лекарственные препараты, такие как суспензия или сироп для перорального введения, могут содержать, например, от 0,1 до 10% масс. активного ингредиента, исходя из общей массы лекарственного препарата.
Лекарственные препараты могут быть получены средним специалистом в данной области в соответствии с обычными способами или общепринятым оборудованием. Например, получение может быть осуществлено, если лекарственный препарат представляет собой пероральный лекарственный препарат, например, смешиванием соответствующего количества соединения по изобретению с соответствующим количеством лактозы и загрузкой этой смеси в твердые желатиновые капсулы, которые удобны для перорального введения. С другой стороны, получение может быть осуществлено, если лекарственный препарат, содержащий соединение изобретения, представляет собой инъекционный раствор, например, путем смешивания соответствующего количества соединения по изобретению с соответствующим количеством 0,9% физиологического раствора и загрузкой этой смеси в ампулы для инъекционного раствора.
Соединения по изобретению могут быть использованы, необязательно, как комбинированные с каким-либо другим веществом, полезным для лечения различных злокачественных опухолей, или с лучевой терапией. Индивидуальные ингредиенты для такой комбинации могут быть введены в разные моменты времени или в одно время как дробные лекарственные препараты или как один препарат в период лечения. В соответствии с этим изобретение следует интерпретировать таким образом, что оно включает все способы введения в одно время или в разные моменты времени, и введение в этом изобретении, следовательно, должно быть интерпретировано. Возможные комбинации соединения по изобретению и какого-либо другого вещества, применимого в случае вышеупомянутых заболеваний, должны включать, в принципе, какую-либо и каждую комбинацию его с каким-либо и каждым фармацевтическим веществом, пригодным для лечения вышеупомянутых заболеваний.
Лучевая терапия сама по себе подразумевает обычный способ в области лечения злокачественной опухоли. Для лучевой терапии реально применимыми являются различные излучения, такие как рентгеновские лучи, γ-излучение, нейтронное излучение, электронный пучок, протонный пучок; и источники излучения. В наиболее распространенной лучевой терапии используется линейный ускоритель для облучения с внешними излучениями, γ-излучение.
Соединения по изобретению могут быть скомбинированы с лучевой терапией для усиления терапевтического воздействия в лучевой терапии; и вследствие этого соединения могут быть применимы в качестве радиосенсибилизаторов в области лечения злокачественной опухоли.
Другой аспект соединений по изобретению заключается в том, что соединения также применимы в качестве сенсибилизаторов для каких-либо других противораковых агентов в области лечения злокачественной опухоли.
Соединения по изобретению можно комбинировать с лучевой терапией и/или комбинировать с какими-либо другими противораковыми агентами, описанными ниже, при их использовании для лечения злокачественной опухоли.
"Сенсибилизатор" для лучевой терапии или противораковый агент, как упоминается в данном документе, предназначен для обозначения лекарственного средства, которое при использовании в качестве комбинированного с лучевой терапией и/или химиотерапией с противораковым агентом, может аддитивно или синергитически усиливать терапевтическое воздействие этой лучевой терапии и/или химиотерапии.
Вещества для комбинированных лекарственных препаратов в изобретении могут быть в каких-либо формах, выбранных в каком-либо способе, и они могут быть получены таким же способом, как для вышеупомянутых лекарственных препаратов. Комбинированный агент, включающий соединение по изобретению и какой-то другой противораковый агент, может быть легко получен специалистом в данной области обычными способами или при помощи общепринятого оборудования.
Вышеупомянутая комбинация включают не только композиции по изобретению, которые содержат некое единственное активное вещество, но также такие, которые содержат два или более других активных веществ. Существует множество примеров комбинации композиции по изобретению и одного, или двух, или нескольких активных веществ, выбранных из лечебных средств для вышеупомянутых заболеваний.
Агенты для комбинирования с композициями включают, например, противораковый агент, выбранный из группы, состоящей из противораковых алкилирующих агентов, противораковых антиметаболитов, противораковых антибиотиков, противораковых агентов растительного происхождения, противораковых координационных соединений платины, противораковых производных камптотецина, противораковых ингибиторов тирозин-киназы, моноклональных антител, интерферонов, модификаторов биологической чувствительности и других противораковых агентов, а также их фармацевтически приемлемой(мых) соли(ей) или эфира(ов).
Термин "противораковый алкилирующий агент", как он используется в настоящем описании изобретения, относится к алкилирующему агенту, имеющему противораковую активность, и термин "алкилирующий агент" в этом документе обычно относится к агенту, дающему алкильную группу в реакции алкилирования, в которой атом водорода органического соединения замещают алкильной группой. Термин "противораковый алкилирующий агент" может быть представлен, в качестве примера, N-оксидом азотистого иприта, циклофосфамидом, ифосфамидом, мелфаланом, бусульфаном, митобронитолом, карбоквоном, тиотепой, ранимустином, нимустином, темозоломидом или кармустином.
Термин "противораковый антиметаболит", как он используется в настоящем описании изобретения, относится к антиметаболиту, имеющему противораковую активность, и термин "антиметаболит" в этом документе включает, в широком смысле, вещества, которые нарушают нормальный метаболизм, и вещества, которые ингибируют электрон-транспортную систему, препятствуя образованию богатых энергией интермедиатов, за счет их структурного или функционального сходства с метаболитами, которые важны для живых организмов (такими как витамины, коферменты, аминокислоты и сахариды). Термин "противораковые антиметаболиты" может быть представлен, в качестве примера, метотрексатом, 6-меркаптопуринрибозидом, меркаптопурином, 5-фторурацилом, тегафуром, доксифлуридином, кармофуром, цитарабином, цитарабина окфосфатом, эноцитабином, S-1, гемцитабином, флударабином или пеметрекседом динатрия, и предпочтительными являются цитарабин, гемцитабин и им подобные.
Термин "противораковый антибиотик", как он используется в настоящем описании изобретения, относится к антибиотику, имеющему противораковую активность, и "антибиотик" в этом документе включает вещества, которые продуцируются микроорганизмами и ингибируют клеточный рост и другие функции микроорганизмов и других живых организмов. Термин "противораковый антибиотик" может быть представлен, в качестве примера, актиномицином D, доксорубицином, даунорубицином, неокарциностатином, блеомицином, пепломицином, митомицином C, акларубицином, пирарубицином, эпирубицином, зиностатина стималамером, идарубицином, сиролимусом или вальрубицином, и предпочтительными являются доксорубицин, митомицин C и им подобные.
Термин "противораковый агент растительного происхождения", как он используется в настоящем описании изобретения, включает соединения, имеющие противораковые активности, которые происходят из растений, или соединения, полученные посредством применения химической модификации к вышеупомянутым соединениям. Термин "противораковый агент растительного происхождения" может быть представлен, в качестве примера, винкристином, винбластином, виндезином, этопозидом, собузоксаном, доцетакселом, паклитакселом и винорелбином, и предпочтительными являются этопозид и ему подобные.
Термин "противораковое производное камптотецина", как он используется в настоящем описании изобретения, относится к соединениям, которые структурно родственные камптотецину и ингибируют рост раковой клетки, включая камптотецин как таковой. Термин "противораковое производное камптотецина" не является особо ограниченным, но может быть представлен, в качестве примера, камптотецином, 10-гидроксикамптотецином, топотеканом, иринотеканом или 9-аминокамптотецином, причем камптотецин предпочтителен. Кроме того, иринотекан подвергается метаболизму in vivo и проявляет противораковое воздействие, как, например, SN-38. Механизм действия и активность производных камптотецина, как полагают, по существу такие же, как для камптотецина (например, Nitta, et al., Gan to Kagaku Ryoho, 14, 850-857 (1987)).
Термин "противораковое координационное соединение платины", как он используется в настоящем описании изобретения, относится к координационному соединению платины, имеющему противораковую активность, и термин "координационное соединение платины" в этом документе относится к координационному соединению платины, которое обеспечивает существование платины в ионной форме. Предпочтительные соединения платины включают цисплатин; ион цис-диамминдиаквоплатины (II); хлор(диэтилентриамин)платины (II) хлорид; дихлор(этилендиамин)платину (II); диаммин(1,1-циклобутандикарбоксилато)платину (II) (карбоплатин); спироплатин; ипроплатин; диаммин(2-этилмалонато)платину (II); этилендиаминмалонатоплатину (II); аква(1,2-диаминодициклогексан)сульфатоплатину (II); аква(1,2-диаминодициклогексан)малонатоплатину (II); (1,2-диаминоциклогексан)малонатоплатину (II); (4-карбоксифталато)(1,2-диаминоциклогексан)платину (II); (1,2-диаминоциклогексан)(изоцитрато)платину (II); (1,2- диаминоциклогексан)оксалатоплатину (II); ормаплатин; тетраплатин; карбоплатин, недаплатин и оксалиплатин, и предпочтительным является цисплатин. Кроме того, другие противораковые координационные соединения платины, упомянутые в описании изобретения, известны и коммерчески доступны и/или могут быть получены средним специалистом в данной области посредством использования обычного оборудования.
Термин "противораковый ингибитор тирозин-киназы", как он используется в описании изобретения, относится к ингибитору тирозин-киназы, имеющему противораковую активность, и термин "ингибитор тирозин-киназы" в этом документе относится к химическому веществу, ингибирующему "тирозин-киназу", которая перемещает γ-фосфатную группу ATФ к гидроксильной группе специфического тирозина в белке. Термин "противораковый ингибитор тирозин-киназы" может быть представлен, в качестве примера, гефитинибом, иматинибом или эрлотинибом.
Термин "моноклональное антитело", как он используется в описании изобретения, который также известен как антитело одного клона, относится к антителу, полученному с помощью моноклональной антитело-продуцирующей клетки, и примеры такового включают цетуксимаб, бевацизумаб, ритуксимаб, алемтузумаб и трастузумаб.
Термин "интерферон", как он используется в описании изобретения, относится к интерферону, имеющему противораковую активность, и он представляет собой гликопротеин, имеющий молекулярную массу, приблизительно, 20000, который продуцируется и секретируется большинством животных клеток при вирусной инфекции. Он имеет не только воздействие ингибирования развития вируса, но также различные иммуноэффекторные механизмы действия, включающие ингибирование клеточного роста (а именно опухолевых клеток) и усиление естественной активности клеток-киллеров, вследствие этого обозначаемый как один из типов цитокинов. Примеры "интерферона" включают интерферон α, интерферон α-2a, интерферон α-2b, интерферон β, интерферон γ-1a и интерферон γ-n1.
Термин "модификатор биологической чувствительности", как он используется в описании изобретения, представляет собой так называемый модификатор биологической чувствительности или BRM, и обычно представляет собой общий термин для веществ или лекарств, направленных на изменение защитных механизмов живых организмов или биологических реакций, таких как выживаемость, рост или дифференциация клеток ткани с целью ориентировать их быть полезными индивидууму в противостоянии опухоли, инфекции или другим заболеваниям. Примеры "модификатора биологической чувствительности" включают крестин, лентинан, сизофиран, пицибанил и убенимекс.
Термин "другой противораковый агент", как он применяется в настоящем описании изобретения, относится к противораковому агенту, который не принадлежит к какому-либо из вышеописанных агентов, имеющих противораковые активности. Примеры "другого противоракового агента" включают митоксантрон, L-аспарагиназу, прокарбазин, дакарбазин, гидроксикарбамид, пентостатин, третиноин, алефасепт, дарбепоэтин альфа, анастрозол, экземестан, бикалутамид, леупрорелин, флутамид, фулвестрант, пегаптаниб октанатрия, денилейкин дифтитокс, алдеслейкин, тиротропин альфа, триоксид мышьяка, бортезомиб, капецитабин и гозерелин.
Вышеописанные термины "противораковый алкилирующий агент", "противораковый антиметаболит", "противораковый антибиотик", "противораковый агент растительного происхождения", "противораковое координационное соединение платины", "противораковое производное камптотецина", "противораковый ингибитор тирозин-киназы", "моноклональное антитело", "интерферон", "модификатор биологической чувствительности" и "другой противораковый агент" все являются известными и являются либо коммерчески доступными, либо получаемыми средним специалистом в данной области способами, как таковыми известными, посредством хорошо известных или общепринятых методов. Процесс получения гефитиниба описан, например, в USP No. 5770599; процесс получения цетуксимаба описан, например, в WO 96/40210; процесс получения бевацизумаба описан, например, в WO 94/10202; процесс получения оксалиплатина описан, например, в USP No. 5420319 и 5959133; процесс получения гемцитабина описан, например, в USP No. 5434254 и 5223608 и процесс получения камптотецина описан в USP No. 5162532, 5247089, 5191082, 5200524, 5243050 и 5321140; процесс получения иринотекана описан, например, в USP No. 4604463; процесс получения топотекана описан, например, в USP No. 5734056; процесс получения темозоломида описан, например, в JP-B No. 4-5029 и процесс получения ритуксимаба описан, например, в JP-W No. 2-503143.
Вышеупомянутые противораковые алкилирующие агенты являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: N-оксид азотистого иприта от фирмы Mitsubishi Pharma Corp. как Nitromin (торговое наименование); циклофосфамид от фирмы Shionogi & Co., Ltd. как Endoxan (торговое наименование); ифосфамид от фирмы Shionogi & Co., Ltd. как Ifomide (торговое наименование); мелфалан от фирмы GlaxoSmithKline Corp. как Alkeran (торговое наименование); бусульфан от фирмы Takeda Pharmaceutical Co., Ltd. как Mablin (торговое наименование); митобронитол от Kyorin Pharmaceutical Co., Ltd. как Myebrol (торговое наименование); карбоквон от фирмы Sankyo Co., Ltd. как Esquinon (торговое наименование); тиотепа от фирмы Sumitomo Pharmaceutical Co., Ltd. как Tespamin (торговое наименование); ранимустин от фирмы Mitsubishi Pharma Corp. как Cymerin (торговое наименование); нимустин от фирмы Sankyo Co., Ltd. как Nidran (торговое наименование); темозоломид от фирмы Schering Corp. как Temodar (торговое наименование) и кармустин от фирмы Guilford Pharmaceuticals Inc. как Gliadel Wafer (торговое наименование).
Вышеупомянутые противораковые антиметаболиты являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: метотрексат от фирмы Takeda Pharmaceutical Co., Ltd. как Methotrexate (торговое наименование); 6-меркаптопуринрибозид от фирмы Aventis Corp. как Thioinosine (торговое наименование); меркаптопурин от фирмы Takeda Pharmaceutical Co., Ltd. как Leukerin (торговое наименование); 5-фторурацил от фирмы Kyowa Hakko Kogyo Co., Ltd. как 5-FU (торговое наименование); тегафур от фирмы Taiho Pharmaceutical Co., Ltd. как Futraful (торговое наименование); доксифлуридин от фирмы Nippon Roche Co., Ltd. как Furutulon (торговое наименование); кармофур от фирмы Yamanouchi Pharmaceutical Co., Ltd. как Yamafur (торговое наименование); цитарабин от фирмы Nippon Shinyaku Co., Ltd. как Cylocide (торговое наименование); цитарабин окфосфат от фирмы Nippon Kayaku Co., Ltd. как Strasid (торговое наименование); эноцитабин от фирмы Asahi Kasei Corp. как Sanrabin (торговое наименование); S-1 от фирмы Taiho Pharmaceutical Co., Ltd. как TS-1 (торговое наименование); гемцитабин от фирмы Eli Lilly & Co. как Gemzar (торговое наименование); флударабин от фирмы Nippon Schering Co., Ltd. как Fludara (торговое наименование) и пеметрексед динатрий от фирмы Eli Lilly & Co. как Alimta (торговое наименование).
Вышеупомянутые противораковые антибиотики являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: актиномицин D от фирмы Banyu Pharmaceutical Co., Ltd. как Cosmegen (торговое наименование); доксорубицин от фирмы Kyowa Hakko Kogyo Co., Ltd. как adriacin (торговое наименование); даунорубицин от фирмы Meiji Seika Kaisha Ltd. как Daunomycin; неокарциностатин от фирмы Yamanouchi Pharmaceutical Co., Ltd. как Neocarzinostatin (торговое наименование); блеомицин от фирмы Nippon Kayaku Co., Ltd. как Bleo (торговое наименование); пепромицин от фирмы Nippon Kayaku Co, Ltd. как Pepro (торговое наименование); митомицин C от фирмы Kyowa Hakko Kogyo Co., Ltd. как Mitomycin (торговое наименование); акларубицин от фирмы Yamanouchi Pharmaceutical Co., Ltd. как Aclacinon (торговое наименование); пирарубицин от фирмы Nippon Kayaku Co., Ltd. как Pinorubicin (торговое наименование); эпирубицин от фирмы Pharmacia Corp. как Pharmorubicin (торговое наименование); зиностатин стималамер от фирмы Yamanouchi Pharmaceutical Co., Ltd. как Smancs (торговое наименование); идарубицин от фирмы Pharmacia Corp. как Idamycin (торговое наименование); сиролимус от фирмы Wyeth Corp. как Rapamune (торговое наименование) и вальрубицин от фирмы Anthra Pharmaceuticals Inc. как Valstar (торговое наименование).
Вышеупомянутые противораковые агенты растительного происхождения являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: винкристин от фирмы Shionogi & Co., Ltd. как Oncovin (торговое наименование); винбластин от фирмы Kyorin Pharmaceutical Co., Ltd. как Vinblastine (торговое наименование); виндезин от фирмы Shionogi & Co., Ltd. как Fildesin (торговое наименование); этопозид от фирмы Nippon Kayaku Co., Ltd. как Lastet (торговое наименование); собузоксан от фирмы Zenyaku Kogyo Co., Ltd. как Perazolin (торговое наименование); доцетаксел от фирмы Aventis Corp. как Taxsotere (торговое наименование); паклитаксел от фирмы Bristol-Myers Squibb Co. как Taxol (торговое наименование) и винорелбин от фирмы Kyowa Hakko Kogyo Co., Ltd. как Navelbine (торговое наименование).
Вышеупомянутые противораковые координационные соединения платины являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: цисплатин от фирмы Nippon Kayaku Co., Ltd. как Randa (торговое наименование); карбоплатин от фирмы Bristol-Myers Squibb Co. как Paraplatin (торговое наименование); недаплатин от фирмы Shionogi & Co., Ltd. как Aqupla (торговое наименование) и оксалиплатин от фирмы Sanofi- Synthelabo Co. как Eloxatin (торговое наименование).
Вышеупомянутые противораковые производные камптотецина являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: иринотекан от фирмы Yakult Honsha Co., Ltd. как Campto (торговое наименование); топотекан от фирмы GlaxoSmithKline Corp. как Hycamtin (торговое наименование) и камптотецин от фирмы Aldrich Chemical Co., Inc., U.S.A.
Вышеупомянутые противораковые ингибиторы тирозин-киназы являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: гефитиниб от фирмы AstraZeneca Corp. как Iressa (торговое наименование); иматиниб от фирмы Novartis AG как Gleevec (торговое наименование) и эрлотиниб от фирмы OSI Pharmaceuticals Inc. как Tarceva (торговое наименование).
Вышеупомянутые моноклональные антитела являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: цетуксимаб от фирмы Bristol-Myers Squibb Co. как Erbitux (торговое наименование); бевацизумаб от фирмы Genentech, Inc. как Avastin (торговое наименование); ритуксимаб от фирмы Biogen Ideс Inc. как Rituxan (торговое наименование); алемтузумаб от фирмы Berlex Inc. как Campath (торговое наименование) и трастузумаб от фирмы Chugai Pharmaceutical Co., Ltd. как Herceptin (торговое наименование).
Вышеупомянутые интерфероны являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: интерферон α от фирмы Sumitomo Pharmaceutical Co., Ltd. как Sumiferon (торговое наименование); интерферон α-2a от фирмы Takeda Pharmaceutical Co., Ltd. как Canferon-A (торговое наименование); интерферон α-2b от фирмы Schering-Plough Corp. как Intron A (торговое наименование); интерферон β от фирмы Mochida Pharmaceutical Co., Ltd. как IFNβ (торговое наименование); интерферон γ-1a от фирмы Shionogi & Co., Ltd. как Imunomax-γ (торговое наименование) и интерферон γ-n1 от фирмы Otsuka Pharmaceutical Co., Ltd. как Ogamma (торговое наименование).
Вышеупомянутые модификаторы биологической чувствительности являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: крестин от фирмы Sankyo Co., Ltd. как krestin (торговое наименование); лентинан от фирмы Aventis Corp. как Lentinan (торговое наименование); сизофиран от фирмы Kaken Seiyaku Co., Ltd. как Sonifiran (торговое наименование); пицибанил от фирмы Chugai Pharmaceutical Co., Ltd. как Picibanil (торговое наименование) и убенимекс от фирмы Nippon Kayaku Co., Ltd. как Bestatin (торговое наименование).
Вышеупомянутые другие противораковые агенты являются коммерчески доступными, что представлено, в качестве примера, нижеследующим: митоксантрон от фирмы Wyeth Lederle Japan, Ltd. как Novantrone (торговое наименование); L-аспарагиназа от фирмы Kyowa Hakko Kogyo Co., Ltd. как Leunase (торговое наименование); прокарбазин от фирмы Nippon Roche Co., Ltd. как Natulan (торговое наименование); дакарбазин от фирмы Kyowa Hakko Kogyo Co., Ltd. как Dacarbazine (торговое наименование); гидроксикарбамид от фирмы Bristol-Myers Squibb Co. как Hydrea (торговое наименование); пентостатин от фирмы Kagaku Oyobi Kessei Ryoho Kenkyusho как Coforin (торговое наименование); третиноин от фирмы Nippon Roche Co., Ltd. как Vesanoid (торговое наименование); алефасепт от фирмы Biogen Idec Inc. как Amevive (торговое наименование); дарбепоэтин альфа от фирмы Amgen Inc. как Aranesp (торговое наименование); анастрозол от фирмы AstraZeneca Corp. как Arimidex (торговое наименование); эксеместан от фирмы Pfizer Inc. как Aromasin (торговое наименование); бикалутамид от фирмы AstraZeneca Corp. как Casodex (торговое наименование); леупрорелин от фирмы Takeda Pharmaceutical Co., Ltd. как Leuplin (торговое наименование); флутамид от фирмы Schering-Plough Corp. как Eulexin (торговое наименование); фулвестрант от фирмы AstraZeneca Corp. как Faslodex (торговое наименование); пегаптаниб октанатрия от фирмы Gilead Sciences, Inc. как Macugen (торговое наименование); денилейкин дифтитокс от фирмы Ligand Pharmaceuticals Inc. как Ontak (торговое наименование); альдеслейкин от фирмы Chiron Corp. как Proleukin (торговое наименование); тиротропин альфа от фирмы Genzyme Corp. как Thyrogen (торговое наименование); триоксид мышьяка от фирмы Cell Therapeutics, Inc. как Trisenox (торговое наименование); бортезомиб от фирмы Millennium Pharmaceuticals, Inc. как Velcade (торговое наименование); капецитабин от фирмы Hoffmann-La Roche, Ltd. как Xeloda (торговое наименование) и гозерелин от фирмы AstraZeneca Corp. как Zoladex (торговое наименование).
Изобретение также относится к способу лечения злокачественной опухоли, который включает в себя введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соединения по изобретению, или его соли, или его эфира.
В ходе процесса, согласно изобретению, предпочтительная терапевтическая единичная доза может меняться в соответствии с, например, способом введения соединения по изобретению, типом используемого соединения по изобретению и используемой лекарственной формой соединения по изобретению; типом, способом введения и лекарственной формой другого противоракового агента, используемого в комбинации; и типом клеток, подвергаемых лечению, состоянием пациента и т.п. Оптимальное лечение при данных условиях может быть установлено средним специалистом в данной области на основании обычно принятой терапевтической единичной дозы и/или на основании содержания настоящего описания изобретения.
В ходе процесса, согласно изобретению, терапевтическая единичная доза соединения по изобретению может меняться в соответствии с, как правило, типом используемого соединения, типом составленной композиции, частотой применения и специфической областью лечения, тяжестью заболевания, возрастом пациента, врачебным диагнозом, типом злокачественной опухоли и т.п. Однако же, в качестве иллюстративной ссылки, дневная доза для взрослого может быть в диапазоне, например, от 1 до 1000 мг в случае перорального введения. В случае парентерального введения, предпочтительно внутривенного введения, и более предпочтительно внутривенной капельной инфузии дневная доза может быть в диапазоне, например, от 1 до 100 мг/м2 (площади поверхности тела). В этом документе, в случае внутривенной капельной инфузии, введение может непрерывно осуществляться в течение, например, от 1 до 48 часов. Кроме того, частота введения может меняться в зависимости от способа введения и симптомов, но она составляет, например, от одного до пяти раз в день. Альтернативно, периодически прерывающееся введение, такое как введение через день, введение через два дня или им подобные также могут применяться в способе введения. Период прекращения назначения лекарственного средства в случае парентерального введения составляет, например, от 1 до 6 недель.
Несмотря на то, что терапевтическая единичная доза для другого противоракового агента, используемого в комбинации с соединением по изобретению, особенно не ограничена, она может быть установлена, если необходимо, средним специалистом в данной области в соответствии с известными в литературе данными. Примеры могут быть, как нижеследующие.
Терапевтическая единичная доза для 5-фторурацила (5-FU) является такой, что в случае перорального введения, например, от 200 до 300 мг в день вводятся от одного до трех раз последовательно и в случае инъекции, например, от 5 до 15 мг/кг в день вводятся один раз в день в течение первых пяти последующих дней посредством внутривенной инъекции или внутривенной капельной инфузии, и затем от 5 до 7,5 мг/кг вводятся один раз в день через день посредством внутривенной инъекции или внутривенной капельной инфузии (доза может быть соответствующим образом повышена или понижена).
Терапевтическая единичная доза S-1 (Тегафур, Гиместат и Остат калия) такова, что, например, начальная доза (однократная доза) устанавливается в нижеследующем стандартном количестве в соответствии с площадью поверхности тела, и она перорально вводится дважды в день, после завтрака и после обеда, в течение 28 последующих дней, за которыми следует прекращение назначения лекарственного средства в течение 14 дней. Это устанавливается как один курс введения, который повторяется. Начальное стандартное количество на единицу площади поверхности тела (эквивалент Тегафура) составляет 40 мг за одно введение для площади менее чем 1,25 м2; 50 мг за одно введение - для площади от 1,25 м2 до менее чем 1,5 м2; 60 мг за одно введение - для площади от 1,5 м2 или более. Эта доза соответствующим образом повышается или понижается в зависимости от состояния пациента.
Терапевтическая единичная доза для гемцитабина составляет, например, 1 г в виде гемцитабина/м2 за одно введение, которая вводится внутривенной капельной инфузией за период времени 30 минут, и одно введение в неделю продолжается в течение 3 недель, за которыми следует прекращение назначения лекарственного средства в течение четвертой недели. Это устанавливается как один курс введения, который повторяется. Доза соответствующим образом понижается в соответствии с возрастом, симптомом или развитием побочных эффектов.
Терапевтическая единичная доза для доксорубицина (например, доксорубицина гидрохлорида) такова, что, например, в случае внутривенной инъекции, 10 мг (0,2 мг/кг) (титр) вводится один раз в день внутривенно одноразовым введением в течение от 4 до 6 последующих дней, за которыми следует прекращение назначения лекарственного средства в течение от 7 до 10 дней. Это устанавливается как один курс введения, который повторяется два или три раза. В этом документе общая доза составляет, предпочтительно, 500 мг (титр)/м2 (площади поверхности тела) или менее, и она может быть соответствующим образом повышена или понижена в диапазоне.
Терапевтическая единичная доза для этопозида такова, что, например, в случае внутривенной инъекции, от 60 до 100 мг/м2 (площади поверхности тела) в день вводится в течении 5 последующих дней, за которыми следует прекращение назначения лекарственного средства в течение трех недель (доза может быть соответствующим образом повышена или понижена). Это устанавливается как один курс введения, который повторяется. Между тем, в случае перорального введения, например, от 175 до 200 мг в день вводится в течение 5 последующих дней, за которыми следует прекращение назначения лекарственного средства в течение трех недель (доза может быть соответствующим образом повышена или понижена). Это устанавливается как один курс введения, который повторяется.
Терапевтическая единичная доза для доцетаксела (доцетаксел гидрат) такова, что, например, 60 мг в виде доцетаксела/м2 (площади поверхности тела) вводится один раз в день посредством внутривенной капельной инфузии в течение периода времени от 1 часа или более с интервалом от 3 до 4 недель (доза может быть соответствующим образом повышена или понижена).
Терапевтическая единичная доза для паклитаксела такова, что, например, 210 мг/м2 (площади поверхности тела) вводится один раз в день посредством внутривенной капельной инфузии в течение периода времени 3 часа, за которыми следует прекращение назначения лекарственного средства в течение, по меньшей мере, 3 недель. Это устанавливается как один курс введения, который повторяется. Доза может быть соответствующим образом повышена или понижена.
Терапевтическая единичная доза для цисплатина такова, что, например, в случае внутривенной инъекции, от 50 до 70 мг/м2 (площади поверхности тела) вводится один раз в день, за которым следует прекращение назначения лекарственного средства в течение 3 недель или дольше (доза может быть соответствующим образом повышена или понижена). Это устанавливается как один курс введения, который повторяется.
Терапевтическая единичная доза для карбоплатина такова, что, например, от 300 до 400 мг/м2 вводится один раз в день посредством внутривенной капельной инфузии в течение периода времени 30 минут или дольше, за которыми следует прекращение назначения лекарственного средства в течение, по меньшей мере, 4 недель (доза может быть соответствующим образом повышена или понижена). Это устанавливается как один курс введения, который повторяется.
Терапевтическая единичная доза для оксалиплатина такова, что 85 мг/м2 вводится один раз в день посредством внутривенной инъекции, за которой следует прекращение назначения лекарственного средства в течение двух недель. Это устанавливается как один курс введения, который повторяется.
Терапевтическая единичная доза для иринотекана (например, иринотекан гидрохлорид) такова, что, например, 100 мг/м2 вводится один раз в день посредством внутривенной капельной инфузии, 3 или 4 раза с интервалом одна неделя, за которыми следует прекращение назначения лекарственного средства в течение, по меньшей мере, двух недель.
Терапевтическая единичная доза для топотекана такова, что, например, 1,5 мг/м2 вводится один раз в день посредством внутривенной капельной инфузии в течение 5 дней, за которыми следует прекращение назначения лекарственного средства в течение, по меньшей мере, 3 недель.
Терапевтическая единичная доза для циклофосфамида такова, что, например, в случае внутривенной инъекции, 100 мг вводятся один раз в день посредством внутривенной инъекции в течение последовательных дней. Если пациент может переносить, дневная доза может быть увеличена до 200 мг. Общая доза составляет от 3000 до 8000 мг, которая может быть соответствующим образом повышена или понижена. Если необходимо, он может быть введен инъекцией или инфузией внутримышечно, интраторакально или внутрь опухоли. С другой стороны, в случае перорального введения, например, от 100 до 200 мг вводятся в день.
Терапевтическая единичная доза для гефитиниба такова, что 250 мг перорально вводятся один раз в день.
Терапевтическая единичная доза для цетуксимаба такова, что, например, 400 мг/м2 вводится в первый день посредством внутривенной капельной инфузии, и затем 250 мг/м2 вводится каждую неделю посредством внутривенной капельной инфузии.
Терапевтическая единичная доза для бевацизумаба такова, что, например, 3 мг/кг вводятся каждую неделю посредством внутривенной капельной инфузии.
Терапевтическая единичная доза для трастузумаба такова, что, например, обычно для взрослого, один раз в день, 4 мг в виде трастузумаба/кг (массы тела) вводится первично, за этим следует внутривенная капельная инфузия 2 мг/кг в течение периода времени 90 минут или дольше, каждую неделю от второго введения.
Терапевтическая единичная доза для эксеместана такова, что, например, обычно для взрослого, 25 мг перорально вводятся один раз в день после еды.
Терапевтическая единичная доза для леупрорелина (например, леупрорелина ацетата) такова, что, например, обычно для взрослого, 11,25 мг вводятся подкожно один раз в 12 недель.
Терапевтическая единичная доза для иматиниба такова, что, например, обычно для взрослых в хронической фазе хронического миелолейкоза, 400 мг вводится перорально один раз в день после еды.
Терапевтическая единичная доза для комбинации 5-FU и лейковорина такова, что, например, 425 мг/м2 5-FU и 200 мг/м2 лейковорина вводятся с первого по пятый день посредством внутривенной капельной инфузии и этот курс повторяется с интервалом в 4 недели.
Изобретение описывается более конкретно со ссылкой на нижеследующие примеры и примеры получения, которые, тем не менее, не предназначены для ограничения объема изобретения.
Для тонкослойной хроматографии в примерах и примерах получения силикагель Silica gel60F254 (Merck) использовали для пластинки и УФ-детектор использовали для детектирования. WakogelTM C-300, или C-200 (Wako Pure Chemical Industries), или NH (Fuji Silysia Chemical) использовали в качестве силикагеля для колоночной хроматографии. Для масс-спектрометрии использовали JMS-SX102A (JEOL) или QUATTROII (Micromass). Для ЯМР-спектрометрии использовали диметилсульфоксид в качестве внутреннего стандарта в виде раствора тяжелого дитметилсульфоксида; спектрометр Gemini-300 (300 МГц; Varian), VXR-300 (300 МГц; Varian), Mercury 400 (400 МГц; Varian) или Inova 400 (400 МГц; Varian) использовали; и все δ величины даны в м.д.
Обозначения сокращений в ЯМР указаны ниже.
с: синглет
д: дублет
дд: дублет дублета
т: триплет
дт: дублет триплета
кв: квартет
м: мультиплет
ушир: уширенный
J: константа связывания
Гц: герц
ДМСО-d6: тяжелый диметилсульфоксид
Пример получения 1
Получение 2-аллил-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1) Трет-бутил 1-аллилгидразинкарбоксилат:
250 г трет-бутилгидразинкарбоксилата добавляли к раствору 280 г фталиевого ангидрида в толуоле (3 л). Используя насадку Дина-Старка для отделения воды, реакционную смесь нагревали с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, образовавшийся осадок отделяли фильтрованием, получая 516 г неочищенного трет-бутил(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)карбамата.
520 г карбоната калия, 43,3 г хлорида бензилтриэтиламмония и 250 мл аллилбромида добавляли в этом порядке к раствору вышеуказанного соединения в ацетонитриле (3,5 л) и перемешивали при комнатной температуре в течение 18 часов. 1,5 л воды добавляли к реакционному раствору и отделяли слой ацетонитрила и концентрировали. Один литр воды добавляли к остатку и водный слой экстрагировали этилацетатом и этилацетатный слой промывали насыщенным водным солевым раствором и затем сушили над безводным сульфатом натрия. Растворитель упаривали при пониженном давлении и выпавшее в осадок бесцветное твердое вещество промывали гексаном и сушили, получая 460 г неочищенного трет-бутилаллил(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)карбамата.
При охлаждении на ледяной бане 100 мл метилгидразина добавляли к раствору вышеуказанного соединения в тетрагидрофуране (3,0 л), затем возвращались к комнатной температуре и перемешивали в течение 18 часов. Выпавшее в осадок нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Смешанный растворитель, представляющий собой гексан/этилацетат (3/1), добавляли к остатку и выпавшее в осадок нерастворимое вещество отделяли фильтрованием. Эту операцию повторяли пять раз, затем фильтрат концентрировали при пониженном давлении, полученный в результате остаток перегоняли при пониженном давлении, получая 211 г указанного в заголовке соединения в виде бледно-желтого маслообразного вещества.
ESI-MS (масс-спектр с ионизацией электрораспылением). Найдено: m/z[M+H]+ 173,4.
2) Получение 2-аллил-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
260 мл N,N-диизопропилэтиламина и 106 г гидразина, полученного на вышеуказанной стадии 1), добавляли к раствору 142 г этил 4-хлор-2-(метилтио)пиридин-5-карбоксилата в тетрагидрофуране (1,5 л) и перемешивали при нагревании с обратным холодильником в течение 18 часов. После охлаждения до комнатной температуры реакционный раствор упаривали при пониженном давлении и к остатку добавляли 500 мл диэтилового эфира и выпавшее в осадок твердое вещество отделяли фильтрованием. Фильтрат упаривали при пониженном давлении, остаток охлаждали на ледяной бане и к нему постепенно добавляли 400 мл трифторуксусной кислоты и перемешивали при комнатной температуре в течение 1 часа и затем при 70°C - в течение 1 часа. Реакционный раствор упаривали при пониженном давлении, к нему добавляли 500 мл этанола и охлаждали на ледяной бане и туда же добавляли 1 литр 6 н. раствора гидроксида натрия и перемешивали при комнатной температуре в течение 15 минут. Охлажденный на ледяной бане реакционный раствор подкисляли с помощью 400 мл концентрированной хлористоводородной кислоты и затем упаривали при пониженном давлении. Остаток распределяли в хлороформе и воде, хлороформенный слой экстрагировали, промывали насыщенным солевым водным раствором и сушили над безводным сульфатом натрия. Растворитель удаляли упариванием при пониженном давлении и образовавшееся желтое твердое вещество отделяли фильтрованием, промывали этанолом и диэтиловым эфиром и сушили, получая 99,1 г указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6) δ: 8,66 (1,0H, ушир.с), 5,83 (1,0H, ддт, J=17,1, 9,8, 5,4 Гц), 5,13 (1,0H, д, J=9,8 Гц), 5,06 (1,0H, д, J=17,1 Гц), 4,34 (2,0H, д, J=5,4 Гц), 2,51 (3,0H, с).
ESI-MS. Найдено: m/z[M+H]+ 223,3.
Пример получения 2
Получение 6-(метилтио)-1-фенил-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
60 мл триэтиламина добавляли к раствору 25 г этил 4-хлор-2-(метилтио)пиримидин-5-карбоксилата и 12,7 мл фенилгидразина в тетрагидрофуране (200 мл) и перемешивали при комнатной температуре в течение 18 часов. Растворитель концентрировали при пониженном давлении, к остатку добавляли воду, затем промывали эфиром и подкисляли с помощью водного 5 н. раствора хлористоводородной кислоты, добавленного туда же. Выпавшее в осадок твердое вещество отделяли фильтрованием и промывали водой и 2-пропанолом, получая 10,8 г указанного в заголовке соединения в виде твердого белого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 12,18 (1H, c), 9,02 (1H, c), 8,13 (2H, дд, J=8,8, 1,0 Гц), 7,52 (2H, тд, J=7,1, 1,6 Гц), 7,26 (1H, тт, J=7,1, 1,0 Гц), 2,61 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 259,1.
Пример 1
Получение 3-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил)-N,N-диметилбензамида

1) Получение метил 3-[2-аллил-6-(метилтио)-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил]бензоата:
20 мл пиридина добавляли к раствору в хлороформе 7,5 г 2-аллил-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, 6,1 г ацетата меди(II) и 10 г [3-(метоксикарбонил)]фенилбороновой кислоты и перемешивали при комнатной температуре в течение 3 дней. Водный 30%-ный раствор аммиака и насыщенный солевой водный раствор добавляли в реакционную жидкую среду в указанном порядке и экстрагировали хлороформом. Органический слой промывали насыщенным солевым водным раствором, затем сушили над безводным сульфатом магния и растворитель удаляли упариванием. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат), получая 6,7 г метил 3-[2-аллил-6-(метилтио)-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил]бензоата в виде желтого маслообразного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,92 (1H, с), 8,11-8,06 (2H, м), 7,65-7,59 (2H, м), 5,68 (1H, ддд, J=17,1, 10,2, 5,9 Гц), 5,13 (1H, дд, J=10,2, 1,0 Гц), 4,97 (1H, дд, J=17,1, 1,0 Гц), 4,45 (2H, д, J=5,9 Гц), 3,96 (3H, с), 2,51 (3H, с).
2) Получение метил 3-[2-аллил-6-(метилсульфинил)-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил]бензоата:
При 0°C добавляли 6,5 г м-хлорпербензойной кислоты к раствору в хлороформе 6,7 г соединения, полученного в вышеприведенной реакции, и перемешивали в течение 30 минут. Насыщенный водный раствор гидрокарбоната натрия добавляли к жидкой реакционной среде и экстрагировали хлороформом/изопропанолом (80/20). Органический слой сушили над безводным сульфатом магния и растворитель удаляли упариванием, получая 5,6 г неочищенного метил 3-[2-аллил-6-(метилсульфинил)-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил]бензоата.
3) Получение метил 3-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил]бензоата:
0,87 г 4-(4-метил-1-пиперазинил)анилина и 2 мл N,N- диизопропилэтиламина добавляли к раствору в толуоле 1,7 г неочищенного продукта, полученного в вышеприведенной реакции, и перемешивали при 70°C в течение 12 часов. Растворитель удаляли упариванием, продукт очищали с помощью колоночной хроматографии на силикагеле (хлороформ/метанол), получая 2,2 г метил 3-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил]бензоата в виде бледно-желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,81 (1H, с), 8,18-8,13 (1H, м), 8,04 (1H, д, J=7,8 Гц), 7,66-7,56 (2H, м), 7,45 (2H, д, J=8,5 Гц), 6,88 (2H, д, J=8,5 Гц), 5,68 (1H, ддд, J=17,1, 10,2, 6,3 Гц), 5,10 (1H, дд, J=10,2, 1,0 Гц), 4,98 (1H, дд, J=17,1, 1,0 Гц), 4,40 (2H, д, J=6,3 Гц), 3,97 (3H, с), 3,26-3,21 (4H, м), 2,72-2,64 (4H, м), 2,43 (3H, ушир.с).
4) Получение 3-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил)-N,N-диметилбензамида:
Водный 1 н. раствор гидроксида натрия добавляли к раствору в 1,4-диоксане/метаноле (50/50) 2,2 г метил 3-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил]бензоата и перемешивали при комнатной температуре в течение 2,5 часов. Эту реакционную смесь нейтрализовали 1 н. хлористоводородной кислотой и растворитель удаляли упариванием, получая свободную карбоновую кислоту исходного сложного эфира. К раствору в N,N-диметилформамиде полученной в результате карбоновой кислоты добавляли 1,67 г гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида, 1,18 г 1-гидроксибензотриазола и 11 мл 1,0M раствора диметиламина в тетрагидрофуране и перемешивали при комнатной температуре в течение 6 часов. Водный насыщенный раствор гидрокарбоната натрия и воду добавляли к реакционной жидкой среде, экстрагировали хлороформом/изопропанолом (80/20) и очищали с помощью колоночной хроматографии на силикагеле (хлороформ/метанол), получая 560 мг 3-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил)-N,N-диметилбензамида в виде бледно-желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,81 (1H, с), 7,57-7,51 (2H, м), 7,49-7,38 (4H, м), 6,90 (2H, д, J=8,8 Гц), 5,69 (1H, ддт, J=17,1, 10,2, 6,3 Гц), 5,10 (1H, дд, J=10,2, 1,0 Гц), 5,00 (1H, дд, J=17,1, 1,0 Гц), 4,40 (2H, д, J=6,3 Гц), 3,32 (3H, с), 3,14 (3H, с), 2,99-2,92 (4H, м), 2,84-2,71 (4H, м), 2,50 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 513.
Пример 2
Получение 2-аллил-6-{[3-(гидроксиметил)-4-(4-метилпиперазин-1-ил)фенил]амино}-1-(3-тиенил)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
17,5 мг указанного в заголовке соединения получали в виде желтого твердого вещества тем же способом, как в примере от 1-1 до 1-3, для которого, однако, 3-тиенилбороновую кислоту использовали вместо [3-(метоксикарбонил)]фенилбороновой кислоты, использованной в примере 1-1, и [5-амино-2-(4-метилпиперазин-1-ил)фенил]метанол использовали вместо 4-(4-метилпиперазин-1-ил)анилина, использованного в примере 1-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,82 (1H, с), 7,17-7,63 (6H, м), 5,65-5,77 (1H, м), 5,13 (1H, д, J=10,2 Гц), 5,04 (1H, д, J=17,1 Гц), 4,76 (2H, с), 4,42 (2H, д, J=6,3 Гц), 2,98-3,06 (4H, м), 2,50-2,76 (4H, м), 2,39 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 478.
Пример 3
Получение 2-аллил-1-[3-(1-гидрокси-1-метилэтил)фенил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1) Получение 3-(1-гидрокси-1-метилэтил)фенилбороновой кислоты:
В атмосфере азота при охлаждении льдом, 5,29 мл 3'-бромацетофенона добавляли к 25 мл 2M раствора метилмагнийиодида в диэтиловом эфире и 100 мл диэтилового эфира и перемешивали в течение 20 минут. Воду и 2 н. хлористоводородную кислоту добавляли к реакционной жидкой среде, экстрагировали этилацетатом, промывали водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым водным раствором и сушили над безводным сульфатом магния. Растворитель удаляли упариванием при пониженном давлении, получая неочищенный 2-(3-бромфенил)пропан-2-ол.
В атмосфере азота, 33 мл 1,66M раствора н-бутиллития в гексане по каплям добавляли к раствору полученного вещества в тетрагидрофуране (200 мл) при -60°C или ниже и перемешивали 20 минут. 11,08 мл триизопропоксиборана добавляли к реакционной жидкой среде и перемешивали в течение 30 минут. Воду добавляли к реакционной жидкой среде, промывали диэтиловым эфиром и полученный в результате водный слой подкисляли водным 10%-ным раствором фосфорной кислоты. Его экстрагировали этилацетатом, промывали водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым водным раствором и сушили над безводным сульфатом магния. Растворитель удаляли упариванием при пониженном давлении и полученные в результате кристаллы собирали, получая 3,13 г указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,96 (2H, с), 7,88 (1H, ушир.с), 7,60 (1H, д, J=7,3 Гц), 7,50 (1H, д, J=8,3 Гц), 7,24 (1H, т, J=7,6 Гц), 4,93 (1H, с), 1,43 (6H, д, J=13,7 Гц).
2) Получение 2-аллил-1-[3-(1-гидрокси-1-метилэтил)фенил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
35,2 мг указанного в заголовке соединения получали в виде желтого твердого вещества тем же способом, как и в примере от 1-1 до 1-3, для которого, однако, вышеуказанная бороновая кислота применялась вместо [3-(метоксикарбонил)]фенилбороновой кислоты, использованной в примере 1-1.
1H-ЯМР (400 МГц, CDCl3) δ: 8,80 (1H, с), 7,57 (1H, с), 7,47 (2H, д, J=4,9 Гц), 7,43 (2H, д, J=8,8 Гц), 7,31-7,28 (1H, м), 6,88 (2H, д, J=8,8 Гц), 5,70 (1H, ддт, J=17,1, 10,0, 6,3 Гц), 5,10 (1H, дд, J=10,0, 1,2 Гц), 4,98 (1H, дд, J=17,1, 1,5 Гц), 4,38 (2H, д, J=6,3 Гц), 3,21 (4H, т, J=4,1 Гц), 2,66 (4H, с), 2,41 (3H, с), 1,62 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 500.
Пример 4
Получение 2-аллил-1-[3-(диметиламинометил)фенил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он
1) Получение 2-аллил-1-[3-(диметиламинометил)фенил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
2,9 мл метансульфонилхлорида и 11 мл N,N-диизопропилэтиламина добавляли в этом порядке к раствору в хлороформе (50 мл) 3,0 г 2-аллил-1-[3-(гидроксиметил)фенил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, который получали при использовании [3-(гидроксиметил)фенил]бороновой кислоты вместо [3- (метоксикарбонил)]фенилбороновой кислоты, использованной в примере 1-1, и перемешивали при комнатной температуре в течение 1 часа. Реакционную жидкую среду промывали 0,5н хлористоводородной кислотой и сушили над безводным сульфатом натрия. Растворитель удаляли упариванием при пониженном давлении, получая неочищенный 2-аллил-1-[3-(метилсульфонилоксиметил)фенил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он в виде желтого маслообразного вещества.
20 мл 2M раствора диметиламина в тетрагидрофуране добавляли к раствору 1,5 г вышеуказанного соединения в тетрагидрофуране (100 мл) и перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли упариванием при пониженном давлении, и остаток разделяли и очищали с помощью колоночной хроматографии на силикагеле (этилацетат), получая 2,5 г указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,90 (1H, с), 7,53-7,26 (4H, м), 5,73-5,62 (1H, м), 5,11 (1H, дд, J=10,2, 1,0 Гц), 4,95 (1H, дд, J=17,1, 1,0 Гц), 4,44 (2H, д, J=3,7 Гц), 3,49 (2H, с), 2,48 (3H, с), 2,27 (6H, с).
ESI-MS. Найдено: m/z M+H]+ 356,1.
2) Получение 2-аллил-1-[3-(диметиламинометил)фенил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
4 н. раствор хлористоводородной кислоты в этилацетате добавляли к 100 мг соединения, полученного на вышеприведенной стадии 1), перемешивали при комнатной температуре и растворитель удаляли упариванием при пониженном давлении, получая гидрохлорид 2-аллил-1-[3-(диметиламинометил)фенил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она.
70 мг м-хлорпербензойной кислоты добавляли к раствору вышеуказанного соединения в N,N-диметилформамиде (2 мл) и перемешивали при комнатной температуре в течение 15 минут. Реакционную жидкую среду промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом натрия. Растворитель удаляли упариванием при пониженном давлении, получая неочищенный 2-аллил-1-[3-(диметиламинометил)фенил]-6-(метилсульфинил)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он в виде белого твердого вещества.
50 мг 4-(4-метилпиперазин-1-ил)анилина и 0,1 мл N,N- диизопропилэтиламина добавляли в этом порядке к раствору вышеуказанного соединения в диметилсульфоксиде/толуоле (1/10, 10 мл) и перемешивали при 120°C в течение 15 часов. Растворитель удаляли упариванием при пониженном давлении, добавляли туда воду и экстрагировали этилацетатом и сушили над безводным сульфатом натрия. Растворитель удаляли упариванием при пониженном давлении и остаток разделяли и очищали с помощью стандартной колоночной хроматографии на силикагеле (этилацетат), получая 11,4 мг указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,80 (1H, c), 7,48-7,33 (6H, м), 6,87 (2H, д, J=8,8 Гц), 5,80-5,60 (1H, м), 5,09 (1H, дд, J=10,2, 1,0 Гц), 4,97 (1H, дд, J=17,1, 1,5 Гц), 4,38 (1H, д, J=5,9 Гц), 3,51 (2H, с), 3,18 (4H, т, J=4,9 Гц), 2,60 (4H, т, J=4,9 Гц), 2,37 (3H, с), 2,28 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 499.
Пример 5
Получение 2-аллил-6-{[3-гидроксиметил-4-(4-метилпиперазин-1-ил)фенил]амино}-1-пиридин-2-ил-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она

1) Получение 2-аллил-6-(метилтио)-1-пиридин-2-ил-3H-пиразоло[3,4-d]пиримидин-3-она:
2,4 мл N,N'-диметилэтилендиамина добавляли к раствору в 1,4-диоксане (50 мл) 4,44 г 2-аллил-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, 3,80 г иодида меди(I), 5,33 г 2-иодпиридина и 3,80 г карбоната калия и перемешивали в течение ночи при 95°C. Реакционную жидкую среду охлаждали, к ней добавляли водный раствор аммиака и экстрагировали этилацетатом, промывали насыщенным солевым водным раствором и сушили над безводным сульфатом магния. Растворитель удаляли упариванием при пониженном давлении и кристаллизовали из этилацетата, получая 5,15 г указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,94 (1H, с), 8,52 (1H, д, J=5,1 Гц), 7,90 (2H, д, J=3,5 Гц), 7,29-7,25 (1H, м), 5,68 (1H, ддт, J=17,0, 10,2, 6,3 Гц), 5,05 (1H, д, J=10,2 Гц), 4,91 (1H, д, J=17,0 Гц), 4,85 (1H, д, J=6,3 Гц), 2,58 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 300.
2) Получение 2-аллил-6-{[3-гидроксиметил-4-(4-метилпиперазин-1-ил)фенил]амино}-1-пиридин-2-ил-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
796 мг м-хлорпербензойной кислоты (>65%) добавляли к раствору в толуоле (20 мл) 898 мг 2-аллил-6-(метилтио)-1-пиридин-2-ил-3H-пиразоло[3,4-d]пиримидин-3-она и перемешивали в течение 30 минут. 1,60 мл N,N-диизопропилэтиламина, 800 мг [5-амино-2-(4-метилпиперазин-1-ил)фенил]метанола и 10 мл тетрагидрофурана добавляли к реакционной жидкой среде и перемешивали в течение ночи. Водный насыщенный раствор гидрокарбоната натрия добавляли к реакционной жидкой среде и экстрагировали смешанным раствором хлороформ/изопропанол (80/20). Этот экстракт сушили над безводным сульфатом магния, растворитель удаляли упариванием, и остаток очищали с помощью стандартной колоночной хроматографии на силикагеле (гексан/этилацетат = от 50/50 до 0/100). Полученное в результате кристаллическое вещество перекристаллизовывали из этанола, получая 941 мг указанного в заголовке соединения в виде белого кристаллического вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,84 (1H, c), 8,53 (1H, д, J=4,8 Гц), 7,91 (1H, дд), 7,88 (1H, дд, J=8,8, 7,6 Гц), 7,87 (1H, д, J=7,6 Гц), 7,64 (1H, с), 7,33 (1H, д, J=8,8 Гц), 7,26 (1H, дд, J=8,8, 4,8 Гц), 7,19 (1H, д, J=8,8 Гц), 5,68 (1H, ддд, J=17,2, 10,4, 5,6 Гц), 5,50 (1H, с), 5,01 (1H, д, 10,4 Гц), 4,91 (1H, д, J=17,2 Гц), 4,79 (2H, с), 4,79 (2H, д, J=5,6 Гц), 3,01 (4H, м), 2,62 (4H, м), 2,37 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 472.
Пример 6
Получение 2-аллил-1-(6-аминопиридин-2-ил)-6-{[3-метил-4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1) Получение ди-трет-бутил {6-[2-аллил-6-(метилтио)-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил]-2-пиридинил}имиддикарбоксилата:
2,00 г указанного в заголовке соединения получали в виде белого твердого вещества таким же способом, как и в примере 5-1, для которого, однако, ди-трет-бутил (6-бромпиридин-2-ил)имиддикарбоксилат использовали вместо 2-иодпиридина, использованного в примере 5-1.
1H-ЯМР (400 МГц, CDCl3) δ: 8,92 (1H, с), 7,92 (1H, т, J=8,0 Гц), 7,80 (1H, д, J=8,8 Гц), 7,35 (1H, д, J=7,8 Гц), 5,63 (1H, ддт, J=17,1, 10,2, 6,3 Гц), 5,03 (1H, дд, J=10,2, 1,0 Гц), 5,00 (1H, дд, J=17,1, 1,2 Гц), 4,82 (2H, д, J=6,3 Гц), 2,58 (3H, с), 1,51 (18H, с).
ESI-MS. Найдено: m/z[M+H]+ 515.
2) Получение 2-аллил-1-(6-аминопиридин-2-ил)-6-{[3-метил-4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
53 мг м-хлорпербензойной кислоты (>65%) добавляли к раствору в толуоле (2 мл) 103 мг ди-трет-бутил {6-[2-аллил-6-(метилтио)-3-оксо-2,3-дигидро-1H-пиразоло[3,4-d]пиримидин-1-ил]-2-пиридинил}имиддикарбоксилата и перемешивали в течение 30 минут. 0,105 мл N,N-диизопропилэтиламина и 49 мг 3-метил-4-(4-метилпиперазин-1-ил)анилина добавляли к реакционной жидкой среде и перемешивали в течение ночи. Водный насыщенный раствор гидрокарбоната натрия добавляли к реакционной жидкой среде, туда же добавляли этилацетат для экстракции, полученный в результате экстракт промывали насыщенным солевым водным раствором и сушили над безводным сульфатом магния. Растворитель удаляли упариванием, остаток очищали с помощью стандартной колоночной хроматографии на силикагеле (гексан/этилацетат = от 1/1 до 0/1). После концентрирования получали 93,2 мг белого твердого вещества.
2 мл трифторуксусной кислоты добавляли к полученному соединению, перемешивали и туда же добавляли насыщенный раствор гидрокарбоната натрия, экстрагировали этилацетатом, промывали солевым водным раствором и сушили над безводным сульфатом магния. Растворитель удаляли упариванием при пониженном давлении, получая 51,8 мг указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,81 (1H, с), 7,60 (1H, т, J=7,8 Гц), 7,52 (1H, с), 7,34 (1H, дд, J=8,8, 2,4 Гц), 7,00 (1H, д, J=8,3 Гц), 6,43 (1H, д, J=7,8 Гц), 5,71 (1H, ддт, J=16,8, 10,2, 5,9 Гц), 5,06 (1H, дд, J=10,2, 1,0 Гц), 5,00 (1H, дд, J=16,8, 1,2 Гц), 4,71 (2H, д, J=5,9 Гц), 4,58 (2H, с), 2,95 (4H, т, J=4,6 Гц), 2,66 (4H, с), 2,42 (3H, с), 2,31 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 412.
Пример 7
Получение 2-аллил-1-(6-аминопиридин-2-ил)-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
966 мг указанного в заголовке соединения получали в виде желтого твердого вещества тем же способом, как и в примере от 6-1 до 6-2, для которого, однако, 4-(4-метилпиперазин-1-ил)анилин использовали вместо 3-метил-4-(4-метилпиперазин-1-ил)анилина, использованного в примере 6-2.
1H-ЯМР (400 МГц, CDCl3) δ: 8,80 (1H, с), 7,59 (1H, т, J=7,8 Гц), 7,39 (1H, ушир.с), 6,91 (2H, д, J=8,8 Гц), 6,42 (1H, д, J=8,3 Гц), 5,71 (1H, ддт, J=17,1, 10,2, 5,9 Гц), 5,06 (1H, дд, J=10,2, 1,0 Гц), 5,00 (1H, дд, J=17,1, 1,0 Гц), 4,70 (2H, д, J=5,9 Гц), 4,57 (2H, с), 3,20 (4H, т, J=5,1 Гц), 2,61 (4H, т, J=4,9 Гц), 2,38 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 458.
Пример 8
Получение 2-аллил-1-{6-[(диметиламино)метил]пиридин-2-ил}-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1) Получение 2-аллил-1-[6-(гидроксиметил)-2-пиридинил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
3,40 г указанного в заголовке соединения получали в виде белого твердого вещества тем же способом, как и в примере 5-1, для которого, однако, (6-бромпиридин-2-ил)метанол использовали вместо 2-иодопиридина, использованного в примере 5-1.
1H-ЯМР (400 МГц, CDCl3) δ: 8,94 (1H, с), 7,91 (1H, т, J=7,8 Гц), 7,78 (1H, дд, J=8,0, 0,7 Гц), 7,27 (1H, д, J=7,8 Гц), 5,76-5,66 (1H, м), 5,07 (1H, дд, J=10,2, 1,0 Гц), 4,95 (1H, дд, J=17,1, 1,0 Гц), 4,84-4,77 (4H, м), 2,58 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 330.
2) Получение гидрохлорида 2-аллил-1-{6-[(диметиламино)метил]-2-пиридинил}-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
1,16 мл триэтиламина и 0,451 мл метансульфонилхлорида добавляли к раствору в тетрагидрофуране (20 мл) 1,37 г соединения, полученного на вышеприведенной стадии 1), и перемешивали в течение 30 минут, и затем 6 мл 2,0M раствора диметиламина в тетрагидрофуране добавляли к реакционной жидкой среде и перемешивали в течение 8 часов. Воду добавляли к реакционной жидкой среде и экстрагировали этилацетатом. Этот экстракт промывали насыщенным солевым водным раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. 10 мл этилацетата и 1,5 мл 4 н. раствора хлористоводородной кислоты в диоксане добавляли к полученному в результате остатку, затем растворитель концентрировали при пониженном давлении и остаток кристаллизовали из метанола/диэтилового эфира, получая 1,50 г указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6) δ: 8,17 (1H, с), 7,36 (1H, т, J=7,8 Гц), 7,21 (1H, д, J=7,8 Гц), 6,76 (1H, д, J=7,3 Гц), 4,92 (1H, ддт, J=17,1, 10,2, 6,0 Гц), 4,26 (1H, дд, J=10,2, 1,5 Гц), 4,14 (1H, дд, J=17,1, 1,5 Гц), 4,00 (2H, дт, J=6,0, 1,3 Гц), 3,75 (2H, с), 2,14 (6H, с), 1,78 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 357.
3) Получение 2-аллил-1-{6-[(диметиламино)метил]пиридин-2-ил}-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
65 мг м-хлорпербензойной кислоты добавляли к раствору в N,N-диметилформамиде (2 мл) 100 мг соединения, полученного на вышеприведенной стадии 2), и перемешивали при комнатной температуре в течение 15 минут. Реакционную жидкую среду промывали водным насыщенным раствором гидрокарбоната натрия и сушили над безводным сульфатом натрия. Растворитель удаляли упариванием при пониженном давлении, получая неочищенный 2-аллил-1-{6-[(диметиламино)метил]пиридин-2-ил}-6- (метилсульфинил)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он в виде белого твердого вещества.
40 мг 4-(4-метилпиперазин-1-ил)анилина и 0,1 мл N,N- диизопропилэтиламина добавляли в этом порядке к раствору в диметилсульфоксиде/толуоле (1/10, 10 мл) 40 мг вышеупомянутого соединения и перемешивали при 120°C в течение 15 часов. Растворитель удаляли упариванием при пониженном давлении, туда же добавляли воду, экстрагировали этилацетатом и сушили над безводным сульфатом натрия. Растворитель удаляли упариванием при пониженном давлении, остаток разделяли и очищали с помощью стандартной колоночной хроматографии на силикагеле (этилацетат), получая 8,4 мг указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,82 (1H, с), 7,82 (1H, т, J=7,8 Гц), 7,74 (1H, д, J=7,8 Гц), 7,47 (2H, д, J=8,8 Гц), 7,39 (1H, д, J=7,3 Гц), 6,92 (2H, д, J=6,3 Гц), 5,74-5,63 (1H, м), 5,00 (1H, дд, J=10,2, 1,0 Гц), 4,89 (1H, дд, J=17,1, 1,0 Гц), 4,80 (2H, д, J=5,9 Гц), 3,64 (2H, с), 3,22 (4H, т, J=4,9 Гц), 2,64 (4H, д, J=4,4 Гц), 2,39 (3H, с), 2,34 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 500.
Пример 9
Получение 2-аллил-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она

1) Получение 2-(6-бром-2-пиридинил)-2-пропанола:
В атмосфере азота 30 мл 3M метилмагнийиодида/в диэтиловом эфире добавляли к 300 мл раствора в диэтиловом эфире 8,72 г метил 6-бромпиридин-2-карбоксилата. Воду и 2 н. хлористоводородную кислоту добавляли к реакционной жидкой среде и экстрагировали этилацетатом. Этот экстракт промывали водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым водным раствором и сушили над безводным сульфатом магния. Растворитель удаляли упариванием при пониженном давлении, получая 8,51 г неочищенного 2-(6-бром-2-пиридинил)-2-пропанола в виде желтого маслообразного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,56 (1H, т, J=7,8 Гц), 7,38 (1H, дд, J=7,8, 1,0 Гц), 7,36 (1H, дд, J=7,8, 1,0 Гц), 1,55 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 216, 218.
2) Получение 2-аллил-1-[6-(1-гидрокси-1-метилэтил)-2-пиридинил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
12,89 г указанного в заголовке соединения получали тем же способом, как в примере 5-1, для которого, однако, соединение, полученное в вышеприведенной реакции, использовали вместо 2- иодпиридина, использованного в примере 5-1.
1H-ЯМР (400 МГц, CDCl3) δ: 8,95 (1H, с), 7,91 (1H, т, J=8,0 Гц), 7,76 (1H, д, J=7,3 Гц), 7,40 (1H, дд, J=7,8, 1,0 Гц), 5,70 (1H, ддт, J=17,1, 10,2, 6,3 Гц), 5,06 (1H, дд, J=10,2, 1,0 Гц), 4,93 (1H, дд, J=17,1, 1,2 Гц), 4,81 (2H, д, J=6,3 Гц), 2,59 (4H, с), 1,59 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 358.
3) Получение 2-аллил-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
817 мг м-хлорпербензойной кислоты (>65%) добавляли к раствору в толуоле (20 мл) 1,10 г полученного выше продукта и перемешивали в течение 20 минут. 1,61 мл N,N- диизопропилэтиламина и 706 мг 4-(4-метилпиперазин-1-ил)анилина добавляли к реакционной жидкой среде и перемешивали в течение ночи. Водный насыщенный раствор гидрокарбоната натрия добавляли к реакционной жидкой среде, экстрагировали этилацетатом, промывали насыщенным солевым водным раствором и сушили над безводным сульфатом магния. Растворитель удаляли упариванием и остаток очищали с помощью стандартной колоночной хроматографии на силикагеле (гексан/этилацетат = от 1/1 до 0/1, этилацетат/этанол = 98/2). После концентрирования проводили перекристаллизацию из этилацетата, получая 1,20 г указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,83 (1H, с), 7,86 (1H, дд, J=8,0, 7,8 Гц), 7,75 (1H, д, J=7,3 Гц), 7,49 (1H, ушир.с), 7,48 (2H, д, J=9,0 Гц), 7,34 (1H, д, J=7,4 Гц), 6,93 (2H, д, J=9,0 Гц), 5,70 (1H, ддт, J=17,2, 10,0, 6,5 Гц), 5,04 (1H, д, J=10,0 Гц), 4,94 (1H, д, J=17,2 Гц), 4,74 (2H, д, J=6,5 Гц), 3,26 (4H, т, J=4,8 Гц), 2,73 (4H, ушир.с), 2,44 (3H, с), 1,59 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 501.
Пример 10
Получение 2-аллил-6-{[4-(4-этилпиперазин-1-ил)фенил]амино}-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
50,3 мг указанного в заголовке соединения получали в виде желтого твердого вещества тем же способом, как и в примере от 9-1 до 9-3, для которого, однако, 4-(4-этил-1-пиперазинил)анилин использовали вместо 4-(4-метилпиперазин-1-ил)анилина, использованного в примере 9-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,83 (1H, с), 7,85 (1H, т, J=7,8 Гц), 7,76 (1H, д, J=7,8 Гц), 7,46 (2H, д, J=8,8 Гц), 7,34 (1H, д, J=8,3 Гц), 6,93 (2H, д, J=8,8 Гц), 5,76-5,64 (1H, м), 5,04 (1H, дд, J=10,2, 1,0 Гц), 4,94 (1H, дд, J=17,1, 1,0 Гц), 4,75 (2H, д, J=6,3 Гц), 4,00 (1H, ушир.с), 3,23 (4H, т, J=4,9 Гц), 2,65 (4H, т, J=4,9 Гц), 2,51 (2H, кв, J=7,3 Гц), 1,59 (6H, с), 1,16 (3H, т, J=7,3 Гц).
ESI-MS. Найдено: m/z[M+H]+ 515.
Пример 11
Получение 6-{[4-(4-ацетилпиперазин-1-ил)фенил]амино}-2-аллил-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
66,4 мг указанного в заголовке соединения получали в виде желтого твердого вещества тем же способом, как и в примере от 9-1 до 9-3, для которого, однако, 4-(4-ацетил-1-пиперазинил)анилин использовали вместо 4-(4-метилпиперазин-1-ил)анилина, использованного в примере 9-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,84 (1H, с), 7,87 (1H, т, J=7,8 Гц), 7,74 (1H, д, J=8,8 Гц), 7,50 (2H, д, J=8,8 Гц), 7,36 (1H, д, J=8,3 Гц), 6,94 (2H, д, J=8,8 Гц), 5,76-5,65 (1H, м), 5,04 (1H, д, J=10,2 Гц), 4,94 (1H, д, J=17,1 Гц), 4,74 (2H, д, J=5,9 Гц), 4,03-3,95 (1H, м), 3,80 (2H, т, J=4,9 Гц), 3,65 (2H, т, J=5,1 Гц), 3,17 (2H, т, J=4,9 Гц), 3,14 (2H, т, J=5,1 Гц), 2,16 (3H, с), 1,59 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 529.
Пример 12
Получение 2-аллил-6-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]фенил}амино)-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
40 мг указанного в заголовке соединения получали в виде желтого твердого вещества тем же способом, как и в примере от 9-1 до 9-3, для которого, однако, 2-[4-(4-аминофенил)-1-пиперазинил]этанол использовали вместо 4-(4-метилпиперазин-1-ил)анилина, использованного в примере 9-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,83 (1H, с), 7,86 (1H, т, J=7,8 Гц), 7,75 (1H, д, J=7,8 Гц), 7,47 (2H, д, J=8,8 Гц), 7,34 (1H, д, J=8,3 Гц), 6,93 (2H, д, J=8,8 Гц), 5,76-5,65 (1H, м), 5,04 (1H, д, J=10,2 Гц), 4,94 (1H, д, J=17,1 Гц), 4,74 (2H, д, J=6,3 Гц), 4,03-3,95 (1H, м), 3,69 (2H, т, J=5,1 Гц), 3,22 (4H, т, J=4,9 Гц), 2,73 (4H, т, J=4,6 Гц), 2,65 (2H, т, J=5,4 Гц), 1,59 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 531.
Пример 13
Получение 2-аллил-1-[6-(1-гидроксициклобутил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1) Получение 1-(6-бром-2-пиримидинил)циклобутанола:
В атмосфере азота при -10°C добавляли по каплям 10,8 мл 2,66M раствора н-бутиллития в гексане к 16 мл 0,9M раствора н-бутилмагнийхлорида в тетрагидрофуране и туда же добавляли раствор в толуоле (60 мл) 9,48 г 2,6-дибромпиридина по каплям при 0°C или ниже. Реакционную жидкую среду перемешивали в течение 1,5 часов, затем охлаждали в бане с сухим льдом в ацетоне и 5,0 г циклобутанона добавляли туда же при -50°C или ниже. После перемешивания в течение 10 минут добавляли воду и 2 н. хлористоводородную кислоту к реакционной жидкой среде и органический слой отделяли, промывали водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым водным раствором и затем сушили над безводным сульфатом магния. После концентрирования при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат = от 20/1 до 4/1), получая 5,30 г указанного в заголовке соединения в виде желтого маслообразного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,60 (1H, т, J=7,8 Гц), 7,52 (1H, дд, J=7,8, 1,0 Гц), 7,40 (1H, дд, J=7,8, 1,0 Гц), 2,53-2,48 (4H, м), 2,12-2,01 (1H, м), 1,91-1,82 (1H, м).
ESI-MS. Найдено: m/z[M+H]+ 228, 230.
2) Получение 2-аллил-1-[6-(1-гидроксициклобутил)-2-пиридинил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
1,44 г указанного в заголовке соединения получали тем же способом, как и в примере 9-2, для которого, однако, соединение, полученное в вышеприведенной реакции, использовали вместо 2-(6- бром-2-пиридинил)-2-пропанола, использованного в примере 9-2.
1H-ЯМР (400 МГц, CDCl3) δ: 8,94 (1H, с), 7,95 (1H, т, J=8,0 Гц), 7,77 (1H, д, J=7,8 Гц), 7,54 (1H, д, J=7,8 Гц), 5,70 (1H, ддт, J=17,1, 10,2, 6,3 Гц), 5,07 (1H, д, J=10,2 Гц), 4,94 (1H, д, J=17,1 Гц), 4,80 (2H, д, J=6,3 Гц), 2,58 (3H, с), 2,56-2,50 (4H, м), 2,15-2,03 (1H, м), 1,97-1,84 (1H, м).
ESI-MS. Найдено: m/z[M+H]+ 370.
3) Получение 2-аллил-1-[6-(1-гидроксициклобутил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
80,8 мг указанного в заголовке соединения получали в виде желтого твердого вещества таким же способом, как и в примере 9-3, для которого, однако, соединение, полученное в вышеприведенной реакции, использовали вместо 2-аллил-1-[6-(1-гидрокси-1-метилэтил)-2-пиридинил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, использованного в примере 9-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,83 (1H, с), 7,90 (1H, т, J=7,8 Гц), 7,77 (1H, д, J=7,8 Гц), 7,48 (2H, дд, J=12,2, 8,3 Гц), 7,48 (1H, ушир.с), 6,93 (2H, д, J=9,3 Гц), 5,70 (1H, тдд, J=5,9, 17,1, 10,0 Гц), 5,04 (1H, дд, J=10,0, 1,2 Гц), 4,94 (1H, дд, J=17,1, 1,0 Гц), 4,73 (2H, д, J=5,9 Гц), 4,20 (1H, с), 3,24 (4H, т, J=4,6 Гц), 2,65 (4H, ушир.с), 2,53 (4H, т, J=8,0 Гц), 2,41 (3H, с), 2,14-2,06 (1H, м), 1,96-1,84 (1H, м).
ESI-MS. Найдено: m/z[M+H]+ 513.
Пример 14
Получение 2-аллил-1-[6-(2-гидрокси-2-метилпропил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1) Получение 1-(6-бромпиридин-2-ил)-2-метилпропан-2-ола:
В атмосфере азота 400 мл тетрагидрофурана, содержащего 31 мл диизопропиламина, охлаждали в бане с сухим льдом в ацетоне и 82,7 мл 2,66M раствора н-бутиллития в гексане добавляли туда же, и 50 мл тетрагидрофурана, содержащего 34,4 г 6-бромпиколина, по каплям добавляли туда же при -70°C или ниже. После добавления 29,4 мл ацетона добавляли туда же при -60°C или ниже. После перемешивания в течение 35 минут добавляли воду к реакционной жидкой среде и органический растворитель концентрировали при пониженном давлении. Этот концентрат экстрагировали диэтиловым эфиром, промывали насыщенным солевым водным раствором и сушили над безводным сульфатом магния. После концентрирования при пониженном давлении остаток очищали с помощью перегонки, получая 27,60 г указанного в заголовке соединения в виде бесцветного маслообразного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,50 (1H, т, J=7,6 Гц), 7,37 (1H, д, J=7,8 Гц), 7,12 (1H, д, J=7,8 Гц), 2,91 (2H, с), 1,23 (6H, с).
ESI-MS. Найдено: m/z[M+H]+: 230, 232.
2) Получение 2-аллил-1-[6-(2-гидрокси-2-метилпропил)пиридин-2-ил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
20,70 г указанного в заголовке соединения получали тем же способом, как и в примере 9-2, для которого, однако, соединение, полученное в вышеуказанной реакции, использовали вместо 2-(6- бром-2-пиридинил)-2-пропанола, использованного в примере 9-2.
1H-ЯМР (400 МГц, CDCl3) δ: 8,93 (1H, с), 7,84 (1H, т, J=7,8 Гц), 7,71 (1H, д, J=8,3 Гц), 7,15 (1H, д, J=7,3 Гц), 5,67 (1H, ддт, J=16,8, 10,2, 6,3 Гц), 5,05 (1H, дд, J=10,2, 1,0 Гц), 4,93 (1H, дд, J=16,8, 1,2 Гц), 4,77 (2H, д, J=6,3 Гц), 2,97 (2H, с), 2,58 (3H, с), 1,25 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 372.
3) Получение 2-аллил-1-[6-(2-гидрокси-2-метилпропил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1,06 г указанного в заголовке соединения получали в виде желтого твердого вещества таким же способом, как и в примере 9-3, для которого, однако, соединение, полученное в вышеприведенной реакции, использовали вместо 2-аллил-1-[6-(1-гидрокси-1-метилэтил)-2-пиридинил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, использованного в примере 9-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,82 (1H, с), 7,79 (1H, т, J=7,8 Гц), 7,66 (1H, ушир.с), 7,45 (2H, д, J=8,8 Гц), 7,08 (1H, д, J=7,8 Гц), 6,93 (2H, д, J=8,8 Гц), 5,78-5,62 (1H, м), 5,13-4,94 (2H, м), 4,63 (2H, с), 3,23 (4H, т, J=4,6 Гц), 2,98 (2H, с), 2,64 (4H, с), 2,40 (3H, с), 1,24 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 515.
Пример 15
Получение 2-аллил-1-[6-(2-гидрокси-2-метилпропил)пиридин-2-ил]-6-{[4-(1-метилпиперидин-4-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
36,8 мг указанного в заголовке соединения получали в виде желтого твердого вещества таким же способом, как и в примере с 14-1 по 14-3, для которого, однако, 4-(1-метилпиперидин-4-ил)анилин использовали вместо 4-(4-метилпиперазин-1-ил)анилина, использованного в примере 14-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,85 (1H, с), 7,89-7,76 (2H, ушир.м), 7,80 (1H, т, J=7,8 Гц), 7,52 (2H, д, J=8,3 Гц), 7,22 (2H, д, J=8,3 Гц), 7,10 (1H, д, J=7,8 Гц), 5,77-5,64 (1H, ушир.м), 5,08 (1H, д, J=9,8 Гц), 5,01 (1H, д, J=17,6 Гц), 4,71-4,58 (2H, ушир.м), 3,05 (2H, д, J=11,2 Гц), 2,99 (2H, с), 2,56-2,45 (1H, м), 2,38 (3H, с), 2,21-2,07 (2H, м), 1,95-1,81 (4H, м), 1,24 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 514.
Пример 16
Получение 2-аллил-1-[6-(2-гидрокси-1,1,2-триметилпропил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1) Получение 2-(6-бромпиридин-2-ил)-2-метилпропионата:
В атмосфере азота 100 мл тетрагидрофурана, содержащего 14 мл диизопропиламина, охлаждали в бане с сухим льдом в ацетоне, и 38 мл 2,66M раствора н-бутиллития в гексане добавляли туда же для получения диизопропиламида лития. Этот полученный раствор по каплям добавляли к 100 мл тетрагидрофурана, содержащего 4,55 мл 6-бромпиколина и 6,06 мл диэтилкарбоната, при -60°C или ниже. После перемешивания в течение 20 минут 6,23 мл метилиодида добавляли туда же и подогревали до комнатной температуры. Воду добавляли к реакционной жидкой среде, экстрагировали диэтиловым эфиром, промывали насыщенным солевым водным раствором и сушили над безводным сульфатом магния. После концентрирования при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат = от 100/0 до 8/1), получая 10,72 г указанного в заголовке соединения в виде бесцветного маслообразного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,49 (1H, т, J=7,8 Гц), 7,33 (1H, дд, J=7,8, 1,0 Гц), 7,22 (1H, дд, J=7,8, 1,0 Гц), 4,16 (2H, кв, J=7,0 Гц), 1,59 (6H, с), 1,20 (3H, т, J=7,1 Гц), 0,00 (1H, д, J=3,4 Гц).
ESI-MS. Найдено: m/z[M+H]+ 272, 274.
2) Получение 3-(6-бромпиридин-2-ил)-2,3-диметилбутан-2-ола:
В атмосфере азота 13 мл 2M раствора метилмагнийиодида в диэтиловом эфире добавляли к раствору в диэтиловом эфире (20 мл) 2,72 г этил 2-(6-бромпиридин-2-ил)-2-метилпропионата, при этом используя охлаждение ледяной баней. Реакционную жидкую среду перемешивали при комнатной температуре в течение 3 часов и затем туда добавляли воду и водный 10% раствор фосфорной кислоты, экстрагировали диэтиловым эфиром, промывали водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым водным раствором, затем сушили над безводным сульфатом магния. После концентрирования при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат = от 9/1 до 8/1), получая 1,47 г указанного в заголовке соединения в виде бесцветного маслообразного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,53 (1H, т, J=7,8 Гц), 7,35 (1H, д, J=7,3 Гц), 7,31 (1H, д, J=7,8 Гц), 1,38 (6H, с), 1,09 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 258, 260.
3) Получение 2-аллил-1-[6-(2-гидрокси-1,1,2-триметилпропил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
74,5 мг указанного в заголовке соединения получали в виде желтого твердого вещества таким же способом, как и в примере с 9-2 по 9-3, в котором, однако, соединение, полученное в вышеуказанной реакции, использовали вместо 2-(6-бром-2-пиридил)-2-пропанола, использованного в примере 9-2.
1H-ЯМР (400 МГц, CDCl3) δ: 8,82 (1H, с), 7,81 (1H, т, J=7,9 Гц), 7,81 (1H, ушир.с), 7,45 (2H, д, J=8,4 Гц), 7,30 (1H, с), 6,93 (2H, д, J=9,2 Гц), 5,71 (1H, с), 5,08 (2H, с), 4,63 (2H, с), 3,24 (4H, с), 2,66 (4H, с), 2,41 (3H, с), 1,47 (6H, с), 1,09 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 543.
Пример 17
Получение 2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1-[6-(2-оксопирролидин-1-ил)пиридин-2-ил]-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
При охлаждении льдом 0,012 мл триэтиламина и 12,4 мг хлорангидрида 4-хлормасляной кислоты добавляли к раствору в тетрагидрофуране (1 мл) 20 мг соединения, полученного в примере 7, 2-аллил-1-(6-аминопиридин-2-ил)-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, и перемешивали при комнатной температуре в течение 1 часа. Воду добавляли к реакционной смеси, экстрагировали хлороформом и органический слой промывали насыщенным солевым водным раствором и сушили над безводным сульфатом натрия. Растворитель удаляли упариванием при пониженном давлении, полученный в результате остаток растворяли в 1 мл N,N-диметилформамида и туда же добавляли 5 мг трет-бутоксида калия и перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси добавляли насыщенный раствор хлорида аммония, экстрагировали этилацетатом и органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель удаляли упариванием при пониженном давлении и полученный в результате остаток очищали с помощью препаративной тонкослойной хроматографии (хлороформ/метанол = 10/1), получая 5 мг указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,82 (1H, с), 8,34 (1H, д, J=8,0 Гц), 7,85 (1H, т, J=8,0 Гц), 7,58 (1H, д, J=8,0 Гц), 7,49-7,34 (1H, ушир.м), 7,46 (2H, д, J=8,8 Гц), 6,92 (2H, д, J=8,8 Гц), 5,68 (1H, ддт, J=17,1, 10,2, 5,9 Гц), 5,04 (1H, дд, J=10,2, 1,0 Гц), 4,94 (1H, дд, J=17,1, 1,0 Гц), 4,76 (2H, д, J=5,9 Гц), 4,13-4,04 (2H, м), 3,30-3,20 (4H, м), 2,76-2,61 (6H, м), 2,42 (3H, с), 2,21-2,09 (2H, м).
ESI-MS. Найдено: m/z[M+H]+ 526.
Пример 18
Получение N-{[6-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил)пиридин-2-ил]метил}-N-метилметансульфонамида
1) Получение 2-аллил-1-[6-(гидроксиметил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
1,36 г имидазола и 1,81 г трет-бутил(хлор)диметилсилана добавляли к раствору в N,N-диметилформамиде (30 мл) 3,29 г соединения, полученного в примере 8-1, и перемешивали в течение ночи. Воду добавляли к реакционной жидкой среде и экстрагировали диэтиловым эфиром. Этот экстракт промывали насыщенным солевым водным раствором и сушили над безводным сульфатом магния. После концентрирования при пониженном давлении полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат = от 9/1 до 4/1) и растворитель удаляли упариванием при пониженном давлении. 40 мл толуола и 3,20 г м-хлорпербензойной кислоты (>65%) добавляли к остатку и перемешивали в течение 30 минут. 5,20 мл N,N-диизопропилэтиламина и 2,29 г 4-(4-метилпиперазин-1-ил)анилина добавляли к реакционной жидкой среде и перемешивали в течение ночи. Водный насыщенный раствор гидрокарбоната натрия добавляли к реакционной жидкой среде и экстрагировали этилацетатом. Этот экстракт сушили над безводным сульфатом магния, растворитель удаляли упариванием, остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ/этанол = от 100/1 до 100/3) и растворитель удаляли упариванием при пониженном давлении. 50 мл 4 н. хлористоводородной кислоты добавляли к остатку и перемешивали и затем раствор подщелачивали с помощью 4 н. водного раствора гидроксида натрия. Этот раствор экстрагировали смешанным раствором хлороформ/изопропанол (80/20), сушили над безводным сульфатом магния, растворитель удаляли упариванием и остаток кристаллизовали из этилацетата, получая 3,78 г указанного в заголовке соединения в виде желтого кристаллического вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,83 (1H, с), 7,86 (1H, т, J=6,0 Гц), 7,75 (1H, д, J=8,2 Гц), 7,46 (2H, д, J=8,6 Гц), 7,40 (1H, ушир.с), 7,22 (1H, д, J=7,6 Гц), 6,92 (2H, д, J=9,0 Гц), 5,71 (1H, ддт, J=16,8, 10,2, 5,9 Гц), 5,06 (1H, д, J=10,2 Гц), 4,96 (1H, д, J=16,8 Гц), 4,81 (2H, д, J=5,5 Гц), 4,71 (1H, д, J=5,9 Гц), 3,23 (4H, ушир.с), 3,14 (1H, т, J=5,5 Гц), 2,64 (4H, ушир.с), 2,40 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 473.
2) Получение 2-аллил-1-{6-[(метиламино)метил]пиридин-2-ил}-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
4,46 мл триэтиламина и 1,0 мл метансульфонилхлорида добавляли к раствору в тетрагидрофуране (120 мл) 3,78 г соединения, полученного в вышеприведенном примере 1, и перемешивали. 20 мл 2,0M раствора метиламина в тетрагидрофуране добавляли к реакционной жидкой среде и перемешивали в течении ночи. Воду добавляли к реакционной жидкой среде и экстрагировали этилацетатом. Этот экстракт промывали насыщенным солевым водным раствором, сушили над безводным сульфатом магния, концентрировали при пониженном давлении и полученный в результате остаток очищали с помощью стандартной колоночной хроматографии на силикагеле (гексан/этилацетат = от 50/50 до 0/100, далее хлороформ), получая 3,38 г указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,82 (1H, с), 7,81 (1H, т, J=7,8 Гц), 7,72 (1H, д, J=7,8 Гц), 7,46 (2H, д, J=8,8 Гц), 7,25 (1H, д, J=7,3 Гц), 6,92 (2H, д, J=9,3 Гц), 5,69 (1H, ддт, J=17,1, 10,2, 6,3 Гц), 5,02 (1H, дд, J=10,2, 1,5 Гц), 4,92 (1H, дд, J=17,1, 1,5 Гц), 4,75 (2H, д, J=6,3 Гц), 3,91 (2H, с), 3,21 (4H, т, J=4,9 Гц), 2,62 (4H, т, J=4,9 Гц), 2,51 (3H, с), 2,38 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 486.
3) Получение N-{[6-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-1-ил)пиридин-2-ил]метил}-N-метилметансульфонамида:
1,50 мл триэтиламина и 0,4 мл метансульфонилхлорида добавляли к раствору в тетрагидрофуране (50 мл) 1,70 г соединения, полученного на вышеприведенной стадии 2), и перемешивали. Воду добавляли к реакционной жидкой среде и экстрагировали этилацетатом. Этот экстракт промывали насыщенным солевым водным раствором, сушили над безводным сульфатом магния, концентрировали при пониженном давлении и получившийся в результате остаток кристаллизовали из 15 мл этилацетата и 10 мл этанола, получая 849 мг указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,83 (1H, с), 7,89 (1H, т, J=7,8 Гц), 7,81 (1H, д, J=8,3 Гц), 7,48 (1H, д, J=9,3 Гц), 7,47 (1H, ушир.с), 7,41 (2H, д, J=7,3 Гц), 6,93 (2H, д, J=8,8 Гц), 5,68 (1H, ддт, J=17,1, 10,2, 6,3 Гц), 5,03 (1H, д, J=10,2 Гц), 4,92 (1H, д, J=18,0 Гц), 4,75 (2H, д, J=6,3 Гц), 4,50 (2H, с), 3,38 (4H, ушир.с), 2,95 (3H, с), 2,92 (4H, ушир.с), 2,91 (3H, с), 2,58 (3H, с).
ESI-MS. Найдено: m/z[M+H]+ 564.
Пример 19
Получение 2-аллил-1-[6-(2-гидрокси-1,1-диметилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она

1) Получение 2-(6-бромпиридин-2-ил)-2-метилпропан-1-ола:
В бане с сухим льдом/в ацетоне, 100 мл 1,01M раствора диизобутилалюмогидрида в толуоле добавляли к раствору в толуоле (50 мл) 10,72 г соединения, полученного в примере 16-1, подогревали до комнатной температуры и перемешивали в течение 40 минут. При охлаждении льдом к реакционной жидкой среде добавляли водный насыщенный раствор хлорида аммония и органический слой отделяли. Этот слой промывали водным насыщенным раствором гидрокарбоната натрия и насыщенным солевым водным раствором, сушили над безводным сульфатом магния, концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат), получая 8,74 г указанного в заголовке соединения в виде бесцветного маслообразного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,52 (1H, т, J=7,8 Гц), 7,33 (1H, дд, J=7,8, 1,0 Гц), 7,27 (1H, д, J=7,8 Гц), 3,74 (2H, с), 1,32 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 230, 232.
2) Получение 2-аллил-1-[6-(2-гидрокси-1,1-диметилэтил)пиридин-2-ил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
7,45 г указанного в заголовке соединения получали таким же способом, как и в примере 9-2, для которого, однако, соединение, полученное в вышеприведенной реакции, использовали вместо 2-(6-бром-2-пиридинил)-2-пропанола, использованного в примере 9-2.
1H-ЯМР (400 МГц, CDCl3) δ: 8,93 (1H, с), 7,86 (1H, т, J=8,0 Гц), 7,60 (1H, д, J=8,8 Гц), 7,31 (1H, д, J=7,8 Гц), 5,67 (1H, ддт, J=17,1, 10,2, 6,3 Гц), 5,05 (1H, дд, J=10,2, 1,0 Гц), 4,92 (1H, дд, J=17,1, 1,5 Гц), 4,79 (2H, д, J=6,3 Гц), 3,78 (2H, с), 2,58 (3H, с), 1,37 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 372.
3) Получение 2-аллил-1-[6-(2-гидрокси-1,1-диметилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
1,4 г указанного в заголовке соединения получали в виде желтого твердого вещества тем же способом, как и в примере 9-3, для которого, однако, соединение, полученное в вышеприведенной реакции, использовали вместо 2-аллил-1-[6-(1-гидрокси-1-метилэтил)-2-пиридинил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, использованного в примере 9-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,83 (1H, с), 7,81 (1H, т, J=7,8 Гц), 7,52-7,41 (3H, м), 7,25 (1H, д, J=9,3 Гц), 6,92 (2H, дд, J=6,8, 2,4 Гц), 5,72 (1H, ушир.с), 5,14-4,96 (2H, ушир.м), 4,64 (2H, ушир.с), 3,79 (2H, д, J=6,3 Гц), 3,24 (4H, т, J=5,0 Гц), 2,65 (4H, ушир.с), 2,41 (3H, с), 1,38 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 515.
Пример 20
Получение 1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-2-(2-пропинил)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
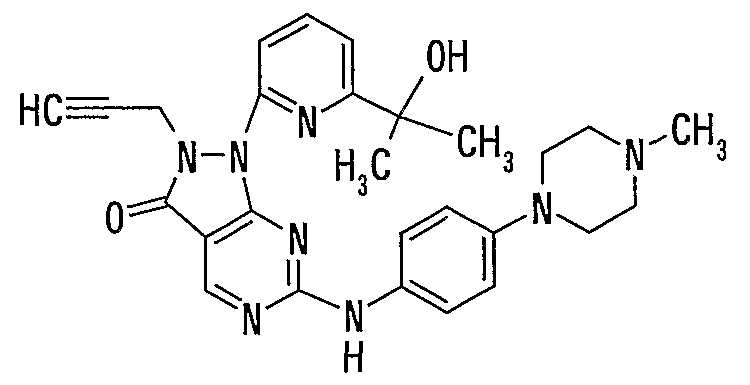
1) Получение 1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-6-(метилтио)-2-(2-пропинил)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
440 мг формиата аммония и 230 мг дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) добавляли к раствору в тетрагидрофуране (13,6 мл) 500 мг 2-аллил-1-[6-(1-гидрокси-1-метилэтил)-2-пиридинил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, полученного в примере 9, и перемешивали при 90°C в течение 3 часов. Реакционную жидкую среду охлаждали до комнатной температуры, к ней добавляли дистиллированную воду и экстрагировали смешанным раствором хлороформ/изопропанол (80/20). Этот экстракт сушили над безводным сульфатом натрия и растворитель удаляли упариванием при пониженном давлении, получали 770 мг черного аморфного вещества. 61,0 мг гидрида натрия добавляли к раствору полученного в результате соединения в N,N-диметилформамиде (14,0 мл) и перемешивали в течение 30 минут. 0,316 мл пропаргилбромида добавляли к реакционному раствору и перемешивали в течение 3,5 часов. Водный насыщенный раствор гидрокарбоната натрия и насыщенный солевой водный раствор добавляли к реакционной жидкой среде и экстрагировали смешанным раствором хлороформ/изопропанол (80/20). Этот экстракт сушили над безводным сульфатом натрия и растворитель удаляли упариванием при пониженном давлении, получая черное аморфное вещество. Полученное в результате аморфное вещество очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат), получая 254 мг указанного в заголовке соединения в виде белого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 8,94 (1H, с), 7,94 (2H, д, J=3,6 Гц), 7,43 (1H, т, J=3,6 Гц), 4,97 (2H, д, J=2,4 Гц), 2,62 (3H, с), 2,16 (1H, т, J=2,4 Гц), 1,59 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 357.
2) Получение 1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-2-(2-пропинил)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
7,3 мг указанного в заголовке соединения получали в виде желтого твердого вещества тем же способом, как и в примере 9-3, для которого, однако, соединение, полученное в вышеприведенной реакции, использовали вместо 2-аллил-1-[6-(1-гидрокси-1-метилэтил)-2-пиридинил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она, использованного в примере 9-3.
1H-ЯМР (400 МГц, CDCl3) δ: 8,83 (1H, с), 7,92 (1H, д, J=8,0 Гц), 7,87 (1H, дд, J=8,0, 8,0 Гц), 7,47 (2H, д, J=7,6 Гц), 7,35 (1H, д, J=8,0 Гц), 6,94 (2H, д, J=7,6 Гц), 4,89 (2H, д, J=2,0 Гц), 3,23 (4H, м), 2,63 (4H, м), 2,39 (3H, с), 2,13 (1H, т, J=2,0 Гц), 1,59 (6H, с).
ESI-MS. Найдено: m/z[M+H]+ 499.
Пример 21
Получение 2-аллил-1-[6-(3-метил-2-оксоимидазолидин-1-ил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она
1) Получение 2-аллил-1-(6-бромпиридин-2-ил)-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
Тем же способом, как и в примере 5-1, но используя 2,6-дибромпиридин вместо 2-иодпиридина, использованного в примере 5-1, 2,94 г указанного в заголовке соединения получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,94 (1H, с), 7,95 (1H, д, J=7,8 Гц), 7,73 (1H, т, J=8,0 Гц), 7,43 (1H, д, J=7,8 Гц), 5,69 (1H, ддт, J=17,1, 10,2, 6,3 Гц), 5,06 (1H, дд, J=10,2, 1,2 Гц), 5,00 (1H, д, J=17,1 Гц), 4,88 (2H, д, J=6,3 Гц), 2,60 (3H, с).
2) Получение 2-аллил-1-[6-(3-метил-2-оксоимидазолидин-1-ил)пиридин-2-ил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
1-Метилимидазолидин-2-он (96 мг), иодид меди (76 мг), карбонат калия (110 мг) и N,N'-диметилэтан-1,2-диамин (85 мкл) добавляли к раствору в диоксане (5 мл) 2-аллил-1-(6-бромпиридин-2-ил)-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она (150 мг) и перемешивали в течение ночи в герметичном сосуде при нагревании до 100°C.
Реакционную жидкую среду охлаждали, к ней добавляли водный раствор аммиака и трижды экстрагировали хлороформом. Органический слой промывали насыщенным солевым водным раствором, сушили над безводным сульфатом магния, фильтровали и растворитель удаляли упариванием. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, получая 136,4 мг указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,92 (1H, с), 8,26 (1H, д, J=8,4 Гц), 7,81 (1H, дд, J=8,4, 7,6 Гц), 7,41 (1H, д, J=7,6 Гц), 5,66 (1H, ддд, J=16,8, 10,0, 6,4 Гц), 5,06 (1H, д, J=10,0 Гц), 4,95 (1H, д, J=16,8 Гц), 4,80 (2H, д, J=6,4 Гц), 4,01 (2H, д, J=8,0 Гц), 3,51 (1H, т, J=8,0 Гц), 2,94 (3H, с), 2,57 (3H, с).
ESI-MS. Найдено: m/z[M+H] 398.
3) Получение 2-аллил-1-[6-(3-метил-2-оксоимидазолидин-1-ил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-она:
Тем же способом, что и в примере 5-2, но используя 4-(4-метилпиперазин-1-ил)анилин вместо [5-амино-2-(4-метилпиперазин-1-ил)фенил]метанола, использованного в примере 5-2, и используя 2-аллил-1-[6-(3-метил-2-оксоимидазолидин-1-ил)пиридин-2-ил]-6-(метилтио)-1,2-дигидро-3H-пиразоло[3,4-d]пиримидин-3-он вместо 2-аллил-6-(метилтио)-1-пиридин-2-ил-3H-пиразоло[3,4-d]пиримидин-3-она, 115,6 мг указанного в заголовке соединения получали в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 8,81 (1H, с), 8,22 (1H, д, J=8,4 Гц), 7,78 (1H, дд, J=8,4, 8,0 Гц), 7,46 (2H, д, J=8,0 Гц), 7,40 (1H, д, J=8,0 Гц), 6,90 (2H, д, J=8,0 Гц), 5,68 (1H, ддд, J=16,8, 10,4, 6,0 Гц), 5,04 (1H, д, J=10,4 Гц), 4,95 (1H, д, J=16,8 Гц), 4,74 (2H, д, J=6,0 Гц), 4,02 (2H, т, J=8,4 Гц), 3,49 (2H, т, J=8,4 Гц), 3,02 (4H, м), 2,94 (3H, с), 2,60 (4H, м), 2,37 (3H, с).
Масс-сп. с ионизацией электрораспылением, найдено: m/z[M+H] 541.
Применяемость в производственных условиях
Соединения по изобретению имеют превосходное ингибирующее воздействие на Weel-киназу и поэтому применимы в области медицины, особенно в лечении различных злокачественных опухолей.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПИРИМИДИНА, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩЕЕ ДЕЙСТВИЕ НА РОСТ РАКОВЫХ КЛЕТОК, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2834201C1 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСОИЗОХИНОЛИНА | 2017 |
|
RU2772226C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK II ТИПА И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2810338C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2525116C2 |
| 1Н-ПИРАЗОЛО[3,4-B]ПИРИДИНЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2013 |
|
RU2689141C2 |
| АНТАГОНИСТЫ ЕР4 | 2016 |
|
RU2761341C2 |
| ПРОИЗВОДНОЕ СУЛЬФОНАМИДА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2014 |
|
RU2667520C9 |
Изобретение описывает соединение общей формулы (I)  где А1 выбирают из следующей формулы (аа1):
где А1 выбирают из следующей формулы (аа1): 
R1c представляет собой атом водорода, низшую алкенильную группу или группу -Q3-A3(R1d)R1e; А3 представляет собой метин или низшую алкильную группу; Q3 представляет собой одинарную связь; R1d и R1e представляют собой независимо атом водорода, гидроксильную группу, низшую алкильную группу или гидроксилсодержащую низшую алкильную группу, или вместе образуют низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода; R1 представляет собой низшую алкенильную группу или низшую алкинильную группу; R2 представляет собой фенильную, пиридильную или тиенильную группу, которая может содержать группу -Q4-A4(R1g)R1h; А4 представляет собой атом азота, низшую алкильную группу, необязательно замещенную гидрокси-низшей алкильной группой, или метиновую группу, необязательно замещенную атомом галогена, гидроксильной группой, низшей алкильной группой или гидрокси-низшей алкильной группой; Q4 представляет собой одинарную связь или низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода; R1g и R1h представляют собой независимо атом водорода, низшую алкильную группу или низшую алкилсульфонильную группу; R5 и R6 представляют собой независимо атом водорода, низшую алкильную группу или гидроксилсодержащую низшую алкильную группу, или его фармацевтически приемлемая соль. Также изобретение описывает фармацевтическую композицию на основе соединений формулы I, обладающую противораковой активностью, противораковый агент, комбинированное лекарственное средство, а кроме того, сенсибилизатор облучения, включающий фармацевтическую композицию. Технический результат: получены и описаны новые соединения, которые обладают превосходным ингибирующим воздействием на Well-киназу и вследствие этого применимы в области медицины, особенно при лечении различных злокачественных опухолей. 8 н. и 5 з.п. ф-лы, 3 табл.
1. Соединение общей формулы (I):
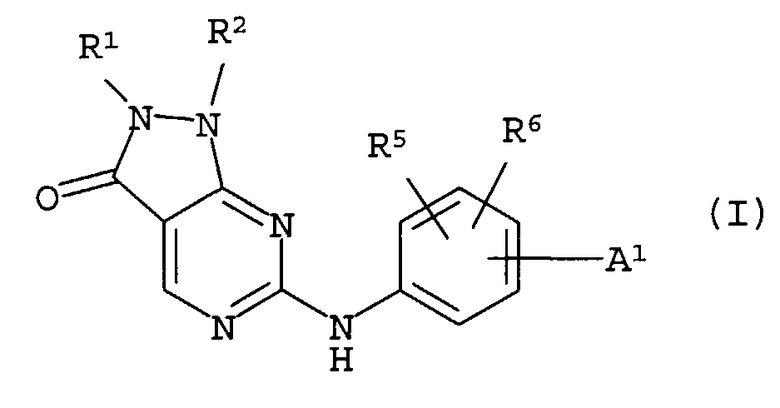
где А1 выбирают из следующей формулы (аа1):

R1c представляет собой атом водорода, низшую алкенильную группу или группу -Q3-A3(R1d)R1e;
А3 представляет собой метин или низшую алкильную группу;
Q3 представляет собой одинарную связь;
R1d и R1e представляют собой независимо атом водорода, гидроксильную группу, низшую алкильную группу или гидроксилсодержащую низшую алкильную группу, или вместе образуют низшую алкиленовую группу, в которой одна или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода;
R1 представляет собой низшую алкенильную группу или низшую алкинильную группу;
R2 представляет собой фенильную, пиридильную или тиенильную группу, которая может содержать группу -Q4-A4(R1g)R1h;
А4 представляет собой атом азота, низшую алкильную группу, необязательно замещенную гидрокси-низшей алкильной группой, или метиновую группу, необязательно замещенную атомом галогена, гидроксильной группой, низшей алкильной группой или гидрокси-низшей алкильной группой;
Q4 представляет собой одинарную связь или низшую алкиленовую группу, в которой одна, или две, или несколько метиленовых групп, составляющих низшую алкиленовую группу, могут быть независимо заменены атомом кислорода;
R1g и R1h представляют собой независимо атом водорода, низшую алкильную группу или низшую алкилсульфонильную группу;
R5 и R6 представляют собой независимо атом водорода, низшую алкильную группу или гидроксилсодержащую низшую алкильную группу, или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 представляет собой низшую алкенильную группу.
3. Соединение по п.2 или его фармацевтически приемлемая соль, где R1 представляет собой аллильную группу.
4. Соединение по п.1 или его фармацевтически приемлемая соль, где R2 представляет собой фенильную или пиридильную группу, содержащую группу -Q4-A4(R1g)R1h.
5. Соединение по п.1 или его фармацевтически приемлемая соль, где R1c представляет собой атом водорода или группу -Q3-A3(R1d)R1e, и в группе -Q3-A3(R1d)R1e,
А3 представляет собой метиновую группу, Q3 представляет собой одинарную связь, и R1d и R1e представляет собой независимо атом водорода или низшую алкильную группу.
6. Соединение по п.1 или его фармацевтически приемлемая соль, представляющее собой:
3-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-1-ил)-N,N-диметилбензамид, 2-аллил-1-[3-(1-гидрокси-3-метилэтил)фенил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он, 2-аллил-1-[3-(диметиламинометил)фенил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он, 2-аллил-6-{[3-гидроксиметил-4-(4-метилпиперазин-1-ил)фенил]амино}-1-пиридин-2-ил-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он, 2-аллил-1-(6-аминопиридин-2-ил)-6-[{4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он, 2-аллил-1-[6-(3-гидрокси-1-метилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-6-{[4-(4-этилпиперазин-1-ил)фенил]амино}-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он, 6-{[4-(4-ацетилпиперазин-1-ил)фенил]амино}-2-аллил-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он, 2-аллил-6-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]фенил}амино)-1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-1,2-дигидро-3Н-пиразоло[3,4-4]пиримидин-3-он,
2-аллил-1-[6-(2-гидрокси-2-метилпропил)пиридин-2-ил]-6-{[4-(1-метилпиперидин-4-ил)фенил]амино}-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он,
2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-1-[6-(2-оксопирролидин-1-ил)пиридин-2-ил]-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он,
N-{[6-(2-аллил-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-3-оксо-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-1-ил)пиридин-2-ил]метил}-N-метилметансульфонамид или
1-[6-(1-гидрокси-1-метилэтил)пиридин-2-ил]-6-{[4-(4-метилпиперазин-1-ил)фенил]амино}-2-(2-пропинил)-1,2-дигидро-3Н-пиразоло[3,4-d]пиримидин-3-он.
7. Фармацевтическая композиция, обладающая противораковой активностью, включающая терапевтически эффективное количество соединения по п.1 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель или разбавитель.
8. Противораковый агент, включающий фармацевтическую композицию по п.7.
9. Комбинированное лекарственное средство для одновременного, раздельного или последовательного введения при лечении злокачественной опухоли, включающее два отдельных лекарственных средства (а) и (b):
(a) лекарственное средство, включающее, совместно с фармацевтически приемлемым носителем или разбавителем, соединение указанной выше формулы (I) или его фармацевтически приемлемую соль; и
(b) лекарственное средство, включающее, совместно с фармацевтически приемлемым носителем или разбавителем, один противораковый агент, выбранный из группы, состоящей из противораковых алкилирующих агентов, противораковых антиметаболитов, противораковых антибиотиков, противораковых агентов растительного происхождения, противораковых координационных комплексных соединений платины, противораковых производных камптотецина, противораковых ингибиторов тирозин-киназы, моноклональных антител, интерферонов, модификаторов биологической чувствительности и других противораковых агентов или их фармацевтически приемлемых солей, в числе которых противораковые алкилирующие агенты представляют собой N-оксид азотистого иприта, циклофосфамид, ифосфамид, мелфалан, бусульфан, митобронитол, карбоквон, тиотепу, ранимустин, нимустин, темозоломид и кармустин;
противораковые антиметаболиты представляют собой метотрексат, 6-меркаптопуринрибозид, меркаптопурин, 5-фторурацил, тегафур, доксифлуридин, кармофур, цитарабин, цитарабина окфосфат, эноцитабин, S-1, гемцитабин, флударабин и пеметрексед динатрия;
противораковые антибиотики представляют собой актиномицин D, доксорубицин, даунорубицин, неокарциностатин, блеомицин, пепломицин, митомицин С, акларубицин, пирарубицин, эпирубицин, зиностатин стималамер, идарубицин, сиролимус и вальрубицин;
противораковые агенты растительного происхождения представляют собой винкристин, винбластин, виндезин, этопозид, собузоксан, доцетаксел, паклитаксел и винорелбин;
противораковые координационные комплексные соединения платины представляют собой цисплатин, карбоплатин, недаплатин и оксалиплатин;
противораковые производные камптотецина представляют собой иринотекан, топотекан и камптотецин;
противораковые ингибиторы тирозин-киназы представляют собой гефитиниб, иматиниб и эрлотиниб;
моноклональные антитела представляют собой цетуксимаб, бевацизумаб, ритуксимаб, алемтузумаб и трастузумаб;
интерфероны представляют собой интерферон α, интерферон α-2а, интерферон α-2b, интерферон β, интерферон γ-1а и интерферон γ-n1, модификаторы биологической чувствительности представляют собой крестин, лентинан, сизофиран, пицибанил или убенимекс, и
другие противораковые агенты представляют собой митоксантрон, L-аспарагиназу, прокарбазин, дакарбазин, гидроксикарбамид, пентостатин, третиноин, алефасепт, дарбепоэтин альфа, анастрозол, экземестан, бикалутамид, леупрорелин, флутамид, фулвестрант, пегаптаниб октанатрия, денилейкин дифтитокс, алдеслейкин, тиротропин альфа, триоксид мышьяка, бортезомиб, капецитабин и гозерелин.
10. Фармацевтическая композиция, включающая, совместно с фармацевтически приемлемым носителем или разбавителем, соединение по п.1 или его фармацевтически приемлемую соль; и противораковый агент, выбранный из группы, состоящей из противораковых алкилирующих агентов, противораковых антиметаболитов, противораковых антибиотиков, противораковых агентов растительного происхождения, противораковых координационных комплексных соединений платины, противораковых производных камптотецина, противораковых ингибиторов тирозин-киназы, моноклональных антител, модификаторов биологической чувствительности и других противораковых агентов, для которых определение каждого противоракового агента такое же, как определено в п.9, или их фармацевтически приемлемых солей.
11. Сенсибилизатор облучения, включающий фармацевтическую композицию по п.7.
12. Сенсибилизатор для противоракового агента, включающий фармацевтическую композицию по п.7, в которой противораковый агент выбирают из группы, состоящей из противораковых алкилирующих агентов, противораковых антиметаболитов, противораковых антибиотиков, противораковых агентов растительного происхождения, противораковых координационных комплексных соединений платины, противораковых производных камптотецина, противораковых ингибиторов тирозинкиназы, моноклональных антител, модификаторов биологической чувствительности и других противораковых агентов, для которых определение каждого противоракового агента такое же, как определено в п.9, или их фармацевтически приемлемых солей.
13. Применение соединения по п.1 или его фармацевтически приемлемой соли для получения противоракового агента.
| MACHON А | |||
| ЕТ AL | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| POLISH JOURNAL OF PHARMACOLOGY AND PHARMACY, vol.28, no.5, 1976, pages 511-520 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| WANG Y | |||
| ЕТ AL | |||
| Веникодробильный станок | 1921 |
|
SU53A1 |
| CANCER RESEARCH, vol.61, 2001, | |||

