Область техники, к которой относится изобретение
[0001]
Настоящее изобретение относится к фармацевтическому препарату, в частности, к новому производному оксоизохинолина, обладающему BTK-ингибирующим действием, или его фармацевтически приемлемой соли.
Уровень техники
[0002]
Тирозинкиназа Брутона (BTK) является членом семейства Tec нерецепторных тирозинкиназ, и является важным сигнальным ферментом, который экспрессируется во всех типах гемопоэтических клеток, за исключением T лимфоцитов и натуральных киллерных клеток.
BTK является важным регулирующим фактором, связанным с выживанием, дифференцировкой, пролиферацией и активацией B-клеток, и играет важную роль в передаче сигналов B-клеток (непатентные документы 1 и 2). В-клеточный рецептор (BCR) клеточной поверхности передает сигналы в клетки через BTK, находящийся ниже BCR, и поэтому считается, что аномальная активация сигнального пути B-клеток ускоряет пролиферацию и выживание раковых клеток В-клеточной лимфомы, хронического лимфоцитарного лейкоза и тому подобное (непатентный документ 3). Известно, что BTK также играет важную роль в сигнальном пути большого числа других клеток, считается, что BTK вовлечена в аллергические заболевания, аутоиммунные заболевания, воспалительные заболевания и тому подобное (непатентный документ 1).
Например, известно, что BTK играет важную роль в передаче сигнала высокоаффинного IgE-рецептора (FcεRI) в тучных клетках, и дегрануляция уменьшается, а продукция провоспалительных цитокинов снижается в тучных клетках с BTK-дефицитом (непатентный документ 4). Предполагается, что BTK участвует в системной красной волчанке (SLE) в тесте на BTK-дефицитных мышах (непатентный документ 5). Кроме того, BTK мутантные мыши проявляют устойчивость к возникновению коллаген-индуцированного артрита (непатентный документ 6).
Ибрутиниб является необратимым BTK-ингибитором и используется для лечения В-клеточной опухоли в качестве противоопухолевого лекарственного средства. Недавно было обнаружено, что толерантность к ибрутинибу была получена благодаря C481S-мутации BTK при лечении ибрутинибом (непатентный документ 7). Также сообщается, что p65 BTK, который является изоформой BTK, экспрессируется ниже RAS сигнального пути при солидном раке, отличном от рака крови, и вовлечен в пролиферацию солидного рака, такого как клетка рака толстой кишки (непатентный документ 8). Следовательно, соединение, обладающее BTK ингибирующей активностью, является полезным для лечения заболеваний с вовлечением сигнального пути BTK, например, рака, В-клеточной лимфомы и хронического лимфоцитарного лейкоза, а также солидного рака, при котором экспрессируется p65BTK. Кроме того, оно является полезным для лечения аллергических заболеваний, аутоиммунных заболеваний и воспалительных заболеваний.
Кроме того, BTK-ингибитор, который является эффективным для лечения рака с BTK-мутацией и толерантен к необратимому BTK-ингибитору, такому как ибрутиниб, является необходимым.
[0003]
Авторы настоящего изобретения сообщили о производном триазина в виде соединения, обладающего BTK-ингибирующей активностью (патентный документ 1 и 2). Соединения, аналогичные соединениям по настоящему изобретению, также раскрыты (патентный документ 3 и 4). Но производное оксоизохинолина настоящего изобретения здесь не раскрыто.
Документ(ы) предшествующего уровня техники
Патентный документ(ы)
[0004]
[Патентный документ 1] WO2013/133367.
[Патентный документ 2] WO2015/012149.
[Патентный документ 3] WO2013/157022.
[Патентный документ 4] CN104211703.
Непатентный документ(ы)
[0005]
[Непатентный документ 1] Satterthwaite,A.B. and Witte, O.N., Immunol.Rev., 2000, 175, 120-127.
[Непатентный документ 2] Kurosaki,T., Curr.Opin.Immunol., 2000, 12, 276-281.
[Непатентный документ 3] Davis,R.E.et al., Nature, 2010, 463, 88-92.
[Непатентный документ 4] Ellmeier,W., et al., FEBS J., (2011), 278, 1990-2000.
[Непатентный документ 5] Halcomb,K.E., Mol.Immunol., 2008, 46(2), 233-241.
[Непатентный документ 6] Jansson, L. and Holmdahl, R., Clin. Exp. Immunol., 1993, 94, 459-465.
[Непатентный документ 7] Cheng,S. et al., Leukemia, 2015, 29, 895-900.
[Непатентный документ 8] Grassili.E., et al., Oncogene, 2016, 35, 4368-4378.
Описание изобретения
Задача, решаемая изобретением
[0006]
Задачей настоящего изобретения является создание фармацевтического препарата, в частности нового производного оксоизихинолина, обладающего BTK-ингибирующим действием, или его фармацевтически приемлемой соли.
Средства для решения задачи
[0007]
Настоящее изобретение достигается посредством (1)-(6):
(1) производное оксоизохинолина формулы (I):
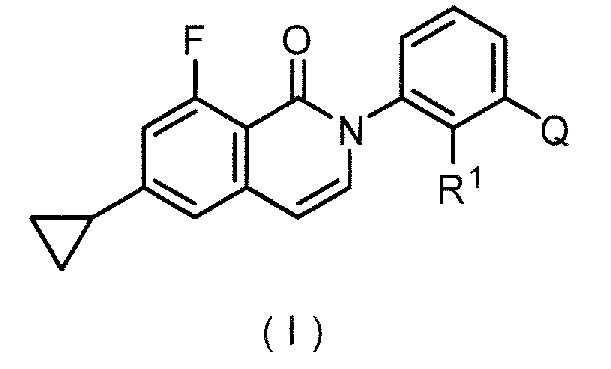 ,
,
где R1 представляет собой необязательно замещенный низший алкил,
Q представляет собой структуру, выбранную из следующих структур (а), (b) и (с);
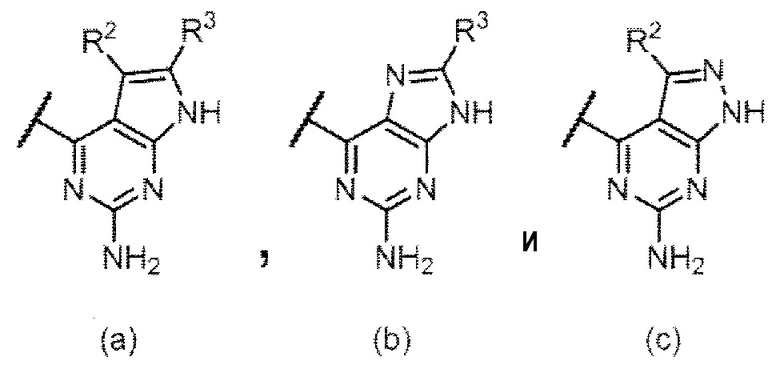 ,
,
где R2 и R3 независимо представляют собой атом водорода, необязательно замещенную низшую алкильную группу, необязательно замещенную циклоалкильную группу, необязательно замещенную арильную группу, необязательно замещенную гетероарильную группу или необязательно замещенную гетероциклическую группу,
или его фармацевтически приемлемая соль;
(2) производное оксоизохинолина по (1) выше, где Q представляет собой структуру (а) и R1 представляет собой гидроксиметильную группу,
[0008]
(3) производное оксоизохинолина формулы (Ia):
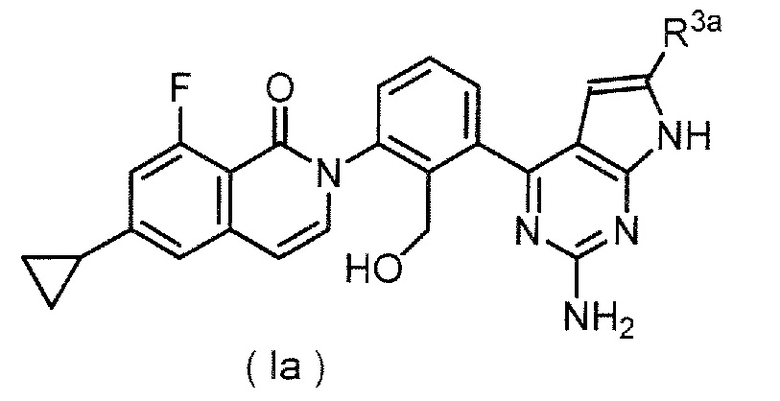 ,
,
где R3a представляет собой необязательно замещенную тетрагидропиридильную группу,
или его фармацевтически приемлемая соль;
(4) производное оксоизохинолина формулы (Ia), где заместитель тетрагидропиридиновой группы выбран из группы, состоящей из оксетанильной группы, ацетильной группы, пропионильной группы, морфолиноацетильной группы, диметилкарбамоильной группы, пирролидинкарбонильной группы, метилсульфонильной группы и изопропилсульфонильной группы,
или его фармацевтически приемлемая соль;
(5) производное оксоизохинолина формулы (Ib);
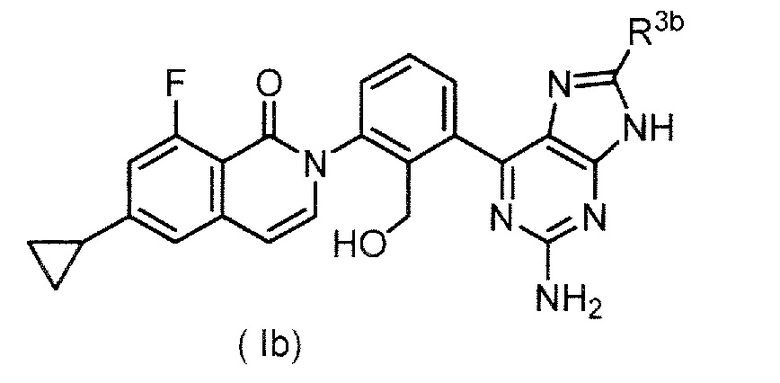 ,
,
где R3b представляет собой фенильную группу, необязательно замещенную низшей алкильной группой
или его фармацевтически приемлемая соль;
[0009]
(6) соединение, выбранное из группы, состоящей из следующих соединений;
2-[3-(2-амино-6-фенил-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 1)
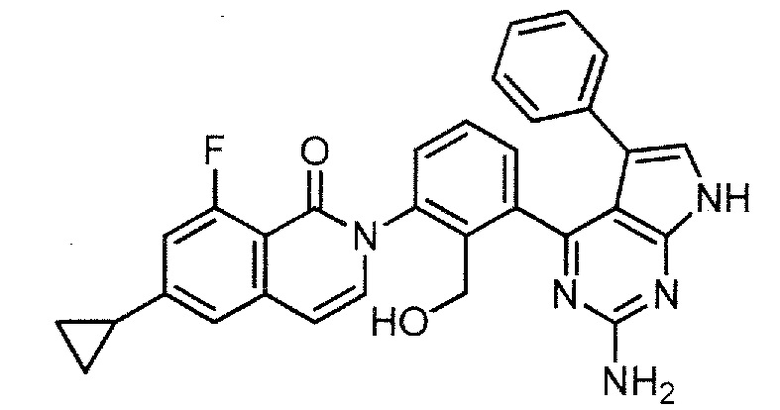
2-[3-(2-амино-8-фенил-9H-пурин-6-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 2)
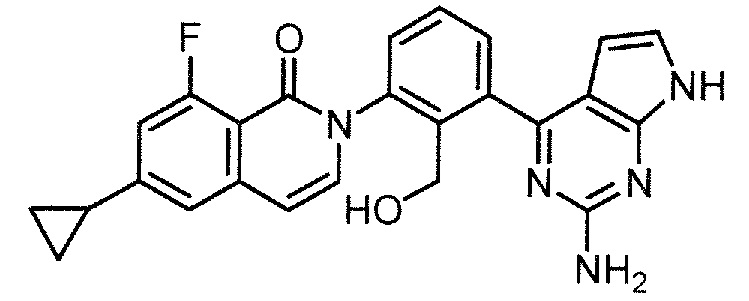
2-[3-(6-амино-3-фенил-1H-пиразоло[3,4-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 3)
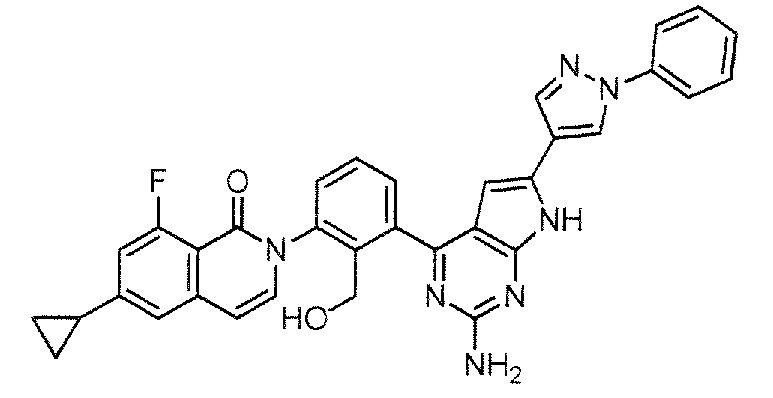
2-[3-(2-амино-9H-пурин-6-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 4)
2-[3-(6-амино-1H-пиразоло[3,4-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 5)
[0010]
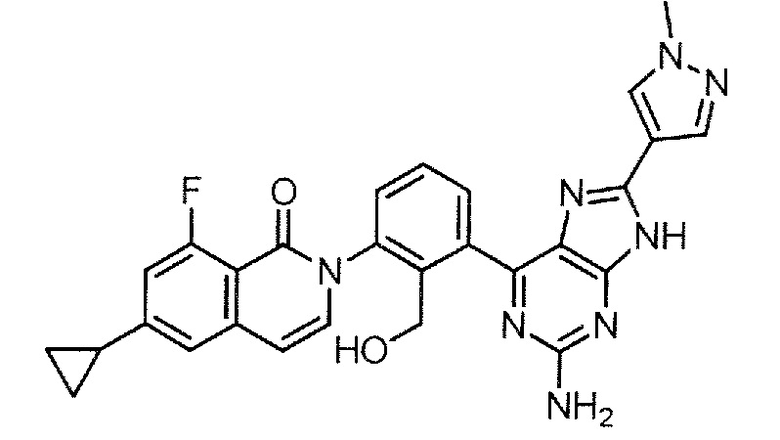
2-[3-(2-амино-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 6)
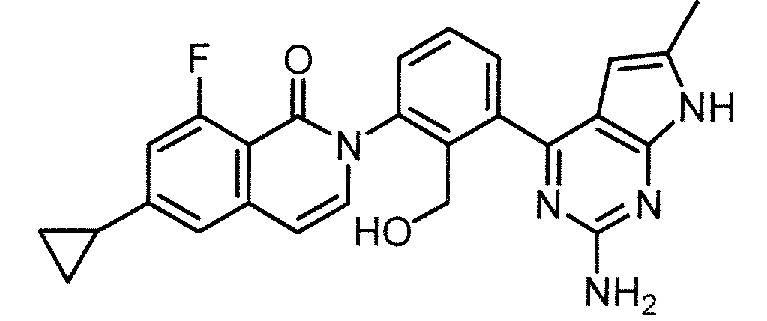
2-[3-(2-амино-6-метил-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 7)
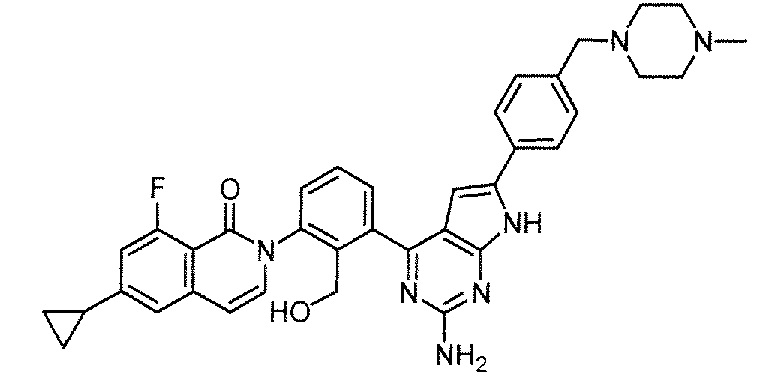
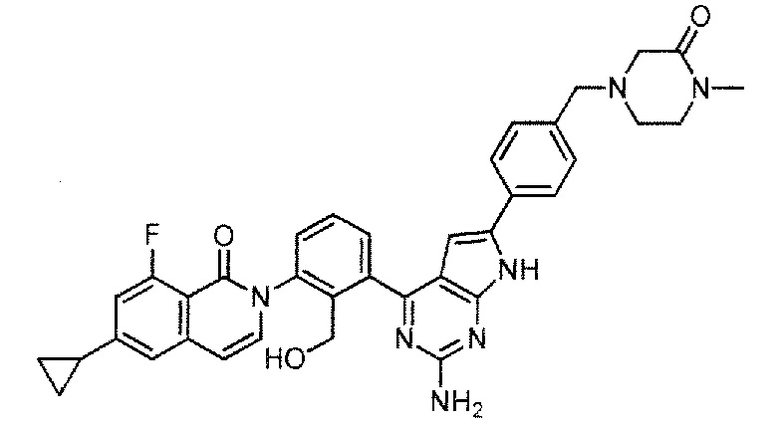
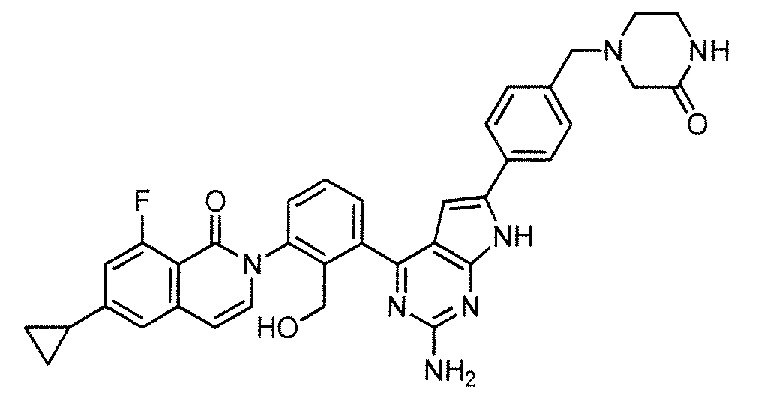
2-[3-(2-амино-6-{4-[(4-метилпиперазин-1-ил)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 8)
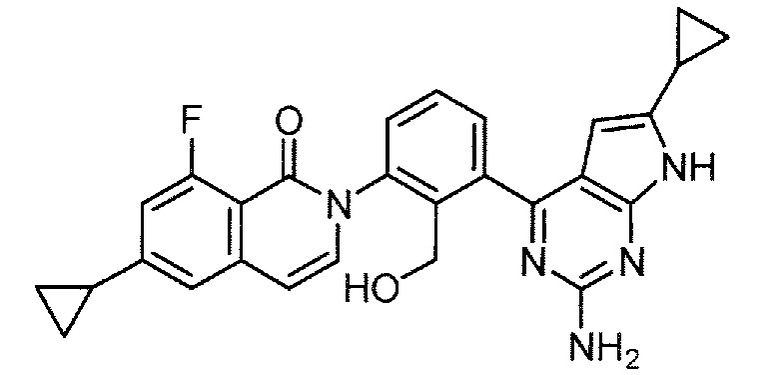
2-[3-(2-амино-6-циклопропил-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 9)
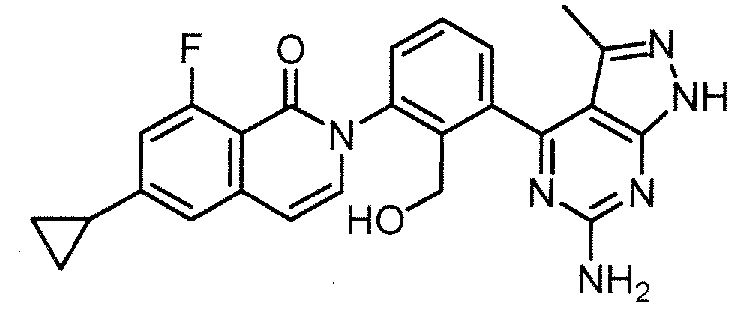
2-[3-(6-амино-3-метил-1H-пиразоло[3,4-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 10)
[0011]
2-{3-[2-амино-6-(гидроксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 11)
2-[3-(2-амино-8-циклопропил-9H-пурин-6-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 12)
2-{3-[2-амино-6-(1-метил-1H-пиразол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 13)
2-{3-[2-амино-6-(2-метоксифенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 14)
2-{3-[2-амино-6-(3-метоксифенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 15)
[0012]
2-{3-[2-амино-6-(4-метоксифенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 16)
2-{3-[2-амино-6-(пиридин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 17)
2-{3-[2-амино-8-(3-метоксифенил)-9H-пурин-6-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 18)
2-{3-[2-амино-6-(пиридин-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 19)
2-{3-[6-амино-3-(4-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 20)
[0013]
2-{3-[6-амино-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 21)
2-{3-[6-амино-3-(3-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 22)
2-(3-{2-амино-6-[1-(оксетан-3-ил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 23)
2-{3-[2-амино-8-(2-метоксифенил)-9H-пурин-6-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 24)
2-{3-[2-амино-8-(пиридин-3-ил)-9H-пурин-6-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 25)
[0014]
2-(3-{2-амино-6-[4-(морфолинометил)фенил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 26)
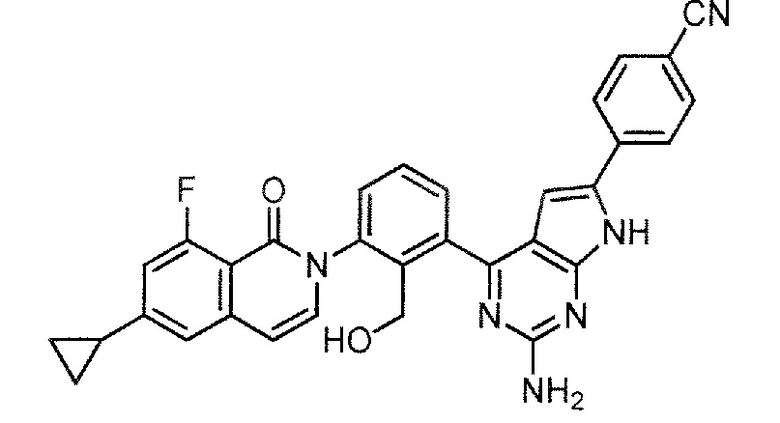
4-{2-амино-4-[3-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-2-(гидроксиметил)фенил]-7H-пирроло[2,3-d]пиримидин-6-ил}бензонитрил (Пример 27)
2-[3-(2-амино-5-фенил-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 28)
2-{3-[2-амино-6-(3-фторфенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 29)
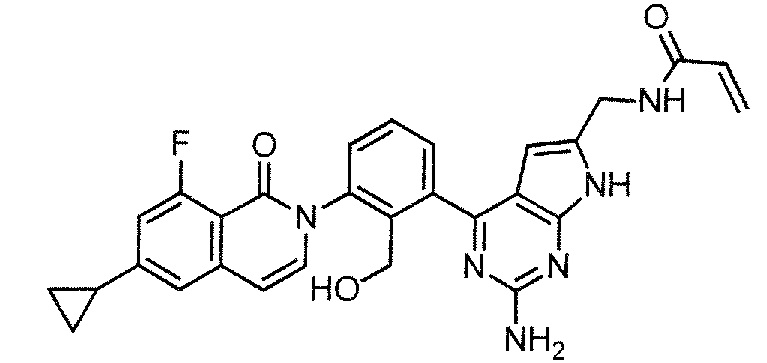
N-({2-амино-4-[3-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-2-(гидроксиметил)фенил]-7H-пирроло[2,3-d]пиримидин-6-ил}метил)акриламид (Пример 30)
[0015]
2-{3-[2-амино-8-(1-метил-1H-пиразол-4-ил)-9H-пурин-6-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 31)
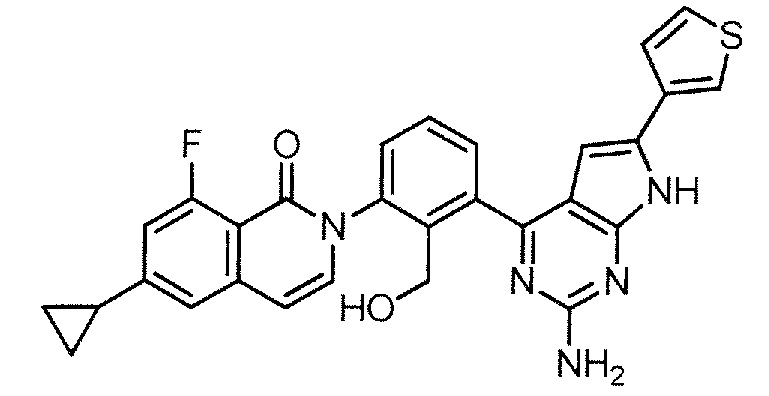
2-{3-[2-амино-6-(тиофен-3-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 32)
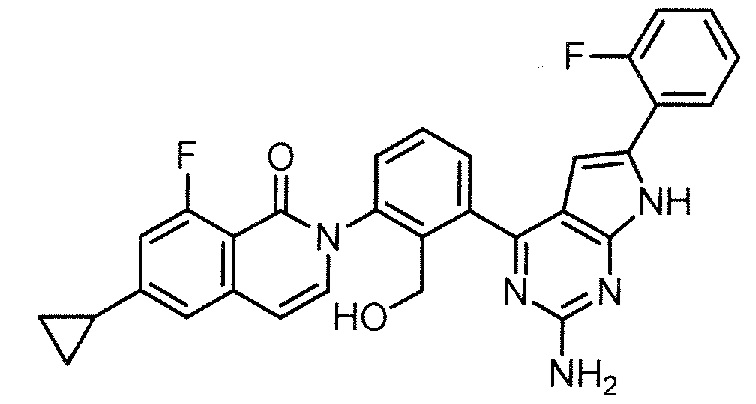
2-{3-[2-амино-6-(2-фторфенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 33)
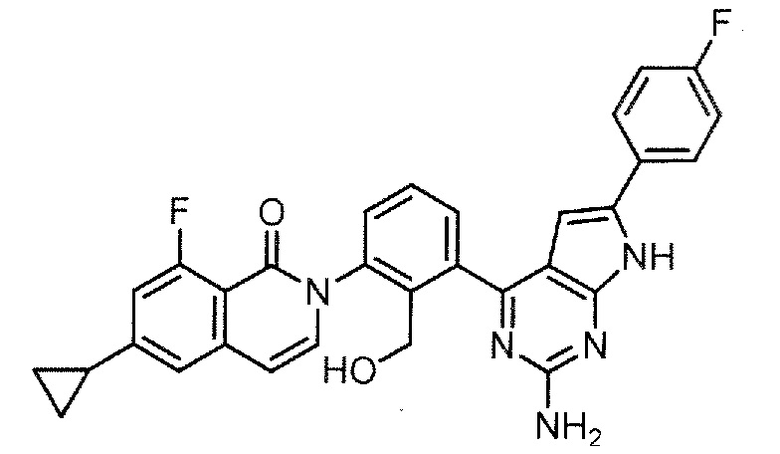
2-{3-[2-амино-6-(4-фторфенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 34)
2-{3-[2-амино-6-(2,4-дифторфенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 35)
[0016]
2-{3-[2-амино-6-(3,4-дифторфенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 36)
2-(3-{2-амино-6-[4-(трифторметил)фенил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 37)
2-(3-{2-амино-6-[4-(трифторметокси)фенил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 38)
2-{3-[2-амино-6-(аминометил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 39)
2-(3-{2-амино-6-[3-(трифторметил)фенил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 40)
[0017]
2-(3-{2-амино-6-[4-(метилсульфонил)фенил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 41)
2-{3-[2-амино-6-(6-фторпиридин-2-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 42)
2-{3-[2-амино-6-(2-фторпиридин-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 43)
2-{3-[2-амино-6-(3,5-дифторфенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 44)
2-{3-[2-амино-6-(5-фторпиридин-2-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 45)
[0018]
2-{3-[2-амино-6-(5-фторпиридин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 46)
2-(3-{2-амино-6-[6-(метиламино)пиридин-3-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 47)
2-{3-[2-амино-6-(6-морфолинопиридин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 48)
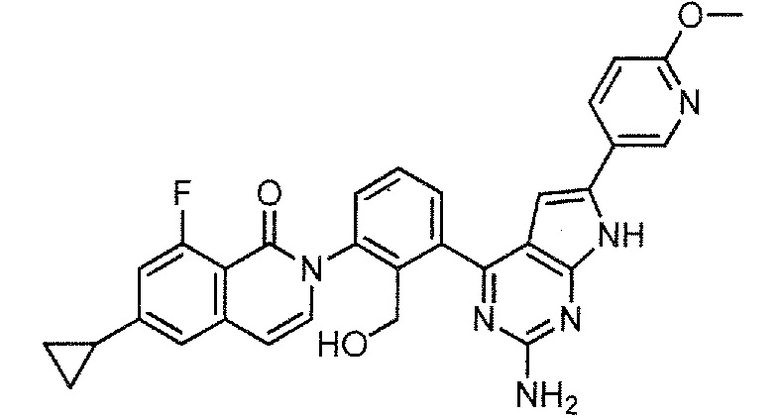
2-{3-[2-амино-6-(2-метоксипиридин-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 49)
2-(3-{2-амино-6-[2-(метиламино)пиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 50)
[0019]
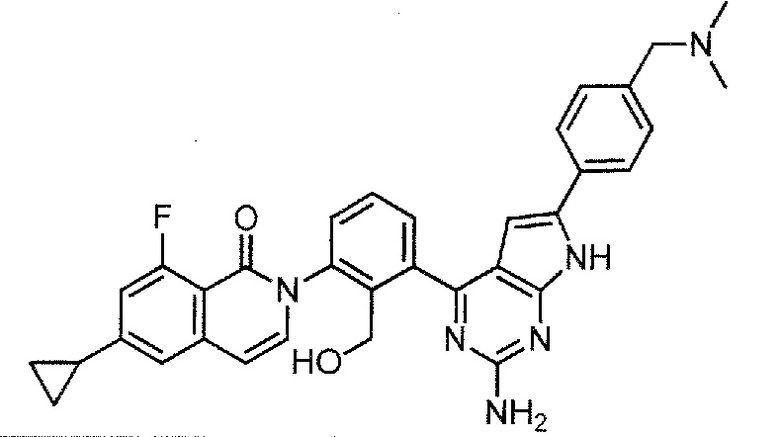
2-[3-(2-амино-6-{4-[(диметиламино)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 51)
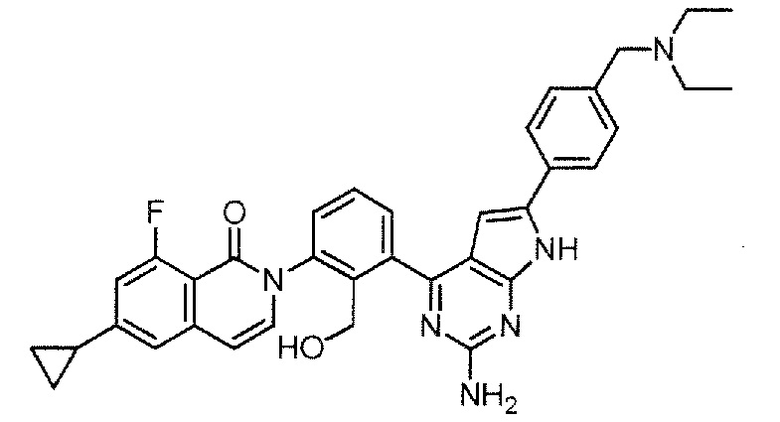
2-[3-(2-амино-6-{4-[(диэтиламино)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 52)
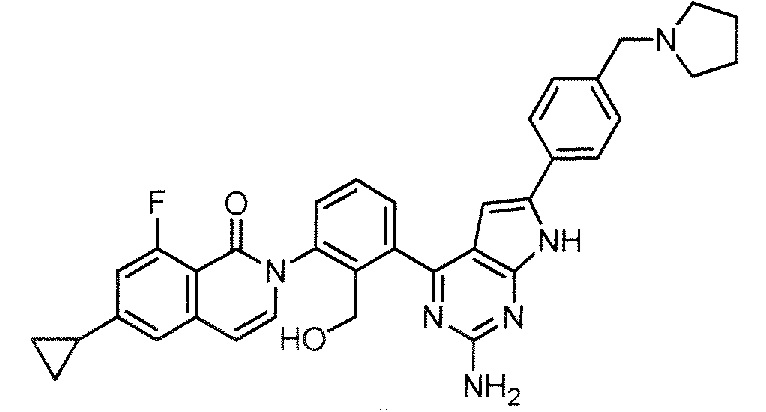
2-(3-{2-амино-6-[4-(пирролидин-1-илметил)фенил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 53)
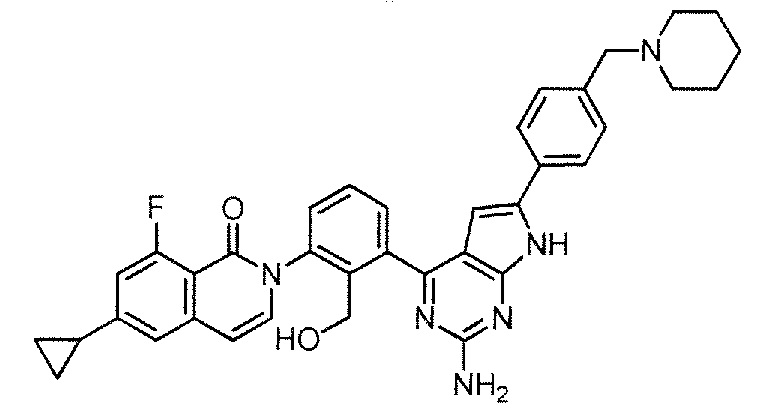
2-(3-{2-амино-6-[4-(пиперидин-1-илметил)фенил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 54)
2-[3-(2-амино-6-{4-[(4-метил-3-оксопиперазин-1-ил)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 55)
[0020]
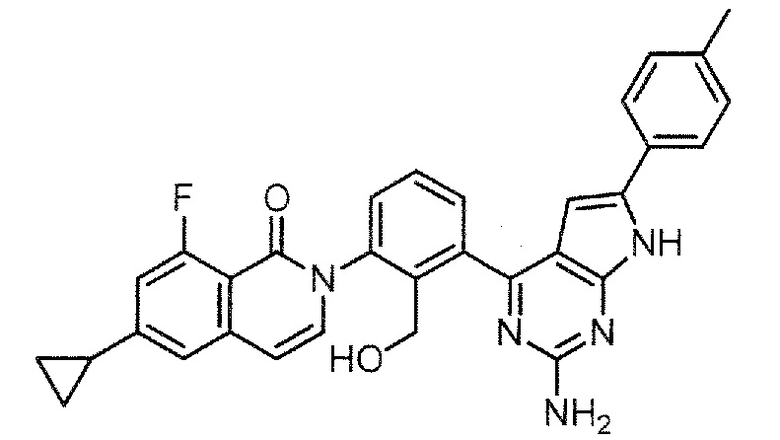
2-{3-[2-амино-6-(п-толил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 56)
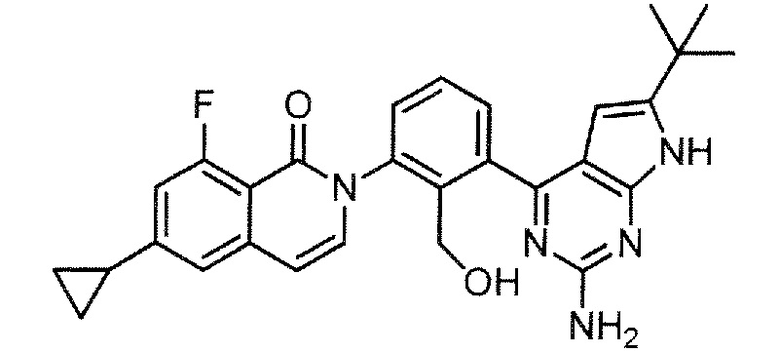
2-{3-[2-амино-6-(трет-бутил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 57)
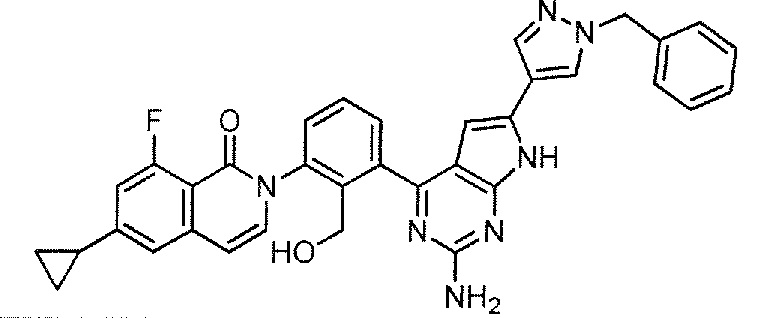
2-{3-[2-амино-6-(1-бензил-1H-пиразол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 58)
2-(3-{2-амино-6-[6-(диметиламино)пиридин-3-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 59)
2-[3-(2-амино-6-{5-[(2-метоксиэтил)амино]пиридин-3-ил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 60)
[0021]
2-{3-[2-амино-6-(4-{[4-(2-гидроксиэтил)пиперазин-1-ил]метил}фенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 61)
2-{3-[2-амино-6-(1-этил-1H-пиразол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 62)
2-{3-[2-амино-6-(1-изопропил-1H-пиразол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 63)
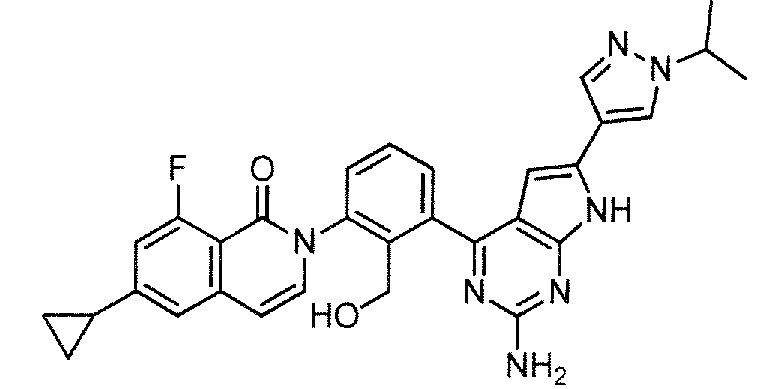
2-{3-[2-амино-6-(1-фенил-1H-пиразол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 64)
2-{3-[2-амино-6-(6-метоксипиридин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 65)
[0022]
2-[3-(2-амино-6-{4-[(3-оксопиперазин-1-ил)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 66)
2-(3-{2-амино-6-[4-(тиазолидин-3-илметил)фенил]-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 67)
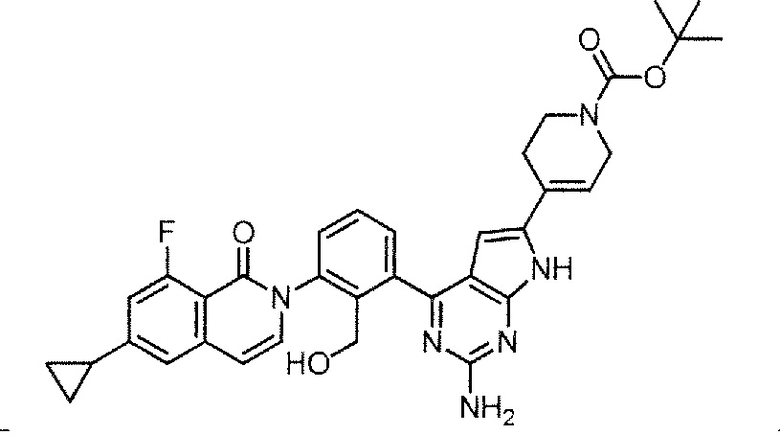
трет-бутил 4-{2-амино-4-[3-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-2-(гидроксиметил)фенил]-7H-пирроло[2,3-d]пиримидин-6-ил}-5,6-дигидропиридин-1(2H)-карбоксилат (Пример 68)
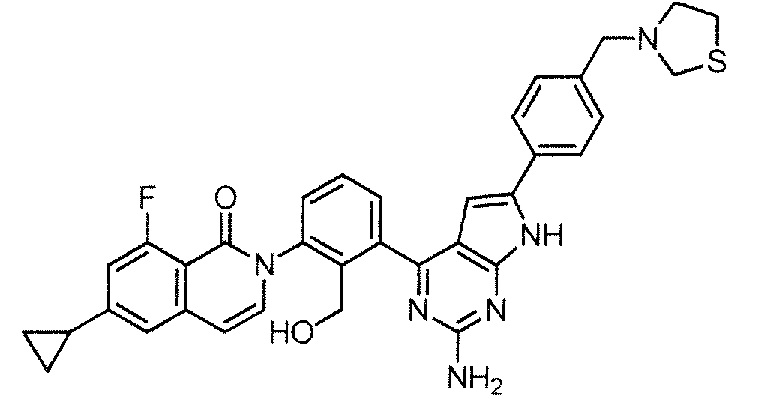
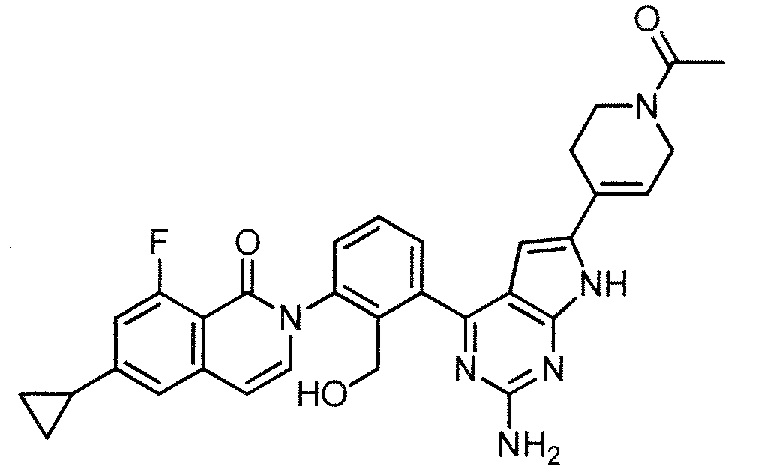
2-{3-[6-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-2-амино-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 69)
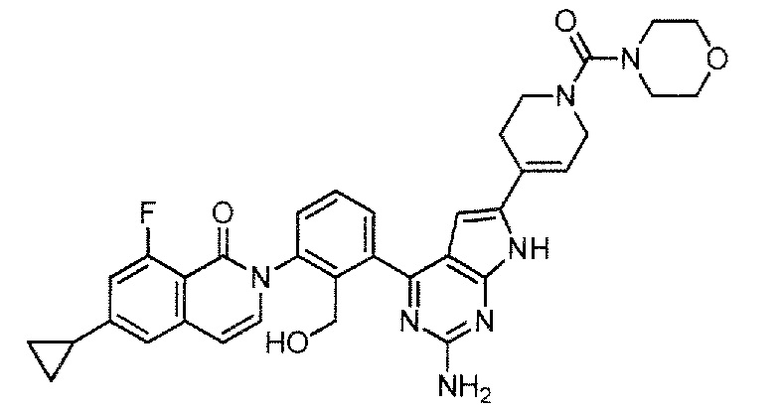
2-(3-{2-амино-6-[1-(морфолин-4-карбонил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 70)
[0023]
2-(3-{2-амино-6-[1-(4-метилпиперазин-1-карбонил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 71)
2-(3-{2-амино-6-[1-(трет-бутил)-1H-пиразол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 72)
2-[3-(2-амино-6-{4-[(4-гидроксипиперидин-1-ил)метил)фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 73)
2-[3-(2-амино-6-{4-[(4-метоксипиперидин-1-ил)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 74)
2-[3-(6-{4-[(4-ацетилпиперазин-1-ил)метил]фенил}-2-амино-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 75)
[0024]
2-[3-(2-амино-6-{4-[(2,6-диметилморфолино)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 76)
2-[3-(2-амино-6-{4-[(4,4-дифторпиперидин-1-ил)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 77)
2-{3-[2-амино-6-(1-метил-1H-пиразол-3-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 78)
2-{3-[2-амино-6-(4-{[4-(2,2,2-трифторэтил)пиперазин-1-ил]метил}фенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 79)
2-[3-(2-амино-6-{4-[(3,3-диметилпиперидин-1-ил)метил]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 80)
[0025]
2-{3-[2-амино-6-(циклогексен-1-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 81)
2-{3-[2-амино-6-(3,6-дигидро-2H-тиопиран-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 82)
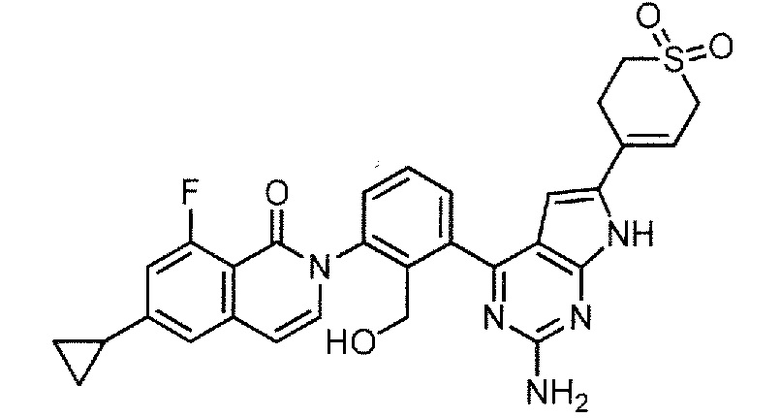
2-{3-[2-амино-6-(1,1-диоксидо-3,6-дигидро-2H-тиопиран-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 83)
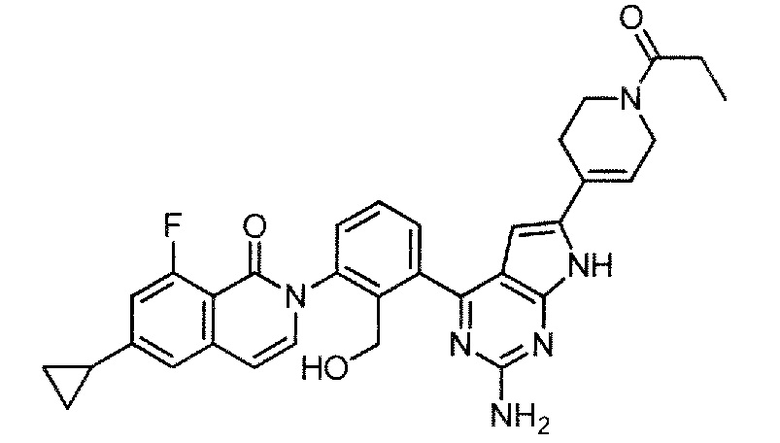
2-{3-[2-амино-6-(1-пропионил-1,2,3,6-тетрагидропиридин-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 84)
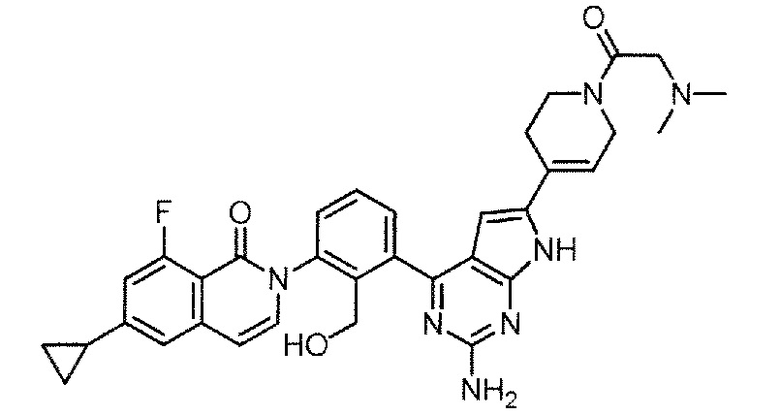
2-[3-(2-амино-6-{1-[2-(диметиламино)ацетил]-1,2,3,6-тетрагидропиридин-4-ил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 85)
[0026]
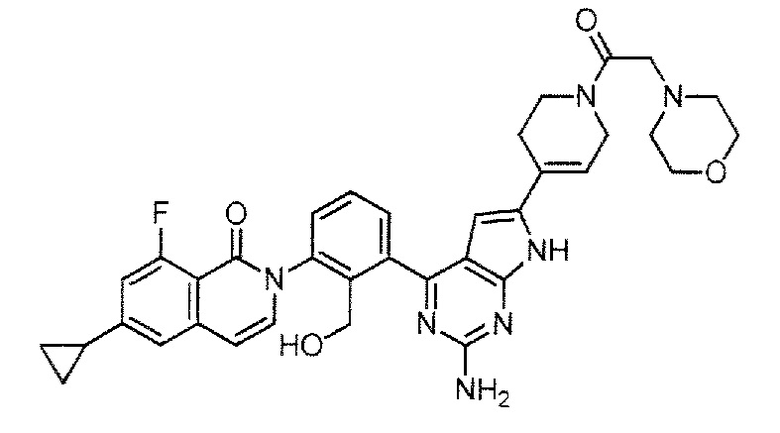
2-(3-{2-амино-6-[1-(2-морфолиноацетил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 86)
4-{2-амино-4-[3-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-2-(гидроксиметил)фенил]-7H-пирроло[2,3-d]пиримидин-6-ил}-N,N-диметил-5,6-дигидропиридин-1(2H)-карбоксамид (Пример 87)
2-(3-{2-амино-6-[1-(пирролидин-1-карбонил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 88)
2-(3-{2-амино-6-[1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 89)
2-(3-{2-амино-6-[1-(изопропилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 90)
[0027]
2-{3-[2-амино-6-(1-этил-1,2,3,6-тетрагидропиридин-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 91)
2-(3-{2-амино-6-[1-(циклопропилметил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 92)
2-{3-[2-амино-6-(1,2,3,6-тетрагидропиридин-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2-(гидроксиметил)фенил}-6-циклопропил-8-фторизохинолин-1(2H)-он (Пример 93)
Эффект изобретения
[0028]
Авторы настоящего изобретения усиленно проводили исследования, для того, чтобы решить вышеупомянутые задачи, и обнаружили, что производное оксоизохинолина формулы (I), описанное ранее, или его фармацевтически приемлемая соль обладает превосходной BTK ингибирующей активностью, и дополнительно подтвердили сильное противоопухолевое действие, когда указанное производное оксоизохинолина или его фармацевтически приемлемую соль перорально вводили на мышиным моделям с раком с использованием штамма OCI-Ly10 для завершения настоящего изобретения.
[0029]
Настоящее изобретение относится к соединению, которое является полезным для профилактики или лечения заболеваний, в которых, как известно, вовлечен аномальной клеточный ответ посредством BTK, например, аутоиммунных заболеваний, воспалительных заболеваний, заболеваний костей и раковых заболеваний, таких как лимфома, и фармацевтической композиции, включающей указанное соединение в качестве активного ингредиента, предпочтительно используется, особенно при пероральном введении.
Соединение, предлагаемое настоящим изобретением, также является полезным в качестве ингибитора BTK, для реагентов, используемых в тестах и исследованиях.
ЛУЧШИЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
[0030]
Настоящее изобретение подробно описано ниже.
[0031]
Новое производное оксоизохинолина по настоящему изобретению представляет собой соединение формулы (I):
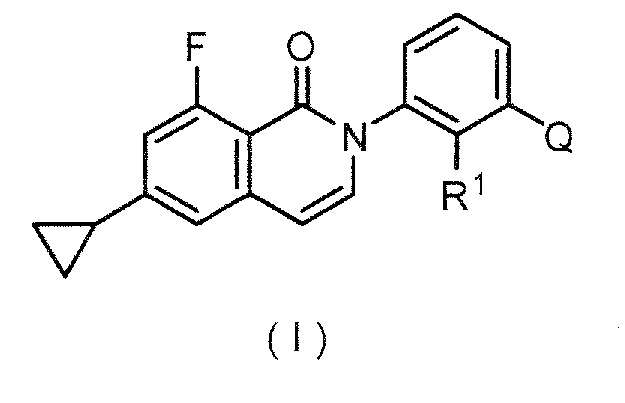 ,
,
где R1 представляет собой необязательно замещенную низшую алкильную группу и
Q представляет собой структуру, выбранную из (а), (b) и (с) ниже:
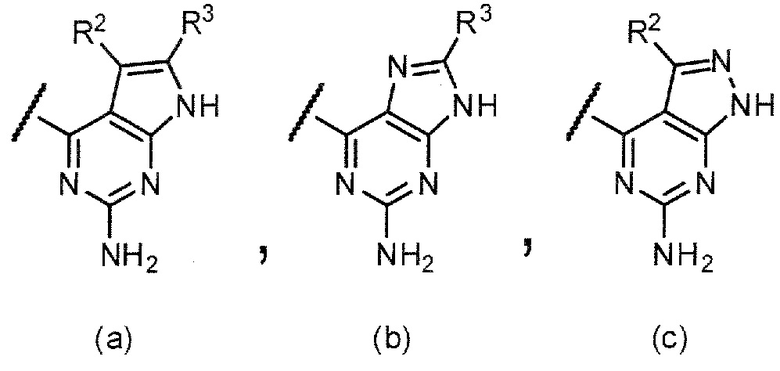 ,
,
R2 и R3 независимо представляют собой атом водорода, необязательно замещенную низшую алкильную группу, необязательно замещенную циклоалкильную группу, необязательно замещенную арильную группу, необязательно замещенную гетероарильную группу и необязательно замещенную гетероциклическую группу.
Структура (а) является предпочтительной как структура Q.
[0032]
В описании настоящей заявки фрагмент низшей алкильной группы в «необязательно замещенной низшей алкильной группе» может быть любой из линейной или разветвленной алкильной группы, содержащей от одного до трех атомов углерода, и, в частности, в качестве примера может быть представлена метильная группа, этильная группа и изопропильная группа и тому подобное.
[0033]
Фрагментом циклоалкильной группы в «необязательно замещенной циклоалкильной группе» может быть любая из циклической алкильной группы, имеющей от трех до шести атомов углерода, и, в частности, в качестве примера может быть представлена циклопропильная группа, циклобутильная группа, циклогексильная группа и тому подобное.
Фрагментом арильной группы в «необязательно замещенной арильной группе» может быть любая из моноциклической или бициклической арильной группы, имеющей от 6 до 14 атомов углерода, и бициклическая арильная группа может быть частично гидрирована. Конкретно, в качестве примера может быть представлена фенильная группа, нафтильная группа, тетрагидронафтильная группа, инденильная группа и тому подобное.
[0034]
Фрагмент гетероарильной группы в «необязательно замещенной гетероарильной группе» включает моноциклическую ароматическую гетероциклическую группу и конденсированную ароматическую гетероциклическую группу, и 5- или 6-членную моноциклическую ароматическую гетероциклическую группу, содержащую один гетероатом, по меньшей мере, выбранный из атома азота, атома серы и атома кислорода, в качестве моноциклической ароматической гетероциклической группы. В частности, в качестве примера может быть представлен пирролил, имидазолил, пиразолил, тиенил, тиазолил, фуранил, пиридил, пиримидил, пиридазил и тому подобное, и примеры конденсированной ароматической гетероциклической группы включают конденсированную бициклическую гетероциклическую группу, в которой 3-8-членное кольцо конденсировано, содержащую один гетероатом, по меньшей мере, выбранный из атома азота, атома серы и атома кислорода. Конкретно, в качестве примера может быть представлен тетрагидроизохинолил, бензотиофенил, бензимидазолил, бензооксазолил, бензотиазолил, индолил и изохинолил.
Фрагмент гетероциклической группы в «необязательно замещенной гетероциклической группе» представляет собой 4-6-членную моноциклическую насыщенную гетероциклическую группу, содержащую один гетероатом, по меньшей мере, выбранный из атома азота, атома серы и атома кислорода, и может включать частично ненасыщенную связь в кольце. В частности, дигидротиопиранильная группа, 1,1-диоксо-дигидротиопиранильная группа и тетрагидропиридильная группа могут быть приведены в качестве примера, и тетрагидропиридильная группа является особенно предпочтительным примером.
[0035]
Заместитель в термине «необязательно замещенный» в необязательно замещенной низшей алкильной группе, необязательно замещенной циклоалкильной группе, необязательно замещенной арильной группе, необязательно замещенной гетероарильной группе и необязательно замещенной гетероциклической группе может быть одинаковым или различным, когда вышеуказанная группа имеет два или более заместителей, и группа может быть замещена одним или двумя или более заместителями любого типа в любом положении, которое является химически допустимым.
Заместитель в термине «необязательно замещенный» в необязательно замещенной низшей алкильной группе, необязательно замещенной циклоалкильной группе, необязательно замещенной арильной группе, необязательно замещенной гетероарильной группе и необязательно замещенной гетероциклической группе может быть одинаковым или различным, когда вышеуказанная группа имеет два или более заместителей, и группа может быть замещена одним, или двумя или более заместителем(ями) любого типа в любом положении, которое является химически допустимым.
[0036]
Примеры заместителя в необязательно замещенной низшей алкильной группе включают, например, атом галогена, C1-C4 алкильную группу, аминогруппу, необязательно замещенную одной или двумя C1-C4 алкильной группой, нитрогруппу, цианогруппу, гидроксигруппу, карбамоильную группу, необязательно замещенную одной или двумя C1-C4 алкильными группами, карбоксильную группу, формильную группу, ацетильную группу, мезильную группу, бензоильную группу, C1-C6 ациламиногруппу, C1-C6 ацилоксигруппу и тому подобное. В качестве примера необязательно замещенной низшей алкильной группы может быть приведена гидроксиметильная группа.
[0037]
Примеры заместителя, относящегося к термину «необязательно замещенный» в необязательно замещенной циклоалкильной группе, необязательно замещенной арильной группе, необязательно замещенной гетероарильной группе и необязательно замещенной гетероциклической группе, включают атом галогена, атом кислорода, C1-C4 алкильную группу, C1-C4 алкоксигруппу, аминогруппу, необязательно замещенную одной или двумя C1-C4 алкильными группами, нитрогруппу, цианогруппу, гидроксигруппу, карбамоильную группу, необязательно замещенную одной или двумя C1-C4 алкильными группами, сульфонильную группу, необязательно замещенную С1-С4 алкильной группой, карбоксигруппу, формильную группу, ацетильную группу, мезильную группу, бензоильную группу, оксетанильную группу, С1-С6 ациламиногруппу и С1-C6 ацилоксигруппу и тому подобное.
[0038]
Изомеры могут иметь место в соединении (I) по настоящему изобретению, в зависимости от типа заместителя. В настоящем описании изомеры могут быть описаны химической структурой только одной их формы, но настоящее изобретение включает все изомеры (геометрический изомер, оптический изомер, таутомер и т.д.), которые могут быть структурно образованы, а также включает только изомеры или их смесь.
[0039]
Примеры фармацевтически приемлемой соли соединения (I) по настоящему изобретению включают соли неорганических кислот с хлористоводородной кислотой, серной кислотой, угольной кислотой и фосфорной кислотой и тому подобное; и соли органических кислот с фумаровой кислотой, малеиновой кислотой, метансульфоновой кислотой и п-толуолсульфоновой кислотой и тому подобное. Настоящее изобретение также включает соли аммония, в дополнение к солям щелочных металлов с натрием и калием; соли щелочноземельных металлов с магнием и кальцием; соли органических аминов с триэтиламином и этаноламином; и соли основных аминокислот с лизином, аргинином, орнитином и тому подобное.
[0040]
Если не указано иное, «соединение (I) по настоящему изобретению» также включает его пролекарство.
[0041]
Соединение (I) и его фармацевтически приемлемая соль в настоящем изобретении могут быть получены, например, способами, показанными ниже. Когда определенная группа может подвергнуться химическому воздействию в условиях приведенного в качестве примера способа в способе получения, показанном ниже, или не подходит для использования для осуществления способа, их можно легко получить способом, который обычно используется в синтетической органической химии, например, способом применения средств, таких как защита функциональной группы или снятие защиты с функциональной группы [T. W. Greene, Protective Groups in Organic Synthesis 3rd Edition, John Wiley&Sons, Inc., 1999]. При необходимости порядок стадии реакции, такой как введение заместителей, также может быть изменен.
[0042]
Значения аббревиатур и символов, используемых в нижеследующем описании, являются следующими.
DCM: дихлорметан
THF: тетрагидрофуран
DIEA: N,N-диизопропилэтиламин
DMF: N,N-диметилформамид
DMSO: диметилсульфоксид
Pd(PPh3)4: тетракис[трифенилфосфин]палладий(0)
[0043]
[Способ получения соединения (I) по настоящему изобретению]
Соединение (I) по настоящему изобретению может быть получено согласно схеме 1, например.
Схема 1
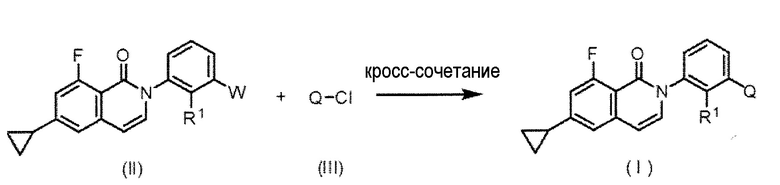 ,
,
где W представляет собой боронильную группу или группу боронатного эфира, и R1 и Q имеют значения, как определено выше.
[0044]
Соединение (I) по настоящему изобретению может быть получено с помощью реакции кросс-сочетания, такой как реакция сочетания Сузуки, с использованием соединения (II) и соединения (III) (относительно условий реакции сочетания Сузуки, см. литературу, например, N. Miyaura et al., J. Am. Chem. Soc., 107, 972 (1985)., N. Miyaura, A. Suzuki, Chem. Rev. 95, 2457 (1995)). То есть, реакция может быть проведена в присутствии металлического катализатора, такого как палладий или никель, при необходимости, с использованием основания и добавок.
[0045]
Примеры растворителя, используемого в реакции, включают THF, диоксан, толуол, диметоксиэтан, метанол, этанол и ацетонитрил. Также целесообразно использовать два или более вида этих растворителей или использовать их в сочетании с водой. Растворитель предпочтительно представляет собой смешанный растворитель из THF и воды или смешанный растворитель из толуола, метанола и воды или диоксан.
[0046]
Соединение (II) предпочтительно используют в эквивалентном или избыточном количестве и более предпочтительно в количестве от 1 эквивалента до 5 эквивалентов, на основе соединения (III). При необходимости можно добавить основание для ускорения реакции, и в качестве основания обычно используют карбонат натрия, карбонат цезия и карбонат калия. Количество используемого основания составляет от 1 эквивалента до 10 эквивалентов, предпочтительно от 1 эквивалента до 5 эквивалентов, на основе соединения (III). В качестве металлического катализатора можно использовать коммерчески доступный палладиевый катализатор (например, PdCl2(dppf), Pd2(dba)3, Pd(PPh3)4, и тому подобное), который используют в реакции кросс-сочетания, и катализатор предпочтительно используют в каталитическом количестве, то есть в количестве от 0,1 эквивалента до 0,5 эквивалента, на основе соединения (III).
[0047]
При необходимости могут быть добавлены добавки для ускорения реакции. Добавка включает, например, rac-BINAP и может использоваться в количестве от 0,01 эквивалента до 1 эквивалента на основе соединения (III). Можно синтезировать продукт путем осуществления реакции при температуре в диапазоне от 0°С до 200°С в течение от нескольких минут до нескольких дней и предпочтительно от 10°С до 100°С в течение от 1 часа до 36 часов. Также возможно синтезировать продукт, путем осуществления реакции при температуре от 60°С до 150°С в течение от нескольких минут до нескольких часов, используя оборудование для микроволнового синтеза.
[0048]
Также соединение (I) по настоящему изобретению может быть получено путем защиты функциональных групп соединения (II) и (III), при необходимости, с использованием общей методики, которая используется в синтетической органической химии, и снятия защиты с них после реакции сочетания.
[0049]
Кроме того, соединение (II), используемое на схеме 1 в качестве исходного вещества, является доступным в соответствии со способом, описанным в патентном документе 2.
[0050]
Соединение (III-a), в котором Q представляет собой структуру (а), является одним из соединений (III), используемых в качестве исходного вещества на схеме 1, и может быть получено по схеме 2, например;
Схема 2
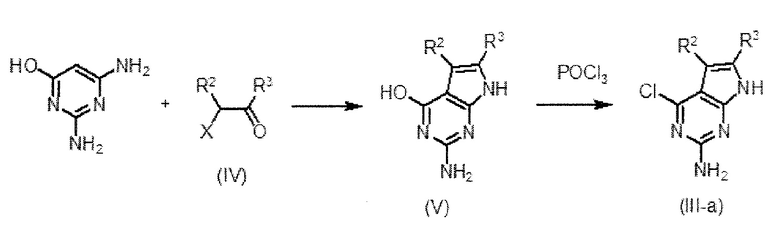 ,
,
где X представляет собой атом галогена и R2 и R3 имеют значения, как определено выше.
[0051]
Соединение (III-a) получают путем циклоконденсации 2,4-диамино-6-гидроксипиримидина и соединения (IV) и последующей реакции хлорирования оксихлоридом фосфора. То есть соединение (V) получают путем взаимодействия 1-5 эквивалентов, предпочтительно 1-1,5 эквивалента соединения (IV), с 2,4-диамино-6-гидроксипиримидином в полярном растворителе и, при необходимости, в присутствии основного катализатора.
[0052]
Может быть использован любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать воду или DMF. Температура реакции обычно составляет от 0°C до 200°С, предпочтительно от комнатной температуры до 150°С. Время реакции не ограничено, но как правило можно указать от 0,2 до 48 часов в качестве примера, и предпочтительно от 1 до 24 часов в качестве примера.
[0053]
Соединение (III-a) получают путем взаимодействия от 1 до 50 эквивалентов, предпочтительно от 5 до 20 эквивалентов оксихлорида фосфора, с соединением (V). Температура реакции обычно составляет от комнатной температуры до 200°С, предпочтительно от 50°С до 150°С. Время реакции не ограничено, но как правило можно указать от 1 до 48 часов в качестве примера, и предпочтительно от 5 до 24 часов в качестве примера.
[0054]
Одно из исходных веществ на схеме 2, 2,4-диамино-6-гидроксипиримидин является коммерчески доступным, и соединение (IV) также является коммерчески доступным или его получают хорошо известным способом или способом в соответствии с ним.
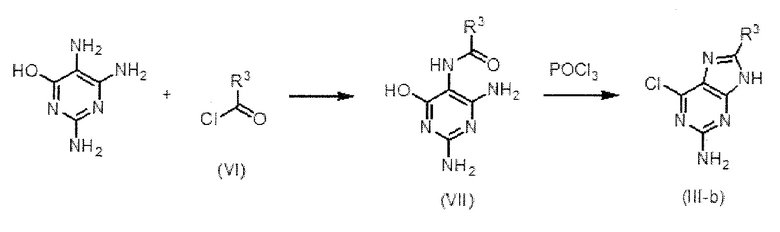
[0055]
Соединение (III-b), в котором Q представляет собой структуру (b), является одним из соединений (III), используемых в качестве исходного вещества на схеме 1, и может быть получено в соответствии со схемой 3, например;
Схема 3
 ,
,
где R3 имеет значение, как определено выше.
[0056]
Соединение (III-b) получают конденсацией 2,5,6-триаминопиримидин-4(3H)-она и хлорида кислоты (VI) и последующей реакцией дегидроциклизации и хлорирования оксихлоридом фосфора. То есть соединение (VII) получают взаимодействием 1-10 эквивалента, предпочтительно 1-3 эквивалента хлорида кислоты (VI) с 2,5,6-триаминопиримидин-4(3H)-оном в растворителе в присутствии основания.
[0057]
Обычно используют органическое основание, такое как DIEA, триэтиламин и тому подобное, или неорганическое основание, такое как гидроксид натрия или карбонат калия и тому подобное, и добавляют от 1 до 10 эквивалентов, предпочтительно от 1 до 5 эквивалентов основания в расчете на хлорид кислоты (VI). Может быть использован любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать воду, DMF и THF. Температура реакции обычно составляет от -20°C до 100°C, предпочтительно от 0°C до 80°C. Время реакции не ограничено, но как правило можно указать от 0,2 до 48 часов в качестве примера, и предпочтительно от 1 до 24 часов в качестве примера.
[0058]
Соединение (III-b) получают путем взаимодействия от 1 до 100 эквивалентов, предпочтительно от 10 до 50 эквивалентов оксихлорида фосфора, с соединением (VII). Температура реакции обычно составляет от комнатной температуры до 200°С, предпочтительно от 50°С до 150°С. Время реакции не ограничено, но как правило можно указать от 1 до 48 часов в качестве примера, и предпочтительно от 5 до 24 часов в качестве примера.
[0059]
Одно из исходных веществ на схеме 3, 2,5,6-триаминопиримидин-4(3H)-он является коммерчески доступным, и соединение (VI) также является коммерчески доступным или его получают хорошо известным способом или способом в соответствии с ним.
[0060]
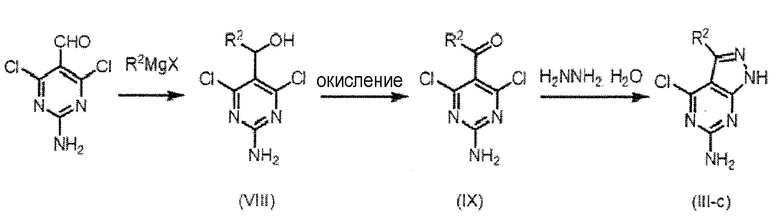
Соединение (III-c), в котором Q представляет собой структуру (c), является одним из соединений (III), используемых в качестве исходного вещества на схеме 1, и может быть получено согласно схеме 4, например;
Схема 4
 ,
,
где R2 и X имеют значения, как определено выше.
[0061]
Соединение (III-c) получают путем конденсации 2-амино-4,6-дихлорпиримидин-5-карбальдегида и R2MgX, и последующей реакцией окисления и циклизации моногидратом гидразина. То есть соединение (VIII) получают взаимодействием 1-10 эквивалента, предпочтительно 1-5 эквивалента R2MgX с 2-амино-4,6-дихлорпиримидин-5-карбальдегидом в растворителе.
[0062]
Может быть использован любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать THF. Температура реакции обычно составляет от -100°C до -30°C, предпочтительно от -80°C до -60°C. Время реакции не ограничено, но как правило можно указать от 0,1 до 12 часов в качестве примера, и предпочтительно от 0,2 до 6 часов в качестве примера.
[0063]
Соединение (IX) получают окислением соединения (VIII) с помощью 1-50 эквивалента, предпочтительно 2-20 эквивалента окислителя и металлсодержащего окислителя, такого как оксид хрома(VI) и диоксид марганца и тому подобное, или гипервалентного йодированного окислителя, такой как периодинан Десса-Мартина и тому подобное. Может быть использован любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать ацетон, DCM и 1,2-дихлорэтан. Температура реакции обычно составляет от -20°C до 100°C, предпочтительно от 0°C до 80°C. Время реакции не ограничено, но как правило можно указать от 0,2 до 24 часов в качестве примера, и предпочтительно от 1 до 12 часов в качестве примера.
[0064]
Соединение (III-c) получают взаимодействием 1-10 эквивалента, предпочтительно 1-5 эквивалента, моногидрата гидразина с соединением (IX) в растворителе и, если необходимо, в присутствии основного катализатора. Может быть использован любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать 1,4-диоксан или THF. Температура реакции обычно составляет от 0°C до 100°С, предпочтительно от комнатной температуры до 60°С. Время реакции не ограничено, но как правило можно указать от 0,2 до 48 часов в качестве примера, и предпочтительно от 0,5 до 24 часов в качестве примера.
[0065]
2-Амино-4,6-дихлорпиримидин-5-карбальдегид, который является исходным веществом на схеме 4, является коммерчески доступным, и R2MgX также является коммерчески доступным или получают хорошо известным способом или способом в соответствии с ним.
[0066]
Соединение (III-a'), в котором Q представляет собой структуру (а) и R2 представляет собой атом водорода, представляет собой одно из соединений (III), используемых в качестве исходного вещества на схеме 1, и может быть получено согласно схеме 5, например.
Схема 5
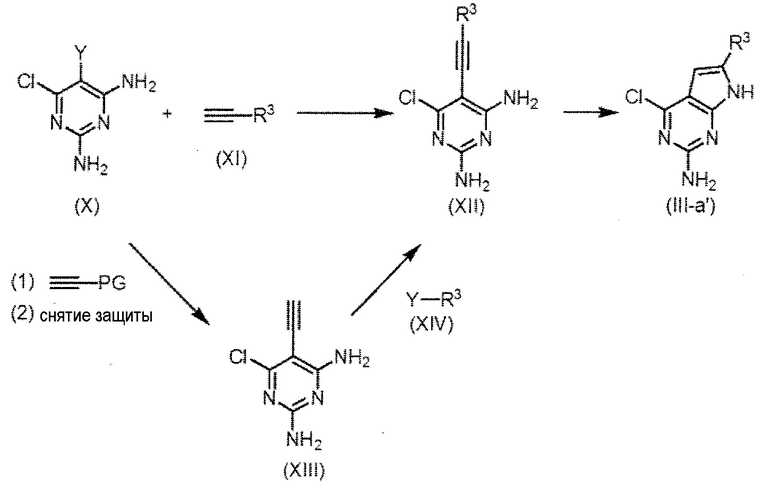 ,
,
где PG представляет собой защитную группу, Y представляет собой атом брома, атом йода или трифторметансульфонильную группу и R3 имеет значение, как определено выше.
Соединение (III-a') получают реакцией циклизации соединения (XII), которое получают реакцией сочетания Соногашира между диаминопиримидином (X) и соединением (XI). В частности, соединение (XII) получают взаимодействием 1-10 эквивалента, предпочтительно 2-5 эквивалента соединения (XI) с диаминопиримидином (X) в полярном растворителе в присутствии йодида меди, палладиевого катализатора и основания, и последующей обработкой водным раствором гидроксида натрия и тетрабутиламмоний фторида.
[0067]
Количество йодида меди, добавляемого в реакцию, составляет 0,01-2 эквивалента, предпочтительно 0,05-0,5 эквивалента, в расчете на диаминопиримидин (X), и палладиевый(0) катализатор, такой как Pd(PPh3)4 и PDCl2(PPh3)2 и тому подобное, используется в качестве палладиевого катализатора. Количество палладиевого катализатора, добавленное в реакцию, составляет 0,01-2 эквивалента, предпочтительно 0,05-0,5 эквивалента, в расчете на диаминопиримидин (X). Органическое основание, такое как DIEA и триэтиламин, обычно используют в реакции в качестве основания, и в реакцию добавляют 1-10 эквивалентов, предпочтительно 1-5 эквивалентов основания, в расчете на диаминопиримидин (X). Можно использовать любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать 1,4-диоксан или DMF. Температура реакции обычно составляет от 0°C до 200°С, предпочтительно от комнатной температуры до 80°С. Время реакции не ограничено, но как правило можно указать от 0,2 до 5 часов в качестве примера, и предпочтительно от 0,5 до 2 часов в качестве примера.
Соединение (III-a') получают путем добавления 1-50 эквивалентов, предпочтительно 2-20 эквивалентов основания, к соединению (XII) и проведения реакции циклизации. В качестве основания можно использовать трет-бутоксид калия или карбонат цезия. Может быть использован любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать N-метилпирролидон, 1,4-диоксан и DMF. Температура реакции обычно составляет от -20°C до 100°С, предпочтительно от°C до 80°С. Время реакции не ограничено, но как правило можно указать от 0,2 до 10 часов в качестве примера, и предпочтительно от 0,5 до 2 часов в качестве примера.
Также соединение (XII) схемы 5 можно синтезировать путем введения фрагмента R3 посредством сочетания Соногашира соединения (XIV) с соединением (XIII), которое получают сочетанием Соногашира между диаминопиридином (X) и терминально-защищенным ацетиленом, и последующим снятием защиты. То есть соединение (XIII) получают взаимодействием 1-10 эквивалентов, предпочтительно 2-5 эквивалентов терминально-защищенного ацетилена, такого как триметилсилилацетилен, с диаминопиримидином (X) в полярном растворителе в присутствии йодида меди, палладиевого катализатора и основания и последующей обработкой его водным раствором гидроксида натрия и тетра-н-бутиламмоний фторида.
[0068]
Количество йодида меди, добавляемого в реакцию, составляет 0,01-2 эквивалента, предпочтительно 0,05-0,5 эквивалента, в расчете на диаминопиримидин (X), и палладиевый(0) катализатор, такой как Pd(PPh3)4 и PDCl2(PPh3)2 и тому подобное, используется в качестве палладиевого катализатора. Количество палладиевого катализатора, добавленное в реакцию, составляет 0,01-2 эквивалента, предпочтительно 0,05-0,5 эквивалента, в расчете на диаминопиримидин (X). Органическое основание, такое как DIEA и триэтиламин, обычно используют в реакции в качестве основания, и в реакцию добавляют 1-10 эквивалентов, предпочтительно 1-5 эквивалентов основания, в расчете на диаминопиримидин (X). Можно использовать любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать 1,4-диоксан или DMF. Температура реакции обычно составляет от 0°C до 200°C, предпочтительно от комнатной температуры до 80°C. Время реакции не ограничено, но как правило можно указать от 0,2 до 5 часов в качестве примера, и предпочтительно от 0,5 до 2 часов в качестве примера.
[0069]
Соединение (XII) получают взаимодействием 1-10 эквивалента, предпочтительно 1-5 эквивалента соединения (XIV) с соединением (XIII) в полярном растворителе в присутствии иодида меди, палладиевого катализатора и основания. Количество йодида меди, добавляемого в реакцию, составляет 0,01-2 эквивалента, предпочтительно 0,05-0,5 эквивалента, в расчете на соединение (XIII), и палладиевый(0) катализатор, такой как Pd(PPh3)4 и PdCl2(PPh3)2 и тому подобное, используется в качестве палладиевого катализатора. Количество палладиевого катализатора, добавленное в реакцию, составляет 0,01-2 эквивалента, предпочтительно 0,05-0,5 эквивалента, в расчете на диаминопиримидин (XIII). Органическое основание, такое как DIEA и триэтиламин, обычно используют в реакции в качестве основания, и в реакцию добавляют 1-10 эквивалентов, предпочтительно 1-5 эквивалентов основания, в расчете на соединение (XIII). Можно использовать любой растворитель, без ограничения, если реакция не нарушается, но предпочтительно использовать 1,4-диоксан или DMF. Температура реакции обычно составляет от 0°C до 200°C, от комнатной температуры до 120°C. Время реакции не ограничено, но как правило можно указать от 0,2 до 5 часов в качестве примера, и предпочтительно от 0,5 до 2 часов в качестве примера.
[0070]
Диаминопиримидин (X) исходного вещества на схеме 5, терминально-защищенный ацетилен (XI) и соединение (XIV) также являются коммерчески доступными или их получают хорошо известным способом или способом в соответствии с ним.
[0071]
В приведенной выше схеме W представляет собой боронильную группу и соль щелочного металла или щелочно-земельного металла. Примеры групп боронатного эфира включают группу боронат диметилового эфира, группу боронат диэтилового эфира, группу боронат дибутилового эфира, боронат дициклогексильную группу, группу боронат этиленгликолевого эфира, группу боронат пропиленгликолевого эфира (группа боронат 1,2-пропандиол эфира, группа боронат 1,3-пропандиол эфира), группа боронат неопентилгликолевого эфира, группа боронатного эфира катехола, группа боронат глицеринового эфира, группа боронат триметиролэтановый эфира, группа боронат диэтаноламинового эфира, сложный эфир боронат-триэтаноламина и тому подобное и ангидрид бороновой кислоты.
[0072]
Можно получить соединение (I), имеющее желаемую функциональную группу, в желаемом положении, настоящего изобретения, соответствующим образом используя вышеуказанные способы в комбинации, а затем осуществляя способ, обычно используемый в органической синтетической химии (например, реакция алкилирования аминогруппы, реакция окисления алкилтиогруппы в сульфоксидную группу или сульфоновую группу, реакция превращения алкоксигруппы в гидроксильную группу или реакция обратного превращения группы).
[0073]
Соединение (I) или его фармацевтически приемлемая соль по настоящему изобретению может быть составлено в виде обычного фармацевтического состава (фармацевтической композиции), который подходит для перорального введения, парентерального введения или местного применения.
[0074]
Лекарственные формы для перорального введения включают твердые лекарственные формы, такие как таблетки, гранулы, порошки и капсулы; и жидкие лекарственные формы, такие как сиропы. Эти лекарственные формы могут быть получены обычным способом. Твердые лекарственные формы могут быть получены с использованием обычных фармацевтических носителей, например, лактозы; крахмалов, таких как кукурузный крахмал; кристаллической целлюлозы, такой как микрокристаллическая целлюлоза; и гидроксипропилцеллюлозы, кальций карбоксиметилцеллюлозы, талька и стеарата магния. Капсулы могут быть получены путем инкапсулирования полученных таким образом гранул или порошков. Сиропы могут быть получены путем растворения или суспендирования соединения (I) или его фармацевтически приемлемой соли по настоящему изобретению в водном растворе, содержащем сахарозу и карбоксиметилцеллюлозу.
[0075]
Лекарственные формы для парентерального введения включают инъекции, такие как инстилляция. Инъекционные составы также могут быть получены обычным способом и могут быть соответствующим образом включать в себя изотонические агенты (например, маннит, хлорид натрия, глюкозу, сорбитол, глицерин, ксилит, фруктоза, мальтоза, манноза), стабилизаторы (например, сульфит натрия, альбумин) и антисептики (например, бензиловый спирт, метил п-оксибензоат).
[0076]
Доза соединения (I) или его фармацевтически приемлемой соли по настоящему изобретению может варьироваться в зависимости от тяжести заболевания, возраста и массы тела пациента и лекарственной формы и обычно находится в диапазоне от 1 мг до 1000 мг в день для взрослых. Соединение или его фармацевтически приемлемую соль можно вводить один раз в день или раздельно вводить два или три раза в день в соответствии с пероральным или парентеральным путем введения.
[0077]
Соединение (I) или его фармацевтически приемлемую соль по настоящему изобретению можно также использовать в качестве ингибитора BTK, для реагентов, которые будут использоваться в экспериментальных испытаниях и/или исследованиях.
Примеры
[0078]
Настоящее изобретение будет более конкретно описано ниже посредством примеров и примеров испытаний, но настоящее изобретение не ограничивается этими примерами.
[0079]
Идентификацию соединения осуществляли по спектру ядерного магнитного резонанса водорода (1H-ЯМР) и масс-спектру (MS). 1H-ЯМР измеряли при 400 МГц или 500 МГц, если не указано иное, и обменный водород иногда нельзя четко наблюдать в зависимости от соединения и условий измерения. Кроме того, шир. означает широкий сигнал (широкий).
[0080]
Препаративную ВЭЖХ хроматографию осуществляли с помощью коммерчески доступной колонки ODS в градиентном режиме, используя воду/метанол (содержащий муравьиную кислоту) или воду/ацетонитрил (содержащий гидрокарбонат аммония) в качестве элюентов, если не указано иное.
[0081]
Пример 1
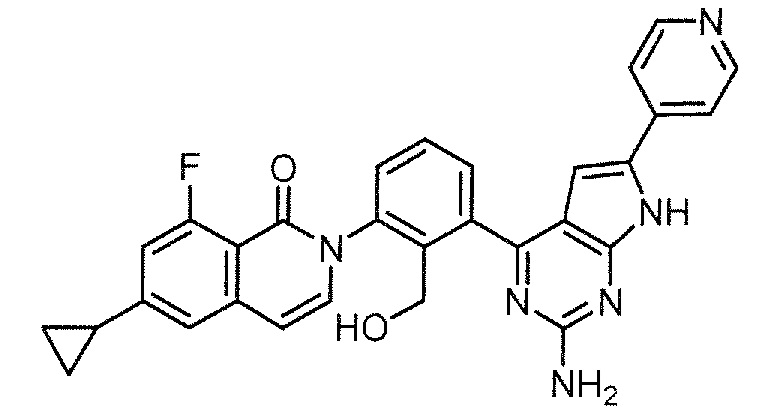
2-[3-(2-амино-6-фенил-7H-пирроло[2,3-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он
(первая стадия)
Ацетат натрия (0,65 г, 7,93 ммоль) добавляли к водному раствору (20 мл) 2,4-диамино-6-гидроксипиримидина (1,0 г, 7,93 ммоль) и смесь перемешивали при 100°C в течение часа. Добавляли 2-бром-ацетофенон (1,89 г, 9,51 ммоль) и смесь перемешивали при 100°C в течение 8 часов. Осажденное твердое вещество собирали с получением 2-амино-6-фенил-7H-пирроло[2,3-d]-пиримидин-4-ола (1,5 г) в виде неочищенного продукта.
LCMS (m/z): 227,11 [M+H]+.
[0082]
(вторая стадия)
Смесь 2-амино-6-фенил-7H-пирро[2,3-d]-пиримидин-4-ола (1,5 г, 6,64 ммоль) и ангидрида пивалевой кислоты (5 мл) перемешивали при 190°C в течение 5 часов. В реакционную смесь добавляли н-пентан и перемешивали при комнатной температуре в течение получаса. Осажденное твердое вещество собирали фильтрованием с получением N-(4-гидрокси-6-фенил-7H-пирроло[2,3-d]пиримидин-2-ил)пиваламида (1,4 г).
LCMS (m/z): 311,33 [M+H]+.
[0083]
(третья стадия)
Смесь N-(4-гидрокси-6-фенил-7H-пирро[2,3-d]пиримидин-2-ил)пиваламида (1,4 г, 4,52 ммоль) и оксихлорид фосфора (5 мл) перемешивали при 100°C в течение 10 часов. Избыток оксихлорида фосфора выпаривали при пониженном давлении, к остатку добавляли насыщенный водный раствор бикарбоната натрия и экстрагировали этилацетатом. Полученный органический слой промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и полученный остаток очищали колоночной хроматографией (силикагель, петролейный эфир/этилацетат) с получением N-(4-хлор-6-фенил-7H-пирроло[2,3-d]пиримидин-2-ил)пиваламида (0,4 г).
1H ЯМР (500 MГц, DMSO-d6) δ=12,93 (с, 1H), 10,06 (с, 1H), 7,99-7,97 (м, 2H), 7,53-7,47 (м, 2H), 7,41-7,38 (м, 1H), 7,04 (д, J=2,0 Гц, 1H), 1,25 (с, 9H);
LCMS (m/z): 329,26 [M+H]+.
[0084]
(четвертая стадия)
Смешанный растворитель DME-вода (5:1, 12 мл) добавляли к 2-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил)бензил ацетату (0,392 г, 0,82 ммоль), N-(4-хлор-6-фенил-7H-пирроло[2,3-d]пиримидин-2-ил)пиваламиду (0,27 г, 0,82 ммоль) и карбонату калия (0,227 г, 1,65 ммоль), и смесь дегазировали в течение 30 минут в атмосфере газа аргона. К смеси добавляли Pd(PPh3)4(95 мг, 0,08 ммоль) и осуществляли реакцию в микроволновом аппарате для реакций при 100°C в течение 10 минут. Реакционную смесь фильтровали через целит, к фильтрату добавляли воду и продукт экстрагировали этилацетатом. Полученный органический слой последовательно промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении с получением 2-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-6-(6-фенил-2-пиваламид-7H-пирроло[2,3-d]пиримидин-4-ил)бензилацетата в виде неочищенного продукта. Полученный неочищенный продукт растворяли в метаноле (2 мл), добавляли 5% водный раствор гидроксида натрия и смесь перемешивали при 70°C в течение 30 минут. Растворитель выпаривали при пониженном давлении и полученный остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (35 мг).
1H ЯМР (400 MГц, DMSO-d6) δ 11,89 (с, 1H), 7,91-7,83 (м, 2H), 7,80 (дд, J=7,7, 1,3 Гц, 1H), 7,62 (т, J=7,8 Гц, 1H), 7,52-7,33 (м, 4H), 7,34-7,25 (м, 2H), 7,00 (дд, J=13,3, 1,7 Гц, 1H), 6,76 (с, 1H), 6,63 (дд, J=7,5, 2,1 Гц, 1H), 6,42 (с, 2H), 5,18 (с, 1H), 4,33-4,25 (м, 1H), 4,12-4,04 (м, 1H), 2,14-2,02 (м, 1H), 1,15-1,01 (м, 2H), 0,96-0,81 (м, 2H);
LCMS (m/z): 518,42 [M+H]+.
[0085]
Пример 2
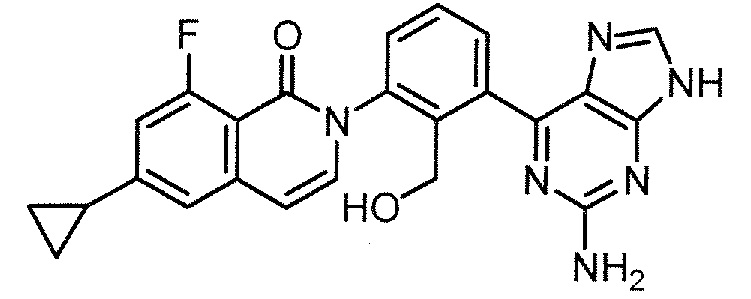
2-[3-(2-амино-8-фенил-9H-пурин-6-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он
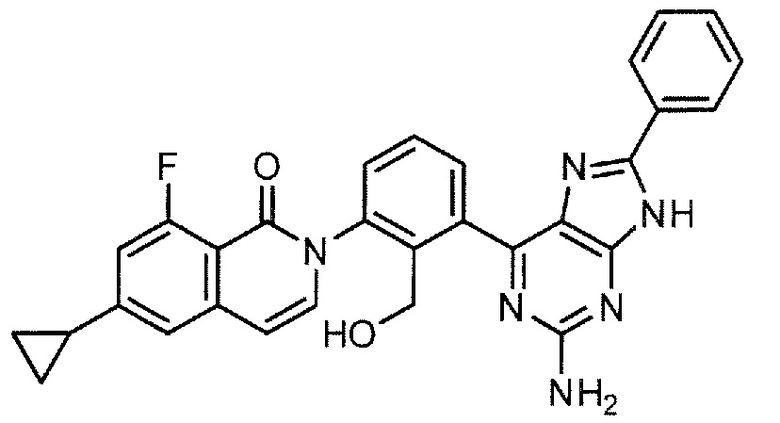
(первая стадия)
2,5,6-Триаминопиримидин-4(3H)-он (1 г, 7,092 ммоль) и бензоилхлорид (1,63 мл, 14,18 ммоль) добавляли к 2 н водному раствору гидроксида натрия (25 мл) при охлаждении льдом, и смесь перемешивали в течение часа. Уксусную кислоту добавляли к реакционной смеси, чтобы довести ее кислотность до pH 5, и осажденное твердое вещество собирали фильтрованием с получением N-(2,4-диамино-6-гидроксипиримидин-5-ил)бензамида (1,7 г).
1H ЯМР (400 MГц, DMSO-d6) δ 10,07 (с, 1H), 8,74 (с, 1H), 7,96-7,92 (м, 2H), 7,53-7,43 (м, 3H), 6,18 (шир. с, 2H), 5,79 (шир. с, 2H);
LCMS (m/z): 246,08 [M+H]+.
[0086]
(вторая стадия)
Смесь N-(2,4-диамино-6-гидроксипиримидин-5-ил)бензамида (2,5 г, 10,2 ммоль) и оксихлорида фосфора (50 мл) перемешивали с обратным холодильником в течение 24 часов. Избыточное количество оксихлорида фосфора выпаривали при пониженном давлении, полученный остаток подщелачивали путем добавления водного аммиака и продукт экстрагировали 10% MeOH-DCM. Полученный органический слой промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии (силикагель, DCM/метанол) с получением 6-хлор-8-фенил-9H-пурин-2-амина (0,25 г)
LCMS (m/z): 245,87 [M+H]+.
[0087]
(третья стадия)
Смешанный растворитель DME-вода (3:1, 13 мл) добавляли к 2-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил)бензил ацетату (0,25 г, 0,52 ммоль), 6-хлор-8-фенил-9H-пурин-2-амину (0,128 г, 0,524 ммоль) и карбонату калия (0,216 г, 1,57 ммоль) и смесь дегазировали в атмосфере аргона в течение 30 минут. Добавляли Pd(PPh3)4 (60 мг, 0,05 ммоль) и осуществляли взаимодействие в микроволновом аппарате при 110°C в течение 30 минут. Реакционную смесь разбавляли этилацетатом, последовательно промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и полученный остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (12 мг).
1H ЯМР (500 MГц, DMSO-d6) δ 13,30 (с, 1H), 8,07 (д, J=6,7 Гц, 2H), 7,91 (д, J=7,0 Гц, 1H), 7,63 (т, J=7,8 Гц, 1H), 7,54-7,48 (м, 4H), 7,41 (д, J=7,3 Гц, 1H), 7,28 (д, J=1,2 Гц, 1H), 7,00 (д, J=13,1 Гц, 1H), 6,68 (шир. с, 2H), 6,63 (дд, J=1,5, 7,3 Гц, 1H), 5,49 (шир. с, 1H), 4,36 (д, J=9,5 Гц, 1H), 4,13-4,09 (м, 1H), 2,10-2,05 (м, 1H), 1,12-1,08 (м, 2H), 0,89-0,86 (м, 2H);
LCMS (m/z): 519,39 [M+H]+.
[0088]
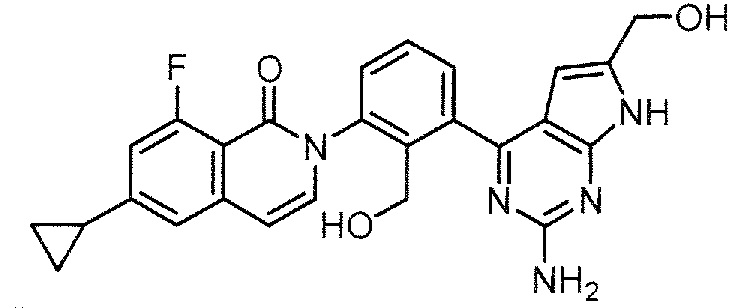
Пример 3
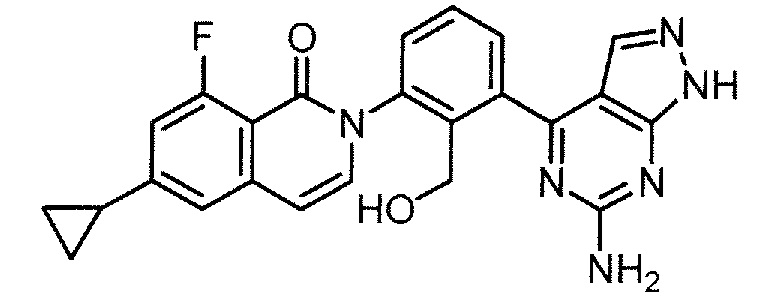
2-[3-(6-амино-3-фенил-1H-пиразоло[3,4-d]пиримидин-4-ил)-2-(гидроксиметил)фенил]-6-циклопропил-8-фторизохинолин-1(2H)-он
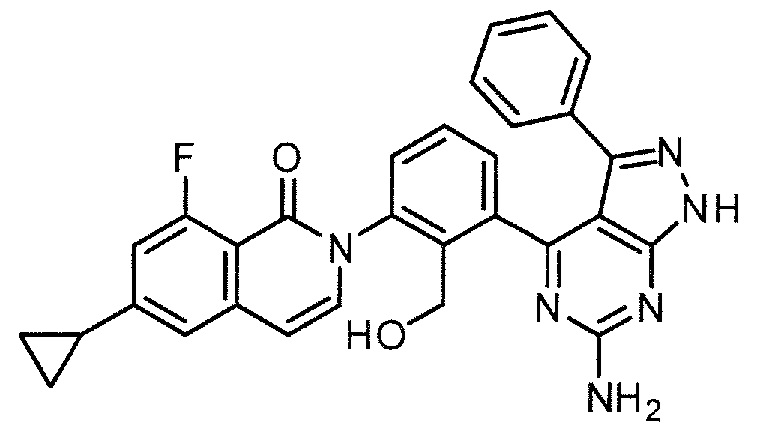
(первая стадия)
Фенил магний бромид (1М раствор THF, 26 мл, 26 ммоль) медленно добавляли к раствору THF (100 мл) 2-амино-4,6-дихлорпиримидин-5-карбальдегида (1,0 г, 5,2 ммоль) при -78°C, и перемешивали в течение 2 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, фильтровали через целит и фильтрат экстрагировали 10%MeOH-DCM. Полученный органический слой промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении, и остаток очищали колоночной хроматографией (силикагель, петролейный эфир/этилацетат) с получением (2-амино-4,6-дихлорпиримидин-5-ил)(фенил)метанола (0,6 г).
1H ЯМР (400MГц, DMSO-d6) δ=7,52 (шир. с, 2H), 7,33-7,30 (м, 4H), 7,26-7,18 (м, 1H), 6,20 (д, J=4,4 Гц, 1H), 6,03 (д, J=4,9 Гц, 1H);
LCMS (m/z): 270,05 [M+H]+.
[0089]
(вторая стадия)
Диоксид марганца (3,88 г, 44,6 ммоль) добавляли к раствору 1,2-дихлорэтана (15 мл) (2-амино-4,6-дихлорпиримидин-5-ил)(фенил)метанола (0,6 г, 2,2 ммоль) при охлаждении льдом, и перемешивали при 80°С в течение 3 часов. Реакционную смесь фильтровали через целит и растворитель выпаривали при пониженном давлении с получением (2-амино-4,6-дихлорпиримидин-5-ил)(фенил)метанона (0,5 г).
1H ЯМР (400 MГц, DMSO-d6) δ 7,97-7,92 (м, 4H), 7,75-7,72 (м, 1H), 7,60-7,56 (м, 2H);
LCMS (m/z): 267,94 [M+H]+.
[0090]
(третья стадия)
Гидразин моногидрат (0,1 мл, 1,87 ммоль) добавляли к THF-раствору (15 мл) (2-амино-4,6-дихлорпиримидин-5-ил)(фенил)метанона (0,5 г, 1,87 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Растворитель реакционной смеси выпаривали при пониженном давлении, к полученному остатку добавляли воду и осажденное твердое вещество собирали фильтрованием с получением 4-хлор-3-фенил-1H-пиразоло[3,4-d]пиримидин-6-амина (0,35 г).
1H ЯМР (400 MГц, DMSO-d6) δ 13,38 (шир. с, 1H), 7,70-7,68 (м, 2H), 7,50-7,44 (м, 3H), 7,18 (шир. с, 2H);
LCMS (m/z): 246,1 [M+H]+.
[0091]
(четвертая стадия)
Смешанный растворитель DME-вода (4:1, 10 мл) добавляли к 2-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил)бензил ацетату (0,193 г, 0,4 ммоль), 4-хлор-3-фенил-1H-пиразоло[3,4-d]пиримидин-6-амину (0,1 г, 0,4 ммоль) и карбонату калия (0,11 г, 0,8 ммоль) и смесь дегазировали в атмосфере аргона в течение 30 минут. Добавляли Pd(PPh3)4(23 мг, 0,02 ммоль) и осуществляли взаимодействие в микроволновом аппарате при 110°C в течение 15 минут. К реакционной смеси добавляли воду, выпавшее в осадок твердое вещество собирали фильтрацией с получением 2-(6-амино-3-фенил-1H-пиразоло[3,4-d]пиримидин-4-ил)-6-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)бензил ацетата (0,3 г) в виде неочищенного продукта. Неочищенный продукт растворяли в метаноле (20 мл), добавляли карбонат калия (0,4 г) и перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали при пониженном давлении, к полученному остатку добавляли воду, осажденное твердое вещество собирали фильтрацией и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (45 мг).
1H ЯМР (400 MГц, DMSO-d6) δ 13,18 (шир. с, 1H), 7,28-6,94 (м, 13H), 6,61 (д, J=5,9 Гц, 1H), 4,69 (шир. с, 1H), 4,49-4,12 (м, 2H), 2,11-2,04 (м, 1H), 1,12-1,07 (м, 2H), 0,89-0,85 (м, 2H);
LCMS (m/z): 519,39 [M+H]+.
Примеры 4-22 и 24-93
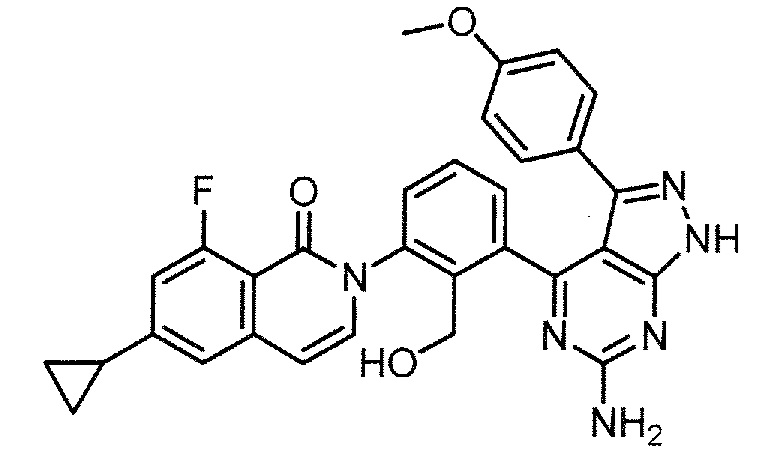
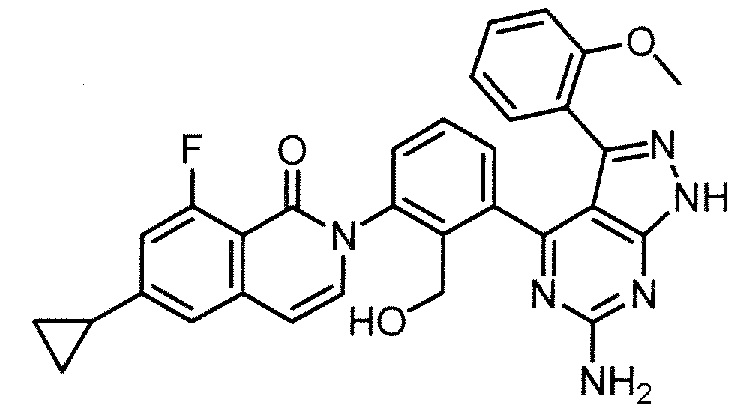
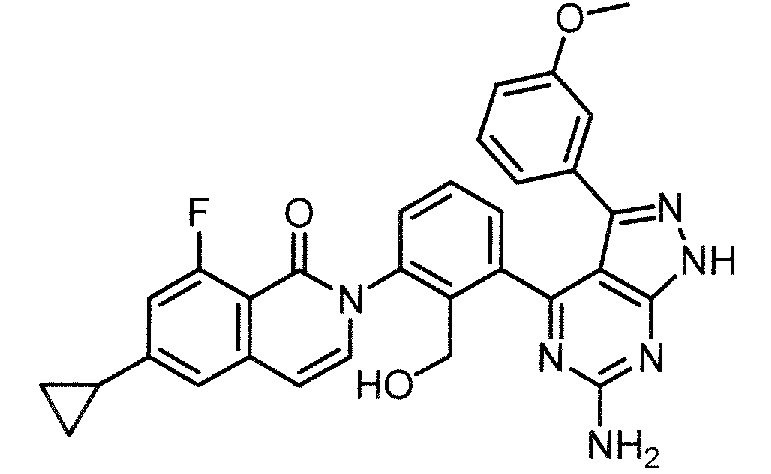
Каждое из соединений примера в следующих [таблица 1-1] и [таблица 1-2] было получено в соответствии со способом, описанным в примере выше, или указанным способом в сочетании с обычным методом, хорошо известным в области органической химии, если необходимо с использованием подходящего исходного вещества (его получают из коммерческого источника или получают с помощью способов, описанных в литературе, или модификаций способов, описанных в литературе, известных специалистам в данной области).
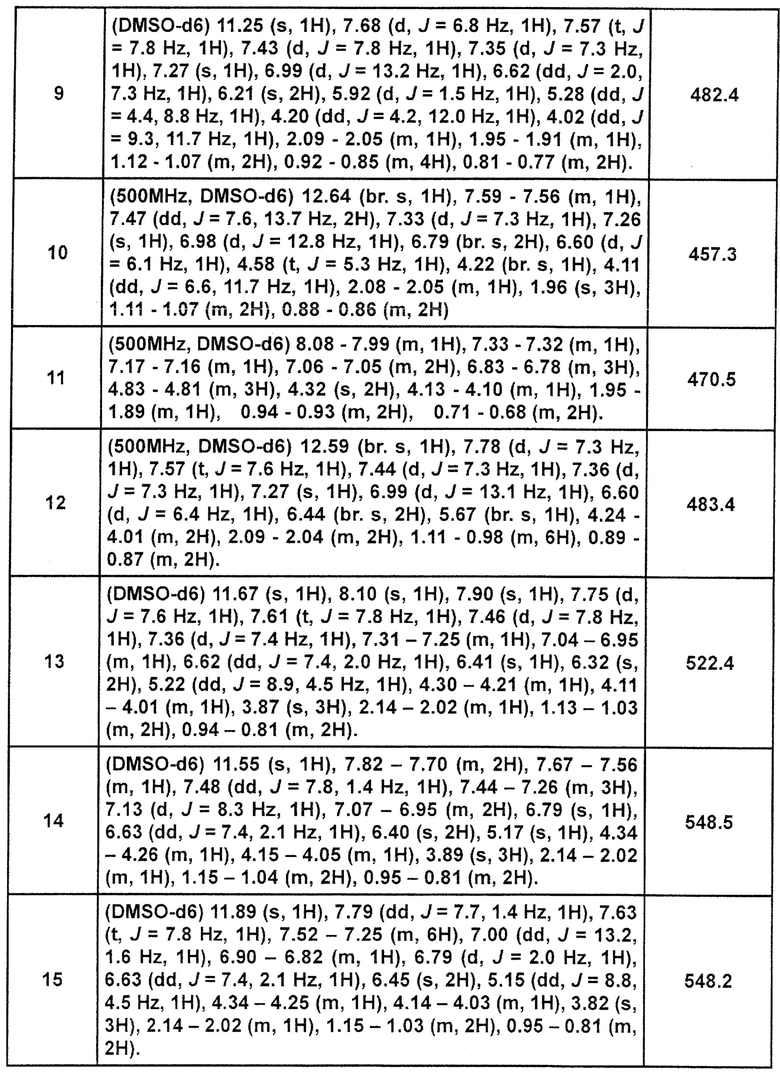
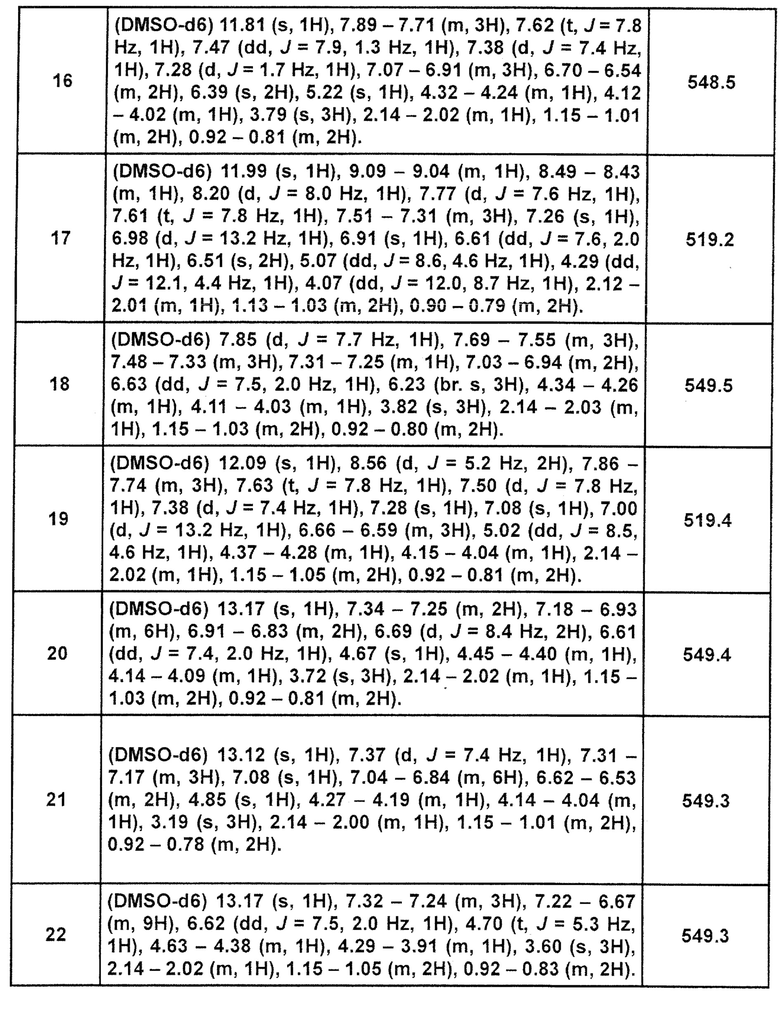
Физико-химические данные каждого соединения представлены в следующих таблицах [Таблица 2-1] и [Таблица 2-2].
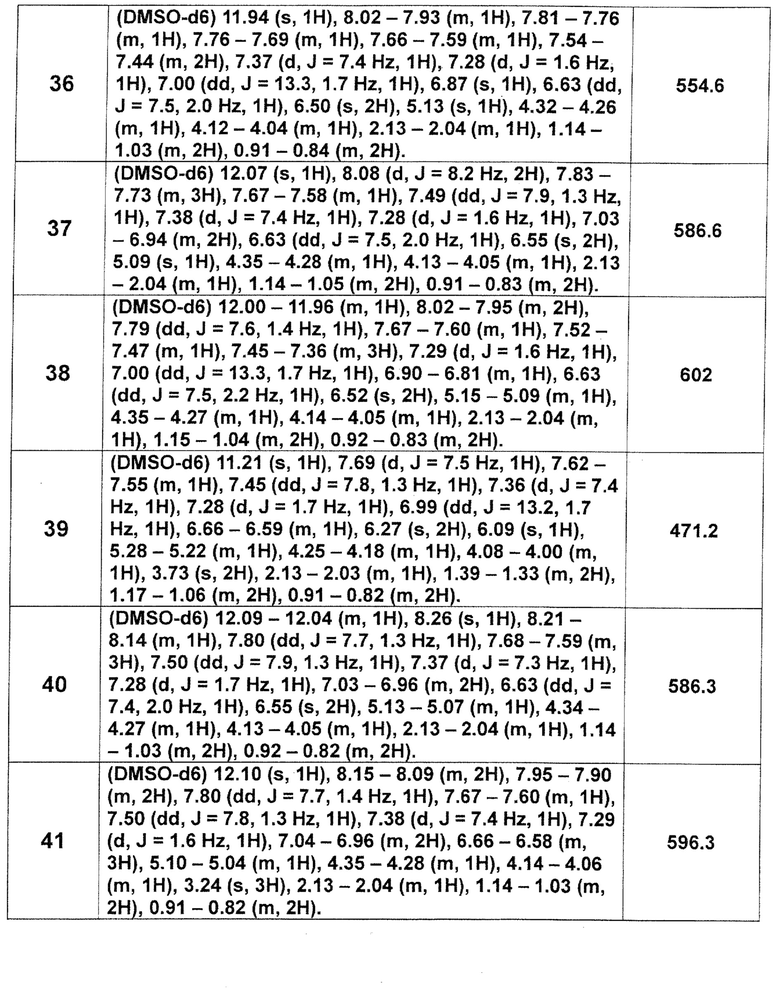
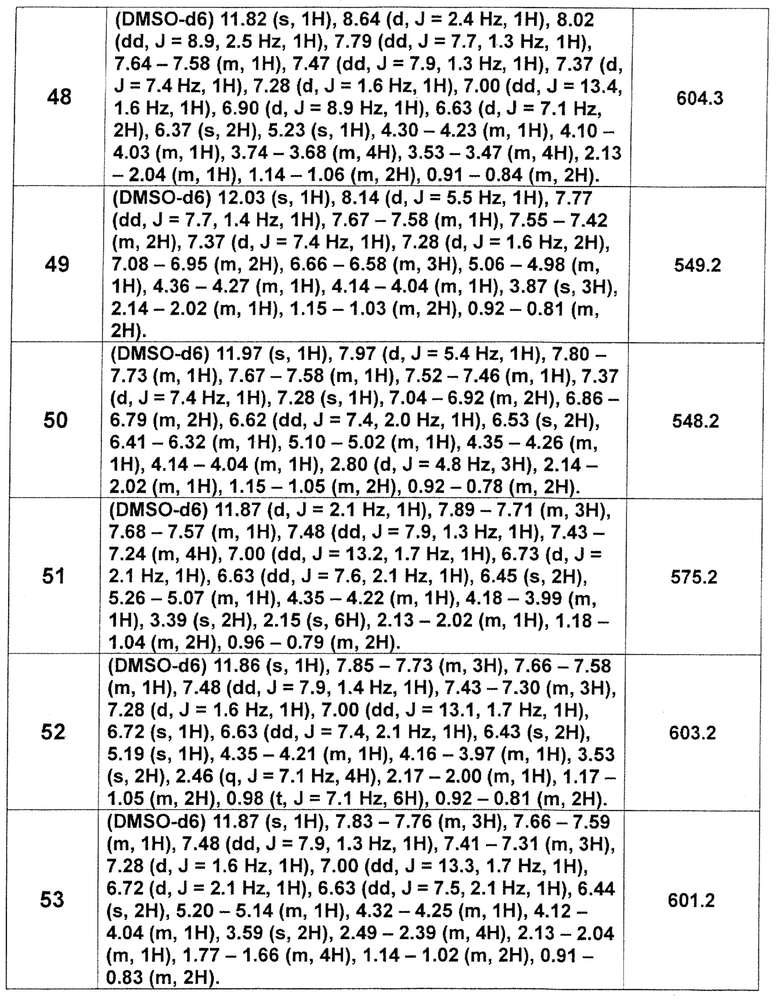
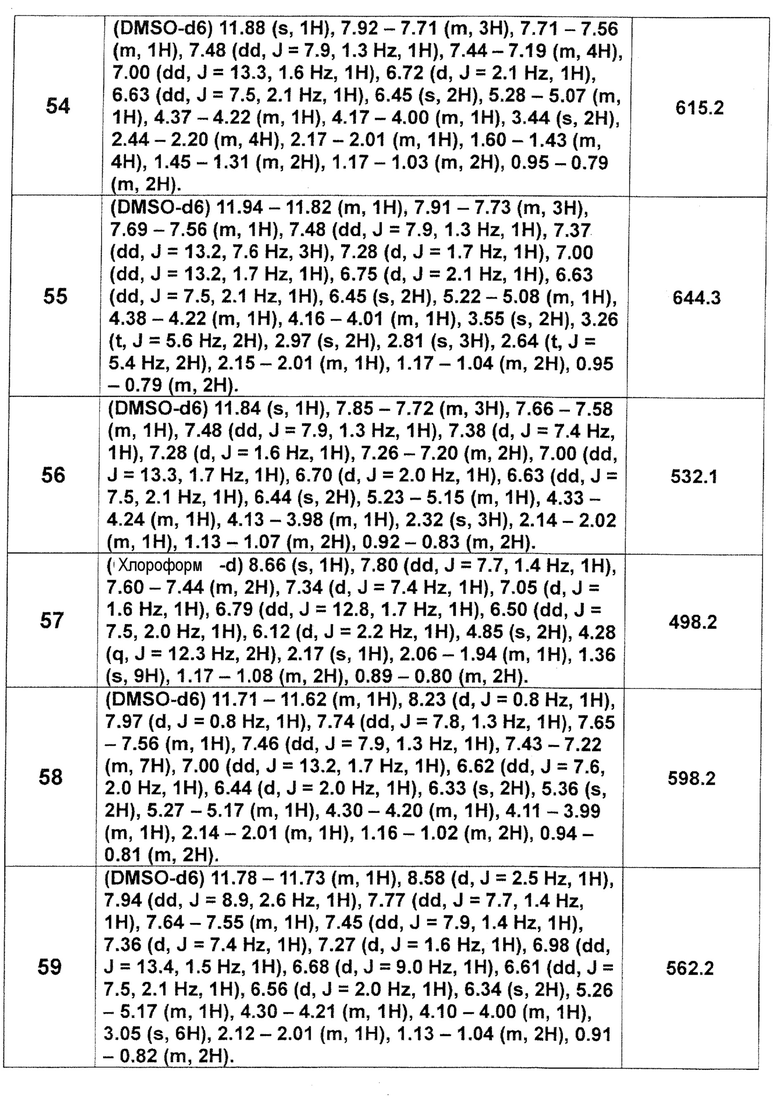
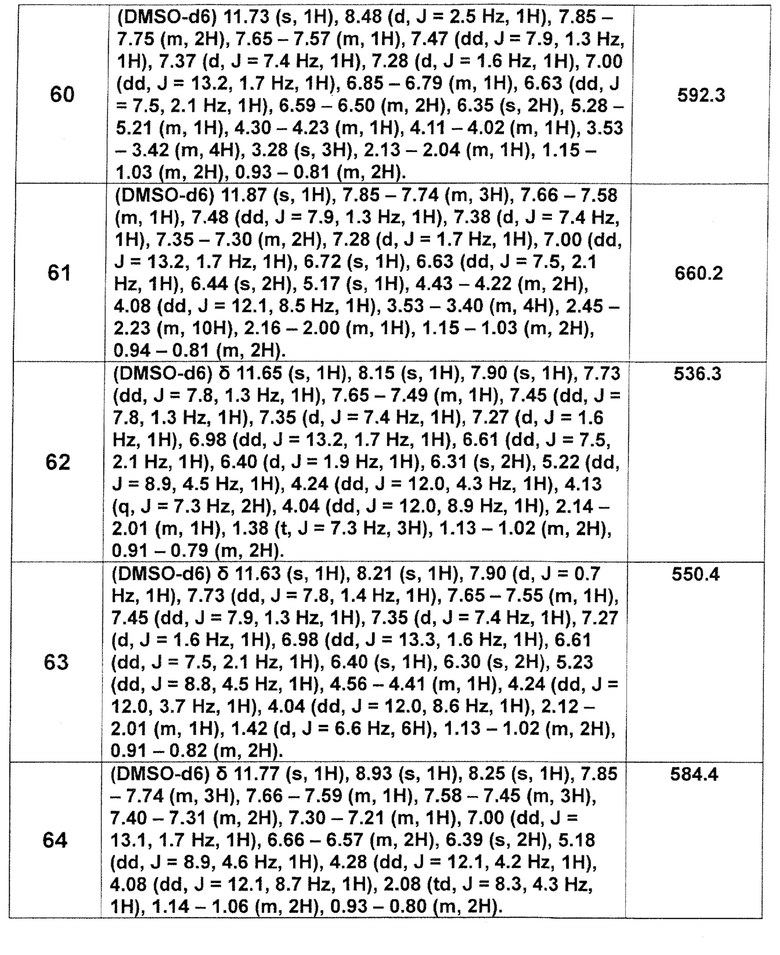
[0093]
[Таблица 1-1]

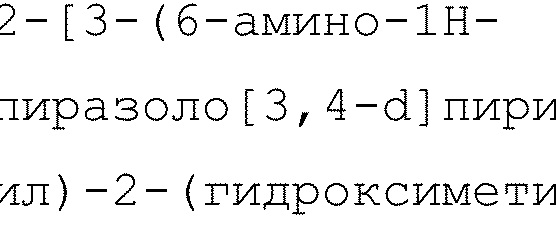
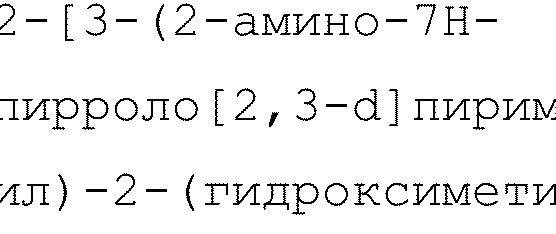
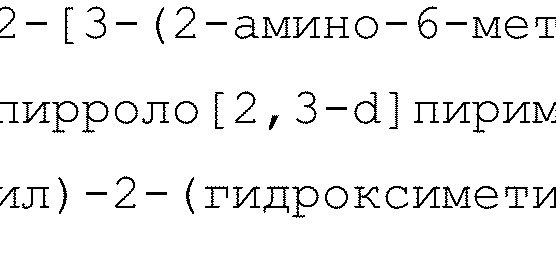


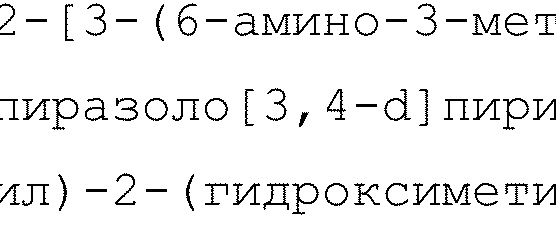



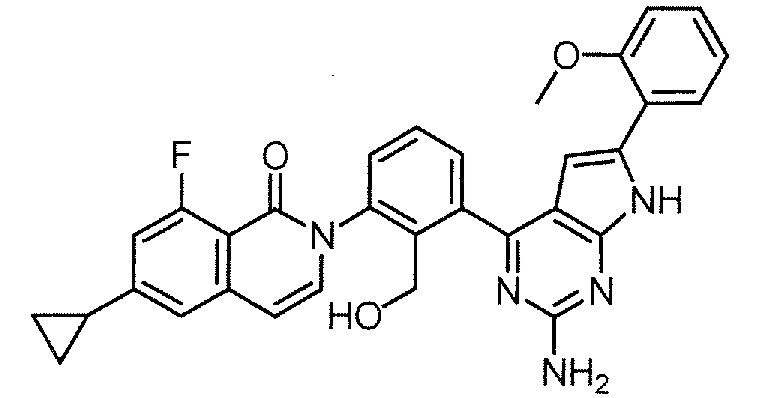

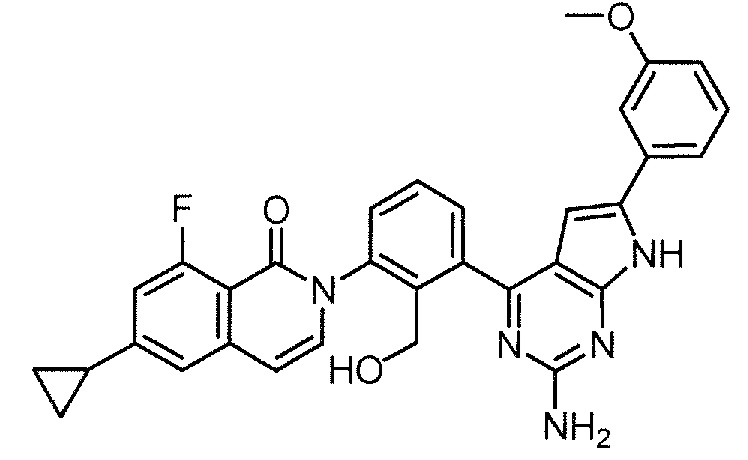



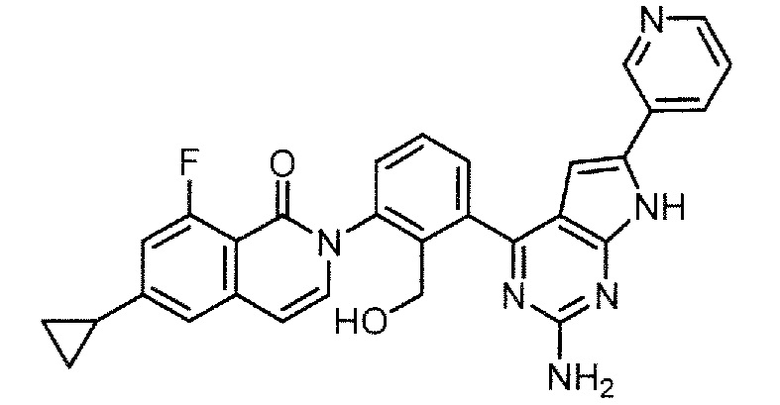

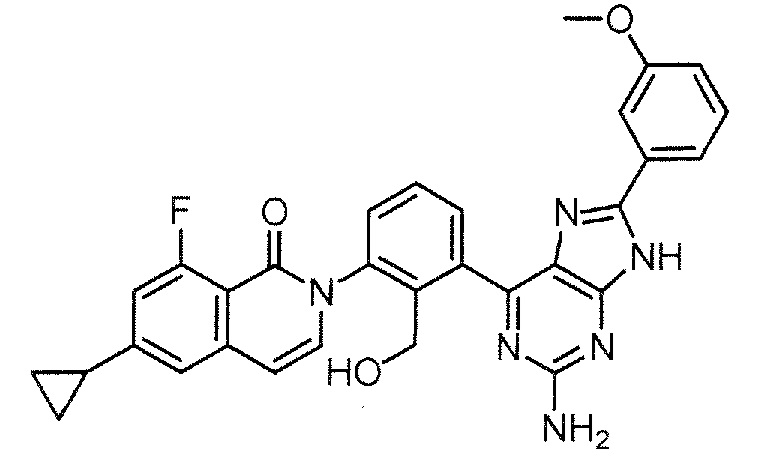



[0094]
Пример 23
2-(3-{2-амино-6-[1-(оксетан-3-ил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-он
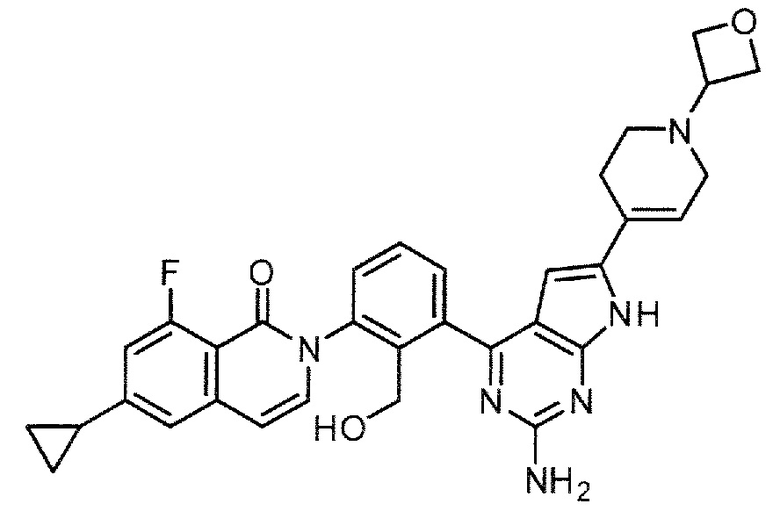
(первая стадия)
Иодид меди (0,25 г, 1,31 ммоль), PdCl2(PPh3)2 (0,92 г, 1,31 ммоль), триметилсилилацетилен (3,87 г, 39,4 ммоль) и триэтиламин (7,32 мл, 52,5 ммоль) добавляли к DMF-раствору (52,5 мл) 6-хлор-5-йод-пиримидин-2,4-диамина (7,1 г, 26,3 ммоль), и перемешивали при 45°C в течение 30 минут. Триметилсилилацетилен (3,87 г, 39,4 ммоль) дополнительно добавляли к реакционному раствору и смесь перемешивали при 45°C в течение 30 минут. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный органический слой последовательно промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали флэш-хроматографией с получением 6-хлор-5-((триметилсилил)этинил)пиримидин-2,4-диамина (6,3 г).
1H ЯМР (400 MГц, DMSO-d6) δ 6,78 (с, 2H), 0,21 (с, 9H);
LCMS (m/z): 241,14 [M+H]+.
[0095]
(вторая стадия)
0,1 М водный раствор гидроксида натрия (58,1 мл, 5,81 ммоль) добавляли к THF-раствору (291 мл) 6-хлор-5-((триметилсилил)этинил)пиримидин-2,4-диамина (7,0 г, 29,1 ммоль), и перемешивали при комнатной температуре в течение часа. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный органический слой последовательно промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении с получением 6-хлор-5-этинилпиримидин-2,4-диамина (4,85 г).
1H ЯМР (400 MГц, DMSO-d6) δ 6,75 (с, 2H), 4,50 (с, 1H);
LCMS (m/z): 169,01 [M+H]+.
[0096]
(третья стадия)
Иодид меди (0,215 г, 1,13 ммоль), PdCl2(PPh3)2 (1,58 г, 2,25 ммоль), трет-бутил 4-{[(трифторметил)сульфонил]окси}-5,6-дигидропиридин-1(2H)-карбоксилат (7,47 г, 22,5 ммоль) и триэтиламин (6,28 мл, 45,1 ммоль) добавляли к DMF-раствору (225 мл) 6-хлор-5-этинилпиримидин-2,4-диамина (3,8 г, 22,5 ммоль), и перемешивали при 90°C в течение 30 минут. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный органический слой последовательно промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали флэш-хроматографией с получением трет-бутил 4-[(2,4-диамино-6-хлорпиридин-5-ил)этинил]-5,6-дигидропиридин-1(2H)-карбоксилата (4,92 г).
1H ЯМР (400 MГц, DMSO-d6) δ 6,73 (с, 2H), 6,14 (с, 1H), 3,97-3,90 (м, 2H), 3,44 (т, J=5,7 Гц, 2H), 2,28-2,24 (м, 2H), 1,41 (с, 9H);
LCMS (m/z): 350,13 [M+H]+.
[0097]
(четвертая стадия)
Трет-бутоксид калия (4,72 г, 42 ммоль) добавляли к раствору N-метилпирролидона (140 мл) трет-бутил 4-[(2,4-диамино-6-хлорпиримидин-5-ил)этинил]-5,6-дигидропиридин-1(2H)-карбоксилата (4,9 г, 14 ммоль) и перемешивали при комнатной температуре в течение часа. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный органический слой последовательно промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали флэш-хроматографией с получением трет бутил 4-(2-амино-4-хлор-7H-пирроло[2,3-d]пиримидин-6-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (2,93 г).
1H ЯМР (400 MГц, DMSO-d6) δ 11,73-11,55 (м, 1H), 6,57 (с, 2H), 6,32 (с, 1H), 6,29-6,16 (м, 1H), 4,14-3,90 (м, 2H), 3,61-3,43 (м, 2H), 2,49-2,35 (м, 2H), 1,42 (с, 9H);
LCMS (m/z): 350,18 [M+H]+.
[0098]
(пятая стадия)
Смешанный растворитель DMF-вода (5:1, 165 мл) добавляли к 2-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил)бензил ацетату (3,96 г, 8,29 ммоль), трет-бутил 4-(2-амино-4-хлор-7H-пирроло[2,3-d]пиримидин--6-ил)-5,6-дигидропиридин-1(2H)-карбоксилату (2,9 г, 8,29 ммоль) и трикалий фосфату (3,52 г, 16,6 ммоль) и смесь дегазировали в атмосфере аргона в течение 30 минут. Добавляли Pd(PPh3)4 (0,96 г, 0,829 ммоль) и перемешивали при 110°С в течение 20 минут. Реакционную смесь разбавляли этилацетатом, последовательно промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении с получением трет бутил 4-{4-[2-(ацетоксиметил)-3-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)фенил]-2-амино-7H-пирроло[2,3-d]пиримидин-6-ил}-5,6-дигидропиридин-1(2H)-карбоксилата (5,43 г).
LCMS (m/z): 665,37 [M+H]+.
[0099]
(шестая стадия)
Триэтиламин (4,45 мл, 32 ммоль) и ацетилхлорид (1,89 мл, 26,6ммоль) добавляли к THF-раствору (53 мл) трет-бутил 4-{4-[2-(ацетоксиметил)-3-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)фенил]-2-амино-7H-пирроло[2,3-d]пиримидин-6-ил}-5,6-дигидропиридин-1(2H)-карбоксилата (3,54 г, 5,33 ммоль), и перемешивали при комнатной температуре в течение часа. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный органический слой последовательно промывали водой, 1М раствором гидроксида натрия и насыщенным солевым раствором последовательно и сушили над безводным сульфатом натрия. Растворитель выпаривали с получением трет-бутил 4-{2-ацетамид-4-[2-(ацетоксиметил)-3-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)фенил]-7H-пирроло[2,3-d]пиримидин-6-ил}-5,6-дигидропиридин-1(2H)-карбоксилата (5,44 г) в виде неочищенного продукта.
LCMS (m/z): 707,43 [M+H]+.
[0100]
[седьмая стадия]
4M хлорид водорода в растворе 1,4-диоксана (50 мл) добавляли к раствору DCM (150 мл) трет-бутил 4-{2-ацетамид-4-[2-(ацетоксиметил)-3-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)фенил]-7H-пирроло[2,3-d]пиримидин-6-ил}-5,6-дигидропиридин-1(2H)-карбоксилата (6,66 г, 9,42 ммоль) и перемешивали при комнатной температуре в течение 6 часов. К реакционной смеси добавляли 4М водный раствор гидроксида натрия (50 мл), добавляли воду и экстрагировали хлороформом. Полученный органический слой последовательно промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и полученный остаток очищали флэш-хроматографией с получением 2-[2-ацетамид-6-(1,2,3,6-тетрагидропиридин-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-6-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)бензил ацетата (2,66 г).
1H ЯМР (400 MГц, DMSO-d6) δ 12,47 (с, 1H), 10,53 (с, 1H), 7,77-7,66 (м, 2H), 7,54 (дд, J=6,8, 2,4 Гц, 1H), 7,40 (д, J=7,4 Гц, 1H), 7,28 (д, J=1,7 Гц, 1H), 7,00 (дд, J=13,3, 1,7 Гц, 1H), 6,64 (дд, J=7,5, 2,1 Гц, 1H), 6,57-6,50 (м, 1H), 6,41 (с, 1H), 5,21 (д, J=12,6 Гц, 1H), 4,99 (д, J=12,6 Гц, 1H), 3,71-3,66 (м, 2H), 3,18 (т, J=6,0 Гц, 2H), 2,62-2,57 (м, 2H), 2,15 (с, 3H), 2,13-2,02 (м, 1H), 1,51 (с, 3H), 1,15-1,05 (м, 2H), 0,94-0,86 (м, 2H);
LCMS (m/z): 607,31 [M+H]+.
[0101]
(восьмая стадия)
Оксетан-3-он (1,25 г, 17,3 ммоль) и триацетоксиборгидрид натрия (3,67 г, 17,3 ммоль) добавляли к раствору DCM (69 ммоль) 2-[2-ацетамид-6-(1,2,3,6-тетрагидропиридин-4-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-6-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)бензил ацетата (2,1 г, 3,46 ммоль), и перемешивали при комнатной температуре в течение часа. Оксетан-3-он (1,25 г, 17,3 ммоль) и триацетоксиборгидрид натрия (3,67 г, 17,3 ммоль) снова добавляли к реакционной смеси и перемешивали при комнатной температуре еще час. К реакционной смеси добавляли воду, и экстрагировали этилацетатом. Полученный органический слой последовательно промывали водой, 1 М водным раствором гидроксида натрия и насыщенным солевым раствором. и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении с получением 2-{2-ацетамид-6-[1-(оксетан-3-ил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-6-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)бензил ацетата (2,29 г).
1H ЯМР (400 MГц, DMSO-d6) δ 12,48 (с, 1H), 10,52 (с, 1H), 7,77-7,66 (м, 2H), 7,53 (дд, J=7,0, 2,2 Гц, 1H), 7,44-7,35 (м, 1H), 7,28 (д, J=1,7 Гц, 1H), 7,00 (дд, J=13,3, 1,7 Гц, 1H), 6,63 (дд, J=7,6, 2,1 Гц, 1H), 6,59-6,52 (м, 1H), 6,31 (с, 1H), 5,22 (д, J=12,6 Гц, 1H), 4,99 (д, J=12,6 Гц, 1H), 4,67-4,46 (м, 4H), 3,61-3,50 (м, 1H), 3,06-3,01 (м, 2H), 2,51-2,43 (м, 4H), 2,16 (с, 3H), 2,12-2,02 (м, 1H), 1,51 (с, 3H), 1,15-1,01 (м, 2H), 0,94-0,80 (м, 2H);
LCMS (m/z): 663,37 [M+H]+.
[0102]
(девятая стадия)
2М водный раствор гидроксида натрия (50 мл) добавляли к метанольному раствору (100 мл) 2-{2-ацетамид-6-[1-(оксетан-3-ил)-1,2,3,6-тетрагидропиридин-4-ил]-7H-пирроло[2,3-d]пиримидин-4-ил}-6-(6-циклопропил-8-фтор-1-оксоизохинолин-2(1H)-ил)бензил ацетата (2,3 г, 3,47 ммоль), и перемешивали при 70°С в течение 2 часов. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный органический слой последовательно промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении с получением указанного в заголовке продукта (1,4 г).
1H ЯМР (400 MГц, DMSO-d6) δ 11,52 (с, 1H), 7,72 (дд, J=7,8, 1,3 Гц, 1H), 7,64-7,53 (м, 1H), 7,46 (дд, J=7,8, 1,3 Гц, 1H), 7,37 (д, J=7,4 Гц, 1H), 7,28 (д, J=1,6 Гц, 1H), 6,99 (дд, J=13,2, 1,7 Гц, 1H), 6,62 (дд, J=7,4, 2,1 Гц, 1H), 6,41 (с, 2H), 6,38-6,32 (м, 1H), 6,27-6,18 (м, 1H), 5,20 (дд, J=8,8, 4,5 Гц, 1H), 4,61-4,46 (м, 4H), 4,25 (дд, J=12,0, 4,2 Гц, 1H), 4,06 (дд, J=12,0, 8,8 Гц, 1H), 3,60-3,48 (м, 1H), 3,04-2,98 (м, 2H), 2,48-2,43 (м, 4H), 2,14-2,00 (м, 1H), 1,15-1,01 (м, 2H), 0,92-0,82 (м, 2H);
LCMS (m/z): 579,60 [M+H]+.
[0103]
[Таблица 1-2]
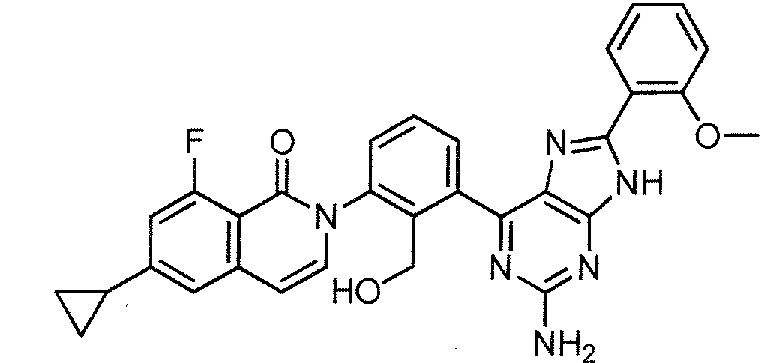
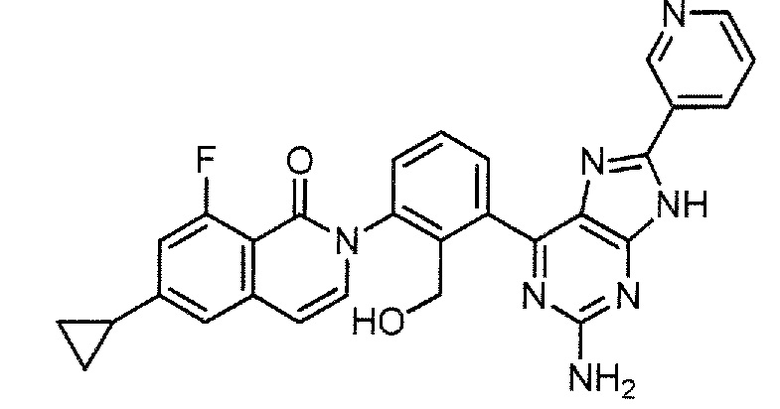
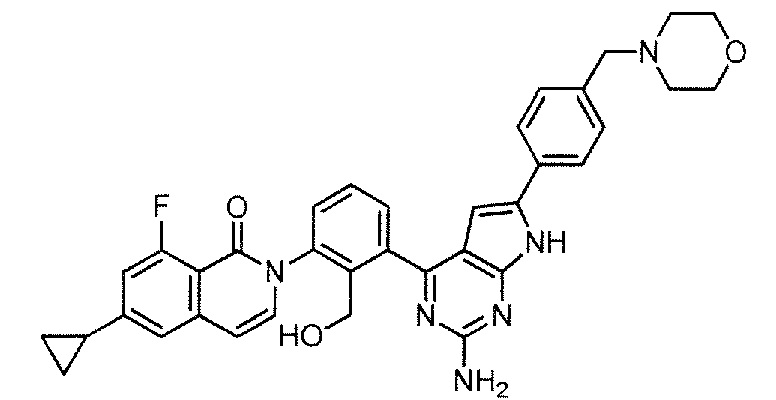
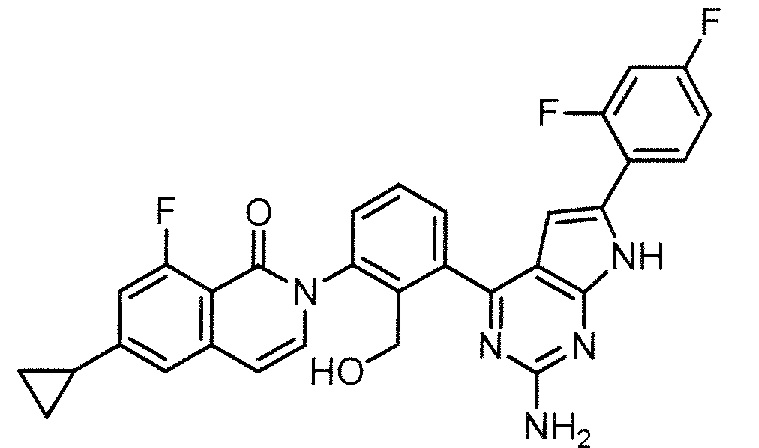
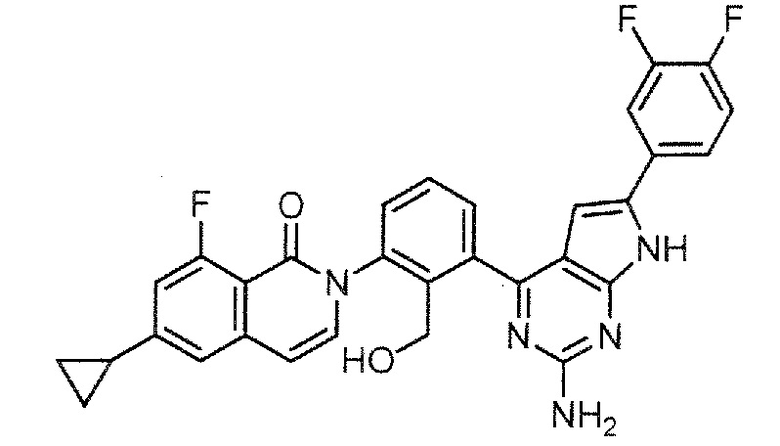
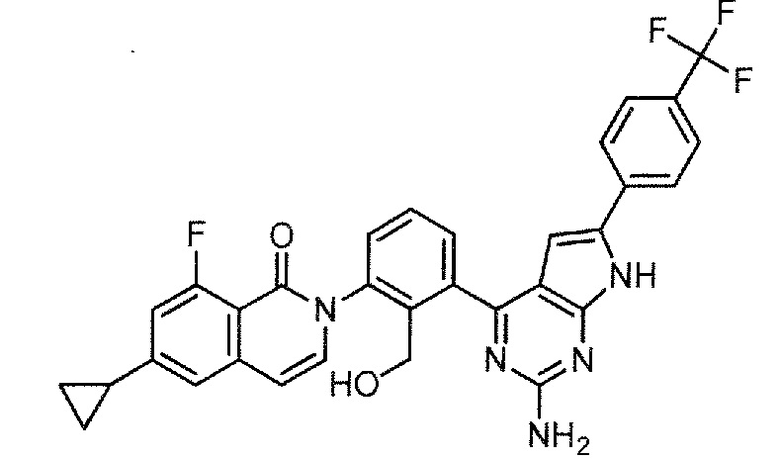
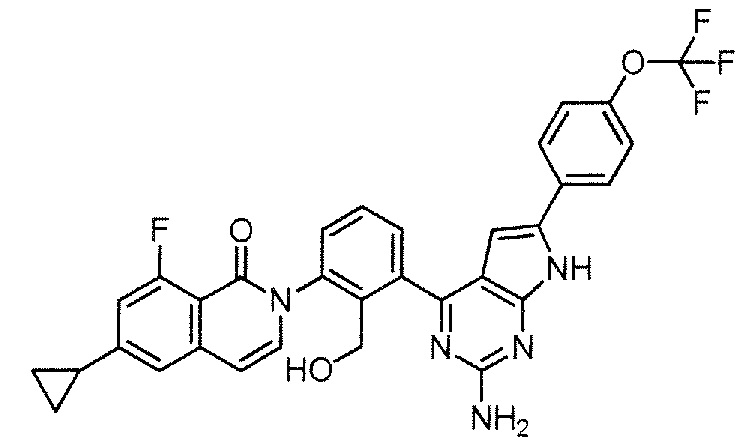
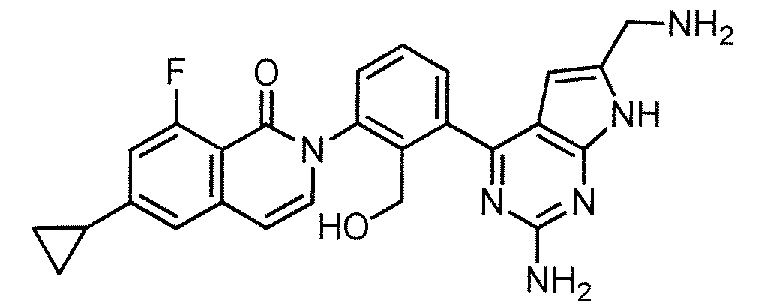
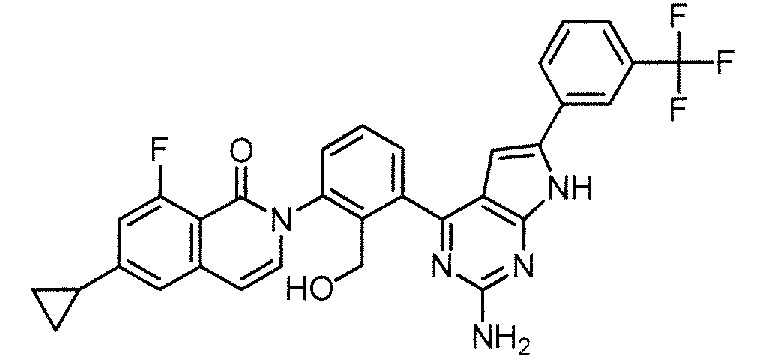
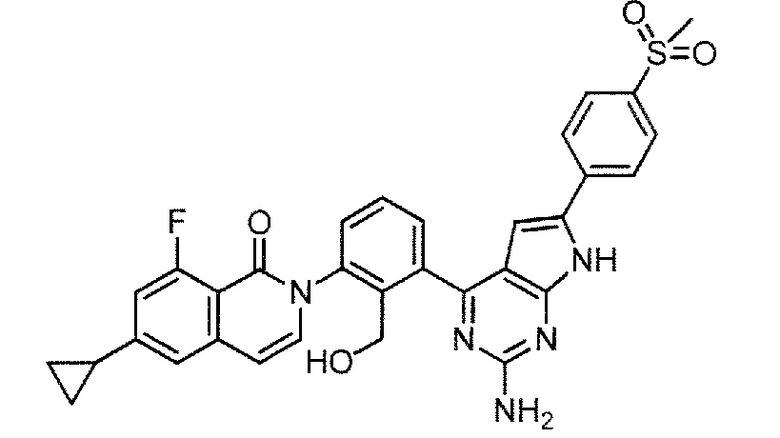
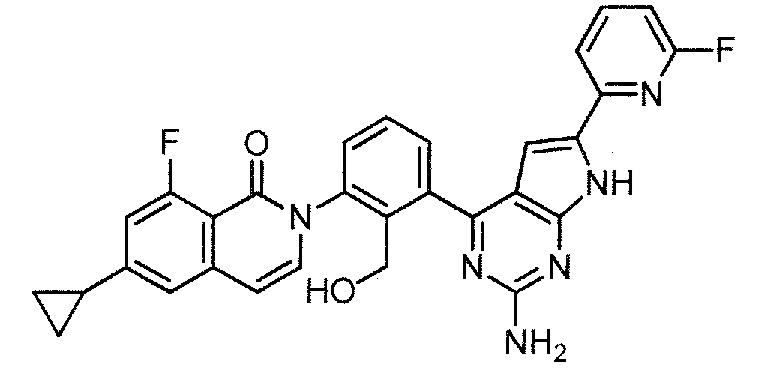
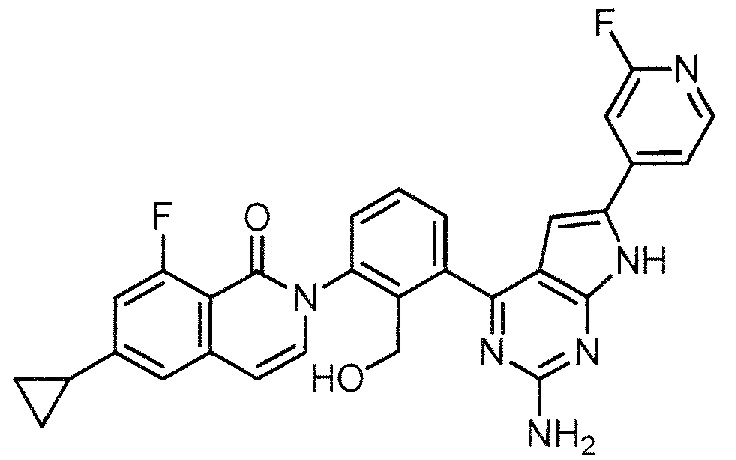
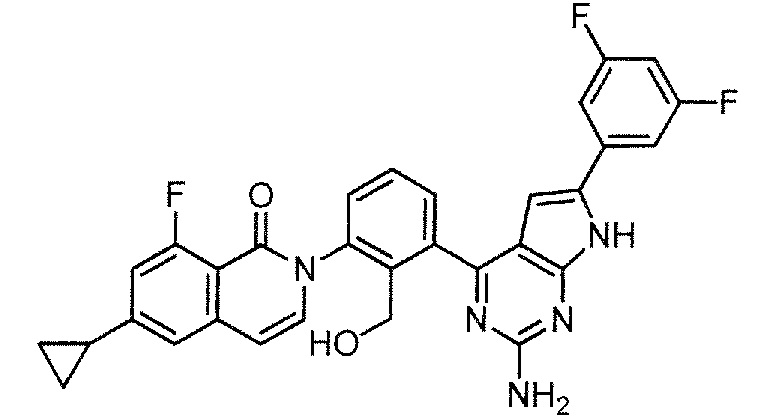
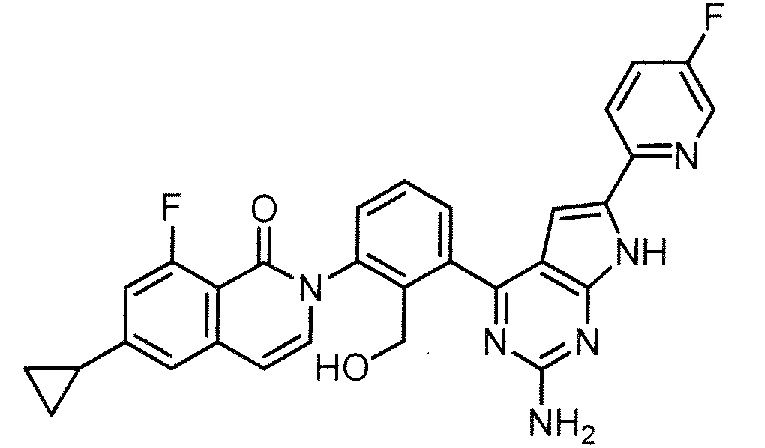
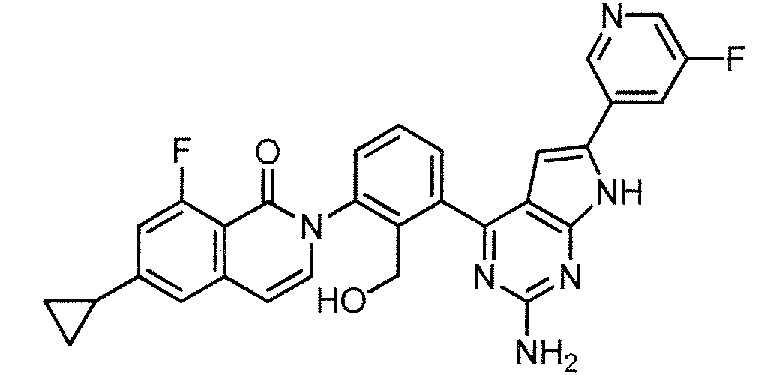
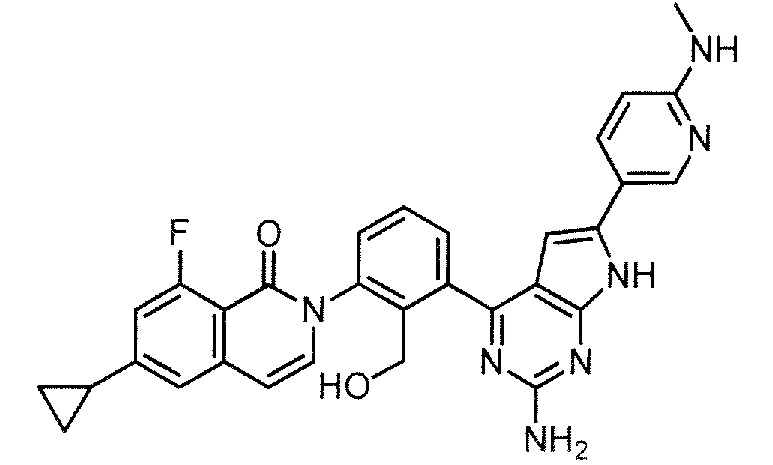
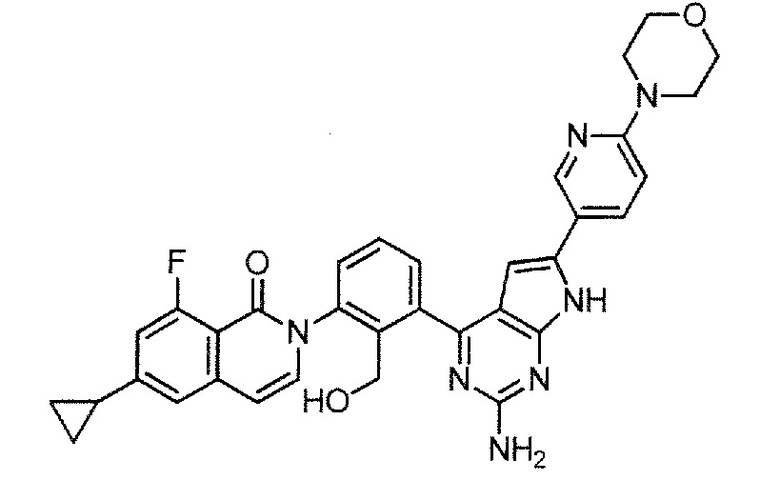
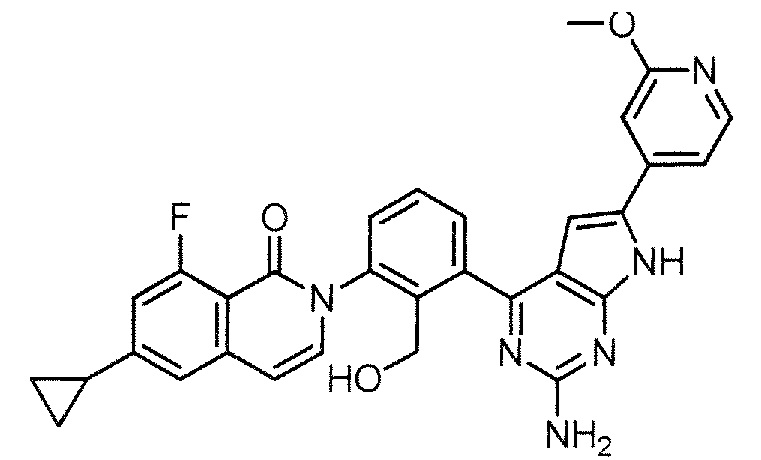
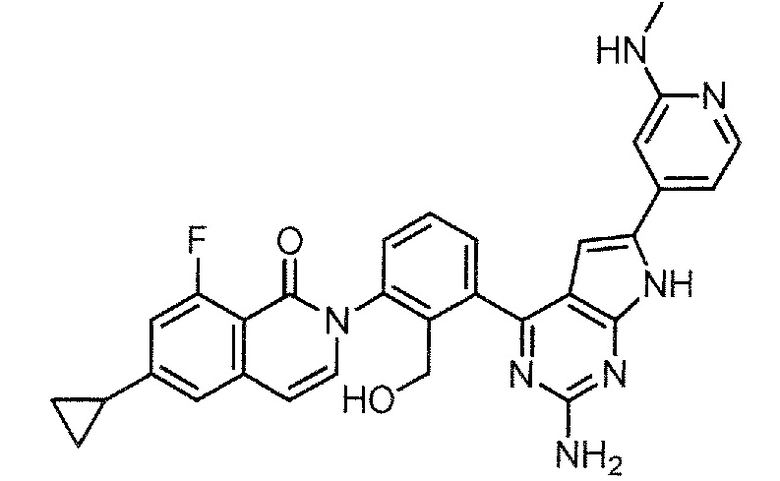
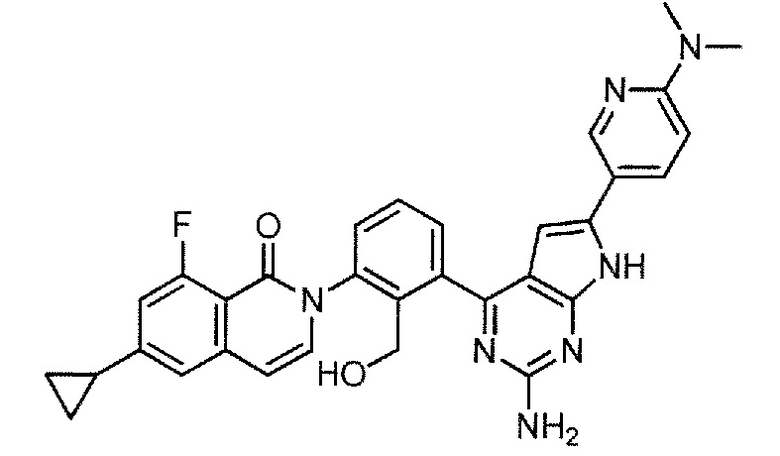
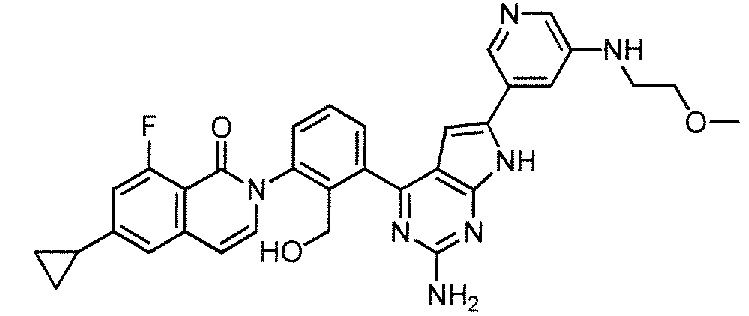
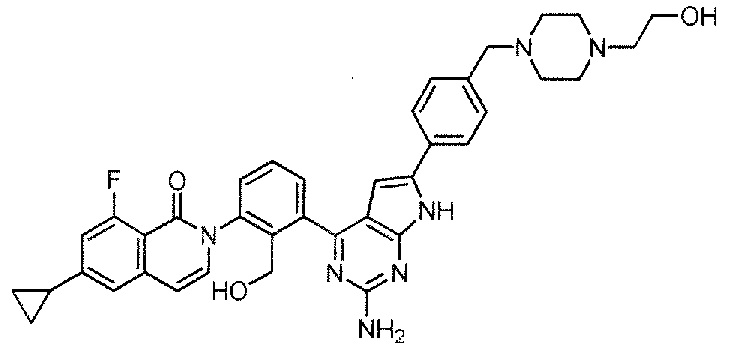
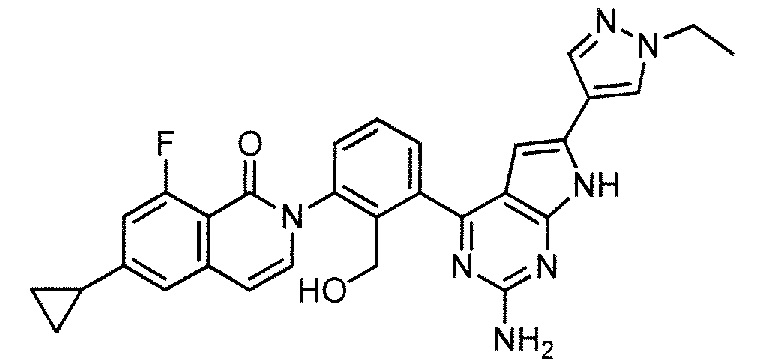
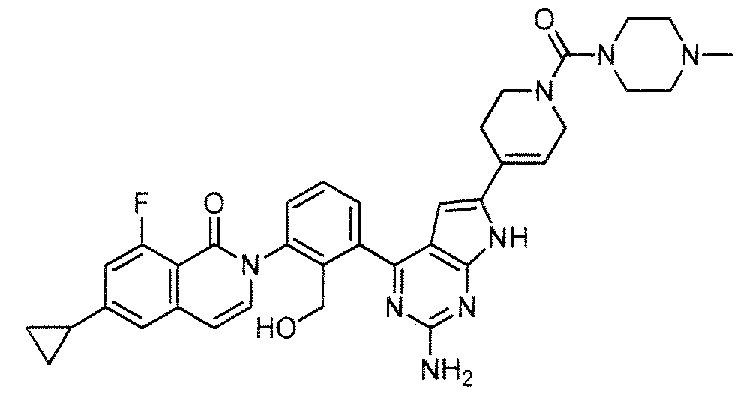
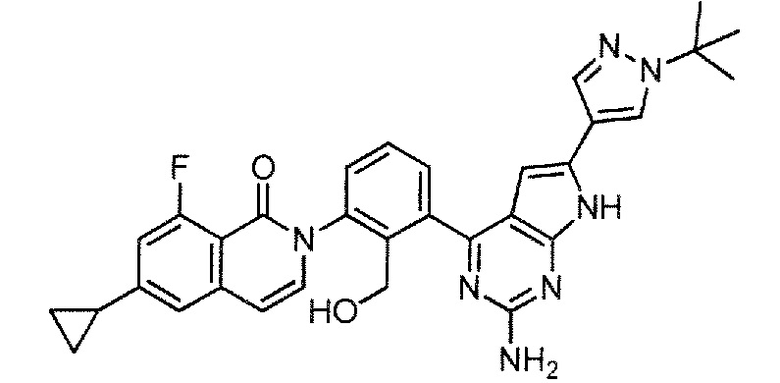
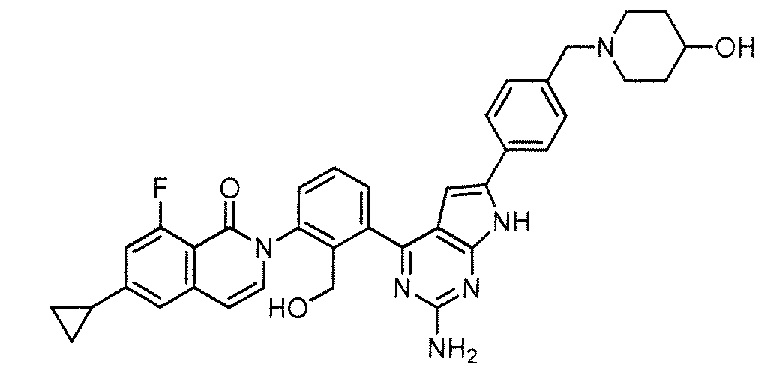
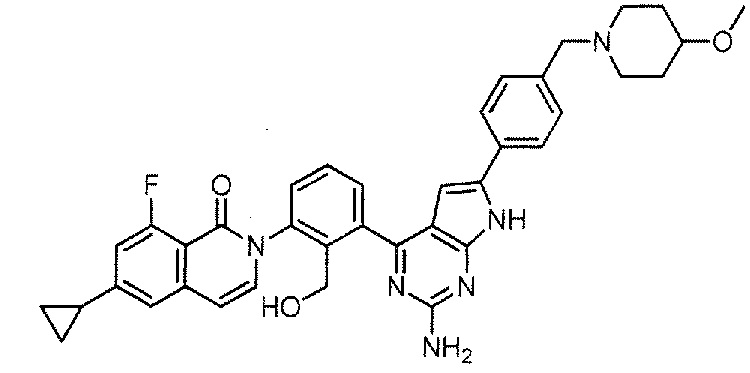
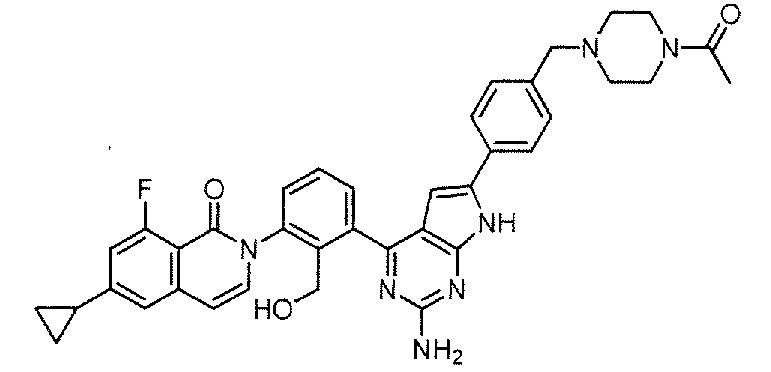
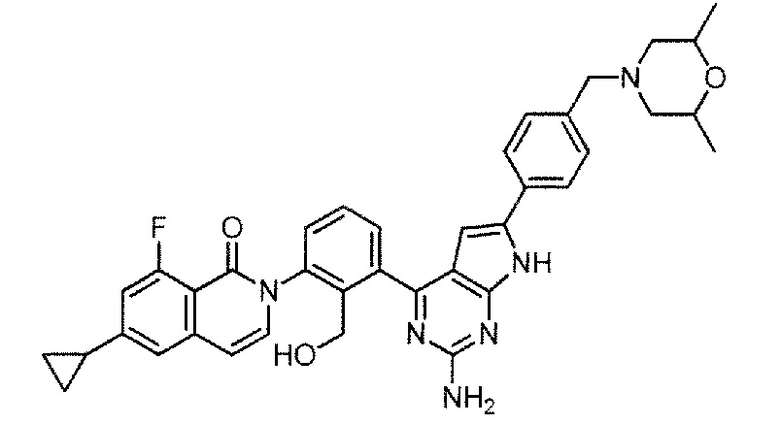
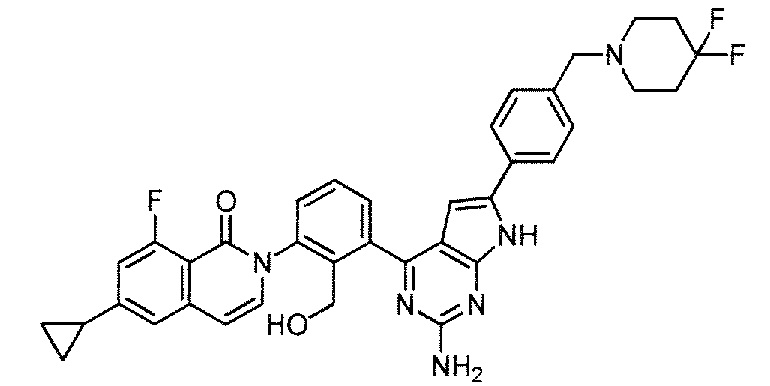
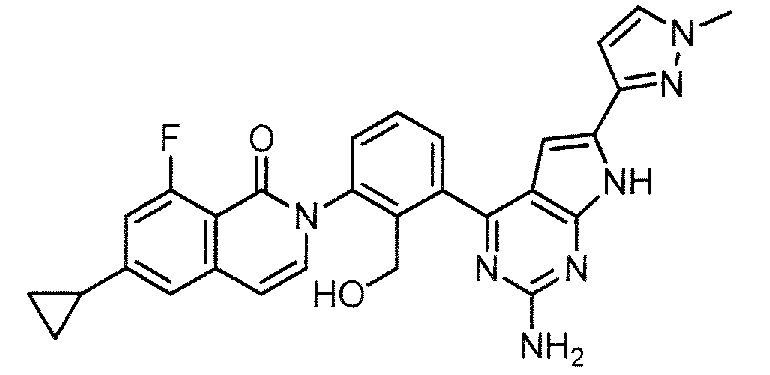
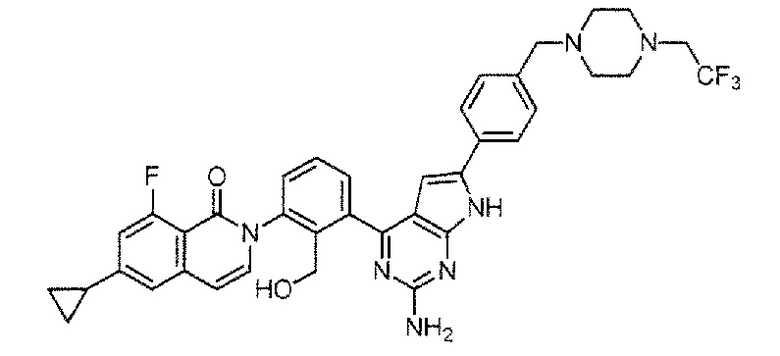
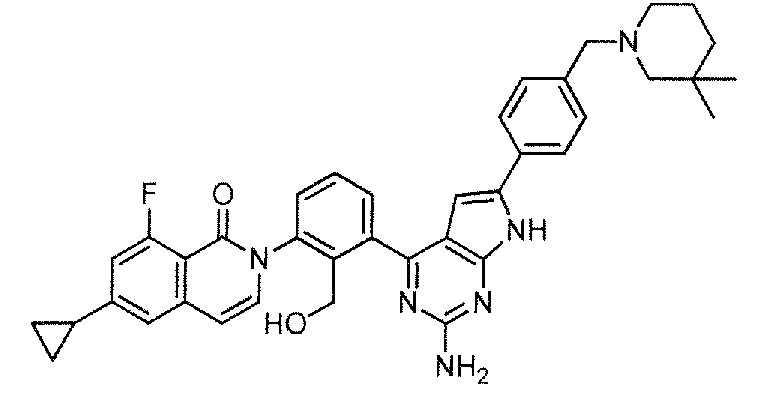
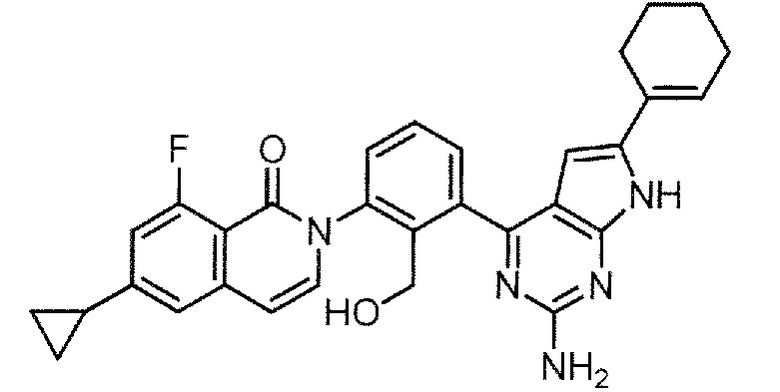
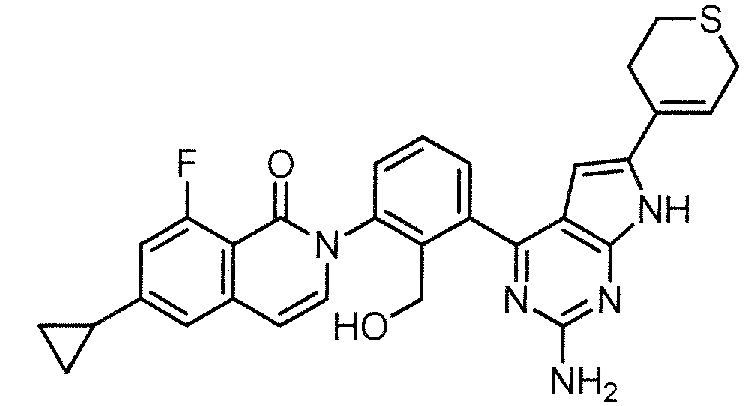
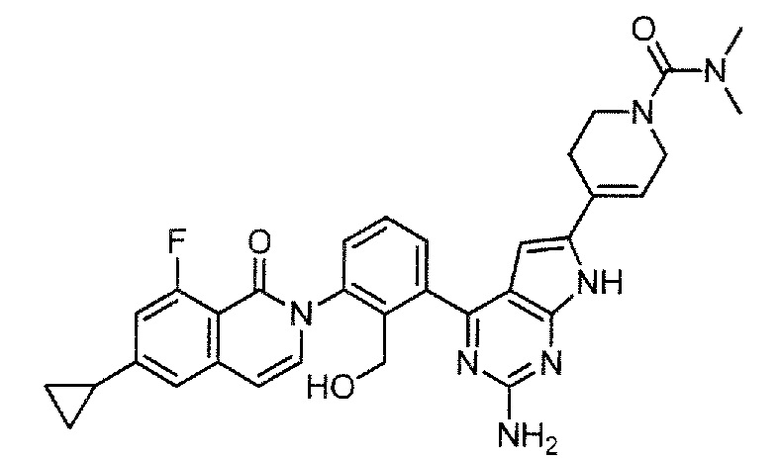
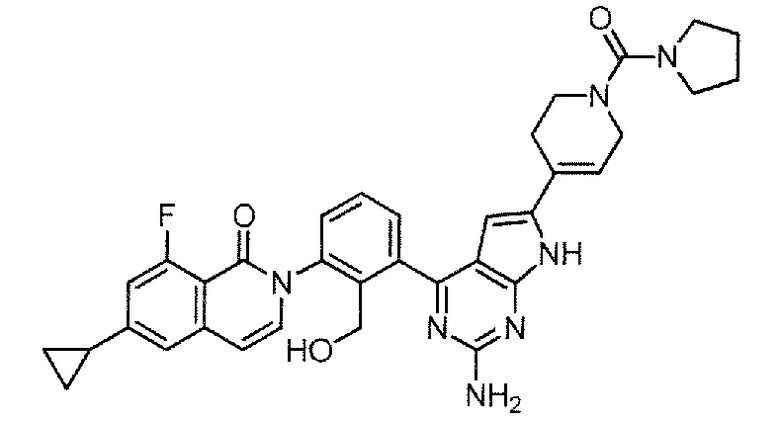
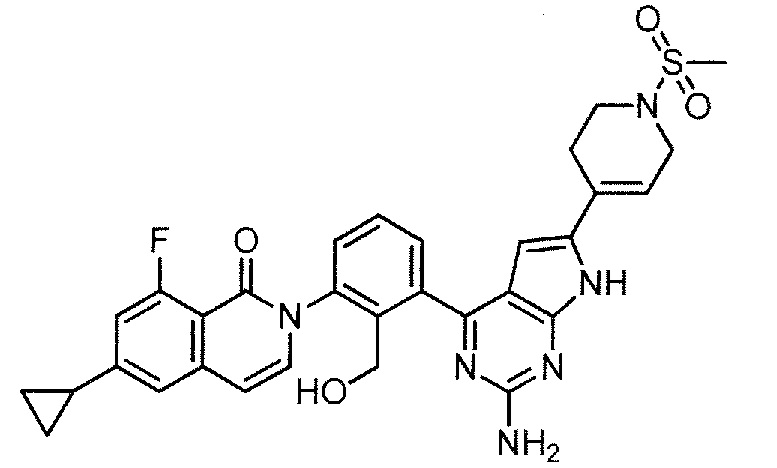
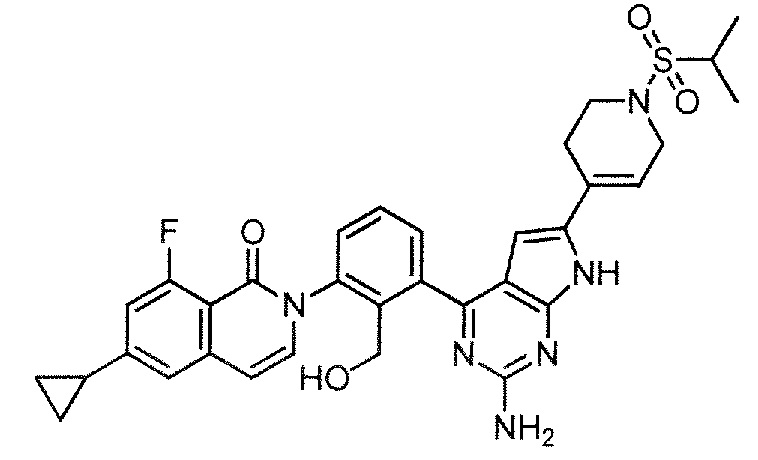
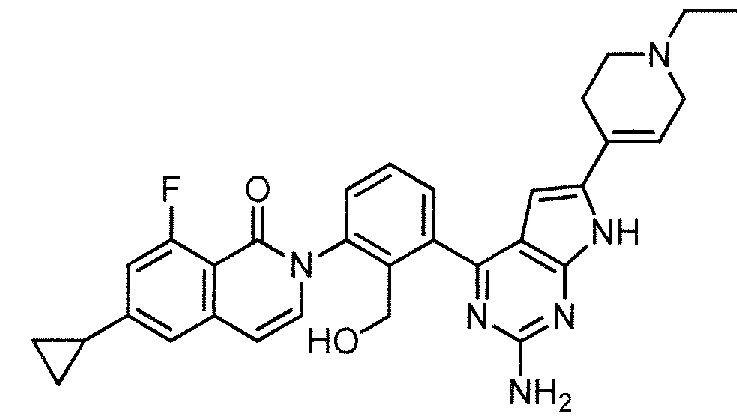
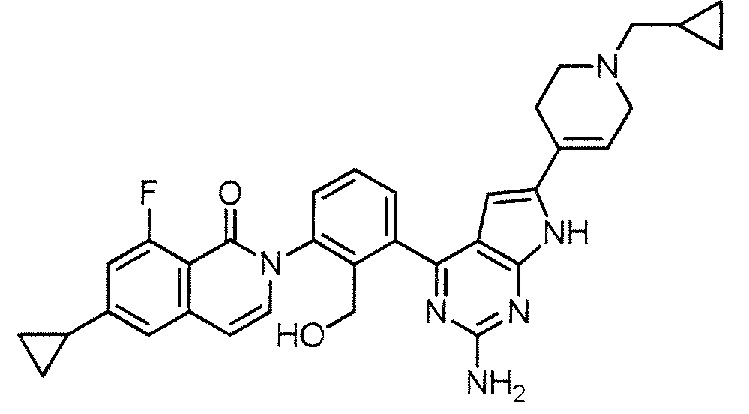
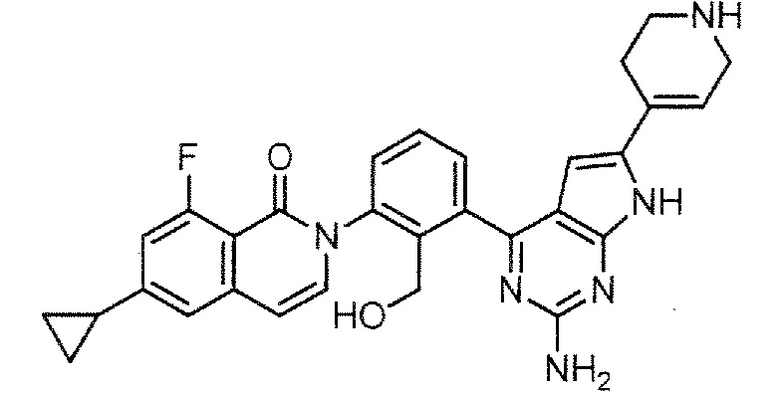
[0104]
[Таблица 2-1]
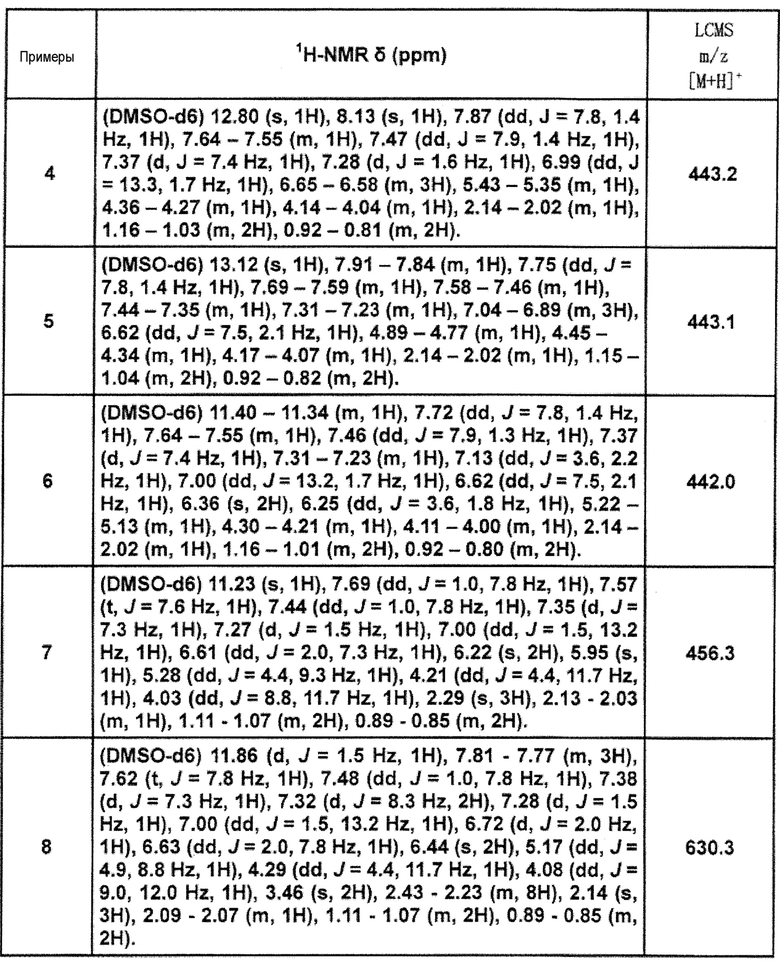
[0105]
[Таблица 2-2]
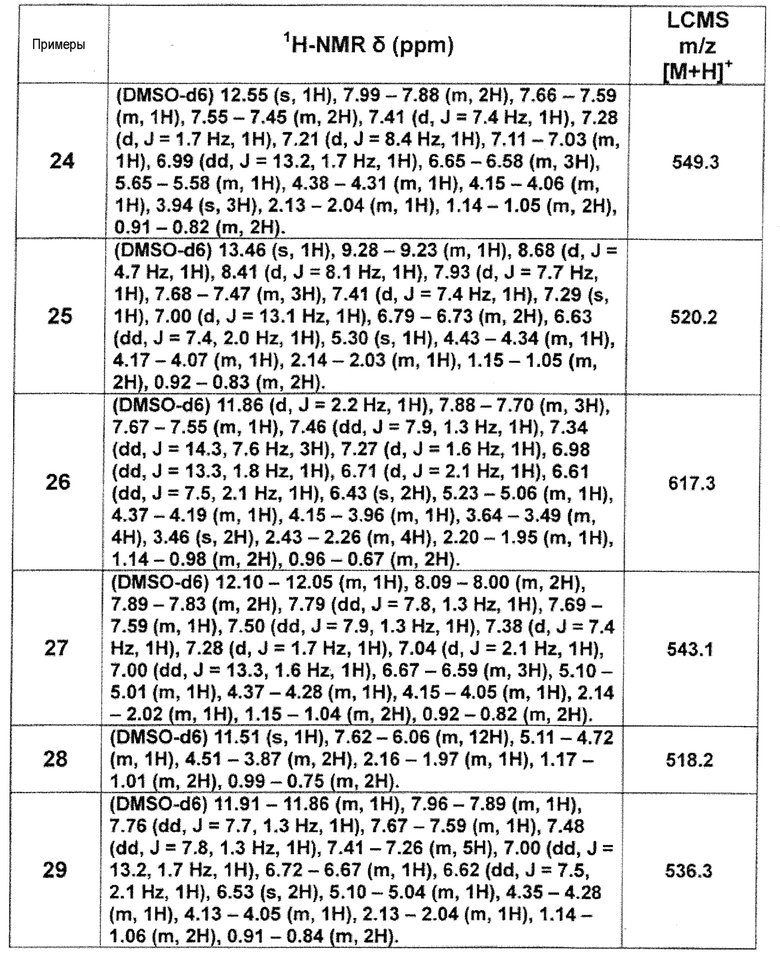
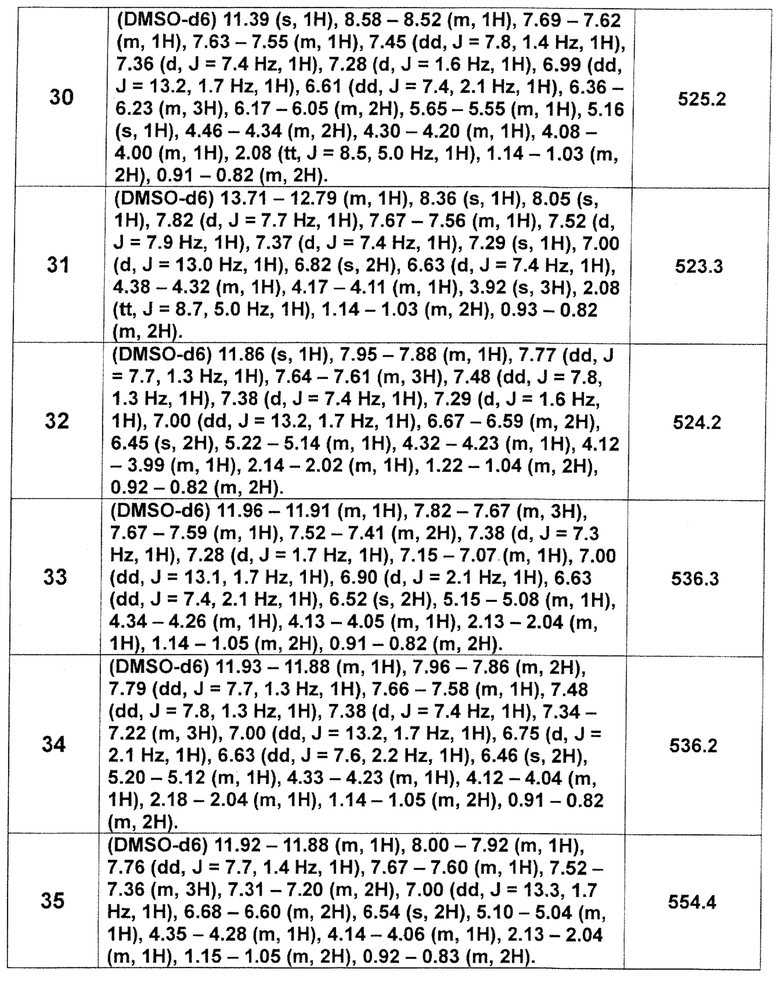

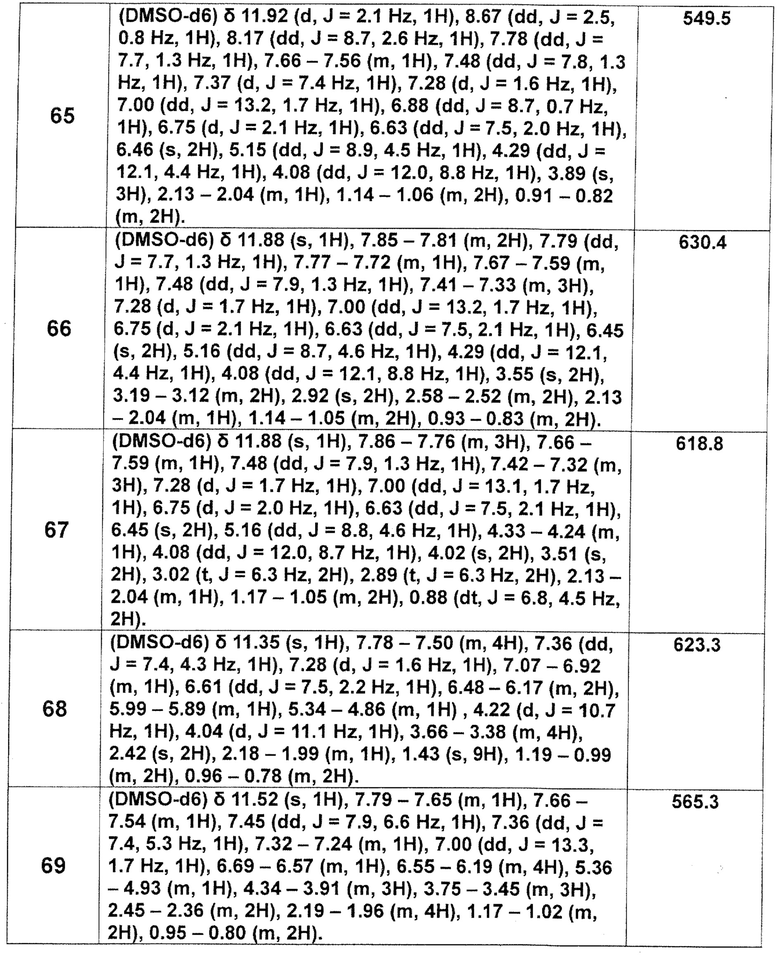
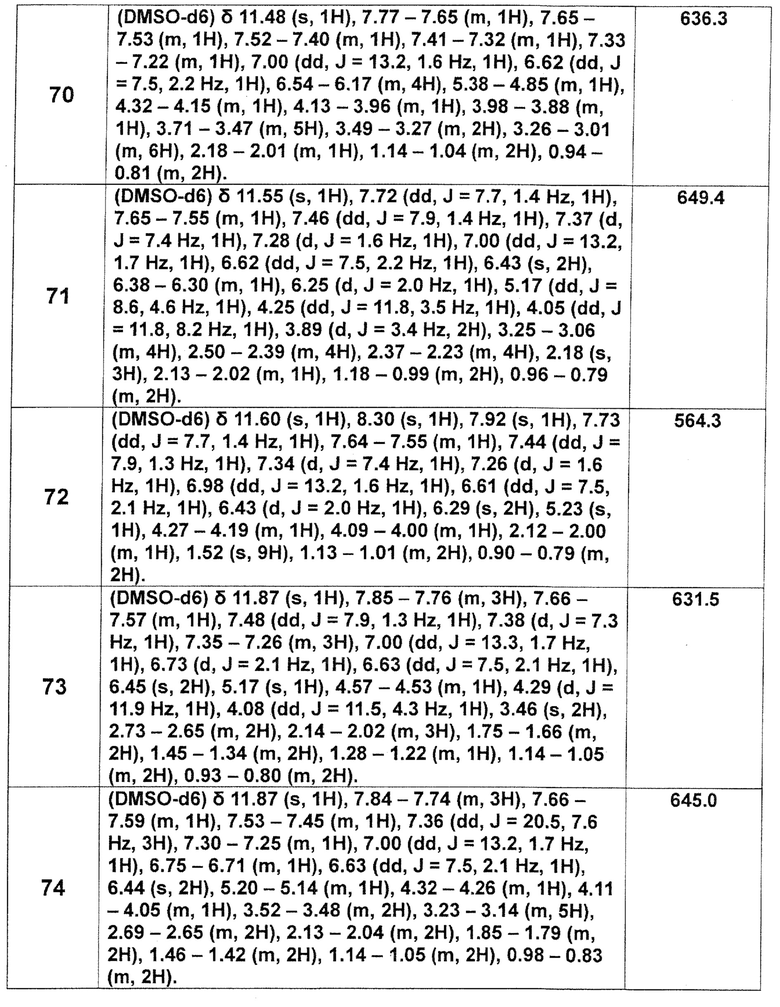
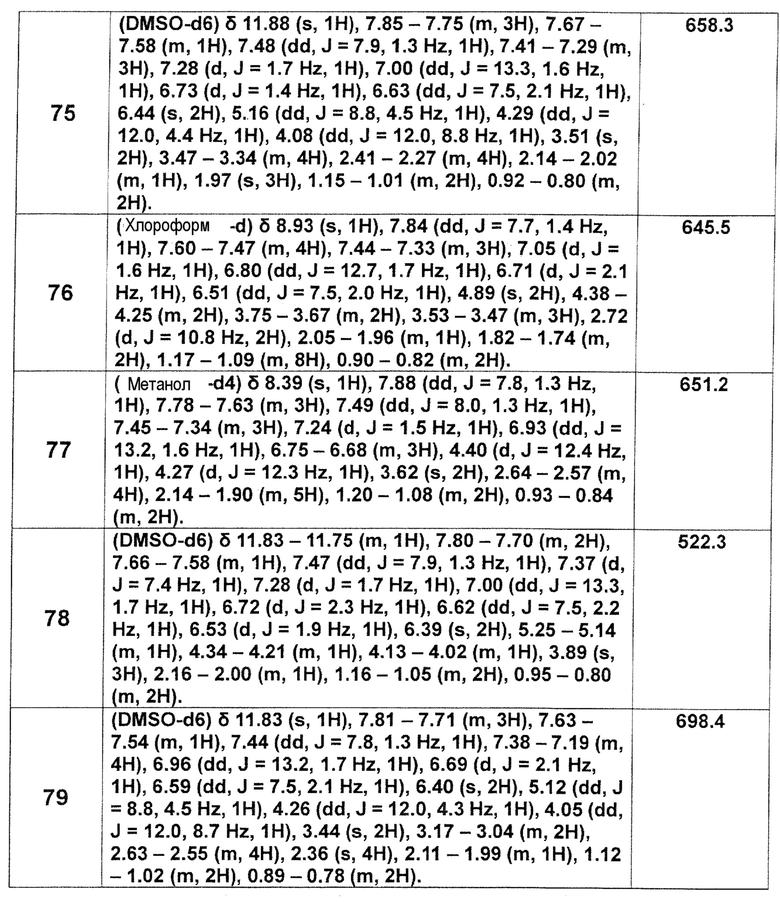
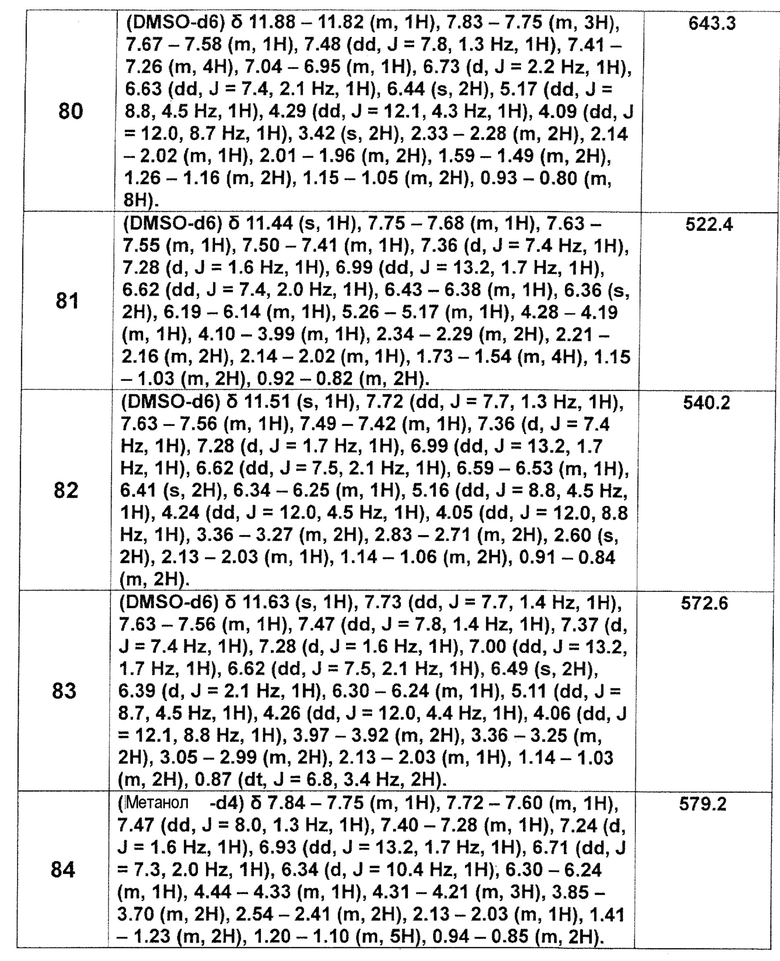

[0106]
Пример испытаний 1
Тест на ингибирование активности BTK
(Получение дефосфорилированного BTK)
Дефосфорилированный BTK получали путем добавления λ протеинфосфатазы (производства New England BioLabs Inc., Code No.P0753S) и MnCl2 при 10 ед./мкг и 2 мМ, соответственно, к биотинилированному BTK-белку BTN-BTK (производства Carna Biosciences, Inc.) ферментного раствора, реакции смеси при 4°С в течение ночи и удаления λ протеинфосфатазы с помощью хроматографии на агарозном геле с анти DYKDDDDK-tag антителом с последующим обменом буфера с использованием колонки для обессаливания 10DG Desalting Column.
[0107]
(Метод измерения киназной активности)
Активность киназы измеряли с использованием QuickScout Screening Assist (торговая марка) MSA (коммерчески доступный набор, выпускаемый Carna Biosciences, Inc.) методом анализа изменения подвижности (MSA). Субстратом киназной реакции был FITC-меченный SRCtide пептид, включенный в набор. Использовали буфер для анализа [20 мМ HEPES, 0,01% Triton X-100 (торговая марка), 2 мМ дитиотреитол, pH 7,5] и доводили до 4 мкМ субстрата, 20 мМ MgCl2 и 200 мкм ATP для получения раствора субстратной смеси. Ферментный раствор также получали разбавлением дефосфорилированного BTK до 0,46 нМ с использованием буфера для анализа. 10 мМ раствор испытуемого соединения в DMSO дополнительно разбавляли DMSO до 10 уровней концентрации (0,00003 мM, 0,0001 мM, 0,0003 мM, 0,001 мM, 0,003 мM, 0,01 мM, 0,03 мM, 0,1 мM, 0,3 мM, 1 мM), каждый из которых подвергали 25-кратному разбавлению буфером для анализа для получения растворов лекарственного средства (4% растворы DMSO). 5 мкл раствора лекарственного средства или контрольного раствора (4% DMSO-буфер для анализа), 5 мкл раствора субстратной смеси и 10 мкл ферментного раствора смешивали в лунках полипропиленового 384-луночного планшета и давали возможность взаимодействовать при комнатной температуре в течение 2 часов, а затем гасили путем добавления 60 мкл терминирующего буфера, включенного в набор. Затем количество субстратов (S) и фосфорилированного субстрата (P) в реакционном растворе измеряли с использованием системы LabChip EZ Reader II (производства Caliper Life Sciences) в соответствии с протоколом набора для анализа.
[0108]
(Способ оценки BTK ингибирующей активности)
Высоты пиков изолированного субстрата и фосфорилированного субстрата были представлены как S и P, соответственно, и также был измерен холостой раствор, который содержал буфер для анализа вместо ферментного раствора.
Степень ингибирования (%) тестируемого соединения рассчитывали в соответствии со следующим уравнением;
Степень ингибирования (%)=(1-(C-A)/(B-A))×100
где, A, B и C представляют P/(P+S) холостой лунки, P/(P+S) контрольной лунки и P/(P+S) лунки, содержащей соединение, соответственно.
[0109]
Значение IC50 рассчитывали с помощью регрессионного анализа степени ингибирования (%) и концентрации тестируемого соединения (логарифмическая величина).
[0110]
(Результаты анализа)
Поскольку группа соединений из примеров показала значения IC50 от 10 нМ или менее до 100 нМ или менее в отношении дефосфорилированного BTK, было обнаружено, что соединение (I) по настоящему изобретению обладает сильным BTK ингибирующим эффектом. Ингибирующая активность в отношении дефосфорилированного BTK типичных соединений по настоящему изобретению показана в таблице 3. Ингибирующая активность была обозначена знаком «***», когда значение IC50 составляет менее 0,01 мкМ, знаком «**», когда значение IC50 составляет от 0,01 мкМ до менее 0,1 мкМ, и знаком «*», когда значение IC50 составляет от 0,1 мкМ до менее 1,0 мкМ.
[Таблица 3]
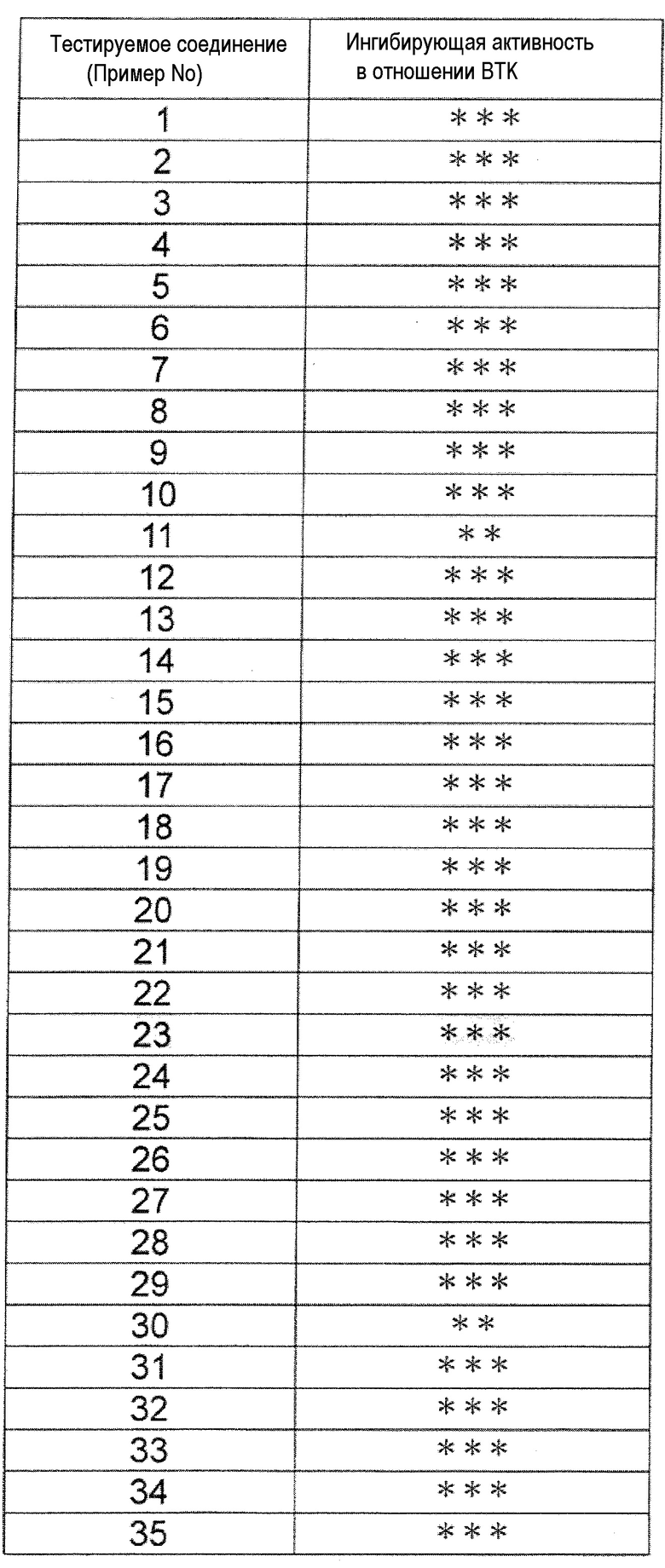
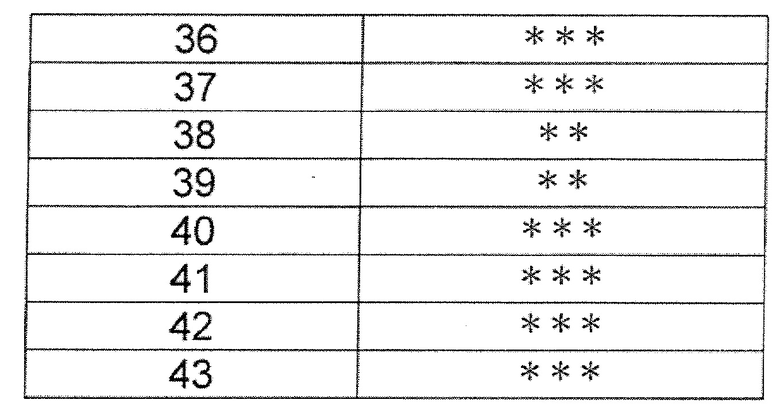
[0111]
Пример испытаний 2
Анализ на ингибирование внутриклеточной BTK аутофосфорилирующей активности
(Культура клеток для использования)
Клетки Ramos ((2G6.4C10, ATCC No.CRL-1923) культивировали в колбе T75, содержащей среду RPMI-1640 (GIBCO, #A10491-01), дополненную 10% FBS (AusGene) и 1% пенициллин-стрептомицина (Nacalai Tesque, Inc.) (далее называемая как питательная среда) в инкубаторе с 5% CO2.
[0112]
(Добавление соединения для тестирования)
Культивируемые клетки Ramos разбавляли до плотности клеток 7,5×106 клеток/мл с помощью бессывороточной RPMI-1640 (далее называемого средой) и хранили при 37°C в течение 45 минут. Суспензию клеток распределяли по 1 мл аликвотам в 2,0 мл пробирки. 0,03 мМ раствор тестируемого вещества в DMSO разбавляли средой для получения 0,09 мкМ раствора тестируемого соединения, 500 мкл которого затем добавляли в пробирки, и инкубацию проводили при 37°С в течение 1 часа в присутствии тестируемого соединения в конечной концентрации 0,03 мкМ. После этого добавляли анти-IgM антитело (Invitrogen, H15100), которое было разбавлено средой, до конечной концентрации 10 мкг/мл, и инкубацию проводили при 37°С в течение 10 минут.
[0113]
(Экстракция белков)
К осадкам, полученным путем восстановления клеток посредством центрифугирования, добавляли 100 мкл буфера для лизиса [буфер RIPA(×1)(Cell Signaling Technology, Inc.), дополненный 1% коктейлем ингибиторов фосфатазы 3 (Sigma Corporation, No.P0044), 1% коктейлем ингибиторов фосфатазы (Nacalai Tesque, Inc., No.07575) и 1 мМ фенилметилсульфонилфторидом (PMSF)] и осторожно перемешивали и затем давали отстояться в течение 10 минут. Супернатант извлекали центрифугированием (15000 об/мин, 15 минут) и количественно определяли уровень белка. Порцию смешивали с буфером SDS-образца, давая взаимодействовать в течение 5 минут при 95°C для денатурации белка, таким образом получая раствор образца. Каждые 5 мкл растворов образцов вносили в каждую лунку, содержащую 5-20% градиентный акриламидный гель (Nacalai Tesque, Inc., No.13064-04), и проводили электрофорез. После этого систему переноса геля iBlot (Life Technologies Corporation) использовали для переноса белков в геле на мембрану PVDF.
[0114]
(Обнаружение BTK или фосфорилированного BTK)
Мембрану PVDF после переноса блокировали блокирующим реагентом 2% ECL prime blocking Reagent (GE Healthcare), и после этого реакцию осуществляли в течение ночи при 4°C с использованием мышиного анти-BTK антитела (BD transduction laboratory, No.611116) или кроличьего анти-фосфорилированной BTK антитела (pY223, EPITOMICS, No.2207-1) в качестве первичного антитела. Непрореагировавшее первичное антитело промывали буфером TBST (10 мM Tris-HCl (pH7,5), 150 мM NaCl, 0,1% Tween 20) и затем реакцию проводили в течение 1 часа при комнатной температуре в буфере TBST, дополненном блокирующим реагентом 2% ECL prime blocking Reagent, используя HRP-меченное лошадиное анти-мышиное IgG антитело (Cell Signaling Technology, No.7076) или козье анти-кроличье IgG антитело (Cell Signaling Technology, No.7074) в качестве вторичного антитела. После промывки непрореагировавшего вторичного антитела буфером TBST, Chemi-Lumi One Super (Nacalai Tesque, Inc.) использовали для проведения реакции в соответствии с прилагаемым протоколом, а затем соответствующие полосы в виде хемилюминесценции детектировали с помощью CCD-камеры (GE Healthcare ImageQuant LAS 500). Обнаруженные полосы подвергали денситометрии (программное обеспечение для анализа ImageQuant TL v8.1), чтобы представить их в виде числовых значений, и степень ингибирования (%) рассчитывали на основе интенсивности полосы в каждой группе, принимая при этом люминесценцию полосы фосфорилированной BTK в группе без добавленного соединения с IgM стимуляцией как 100% и люминесценцию полосы фосфорилированной BTK в группе без добавленного соединения без IgM стимуляции как 0%. Каждая полоса фосфорилированной BTK была скорректирована на основе общей BTK.
[0115]
Комбинации первичных антител и вторичных антител, использованные в этом тесте, и величины их разведения показаны ниже.
[Таблица 4]
(величина разведения)
(величина разведения)
(1/4000)
(1/5000)
(1/500)
(1/5000)
Результаты, полученные при концентрации испытуемого соединения 0,03 мкМ, приведены в таблице 5. Ингибирующая активность внутриклеточного BTK-аутофосфорилирования была обозначена знаком «***», когда 90% или более, знаком «**», когда 70% или более и менее чем 90%, и знаком «*», когда 50% или более и менее чем 70%.
[0116]
Ингибирующее действие типичных соединений по настоящему изобретению на внутриклеточную аутофосфоризацию показано в таблице 5. Как показано в таблице, активность внутриклеточной аутофосфоризации была сильно ингибирована соединениями (I) по настоящему изобретению в концентрации 0,03 мкМ.
[Таблица 5]

Результаты примера испытаний 2 показывают, что соединения по настоящему изобретению оказывают сильное ингибирующее действие также на ʺактивность внутриклеточного BTK-аутофосфорилированияʺ.
[0117]
Пример испытаний 3
Тест на ингибирующую активность в отношении мутанта C481S BTK
(Метод измерения киназной активности)
Активность киназы измеряли с использованием QuickScout Screening Assist (торговая марка) MSA (коммерчески доступный набор, выпускаемый Carna Biosciences, Inc.) методом анализа изменения подвижности (MSA). Субстратом киназной реакции был FITC-меченный SRCtide пептид, включенный в набор. Использовали буфер для анализа [20 мМ HEPES, 0,01% Triton X-100 (торговая марка), 2 мМ дитиотреитол, pH 7,5] и доводили до 4 мкМ субстрата, 20 мМ MgCl2 и 120 мкМ и 100 мкМ ATP, которые являются концентрациями ATP, близкими к значению Km дикого типа и мутанта C481S BTK, соответственно, для получения раствора субстратной смеси. Раствор фермента также получали путем разбавления BTK дикого типа или мутанта C481S BTK до 0,28 нМ с использованием буфера для анализа. 10 мМ раствор испытуемого соединения в DMSO дополнительно разбавляли DMSO до 10 уровней концентрации (0,00003 мM, 0,0001 мM, 0,0003 мM, 0,001 мM, 0,003 мM, 0,01 мM, 0,03 мM, 0,1 мM, 0,3 мM, 1 мM), каждый из которых подвергали 25-кратному разбавлению буфером для анализа для получения растворов лекарственного средства (4% растворы DMSO).
5 мкл раствора лекарственного средства или контрольного раствора (4% DMSO-буфер для анализа), 5 мкл раствора субстратной смеси и 10 мкл ферментного раствора смешивали в лунках полипропиленового 384-луночного планшета и давали возможность взаимодействовать при комнатной температуре в течение 1 часа, а затем гасили путем добавления 60 мкл терминирующего буфера, включенного в набор. Затем количество субстратов (S) и фосфорилированного субстрата (P) в реакционном растворе измеряли с использованием системы LabChip EZ Reader II (производства Caliper Life Sciences) в соответствии с протоколом набора для анализа.
[0118]
(Способ оценки BTK ингибирующей активности)
Высоты пиков изолированного субстрата и фосфорилированного субстрата были представлены как S и P, соответственно, и также был измерен холостой раствор, который содержал буфер для анализа вместо ферментного раствора.
Степень ингибирования (%) тестируемого соединения рассчитывали в соответствии со следующим уравнением;
Степень ингибирования (%)=(1-(C-A)/(B-A))×100
где, A, B и C представляют P/(P+S) холостой лунки, P/(P+S) контрольной лунки и P/(P+S) лунки, содержащей соединение, соответственно.
[0119]
Значение IC50 рассчитывали с помощью регрессионного анализа степени ингибирования (%) и концентрации тестируемого соединения (логарифмическая величина).
[0120]
Ингибирующая активность в отношении BTK дикого типа [BTK(WT)] и C481S мутанта BTK [BTK(C481S)] типичных соединений по настоящему изобретению показана в таблице 6. Значения IC50[BTK(C481S)]/IC50[BTK(WT)] также перечислены в таблице как приближенное показание для C481S мутация-резистентного.
[Таблица 6]
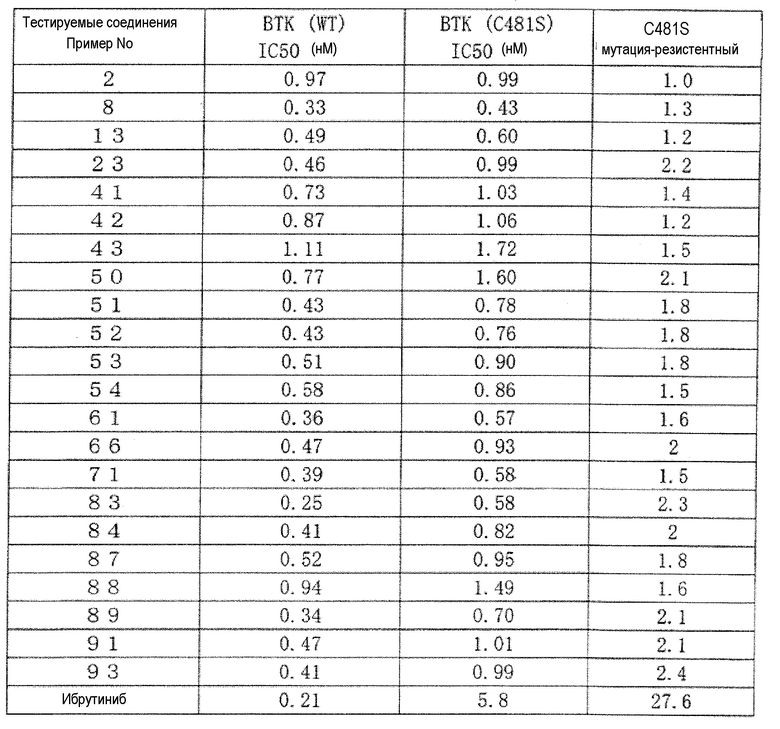
Как показано в примере испытаний 3, соединения (I) по настоящему изобретению обладают сильной ингибирующей активностью также против мутанта C481S BTK.
[0121]
Пример испытаний 4
Тест на ингибирование пролиферации в отношении диффузной крупноклеточной В-клеточной лимфомы штамма OCI-Ly10
Клетки OCI-Ly10 культивировали в среде IMDM (среда Дульбекко в модификации Искова, производства Thermo Fisher Scientific Inc., далее «среда»), содержащей 20% фетальной бычьей сыворотки и 1% пенициллин-стрептомицина (Nacalai Tesque, Inc.) в 5% СО2 инкубаторе. Клетки OCI-Ly10 высевали в 96-луночный планшет (20000 клеток/лунку), добавляли разбавленные средой соединения с конечными концентрациями от 0,9 нМ до 30000 нМ (конечная концентрация DMSO, 0,3%), инкубировали в течение 96 часов и добавляли реагент alamorBlue (Thermo Fisher Scientific Inc.). Абсорбцию при 570-600 нм измеряли через 3 часа, и значение IC50 ингибирующей активности рассчитывали при условиях, когда абсорбция лунки, не содержащей соединения и клетки, составляла 100%, и абсорбция лунки, содержащей клетку, и не содержащей соединение, составляла 0%.
[0122]
Активность ингибирования пролиферации типичных соединений по настоящему изобретению в отношении штамма OCI-Ly10 показана в таблице 7.
[Таблица 7]
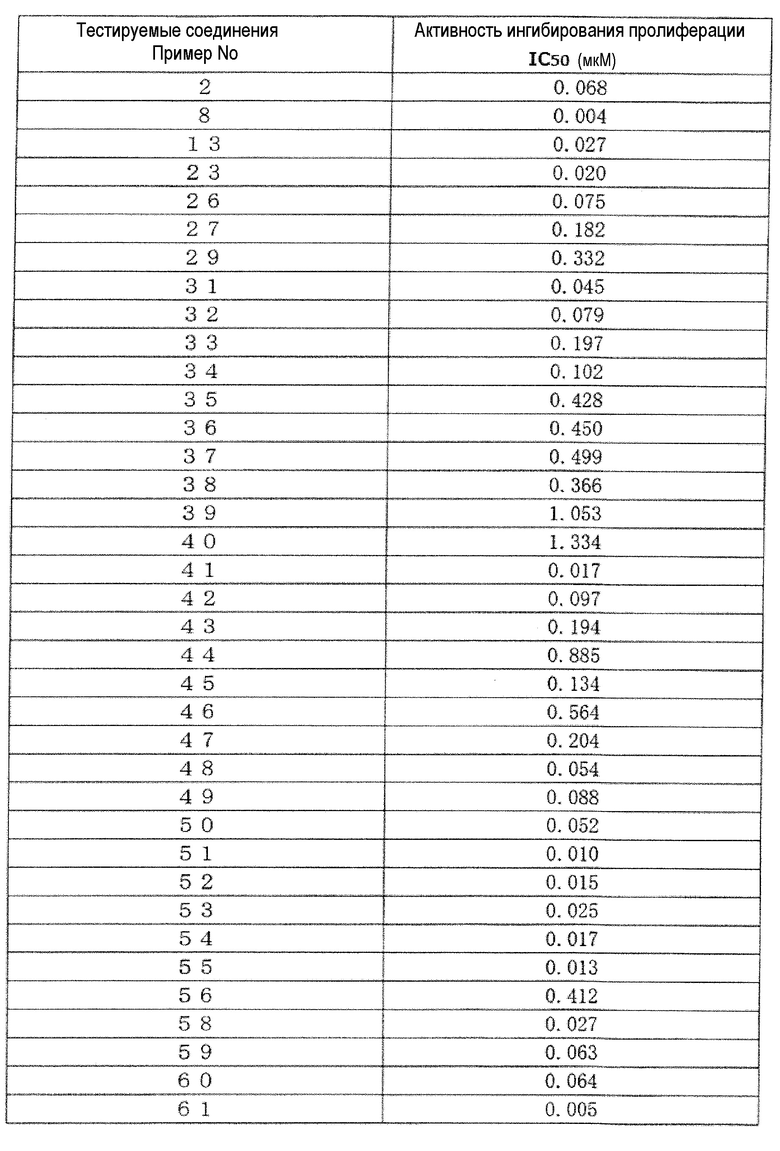
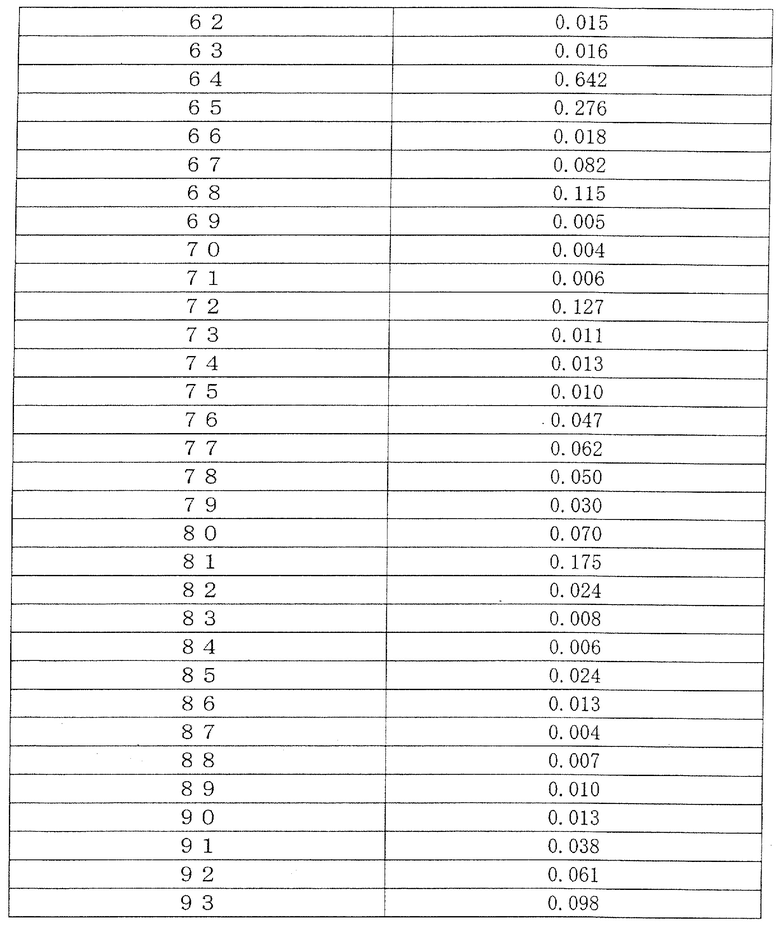
Как показано в результатах примера испытаний 4, соединения (I) по настоящему изобретению обладают активностью ингибирования пролиферации в отношении штамма OCI-Ly10.
Промышленная применимость
[0123]
Настоящее изобретение относится к соединению, полезному для профилактики или лечения заболеваний, в которые, как известно, вовлечен аномальной клеточный ответ посредством BTK, например, аутоиммунных заболеваний, воспалительных заболеваний, заболеваний костей и раковых заболеваний, таких как лимфома. Соединение также является полезным в качестве ингибитора БТК для реагентов, используемых в тестах и исследованиях.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| ПРОИЗВОДНЫЕ ОКСИФТОРПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2758370C1 |
| ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2761626C2 |
| ПРОИЗВОДНЫЕ АМИНОФТОРПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2762637C1 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2756505C1 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ПИРИМИДО[5,4-b]ИНДОЛИЗИНОВОЕ ИЛИ ПИРИМИДО[5,4-b]ПИРРОЛИЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2721774C1 |
| ПИРРОЛОПИРИМИДИНЫ В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ МВТР | 2017 |
|
RU2757457C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2014 |
|
RU2677653C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
Изобретение относится к области органической химии, а именно к производному оксоизохинолина указанной ниже формулы. Также изобретение относится к способу лечения В-клеточной лимфомы, основанному на использовании соединения указанной ниже структуры. Технический результат: получено новое гетероциклическое соединение, которое является полезным в качестве ингибитора киназы Брутона. 2 н.п. ф-лы, 9 табл., 97 пр.
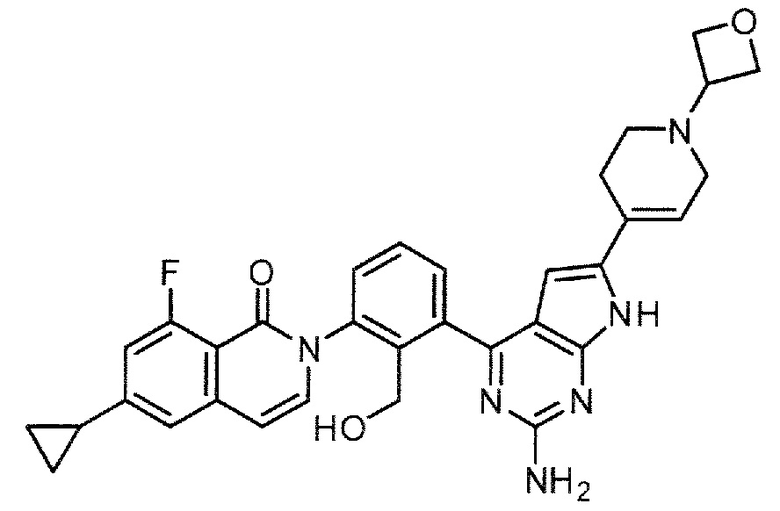
1. Производное оксоизохинолина следующей формулы:
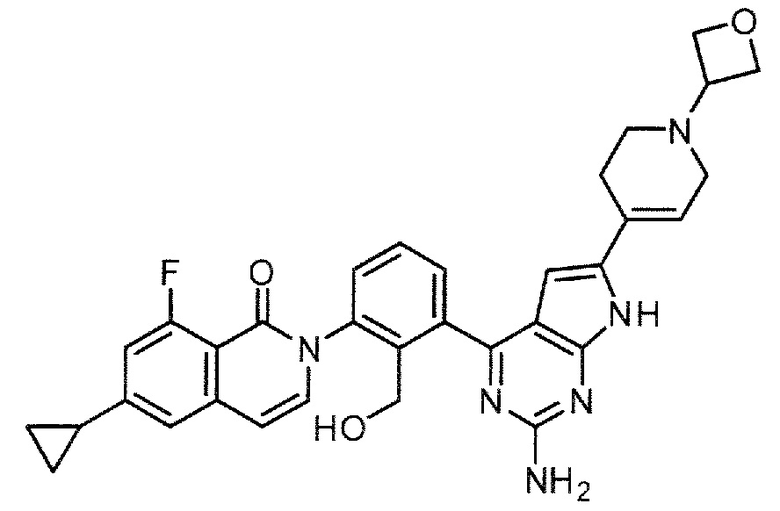
2. Способ лечения В-клеточной лимфомы, включающий введение терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
| WO 2013157022 A1, 24.10.2013 | |||
| RU 2012152548 A, 20.06.2014. |

