Настоящее изобретение относится к способу гетерогенного каталитического газофазного парциального окисления по меньшей мере одного органического исходного соединения молекулярным кислородом на свежем внесенном в реакционное пространство неподвижном слое катализатора, согласно которому с целью парциального окисления реакционную смесь, содержащую по меньшей мере одно исходное органическое соединение и молекулярный кислород, пропускают через слой неподвижного катализатора, а также отводят тепло реакции посредством непрямого теплообмена с подаваемым вне реакционного пространства жидким теплоносителем, а затем, когда с увеличением продолжительности работы происходит нарастающее ухудшение качества неподвижного слоя катализатора, то для восстановления качества неподвижного слоя катализатора не весь, а лишь часть неподвижного слоя катализатора заменяют частью неподвижного слоя заменяющего катализатора.
При этом под полным окислением органического соединения молекулярным кислородом подразумевают, что органическое соединение вступает в реакцию с молекулярным кислородом таким образом, что весь содержащийся в органическом соединении углерод превращается в оксиды углерода, а весь содержащийся в органическом соединении водород превращается в оксиды водорода. Все отличающиеся от этого типы взаимодействия органического соединения под реакционным воздействием молекулярного кислорода охватываются здесь термином «парциальное окисление» органического соединения.
В особенности, под парциальным окислением здесь понимают такое взаимодействие органических соединений под реакционным воздействием молекулярного кислорода, при котором по окончании взаимодействия подвергнутое парциальному окислению органическое соединение содержит, по меньшей мере, один атом кислорода с более прочной химической связью, чем перед проведением парциального окисления.
Под содержащимся в условиях гетерогенного каталитического газофазного парциального окисления, по существу, инертным газом-разбавителем подразумевают такие газы-разбавители, компоненты которых в условиях гетерогенного каталитического газофазного парциального окисления, принимая во внимание каждый их компонент, остаются содержащимися без изменения более чем на 95 мол.%, предпочтительно, более чем на 99 мол.%.
Под нагрузкой реакционной смесью катализирующего реакционную стадию неподвижного слоя катализатора подразумевают количество реакционной газовой смеси в нормальных литрах (= NI, = нл, объемы в литрах, занимаемые соответствующими количествами реакционных газовых смесей при нормальных условиях, т.е. при температуре 25°С и давлении 1 атм), которые пропускают через неподвижный слой катализатора в час в расчете на объемы его насыпной массы (при этом отрезки чисто инертного материала не учитываются) (→ размерность = нл/л·ч). Нагрузка может также относиться только лишь к одному компоненту реакционной газовой смеси. В этом случае оно равно объемному количеству этого компонента, которое пропускают в час через неподвижный слой катализатора в расчете на объем его засыпанной насыпной массы.
Общеизвестно, что посредством парциального и гетерогенного каталитического окисления различных исходных органических соединений молекулярным кислородом в газовой фазе на неподвижном слое катализатора могут быть получены разнообразные основные (целевые) продукты. В качестве примера можно назвать превращение пропилена в акролеин и/или акриловую кислоту (сравни, например, патент DE-A-2351151); превращение трет.бутанола, изобутена, изобутана, изобутиральдегида или метилового эфира бутанола в метакролеин и/или метакриловую кислоту (сравни, например, патент DE-A-2526238, европейские патенты ЕР-А 092097 и ЕР-А 058927; патенты DE-A-4132263, DE-A-4132684 и DE-A-4022212); превращение акролеина в акриловую кислоту, превращение метакролеина в метакриловую кислоту (сравни, например, DE-A-2526238); превращение о-ксилола, п-ксилола или нафталина во фталевый ангидрид (сравни, например, ЕР-А 522871) или соответствующие кислоты, а также превращение бутадиена в малеиновый ангидрид (сравни, например, патенты Великобритании GB-А 1464198 и GB-A 1291354); превращение инданов, например, в антрахинон (сравни, например, DE-A-2025430); превращение этилена в окись этилена или пропилена в окись пропилена (сравни, например, акцептованную заявку DE-AS 1254137, патент DE-A 2159346, европейский патент ЕР-А 372972, международную заявку на патент WO 89/07101, патент DE-A 4311608 и учебник Beyer, Lehrbuch der organischen Chemie, 17. Auflage (1973), Hirzel Verlag Stuttgart, Seite 261); превращение пропилена и/или акролеина в акрилонитрил (сравни, например, DE-A 2351151); превращение изобутена и/или метакролеина в метакрилонитрил (т.е. термин «парциальное окисление» в этом описании должен охватывать также парциальное аммоокисление, т.е. парциальное окисление в присутствии аммиака); окислительное дегидрирование углеводородов (сравни, например, DE-A 2351151); превращение пропана в акрилонитрил или акролеин и/или акриловую кислоту (сравни, например, патент DE-A 10131297, европейский патент ЕР-А 1090684, европейский ЕР-А 608838, DE-A 10046672, ЕР-А 529853, международную заявку на патент WO 01/96270 и DE-A 10028582); превращения изобутана в метакролеин и/или метакриловую кислоту, а также превращения этана в уксусную кислоту, этилена в окись этилена, бензола в фенол, а также бутена-1 или бутена-2 в соответствующие бутандиолы и т.п.
При этом задачей неподвижного слоя катализатора является способствовать предпочтительному протеканию желаемого парциального газофазного окисления по сравнению с полным окислением.
Химическое взаимодействие осуществляют при пропускании потока реакционной газовой смеси через неподвижный слой катализатора во время нахождения в нем реакционной газовой смеси.
Что касается твердых катализаторов, то речь часто идет об оксидных массах или о благородных металлах (например, о серебре). Помимо кислорода, каталитически активная оксидная масса может содержать только один другой элемент или более одного другого элемента (в случае так называемых многоэлементных оксидных масс).
В качестве каталитически активных оксидных масс особенно часто используют такие каталитически активные оксидные массы, которые содержат более одного металлического элемента, в особенности элемента переходных металлов. В этом случае говорят о многометаллических оксидных массах. Обычно они не представляют собой каких-либо простых физических смесей оксидов их элементарных компонентов, а являются смесями комплексных полисоединений этих элементов. На практике названные ранее каталитически активные твердые массы, как правило, формуют различной геометрической формы (колец, полнотельного цилиндра, шариков и т.д.). При этом формование (в формованное изделие) может осуществляться таким образом, что каталитически активную массу формуют как таковую (например, в экструдере или в устройстве для таблетирования), в результате чего получают так называемый цельный катализатор, или активную массу наносят на предварительно сформованный носитель (сравни, например, международные заявки на патент WO 2004/009525 и WO 2005/113127).
Примеры катализаторов, пригодных для гетерогенного каталитического газофазного парциального окисления на неподвижном слое катализатора, по меньшей мере одного исходного органического соединения по изобретению, можно найти, например, в DE-A 10046957, ЕР-А 1097745, DE-A 4431957, DE-A 10046928, DE-A 19910506, DE-A 19622331, DE-A 10121592, ЕР-А 700714, DE-A 19910508 и в европейских заявках ЕР-А 415347, ЕР-А 471853 и ЕР-А 700893.
Обычно гетерогенное каталитическое газофазное парциальное окисление является сильно экзотермическим. Вследствие многообразия возможных параллельных и/или последовательных реакций, с точки зрения как можно более селективного взаимодействия подлежащего частичному окислению, по меньшей мере, одного исходного органического соединения для получения желаемого целевого продукта одного только совместного использования катализатора обычно является недостаточным. Гораздо чаще для как можно более селективного проведения гетерогенного каталитического газофазного парциального окисления на неподвижном слое катализатора требуется еще дополнительно контролировать подъем реакционной температуры или подъем температуры неподвижного слоя катализатора в направлении потока реакционной газовой смеси в некотором определенном интервале.
Поэтому вследствие необходимости отвода тепла такое парциальное окисление, как правило, проводят в «изотермических» реакторах с неподвижным слоем катализатора, в которых неподвижный слой катализатора является внесенным в реакционное пространство, чтобы с целью непрямого теплообмена вне реакционного пространства провести жидкий теплоноситель (средство для теплообмена), который соприкасается с материалом оболочки реакционного пространства (стенкой реакционного пространства) (находится в контакте с ней). Например, неподвижный слой катализатора может находиться в контактных трубах секционного трубчатого реактора, чтобы для отвода тепла провести (направить) расплав соли или металла.
Дополнительно реакционный компонент в условиях гетерогенного каталитического газофазного парциального окисления обычно разбавляют по существу инертным газом, который благодаря своей теплоемкости может абсорбировать выделяющееся реакционное тепло.
Поэтому в реакционную газовую смесь для гетерогенного каталитического газофазного парциального окисления по меньшей мере одного исходного органического соединения, помимо по меньшей мере одного исходного органического соединения и молекулярного кислорода, как правило, дополнительно включают по меньшей мере один инертный газ-разбавитель.
Наиболее часто используемым совместно инертным газом-разбавителем является молекулярный азот, который подают автоматически всегда, когда в качестве источника кислорода для гетерогенного каталитического газофазного парциального окисления используют воздух.
Другим часто используемым совместно инертным газом-разбавителем благодаря его общедоступности является водяной пар.
Другими обычно используемыми газами-разбавителями являются благородные газы (например, гелий, аргон, неон) или окислы углерода (двуокись и/или моноокись углерода).
Особенно благоприятным является обычно использование газов-разбавителей с как можно более высокой молярной теплоемкостью (сравни, например, ЕР-А 253409). К ним часто относятся, например, в случае парциального окисления ненасыщенного исходного органического соединения и прочих, насыщенные углеводороды, такие, например, как пропан в случае парциального окисления пропилена в акролеин и/или акриловую кислоту.
В качестве инертного газа-разбавителя часто используют совместно также циркуляционный (контурный) газ (сравни, например, европейский патент ЕР-А 1180508). Циркуляционным газом называют остаточный газ после одноступенчатого или многоступенчатого гетерогенного каталитического газофазного парциального окисления по меньшей мере одного исходного органического соединения (при многоступенчатом гетерогенном каталитическом газофазным парциальном окислении, по меньшей мере, одного исходного органического соединения в отличие от одноступенчатого гетерогенное каталитическое газофазное парциальное окисление проводят не в одной, а, по меньшей мере, в двух последовательно расположенных друг за другом секциях реактора (реакционных пространствах) (которые могут переходить одна в другую в одном общем корпусе без стыка или могут помещаться в двух пространственно разделенных расположенных последовательно друг за другом реакторах), причем между двумя следующими друг за другом реакционными секциями или реакторами, при необходимости, добавляют инертный газ и/или окислитель. Многоступенчатость используют, в особенности, тогда, когда парциальное окисление совершается на следующих друг за другом стадиях. В этих случаях часто целесообразно оптимизировать как неподвижный слой катализатора, так и прочие условия реакции применительно к конкретной реакционной стадии и проводить реакционную стадию в одной специальной реакционной секции или в одном специальном реакторе, то есть как одну, или в отдельных реакционных стадиях. Но многоступенчатость может также использоваться и тогда, когда из-за необходимости отвода тепла или по другим причинам (сравни, например, патент Германии DE-A 19902562) взаимодействие переносят на несколько последовательно расположенных друг за другом реакторных секций или реакторов. Примером часто проводимого в две стадии гетерогенного каталитического парциального окисления является парциальное окисление пропилена в акриловую кислоту. На первой реакционной стадии пропилен подвергают парциальному окислению в акролеин, а на второй реакционной стадии акролеин подвергают парциальному окислению в акриловую кислоту. Соответствующим образом часто также получение метакриловой кислоты, главным образом, из изобутана проводят двухстадийно. Но оба вышеназванные парциальные окисления могут также проводиться при использовании загрузок соответствующего катализатора одностадийно (обе стадии в одной реакторной секции на неподвижном слое катализатора, катализирующего обе стадии), как, например, описано для парциального окисления пропилена в акриловую кислоту в патенте Германии DE-A 10121592). Гетерогенное каталитическое газофазное парциального окисление, по меньшей мере, одного исходного органического соединения проводят тогда, когда из газовой смеси целевого продукта отделяется более менее селективно (например, абсорбцией в соответствующем растворителе или фракционной конденсацией). Как правило, он состоит, преимущественно, из использованных для парциального окисления инертных газов-разбавителей, а также из водяного пара, обычно образующегося при парциальном окислении в качестве побочного продукта или использованного в качестве газа-разбавителя, и образующихся при нежелательном полном окислении окислов углерода. Отчасти он содержит еще небольшое количество не использованного при парциальном окислении молекулярного кислорода (остаточного кислорода) и/или не превращенного органического исходного соединения.
При этом используемые совместно газы-разбавители оказываются полезными не только потому, что они отводят тепло реакции, но и, как правило, одновременно они гарантируют безопасность процесса гетерогенного каталитического газофазного парциального окисления, по меньшей мере, одного исходного органического соединения, когда они поддерживают реакционную смесь либо вне области взрыва, либо в еще безопасной зоне взрывоопасной области.
Несмотря на описанные внешние и внутренние меры, направленные на то, чтобы снизить (контролировать) температуру реакции или температуру неподвижного слоя катализатора, следует распознавать все же существующее обычно различие между «температурой неподвижного слоя катализатора» и «эффективной температурой неподвижного слоя катализатора».
Под температурой неподвижного слоя катализатора подразумевают температуру неподвижного слоя катализатора при осуществлении способа парциального окисления, однако в фиктивном отсутствии химической реакции (т.е. без влияния реакционного тепла) (это означает, что влияние направляемого вне реакционного пространства жидкого теплоносителя учитывается так же, как и при осуществлении способа парциального окисления). Под «эффективной температурой» неподвижного слоя катализатора подразумевают, напротив, действительную температуру неподвижного слоя катализатора при дополнительном учете парциального окисления. Если же температура неподвижного слоя катализатора вдоль этого слоя непостоянна (например, в случае нескольких температурных зон), то термин «температура неподвижного слоя катализатора» в этом описании означает (числовое) среднее значение температуры вдоль неподвижного слоя катализатора. Однако способ по изобретению особенно пригоден тогда, когда температура неподвижного слоя катализатора в направлении потока реакционной газовой смеси вдоль неподвижного слоя катализатора является постоянной.
В связи с вышеизложенным имеет значение, что температура реакционной газовой смеси (и, следовательно, также и эффективная температура неподвижного слоя катализатора) при прохождении неподвижного слоя катализатора в направлении потока реакционной газовой смеси на каждой реакционной стадии обычно проходит через максимальное значение (так называемое значение точки перегрева). Разность между значением температуры точки перегрева и температурой неподвижного слоя катализатора в месте точки перегрева называется протяженностью (расширением) точки перегрева ΔTHB. Среди прочего это опять приводит к тому, что концентрация реагентов в реакционной газовой смеси на входе (при поступлении) реакционной газовой смеси в неподвижный слой катализатора является максимальной, что обусловливает там особенно высокие скорости реакции, при которых происходит особенно высокое выделение реакционного тепла в единицу времени (при поступлении в неподвижный слой катализатора реакционная газовая смесь, как правило, имеет, по существу, температуру неподвижного слоя катализатора).
Гетерогенное каталитическое газофазное парциальное окисление при однократном пропуске реакционной газовой смеси через неподвижный слой катализатора чаще всего требует повышенных температур неподвижного слоя катализатора для экономичного превращения исходных реагентов в реакции парциального окисления. Как правило, они составляют несколько сотен градусов Цельсия, обычно от 100 до 600°С, часто от 150 до 500°С, чаще всего от 200 или 250 до 450°С.
Рабочее давление при гетерогенном каталитическом газофазном парциальном окислении на неподвижном слое катализатора может быть ниже 1 атм или выше 1 атм. Как правило, оно находится в области от ≥1 до 20 атм или до 10 атм. Общеизвестно, что гетерогенное каталитическое газофазное парциальное окисление, по меньшей мере, одного исходного органического соединения может осуществляться на свежевнесенном в реакционное пространство неподвижном слое катализатора, по существу, непрерывно в течение более продолжительного периода времени на том же самом неподвижном слое катализатора. Реакционные условия при этом, как правило, поддерживаются, по существу, постоянными.
Правда, при этом с течением рабочего времени реакции неподвижный слой катализатора обычно теряет свое качество. Как правило, прежде всего ухудшается удельно-объемная активность неподвижного слоя катализатора (чем выше при прочих неизменных реакционных условиях температура, требуемая для определенной степени превращения исходных реагентов при одноразовом пропуске реакционной газовой смеси через неподвижный слой катализатора, тем ниже удельно-объемная активность неподвижного слоя катализатора). Однако чаще всего страдает также и селективность образования целевого продукта.
Снижение удельно-объемной активности неподвижного слоя катализатора является недостатком прежде всего потому, что при этом с увеличением продолжительности работы неподвижного слоя катализатора при прочих постоянных условиях технологического процесса снижается степень конверсии исходных реагентов в расчете на одноразовый пропуск реакционной газовой смеси через неподвижный слой катализатора, что снижает намеченный пространственно-временной выход целевого продукта производственной установки (выход в расчете на единицу времени и объема).
В европейских патентах ЕР-А 990636 и ЕР-А 1106598 пытались исправить названный недостаток в долговременном производственном цикле процесса гетерогенного каталитического газофазного парциального окисления, по меньшей мере, одного исходного органического соединения на том же самом неподвижном слое катализатора благодаря тому, что температуру неподвижного слоя катализатора с течением рабочего времени при прочих остающихся в значительной степени одинаковыми условиях процесса все больше и больше повышают, чтобы, по существу, сохранить степень превращения исходных соединений при одноразовом проходе реакционной газовой смеси через неподвижный слой катализатора.
Под скоростью деактивации неподвижного слоя катализатора в этом тексте описания называют точно рассчитанное при продолжительности работы в один год (365 дней) повышение температуры неподвижного слоя катализатора, необходимое для сохранения степени конверсии исходного соединения (эдукта) при одноразовом пропуске реакционной газовой смеси через неподвижный слой катализатора (при прочих неизменных условиях процесса).
Однако недостатком рекомендованного в европейском патенте ЕР-А 990636, а также в европейском патенте ЕР-А 1106598 способа является то, что со все возрастающим повышением температуры неподвижного слоя катализатора, как правило, ускоряется процесс его старения, отчего при достижении максимального значения температуры неподвижного слоя катализатора его обычно полностью заменяют и в реакционное пространство вносят свежий совершенно не использованный неподвижный слой катализатора (деактивацию уже нельзя больше компенсировать повышением температуры неподвижного слоя катализатора).
В патентах Германии DE-A 10351269, DE-A 10350812, DE-A 10350822, в европейском патенте ЕР-А 614872 и в патенте Германии DE-A 10350822 рекомендуется устранить необходимость полной замены неподвижного слоя катализатора благодаря периодической регенерации неподвижного слоя катализатора (т.е. периодическим прекращением процесса гетерогенного каталитического газофазного парциального окисления на неподвижном слое катализатора и, например, пропуском через неподвижный слой катализатора горячей смеси молекулярного кислорода и инертного газа). Недостатком этого способа, однако, является то, что с возрастанием общей продолжительности работы его эффективность истощается.
В патенте Германии DE-A 102004025445 предлагается в качестве другой меры для устранения необходимости полной замены неподвижного слоя катализатора повышать рабочее давление в газовой фазе. Недостатком этой меры является, однако, то, что ее эффективность также истощается с возрастанием продолжительности общего производственного процесса и одновременно требуется увеличивающаяся компрессорная мощность.
В патенте Германии DE-A 10232748 и в международной заявке на патент WO 2004/009525 предлагается в качестве другой возможности устранить полную замену неподвижного слоя катализатора, заменять лишь часть последнего заменяющим (резервным) неподвижным слоем катализатора, удельная объемная активность которого должна быть равной удельной объемной активности заменяемого неподвижного слоя катализатора в его свежевнесенном в реакционное пространство состоянии.
Таким образом можно вновь достигнуть требуемой степени конверсии исходного соединения (эдукта) (в расчете на одноразовый пропуск реакционной газовой смеси через неподвижный слой катализатора) при прочих неизменных условиях процесса сравнительно ограниченно повышенной температурой неподвижного слоя катализатора (в сравнении с температурой неподвижного слоя катализатора, требуемой для той же самой степени конверсии исходного соединения с первоначальным свежевведенным в реакционное пространство неподвижным слоем катализатора).
Однако недостатком способа, описанного в патенте Германии DE-A 10232748 и в международной заявке на патент WO 2004/009525, является то, что после замены части неподвижного слоя катализатора скорость деактивации неподвижного слоя катализатора, полученного после частичной его замены, при работе при температуре, при которой обеспечивается требуемая степень конверсии исходного соединения (эдукта), повышается (в сравнении со скоростью деактивации свежевнесенного в реакционное пространство неподвижного слоя катализатора при прочих соответствующих и нацеленных на получение той же самой степени конверсии исходного соединения (эдукта) технологических условиях процесса, вследствие чего после частичной замены имеющийся в распоряжении период времени до необходимой полной замены неподвижного слоя катализатора сравнительно сокращается.
Поэтому задача настоящего изобретения состояла в том, чтобы создать улучшенный вариант осуществления частичной замены используемого неподвижного слоя катализатора, направленный на невысокую скорость деактивации полученного после частичной замены неподвижного слоя катализатора при одинаковой степени конверсии исходного соединения (эдукта) и также при соответствующем в остальном таком же образе действия, что и при частичной замене катализатора согласно патенту Германии DE-A 10232748 и международной заявке на патент WO 2004/009525.
В соответствии с этим был найден способ гетерогенного каталитического газофазного парциального окисления, по меньшей мере, одного исходного органического соединения молекулярным кислородом на свежевнесенном в реакционное пространство неподвижном слое катализатора, при котором с целью парциалього окисления реакционную смесь, содержащую, по меньшей мере, одно исходное органическое соединение и молекулярный кислород, пропускают через неподвижный слой катализатора, а также отводят реакционное тепло посредством непрямого теплообмена с пропускаемым вне реакционного пространства жидким теплоносителем и затем, когда с увеличением продолжительности работы происходит нарастающее снижение качества неподвижного слоя катализатора, заменяют не весь, а лишь часть неподвижного слоя катализатора заменяющей (резервной) частью неподвижного слоя катализатора (как правило, свежеполученного катализатора), который характеризуется тем, что удельная объемная активность заменяющей части неподвижного слоя катализатора меньше, чем удельная объемная активность заменяемой части неподвижного слоя катализатора в его свежевнесенном состоянии.
В качестве меры удельной объемной активности засыпки неподвижного слоя катализатора (или части его), как уже указывалось, брали температуру засыпки неподвижного слоя катализатора при идентичных объемах засыпки, требуемую для того, чтобы при прочих равных условиях способа (идентичном составе реакционной газовой смеси, идентичном питании введенной насыпной массы неподвижного слоя катализатора реакционной газовой смесью) в расчете на одноразовый пропуск реакционной газовой смеси через насыпную массу неподвижного слоя катализатора достигнуть (в промышленном производстве) желаемой степени конверсии исходного соединения (эдукта). Чем выше требуемая температура, тем ниже удельная объемная активность. Альтернативно, в качестве меры удельной объемной активности можно взять степень конверсии исходного соединения (эдукта), полученную при идентичной температуре засыпки неподвижного слоя катализатора и прочих идентичных условиях способа и условиях работы (идентичном составе реакционной газовой смеси, идентичном питании введенной насыпной массы неподвижного слоя катализатора реакционной газовой смесью) в расчете на одноразовый пропуск через неподвижный слой катализатора. Чем выше достигнутая степень конверсии исходного соединения (эдукта), тем выше удельная объемная активность.
Удельную объемную активность (т.е. отнесенную к единице объема засыпки) можно снизить простым способом, например гомогенным разбавлением основного количества однородно полученных формованных тел катализатора формованными телами инертного разбавителя. Чем выше содержание формованных тел инертного разбавителя, тем меньше содержащаяся в определенном объеме засыпки активная масса, то есть тем меньше активность катализатора. При этом под инертными формованными телами разбавителя подразумевают формованное тело из таких веществ, которые являются, по существу, инертными по отношению к гетерогенному каталитическому газофазному парциальному окислению, т.е. как можно в большей степени не обусловливающими какого-либо значительного превращения исходного соединения (эдукта). В качестве таких веществ для многочисленных способов гетерогенного каталитического газофазного парциального окисления исходных органических соединений могут использоваться, например, пористые и непористые окислы алюминия, двуокись кремния, двуокись тория, двуокись циркония, карбид кремния, силикаты, такие как силикат магния или силикат алюминия, или стеатит.
Геометрическая форма такого инертного формованного тела разбавителя, в принципе, может быть любой. Она может быть, например, шарообразной, многоугольной, в виде полнотелого цилиндра или колец. Согласно изобретению в качестве инертного формованного тела разбавителя, предпочтительно, выбирают такое, геометрия которого соответствует геометрии подлежащего разбавлению им формованного тела катализатора. Но снижение удельной объемной активности катализатора возможно также, например, благодаря и тому, что при остающейся одинаковой геометрической форме и виде активной массы формованного тела катализаторов с оболочками толщина нанесенного на носитель слоя активной массы уменьшается или увеличивается содержание формованных тел с меньшим массовым содержанием активных масс в смеси из катализаторов с оболочкой с одинаковой геометрической формой, но с различным массовым содержанием активной массы. Аналогичный эффект достигается также, например, благодаря тому, что в смесях из цельных катализаторов и из катализаторов с оболочками (при идентичной активной массе) соответственно изменяются их соотношения в смеси. Разумеется, что можно также комбинировать описанные варианты. Но удельную объемную активность катализатора можно также уменьшить благодаря тому, что при одинаковом элементарном составе активной массы и при идентичном способе формования удельная поверхность активной массы уменьшается, например, в результате обработки активной массы при повышенной температуре и/или в течение более продолжительного времени термической обработки.
Разумеется, что можно также повлиять на удельную объемную активность катализатора, например, тем, что при идентичном формовании изменить элементарный состав активной массы и, например, уменьшить содержание тех ее элементарных компонентов, которые особенно необходимы для повышенной активности. Альтернативно, можно разбавить также сами активные массы, если при их получении, например, в термически обрабатываемую сухую смесь из исходных соединений ввести инертные вещества-разбавители, такие как высоко обожженная двуокись кремния. Различные количества добавки вещества-разбавителя автоматически приводят к различным активностям. Чем больше вещества-разбавителя добавляют, тем меньше полученная активность. Согласно изобретению для регулирования удельной объемной активности заменяющей части неподвижного слоя катализатора может использоваться каждая из названных мер как в отдельности, так и в любой их комбинации. К этому относится также, не в последнюю очередь, возможность увеличить протяженность частиц носителя (например, диаметр шариков носителя) при одинаковой геометрической форме катализатора с оболочкой, а также при одинаковой толщине оболочки и одинаковой оболочке активных масс.
Предпосылкой к идее изобретения является то обстоятельство, что свежевнесенный в реакционное пространство неподвижный слой катализатора с увеличением продолжительности проводимого на нем процесса гетерогенного каталитического газофазного парциального окисления, по меньшей мере, одного исходного органического соединения теряет свое качество не гомогенно и не равномерно по неподвижному слою катализатора (сравни, например, международную заявку на патент WO 2004/009525). Причиной этого может являться, например, появление точки перегрева и/или негомогенная концентрация содержащихся в реакционной газовой смеси катализаторных ядов (промышленная реакционная газовая смесь получается из сырья (исходных веществ) невысокой степени чистоты). Независимо от конкретной причины деактивация происходит в каждом участке неподвижного слоя катализатора, но тем быстрее, чем выше эффективная температура неподвижного слоя катализатора на данном участке неподвижного слоя катализатора.
Если теперь после более продолжительного времени работы такой сверхпропорциональный деактивированный участок неподвижного слоя катализатора заменяют на часть заменяющего (резервного) неподвижного слоя катализатора, удельная объемная активность которого соответствует удельной объемной активности заменяемого неподвижного слоя катализатора в его свежевнесенном состоянии, то весь неподвижный слой катализатора состоит после этого из двух частичных участков. Один из них находится в свежем первоначальном состоянии, а другой находится в деактивированном состоянии пропорционально времени работы. Для достижения с таким неподвижным слоем катализатора в расчете на одноразовый пропуск реакционной газовой смеси при прочих неизменных условиях процесса такой же степени превращения исходного соединения (эдукта), как и при внесенном в реакционное пространство целиком свежевнесенном неподвижном слое катализатора, в первом случае потребуется более высокая температура неподвижного слоя катализатора, чем в последнем случае. Но это обусловливает сверхпропорционально повышенную эффективную температуру неподвижного слоя катализатора в свежей заменяющей части неподвижного слоя катализатора, так как активность засыпанной свежей насыпной массы неподвижного слоя катализатора с повышением температуры процесса возрастает быстрее, чем линейное возрастание (сравни европейские патенты ЕР-А 099636 и ЕР-А 1106598). На незамененный участок неподвижного слоя катализатора выпадает еще более низкая часть степени превращения, чем до замены части неподвижного слоя катализатора. Это обусловливает в совокупности более высокую скорость деактивации, чем в случае работы свежевнесенного в реакционное пространство неподвижного слоя катализатора в его целостности.
Если же заменяющая часть неподвижного слоя катализатора, напротив, обладает более низкой удельной объемной активностью, чем заменяемый участок неподвижного слоя катализатора в его свежевнесенном состоянии, то хотя для достижения желаемой степени конверсии исходного соединения (эдукта) потребуется еще более высокая температура неподвижного слоя катализатора, однако в этом случае незамененный участок неподвижного слоя катализатора должен способствовать сравнительно более высокой доле превращения (разумеется, что вышеуказанное рассмотрение всегда предполагает сохранение состава реакционной газовой смеси, а также питания неподвижного слоя катализатора реакционной газовой смесью). Поэтому, как правило, происходит меньшая протяженность (расширение) точки перегрева ΔTHB и последнее приводит обычно к более низким скоростям деактивации, чем в случае части заменяющего неподвижного слоя катализатора с удельной объемной активностью, соответствующей его первоначальному свежевнесенному состоянию. Однако следствием такой сравнительно пониженной скорости деактивации является обычно увеличенная общая продолжительность процесса, требующаяся до полной замены неподвижного слоя катализатора. Это означает, что привлекательность способа по изобретению состоит в том, чтобы как можно в большей степени активизировать каталитический потенциал незамененного ранее востребованного участка неподвижного слоя катализатора и впоследствии его заменить. Этот образ действия с очевидностью включает неожиданно и замену части неподвижного слоя катализатора, при которой удельная объемная активность заменяющей части неподвижного слоя катализатора меньше, чем удельная объемная активность заменяемой части неподвижного слоя катализатора к моменту его замены. Конечно, формованное тело катализатора в заменяющей части неподвижного слоя катализатора является свежеизготовленным формованным телом катализатора или включает его. Другое экономическое преимущество способа по изобретению по сравнению со способом из известного уровня техники состоит в том, что с уменьшением удельной объемной активности заменяемой части неподвижного слоя катализатора, например при повышенном содержании в нем формованного инертного тела разбавителя, уменьшаются финансовые расходы на заменяющую часть неподвижного слоя катализатора. В качестве альтернативы соблюсти экономическое преимущество снижения скорости деактивации катализатора можно также, разумеется, увеличив питание неподвижного слоя катализатора реакционной газовой смесью, и таким образом, при сохранении формальной скорости деактивации катализатора повысить пространственно-временной выход целевого продукта (в единицу времени и в единице объема).
Согласно изобретению, предпочтительно, удельная объемная активность заменяющей части неподвижного слоя катализатора выбирается такой, чтобы в расчете на равную степень превращения исходного соединения (эдукта) при одноразовом пропуске реакционной газовой смеси через неподвижный слой катализатора, а также при одинаковом составе реакционной газовой смеси и одинаковой нагрузке неподвижного слоя катализатора реакционной газовой смесью, разность dΔТ между расширением (протяженностью) точки перегрева ΔTHB n после частичной замены неподвижного слоя катализатора и расширением (протяженностью) точки перегрева (непосредственно) перед частичной заменой неподвижного слоя катализатора ΔTHB v составляла: (dΔТ=ΔTHB n-ΔTHB v)≤30°С. Предпочтительно, удельную объемную активность заменяющей части неподвижного слоя катализатора выбирают такой, чтобы разность dΔТ составляла ≤25°С, или ≤20°С, или ≤15°С, лучше ≤10°С или ≤15°С, особенно предпочтительно ≤0°С или ≤-5°С, чаще ≤-15°С или до -20°С. Как правило, dΔT не составляет <-20°С. Предпочтительным является такой способ по изобретению, в котором dΔТ составляет от -15°С до +10°С. Особенно предпочтительным является такой способ по изобретению, в котором dΔТ составляет от -10°С до 0°С. Благоприятным является также dΔT от -5°С до 0°С.
Примером реакционного пространства в способе по изобретению может являться внутреннее пространство (контактной или реакционной) трубы, в которую внесен неподвижный слой катализатора и у наружной стены которой пропускают жидкий теплоноситель. Это может осуществляться, в принципе, прямотоком, противотоком или поперечным потоком по отношению к пропускаемой через реакционную трубу реакционной газовой смеси. Целесообразно, если контактная труба находится в секционном трубчатом реакторе.
Таким образом, эксплуатационно-технически целесообразно проводить гетерогенное каталитическое газофазное парциальное окисление, по меньшей мере, одного исходного органического соединения согласно изобретению в промышленном масштабе в многоконтактном трубчатом реакторе с неподвижным слоем катализатора (трубчатом секционном реакторе). Такие реакторы по своему типу соответствуют кожухо-трубчатым теплообменникам (но для способа по изобретению, в принципе, могут использоваться любые иные типы известных непрямых теплообменников для приема неподвижного слоя катализатора, то есть их обычная конструкция состоит, как правило, из цилиндрического резервуара, в котором размещается множество (обычно одинаковых) (реакционных) труб, обычно установленных вертикально, соответственно охлаждающим трубам кожухо-трубчатого теплообменника. Эти контактные трубы, каждая из которых содержит (обычно, главным образом, идентичную) насыпную массу засыпанного в нее используемого неподвижного слоя катализатора (размещение неподвижного слоя соответствующей засыпки катализатора), обычно уплотненно (герметично) закреплены своими концами в корпусе трубы и, целесообразно, входят каждым верхним или нижним концом в соединенную с резервуаром крышку (колпак). Через эту крышку (колпак) подводят и отводят поток реакционной газовой смеси, направляемый по контактным трубам, в результате чего внутреннее пространство каждой контактной трубы соответствует продольному реакционному пространству (в значительной степени однородному).
Через окружающее контактные трубы пространство направляют жидкий теплоноситель (жидкое средство для теплообмена) для отвода (удаления) реакционного тепла (тепла реакции). После выхода из резервуара нагретый жидкий теплоноситель опять доводят до его первоначальной температуры перед возвращением его в реакционный резервуар (сравни, например, патент ФРГ DE-A 3042468).
Если теплоноситель (средство для теплообмена) поступает в реактор вдоль контактных труб (реакционных труб) на различной их высоте (на нескольких высотах), то в этом описании говорят об использовании многозонного реактора (реакторного пространства), имеющего несколько циркуляционных контуров теплообменника или также несколько температурных зон. Если же теплоноситель (средство для теплообмена) поступает в реактор только на одной высоте (способ по изобретению является предпочтительным для этого случая), то при этом говорят об одном циркуляционном контуре теплоносителя или также о реакторе с одной реакционной зоной, даже когда этот циркуляционный контур работает не с одним, а, по соображениям целесообразности, с несколькими насосами.
Таким образом, способ по изобретению включает в качестве варианта его осуществления, в особенности, способ гетерогенного каталитического газофазного парциального окисления, по меньшей мере, одного исходного органического соединения молекулярным кислородом на неподвижном слое катализатора, свежевведенном в реакционное пространство (в контактные трубы) многоконтрактного трубчатого реактора с множеством контактных труб и неподвижным слоем катализатора, в котором с целью парциального окисления реакционную смесь, содержащую, по меньшей мере, одно исходное органическое соединение и молекулярный кислород, пропускают через неподвижный слой катализатора, а также отводят тепло реакции посредством непрямого теплообмена с направляемым вне реакционного пространства (вне контактных труб) жидким теплоносителем, а затем, когда с увеличением продолжительности работы происходит нарастающее ухудшение качества неподвижного слоя катализатора, то с целью восстановления качества неподвижного слоя катализатора в каждой контактной трубе заменяют не весь, а лишь часть неподвижного слоя катализатора заменяющей (резервной) частью неподвижного слоя катализатора, отличающийся тем, что удельная объемная активность заменяющей части неподвижного слоя катализатора ниже, чем удельная объемная активность заменяемой части неподвижного слоя катализатора в его свежевнесенном состоянии. Это особенно относится к тому случаю, когда теплоноситель направляют в реактор вдоль контактных труб только на одной высоте, и следовательно, речь идет о реакторе с одной зоной. В частности, к этим обоим названным вариантам осуществления способа относятся все сделанные в этом тексте высказывания относительно способа по изобретению, в частности количественное определение dΔТ.
Примеры используемых однозонных и многозонных реакторов со множеством контактных труб и неподвижным слоем катализатора находятся, например, в описаниях патентов Германии DE-A 10024348, DE-A 19836792, DE-A 10032304, международной заявки на патент WO 01/87476, в патентах Германии DE-A 19910508, DE-A 19910506, DE-A 19927624, DE-A 19948241, DE-A 19948248, DE-A 19948523, DE-A 19955168, DE-A 10134026, DE-A 10101695, патенте США US-A 5442108, в европейских патентах ЕР-А 911313, ЕР-А 1097745 и в патентах Германии DE-A 10137768, DE-A 10135498 и DE-A 10040781.
Обычно контактные трубы изготавливают из ферритной стали и часто они имеют толщину стенки от 1 до 3 мм. Их внутренний диаметр часто составляет от 20 до 30 мм, обычно от 21 до 26 мм. Длина труб обычно достигает нескольких метров (обычно длина контактных труб находится в пределах от 2 до 4 м, часто от 2,5 до 3,5 м). Из этой длины, как правило, по меньшей мере, 60%, часто, по меньшей мере, 75% занимает неподвижный слой катализатора. Эксплуатационно-технически целесообразно, если число введенных в резервуар контактных труб составляет, по меньшей мере, 5000, предпочтительно, по меньшей мере, 10000. Обычно число введенных в резервуар контактных труб составляет от 15000 до 30000 или до 40000. Секционный трубчатый реактор с числом контактных труб выше 50000 образует скорее исключение. Внутри резервуара контактные трубы обычно расположены распределенными равномерно, причем распределение целесообразно выбирать таким образом, что расстояние центральных внутренних осей между соседними контактными трубами (так называемое распределение контактных труб) составляет от 30 до 50 мм, часто от 35 до 45 мм (сравни, например, европейский патент ЕР-А 468290).
В качестве жидкого теплоносителя для способа по изобретению можно использовать, главным образом, но особенно в случае использования реактора со множеством контактных труб и с неподвижным слоем катализатора солевые расплавы, например, таких солей, как нитрат калия, нитрит калия, нитрит натрия и/или нитрат натрия. Но, отчасти, в зависимости от точки плавления расплавов могут также использоваться расплавы низкоплавких металлов, таких как натрий, ртуть, а также сплавы различных металлов. Средство для теплообмена, теплоноситель, может направляться, главным образом, просто непосредственно вдоль контактных труб (прямотоком или противотоком к потоку реакционной газовой смеси). Однако можно также этот продольный поток (прямотоком или противотоком к потоку реакционной газовой смеси) осуществлять через весь упомянутый резервуар, и этот продольный поток внутри резервуара посредством расположения последовательно друг за другом по длине контактных труб, освобождающих проходное сечение поворотных дисков, располагать в виде поперечного потока таким образом, что в продольном сечении получается меандровое течение потока средства для отвода тепла через секцию труб. Как правило, средство для теплообмена покидает резервуар (реактор) с температурой, которая (вследствие экзотермичности реакции) превышает его температуру на входе в реактор (зачастую на величину от ≥0 до 10°С, часто от ≥2 до 8°С, во многих случаях от ≥3 до 6°С).
Указанные выше и все другие высказывания в отношении способа по изобретению в этом тексте описания, в особенности, действенны для гетерогенного каталитического газофазного парциального окисления на неподвижном слое катализатора пропилена в акролеин и/или акриловую кислоту, изобутена в метакролеин и/или метакриловую кислоту, (мет)акролеина в (мет)акриловую кислоту, пропана в акролеин и/или акриловую кислоту, а также изобутана в метакролеин и/или метакриловую кислоту. Однако, разумеется, что они справедливы также для всех других названных в начале этого текста описания процессов парциального окисления.
Согласно изобретению благоприятно, если в способе по изобретению свежевнесенный в реакционное пространство неподвижный слой катализатора преимущественно оформляют таким образом, что его удельная объемная активность изменяется в направлении потока реакционной газовой смеси. Особенно полезно, если его оформляют таким образом, что его удельная объемная активность в направлении потока реакционной газовой смеси, по меньшей мере однократно, мгновенно или ступенчато, или непрерывно возрастает.
Особенно благоприятно, если свежевнесенный в реакционное пространство неподвижный слой катализатора не снижает своей удельной объемной активности в направлении потока реакционной газовой смеси. Кроме того, согласно изобретению благоприятно, когда катализаторы свежевнесенного в реакционное пространство неподвижного слоя катализатора содержат только одну активную массу, которая с особым преимуществом сформована в теле с единственной используемой в этом неподвижном слое катализатора геометрией формованного тела. Помимо этого, согласно изобретению благоприятно, если этот ранее названный тип катализатора используют в его свежеизготовленной форме также в качестве единственного катализатора для замещающей части неподвижного слоя катализатора.
Кроме того, согласно изобретению благоприятно, если внутри свежевнесенного в реакционное пространство неподвижного слоя катализатора совместно используют только один вид инертного формованного тела разбавителя. Это формованное тело разбавителя целесообразно тогда использовать также для заменяющей части неподвижного слоя катализатора. Таким образом, особенно благоприятно для способа по изобретению, когда заменяющая часть неподвижного слоя катализатора и замененный им участок неподвижного слоя катализатора в его свежевнесенном в реакционное пространство состоянии отличаются друг от друга только повышенным содержанием формованных тел разбавителя в заменяющей части неподвижного слоя катализатора.
В нижеследующем воплощении способа по изобретению приводится более подробно, без каких-либо ограничений его общей применимости и исключительно в качестве иллюстрации, на примере способа гетерогенного каталитического газофазного парциального окисления на неподвижном слое катализатора пропилена в акролеин и/или акриловую кислоту (это воплощение, однако, соответствующим образом переносится на другие возможные по изобретению способы гетерогенного каталитического газофазного парциального окисления других исходных органических соединений на неподвижном слое катализатора и на полученные при этом целевые продукты). Все сделанные в данном тексте описания высказывания относятся, в особенности, к этим обоим способам. Что касается требуемого исходного сырья - пропилена, то при этом, как правило, в используемую реакционную газовую смесь вводят пропилен в виде компонента пропилена сорта «полимерный» или «химический» (сравни международную заявку на патент WO 2004/009525). Разумеется, что источником пропилена может являться также гетерогенно каталитически частично дегидратированный или окисногидратированный пропан, например, как описано в международной заявке на патент WO 01/96270 и в патенте Германии DE-A 10316039, международной заявке на патент WO 01/95271, патенте ФРГ DE-A 3313573, международной заявке на патент WO 03/011804, патенте Германии DE-A 10245585, а также в патентах Германии DE-A 102004032129 и DE-A 102005013039.
Так как гетерогенное каталитическое газофазное парциальное окисление на неподвижном слое катализатора пропилена в акриловую кислоту протекает в две последовательные стадии с образованием акролеина в качестве промежуточного соединения, то, как уже отмечалось, оно может проводиться в одну или в две стадии.
Помимо частичной замены неподвижного слоя катализатора, согласно изобретению двухстадийное гетерогенное каталитическое газофазное парциальное окисление согласно изобретению пропилена в акролеин с использованием содержащей пропилен исходной реакционной газовой смеси можно проводить, например, как описано в описании к европейскому патенту ЕР-А 700714 (первая реакционная стадия, как описано там, но также при соответствующем противотоке солевой ванны и исходной реакционной газовой смеси через секционный трубчатый реактор); в европейском патенте ЕР-А 700893 (вторая реакционная стадия, как там описано, но также при соответствующем противотоке); международной заявке на патент WO 04/085369 (этот текст в особенности рассматривается как составная часть этой публикации) (в виде двухстадийного способа), международной заявке на патент WO 04/085363, патенте Германии DE-A 10313212 (первая реакционная стадия), европейском патенте ЕР-А 1159248 (в виде двухстадийного способа), европейском патенте ЕР-А 1159246 (вторая реакционная стадия), европейском патенте ЕР-А 1159247 (в виде двухстадийного способа), патенте Германии DE-A 19948248 (в виде двухстадийного способа), патенте Германии DE-A 10101695 (двухстадийно), международной заявке на патент WO 04/085368 (в виде двухстадийного способа), патенте Германии DE-А 10351269 (двухстадийно), патенте Германии DE-А 102004021764 (двухстадийно), международной заявке на патент WO 04/085362 (первая реакционная стадия), международной заявке на патент WO 04/085370 (вторая реакционная стадия), международной заявке на патент WO 04/085365 (вторая реакционная стадия), международной заявке на патент WO 04/085367 (двухстадийно), международной заявке на патент WO 2004/009525 (двухстадийно), европейских патентах ЕР-А 990636, ЕР-А 1007007 и ЕР-А 1106598.
В частности, это справедливо для всех содержащихся в этих публикациях примеров осуществления. Если при двухстадийном способе между обеими реакционными стадиями осуществляют подвод вторичного молекулярного кислорода, то это осуществляют, преимущественно, в форме воздуха. Однако он может осуществляться также в виде чистого молекулярного кислорода или также в виде иной смеси из молекулярного кислорода и инертного газа. Подвод вторичного кислорода, предпочтительно, осуществляют в таком количестве, чтобы газовая смесь продукта второй реакционной стадии (акролеин → акриловая кислота) содержала еще не вступивший в реакцию молекулярный кислород. Конечно, необходимое для всего способа в целом количество молекулярного кислорода может добавляться также уже в реакционную смесь для первой стадии реакции (пропилен → акролеин). Как правило, молярное отношение содержащей молекулярный кислород реакционной газовой смеси, подводимой к неподвижному слою катализатора первой реакционной стадии, к содержащемуся в этой смеси пропилену составляет ≥1 и ≤3.
Пригодные для каждой из обеих реакционных стадий мультиметаллооксидные катализаторы многократно описаны ранее и хорошо известны специалисту. В качестве примера европейский патент ЕР-А 253409, с.5, ссылается на соответствующий патент США. Пригодные для каждой стадии окисления (реакционной стадии) катализаторы описаны также в патентах Германии DE-A4431957, DE-A 102004025445 и DE-A 4431949. Это справедливо также для каждой общей формулы I в обоих названных более ранних описаниях. Используемые для каждой стадии окисления (реакционной стадии) катализаторы описаны также в описаниях к патентам Германии DE-A 10325488, DE-A 10325487, DE-A 10353954, DE-A 10344149, DE-A 10351269, DE-A 10350812 и DE-A 10350822.
Таким образом, для первой стадии реакции (пропилен → акролеин), в особенности, используют такие катализаторы, активная масса которых представляет собой мультиметаллическую окись, по меньшей мере, одного элемента - молибдена и/или вольфрама, а также, по меньшей мере, одного элемента из группы висмут, теллур, сурьма, олово и медь. Среди них предпочтительными являются такие, активная масса которых представляет собой мультиметаллическую окись, содержащую молибден, висмут и железо.
На первой реакционной стадии мультиметаллоксидные активные массы, содержащие молибден, железо и висмут, могут являться, например, мультиметаллоксидными массами общей формулы I по патенту Германии DE-A 19955176, мультиметаллоксидными массами общей формулы I по патенту Германии DE-A 19948523, мультиметаллоксидными массами общих формул I, II и III по патенту Германии DE-A 10101695, мультиметаллоксидными массами общих формул I, II и III по патенту Германии DE-A 19948248 и мультиметаллоксидными массами общих формул I, II и III по патенту Германии DE-A 19955168, а также мультиметаллоксидными массами, названными в европейском патенте ЕР-А 700714. Могут также использоваться все названные в международной заявке на патент WO 2004/009525 для первой реакционной стадии мультиметаллоксидные массы, содержащие молибден, висмут и железо.
Кроме того, для первой стадии реакции по изобретению пригодны мультиметаллоксидные катализаторы, содержащие молибден, висмут и железо, описанные в опубликованном научном открытии №497012 от 29.08.2005, в патентах Германии DE-A 10046957, DE-A 10063162, DE-C 3338380, DE-A 19902562, в европейском патенте ЕР-А 15565, в патенте Германии DE-C 2380765, в европейском патенте ЕР-А 807465, в европейском патенте ЕР-А 279374, в патенте Германии DE-A 3300044, в европейском патенте ЕР-А 575897, в патенте США US-A 4438217, в патенте Германии DE-A 19855913, в международной заявке на патент WO 98/24746, в патенте Германии DE-A 19746210 (катализаторы общей формулы II), в патенте Японии JP-A 91/294239, в европейских патентах ЕР-А 293224 и ЕР-А 700714. Это особенно относится к примерам осуществления изобретения в этих опубликованных источниках, среди которых особенно предпочтительными являются описанные в европейских патентах ЕР-А 15565 и ЕР-А 575897, а также в патентах Германии DE-A 19746210 и DE-A 19855913. Особенно выделяется в этой связи катализатор согласно примеру 1с из европейского патента ЕР-А 15565, а также соответствующим образом подаваемый катализатор, активная масса которого имеет, однако, состав Mo12Ni6,5Zn2Fe2Bi1P0,0065K0,06Ox·10SiO2. Кроме того, выделяется пример под порядковым номером 3 из патента Германии DE-A 19855913 (стехиометрия Mo12Co7Fe3Bi0,6К0,08SiO1,6Ох) в качестве полого цилиндрического цельного катализатора с размерами 5 мм × 3 мм × 2 мм (внешний диаметр × высота × внутренний диаметр), а также мультиметаллоксидный II - цельный катализатор согласно примеру 1 из патента Германии DE-A 19746210. Кроме того, можно назвать мультиметаллоксидные катализаторы по патенту США US-A 4438217. Последний имеет значение особенно тогда, когда этот полый цилиндр имеет размеры 5,5 мм × 3 мм × 3,5 мм, либо 5 мм × 2 мм × 2 мм, либо 5 мм × 3 мм × 2 мм, либо 6 мм × 3 мм × 3 мм, либо 7 мм × 3 мм × 4 мм (в каждом случае внешний диаметр × высота × внутренний диаметр). Другой возможной формой катализатора в этой связи являются прутки (например, длиной 7,7 мм и диаметром 7 мм либо длиной 6,4 мм и диаметром 5,7 мм).
Множество пригодных для первой стадии реакции мультиметаллоксидных активных масс, содержащих молибден, железо и висмут, могут быть объединены общей формулой IV:
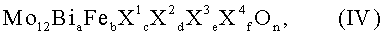
в которой переменные члены имеют следующие значения:
X1 = никель и/или кобальт,
X2 = таллий, щелочной металл и/или щелочноземельный металл,
X3 = цинк, фосфор, мышьяк, бор, сурьма, олово, германий, свинец и/или висмут,
X4 = кремний, алюминий, титан и/или цирконий,
а = от 0,5 до 5,
b = от 0,01 до 5, предпочтительно от 2 до 4,
с = от 0 до 10, предпочтительно от 3 до 10,
d = от 0 до 2, предпочтительно от 0,02 до 2,
е = от 0 до 8, предпочтительно от 0 до 5,
f = от 0 до 10 и
n означает число, которое определяется валентностью и частотой повторения элемента, отличающегося от кислорода.
Вышеназванное, прежде всего, справедливо в том случае, когда Х получают известным способом (см., например, патент ФРГ DE-A 4023239) и согласно изобретению используют в виде веществ, сформованных, например, в виде шариков, колец или цилиндров, или также в виде катализаторов с оболочкой, то есть покрытых активной массой предварительно сформованных инертных тел носителей. Разумеется, что сказанное справедливо также, когда их используют в порошкообразной форме в качестве катализаторов для первой стадии реакции (пропилен → акролеин).
В принципе, активные массы формулы IV получают простым способом, как правило, благодаря тому, что из соответствующих источников их элементарных компонентов получают как можно более тщательно смешанную, преимущественно тонкодисперсную составленную из их стехиометрического количества, сухую смесь и кальцинируют ее при температуре от 350 до 650°С. Кальцинирование может осуществляться как под инертным газом, так и в окислительной атмосфере, такой как воздух (смесь инертного газа и кислорода), а также в восстановительной атмосфере (например, в смеси инертного газа, аммиака, окиси углерода и/или водорода). Продолжительность кальцинирования может составлять от нескольких минут до нескольких часов и обычно увеличивается с повышением температуры. В качестве источников элементарных компонентов мультиметаллоксидных активных масс IV могут использоваться такие соединения, которые уже являются окислами, и/или такие соединения, которые могут превращаться в окислы при нагревании, по меньшей мере, в присутствии кислорода.
Кроме окислов, в качестве исходных соединений могут быть использованы, прежде всего, галогениды, нитраты, формиаты, оксалаты, цитраты, ацетаты, карбонаты, аминовые комплексы, соли аммония и/или гидроокиси (в сухую тщательно смешанную смесь дополнительно могут быть введены такие соединения, как гидроокись аммония, карбонат аммония, нитрат аммония, муравьинокислый аммоний, уксусная кислота, уксуснокислый аммоний и/или оксолат аммония, которые при последующем кальцинировании могут распадаться и/или разлагаться с выделением газообразных соединений).
Тщательное смешение исходных соединений для получения мультиметаллоксидных активных масс IV может осуществляться в сухой или мокрой форме. Если его осуществляют в сухой форме, то целесообразно использовать исходные соединения в виде высокодисперсного порошка и после смешения и, при необходимости, прессования осуществлять кальцинирование. Однако, преимущественно, тщательное смешение осуществляют в мокрой форме. При этом обычно исходные соединения смешивают друг с другом в форме водного раствора и/или суспензии. Особенно тщательное сухое смешение при описанных способах смешения получают в том случае, когда исходят исключительно из находящихся в растворенной форме источников элементарных компонентов. В качестве растворителя, предпочтительно, используют воду. После этого полученную водную массу сушат, причем процесс сушки проводят, предпочтительно, в виде распылительной сушки водной смеси с температурой на выходе от 100 до 150°С.
Мультиметаллоксидные активные массы IV могут использоваться для первой стадии реакции парциального окисления по изобретению пропилена в акриловую кислоту как в форме порошка, так и сформованными в определенную геометрическую форму катализатора, причем формование может осуществляться перед или после заключительного кальцинирования. Например, можно получить цельные катализаторы из порошка активной массы или из ее некальцинированной и/или частично кальцинированной предварительной массы посредством прессования для получения желаемой геометрической формы катализатора (например, таблетированием, экструдированием или штренг-прессованием), причем, при необходимости, могут добавляться вспомогательные средства, такие, например, как графит или стеариновая кислота, в качестве смазки и/или средство, облегчающее выемку из пресс-формы, и армирующий наполнитель (усилитель), такой как микроволокна из стекла, асбест, карбид кремния или титанат калия. Однако в качестве вспомогательного вещества при формовании вместо графита может также использоваться гексагональный нитрид бора, такой как рекомендуется в патенте Германии DE-A 102005037678. Пригодными формами цельного катализатора являются, например, полнотелый или полый цилиндр с внешним диаметром и длиной от 2 до 10 мм. В случае полого цилиндра целесообразной является толщина его стенки от 1 до 3 мм. Разумеется, цельный катализатор может также иметь форму шариков, при этом диаметр шариков составляет от 2 до 10 мм. Особенно релевантные размеры полого цилиндра, в особенности в случае цельных катализаторов, составляют 5 мм × 3 мм × 2 мм (внешний диаметр × высота × внутренний диаметр).
Разумеется, формование пригодных порошкообразных активных масс по изобретению или порошкообразной еще некальцинированной и/или частично кальцинированной предварительной массы может осуществляться также нанесением катализатора на предварительно сформованный инертный носитель. Покрытие тела носителя для получения катализаторов с оболочкой, как правило, проводят в соответствующем вращающемся контейнере, таком, например, как известен из патента Германии DE-A 2909671, европейского патента ЕР-А 293859 или из европейского патента ЕР-А 714700. Для покрытия тела носителя целесообразно наносимую порошкообразную массу увлажнять и после нанесения вновь высушить, например, горячим воздухом. Толщину слоя наносимой на тело носителя порошкообразной массы часто выбирают в пределах от 10 до 1000 мкм, предпочтительно в пределах от 50 до 500 мкм и особенно предпочтительно в пределах от 150 до 250 мкм.
В качестве веществ для носителя могут использоваться обычные пористые или непористые окислы алюминия, двуокись кремния, двуокись тория, двуокись циркония, карбид кремния или силикаты, такие как силикат магния или силикат алюминия. Они, по существу, инертны в отношении парциального окисления пропилена. Тело носителя может быть сформовано с правильной или неправильной структурой, причем предпочтительным является тело носителя, сформованное с правильной структурой и с явно образованной поверхностной шероховатостью, например такое, как шарики или полый цилиндр. Согласно изобретению возможно использовать, по существу, непористые с шероховатой поверхностью носители из стеатита, имеющие форму шариков, диаметр которых составляет от 1 до 10 мм или до 8 мм, предпочтительно от 4 до 5 мм. Однако согласно изобретению можно использовать также в качестве тела носителя цилиндры, длина которых составляет от 2 до 10 мм, а внешний диаметр равен от 4 до 10 мм. Если в качестве тела носителя используют кольца, то, кроме того, толщина их стенки составляет обычно от 1 до 4 мм. Используемое согласно изобретению кольцеобразное тело носителя имеет длину от 2 до 6 мм, внешний диаметр от 4 до 8 мм и толщину стенки от 1 до 2 мм. Согласно изобретению в качестве тела носителя пригодны также кольца размером 7 мм × 3 мм × 4 мм (внешний диаметр × высота × внутренний диаметр). Толщину наносимой на поверхность тела носителя каталитически активной оксидной массы, разумеется, выбирают соответственно желаемой толщине оболочки (сравни европейский патент ЕР-А 714700).
Пригодными для стадии получения акролеина из пропилена мультиметаллоксидными активными массами согласно изобретению являются, кроме того, массы общей формулы V:
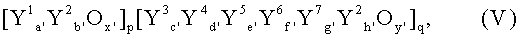
в которой переменные члены имеют следующие значения:
Y1 = только висмут или висмут и, по меньшей мере, один элемент из группы: теллур, сурьма, олово и медь;
Y2 = молибден или вольфрам либо молибден и вольфрам;
Y3 = щелочной металл, таллий или самарий;
Y4 = щелочноземельный металл, никель, кобальт, медь, марганец, цинк, олово, кадмий или ртуть;
Y5 = железо либо железо и, по меньшей мере, один из элементов хром и церий;
Y6 = фосфор, мышьяк, бор и/или сурьма;
Y7 = редкоземельный металл, титан, цирконий, ниобий, тантал, рений, рутений; родий, серебро, золото, алюминий, галлий, индий, кремний, германий, свинец, торий и/или уран;
а' = от 0,01 до 8,
b' = от 0,1 до 30,
с' = от 0 до 20,
d' = от 0 до 20,
е' > от 0 до 20,
f' = от 0 до 6,
g' = от 0 до 15,
h' = от 8 до 16,
x', y' = числа, которые определяются валентностью и повторяемостью отличающихся от кислорода элементов, а
р, q = числа, отношение которых p/q составляет от 0,1 до 10,
содержащие трехмерные пространственные протяженные ограниченные их локальным окружением благодаря их составу, отличающемуся от локального окружения, области химического состава Y1 a'Y2 b'Ox', наибольший диаметр которых (самая длинная, проходящая через центр тяжести области прямая протяженность соединения двух точек, находящихся на поверхности (поверхности раздела) области) составляет от 1 нм до 100 мкм, часто от 10 нм до 500 нм или от 1 мкм до 50 или 25 мкм.
Особенно пригодными мультиметаллоксидными массами V согласно изобретению являются такие, в которых Y1 является только висмутом.
Среди этих соединений опять особенно пригодными являются те из них, которые соответствуют общей формуле VI:
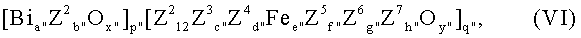
где переменные члены имеют следующие значения:
Z2 = молибден или вольфрам либо молибден и вольфрам;
Z3 = никель и/или кобальт;
Z4 = таллий, щелочной металл и/или щелочноземельный металл;;
Z5 = фосфор, мышьяк, бор, олово, сурьма; церий и/или свинец;
Z6 = кремний, алюминий, титан и/или цирконий;
Z7 = медь, серебро и/или золото;
а” = от 0,1 до 1,
b” = от 0,2 до 2,
с” = от 3 до 10,
d” = от 0,02 до 2,
е” = от 0,01 до 5, предпочтительно от 0,1 до 3,
f” = от 0 до 5,
g” = от 0 до 10,
h” = от 0 до 1,
x”, y” = числа, значения которых определяются валентностью и частотой повторяемости отличающихся от кислорода элементов в VI,
р”, q” = числа, отношение которых p”/q” составляет от 0,1 до 5, предпочтительно от 0,5 до 2,
причем более всего предпочтительными являются те массы VI, в которых Z2 b”=(вольфрам)b”, a Z2 12=(молибден)12.
Кроме того, согласно изобретению имеет также значение, когда, по меньшей мере, 25 мол.% (предпочтительно, по меньшей мере, 50 мол.% и особенно предпочтительно 100 мол.%) от общего содержания [Y1 a'Y2 b'Ox']p([Bia”Z2 b”Ox”]p”) пригодных по изобретению мультиметаллоксидных масс V (мультиметаллоксидных масс VI) находится в пригодных по изобретению мультиметаллоксидных массах V (мультиметаллоксидных массах VI) в форме трехмерной (пространственной) протяженной (вытянутой) ограниченной ее локальным окружением вследствие отличающегося от нее локального окружения химического состава области химического состава Y1 a'Y2 b'Ox'[Bia”Z2 b”Ox”], наибольший диаметр которой находится в пределах от 1 нм до 100 мкм.
Что касается формования, то в отношении мультиметаллоксидных масс V-катализаторов справедливо сказанное о мультиметаллоксидных массах IV-катализаторов.
Получение мультиметаллоксидных масс V-активных масс описано, например, в европейском патенте ЕР-А 575897, а также в патенте Германии DE-A 19855913.
Рекомендуемые выше инертные вещества для носителя, кроме прочего, могут также использоваться в качестве инертных веществ для разбавления и/или ограничения соответствующего неподвижного слоя катализатора или в качестве их защитной и/или нагревающей подводимую реакционную газовую смесь предварительной сыпучей массы.
Для второй стадии (второй реакционной стадии), то есть для гетерогенного каталитического газофазного парциального окисления акролеина в акриловую кислоту согласно изобретению в качестве активных масс для необходимых катализаторов могут использоваться, в принципе, все содержащие молибден и ванадий мультиметаллоксидные массы, например такие, как описаны в патентах Германии DE-A 10046928 и DE-А 19815281.
Многие из них являющиеся согласно изобретению благоприятными, могут охватываться общей формулой VII:
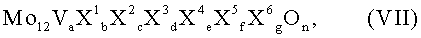
в которой переменные члены имеют следующие значения:
X1 = вольфрам, ниобий, тантал, хром и/или церий;
X2 = медь, никель, кобальт, железо, марганец и/или цинк;
X3 = сурьма и/или висмут;
X4 = один или несколько щелочных металлов;
X5 = один или несколько щелочноземельных металлов;
X6 = кремний, алюминий, титан и/или цирконий;
а = от 1 до 6,
b = от 0,2 до 4,
с = от 0,5 до 18,
d = от 0 до 40,
е = от 0 до 2,
f = от 0 до 4,
g = 0 до 40 и
n = число, значение которого определяется валентностью и повторяемостью отличных от кислорода элементов в VII.
Согласно изобретению особенно благоприятными формами воплощения среди активных мультиметаллоксидов VII являются такие, которые включают нижеследующие значения переменных членов общей формулы VII:
X1 = вольфрам, ниобий и/или хром;
X2 = медь, никель, кобальт и/или железо;
X3 = сурьма;
X4 = натрий и/или калий;
X5 = кальций, стронций и/или барий;
X6 = кремний, алюминий и/или титан;
а = от 1,5 до 5,
b = от 0,5 до 2,
с = от 0,5 до 3,
d = от 0 до 2,
е = от 0 до 0,2,
f = от 0 до 1 и
n = число, значение которого определяется валентностью и повторяемостью отличных от кислорода элементов в VII.
Согласно изобретению наиболее благоприятными мультиметаллоксидами VII являются мультиметаллоксиды общей формулы VIII;
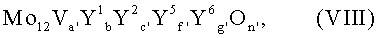
в которой
Y1 = вольфрам и/или ниобий;
Y2 = медь и/или никель;
Y5 = кальций и/или стронций;
Y6 = кремний и/или алюминий;
а' = от 2 до 4,
b' = от 1 до 1,5,
с' = от 1 до 3,
f' = от 0 до 0,5,
g' = от 0 до 8 и
n' = число, значение которого определяется валентностью и повторяемостью отличных от кислорода элементов в VIII.
Пригодные по изобретению мультиметаллоксидные активные массы (VII) получают известным способом, описанным, например, в патенте ФРГ DE-A 4335973 или в европейском патенте ЕР-А 714700.
Пригодные для стадии получения «акролеин → акриловая кислота» мультиметаллоксидные активные массы по изобретению, особенно общей формулы VII, получают простым способом в результате того, что как можно более тщательно смешивают соответствующие источники их элементарных компонентов в их стехиометрическом количестве, предпочтительно в высокодисперсной форме, получают сухую смесь и кальцинируют ее при температуре от 350 до 600°С. Кальцинирование можно проводить как под инертным газом, так также и в окислительной атмосфере (например, в смеси инертного газа и восстановительных газов, таких как водород, аммиак, моноокись углерода, метан и/или акролеин или самих названных восстановительных газов). Продолжительность кальцинирования может составлять от нескольких минут до нескольких часов и обычно увеличивается с повышением температуры. В качестве источников для элементарных компонентов мультиметаллоксидных активных масс VII можно использовать такие соединения, о которых уже говорилось в отношении окислов, и/или такие соединения, которые переводят в окислы нагреванием, по меньшей мере, в присутствии кислорода.
Тщательное смешение исходных соединений для получения мультиметаллоксидных масс VII может осуществляться в сухой или мокрой форме. Если это осуществляют в сухой форме, то целесообразно использовать исходные соединения в виде высокодисперсного порошка и после смешения и, при необходимости, прессования осуществлять кальцинирование. Однако, предпочтительно, тщательное смешение осуществляют в мокрой форме. При этом исходные соединения обычно смешивают друг с другом в форме водного раствора и/или суспензии. Особенно тщательно смешенные сухие смеси при описанном способе смешения получают в том случае, когда исходят исключительно из находящихся в растворенной форме имеющихся источников элементарных компонентов. В качестве растворителя, предпочтительно, используют воду. После этого полученную водную массу сушат, причем, предпочтительно, методом распылительной сушки водной смеси с температурой на выходе от 100 до 150°С.
Полученные мультиметаллоксидные массы, особенно общей формулы VII, могут использоваться для парциального окисления акролеина по изобретению как в форме порошка, так и сформованными в катализаторы определенной формы, причем формование может осуществляться как перед кальцинированием, так и после заключительного кальцинирования. Например, из порошкообразной активной массы или из ее некальцинированной предварительной (предшествующей) массы цельные катализаторы могут быть получены посредством прессования для получения желаемой формы катализатора (например, таблетированием, экструдированием или прессованием с получением прутков, штренг-прессованием), причем, при необходимости, могут добавляться вспомогательные средства, например такие, как графит или стеариновая кислота, в качестве смазки и/или вспомогательное средство, облегчающее выемку из пресс-формы, или армирующий наполнитель, такой как микроволокна из стекла, асбеста, карбида кремния или титаната калия. Благоприятной геометрической формой цельного катализатора является, например, полый цилиндр или полнотелый цилиндр с внешним диаметром и длиной от 2 до 10 мм. В случае полого цилиндра целесообразно, если толщина его стенок составляет от 1 до 3 мм. Разумеется, цельный катализатор может также иметь шаровую геометрическую форму, причем диаметр шариков может составлять от 2 до 10 мм (например, 8,2 мм или 5,1 мм).
Конечно, формование порошкообразной активной массы или ее порошкообразной еще не кальцинированной предшествующей массы может также осуществляться нанесением катализатора на предварительно сформованный инертный носитель. Покрытие тела носителя для получения катализаторов с оболочками, как правило, проводят в соответствующем вращающемся контейнере, например, как известно из патента ФРГ DE-A 2909671 и европейских патентов ЕР-А 293859 и ЕР-А 714700.
Для нанесения покрытия на тело носителя целесообразно увлажнять наносимую порошкообразную массу и после нанесения вновь высушить, например, горячим воздухом. Толщина слоя нанесенной на тело носителя порошкообразной массы по изобретению часто находится в пределах от 10 до 1000 мкм, предпочтительно в пределах от 50 до 500 мкм и наиболее предпочтительно в пределах от 150 до 250 мкм.
В качестве веществ носителя могут применяться обычно используемые пористые или непористые окислы алюминия, двуокись кремния, двуокись тория, двуокись циркония, карбид кремния или силикаты, такие как силикат магния или силикат алюминия. Тело носителя может быть сформовано правильной или неправильной формы, причем предпочтительным является сформованное тело носителя правильной формы с явно образованной поверхностной шероховатостью, например шарики или полый цилиндр с расщепленным (шероховатым) поверхностным слоем. Можно использовать, по существу, непористые шарообразные носители с шероховатой поверхностью из стеатита, диаметр которых составляет от 1 до 10 мм или до 8 мм, предпочтительно от 4 до 5 мм. Следовательно, пригодная шаровая геометрическая форма может иметь диаметр от 8,2 до 5,1 мм. Однако можно также использовать в качестве тела носителя цилиндры, длина которых составляет от 2 до 10 мм, а внешний диаметр равен от 4 до 10 мм. Кроме того, в случае использования в качестве тела носителя колец толщина их стенки составляет обычно от 1 до 4 мм. Предпочтительно, используемое кольцеобразное тело носителя имеет длину от 2 до 6 мм, внешний диаметр от 4 до 8 мм и толщину стенки от 1 до 2 мм. В качестве носителя прежде всего могут использоваться также кольца размером 7 мм × 3 мм × 4 мм (внешний диаметр × высота × внутренний диаметр). Толщина слоя наносимых на поверхность тела носителя каталитически активных оксидных масс, разумеется, соответствует желаемой толщине оболочки (сравни европейский патент ЕР-А 714700).
Пригодными для стадии парциального окисления «акролеин → акриловая кислота», кроме того, являются мультиметаллоксидные активные массы общей формулы IX:
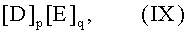
в которой переменные члены имеют следующие значения:
D=Mo12Va”Z1 b”Z2 c”Z3 d”Z4 e”Z5 f”Z6 q”Ox”,
E=Z7 12Cuh”Hi”Oy”,
Z1 = вольфрам, ниобий, тантал, хром и/или церий;
Z2 = медь, никель, кобальт, железо, марганец и/или цинк;
Z3 = сурьма и/или висмут;
Z4 = литий, натрий, калий, рубидий, цезий и/или водород;
Z5 = магний, кальций, стронций и/или барий;
Z6 = кремний, алюминий, титан и/или цирконий;
Z7 = молибден, вольфрам, ванадий, ниобий и/или тантал, предпочтительно молибден и/или вольфрам;
а” = от 1 до 8,
b” = от 0,2 до 5,
с” = от 0 до 23,
d” = от 0 до 50,
е” = от 0 до 2,
f” = от 0 до 5,
g” = от 0 до 50,
h” = от 4 до 30,
i” = от 0 до 20, и
x”, y” = числа, значения которых определяются валентностью и повторяемостью отличающихся от кислорода элементов в IX, и
p, q = отличные от нуля числа, отношение которых p/q составляет от 160:1 до 1:1,
и которые получают в результате того, что предварительно получают отдельно мультиметаллоксидную массу Е:
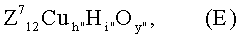
в тонкодисперсной форме (исходная масса 1) и непосредственно после этого предварительно образованную твердую исходную массу 1 вводят в водный раствор, водную суспензию или в высокодисперсную сухую смесь источников элементов молибден, вольфрам, Z1, Z2, Z3, Z4, Z5, Z6, которая содержит названные элементы в стехиометрии D:
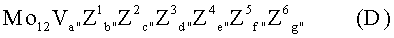
(исходная масса 2), в желаемом количественном соотношении p:q, образованную при этом водную смесь, при необходимости, сушат и полученную таким образом сухую предварительную массу перед или после ее сушки кальцинируют для получения катализатора желаемой геометрической формы при температуре от 250 до 600°С.
Особенно пригодными являются мультиметаллоксидные массы IX, у которых введение предварительно образованной твердой исходной массы 1 в водную исходную массу 2 осуществляют при температуре <70°С. Подробное описание получения мультиметаллоксидных масс VI-катализаторов содержится, например, в европейском патенте ЕР-А 668104, патентах Германии DE-A 19736105, DE-A 10046928, DE-A 19740493 и DE-A 19528646.
Что касается формования, то для него справедливо сказанное относительно мультиметаллоксидных масс IX-катализаторов. Особенно пригодными мультиметаллоксидными катализаторами по изобретению для стадии получения «акролеин → акриловая кислота» являются также описанные в патенте Германии DE-A 19815281, особенно мультиметаллоксидные активные массы общей формулы I согласно патенту Германии DE-A 19815281.
Для стадии реакции получения акролеина из пропилена преимущественными являются кольца цельного катализатора, для стадии реакции получения из акролеина акриловой кислоты - кольца катализатора с оболочкой.
Целесообразно, если температура неподвижного слоя катализатора для первой стадии реакции (пропилен → акролеин) составляет от 270 до 450°С или от 280 до 420°С, предпочтительно от 300 до 380°С. Для второй стадии реакции (акролеин → акриловая кислота) целесообразной является температура неподвижного слоя катализатора от 200 до 370°С или от 200 до 320°С, предпочтительно от 220 до 380°С. При проведении способа по изобретению в однозонных многоконтактных трубчатых реакторах с неподвижным слоем и множеством контактных труб названные температуры соответствуют температурам теплоносителя (солевой расплав) на входе в окружающий контактную трубу резервуар.
В принципе, в способе по изобретению внутри свежевнесенного в реакционное пространство неподвижного слоя катализатора для первой стадии реакции его удельная объемная активность в направлении потока реакционной газовой смеси по длине пути потока (то есть по длине неподвижного слоя катализатора) может быть постоянной или, преимущественно, по меньшей мере, однократно она может повышаться (непрерывно, или внезапно, или ступенчато). Во всех вышеназванных случаях, кроме того, полезно, если состав активной массы по длине пути потока (то есть внутри неподвижного слоя катализатора) не изменяется. Названное для первой стадии реакции действительно также и для второй стадии реакции гетерогенного каталитического газофазного парциального окисления на неподвижном слое катализатора пропилена в акриловую кислоту.
Для приготовления свежевносимого в реакционное пространство неподвижного слоя катализатора для первой реакционной стадии могут использоваться содержащие только лишь мультиметаллоксидную массу тела катализатора или также в значительной мере гомогенная смесь из содержащих мультиметаллоксидную массу тел катализатора и не содержащих никакой мультиметаллоксидной массы, по существу, инертных по отношению к гетерогенному каталитическому газофазному парциальному окислению формованных тел (тел для разбавления). В качестве веществ для таких инертных тел, в принципе, могут использоваться все инертные тела, которые пригодны также в качестве вещества носителя для пригодных по изобретению катализаторов с оболочкой. В качестве таких веществ могут использоваться, например, пористые или непористые окислы алюминия, двуокись кремния, двуокись тория, двуокись циркония, карбид кремния, силикаты, такие как силикат магния или алюминия, или уже упомянутый стеатит.
Геометрия таких инертных формованных тел разбавителя, в принципе, может быть любой, т.е. они могут быть, например, шарами, многоугольниками, полнотелым цилиндром или так же, как содержащее активную массу тело катализатора, кольцами. В качестве инертных формованных тел разбавителя согласно изобретению, предпочтительно, выбирают такие, геометрия которых, в основном, соответствует геометрии разбавляемых ими формованных тел катализатора (ранее приведенные высказывания действительны также в отношении приготовления неподвижного слоя катализатора для используемой на второй реакционной стадии в значительной степени гомогенной смеси из содержащих мультиметаллоксидную активную массу формованных тел катализатора и формованных тел носителя).
Полезно, если химический состав используемой активной массы в свежевнесенном в реакционное пространство неподвижном слое катализатора для первой реакционной стадии (насыпной массы неподвижного слоя катализатора) не изменяется. Это означает, что используемая для отдельных формованных тел катализатора активная масса может, конечно, являться смесью из различных мультиметаллоксидов, однако тогда для сыпучей массы неподвижного слоя катализатора 1 всех формованных тел катализатора, предпочтительно, использовать одинаковую смесь.
Удельную объемную активность (т.е. рассчитанную на единицу объема), как уже говорилось, можно понизить простым способом благодаря тому, что основное количество однородно изготовленных формованных тел катализатора гомогенно разбавляют формованными телами разбавителя. Чем выше содержание формованных тел разбавителя, тем меньше содержание активной массы, содержащейся в определенном объеме сыпучей массы, т.е. тем меньше катализаторная активность.
По меньшей мере, однократное повышение удельной объемной активности катализатора в направлении потока реакционной газовой смеси через насыпную массу неподвижного слоя катализатора может достигаться в способе по изобретению простым способом в результате того, что начинают засыпку с высоким содержанием инертных формованных тел разбавителя в расчете на вид (сорт) формованных тел катализатора и затем это содержание формованных тел разбавителя уменьшают в направлении потока либо непрерывно, либо, по меньшей мере, однократно или многократно прерывисто (например, ступенчато). Если содержание формованных тел разбавителя оставляют постоянным или вообще не используют никаких формованных тел разбавителя в сыпучей массе неподвижного слоя катализатора 1, то это приводит к постоянной удельной объемной активности в направлении потока реакционной газовой смеси через насыпную массу неподвижного слоя катализатора 1. Однако нарастание удельной объемной активности возможно также, например, в результате того, что увеличивают толщину слоя нанесенных на носитель активных масс при остающихся одинаковыми геометрии и виде активных масс формованных тел катализатора с оболочкой и повышают или толщину нанесенного на носитель слоя активной массы, или содержание формованных тел катализатора с повышенным массовым содержанием активных масс в смеси из катализаторов с оболочками с одинаковой геометрией, но с различным массовым содержанием активной массы. Аналогичного эффекта можно также достигнуть, например, в результате того, что изменяют соответствующим образом соотношение компонентов в смеси из цельных катализаторов и катализаторов с оболочками (при идентичной активной массе). Разумеется, что можно также комбинировать описанные варианты.
Обычно при двухстадийном парциальном окислении пропилена в акриловую кислоту по изобретению удельную объемную активность внутри насыпной массы неподвижного слоя катализатора 1 еще внутри насыпной массы неподвижного слоя катализатора 2 (т.е. в реакционном пространстве свежевведенного неподвижного слоя катализатора для второй стадии реакции) однократно снижают в направлении потока реакционной газовой смеси.
Без и/или в сочетании с насыпной массой неподвижного слоя катализатора 1 могут находиться сыпучие массы, состоящие исключительно из инертного вещества (например, только из формованных тел разбавителя). (В этом тексте описания их, понятно, не причисляют к насыпке неподвижного слоя катализатора 1, так как они не содержат никаких формованных тел, содержащих мультиметаллоксидную активную массу). При этом используемые для инертной насыпной массы формованные тела могут иметь такую же геометрию, как и используемые формованные тела катализатора в сыпучей массе неподвижного слоя катализатора 1.
Однако геометрия используемых для инертной насыпной массы формованных тел катализатора может также отличаться от ранее названной геометрии формованных тел катализатора (например, может иметь шарообразную форму вместо кольцеобразной).
Согласно изобретению предпочтительно, если в способе по изобретению насыпная масса неподвижного слоя катализатора 1 в направлении потока реакционной газовой смеси является структурированной следующим образом.
На длине участка от 10 до 60%, предпочтительно от 10 до 50%, особенно предпочтительно от 20 до 40% и наиболее предпочтительно от 25 до 35% (т.е., например, на длине от 0,70 до 1,50 м, предпочтительно от 0,90 до 1,20 м), в каждом случае, от общей длины насыпной массы неподвижного слоя катализатора 1 либо только формованные тела катализатора, либо гомогенная смесь (либо две такие следующие друг за другом с пониженным разбавлением гомогенные смеси) из формованных тел катализатора и формованных тел разбавителя (причем оба имеют, преимущественно, в основном, одинаковую геометрическую форму), при этом массовое содержание формованных тел разбавителя (массовая плотность формованных тел катализатора и формованных тел разбавителя, как правило, отличаются лишь незначительно) в нормальных условиях обычно составляет от 5 до 40 мас.%, или от 10 до 40 мас.%, или от 20 до 40 мас.%, или от 25 до 35 мас.%. К этой первой зоне неподвижного слоя катализатора затем присоединяется согласно изобретению, преимущественно, до конца длины насыпной массы неподвижного слоя катализатора 1 (т.е., например, на длину участка от 2,00 до 3,50 м, предпочтительно от 2,50 до 3,00 м) либо лишь в меньшем объеме (чем в первой зоне) разбавленная сыпучая масса формованных тел катализатора, либо, наиболее предпочтительно, только одна (неразбавленная) сыпучая масса тех же самых формованных тел катализатора, которую использовали также и в первой зоне.
Вышесказанное, в особенности, относится к тому случаю, когда в насыпной массе неподвижного слоя катализатора 1 в качестве формованных тел катализатора используют цельный катализатор в форме колец или катализатор с оболочкой в форме колец (особенно названные в этом тексте описания в качестве предпочтительных). Названные в рамках вышеназванного структурирования как формованные тела катализатора, так и формованные тела разбавителя в способе по изобретению, преимущественно, имеют, в основном, размеры колец 5 мм × 3 мм × 2 мм (внешний диаметр × длина × внутренний диаметр).
Как может варьироваться удельная объемная активность насыпной массы неподвижного слоя катализатора 1, так соответствующим образом может также варьироваться и удельная объемная активность сыпучей массы неподвижного слоя катализатора 2. При этом без и/или в присоединении к соответствующей сыпучей массе неподвижного слоя катализатора 2 опять может находиться соответствующая инертная насыпная масса (понятно, что в этом тексте описания она не причислялась к сыпучей массе неподвижного слоя катализатора 2, так как не содержит формованных тел, содержащих мультиметаллоксидную активную массу).
В способе согласно изобретению является предпочтительным, если сыпучую массу неподвижного слоя катализатора 2 в направлении потока реакционной газовой смеси структурируют следующим образом.
На длине участка от 10 до 60%, предпочтительно от 10 до 50%, особенно предпочтительно от 20 до 40% и наиболее предпочтительно от 25 до 35% (т.е., например, на длине от 0,70 до 1,50 м, предпочтительно от 0,90 до 1,20 м), в каждом случае, от общей длины насыпной массы неподвижного слоя катализатора 2 либо только формованные тела катализатора, либо гомогенная смесь (либо две такие следующие друг за другом с пониженным разбавлением гомогенные смеси) из формованных тел катализатора и формованных тел разбавителя (причем, преимущественно, оба имеют, в основном, одинаковую геометрическую форму), при этом массовое содержание формованных тел разбавителя (массовая плотность формованных тел катализатора и формованных тел разбавителя, как правило, отличаются лишь незначительно) в нормальных условиях обычно составляют от 10 до 50 мас.%, преимущественно от 20 до 45 мас.% и особенно предпочтительно от 25 до 35 мас.%. К этой первой зоне неподвижного слоя катализатора затем присоединяется согласно изобретению, преимущественно, до конца длины насыпной массы неподвижного слоя катализатора 2 (т.е., например, на длину участка от 2,00 до 3,50 м, предпочтительно от 2,50 до 3,00 м) либо лишь в меньшем объеме (чем в первой зоне) разбавленная сыпучая масса формованных тел катализатора, либо, наиболее предпочтительно, только одна (неразбавленная) сыпучая масса тех же самых формованных тел катализатора, которую использовали также и в первой зоне.
Вышесказанное, в особенности, относится к тому случаю, когда в насыпной массе неподвижного слоя катализатора 2 в качестве формованных тел катализатора используют катализатор с оболочкой в форме колец (особенно названный в этом тексте описания в качестве предпочтительного). Указанные в рамках вышеназванного структурирования как формованные тела катализатора или их носитель в форме колец, так и формованные тела разбавителя в способе по изобретению имеют, преимущественно, в основном, размеры колец 7 мм × 3 мм × 4 мм (внешний диаметр × длина × внутренний диаметр).
Обычно свежевносимый в реакционное пространство неподвижный слой катализатора и прочие предельные условия для обеих стадий реакции выбирают такими, как описано в европейских патентах ЕР-А 990636 и ЕР-А 1106598, как протяженность (расширение) точки перегрева, так и температура ее чувствительности являются как можно более низкими (ΔTHB, как правило, составляет ≤80°С, чаще всего ≤70°С, часто от 20 до 70°С; преимущественно, ΔTHB является низкой; «peak-to-salt» максимальная температура чувствительности к соли составляет ≤9°С, или ≤7°С, или ≤5°С, или ≤3°С).
Разумеется, до замены части неподвижного слоя катализатора могут использоваться все известные в технике способы, в каждом случае, сами по себе или в их сочетании, которые пригодны для того, чтобы отсрочить на некоторое время требование частичной замены части неподвижного слоя катализатора по изобретению.
Технологически целесообразно проводить как первую, так и вторую стадии реакции, в каждом случае, в свежевнесенных в реакционное пространство неподвижных слоях катализатора в уже описанном в этом тексте как пригодном для этого однозонном многоконтактном трубчатом реакторе с неподвижным слоем катализатора. Для этого, предпочтительно, пригодны однозонные многоконтактные трубчатые реакторы с неподвижным слоем катализатора, описанные в европейских патентах ЕР-А 700714 и ЕР-А 700893. Однако, в принципе, можно также использовать для обеих реакционных стадий технологические процессы в многозонном многоконтактном трубчатом реакторе с неподвижным слоем катализатора, например, такие, как описаны в патентах Германии DE-A 10313213 и DE-A 102005062026, в международной заявке на патент WO 2004/009525 и в патенте Германии DE-A 10351269.
Как на первой, так и на второй стадии реакции рабочее давление может быть как ниже атмосферного давления (например, до 0,5 бар; реакционная смесь просасывается через неподвижный слой катализатора), так и выше атмосферного давления. Обычно рабочее давление на обеих стадиях составляет от 1 до 5 бар, часто от 1,5 до 3,5 бар. Обычно рабочее давление на обеих стадиях не превышает 100 бар.
Питание пропиленом насыпной массы неподвижного слоя катализатора 1 может составлять ≥80 нл/л·ч, или ≥100 нл/л·ч, или ≥120 нл/л·ч, или ≥140 нл/л·ч, или ≥165 нл/л·ч, или ≥170 нл/л·ч, или ≥175 нл/л·ч, или ≥180 нл/л·ч, или ≥185 нл/л·ч, или ≥190 нл/л·ч, или ≥200 нл/л·ч, или ≥210 нл/л·ч, или ≥220 нл/л·ч, или ≥230 нл/л·ч, или ≥240 нл/л·ч, или ≥250 нл/л·ч. Обычно питание пропиленом насыпной массы неподвижного слоя катализатора 1 не превышает 600 нл/л·ч. Обычно загрузка пропиленом насыпки неподвижного слоя катализатора 1 составляет ≤300 нл/л·ч, часто ≤250 нл/л·ч.
При этом общее питание на обеих реакционных стадиях может составлять, например, от 1000 до 3000 нл/л·ч. Вышесказанное относится также и к питанию акролеином насыпной массы неподвижного слоя катализатора 2.
В качестве источника необходимого для обеих стадий реакции молекулярного кислорода может использоваться как воздух, так и обедненный азотом и молекулярным кислородом воздух или чистый молекулярный кислород. Молярное отношение молекулярного кислорода к пропилену в реакционной газовой смеси для первой стадии реакции, как правило, составляет ≥1. Молярное отношение молекулярного кислорода к акролеину в реакционной газовой смеси для второй стадии реакции, как правило, составляет ≥0,5. На обеих стадиях это молярное отношение обычно ≤3.
Реакционная газовая смесь, которой питают насыпную массу неподвижного слоя катализатора 1 (в этом тексте описания названная также реакционной газовой смесью 1), как правило, имеет следующее объемное соотношение компонентов (нл/л·ч) пропилен: кислород: инертный газ (включая водяной пар) = 1:(от 0,1 до 3,0):(от 5 до 25), предпочтительно 1:(от 1,7 до 2,3):(от 10 до 15).
Реакционная газовая смесь, которой питают сыпучую массу неподвижного слоя катализатора 2 (в этом тексте описания называемая также реакционной газовой смесью 2), как правило, имеет следующее объемное соотношение компонентов (нл/л·ч) акролеин: кислород: водяной пар: инертные газы (за исключением водяного пара) = 1:(от 0,5 до 3,0):(от 0 до 20):(от 3 до 30), предпочтительно 1:(от 1 до 3):(от 0,5 до 10):(от 7 до 18).
В принципе, обе реакционные стадии могут действовать независимо друг от друга. Однако газовую смесь продукта первой стадии часто используют для питания второй реакционной стадии. При этом, как целесообразное, рекомендуется покидающую первую реакционную стадию газовую смесь продукта охлаждать перед поступлением на вторую реакционную стадию для подавления последующего сгорания части акролеина, образованного на первой реакционной стадии. Для этого между обеими реакционными стадиями обычно устанавливают вторичный холодильник. В простейшем случае это может быть непрямой секционный трубчатый теплообменник. Между обеими реакционными стадиями может дозироваться вторичный газ (молекулярный кислород и/или инертный газ). Часто к газовой смеси продукта первой реакционной стадии дозируют воздух перед его использованием для питания на второй реакционной стадии. Технологически целесообразно подводить реакционную газовую смесь к насыпной массе неподвижного слоя катализатора 1, предварительно нагрев ее до температуры неподвижного слоя катализатора на первой реакционной стадии.
В названном выше вторичном холодильнике газовую смесь продукта первой реакционной стадии, как правило, охлаждают до температуры от 210°С до 290°С, часто от 230 до 280°С или от 250°C до 270°С. При этом охлаждение непременно осуществляют до температуры ниже температуры неподвижного слоя катализатора второй стадии реакции. Благоприятно, если газовая смесь продукта первой стадии реакции, а также и второй стадии реакции содержит до 5 об.%, часто до 3 об.% остаточного молекулярного кислорода.
Описанное повторное охлаждение ни в коем случае не является обязательным и может быть исключено, особенно, в случае короткого пути прохождения газовой смеси продукта с первой реакционной стадии на вторую реакционную стадию. Также не является обязательным добавление вторичного газа между обеими стадиями реакции. Обычно оно отпадает, в особенности, когда два однозонных многоконтактных трубчатых реактора для обеих реакционных стадий сливаются в один, в этом случае уже двухзонный многоконтактный трубчатый реактор с неподвижным слоем катализатора (называемый также «единым реактором»), такой как описан, например, в патенте Германии DE-C 2830765 и в европейских патентах ЕР-А 911313 и ЕР-А 383224. В этом случае первую реакционную стадию осуществляют в первой температурной зоне, а вторую реакционную стадию - во второй температурной зоне двухзоного многоконтактного трубчатого реактора с неподвижным слоем катализатора, и реакционная газовая смесь для первой реакционной стадии содержит весь добавляемый необходимый молекулярный кислород. При этом теплоноситель для обеих зон, как правило, отделен друг от друга разделяющей жестью. Длина реакционных труб в случае слияния реакторов часто соответствует длинам труб в соответствующих не слитых друг с другом секционных трубчатых реакторах.
Как правило первая реакционная стадия протекает так, что степень конверсии пропилена UP при одноразовом пропуске реакционной газовой смеси составляет ≥90 мол.%, а селективность образования акролеина, а также побочных продуктов образования акриловой кислоты (в расчете на вступивший в реакцию пропилен) составляет в сумме (SAC)≥80 мол.%. Предпочтительно, UP составляет ≥93 мол.%, a (SAC), преимущественно, составляет ≥85 мол.%, или ≥90 мол.%, или ≥95 мол.%.
Соответственно, вторая реакционная стадия протекает, как правило, таким образом, что степень конверсии акролеина UA при одноразовом пропуске реакционной газовой смеси составляет ≥90 мол.%, часто ≥93 мол.%, чаще ≥95 мол.%, или ≥97 мол.%, или ≥99 мол.%.
Селективность образования акриловой кислоты (в расчете на вступивший в реакцию акролеин) составляет обычно ≥90 мол.%, часто ≥93 мол.% и чаще всего ≥96 мол.%.
Благоприятно, если первая реакционная стадия протекает таким образом, что содержание пропилена в газовой смеси продукта этой стадии не превышает величину 10000 мас.ч. на млн.ч., предпочтительно 6000 мас.ч. на млн.ч. и особенно предпочтительно 4000 или 2000 мас.ч. на млн.ч.
Полезно, если вторая реакционная стадия протекает таким образом, что содержание акролеина в газовой смеси продукта этой стадии не превышает величины 1500 мас.ч. на млн.ч., предпочтительно 600 мас.ч. на млн.ч. и особенно предпочтительно 350 мас.ч. на млн.ч.
Как правило, реакционная газовая смесь для первой реакционной стадии (называемая здесь также исходной реакционной газовой смесью 1) в способе по изобретению содержит от 3 до 25 об.%, часто от 5 до 20 об.% и чаще всего от 6 до 13 об.% пропилена.
Соответственное содержание акролеина имеет, как правило, реакционная газовая смесь 2 для питания второй реакционной стадии.
Содержание молекулярного кислорода в исходной реакционной газовой смеси 1 согласно изобретению (как уже указывалось) обычно определяется таким, что молярное отношение V1 содержащегося в исходной реакционной газовой смеси 1 кислорода к содержащемуся в исходной реакционной газовой смеси 1 пропилену составляет ≥1. Обычно V1 составляет в способе по изобретению ≥1 и ≤3, чаще всего ≥1,3 и ≤2,5, часто от ≥1,5 до ≤2,3. Количество молекулярного кислорода в исходной реакционной газовой смеси 2 (реакционная газовая смесь, которой питают неподвижный слой катализатора второй реакционной стадии), как уже отмечалось, обычно определяют так, чтобы молярное отношение содержащегося в исходной реакционной газовой смеси 2 кислорода к содержащемуся в исходной реакционной газовой смеси 2 акролеину составляло от ≥0,5 до ≤3 или ≤2, часто от ≥0,75 до ≤1,5.
Исходная реакционная газовая смесь 1 также может содержать ≥0,01 об.%, или ≥0,1 об.%, или ≥0,5 об.%, или ≥2 об.% двуокиси углерода. Чаще всего названное содержание двуокиси углерода ≤25 об.%.
Исходная реакционная газовая смесь 1 может содержать в качестве другого инертного газа-разбавителя молекулярный азот, в особенности, когда в качестве источника молекулярного кислорода в способе по изобретению используют воздух. В принципе, исходная реакционная газовая смесь 1 в способе по изобретению может содержать ≥1 об.%, или ≥5 об.%, или ≥10 об.%, или ≥20 об.%, или ≥30 об.%, или ≥40 об.% молекулярного азота. Как правило, однако, содержание молекулярного азота в исходной реакционной газовой смеси 1 составляет величину ≤80 мол.%, или ≤70 мол.%, или ≤60 мол.%.
Исходная реакционная газовая смесь 1 может также содержать в качестве инертного газа-разбавителя пропан. Это содержание пропана в исходной реакционной газовой смеси 1 может составлять до 70 об.% (например, от 5 до 70 об.%), или до 60 об.%, или до 50 об.%, или до 40 об.%, или до 30 об.%, или до 20 об.%, или до 10 об.%. Часто это содержание пропана составляет ≥0,5 об.% или ≥1 об.%. Однако оно может составлять величину ≥0,01 об.%, или ≥0,02 об.%, или ≥0,03 об.%. Как правило, исходная реакционная газовая смесь 1 содержит ≤10 об.%, часто ≤5 об.% пропана.
Этот пропан в способе по изобретению может добавляться, например, известным способом в качестве отдельно подводимого к исходной реакционной газовой смеси 1 известного газа-разбавителя.
Однако, разумеется, пропан может также являться компонентом исходной реакционной газовой смеси 1 вследствие того, что в качестве источника пропилена для нее служит частично дегидрированный или оксидированный пропан (как правило, это осуществляют гетерогенно каталитически). Это означает, что содержащийся в исходной реакционной газовой смеси 1 пропилен, по меньшей мере, частично может подводиться к исходной реакционной газовой смеси 1 в сочетании с не вступившим в реакцию пропаном после парциального дегидрирования (например, гетерогенно и/или гетерогенно катализируемого в присутствии и/или при исключении молекулярного кислорода).
Способ по изобретению включает также и такие варианты его осуществления, при которых исходная реакционная газовая смесь 1 содержит > от 0 до 35 об.%, часто от 1 до 25 об.%, или от 5 до 15 об.%, или до 10 об.% воды.
Типичной исходной реакционной газовой смесью 1 является, например, такая смесь, которая содержит:
Исходная реакционная газовая смесь 1 по изобретению может также содержать:
Исходная реакционная газовая смесь 2 может, например, содержать:
Однако исходная реакционная газовая смесь 1 может также содержать до 20 об.% водорода.
Таким образом, исходная реакционная газовая смесь 1 в способе по изобретению может также содержать:
Для способа по изобретению полезно также, когда исходная реакционная газовая смесь 1 содержит от 0,1 до 30 об.% двуокиси углерода.
Согласно изобретению исходная реакционная газовая смесь 2 может также содержать:
Для изобретения является важным, что с учетом всей ранее названной информации способ по изобретению может использоваться для обеих стадий, в каждом случае, как тогда, когда обе стадии протекают независимо друг от друга, так также и тогда, когда они, как уже ранее отмечалось, протекают последовательно друг за другом. Однако может быть также успешным, когда обе стадии, как описано в патенте Германии DE-A 10121592, осуществляют в одном реакторе с одним питанием.
При этом частичная замена неподвижного слоя катализатора (как общая в способе по изобретению в этом тексте описания), во всех случаях, может распространяться в направлении потока (реакционной газовой смеси) на длину до 80%, или лишь на длину до 70%, или лишь на длину до 60%, или лишь на длину до 50%, или лишь на длину до 40%, или лишь на длину до 30%, или, предпочтительно, на длину до 25%, особенно предпочтительно на длину от 30 до 50% и наиболее предпочтительно на длину от 35 до 45% от длины засыпанной насыпной массы каждого неподвижного слоя катализатора (состоящий на 100% из инертного вещества слой засыпки (первой по виду засыпки потока) при этом не рассматривается как относящийся к неподвижному слою катализатора. Однако из соображений целесообразности этот слой засыпки заменяют. Соответственно, состоящий на 100% из инертного вещества заключительный слой засыпки (конечный слой засыпки по виду потока) не рассматривается как относящийся к неподвижному слою катализатора. Однако состоящий на 100% из инертного материала промежуточный слой засыпки рассматривается как относящийся к неподвижному слою катализатора. Целесообразно, если названный процентный прирост для замены части катализатора составляет часто не более чем 10% или 20%.
Наконец, еще раз следует упомянуть, что, в частности, может существовать часть газообразной смеси питания первой стадии реакции (пропилен → акролеин) так называемый циркуляционный газ. При этом речь идет о газе, который остается после отделения продукта реакции (отделения акриловой кислоты из газообразной смеси продукта второй стадии и, как правило, частично возвращается в виде инертного газа-разбавителя для питания первой и/или второй стадии при последовательном расположении обеих стадий друг за другом.
Типичный состав циркуляционного газа следующий:
При этом отделение акриловой кислоты может осуществляться, например, как описано в европейских патентах ЕР-А 982287 и ЕР-А 982289, в патентах Германии DE-A 19924532, DE-A 10115277, DE-A 19606877, DE-A 19740252, DE-A 19627847, DE-A 10053086, в европейском патенте ЕР-А 982288 и в патенте Германии DE-A 19627847.
В принципе, замена части неподвижного слоя катализатора может проводиться в любое время, то есть, например, после одногодичной, двухгодичной, трехгодичной или многогодичной продолжительности производственного процесса. Как правило, ее проводят с учетом экономических соображений.
Наконец, необходимо упомянуть, что замена части неподвижного слоя катализатора согласно изобретению, как правило, благоприятно влияет также на падение давления при пропуске реакционной газообразной смеси через загруженную насыпную массу катализатора.
Кроме того, следует еще раз упомянуть, что средство для теплообмена (теплоноситель, расплавы соли), предпочтительно, пропускают через соответствующие многоконтактные трубчатые реакторы с неподвижным слоем в таком количестве, что разница между их температурой на входе и выходе составляет ≤5°С.
Примеры и сравнительный пример
I. Общее описание первой стадии реакции
II. Промежуточное охлаждение и подача вторичного газа
Газообразную смесь продукта первой реакционной стадии охлаждали до 250°С в секционном трубчатом теплообменнике посредством непрямого теплообмена с расплавом соли из 60 мас.% нитрата калия и 40 мас.% нитрата натрия, по существу, без потерь акролеина. Затем подавали сжатый воздух с температурой 140°С в таком количестве, что молярное соотношение кислород:акролеин в полученной смеси составляло примерно 1,28. Эту смесь подавали с температурой 220°С на вторую стадию реакции.
III. Общее описание второй реакционной стадии
Контактная труба соответствовала контактной трубе первой стадии реакции. Солевой расплав (того же состава, что и в первой реакционной стадии) и реакционную газовую смесь подавали противотоком. Солевой расплав подавали при свежей загрузке контактной трубы неподвижным слоем катализатора с температурой 260°С и отводили с температурой 260°С.
IV. Результаты (селективность образования акриловой кислоты оставалась, по существу постоянной)
А) Пример
Анализ газовой смеси продукта второй стадии реакции показал следующие результаты.
Степень конверсии образования акролеина на второй стадии реакции со свежевнесенным на второй стадии реакции неподвижным слоем катализатора (после завершения его образования) при температуре расплава соли на входе Tein на второй стадии реакции 260°С составила 99,3 мол.% при селективности образования акриловой кислоты 88,9 мол.% (эти данные относятся так же, как и последующие данные, к одноразовому пропуску).
С увеличением длительности производственного процесса степень превращения акролеина на второй реакционной стадии снизилась. Постепенным повышением температуры на входе солевого расплава на вторую стадию реакции эта потеря активности могла выровняться (степень деактивации составляла стабильно 8°С/год).
При достижении при температуре на входе Tein 283°С разность ΔTHB v составляла 33°С. В это время процесс прерывали и на второй стадии реакции всю зону А, а также всю зону В отсасывали и заменяли свежей зной А, а также свежей зоной В, причем свежая зона В все же содержала 50 мас.% колец из стеатита размером 7 мм × 3 мм × 4 мм и лишь еще 50 мас.% свежего катализатора с оболочками согласно примеру 5 из патента Германии DE-A 10046928.
При температуре на входе Tein 275°С при скорости деактивации катализатора 12°С/год и разности ΔTHB n 32°С способ мог все-таки затем продолжаться без изменения при степени конверсии акролеина 99,3 мол.%.
В) Сравнительный пример
Действовали, как в Примере. Однако после отсоса зону В заменяли свежей зоной В, которая как и первоначальная зона В содержала лишь 30 мас.% колец из стеатита размером 7 мм × 3 мм × 4 мм и 70 мас.% свежего катализатора с оболочкой согласно примеру получения 5 из патента Германии DE-A 10046928.
Хотя процесс мог затем продолжаться со степенью конверсии акролеина 99,3 мол.% при температуре на входе Tein лишь 270°С, однако разность ΔTHB n составляла 48°С, а скорость деактивации катализатора составляла 20°С/год.
Заявка на временный патент США US №60/756207, поданная 05.01.2006, приведена в настоящей заявке на патент в качестве литературной ссылки.
С точки зрения вышеназванных рекомендаций возможны многочисленные изменения и отклонения от настоящего изобретения.
Поэтому можно исходить из того, что изобретение, в рамках прилагаемой его формулы, может быть осуществлено по-иному, чем конкретно описано здесь.
Изобретение относится к усовершенствованному способу гетерогенного каталитического газофазного парциального окисления по меньшей мере одного исходного органического соединения, выбранного из пропилена, изобутена, акролеина, метакролеина, пропана или изобутана, молекулярным кислородом на свежевнесенном в реакционное пространство неподвижном слое катализатора, в котором с целью парциального окисления реакционную газовую смесь, содержащую по меньшей мере одно исходное органическое соединение и молекулярный кислород, пропускают через неподвижный слой катализатора, а также отводят тепло реакции посредством непрямого теплообмена с направляемым вне реакционного пространства жидким теплоносителем, а затем, когда с увеличением продолжительности работы происходит нарастающее снижение качества неподвижного слоя катализатора, то для восстановления качества неподвижного слоя катализатора не весь, а лишь часть неподвижного слоя катализатора заменяют частью заменяющего неподвижного слоя катализатора, причем удельно-объемная активность заменяющей части неподвижного слоя катализатора ниже, чем удельно-объемная активность заменяемой части неподвижного слоя катализатора в его свежевнесенном состоянии. В данном способе снижение качества неподвижного слоя катализатора с увеличением продолжительности работы компенсируют в результате замены части неподвижного слоя катализатора заменяющей частью неподвижного слоя катализатора, при этом скорость деактивирования катализатора является самой низкой в заявленном способе. 9 з.п. ф-лы.
1. Способ гетерогенного каталитического газофазного парциального окисления по меньшей мере одного исходного органического соединения, выбранного из пропилена, изобутена, акролеина, метакролеина, пропана или изобутана, молекулярным кислородом на свежевнесенном в реакционное пространство неподвижном слое катализатора, в котором с целью парциального окисления реакционную газовую смесь, содержащую по меньшей мере одно исходное органическое соединение и молекулярный кислород, пропускают через неподвижный слой катализатора, а также отводят тепло реакции посредством непрямого теплообмена с направляемым вне реакционного пространства жидким теплоносителем, а затем, когда с увеличением продолжительности работы происходит нарастающее снижение качества неподвижного слоя катализатора, то для восстановления качества неподвижного слоя катализатора не весь, а лишь часть неподвижного слоя катализатора заменяют частью заменяющего неподвижного слоя катализатора, отличающийся тем, что удельно-объемная активность заменяющей части неподвижного слоя катализатора ниже, чем удельно-объемная активность заменяемой части неподвижного слоя катализатора в его свежевнесенном состоянии.
2. Способ по п.1, отличающийся тем, что газофазным парциальным окислением является газофазное парциальное окисление пропилена в акролеин и/или акриловую кислоту, или газофазное парциальное окисление изобутена в метакролеин и/или в метакриловую кислоту, или газофазное парциальное окисление акролеина в акриловую кислоту, или газофазное парциальное окисление метакролеина в метакриловую кислоту, или газофазное парциальное окисление пропана в акриловую кислоту, или газофазное парциальное окисление изобутана в метакриловую кислоту.
3. Способ по п.1, отличающийся тем, что газофазным парциальным окислением является вторая стадия двухстадийного газофазного парциального окисления.
4. Способ по п.3, отличающийся тем, что газофазным парциальным окислением является парциальное окисление акролеина в акриловую кислоту в двухстадийном газофазном парциальном окислении пропилена в акриловую кислоту.
5. Способ по п.1, отличающийся тем, что удельно-объемную активность заменяющей части неподвижного слоя катализатора рассчитывают таким образом, чтобы при одинаковой степени превращения исходного органического соединения в результате одноразового пропуска реакционной газовой смеси через неподвижный слой катализатора, а также при одинаковом составе реакционной газовой смеси и одинаковой нагрузке неподвижного слоя катализатора реакционной газовой смесью разность dΔT между расширением точки перегрева ΔTHB n неподвижного слоя катализатора после проведенной замены заменяющей частью неподвижного слоя катализатора и расширением точки перегрева ΔTHB v перед проведением замены заменяющей частью неподвижного слоя катализатора, образованная как: dΔT=ΔTHB n-ΔTHB v, составляла ≤30°С.
6. Способ по п.5, отличающийся тем, что dΔT составляет от -15 до +10°С.
7. Способ по п.5, отличающийся тем, что dΔT составляет от -10 до 0°С.
8. Способ по п.1, отличающийся тем, что реакционным пространством является внутреннее пространство реакционной трубы.
9. Способ по п.8, отличающийся тем, что реакционная труба находится в секционном трубчатом реакторе.
10. Способ по одному из пп.1-9, отличающийся тем, что часть неподвижного слоя катализатора, заменяемая заменяющей частью неподвижного слоя катализатора, простирается в направлении потока реакционной газовой смеси на до 80% насыпной длины неподвижного слоя катализатора.
| US 20040015013 A1, 22.01.2004 | |||
| DE 102004025445 A1, 29.01.2004 | |||
| DE 19902562 A, 27.07.2000 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| RU 96104337 A, 10.08.1996. | |||

