Настоящее изобретение относится к производным циннамоил-пиперазина, к способу их получения, к фармацевтическим композициям, содержащим эти производные, и к применению этих производных в качестве лекарств для лечения и/или предотвращения артериального и/или венозного тромбоза, острых коронарных синдромов, рестеноза, стабильной стенокардии, нарушений сердечного ритма, инфаркта миокарда, гипертензии, сердечной недостаточности, инсульта, воспалительных расстройств, легочных заболеваний, заболеваний желудочно-кишечного тракта, развития фиброза у пациентов с хроническим заболеванием печени, рака и заболеваний кожи. Настоящее изобретение относится также к комбинациям соединений, составляющих суть настоящего изобретения, с другими сердечно-сосудистыми агентами.
Тромбоз считается первичным фактором в закупорке сосудов, которая является причиной большого числа патофизиологических осложнений. Поэтому противотромбозная терапия необычайно важна, так как она может снизить риск смерти от сердечно-сосудистых причин и сердечных нарушений. Хотя было установлено, что несколько типов молекул обладают высокой противотромбозной активностью у человека, сохраняется необходимость в поиске новых молекул. Действительно, можно усовершенствовать существующие соединения, некоторые из которых отрицательно влияют на длительность кровотечения или связаны с другими нежелательными побочными эффектами (такими, как, например, риск образования язвы при применении аспирина).
Недавно был клонирован активируемый протеазой рецептор-1 (protease-activated receptor-1, PAR-1) (Vu et al. // Cell. 1991. T.64. C.1057-1068), и был установлен механизм его действия (Coughlin et аl. // J. Clin. Invest. 1992. Т.89, №2. С.351-355). Этот рецептор, присутствующий, прежде всего, на поверхности тромбоцитов, на поверхности клеток эндотелия (O'Brien et al. // J. Biol. Chem. 2000. Т.275. С.13502-13509), клеток гладких мышц (Hamilton et al. // Br. J. Pharmacol. 2000. T.130. C.181-188) и фибробластов (Hung et al. // J. Cell. Biol. 1992. T.116, №3. C.827-832), активируется тромбином и поэтому называется также тромбиновым рецептором. N-терминаль белка расщепляется тромбином между аргинином в положении 41 и серином в положении 42 с высвобождением нового конца, который после фолдинга будет действовать на активный центр как агонист рецептора (Vu et al. // Nature. 1991. Т.353. С.674-677). Что касается тромбоцитов, этот специфический механизм активации рецептора PAR-1 приводит к направляемой тромбином агрегации тромбоцитов.
Блокирование этой активации, например, антагонистами рецептора PAR-1, может ингибировать опосредованную тромбином агрегацию тромбоцитов (Ahn et al. // Drug of the Future. 2001. T.26. C.1065-1085). Поэтому блокирование этих рецепторов может привести к лечению или предотвращению тромбоза (Derian et al. // J. Pharmacol. Exp. Ther. 2003. C.855-861), острого коронарного синдрома (Ossovskaya et al. // Physiol. Rev. 2004. T.84. C.579-621) и рестеноза (Maryanoff et al. // Curr. Med. Chem. Cardiovasc. Hematol. Agents. 2003. С.13-36) и может уменьшать некроз миокарда при инфаркте или повторной перфузии (Steinberg et al. // Molec. Pharmacol. 2005. Т.67. С.2-11). Активность антагонистов PAR-1 может предотвращать некоторые воспалительные заболевания легочной системы (Moffatt et al. // Curr. Op. Pharmacol. 2004. C.221-229) и желудочно-кишечной системы (Vergnolle et al. // J. Clin. Invest. 2004. C.1444-1456). Антагонисты PAR-1 могут быть также полезны в лечении фиброзов у пациентов с хроническим заболеванием печени (Fiorucci et al. // Hepatology. 2004. Т.39. С.365-375). Они могут быть также полезны как противораковые средства, поскольку они способны контролировать пролиферацию и метастазирование (Evan-Ram et al. // Nat. Med. 1998. С.909-914; Boire et al. // Cell. 2005. T.120. C. 303-313). Наконец, антагонисты PAR-1 могут представлять интерес в дерматологии для лечения некоторых кожных заболеваний (Schechter et al. // J. Cell. Physiol. 1998. T.176. C.365-373; Algermissen et al. // Arch. Dermatol. Res. 2000. T.292. C.488-495; Meyer-Hoffert et al. // Exp. Dermatol. 2004. T.13. C.234-241).
Настоящее изобретение относится к новому классу антагонистов PAR-1, которые отличаются от антагонистов предшествующего уровня техники другой химической структурой и своими замечательными биологическими свойствами.
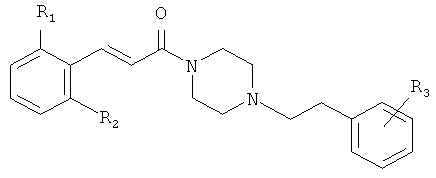
Соединения согласно настоящему изобретению имеют общую формулу (I):
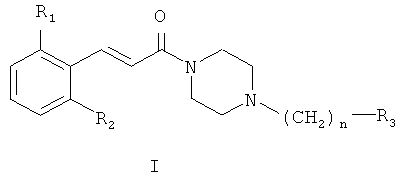
где
R1 представляет собой галоген, CN или NO2;
R2 представляет собой водород или галоген;
n равно 1 или 2;
R3 представляет собой фенил, замещенный одним или более галогенами или С1-С6 алкилами, или циклогексил;
или представляют собой их терапевтически приемлемые соли или сольваты.
В предшествующих определениях:
Все комбинации заместителей или вариантов возможны постольку, поскольку они дают стабильные соединения.
Термин «галоген» обозначает фтор, хлор, бром или йод.
Термин «алкил» обозначает линейные или разветвленные, насыщенные или ненасыщенные алифатические углеводородные цепи, содержащие определенное число атомов углерода.
Терапевтически приемлемые соли соединений согласно настоящему изобретению включают обычно употребляемые нетоксичные соли соединений согласно настоящему изобретению, такие как соли, образуемые органическими или неорганическими кислотами. В качестве примера можно указать следующее: соли неорганических кислот, таких как соляная, бромистоводородная, фосфорная и серная кислоты, а также соли органических кислот, таких как уксусная, трифторуксусная, пропионовая, янтарная, фумаровая, яблочная, винная, лимонная, аскорбиновая, малеиновая, глутаминовая, бензойная, салициловая, толуолсульфоновая, метансульфоновая, стеариновая и молочная кислоты.
Эти соли могут быть синтезированы из соединений согласно настоящему изобретению, содержащих основную часть, и соответствующих кислот с использованием стандартных химических способов.
Терапевтически приемлемые сольваты соединений настоящего изобретения включают обычные сольваты, такие как сольваты, образованные в ходе конечного этапа получения соединений согласно настоящему изобретению как результат присутствия растворителей. В качестве примера могут быть упомянуты сольваты, образующиеся благодаря присутствию воды или этанола.
Среди соединений общей формулы (I) согласно настоящему изобретению один особенно полезный класс соединений включает соединения общей формулы (I), где R1 - галоген, R2 - водород, n равно 1 или 2, R3 - фенил, замещенный одним или более галогенами или С1-С6 алкилами.
Другой особенно полезный класс соединений согласно настоящему изобретению включает соединения общей формулы (I), где R1 - цианогруппа, R2 - водород, n равно 1, a R3 - фенил, замещенный одним или более галогенами или С1-С6 алкилами.
Другой особенно полезный класс соединений согласно настоящему изобретению включает соединения общей формулы (I), где R1 - галоген, R2 - водород, n равно 1, a R3 - циклогексил.
Другой особенно полезный класс соединений согласно настоящему изобретению включает соединения общей формулы (I), где R1 - цианогруппа, R2 - водород, n равно 1, a R3 - циклогексил.
Настоящее изобретение относится также к получению соединений общей формулы (I) обычными методами, описанными в следующих схемах синтеза, дополненных в нужных случаях любыми стандартными методиками, описанными в литературе, известными специалистам в данной области или представленными в экспериментальном разделе.
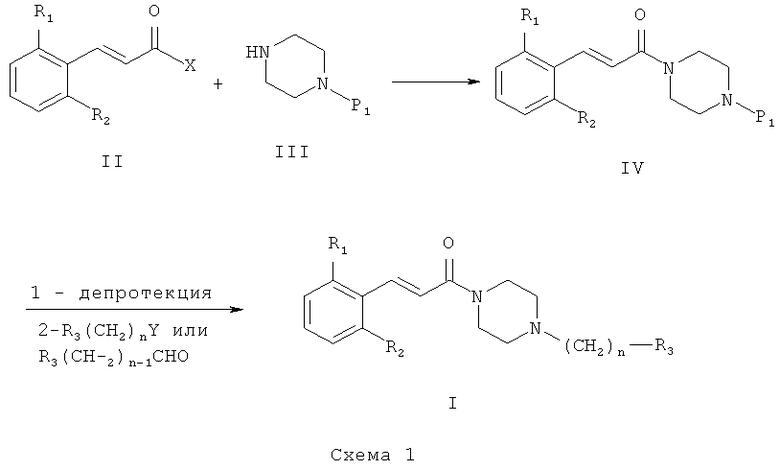
Схема 1 иллюстрирует первый общий метод, который может быть применен для синтеза соединений общей формулы (I). В представленных выше общих формулах R1, R2, R3 и n определены так, как в предшествующем описании общей формулы (I). P1 - это защитная группа. X может представлять собой уходящую группу, такую как хлор. В этом случае первый этап состоит из реакции между кислотным хлором и амином. Эта реакция может быть осуществлена с помощью методов и аппаратуры, известных специалистам в данной области. Особенно удобный метод состоит в осуществлении реакции двух участников в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, пиридин, NaH, Cs2CO3 или K2CO3, в растворителе, таком как ТГФ, дихлорметан, ДМФ или ДМСО при температуре от -20°C до 100°C. X может также представлять собой гидроксил. В этом случае первый этап состоит в реакции конденсации между карбоновой кислотой (II) и амином (III). Эта реакция может быть осуществлена с помощью методов и аппаратуры, известных специалистам в данной области. Особенно удобный метод состоит в осуществлении реакции этих двух молекул в присутствии 1-(3-диметиламинопропил)-3-этил-карбодиимида (EDC), 3-окси-1,2,3-бензотриазин-4(3Н)-она и третичного амина, такого как диизопропилтриэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре от -15°C до 40°C.
После снятия защиты с промежуточного соединения (IV) с помощью методов и аппаратуры, известных опытным специалистам в данной области (см. T.W.Greene «Protective Groups in Organic Synthesis» / John Wiley & Sons, 1981 и P.J.Kocienski «Protecting Groups» / Thieme Verlag, 1994), полученное промежуточное соединение может прореагировать с реагентом формулы R3(CH2)nY, где Y представляет собой уходящую группу, такую как Cl, Br, I, OSO2CH3, OSO2CF3 или О-тозил. В этом случае реакция может быть проведена в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, NaH, Cs2CO3 или K2CO3, которые могут быть прикреплены к смоле, такой как PS-DIEA или МР-карбонат, в полярном безводном растворителе, таком как дихлорметан, ТГФ, ДМФ или ДМСО, при температуре от -20°C до 100°C. Другой метод получения состоит в проведении реакции восстановительного аминирования с использованием альдегида формулы R3-(CH2)n-1-CHO, в которой R3 и n таковы, как они определены выше, с освобожденным от защиты амином общей формулы (IV) и восстанавливающим агентом, таким как NaBH4, NaBH3CN или NaBH(OAc)3, которые могут быть прикреплены к смоле, такой как MP-BH3CN, в полярном растворителе, таком как 1,2-дихлорэтан, дихлорметан, ТГФ, ДМФ или МеОН, при pH, который можно устанавливать добавлением кислоты, такой как уксусная кислота, при температуре от -20°C до 100°C.
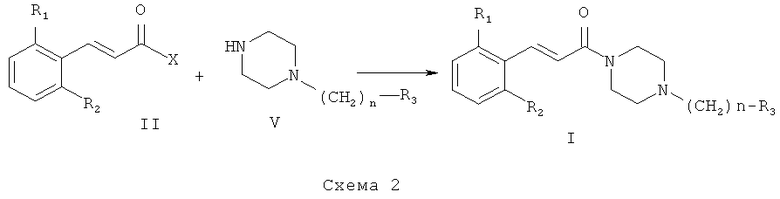
Схема 2 иллюстрирует второй общий метод, который можно использовать для получения соединений общей формулы (I). В представленных выше общих формулах R1, R2, R3 и n определены так, как в предшествующем описании общей формулы (I). X может представлять собой уходящую группу, такую как хлор. В этом случае синтез заключается в проведении реакции между хлоридом кислоты и амином. Эта реакция может быть осуществлена с помощью методов и аппаратуры, известных специалистам в данной области. Особенно удобный метод состоит в осуществлении реакции этих двух молекул в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, пиридин, NaH, Cs2CO3 или K2CO3, в растворителе, таком как ТГФ, дихлорметан, ДМФ или ДМСО, при температуре от -20°C до 100°C.
X может представлять собой также гидроксильную группу. В этом случае синтез состоит в проведении реакции конденсации между карбоновой кислотой (II) и амином (V). Реакция может быть осуществлена с помощью методов и аппаратуры, известных специалистам в данной области. Особенно удобный метод состоит в конденсировании карбоновой кислоты общей формулы (II) с амином общей формулы (V) в присутствии 1-(3-диметиламинопропил)-3-этил-карбодиимида (EDC), 3-окси-1,2,3-бензотриазин-4(3H)-она и третичного амина, такого как диизопропилтриэтиламин, в полярном апротонном растворителе, таком как дихлорметан, при температуре от -15°C до 40°C.
Если необходимо выделить соединение общей формулы (I), содержащее по меньшей мере одну основную функциональную группу, в состоянии соли путем добавления кислоты, такой результат может быть достигнут обработкой свободного основания общей формулы (I) (в котором присутствует по меньшей мере одна основная функциональная группа) подходящей кислотой, предпочтительно добавляемой в эквивалентном количестве.
Приведенные ниже примеры иллюстрируют настоящее изобретение, ни в коей мере не ограничивая его объем.
Пример 1
3-(2-Хлорфенил)-1-[4-(4-фторбензил)-пиперазин-1-ил]-пропенон
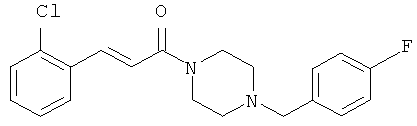
Пример 1А
Трет-бутиловый эфир 4-(4-фторбензил)-пиперазин-1-карбоновой кислоты
Раствор трет-бутилового эфира пиперазин-1-карбоновой кислоты (5,0 г, 26,8 ммоль) в дихлорметане (100 мл) в присутствии диизопропилэтиламина (DIEA) (5,59 мл, 40,2 ммоль) обрабатывают 4-фторбензилбромидом (3,68 мл, 29,5 ммоль) при комнатной температуре. После встряхивания в течение 16 ч реакционную смесь разбавляют дихлорметаном и промывают водой. Органическую фазу сушат над MgSO4, фильтруют и упаривают досуха. Полученный сироп очищают хроматографией на колонке с двуокисью кремния и элюируют смесью CH2Cl2/MeOH с соотношением от 98/2 до 95/5. Продукт 1А выделяют в форме белого твердого вещества (выход 7,03 г, 88%).
1Н-ЯМР в ДМCO-d6 (млн-1): 1,38 (s, 9Н); 2,29 (t, 4Н); 3,30 (уширенный s, 4Н); 3,45 (s, 2Н); 7,14 (t, 2Н); 7,32 (dd, 2Н).
Пример 1Б
4-(4-Фторбензил)-пиперазин
Раствор трет-бутилового эфира 4-(4-фторбензил)-пиперазин-1-карбоновой кислоты (7,03 г, 23,8 ммоль) в толуоле (300 мл) обрабатывают трифторуксусной кислотой (53,2 мл, 716 ммоль) при комнатной температуре. После встряхивания в течение 2 ч реакционную смесь разбавляют дихлорметаном, промывают 1 н. содой и затем водой. Органическую фазу сушат над MgSO4, фильтруют и выпаривают досуха. Неочищенный продукт выделяют для проведения последующей реакции (4,2 г, 90%).
Пример 1
3-(2-Хлорфенил)-1-[4-(4-фторбензил)-пиперазин-1-ил]-пропенон
Раствор смеси 2-хлор-коричной кислоты (2,43 г, 13,3 ммоль) и 4-(4-фторбензил)-пиперазина (2,16 г, 11,1 ммоль) в дихлорметане (70 мл) обрабатывают гидрохлоридом 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDCI) (2,55 г, 13,3 ммоль) и 3-окси-1,2,3-бензотриазин-4(3Н)-оном (НООВТ) в присутствии DIEA (3,86 мл, 22,2 ммоль) при комнатной температуре. После встряхивания в течение 48 ч реакционную смесь разбавляют этилацетатом и промывают 1 н. содой, затем водой. Органическую фазу сушат над MgSO4, фильтруют и выпаривают досуха. Полученный сироп очищают хроматографией на колонке с двуокисью кремния и элюируют смесью CH2Cl2/MeOH/NH4OH с соотношением 97,75/2/0,25. Продукт 1 выделяют в форме желтого масла (3,77 г, 95%). Этот продукт переводят в этилацетат и получают его соль добавлением раствора HCl в эфире, получая соответствующий гидрохлорид в виде желтого твердого вещества (4,14 г).
1Н-ЯМР в ДМСО-d6 (млн-1): 3,02 (m, 2Н); 3,21 (t, 1Н); 3,63 (t, 1Н); 4,05 (уширенный s, 2Н); 4,34 (s, 2Н); 4,52 (t, 2Н); 7,32 (m, 3Н); 7,43 (m, 2Н); 7,53 (m, 1Н); 7,66 (m, 2Н); 7,92 (d, 1Н); 8,00 (m 1Н); 11,49 (s, 1Н).
Масс-спектр (ESI+): m/z 359 (М+Н+)
Элементный анализ: C20H20N2O1·HCl и 0,5 Н2O
Рассчитанное соотношение (в %): С - 59,41; Н - 5,48; N - 6,93
Истинное соотношение (в %): С - 59,39; Н - 5,56; N - 6,92
Примеры с 2 по 4
Соединения с 2-го по 4-е были синтезированы из коричных кислот и соответствующих аминов в соответствии с условиями, описанными для синтеза соединения 1.
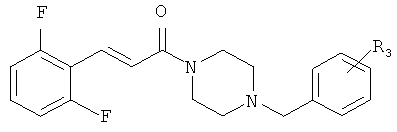
Пример 5
3-(2,6-Дифторфенил)-1-[4-(4-фторбензил)-пиперазин-1-ил]-пропенон
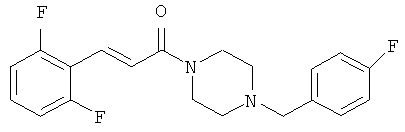
Пример 5А
Трет-бутиловый эфир 4-[3-(2,6-дифторфенил)-акрилоил]-пиперазин-1 -карбоновой кислоты
Раствор 3-(2,6-дифторфенил)-акрилоил-хлорида (3,0 г, 14,8 ммоль) в дихлорметане (70 мл) в присутствии PS-DIEA (4,07 г, 13,5 ммоль, 3,33 ммоль/г) обрабатывают трет-бутиловым эфиром пиперазин-1-карбоновой кислоты (2,3 г, 12,3 ммоль) при комнатной температуре. После встряхивания в течение 6 ч реакционную смесь фильтруют, переводят в дихлорметан и промывают 1 н. содой и водой. Органическую фазу сушат над MgSO4, фильтруют и выпаривают досуха. Полученный сироп очищают хроматографией на колонке с двуокисью кремния и элюируют смесью CH2Cl2/MeOH/NH4OH с соотношением от 95/4,5/0,5 до 90/9,5/0,5. Продукт 5А выделяют в виде окрашенного твердого вещества (3,87 г, 89%).
1Н-ЯМР в ДМСО-d6 (млн-1): 1,42 (s, 9Н); 3,37 (уширенный s, 4Н); 3,58 (уширенный s, 4Н); 7,22 (m, 2Н); 7,50 (m, 1Н).
Пример 5Б
3-(2,6-Дифторфенил)-1 -пиперазин-1-ил-пропенон
Раствор трет-бутилового эфира 4-[3-(2,6-дифторфенил)-акрилоил]-пиперазин-1-карбоновой кислоты (3,87 г, 10,97 ммоль) в толуоле (50 мл) обрабатывают трифторуксусной кислотой (30 мл, 395 ммоль) при комнатной температуре. После встряхивания в течение 2 ч реакционную смесь выпаривают досуха, переводят в дихлорметан и промывают 1 н содой и затем водой. Органическую фазу сушат над MgSO4, фильтруют и выпаривают досуха. Неочищенный продукт выделяют для проведения последующей реакции (2,3 г, 88%).
Пример 5
3-(2,6-Дифторфенил)-1-[4-(4-фторбензил)-пиперазин-1-ил]-пропенон
Раствор соединения 5Б (100 мг, 0,42 ммоль) в дихлорметане (5 мл) в присутствии триэтиламина (Et3N) (0,088 мл, 0,63 ммоль) обрабатывают 4-фторбензил-бромидом (0,078 мл, 0,63 ммоль) при комнатной температуре. После встряхивания в течение 15 ч реакционную смесь разбавляют дихлорметаном и промывают водой. Органическую фазу сушат над MgSO4, фильтруют и выпаривают досуха. Полученный сироп очищают хроматографией на колонке с двуокисью кремния и элюируют смесью CH2Cl2/MeOH с соотношением от 100/0 до 90/10. Продукт 5 выделяют в виде светлобежевого твердого вещества (72 мг, 48%).
Масс-спектр (ESI+): m/z 361 (М+Н+)
Примеры с 6 по 12
Соединения с 6-го по 12-е были синтезированы из промежуточного соединения 5Б и соответствующих бензил-хлоридов или бромидов в соответствии с условиями, описанными для синтеза соединения 5.
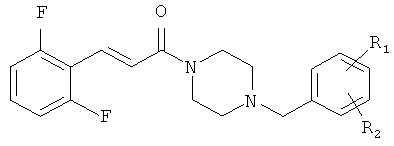
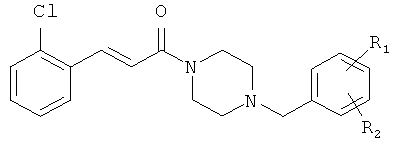
Примеры с 13 по 21
Пример13А
3-(2-хлорфенил)-1-пиперазин-1-ил-пропенон
Соединение 13А было получено в 2 стадии из 3-(2-хлорфенил)-акрилоил-хлорида согласно условиям, описанным для синтеза соединения 5Б.
Примеры с 13 по 21
Соединения с 13-го по 21-е были синтезированы из промежуточного соединения 13А согласно условиям, описанным для синтеза соединения 5.
Пример 22
3-(2,6-Дифторфенил)-1-[4-(2-фторбензил)-пиперазин-1-ил]-пропенон
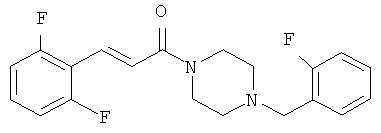
Раствор смеси промежуточного соединения 5Б (60 мг, 0,25 ммоль) и 2-фтор-бензальдегида (0,031 мл, 0,3 ммоль) в дихлорметане (3 мл) в присутствии уксусной кислоты (0,057 мл, 1,0 ммоль) обрабатывают MP-BH3CN (117 мг, 0,275 ммоль, 2,35 ммоль/г) при комнатной температуре. После встряхивания в течение 24 ч реакционную смесь фильтруют через картридж ChemElut, предварительно пропитанный 1 н NaOH, и затем выпаривают досуха. Полученный сироп очищают хроматографией на колонке с двуокисью кремния и элюируют смесью CH2Cl2/MeOH с соотношением от 100/0 до 95/5. Продукт 22 выделяют в виде желтого сиропа (23 мг, 25%).
Масс-спектр (ESI+): m/z 361 (М+Н+)
Пример 23
1-[4-(2-Фторбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенон
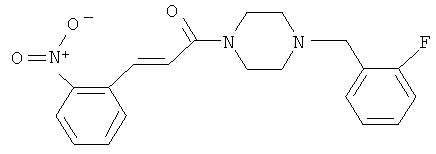
Пример 23А
3-(2-Нитрофенил)-1-пиперазин-1-ил-пропенон
Соединение 23А было получено в 2 стадии из 3-(2-нитрофенил)-акрилоил-хлорида согласно условиям, описанным для синтеза соединения 5Б.
Пример 23
1-[4-(2-Фторбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенон
Соединение 23 было синтезировано из соединения 23А согласно условиям, описанным для синтеза соединения 22.
Масс-спектр (ESI+): m/z 370 (М+Н+)
Пример 24
1-(4-Циклогексилметил-пиперазин-1-ил)-3-(2,6-дифторфенил)-пропенон
Соединение 24 было синтезировано из соединения 5Б согласно условиям, описанным для синтеза соединения 22.
Масс-спектр (ESI+): m/z 349 (М+Н+)
Примеры с 25 по 28
Соединения с 25-го по 28-е были синтезированы из 1-циклогексилметил-пиперазина и соответствующих коричных кислот согласно условиям, описанным для синтеза соединения 1.
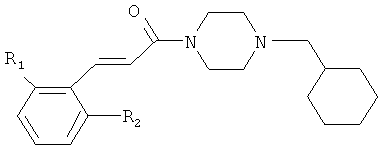
Примеры с 29 по 33
Соединения с 29-го по 33-е были синтезированы из соединения 23А и соответствующих бензил-хлоридов или бромидов согласно условиям, описанным для синтеза соединения 5.
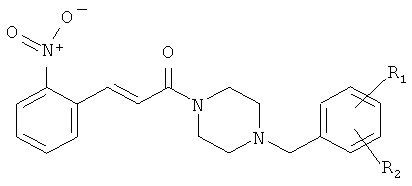
Пример 34
1-[4-(2,6-Диметилбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенон
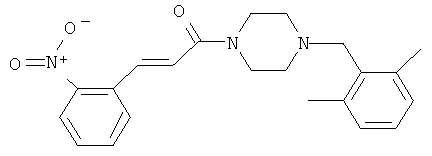
Раствор смеси соединения 23А (70 мг, 0,27 ммоль) и 2,5-диметил-бензальдегида (40 мг, 0,32 ммоль) в дихлорэтане (3 мл) в присутствии уксусной кислоты (0,092 мл, 1,6 ммоль) обрабатывают NaBH(OAc)3 (63 мг, 0,297 ммоль) при комнатной температуре. После встряхивания в течение 24 ч реакционную смесь обрабатывают насыщенным раствором NaHCO3 (2 мл), фильтруют через картридж ChemElut и выпаривают досуха. Полученный сироп очищают хроматографией на колонке с двуокисью кремния и элюируют смесью CH2Cl2/MeOH с соотношением от 100/0 до 90/10 (+10% NH4OH). Продукт 34 выделяют и затем получают его соль добавлением HCl в эфире, что дает белое твердое вещество (40 мг, 40%).
Масс-спектр (ESI+): m/z 380 (М+Н+)
Примеры с 35 по 38
Соединения с 35-го по 38-е были синтезированы из соединений 5Б, 13А и 23А и соответствующих фенэтил-хлоридов или бромидов согласно условиям, описанным для синтеза соединения 5.
Производные согласно настоящему изобретению являются антагонистами рецепторов PAR-1, поскольку описанные ниже результаты опытов на моделях показывают следующее.
Во многих типах клеток активация рецепторов PAR-1 пептидом SFLLR (селективным агонистом PAR-1) запускает внутриклеточный сигнальный каскад, приводящий к высвобождению кальция эндоплазматическим ретикулумом. Клетки яичников китайского хомячка (СНО) устойчиво экспрессируют рецептор PAR-1. В этой линии клеток высвобождение кальция вслед за активацией рецепторов PAR-1 пептидом SFLLR измеряют методом флуориметрической методики (флуориметрическое визуализационное устройство для чтения планшетов, или FLIPR) с использованием селективного датчика кальция (Fluo-3АМ). Излучение флуоресценции с фармакологической точки зрения пропорционально эффективности PAR-1 агониста и его концентрации. Соединения, описанные в настоящем изобретении, показали, что они способны выступать в качестве антагонистов PAR-1 рецепторов и таким образом снижать выделение кальция, вызванное агонистом.
Материалы:
Культуральная среда: среда Хэма F-12 (Ham R.G. // Proc. Natl. Acad. Sci. 1965. Т.53. С.288) с добавкой 10% сыворотки коровьего плода и антибиотика (пробенецид, 2,5 мМ).
Флуоресцентный зонд: Fluo-ЗАМ 4 мкМ (Teflabs, Austin, Texas, USA).
Агонист: SFLLR-NH2 (серии, фенилаланин, лейцин, лейцин, аргинин).
Методы:
Клетки СНО инокулировали в планшеты на 96 ячеек (60000 клеток в каждую ячейку) в присутствии 200 мкл культуральной среды и инкубировали в течение 24 ч. Клетки инкубировали с флуоресцентным зондом на кальций в течение 1 ч при 37°C. Затем клетки промывали в течение 10 мин, после чего измеряли сигнал. После этого вводили антагонист PAR-1 (концентрации от 0,01 мкМ до 10 мкМ). Планшеты помещали в FLIPR (Molecular Devices, UK) для измерения флуоресценции кальция при двух длинах волн (488 нм и 540 нм: Sullivan et al. // Calcium Signaling Protocols. 1999. C.125-136). Измерения проводили за 5 мин до добавления антагониста и в течение 10 мин после его введения. В 4-х различных ячейках измеряли максимум флуоресценции, вычитая базовую линию. Тест проводили при двух повторностях. При этих условиях производные согласно настоящему изобретению были идентифицированы как антагонисты рецепторов PAR-1 (подавление сигнала кальция более 60% при 10 мкМ). Для агониста SFLLR были получены дозовые зависимости (в диапазоне от 0,01 мкМ до 32 мкМ), что позволило определить эффективную концентрацию, дающую 50% от максимального эффекта (ЕС50). Сила (рА2) некоторых из антагонистов PAR-1, описанных в настоящем изобретении, была рассчитана по методу Arunlakshana and Schild (Brit. J. Pharmacol. 1959. T.14. C.48-58) по сдвигам EC50, наблюдаемым при трех концентрациях.
Результаты:
Несколько приведенных далее примеров, выбранных из соединений согласно настоящему изобретению, иллюстрируют полностью неожиданную способность этих соединений выступать антагонистами рецепторов PAR-1.
Подавление агрегации тромбоцитов in vivo и противотромбозная активность антагонистов PAR-1 была продемонстрирована у морских свинок на модели артериального тромбоза, который дает очень высокий стресс вследствие гемодинамического сдвига. В выстилке сосудов повреждение эндотелия вызывает внутрисосудистое образование богатого тромбоцитами тромба, который постепенно перекрывает весь просвет сосуда. Процесс агрегации тромбоцитов в сильной степени активируется тромбином при участии рецепторов PAR-1. Было продемонстрировано, что описанные в настоящем изобретении соединения способны быть антагонистами рецепторов PAR-1 и таким путем задерживать образование тромба.
Материалы:
Исследования проведены на морских свинках (их рецепторы PAR-1 подобны рецепторам человека). Облучение зеленым светом лазера в присутствии фотосенсибилизирующего агента (внутривенно введенный краситель Бенгальская роза) повреждает эпителий сонной артерии. Скорость протока крови через сонную артерию определяют количественно с помощью зонда Transonic flow. Измеряется время, необходимое для полного перекрывания сонной артерии (скорость протока равна 0).
Методы:
После усыпления животного (фенобарбитал 60 мг/кг) иссекают 5 мм участок сонной артерии и в 4 мм над артерией помещают лазер. Зонд для определения скорости протока, помещенный в направлении вверх, позволяет измерить время перекрывания сосуда. Бенгальскую розу (20 мг/кг) вводят внутривенно и сосуд облучают в течение 3 мин при длине волны 514 нм. Антагонисты PAR-1 вводят внутривенно болюсно (в течение 2 мин непосредственно перед введением Бенгальской розы), за этим следует 15-минутная перфузия, которая начинается при включении лазера.
Результаты:
Было установлено, что некоторые соединения, описанные в настоящем изобретении, способны после внутривенного введения в дозах от 0,16 мг/кг до 2,5 мг/кг увеличивать время до образования тромба на величину от 5% до 135% по сравнению с животными, которым вводили плацебо.
Производные согласно настоящему изобретению полезны также для лечения фибрилляции предсердий.
В случае постинфарктного переполнения сердечной полости, правое и левое ушко предсердия расширяются, и это создает основу для возникновения фибрилляции предсердий. Нарушение гомеостаза в полости расширенного ушка предсердия пациента, страдающего фибрилляцией предсердий, приводит к повышенной концентрации тромбина. Заявители продемонстрировали, что накопление тромбина ответственно за регуляцию со стимуляцией PAR-1, которые могут запускать пролиферацию фибробластов, а также образование тромбоцитного тромба.
Поэтому в соответствии с механизмом их действия, антагонисты PAR-1 могут предотвращать расширение предсердия, пролиферацию фибробластов и образование тромбов в ушке предсердия пациента, страдающего фибрилляцией предсердий.
В результате антагонист PAR-1 обеспечивает эффективное профилактические и/или лечебное действие в случае фибрилляции предсердий. Описанные в настоящем изобретении соединения показали, что они способны служить антагонистами рецепторов PAR-1 и предотвращать расширение предсердия.
Материалы:
Исследования проводят на крысах-самцах. Для опытов были выбраны крысы с весом в диапазоне 180-200 г при поступлении, поскольку они лучше переносят хирургическую операцию. Измерения различных полостей миокарда выполняют с помощью эхокардиографии у усыпленного животного.
Методы:
Животных усыпляют смесью 3,5% изофлурана в кислороде (Aerrane, Baxter Laboratories). На уровне четвертого межпозвоночного просвета в сторону левой передней лапы проводят перпендикулярно грудине торакотомию размером приблизительно 2 см. Вокруг левой венечной артерии на расстоянии 1 мм от ее начала накладывают лигатуру (шелк 4-0, игла CC1). Хирургическим способом вокруг левой венечной артерии завязывают узел, достаточно тугой для полного перекрывания сосуда. Непрерывно регистрируемая электрокардиограмма позволяет проверять правильное размещение лигатуры. Через 2 месяца после операции животных снова усыпляют для эхокардиографического измерения методом импульсной допплерографии сердечных полостей и скорости протока крови в миокарде. В заключение животных умерщвляют передозировкой фенобарбитала натрия (160 мг/кг внутрибрюшинно) для проведения различных гистологических исследований. Животным насильно скармливали ежедневно продукты-антагонисты PAR-1, начиная с 24 ч после образования инфаркта и до сакрификации.
Результаты:
Было установлено, что некоторые соединения, описанные в настоящем изобретении, способны при пероральном введении в дозах от 10 до 100 мг/кг/день в течение 60 дней снижать на 20-90% поверхность ушка предсердия по сравнению с не подвергавшимися воздействию животными.
Настоящее изобретение относится также к фармацевтическим композициям, содержащим в качестве активного ингредиента соединение общей формулы (I) или его фармацевтически приемлемую соль, смешанные или объединенные с подходящим наполнителем. Такие композиции могут, например, иметь форму твердых или жидких композиций, эмульсий, лосьонов или кремов.
В качестве твердых композиций для перорального введения могут быть использованы таблетки, пилюли, порошки (в желатиновых капсулах или в пакетиках) или гранулы. В таких композициях активный ингредиент согласно настоящему изобретению смешивают с одним или более из инертных разбавителей - таких, как крахмал, целлюлоза, сахароза, лактоза или окись кремния, в потоке аргона. Такие композиции могут также включать иные, чем разбавители, вещества, например: одно или более из смазывающих веществ - таких, как стеарат магния или тальк, краситель, покрытие (для покрытых сахаром пилюль) или глазурь.
В качестве жидких композиций для перорального введения могут быть использованы следующие: фармацевтически приемлемые растворы, суспензии, эмульсии, сиропы и эликсиры, содержащие инертные разбавители - такие, как вода, этанол, глицерин, растительные масла или жидкий парафин. Такие композиции могут включать иные, чем разбавители, вещества, например: увлажняющие, подслащивающие, загущающие, ароматизирующие или стабилизирующие средства.
Стерильные композиции для парентерального введения могут предпочтительно быть водными или неводными растворами, суспензиями или эмульсиями. В качестве растворителя или носителя может быть использовано следующее: вода, пропилен-гликоль, полиэтилен-гликоль, растительные масла, в частности оливковое масло, пригодные для инъекции органические эфиры, например этилолеат, или другие подходящие органические растворители. Такие композиции могут также содержать добавки, в частности увлажняющие средства, изотонические средства, эмульгаторы, диспергирующие средства и стабилизаторы. Стерилизацию можно также осуществлять различными путями, например: стерилизующей фильтрацией, введением в композицию стерилизующих средств, облучением или нагреванием. Такие композиции могут быть также приготовлены в форме стерильных твердых композиций, которые могут быть непосредственно перед употреблением растворены в стерильной воде или в любой другой пригодной для инъекций стерильной среде.
Композиции для ректального введения представляют собой суппозитории или ректальные капсулы, которые содержат, кроме активного продукта, наполнители, такие как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Композиции для местного введения могут представлять собой, например, кремы, лосьоны, глазные капли, ополаскиватели для рта, капли в нос или аэрозоли.
Дозы зависят от желаемого эффекта, длительности лечения и пути введения и обычно лежат в диапазоне от 0,001 г до 1 г (предпочтительно - от 0,005 г до 0,75 г) в день, предпочтительно - при оральном пути введения взрослым людям, при этом разовые дозы варьируют от 0,1 мг до 500 мг активного вещества.
Обычно подходящую дозировку определяет лечащий врач в зависимости от возраста, веса пациента и других специфических факторов в каждом конкретном случае.
Согласно особому варианту осуществления настоящее изобретение также относится к продуктам, содержащим соединение общей формулы (I) и другое сердечно-сосудистый агент, то есть к комбинированным продуктам для одновременного, раздельного или отсроченного по времени использования для лечения сердечно-сосудистых заболеваний, причем другой сердечно-сосудистый агент может быть антитромбоцитарным средством, таким как аспирин, клопидогрел, тиклопидин, абциксимаб, тирофибан или эптифибатид.
Согласно дополнительным признакам настоящего изобретения соединения общей формулы (I) пригодны для применения для изготовления лекарства, предназначенного для ингибирования пролиферации клеток гладких мышц (рестеноза) и/или для лечебного и/или профилактического воздействия на пролиферацию эндотелиальных, фибробластных, кардиофибробластных, глиальных клеток, клеток гладких мышц или раковых клеток.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ФЕНИЛПЕНТАДИЕНОИЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ PAR 1 | 2007 |
|
RU2440985C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 4,5-ДИГИДРО-1H-ПИРАЗОЛА, ИМЕЮЩИЕ CB-АНТАГОНИСТИЧЕСКУЮ АКТИВНОСТЬ | 2002 |
|
RU2299199C2 |
| НОВЫЕ ЛИГАНДЫ КАННАБИОИДНЫХ РЕЦЕПТОРОВ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2420518C2 |
| ЗАМЕЩЕННЫЕ БЕНЗО[d]ИЗОКСАЗОЛ-3-ИЛАМИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНАЛЬГЕТИКОВ | 2006 |
|
RU2416607C2 |
| ПРОИЗВОДНЫЕ 4-(4-АЛКОКСИ-3-ГИДРОКСИФЕНИЛ)-2-ПИРРОЛИДОНА В КАЧЕСТВЕ ИНГИБИТОРОВ PDE-4 ДЛЯ ЛЕЧЕНИЯ НЕВРОЛОГИЧЕСКИХ СИНДРОМОВ | 2002 |
|
RU2340600C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ С-ЦИКЛОГЕКСИЛМЕТИЛАМИНА, ЛЕКАРСТВЕННОЕ СРЕДСТВО И ПРИМЕНЕНИЕ | 2001 |
|
RU2295515C2 |
| ПИПЕРАЗИНИЛ- И ПИПЕРИДИНИЛЦИКЛОГЕКСАНОЛЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОВЫШЕНИЯ СЕРОТОНИНЕРГИЧЕСКОЙ НЕЙРОТРАНСМИССИИ | 1992 |
|
RU2088574C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ КОЛИТА | 2010 |
|
RU2518416C2 |
| ПИРРОЛОХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ 5-НТ6 АНТАГОНИСТОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2013 |
|
RU2688161C2 |
| ЗАМЕЩЕННЫЕ НИКОТИНАМИДНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 2008 |
|
RU2489425C2 |
Настоящее изобретение относится к соединениям общей формулы (I)
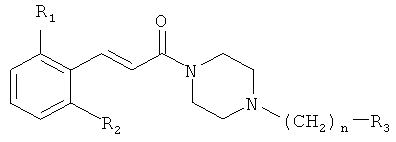
где R1 представляет собой галоген, CN или NO2; R2 представляет собой водород или галоген; n равно 1 или 2; R3 представляет собой фенил, замещенный одним или более галогенами или C1-C6 алкилами, или циклогексил; а также к их терапевтически приемлемым солям или сольватам. Соединения полезны как антагонисты активируемых протеазой рецепторов-1 (PAR-1), особенно для лечения тромбоза. 13 н. и 6 з,п. ф-лы, 6 табл.
1. Соединения общей формулы (I):
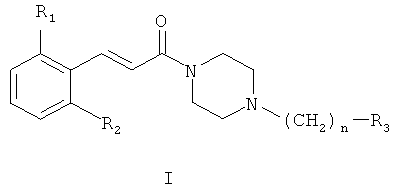
где R1 представляет собой галоген, CN или NO2;
R2 представляет собой водород или галоген;
n равно 1 или 2;
R3 представляет собой фенил, замещенный одним или более галогенами или C1-C6 алкилами; или циклогексил;
и их терапевтически приемлемые соли или сольваты.
2. Соединения по п.1, где R1 - галоген, R2 - водород, n равно 1, а R3 - фенил, замещенный одним или более галогенами или C1-C6 алкилами.
3. Соединения по п.1, где R1 - цианогруппа, R2 - водород, n равно 1, а R3 - фенил, замещенный одним или более галогенами или C1-C6 алкилами.
4. Соединения по п.1, где R1 - галоген, R2 - водород, n равно 1, а R3 - циклогексил.
5. Соединения по п.1, где R1 - цианогруппа, R2 - водород, n равно 1, а R3 - циклогексил.
6. Соединение по п.1, выбранное из:
3-(2-хлорфенил)-1-[4-(4-фторбензил)-пиперазин-1-ил]-пропенона;
1-[4-(4-фторбензил)-пиперазин-1-ил]-3-(2-фторфенил)-пропенона;
3-(2-бромфенил)-1-[4-(4-фторбензил)-пиперазин-1-ил]-пропенона;
3-(2-хлорфенил)-1-[4-(4-метилбензил)-пиперазин-1-ил]-пропенона;
3-(2,6-дифторфенил)-1-[4-(4-фторбензил)-пиперазин-1-ил]-пропенона;
3-(2,6-дифторфенил)-1-[4-(4-метилбензил)-пиперазин-1-ил]-пропенона;
3-(2,6-дифторфенил)-1-[4-(3,4-диметилбензил)-пиперазин-1-ил]-пропенона;
1-[4-(3,4-дифторбензил)-пиперазин-1-ил]-3-(2,6-дифторфенил)-пропенона;
1-[4-(4-хлорбензил)-пиперазин-1-ил]-3-(2,6-дифторфенил)-пропенона;
3-(2,6-дифторфенил)-1-[4-(3-метилбензил)-пиперазин-1-ил]-пропенона;
1-[4-(3-хлорбензил)-пиперазин-1-ил]-3-(2,6-дифторфенил)-пропенона;
3-(2,6-дифторфенил)-1-[4-(2-метилбензил)-пиперазин-1-ил]-пропенона;
3-(2-хлорфенил)-1-[4-(3-метилбензил)-пиперазин-1-ил]-пропенона;
1-[4-(4-хлорбензил)-пиперазин-1-ил]-3-(2-хлорфенил)-пропенона;
3-(2-хлорфенил)-1-[4-(2-фторбензил)-пиперазин-1-ил]-пропенона;
3-(2-хлорфенил)-1-[4-(2-метилбензил)-пиперазин-1-ил]-пропенона;
1-[4-(2-хлорбензил)-пиперазин-1-ил]-3-(2-хлорфенил)-пропенона;
3-(2-хлорфенил)-1-[4-(3-фторбензил)-пиперазин-1-ил]-пропенона;
1-[4-(3-хлорбензил)-пиперазин-1-ил]-3-(2-хлорфенил)-пропенона;
3-(2-хлорфенил)-1-[4-(2,3-дифторбензил)-пиперазин-1-ил]-пропенона;
3-(2-хлорфенил)-1-[4-(3,4-дифторбензил)-пиперазин-1-ил]-пропенона;
3-(2,6-дифторфенил)-1-[4-(2-фторбензил)-пиперазин-1-ил]-пропенона;
1-[4-(2-фторбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенона;
1-(4-циклогексилметил-пиперазин-1-ил)-3-(2,6-дифторфенил)-пропенона;
2-[3-(4-циклогексилметил-пиперазин-1-ил)-3-оксо-пропенил]-бензонитрила;
1-(4-циклогексилметил-пиперазин-1-ил)-3-(2-нитрофенил)-пропенона;
1-(4-циклогексилметил-пиперазин-1-ил)-3-(2-фторфенил)-пропенона;
3-(2-хлорфенил)-1-(4-циклогексилметил-пиперазин-1-ил)-пропенона;
1-[4-(4-фторбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенона;
1-[4-(4-метилбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенона;
1-[4-(3,4-дифторбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенона;
1-[4-(4-хлорбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенона;
1-[4-(3-метилбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенона;
1-[4-(2,6-диметилбензил)-пиперазин-1-ил]-3-(2-нитрофенил)-пропенона;
3-(2,6-дифторфенил)-1-(4-фенэтил-пиперазин-1-ил)-пропенона;
3-(2-хлорфенил)-1-(4-фенэтил-пиперазин-1-ил)-пропенона;
3-(2-хлорфенил)-1-{4-[2-(4-фторфенил)-этил]-пиперазин-1-ил}-пропенона;
а также их терапевтически приемлемых солей и сольватов.
7. Соединения по любому из пп.1-6 для применения в качестве лекарства для профилактики и/или лечения заболеваний, связанных с активацией рецептора PAR-1.
8. Способ получения соединений общей формулы (I) по любому из пп.1-6, включающий конденсацию промежуточного соединения общей формулы (II)
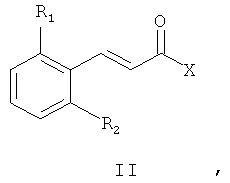
где R1 и R2 определены так, как в описании общей формулы (I) в п.1, Х может представлять собой уходящую группу - такую, как хлор, или Х может представлять собой гидроксил, с амином общей формулы (III)
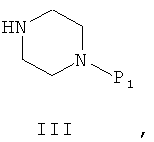
где P1 представляет собой защитную группу, полученное промежуточное соединение общей формулы (IV)
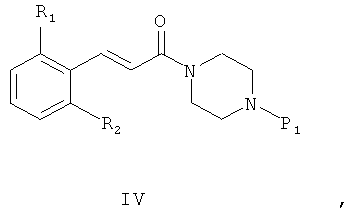
где R1, R2 и P1 определены выше, дает соединения общей формулы (I) после снятия защиты и проведения реакции амина либо с реагентом общей формулы R3(CH2)nY, где R3 определен выше, а Y представляет собой уходящую группу - такую, как, например, Cl, Br, I, OSO2CH3, OSO2CF3 или O-тозил, либо с альдегидом формулы R3-(CH2)n-1-СНО, где R3 и n определены выше.
9. Фармацевтическая композиция, обладающая антагонистической активностью в отношении рецептора PAR-1, содержащая в качестве активного ингредиента по меньшей мере одно соединение по любому из пп.1-6 в комбинации с фармацевтически приемлемым носителем.
10. Применение соединения по любому из пп.1-6 для изготовления лекарства, являющегося антагонистом тромбинового рецептора.
11. Применение соединения по любому из пп.1-6 для изготовления лекарства для лечения и/или профилактики расстройств, связанных с активацией рецептора PAR-1 (активируемого протеазой рецептора-1).
12. Применение соединения по любому из пп.1-6 для изготовления лекарства против агрегации тромбоцитов.
13. Применение соединения по любому из пп.1-6 для изготовления лекарства для лечения и/или профилактики артериального и/или венозного тромбоза.
14. Применение соединения по любому из пп.1-6 для изготовления лекарства для лечения и/или профилактики стабильной стенокардии, нарушений сердечного ритма, инсультов, сердечной недостаточности, гипертензии или инфаркта миокарда.
15. Применение соединения по любому из пп.1-6 для изготовления лекарства для лечения и/или профилактики фибрилляции предсердий и ремоделирования миокарда.
16. Применение соединения по любому из пп.1-6 для изготовления лекарства для лечения и/или профилактики острых коронарных синдромов.
17. Применение соединения по любому из пп.1-6 для изготовления лекарства для ингибирования пролиферации клеток гладких мышц (рестеноза).
18. Применение соединения по любому из пп.1-6 для изготовления лекарства для лечения и/или профилактики воспалительных расстройств, легочных заболеваний, желудочно-кишечных заболеваний, развития фиброза у пациентов с хроническим заболеванием печени или кожных заболеваний.
19. Применение соединения по любому из пп.1-6 для изготовления лекарства для лечения и/или профилактики пролиферации эндотелиальных, фибробластных, кардиофибробластных, глиальных клеток, клеток гладкой мускулатуры или раковых клеток.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ ОТСЧЕТА ОТ ОДНОГО НАЧАЛА В ЦИФРОВЫХ ИНТЕГРАТОРАХ ИНТЕРВАЛОВВРЕМЕНИ | 0 |
|
SU209843A1 |
| RU 2005115964 A, 27.01.2006 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЭФИРЫ БИС-ФЕНИЛПИПЕРАЗИННИКОТИНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 1993 |
|
RU2127732C1 |
| RU 2005117383 A, 20.01.2006. | |||

