Изобретение относится к производным фенилпентадиеноила, к способу их получения, к фармацевтическим композициям, содержащим эти производные, и к применению этих производных в качестве лекарственных средств для лечения и/или профилактики артериальных и венозных тромбозов, острых коронарных синдромов, рестеноза, стабильной стенокардии, нарушений сердечного ритма, инфаркта миокарда, гипертензии, сердечной недостаточности, инсульта, воспалительных заболеваний, легочных заболеваний, желудочно-кишечных заболеваний, развития фиброза у больных с хроническими заболеваниями печени, рака и кожных болезней. Настоящее изобретение также относится к комбинациям соединений согласно настоящему изобретению с другими сердечно-сосудистыми средствами.
Тромбоз считается первичным фактором окклюзии сосудов, которая является причиной многих патофизиологических осложнений. Поэтому антитромботическая терапия имеет исключительную важность, так как она способна снизить риск смертности от сердечно-сосудистых заболеваний и коронарных явлений. Хотя было показано, что значительной антитромботической активностью для человека обладает несколько типов молекул, все же сохраняется потребность в новых молекулах, которые обладали бы преимуществами по сравнению с существующими соединениями, часть из которых оказывает негативное влияние на время кровотечения или имеет другие нежелательные побочные эффекты (например, риск возникновения язв желудка при приеме аспирина).
Недавно был клонирован активируемый протеазой рецептор 1 типа (PAR-1) (Vu et al., Cell, 1991, 64:1057-1068) и выяснен его механизм действия (Coughlin et al., J. Clin. Invest. 1992, 89(2):351-355). Этот рецептор, присутствующий, прежде всего, на поверхности тромбоцитов, а также на поверхности эндотелиальных клеток (O'Brien et al., J. Biol. Chem. 2000, 275:13502-13509), гладкомышечных клеток (Hamilton et al., Br. J. Pharmacol. 2000, 130:181-188) и фибробластов (Hung et al., J. Cell. Biol. 1992, 116(3):827-832), активируется тромбином, поэтому его также называют тромбиновым рецептором. N-терминаль белка расщепляется тромбином между аргинином в положении 41 и серином в положении 42 с высвобождением нового конца, который после фолдинга будет действовать на активный центр как агонист рецептора (Vu et al., Nature, 1991, 353, 674-677). Что касается тромбоцитов, то этот специфический механизм активации PAR-1 рецептора приводит к опосредованной тромбином агрегации тромбоцитов.
Блокирование этой активации, например, антагонистами PAR-1 рецептора может ингибировать опосредованную тромбином агрегацию тромбоцитов (Ann et al., Drug of the Future, 2001, 26:1065-1085). Поэтому блокирование этих рецепторов может быть использовано для лечения или профилактики тромбоза (Derian et al., J. Pharmacol. Exp. Ther., 2003, 855-861), острых коронарных синдромов (Ossovskaya et al., Physiol. Rev., 2004, 84:579-621) и рестеноза (Maryanoff et al., Curr. Med. Chem. Cardiovasc. Hematol. Agents, 2003, 13-36) и может уменьшить некрозы миокарда во время инфаркта или реперфузии (Steinberg et al., Mol. Pharmacol., 2005, 67:2-11). На уровне легких активность антагониста PAR-1 может предотвратить возникновение некоторых воспалительных заболеваний (Moffatt et al., Curr. Op. Pharmacol., 2004, 221-229). На уровне желудочно-кишечного тракта активность антагониста PAR-1 может предотвратить возникновение некоторых воспалительных заболеваний (Vergnolle et al., J. Clin. Invest., 2004, 1444-1456). Антагонисты PAR-1 рецептора могут также быть использованы для лечения фиброзов у пациентов с хроническим заболеванием печени (Fiorucci et al., Hepatology, 2004, 39:365-375). Их можно также использовать в качестве противораковых агентов, так как они регулируют пролиферацию клеток и образование метастазов (Evan-Rаm et al., Nat. Med., 1998, 909-914; Boire et al., Cell., 2005, 120:303-313). Наконец, антагонисты PAR-1 могут представлять интерес в дерматологии для лечения некоторых кожных болезней (Schechter et al., J. Cell. Physiol., 1998, 176:365-373; Algermissen et al., Arch. Dermatol. Res., 200, 292: 488-495; Meyer-Hoffert et al., Exp. Dermatol., 2004, 13:234-241).
Настоящее изобретение относится к новому классу антагонистов PAR-1, которые отличаются от антагонистов предшествующего уровня техники другой химической структурой и явными биологическими особенностями.
Соединения согласно настоящему изобретению имеют общую формулу (I):
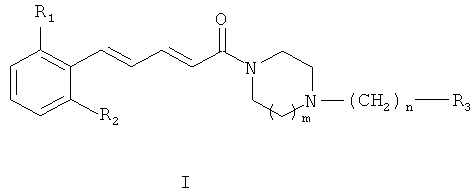
где R1 и R2, идентичные или различные, представляют собой:
атом водорода или атом галогена, CN или NO2, причем R1 и R2 не являются
атомами водорода одновременно;
m означает:
1 или 2;
n означает:
0, 1 или 2;
R3 представляет собой:
фенильный радикал, замещенный или не замещенный одним или несколькими радикалами, выбранными из атома галогена, гидроксильной группы или C1-C6 алкильной группы; С2-С6 алкильную группу, замещенную или не замещенную одним или несколькими радикалами, выбранными из атома галогена или гидроксильной группы; циклоалкильную группу; пиридин; тиофен; пиррол, замещенный или не замещенный C1-C6 алкильной группой; тиазол или фуран;
или их терапевтически приемлемые соли или сольваты.
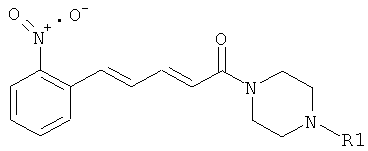
В приведенных выше определениях все комбинации заместителей или переменных возможны в тех случаях, когда они дают стабильные соединения.
Термин «галоген» означает фтор, хлор, бром или йод.
Термин «алкильная группа» означает линейные или разветвленные, насыщенные или ненасыщенные алифатические углеводородные цепи, содержащие заданное число атомов углерода.
Термин «циклоалкильная группа» означает циклические углеводородные цепи, содержащие от 3 до 10 атомов углерода.
Терапевтически приемлемые соли соединений согласно настоящему изобретению включают в себя стандартные нетоксичные соли соединений согласно настоящему изобретению, например соли, образованные органическими или неорганическими кислотами. В качестве примера можно назвать следующие соли: соли неорганических кислот, таких как хлористоводородная, бромистоводородная, фосфорная и серная кислоты, а также соли органических кислот, таких как уксусная, трифторуксусная, пропионовая, янтарная, фумаровая, яблочная, винная, лимонная, аскорбиновая, малеиновая, глутаминовая, бензойная, салициловая, толуолсульфоновая, метансульфоновая, стеариновая и молочная кислоты.
Эти соли могут быть синтезированы из соединений согласно настоящему изобретению, содержащих основную часть, и соответствующих кислот с использованием стандартных химических способов.
Терапевтически приемлемые сольваты соединений согласно настоящему изобретению включают в себя стандартные сольваты, например сольваты, полученные во время последней стадии получения соединений согласно настоящему изобретению вследствие присутствия растворителей. В качестве примера можно привести сольваты вследствие присутствия воды или этанола.
Среди соединений общей формулы (I) согласно настоящему изобретению особенно полезным классом соединений являются соединения общей формулы (I), где R1 является нитрогруппой, R2 является водородом, m равно 1, n равно 0, a R3 является фенильной группой, замещенной одним или несколькими атомами галогенов или одной или несколькими C1-C6 алкильными группами, циклоалкильной группой или пиридином.
Среди соединений общей формулы (I) согласно настоящему изобретению второй особенно полезный класс соединений соответствует соединениям общей формулы (I), где R1 является цианогруппой, R2 является атомом водорода, m равно 1, n равно 0, a R3 является фенильной группой, замещенной одним или несколькими атомами галогенов или одной или несколькими C1-C6 алкильными группами, циклоалкильной группой или пиридином.
Настоящее изобретение также относится к получению соединений общей формулы (I) стандартными способами, описанными в приведенных ниже схемах синтеза и дополненными, если это необходимо, любой стандартной техникой, описанной в литературе, известной специалистам в данной области техники или представленной в разделе, посвященном экспериментам.
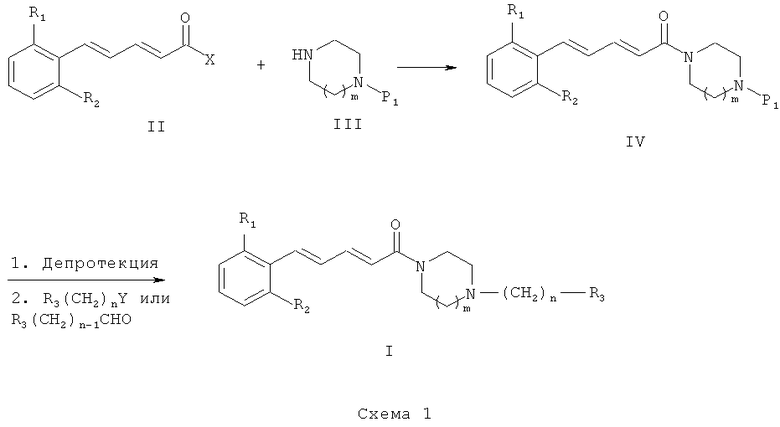
Схема 1 иллюстрирует первый общий способ, который может быть использован для получения соединений общей формулы (I). В общих формулах, приведенных выше, R1, R2, R3 являются такими же, как в приведенном ранее описании общей формулы (I). Однако на Схеме 1, приведенной выше, n означает только 1 или 2, P1 обозначает защитную группу, а Х может обозначать такую группу, как атом хлора или гидроксильная группа. Исходное соединение общей формулы (II) может быть получено с использованием способов и методик, известных специалистам в данной области техники. Особо предпочтительный способ состоит в реакции бензилгалида с трифенилфосфином в полярном растворителе, таком как диметилформамид (ДМФ) или диметилсульфоксид (ДМСО), при температуре в диапазоне от 20 до 100°С с получением фосфониевой соли. Фосфониевую соль можно затем депротонировать с использованием основания, например NaH, в растворителе, таком как диметилформамид (ДМФ) или тетрагидрофуран (ТГФ), при температуре в диапазоне от -20 до 40°С, после чего провести реакцию с α,β-ненасыщенным альдегидом, несущим сложный эфир, например этильный эфир (2Е)-4-оксобут-2-еноата. Полученный сложный эфир (смесь Z/E и Е/Е изомеров) вначале изомеризуют посредством обработки йодом в полярном растворителе, например в ацетонитриле, с получением исключительно Е/Е изомера, а затем омыляют посредством обработки минеральным основанием, таким как KОН, NaOH или LiOH, в полярном растворителе, например в воде, этаноле или ТГФ, при температуре в диапазоне от 20 до 100°С с получением соединений (II), в которых Х является гидроксильной группой. Второй особо предпочтительный способ состоит в реакции ароматического альдегида с фосфонатом, например с этильным эфиром 4-(диэтоксифосфорил)бут-2-еноата, в присутствии основания, например NaH, Cs2CO3 или K2CO3, в растворителе, например в ТГФ, дихлорметане или дихлорэтане, при температуре в диапазоне от 20 до 100°С. Полученный сложный эфир (смесь Z/E и Е/Е изомеров) вначале изомеризуют посредством обработки йодом в полярном растворителе, например в ацетонитриле, с получением исключительно Е/Е изомера, а затем омыляют посредством обработки минеральным основанием, таким как KОН, NaOH или LiOH, в полярном растворителе, например в воде, этаноле или ТГФ, при температуре в диапазоне от -20 до 100°С с получением соединений (II), в которых Х является гидроксильной группой. Третий особо предпочтительный способ состоит в реакции ароматического α,β-ненасыщенного альдегида с этильным эфиром (диэтоксифосфорил)ацетата в присутствии основания, например NaH, Cs2CO3 или K2CO3, в растворителе, например в ТГФ, дихлорметане или дихлорэтане, при температуре в диапазоне от -20 до 100°С. Полученный сложный эфир можно омылить посредством обработки минеральным основанием, таким как KОН, NaOH или LiOH, в растворителе, например в воде, этаноле или ТГФ, при температуре в диапазоне от 20 до 100°С с получением соединений (II), в которых Х является гидроксильной группой. Четвертый особо предпочтительный способ состоит в реакции ароматического соединения, несущего на себе галоген, например бром или йод, с (Е)-пента-2,4-диеноильным сложным эфиром, например метальным или этильным сложным эфиром (Е)-пента-2,4-диеноата, в присутствии палладиевого катализатора, например палладия ацетата, фосфина, например три-о-толилфосфина или трифенилфосфина, в присутствии основания, например Et3N или iPr2NEt, в открытом или герметично закрытом реакторе, без растворителя или в растворителе, таком как ДМФ, ДМСО или ДМА, при температуре в диапазоне от 20 до 120°С. Полученный таким образом сложный эфир (преимущественно Е/Е изомер) можно омылить посредством обработки минеральным основанием, таким как KОН, NaOH или LiOH, в растворителе, например в воде, этаноле или ТГФ, при температуре в диапазоне от 20 до 100°С с получением соединений (II), в которых Х является гидроксильной группой. В этом случае первой стадией является реакция конденсации между карбоновой кислотой (II) и амином (III). Эту реакцию можно провести с использованием способов и методик, известных специалистам в данной области техники. Особо предпочтительный способ состоит в реакции этих двух молекул в присутствии 1-(3-диметиламинопропил)-3-этилкарбодиимида (ЭДК), 3-гидрокси-1,2,3-бензотриазин-4(3H)-она и третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан, при температуре в диапазоне от -15 до 40°С. Карбоновую кислоту можно также преобразовать в хлорид кислоты (тогда Х соответствует хлору) посредством обработки реагентом, например тионилхлоридом, при температуре в диапазоне от 20 до 100°С. В этом случае первая стадия состоит в реакции между хлоридом кислоты и амином. Эту реакцию можно провести с использованием способов и методик, известных специалистам в данной области техники. Особо предпочтительный способ состоит в реакции этих двух молекул в присутствии органического или неорганического основания, например Et3N, iPr2NEt, пиридина, NaH, Cs2CO3 или K2СО3, в растворителе, таком как ТГФ, дихлорметан, ДМФ или ДМСО, при температуре в диапазоне от -20 до 100°С.
После депротекции промежуточного соединения (IV) с использованием способов и методик, известных специалистам в данной области техники («Protective Groups in Organic Synthesis», T.W. Greene, John Wiley & Sons, 1981, и «Protective Groups», P.J. Kocienski, Thieme Verlag, 1994), полученное промежуточное соединение может реагировать с реагентом, имеющим формулу R3(СН2)nY, где Y представляет собой уходящую группу, например Cl, Br, l, OSO2CH3, OSO2CF3 или O-тозильную группу. В этом случае реакцию проводят в присутствии органического или неорганического основания, например Et3N, iPr2NEt, NaH, Cs2CO3 или K2CO3, способного удерживаться на смоле, такой как PS-DIEA или МР-карбонат, в полярном безводном растворителе, таком как дихлорметан, ТГФ, ДМФ или ДМСО, при температуре в диапазоне от -20 до 100°С. Другой способ получения состоит в проведении реакции аминирования с использованием альдегида, имеющего формулу R3-(СН2)n-1-СНО, в которой R3 и n являются такими, как определено ранее, с депротектированным амином общей формулы (IV) и восстановителем, таким как NaBH4, NaBH3CN или NaBH(ОАс)3, способным удерживаться на смоле, такой как МР-ВН3СМ, в полярном растворителе, таком как 1,2-дихлорэтан, дихлорметан, ТГФ, ДМФ или МеОН, при рН, который можно регулировать добавлением кислоты, такой как уксусная кислота, и при температуре в диапазоне от -20 до 100°С.
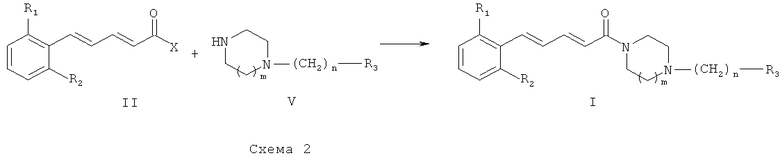
Схема 2 иллюстрирует второй общий способ, который можно использовать для получения соединений общей формулы (I). В общих формулах, приведенных выше, R1, R2, R3 и n являются такими же, как в описании общей формулы (I). X может обозначать функциональную группу, такую как хлор или гидроксил. Исходное соединение общей формулы (II) можно получить с использованием способов и методик, известных специалистам в данной области техники, в частности способов и методик, описанных выше. В случае если Х является хлором, синтез состоит в реакции между хлоридом кислоты и амином. Эту реакцию можно провести с использованием способов и методик, известных специалистам в данной области техники. Особо предпочтительный способ состоит в реакции этих двух молекул в присутствии органического или неорганического основания, например Et3N, iPr2NEt, пиридина, NaH, Cs2CO3 или K2CO3, в растворителе, таком как ТГФ, дихлорметан, ДМФ или ДМСО, при температуре в диапазоне от -20 до 100°С.
В случае если Х является гидроксильной группой, синтез состоит в конденсации карбоновой кислоты (II) и амина (V). Реакцию можно провести с использованием способов и методик, известных специалистам в данной области техники. Особо предпочтительный способ состоит в конденсации карбоновой кислоты общей формулы (II) с амином общей формулы (III) в присутствии 1-(3-диметиламинопропил)-3-этилкарбодиимида (ЭДК), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-она и третичного амина, например диизопропилэтиламина, в полярном апротонном растворителе, таком как дихлорметан, при температуре в диапазоне от -15 до 40°С.
Если желательно выделить соединение общей формулы (I), содержащее по меньшей мере одну основную функциональную группу в состоянии соли за счет добавления кислоты, такой результат можно получить посредством обработки свободного основания общей формулы (I) (в котором присутствует по меньшей мере одна функциональная группа с основными свойствами) подходящей кислотой, предпочтительно в эквивалентном количестве.
Приведенные ниже примеры иллюстрируют настоящее изобретение, ни в коей мере не ограничивая его объем.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Пример 1
2[5-Оксо-5-(4-пиридин-2-ил-пиперазин-1-ил)-пента-1,3-диенил]-бензонитрил
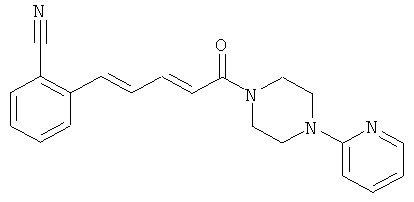
Пример 1А: Этил-5-(2-цианофенил)-пента-2,4-диеноат
2-Бромметилбензонитрил (3 г, 15,3 ммоль) в виде раствора в ДМФ (50 мл) при 80°С обработали трифенилфосфином (4,42 г, 16,83 ммоль). После перемешивания в течение 3 часов температуру смеси снизили до комнатной температуры и добавили гидрид натрия (60% в масле) (673 мг, 16,83 ммоль) и этильный эфир 4-оксобут-2-еноата (2,16 г, 16,83 ммоль). После 16 часов перемешивания при комнатной температуре смесь выпарили досуха, перенесли в этилацетат и промыли водой. Органическую фазу высушили над Na2SO4, профильтровали и выпарили досуха. Полученный сироп очистили посредством хроматографии на колонке с диоксидом кремния и элюировали с использованием смеси EDP/AcOEt в соотношении 9/1. Продукт 1А выделили в виде желтого сиропа (2,73 г, 71%) в виде смеси Е/Е и Z/E изомеров.
Масс-спектр (ESI+): m/z 228 (M+H+).
Пример 1В: Этил-(2Е,4Е)-5(2-цианофенил)-пента-2,4-диеноат
Соединение 1А (2,34 г, 10,3 ммоль) в виде раствора в ацетонитриле (14 мл) обработали при комнатной температуре йодом (15,0 мг, 0,06 ммоль). После перемешивания в течение 3 часов смесь выпарили досуха, перенесли в дихлорметан и промыли раствором Na2SO3, профильтровали и выпарили досуха. Продукт 1В выделили в виде твердого вещества (2,26 г, 97%) и использовали без дальнейшей обработки для следующей стадии.
1H-ЯМР, ДМСО-d6 (млн-1): 1,25 (t, 3Н); 4,16 (q, 2Н); 6,21 (d, 1H); 7,34 (m, 2H); 7,50 (m, 2H); 7,74 (t, 1H); 7,87 (d, 1H); 7,96 (d, 1H).
Пример 1C: 5-(2-Цианофенил)-пента-2,4-диеновая кислота
Соединение 1В (2,0 г, 8,82 ммоль) в виде раствора в этаноле (50 мл) обработали 1N раствором поташа (13,2 мл, 13,2 ммоль). После 1,5 часов перемешивания с обратным холодильником смесь выпарили досуха, перенесли в воду и обработали 1N HCl до получения кислотного рН. Образовавшийся осадок отфильтровали, промыли водой и высушили под вакуумом до получения чистого продукта 1C (1,63 г, 93%).
Масс-спектр (ESI-): m/z 198 (М-Н-).
Пример 1: 2-[5-Оксо-5-(4-пиридин-2-ил-пиперазин-1-ил)-пента-1,3-диенил]-бензонитрил
Кислоту 1C (700 мг, 3,51 ммоль) в виде раствора в дихлорметане (10 мл) в присутствии диизопропилэтиламина (DIEA) (1,2 мл, 7,02 ммоль) обработали при комнатной температуре 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDCl) гидрохлоридом (807 мг, 4,21 ммоль), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-оном (НООВТ) (686 мг, 4,21 ммоль) и затем 1-пиридин-2-ил-пиперазином (642 мкл, 4,21 ммоль). После перемешивания в течение 16 часов реакционную смесь разбавили дихлорметаном и промыли 1N раствором соды и водой. Органическую фазу высушили над MgSO4, профильтровали и выпарили досуха. Полученный сироп очистили посредством хроматографии на колонке с диоксидом кремния и элюировали смесью петролейного эфира/AcOEt в соотношении ½. Продукт 1 выделили в виде желтого твердого вещества (910 мг, 75%). Этот продукт перенесли в этилацетат, затем получили соль посредством добавления раствора HCl в эфире с получением соответствующего гидрохлорида в виде желтого твердого вещества.
1H-ЯМР, ДМСО-d6 (млн-1): 3,81 (широкая полоса S, 8H); 6,94 (m, 2H); 7,18 (m, 1Н); 7,38 (m, 3H); 7,51 (t, 1H); 7,74 (t, 1H); 7,86 (d, 1H); 7,91 (d, 1H); 7,96 (t, 1H); 8,06 (d, 1H).
Масс-спектр (ESI+): m/z 345 (M+H+).
Примеры 2-8
Соединения 2-8 были синтезированы из промежуточного продукта 1C и соответствующих аминов в условиях, описанных при получении соединения 1.
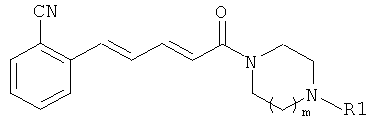
Пример 9
5-(2-Хлорфенил)-1-(4-цикпопентилпиперазин-1-ил)-пента-2,4-диен-1-он
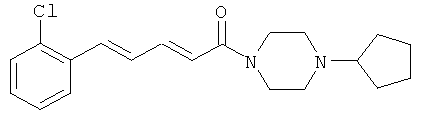
Пример 9А: Этил-5-(2-хлорфенил)-пента-2,4-диеноат
Этил-4-(диэтоксифосфорил)-бут-2-еноат (3,92 г, 15,65 ммоль) в растворе ТГФ (79 мл) при 0°С обработали гидридом натрия (60% в масле) (630 мг, 15,7 ммоль). После 30 минут перемешивания при 0°С добавили 2-хлорбензальдегид (2,0 г, 14,22 ммоль) и взбалтывали смесь в течение 16 часов, постепенно повышая температуру от 0°С до комнатной температуры. Затем смесь выпарили досуха, перенесли в AcOEt и промыли водой. Органическую фазу высушили над MgSO4, профильтровали и выпарили досуха. Полученный сироп очистили посредством хроматографии на колонке с диоксидом кремния и элюировали смесью EDP/CH2Cl2 в соотношении 2/1. Продукт 9А был выделен в виде желтого масла (1,1 г, 33%).
Пример 9В: 5-(2-Хлорфенил)-пента-2,4-диеновая кислота
Соединение 9А (2,1 г, 8,87 ммоль) в виде раствора в ТГФ (20 мл) обработали 1N раствором LiOH (35 мл, 35,4 ммоль). После перемешивания в течение 2 часов при комнатной температуре и в течение 1 часа с обратным холодильником смесь выпарили досуха, перенесли в воду и обработали 4N раствором HCl до кислотного рН. Образовавшийся осадок отфильтровали, промыли водой и затем высушили под вакуумом с получением чистого продукта 9В (1,70 г, 92%).
Масс-спектр (ESI-): m/z 207 (М-Н-).
Пример 9: 5-(2-Хлорфенил)-1-(4-циклопентилпиперазин-1-ил)-пента-2,4-диен-1-он
Соединение 9 получили из промежуточного продукта 9В (67,0 мг, 0,32 ммоль) и циклопентилпиперазина (101,3 мг, 0,45 ммоль) в условиях, описанных для получения соединения 1 из 1C. Чистый продукт был выделен в виде гидрохлорида (99 мг, 81%).
Масс-спектр (ESI+): m/z 345 (М+Н+).
Примеры 10-15
Соединения 10-15 были синтезированы из промежуточного продукта 9В и соответствующих аминов в условиях, описанных для получения соединения 1 из 1C.
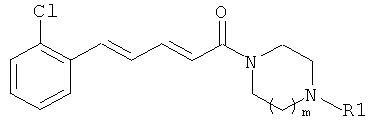
Пример 16
5-(2-Нитрофенил)-1-(4-фенилпиперазин-1-ил)-пента-2,4-диен-1-он
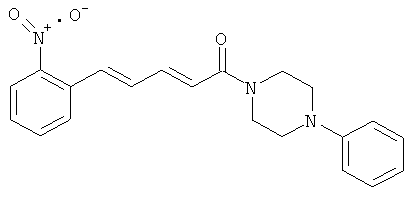
Пример 16А: Этил-5-(2-нитрофенил)-пента-2,4-диеноат
3-(2-Нитрофенил)-пропеналь (4,0 г, 22,5 ммоль) в виде раствора в толуоле (67 мл) обработали этил-(трифенилфосфоранил)-ацетатом (8,25 г, 23,7 ммоль). После перемешивания в течение 2 дней с обратным холодильником смесь выпарили досуха, очистили посредством хроматографии на колонке с диоксидом кремния и элюировали смесью EDP/AcOEt в соотношении 2/1. Продукт 16А был выделен в виде желтого твердого вещества (4,96 г, 90%).
1H-ЯМР, ДМСО-d6 (млн-1): 1,24 (t, 3H); 4,16 (q, 2H); 6,19 (d, 1H); 7,17(dd, 1H); 7,35 (d, 1H); 7,45 (dd, 1H); 7,59 (t, 1H); 7,77 (t, 1H); 7,89 (d, 1H); 8,00 (d, 1H).
Масс-спектр (ESI+): m/z 248 (M+H+) 15.
Пример 16В: 5-(2-Нитрофенил)-пента-2,4-диеновая кислота
Промежуточное соединение 16А (2,59 г, 10,5 ммоль) омылили в условиях, описанных для получения соединения 1C из 1В. Чистый продукт был выделен в виде белого твердого вещества (2,27 г, 99%).
Масс-спектр (ESI-): m/z 218 (М-Н-).
Пример 16: 5-(2-Нитрофенил)-1-(4-фенилпиперазин-1-ил)-пента-2,4-диен-1-он
Соединение 16 было получено из промежуточного соединения 16В (404 мг, 1,84 ммоль) и фенилпиперазина (415 мкл, 2,20 ммоль) в условиях, описанных для получения соединения 1 из 1C. Чистый продукт был выделен в виде гидрохлорида (621 мг, 87%).
1H-ЯМР, ДМСО-d6 (млн-1): 3,29 (широкая полоса s, 4H); 3,82 (широкая полоса s, 4H); 6,94 (d, 1H); 7,00 (t, 1H); 7,20 (m, 4H); 7,32 (m, 3H); 7,57 (t, 1H); 7,75 (t, 1H); 7,87 (d, 1H); 7,99(d,1H).
Масс-спектр (ESI+): m/z 364 (M+H-).
Примеры 17-26
Соединения 17-26 были синтезированы из промежуточного соединения 16В и соответствующих аминов в условиях, описанных для получения соединения 1 из 1C.
Пример 27
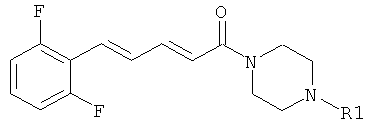
5-(2,6-Дифторфенил)-1-(4-фенилпиперазин-1-ил)-пента-2,4-диен-1-он
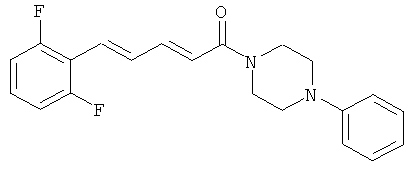
Пример 27А: Этил-5-(2.6-дифторфенил)-пента-2,4-диеноат
Этил-(диэтоксифосфорил)-ацетат (3,72 мл, 18,7 ммоль) в виде раствора в ТГФ (114 мл) обработали гидридом натрия (60% в масле) (819 мг, 20,4 ммоль) при комнатной температуре в течение 5 минут. Затем по каплям добавили 3-(2,6-дифторфенил)-пропеналь (2,87 г, 17,0 ммоль) в виде раствора в ТГФ (29 мл). После 3 часов перемешивания при комнатной температуре смесь выпарили досуха, перенесли в этилацетат и промыли водой. Органическую фазу высушили над Na2SO4, профильтровали и выпарили досуха. Полученное желтое твердое вещество без дальнейшей обработки использовали в следующей реакции.
Пример 27В: 5-(2,6-Дифторфенил)-пента-2,4-диеновая кислота
Промежуточное соединение 27В (3,28 г, 13,76 ммоль) омылили в условиях, описанных для получения соединения 1C из 1В. Чистый продукт был выделен в виде бежевого твердого вещества (2,56 г, 88%).
Масс-спектр (ESI-): m/z 209 (М-Н-).
Пример 27: 5-(2,6-Дифторфенил)-пента-2,4-диеновая кислота
Соединение 27 было получено из промежуточного соединения 27В (60 мг, 0,285 ммоль) и фенилпиперазина (68,1 мкл, 0,342 ммоль) в условиях, описанных для получения соединения 1 из 1C. Чистый продукт был выделен в виде бежевого порошка (72 мг, 79%),
Масс-спектр (ESI+): m/z 355 (M+H+).
Примеры 28-31
Соединения 28-31 были синтезированы из промежуточного соединения 27В и соответствующих аминов в условиях, описанных для получения соединения 1 из 1C.
Пример 32
1-(4-Цикпопентилпиперазин-1-ил)-5-(2-фторфенил)-пента-2,4-диен-1-он
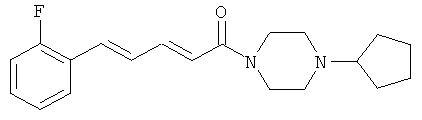
Пример 32А: Этил-5-(2-фторфенил)-пента-2,4-диеноат
Промежуточное соединение 32А получено из 3-(2-дифторфенил)-пропеналя и этил-(диэтоксифосфорил)-ацетата в условиях, описанных для получения соединения 27А.
Масс-спектр (ESI+): m/z 221 (M+H+).
Пример 32В: 5-(2-Фторфенил)-пента-2,4-диеновая кислота
Промежуточное соединение 32В было получено из соединения 32А в условиях, описанных для получения соединения 27В.
Масс-спектр (ESI-): m/z 191 (М-Н-).
Пример 32: 1-(4-Циклопентилпиперазин-1-ил)-5-(2-фторфенил)-пента-2,4-диен-1-он
Соединение 32 было получено из промежуточного соединения 32В (100,0 мг, 0,52 ммоль) и цикпопентилпиперазина (165,3 мг, 0,73 ммоль) в условиях, описанных для получения соединения 1 из 1C. Чистый продукт был выделен в виде белого порошка (122 мг, 64%).
Масс-спектр (ESI+): m/z 329 (M+H+).
Примеры 33-36
Соединения 33-36 были синтезированы из промежуточного соединения 32В и соответствующих аминов в условиях, описанных для получения соединения 1 из 1C.
Производные согласно настоящему изобретению являются антагонистами PAR-1 рецептора, поскольку результаты, полученные на моделях, описанных ниже, демонстрируют следующее.
В различных типах клеток активация PAR-1 рецептора пептидом SFLLR (селективным агонистом PAR-1) запускает внутриклеточный сигнальный каскад, приводящий к выделению кальция из эндоплазматического ретикулума. Клетки яичника китайского хомячка (СНО) устойчиво экспрессируют PAR-1 рецептор. В этой клеточной линии выделение кальция вследствие активации рецепторов SFLLR измеряли посредством флуориметрической методики (флуориметрическое визуализационное устройство для чтения планшетов, или FLIPR) с использованием селективного датчика кальция (Fluo-3АМ). Излучение флуоресценции с фармакологической точки зрения пропорционально эффективности PAR-1 агониста и его концентрации. Соединения, описанные в настоящем изобретении, показали, что они способны выступать в качестве антагонистов PAR-1 рецепторов и таким образом снижать выделение кальция, вызванное агонистом.
Материалы
Культуральная среда: F-12 по Хэму (Ham, R.G., Proc. Natl. Acad. Sci., 1965, 53: 288) с добавлением 10% плодной сыворотки коров и антибиотика (Пробеницид, 2,5 мМ).
Флуоресцентный зонд: Fluo-3АМ (4 мкМ; Teflabs, Остин, Техас, США).
Агонист: SFLLR-NH2 (серин, фенилаланин, лейцин, лейцин, аргинин).
Способы: СНО-клетки культивировали в 96-луночном планшете (по 60000 клеток на ячейку) в присутствии 200 мкл культуральной среды в течение 24 часов. Клетки инкубировали с флуоресцентным зондом на кальций в течение 1 часа при 37°С. Затем клетки промывали в течение 10 минут перед измерением сигнала. Затем инъецировали PAR-1 антагонист (от 0,01 до 10 мкМ). Планшеты помещали в FLIPR (Molecular Devices, Соединенное Королевство) для измерения флуоресценции на двух длинах волн (488 нм и 540 нм: Sullivan et al., Calcium Signaling Protocols, 1999, 125-136). Измерения проводили за 5 минут до добавления антагониста и через 10 минут после его введения. Максимальную флуоресценцию за вычетом исходной флуоресценции измеряли на 4 различных длинах волн. Анализ выполняли дважды. В этих условиях производные согласно настоящему изобретению были идентифицированы как антагонисты PAR-1 рецепторов (антагонизм >60% кальциевого сигнала при 10 мкМ). Кривые «доза-реакция» (от 0,01 до 32 мкМ), полученные с использованием SFLLR агониста, дали возможность определить эффективную концентрацию, вызывающую эффект, равный 50% от максимального эффекта (ЕС50). Эффективность (рА2) некоторых из антагонистов PAR-1, описанных в настоящем изобретении, была рассчитана с использованием способа Arunlakshana и Schild (Brit. J. Pharmacol., 1959, 14: 48-58) по сдвигам EC50, наблюдавшимся при трех концентрациях.
Результаты: несколько приведенных ниже примеров, выбранных из соединений согласно настоящему изобретению, иллюстрируют абсолютно неожиданную способность этих соединений быть антагонистами PAR-1 рецепторов.
Активность против агрегации тромбоцитов и антитромботическая активность антагонистов PAR-1 была показана на модели артериального тромбоза у морской свинки, которая была подвергнута очень сильному гемодинамическому сдвиговому напряжению. В сосудистом русле повреждение эндотелия вызывает внутрисосудистое образование обогащенного тромбоцитами тромба, который постепенно закупоривает весь просвет сосудов. Процесс агрегации тромбоцитов эффективно активируется тромбином через PAR-1 рецепторы. Соединения, описанные в настоящем изобретении, показали, что они способны оказывать антагонистический эффект на PAR-1 рецепторы и за счет этого задерживать образование тромба.
Материалы: исследования были выполнены на морских свинках (PAR-1 рецепторы которых сходны с рецепторами человека). Облучение светом зеленого лазера в присутствии фотосенсибилизирующего агента (бенгальский розовый, введенный внутривенно) повреждает эпителий сонных артерий. Кровоток через сонные артерии количественно оценивали с использованием датчика потока Transonic. Измеряли время, необходимое для полной закупорки сонной артерии (объемная скорость кровотока равна 0).
Способы: после обезболивания животного (60 мг/кг пентобарбитала) иссекали 5 мм сонной артерии и на 4 мм выше артерии помещали лазер. Датчик потока, размещенный выше по течению, измеряет время окклюзии. Бенгальский розовый (20 мг/кг) вводили внутривенно, а сосуд облучали излучением с длиной волны 514 нм (в течение 3 минут). Антагонисты PAR-1 вводили внутривенно болюсно (в течение 2 минут непосредственно перед введением бенгальского розового) с последующей 15-минутной перфузией, которую начинали одновременно с включением лазера.
Результаты: некоторые соединения, описанные в настоящем изобретении, продемонстрировали, что они способны после внутривенного введения в дозах от 0,16 до 2,5 мг/кг увеличивать время до образования тромба на 10-90% по сравнению с животными, получавшими только разбавитель.
Производные согласно настоящему изобретению можно также использовать для лечения фибрилляции предсердий.
В случае постинфарктной объемной перегрузки полости сердца правое и левое ушки предсердий расширяются, что образует основу для возникновения фибрилляции предсердий. Нарушение гомеостаза в полости расширенного ушка у пациента, страдающего фибрилляцией предсердий, приводит к аномальной концентрации тромбина. Авторы изобретения продемонстрировали, что эта аккумуляция тромбина ответственна за апрегуляцию PAR-1, который может запустить пролиферацию фибробластов и образование тромбоцитарного тромба.
В соответствии со своим механизмом действия антагонисты PAR-1 могут предотвратить дилатацию предсердий, пролиферацию фибробластов и образование тромба в ушке предсердия у пациента, страдающего фибрилляцией предсердий.
В результате антагонист PAR-1 представляет собой эффективное профилактическое и/или лечебное средство для фибрилляции предсердий. Соединения, описанные в настоящем изобретении, продемонстрировали, что они способны оказывать антагонистический эффект на PAR-1 рецепторы и предотвращать дилатацию предсердий.
Материалы: исследования были выполнены на крысах-самцах. Для эксперимента были выбраны крысы с массой тела 180-200 г, поскольку они лучше всего переносили хирургическую операцию. Измерения различных полостей в миокарде были выполнены с использованием эхокардиографии на наркотизированных животных.
Методы: животное наркотизировали 3,5%-ной смесью изофлурана с кислородом (Aerrane, Baxter Laboratories). Торакотомию размером около 2 см выполняли перпендикулярно грудине на уровне четвертого межреберного промежутка по направлению к левой передней лапе. Лигатуру (шелк 4-0, игла СС1, Ethicon) накладывали вокруг левой коронарной артерии на расстоянии 1 мм от ее начала. Вокруг левой коронарной артерии завязывали хирургический узел, достаточно тугой для полного пережатия сосуда. Непрерывная регистрация электрокардиограммы давала возможность подтвердить правильность размещения лигатуры. Через два месяца после процедуры животных снова наркотизировали для эхокардиографического измерения размеров полостей сердца и измерения скорости кровотока в миокарде с использованием импульсной доплерографии. После этого животных подвергали эвтаназии посредством передозировки пентобарбитала натрия (160 мг/кг, внутрибрюшинно) для выполнения различных гистологических измерений. Животным ежедневно перорально вводили продукты, являющиеся антагонистами PAR-1, начиная с 24 часов после инфаркта и до умерщвления животного.
Результаты: было продемонстрировано, что некоторые соединения, описанные в настоящем изобретении, способны после перорального введения в дозах 10-100 мг/кг/день в течение 60 дней уменьшать на 20-90% поверхность ушка предсердия (измеренную посредством эхокардиографии) по сравнению с животными, не получавшими лечения.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим в качестве активного ингредиента соединение общей формулы (I) или его фармацевтически приемлемую соль в смеси или в сочетании с подходящим наполнителем. Такие композиции могут, например, иметь форму твердых или жидких композиций, эмульсий, лосьонов или кремов.
В качестве твердых композиций для перорального введения могут быть использованы таблетки, пилюли, порошки (в желатиновых капсулах или в пакетиках) или гранулы. В таких композициях активный ингредиент согласно настоящему изобретению смешивают с одним или несколькими инертными разбавителями, такими как крахмал, целлюлоза, сахароза, лактоза или диоксид кремния, под потоком аргона. Такие композиции могут также включать в себя другие вещества, кроме разбавителей, например одно или несколько смазывающих веществ, таких как стеарат магния или тальк, краситель, покрытие (для пилюль, покрытых сахаром) или лак.
В качестве жидких композиций для орального введения можно использовать следующее: фармацевтически приемлемые растворы, суспензии, эмульсии, сиропы и эликсиры, содержащие инертные разбавители, такие как вода, этанол, глицерин, растительные масла или жидкий парафин. Такие композиции могут, кроме разбавителей, содержать другие вещества, например увлажнители, подсластители, загустители, вкусовые или стабилизирующие добавки.
Стерильные композиции для парентерального введения могут быть, предпочтительно, водными или неводными растворами, суспензиями или эмульсиями. В качестве растворителя или носителя можно использовать следующее: воду, пропиленгликоль, полиэтиленгликоль, растительные масла, более конкретно оливковое масло, пригодные для инъекций сложные органические эфиры, например этилолеат, или другие подходящие органические растворители. Такие композиции могут также содержать добавки, более конкретно смачивающие агенты, изотонические агенты, эмульгаторы, дисперганты и стабилизаторы. Стерилизацию можно выполнить несколькими способами, например посредством стерилизующей фильтрации, включения в композицию стерилизующих средств, облучения или нагревания. Такие композиции можно также приготовить в виде стерильных твердых композиций, которые можно растворить в стерильной воде или в любой другой стерильной среде, подходящей для инъекций, непосредственно перед употреблением.
Композиции для ректального введения представляют собой суппозитории или ректальные капсулы, которые содержат, кроме активного продукта, наполнители, такие как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Композиции для местного введения могут представлять собой, например, кремы, лосьоны, глазные капли, ополаскиватели для рта, капли в нос или аэрозоли.
Дозы зависят от желаемого эффекта, длительности лечения и пути введения и обычно лежат в диапазоне от 0,001 до 1 г (предпочтительно от 0,005 до 0,75 г) в день, предпочтительно при оральном пути введения взрослым людям, при этом разовые дозы варьируют от 0,1 до 500 мг активного вещества.
Обычно подходящую дозировку определяет лечащий врач в зависимости от возраста, веса пациента и других специфических факторов в каждом конкретном случае.
Согласно особому варианту осуществления настоящее изобретение также относится к продуктам, содержащим соединение общей формулы (I) и другой сердечно-сосудистый агент, то есть к комбинированным продуктам для одновременного, раздельного или отсроченного по времени использования для лечения сердечно-сосудистых заболеваний, причем другой сердечно-сосудистый агент может быть антитромбоцитарным средством, таким как аспирин, клопидогрел, тиклопидин, абциксимаб, тирофибан или эптифибатид.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЦИННАМОИЛ-ПИПЕРАЗИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ PAR-1 | 2007 |
|
RU2440997C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ | 2001 |
|
RU2286343C2 |
| НОВЫЕ АРИЛ- И ГЕТЕРОАРИЛПИПЕРАЗИНЫ | 2003 |
|
RU2361869C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2277909C2 |
| АНТАГОНИСТЫ АРИЛСУЛЬФОНАМИДА CCR3 | 2010 |
|
RU2539591C2 |
| ГЕТЕРОПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, АССОЦИИРОВАННЫХ С МЕТАБОТРОПНЫМИ ГЛУТАМАТНЫМИ РЕЦЕПТОРАМИ | 2000 |
|
RU2296127C9 |
| ХИНОКСАЛИНЫ И АЗАХИНОКСАЛИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА CRTH | 2011 |
|
RU2589709C2 |
| ДИАРИЛ-ЗАМЕЩЕННЫЕ 6,5-СОПРЯЖЕННЫЕ ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ C5aR | 2018 |
|
RU2796983C2 |
| ПРОИЗВОДНЫЕ ХРОМОНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2010 |
|
RU2545214C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗИН-3-АМИНА | 2011 |
|
RU2771819C2 |
Настоящее изобретение относится к соединениям общей формулы (I),
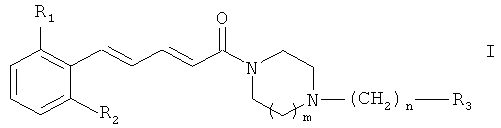
где R1 и R2, идентичные или различные, представляют собой: атом водорода или атом галогена, CN или NO2, причем R1 и R2 не являются атомами водорода одновременно; m означает: 1 или 2; n означает: 0, 1 или 2; R3 представляет собой: фенильный радикал, замещенный или не замещенный одним или несколькими радикалами, выбранными из атома галогена, гидроксильной группы или С1-С6 алкильной группы; С2-С6 алкильную группу, замещенную или не замещенную одним или несколькими радикалами, выбранными из атома галогена или гидроксильной группы; циклоалкильную группу; пиридин; тиофен; пиррол, замещенный или не замещенный C1-С6 алкильной группой; тиазол или фуран; или к их терапевтически приемлемым солям или сольватам. Кроме того, изобретение относится к способам получения соединений формулы I, к фармацевтическим композициям на основе этих соединений, обладающим антитромботической активностью, а также к применению данных соединений для изготовления лекарственного средства против агрегации тромбоцитов. 6 н. и 5 з.п. ф-лы.
1. Соединения общей формулы (I):
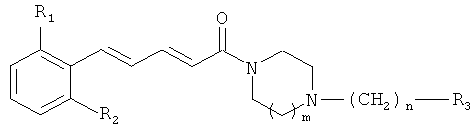
где
R1 и R2, идентичные или различные, представляют собой
атом водорода или атом галогена, CN или NO2, причем R1 и R2 не являются атомами водорода одновременно;
m означает 1 или 2;
n означает 0, 1 или 2;
R3 представляет собой
фенильный радикал, замещенный или не замещенный одним или несколькими радикалами, выбранными из атома галогена, гидроксильной группы или С1-С6 алкильной группы; С2-С6 алкильную группу, замещенную или не замещенную одним или несколькими радикалами, выбранными из атома галогена или гидроксильной группы; циклоалкильную группу; пиридин; тиофен; пиррол, замещенный или не замещенный C1-С6 алкильной группой; тиазол или фуран;
или их терапевтически приемлемые соли или сольваты.
2. Соединения по п.1, отличающиеся тем, что R1 является нитрогруппой, R2 является атомом водорода, m равно 1, n равно 0, и R3 является фенильной группой, замещенной одним или несколькими атомами галогенов или одной или несколькими С1-С6 алкильными группами, циклоалкильной группой или пиридином.
3. Соединения по п.1, отличающиеся тем, что R1 является цианогруппой, R2 является атомом водорода, m равно 1, n равно 0, и R3 является фенильной группой, замещенной одним или несколькими атомами галогенов или одной или несколькими C1-С6 алкильными группами, циклоалкильной группой или пиридином.
4. Соединения по п.1, отличающиеся тем, что они выбраны из:
2-[5-оксо-5-(4-пиридин-2-ил-пиперазин-1-ил)-пента-1,3-диенил]-бензонитрила;
2-[5-(4-циклопентилпиперазин-1-ил)-5-оксопента-1,3-диенил]-5-бензонитрила;
2-[5-(4-циклогексилпиперазин-1-ил)-5-оксопента-1,3-диенил]-бензонитрила;
2-{5-[4-(3-хлорпропил)-пиперазин-1-ил]-5-оксопента-1,3-диенил}-бензонитрила;
2-{5-[4-(3-хлорфенил)-пиперазин-1-ил]-5-оксопента-1,3-диенил}-бензонитрила;
2-{5-[4-(2-гидроксифенил)-пиперазин-1-ил]-5-оксопента-1,3-диенил}-бензонитрила;
2-{5-[4-(2,4-диметилбензил)-[1,4]диазепан-1-ил]-5-оксопента-1,3-диенил}-бензонитрила;
2-{5-[4-(2-метилбензил)-[1,4]диазепан-1-ил]-5-оксопента-1,3-диенил}-бензонитрила;
5-(2-хлорфенил)-1-(4-циклопентилпиперазин-1-ил)-пента-2,4-диен-1-она;
5-(2-хлорфенил)-1-(4-циклогексилпиперазин-1-ил)-пента-2,4-диен-1-она;
5-(2-хлорфенил)-1-(4-циклогептилпиперазин-1-ил)-пента-2,4-диен-1-она;
5-(2-хлорфенил)-1-[4-(3-хлорпропил)-пиперазин-1-ил]-пента-2,4-диен-1-она;
5-(2-хлорфенил)-1-(4-пиридин-2-ил-пиперазин-1-ил)-пента-2,4-диен-1-она;
5-(2-хлорфенил)-1-[4-(2-метилбензил)-[1,4]диазепан-1-ил]-пента-2,4-диен-1-она;
5-(2-хлорфенил)-1-[4-(2-фторбензил)-[1,4]диазепан-1-ил]-пента-2,4-диен-1-она;
5-(2-нитрофенил)-1-(4-фенилпиперазин-1-ил)-пента-2,4-диен-1-она;
1-(4-циклогексилпиперазин-1-ил)-5-(2-нитрофенил)-пента-2,4-диен-1-она;
1-(4-циклопентилпиперазин-1-ил)-5-(2-нитрофенил)-пента-2,4-диен-1-она;
1-[4-(4-фторфенил)-пиперазин-1-ил]-5-(2-нитрофенил)-пента-2,4-диен-1-она;
1-[4-(3-хлорпропил)-пиперазин-1-ил]-5-(2-нитрофенил)-пента-2,4-диен-1-она;
5-(2-нитрофенил)-1-(4-пиридин-2-ил-пиперазин-1-ил)-пента-2,4-диен-1-она;
1-(4-циклопентилметилпиперазин-1-ил)-5-(2-нитрофенил)-пента-2,4-диен-1-она;
5-(2-нитрофенил)-1-(4-тиофен-3-ил-метилпиперазин-1-ил)-пента-2,4-диен-1-она;
1-[4-(4-фторбензил)-пиперазин-1-ил]-5-(2-нитрофенил)-пента-2,4-диен-1-она;
1-(4-бутил-пиперазин-1-ил)-5-(2-нитрофенил)-пента-2,4-диен-1-она;
1-[4-(3-хлорфенил)-пиперазин-1-ил]-5-(2-нитрофенил)-пента-2,4-диен-1-она;
5-(2,6-дифторфенил)-1-(4-фенилпиперазин-1-ил)-пента-2,4-диен-1-она;
1-(4-циклогексилпиперазин-1-ил)-5-(2,6-дифторфенил)-пента-2,4-диен-1-она;
1-[4-(3-хлорпропил)-пиперазин-1-ил]-5-(2,6-дифторфенил)-пента-2,4-диен-1-она;
1-(4-циклопентилпиперазин-1-ил)-5-(2,6-дифторфенил)-пента-2,4-диен-1-она;
5-(2,6-дифторфенил)-1-[4-(4-фторбензил)-пиперазин-1-ил]-пента-2,4-диен-1-она;
1-(4-циклопентилпиперазин-1-ил)-5-(2-фторфенил)-пента-2,4-диен-1-она;
1-(4-циклогексилпиперазин-1-ил)-5-(2-фторфенил)-пента-2,4-диен-1-она;
5-(2-фторфенил)-1-(4-пиридин-2-ил-пиперазин-1-ил)-пента-2,4-диен-1-она;
5-(2-фторфенил)-1-(4-фенилпиперазин-1-ил)-пента-2,4-диен-1-она;
1-[4-(3-хлорпропил)-пиперазин-1-ил]-5-(2-фторфенил)-пента-2,4-диен-1-она;
и их терапевтически приемлемых солей и сольватов.
5. Соединения по любому из пп.1-4, отличающиеся тем, что их используют в качестве лекарственных средств для лечения и/или профилактики заболеваний, опосредованных активацией рецептора PAR-1.
6. Способ получения соединений общей формулы (I) по любому из пп.1-4, включающий конденсацию промежуточного соединения общей формулы (II):
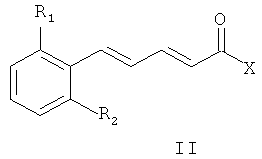
где R1 и R2 являются такими же, как в описании общей формулы (I) в п.1, X может представлять собой уходящую группу, такую как хлор, или X может быть гидроксильной группой, с амином общей формулы (III)
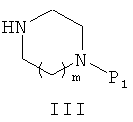
где P1 представляет собой защитную группу;
полученное промежуточное соединение общей формулы (IV)
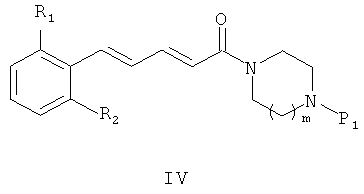
где R1, R2 и P1 являются такими же, как определено выше, дает соединения общей формулы (I) после депротекции и реакции полученного амина либо с реагентом общей формулы R3(CH2)nY, где R3 и n являются такими же, как определено в описании общей формулы (I) в п.1, a Y представляет собой уходящую группу, например - Cl, Br, I, OSO2CH3, OSO2CF3 или О-тозил, либо с альдегидом формулы R3-(CH2)n-1-CHO, где R3 и n являются такими же, как определено выше.
7. Способ получения соединений общей формулы (I) по любому из пп.1-4, включающий конденсацию промежуточного соединения общей формулы (II):
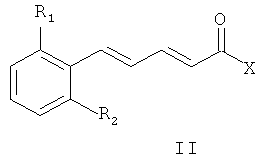
где R1 и R2 являются такими же, как в описании общей формулы (I) в п.1, X может представлять собой уходящую группу, такую как хлор, или X может быть гидроксильной группой, с амином общей формулы (V)
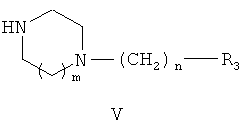
где m, n и R3 являются такими же, как в описании общей формулы (I) п.1, в результате которой получают соединения общей формулы (I).
8. Фармацевтические композиции, обладающие антитромботической активностью, содержащие в качестве активного продукта по меньшей мере одно соединение по любому из пп.1-4 в комбинации с фармацевтически приемлемым носителем.
9. Применение соединения по любому из пп.1-4 для изготовления лекарственного средства против агрегации тромбоцитов, для лечения и/или профилактики артериальных или венозных тромбозов, стабильной стенокардии, нарушений сердечного ритма, церебральных сосудистых явлений, сердечной недостаточности, гипертензии или инфаркта миокарда, острых коронарных синдромов, для лечения рестеноза, для лечения и/или профилактики фибрилляции предсердий и ремоделирования миокарда, для лечения и/или профилактики воспалительных заболеваний, легочных заболеваний, желудочно-кишечных заболеваний, развития фиброза у пациентов с хроническим заболеванием печени или кожных болезней, или для лечения и/или профилактики рака.
10. Продукт, содержащий по меньшей мере одно соединение по любому из пп.1-4 и другой сердечно-сосудистый агент, в виде комбинированного продукта для одновременного, раздельного или отсроченного применения при лечении сердечно-сосудистых заболеваний.
11. Продукт по п.10, отличающийся тем, что другой сердечно-сосудистый агент представляет собой агент против агрегации тромбоцитов, такой как аспирин, клопидогрел, тиклопидин, абциксимаб, тирофибан или эптифибатид.
| Derwent Publications Ltd., London, GB; AN 1986-207178 Derwent Publications Ltd., London, GB; AN 1986-173349 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Med | |||
| Chem, 2003, 1, 37-45 | |||
| RU 2002122335 A, 10.01.2004. | |||

