Соль-мезилат N-{5-[4-(4-метилпиперазинометил)-бензоламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина (соединение формулы I, Иматиниб) продается на рынке под торговой маркой Гливек (англ. Glivec®, Gleevec®). Гливек является ингибитором тирозинкиназы, подходящим для лечения хронической миелоидной лейкемии и злокачественной стромальной опухоли желудочно-кишечного тракта. N-{5-[4-(4-метилпиперазинометил)-бензоламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамин может быть получен, например, как описано в патентной заявке США №5521184. Соль-мономезилат N-{5-[4-(4-метил-пиперазино-метил)-бензоламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина может быть получена и охарактеризована, например, как описано в примерах 4 и 6 патентной заявки № WO 99/03854 или как описано в патентной заявке № WO 03/090720.
Настоящее изобретение относится к новым способам получения N-{5-[4-(4-метилпиперазинометил)бензоламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина (соединение формулы I, Иматиниб), новым способам получения метаболитов N-{5-[4-(4-метил-пиперазино-метил)-бензоламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина, обнаруженным после введения соединения согласно настоящему изобретению теплокровным животным, также как и к промежуточным соединениям, получающимся согласно данным способам. Новые исходные вещества, как и способы их получения, также являются предметом настоящего изобретения. Способы, описанные в настоящей заявке, являются особенно подходящими для получения вышеописанных соединений, меченных радиоактивными изотопами. Полученные указанным выше образом меченные соединения являются, в частности, подходящими для отслеживания и исследования метаболизма N-{5-[4-(4-метилпиперазинометил)-бензоламидо]-2-метилфенил}-4-(3-пиридил)-2-пиримидинамина и его фармацевтически приемлемых солей при клиническом и доклиническом исследованиях.
Основываясь на описании настоящей патентной заявки, специалист в области техники способен получить N-оксидные производные соединений, описанных в настоящей заявке, отличные от явно упомянутых N-оксидных производных, производные-пролекарства, защищенные производные, индивидуальные изомеры и смеси таких изомеров, также как и их фармацевтически приемлемые соли.
Согласно первому варианту осуществления настоящего изобретения соль-мезилат соединения формулы I
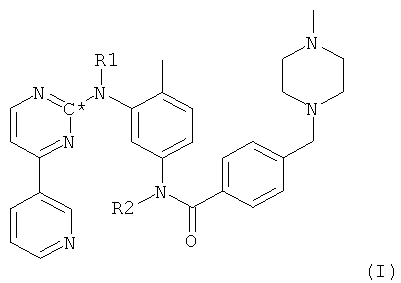
в которой R1 и R2 оба представляют собой водород и С* означает атом углерода, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся меченным более высокой концентрацией одного из изотопов углерода, например 14С, получают путем взаимодействия 1-метилпиперазина с соединением формулы II,
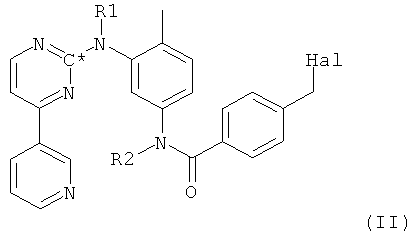
в которой Hal представляет собой галоген, предпочтительно хлор, и в которой остальные радикалы и символы имеют значения, определенные выше для соединения формулы I.
Соединение формулы II, в которой Hal представляет собой галоген, предпочтительно хлор, и в которой остальные радикалы и символы имеют значения, определенные выше для соединения формулы I, может быть получено посредством взаимодействия на первой стадии соли-продукта присоединения хлороводородной кислоты, имеющей общую формулу III,
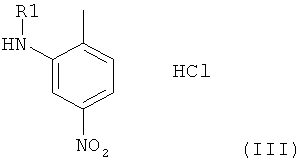
в которой R1 имеет значение, определенное выше для соединения формулы I, с соединением H2NC*N, в котором С* означает атом углерода, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся меченным более высокой концентрацией одного из изотопов, например 14С, с получением при этом гуанидинилзамещенного нитрофенильного производного формулы IV
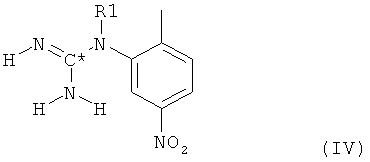
в которой радикалы и символы имеют значения, определенные выше для соединения формулы I. На второй стадии гуанидинилзамещенное нитрофенильное производное формулы IV далее восстанавливают до соответствующего гуанидинилзамещенного аминофенильного производного формулы V
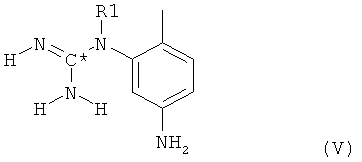
в которой радикалы и символы имеют значения, определенные выше для соединения формулы I. На третьей стадии посредством взаимодействия такого гуанидинилзамещенного аминофенильного производного формулы V с пиридильным производным формулы VI,
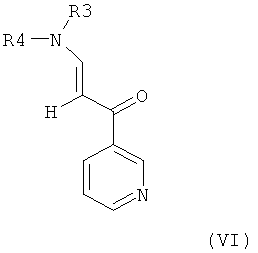
в которой R3 и R4 оба представляют собой С1-4алкил, получают пиридилпиримидин формулы VII,
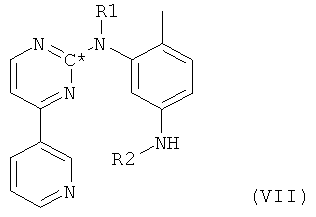
в которой радикалы и символы имеют значения, определенные выше для соединения формулы I. На последней стадии такой пиридилпиримидин формулы VII реагирует с 4-галогенметилбензойной кислотой с получением при этом амида формулы II.
Пиридилпиримидин формулы VII, в котором радикалы и символы имеют значения, определенные выше для соединения формулы I, может также быть получен согласно следующей методике. На первой стадии соединение формулы I, в которой радикалы и символы имеют значения, определенные выше, взаимодействует с реагентом, замещающим атомы водорода, связанные с атомами азота, на защитные группы с получением при этом соединения формулы I
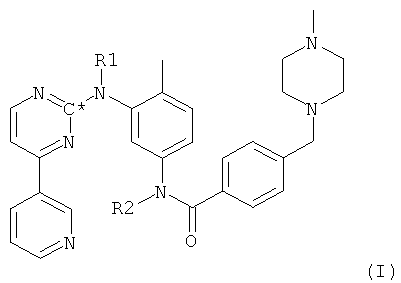
в которой оба R1 и R2 представляют собой защитные группы и в которой С* означает атом углерода, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся меченным более высокой концентрацией одного из изотопов, например 14С. На второй стадии из молекулы посредством гидролиза удаляют часть, соответствующую бензойной кислоте, получая при этом свободный амин формулы XII,
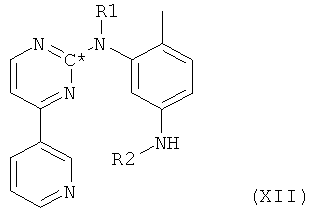
в которой оба R1 и R2 представляют собой защитные группы и в которой С* означает атом углерода, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся помеченным более высокой концентрацией одного из изотопов, например 14С, с последующим замещением защитных групп R1 и R2 на водород, т.е. снятием защитных групп, с получением при этом соединения формулы II, в которой радикалы и символы имеют значения, определенные выше для соединения формулы I.
Согласно второму воплощению настоящего изобретения, соединение формулы VIII
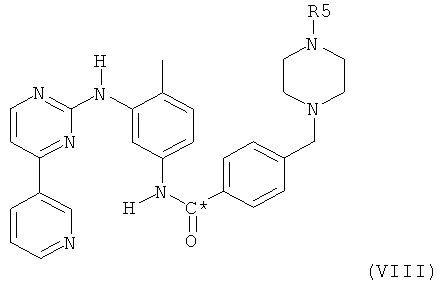
в которой R5 представляет собой водород или C1-4алкил и в которой С* означает атом углерода, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся меченным более высокой концентрацией одного из изотопов, например 14С, может быть получено посредством взаимодействия соединения формулы X,
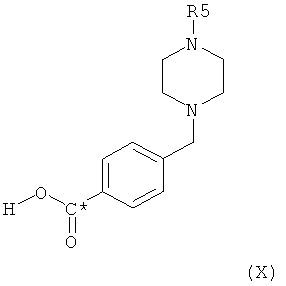
в которой R5 представляет собой защитную группу, например трет-бутоксикарбонил, или С1-4алкил, и С* означает атом углерода, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся меченным более высокой концентрацией одного из изотопов, например 14С, с соединением формулы XI,
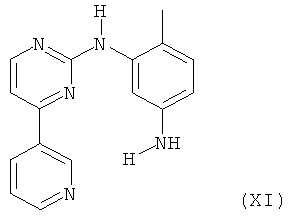
с последующим отделением защитной группы R5 в том случае, когда R5 представляет собой защитную группу.
Соединение формулы X,
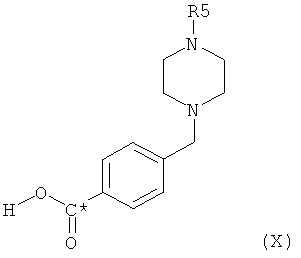
в котором радикалы и символы имеют значения, определенные выше, может быть получено посредством взаимодействия на первой стадии соединения формулы IX
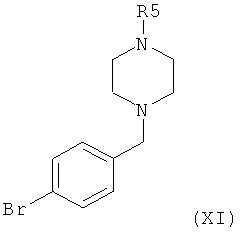
в которой R5 представляет собой защитную группу, например трет-бутоксикарбонил, или С1-4алкил, с трет-бутиллитием, с получением при этом соответствующего литиопроизводного, и затем взаимодействия этого литиопроизводного с соединением формулы С*O2, в которой С* означает атом углерода, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся меченным более высокой концентрацией одного из изотопов, например 14С.
Согласно другому варианту осуществления настоящее изобретение относится к способу получения соединения формулы XIII
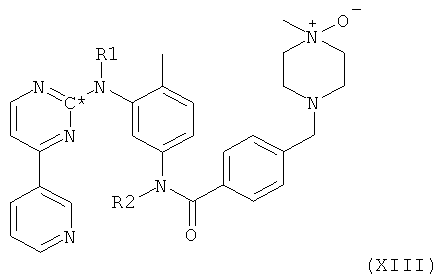
в которой радикалы и символы имеют значения, определенные выше для соединения формулы I, посредством окисления соединения формулы I, в которой радикалы и символы имеют значения, определенные выше.
Согласно своему дополнительному аспекту настоящее изобретение относится к получению соединения формулы XIV
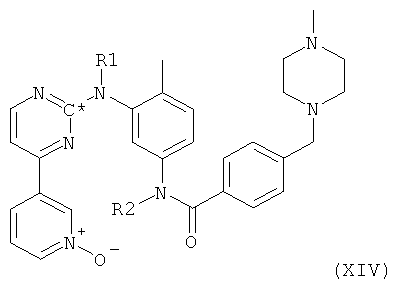
в которой радикалы и символы имеют значения, определенные выше для соединения формулы I, посредством взаимодействия соединения формулы XVI,
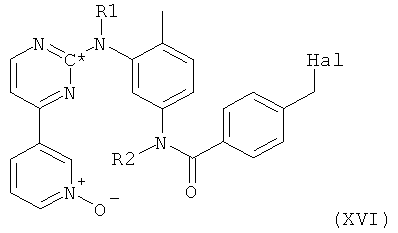
в которой Hal представляет собой галоген, предпочтительно хлор, и в которой все остальные радикалы и символы имеют значения, определенные выше для соединения формулы I, с 1-метилпиперазином.
Исходное вещество - соединение формулы XVI, в которой Hal представляет собой галоген и в которой все остальные радикалы и символы имеют значения, определенные выше для соединения формулы I, может быть получено согласно следующей методике. На первой стадии свободный амин формулы VII, в которой R1 и R2 оба представляют собой водород, реагирует с агентом, вводящим защитную группу, в таких условиях, что происходит селективное замещение одного атома водорода в первичной аминогруппе в присутствии вторичной аминогруппы, с последующим окислением атома азота, входящего в пиридильную группу, например, с таким агентом, как МСРВА, с получением при этом N-оксида формулы XV,
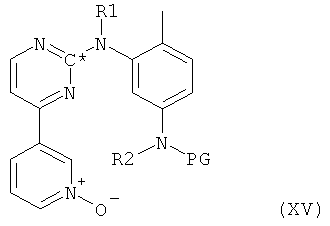
в которой оба R1 и R2 представляют собой водород, PG представляет собой защитную группу и все остальные символы имеют значения, определенные выше для соединения формулы I. Такой N-оксид формулы XV сначала помещают в условия, способствующие снятию защитной группы PG, и затем полученный свободный амин подвергают взаимодействию с 4-галогенметилбензойной кислотой с получением при этом соединения формулы XVI, в которой Hal представляет собой галоген, и все остальные радикалы и символы имеют значения, определенные выше для соединения формулы I.
Согласно другому варианту осуществления настоящего изобретения представлено соединение формулы XVIII
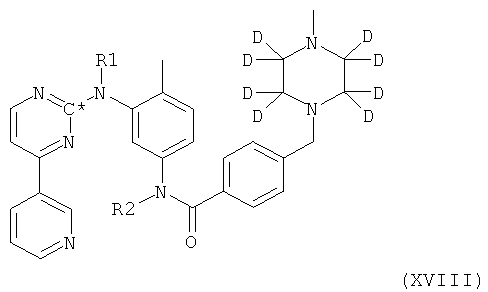
в которой радикалы и символы имеют значения, определенные выше для соединения формулы I, полученное посредством взаимодействия соединения формулы II,
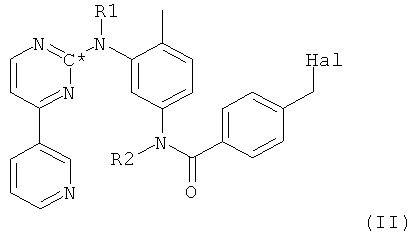
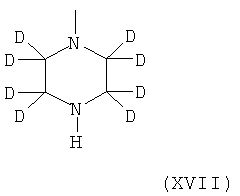
в которой Hal представляет собой галоген, предпочтительно хлор, и все остальные радикалы и символы имеют значения, определенные выше для соединения формулы I, с пиперазиновым производным формулы XVII.
Согласно своему последнему воплощению настоящее изобретение относится к получению соединения формулы XX
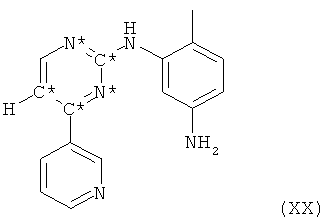
в которой С* означает атом углерода, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся меченным более высокой концентрацией одного из изотопов, например 13С, и N* означает атом азота, имеющий естественное распределение изотопов или, в качестве альтернативы, являющийся меченным более высокой концентрацией одного из изотопов, например 15N, включающему взаимодействие пиридинового производного формулы XXI
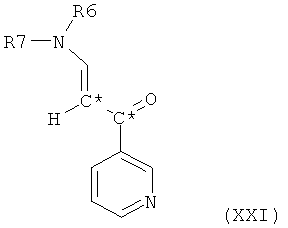
в которой оба R6 и R7 представляют собой С1-4алкил, и все остальные символы имеют значения, определенные выше для соединения формулы XX, с фенилгуанидиновым производным формулы XXII,
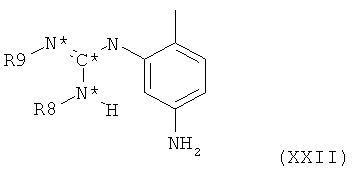
в которой оба R8 и R9 представляют собой водород и все остальные символы имеют значения, определенные выше для соединения формулы XX.
Пиридиновое производное формулы XXI, в которой оба R6 и R7 представляют собой С1-4алкил и все остальные символы имеют значения, определенные выше для соединения формулы XX, может быть получено, исходя из 3-триметилстаннилпиридина, посредством его взаимодействия с ацетилгалогенидом формулы XXIV,
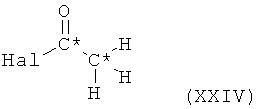
в которой Hal означает галоген и все остальные символы имеют значения, определенные выше для соединения формулы XX, с получением при этом ацетилпиридина формулы XXIII
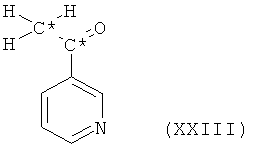
в которой все символы имеют значения, определенные выше для соединения формулы XX. Такой ацетилпиридин формулы XXIII подвергают дальнейшему взаимодействию с ди-С1-4алкилацеталем ди-С1-4алкилформамида, с получением при этом требуемого пиридинового производного формулы XXI, в которой R6 и R7 оба представляют собой С1-4алкил и все остальные символы имеют значения, определенные выше для соединения формулы XX.
Аминофенилгуанидиновое производное формулы XXII, в которой оба R8 и R9 представляют собой водород и все остальные символы имеют значения, определенные выше для соединения формулы XX, может быть получено, исходя из тиомочевинового производного формулы XXV,
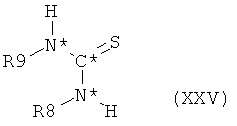
в которой R8 и R9 оба представляют собой водород и все остальные символы имеют значения, определенные выше для соединения формулы XX, посредством взаимодействия такого тиомочевинового производного формулы XXV с реагентом, осуществляющим замещение одного атома водорода каждой аминогруппы на защитную группу, например, такую как трет-бутилоксикарбонил, с получением при этом тиомочевинового производного формулы XXV, в которой R8 и R9 оба представляют собой защитную группу. Такое тиомочевиновое производное формулы XXV затем подвергают взаимодействию с 2-метил-5-нитроанилином с получением при этом нитрофенилгуанидинового производного формулы XXVI,
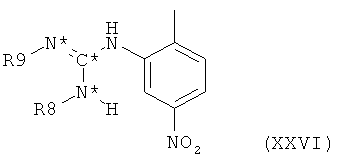
в которой оба R8 и R9 представляют собой защитную группу и все остальные символы имеют значения, определенные выше для соединения формулы XX. Отщепление защитных групп с последующим восстановлением нитрогруппы приводит к получению аминофенилгуанидинового производного формулы XXII, в которой оба R8 и R9 представляют собой водород и все остальные символы имеют значения, определенные выше для соединения формулы XX.
Все остальные исходные вещества или известны, или могут быть получены согласно известным методикам, или являются коммерчески доступными, в частности они могут быть получены способами, описанными в примерах.
Все стадии способов, описанные в настоящей заявке, могут быть проведены в известных условиях реакций, предпочтительно, в тех условиях, которые явно указаны, в отсутствие или обычно в присутствии растворителей или разбавителей, предпочтительно таких, которые инертны по отношению к используемым реагентам и способны их растворять, в отсутствие или в присутствии катализаторов, конденсирующих или нейтрализующих агентов, например ионообменников, например, в форме Н+, в зависимости от типа реакции и/или реагентов, при пониженной, нормальной или повышенной температуре, например, в диапазоне от -100°С до приблизительно 190°С, предпочтительно от приблизительно -80°С до приблизительно 150°С, например, от -80 до -60°С, при комнатной температуре, от - 20 до 40°С или при температуре кипения используемого растворителя, при атмосферном давлении или в закрытом сосуде, в подходящих случаях под давлением и/или в инертной атмосфере, например в атмосфере аргона или азота. Растворители, из которых осуществляют выбор тех, которые подходят для рассматриваемой реакции, включают, например, воду, сложные эфиры, обычно низший алкил-низший алканоат, например, диэтилацетат, простые эфиры, обычно алифатические простые эфиры, например, диэтиловый эфир, или циклические эфиры, например, тетрагидрофуран, жидкие ароматические углеводороды, обычно бензол или толуол, спирты, обычно метанол, этанол или 1- или 2-пропанол, нитрилы, обычно ацетонитрил, галогенированные углеводороды, обычно дихлорметан, амиды кислот, обычно диметилформамид, основания, обычно гетероциклические азотистые основания, например, пиридин, карбоновые кислоты, обычно низшие алканкарбоновые кислоты, например, уксусную кислоту, ангидриды карбоновых кислот, обычно ангидриды низших алканкарбоновых кислот, например, уксусный ангидрид, циклические, линейные или разветвленные углеводороды, обычно циклогексан, гексан, или изопентан, или смеси таких растворителей, например, водные растворы, если иное не указано в описании способа. Такие смеси растворителей могут также быть использованы при дальнейшей обработке, например при хроматографии или распространении.
Соединения согласно настоящему изобретению могут быть получены в виде фармацевтически приемлемых солей-продуктов присоединения кислоты посредством взаимодействия соединений в форме свободных оснований с фармацевтически приемлемыми неорганическими или органическими кислотами. В качестве альтернативы, фармацевтически приемлемая соль-продукт присоединения основания может быть получена посредством взаимодействия соединения в форме свободной кислоты с фармацевтически приемлемым неорганическим или органическим основанием. В качестве альтернативы, соединения согласно настоящему изобретению в форме солей могут быть получены посредством использования солей исходных веществ или промежуточных соединений.
Соединения согласно настоящему изобретению в форме свободной кислоты или свободного основания могут быть получены из соответствующих солей-продуктов присоединения основания или солей-продуктов присоединения основания, соответственно. Например, соединение согласно настоящему изобретению в форме соли-продукта присоединения кислоты может быть превращено в соответствующую форму свободного основания посредством обработки подходящим основанием (например, раствором гидроксида аммония, гидроксида натрия и подобными им). Соединение согласно настоящему изобретению в форме соли-продукта присоединения основания может быть превращено в соответствующую свободную кислоту посредством обработки подходящей кислотой (например, хлороводородной кислотой, и подобной ей).
Соединения согласно настоящему изобретению в неокисленной форме могут быть получены из N-оксидов соединений согласно настоящему изобретению посредством обработки восстанавливающим агентом (например, серой, диоксидом серы, трифенилфосфином, боргидридом лития, боргидридом натрия, трихлоридом фосфора, трибромидом фосфора и им подобными) в подходящем инертном органическом растворителе (например, ацетонитриле, этаноле, водном диоксане, и им подобными) при температуре от 0 до 80°С.
Производные-пролекарства соединений согласно настоящему изобретению могут быть получены способами, известными специалисту в данной области техники (например, для дополнительной информации см. Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol.4, p.1985). Например, подходящие пролекарства могут быть получены посредством взаимодействия неизмененного соединения согласно настоящему изобретению с подходящим карбамилирующим агентом (например, с 1,1-ацилоксиалкилкарбанохлоридатом, пара-нитрофенилкарбонатом и им подобными).
Защищенные производные соединений согласно настоящему изобретению могут быть получены способами, известными специалисту в данной области техники. Подробное описание способов, подходящих для получения защитных групп и их удаления может быть найдено в книге Т. W. Greene, "Protecting Groups in Organic Chemistry", 3rd edition, John Wiley and Sons, Inc., 1999.
Соединения согласно настоящему изобретению могут быть легко получены или преобразованы в процессе осуществления способов согласно настоящему изобретению в виде сольватов (например, гидратов). Гидраты соединений согласно настоящему изобретению могут быть легко получены посредством перекристаллизации из смеси водных/органических растворителей, с использованием органических растворителей, таких как диоксин, тетрагидрофуран или метанол.
Соединения согласно настоящему изобретению могут быть получены в виде индивидуальных стереоизомеров посредством взаимодействия рацемической смеси соединения согласно настоящему изобретению с оптически активным разделяющим агентом с получением при этом пары диастереомерных соединений, разделением диастереометров и получением оптически чистых энантиомеров. В то время как разделение энантиомеров может быть проведено с использованием ковалентных диастереомерных производных соединений согласно настоящему изобретению, предпочтительными являются диссоциирующие комплексы (например, кристаллические диастереомерные соли). Диастереомеры имеют различные физические характеристики (например, температуры плавления, температуры кипения, растворимости, реакционную способность, и так далее) и могут быть легко разделены с использованием таких различий. Диастереомеры могут быть разделены посредством хроматографии, или, предпочтительно, посредством методик разделения, основанных на различиях в растворимости. После этого любым подходящим способом, не приводящим к рацемизации, восстанавливают оптически чистый энантиомер вместе с разделяющим агентом. Более подробное описание методик, подходящих для разделения стереоизомеров из рацемической смеси может быть найдено в книге Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley And Sons, Inc., 1981.
Подробное описание синтеза соединений, описанных выше, приведено в примерах. Согласно предпочтительному воплощению, соединения, описанные выше, получают согласно или по аналогии со способами и их стадиями, определенными в примерах.
Примеры
Далее настоящее изобретение более подробно описывается, но не ограничивается, примерами, которые иллюстрируют получение соединений, описанных выше.
Сокращения
ДМАП диметиламинопиридин
ДМСО диметилсульфоксид
МСРВА мета-хлорпербензойная кислота
МеОН метанол
ЯМР ядерный магнитный резонанс
RT комнатная температура
ТГФ тетрагидрофуран
ТСХ тонкослойная хроматография
м/о мас./об.
Пример 1: 4-(4-метилпиперазин-1-илметил)-N-[4-метил-3-(4-пиридин-3-ил[2-14С]пиримидин-2-иламино)фенил]бензамид мезилат
1-Метилпиперазин (3 мл, 27 ммоль) добавляют к раствору соли-гидрохлорида 4-хлорметил-N-{4-метил-3-[4-(1-оксипиридин-3-ил)-[2-14С]пиримидин-2-иламино]фенил}бензамида (стадия 1,5, 335 мг, 0,71 ммоль) в этаноле (20 мл) и полученную смесь перемешивают в течение 5 ч при 45°С. Суспензию охлаждают до комнатной температуры и добавляют воду (4,4 мл), а затем 20% м/о гидроксида натрия (130 мкл). Полученную суспензию перемешивают 30 минут при 90°С, затем охлаждают до комнатной температуры, центрифугируют и удаляют водный растворитель. Бледноватые кристаллы дополнительно промывают 2 раза водой и высушивают под глубоким вакуумом. Твердое вещество окончательно очищают флеш-хроматографией на силикагеле, элюируя дихлорметаном:МеОН 90:10 и 1% аммония с получением при этом промежуточного соединения в виде желтого маслянистого вещества. Наконец, полученное промежуточное вещество растворяют в МеОН (4 мл), добавляют метансульфоновую кислоту (21 мкл) и полученные растворы перемешивают. Полученный продукт перекристаллизовывают из раствора посредством внесения затравки неактивного контрольного продукта с получением при этом указанного в заголовке соединения с удельной радиоактивностью 1,73 ГБк/ммоль.
1Н-ЯМР 400 МГц (ДМСО-d6) δ: 2,21 (s, 3Н); 2,3 (s, 3H); 2,8 (s, 3H); 2,85-3,1, m, 4H); 3,25-3,4 (m, 4H); 3,62 (br.s, 3H); 7,18 (d, J=8 Гц; 1Н); 7,38-7,51 (m, 4H); 7,91 (d, 7,6 Гц, 1Н); 8,5 (d, J=1,4 Гц, 1Н); 8,45 (d, J=7,8, 1Н); 8,49 (d, J=5 Гц, 1Н); 8,65 (dd, J=1,5 и 4,6 Гц, 1Н); 8,95 (s, 1Н); 9,22 (d, J=1,5 Гц, 1H); 9,29 (br.s, 1Н), 10,12 (s, 1Н); MS: (MH+, 496,4).
Стадия 1,1: [14С]цианамид
[14С]карбонат бария (1,18 г, 6 ммоль, 10,4 ГБк, 1,79 ГБк/ммоль) помещают в цилиндрическую трубку из кварцевого стекла и нагревают до 800°С при пропускании небольшого потока газообразного аммиака. Через 3 часа необработанный продукт- [14С]цианамид бария оставляют для охлаждения до комнатной температуры и прекращают подачу аммиака. Необработанный [14С]цианамид бария (1,063 г, 6 ммоль) суспендируют в воде (10 мл) и растворяют посредством обработки ультразвуком. Осторожно по каплям при перемешивании добавляют 2М серную кислоту (2,6 мл) до тех пор, пока значение рН не достигнет 5,5. Удаляют воду при 60°С и давлении 23 Торр. Остаток охлаждают на сухом льде/ацетоне и остаточную воду лиофилизуют с получением при этом кристаллического твердого вещества. Затем его помещают в диэтиловый эфир и отфильтровывают для удаления нерастворимого твердого остатка. Удаляют эфир и полученное твердое вещество растворяют в трет-бутаноле.
Стадия 1,2: N-(2-метил-5-нитрофенил)-[14С]гуанидин
К перемешиваемому раствору 2-метил-4-нитроанилина (1,52 г, 19 ммоль) в трет-бутаноле при 40°С добавляют 4М хлороводородную кислоту в диоксане (5 мл) с получением при этом желтой суспензии. Растворитель удаляют и сухой остаток добавляют к раствору необработанного [14С]цианамида (стадия 1,2) в трет-бутаноле (5 мл), получая при этом желтую суспензию. После нагревания до 100°С в течение 1 часа, получают раствор. Еще через 5 часов удаляют растворитель при 45°С и остаток обрабатывают 3% м/о гидроксидом натрия (10 мл) и экстрагируют дихлорметаном (3×30 мл). Органические фазы объединяют и выпаривают, и хроматографируют на силикагеле, элюируя смесью 75:25:0,5:0,5 дихлорметан:МеОН:вода:уксусная кислота, получая при этом указанное в заголовке соединение, 6,07 ГБк.
Стадия 1,3: N-(5-амино-2-метил-фенил')-[14С]гуанидин
Смесь N-(2-метил-5-нитрофенил)[14С]гуанидина (стадия 1,2) (760 мг, 3,91 ммоль) в н-бутаноле (25 мл), содержащую 10% Pd/C (125 мг), перемешивают в атмосфере водорода под давлением 1 атмосфера при комнатной температуре в течение 4,5 часов. Полученную смесь фильтруют через целит Хайфло (Hyflo), промывают дополнительной порцией н-бутанола (3×10 мл), объединяют и удаляют н-бутанол на роторном испарителе при 45°С, получая при этом необработанный продукт в виде бесцветного маслянистого вещества. Радио ТСХ: 80:25:0,5:0,5 дихлорметан:МеОН:вода:уксусная кислота, силикагелевые пластинки F254, подвижность RF=0,34; общая радиоактивность 3,7 ГБк.
Стадия 1.4: 4-метил-N*3*-(4-пиридин-3-ил-[2-14С]пиримидин-2-ил)-бензол-1,3-диамин
Раствор необработанного N-(5-амино-2-метил-фенил)-[14С]гуанидина (стадия 1,4, 476 мг, 2,89 ммоль) и 3-диметиламино-1-пиридин-3-илпропенона (509 мг, 2,89 ммоль) в н-бутаноле (30 мл) нагревают с обратным холодильником (130°С) в течение 3,5 часов. После удаления растворителя и очистки флеш-хроматографией с элюированием смесью 2:98 пентан:ацетон получают указанное в заголовке соединение в виде желтого вспененного вещества. РадиоТСХ, 2:98 пентан:ацетон, силикагелевые пластинки F254, подвижность RF=0,5. Общая радиоактивность 2,3 ГБк.
Стадия 1,5: 4-хлорметил-N-[4-метил-3-(4-пиридин-3-ил-[2-14С]пиримидин-2-иламино)фенил]бензамид
К раствору 4-метил-N*3*-(4-пиридин-3-ил-[2-14С]пиримидин-2-ил)-бензол-1,3-диамина (стадия 1,4, 655 мг, 2,36 ммоль) в сухом ТГФ (20 мл) в атмосфере аргона при 0°С по каплям добавляют раствор 4-(хлорметил)бензоилхлорида (2,646 г, 14 ммоль) в ТГФ (10 мл) и соответствующую смесь перемешивают при комнатной температуре в течение 16 ч. Полученную суспензию центрифугируют, продукт-осадок промывают эфиром, снова центрифугируют, удаляют эфир и оставшееся твердое вещество высушивают с получением при этом соль-гидрохлорид указанного в заголовке соединения.
Пример 2: гидрохлорид 4-(пиперазин-1-илметил)-N-[4-метил-3-(4-пиридин-3-ил-[2-14С]пиримидин-2-иламино)фенил]бензамида
К трет-бутиловому эфиру 4-{4-[4-метил-3-(4-пиридин-3-ил-пиримидин-2-иламино)-фенил[14С]карбамоил]бензил}пиперазин-1-карбоновой кислоты (стадия 2,3, 200,5 мг, 0,536 ммоль, 486,8 МБк) добавляют 3М НСl в МеОН (20 мл). Через 30 минут добавляют дополнительно 10 мл 3М НСl в МеОН и реакционную смесь перемешивают еще в течение 60 минут. Продукт отфильтровывают посредством вакуумного отсасывания через стеклокерамический фильтр и промывают сначала холодным МеОН (5 мл), а затем этилацетатом (100 мл) и оставляют сушиться под глубоким вакуумом с получением при этом указанного в заголовке соединения в виде оранжевых кристаллов.
Стадия 2,1: трет-бутиловый эфир 4-(4-бромбензил)-пиперазин-1-карбоновой кислоты
Смесь N-boc-пиперазина (4,94 г, 26,5 ммоль), пара-бромбензилбромида (5,52 г, 22 ммоль) и К2СО3 (6,4 г, 46 ммоль) в МеОН (50 мл) перемешивают в течение 90 минут при комнатной температуре. Смесь отфильтровывают через стеклокерамический фильтр и растворитель выпаривают. Остаток помещают в дихлорметан, второй раз отфильтровывают через стеклокерамический фильтр и промывают дихлорметаном. Фильтрат упаривают и необработанное бесцветное маслянистое вещество хроматографируют на силикагеле, элюируя смесью диэтиловый эфир/н-гексан (от 10:90 до 50:50) с получением при этом указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2,2: трет-бутиловый эфир 4-(4-14С]карбоксибензил)пиперазин-1-карбоновой кислоты
1,5 М Раствор трет-бутиллития в гексане (0,97 мл, 1,46 ммоль) добавляют при -78°С к раствору трет-бутилового эфира 4-(4-бромбензил)пиперазин-1-карбоновой кислоты (стадия 2,1, 285 мг, 0,8 ммоль) в безводном ТГФ (5 мл) и смесь перемешивают при -78°С в течение 4 минут. Полученное литиопроизводное охлаждают до -192°С и подвергают взаимодействию с диоксидом [14С]углерода, полученного добавлением концентрированной серной кислоты к [14С]карбонату бария (0,85 экв, 122 мг, 0,62 ммоль, 1378 МБк, произведен фирмой «Amersham Pharmacia Biotech») и затем подогревают до -78°С и перемешивают в течение 30 минут. Реакцию гасят добавлением МеОН (1 мл) при -78°С, и затем дают нагреться до комнатной температуры. Растворитель удаляют в вакууме и добавляют насыщенный раствор хлорида аммония. Смесь экстрагируют этилацетатом (3×15 мл), объединяют и высушивают над сульфатом натрия. После фильтрации и удаления растворителя получают необработанное твердое белое вещество. Необработанный продукт хроматографируют на силикагеле, элюируя сначала диэтиловым эфиром, а затем смесью 10% МеОН и 90% диэтилового эфира. Фракции объединяют, выпаривают сначала на роторном испарителе, и затем под глубоким вакуумом, получая указанное в заголовке соединение в виде белого твердого вещества.
Стадия 2,3: трет-бутиловый эфир 4-{4-[4-Метил-3-(4-пиридин-3-ил-пиримидин-2-иламино)-фенил[14С]карбамоил]бензил}пиперазин-1-карбоновой кислоты
К перемешиваемому раствору трет-бутилового эфира 4-(4-[14С]карбоксибензил)пиперазин-1-карбоновой кислоты (стадия 2,2, 171,5 мг, 0,536 ммоль, 1114,26 МБк) в ТГФ (5 мл) при комнатной температуре по каплям добавляют триэтиламин (332 мкл, 2,4 ммоль) и затем по каплям добавляют изобутилхлорформиат (78 мкл, 1,0 ммоль). Смесь дополнительно перемешивают в течение 3 минут, и затем добавляют 4-метил-N-3-(4-пиридин-3-ил-пиримидин-2-ил)-бензол-1,3-диамин (165 мг, 1,0 ммоль). Через 18 часов раствор упаривают и остаток подвергают флеш-хроматографии на силикагеле, элюируя сначала этилацетатом, а затем смесью 10% МеОН / 90% этилацетат, затем смесью 35% МеОН / 65% этилацетат, выделяя указанное в заголовке соединение в виде светло-желтого твердого вещества.
Пример 3: 4-метил-N*3*-(4-пиридин-3-ил-[2-14C]пиримидин-2-ил)-бензол-1,3-диамин
Раствор трет-бутилового эфира (5-трет-бутоксикарбониламино-2-метил-фенил)-(4-пиридин-3-ил-[2-14С]пиримидин-2-ил)-карбаминовой кислоты (стадия 3,2, 86 мг, 0,18 ммоль) и трифторуксусной кислоты (463 мг, 310 мкл, 4,06 ммоль) перемешивают в течение ночи при комнатной температуре. На основании активного контроля (РТСХ, силикагель, CH2Cl2:МеОН 95:5) делают вывод, что исходные вещества не обнаруживаются. Растворитель удаляют в вакууме и необработанный материал разделяют между 1 н. водным раствором НСl и CH2Cl2. После переэкстрагирования органической фазы объединяют кислые водные фракции, добавляют 2 н. водный раствор NaOH и три раза экстрагируют CH2Cl2. После промывания органической фазы насыщенным соляным раствором органическую фазу высушивают над Na2SO4 и выпаривают растворитель. Необработанное вещество (48 мг) очищают флеш-хроматографией (силикагель, СН2Сl2:МеОН 98,5:1,5), получая при этом указанное в заголовке соединение. Удельная радиоактивность 1,934 ГБк/ммоль. MS: +cESI: 279,89 М+.
1Н-ЯМР (500,1 МГц, d6-ДМСО): 2.08 (s, 3Н), 4,88 (s, 2Н), 6,35 (dd, 1H, J=2 Гц, J=7,9 Гц), 6,8 (d, 1H, J=1,7 Гц). 6,80 (d, 1H, 8,1 Гц), 7,37 (d, 1H, 5,3 Гц), 7,55 (dd, 1H, J=4,8 Гц, J=8 Гц), 8,41 (m, 1H), 8,47 (d, 1H, J=5,2 Гц), 8,67 (s, 1H), 8,70 (dd, 1H, J=1,5 Гц, J=4,9 Гц), 9,25 (d, 1H, J=1,9 Гц).
Стадия 3,1: трет-бутиловый эфир (5-{трет-бутоксикарбонил-[4-(4-метил-пиперазин-1-илметил)бензоил]амино}-2-метилфенил)(4-пиридин-3-ил-[2-14С]пиримидин-2-ил)-карбаминовой кислоты
После добавления 4-диметиламинопиридина (30 мг, 0,24 ммоль) к раствору 4-(4-метилпиперазин-1-илметил)-N-[4-метил-3-(4-пиридин-3-ил-[2-14С]пиримидин-2-иламино)фенил]бензамида (пример 1, 120 мг, 0,24 ммоль) и ди-трет-бутилдикарбоната (265 мг, 1,22 ммоль) в CH2Cl2 (2 мл) в атмосфере аргона при комнатной температуре образуется оранжевый раствор, который перемешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме и необработанный продукт хроматографируют на силикагеле, элюируя смесью СН2Сl2:МеОН 93:7. Фракции объединяют, выпаривают сначала на роторном испарителе, а затем под глубоким вакуумом, получая при этом указанное в заголовке соединение.
Стадия 3,2: трет-бутиловый эфир (5-трет-бутоксикарбониламино-2-метилфенил)-(4-пиридин-3-ил-[2-14С]пиримидин-2-ил)-карбаминовой кислоты
Раствор трет-бутилового эфира (5-{трет-бутоксикарбонил-[4-(4-метил-пиперазин-1-илметил)-бензоил]амино}-2-метил-фенил)-(4-пиридин-3-ил-[2-14С]пиримидин-2-ил)-карбаминовой кислоты (стадия 3,1, 153 мг, 0,22 ммоль) и 2-диэтиламиноэтиламина (53 мг, 64 мкл, 0,45 ммоль) в ТГФ (0,8 мл) перемешивают в течение ночи при комнатной температуре. На основании активного контроля (РТСХ, силикагель, CH2Cl2:МеОН 95:5) добавляют дополнительную порцию 2-диэтиламиноэтиламина (159 мг, 192 мкл, 1,35 ммоль) до тех пор, пока исходные вещества практически не будут обнаруживаться. После удаления растворителя в вакууме необработанный продукт очищают флеш-хроматографией (силикагель, СН2Сl2:МеОН 98:2), получая при этом указанное в заголовке соединение.
Пример 4: 4-(4-Метил-4-оксипиперазин-1-илметил)-N-[4-метил-3-(4-пиридин-3-ил-пиримидин-2-иламино)фенил]бензамид
Раствор 4-(4-метил-пиперазин-1-илметил)-N-[4-метил-3-(4-пиридин-3-ил-[2-14С]пиримидин-2-иламино)фенил]бензамида (пример 1, 414 мг, 43,4 мКи, 1605,8 МБк) в безводном дихлорметане (10 мл) обрабатывают МСРВА (260 мг, 1,5 ммоль) при комнатной температуре в течение 2 ч. Образовавшийся осадок отфильтровывают, фильтрат упаривают и необработанный продукт очищают флеш-хроматографией на силикагеле, элюируя смесью дихлорметан:МеОН 70:35, получая при этом указанное в заголовке соединение в виде белого твердого вещества. Продукт перекристаллизовывают из этанола/этилацетата.
1Н-ЯМР 400 МГц (ДМСО-d6) δ: 2,21 (s, 3Н), 2,78-2,89 (m, 4H); 3,01 (s, 3H); 3,28-3,38 (m, 4H); 3,6 (s, 2H); 7,18 (d, J=8,3 Гц, 1Н); 7,38-7,51 (m, 5H); 7,89 (s, 1H); 7,91 (s, 1Н); 8,03 (s, 1Н); 8,45 (d, J=7,9 Гц, 1Н); 8,49 (d, J=5,4 Гц, 1Н); 8,66 (dd, J=2, 4,2, 1Н); 8,98 (s, 1Н); 9,25 (d, J=2; 1Н); 10,2 (s, 1H); MS: (MH+=512).
Пример 5: N-{4-Метил-3-[4-(1-оксипиридин-3-ил)-[2-14С]пиримидин-2-иламино]-фенил}-4-(4-метил-пиперазин-1-илметил)-бензамид
1-Метилпиперазин (2 мл, 18 ммоль) добавляют к раствору 4-хлорметил-N-{4-метил-3-[4-(1-оксипиридин-3-ил)-[2-14С] пиримидин-2-иламино]-фенил}-бензамида (стадия 5,2, 151 мг, 0,34 ммоль) в этаноле (20 мл) и полученную смесь перемешивают в течение 1 ч при 60°С. Раствор упаривают и остаток очищают флеш-хроматографией на силикагеле, элюируя смесью дихлорметан:МеОН 90:10 и 1% гидроксидом аммония, получая при этом желтое маслянистое вещество. Это вещество перекристаллизовывают из смеси дихлорметан:МеОН, получая при этом продукт-N-оксид в виде кристаллического твердого вещества, 175,3 МБк.
1Н-ЯМР 400 МГц (ДМСО-d6) δ: 2,15 (s, 3Н); 2,21 (s, 3H); 2,25-2,42 (m, 4H); 3,28-3,34 (m, 4H); 3,52 (s, 2H); 7,19 (d, J=9,1 Гц; 1H); 7,38-7,42 (m, 3Н); 7,45-7,52 (m, 2H); 7,88 (s, 1H); 7,89 (s, 1H); 7,99-8,2 (m, 2H); 8,28 (d, J=6,8 Гц, 1H); 8,5 (d, J=5,4 Гц, 1H); 8,8 (2, 1H); 9,2 (s, 1H), 10,1 (s, 1H).
Стадия 5,1: трет-бутиловый эфир {4-метил-3-[4-(1-окси-пиридин-3-ил)-[2-14С]пиримидин-2-иламино]фенил}карбаминовой кислоты
Суспензию 4-метил-N-3-(4-пиридин-3-ил-[2-14С]пиримидин-2-ил)-бензол-1,3-диамина (пример 3, 286 мг, 1,03 ммоль, 1890 МБк) в ТГФ (10 мл) обрабатывают ди-трет-бутилдикарбонатом (467 мг, 2,33 ммоль) и каталитическими количествами ДМАП и нагревают с обратным холодильником в течение 2 ч. Растворитель удаляют в вакууме и необработанный продукт очищают флеш-хроматографией на силикагеле, элюируя смесью дихлорметан:МеОН, 95:5, с получением при этом защищенного продукта в виде желтого вспененного вещества. Это вещество растворяют в дихлорметане, охлаждают до -10°С, обрабатывают МСРВА (285 мг, 1,65 ммоль) и перемешивают при 0°С в течение 4 ч. Осадок отфильтровывают, растворитель выпаривают и необработанный продукт далее очищают флеш-хроматографией на силикагеле, элюируя смесью дихлорметан: МеОН 95:5, с получением при этом указанного в заголовке соединения в виде желтого вспененного вещества.
Стадия 5,2: 4-хлорметил-N-{4-метил-3-[4-(1-оксипиридин-3-ил)-[2-14С]пиримидин-2-иламино]фенил}бензамид
Хлороводородную кислоту (4 мл 4 н. раствора) добавляют к суспензии трет-бутилового эфира {4-метил-3-[4-(1-оксипиридин-3-ил)-[2-14С]пиримидин-2-иламино]фенил}карбаминовой кислоты (стадия 5,1, 187 мг, 0,48 ммоль) в ТГФ (10 мл). Полученную смесь выдерживают при нагревании до 70°С в течение 1 ч. Охлажденную смесь упаривают с получением при этом желтого твердого вещества, которое очищают флеш-хроматографией на силикагеле, элюируя смесью дихлорметан: МеОН 75:35, с получением при этом желтого кристаллического вещества. Кристаллы суспендируют в ТГФ (10 мл), обрабатывают 25%-ным водным раствором гидроксида аммония (1 капля) и полученную смесь обрабатывают ультразвуком. Суспензию отфильтровывают, и растворитель упаривают, получая при этом желтое вспененное вещество. Это вспененное вещество растворяют в ТГФ (10 мл), и по каплям добавляют раствор 4-(хлорметил)бензоилхлорида (149 мг, 0,8 ммоль) в ТГФ (2 мл), и соответствующую смесь перемешивают при комнатной температуре в течение 2 ч. Полученную суспензию центрифугируют, осадок дополнительно промывают эфиром, заново центрифугируют, удаляют эфир и остаток высушивают с получением при этом указанного в заголовке соединения в виде коричневых кристаллов.
Пример 6: 4-(4-Метил-[2,2,3,3,5,5,6,6-D8]пиперазин-1-илметил)-N-[4-метил-3-(4-пиридин-3-ил-пиримидин-2-иламино)фенил]бензамид
4-Хлорметил-N-[4-метил-3-(4-пиридин-3-ил-пиримидин-2-иламино)-фенил]бензамид (3,57 г, 8,30 ммоль; получают в соответствии с примером 1, используя немеченные исходные вещества) растворяют в диметилформамиде (50 мл). К этому раствору добавляют карбонат калия (5,74 г, 41,5 ммоль) и N-метилпиперазин-2,2,3,3,5,5,6,6-d8 (815 мг, 7,54 ммоль, произведен фирмой «Isotec, Inc», Майамисбург, Огайо, США). Смесь нагревают до 45-50°С в течение приблизительно 12-14 часов. Реакцию контролируют посредством ТСХ, используя систему растворителей, состоящую из ДХМ/ EtOAc/ MeOH/ NH4OH (25%) (60/10/10/2), подвижность продукта Rf составляет приблизительно 0,30. Диметилформамид удаляют в вакууме, и остаток перетирают с водой. После такого водного перетирания получают очень вязкое вещество, которое растворяют в этилацетате, водную фазу концентрируют в вакууме, и полученный от нее остаток растворяют в этилацетате. Объединенные вещества четырежды очищают с использованием флеш-хроматографии с системой растворителей, описанной выше. Продукт помещают в емкость и помещают эту емкость в сушильный аппарат Абдерхальдена под вакуумом (0,01 мм рт.ст.), температура сушки составляет 65°С (в кипящем метаноле с обратным холодильником). MS ES+502,4 (100%) [М+Н]+, 503,5 (40%), 504,5 (4%); 1Н ЯМР (d6-ДМСО) δ: 2,1 (3Н, s), 2,4 (3Н, s), 3,6 (2Н, s), 7,2 (1Н, d), 7,5 (3H, m), 7,8 (2H, d), 8,1 (1Н, s), 8,5 (2H, m), 8,8 (1Н, s), 9,0 (1Н, s), 9,2 (1Н, s), 10,1 (1Н, s).
Пример 7: 4-мeтил-N-(4-пиpидин-3-ил-[1,3-15N2,2,4,5-13С]пиримидин-2-ил)-бензол-1,3-диамин
Раствор неочищенного N-(5-амино-2-метил-фенил)-[14С,15N2]гуанидина (стадия 7,7, 157 мг, 0,96 ммоль) и 3-диметиламино-1-пиридин-3-ил-[1,2-13С2]пропенона (стадия 7,3, 169 мг, 0,96 ммоль) в н-бутаноле нагревают с обратным холодильником (130°С) в течение 5 часов. После удаления растворителя и очистки флеш-хроматографией с элюированием смесью 5:95 пентан:ацетон получают указанное в заголовке соединение в виде желтого твердого вещества, 1Н-ЯМР 400 МГц (CDCl3) δ: 9,14 (1Н, s, ArH), 8,73 (1Н, d, J4, ArH), 8,50 (1Н, d октет, J 7,1,2, ArH), 8,37 (1Н, m, ArH), 7,70 (1Н, s, NH), 7,45 (1Н, dd, J8, 5), 7,17 (1Н, dm, J 168, ArH), 7,03-6,99 (2H, m, ArH), 6,46 (1Н, dd, J 8,1,2, ArH), 2,27 (3Н, s, СН3): MS (c+ESI): 283,3 (100%) [M+H]+, 284,4 (15%).
Стадия 7,1: 3-триметилстаннанилпиридин
В двугорлую колбу в атмосфере азота помещают 3-бромпиридин (4 г, 25,3 ммоль), затем диэтиловый эфир (40 мл). Раствор охлаждают до -78°С, затем добавляют 1,5 М н-бутиллитий в гексане (20 мл, 30,4 ммоль). Полученный желтый раствор перемешивают в течение 10 минут, затем добавляют раствор хлорида триметилолова (5,04 г, 25,3 ммоль) в диэтиловом эфире (2 мл). Полученный раствор перемешивают при -78°С в течение 20 минут, затем медленно дают нагреться до комнатной температуры в течение 1 часа. Раствор разделяют между гексаном (50 мл) и водой (100 мл), встряхивают и отделяют органический слой. Водную фазу дважды экстрагируют гексаном (50 мл), объединяют органические фазы и промывают водой (100 мл). Гексановую фазу высушивают над сульфатом натрия, отфильтровывают и фильтрат упаривают с получением неочищенного маслянистого вещества, которое пропускают через силикагельную колонку, элюируя смесью 9:1 гексан:этилацетат, и собирают фракции, содержащие продукт. Фракции объединяют и упаривают с получением при этом светло-желтого маслянистого вещества, которое дополнительно очищают посредством дистилляции в печи Кюгельрора с получением при этом указанного в заголовке соединения в виде бесцветного маслянистого вещества, которое перегоняют при давлении 50 мбар и температуре 100°С.
Стадия 7,2: 1-пиридин-3-ил-[13С2]этанон
В ампулу объемом 30 мл помещают 3-триметилстаннилпиридин (стадия 7,1, 1,83 г, 7,56 ммоль), свежеперегнанный [13С2]ацетилхлорид (0,67 г, 8,32 ммоль), РdСl2(РРh3)2 (0,3 г, 0,44 ммоль) и сухой бензол (20 мл). Ампулу запаивают и помещают на ночь в масляную ванну при температуре 95°С. Ампулу открывают, содержимое удаляют и разделяют между водой (30 мл) и этилацетатом (30 мл), после чего встряхивают. Отделяют органическую фазу и дополнительно дважды переэкстрагируют водную фазу этилацетатом (2×30 мл). Органические фазы объединяют и промывают водой (30 мл), затем высушивают над сульфатом натрия, отфильтровывают, и фильтрат осторожно выпаривают. Неочищенное маслянистое вещество очищают на силикагеле, элюируя смесью 1:1 гексан:этилацетат, с получением при этом указанного в заголовке соединения после осторожной лиофилизации.
Стадия 7,3: (Е/Z)-3-диметиламино-1-пиридин-3-ил-[1,2-13С2]пропенон
1-Пиридин-3-ил-[13С2]этанон (стадия 7,2, 500 мг, 4,06 ммоль) и диметилацеталь N,N-диметилформамида (628 мг, 5,28 ммоль) нагревают до 100°С в запаянной ампуле в течение ночи. Необработанный продукт очищают хроматографией на силикагеле, элюируя смесью 9:1 дихлорметан:метанол, с получением при этом указанного в заголовке соединения в виде желтого кристаллического вещества.
Стадия 7,4: трет-бутиловый эфир [13С,15N2]тиомочевина-N,N'-дикарбоновой кислоты
К суспензии гидрида натрия (60% дисперсия в минеральном масле, 1,18 г, 29,61 ммоль) в сухом ТГФ (50 мл) при 0°С в атмосфере азота по каплям добавляют раствор [13С, 15N2]тиомочевины (500 мг, 6,58 ммоль), полученной от фирмы «Aldrich Chem. Со», в сухом ТГФ (82 мл). После завершения добавления раствор перемешивают в течение 5 минут с последующим добавлением ди-трет-бутилдикарбоната (3,16 г, 14,48 ммоль) и полученный раствор перемешивают в течение 1 часа. По каплям добавляют сначала насыщенный раствор NаНСО3 (13 мл), затем воду (230 мл). Продукт экстрагируют этилацетатом (3×150 мл), объединяют органические фазы, промывают насыщенным соляным раствором и высушивают над сульфатом натрия, отфильтровывают и фильтрат упаривают, получая при этом неочищенный продукт в виде маслянистого вещества.
Стадия 7,5: трет-бутиловый эфир N-(2-Метил-5-нитрофенил)-[13С,15N]гуанидин-N,N'-кислоты
К перемешиваемому оранжевому раствору неочищенного трет-бутилового эфира [13С, 15N2]тиомочевина-N,N'-дикарбоновой кислоты (стадия 7,4, 2,35 г, 6,58 ммоль), 2-метил-5-нитроанилина (1,00 г, 6,58 ммоль), триэтиламина (3,0 мл, 21,71 ммоль) в ДМФ при 0°С добавляют твердый хлорид ртути (II) (1,97 г, 7,24 ммоль). Через 15 минут перемешивания при комнатной температуре, добавляют этилацетат (50 мл) и смесь отфильтровывают через целит Хайфло (Hyflo). Этилацетатный раствор промывают сначала водой (50 мл), затем насыщенным соляным раствором (50 мл) и высушивают над сульфатом натрия. Высушивающий агент отфильтровывают и фильтрат выпаривают с получением при этом маслянистого вещества, которое очищают флеш-хроматографией на силикагеле, элюируя сначала 100%-ным гексаном, затем смесью 90:10 гексан:этилацетат, затем смесью 80:20 гексан:этилацетат, с получением при этом указанного в заголовке соединения.
Стадия 7,6: N-(2-метил-5-нитрофенил)-[15N2,13С]гуанидин
Раствор трет-бутилового эфира N-(2-метил-5-нитрофенил)-[13С, 15N2]гуанидин-N,N'-кислоты (стадия 7,5, 445 мг, 1,13 ммоль) в смеси 1:1 трифторуксусная кислота (9 мл):дихлорметан (9 мл) перемешивают при комнатной температуре в течение 1 часа. Добавляют 2 М гидроксид натрия (50 мл) и полученный желтый раствор экстрагируют этилацетатом (3×30 мл), объединяют и промывают водой. Органические фазы высушивают над сульфатом натрия, отфильтровывают и фильтрат упаривают с получением при этом желтого маслянистого вещества, которое очищают флеш-хроматографией на обычном силикагеле, элюируя смесью 90:10:0,5:0,5 дихлорметан:метанол:вода:уксусная кислота, получая при этом указанное в заголовке соединение.
Стадия 7,7: N-(5-амино-2-метилфенил)-[15N2,13С]гуанидин
Смесь N-(2-метил-5-нитрофенил)[13С,15N2]гуанидина (стадия 7,6, 121 мг, 0,62 ммоль) в н-бутаноле (3 мл), содержащую 10% Pd/C (12 мг), перемешивают в атмосфере газообразного водорода под давлением в 1 атмосферу при комнатной температуре в течение 16 часов. Добавляют дополнительно 12 мг 10% Pd/C и смесь перемешивают в течение дополнительных 16 часов. Полученную смесь отфильтровывают через целит Хайфло (Hyflo), промывают дополнительной порцией н-бутанола (3×2 мл), объединяют и удаляют н-бутанол на роторном испарителе, получая при этом неочищенное соединение, указанное в заголовке.
| название | год | авторы | номер документа |
|---|---|---|---|
| Триазолпиримидиновые соединения и их применение в лечении рака | 2019 |
|
RU2793249C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| АЗОТОСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛОВ | 2011 |
|
RU2559895C2 |
| ПРОИЗВОДНЫЕ ПРОПИОНОВЫХ КИСЛОТ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АКТИВАТОРОВ hPPARs. | 2003 |
|
RU2316539C2 |
| ИНГИБИТОРЫ КАНАЛОВ С ТРАНЗИТОРНЫМ ПОТЕНЦИАЛОМ НА ОСНОВЕ ОКСАДИАЗОЛОВ | 2019 |
|
RU2818244C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ 637: ПИРИДОПИРИМИДИНДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2008 |
|
RU2479584C2 |
| НОВЫЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННЫЕ БЕНЗОТИОФЕНЫ И БЕНЗОФУРАНЫ 709 | 2008 |
|
RU2472789C2 |
| ИМИДАЗОПИРАЗИНЫ | 2012 |
|
RU2600327C2 |
| N-ГИДРОКСИ-БЕНЗАМИДЫ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2577861C2 |
| ПРОИЗВОДНЫЕ ПИРИДИН-2-КАРБОКСАМИДА В КАЧЕСТВЕ АНТАГОНИСТОВ mGluR5 | 2006 |
|
RU2411237C2 |
Изобретение относится к соединению формулы (XVIII):
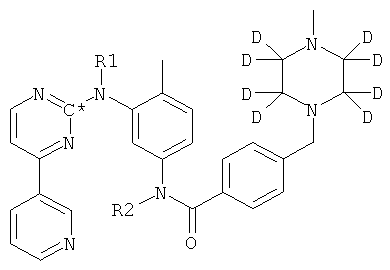
которое может быть использовано для наблюдения и исследования метаболизма в клинических и доклинических обследованиях. Изобретение также относится к способу получения указанного соединения. 2 н.п. ф-лы.
1. Соединение формулы XVIII
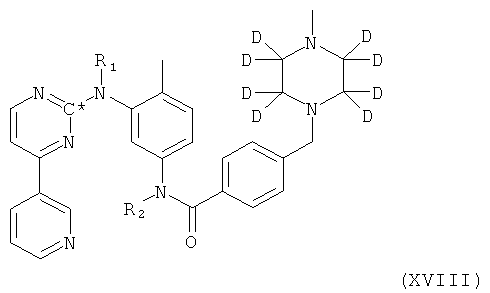
в которой R1 и R2 оба представляют собой водород и С* представляет собой 14С.
2. Способ получения соединения формулы XVIII
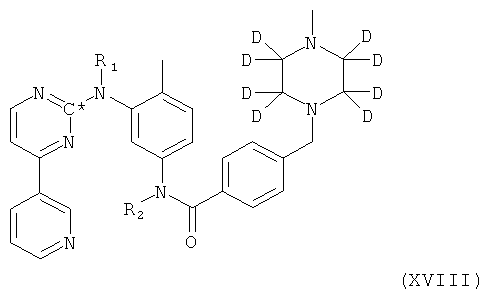
в которой R1 и R2 оба представляют собой водород и С* представляет собой 14С, посредством взаимодействия соединения формулы II,
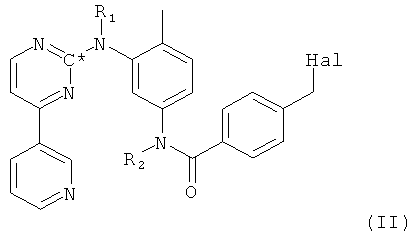
в которой Hal представляет собой галоген, R1 и R2 оба представляют собой водород и С* представляет собой 14С, с пиперазиновым производным формулы XVII.
| US 2003198594 A1, 23.10.2003 | |||
| WO 2002022597 A1, 21.03.2002 | |||
| RU 2004134355 A, 10.06.2005 | |||
| SZAKACS Z | |||
| et al., Journal of medicinal chemistry, 2005, vol.48(1), pp.249-255 | |||
| GSCHWIND H.P | |||
| et al., Drug metabolism and disposition, 2005, vol.33(10), pp.1503-1512. |

