ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Это описание главным образом относится к триазолпиримидиновым соединениям и их фармацевтически приемлемым солям. Эти соединения и их фармацевтически приемлемые соли селективно ингибируют МСТ4 (транспортер монокарбоксилата 4) и, поэтому, описание также относится к применению таких соединений и их солей для лечения или предупреждения МСТ4-опосредованного заболевания, включая рак. Описание также относится к кристаллическим формам триазолпиримидиновых соединений и их фармацевтически приемлемых солей; фармацевтическим композициям, содержащим такие соединения и соли; наборам, содержащим такие соединения и соли; способам получения таких соединений и солей; и к способам лечения МСТ4-опосредованного заболевания, включая рак, с использованием таких соединений и солей.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Транспортеры монокарбоксилатов кодируются генами семейства SLC16. Семейство также известно как семейство транспортеров монокарбоксилатов (МСТ), поскольку было продемонстрировано, что первые идентифицированные члены ответственны за протон-связанный транспорт монокарбоксилатов, таких как L-лактат, пируват и кетоновые тела, через цитоплазматическую мембрану. Прямое проявление протон-связанного транспорта лактата и пирувата было продемонстрировано для МСТ1 (SLC16A1), МСТ2 (SLC16A7), МСТ3 (SLC16A8) и МСТ4 (SLC16A3). Номенклатура семейства МСТ взята из Halestrap and Price, Biochemical Journal (1999) 343: 281-299.
МСТ имеют 12 трансмембранных спиралей, и функциональная экспрессия требует взаимодействия с шаперонами с одним трансмембранным доменом, известными как CD 147 (также известным как базигин и EMMPRIN (внеклеточный индуктор матриксных металлопротеиназ)) и эмбигин. CD 147 действует как важный шаперон для доставки МСТ1 и МСТ4 к цитоплазматической мембране, где переносчик и CD147 остаются тесно связанными (Kirk et al. (2000) EMBO J. 19: 3896-3904). При правильной экспрессии МСТ2 в цито плазматической мембране строгое предпочтение перед CD147 отдается эмбигину (Wilson et al (2005) J. Biol. Chem. 280: 27213-27221).
Хорошо известно, что опухоли демонстрируют измененный метаболизм (Vander Heiden (2011) Nat. Drag Dis. 10:671-684). Опухоли состоят из хорошо оксигенированных (аэробных) и плохо оксигенированных (гипоксических) участков. По сравнению с нормальными клетками опухолевые клетки имеют повышенную зависимость от гликолитического пути образования АТФ либо посредством аэробного гликолиза (эффект Варбурга), либо анаэробного гликолиза как следствия гипоксии опухоли. Оказывается, быстро пролиферирующие опухоли и гипоксические опухоли особенно зависят от гликолиза для удовлетворения энергетических и биосинтетических потребностей. Широкое клиническое использование ФДГ-ПЭТ (позитронная эмиссионная томография с фтордезоксиглюкозой) - ПЭТ-сканирования с индикатором фтор-18-фтордезоксиглюкозой (F-18-ФДГ) продемонстрировало, что этот гликолитический фенотип наблюдается в целом ряде солидных и гематологических опухолей. В результате ФДГ-ПЭТ может быть использована для диагностики, определения стадии и мониторинга лечения рака. ФДГ-ПЭТ в сочетании с компьютерной томографией имеет чувствительность и специфичность более 90% относительно обнаружения метастазов большинства эпителиальных опухолей (Mankoffetal. (2007) Clin. Cancer Res. 13:3460-3469).
Побочным продуктом повышенных скоростей гликолиза в опухолях является накопление лактата. Внутриклеточный лактат может транспортироваться из опухолевых клеток с помощью транспортеров монокарбоксилатов (МСТ 1, 2, 3 и 4) (Halestrap and Price, Biochem J. (1999) 343; 291-299). Лактат, продуцируемый опухолевыми клетками, может поглощаться стромальными и оксигенированными опухолевыми клетками (посредством транспортеров монокарбоксилатов МСТ1 и МСТ2) для регенерации пирувата, который может быть использован для обеспечения окислительного фосфорилирования (OXPHOS) (Koukouris et al., Cancer Res. (2006) 66; 632-637; Sonveaux et al., J. Clin. Invest. (2008) 118; 3930-3942). Одним из ключевых факторов в развитии гликолитического фенотипа опухолей является активация фактора, индуцируемого гипоксией (HIF), фактора транскрипции, который активируется гипоксическим стрессом. МСТ4 представляет собой ген-мишень HIF и активируется гипоксией, и необходим для экспорта лактата из гликолитических опухолей (Ullah et al. (2006) J. Biol. Chem. 281:9030- 9037). Кинетические свойства MCT4 направлены на его роль в экспорте молочной кислоты, полученной в результате гликолиза, поскольку его очень высокая Km (константа Михаэлиса) для пирувата (150 мМ) гарантирует, что пируват не будет выводиться из клетки. Это важно, потому что НАДН (восстановленный никотинамидадениндинуклеотид), полученный в результате восстановления пирувата до лактата, необходим для управления гликолитическим потоком (Halestrap and Wilson (2012) IUMBM Life 64: 109-119).
В отличие от нормального эпителия в ряде солидных опухолей сверхэкспрессируется МСТ4, включая опухоли почек (Fisel et al. (2013) Clin. Cancer Res. 19: 5170-5181; Gerlinger et al. J. Pathol. 227: 146-156), опухоли поджелудочной железы (Baek et al. (2014) Cell Rep.9:2233-2249), колоректальные опухоли (Pinheiro et al., Virchows Arch. (2008) 452; 139-146), HNSCC (плоскоклеточный рак головы и шеи) (Zhu et al. (2014) PLoS One 9:e87904), рак молочной железы (Doyen et al. (2014) Biochem. Biophys. Res. Commun. 451:54-61), рак предстательной железы (Pertega-Gomes et al. BMC Cancer (2011) H:312) и рак печени (Gao et al. (2015) J Cancer Res. 141: 1151-1162).
Последние данные показали, что лактат играет важную роль в регулировании функции иммунных клеток. Было показано, что лактат подавляет активность иммунных эффекторных клеток, таких как Т-клетки и NK-клетки. Молочная кислота подавляет пролиферацию и активацию Т-клеток человека ex vivo (Fisher et al. (2007) Blood 109:3812- 3819; Haas et al. (2015) PLoS Biol 13). Husain et al. продемонстрировали, что NK-клетки из опухолей, лишенных LDHA (лактатдегидрогеназы А), показали улучшенную цитолитическую функцию, а обработка NK-клеток лактатом снизила их цитотоксическую активность (Husain et al. (2013) J. Immunol. 191:1486-1495). Кроме того, Brand et al. продемонстрировали, что у иммунокомпетентных мышей нокдаун LDHA снижал продукцию молочной кислоты, а в опухолях наблюдали повышенную инфильтрацию IFN- у (интерферон у)-продуцирующих Т- и NK-клеток (Brand et al. (2016) Cell Metab. 24:657- 671). Также было показано, что лактат подавляет активацию моноцитов и дифференцировку дендритных клеток (Gottfried et al. (2006) Blood 107:2013-2021; Dietl et al. (2010) J. Immunol. 184:1200-1209) и также индуцирует M2 (иммуносупрессивную) поляризацию связанных с опухолью макрофагов (Colegio et al. (2014) Nature 513:559-563). Взятые вместе, эти данные подтверждают гипотезу о том, что лактат, продуцируемый как побочный продукт гликолитического фенотипа опухолей, оказывает иммуносупрессивное действие в микроокружении опухоли.
Аккумуляция лактата в микроокружении опухоли сопровождается ацидозом (за счет совместного транспорта с протонами). Низкий рН в микроокружении опухоли был связан с деградацией внеклеточного матрикса и миграцией опухолевых клеток (Gillies and Gatenby (2015) Cancer J. 21: 88-96).
Потенциальные ингибиторы МСТ1/2 были описаны в WO 2004/065394, где показано, что в Т-лимфоцитах отток лактата происходит через МСТ1, поскольку низкомолекулярные ингибиторы МСТ1 приводят к аккумуляции внутриклеточного лактата (Murray et al., Nat. Chem Biol. (2005) 1; 371-376).
Однако при этом существует потребность в ингибиторах МСТ4, которые подавляют транспорт лактата в МСТ4-зависимые клетки, демонстрируют хорошую биодоступность и подходят для введения.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Кратко, в этом описании представлено, в том числе, соединение формулы (I):
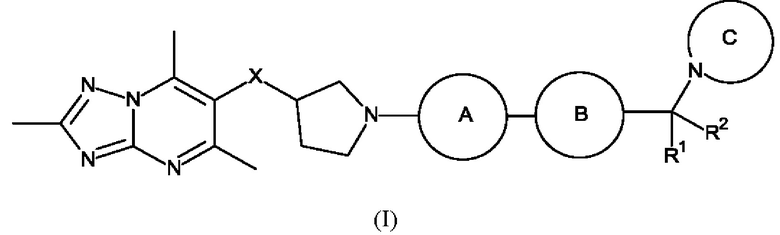
или его фармацевтически приемлемая соль, где:
каждый R1 и R2 независимо представляет собой водород или метил;
X представляет собой СН2 или О;
каждое кольцо А и кольцо В независимо представляет собой кольцо, выбранное из фенила, пиридинила, пиразинила, пиримидинила и пиридазинила, где каждое кольцо А и кольцо В независимо возможно замещено одним или более чем одним заместителем, выбранным из С1-3алкила и С1-3алкокси;
кольцо С представляет собой 5-9-членный моноциклический или бициклический насыщенный гетероциклоалкил, возможно содержащий один или более чем один дополнительный гетероатом, независимо выбранный из О, N и S, где кольцо С возможно замещено одним или более чем одним заместителем, выбранным из С1-3алкила, возможно замещенного метокси или гидроксилом; диоксо, С0-2алкил-С(O)N(Ме)2, С(O)С1-2алкила и S(O)2С1-2алкила.
В этом описании также представлена, в том числе, фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый эксципиент.
В этом описании также представлено, в том числе, соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
В этом описании также представлено, в том числе, соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака.
В этом описании также представлено, в том числе, соединение формулы (I) или его фармацевтически приемлемая соль для получения лекарственного средства для лечения рака.
В этом описании также представлен, в том числе, способ лечения рака у теплокровного животного, нуждающегося в таком лечении, включающий введение теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 показана картина дифракции рентгеновских лучей на порошке (ДРЛП) формы А (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина (соединение А, пример 62).
На Фиг. 2 показана термограмма ДСК (дифференциальная сканирующая калориметрия) формы А (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина (соединение А, пример 62).
На Фиг. 3 показана in vivo активность ингибитора МСТ4 (пример 62) в сочетании с VEGFR TKI (тирозинкиназа рецептора фактора роста эндотелия сосудов 1) (AZD2171, также известным как цедираниб) в модели ксенотрансплантата рака легких.
На Фиг. 4 показана in vivo активность ингибитора МСТ4 (пример 62) в сочетании с анти-CTLA4 (антиген-4 цитотоксических Т-лимфоцитов) антителом в сингенной мышиной модели.
На Фиг. 5 показана in vivo активность ингибитора МСТ4 (пример 62) в сочетании с анти-PD-1 (белок программируемой клеточной смерти 1) антителом в сингенной мышиной модели.
ОПИСАНИЕ ИЛЛЮСТРАТИВНЫХ ВОПЛОЩЕНИЙ
Многие воплощения изобретения подробно приведены в описании и будут очевидны специалисту в данной области техники. Изобретение не следует толковать как ограниченное каким-либо конкретным его воплощением(воплощениями).
В первом воплощении предложено соединение формулы (I):
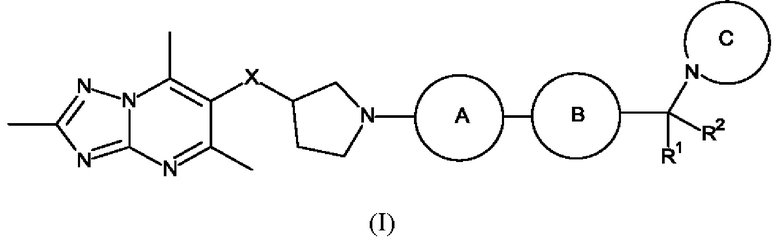
или его фармацевтически приемлемая соль, где:
каждый R1 и R2 независимо представляет собой водород или метил;
X представляет собой СН2 или О;
каждое кольцо А и кольцо В независимо представляет собой кольцо, выбранное из фенила, пиридинила, пиразинила, пиримидинила и пиридазинил, где каждое кольцо А и кольцо В независимо возможно замещено одним или более чем одним заместителем, выбранным из С1-3алкила и С1-3алкокси;
кольцо С представляет собой 5-9-членный моноциклический или бициклический насыщенный гетероциклоалкил, возможно содержащий один или более чем один дополнительный гетероатом, независимо выбранный из О, N и S, где гетероциклоалкил возможно замещен одним или более чем одним заместителем, выбранным из С1-3алкила, возможно замещенного метокси или гидроксилом; диоксо, С0-2алкил-С(O)N(Ме)2, C(O)C1-2галкила и S(O)2C1-2алкила.
Термин "гетероциклоалкил" означает 5-9-членное насыщенное азотсодержащее неароматическое кольцо (кольцо С в формуле (I)), содержащее один или более чем один дополнительный гетероатом, независимо выбранный из азота, кислорода и серы. Примеры подходящих гетероциклоалкильных групп включают морфолинил, пиперазинил, пиперидинил, тиоморфолинил, диазабициклооктанил, октагидропирроло[1,2-а]пиразинил, пирролидинил, диазепанил, оксазепанил и азепанил. Во избежание разночтений, заместители в гетероциклоалкильном кольце могут быть связаны либо через атом углерода, либо через гетероатом.
Термин "диоксо" означает два оксозаместителя, которые присоединены к одному и тому же атому. Примеры диоксо-замещения включают случаи, когда кольцо С представляет собой тиоморфолинил, где атом серы замещен двумя оксогруппами, то есть где кольцо С представляет собой тиоморфолин-1,1-диоксид.
Префикс Cp-q в Ср-qалкиле и других терминах (где р и q представляют собой целые числа) указывает количество атомов углерода, которые присутствуют в группе, например C1-3алкил включает С1алкил (метил), С2алкил (этил) и С3алкил (пропил, такой как н-пропил и изопропил). В одном воплощении С1-3алкил представляет собой метил.
Термин Ср-qалкокси включает -O-Ср-qалкильные группы. Например С1-3алкокси включает С1алкокси (метокси), С2алкокси (этокси) и С3алкокси (пропокси, такой как н-пропокси и изопропокси). В одном воплощении C1-3алкокси представляет собой метокси.
В случаях, когда используется термин "возможно", подразумевают, что последующая особенность может проявиться, а может не проявляться. Таким образом, использование термина "возможно" включает случаи, когда функция присутствует, а также случаи, когда функция отсутствует. Например, группа "возможно замещенная одной метоксигруппой" включает группы с метоксизаместителем и без него.
Термин "замещенный" означает, что один или более чем один атом водорода (например один или два атома водорода или, альтернативно, один атом водорода) в обозначенной группе заменен указанным заместителем(заместителями) (например одним или двумя заместителями или, альтернативно, одним заместителем) при условии, что любой атом(ы), несущий заместитель, сохраняет допустимую валентность. Комбинации заместителей включают только стабильные соединения и стабильные промежуточные соединения синтеза. "Стабильный" означает, что соответствующее соединение или промежуточное соединение является достаточно устойчивым при выделении и может быть использовано либо в качестве промежуточного соединения синтеза, либо в качестве агента, имеющего потенциальную терапевтическую пользу. Если группа не описана как "замещенная" или "возможно замещенная", ее следует рассматривать как незамещенную (т.е. что ни один из атомов водорода указанной группы не был заменен).
Термин "фармацевтически приемлемый" используется для обозначения того, что объект (например, соль, дозированная форма, эксципиент) подходит для применения пациентами. Список примеров фармацевтически приемлемых солей можно найти в Handbook of Pharmaceutical Salts: Properties, Selection and Use, P.H.
В дополнительном воплощении предложено любое из воплощений, определенных здесь (например воплощение пункта 1 формулы изобретения), при условии, что один или более чем один конкретный пример (например, один, два или три конкретных примера), выбранный из группы, состоящей из примеров 1, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86 и 87, индивидуально отклонен.
В одном воплощении X представляет собой СН2. В другом воплощении X представляет собой О.
В одном воплощении R1 и R2 оба представляют собой водород. В другом воплощении R1 и R2 оба представляют собой метил. В другом воплощении R1 представляет собой водород, и R2 представляет собой метил. В одном воплощении R1 и R2 оба представляют собой водород, или R1 представляет собой водород, и R2 представляет собой метил.
Кольцо А выбрано из фенила, пиридинила, пиразинила, пиримидинила и пиридазинила.
Кольцо В выбрано из фенила, пиридинила, пиразинила, пиримидинила и пиридазинила.
В формуле (I) кольцо А присоединено к атому азота пирролидинового кольца и к кольцу В, и кольцо В присоединено к кольцу А и к группе -С(R1R2)-кольцо С. Кольцо А и кольцо В могут быть возможно дополнительно замещены, как определено здесь. В одном воплощении пирролидиновое кольцо и кольцо В ориентированы в пара-положение (то есть 1,4) в кольце А. В другом воплощении кольцо А и группа -C(R1R2)-кольцо С ориентированы в пара-положение (то есть 1,4) в кольце В. В одном воплощении пирролидиновое кольцо и кольцо В ориентированы в пара-положение (то есть 1,4) в кольце А, и кольцо А и группа -C(R1R2)-кольцо С ориентированы в пара-положение (то есть 1,4) в кольце В. В другом воплощении пирролидиновое кольцо и кольцо В ориентированы в пара-положение (то есть 1,4) в кольце А, и кольцо А и группа -C(R1R2)- кольцо С ориентированы в пара-положение (то есть 1,4) в кольце В, и кольцо А и кольцо В связаны друг с другом через атом углерода кольца и связаны с остальной частью молекулы через атом углерода кольца.
В одном воплощении по меньшей мере одно кольцо А или кольцо В выбрано из пиридинила, пиразинила, пиримидинила и пиридазинила.
В одном воплощении каждое кольцо А и кольцо В независимо выбрано из фенила, пиридазинила и пиразинила. В другом воплощении каждое кольцо А и кольцо В независимо выбрано из фенила и пиридазинила. В другом воплощении кольцо А представляет собой фенил, и кольцо В представляет собой пиридазинил. В другом воплощении кольцо А представляет собой пиразинил, и кольцо В представляет собой фенил.
В одном воплощении кольцо А и кольцо В возможно замещены одним или двумя заместителями, выбранными из С1-3алкила и С1-3алкокси. В одном воплощении каждое кольцо А и кольцо В независимо возможно замещено одним заместителем, выбранным из C1-3алкила и С1-3залкокси. В одном воплощении каждое кольцо А и кольцо В независимо возможно замещено одним заместителем, выбранным из метила и метокси.
В одном воплощении кольцо С представляет собой 5-7 членное моноциклическое насыщенное гетероциклоалкильное кольцо. В другом воплощении кольцо С представляет собой 8- или 9-членное бициклическое насыщенное гетероциклоалкильное кольцо. Бициклическое гетероциклоалкильное кольцо может представлять собой мостиковое или конденсированное бициклическое кольцо.
В одном воплощении кольцо С выбрано из группы, состоящей из морфолинила, пиперазинила, пиперидинила, тиоморфолинила, диазабициклооктанила, октагидропирроло[1,2-а]пиразинила, пирролидинила, диазепанила, оксазепанила и азепанила.
В одном воплощении кольцо С выбрано из группы, состоящей из морфолин-4-ила, пиперазин-4-ила, пиперидин-1-ила, тиоморфолин-4-ила, 3,8-диазабицикло [3.2.1] октан-8-ила, октагидропирроло[1,2-а]пиразин-2-ила, пирролидин-1-ила, 1,4-диазепан-1-ила, 1,4- оксазепан-4-ила, азепанил-1-ила.
В одном воплощении кольцо С представляет собой морфолинил или пиперазинил. В одном воплощении кольцо С представляет собой морфолин-4-ил или пиперазин-4-ил.
В одном воплощении кольцо С представляет собой морфолинил. В одном воплощении кольцо С представляет собой морфолин-4-ил.
В одном воплощении кольцо С представляет собой пиперазинил. В одном воплощении кольцо С представляет собой пиперазин-4-ил.
В одном воплощении кольцо С возможно замещено одним или более чем одним (например одним, двумя или тремя) заместителями, независимо выбранными из гидроксила; этила, возможно замещенного метокси или гидроксилом; диоксо, C(O)N(Me)2, CH2C(O)N(Me)2, С(O)Ме и S(O)2Me. В одном воплощении кольцо С замещено С(O)Ме или метилом.
В одном воплощении кольцо С представляет собой пиперазин-4-ил-этанон. В другом воплощении кольцо С представляет собой 4-метил-1-пиперазинил.
В одном воплощении:
R1 и R2 оба представляют собой водород;
X представляет собой СН2 или О;
каждое кольцо А и кольцо В независимо представляет собой кольцо, выбранное из фенила, пиридинила, пиразинила, пиримидинила и пиридазинила, где каждое кольцо А и кольцо В независимо возможно замещено одним или более чем одним заместителем, выбранным из С1-3алкила и С1-3алкокси;
кольцо С представляет собой 5-9-членный моноциклический или бициклический насыщенный гетероциклоалкил, возможно содержащий один или более чем один дополнительный гетероатом, независимо выбранный из О, N и S, где гетероциклоалкил возможно замещен одним или более чем одним заместителем, выбранным из С1-3алкила, возможно замещенного метокси или гидроксилом; диоксо, С0-2алкил-С(O)N(Ме)2, C(O)C1-2алкила и S(O)2С1-2алкила.
В одном воплощении:
R1 и R2 оба представляют собой водород;
X представляет собой О;
каждое кольцо А и кольцо В независимо выбрано из фенила и пиридазинила;
кольцо С представляет собой морфолинил или пиперазинил, где пиперазинил возможно замещен С1-3алкилом, возможно замещенным метокси или гидроксилом; С0-2алкил-С(O)N(Ме)2, С(O)С1-2алкилом и S(O)2С1-2алкилом.
В одном воплощении:
R1 и R2 оба представляют собой водород;
X представляет собой СН2;
каждое кольцо А и кольцо В независимо выбрано из фенила, пиридазинила и пиразинила;
кольцо С представляет собой морфолинил, пиперазинил или пиразинил, где пиразинил или пиперазинил возможно замещен С1-3алкилом, возможно замещенным метокси или гидроксилом; С0-2алкил-С(O)N(Ме)2, С(O)С1-2алкилом и S(O)2С1-2алкилом.
Согласно дополнительному воплощению предложено соединение формулы (Ia):
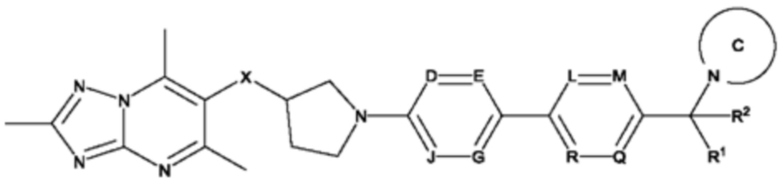
или его фармацевтически приемлемая соль, где
каждый R1 и R2 независимо представляет собой водород или метил;
X представляет собой СН2 или О;
каждый D, Е, G, J, L, М, Q и R независимо представляет собой N или CR3, где не более двух из D, Е, G и J представляют собой N, и где не более двух из L, М, Q и R представляют собой N;
R3 представляет собой водород, С1-3алкил или С1-3алкокси;
кольцо С представляет собой 5-9-членный моноциклический или бициклический насыщенный гетероциклоалкил, возможно содержащий один или более чем один дополнительный гетероатом, независимо выбранный из О, N и S, где гетероциклоалкил возможно замещен одним или более чем одним заместителем, выбранным из С1-3алкила, возможно замещенного метокси или гидроксилом; диоксо, С0-2алкил-С(O)N(Ме)2, C(O)C1-2алкила и S(O)2C1-2алкила.
В одном воплощении R1 и R2 оба представляют собой водород. В другом воплощении R1 и R2 оба представляют собой метил. В другом воплощении R1 представляет собой водород, и R2 представляет собой метил. В одном воплощении R1 и R2 оба представляют собой водород, или R1 представляет собой водород, и R2 представляет собой метил.
D, Е, G и J каждый независимо представляет собой N или CR3, где не более двух из D, Е, G and J представляют собой N.
L, М, Q и R каждый независимо представляет собой N или CR3, где не более двух из L, М, Q и R представляют собой N.
В одном воплощении D, Е, G и J каждый независимо представляет собой CR3. В другом воплощении один из D, Е, G и J представляет собой N, а каждый оставшийся независимо представляет собой CR3. В другом воплощении два из D, Е, G и J представляют собой N, а оба оставшихся независимо представляют собой CR3.
В одном воплощении L, М, Q и R каждый независимо представляет собой CR3. В другом воплощении один из L, М, Q и R представляет собой N, а каждый оставшийся независимо представляет собой CR3. В другом воплощении два из L, М, Q и R представляют собой N, а оба оставшихся независимо представляют собой CR3.
В одном воплощении D, Е, G и J каждый независимо представляет собой CR3, a L, М, Q и R каждый независимо представляет собой N или CR3, где не более двух из L, М, Q и R представляют собой N. В одном воплощении один из D, Е, G и J представляет собой N, а каждый оставшийся независимо представляет собой CR3, и L, М, Q и R каждый независимо представляет собой N или CR3, где не более двух из L, М, Q и R представляют собой N. В одном воплощении два из D, Е, G и J представляют собой N, а оба оставшихся независимо представляют собой CR3, и L, М, Q и R каждый независимо представляет собой N или CR, где не более двух из L, М, Q и R представляют собой N.
В одном воплощении D, Е, G и J представляют собой N или CR3, где не более двух из D, Е, G и J представляют собой N, а каждый L, М, Q и R независимо представляет собой CR3. В одном воплощении D, Е, G и J представляют собой N или CR3, где не более двух из D, Е, G и J представляют собой N, и один из L, М, Q и R представляет собой N, а каждый оставшийся независимо представляет собой CR3. В одном воплощении D, Е, G и J представляют собой N или CR3, где не более двух из D, Е, G и J представляют собой N, и два из L, М, Q и R представляют собой N, а оба оставшихся независимо представляют собой CR3.
В одном воплощении D, Е, G и J каждый независимо представляет собой CR3, и L, М, Q и R каждый независимо представляет собой CR3.
В одном воплощении D, Е, G и J каждый независимо представляет собой CR3, и один из L, М, Q и R представляет собой N, а каждый оставшийся независимо представляет собой CR3.
В одном воплощении D, Е, G и J каждый независимо представляет собой CR3, и два из L, М, Q и R представляют собой N, а оба оставшихся независимо представляют собой CR3.
В одном воплощении один из D, Е, G и J представляет собой N, а каждый оставшийся независимо представляет собой CR3, и L, М, Q и R каждый независимо представляет собой CR3.
В одном воплощении один из D, Е, G и J представляет собой N, а каждый оставшийся независимо представляет собой CR3, и один из L, М, Q и R представляет собой N, а каждый оставшийся независимо представляет собой CR3.
В одном воплощении один из D, Е, G и J представляет собой N, а каждый оставшийся независимо представляет собой CR3, и два из L, М, Q и R представляют собой N, а оба оставшихся независимо представляют собой CR3.
В одном воплощении два из D, Е, G и J представляют собой N, а оба оставшихся независимо представляют собой CR3, и каждый L, М, Q и R независимо представляет собой CR3.
В одном воплощении два из D, Е, G и J представляют собой N, а оба оставшихся независимо представляют собой CR3, и один из L, М, Q и R представляет собой N, а каждый оставшийся независимо представляет собой CR3.
В одном воплощении два из D, Е, G и J представляют собой N, а оба оставшихся независимо представляют собой CR3, и два из L, М, Q и R представляют собой N, а оба оставшихся независимо представляют собой CR3.
В одном воплощении кольцо, содержащее D, Е, G и J, представляет собой кольцо, выбранное из фенила, пиридинила, пиразинила, пиримидинила и пиридазинила. В одном воплощении кольцо, содержащее D, Е, G и J, представляет собой кольцо, выбранное из фенила, пиридазинила и пиразинила. В одном воплощении кольцо выбрано из фенила и пиридазинила.
В одном воплощении кольцо, содержащее L, М, Q и R, представляет собой кольцо, выбранное из фенила, пиридинила, пиразинила, пиримидинила и пиридазинила. В одном воплощении кольцо, содержащее L, М, Q и R, представляет собой кольцо, выбранное из фенила, пиридазинила и пиразинила. В одном воплощении кольцо выбрано из фенила и пиридазинила.
В другом воплощении кольцо, содержащее D, Е, G и J, представляет собой фенил, а кольцо, содержащее L, М, Q и R представляет собой пиридазинил. В другом воплощении кольцо, содержащее D, Е, G и J, представляет собой пиразинил, а кольцо, содержащее L, М, Q и R, представляет собой фенил.
В одном воплощении каждое кольцо, содержащее D, Е, G и J, и кольцо, содержащее L, М, Q и R, независимо возможно замещено одним или двумя заместителями, то есть где R3 представляет собой С1-3алкил и С1-3алкокси. В одном воплощении каждое кольцо, содержащее D, Е, G и J, и кольцо, содержащее L, М, Q и R, независимо возможно замещено одним заместителем, то есть где R3 представляет собой С1-3алкил и С1-3алкокси. В одном воплощении каждое кольцо, содержащее D, Е, G и J, и кольцо, содержащее L, М, Q и R, независимо возможно замещено одним заместителем, где R3 выбран из метила и метокси.
В одном воплощении кольцо С представляет собой 5-7-членное моноциклическое насыщенное гетероциклоалкильное кольцо. В другом воплощении кольцо С представляет собой 8- или 9-членное бициклическое насыщенное гетероциклоалкильное кольцо. Бициклическое гетероциклоалкильное кольцо может представлять собой мостиковое или конденсированное бициклическое кольцо.
В одном воплощении кольцо С выбрано из группы, состоящей из морфолинила, пиперазинила, пиперидинила, тиоморфолинила, диазабициклооктанила, октагидропирроло[1,2-а]пиразинила, пирролидинила, диазепанила, оксазепанила и азепанила.
В одном воплощении кольцо С выбрано из группы, состоящей из морфолин-4-ила, пиперазин-4-ила, пиперидин-1-ила, тиоморфолин-4-ила, 3,8-диазабицикло [3.2.1] октан-8-ила, октагидропирроло[1,2-а]пиразин-2-ила, пирролидин-1-ила, 1,4-диазепан-1-ила, 1,4- оксазепан-4-ила, азепанил-1-ила.
В одном воплощении кольцо С представляет собой морфолинил или пиперазинил. В одном воплощении кольцо С представляет собой морфолин-4-ил или пиперазин-4-ил.
В одном воплощении кольцо С представляет собой морфолинил. В одном воплощении кольцо С представляет собой морфолин-4-ил.
В одном воплощении кольцо С представляет собой пиперазинил. В одном воплощении кольцо С представляет собой пиперазин-4-ил.
В одном воплощении кольцо С возможно замещено одним или более чем одним (например одним, двумя или тремя) заместителями, независимо выбранными из гидроксила; этила, возможно замещенного метокси или гидроксилом; диоксо, C(O)N(Me)2, CH2C(O)N(Me)2, С(O)Ме и S(O)2Me. В одном воплощении кольцо С замещено С(O)Ме или метилом.
В одном воплощении кольцо С представляет собой пиперазин-4-ил-этанон. В другом воплощении кольцо С представляет собой 4-метил-1-пиперазинил.
В одном воплощении:
R1 и R2 оба представляют собой водород;
X представляет собой СН2 или О;
D, Е, G, J, L, М, Q и R каждый независимо представляет собой N или CR3, где не более двух из D, Е, G и J представляют собой N, и где не более двух из L, М, Q и R представляют собой N;
R3 представляет собой водород, С1-3алкил или С1-3алкокси;
кольцо С представляет собой 5-9-членный моноциклический или бициклический насыщенный гетероциклоалкил, возможно содержащий один или более чем один дополнительный гетероатом, независимо выбранный из О, N и S, где гетероциклоалкил возможно замещен одним или более чем одним заместителем, выбранным из С1-3алкила, возможно замещенного метокси или гидроксилом; диоксо, С0-2алкил-С(O)N(Ме)2, C(O)C1-2алкила и S(O)2С1-2алкила.
В одном воплощении:
R1 и R2 оба представляют собой водород;
X представляет собой О;
D, Е, G, J, L, М, Q и R каждый независимо представляет собой N или CR3, где не более двух из D, Е, G и J представляют собой N, и где не более двух из L, М, Q и R представляют собой N;
R3 представляет собой водород, метил или метокси;
кольцо С представляет собой морфолинил или пиперазинил, где пиперазинил возможно замещен С1-3алкилом, возможно замещенным метокси или гидроксилом; С0-2алкил-С(O)N(Ме)2, С(O)С1-2алкилом и S(O)2С1-2алкилом.
В одном воплощении:
R1 и R2 оба представляют собой водород;
X представляет собой СН2;
D, Е, G, J, L, М, Q и R каждый независимо представляет собой N или CR3, где не более двух из D, Е, G и J представляют собой N, и где не более двух из L, М, Q и R представляют собой N;
R3 выбран из водорода, метила или метокси;
кольцо С представляет собой морфолинил, пиперазинил или пиразинил, где пиразинил или пиперазинил возможно замещен С1-3алкилом, возможно замещенным метокси или гидроксилом; С0-2алкил-С(O)N(Ме)2, С(O)С1-2алкилом и S(O)2С1-2алкилом.
Согласно дополнительному воплощению предложено соединение формулы (Ib):
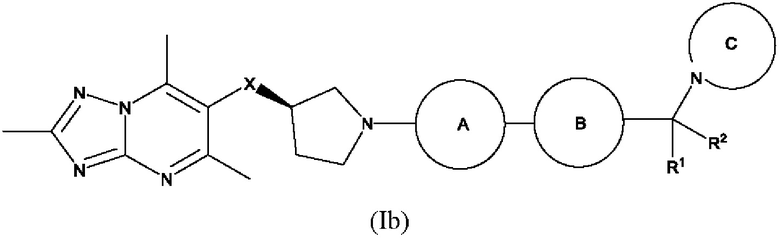
или его фармацевтически приемлемая соль, где R1, R2, X, кольцо А, кольцо В и кольцо С являются такими, как определено здесь.
Согласно дополнительному воплощению предложено соединение формулы (Ic):
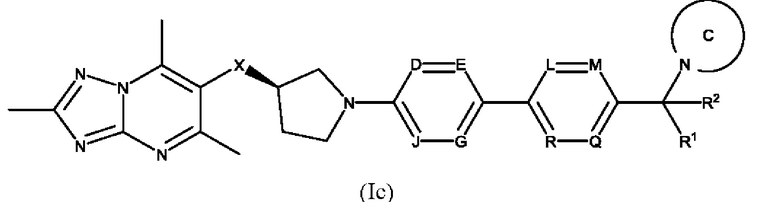
или его фармацевтически приемлемая соль, где R1, R2, X, D, Е, G, J, L, М, Q, R и кольцо С являются такими, как определено здесь.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение выбрано из группы, состоящей из:
(R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)-[3,3'-бипиридин]-6-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(6'-((4-метилпиперазин-1-ил)метил)-[3,3'-бипиридин]-6-ил)пирролидин-3-ил)метил)- [1,2,4]триазоло [1,5-а] пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиридин-2-ил)пирролидин-3-ил)метил)- [1,2,4]триазоло [1,5-а] пиримидина;
(R)-4-(4-(2-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6- ил)метил)пирролидин-1-ил)пиримидин-5-ил)бензил)морфолина;
(R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6- ил)метил)пирролидин-1-ил)пиридин-2-ил)пиразин-2-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(6-(4-((4-метилпиперазин-1-ил)метил)фенил)пиридин-3- ил)пирролидин-3-ил)метил)- [1,2,4]триазоло [1,5-а] пиримидина;
(R)-4-((6-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6- ил)метил)пирролидин-1-ил)пиридин-2-ил)пиридазин-3-ил)метил)морфолина;
6-(((R)-1-(2-(4-(((S)-2,4-диметилпиперазин-1-ил)метил)фенил)пиримидин-5- ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6- ил)метил)пирролидин-1-ил)пиримидин-2-ил)пиридин-2-ил)метил)морфолина;
(R)-6-((1-(2-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5- ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(2-(2-метил-4-((4-метилпиперазин-1- ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а] пиримидина;
6-(((R)-1-(2-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)пиримидин-5- ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-N,N-диметил-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6- ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)ацетамида;
(R)-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6- ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)этанола;
(R)-1-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6- ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)этенона;
(R)-6-((1-(2-(4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)фенил)пиримидин-5- ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6- ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)морфолина;
(R)-2,5,7-триметил-6-((1-(2-(4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло [1,5-а] пиримидина;
(R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиразин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(4-(2-((4-метилпиперазин-1-ил)метил)пиримидин-5-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридин-3-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(4-(6-((4-метилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиримидин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1 -ил)фенил)пиразин-2-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиридин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1 -ил)фенил)пиримидин-2-ил)метил)морфолина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(R)-6-((1-(5-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)-6-метилпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
2,5,7-триметил-6-(((R)-1-(5-(4-(((3R,5S)-3,4,5-триметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
6-(((R)-1-(5-(4-(((R)-3,4-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
6-(((R)-1-(5-(4-(((R)-2,4-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
2,5,7-триметил-6-(((R)-1-(5-(4-(((2R,5R)-2,4,5-триметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
2,5,7-триметил-6-(((R)-1-(5-(4-(((2S,5R)-2,4,5-триметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
2,5,7-триметил-6-[[(3R)-1-[5-[4-(1-пиперидилметил)фенил]пиразин-2-ил]пирролидин-3-ил]метил]-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-6-((1-(5-(4-((4-этилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(5-((4-метилпиперазин-1-ил)метил)пиридин-2-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
6-(((R)-1-(5-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
2-{4-[4-(5-{(3R)-3-[(2,5,7-триметил[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил]пирролидин-1-ил}пиразин-2-ил)бензил]пиперазин-1-ил}этанола;
(R)-6-((1-(5-(4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-6-((1-(5-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
{1-[4-(5-{(3R)-3-[(2,5,7-триметил[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил]пирролидин-1-ил}пиразин-2-ил)бензил]пиперидин-4-ил}метанола;
6-{[(3R)-1-(5-{4-[(1,1-диоксидотиоморфолин-4-ил)метил]фенил}пиразин-2-ил)пирролидин-3-ил]метил}-2,5,7-триметил[1,2,4]триазоло[1,5-а]пиримидина;
2,5,7-триметил-6-({(3R)-1-[5-(4-{[4-(метилсульфонил)пиперидин-1-ил]метил}фенил)пиразин-2-ил]пирролидин-3-ил}метил)[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((6-(3-метил-5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)пиридин-3-ил)метил)морфолина;
6-(((R)-1-(5-(4-(((S)-3,4-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)пиридин-2-ил)метил)морфолина;
6-(((R)-1-(5-(4-(((S)-2,4-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-(4-(3-метил-5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)морфолина;
2,5,7-триметил-6-({(3R)-1-[5-(4-{[4-(метилсульфонил)пиперазин-1-ил]метил}фенил)пиразин-2-ил]пирролидин-3-ил}метил)[1,2,4]триазоло[1,5-а]пиримидина;
(R)-N,N-диметил-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)пиперазин-1-ил)ацетамида;
(R)-2,5,7-триметил-6-((1-(6-метил-5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
N,N-диметил-4-[4-(5-{(3R)-3-[(2,5,7-триметил[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил]пирролидин-1-ил}пиразин-2-ил)бензил]пиперазин-1-карбоксамида;
(R)-2,5,7-триметил-6-((1-(5-(3-метил-4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-1-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)пиперазин-1-ил)этенона;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1 -ил)пиразин-2-ил)бензил)морфолина;
(R)-4-(4-(6-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиридазин-3-ил)бензил)морфолина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(4-(6-(пиперидин-1-илметил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(4-(6-((4-метилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидина;
2,5,7-триметил-6-(((3R)-1-(4-(6-((3-метил-3,8-диазабицикло[3.2.1]октан-8-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидина;
6-(((R)-1-(4-(6-(((R)-гексагидропирроло[1,2-а]пиразин-2(1H)-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
6-(((R)-1-(4-(6-(((S)-гексагидропирроло[1,2-а]пиразин-2(1H)-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиримидин-2-ил)метил)морфолина;
(R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиразин-2-ил)метил)морфолина;
(R)-1-(4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)пиперазин-1-ил)этан-1-она;
(R)-6-((1-(4-(6-((4-этилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-6-((1-(4-(6-((4-(2-метоксиэтил)пиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)-[2,3'-бипиридин]-5-ил)метил)морфолина;
(R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)-[3,3'-бипиридин]-6-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(2-(4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)пиразин-2-ил)бензил)морфолина;
1-[4-[[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил]пирролидин-1-ил]пиразин-2-ил]фенил]метил]-1,4-диазепан-1-ил]этенона;
(R)-2,5,7-триметил-6-((1-(5-(4-(пирролидин-1-илметил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(5-(пирролидин-1-илметил)пиридин-2-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(6-(пирролидин-1-илметил)пиридин-3-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина;
(R-2,5,7-триметил-6-((1-(5-(4-((4-метил-1,4-диазепан-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)- [1,2,4]триазоло[1,5-а]пиримидина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)-1,4-оксазепана;
(R)-6-((1-(4-(6-(азепан-1-илметил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина;
4-[(1R1-[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси]пирролидин-1-ил]пиразин-2-ил]фенил]этил]морфолина; и
4-[(1S-1-[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси]пирролидин-1-ил]пиразин-2-ил]фенил]этил]морфолина.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение выбрано из группы, состоящей из:
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(R)-1-(4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)пиперазин-1-ил)этан-1-она;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидина; и
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение представляет собой (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1- ил)фенил)пиридазин-3-ил)метил)морфолин (также именуемое как соединение А).
В одном воплощении предложено соединение формулы (I), или его фармацевтически приемлемая соль, где соединение представляет собой (R)-1-(4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)пиперазин-1-ил)этан-1-он.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение представляет собой (R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)- [1,2,4]триазоло [1,5-а]пиримидин.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение представляет собой (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин.
В одном воплощении соединение формулы (I) находится в форме свободного основания.
Соединения и соли, описанные в этом описании, могут существовать в сольватированных и несольватированных формах. Например, сольватированная форма может представлять собой гидратированную форму, такую как полугидрат, моногидрат, дигидрат, тригидрат или их альтернативное количество. Изобретением охватываются все такие сольватированные и несольватированные формы соединений формулы (I), особенно при условии, что такие формы обладают ингибирующей активностью в отношении МСТ4, такой как, например, измеренной с использованием описанных здесь тестов.
Атомы соединений и солей, описанных в этом описании, могут существовать в виде изотопов. Изобретением охватываются все соединения формулы (I), в которых атом заменен одним или более чем одним изотопом (например соединение формулы (I), где один или более чем один атом углерода представляет собой изотоп углерода 11C или 13С, или где один или более чем один атом водорода представляет собой изотоп 2Н или 3Н, или где один или более чем один атом азота представляет собой изотоп 15N, или где один или более чем один атом кислорода представляет собой изотоп 17O или 18O).
Соединения и соли, описанные в этом описании, могут существовать в оптически активных или рацемических формах благодаря одному или более чем одному асимметричному атому углерода. Изобретение включает любую оптически активную или рацемическую форму соединения формулы (I), которая обладает ингибирующей активностью в отношении МСТ4, например измеренной с использованием описанных здесь тестов. Синтез оптически активных форм может быть проведен посредством стандартных методов органической химии, хорошо известных в данной области техники, например посредством синтеза с использованием оптически активных веществ или посредством разделения рацемической формы.
Поэтому, в одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль, которая представляет собой отдельный оптический изомер, присутствующий в энантиомерном избытке (% е.е.) не менее 95%, не менее 98% или не менее 99%. В одном воплощении отдельный оптический изомер присутствует в энантиомерном избытке (% е.е.) не менее 99%.
Некоторые из соединений формулы (I) могут быть кристаллическими и могут иметь более одной кристаллической формы. Следует понимать, что изобретением охватывается любая кристаллическая или аморфная форма или ее смеси, которые обладают свойствами, полезными для ингибирующей активности в отношении МСТ4. Хорошо известно, как определить эффективность кристаллической или аморфной формы с помощью стандартных тестов, описанных ниже.
Общеизвестно, что кристаллические вещества могут быть проанализированы с использованием обычных методов, таких как, например анализ дифракции рентгеновских лучей на порошке (здесь и далее ДРЛП) и дифференциальная сканирующая калориметрия (здесь и далее ДСК).
Например, соединение из примера 62 проявляет кристалличность, и была идентифицирована кристаллическая форма, форма А.
Следовательно, дополнительным аспектом раскрытия является форма А соединения А (пример 62).
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП с по меньшей мере одним характерным пиком при угле 2- тета, равном примерно 7,8°, измеренном при использовании CuKα-излучения.
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП с по меньшей мере одним характерным пиком при угле 2- тета, равном примерно 19,0°, измеренном при использовании CuKα-излучения.
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП с по меньшей мере двумя характерными пиками при углах 2-тета, равных примерно 7,8° и 19,0°, измеренных с помощью CuKα-излучения.
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП с характерными пиками при углах 2-тета, равных примерно 7,8; 9,2; 9,5; 10,4; 11,6; 15,8; 18,3; 19,0; 22,3 и 25,3°, измеренных с помощью CuKα-излучения.
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП, по существу аналогичную картине ДРЛП, показанной на Фиг. 1, установленной с помощью CuKα-излучения.
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП с по меньшей мере одним характерным пиком при угле 2- тета, равном 7,8° плюс или минус 0,2°, измеренном с помощью CuKα-излучения.
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП с по меньшей мере одним характерным пиком при угле 2- тета, равном 19,0° плюс или минус 0,2°, измеренном с помощью CuKα-излучения.
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП с по меньшей мере двумя характерными пиками при углах 2-тета, равных 7,8° и 19,0°, где указанные значения могут быть больше или меньше на 0,2°, которые измерены с помощью CuKα-излучения.
Согласно раскрытию предложена кристаллическая форма, форма А, соединения А, которая имеет картину ДРЛП с характерными пиками при углах 2-тета, равных 7,8; 9,2; 9,5; 10,4; 11,6; 15,8; 18,3; 19,0; 22,3 и 25,3°, где указанные значения могут быть больше или меньше на 0,2°, которые измерены с помощью CuKα-излучения.
Анализ ДСК соединения А, форма А, показывает эндотерму плавления с началом при температуре примерно 210,2°С и пиком при температуре примерно 213,2°С (Фиг. 2).
Таким образом, анализ ДСК показывает, что соединение А, форма А, представляет собой высокоплавкое твердое вещество с началом плавления при температуре примерно 210,2°С и пиком при температуре примерно 213,2°С.
Когда указывается, что настоящее раскрытие относится к кристаллической форме формы А соединения А, степень кристалличности подходяще составляет более чем примерно 60%, более подходяще более чем примерно 80%, предпочтительно более чем примерно 90% и более предпочтительно более чем примерно 95%. Наиболее предпочтительно степень кристалличности составляет более чем примерно 98%.
Следует понимать, что значения углов 2-тета картины ДРЛП могут незначительно отличаться от одного прибора к другому или от одного образца к другому, и поэтому приведенные значения не следует расценивать как абсолютные.
Известно, что может быть получена картина ДРЛП, которая имеет одну или более чем одну ошибку измерения в зависимости от условий измерения (таких как используемое оборудование или прибор). В частности, общеизвестно, что интенсивность картины ДРЛП может колебаться в зависимости от условий измерения. Поэтому, следует понимать, что соединение А, форма А, настоящего раскрытия не ограничивается кристаллами, которые дают картины ДРЛП, идентичные картине ДРЛП, показанной на Фиг. 1, и любые кристаллы, дающие картины ДРЛП, по существу такие же, как та, которая показана на Фиг. 1. входят в объем настоящего раскрытия. Специалист в области ДРЛП способен оценить существующую идентичность картин ДРЛП.
Специалистам в области ДРЛП будет понятно, что на относительную интенсивность пиков могут оказывать влияние, например зерна размером более 30 микрон и неунитарные соотношения сторон, которые могут влиять на анализ образцов. Специалист также поймет, что на положение отражений может влиять точная высота, на которой образец находится в дифрактометре, и калибровка нуля дифрактометра. Планарность поверхности образца также может оказывать небольшое влияние. Следовательно, представленные данные дифракционной картины не следует принимать за абсолютные значения. (Jenkins, R & Snyder, R.L. 'Introduction to X-Ray Powder Diffractometry' John Wiley & Sons 1996; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; Klug, H.P. & Alexander, L.E. (1974), X-Ray Diffraction Procedures).
Как правило, погрешность измерения угла дифракции на дифрактограмме рентгеновских лучей на порошке составляет приблизительно плюс или минус 0,2° угла 2-тета, и такую степень погрешности измерения следует принимать во внимание, анализируя картину ДРЛП на Фиг. 1, и при рассмотрении таблицы А. Кроме того, следует понимать, что интенсивности могут колебаться в зависимости от условий эксперимента и подготовки образца (предпочтительной ориентации).
Соединения формулы (I) содержат один или более чем один хиральный центр. В случаях, когда структура или химическое название в этом описании не указывает на хиральность, структура или название предназначены включать любой отдельный стереоизомер (т.е. любой отдельный хиральный изомер), соответствующий этой структуре или названию, а также любую смесь стереоизомеров (например рацемат). В данной области техники хорошо известно, как можно получить такие оптически активные формы. Например отдельный стереоизомер может быть получен посредством выделения его из смеси изомеров (например рацемата) при использовании, например хирального хроматографического разделения. В других воплощениях отдельный стереоизомер получают посредством прямого синтеза, например из хирального исходного вещества.
Соединения формулы (I), где X представляет собой О, могут быть получены, например, посредством взаимодействия соединения формулы (II):
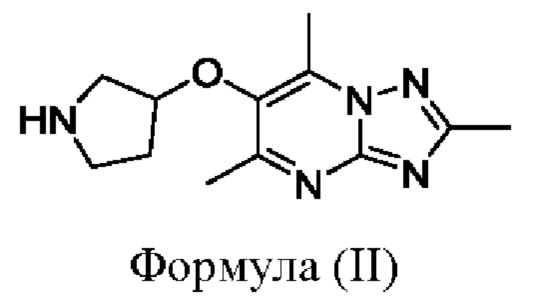
или его соли с соединением формулы (III):
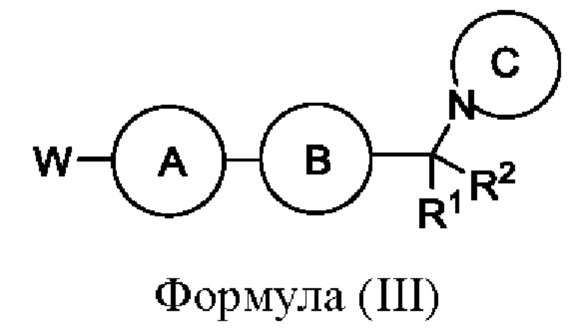
или его солью, где W представляет собой подходящую уходящую группу, например Cl, Br, I или OTf, и R1, R2, кольцо А, кольцо В и кольцо С являются такими, как определено в любом из воплощений здесь. Взаимодействие удобно проводить в подходящем растворителе (например 2-метилтетрагидрофуране) в присутствии основания (например карбоната цезия) и в присутствии подходящего катализатора (например Pd Ruphos 3-го поколения) и лиганда (например Ruphos) при подходящей температуре (например в диапазоне от 60°С до 80°С).
Альтернативно, в зависимости от природы кольца А, соединения формулы (I) или их фармацевтически приемлемые соли также могут быть получены посредством взаимодействия соединения формулы (II) или его соли и соединения формулы (III) или его соли с использованием стандартных условий ароматического замещения, хорошо известных специалистам в данной области техники, например, с подходящим основанием (например диизопропилэтиламином) в подходящем растворителе (например 1-бутаноле) при подходящей температуре (например в диапазоне от 60°С до 120°С).
Таким образом, соединения формулы (II) и (III) и их соли используются в качестве промежуточных соединений при получении соединений формулы (I) и представляют собой дополнительное воплощение.
Соединения формулы (II) могут быть получены, например, по следующей схеме 1:
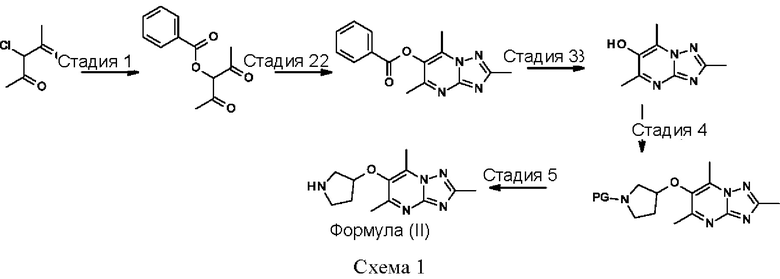
где PG представляет собой подходящую защитную группу азота, например Boc.
Стадия 1: осуществляют с использованием подходящего нуклеофила (например бензойной кислоты) в присутствии подходящего основания (например КОН) в подходящем растворителе (например DMF (диметилформамид)) при подходящей температуре (например в диапазоне от 20°С до 50°С).
Стадия 2: осуществляют с использованием подходящего субстрата циклизации (например 3-метил-1H-1,2,4-триазол-5-амина) в подходящем растворителе (например АсОН) при подходящей температуре (например в диапазоне от 70°С до 90°С).
Стадия 3: осуществляют в стандартных условиях гидролиза, хорошо известных специалистам в данной области техники, например 1 М NaOH (водн.) в ЕЮН при комнатной температуре.
Стадия 4: осуществляют в стандартных условиях, хорошо известных специалистам в данной области техники. Взаимодействие может быть проведено в стандартных условиях реакции Мицунобу с подходящим спиртовым субстратом (например трет- бутил-3-гидроксипирролидин-1-карбоксилатом) с трифенилфосфином и DIAD (диизопропилазодикарбоксилатом) в THF (тетрагидрофуране) при комнатной температуре.
Стадия 5: осуществляют в стандартных условиях снятия защиты, хорошо известных специалистам в данной области техники. Например, когда PG (защитная группа) представляет собой Boc, взаимодействие может быть проведено при использовании 4 М НС1 в 1,4-диоксане в МеОН при комнатной температуре.
Соединения формулы (III), где R1 и R2 представляют собой водород, могут быть получены, например, посредством взаимодействия соединения формулы (IV)
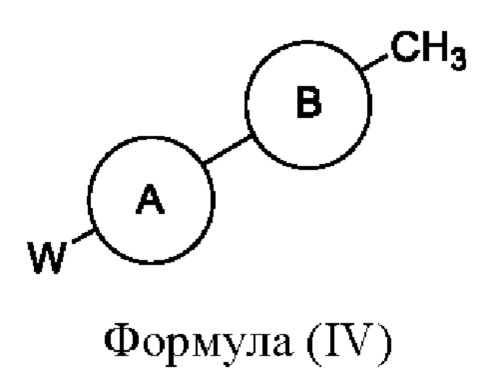
или его соли с подходящим хлорирующим реагентом (например трихлоризоциануровой кислотой) в подходящем растворителе (например дихлорэтане) с последующим взаимодействием с подходящим амином на основе кольца С (например морфолином) при подходящей температуре (например при комнатной температуре), где W представляет собой подходящую уходящую группу, например Cl, Br, I или OTf, а кольцо А, кольцо В и кольцо С являются такими, как определено в любом из воплощений здесь. В одном воплощении кольцо А представляет собой фенил, а кольцо В представляет собой пирадазинил.
Соединения формулы (I), где X представляет собой СН2, могут быть получены, например, посредством взаимодействия соединений формулы (III) или их солей, где R1, R2, кольцо А, кольцо В и кольцо С являются такими, как определено в любом из воплощений здесь, с соединением формулы (V)
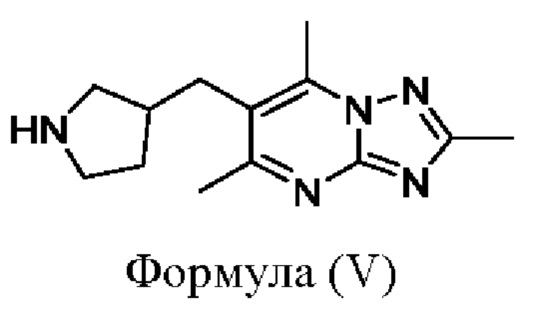
или его солью. Взаимодействие удобно проводить в подходящем растворителе (например 1,4-диоксане) в присутствии основания (например карбоната цезия) и в присутствии подходящего катализатора (например Pd Ruphos 3-го поколения) и лиганда (например Ruphos) при подходящей температуре (например в диапазоне от 60°С до 90°С).
Следовательно, соединения формулы (V) также используются в качестве промежуточных соединений при получении соединений формулы (I) и представляют собой дополнительное воплощение.
Соединения формулы (V) могут быть получены, например, по следующей схеме 2:
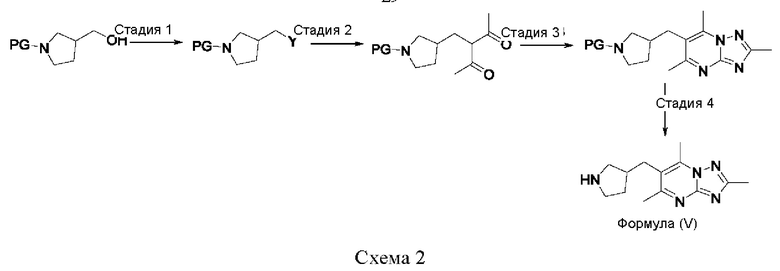
где PG представляет собой подходящую защитную группу азота, например Boc, а Y представляет собой подходящую уходящую группу, например Cl, Br, I или OMs.
Стадия 1: осуществляют в условиях, которые превращают ОН в подходящую уходящую группу, например, если Br выбран в качестве уходящей группы, тогда взаимодействие может быть проведено в условиях, хорошо известных специалистам в данной области техники, например, с трифенилфосфином, тетрабромметаном в DCM (дихлорметане) при температуре в диапазоне от 0°С до комнатной температуры.
Стадия 2: осуществляют с использованием подходящего нуклеофила (например пентан-2,4-диона) в присутствии подходящего основания (например карбоната калия) в подходящем растворителе (например DMF) при подходящей температуре (например в диапазоне от 60°С до 80°С).
Стадия 3: осуществляют с использованием подходящего субстрата циклизации (например 3-метил-1H-1,2,4-триазол-5-амина) в подходящем растворителе (например АсОН) при подходящей температуре (например в диапазоне от 70°С до 90°С).
Стадия 4: осуществляют с использованием стандартных условий снятия защиты, хорошо известных специалистам в данной области техники. Например, когда PG представляет собой Boc, взаимодействие может быть проведено при использовании 4 М HCl в 1,4-диоксане в МеОН при комнатной температуре.
Альтернативно, соединения формулы (I), где X представляет собой СН2, могут быть получены посредством взаимодействия соединения формулы (VI)
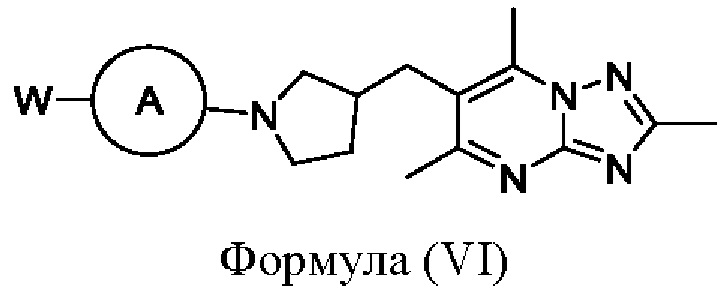
или его соли, где W представляет собой подходящую уходящую группу, например Cl, Br, I или OTf, и кольцо А является таким, как определено в любом из воплощений здесь, с соединением формулы (VII)
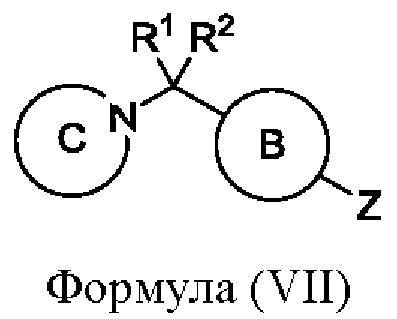
или его солью, где Z представляет собой подходящую уходящую группу, например пинаколборонат, бороновую кислоту или оловоорганическое соединение, a R1, R2, кольцо В и кольцо С являются такими, как определено в любом из воплощений здесь. Когда Z представляет собой пинаколборонат, тогда взаимодействие удобно проводить в подходящем растворителе (например смеси 1,4-диоксана и воды) в присутствии основания (например карбоната цезия) и в присутствии подходящего катализатора (например Pd Xphos 2-го поколения) при подходящей температуре (например в диапазоне от 60°С до 90°С). В одном воплощении R1 и R2 представляют собой водород, кольцо А представляет собой пиразинил, кольцо В представляет собой фенил, и кольцо С представляет собой 4-метил-1-пиперазинил.
Соединения формулы (VI) или их соли могут быть получены посредством взаимодействия соединения формулы (V) или его соли с кольцом А при использовании стандартных химических механизмов ароматического замещения. Кольцо А будет иметь подходящую уходящую группу (например хлор или бром), и взаимодействие удобно проводить в подходящем растворителе (например 1-бутаноле) в присутствии подходящего основания (например диизопропилэтиламина) при подходящей температуре (например от 60°С до 120°С). Альтернативно, соединения формулы (VI) или их соли могут быть получены посредством взаимодействия соединения формулы (V) или его соли с кольцом А при использовании стандартных условий кросс-сочетания. Кольцо А будет иметь подходящую уходящую группу (например хлор, бром или йод) в подходящем растворителе (например 1,4-диоксане) в присутствии основания (например карбоната цезия) и в присутствии подходящего катализатора, (например Pd Ruphos 3-го поколения) при подходящей температуре (например от 60°С до 90°С).
Альтернативно, соединения формулы (I), где X представляет собой СН2, могут быть получены посредством взаимодействия соединения формулы (VIII)
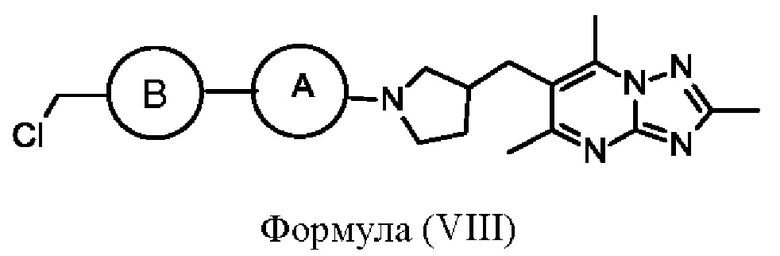
или его соли с подходящим амином на основе кольца С, где кольцо А, кольцо В и кольцо С являются такими, как определено в любом из воплощений здесь. Взаимодействие может быть проведено в стандартных условиях, хорошо известных специалистам в данной области техники, например с триэтиламином в THF при 60°С.
Альтернативно, соединения формулы (I), где X представляет собой СН2, могут быть получены посредством взаимодействия соединения формулы (IX)
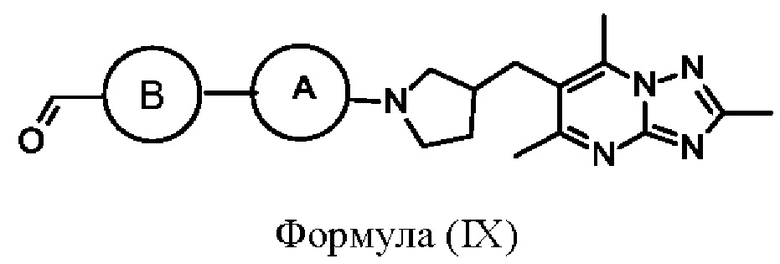
или его соли с подходящим амином на основе кольца С, где кольцо А, кольцо В и кольцо С являются такими, как определено в любом из воплощений здесь. Взаимодействие может быть проведено в стандартных условиях, хорошо известных специалистам в данной области техники, например с триацетоксиборгидридом натрия, АсОН в DCM при комнатной температуре.
Следует понимать, что некоторые из различных кольцевых заместителей в соединениях по настоящему раскрытию могут быть введены посредством стандартных реакций ароматического замещения или образованы посредством обычных модификаций функциональных групп либо до, либо сразу после способов, упомянутых выше, и как таковые включены в аспект способа раскрытия. Например, соединения формулы (I) могут быть превращены в другие соединения формулы (I) посредством стандартных реакций ароматического замещения или посредством обычных модификаций функциональных групп. Такие реакции и модификации включают, например, введение заместителя посредством реакции ароматического замещения, восстановление заместителей, алкилирование заместителей и окисление заместителей. Реагенты и реакционные условия для таких методик хорошо известны в области химии.
Также следует понимать, что в некоторых взаимодействиях, упомянутых здесь, может быть необходимым/желательным защитить любые чувствительные группы в соединениях. Случаи, когда защита необходима или желательна, и подходящие способы защиты известны специалистам в данной области техники. Могут быть использованы обычные защитные группы в соответствии со стандартной практикой (для иллюстрации смотри T.W. Green, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991). Таким образом, если реагенты включают группы, такие как амино, карбокси или гидрокси, может быть желательно защитить группу в некоторых из упомянутых здесь взаимодействий.
В любом из воплощений, где упоминается соединение формулы (II)-(ГХ) или его соль, следует понимать, что такие соли необязательно должны быть фармацевтически приемлемыми солями.
Соединения формулы (I) и любые промежуточные соединения, используемые для их получения, могут быть получены посредством способов, аналогичных тем, которые показаны в разделе «Примеры».
Биологические анализы
Следующие анализы in vitro использовали для измерения эффектов описанных здесь соединений.
В описании анализов использовали следующие сокращения: BCECF AM - 2',7'- ацетоксиметиловый эфир бис-(2-карбоксиэтил)-5-(и-6)-карбоксифлуоресцеина; DMSO - диметилсульфоксид; FBS - фетальная бычья сыворотка; HBSS - сбалансированный солевой раствор Хэнкса; HEPES - 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота; OD - оптическая плотность; RPMI - среда 1640, разработанная в Roswell Park Memorial Institute.
Эффективность соединения в анализах определяли посредством измерения значения IC50 (концентрация тестируемого соединения, при которой происходит ингибирование 50% биологической активности). Значения IC50 рассчитывали с использованием интеллектуальной модели подбора в пакете программ для анализа Screener (Genedata AG).
Обоснование:
МСТ4 преимущественно экспрессируется в нормальных скелетных мышцах и активируется в ряде солидных опухолей, где он способствует оттоку лактата из клетки, тем самым предотвращая внутриклеточное закисление. Таким образом, ингибирование активности МСТ4 представляет собой потенциальное повышение терапевтической эффективности при наличии онкологии. МСТ1 является близкородственным транспортером монокарбоксилата и, как таковой, представляет собой ключевую мишень для селективного воздействия ингибиторов МСТ4. Несмотря на то, что МСТ4 в первую очередь участвует в оттоке лактата из клеток, можно управлять притоком лактата в in vitro системе, как описано для систем анализа (b) и (d) ниже.
Целью этих тестов является идентификация соединений, которые влияют на отток лактата в линии нативных клеток с гликолитическим фенотипом, преимущественно экспрессирующих МСТ4, и транспорт лактата в клетки в следующих системах анализа in vitro:
a) клетки SK-Br-3 (АТСС НТВ-30) - линия аденокарциномы молочной железы человека с нативным гликолитическим фенотипом, преимущественно экспрессирующая МСТ4 (подтверждено вестерн-блоттингом)
клетки NCI-H358 (АТСС CRL-5807) - линия аденокарциномы легких человека, преимущественно экспрессирующая МСТ4, небольшое количество МСТ2 и не экспрессирующая МСТ1 (подтверждено вестерн-блоттингом)
c) клетки K562 (АТСС CCL-243) - линия эритролейкемии человека, преимущественно экспрессирующая МСТ1, небольшое количество МСТ2 и не экспрессирующая МСТ4 (подтверждено вестерн-блоттингом)
d) клетки INS-1 МСТ4 (родительская линия INS-1, подаренная AstraZeneca Женевским университетом, Швейцария) - линия бета-клеток поджелудочной железы крысы, изначально не содержащая ни одной изоформы МСТ и модифицированная собственными силами для стабильной экспрессии человеческого МСТ4 (подтверждено вестерн-блоттингом).
Система анализа (а):
Соединения формулы I в 100% DMSO добавляли в пустые 384-луночные планшеты для анализа (Costar #3712) посредством акустического дозирования (120 нл/лунка в диапазоне концентраций). Клетки SK-Br-3 (АТСС НТВ-30) высевали в аналитические планшеты (непосредственно на тестируемое соединение) при плотности 4000 клеток/лунка в 40 мкл среды RPMI (Sigma #R0883), содержащей 1% L-глутамина (Sigma #G7513) и 20% FBS (Sigma #F7524). Планшеты для анализа закрывали крышками и инкубировали в течение 4 часов при 37°С и 5% СО2.
После инкубирования с тестируемым соединением 5 мкл среды переносили во второй 384-луночный планшет для анализа (Costar #3712), используя платформу для автоматического дозирования с помощью наконечников (CyBio Felix). Количество молочной кислоты, присутствующей в среде, количественно определяли с помощью коммерческого набора для определения лактата (Trinity Biotech #735-10), используя следующий принцип сопряженных ферментов: молочная кислота превращается в пируват и Н2О2 с помощью лактатоксидазы; в присутствии образовавшегося Н2О2 пероксидаза катализирует окислительную конденсацию предшественника хромогена с образованием окрашенного красителя с максимумом поглощения при 540 нм (увеличение поглощения прямо пропорционально увеличению лактата в образце). Затем оптическую плотность измеряли на автоматическом считывающем устройстве для микропланшетов (PerkinElmer EnVisiori) с использованием фильтра 535 нм, и значения OD нормализовали к контрольным лункам (обработаны либо DMSO (максимальный анализируемый сигнал), либо химическим веществом, относящимся к описанным в этом патенте (минимальный анализируемый сигнал)) перед построением кривых зависимости эффекта от концентрации для определения значения IC50.
Системы анализа (b), (с):
Соединения формулы I в 100% DMSO добавляли в пустые 384-луночные планшеты для анализа (Costar #3683) посредством акустического дозирования (90 нл/лунка в диапазоне концентраций).
Клетки NCI-H358 (АТСС CRL-5807) или клетки K562 (АТСС CCL-243) размораживали непосредственно после криоконсервации, промывали и ресуспендировали в HBSS (Gibco #14170) с 1 мМ [конечный] HEPES (Gibco #15630).
Клетки нагружали рН-чувствительным красителем BCECF AM (Invitrogen #В1150) перед промывкой для удаления избытка красителя и высевали в планшеты для анализа (непосредственно на тестируемое соединение) при плотности 15000 клеток/лунка (NCI- Н358) или 30000 клеток/лунка (K562) в 30 мкл HBSS с 1 мМ [конечный] HEPES. Планшеты для анализа закрывали крышками и центрифугировали в центрифуге для планшетов при 170 g в течение 1 минуты. Затем планшеты заворачивали в фольгу и инкубировали в темноте в течение 1 часа при комнатной температуре.
После инкубирования с тестируемым соединением 10 мкл 25 мМ L-лактата натрия (Sigma #L7022), приготовленного в HBSS с 1 мМ [конечный] HEPES, непосредственно добавляли в 30 мкл среды в тест-лунках, получая 6,25 мМ [конечный]. Это добавление проводили непосредственно на платформе FLIPR Tetra (Molecular Devices) как часть протокола кинетического считывания в реальном времени. Флуоресценцию клеток с использованием набора фильтров Ех470-495_Em515-575 измеряли в динамике и регистрировали изменение относительно исходного уровня (добавление до лактата) до «добавления лактата плюс 80 секунд». Процентное изменение от исходного уровня рассчитывали для каждой тест-лунки, и значения нормализовали к контрольным лункам (обработаны либо DMSO (максимальный анализируемый сигнал), либо химическим веществом, относящимся к описанным в этом патенте (минимальный анализируемый сигнал)) перед построением кривых зависимости эффекта от концентрации для определения значения IC50.
Система анализа (d):
Соединения формулы I в 100% DMSO добавляли в пустые 384-луночные планшеты для анализа (Costar #3683) посредством акустического дозирования (90 нл/лунка в диапазоне концентраций).
Клетки INS-1 МСТ4 были получены посредством трансфекции родительских клеток следующей последовательностью ДНК (дезоксирибонуклеиновая кислота), вставленной в вектор экспрессии млекопитающих pcDNA3.1 (ThermoFisher #V79020):
 .
.
Полученный пул клеток непрерывно культивировали при 37°С и 5% СО2 с селективным антибиотиком в среде RPMI (Sigma #R0883), содержащей 1% L-глутамина (Sigma #G7513), 10% FBS (Sigma #F7524), 10 мМ [конечный] HEPES (Gibco #15630), 0,004% р-меркаптоэтанола (Sigma #M6250) и 100 мкг/мл генетицина (Thermo Fisher #10131027). Отдельный клон был отобран и размножен перед криоконсервацией в нескольких отдельных флаконах для дальнейшего использования. Перед тестированием в биологическом анализе отдельный криофлакон с INS-1 МСТ4 размораживали и непрерывно пересевали в течение нескольких недель (в среде с селективным антибиотиком генетицином, описанной ранее) для получения необходимого количества клеток. Во время тестирования клетки отбирали с поверхности колб с культурой, объединяли, промывали и ресуспендировали в HBSS (Gibco #14170) с 1 мМ [конечный] HEPES (Gibco #15630).
Клетки нагружали рН-чувствительным красителем BCECF AM (Invitrogen #В1150) перед промывкой для удаления избытка красителя и высевали в планшеты для анализа (непосредственно на тестируемое соединение) при плотности 15000 клеток/лунка в 30 мкл HBSS с 1 мМ [конечный] HEPES. Планшеты для анализа закрывали крышками и центрифугировали в центрифуге для планшетов при 170 g в течение 1 минуты. Затем планшеты заворачивали в фольгу и инкубировали в темноте в течение 1 часа при комнатной температуре.
После инкубирования с тестируемым соединением 10 мкл 25 мМ L-лактата натрия (Sigma #L7022), приготовленного в HBSS с 1 мМ [конечный] HEPES, непосредственно добавляли в 30 мкл среды в тест-лунках, получая 6,25 мМ [конечный]. Это добавление проводили непосредственно на платформе FLIPR Tetra (Molecular Devices) как часть протокола кинетического считывания в реальном времени. Флуоресценцию клеток с использованием набора фильтров Ех470-495_Em515-575 измеряли в динамике и регистрировали изменение относительно исходного уровня (добавление до лактата) до «добавления лактата плюс 80 секунд». Процентное изменение от исходного уровня рассчитывали для каждой тест-лунки, и значения нормализовали к контрольным лункам (обработаны либо DMSO (максимальный анализируемый сигнал), либо химическим веществом, относящимся к описанным в этом патенте (минимальный анализируемый сигнал)) перед построением кривых зависимости эффекта от концентрации для определения значения IC50.
Следующие анализы in vivo использовали для измерения эффектов описанных здесь соединений в комбинации с другими агентами.
Модель ксенотрансплантата рака легкого человека - комбинация с VEGFR TKI
Эффективность ингибиторов МСТ4 in vivo была протестирована на человеческих моделях ксенотрансплантатов. NSCLC клеточная линия NCI Н358 может быть выращена в виде подкожного ксенотрансплантата у самок голых мышей, а объем опухоли рассчитан на основе билатеральных измерений с помощью калипера. Для исследований эффективности три миллиона клеток NCI Н358 инокулировали подкожно на левый бок животного в объеме 0,1 мл бессывороточной среды (RPMI) и матригеля. Животные были распределены в группы лечения через 14 суток после имплантации клеток и получали либо AZD2171 (3 мг/кг один раз в сутки) (Wedge et al., (2005) Cancer Res. 65: 4389-4400), либо соединение формулы (I) (100 мг/кг дважды в сутки), либо их комбинацию через желудочный зонд. Контрольный носитель 0,5% гидроксипропилметилцеллюлоза/0,1% Tween 80 (полиоксиэтилен (20) сорбитан моноолеат) вводили дважды в сутки перорально. Дозирование продолжали в течение 17 суток, и объем опухоли, массу тела и состояние опухоли регистрировали дважды в неделю на протяжении всего исследования.
Результаты тестирования соединения из примера 62 показаны на Фиг. 3. Ингибитор VEGF AZD2171 был эффективен, демонстрируя 33% (р не более 0,001) регресс опухоли по сравнению с группой носителя. Комбинация AZD2171 с ингибитором МСТ4 из примера 62 продемонстрировала более существенное изменение, составляющее 72%-й регресс опухоли (р не более 0,001 по сравнению с контролем), которое также значительно отличалось (р равно 0,013) от AZD2171, вводимого отдельно. Размеры групп на момент завершения исследования: носитель (n равно 11); соединение из примера 62 (n равно 4); AZD2171 (n равно 10); AZD2171 плюс соединение из примера 62 (n равно 9).
Мышиная сингенная модель - комбинация с анти-CTLA4 антителами и комбинации с анти-PD-1 антителами
Эффективность ингибиторов МСТ4 in vivo была протестирована на сингенных моделях мышей. Чтобы протестировать селективное воздействие ингибиторов МСТ4 формулы (I), МСТ1 нокаутировали (КО) в сингенной модели на основе клеточной линии МС38 при использовании точного редактирования генома с помощью CRISPR (короткие палиндромные повторы, регулярно расположенные группами). Линия колоректальных клеток мышей МС38 МСТ1 КО может быть выращена подкожно у самок мышей С57.В16, а объем опухоли рассчитан на основе билатеральных измерений с помощью калипера. Для исследований эффективности десять миллионов клеток инокулировали подкожно на левый бок животного в объеме 0,1 мл бессывороточной среды DMEM (модифицированная по способу Дульбекко среда Игла). Животные были рандомизированы по массе тела во время имплантации клеток, и лечение начинали на следующие сутки. В первом из этих исследований, показанных на Фиг. 4, группы монотерапии получали либо соединение формулы (I) (100 мг/кг дважды в сутки) через желудочный зонд, либо анти-CTLA4 антитело (aCTLA4, антитело против CTLA-4 9D9 mlgGl, описанное в WO 200712373) (10 мг/кг дважды в неделю) внутрибрюшинно. Комбинированные группы состояли из соединения формулы (I) (100 или 10 мг/кг перорально дважды в сутки) с анти-CTLA4 антителом (10 мг/кг внутрибрюшинно дважды в неделю). Во втором из этих исследований, показанных на Фиг. 5, группы монотерапии получали либо соединение формулы (I) (30 мг/кг перорально дважды в сутки) с анти-PD-1 антителом (от Bio X Cell, номер по каталогу ВЕ0146, партия 665417s1, 6,78 мг/мл) (10 мг/кг внутрибрюшинно дважды в неделю). В обоих исследованиях дозирование продолжалось до 6 недель, и объем опухоли, массу тела и состояние опухоли регистрировали три раза в неделю. В обоих исследованиях носителем являлась смесь 0,5% гидроксипропилметилцеллюлоза/0,1% Tween 80, вводимая дважды в сутки через желудочный зонд, с PBS/а, вводимым дважды в неделю внутрибрюшинно.
Результаты тестирования соединения из примера 62 в первом исследовании сингенных моделей показаны на Фиг. 4. Индивидуальные профили роста опухоли показаны на животных, изъятых из исследования с использованием критериев времени до наступления события, основанных на максимальном объеме опухоли 1,5 см, состоянии опухоли или пределах благополучия животных. Из-за изъятия животных из исследования во время более поздних стадий эксперимента геометрические средние значения процентного ингибирования были рассчитаны на 15 сутки. По сравнению с контрольным носителем монотерапия соединением из примера 62 (100 мг/кг) обеспечивала 60,0%-ное (р не более 0,05) ингибирование роста на эти сутки. Монотерапия aCTLA4 (10 мг/кг) оказалась неэффективной с 6,1% (незначим). Комбинация соединения из примера 62 (10 и 100 мг/кг) с анти-CTLA4 антителом показала 67,4%-ное (р не более 0,01) и 82,2%-ное (р не более 0,001) ингибирование соответственно, при этом комбинация соединения из примера 62 (100 мг/кг дважды в сутки) плюс анти-CTLA4 антитело достигла статистической значимости (р равно 0,0311) по сравнению с соединением из примера 62 (100 мг/кг дважды в сутки), введенным отдельно. Размеры групп на 15 сутки: носитель (n равно 12); соединение из примера 62 (100 мг/кг) (n равно 11); анти-CTLA4 антитело (10 мг/кг) (n равно 11); соединение из примера 62 (100 мг/кг) плюс анти-CTLA4 антитело (n равно 12); соединение из примера 62 (10 мг/кг) плюс анти-CTLA4 антитело (n равно 9).
Для тестирования соединения из примера 62 во втором исследовании сингенных моделей результаты показаны на Фиг. 5. Снова индивидуальные профили роста опухоли показаны на животных, изъятых из исследования с использованием критериев времени до наступления события, основанных на максимальном объеме опухоли 1,5 см, состоянии опухоли или пределах благополучия животных. Из-за того, что животные достигли конечной точки события в разное время на более поздних стадиях эксперимента, геометрические средние значения процентного ингибирования были рассчитаны на 13 сутки. По сравнению с контрольным носителем монотерапия соединением из примера 62 (30 мг/кг) обеспечивала 30,1%-ное (незначим) ингибирование роста на эти сутки. Эффективность монотерапии αPD-1 (10 мг/кг) была аналогичной, обеспечивая 33,4%-ное (незначим) ингибирование. Комбинация соединения из примера 62 (30 мг/кг) с анти-PD-1 антителом была более эффективной чем монотерапия, демонстрируя 82,3%-ное (р не более 0,001) ингибирование роста. Размеры групп на 13 сутки: носитель (n равно 8); соединение из примера 62 (30 мг/кг) (n равно 10); анти-PD-1 антитело (10 мг/кг) (n равно 11); соединение из примера 62 (30 мг/кг) плюс анти-PD-1 антитело (n равно 10).
Следует отметить, что диаграммы, показанные на Фиг. 4 и 5, представляют данные о росте опухоли у отдельных животных, при сравнении роста у групп с контрольным носителем с группами лечения.
Для примеров были получены следующие данные анализов (a)-(d) (представленные значения pIC50 являются вычисленным средним результатом экспериментов, проведенных по меньшей мере в трех повторностях, за исключением примера 32, отмеченного *, где значение pIC50 является вычисленным средним результатом экспериментов, проведенных в двух повторностях):
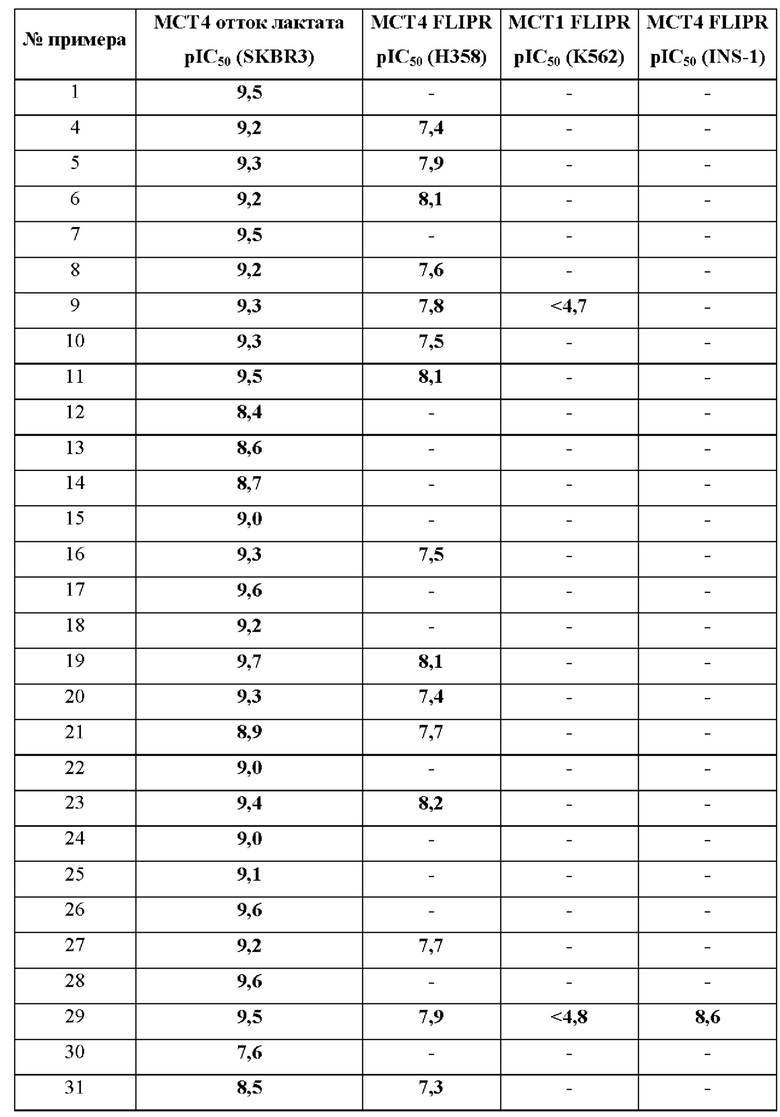
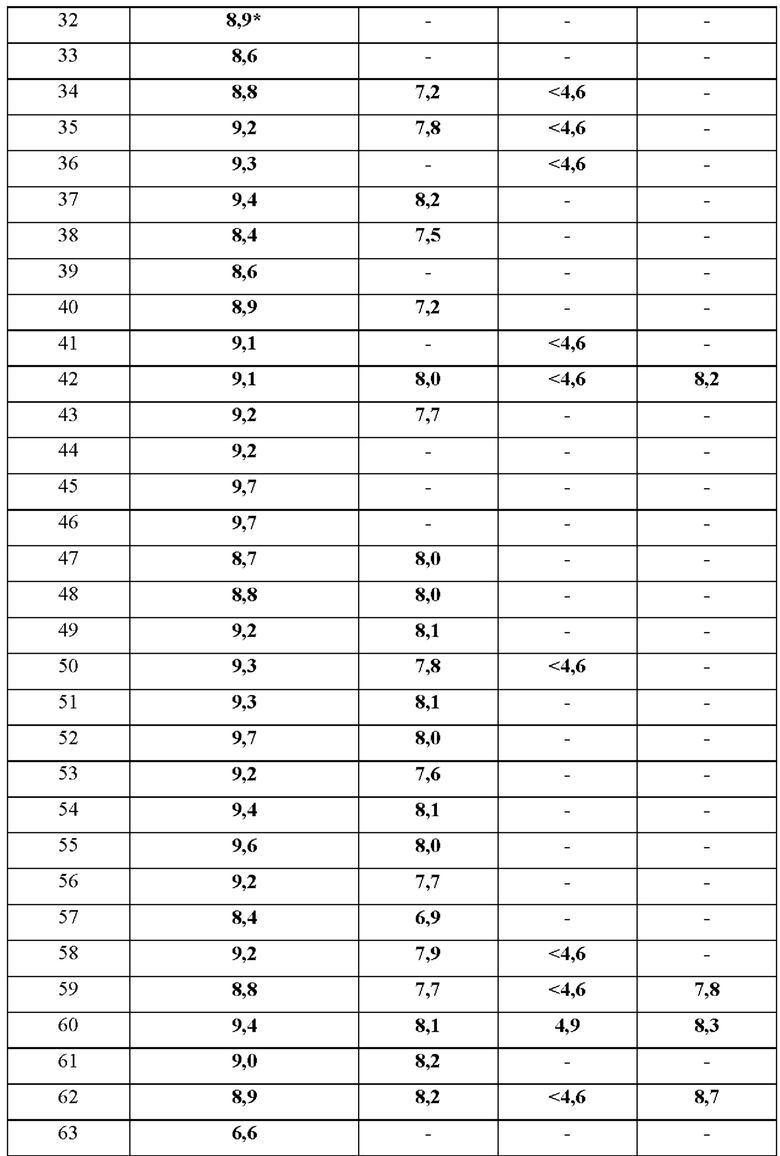
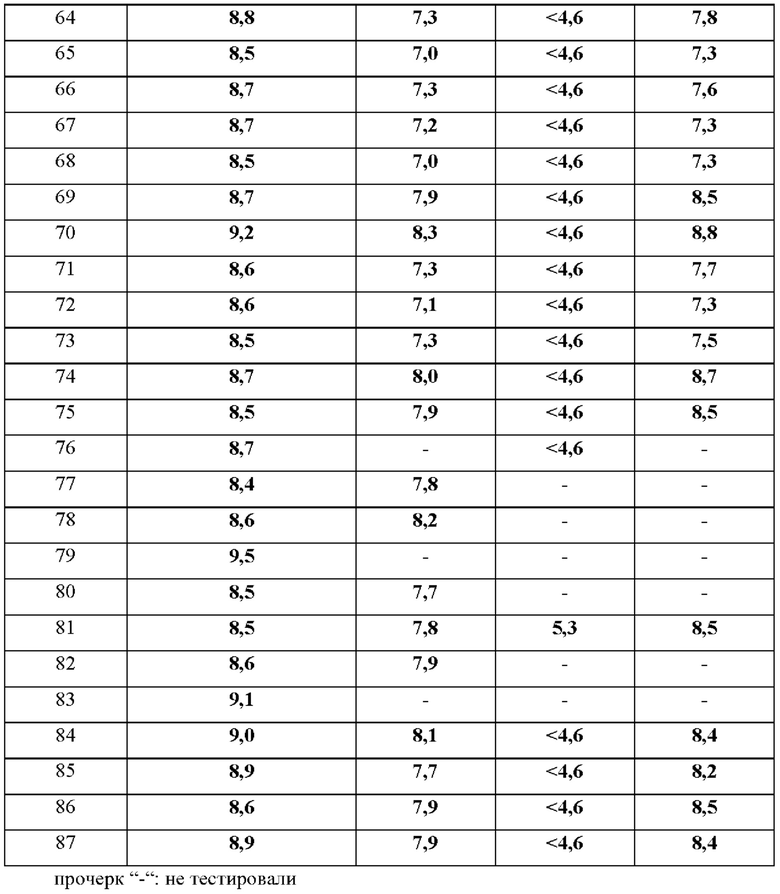
Данные показывают, что описанные здесь соединения влияют как на отток лактата из линии опухолевых клеток, преимущественно экспрессирующих МСТ4, так и в соответствующих условиях ингибируют транспорт лактата в МСТ4-зависимые клетки и, следовательно, полезны в качестве ингибиторов МСТ4.
Соединения могут быть дополнительно выбраны на основе дополнительных биологических или физических свойств, которые могут быть измерены посредством методов, известных в данной области техники, и которые могут быть использованы при оценке или выборе соединений для терапевтического или профилактического применения.
Ожидается, что благодаря их ингибирующей активности в отношении МСТ4 соединения формулы (I) и их фармацевтически приемлемые соли будут полезны в терапии.
Подразумевается, что термин "терапия" имеет свое обычное значение в отношении заболевания с ориентировкой на полное или частичное облегчение одного, нескольких или всех его симптомов, или корректировку или компенсацию лежащей в основе патологии. Термин "терапия" также включает "профилактику", если не оговорено особо. Термины "терапевтический" и "терапевтически" следует интерпретировать соответствующим образом.
Подразумевается, что термин "профилактика" имеет свое обычное значение и включает первичную профилактику для предупреждения развития заболевания и вторичную профилактику, при которой заболевание уже развилось, и пациент временно или постоянно защищен от обострения или ухудшения заболевания или развития, новых симптомов, связанных с заболеванием.
Термин "лечение" используется как синоним "терапии". Точно так же термин "лечить" можно рассматривать как "применение терапии", где "терапия" имеет значение, определенное здесь.
Когда упоминается термин "рак", он включает и неметастатический рак, а также и метастатический рак, так что лечение рака включает лечение как первичных опухолей, так и метастазов опухолей.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
В одном воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении заболевания, опосредованного МСТ4. В одном воплощении заболевание, опосредованное МСТ4, представляет собой рак. В одном воплощении рак выбран из группы, состоящий из колоректального рака, глиобластомы, рака желудка, рака яичников, диффузной В- клеточной крупноклеточной лимфомы, хронического лимфолейкоза, острого миелоидного лейкоза, плоскоклеточного рака головы и шеи, рака молочной железы, рака предстательной железы, рака мочевого пузыря, гепатоцеллюлярной карциномы, рака почки, рака щитовидной железы, рака поджелудочной железы, мелкоклеточного рака легкого и немелкоклеточного рака легкого.
В одном воплощении рак представляет собой немелкоклеточный рак легкого.
В одном воплощении рак представляет собой аденокарциному легкого.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака.
В одном воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения заболевания, опосредованного МСТ4. В одном воплощении заболевание, опосредованное МСТ4, представляет собой рак. В одном воплощении рак выбран из группы, состоящей из колоректального рака, глиобластомы, рака желудка, рака яичников, диффузной В-клеточной крупноклеточной лимфомы, хронического лимфолейкоза, острого миелоидного лейкоза, плоскоклеточного рака головы и шеи, рака молочной железы, рака предстательной железы, рака мочевого пузыря, гепатоцеллюлярной карциномы, рака почки, рака щитовидной железы, рака поджелудочной железы, мелкоклеточного рака легкого и немелкоклеточного рака легкого.
В одном воплощении рак представляет собой немелкоклеточный рак легкого.
В одном воплощении рак представляет собой аденокарциному легкого.
В одном воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака.
В одном воплощении предложен способ лечения заболевания, при котором ингибирование МСТ4 является полезным, у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном воплощении заболевание представляет собой рак. В одном воплощении рак выбран из группы, состоящей из колоректального рака, глиобластомы, рака желудка, рака яичников, диффузной В-клеточной крупноклеточной лимфомы, хронического лимфолейкоза, острого миелоидного лейкоза, плоскоклеточного рака головы и шеи, рака молочной железы, рака предстательной железы, рака мочевого пузыря, гепатоцеллюлярной карциномы, рака почки, рака щитовидной железы, рака поджелудочной железы, мелкоклеточного рака легкого и немелкоклеточного рака легкого.
В одном воплощении рак представляет собой немелкоклеточный рак легкого.
В одном воплощении рак представляет собой аде но карциному легкого.
В одном воплощении предложен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Опухоли, которые избирательно экспрессируют МСТ4 по сравнению с МСТ1, имеют повышенную вероятность ответа на лечение ингибитором МСТ4. Следовательно, пациенты, опухоли которых экспрессируют МСТ1 на низком уровне, вероятно, лучше будут реагировать на ингибитор МСТ4, чем те пациенты, опухоли которых экспрессируют МСТ1 на более высоком уровне. В большинстве случаев, пациенты, опухоли которых имеют высокое соотношение экспрессии МСТ4 : МСТ1 (из-за низкого уровня экспрессии МСТ1), вероятно, демонстрируют лучший ответ, поэтому оценка относительных уровней экспрессии как МСТ1, так и МСТ4 предоставляет возможность отбора пациентов для лечения ингибитором МСТ4. Способы определения относительных уровней экспрессии МСТ1 и МСТ4 известны в данной области техники и описаны в WO 2010/089580, включенной здесь посредством ссылки.
В одном воплощении предложен способ лечения рака, включающий (I) тестирование образца опухоли, полученного от пациента, страдающего или вероятно страдающего от рака, на селективную экспрессию МСТ4 по сравнению с МСТ1 и (II) введение пациенту, имеющему опухоль, которая селективно экспрессирует МСТ4 по сравнению с МСТ1, соединения формулы (I) или его фармацевтически приемлемой соли.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака у пациента, при котором злокачественная опухоль селективно экспрессирует МСТ4 по сравнению с МСТ1.
В одном воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака у пациента, при котором злокачественная опухоль селективно экспрессирует МСТ4 по сравнению с МСТ1.
В одном воплощении предложен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, где злокачественная опухоль селективно экспрессирует МСТ4 по сравнению с МСТ1.
Термин "терапевтически эффективное количество" относится к количеству соединения формулы (I), как описано в любом из воплощений здесь, которое является эффективным для обеспечения "терапии" у субъекта или для того, чтобы "лечить" заболевание или расстройство у субъекта. В случае рака терапевтически эффективное количество может вызвать любое из наблюдаемых или измеримых изменений у субъекта, как описано выше в определении "терапия", "лечение" и "профилактика". Например, эффективное количество может снижать число раковых или опухолевых клеток; уменьшать общий размер опухоли; подавлять или останавливать инфильтрацию опухолевых клеток в периферические органы, включая, например, мягкие ткани и кости; подавлять и останавливать метастазирование опухоли; подавлять и останавливать рост опухоли; облегчать до некоторой степени один или более чем один симптом, связанный с раком; снижать заболеваемость и смертность; улучшать качество жизни; или вызывать комбинацию таких эффектов. Эффективное количество может представлять собой количество, достаточное для уменьшения симптомов заболевания, реагирующего на ингибирование активности МСТ4. При лечении рака эффективность in vivo может быть измерена, например, посредством оценки продолжительности выживания, времени до прогрессирования заболевания (ВПЗ), частоты ответа (ЧО), продолжительности ответа и/или качества жизни. Как известно специалистам в данной области техники, эффективные количества могут варьироваться в зависимости от пути введения, использования эксципиента и совместного использования с другими агентами. Например, когда используют комбинированную терапию, количество соединения формулы (I) или фармацевтически приемлемой соли, описанное в этом описании, и количество другого фармацевтически активного агента(агентов), когда они объединены, в совокупности являются эффективными для лечения являющегося мишенью расстройства у больного животного. В этом контексте объединенные количества находятся в «терапевтически эффективном количестве», если они при объединении достаточны для уменьшения симптомов заболевания, реагирующего на ингибирование активности МСТ4, как описано выше. Как правило, такие количества могут быть определены специалистом в данной области техники, например исходя из диапазона доз, описанного в данном описании для соединения формулы (I) или его фармацевтически приемлемой соли, и утвержденного или объявленного иным образом диапазона(диапазонов) доз другого фармацевтически активного соединения(соединений).
Термин "теплокровные животные" включает, например, людей.
Противоопухолевое лечение, описанное в этом описании, может быть полезным в качестве монотерапии или может включать, помимо введения соединения формулы (I) или его фармацевтически приемлемой соли, традиционное хирургическое вмешательство, лучевую терапию или химиотерапию; или комбинацию таких дополнительных методов лечения. Такое традиционное хирургическое вмешательство, лучевую терапию или химиотерапию можно проводить одновременно, последовательно или раздельно с лечением соединением формулы (I) или его фармацевтически приемлемой солью.
Когда комбинированную терапию применяют «одновременно», она включает лечение пациента одной дозированной формой (например таблеткой), содержащей как формулу соединения (I) или его фармацевтически приемлемую соль, так и дополнительное противоопухолевое вещество; а также одновременное введение отдельных дозированных форм, при этом каждая по отдельности содержит один из соответствующих компонентов комбинации.
Когда комбинированную терапию применяют «последовательно» или «раздельно», она включает лечение пациента первой дозированной формой (например таблеткой), содержащей соединение формулы (I) или его фармацевтически приемлемую соль с последующим лечением того же пациента второй дозированной формой, содержащей дополнительное противоопухолевое вещество; или лечение пациента одной дозированной формой (например таблеткой), содержащей конкретное противоопухолевое вещество с последующим лечением того же пациента второй дозированной формой, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Интервал между последовательными или отдельными дозами может оценить квалифицированный лечащий врач со ссылкой на информацию в этом описании.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят перед хирургическим вмешательством.
Введение соединения формулы (I) или его фармацевтически приемлемой соли перед хирургическим вмешательством с целью полного или частичного удаления рака можно назвать «неоадъювантной терапией». При таком сценарии цель введения соединения формулы (I) или его фармацевтически приемлемой соли, как правило, состоит в том, чтобы уменьшить размер опухоли-мишени, чтобы увеличить шансы на успешную резекцию. Таким образом, продолжительность введения соединения формулы (I) или его фармацевтически приемлемой соли перед хирургическим вмешательством может оценить квалифицированный лечащий врач со ссылкой на информацию в этом описании.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят после хирургического вмешательства.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят в комбинации с по меньшей мере одним дополнительным противоопухолевым веществом.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, последовательно или раздельно с по меньшей мере одним дополнительным противоопухолевым веществом.
Противоопухолевое лечение, определяемое здесь, может быть применено в качестве монотерапии или может включать помимо соединений, указанных в описании, традиционное хирургическое вмешательство, лучевую терапию, или химиотерапию. Такая химиотерапия может включать одну или более чем одну из следующих категорий противоопухолевых агентов:
I) другие антипролиферативные/противоопухолевые лекарственные средства и их комбинации, используемые в медицинской онкологии, такие как алкилирующие агенты (например цисплатин, оксалиплатин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан, темозоломид и нитрозомочевины); антиметаболиты (например гемцитабин и антифолаты, такие как фторпиримидины, такие как 5-фторурацил и тегафур, ралтитрексед, метотрексат, пеметрексед, цитозинарабинозид и гидроксимочевина); противоопухолевые антибиотики (например антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин и митрамицин); антимитотические агенты (например, алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин, и таксаны, такие как таксол и таксотер, и ингибиторы полокиназы); и ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан и камптотецин);
II) антиангиогенные агенты, такие как агенты, ингибирующие действия фактора роста эндотелия сосудов, например антитело против фактора роста эндотелиальных клеток сосудов, бевацизумаб (Avastin™), и, например ингибитор тирозинкиназы рецептора VEGF, такой как сорафениб, акситиниб, пазопаниб, сунитиниб и цедираниб;
III) иммуннотерапевтические подходы, включая, например ex vivo и in vivo подходы для повышения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4, или гранулоцитарно-макрофагальным колониестимулирующим фактором, подходы для уменьшения анергии Т-клеток, подходы, использующие трансфицированные иммунные клетки, такие как цитокин-трансфицированные дендритные клетки, подходы, использующие цитокин-трансфицированные опухолевые клеточные линии, и подходы, использующие антиидиотипические антитела. Конкретные примеры включают моноклональные антитела, нацеленные на PD-1 (например ниволумаб и пембролизумаб), PD-L1 (например дурвалумаб и атезолизумаб), CTLA4 (например тремелимумаб и ипилимумаб) или CD (кластер дифференцировки) 73 (например олеклумаб), а также лиганд CD40-слитые белки и лиганд GITR (глюкокортикоид-индуцированный рецептор фактора некроза опухолей)-слитые белки;
IV) ингибиторы транспортеров лактата, включая, например ингибиторы МСТ1, такие как AZD3965;
V) агенты, нацеленные на метаболизм опухоли, включая агенты, которые ингибируют GLS1, комплекс I, ингибиторы митохондриального переносчика пирувата.
Поэтому, в одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное противоопухолевое вещество для применения в лечении рака. В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят в комбинации с дополнительным противоопухолевым веществом. В одном воплощении присутствует одно дополнительное противоопухолевое вещество. В одном воплощении присутствуют два дополнительных противоопухолевых вещества. В одном воплощении присутствуют три или более дополнительных противоопухолевых вещества.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное противоопухолевое вещество для применения в одновременном, раздельном или последовательном лечении рака. В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, раздельно или последовательно с дополнительным противоопухолевым веществом.
В одном воплощении предложен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного дополнительного противоопухолевого вещества, где количества соединения формулы (I) или его фармацевтически приемлемой соли и дополнительного противоопухолевого вещества вместе оказывают эффективное противоопухолевое действие.
В одном воплощении предложен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному соединения формулы (I) или его фармацевтически приемлемой соли и одновременное, раздельное или последовательное введение по меньшей мере одного дополнительного противоопухолевого вещества указанному теплокровному животному, где количества соединения формулы (I) или его фармацевтически приемлемой соли и дополнительного противоопухолевого вещества вместе оказывают эффективное противоопухолевое действие.
В любом воплощении дополнительное противоопухолевое вещество выбрано из группы, состоящей из одного или более чем одного из противоопухолевых веществ, перечисленных выше в пунктах (I)-(V).
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное противоопухолевое вещество, выбранное из группы, состоящей из антител к PD-1 (например ниволумаба и пембролизумаба), антител к PD-L1 (например дурвалумаба и атезолизумаба), антител к CTLA4 (например тремелимумаба и ипилимумаба), антител к CD73 (например олеклумаба), лиганд CD40-слитых белков и лиганд GITR-слитых белков, для применения в лечении рака.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное противоопухолевое вещество, выбранное из группы, состоящей из антител к PD-L1 (например дурвалумаба и атезолизумаба), для применения в лечении рака.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль и дурвалумаба для применения в лечении рака.
В одном воплощении рак представляет собой немелкоклеточный рак легкого. В одном воплощении рак представляет собой аденокарциному легкого.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, раздельно или последовательно с по меньшей мере одним дополнительным противоопухолевым веществом, выбранным из группы, состоящей из антител к PD-1 (например ниволумаба и пембролизумаба), антител к PD-L1 (например дурвалумаба и атезолизумаба), антител к CTLA4 (например тремелимумаба и ипилимумаба), антител к CD73 (например олеклумаба), лиганд CD40-слитых белков и лиганд GITR-слитых белков.
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, раздельно или последовательно с по меньшей мере одним дополнительным противоопухолевым веществом, выбранным из группы, состоящей из антител к PD-L1 (например дурвалумаба и атезолизумаба).
В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, раздельно или последовательно с дурвалумабом.
В одном воплощении рак представляет собой немелкоклеточный рак легкого. В одном воплощении рак представляет собой аденокарциному легкого.
В одном воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, раздельно или последовательно с по меньшей мере одним дополнительным противоопухолевым веществом, выбранным из группы, состоящей из антител к PD-1 (например ниволумаба и пембролизумаба), антител к PD-L1 (например дурвалумаба и атезолизумаба), антител к CTLA4 (например тремелимумаба и ипилимумаба), антител к CD73 (например олеклумаба), лиганд CD40-слитых белков и лиганд GITR-слитых белков. В одном воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, раздельно или последовательно с по меньшей мере одним дополнительным противоопухолевым веществом, выбранным из группы, состоящей из антител к PD-L1 (например дурвалумаба и атезолизумаба).
В одном воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, раздельно или последовательно с дурвалумабом.
В одном воплощении рак представляет собой немелкоклеточный рак легкого. В одном воплощении рак представляет собой аденокарциному легкого.
В одном воплощении предложен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли одновременно, раздельно или последовательно с по меньшей мере одним дополнительным противоопухолевым веществом, выбранным из группы, состоящей из антител к PD-1 (например ниволумаба и пембролизумаба), антител к PD-L1 (например дурвалумаба и атезолизумаба), антител к CTLA4 (например тремелимумаба и ипилимумаба), антител к CD73 (например олеклумаба), лиганд CD40-слитых белков и лиганд GITR-слитых белков.
В одном воплощении предложен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли одновременно, раздельно или последовательно с по меньшей мере одним дополнительным противоопухолевым веществом, выбранным из группы, состоящей из антител к PD-L1 (например дурвалумаба и атезолизумаба).
В одном воплощении предложен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли одновременно, раздельно или последовательно с дурвалумабом.
В одном воплощении рак представляет собой немелкоклеточный рак легкого. В одном воплощении рак представляет собой аденокарциному легкого.
В одном воплощении предложена фармацевтическая композиция, содержащая соединение формулы (I) и по меньшей мере одно дополнительное противоопухолевое вещество, для применения в лечении рака. В одном воплощении фармацевтическая композиция также содержит по меньшей мере один фармацевтически приемлемый эксципиент. В одном воплощении противоопухолевое вещество представляет собой антинеопластический агент. Согласно дополнительному воплощению предложен набор, содержащий:
a) соединение формулы (I) или его фармацевтически приемлемую соль в первой единичной дозированной форме;
b) другое дополнительное противоопухолевое вещество в дополнительной единичной дозированной форме;
c) контейнер, предназначенный для того, чтобы вмещать указанную первую и дополнительную единичные дозированные формы; и возможно
d) инструкцию по применению.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть введены в виде фармацевтических композиций, содержащих один или более чем один фармацевтически приемлемый эксципиент.
Поэтому, в одном воплощении предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый эксципиент. Композиции могут находиться в форме, подходящей для перорального применения (например в виде таблеток, лепешек, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного применения (например в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения посредством ингаляции (например в виде тонкоизмельченного порошка или аэрозоля в виде жидкости), для введения посредством инсуффляции (например в виде тонкоизмельченного порошка) или для парентерального введения (например в виде стерильного водного или масляного раствора для внутривенного, подкожного или внутримышечного введения) или в виде суппозитория для ректального введения. Композиции могут быть получены посредством общеизвестных способов с использованием общеизвестных фармацевтических эксципиентов, хорошо известных в данной области техники. Таким образом, композиции, предназначенные для перорального применения, могут содержать, например, один или более чем один краситель, подсластитель, ароматизатор и/или консервант.
В одном воплощении предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый эксципиент, для применения в терапии.
В одном воплощении предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый эксципиент, для применения в лечении рака. В одном воплощении указанный рак выбран из группы, состоящей из колоректального рака, глиобластомы, рака желудка, рака яичников, диффузной В-клеточной крупноклеточной лимфомы, хронического лимфолейкоза, острого миелоидного лейкоза, плоскоклеточного рака головы и шеи, рака молочной железы, рака предстательной железы, рака мочевого пузыря, гепатоцеллюлярной карциномы, рака почки, рака щитовидной железы, рака поджелудочной железы, мелкоклеточного рака легкого и немелкоклеточного рака легкого.
В одном воплощении рак представляет собой немелкоклеточный рак легкого.
В одном воплощении рак представляет собой аденокарциному легкого.
Соединение формулы (I) обычно будет введено теплокровному животному в стандартной дозе в диапазоне 2,5-5000 мг/м2 площади тела животного или приблизительно 0,05-100 мг/кг, и это обычно обеспечивает терапевтически эффективную дозу. Единичная дозированная форма, такая как таблетка или капсула, обычно будет содержать, например 0,1-500 мг активного ингредиента. Суточная доза обязательно будет варьироваться в зависимости от пациента, которого лечат, конкретного пути введения, любых методов лечения, применяемых совместно, и тяжести заболевания, подлежащего лечению.
ПРИМЕРЫ
Аспекты настоящего раскрытия могут быть дополнительно определены посредством ссылки на следующие неограничивающие примеры, в которых подробно описано получение определенных соединений и промежуточных соединений по настоящему раскрытию и способы применения соединений по настоящему раскрытию. Специалистам в данной области техники будет очевидно, что многие модификации, как в веществах, так и в способах, могут быть осуществлены в пределах объема настоящего раскрытия.
Если не оговорено особо, исходные вещества имелись в продаже. Все растворители и коммерческие реагенты были лабораторного качества и были использованы в состоянии поставки.
Общие подробности экспериментов
Теперь изобретение будет проиллюстрировано следующими примерами, в которых, как правило:
I) процессы проводили при комнатной температуре (rt), то есть в диапазоне от 17 до 25°С и в атмосфере инертного газа, такого как N2 или Ar, если не оговорено особо;
II) как правило, за ходом реакций следили с помощью тонкослойной хроматографии (ТСХ) и/или аналитической высокоэффективной жидкостной хроматографии (ВЭЖХ или СВЭЖХ), которые обычно сочетали с масс-спектрометрией (ЖХ/МС). Приведенное время реакции не обязательно является минимально достижимым;
III) при необходимости органические растворы сушили над безводным MgSO4 или Na2SO4, процедуры обработки проводили с использованием традиционных методик разделения фаз или с использованием СКО (сильного катионообменного сорбента), как описано в (XIII), выпаривание проводили либо посредством роторного испарения под вакуумом, либо в испарителях Genevac HT-4/EZ-2 или Biotage V10;
IV) выходы, если они есть, не обязательно являются максимально достижимыми, и при необходимости взаимодействия повторяли, если требовалось большее количество продукта реакции;
V) как правило, структуры конечных продуктов формулы (I) подтверждали посредством методов ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии; данные масс-спектрометрии с электрораспылением получали при использовании системы СВЭЖХ Waters Acquity, соединенной с одноквадрупольным масс-спектрометром Waters, регистрирующим данные как для положительных, так и для отрицательных ионов, и, как правило, указываются только ионы, относящиеся к исходной структуре; величины химического сдвига протонного ЯМР измеряли по дельта-шкале при использовании либо спектрометра Bruker AV500, работающего при напряженности поля на частоте 500 МГц, либо Bruker AV400, работающего на 400 МГц, либо Bruker AV300, работающего на 300 МГц. Если не оговорено особо, ЯМР-спектры получали при 500 МГц в d6-диметилсульфоксиде. Были использованы следующие сокращения (и их производные, например dd, дублет дублетов и так далее): s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; br, широкий; qn, квинтет; р, пентет;
VI) если не оговорено особо, соединения, содержащие асимметричный атом углерода и/или серы, не разделяли;
VII) промежуточные продукты не обязательно были полностью очищены, но их структуры и чистоту оценивали с помощью ТСХ, аналитической ВЭЖХ/СВЭЖХ, и/или анализа ЯМР, и/или масс-спектрометрии;
VIII) если не оговорено особо, колоночную флэш-хроматографию (КФХ) проводили на диоксиде кремния Merck Kieselgel (артикул 9385), или на диоксиде кремния с обращенной фазой (силикагель Fluka 90 С18), или на картриджах Silicycle (диоксид кремния 40-63 мкм, масса от 4 до 330 г) или на картриджах Puriflash (диоксид кремния 50 мкм, масса от 4 до 330 г) или на картриджах Grace Resolv (от 4 до 120 г), или на колонках RediSep Rf 1.5 Flash, или на высокоэффективных колонках RediSep Rf Gold Flash (масса от 150 до 415 г), или на колонках RediSep Rf Gold с обращенной фазой С18 (диоксид кремния 20-40 мкм) либо вручную, либо автоматически с использованием системы Isco CombiFlash Companion или аналогичной системы;
IX) препаративную обращенно-фазовую ВЭЖХ (ОФ-ВЭЖХ) проводили на обращенно-фазовом диоксиде кремния С18 обычно при использовании колонки Waters XSelect CSH С18 или Phenomenex Gemini-NX axia Prep C18 OBD (диоксид кремния 5 мкм, диаметр 30 мм, длина 100 мм) с использованием смесей с уменьшающейся полярностью в качестве элюента, например [содержащих 0,1% муравьиной кислоты или 0,3-0,5% водного гидроксида аммония (d составляет 0,91)] в качестве растворителя А и ацетонитрила в качестве растворителя В; типичная методика будет следующей: градиент растворителя в течение 10-20 минут, со скоростью 40-50 мл в минуту, от смеси растворителей А и В, 95:5 соответственно, до смеси растворителей А и В, 5:95 (или алтернативного соотношения, если целесообразно);
X) использовали следующие аналитические методы СВЭЖХ; как правило, использовали диоксид кремния с обращенной фазой С18 при скорости потока 1 мл/мин, и детектирование осуществляли посредством масс-спектрометрии с электрораспылением и посредством регистрации УФ-поглощения в диапазоне длин волн 220-320 нм. Аналитическую СВЭЖХ проводили на диоксиде кремния CSH с обращенной фазой С18 при использовании колонки Waters XSelect CSH C18 (размеры 2,1×50 мм и размер частиц 1,7 микрон). Градиентный анализ применяли с использованием смесей с уменьшающейся полярностью в качестве элюента, например смесей с уменьшающейся полярностью, таких как смеси воды (содержащей 0,1% муравьиной кислоты или 0,1% аммиака) в качестве растворителя А и ацетонитрила в качестве растворителя В. В обычном 2-минутном аналитическом методе СВЭЖХ будет использован градиент растворителя в течение 1,3 минуты со скоростью приблизительно 1 мл в минуту от смеси растворителей А и В, 97:3 соответственно, до смеси растворителей А и В, 3:97.
XI) в случаях, когда определенные соединения были получены в виде соли присоединения кислоты, например гидрохлорида или дигидрохлорида, стехиометрия соли была основана на количестве и природе основных групп в соединении, точную стехиометрия соли обычно не определялась, например, с помощью данных элементного анализа;
XII) в случаях, когда для взаимодействий требуется использование микроволновой печи, использовали один из следующих микроволновых реакторов: Biotage Initiator, Personal Chemistry Emrys Optimizer, Personal Chemistry Smithcreator или СЕМ Explorer;
XIII) соединения очищали посредством хроматографии на сильном катионообменном сорбенте (СКО) с использованием колонок Isolute SPE Flash SCX-2 или SCX-3 (International Sorbent Technology Limited, Mid Glamorgan, UK);
XIV) препаративную хиральную хроматографию проводили с использованием ВЭЖХ или СФХ при использовании системы Waters SFC 100 или аналогичной. Хиральную стационарную фазу, такую как в хиральной колонке Daicel с целлюлозой или амилозой или ее эквивалент, выбирали для оптимизации разделения изомеров в образце. Для полупрепаративного разделения обычно использовали хиральную колонку размером 30×250 мм, 5 микрон, со скоростью потока 100 мл/мин в СФХ или скоростью потока 40 мл/мин в ВЭЖХ. Детектирование проводили по поглощению в УФ области спектра или посредством масс-спектрометрического детектирования. Для УФ-детектирования использовали общую длину волны, обычно 220 нм или 254 нм, или длину волны выбирали так, чтобы максимизировать сигнал продукта. Для масс-детектирования использовали метод мягкой ионизации, такой как ионизация электрораспылением, позволяющий детектировать продукт с помощью направленного МН+ сигнала. Образцы растворяли в совместимом растворителе для ввода в хроматографическую систему. Для разделения путем СФХ температуру колонки поддерживали на постоянном уровне приблизительно 40°С, а противодавление регулировали до постоянного давления приблизительно 100-150 бар (10-15 МПа).
XV) хиральный анализ проводили с использованием СФХ или ВЭЖХ при использовании системы Waters UPC2 SFC, Agilent 1200 HPLC или аналогичных. Хиральную стационарную фазу, такую как в хиральных колонках Daicel с целлюлозой или амилозой или ее эквивалент, выбирали для оптимизации разделения изомеров в образце. Для аналитического разделения обычно использовали хиральную колонку размером 3,5×150 мм, 3 микрона, со скоростью потока 2 мл/мин в СФХ или скоростью потока 0,5 мл/мин в ВЭЖХ. Детектирование проводили по поглощению в УФ области спектра (ДМД (диодно-матричный детектор)) и/или посредством масс-спектрометрии (полное сканирование). Образец растворяли в совместимом растворителе в концентрации приблизительно 0,5 мг/мл и вводили непосредственно в хроматографическую систему. Для разделения путем СФХ температуру колонки поддерживали на постоянном уровне приблизительно 40°С, а противодавление регулировали до постоянного давления приблизительно 100-150 бар (10-15 МПа).
XVI) в большинстве случаев соединения из примеров и промежуточные соединения были названы с использованием программы ACD Name, пункта в меню "Structure to Name" (Преобразование структуры в название) программы ChemDraw Ultra (CambridgeSoft) или Biovia Draw 2016;
XVII) в случаях, когда подразумевают, что реакционные смеси дегазированы, это может быть проведено, например, посредством продувки реакционного растворителя постоянным потоком азота в течение подходящего периода времени (например, от 5 до 10 минут).
XVIII) в дополнение к упомянутым выше, были использованы следующие сокращения:
XIX) для анализа ДРЛП использовали прибор Bruker D4. Дифрактограмму рентгеновских лучей на порошке получали посредством размещения образца кристаллического вещества на подложке из монокристалла кремния (SSC) Bruker и распределения образца тонким слоем с помощью предметного стекла. Образец вращали со скоростью 30 оборотов в минуту (для улучшения статистики измерений) и облучали рентгеновскими лучами, генерируемыми острофокусной трубкой с медным анодом, работающей при 40 кВ и 40 мА, с длиной волны 1,5418 ангстрем. Коллимированный пучок рентгеновских лучей пропускали через установку с автоматически регулируемой расходимостью щелей при V20, и отраженное излучение направляли через антирассеивающую щель 5,89 мм и щель детектора 9,55 мм. Образцы измеряли с использованием геометрии отражений в режиме θ - 2θ в интервале сканирования от 2° до 40° угла 2θ с номинальной экспозицией 0,12 секунды на шаг сканирования 0,02°. Прибор был оснащен позиционно-чувствительным детектором (Lynxeye). Специалистам в области дифракции рентгеновских лучей на порошке будет понятно, что на относительную интенсивность пиков могут оказывать влияние, например зерна размером более 30 микрон и неунитарные соотношения сторон, которые могут влиять на анализ образцов. Специалист также поймет, что на положение отражений может влиять точная высота, на которой образец находится в дифрактометре, и калибровка нуля дифрактометра. Планарность поверхности образца также может оказывать небольшое влияние. Следовательно, представленные данные дифракционной картины не следует принимать за абсолютные значения;
XX) для дифференциальной сканирующей калориметрии использовали прибор ТА Instruments Q2000 DSC. Обычно менее 3 мг вещества, помещенного в стандартный алюминиевый тигель с крышкой, нагревали в диапазоне температур от 25°С до 300°С с постоянной скоростью нагревания 10°С в минуту. Использовали продувку газообразным азотом со скоростью потока 50 мл в минуту. Температурные данные анализировали при использовании стандартного программного обеспечения, например Universal V.4.5A от ТА INSTRUMENTS®.
Промежуточное соединение 1: (R)-трет-бутил-3-(бромметил)пирролидин-1-карбоксилат
Трифенилфосфин (78 г; 298,1 ммоль) порциями добавляли к (R)-трет-бутил-3-(гидроксиметил)пирролидин-1-карбоксилату (50 г; 248,4 ммоль) и CBr4 (115 г; 347,8 ммоль) в DCM (1 л) при 0°С в течение 5 минут. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали при конатной температуре в течение 16 часов, затем концентрировали под вакуумом и очищали посредством КФХ, элюируя 0-16% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (61,6 г; 94%) в виде бесцветного масла; 1Н ЯМР (300 МГц, CDCl3) 1.47 (9Н, s), 1.72 (1H, dq), 1.99-2.16 (1H, m), 2.52-2.68 (1H, m), 3.10 (1H, dd), 3.26-3.44 (3Н, m), 3.49 (1H, ddd), 3.59 (1H, dd).
Промежуточное соединение 2: (R)-трет-бутил-3-(2-ацетил-3-оксобутил)пирролидин-1-карбоксилат
K2CO3 (48,3 г; 349,8 ммоль) добавляли к (R)-трет-бутил-3-(бромметил)пирролидин-1-карбоксилату (61,6 г; 233,2 ммоль) и пентан-2,4-диону (23,35 г; 233,2 ммоль) в DMF (600 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры, разбавляли EtOAc (500 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-30% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (43,9 г; 66%) в виде бледно-желтого масла; m/z МН+ 284.
Промежуточное соединение 3: (R)-трет-бутил-3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-карбоксилат
3-Метил-1Н-1,2,4-триазол-5-амин (15,20 г; 154,9 ммоль) добавляли одной порцией к (R)-трет-бутил-3-(2-ацетил-3-оксобутил)пирролидин-1-карбоксилату (43,9 г; 154,9 ммоль) в АсОН (500 мл) при комнатной температуре. Реакционную смесь нагревали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-80% МеОН в воде, с получением указанного в заголовке соединения (18,00 г; 34%) в виде бледно-желтой смолы; 1Н ЯМР (300 МГц, CDCl3) 1.45 (9Н, s), 1.70 (1H, dt), 1.91-2.07 (1H, m), 2.32-2.44 (1H, m), 2.60 (3Н, s), 2.67 (3Н, s), 2.78 (5Н, s), 3.06 (1H, dt), 3.23-3.54 (3Н, dt); m/z МН+ 346.
Промежуточное соединение 4: (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидин (соль дигидрохлорид)
4 М HCl в 1,4-диоксане (200 мл; 800 ммоль) добавляли к (R)-трет-бутил-3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-карбоксилату (18 г; 52,11 ммоль) в 1,4-диоксане (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученный осадок собирали посредством фильтрования, промывали 1,4-диоксаном (100 мл) и сушили под вакуумом с получением указанного в заголовке соединения (13,80 г; 83%) в виде белого твердого вещества, которое использовали без дальнейшей очистки; 1Н ЯМР (300 МГц, DMSO) 1.63 (1Н, dq), 1.90-2.05 (1H, m), 2.51 (4Н, s), 2.65 (3Н, s), 2.75-3.13 (7Н, m), 3.26 (2Н, ddt), 7.30 (1H, br s) 9.28 (1H, s), 9.52 (1H, s); m/z МН+ 246.
Промежуточное соединение 4В (свободное основание промежуточного соединения 4): (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидин
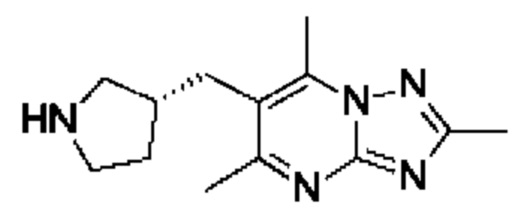
1,25 М HCl в EtOAc (36,4 мл; 45,51 ммоль) добавляли одной порцией к (R)-трет-бутил-3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-карбоксилату (2,62 г; 7,58 ммоль) в EtOAc (15 мл) при комнатной температуре. Полученный раствор перемешивали при комнатной температуре в течение 5 суток. Растворитель выпаривали под вакуумом с получением неочищенной соли гидрохлорид в виде оранжевой смолы. Неочищенный продукт превращали в свободное основание с применением ионообменной хроматографии, используя колонку с СКО. Требуемый продукт элюировали из колонки при использовании 1 М NH3/МеОН и чистые фракции упаривали досуха с получением указанного в заголовке соединения (1,77 г; 95%) в виде оранжевой смолы; 1Н ЯМР (500 МГц, CDCl3) 1.51 (1H, dq), 1.92 (1H, ddd), 2.32 (1H, р), 2.60 (3Н, s), 2.64 (1H, dd), 2.69 (3Н, s), 2.80 (3Н, s), 2.82 (2Н, dd), 2.98 (1H, dt), 3.03-3.13 (2Н, m), NH не обнаружен; m/z МН+ 246.
Промежуточное соединение 5: (R)-6-((1-(5-бромпиридин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
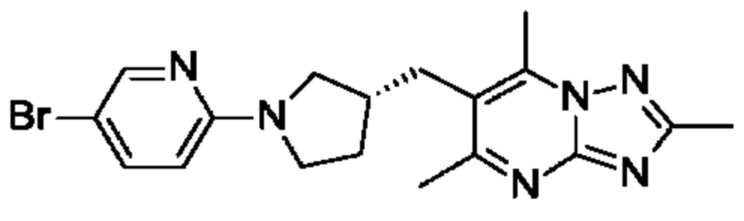
DIPEA (4,39 мл; 25,14 ммоль), дигидрохлорид (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (2 г; 6,28 ммоль) и 5-бром-2-фторпиридин (1,66 г; 9,43 ммоль) растворяли в н-пропаноле (10 мл) и герметизировали в пробирке для микроволновой печи. Реакционную смесь нагревали при 150°С в течение 2,5 часа в микроволновом реакторе и охлаждали до комнатной температуры. Растворитель удаляли при пониженном давлении. Реакционную смесь выливали в воду (100 мл), экстрагировали EtOAc (3×100 мл), органический слой сушили над Na2SO4, фильтровали и упаривали с получением желтого твердого вещества. Неочищенный продукт очищали посредством кристаллизации из смеси EtOAc/петролейный эфир с получением указанного в заголовке соединения (1,30 г; 52%) в виде бледно-желтого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.90 (1Н, dq), 2.19 (1H, dq), 2.63 (4Н, s), 2.71 (3Н, s), 2.80 (3Н, s), 2.93 (2Н, qd), 3.24 (1H, dd), 3.45 (1H, q), 3.63 (2Н, q), 6.28 (1H, d), 7.54 (1H, dd), 8.19 (1H, d); m/z МН+ 401.
Промежуточное соединение 7: 1-((6-бромпиридин-3-ил)метил)-4-метилпиперазин
6-Бромникотинальдегид (3 г; 16,13 ммоль) и 1-метилпиперазин (4,85 г; 48,39 ммоль) перемешивали в DCM (200 мл) при комнатной температуре в течение 2 часов. Добавляли АсОН (0,097 г; 1,61 ммоль) и триацетоксиборгидрид натрия (6,84 г; 32,26 ммоль) и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили насыщ. водн. NaHCO3 и органический слой отделяли. Водный слой экстрагировали EtOAc и объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-80% МеОН в воде, с получением указанного в заголовке соединения (2,00 г; 46%) в виде желтого масла; 1Н ЯМР (300 МГц, DMSO) 2.55 (3Н, s), 2.65 (4Н, d), 2.91 (4Н, s), 3.49 (2Н, s), 7.37-7.54 (2Н, m), 8.27 (1H, d); m/z МН+ 270.
Промежуточное соединение 8: 1-((5-бромпиридин-2-ил)метил)-4-метилпиперазин
5-Бромпиколинальдегид (3,5 г; 18,82 ммоль) и 1-метилпиперазин (5,65 г; 56,45 ммоль) в DCM (20 мл) перемешивали при комнатной температуре в течение 2 часов. Добавляли АсОН (0,113 г; 1,88 ммоль) и триацетоксиборгидрид натрия (7,98 г; 37,63 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали под вакуумом и полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (2,40 г; 47%) в виде желтого масла; 1Н ЯМР (300 МГц, DMSO) 2.26 (3Н, s), 2.47 (8Н, s), 3.58 (2Н, s), 7.42 (1Н, m), 8.03 (1H, dd), 8.62 (1H, dd); m/z МН+ 270.
Промежуточное соединение 9: (6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)бороновая кислота
PdCl2(dppf) (21,67 мг; 0,03 ммоль) добавляли в 1-((5-бромпиридин-2-ил)метил)-4-метилпиперазин (160 мг; 0,59 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (165 мг; 0,65 ммоль) и ацетат калия (116 мг; 1,18 ммоль) в 1,4-диоксане (4 мл) при комнатной температуре в атмосфере азота. Полученную смесь перемешивали при 90°С в течение 16 часов.
Смесь очищали посредством флэш-хроматографии на сорбенте С18 с градиентом элюирования от 5 до 100% MeCN в воде с получением неочищенного соединения, указанного в заголовке (80 мг; 58%) в виде желтой смолы; m/z МН+ 236 (на основании данных ЖХ/МС полагали, что соединение является бороновой кислотой).
Промежуточное соединение 10: рац-6-((1-(5-бромпиридин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
DIPEA (0,878 мл; 5,03 ммоль) добавляли к 5-бром-2-хлорпиримидину (486 мг; 2,51 ммоль) и дигидрохлориду рац-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (800 мг; 2,51 ммоль) (полученному в 4 стадии посредством способа, аналогичного способу получения промежуточного соединения 4, с рац-трет-бутил-3-(гидроксиметил)пирролидин-1-карбоксилатом в качестве исходного вещества) в EtOAc (10 мл) при комнатной температуре. Реакционную смесь нагревали при 70°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 5-10% МеОН в DCM, с получением указанного в заголовке соединения (350 мг; 35%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.79-1.93 (1H, m), 2.09-2.27 (1H, m), 2.49-2.62 (1H, m), 2.65 (3Н, s), 2.72 (3Н, s), 2.80 (3Н, s), 2.92 (2Н, dd), 3.30 (1H, dd), 3.46-3.61 (1H, m), 3.64-3.81 (2Н, m), 8.31 (2Н, s); m/z МН+ 402.
Промежуточное соединение 11: (R)-2,5,7-триметил-6-((1-(пиридин-3-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а] пиримидин
Cs2CO3 (1,97 г; 6,03 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидину (0,74 г; 3,02 ммоль) и 3-иодпиридину (0,68 г; 3,32 ммоль) в 1,4-диоксане (10 мл). Реакционную смесь дегазировали и добавляли пред катализатор RuPhos 3-го поколения (0,25 г; 0,30 ммоль). Реакционную смесь нагревали при 90°С в течение 5 часов, затем оставляли для охлаждения до комнатной температуры и разбавляли DCM и фильтровали. Фильтрат концентрировали под вакуумом и очищали посредством КФХ, элюируя смесью 0-5% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (0,413 г; 43%) в виде желтого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.88 (1H, dq), 2.18 (1Н, dtd), 2.58-2.67 (4Н, m), 2.70 (3Н, s), 2.78 (3Н, s), 2.89 (1H, dd), 2.93-3 (1H, m), 3.09 (1Н, dd), 3.31-3.43 (2Н, m), 3.49-3.56 (1H, m), 6.78 (1H, ddd), 7.11 (1H, ddd), 7.92-8 (2Н, m); m/z МН+ 323.
Промежуточное соединение 12: (R)-6-((1-(6-бромпиридин-3-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4] триазоло[1,5-а]пиримидин
NBS (229 мг; 1,28 ммоль) добавляли одной порцией к (R)-2,5,7-триметил-6-((1-(пиридин-3-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидину (414 мг; 1,28 ммоль) в MeCN (5 мл) при 0°С в воздушной атмосфере. Реакционную смесь перемешивали при 0°С и оставляли для медленного нагревания до комнатной температуры в течение 3 часов. Полученный осадок выделяли посредством фильтрования и промывали MeCN (5 мл) с получением указанного в заголовке соединения (298 мг; 58%) в виде белого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.88 (1H, dq), 2.11-2.24 (1H, m), 2.61 (3Н, s), 2.62-2.67 (1H, m), 2.69 (3Н, s), 2.78 (3Н, s), 2.88 (1H, dd), 2.95 (1H, dd), 3.06 (1H, dd), 3.28-3.41 (2Н, m), 3.48 (1H, td), 6.69 (1Н, dd), 7.25 (1H, dd), 7.69 (1Н, d); m/z МН+ 401.
Промежуточное соединение 13: 3-хлор-6-(хлорметил)пиридазин
1,3,5-Трихлор-1,3,5-триазинан-2,4,6-трион (1,808 г; 7,78 ммоль) добавляли одной порцией к 3-хлор-6-метилпиридазину (2,00 г; 15,56 ммоль) в DCE (100 мл) при комнатной температуре. Реакционную смесь перемешивали при 60°С в течение 2 часов, затем оставляли для охлаждения до комнатной температуры и фильтровали. Фильтрат концентрировали под вакуумом и очищали посредством КФХ, элюируя 10-50% EtOAc в гептане, с получением указанного в заголовке соединения (1,60 г; 63%) в виде бледно-желтого масла; 1Н ЯМР (500 МГц, CDCl3) 4.88 (2Н, s), 7.58 (1H, d), 7.70 (1H, d); m/z МН+ 163.
Промежуточное соединение 14: 4-((6-хлорпиридазин-3-ил)метил)морфолин
Морфолин (0,86 мл; 9,82 ммоль) добавляли одной порцией к 3-хлор-6-(хлорметил)пиридазину (1,6 г; 9,82 ммоль) и DIPEA (2,05 мл; 11,8 ммоль) в THF (16 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 3 суток, затем фильтровали и промывали THF. Объединенный фильтрат концентрировали под вакуумом и полученный неочищенный продукт очищали посредством КФХ, элюируя 0-5% МеОН в DCM, с получением указанного в заголовке соединения (1,380 г; 66%) в виде белого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 2.47-2.56 (4Н, m), 3.68-3.76 (4Н, m), 3.85 (2Н, s), 7.50 (1Н, d), 7.68 (1H, d); m/z МН+ 214.
Промежуточное соединение 15: (R)-2,5,7-триметил-6-((1-(пиримидин-5-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
Cs2CO3 (6,16 г; 18,91 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидину (2,32 г; 9,46 ммоль) и 5-бромпиридину (1,503 г; 9,46 ммоль) в 1,4-диоксане (64 мл). Реакционную смесь дегазировали и добавляли пред катализатор RuPhos 3-го поколения (0,395 г; 0,47 ммоль) и RuPhos (0,221 г; 0,47 ммоль). Реакционную смесь перемешивали при 90°С в течение 18 часов, затем оставляли для охлаждения до комнатной температуры, разбавляли DCM и фильтровали. Фильтрат концентрировали под вакуумом и очищали посредством КФХ, элюируя 0-5% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (1,160 г; 38%) в виде желтого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.88-1.95 (1H, m), 2.20 (1H, dtd), 2.61 (3Н, s), 2.63-2.68 (1H, m), 2.70 (3Н, s), 2.80 (3Н, s), 2.91 (1H, dd), 2.98 (1H, dd), 3.12 (1H, dd), 3.37 (1H, dt), 3.47 (1H, s), 3.54 (1H, td), 8.05 (2Н, s), 8.60 (1H, s); m/z МН+ 324.
Промежуточное соединение 16: (R)-6-((1-(2-бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
(R)-2,5,7-Триметил-6-((1-(пиримидин-5-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин (1,59 г; 4,92 ммоль) добавляли к MeCN (80 мл) и реакционную смесь охлаждали до 0°С. Затем добавляли NBS (1,050 г; 5,90 ммоль) одной порцией. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 2 часов. Полученную суспензию фильтровали и неочищенное твердое вещество собирали и суспендировали в MeCN (10 мл) и нагревали при температуре образования флегмы. Суспензию оставляли для охлаждения до комнатной температуры и твердое вещество собирали посредством фильтрования с получением указанного в заголовке соединения (0,652 г; 33%) в виде желтого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.90 (1H, dq), 2.16-2.25 (1H, m), 2.58-2.67 (4Н, m), 2.70 (3Н, s), 2.80 (3Н, s), 2.88-2.94 (1H, m), 2.98 (1H, dd), 3.10 (1H, dd), 3.34 (1H, dt), 3.44 (1H, dd), 3.51 (1Н, td), 7.87 (2Н, s); m/z МН+ 402.
Промежуточное соединение 17: (S)-2,4-диметил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазин
Карбонат калия (2,43 г; 17,64 ммоль) добавляли одной порцией к 2-(4-(бромметил)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолану (1,31 г; 4,41 ммоль) и дигидрохлориду (S)-1,3-диметилпиперазина (0,83 г; 4,41 ммоль) в MeCN (40 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 18 часов, затем оставляли для охлаждения до комнатной температуры и фильтровали. Фильтрат концентрировали под вакуумом и полученный неочищенный продукт очищали посредством КФХ, элюируя 0-10% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (506 мг; 35%) в виде бесцветного масла; m/z МИ 331.
Промежуточное соединение 18: 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегид
Аддукт PdCl2(dppf)-CH2Cl2 (92 мг; 0,13 ммоль) добавляли в 4-бром-3-метилбензальдегид (500 мг; 2,51 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (638 мг; 2,51 ммоль) и ацетат калия (493 мг; 5,02 ммоль) в 1,4-диоксане (10 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 10% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (580 мг; 94%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.39 (12Н, s), 2.63 (3Н, s), 7.64-7.70 (2Н, m), 7.92 (1Н, d), 10.03 (1H, s); m/z МН+ 247.
Промежуточное соединение 19: (R)-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензальдегид
Pd(Ph3P)4 (0,287 г; 0,25 ммоль) добавляли к (R)-6-((1-(2-бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (1,00 г; 2,49 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегиду (0,692 г; 2,98 ммоль) и 2 М водн. раствору карбоната натрия (2,49 мл; 4,97 ммоль) в дегазированном 1,4-диоксане (40 мл) и воде (7,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный остаток помещали в DCM и пропускали через фильтровальную бумагу для разделения фаз, затем концентрировали под вакуумом и очищали посредством КФХ, элюируя 0-10% МеОН в DCM, с получением указанного в заголовке соединения (0,510 г; 48%) в виде желтого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.88-1.96 (1H, m), 2.23 (1H, ddd), 2.62 (3Н, s), 2.72 (4Н, s), 2.81 (3Н, s), 2.88-3.04 (2Н, m), 3.20 (1H, dd), 3.45 (1H, dt), 3.54 (1H, dd), 3.63 (1H, td), 7.9-7.99 (2Н, m), 8.16 (2Н, s), 8.42-8.53 (2Н, m), 10.06 (1H, s); m/z МН+ 428.
Промежуточное соединение 20: трет-бутил-4-(2-(диметиламино)-2-оксоэтил)пиперазин-1-карбоксилат
HATU (3,74 г; 9,82 ммоль) добавляли к 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)уксусной кислоте (2 г; 8,19 ммоль) в DMF (25 мл) при комнатной температуре в воздушной атмосфере и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Диметиламин (12,28 мл; 24,56 ммоль) и DIPEA (4,29 мл; 24,56 ммоль) добавляли к смеси при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем выливали в насыщ. водн. NaHCO3 (200 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (1,65 г; 74%) в виде бледно-желтого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 1.47 (9Н, s), 2.49 (4Н, t), 2.95 (3Н, s), 3.11 (3Н, s), 3.27 (2Н, s), 3.43-3.50 (4Н, m); m/z МН+ 272.
Промежуточное соединение 21: дигидрохлорид N,N-диметил-2-(пиперазин-1-ил)ацетамида
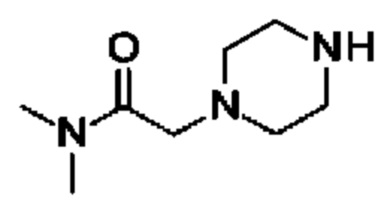
4 М HCl в EtOAc (25 мл; 100 ммоль) добавляли к трет-бутил-4-(2-(диметиламино)-2-оксоэтил)пиперазин-1-карбоксилату (1,6 г; 5,9 ммоль) в EtOAc (25 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали под вакуумом с получением указанного в заголовке соединения (1,4 г; 100%) в виде белого твердого вещества; m/z МН+ 172.
Промежуточное соединение 22: 2-(4-(4-бромбензил)пиперазин-1-ил)-N,N-диметилацетамид
Дигидрохлорид N,N-диметил-2-(пиперазин-1-ил)ацетамида (800 мг; 3,28 ммоль) добавляли к 4-бромбензальдегиду (606 мг; 3,28 ммоль) в THF (25 мл) при комнатной температуре в воздушной атмосфере и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Триацетоксиборгидрид натрия (1389 мг; 6,55 ммоль) и АсОН (2 капли) добавляли к смеси и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли водой и концентрировали под вакуумом почти досуха. Неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (704 мг; 63%) в виде коричневого масла; 1H ЯМР (400 МГц, CDCl3) 2.49 (4Н, m), 2.56 (4Н, m), 2.95 (3Н, s), 3.08 (3Н, s), 3.18 (2Н, s), 3.46 (2Н, s), 7.17-7.25 (2Н, m), 7.40-7.49 (2Н, m); m/z МН+ 340.
Промежуточное соединение 23: 2-(4-(4-бромбензил)пиперазин-1-ил)этанол
2-(Пиперазин-1-ил)этанол (1,407 г; 10,81 ммоль) добавляли к 4-бромбензальдегиду (1 г; 5,40 ммоль) в THF (25 мл) при комнатной температуре и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. В смесь добавляли триацетоксиборгидрид натрия (2,291 г; 10,81 ммоль) и АсОН (2 капли) при комнатной температуре в воздушной атмосфере и реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем выливали в воду (20 мл) и концентрировали под вакуумом почти досуха. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (1,20 г; 74%) в виде коричневого масла; 1Н ЯМР (400 МГц, CDCl3) 2.23-2.57 (11H, m), 3.48 (2Н, s), 3.63 (2Н, t), 7.22 (2Н, d), 7.46 (2Н, d); m/z МН+ 299.
Промежуточное соединение 24: 4-(4-(5-бромпиримидин-2-ил)бензил)морфолин
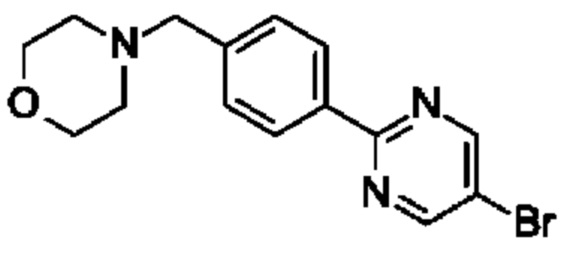
Pd(Ph3P)4 (578 мг; 0,50 ммоль) добавляли к 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)морфолину (1516 мг; 5,00 ммоль), 5-бром-2-иодпиримидину (1424 мг; 5 ммоль) и Na2CO3 (1060 мг; 10,00 ммоль) в толуоле (20 мл) и воде (4 мл) и реакционную смесь перемешивали при 120°С в течение 3 суток, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Остаток помещали в EtOAc (100 мл) и последовательно промывали водой (2×20 мл). Органический слой выделяли и сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-50% МеОН в воде, с получением указанного в заголовке соединения (510 мг; 31%) в виде бледно-желтого твердого вещества; m/z МН+ 334.
Промежуточное соединение 25: (R)-2,5,7-триметил-6-((1-фенилпирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
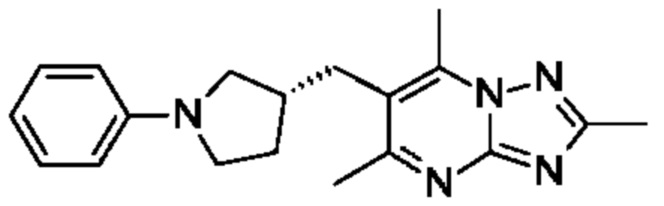
Дегазированный трет-бутанол (5 мл) добавляли к бромбензолу (74 мкл; 0,7 ммоль), дигидрохлориду (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (267 мг; 0,84 ммоль), Cs2CO3 (1368 мг; 4,20 ммоль), RuPhos (65 мг; 0,14 ммоль) и пред катализатору RuPhos 3-го поколения (59 мг; 0,07 ммоль). Реакционную смесь нагревали при 90°С в течение ночи, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Остаток растворяли в DCM (30 мл) и промывали водой (30 мл). Водный слой повторно экстрагировали DCM (30 мл) и объединенные органические слои пропускали через гидрофобную (разделение фаз) фритту и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (72 мг; 32%) в виде белого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.78-1.9 (1H, m), 2.1-2.21 (1H, m), 2.55-2.64 (4Н, m), 2.69 (3Н, s), 2.78 (3Н, s), 2.83-2.98 (2Н, m), 3.08 (1H, dd), 3.28-3.39 (2Н, m), 3.45-3.55 (1H, m), 6.54 (2Н, dd), 6.70 (1H, tt), 7.23 (2Н, dd); m/z МН+ 322.
Промежуточное соединение 26: (R)-6-((1-(4-бромфенил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
NBS (0,460 г; 2,58 ммоль) добавляли к (R)-2,5,7-триметил-6-((1-фенилпирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидину (0,83 г; 2,58 ммоль) в THF (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-90% МеОН в воде, с получением указанного в заголовке соединения (0,750 г; 73%) в виде желтого твердого вещества; m/z МН+ 400.
Промежуточное соединение 27: (R)-2,5,7-триметил-6-((1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
Аддукт PdCl2(dppf)-CH2Cl2 (94 мг; 0,11 ммоль) добавляли в (R)-6-((1-(4-бромфенил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (460 мг; 1,15 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (292 мг; 1,15 ммоль) и ацетат калия (226 мг; 2,30 ммоль) в 1,4-диоксане (5 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя EtOAc, с получением указанного в заголовке соединения (350 мг; 68%) в виде бледно-желтого твердого вещества; m/z МН+ 448.
Промежуточное соединение 28: 2-бром-5-((4-метилпиперазин-1-ил)метил)пиразин
Иодид калия (0,659 г; 3,97 ммоль) добавляли к 1-метилпиперазину (0,398 г; 3,97 ммоль), 2-бром-5-(бромметил)пиразину (1 г; 3,97 ммоль) и K2CO3 (0,549 г; 3,97 ммоль) в DMF (30 мл) в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (0,520 г; 48%) в виде желтого масла; m/z МН+ 271.
Промежуточное соединение 29: 5-бром-2-((4-метилпиперазин-1-ил)метил)пиримидин
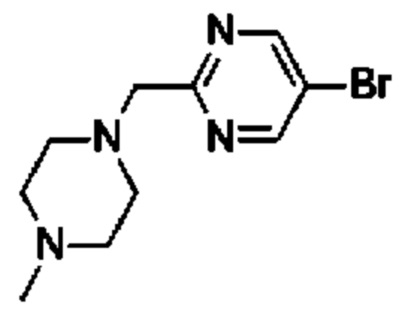
Иодид калия (758 мг; 4,57 ммоль) добавляли к 5-бром-2-(бромметил)пиримидину (767 мг; 3,04 ммоль), 1-метилпиперазину (457 мг; 4,57 ммоль) и K2CO3 (631 мг; 4,57 ммоль) в DMF (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (340 мг; 41%) в виде белого твердого вещества; m/z МН+ 271.
Промежуточное соединение 30: 4-((6-бромпиридин-3-ил)метил)морфолин
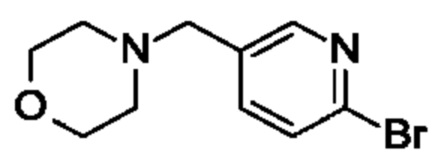
Иодид калия (1,985 г; 11,96 ммоль) добавляли к 2-бром-5-(бромметил)пиридину (3 г; 11,96 ммоль), морфолину (1,042 г; 11,96 ммоль) и K2CO3 (1,652 г; 11,96 ммоль) в DMF (25 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем выливали в воду (150 мл) и экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-75% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (2,60 г; 85%) в виде белого твердого вещества; 1H ЯМР (400 МГц, CDCl3) 2.41-2.48 (4Н, m), 3.48 (2Н, s), 3.68-3.75 (4Н, m), 7.46 (1H, d), 7.58 (1H, dd), 8.31 (1H, d); m/z МН+ 257.
Промежуточное соединение 31: 4-((6-(4-бромфенил)пиридин-3-ил)метил)морфолин
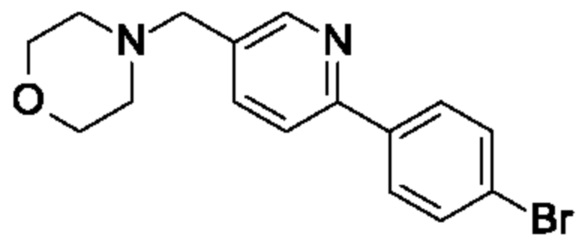
Pd(Ph3P)4 (0,674 г; 0,58 ммоль) добавляли к 4-((6-бромпиридин-3-ил)метил)морфолину (1,5 г; 5,83 ммоль), (4-бромфенил)бороновой кислоте (1,289 г; 6,42 ммоль) и Na2CO3 (1,237 г; 11,67 ммоль) в 1,4-диоксане (14 мл) и воде (7,00 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 5 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 50% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (1,28 г; 66%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 2.39 (4Н, dd), 3.49-3.64 (6Н, m), 7.58-7.76 (2Н, m), 7.82 (1H, dd), 7.93-8.01 (1Н, m), 8.01-8.09 (2Н, m), 8.59 (1Н, d); m/z МН+ 333.
Промежуточное соединение 32: 3-хлор-6-((4-метилпиперазин-1-ил)метил)пиридазин
Иодид калия (1,200 г; 7,23 ммоль) добавляли к 3-(бромметил)-6-хлорпиридазину (1 г; 4,82 ммоль), 1-метилпиперазину (0,724 г; 7,23 ммоль) и K2CO3 (0,999 г; 7,23 ммоль) в DMF (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем разбавляли водой и полученный раствор очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (300 мг; 28%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, DMSO) 2.19 (3Н, s), 2.40 (8Н, m), 3.77 (2Н, s), 7.76 (1Н, d), 7.88 (1H, d); m/z МН+ 227.
Промежуточное соединение 33: 2-(4-бромфенил)-5-метилпиримидин
Pd(Ph3P)4 (1,002 г; 0,87 ммоль) добавляли к 2-бром-5-метилпиримидину (1,5 г; 8,67 ммоль), (4-бромфенил)бороновой кислоте (1,74 г; 8,7 ммоль) и Na2CO3 (1,84 г; 17,3 ммоль) в 1,4-диоксане (35 мл) и воде (7 мл) в воздушной атмосфере. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 2-5% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (1,00 г; 46%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 2.37 (3Н, s), 7.55 (2Н, d), 8.30 (2Н, d), 8.64 (2Н, s); m/z МН+ 249.
Промежуточное соединение 34: 5-(бромметил)-2-(4-бромфенил)пиримидин
Бензоилпероксид (58,3 мг; 0,24 ммоль) добавляли к 2-(4-бромфенил)-5-метилпиримидину (600 мг; 2,41 ммоль) и NBS (429 мг; 2,41 ммоль) в CCl4 (12 мл) в воздушной атмосфере. Реакционную смесь перемешивали при 80°С в течение 4 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% МеОН в воде, с получением указанного в заголовке соединения (310 мг; 39%) в виде желтого масла; m/z МН+ 327.
Промежуточное соединение 35: 2-(4-бромфенил)-5-((4-метилпиперазин-1-ил)метил)пиримидин
Иодид калия (152 мг; 0,91 ммоль) добавляли к 5-(бромметил)-2-(4-бромфенил)пиримидину (300 мг; 0,91 ммоль), 1-метилпиперазину (82 мг; 0,82 ммоль) и K2CO3 (126 мг; 0,91 ммоль) в DMF (10 мл) в воздушной атмосфере. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (195 мг; 61%) в виде желтого масла; 1Н ЯМР (300 МГц, CDCl3) 2.61 (3Н, s), 2.76-3.20 (8Н, m), 3.61 (2Н, s), 7.54 (2Н, d), 8.33 (2Н, d), 8.73 (2Н, s); m/z МН+ 347.
Промежуточное соединение 36: трет-бутил-4-((6-бромпиридин-3-ил)метил)пиперазин-1-карбоксилат
Триацетоксиборгидрид натрия (4,56 г; 21,50 ммоль) добавляли к 6-бромникотинальдегиду (2,00 г; 10,75 ммоль), трет-бутил-пиперазин-1-карбоксилату (4,01 г; 21,50 ммоль) и АсОН (0,062 мл; 1,08 ммоль) в DCM (100 мл) при 20°С.Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь промывали насыщ. NaHCO3, пропускали через фильтровальную бумагу для разделения фаз и растворитель удаляли под вакуумом. Неочищенный продукт очищали посредством КФХ, элюируя от 0 до 70% EtOAc в гептане, с получением указанного в заголовке соединения (2,80 г; 73,1%) в виде бесцветного масла, которое кристаллизовалось при выстаивании; 1Н ЯМР (500 МГц, CDCl3) 1.45 (9Н, s), 2.31-2.43 (4Н, m), 3.39-3.44 (4Н, m), 3.47 (2Н, s), 7.45 (1H, dd), 7.55 (1H, dd), 8.29 (1H, dd); m/z МН+ 356.
Промежуточное соединение 37: трет-бутил-4-((6-(4-бромфенил)пиридин-3-ил)метил)пиперазин-1-карбоксилат
Pd(Ph3P)4 (0,389 г; 0,34 ммоль) добавляли к трет-бутил-4-((6-бромпиридин-3-ил)метил)пиперазин-1-карбоксилату (2,40 г; 6,74 ммоль), (4-бромфенил)бороновой кислоте (1,35 г; 6,74 ммоль) и карбонату калия (2,79 г; 20,2 ммоль) в дегазированном 1,4-диоксане (12 мл) и воде (3 мл) при комнатной температуре. Реакционную смесь нагревали при 85°С в микроволновом реакторе в течение 3 часов, затем оставляли для охлаждения до комнатной температуры и разбавляли EtOAc. Органическую фазу пропускали через фазоразделитель и затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-5% МеОН в DCM, с получением указанного в заголовке соединения (1,20 г; 41%) в виде желтого масла, которое затвердевало при выстаивании; 1Н ЯМР (500 МГц, CDCl3) 1.46 (9Н, s), 2.36-2.47 (4Н, m), 3.4-3.47 (4Н, m), 3.55 (2Н, s), 7.56-7.62 (2Н, m), 7.67 (1H, dd), 7.74 (1H, dd), 7.84-7.91 (2Н, m), 8.57-8.63 (1H, m); m/z MH+ 432.
Промежуточное соединение 38: 4-((5-бромпиримидин-2-ил)метил)морфолин
Иодид калия (758 мг; 4,57 ммоль) добавляли к 5-бром-2-(бромметил)пиримидину (767 мг; 3,04 ммоль), морфолину (398 мг; 4,57 ммоль) и K2CO3 (631 мг; 4,57 ммоль) в DMF (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем разбавляли водой и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (720 мг; 92%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.55-2.63 (4Н, m), 3.74-3.82 (6Н, m), 8.79 (2Н, s); m/z МН+ 258.
Промежуточное соединение 39: 4-((5-(4-бромфенил)пиримидин-2-ил)метил)морфолин
Pd(Ph3P)4 (179 мг; 0,15 ммоль) добавляли к 4-((5-бромпиримидин-2-ил)метил)морфолину (400 мг; 1,55 ммоль), (4-бромфенил)бороновой кислоте (311 мг; 1,55 ммоль) и Na2CO3 (328 мг; 3,10 ммоль) в 1,4-диоксане (5 мл) и воде (2,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением указанного в заголовке соединения (330 мг; 64%) в виде белого твердого вещества; Н ЯМР (400 МГц, CDCl3) 2.62-2.69 (4Н, m), 3.78-3.86 (4Н, m), 3.90 (2Н, s), 7.42-7.53 (2Н, m), 7.63-7.74 (2Н, m), 8.93 (2Н, s); m/z МН+ 334.
Промежуточное соединение 40: 1-бром-4-(бромметил)-2-метоксибензол
Трифенилфосфин (5,53 мл; 23,9 ммоль) порциями добавляли к (4-бром-3-метоксифенил)метанолу (4,32 г; 20,0 ммоль) и тетрабромметану (7,26 г; 21,9 ммоль) в DCM (100 мл) при 0°С в течение 2 минут. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали при конатной температуре в течение 18 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-50% DCM в гептане, с получением указанного в заголовке соединения (5,57 г; 100%) в виде бесцветной смолы; 1Н ЯМР (400 МГц, CDCl3) 3.91 (3Н, s), 4.44 (2Н, s), 6.86 (1Н, dd), 6.92 (1H, d), 7.49 (1H, d).
Промежуточное соединение 41: 1-(4-бром-3-метоксибензил)-4-метилпиперазин
Иодид калия (2,135 г; 12,86 ммоль) добавляли к 1-бром-4-(бромметил)-2-метоксибензолу (3 г; 10,7 ммоль), 1-метилпиперазину (1,29 г; 12,9 ммоль) и K2CO3 (1,78 г; 12,9 ммоль) в DMF (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем выливали в воду (200 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-5% МеОН в DCM, с получением указанного в заголовке соединения (1,70 г; 53%) в виде бледно-желтого масла; 1Н ЯМР (400 МГц, CDCl3) 2.34 (3Н, s), 2.50 (8Н, br m), 3.48 (2Н, s), 3.91 (3Н, s), 6.82 (1H, dd), 6.91 (1H, d), 7.46 (1H, dd); m/z МН+ 299.
Промежуточное соединение 42: (2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)бороновая кислота
Аддукт PdCl2(dppf)-CH2Cl2 (0,464 г; 0,57 ммоль) добавляли в 1-(4-бром-3-метоксибензил)-4-метилпиперазин (1,7 г; 5,68 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (2,89 г; 11,4 ммоль) и ацетат калия (1,67 г; 17,1 ммоль) в 1,4-диоксане (25 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 48 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде (с 0,5% NH4HCO3), с получением указанного в заголовке соединения (1,10 г; 73%) в виде красного твердого вещества; m/z МН+ 265.
Промежуточное соединение 43: (R)-6-((1-(6-хлорпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
Триэтиламин (2,10 мл; 15,1 ммоль) добавляли к дигидрохлориду (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (1,2 г; 3,77 ммоль) и 2,6-дихлорпиразину (0,562 г; 3,77 ммоль) в н-пропаноле (20 мл). Реакционную смесь перемешивали при 90°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. К остатку добавляли воду (50 мл) и экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-5% МеОН в DCM, с получением указанного в заголовке соединения (1,10 г; 82%) в виде бледно-желтого твердого вещества; 1Н ЯМР (300 МГц, DMSO) 1.81 (1Н, dq), 2.07 (1H, m), 2.46 (3Н, s), 2.62 (4Н, s), 2.73 (3Н, s), 2.91 (2Н, d), 3.18 (1H, m), 3.35 (1H, q), 3.61 (2Н, td), 7.75 (1H, m), 7.90 (1Н, m); m/z MH+ 358.
Промежуточное соединение 44: (R)-2,5,7-триметил-6-((1-(6-метилпиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
Pd(Ph3P)4 (0,355 г; 0,31 ммоль) добавляли к (R)-6-((1-(6-хлорпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (1,1 г; 3,07 ммоль), 2,4,6-триметил-1,3,5,2,4,6-триоксатриборинану в THF (50 мас. %) (1,54 г; 6,15 ммоль) и Na2CO3 (1,30 г; 12,3 ммоль) в 1,4-диоксане (6 мл) и воде (3 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 1-5% МеОН в DCM, с получением указанного в заголовке соединения (1,00 г; 96%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, DMSO) 1.81 (1H, dq), 2.06 (1H, dq), 2.26 (3Н, s), 2.54 (6Н, d), 2.73 (3Н, s), 2.92 (2Н, m), 3.17 (1H, m), 3.36 (2Н, m), 3.59 (2Н, tt), 7.63 (1H, s), 7.73 (1H, s); m/z МН+ 338.
Промежуточное соединение 45: (R)-6-((1-(5-бром-6-метилпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
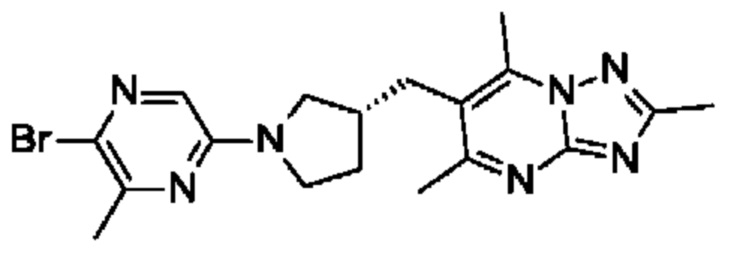
NBS (0,527 г; 2,96 ммоль) добавляли к (R)-2,5,7-триметил-6-((1-(6-метилпиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидину (1 г; 2,96 ммоль) в DCM (10 мл) при 0°С в воздушной атмосфере. Реакционную смесь перемешивали при 0°С в течение 1 часа, затем добавляли воду (25 мл), и смесь экстрагировали DCM (3×25 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 1-2% МеОН в DCM, с получением указанного в заголовке соединения (0,90 г; 73%) в виде бледно-желтого твердого вещества; 1Н ЯМР (300 МГц, DMSO) 1.81 (1Н, dq), 2.06 (1Н, dq), 2.39 (3Н, s), 2.48 (3Н, s), 2.55 (1H, m), 2.62 (3Н, s), 2.73 (3Н, s), 2.92 (2Н, m), 3.15 (1Н, m), 3.33 (1H, q), 3.58 (2Н, ddd), 7.58 (1Н, s); m/z МН+ 416.
Промежуточное соединение 46: (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
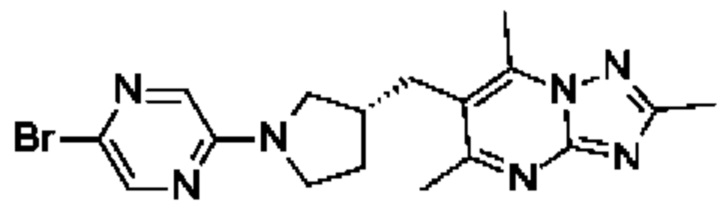
2,5-Дибромпиразин (3,74 г; 15,7 ммоль) добавляли к дигидрохлориду (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (5,00 г; 15,7 ммоль) и DIPEA (13,7 мл; 78,6 ммоль) в н-бутаноле (60 мл) при комнатной температуре. Реакционную смесь нагревали при температуре образования флегмы в течение 2 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт растирали с водой и фильтровали с получением указанного в заголовке соединения (4,86 г; 77%) в виде кремового твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.89 (1H, dq), 2.16-2.24 (1H, m), 2.61 (4Н, s), 2.70 (3Н, s), 2.79 (3Н, s), 2.93 (2Н, tt), 3.23 (1H, dd), 3.45 (1H, dt), 3.6-3.68 (2Н, m), 7.61 (1H, d), 8.10 (1H, d);m/z MH+ 402.
Промежуточное соединение 47: (R)-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)фенил)метанол
(4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метанол (165 мг; 0,71 ммоль), (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (237 мг; 0,59 ммоль) и Cs2CO3 (384 мг; 1,18 ммоль) добавляли в 1,4-диоксан (6,2 мл) и воду (3,1 мл) и реакционную смесь дегазировали в течение 10 минут. Добавляли пред катализатор XPhos 2-го поколения (23 мг; 0,03 ммоль) и реакционную смесь перемешивали в течение 2 часов при 90°С, затем оставляли для охлаждения до комнатной температуры. Реакционную смесь разбавляли EtOAc (50 мл). Органический слой отделяли и промывали насыщ. рассолом и сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (200 мг; 79%) в виде белой пены; 1Н ЯМР (500 МГц, CDCl3) 1.74 (1H, t), 1.92 (1H, dq), 2.18-2.27 (1H, m), 2.62 (4Н, s), 2.71 (3Н, s), 2.80 (3Н, s), 2.87-3.02 (2Н, m), 3.33 (1Н, dd), 3.55 (1H, dt), 3.68-3.8 (2Н, m), 4.74 (2Н, d), 7.41-7.47 (2Н, m), 7.84-7.91 (2Н, m), 7.95 (1H, d), 8.51 (1H, d); m/z MH+ 430.
Промежуточное соединение 48: (R)-6-((1-(5-(4-(хлорметил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
(R)-(4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)фенил)метанол (200 мг; 0,47 ммоль) растворяли в DCM (7 мл) и DMF (0,34 мг; 4,66 мкмоль). Реакционную смесь охлаждали до 0°С и по каплям добавляли тионилхлорид (0,037 мл; 0,51 ммоль). Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали при конатной температуре в течение 3 часов, затем разбавляли DCM (50 мл) и доводили до рН 8-9 с помощью Na2CO3 (2 М водн. раствор). Органический слой отделяли и промывали насыщ. рассолом, пропускали через картридж для разделения фаз и концентрировали под вакуумом с получением указанного в заголовке соединения (181 мг; 87%) в виде желтого твердого вещества, которое использовали без дальнейшей очистки; m/z МН+ 448.
Промежуточное соединение 49: (R)-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)-бензальдегид
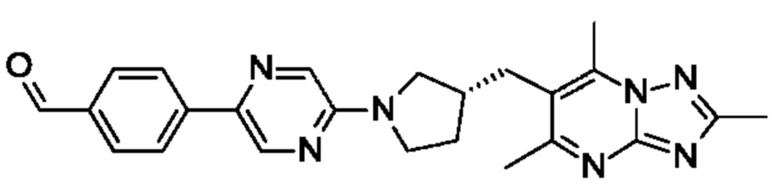
Pd(Ph3P)4 (0,373 г; 0,32 ммоль) добавляли к (4-формилфенил)бороновой кислоте (0,485 г; 3,23 ммоль), (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (1,30 г; 3,23 ммоль) и Na2CO3 (0,685 г; 6,46 ммоль) в 1,4-диоксане (10 мл) и воде (2 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-80% МеОН в воде, с получением указанного в заголовке соединения (1,20 г; 87%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.94 (1H, dq), 2.25 (1H, dq), 2.57-3.07 (12Н, m), 3.24-3.42 (1H, m), 3.45-3.87 (3Н, m), 7.79-8.18 (5Н, m), 8.60 (1H, d), 10.04 (1H, s); m/z MH+ 428.
Промежуточное соединение 50: 1-(4-бром-2-метилбензил)-4-метилпиперазин
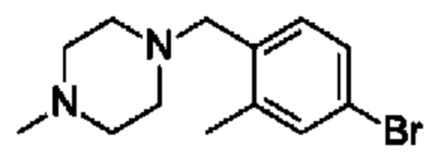
4-Бром-2-метилбензальдегид (1 г; 5,02 ммоль) и 1-метилпиперазин (0,503 г; 5,02 ммоль) объединяли с МеОН (20 мл) и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Добавляли NaCNBH4 (0,631 г; 10,1 ммоль) и АсОН (0,030 г; 0,50 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 1-70% МеОН в воде, с получением указанного в заголовке соединения (1,00 г; 70%) в виде желтого твердого вещества; m/z МН+ 283.
Промежуточное соединение 51: 1-метил-4-(2-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазин
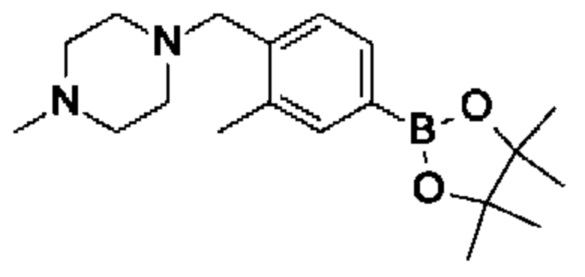
Аддукт PdCl2(dppf)-CH2Cl2 (0,058 г; 0,07 ммоль) добавляли в 1-(4-бром-2-метилбензил)-4-метилпиперазин (0,2 г; 0,71 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (0,215 г; 0,85 ммоль) и ацетат калия (0,139 г; 1,41 ммоль) в 1,4-диоксане (10 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-50% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (70 мг; 30%) в виде желтого твердого вещества; m/z МН+ 331.
Промежуточное соединение 52: 1-(4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазин-1-ил)этанон
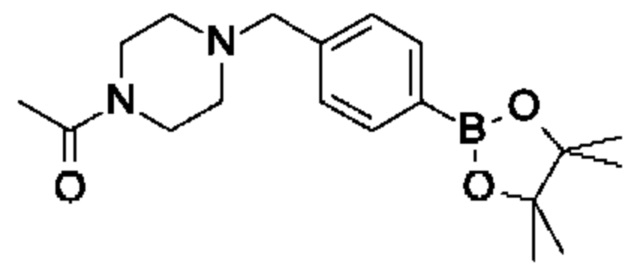
Карбонат калия (2,23 г; 16,2 ммоль) добавляли одной порцией к 2-(4-(бромметил)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолану (2,4 г; 8,08 ммоль) и 1-(пиперазин-1-ил)этанону (1.04 г; 8,08 ммоль) в MeCN (70 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 18 часов, затем оставляли для охлаждения до комнатной температуры и фильтровали. Фильтрат концентрировали под вакуумом и полученный неочищенный продукт очищали посредством КФХ, элюируя смесью 0-5% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (2,73 г; 98%) в виде бесцветного масла; 1Н ЯМР (500 МГц, CDCl3) 1.34 (12Н, s), 2.07 (3Н, s), 2.41 (4Н, dq), 3.42-3.46 (2Н, m), 3.53 (2Н, s), 3.59-3.64 (2Н, m), 7.33 (2Н, d), 7.72-7.81 (2Н, m); m/z МН+ 345.
Промежуточное соединение 53: (R)-6-((1-(6-хлорпиридазин-3-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
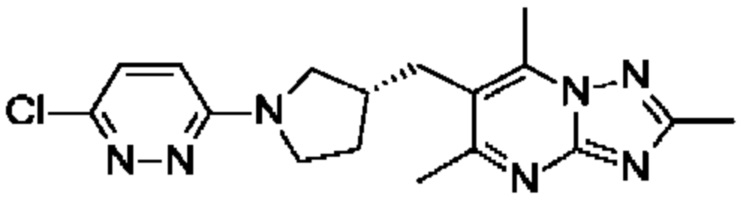
Триэтиламин (1,75 мл; 12,6 ммоль) добавляли к дигидрохлориду (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (1 г; 3,14 ммоль), 3,6-дихлорпиридазину (0,468 г; 3,14 ммоль) в н-пропаноле (10 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при 90°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и частично концентрировали под вакуумом. Осадок собирали посредством фильтрования, промывали водой (50 мл) и МеОН (10 мл) и сушили под вакуумом с получением указанного в заголовке соединения (0,610 г; 54%) в виде коричневого твердого вещества, которое использовали без дальнейшей очистки; 1Н ЯМР (400 МГц, DMSO) 1.85 (1Н, dq), 2.11 (1H, ddd), 2.48 (3Н, s), 2.53-2.63 (1H, m), 2.64 (3Н, s), 2.75 (3Н, s), 2.94 (2Н, d), 3.14-3.23 (1H, m), 3.40 (1H, dt), 3.49-3.93 (2Н, m), 7.00 (1H, d), 7.47 (1H, d); m/z МН+ 358.
Промежуточное соединение 54: 2,4-диоксопентан-3-ил-бензоат
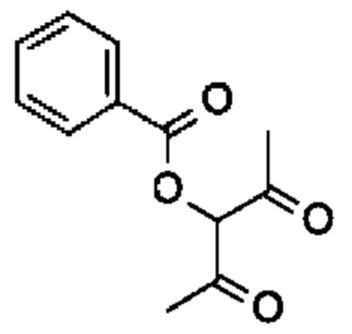
Гидроксид калия (50,5 г; 901 ммоль) добавляли к бензойной кислоте (100 г; 819 ммоль) в DMF (1 л) при комнатной температуре. Реакционную смесь перемешивали при 50°С в течение 1 часа, затем добавляли 3-хлорпентан-2,4-дион (110 г; 819 ммоль) и реакционную смесь перемешивали при 50°С в течение ночи. Реакционную смесь оставляли для охлаждения до комнатной температуры, разбавляли EtOAc (3 л) и последовательно промывали водой (1 л × 2), насыщ. водн. NH4Cl (500 мл) и насыщ. рассолом (500 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-20% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (130 г; 72%) в виде желтого масла; m/z МН+ 221.
Промежуточное соединение 55: 2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил-бензоат
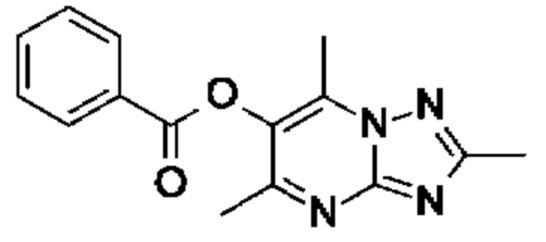
2,4-Диоксопентан-3-ил-бензоат (58 г; 263 ммоль) добавляли к 3-метил-1H-1,2,4-триазол-5-амину (27,1 г; 277 ммоль) в АсОН (300 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 10 часов, затем оставляли для охлаждения до комнатной температуры, концентрировали под вакуумом и доводили до рН более 7 с помощью насыщ. водн. NaHCO3. Смесь экстрагировали EtOAc (3×500 мл). Объединенные органические слои промывали насыщ. рассолом, пропускали через фильтровальную бумагу для разделения фаз и концентрировали под вакуумом с получением указанного в заголовке соединения (52,0 г; 70%) в виде бежевого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.57 (3Н, s), 2.66 (3Н, s), 2.70 (3Н, s), 7.54-7.65 (2Н, m), 7.71-7.80 (1H, m), 8.24-8.31 (2Н, m); m/z МН+ 283.
Промежуточное соединение 56: 2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ол
1 М водн. NaOH (177 мл; 177,11 ммоль) добавляли к 2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил-бензоату (50 г; 177 ммоль) в EtOAc (500 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем частично концентрировали под вакуумом и раствор медленно подкисляли с помощью 6 М водн. HCl до образования осадка (рН приблизительно 7). Осадок отделяли посредством фильтрования, промывали водой и сушили под вакуумом с получением указанного в заголовке соединения (24 г; 76%) в виде кремового твердого вещества; m/z МН+ 179.
Промежуточное соединение 57: трет-бутил-(R)-3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-карбоксилат
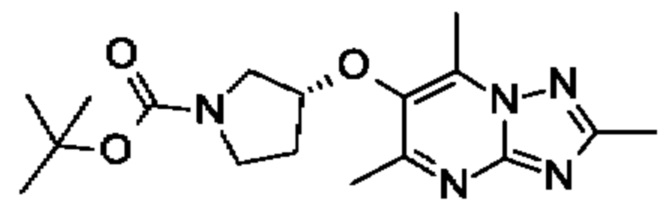
Диизопропилазодикарбоксилат по каплям добавляли (52,4 мл; 269 ммоль) к 2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-олу (40 г; 224 ммоль), трет-бутил-(S)-3-гидроксипирролидин-1-карбоксилату (46,2 г; 247 ммоль) и трифенилфосфину (70,7 г; 269 ммоль) в THF (800 мл) при комнатной температуре в течение 2 часов, и реакционную смесь перемешивали при комнатной температуре в течение 10 минут и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-100% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (62,0 г; 80%) в виде светло-желтого масла; 1H ЯМР (400 МГц, DMSO) 1.42 (9Н, d), 2.07-2.14 (1H, m), 2.20-2.23 (1H, m), 2.47 (3Н, s), 2.51-2.54 (3Н, m), 2.63 (3Н, d), 3.34 (2Н, s), 3.43-3.56 (2Н, m), 4.74 (1H, s); m/z МН+ 348.
Промежуточное соединение 58: дигидрохлорид (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидина
4 М HCl в 1,4-диоксане (150 мл; 600 ммоль) добавляли к трет-бутил-(R)-3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-карбоксилату (52 г; 149,7 ммоль) в МеОН (200 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали под вакуумом. К остатку добавляли ацетон и перемешивали в течение 30 минут. Осадок собирали посредством фильтрования с получением указанного в заголовке соединения (26 г; 55%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 2.03-2.13 (1Н, m), 2.22-2.28 (1H, m), 2.51-2.54 (3Н, m), 2.62 (3Н, d), 2.74 (3Н, s), 3.32-3.45 (3Н, m), 3.46-3.56 (1H, m), 4.90 (1H, s), 9.91 (2Н, s); m/z МН+ 248.
Промежуточное соединение 58В (свободное основание промежуточного соединения 58): (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидин
Дигидрохлорид (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло-[1,5-а]пиримидина (4,12 г; 16,7 ммоль) растворяли в МеОН и загружали на колонку SCX (50 г). Колонку промывали МеОН, затем элюировали 1 М NH3 в МеОН. Растворитель удаляли под вакуумом с получением указанного в заголовке соединения (2,73 г; 66%) в виде оранжевой смолы, которую использовали на следующей стадии без очистки; 1Н ЯМР (400 МГц, CDCl3) 1.97-2.16 (2Н, m, 2.60 (3Н, s), 2.65 (3Н, s), 2.76 (3Н, s), 3.08 (2Н, ddd), 3.19 (1H, s), 3.24-3.37 (2Н, m), 4.62 (1H, ddt); m/z МН+ 248.
Промежуточное соединение 59: (R)-6-((1-(5-бромпиридин-2-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
DIPEA (0,282 мл; 1,62 ммоль) добавляли одной порцией к 5-бром-2-фторпиридину (0,166 мл; 1,62 ммоль) и (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (400 мг; 1,62 ммоль) в н-бутаноле (4 мл) при комнатной температуре. Реакционную смесь нагревали при 150°С в микроволновом реакторе в течение 2 часов, затем оставляли для охлаждения до комнатной температуры, разбавляли EtOAc (50 мл) и последовательно промывали водой (20 мл) и насыщ. рассолом (20 мл). Органический слой сушили MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-5% МеОН в DCM, с получением указанного в заголовке соединения (346 мг; 53%) в виде желтого твердого вещества; m/z МН+ 403.
Промежуточное соединение 60: (R)-2,5,7-триметил-6-((1-(пиримидин-5-ил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидин
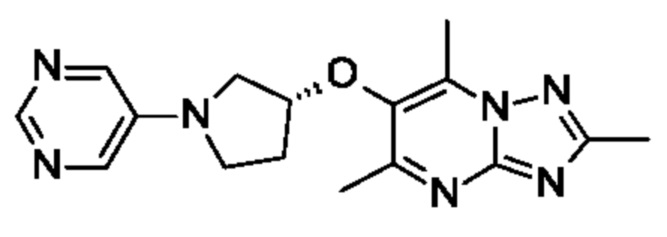
Cs2CO3 (1,98 г; 6,07 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (500 мг; 2,02 ммоль) и 5-бромпиримидину (321 мг; 2,02 ммоль) в 1,4-диоксане (20 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (169 мг; 0,20 ммоль) и RuPhos (189 мг; 0,40 ммоль). Полученную суспензию перемешивали при 90°С в течение 18 часов, затем оставляли для охлаждения до комнатной температуры. Реакционную смесь разбавляли EtOAc (50 мл) и промывали водой (30 мл) и насыщ. рассолом (25 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали под вакуумом.
Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-5% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (291 мг; 44%) в виде оранжевой смолы; 1Н ЯМР (500 МГц, CDCl3) 2.32 (1H, dtd), 2.46-2.55 (1Н, m), 2.60 (3Н, s), 2.62 (3Н, s), 2.67 (3Н, s), 3.5-3.62 (3Н, m), 3.7-3.81 (1H, m), 4.86 (1H, s), 8.12 (2Н, s), 8.66 (1H, s); m/z МН+ 326.
Промежуточное соединение 61: (R)-6-((1-(2-бромпиримидин-5-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
NBS (159 мг; 0,89 ммоль) порциями добавляли к (R)-2,5,7-триметил-6-((1-(пиримидин-5-ил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидину (291 мг; 0,89 ммоль) в MeCN (10 мл) при 0°С в течение 10 минут в воздушной атмосфере. Реакционную смесь перемешивали при 0°С в течение 3 часов, затем разбавляли DCM (50 мл) и последовательно промывали водой (20 мл) и насыщ. рассолом (20 мл). Органический слой отделяли и сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (140 мг; 39%) в виде бесцветной смолы; m/z МН+ 404.
Промежуточное соединение 62: (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
DIPEA (1,55 мл; 8,92 ммоль) добавляли одной порцией к 2,5-дибромпиразину (1,06 г; 4,46 ммоль) и (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (735 мг; 2,97 ммоль) в н-бутаноле (7 мл) при комнатной температуре. Реакционную смесь перемешивали при 120°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% МеОН в DCM, с получением указанного в заголовке соединения (740 мг; 62%) в виде желтой пены; 1Н ЯМР (500 МГц, CDCl3) 2.33 (1Н, dtd), 2.52 (1Н, ddq), 2.60 (3Н, s), 2.61 (3Н, s), 2.65 (3Н, s), 3.55 (1H, dd), 3.72 (1H, td), 3.75-3.88 (2Н, m), 4.81 (1H, tt), 7.70 (1H, d), 8.14 (1H, d); m/z МН+ 404.
Промежуточное соединение 63: 1-(4-бромфенил)-1,4-пентандион
Указанное в заголовке соединение синтезировали согласно ссылочному документу Ryzhkov, I.О. et al. Chemistry of Heterocyclic Compounds, 47(2), 182-193; 2011. Промежуточное соединение 64: 3-(4-бромфенил)-6-метилпиридазин
1-(4-Бромфенил)-1,4-пентандион (183 г; 0,717 моль) растворяли в АсОН (1,8 л) и охлаждали до 0°С. Затем добавляли моногидрат гидразина (35 мл; 0,717 моль) и смесь перемешивали в течение 30 минут. 2,3-Дихлор-5,6-дицианобензохинон (244 г; 1,08 моль) добавляли порциями в течение 30 минут и реакционную смесь перемешивали при комнатной температуре в течение 40 минут, затем концентрировали под вакуумом. Остаток распределяли между EtOAc (4 л) и насыщ. водн. NaHCO3 (4 л). Органический слой отделяли и водный слой экстрагировали EtOAc (2 л). Объединенные органические слои промывали насыщ. водн. NaHCO3 (10×2 л) и сушили над MgSO4, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали с помощью колонки под вакуумом (воронка со спеченым стеклом на 4 л, приблизительно наполовину заполненная диоксидом кремния, фракции по 1 л, продукт загружали на колонку с помощью растворения в DCM), элюируя 40-60% EtOAc/петролейный эфир. Полученный неочищенный продукт перемешивали в диэтиловом эфире (340 мл) в течение 30 минут, затем фильтровали и промывали Et2O (2×75 мл), затем петролейным эфиром (2×75 мл) и сушили на воздухе с получением указанного в заголовке соединения (91,2 г; 51%) в виде бежевого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.75 (3Н, s), 7.38 (1H, d), 7.65-7.60 (2Н, m), 7.71 (1H, d), 7.96-7.91 (2Н, m); 13С ЯМР (400 МГц, CDCl3) 22.1, 123.5, 124.4, 127.3, 128.4, 132.2, 135.4, 156.2, 158.6.
Промежуточное соединение 65: 4-((6-(4-бромфенил)пиридазин-3-ил)метил)морфолин
1,3,5-Трихлор-1,3,5-триазинан-2,4,6-трион (20,4 г; 87,8 ммоль) порциями добавляли к 3-(4-бромфенил)-6-метилпиридазину (промежуточное соединение 64) (50 г; 200,7 ммоль) в DCM (600 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли дополнительную порцию 1,3,5-трихлор-1,3,5-триазинан-2,4,6-триона (0,7 г) и реакционную смесь перемешивали в течение 30 минут, затем добавляли морфолин (87 мл; 1003,6 ммоль) и реакционную смесь перемешивали в течение 18 часов. Реакционную смесь разбавляли 300 мл воды. Органический слой отделяли и промывали водой (300 мл), и концентрировали под вакуумом. Полученный остаток суспендировали в кипящем при температуре образования флегмы EtOAc (150 мл), охлаждали до комнатной температуры и фильтровали. Отфильтрованное твердое вещество промывали 200 мл МТВЕ и сушили под вакуумом с получением указанного в заголовке соединения (53,4 г; 80%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.56 (4Н, m), 3.73 (4Н, m), 3.91 (2Н, s), 7.64 (2Н, m), 7.73 (1H, d), 7.81 (1H, d), 7.96 (2Н, m); m/z МН+ 334.
Промежуточное соединение 66: 8-((6-(4-бромфенил)пиридазин-3-ил)метил)-3-метил-3,8-диазабицикло [3.2.1] октан
Трихлоризоциануровую кислоту (65,3 мг; 0,28 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (200 мг; 0,80 ммоль) в DCE (5,9 мл) при комнатной температуре в воздушной атмосфере и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Посредством ЖХ/МС подтвердили, что было образовано хлорсодержащее промежуточное соединение. Добавляли 3-метил-3,8-диазабицикло[3.2.1]октан (101 мг; 0,80 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли DCM (50 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-10% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (193 мг; 64%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.93 (2Н, q), 2.12 (2Н, dd), 2.32 (3Н, s), 2.37 (2Н, d), 3.29 (2Н, s), 4.01 (2Н, s), 7.92-7.97 (2Н, m), 8.08 (1H, d), 8.29 (2Н, d), 8.41 (1H, d), предполагалось, что недостающие 2Н находятся под пиком DMSO; m/z МН+ 372.
Промежуточное соединение 67: (R)-2-((6-(4-бромфенил)пиридазин-3-ил)метил)октагидропирроло[1,2-а]пиразин
Трихлоризоциануровую кислоту (65,3 мг; 0,28 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (200 мг; 0,80 ммоль) в DCE (5,2 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Посредством ЖХ/МС подтвердили, что было образовано хлорсодержащее промежуточное соединение. Добавляли DIPEA (0,7 мл; 4,01 ммоль) и дигидрохлорид (R)-октагидропирроло[1,2-а]пиразина (240 мг; 1,20 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем разбавляли DCM (50 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-10% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (189 мг; 63%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.19-1.31 (1H, m), 1.66 (3Н, ddt), 1.89-2.09 (3Н, m), 2.14 (1H, td), 2.27 (1H, td), 2.73 (1Н, d), 2.86-2.98 (3Н, m), 3.83-3.94 (2Н, m), 7.74-7.8 (3Н, m), 8.09-8.14 (2Н, m), 8.23 (1H, d); m/z МН+ 373.
Промежуточное соединение 68: (S)-2-((6-(4-бромфенил)пиридазин-3-ил)метил)октагидропирроло[1,2-а]пиразин
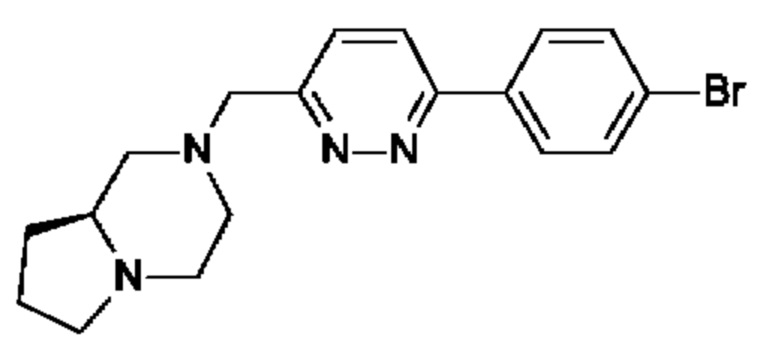
Трихлоризоциануровую кислоту (65,3 мг; 0,28 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (200 мг; 0,80 ммоль) в DCE (5,3 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Посредством ЖХ/МС подтвердили, что было образовано хлорсодержащее промежуточное соединение. Добавляли DIPEA (0,6 мл; 3,21 ммоль) и гидробромид (S)-октагидропирроло[1,2-а]пиразина (166 мг; 0,80 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 суток, затем разбавляли DCM (50 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-10% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (299 мг; 100%) в виде желтого твердого вещества; m/z МН+ 373.
Промежуточное соединение 69: 1-(4-((6-(4-бромфенил)пиридазин-3-ил)метил)пиперазин-1-ил)этан-1-он
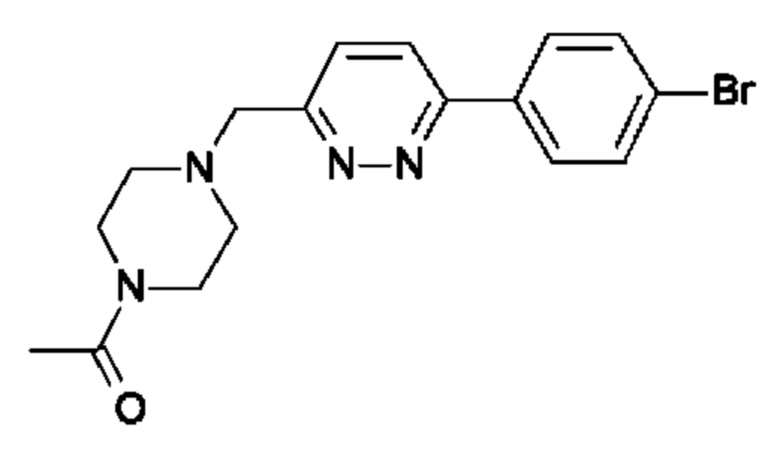
Трихлоризоциануровую кислоту (65,3 мг; 0,28 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (200 мг; 0,80 ммоль) в DCE (5,9 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли дополнительные 0,15 эквив. трихлоризоциануровой кислоты и реакционную смесь перемешивали в течение 30 минут. Добавляли 1-(пиперазин-1-ил)этан-1-он (515 мг; 4,01 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем разбавляли DCM (20 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (246 мг; 82%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.09 (3Н, s), 2.56 (4Н, dt), 3.45-3.51 (2Н, m), 3.62-3.68 (2Н, m), 3.94 (2Н, s), 7.63-7.69 (2Н, m), 7.71 (1H, d), 7.83 (1H, d), 7.94-8.02 (2Н, m); m/z МН+ 375.
Промежуточное соединение 70: 3-(4-бромфенил)-6-(пиперидин-1-илметил)пиридазин
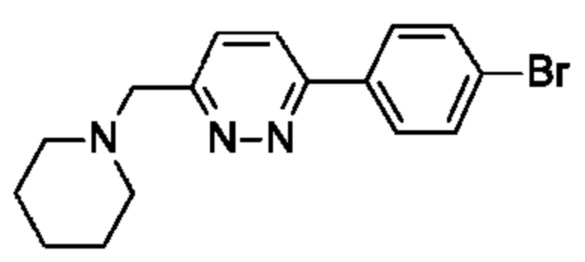
Трихлоризоциануровую кислоту (65,3 мг; 0,28 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (200 мг; 0,80 ммоль) в DCE (5,5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли дополнительные 0,15 эквив. трихлоризоциануровой кислоты и реакционную смесь перемешивали в течение 30 минут. Добавляли пиперидин (397 мкл; 4,01 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем разбавляли DCM (20 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (246 мг; 92%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.43 (2Н, q), 1.54 (4Н, р), 2.38-2.48 (4Н, m), 3.81 (2Н, s), 7.76-7.81 (3Н, m), 8.09-8.16 (2Н, m), 8.23 (1H, d); m/z МН+ 332.
Промежуточное соединение 71: 3-(4-бромфенил)-6-((4-метилпиперазин-1-ил)метил)пиридазин
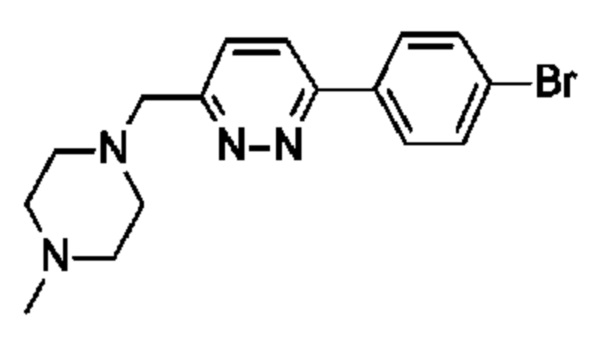
Трихлоризоциануровую кислоту (65,3 мг; 0,28 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (200 мг; 0,80 ммоль) в DCE (5,5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли дополнительные 0,15 эквив. трихлоризоциануровой кислоты и реакционную смесь перемешивали в течение 30 минут. Добавляли 1-метилпиперазин (445 мкл; 4,01 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем разбавляли DCM (50 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя смесью 0-10% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (237 мг; 85%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 2.16 (3Н, s), 2.28-2.4 (4Н, m), 2.47 (4Н, s), 3.83 (2Н, s), 7.74-7.8 (3Н, m), 8.09-8.14 (2Н, m), 8.23 (1H, d); m/z МН+ 347.
Промежуточное соединение 72: 3-(4-бромфенил)-6-((4-этилпиперазин-1-ил)метил)пиридазин
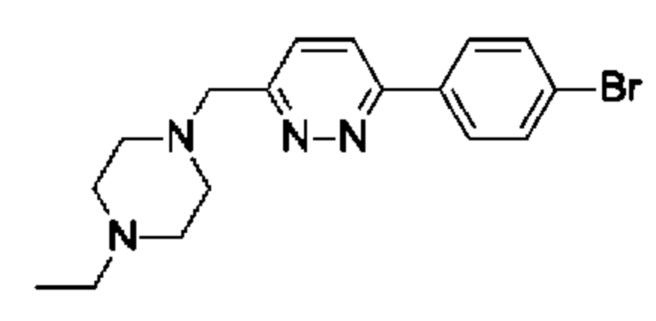
Трихлоризоциануровую кислоту (0,327 г; 1,40 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (1 г; 4,01 ммоль) в DCE (27,0 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли 1-этилпиперазин (2,55 мл; 20,1 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем разбавляли DCM (20 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-10% 1 М NH3/МеОН, с получением указанного в заголовке соединения (125 мг; 9%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.08 (3Н, t), 2.42 (2Н, q), 2.50 (4Н, s), 2.62 (4Н, s), 3.93 (2Н, s), 7.63-7.68 (2Н, m), 7.71 (1H, d), 7.80 (1H, d), 7.94 - 8.00 (2Н, m); m/z МН+ 361.
Промежуточное соединение 73: 3-(4-бромфенил)-6-((4-(2-метоксиэтил)пиперазин-1-ил)метил)пиридазин
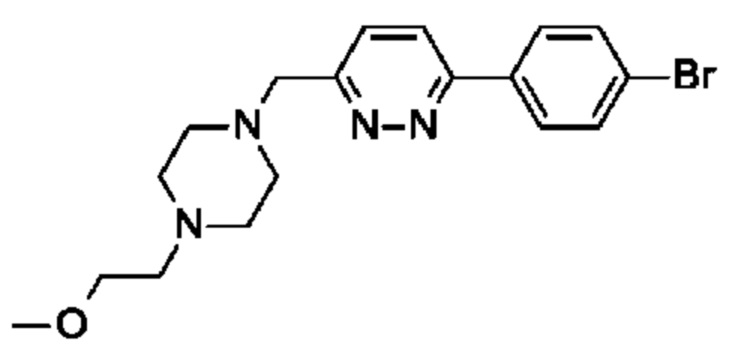
Трихлоризоциануровую кислоту (0,327 г; 1,40 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (1 г; 4,01 ммоль) в DCE (26,5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли 1-(2-метоксиэтил)пиперазин (2,98 мл; 20,1 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем разбавляли DCM (20 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-10% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (525 мг; 33%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.61 (10Н, dt), 3.35 (3Н, s), 3.5-3.55 (2Н, m), 3.93 (2Н, s), 7.62-7.67 (2Н, m), 7.70 (1H, d), 7.80 (1H, d), 7.94-8 (2Н, m); m/z МН+ 391.
Промежуточное соединение 74: 4-((6-(4-бромфенил)пиридазин-3-ил)метил)-1,4-оксазепан
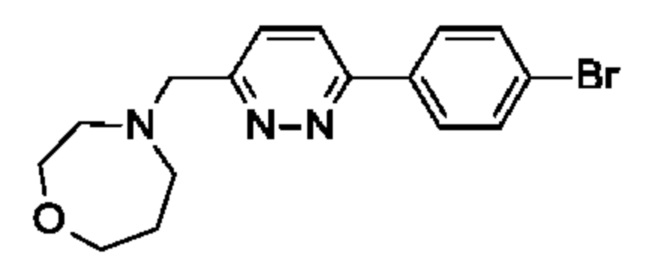
Трихлоризоциануровую кислоту (65,3 мг; 0,28 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (200 мг; 0,80 ммоль) в DCE (5,9 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли дополнительные 0,15 эквив. трихлоризоциануровой кислоты и реакционную смесь перемешивали в течение 30 минут. Добавляли 1,4-оксазепан (406 мг; 4,01 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем разбавляли DCM (50 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (200 мг; 72%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.83 (2Н, р), 2.72 (4Н, q), 3.6-3.67 (2Н, m), 3.72 (2Н, t), 4.01 (2Н, s), 7.73-7.81 (2Н, m), 7.83 (1H, d), 8.07-8.16 (2Н, m), 8.24 (1H, d); m/z МН+ 348.
Промежуточное соединение 75: 1-((6-(4-бромфенил)пиридазин-3-ил)метил)азепан
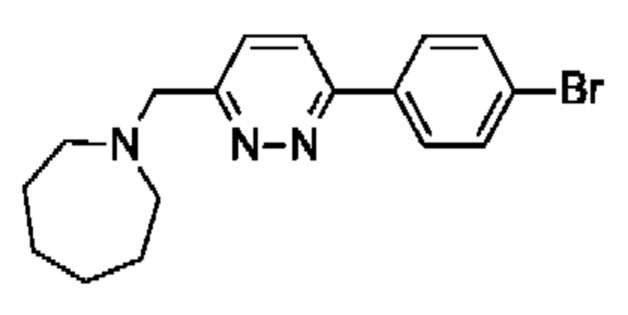
Трихлоризоциануровую кислоту (65,3 мг; 0,28 ммоль) добавляли одной порцией к раствору 3-(4-бромфенил)-6-метилпиридазина (промежуточное соединение 64) (200 мг; 0,80 ммоль) в DCE (5,5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли дополнительные 0,15 эквив. трихлоризоциануровой кислоты и реакционную смесь перемешивали в течение 30 минут. Добавляли азепан (452 мкл; 4,01 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 суток, затем разбавляли DCM (50 мл), фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (88 мг; 32%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.59 (8Н, s), 2.64-2.7 (4Н, m), 3.98 (2Н, s), 7.74-7.79 (2Н, m), 7.82 (1H, d), 8.09-8.14 (2Н, m), 8.23 (1H, d); m/z MH+ 346.
Промежуточное соединение 76: 4-((5-хлорпиразин-2-ил)метил)морфолин
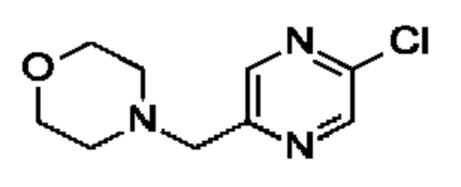
Триацетоксиборгидрид натрия (4,46 г; 21,1 ммоль) добавляли к 5-хлорпиразин-2-карбальдегиду (1 г; 7,02 ммоль) и морфолину (0,614 мл; 7,02 ммоль) в DCM (10 мл) при комнатной температуре и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли насыщ. водн. NaHCO3 и перемешивали в течение 10 минут, затем пропускали через фильтровальную бумагу для разделения фаз и концентрировали под вакуумом с получением указанного в заголовке соединения (1,180 г; 79%) в виде бесцветного масла; 1Н ЯМР (400 МГц, CDCl3) 2.49-2.55 (4Н, m), 3.69 (2Н, s), 3.71-3.77 (4Н, m), 8.47 (1H, d), 8.55 (1H, d); m/z МН+ 214.
Промежуточное соединение 77: 4-((5-(4-бромфенил)пиразин-2-ил)метил)морфолин
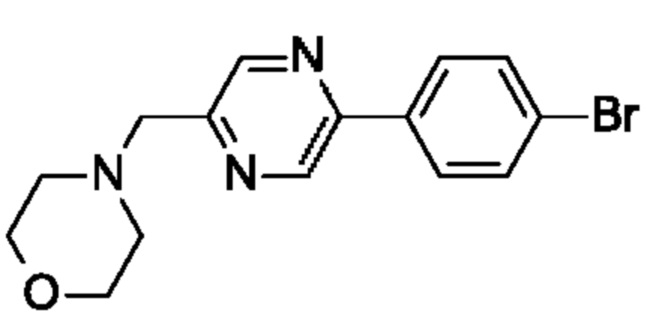
Pd(Ph3P)4 (135 мг; 0,12 ммоль) добавляли одной порцией к дегазированной смеси (4-бромфенил)бороновой кислоты (470 мг; 2,34 ммоль), 4-((5-хлорпиразин-2-ил)метил)морфолина (500 мг; 2,34 ммоль) и 2 М водн. Na2CO3 (2,34 мл; 4,68 ммоль) в 1,4-диоксане (7,8 мл) и воде (1.5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 2 часов, затем оставляли для охлаждения до комнатной температуры, разбавляли EtOAc (75 мл) и последовательно промывали водой (20 мл) и насыщ. рассолом (20 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-3% МеОН в DCM, с получением указанного в заголовке соединения (730 мг; 93%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.57 (4Н, q), 3.72-3.79 (6Н, m), 7.62-7.66 (2Н, m), 7.88-7.93 (2Н, m), 8.71 (1H, d), 8.95 (1H, d); m/z МН+ 334.
Промежуточное соединение 78: 2-бром-5-(пирролидин-1-илметил)пиридин
Иодид калия (331 мг; 1,99 ммоль) добавляли к 2-бром-5-(бромметил)пиридину (500 мг; 1,99 ммоль), пирролидину (142 мг; 1,99 ммоль) и K2CO3 (275 мг; 1,99 ммоль) в DMF (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем разбавляли водой (50 мл), экстрагировали EtOAc (3×50 мл), и объединенные органические слои промывали насыщ. рассолом (3×50 мл) и сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением указанного в заголовке соединения (470 мг; 98%) в виде желтого масла; m/z МН+ 241.
Промежуточное соединение 79: 5-бром-2-(бромметил)пиридин
Бензоилпероксид (0,063 г; 0,26 ммоль) добавляли к 5-бром-2-метилпиридину (0,9 г; 5,23 ммоль) и NBS (0,978 г; 5,49 ммоль) в CCl4 (10 мл) при 15°С. Реакционную смесь нагревали при температуре образования флегмы в течение 15 часов, затем оставляли для охлаждения до комнатной температуры. Реакционную смесь разбавляли DCM (100 мл) и последовательно промывали водой (50 мл) и насыщ. рассолом (50 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 5% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (0,440 г; 34% в случае чистого) в виде неочищенного фиолетового масла; m/z МН+ 250.
Промежуточное соединение 80: 5-бром-2-(пирролидин-1-илметил)пиридин
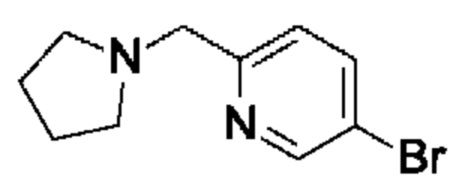
Иодид калия (437 мг; 2,63 ммоль) добавляли к 5-бром-2-(бромметил)пиридину (660 мг; 2,63 ммоль), пирролидину (187 мг; 2,63 ммоль) и K2CO3 (364 мг; 2,63 ммоль) в DMF (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем гасили водой (50 мл) и экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали насыщ. рассолом (3×50 мл) и сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением указанного в заголовке соединения (570 мг; 90%) в виде черного масла; m/z МН+ 241.
Промежуточное соединение 81: (R)-3-метил-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензальдегид
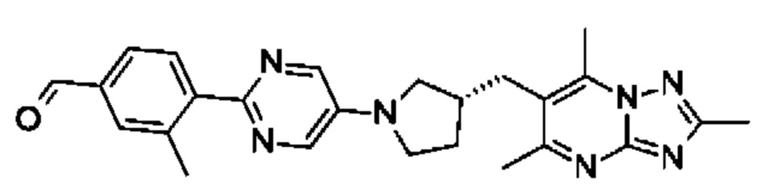
Бифенил-2-аминохлорид XPhos-палладия(II) (20 мг; 0,02 ммоль) добавляли к (R)-6-((1-(2-бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]-триазоло-[1,5-а]пиримидину (промежуточное соединение 16) (200 мг; 0,50 ммоль), 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегиду (промежуточное соединение 18) (122 мг; 0,50 ммоль) и Cs2CO3 (324 мг; 0,99 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде, с получением указанного в заголовке соединения (75 мг; 34%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.90 (1Н, s), 2.27 (1H, s), 2.66 (7Н, d), 2.77 (3Н, s), 2.86 (3Н, s), 2.92-3.11 (2Н, m), 3.24 (1H, s), 3.45-3.54 (1H, m), 3.59 (1H, s), 3.67 (1H, s), 7.82 (2Н, dt), 7.98 (1H, d), 8.24 (2Н, s), 10.06 (1H, s); m/z МН+ 442.
Промежуточное соединение 82: (R)-трет-бутил-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридин-3-ил)метил)пиперазин-1-карбоксилат
Дегазированный 1,4-диоксан (15 мл) добавляли к дигидрохлориду (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (промежуточное соединение 4) (500 мг; 2,04 ммоль) и трет-бутил-4-((6-(4-бромфенил)пиридин-3-ил)метил)пиперазин-1-карбоксилату (промежуточное соединение 37) (881 мг; 2,04 ммоль). Затем добавляли Cs2CO3 (1,99 г; 6,11 ммоль) с последующим добавлением RuPhos (190 мг; 0,41 ммоль) и предкатализатора RuPhos 3-го поколения (341 мг; 0,41 ммоль). Реакционную смесь нагревали при 90°С в течение 24 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Остаток помещали в DCM, фильтровали через целит, концентрировали под вакуумом и очищали посредством КФХ, элюируя 0-10% МеОН в DCM, с получением указанного в заголовке соединения (290 мг; 24%) в виде бежевого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.45 (9Н, s), 1.82-1.94 (1H, m), 2.13-2.23 (1H, m), 2.41 (4Н, s), 2.62 (4Н, s), 2.70 (3Н, s), 2.78 (3Н, s), 2.86-2.99 (2Н, m), 3.14 (1H, dd), 3.37-3.47 (6Н, m), 3.52 (2Н, s), 3.57 (1H, td), 6.58-6.64 (2Н, m), 7.60 (1H, dd), 7.64 (1H, dd), 7.86-7.96 (2Н, m), 8.51 (1Н, d); m/z МН+ 597.
Пример 1: (R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)-[3,3'-бипиридин] -6-ил)метил)морфолин
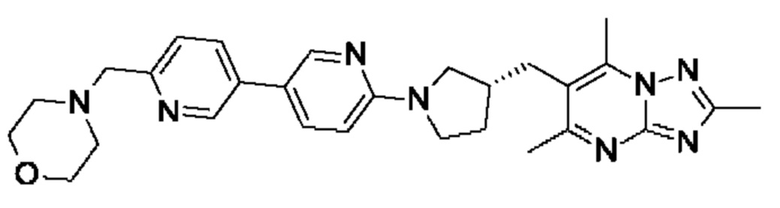
Пред катализатор XPhos 2-го поколения (29,4 мг; 0,04 ммоль) добавляли к (R)-6-((l-(5-бромпиридин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 5) (150 мг; 0,37 ммоль), (6-(морфолинометил)пиридин-3-ил)бороновой кислоте (из коммерческих источников) (83 мг; 0,37 ммоль) и Cs2CO3 (244 мг; 0,75 ммоль) в 1,4-диоксане (2 мл) и воде (1 мл). Реакционную смесь перемешивали при 80°С в течение 3 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением формиата указанного в заголовке соединения (80 мг; 36%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.89-2.08 (1H, m), 2.19-2.33 (1H, m), 2.56 (3Н, s), 2.71-2.73 (4Н, m), 2.77-2.89 (7Н, m), 3.04 (2Н, d), 3.24-3.33 (1Н, m), 3.44-3.57 (1Н, m), 3.57-3.75 (1H, m), 3.71-3.85 (5Н, m), 3.98 (2Н, d), 6.67 (1H, d), 7.58 (1Н, d), 7.89 (1Н, dd), 8.04 (1H, dd), 8.18 (2Н, s, соответствует 2 эквивалентам формиата), 8.33 (1Н, d), 8.76 (1Н, d) m/z МН+ 499.
Пример 4: (R)-2,5,7-триметил-6-((1-(6,-((4-метилпиперазин-1-ил)метил)-[3,3,-бипиридин]-6-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
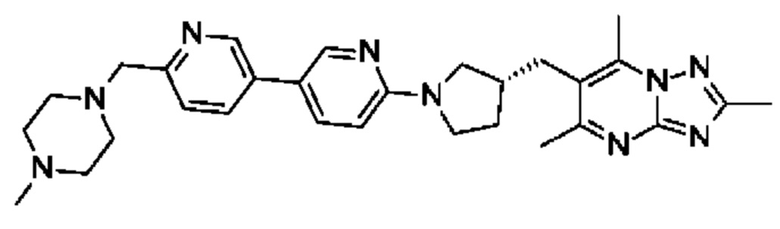
Pd(Ph3P)4 (72,9 мг; 0,06 ммоль) добавляли к (R)-6-((1-(5-бромпиридин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 5) (253 мг; 0,63 ммоль), (6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)бороновой кислоте (промежуточное соединение 9) (200 мг; 0,85 ммоль) и Na2CO3 (134 мг; 1,26 ммоль) в толуоле (10 мл) и воде (2 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением формиата указанного в заголовке соединения (60 мг; 17%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, DMSO) 1.84 (1H, m), 2.10 (1H, m), 2.20 (3Н, s), 2.30-2.50 (12Н, m), 2.63 (3Н, s), 2.74 (3Н, s), 2.92 (2Н, d), 3.18 (1H, dd), 3.38 (1H, dt), 3.57 (4Н, m), 6.55 (1H, d), 7.42 (1H, d), 7.84 (1H, dd), 7.96 (1H, dd), 8.20 (1H, s), 8.43 (1H, d), 8.72 (1H, dd); m/z МН+ 512.
Пример 5: (R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиридин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
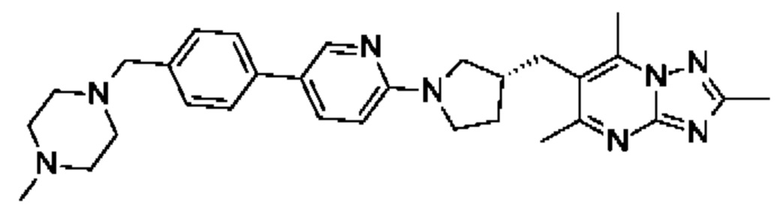
Pd(Ph3P)4 (17 мг; 0,01 ммоль) добавляли к 1-метил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазину (из коммерческих источников) (95 мг; 0,30 ммоль), (R)-6-((1-(5-бромпиридин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 5) (120 мг; 0,30 ммоль) и Na2CO3 (63,4 мг; 0,60 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением формиата указанного в заголовке соединения (55 мг; 33%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 1.99 (1H, dq), 2.26 (1H, dq), 2.57 (3Н, s), 2.70 (11Н, m), 2.85 (3Н, s), 3.05 (6Н, m), 3.25-3.34 (2Н, m), 3.50 (1H, m), 3.60 (1H, m), 3.70 (3Н, m), 6.63 (1H, d), 7.42 (2Н, d), 7.55(2Н, d), 7.84 (1H, dd), 8.28 (1H, d), 8.47 (1H, s); m/z МН+ 511.
Пример 6: (R)-4-(4-(2-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-5-ил)бензил)морфолин
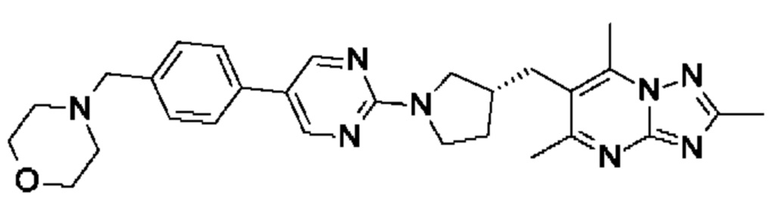
Предкатализатор XPhos 2-го поколения (34 мг; 0,04 ммоль) добавляли к рац-6-((1-(5-бромпиримидин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло-[1,5-а]пиримидину (промежуточное соединение 10) (350 мг; 0,87 ммоль), 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)морфолину (из коммерческих источников) (264 мг; 0,87 ммоль) и Cs2CO3 (567 мг; 1,74 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,08% NH4CO3). Полученный рацемический продукт (390 мг) разделяли посредством препаративной хиральной ВЭЖХ на колонке Chiralpak IA, элюируя 50% EtOH в ТВМЕ (модифицированный DEA) в качестве элюента, с получением указанного в заголовке соединения (120 мг; 31%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 1.90-2.04 (1H, m), 2.23 (1H, dt), 2.50 (4Н, t), 2.57 (3Н, s), 2.74 (4Н, s), 2.86 (3Н, s), 3.05 (2Н, d), 3.39 (1H, dd), 3.60 (3Н, d), 3.68-3.79 (5Н, m), 3.86 (1H, ddd), 7.45 (2Н, d), 7.55 (2Н, d), 8.61 (2Н, s); m/zMH+ 499.
Пример 7: (R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиридин-2-ил)пиразин-2-ил)метил)морфолин
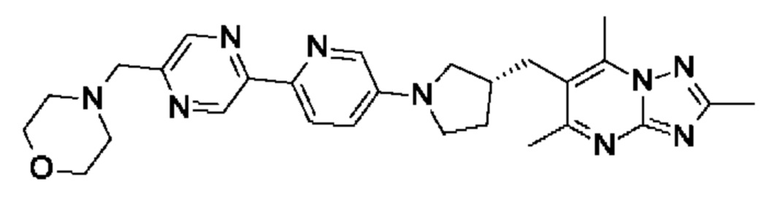
Pd(Ph3P)4 (46,1 мг; 0,04 ммоль) добавляли к 4-((5-хлорпиразин-2-ил)метил)морфолину (промежуточное соединение 76) (85 мг; 0,40 ммоль), (R)-6-((1-(6-бромпиридин-3-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]-пиримидину (промежуточное соединение 12) (160 мг; 0,40 ммоль) и 1,1,1,2,2,2-гексаметилдистаннану (131 мг; 0,40 ммоль) в 1,4-диоксане (5 мл) при комнатной температуре. Реакционную смесь перемешивали при 110°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением указанного в заголовке соединения (87 мг; 43%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.89-2.08 (1H, m), 2.18-2.31 (1H, m), 2.56 (3Н, s), 2.60-2.87 (10Н, m), 3.05 (2Н, dd), 3.23 (1H, dd), 3.37-3.85 (10Н, m), 7.10 (1H, dd), 8.03 (1H, d), 8.20 (1H, d), 8.66 (1H, d), 9.31 (1H, d); m/zMH+ 500.
Пример 8: (R)-2,5,7-триметил-6-((1-(6-(4-((4-метилпиперазин-1-ил)метил)фенил)пиридин-3-ил)пирр о лидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
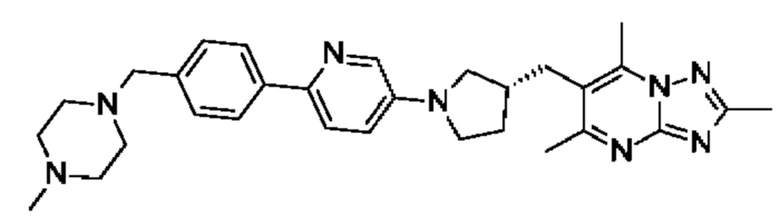
Pd(Ph3P)4 (202 мг; 0,17 ммоль) добавляли к (R)-6-((1-(6-бромпиридин-3-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 12) (700 мг; 1,74 ммоль), 1-метил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазину (из коммерческих источников) (607 мг; 1,92 ммоль) и 1 М водн. Na2CO3 (3,49 мл; 3,49 ммоль) в дегазированном 1,4-диоксане (7 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 18 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Остаток помещали в DCM, фильтровали и полученный неочищенный продукт очищали посредством КФХ, элюируя смесью 0-10% 1 М NH3/МеОН в DCM, затем дополнительно очищали посредством перекристаллизации из EtOAc (с небольшим количеством МеОН) с получением указанного в заголовке соединения (240 мг; 27%) в виде бледно-желтого кристаллического твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.86-1.94 (1H, m), 2.14-2.23 (1H, m), 2.28 (3Н, s), 2.49 (8Н, s), 2.62 (4Н, s), 2.71 (3Н, s), 2.79 (3Н, s), 2.86-3.01 (2Н, m), 3.14 (1H, dd), 3.37-3.48 (2Н, m), 3.54 (2Н, s), 3.55-3.61 (1Н, m), 6.86 (1H, dd), 7.37 (2Н, d), 7.59 (1Н, dd), 7.82-7.88 (2Н, m), 8.06 (1H, d); m/z МН+ 511.
Пример 9: (R)-4-((6-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиридин-2-ил)пиридазин-3-ил)метил)морфолин
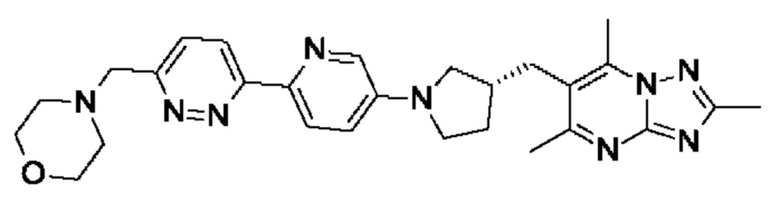
Pd(Ph3P)4 (343 мг; 0,30 ммоль) добавляли одной порцией к 4-((6-хлорпиридазин-3-ил)метил)морфолину (промежуточное соединение 14) (634 мг; 2,97 ммоль), (R)-6-((1-(6-бромпиридин-3-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 12) (1,19 г; 2,97 ммоль) и 1,1,1,2,2,2-гексаметилдистаннану (0,615 мл; 2,97 ммоль) в 1,4-диоксане (30 мл) при комнатной температуре. Реакционную смесь перемешивали при 100°С в течение 20 часов, затем оставляли для охлаждения до комнатной температуры, фильтровали и промывали DCM (20 мл). Фильтрат концентрировали под вакуумом, и полученный неочищенный продукт очищали посредством КФХ, элюируя смесью 0-10% 1 М NH3/МеОН в DCM, затем дополнительно очищали посредством препаративной ВЭЖХ, затем растворяли в МеОН и загружали на колонку SCX (50 г). Колонку промывали МеОН (2хобъем колонки), затем элюировали смесью 1 М NH3/МеОН. Полученную смолу растирали с Et2O с получением твердого вещества, которое отфильтровывали и сушили под вакуумом с получением указанного в заголовке соединения (608 мг; 41%) в виде бледно-желтого твердого вещества; 1Н ЯМР (500 МГц, DMSO) 1.87 (1H, dq), 2.06-2.22 (1H, m), 2.45 (7Н, d), 2.64 (4Н, s), 2.74 (3Н, s), 2.93 (2Н, d), 3.12 (1Н, dd), 3.33-3.38 (1H, m), 3.48 (1Н, dd), 3.51-3.64 (5Н, m), 3.79 (2Н, s), 7.06 (1Н, dd), 7.71 (1Н, d), 8.05 (1H, d), 8.33 (2Н, dd); m/z МН+ 500.
Пример 10: 6-((R)-1-(2-(4-(((5)-2,4-диметилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4] триазоло [1,5-а]пиримидин
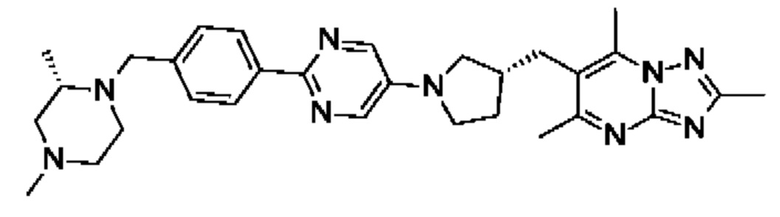
(R)-6-((1-(2-Бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (промежуточное соединение 16) (300 мг; 0,75 ммоль), (S)-2,4-диметил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазин (промежуточное соединение 17) (296 мг; 0,89 ммоль), Pd(Ph3P)4 (86 мг; 0,07 ммоль) и карбонат калия (206 мг; 1,49 ммоль) растворяли в 1,4-диоксане (12 мл) и воде (4 мл) и нагревали при 80°С в течение 18 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, затем выливали в DCM (50 мл). Водный слой отделяли, затем экстрагировали 20% MeOH/DCM. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-5% 1 М NH3/МеОН в DCM. Полученную смолу растирали с EtOAc и полученное твердое вещество отфильтровывали и сушили под вакуумом с получением указанного в заголовке соединения (52 мг; 13%) в виде белого твердого вещества; 1Н ЯМР (500 МГц, DMSO) 1.06 (3Н, d), 1.78-1.9 (2Н, m), 1.98 (1H, d), 2.10 (5Н, s), 2.41 (1H, s), 2.47 (4Н, s), 2.55 (2Н, d), 2.64 (4Н, s), 2.74 (3Н, s), 2.92 (2Н, d), 3.04-3.18 (2Н, m), 3.34 (1H, d), 3.46 (1H, dd), 3.5-3.58 (1H, m), 3.97 (1H, d), 7.33 (2Н, d), 8.16 (2Н, d), 8.20 (2Н, s); m/z МН+526.
Пример 11: (R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)га1рролидин-1-ил)пиримидин-2-ил)пиридин-2-ил)метил)морфолин
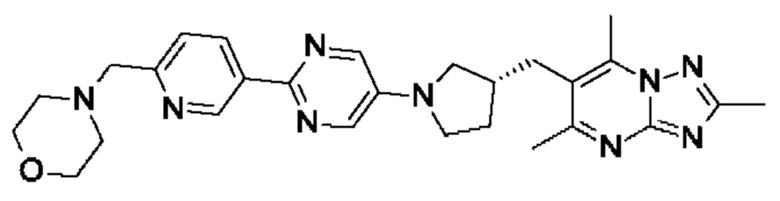
Предкатализатор XPhos 2-го поколения (9 мг; 0,01 ммоль) добавляли к (R)-6-((l-(2-бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 16) (90 мг; 0,22 ммоль), (6-(морфолинометил)пиридин-3-ил)бороновой кислоте (из коммерческих источников) (50 мг; 0,22 ммоль) и Cs2CO3 (146 мг; 0,45 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде, с получением указанного в заголовке соединения (22 мг; 20%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 2.00 (1H, dq), 2.26 (1H, dq), 2.57 (3Н, s), 2.64 (4Н, s), 2.75 (4Н, s), 2.86 (3Н, s), 3.08 (2Н, h), 3.25 (1H, dd), 3.46 (1H, dt), 3.54-3.72 (2Н, m), 3.72-3.83 (6Н, m), 7.63 (1H, d), 8.24 (2Н, s), 8.61 (1H, dd), 9.34 (1H, d); m/z МН+ 500.
Пример 12: (R)-6-((1-(2-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
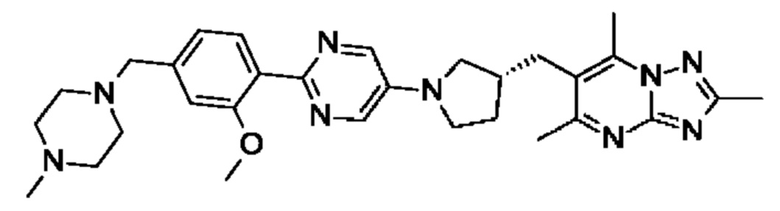
Предкатализатор XPhos 2-го поколения (20 мг; 0,02 ммоль) добавляли к (R)-6-((l-(2-бромпиридин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 16) (200 мг; 0,50 ммоль), (2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)бороновой кислоте (промежуточное соединение 42) (131 мг; 0,50 ммоль) и Cs2CO3 (324 мг; 0,99 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде, с получением формиата указанного в заголовке соединения (56 мг; 19%) в виде бледно-желтого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 2.00 (1H, dq), 2.25 (1H, ddd), 2.57 (3Н, s), 2.72 (10Н, m), 2.87 (3Н, s), 2.99-3.15 (7Н, m), 3.22 (1H, dd), 3.44 (1H, dt), 3.51-3.72 (4Н, m), 3.83 (3Н, s), 7.04 (1H, dd), 7.14 (1H, d), 7.49 (1H, d), 8.19 (2Н, s), 8.49 (1H, s); m/z МН+ 542.
Пример 13: (R)-2,5,7-триметил-6-((1-(2-(2-метил-4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
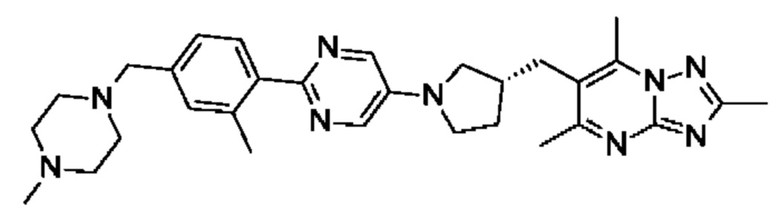
1-Метилпиперазин (34 мг; 0,34 ммоль) добавляли к (R)-3-метил-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензальдегиду (75 мг; 0,17 ммоль) в DCM (0,5 мл) при комнатной температуре в воздушной атмосфере, и реакционную смесь перемешивали в течение 2 часов. Добавляли триацетоксиборгидрид натрия (180 мг; 0,85 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 50 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде, с получением указанного в заголовке соединения (60 мг; 67%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 2.00 (1H, dq), 2.26 (1H, ddd), 2.42 (3Н, s), 2.57 (3Н, s), 2.72 (10Н, m), 2.87 (3Н, s), 3.06 (7Н, m), 3.23 (1H, dd), 3.44 (1H, dt), 3.52-3.70 (4Н, m), 7.24-7.32 (2Н, m), 7.54 (1H, d), 8.23 (2Н, s); m/z МН+ 526.
Пример 14: 6-(((R)-1-(2-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
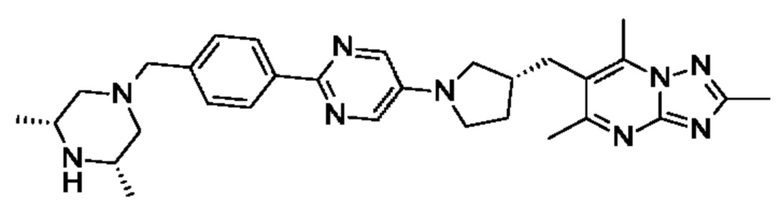
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиримидин-2-ил)бензальдегид (промежуточное соединение 19) (120 мг; 0,28 ммоль) добавляли к (2R,6S)-2,6-диметилпиперазину (из коммерческих источников) (32 мг; 0,28 ммоль) в DCM (3 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли АсОН (1,607 мкл; 0,03 ммоль) и триацетоксиборгидрид натрия (178 мг; 0,84 ммоль) и реакционную смесь перемешивали в течение 2 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (16 мг; 11%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.18 (6Н, s), 1.94 (3Н, ddd), 2.21 (1H, m), 2.56 (3Н, s), 2.73 (4Н, m), 2.84 (3Н, s), 3.08 (4Н, m), 3.18 (3Н, m), 3.58 (2Н, m), 3.62 (4Н, m), 7.41 (2Н, dd), 8.17 (4Н, dt); m/z МН+ 526.
Пример 15: (R)-N,N-диметил-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)ацетамид
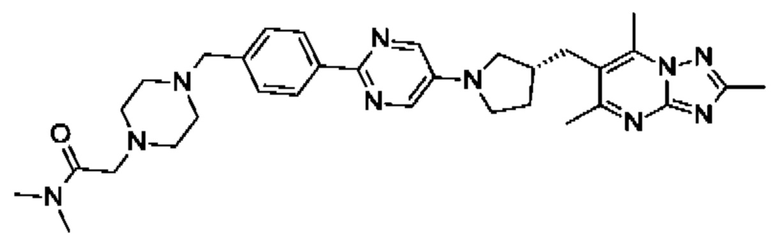
Аддукт PdCl2(dppf)-CH2Cl2 (168 мг; 0,21 ммоль) добавляли в 2-(4-(4-бромбензил)пиперазин-1-ил)-N,N-диметилацетамид (промежуточное соединение 22) (700 мг; 2,06 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (627 мг; 2,47 ммоль) и ацетат калия (404 мг; 4,11 ммоль) в 1,4-диоксане (10 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением (4-((4-(2-(диметиламино)-2-оксоэтил)пиперазин-1-ил)метил)фенил)бороновой кислоты (186 мг; 30%), которую использовали немедленно без описания свойств. Pd(Ph3P)4 (31,6 мг; 0,03 ммоль) добавляли к (4-((4-(2-(диметиламино)-2-оксоэтил)пиперазин-1-ил)метил)-фенил)бороновой кислоте (92 мг; 0,30 ммоль), (R)-6-((1-(2-бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 16) (110 мг; 0,27 ммоль) и Na2CO3 (58,0 мг; 0,55 ммоль) в 1,4-диоксане (2 мл) и воде (1 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% MeCN в воде (плюс 0,1% FA), затем дополнительно очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (50 мг; 31%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.98 (1Н, dt), 2.16-2.34 (1H, m), 2.43-2.64 (10Н, m), 2.73 (4Н, s), 2.84 (3Н, s), 2.93 (3Н, s), 2.97-3.15 (5Н, m), 3.22 (3Н, d), 3.33-3.49 (2Н, m), 3.48-3.70 (4Н, m), 7.41 (2Н, d), 8.11-8.21 (4Н, m); m/z МН+ 583.
Пример 16: (R)-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)этанол
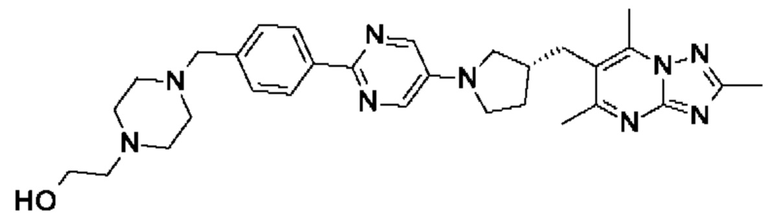
Аддукт PdCl2(dppf)-CH2Cl2 (164 мг; 0,20 ммоль) добавляли в 2-(4-(4-бромбензил)пиперазин-1-ил)этанол (промежуточное соединение 23) (600 мг; 2,01 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (611 мг; 2,41 ммоль) и ацетат калия (394 мг; 4,01 ммоль) в 1,4-диоксане (10 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде, с получением (4-((4-(2-гидроксиэтил)пиперазин-1-ил)метил)фенил)бороновой кислоты (143 мг; 27%) в виде коричневого твердого вещества, которое использовали немедленно без описания свойств. Pd(Ph3P)4 (31,6 мг; 0,03 ммоль) добавляли к (4-((4-(2-гидроксиэтил)пиперазин-1-ил)метил)-фенил)бороновой кислоте (87 мг; 0,33 ммоль), (R)-6-((1-(2-бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 16) (110 мг; 0,27 ммоль) и Na2CO3 (58,0 мг; 0,55 ммоль) в 1,4-диоксане (2 мл) и воде (1 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% MeCN в воде (плюс 0,1% FA), затем дополнительно очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (50 мг; 34%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.88-2.02 (1H, m), 2.15-2.26 (1H, m), 2.68 (18Н, m), 2.82 (3Н, s), 2.87-3.05 (2Н, m), 3.18 (1H, dd), 3.36-3.58 (2Н, m), 3.58-3.71 (5Н, m), 7.41 (2Н, d), 8.14 (2Н, s), 8.21-8.30 (2Н, m); m/z МН+ 542.
Пример 17: (R)-1-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)этанон
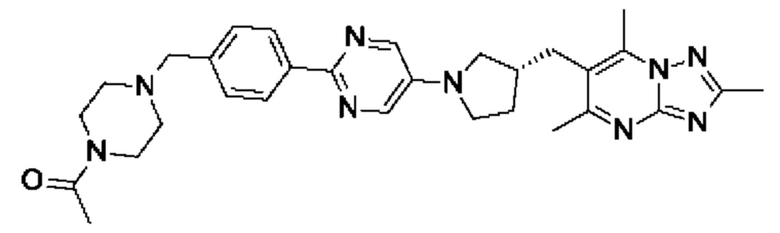
Pd(Ph3P)4 (28,7 мг; 0,02 ммоль) добавляли к 1-(4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазин-1-ил)этанону (промежуточное соединение 52) (86 мг; 0,25 ммоль), (R)-6-((1-(2-бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 16) (100 мг; 0,25 ммоль) и Na2CO3 (53 мг; 0,50 ммоль) в 1,4-диоксане (5 мл) и воде (1 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (50 мг; 37%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.96 (1H, dq), 2.10 (3Н, s), 2.22 (1H, m), 2.64 (11Н, m), 2.84 (3Н, s), 3.03 (2Н, dd), 3.20 (1H, dd), 3.60 (9Н, m), 7.43 (2Н, m), 8.19 (4Н, d); m/z МН+540.
Пример 18: (R)-6-((1-(2-(4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
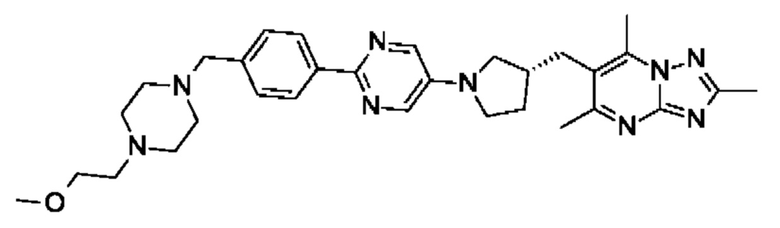
Триацетоксиборгидрид натрия (283 мг; 1,33 ммоль) добавляли к (R)-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)-пиримидин-2-ил)бензальдегиду (промежуточное соединение 19) (190 мг; 0,44 ммоль), и 1-(2-метоксиэтил)пиперазину (77 мг; 0,53 ммоль), и АсОН (0,013 мл; 0,22 ммоль) в DCM (20 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем концентрировали под вакуумом, нейтрализовали смесью 1 М NH3/MeOH и концентрировали под вакуумом. Полученный остаток помещали в DCM, фильтровали и очищали посредством КФХ, элюируя 0-5% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (175 мг; 71%) в виде бледно-желтого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.86-1.96 (1H, m), 2.14-2.25 (1H, m), 2.39-2.6 (10Н, m), 2.62 (3Н, s), 2.64-2.69 (1H, m), 2.71 (3Н, s), 2.80 (3Н, s), 2.87-3.02 (2Н, m), 3.17 (1H, dd), 3.34 (3Н, s), 3.39-3.46 (1H, m), 3.47-3.53 (3Н, m), 3.56 (2H, s), 3.59 (1H, td), 7.39 (2H, d), 8.13 (2H, s), 8.23 (2H, d); m/z MH+ 556.
Пример 19: (R)-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2 -ил)бензил)морфолин
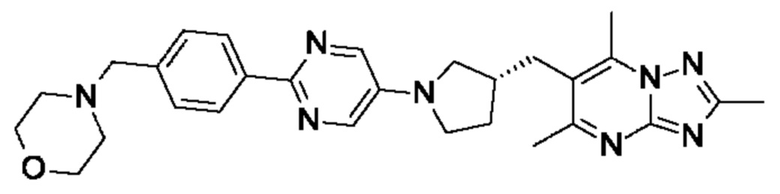
Предкатализатор RuPhos 3-го поколения (50 мг; 0,06 ммоль) добавляли в 4-(4-(5-бромпиримидин-2-ил)бензил)морфолин (промежуточное соединение 24) (200 мг; 0,60 ммоль), (R)_2,5,7-триметил-6-(пирролидин-3-илметил)- [1,2,4]триазоло[1,5-а]пиримидин⋅2HCl (промежуточное соединение 4) (190 мг; 0,60 ммоль), Cs2CO3 (780 мг; 2,39 ммоль) и RuPhos (55,8 мг; 0,12 ммоль) в 1,4-диоксане (8 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 6 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% МеОН в воде (плюс 0,1% FA), с получением указанного в заголовке соединения (180 мг; 60%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.87-2.06 (1H, m), 2.16-2.27 (1Н, m), 2.53-2.62 (7Н, m), 2.73 (4Н, s), 2.84 (3Н, s), 3.04 (2Н, dd), 3.20 (1H, dd), 3.41 (1H, dt), 3.49-3.71 (4Н, m), 3.71-3.77 (4Н, m), 7.43 (2Н, d), 8.13-8.22 (4Н, m); m/z МН+ 499.
Пример 20: (R)-2,5,7-триметил-6-((1-(2-(4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
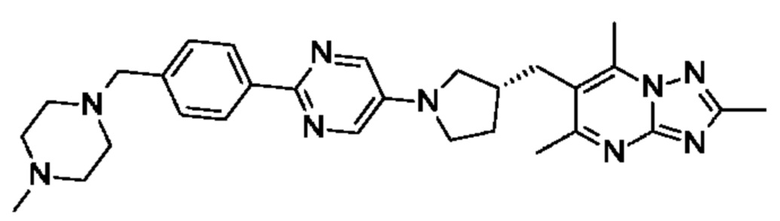
Pd(Ph3P)4 (43,1 мг; 0,04 ммоль) добавляли к (R)-6-((1-(2-бромпиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 16) (300 мг; 0,75 ммоль), 1-метил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазину (из коммерческих источников) (236 мг; 0,75 ммоль) и Na2CO3 (158 мг; 1,49 ммоль) в 1,4-диоксане (10 мл) и воде (5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1 ммоль/л NH4HCO3), с получением указанного в заголовке соединения (285 мг; 75%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 1.94-2.05 (1H, m), 2.31 (4Н, s), 2.57 (11Н, s), 2.75 (4Н, s), 2.86 (3Н, s), 3.06 (2Н, р), 3.22 (1H, dd), 3.38-3.49 (1Н, m), 3.51-3.69 (4Н, m), 7.42 (2Н, d), 8.14-8.22 (4Н, m); m/z МН+ 512.
Пример 21: (R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиразин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]-пиримидин
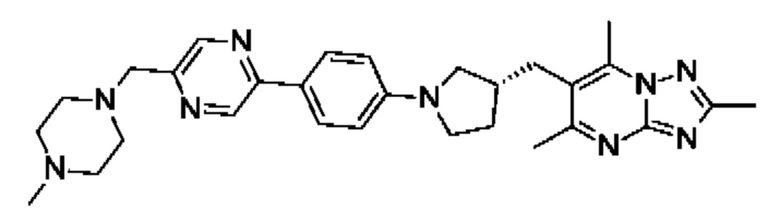
Pd(Ph3P)4 (51,7 мг; 0,04 ммоль) добавляли к 2-бром-5-((4-метилпиперазин-1-ил)метил)пиразину (промежуточное соединение 28) (121 мг; 0,45 ммоль), (R)-2,5,7-триметил-6-((1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 27) (200 мг; 0,45 ммоль) и Na2CO3 (95 мг; 0,89 ммоль) в толуоле (0,2 мл) и воде (1 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% MeCN в воде, с получением указанного в заголовке соединения (11 мг; 5%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.91 (1H, dq), 2.21 (1H, dq), 2.40 (3Н, s), 2.67 (14Н, d), 2.80 (3Н, s), 2.95 (3Н, s), 3.16 (1H, dd), 3.35-3.67 (3Н, m), 3.73 (2Н, s), 6.59-6.68 (2Н, m), 7.87-7.99 (2Н, m), 8.56 (1H, d), 8.90 (1H, d); m/zMH+ 512.
Пример 22: (R)-2,5,7-триметил-6-((1-(4-(2-((4-метилпиперазин-1-ил)метил)пиримидин-5-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]-пиримидин
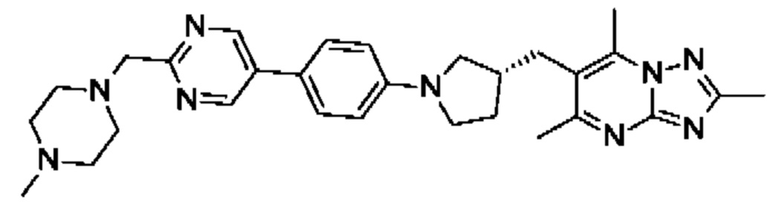
Pd(Ph3P)4 (63,9 мг; 0,06 ммоль) добавляли к (R)-2,5,7-триметил-6-((1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло-[1,5-а]пиримидину (промежуточное соединение 27) (247 мг; 0,55 ммоль), 5-бром-2-((4-метилпиперазин-1-ил)метил)пиримидину (промежуточное соединение 29) (150 мг; 0,55 ммоль) и Na2CO3 (117 мг; 1,11 ммоль) в 1,4-диоксане (2 мл) и воде (1 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (40 мг; 14%) в виде желтого твердого вещества; 1Н ЯМР (300 МГц, DMSO) 1.85 (1H, dq), 2.10 (1H, dq), 2.26 (3Н, s), 2.49 (4Н, m), 2.63 (8Н, m), 2.74 (3Н, s), 2.92 (2Н, d), 3.06 (1H, dd), 3.26 (6Н, m), 3.70 (2Н, s), 6.64 (2Н, d), 7.62 (2Н, d), 8.99 (2Н, s); m/z МН+ 512.
Пример 23: (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридин-3-ил)метил)морфолин
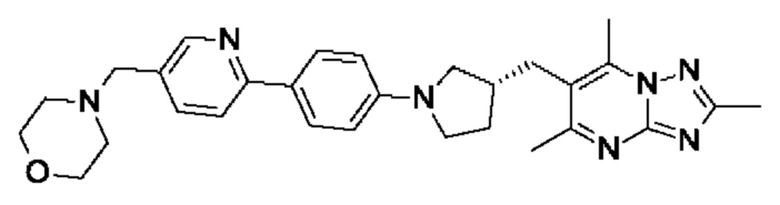
Предкатализатор RuPhos 3-го поколения (39 мг; 0,05 ммоль) добавляли к дигидрохлориду (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (промежуточное соединение 4) (150 мг; 0,47 ммоль), 4-((6-(4-бромфенил)пиридин-3-ил)метил)морфолину (промежуточное соединение 31) (157 мг; 0,47 ммоль), Cs2CO3 (768 мг; 2,36 ммоль) и RuPhos (44 мг; 0,09 ммоль) в 1,4-диоксане (3 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (54 мг; 23%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.87-2.05 (1H, m), 2.22 (1H, dq), 2.50 (4Н, dd), 2.55 (3Н, s), 2.71 (4Н, s), 2.82 (3Н, s), 2.94-3.09 (2Н, m), 3.16 (1H, dd), 3.32-3.51 (2Н, m), 3.54-3.63 (3Н, m), 3.63-3.76 (4Н, m), 6.61-6.73 (2Н, m), 7.67-7.87 (4Н, m), 8.44 (1H, d); m/z MH+ 498.
Пример 24: (R)-2,5,7-триметил-6-((1-(4-(6-((4-метилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]-пиримидин
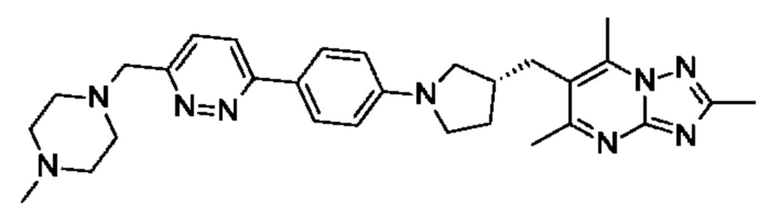
Pd(Ph3P)4 (26 мг; 0,02 ммоль) добавляли к (R)-2,5,7-триметил-6-((1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло-[1,5-а]пиримидину (промежуточное соединение 27) (100 мг; 0,22 ммоль), 3-хлор-6-((4-метилпиперазин-1-ил)метил)пиридазину (промежуточное соединение 32) (51 мг; 0,22 ммоль) и Na2CO3 (47 мг; 0,45 ммоль) в 1,4-диоксане (5 мл) и воде (1 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (35 мг; 31%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, DMSO) 1.87 (1H, m), 2.15 (4Н, m), 2.20-2.50 (11Н, m), 2.64 (4Н, m), 2.75 (3Н, s), 2.93 (2Н, d), 3.09 (1H, dd), 3.30-3.60 (3Н, m), 3.74 (2Н, s), 6.65 (2Н, m), 7.59 (1H, d), 8.00 (3Н, m); m/z MH+ 512.
Пример 25: (R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиримидин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]-пиримидин
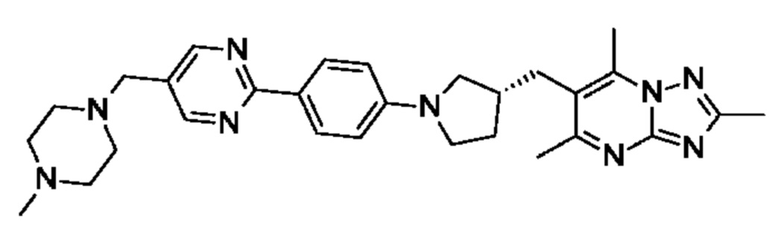
Предкатализатор RuPhos 3-го поколения (36 мг; 0,04 ммоль) и RuPhos (20 мг; 0,04 ммоль) добавляли к 2-(4-бромфенил)-5-((4-метилпиперазин-1-ил)метил)пиримидину (промежуточное соединение 35) (150 мг; 0,43 ммоль), дигидрохлориду (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (промежуточное соединение 4) (137 мг; 0,43 ммоль) и Cs2CO3 (563 мг; 1,73 ммоль) в 1,4-диоксане (5 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% MeCN в воде, с получением указанного в заголовке соединения (58 мг; 26%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.81-1.99 (2Н, m), 2.19 (1H, dt), 2.35 (3Н, s), 2.67 (14Н, m), 2.76-3.04 (5Н, m), 3.18 (1H, dd), 3.36-3.68 (5Н, m), 6.55-6.66 (2Н, m), 8.26-8.38 (2Н, m), 8.64 (2Н, s); m/z МН+ 512.
Пример 26: (R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиразин-2-ил)метил)морфолин
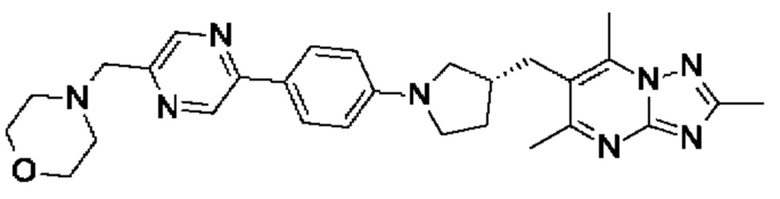
Pd(Ph3P)4 (34 мг; 0,03 ммоль) добавляли к (R)-2,5,7-триметил-6-((1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло-[1,5-а] пиримидину (промежуточное соединение 27) (130 мг; 0,29 ммоль), 4-((5-хлорпиразин-2-ил)метил)морфолину (промежуточное соединение 76) (62 мг; 0,29 ммоль) и Na2CO3 (62 мг; 0,58 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл). Реакционную смесь перемешивали при 90°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% MeCN в воде (плюс 0,1% FA), с получением указанного в заголовке соединения (70 мг; 48%) в виде бледно-желтого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.91 (1H, dq), 2.21 (1H, dq), 2.67 (11Н, m), 2.80 (3Н, s), 2.95 (2Н, m), 3.16 (1Н, dd), 3.35-3.54 (2Н, m), 3.60 (1Н, td), 3.71 (2Н, s), 3.77 (4Н, t), 6.58-6.69 (2Н, m), 7.88-8.00 (2Н, m), 8.60 (1H, s), 8.91 (1H, d); m/z МН+ 499.
Пример 27: (R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиридин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
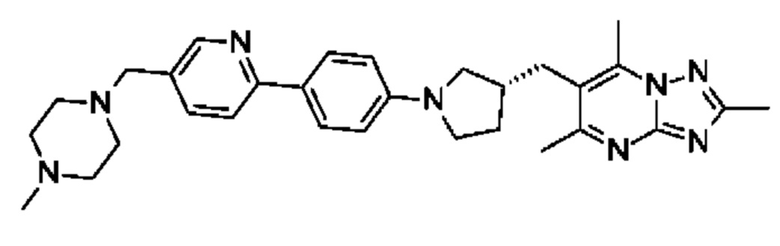
Формальдегид (37% в воде) (5 мл; 67,2 ммоль) добавляли одной порцией к (R)-трет-бутил-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)фенил)пиридин-3-ил)метил)пиперазин-1-карбоксилату (промежуточное соединение 82) (290 мг; 0,49 ммоль) в муравьиной кислоте (10 мл) при комнатной температуре. Реакционную смесь перемешивали при 55°С в течение 4 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Остаток повторно растворяли в 2 М NH3/MeOH и очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (58 мг; 26%) в виде белого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.83-1.93 (1H, m), 2.12-2.22 (1H, m), 2.29 (3Н, s), 2.53 (8Н, d), 2.62 (4Н, s), 2.70 (3Н, s), 2.79 (3Н, s), 2.85-2.99 (2Н, m), 3.14 (1H, dd), 3.37-3.47 (2Н, m), 3.52 (2Н, s), 3.57 (1H, td), 6.55-6.65 (2Н, m), 7.59 (1H, dd), 7.65 (1H, dd), 7.87-7.95 (2Н, m), 8.51 (1H, d); m/z МН+ 511.
Пример 28: (R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиримидин-2-ил)метил)морфолин
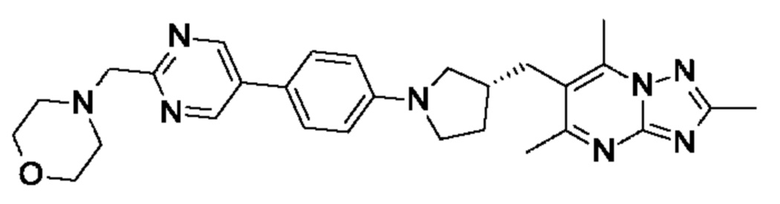
Предкатализатор RuPhos 3-го поколения (53 мг; 0,06 ммоль) добавляли к дигидрохлориду (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидина (промежуточное соединение 4) (200 мг; 0,63 ммоль), 4-((5-(4-бромфенил)пиримидин-2-ил)метил)морфолину (промежуточное соединение 39) (231 мг; 0,69 ммоль), Cs2CO3 (819 мг; 2,51 ммоль) и RuPhos (59 мг; 0,13 ммоль) в 1,4-диоксане (8 мл). Реакционную смесь перемешивали при 90°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 0-100% MeCN в воде (плюс 0,1% FA), с получением указанного в заголовке соединения (180 мг; 57%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.88-2.06 (1H, m), 2.22 (1H, dq), 2.55 (3Н, s), 2.60-2.69 (4Н, m), 2.71 (4Н, m), 2.82 (3Н, s), 3.02 (2Н, dd), 3.15 (1H, dd), 3.31-3.50 (2Н, m), 3.52-3.66 (1Н, m), 3.69-3.79 (4Н, m), 3.83 (2Н, s), 6.67-6.77 (2Н, m), 7.52-7.63 (2Н, m), 8.97 (2Н, s); m/z МН+ 499.
Пример 29: (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин
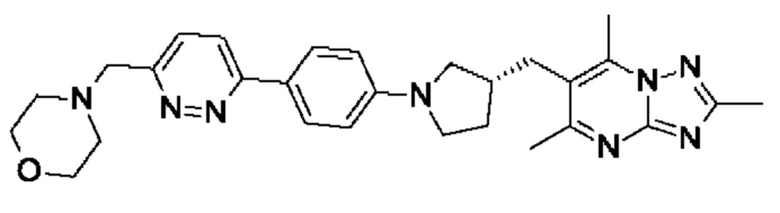
Дегазированный 1,4-диоксан (10 мл) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илметил)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 4) (257 мг; 1,05 ммоль) и 4-((6-(4-бромфенил)пиридазин-3-ил)метил)морфолину (промежуточное соединение 65) (350 мг; 1,05 ммоль). Затем добавляли Cs2CO3 (1,02 г; 3,14 ммоль) с последующим добавлением RuPhos (24 мг; 0,05 ммоль) и предкатализатора RuPhos 3-го поколения (44 мг; 0,05 ммоль). Реакционную смесь нагревали при 90°С в течение 6 часов, оставляли для охлаждения до комнатной температуры, разбавляли DCM (50 мл) и фильтровали. Фильтрат концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-5% 1 М NH3/MeOH в DCM. Полученное масло растирали со смесью EtOAc:гептан (4:1, 10 мл), и осадок отделяли посредством фильтрования и сушили под вакуумом с получением указанного в заголовке соединения (270 мг; 52%) в виде желтого твердого вещества; 1Н ЯМР (500 МГц, CDCl3) 1.90 (1H, dq), 2.20 (1H, td), 2.56 (4Н, q), 2.62 (4Н, s), 2.70 (3Н, s), 2.79 (3Н, s), 2.93 (2Н, qd), 3.15 (1H, dd), 3.37-3.49 (2Н, m), 3.55-3.63 (1H, m), 3.69-3.77 (4Н, m), 3.87 (2Н, s), 6.59-6.68 (2Н, m), 7.59 (1H, d), 7.75 (1H, d), 7.98-8.06 (2Н, m); m/z МН+ 499.
Пример 30: (S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин
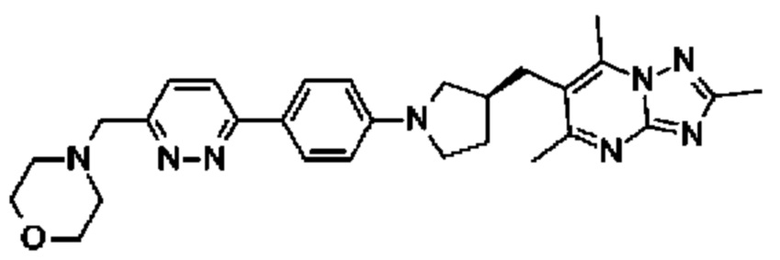
Соединение по примеру 30 получали посредством способа, аналогичного способу получения по примеру 29, за 5 стадий из имеющегося в продаже (S)-трет-бутил-3-(гидроксиметил)пирролидин-1-карбоксилата с получением указанного в заголовке соединения (122 мг; 30% на конечной стадии) в виде белого твердого вещества; 1Н ЯМР (500 МГц, DMSO) 1.86 (1Н, dq), 2.07-2.16 (1H, m), 2.38-2.45 (4Н, m), 2.47 (3Н, s), 2.59 (1H, d), 2.64 (3Н, s), 2.74 (3Н, s), 2.93 (2Н, d), 3.09 (1H, dd), 3.32 (1H, d), 3.43 (1H, dd), 3.46-3.53 (1H, m), 3.54-3.63 (4Н, m), 3.75 (2Н, s), 6.65 (2Н, d), 7.62 (1H, d), 7.98 (2Н, d), 8.02 (1H, d); m/z МН+ 499.
Пример 31: (R)-6-((1-(5-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)-6-метилпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а] пиримидин
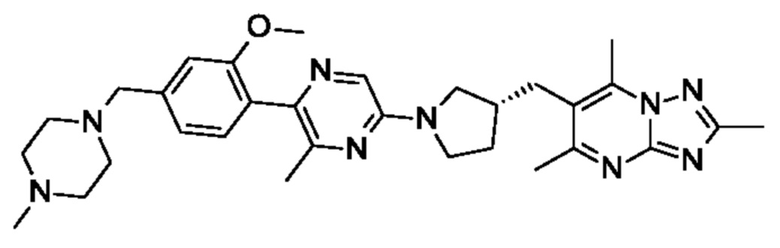
Предкатализатор XPhos 2-го поколения (30 мг; 0,04 ммоль) добавляли к (2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)бороновой кислоте (промежуточное соединение 42) (112 мг; 0,42 ммоль), (R)-6-((1-(5-бром-6-метилпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 45) (160 мг; 0,38 ммоль) и Cs2CO3 (250 мг; 0,77 ммоль) в 1,4-диоксане (2 мл) и воде (1 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (90 мг; 42%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.78-2.24 (3Н, m), 2.27 (3Н, s), 2.31-2.77 (17Н, m), 2.82 (3Н, s), 2.87-3.06 (2Н, m), 3.34 (1H, dd), 3.45-3.61 (3Н, m), 3.66-3.83 (5Н, m), 6.93-7.04 (2Н, m), 7.22 (1H, d), 7.77 (1H, s); m/z МН+ 556.
Пример 32: 2,5,7-триметил-6-(((R)-1-(5-(4-(((3R,5S)-3,4,5-триметилпиперазин-1-ил)метил)фенил)пир азин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]-пиримидин
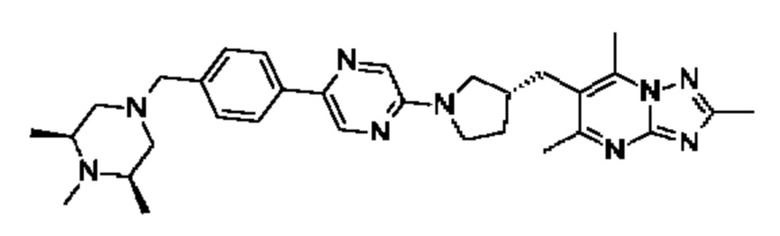
(2S,6R)-1,2,6-Триметилпиперазин (из коммерческих источников) (104 мг; 0,81 ммоль) добавляли одной порцией к (R)-6-((1-(5-(4-(хлорметил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 48) (181 мг; 0,40 ммоль) и триэтиламину (0,28 мл; 2,02 ммоль) в THF (5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при 60°С в течение 24 часов, оставляли для охлаждения до комнатной температуры, и разбавляли EtOAc (50 мл) и водой (15 мл). Органический слой отделяли и промывали насыщ. рассолом (15 мл), сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (115 мг; 53%) в виде бледно-желтого твердого вещества; 1Н ЯМР (500 МГц, DMSO) 0.92 (6Н, d), 1.75 (2Н, m), 1.8-1.91 (1H, m), 2.06-2.16 (5Н, m), 2.46 (3Н, s), 2.5-2.52 (1H, m), 2.63 (6Н, d), 2.74 (3Н, s), 2.93 (2Н, d), 3.2-3.26 (1H, m), 3.37-3.47 (3Н, m), 3.63 (1H, dd), 3.66-3.73 (1H, m), 7.32 (2Н, d), 7.87 (2Н, d), 8.02 (1H, d), 8.61 (1Н, d); m/z MH+ 540.
Пример 33: 6-((R)-1-(5-(4-((R)-3,4-диметилпиперазин-1-ил)метил)фенил)-пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]-пиримидин
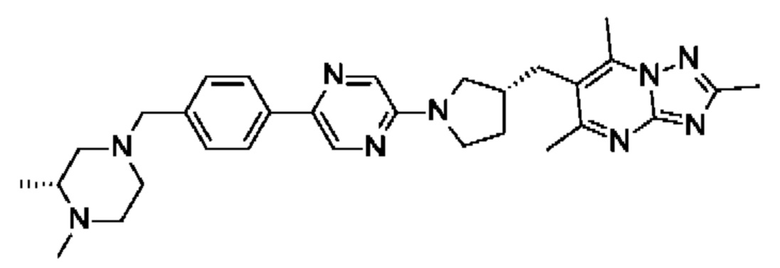
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (200 мг; 0,47 ммоль) и (R)-1,2-диметилпиперазин (из коммерческих источников) (267 мг; 2,34 ммоль) в МеОН (10 мл) перемешивали при комнатной температуре в течение 1 часа, затем добавляли триацетоксиборгидрид натрия (397 мг; 1,87 ммоль) и АсОН (2,81 мг; 0,05 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (126 мг; 51%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.06 (3Н, d), 1.93 (2Н, dq), 2.22 (3Н, dt), 2.33 (4Н, s), 2.56-3.06 (15Н, m), 3.34 (1Н, dd), 3.56 (3Н, d), 3.67-3.84 (2H, m), 7.40 (2H, d), 7.78-7.88 (2H, m), 7.95 (1H, d), 8.51 (1H, d); m/z MH+ 526.
Пример 34: 6-((R)-1-(5-(4-((R)-2,4-диметилпиперазин-1-ил)метил)фенил)-пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]-пиримидин
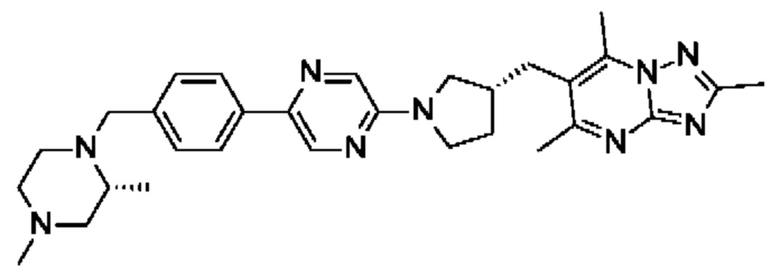
Дигидрохлорид (R)-1,3-диметилпиперазина (из коммерческих источников) (167 мг; 0,89 ммоль) добавляли одной порцией к (R)-6-((1-(5-(4-(хлорметил)фенил)-пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 48) (200 мг; 0,45 ммоль) и триэтиламину (0,31 мл; 2,23 ммоль) в 1,4-диоксане (5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при 80°С в течение 24 часов, оставляли для охлаждения до комнатной температуры и разбавляли EtOAc (50 мл) и водой (15 мл). Органический слой отделяли и промывали насыщ. рассолом (15 мл), сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/МеОН в DCM, затем дополнительно очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (32 мг; 14%) в виде желтой пены; 1Н ЯМР (500 МГц, DMSO) 1.06 (3Н, d), 1.79-1.91 (2Н, m), 1.99 (1H, s), 2.10 (5Н, s), 2.41 (1H, s), 2.46 (4Н, s), 2.52-2.62 (3Н, m), 2.63 (3Н, s), 2.74 (3Н, s), 2.93 (2Н, d), 3.12 (1Н, d), 3.23 (1Н, dd), 3.4-3.49 (1Н, m), 3.63 (1H, dd), 3.65-3.74 (1H, m), 3.95 (1H, d), 7.32 (2Н, d), 7.82-7.90 (2Н, m), 8.02 (1H, d), 8.60 (1H, d); m/z МН+ 526.
Пример 35: 2,5,7-триметил-6-(((R)-l-(5-(4-(((2R,5R)-2,4,5-триметилпиперазин-l-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
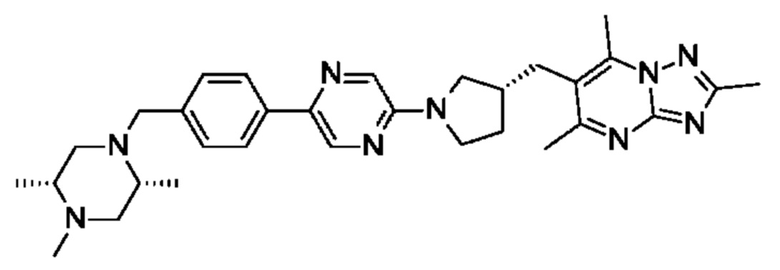
Гидрохлорид (2R,5R)-1,2,5-триметилпиперазина (из коммерческих источников) (147 мг; 0,89 ммоль) добавляли одной порцией к (R)-6-((1-(5-(4-(хлорметил)фенил)-пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 48) (200 мг; 0,45 ммоль) и триэтиламину (0,31 мл; 2,23 ммоль) в 1,4-диоксане (5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при 80°С в течение 24 часов, оставляли для охлаждения до комнатной температуры и разбавляли EtOAc (50 мл) и водой (15 мл). Органический слой отделяли и промывали насыщ. рассолом (15 мл), сушили над MgSO4, фильтровали и сушили под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/МеОН в DCM, затем дополнительно очищали посредством растирания с МеОН с получением указанного в заголовке соединения (78 мг; 32%) в виде бледно- желтого твердого вещества; 1Н ЯМР (500 МГц, DMSO) 0.90 (3Н, d), 1.03 (3Н, d), 1.8-1.91 (1H, m), 2.10 (5Н, s), 2.22-2.32 (3Н, m), 2.46 (4Н, s), 2.57-2.61 (1H, m), 2.63 (3Н, s), 2.74 (4Н, s), 2.93 (2Н, d), 3.23 (1H, dd), 3.41-3.48 (2Н, m), 3.55-3.66 (2Н, m), 3.66-3.73 (1H, m), 7.35 (2Н, d), 7.86 (2Н, d), 8.02 (1H, d), 8.60 (1H, d); m/z МН+ 540.
Пример 36: 2,5,7-триметил-6-(((R)-1-(5-(4-(((2S,5R)-2,4,5-триметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
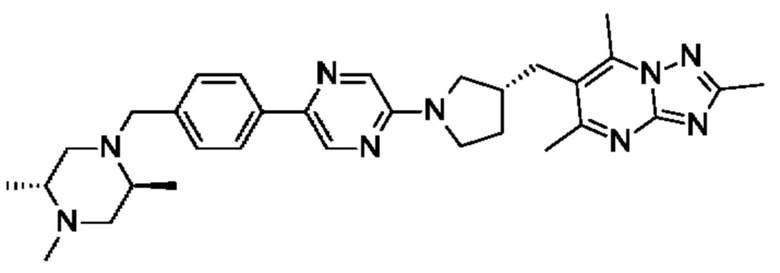
Дигидрохлорид (2R,5S)-1,2,5-триметилпиперазина (из коммерческих источников) (180 мг; 0,89 ммоль) добавляли одной порцией к (R)-6-((l-(5-(4-(хлорметил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]-триазоло[1,5-а]пиримидину (промежуточное соединение 48) (200 мг; 0,45 ммоль) и триэтиламину (0,31 мл; 2,23 ммоль) в 1,4-диоксане (5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при 80°С в течение 24 часов, оставляли для охлаждения до комнатной температуры и разбавляли EtOAc (50 мл) и водой (15 мл). Органический слой отделяли и промывали насыщ. рассолом (15 мл), сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/МеОН в DCM, затем дополнительно очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (81 мг; 34%) в виде бледно-желтого твердого вещества; 1Н ЯМР (500 МГц, DMSO) 0.83 (3Н, d), 1.07 (3Н, d), 1.73 (1H, t), 1.80-1.93 (3Н, m), 2.09 (4Н, s), 2.35 (1H, s), 2.46 (3Н, s), 2.63 (5Н, s), 2.74 (3Н, s), 2.93 (2Н, d), 3.01 (1H, d), 3.20-3.26 (1H, m), 3.43 (1Н, d), 3.59-3.74 (2Н, m), 4.02 (1H, d), 7.32 (2Н, d), 7.86 (2Н, d), 8.02 (1H, s), 8.61 (1H, s), полагали, что недостающий 1H находится под пиком DMSO; m/z МН+ 540.
Пример 37: 2,5,7-триметил-6-[[(3R)-1-[5-[4-(1-пиперидилметил)фенил]-пиразин-2-ил]пирролидин-3-ил]метил]-[1,2,4]триазоло[1,5-а]пиримидин
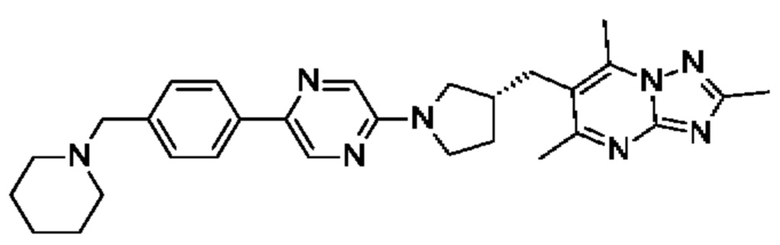
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (30 мг; 0,07 ммоль), пиперидин (12 мг; 0,14 ммоль), DCM (2 мл) и АсОН (2 капли) объединяли, и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Добавляли триацетоксиборгидрид натрия (45 мг; 0,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (15 мг; 44%); 1Н ЯМР (400 МГц, DMSO) 1.47 (6Н, d), 1.82-1.92 (1H, m), 2.12 (1H, dd), 2.36 (4Н, s), 2.48 (3Н, s), 2.65 (4Н, s), 2.76 (3Н, s), 2.95 (2Н, d), 3.25 (2Н, dd), 3.41-3.51 (2Н, m), 3.62-3.75 (2Н, m), 7.37 (2Н, s), 7.90 (2Н, s), 8.04 (1H, d), 8.64 (1H, s); m/z МН+ 497.
Пример 38: (R)-6-((1-(5-(4-((4-этилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
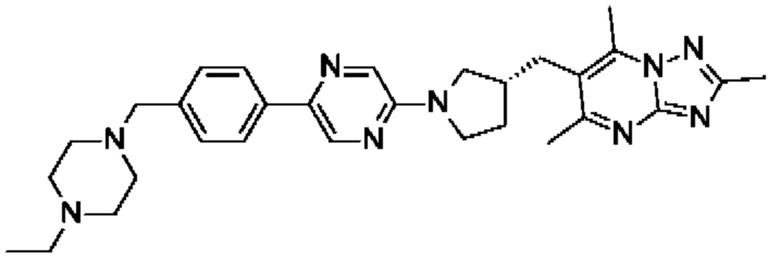
1-Этилпиперазин (136 мг; 1,19 ммоль) добавляли к (R)-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)-бензальдегиду (промежуточное соединение 49) (170 мг; 0,40 ммоль) в МеОН (5 мл) при комнатной температуре и реакционную смесь перемешивали в течение 1 часа. Добавляли триацетоксиборгидрид натрия (421 мг; 1,99 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 60 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением формиата указанного в заголовке соединения (114 мг; 51%) в виде бледно-желтого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.25 (3Н, t), 1.93 (1Н, dq), 2.15-2.32 (1Н, m), 2.56-3.06 (22Н, m), 3.34 (1Н, dd), 3.48-3.65 (3Н, m), 3.76 (2Н, ddt), 7.34-7.44 (2Н, m), 7.78-7.89 (2Н, m), 7.95 (1H, d), 8.47-8.55 (2Н, m); m/z МН+ 526.
Пример 39: (R)-2,5,7-триметил-6-((1-(5-(5-((4-метилпиперазин-1-ил)метил)пиридин-2-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло-[1,5-а] пиримидин
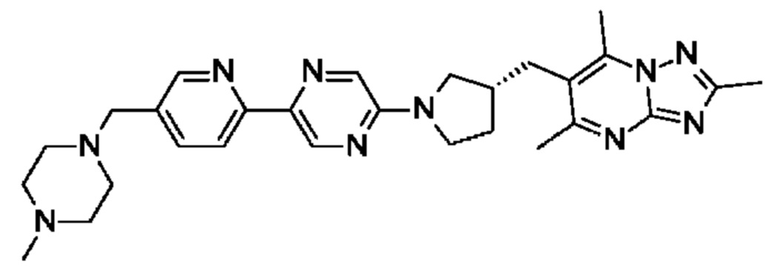
Хлорид бис(трифенилфосфоранил)палладия(IV) (35 мг; 0,05 ммоль) добавляли к 1-((6-бромпиридин-3-ил)метил)-4-метилпиперазину (промежуточное соединение 7) (135 мг; 0,50 ммоль), 1,1,1,2,2,2-гексаметилдистаннану (180 мг; 0,55 ммоль) в THF (3 мл) при комнатной температуре. Реакционную смесь перемешивали при 85°С в течение 16 часов. Добавляли (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (промежуточное соединение 46) (200 мг; 0,50 ммоль), и Pd(Ph3P)4 (57,4 мг; 0,05 ммоль), и THF (5 мл). Реакционную смесь переносили в пробирку для микроволновой печи и нагревали при 100°С в течение 10 часов в микроволновом реакторе и затем оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением указанного в заголовке соединения (59 мг; 23%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.95 (1Н, dq), 2.25 (1Н, tt), 2.47 (3Н, s), 2.60-2.75 (15Н, m), 2.82 (3Н, s), 2.97 (2Н, qd), 3.38 (1Н, dd), 3.54-3.65 (3Н, m), 3.80 (2H, td), 7.73 (1H, dd), 7.92 (1H, d), 8.11 (1H, d), 8.53-8.59 (1H, m), 9.09 (1H, d); m/z MH+ 513.
Пример 40: 6-(((R)-1-(5-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]-триазоло[1,5-а]пиримидин
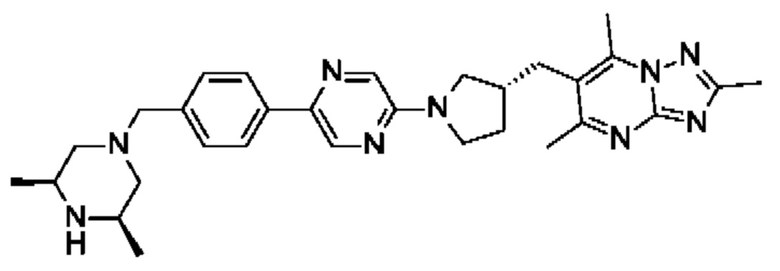
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (200 мг; 0,47 ммоль) добавляли к (2R,6S)-1,2,6-триметилпиперазину (из коммерческих источников) (120 мг; 0,94 ммоль) в DCM (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли АсОН (2,7 мкл; 0,05 ммоль) и цианоборгидрид натрия (118 мг; 1,87 ммоль) и реакционную смесь перемешивали в течение 16 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (75 мг; 31%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.06 (6Н, d), 1.73 (2Н, t), 1.93 (1Н, dq), 2.23 (1Н, dq), 2.82 (17Н, m), 3.34 (1H, dd), 3.55 (3Н, d), 3.75 (2Н, m), 7.40 (2Н, m), 7.83 (2Н, m), 7.95 (1H, d), 8.51 (1Н, d); m/z MH+ 526.
Пример 41: 2-{4-[4-(5-{(3R)-3-[(2,5,7-триметил[l,2,4]триазоло[1,5-a]пиримидин-6-ил)метил]пирролидин-1-ил}пиразин-2-ил)бензил]пиперазин-1-ил}этанол
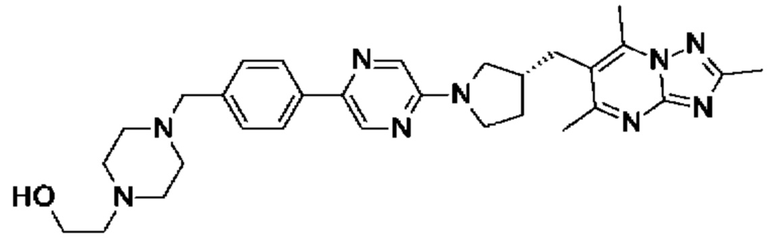
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (30 мг; 0,07 ммоль), 2-пиперазин-1-илэтанол (18,23 мг; 0,14 ммоль), DCM (2 мл) и АсОН (2 капли) объединяли и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Затем добавляли триацетоксиборгидрид натрия (45 мг; 0,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (8 мг; 20%); 1Н ЯМР (400 МГц, DMSO) 1.81-1.93 (1H, m), 2.12 (1Н, d), 2.31-2.44 (10Н, m), 2.48 (3Н, s), 2.65 (4Н, s), 2.76 (3Н, s), 2.95 (2Н, d), 3.22-3.27 (2Н, m), 3.47 (4Н, s), 3.62-3.76 (2Н, m), 4.32 (1H, s), 7.34 (2H, d), 7.88 (2H, d), 8.04 (1H, d), 8.62 (1H, d); m/z MH+ 542.
Пример 42: (R)-6-((1-(5-(4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло-[1,5-а]пиримидин
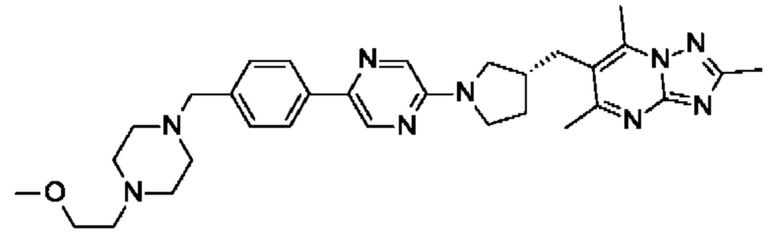
1-(2-Метоксиэтил)пиперазин (202 мг; 1,40 ммоль) добавляли к (R)-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)-бензальдегиду (промежуточное соединение 49) (200 мг; 0,47 ммоль) в DCM (5 мл) при комнатной температуре в воздушной атмосфере и реакционную смесь перемешивали в течение 1 часа. Добавляли триацетоксиборгидрид натрия (496 мг; 2,34 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением формиата указанного в заголовке соединения (194 мг; 70%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.89-2.01 (1H, m), 2.18-2.31 (1H, m), 2.59-3.05 (22Н, m), 3.35 (4Н, s), 3.52-3.67 (3Н, m), 3.67-3.83 (4Н, m), 7.38-7.46 (2Н, m), 7.82-7.90 (2Н, m), 7.97 (1H, s), 8.45 (1H, s), 8.54 (1H, d); m/z МН+ 556.
Пример 43: (R)-6-((1-(5-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
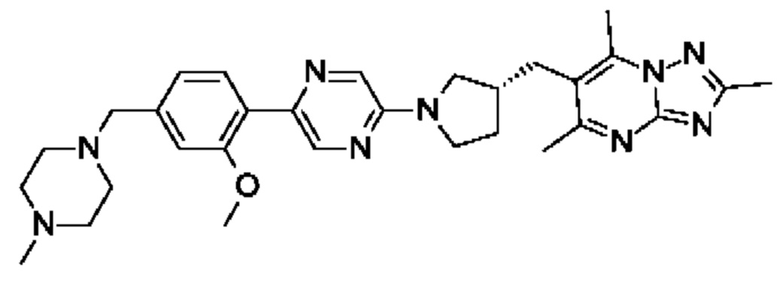
Предкатализатор XPhos 2-го поколения (14,7 мг; 0,02 ммоль) добавляли к (R)-6-((l-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 46) (150 мг; 0,37 ммоль), (2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)бороновой кислоте (промежуточное соединение 42) (108 мг; 0,41 ммоль) и Cs2CO3 (243 мг; 0,75 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 4 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением формиата указанного в заголовке соединения (168 мг; 79%) в виде бледно-желтого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.93 (1H, dq), 2.23 (1Н, dq), 2.54-3.05 (24Н, m), 3.34 (1H, dd), 3.51-3.65 (3Н, m), 3.76 (2Н, ddd), 3.89 (3Н, s), 6.96-7.06 (2Н, m), 7.73 (1H, d), 7.98 (1H, d), 8.69 (1H, d); m/z МН+ 542.
Пример 44: {1-[4-(5-{(3R)-3-[(2,5,7-триметил[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил]пирролидин-1-ил}пиразин-2-ил)бензил]пиперидин-4-ил}метанол
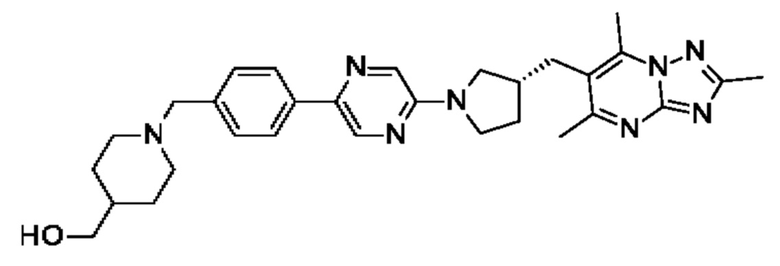
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (30 мг; 0,07 ммоль), 4-пиперидилметанол (16,12 мг; 0,14 ммоль), DCM (2 мл) и АсОН (2 капли) объединяли и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Затем добавляли триацетоксиборгидрид натрия (45 мг; 0,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (17,4 мг; 47%); 1Н ЯМР (400 МГц, DMSO) 1.07-1.2 (2Н, m), 1.34 (1H, s), 1.62 (2Н, d), 1.85-1.95 (3Н, m), 2.12 (1H, dd), 2.48 (3Н, s), 2.65 (4Н, s), 2.76 (3Н, s), 2.82 (2Н, d), 2.95 (2Н, d), 3.21-3.28 (4Н, m), 3.45 (2Н, s), 3.62-3.75 (2Н, m), 4.37 (1H, s), 7.34 (2Н, d), 7.88 (2Н, d), 8.04 (1H, d), 8.62 (1H, d); m/z МН+ 527.
Пример 45: 6-{[(3R)-1-(5-{4-[(l,l-диоксидотиоморфолин-4-ил)метил]фенил}-пиразин-2-ил)пирролидин-3-ил]метил}-2,5,7-триметил[1,2,4]триазоло[1,5-а] пиримидин
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (30 мг; 0,07 ммоль), гидрохлорид 1,4-тиазинан-1,1-диоксида (24,03 мг; 0,14 ммоль), DCM (2 мл) и АсОН (2 капли) объединяли и смесь перемешивали при комнатной температуре в течение 5 часов. Затем добавляли триацетоксиборгидрид натрия (45 мг; 0,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (12 мг; 30%); 1Н ЯМР (400 МГц, DMSO) 1.87 (1H, dd), 2.12 (1H, dd), 2.48 (3Н, s), 2.65 (4Н, s), 2.76 (3Н, s), 2.86-2.93 (4Н, m), 2.95 (2Н, d), 3.09-3.14 (4Н, m), 3.25 (1H, dd), 3.45 (1H, dt), 3.62-3.76 (4Н, m), 7.39 (2Н, d), 7.91 (2Н, d), 8.04 (1H, d), 8.64 (1H, d); m/z МН+ 547.
Пример 46: 2,5,7-триметил-6-({(3R)-1-[5-(4-{[4-(метилсульфонил)пиперидин-1-ил]метил}фенил)пиразин-2-ил]пирролидин-3-ил}метил)[1,2,4]триазоло[1,5-а]пиримидин
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (30 мг; 0,07 ммоль), 4-метилсульфонилпиперидин (22,9 мг; 0,14 ммоль), DCM (2 мл) и АсОН (2 капли) объединяли и смесь перемешивали при комнатной температуре в течение 5 часов. Затем добавляли триацетоксиборгидрид натрия (45 мг; 0,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь очищали посредством препаративной ВЭЖХ и лиофилизировали с получением указанного в заголовке соединения (15 мг; 38%); 1Н ЯМР (500 МГц, DMSO) 1.56 (2Н, tt), 1.76-1.85 (1Н, m), 1.92 (4H, t), 2.05 (1H, dd), 2.41 (3H, s), 2.58 (4H, s), 2.69 (3H, s), 2.84 (3H, s), 2.88 (4H, d), 2.98 (1H, ddt), 3.18 (1H, dd), 3.38 (1H, dt), 3.44 (2H, s), 3.55-3.68 (2H, m), 7.28 (2H, d), 7.82 (2H, d), 7.97 (1H, d), 8.55 (1H, d); m/z MH+ 575.
Пример 47: (R)-4-((6-(3 -метил-5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)пиридин-3-ил)метил)-морфолин
Хлорид бис(трифенилфосфоранил)палладия(IV) (164 мг; 0,23 ммоль) добавляли к 4-((6-бромпиридин-3-ил)метил)морфолину (промежуточное соединение 30) (600 мг; 2,33 ммоль), 1,1,1,2,2,2-гексаметилдистаннану (841 мг; 2,57 ммоль) в THF (8 мл) при комнатной температуре. Реакционную смесь перемешивали при 85°С в течение 16 часов. Добавляли (R)-6-((1-(5-бром-6-метилпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (промежуточное соединение 45) (300 мг; 0,72 ммоль) и Pd(Ph3P)4 (83 мг; 0,07 ммоль) и смесь переносили в пробирку для микроволновой печи с THF (10 мл). Реакционную смесь нагревали при 100°С в течение 5 часов в микроволновом реакторе и оставляли для охлаждения до комнатной температуры, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), затем дополнительно очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (160 мг; 43%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.97 (1H, dq), 2.24 (1H, dq), 2.47-2.59 (10Н, m), 2.73 (4Н, s), 2.84 (3Н, s), 3.04 (2Н, dd), 3.32-3.42 (1H, m), 3.47-3.66 (3Н, m), 3.66-3.87 (6Н, m), 7.72 (1H, dd), 7.81 (1H, s), 7.91 (1H, dd), 8.53-8.60 (1H, m); m/z МН+ 514.
Пример 48: 6-(((R)-1-(5-(4-(((S)-3,4-диметилпиперазин-1-ил)метил)фенил)-пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а] пиримидин
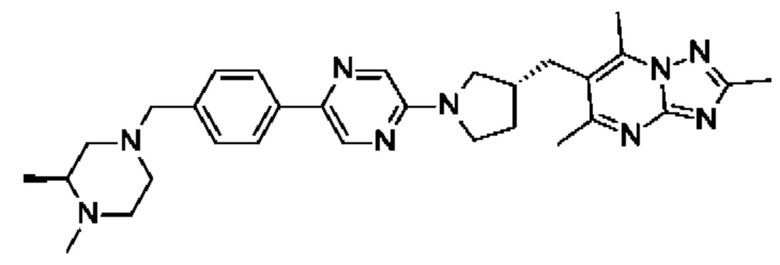
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (200 мг; 0,47 ммоль) добавляли к дигидрохлориду (S)-1,2-диметилпиперазина (350 мг; 1,87 ммоль) в МеОН (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли АсОН (2,81 мг; 0,05 ммоль) и триацетоксиборгидрид натрия (397 мг; 1,87 ммоль) и реакционную смесь перемешивали в течение 16 часов. Реакционную смесь гасили водой и полученный раствор очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (132 мг; 54%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.07 (3Н, d), 1.94 (2Н, dq), 2.18-2.35 (6Н, m), 2.39 (1H, s), 2.59-2.85 (13Н, m), 2.97 (2Н, qd), 3.35 (1H, dd), 3.56 (3Н, d), 3.76 (2Н, ddt), 7.38-7.45 (2Н, m), 7.80-7.88 (2Н, m), 7.96 (1Н, d), 8.52 (1H, d); m/z МН+ 526.
Пример 49: (R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)пиридин-2-ил)метил)морфолин
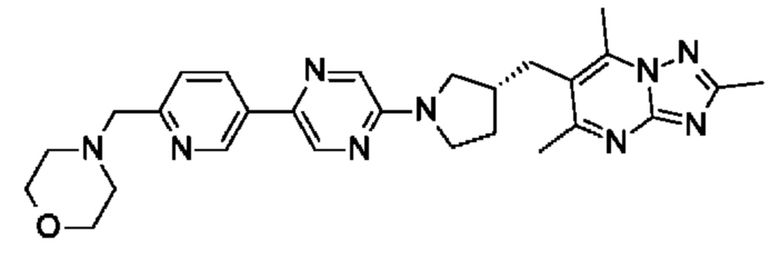
Предкатализатор XPhos 2-го поколения (19,6 мг; 0,02 ммоль) добавляли к (R)-6-((l-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 46) (200 мг; 0,50 ммоль), (6-(морфолинометил)пиридин-3-ил)бороновой кислоте (из коммерческих источников) (110 мг; 0,50 ммоль) и Cs2CO3 (324 мг; 0,99 ммоль) в 1,4-диоксане (4 мл) и воде (2 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (172 мг; 69%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 1.91-2.06 (1H, m), 2.23-2.29 (1H, m), 2.53-2.57 (7Н, m), 2.66-2.74 (4Н, m), 2.85 (3Н, d), 2.97-3.11 (2Н, m), 3.35-3.38 (1Н, m), 3.53-3.59 (1Н, m), 3.67-3.85 (8Н, m), 7.61 (1H, d), 8.05 (1H, s), 8.30 (1H, dd), 8.56 (1H, s), 9.01-9.02 (1H, m); m/z MH+ 500.
Пример 50: 6-(((R)-1-(5-(4-(((S)-2,4-диметилпиперазин-1-ил)метил)фенил)-пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а] пиримидин
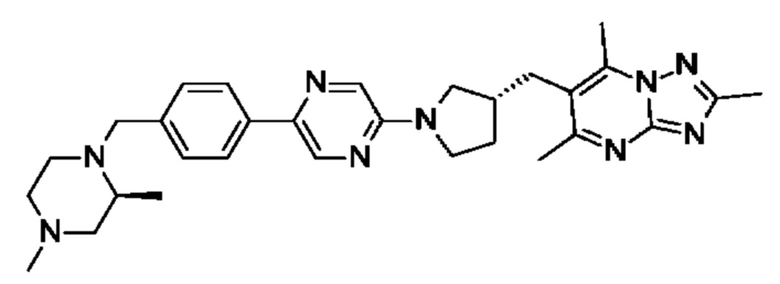
Дигидрохлорид (S)-1,3-диметилпиперазина (167 мг; 0,89 ммоль) добавляли одной порцией к (R)-6-((1-(5-(4-(хлорметил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 48) (200 мг; 0,45 ммоль) и триэтиламину (0,31 мл; 2,23 ммоль) в 1,4-диоксане (5 мл) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали при 80°С в течение 24 часов, оставляли для охлаждения до комнатной температуры и разбавляли EtOAc (50 мл) и водой (15 мл). Органический слой отделяли и промывали насыщ. рассолом (15 мл), сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (54 мг; 23%) в виде бледно-желтого твердого вещества; 1Н ЯМР (500 МГц, DMSO) 1.06 (3Н, d), 1.80-1.91 (2Н, m), 1.94-2.04 (1H, m), 2.05-2.14 (5Н, m), 2.37-2.43 (1H, m), 2.46 (4Н, s), 2.52-2.61 (3Н, m), 2.63 (3Н, s), 2.74 (3Н, s), 2.93 (2Н, d), 3.12 (1Н, d), 3.23 (1H, dd), 3.43 (1Н, dt), 3.63 (1H, dd), 3.66-3.73 (1Н, m), 3.95 (1Н, d), 7.33 (2Н, d), 7.82-7.91 (2Н, m), 8.02 (1H, d), 8.60 (1Н, d); m/z МН+ 526.
Пример 51: (R)-4-(4-(3-метил-5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)морфолин
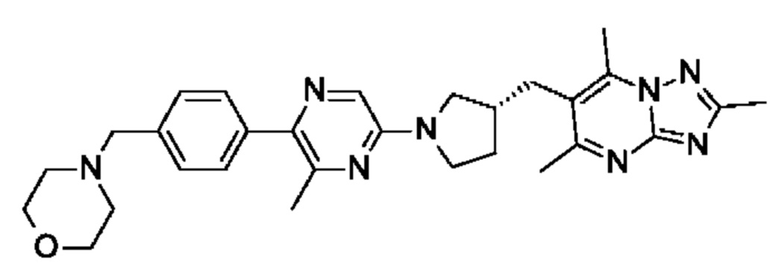
Предкатализатор XPhos 2-го поколения (11,3 мг; 0,01 ммоль) добавляли к 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)морфолину (из коммерческих источников) (87 мг; 0,29 ммоль), (R)-6-((1-(5-бром-6-метилпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 45) (120 мг; 0,29 ммоль) и Cs2CO3 (188 мг; 0,58 ммоль) в 1,4-диоксане (4 мл) и воде (1,5 мл). Реакционную смесь перемешивали при 90°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и фильтровали через целит.Фильтрат концентрировали под вакуумом, и полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (49 мг; 31%) в виде бледно-желтого твердого вещества; 1Н ЯМР (300 МГц, MeOD) 1.92-1.99 (1H, m), 2.17-2.27 (1H, m), 2.41 (3H, s), 2.55 (3H, s), 2.61-2.83 (11Н, m), 2.96-3.03 (2Н, m), 3.29-3.35 (1H, m), 3.47-3.55 (1H, m), 3.65-3.79 (6Н, m), 3.86 (2Н, s), 7.47-7.53 (4Н, m), 7.75 (1H, s); m/z MH+ 513.
Пример 52: 2,5,7-триметил-6-({(3R)-1-[5-(4-{[4-(метилсульфонил)пиперазин-1-ил]метил}фенил)пиразин-2-ил]пирролидин-3-ил}метил)[1,2,4]триазоло[1,5-а]пиримидин
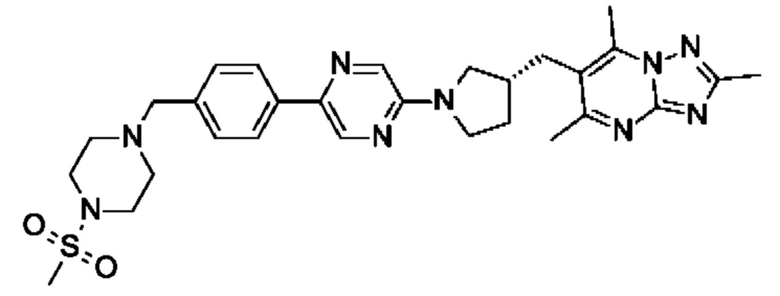
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (30 мг; 0,07 ммоль), 1-метилсульфонилпиперазин (22,99 мг; 0,14 ммоль), DCM (2 мл) и АсОН (2 капли) объединяли и смесь перемешивали при комнатной температуре в течение 5 часов. Затем добавляли триацетоксиборгидрид натрия (45 мг; 0,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (11,9 мг; 30%); 1Н ЯМР (400 МГц, DMSO) 1.87 (1H, dd), 2.12 (1H, dd), 2.48 (7Н, s), 2.65 (4Н, s), 2.76 (3H, s), 2.87 (3H, s), 2.95 (2Н, d), 3.09-3.16 (4Н, m), 3.25 (1H, dd), 3.41-3.50 (1H, m), 3.55 (2Н, s), 3.62-3.75 (2Н, m), 7.37 (2Н, d), 7.90 (2Н, d), 8.04 (1Н, d), 8.63 (1H, d); m/z MH+ 576.
Пример 53: (R)-N,N-диметл-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло-[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)пиперазин-1-ил)ацетамид
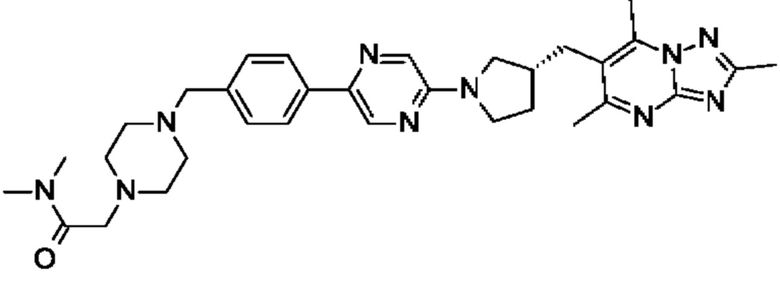
N,N-Диметил-2-(пиперазин-1-ил)ацетамид (промежуточное соединение 21) (240 мг; 1,40 ммоль) добавляли к (R)-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензальдегиду (промежуточное соединение 49) (200 мг; 0,47 ммоль) в DCM (5 мл) при комнатной температуре и реакционную смесь перемешивали в течение 1 часа. Добавляли триацетоксиборгидрид натрия (496 мг; 2,34 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением неполного (0,6 эквивалента) формиата указанного в заголовке соединения (172 мг; 60%) в виде желтого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.87-2.01 (1H, m), 2.18-2.30 (1Н, m), 2.64 (3H, s), 2.60-2.71 (1H, m), 2.72-2.78 (11Н, m), 2.83 (3H, s), 2.96 (3H, s), 2.88-3.05 (2Н, m), 3.05 (3H, s), 3.24 (2Н, s), 3.35 (1H, dd), 3.52-3.63 (1H, m), 3.72-3.81 (1H, m), 3.78 (3H, s), 7.45 (2Н, d), 7.87 (2Н, d), 7.97 (1Н, d), 8.42 (0.6Н, s, соответствует 0,6 эквив. формиата), 8.52 (1H, d); m/z МН+ 583.
Пример 54: (R)-2,5,7-триметил-6-((1-(6-метил-5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
Пред катализатор XPhos 2-го поколения (28 мг; 0,04 ммоль) добавляли к 1-метил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазину (125 мг; 0,40 ммоль), (R)-6-((1-(5-бром-6-метилпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 45) (150 мг; 0,36 ммоль) и Cs2CO3 (235 мг; 0,72 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (110 мг; 58%) в виде белого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) 1.81-2.00 (1H, m), 2.20 (1H, dq), 2.31-2.76 (21Н, m), 2.82 (3H, s), 2.94 (2Н, qd), 3.34 (1H, dd), 3.46-3.61 (3H, m), 3.74 (2Н, ddd), 7.38 (2Н, d), 7.43-7.53 (2Н, m), 7.79 (1H, s); m/z МН+ 526.
Пример 55: N,N-диметил-4-[4-(5-{(3R)-3-[(2,5,7-триметил[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил}пиразин-2-ил)бензил]пиперазин-1-карбоксамид
(R)-4-(5-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)-пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (30 мг; 0,07 ммоль), N,N-диметилпиперазин-1-карбоксамид (23 мг; 0,14 ммоль), DCM (2 мл) и АсОН (2 капли) объединяли и смесь перемешивали при комнатной температуре в течение 5 часов. Затем добавляли триацетоксиборгидрид натрия (45 мг; 0,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (13 мг; 33%); 1Н ЯМР (400 МГц, DMSO) 1.82-1.92 (1H, m), 2.07-2.17 (1Н, m), 2.35-2.41 (4H, m), 2.48 (3H, s), 2.65 (4H, s), 2.72 (6H, s), 2.76 (3H, s), 2.95 (2H, d), 3.07-3.15 (4H, m), 3.22-3.27 (1H, m), 3.41-3.54 (3H, m), 3.68 (2H, ddd), 7.36 (2H, d), 7.89 (2H, d), 8.04 (1H, d), 8.63 (1H, d); m/zMH+ 569.
Пример 56: (R)-2,5,7-триметил-6-((1-(5-(3-метил-4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
Pd(Ph3P)4 (35 мг; 0,03 ммоль) добавляли к (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 46) (122 мг; 0,30 ммоль), 1-метил-4-(2-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазину (промежуточное соединение 51) (100 мг; 0,30 ммоль) и Na2CO3 (64,2 мг; 0,61 ммоль) в толуоле (4 мл) и воде (1 мл). Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (15 мг; 9%) в виде белого твердого вещества; 1H ЯМР (300 МГц, CDCl3) 1.94 (1Н, m), 2.22 (1H, dt), 2.30-2.75 (21Н, m), 2.81 (3H, s), 2.95 (2Н, t), 3.34 (1H, dd), 3.54 (3H, m), 3.75 (2Н, m), 7.34 (1H, m), 7.65 (2Н, m), 7.94 (1H, d), 8.50 (1H, s); m/z МН+ 526.
Пример 57:(R)-2,5,7-триметил-6-((1-(5-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)1 шразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло-[1,5-а]пиримидин
Пред катализатор XPhos 2-го поколения (10,76 мг; 0,01 ммоль) добавляли к (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 46) (110 мг; 0,27 ммоль), (6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)бороновой кислоте (промежуточное соединение 9) (71 мг; 0,30 ммоль) и Cs2CO3 (178 мг; 0,55 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 4 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением неполного (0,5 эквив.) формиата указанного в заголовке соединения (141 мг; 96%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.96 (1H, dq), 2.25 (1Н, dt), 2.62-2.76 (10Н, m), 2.83 (3H, s), 2.89-3.06 (10Н, m), 3.36 (1H, dd), 3.59 (1H, dt), 3.71-3.84 (4Н, m), 7.41 (1Н, d), 7.99 (1H, d), 8.18 (1H, dd), 8.40 (0.5Н, s, от 0,5 эквив. формиата), 8.53 (1H, d), 9.11 (1H, d); m/z МН+ 513.
Пример 58: (R)-1-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)пиперазин-1-ил)этанон
1-(4-(4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазин-1-ил)-этанон (промежуточное соединение 52) (667 мг; 1,94 ммоль), (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (промежуточное соединение 46) (600 мг; 1,49 ммоль) и Cs2CO3 (972 мг; 2,98 ммоль) добавляли к 1,4-диоксану (30 мл) и воде (15 мл) и смесь дегазировали в течение 10 минут. Добавляли пред катализатор XPhos 2-го поколения (59 мг; 0,07 ммоль) и реакционную смесь перемешивали в течение 18 часов при 90°С, затем оставляли для охлаждения до комнатной температуры и разбавляли EtOAc (50 мл). Органический слой отделяли, промывали насыщ. рассолом и сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-4% 1 NH3/МеОН в DCM. Продукт растворяли в EtOAc (2 мл) и перемешивали в течение 2 часов, и полученный осадок выделяли посредством фильтрования и сушили под вакуумом с получением указанного в заголовке соединения (209 мг; 26%) в виде белого твердого вещества; 1H ЯМР (500 МГц, DMSO) 1.78-1.91 (1Н, m), 1.96 (3H, s), 2.03-2.16 (1H, m), 2.26-2.33 (2Н, m), 2.34-2.4 (2Н, m), 2.46 (3H, s), 2.55-2.67 (4Н, m), 2.74 (3H, s), 2.93 (2Н, d), 3.2-3.26 (1H, m), 3.35-3.47 (5Н, m), 3.50 (2Н, s), 3.59-3.75 (2Н, m), 7.34 (2Н, d), 7.88 (2Н, d), 8.02 (1H, s), 8.62 (1H, s); m/z МН+ 540.
Пример 59: (R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
Пред катализатор XPhos 2-го поколения (0,780 г; 0,99 ммоль) добавляли к смеси (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло-[1,5-а]пиримидина (промежуточное соединение 46) (7,98 г; 19,84 ммоль), 1-метил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазина (из коммерческих источников) (7,53 г; 23,8 ммоль) и Cs2CO3 (12,93 г; 39,7 ммоль) в 1,4-диоксане (187 мл) и воде (75 мл). Реакционную смесь нагревали при 90°С в течение 3 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Остаток помещали в воду (1 л) и экстрагировали DCM (3×800 мл). Объединенные органические слои промывали водой, насыщ. рассолом, сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя 0-15% МеОН в DCM. Продукт помещали в MeCN (150 мл) и перемешивали в течение 2 часов после обработки ультразвуком. Полученный осадок выделяли посредством фильтрации, промывали диэтиловым эфиром и затем н-гептаном, и сушили под вакуумом с получением указанного в заголовке соединения (6,30 г; 62%) в виде бледно- кремового твердого вещества; 1H ЯМР (500 МГц, DMSO) 1.82-1.88 (1H, m), 2.10-2.14 (1H, m), 2.15 (3H, s), 2.30-2.34 (4H, m), 2.37-2.41 (4H, m), 2.47 (3H, s), 2.58-2.64 (1H, m), 2.64 (3H, s), 2.75 (3H, s), 2.91-2.95 (2H, m), 3.23-3.26 (1H, m), 3.43-3.46 (1H, m), 3.47 (2H, s), 3.64-3.67 (1H, m), 3.68-3.72 (1H, m), 7.32 (2H, d), 7.85 (2H, d), 8.00 (1H, s), 8.56 (1H, s); m/z MH+ 512.
Пример 60: (R-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)морфолин
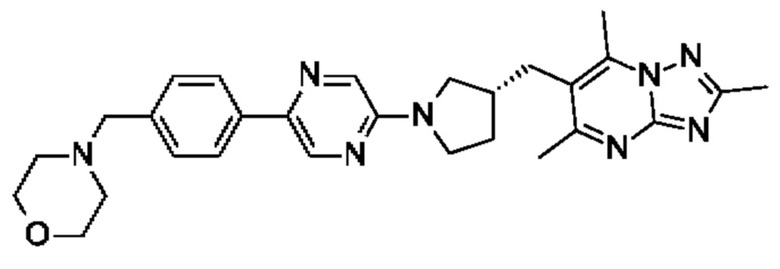
Пред катализатор XPhos 2-го поколения (24,45 мг; 0,03 ммоль) добавляли к (R-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 46) (250 мг; 0,62 ммоль), 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)морфолину (из коммерческих источников) (188 мг; 0,62 ммоль) и Cs2CO3 (405 мг; 1,24 ммоль) в 1,4-диоксане (5 мл) и воде (2,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 16 часов, оставляли для охлаждения до комнатной температуры, фильтровали через целит и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (191 мг; 56%) в виде бледно-желтого твердого вещества; 1 ЯМР (300 МГц, MeOD) 1.93-2.02 (1H, m), 2.18-2.28 (1H, m), 2.55 (3H, s), 2.65-2.74 (4Н, m), 2.78-2.83 (7Н, m), 3.02-3.04 (2Н, m), 3.29-3.35 (1H, m), 3.49-3.58 (1H, m), 3.66-3.81 (6Н, m), 3.86 (2Н, s), 7.47 (2Н, d), 7.90 (2Н, d), 7.99 (1H, s), 8.49 (1H, s); m/z NH+ 499.
Пример 61: (R)-4-(4-(6-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиридазин-3-ил)бензил)морфолин
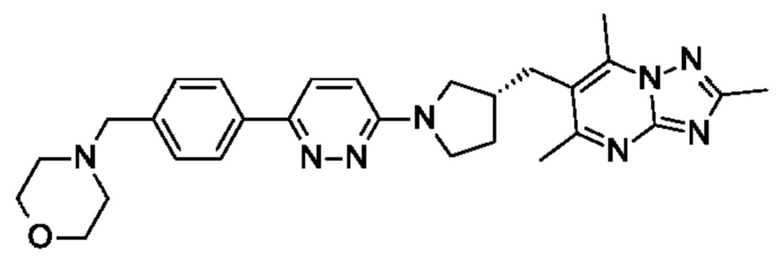
Пред катализатор XPhos 2-го поколения (11 мг; 0,01 ммоль) добавляли к (R)-6-((l-(6-хлорпиридазин-3-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 53) (100 мг; 0,28 ммоль), 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)морфолину (из коммерческих источников) (85 мг; 0,28 ммоль) и Cs2CO3 (182 мг; 0,56 ммоль) в 1,4-диоксане (2 мл) и воде (1 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 2 часов, оставляли для охлаждения до комнатной температуры и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,08% NH4HCO3), с получением указанного в заголовке соединения (101 мг; 73%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 2.01 (1H, dq), 2.26 (1H, dt), 2.54 (7Н, d), 2.75 (4Н, s), 2.86 (3H, s), 3.07 (2Н, dd), 3.38 (1Н, dd), 3.52-3.63 (3H, m), 3.68-3.86 (6Н, m), 7.04 (1H, d), 7.44-7.51 (2Н, m), 7.82-7.95 (3H, m); m/z МН+ 499.
Пример 62: (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин
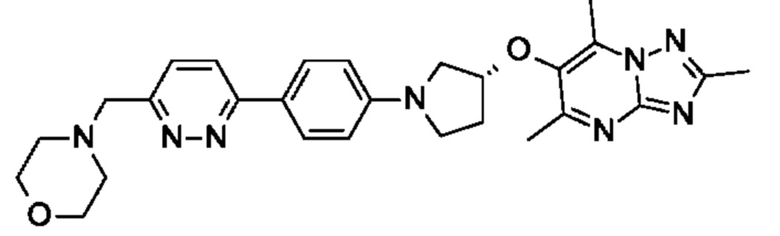
Объединяли Cs2CO3 (50 г; 153 ммоль), 4-((6-(4-бромфенил)пиридазин-3-ил)метил)морфолин (10 г; 29,80 ммоль) (промежуточное соединение 65) и дигидрохлорид (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а] пиримидина (промежуточное соединение 58) (10 г; 29,80 ммоль), пред катализатор RuPhos 3-го поколения (1,87 г; 2,24 ммоль) и RuPhos (1,04 г; 2,24 ммоль). Добавляли дегазированную смесь 2-метилтетрагидрофурана (80 мл) и воды (40 мл) и реакционную смесь нагревали при температуре образования флегмы в течение 17 часов, затем оставляли для охлаждения до комнатной температуры и разбавляли DCM (250 мл) и водой (250 мл). Органический слой отделяли и промывали смесью насыщ. рассол : вода, 1:1 (100 мл), затем фильтровали через набивку из диоксида кремния, затем концентрировали под вакуумом с получением неочищенного продукта. Затем взаимодействие повторяли, используя следующее: Cs2CO3 (75 г; 230 ммоль), 4-((6-(4-бромфенил)пиридазин-3-ил)метил)морфолин (15 г; 44,70 ммоль), дигидрохлорид (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а] пиримидина (15 г; 44,7 ммоль), пред катализатор RuPhos 3-го поколения (2,80 г; 3,35 ммоль) и RuPhos (1,56 г; 3,35 ммоль) в дегазированной смеси 2-метилтетрагидрофурана (120 мл) и воды (60 мл). Второе взаимодействие проводили посредством способа, аналогичного способу, описанному для первого взаимодействия. Две порции неочищенного продукта объединяли и очищали посредством СФХ (колонка Kromasil SIL 250×50 мм, 10 мкм, при скорости потока 450 мл/мин при использовании 33% МеОН в сверхкритическом СО2 при давлении 130 бар (13 МПа) и температуре термостата 30°С) с получением указанного в заголовке соединения (29 г; выход 80% с учетом двух объединенных порций) в виде белого твердого вещества; 1Н ЯМР (600 МГц, DMSO) 2.31 (1H, dtd), 2.42 (5Н, d), 2.46 (3H, s), 2.52 (3H, s), 2.58 (3H, s), 3.45 (1Н, dd), 3.54 (1H, td), 3.58 (5H, q), 3.62-3.68 (1H, m), 3.77 (2H, s), 4.96 (1H, s), 6.74 (2H, d), 7.65 (1H, d), 8.03 (2H, d), 8.07 (1H, d); m/z MH+ 501.
Форма A
Конечный продукт, (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин, анализировали посредством ДРЛП и ДСК и установили, что он является кристаллическим. ДРЛП образца вещества давала дифракционную картину, которая показана на Фиг. 1. (R)-4-((6-(4-(3-((2,5,7-Триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин, форма А, характеризуется по меньшей мере одним пиком при значении угла 2θ 7,8° или 19,0°, измеренном при использовании CuKα-излучения. Десять самых главных пиков ДРЛП показаны в таблице А.

Пример 62.1: масштабирование получения (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4] триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина
Путь синтеза 1 части соединения:
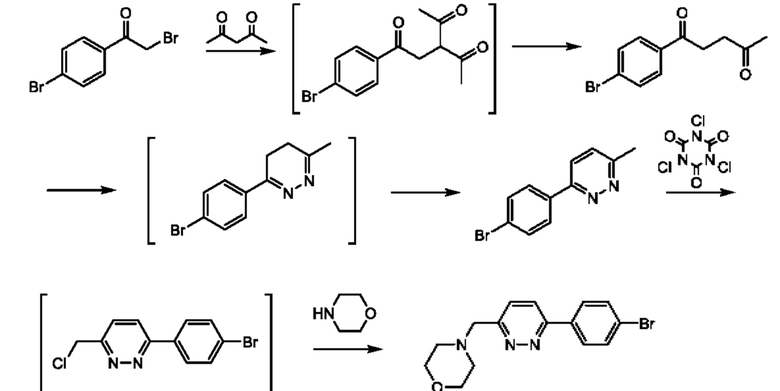
Стадия 1: 1-(4-бромфенил)-1,4-пентандион (промежуточное соединение 63)
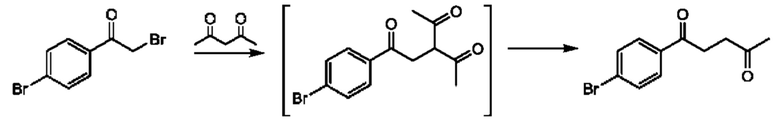
2-Бром-1-(4-бромфенил)этенон (12,2 кг; 43,89 моль), 2,4-пентандион (9,2 кг; 91,89 моль) и карбонат калия (12,2 кг; 88,27 моль) суспендировали в этаноле (72,25 л) в течение приблизительно 24 часов при комнатной температуре. Добавляли этилацетат (84,26 л) и воду (74 л) для осуществления разделения фаз, при котором нижнюю фазу отбрасывали. Замена растворителя органической фазы на н-гептан (2×84,8 л и 1×73,1 л) приводила к образованию твердого вещества, которое отделяли посредством фильтрования и промывали н-гептаном (17,54 л) с получением указанного в заголовке соединения (7,26 кг; выход 59,4% исходя из анализируемой величины 91,6% (мас./мас.)). 1Н ЯМР (400 МГц, CDCl3) 2.15 (3H, s), 2.56 (2Н, t), 3.07 (2Н, t), 7.44 (2Н, d), 7.89 (2Н, d).
Стадия 2: 3-(4-бромфенил)-6-метилпиридазин (промежуточное соединение 64)
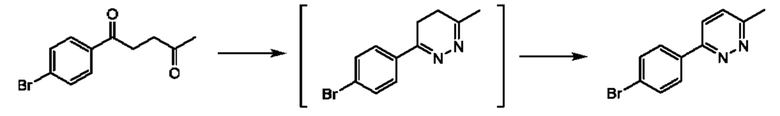
1-(4-Бромфенил)-1,4-пентадион (13,2 кг; 51,74 моль) и гидрат гидразина (10,9 кг; 217,74 моль) смешивали в этаноле (54,5 л) в течение приблизительно 6 часов при комнатной температуре. Добавление воды давало твердое вещество, которое отделяли посредством фильтрования и промывали водой (3×66 л) и н-гептаном (2×36,5 л). Твердое вещество смешивали с тетрахлор-1,4-бензохиноном (14,5 кг; 58,97 моль) и ацетонитрилом (137,4 л) в течение приблизительно 6 часов при комнатной температуре. Реакцию останавливали посредством добавления 12,5% (мас./мас.) раствора сульфита натрия (85 л), и ацетонитрил удаляли посредством перегонки. Добавление воды (66 л) и 2 М раствора гидроксида натрия (66 л) давало твердое вещество, которое отделяли посредством фильтрования и промывали 2 М гидроксидом натрия (2×40 л) и водой (2×145 л). Твердое вещество смешивали с дихлорметаном (140 л) и снова фильтровали перед промыванием водой (3×145 л). Органическую фазу затем обрабатывали активированным углем с последующей заменой растворителя на метил-трет-бутиловый эфир (100 л) и окончательной промывкой метил-трет-бутиловым эфиром (2×55 л). Это давало указанное в заголовке соединение (8,25 кг; выход 61% исходя из анализируемой величины 95,2% (мас./мас.)). 1Н ЯМР (400 МГц, CDCl3) 2.75 (3H, s), 7.38 (1H, d), 7.65-7.60 (2Н, m), 7.71 (1Н, d), 7.96-7.91 (2Н, m); 13С ЯМР (400 МГц, CDCl3) 22.1, 123.5, 124.4, 127.3, 128.4, 132.2, 135.4, 156.2, 158.6.
Стадия 3: 4-((6-(4-бромфенил)пиридазин-3-ил)метил)морфолин (промежуточное соединение 65)
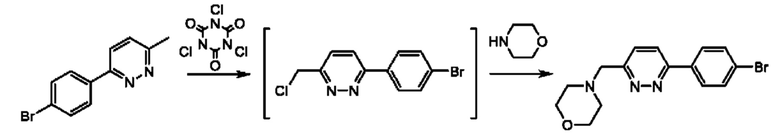
1,3,5-Трихлор-1,3,5-триазинан-2,4,6-трион (2,8 кг; 12,05 моль) порциями добавляли к 3-(4-бромфенил)-6-метилпиридазину (промежуточное соединение 64) (7,15 кг; 27,32 моль) в DCM (338,35 л) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем добавляли дополнительную порцию 1,3,5-трихлор-1,3,5-триазинан-2,4,6-триона (0,31 кг; 1,33 моль) и реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь фильтровали и органический фильтрат промывали водой (68 л) перед заменой растворителя на ацетонитрил (55 л). Добавляли морфолин (4,75 кг; 54,52 моль) и смесь нагревали в течение 4 часов перед концентрированием и фильтрованием с получением твердого вещества, которое промывали ацетонитрилом (6 л) и водой (6 л). Твердое вещество растворяли, используя 4 н соляную кислоту (7,5 кг) и воду (45 л). Кислый раствор промывали этилацетатом (2×2,5 л) перед доведением рН с помощью 15% (мас./мас.) гидроксида натрия (11 кг) с получением твердого вещества, которое выделяли посредством фильтрования. Это давало указанное в заголовке соединение (6,2 кг; выход 67% исходя из анализируемой величины 98,7% (мас./мас.)). 1Н ЯМР (400 МГц, CDCl3) 2.56 (4Н, m), 3.73 (4Н, m), 3.91 (2Н, s), 7.64 (2Н, m), 7.73 (1H, d), 7.81 (1H, d), 7.96 (2Н, m); m/z МН+ 334.
Путь синтеза 2 части соединения:
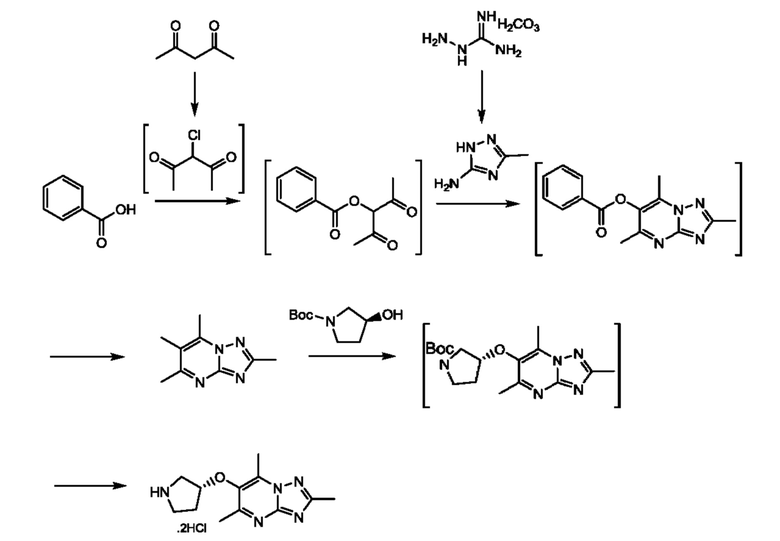
Стадия 4: 3-метил-1H-1,2,4-триазол-5-амин
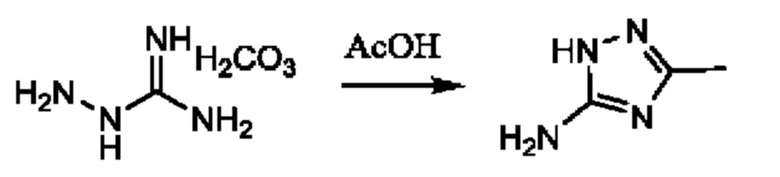
Бикарбонат аминогуанидина (17,7 кг; 130,04 моль) смешивали с азотной кислотой (165 г; 2,62 моль) в уксусной кислоте (29,8 L; 521,21 моль) в течение 64 часов при 90°С. Смесь охлаждали и разбавляли изопропилацетатом (30 л) и фильтровали с получением указанного в заголовке соединения. Неочищенное вещество (13,9 кг; выход 36,7% исходя из анализируемой величины 33,7% (мас./мас.)) использовали непосредственно на стадии 5.
Стадия 5: 2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ол (промежуточное соединение 56)
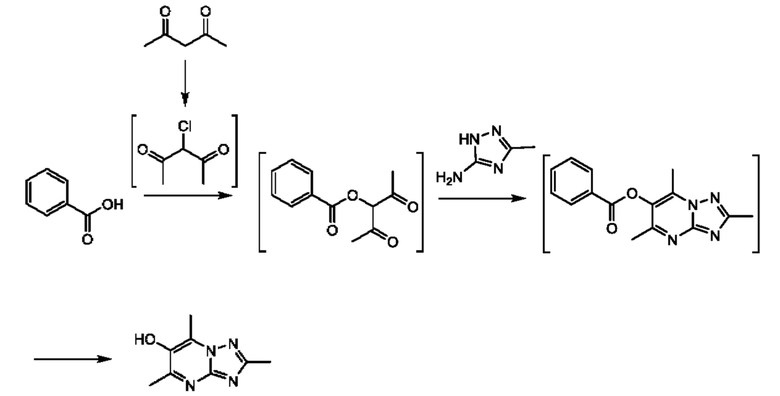
2,4-Пентандион (5,6 кг; 55,93 моль) и бромид тетра-н-бутиламмония (0,9 кг; 2,79 моль) охлаждали в ацетонитриле (39,44 л). Медленное добавление триметилсилилхлорида (7,3 кг; 67,19 моль) и диметилсульфоксида (4,4 кг; 56,32 моль) давало промежуточное соединение 3-хлор-2,4-пентандион в виде ацетонитрильного раствора. В этот раствор загружали бензойную кислоту (4,9 кг; 40,12 моль) и диизопропилэтиламин (8,6 кг; 66,54 моль) с получением 2,4-диоксопентан-3-илбензоата (промежуточное соединение 54). За заменой растворителя на 2-метилтетрагидрофуран (28,1 л) с последующей промывкой 5%-ным сульфатом натрия следовало добавление 3-метил-1Н-1,2,4-триазол-5-амина (12,8 кг; 43,97 моль). Добавление уксусной кислоты (32,19 л) и нагревание при 90°С в течение 20 часов давали 2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-илбензоат (промежуточное соединение 55). Замена растворителя на толуол (25 л) и перемешивание при комнатной температуре с 5 М гидроксидом натрия (63 л) в течение 20 часов при комнатной температуре давали указанное в заголовке соединение. Добавление 6 М соляной кислоты приводило к получению твердого вещества, которое выделяли посредством фильтрования и промывали водой (16 л). Твердое вещество суспендировали в воде (40 л) и толуоле (15 л) и фильтровали, затем промывали водой (16 л) и сушили с получением 5,8 кг (анализ 94,6% (мас./мас.); 30,79 моль; выход 76,7% по бензойной кислоте).
Стадия 6: промежуточное соединение 58: дигидрохлорид (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидина (промежуточное соединение 58)
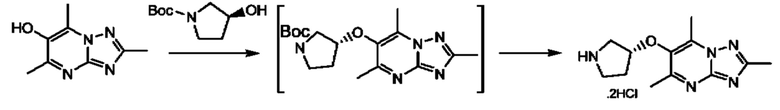
Диизопропилазодикарбоксилат (6,9 кг; 34,12 моль) в THF (14,62) по каплям добавляли к 2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-олу (4,55 кг; 24,16 моль), трет-бутил-(S)-3-гидроксипирролидин-1-карбоксилату (5,3 кг; 28,31 моль) и трифенилфосфину (7,3 кг; 27,83 моль) в THF (85,49 л) при 0°С и затем перемешивали при комнатной температуре в течение 20 часов. После этого следовала замена растворителя на 2-метил-ТНР (46,84 л) и промывка 25%-ным (мас./мас.) хлоридом натрия (2×22 кг). 2-Метил-THF удаляли посредством замены растворителя на изопропиловый спирт (45 л), и Вос-группу удаляли посредством добавления 15%-ной соляной кислоты в изопропиловом спирте (73,8 кг) и перемешивания в течение 15 часов при комнатной температуре. Охлаждение и добавление затравки дигидрохлорида (11)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1.5-а]пиримидина (120 г; 370 ммоль) давало твердое вещество, которое выделяли посредством фильтрования и промывали изопропиловым спиртом. В заключении твердое вещество суспендировали в этилацетате (45 л) перед окончательными фильтрованием и промывкой этилацетатом (45 л) с получением 5,35 кг (анализ* 74% (мас./мас); 16,01 моль; выход 66%). 1Н ЯМР (400 МГц, DMSO) 2.03-2.13 (1H, m), 2.22-2.28 (1H, m), 2.51-2.54 (3H, m), 2.62 (3H, d), 2.74 (3H, s), 3.32-3.45 (3H, m), 3.46-3.56 (1H, m), 4.90 (1H, s), 9.91 (2Н, s); m/z МН+ 248. * Анализ выполняют в расчете на эквивалент свободного основания.
Объединение:
Стадия 7: (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин (пример 62)
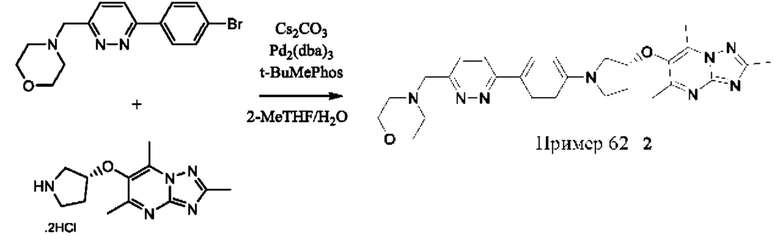
Объединяли Cs2CO3 (23,8 кг; 73,0 моль), 4-((6-(4-бромфенил)пиридазин-3-ил)метил)морфолин (4,89 кг; 14,63 моль) (промежуточное соединение 65), и дигидрохлорид (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидина (промежуточное соединение 58) (4,33 кг; 17,51 моль*), 2-дитрет-бутилфосфино-2'-метилбифенил (0,23 кг; 736 ммоль), и трис(дибензилиденацетон)дипалладий(0) (0,33 кг; 360 ммоль). Добавляли дегазированную смесь 2-метилтетрагидрофурана (80 л) и воды (40 л), и реакционную смесь нагревали при температуре образования флегмы в течение 24 часов, затем оставляли для охлаждения до комнатной температуры и разбавляли DCM (100 л) и водой (50 л). Реакционную смесь фильтровали, и в органической фазе заменяли растворитель на этилацетат с получением 8,4 кг неочищенного вещества. Его растворяли в DCM (200 л) и промывали последовательно 2 М соляной кислотой (2×90 л), 2 М гидроксидом натрия (140 л) и 5%-ным тиосульфатом натрия (2×50 л). Полученный DCM раствор пропускали через силикагелевую смолу с тиоловыми группами (0,5 кг) перед заменой растворителя на этилацетат с получением 5 кг неочищенного вещества. Его растворяли в DCM (25 л) и получали истинную форму А посредством добавления н-гептана (25 л) с затравкой (форма А (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)-пирролидин-1-ил)фенил)-пиридазин-3-ил)метил)морфолина), используемой для контроля точки нуклеации. Фильтрование и сушка (30°С) давали указанное в заголовке соединение (4,06 кг; 8,11 моль; выход 55%) в виде белого твердого вещества; 1Н ЯМР (600 МГц, DMSO) 2.31 (1H, dtd), 2.42 (5Н, d), 2.46 (3H, s), 2.52 (3H, s), 2.58 (3H, s), 3.45 (1H, dd), 3.54 (1H, td), 3.58 (5Н, q), 3.62-3.68 (1H, m), 3.77 (2Н, s), 4.96 (1H, s), 6.74 (2Н, d), 7.65 (1H, d), 8.03 (2Н, d), 8.07 (1Н, d); m/z МН+ 501. *3агрузка представляет собой количество основного вещества, скорректированное на свободное основание промежуточного соединения 58, за вычетом общей загрузки и составляет 5,85 кг соли дигидрохлорида.
Пример 63: (S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин
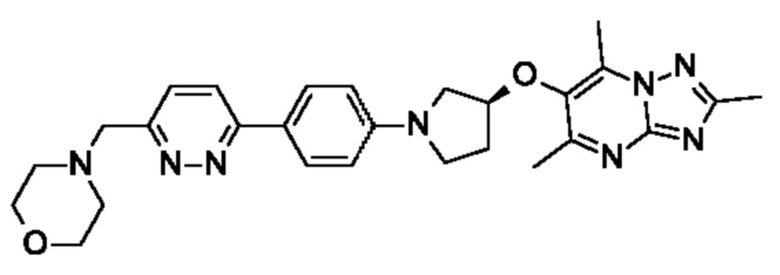
Соединение по примеру 63 получали посредством способа, аналогичного способу получения по примеру 62, за 3 стадии из имеющегося в продаже трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилата с получением указанного в заголовке соединения (125 мг; 31% на конечной стадии) в виде белого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 2.26-2.47 (9H, m), 2.52 (3H, s), 2.58 (3H, s), 3.45 (1H, dd), 3.5-3.61 (6H, m), 3.61-3.69 (1H, m), 3.77 (2H, s), 4.96 (1H, s), 6.73 (2H, d), 7.64 (1H, d), 8.04 (3H, dd); m/z MH+ 501.
Пример 64: (R)-2,5,7-триметил-6-((1-(4-(6-(пиперидин-1-илметил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4] триазоло[1,5-а]пиримидин
Cs2CO3 (593 мг; 1,82 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58В) (150 мг; 0,61 ммоль) и 3-(4-бромфенил)-6-(пиперидин-1-илметил)пиридазину (промежуточное соединение 70) (202 мг; 0,61 ммоль) в 2-метилтетрагидрофуране (1,6 мл) и воде (0,8 мл). Реакционную смесь дегазировали и добавляли пред катализатор RuPhos 3-го поколения (38 мг; 0,05 ммоль) и RuPhos (21 мг; 0,05 ммоль). Реакционную смесь нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, и затем разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (83 мг; 27%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.37-1.45 (2Н, m), 1.52 (4Н, р), 2.32 (1H, dt), 2.40 (5Н, s), 2.47 (3H, s), 2.53 (3H, s), 2.59 (3H, s), 3.46 (1Н, dd), 3.51-3.61 (2Н, m), 3.66 (1H, q), 3.74 (2Н, s), 4.98 (1H, s), 6.75 (2Н, d), 7.63 (1H, d), 8-8.07 (3H, m); m/z МН+ 499.
Пример 65: (R)-2,5,7-триметил-6-((1-(4-(6-((4-метилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидин
Cs2CO3 (593 мг; 1,82 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58 В) (150 мг; 0,61 ммоль) и 3-(4-бромфенил)-6-((4-метилпиперазин-1-ил)метил)пиридазину (промежуточное соединение 71) (211 мг; 0,61 ммоль) в 2-метилтетрагидрофуране (1,6 мл) и воде (0,8 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (38 мг; 0,05 ммоль) и RuPhos (21 мг; 0,05 ммоль). Реакционную смесь нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, и затем разбавляли DCM (10 мл) и водой (5 мл), и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (154 мг; 49%) в виде кремового твердого вещества; 1Н ЯМР (400 МГц, DMSO) 2.16 (3H, s), 2.27-2.38 (5Н, m), 2.38-2.48 (8Н, m), 2.53 (3H, s), 2.59 (3H, s), 3.46 (1H, dd), 3.51-3.61 (2Н, m), 3.66 (1H, q), 3.77 (2Н, s), 4.97 (1H, s), 6.74 (2Н, d), 7.62 (1H, d), 8.01-8.09 (3H, m); m/z МН+ 514.
Пример 66: 2,5,7-триметил-6-(((3R)-1-(4-(6-((3-метил-3,8-диазабицикло[3.2.1]октан-8-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидин
Cs2CO3 (494 мг; 1,52 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58В) (125 мг; 0,51 ммоль) и 8-((6-(4-бромфенил)пиридазин-3-ил)метил)-3-метил-3,8-диазабицикло[3.2.1]октану (промежуточное соединение 66) (189 мг; 0,51 ммоль) в 2-метилтетрагидрофуране (1,6 мл) и воде (0,8 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (31,7 мг; 0,04 ммоль) и RuPhos (17,69 мг; 0,04 ммоль). Реакционную смесь нагревали при температуре образования флегмаы в течение 18 часов, оставляли для охлаждения до комнатной температуры, затем разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (121 мг; 44%) в виде кремового твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.86 (2Н, q), 2.05 (2Н, dd), 2.23 (3H, s), 2.27-2.39 (3H, m), 2.46-2.54 (1H, m), 2.57-2.63 (8Н, m), 2.65 (3H, s), 3.13 (2Н, s), 3.48-3.65 (3H, m), 3.73-3.82 (1H, m), 3.87 (2Н, s), 4.85 (1H, s), 6.68 (2Н, d), 7.74-7.82 (2Н, m), 8.04 (2Н, d); m/z МН+ 540.
Пример 67: 6-(((R)-1-(4-(6-(((R)-гексагидропирроло[1,2-а]пиразин-2(1H)-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]-триазоло[1,5-а]пиримидин
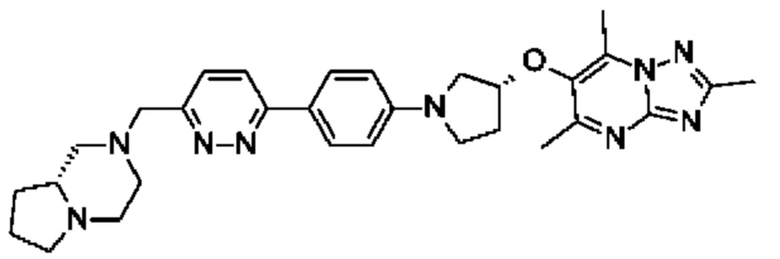
Cs2CO3 (494 мг; 1,52 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58В) (125 мг; 0,51 ммоль) и (R)-2-((6-(4-бромфенил)пиридазин-3-ил)метил)октагидропирроло[1,2-а]пиразину (промежуточное соединение 67) (189 мг; 0,51 ммоль) в 2-метилтетрагидрофуране (1,6 мл) и воде (0,8 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (31,7 мг; 0,04 ммоль) и RuPhos (17,7 мг; 0,04 ммоль). Реакционную смесь нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, и затем разбавляли DCM (10 мл) и водой (5 мл), и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (33 мг; 12%) в виде кремового твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.20-1.31 (1H, m), 1.66 (3H, ttd), 1.87-2.06 (3H, m), 2.14 (1H, td), 2.25 (1H, td), 2.33 (1H, dtd), 2.42 (1H, dd), 2.47 (3H, s), 2.53 (3H, s), 2.59 (3H, s), 2.73 (1H, d), 2.86-2.98 (3H, m), 3.46 (1H, dd), 3.51-3.62 (2Н, m), 3.62-3.71 (1H, m), 3.77-3.87 (2Н, m), 4.97 (1Н, s), 6.75 (2Н, d), 7.62 (1H, d), 8.05 (3H, t); m/z МН+ 540.
Пример 68: 6-(((R)-1-(4-(6-(((S)-гексагидропирроло[1,2-а]пиразин-2(1H)-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]-триазоло[1,5-а]пиримидин
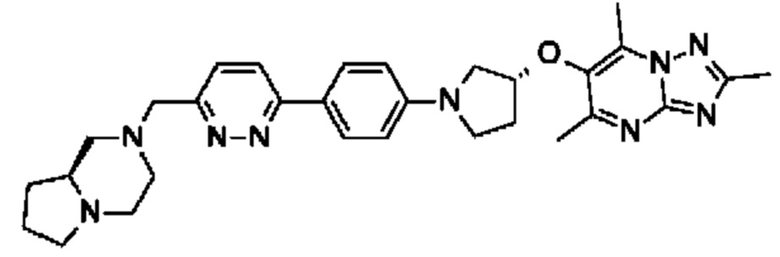
Cs2CO3 (494 мг; 1,52 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58В) (125 мг; 0,51 ммоль) и (S)-2-((6-(4-бромфенил)пиридазин-3-ил)метил)октагидропирроло[1,2-а]пиразину (промежуточное соединение 68) (189 мг; 0,51 ммоль) в 2-метилтетрагидро-фуране (1,6 мл) и воде (0,8 мл). Реакционную смесь дегазировали и добавляли пред катализатор RuPhos 3-го поколения (31,7 мг; 0,04 ммоль) и RuPhos (17,7 мг; 0,04 ммоль). Реакционную смесь нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, затем разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (76 мг; 28%) в виде кремового твердого вещества; 1H ЯМР (400 МГц, DMSO) 1.20-1.32 (1H, m), 1.66 (3H, dqd), 1.88-2.06 (3H, m), 2.14 (1H, td), 2.25 (1H, td), 2.3-2.37 (1H, m), 2.42 (1H, d), 2.47 (3H, s), 2.53 (3H, d), 2.60 (3H, s), 2.73 (1H, d), 2.85-2.98 (3H, m), 3.47 (1H, dd), 3.51-3.62 (2Н, m), 3.66 (1H, q), 3.77-3.87 (2Н, m), 4.98 (1H, s), 6.75 (2Н, d), 7.62 (1H, d), 8.01-8.09 (3H, m); m/z МН+ 540.
Пример 69: (R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиримидин-2-ил)метил)морфолин
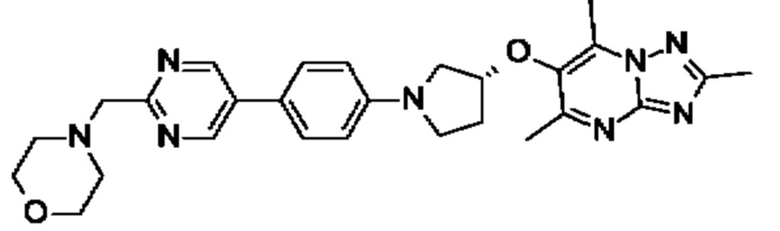
Cs2CO3 (401 мг; 1,23 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58В) (101 мг; 0,41 ммоль) и 4-((5-(4-бромфенил)пиримидин-2-ил)метил)морфолину (промежуточное соединение 39) (137 мг; 0,41 ммоль) в 2-метилтетрагидрофуране (1,4 мл) и воде (0,7 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (25,7 мг; 0,03 ммоль) и RuPhos (14,4 мг; 0,03 ммоль). Реакционную смесь нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, затем разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (82 мг; 40%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.34 (1H, dp), 2.47-2.55 (1H, m), 2.60 (3H, s), 2.61-2.65 (7Н, m), 2.65 (3H, s), 3.45-3.54 (2Н, m), 3.58 (1Н, td), 3.72-3.78 (1H, m), 3.78-3.83 (4Н, m), 3.85 (2Н, s), 4.83-4.89 (1H, m), 6.65-6.72 (2Н, m), 7.46-7.53 (2Н, m), 8.89 (2Н, s); m/z MH+ 501.
Пример 70: (R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиразин-2-ил)метил)морфолин
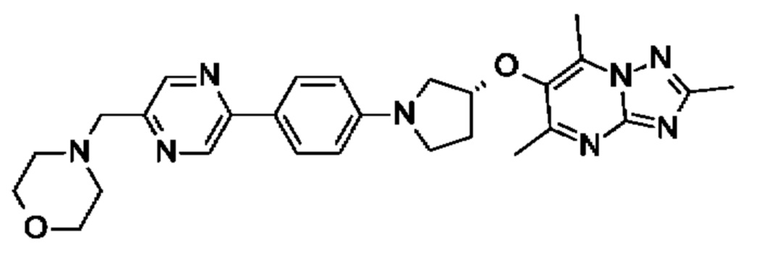
Cs2CO3 (668 мг; 2,05 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58В) (169 мг; 0,68 ммоль) и 4-((5-(4-бромфенил)пиразин-2-ил)метил)морфолину (промежуточное соединение 77) (228 мг; 0,68 ммоль) в 2-метилтетрагидрофуране (2,3 мл) и воде (1,1 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (42,9 мг; 0,05 ммоль) и RuPhos (23,9 мг; 0,05 ммоль). Реакционную смесь нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, затем разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (104 мг; 30%) в виде кремового твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.33 (1H, dtd), 2.50 (1H, dd), 2.53-2.57 (4Н, m), 2.60 (3H, s), 2.61 (3H, s), 2.64 (3H, s), 3.46-3.6 (2Н, m), 3.61 (1H, td), 3.70 (2Н, s), 3.72-3.83 (5Н, m), 4.85 (1H, dt), 6.63-6.71 (2Н, m), 7.91-8.00 (2Н, m), 8.60 (1H, d), 8.91 (1H, d); m/z MH+ 501.
Пример 71: (R)-1-(4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)-пиперазин-1-ил)этан-1-он
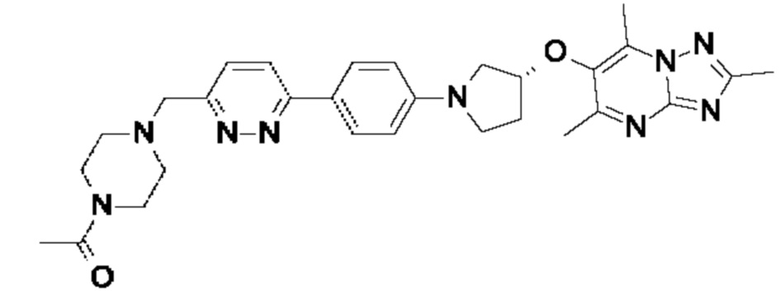
Cs2CO3 (632 мг; 1,94 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58В) (160 мг; 0,65 ммоль) и 1 -(4-((6-(4-бромфенил)пиридазин-3-ил)метил)пиперазин-1-ил)этан-1-ону (промежуточное соединение 69) (243 мг; 0,65 ммоль) в 2-метилтетрагидрофуране (1,7 мл) и воде (0,9 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (40,6 мг; 0,05 ммоль) и RuPhos (22,6 мг; 0,05 ммоль). Полученную суспензию нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (154 мг; 44%) в виде бледно-желтого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.99 (3H, s), 2.33 (1H, d), 2.37-2.43 (3H, m), 2.47 (5Н, s), 2.53 (3H, s), 2.59 (3H, s), 3.46 (5Н, dd), 3.56 (2Н, q), 3.66 (1H, q), 3.82 (2Н, s), 4.98 (1H, s), 6.75 (2Н, d), 7.66 (1H, d), 8.06 (3H, dd); m/z МН+ 542.
Пример 72: (R)-6-((1-(4-(6-((4-этилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
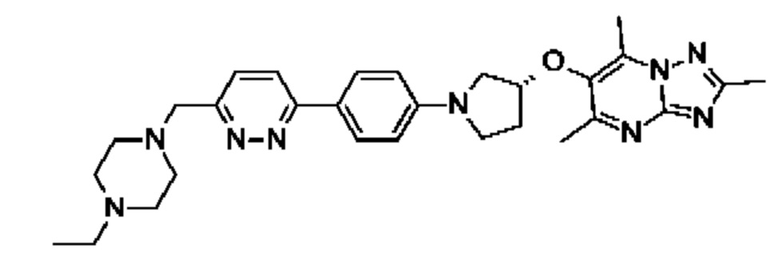
Cs2CO3 (336 мг; 1,03 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58 В) (85 мг; 0,34 ммоль) и 3-(4-бромфенил)-6-((4-этилпиперазин-1-ил)метил)пиридазину (промежуточное соединение 72) (124 мг; 0,34 ммоль) в 2-метилтетрагидрофуране (0,9 мл) и воде (0,5 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (21,6 мг; 0,03 ммоль) и RuPhos (12,0 мг; 0,03 ммоль). Реакционную смесь нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (65 мг; 36%) в виде бледно-желтого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.08 (3H, t), 2.33 (1H, dtd), 2.42 (3H, q), 2.45-2.54 (4Н, m), 2.57-2.64 (ЮН, m), 2.65 (3H, s), 3.52 (2Н, dd), 3.57-3.65 (1H, m), 3.78 (1H, td), 3.89 (2Н, s), 4.85 (1H, dt), 6.68 (2Н, d), 7.60 (1H, d), 7.74 (1Н, d), 8-8.08 (2Н, m); m/z МН+ 528.
Пример 73: (R)-6-((1-(4-(6-((4-(2-метоксиэтил)пиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]-триазоло[1,5-а]пиримидин
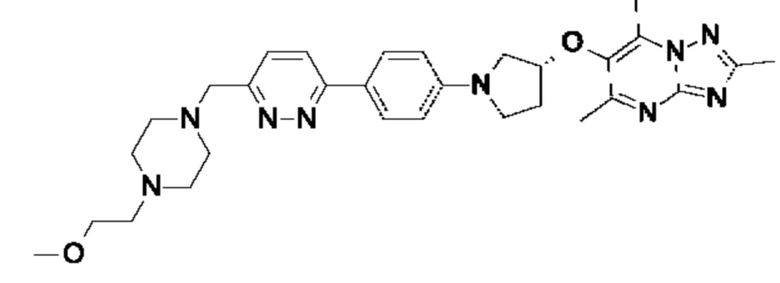
Cs2CO3 (870 мг; 2,67 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58В) (220 мг; 0,89 ммоль) и 3-(4-бромфенил)-6-((4-(2-метоксиэтил)пиперазин-1-ил)метил)пиридазину (промежуточное соединение 73) (348 мг; 0,89 ммоль) в 2-метилтетрагидрофуране (2,4 мл) и воде (1,1 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (55,8 мг; 0,07 ммоль) и RuPhos (31,1 мг; 0,07 ммоль). Реакционную смесь нагревали при температуре образования флегмы в течение 18 часов, оставляли для охлаждения до комнатной температуры, разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (211 мг; 43%) в виде бледно-желтого пенистого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 2.33 (1H, dtd), 2.49 (1H, dd), 2.51-2.64 (16Н, m), 2.65 (3H, s), 3.35 (3H, s), 3.46-3.56 (4Н, m), 3.57-3.64 (1H, m), 3.78 (1H, td), 3.89 (2Н, s), 4.84 (1H, dd), 6.68 (2Н, d), 7.59 (1H, d), 7.74 (1H, d), 8.00-8.07 (2H, m); m/zMH+ 558.
Пример 74: (R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)-[2,3'-бипиридин]-5-ил)метил)морфолин
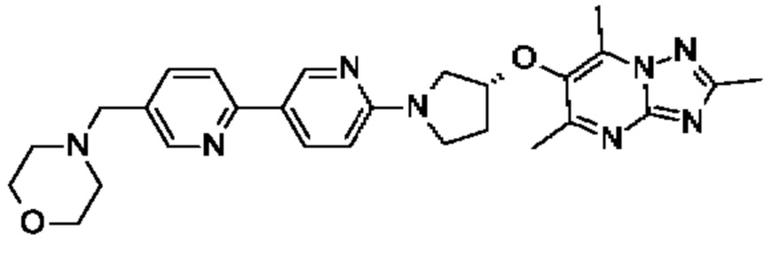
В (R)-6-((1-(5-бромпиридин-2-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (промежуточное соединение 59) (190 мг; 0,47 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (144 мг; 0,57 ммоль) и ацетат калия (92 мг; 0,94 ммоль) в 1,4-диоксане (5 мл) в пробирке для микроволновой печи добавляли аддукт PdCl2(dppf)-CH2Cl2 (19 мг; 0,02 ммоль) и реакционную смесь нагревали при 90°С в течение ночи, затем оставляли для охлаждения до комнатной температуры. Добавляли 4-((6-бромпиридин-3-ил)метил)морфолин (промежуточное соединение 30) (121 мг; 0,47 ммоль) с последующим добавлением Cs2CO3 (307 мг; 0,94 ммоль), предкатализатора XPhos 2-го поколения (74 мг; 0,05 ммоль) и воды (3 мл). Реакционную смесь нагревали при 90°С в течение 18 часов, затем оставляли для охлаждения до комнатной температуры и разбавляли EtOAc (50 мл) и водой (10 мл) и слои разделяли. Водный слой дополнительно экстрагировали DCM (50 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (44 мг; 19%) в виде бледно-коричневой пены; 1Н ЯМР (400 МГц, CDCl3) 2.34 (1H, dtd), 2.42-2.56 (5Н, m), 2.60 (3H, s), 2.61 (3H, s), 2.64 (3H, s), 3.53 (2Н, s), 3.61 (1H, dd), 3.68-3.73 (4Н, m), 3.74-3.92 (3H, m), 4.82 (1H, s), 6.51 (1H, d), 7.60 (1Н, d), 7.70 (1H, dd), 8.21 (1H, dd), 8.55 (1H, d), 8.76 (1Н, d); m/z МН+ 501.
Пример 75: (R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)-[3,3'-бипиридин]-6-ил)метил)морфолин
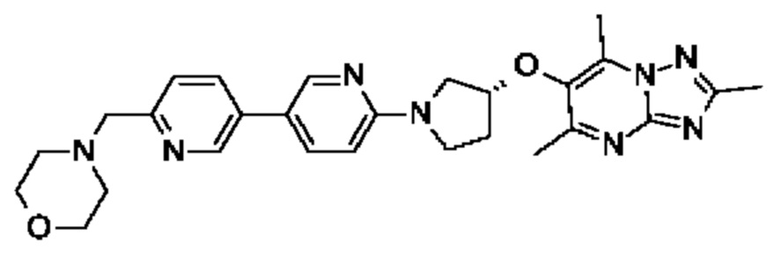
Cs2CO3 (242 мг; 0,74 ммоль) добавляли к (6-(морфолинометил)пиридин-3-ил)бороновой кислоте (83 мг; 0,37 ммоль) и (R)-6-((1-(5-бромпиридин-2-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 59) (150 мг; 0,37 ммоль) в 1,4-диоксане (2,5 мл) и воде (1,2 мл). Реакционную смесь дегазировали и добавляли предкатализатор XPhos 2-го поколения (59 мг; 0,04 ммоль). Реакционную смесь перемешивали при 80°С в течение 18 часов, оставляли для охлаждения до комнатной температуры, разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт растворяли в DMSO (2 мл) и очищали посредством препаративной ВЭЖХ, затем дополнительно очищали посредством КФХ, элюируя 0-3% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (18 мг; 10%) в виде бесцветной смолы; 1Н ЯМР (400 МГц, CDCI3) 2.34 (1H, dtd), 2.48-2.53 (1H, m), 2.52-2.57 (4Н, m), 2.60 (3H, s), 2.62 (3H, s), 2.65 (3H, s), 3.60 (1H, dd), 3.69 (2Н, s), 3.76 (5Н, h), 3.8-3.89 (2Н, m), 4.83 (1H, dt), 6.48-6.54 (1H, m), 7.46 (1H, d), 7.72 (1H, dd), 7.78 (1H, dd), 8.41 (1H, dd), 8.74 (1H, dd); m/z MH+ 501.
Пример 76: (R)-2,5,7-триметил-6-((1-(2-(4-((4-метилпиперазин-1-ил)метил)-фенил)пиримидин-5-ил)пирролидин-3-ил)окси)-[1,2,4] триазоло[1,5-а]пиримидин
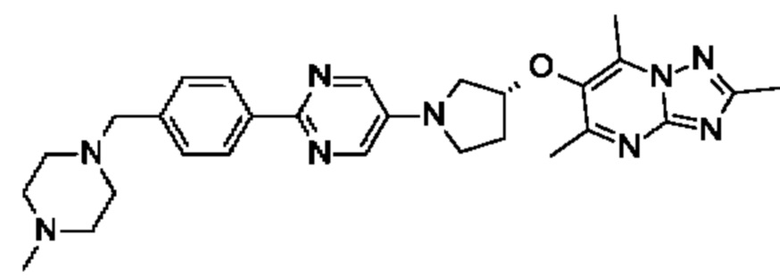
1-Метил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазин (131 мг; 0,42 ммоль), (R)-6-((1-(2-бромпиридин-5-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (промежуточное соединение 61) (140 мг; 0,35 ммоль) и Cs2CO3 (226 мг; 0,69 ммоль) добавляли к 1,4-диоксану (3,7 мл) и воде (1,8 мл) и смесь дегазировали в течение 10 минут. Добавляли предкатализатор XPhos 2-го поколения (13,62 мг; 0,02 ммоль) и реакционную смесь перемешивали в течение 2 часов при 85°С, оставляли для охлаждения до комнатной температуры, и выливали в воду (25 мл) и экстрагировали EtOAc(2x50 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством препаративной ВЭЖХ, затем дополнительно очищали посредством КФХ, элюируя 0-5% 1 М NH3/МеОН в DCM, с получением указанного в заголовке соединения (54 мг; 30%) в виде белой пены; 1Н ЯМР (500 МГц, DMSO) 2.16 (3H, s), 2.24-2.46 (9Н, m), 2.48 (3H, s), 2.52-2.54 (1H, m), 2.55 (3H, s), 2.60 (3H, s), 3.44-3.52 (3H, m), 3.57-3.64 (1H, m), 3.69 (2Н, dd), 5.00 (1H, s), 7.37 (2Н, d), 8.18-8.24 (2Н, m), 8.31 (2Н, s); m/z МН+ 514.
Пример 77: (R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)-фенил)пиразин-2-ил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-а]пиримидин
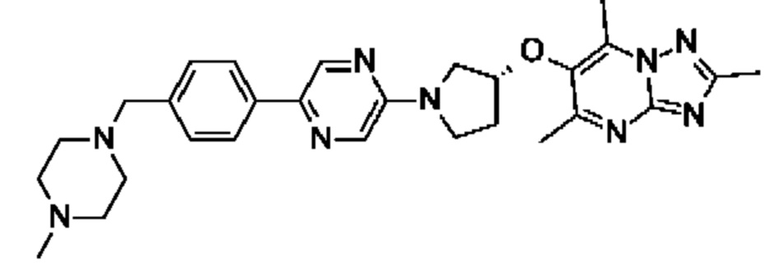
Ацетат палладия(II) (3,2 мг; 0,01 ммоль) добавляли к (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 62) (115 мг; 0,28 ммоль), 1-метил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пиперазину (117 мг; 0,37 ммоль), фосфату калия (151 мг; 0,71 ммоль) и дициклогексил(2',4',6'-триизопропил-[1,1'-бифенил]-2-ил)фосфину (13,6 мг; 0,03 ммоль) в дегазированной воде (1,6 мл) и THF (5 мл) при комнатной температуре. Реакционную смесь нагревали при температуре образования флегмы в течение 1 часа, оставляли для охлаждения до комнатной температуры, разбавляли EtOAc (50 мл) и промывали водой (50 мл). Органический слой отделяли и сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт растворяли в DCM (1 мл) и очищали посредством КФХ, элюируя 0-5% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (83 мг; 57%) в виде желтой пены; 1Н ЯМР (500 МГц, CDCl3) 2.29 (3H, s), 2.3-2.59 (10Н, m), 2.60 (3H, s), 2.63 (3H, s), 2.66 (3H, s), 3.55 (2Н, s), 3.63 (1H, dd), 3.78-3.93 (3H, m), 4.84 (1H, s), 7.41 (2Н, d), 7.79-7.88 (2Н, m), 8.02 (1H, d), 8.53 (1H, d); m/z МН+ 514.
Пример 78: (R)-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)пиразин-2-ил)бензил)морфолин
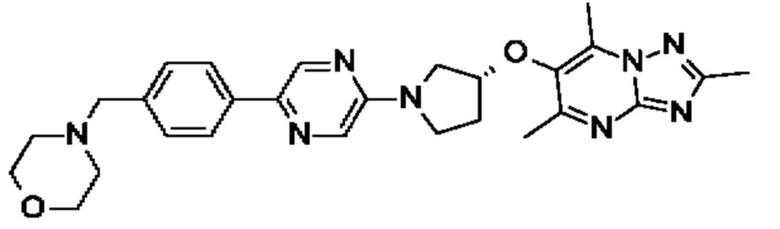
Ацетат палладия(II) (6,9 мг; 0,03 ммоль) добавляли к (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 62) (250 мг; 0,62 ммоль), 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)морфолину (225 мг; 0,74 ммоль), фосфату калия (328 мг; 1,55 ммоль) и дициклогексил(2',4',6'-триизопропил-[1,1'-бифенил]-2-ил)фосфину (29,5 мг; 0,06 ммоль) в дегазированной воде (3,33 мл) и THF (10 мл) при комнатной температуре. Реакционную смесь нагревали при температуре образования флегмы в течение 1 часа, оставляли для охлаждения до комнатной температуры, разбавляли EtOAc (50 мл) и промывали водой (50 мл). Органический слой выделяли и сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт растворяли в DCM (1 мл) и очищали посредством КФХ, элюируя 0-5% 1 М NH3/MeOH в DCM, с получением указанного в заголовке соединения (58 мг; 19%) в виде желтой пены; 1Н ЯМР (500 МГц, CDCl3) 2.35 (1H, dtd), 2.44-2.5 (4Н, m), 2.50-2.58 (1H, m), 2.60 (3H, s), 2.62 (3H, s), 2.66 (3H, s), 3.54 (2Н, s), 3.63 (1H, dd), 3.70-3.75 (4Н, m), 3.82 (1H, td), 3.85-3.93 (2Н, m), 4.83 (1H, d), 7.42 (2Н, d), 7.81-7.88 (2Н, m), 8.02 (1H, d), 8.53 (1H, d); m/z MH+ 501.
Пример 79: 1-[4-[[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил]пирролидин-1-ил]пиразин-2-ил]фенил]метил]-1,4-диазепан-1-ил]этанон
Объединяли (R)-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензальдегид (промежуточное соединение 49) (30 мг; 0,07 ммоль), 1-(1,4-диазепан-1-ил)этанон (19,9 мг; 0,14 ммоль), DCM (2 мл) и АсОН (2 капли) и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Добавляли триацетоксиборгидрид натрия (45 мг; 0,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученную смесь очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (18 мг; 47%); 1Н ЯМР (400 МГц, DMSO) 1.71 (1H, s), 1.81 (1H, d), 1.87 (1H, dd), 2.00 (3H, d), 2.14 (1H, dd), 2.48 (3H, s), 2.54-2.62 (4H, m), 2.65-2.69 (4H, m), 2.76 (3H, s), 2.95 (2H, d), 3.25 (1H, dd), 3.41-3.52 (5H, m), 3.61-3.75 (4H, m), 7.37 (2H, dd), 7.89 (2H, dd), 8.04 (1H, d), 8.63 (1H, d); m/z MH+ 554.
Пример 80: (R)-2,5,7-триметил-6-((1-(5-(4-(пирролидин-1-илметил)фенил)-пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
Пред катализатор XPhos 2-го поколения (19,56 мг; 0,02 ммоль) добавляли к (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидину (200 мг; 0,50 ммоль), 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пирролидину (143 мг; 0,50 ммоль) и Cs2CO3 (324 мг; 0,99 ммоль) в дегазированном 1,4-диоксане (4 мл) и воде (2 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов, оставляли для охлаждения до комнатной температуры и очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,08% NH4HCO3), с получением указанного в заголовке соединения (63 мг; 26%) в виде бледно-желтого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 1.85 (4Н, р), 1.99 (1Н, dq), 2.26 (1H, dq), 2.59 (7Н, d), 2.75 (4Н, s), 2.86 (3H, s), 3.05 (2Н, dt), 3.33-3.42 (1H, m), 3.56 (1H, dt), 3.66-3.85 (4Н, m), 7.41-7.48 (2Н, m), 7.81-7.90 (2Н, m), 8.01 (1H, d), 8.50 (1H, d); m/z МН+ 483.
Пример 81: (R)-2,5,7-триметил-6-((1-(5-(5-(пирролидин-1-илметил)пиридин-2-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4] триазоло[1,5-а]пиримидин
Хлорид бис(трифенилфосфин)палладия(II) (137 мг; 0,19 ммоль) добавляли к 2-бром-5-(пирролидин-1-илметил)пиридину (промежуточное соединение 78) (470 мг; 1,95 ммоль), 1,1,1,2,2,2-гексаметилдистаннану (702 мг; 2,14 ммоль) в THF (6 мл) при комнатной температуре. Реакционную смесь перемешивали при 85°С в течение 16 часов. Реакционную смесь переносили в пробирку для микроволновой печи и добавляли (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин (300 мг; 0,75 ммоль) и Pd(Ph3P)4 (86 мг; 0,07 ммоль). Реакционную смесь нагревали при 100°С в течение 5 часов в микроволновом реакторе, затем оставляли для охлаждения до комнатной температуры. Полученную смесь очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), затем дополнительно очищали посредством препаративной ВЭЖХ, элюируя смесями воды (содержащей 10 ммоль NH4HCO3) и MeCN с уменьшающейся полярностью, с получением указанного в заголовке соединения (115 мг; 32%) в виде бледно-желтого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 1.86 (4Н, р), 2.00 (1H, dq), 2.26 (1H, dq), 2.60 (7Н, d), 2.75 (4Н, s), 2.86 (3H, s), 3.07 (2Н, dd), 3.38 (1H, dd), 3.58 (1Н, dt), 3.71-3.88 (4Н, m), 7.87 (1H, dd), 8.04 (1H, d), 8.14 (1Н, d), 8.51-8.57 (1H, m), 8.92 (1H, d); m/z МН+ 484.
Пример 82: (R)-2,5,7-триметил-6-((1-(5-(6-(пирролидин-1-илметил)пиридин-3-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
Аддукт PdCl2(dppf)-CH2Cl2 (85 мг; 0,12 ммоль) добавляли в 5-бром-2-(пирролидин-1-илметил)пиридин (промежуточное соединение 80) (560 мг; 2,32 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (590 мг; 2,32 ммоль) и ацетат калия (456 мг; 4,64 ммоль) в 1,4-диоксане (10 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов и оставляли для охлаждения до комнатной температуры. Реакционную смесь очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% МеОН в воде, с получением неочищенного 2-(пирролидин-1-илметил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (280 мг; 42% в случае чистого) в виде желтого твердого вещества. Предкатализатор XPhos 2-го поколения (29,3 мг; 0,04 ммоль) добавляли к (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]-пиримидину (300 мг; 0,75 ммоль), неочищенному 2-(пирролидин-1-илметил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридину с первой стадии (258 мг; 0,89 ммоль в случае чистого) и Cs2CO3 (486 мг; 1,49 ммоль) в 1,4-диоксане (4 мл) и воде (2 мл) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение 16 часов. Неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением указанного в заголовке соединения (54 мг; 15%) в виде бледно-желтого твердого вещества; 1Н ЯМР (400 МГц, MeOD) 1.87-2.07 (5Н, m), 2.27 (1H, dq), 2.57 (3H, s), 2.67-2.89 (11Н, m), 3.07 (2Н, dd), 3.34-3.42 (1H, m), 3.54-3.64 (1H, m), 3.69-3.88 (2Н, m), 3.98 (2Н, s), 7.58 (1H, d), 8.08 (1H, d), 8.33 (1H, dd), 8.54-8.61 (1Н, m), 9.06 (1Н, dd); m/z МН+ 484.
Пример 83: (R)-2,5,7-триметил-6-((1-(5-(4-((4-метил-1,4-диазепан-1-ил)метил)фенил)пир азин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-а]пиримидин
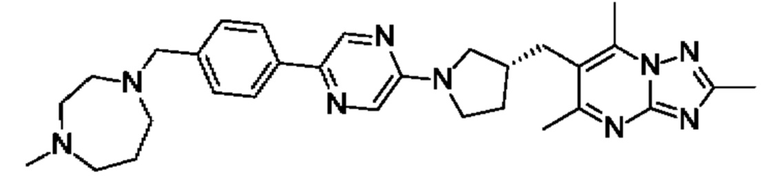
1-Метил-1,4-диазепан (53 мг; 0,47 ммоль) добавляли к (R)-4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензальдегиду (промежуточное соединение 49) (200 мг; 0,47 ммоль) в DCM (5 мл) при комнатной температуре в воздушной атмосфере, и реакционную смесь перемешивали в течение 1 часа. Добавляли триацетоксиборгидрид натрия (496 мг; 2,34 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством флэш-хроматографии на сорбенте С18, элюируя 5-100% MeCN в воде (плюс 0,1% FA), с получением формиата указанного в заголовке соединения (129 мг; 48%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, CDCl3) 1.88-2.02 (1H, m), 2.08 (2Н, dt), 2.25 (1H, dq), 2.60-2.76 (10Н, m), 2.79-3.06 (11Н, m), 3.14-3.22 (2Н, m), 3.35 (1H, dd), 3.58 (1H, dt), 3.69-3.84 (4Н, m), 7.38-7.45 (2Н, m), 7.81-7.89 (2Н, m), 7.97 (1Н, d), 8.52 (1H, d), 8.60 (1H, s); m/z МН+ 526.
Пример 84: (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)-1,4-оксазепан
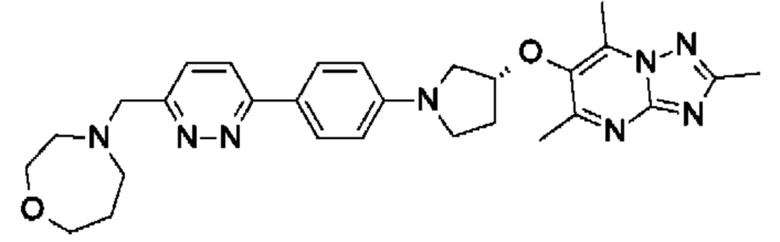
Cs2CO3 (553 мг; 1,70 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58 В) (140 мг; 0,57 ммоль) и 4-((6-(4-бромфенил)пиридазин-3-ил)метил)-1,4-оксазепану (промежуточное соединение 74) (197 мг; 0,57 ммоль) в 2-метилтетрагидрофуране (1,5 мл) и воде (0,76 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (35,5 мг; 0,04 ммоль) и RuPhos (19,8 мг; 0,04 ммоль). Реакционную смесь перемешивали при 88°С в течение 18 часов, оставляли для охлаждения до комнатной температуры, разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт растворяли в DMSO (2 мл) и очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения (136 мг; 47%) в виде бледно-желтого твердого вещества; 1H ЯМР (400 МГц, DMSO) 1.84 (2Н, р), 2.35 (1Н, tt), 2.43 (1Н, dd), 2.48 (3H, s), 2.54 (3H, s), 2.60 (3H, s), 2.72 (4Н, q), 3.47 (1H, dd), 3.56 (2Н, dd), 3.60-3.69 (3H, m), 3.72 (2Н, t), 3.95 (2Н, s), 4.98 (1H, s), 6.75 (2Н, d), 7.69 (1H, d), 8.02-8.11 (3H, m); m/z МН+ 515.
Пример 85: (R)-6-((1-(4-(6-(азепан-1-илметил)пиридазин-3-ил)фенил)-пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин
Cs2CO3 (249 мг; 0,76 ммоль) добавляли к (R)-2,5,7-триметил-6-(пирролидин-3-илокси)-[1,2,4]триазоло[1,5-а]пиримидину (промежуточное соединение 58 В) (63 мг; 0,25 ммоль) и 1-((6-(4-бромфенил)пиридазин-3-ил)метил)азепану (промежуточное соединение 75) (88 мг; 0,25 ммоль) в 2-метилтетрагидрофуране (0,6 мл) и воде (0,3 мл). Реакционную смесь дегазировали и добавляли предкатализатор RuPhos 3-го поколения (16 мг; 0,02 ммоль) и RuPhos (9 мг; 0,02 ммоль). Реакционную смесь перемешивали при 88°С в течение 18 часов, оставляли для охлаждения до комнатной температуры, разбавляли DCM (10 мл) и водой (5 мл) и пропускали через картридж для разделения фаз. Полученный DCM слой концентрировали под вакуумом. Полученный неочищенный продукт растворяли в DMSO (2 мл) и очищали посредством препаративной ВЭЖХ. Полученный продукт загружали на колонку SCX (5 г), промывая МеОН (2×объем колонки), затем элюируя 1 М NH3 в МеОН (2×объем колонки), и концентрировали под вакуумом с получением указанного в заголовке соединения (35 мг; 27%) в виде белого твердого вещества; 1Н ЯМР (400 МГц, DMSO) 1.60 (8Н, s), 2.3-2.37 (1H, m), 2.43 (1H, d), 2.48 (3H, s), 2.54 (3H, d), 2.61 (3H, s), 2.66 (4Н, d), 3.48 (1H, dd), 3.52-3.63 (2Н, m), 3.67 (1H, q), 3.92 (2Н, s), 4.99 (1H, s), 6.76 (2Н, d), 7.68 (1H, d), 8.01-8.11 (3H, m); m/z МН+ 513.
Примере 86 и 87: 4-[(1R)-1-[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси]пирролидин-1-ил]пиразин-2-ил]фенил]этил]морфолин и 4-[(1S)-1-[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси]-пирролидин-1-ил]пиразин-2-ил]фенил]этил]морфолин
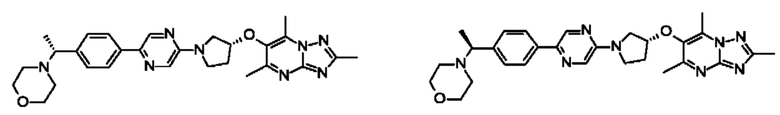
Тетракис(трифенилфосфин)палладий(0) (157 мг; 0,14 ммоль) добавляли к дегазированному раствору 4-(1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-фенил)этил)морфолина (из коммерческих источников) (432 мг; 1,36 ммоль), (R)-6-((1-(5-бромпиразин-2-ил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидина (промежуточное соединение 62) (550 мг; 1,36 ммоль) и карбоната натрия (2,0 мл; 4,1 ммоль) в 1,4-диоксане (7,7 мл) и воде (3,9 мл) в атмосфере азота. Полученный раствор перемешивали при 80°С в течение 2 часов, затем оставляли для охлаждения до комнатной температуры. Реакционную смесь разбавляли EtOAc (50 мл) и последовательно промывали водой (25 мл) и насыщ. рассолом (25 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали под вакуумом. Полученный неочищенный продукт очищали посредством КФХ, элюируя градиентом 0-5% МеОН в DCM, с получением указанного в заголовке соединения в виде смеси двух диастереомеров (355 мг). Изомеры разделяли посредством хиральной СФХ (колонка Phenomonex С1, 30×250 мм, 5 микрон, при скорости потока 80 мл/мин при использовании 40% IPA плюс 0,1% DEA/60% сверхкритический СО2 при 120 бар (12 МПа) и температуре колонки 40°С) с получением одного диастереомера (неизвестная хиральность альфа в отношении морфолина) 4-(1-(4-(5-((R)-3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)пиразин-2-ил)фенил)-этил)морфолина (62 мг; 18%); 1Н ЯМР (400 МГц, CDCl3) 1.38 (3H, d), 2.28-2.47 (3H, m), 2.47-2.58 (3H, m), 2.60 (3H, s), 2.62 (3H, s), 2.66 (3H, s), 3.35 (1H, q), 3.59-3.75 (5Н, m), 3.77-3.96 (3H, m), 4.84 (1H, s), 7.40 (2Н, d), 7.84 (2Н, d), 8.01 (1Н, d), 8.52 (1H, d); m/z МН+ 515; а затем второго диастереомера (неизвестная хиральность альфа в отношении морфолина) 4-(1-(4-(5-((R)-3-((2,5,7-триметил-[1,2,4]триазоло[1,5-а]пиримидин-6-ил)окси)пирролидин-1-ил)пиразин-2-ил)фенил)-этил)морфолина (68 мг; 19%); 1Н ЯМР (400 МГц, CDCl3) 1.38 (3H, d), 2.27-2.46 (3H, m), 2.46-2.58 (3H, m), 2.60 (3H, s), 2.62 (3H, s), 2.66 (3H, s), 3.35 (1H, q), 3.57-3.76 (5Н, m), 3.77-3.96 (3H, m), 4.84 (1Н, s), 7.40 (2Н, d), 7.84 (2Н, d), 8.01 (1H, d), 8.52 (1H, d); m/z МН+ 515.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| 4-(ИМИДАЗО[1,2-а]ПИРИДИН-3-ИЛ)-ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2822388C2 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРИДИН АЗОЛОПИРИМИДИН-5-(6Н)-ОНА | 2013 |
|
RU2653054C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| ПОЛИГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МЕТАБОТРОПНОГО РЕЦЕПТОРА ГЛУТАМАТА | 2005 |
|
RU2381226C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| АНТАГОНИСТЫ CXCR7 | 2013 |
|
RU2649004C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТРИАЗОЛОПИРАЗИНА И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2643361C2 |
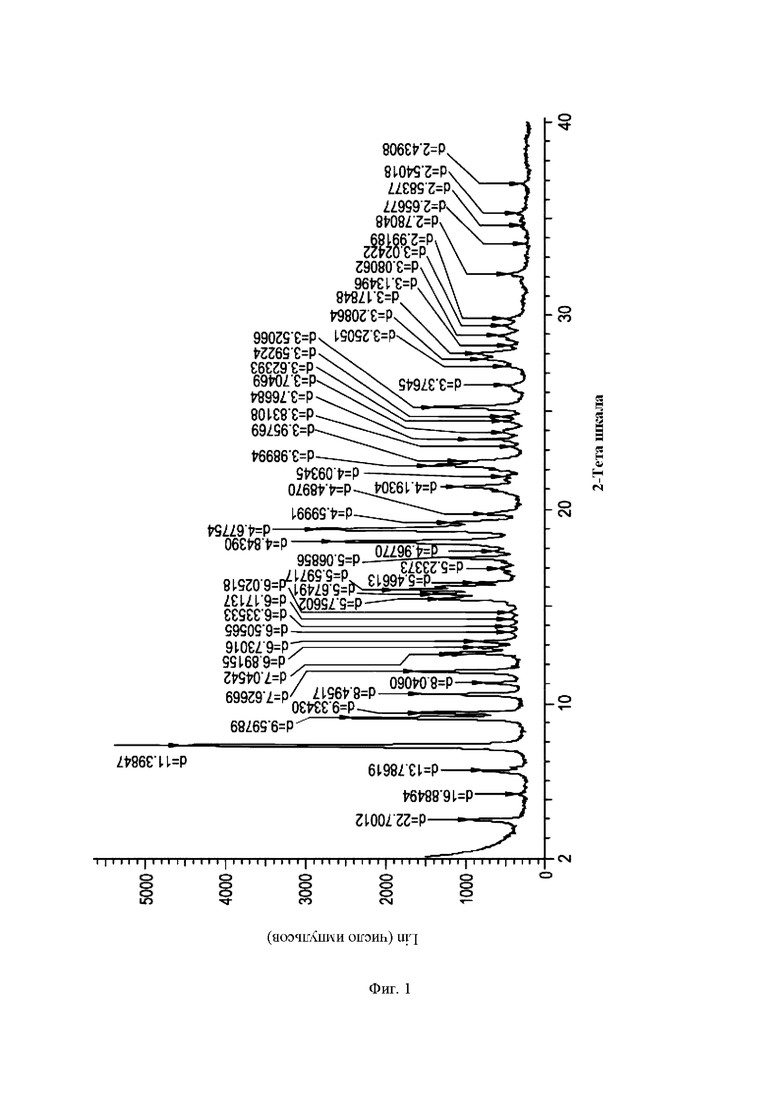
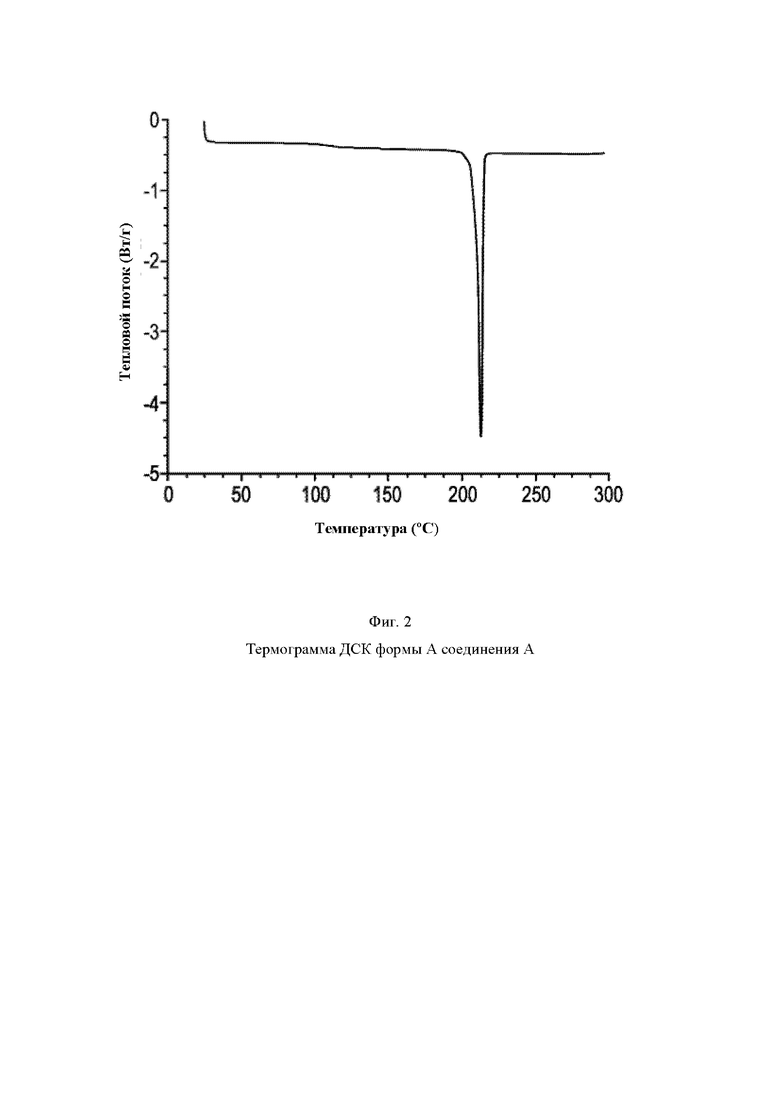
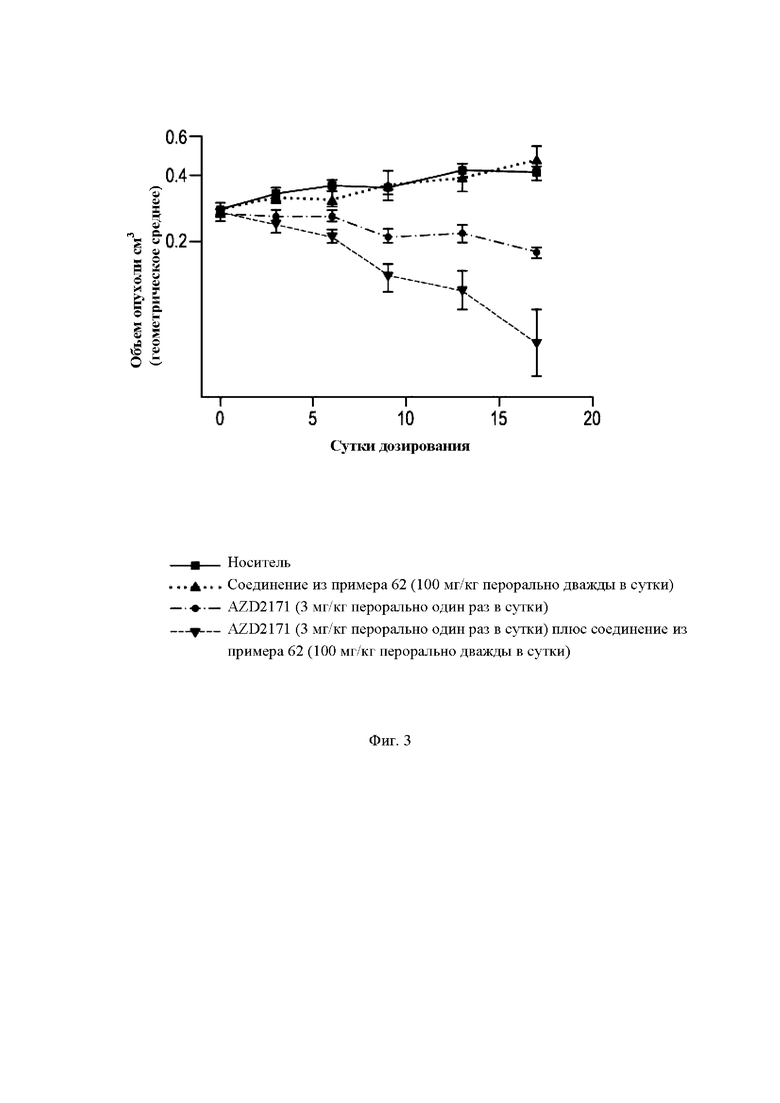
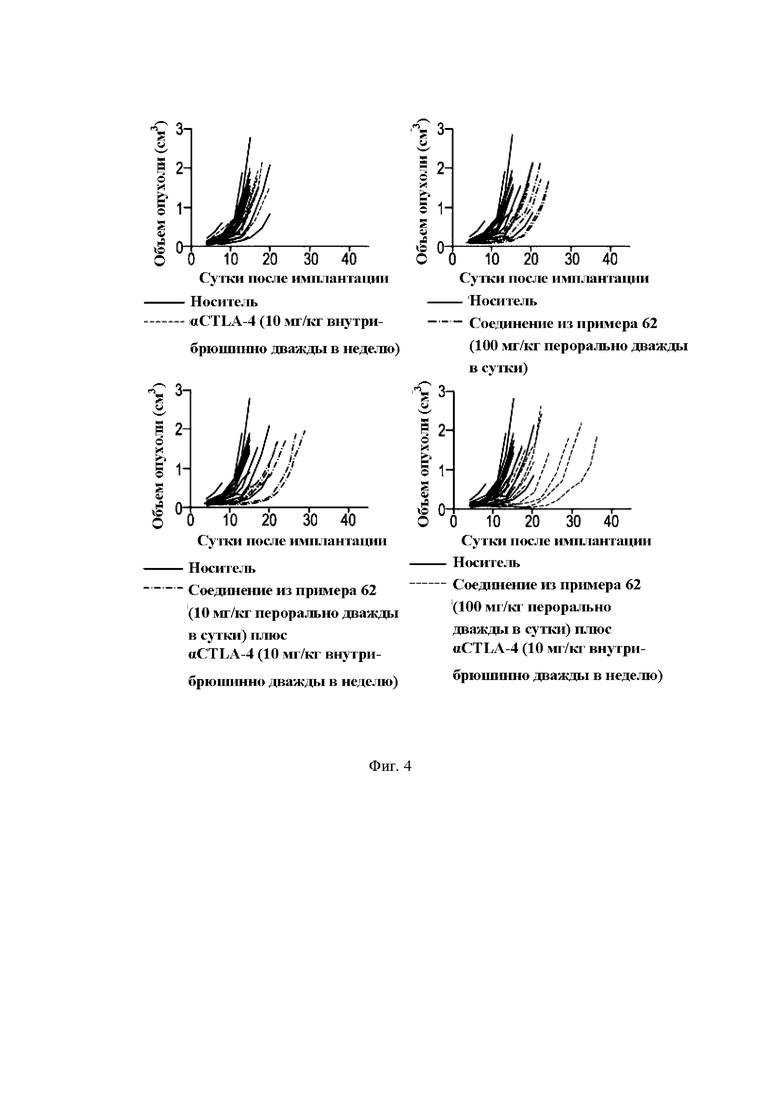
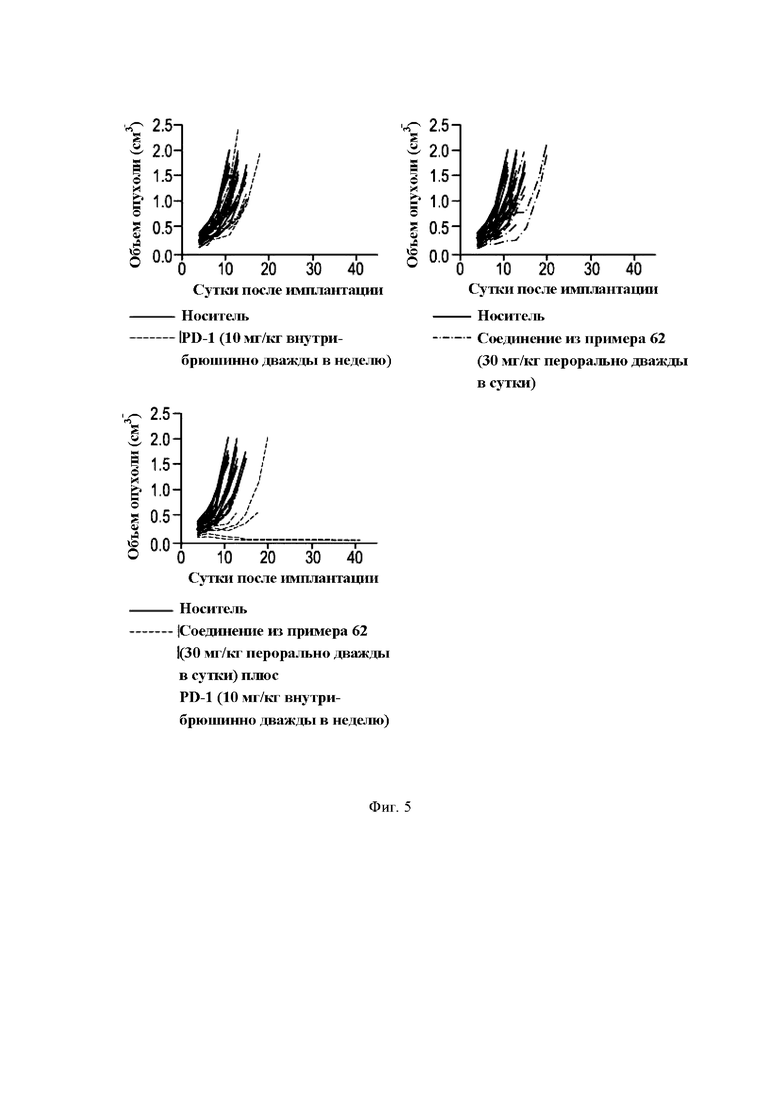
Изобретение относится к соединениям формулы (I) и их фармацевтически приемлемым солям, селективно ингибирующим MCT4 (транспортер монокарбоксилата 4), фармацевтическим композициям на их основе, их применение для получения лекарственного средства и способу лечения рака, опосредованного MCT4. Технический результат: разработка соединений, которые селективно ингибируют МСТ4 (транспортер монокарбоксилата 4). В общей формуле (I) R1 и R2 каждый независимо представляет собой водород или метил; X представляет собой CH2 или O; каждое кольцо A и кольцо B независимо представляет собой кольцо, выбранное из фенила, пиридинила, пиразинила, пиримидинила и пиридазинила, где каждое кольцо А и кольцо В независимо возможно замещено одним или более чем одним заместителем, выбранным из C1-3алкила и C1-3алкокси; кольцо C представляет собой 5-9-членный моноциклический или бициклический насыщенный гетероциклоалкил, возможно содержащий один или более чем один дополнительный гетероатом, независимо выбранный из O, N и S, где кольцо С возможно замещено одним или более чем одним заместителем, выбранным из C1-3алкила, возможно замещенного метокси или гидроксилом; диоксо, C0-2алкил-C(O)N(Me)2, C(O)C1-2алкила и S(O)2C1-2алкила. 4 н. и 19 з.п. ф-лы, 5 ил., 3 табл., 87 пр.
1. Соединение формулы (I):
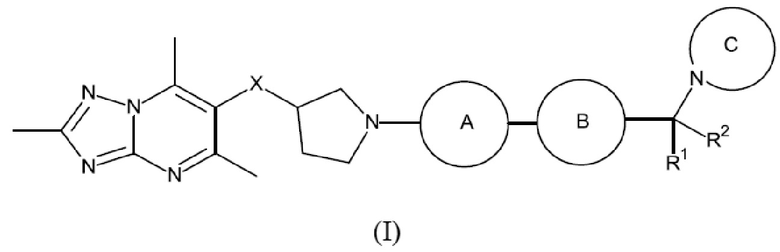
или его фармацевтически приемлемая соль, где:
R1 и R2 каждый независимо представляет собой водород или метил;
X представляет собой CH2 или O;
каждое кольцо A и кольцо B независимо представляет собой кольцо, выбранное из фенила, пиридинила, пиразинила, пиримидинила и пиридазинила, где каждое кольцо А и кольцо В независимо возможно замещено одним или более чем одним заместителем, выбранным из C1-3алкила и C1-3алкокси;
кольцо C представляет собой 5-9-членный моноциклический или бициклический насыщенный гетероциклоалкил, возможно содержащий один или более чем один дополнительный гетероатом, независимо выбранный из O, N и S, где кольцо С возможно замещено одним или более чем одним заместителем, выбранным из C1-3алкила, возможно замещенного метокси или гидроксилом; диоксо, C0-2алкил-C(O)N(Me)2, C(O)C1-2алкила и S(O)2C1-2алкила.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где X представляет собой CH2.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где X представляет собой O.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-3, где R1 и R2 оба представляют собой водород.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-4, где кольцо C выбрано из морфолинила, пиперазинила, пиперидинила, тиоморфолинила, диазабициклооктанила, октагидропирроло[1,2-a]пиразинила, пирролидинила, диазепанила, оксазепанила и азепанила.
6. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-5, где каждое кольцо A и кольцо B независимо возможно замещено одним или более чем одним заместителем, выбранным из метила и метокси.
7. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-6, где кольцо С возможно замещено одним или более чем одним заместителем, выбранным из метила, возможно замещенного гидроксилом; этила, возможно замещенного метокси или гидроксилом; диоксо, C(O)N(Me)2, CH2C(O)N(Me)2, C(O)Me и S(O)2Me.
8. Соединение формулы (I) по любому из пп. 1-7, где соединение формулы (I) представляет собой соединение формулы (Ia):
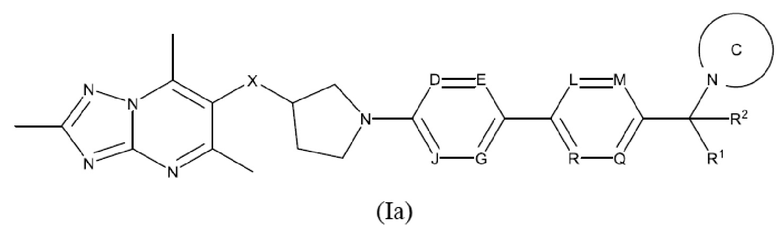
или его фармацевтически приемлемую соль, где X, R1, R2 и кольцо C являются такими, как определено в любом из пп. 1-5 или 7, и где каждый D, E, G, J, L, M, Q и R независимо представляет собой N или CR3, где не более двух из D, E, G и J представляют собой N и где не более двух из L, M, Q и R представляют собой N, и R3 представляет собой водород, C1-3алкил или C1-3алкокси.
9. Соединение формулы (I) по любому из пп. 1-7, где соединение формулы (I) представляет собой соединение формулы (Ib):
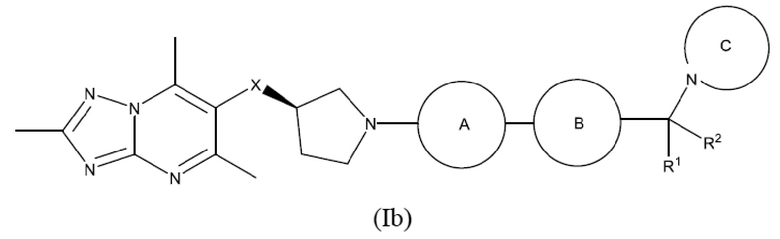
или его фармацевтически приемлемую соль, где R1, R2, X, кольцо A, кольцо B и кольцо C являются такими, как определено в любом из пп. 1-7.
10. Соединение формулы (I) по п. 8, где соединение формулы (I) представляет собой соединение формулы (Ic):
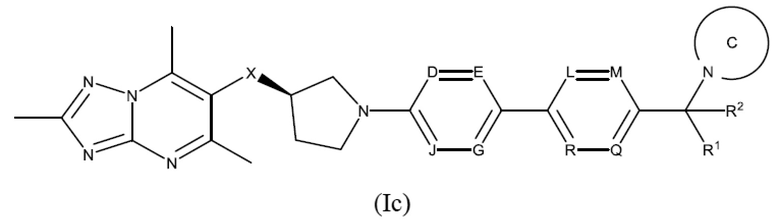
или его фармацевтически приемлемую соль, где R1, R2, X, D, E, G, J, L, M, Q, R, кольцо A, кольцо B и кольцо C являются такими, как определено в п. 8.
11. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где соединение выбрано из группы, состоящей из:
(R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)-[3,3'-бипиридин]-6-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(6'-((4-метилпиперазин-1-ил)метил)-[3,3'-бипиридин]-6- ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиридин-2- ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-(4-(2-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6- ил)метил)пирролидин-1-ил)пиримидин-5-ил)бензил)морфолина;
(R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6- ил)метил)пирролидин-1-ил)пиридин-2-ил)пиразин-2-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(6-(4-((4-метилпиперазин-1-ил)метил)фенил)пиридин-3- ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((6-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6- ил)метил)пирролидин-1-ил)пиридин-2-ил)пиридазин-3-ил)метил)морфолина;
6-(((R)-1-(2-(4-(((S)-2,4-диметилпиперазин-1-ил)метил)фенил)пиримидин-5- ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6- ил)метил)пирролидин-1-ил)пиримидин-2-ил)пиридин-2-ил)метил)морфолина;
(R)-6-((1-(2-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5- ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(2-(2-метил-4-((4-метилпиперазин-1- ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5- a]пиримидина;
6-(((R)-1-(2-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-N,N-диметил-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)ацетамида;
(R)-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)этанола;
(R)-1-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)пиперазин-1-ил)этенона;
(R)-6-((1-(2-(4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиримидин-2-ил)бензил)морфолина;
(R)-2,5,7-триметил-6-((1-(2-(4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиразин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(4-(2-((4-метилпиперазин-1-ил)метил)пиримидин-5-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридин-3-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(4-(6-((4-метилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиримидин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиразин-2-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(4-(5-((4-метилпиперазин-1-ил)метил)пиридин-2-ил)фенил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиримидин-2-ил)метил)морфолина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(R)-6-((1-(5-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)-6-метилпиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
2,5,7-триметил-6-(((R)-1-(5-(4-(((3R,5S)-3,4,5-триметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
6-(((R)-1-(5-(4-(((R)-3,4-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
6-(((R)-1-(5-(4-(((R)-2,4-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
2,5,7-триметил-6-(((R)-1-(5-(4-(((2R,5R)-2,4,5-триметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
2,5,7-триметил-6-(((R)-1-(5-(4-(((2S,5R)-2,4,5-триметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
2,5,7-триметил-6-[[(3R)-1-[5-[4-(1-пиперидилметил)фенил]пиразин-2-ил]пирролидин-3-ил]метил]-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-6-((1-(5-(4-((4-этилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(5-((4-метилпиперазин-1-ил)метил)пиридин-2-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
6-(((R)-1-(5-(4-(((3R,5S)-3,5-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
2-{4-[4-(5-{(3R)-3-[(2,5,7-триметил[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил]пирролидин-1-ил}пиразин-2-ил)бензил]пиперазин-1-ил}этанола;
(R)-6-((1-(5-(4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-6-((1-(5-(2-метокси-4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
{1-[4-(5-{(3R)-3-[(2,5,7-триметил[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил]пирролидин-1-ил}пиразин-2-ил)бензил]пиперидин-4-ил}метанола;
6-{[(3R)-1-(5-{4-[(1,1-диоксидотиоморфолин-4-ил)метил]фенил}пиразин-2-ил)пирролидин-3-ил]метил}-2,5,7-триметил[1,2,4]триазоло[1,5-a]пиримидина;
2,5,7-триметил-6-({(3R)-1-[5-(4-{[4-(метилсульфонил)пиперидин-1-ил]метил}фенил)пиразин-2-ил]пирролидин-3-ил}метил)[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((6-(3-метил-5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)пиридин-3-ил)метил)морфолина;
6-(((R)-1-(5-(4-(((S)-3,4-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((5-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)пиридин-2-ил)метил)морфолина;
6-(((R)-1-(5-(4-(((S)-2,4-диметилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-(4-(3-метил-5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)морфолина;
2,5,7-триметил-6-({(3R)-1-[5-(4-{[4-(метилсульфонил)пиперазин-1-ил]метил}фенил)пиразин-2-ил]пирролидин-3-ил}метил)[1,2,4]триазоло[1,5-a]пиримидина;
(R)-N,N-диметил-2-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)пиперазин-1-ил)ацетамида;
(R)-2,5,7-триметил-6-((1-(6-метил-5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
N,N-диметил-4-[4-(5-{(3R)-3-[(2,5,7-триметил[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил]пирролидин-1-ил}пиразин-2-ил)бензил]пиперазин-1-карбоксамида;
(R)-2,5,7-триметил-6-((1-(5-(3-метил-4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-1-(4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)пиперазин-1-ил)этенона;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиразин-2-ил)бензил)морфолина;
(R)-4-(4-(6-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)пиридазин-3-ил)бензил)морфолина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(4-(6-(пиперидин-1-илметил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(4-(6-((4-метилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-a]пиримидина;
2,5,7-триметил-6-(((3R)-1-(4-(6-((3-метил-3,8-диазабицикло[3.2.1]октан-8-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-a]пиримидина;
6-(((R)-1-(4-(6-(((R)-гексагидропирроло[1,2-a]пиразин-2(1H)-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
6-(((R)-1-(4-(6-(((S)-гексагидропирроло[1,2-a]пиразин-2(1H)-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиримидин-2-ил)метил)морфолина;
(R)-4-((5-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиразин-2-ил)метил)морфолина;
(R)-1-(4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)пиперазин-1-ил)этан-1-она;
(R)-6-((1-(4-(6-((4-этилпиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-6-((1-(4-(6-((4-(2-метоксиэтил)пиперазин-1-ил)метил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)-[2,3'-бипиридин]-5-ил)метил)морфолина;
(R)-4-((6'-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)-[3,3'-бипиридин]-6-ил)метил)морфолина;
(R)-2,5,7-триметил-6-((1-(2-(4-((4-метилпиперазин-1-ил)метил)фенил)пиримидин-5-ил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)окси)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-(4-(5-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)пиразин-2-ил)бензил)морфолина;
1-[4-[[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил]пирролидин-1-ил]пиразин-2-ил]фенил]метил]-1,4-диазепан-1-ил]этенона;
(R)-2,5,7-триметил-6-((1-(5-(4-(пирролидин-1-илметил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(5-(пирролидин-1-илметил)пиридин-2-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(6-(пирролидин-1-илметил)пиридин-3-ил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метил-1,4-диазепан-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)-1,4-оксазепана;
(R)-6-((1-(4-(6-(азепан-1-илметил)пиридазин-3-ил)фенил)пирролидин-3-ил)окси)-2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидина;
4-[(1R)-1-[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси]пирролидин-1-ил]пиразин-2-ил]фенил]этил]морфолина; и
4-[(1S)-1-[4-[5-[(3R)-3-[(2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси]пирролидин-1-ил]пиразин-2-ил]фенил]этил]морфолина.
12. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где соединение выбрано из:
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина;
(R)-1-(4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)пиперазин-1-ил)этан-1-она;
(R)-2,5,7-триметил-6-((1-(5-(4-((4-метилпиперазин-1-ил)метил)фенил)пиразин-2-ил)пирролидин-3-ил)метил)-[1,2,4]триазоло[1,5-a]пиримидина;
(R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)метил)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина; и
(S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолина.
13. Соединение формулы (I) по любому из пп. 1-12, где соединение представляет собой (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)-пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин или его фармацевтически приемлемую соль.
14. Соединение формулы (I) по любому из пп. 1-12, где соединение представляет собой (S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)-пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин или его фармацевтически приемлемую соль.
15. Соединение формулы (I) по любому из пп. 1-12, где соединение представляет собой (R)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)-пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин.
16. Соединение формулы (I) по любому из пп. 1-12, где соединение представляет собой (S)-4-((6-(4-(3-((2,5,7-триметил-[1,2,4]триазоло[1,5-a]пиримидин-6-ил)окси)-пирролидин-1-ил)фенил)пиридазин-3-ил)метил)морфолин.
17. Соединение формулы (I) по п. 15, где соединение находится в кристаллической форме с картиной дифракции рентгеновских лучей на порошке (ДРЛП) по существу такой, как показано на Фиг. 1, установленной при использовании CuKα-излучения.
18. Фармацевтическая композиция, селективно ингибирующая MCT4 (транспортер монокарбоксилата 4), которая содержит эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-17 и по меньшей мере один фармацевтически приемлемый эксципиент.
19. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-17 для применения в качестве ингибитора MCT4.
20. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-17 для применения в лечении рака, опосредованного MCT4.
21. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-17 для получения лекарственного средства для лечения рака, опосредованного MCT4.
22. Способ лечения рака, опосредованного MCT4, у теплокровного животного, нуждающегося в таком лечении, включающий введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-17.
23. Соединение для применения, применение или способ по любому из пп. 20-22, где рак представляет собой немелкоклеточный рак легкого, например аденокарциному легкого.
| WO 2017196936 A1, 16.11.2017 | |||
| US 20180008605 A1, 11.01.2018 | |||
| US 20040072746 A1, 15.04.2004 | |||
| ВИНТОВОЙ РЕЖУЩИЙ АППАРАТ ДЛЯ ЖАТВЕННЫХ МАШИН | 1924 |
|
SU3399A1 |

