ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка испрашивает приоритет согласно предварительной заявке на патент США No. 62/644987, поданной 19 марта, 2018, предварительной заявке на патент США No. 62/676057, поданной 24 мая, 2018, предварительной заявке на патент США No. 62/725488, поданной 31 августа, 2018, и предварительной заявке на патент США No. 62/812806, поданной 1 марта, 2019, содержание которых полностью включено в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к соединениям оксадиазолов, их получению, содержащим их фармацевтическим композициям и к их применению в качестве антагонистов катионных каналов, действующих по механизму транзиторного рецепторного потенциала (англ. TRP, Transient Receptor Potential).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Каналы TRP представляют собой класс ионных каналов, расположенных в цитоплазматической мембране различных типов клеток человека (и других животных). Существует по меньшей мере 28 известных каналов TRP человека, которые подразделяются на множество семейств или групп, исходя из гомологии и функции последовательностей. Катионный канал с транзиторным рецепторным потенциалом, подсемейство А, член 1 (TRPA1), представляет собой неселективный катионный проводящий канал, модулирующий мембранный потенциал через поток натрия, калия и кальция. Было показано, что экспрессия TRPA1 в большей степени выражена в нейронах ганглия задних корешков спинного мозга и периферических сенсорных нервах человека. У человека TRPA1 активируется рядом реакционноспособных соединений, таких как акролеин, аллилизотиоцианат, озон, и нереакционноспособных соединений, таких как никотин и ментол, и в связи с этим полагают, что он выполняет функцию химического датчика (хемосенсора).
Многие известные агонисты TRPA1 обладают раздражающим действием и вызывают боль, раздражение и нейрогенное воспаление у человека и других животных. Вследствие этого можно было бы ожидать, что антагонисты TRPA1 или агенты, блокирующие биологическое действие активаторов TRPA1, можно использовать при лечении таких заболеваний, как астма и ее обострения, хронический кашель и связанные с ним заболевания, а также при лечении острой и хронической боли. Кроме того, недавно было показано, что продукты повреждения тканей и окислительного стресса (например, 4-гидроксиноненаль и родственные соединения) активируют канал TRPA1. Это открытие дает дополнительное обоснование целесообразности использования низкомолекулярных антагонистов TRPA1 при лечении заболеваний, связанных с повреждением тканей, окислительным стрессом и сокращением гладкой мускулатуры бронхов, таких как астма, хроническое обструктивное заболевание легких (ХОЗЛ, англ. COPD), профессиональная бронхиальная астма и вирусное воспаление легких. Более того, недавно полученные данные скоррелировали активацию каналов TRPA1 с повышенным восприятием боли (Kosugi et al., J. Neurosci. 27, (2007) 4443-4451; Kremayer et al., Neuron 66 (2010) 671-680; Wei et al., Боль 152 (2011) 582-591); Wei et al., Neurosci. Lett. 479 (2010) 253-256)), что обеспечивает еще одно обоснование целесообразности использования низкомолекулярных ингибиторов TRPA1 при лечении болевых расстройств.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Согласно некоторым вариантам осуществления, предложено соединение формулы (I), его стереоизомеры, таутомеры и соли:
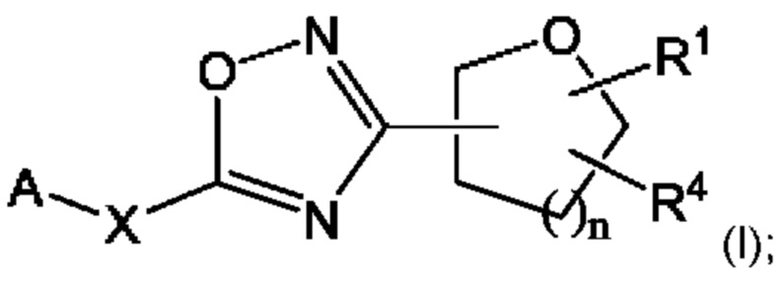
где:
А представляет собой замещенный или незамещенный 6-6 конденсированный бициклический гетероарил; замещенный или незамещенный 5-6 конденсированный бициклический гетероарил; или замещенный и незамещенный 6-5 конденсированный бициклический гетероарил;
X представляет собой связь; С1-4алкилен; -О-; -S-; -SO2-; или -N(Ra)-;
n равно 0, 1, 2 или 3;
Ra представляет собой Н или С1-6алкил, который может быть незамещенным или замещенным одним или более атомами галогена;
R1 представляет собой Н; или С1-6алкил; и
R4 представляет собой замещенный или незамещенный фенил; замещенный или незамещенный гетероарил; или замещенный или незамещенный нафтил;
или R1 и R4 могут вместе образовывать незамещенный или замещенный С3-6циклоалкил, сконденсированный с замещенным или незамещенным фенилом; замещенным или незамещенным гетероарилом; или замещенным или незамещенным нафтилом.
Согласно другим вариантам осуществления, предложены следующие соединения, их стереоизомеры и фармацевтически приемлемые соли.
Некоторые другие варианты осуществления предлагают фармацевтические композиции, содержащие описанное выше соединение или его фармацевтически приемлемую соль, и фармацевтически приемлемые носитель, разбавитель или вспомогательное вещество.
Некоторые другие варианты осуществления предлагают соединение, такое как описано выше, или его фармацевтически приемлемую соль для применения в лекарственной терапии.
Некоторые другие варианты осуществления предлагают соединение, такое как описано выше, или его фармацевтически приемлемую соль для лечения или профилактики респираторного расстройства.
Некоторые другие варианты осуществления предлагают соединение, такое как описано выше, или его фармацевтически приемлемую соль для получения лекарственного средства для лечения или профилактики респираторного расстройства.
Некоторые другие варианты осуществления предлагают способ лечения респираторного расстройства у млекопитающих, включающий введение млекопитающему терапевтически эффективного количества соединения, такого как описано выше, или его фармацевтически приемлемой соли.
Некоторые другие варианты осуществления предлагают соединение, такое как описано выше, или его фармацевтически приемлемую соль для модулирования активности TRPA1.
Некоторые другие варианты осуществления предлагают соединение, такое как описано выше, или его фармацевтически приемлемую соль для лечения или профилактики заболевания или состояния, опосредованного активностью TRPA1.
Некоторые другие варианты осуществления предлагают применение соединения, такого как описано выше, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики заболевания или состояния, опосредованного активностью TRPA1.
Некоторые другие варианты осуществления предлагают способ лечения заболевания или состояния, опосредованного активностью TRPA1, у млекопитающих, включающий введение терапевтически эффективного количества соединения, такого как описано выше, или его фармацевтически приемлемой соли млекопитающему.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Если не указано иное, следующие конкретные термины и фразы, используемые в разделах Описание и Формула изобретения, опеределяются следующим образом.
Термины "фрагмент" и "заместитель" относятся к атому или группе химически связанных атомов, которые присоединены к другому атому или молекуле при помощи одной или более химических связей, образуя посредством этого часть молекулы.
Термин "замещенный" относится к замене по меньшей мере одного атома водорода соединения или фрагмента другим заместителем или фрагментом. Примеры таких заместителей включают, не ограничиваясь перечнем, галоген, -ОН, -CN, оксо, алкокси, алкил, алкилен, арил, гетероарил, галогеналкил, галогеналкокси, циклоалкил и гетероцикл. Например, термин "алкил, замещенный галогеном" относится к случаю, когда один или более атомов водорода алкила (как определен ниже) замещены одним или более атомами галогена (например, трифторметил, дифторметил, фторметил, хлорметил и т.д.).
Термин "алкил" относится к алифатическому насыщенному углеводородному фрагменту с прямой или разветвленной цепью, содержащему от 1 до 20 атомов углерода. Согласно частным вариантам осуществления, алкил содержит от 1 до 10 атомов углерода. Согласно частным вариантам осуществления, алкил содержит от 1 до 6 атомов углерода. Алкильные группы необязательно могут быть независимо замещены одним или более заместителями, описанными в контексте данного изобретения.
Термин "алкилен" при использовании в контексте данного изобретения относится к линейному или разветвленному насыщенному двухвалентному углеводородному радикалу, содержащему от одного до двенадцати атомов углерода, и согласно другому варианту осуществления, от одного до шести атомов углерода, где алкиленовый радикал может быть необязательно независимо замещен одним или более заместителями, описанными в данном контексте. Примеры включают, не ограничиваясь перечнем, метилен, этилен, пропилен, 2-метилпропилен, пентилен и т.п.
Термин "алкенилен" относится к линейному или имеющему разветвленную цепь двухвалентному углеводородному радикалу, содержащему от двух до восьми атомов углерода (С2-8) с по меньшей мере одним центром ненасыщенности, т.е. двойной углерод-углеродной связью, где алкениленовый радикал может быть необязательно замещенным. Примеры включают, не ограничиваясь перечнем, этиленилен или винилен (-СН=СН-), аллил (-СН2СН=СН-) и т.п.
Термин "алкокси" означает группу формулы -O-R', где R' является алкильной группой. Алкоксильные группы могут быть необязательно независимо замещены одним или более заместителями, описанными в данном контексте. Примеры алкоксильных фрагментов включают метокси, этокси, изопропокси и трет-бутокси.
"Арил" означает циклический ароматический углеводородный фрагмент, содержащий моно-, би- и трициклическое ароматическое кольцо, состоящее из от 5 до 16 атомов углерода в кольце. Бициклические арильные кольцевые системы включают конденсированные бициклические системы, содержащие два конденсированных пятичленных арильных кольца (обоначаемые как 5-5), содержащие пятичленное арильное кольцо и конденсированное шестичленное арильное кольцо (обозначаемые как 5-6 и 6-5), и содержащие два конденсированных шестичленных арильных кольца (обозначаемые как 6-6). Арильная группа может быть необязательно замещена, как указано в данном контексте. Примеры арильных фрагментов включают, не ограничиваясь перечнем, фенил, нафтил, фенантрил, флуоренил, инденил, пенталенил, азуленил и т.п. Термин "арил" также включает частично гидрированные производные циклического ароматического углеводородного фрагмента при условии, что по меньшей мере одно кольцо циклического ароматического углеводородного фрагмента является ароматическим, причем каждое из них необязательно замещено.
Термин "гетероарил" означает ароматическую гетероциклическую моно-, би или трициклическую кольцевую систему, содержащую от 5 до 16 кольцевых атомов, включающую 1, 2, 3 или 4 гетероатома, выбранных из N, О и S, при этом остальные атомы в кольце представляют собой углерод. Согласно некоторым аспектам, моноциклические гетероарильные кольца могут быть 5-6 членными. Бициклические гетероарильные кольцевые системы включают в себя конденсированные бициклические системы, содержащие два конденсированных пятичленных гетероарильных кольца (обозначаемые как 5-5), содержащие пятичленное гетероарильное кольцо и конденсированное шестичленное гетероарильное кольцо (обозначаемые как 5-6 и 6-5), и содержащие два конденсированных шестичленных гетероарильных кольца (обозначаемые как 6-6). Гетероарильная группа может быть необязательно замещена, как указано в данном контексте. Примеры гетероарильных фрагментов включают пирролил, фуранил, тиенил, имидазолил, оксазолил, тиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиразинил, пиразолил, пиридазинил, пиримидинил, триазинил, изоксазолил, бензофуранил, изотиазолил, бензотиенил, индолил, изоиндолил, изобензофуранил, бензимидазолил, бензоксазолил, бензоизоксазолил, бензотиазолил, бензоизотиазолил, бензоксадиазолил, бензотиадиазолил, бензотриазолил, пуринил, хинолинил, изохинолинил, хиназолинил или хиноксалинил.
Термины "гало", "галоген" и "галоид", которые могут употребляться как синонимы, относятся к таким заместителям, как фтор, хлор, бром или йод.
Термин "галогеналкил" означает алкильную группу, в которой один или более атомов водорода алкильной группы заменен одинаковыми или разными атомами галогенов, в частности, атомами фтора. Примеры галогеналкила включают монофтор-, дифтор- или трифторметил, -этил или -пропил, например, 3,3,3-трифторпропил, 2-фторэтил, 2,2,2-трифторэтил, фторметил, дифторметил или трифторметил.
Термин "гетероалкил" относится к прямой или разветвленной алкильной цепи, как указана в данном контексте, содержащей от 2 до 14 атомов углерода, от 2 до 10 атомов углерода или от 2 до 6 атомов углерода в цепи, один или более из которых заменен гетероатомом, выбранным из S, О, Р и N. Неограничивающие примеры гетероалкилов включают простые алкиловые эфиры, вторичные и третичные алкиламины, амиды и алкил сульфиды.
"Циклоалкил" одначает насыщенный или частично ненасыщенный карбоциклический фрагмент, содержащий моно-, би- (включая бициклические с внутренним мостиком) или трициклические кольца с от 3 до 10 атомами углерода в кольце. Циклоалкильный фрагмент может быть необязательно замещен одним или более заместителями. Согласно частным вариантам осуществления, циклоалкил содержит от 3 до 8 атомов углерода (т.е. (С3-С8)циклоалкил). Согласно другим частным вариантам осуществления, циклоалкил содержит от 3 до 6 атомов углерода (т.е. (С3-С6)циклоалкил). Примеры циклоалкильных фрагментов включают, не ограничиваясь перечнем, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, и их частично ненасыщенные (циклоалкенильные) производные (например, циклопентенил, циклогексенил и циклогептенил), бицикло[3,1,0]гексанил, бицикло[3,1,0]гексенил, бицикло[3,1,1]гептанил и бицикло[3,1,1]гептенил. Циклоалкильный фрагмент может быть присоединен "спироциклоалкильным" способом, как, например, "спироциклопропил": 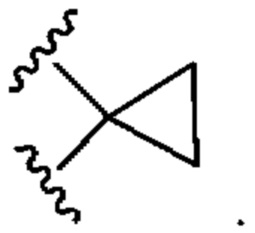
"Гетероцикл" или "гетероциклил" относится к 4-, 5-, 6- и 7-членному моноциклическому, 7-, 8-, 9- и 10-членному бициклическому (включая бициклический с внутренним мостиком) или 10-, 11-, 12-, 13-, 14- и 15-членному бициклическому гетероциклическому фрагменту, являющемуся насыщенным или частично ненасыщенным и содержащему один или более (например, 1, 2, 3 или 4) гетероатомов, выбранных из кислорода, азота и серы, в кольце, при этом остальные атомы в кольце представляют собой углерод. Согласно некоторым аспектам, гетероцикл является гетероциклоалкилом. Согласно частным вариантам осуществления, гетероцикл или гетероциклил относится к 4-, 5-, 6- или 7-членному гетероциклу. Являясь атомами кольца гетероцикла, азот или сера также могут иметь окисленную форму, азот при этом может быть замещен одной или более (С1-С6)алкильными группами. Гетероцикл может быть соединен со своей боковой группой у любого гетероатома или атома углерода, что приводит к стабильной структуре. Любой из кольцевых атомов гетероцикла может быть необязательно замещен одним или более заместителями, описанными в данном контексте. Примеры таких насыщенных или частично ненасыщенных гетероциклов включают, не ограничиваясь перечнем, тетрагидрофуранил, тетрагидротиенил, пирролидинил, пирролидонил, пиперидинил, пирролинил, тетрагидрохинолинил, тетрагидроизохинолинил, декагидрохинолинил, оксазолидинил, пиперазинил, диоксанил, диоксоланил, диазепинил, оксазепинил, тиазепинил, морфолинил и хинуклидинил. Термин «гетероцикл» также включает группы, в которых гетероцикл сконденсирован с одним или более арильными, гетероарильными или циклоалкильными кольцами, такие как индолинил, 3Н-индолил, хроманил, азабицикло[2,2,1]гептанил, азабицикло[3,1,0]гексанил, азабицикло[3,1,1]гептанил, октагидроиндолил или тетрагидрохинолинил.
Термин "конденсированный бициклический" относится к кольцевой системе, содержащей два конденсированных кольца, включая циклоалкил и гетероциклоалкил с внутренним мостиком, как определены в другом месте данного документа. Каждое из колец независимо представляет собой арил, гетероарил, циклоалкил и гетероцикл. Согласно некоторым аспектам, каждое из колец независимо представляет собой С5-6арил, 5-6-членный гетероарил, С3-6циклоалкил и 4-6-членный гетероцикл. Неограничивающие примеры конденсированных бициклических кольцевых систем включают С5-6арил-С5-6арил, С5-6арил-4-6-членный гетероарил и С5-6арил-С5-6циклоалкил.
Термин "конденсированная трициклическая" означает кольцевую систему, содержащую три конденсированных кольца. Каждое из колец независимо представляет собой арил, гетероарил, циклоалкил и гетероцикл. Согласно некоторым аспектам, каждое из колец независимо представляет собой С5-6арил, 5-6-членный гетероарил, С3-6циклоалкил, и 4-6-членый гетероцикл. Неограничивающим примером конденсированной трициклической кольцевой системы является C3-6циклоалкил-С3-6циклоалкил-С5-6арил, например, С3-циклоалкил-С5-циклоалкил-С6-арил.
Если не указано иное, термин "водород" или "гидро" относится к фрагменту, являющемуся атомом водорода (-Н), а не к молекуле Н2.
При описании в данном контексте, если имеется расхождение между изображенной структурой и названием, данным этой структуре, изображенная структура является определяющей. Кроме того, если стереохимия структуры или части структуры не обозначена, например, жирными клиновидными или пунктирными линиями, структуру или часть структуры следует интерпретировать, как охватывающую все ее стереоизомеры. Однако в некоторых случаях, при наличии более одного хирального центра, для облегчения описания относительной стереохимии структура и название могут быть представлены в виде отдельных энантиомеров.
Если не указано иное, термин "соединение конкретной формулы" или "соединение формулы", или "соединения конкретной формулы", или "соединения формулы" относится к любому соединению, выбранному из такого рода соединений, как те, что определены формулой (включая любую фармацевтически приемлемую соль или сложный эфир любого такого соединения, если не указано иное).
Термин "фармацевтически приемлемые соли" относится к солям, сохраняющим биологическую эффективность и свойства свободных оснований или свободных кислот и не являющимся нежелательными с биологической или иной точек зрения. При использовании в данном контексте, "фармацевтически приемлемый" относится к носителю, разбавителю или вспомогательному веществу, которые совместимы с другими ингредиентами композиции и не наносят вреда их реципиенту. Соли могут быть образованы с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., предпочтительно, с соляной кислотой, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, салициловая кислота, сукциновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфокислота, N-ацетилцистеин и т.п. Кроме того, соли могут быть получены добавлением неорганического или органического основания к свободной кислоте. Соли, полученные из неорганического основания, включают, не ограничиваясь перечнем, соли натрия, калия, лития, аммония, кальция и магния и т.п. Соли, полученные из органических оснований, включают, не ограничиваясь перечнем, соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как смолы на основе изопропиламина, триметиламина, диэтиламина, триэтиламина, трипропиламина, этаноламина, лизина, аргинина, N-этилпиперидина, пиперидина, полиаминовые смолы и т.п.
Соединения по настоящему изобретению могут быть представлены в форме фармацевтически приемлемых солей. Другой вариант осуществления предлагает фармацевтически неприемлемые соли соединения формулы I, которые могут использоваться в качестве промежуточных продуктов для выделения или очистки соединения формулы I. Соединения по настоящему изобретению также могут присутствовать в форме фармацевтически приемлемых сложных эфиров (т.е. метиловых и этиловых сложных эфиров кислот формулы I, используемых в качестве пролекарств). Соединения по настоящему изобретению также могут быть сольватированы, т.е. гидратированы. Сольватация может происходить во время производственного процесса или может протекать, например, вследствие гигроскопических свойств изначально безводного соединения формулы I.
Соединения, имеющие одинаковую молекулярную формулу, но различающиеся характером или расположением атомов молекулы в пространстве, называют "изомерами". Изомеры, различающиеся расположением атомов в пространстве, называются "стереоизомерами". Диастереомеры представляют собой стереоизомеры с противоположной конфигурацией при одном или более хиральных центрах, не являющиеся энантиомерами. Стереоизомеры, несущие один или более асимметрических центров и являющиеся несовпадающим при наложении зеркальным отображением друг друга, называются "энантиомерами". Если соединение имеет несколько асимметрических центров, например, если атом углерода связан с четырьмя разными группами, может существовать пара энантимеров. Энантиомер может быть охарактеризован абсолютной конфигурацией его асимметрического центра и описан правилами R- и S-секвенирования Кана-Ингольда-Прелога или при помощи способа, которым молекула вращает плоскость поляризованного света, и, соответственно, обозначен как правовращающий или левовращающий (т.е. (+) или (-)) изомер. Хиральное соединение может существовать в виде отдельного энантиомера либо в виде смеси энантиомеров. Смесь, содержащую равные доли энантиомеров, называют "рацемической смесью". Согласно некоторым вариантам осуществления, соединение обогащено по меньшей мере приблизительно на 90 мас. % одним диастереомером или энантиомером. Согласно другим вариантам осуществления, соединение обогащено по меньшей мере приблизительно на 95%, 98% или 99 мас. % одним диастереомером или энантиомером.
Некоторые соединения по настоящему изобретению содержат асимметрические атомы углерода (оптические центры) или двойные связи; рацематы, диастереомеры, региоизомеры и отдельные изомеры (например, отдельные энантиомеры) входят в объем настоящего изобретения.
Соединения по изобретению могут содержать асимметрические или хиральные центры, поэтому они существуют в различных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь перечнем, диастереомеры, энантиомеры и атропизомеры, а также их смеси, такие как рацемические смеси, входят в состав настоящего изобретения. В некоторых случаях стереохимия не определена или обозначена ориентироочно. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость поляризованного света. При описании оптически активного соединения для обозначения абсолютной конфигурации молекулы в области ее хирального центра (центров) используют префиксы D и L или R и S. Для обозначения направления вращения соединением плоскополяризованного света используют префиксы d и l или (+) и (-), при этом (-) или l означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Для данной химической структуры такие стереоизомеры являются идентичными, за исключением того, что они представляют собой зеркальное отображение друг друга. Конкретный стереоизомер также может называться энатиомером, а смесь таких изомеров часто называют энантиомерной смесью. Смесь, содержащая энантиомеры в соотношении 50:50, называется рацемической смесью или рацематом, она может образоваться, если во время химической реакции или процесса отсутствовала стереоселективность или стереоспецифичность. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомерных частиц, лишенной оптической активности. Энантиомеры могут быть выделены из рацемической смеси при помощи метода хирального разделения, такого как хроматография со сверхкритической подвижной фазой (англ. SFC, supercritical fluid chromatography). Присваивание конфигурации хиральным центрам в отдельных энантиомерах может быть ориентировочным и обозначаться в соединениях как (l), (m) и (n) в иллюстративных целях, окончательно же стереохимию устанавливают, например, на основании данных рентгеноструктурной кристаллографии.
Термин "терапевтически эффективное количество" соединения означает количество соединения, являющееся эффективным для предотвращения, облегчения или улучшения симптомов заболевания или увеличения продолжительности жизни субъекта, подвергаемого лечению. Определение терапевтически эффективного количества находится в компетенции специалиста в данной области техники. Терапевтически эффективное количество или дозировка соединения в соответствии с настоящим изобретением может варьироваться в широких пределах и может быть определена способом, известным в данной области. В каждом конкретном случае такая дозировка будет скорректирована в соответствии с индивидуальными потребностями, включая конкретное вводимое соединение (соединения), способ введения, состояние, подвергаемое лечению, а также конкретного пациента, проходящего лечение. Как правило, в случае перорального или парентерального введения взрослому человеку с массой тела приблизительно 70 кг суточная дозировка может составлять приблизительно от 0,1 мг до 5000 мг, от 1 мг до приблизительно 1000 мг или от 1 мг до 100 мг, хотя при наличии показаний верхний и нижний пределы могут быть превышены. Суточную дозировку можно вводить в виде разовой дозы либо раздельными дозами, или, при парентеральном введении, ее можно вводить в виде непрерывной инфузии.
Термин "фармацевтически приемлемый носитель" рассчитан на включение любого возможного материала, совместимого с фармацевтическим введением, включая растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические агенты и агенты, замедляющие абсорбцию, а также другие материалы и соединения, совместимые с фармацевтическим введением. Предполагается, что за исключением случаев, когда какая-либо обычная среда или агент являются несовместимыми с активным соединением, их можно использовать в композициях по изобретению. В композицию также могут быть включены дополнительные активные соединения.
Подходящие фармацевтические носители для приготовления композиций по настоящему изобретению могут быть твердыми, жидкими или газообразными; благодаря этому композиции могут иметь форму таблеток, пилюль, капсул, суппозиториев, порошков, составов, покрытых кишечнорастворимой оболочкой или иным образом защищенных (например, связывающихся с ионообменными смолами или упакованных в липидно-белковые везикулы), составов с замедленным высвобождением, растворов, суспензий, эликсиров, аэрозолей и т.п. Носитель может быть выбран из различных масел, включая масла минерального, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло, минеральное масло, кунжутное и т.п. Предпочтительными жидкими носителями, в частности, для инъекционных растворов (когда они изотоничны крови), являются вода, водные растворы декстрозы или гликоли. Например, препараты для внутривенного введения включают в себя стерильные водные растворы активного ингредиента (ингредиентов), приготавливаемые растворением твердого активного ингредиента (ингредиентов) в воде с получением водного раствора и приданием раствору стерильности. Подходящие фармацевтические вспомогательные вещества включают крахмал, целлюлозу, тальк, глюкозу, лактозу, тальк, желатин, солод, рис, муку, мел, диоксид кремния, стеарат магния, стеарат натрия, моностеарат глицерина, хлорид натрия, сухое обезжиренное молоко, глицерин, пропиленгликоль, воду, этанол и т.п. В состав композиций могут быть добавлены обычные фармацевтические добавки, такие как консерванты, стабилизирующие агенты, увлажняющие или эмульгирующие агенты, соли для регулирования осмотического давления, буферы и т.п. Примеры подходящих фармацевтических носителей и их состав описаны в «Remington's Pharmaceutical Sciences» под ред. Е.W. Martin. Такие композиции в любом случае будут содержать эффективное количество активного соединения вместе с подходящим носителем так, чтобы приготовить надлежащую дозированную лекарственную форму, подходящую для введения реципиенту.
При практическом применении способа по настоящему изобретению терапевтически эффективное количество любого из соединений по данному изобретению или комбинации любого из соединений по данному изобретению, или его фармацевтически приемлемой соли, или сложного эфира вводят любым обычным и приемлемым способом, известным в данной области, по отдельности или в комбинации. Таким образом, соединения или композиции можно вводить перорально (например, в щечную полость), подъязычно, парентерально (например, внутримышечно, внутривенно или подкожно), ректально (например, при помощи суппозиториев или промываний), трансдермально (например, электропорацией кожи), путем ингаляции (например, в виде аэрозоля) и в форме твердых, жидких или газообразных дозировок, включая таблетки и суспензии. Можно осуществлять введение в виде единичной дозированной лекарственной формы при непрерывной терапии или в виде терапии разовой дозой ad libitum (по потребности). Терапевтическая композиция также может иметь форму масляной эмульсии или дисперсии в сочетании с липофильной солью, такой как памовая кислота, или форму биоразлагаемой композиции с замедленным высвобождением для подкожного или внутримышечного введения.
Соединения
Согласно одному из вариантов осуществления настоящего изобретения, предложено соединение формулы I, его стереоизомеры, таутомеры и соли:

или его фармацевтически приемлемая соль,
где:
А представляет собой замещенный или незамещенный 6-6 конденсированный бициклический гетероарил, который может быть частично насыщенным; замещенный или незамещенный 5-6 конденсированный бициклический гетероарил, который может быть частично насыщенным; или замещенный и незамещенный 6-5 конденсированный бициклический гетероарил, который может быть частично насыщенным;
X представляет собой связь; С1-4алкилен; -О-; -S-; -SO2-; или -N(Ra)-;
n равно 0, 1, 2 или 3;
Ra представляет собой Н или С1-6алкил, который может быть незамещенным или замещенным одним или несколькими атомами галогена;
R1 представляет собой Н; или С1-6алкил; и
R4 представляет собой замещенный или незамещенный фенил; замещенный или незамещенный гетероарил; или замещенный или незамещенный нафтил;
или R1 и R4 могут вместе образовывать незамещенный или замещенный С3-6циклоалкил, сконденсированный с замещенным или незамещенным фенилом; замещенным или незамещенным гетероарилом; или замещенным или незамещенным нафтилом.
Согласно некоторым аспектам, n равно 0, 1 или 2. Согласно некоторым аспектам, n равно 0 или 1. Согласно некоторым аспектам, n равно 0. Согласно некоторым аспектам, n равно 1.
Согласно некоторым аспектам, А выбран из:
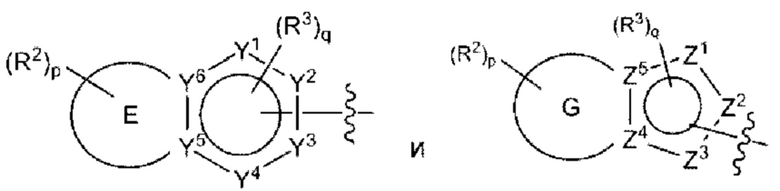
где:
Е представляет собой пятичленное или шестичленное гетероарильное кольцо, в котором один кольцевой атом углерода необязательно замещен оксогруппой;
G представляет собой шестичленное гетероарильное кольцо, содержащее один кольцевой атом углерода, замещенный оксогруппой;
от одного до трех из Y1, Y2, Y3, Y4, Y5 и Y6 представляют собой азот, а остальные Y1, Y2, Y3, Y4, Y5 и Y6 являются углеродом, при этом один из Y1, Y2, Y3 и Y4 может быть -С(O)- или -C(S)-;
один или два из Z1, Z2, Z3, Z4 и Z5 представляют собой азот, а остальные Z1, Z2, Z3, Z4 и Z5 являются углеродом;
каждый R2 независимо представляет собой Н, -С1-4алкил; -С1-4галогеналкил; -CN; галоген; галоген-С1-4алкокси; С1-6алкокси; -ОН; -SO2-С1-4алкил; -C1-4CN, С1-4альдегид; C1-4кетон; бензиламино; или NR14R15;
р равно 0, 1 или 2;
каждый R3 независимо представляет собой: Н; -С1-4алкил; -С1-4галогеналкил; -CN; галоген; или -NR14R15;
q равно 0 или 1;
каждый из R14 и R15 независимо друг от друга представляет собой: Н; замещенный или незамещенный -С1-4алкил; замещенный или незамещенный -С(O)-С1-4алкил; замещенный или незамещенный С3-6циклоалкил; замещенный или незамещенный от 3- до 6-членный гетероциклоалкил; замещенный или незамещенный от 3- до 6-членный -С1-4алкилгетероциклоалкил; замещенный или незамещенный -C1-4гетероалкил; -C(O)NR16R17; замещенный или незамещенный -C1-4алкил-C(O)NR16R17; замещенный или незамещенный фенил; или замещенный или незамещенный бензил;
или R14 и R15 вместе с атомами, к которым они присоединены, могут образовывать 4-, 5-, 6- или 7-членное кольцо, необязательно включающее один дополнительный гетероатом, выбранный из О, N и S; и
каждый из R16 и R17 независимо представляет собой Н и С1-4алкил.
Согласно некоторым аспектам, А представляет собой конденсированный гетероарильный фрагмент, выбранный из:

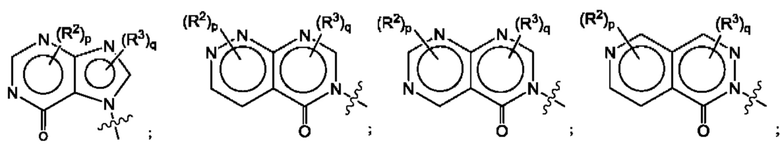
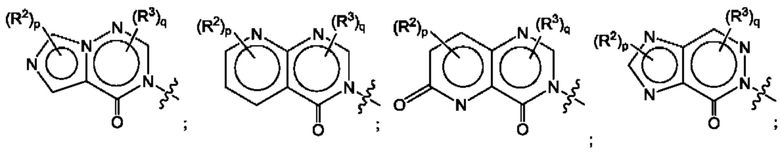

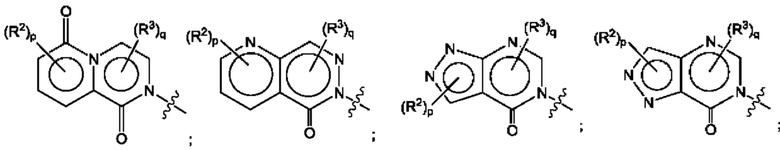
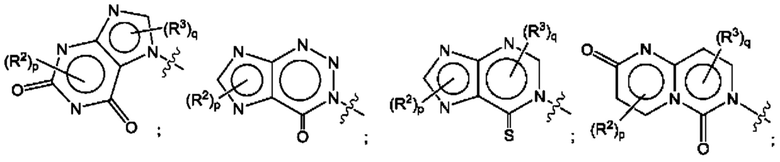
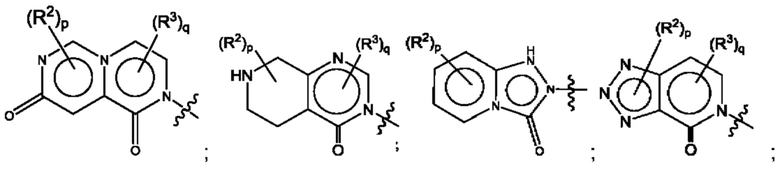


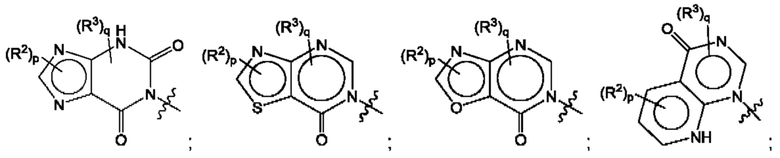
где:
каждый R2 независимо представляет собой: Н; D; -С1-4алкил; -С1-4галогеналкил; С1-4алкокси; -CN; галоген; -C(O)CH3; -C(O)NR16R17; -NH2; NHC1-4алкил, где С1-4алкил необязательно содержит атом кислорода или заместитель -ОН; -NHC(O)-С1-4алкил; -NHCH2C(O)М(С1-4алкил)2; бензиламино; и -NH-С4-6гетероцикл, содержащий гетероатом кислорода;
каждый R3 независимо представляет собой: Н; D: -С1-4алкил; -С1-4галогеналкил; -CN; галоген; или NR14R15;
р равно 0, 1 или 2; и
q равно 0 или 1.
Согласно некоторым аспектам, каждый R2 независимо выбран из Н, -D, -С1-4алкила, -С1-4галогеналкила, -CN, галогена, -C(O)CH3, -NH2, NHC1-4алкила, где С1-4алкил необязательно содержит атом кислорода или заместитель -ОН, -NHC(O)-С1-4алкила, -NHCH2C(O)N(С1-4алкил)2, -C(O)-NH2; и -NH-С4-6гетероцикла, содержащего гетероатом кислорода. Согласно некоторым аспектам, каждый R2 независимо выбран из Н, D, -СН3, -CN, -галогена, -NH2, -NHCH3, NHCH2CH3, -NHCH2CH2CH2OH, -NHCH2CH2CH3, -NHC(O)CH3, -NHCH2C(O)N(CH3)2,  a p равно 0 или 1. R3 выбран из H, -D, -С1-4алкила, -С1-4галогеналкила, -CN и галогена.
a p равно 0 или 1. R3 выбран из H, -D, -С1-4алкила, -С1-4галогеналкила, -CN и галогена.
Согласно некоторым аспектам, R3 выбран из Н, -D и -CN.
Согласно некоторым аспектам, каждый R2 независимо выбран из Н, -D, -С1-4алкила или -NH2.
Согласно некоторым аспектам, каждый R3 независимо выбран из Н, -D, -С1-4алкила или -NH2.
Согласно некоторым аспектам, каждый R2 независимо выбран из Н, -D или -С1-4алкила.
Согласно некоторым аспектам, каждый R3 независимо выбран из Н, -D или -С1-4алкила.
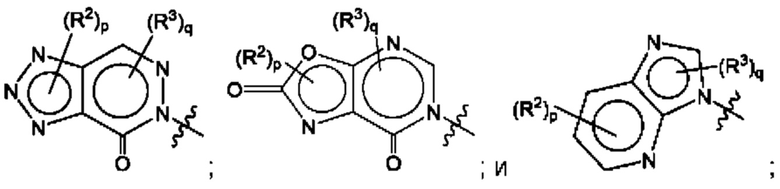
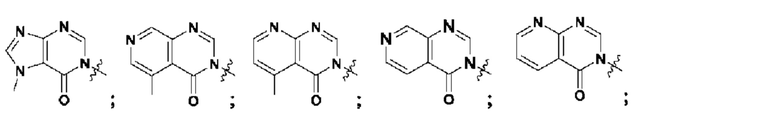
Согласно некоторым аспектам, А представляет собой конденсированный гетероарильный фрагмент, выбранный из:


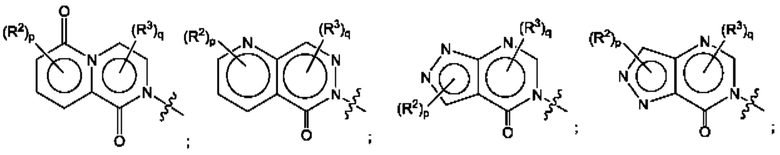
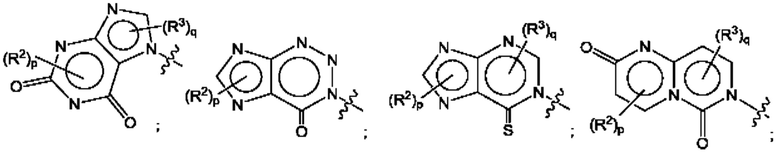
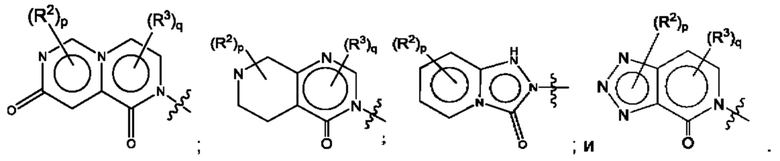
Согласно некоторым аспектам, А представляет собой конденсированный гетероарильный фрагмент, выбранный из:
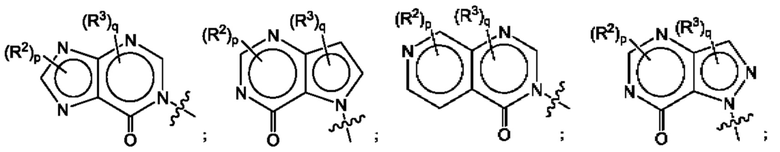
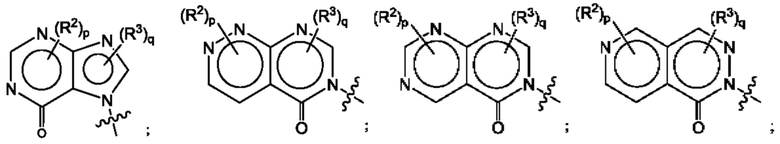
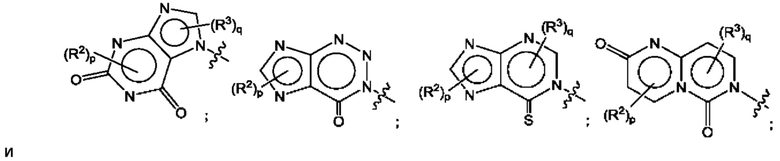
где R2, R3, р и q такие, как определены в другом месте данного документа.
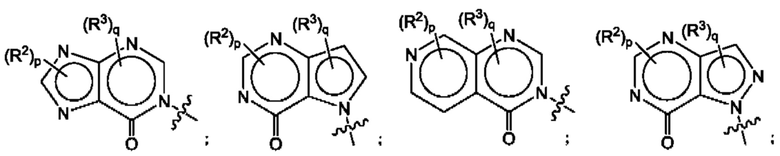
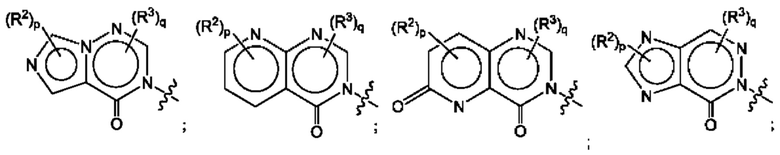
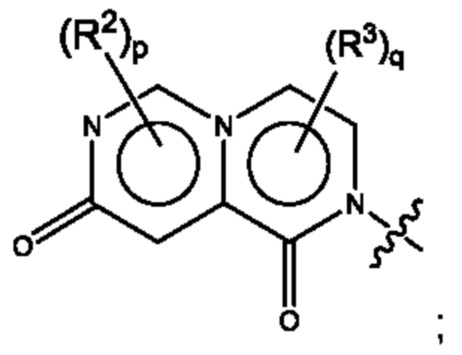
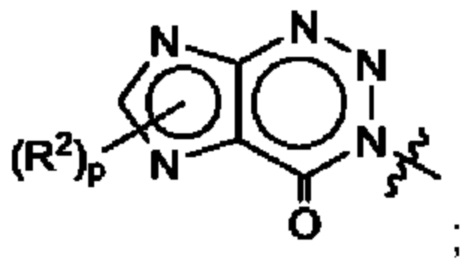
Согласно некоторым аспектам, А выбран из:
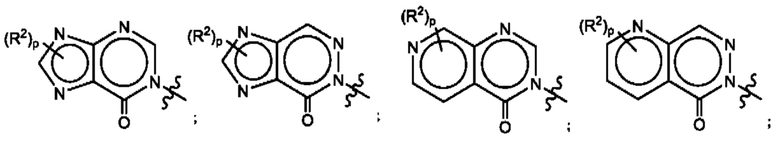

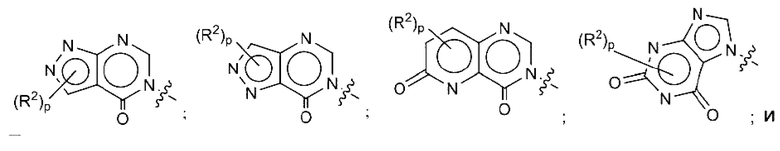
где R2, R3, р и q такие, как определены в другом месте данного документа.
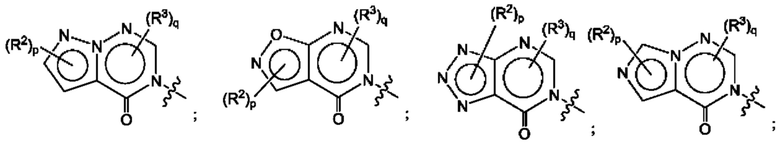
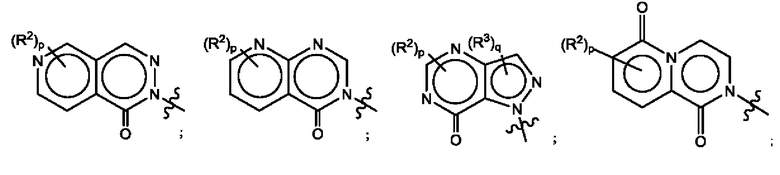
Согласно некоторым аспектам, А выбран из:
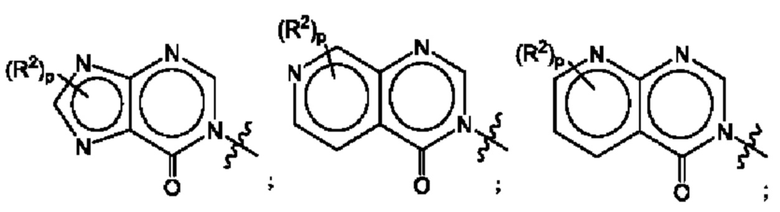
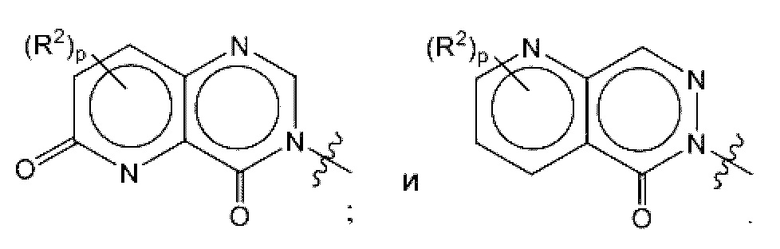
где R2, R3, р и q такие, как определены в другом месте данного документа.
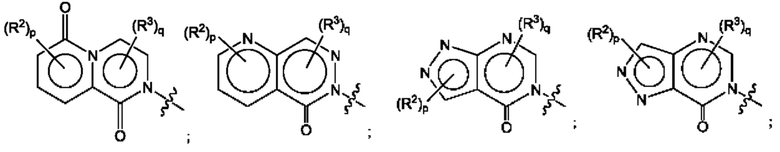
Согласно некоторым аспектам, А представляет собой:
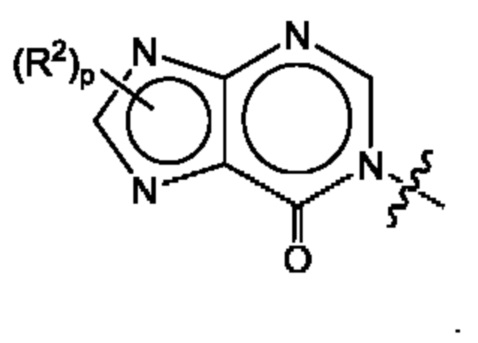
Согласно некоторым аспектам, А представляет собой:
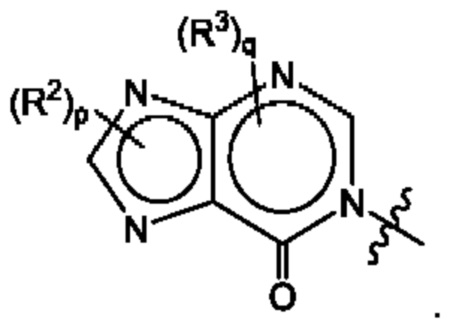
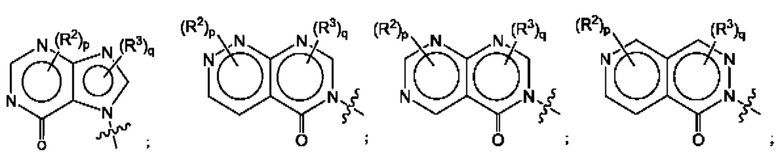
Согласно некоторым аспектам, А выбран из:

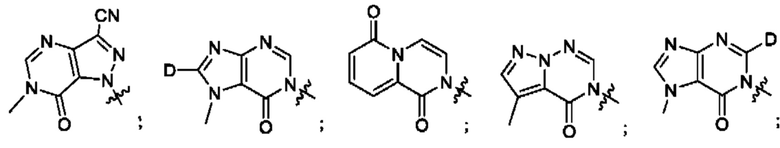
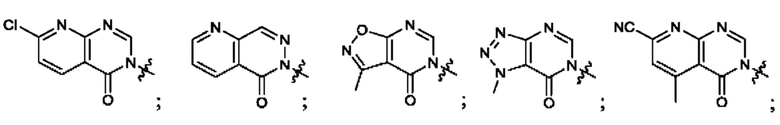
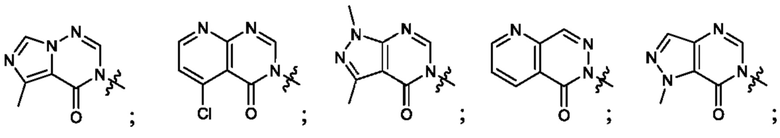

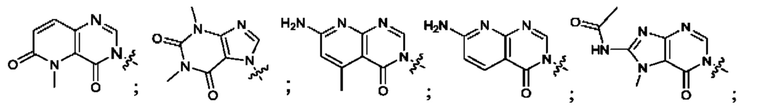
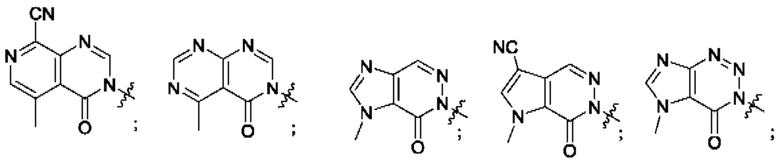

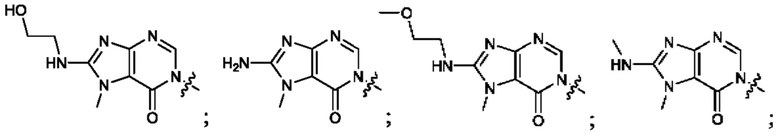


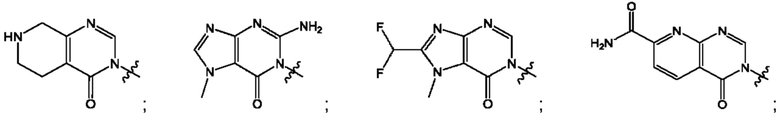

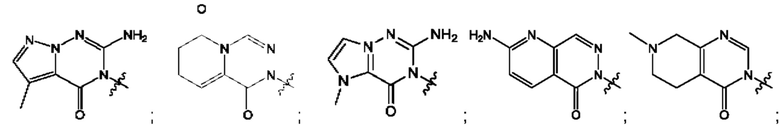


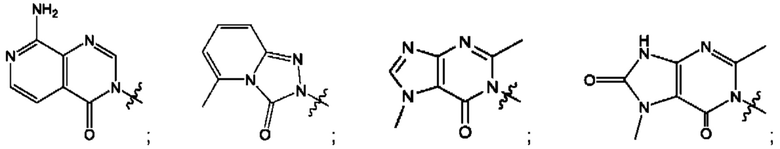
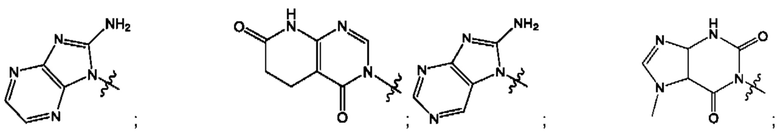
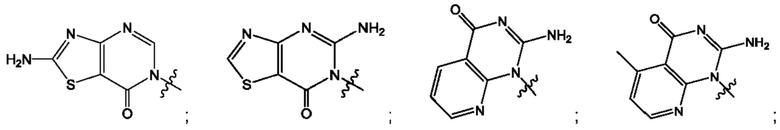
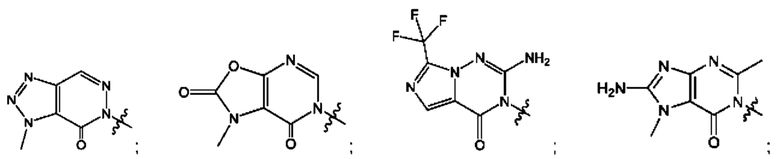

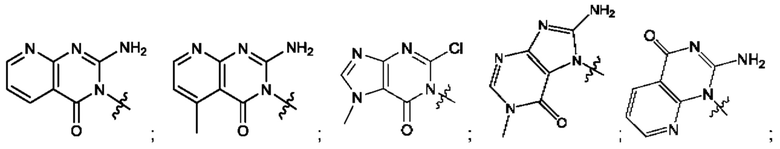
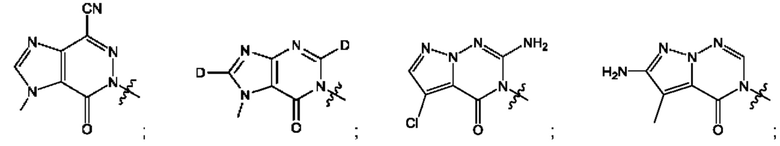
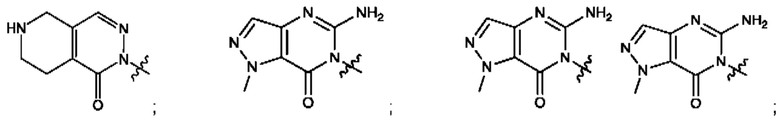
Согласно некоторым аспектам, А выбран из:


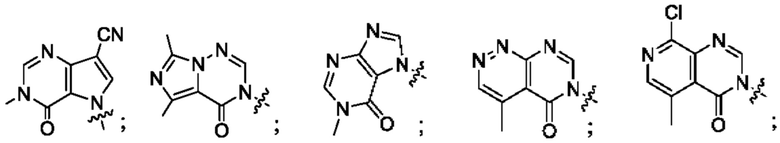

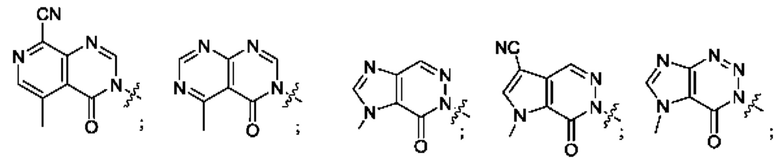
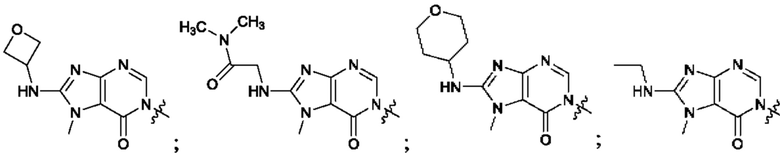

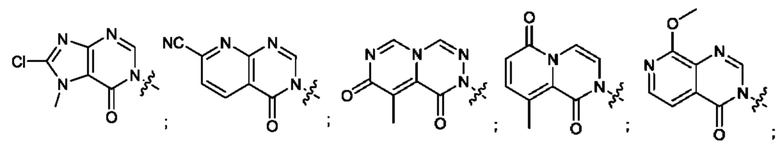
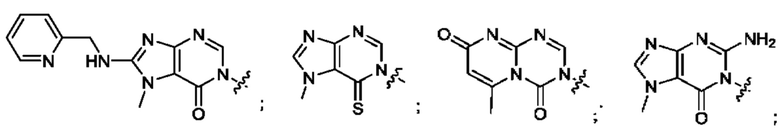
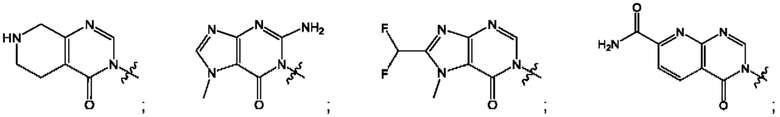
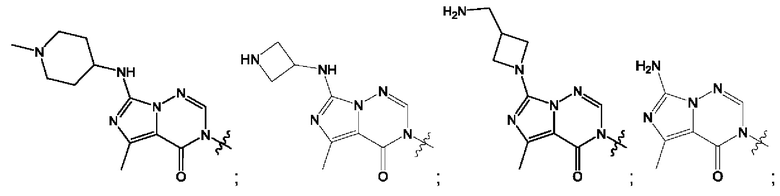
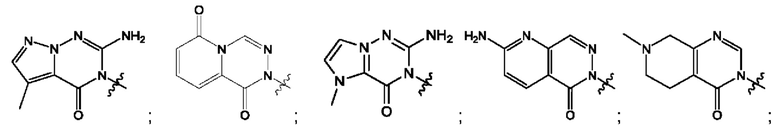
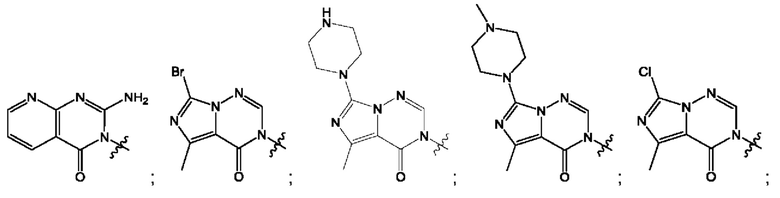

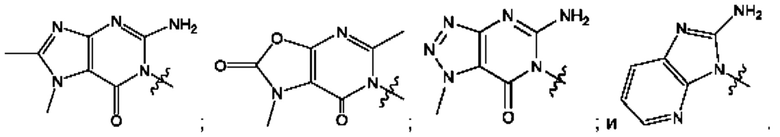
Согласно некоторым аспектам, А выбран из:
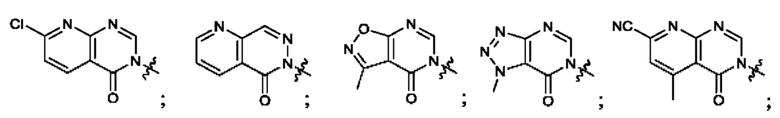

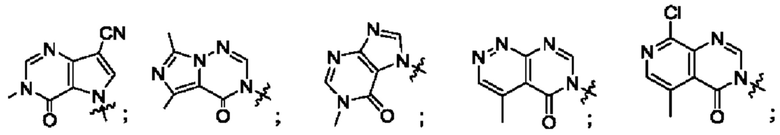
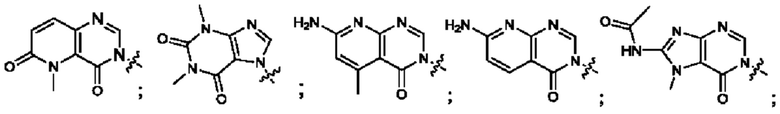
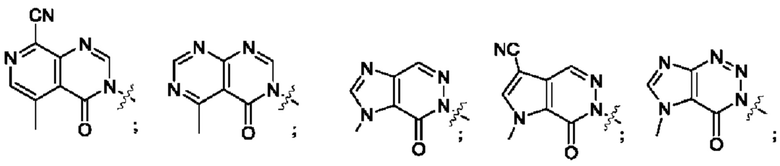

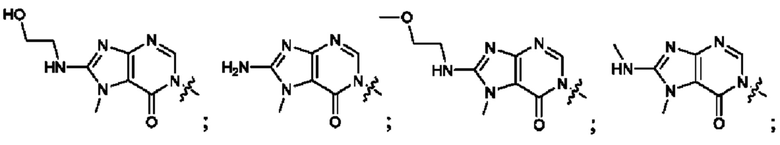
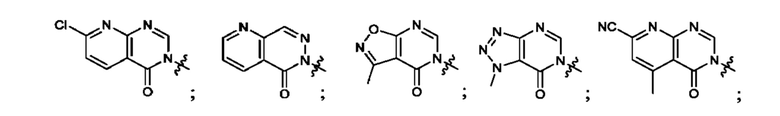
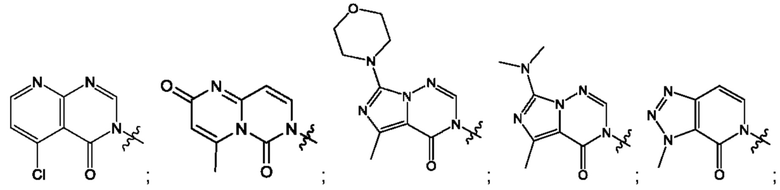
Согласно некоторым аспектам, А выбран из:
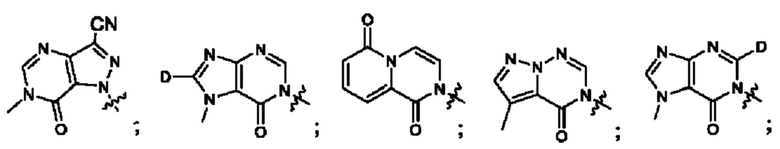


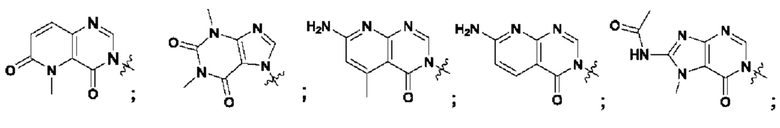

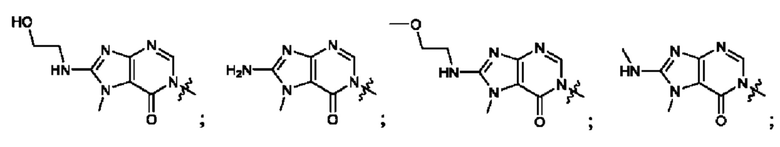
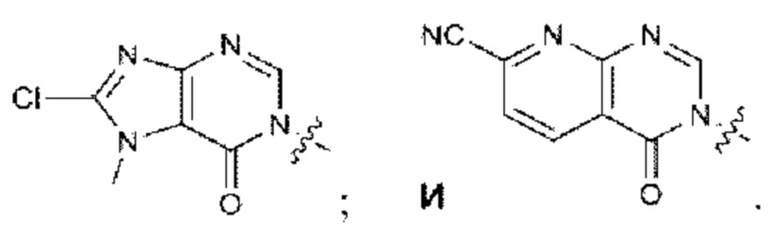
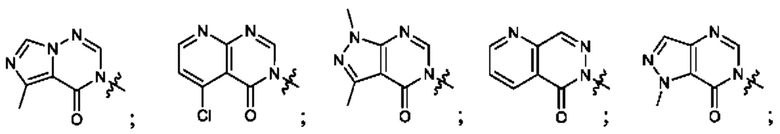
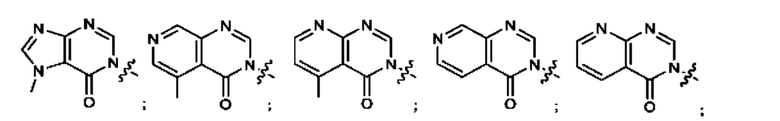
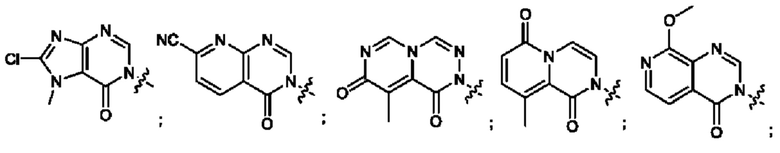
Согласно некоторым аспектам, А выбран из:
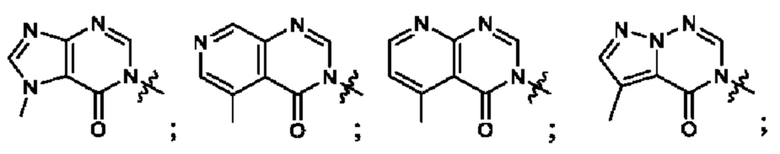
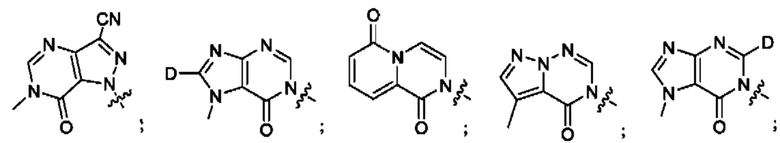
Согласно некоторым аспектам, А представляет собой:

Согласно некоторым аспектам, А представляет собой:
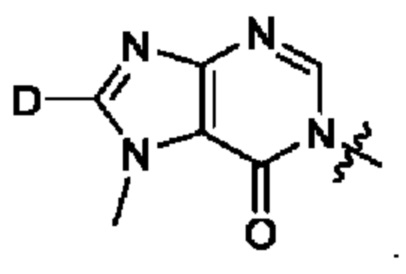
Согласно некоторым аспектам, А представляет собой:
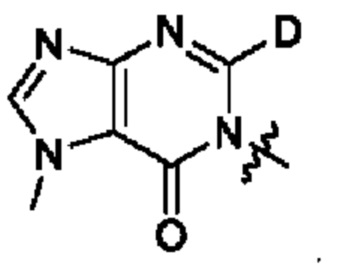
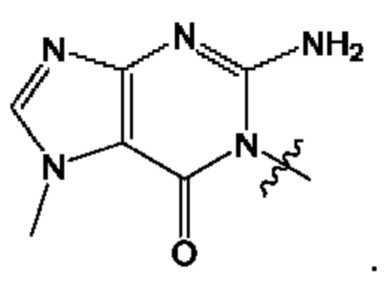
Согласно некоторым аспектам, А представляет собой:
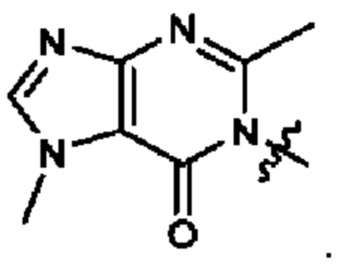
Согласно некоторым аспектам, А представляет собой:
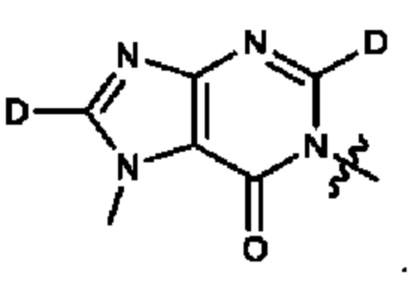
Согласно некоторым аспектам, А представляет собой:
Согласно некоторым аспектам, X представляет собой метилен.
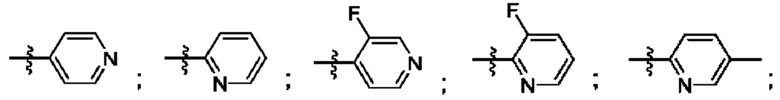
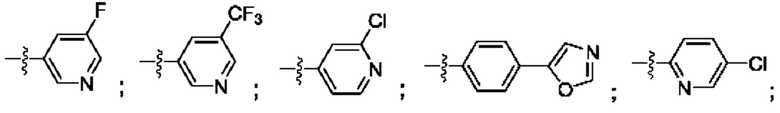
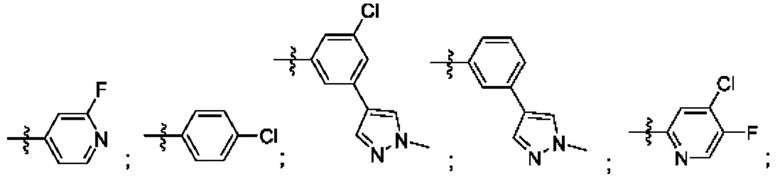
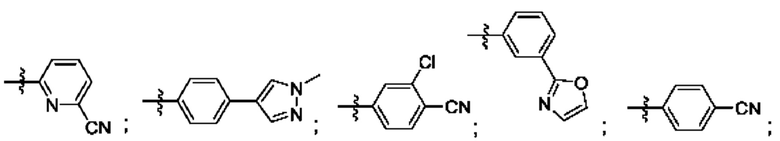
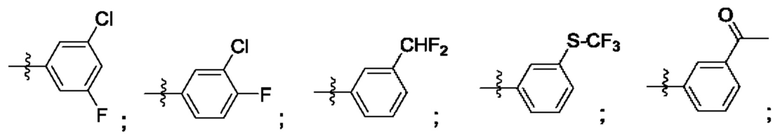
R4 выбран из замещенного или незамещенного фенила, замещенного или незамещенного гетероарила и замещенного или незамещенного нафтила. Согласно некоторым аспектам, R4 представляет собой:
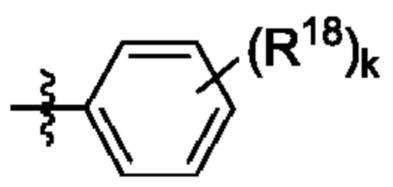
где каждый R18 независимо выбран из Н, галогена, -ОН, -С1-4алкила, -C1-4галогеналкила, -CN, галогена, С1-4галогеналкокси, С1-4алкокси, -SO2-С1-4алкила, -C1-4CN, С1-4альдегида, С1-4кетона, пентафторсульфанила, незамещенного или замещенного С3-6циклоалкила, незамещенного или замещенного фенила, незамещенного или замещенного гетероарила, конденсированного арила и конденсированного гетероарила; а k равно от 0 до 3. Согласно некоторым аспектам, каждый R18 независимо выбран из Н, Cl, -OCHF2, -OCF3, -ОСН3 и -CN. Согласно некоторым вариантам осуществления, R18 представляет собой галоген. Согласно некоторым вариантам осуществления, R18 представляет собой хлор или фтор. Согласно некоторым вариантам осуществления, k равно 0, 1 или 2.
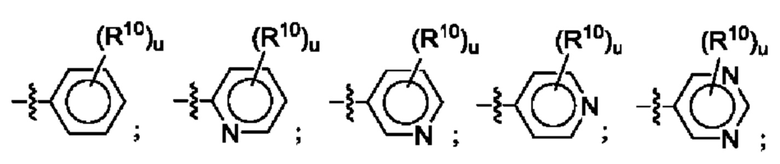
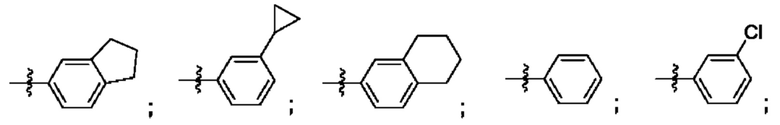
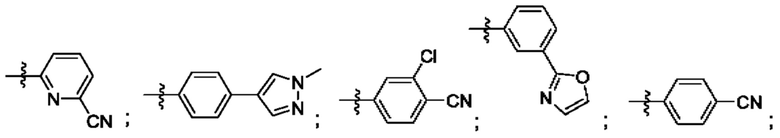
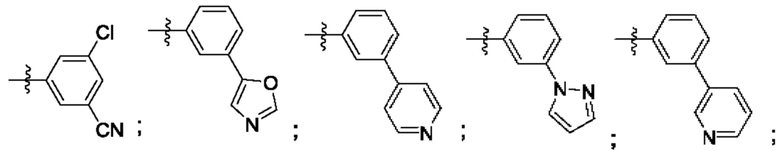
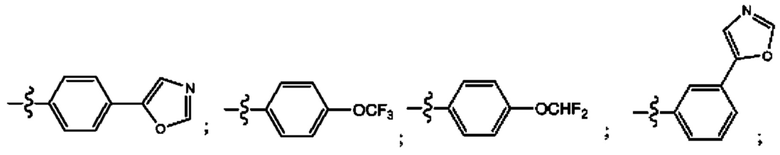
Согласно некоторым аспектам, R4 выбран из:
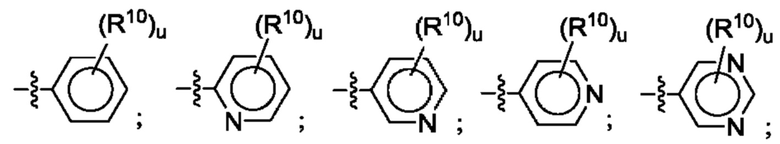




Согласно некоторым аспектам, R4 выбран из:
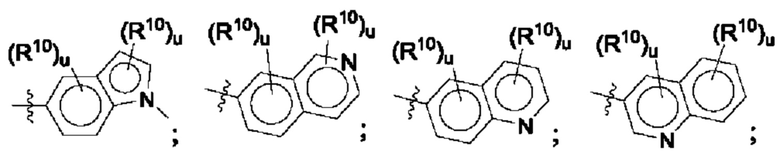
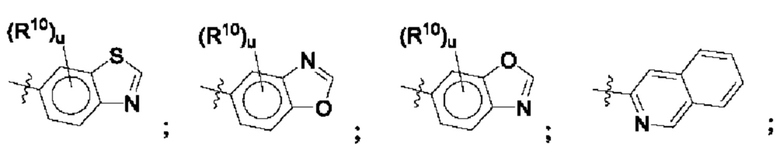

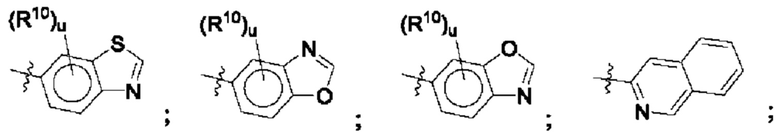
Согласно этим аспектам, каждый R10 независимо выбран из Н, галогена, -CN, -ОН, С1-4алкила, замещенного или незамещенного С3-6циклоалкила, С1-4галогеналкила, С1-4галогеналкокси, С1-4алкокси, -SO2-С1-4алкила, C1-4CN, C1-4альдегида, С1-4кетона, -S-С1-4галогеналкила, пентафторсульфанила, незамещенного или замещенного С3-6циклоалкила, незамещенного или замещенного фенила, замещенного или незамещенного от 5- до 6-членного гетероарила, замещенного или незамещенного от 4-до 6-членного гетероцикпоалкила, замещенного или незамещенного фенила и замещенного или незамещенного нафтила. Каждый и независимо выбран из 0, 1, 2 и 3. Согласно некоторым аспектам, каждый R10 независимо выбран из галогена, С1галогеналкокси и С1алкокси. Согласно некоторым вариантам осуществления, каждый R10 представляет собой галоген.
Согласно некоторым аспектам, в случае когда R1 и R4 вместе образуют незамещенный или замещенный С3-6циклоалкил, сконденсированный с замещенным или незамещенным фенилом; замещенным или незамещенным гетероарилом; или замещенным или незамещенным нафтилом, такие комбинированные R1 и R4 могут иметь формулу
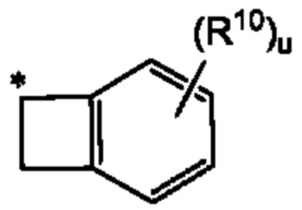
где * представляет собой спиро-точку присоединения, а u и R10 такие, как указаны в данном контексте.
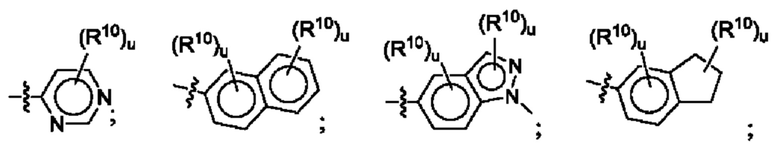
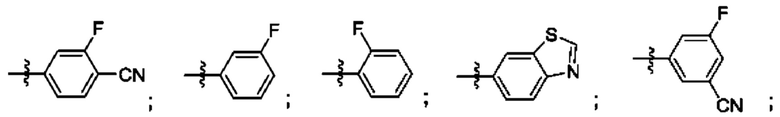
Согласно некоторым аспектам, R4 выбран из:

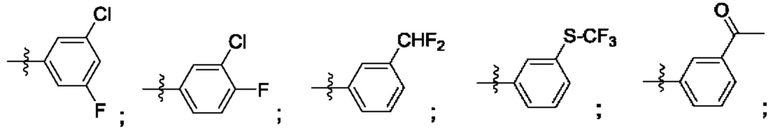
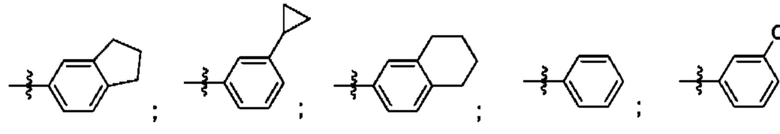
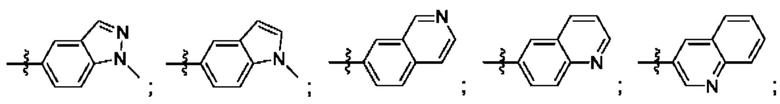
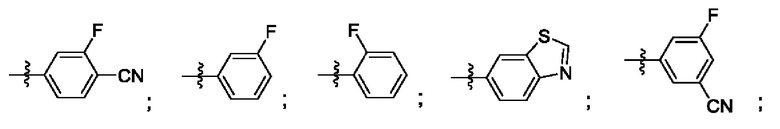
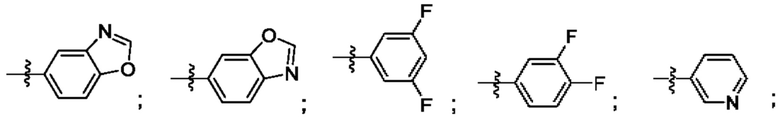


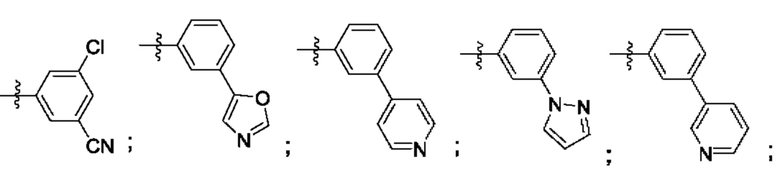
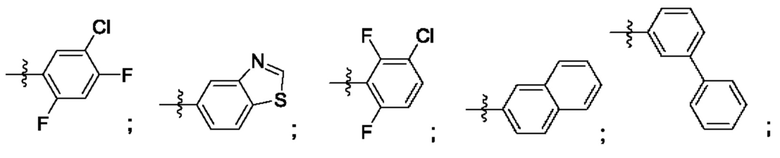
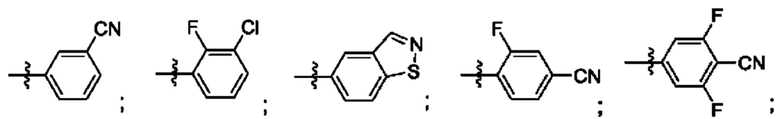
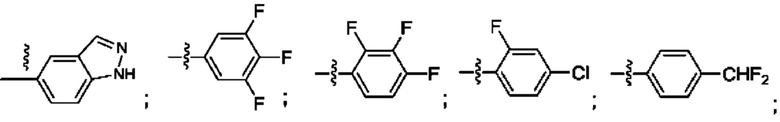
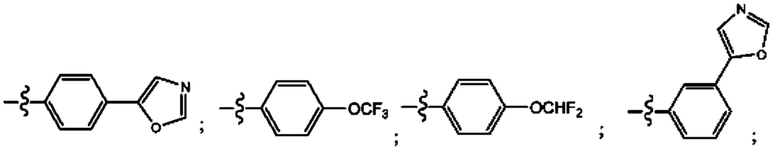
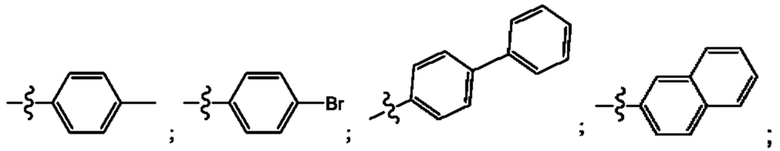
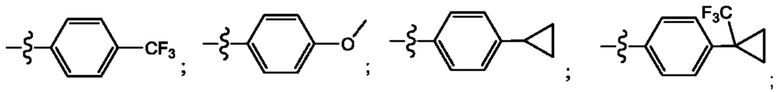
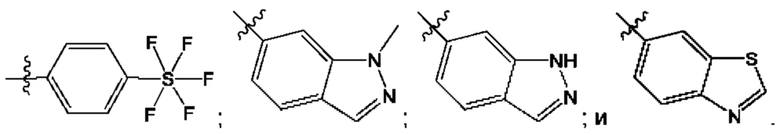
Согласно некоторым аспектам, R4 выбран из:


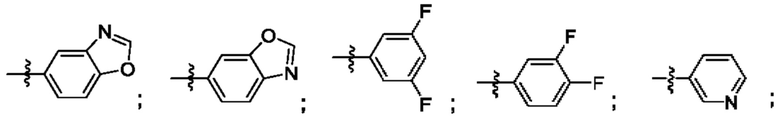
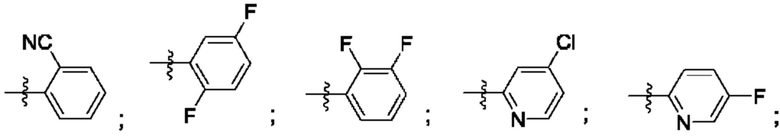
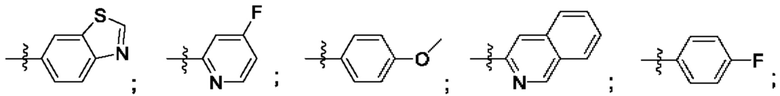

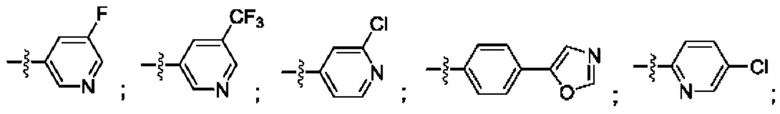
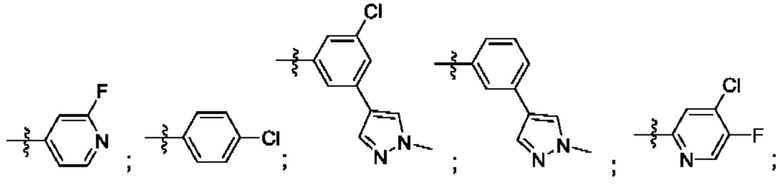



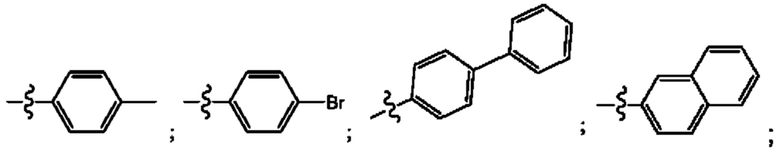

Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (II):

где А, X, R1 и R4 такие, как указано в данном контексте.
Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (IIIa) или формулу (IIIb)


где Е, G, R1, R2, R3, R4, X Y1, Y2, Y3, Y4, Y5, Y6, f Z1, Z2, Z3, Z4, Z5, p и q такие, как указано в данном контексте.
Согласно некоторым аспектам, X представляет собой С1-4алкилен.
Согласно некоторым аспектам, X представляет собой метилен.
Согласно некоторым аспектам, R1 представляет собой Н.
Согласно некоторым аспектам, R4 представляет собой:

Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (IVa) или формулу (IVb)
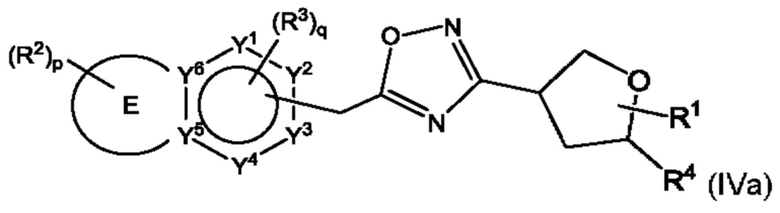

где Е, G, R1, R2, R3, R4, Y1, Y2, Y3, Y4, Y5, Y6, f Z1, Z2, Z3, Z4, Z5, р и q такие, как указано в данном контексте.
Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (Va)
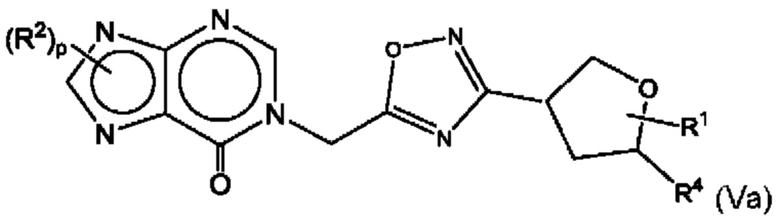
где R1, R2, R4 и р такие, как указано в данном контексте.
Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (Vb)
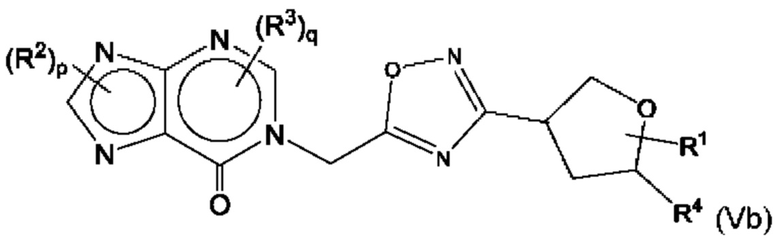
где R1, R2, R3, R4, р и q такие, как указано в данном контексте.
Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (VIa) или формулу (VIb)
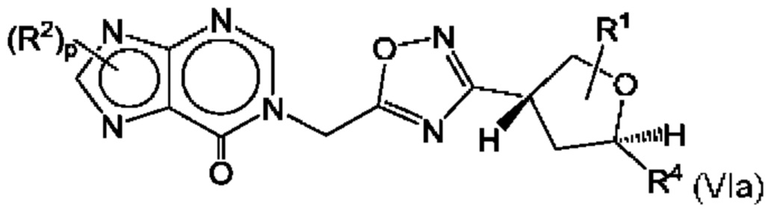

где R1, R2, R4 и p такие, как указано в данном контексте.
Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (VIc) или формулу (VId)
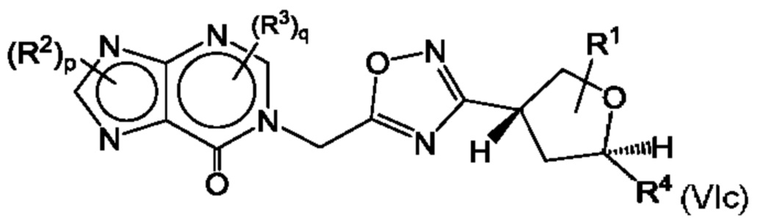
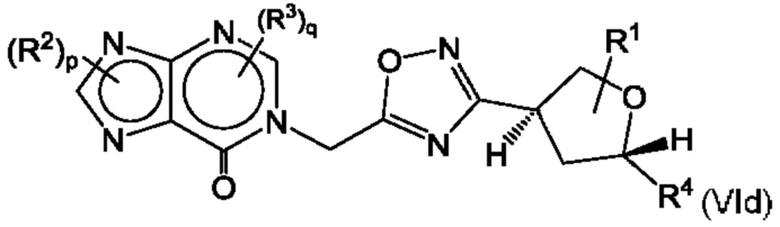
где R1, R2, R3, R4, р и q такие, как указано в данном контексте.
Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (VIIa) или формулу (VIIb)
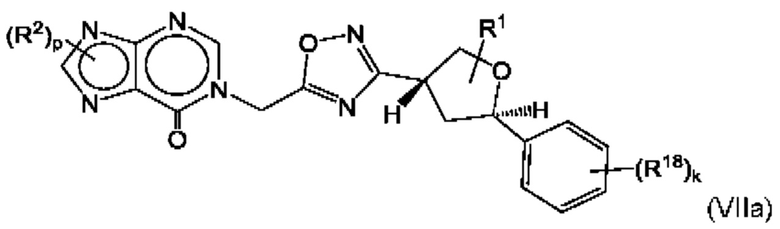
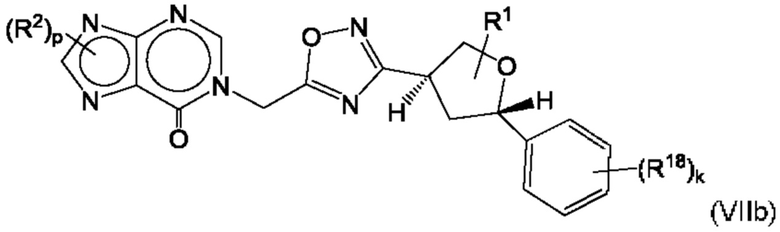
где R1, R2, R4, R18, р и k такие, как указано в данном контексте.
Согласно некоторым аспектам, соединение формулы (I) может иметь формулу (VIIc) или формулу (VIIc)
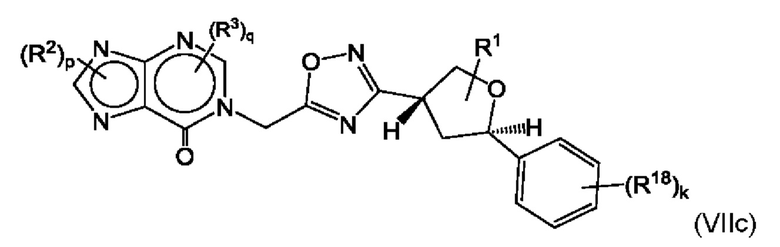
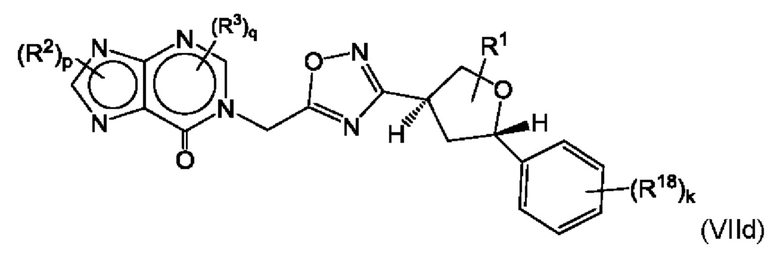
где R1, R2, R3, R18, p, q и k такие, как указано в данном контексте.
Согласно некоторым вариантам осуществления формулы (I), группа А может быть группой формулы:
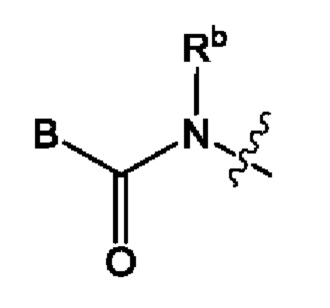
где:
В представляет собой пятичленный гетероарил, выбранный из пиррола, пиразола, пиразола, имидазола или триазола, каждый из которых может быть незамещенным или однократно замещенным Ra, и где каждый из пиррола, пиразола и имидазола может быть частично насыщенным; а
Rb представляет собой водород, C1-6алкил, который может быть незамещенным или однократно замещенным -NR16R17.
Согласно некоторым вариантам осуществления, В представляет собой имидазолил.
Согласно некоторым вариантам осуществления, В представляет собой 1-метилимидазол-5-ил-.
Согласно некоторым вариантам осуществления, В является триазолилом.
Согласно некоторым вариантам осуществления, В представляет собой 5-метил-1Н-1,2,3-триазол-1-ил.
Согласно некоторым вариантам осуществления, В представляет собой 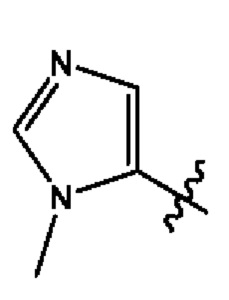 .
.
Согласно некоторым вариантам осуществления, В представляет собой 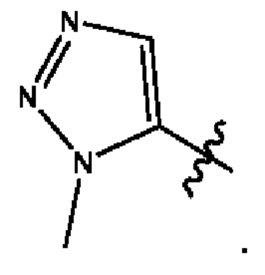
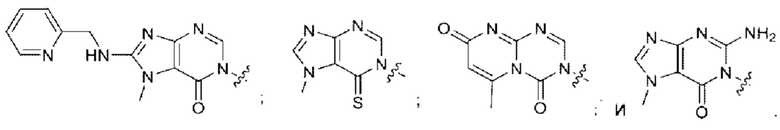
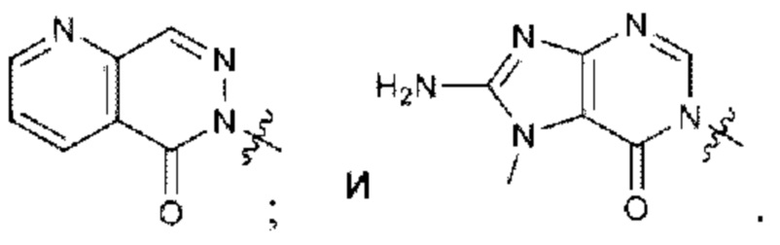
Согласно некоторым вариантам осуществления, А представляет собой 2-амино-3-метилпиримидин-4(3Н)-он-5-ил.
Согласно некоторым вариантам осуществления, А представляет собой 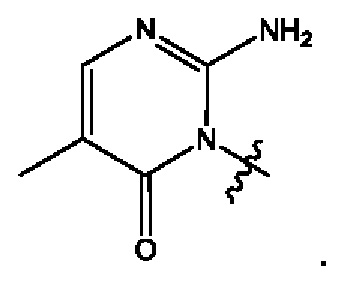
Согласно некоторым вариантам осуществления, Rb представляет собой водород.
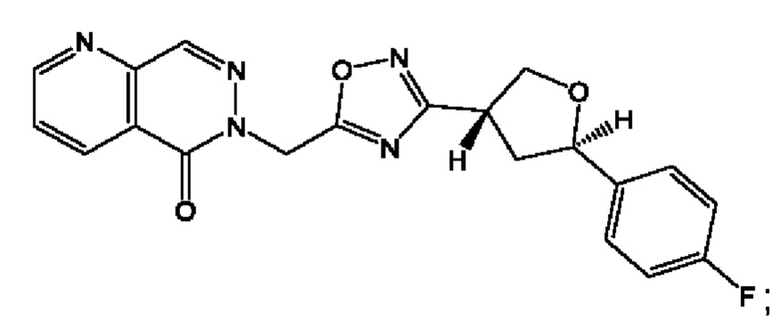
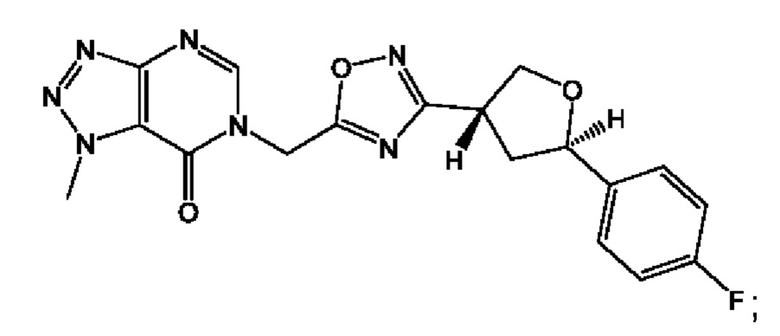
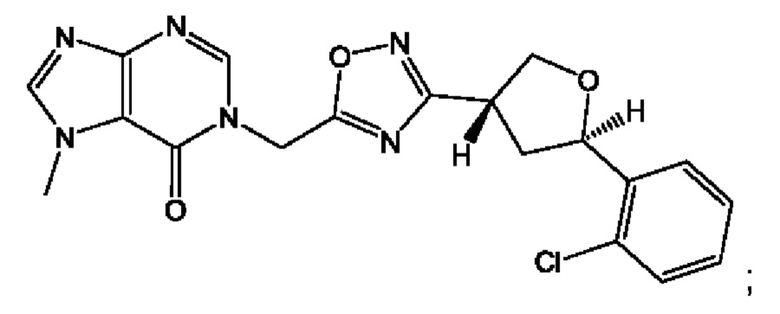
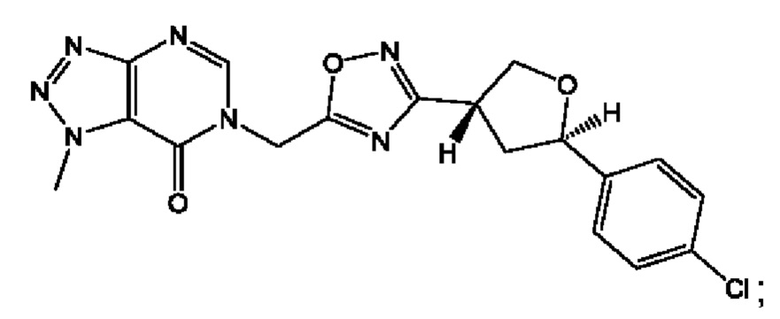
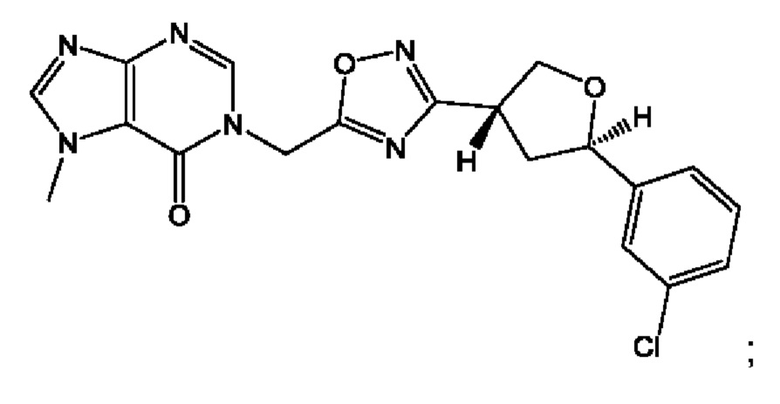
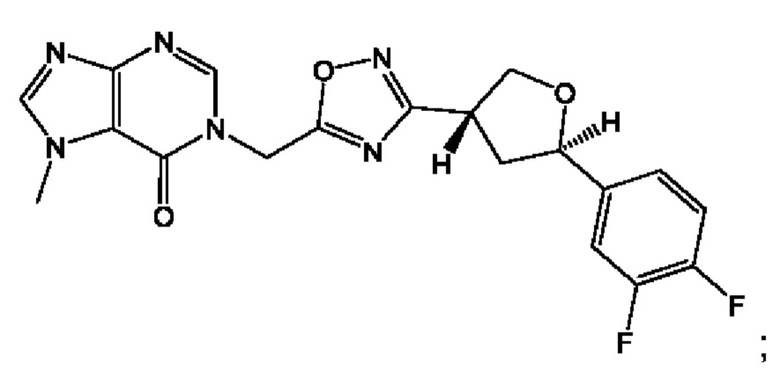
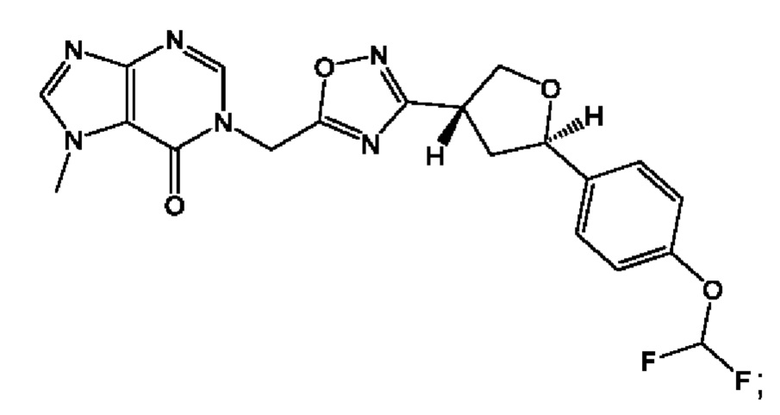
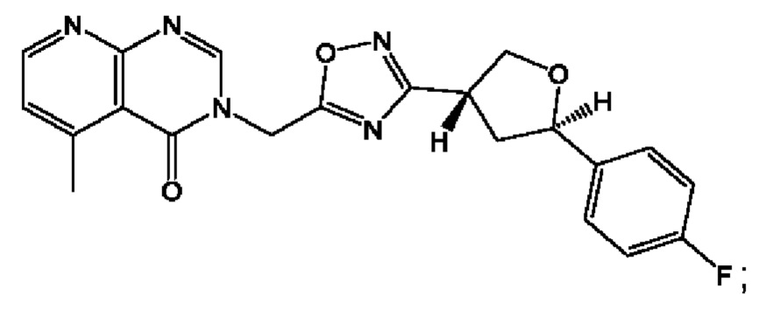
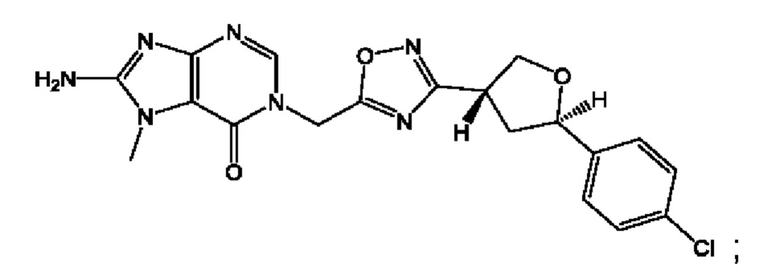
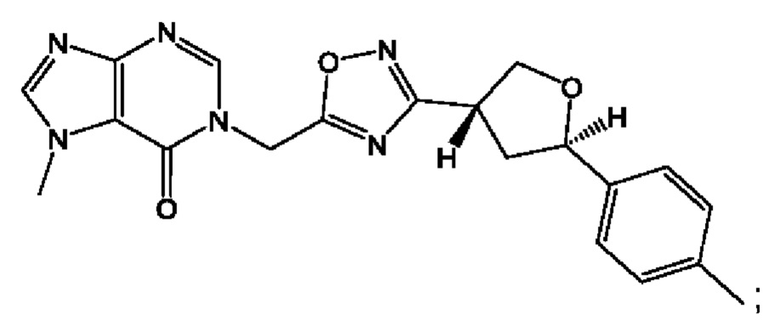
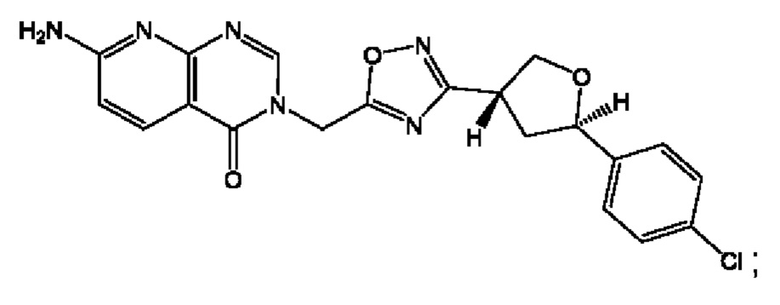
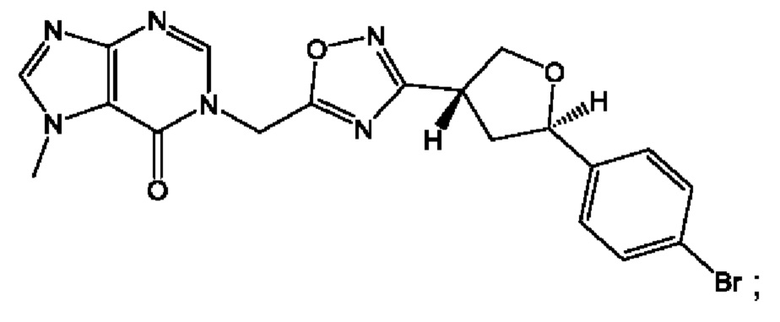
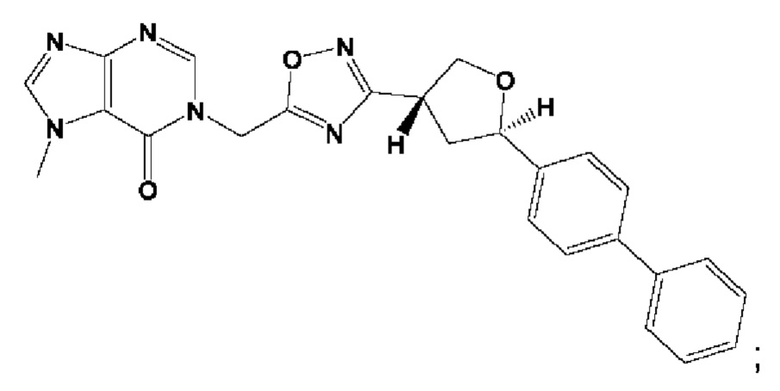
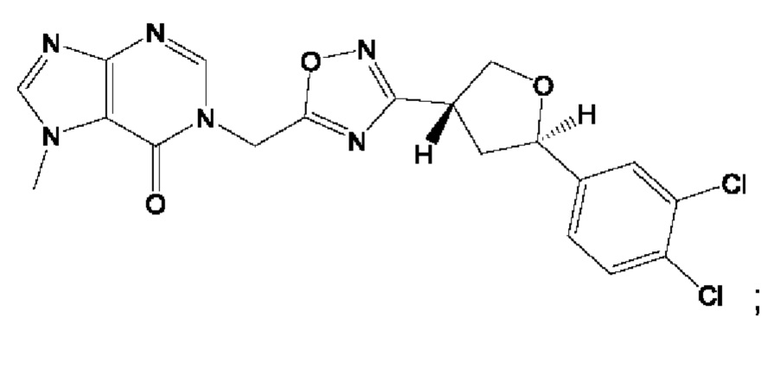
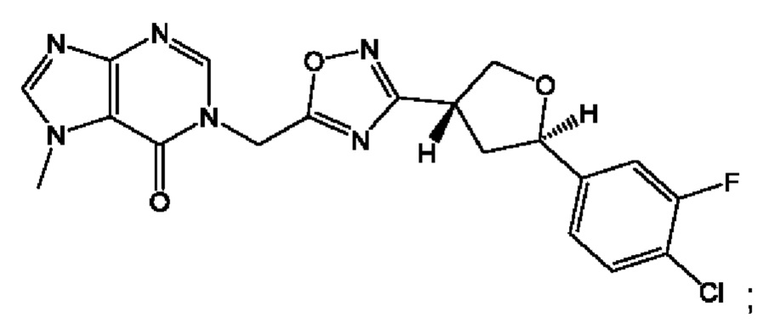
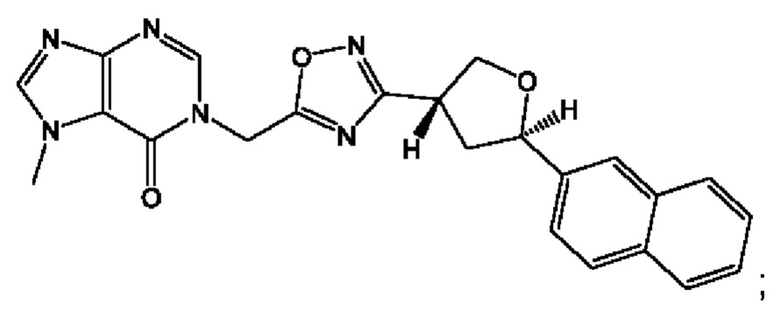
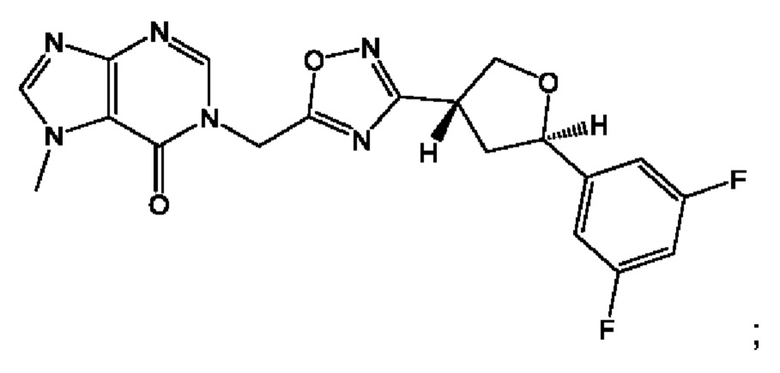
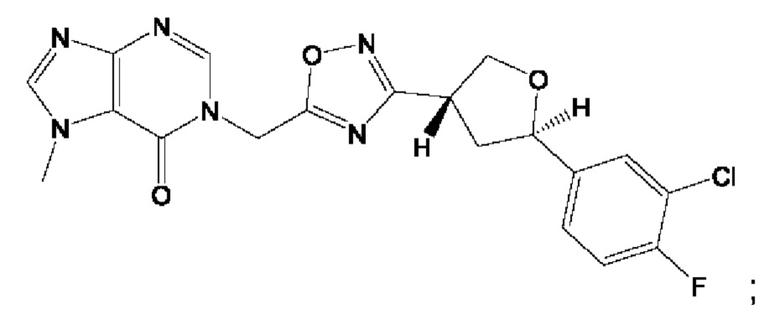
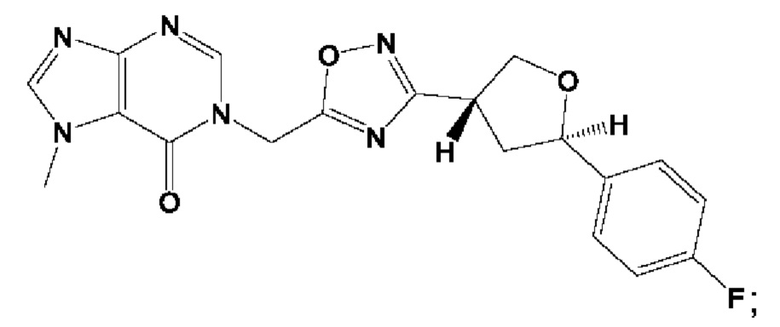
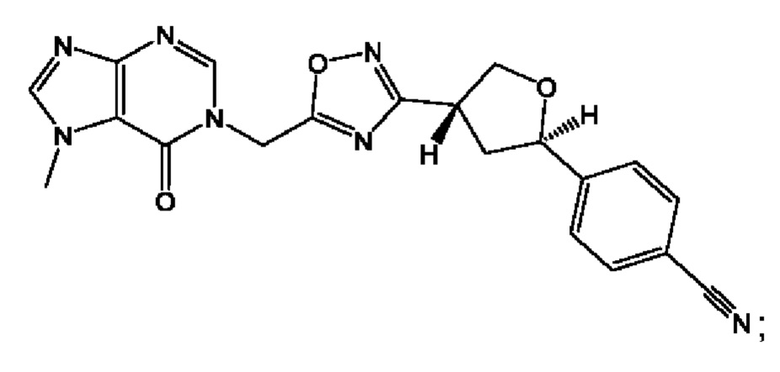
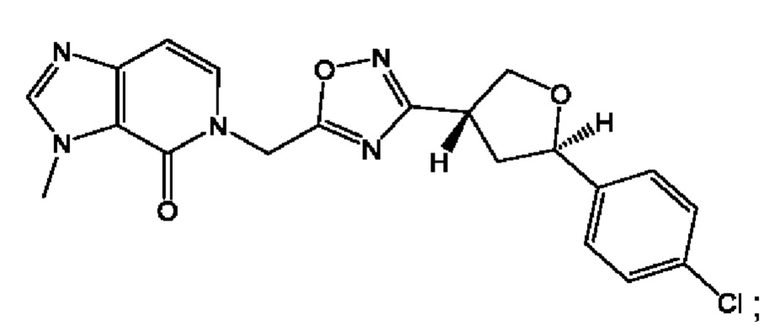
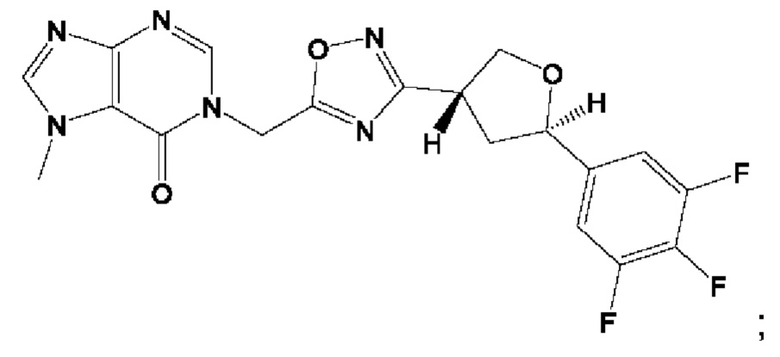
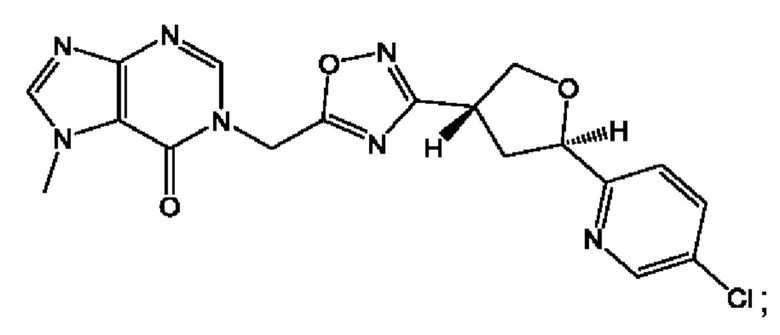
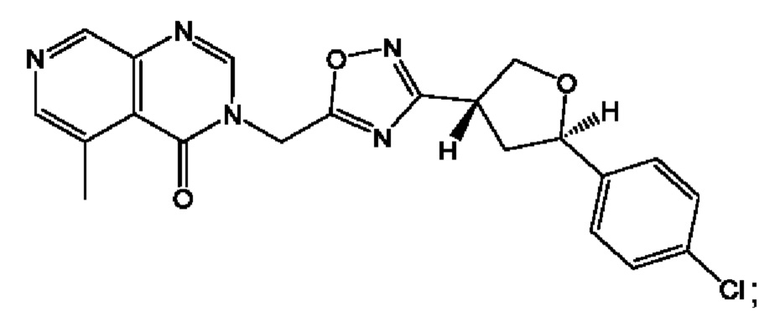
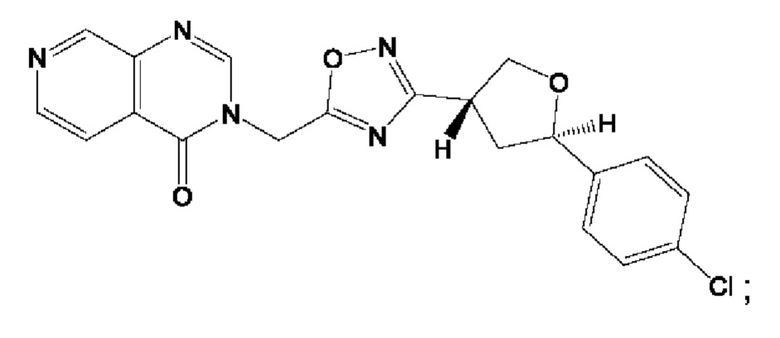
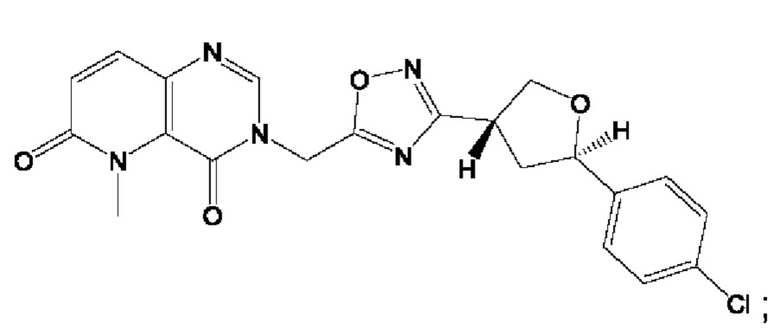
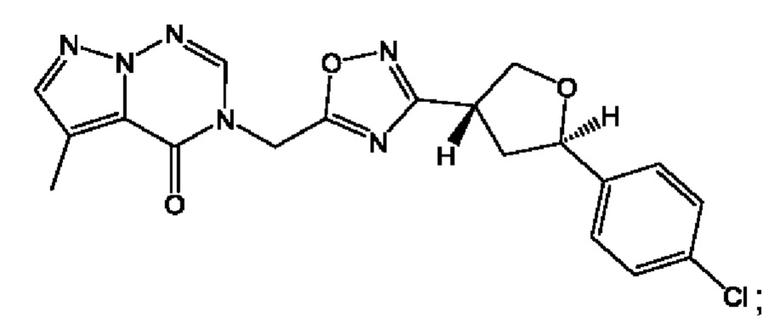
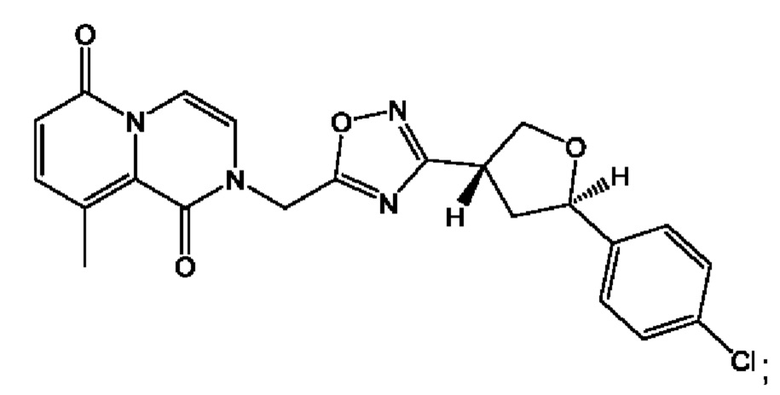
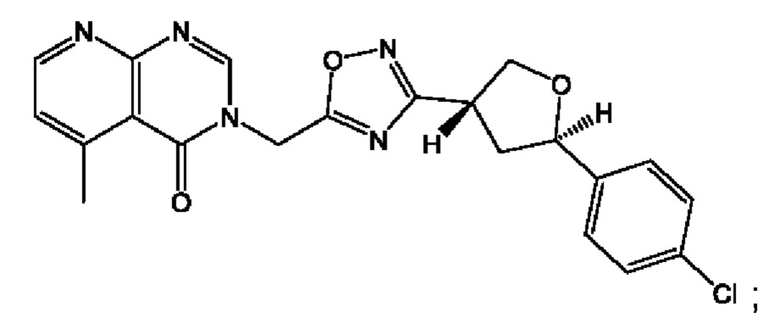
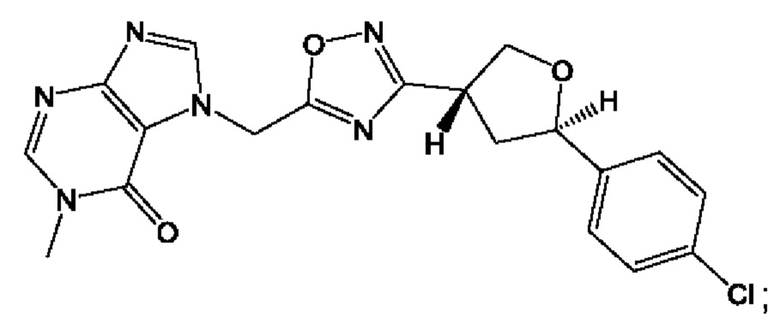
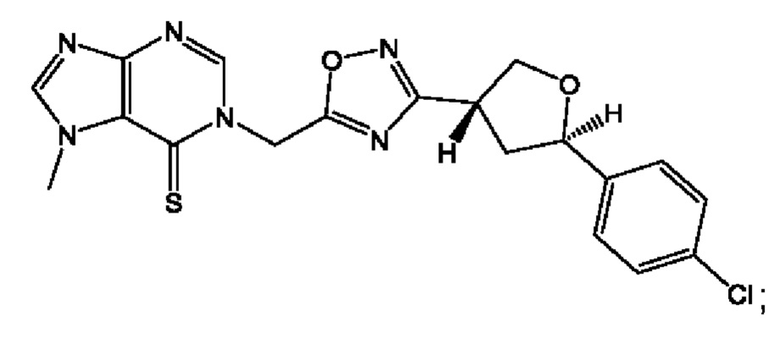
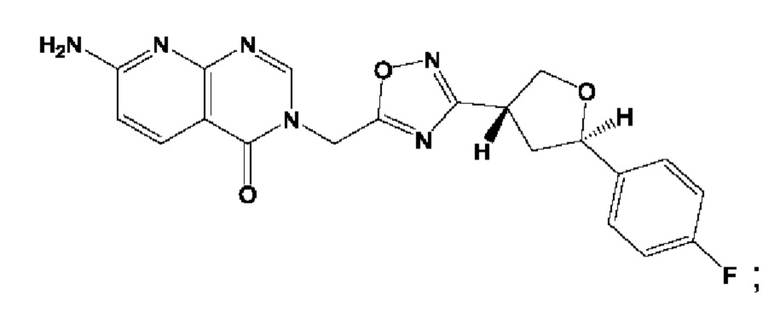
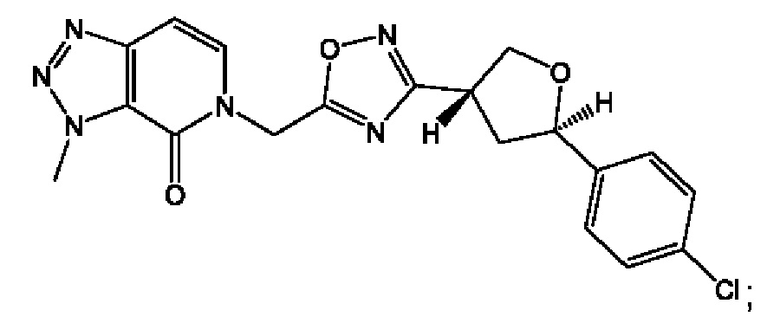
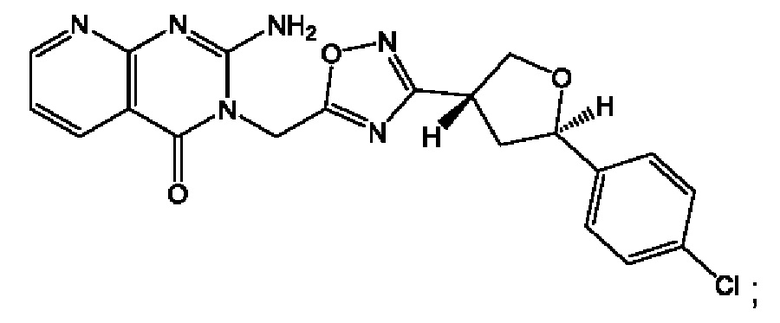
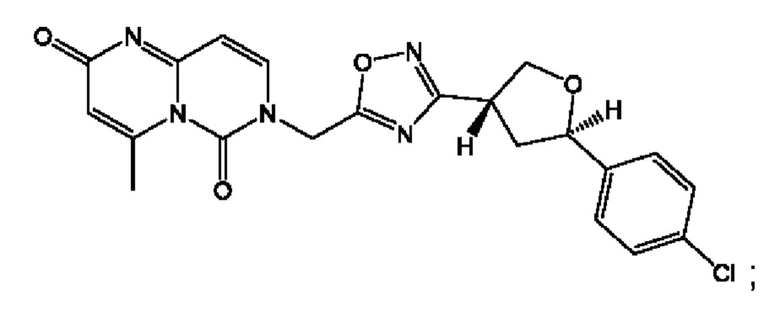
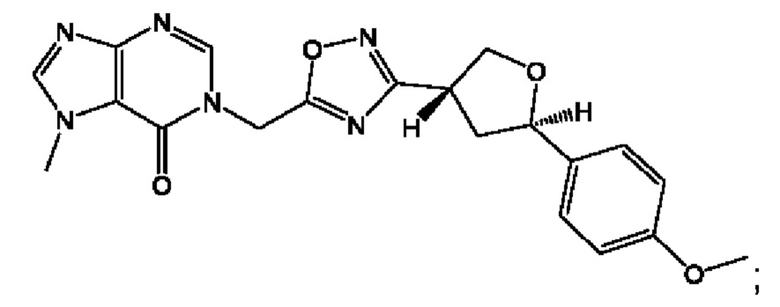
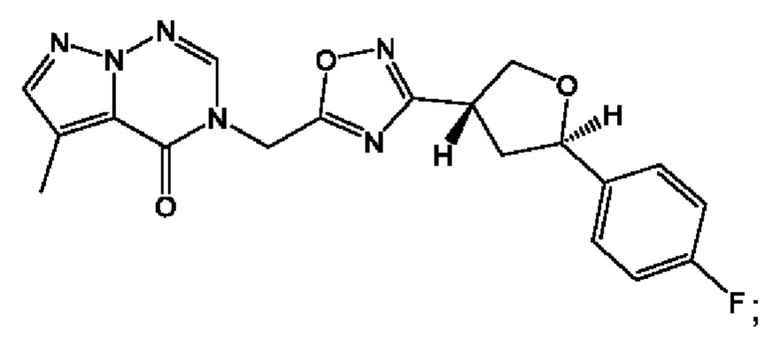
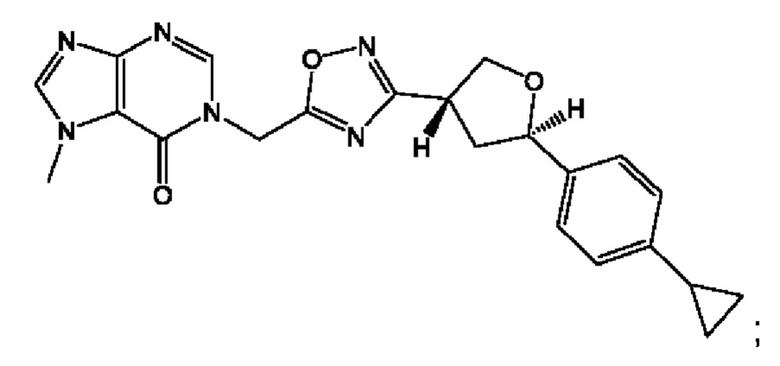
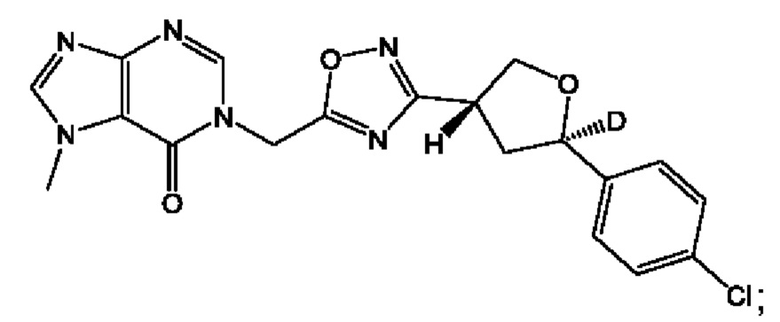
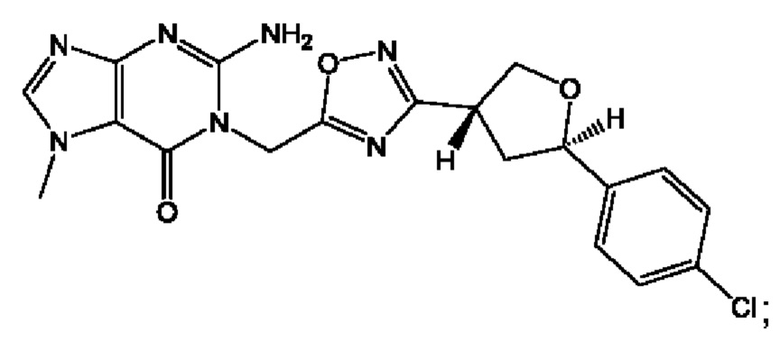
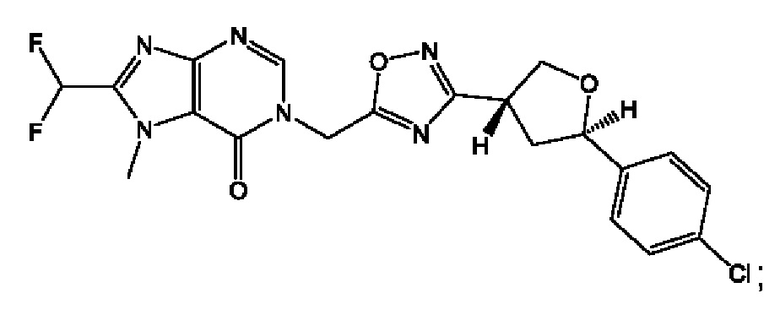
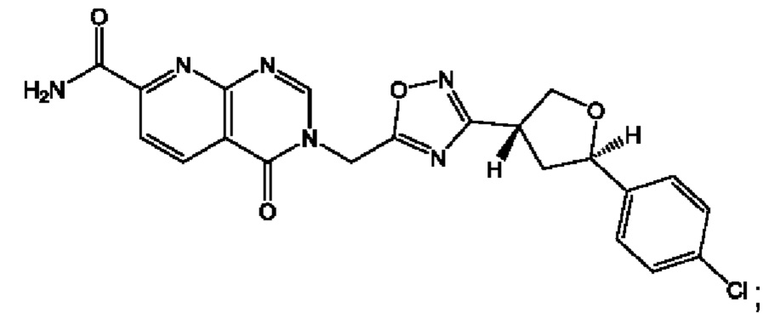
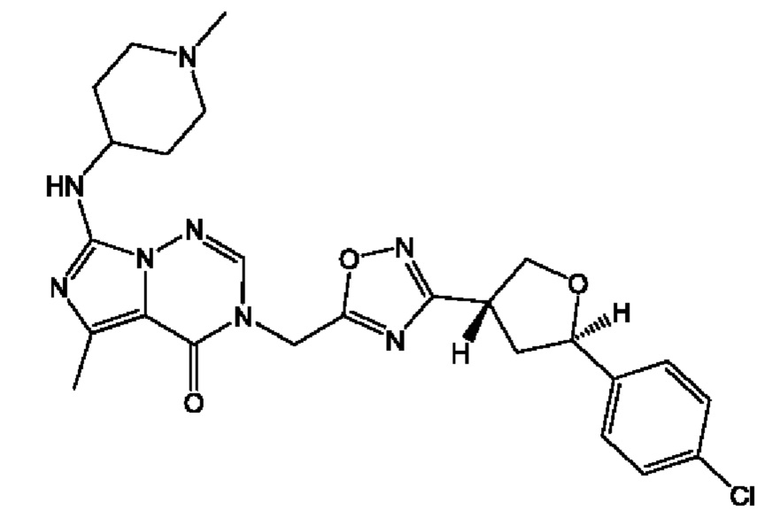
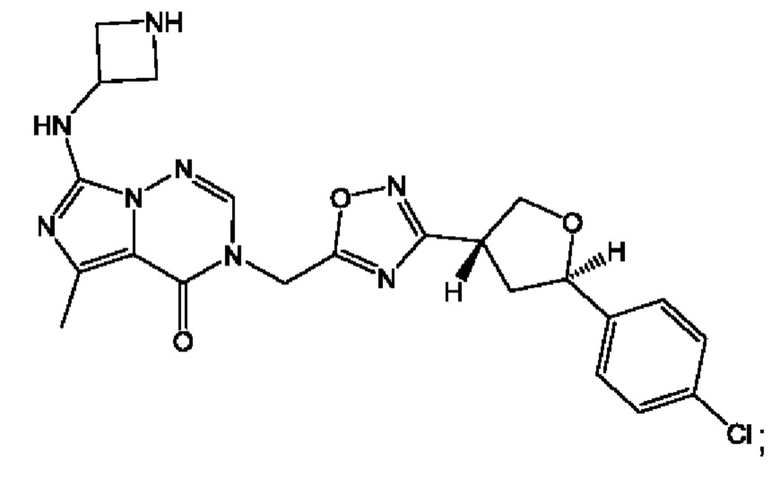
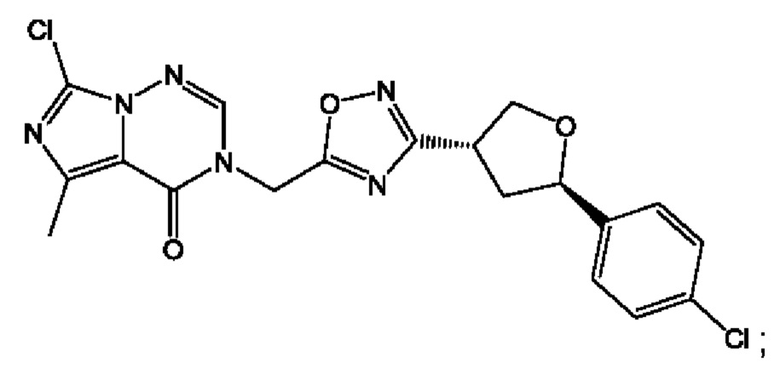
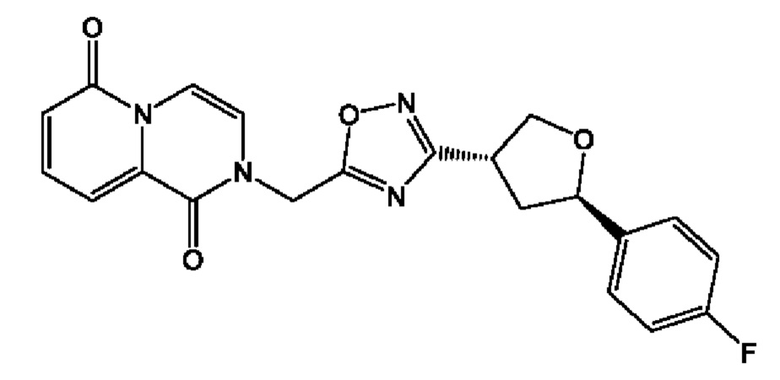
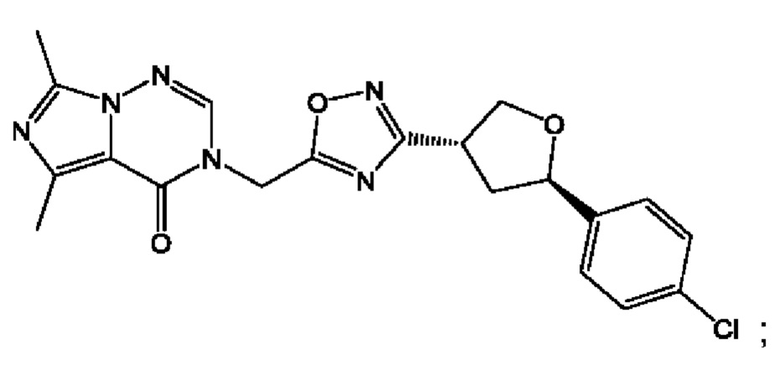
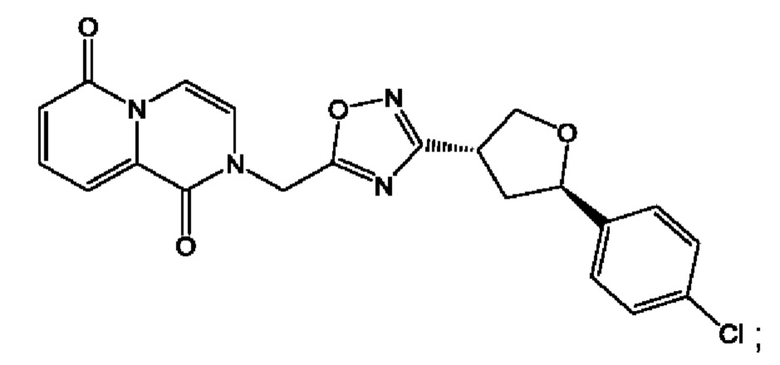
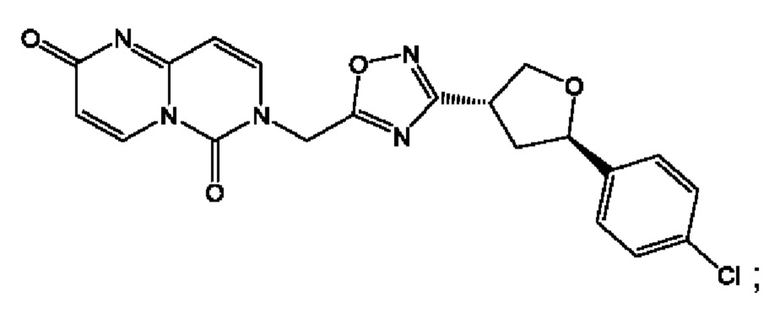
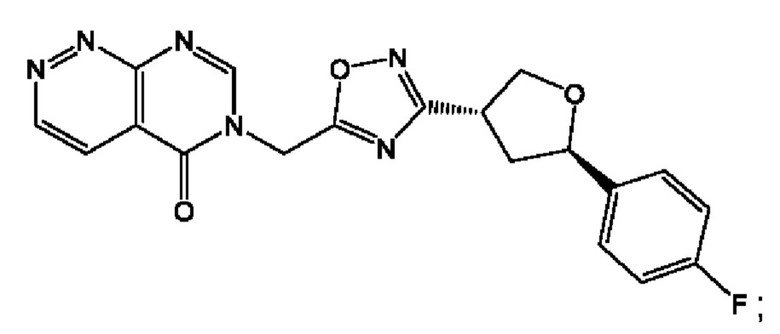
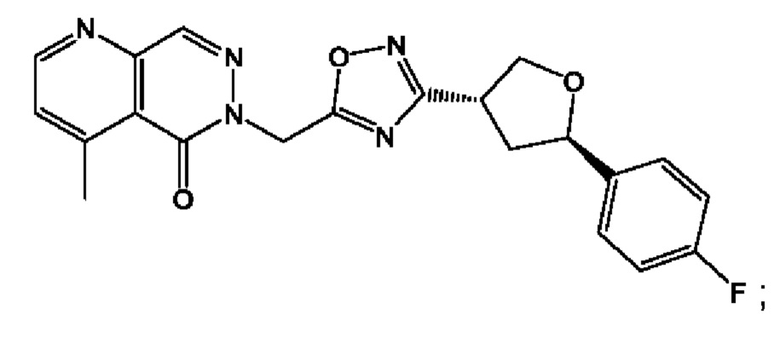
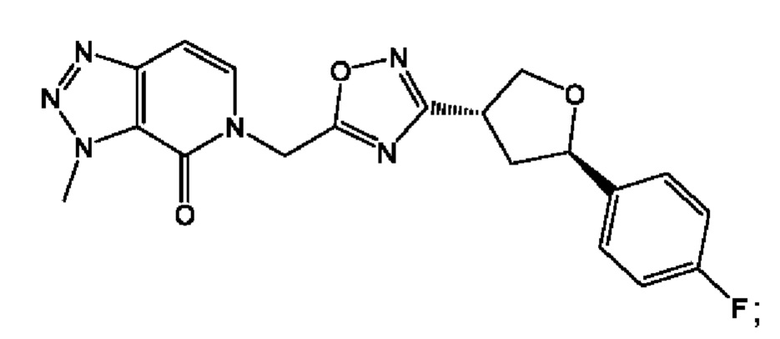
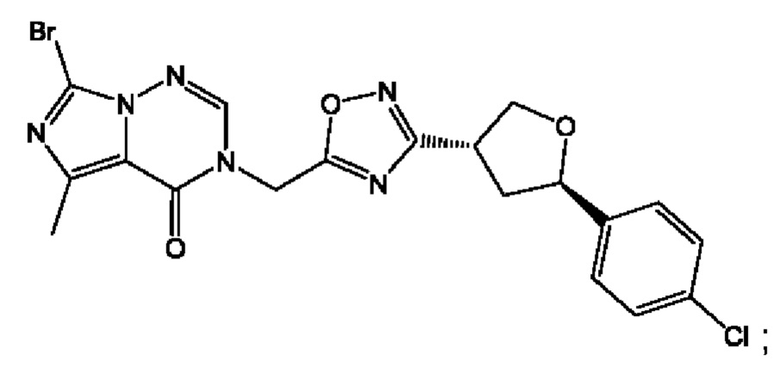
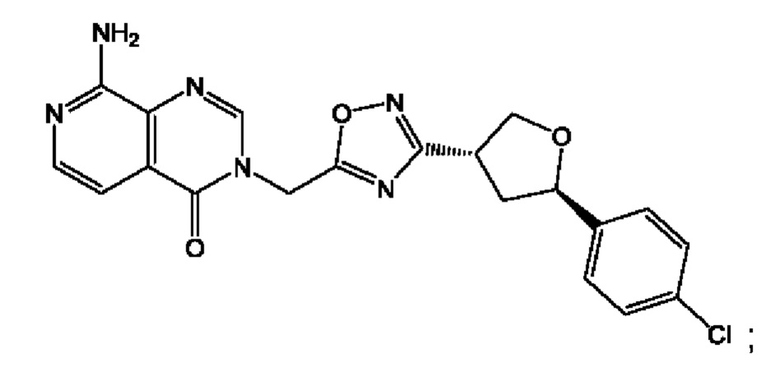
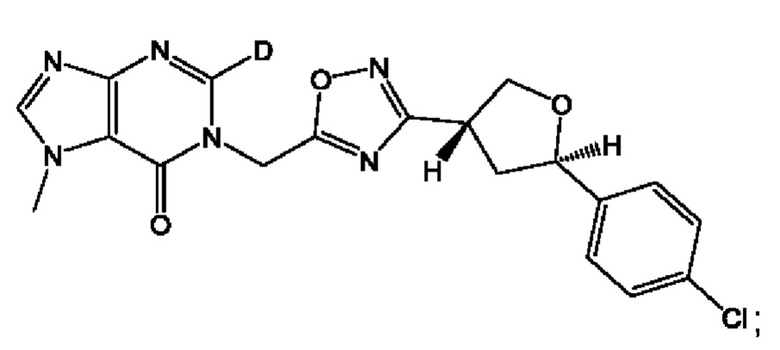
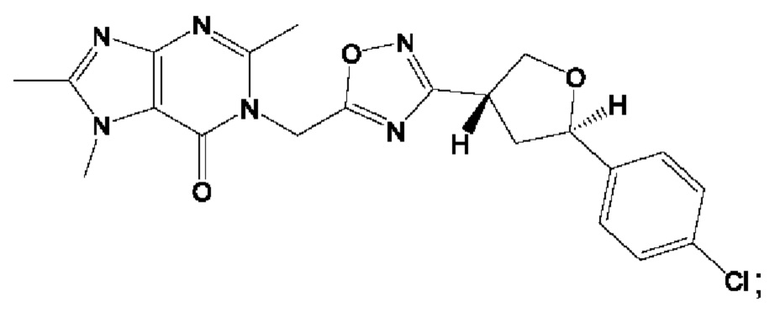
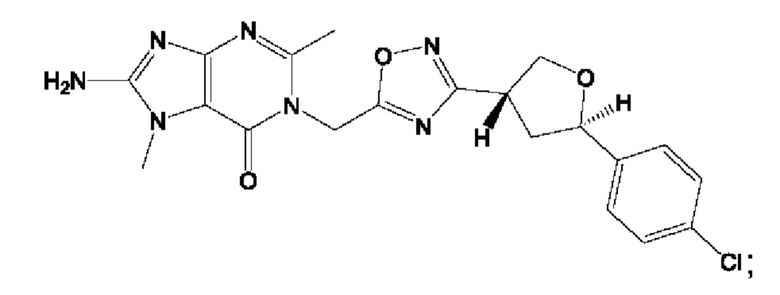
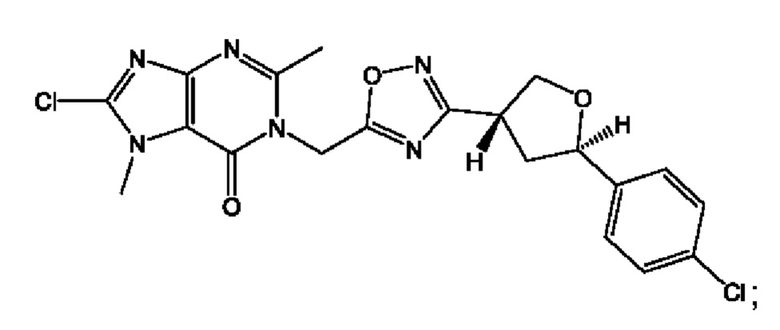
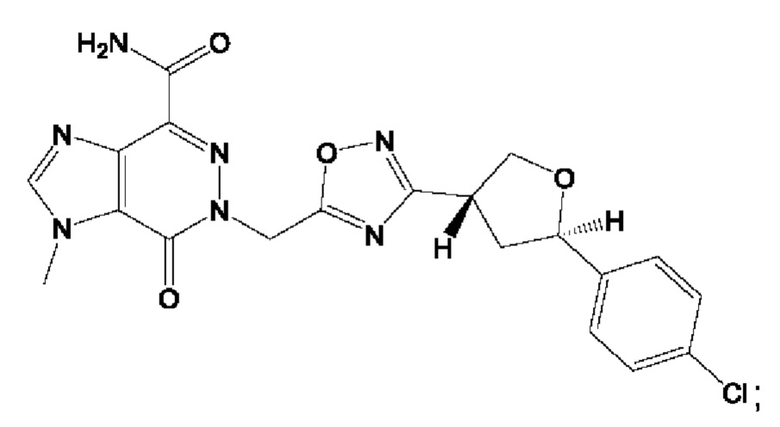
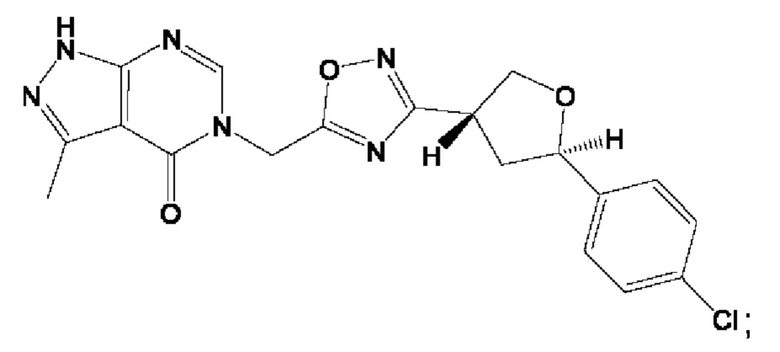
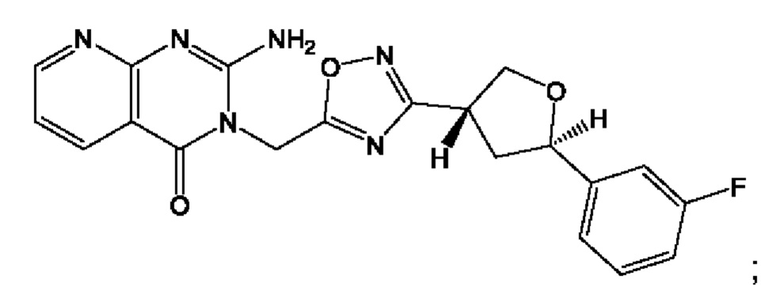
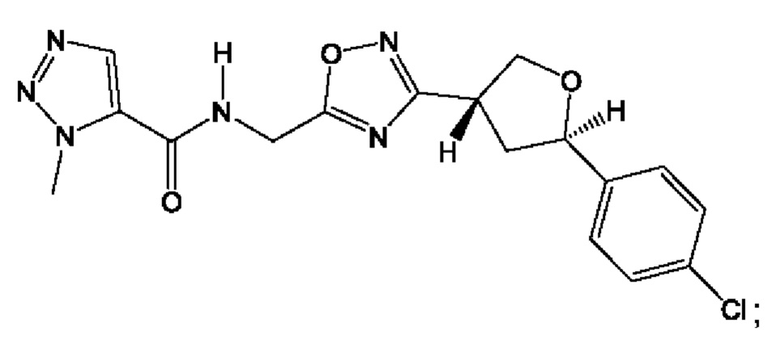
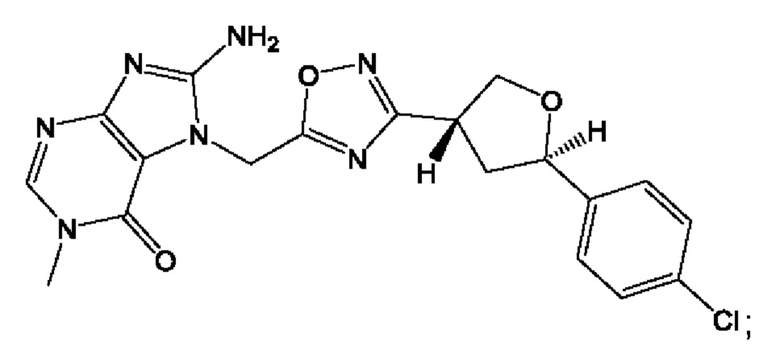
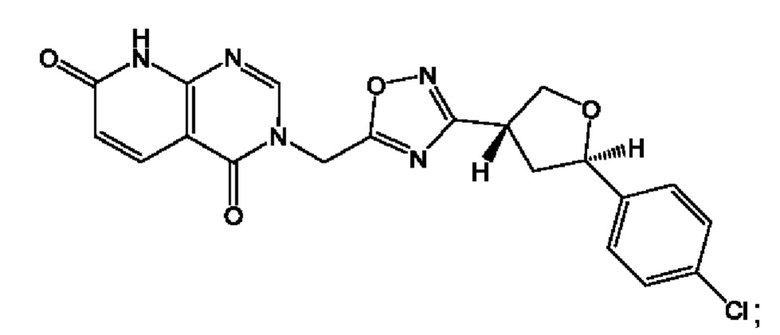
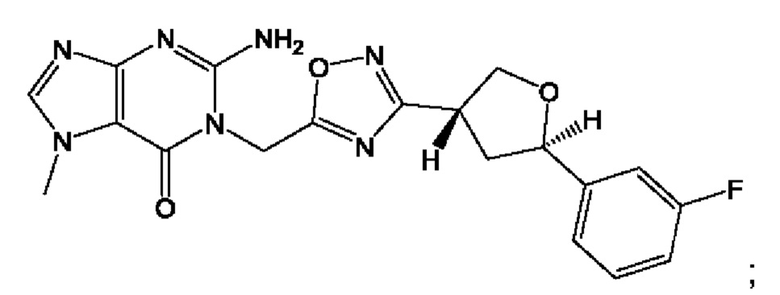
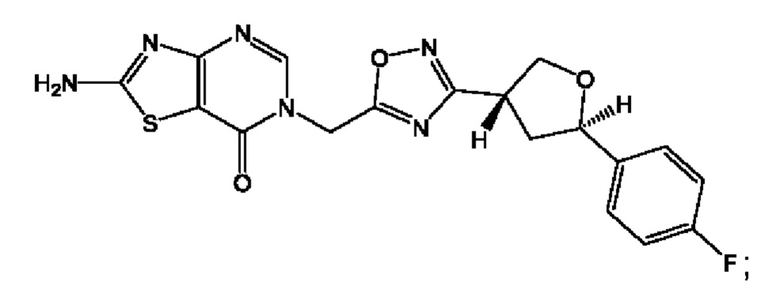
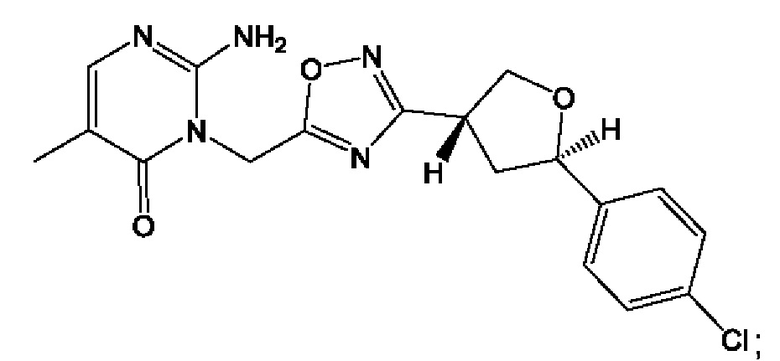
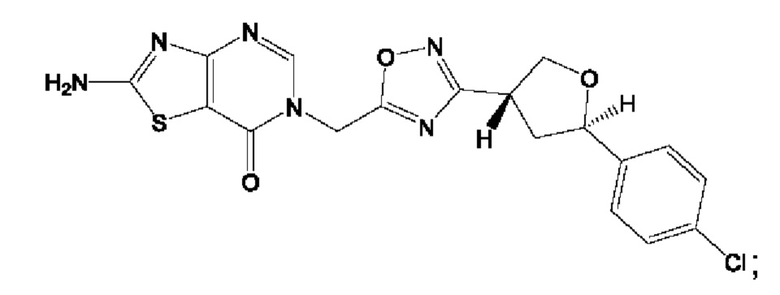
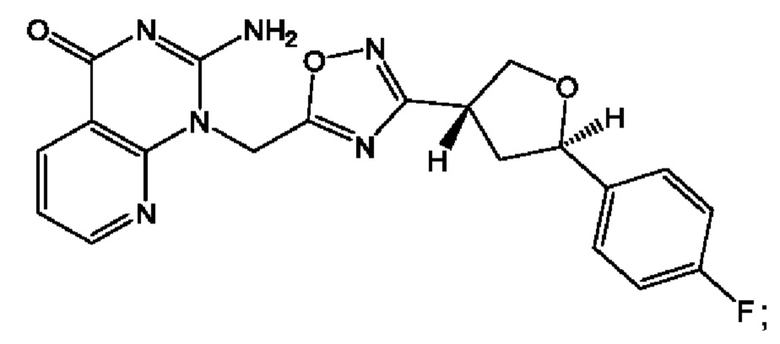
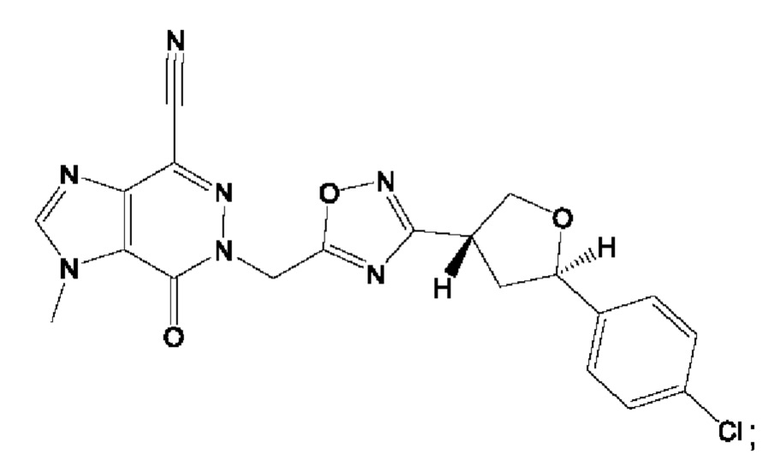
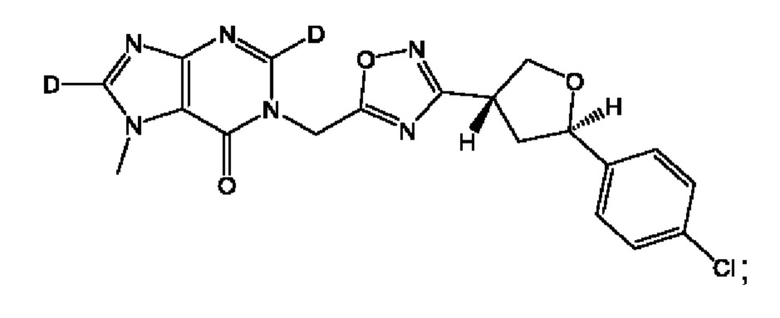
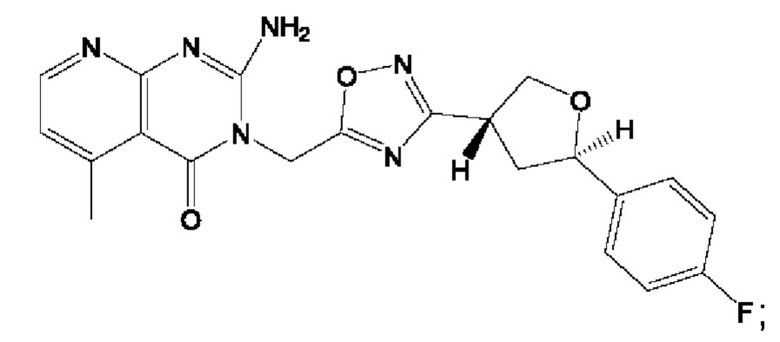
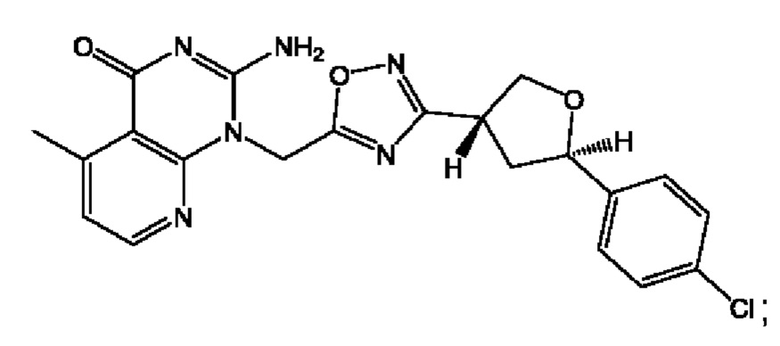
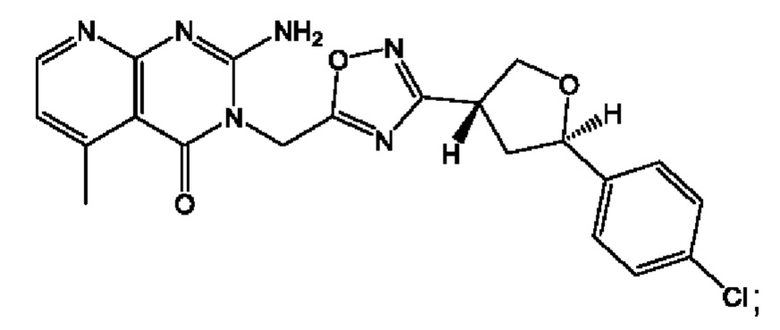
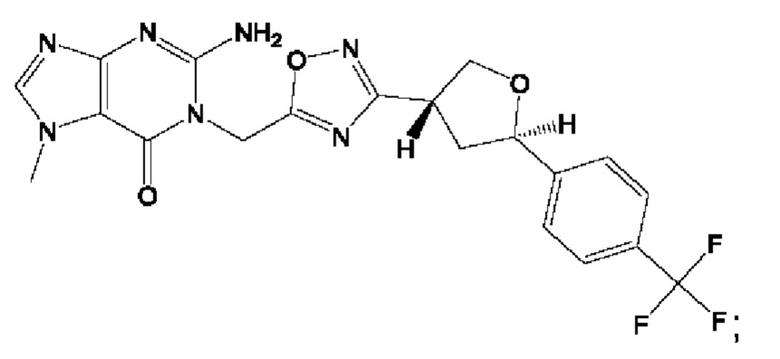
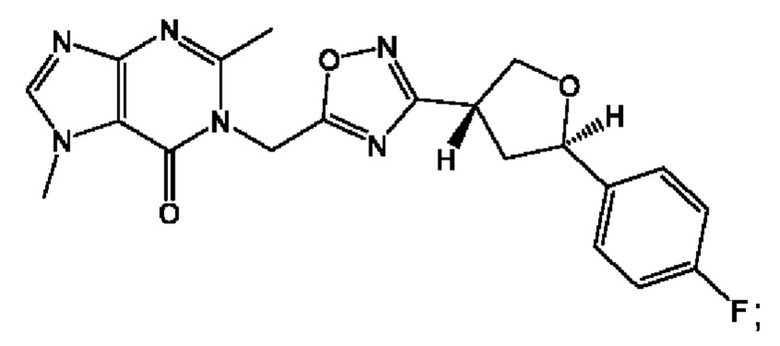
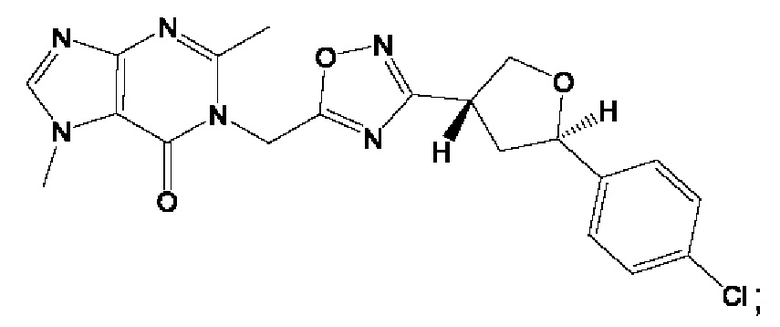
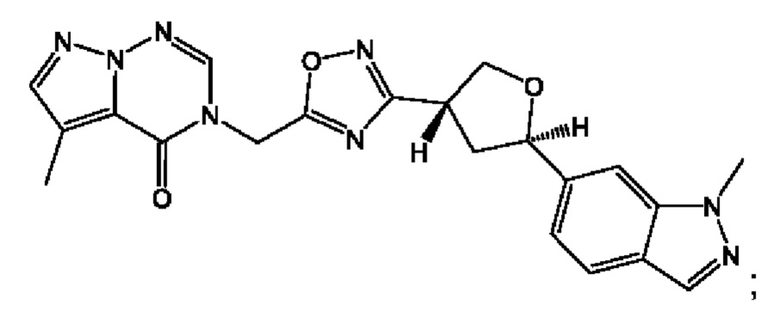
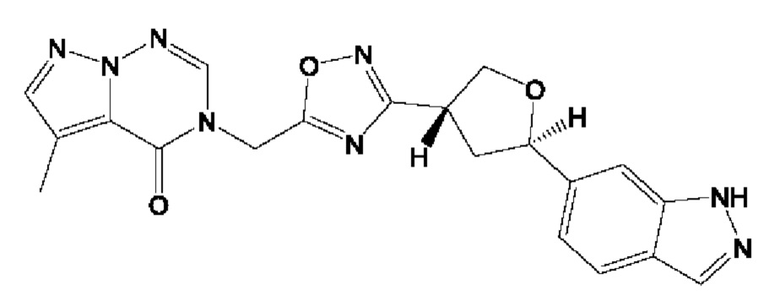
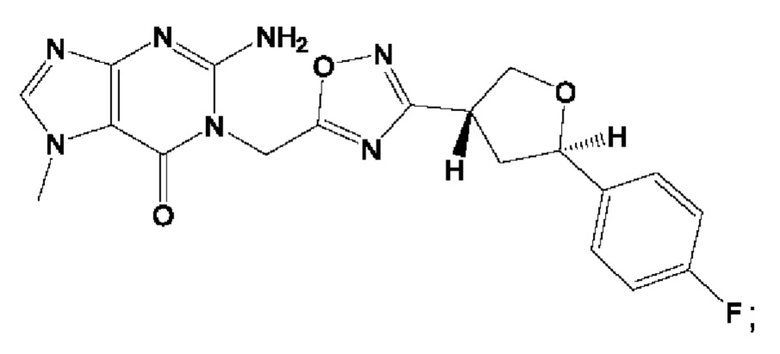
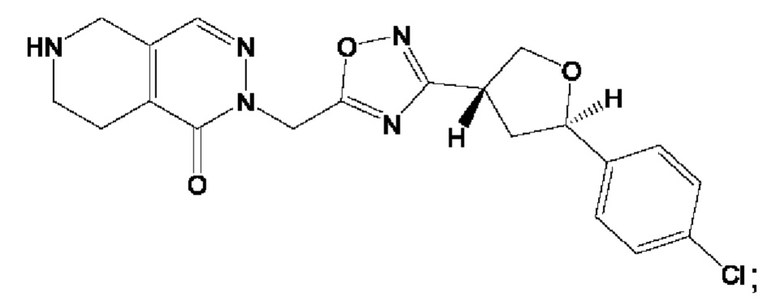
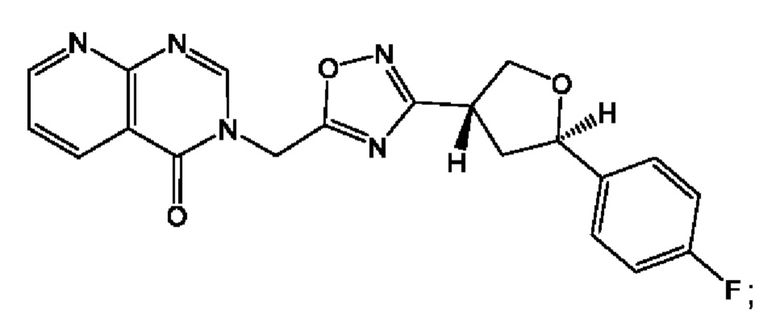
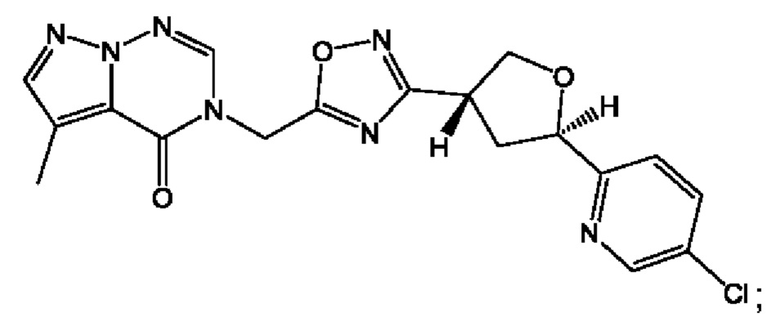
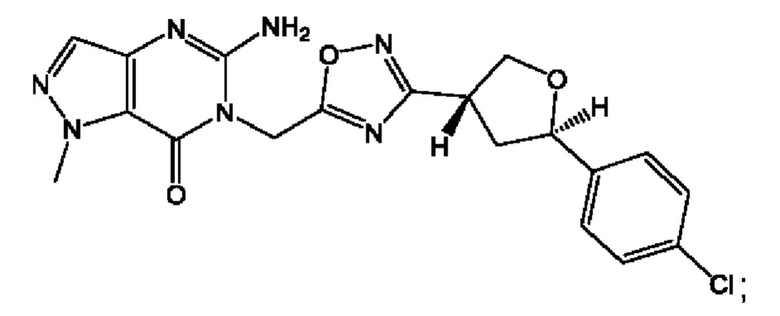
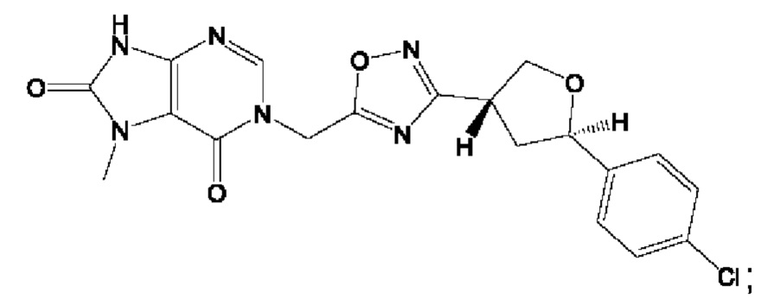
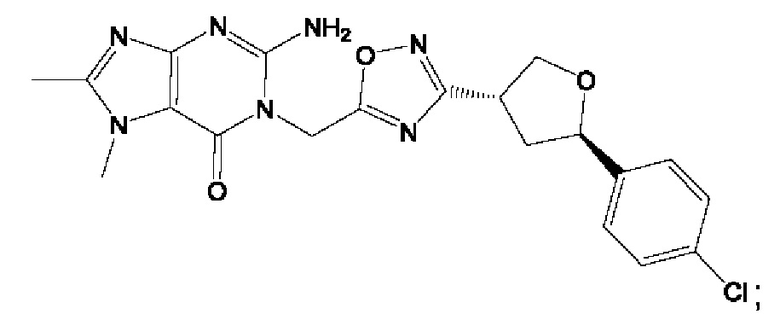
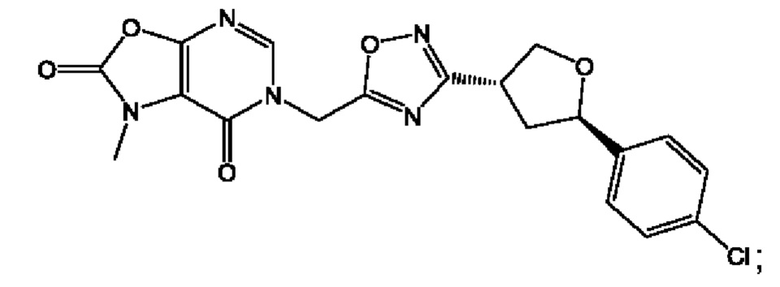
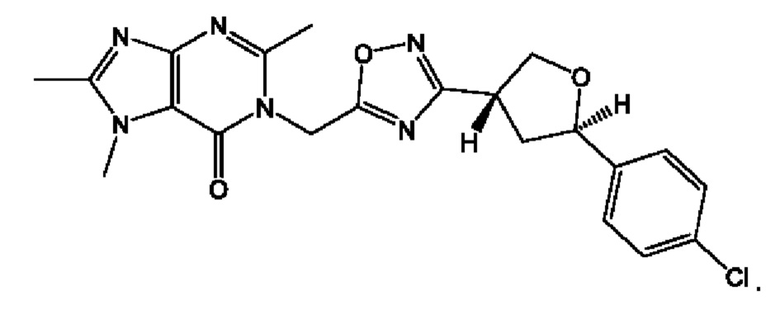
Согласно некоторым аспектам, соединение формулы (I) или его фармацевтически приемлемые соли и стереоизомеры выбраны из следующего перечня:




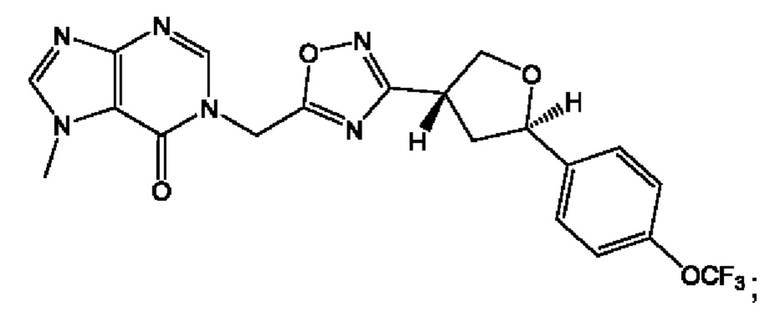
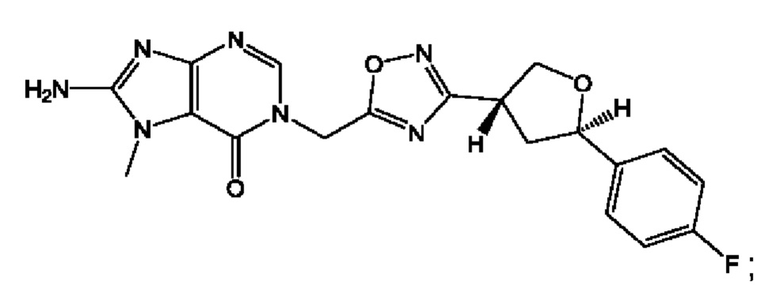
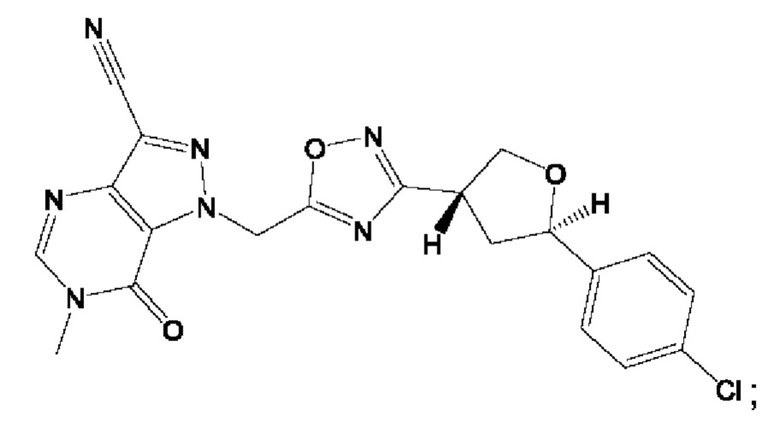

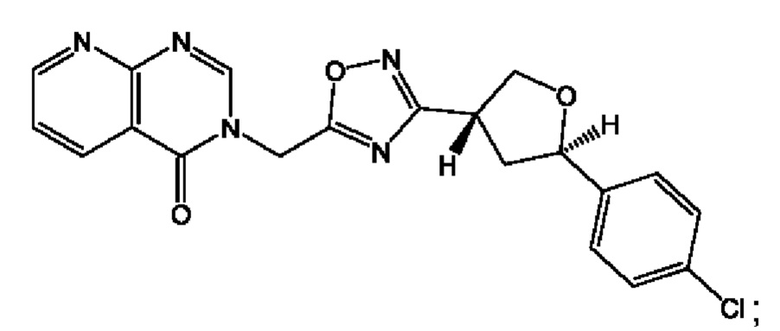




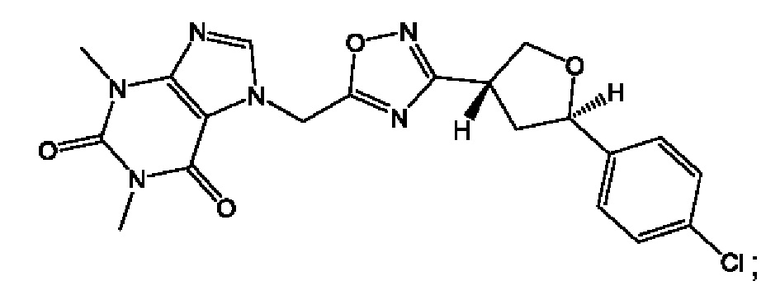
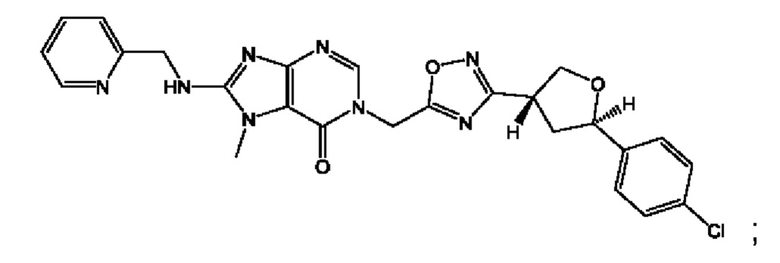
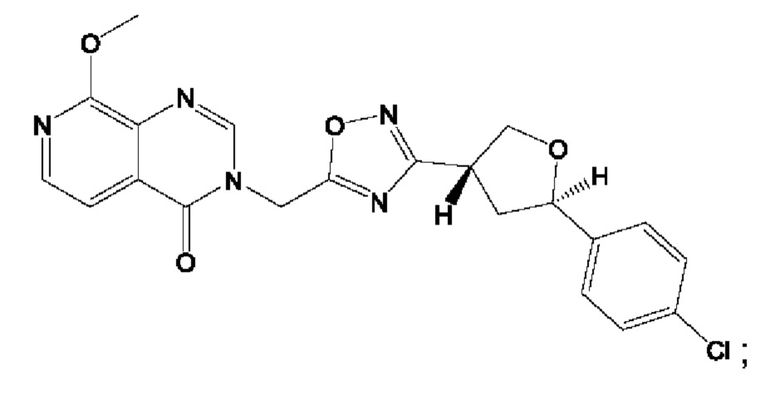
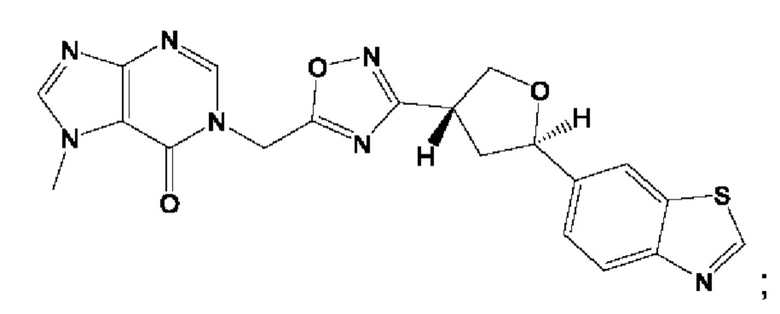





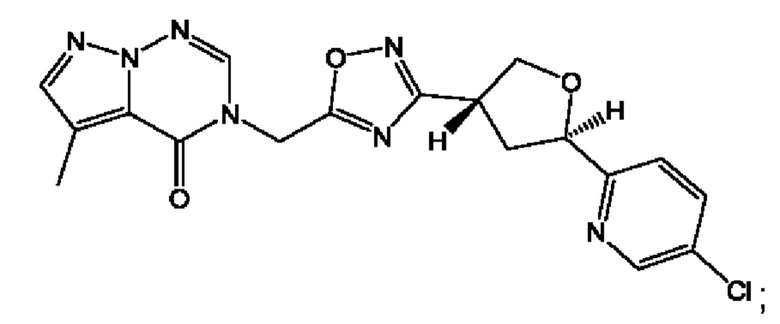
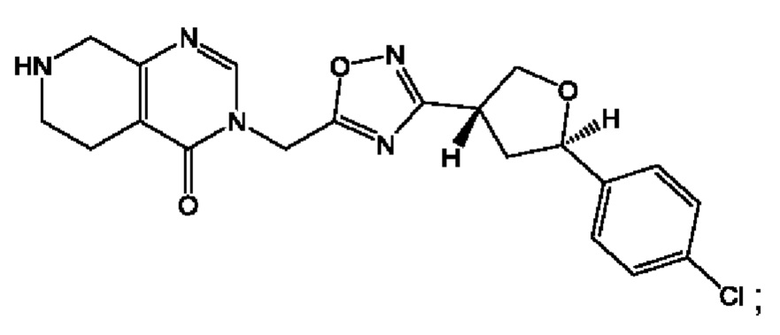
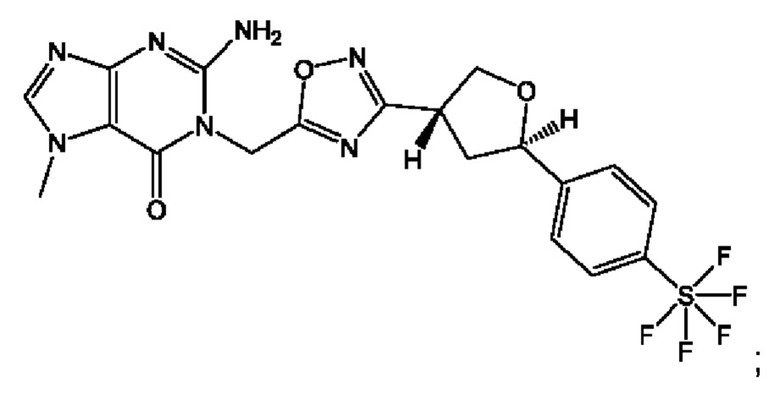
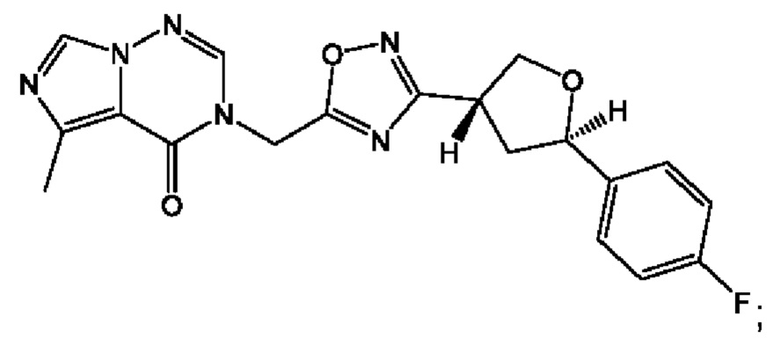
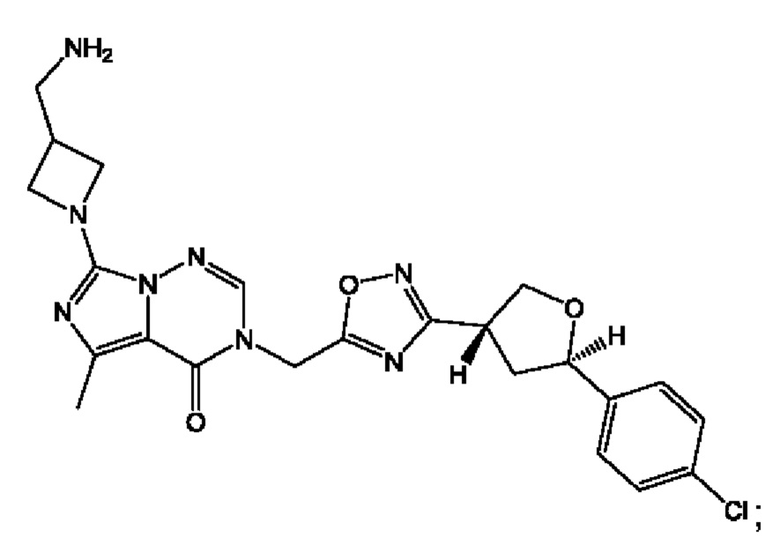
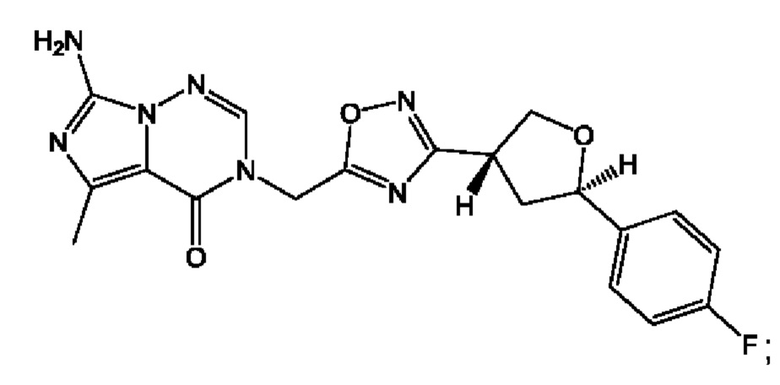
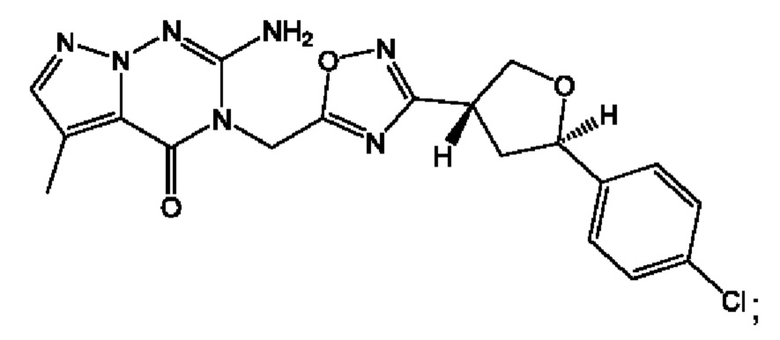

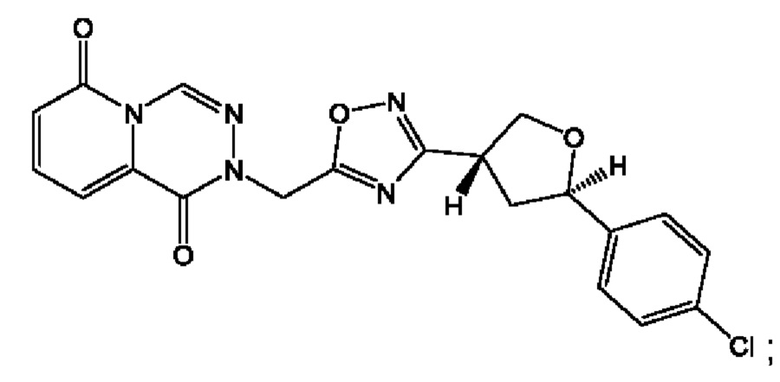
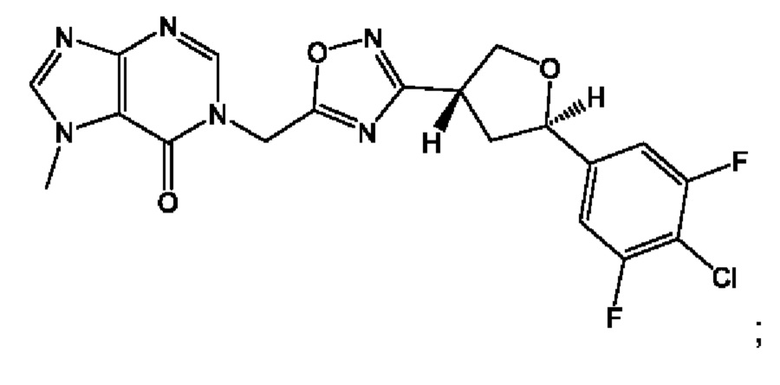
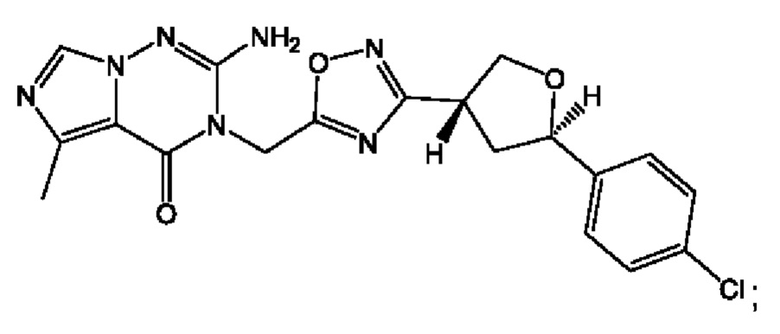
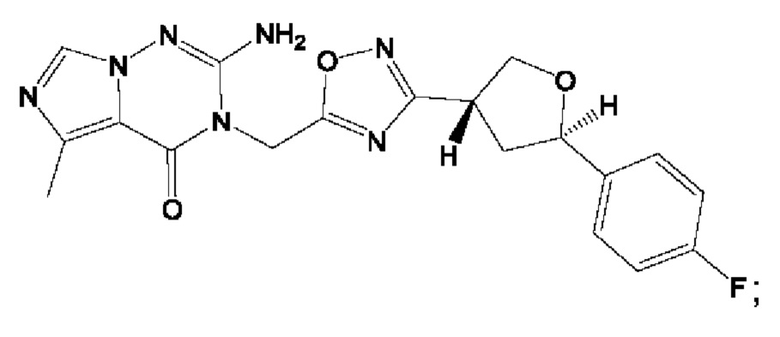
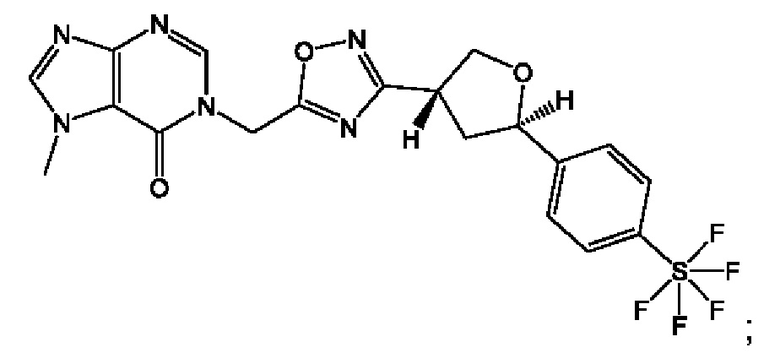
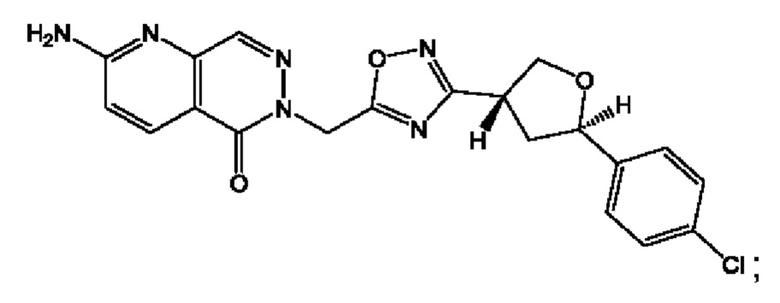
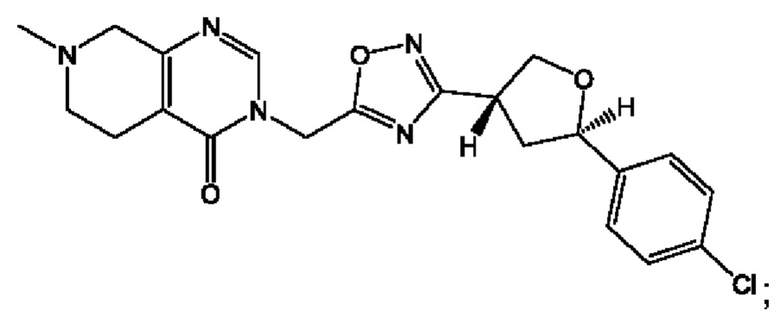
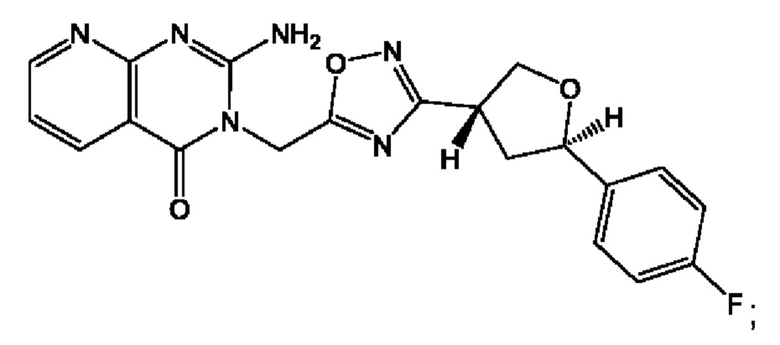
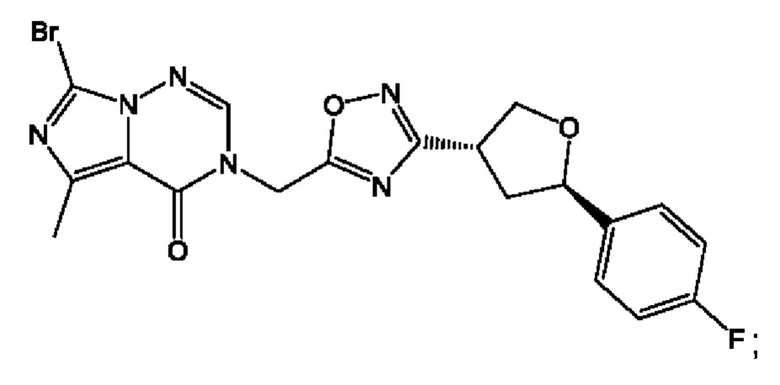



















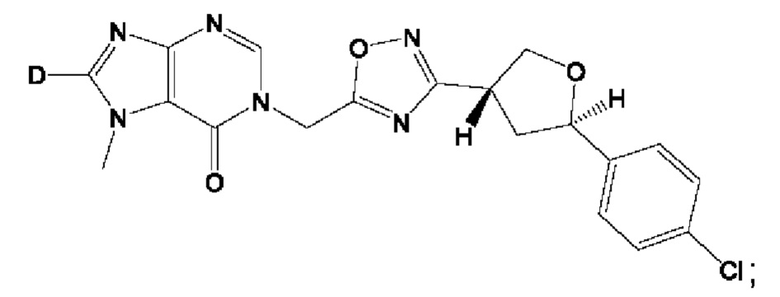
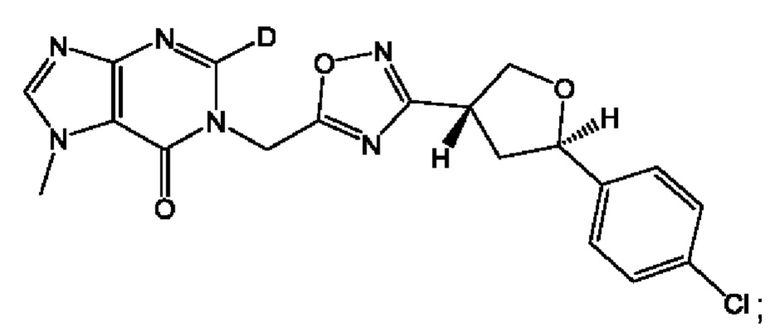
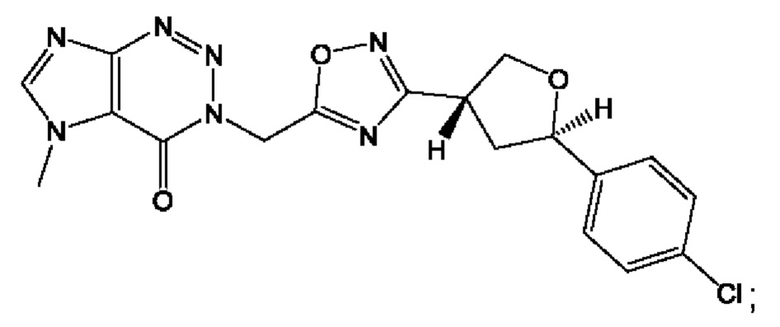
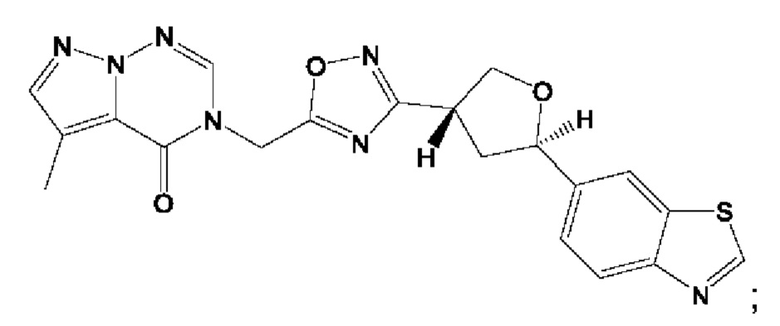
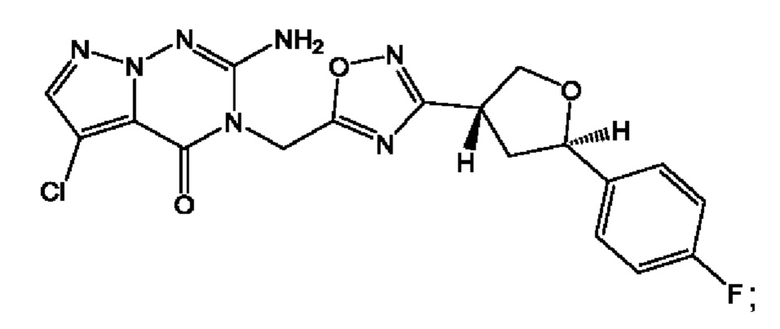





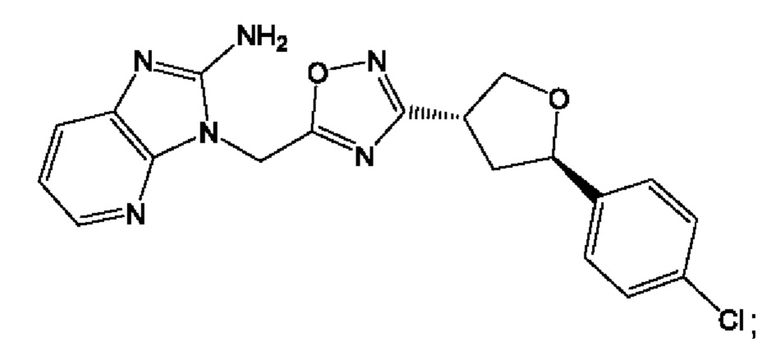
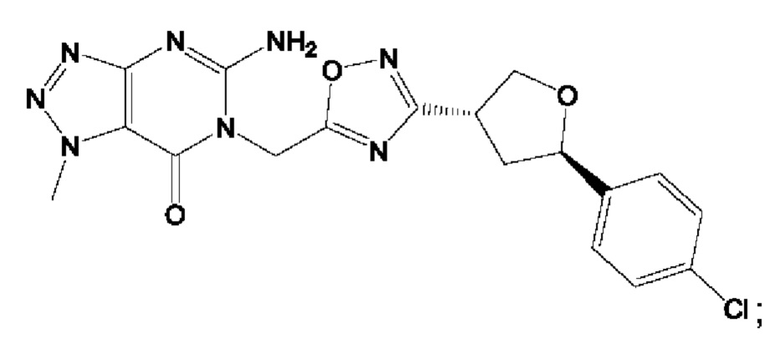
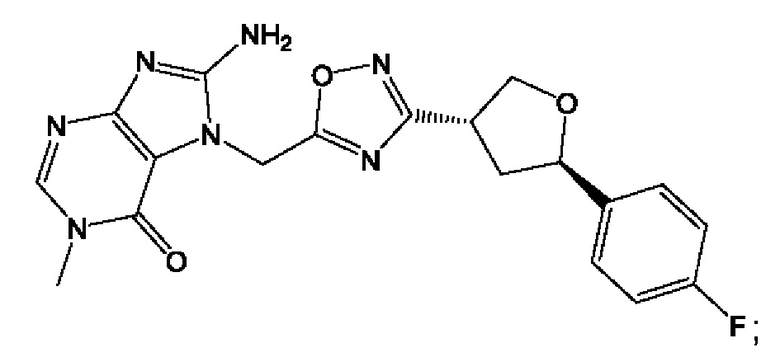
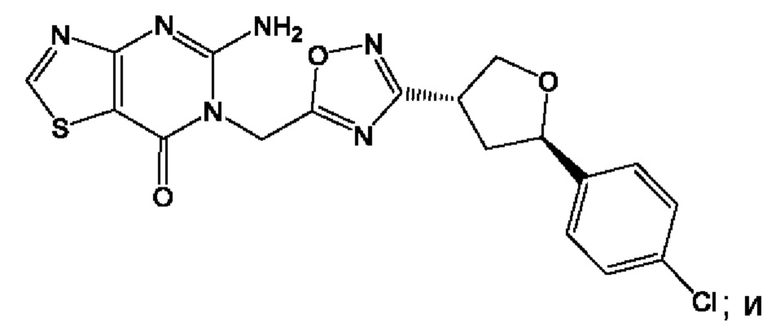
Согласно другому варианту осуществления изобретения, соединения формулы (I) являются изотопно меченными, так как содержат один или более атомов, замененных атомом, имеющим другую атомную массу или массовое число. Такие изотопно меченные (т.е. меченные радиоактивным изотопом) соединения формулы I считаются подпадающими под объем настоящего изобретения. Примеры изотопов, которые могут быть введены в соединения формулы I, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и йода, такие, как, не ограничиваясь перечнем, 2Н, 3H, 11С, 13С, 14С, 13N, 15N, 15О, 17О, 18О, 31P, 32Р, 35S, 18F, 36Cl, 123I и 125I, соответственно. Такие изотопно меченные соединения могут применяться для определения или измерения эффективности соединений, позволяя определять, например, место приложения или способ воздействия на ионные каналы, или аффинность связывания с фармакологически важным местом воздействия на ионных каналах, в частности, TRPA1. Некоторые изотопно меченные соединения формулы I, например, соединения, содержащие радиоактивный изотоп, могут применяться при изучении распределения лекарственного средства и/или субстрата в тканях. Особенно хорошо для этой цели подходят радиоактивные изотопы тритий, т.е. 3H, и углерод-14, т.е. 14С, благодаря легкости их введения и имеющихся в наличии средств их обнаружения. Например, соединение формулы I может быть обогащено такими изотопом на 1, 2, 5, 10, 25, 50, 75, 90, 95 или 99%.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечить определенные терапевтические преимущества, обусловленные более высокой метаболической стабильностью, например, увеличенным периодом полураспада, или снижением требуемой дозы. Соответственно, термин "водород" или -Н при использовании в данном контексте следует понимать, как включающий в себя дейтерий и тритий.
Замещение изотопами, испускающими позитроны, такими как 11С, 18F, 15О и 13N, может применяться при исследованиях методом позитронно-эмиссионной томографии (ПЭТ, англ. PET) для изучения степени занятости рецептора субстрата. Меченные изотопом соединения формулы I обычно получают по общепринятым методикам, известным специалисту в данной области техники, или с помощью способов, аналогичных описанным в представленных ниже Примерах, используя подходящий меченный изотопом реагент вместо использовавшегося ранее немеченного реагента.
Согласно другому варианту осуществления, изобретение предлагает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения в соответствии с формулой I и фармацевтически приемлемые носитель, разбавитель и/или вспомогательное вещество.
Наряду с солевыми формами настоящее изобретение предлагает соединения в форме пролекарств. При использовании в данном контексте термин "пролекарство" относится к соединениям, легко претерпевающим химические изменения в физиологических условиях с образованием соединений по настоящему изобретению. Кроме того, пролекарства могут быть преобразованы в соединения по настоящему изобретению при помощи химических или биохимических методов в среде ex vivo (лат. вне организма). Например, пролекарства могут медленно превращаться в соединения по настоящему изобретению, будучи помещенными в резервуар трансдермального пластыря с подходящими ферментом или химическим реагентом.
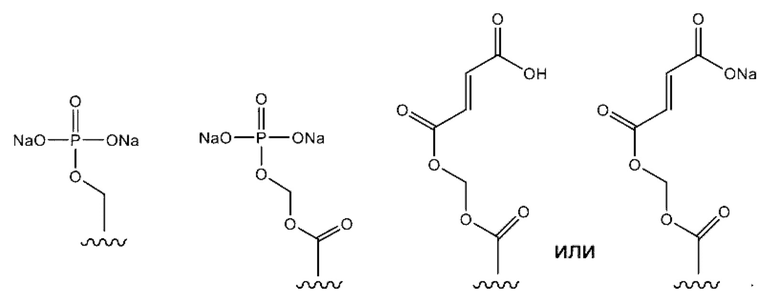
Пролекарства по изобретению могут включать фосфаты, сложные эфиры фосфатов, алкилфосфаты, сложные эфиры алкилфосфатов, простые ациловые эфиры или другие фрагменты пролекарств, такие, как будут описаны ниже. Согласно некоторым вариантам осуществления, фрагмент пролекарства представляет собой:
Также включены дополнительные типы пролекарств, например, такие, в которых аминокислотный остаток или полипептидная цепь, состоящая из двух или более (например, из двух, трех или четырех) аминокислотных остатков, ковалентно соединены посредством амидной или сложноэфирной связи со свободной аминогруппой, гидроксильной группой или карбоксильной группой соединения по настоящему изобретению. Аминокислотные остатки включают, не ограничиваясь перечнем, 20 природных аминокислот, обычно обозначаемых трехбуквенными символами, а также фосфосерин, фосфотреонин, фосфотирозин, 4-гидроксипролин, гидроксилизин, демозин, изодемозин, гамма-карбоксиглутамат, гиппуровую кислоту, окта гидроиндол-2-карбоновую кислоту, статин, 1,2,3,4-тетрагидроизохинолин-3-карбоновую кислоту, пеницилламин, орнитин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитруллин, гомоцистеин, гомосерин, метилаланин, пара-бензоилфенилаланин, фенилглицин, пропаргилглицин, сакрозин, метионинсульфон и трет-бутилглицин.
Также включены дополнительные типы пролекарств. Например, могут быть получены соединения по изобретению со свободной карбоксильной группой в виде амида или сложного алкилового эфира. В качестве другого примера, соединения по настоящему изобретению, содержащие свободные гидроксильные группы, могут быть получены в виде пролекарств путем преобразования гидроксильной группы в такие группы, как, не ограничиваясь перечнем, фосфатная сложноэфирная группа, гемисукцинатная группа, диметиламиноацетатная или фосфорилоксиметилоксикарбонильная группа, как описано в работе Fleisher, D. et al., (1996). Improved oral drug delivery: solubility limitations overcome by the use of prodrugs (Улучшенная пероральная доставка лекарственных средств: ограничения растворимости преодолеваются за счет использования пролекарств), Advanced Drug Delivery Reviews, 19:115. Кроме того, включены карбаматные пролекарства гидроксильных групп и аминогрупп, а также карбонатные пролекарства, сульфонатные эфиры и сульфатные эфиры гидроксильных групп. Также включено получение производных гидроксильных групп в виде (ацилокси)метиловых и (ацилокси)этиловых простых эфиров, где ацильная группа может быть алкиловым сложным эфиром, необязательно замещенным группами, включающими, не ограничиваясь перечнем, функциональные группы простого эфира, амина и карбоновой кислоты, или где ацильная группа представляет собой сложный эфир аминокислоты, как описано выше. Пролекарства такого типа описаны в работе J. Med. Chem., (1996), 39:10. Более конкретные примеры включают замену атома водорода спиртовой группы такой группой, как (C1-6)алканоилоксиметил, 1-((C1-6)алканоилокси)этил, 1-метил-1-((C1-6)алканоилокси)этил, (C1-6)алкоксикарбонилоксиметил, N-(C1-6)алкоксикарбониламинометил, сукциноил, (C1-6)алканоил, альфа-амино(С1-4)алканоил, арилацил и альфа-аминоацил или альфа-аминоацил-альфа-аминоацил, где каждая альфа-аминоацильная группа независимо выбрана из природных L-аминокислот, Р(O)(ОН)2, -Р(O)(O(C1-6)алкил)2 или гликозила (радикала, образующегося в результате удаления гидроксильной группы у полуацетальной формы углевода).
Дополнительные примеры производных пролекарств можно найти, например, в работах a) Design of Prodrugs (Создание пролекарств), под ред. Н. Bundgaard, (Elsevier, 1985), и Methods in Enzymology (Методы энзимологии), vol. 42, p. 309-396, под ред. K. Widder и др. (Academic Press, 1985); b) A Textbook of Drug Design and Development (Пособие по разработке и созданию лекарственных препаратов), под ред. Krogsgaard-Larsen и Н. Bundgaard, глава 5 "Design and Application of Prodrugs (Разработка и применение пролекарств)", Н. Bundgaard p. 113-191 (1991); с) Н. Bundgaard, Advanced Drug Delivery Reviews, 8:1-38 (1992); d) Н. Bundgaard, et al., Journal of Pharmaceutical Sciences, 77:285 (1988); и е) N. Kakeya, et al., Chem. Pharm. Bull., 32:692 (1984), каждая из которых определенным образом включена в настоящий документ посредством ссылки.
Кроме того, настоящее изобретение предлагает метаболиты соединений по изобретению. При использовании в данном контексте термин "метаболит" относится к продукту, образующемуся в результате метаболизма определенного соединения или его соли в организме. Такие продукты могут быть получены, например, в результате окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, дезэтерификации, ферментного расщепления и т.п., вводимого соединения.
Продукты метаболизма обычно идентифицируют с помощью получения меченного радиоактивным изотопом (например, 14С или 3H) соединения по изобретению, введения его парентерально в обнаруживаемой дозе (например, больше, чем приблизительно 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, обеспечивая при этом достаточное время для протекания метаболизма (обычно, приблизительно от 30 секунд до 30 часов), и выделения продуктов его превращения из мочи, крови или других биологических образцов. Эти продукты легко выделить, поскольку они являются мечеными (остальные выделяют с использованием антител, способных связывать эпитопы, выживающие при метаболизме). Структуру метаболитов идентифицируют обычным способом, например, при помощи анализа методом МС (англ. MS, mass spectrometry - масс-спектроскопии), ЖХ/МС (англ. LC/MS, Liquid Chromatograph Mass Spectrometer - жидкостная хроматография с масс-спектроскопией) или ЯМР (англ. NMR, nuclear magnetic resonance - ядерно-магнитный резонанс). Как правило, анализ метаболитов проводят таким же образом, как и обычные исследования метаболизма лекарственных средств, хорошо известные специалисту в данной области техники. Продукты метаболизма, если они не обнаруживаются иным образом in vivo, можно использовать в диагностических анализах для терапевтического дизирования соединений по изобретению.
Некоторые соединения по настоящему изобретению могут существовать как в несольватированных формах, так и в форме сольватов, включая гидраты. Как правило, сольватированные формы эквивалентны несольватированным и предполагается, что они входят в объем настоящего изобретения. Некоторые соединения по настоящему изобретению могут существовать в множестве кристаллических или аморфных форм. Как правило, все физические формы являются эквивалентными для применения, предусмотренного настоящим изобретением, и предполагается, что они входят в объем настоящего изобретения.
Согласно другому варианту осуществления изобретения, предложены способы получения рассматриваемых соединений. На Схеме 1 изображена общая схема синтеза соединений по изобретению, где Xa представляет собой галоген и может быть одинаковым или различным в каждом отдельном случае, Ra является арилом или C1-6алкилом и может быть одинаковым или различным в каждом отдельном случае, a R1, R18 и k такие, как указано в данном контексте.
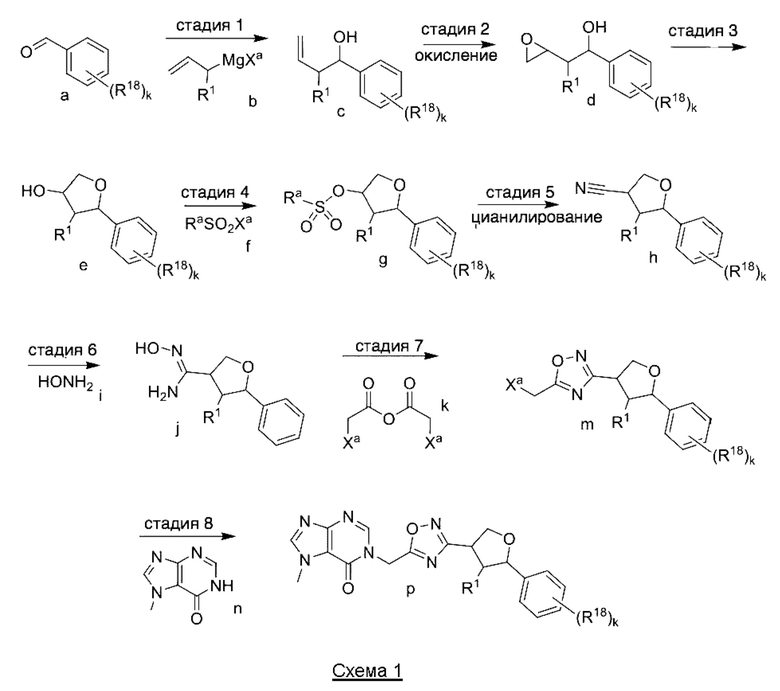
На стадии 1 Схемы 1 арилальдегид а реагирует с аллильным реактивом Гриньяра b с образованием аллиларилового спирта с. Согласно некоторым вариантам осуществления, Xa может быть галогеном, a R1 - водородом. Реакцию на стадии 1 можно проводить в полярном апротонном растворителе, например, ТГФ (англ. THF, Тетра hydro фуран - тетрагидрофуран) или т.п.
На стадии 2 выполняют окисление ненасыщенной связи соединения с, в результате получают эпоксиариловый спирт d. Эту реакцию можно осуществлять с помощью мягкого окислителя, такого как мета-хлорнадбензойная кислота, в полярном апротонном растворителе, таком как дихлорметан.
На стадии 3 выполняют перегруппировку путем обработки эпоксидного соединения d сильной кислотой, такой как серная кислота, с образованием арилгидроксилтетрагидрофурана е. Эту реакцию можно проводить в водорастворимом полярном растворителе, таком как диоксан.
На стадии 4 соединение е реагирует с сульфонилгалогенидом - реагентом f - с получением арилтетрагидрофурансульфоната g. Реакцию на стадии 4 можно проводить в присутствии катализатора амина в полярном апротонном растворителе, таком как дихлорметан. Согласно некоторым вариантам осуществления, Ra может быть метилом, а Xa - хлором.
На стадии 5 соединение g обрабатывают цианидным реагентом, таким как цианид натрия или цианид калия, для замещения сульфонатной группы и получения арилтетрагидрофураннитрила - соединения h. Эту реакцию можно проводить в полярном смешивающемся с водой растворителе, таком как диметилсульфоксид.
На стадии 6 нитрильное производное h обрабатывают гидроксиламином и получают арилтетрагидрофурангидроксилкарбоксимидамид - соединение i. Реакцию на стадии 6 можно проводить в спиртовом растворителе, таком как этанол.
На стадии 7 происходит образование цикла в ходе реакции соединения i с бис-галогенуксусным ангидридом - реагентом k, в результате образуется арилоксадиазолилтетрагидрофуран - соединение m. Эту реакцию можно проводить в полярном апротонном растворителе, таком как дихлорэтан. Согласно некоторым вариантам осуществления, Xa представляет собой хлор.
На стадии 8 выполняют N-алкилирование по реакции имидазопиримидона - соединения n - с соединением m с образованием тетрагидрофураноксадиазола - соединения р, которое представляет собой соединение формулы I в соответствии с изобретением. Реакцию на стадии 8 можно проводить в присутствии карбоната калия и йодида триалкиламмония в растворителе, таком как ДМФА (англ. DMF, dimethyl form амид - диметилформамид).
На Схеме 2 изображен другой способ получения соединений по изобретению.
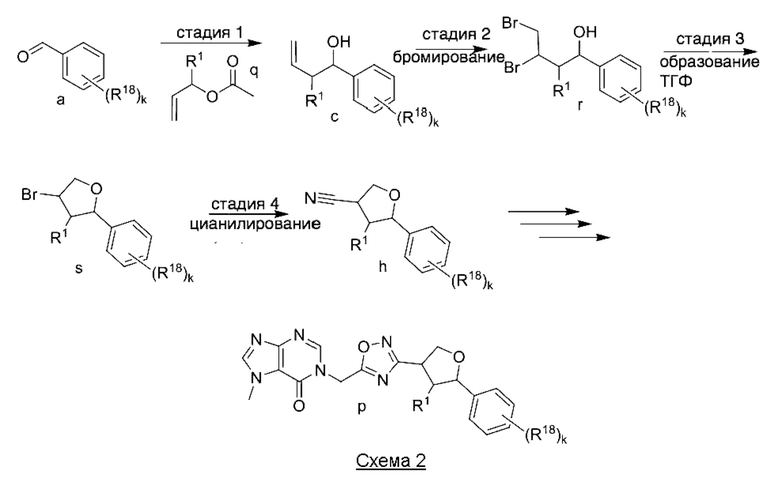
На стадии 1 Схемы 2 арилальдегид а взаимодействует с аллилацетатным реагентом q с получением арилаллилового спирта - соединения с. Для придания соединению с требуемой стереохимии на стадии 1 может быть использован реагент для хирального или асимметричного синтеза, такой как (R) или (S) BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтрил). В этом случае реакцию на стадии 1 можно проводить в присутствии карбоната цезия, 4-хлор-3-нитробензойной кислоты и/или димера галоген(циклооктадиен)иридия (I) в спиртовом растворителе, таком как изопропанол.
На стадии 2 соединение c бромируют с образованием дибромпроизводного r. Реакцию на стадии 2 можно проводить путем обработки соединения c непосредственно бромом в полярном апротонном растворителе, таком как дихлорметан.
На стадии 3 выполняют циклизацию с получением арилбромтетрагидрофурана - соединения s. Реакцию на стадии 3 можно проводить путем обработки соединения r карбонатом калия в спиртовом растворителе, таком как метанол.
На стадии 4 арилбромтетрагидрофуран - соединение s - обрабатывают цианатом с образованием арилтетрагидрофураннитрила - соединения h. Для этой реакции можно использовать цианат калия в диметилсульфоксиде или подобном растворителе.
Из соединения h, выполняя стадии 6, 7 и 8 Схемы 1, можно получить тетрагидрофураноксадиазол - соединение p, представляющее собой соединение формулы I в соответствии с изобретением.
В рамках данного изобретения возможны различные варианты описанных выше процедур, их смогут порекомендовать специалисты в данной области техники. Вместо имидазопиримидона n можно использовать некоторые другие бициклические гетероарильные соединения, как будет показано в приведенных ниже экспериментальных примерах. Для получения конкретных требуемых стереоизомеров конечного соединения p и некоторых промежуточных соединений можно использовать разделение с помощью хиральной колонки, как будет описано ниже в экспериментальных примерах.
Фармацевтические композиции и введение
Наряду с одним или более соединениями, представленными выше (включая их стереоизомеры, таутомеры, сольваты, метаболиты, изотопы, фармацевтически приемлемые соли или пролекарства), изобретение также предлагает композиции и лекарственные средства, содержащие соединение формулы I или/и его вариант осуществления и по меньшей мере один фармацевтически приемлемый носитель. Композиции по изобретению могут применяться для селективного ингибирования TRPA1 у пациентов (например, людей).
Термин "композиция" при использовании в данном контексте, означает продукт, содержащий определенные ингредиенты в определенных количествах, а также любой продукт, прямо или косвенно являющийся результатом комбинации определенных ингредиентов в определенных количествах.
Согласно одному из вариантов осуществления, изобретение предлагает фармацевтические композиции или лекарственные средства, содержащие соединение формулы I или его вариант осуществления и его стереоизомеры, таутомеры, сольваты, метаболиты, изотопы, фармацевтически приемлемые соли или их пролекарства и фармацевтически приемлемые носитель, разбавитель или вспомогательное вещество. Согласно другому варианту осуществления, изобретение предлагает приготовление композиций (или лекарственных средств), содержащих соединения по изобретению. Согласно другому варианту осуществления, изобретение предлагает введение соединения формулы I или его вариантов осуществления и композиций, содержащих соединение формулы I или его вариант осуществления, пациенту (например, человеку), нуждающемуся в этом.
Композиции готовят, дозируют и вводят способом, не противоречащим надлежащей медицинской практике. Факторы, которые следует учитывать в данном контексте, включают конкретное заболевание, подлежащее лечению, конкретное млекопитающее, подлежащее лечению, клиническое состояние отдельного пациента, причину заболевания, место доставки агента, способ введения, схему введения и другие факторы, известные практикующему врачу. Эффективное количество соединения, подлежащего введению, будет определяться этими факторами и представляет собой минимальное количество, необходимое для ингибирования активности TRPA1, требующееся для предотвращения или лечения нежелательного заболевания или расстройства, такого как, например, боль. Например, это количество может быть меньше количества, токсичного для нормальных клеток или млекопитающего в целом.
Согласно одному из примеров, терапевтически эффективное количество соединения по изобретению, вводимое парентерально на одну дозу будет составлять приблизительно от 0,01 до 100 мг/кг, в качестве альтернативы, например, приблизительно от 0,1 до 20 мг/кг массы тела пациента в день, при этом ориентировочный начальный диапазон используемого соединения составляет от 0,3 до 15 мг/кг/день. Согласно некоторым вариантам осуществления, суточные дозы вводят в виде однократной суточной дозы или разделенными дозами от двух до шести раз в день либо в форме с замедленным высвобождением. В случае взрослого человека с массой тела 70 кг общая суточная доза обычно составляет приблизительно от 7 мг до 1400 мг. Этот режим дозирования может быть скорректирован для обеспечения оптимального терапевтического ответа. Соединения можно вводить по схеме от 1 до 4 раз в день, предпочтительно, один или два раза в день.
Соединения по настоящему изобретению можно вводить в любой удобной для введения форме, например, в виде таблеток, порошков, капсул, растворов, дисперсий, суспензий, сиропов, спреев, суппозиториев, гелей, эмульсий, пластырей и т.д. Такие композиции могут содержать компоненты, стандартные для фармацевтических препаратов, например, разбавители, носители, модификаторы рН, подсластители, объемообразующие агенты и добавочные активные агенты.
Соединения по настоящему изобретению можно вводить любыми подходящими способами, включая пероральное, местное (в том числе защечное (буккальное) и подъязычное (сублингвальное)), ректальное, вагинальное, чрескожное (трансдермальное), парентеральное, подкожное, внутрибрюшинное (интраперитонеальное), внутрилегочное, внутрикожное, подоболочечное (интратекальное), эпидуральное и интраназальное введение, а также, если это необходимо для местного лечения, внутриочаговое введение. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное, внутрибрюшинное, внутримозговое, внутриглазное, внутричерепное или подкожное введение.
Композиции, содержащие соединение формулы I или его вариант осуществления, обычно готовят в соответствии со стандартной фармацевтической практикой в виде фармацевтической композиции. Типичный состав получают путем смешивания соединения по настоящему изобретению и разбавителя, носителя или вспомогательного вещества. Подходящие разбавители, носители и вспомогательные вещества хорошо известны специалистам в данной области и подробно описаны, например, в работах Ansel, Howard С., et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems (Лекарственные формы и системы доставки лекарственных средств). Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy (Наука и практика фармации). Philadelphia: Lippincott, Williams & Wilkins, 2000; и Rowe, Raymond C. Handbook of of Pharmaceutical Excipients (Справочник по фармацевтическим вспомогательным веществам). Chicago, Pharmaceutical Press, 2005. Рецептуры могут также содержать один или более буферов, стабилизирующих агентов, поверхностно-активных веществ, увлажняющих агентов, смазывающих агентов, эмульгаторов, суспендирующих агентов, консервантов, антиокислителей, кроющих агентов, веществ, способствующих скольжению, технологических добавок, красителей, подсластителей, ароматизирующих добавок, вкусо-ароматических добавок, разбавителей и других известных добавок для обеспечения эстетичной формы лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или для помощи при изготовлении фармацевтического продукта (т.е. лекарственного средства). Подходящие носители, разбавители и вспомогательные вещества хорошо известны специалистам в данной области и включают в себя буферы, такие как фосфатный буфер, цитратный буфер и буфер на основе других органических кислот; антиокислители, включая аскорбиновую кислоту и метионин; консерванты (такие как хлорид октадецилдиметилбензиламмония; хлорид гексаметония; хлорид бензалкония, хлорид бензетония; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; катехол; резорцин; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как EDTA (англ. ethylene diamine тетра acetic acid - этилендиаминтетрауксусная кислота); сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексные соединения металлов (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (PEG). Активный фармацевтический ингредиент по изобретению (например, соединение формулы I или его вариант осуществления) также может быть заключен в микрокапсулу, полученную, например, методом коацервации или межфазной полимеризации, например, в гидроксиметилцеллюлозную или желатиновую микрокапсулу и капсулу из поли(метилметацилата), соответственно, в коллоидные системы доставки лекарственного средства (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие способы раскрыты в работе Remington: The Science and Practice of Pharmacy: Remington the Science and Practice of Pharmacy (2005) 21st Edition, Lippincott Williams & Wilkins, Philadelphia, PA. Конкретные используемые носитель, разбавитель или вспомогательное вещество будут зависеть от средств и цели, для которой применяется соединение по настоящему изобретению. Растворители обычно выбирают на основе растворителей, признанных специалистами в данной области безопасными (GRAS, англ. Generally Recognised as Safe - признанные полностью безопасными) для введения млекопитающему. Как правило, безопасные растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, являющиеся растворимыми в воде или смешивающимися с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, PEG 400, PEG 300) и т.д. и их смеси. Приемлемые разбавители, носители, вспомогательные вещества и стабилизаторы являются нетоксичными для реципиентов в используемых дозировках и концентрациях.
Могут быть получены препараты с замедленным высвобождением соединения по изобретению (например, соединения формулы I или его варианта осуществления). Подходящие примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение формулы I или его вариант осуществления, при этом матрицы могут иметь форму профилированных изделий, например, пленок или микрокапсул. Примеры матриц с замедленным высвобождением включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)), полилактиды (патентный документ США No. 3773919), сополимеры L-глутаминовой кислоты и гамма-этил-L-глутамата (Sidman et al., Biopolymers. 22:547, 1983), неразлагаемый этилен-винилацетат (Langer et al., J. Biomed. Mater. Res. 15:167, 1981), разлагаемые сополимеры молочной кислоты и гликолевой кислоты, такие как LUPRON DEPOT™ (микросферы для инъекций, состоящие из сополимера молочной кислоты-гликолевой кислоты и ацетата лейпролида) и поли-D-(-)-3-гидроксимасляная кислота (ЕР 133988 А). Композиции с замедленным высвобождением также включают соединения, заключенные в липосому, которые могут быть получены известными способами (Epstein et al., Proc. Natl. Acad. Sci. U.S.A. 82:3688, 1985; Hwang et al., Proc. Natl. Acad. Sci. U.S.A. 77:4030, 1980; патентные документы США No. 4485045 и 4544545; и ЕР 102324 А). Обычно липосомы имеют небольшой размер (приблизительно от 200 до 800 ангстрем) и относятся к однослойному типу с содержанием липидов более, чем приблизительно 30 мол. % холестерина, при этом выбранное содержание корректируется для оптимальной терапии.
Согласно одному из примеров, соединения формулы I или его вариант осуществления могут быть получены путем смешивания при температуре окружающей среды, соответствующей величине рН и требуемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, нетоксичными для реципиентов в дозировках и концентрациях, используемых в форме галенового введения. Величина рН рецептуры зависит главным образом от конкретного применения и концентрации соединения, но предпочтительно находится в диапазоне приблизительно от 3 до 8. Согласно одному из примеров, соединение формулы I (или его вариант осуществления) готовят в ацетатном буфере при рН 5. Согласно другому варианту осуществления, соединения формулы I или его вариант осуществления являются стерильными. Соединение может храниться, например, в виде твердой или аморфной композиции, в виде лиофилизированной композиции либо в виде водного раствора.
Рецептуры соединения по изобретению (например, соединения формулы I или его варианта осуществления), подходящие для перорального введения, могут быть приготовлены в виде дискретных компонентов, таких как пилюли, капсулы, крахмальные капсулы или таблетки, каждая из которых содержит определенное количество соединения по изобретению.
Прессованные таблетки могут быть изготовлены путем прессования на соответствующем устройстве активного ингредиента в сыпучей форме, такой как порошок или гранулы, необязательно в смеси со связующим, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены путем формования на соответствующем устройстве смеси порошкообразного активного ингредиента, увлажненного жидким инертным разбавителем. Таблетки необязательно могут иметь покрытие или иметь насечки и необязательно могут быть приготовлены таким образом, чтобы обеспечивать медленное или контролируемое высвобождение из них активного ингредиента.
Для перорального применения могут быть приготовлены таблетки, пастилки, таблетки для рассасывания, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, например, желатиновые капсулы, сиропы или эликсиры. Рецептуры соединения по изобретению (например, соединения формулы I или его варианта осуществления), предназначенные для перорального применения, могут быть приготовлены в соответствии с любым известным способом приготовления фармацевтических композиций, при этом такие композиции могут содержать один или более агентов, включая подсластители, вкусо-ароматические добавки, красители и консерванты, для получения приятного на вкус препарата. Допустимы таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым вспомогательным веществом, подходящим для приготовления таблеток. Такими вспомогательными веществами могут быть, например, инертные разбавители, такие как карбонат кальция или натрия, лактоза, фосфат кальция или натрия; гранулирующие агенты и вещества для улучшения распадаемости таблеток, такие как кукурузный крахмал или альгиновая кислота; связующие агенты, такие как крахмал, желатин или аравийская камедь; и смазывающие агенты, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или на них могут быть нанесены покрытия при помощи известных способов, включая микрокапсулирование для задержки распада и адсорбции в желудочно-кишечном тракте и, посредством этого, обеспечения пролонгированного действия в течение более длительного периода. Например, может быть использован материал с задержкой по времени, такой как глицерилмоностеарат или глицерилдистеарат, отдельно или вместе с воском.
Примером подходящей формы для перорального применения является таблетка, содержащая приблизительно 1 мг, 5 мг, 10 мг, 25 мг, 30 мг, 50 мг, 80 мг, 100 мг, 150 мг, 250 мг, 300 мг и 500 мг соединения по изобретению, смешанного с приблизительно от 90 до 30 мг безводной лактозы, приблизительно от 5 до 40 мг кроскармеллозы натрия, приблизительно от 5 до 30 мг поливинилпирролидона (PVP) K30 и приблизительно от 1 до 10 мг стеарата магния. Порошкообразные ингредиенты сначала смешивают друг с другом, а затем - с раствором PVP. Полученная в результате композиция может быть высушена, гранулирована, смешана со стеаратом магния и спрессована с получением таблетированной формы при помощи стандартного оборудования. Пример аэрозольной рецептуры можно получить растворением соединения по изобретению в количестве, например, от 5 до 400 мг, в приемлемом буферном растворе, например, в фосфатном буфере, добавлением, при необходимости, модификатора тоничности, например, соли, такой как хлорид натрия. Раствор может быть профильтрован, например, с помощью фильтра 0,2 микрон, для удаления примесей и загрязнений.
Для лечения глаз и других внешних тканей, например, полости рта и кожи, предпочтительно применяют рецептуры в виде мази или крема для местного применения, содержащие активный ингредиент (ингредиенты) в количестве, например, от 0,075 до 20 мас. %. В составе мази активный ингредиент может использоваться вместе с парафиновой или смешивающейся с водой мазевой основой. В качестве альтернативы, активные ингредиенты могут быть приготовлены в виде крема с кремовой основой типа «масло в воде». При необходимости водная фаза основы для крема может включать многоатомный спирт, т.е. спирт, содержащий две или более гидроксильных групп, такой как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая PEG 400) и их смеси. Составы для местного применения могут при необходимости содержать соединение, усиливающее абсорбцию или проникновение активного ингредиента через кожу или другие пораженные участки. Примеры таких усилителей проникновения через кожу включают диметилсульфоксид и родственные аналоги.
В случае составов для местного применения желательно вводить эффективное количество фармацевтической композиции в соответствии с изобретением в целевую область, например, поверхность кожи, слизистую оболочку и т.п., примыкающую к периферическим нейронам, подлежащим лечению. Это количество обычно составляет приблизительно от 0,0001 мг до 1 г соединения по изобретению на одно применение в зависимости от области, подлежащей лечению, того, является ли применение диагностическим, профилактическим или терапевтическим, тяжести симптомов и природы используемого местного носителя. Предпочтительным препаратом для местного применения является мазь, содержащая приблизительно от 0,001 до 50 мг активного ингредиента на кубический сантиметр мазевой основы. Фармацевтическая композиция может быть приготовлена в виде трансдермальных композиций или средств трансдермальной доставки ("пластырей"). Такие композиции включают, например, подложку, резервуар с активным соединением, регулирующую мембрану, вкладыш и контактный клей. Такие трансдермальные пластыри могут применяться для обеспечения непрерывной пульсирующей доставки или доставки по требованию соединений по настоящему изобретению.
Составы могут быть упакованы в однодозовые упаковки или многодозовые контейнеры, например, запаянные ампулы и флаконы, и могут храниться в высушенном замораживанием (лиофилизированном) состоянии, требующем лишь добавления стерильного жидкого носителя, например, воды, для инъекций непосредственно перед применением. Экстемпоральные инъекционные растворы и суспензии готовят из стерильных порошков, гранул и таблеток описанного ранее вида. Предпочтительными составами единичных дозированных форм являются те из них, которые содержат суточную дозу или часть единичной суточной дозы, как указано выше в данном документе, либо ее соответствующую фракцию, активного ингредиента.
В случае, если мишень для связывания локализована в головном мозге, некоторые варианты осуществления изобретения предлагают соединение формулы I (или его вариант осуществления) для преодоления гематоэнцефалического барьера. Некоторые нейродегенеративные расстройства связаны с повышением проницаемости гематоэнцефалического барьера, так что соединение формулы I (или его вариант осуществления) может быть легко индуцировано в головной мозг. Если гематоэнцефалический барьер остается неизмененным, существует несколько известных в данной области техники подходов для переноса молекул через него, включая, но не ограничиваясь перечнем, физические методы, методы, основанные на применении липидов, и методы, основанные на применении рецепторов и каналов.
Физические методы переноса соединения формулы I (или его варианта осуществления) через гематоэнцефалический барьер включают, не ограничиваясь перечнем, полный обход гематоэнцефалического барьера или создание отверстий в гематоэнцефалическом барьере.
Методы обхода включают, не ограничиваясь перечнем, непосредственное введение в головной мозг (см., например, Papanastassiou et al., Gene Therapy. 9:398-406, 2002), внутритканевую инфузию/конвекционную усовершенствованную доставку (см., например, Bobo et al., Proc. Natl. Acad. Sci. U.S.A. 91:2076-2080, 1994) и имплантацию устройства доставки в головной мозг (см., например, Gill et al., Nature Med. 9:589-595, 2003; и Gliadel Wafers™, Guildford).
Методы создания отверстий в барьере включают, не ограничиваясь перечнем, ультразвук (см., например, публикацию заявки на патент США No. 2002/0038086), осмотическое давление (например, посредством введения гипертонического раствора маннита (Neuwelt, E.A., Implication of the Blood-Brain Barrier and its Manipulation (Роль гематоэнцефалического барьера и операции с ним), Volumes 1 и 2, Plenum Press, N.Y., 1989)) и изменение проницаемости с помощью, например, брадикинина или пермеабилизатора А-7 (см., например, патентные документы США No. 5,112,596, 5,268,164, 5,506,206 и 5,686,416).
Основанные на применении липидов методы переноса соединения формулы I (или его варианта осуществления) через гематоэнцефалический барьер включают, не ограничиваясь перечнем, инкапсулирование соединения формулы I (или его варианта осуществления) в липосомы, которые соединены с антителосвязывающими фрагментами, связывающимися с рецепторами на эндотелии сосудов гематоэнцефалического барьера (см., например, публикацию заявки на патент США No. 2002/0025313), и покрытие соединения формулы I (или его варианта осуществления) частицами липопротеинов низкой плотности (см., например, публикацию заявки на патент США No. 2004/0204354) или аполипротеинами Е (см., например, публикацию заявки на патент США No. 2004/0131692).
Основанные на применении рецепторов и каналов методы переноса соединения формулы I (или его варианта осуществления) через гематоэнцефалический барьер включают, не ограничиваясь перечнем, использование глюкокортикоидных блокаторов для повышения проницаемости гематоэнцефалического барьера (см., например, публикации заявок на патент США No. 2002/0065259, 2003/0162695 и 2005/0124533); активацию калиевых каналов (см., например, публикацию заявки на патент США No. 2005/0089473), ингибирование транспортеров лекарственных средств АВС (см., например, публикацию заявки на патент США No. 2003/0073713); покрытие соединения формулы I (или его варианта осуществления) трансферрином и модуляцию активности одного или более трансферриновых рецепторов (см., например, публикацию заявки на патент США No. 2003/0129186) и катионизацию антител (см., например, патентный документ США No. 5,004,697).
В случае внутримозгового применения, согласно некоторым вариантам осуществления, соединения можно вводить непрерывно путем инфузии в резервуары жидкостей ЦНС, но может применяться и болюсная инъекция. Ингибиторы можно подавать в желудочки головного мозга или вводить иным образом в ЦНС или спинномозговую жидкость. Введение можно проводить с помощью постоянного катетера и средства непрерывного введения, такого как помпа, или же соединение можно вводить путем имплантации, например, внутримозговой имплантации основы с замедленным высвобождением. Более конкретно, ингибиторы можно вводить через постоянно имплантированные канюли либо постоянно подавать с помощью осмотических мини-насосов. Существуют подкожные насосы, доставляющие белки через небольшую трубку в желудочки головного мозга. Высокотехнологичные насосы можно перезаполнять через кожу, при этом их скорость подачи может быть отрегулирована без хирургического вмешательства. Примерами подходящих протоколов введения и систем доставки, включающих в себя подкожное насосное устройство или непрерывную интрацеребровентрикулярную инфузию через полностью имплантированную систему доставки лекарственного средства являются протоколы, используемые для введения допамина, агонистов допамина и холинергических агонистов пациентам с болезнью Альцгеймера и моделям болезни Паркинсона на животных, как описано в работах Harbaugh, J. Neural Transm. Suppl. 24:271, 1987; и DeYebenes et al., Mov. Disord. 2: 143, 1987.
Показания и способы лечения
Было показано, что типичные соединения по изобретению модулируют активность TRPA1. Соответственно, соединения по изобретению могут применяться для лечения заболеваний и состояний, опосредованных активностью TRPA1. Такие заболевания и состояния включают, не ограничиваясь перечнем: боль (острую, хроническую, воспалительную или нейропатическую боль); зуд или различные воспалительные заболевания; заболевания внутреннего уха; лихорадочное состояние или другие расстройства терморегуляции; трахеобронхиальную или диафрагмальную дисфункцию; заболевания желудочно-кишечного тракта или мочевых путей; хроническое обструктивное заболевание легких; недержание; и расстройства, связанные со снижением кровотока в ЦНС или гипоксией ЦНС.
Согласно одному из конкретных вариантов осуществления, соединения по изобретению можно вводить для лечения боли, включая, но не ограничиваясь перечнем, в том числе нейропатическую и воспалительную боль. Некоторые типы боли можно рассматривать как заболевание или расстройство, тогда как другие типы могут считаться симптомами различных заболеваний или расстройств, а боль может иметь различную этиологию. Типичные виды боли, подлежащей лечению TRPA1 -модулирующим агентом в соответствии с изобретением, включают боль, связанную с, возникающую при или вызванную: остеоартритами, расстройствами мышц-вращателей плеча, артритами (например, ревматоидным артритом или воспалительным артритом; см., Barton et al. Exp. Mol. Pathol. 2006, 81(2), 166-170), фибромиалгией, мигренью и головной болью (например, кластерной головной болью, синусовой головной болью или головной болью напряжения; см., Goadsby Curr. Pain Headache Reports 2004, 8, 393), синуситами, мукозитами слизистой оболочки полости рта, зубной болью, травмой зуба, болью после удаления зуба, зубной инфекцией, ожогом (Bolcskei et al., Pain 2005, 117(3), 368-376), солнечным ожогом, дерматитом, псориазом, экземой, укусом насекомого, мышечно-скелетными повреждениями, переломами костей, растяжениями связок, подошвенным фасцитом, реберным хондритом, тендинитом, бурситом, лучеплечевым бурситом (локтем теннисиста), медиальным эпикондилитом локтевого сустава (локтем гольфиста), тендинитом собственной связки надколенника (коленом прыгуна), травмой от повторяющихся нагрузок, миофасциальным синдромом, растяжением мышц, миозитом, дисфункцией височно-нижнечелюстного сустава, ампутацией, болью в пояснично-крестцовой области, повреждением спинного мозга, болью в шее, хлыстовой травмой, спазмами мочевого пузыря, расстройствами желудочно-кишечного тракта, циститом, интерстециальным циститом, холециститом, инфекцией мочевых путей, уретральной коликой, почечной коликой, фарингитом, простым герпесом, стоматитом, наружным отитом, отитом среднего уха (Chan et al., Lancet, 2003, 361, 385), синдромом жжения полости рта, мукозитом, болью в пищеводе, спазмами пищевода, заболеваниями брюшной полости, гастроэзофагиальным рефлюксом, панкреатитом, энтеритом, синдромом раздраженного кишечника, воспалительным заболеванием кишечника, болезнью Крона, язвенным колитом, растяжением толстой кишки, сжимающей болью брюшной полости, дивертикулезом, дивертикулитом, газообразованием в кишечнике, геморроем, анальными трещинами, поражением аноректальной области, простатитом, эпидимитом, болью в яичках, проктитом, ректальной болью, болью при схватках, родах, эндометриозом, менструальным спазмом, болью в области таза, вульфодинией, вагинитом, оролабиальными и генитальными инфекциями (например, простым герпесом), плевритом, перикардитом, болью в груди несердечного происхождения, ушибами, ссадинами, порезами кожи (Honore, P. et al., J. Pharmacal Exp.Ther., 2005, 314, 410-21), послеоперационной болью, периферической нейропатией, центральной нейропатией, диабетической нейропатией, острой герпетической невралгией, постгерпетической невралгией, невралгией тройничного нерва, глоссофарингеальной невралгией, атипичной болью в области лица, градикулопатией, ВИЧ-ассоциированной нейропатией, физическим повреждением нерва, каузалгией, рефлекторной симпатической дистрофией, ишиалгией, шейной, грудной или поясничной радикулопатией, плексопатией плечевого сплетения, поясничной плексопатией, нейродегенеративными расстройствами, затылочной невралгией, межреберной невралгией, невралгией надглазничного нерва, паховой невралгией, невралгией латерального кожного нерва бедра, генитофеморальной невралгией, туннельным синдромом запястья, невромой Мортона, постмастэктомическим синдромом, постторакотомическим синдромом, постполиомиелитным синдромом, синдромом Гийена-Барре, синдромом Рейно, спазмом коронарной артерии (стенокардией Принцметалла или изменчивой стенокардией), висцеральной гипералгезией (Pomonis, J.D. et al. J. Pharmacal. Exp. Ther. 2003, 306, 387; Walker, K.M. et al., J. Pharmacal. Exp.Ther. 2003, 304(1), 56-62), таламической болью, раком (например, болью, вызванной раком, включая остеолитическую саркому, болью при лечении рака лучевой терапией или химиотерапией, либо болью при поражениях нервов или костей, обусловленных раком (см., Menendez, L. et al., Neurosci. Lett. 2005, 393 (1), 70-73; Asai, ч. et al., Боль 2005, 117, 19-29), или болью, вызванной разрушением кости (см., Ghilardi, J.R. et al., J. Neurosci. 2005, 25, 3126-31)), инфекцией или болезнью обмена веществ. Кроме того, соединения могут применяться для лечения болевых симптомов, таких как висцеральная боль, глазная боль, боль, вызванная термическим ожогом, зубная боль, боль, вызванная капсаицином (а также другие симптоматические состояния, вызванные капсаицином, такие как кашель, слезотечение и бронхоспазм).
Согласно другому конкретному варианту осуществления, соединения по изобретению можно вводить для лечения зуда различного происхождения, как, например, вызванного дерматологическими или воспалительными заболеваниями.
Согласно другому конкретному варианту осуществления, соединения по изобретению можно вводить для лечения воспалительных заболеваний, в том числе заболеваний, выбранных из группы, включающей: нарушения функции почек или печени, иммунологические нарушения, реакции на лекарственные средства и неизвестные/идиопатические состояния. Воспалительные заболевания, поддающиеся лечению с помощью агента по настоящему изобретению, включают, например, воспалительные заболевания кишечника (IBO), болезнь Крона и язвенный колит (Geppetti, P. et al., Br. J. Pharmacal. 2004, 141, 1313-20; Yiangou, Y. et al., Lancet2001, 357, 1338-39; Kimball, E.S. et al., Neurogastroenterol. Motif., 2004, 16, 811), остеоартриты (Szabo, A. et al., J. Pharmacal. Exp. Ther. 2005, 314, 111-119), псориаз, псориатический артрит, ревматоидный артрит, тяжелую миастению, множественный склероз, склеродермию, гломерулонефрит, панкреатит, воспалительный гепатит, астму, хроническое обструктивное заболевание легких, аллергический ринит, увеит и сердечно-сосудистые проявления воспаления, включая атеросклероз, миокрадит, перикардит и васкулит.
Согласно другому конкретному варианту осуществления, соединения по изобретению можно вводить для лечения заболевания внутреннего уха. Такие заболевания включают, например, гиперакузию, шум в ушах, повышенную вестибулярную чувствительность и периодические головокружения.
Например, соединения по изобретению можно вводить для лечения трахеобронхиальной и диафрагмальной дисфункций, включая, например, астму и обусловленные аллергией иммунные реакции (Agopyan, N. et al., Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L563-72; Agopyan, N. et al., Toxicol. Appl. Pharmacal. 2003, 192, 21-35), кашель (например, острый или хронический кашель или кашель, вызванный раздражением вследствие гастроэзофагеальной рефлюксной болезни; см., Lalloo, U.G. et al., J. Appl. Physiol. 1995, 79(4), 1082-7), бронхоспазм, хроническое обструктивное заболевание легких, хронический бронхит, эмфизему и икоту (hiccoughs, singultus).
Согласно другому конкретному варианту осуществления, соединения по изобретению можно вводить для лечения заболеваний желудочно-кишечного тракта и мочевых путей, таких как повышенная активность мочевого пузыря, воспалительная гипералгезия, висцеральная гиперрефлексия мочевого пузыря, геморрагический цистит (Dinis, P. et al., J Neurosci., 2004, 24, 11253-11263), интерстициальный цистит (Sculptoreanu, A. et al., Neurosci Lett., 2005, 381, 42-46), воспалительное заболевание предстательной железы, простатит (Sanchez, M. et al., Eur J Pharmacal., 2005, 515, 20-27), тошнота, рвота, кишечные спазмы, вздутие кишечника, спазм мочевого пузыря, неотложный позыв к мочеиспусканию, неотложный позыв к дефекации и недержание мочи.
Согласно другому конкретному варианту осуществления, соединения по изобретению можно вводить для лечения расстройств, связанных со снижением кровотока в ЦНС или гипоксией ЦНС. Такие расстройства включают, например, травму головы, повреждение спинного мозга, тромболический или геморрагический инсульт, преходящее ишемическое нарушение мозгового кровообращения, спазм сосудов головного мозга, гипогликемию, остановку сердца, эпилептическое состояние, перинатальную асфиксию, болезнь Альцгеймера и болезнь Гентингтона.
Согласно другим вариантам осуществления, соединения по изобретению можно вводить для лечения других заболеваний, расстройств или состояний, опосредованных активностью TRPA1, таких как тревожное расстройство; нарушение познавательной способности или расстройство памяти; заболевания глаз (такие как глаукома, потеря зрения, повышенное глазное давление и конъюнктивит); алопеция (например, для стимуляции роста волос); диабет (включая инсулинорезистентный диабет или диабетические состояния, опосредованные чувствительностью к инсулину или секрецией инсулина); ожирение (например, посредством подавления аппетита); диспепсия; желчная колика; почечная колика; синдром раздраженного мочевого пузыря; воспаление пищевода; заболевание верхних дыхательных путей; недержание мочи; острый цистит; и интоксикация, обусловленная нападением ядовитого животного (такого как морское животное, змея или жалящие насекомые, включая отравление ядом медузы, паука или шипохвостого ската).
Согласно одному из конкретных вариантов осуществления, соединения по изобретению вводят для лечения боли (включая, но не ограничиваясь перечнем, острую, хроническую, нейропатическую и воспалительную боль), артрита, зуда, кашля, астмы или воспалительного заболевания кишечника.
Согласно другому варианту осуществления, изобретение предлагает способ лечения нейропатической боли или воспалительной боли, включающий в себя стадию введения терапевтически эффективного количества соединения, такого как описано в данном контексте, субъекту, нуждающемуся в этом.
Согласно другому варианту осуществления, в изобретении предложено соединение, такое как описано в данном контексте, или его фармацевтически приемлемая соль для модулирования активности TRPA1.
Согласно другому варианту осуществления, в изобретении предложено соединение, такое как описано в данном контексте, или его фармацевтически приемлемая соль для применения при лекарственной терапии.
Согласно другому варианту осуществления, в изобретении предложен способ лечения респираторного расстройства, выбранного из хронического обструктивного заболевания легких (ХОЗЛ), астмы, аллергического ринита и бронхоспазма, включающий в себя стадию введения терапевтически эффективного количества соединения, такого как описано в данном контексте, субъекту, нуждающемуся в этом.
Согласно другому варианту осуществления, в изобретении предложено соединение, такое как описано в данном контексте, или его фармацевтически приемлемая соль для лечения или профилактики респираторного расстройства.
Согласно другому варианту осуществления, в изобретении предложено применение соединения, такого как описано в данном контексте, или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения или профилактики респираторного расстройства.
Согласно другому варианту осуществления, в изобретении предложен способ лечения респираторного расстройства у млекопитающих (например, у человека), включающий в себя введение соединения, такого как описано в данном контексте, или его фармацевтически приемлемой соли млекопитающему.
Согласно другому варианту осуществления, в изобретении предложен способ модулирования активности TRPA1, включающий в себя контактирование TRPA1 с соединением, таким как описано в данном контексте, или его фармацевтически приемлемой солью.
Согласно другому варианту осуществления, в изобретении предложено соединение, такое как описано в данном контексте, или его фармацевтически приемлемая соль для лечения или профилактики заболевания или состояния, опосредованного активностью TRPA1. В рамках аспектов данного варианта осуществления заболевание или состояние представляет собой боль (включая, но не ограничиваясь перечнем, острую, хроническую, нейропатическую и воспалительную боль), зуд, воспалительное расстройство, заболевание внутреннего уха, лихорадочное состояние или другое нарушение терморегуляции, трахеобронхиальную или диафрагмальную дисфункцию, расстройство желудочно-кишечного тракта или мочевых путей, хроническое обструктивное заболевание легких, недержание или расстройство, обусловленное снижением кровотока в ЦНС или гипоксией ЦНС. В рамках определенных аспектов данного варианта осуществления заболевание или состояние представляет собой боль (включая, но не ограничиваясь перечнем, острую, хроническую, нейропатическую и воспалительную боль), артрит, зуд, кашель, астму, воспалительное заболевание кишечника или заболевание внутреннего уха.
Согласно другому варианту осуществления, в изобретении предложено применение соединения, такого как описано в данном контексте, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики заболевания или состояния, опосредованного активностью TRPA1. В рамках аспектов данного варианта осуществления заболевание или состояние представляет собой боль (включая, но не ограничиваясь перечнем, острую, хроническую, нейропатическую и воспалительную боль), зуд, воспалительное расстройство, заболевание внутреннего уха, лихорадочное состояние или другое нарушение терморегуляции, трахеобронхиальную или диафрагмальную дисфункцию, расстройство желудочно-кишечного тракта или мочевых путей, хроническое обструктивное заболевание легких, недержание или расстройство, обусловленное снижением кровотока в ЦНС или гипоксией ЦНС. В рамках аспектов данного варианта осуществления заболевание или состояние представляет собой боль (включая, но не ограничиваясь перечнем, острую, хроническую, нейропатическую и воспалительную боль), артрит, зуд, кашель, астму, воспалительное заболевание кишечника или заболевание внутреннего уха.
Согласно другому варианту осуществления, в изобретении предложен способ лечения заболевания или состояния, опосредованного активностью TRPA1, у млекопитающих (например, у человека), включающий введение соединения, такого, как описано в данном контексте, или его фармацевтически приемлемой соли млекопитающему. В рамках определенных аспектов данного варианта осуществления заболевание или состояние представляет собой боль (включая, но не ограничиваясь перечнем, острую, хроническую, нейропатическую и воспалительную боль), зуд, воспалительное расстройство, заболевание внутреннего уха, лихорадочное состояние или другое нарушение терморегуляции, трахеобронхиальную или диафрагмальную дисфункцию, расстройство желудочно-кишечного тракта или мочевых путей, хроническое обструктивное заболевание легких, недержание или расстройство, обусловленное снижением кровотока в ЦНС или гипоксией ЦНС. В рамках определенных аспектов данного варианта осуществления, заболевание или состояние представляет собой боль (включая, но не ограничиваясь перечнем, острую, хроническую, нейропатическую и воспалительную боль), артрит, зуд, кашель, астму, воспалительное заболевание кишечника или заболевание внутреннего уха. Согласно некоторым вариантам осуществления, заболевание или состояние представляет собой астму.
Комбинированная терапия
Соединения по изобретению можно использовать вместе с одним или более другими соединениями по изобретению, или одним или более другими терапевтическими агентами, или в виде любого их сочетания при лечении заболеваний и состояний, опосредованных ионными каналами. Например, соединение по изобретению можно вводить одновременно, последовательно или отдельно в сочетании с другими терапевтическими агентами, включая, но не ограничиваясь перечнем, следующие:
- опиоидные анальгетики, например, морфин, героин, кокаин, оксиморфин, леворфанол, леваллорфан, оксикодон, кодеин, дигидрокодеин, пропоксифен, налмефен, фентанил, гидрокодон, гидроморфон, мерипидин, метадон, налорфин, налоксон, налтрексон, бупренорфин, буторфанол, налбуфин и пентазоцин;
- неопиоидные анальгетики, например, ацетаминофен и салицилаты (например, аспирин);
- нестероидные противовоспалительные лекарственные средства (англ. NSAID), например, ибупрофен, напроксен, фенопрофен, кетопрофен, целекоксиб, дикпофенак, дифлузинал, этодолак, фенбуфен, фенопрофен, флуфенизал, флурбипрофен, ибупрофен, индометацин, кетопрофен, кеторолак, мекпофенамовая кислота, мефенамовая кислота, мелоксикам, набуметон, напроксен, нимесулид, нитрофлурбипрофен, олсалазин, оксапрозин, фенилбутазон, пироксикам, сульфасалазин, сулиндак, толметин и зомепирак;
- противосудорожные средства, например, карбамазепин, окскарбазепин, ламотригин, вальпроат, топирамат, габапентин и прегабалин;
- антидепрессанты, такие как трицикпические антидепрессанты, например, амитриптилин, кпомипрамин, дезпрамин, имипрамин и нортриптилин;
- селективные ингибиторы СОХ-2, например, целекоксиб, рофекоксиб, парекоксиб, валдекоксиб, деракоксиб, эторикоксиб и лумиракоксиб;
- альфа-адренергические агенты, например, доксазосин, тамсулозин, клонидин, гуанфацин, дексметатомидин, модафинил и 4-амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хиназолин;
- седативные средства на основе барбитуратов, например, амобарбитал, апробарбитал, бутабарбитал, бутабитал, мефобарбитал, метарбитал, метогекситал, пентобарбитал, фенобарбитал, секобарбитал, талбутал, теамилал и тиопентал;
- антагонист тахикинина (NK), в частности, антагонист NK-3, NK-2 или NK-1, например, (aR, 9R)-7-[3,5-бис(трифторметил)бензил)]-8,9,10,11-тетрагидро-9-метил-5-(4-метилфенил)-7Н-[1,4]диазоцино[2,1-g][1,7]-нафтиридин-6-13-дион (TAK-637), 5-[[2R,3S)-2-[(1R)-1-[3,5-бис(трифторметилфенил]этокси-3-(4-фторфенил)-4-морфолинил]-метил]-1,2-дигидро-3Н-1,2,4-триазол-3-он (MK-869), апрепитант, ланепитант, дапитант или 3-[[2-метокси5-(трифторметокси)фенил]-метиламино]-2-фенилпиперидин (2S,3S);
- анальгетики, являющиеся производными анилина, например, парацетамол;
- ингибиторы обратного захвата серотонина, например, пароксетин, сертралин, норфлуоксетин (дезметилметаболит флуоксетина), метаболит деметилсертралина, флувоксамин, пароксетин, циталопрам, метаболит циталопрама дезметилциталопрам, эсциталопрам, d,l-фенфлурамин, фемоксетин, ифоксетин, цианодитиепин, литоксетин, дапоксетин, нефазодон, церикламин, тразодон и флуоксетин;
- ингибиторы обратного захвата норадреналина (норэпинефрина), например, мапротилин, лофепрамин, миртазепин, оксапротилин, фезоламин, томоксетин, миансерин, бупроприон, метаболит бупроприона гидроксибупроприон, номифензин и вилоксазин (Vivalan®)), в особенности, селективный ингибитор обратного захвата норадреналина, такой как ребоксетин, в частности, (S,S)-ребоксетин и нейролептические седативные/анксиолитические средства венлафаксин и дулоксетин;
- ингибиторы обратного захвата серотонина-норадреналина двойного действия, такие как венлафаксин, метаболит венлафаксина O-дезметилвенлафаксин, кломипрамин, метаболит кломипрамина дезметилкломипрамин, дулоксетин, милнаципран и имипрамин;
- ингибиторы ацетилхолинэстеразы, например, донепезил;
- антагонисты 5-НТЗ, например, ондансетрон;
-антагонисты метаботропного рецептора глутамата (mGluR);
- местные анестетики, например, мексилетин и лидокаин;
- кортикостероиды, например, дексаметазон;
- антиаритмические средства, например, мексилетин и фенитоин;
- антагонисты мускариновых рецепторов, например, толтеродин, пропиверин, хлорид троспия, дарифенацин, солифенацин, темиверин и ипратропий;
- каннабиноиды;
- агонисты (например, резинифератоксин) или антагонисты (например, капсазепин) ванилоидных рецепторов;
- седативные средства, например, глутетимид, мепробамат, метаквалон и дихлоралфеназон;
- анксиолитические средства, например, бензодиазепины.
- антидепрессанты, например, миртазапин.
- препараты местного применения, например, лидокаин, капсацин и резинифератоксин;
- миорелаксанты, например, бензодиазепины, бакпофен, карисопродол, хлорзоксазон, циклобензаприн, метокарбамол и орфенадрин;
- антигистаминные средства или антагонисты Н1;
- антагонисты NMDA-рецепторов (англ. NMDA-N-метил-D-аспартат);
- агонисты/антагонисты 5-НТ рецепторов;
- ингибиторы PDEV (англ. PDE 5 inhibitors - ингибиторы фосфодиэстеразы 5-го типа);
- трамадол (Tramadol®);
- холинергические (никотиновые) анальгетики;
- альфа-2-дельта лиганды;
- антагонисты простагландина подтипа Е2;
- антагонисты лейкотриена В4;
- ингибиторы 5-липоксигеназы;
- антагонисты 5-НТЗ.
При использовании в данном контексте термин "комбинация" относится к любой смеси или сочетанию одного или более соединений по изобретению и одного или более других соединений по изобретению или одного или более дополнительных терапевтических агентов. Если из контекста явным образом не следует иное, термин "комбинация" может включать в себя одновременную или последовательную доставку соединения по изобретению вместе с одним или более терапевтическими агентами. Если из контекста явным образом не следует иное, термин "комбинация" может включать в себя дозированные лекарственные формы соединения по изобретению вместе с другим терапевтическим агентом. Если из контекста явным образом не следует иное, термин "комбинация" может включать в себя пути введения соединения по изобретению вместе с другим терапевтически агентом. Если из контекста явным образом не следует иное, термин "комбинация" может включать в себя рецептуры соединения по изобретению вместе с другим терапевтически агентом. Дозированные лекарственные формы, пути введения и фармацевтические композиции включают, не ограничиваясь перечнем, описанные в данной работе дозированные лекарственные формы, пути введения и фармацевтические композиции.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Общее получение соединений формулы 1
Исходные материалы и реагенты, используемые при синтезе этих соединений, обычно являются доступными от коммерческих поставщиков, таких как Aldrich Chemical Co., или могут быть получены известными способами в соответствии с методиками, изложенными в литературе по ссылкам, как, например, Fieser, Reagents for Organic Synthesis (Реагенты для органического синтеза); Wiley & Sons: New York, 1991, V. 1-15; Rodd's Chemistry of Carbon Compounds (Химия углеродсодержащих соединений), Elsevier Science Publishers, 1989, V. 1-5, и дополнения; и Organic Reactions (Органические реакции), Wiley & Sons: New York, 1991, V. 1-40.
Приведенные ниже схемы реакций синтеза являются лишь иллюстрацией некоторых способов, с помощью которых можно синтезировать соединения по настоящему изобретению, при этом возможны различные модификации данных схем синтеза, которые могут быть предложены специалистом в данной области техники со ссылкой на раскрытие, содержащееся в данной заявке.
Исходные материалы и промежуточные продукты для схем синтеза могут быть выделены и при необходимости очищены с помощью обычных методов, включая, но не ограничиваясь перечнем, фильтрацию, перегонку, кристаллизацию, хроматографию и т.п. Такие материалы могут быть описаны с помощью обычных средств, включая физические константы и спектральные данные.
Хотя в данной работе изображены и описаны некоторые примеры вариантов осуществления, соединения по настоящему изобретению могут быть получены с использованием подходящих исходных материалов в соответствии со способами, описанными в общих чертах в данной работе, и/или с помощью способов, доступных специалисту средней квалификации в данной области.
Промежуточные и конечные соединения могут быть очищены при помощи флэш-хроматографии и/или препаративной обращенно-фазовой ВЭЖХ (англ. HPLC, high performance liquid chromatography - высокоэффективная жидкостная хроматография), и/или при помощи хроматографии со сверхкритической подвижной фазой. Если не указано иное, флэш-хроматографию можно проводить при использовании предварительно укомплектованных картриджей с силикагелем ISCO или SiliCycle на хроматографическом приборе ISCO CombiFlash® (компания Teledyne Isco, Inc.). Препаративную обращенно-фазовую ВЭЖХ можно выполнять с использованием (1) колонки Polaris C-18 5 мкм (50×21 мм) или (2) колонки XBridge Prep C-18 OBD 5 мкм (19×150 мм). Хроматографию со сверхкритической подвижной фазой можно выполнять с использованием насадочных колонок Chiral Technologies, Chiralpak AD, Chiralpak AS, Chiralpak IA, Chiralpak IB, Chiralpak IC, Chiralcel OD или Chiralcel OJ при размерах колонок, таких как (1) 4,6 см × 5 см, 3 мкм, (2) 4,6 см × 5 см, 5 мкм или (3) 15 см × 21,2 мм, 5 мкм.
Масс-спектрометрию (МС) можно выполнять на (1) масс-спектрометре Sciex 15 в режиме ES+или (2) масс-спектрометре Shimadzu LCMS 2020 в режиме ESI+. Данные масс-спектров обычно показывают только ионы-предшественники, если не оговорено иное. Данные МС или МСВР (англ. HRMS, high-resolution mass spectrometry - масс-спектрометрия высокого разрешения) приведены для конкретного промежуточного продукта или соединения там, где это указано.
Спектроскопию ядерного магнитного резонанса (ЯМР, англ. NMR) можно проводить с использованием (1) спектрометра ЯМР Bruker AV III 300, (2) спектрометра ЯМР Bruker AV III 400 или (3) спектрометра ЯМР Bruker AV III 500, с внутренним стандартом - тетраметилсиланом. Данные ЯМР приведены для конкретного промежуточного продукта или соединения там, где это указано.
Все реакции с участием чувствительных к воздействию воздуха реагентов проводили в инертной атмосфере. Реагенты использовали в том виде, в котором они были получены от поставщиков, если не указано иное.
В представленных ниже примерах могут быть использованы следующие сокращения:
Получение 1
7-Метил-1Н-пурин-6(7Н)-он
Схема реакций для Получения 1 имеет следующий вид:
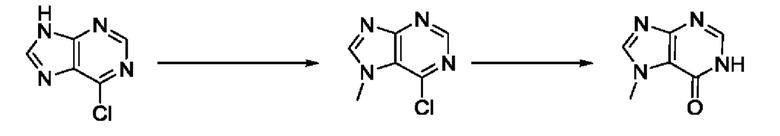
Стадия 1
Получение 6-хлор-7-метил-7Н-пурина
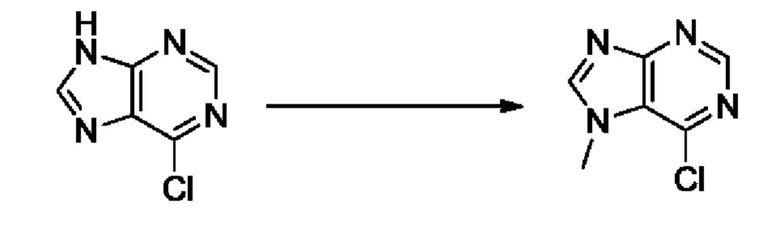
В трехгорлую круглодонную колбу емкостью 1 л после продувки азотом и при поддержании в ней инертной атмосферы азота помещали 6-хлор-9Н-пурин (15,4 г, 0,1 моль, 1 экв.) и тетрагидрофуран (155 мл) при температуре 0°С, после чего по каплям при перемешивании прибавляли MeMgCl (36,6 мл, 1,0 М раствор в ТГФ, 1,1 экв.). Смесь перемешивали при температуре 0°С в течение 30 мин. К полученной смеси по каплям при перемешивании прибавляли йодметан (42,6 г, 3 экв.). Полученный раствор перемешивали при температуре 50°С на масляной бане в течение 5 ч, гасили добавлением 50 мл водного раствора NH4Cl и экстрагировали дихлорметаном (3×50 мл). Объединенные органические фракции промывали солевым раствором (2×50 мл) и концентрировали в вакууме. Неочищенный продукт перекристаллизовывали из смеси CH2Cl2/петролейный эфир в соотношении 1:10, и получали требуемый продукт в виде твердого вещества зеленоватого цвета (7 г, 42%).
Стадия 2
Получение 7-метил-1Н-пурин-6(7Н)-она
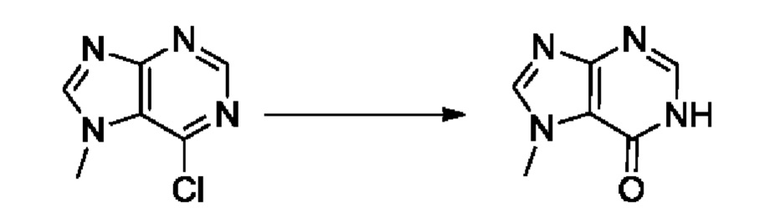
В трехгорлую круглодонную колбу емкостью 1 л помещали 6-хлор-7-метил-7Н-пурин (100 г, 590 ммоль, 1 экв.) и муравьиную кислоту (1 л). Образовавшийся раствор перемешивали при температуре 70°С в течение 3 ч. Полученную смесь концентрировали в вакууме, остаток разбавляли 500 мл воды. Полученный в результате раствор экстрагировали 3×250 мл смеси диэтиловый эфир/этил ацетат (20:1), водные фракции концентрировали в вакууме с толуолом для удаления воды и муравьиной кислоты. Остаток растворяли в воде. Величину рН раствора доводили до 9 с помощью NH3⋅Н2О (25%), водный слой концентрировали в вакууме. Твердый осадок отфильтровывали, дважды промывали водой и сушили с получением требуемого продукта (55 г, 62%) в виде твердого вещества желтого цвета.
Получение 2
5-Метилпиридо[3,4-d]пиримидин-4(3Н)-он
Схема реакций для Получения 2 имеет следующий вид:
Стадия 1
Получение 3-бром-5-фторизоникотиновой кислоты
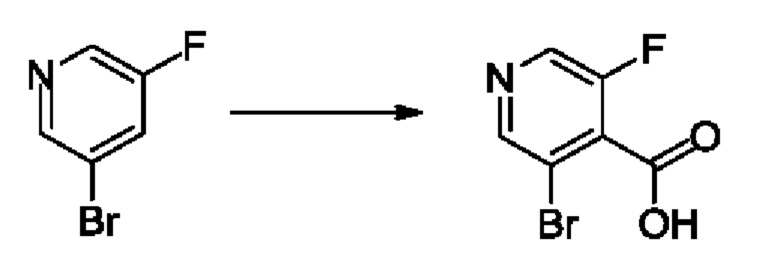
н-BuLi (250 мл, 0,62 моль, 2,5 экв.) прибавляли по каплям к раствору бис(пропан-2-ил)амина (76 г, 0,75 ммоль, 3 экв.) и тетрагидрофурана (1 л) при температуре 0°С в атмосфере азота. Смесь перемешивали в течение 30 мин при температуре 0°С. К полученной смеси по каплям прибавляли 3-бром-5-фторпиридин (44 г, 0,25 моль, 1 экв.) при перемешивании при температуре -70°С. Полученный раствор перемешивали в течение 1 ч при температуре -70°С. Затем реакционную массу выливали в смесь сухого льда с 500 мл ТГФ. Полученную массу перемешивали в течение 30 мин и затем концентрировали в вакууме. Остаток растворяли в воде. Величину рН раствора доводили до 3 с помощью соляной кислоты (1 моль/л). Смесь экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали в вакууме с получением продукта (40 г, 72%) в виде твердого вещества желтого цвета.
Стадия 2
Получение метил-3-бром-5-фторизоникотината
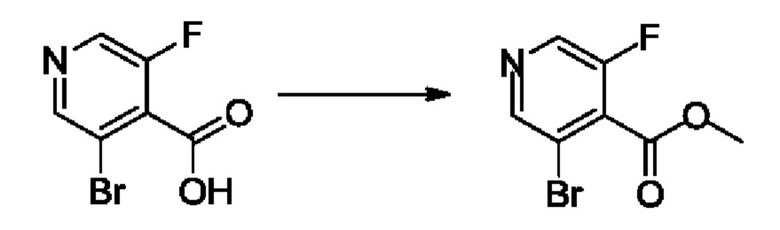
TMSCHN2 (180 мл, 360 ммоль, 2 экв.) по каплям прибавляли к раствору 3-бром-5-фторизоникотиновой кислоты (40 г, 182 ммоль, 1 экв.), ТГФ (240 мл) и МеОН (80 мл) при перемешивании при температуре 0°С в атмосфере азота. Полученный в результате раствор перемешивали в течение 3 ч при комнатной температуре. Полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью этилацетат/петролейный эфир (1/9), и получали указанное в заголовке соединение (35 г, 83%) в виде масла желтого цвета.
Стадия 3
Получение метил-3-фтор-5-метилизоникотината
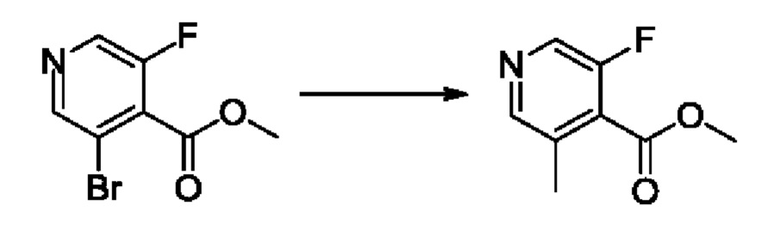
Zn(СН3)2 (225 мл, 0,22 моль, 1,5 экв.) добавляли в смесь 3-бром-5-фторизоникотината (35 г, 0,15 моль, 1 экв.), диоксана (1 л) и Pd(dppf)Cl2 (11 г, 15 ммоль, 0,1 экв.) при комнатной температуре в атмосфере азота. Полученный в результате раствор перемешивали в течение 3 ч при температуре 50°С. Затем гасили реакцию добавлением метанола. Осадок отфильтровывали. Полученную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии с получением продукта (17 г, 69%).
Стадия 4
Получение 3-фтор-4-(метоксикарбонил)-5-метилпиридин-1-оксида
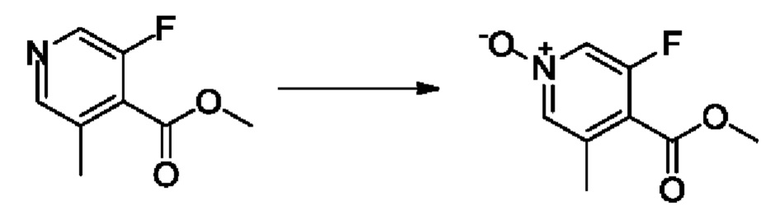
м-СРВА (96 г, 0,56 моль, 1,5 экв.) добавляли в раствор метил-5-фтор-3-метилпиридин-4-карбоксилата (63 г, 0,37 моль, 1 экв.) в дихлорметане (1,7 л) при температуре 0°С в атмосфере азота. Полученную смесь перемешивали в течение 15 ч при комнатной температуре. Реакцию гасили добавлением насыщенного раствора бикарбоната натрия, экстрагировали этилацетатом, промывали насыщенным раствором Na2S2O3 и солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью этилацетат/гептан (9/1), и получали указанное в заголовке соединение (62 г, 89%) в виде твердого вещества желтого цвета.
Стадия 5
Получение 3-амино-4-(метоксикарбонил)-5-метилпиридин-1-оксида

Через смесь 3-фтор-4-(метоксикарбонил)-5-метилпиридин-1-оксида (62 г, 0,34 моль, 1 экв.) в ДМСО (600 мл) барботировали NH3 (г), реакционную массу перемешивали в течение 12 ч при температуре 80°С. По окончании смесь разбавляли водой (1500 мл) и экстрагировали этилацетатом (800 мл × 3). Объединенную органическую фракцию дважды промывали солевым раствором. Полученную смесь концентрировали в вакууме, и получали указанное в заголовке соединение (62 г, неочищенное) в виде твердого вещества желтого цвета, которое использовали на следующей стадии без дополнительной очистки.
Стадия 6
Получение метил-3-амино-5-метилизоникотината
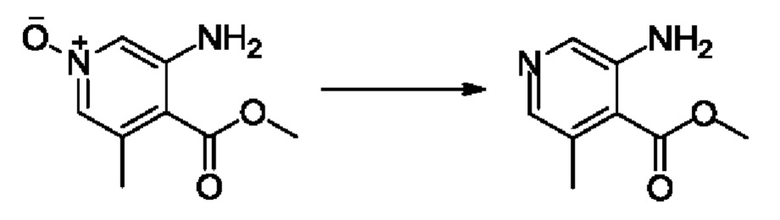
Смесь 3-амино-4-(метоксикарбонил)-5-метилпиридин-1-оксида (62 г, 0,34 моль, 1 экв.), метанола (400 мл) и никеля Ренея (10 г) перемешивали в течение 30 мин при комнатной температуре в атмосфере водорода. Осадок отфильтровывали. Полученный в результате раствор концентрировали в вакууме, и получали указанное в заголовке соединение (40 г, 71% для 2 стадий) в виде твердого вещества желтого цвета.
Стадия 7
Получение 3-амино-5-метилизоникотиновой кислоты
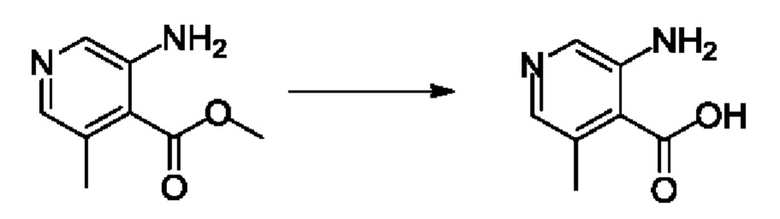
Смесь метил-3-амино-5-метилпиридин-4-карбоксилата (40 г, 0,24 моль, 1 экв.), метанола (450 мл), воды (90 мл) и гидроксида натрия (38 г, 0,96 моль, 4 экв.) перемешивали в течение 12 ч при комнатной температуре. Величину рН раствора доводили до 3 с помощью соляной кислоты (1 моль/л). Полученную смесь концентрировали в вакууме. Остаток растворяли в этаноле. Осадок отфильтровывали. Полученный фильтрат концентрировали в вакууме, и получали указанное в заголовке соединение (35 г, 95%) в виде твердого вещества желтого цвета.
Стадия 8
Получение 5-метилпиридо[3,4-d]пиримидин-4(3Н)-она
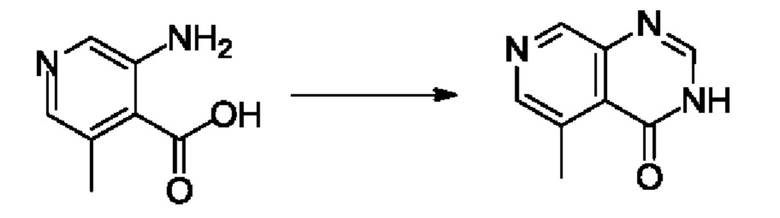
Смесь 3-амино-5-метилпиридин-4-карбоновой кислоты (35 г, 0,23 моль, 1 экв.), этанола (450 мл), уксусной кислоты и метанимидамида (35 г, 0,34 моль, 1,5 экв.) перемешивали в течение 3 ч при температуре 80°С. Полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью дихлорметан/метанол (5/1), и получали указанное в заголовке соединение (22 г, 59%) в виде твердого вещества желтого цвета.
Получение 3
5-Метилпиридо[2,3-d]пиримидин-4(3Н)-он
Схема реакций для Получения 3 изображена ниже.
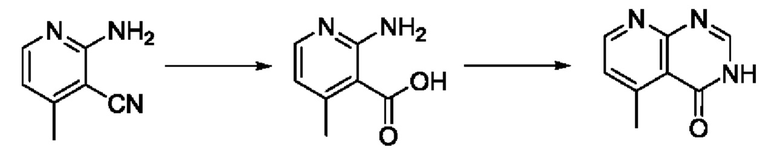
Стадия 1
Получение 2-амино-4-метилникотиновой кислоты
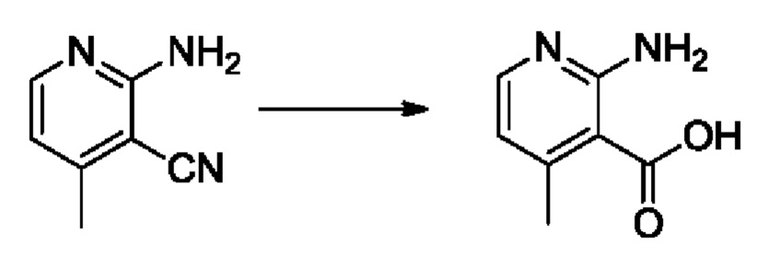
В круглодонную колбу емкостью 2 л помещали 2-амино-4-метилпиридин-3-карбонитрил (50 г, 375,51 ммоль, 1,00 экв.) и водный раствор гидроксида калия (20%, 700 мл). Полученный раствор перемешивали при температуре 110°С на масляной бане в течение ночи и охлаждали до комнатной температуры. Величину рН смеси доводили до 3 с помощью водного раствора HCl (2 N). Смесь концентрировали в вакууме. Остаток промывали 2×400 мл этанола. Твердое вещество отфильтровывали. Фильтрат концентрировали в вакууме с получением 40 г (неочищенной) 2-амино-4-метилпиридин-3-карбоновой кислоты в виде твердого вещества желтого цвета, которое сразу использовали на следующей стадии.
Стадия 2
Получение 5-метилпиридо[2,3-d]пиримидин-4(3Н)-она
В круглодонную колбу емкостью 1 л помещали раствор 2-амино-4-метилпиридин-3-карбоновой кислоты (40 г, 262,90 ммоль, 1,00 экв.) в этаноле (500 мл) и формамидинацетат (82,11 г, 788,69 ммоль, 3,00 экв.). Полученный раствор перемешивали при температуре 100°С на масляной бане в течение ночи и охлаждали до комнатной температуры. Твердый осадок отфильтровывали, промывали 3×100 мл МеОН и сушили в вакууме с получением 21 г (50%) 5-метил-3Н,4Н-пиридо[2,3-d]пиримидин-4-она в виде твердого вещества белого цвета.
Получение 4
5,7-Диметил-3H,4H-имидазо[4,3-f][1,2,4]триазин-4-он
Схема реакций для Получения 4 изображена ниже.
Стадия 1
Получение этил-2-(гидроксиимино)-3-оксобутаноата
Раствор NaNO2 (3,6 г, 52,18 ммоль) в воде (6 мл) прибавляли по каплям к смеси этил-3-оксобутаноата (5,2 г, 39,96 ммоль) и АсОН (6 мл) при перемешивании при температуре 0°С. Полученный раствор перемешивали в течение 12 ч при комнатной температуре. Величину рН раствора доводили до значения от 7 до 8 с помощью бикарбоната натрия (насыщенного раствора). Полученный в результате раствор экстрагировали этилацетатом, промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. В результате получали указанное в заголовке соединение (5,2 г, 82%) в виде бесцветного масла, которое использовали на следующей стадии без дополнительной очистки.
ЖХМС [М+H+] 160.
Стадия 2
Получение гидрохлорида этил-2-амино-3-оксобутаноата
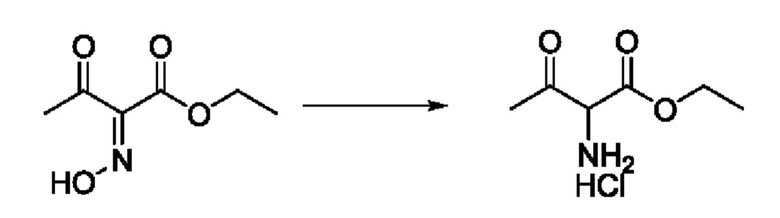
Смесь этил-2-(гидроксиимино)-3-оксобутаноата (5,2 г, 32,68 ммоль), этанола (50 мл), концентрированной соляной кислоты (5 мл) и Pd/C (1 г, 10%) перемешивали в течение 48 ч при комнатной температуре в атмосфере водорода. Осадок отфильтровывали. Полученную смесь концентрировали в вакууме. В результате получали указанное в заголовке соединение (5 г, 84%) в виде твердого вещества грязно-белого цвета, которое использовали на следующей стадии без дополнительной очистки.
ЖХМС [М+H+] 146.
Стадия 3
Получение этил-2,4-диметил-1Н-имидазол-5-карбоксилата
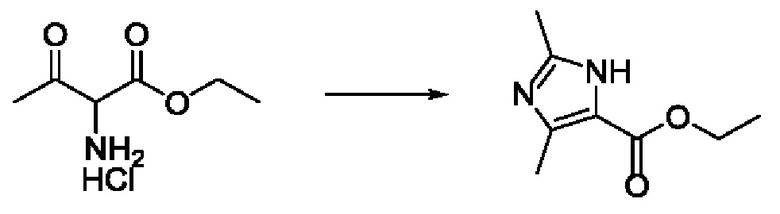
Раствор гидрохлорида этил-2-амино-3-оксобутаноата (4,6 г, 25,33 ммоль) в этаноле (10 мл) прибавляли по каплям к смеси гидрохлорида этилэтанкарбоксимидата (8,1 г, 65,54 ммоль), этанола (100 мл) и ТЭА (8,4 г, 83,01 ммоль) при перемешивании при комнатной температуре. Полученный раствор перемешивали в течение 12 ч при комнатной температуре. Полученный в результате раствор разбавляли этилацетатом, промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью дихлорметан/метанол (10/1), и получали указанное в заголовке соединение (1,3 г, 31%) в виде твердого вещества светло-желтого цвета.
ЖХМС [М+H+] 169.
Стадия 4
Получение этил-1-амино-2,4-диметил-1Н-имидазол-5-карбоксилата
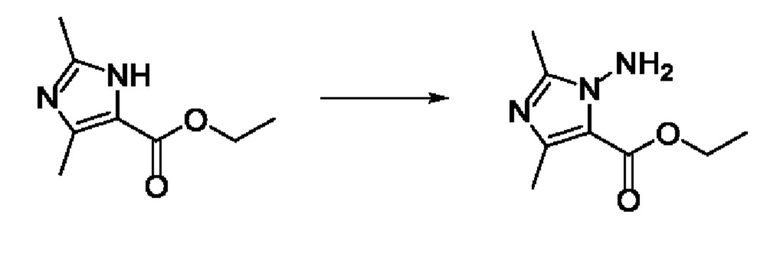
LiHMDS (8,5 мл, 1М в ТГФ) прибавляли по каплям к смеси этил-2,4-диметил-1Н-имидазол-5-карбоксилата (1,3 г, 7,73 ммоль) и N,N-диметилформамида (200 мл) в атмосфере азота при перемешивании при температуре -10°С на бане с сухим льдом. Полученный в результате раствор перемешивали в течение 30 мин при температуре -10°С. К полученной смеси порциями прибавляли аминодифенилфосфинат (2,2 г, 9,43 ммоль) при температуре 0°С. Полученный в результате раствор оставляли для протекания реакции перемешиваться в течение еще 12 ч при комнатной температуре. Полученный в результате раствор разбавляли водой, экстрагировали этилацетатом, промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью дихлорметан/метанол (10/1), и получали указанное в заголовке соединение (1,0 г, 71%) в виде твердого вещества светло-желтого цвета.
ЖХМС [М+H+] 184.
Стадия 5
Получение 5,7-диметил-3H,4H-имидазо[4,3-f][1,2,4]триазин-4-она

Смесь этил-1-амино-2,4-диметил-1H-имидазол-5-карбоксилата (1,0 г, 5,46 ммоль), формамида (10 мл) и MeONa (3,0 мл, 5,4М в МеОН) перемешивали в течение 1 ч при температуре 100°С на масляной бане. Полученный в результате раствор разбавляли водой. Величину рН раствора доводили до 5 с помощью соляной кислоты (1N). Твердый осадок отфильтровывали и сушили в вакууме с получением 130 мг твердого вещества белого цвета. Фильтрат очищали на колонке с силикагелем С18, элюируя смесью CH3CN/Н2О (10 ммоль/л NH4HCO3), увеличивая от 5% до 95% в течение 30 мин. Фракции собирали и концентрировали с получением 370 мг твердого вещества белого цвета. В результате получали указанное в заголовке соединение (всего 500 мг, 56%) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 165.
Получение 5
Получение 6-метил-7-оксо-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-3-карбонитрила
Схема реакций для Получения 5 изображена ниже.
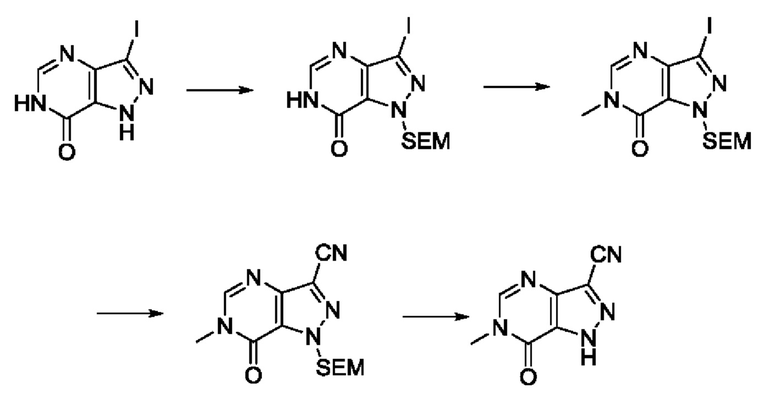
Стадия 1
Получение 3-йод-1-[[2-(триметилсилил)этокси]метил]-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-7-она
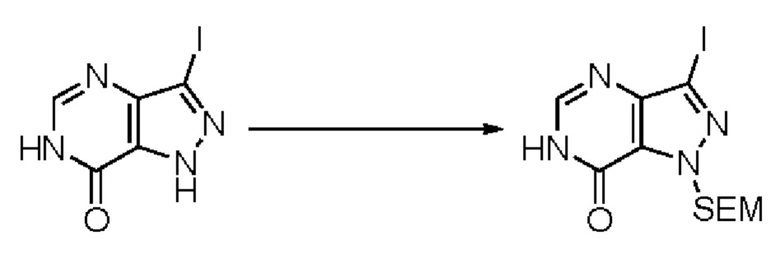
Гидрид натрия (687 мг, 28,62 ммоль) порциями прибавляли к раствору 3-йод-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-7-она (1,5 г, 5,72 ммоль) в N,N-диметилформамиде (50 мл) при температуре 0°С. К полученному раствору через 20 мин по каплям прибавляли SEM-Cl (950 мг, 6,22 ммоль). Полученный в результате раствор перемешивали в течение 12 ч при комнатной температуре и использовали на следующей стадии без дополнительной очистки.
ЖХМС [M+H+] 393.
Стадия 2
Получение 3-йод-6-метил-1-[[2-(триметилсилил)этокси]метил]-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-7-она
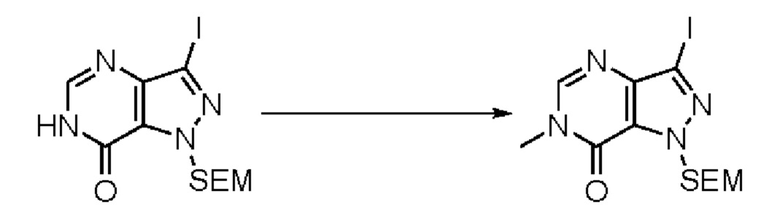
Гидрид натрия (60 мг 2,50 ммоль) добавляли в раствор 3-йод-1-[[2-(триметилсилил)этокси]метил]-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-7-она (~ 0,11М в ДМФА, 50 мл, полученный со стадии 1) при температуре 0°С. Через 20 мин прибавляли по каплям CH3I (430 мг, 3,02 ммоль), и полученную смесь перемешивали в течение 3 ч при комнатной температуре. Реакционный раствор очищали на колонке с силикагелем С18, элюируя смесью CH3CN/H2O (10 ммоль/л NH4HCO3, от 5% до 95% в течение 30 мин). В результате получали указанное в заголовке соединение (250 мг, 24%) в виде твердого вещества белого цвета.
ЖХМС [M+H+] 407.
Стадия 3
Получение 6-метил-7-оксо-1-[[2-(триметилсилил)этокси]метил]-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-3-карбонитрила

Смесь 3-йод-6-метил-1-[[2-(триметилсилил)этокси]метил]-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-7-она (260 мг, 0,64 ммоль), Zn(CN)2 (148 мг, 1,26 ммоль), Pd2(dba)3⋅CHCl3 (66 мг, 0,06 ммоль), dppf (71 мг, 0,13 ммоль) и N,N-диметилформамида (5 мл) подвергали воздействию микроволнового излучения в течение 1 ч при температуре 100°С в атмосфере азота. Осадок отфильтровывали. Фильтрат очищали на колонке с силикагелем С18, элюируя смесью CH3CN/H2O (10 ммоль/л NH4HCO3, от 5% до 95% в течение 30 мин). В результате получали указанное в заголовке соединение (150 мг, 77%) в виде твердого вещества белого цвета.
ЖХМС [M+H+] 306.
Стадия 4 Получение 6-метил-7-оксо-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-3-карбонитрила
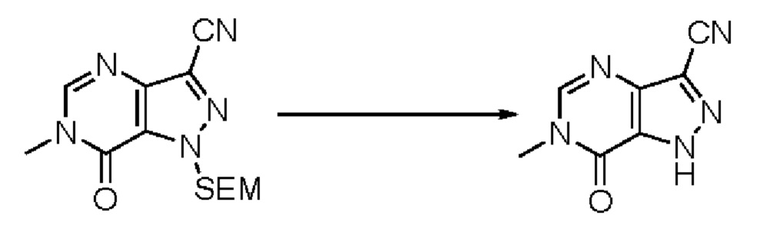
Смесь 6-метил-7-оксо-1-[[2-(триметилсилил)этокси]метил]-1Н,6Н,7Н-пиразоло[4,3-d]пиримидин-3-карбонитрила (120 мг, 0,39 ммоль) и трифторуксусной кислоты (3 мл) перемешивали в течение 18 ч при температуре 60°С. Полученную смесь концентрировали в вакууме. В результате получали указанное в заголовке соединение (60 мг, 87%) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 176.
Получение 6
1-Метил-6,7-дигидро-1Н-пурин-6-он
Общая схема реакций для Получения 6 имеет следующий вид:
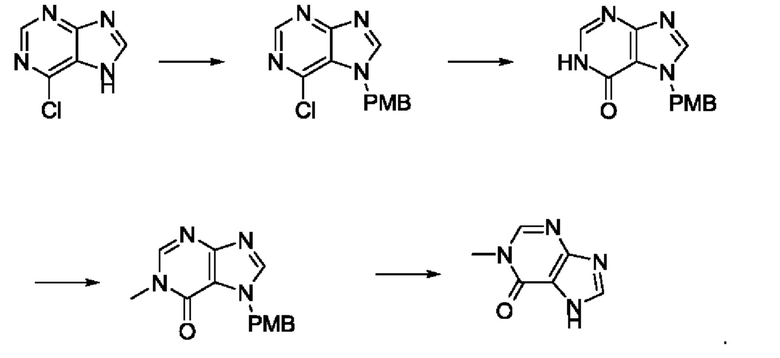
Стадия 1
Получение 6-хлор-7-[(4-метоксифенил)метил]-7Н-пурина
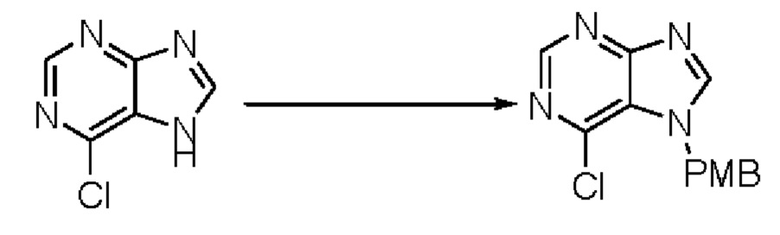
Гидрид натрия (858 мг, 35,75 ммоль) порциями прибавляли к раствору 6-хлор-7Н-пурина (3 г, 19,41 ммоль) в N,N-диметилформамиде (30 мл). Через 20 мин к полученной смеси по каплям прибавляли PMBCl (6,1 г, 38,81 ммоль). Полученный раствор перемешивали в течение 4 ч при комнатной температуре, разбавляли этилацетатом, промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью этилацетат/петролейный эфир (1:1), и получали указанное в заголовке соединение (2,3 г, 43%) в виде масла светло-желтого цвета.
ЖХМС [M+H+] 275.
Стадия 2
Получение 7-[(4-метоксифенил)метил]-6,7-дигидро-1Н-пурин-6-она

Смесь 6-хлор-7-[(4-метоксифенил)метил]-7Н-пурина (2,3 г, 8,37 ммоль), 1,4-диоксана (3 мл), гидроксида натрия (1 г, 25,00 ммоль) и воды (25 мл) перемешивали в течение 1,5 ч при температуре 90°С. Величину рН раствора доводили до 7 с помощью HCl (2 М). Твердый осадок отфильтровывали, и получали указанное в заголовке соединение (1,95 г, 91%) в виде твердого вещества белого цвета.
ЖХМС [M+H+] 257.
Стадия 3
Получение 7-[(4-метоксифенил)метил]-1-метил-6,7-дигидро-1Н-пурин-6-она
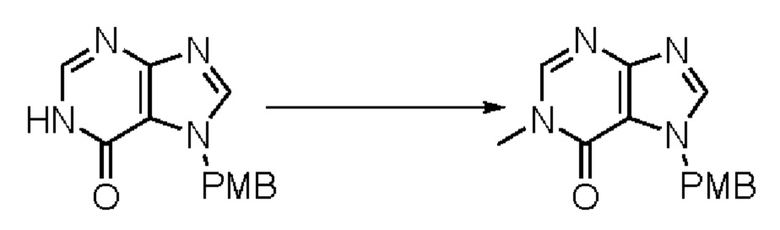
Смесь 7-[(4-метоксифенил)метил]-6,7-дигидро-1Н-пурин-6-она (1 г, 3,90 ммоль), карбоната калия (1,1 г, 7,80 ммоль), N,N-диметилформамида (12 мл) и CH3I (666 мг, 4,69 ммоль) перемешивали в течение 1,5 ч при комнатной температуре. Осадок отфильтровывали. Реакционную смесь очищали на колонке с силикагелем С18, элюируя смесью CH3CN/H2O (10 ммоль/л NH4HCO3, от 5% до 95% в течение 30 мин). В результате получали указанное в заголовке соединение (700 мг, 66%) в виде твердого вещества белого цвета.
ЖХМС [M+H+] 271.
Стадия 4
Получение 1-метил-6,7-дигидро-1Н-пурин-6-она
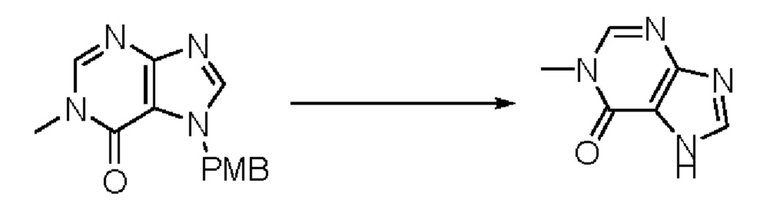
Смесь 7-[(4-метоксифенил)метил]-1-метил-6,7-дигидро-1Н-пурин-6-она (700 мг, 2,590 ммоль) и трифторуксусной кислоты (10 мл) перемешивали в течение 15 ч при температуре 70°С. Полученную смесь концентрировали в вакууме. В результате получали указанное в заголовке соединение (700 мг, неочищенное) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 151.
Получение 7
4-Метилпиримидо[4,5-с]пиридазин-5(6Н)-он
Общая схема реакций для Получения 7 имеет следующий вид:

Стадия 1
Получение 4,6-дибром-5-метилпиридазин-3-амина
Раствор Br2 (9,6 г, 60,07 ммоль) в метаноле (30 мл) прибавляли по каплям к смеси 5-метилпиридазин-3-амина (3 г, 27,49 ммоль), метанола (100 мл) и бикарбоната натрия (11,5 г, 136,89 ммоль) при температуре 0°С. Полученный раствор перемешивали в течение 2 ч при комнатной температуре, разбавляли водой, экстрагировали этилацетатом, сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью этилацетат/петролейный эфир (1/4), и получали указанное в заголовке соединение (4,0 г, 55%) в виде твердого вещества коричневого цвета.
ЖХМС [M+H+] 266.
Стадия 2
Получение 4-бром-5-метилпиридазин-3-амина

EtMgBr (2 мл, 15,15 ммоль, 3М в ТГФ) прибавляли по каплям к раствору 4,6-дибром-5-метилпиридазин-3-амина (400 мг, 1,49 ммоль) в тетрагидрофуране (8 мл) при температуре от 0 до 10°С в атмосфере азота. Полученный раствор перемешивали в течение 35 мин при температуре 63°С. Реакцию гасили водой и концентрировали в вакууме. Остаток очищали на колонке с силикагелем С18, элюируя смесью CH3CN/Н2О (10 ммоль/л NH4HCO3, от 5% до 95% в течение 30 мин). В результате получали указанное в заголовке соединение (36 мг, 13%) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 188.
Стадия 3
Получение 3-амино-5-метилпиридазин-4-карбоксамида

Смесь 4-бром-5-метилпиридазин-3-амина (80 мг, 425,47 ммоль), NH3/МеОН (7М) (4 мл), Pd(dppf)Cl2 (31 мг, 0,04 ммоль), ТЭА (128 мг, 1,26 ммоль) и окиси углерода перемешивали в течение ночи при температуре 100°С под давлением 10 атм. Реакционный раствор очищали на колонке с силикагелем С18, элюируя смесью CH3CN/H2O (10 ммоль/л NH4HCO3, от 5% до 95% в течение 30 мин). В результате получали указанное в заголовке соединение (85 мг, неочищенное) в виде твердого вещества светло-желтого цвета.
ЖХМС [М+H+] 153.
Стадия 4
Получение 4-метилпиримидо[4,5-с]пиридазин-5(6Н)-она

Смесь 3-амино-5-метилпиридазин-4-карбоксамида (150 мг, 0,98 ммоль), этанола (3 мл), EtONa (21%) (3,2 г, 0,04 ммоль), этилформиата (360 мг, 4,86 ммоль) перемешивали в течение 1 ч при температуре 80°С в атмосфере азота. Полученную смесь концентрировали в вакууме. Величину рН раствора доводили до 8 с помощью соляной кислоты/Н2О (5%). Полученную смесь концентрировали в вакууме и разбавляли этанолом. Осадок отфильтровывали, фильтрат концентрировали в вакууме, и получали указанное в заголовке соединение (120 мг, 75%) в виде твердого вещества коричневого цвета.
ЖХМС [М+H+] 163.
Получение 8
5-Метил-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-7-карбонитрил
Общая схема реакций для Получения 8 имеет следующий вид:
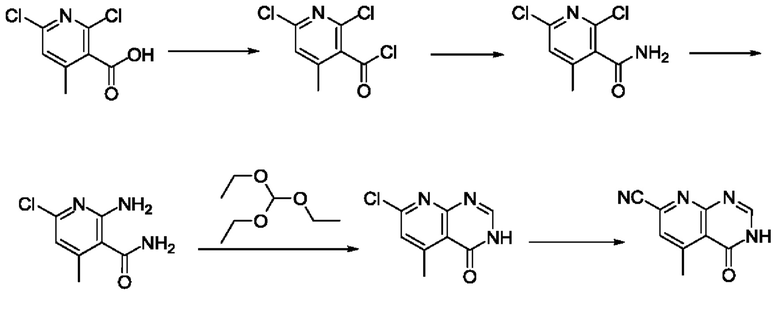
Стадия 1
Получение 2,6-дихлор-4-метилникотиноилхлорида

Оксалилхлорид (5,5 г, 43,33 ммоль) прибавляли по каплям к раствору 2,6-дихлор-4-метилпиридин-3-карбоновой кислоты (3 г, 14,56 ммоль), N,N-диметилформамида (50 мг, 0,68 ммоль) и дихлорметана (100 мл) при температуре 0°С. Полученный в результате раствор перемешивали в течение ночи при комнатной температуре и концентрировали в вакууме. В результате получали указанное в заголовке соединение (3,1 г, неочищенное) в виде жидкости светло-желтого цвета.
Стадия 2
Получение 2,6-дихлор-4-метилникотинамид
Раствор 2,6-дихлор-4-метилпиридин-3-карбонилхлорида (3,1 г, 13,81 ммоль) в дихлорметане (15 мл) прибавляли по каплям к перемешиваемому раствору NH3/ТГФ (0,5М) (42 мл) при температуре 25°С. После перемешивания в течение 1 ч при комнатной температуре полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью этилацетат/петролейны и эфир (1/1), и получали указанное в заголовке соединение (1,5 г, 53%) в виде твердого вещества белого цвета.
ЖХМС [M+H+] 205.
Стадия 3
Получение 2-амино-6-хлор-4-метилникотинамида

Смесь 2,6-дихлор-4-метилпиридин-3-карбоксамида (50 мг, 0,24 ммоль), диоксана (2 мл, 23,60 ммоль) и аммиака (30%, 0,5 мл) перемешивали в течение ночи при температуре 130°С. Полученную смесь концентрировали в вакууме. Неочищенный продукт очищали на колонке с силикагелем С18, элюируя смесью CH3CN/Н2О (10 ммоль/л NH4HCO3, от 5% до 95% в течение 30 мин). В результате получали указанное в заголовке соединение (25 мг, 55%) в виде твердого вещества белого цвета.
ЖХМС [M+H+] 186.
Стадия 4
Получение 7-хлор-5-метилпиридо[2,3-d]пиримидин-4(3Н)-

Смесь 2-амино-6-хлор-4-метилпиридин-3-карбоксамида (320 мг, 1,72 ммоль) и (диэтоксиметокси)этана (5 мл) перемешивали в течение ночи при температуре 140°С. Твердый осадок отфильтровывали. В результате получали указанное в заголовке соединение (180 мг, 53%) в виде твердого вещества серого цвета.
ЖХМС [М+H+] 211.
Стадия 5
Получение 5-метил-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-7-карбонитрила

Смесь 7-хлор-5-метил-3Н,4Н-пиридо[2,3-d]пиримидин-4-она (170 мг, 0,86 ммоль), N,N-диметилформамида (5 мл), Zn(CN)2 (151 мг, 1,28 ммоль), Pd2(dba)3⋅CHCl3 (90 мг, 0,08 ммоль) и dppf (96 мг, 0,17 ммоль) перемешивали в течение 3 ч при температуре 100°С в атмосфере азота. Осадок отфильтровывали. Неочищенный продукт очищали на колонке с силикагелем С18, элюируя смесью CH2CN/Н2О (10 ммоль/л NH4HCO3, от 5% до 95% в течение 30 мин). В результате получали указанное в заголовке соединение (100 мг, 62%) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 187.
Получение 9
1-[(4-Метоксифенил)метил]-1Н,6Н,7Н-имидазо[4,5-d]пиридазин-7-он
Общая схема реакций для Получения 9 имеет следующий вид:
Стадия 1
Получение метил-4-(гидроксиметил)-1-(4-метоксибензил)-1Н-имидазол-5-карбоксилата
Смесь метил-4-(гидроксиметил)-1Н-имидазол-5-карбоксилата (500 мг, 3,20 ммоль), N,N-диметилформамида (10 мл), карбоната калия (885 мг, 6,40 ммоль), PMBCl (пара-метоксибензилхлорид, 550 мг, 3,51 ммоль) перемешивали в течение ночи при комнатной температуре. Осадок отфильтровывали. Полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью дихлорметан/метанол (10/1), и получали указанное в заголовке соединение (600 мг, 68%) в виде твердого вещества зеленоватого цвета.
ЖХМС [M+H+] 277.
Стадия 2
Получение метил-4-формил-1-(4-метоксибензил)-1Н-имидазол-5-карбоксилата
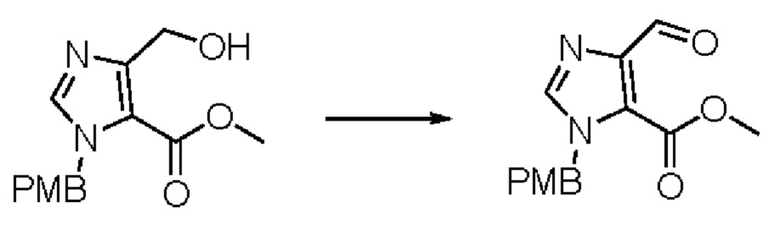
Смесь метил-4-(гидроксиметил)-1-[(4-метоксифенил)метил]-1Н-имидазол-5-карбоксилата (580 мг, 2,10 ммоль), дихлорметана (20 мл) и реактива Демма-Мартина (888 мг, 2,09 ммоль) перемешивали в течение 1 ч при комнатной температуре. Осадок отфильтровывали. Полученную смесь концентрировали в вакууме, и получали указанное в заголовке соединение (460 мг, 80%) в виде масла зеленоватого цвета.
ЖХМС [M+H+] 275.
Стадия 3
Получение 1-[(4-метоксифенил)метил]-1Н,6Н,7Н-имидазо[4,5-d]пиридазин-7-она
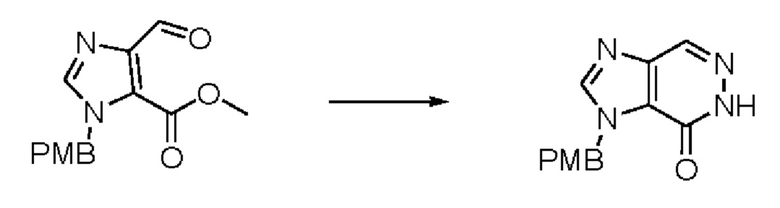
Смесь метил-4-формил-1-[(4-метоксифенил)метил]-1Н-имидазол-5-карбоксилата (460 мг, 1,68 ммоль), этанола (20 мл) и NH2NH2⋅H2O (1,045 г, 20,88 ммоль) перемешивали в течение 1 ч при температуре 80°С. Полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью дихлорметан/метанол (20/1), и получали указанное в заголовке соединение (400 мг, 93%) в виде твердого вещества белого цвета.
ЖХМС [M+H+] 257.
Получение 10
5-Метил-4-оксо-3,4-дигидропиридо[3,4-d]пиримидин-8-карбонитрил
Общая схема реакций для Получения 10 имеет следующий вид:
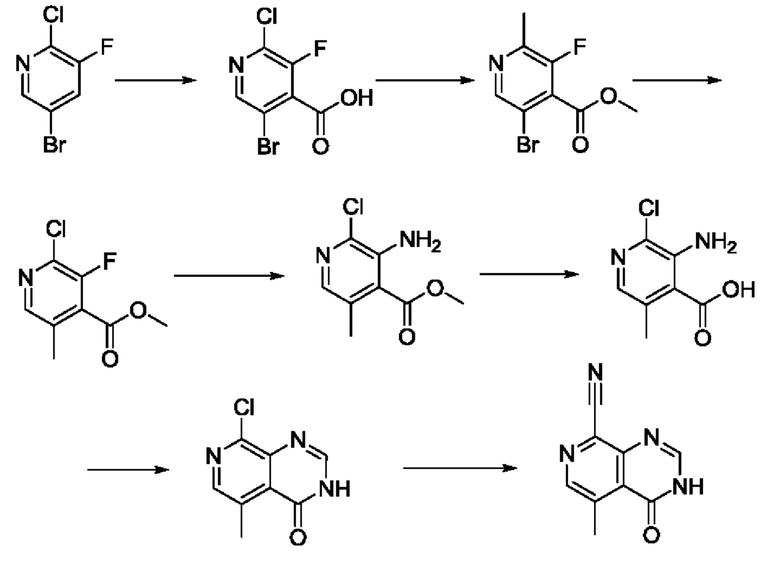
Стадия 1
Получение 5-бром-2-хлор-3-фторизоникотиновой кислоты
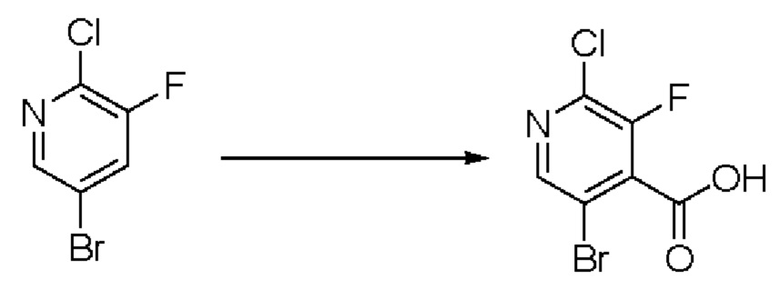
LDA (47,5 мл, 2,0 моль в ТГФ) прибавляли по каплям к раствору 5-бром-2-хлор-3-фторпиридина (10 г, 47,52 ммоль) в тетрагидрофуране (300 мл) при температуре -78°С в атмосфере азота. Полученный раствор перемешивали в течение 2 ч при температуре -78°С. Полученную смесь выливали в CO2 (твердый) в ТГФ. Реакционную смесь концентрировали в вакууме. Величину рН остатка доводили до <7 с помощью соляной кислоты (2 М). Полученную смесь экстрагировали этилацетатом, промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. В результате получали указанное в заголовке соединение (15 г, неочищенное) в виде твердого вещества белого цвета.
ЖХМС [M-H+] 254.
Стадия 2
Получение метил-5-бром-3-фтор-2-метилизоникотината
Смесь 5-бром-2-хлор-3-фторпиридин-4-карбоновой кислоты (15 г, 59,0 ммоль), тетрагидрофурана (100 мл), метанола (20 мл), TMSCHN2 (60 мл, 2М в гексане) перемешивали в течение 2 ч при комнатной температуре. Полученную смесь концентрировали в вакууме. В результате получали указанное в заголовке соединение (12 г, неочищенное) в виде масла, которое использовали на следующей стадии без дополнительной очистки.
ЖХМС [М+Н+] 248.
Стадия 3
Получение метил-2-хлор-3-фтор-5-метилизоникотината

Смесь метил-5-бром-2-хлор-3-фторпиридин-4-карбоксилата (5 г, 18,62 ммоль), трициклогексилфосфана (1,3 г, 4,60 ммоль), ацетата палладия (147 мг, 0,66 ммоль) и толуола (60 мл) перемешивали в течение 12 ч при температуре 100°С в атмосфере азота. Полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью этилацетат/петролейный эфир (1:5), и получали указанное в заголовке соединение (2,5 г, 66%) в виде твердого вещества белого цвета.
ЖХМС [М+Н+] 204.
Стадия 4
Получение метил-3-амино-2-хлор-5-метилизоникотината

NH3(г) (17,5 мл, 7 М в СН3ОН) прибавляли по каплям к раствору метил-2-хлор-3-фтор-5-метилпиридин-4-карбоксилата (2,5 г, 12,28 ммоль) в СН3ОН (50 мл). Полученный раствор перемешивали в течение 12 ч при температуре 100°С и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью этилацетат/петролейный эфир (1:5), и получали указанное в заголовке соединение (300 мг, 12%) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 201.
Стадия 5
Получение 3-амино-2-хлор-5-метилизоникотиновой кислоты
Смесь метил-3-амино-2-хлор-5-метилпиридин-4-карбоксилата (300 мг, 1,5 ммоль), воды (2 мл), гидроксида натрия (200 мг, 5,00 ммоль) и метанола (10 мл) перемешивали в течение 2 ч при температуре 50°С. Полученную смесь концентрировали в вакууме. В результате получали указанное в заголовке соединение (250 мг, неочищенное) в виде твердого вещества белого цвета, которое использовали на следующей стадии без дополнительной очистки.
ЖХМС [М+H+] 187.
Стадия 6
Получение 8-хлор-5-метилпиридо[3,4-d]пиримидин-4(3Н)-она
Смесь 3-амино-2-хлор-5-метилпиридин-4-карбоновой кислоты (252 мг, 1,35 ммоль), уксусной кислоты, метанимидамида (600 мг, 5,80 ммоль) и BuOH (15 мл) перемешивали в течение 12 ч при температуре 120°С. Полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью дихлорметан/метанол (20:1), и получали указанное в заголовке соединение (140 мг, 53%) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 196.
Стадия 7
Получение 5-метил-4-оксо-3,4-дигидропиридо[3,4-d]пиримидин-8-карбонитрила

Смесь 8-хлор-5-метил-3Н,4Н-пиридо[3,4-d]пиримидин-4-она (200 мг, 1,02 ммоль), Pd2(dba)3 (100 мг, 0,10 ммоль), dppf (200 мг, 0,36 ммоль), цианида цинка (120 мг, 1,00 ммоль) и N,N-диметилформамида (5 мл) подвергали воздействию микроволнового излучения в течение 1 ч при температуре 130°С в атмосфере азота. Полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью дихлорметан/метанол (10/1), и получали указанное в заголовке соединение (140 мг, 74%) в виде твердого вещества белого цвета.
ЖХМС [М+Н+] 187.
Получение 11
5-Метилпиримидо[4,5-d]пиримидин-4(3Н)-он
Общая схема реакций для Получения 11 имеет следующий вид:
Стадия 1
Получение этил-4-амино-6-метилпиримидин-5-карбоксилата
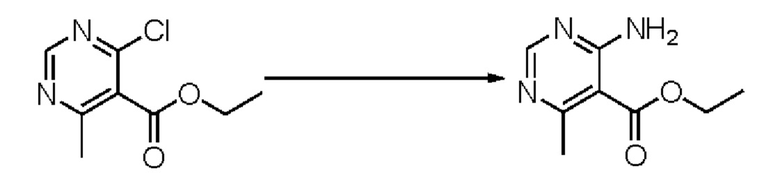
NH3 (г) (8 мл, ~14% в этаноле) прибавляли по каплям к раствору этил-4-хлор-6-метилпиримидин-5-карбоксилата (800 мг, 4,00 ммоль) в этаноле (10 мл). Образовавшийся раствор перемешивали в течение 16 ч при температуре 120°С. Полученную смесь концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью этилацетат/петролейный эфир (1:5), и получали указанное в заголовке соединение (700 мг, 97%) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 182.
Стадия 2
Получение 4-амино-6-метилпиримидин-5-карбоновой кислоты
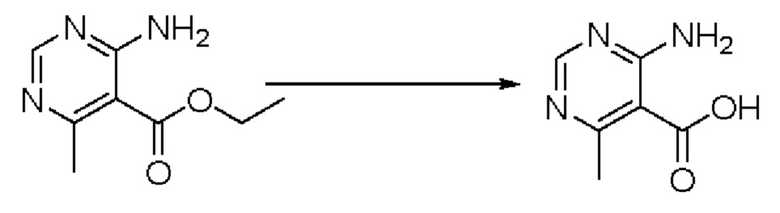
Смесь этил-4-амино-6-метилпиримидин-5-карбоксилата (700 мг, 3,90 ммоль), гидроксида натрия (464,4 мг, 11,60 ммоль), воды (6 мл) и метанола (30 мл) перемешивали в течение 3 ч при температуре 50°С. Величину рН раствора доводили до 3 с помощью соляной кислоты (2М). Полученную смесь концентрировали в вакууме, и получали указанное в заголовке соединение (700 мг, неочищенное) в виде твердого вещества белого цвета.
ЖХМС [М+H+] 154.
Стадия 3
Получение 5-метилпиримидо[4,5-d]пиримидин-4(3Н)-она

Смесь 4-амино-6-метилпиримидин-5-карбоновой кислоты (700 мг, 4,6 ммоль), формамидинацетата (2 г, 19,40 ммоль) и бутан-1-ола (35 мл) перемешивали в течение 3 дней при температуре 130°С. Реакционную массу разбавляли водой, экстрагировали этилацетатом, промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя смесью дихлорметан/метанол (20/1), и получали указанное в заголовке соединение (300 мг) в виде твердого вещества светло-желтого цвета.
ЖХМС [М+H+] 163.
Получение 12
5-(Хлорметил)-3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол
Общая схема реакций для Получения 12 имеет следующий вид:

Стадия 1
Получение (R)-1-(4-хлорфенил)бут-3-ен-1-ола
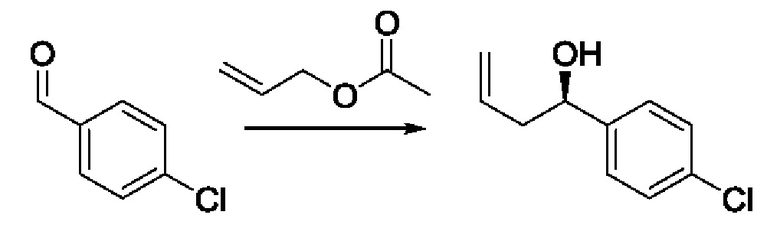
Проводили параллельно четыре реакции: в реактор высокого давления помещали 4-фторбензальдегид (100,0 г, 711,4 ммоль, 1,00 экв.), хлорид иридия, (1Z,5Z)-циклоокта-1,5-диен (14,3 г, 21,3 ммоль, 0,03 экв.), Cs2CO3 (46,3 г, 142,2 ммоль, 0,2 экв.) и 4-хлор-3-нитро-бензойную кислоту (14,3 г, 71,4 ммоль, 0,1 экв.), (R)-BINAP (22,1 г, 35,5 ммоль, 0,05 экв.). Реактор продували азотом перед добавлением 1,4-диоксана (700,0 мл), аллилацетата (712,2 г, 7,11 моль, 10,0 экв.) и i-PrOH (85,5 г, 1,42 моль, 108,9 мл, 2,00 экв.). Реакционную смесь выдерживали при перемешивании при температуре 112°С на масляной бане в течение 20 ч. Анализ методом ТСХ (англ. TLC, thin-layer chroniatography - тонкослойная хроматография) (в системе петролейный эфир : этилацетат = 5:1, Rf = 0,34) показал появление одного нового основного пятна. Реакционным смесям давали возможность охладиться, объединяли и затем фильтровали для удаления твердой фракции. Твердую фракцию промывали EtOAc (2×400,0 мл), и фильтрат концентрировали при пониженном давлении. Неочищенный остаток очищали при помощи колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1), и получали (R)-1-(4-хлорфенил)бут-3-ен-1-ол (414,0 г, 2,27 моль, выход 79,6%) в виде масла коричневого цвета.
Стадия 2
Получение (1R)-3,4-дибром-1-(4-хлорфенил)бутан-1-ола

Проводили параллельно три реакции: раствор Br2 (126,7 г, 793,3 ммоль, 40,9 мл, 1,05 экв.) в ДХМ (англ. DCM, dichloromethane - дихлорметан) (480,0 мл) по каплям прибавляли к раствору (R)-1-(4-хлорфенил)бут-3-ен-1-ола (138,0 г, 755,5 ммоль, 1,00 экв.) в ДХМ (480,0 мл) при температуре -30°С. Полученную смесь перемешивали в течение 1 ч при температуре -30°С. Анализ методом ТСХ (в системе петролейный эфир : этилацетат = 5:1, Rf1 = 0,42, Rf2 = 0,31) показал расходование исходного материала и появление двух новых пятен. Затем эти три реакции объединяли вместе для обработки. Реакции гасили добавлением насыщенного водного раствора Na2S2O3 (3,00 л) и воды (3,00 л). Колбу снимали с бани и оставляли интенсивно перемешиваться при температуре 20°С (оранжевая окраска быстро исчезала). Полученную смесь экстрагировали ДХМ (2×2,50 л), объединенные органические фракции промывали солевым раствором (2,00 л × 2), сушили над Na2SO4 и концентрировали при пониженном давлении. Реакционную массу использовали на следующей стадии без дополнительной очистки. (1R)-3,4-Дибром-1-(4-хлорфенил)бутан-1-ол (743,0 г, неочищенный) получали в виде масла коричневого цвета.
Стадия 3
Получение (2R)-4-бром-2-(4-хлорфенил)тетрагидрофурана
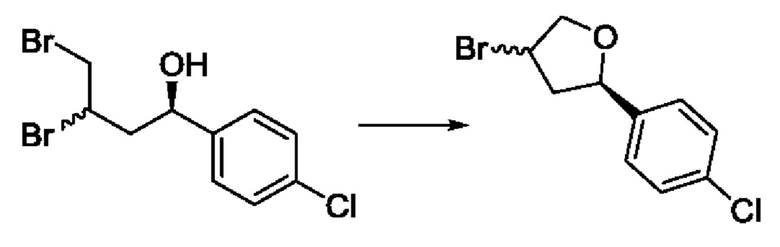
Проводили параллельно три реакции: к раствору (1R)-3,4-дибром-1-(4-хлорфенил)бутан-1-ола (247,0 г, 721,2 ммоль, 1,00 экв.) в МеОН (1,70 л) добавляли K2CO3 (398,7 г, 2,89 моль, 4,00 экв.) при температуре 20°С. Затем реакционную массу перемешивали при температуре 20°С в течение 12 ч. Анализ методом ТСХ (в системе петролейный эфир : этилацетат = 5:1, Rf1 = 0,72, Rf2 = 0,56) показал расходование исходного материала. Затем эти три реакционные массы объединяли для обработки. Реакционные смеси охлаждали и добавляли сначала насыщенный водный раствор NH4Cl (1,50 л), затем воду (1,50 л). Смесь переносили в делительную воронку и экстрагировали EtOAc (2×3,0 л). Затем объединенные органические фракции промывали смесью воды (3,00 л) и солевого раствора (3,00 л), после чего объединенные органические фракции сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Реакционную массу использовали на следующей стадии без дополнительной очистки. (2R)-4-Бром-2-(4-хлорфенил)тетрагидрофуран (555,0 г, неочищенный) получали в виде масла коричневого цвета.
Стадия 4
Получение (3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-карбонитрила
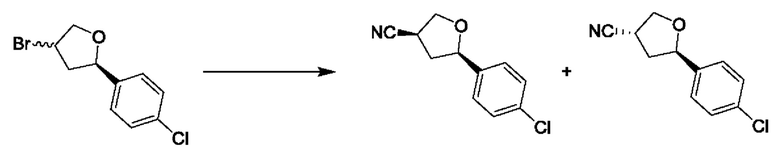
Проводили параллельно три реакции: смесь (2R)-4-бром-2-(4-хлорфенил)тетрагидрофурана (170,0 г, 649,9 ммоль, 1,00 экв.) и KCN (112,3 г, 1,62 моль, 2,50 экв.) в ДМСО (1,20 мл) дегазировали и трижды продували азотом, после чего массу перемешивали при температуре 60°С в течение 20 часов в атмосфере азота. Анализ методом ТСХ (в системе петролейный эфир : этилацетат = 5/1, Rf1 = 0,51, Rf2 = 0,21) показал два новых пятна. Реакцию останавливали удалением масляной бани, три реакционные массы обрабатывали вместе и объединяли. После охлаждения до температуры 20°С смесь разбавляли EtOAc (200,0 мл), осадок отфильтровывали на пористом фильтре и дополнительно промывали EtOAc (140,0 мл). Фильтрат переносили в делительную воронку объемом 10,0 л, затем добавляли воду (4,00 л) и солевой раствор (800,0 мл). Слои разделяли, и водный слой дополнительно экстрагировали EtOAc (2×800,0 мл). После этого объединенные органические фракции промывали смесью воды (800,0 мл) и солевого раствора (800,0 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением масла оранжевого цвета. Остаток очищали при помощи колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 50/1 до 5:1). (3R,5R)-5-(4-Хлорфенил)тетрагидрофуран-3-карбонитрил (102,0 г, 392,9 ммоль, 20,1% выход, чистота 80,0%) получали в виде твердого вещества желтого цвета вместе с диастереомером (120,0 г, 577,8 ммоль, 29,6% выход) в виде твердого вещества желтого цвета.
Стадия 5
Получение (3R,5R,Z)-5-(4-хлорфенил)-N'-гидрокситетрагидрофуран-3-карбоксимидамида
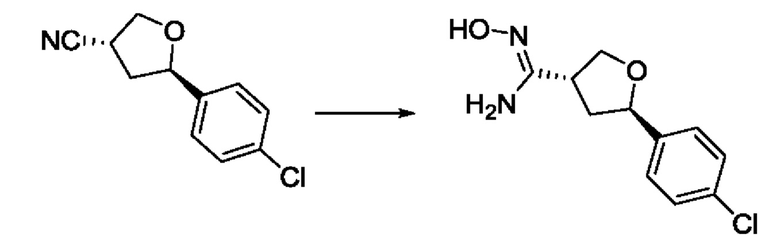
Смесь (3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-карбонитрила (102,0 г, 491,2 ммоль, 1,00 экв.) и гидроксиламина (40,5 г, 1,23 моль, 2,50 экв.) в EtOH (600,0 мл) дегазировали и трижды продували N2, затем реакционную массу перемешивали при температуре 83°С в течение 2 ч в атмосфере N2, после чего анализ методом ВЭЖХ показал, что реакция завершена. Реакционную смесь концентрировали при пониженном давлении, и получали неочищенный остаток, который обрабатывали 100,0 мл МТВЕ и перемешивали в течение 1 ч, после чего появлялся осадок белого цвета. Смесь фильтровали, и фильтрат концентрировали с получением (3R,5R,Z)-5-(4-хлорфенил)-N'-гидрокситетрагидрофуран-3-карбоксимидамида (107,0 г, 444,5 ммоль, выход 90,5%) в виде твердого вещества белого цвета.
Стадия 6
Получение (3R,5R)-N'-(2-хлорацетокси)-5-(4-хлорфенил)тетрагидрофуран-3-карбоксимидамида
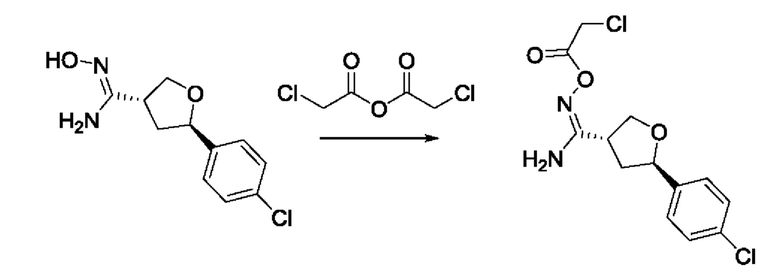
В смесь (3R,5R,Z)-5-(4-хлорфенил)-N'-гидрокситетрагидрофуран-3-карбоксимидамида (89,8 г, 525,5 ммоль, 21,0 мл, 1,10 экв.) в МТВЕ (800,0 мл) добавляли 2-хлоруксусный ангидрид (115,0 г, 477,8 ммоль, 1 экв.) при температуре 0°С, и трижды продували N2, после чего смесь перемешивали при температуре 25°С в течение 0,5 ч в атмосфере N2. Анализ методом ВЭЖХ показал, что реакция завершена. Реакционную смесь выливали в 500,0 мл насыщенного водного раствора NaHCO3. Полученную массу экстрагировали EtOAc (3х250,0 мл), объединенные органические фракции сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. В масло желтого цвета добавляли 300,0 мл МТВЕ, смесь перемешивали в течение 5 мин, затем охлаждали до температуры -20°С. Образовавшийся осадок отфильтровывали на воронке Бюхнера, промывали 150,0 мл холодного МТВЕ (-20°С), и получали требуемый продукт (3R,5R)-N'-(2-хлорацетокси)-5-(4-хлорфенил)тетрагидрофуран-3-карбоксимидамид (120,0 г, 378,3 ммоль, выход 79,1%) в виде твердого вещества белого цвета.
Стадия 7
Получение 5-(хлорметил)-3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазола
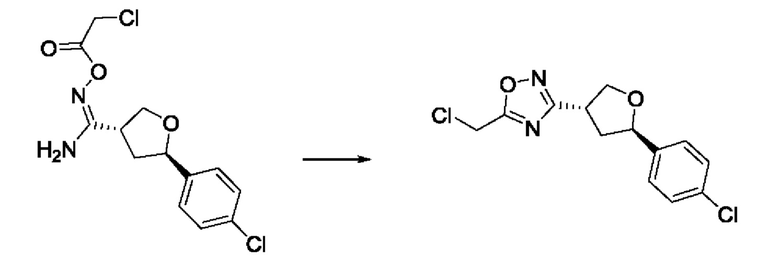
Смесь (3R,5R)-N'-(2-хлорацетокси)-5-(4-хлорфенил)тетрагидрофуран-3-карбоксимидамида (135,0 г, 425,6 ммоль, 1,00 экв.) и активированных молекулярных сит  (135,0 г) в толуоле (675,0 мл) дегазировали и трижды продували N2, после чего массу перемешивали при температуре 120°С в течение 2 ч в атмосфере N2. Анализ методами ВЭЖХ и ТСХ (в системе петролейный эфир: этилацетат = 1/1, Rf = 0,60) показал, что реакция завершена. Реакционную смесь фильтровали, фильтрат концентрировали. Неочищенный остаток очищали при помощи колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 25/1 до 5:1), и получали 5-(хлорметил)-3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол (82,0 г, 274,1 ммоль, 64,4% выход) в виде масла желтого цвета.
(135,0 г) в толуоле (675,0 мл) дегазировали и трижды продували N2, после чего массу перемешивали при температуре 120°С в течение 2 ч в атмосфере N2. Анализ методами ВЭЖХ и ТСХ (в системе петролейный эфир: этилацетат = 1/1, Rf = 0,60) показал, что реакция завершена. Реакционную смесь фильтровали, фильтрат концентрировали. Неочищенный остаток очищали при помощи колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 25/1 до 5:1), и получали 5-(хлорметил)-3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол (82,0 г, 274,1 ммоль, 64,4% выход) в виде масла желтого цвета.
Пример 1
Получение 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6(7H)-она
Общая схема реакций Примера 1 имеет следующий вид:
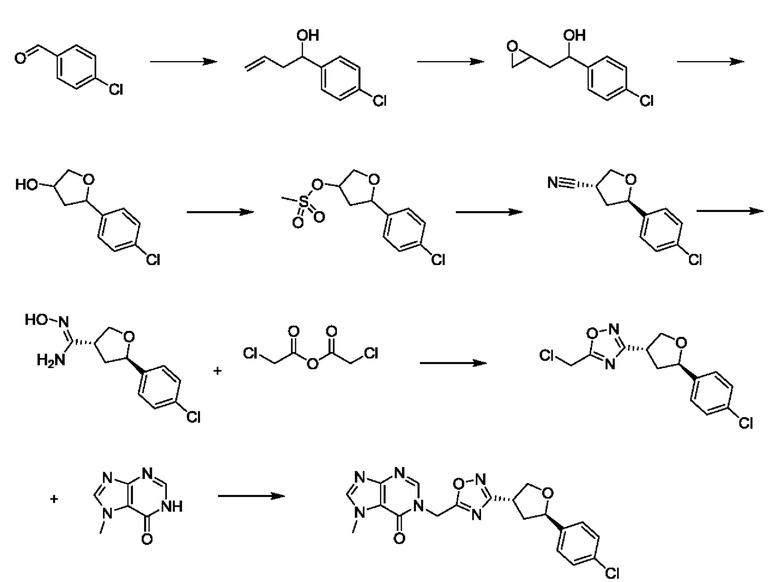
Стадия 1
Получение 1-(4-хлорфенил)бут-3-ен-1-ола
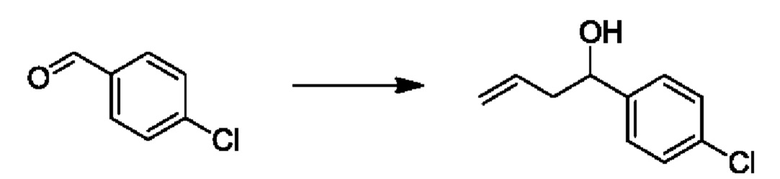
Хлористый аллилмагний (2,0 М в ТГФ, 15,5 мл, 31,0 ммоль) прибавляли в течение 30 мин к раствору 4-хлорбензальдегида (3,43 мл, 28,2 ммоль) в ТГФ (28 мл) при температуре 0°С. Полученную смесь перемешивали при температуре 0°С в течение 30 мин, разбавляли диэтиловым эфиром (25 мл), и гасили реакцию насыщенным раствором NH4Cl (25 мл) и Н2О (25 мл). Слои разделяли, водный слой экстрагировали диэтиловым эфиром (3 х 50 мл). Объединенные органические фракции промывали солевым раствором (50 мл), сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи хроматографии на SiO2 (градиент от 5 до 20% EtOAc в гексане), и получали указанное в заголовке соединение (2,71 г, 53%) в виде бесцветного масла.
1H ЯМР (500 МГц, CDCl3): δ 7,34 - 7,27 (м, 4Н), 5,84 - 5,73 (м, 1Н), 5,21 - 5,16 (м, 1Н), 5,16-5,14 (м, 1Н), 4,75-4,71 (м, 1Н), 2,57 - 2,40 (м, 2Н), 2,03 (d, J = 3,3 Гц, 1Н).
Стадия 2
Получение 1-(4-хлорфенил)-2-(оксиран-2-ил)этанола
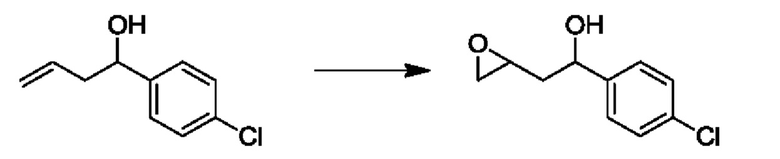
м-СРВА (77%, 3,31 г, 14,8 ммоль) прибавляли порциями к раствору 1-(4-хлорфенил)бут-3-ен-1-ола (2,71 г, 14,8 ммоль) в ДХМ (48 мл) при температуре 0°С. Полученную смесь оставляли нагреваться до температуры 20°С и перемешивали в течение 18 ч. Реакционную смесь охлаждали до температуры 0°С, разбавляли ДХМ и гасили, добавляя порциями Са(ОН)2 (2,6 г, 29,6 ммоль). Полученную смесь перемешивали в течение 2 ч при температуре 20°С, осадок отфильтровывали, фильтрат концентрировали при пониженном давлении, и получали указанное в заголовке соединение (2,79 г, 95%) в виде бесцветного масла.
1H ЯМР (500 МГц, CDCl3, смесь диастереоизомеров): δ 7,35 - 7,29 (м, 4Н), 4,96 (дд, J = 8,3, 5,0 Гц, 0,5Н), 4,92 (дд, J = 9,0, 3,4 Гц, 0,5Н), 3,19 - 3,13 (м, 0,5Н), 3,05 - 2,99 (м, 0,5Н), 2,83 (дд, J = 4,7, 4,1 Гц, 0,5Н), 2,77 (дд, J = 4,8, 4,1 Гц, 0,5Н), 2,62 (дд, J = 4,8, 2,8 Гц, 0,5Н), 2,54 - 2,46 (м, 1,5Н), 2,14 (ддд, J = 14,6, 9,0, 3,8 Гц, 0,5Н), 2,06 (ддд, J = 14,3, 4,9, 4,0 Гц, 0,5Н), 1,85 - 1,74 (м, 1Н).
Стадия 3
Получение 5-(4-хлорфенил)тетрагидрофуран-3-ола
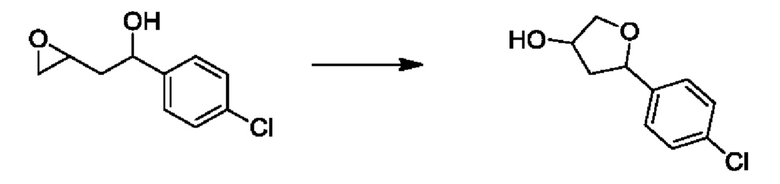
В смесь 1-(4-хлорфенил)-2-(оксиран-2-ил)этанола (2,59 г, 13,0 ммоль) в 1,4-диоксане (130 мл) добавляли серную кислоту (98%, 0,68 мл, 12,5 ммоль). Полученную массу перемешивали при температуре 50°С в течение 16 ч. Реакционную массу выливали на колотый лед, нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали ДХМ (3 х 150 мл). Объединенные органические фракции сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи хроматографии на SiO2 (градиент от 10 до 50% EtOAc в гексане), и получали указанное в заголовке соединение (1,38 г, 53%) в виде масла желтого цвета.
1H ЯМР (500 МГц, CDCl3, смесь диастереоизомеров): δ 7,37 - 7,26 (м, 4Н), 5,13 (дд, J = 10,2, 5,7 Гц, 0,56Н), 4,91 - 4,86 (м, 0,44Н), 4,67 - 4,55 (м, 1Н), 4,24 (дд, J = 9,9, 4,4 Гц, 0,56Н), 4,07 - 4,03 (м, 0.44Н), 3,93 - 3,87 (м, 1Н), 2,70 - 2,62 (м, 0.44Н), 2,35 - 2,29 (м, 0,56Н), 1,93 - 1,84 (м, 1Н), 1,75 (д, J = 4,1 Гц, 0,56Н), 1,63(д, J = 5,7 Гц, 0,44Н)
Стадия 4
Получение 5-(4-хлорфенил)тетрагидрофуран-3-илметансульфоната
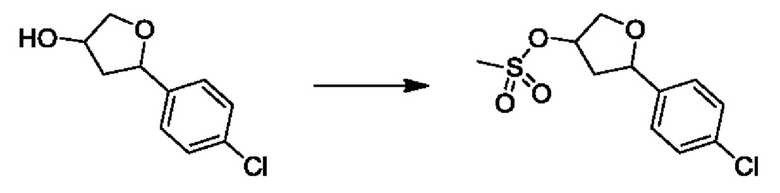
Метансульфонилхлорид (0,50 мл, 6,41 ммоль) добавляли в раствор 5-(4-хлорфенил)тетрагидрофуран-3-ола (980 мг, 4,93 ммоль) и триэтиламина (2,06 мл, 14,8 ммоль) в ДХМ (25 мл) при температуре 0°С. Полученную смесь перемешивали в течение 15 мин, после чего добавляли насыщенный водный раствор NaHCO3 (25 мл). Слои разделяли, водный слой экстрагировали ДХМ (25 мл). Объединенные органические фракции промывали солевым раствором (25 мл), сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и получали указанное в заголовке соединение (1,36 г, 99%) в виде бесцветного масла.
1H ЯМР (500 МГц, CDCl3, смесь диастереоизомеров): δ 7,35 - 7,24 (м, 4Н), 5,44 - 5,36 (м, 1Н), 5,10 (дд, J = 10,3, 5,5 Гц, 0,56Н), 4,88 (т, J = 7,5 Гц, 0,44Н), 4,38 - 4,33 (м, 1Н), 4,16 - 4,13 (м, 0,56Н), 3,97 (дд, J = 11,1, 4,5 Гц, 0,44Н), 3,09 (с, 1,68Н), 2,98 (с, 1,32Н), 2,84 - 2,75 (м, 0,44Н), 2,68 - 2,63 (м, 0,56Н), 2,20 - 2,14 (м, 0,44Н), 2,08 - 2,00 (м, 0,56Н).
Стадия 5
Получение (3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-карбонитрила
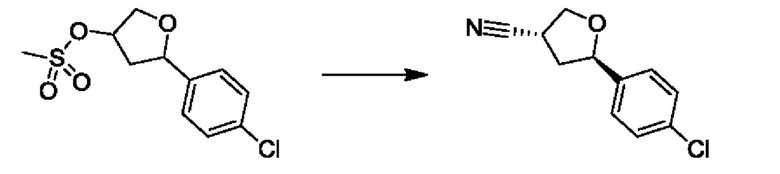
Смесь 5-(4-хлорфенил)тетрагидрофуран-3-илметансульфоната (1,36 г, 4,91 ммоль) и цианида калия (1,60 г, 24,6 ммоль) в ДМСО (12 мл) перемешивали в течение 1 ч при температуре 105°С. Реакционную массу разбавляли EtOAc и промывали насыщенным водным раствором NaHCO3, водой и солевым раствором. Органическую фракцию сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи хроматографии на SiO2 (градиент от 0 до 40% EtOAc в гексане), и получали указанное в заголовке соединение (213 мг, 21%) в виде масла желтого цвета (первый элюируемый диастереоизомер) и нежелательный цис-диастереоизомер (224 мг, 22%) в виде масла желтого цвета (второй элюируемый диастереоизомер).
Указанное в заголовке соединение: 1H ЯМР (500 МГц, CDCl3): δ 7,35 - 7,32 (м, 2Н), 7,26 - 7,23 (м, 2Н), 5,09 - 5,02 (м, 1Н), 4,38 (дд, J = 8,9, 7,6 Гц, 1Н), 4,09 - 4,03 (м, 1Н), 3,28 - 3,19 (м, 1Н), 2,68 - 2,61 (м, 1Н), 2,18 - 2,11 (м, 1Н).
цис-Диастереоизомер: 1H ЯМР (500 МГц, CDCl3): δ 7,37 - 7,33 (м, 2Н), 7,33 - 7,28 (м, 2Н), 4,86 (дд, J = 8,7, 6,7 Гц, 1Н), 4,29 (дд, J = 9,0, 5,4 Гц, 1Н), 4,12 (дд, J = 9,0, 7,7 Гц, 1Н), 3,31 - 3,22 (м, 1Н), 2,80 - 2,72 (м, 1Н), 2,17 - 2,08 (м, 1Н).
Стадия 6
Получение (3R,5R)-5-(4-хлорфенил)-N'-гидрокситетрагидрофуран-3-карбоксимидамида
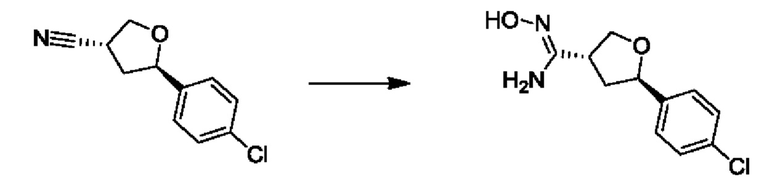
Гидроксиламин (50% в воде, 0,31 мл, 5,13 ммоль) добавляли в раствор (3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-карбонитрила (213 мг, 1,03 ммоль) в EtOH (5,1 мл), и полученную смесь перемешивали при температуре 80°С в течение 1 ч. Растворитель отгоняли при пониженном давлении, остаток упаривали вместе с EtOH (2х) и ДХМ (1х), и получали указанное в заголовке соединение (231 мг, 94%) в виде густого бесцветного масла.
ЖХМС [М+Н+] 241.
Стадия 7
Получение 5-(хлорметил)-3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазола

Хлоруксусный ангидрид (212 мг, 1,24 ммоль) добавляли в раствор (3R,5R)-5-(4-хлорфенил)-N'-гидрокситетрагидрофуран-3-карбоксимидамида (231 мг, 0,960 ммоль) в ДХЭ (5 мл) при температуре 20°С. Полученную массу перемешивали в течение 15 мин, добавляли диизопропилэтиламин (0,25 мл, 1,44 ммоль), и реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли толуолом (5 мл), и реакционную смесь перемешивали при температуре 100°С в течение 1 ч. Реакционную смесь разбавляли EtOAc (25 мл) и промывали насыщенным водным раствором NaHCO3, водой и солевым раствором. Органическую фракцию сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи хроматографии на SiO2 (градиент от 0 до 25% EtOAc в гексане), и получали указанное в заголовке соединение (235 мг, 82%) в виде масла желтого цвета.
ЖХМС [M+H+] 299.
1H ЯМР (500 МГц, CDCl3): δ 7,35 - 7,29 (м, 4Н), 5,14 (т, J = 7,3 Гц, 1Н), 4,69 (с, 2Н), 4,47 (дд, J = 8,8, 7,6 Гц, 1Н), 4,10 (дд, J = 8,8, 6,6 Гц, 1Н), 3,79 - 3,73 (м, 1Н), 2,72 (ддд, J = 12,5, 7,0, 5,3 Гц, 1Н), 2,23 (ддд, J = 12,8, 9,0, 7,6 Гц, 1Н).
Стадия 8
Получение 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6(7Н)-она

Смесь рацематов 5-(хлорметил)-3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазола (130 мг, 0,430 ммоль), 7-метил-1H-пурин-6-она (98 мг, 0,650 ммоль), йодистого тетрабутиламмония (4 мг, 0,010 ммоль) и карбоната калия (180 мг, 1,30 ммоль) в ДМФА (2 мл) перемешивали при температуре 20°С в течение 2 ч. В реакционную массу добавляли воду (2 мл), твердую фракцию отфильтровывали, промывали водой, сушили в вакууме, и получали указанное в заголовке соединение (152 мг, 85%) в виде рацемической смеси. Энантиомеры разделяли при помощи SFC (англ. Supercritical Fluid Chromatography - хроматография со сверхкритической подвижной фазой) (колонка: Lux Cel-3, 10 х 250 мм, 5 мкм, 40% МеОН, 10 мл/мин, 150 бар, температура колонки: 40°С, время хроматографирования 16 мин), и получали указанное в заголовке соединение (43 мг, 24%) в виде твердого вещества белого цвета (первый элюируемый энантиомер, RT = 11,5 мин) и энантиомер указанного в заголовке соединения (43 мг, 24%) в виде твердого вещества белого цвета (второй элюируемый энантиомер, RT = 13,5 мин).
Указанное в заголовке соединение: ЖХМС [М+Н+] 413.
1H ЯМР (500 МГц, ДМСО-d6): δ 8,46 (с, 1Н), 8,24 (с, 1Н), 7,42 - 7,36 (м, 4Н), 5,57 (с, 2Н), 5,01 (т, J = 7,3 Гц, 1Н), 4,35 (дд, J = 8,6, 7,4 Гц, 1Н), 3,96 (с, 3Н), 3,90 (дд, J = 8,6, 6,3 Гц, 1Н), 3,80 - 3,71 (м, 1Н), 2,60 - 2,54 (м, 1Н), 2,17 - 2,10 (м, 1Н).
Пример 2
Синтез 1-((3-((3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6(7Н)-она
Общая схема реакций Примера 2 имеет следующий вид:

Стадия 1
Получение (R)-1-(4-фторфенил)бут-3-ен-1-ола
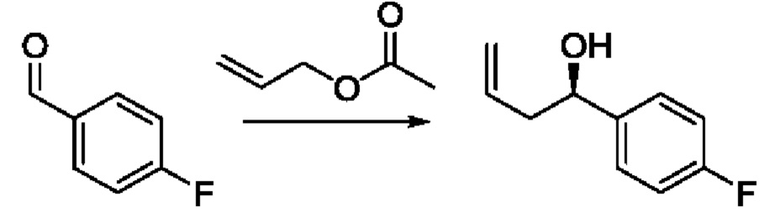
(R)-(+)-BINAP (12,9 г, 20,1 ммоль), 4-хлор-3-нитробензойную кислоту (8,12 г, 40,3 ммоль) и Cs2CO3 (26,3 г, 80,6 ммоль) загружали в двухгорлую колбу емкостью 2 л. Добавляли 1,4-диоксан (671 мл), аллилацетат (435 мл, 4028 ммоль), изопропанол (62 мл, 806 ммоль) и 4-фторбензальдегид (43,2 мл, 4023 ммоль). На колбу устанавливали холодильник и мембрану. Через реакционную смесь барботировали азот. В раствор при барботировании добавляли димер хлор(1,5-циклооктадиен)иридия(i) (6,83 г, 10,1 ммоль), и барботирование через реакционную смесь продолжали в течение еще 10 мин. Реакционную смесь перемешивали при температуре 112°С на масляной бане в течение 27 ч. Затем реакционную смесь охлаждали до комнатной температуры, и отфильтровывали твердую фракцию. Фильтрат концентрировали на роторном испарителе.
Неочищенную смесь упаривали с толуолом (2х). Неочищенный продукт очищали на колонке с силикагелем (15W x 15H). Продукт наносили в минимальном количестве толуола и элюировали (по 2 л каждой смеси растворителей 3%, 4%, 6%, 8%, 10%, 15%, 20% iPrOAc/гептан). 23 г смешанных фракций повторно очищали на колонке с силикагелем, элюируя 5% iPrOAc/гептан, затем 20% iPrOAc/гептан. Еще 7,3 г повторно очищали в тех же условиях, и получали указанное в заголовке соединение (58,3 г, выход 87%) в виде масла оранжевого цвета.
Стадия 2
Получение (R)-3,4-дибром-1-(4-фторфенил)бутан-1-ола
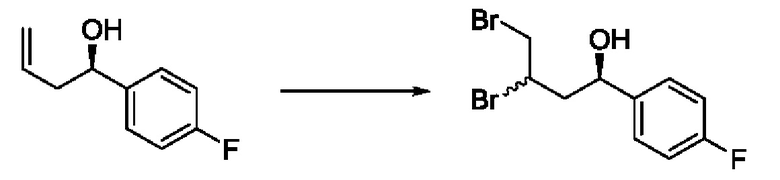
Раствор брома (16,3 мл, 317 ммоль) в ДХМ (302 мл) прибавляли по каплям в течение 1 ч 30 мин к раствору (1R)-1-(4-фторфенил)бут-3-ен-1-ола (50,2 г, 302 ммоль) в ДХМ (755 мл) при температуре от -30°С до -40°С. Реакционную массу перемешивали при температуре -30°С в течение 30 мин. Реакцию гасили насыщенным Na2S2O3 (300 мл) и водой (300 мл) и перемешивали при комнатной температуре в течение 30 мин. Фазы разделяли, водный слой экстрагировали (2х) ДХМ. Объединенные органические фракции сушили над MgSO4, фильтровали и концентрировали, в результате получали указанное в заголовке соединение (96,9 г, 98% выход) в виде неочищенного масла коричневого цвета.
Стадия 3
Получение (R)-4-бром-2-(4-фторфенил)тетрагидрофурана
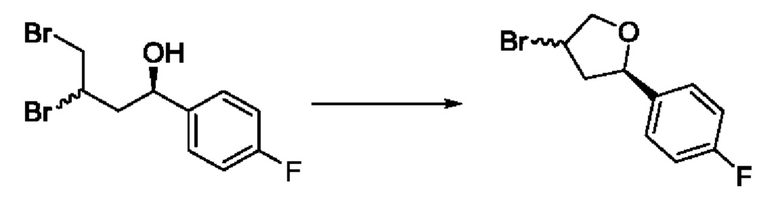
K2CO3 (166 г, 1189 ммоль) добавляли в раствор (1R)-3,4-дибром-1-(4-фторфенил)бутан-1-ола (96,9 г, 297 ммоль) в МеОН (743 мл). Для регулирования экзотермичности реакции использовали водяную баню с температурой 20°. Реакционную массу перемешивали при комнатной температуре в течение ночи. После этого реакционную массу охлаждали до температуры 10°С и добавляли сначала насыщенный NH4Cl (500 мл), затем воду (500 мл). Смесь экстрагировали iPrOAc (3х), промывали водой и солевым раствором. Объединенные органические фракции сушили над MgSO4, фильтровали и концентрировали, в результате получали указанное в заголовке соединение (69,8 г, 96% выход) в виде неочищенного масла коричневого цвета.
Стадия 4
Получение (3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-карбонитрила
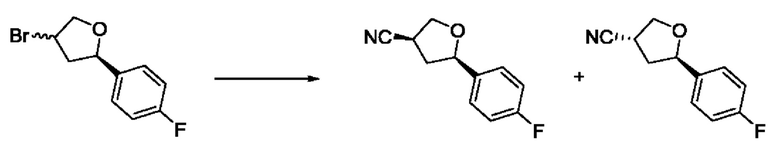
Цианид калия (57,4 г, 855 ммоль) добавляли в раствор (2R)-4-бром-2-(4-фторфенил)тетрагидрофурана (69,8 г, 285 ммоль) в ДМСО (570 мл). Реакционную массу перемешивали при температуре 105°С в течение 15 ч и оставляли охлаждаться до комнатной температуры. Избыток KCN удаляли фильтрацией, осадок промывали iPrOAc. Реакционную массу распределяли в смеси вода/iPrOAc и экстрагировали iPrOAc (3х). Объединенные органические экстракты промывали водой (2х) и солевым раствором, затем сушили над MgSO4, фильтровали и концентрировали. Неочищенную смесь очищали при помощи хроматографии на силикагеле, используя колонку 15 см (ширина) х 18 см (высота), элюируя смесью 5%, 7%, 10%, 12%, 15%, 20%, 25%, 30%, 35%, 40% iPrOAc/гептан с получением указанного в заголовке транс-нитрила (18,4 г, выход 34%) в виде прозрачного масла желтого цвета. Цис-нитрил, (3S,5R)-5-(4-фторфенил)тетрагидрофуран-3-карбонитрил, получали (15,7 г, выход 29%) в виде прозрачного масла желтого цвета.
Стадия 5
Получение (3R,5R,Z)-5-(4-фторфенил)-N'-гидрокситетрагидрофуран-3-карбоксимидамида
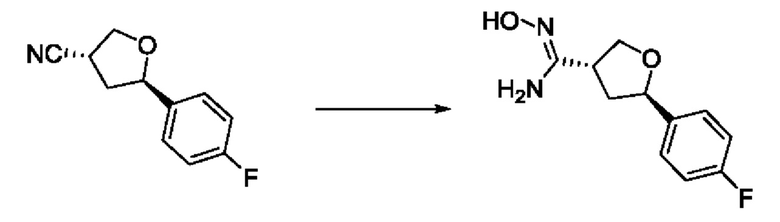
Гидроксиламин (50 мас. % в воде) (49,5 мл, 808 ммоль) добавляли в раствор (3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-карбонитрила (48,3 г, 202 ммоль) в EtOH (505 мл). Смесь перемешивали при температуре 80°С в течение 2 ч. Реакционную смесь концентрировали на роторном испарителе. Смесь очищали на слое силикагеля (10 см ширина х 7 см высота), сначала элюируя примеси 100% ДХМ (1,5 л), затем элюируя продукт 10% МеОН/ДХМ (2 л). Часть продукта выходила в первой фракции, остальные фракции были чистыми. Первую фракцию концентрировали отдельно и повторно очищали. 19,6 г смешанных фракций повторно очищали на колонке с силикагелем сначала 100% ДХМ, затем смесью 10% МеОН/ДХМ, и получали указанное в заголовке соединение (36,4 г, выход 80%) в виде смолы сине-серого цвета.
Стадия 6
Получение (3R,5R,Z)-N'-(2-хлорацетокси)-5-(4-фторфенил)тетрагидрофуран-3-карбоксимидамида
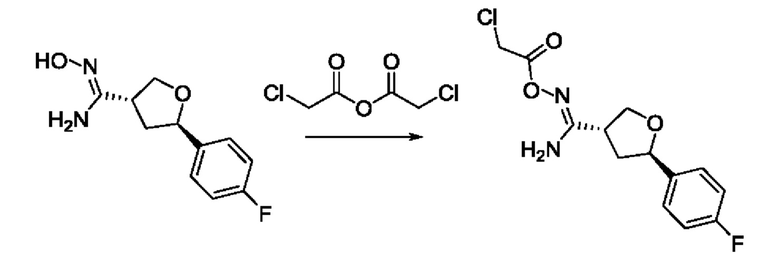
Хлоруксусный ангидрид (10,8 г, 60,1 ммоль) прибавляли порциями к раствору (3R,5R)-5-(4-фторфенил)-N'-гидрокси-тетрагидрофуран-3-карбоксамидина (12,3 г, 54,6 ммоль) в МТВЕ (137 мл) при температуре 0°С. Реакционную массу перемешивали при комнатной температуре в течение 20 мин. Затем реакционную смесь охлаждали до температуры 0°С, разбавляли iPrOAc и разделяли насыщенным раствором NaHCO3. Водный слой экстрагировали iPrOAc (3х). Объединенные органические фракции снова промывали насыщенным раствором NaHCO3 и солевым раствором, сушили над MgSO4, фильтровали и концентрировали не полностью. Растворитель заменяли на МТВЕ и концентрировали до получения маслянистого остатка. Продукт растирали со 150 мл МТВЕ. Раствор охлаждали до температуры -20°С, фильтровали и промывали МТВЕ при температуре -20°С. Продукт сушили в вакууме и получали указанный в заголовке продукт (11,9 г, выход 73%) в виде твердого вещества белого цвета.
Стадия 7
Получение 5-(хлорметил)-3-((3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазола

Смесь [(Z)-[амино-[(3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-ил]метилен]амино]-2-хлорацетата (36,0 г, 120 ммоль) и порошка молекулярных сит (40 г, предварительно активированного в печи) в толуоле (479 мл) перемешивали при температуре 115°С в течение 8 ч. Реакционную массу охлаждали до комнатной температуры. Молекулярные сита удаляли фильтрацией и промывали iPrOAc. Фильтрат концентрировали на роторном испарителе. Неочищенную смесь очищали на колонке с силикагелем с помощью 0-50% iPrOAc/гептан, и получали указанное в заголовке соединение (32,1 г, 95% выход) в виде прозрачного масла.
Стадия 8
Получение 1-((3-((3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6(7Н)-она
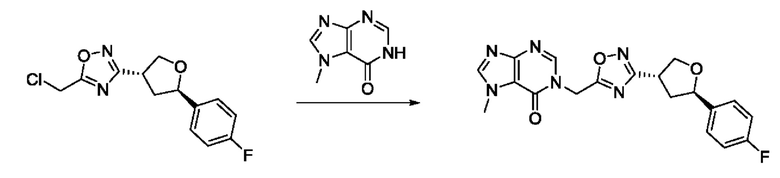
7-Метил-1Н-пурин-6-он (23,9 г, 159 ммоль) и K2CO3 (47,6 г, 341 ммоль) добавляли в раствор 5-(хлорметил)-3-[(3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазола (32,1 г, 114 ммоль) в ДМФА (227 мл) при температуре 0°С. Смесь перемешивали при температуре 0°С в течение 6 ч. Реакционную массу оставляли нагреваться до комнатной температуры в течение ночи. При температуре 0°С добавляли сначала EtOH (95 мл), затем воду (950 мл). Твердую фракцию отфильтровывали на воронке с пористым стеклянным фильтром и промывали водой при температуре 0°С. Осадок сушили в вакууме в течение 15 мин. Твердое вещество растворяли в ДХМ, сушили над MgSO4, фильтровали и концентрировали. После получения пасты на роторном испарителе заменяли ДХМ на iPrOAc и дважды упаривали с ~100 мл iPrOAc. Остаток растирали с 415 мл iPrOAc, охлаждали до температуры 0°С, осадок отфильтровывали, промывали холодным iPrOAc и сушили в вакууме, в результате получали указанное в заголовке соединение (40,5 г, выход 90%) в виде твердого вещества грязно-белого цвета.
Стадия 9
Перекристаллизация 1-((3-((3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6(7Н)-она
1-((3-((3R,5R)-5-(4-Фторфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6(7Н)-он (40 г, 101 ммоль) помещали в реактор емкостью 1 л и добавляли смесь EtOH/вода (7:3, 720 мл) (18 объемов). Реакционную массу нагревали до температуры 75°С. Материал медленно растворялся в течение 40 мин. В это время в реакционную массу в качестве затравки добавляли суспензию неочищенного материала (1 г вещества, суспендированного в EtOH/вода, 1:1, 20 мл). Образовавшуюся суспензию выдерживали при температуре 75°С в течение 30 мин. После этого суспензию медленно охлаждали до температуры 25°С в течение 2 ч и оставляли при температуре 25°С еще на 1 ч. Материал фильтровали через фильтровальную воронку; твердую фракцию промывали смесью EtOH/вода (1:1, 100 мл). Твердую фракцию сначала сушили на воздухе, затем оставляли в вакуумном сушильном шкафу (60°С, в течение ночи) со слабым током азота, и получали указанное в заголовке соединение (36,2 г, 91% выход) в виде кристаллов бегого цвета.
Пример 3
2-Амино-3-((3-((3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-5-метилпиразоло[5,1-f][1,2,4]триазин-4(3Н)-он
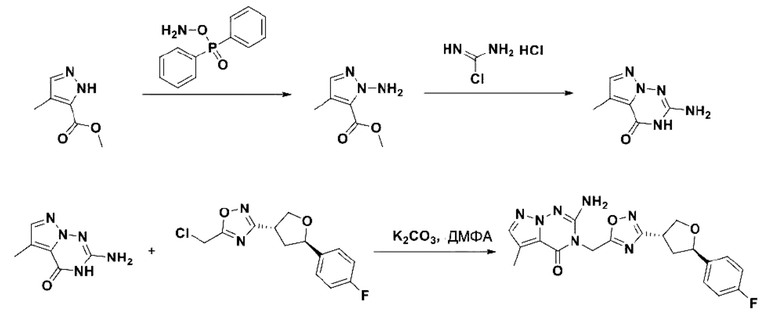
Стадия 1
Получение метил-1-амино-4-метил-1Н-пиразол-5-карбоксилата
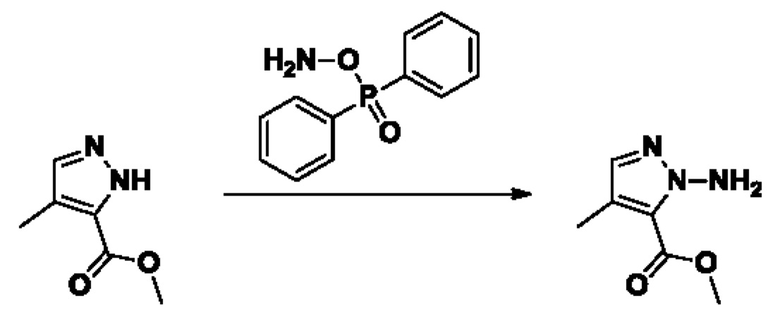
LiHMDS (1M в ТГФ)(14,3 мл, 14,3 ммоль) прибавляли в течение 5 мин к раствору этил-4-метил-1Н-пиразол-5-карбоксилата (2,00 г, 13,0 ммоль) в ДМФА (130 мл) при температуре -20°С. Полученную смесь перемешивали в течение 15 мин при температуре 0°С, после чего в один прием добавляли о-(дифенилфосфинил)гидроксиламин (3,63 г, 15,6 ммоль). После появления белого осадка реакционная масса достаточно быстро сильно загустевала, и ее время от времени перемешивали вручную в течение 1 ч при комнатной температуре. Реакционную массу разбавляли водой до полного растворения осадка и перемешивали при комнатной температуре в течение 15 мин. Затем реакционную массу концентрировали досуха и разбавляли приблизительно 150 мл смеси 2:1 ДХМ/EtOAc. Твердую фракцию отделяли фильтрацией, осадок на фильтре дополнительно промывали смесью 2:1 ДХМ/EtOAc, фильтрат концентрировали при пониженном давлении и дважды упаривали с гептаном. Остаток очищали при помощи колоночной флэш-хроматографии (загрузка в ДХМ, 100 г SiO2 Biotage, градиент 0-4% МеОН в ДХМ, 17 CV (англ. column volume - объем колонки)), и получали этил-2-амино-4-метилпиразол-3-карбоксилат (1,54 г, 9,10 ммоль, 70% выход) в виде масла светло-желтого цвета.
ЖХМС: чистота = 98%, МН+ = 169,8.
1H ЯМР (400 МГц, CDCl3): δ 7,14 (д, J = 0,4 Гц, 1Н), 5,62 (ушир.с, 2Н), 4,39 (кв, J = 7,1 Гц, 2Н), 2,23 (д, J = 0,6 Гц, 3Н), 1,40 (т, J = 7,1 Гц, 3Н).
Стадия 2
Получение 2-амино-5-метилпиразоло[5,1-f][1,2,4]триазин-4(3Н)-она
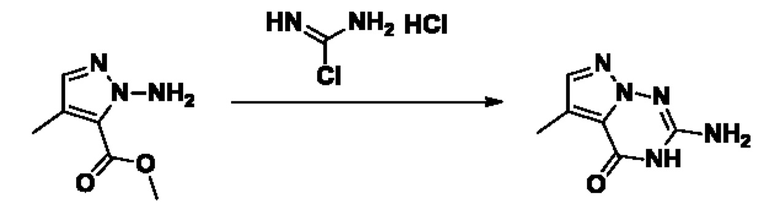
N,N-Диизопропилэтиламин (1,16 мл, 6,65 ммоль) добавляли в смесь этил-2-амино-4-метилпиразол-3-карбоксилата (450 мг, 2,66 ммоль) и гидрохлорида хлорформамидина (611 мг, 5,32 ммоль) в ДХМ (10 мл). Реакционную массу нагревали под действием микроволнового излучения при температуре 150°С в течение 2 ч. Раствор охлаждали до комнатной температуры, осадок отфильтровывали и промывали ДХМ с получением 2-амино-5-метил-3Н-пиразоло[5,1-f][1,2,4]триазин-4-она (432 мг, 2,62 ммоль, выход 98%) в виде порошка бежевого цвета.
ЖХМС: чистота = 85%, МН+ = 166,3.
1H ЯМР (400 МГц, ДМСО-D6): 11,25 (с, 1Н), 7,32 (с, 1Н), 6,13 (с, 1Н), 2,26 (т, 4Н).
Стадия 3
Получение 2-амино-3-((3-((3R,5R)-5-(4-фторфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-5-метилпиразоло[5,1-f][1,2,4]триазин-4(3Н)-она
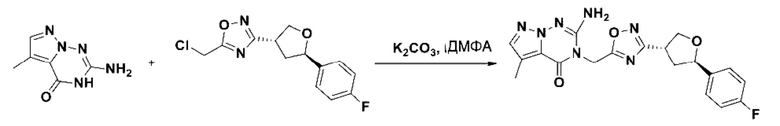
К раствору 5-(хлорметил)-3-[5-(4-фторфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазола (40,0 мг, 0,140 ммоль, полученного из Примера 6) в ДМФА (0,42 мл) прибавляли сначала 2-амино-5-метил-3Н-пиразоло[5,1-f][1,2,4]триазин-4-он (28,0 мг, 0,170 ммоль), затем карбонат калия (39,1 мг, 0,280 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Разбавляли водой, экстрагировали EtOAc, промывали NH4Cl, солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали при помощи нормально-фазовой флэш-хроматографии (12 г, SiO2, 0-10% МеОН в ДХМ). Фракции, содержащие продукт, объединяли и упаривали в вакууме. Соединение растворяли в смеси ацетонитрила и воды, сушили вымораживанием и лиофилизировали с получением 2-амино-3-[[3-[5-(4-фторфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазол-5-ил]метил]-5-метилпиразоло[5,1-f][1,2,4]триазин-4-она (16,2 мг, 0,039 ммоль, выход 28%) в виде твердого вещества белого цвета.
ЖХМС: чистота = 96%, МН+ = 412,0.
1H ЯМР (400 МГц, ДМСО): 7,44 - 7,35 (м, 3Н), 7,21 - 7,11 (м, 2Н), 6,88 (с, 2Н), 5,48 (с, 2Н), 5,00 (т, J = 7,4 Гц, 1Н), 4,36 (дд, J = 8,5, 7,4 Гц, 1Н), 3,89 (дд, J = 8,5, 6,3 Гц, 1Н), 3,77 (дт, J = 13,9, 6,9 Гц, 1Н), 2,56 (ддд, J = 12,4, 7,0, 5,1 Гц, 1Н), 2,29 (д, J = 0,4 Гц, 3Н), 2,15(ддд, J = 12,7, 8,9, 7,8 Гц, 1Н).
Пример 4
Получение 2-амино-3-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-5-метилпиразоло[5,1-f][1,2,4]триазин-4(3Н)-она
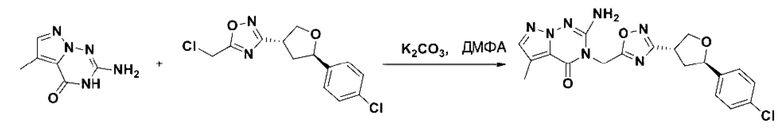
В раствор 5-(хлорметил)-3-[5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазола (40,0 мг, 0,130 ммоль, полученного в Примере 1) в ДМФА (0,42 мл) добавляли 2-амино-5-метил-3Н-пиразоло[5,1-f][1,2,4]триазин-4-он (26,5 мг, 0,160 ммоль, полученный в Примере 105), затем добавляли карбонат калия (37,0 мг, 0,270 ммоль). Реакционную массу перемешивали при комнатной температуре в течение 2 ч. Разбавляли водой, экстрагировали EtOAc, промывали NH4Cl, солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали при помощи флэш-хроматографии (SiO2, 12 г, 0-10% МеОН в ДХМ). Фракции, содержащие продукт, объединяли и упаривали в вакууме. Соединение растворяли в смеси ацетонитрила и воды, сушили вымораживанием и лиофилизировали с получением 2-амино-3-[[3-[5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазол-5-ил]метил]-5-метилпиразоло[5,1-f][1,2,4]триазин-4-она (21,7 мг, 0,051 ммоль, 38% выход) в виде твердого вещества белого цвета.
ЖХМС: чистота = 95%, МН+ = 428,1.
1H ЯМР (400 МГц, ДМСО): 7,44 - 7,35 (м, 5Н), 6,87 (с, 2Н), 5,48 (с, 2Н), 5,01 (т, J = 7,5 Гц, 1Н), 4,36 (дд, J = 8,5, 7,4 Гц, 1Н), 3,90 (дд, J = 8,6, 6,3 Гц, 1Н), 3,77 (дд, J = 14,9, 6,3 Гц, 1Н), 2,57 (ддд, J = 7,3, 6,2, 3,3 Гц, 1Н), 2,29 (с, 3Н), 2,20 - 2,08 (м, 1Н).
Пример 5
1-((3-((3R,5R)-5-(4-Хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1,7-дигидро-6H-пурин-6-он-2-d
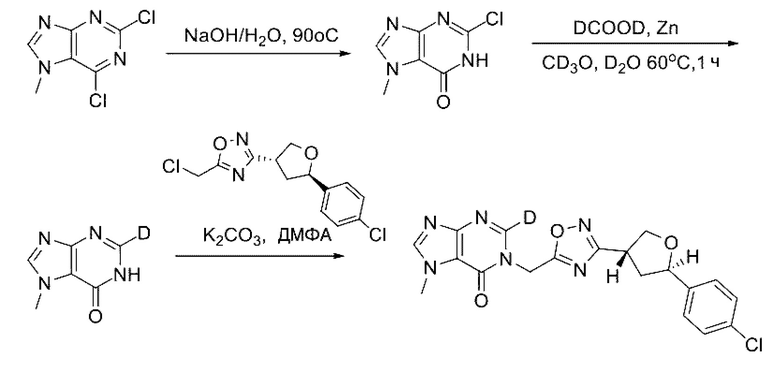
Стадия 1
Получение 2-хлор-7-метил-1,7-дигидро-6Н-пурин-6-она
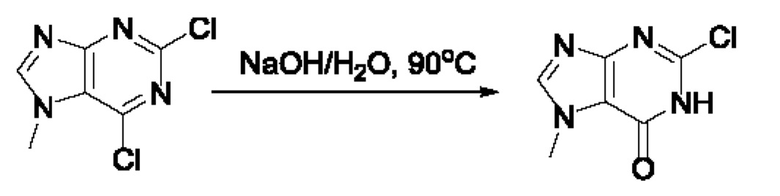
Раствор 2,6-дихлор-7-метилпурина (1,0 г, 4,93 ммоль) и NaOH (0,99 г, 24,63 ммоль) в воде (10 мл) перемешивали при температуре 90°С в течение 1 часа. Величину рН реакционной смеси доводили до 2 с помощью HCl (10%). Твердый осадок отфильтровывали, и получали указанное в заголовке соединение в виде твердого вещества белого цвета.
Стадия 2
Получение 7-метил-1,7-дигидро-6Н-пурин-6-он-2-d

Смесь 2-хлор-7-метил-1Н-пурин-6-она (700,0 мг, 3,79 ммоль), Zn (2465,06 мг, 37,92 ммоль) и DCOOD (1820,35 мг, 37,92 ммоль) в CD3OD (10,0 г, 277,78 ммоль) и D2O (5,0 г, 250 ммоль) перемешивали при температуре 60°С в течение 16 часов. Реакционную смесь разбавляли МеОН (100 мл). Твердую фракцию отфильтровывали, фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колонки с силикагелем С 18, элюируя смесью CH3CN/H2O (10 м моль/л NH4HCO3, от 5% до 95%, в течение 30 мин). В результате получали указанное в заголовке соединение (370 мг) в виде твердого вещества белого цвета.
Стадия 3
Получение 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1,7-дигидро-6Н-пурин-6-он-2-d

В раствор 5-(хлорметил)-3-[5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазола (20,0 мг, 0,070 ммоль, полученного по аналогии с Примером 2) в ДМФА (0,5 мл) добавляли 2-дейтерио-7-метил-1Н-пурин-6-он (12,1 мг, 0,080 ммоль), затем добавляли карбонат калия (18,5 мг, 0,130 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Затем смесь разбавляли водой, экстрагировали EtOAc, промывали NH4Cl, солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали при помощи флэш-хроматографии (SiO2, 12 г, 0-10% МеОН в ДХМ). Фракции, содержащие продукт, объединяли и упаривали в вакууме. Соединение растворяли в смеси ацетонитрила и воды, сушили вымораживанием и лиофилизировали с получением 1-[[3-[5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазол-5-ил]метил]-2-дейтерио-7-метил-пурин-6-она (18,0 мг, 0,044 ммоль, выход 65%) в виде твердого вещества белого цвета.
ЖХМС: чистота = 96%, MH+ = 414,0.
1H ЯМР (400 МГц, ДМСО): 8,22 (с, 1Н), 7,42 - 7,28 (м, 4Н), 5,55 (с, 2Н), 4,99 (т, J = 7,4 Гц, 1Н), 4,33 (дд, J = 8,5, 7,4 Гц, 1Н), 3,93 (с, 3Н), 3,88 (дд, J = 8,6, 6,3 Гц, 1Н), 3,80 - 3,70 (м, 1Н), 2,61 - 2,52 (м, 1Н), 2,19 - 2,08 (м, 1Н).
Пример 6
1-((3-((3R,5R)-5-(4-Хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1,7-дигидро-6H-пурин-6-он-8-d
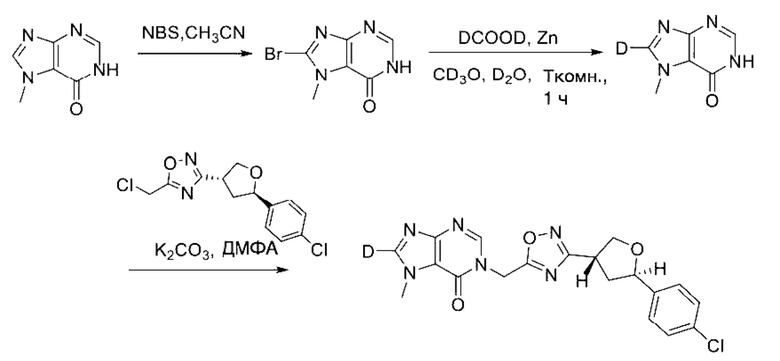
Стадия 1
Получение 8-бром-7-метил-1,7-дигидро-6Н-пурин-6-она
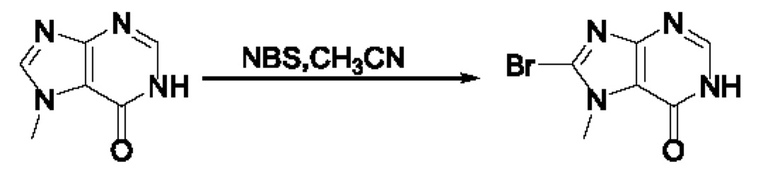
Смесь 7-метил-1Н-пурин-6-она (200,0 мг, 1,33 ммоль) и NBS (284,53 мг, 1,6 ммоль) в ацетонитриле (8 мл) перемешивали в течение ночи при температуре 80°С. Растворитель отгоняли в вакууме. Остаток очищали при помощи флэш-хроматографии на силикагеле, элюируя смесью CH2Cl2/МеОН (10:1), и получали указанное в заголовке соединение в виде твердого вещества белого цвета.
Стадия 2
Получение 7-метил-1,7-дигидро-6Н-пурин-6-он-8-d

Смесь 8-бром-7-метил-1Н-пурин-6-она (1,4 г, 6,11 ммоль), D2O (5 мл, ммоль), CD3OD (10 мл, ммоль), Zn (3,91 г, 61,13 ммоль) и DCOOD (2,93 г, 61,13 ммоль) перемешивали при комнатной температуре в течение 1 ч. Осадок отфильтровывали, фильтрат очищали на колонке с силикагелем С18, элюируя смесью CH3CN/H2O (10 мм оль/л NH4HCO3, от 5% до 95% в течение 30 мин). В результате получали указанное в заголовке соединение (510 мг) в виде твердого вещества белого цвета.
Стадия 3
Получение 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1,7-дигидро-6H-пурин-6-он-8-d

В раствор 5-(хлорметил)-3-[5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазола (20,0 мг, 0,070 ммоль, полученного по аналогии с Примером 2) в ДМФА (0,5 мл) добавляли 8-дейтерио-7-метил-1Н-пурин-6-он (12,1 мг, 0,080 ммоль), затем добавляли карбонат калия (18,5 мг, 0,130 ммоль). Реакционную смесь, перемешивали при комнатной температуре в течение 16 ч. Разбавляли водой, экстрагировали EtOAc, промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали при помощи флэш-хроматографии (SiO2, 12 г, 0-10% МеОН в ДХМ). Фракции, содержащие продукт, объединяли и упаривали в вакууме. Соединение растворяли в смеси ацетонитрила и воды, сушили вымораживанием и лиофилизировали с получением 1-[[3-[5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазол-5-ил]метил]-8-дейтерио-7-метил-пурин-6-она (12,0 мг, 0,029 ммоль, выход 43%) в виде твердого вещества белого цвета.
ЖХМС: чистота = 99%, МН+ = 414,0.
1H ЯМР (400 МГц, ДМСО): 8,44 (с, 1Н), 7,47 - 7,28 (м, 4Н), 5,56 (с, 2Н), 4,99 (т, J = 7,3 Гц, 1Н), 4,33 (т, J = 7,9 Гц, 1Н), 3,93 (с, 3Н), 3,88 (дд, J = 8,5, 6,3 Гц, 1Н), 3,79 - 3,70 (м, 1Н), 2,61 - 2,51 (м, 1Н), 2,19 - 2,08 (м, 1Н).
Пример 7
1-((3-((3R,5R)-5-(4-Хлорфенил)-тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-2,6(3Н,7Н)-дион
Схема реакций Примера 7 имеет следующий вид:
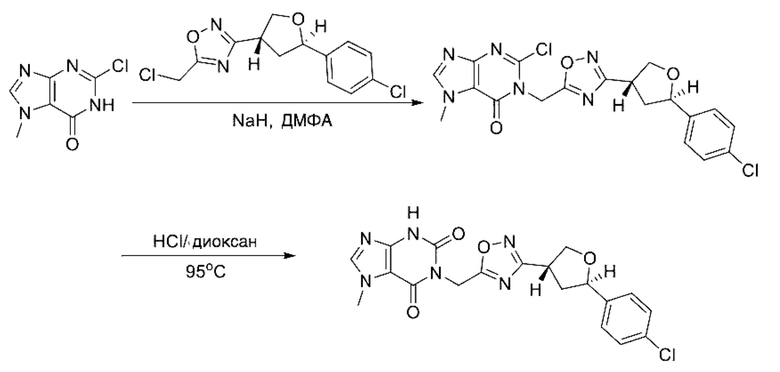
Стадия 1
Получение 2-хлор-1-((3-((3R,5R)-5-(4-хлорфенил)-тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6(7Н)-она
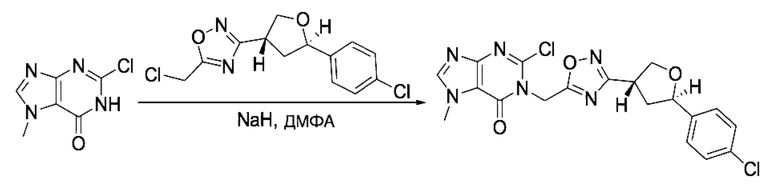
NaH (60%) (39,0 мг, 0,65 ммоль) порциями прибавляли к раствору 2-хлор-7-метил-1Н-пурин-6-она (100,0 мг, 0,54 ммоль) в ДМФА (3 мл) при комнатной температуре в атмосфере азота и перемешивали в течение 1 часа. Затем добавляли раствор 5-(хлорметил)-3-[(3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазола (194,0 мг, 0,65 ммоль, полученного по аналогии с Примером 1) в ДМФА (3 мл). Полученный раствор перемешивали при температуре 50°С в течение ночи. Полученный в результате раствор дополнительно очищали с помощью ОФ-ВЭЖХ (англ. RP-HPLC, Reversed Phase High Performance Liquid Chromatography - обращенно-фазовая высокоэффективная жидкостная хроматография) и получали указанное в заголовке соединение (91 мг, 38%) в виде твердого вещества белого цвета.
Стадия 2
Получение 1-((3-((3R,5R)-5-(4-хлорфенил)-тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-2,6(3Н,7Н)-диона
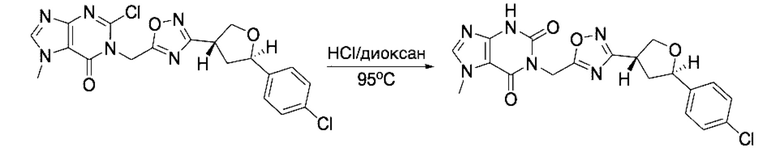
Раствор 2-хлор-1-[[3-[(3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазол-5-ил]метил]-7-метил-пурин-6-она (300,0 мг, 0,67 ммоль) и HCl (36%) (0,5 мл, 0,67 ммоль) в 1, 4-диоксане (10 мл) перемешивали при температуре 95°С в течение 4 часов. Остаток очищали при помощи ОФ-ВЭЖХ, и получали указанное в заголовке соединение (145 мг, 50%) в виде твердого вещества белого цвета.
ЖХМС [М+Н+]: 429,1.
1H ЯМР (400 МГц, ДМСО-d6): δ 12,21 (с, 1Н), 8,03 (с, 1Н), 7,40 (д, J = 1,3 Гц, 4Н), 5,31 (с, 2Н), 5,02 (т, J = 7,3 Гц, 1Н), 4,36 (дд, J = 8,6, 7,4 Гц, 1Н), 3,91 (дд, J = 8,6, 6,3 Гц, 1Н), 3,87 (с, 3Н), 3,77 - 3,73 (м, 1Н), 2,1 - 2,56 (м, 1Н), 2,20 - 2,06 (м, 1Н).
Пример 8
1-((3-((3R,5R)-5-(4-Хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-2,7-диметил-1Н-пурин-6(7Н)-он
Общая схема реакций Примера 8 имеет следующий вид:
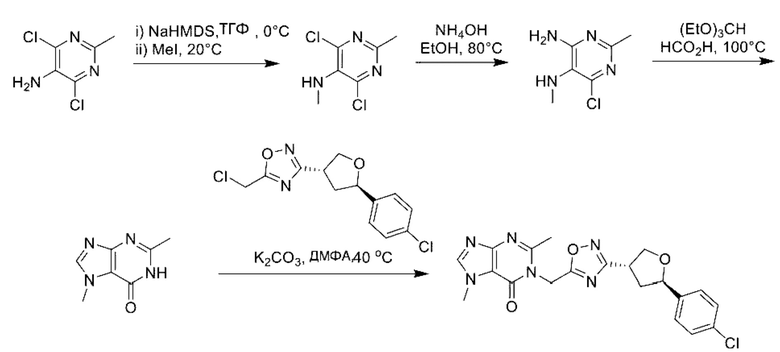
Стадия 1
Получение 4,6-дихлор-N,2-диметилпиримидин-5-амина

К 1 л RBF в атмосфере азота добавляли NaHMDS (1M в ТГФ, 126 мл, 126 ммоль) и ТГФ (120 мл). Раствор охлаждали на ледяной бане, и когда внутренний датчик показывал температуру 2°С, по каплям прибавляли раствор 4,6-дихлор-2-метилпиримидин-5-амина (20,0 г, 112 ммоль) в ТГФ (120 мл) в течение 40 мин, поддерживая внутреннюю температуру в диапазоне от 2 до 4°С. Реакционную смесь оставляли перемешиваться при температуре 2°С в течение 1 часа, затем в течение 5 мин прибавляли йодметан (8,2 мл, 131 ммоль). Реакционную смесь перемешивали при температуре 20°С в течение 2 часов, и останавливали реакцию добавлением насыщенного водного раствора NH4Cl (350 мл), воды (50 мл) и солевого раствора (50 мл). Полученную смесь экстрагировали EtOAc (3х200 мл). Объединенные органические фракции промывали солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного 4,6-дихлор-N,2-диметилпиримидин-5-амина (предположительно, 21,6 г, выход неочищенного продукта 100%) в виде масла коричневого цвета, затвердевающего при стоянии при комнатной температуре. Анализ методом ЖХМС показал чистоту 88%, неочищенный материал в таком виде использовали для следующей реакции.
Стадия 2
Получение 6-хлор-N5,2-диметилпиримидин-4,5-диамина
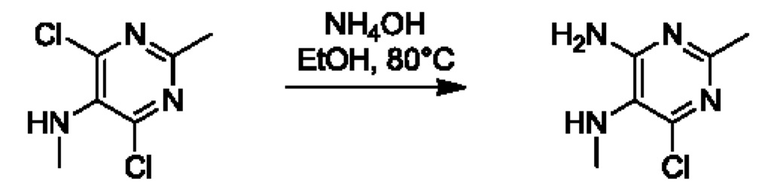
В реактор высокого давления емкостью 500 мл переносили неочищенный 4,6-дихлор-N,2-диметилпиримидин-5-амин (21,6 г, 112 ммоль) в этаноле (70 мл). Добавляли 28% водный NH4OH (130 мл, 1924 ммоль), реактор герметизировали, и смесь перемешивали на масляной бане при температуре 80°С в течение 18 часов. Реакционную смесь оставляли охлаждаться до температуры 20°С и дополнительно охлаждали на ледяной бане. Происходило образование твердого вещества золотистого цвета, смесь перемешивали при температуре 0°С в течение 2 часов. Суспензию фильтровали, твердую фракцию промывали смесью холодного этанола EtOH (30 мл) и воды (60 мл), и получали 6-хлор-N5,2-диметил-пиримидин-4,5-диамин (12,2 г, 70,7 ммоль, 63% выход, чистота в соответствии с ЖХМС 91%) в виде твердого вещества золотистого цвета. Маточный раствор концентрировали, полученное твердое вещество суспендировали в МеОН и воде (-5:1). Смесь нагревали до температуры 70°С и оставляли охлаждаться до температуры 20°С. Полученную в результате суспензию фильтровали, твердую фракцию промывали минимальным количеством холодного МеОН, и получали вторую порцию 6-хлор-N5,2-диметилпиримидин-4,5-диамина (1,40 г, 8,11 ммоль, 7% выход, чистота в соответствии с ЖХМС 97%) в виде твердого вещества бежевого цвета.
Стадия 3
Получение 2,7-диметил-1Н-пурин-6(7Н)-она

В раствор 6-хлор-2,N5-диметилпиримидин-4,5-диамина (3,50 г, 20,3 ммоль) в триэтилортоформиате (15 мл) добавляли муравьиную кислоту (2,80 г, 60,8 ммоль). Полученную смесь перемешивали при температуре 100°С в течение ночи. Затем реакционную массу концентрировали. Неочищенный продукт промывали этилацетатом, и получали 2,7-диметил-1Н-пурин-6(7Н)-он (2,00 г, выход 60%) в виде твердого вещества желтого цвета.
1H ЯМР (300 МГц, ДМСО-d6): δ 12,14 (с, 1Н), 8,07 (с, 1Н), 3,93 (с, 3Н), 2,32 (с, 3Н).
Стадия 4
Получение 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-2,7-диметил-1Н-пурин-6(7Н)-она
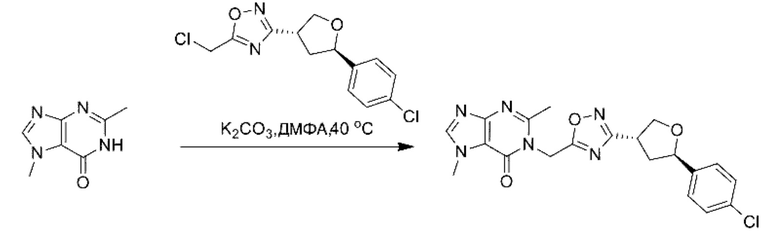
5-(Хлорметил)-3-[(3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазол (8,40 г, 28,1 ммоль, полученный по аналогии с Примером 1), 2,7-диметил-1Н-пурин-6(7Н)-он (5,07 г, 30,9 ммоль) и карбонат калия (11,6 г, 84,2 ммоль) загружали в 200 мл RBF. Добавляли ДМФА (56 мл), и смесь перемешивали на масляной бане при температуре 40°С в течение 2 часов. Нагрев убирали, реакционную массу переносили в делительную воронку, содержащую EtOAc (100 мл) и воду (500 мл). Смесь экстрагировали EtOAc (3 х 100 мл). Объединенные органические фракции промывали смесью воды (40 мл) и солевого раствора (40 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта (чистота в соответствии с ЖХМС 77%). Неочищенное масло коричневого цвета растворяли в EtOAc (100 мл) и по каплям прибавляли гептан (60 мл), при этом происходило медленное образование твердого вещества. Смесь перемешивали при температуре 0°С в течение 30 мин, твердую фракцию отфильтровывали на воронке Бюхнера, промывали гептаном, сушили на воздухе в течение 15 минут и получали первую порцию 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-2,7-диметил-1Н-пурин-6(7Н)-она (8,0 г, 18,7 ммоль, выход 67%, чистота в соответствии с ЖХМС 92%) в виде твердого вещества желтого цвета. Фильтрат концентрировали и очищали при помощи хроматографии с обращенной фазой (С18, MeCN / 10 мМ NH4HCO2 в Н2О, рН 3,8, градиент от 0 до 60%), и получали вторую порцию 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-2,7-диметил-1Н-пурин-6(7Н)-она (950 мг, 2,23 ммоль, выход 8%, чистота в соответствии с ЖХМС > 99%) в виде твердого вещества белого цвета.
Первую порцию требуемого продукта (8,0 г, чистота в соответствии с ЖХМС 92%) объединяли с третьей порцией, полученной после предыдущей серии опытов (1,3 г, чистота в соответствии с ЖХМС 96%). Материал растворяли в EtOAc (250 мл), на роторном испарителе растворитель заменяли на iPrOH (3 цикла добавления iPrOH (50 мл) / отгонки 50 мл растворителя). В ходе этого процесса происходило образование твердого вещества, а количество растворителя на роторном испарителе уменьшалось приблизительно до -100 мл. Суспензию охлаждали до температуры 0°С, твердую фракцию отфильтровывали на воронке Бюхнера, промывали холодным iPrOH, сушили на воздухе в течение 15 минут и получали четвертую порцию 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-2,7-диметил-1Н-пурин-6(7Н)-она (8,50 г, чистота в соответствии с ЖХМС 99%) в виде твердого вещества светло-бежевого цвета. Все еще неудовлетворительный по цвету очищенный материал растворяли в EtOAc (500 мл), и раствор коричневого цвета обрабатывали 8 г активированного угля. Смесь перемешивали в течение 15 мин, суспензию фильтровали через слой целита, осадок на фильтре промывали EtOAc. Бесцветный фильтрат упаривали при пониженном давлении, и получали пятую порцию 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-2,7-диметил-1Н-пурин-6(7Н)-она (7,50 г, чистота в соответствии с ЖХМС 99%) в виде твердого вещества белого цвета. И, наконец, вторую порцию (950 мг, чистота в соответствии с ЖХМС > 99%) и пятую порцию (7,50 г, чистота в соответствии с ЖХМС 99%) объединяли с 200 мл RBF и добавляли iPrOH (100 мл). Суспензию перемешивали на масляной бане при температуре 100°С до полного растворения, и добавляли воду (3 мл). Баню убирали, полученный прозрачный раствор светло-желтого цвета оставляли охлаждаться до комнатной температуры. Суспензию охлаждали на ледяной бане, твердую фракцию отфильтровывали на воронке Бюхнера, промывали холодным iPrOH (20 мл), сушили на воздухе в течение 24 часов и получали указанное в заголовке соединение 1-((3-((3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-2,7-диметил-1Н-пурин-6(7Н)-он (7,3 г) в виде твердого вещества белого цвета.
ЖХМС: чистота = 99,4%, МН+ = 427,2/429,2.
1H ЯМР (400 МГц, ДМСО-d6): δ 8,18 (с, 1Н), 7,46 - 7,33 (м, 4Н), 5,63 (с, 2Н), 5,00 (т, J = 7,4 Гц, 1Н), 4,35 (дд, J = 8,4, 7,5 Гц, 1Н), 3,93 (с, 3Н), 3,95 - 3,88 (м, 1Н), 3,79-3,70 (м, 1Н), 2,60 (с, 3Н), 2,63 - 2,53 (м, 1Н), 2,13 (ддд, J = 12,7, 8,9, 7,8 Гц, 1Н).
Пример 9
1-((3-((3R,3R)-5-(4-Хлорфенил)-тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6,8(7Н,9Н)-дион
Общая схема реакций Примера 9 имеет следующий вид:
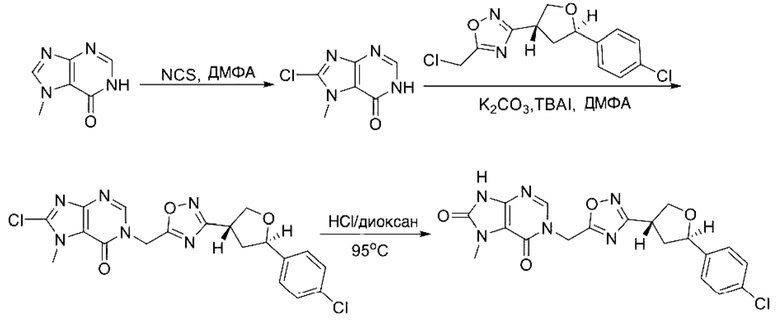
Стадия 1
Получение 8-хлор-7-метил-1Н-пурин-6(7Н)-она
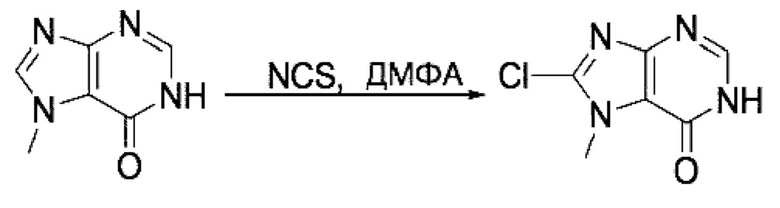
Раствор NCS (6,0 г, 44,93 ммоль) и 7-метил-1Н-пурин-6-она (8,7 г, 57,95 ммоль) в ДМФА (70 мл) перемешивали при комнатной температуре в течение ночи. Полученный в результате раствор дополнительно очищали с помощью ОФ-ВЭЖХ, и получали указанное в заголовке соединение (4,7 г, 65%) в виде твердого вещества грязно-белого цвета.
Стадия 2
Получение 8-хлор-1-((3-((3R,5R)-5-(4-хлорфенил)-тетрагидрофуран-3-ил]-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6(7Н)-она
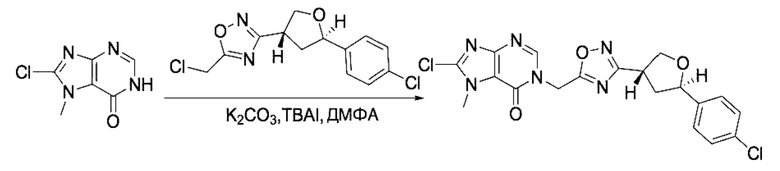
Раствор 5-(хлорметил)-3-[(3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазола (55,0 мг, 0,18 ммоль, полученного по аналогии с Примером 1), 8-хлор-7-метил-1Н-пурин-6(7Н)-она (30,0 мг, 0,19 ммоль), K2CO3 (51,0 мг, 0,37 ммоль) и TBAI (3,4 мг, 0,01 ммоль) в ДМФА (3 мл) перемешивали при комнатной температуре в течение 2 часов. Остаток очищали при помощи ОФ-ВЭЖХ, и получали указанное в заголовке соединение (48 мг, 67%) в виде твердого вещества белого цвета.
Стадия 3
Получение 1-((3-((3R,5R)-5-(4-хлорфенил)-тетрагидрофуран-3-ил)-1,2,4-оксадиазол-5-ил)метил)-7-метил-1Н-пурин-6,8(7Н,9Н)-диона

Раствор 8-хлор-1-[[3-[(3R,5R)-5-(4-хлорфенил)тетрагидрофуран-3-ил]-1,2,4-оксадиазол-5-ил]метил]-7-метил-пурин-6-она (230,0 мг, 0,51 ммоль) в муравьиной кислоте (5 мл) перемешивали при температуре 95°С в течение 2 часов. Полученный в результате раствор дополнительно очищали с помощью ОФ-ВЭЖХ, и получали указанное в заголовке соединение (51 мг, 23%) в виде твердого вещества белого цвета.
ЖХМС [М+Н+]: 429,1.
1H ЯМР (400 МГц, ДМСО-d6): δ 11,88 (с, 1Н), 8,41 (с, 1Н), 7,39 (д, J = 2,1 Гц, 4Н), 5,53 (с, 2Н), 5,01 (т, J = 7,3 Гц, 1Н), 4,36 (т, J = 8,0 Гц, 1Н), 3,90 (дд, J = 8,5, 6,2 Гц, 1Н), 3,81 - 3,75 (м, 1Н), 2,61 - 2,56 (м, 1Н), 2,20 - 2,06 (м, 1Н).
Описанные выше соединения вместе с другими соединениями, полученными при использовании описанных выше методик и соответствующих исходных материалов, представлены в Таблице 1, там же приведены величины hTRPA1 IC50 для каждого из соединений.

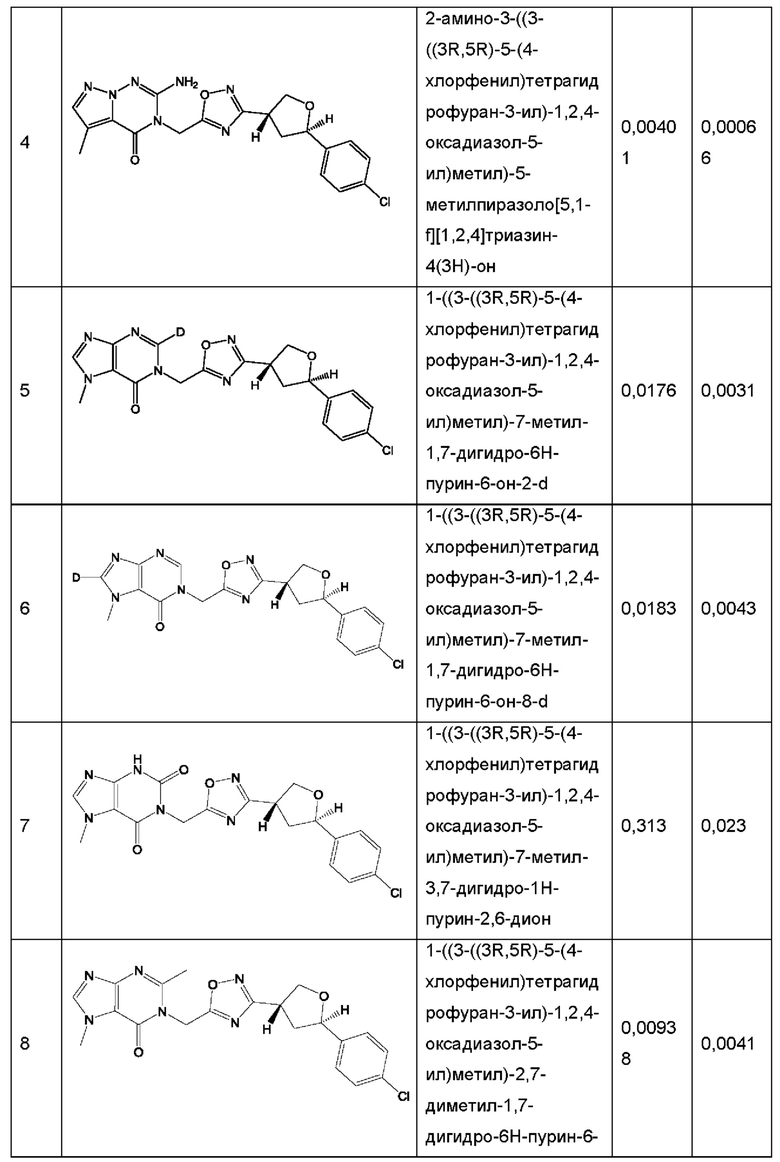
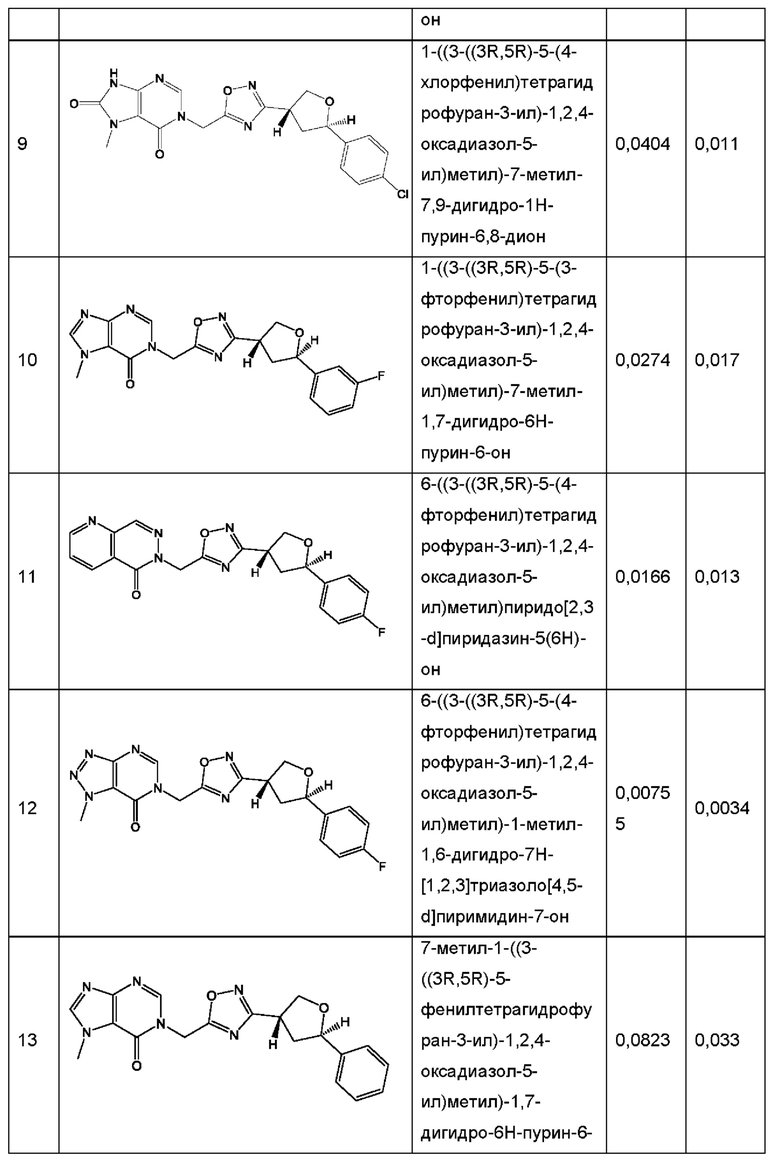

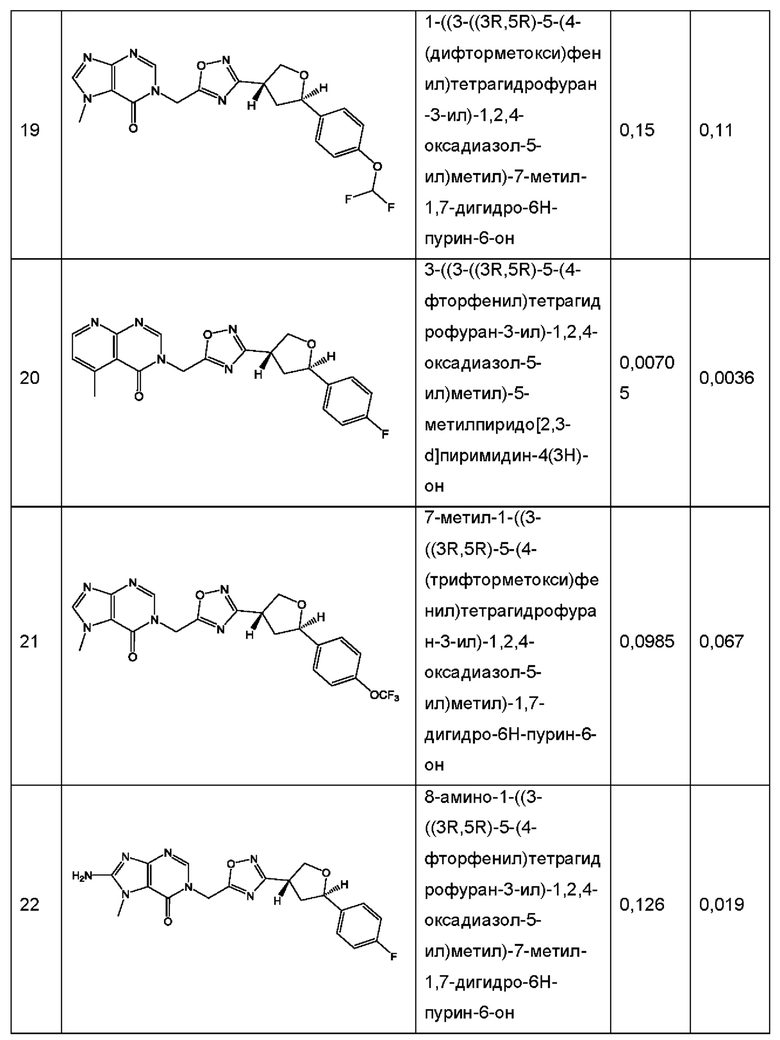
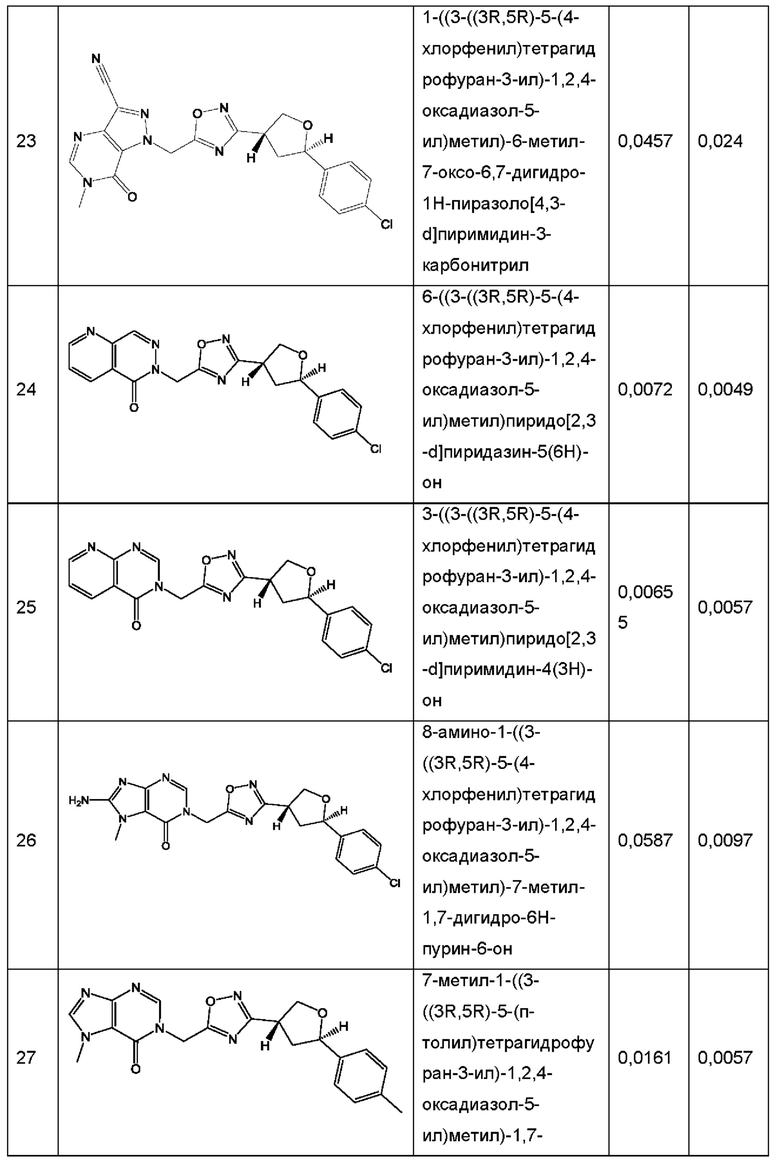
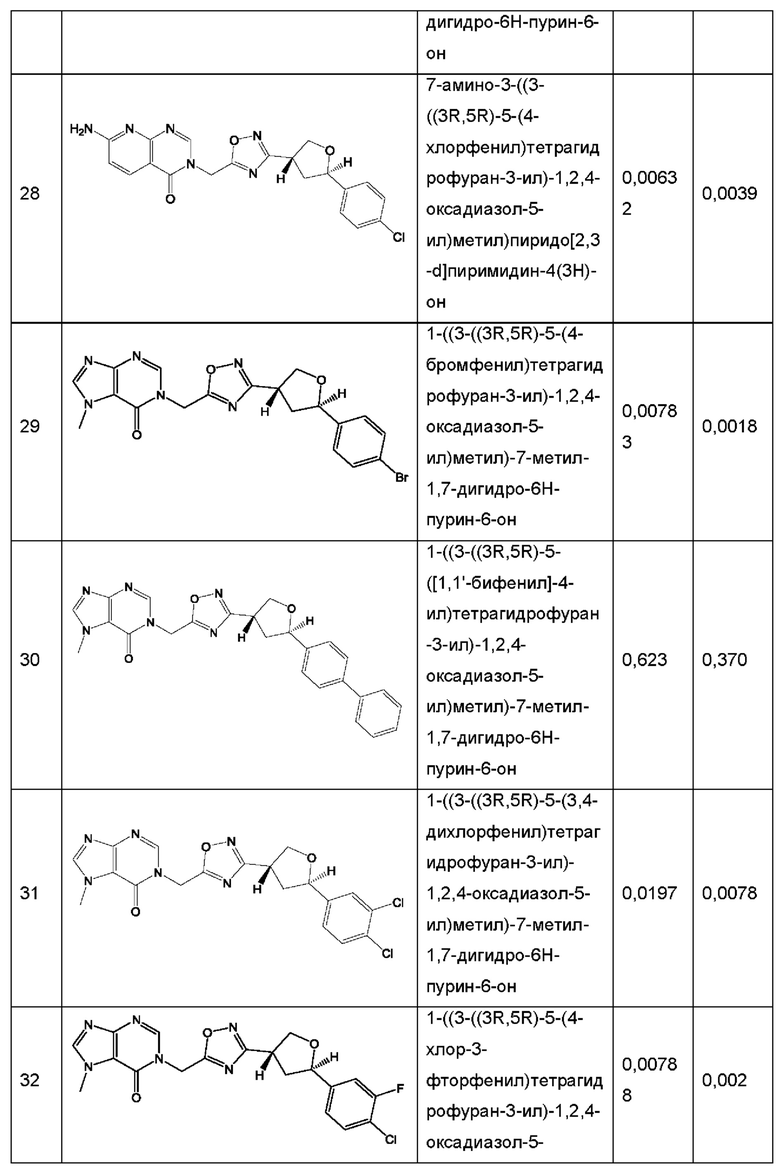
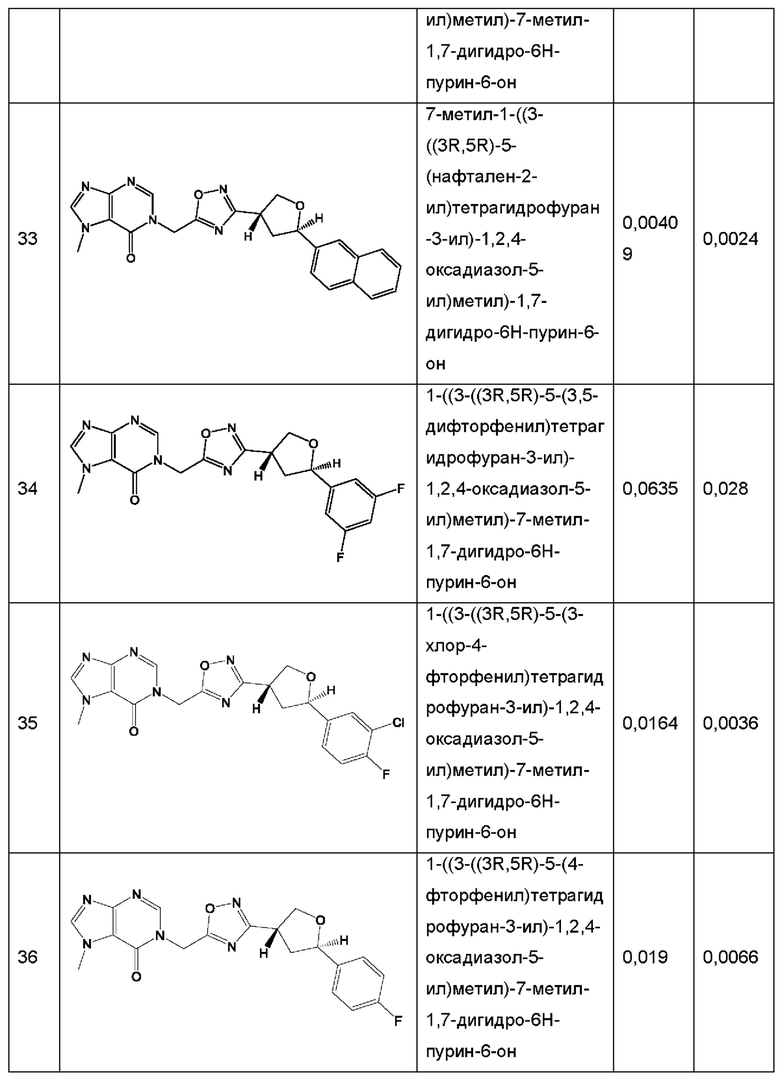
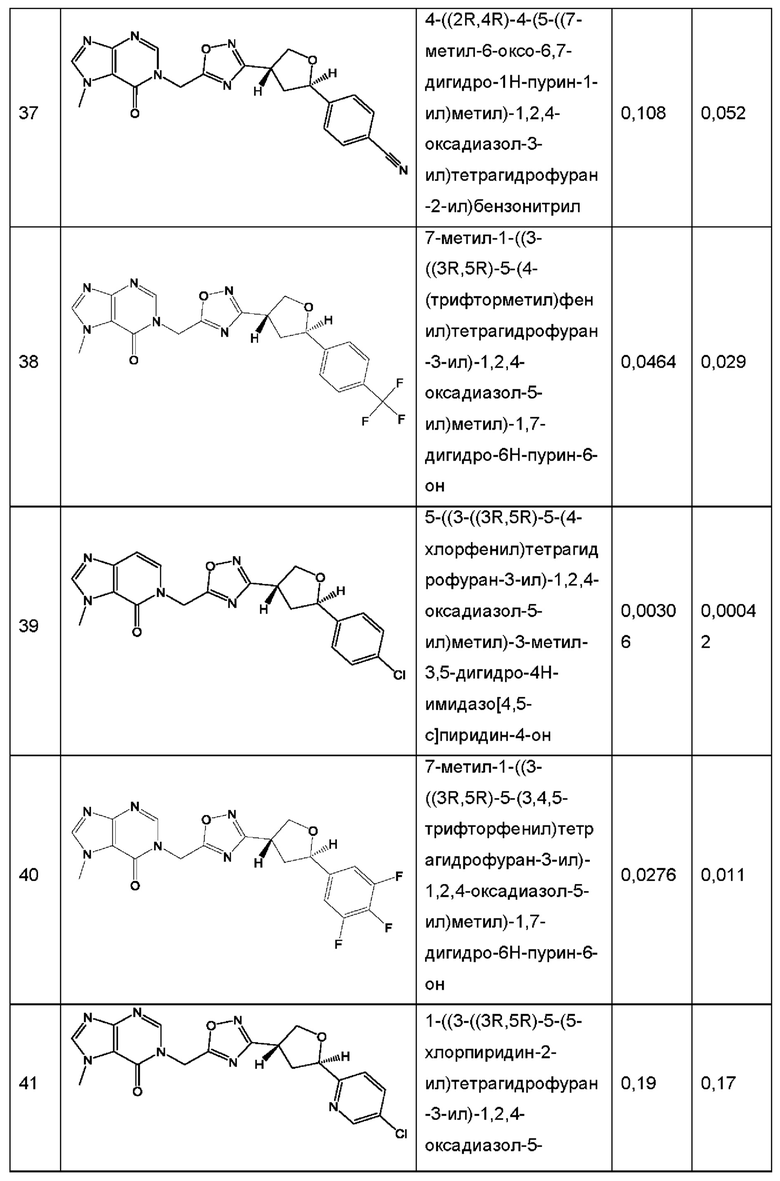
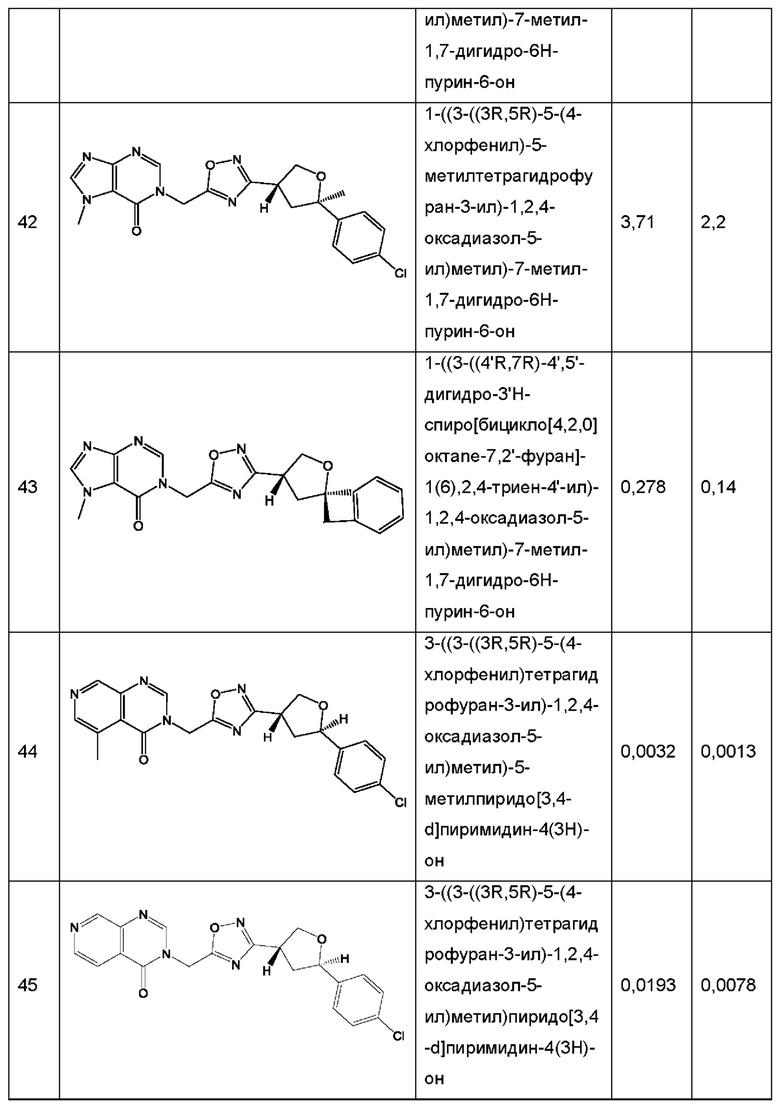
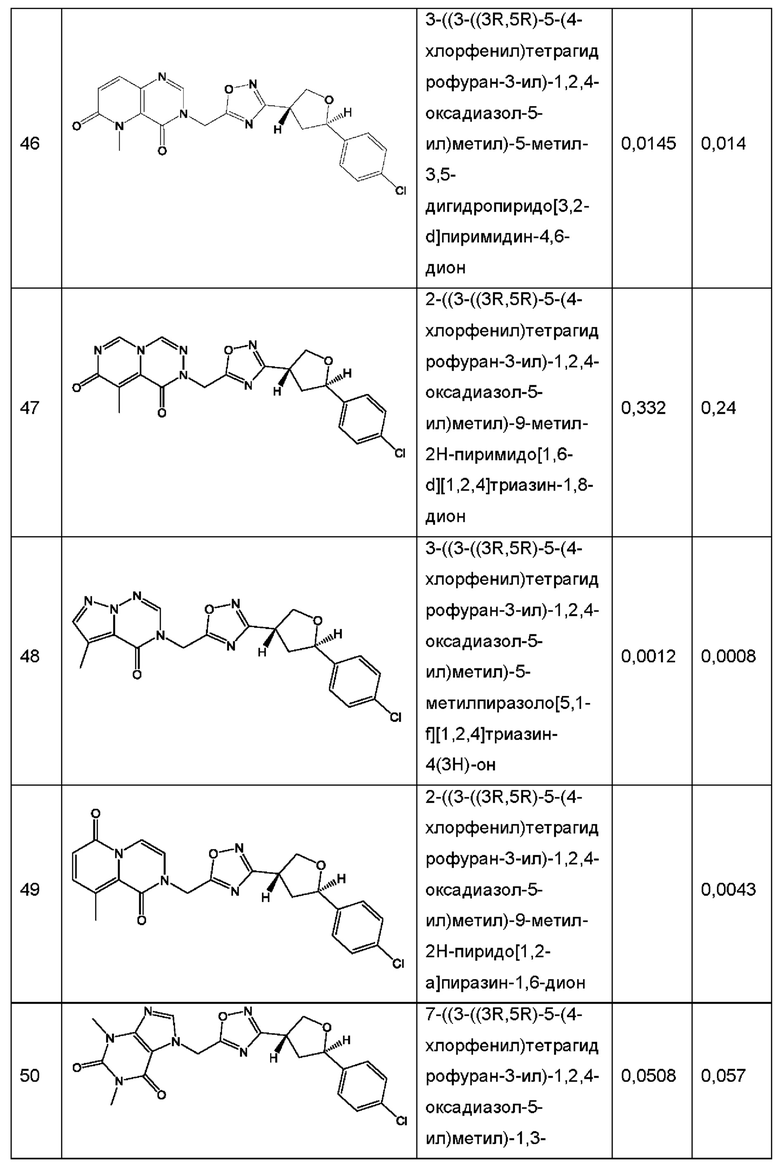
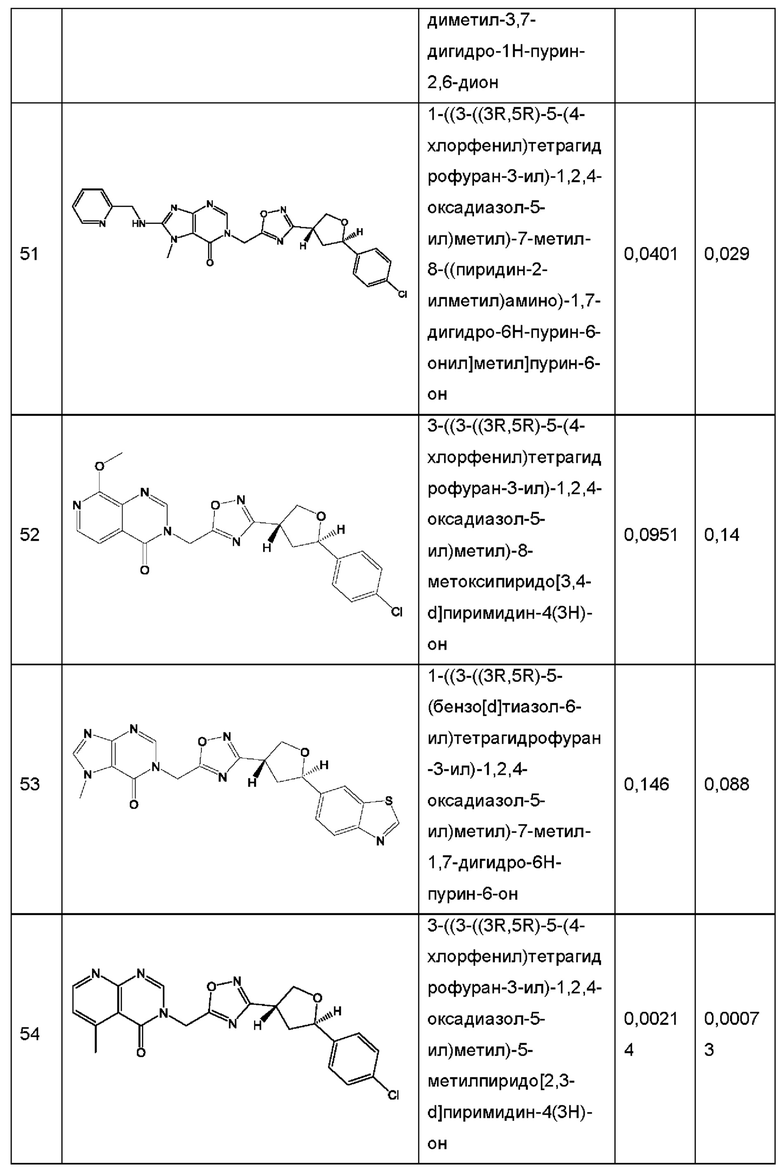
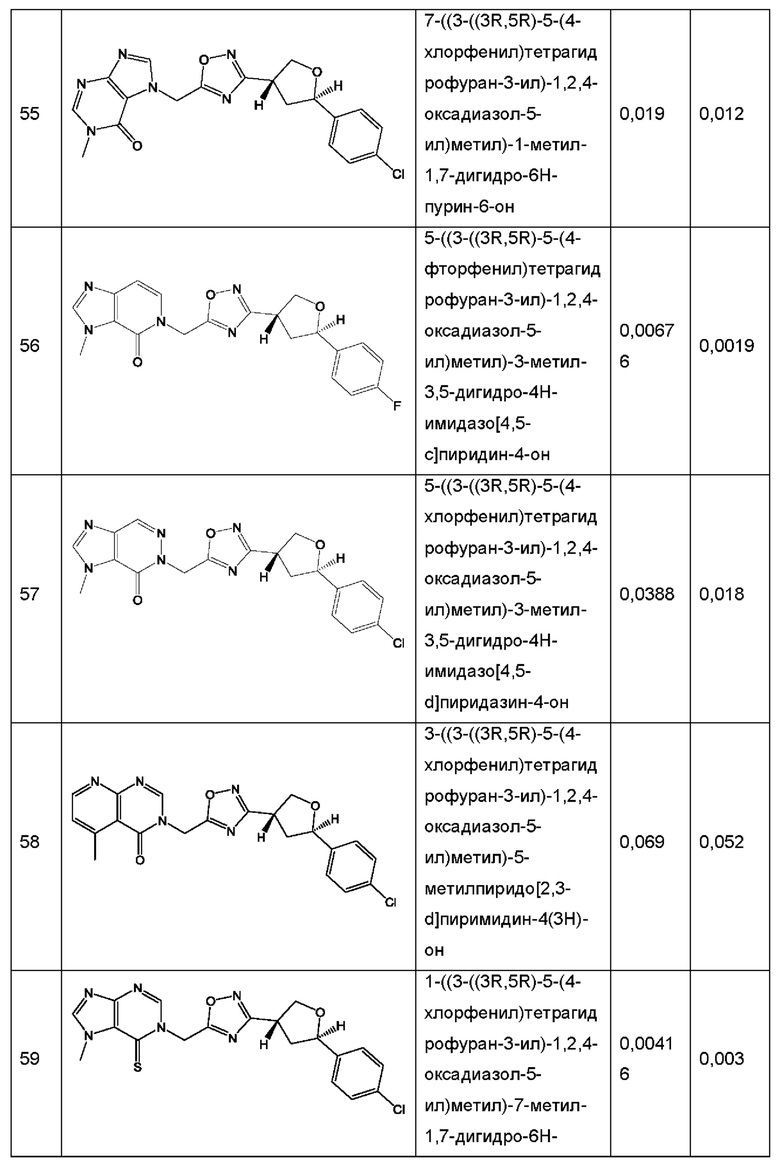
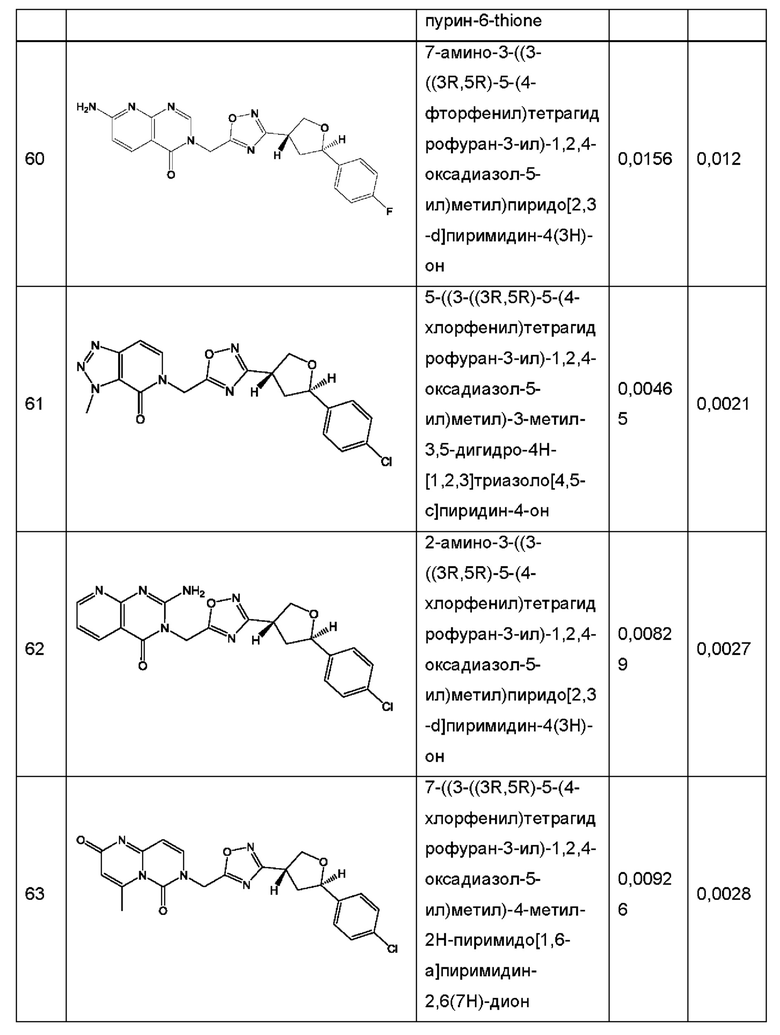
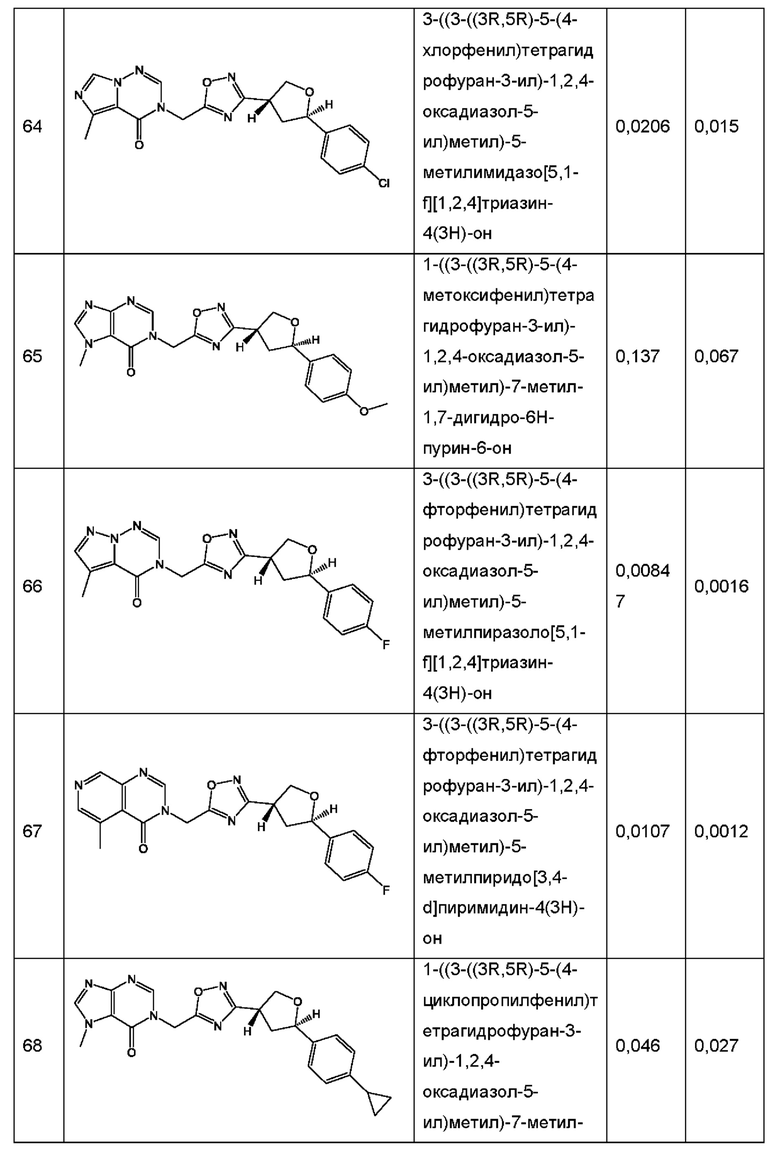
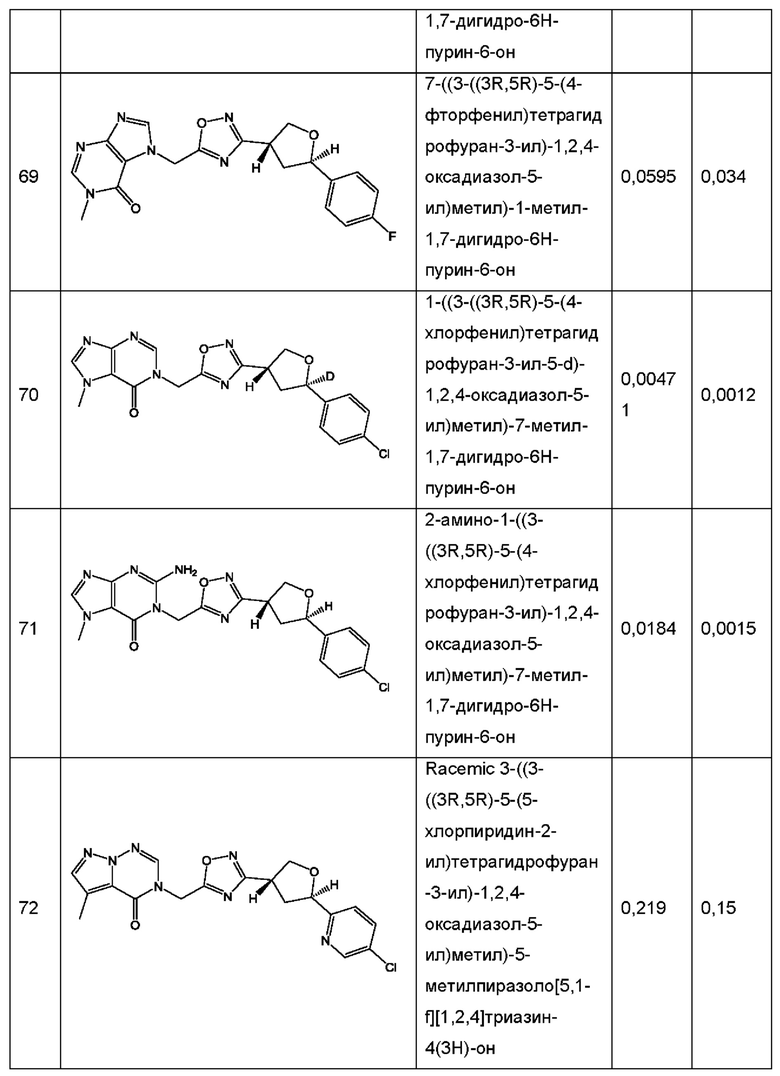
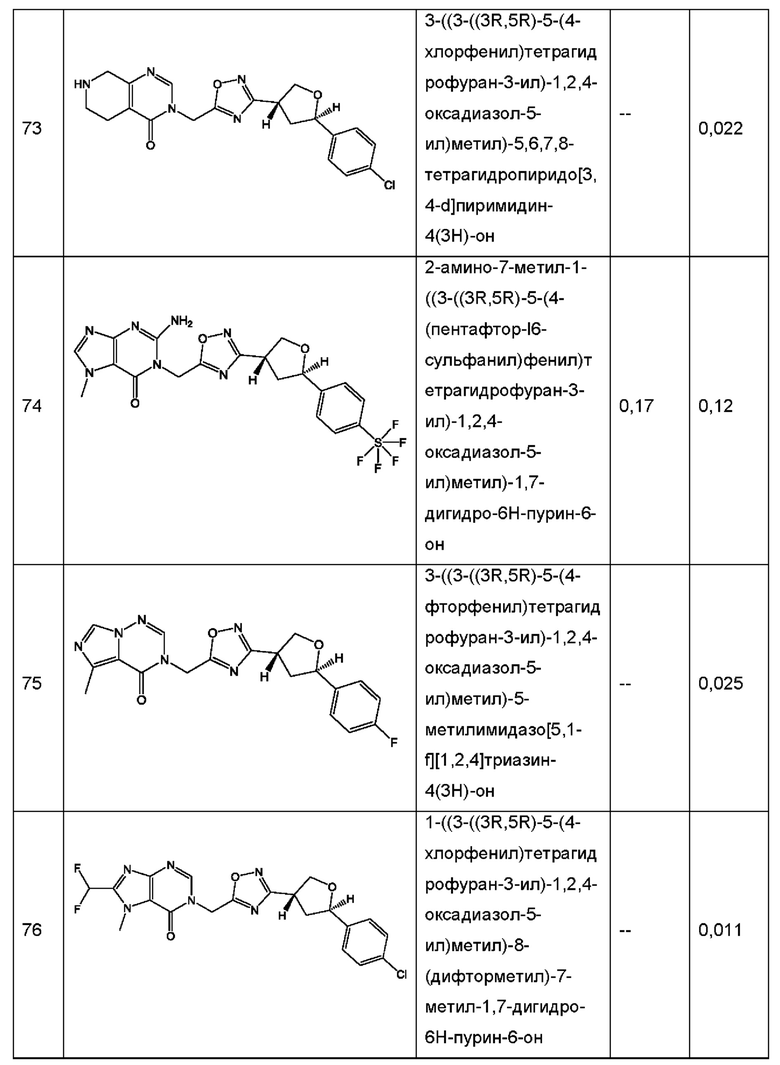
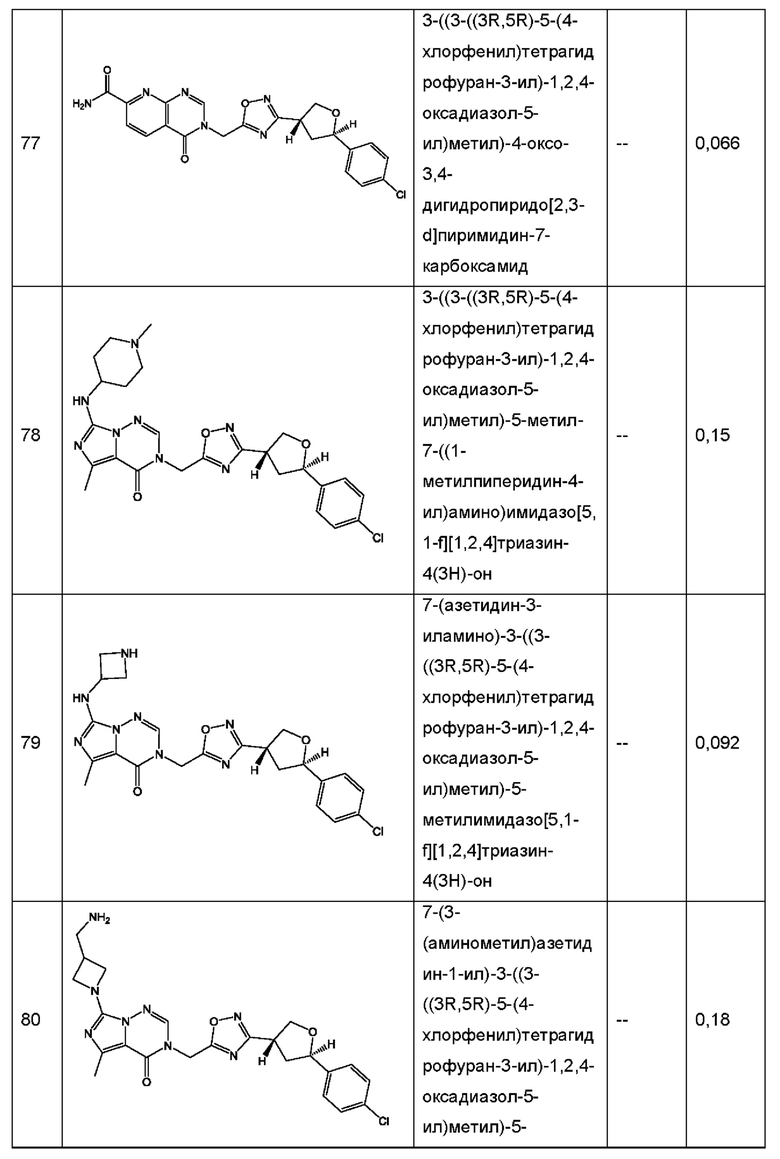
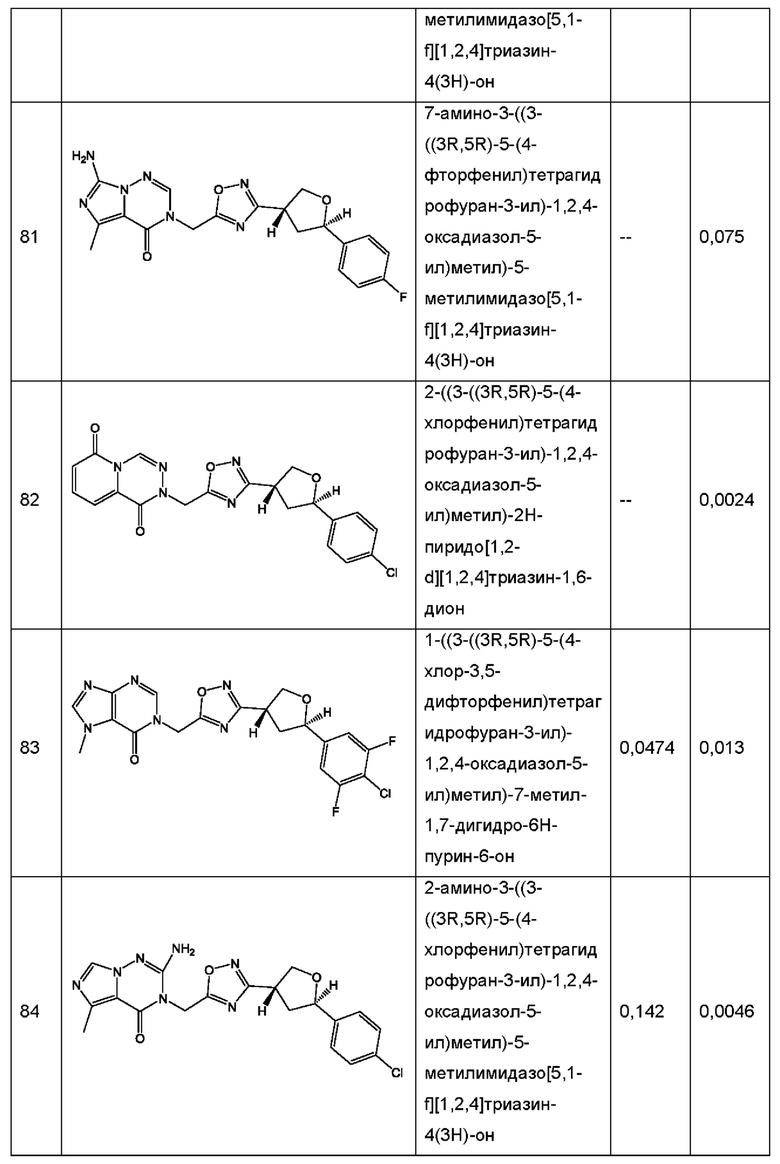

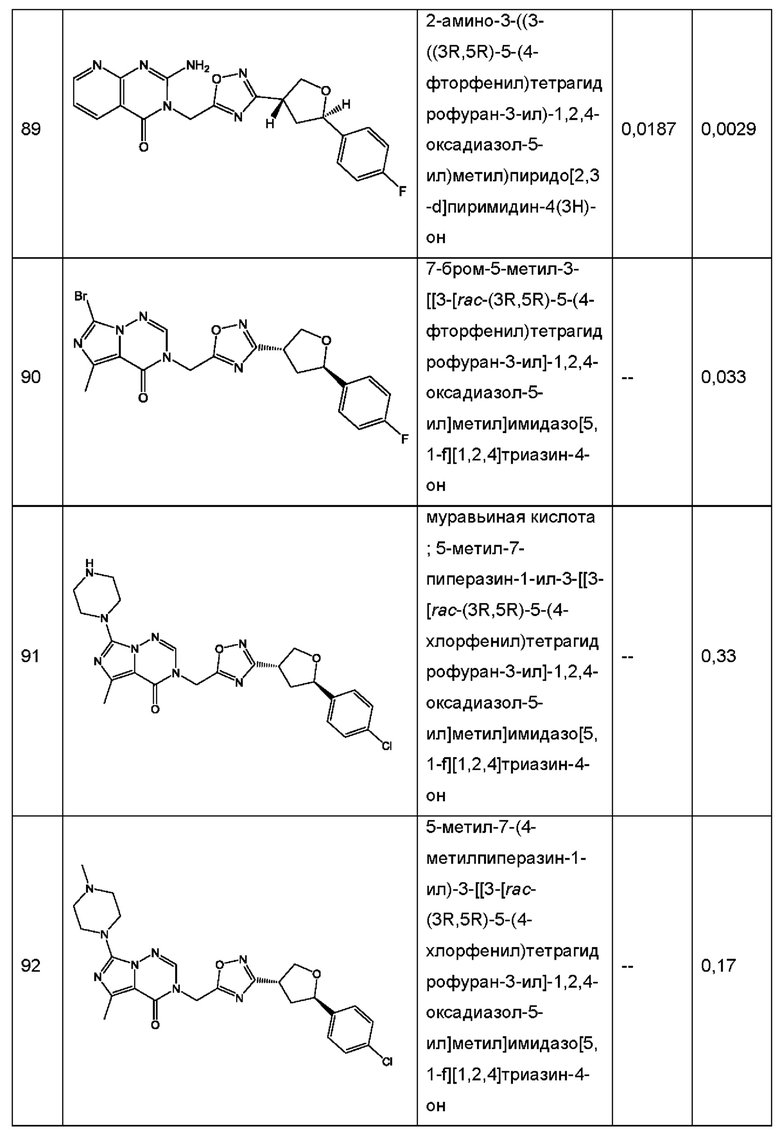
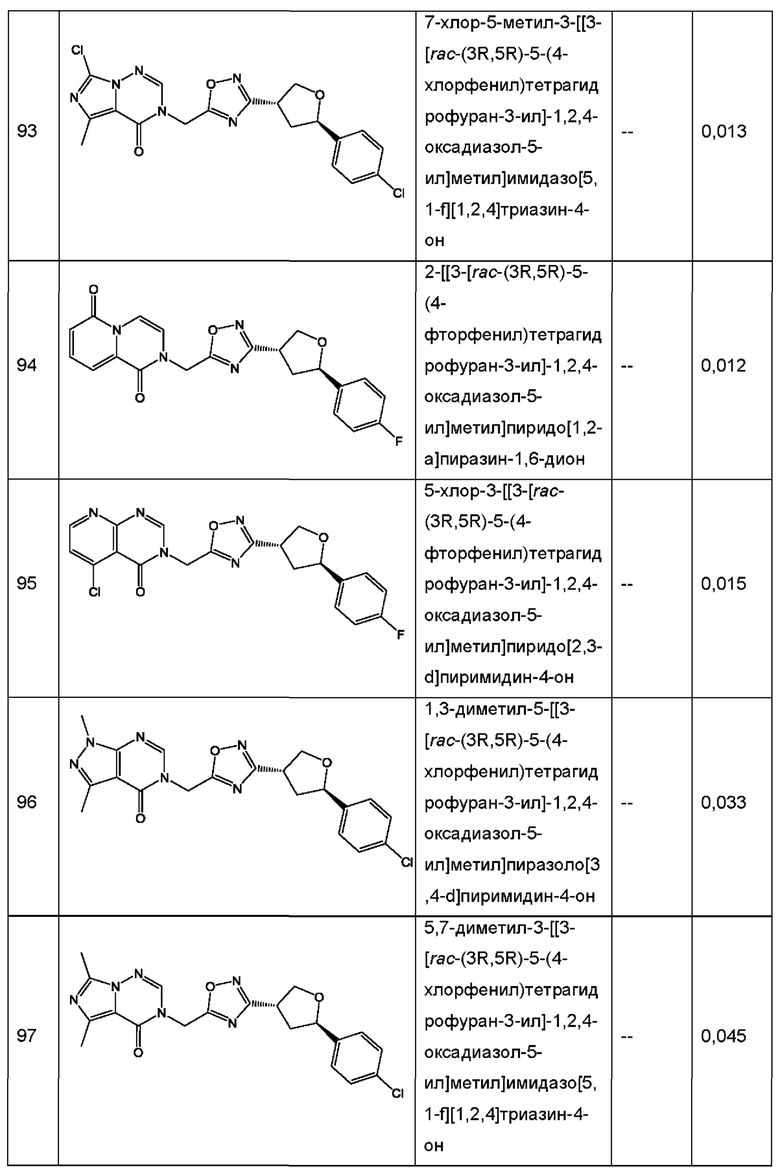
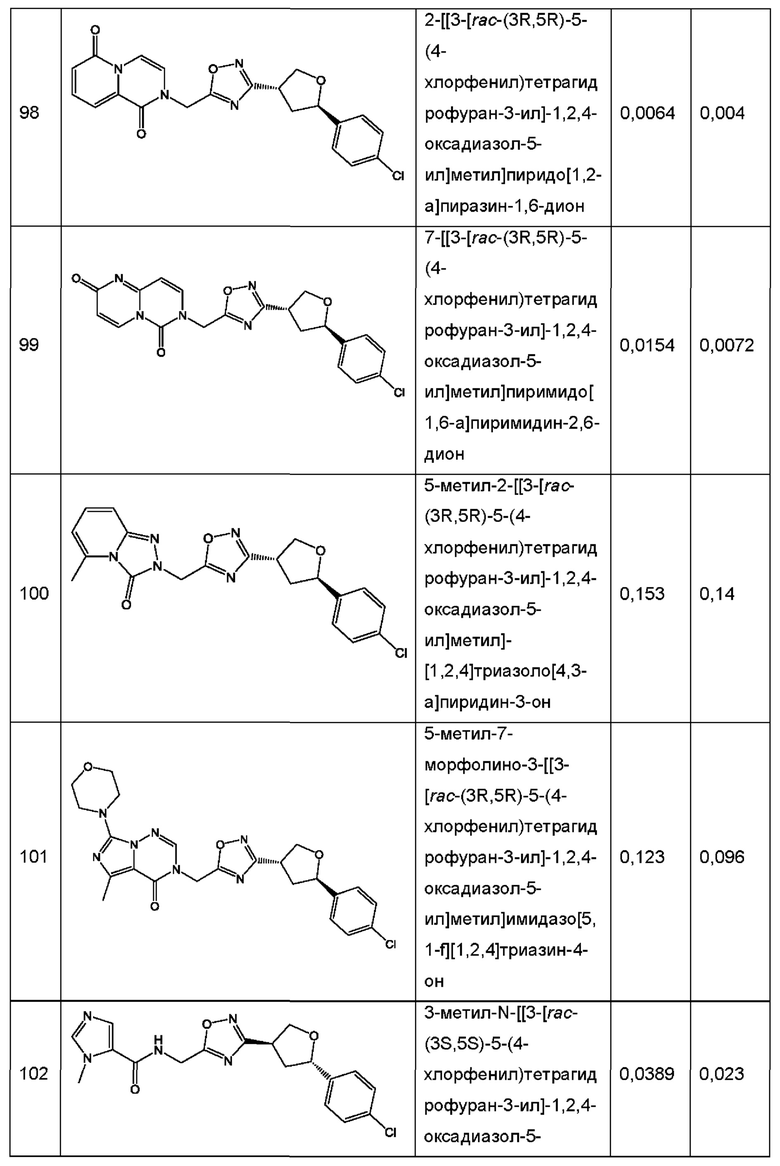

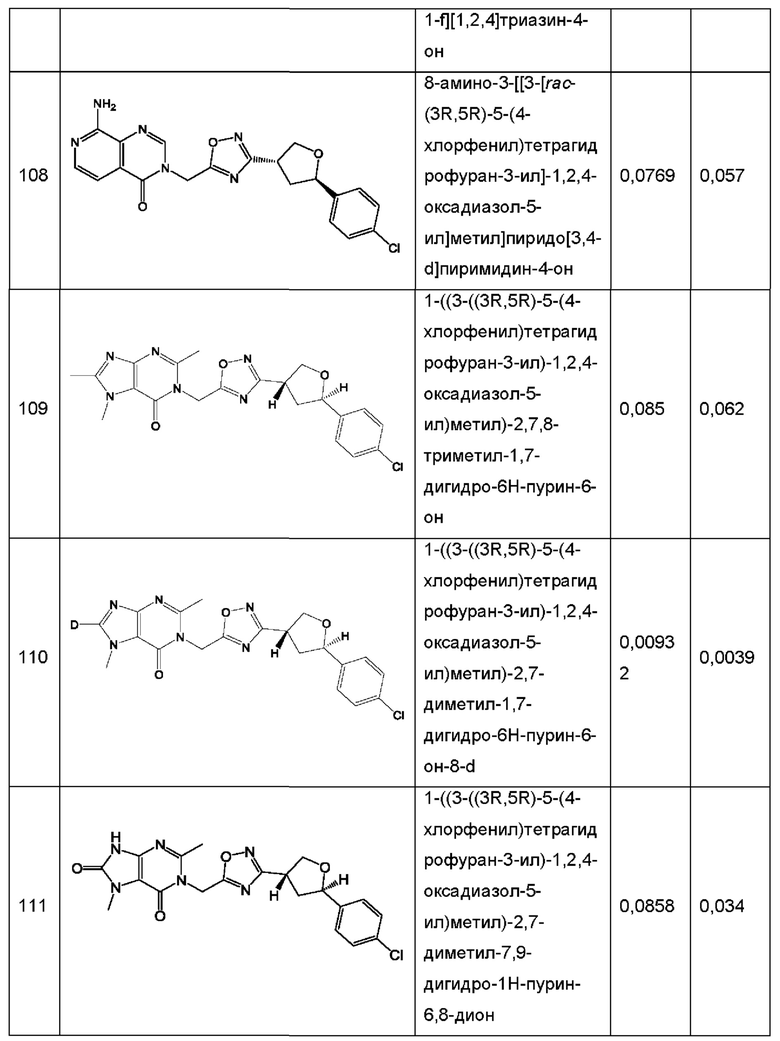
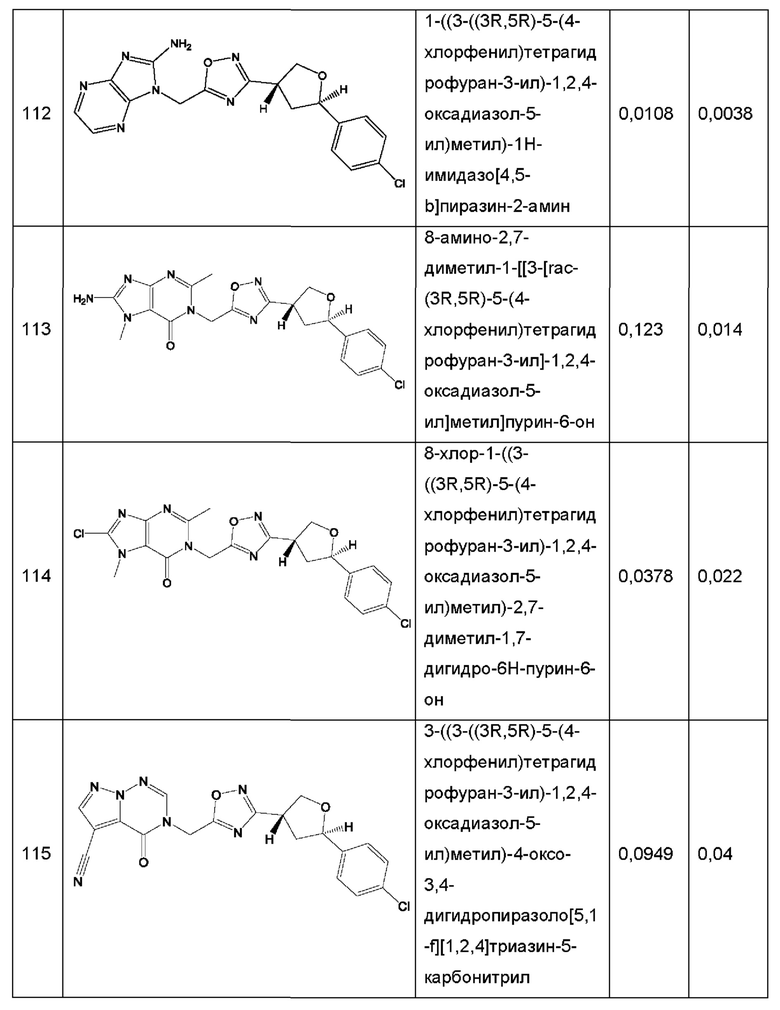
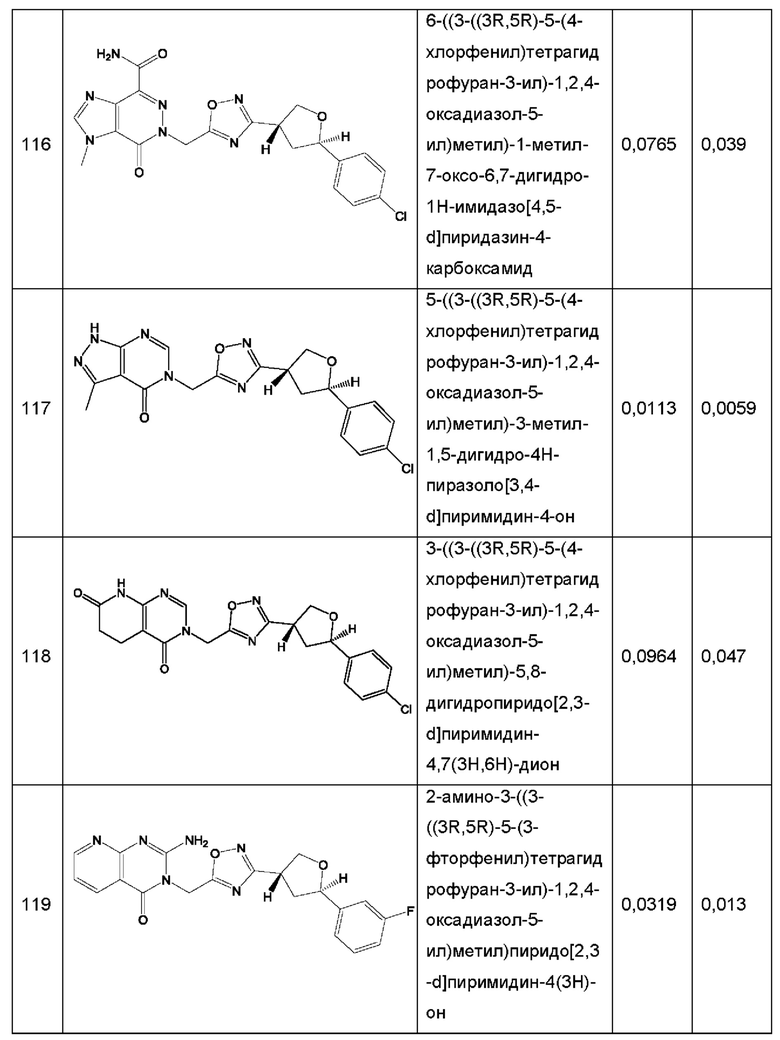
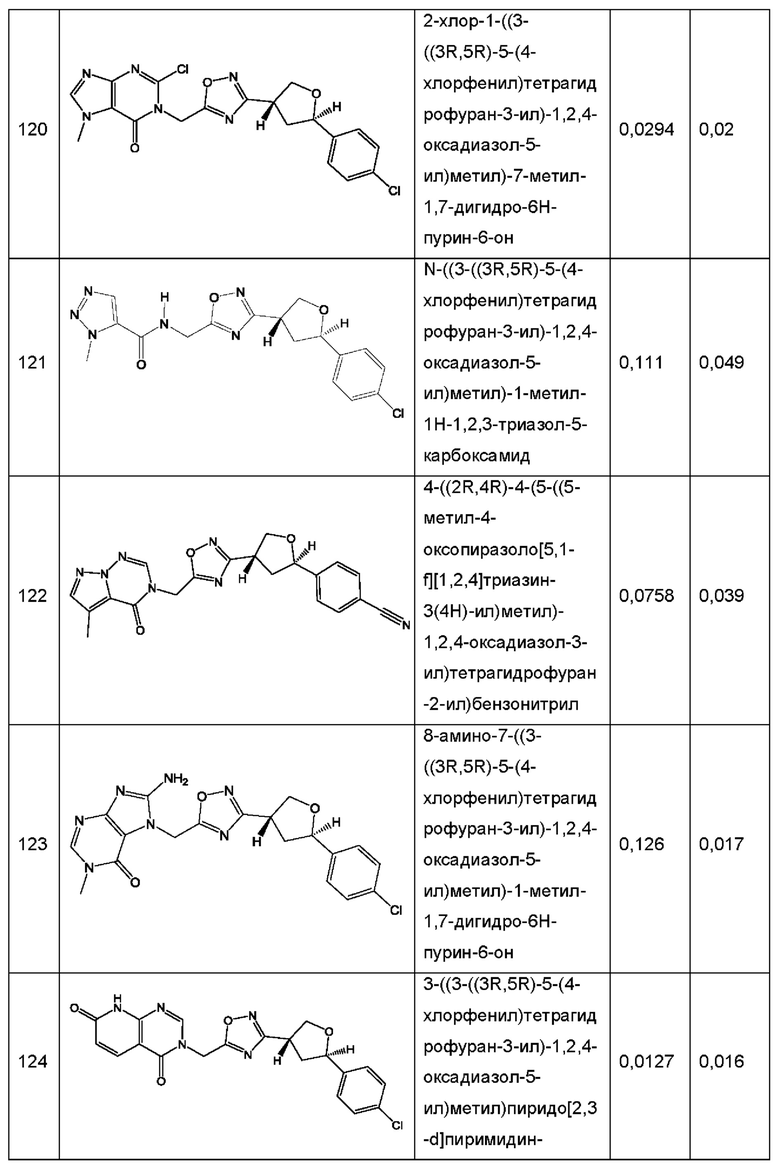

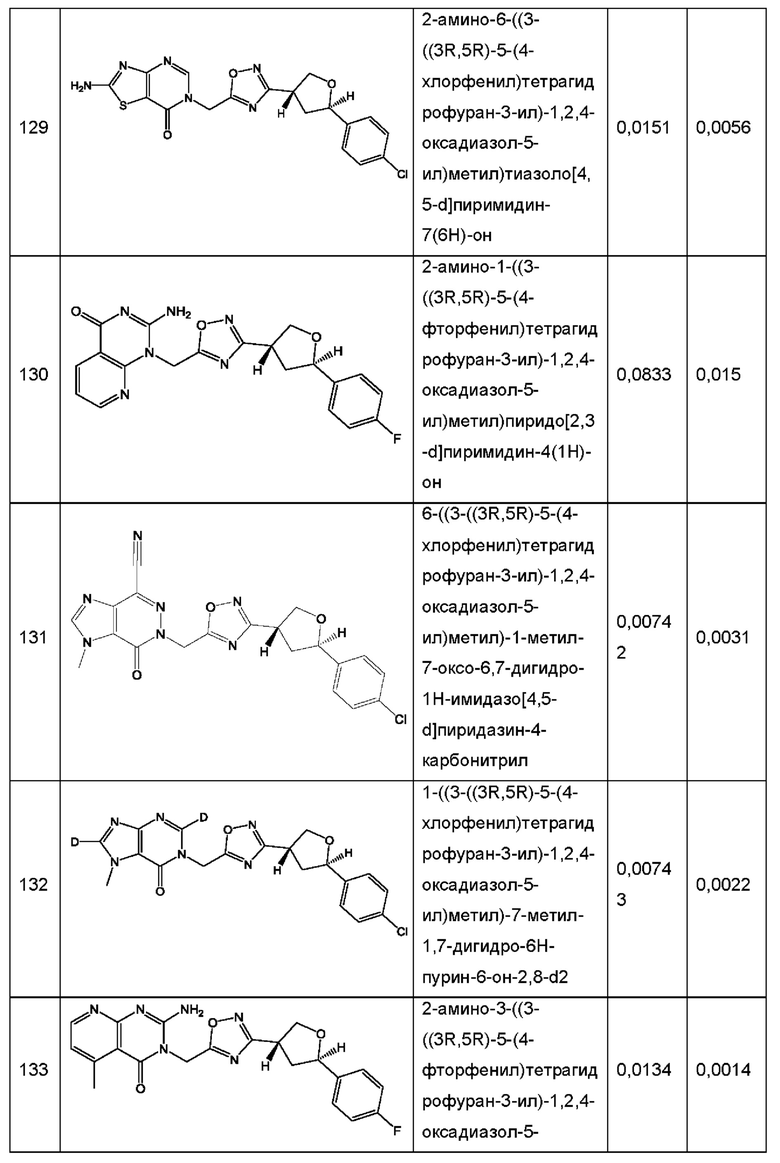

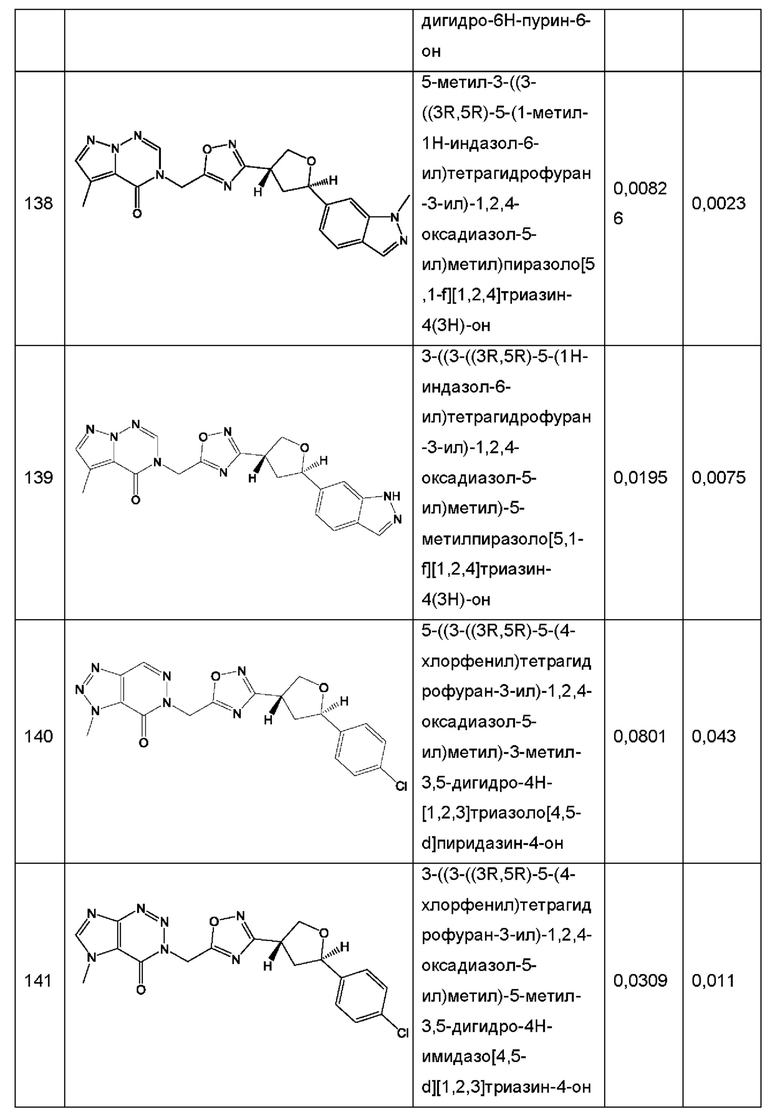
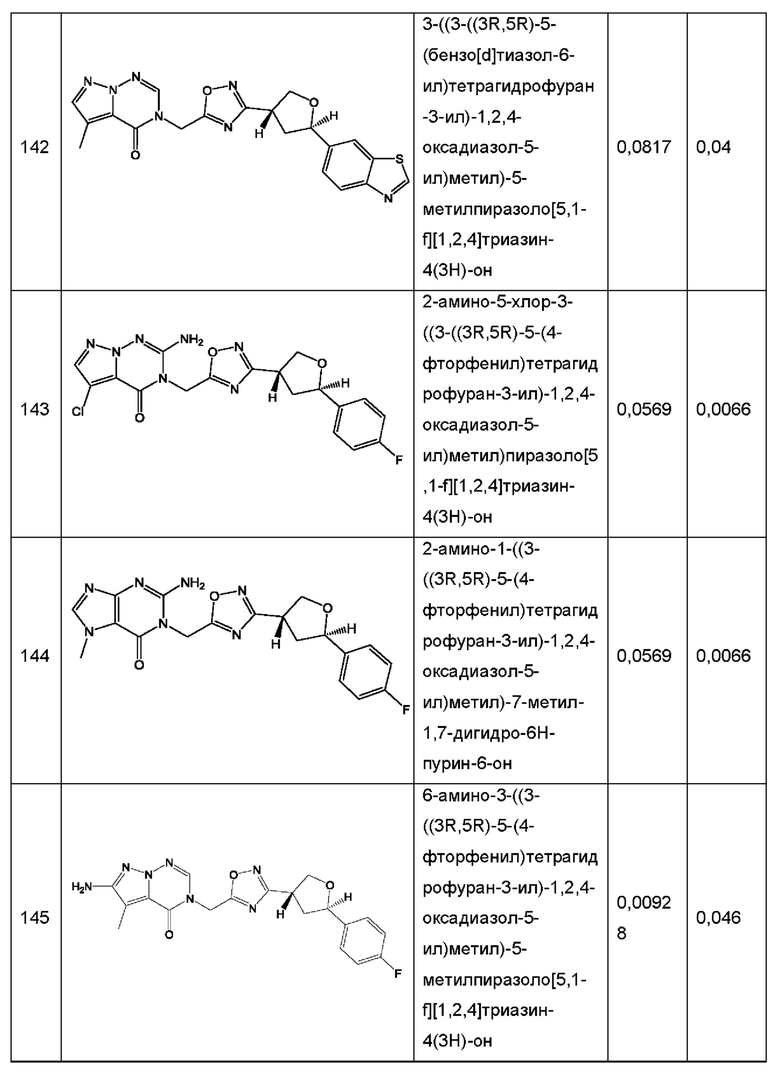

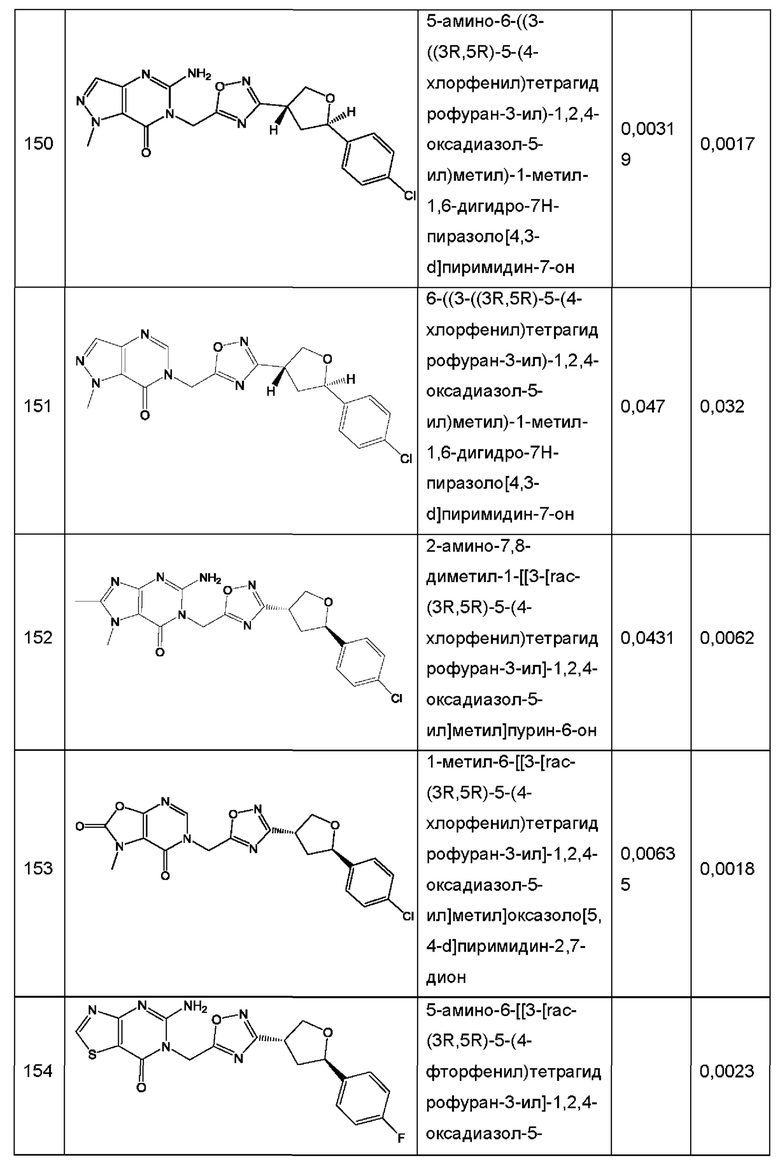
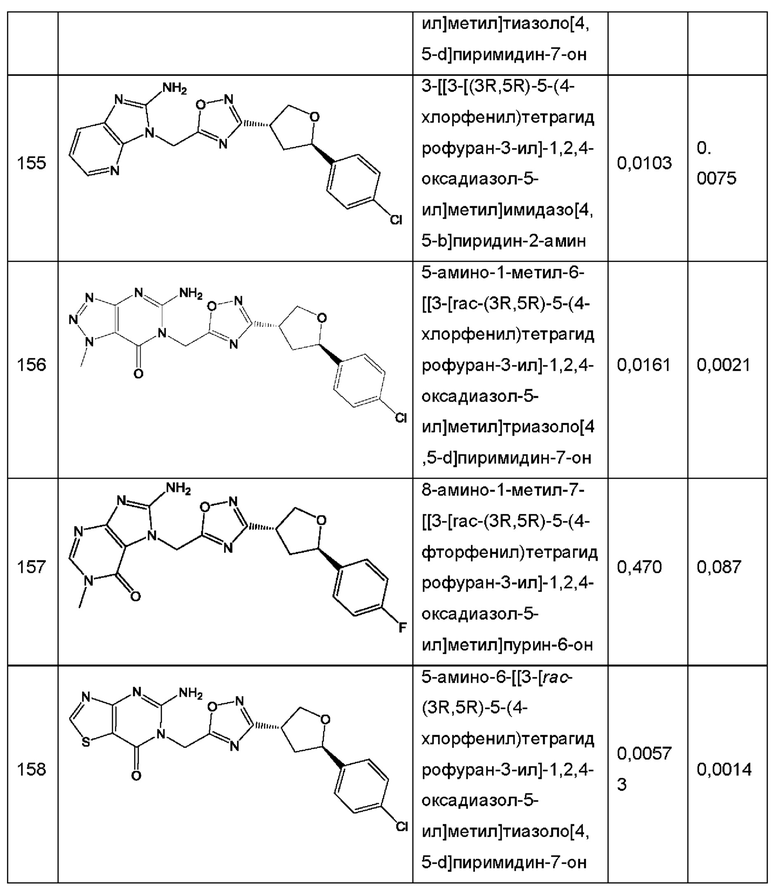
В Таблице 2 ниже представлены данные протонного ЯМР для соединений Таблицы 1.


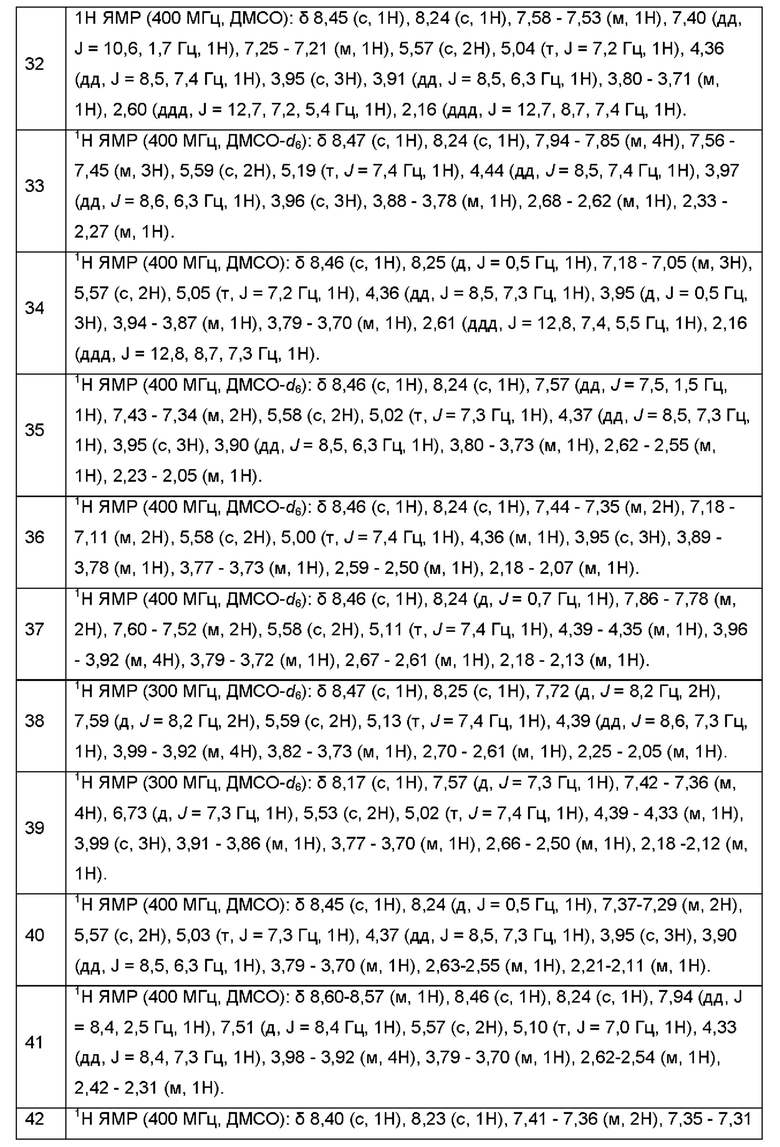
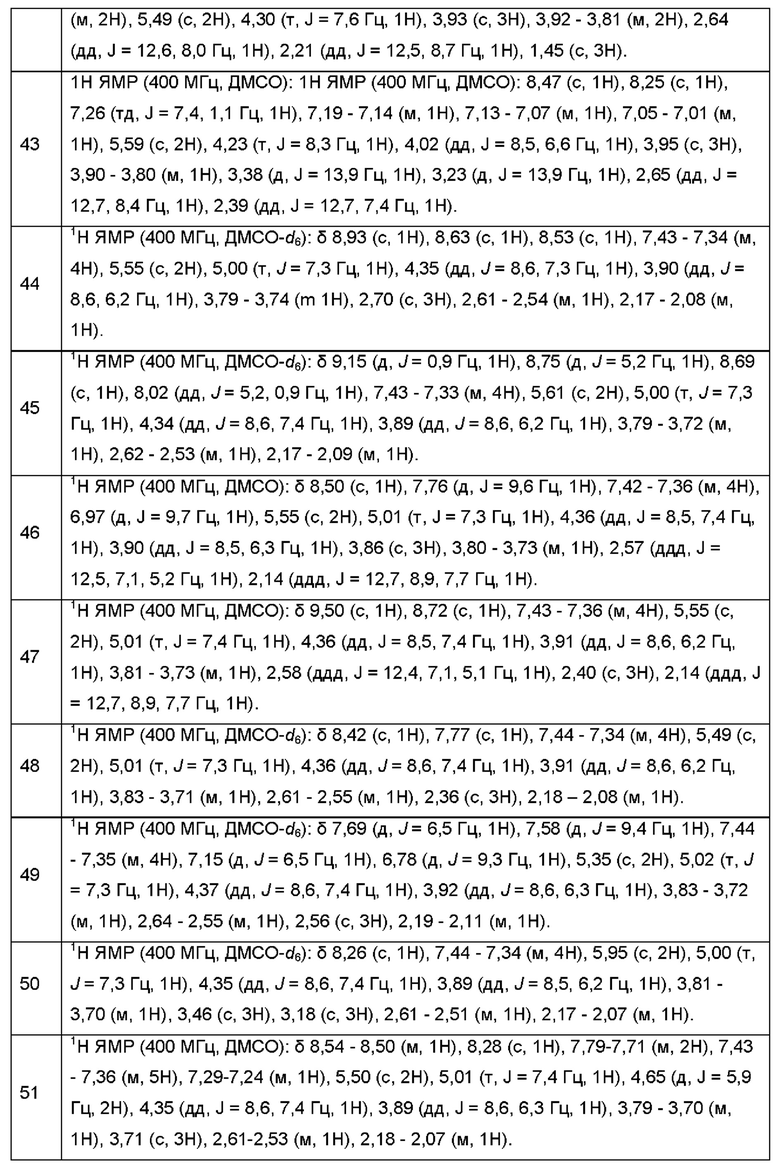
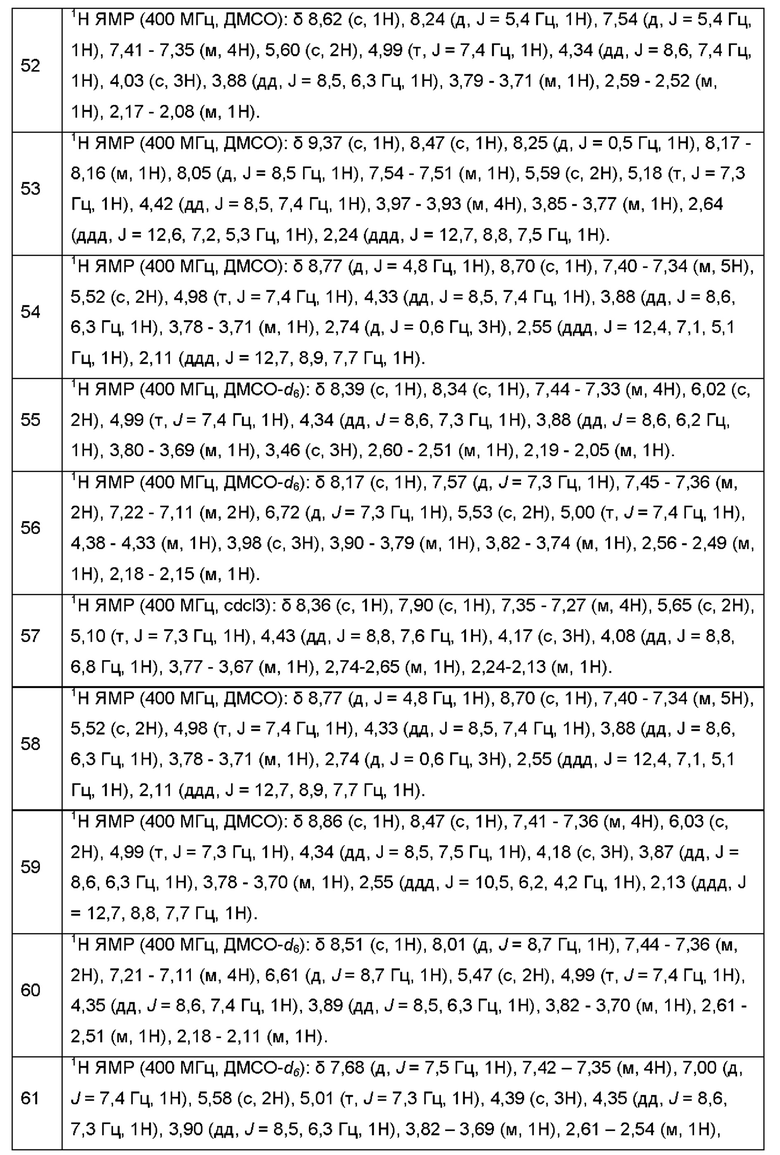

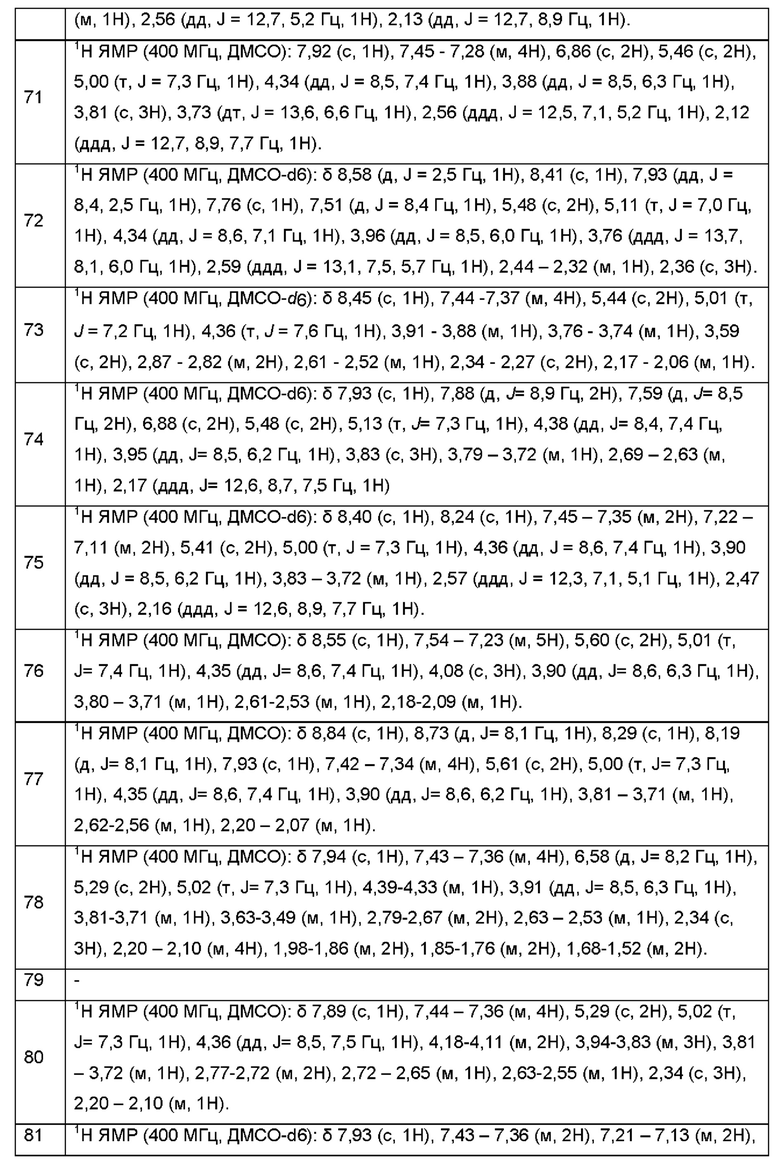
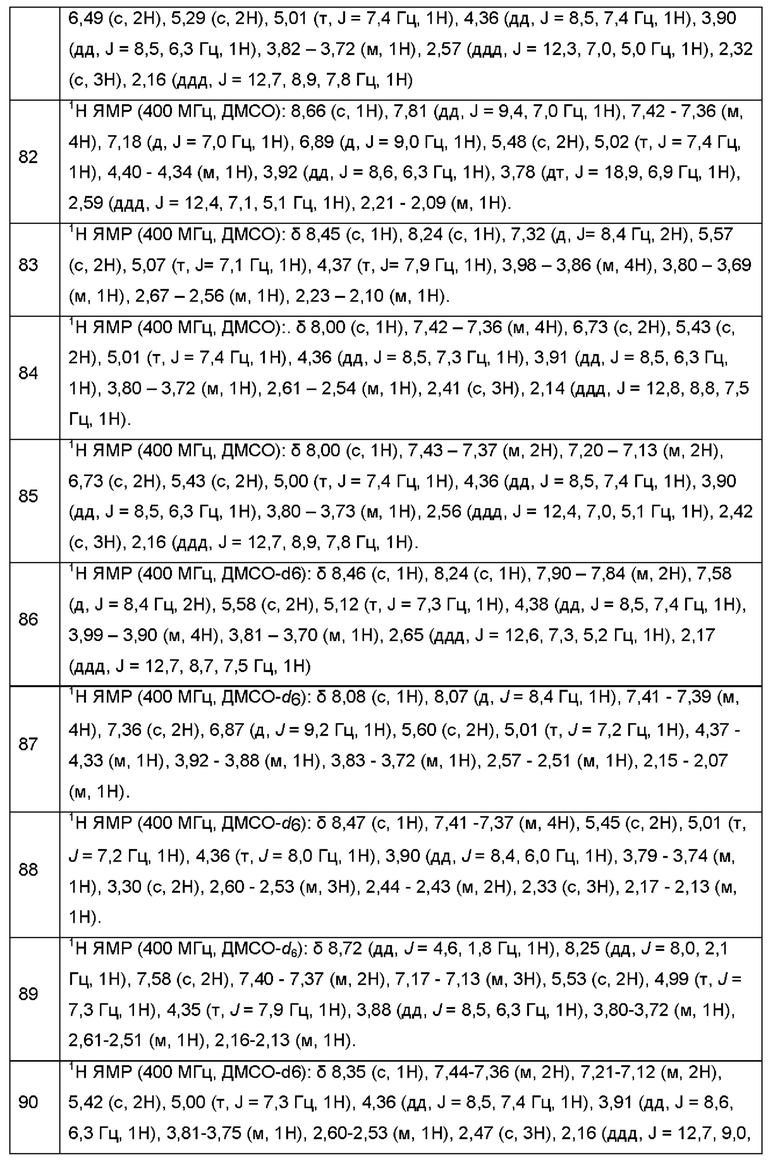
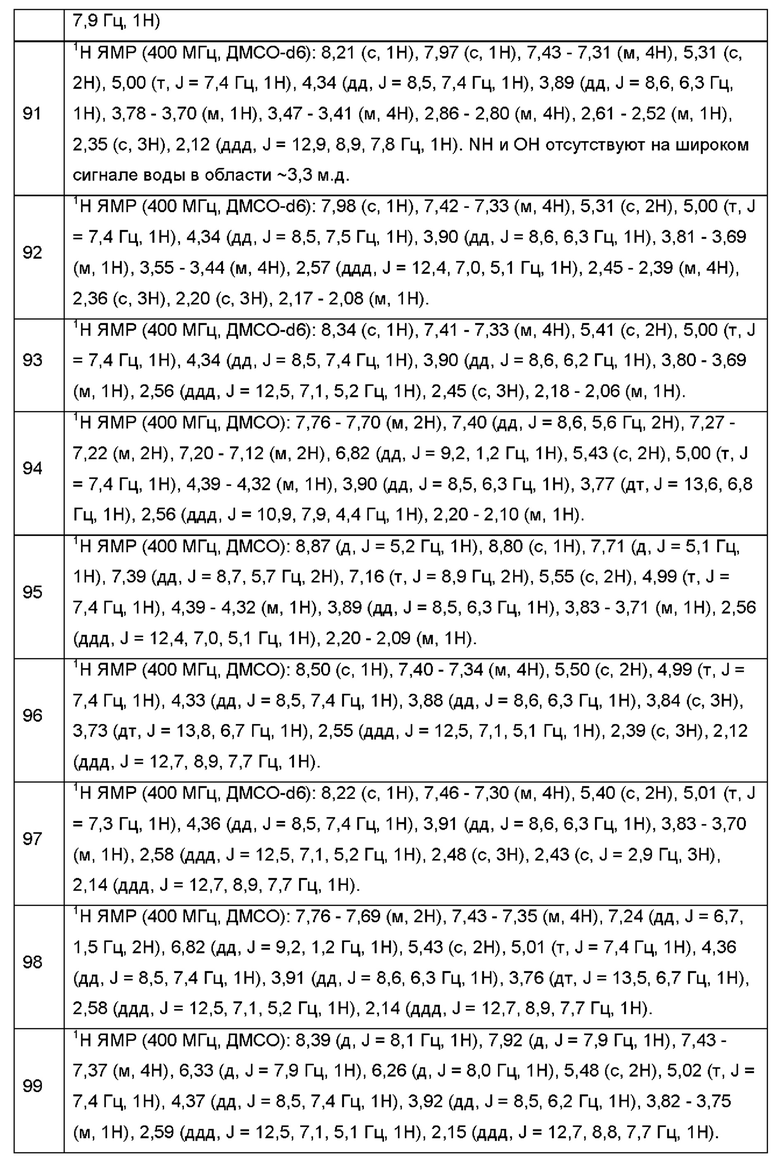
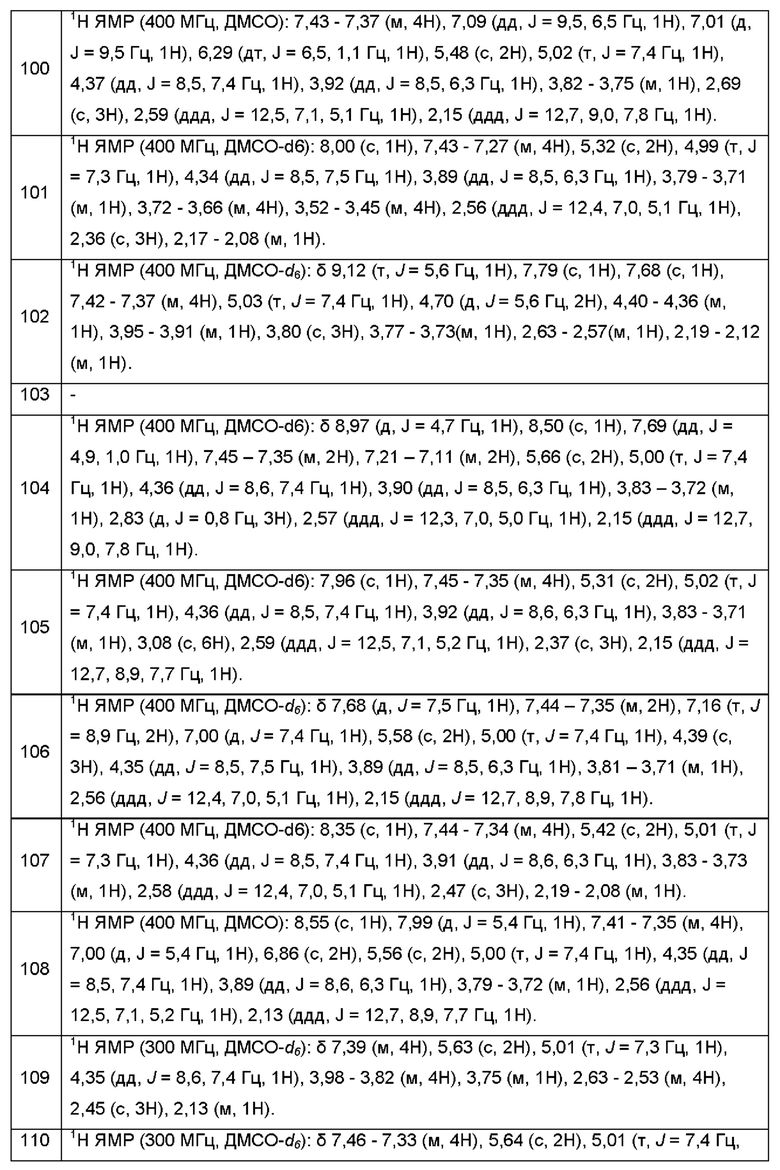


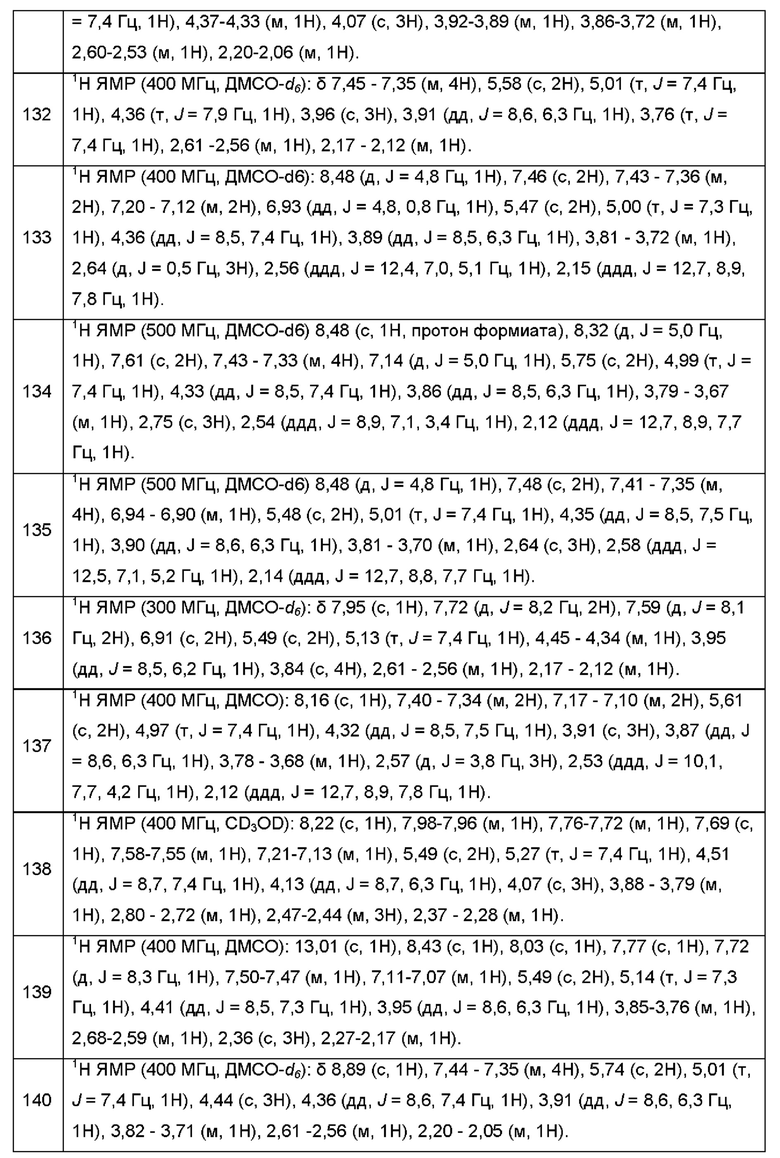
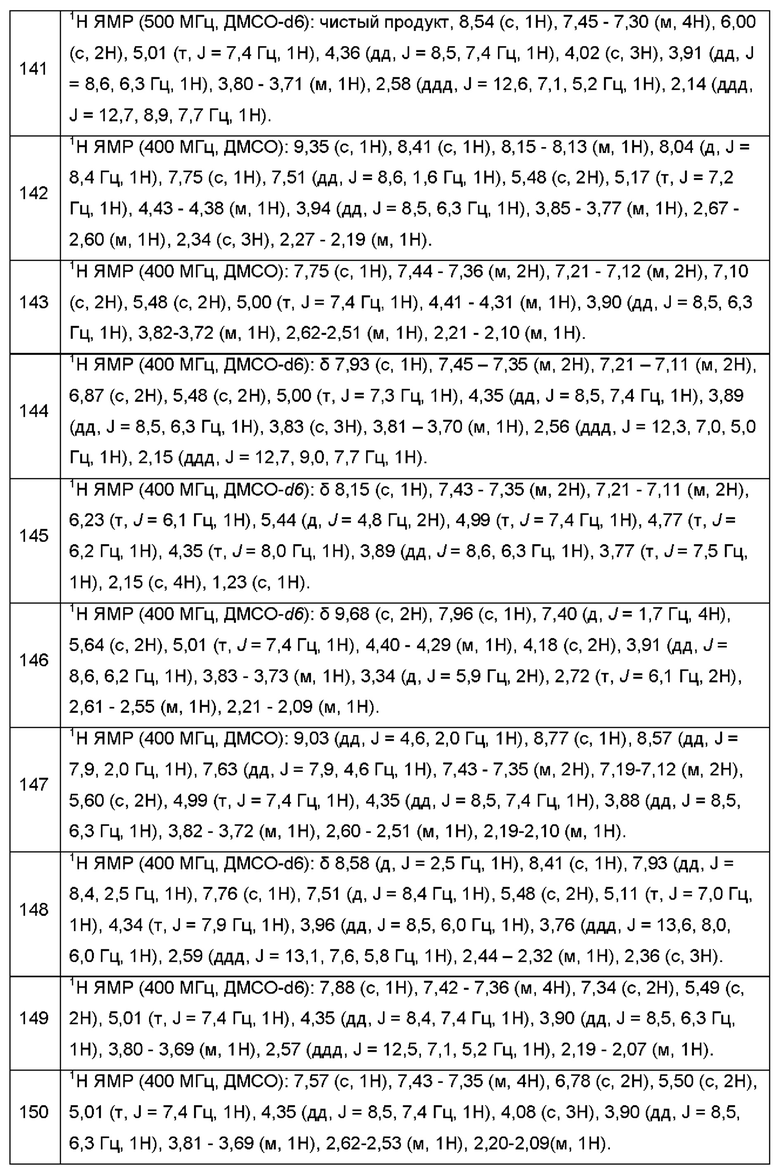
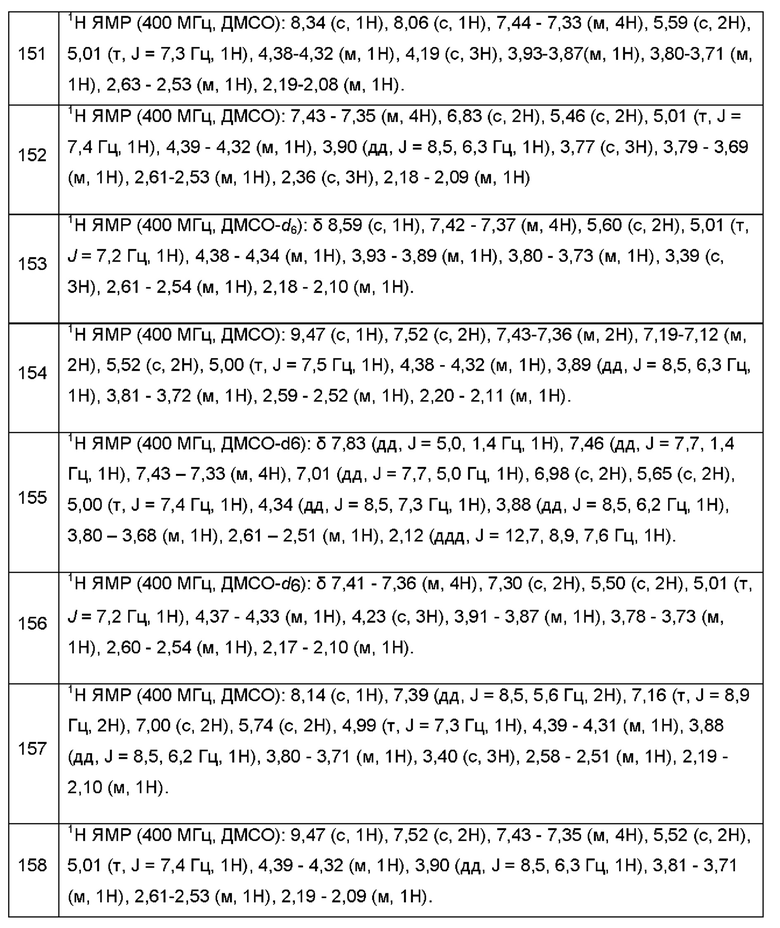
Определение величины IC50 рассматриваемых соединений
IC50 (эффективную концентрацию) соединений в области каналов TRPA1 человека определяли с помощью прибора FLIPR Tetra. Клетки СНО (англ. Chinese hamster ovary - яичник китайского хомячка), экспрессирующие TRPA1, помещали в 384-луночные планшеты, инкубировали в течение ночи при температуре 37°С и нагружали индикаторным красителем компании BD для кальция сначала в течение 1 ч при температуре 37°С, затем в течение 15 минут при комнатной температуре. Аналитический буфер представлял собой сбалансированный солевой раствор Хенкса (англ. HBSS, Hank's Balanced Salt Solution), содержащий 20 мМ HEPES (англ. N-2-hydroxyethyl-piperazine-N-2-ethanesulfonic acid - N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота) (величину рН доводили до 7,4) с 0,02% BSA (англ. bovine serum albumin - бычий сывороточный альбумин).
После загрузки красителя и охлаждения планшета в ячейки с помощью FLIPR Tetra добавляли соединения. Далее планшеты, содержащие соединения, инкубировали в течение 10 минут или 90 минут при комнатной температуре перед добавлением агониста. После инкубирования добавляли коричный альдегид (75) в концентрации приблизительно EC80 для активации каналов и измеряли блокировку индуцированного коричным альдегидом притока кальция.
Результаты IC50 соответствовали стандартной функции Хилла, сохраняя коэффициент Хилла (n) фиксированным на уровне 1,5. Фиксация коэффициента Хилла обычно снижает вариабельность определения IC50. Результаты IC50 изучали независимо друг от друга, чтобы убедиться, что точки MIN и МАХ установлены правильно до проверки результатов.
Результаты IC50 (hTRPA1 1050 (микромолярный)), полученные для соединений по настоящему раскрытию, представлены выше в Таблице 1, где "hTRPA1" относится к hTRPA1 CHO Ca2+ MAX EVO (IC50).
В данном подробном описании использованы примеры, позволяющие раскрыть изобретение, в том числе его лучший вариант осуществления, а также дающие возможность любому специалисту в данной области техники применить изобретение на практике, включая изготовление и использование любых устройств или систем и выполнение любых включенных способов. Патентоспособный объем изобретения определяется Формулой изобретения и может включать в себя другие примеры, с которыми могут столкнуться специалисты в данной области техники. Подразумевается, что такие другие примеры находятся в пределах объема данной Формулы изобретения, если они имеют структурные элементы, не отличающиеся от буквальной формулировки Формулы изобретения, или если они включают эквивалентные структурные элементы, несущественно отличающиеся от буквальной формулировки Формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730508C2 |
| ПОЛИГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МЕТАБОТРОПНОГО РЕЦЕПТОРА ГЛУТАМАТА | 2005 |
|
RU2381226C2 |
| ЧАСТИЧНЫЕ И ПОЛНЫЕ АГОНИСТЫ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ А | 2003 |
|
RU2317994C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2010 |
|
RU2574410C2 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Ха | 2004 |
|
RU2346944C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2821941C2 |
| ПРОИЗВОДНЫЕ С-АРИЛГЛЮКОЗИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2606501C2 |
| β-D-2'-ДЕЗОКСИ-2'-α-ФТОР-2'-β-С-ЗАМЕЩЕННЫЕ-2-МОДИФИЦИРОВАННЫЕ-N6-ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ НУКЛЕОТИДЫ ДЛЯ ЛЕЧЕНИЯ ВЫЗВАННЫХ HCV ЗАБОЛЕВАНИЙ | 2016 |
|
RU2764767C2 |
Изобретение относится к соединениям на основе оксадиазолов и его фармацевтичеcки приемлемым солям, представленным общей формулой I, где A представляет собой гетероциклические структурные фрагменты, приведенные в формуле изобретения; Х представляет собой C1-4алкилен; n равно 1; R1 представляет собой H; каждый R2 независимо представляет собой H, D, -C1-4алкил, -C1-4галогеналкил, C1-4алкокси, -CN, галоген и NR14R15; каждый R3 независимо представляет собой H, D, -C1-4алкил, -CN, NR14R15 или галоген; R4 представляет собой: незамещенный фенил или фенил, замещенный 1-3 заместителями, выбранными из галогена, -CN, -С1-4алкила, незамещенного С3-6циклоалкила, С1-4галогеналкила, С1-4галогеналкокси, С1-4алкокси, пентафторсульфанила или незамещенного фенила; незамещенный гетероарил или гетероарил, замещенный 1 заместителем, выбранным из галогена и С1-4алкила, где гетероарил выбран из пиразола, пиридина, бензотиазола; или незамещенный нафтил; или R1 и R4 могут вместе образовывать незамещенный C3-6циклоалкил, сконденсированный с незамещенным фенилом; p равно 0, 1 или 2; q равно 0 или 1; каждый из R14 и R15 независимо друг от друга представляет собой: H; замещенный одним пиридином или незамещенный -C1-4алкил; замещенный одним C1-4алкилом или незамещенный 4-6-членный гетероциклоалкил, содержащий один атом азота; или R14 и R15 вместе с атомами, к которым они присоединены, могут образовывать 6-членное кольцо, включающее один дополнительный гетероатом, выбранный из O, N. Также изобретение относится к соединениям, представленным структурными формулами IVa и IVb, конкретным соединениям (приведены в формуле изобретения), фармацевтической композиции, применению и способу ингибирования активности TRPA1. Технический результат – соединения, применяемые для лечения заболеваний, опосредованных TRPA1. 6 н. и 30 з.п. ф-лы, 2 табл., 9 пр.
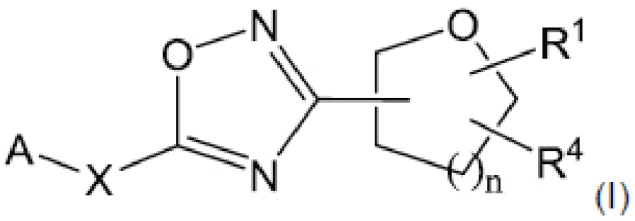
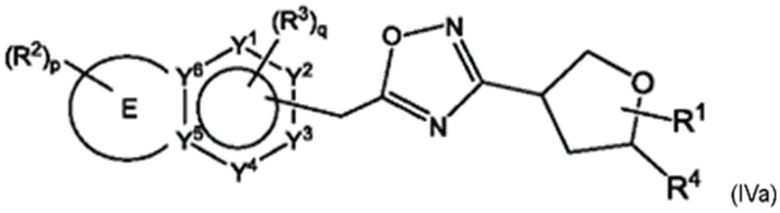
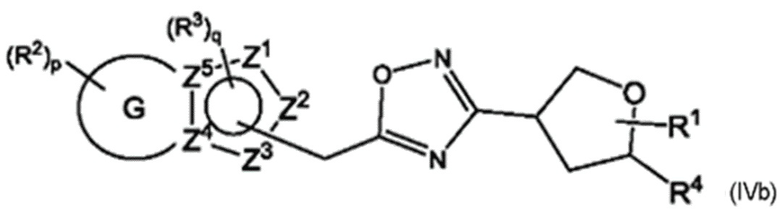
1. Соединение формулы (I)
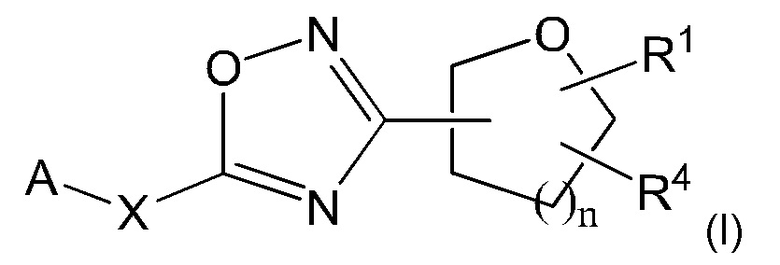
или его фармацевтически приемлемая соль,
где А выбран из:
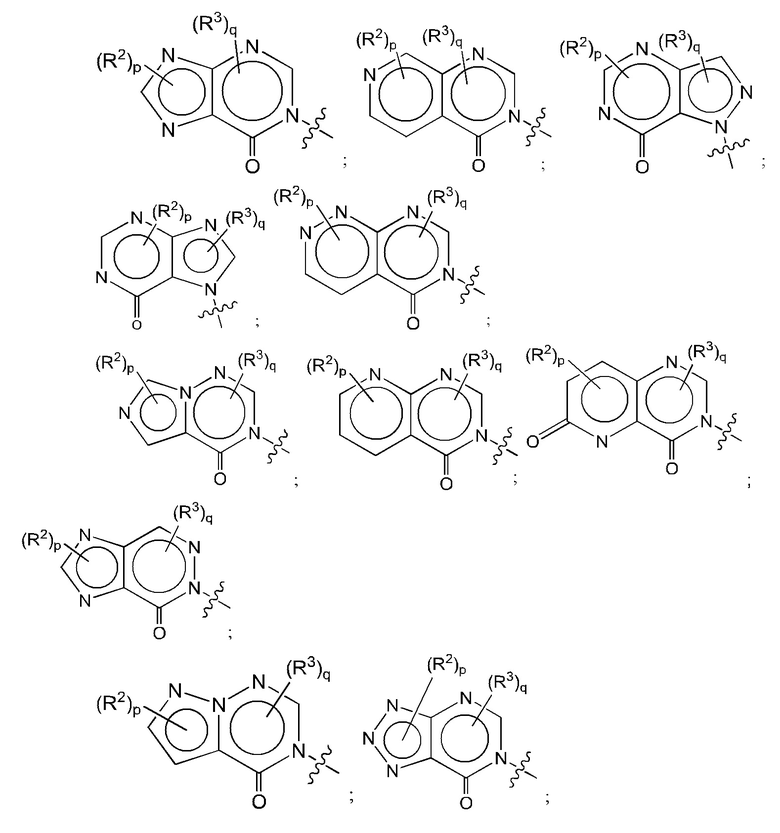
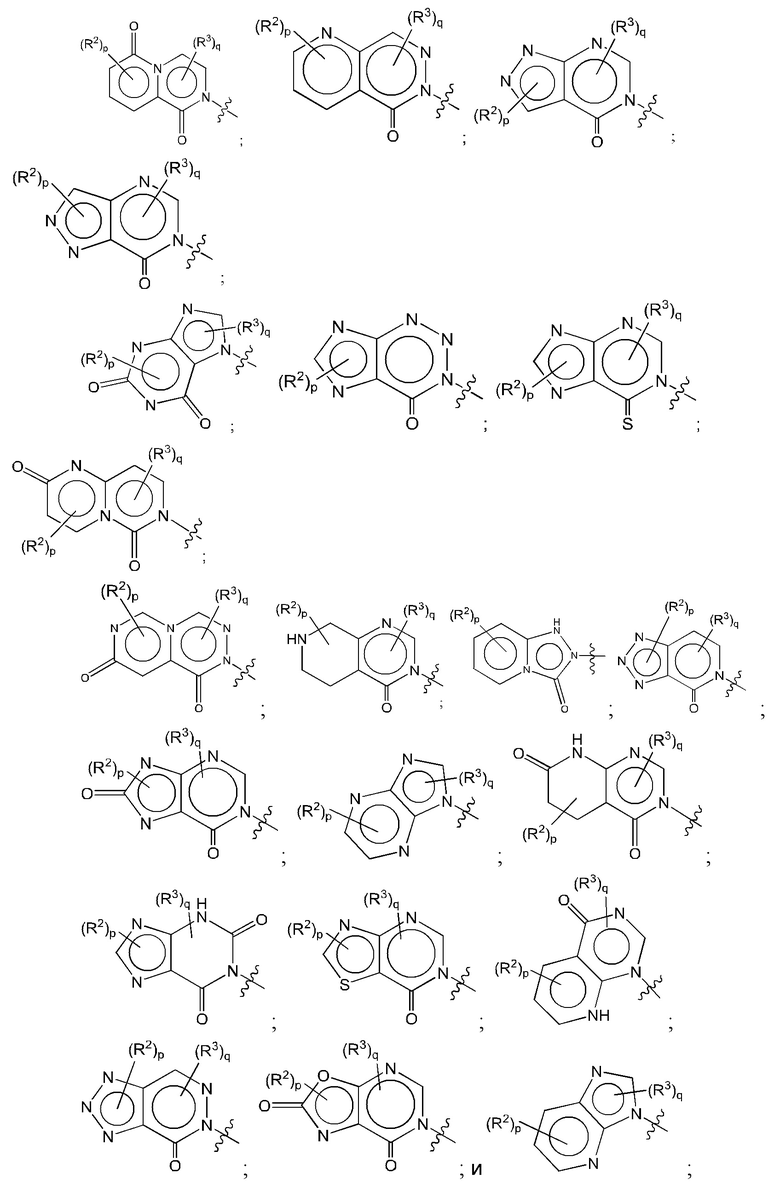
X представляет собой С1-4алкилен;
n равно 1;
R1 представляет собой Н;
каждый R2 независимо представляет собой: Н; D; -С1-4алкил; -С1-4галогеналкил; C1-4алкокси; -CN, галоген; и NR14R15;
каждый R3 независимо представляет собой: Н; D; -С1-4алкил; -CN; NR14R15; или галоген;
R4 представляет собой незамещенный фенил или фенил, замещенный 1-3 заместителями, выбранными из галогена, -CN, -С1-4алкила, незамещенного С3-6циклоалкила, С1-4галогеналкила, С1-4галогеналкокси, С1-4алкокси, пентафторсульфанила или незамещенного фенила; незамещенный гетероарил или гетероарил, замещенный 1 заместителем, выбранным из галогена и С1-4алкила, где гетероарил выбран из пиразола, пиридина, бензотиазола; или незамещенный нафтил;
или R1 и R4 могут вместе образовывать незамещенный С3-6циклоалкил, сконденсированный с незамещенным фенилом;
р равно 0, 1 или 2;
q равно 0 или 1;
каждый из R14 и R15 независимо друг от друга представляет собой: Н; замещенный одним пиридином или незамещенный -С1-4алкил; замещенный одним С1-4алкилом или незамещенный 4-6-членный гетероциклоалкил, содержащий один атом азота; или R14 и R15 вместе с атомами, к которым они присоединены, могут образовывать 6-членное кольцо, включающее один дополнительный гетероатом, выбранный из О, N.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где А представляет собой:
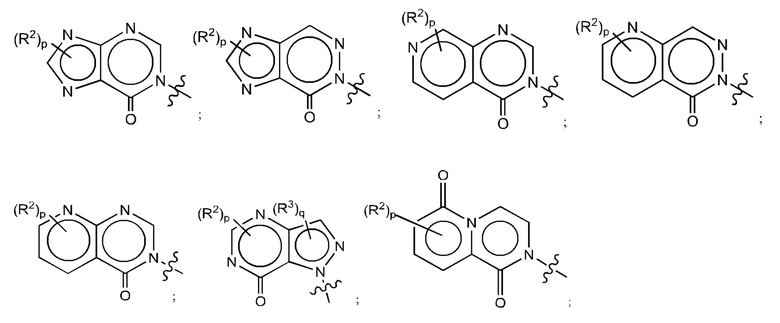

или 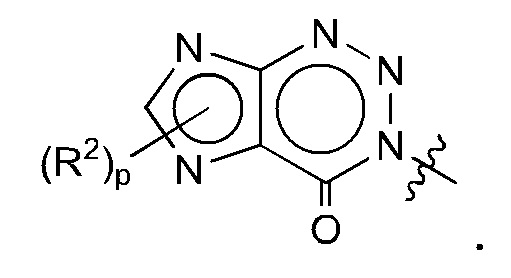
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где А представляет собой

4. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где каждый R2 независимо представляет собой Н; -СН3; -CN; -галоген; -NH2; -NHCH3; и -NHCH2CH3; а р равно 0 или 1.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, где А представляет собой:
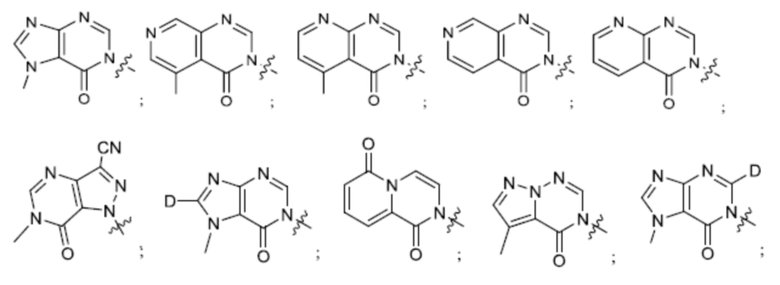
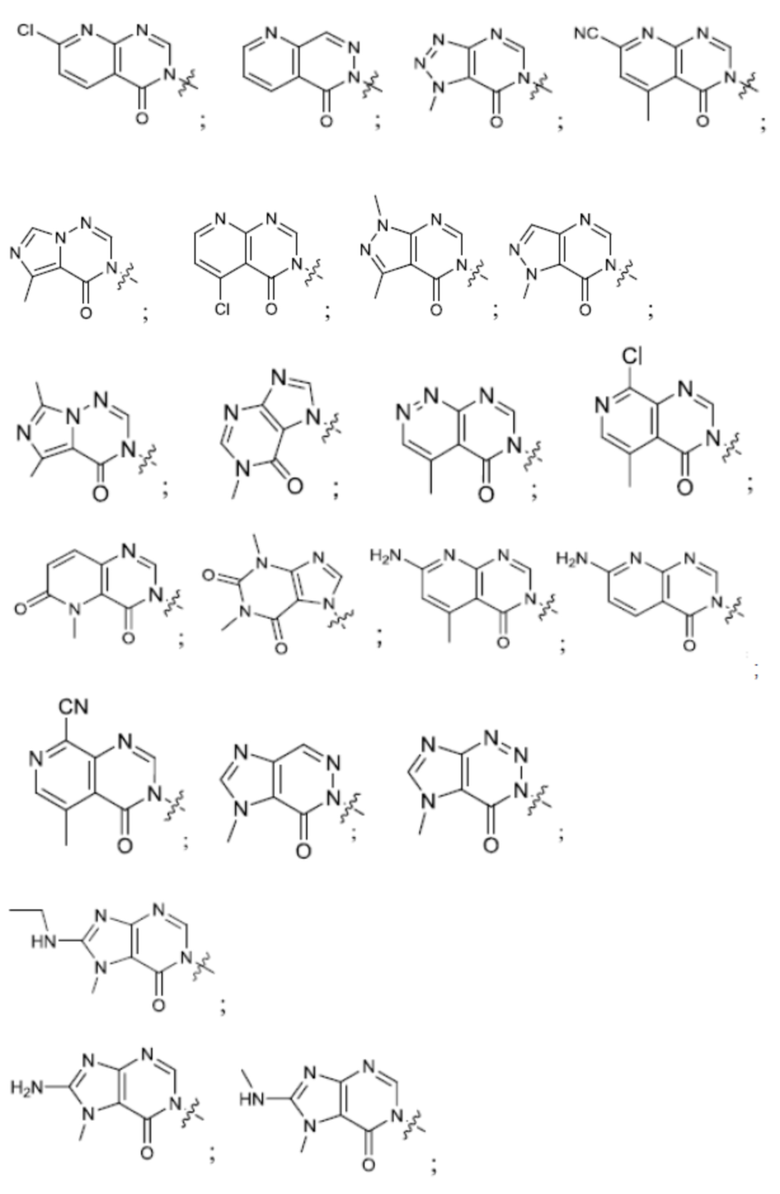
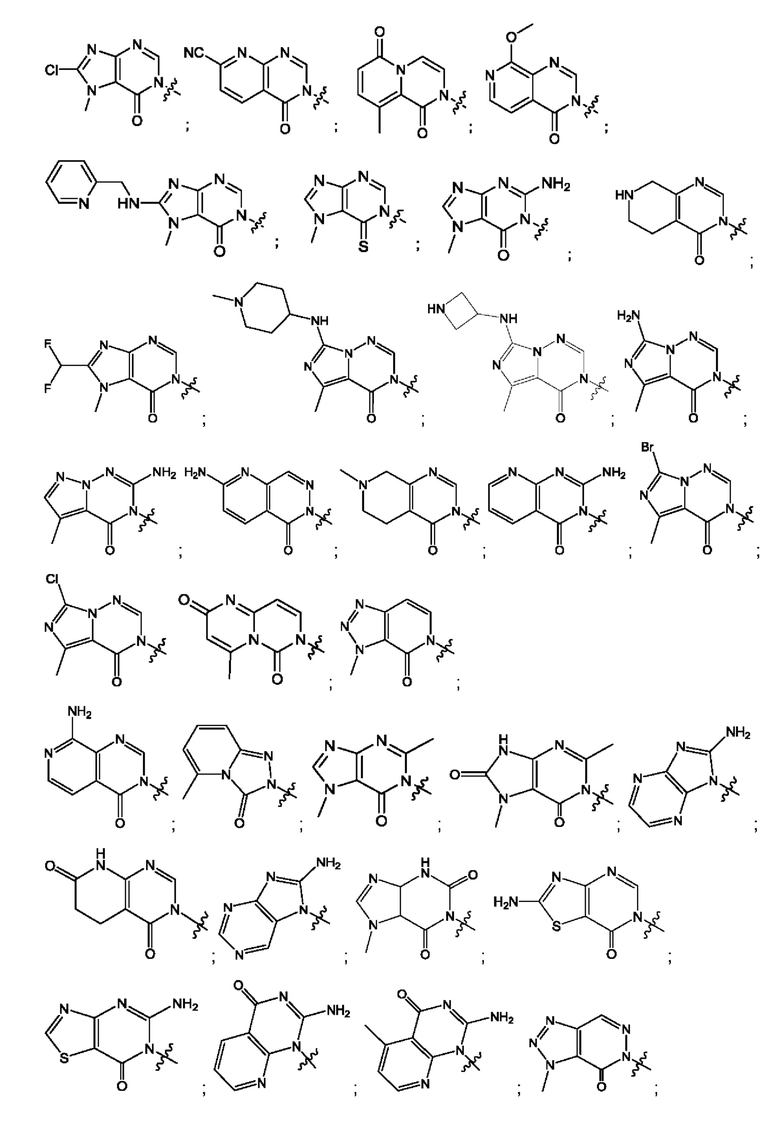
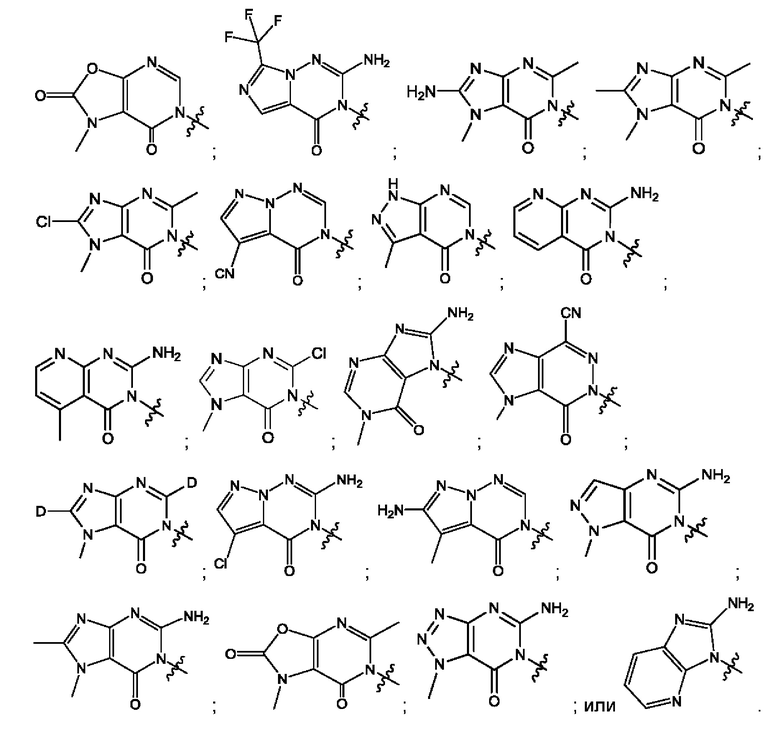
6. Соединение по п. 1 или его фармацевтически приемлемая соль, где А представляет собой:
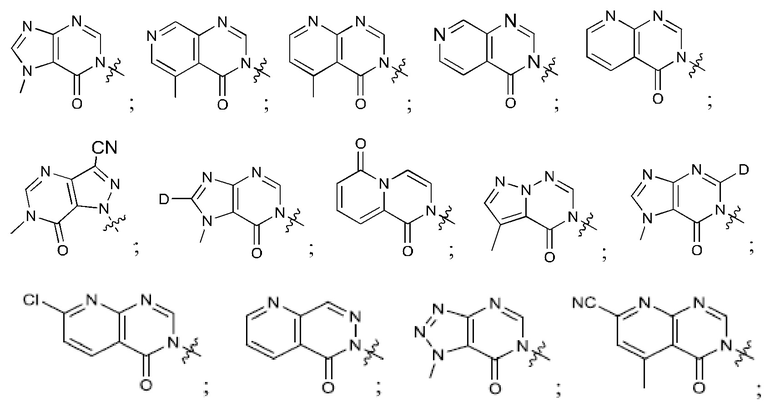
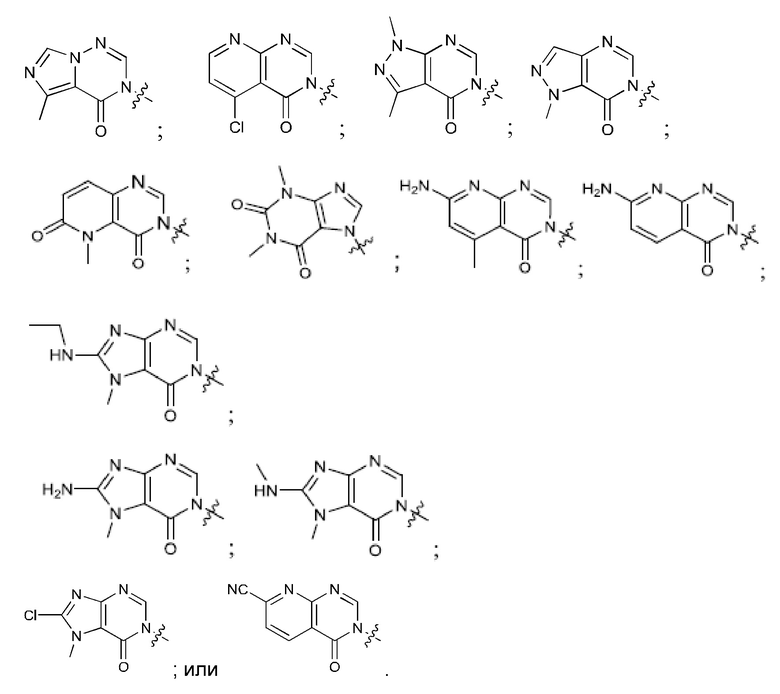
7. Соединение по п. 1 или его фармацевтически приемлемая соль, где А представляет собой:

8. Соединение по п. 1 или его фармацевтически приемлемая соль, где А представляет собой

9. Соединение по любому из пп. 1-8 или его фармацевтически приемлемая соль, где X представляет собой метилен.
10. Соединение по любому из пп. 1-9 или его фармацевтически приемлемая соль, где R4 представляет собой

где каждый R18 независимо выбран из галогена, -С1-4алкила,-С1-4галогеналкила, -CN, С1-4галогеналкокси, С1-4алкокси, -SF6, незамещенного С3-6циклоалкила, незамещенного фенила; и k равно от 0 до 3.
11. Соединение по п. 10 или его фармацевтически приемлемая соль, где каждый R18 независимо представляет собой: Cl; -OCHF2; -ОСН3; или -CN.
12. Соединение по п. 10 или его фармацевтически приемлемая соль, где каждый R18 независимо представляет собой фтор или хлор.
13. Соединение по любому из пп. 1-9 или его фармацевтически приемлемая соль, где R4 представляет собой:
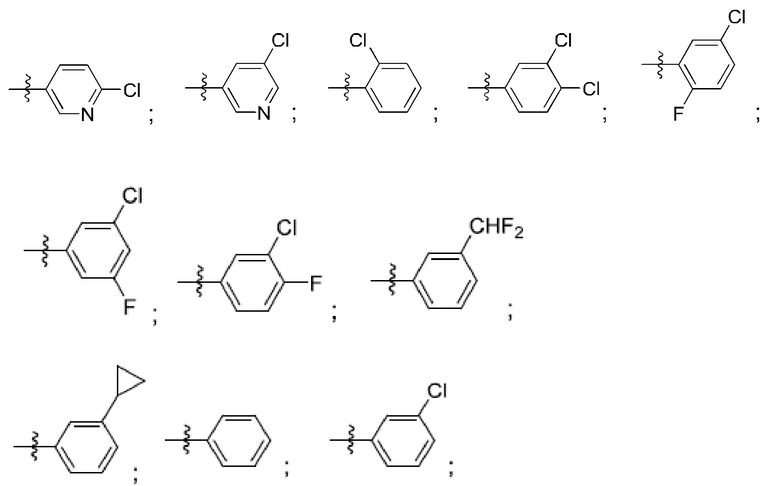

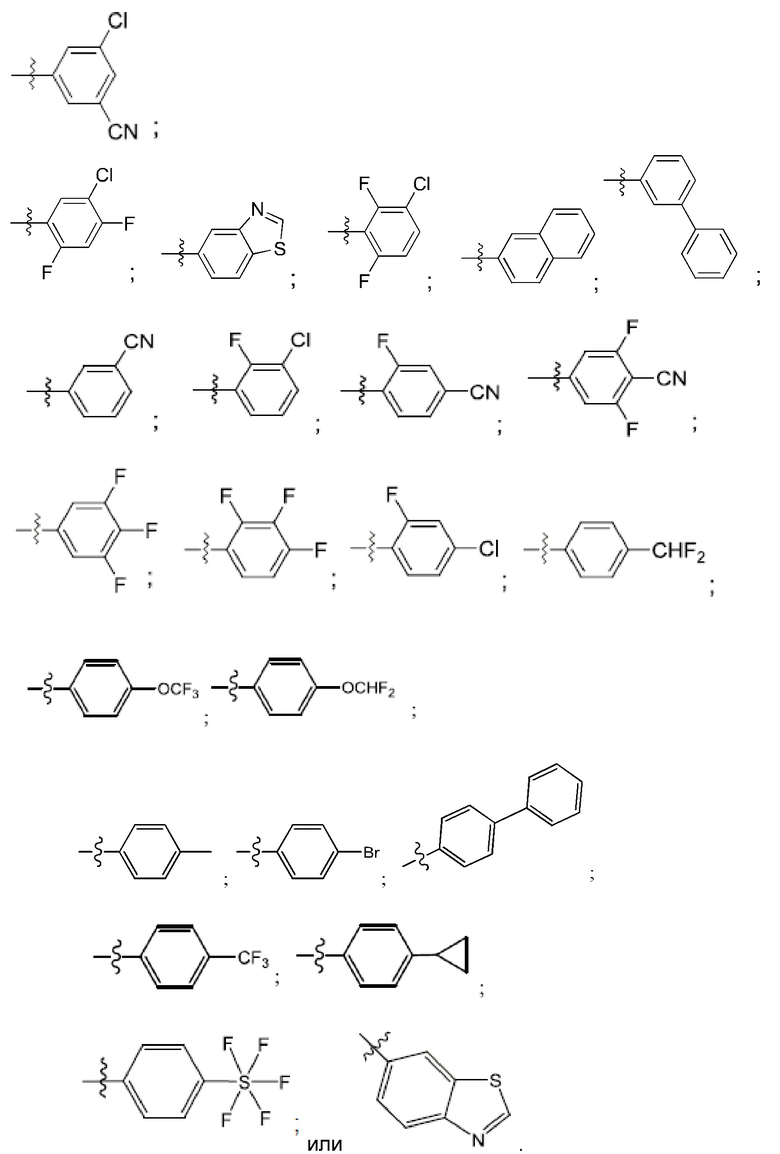
14. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение формулы (I) представляет собой соединение формулы (II)
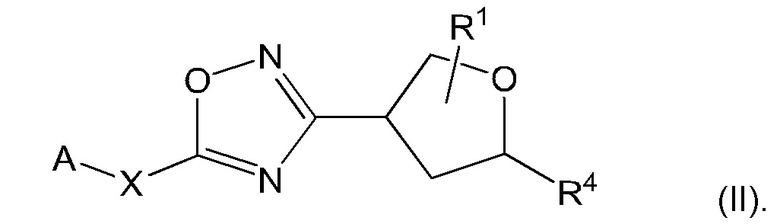
15. Соединение формулы (IVa) или соединение формулы (IVb)
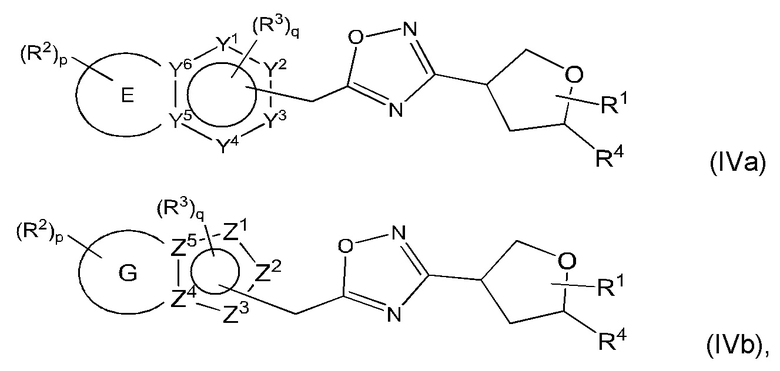
или его фармацевтически приемлемая соль, где:
Е представляет собой пятичленное или шестичленное гетероарильное кольцо, содержащее 1-3 гетероатома, выбранных из S, О, N, в котором один кольцевой атом углерода необязательно замещен оксогруппой;
G представляет собой шестичленное гетероарильное кольцо, содержащее 1-2 гетероатома N, содержащее один кольцевой атом углерода, замещенный оксогруппой;
от одного до трех из Y1, Y2, Y3, Y4, Y5 и Y6 представляют собой азот, а остальные Y1, Y2, Y3, Y4, Y5 и Y6 являются углеродом, где один из Y1, Y2, Y3 и Y4 может быть -С(O)- или -C(S)-;
один или два из Z1, Z2, Z3, Z4 и Z5 представляют собой азот, а остальные Z1, Z2, Z3, Z4 и Z5 являются углеродом; R1 представляет собой Н;
каждый R2 независимо представляет собой Н, -С1-4алкил; -С1-4галогеналкил; -CN; галоген; С1-4алкокси; и NR14R15;
каждый R3 независимо представляет собой: Н; D; -С1-4алкил; -CN; галоген; или -NR14R15;
R4 представляет собой незамещенный фенил или фенил, замещенный 1-3 заместителями, выбранными из галогена, -CN, -С1-4алкила, незамещенного С3-6циклоалкила, С1-4галогеналкила, С1-4галогеналкокси, С1-4алкокси, пентафторсульфанила или незамещенного фенила; незамещенный гетероарил или гетероарил, замещенный 1 заместителем, выбранным из галогена и С1-4алкила, где гетероарил выбран из пиразола, пиридина, бензотиазола; или незамещенный нафтил;
или R1 и R4 могут вместе образовывать незамещенный С3-6циклоалкил, сконденсированный с незамещенным фенилом;
р равно 0, 1 или 2;
q равно 0 или 1;
каждый из R14 и R15 независимо друг от друга представляет собой: Н; замещенный одним пиридином или незамещенный -С1-4алкил; замещенный одним С1-4алкилом или незамещенный 4-6-членный гетероциклоалкил, содержащий один атом азота; или R14 и R15 вместе с атомами, к которым они присоединены, могут образовывать 6-членное кольцо, включающее один дополнительный гетероатом, выбранный из О, N.
16. Соединение по п. 15 или его фармацевтически приемлемая соль, где соединение формулы (IVa) представляет собой соединение формулы (Va)
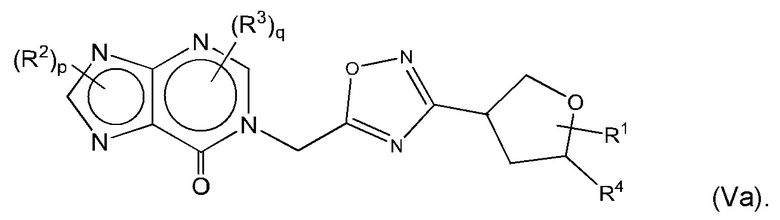
17. Соединение по п. 16 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы (VIc) или соединение формулы (VId)
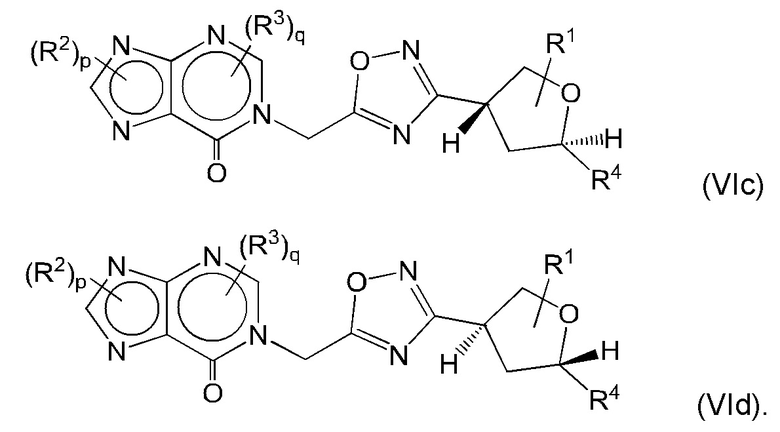
18. Соединение по п. 16 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы (VIIc) или соединение формулы (VIId)
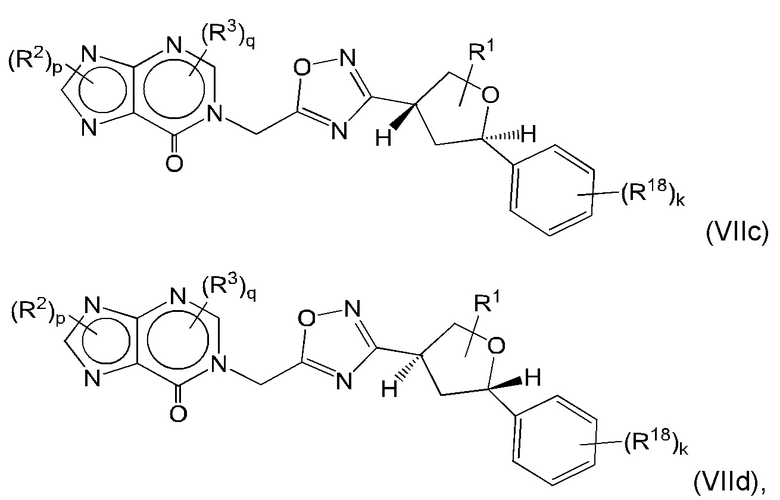
где каждый R18 независимо выбран из галогена, -С1-4алкила, -С1-4галогеналкила, -CN, С1-4галогеналкокси, С1-4алкокси, -SF5, незамещенного С3-6циклоалкила и незамещенного фенила; и k равно от 0 до 3.
19. Соединение, выбранное из:
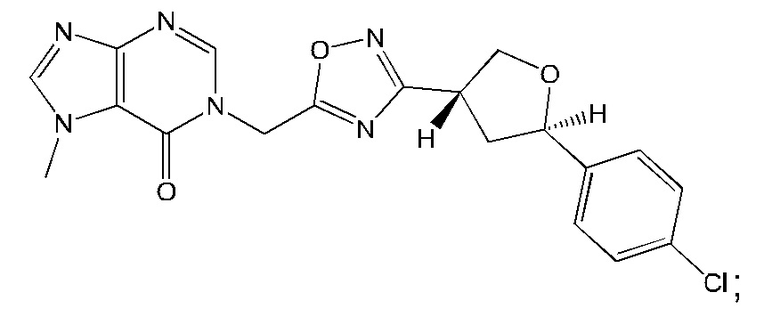
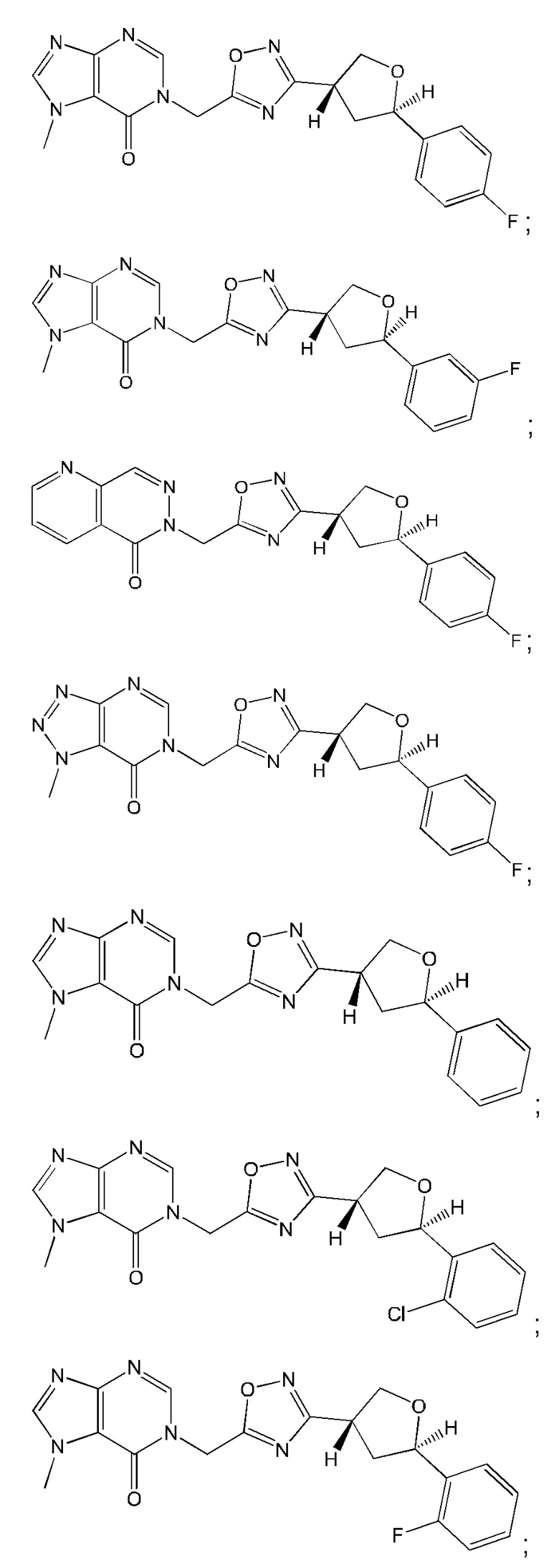
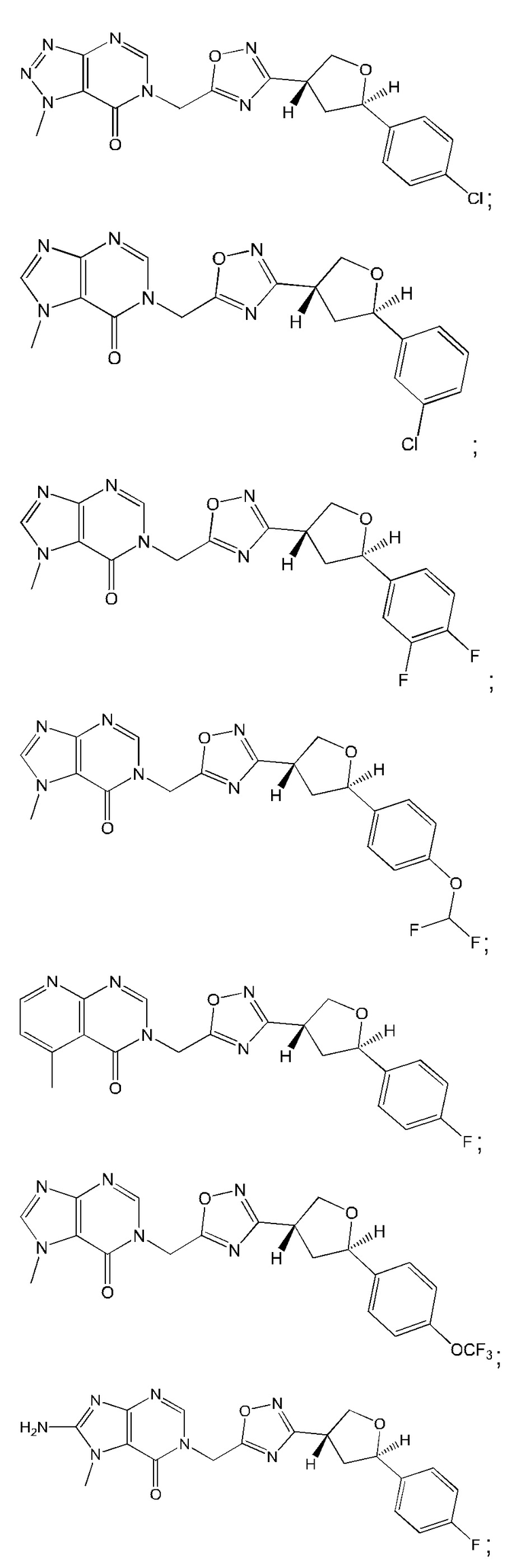
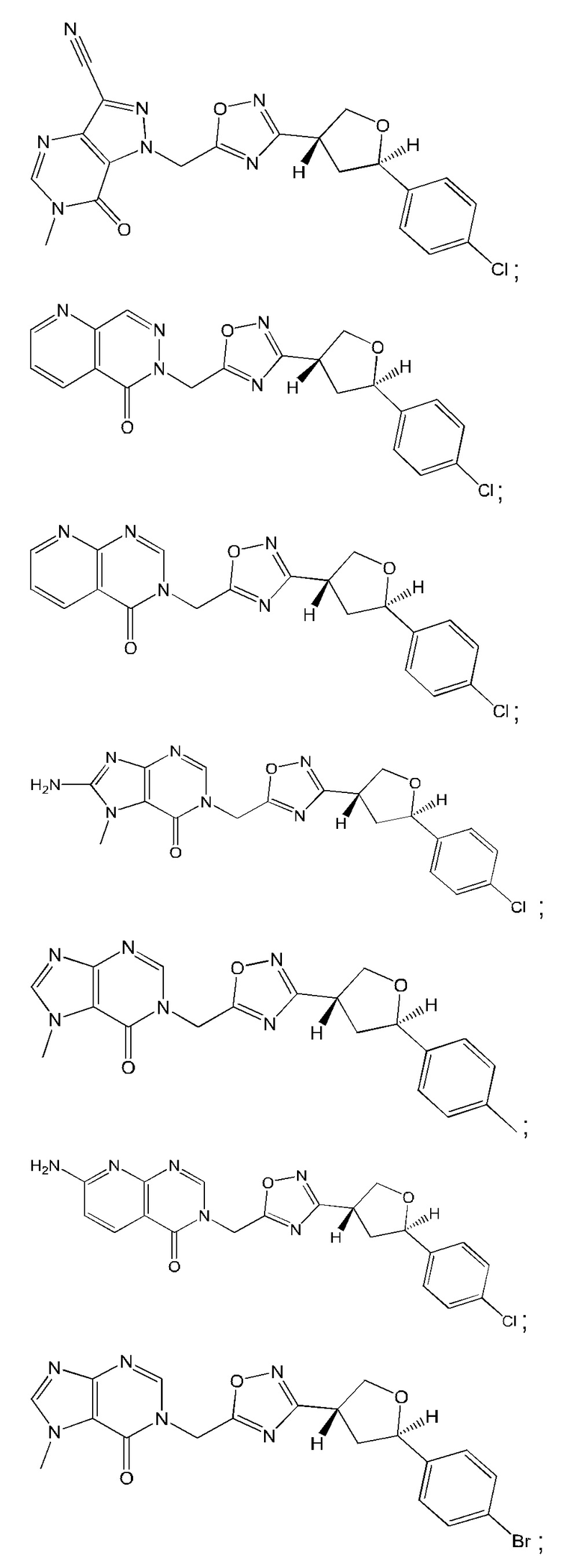

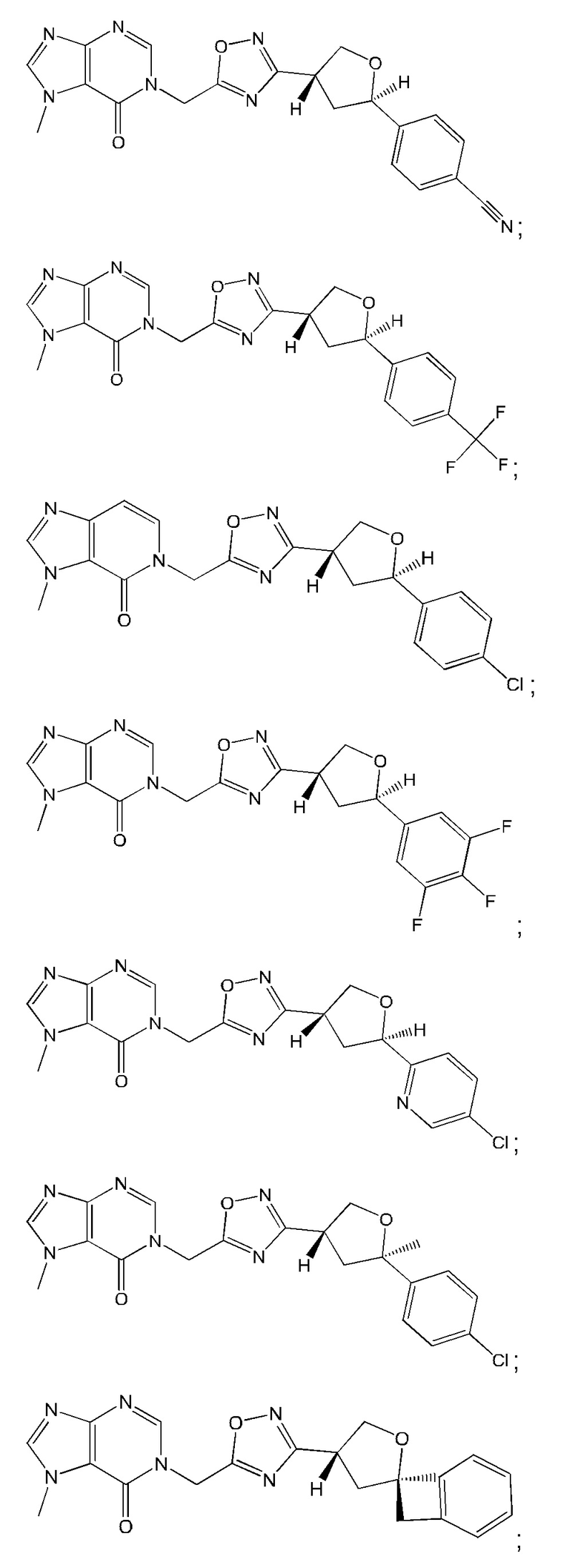

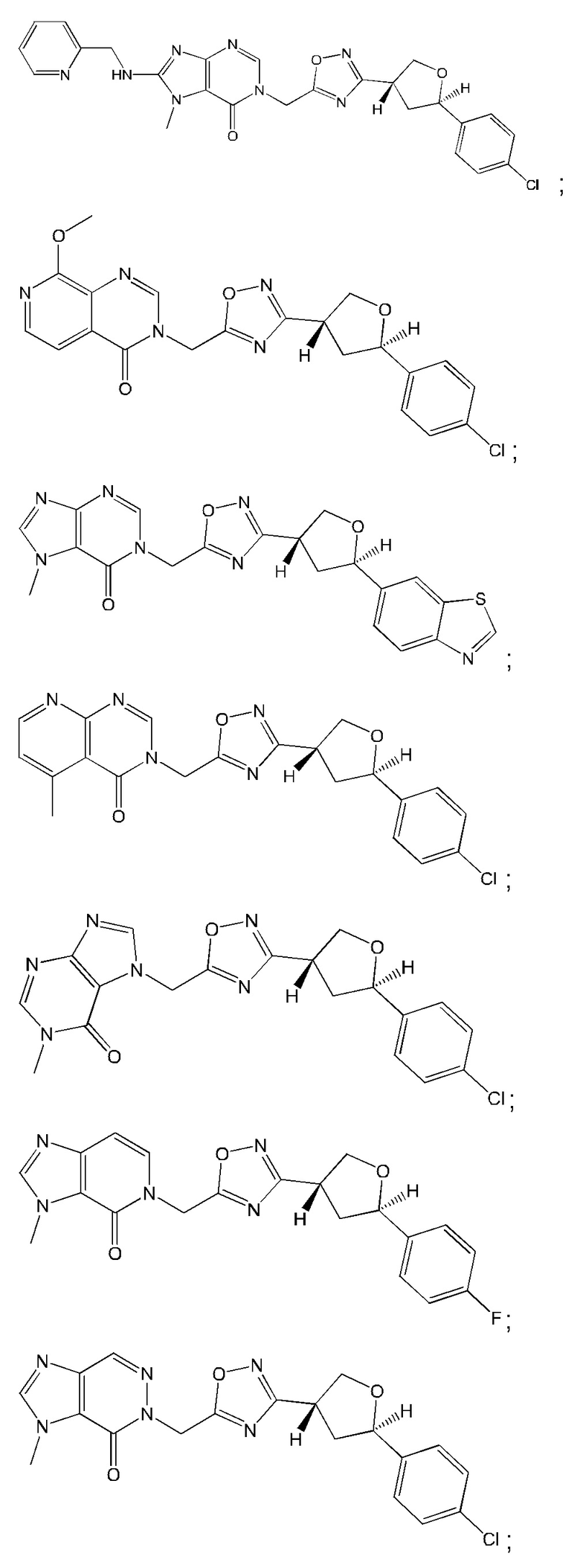
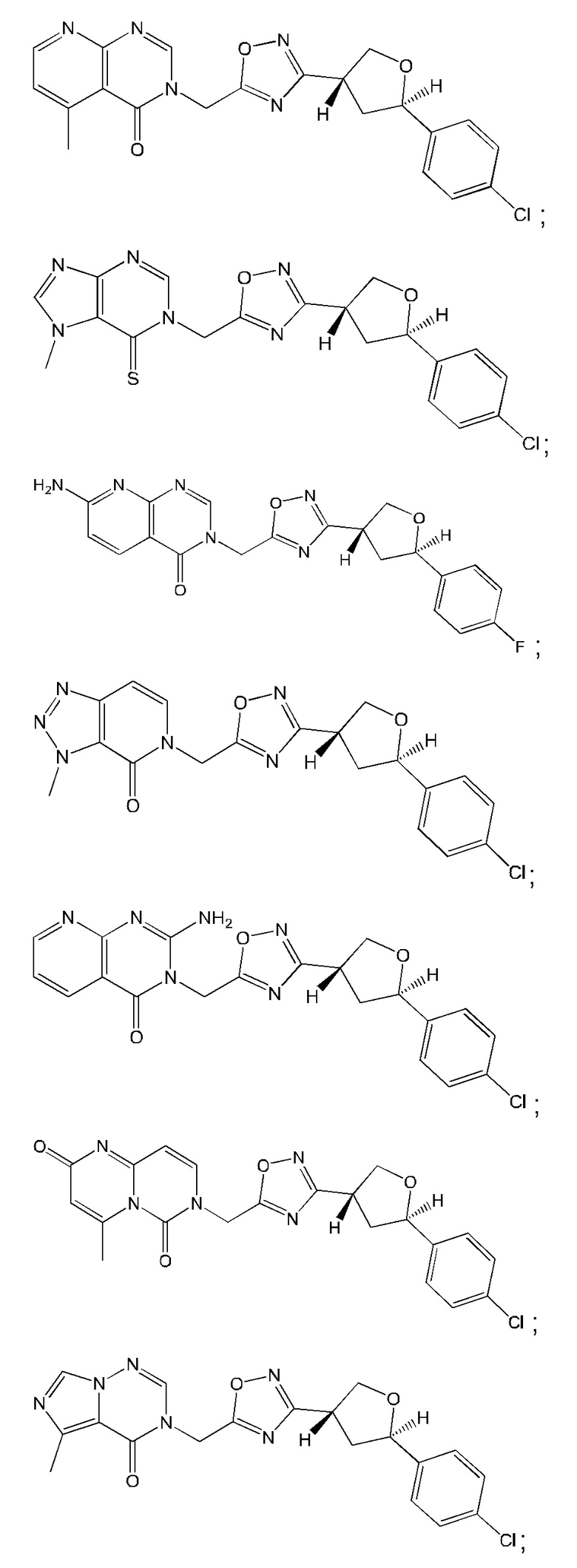
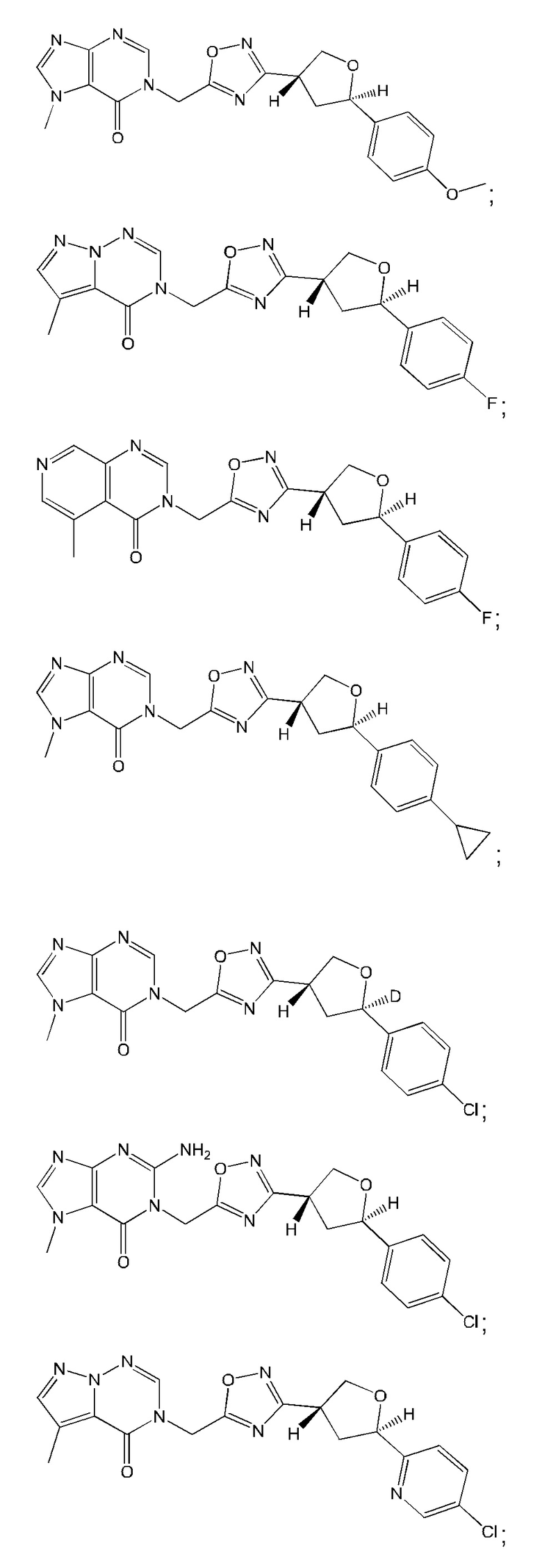

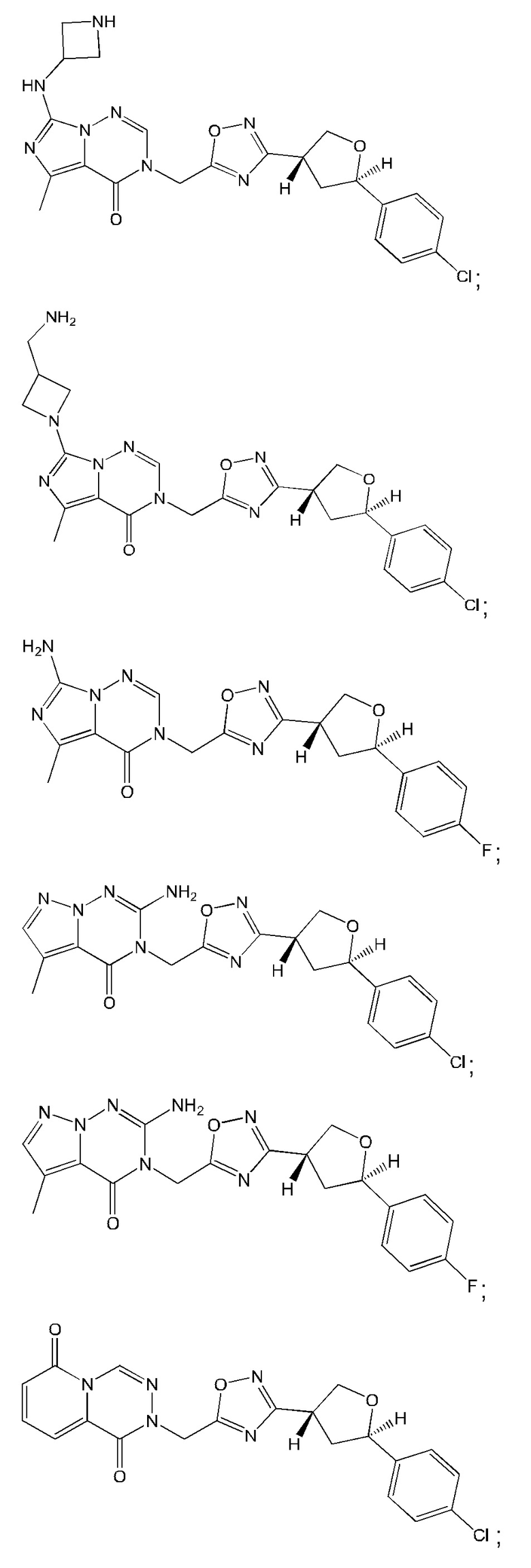

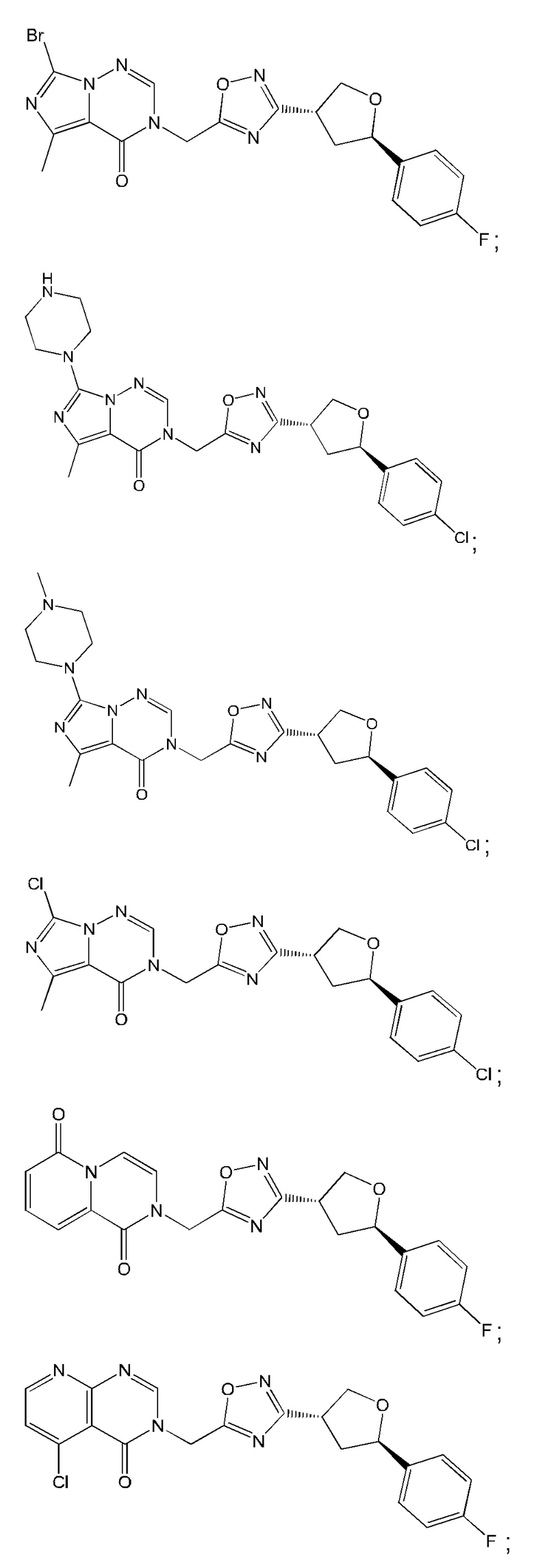

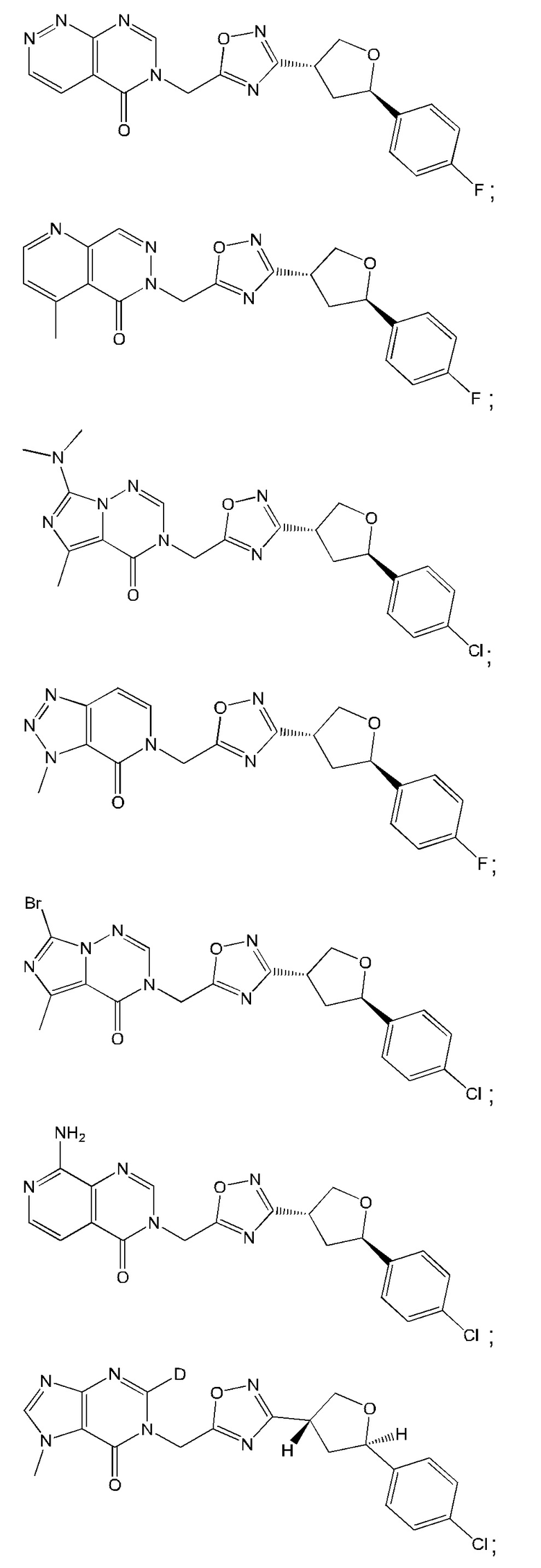
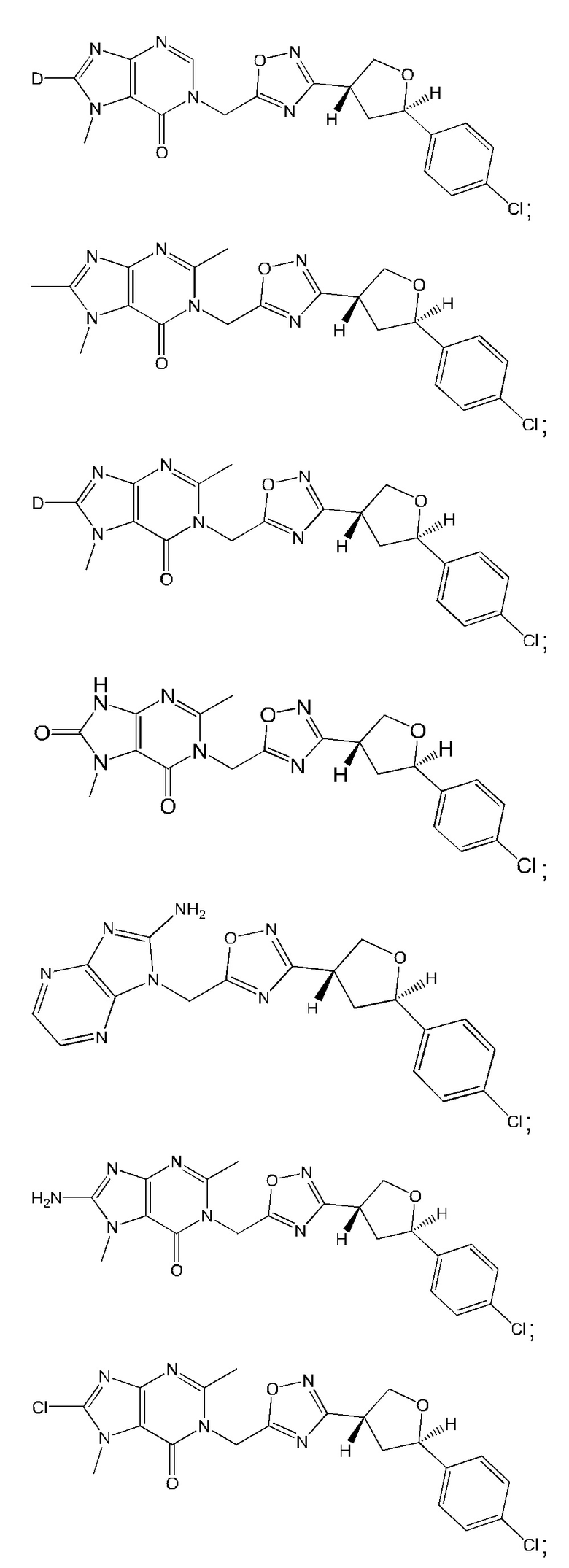
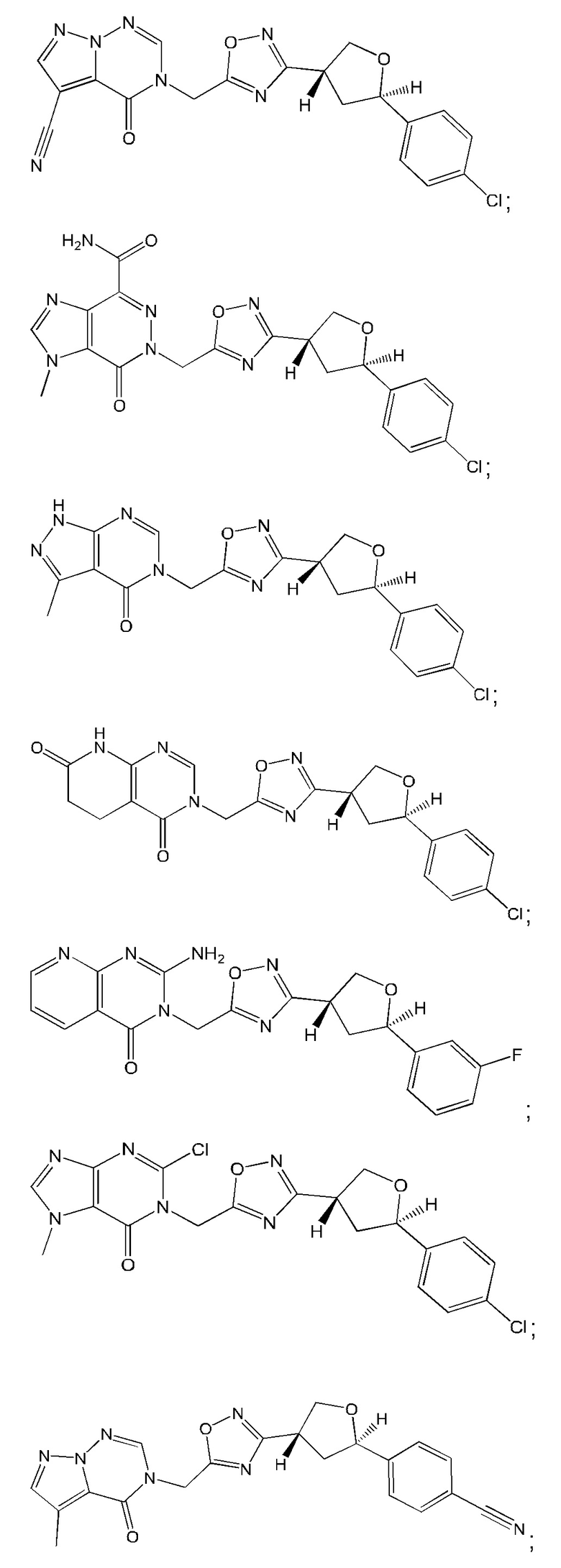
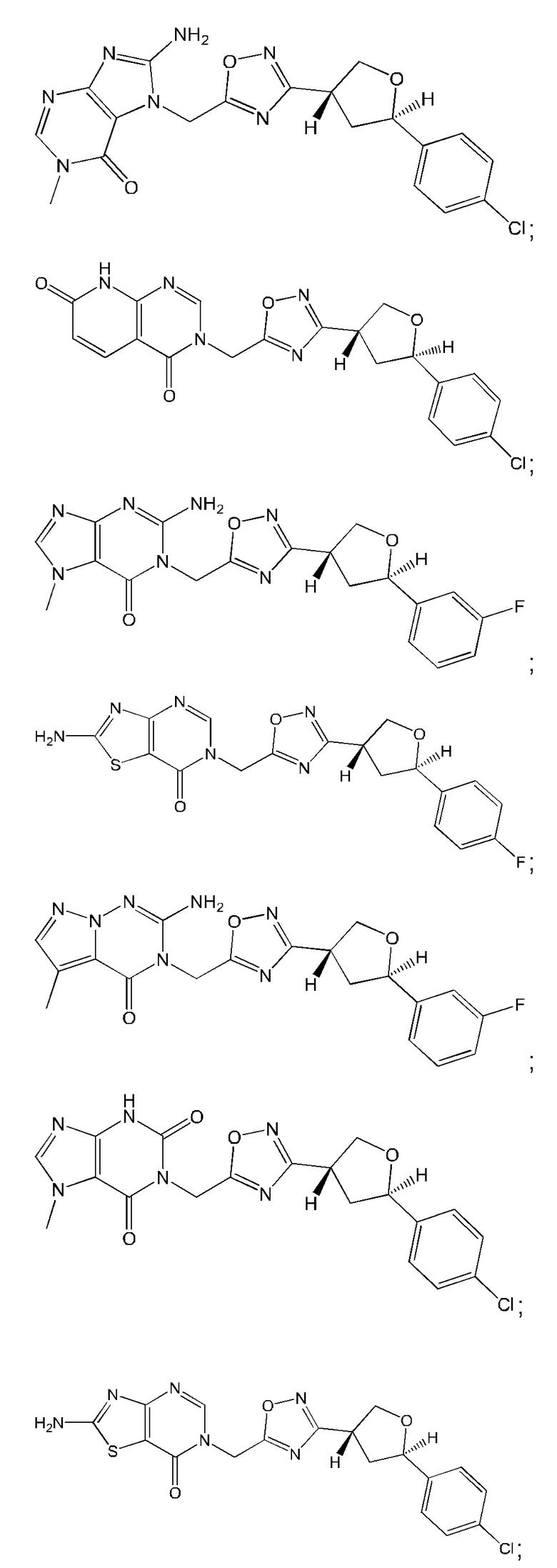
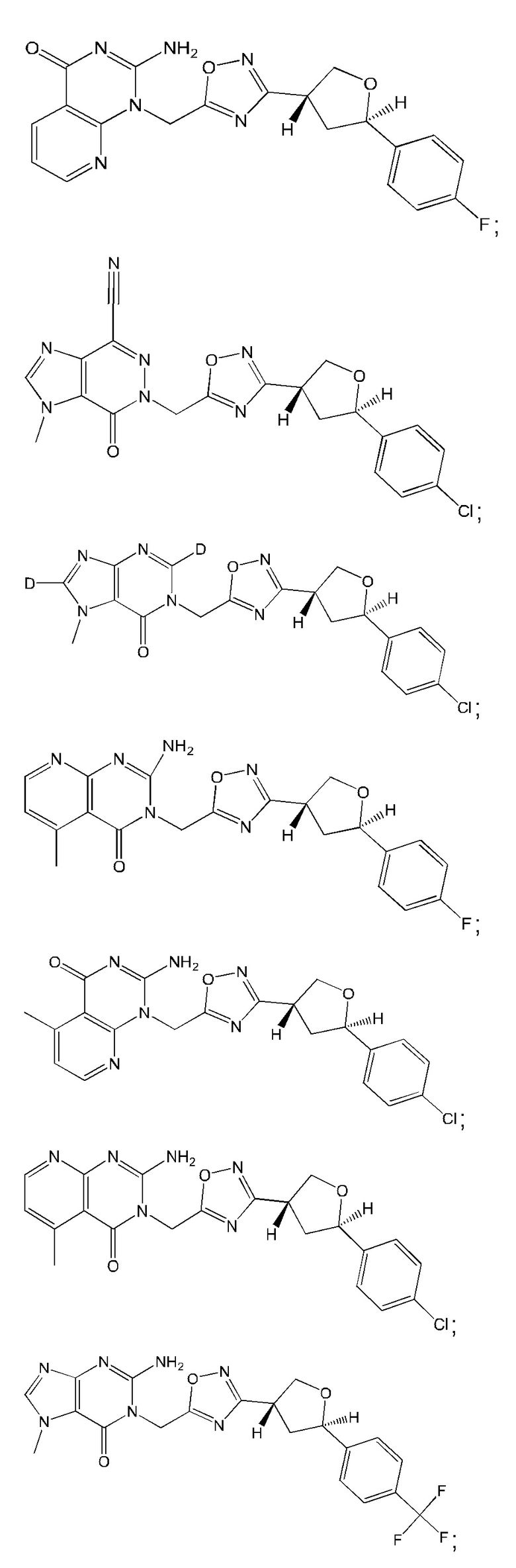
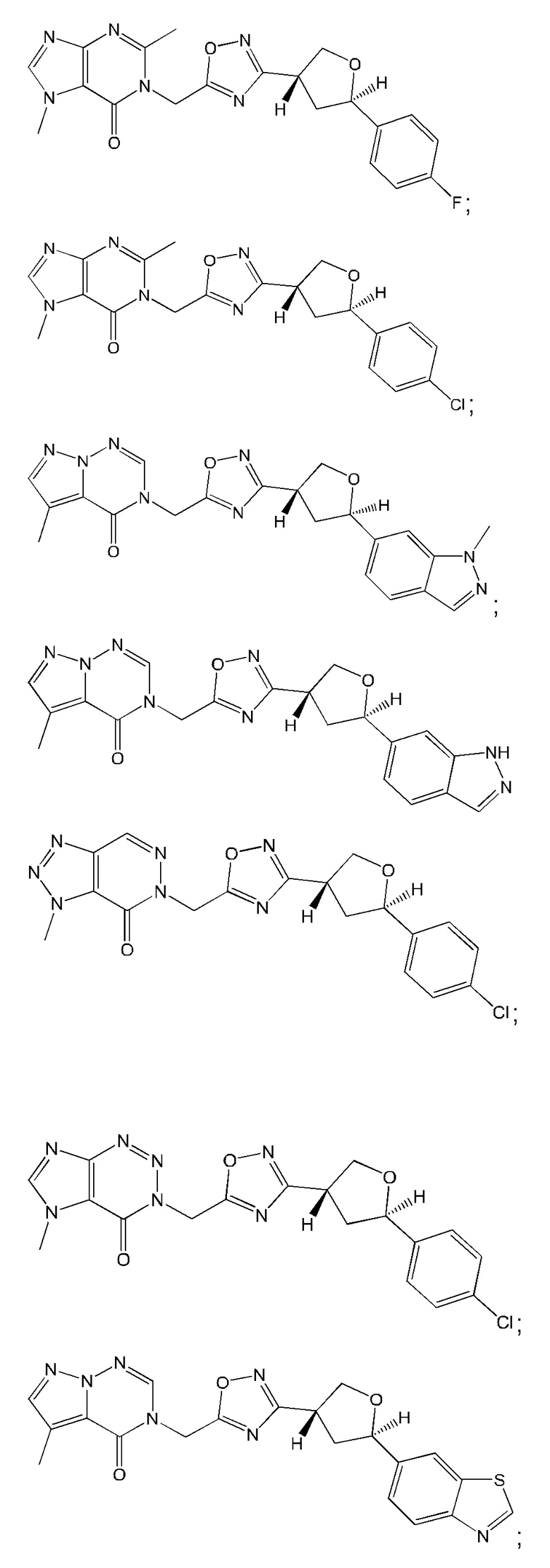
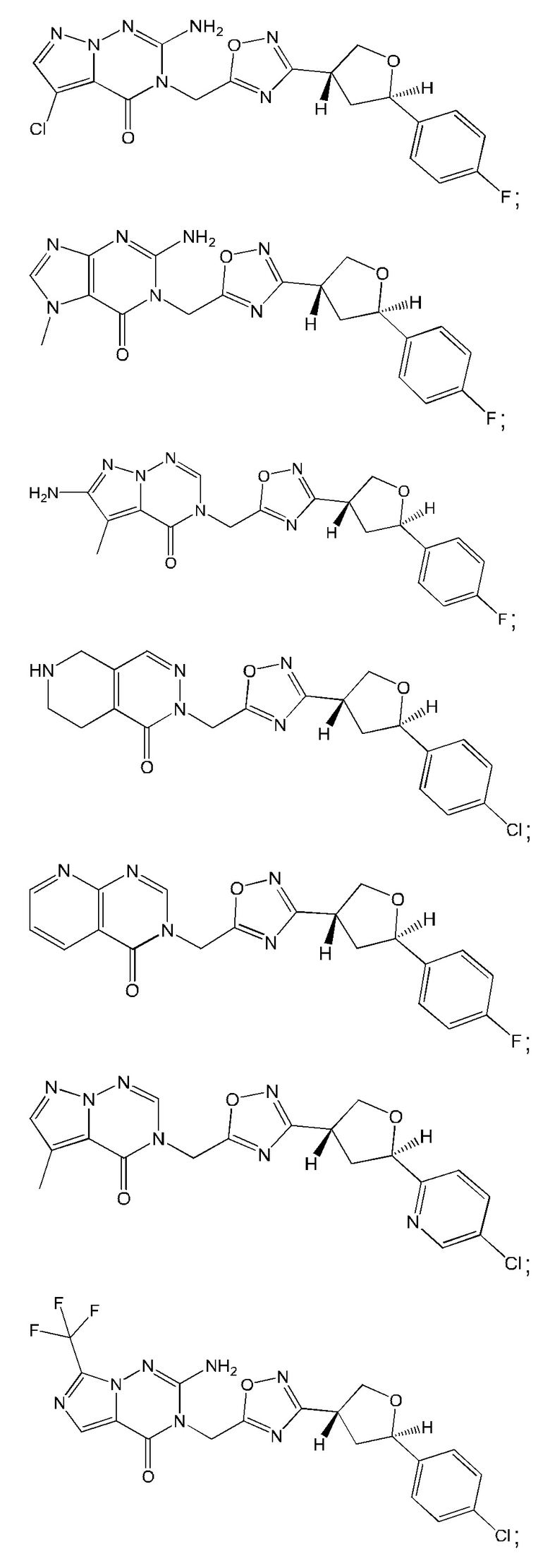
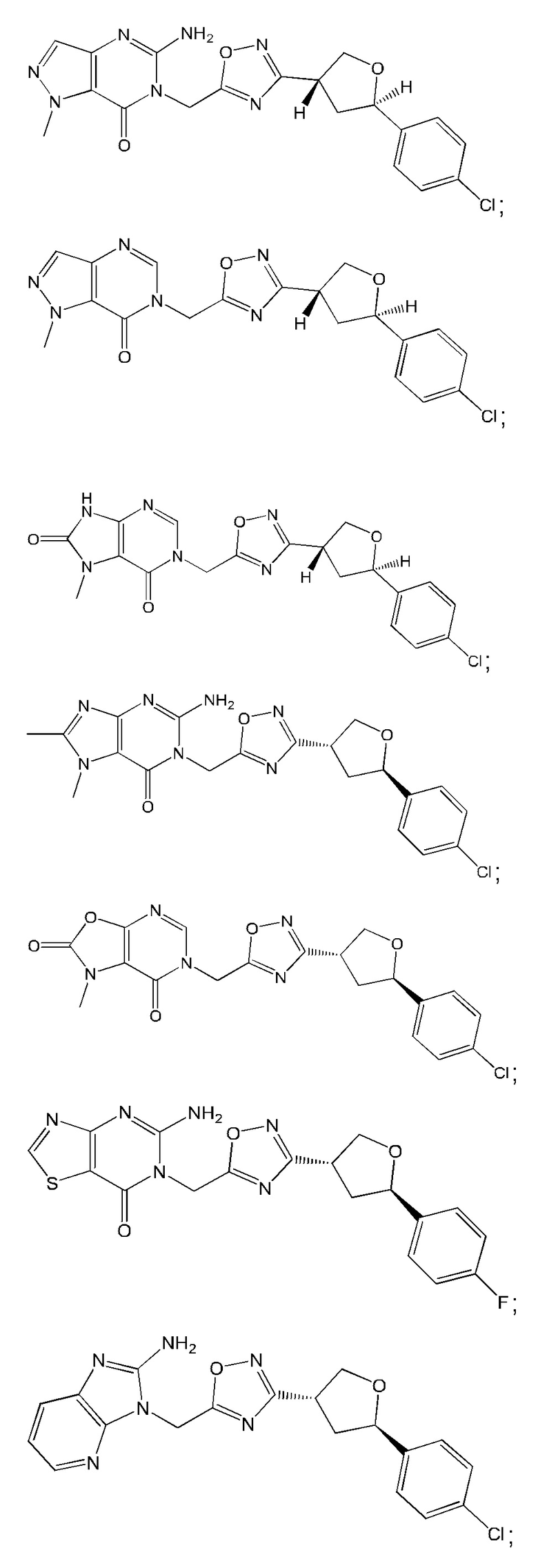
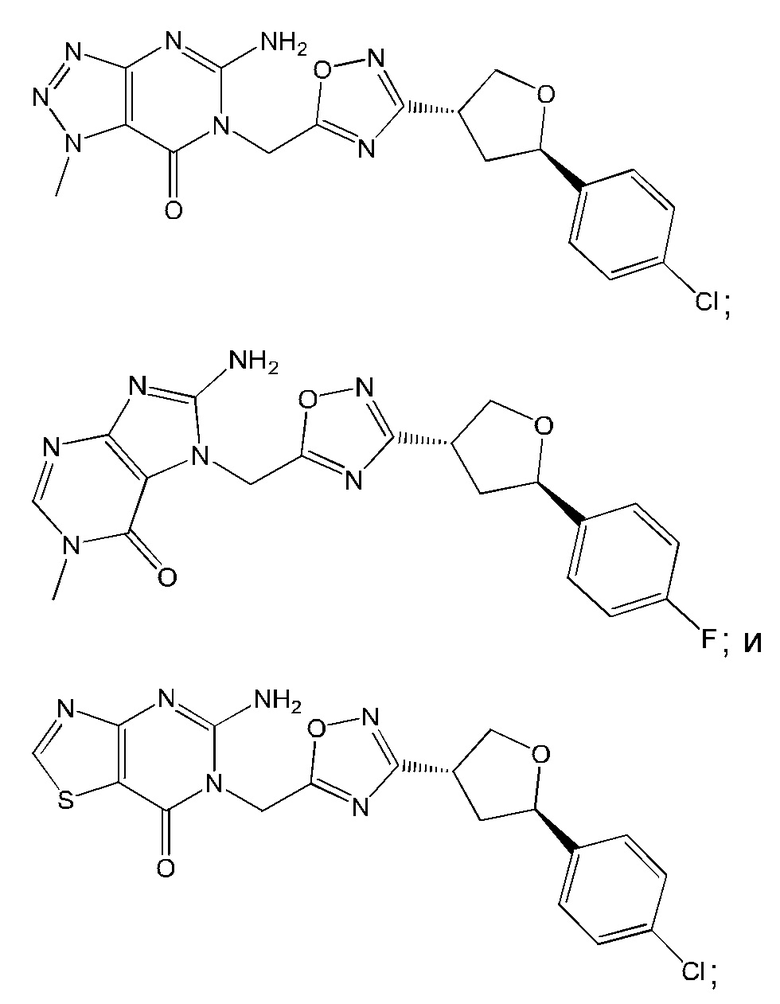
или его фармацевтически приемлемая соль.
20. Соединение по п. 1 или его фармацевтически приемлемая соль, где А представляет собой 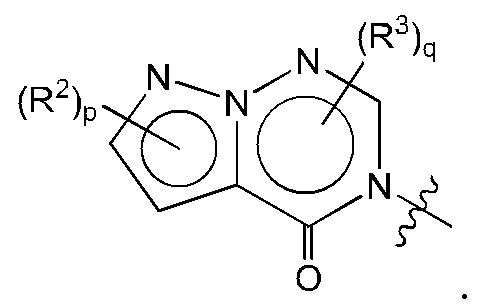
21. Соединение по п. 3 или его фармацевтически приемлемая соль, где А представляет собой 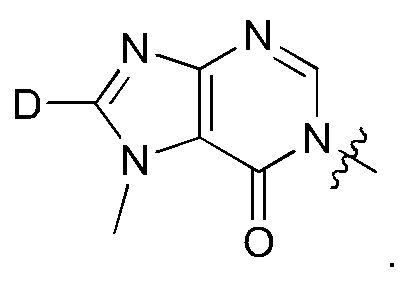
22. Соединение по п. 3 или его фармацевтически приемлемая соль, где А представляет собой 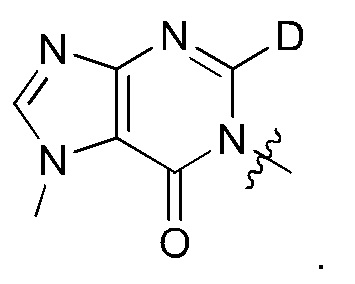
23. Соединение по п. 3 или его фармацевтически приемлемая соль, где А представляет собой 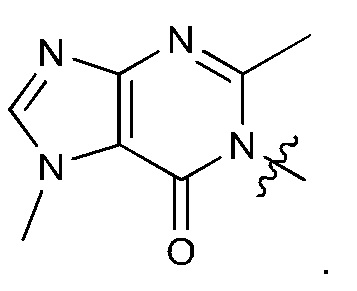
24. Соединение по п. 3 или его фармацевтически приемлемая соль, где А представляет собой 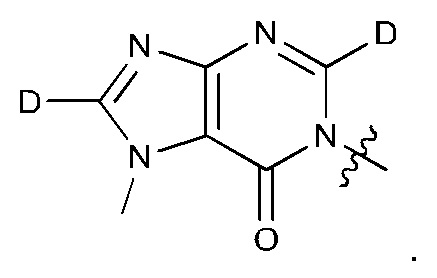
25. Соединение по п. 3 или его фармацевтически приемлемая соль, где А представляет собой 
26. Соединение по п. 20 или его фармацевтически приемлемая соль, где А представляет собой 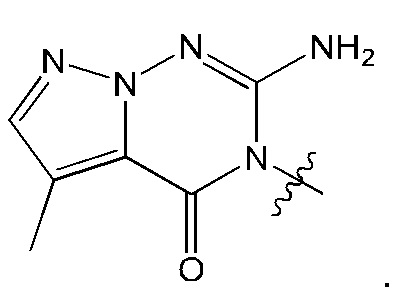
27. Соединение по п. 19, где соединение выбрано из:

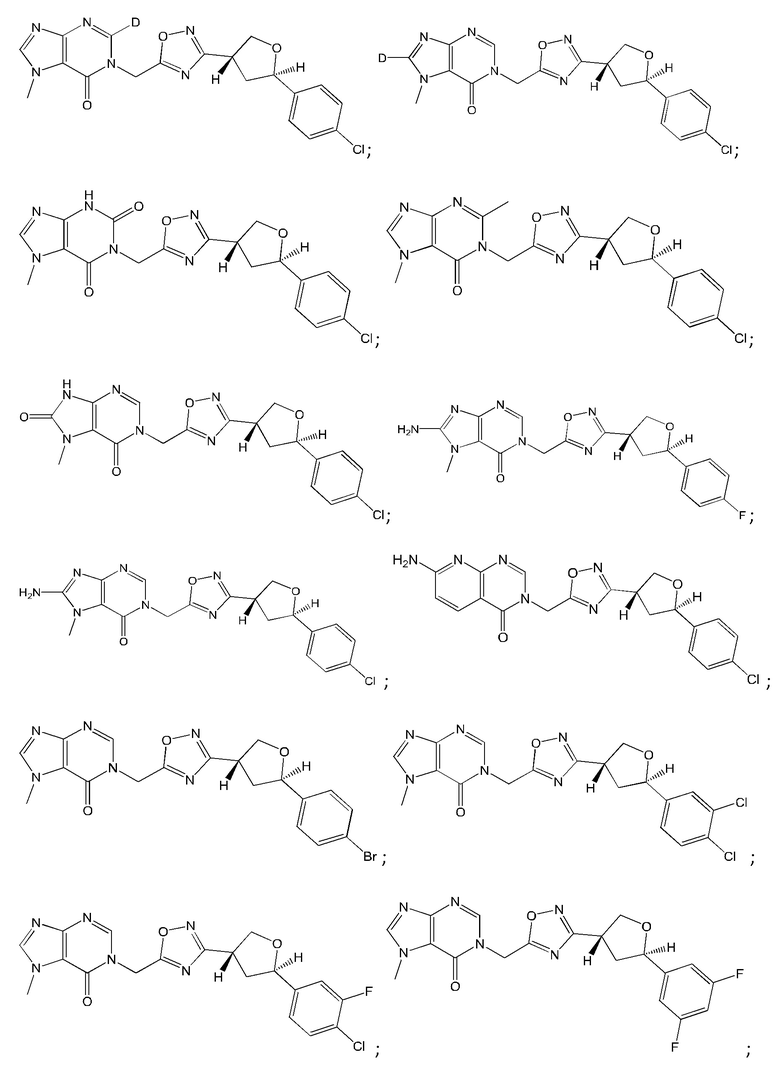
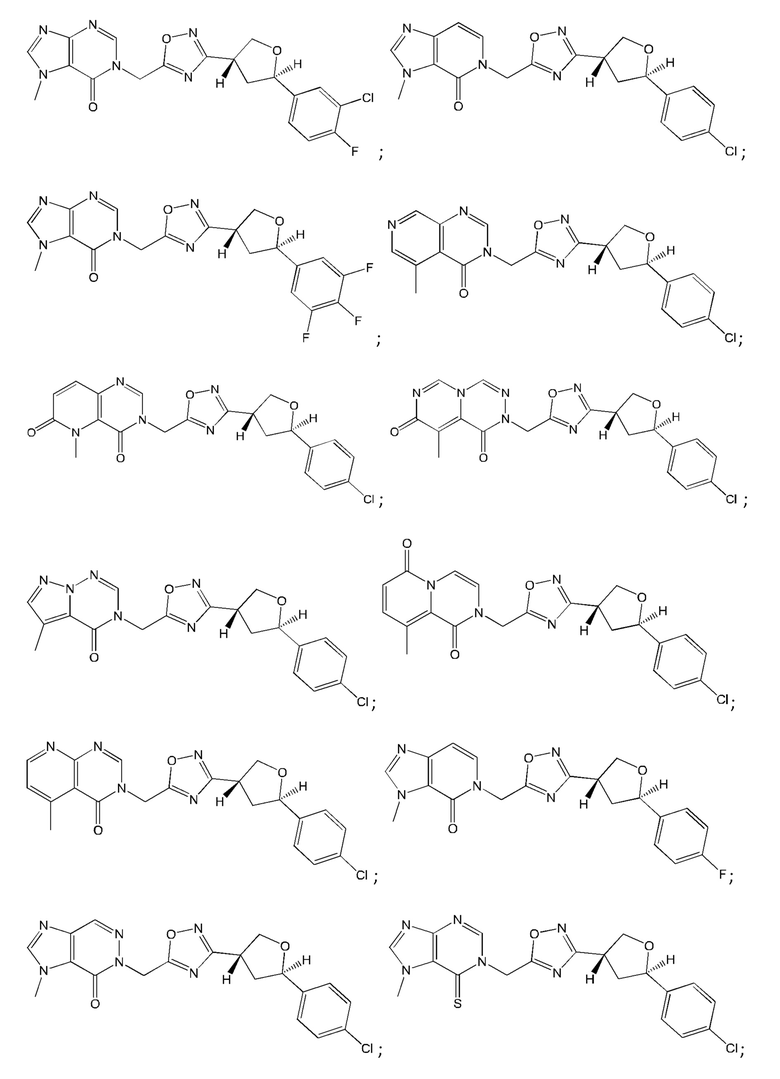
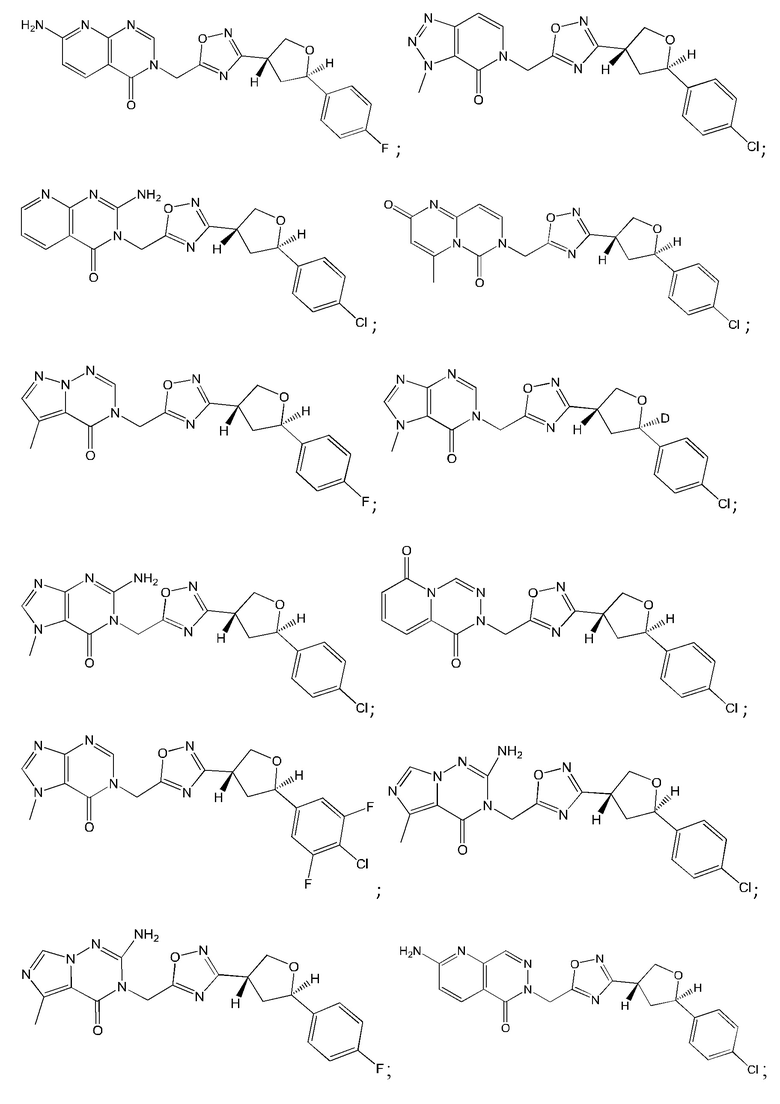
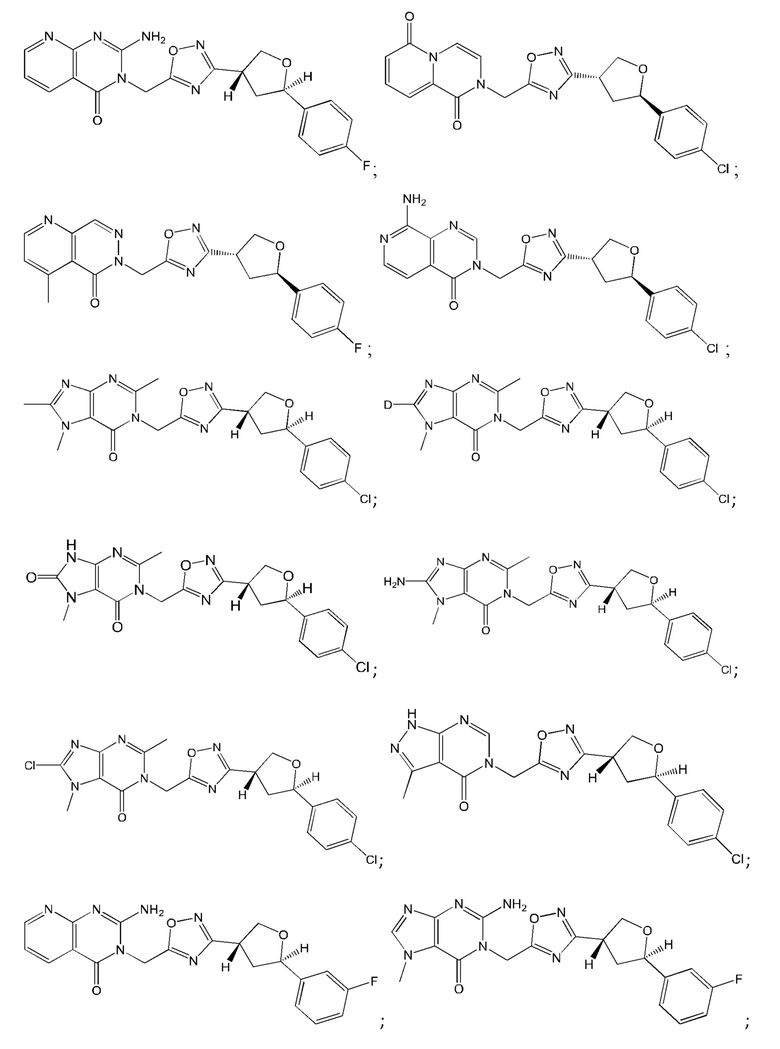
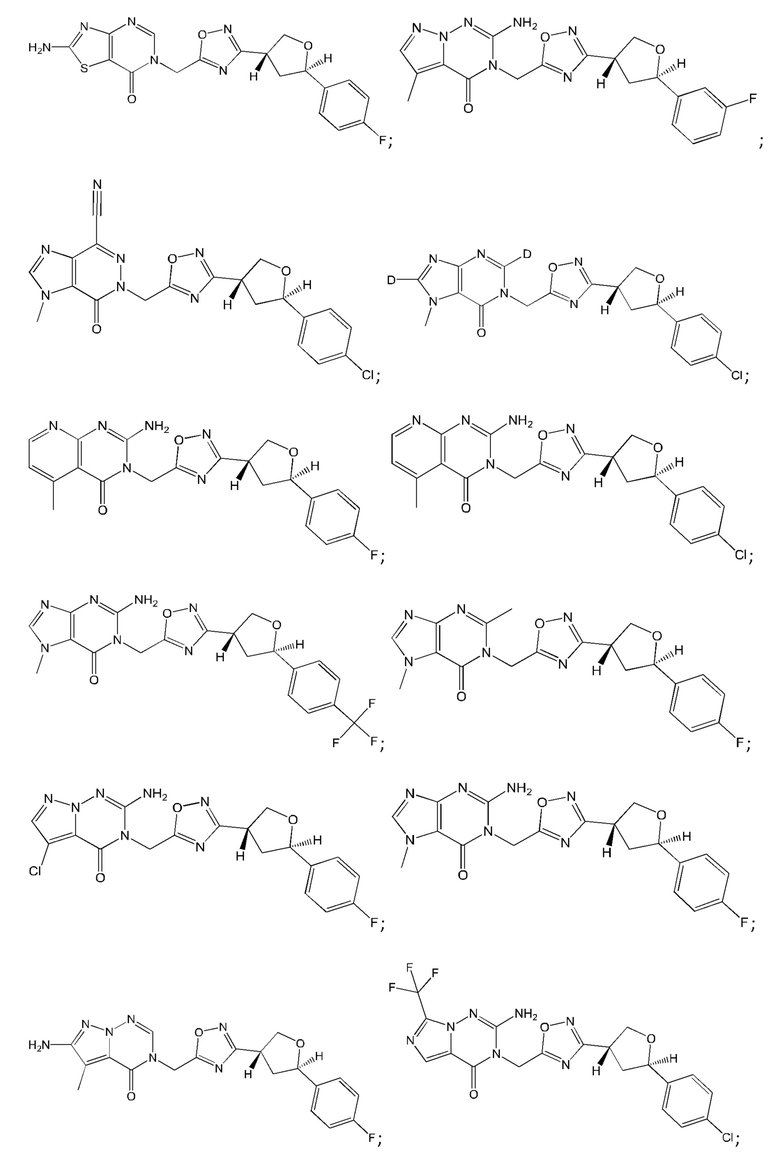
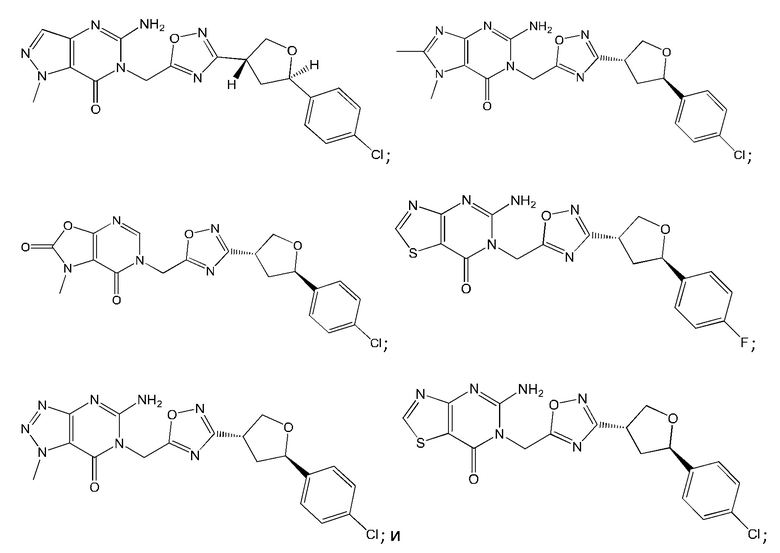
или его фармацевтически приемлемая соль.
28. Соединение по п. 19, где соединение выбрано из:
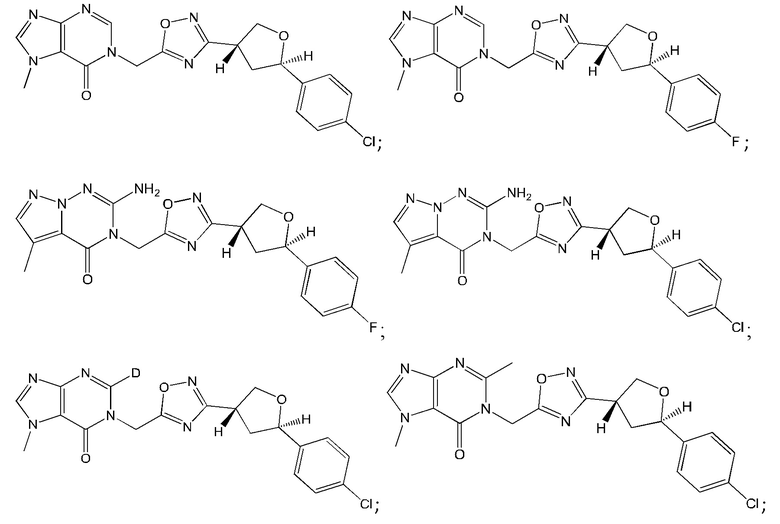

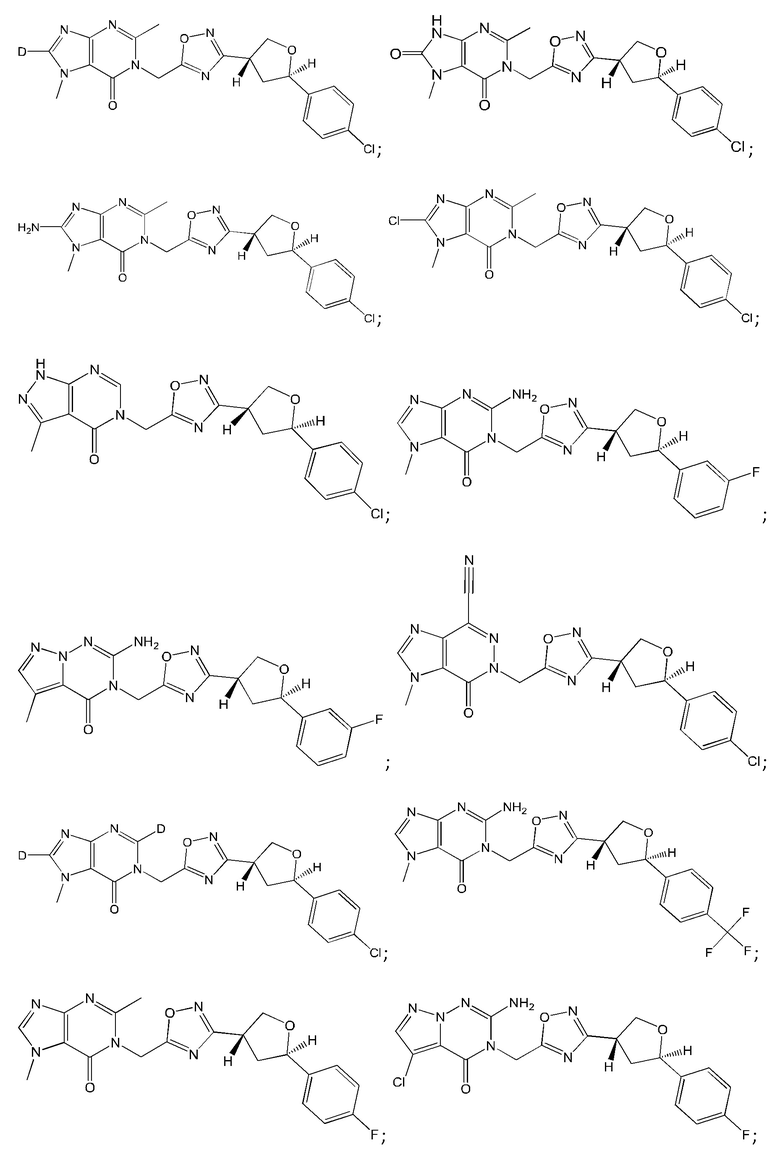
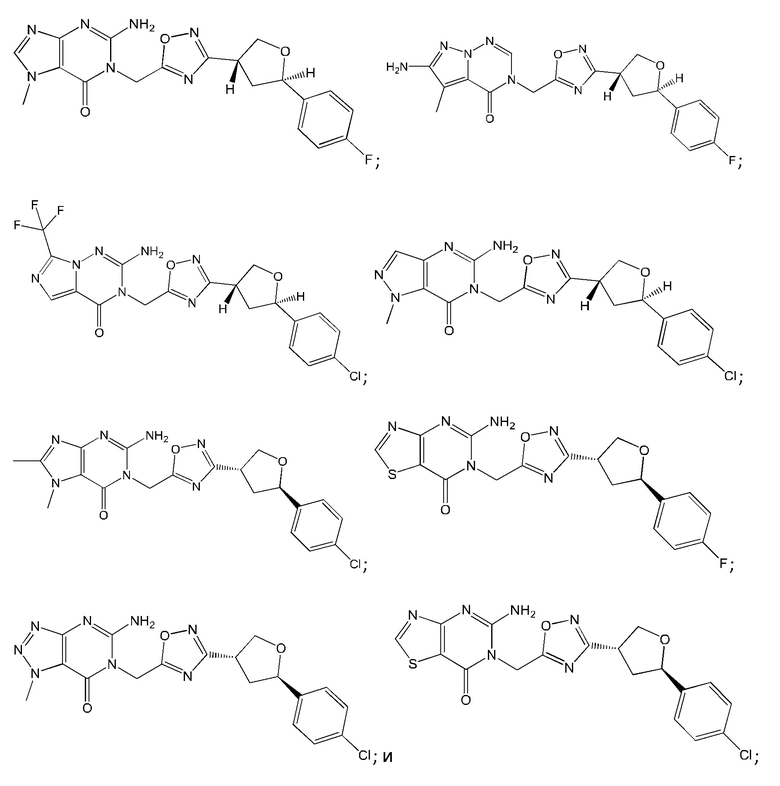
или его фармацевтически приемлемая соль.
29. Соединение по п. 19, где соединение выбрано из:
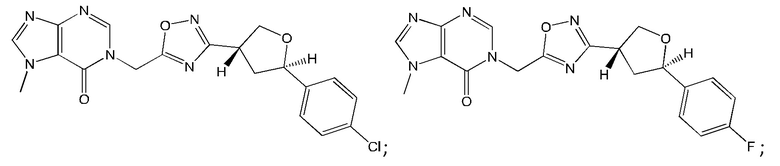
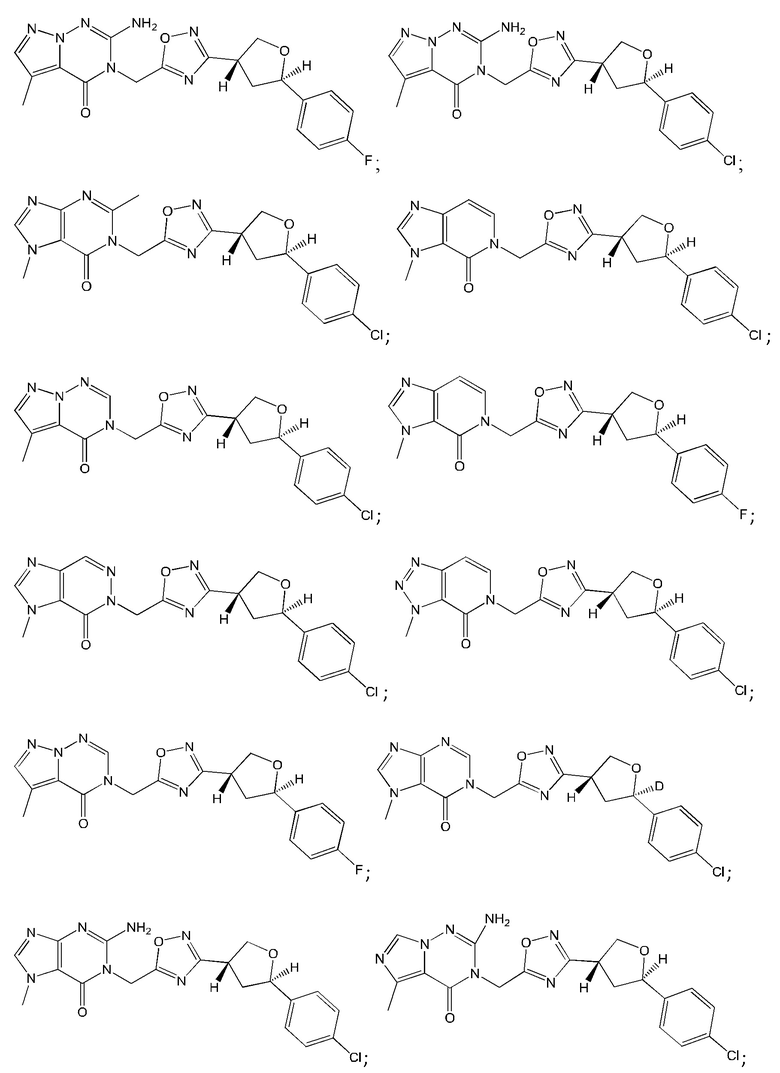
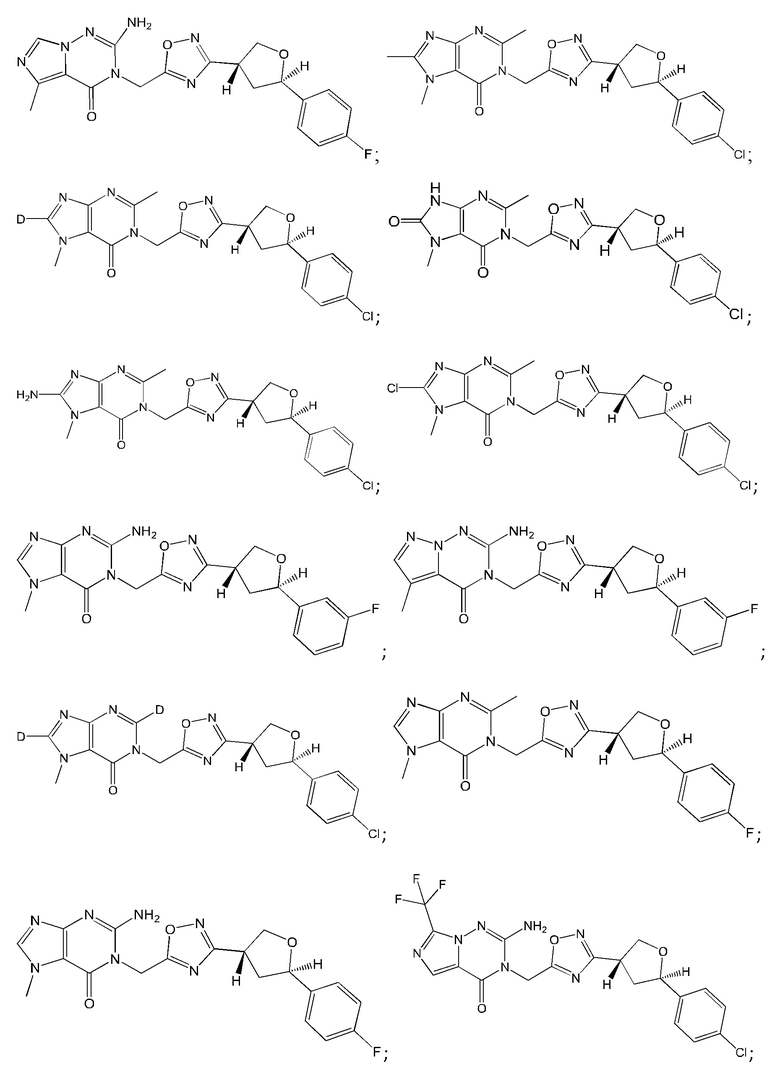

или его фармацевтически приемлемая соль.
30. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении TRPA1, содержащая эффективное количество соединения по любому из пп. 1-29 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
31. Соединение по любому из пп. 1-29 или его фармацевтически приемлемая соль для ингибирования активности TRPA1.
32. Соединение по любому из пп. 1-29 или его фармацевтически приемлемая соль для лечения или профилактики заболевания или состояния, опосредованного активностью TRPA1.
33. Соединение по п. 32, где заболевание или состояние представляет собой боль, зуд, воспалительное расстройство, заболевание внутреннего уха, лихорадочное состояние или другое нарушение терморегуляции, трахеобронхиальную или диафрагмальную дисфункцию, расстройство желудочно-кишечного тракта или мочевых путей, хроническое обструктивное заболевание легких, недержание или расстройство, обусловленное снижением кровотока в ЦНС или гипоксией ЦНС.
34. Соединение по п. 32, где заболевание или состояние представляет собой боль, артрит, зуд, кашель, астму, воспалительное заболевание кишечника или заболевание внутреннего уха.
35. Применение соединения по любому из пп. 1-29 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики заболевания или состояния, опосредованного активностью TRPA1, где заболевание или состояние представляет собой боль, зуд, воспалительное расстройство, заболевание внутреннего уха, лихорадочное состояние или другое нарушение терморегуляции, трахеобронхиальную или диафрагмальную дисфункцию, расстройство желудочно-кишечного тракта или мочевых путей, хроническое обструктивное заболевание легких, недержание, расстройство, обусловленное снижением кровотока в ЦНС или гипоксией ЦНС, артрит, кашель, астму или воспалительное заболевание кишечника.
36. Способ ингибирования активности TRPA1, включающий приведение TRPA1 в контакт с терапевтически эффективным количеством соединения по любому из пп. 1-29 или его фармацевтически приемлемой соли.
| база данных REGISTRY CAS RN 1309344-12-4 и 1309341-59-0 введены 14.06.2011 | |||
| Устройство для пуска колебательного электропривода | 1985 |
|
SU1309229A1 |
| Аэрозольная установка для лечения и профилактики животных | 1982 |
|
SU1069804A1 |
| Устройство для сообщения между разными пунктами с помощью электрических ламп | 1929 |
|
SU23857A1 |
| WO 2017060488 A1, 13.04.2017 | |||
| LAURIE B | |||
| SCHENKEL ET AL, | |||

