Область техники
Настоящее изобретение относится к аза-кольцевому соединению с внутренним мостиком, которое используют в качестве активного ингредиента фармацевтической композиции, в частности фармацевтической композиции для лечения воспалительных заболеваний.
Предпосылки создания изобретения
Ацетилхолин, высвобождаемый из холинергического нерва в периферической и центральной нервной системе, вызывает различные биологические реакции посредством связывания с двумя типами ацетилхолиновых рецепторов, никотиновым рецептором и мускариновым рецептором, соответственно. Из них мускариновый рецептор принадлежит к суперсемейству семиканальных трансмембранных, конъюгированных с G белком рецепторов, и в настоящее время существует пять подтипов этих рецепторов, M1, M2, M3, M4 и M5, каждый из которых кодируется различными последовательностями генов. Эти пять типов рецепторов широко распространены в каждой ткани организма позвоночных. Известно, что мускариновый рецептор обладает как возбуждающим, так и ингибирующим действием, в зависимости от его подтипа. В частности, функциональные роли различных мускариновых рецепторов, например участие рецептора M3, присутствующего в гладкой мышце дыхательных путей, в реакциях сокращения гладкой мускулатуры или другие роли, были описаны в General Overview of Caulfield, et al. (Непатентный документ 1).
Что касается легких, мускариновый рецептор присутствует в гладких мышцах трахей и бронхов, подслизистых железах и парасимпатическом узле. Установлено, что наибольшая плотность распространения мускариновых рецепторов в парасимпатическом узле, а затем в подслизистых железах и гладких мышцах трахей, в указанном порядке, а наименьшая в гладких мышцах бронхов (Непатентный документ 2).
В качестве мускаринового рецептора, который играет важную роль в ткани легких, можно указать три типа M1, M2 и M3. Рецептор M3, который присутствует в гладких мышцах дыхательных путей, участвует в сокращении гладкой мышцы, которое вызывает обструкцию дыхательных путей. Если рецептор M3 активирован, фосфолипаза C в цитоплазме активируется через активацию стимулирующего G-белка, далее происходит диссоциация фосфатидилинозит 3-фосфата в фосфатидилинозитол 4,5-дифосфат и, наконец, происходит фосфорилирование сократительного белка. Рецептор M3 присутствует в подслизистых железах, а также в гладкой мышце, которая присутствует в легочной ткани. Если этот тип рецептора M3 активируется, происходит выделение слизи.
Рецепторы M2 составляют около 50-80% холинергических рецепторов, которые присутствуют в дыхательных путях гладкой мускулатуры. Подробности, касающиеся роли этого подтипа рецептора, все еще не выяснены, но полагают, что снижение количества cAMP, продуцируемого в цитоплазме, ингибирует релаксацию гладкой мускулатуры дыхательных путей из-за симпатической иннервации. Центральные рецепторы M2 распространены в постганглионарных парасимпатических волокнах. В физиологических условиях центральный рецептор M2 играет роль в отрицательной регуляции высвобождения ацетилхолина из парасимпатической ткани. Рецептор M2 экспрессируется в сердечной мышце, что происходит в результате регуляции хронотропного действия. Рецептор M1 обнаружен в парасимпатическом ганглии легочной ткани, и он осуществляет функцию, способствующую нейротрансмиссии. Рецепторы M1 распространены не только в ганглии, но также в периферической легочной паренхиматозной ткани, но их функции не выяснены.
В легочных тканях аномальная функция мускаринового рецептора ощущается при формировании множества патологических условий. В частности, для воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные, продолжительная воспалительная реакция приводит к дисфункции ингибирующего рецептора M2, который присутствует в парасимпатическом нерве, и повышает высвобождение ацетилхолина путем стимуляции блуждающего нерва (Непатентный документ 3). Таким образом, дисфункция этого рецептора обуславливает относительное преимущество функции, опосредованной рецептором M3, что приводит к индукции гиперактивности дыхательных путей. В этой связи считают, что лекарственное средство, которое селективно антагонизирует функцию, опосредованную рецептором M3, не влияя при этом на функцию, опосредованную рецептором M2, является эффективным терапевтическим средством.
ХОЗЛ, при котором наблюдается ограничение проходимости дыхательных путей, обычно в результате органического изменения на фоне длительного воспаления в периферических дыхательных путях и, прежде всего, альвеолах, что приводит к появлению симптомов кашля, мокроты, одышки и подобных, занимает четвертое место среди основных причин смертности в 2005 году и является основной причиной смертности во всем мире. Кроме того, ожидают, что к 2020 году это будет третья из основных причин смертности. Курение является основным фактором риска ХОЗЛ, и в последнее время, помимо этого, загрязнение воздуха из-за пыли или подобные факторы также указывают в качестве факторов риска, среди прочих. Стоимость медицинского лечения ХОЗЛ очень высока, и ожидается увеличение числа пациентов в будущем.
Терапия с антихолинергическим средством для ингаляции рассматривается как лекарственное средство, в первую очередь, выбираемое для лечения описанных выше заболеваний (Непатентный документ 4), и в последние годы, в каждом регионе в странах Запада и в Азии долгодействующее антихолинергическое средство, тиотропийбромид (Spiriva (зарегистрированный товарный знак)), стал поступать на рынок. Однако, принимая во внимание статус лечения, ни одно из обычных антихолинергических средств, включая Spiriva (зарегистрированный товарный знак), не являются полноценными с точки зрения удобства и безопасности, и, таким образом, на данный момент существует пространство для совершенствования. Таким образом, весьма желательно создание антихолинергического средства для перорального введения или ингаляций, усовершенствованного с любой из указанных выше точек зрения.
Например, известно карбаматное соединение, которое обладает антагонистическим действием в отношении рецептора M3 и обладает ингибирующим действием на сокращение дыхательных путей, в котором кольцо А представленной ниже формулы представляет собой незамещенный бензол или незамещенный пиридин (Патентный документ 1). В этом патентном документе не раскрывается и не предлагается соединение по настоящему изобретению.
Химическая формула 1
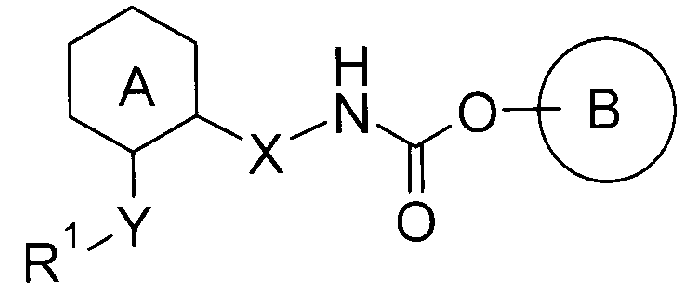
[значения символов в формуле смотри в этой публикации]
Далее известно карбаматное соединение, представленное ниже, которое обладает антагонистическим действием в отношении рецептора M3 и обладает ингибирующим действием на сокращение дыхательных путей (Патентный документ 2). Однако в этом патентном документе не раскрывается и не предлагается соединение по настоящему изобретению.
Химическая формула 2
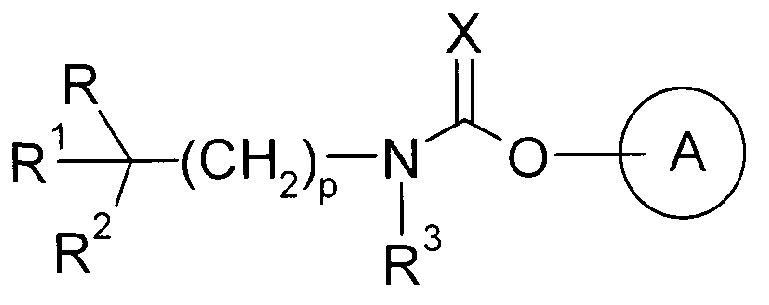
[значения символов в формуле смотри в этой публикации]
[Патентный документ 1] Издание международной публикации № WO 95/21820
[Патентный документ 2] Издание международной публикации № WO 95/06635
[Непатентный документ 1] Pharmacology and Therapeutics, 1993, vol. 58, pp. 319-379
[Непатентный документ 2] American Journal Respiratory and Critical Care Medicine, 1998, 158, pp. 154S-160S
[Непатентный документ 3] Life Science, 1999, vol. 64(6-7), pp. 449-455
[Непатентный документ 4] American Journal Respiratory and Critical Care Medicine, 2001, 163, pp. 1256-1276
Раскрытие изобретения
Задача, решаемая настоящим изобретением
Представлено соединение, которое является полезным в качестве активного ингредиента фармацевтической композиции, в частности фармацевтической композиции для лечения воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные.
Средства решения задачи
Авторы настоящего изобретения провели всесторонние исследования соединения, обладающего антагонистическим действием на мускариновый рецептор M3, и в результате было обнаружено, что аза-кольцевое соединение с внутренним мостиком является полезным для антагонистического действия на мускариновый рецептор M3, таким образом, было создано настоящее изобретение.
В частности, настоящее изобретение относится к соединению формулы (I) или его соли и фармацевтической композиции, включающей соединение формулы (I) или его соль и эксципиент.
Химическая формула 3
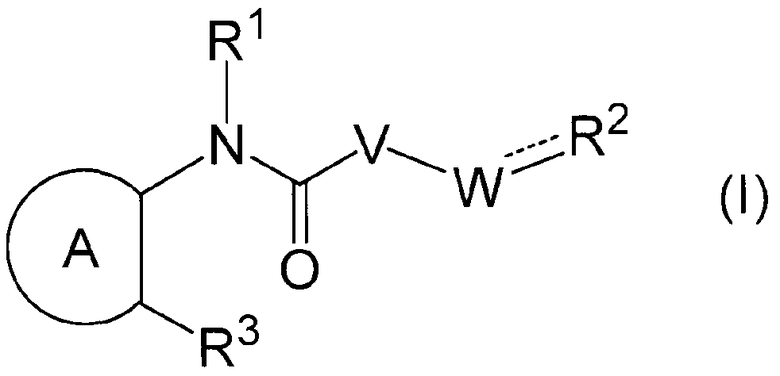
R1 представляет собой -H или C1-6 алкил;
R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, включающей формулы (a), (b), (c) и (d):
Химическая формула 4
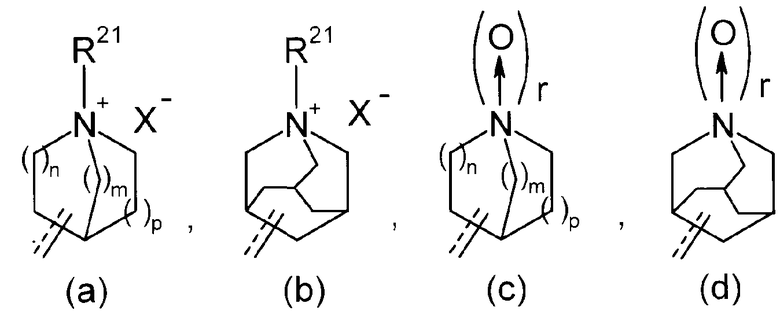
где кольцевой атом углерода в аза-кольце с внутренним мостиком может быть замещенным одной или несколькими группами R22;
m, n и p имеют значение, соответственно, 1 или 2;
r имеет значение 0 или 1;
R21 представляет собой C1-6 алкил, -C1-6 алкил-O-арил или -C1-6 алкил-арил;
R22 представляет собой -C1-6 алкил-циклоалкил или -C1-6 алкил-арил;
R3 представляет собой тиенил, фенил, пиридил, пиразинил, тиазолил или пиразолил, каждый из которых может быть замещен одним или несколькими R31;
где R31 представляет собой галоген, -OH, -CN, -CF3, C1-6 алкил или -O-C1-6 алкил;
кольцо А представляет собой ароматическое углеводородное кольцо, гетерокольцо или циклоалкан,
каждый из которых может быть замещен группой RA;
где RA представляет собой галоген, -CN, -NH2, C1-6 алкил, -O-C1-6 алкил, -CONH2, -NH-C1-6 алкил, -NH-C1-6 алкил-O-C1-6 алкил-арил, -NH-C1-6 алкил-арил или -NH-C1-6 алкил-OH,
где C1-6 алкил может быть замещен одним или несколькими атомами галогена;
V представляет собой -NH- или -O-;
W представляет собой -(CH2)q- или -(CH2)s-CH = ;
q имеет значение 0, 1 или 2;
s имеет значение 1 или 2;
X- представляет собой противоанион; и
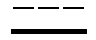 представляет собой простую связь или двойную связь;
представляет собой простую связь или двойную связь;
при условии, что в случае, когда кольцо А представляет собой замещенный бензол, R3 представляет собой фенил, пиридил, пиразинил, тиазолил или пиразолил, каждый из которых может быть замещен одним или несколькими R31; и
в случае, когда кольцо А представляет собой незамещенный бензол, q имеет значение 1 или 2; и
в случае, когда кольцо А представляет собой циклоалкан, R3 представляет собой фенил, который может быть замещен одним или несколькими R31.
Более того, если не указано иное, в случае, когда символы в любых формулах в настоящем описании также использованы в других формулах, одни и те же символы имеют одинаковые значения.
Настоящее изобретение относится к фармацевтической композиции для лечения воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные, которая включает соединение формулы (I) или его соль, то есть терапевтическому средству для лечения воспалительного заболевания, такого как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные, которое включает соединение формулы (I) или его соль.
Далее, настоящее изобретение относится к применению соединения формулы (I) или его соли для получения фармацевтической композиции для лечения воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные, и к способу лечения воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные, включающему введение пациенту эффективного количества соединения формулы (I) или его соли.
Эффект настоящего изобретения
Соединение формулы (I) или его соль обладает антагонистическим действием на связывание мускаринового рецептора M3 и, в связи с этим, может быть использовано в качестве профилактического и/или терапевтического средства для лечения воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные.
Лучший способ осуществления настоящего изобретения
Далее в настоящей заявке, настоящее изобретение будет описано подробно.
В настоящем описании “галоген” относится к F, Cl, Br или I.
В настоящем описании “C1-6 алкил” относится к алкилу с прямой или разветвленной цепью, содержащему 1-6 атомов углерода, например, метилу, этилу, н-пропилу, изопропилу, н-бутилу изобутилу, втор-бутилу, трет-бутилу, н-пентилу, н-гексилу или подобным. В другом варианте воплощения он представляет собой C1-4 алкил. В следующем варианте воплощения он представляет собой метил, этил или изопропил.
В настоящем описании “-C1-6 алкил-” относится к, в случаях, когда он имеет приставку и суффикс с дефисом, C1-6 алкилену с прямой или разветвленной цепью, например, метилену, этилену, триметилену, тетраметилену, пентаметилену, гексаметилену, пропилену, метилметилену, этилэтилену, 1,2-диметилэтилену, 1,1,2,2-тетраметилэтилену или подобным. В следующем варианте воплощения он представляет собой C1-4 алкилен и еще в одном варианте воплощения - метилен, этилен или триметилен.
В настоящем описании “ароматическое углеводородное кольцо” означает ароматическое углеводородное кольцо, содержащее 6-14 атомов углерода. В одном варианте воплощения его примеры включают бензол, нафталин и подобные, а в следующем варианте воплощения - бензол.
В настоящем описании “гетерокольцо” означает 5-6-членное ароматическое гетерокольцо, содержащее один или несколько гетероатомов, которые являются одинаковыми или отличными друг от друга, выбранных из группы, включающей азот, кислород и серу, которое может быть конденсировано с циклоалкильным кольцом или бензольным кольцом. В частности, его примеры включают пиридин, пиразин, пиримидин, пиридазин, пиразол, имидазол, оксазол, тиазол, тиофен, фуран, оксадиазол, изотиазол, изооксазол, тиадиазол, хинолин, изохинолин, бензотиазол, бензотиофен, бензоксазол, индол, индазол, циклопентатиофен и подобные. В следующем варианте воплощения гетерокольцо представляет собой 5-членное гетерокольцо, и еще в одном варианте воплощения его примеры включают оксазол, тиофен, тиазол, тиадиазол и циклопентатиофен. Еще в одном варианте воплощения его примеры включают тиофен и тиазол.
В настоящем описании “циклоалкан” означает C3-9 неароматическое углеродное кольцо, которое может содержать частично ненасыщенную связь и может быть конденсировано с бензольным кольцом. Кроме того, он также включает кольцо с внутренним мостиком. Следовательно, его конкретные примеры включают циклопропан, циклобутан, циклогексан, циклогептан, циклооктан, циклобутен, циклогексен, циклооктадиен, норборан, борнен, индан, тетрагидронафталин и подобные, а еще в одном варианте воплощения - циклогексан.
Далее, примеры противоаниона “X-” включают галогенидный ион, трифторметансульфонат, пара-толуолсульфонат, метансульфонат и подобные, а в следующем варианте воплощения он представляет собой предпочтительно галогенидный ион (например, хлоридный ион, бромидный ион и йодидный ион), но не ограничивается этим. Далее, в следующем варианте воплощения его примеры также включают неорганические анионы, такие как ион азотной кислоты, ион фосфорной кислоты, ион карбоновой кислоты и подобные, карбоксилаты, такие как формиат (HCOO-), ацетат (CH3COO-), пропионат, оксалат и подобные, и анионы аминокислот, например глутаминовой кислоты и т.п.
“Анионообменник” относится к другому соединению “X-”.
В настоящем описании выражение “может быть замещен” означает, что группа является незамещенной, или оно имеет от 1 до 5 заместителей. Более того, в случае нескольких заместителей эти заместители могут быть как одинаковыми, так и отличными друг от друга.
В настоящем описании заместитель, для которого приемлемы выражения “может быть замещен” или “является замещенным”, может быть любым заместителем, который обычно используют в качестве заместителя для каждой группы.
Например, примеры заместителя в кольце A, для которого приемлемы выражения “может быть замещен” или “является замещенным”, включают галоген, -CN, -NH2, C1-6 алкил, -O-C1-6 алкил, -CONH2, -NH-C1-6 алкил, -NH-C1-6 алкил-O-C1-6 алкил-арил, -NH-C1-6 алкил-арил и -NH-C1-6 алкил-OH.
Вариант воплощения [1] в соответствии с настоящим изобретением представляет собой следующий. В отношении формулы (I),
(1) соединение, в котором R1 представляет собой -H,
(2) соединение, в котором R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, состоящей из формул (a), (b), (c) и (d):
Химическая формула 5
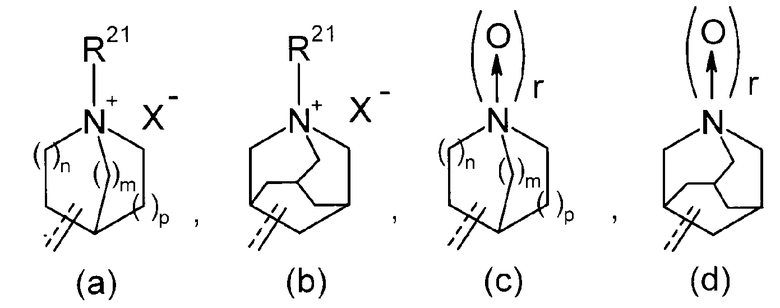
(3) соединение, в котором r имеет значение 0,
(4) соединение, в котором X представляет собой галоген,
(5) соединение, в котором R3 представляет собой фенил, который может быть замещен одним или несколькими R31, и
R31 представляет собой галоген, -OH, -CN, -CF3, -C1-6 алкил или -O-C1-6 алкил,
(6) соединение, в котором кольцо А представляет собой гетерокольцо или циклоалкил,
каждый из которых может быть замещен группой, выбранной из группы, состоящей из одного или нескольких RA,
(7) соединение, в котором V представляет собой O-,
(8) соединение, в котором W представляет собой -(CH2)q- и q имеет значение 0 или 1,
(9) соединение, в котором представляет собой простую связь,
(10) соединение, в котором m, n и p имеют значение, соответственно, 1 или 2, и
(11) соединение, которое представляет собой комбинацию двух или более из указанных выше (1)-(10), или его соль.
Другой вариант воплощения [2] настоящего изобретения представляет собой следующий.
Соединение или его соль согласно варианту воплощения [1], в котором
кольцо А представляет собой гетерокольцо или циклоалкан,
каждый из которых может быть замещен группой, выбранной из группы, состоящей из одного или нескольких RA1;
где RA1 представляет собой галоген, -CN, -NH2, C1-6 алкил, -O-C1-6 алкил, -CONH2, -NH-C1-6 алкил, -NH-C1-6 алкил-O-C1-6 алкил-фенил, -NH-C1-6 алкил-фенил или -NH-C1-6 алкил-OH,
где C1-6 алкил может быть замещен галогеном.
Другой вариант воплощения [3] настоящего изобретения представляет собой следующий.
Соединение или его соль согласно варианту воплощения [2], в котором
кольцо А представляет собой группу, включающую тиофен, тиазол, изотиазол, тиадиазол, оксазол, изооксазол, циклогексан, норборан, бензотиофен и 5,6-дигидро-4H-циклопентатиофен, каждый из которых может быть замещен группой, выбранной из группы, состоящей из одного или нескольких RA1.
Следующий вариант воплощения [4] настоящего изобретения представляет собой следующий.
Соединение или его соль согласно варианту воплощения [3], в котором
кольцо А представляет собой группу, включающую тиофен, тиазол и циклогексан,
каждый из которых может быть замещен группой, выбранной из группы, состоящей из одного или нескольких RA1.
Следующий вариант воплощения [5] настоящего изобретения представляет собой следующий.
Соединение или его соль согласно варианту воплощения [4], в котором R1 представляет собой -H.
Следующий вариант воплощения [6] настоящего изобретения представляет собой следующий.
Соединение или его соль согласно варианту воплощения [5], в котором
R3 представляет собой фенил, который может быть замещен одним или несколькими R31,
и R31 представляет собой галоген, -OH, -CN, -CF3, -C1-6 алкил или -O-C1-6 алкил.
Следующий вариант воплощения [7] настоящего изобретения представляет собой следующий.
Соединение или его соль согласно варианту воплощения [6], в котором
R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, состоящей из формул (a), (b), (c) и (d), где в случае (a) или (c), (m, n, p) имеет значение (2, 1, 1), (1, 1, 2) или (2, 1, 2) для каждой последовательности.
Следующий вариант воплощения [8] настоящего изобретения представляет собой следующий.
Соединение или его соль согласно варианту воплощения [7], в котором
R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, состоящей из формул (a) и (b), и
R21 представляет собой C1-6 алкил, -C1-6 алкил-O-фенил или -C1-6 алкил-фенил.
Примеры конкретных соединений, включенных в настоящее изобретение, включают следующие соединения или их свободные основания.
(1) 1-азабицикло[2.2.2]окт-4-илметил(5-фенил-1,3-тиазол-4-ил)карбамат, гидрохлорид,
(2) 1-азабицикло[3.2.2]нон-5-ил(5-фенил-1,3-тиазол-4-ил)карбамат, гидрохлорид,
(3) (3R)-1-азабицикло[2.2.2]окт-3-ил[(1R,2S)-2-фенилциклогексил]карбамат, гидрохлорид,
(4) 1-азатрицикло[3.3.1.1-3,7-]дец-4-ил(5-фенил-1,3-тиазол-4-ил)карбамат, гидрохлорид,
(5) 1-азабицикло[2.2.2]окт-3-илметил(2-фенил-3-тиенил)карбамат, гидрохлорид,
(6) 1-азабицикло[2.2.2]окт-4-ил[5-(4-фторфенил)-1,3-тиазол-4-ил]карбамат, гидрохлорид,
(7) 1-азабицикло[3.2.1]окт-6-илметил(5-фенил-1,3-тиазол-4-ил)карбамат, гидрохлорид или
(8) 1-азабицикло[2.2.2]окт-3-илметил[5-(4-фторфенил)-1,3-тиазол-4-ил]карбамат, фумарат.
Примеры конкретных соединений, включенных в настоящее изобретение, включают следующие соединения или их анионообменники.
(1) 4-({[5-(3-хлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанйодид,
(2) 1-(3-фенилпропил)-3-({[(2-фенил-3-тиенил)карбамоил]окси}метил)-1-азониабицикло[2.2.2]октанбромид,
(3) 1-(2-фенилэтил)-4-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[2.2.2]октанбромид,
(4) 1-(2-феноксиэтил)-4-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[2.2.2]октанбромид,
(5) 4-({[5-(2,5-дифторфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанйодид,
(6) 1-метил-5-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[3.2.2]нонанйодид,
(7) 4-({[5-(3-хлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-(2-феноксиэтил)-1-азониабицикло[2.2.2]октанйодид,
(8) 4-({[5-(3-хлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанбромид,
(9) 4-({[5-(3,5-дихлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанйодид,
(10) 4-({[5-(2,5-дифторфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанбромид,
(11) 4-({[5-(2,4-дифторфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанбромид,
(12) 1-метил-4-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[2.2.2]октанбромид,
(13) 4-({[5-(3,5-дихлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанбромид или
(14) 1-метил-5-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[3.2.2]нонанбромид.
Соединение формулы (I) может существовать в форме геометрических изомеров, в зависимости от типов заместителей. В настоящем описании соединение формулы (I) может быть описано только в одной форме изомера, но настоящее изобретение включает другие изомеры, выделенные формы изомеров или их смеси.
Далее, соединение формулы (I) может содержать асимметричный атом углерода и, соответственно, может существовать в форме оптических изомеров. Настоящее изобретение включает выделенную форму этих оптических изомеров соединения формулы (I) или их смеси.
Более того, настоящее изобретение также включает фармацевтически приемлемое пролекарство соединения формулы (I). Фармацевтически приемлемое пролекарство относится к соединению, содержащему группу, которая может быть преобразована в аминогруппу, гидроксильную группу, карбоксильную группу и подобные, посредством сольволиза или в физиологических условиях. Примеры групп для образования пролекарства включают группы, описанные в "Prog. Med., 5, 2157-2161 (1985)" или "Iyakuhin no Kaihatsu (Development of Medicines) (Hirokawa Shoten, 1990), vol. 7, Bunshi Sekkei (Molecular Design)", 163-198.
Кроме того, соль соединения формулы (I) относится к фармацевтически приемлемой соли соединения формулы (I), а в некоторых случаях, к формам ее кислотно-аддитивной соли или соли с основанием, в зависимости от типа заместителей. В частности, их примеры включают кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные, и с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, миндальная кислота, винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, аспарагиновая кислота, глутаминовая кислота и подобные, и соли с различными аминокислотами и производными аминокислот, такими как ацетиллейцин и подобные.
Кроме того, настоящее изобретение также включает различные гидраты или сольваты и полиморфные кристаллические вещества соединения формулы (I) и его соли. Также, настоящее изобретение включает соединения, меченные различными радиоактивными или нерадиоактивными изотопами.
В настоящем описании используются следующие аббревиатуры.
BINAP: (R)-(+)- или (S)-(-)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил
CHCl3: хлороформ
Cs2CO3: карбонат цезия
CuCl2: хлорид меди
DCE: 1,2-дихлорэтан
ДХМ: дихлорметан
DEAD: диэтилазодикарбоксилат
DIBOC: ди-трет-бутилдикарбонат
DIPA: диизопропиламин
DIPEA: N,N-диизопропилэтиламин
DMA: диметилацетамид
DMAP: N,N-диметил-4-аминопиридин
ДМФА: N,N-диметилформамид
DMSO: диметилсульфоксид
DPPA: азид дифенилфосфорной кислоты
DPPF: дифенилфосфиноферроцен
EtOAc: этилацетат
EtOH: этанол
HCl/диоксан: раствор хлористый водород/диоксан
HCl/EtOAc: раствор хлористый водород/этилацетат
HCl/MeOH: раствор хлористый водород/метанол
IPE: диизопропиловый эфир
K2CO3: карбонат калия
KOH: гидроксид калия
LAH: литийалюмогидрид
LiBH4: боргидроксид лития
MCPBA: метахлор-пербензойная кислота
MEK: 2-бутанон
MeCN: ацетонитрил
MeOH: метанол
MgSO4: безводный сульфат магния
NBS: н-бромсукцинимид
NCS: н-хлорсукцинимид
NMP: н-метилпирролидон
Na2CO3: карбонат натрия
Na2SO4: безводный сульфат натрия
Na2SO4–10H2O: сульфат натрия, декаангидрид
NaH: гидрид натрия
NaHCO3: гидрокарбонат натрия
NaOAc: ацетат натрия
NaOBut: трет-бутоксид натрия
NaOEt: этоксид натрия
NaOH: гидроксид натрия
NaOMe: метоксид натрия
P(But)3: три(третбутил)фосфин
PPh3: трифенилфосфин
Pd(OAc)2: ацетат палладия
PdCl2(PPh3)2: дихлорбистрифенилфосфинпалладий
Pd2dba3: трис(дибензилиденацетон) дипалладий
Pd(PPh3)4: тетракис(трифенилфосфин)палладий (0)
rel: относительная конфигурация
Rt: время удерживания
TEA: триэтиламин
ТФУК: трифторуксусная кислота
ТГФ: тетрагидрофуран
насыщенный солевой раствор: насыщенный физиологический раствор
tBuOH: трет-бутанол
tBuOK: трет-бутоксид калия
Способы получения
Соединение формулы (I) и его соль можно получить, используя различные известные способы синтеза, с использованием характеристик на основании их основных скелетов или типов заместителей. На данный момент, в зависимости от типов функциональных групп, в некоторых случаях является эффективным, с точки зрения методов получения, замещение функциональной группы подходящей защитной группой (группой, которая может быть легко преобразована в функциональную группу), в ходе стадий от исходных веществ до промежуточных соединений. Примеры такой функциональной группы включают защитные группы, описанные в "Greene's Protective Groups in Organic Synthesis (4th Edition, 2006)", изданном P.G.M. Wuts and T.W. Greene, которые можно подходящим образом выбрать и использовать в зависимости от условий реакций. В этих способах требуемое соединение можно получить путем введения защитной группы для осуществления реакции и затем, если требуется, удаления защитной группы.
Кроме того, пролекарство соединения формулы (I) можно получить путем введения конкретной группы в ходе стадий от исходных веществ до промежуточных соединений, таким же способом, как указано выше в отношении защитных групп, или путем дальнейшего осуществления реакции, используя полученное соединение формулы (I). Реакцию можно осуществить с использованием способа, хорошо известного специалистам в данной области, такого как хорошо известные этерификация, амидирование, дегидрирование и подобные.
Далее в настоящей заявке описаны типичные способы получения соединения формулы (I). Более того, способы получения по настоящему изобретению не ограничиваются представленными ниже примерами.
Способ получения 1
Химическая формула 6
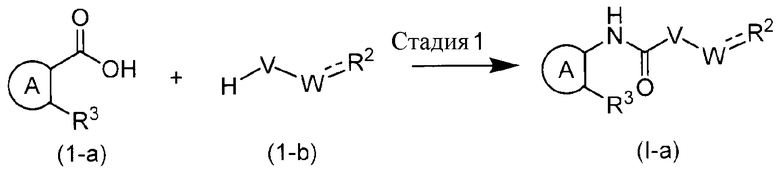
Этот способ получения представляет собой способ получения соединения формулы (I-a) с R1=H из соединения (1-a).
Стадия, указанная как Стадия 1, представляет собой реакцию, обеспечивающую взаимодействие карбоксильной группы соединения (1-a) с агентом азидирования, таким как DPPA, азид натрия и подобными, для образования карбаматной группы или мочевинной группы при помощи так называемой реакции перегруппировки Курциуса, которую предпочтительно осуществляют в присутствии основания. В качестве основания обычно используют TEA, пиридин или подобные, и реакцию можно осуществлять при комнатной температуре, в условиях от комнатной температуры до нагревания или в условиях нагревания при температуре кипения с обратным холодильником. Кроме того, получение также можно осуществлять способом, включающим получение через изоцианат, который получают из производного карбоновой кислоты, с использованием азотистоводородной кислоты в присутствии концентрированной серной кислоты.
Способ получения 2
Химическая формула 7

где Lv представляет собой удаляемую группу.
Этот способ получения является способом получения соединения формулы (I) с использованием соединения (2-a) в качестве исходного вещества.
Стадия 2 представляет собой реакцию, обеспечивающую взаимодействие соединения (2-a) с соединением (2-b), которую предпочтительно осуществляют в присутствии основания. В качестве основания обычно используют TEA, пиридин или подобные, и реакцию можно осуществлять при комнатной температуре, в условиях от комнатной температуры до нагревания или в условиях нагревания при температуре кипения с обратным холодильником. Здесь, примеры удаляемой группы включают галоген; метансульфонилокси, этансульфонилокси, бензолсульфонилокси, пара-толуолсульфонилокси, трифторметансульфонилокси и подобные.
Способ получения 3
Химическая формула 8
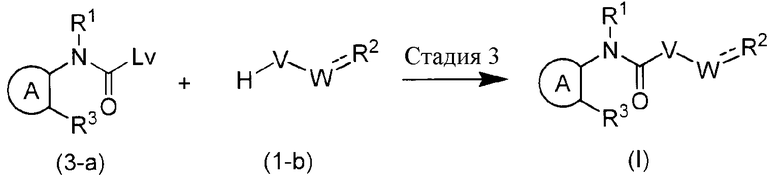
Этот способ получения является способом получения соединения формулы (I) из соединения (3-a).
Стадия, указанная как Стадия 3, представляет собой реакцию, обеспечивающую взаимодействие соединения (3-a) с соединением (1-b), которую предпочтительно осуществляют в присутствии основания (например, NaH, NaOMe, NaOEt, NaOH, KOH и подобных), что способствует лучшему осуществлению реакции. Здесь, примеры удаляемой группы Lv включают галоген, метокси, этокси, фенокси, пара-нитрофенокси и подобные.
Способ получения 4
Химическая формула 9
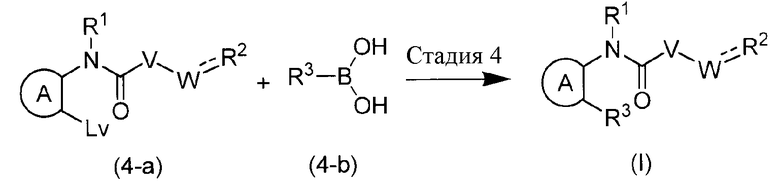
Эта реакция является способом получения соединения формулы (I) путем взаимодействия галогенированного арильного соединения (4-a) с соединением арилборной кислоты (4-b) и с использованием так называемой реакции сочетания Сузуки. Реакцию можно осуществлять без растворителя или в инертном для реакции растворителе, таком как ароматические углеводороды (например, бензол, толуол, ксилол и подобные), простые эфиры (например, ТГФ, диоксан и подобные); галогенированные углеводороды (например, ДХМ, DCE, CHCl3 и подобные); ДМФА, DMA, NMP; EtOAc, MeCN и подобные, при температуре от комнатной до температуры кипения с обратным холодильником. Реакцию осуществляют при одновременном присутствии палладия, фосфиновых лигандов и оснований металлов. В качестве палладия может быть использован дивалентный палладий, такой как Pd(OAc)2 и подобные, или палладий с нулевой валентностью, такой как Pd2dba3 и подобные. В качестве фосфинового лиганда может быть использован бидентатный лиганд, такой как BINAP, DPPF и подобные, монодентатный лиганд, такой как P(But)3 и подобные. В качестве основания металла может быть использован K2CO3, Cs2CO3, фосфат калия, NaOBut и подобные. Здесь, примеры удаляемой группы Lv включают галоген, трифторметансульфонилокси и подобные.
Способ получения 5
Химическая формула 10
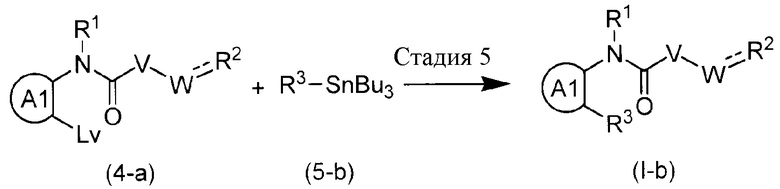
где A1 представляет ароматический углеводород или гетерокольцо.
Эта реакция является способом получения соединения формулы (I-b) путем взаимодействия соединения (4-a) с соединением олова (5-b) и с использованием так называемой реакции сочетания Стилле. Реакцию можно осуществлять без растворителя или в инертном для реакции растворителе, таком как ароматические углеводороды (например, бензол, толуол, ксилол и подобные), простые эфиры (например, ТГФ, диоксан и подобные); галогенированные углеводороды (например, ДХМ, DCE, CHCl3 и подобные); ДМФА, DMA, NMP; EtOAc, MeCN и подобные, при температуре от комнатной до температуры кипения с обратным холодильником. Реакцию осуществляют при одновременном присутствии палладия и фосфиновых лигандов. В качестве палладия может быть использован дивалентный палладий, такой как Pd(OAc)2 и подобные, или палладий с нулевой валентностью, такой как Pd2dba3 и подобные. В качестве фосфинового лиганда может быть использован бидентатный лиганд, такой как BINAP, DPPF и подобные, монодентатный лиганд, такой как P(But)3 и подобные. Здесь, примеры удаляемой группы включают галоген, трифторметансульфонилокси и подобные.
Способ получения 6
Химическая формула 11
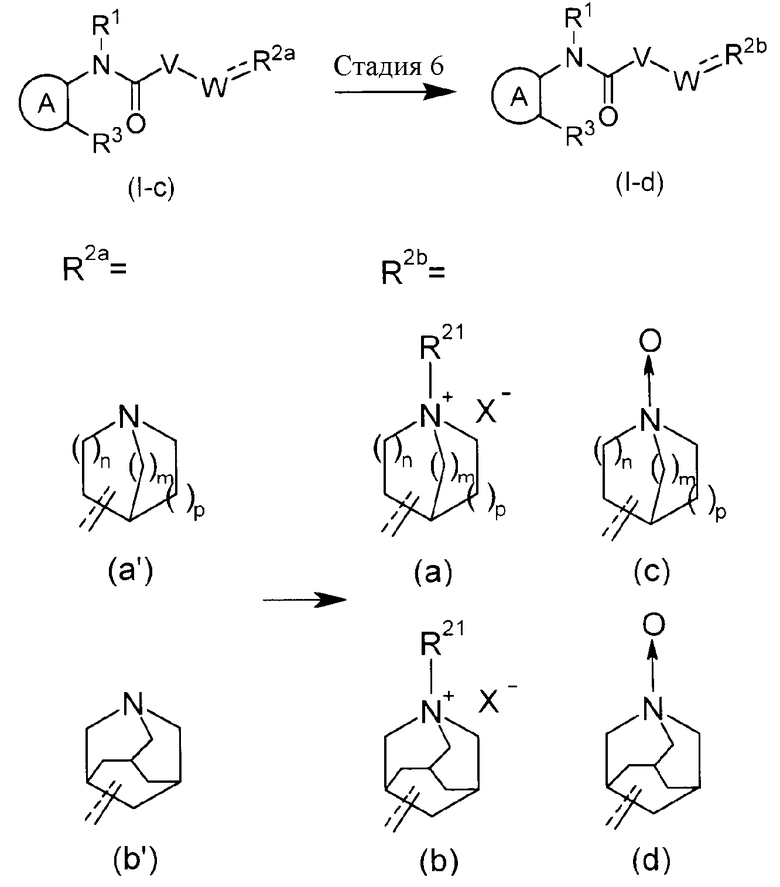
В соединении формулы (I), соединения (a, b), в которых атом азота аза-кольцевой группы с внутренним мостиком образует четвертичную аммониевую соль, или соединения (c, d), в которых указанный атом азота является окисленным, можно получить, подвергая третичное аминовое соединение (1-c) соединения по настоящему изобретению реакции N-алкилирования или N-оксиления, которые обычно осуществляют на конечной стадии.
Реакцию N-алкилирования можно осуществлять при помощи способа, традиционно используемого для реакции N-алкилирования, но в конкретном случае, ее осуществляют путем перемешивания соединения третичного амина по настоящему изобретению с соответствующим количеством алкилирующего агента в инертном растворителе, таком как ДМФА, CHCl3, ацетон, MEK, MeCN, ТГФ и подобные, при температуре от температуры охлаждения льдом до комнатной температуры или, в некотрых случаях, при нагревании до температуры кипения с обратным холодильником.
Примеры алкилирующего агента включают алкилгалогенид, C1-6 алкилтрифторметансульфонат, C1-6 алкил-пара-толуолсульфонат, C1-6 алкилметансульфонат и подобные.
Реакцию N-окисления можно осуществлять при помощи способа, традиционно используемого для реакции окисления, но в конкретном случае, ее осуществляют путем перемешивания соединения третичного амина по настоящему изобретению с соответствующим количеством или избыточным количеством окислителя в инерном растворителе, таком как CHCl3, ДХМ, DCE и подобные, спирты, такие как MeOH, EtOH и подобные, вода, или в смешанном растворителе из вышеперечисленных, от температуры охлаждения до комнатной температуры или, в некоторых случаях, с нагреванием при кипячении с обратным холодильником. Примеры окислителя включают органическую перкислоту, такую как MCPBA и подобные, перйодид натрия, пероксид водорода и подобные.
Способ получения 1 для промежуточного соединения
Химическая формула 12

где Hal представляет галоген и R7 представляет защитную группу карбоновой кислоты.
Соединение (1-a) Стадии 1 можно получить следующим способом.
Стадия 7-1 является способом получения соединения (7-b) с использованием соединения (7-a) в качестве исходного вещества. Галогенированный арил можно получить, обеспечив взаимодействие соли щелочного металла, образованной с азотной кислотой или азотным эфиром, с соединением (7-a) в кислотных условиях, с получением диазониевой соли ариламиносоединения, и добавляя ее в раствор, содержащий гидрогалогенид, в присутствии купритового катализатора. Реакцию можно осуществлять в растворителе, таком как серная кислота, уксусная кислота, фосфорная кислота, ацетон, MeCN, DMSO и подобные. В качестве галогенирующего агента можно использовать галогенированную медь или галогенированное железо, а в качестве куприта можно использовать CuCl2 или CuBr2. Относительно температуры реакции, реакцию осуществляют при температуре от охлаждения льдом до комнатной температуры, или от комнатной температуры до температуры кипения с обратным холодильником.
Стадии, указанные как Стадия 7-2 и Стадия 7-2', можно осуществлять с использованием способа, аналогичного Четвертому способу или Способу получения 5, описанным выше.
Стадия 7-3 представляет собой реакцию получения карбоновой кислоты, подвергая эфирную группу гидролизу или реакции удаления защиты, для чего можно применять условия реакции удаления защиты карбоксильной группы, такие как описанные в “Protective Groups in Organic Synthesis”, как указано выше. Примеры защитной группы включают C1-6 алкил, который может быть замещен; и -C(=O)-C1-6 алкил, который может быть замещен, и, в частности, включают метил, этил, бензил, аллил и трет-бутил.
Способ получения 2 для промежуточного соединения
Химическая формула 13

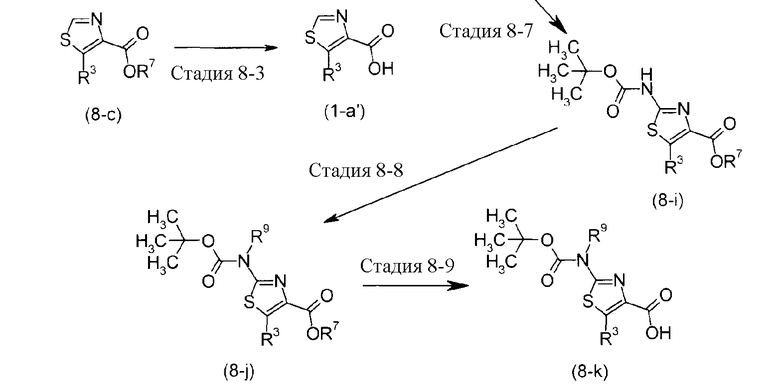
где R8 представляет C1-6 алкильную группу, которая может быть замещена галогеном, и R9 представляет C1-6 алкильную группу.
Кроме того, получение можно осуществить следующим способом в случае, когда кольцо A карбоновокислотного соединения (1-a), которое является исходным веществом Стадии 1 Способа получения 1, представляет собой тиазол.
Стадия 8-1 представляет собой способ получения деаминированного продукта (8-b) с использованием соединения (8-a) в качестве исходного вещества. Получение можно осуществить, осуществляя реакцию соли щелочного металла, образованной с азотной кислотой или сложными эфирами азотной кислоты, в кислотных условиях с получением соли диазония, с удалением азота. Реакцию можно осуществлять в растворителе, таком как серная кислота, уксусная кислота, фосфорная кислота, ацетон, MeCN, DMSO и подобные.
Стадия 8-2 представляет собой способ получения соединения (8-c) путем взаимодействия арилборной кислоты или соединения алкил-олова с соединением (8-b). Взаимодействие можно осуществлять способом, аналогичным Способам получения 4 и 5, описанным выше.
Стадия 8-3 представляет собой реакцию получения карбоновой кислоты соединения (1-a'), подвергая сложноэфирную группу гидролизу или реакции удаления защиты группы, и ее можно осуществить способом, аналогичным Стадии 7-3, описанным выше.
На Стадии 8-4 соединение формулы (8-e) или формулы (8-f) можно получить из соединения (8-d) и альдегида с использованием способа TSUBOI, et al. (Bulletin of Chemical Society of Japan., 1987, Vol. 60, p. 2475), и на Стадии 8-5 тиазольное соединение (8-g) или соединение (8-h) можно получить путем взаимодействия соединения (8-e) или соединения (8-f) с тиомочевиной или тиоамидом. Реакцию осуществляют в спирте или MeCN при нагревании.
Стадия 8-6 представляет собой способ получения деаминированного соединения (8-c) из 2-аминотиазольного соединения (8-g), и ее можно осуществить способом, аналогичным способу Стадии 8-1, описанному выше.
Стадия 8-7 представляет собой способ получения соединения (8-i) из соединения (8-g), содержащего 2-аминотиазольную группу, для чего могут быть применены условия для удаления защиты аминогруппы, как описано в указанном выше “Protective Groups in Organic Synthesis”.
Стадия 8-8 представляет собой способ получения N-алкил N-трет-бутоксикарбонильного продукта из трет-бутоксикарбонильного продукта (8-i) с использованием способа KIM, et al. (Synlett., 1999, pp. 1239).
Стадия 8-9 представляет собой реакцию получения карбоновокислотного соединения (8-k), которую осуществляют, подвергая сложноэфирную группу гидролизу или реакции удаления защиты группы, и ее можно осуществить способом, аналогичным Стадии 7-3, описанным выше.
Способ получения 3 для промежуточного соединения
Кроме того, гетероарилкарбаматное соединение (4-a), используемое на стадии Способов получения 4 и 5, в котором R1 представляет собой -H, может быть получено способом, аналогичным Способу получения 1.
Химическая формула 14

Более того, некоторые соединения формулы (I) можно получить из соединения по настоящему изобретению, полученного, как описано выше, с использованием любой комбинации хорошо известных способов, которые традиционно используют специалисты в данной области, такие как алкилирование, ацилирование, реакция замещения, окисления, восстановления, гидролиза, удаления защитной группы и подобные.
Соединение формулы (I) выделяют и очищают в виде его свободного соединения, соли, гидрата, сольвата или его полиморфного кристаллического вещества. Соль соединения формулы (I) также можно получить в соответствии с обычным способом, используемым для реакции образования соли.
Выделение и очистку осуществляют, используя общепринятые химические приемы, такие как экстракция, фракционированная кристаллизация, различные виды фракционной хроматографии и подобные.
Различные изомеры можно получить, отбирая подходящее исходное соединение, или путем разделения, используя разницу в физико-химических свойствах между изомерами. Например, оптический изомер можно получить методами основного оптического разделения рацемических продуктов (например, фракционированной кристаллизации для индукции диастереомеров с оптически активными основаниями или кислотами, хроматографии с использованием хиральной колонки и т.д., и подобных). Кроме того, изомеры также можно получить из подходящего оптически активного исходного вещества.
Фармакологическую активность соединений формулы (I) подтверждали при помощи следующего испытания.
Пример испытания 1: Испытание сродства с мускариновым рецептором (in vitro)
a. Приготовление образца мембраны
Брали подчелюстную железу/сердце и кору головного мозга самца крысы SD (Japan SLC, Inc.) и к этому добавляли 10-кратный объем 25 мМ Tris буфера (pH 7,4, далее в заявке обозначаемый как Tris буфер), содержащего 3,75 мМ хлорида магния, с последующей гомогенизацией при охлаждении льдом. Центрифугирование осуществляли при 1000 g, 4°C в течение 10 минут и затем осуществляли ультрацентрифугирование при 100000 g, 4°C в течение 30 минут. Полученный осадок суспендировали в Tris буфере и хранили при -80°C. После этого его расплавляли для использования и испытания.
b. Эксперимент связывания мускаринового рецептора
Образец мембраны любого из подчелюстной железы, сердца и коры головного мозга, [3H]-N-метилскополамин (N-метилскополамин) и испытываемое соединение инкубировали в 0,3 мл Tris буфере при 25°C в течение 2 часов и фильтровали с отсосом через стеклянный фильтр (Whatman GF/B) и фильтр промывали 8 раз при помощи 0,3 мл Tris буфера. Радиоактивность [3H]-N-метилскополамина, адсорбированного на фильтре, измеряли при помощи Top Count. Далее рецептор-специфическое связывание определяли путем добавления 1 мкМ N-метилскополамина. Сродство испытываемого соединения в отношении мускаринового рецептора определяли как константу диссоциации (Ki), которую рассчитывали из концентрации (IC50) испытываемого соединения, которая ингибирует связывание [3H]-N-метилскополамина в качестве меченного лиганда на 50%.
Как результат, для любого указанного соединения из соединений формулы (I) результаты экспериментов по определению антагонистической активности в отношении связывания с рецептором (показатели активности, значения Ki, нМ) представлены в Таблице 1.
a. Пример испытания 2) Испытание антагонизма в отношении мускаринового рецептора (in vivo)
a. Испытание сокращения дыхательных путей у крысы
Самцов крыс SD (250-400 г) анестизировали путем интраперитонеального введения пентобарбитала натрия (Nembutal; 50 мг/кг) и иссекали основные дыхательные пути. Дыхательный катетр вводили в трахею и давление в дыхательных путях измеряли при помощи датчика давления. После введения панкуронийбромида (0,2 мг/кг, внутривенно) получали стабильное давление в дыхательных путях перед началом эксперимента. Эксперимент осуществляли путем введения во внешнюю верхнечелюстную вену/введения в двенадцатиперстную кишку/перорального введения/введения в дыхательные пути физиологического раствора (в случае перорального введения/введения в двенадцатиперстную кишку - дистиллированной воды) или вводили испытываемое соединение и через 5-30 минут в случае внутривенного введения, через 0,25-6 часов в случае перорального введения в двенадцатиперстную кишку и через 0,25-72 часов в случае введения в дыхательные пути вводили внутривенно карбахол при дозе (объемной) 30 мкг/кг (1 мл/кг) и затем измеряли показатели давления в дыхательных путях, соответственно, в течение 5 минутного периода. Для испытываемого лекарственного средства измеряли показатель ингибирования карбахол-индуцированного повышения давления в дыхательных путях при введении физиологического раствора, и доза испытываемого соединения, которая ингибировала повышение на 50%, была взята в качестве величины ED50.
b. Испытание слюновыделения у крыс
Самцов крыс SD (250-400 г) анестизировали путем интраперитонеального введения пентобарбитала натрия (Nembutal; 50 мг/кг). Вводили испытываемое соединение (для контрольной группы вводили физиологический раствор) и через 5 минут вводили карбахол при дозе 30 мкг/кг (1 мл/кг). Введение лекарственного средства осуществляли таким же способом, как в описанном выше методе испытания, путем введения во внешнюю верхнечелюстную вену/введения в двенадцатиперстную кишку/перорального введения/введения в дыхательные пути. Выделяемую слюну в течение 5 минут непосредственно после введения карбахола извлекали при помощи ватных тампонов и измеряли ее вес. Определяли показатель ингибирования в отношении количества выделяемой слюны в контрольной группе, и доза испытываемого соединения, которая ингибировала количество выделяемой слюны в контрольной группе на 50%, была взята как значение ID50.
c. Испытание брадикардии у крыс
Самцов крыс SD (250-400 г) анестезировали путем интраперитонеального введения пентобарбитала натрия (50 мг/кг), шею разрезали и дыхательные пути укрепляли путем введения канюли в трахею. В условиях искусственного дыхания (10 мл/кг при 90 раз в минуту) отслеживали частоту сердечных сокращений по всей верхнечелюстной артерии. Вставляли канюлю во внешнюю верхнечелюстную вену, посредством чего осуществляли введение лекарственного средства. Осуществляли введение канюли в трахею и оставляли на 10 минут, после чего испытываемое соединение вводили внутривенно/вводили в двенадцатиперстную кишку/вводили перорально/вводили в дыхательные пути (что касается контрольной группы, вводили физиологический раствор в случае внутривенного введения/перорального введения и вводили дистиллированную воду в случае перорального введения в двенадцатиперстную кишку). Через 5-30 минут вводили карбахол при дозе 30 мкг/кг (1 мл/кг) и определяли эффект снижения частоты сердечных сокращений в контрольной группе в течение 5 минут после введения. Определяли показатель ингибирования в отношении снижения частоты сердечных сокращений, и доза испытываемого соединения, которая ингибировала снижение частоты сердечных сокращений в контрольной группе на 50%, была взята как значение ID50. Для любого указанного соединения из соединений формулы (I) результаты действия по ингибированию карбахол-индуцированного сокращения дыхательных путей представлены в Таблицах 2 и 3. (Таблица 2: Определение через 3 часа после перорального введения, и Таблица 3: Определение через 24 часа после введения в трахею 0,5 мкг/кг лекарственного средства, соответственно).
Пр.
(мкг/кг)
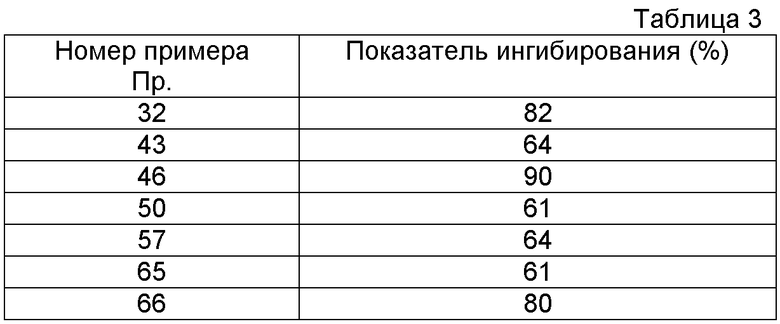
На основании приведенных выше результатов испытаний было подтверждено, что соединение формулы (I) обладает антагонистическим действием в отношении связывания мускаринового рецептора M3. Таким образом, его можно использовать для лечения респираторных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), хронический бронхит, астма, хроническая обструкция дыхательных путей, заболевание легочных волокон, эмфизема, ринит и подобные; заболеваний пищеварительной системы, таких как синдром раздраженной толстой кишки, спастический колит, язвы желудка и двенадцатиперстной кишки, желудочно-кишечные приступы или повышенная моторика, боли из-за дивертикулита и сокращение гладких мышц в пищеварительной системе и подобные; заболеваний мочевой системы, включающих дизурию, такую как недержание мочи, позывы на мочеиспускание, частое мочеиспускание и подобные, при таких заболеваниях, как нейрогенное частое мочеиспускание, нейрогенный мочевой пузырь, ночной энурез, нестабильность мочевого пузыря, спазмы мочевого пузыря, хронический цистит и подобные; заболеваний, связанных с образом жизни, таких как ожирение, диабет и подобные; болезни движения и подобных.
Фармацевтическую композицию, включающую один или два или более типов соединения формулы (I) или его соли в качестве активного ингредиента, можно получить в соответствии со способом, который обычно используют в данной области, с использованием эксципиента, который обычно используют в данной области, то есть фармацевтически приемлемого эксципиента, фармацевтически приемлемого растворимого носителя и т.п.
Введение можно осуществлять в любой форме перорального введения с использованием таблеток, пилюль, капсул, гранул, порошков, жидких препаратов и т.п., или парентерального введения путем инъекций, таких как внутрисуставная, внутривенная, внутримышечная, с использованием суппозиториев, глазных капель, глазных мазей, чрескожных жидких препаратов, мазей, чрескожных пластырей, жидких препаратов для введения через слизистую оболочку, пластырей для введения через слизистую оболочку, ингаляций и т.п.
Что касается твердых композиций для перорального введения, используют таблетки, порошки, гранулы и т.п. В таких твердых композициях один или два или более видов активных ингредиентов смешивают с, по меньшей мере, одним инертным эксципиентом, таким как лактоза, манит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон и/или алюмометасиликат магния и подобными. В соответствии с традиционным способом композиция может содержать инертные добавки, такие как смазывающее вещество, такое как стеарат магния, разрыхлитель, такой как натрий карбоксиметилкрахмал и подобные, стабилизатор и солюбилизирующее вещество. Если это необходимо, таблетки или пилюли могут иметь пленочное покрытие, сахарное покрытие, гастро- или энтеросолюбильное покрытие.
Жидкая композиция для перорального введения включает фармацевтически приемлемую эмульсию, растворимый жидкий препарат, суспензию, сироп, эликсир и т.п. и содержит традиционно используемый инертный разбавитель, например дистиллированную воду или EtOH. Помимо инертного разбавителя такая жидкая композиция может содержать вспомогательное вещество, такое как солюбилизирующее вещество, смачивающее вещество и суспендирующее вещество, подсластитель, отдушку, ароматизатор или антисептик.
Препараты для инъекций для парентерального введения включают водные или не-водные растворимые жидкие препараты, суспензии или эмульсии. Водный растворитель включает, например, дистиллированную воду для инъекций или физиологический раствор. Примеры неводного растворителя включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, спирты, такие как EtOH, Полисорбат 80 (Фармакопея Японии) и подобные. Такая композиция может также включать агент тоничности, антисептик, смачивающее вещество, эмульгатор, диспергирующее вещество, стабилизатор или солюбилизирующее вещество. Такие композиции стерилизуют, например, при помощи фильтрации через удерживающий бактерии фильтр, смешивания с бактерицидом, или облучения. Кроме того, их также можно использовать путем получения стерильной твердой композиции и ее растворения или суспендирования в стерильной воде или стерильном растворителе для инъекций перед применением.
Лекарственные средства для наружного применения включают мази, пластыри, кремы, желе, препараты для нанесения в виде пятна, спреи, лосьоны, глазные капли, глазные мази и подобные. Лекарственные средства содержат традиционно используемые основы для мази, основы для лосьона, водные или неводные растворимые жидкие препараты, суспензии, эмульсии и т.п. Примеры основы для мази или основы для лосьона включают полиэтиленгликоль, пропиленгликоль, белый вазелин, отбеленный пчелиный воск, полиоксиэтилен-гидрированное касторовое масло, глицерилмоностеарат, стеариловый спирт, цетиловый спирт, лауромакроголь, сорбитансесквиолеат и подобные.
Что касается средств для введения через слизистую оболочку, например ингаляций, средств для введения через нос и подобных, используют средства в форме твердого вещества, жидкости или в виде полутвердого вещества, и они могут быть получены в соответствии с традиционными способами. Например, можно добавить соответственно известный эксципиент, а также агент для регулирования pH, антисептик, поверхностно-активное вещество, смазывающее вещество, стабилизатор, загуститель или подобные вещества. Для их введения можно использовать подходящее устройство для ингаляции или вдувания. Например, соединение можно вводить отдельно или в виде порошкообразной сформулированной смеси, или в виде раствора или суспензии в сочетании с фармацевтически приемлемым носителем, с использованием традиционного устройства или распылителя, такого как устройство для ингаляции с отмеренными дозами. Ингалятор сухого порошка и т.п. может быть предназначен для разового или многократного введения, и можно использовать сухой порошок или содержащую порошок капсулу. Альтернативно, это может быть такая форма, как находящийся под давлением аэрозольный спрей, содержащий подходящий пропеллент, например подходящий газ, такой как хлорфторалкан, гидрофторалкан, диоксид углерода и подобные, или другие формы.
При пероральном введении суточная доза обычно составляет от около 0,001 до 100 мг/кг, предпочтительно от 0,1 до 30 мг/кг, и еще более предпочтительно от 0,1 до 10 мг/кг массы тела, вводимая в один прием или в виде 2-4 раздельных доз. В случае внутривенного введения подходящая суточная доза составляет от около 0,01 до 10 мг/кг массы тела, один раз в день или два или более раз в день. Кроме того, лекарственное средство в форме средства для введения через слизистую оболочку вводят при дозе от около 0,001 до 100 мг/кг массы тела, один раз в день или два или более раз в день. В случае введения путем ингаляции суточная доза составляет от около 0,1 до 100 мкг/кг массы тела, один раз в день или два или более раз в день. Дозу соответственно определяют для каждого конкретного случая с учетом симптомов, возраста, пола и подобных факторов.
Соединение формулы (I) можно использовать в комбинации с различными терапевтическими средствами или профилактическими средствами для лечения заболеваний, для которых соединение формулы (I), как считают, будет эффективным, как указано выше. Такие комбинации можно вводить одновременно или отдельно, и непрерывно или с желаемыми интервалами. Препараты для совместного введения могут быть в форме смеси или могут быть в виде отдельных препаратов.
Примеры
Далее в настоящей заявке более подробно описаны способы получения соединения формулы (I) со ссылкой на Примеры. Кроме того, настоящее изобретение не ограничено соединениями, описанными ниже в Примерах. Также, каждый из способов получения исходного соединения описан со ссылкой на Примеры получения. Более того, способы получения соединения формулы (I) не ограничены этими конкретными способами получения Примеров и, таким образом, соединение формулы (I) также можно получить путем сочетания таких способов получения или известным способом получения, который очевиден специалистам в данной области.
Соединения, представленные ниже в Таблицах, были получены при использовании такого способа получения, который очевиден специалистам в данной области или его модификации. Структуры и физико-химические данные этих соединений Примеров и способы их получения представлены в Таблицах. Также, символы в Таблицах имеют следующие значения.
Пр.: № Примера получения
Пр.: № Примера
Синтез: Способ получения (цифра показывает, что соединение Примера получали, используя такой же способ получения, как для соединения, обозначенного таким номером, как номер Примера).
Структура: Структурная формула
данные: Физико-химические данные. Например, ЯМР и MS являются следующими.
– ЯМР: Сигнал δ (м.д.) 1H-ЯМР с использованием DMSO-d6 в качестве растворителя для измерения
– ЯМР (CDCl3): Сигнал δ (м.д.) 1H-ЯМР, при использовании CDCl3 в качестве растворителя для измерения
– ESI (+): Величины, измеренные в положительном режиме
– ESI (-): Величины, измеренные в отрицательном режиме
Пример получения 1
При охлаждении льдом, к суспензии CuBr2 в MeCN добавляли трет-бутилнитрат. Далее к смеси порциями добавляли метил 2-амино-5,6-дигидро-4H-циклопента[b]тиофен-3-карбоксилат с последующим перемешиванием при охлаждении льдом в течение 2 часов и затем при комнатной температуре в течение 3 часов. К реакционной смеси добавляли хлористоводородную кислоту и водный слой экстрагировали при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN, YFLC WPrep2XY, гексан:EtOAc) с получением метил 2-бром-5,6-дигидро-4H-циклопента[b]тиофен-3-карбоксилата в виде бесцветной жидкости.
Соединения Примеров получения 1-1 - 1-2, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 1, используя соответствующие исходные вещества.
Пример получения 2
К раствору этил 2-амино-5-фенилтиазол-4-карбоксилата в MeCN добавляли CuCl2 при 0°C и к смеси по каплям медленно добавляли изоамилнитрат с последующим перемешиванием в течение 1 часа. К реакционной смеси добавляли CHCl3 с последующим перемешиванием при 0°C в течение 2 часов и затем при комнатной температуре в течение 2 часов. Реакционную смесь нейтрализовали путем добавления 1 M водного раствора NaOH и фильтровали через целит. Фильтрат экстрагировали при помощи EtOAc и органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором и затем сушили над MgSO4. После концентрирования при пониженном давлении полученный остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением этил 2-хлор-5-фенилтиазол-4-карбоксилата в виде желтого масла.
Пример получения 3
К суспензии метил 2-бром-5,6-дигидро-4H-циклопента[b]тиофен-3-карбоксилата, фенилбороновой кислоты и Pd(PPh3)4 в диоксане добавляли 2 M водный раствор Na2CO3 с последующим перемешиванием при 90°C в течение 15 часов. К смеси добавляли воду и водный слой экстрагировали при помощи EtOAc. Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением метил 2-фенил-5,6-дигидро-4H-циклопента[b]тиофен-3-карбоксилата в виде бесцветной жидкости.
Соединения Примеров получения 3-1 - 3-24, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 3, используя соответствующие исходные вещества.
Пример получения 4
К раствору метил 2-фенил-5,6-дигидро-4H-циклопента[b]тиофен-3-карбоксилата в EtOH добавляли 1 M водный раствор NaOH с последующим нагреванием при кипячении с обратным холодильником в течение 5 часов. Реакционную смесь доводили до pH 2 путем добавления 1 M раствора хлористоводородной кислоты и выпавшее в осадок твердое вещество собирали при помощи фильтрации и промывали водой с получением 2-фенил-5,6-дигидро-4H-циклопента[b]тиофен-3-карбоновой кислоты в виде твердого белого вещества.
Соединения Примеров получения 4-1 - 4-20, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 4, используя соответствующие исходные вещества.
Пример получения 5
К раствору этил 2-хлор-5-фенилтиазол-4-карбоксилата в ТГФ добавляли 1 M водный раствор KOH с последующим перемешиванием при 60°C в течение 30 минут. К реакционной смеси добавляли 1 M раствор хлористоводородной кислоты и выпавшее в осадок твердое вещество собирали при помощи фильтрации с получением 2-хлор-5-фенилтиазол-4-карбоновой кислоты в виде твердого белого вещества.
Соединения Примеров получения 5-1 - 5-23, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 5, используя соответствующие исходные вещества.
Пример получения 6
К раствору 2-фенил-5,6-дигидро-4H-циклопента[b]тиофен-3-карбоновой кислоты и TEA в толуоле добавляли раствор DPPA в толуоле с последующим перемешиванием при комнатной температуре в течение 40 минут и затем перемешиванием при 90°C в течение 60 минут. Кроме того, к смеси добавляли раствор (3R)-хинуклидинола в ДМФА с последующим нагреванием при кипячении с обратным холодильником в течение 15 часов. К реакционной жидкости добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Полученное маслянистое вещество очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением 2-фенил-5,6-дигидро-4H-циклопента[b]тиофен-3-амина в виде бледно-коричневого масла, но не требуемого (3R)-1-азабицикло[2.2.2]окт-3-ил(2-фенил-5,6-дигидро-4H-циклопента[b]тиен-3-ил)карбамата.
Пример получения 7
К раствору 2-бромтиофен-3-карбоновой кислоты и TEA в толуоле по каплям добавляли раствор DPPA в толуоле. Реакционную жидкость перемешивали при комнатной температуре в течение 40 минут и затем перемешивали при 90°C в течение 60 минут. К этому раствору добавляли раствор 3-хинуклидинола в ДМФА с последующим нагреванием при кипячении с обратным холодильником в течение 15 часов. К реакционной жидкости добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH) с получением 1-азабицикло[2.2.2]окт-3-ил(2-бром-3-тиенил)карбамата в виде бледно-коричневого твердого вещества.
Соединения Примеров получения 7-1 - 7-5, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 7, используя соответствующие исходные вещества.
Пример получения 8
К раствору 2-фенилтиофен-3-карбоновой кислоты и TEA в толуоле по каплям добавляли раствор DPPA в толуоле с последующим перемешиванием при комнатной температуре в течение 40 минут и затем перемешиванием при 90°C в течение 60 минут. К этому раствору добавляли tBuOH с последующим нагреванием при кипячении с обратным холодильником в течение 15 часов. К реакционному раствору добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением трет-бутил (2-фенил-3-тиенил)карбамата в виде бледно-коричневого масла.
Пример получения 9
К суспензии LAH в ТГФ добавляли раствор трет-бутил (2-фенил-3-тиенил)карбамата в ТГФ с последующим перемешиванием при комнатной температуре в течение 30 минут и перемешиванием при 70°C в течение 4 часов. После охлаждения при комнатной температуре к смеси добавляли Na2SO4–10H2O с последующим перемешиванием при комнатной температуре в течение 30 минут. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением 3-метиламино-2-фенилтиофена в виде бесцветного маслянистого вещества.
Пример получения 10
К смеси метил 2-хлоризоникотината, Pd(PPh3)4 и [(E)-2-фенилвинил]борной кислоты в диоксане добавляли 2 M водный раствор Na2CO3 с последующим перемешиванием при 90°C в течение 5 часов. К реакционной смеси добавляли воду и водный слой экстрагировали при помощи EtOAc. Органический слой сушили над MgSO4 и фильтрат затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением метил 2-[(E)-2-фенилвинил]изоникотината в виде бледно-желтого твердого вещества.
Пример получения 11
К раствору метил 2-[(E)-2-фенилвинил]изоникотината в хлористоводородной кислоте и MeOH добавляли оксид платины с последующим перемешиванием при комнатной температуре в течение 8 часов в атмосфере водорода при давлении 3 атм. Реакционную смесь фильтровали через целит и фильтрат затем концентрировали при пониженном давлении. К остатку добавляли водный раствор Na2CO3 и ксилол и водный слой экстрагировали при помощи CHCl3. Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении с получением метил 2-(2-циклогексилэтил)пиперидин-4-карбоксилата в виде бесцветного маслянистого вещества.
Пример получения 12
К раствору метил 2-[(E)-2-фенилвинил]изоникотината в уксусной кислоте добавляли оксид платины с последующим перемешиванием при комнатной температуре в течение 8 часов в атмосфере водорода при давлении 3 атм. Реакционную смесь фильтровали через целит и фильтрат затем концентрировали при пониженном давлении. К остатку добавляли водный раствор Na2CO3 и экстрагировали при помощи EtOAc. Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении с получением метил 2-(2-фенилэтил)пиперидин-4-карбоксилата в виде бесцветного масла.
Пример получения 13
К суспензии метил 2-(2-фенилэтил)пиперидин-4-карбоксилата в ксилоле добавляли этилбромацетат и K2CO3 с последующим нагреванием при кипячении с обратным холодильником в течение 8 часов. Реакционную смесь разбавляли при помощи EtOAc, органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем фильтровали. Фильтрат концентрировали при пониженном давлении. К остатку добавляли гексан и нерастворившиеся вещества удаляли при помощи фильтрации с последующим концентрированием при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением метил 1-(2-этокси-2-оксоэтил)-2-(2-фенилэтил)пиперидин-4-карбоксилата в виде бледно-коричневого масла.
Соединение Примера получения 13-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 13, используя соответствующее исходное вещество.
Пример получения 14
Суспензию tBuOK в толуоле нагревали при кипячении с обратным холодильником и к смеси по каплям добавляли раствор метил 1-(2-этокси-2-оксоэтил)-2-(2-фенилэтил)пиперидин-4-карбоксилата в толуоле с последующим перемешиванием при той же температуре в течение 15 часов. После охлаждения до комнатной температуры к смеси добавляли концентрированную хлористоводородную кислоту. Водный слой отделяли и органический слой экстрагировали при помощи концентрированной хлористоводородной кислоты. Слой хлористоводородной кислоты нагревали при кипячении с обратным холодильником в течение 15 часов. Реакционную смесь нейтрализовали путем добавления K2CO3 и водный слой экстрагировали при помощи CHCl3. Органический слой сушили и затем концентрировали при пониженном давлении. Полученный остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH) с получением 6-(2-фенилэтил)хинуклидин-3-она в виде бледно-коричневого масла.
Соединение Примера получения 14-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 14, используя соответствующее исходное вещество.
Пример получения 15
К суспензии LAH в диэтиловом эфире добавляли раствор 6-(2-фенилэтил)хинуклидин-3-она в ТГФ с последующим нагреванием при кипячении с обратным холодильником в течение 4 часов. К реакционной смеси добавляли Na2SO4–10H2O с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат затем концентрировали при пониженном давлении с получением 6-(2-фенилэтил)хинуклидин-3-ола в виде бесцветного масла.
Соединения Примеров получения 15-1 и 15-2, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 15, используя соответствующие исходные вещества.
Пример получения 16
К 1-азатрицикло[3.3.1.13,7]декан-4-карбонитрилу добавляли концентрированную хлористоводородную кислоту с последующим нагреванием при кипячении с обратным холодильником в течение 3 часов. Реакционную жидкость концентрировали при пониженном давлении с получением гидрохлорида 1-азатрицикло[3.3.1.13,7]декан-4-карбоновой кислоты в виде твердого вещества. Это вещество использовали на следующей стадии реакции без очистки.
Пример получения 17
К раствору гидрохлорида 1-азатрицикло[3.3.1.13,7]декан-4-карбоновой кислоты в EtOH добавляли концентрированную серную кислоту с последующим нагреванием при кипячении с обратным холодильником в течение 18 часов. Реакционную жидкость концентрировали при пониженном давлении и затем разбавляли при помощи EtOAc. Слой EtOAc в указанном порядке промывали водным раствором NaHCO3 и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением желтого маслянистого вещества. Это вещество очищали при помощи колоночной хроматографии на силикагеле (CHCl3:MeOH) с получением этил 1-азатрицикло[3.3.1.13,7]декан-4-карбоксилата в виде желтого твердого вещества.
Пример получения 18
К суспензии LAH в ТГФ по каплям добавляли раствор этил 1-азатрицикло[3.3.1.13,7]декан-4-карбоксилата в ТГФ при температуре от 0 до 5°C с последующим перемешиванием при той же температуре в течение 1 часа. К реакционной смеси при охлаждении льдом в указанном порядке добавляли воду, 15% водный раствор NaOH и затем воду. Нерастворившиеся вещества удаляли при помощи фильтрации через целит с последующим промыванием при помощи EtOAc. Фильтрат сушили над MgSO4 и затем концентрировали при пониженном давлении с получением 1-азатрицикло[3.3.1.13,7]декaн-4-илметанола в виде бесцветного маслянистого вещества.
Соединения Примеров получения 18-1 - 18-11, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 18, используя соответствующие исходные вещества.
Пример получения 19
К раствору метил 2-фенилтиофен-3-карбоксилата и NCS в CHCl3 добавляли перхлорную кислоту с последующим перемешиванием при 50°C в течение ночи. К реакционной смеси добавляли воду и водный слой экстрагировали при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Полученный остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением метил 5-хлор-2-фенилтиофен-3-карбоксилата в виде твердого белого вещества.
Пример получения 20
К 60% масляному раствору NaH в MeOH добавляли этил 2-хлор-5-фенилтиазол-4-карбоксилат с последующим перемешиванием при 70°C в течение 1 часа. К реакционной смеси добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH) с получением метил 2-метокси-5-фенилтиазол-4-карбоксилата в виде твердого белого вещества.
Пример получения 21
К раствору этил 2-метил-5-фенил-1,3-тиазол-4-карбоксилата в тетрахлориде углерода добавляли NBS с последующим нагреванием при кипячении с обратным холодильником. Реакционную жидкость фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc = от 8:1 до 6:1) с получением этил 2-(бромметил)-5-фенил-1,3-тиазол-4-карбоксилата (21a) и этил 2-(дибромметил)-5-фенил-1,3-тиазол-4-карбоксилата (21b) в виде желтого твердого вещества, соответственно.
Соединение Примера получения 21-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 21, используя соответствующее исходное вещество.
Пример получения 22
Суспензию этил 2-(бромметил)-5-фенил-1,3-тиазол-4-карбоксилата, полученную в Примере получения 21, и NaOAc в MeCN нагревали при кипячении с обратным холодильником. К реакционной смеси добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc = 4:1) с получением этил 2-(ацетоксиметил)-5-фенил-1,3-тиазол-4-карбоксилата в виде бледно-желтого твердого вещества.
Пример получения 23
К раствору этил 2-(ацетоксиметил)-5-фенил-1,3-тиазол-4-карбоксилата, полученного в Примере получения 22, в EtOH добавляли 1 M водный раствор NaOH с последующим перемешиванием при комнатной температуре. Реакционную жидкость нейтрализовали путем добавления 1 M раствора хлористоводородной кислоты и экстрагировали при помощи CHCl3. Органический слой промывали насыщенным солевым раствором, сушили и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc = 1:1) с получением этил 2-(гидроксиметил)-5-фенил-1,3-тиазол-4-карбоксилата в виде бледно-желтого твердого вещества.
Пример получения 24
К раствору этил 2-(гидроксиметил)-5-фенил-1,3-тиазол-4-карбоксилата, полученного в Примере получения 23, в ДХМ при 0°C добавляли трифторид бис(2-метоксиэтил)аминосеры (Деоксо-Фтор(R)) с последующим перемешиванием в течение 30 минут. Реакцию останавливали добавлением к реакционной смеси водного раствора NaHCO3 с последующей экстракцией путем добавления EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc) с получением этил 2-(фторметил)-5-фенил-1,3-тиазол-4-карбоксилата в виде бледно-желтого маслянистого вещества.
Соединения Примеров получения 24-1 и 24-2, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 24, используя альдегидные соединения Примеров получения 25 и 25-1, как описано ниже в качестве исходных веществ.
Пример получения 25
К раствору водного раствора дибром-продукта (21b), полученного в Примере получения 21, в EtOH добавляли водный раствор нитрата серебра с последующим нагреванием при кипячении с обратным холодильником в течение 15 минут. К реакционной жидкости добавляли 1 M раствор хлористоводородной кислоты, выпавшее в осадок твердое вещество удаляли при помощи фильтрации и фильтрат затем экстрагировали при помощи CHCl3. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc) с получением этил 2-формил-5-фенил-1,3-тиазол-4-карбоксилата в виде бледно-желтого твердого вещества.
Соединение Примера получения 25-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 25, используя соответствующее исходное вещество.
Пример получения 26
Раствор этил 3-хлор-2-оксо-3-фенилпропаноата и трифтортиоацетамида в MeCN нагревали при кипячении с обратным холодильником. К реакционной смеси добавляли водный раствор Na2CO3 с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан: EtOAc) с получением этил 5-фенил-2-(трифторметил)-1,3-тиазол-4-карбоксилата в виде твердого белого вещества.
Пример получения 27
К суспензии метил (2-бром-4-метилфенил)карбамата и хинуклидин-3-ола и MS4A в толуоле добавляли 60% масляный раствор NaH с последующим нагреванием при кипячении с обратным холодильником в течение 36 часов. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3: MeOH) с получением 1-азабицикло[2.2.2]окт-3-ил-2-бром-4-метилфенил)карбамата в виде твердого белого вещества.
Пример получения 28
К суспензии этил 5-бромтиазол-4-карбоксилата, 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола и Pd(PPh3)4 в диоксане добавляли 2 M водный раствор Na2CO3 с последующим нагреванием при кипячении с обратным холодильником в течение 4 часов. К реакционной смеси добавляли воду и водный слой экстрагировали при помощи EtOAc. Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи хроматографии на силикагеле (гексан:EtOAc) с получением этил 5-(1-метил-1H-пиразол-4-ил)-1,3-тиазол-4-карбоксилата в виде твердого коричневого вещества.
Пример получения 29
Натрий порциями добавляли к EtOH при 60°C при перемешивании. EtOH удаляли при помощи выпаривания и к смеси добавляли диэтиловый эфир, затем к смеси по каплям добавляли этил 2,2-дихлор-3-оксобутанат при 0°C. К реакционной смеси по каплям добавляли 2,6-дифторбензальдегид с последующим перемешиванием при 0°C в течение 30 минут и затем нагреванием при кипячении с обратным холодильником в течение 4 часов. К реакционной смеси добавляли смесь лед-вода с последующей нейтрализацией при помощи 2 M раствора HCl и экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc = от 85:15 до 6:4) с получением этил 2-хлор-3-(2,6-дифторфенил)оксиран-2-карбоксилата в виде желтого масла.
Пример получения 30
К раствору этил 2-хлор-3-(2,6-дифторфенил)оксиран-2-карбоксилата в EtOH добавляли тиомочевину с последующим нагреванием при кипячении с обратным холодильником в течение 4 часов. К реакционной смеси добавляли смесь лед-вода, с последующим добавлением K2CO3 для подщелачивания смеси. Выпавшее в осадок твердое вещество собирали при помощи фильтрации и промывали водой с получением этил 2-амино-5-(2,6-дифторфенил)-1,3-тиазол-4-карбоксилата в виде желтого твердого вещества.
Пример получения 31
К раствору этил 2-амино-5-(2,6-дифторфенил)-1,3-тиазол-4-карбоксилата в ТГФ медленно по каплям добавляли трет-бутилнитрата при нагревании при кипячении с обратным холодильником. После завершения добавления по каплям раствор продолжали перемешивать при той же температуре в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры с последующим добавлением воды и экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc = 9:1-7:3) с получением этил 5-(2,6-дифторфенил)-1,3-тиазол-4-карбоксилата в виде желтого маслянистого вещества.
Пример получения 32
К смешанному раствору этил 2-амино-5-фенил-1,3-тиазол-4-карбоксилата, уксусной кислоты и серной кислоты медленно добавляли водный раствор NaNO2 (5 мл) при 0°C с последующим перемешиванием при той же температуре в течение 1 часа. Этот раствор добавляли к водному раствору (40 мл), содержащему CuCN, NaCN и NaHCO3 (30 г), в течение 30 минут с последующим перемешиванием при той же температуре в течение 1 часа. К смеси добавляли EtOAc и нерастворившиеся вещества фильтровали через целит. Фильтрат экстрагировали при помощи EtOAc, органический слой сушили и растворитель удаляли путем выпаривания при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc = от 5:1 до 3:1 до 2:1) с получением этил 2-циано-5-фенил-1,3-тиазол-4-карбоксилата в виде желтого твердого вещества.
Пример получения 33
К раствору этил 2-циано-5-фенил-1,3-тиазол-4-карбоксилата в ТГФ добавляли 1 M водный раствор KOH с последующим перемешиванием при комнатной температуре в течение 12 часов. К реакционной смеси добавляли 1 M раствор хлористоводородной кислоты и выпавшее в осадок твердое вещество собирали при помощи фильтрации с получением 2-карбамоил-5-фенил-1,3-тиазол-4-карбоновой кислоты в виде бледно-желтого твердого вещества.
Пример получения 34
К раствору этил 5-бром-1,3-тиазол-4-карбоксилата в толуоле добавляли 2-(трибутилстаннил)-1,3-тиазол и PdCl2(PPh3)2 с последующим нагреванием при кипячении с обратным холодильником в течение 6 часов. К смеси добавляли воду и водный слой экстрагировали при помощи EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан: EtOAc) с получением этил 2,5'-бис-1,3-тиазол-4'-карбоксилата в виде желтого твердого вещества.
Соединение Примера получения 34-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 34, используя соответствующее исходное вещество.
Пример получения 35
К раствору этил 2-амино-5-фенилтиазол-4-карбоксилата в ТГФ добавляли DIBOC, DMAP и TEA с последующим перемешиванием при комнатной температуре. К реакционной смеси добавляли 1 M раствор хлористоводородной кислоты с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан: EtOAc) с получением этил 2-[(трет-бутоксикарбонил)амино]-5-фенил-1,3-тиазол-4-карбоксилата в виде желтовато-белого аморфного вещества.
Пример получения 36
К раствору этил 2-[(трет-бутоксикарбонил)амино]-5-фенил-1,3-тиазол-4-карбоксилата в ТГФ добавляли EtOH, PPh3 и DEAD с последующим перемешиванием при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением этил 2-[(трет-бутоксикарбонил)(этил)амино]-5-фенил-1,3-тиазол-4-карбоксилата в виде желтого масла.
Соединения Примеров получения 36-1 и 36-2, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 36, используя соответствующие исходные вещества.
Пример получения 37
К 60% маслянистой суспензии NaH в ТГФ при охлаждении льдом добавляли диэтилфосфоноэтилацетат. Реакционную смесь перемешивали в течение 30 минут при охлаждении льдом и затем при охлаждении льдом добавляли смесь гидрохлорида хинуклидин-3-она и ТГФ с последующим перемешиванием при комнатной температуре в течение 3 часов и затем перемешиванием при 60°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, с последующим добавлением воды и экстракцией при помощи EtOAc. Органический слой экстрагировали при помощи 1 M раствора хлористоводородной кислоты. Водный слой подщелачивали 6 M водным раствором NaOH и экстрагировали при помощи CHCl3. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением смеси этил (2E)-1-азабицикло[2.2.2]окт-3-илиденацетата и этил (2Z)-1-азабицикло[2.2.2]окт-3-илиденацетата в виде бесцветного масла.
Пример получения 38
К хинуклидин-4-илацетонитрилу (500 мг), который синтезировали способом, описанным в опубликованном японском переводе заявки PCT № 2001-521033, добавляли концентрированную хлористоводородную кислоту (3 мл) с последующим нагреванием при кипячении с обратным холодильником. Реакционную смесь концентрировали при пониженном давлении, добавляли толуол с последующим концентрированием при пониженном давлении. К остатку добавляли EtOH (10 мл) и концентрированную серную кислоту (1 мл) с последующим перемешиванием при 100°C. Реакционную смесь концентрировали при пониженном давлении, нейтрализовали путем добавления 1 M водного раствора NaOH и экстрагировали при помощи CHCl3. Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении с получением этил хинуклидин-4-илацетата (500 мг) в виде желтого маслянистого вещества.
Пример получения 39
К 4-метилбензолсульфонату 1-бензил-5-гидрокси-1-азониабицикло[3.2.1]октана (800 мг), который синтезировали способом, описанным в опубликованном японском переводе заявки PCT № 63-290878, добавляли смесь EtOH (8 мл), 10% Pd/углерод с 50% гидратацией (200 мг) и формиат аммония (500 мг) с последующим нагреванием при кипячении с обратным холодильником. Реакционную смесь подщелачивали добавлением 1 M водного раствора NaOH и экстрагировали смешанным растворителем (CHCl3:MeOH = 9:1). Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении с получением 1-азабицикло[3.2.1]октан-5-ола (180 мг) в виде бесцветного маслянистого вещества.
Соединение Примера получения 39-1, представленное ниже в Таблицах, получали способом, аналогичным описанному в Примере получения 39, используя вещество Примера получения 44, описанного далее, в качестве исходного вещества.
Пример получения 40
На бане со льдом к DIPA (4,1 мл) по каплям добавляли 1 M раствор н-бутиллития в гексане (10,1 мл). Реакционную смесь разбавляли при помощи диэтилового эфира (10 мл) с последующим перемешиванием в течение 20 минут. Далее реакционную смесь перемешивали при -78°C и к смеси по каплям добавляли раствор этил 1-бензилпиперидин-3-карбоксилата (6,0 г) в диэтиловом эфире (20 мл) с последующим перемешиванием при -50°C в течение 15 минут. Далее, после добавления при 70°C 1,3-дибромпропана (2,8 мл) смесь медленно возвращали к комнатной температуре. Затем смесь нагревали при кипячении с обратным холодильником в течение 30 минут. Реакционную смесь охлаждали, разбавляли водой и экстрагировали диэтиловым эфиром. Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан: EtOAc) с получением этил 1-бензил-(3-бромпропил)пиперидин-3-карбоксилата (1,08 г) в виде бесцветного маслянистого вещества.
Пример получения 41
Смесь этил 1-бензил-(3-бромпропил)пиперидин-3-карбоксилата (1,08 г), толуола (10 мл) и CHCl3 (2 мл) нагревали при кипячении с обратным холодильником в течение 2 часов. Эту смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли EtOAc и твердое вещество собирали при помощи фильтрации с получением 1-бензил-5-(этоксикарбонил)-1-азониабицикло[3.3.1]нонанбромида (942 мг) в виде бесцветного твердого вещества.
Пример получения 42
Смесь 1-бензил-5-(этоксикарбонил)-1-азониабицикло[3.3.1]нонанбромида (932 мг), 10% Pd/углерод с 50% гидратированием (25 мг) и EtOH (15 мл) перемешивали при комнатной температуре в атмосфере водорода при давлении 3 атм. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением бесцветного твердого вещества. К этому твердому веществу добавляли CHCl3 и насыщенный водный раствор NaHCO3 и органический слой отделяли. Далее, водный слой экстрагировали при помощи CHCl3. Органический слой собирали, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением этил 1-азабицикло[3.3.1]нонан-5-карбоксилата (376 мг).
Соединение Примера получения 42-1 представленное ниже в Таблицах, получали способом, аналогичным описанному в Примере получения 42, используя вещество Примера получения 56, описанного далее, в качестве исходного вещества.
Пример получения 42-2
К раствору 1-бензил-1-азониабицикло[2.2.1]гептан-4-карбоксилата (3,25 г) в MeOH добавляли 10% Pd на углероде с 50% гидратированием (650 мг) с последующим перемешиванием при комнатной температуре в течение 5 часов в атмосфере водорода при давлении 3 атм. Реакционную смесь фильтровали через целит и фильтрат концентрировали. Остаток растворяли в MeOH и добавляли серную кислоту (3 мл) с последующим нагреванием при кипячении с обратным холодильником в течение 1 часа. Реакционную смесь нейтрализовали водным раствором K2CO3 и экстрагировали при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3: MeOH: 28% водный раствор аммиака) с получением метил 1-азабицикло[2.2.1]гептан-4-карбоксилата (890 мг) в виде бесцветного маслянистого вещества.
Пример получения 43
К раствору DIPA (3,1 мл) в ТГФ (30 мл) по каплям добавляли 2,6 M раствор (8,6 мл) н-бутиллития в гексане при -70°C. После перемешивания при 0°C в течение 40 минут к смеси по каплям при -70°C добавляли EtOAc (2,3 мл) с последующим перемешиванием в течение 5 минут. К смеси по каплям добавляли раствор 1-бензилазепан-4-она (3,6 г) в ТГФ (10 мл) с последующим перемешиванием в течение 40 минут. К реакционной смеси добавляли насыщенный водный раствор NH4Cl с последующим добавлением воды и экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH: водный раствор аммиака) с получением этил (1-бензил-4-гидроксиазепан-4-ил)ацетата (5,0 г) в виде желтого маслянистого вещества.
Соединение Примера получения 43-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 43, используя соответствующее исходное вещество.
Пример получения 44
К раствору 1-бензил-4-(2-гидроксиэтил)азепан-4-ола (2,0 г) в ДХМ (20 мл) добавляли пиридин (4 мл) и пара-толуолсульфонилхлорид (1,68 г) при 0°C с последующим перемешиванием при 0°C в течение 3 часов и затем при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, подщелачивали путем добавления насыщенного водного раствора NaHCO3 и затем экстрагировали при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением бледно-красного твердого вещества. К смеси добавляли EtOAc и твердое вещество распыляли и собирали при помощи фильтрации с получением 4-метилбензолсульфоната 1-бензил-5-гидрокси-1-азониабицикло[3.2.2]нонана (1,85 г) в виде бледно-красного порошка.
Пример получения 45
К раствору трет-бутил 3-(2-этокси-2-оксоэтил)-3-гидроксипирролидин-1-карбоксилата (6,35 г) в ТГФ (100 мл) добавляли LiBH4 (1,01 г) с последующим нагреванием при кипячении с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, затем добавляли воду и экстрагировали при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением трет-бутил 3-гидрокси-3-(2-гидроксиэтил)пирролидин-1-карбоксилата (4,72 г) в виде бесцветного маслянистого вещества.
Пример получения 46
При криогенном охлаждении (от -10°C до 0°C), к раствору трет-бутил 3-гидрокси-3-(2-гидроксиэтил)пирролидин-1-карбоксилата (4,72 г) в пиридине (10 мл) добавляли пара-толуолсульфонилхлорид (1,68 г) с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и к смеси добавляли 1 M раствор хлористоводородной кислоты с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, гексан:EtOAc) с получением трет-бутил 3-гидрокси-3-(2-{[(4-метилфенил)сульфонил]окси}этил)пирролидин-1-карбоксилата (3,25 г) в виде бесцветного маслянистого вещества.
Пример получения 47
К раствору трет-бутил 3-гидрокси-3-(2-{[(4-метилфенил)сульфонил]окси}этил)пирролидин-1-карбоксилата (3,24 г) в EtOH добавляли 4 M раствор HCl/диоксан с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, с последующим добавлением водного раствора Na2CO3 и экстракцией при помощи EtOAc. Водный слой промывали при помощи EtOAc и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (CHCl3:MeOH:водный раствор аммиака = 7:3:0,3) с получением 1-азабицикло[2.2.1]гептан-4-ола (950 мг) в виде бесцветного твердого вещества.
Пример получения 48
К DIPA (3,6 мл) по каплям добавляли 2,64 M раствор н-бутиллития в гексане (8,93 мл) при -78°C с последующим перемешиванием в течение 30 минут. К реакционной смеси добавляли этил 1-бензил-пирролидин-3-карбоксилат (5,0 г) с последующим перемешиванием в течение 30 минут. К реакционной смеси по каплям добавляли раствор 1,2-дибромэтана (6,05 г) в ТГФ, с последующим нагреванием до комнатной температуры и перемешиванием в течение 2 часов. К реакционной смеси добавляли водный раствор K2CO3 с последующей экстракцией при помощи EtOAc. Водный слой концентрировали при пониженном давлении и к остатку добавляли EtOH с последующим перемешиванием при комнатной температуре в течение 30 минут. Нерастворившиеся вещества удаляли при помощи фильтрации и фильтрат концентрировали при пониженном давлении. К остатку добавляли EtOH с последующим перемешиванием при комнатной температуре в течение 10 минут. Нерастворившиеся вещества удаляли при помощи фильтрации и фильтрат концентрировали при пониженном давлении с получением 1-бензил-1-азониабицикло[2.2.1]гептан-4-карбоксилата (3,25 г) в виде бесцветного твердого вещества.
Пример получения 49
К раствору этил 2-метил-1,3-тиазол-4-карбоксилата (14,53 г) в MeCN (150 мл) добавляли NBS (22,66 г) с последующим нагреванием при кипячении с обратным холодильником в течение 3 часов. К реакционной смеси добавляли NBS (7,55 г) с последующим нагреванием при кипячении с обратным холодильником в течение 2 часов. При охлаждении льдом к реакционной смеси добавляли насыщенный водный раствор NaHCO3 с последующим перемешиванием в течение 5 минут и затем экстракцией при помощи EtOAc. Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:EtOAc = от 100:0 до 60:40) с получением этил 5-бром-2-метил-1,3-тиазол-4-карбоксилата (8,52 г) в виде желтого твердого вещества.
Пример получения 50
Смесь гидрохлорида 1-азабицикло[2.2.1]гептан-3-она (Journal of Medicinal Chemistry, 1990, 33, pp. 2690-2697) (350 мг), оксида платины (35 мг) и EtOH (5 мл) перемешивали при комнатной температуре в атмосфере водорода. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением гидрохлорида 1-азабицикло[2.2.1]гептан-3-ола в виде бледно-коричневого твердого вещества.
Соединения Примеров получения 50-1 и 50-2, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 50, используя соответствующие исходные вещества.
Пример получения 51
Смесь 1-азабицикло[3.3.1]нонан-4-карбонитрила (US 5834499, Ссылочный Пример 44) (1,7 г) и концентрированной хлористоводородной кислоты (10 мл) нагревали при кипячении с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли EtOH (10 мл) и концентрированную серную кислоту (2 капли) с последующим нагреванием при кипячении с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении и к остатку добавляли CHCl3 и 10% водный раствор K2CO3 и слои разделяли. Водный слой экстрагировали при помощи CHCl3 и органический слой затем объединяли, промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:водный раствор аммиака) с получением этил 1-азабицикло[3.3.1]нонан-4-карбоксилата (2,2 г) в виде бледно-коричневого маслянистого вещества.
Соединение Примера получения 51-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 51, используя соответствующее исходное вещество.
Пример получения 52
К смеси гидрохлорида (2,3 г) 1-азабицикло[3.2.1]октан-4-она (Journal of Medicinal Chemistry, 1993, 36, pp. 683-689), диметоксиэтана (23 мл) и tBuOH (4,6 мл) добавляли tBuOK (1,60 г) с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционной смеси добавляли пара-толуолсульфонилметилизоцианид (3,06 г) с последующим перемешиванием на бане со льдом. К реакционной смеси добавляли tBuOK (3,19 г) двумя отдельными порциями с последующим перемешиванием в течение 30 минут и затем перемешиванием при комнатной температуре в течение 2 часов. К реакционной смеси добавляли воду с последующей экстракцией при помощи CHCl3. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором и затем экстрагировали 1 M раствором хлористоводородной кислоты. Водный слой промывали при помощи EtOAc и концентрировали при пониженном давлении с получением бледно-коричневого твердого вещества (1,9 г). К этому веществу добавляли концентрированную хлористоводородную кислоту (10 мл), нагревали при кипячении с обратным холодильником в течение 4 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли EtOH (20 мл) и концентрированную серную кислоту (2 капли) с последующим нагреванием при кипячении с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении и остаток смеси нейтрализовали добавлением 1 M водного раствора NaOH и экстрагировали при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:водный раствор аммиака) с получением этил 1-азабицикло[3.2.1]октан-4-карбоксилата (850 мг) в виде бесцветного маслянистого вещества.
Соединение Примера получения 52-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 52, используя 1-бензилазепан-4-он.
Пример получения 53
К смеси гидрохлорида (21,63 г) 1-азабицикло[3.2.1]октан-6-она (Journal of Medicinal Chemistry, 1993, 36, pp. 683-689), диметоксиэтана (210 мл) и tBuOH (43 мл) при охлаждении льдом добавляли tBuOK (15 г) с последующим перемешиванием при комнатной температуре в течение 40 минут. Далее при охлаждении льдом к смеси добавляли пара-толуолсульфонилметилизоцианид (28,74 г), к которому двумя отдельными порциями добавляли tBuOK (30 г) с последующим перемешиванием в течение 30 минут и перемешиванием при комнатной температуре в течение 4 часов. К реакционной смеси добавляли воду с последующей экстракцией при помощи CHCl3. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:водный раствор аммиака) с получением продукта с низкой полярностью (6,17 г) и высокополярного продукта (4,9 г), 1-азабицикло[3.2.1]октан-6-карбонитрила в виде желтого маслянистого вещества и разбавленного коричневого твердого вещества, соответственно.
Пример получения 54
Смесь продукта с высокой полярностью (1,44 г) 1-азабицикло[3.2.1]октан-6-карбонитрила, полученного в Примере получения 53, и концентрированной хлористоводородной кислоты (15 мл) нагревали при кипячении с обратным холодильником в течение 4 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Далее к остатку добавляли MeOH с последующим концентрированием при пониженном давлении. К остатку добавляли MeOH (25 мл) и серную кислоту (0,67 мл) с последующим нагреванием при кипячении с обратным холодильником в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и отверждали досуха и к остатку добавляли CHCl3 и 1 M водный раствор K2CO3. Водный слой экстрагировали при помощи CHCl3, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:водный раствор аммиака) с получением метил 1-азабицикло[3.2.1]октан-6-карбоксилата (657 мг) в виде бесцветного маслянистого вещества.
Соединение Примера получения 54-1, представленное ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере получения 54, используя соответствующее исходное вещество.
Пример получения 55
К DIPA (3,22 мл) при -78°C по каплям добавляли 2,64 M раствор н-бутиллития в гексане (7,97 мл) с последующим перемешиванием в течение 30 минут. К реакционной смеси добавляли этил 1-бензилазепан-4-карбоксилат (5,0 г) с последующим перемешиванием в течение 30 минут. Далее к этой смеси по каплям добавляли раствор 1-бром-2-хлорэтана (3,02 г) в ТГФ, с последующим нагреванием до комнатной температуры и перемешиванием в течение 2 часов. К реакционной смеси добавляли водный раствор K2CO3 с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:водный раствор аммиака). Требуемые фракции собирали и концентрировали при пониженном давлении. После концентрирования при пониженном давлении к остатку, часть которого отверждали, добавляли EtOAc с последующей очисткой при помощи колоночной хроматографии на силикагеле (EtOAc) с получением этил 1-бензил-4-(2-хлорэтил)азепан-4-карбоксилата (4,11 г) в виде бесцветного маслянистого вещества.
Пример получения 56
Раствор этил 1-бензил-4-(2-хлорэтил)азепан-4-карбоксилата (4,1 г) в MeCN (200 мл) перемешивали при 40°C в течение 10 часов. Реакционную смесь концентрировали при пониженном давлении с получением 1-бензил-5-(этоксикарбонил)-1-азониабицикло[3.2.2]нонанхлорида (4,1 г) в виде бесцветного маслянистого вещества.
Пример получения 57
Соединения Примеров получения 57 - 57-3, представленные ниже в Таблицах, синтезировали способом, аналогичным описанному в Примере 6, используя соответствующее исходное вещество.
В целях экономии места на бумаге, обозначение SB (представлен ниже) в Таблицах Примеров получения, представленных выше, означает следующее.
Пример 1
К раствору 1-азабицикло[2.2.2]окт-4-ил(2-фенил-3-тиенил)карбамата (120 мг) в ДХМ (10 мл) порциями при охлаждении льдом добавляли MCPBA (90 мг). После перемешивания в течение 30 минут реакционную жидкость концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (CHCl3:MeOH = от 20:1 до 10:1, подвергали обработке для отделения жидкости при помощи 28% водного раствора аммиака) с получением 1-оксид-1-азабицикло[2.2.2]окт-4-ил(2-фенил-3-тиенил)карбамата (105 мг) в виде бесцветного аморфного вещества.
Пример 2
К раствору 2-фенил-5,6-дигидро-4H-циклопента[b]тиофен-3-амина (110 мг) в пиридине (2 мл) добавляли (3R)-хинуклидин-3-ол и (3R)-1-азабицикло[2.2.2]окта-3-илхлоркарбонатгидрохлорид (231 мг), синтезированный из трифосгена, с последующим перемешиванием при комнатной температуре в течение 30 минут и затем перемешиванием при 80°C в течение 15 часов. К реакционной смеси добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, затем сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH) с получением бесцветного маслянистого вещества (135 мг). Это вещество растворяли в EtOH и к смеси добавляли фумаровую кислоту (38 мг). Осажденное кристаллическое вещество собирали при помощи фильтрации с получением (3R)-1-азабицикло[2.2.2]окт-3-ил (2-фенил-5,6-дигидро-4H-циклопента[b]тиен-3-ил)карбаматфумарата (103 мг) в виде твердого белого вещества.
Пример 3
К раствору трифосгена (362 мг) в ДХМ (3 мл) при 0°C добавляли пиридин (162 мг) и затем к смеси по каплям добавляли раствор цис-2-фенилциклогексиламина (300 мг) в ДХМ (3 мл) с последующим перемешиванием при комнатной температуре в течение 40 минут. К 60% маслянистой суспензии NaH (110 мг) в ТГФ (1,5 мл) при 0°C добавляли 1-азабицикло[2.2.2]окт-3-илметанол (363 мг) с последующим перемешиванием при комнатной температуре в течение 20 минут и к смеси по каплям при 0°C добавляли указанный выше раствор ДХМ с последующим перемешиванием при 80°C в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор NaHCO3 с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (CHCl3:MeOH:28% водный раствор аммиака = 80:10:1) с получением бесцветного маслянистого вещества (100 мг). Полученное маслянистое вещество растворяли в смешанном растворителе (EtOAc:EtOH = 1:1) и к смеси добавляли фумаровую кислоту (32 мг). Смесь концентрировали при пониженном давлении, затем к смеси добавляли смешанный растворитель (EtOAc:IPE) и выпавшее в осадок твердое вещество собирали при помощи фильтрации с получением 1-азабицикло[2.2.2]окт-3-илметил rel-[(1R,2R)-2-фенилциклогексил]карбаматфумарата (103 мг) в виде бесцветного твердого вещества.
Пример 4
К раствору 1-азабицикло[2.2.2]окт-3-илметил (2-фенил-3-тиенил)карбамата (100 мг) в EtOAc (2 мл) при комнатной температуре добавляли йодметан (36 мкл) с последующим перемешиванием в течение ночи. Реакционную смесь фильтровали, затем концентрировали при пониженном давлении и сушили вымораживанием с получением 1-метил-3-({[(2-фенил-3-тиенил)карбамоил]окси}метил)-1-азониабицикло[2.2.2]октанйодида (88 мг) в виде желтого аморфного вещества.
Пример 5
В раствор смеси 1-азабицикло[2.2.2]окт-3-ил(2-бром-3-тиенил)карбамата (295 мг), Pd(PPh3)4 (103 мг), 4-фторфенилборной кислоты (187 мг) и 1,4-диоксана (3 мл) добавляли 2 M водный раствор Na2CO3 (0,89 мл) с последующим перемешиванием в течение 5 часов при нагревании при 90°C. Реакционную смесь разбавляли водой и экстрагировали при помощи EtOAc. Органический слой промывали водой и доводили до pH 1 при помощи 1 M раствора хлористоводородной кислоты. Далее, водный слой промывали при помощи EtOAc и доводили до pH 10 водным раствором NaOH и водный слой экстрагировали при помощи CHCl3. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи основной колоночной хроматографии на силикагеле (изготовитель Fuji Silysia Chemical Ltd., YAMAZEN YFLC WPrep2XY, CHCl3). Собранную фракцию концентрировали при пониженном давлении и полученное масло растворяли в EtOAc с последующим добавлением 4 M раствора HCl/EtOAc, концентрированием при пониженном давлении и сушкой с отверждением. К полученному остатку добавляли EtOH и EtOAc для кристаллизации с получением 1-азабицикло[2.2.2]окт-3-ил [2-(4-фторфенил)-3-тиенил]карбамат, гидрохлорида (135 мг) в виде твердого белого вещества.
Пример 6
К раствору 5-фенил-1,3-тиазол-4-карбоновой кислоты (250 мг) в толуоле (6 мл) при комнатной температуре по каплям добавляли TEA (221 мкл) и DPPA (315 мкл) с последующим перемешиванием при той же температуре в течение 40 минут. Затем после перемешивания при 90°C в течение 40 минут к смеси добавляли раствор 1-азабицикло[2.2.2]окт-4-илметанола (241 мг) в смеси ДМФА (2 мл)/толуол (2 мл) с последующим нагреванием при кипячении с обратным холодильником в течение 2 часов. К реакционной жидкости добавляли насыщенный водный раствор NaHCO3 с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (YAMAZEN YFLC WPrep2XY, элюент; CHCl3:MeOH:28% водный раствор аммиака = 80:10:1) с получением желтого маслянистого вещества. К этому раствору в EtOH (2 мл) при комнатной температуре добавляли смесь 4 M раствор HCl/EtOAc (1 мл) с последующим перемешиванием и растворитель концентрировали при пониженном давлении. Полученный остаток повторно кристаллизовали из смеси EtOH/эфир с получением 1-азабицикло[2.2.2]окт-4-илметил(5-фенил-1,3-тиазол-4-ил)карбамата, гидрохлорида (145 мг) в виде желтого твердого вещества.
Пример 7
К раствору 1-азабицикло[2.2.2]окт-4-илметил (2-карбамоил-5-фенил-1,3-тиазол-4-ил)карбамата (100 мг) в ДМФА (5 мл) при 0°C добавляли оксихлорид фосфора (48 мкл) с последующим перемешиванием в течение 1 часа. К реакционной смеси добавляли EtOAc с последующим разбавлением, добавлением водного раствора NaHCO3 и экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, затем сушили и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (CHCl3:MeOH:28% водный раствор аммиака = 9:1:0,1) и полученное коричневое маслянистое вещество растворяли в EtOAc, с последующим добавлением 4 M раствора HCl/EtOAc и перемешиванием. Смесь концентрировали при пониженном давлении, к смеси добавляли EtOAc и выпавшее в осадок твердое вещество собирали при помощи фильтрации с получением 1-азабицикло[2.2.2]окт-4-илметил (2-циано-5-фенил-1,3-тиазол-4-ил)карбамата, гидрохлорида (33 мг) в виде бледно-коричневого твердого вещества.
Пример 8
1-азабицикло[2.2.2]окт-3-илметил [5-(4-фторфенил)-1,3-тиазол-4-ил]карбамат (120 мг) подвергали ВЭЖХ (CHIRALPAK AD-H, 0,46 см ×25 см), чтобы отдельно собрать энантиомер 8a (время удерживания: около 12,8 минут, 51 мг) и энантиомер 8b (время удерживания: около 16,9 минут, 54 мг) в виде твердого белого вещества, соответственно.
Условия: подвижная фаза; гексан:EtOH:диэтиламин = 50:50:0,1, скорость потока; 0,5 мл/минута, температура колонки; 40°C, длина волны детекции; 254 нм.
Каждый из энантиомеров растворяли в EtOH и к смеси добавляли фумаровую кислоту для получения фумарата, соответственно.
Пример 9
В смешанном растворителе (MeOH:EtOAc = 5 мл:5 мл) к 1-азабицикло[2.2.2]окт-4-илметил трет-бутил(5-фенил-1,3-тиазол-2,4-диил)бискарбамату добавляли 4 M HCl/EtOAc (7 мл) с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, к остатку добавляли EtOAc и твердое вещество собирали при помощи фильтрации с получением 1-азабицикло[2.2.2]окт-4-илметил (2-амино-5-фенил-1,3-тиазол-4-ил)карбаматдигидрохлорида в виде твердого белого вещества.
Пример 10
К раствору 5-фенил-1,3-тиазол-4-карбоновой кислоты (300 мг) в толуоле (6 мл) при комнатной температуре по каплям добавляли TEA (672 мкл) и DPPA (409 мкл) с последующим перемешиванием при той же температуре в течение 20 минут. Затем после перемешивания при 90°C в течение 5 минут к смеси добавляли смесь 3-аминохинуклидиндигидрохлорида (407 мг) и ДМФА (2 мл) с последующим нагреванием при кипячении с обратным холодильником в течение 1 часа. К реакционной жидкости добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (YAMAZEN YFLC WPrep2XY, элюент; CHCl3:MeOH:28% водный раствор аммиака = 80:10:1) с получением маслянистого вещества. Это вещество растворяли в EtOH с последующим добавлением при комнатной температуре 10% HCl/MeOH и концентрированием при пониженном давлении. К полученному остатку добавляли EtOH и EtOAc и оставляли выстаиваться, выпавшее в осадок твердое вещество собирали при помощи фильтрации и промывали при помощи EtOAc с получением гидрохлорида 1-(1-азабицикло[2.2.2]окт-3-ил)-3-(5-фенил-1,3-тиазол-4-ил)мочевины (15 мг) в виде твердого белого вещества.
Пример 11
К раствору 5-фенил-1,3-тиазол-4-карбоновой кислоты (400 мг) в толуоле (5 мл) при комнатной температуре добавляли TEA (380 мкл). К реакционной жидкости по каплям добавляли раствор DPPA (546 мкл) в толуоле (5 мл) с последующим перемешиванием при той же температуре в течение 20 минут. После перемешивания при 90°C в течение 5 минут к смеси добавляли смесь 1-азабицикло[3.2.1]окт-5-илметанола (357 мг) и ДМФА (2 мл) с последующим нагреванием при кипячении с обратным холодильником в течение 1 часа. К реакционной жидкости добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:28% водный раствор аммиака = 80:10:1) с получением маслянистого вещества. Это вещество растворяли в EtOAc с последующим добавлением при комнатной температуре 4 M HCl/диоксан. Выпавшее в осадок твердое вещество собирали при помощи фильтрации и промывали при помощи EtOAc с получением 1-азабицикло[3.2.1]окт-5-илметил (5-фенил-1,3-тиазол-4-ил)карбамата, гидрохлорида (174 мг) в виде твердого белого вещества.
Пример 12
К раствору 5-фенил-1,3-тиазол-4-карбоновой кислоты (385 мг) в толуоле (5 мл) при комнатной температуре добавляли TEA (314 мкл). Далее по каплям добавляли DPPA (486 мкл) с последующим перемешиванием при той же температуре в течение 30 минут. После перемешивания при 90°C в течение 5 минут к смеси добавляли смесь 1-азабицикло[3.3.1]нон-5-илметанола (291 мг), ДМФА (1 мл) и толуола (4 мл) с последующим перемешиванием при 90°C в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры и затем к смеси добавляли CHCl3 и воду для отделения органического слоя. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:водный раствор аммиака). Собранную фракцию концентрировали при пониженном давлении с последующим добавлением 4 M HCl/EtOAc и концентрированием при пониженном давлении. Для отделения водного слоя к остатку добавляли EtOAc и воду. Водный слой нейтрализовали при помощи NaHCO3 и экстрагировали при помощи CHCl3. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением клейкого вещества (129 мг). Это вещество растворяли в EtOH с последующим добавлением раствора фумаровой кислоты (40 мг) в EtOH и концентрировали при пониженном давлении с получением 1-азабицикло[3.3.1]нон-5-илметил(5-фенил-1,3-тиазол-4-ил)карбаматфумарата (161 мг) в виде бесцветного аморфного флуоресцентного вещества.
Пример 13
К смеси 1-азабицикло[2.2.2]окт-4-илметил(2-{[2-(бензилокси)этил]амино}-5-фенил-1,3-тиазол-4-ил)карбаматдигидрохлорида (350 мг) и ТФУК (3,5 мл) в атмосфере азота добавляли тиоанизол (362 мкл) с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, подщелачивали добавлением 1 M водного раствора NaOH и экстрагировали смешанным растворителем (CHCl3:MeOH = 8:1). Органический слой сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:водный раствор аммиака). Собранную фракцию концентрировали при пониженном давлении, затем к остатку добавляли EtOAc и выпавшее в осадок твердое вещество распыляли, собирали при помощи фильтрации и промывали при помощи EtOAc с получением 1-азабицикло[2.2.2]окт-4-илметил {2-[(2-гидроксиэтил)амино]-5-фенил-1,3-тиазол-4-ил}карбамата (110 мг) в виде бесцветного твердого вещества.
Пример 14
К раствору 5-фенил-1,3-тиазол-4-карбоновой кислоты (385 мг) в толуоле (8 мл) при комнатной температуре добавляли TEA (285 мкл). К реакционной смеси по каплям добавляли DPPA (404 мкл) с последующим перемешиванием при той же температуре в течение 40 минут. Кроме того, после перемешивания при 90°C в течение 5 минут к смеси добавляли смесь 1-азабицикло[3.2.2]нонан-5-ола (313 мг) и ДМФА (3 мл) с последующим перемешиванием при 110°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, с последующим добавлением для подкисления 1 M раствора хлористоводородной кислоты и экстракцией при помощи EtOAc. Водный слой подщелачивали добавлением 1 M водного раствора NaOH и экстрагировали при помощи CHCl3. Органический слой, в указанном порядке, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи препаративной жидкостной хроматографии среднего давления (силикагель, YAMAZEN YFLC WPrep2XY, CHCl3:MeOH:водный раствор аммиака) с получением желтого аморфного флуоресцентного вещества. К этому веществу добавляли EtOAc и полученное твердое вещество собирали при помощи фильтрации и промывали при помощи EtOAc с получением 1-азабицикло[3.2.2]нон-5-ил(5-фенил-1,3-тиазол-4-ил)карбамата (238 мг) в виде бесцветного порошка.
Пример 15
Получали раствор 1-азабицикло[3.2.1]окт-6-илметил(5-фенил-1,3-тиазол-4-ил)карбамата в EtOH (1 мл) и подвергали препаративной ВЭЖХ (CHIRALCEL OD, 0,46 см ×25 см), чтобы отдельно собрать энантиомер 15a (время удерживания: около 6,2 минут) и энантиомер 15b (время удерживания: около 7,9 минут). Каждый из энантиомеров подвергали перекристаллизации из смешанного растворителя (гексан:EtOH) с получением бесцветного твердого вещества.
Условия: подвижная фаза; гексан:EtOH:диэтиламин = 50:50:0,1, скорость потока; 0,9 мл/минута, температура колонки; 40°C, длина волны детекции; 254 нм.
Соединения, представленные далее в Таблицах 18-41, получали с использованием описанного выше способа получения, способа, который очевиден для специалистов в данной области, или модификации этого способа. В Таблицах представлены номера Примеров (Синт.), указывающие, что соединения можно получить тем способом, что и структуру соединений Примеров. Физико-химические данные представлены далее в Таблицах 42-57.
Промышленная применимость
Соединение формулы (I) или его соль обладает антагонистическим действием на связывание мускаринового рецептора M3 и поэтому может быть использовано в качестве профилактического и/или терапевтического средства для лечения воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИНУКЛИДИНОВЫЕ ПРОИЗВОДНЫЕ (ГЕТЕРО)АРИЛЦИКЛОГЕПТАНКАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2456286C2 |
| АЛКАЛОИДНЫЙ ЭФИР И КАРБАМАТНЫЕ ПРОИЗВОДНЫЕ И ИХ МЕДИЦИНСКИЕ КОМПОЗИЦИИ | 2012 |
|
RU2611627C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2022 |
|
RU2803084C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2777979C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2023 |
|
RU2826177C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2747318C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ KDM5, И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2840256C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |

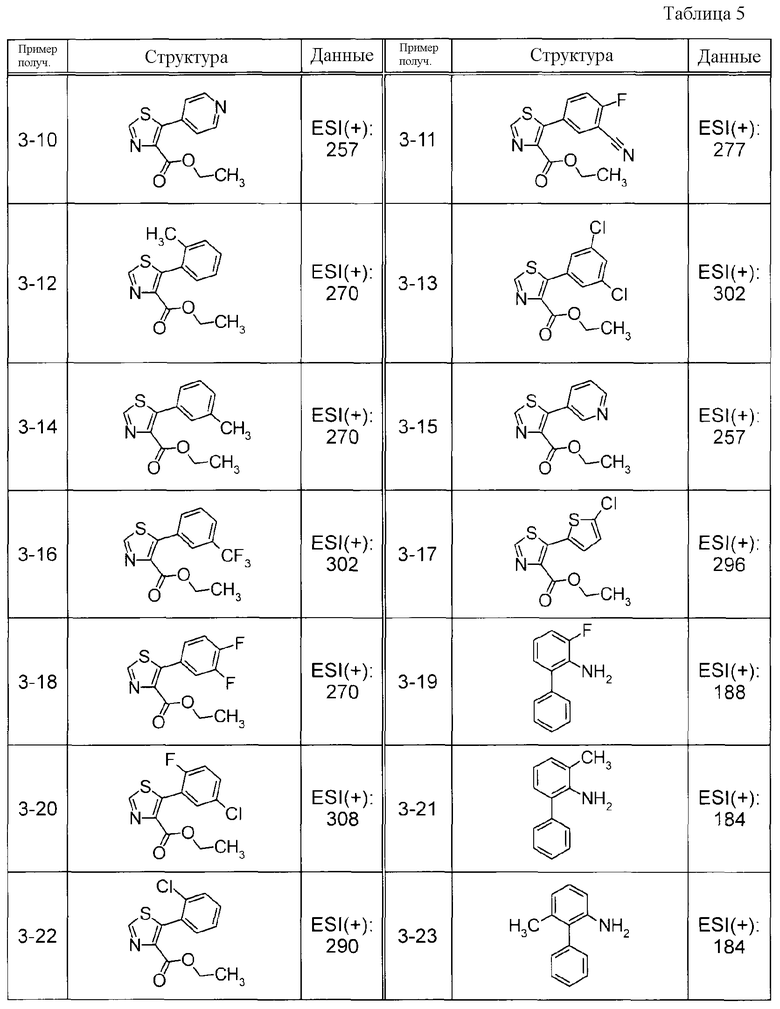
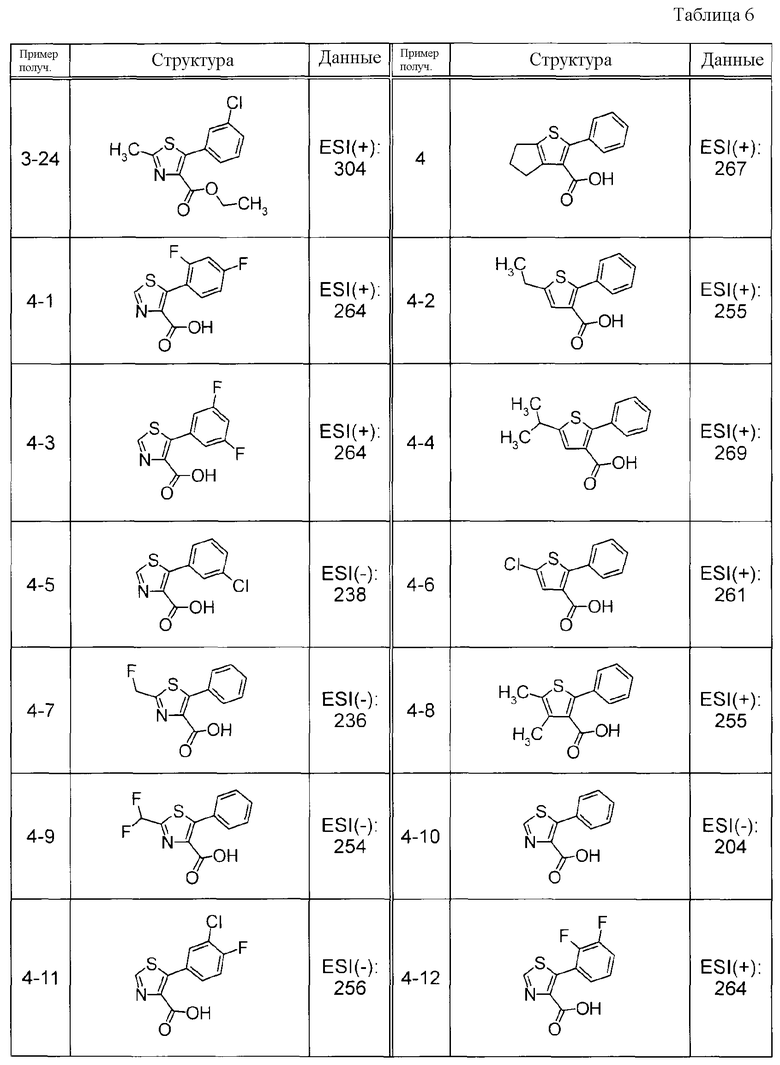
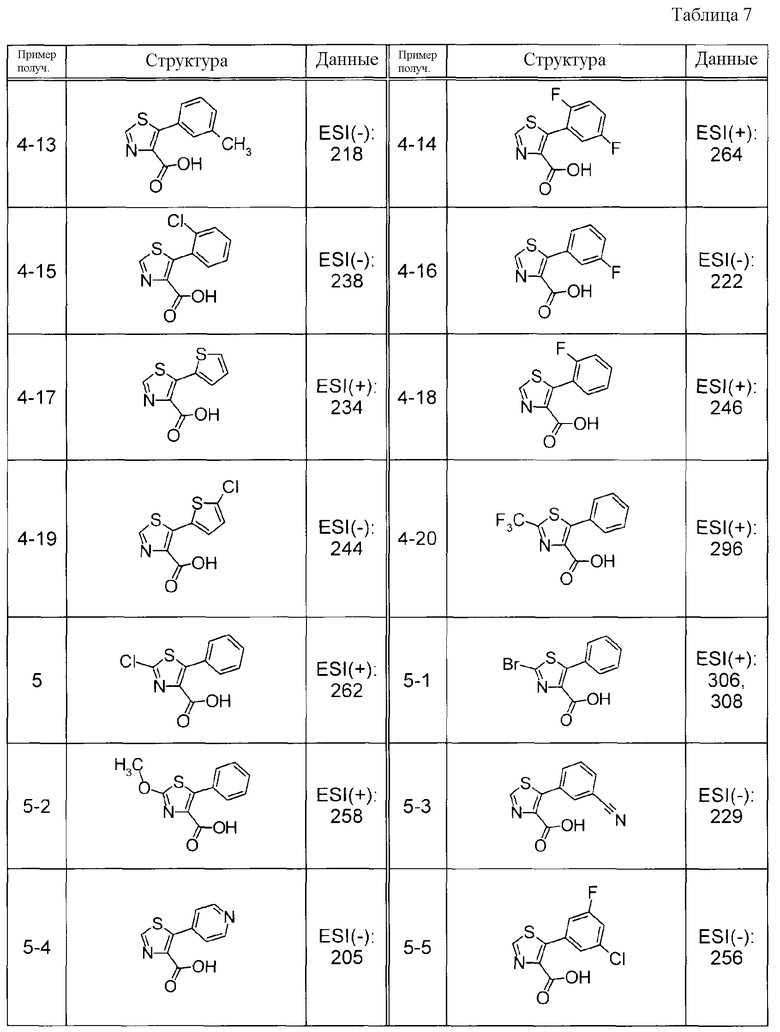


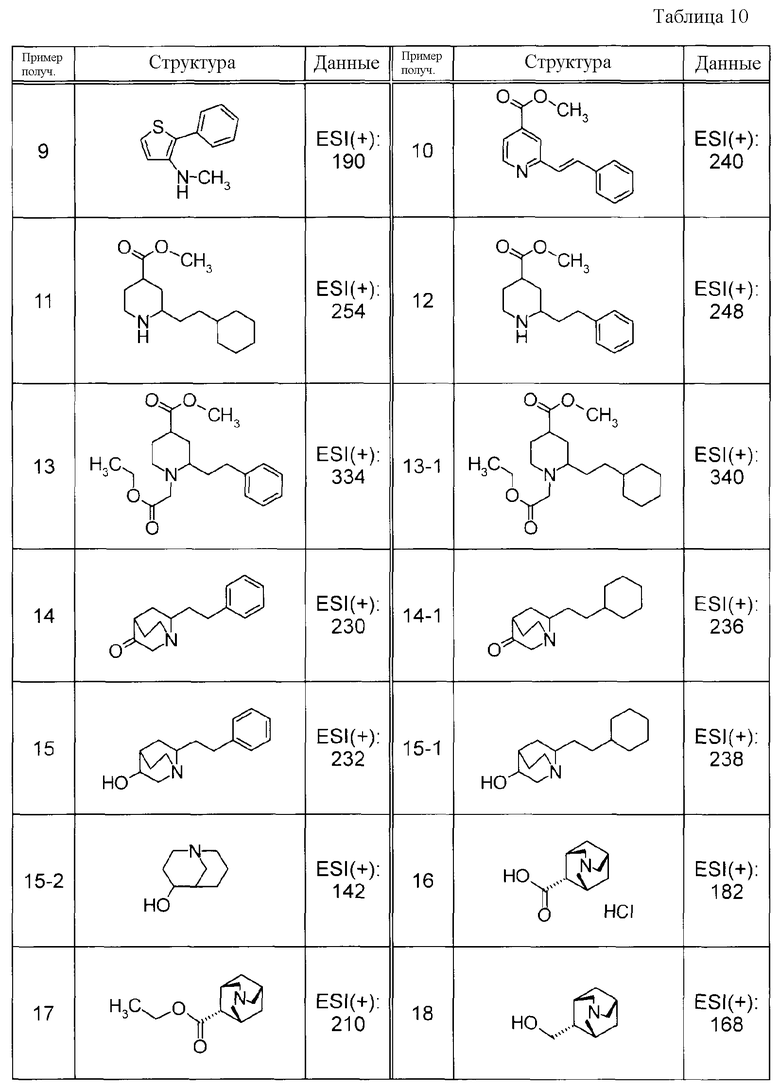

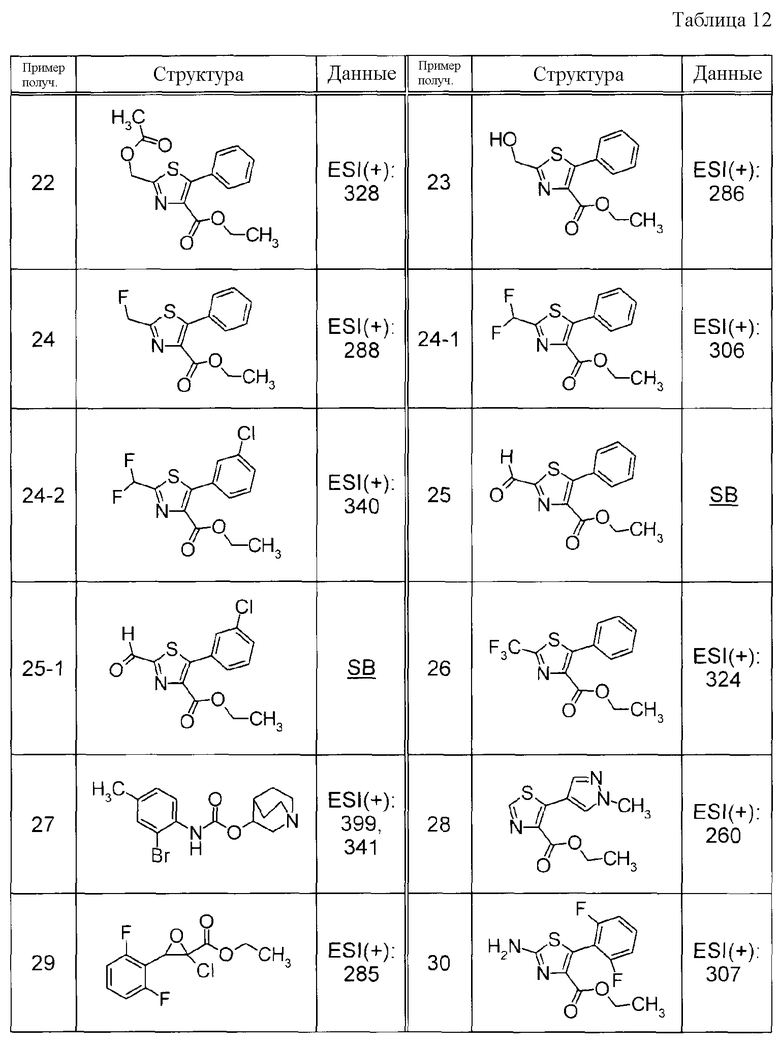
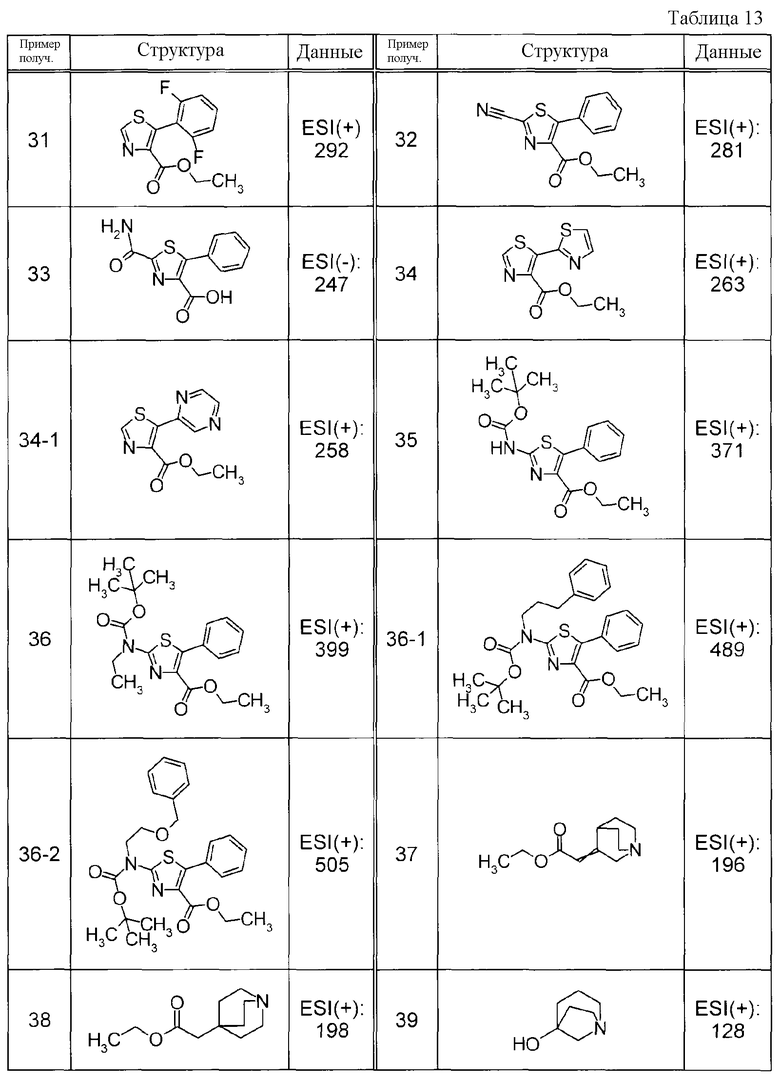
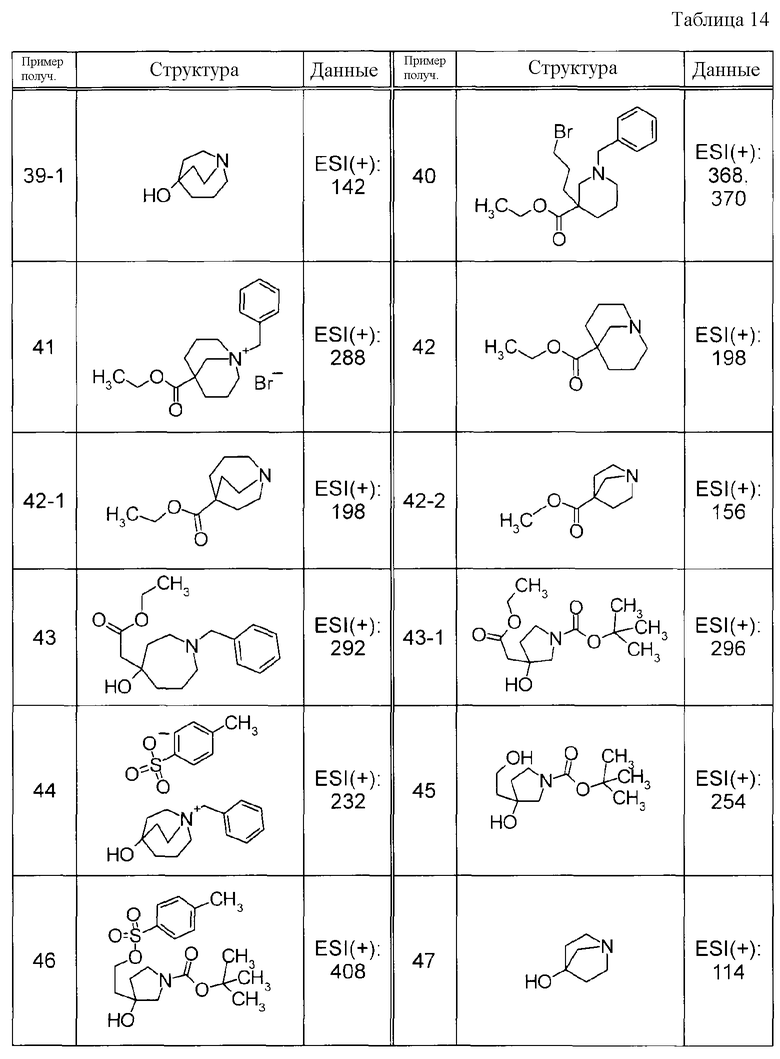



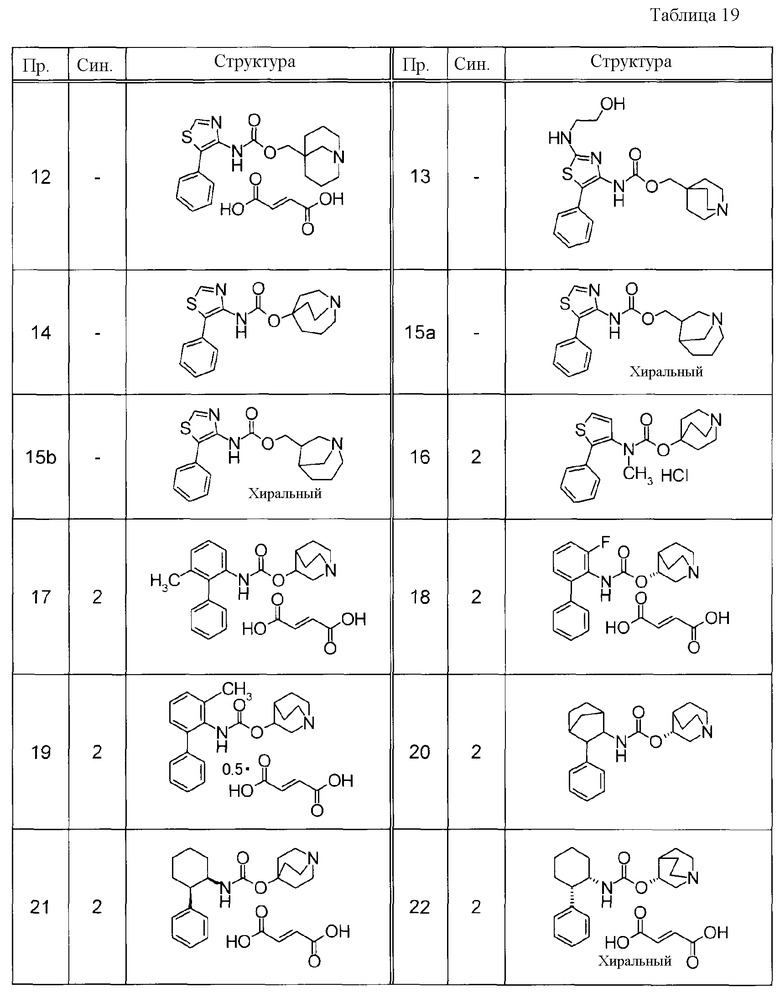
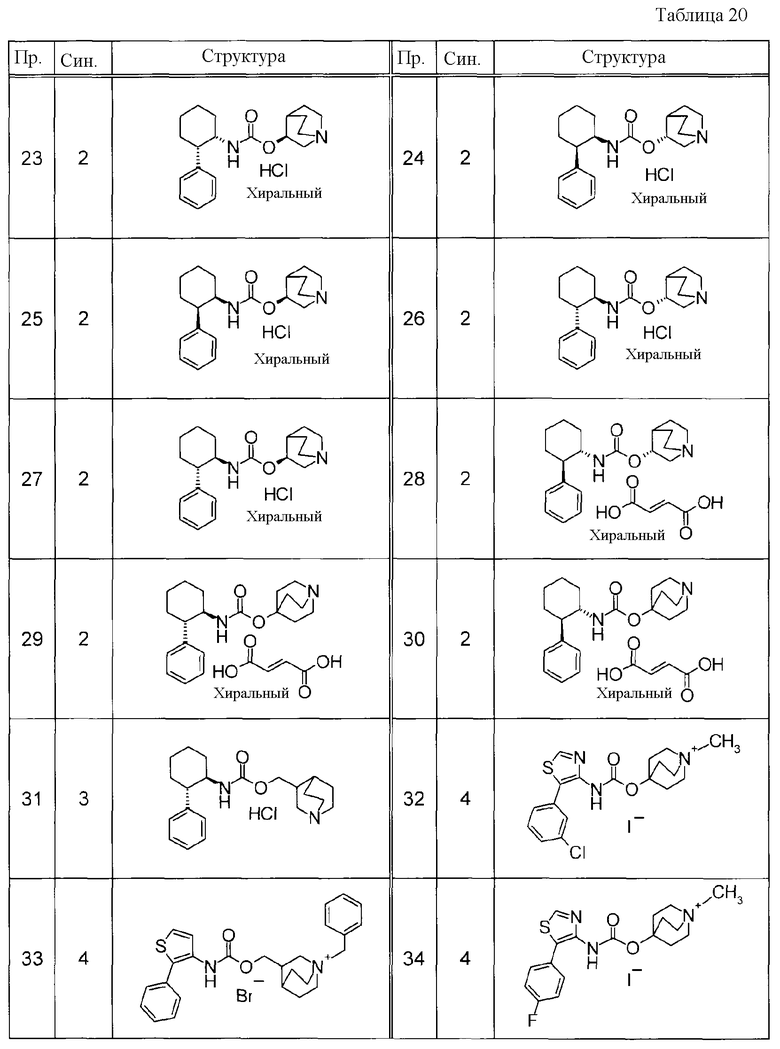
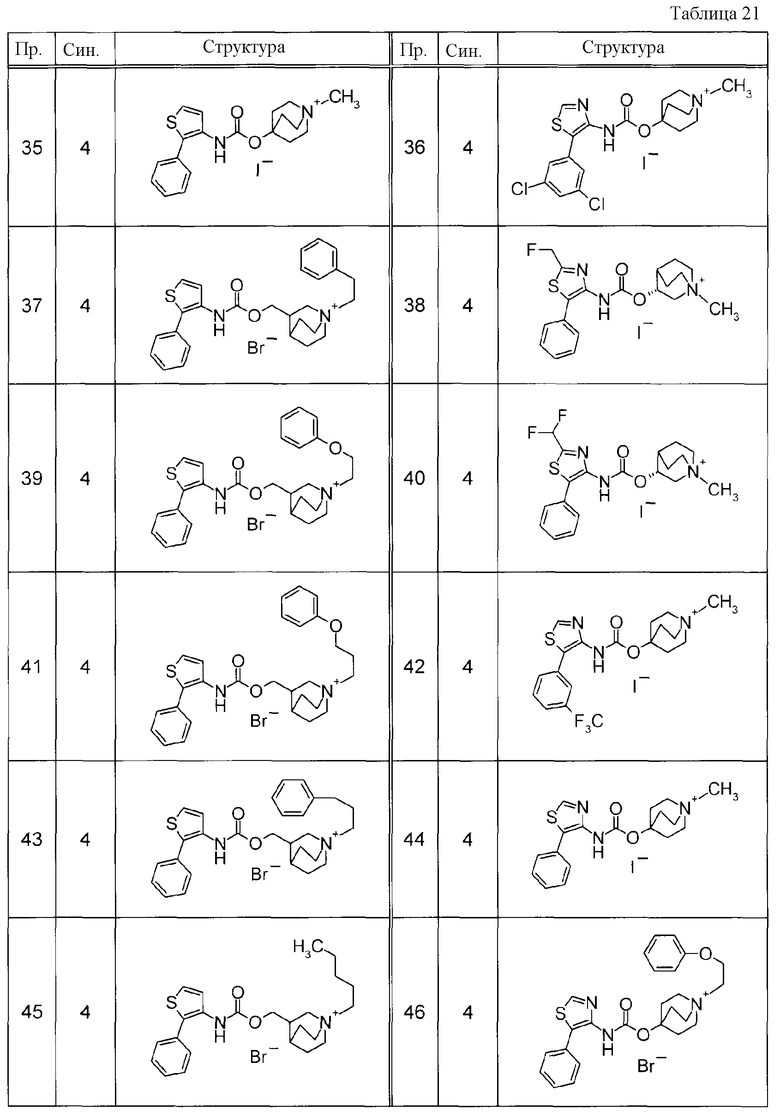
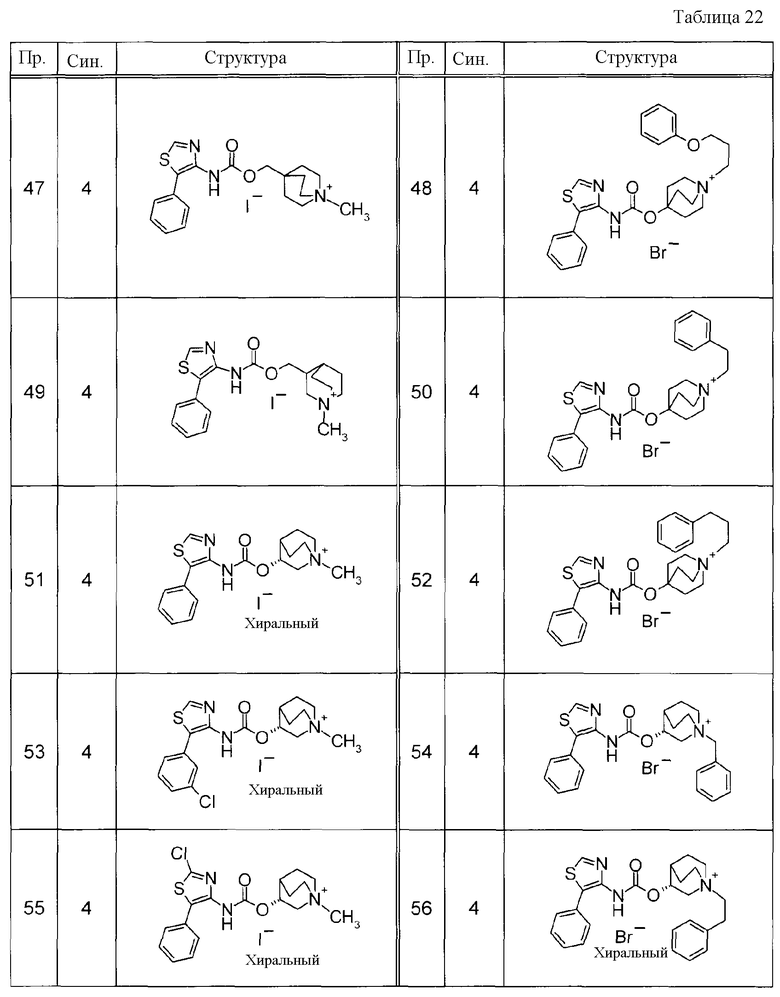
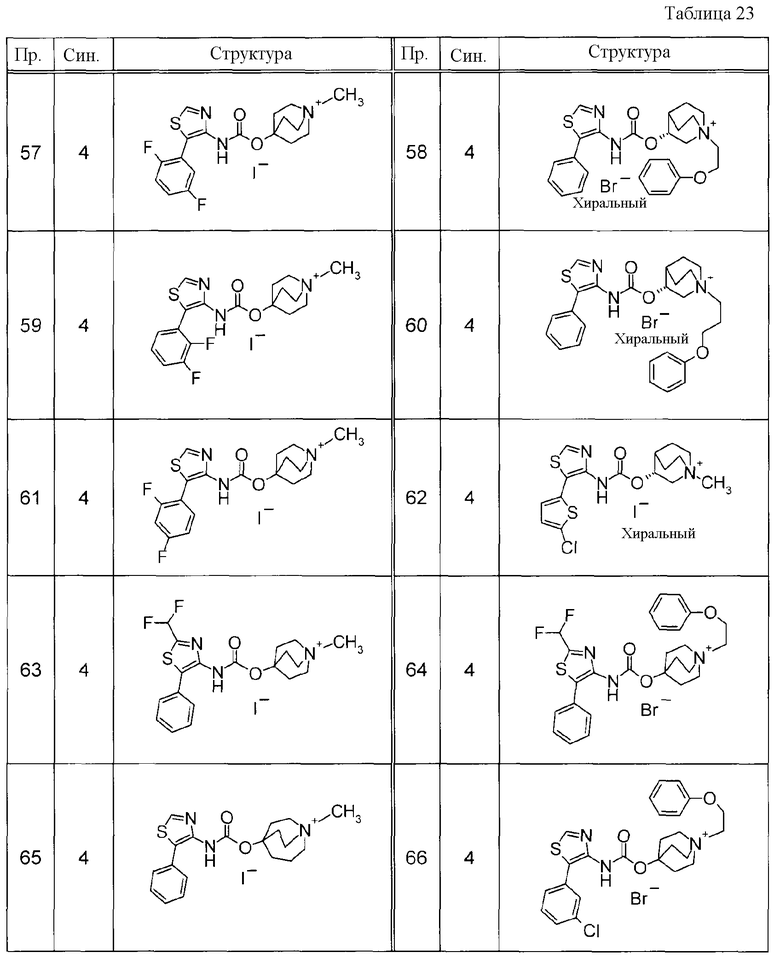
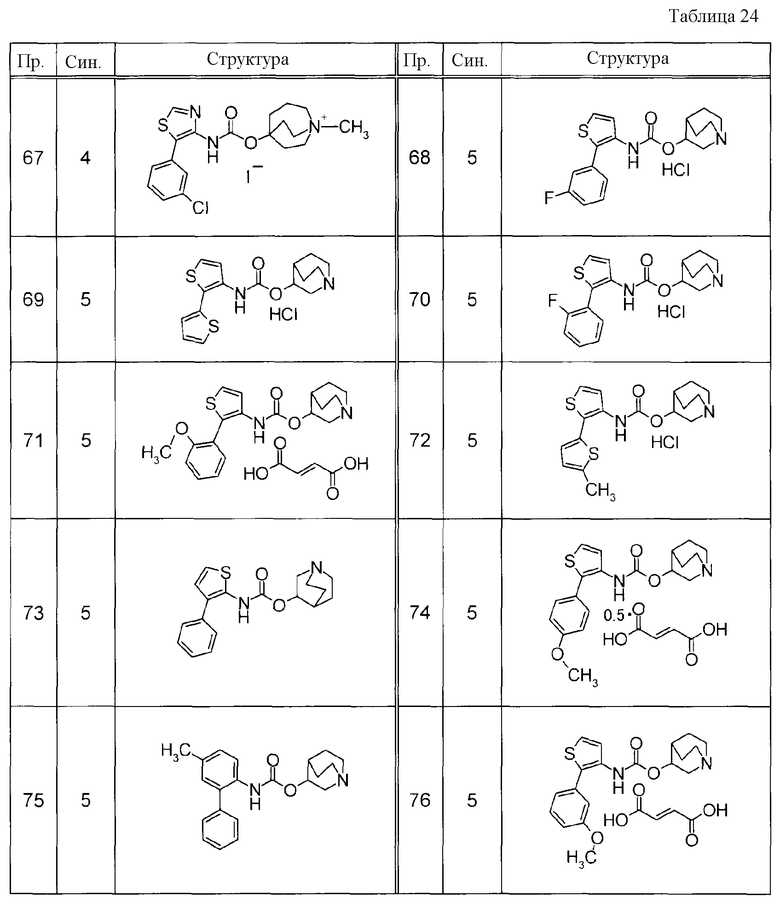
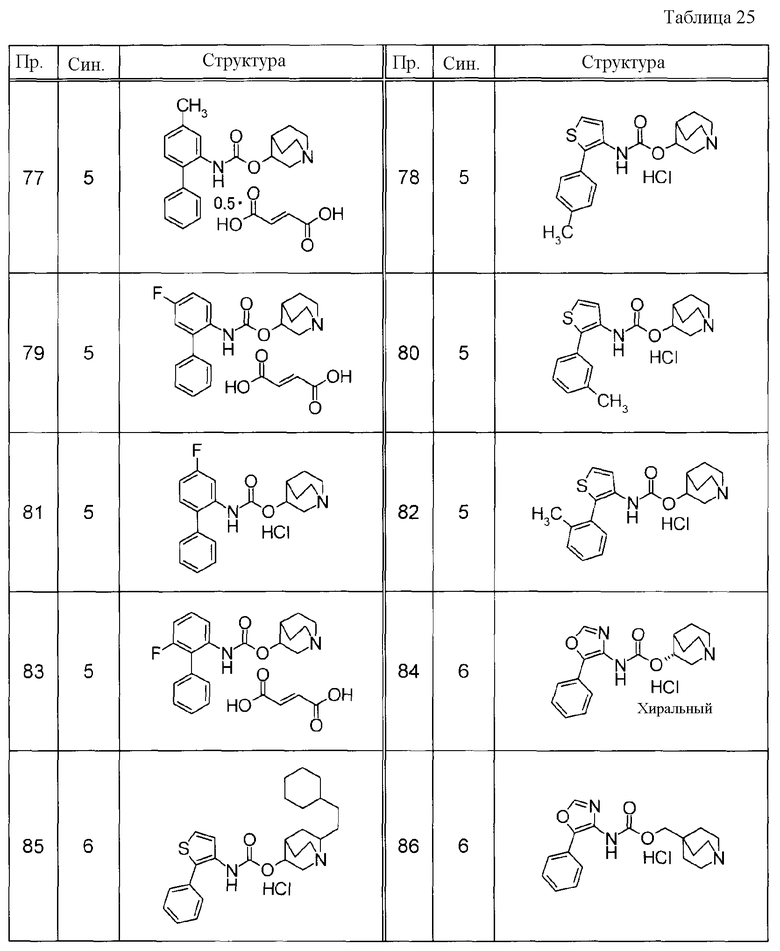
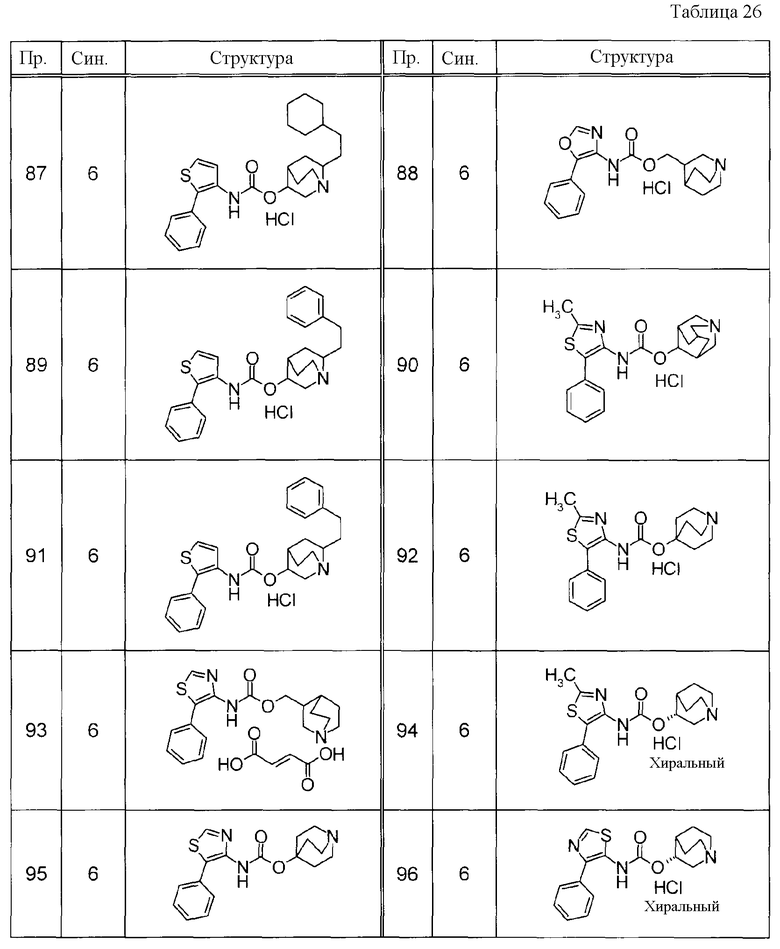
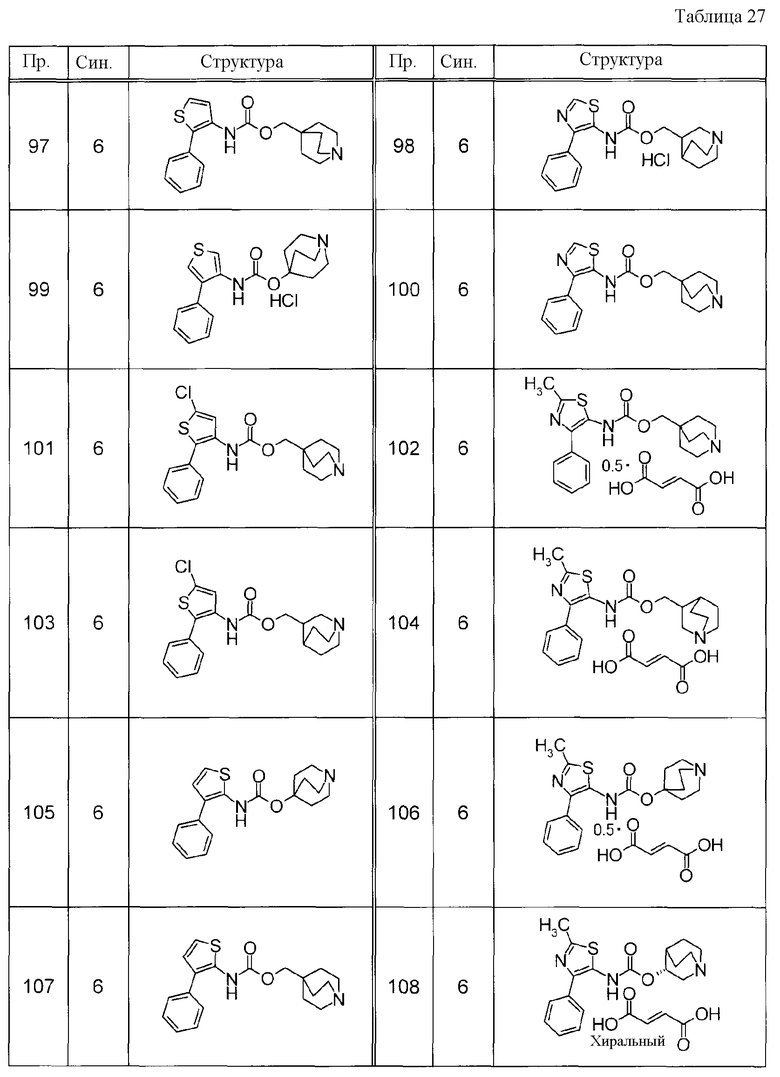
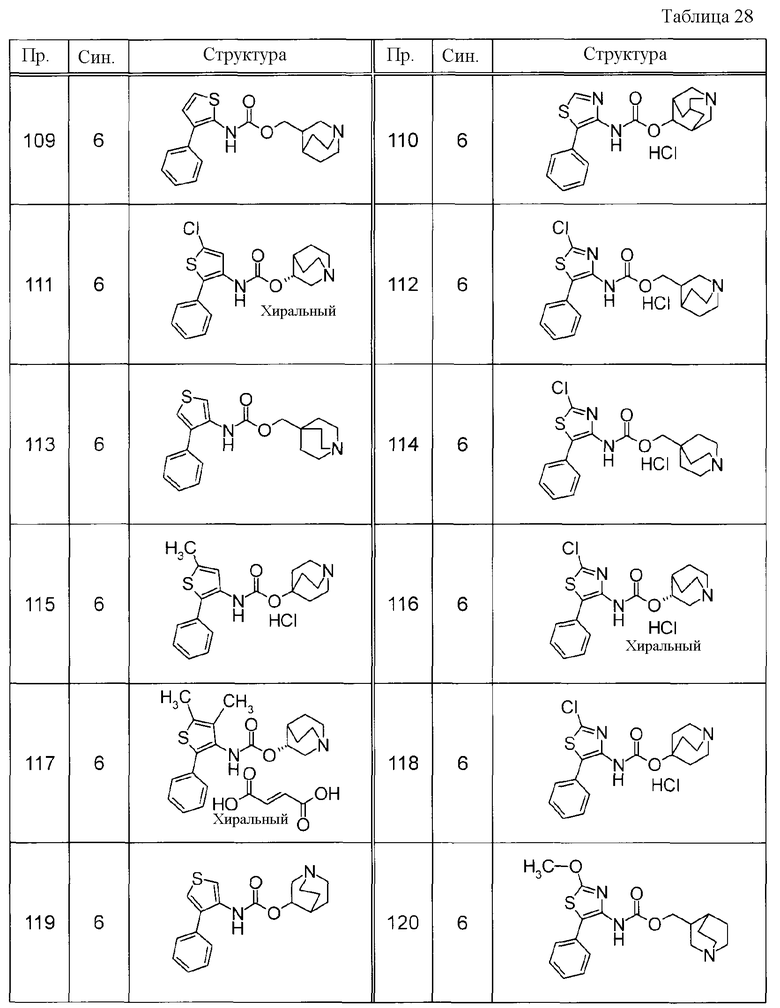
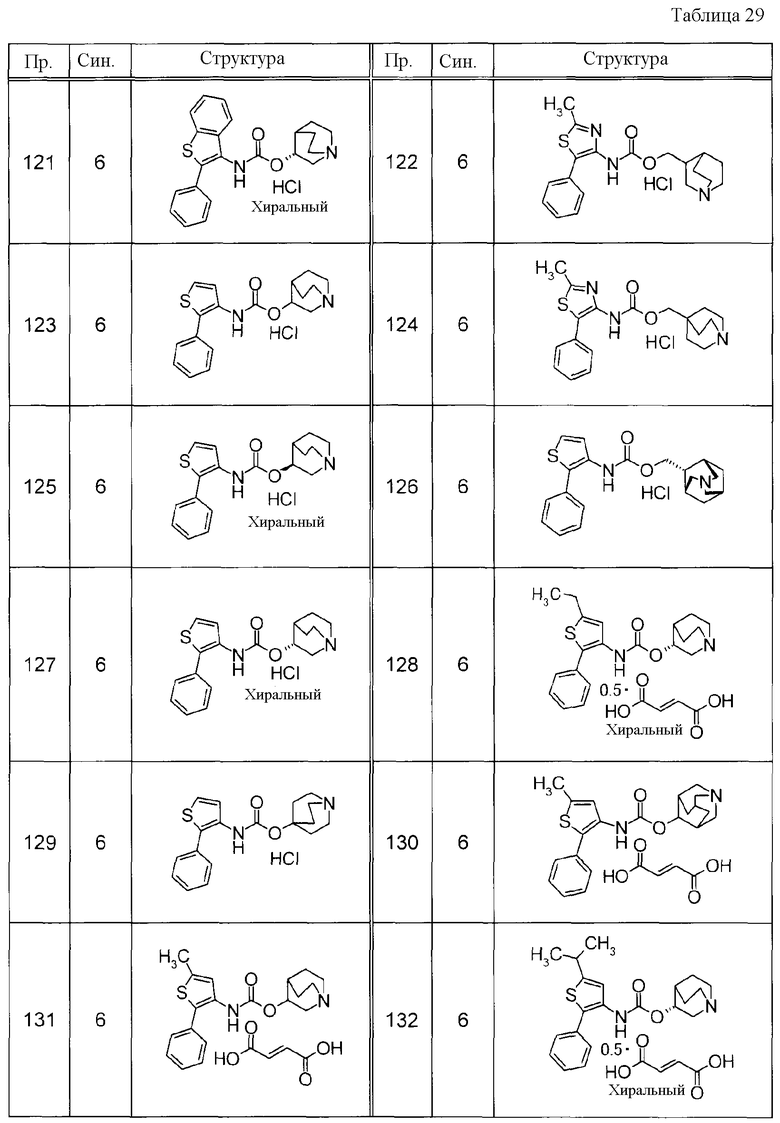
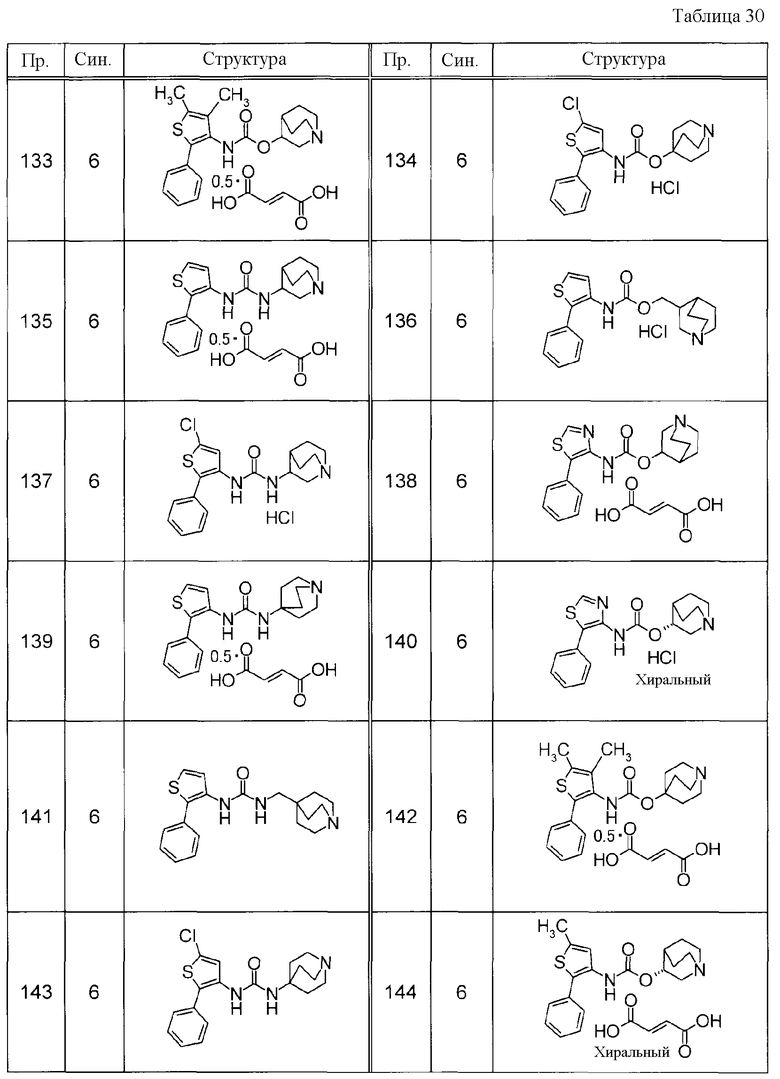
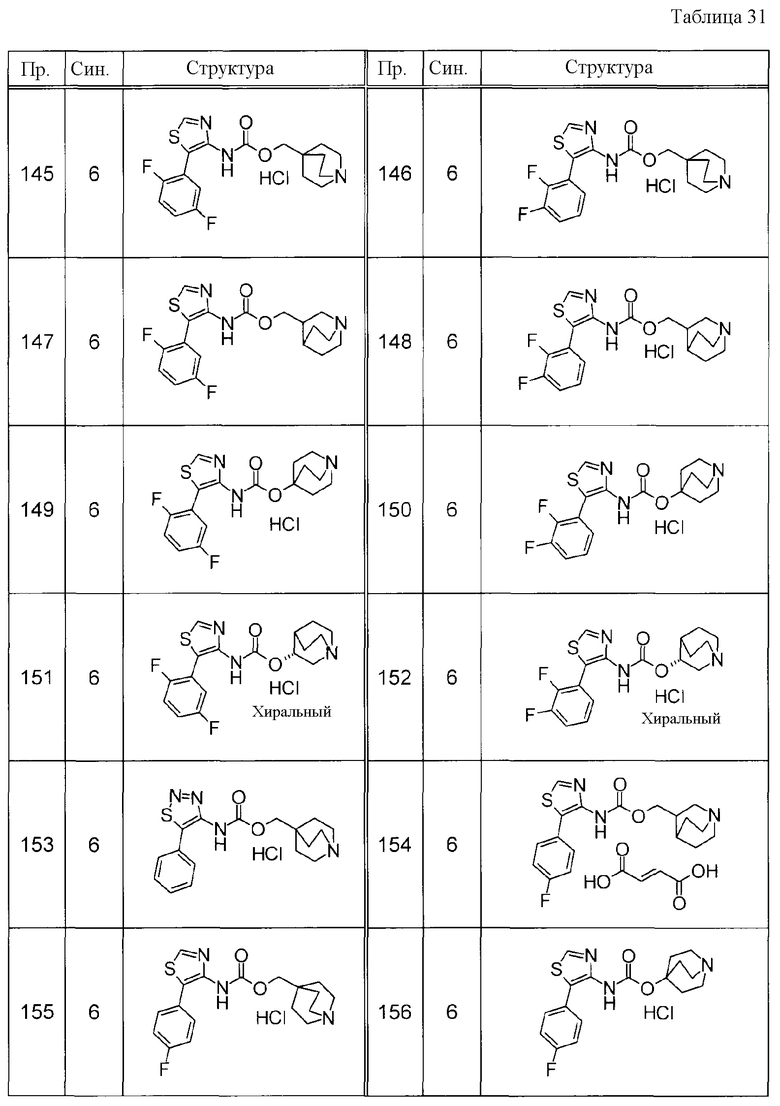
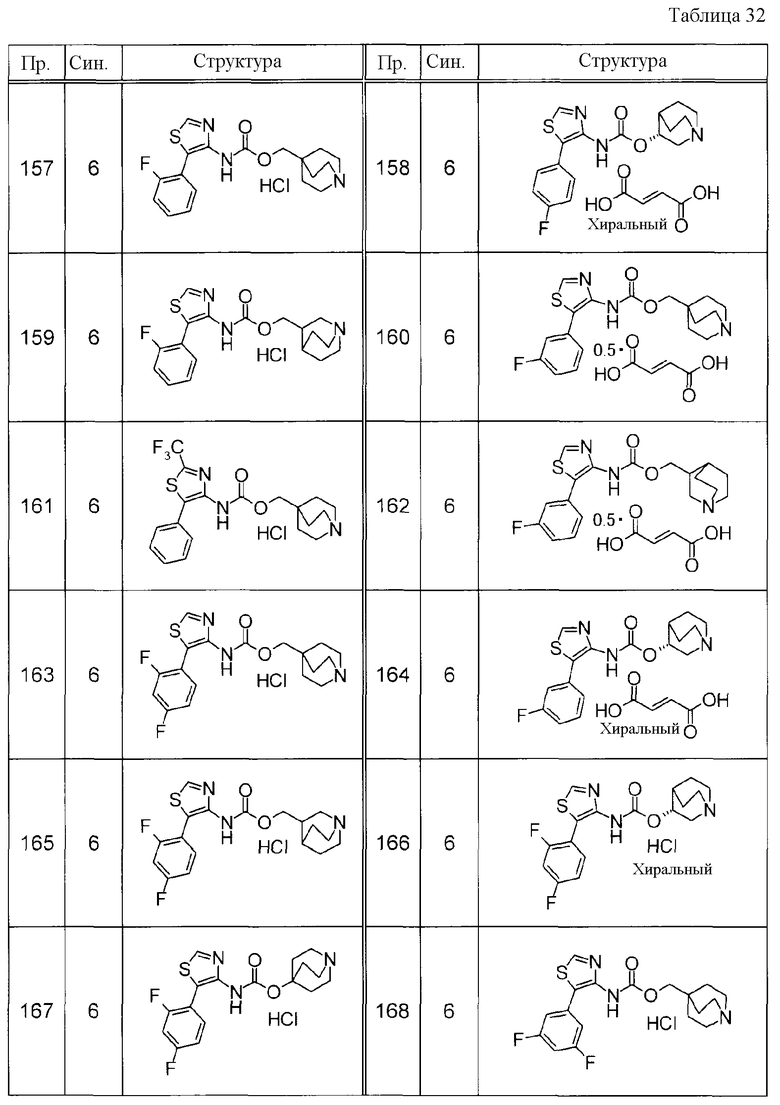
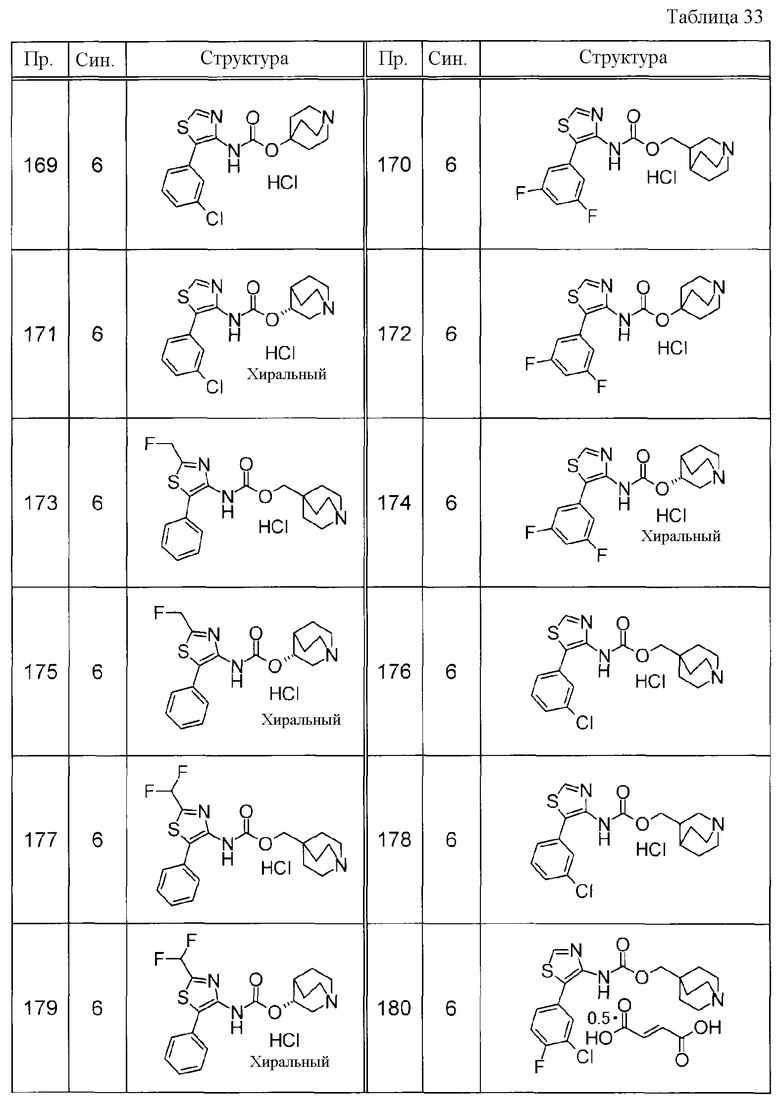
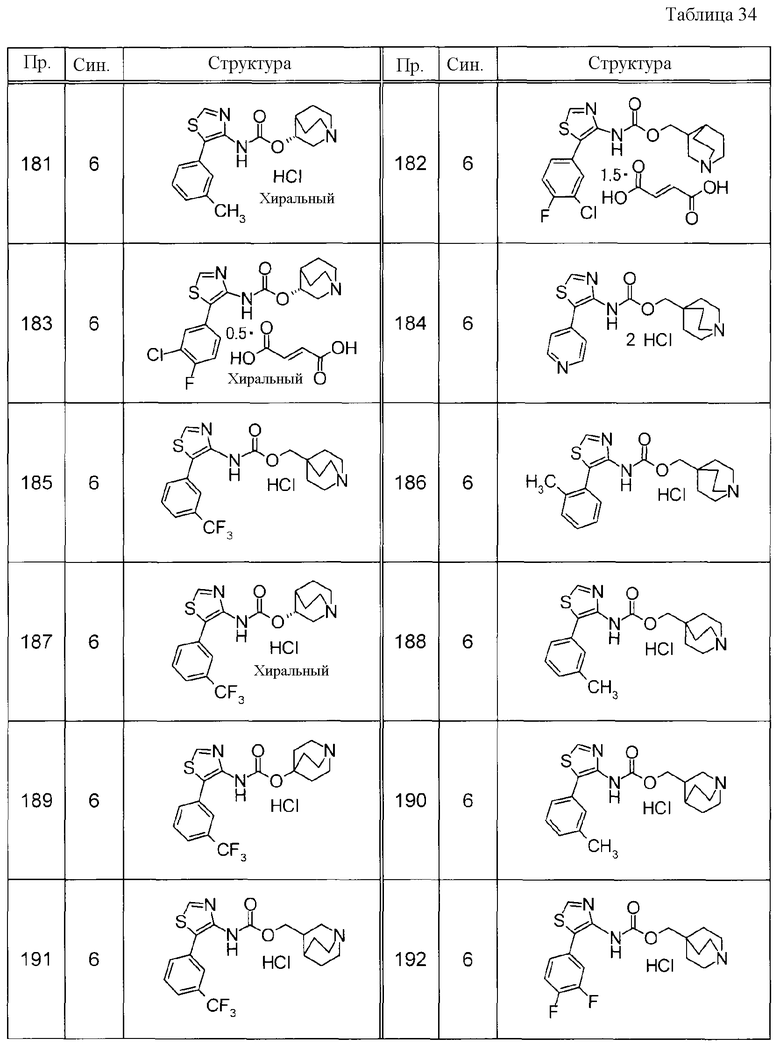
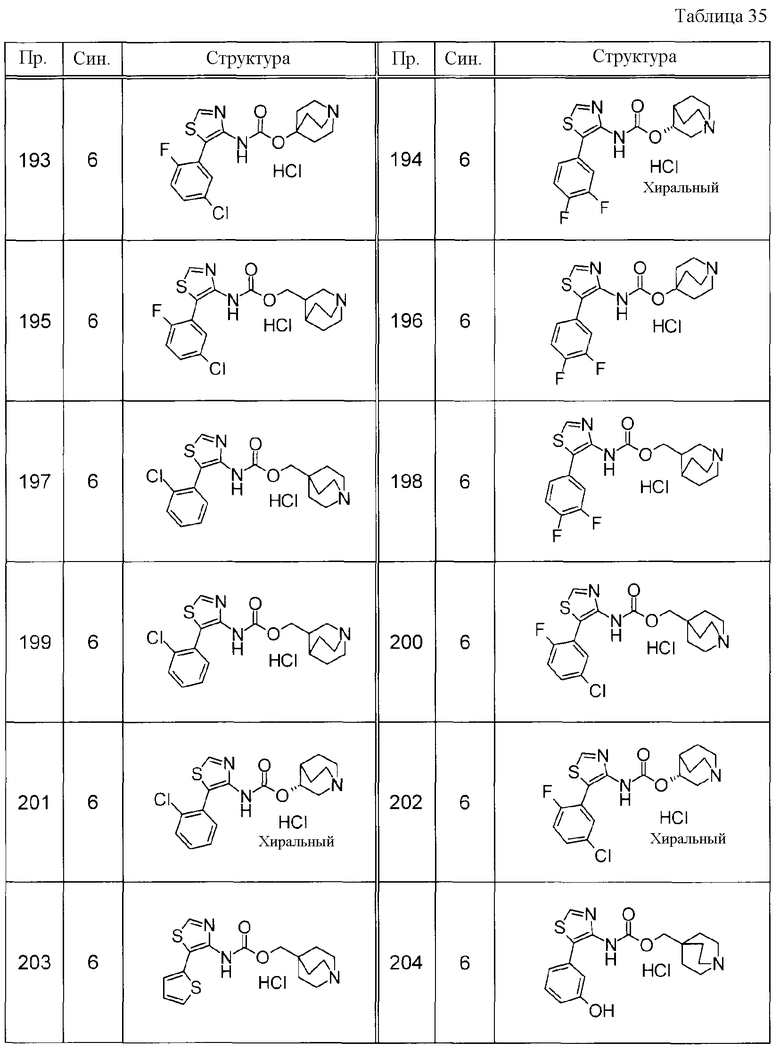
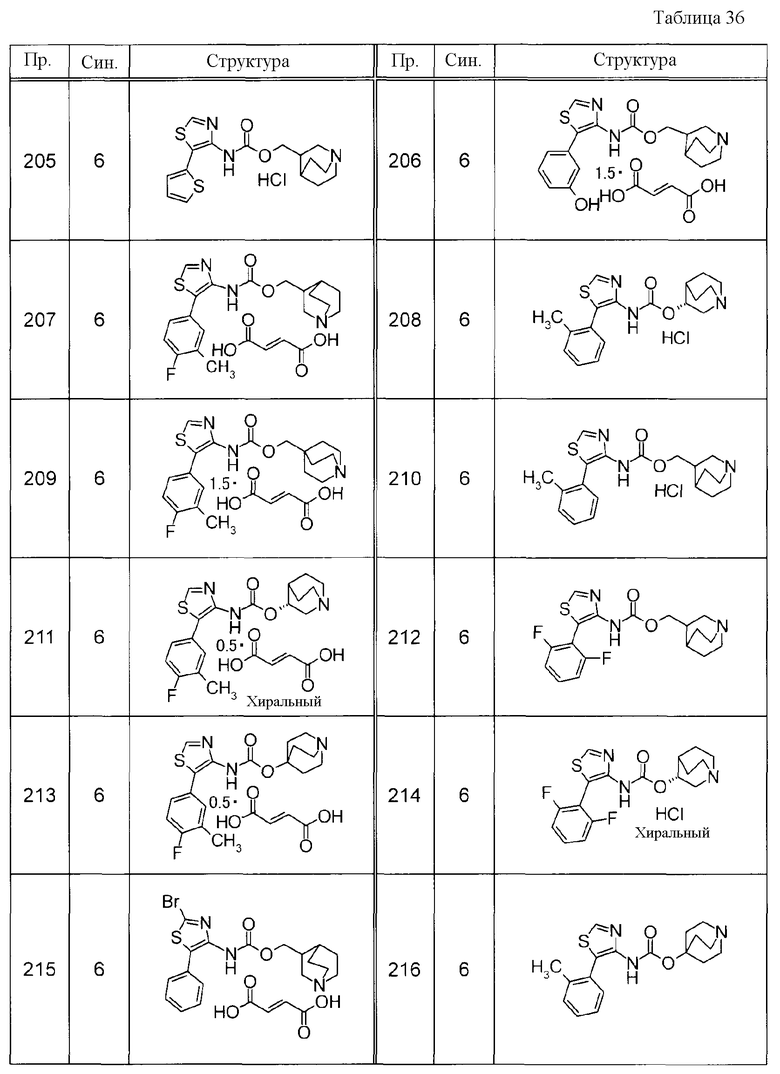

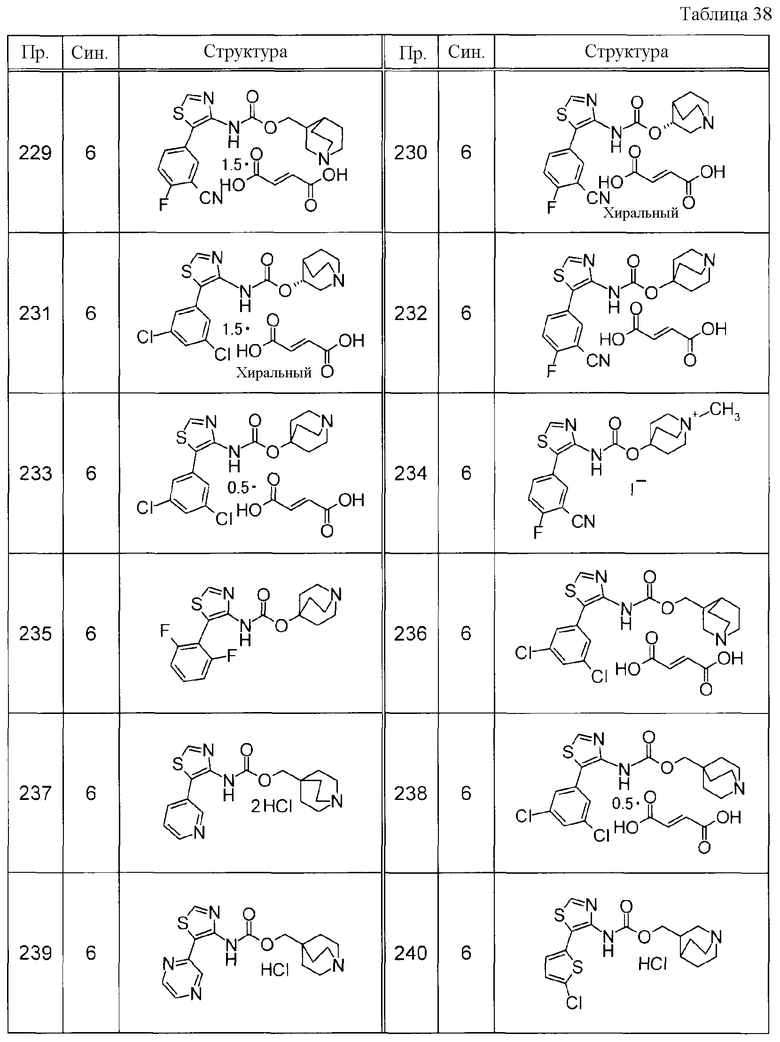
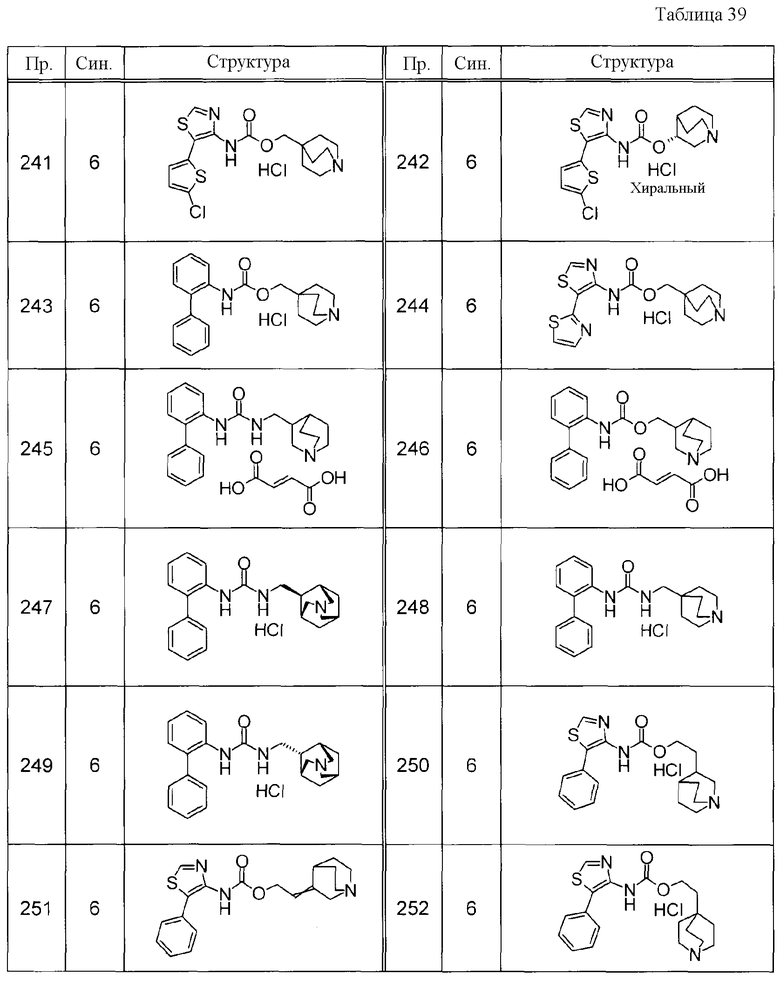
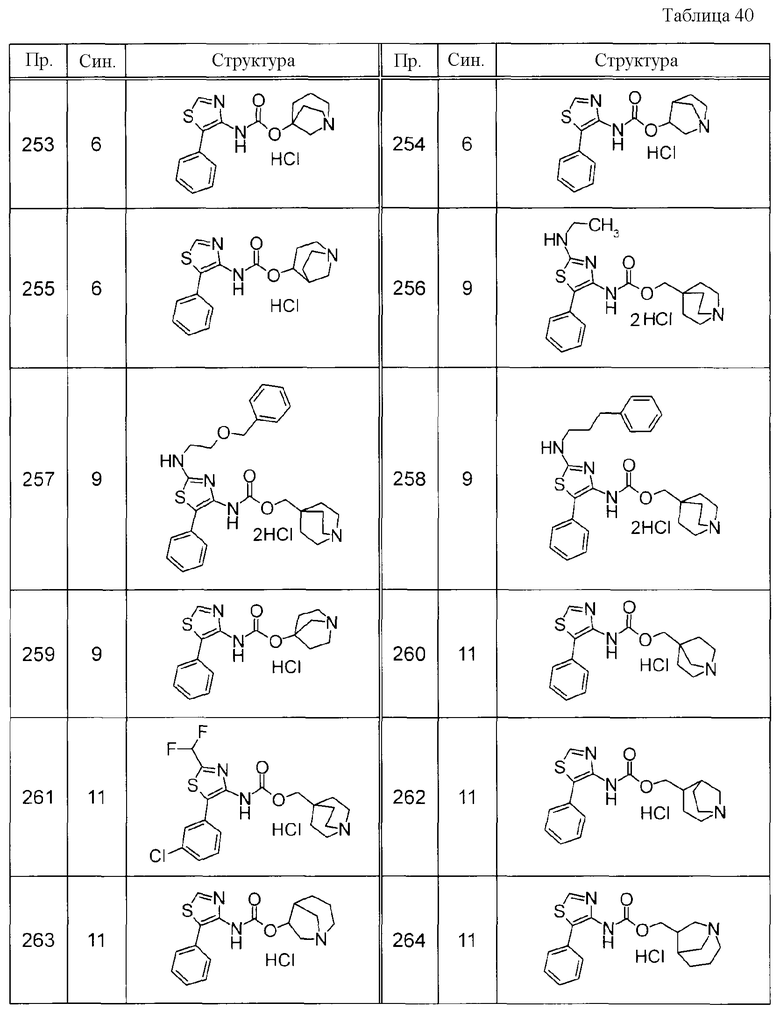












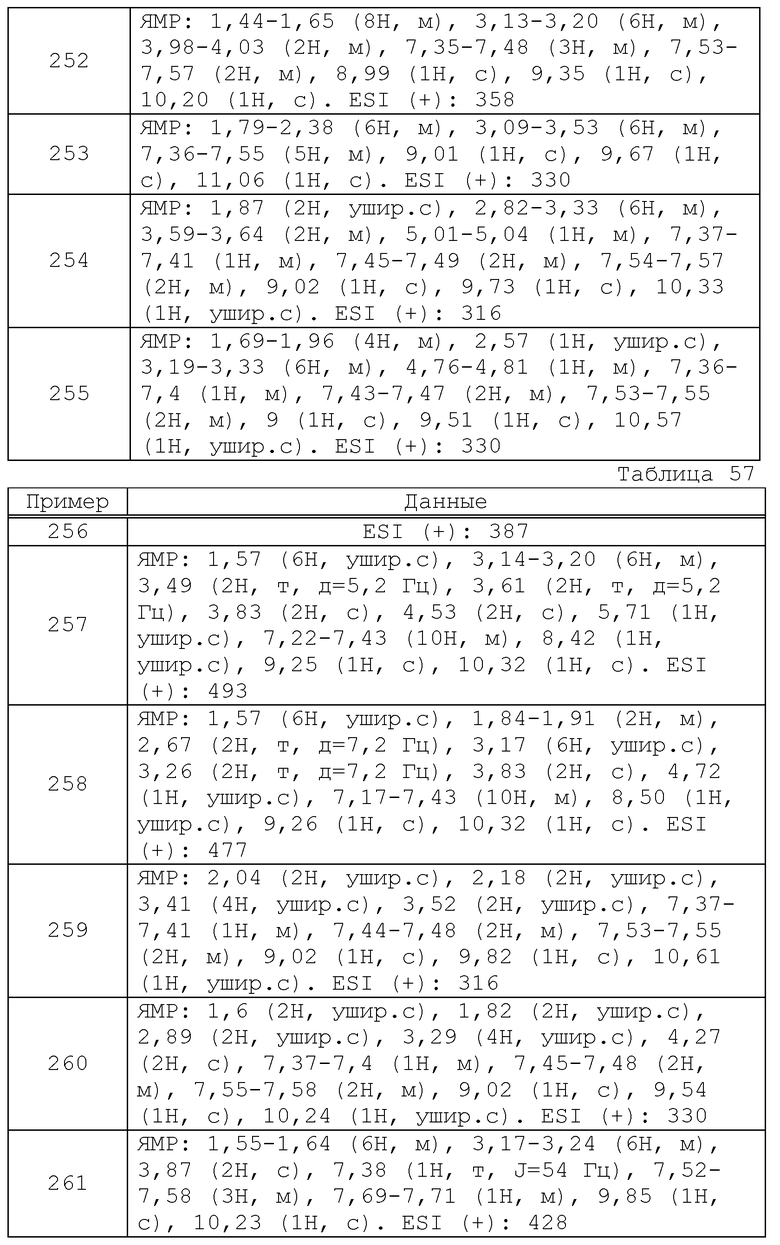

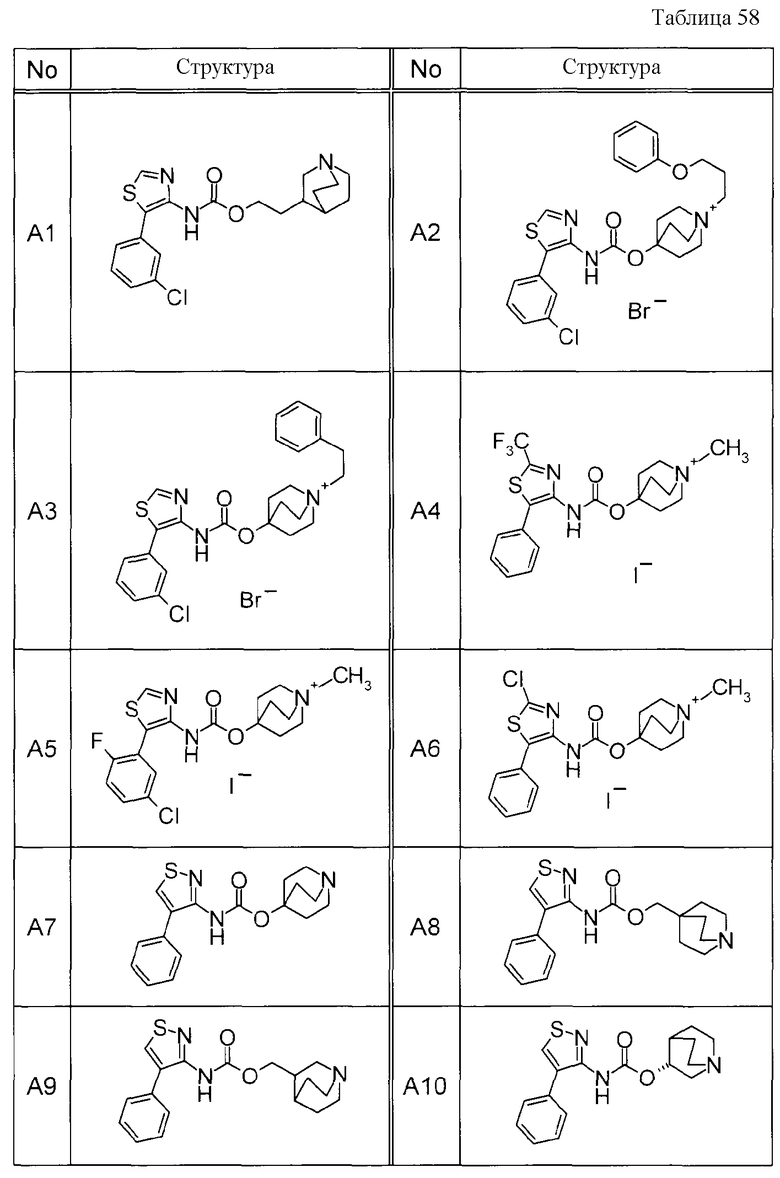
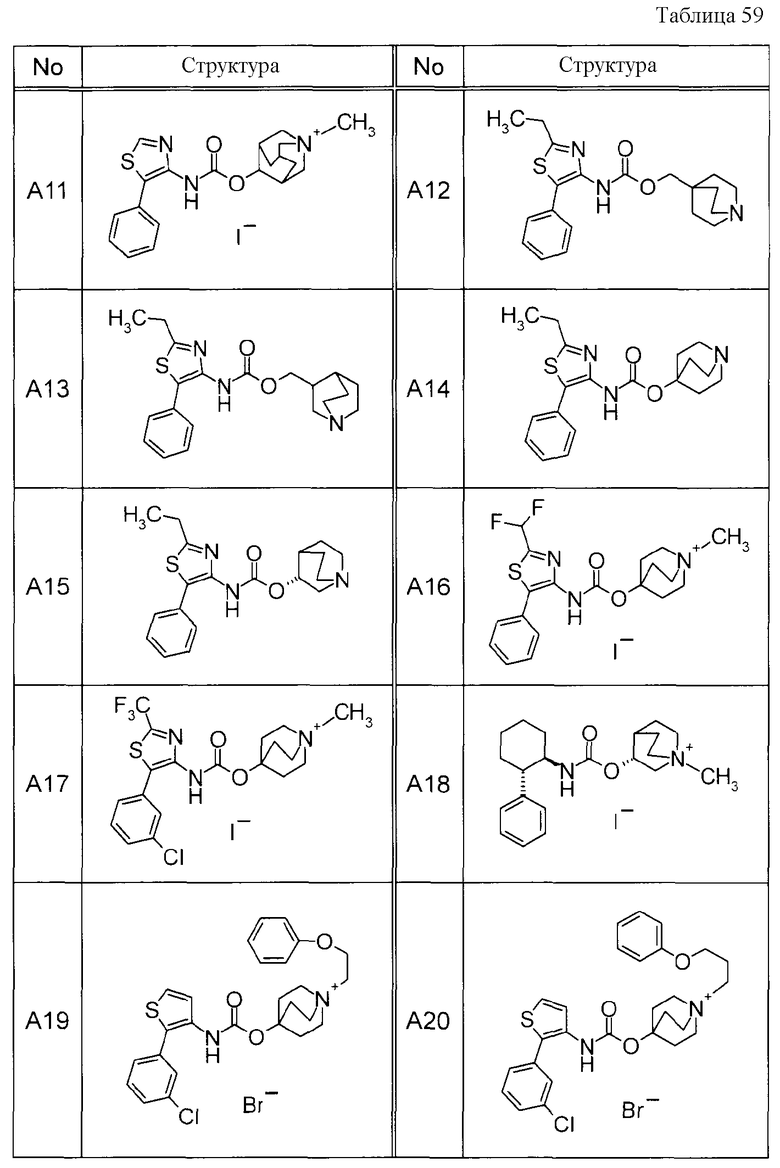
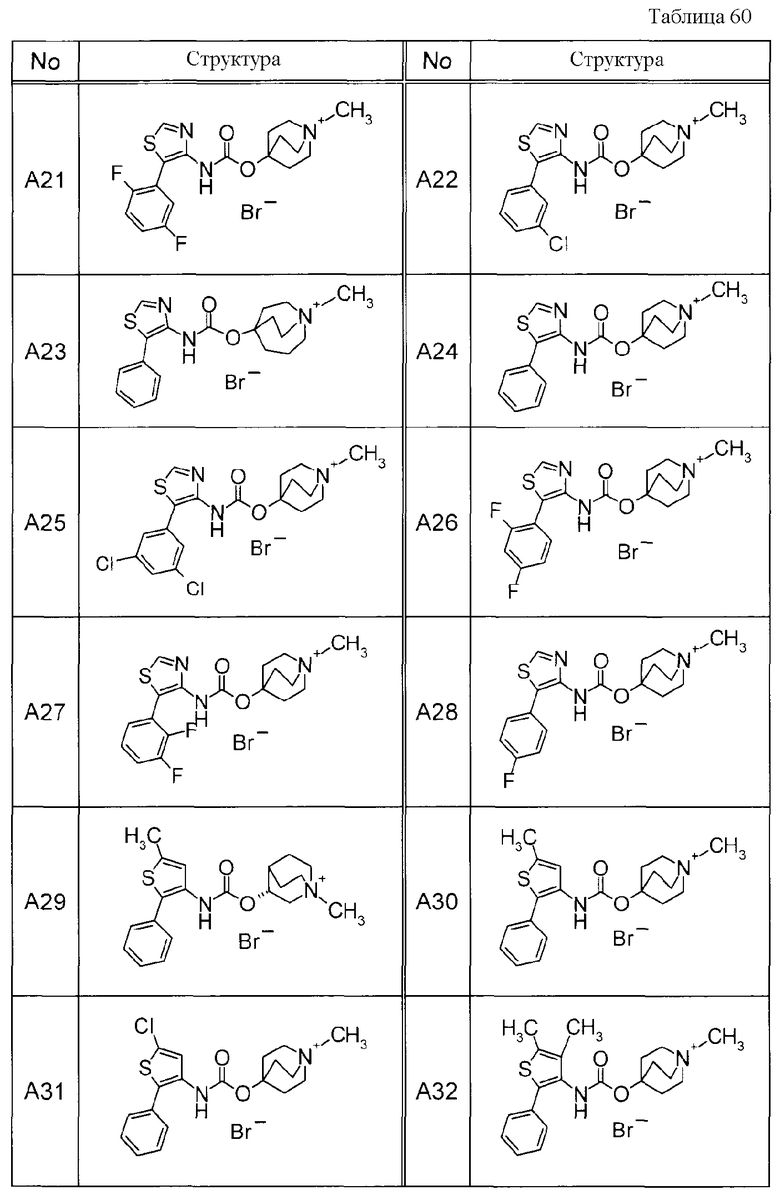
Изобретение относится к соединению формулы (I) или к его соли
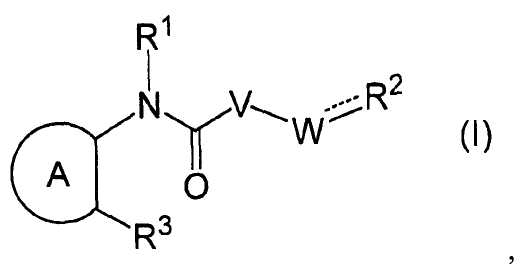 где R1 представляет собой -Н или С1-6алкил; R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, включающей формулы (а), (b), (с) и (d):
где R1 представляет собой -Н или С1-6алкил; R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, включающей формулы (а), (b), (с) и (d):
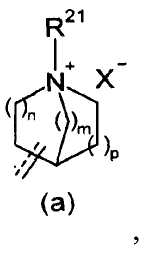
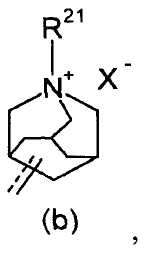
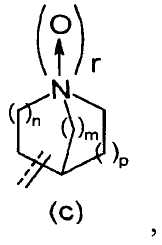
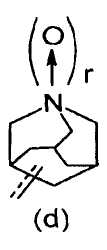
где кольцевой атом углерода в аза-кольце с внутренним мостиком может быть замещенным одной или несколькими группами R22; m, n и р имеют значение, соответственно, 1 или 2; r имеет значение 0 или 1; R21 представляет собой C1-6алкил, -C1-6алкил-О-фенил или -С1-6алкил-фенил; R22 представляет собой -C1-6алкил-циклоалкил или -С1-6алкил-фенил; R3 представляет собой тиенил, фенил, пиридил, пиразинил, тиазолил или пиразолил, каждый из которых может быть замещен одним или несколькими R31; R31 представляет собой галоген, -ОН, -CN, -CF3, С1-6алкил или -O-C1-6алкил; кольцо А представляет собой группу, состоящую из тиофена, тиазола, изотиазола, тиадиазола, оксазола, изо-оксазола, циклогексана, норборана, бензотиофена и 5,6-дигидро-4Н-циклопентатиофена, каждый из которых может быть замещен группой, выбранной из группы, состоящей из одного или нескольких RA1; где RA1 представляет собой галоген, -CN, -NH2, C1-6алкил, -O-C1-6алкил, -CONH2, -NH-C1-6алкил, -NH-C1-6алкил-О-С1-6алкил-фенил, -NH-C1-6алкил-фенил или -NH-С1-6алкил-ОН, где C1-6алкил может быть замещен одним или несколькими атомами галогена; V представляет собой -NH- или -О-; W представляет собой -(CH2)q-; q имеет значение 0, 1 или 2; X- представляет собой противоанион; и  представляет собой простую связь; при условии, что в случае, когда кольцо А представляет собой циклогексан, R3 представляет собой фенил, который может быть замещен одним или несколькими R31. Изобретение также относится к фармацевтической композиции, обладающей антагонистическим действием на мускариновый рецептор М3, на основе указанного соединения. Технический результат - получены новое соединение и фармацевтическая композиция на его основе, которые могут найти применение в медицине в качестве активного ингредиента профилактического и/или терапевтического средства для лечения воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные. 6 н. и 8 з.п. ф-лы, 60 табл.
представляет собой простую связь; при условии, что в случае, когда кольцо А представляет собой циклогексан, R3 представляет собой фенил, который может быть замещен одним или несколькими R31. Изобретение также относится к фармацевтической композиции, обладающей антагонистическим действием на мускариновый рецептор М3, на основе указанного соединения. Технический результат - получены новое соединение и фармацевтическая композиция на его основе, которые могут найти применение в медицине в качестве активного ингредиента профилактического и/или терапевтического средства для лечения воспалительных заболеваний, таких как хроническое обструктивное заболевание легких (ХОЗЛ), астма и подобные. 6 н. и 8 з.п. ф-лы, 60 табл.
1. Соединение формулы (I) или его соль
[Химическая формула 15]
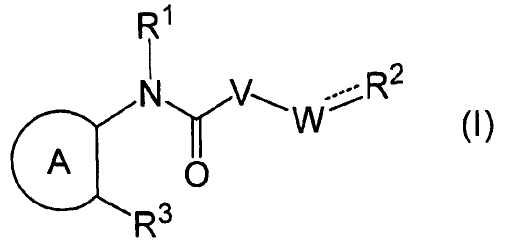
R1 представляет собой -Н или C1-6алкил;
R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, включающей формулы (а), (b), (с) и (d):
[Химическая формула 16]
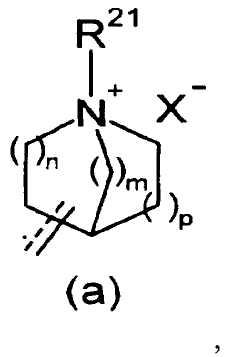
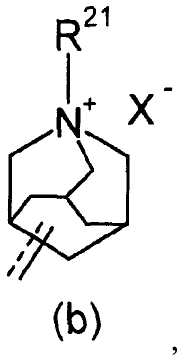
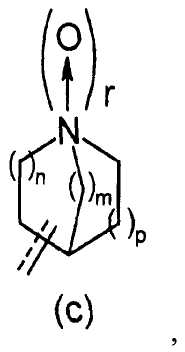
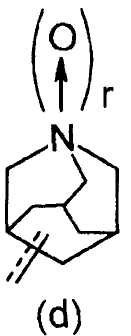
где кольцевой атом углерода в аза-кольце с внутренним мостиком может быть замещенным одной или несколькими группами R22;
m, n и р имеют значение соответственно 1 или 2;
r имеет значение 0 или 1;
R21 представляет собой C1-6алкил, -C1-6алкил-О-фенил или -C1-6алкил-фенил;
R22 представляет собой -C1-6алкил-циклоалкил или -С1-6алкил-фенил;
R3 представляет собой тиенил, фенил, пиридил, пиразинил, тиазолил или пиразолил,
каждый из которых может быть замещен одним или несколькими R31;
R31 представляет собой галоген, -ОН, -CN, -CF3, C1-6алкил или -O-C1-6алкил;
кольцо А представляет собой группу, состоящую из тиофена, тиазола, изотиазола, тиадиазола, оксазола, изо-оксазола, циклогексана, норборана, бензотиофена и 5,6-дигидро-4Н-циклопентатиофена, каждый из которых может быть замещен группой, выбранной из группы, состоящей из одного или нескольких RA1;
где RA1 представляет собой галоген, -CN, -NH2, С1-6алкил, -O-C1-6алкил, -CONH2, -NH-C1-6алкил, -NH-C1-6алкил-О-С1-6алкил-фенил, -NH-C1-6алкил-фенил или -NH-C1-6алкил-ОН,
где С1-6алкил может быть замещен одним или несколькими атомами галогена;
V представляет собой -NH- или -О-;
W представляет собой -(CH2)q-;
q имеет значение 0, 1 или 2;
X- представляет собой противоанион; и
представляет собой простую связь;
при условии, что в случае, когда кольцо А представляет собой циклогексан, R3 представляет собой фенил, который может быть замещен одним или несколькими R31.
2. Соединение или его соль по п.1, в котором кольцо А представляет собой группу, выбранную из группы, включающей тиофен, тиазол и циклогексан, каждый из которых может быть замещен группой, выбранной из группы, состоящей из одного или нескольких RA1.
3. Соединение или его соль по п.2, в котором R1 представляет собой -Н.
4. Соединение или его соль по п.3, в котором
R3 представляет собой фенил, который может быть замещен одним или несколькими R31,
где R31 представляет собой галоген, -ОН, -CN, -CF3, -C1-6алкил или -O-C1-6алкил.
5. Соединение или его соль по п.4, в котором R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, состоящей из формул (а), (b), (с) и (d), где в случае (а) или (с), m, n, p имеют значение (2, 1, 1), (1, 1, 2) или (2, 1, 2) для каждой последовательности.
6. Соединение или его соль по п.5, в котором R2 представляет собой аза-кольцо с внутренним мостиком, выбранное из группы, состоящей из формул (а) и (b), и
R21 представляет собой C1-6алкил, -С1-6алкил-О-фенил или -C1-6алкил-фенил.
7. Соединение или его соль по п.6, в котором кольцо А представляет собой тиазол, который может быть замещен группой, выбранной из группы, состоящей из одного или нескольких RA1.
8. Соединение или его свободное основание по п.7 которое представляет собой
(1) 1-азабицикло[2.2.2]окт-4-илметил(5-фенил-1,3-тиазол-4-ил)карбамат, гидрохлорид,
(2) 1-азабицикло[3.2.2]нон-5-ил(5-фенил-1,3-тиазол-4-ил)карбамат, гидрохлорид,
(3) (3R)-l-азабицикло[2.2.2]окт-3-ил[(1R,2S)-2-фенилциклогексил]карбамат, гидрохлорид,
(4) 1-азатрицикло[3.3.1.1-3,7-]дец-4-ил(5-фенил-1,3-тиазол-4-ил)карбамат, гидрохлорид,
(5) 1-азабицикло[2.2.2]окт-3-илметил(2-фенил-3-тиенил)карбамат, гидрохлорид,
(6) 1-азабицикло[2.2.2]окт-4-ил[5-(4-фторфенил)-1,3-тиазол-4-ил]карбамат, гидрохлорид,
(7) 1-азабицикло[3.2.1]окт-6-илметил(5-фенил-1,3-тиазол-4-ил)карбамат, гидрохлорид или
(8) 1-азабицикло[2.2.2]окт-3-илметил[5-(4-фторфенил)-1,3-тиазол-4-ил]карбамат, фумарат.
9. Соединение или его анионообменник по п.2, которое представляет собой
(1) 4-({[5-(3-хлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанйодид,
(2) 1-(3-фенилпропил)-3-({[(2-фенил-3-тиенил)карбамоил]окси}метил)-1-азониабицикло[2.2.2]октанбромид,
(3) 1-(2-фенилэтил)-4-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[2.2.2]октанбромид,
(4) 1-(2-феноксиэтил)-4-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[2.2.2]октанбромид,
(5) 4-({[5-(2,5-дифторфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанйодид,
(6) 1-метил-5-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[3.2.2]нонанйодид,
(7) 4-({[5-(3-хлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-(2-феноксиэтил)-1-азониабицикло[2.2.2]октанйодид,
(8) 4-({[5-(3-хлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанбромид,
(9) 4-({[5-(3,5-дихлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанйодид,
(10) 4-({[5-(2,5-дифторфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанбромид,
(11) 4-({[5-(2,4-дифторфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанбромид,
(12) 1-метил-4-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[2.2.2]октанбромид,
(13) 4-({[5-(3,5-дихлорфенил)-1,3-тиазол-4-ил]карбамоил}окси)-1-метил-1-азониабицикло[2.2.2]октанбромид или
(14) 1-метил-5-{[(5-фенил-1,3-тиазол-4-ил)карбамоил]окси}-1-азониабицикло[3.2.2]нонанбромид.
10. Фармацевтическая композиция, обладающая антагонистическим действием на мускариновый рецептор М3, содержащая эффективное количество соединения или его соли по п.1, и фармацевтически приемлемый носитель или эксципиент.
11. Фармацевтическая композиция для предупреждения или лечения воспалительного заболевания, включающая соединение или его соль по п.1.
12. Применение соединения или его соли по п.1 для получения фармацевтической композиции для предупреждения или лечения воспалительного заболевания.
13. Применение соединения или его соли по п.1 для предупреждения или лечения воспалительного заболевания.
14. Способ предупреждения или лечения воспалительного заболевания, включающий введение пациенту эффективного количества соединения или его соли по п.1.
| ЕР 0747355 А1, 11.12.1996 | |||
| JP 8198751 А, 06.08.1996 | |||
| Инвентарный блок, используемый в качестве временной опоры под пролетные фермы при строительстве морских эстакад | 1960 |
|
SU142213A1 |
| RU 2003122341 А, 27.12.2004 | |||
| ПРОИЗВОДНЫЕ ХИНУКЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2282629C2 |
| Способ измерения диэлектрического коэффициента веществ | 1941 |
|
SU72632A1 |

