ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, действующим в качестве антагонистов мускариновых рецепторов, способам получения данных производных, к содержащим их композициям и их терапевтическому применению.
УРОВЕНЬ ТЕХНИКИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Четвертичные аммониевые соли, действующие в качестве лекарственных средств, являющихся антагонистами мускариновых (M) рецепторов, в настоящее время применяют в терапии для индуцирования бронходилатации для лечения респираторных заболеваний. Примеры хорошо известных антагонистов M рецепторов представлены, например, бромидом ипратропия и бромидом тиотропия.
Некоторые химические классы, действующие в качестве лекарственных средств, являющихся селективными антагонистами M3 рецепторов, разрабатывают для лечения воспалительных или обструктивных заболеваний дыхательных путей, таких как астма и хроническое обструктивное заболевание легких (COPD).
Хинуклидинкарбаматные производные и их применение в качестве M3 антагонистов описаны, например, в WO 02/051841, WO 03/053966 и WO 2008/012290.
WO 2010/015324 описывает карбонатные производные и их применение в качестве M3 антагонистов.
Соединения настоящего изобретения характеризуются хорошей активностью в качестве M3 антагонистов и повышенной стабильностью в легких.
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
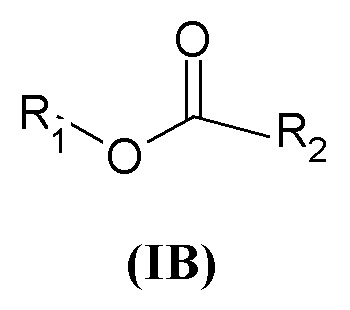
Настоящее изобретение относится к соединениям общей формулы (I), действующим в качестве антагонистов мускариновых рецепторов, способам их получения, содержащим их композициям, терапевтическому применению и комбинациям с другими фармацевтически активными ингредиентами, среди которых присутствуют, например, активные агенты, применяемые в настоящее время для лечения респираторных заболеваний, например, бета2-агонисты, кортикостероиды, P38 MAP киназные ингибиторы, IKK2, HNE ингибиторы, PDE4 ингибитор, лейкотриеновые модуляторы, NSAID и регуляторы слизистых.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
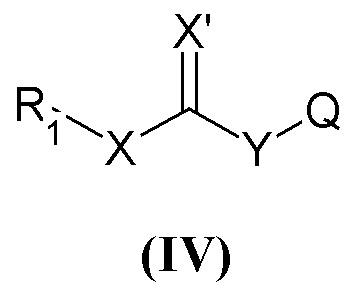
В частности, настоящее изобретение относится к соединениям общей формулы
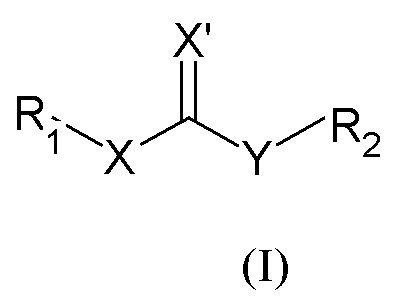
где
X может представлять собой O или S;
X' может представлять собой O или S;
Y может представлять собой NH или отсутствовать;
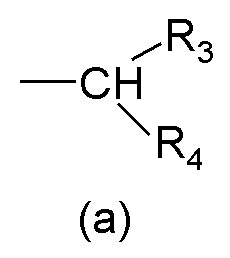
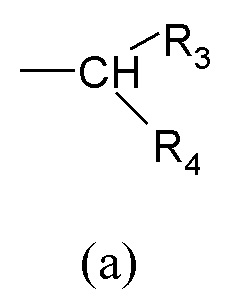
R1 выбран из группы, состоящей из арила, гетероарила, арил(C1-C6)алкила, гетероарил(C1-C6)алкила и группы формулы (a) или (b)
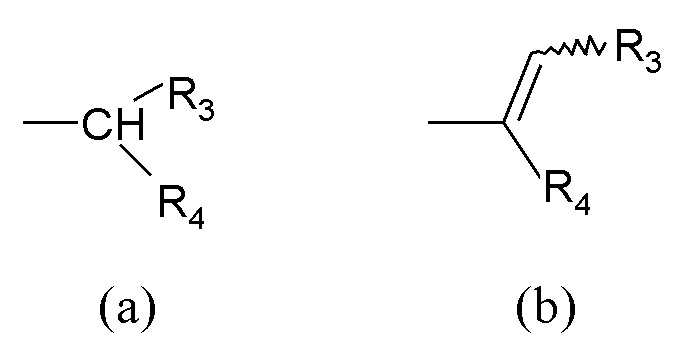
где
R3 и R4 являются одинаковыми или различными и могут независимо представлять собой H или выбраны из группы, состоящей из (C3-C8)циклоалкила, арила, арил(C1-C6)алкила, гетероарила и гетероарил(C1-C6)алкила, которые могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -OH, (C1-C6)галогеналкила, (C1-C6)алкокси, оксо (=O), -SH, -NO2, -CN, -CONH2, -COOH, (C1-C6)алкоксикарбонила, (C1-C6)алкилсульфанила, (C1-C6)алкилсульфинила, (C1-C6)алкилсульфонила и (C1-C6)алкила, или когда оба R3 и R4 независимо представляют собой арил или гетероарил, они могут быть соединены друг с другом через (CH2)r с r=0-2, образуя трициклическую кольцевую систему, где любая из метиленовых (CH2)r групп может быть необязательно замещена гетероатомом или гетероароматической группой, выбранной из O, S, N и NH, и при условии, что R3 и R4 не являются одновременно H;
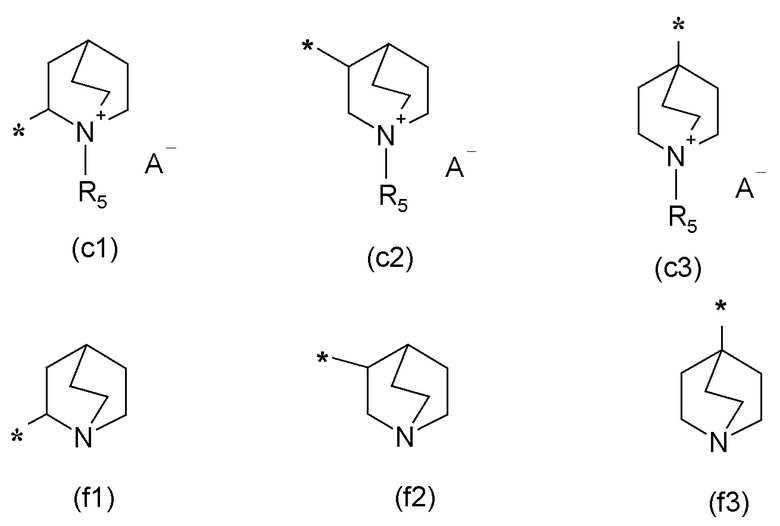
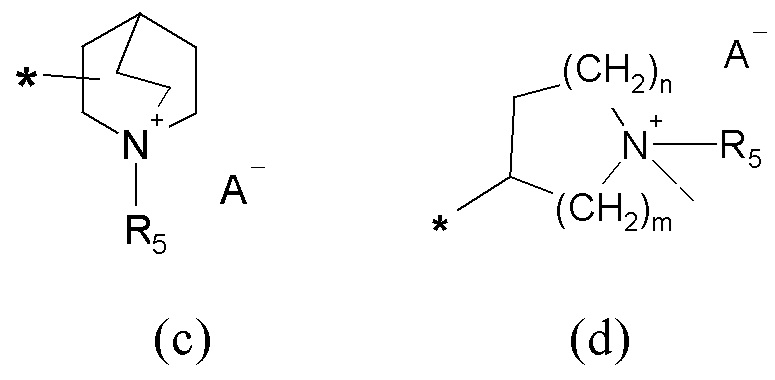
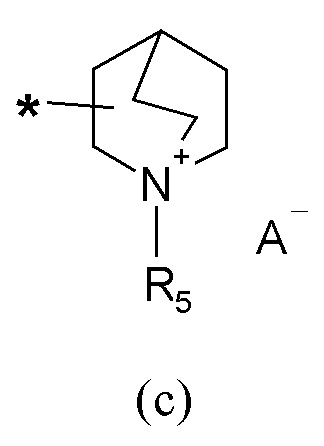
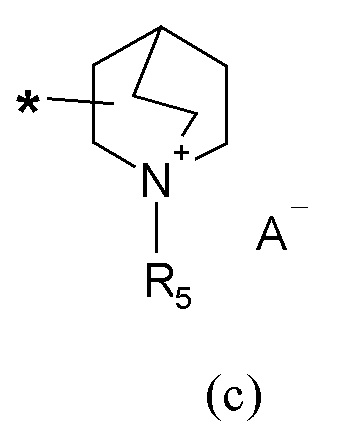
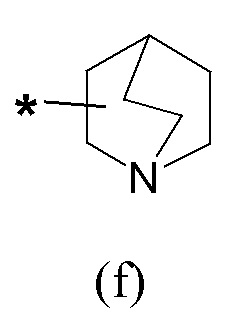
R2 представляет собой группу формулы (c) или (d):
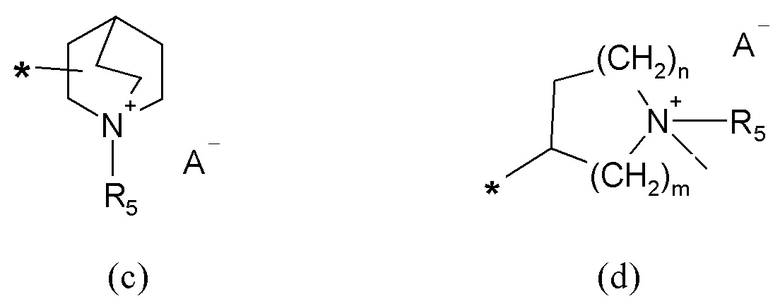
где
m=1, 2 или 3;
n=1, 2 или 3;
A- представляет собой физиологически приемлемый анион;
R5 представляет собой группу формулы (e):
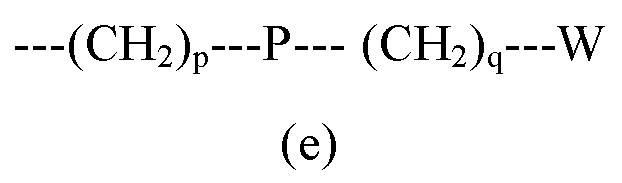
где
p равен 0 или целому числу от 1 до 4;
q равен 0 или целому числу от 1 до 4;
P отсутствует или выбран из группы, состоящей из -O-, -S-, -S(O)-, -S(O2)-, -C(O)-, -CO(O)-, -N(R6)-, -CH=CH-, -N(R6)(SO2)-, -N(R6)CO(O)-, -N(R6)C(O)-, -SO2N(R6)-, -CO(O)N(R6)- и -C(O)N(R6)-;
W выбран из группы, состоящей из H, (C1-C6)алкила, (C2-C6)алкенила, (C3-C8)циклоалкила, (C3-C8)гетероциклоалкила, арила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -N(R6)2, -CN, -CON(R6)2, -COOH, -NHCOR6, -CO2R6, (C1-C6)алкоксикарбонила, (C1-C6)алкилсульфанила, (C1-C6)алкилсульфинила, (C1-C6)алкилсульфонила, (C!-C6)алкила, (C1-C6)алкокси, арила и гетероарила;
R6 независимо представляет собой в каждом случае H или выбран из группы, состоящей из (C1-C6)алкила, (C1-C6)галогеналкила, (C2-C6)алкинила, (C2-C6)алкенила, (C3-C8)циклоалкила, гетероарила и арила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -CN, -CONH2, -COOH, (C1-C6)алкоксикарбонила, (C1-C6)алкилсульфанила, (C1-C6)алкилсульфинила, (C1-C6)алкилсульфонила, (C3-C8)циклоалкила, (C!-C6)алкила и (C1-C6)алкокси;
и их фармацевтически приемлемым солям.
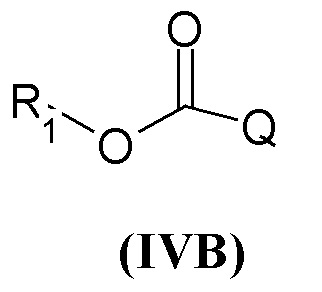
Настоящее изобретение также относится к соединениям общей формулы
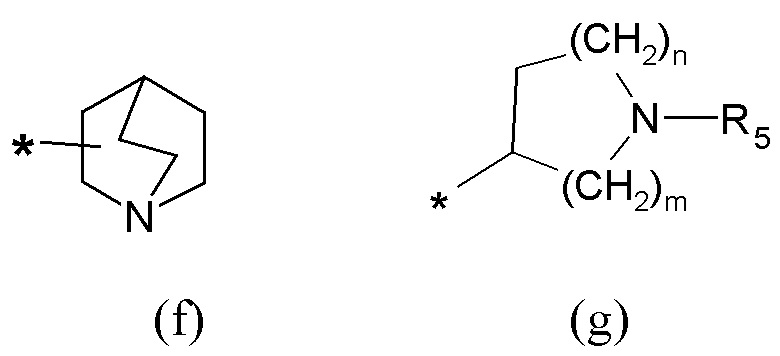
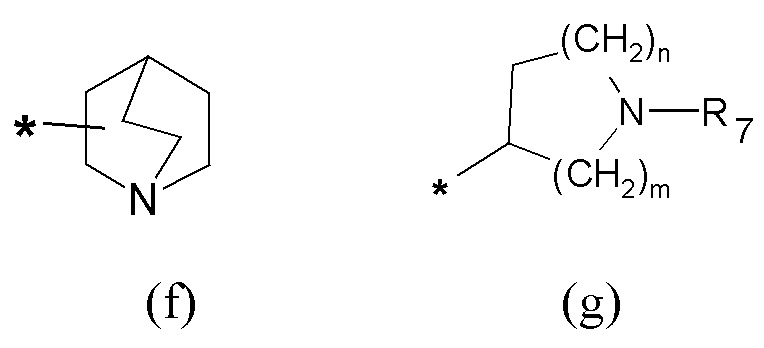
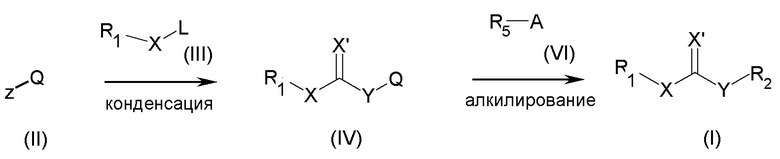
где Q представляет собой группу формулы (f) или (g)

R7 выбран из группы, состоящей из (C1-C6)алкила и арил(C1-C6)алкила, и R1, X, X', n, m и Y имеют указанные выше значения в формуле (I).
Термин "атомы галогена", как применяют в настоящем изобретении, включает фтор, хлор, бром и йод.
Выражение "(C1-C6)алкил" относится к линейным или разветвленным алкильным группам, где количество атомов углерода составляет от 1 до 6. Примерами данных групп являются метил, этил, н-пропил, изопропил, трет-бутил, пентил, гексил и подобные.
Термин "(C1-C6)алкокси" относится к алкил-окси (например, алкокси) группам, причем алкильная часть определена выше. Таким образом, примеры указанных групп включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси, гексокси и подобные.
Производное выражение "(C1-C6)алкоксикарбонил" относится к алкокси-CO-группам, где алкокси определен выше.
Выражения "(C1-C6)галогеналкил" и "(C1-C6)галогеналкокси" относятся к приведенным выше "(C1-C6)алкильным" и "(C1-C6)алкокси" группам, где один или более атомов водорода замещены одним или более атомами галогена, которые могут быть одинаковыми или отличными друг от друга.
Выражения "(C1-C6)алкилсульфанил", "(C1-C6)алкилсульфинил" и "(C1-C6)алкилсульфонил" относятся, соответственно, к алкил-S-, алкил-SO- и алкил-SO2-группам.
Выражение "(C2-C6)алкенил" относится к линейной или разветвленной углеродной цепи с одной или более двойными связями. Таким образом, примеры указанных групп включают этенил, пропенил, бутенил, пентенил, гексенил и подобные.
Выражение "(C2-C6)алкинил" относится к линейной или разветвленной углеродной цепи с одной или более тройными связями. Таким образом, примеры указанных групп включают этинил, пропинил, бутинил, пентинил, гексинил и подобные.
Выражение "(C3-C8)циклоалкил" относится к моно- или бициклоалифатическим углеводородным группам с 3-8 атомами углерода. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, бицикло[2.2.1]гепт-2-ил и подобные.
Производное выражение "(C3-C8)гетероциклоалкил" относится к (C3-C8)циклоалкильным группам, в которых по меньшей мере один атом углерода в кольце замещен гетероатомом или гетероароматической группой (например, N, NH, S или O). Примеры включают хинуклидинил, пирролидинил, пиперидинил и подобные.
Выражение "арил" относится к моно-, би- или трицклическим кольцевым системам, содержащим 5-20, предпочтительно 5-15, атомов в кольце, и где по меньшей мере одно кольцо является ароматическим.
Выражение "гетероарил" относится к моно, би- или трициклическим кольцевым системам с 5-20 атомами в кольце, предпочтительно 5-15, в которых по меньшей мере одно кольцо является ароматическим и в которых по меньшей мере один атом углерода в кольце представляет собой гетероатом или гетероароматическую группу (например, N, NH, S или O).
Примеры подходящих арильных или гетероарильных моноциклических систем включают, например, тиофеновый, бензольный, пиррольный, пиразольный, имидазольный, изоксазольный, оксазольный, изотиазольный, тиазольный, пиридиновый, имидазолидиновый, фурановый радикалы и подобные.
Примеры подходящих арильных или гетероарильных бициклических систем включают нафталиновый, бифениленовый, пуриновый, птеридиновый, бензотриазольный, хинолиновый, изохинолиновый, индольный, изоиндольный, бензотиофеновый, дигидробензодиоксиновый, дигидробензодиоксепиновый, бензооксазиновый радикалы и подобные.
Примеры подходящих арильных или гетероарильных трициклических систем включают флуореновые радикалы, а также бензоконденсированные производные приведенных выше гетероарильных бициклических систем.
Выражения "арил(C1-C6)алкил" и "гетероарил(C1-C6)алкил" относятся к "(C1-C6)алкилу", соответственно замещенному одним или более арильными или гетероарильными группами, как определено выше.
Предпочтительно физиологически приемлемые анионы A- включают анионы, выбранные из хлорида, бромида, йодида, трифторацетата, формиата, сульфата, фосфата, метансульфоната, нитрата, малеата, ацетата, цитрата, фумарата, тартрата, оксалата, сукцината, бензоата и п-толуолсульфоната, предпочтительно хлорида, бромида и трифторацетата.
Помимо наличия A- аниона, всякий раз, когда в соединениях формулы (I) присутствуют дополнительные основные аминогруппы, могут присутствовать дополнительные физиологически приемлемые анионы, среди тех, что указаны выше. Аналогично, в присутствии кислотных групп, таких как COOH группы, могут присутствовать соответствующие физиологические катионные соли, например, содержащие ионы щелочных или щелочноземельных металлов.
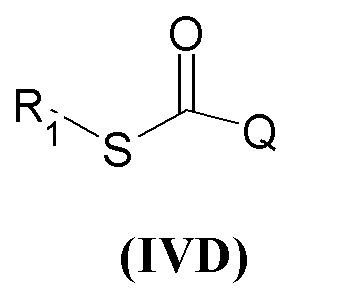
В настоящем описании и если не указано иначе, в формуле (I) или (IV) Y может представлять собой двухвалентную NH группу, или, когда она отсутствует, она очевидно представляет собой единичную связь так, чтобы получить соединения R1-X-C(=X')R2 или R1-X-C(=X')Q.
Кроме того, когда оба R3 и R4 независимо выбраны из арильных или гетероарильных групп, указанные группы могут быть соединены друг с другом через (CH2)r группы так, чтобы образовывать трициклическую кольцевую систему.
Из всего приведенного выше специалисту в данной области техники ясно, что когда r равен 0, указанные R3 и R4 группы соединены друг с другом связью.
Если не указано иначе, в формуле (I) и (IV) R2 и Q группы представлены группами (c), (d) и (f), (g), причем звездочка в последних представляет собой место присоединения в остатке молекулы.
В качестве примера можно указать следующие группы:
Ясно, что соединения общей формулы (I) и (IV) могут содержать асимметрические центры. Следовательно, настоящее изобретение также включает оптические стереоизомеры и их смеси.
Когда соединения согласно настоящему изобретению содержат два или более асимметрических центров, они могут дополнительно существовать в виде диастереомеров. Ясно, что все данные изомеры и их смеси в любом соотношении включены в объем настоящего изобретения.
Более конкретно, активные соединения формулы (I) и (IV) содержат по меньшей мере один хиральный центр, который представлен атомом углерода, содержащимся в Q или R2, и который связан непосредственно с группой Y.
Следовательно, согласно конкретному варианту осуществления в соединении (I) атом углерода R2 группы, соединенный с Y, находится в форме (S)-энантиомера, когда R2 представляет собой группу формулы (c).
Согласно предпочтительному варианту осуществления в соединении (I) атом углерода R2 группы, соединенный с Y, находится в форме (R)-энантиомера, когда R2 представляет собой группу формулы (c).
Согласно другому варианту осуществления в соединении (IV) атом углерода Q группы, соединенный с Y, находится в форме (S)-энантиомера, когда Q представляет собой группу формулы (f) или группу формулы (g), в последнем случае явно исключая вариант, когда m равен 2 и n равен 1, m равен 3 и n равен 2.
Согласно предпочтительному варианту осуществления в соединении (IV) атом углерода Q группы, соединенный с Y, находится в форме (R)-энантиомера, когда Q представляет собой группу формулы (f) или группу формулы (g), в последнем случае явно исключая вариант, когда m равен 2 и n равен 1, m равен 3 и n равен 2.
В соединениях общей формулы (I) настоящего изобретения, когда R2 представляет собой группу формулы (a), и R3 и R4 имеют разные значения, атом углерода, несущий R3 и R4, представляет собой хиральный центр.
Более того, в соединениях формулы (I), когда R1 представляет собой группу формулы (b), R3 соединен с атомом углерода, несущим двойную связь в любой из возможных (Z) или (E) конфигураций и в настоящий момент указан символом

Первая предпочтительная группа соединений представляет собой группу общей формулы (IVA)
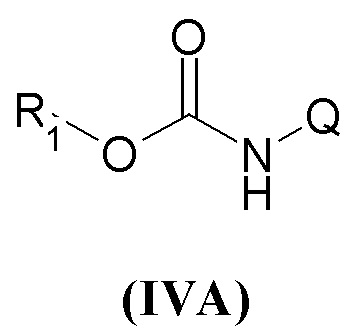
где R1 представляет собой группу формулы (a)
где R3 и R4 являются одинаковыми или различными и представляют собой H или выбраны из группы, состоящей из арила и гетероарила, которые могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из атомов галогена, (C1-C6)алкокси и (C1-C6)галогеналкила, Q представляет собой группу формулы (f) или (g)
где n=m=1, и R5 представляет собой группу формулы (e)
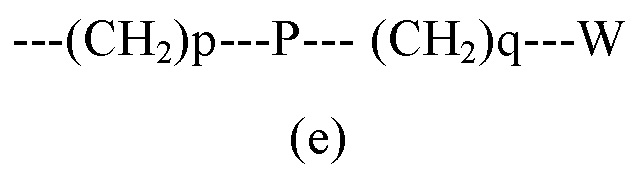
где p=0, P отсутствует, q=1, и W представляет собой арил.
Еще более предпочтительно, в данном классе есть соединения общей формулы (IVA), где R1 выбран из бис(3-фторфенил)метила, бензгидрила, (4-метоксифенил)(фенил)метила, (2-фторфенил)(4-фторфенил)метила, (2-фторфенил)(3-фторфенил)метила, ((3,4-дифторфенил)(фенил)метила, 4-(трифторметил)фенил)метила, (2-хлорфенил)(4-хлорфенил)метила и тиофен-2-илметила, и Q выбран из хинуклидинила и бензилпирролидинила.
Вторая предпочтительная группа соединений представляет собой группу общей формулы (IA)
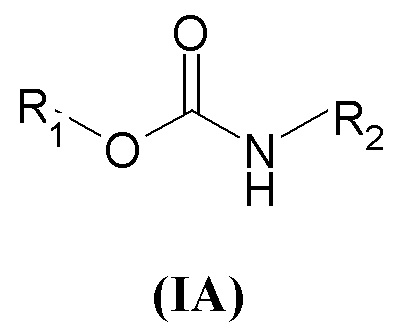
где R1 представляет собой группу формулы (a)
где R3 и R4 являются одинаковыми или различными и представляют собой H или выбраны из группы, состоящей из арила и гетероарила, которые могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из атомов галогена, (C1-C6)алкокси и (C1-C6)галогеналкила, R2 представляет собой группу формулы (c) или (d)
где n=m=1, и R5 представляет собой группу формулы (e), где p равен 0, 1, 2 или 3, P отсутствует или выбран из группы, состоящей из -O-, -CO и -CONH, q равен 0, 1 или 2, и W выбран из группы, состоящей из (C1-C6)алкила, (C2-C6)алкенила, арила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, OH, CN, (C1-C6)алкила и (C1-C6)алкокси.
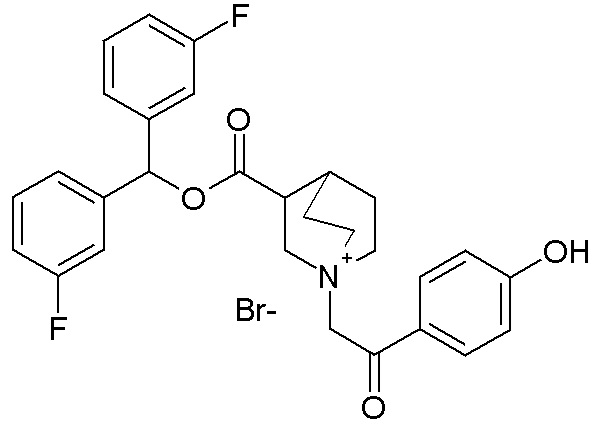
Еще более предпочтительно, в данном классе есть соединения общей формулы (IA), где R1 выбран из бис(3-фторфенил)метила, бензгидрила, бис(4-фторфенил)метила, (4-метоксифенил)(фенил)метила, (2-фторфенил)(4-фторфенил)метила), (3,4-дифторфенил)(фенил)метила, (4-(трифторметил)фенил)метила, (2-хлорфенил)(4-хлорфенил)метила и тиофен-2-илметила, и R2 выбран из (2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октанила, (2-(4-хлорфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-п-толилэтил)-1-азониабицикло[2.2.2]октанила, (2-(4-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(3-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(2-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(4-метоксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(4-гидроксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (бис(3-фторфенил)метокси)карбониламино)-1-азониабицикло[2.2.2]октанила, (2-(5-хлортиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-(тиазол-2-ил)этил)-1-азониабицикло[2.2.2]октанила, (2-оксопропил)-1-азониабицикло[2.2.2]октанила, (3-метилбут-2-енил)-1-азониабицикло[2.2.2]октанила, бензил-1-азониабицикло[2.2.2]октанила, (3-феноксипропил)-1-азониабицикло[2.2.2]октанила, (2-(5-цианотиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октанила, (2-(изоксазол-3-иламино)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанила, (2-феноксиэтил)-1-азониабицикло[2.2.2]октанила, (2,3-дигидробензофуран-5-ил)этил)-1-азониабицикло[2.2.2]октанила, (4-фторфенэтил)-1-азониабицикло[2.2.2]октанила и бензилметилпирролидинила.
Третья предпочтительная группа соединений представляет собой группу общей формулы (IVB)
где R1 представляет собой арил или группу формулы (a)

R3 и R4 являются одинаковыми или различными и представляют собой H или выбраны из группы, состоящей из арила, арил(C1-C6)алкила и гетероарила, которые могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из атомов галогена, (C1-C6)алкокси и (C1-C6)галогеналкила, или когда оба R3 и R4 независимо представляют собой арил или гетероарил, они могут быть соединены друг с другом через (CH2)r с r=0-2, образуя трициклическую кольцевую систему, где любая из метиленовых (CH2)r групп может представлять собой гетероатом или гетероароматическую группу, выбранную из O, S, N и NH, и при условии, что R3 и R4 не являются одновременно H; Q представляет собой группу формулы (f) или (g)
где n равен 1, m равен 2, и R7 представляет собой группу формулы (f) где p=0, P отсутствует, q равен 0 или 1, и W представляет собой (C1-C6)алкил.
Еще более предпочтительно, в данном классе есть соединения общей формулы (IVB), где R1 выбран из (3,4-дифторфенил)(фенил)метила, бис(3-фторфенил)метила, 1,2-дифенилэтила, бис(4-хлорфенил)метила, бис(4-фторфенил)метила, бензгидрила, (4-метоксифенил)(фенил)метила, (2-хлорфенил)(4-хлорфенил)метила, 1,2-дифенилвинила, 3-фторбензила, бензила, флуоренила, и Q выбран из хинуклидин-3-ила и метилпиперидинила.
Четвертая предпочтительная группа соединений представляет собой группу общей формулы (IB)
где R1 представляет собой арил или группу формулы (a) или (b)

где R3 и R4 являются одинаковыми или различными и представляют собой H или выбраны из группы, состоящей из арила, арил(C1-C6)алкила и гетероарила, которые могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из атомов галогена, (C1-C6)алкокси и (C1-C6)галогеналкила, или когда оба R3 и R4 независимо представляют собой арил или гетероарил, они могут быть соединены друг с другом через (CH2)r с r=0-2, где когда n=0, образуя трициклическую кольцевую систему, где любая из метиленовых (CH2)r групп может представлять собой гетероатом или гетероароматическую группу, выбранную из O, S, N и NH, и при условии, что R3 и R4 не являются одновременно H, R2 представляет собой группу формулы (c) или (d)

где n=1 или 2, m=1, и R5 представляет собой группу формулы (e), где p=1 или 3, P отсутствует или выбран из группы, состоящей из O, CO и CO(O), q=1, и W выбран из группы, состоящей из (C1-C6)алкила, (C2-C6)алкенила, арила, гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, OH, CN, (C1-C6)алкила, (C1-C6)алкоксикарбонила и (C1-C6)алкокси.
Еще более предпочтительно, в данном классе есть соединения общей формулы (IB), где R1 выбран из бис(3-фторфенил)метила, 1,2-дифенилэтила, бис(4-хлорфенил)метила, бис(4-фторфенил)метила, (4-метоксифенил)(фенил)метила, 3-фторбензила, 1,2-дифенилвинила, 3-фторбензила, бензила, (3,4-дифторфенил)(фенил)метила, флуоренила и дифенилвинила, и R2 выбран из 2-оксо-2-фенилэтил-1-азониабицикло[2.2.2]октанила, 2-оксо-2-(тиофен-2-ил)этил-1-азониабицикло[2.2.2]октанила, 2-оксо-2-(тиофен-3-ил)этил-1-азониабицикло[2.2.2]октанила, 2-(4-метоксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, 2-(4-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-п-толилэтил)-1-азониабицикло[2.2.2]октанила, 2-(5-хлортиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, 2-(4-хлорфенил)-2-оксоэтил-1-азониабицикло[2.2.2]октанила, 1-(2-оксопропил)-1-азониабицикло[2.2.2]октанила, 1-(2-трет-бутокси-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, 1-(2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октанила, 1-(2-(3-(этоксикарбонил)изоксазол-5-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, 1-(2-(4-гидроксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, 1-(2-(бензотиофен-5-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, 1-бензил-1-азониабицикло[2.2.2]октанила, 1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанила и 1-метил-1-(2-оксо-2-(тиофен-2-ил)этил)пиперидинила.
Пятая предпочтительная группа соединений представляет собой группу общей формулы (IC)
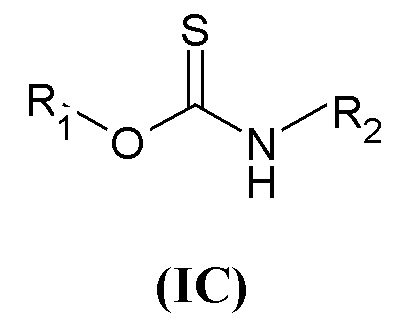
где R1 представляет собой группу формулы (a), где R3 и R4 независимо представляют собой арильные группы, которые могут быть необязательно замещены одним или более атомами галогена, R2 представляет собой группу формулы (c)
где R5 представляет собой группу формулы (e) с p=1, P представляет собой CO, q равен 0, и W представляет собой гетероарил.
Еще более предпочтительно, в данном классе есть соединения общей формулы (IC), где R1 представляет собой бис(3-фторфенил)метил, и R2 представляет собой (2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанил.
Шестая предпочтительная группа соединений представляет собой группу общей формулы (IVC)

где R1 представляет собой группу формулы (a), где R3 и R4 независимо представляют собой арильные группы, необязательно замещенные одним или более атомами галогена, Q представляет собой группу формулы (f)
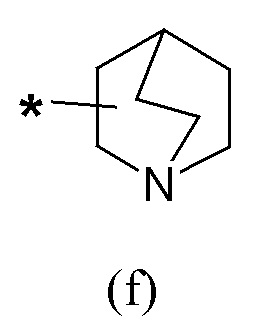
Еще более предпочтительно, в данном классе есть соединения общей формулы (IVC), где R1 представляет собой бис(3-фторфенил)метил, и Q представляет собой хинуклидин-3-ил.
Седьмая предпочтительная группа соединений представляет собой группу общей формулы (ID)
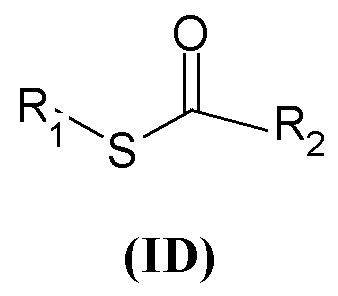
где R1 представляет собой группу формулы (a), где R3 и R4 независимо представляют собой арильные группы, которые могут быть необязательно замещены одним или более атомами галогена, R2 представляет собой группу формулы (c)
где R5 представляет собой группу формулы (e) с p=1, P представляет собой CO, q равен 0, и W представляет собой гетероарил.
Еще более предпочтительно, в данном классе есть соединения общей формулы (ID), где R1 представляет собой бензил, и R2 представляет собой (2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанил.
Восьмая предпочтительная группа соединений представляет собой группу общей формулы (IVD)
где R1 представляет собой группу формулы (a), где R3 и R4 независимо представляют собой арильные группы, необязательно замещенные одним или более атомами галогена, Q представляет собой группу формулы (f)
Еще более предпочтительно, в данном классе есть соединения общей формулы (IVC), где R1 представляет собой бензил, и Q представляет собой хинуклидинил.
Настоящее изобретение также относится к фармацевтическим композициям соединений формулы (I) или (IV) отдельно или в комбинации с или в смеси с одним или более фармацевтически приемлемыми носителями и/или эксципиентами.
Настоящее изобретение также относится к применению соединений формулы (I) или (IV) для получения лекарственного средства.
В следующем аспекте настоящее изобретение относится к применению соединений формулы (I) или (IV) для предотвращения и/или лечения любого бронхообструктивного или воспалительного заболевания, предпочтительно астмы или хронического бронхита или хронического обструктивного заболевания легких (COPD).
В следующем аспекте настоящее изобретение относится к применению соединений формулы (I) или (IV) для получения лекарственного средства для предотвращения и/или лечения любого бронхообструктивного или воспалительного заболевания, предпочтительно астмы или хронического бронхита или хронического обструктивного заболевания легких (COPD).
Кроме того, настоящее изобретение относится к способу предотвращения и/или лечения любого бронхообструктивного или воспалительного заболевания, предпочтительно астмы или хронического бронхита или хронического обструктивного заболевания легких (COPD), который включает введение нуждающемуся в этом пациенту терапевтически эффективного количества соединений общей формулы (I) или (IV).
Настоящее изобретение также относится к фармацевтическим композициям, пригодным для введения ингаляцией.
Препараты для введения ингаляцией включают порошки для ингаляции, содержащие пропеллент дозированные аэрозоли или не содержащие пропеллент составы для ингаляции.
Настоящее изобретение также относится к устройству, которое может представлять собой одно- или многодозовый порошковый ингалятор, ингалятор отмеренных доз и ингалятор мягкого тумана, содержащий соединения формулы (I) или (IV).
Настоящее изобретение также относится к набору, содержащему фармацевтические композиции соединений формулы (I) или (IV) отдельно или в комбинации с или в смеси с одним или более фармацевтически приемлемыми носителями и/или эксципиентами и устройство, которое может представлять собой одно- или многодозовый порошковый ингалятор, ингалятор отмеренных доз и ингалятор мягкого тумана, содержащий соединения формулы (I) или (IV).
Настоящее изобретение также относится к способу получения соединений общей формулы (IV) и (I), где способ включает:
(a) взаимодействие соединения общей формулы (II)
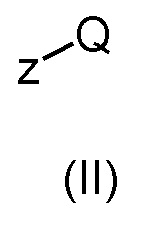
где z представляет собой карбоксильную группу или ее соответствующее ацилхлоридное производное или аминогруппу, и Q определен выше, с соединением общей формулы (III)
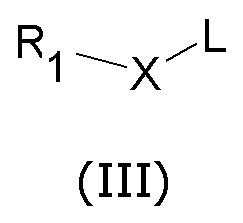
где L представляет собой H или щелочной или щелочноземельный металл, R1 и X определены выше, с получением соединения общей формулы (IV)
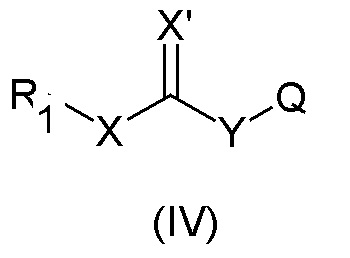
где Χ', Q и Y описаны выше, причем указанное взаимодействие проводят в присутствии подходящих количеств конденсирующих агентов; и необязательно (b) алкилирование соединений общей формулы (IV) агентом общей формулы (VI)
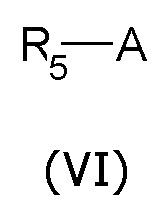
где R5 и A описаны выше, с получением соединения общей формулы (I)
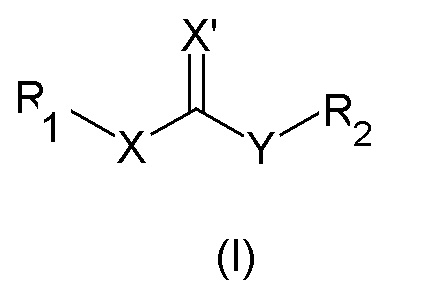
Согласно конкретным вариантам осуществления, настоящее изобретение относится к соединениям, приведенным ниже:

Соединения общей формулы (I) и (IV) можно получить согласно следующей схеме получения 1.
Схема 1
Если не указано иначе, R1, R2, Q, R5, X, X', Y и A определены выше. Ясно, что защитные группы для чувствительных или реакционноспособных групп применяют при необходимости согласно общим принципам химии. С защитными группами манипулируют согласно стандартным способам органического синтеза (Green T.W. and Wuts P.G.M. (1991) Protecting Groups in Organic Synthesis, John Wiley et Sons).
Общий способ получения соединений формулы (I) и (IV)
Соединения общей формулы (IV), относящиеся к настоящему изобретению, можно получить, исходя из соединений общей формулы (II), в которой z может представлять собой либо карбоксильную, либо аминогруппу, и Q может представлять собой группу общей формулы (f) или (g):
где m, n и R5 описаны выше.
Данные соединения (II) могут реагировать с соединениями общей формулы (III), в которой R1 и X представляют собой, как описано выше, и L может представлять собой H или щелочной или щелочноземельный металл (например, калий, литий, натрий, кальций и так далее).
Исходные вещества общей формулы (II) и (III) являются коммерчески доступными или их легко получить согласно стандартным способам, подробно освещенным в литературе.
- В случае, когда z представляет собой карбоксильную группу, конденсацию между соединениями общей формулы (II) и (III) можно осуществлять, применяя стандартные условия амидирования или пептидной конденсации. Рабочие условия выбирают на основе реакционной способности кислоты (II) относительно спирта/тиоспирта (III) и совместимости других групп, присутствующих в обоих соединениях (что касается общего рассмотрения приведенной выше реакции и ее рабочих условий, см., например, Carey, F.A. and Sundeberg, R.J. Advanced Organic Chemistry, Third Edition (1990), Plenum Press, New York и London, стр. 145).
Указанные условия включают, например, активацию кислоты (II) посредством одного или более эквивалентов коммерчески доступного конденсирующего агента, такого как карбодиимид (например, 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC) и подобных), например, в присутствии N-гидроксибензотриазола (HOBt), с последующей реакцией активированного промежуточного соединения со спиртом или тиоспиртом (III). Органическое основание, такое как триэтиламин и подобные, может также присутствовать в реакционной смеси. Активированное промежуточное соединение можно либо выделить, либо предварительно получить, либо синтезировать in situ, а затем надлежащим образом подвергать реакции с соединениями формулы (III). Подходящие растворители для реакции конденсации включают, но не ограничиваясь этим, галогенуглеродные растворители (например, дихлорметан (DCM)), тетрагидрофуран (THF), диоксан, диметилформамид (DMF) и ацетонитрил. Реакция протекает при температуре в диапазоне от приблизительно 0°C вплоть до приблизительно 170°C, в течение периода времени в диапазоне от приблизительно 1 часа вплоть до приблизительно 96 часов. Реакцию можно осуществлять при общепринятом нагревании (применяя масляную баню) или при микроволновом облучении. Реакцию можно проводить или в открытой колбе или в герметично закрытой пробирке.
В некоторых вариантах осуществления настоящего изобретения кислоту (II) наиболее удобно активировать в виде ацилгалогенида, такого как ацилхлорид (z=COCl). Данную активацию можно осуществлять в соответствии с одним из нескольких стандартных способов, описанных в литературе. Они включают, например, обработку кислоты (II) одним или более эквивалентами оксалилхлорида или тионилхлорида. Данную реакцию можно проводить в присутствии каталитического количества диметилформамида (DMF) в подходящем растворителе (например, дихлорметане) или без растворителя, при температуре в диапазоне от приблизительно 0°C до приблизительно 120°C. Активированное промежуточное соединение можно либо выделять, либо предварительно получать, либо синтезировать in situ. Затем данное промежуточное соединение можно подвергать реакции со спиртом или тиоспиртом (III), применяя известные способы для получения соединений формулы (IV). Реакции может содействовать основание, такое как триэтиламин, пиридин и 4-диметиламинопиридин, и осуществлять в подходящем растворителе (например, дихлорметане) или без растворителя. Данную реакцию проводят в диапазоне температур от приблизительно 0°C до приблизительно 140°C в течение периода времени от 1 часа до приблизительно 74 часов. Реакцию можно проводить при общепринятом нагревании (применяя масляную баню) или при микроволновом нагревании. Реакцию можно проводить или в открытой колбе или в герметично закрытой пробирке.
Затем полученные в результате эфиры (X, X'=O) и тиоэфиры (X=S; X'=O) общей формулы (IV) можно алкилировать, получая соединения общей формулы (I). Альтернативно, их можно вначале превращать в соответствующие тиеноэфиры (X=O; X'=S) или дитиоэфиры (X, X'=S) общей формулы (IV). Данное превращение можно осуществлять согласно одному из известных стандартных способов. Например, сложные эфиры (IV) можно обрабатывать реагентом Лавессона (Nicolaou, K. C. et al. Journal of the American Chemical Society, 1990, 12/17, 6263-6276) или декасульфидом тетрафосфора (Cho, D. et al. Tetrahedron, 2010, 66/30, 5583-5588), получая соответствующий тиеноэфир (IV). Аналогично, тиоэфиры можно превращать в дитиоэфиры путем обработки реагентом Лавессона (Cohen, O. et al. Tetrahedron, 2010, 66/20, 3579-3582). Затем полученный в результате тиеноэфир (X=O; X'=S) или дитиоэфир (X, X'=S) общей формулы (IV) можно алкилировать, получая соединения общей формулы (I).
- В случае, когда z представляет собой аминогруппу, соединение общей формулы (II) можно конденсировать с соединением общей формулы (III), получая карбамат, тиокарбамат или дитиокарбамат общей формулы (IV). Данную конденсацию можно осуществлять согласно одному из стандартных способов, широко освещенных в литературе (анализ подходящих реакций приводится Chaturvedi, D. Current Organic Synthesis, 2007, 3, 308 или Smith, M.B. and March, J., March's Advanced Organic Chemistry, Fifth Edition (2001), John Wiley & Sons, Inc., New York, appendix B, 1660). Например, амин (II) можно обрабатывать подходящим активирующим агентом, который можно выбрать из, но не ограничиваясь этим, 1,1'-карбонилдиимидазола, 1,1'-тиокарбонилдиимидазола, дифосфгена, трифосгена или п-нитрофенилхлорформиата. Реакции может способствовать основание, выбранное из группы, состоящей из триэтиламина, пиридина, 4-диметиламинопиридина и т.п., в подходящем растворителе (например, в диметилформамиде (DMF), тетрагидрофуране (THF), дихлорметане (DCM)). Активированное промежуточное соединение обычно получают заранее, но его можно либо синтезировать in situ, либо выделить. Затем активированный амин реагирует со спиртом или тиоспиртом формулы (III), удобнее всего, растворенным в том же растворителе, применяемом для активации соединения (II). Предпочтительно, спирт или тиоспирт предварительно обрабатывают основанием, предпочтительно выбранным из NaH, BuLi (бутиллитий) и диизопропиламида лития (LDA).
Соединения общей формулы (IV), где Q представляет собой группу формулы (f) или (g), наконец, алкилируют агентом общей формулы (VI), получая соединения общей формулы (I), где R2 представляет собой группу с описанным выше значением.
Данный тип реакций широко описан в литературе при нескольких различных условиях, например, реакции можно осуществлять без растворителя или в подходящем растворителе, выбранном из группы, состоящей из ацетонитрила, этилацетата, DMF и тетрагидрофурана. Реакция обычно протекает в диапазоне температур от приблизительно 0°C вплоть до приблизительно 170°C, в течение периода времени в диапазоне от нескольких минут вплоть до приблизительно 72 часов. Реакцию можно осуществлять при общепринятом нагревании (применяя масляную баню) или при микроволновом облучении. Реакцию можно проводить или в открытой колбе или в герметично закрытой пробирке.
Соединения общей формулы (I) можно либо считать конечными продуктами, либо их можно подвергать дополнительной реакции, получая другие соединения общей формулы (I). Таким образом, любой подходящий фрагмент R1 или R2 группы в общей формуле (I) может подвергаться ряду реакций, давая другие конечные соединения общей формулы (I).
Аналогично, необязательное солеобразование соединений формулы (I) можно осуществлять превращением подходящим образом любой из свободных кислотных групп (например, карбоксильной) или свободных аминогрупп в соответствующие фармацевтически приемлемые соли.
В данном случае также рабочие условия, применяемые для необязательного солеобразования соединений настоящего изобретения, являются общепринятыми.
Кроме того, в зависимости от любого из значений, обеспечиваемого для R1 и R2, среди тех, что приводили ранее, специалисту в данной области ясно, что в соединениях формулы (I) могут присутствовать асимметрические центры. Следовательно, настоящее изобретение также включает любой из оптических стереоизомеров, диастереомеры и их смеси, в любом соотношении.
Настоящее изобретение также относится к фармацевтическим композициям соединений формулы (I) в смеси с одним или более фармацевтически приемлемыми носителями, например, фармацевтически приемлемыми носителями, описанными в Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Введение соединений настоящего изобретения можно выполнять согласно потребностям пациента, например, перорально, назально, парентерально (подкожно, внутривенно, внутримышечно, внутригрудинно и вливанием), ингаляцией, ректально, вагинально, местно, локально, трансдермально и глазным введением.
Различные твердые пероральные лекарственные формы можно применять для введения соединений настоящего изобретения, включая такие твердые формы, как таблетки, желатиновые капсулы, капсулы, гранулы, леденцы и насыпные порошки. Соединения настоящего изобретения можно вводить отдельно или в комбинации с различными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмал) и известными эксципиентами, включая суспендирующие агенты, солюбилизаторы, буферные агенты, связующие, разрыхлители, консерванты, красители, ароматизаторы, смазывающие агенты и подобные. Капсулы, таблетки и гели с замедленным высвобождением также являются полезными при введении соединений настоящего изобретения.
Различные жидкие пероральные лекарственные формы можно также применять для введения соединений настоящего изобретения, включая водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Данные лекарственные формы могут также содержать подходящие известные инертные разбавители, такие как вода, и подходящие известные эксципиенты, такие как консерванты, смачивающие агенты, подсластители, ароматизаторы, а также агенты для эмульгирования и/или суспендирования соединений настоящего изобретения. Соединения настоящего изобретения можно вводить, например, внутривенно, в виде изотонического стерильного раствора. Другие препараты также являются возможными.
Суппозитории для ректального введения соединений настоящего изобретения могут быть получены смешиванием соединения с подходящим эксципиентом, таким как масло какао, салицилаты и полиэтиленгликоли.
Составы для вагинального введения могут быть в виде крема, геля, пасты, пены или спрея, содержащего, в добавление к активному ингредиенту, подходящие носители, которые являются также известными.
Для местного введения фармацевтическая композиция может быть в виде кремов, мазей, жидких мазей, лосьонов, эмульсий, суспензий, гелей, растворов, паст, порошков, спреев и капель, пригодных для введения на кожу, глаз, ухо или нос. Местное введение может также включать трансдермальное введение посредством трансдермальных пластырей.
Для лечения заболеваний дыхательных путей, соединения согласно настоящему изобретению предпочтительно вводить ингаляцией.
Препараты для ингаляции включают порошки для ингаляции, содержащие пропеллент дозированные аэрозоли или не содержащие пропеллент составы для ингаляции.
Для введения в виде сухого порошка можно применять известные одно- или многодозовые ингаляторы. В данном случае порошок можно заполнять в желатиновые, пластиковые или другие капсулы, картриджи или блистерные упаковки или в емкость.
Разбавитель или носитель, обычно нетоксичный и химически инертный для соединений настоящего изобретения, например, лактоза или любая другая добавка, пригодная для увеличения вдыхаемой фракции, может быть добавлен к порошкообразным соединениям настоящего изобретения.
Аэрозоли для ингаляции, содержащие пропеллентный газ, такой как гидрофторалканы, могут содержать соединения настоящего изобретения либо в виде раствора, либо в диспергируемой форме. Составы на основе пропеллента могут также содержать другие ингредиенты, такие как сорастворители, стабилизаторы и необязательно другие эксципиенты.
Составы для ингаляции, не содержащие пропеллент, содержащие соединения настоящего изобретения, могут быть в виде растворов или суспензий в водной, спиртовой или водно-спиртовой среде, и их можно доставлять известными струйными или ультразвуковыми распылителями или ингаляторами мягкого тумана, такими как Respimat®.
Соединения настоящего изобретения можно вводить в виде единственного активного агента или в комбинации с другими фармацевтически активными ингредиентами, включая ингредиенты, применяемые в настоящее время для лечения респираторных заболеваний, например, бета2-агонисты, кортикостероиды и антихолинергические или антимускариновые агенты.
Дозы соединений настоящего изобретения зависят от ряда факторов, включая конкретное заболевание, которое лечат, тяжесть симптомов, путь введения, частоту интервала дозирования, конкретное применяемое соединение, эффективность, токсикологический профиль и фармакокинетический профиль соединения.
Предпочтительно, соединения формулы (I) можно вводить, например, при дозе, содержащей от 0,001 до 1000 мг/день, предпочтительно от 0,1 до 500 мг/день.
Когда соединения формулы (I) вводят путем ингаляции, их предпочтительно предоставлять в дозе, содержащей от 0,001 до 500 мг/день, предпочтительно от 0,1 до 200 мг/день.
Соединения формулы (I) можно вводить для предотвращения и/или лечения бронхообструктивных или воспалительных заболеваний, таких как астма, хронический бронхит, хроническое обструктивное заболевание легких (COPD), бронхиальная гиперреактивность, кашель, эмфизема или ринит; урологических заболеваний, таких как недержание мочи, поллакиурия, цистоспазм, хронический цистит и гиперактивный мочевой пузырь (OAB); желудочно-кишечных расстройств, таких как синдром раздраженного кишечника (bowel syndrome), спастический колит, дивертикулит, язвенная болезнь, двигательная активность желудочно-кишечного тракта или секреция желудочной кислоты; сухости во рту, расширения зрачков; тахикардии; офтальмологических вмешательств, сердечно-сосудистых заболеваний, таких как вагальная синусовая брадикардия.
Настоящее изобретение будет далее описано посредством следующих примеров. Если не указано иначе, все исходные вещества получали из коммерческих источников и применяли без любой дополнительной очистки; все реакции проводят в инертной атмосфере и в сухих растворителях.
ПРИМЕР 1
Получение (R)-бис(3-фторфенил)метилхинуклидин-3-илкарбамата (соединение 1)
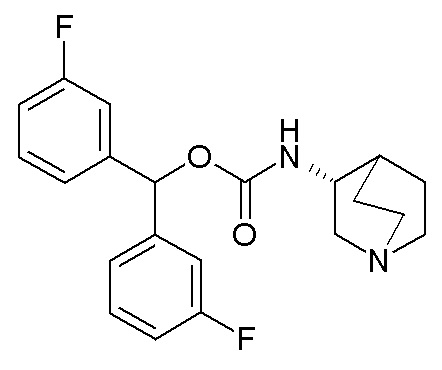
В первой колбе дигидрохлорид (R)-хинуклидин-3-амина (500 мг, 2,51 ммоль) растворяли в MeOH (25 мл) и воде (2,50 мл). Добавляли бикарбонат натрия (211 мг, 2,51 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь упаривали досуха. Остаток растворяли в сухом DMF (25,0 мл) и добавляли CDI (407 мг, 2,51 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов.
Во второй колбе бис(3-фторфенил)метанол (1,11 г, 5,02 ммоль) растворяли в сухом DMF (25 мл) и обрабатывали порциями гидридом натрия (60% суспензия в минеральном масле, 201 мг, 5,02 ммоль) при 0°C. Эту вторую реакционную смесь перемешивали при комнатной температуре в течение 30 минут и затем выливали в первую колбу.
Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Затем реакционную смесь распределяли между Et2O и водой. Органическую фазу сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1 - DCM/MeOH=75/25 + 0,5% TEA), получая (R)-бис(3-фторфенил)метилхинуклидин-3-илкарбамат (419 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,63 (д, 1H), 7,35-7,49 (м, 2H), 7,19-7,32 (м, 4H), 7,04-7,18 (м, 2H), 6,68 (с, 1H), 3,42-3,58 (м, 1H), 2,93-3,15 (м, 1H), 2,71-2,85 (м, 1H), 2,55-2,70 (м, 3H), 2,41-2,47 (м, 1H), 1,65-1,83 (м, 2H), 1,36-1,64 (м, 2H), 1,09-1,35 (м, 1H).
ПРИМЕР 2
Получение (R)-бензгидрилхинуклидин-3-илкарбамата (соединение 2)
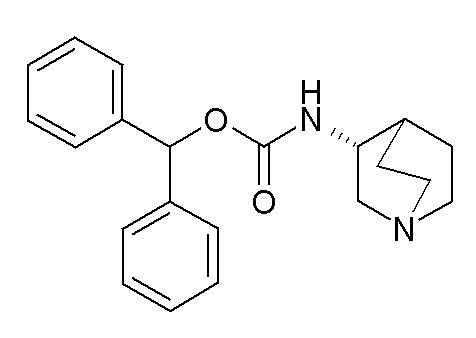
В первой колбе дигидрохлорид (R)-хинуклидин-3-амина (0,10 г, 0,50 ммоль) растворяли в MeOH (5 мл) и воде (0,5 мл). Добавляли бикарбонат натрия (84,0 мг, 1,00 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь упаривали досуха. Твердое вещество растворяли в сухом DMF (5 мл) и добавляли CDI (81,0 мг, 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 7 часов.
Во второй колбе дифенилметанол (0,18 г, 1,00 ммоль) растворяли в сухом DMF (5 мл) и обрабатывали порциями NaH (40,0 мг, 1,00 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 20 минут и затем выливали в первую колбу.
Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь распределяли между Et2O и водой. Органическую фазу промывали соляным раствором, сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1 - DCM/MeOH=75/25 + 0,5% TEA), получая (R)-бензгидрилхинуклидин-3-илкарбамат (105 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,57 (д, 1H), 7,18-7,46 (м, 10H), 6,66 (с, 1H), 3,40-3,59 (м, 1H), 2,96-3,13 (м, 1H), 2,56-2,89 (м, 4H), 2,40-2,48 (м, 1H), 1,67-1,81 (м, 2H), 1,37-1,64 (м, 2H), 1,12-1,35 (м, 1H).
ПРИМЕР 3
Получение (R)-бис(4-фторфенил)метилхинуклидин-3-илкарбамата (соединение 3)
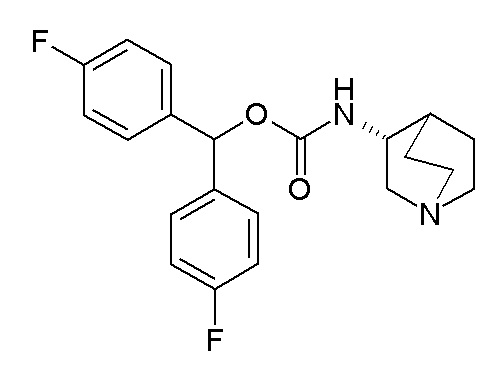
В первой колбе дигидрохлорид (R)-хинуклидин-3-амина (50,0 мг, 0,25 ммоль) растворяли в MeOH (2,5 мл) и воде (0,25 мл). Добавляли бикарбонат натрия (42,0 мг, 0,50 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь упаривали досуха. Твердое вещество растворяли в сухом DMF (2,50 мл) и добавляли CDI (40,5 мг, 0,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 7 часов.
Во второй колбе бис(4-фторфенил)метанол (111 мг, 0,50 ммоль) растворяли в сухом DMF (5 мл) и добавляли порциями при 0°C гидрид натрия (20,0 мг, 0,50 ммоль). Реакционную смесь перемешивали при 0°C в течение 5 минут и затем выливали в первую колбу, охлажденную до 0°C.
Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь распределяли между Et2O и водой. Органическую фазу сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1 - DCM/MeOH=75/25 + 0,5% TEA), получая (R)-бис(4-фторфенил)метилхинуклидин-3-илкарбамат (37,0 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,60 (д, 1H), 7,40 (дд, 4H), 7,03-7,30 (м, 4H), 6,68 (с, 1H), 3,37-3,60 (м, 1H), 2,95-3,16 (м, 1H), 2,74-2,89 (м, 1H), 2,55-2,70 (м, 3H), 2,45 (д, 1H), 1,65-1,84 (м, 2H), 1,36-1,64 (м, 2H), 1,21-1,36 (м, 1H).
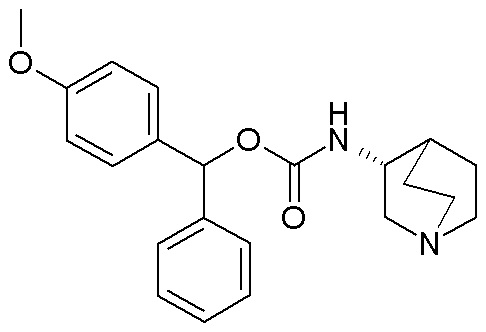
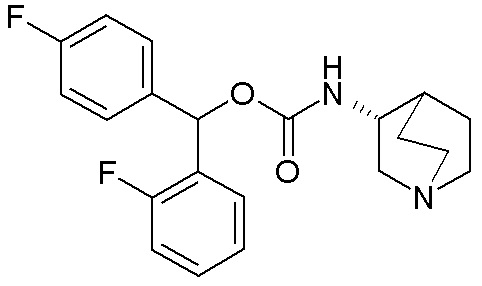
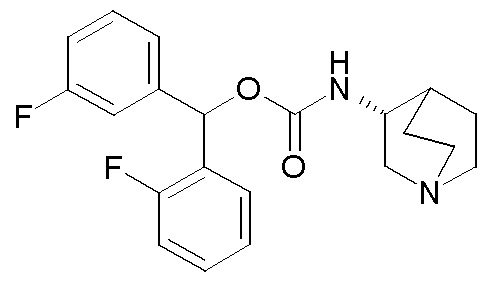
Следующие соединения получали, следуя способу, описанному в примере 3, применяя подходящие спирты вместо бис(4-фторфенил)метанола. Данные соединения получали в виде смеси диастереомеров.
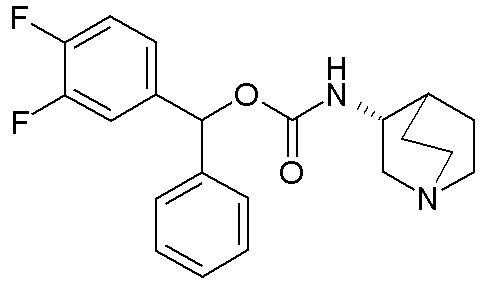
ПРИМЕР 4
Получение бис(3-фторфенил)метилхинуклидин-3-карбоксилата (соединение 8)
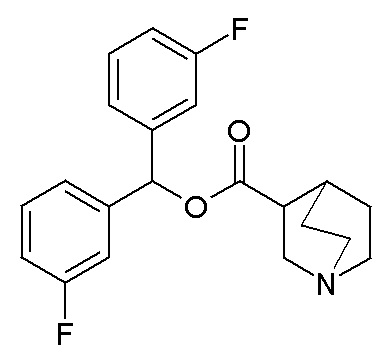
Гидрохлорид хинуклидин-3-карбоновой кислоты (817 мг, 4,26 ммоль), EDC (1,23 г, 6,39 ммоль) и HOBT (979 мг, 6,39 ммоль) растворяли в сухом DMF (40 мл). Добавляли DIPEA (2,61 мл, 14,9 ммоль) и бис(3-фторфенил)метанол (1,03 мг, 4,69 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли водой и экстрагировали несколько раз Et2O. Органические фазы собирали и промывали соляным раствором, сушили (Na2SO4), фильтровали и упаривали. Остаток очищали флэш-хроматографией (DCM/MeOH=9/1), получая бис(3-фторфенил)метилхинуклидин-3-карбоксилат (874 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,42 (тд, 2H), 7,22-7,34 (м, 4H), 7,05-7,19 (м, 2H), 6,85 (с, 1H), 2,99-3,12 (м, 1H), 2,86-2,99 (м, 1H), 2,59-2,83 (м, 5H), 2,13-2,24 (м, 1H), 1,41-1,72 (м, 2H), 1,15-1,40 (м, 2H).
ПРИМЕР 5
Получение 1,2-дифенилэтилхинуклидин-3-карбоксилата (соединение 9)
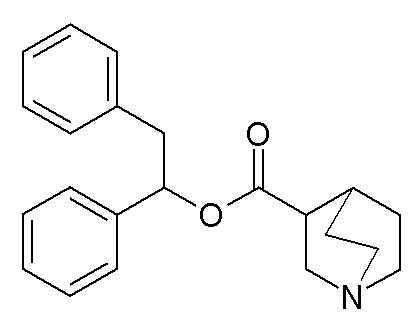
Гидрохлорид хинуклидин-3-карбоновой кислоты (160 мг, 0,83 ммоль), EDC (290 мг, 1,51 ммоль) и HOBT (232 мг, 1,51 ммоль) растворяли в сухом DMF (8 мл). Добавляли 1,2-дифенилэтанол (150 мг, 0,76 ммоль) и TEA (0,32 мл, 2,27 ммоль), и полученную в результате смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли гидрохлорид хинуклидин-3-карбоновой кислоты (72,5 мг, 0,38 ммоль), EDC (87 мг, 0,45 ммоль) и HOBT (57,9 мг, 0,38 ммоль), с последующим добавлением TEA (0,16 мл, 1,13 ммоль), и смесь перемешивали в течение дополнительных 32 часов. Смесь разбавляли водой и экстрагировали три раза Et2O. Органические фазы собирали, промывали соляным раствором, сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1), получая 1,2-дифенилэтилхинуклидин-3-карбоксилат (136 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 6,83-7,61 (м, 10H), 5,68-6,17 (м, 1H), 3,01-3,25 (м, 2H), 2,54-2,98 (м, 7H), 1,76-1,90 (м, 1H), 1,32-1,64 (м, 2H), 0,80-1,22 (м, 2H).
ПРИМЕР 6
Получение бис(4-хлорфенил)метилхинуклидин-3-карбоксилата (соединение 10)
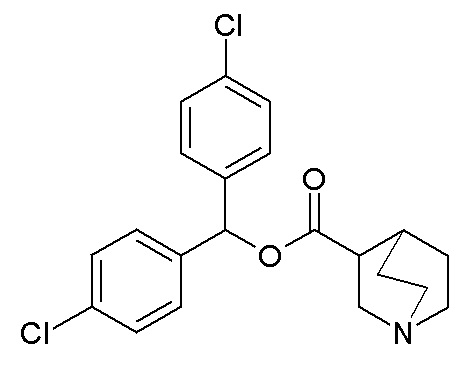
Гидрохлорид хинуклидин-3-карбоновой кислоты (150 мг, 0,78 ммоль), EDC (225 мг, 1,17 ммоль) и HOBT (180 мг, 1,17 ммоль) растворяли в сухом THF (7,5 мл). Добавляли бис(4-хлорфенил)метанол (218 мг, 0,86 ммоль), с последующим добавлением TEA (382 мкл, 2,74 ммоль). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи. THF удаляли в вакууме, и неочищенное вещество распределяли между EtOAc и водой. Органическую фазу промывали насыщенным NaHCO3, сушили над сульфатом натрия, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (EtOAc/MeOH=8/2 - 7:3 + 1% NH4OH), получая бис(4-хлорфенил)метилхинуклидин-3-карбоксилат (85,0 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,30-7,54 (м, 8H), 6,89 (с, 1H), 3,33-3,55 (м, 2H), 2,94-3,26 (м, 5H), 2,54-2,65 (м, 1H), 1,78-2,10 (м, 2H), 1,59-1,78 (м, 1H), 1,31-1,55 (м, 1H).
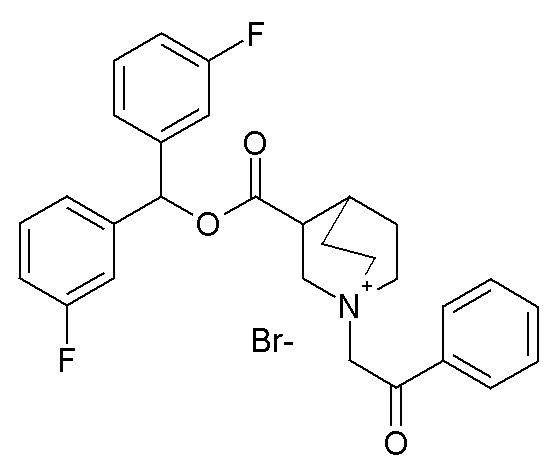
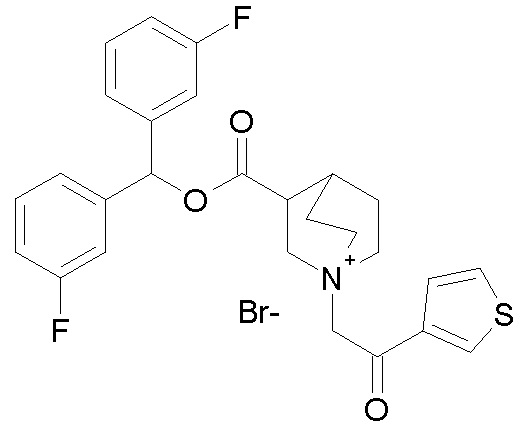
ПРИМЕР 7
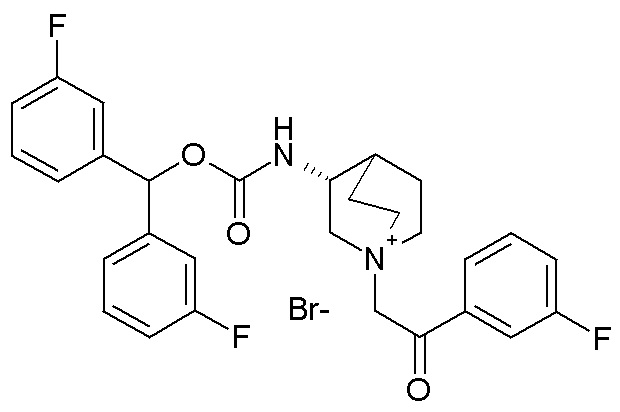
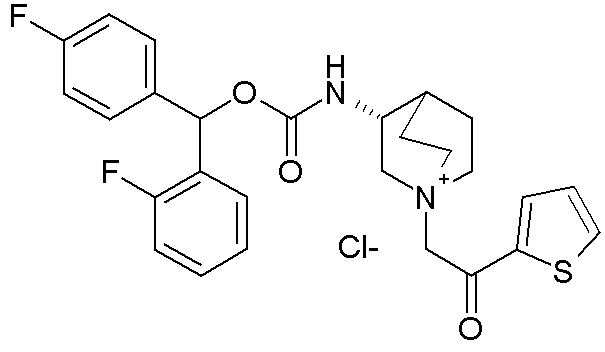
Получение (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октанбромида (соединение 11)
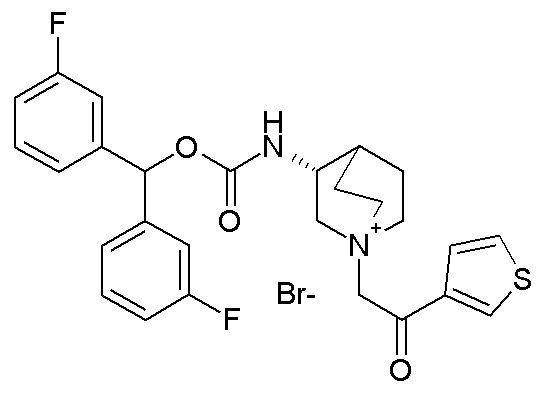
2-Бром-1-(тиофен-3-ил)этанон (27,5 мг, 0,13 ммоль) добавляли к раствору (R)-бис(3-фторфенил)метилхинуклидин-3-илкарбамата (50 мг, 0,13 ммоль, полученного как в примере 1) в этилацетате (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли Et2O (2 мл), и осадок собирали фильтрованием с отсасыванием и сушили в вакууме при 40°C в течение выходных, получая (R)-3-((бис(3-фторфенил)метокси)-карбониламино)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октанбромид (66,3 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,63 (дд, 1H), 8,09-8,25 (м, 1H), 7,74 (дд, 1H), 7,51-7,62 (м, 1H), 7,35-7,50 (м, 2H), 7,20-7,35 (м, 4H), 7,07-7,20 (м, 2H), 6,73 (с, 1H), 4,97 (дд, 2H), 3,92-4,25 (м, 2H), 3,46-3,83 (м, 5H), 1,81-2,25 (м, 5H);
LC-MS (ESI POS): 496,99 (M+).
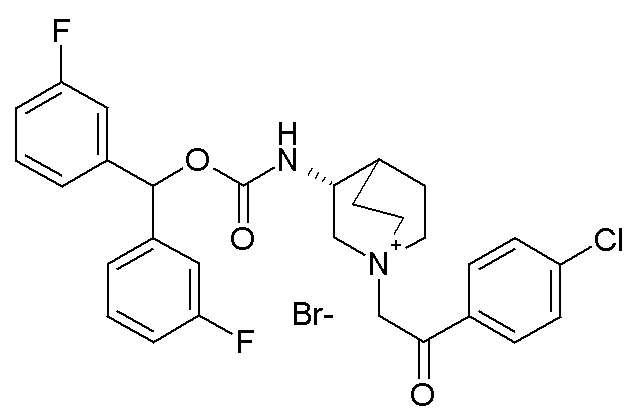
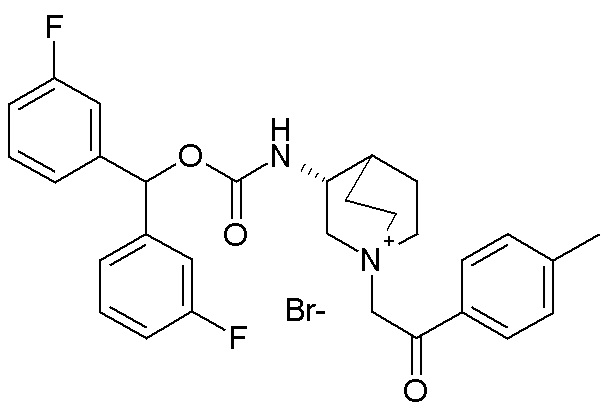
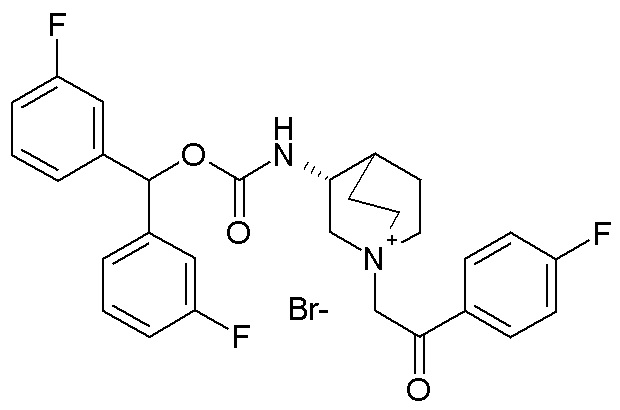
Следующие соединения получали, следуя способу, описанному в примере 7, применяя подходящие алкилирующие агенты вместо 2-бром-1-(тиофен-3-ил)этанона.
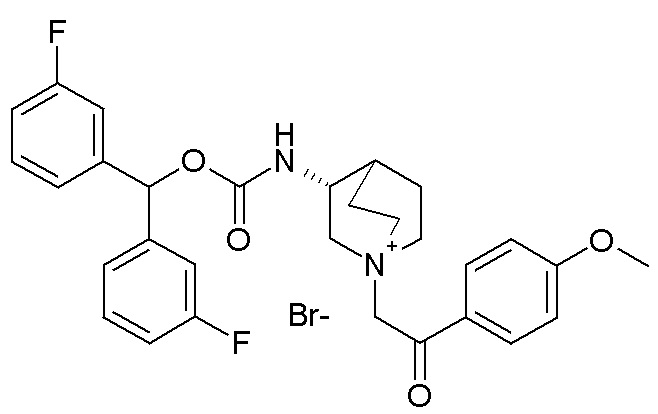
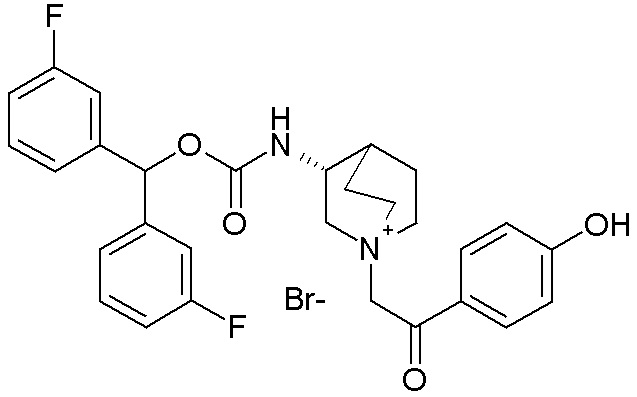
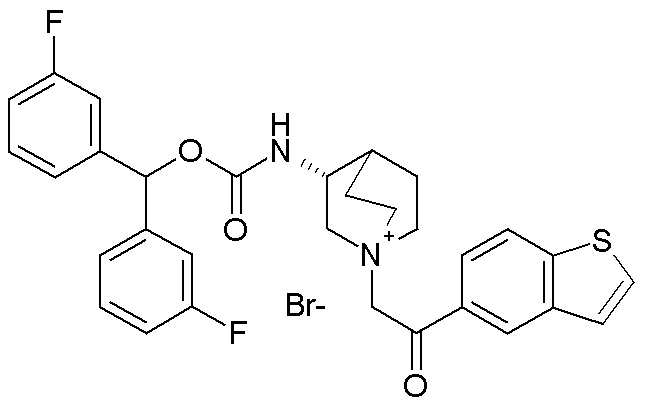
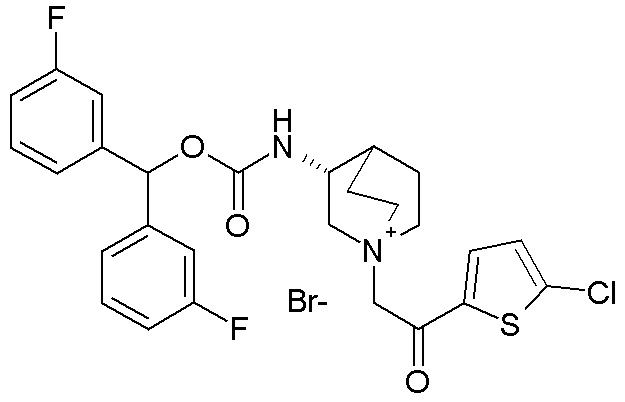
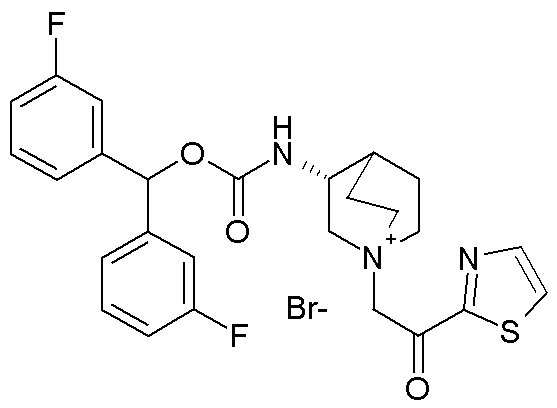
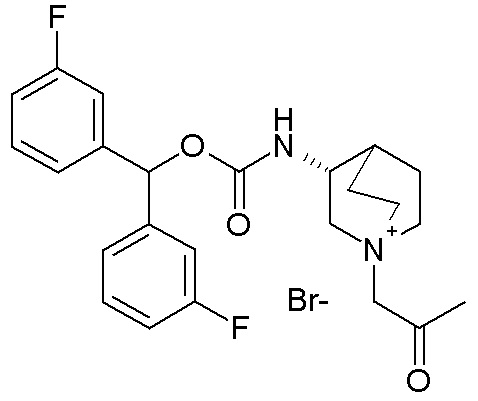

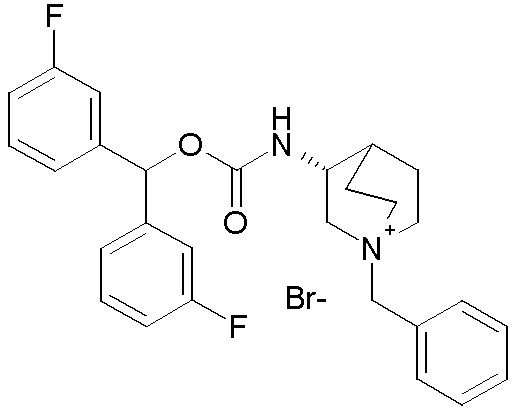
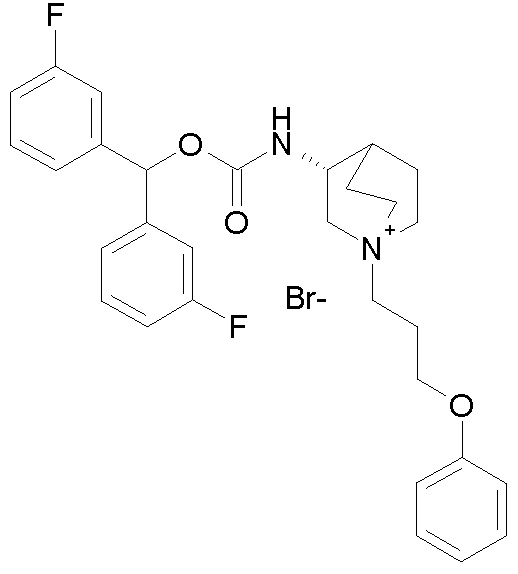
ПРИМЕР 8
Получение (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(5-цианотиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида (соединение 26)
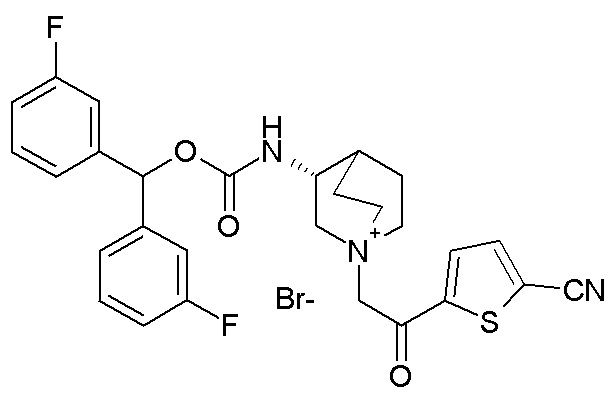
5-(2-Бромацетил)тиофен-2-карбонитрил (33,4 мг, 0,14 ммоль) добавляли к раствору (R)-бис(3-фторфенил)метилхинуклидин-3-илкарбамата (54 мг, 0,14 ммоль, полученного как в примере 1) в этилацетате (2 мл). Полученный в результате раствор перемешивали при комнатной температуре в течение двух дней, затем добавляли 5-(2-бромацетил)тиофен-2-карбонитрил (3,4 мг, 0,015 ммоль), и перемешивание продолжали в течение следующих 16 часов. Добавляли Et2O, и осадок выделяли фильтрацией, получая (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(5-цианотиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромид (68 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,08-8,22 (м, 3H), 7,34-7,58 (м, 2H), 7,20-7,34 (м, 4H), 7,04-7,20 (м, 2H), 6,73 (с, 1H), 5,06 (ушир.с, 2H), 3,86-4,24 (м, 2H), 3,56-3,86 (м, 3H), 3,40-3,56 (м, 2H), 2,10-2,26 (м, 2H), 1,84-2,08 (м, 3H);
LC-MS (ESI POS): 522,14 (M+).
ПРИМЕР 9
Получение (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октанбромида (соединение 27)
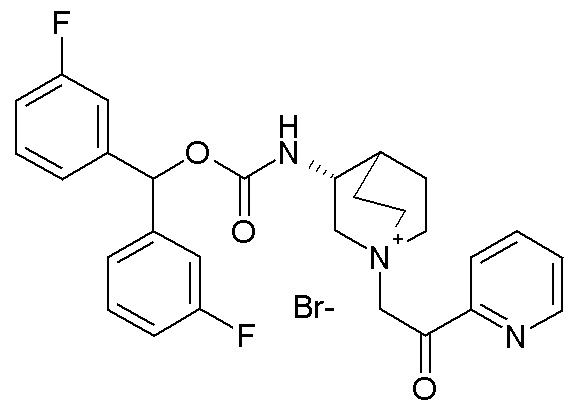
Гидробромид 2-бром-1-(пиридин-2-ил)этанона (39,2 мг, 0,14 ммоль) добавляли к раствору (R)-бис(3-фторфенил)метилхинуклидин-3-илкарбамата (52 мг, 0,14 ммоль, полученного как в примере 1) в этилацетате (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем добавляли Et2O (1 мл), и осадок собирали фильтрованием с отсасыванием и сушили в вакууме при 40°C в течение ночи. Продукт дополнительно очищали препаративной ВЭЖХ, получая (R)-3-((бис(3-фторфенил)метокси)-карбониламино)-1-(2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октанбромид (16,5 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,76 (дт, 1H), 8,15-8,24 (м, 1H), 8,01-8,15 (м, 2H), 7,78 (ддд, 1H), 7,43 (тд, 2H), 7,19-7,36 (м, 4H), 7,07-7,19 (м, 2H), 6,73 (с, 1H), 5,22 (с, 2H), 3,94-4,19 (м, 2H), 3,44-3,90 (м, 5H), 2,17-2,38 (м, 1H), 1,78-2,17 (м, 4H);
LC-MS (ESI POS): 492,20 (M+).
Следующее соединение получали, следуя способу, описанному в примере 9, применяя подходящие алкилирующие агенты вместо 2-гидробромида бром-1-(пиридин-2-ил)этанона.
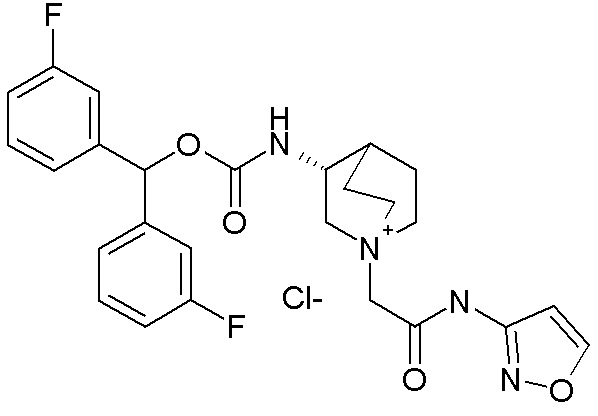
ПРИМЕР 10
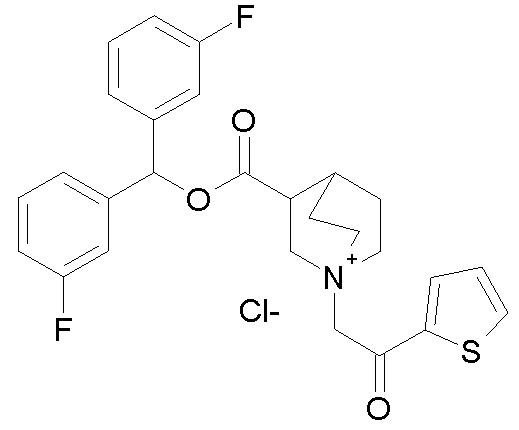
Получение (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 29)
(R)-Бис(3-фторфенил)метилхинуклидин-3-илкарбамат (35 мг, 0,09 ммоль, полученный как в примере 1) и 2-хлор-1-(тиофен-2-ил)этанон (16,6 мг, 0,10 ммоль) растворяли в ацетонитриле (2 мл) и перемешивали при комнатной температуре в течение 16 часов. Ацетонитрил упаривали, и неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1), получая (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (50,1 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,20 (дд, 2H), 8,10 (дд, 1H), 7,38-7,54 (м, 2H), 7,29-7,38 (м, 2H), 7,25 (ушир.с, 3H), 6,95-7,21 (м, 2H), 6,73 (с, 1H), 5,03 (с, 2H), 3,85-4,23 (м, 2H), 3,48-3,80 (м, 5H), 1,95-2,32 (м, 4H), 1,91 (ушир.с, 1H);
LC-MS (ESI POS): 497,33 (M+).
ПРИМЕР 11
Получение (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (соединение 30)
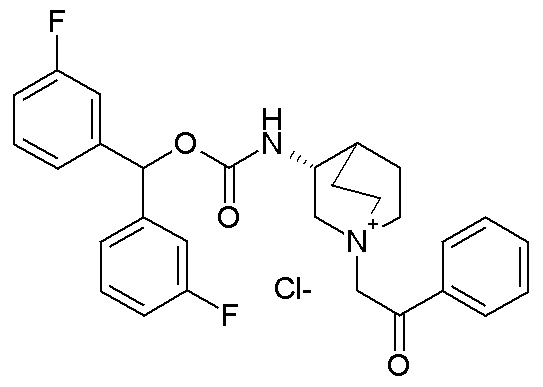
2-Хлор-1-фенилэтанон (20,8 мг, 0,13 ммоль) добавляли к раствору (R)-бис(3-фторфенил)метилхинуклидин-3-илкарбамата (50 мг, 0,13 ммоль, полученного как в примере 1) в этилацетате (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем растворитель упаривали в вакууме. Остаток обрабатывали Et2O (4 мл) и ультразвуком, получая твердое вещество, которое собирали фильтрованием с отсасыванием и очищали флэш-хроматографией (DCM/MeOH=95/5), получая (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорид (37,2 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,22 (д, 1H), 7,91-8,05 (м, 2H), 7,68-7,85 (м, 1H), 7,54-7,68 (м, 2H), 7,43 (тд, 2H), 7,21-7,36 (м, 4H), 7,01-7,20 (м, 2H), 6,73 (с, 1H), 5,15 (с, 2H), 3,95-4,18 (м, 2H), 3,63-3,92 (м, 4H), 3,47-3,63 (м, 1H), 1,98-2,36 (м, 4H), 1,79-1,98 (м, 1H);
LC-MS (ESI POS): 491,21 (M+).
ПРИМЕР 12
Получение (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-феноксиэтил)-1-азониабицикло[2.2.2]октанбромида (соединение 31)

(R)-Бис(3-фторфенил)метилхинуклидин-3-илкарбамат (50 мг, 0,13 ммоль, полученный как в примере 1) добавляли к раствору (2-бромэтокси)бензола (27,0 мг, 0,13 ммоль) в этилацетате (2 мл). Смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли (2-бромэтокси)бензол (27 мг, 0,13 ммоль), и смесь нагревали при 100°C в течение 15 минут при облучении микроволнами. Добавляли каталитическое количество йодида калия, и реакционную смесь нагревали при 120°C в течение 1 часа при облучении микроволнами. Осадок собирали фильтрацией и сушили, получая (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-феноксиэтил)-1-азониабицикло[2.2.2]октанбромид (17 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,05 (д, 1H), 7,42 (тд, 2H), 7,29-7,37 (м, 2H), 7,20-7,29 (м, 4H), 7,08-7,20 (м, 2H), 6,90-7,06 (м, 3H), 6,73 (с, 1H), 4,38-4,46 (м, 2H), 3,95-4,11 (м, 1H), 3,84-3,95 (м, 1H), 3,60-3,72 (м, 2H), 3,44-3,61 (м, 4H), 3,33-3,38 (м, 1H), 2,06-2,22 (м, 2H), 1,73-2,05 (м, 3H);
LC-MS (ESI POS): 493,34 (M+).
ПРИМЕР 13
Получение (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(2,3-дигидробензофуран-5-ил)этил)-1-азониабицикло[2.2.2]октанбромида (соединение 32)
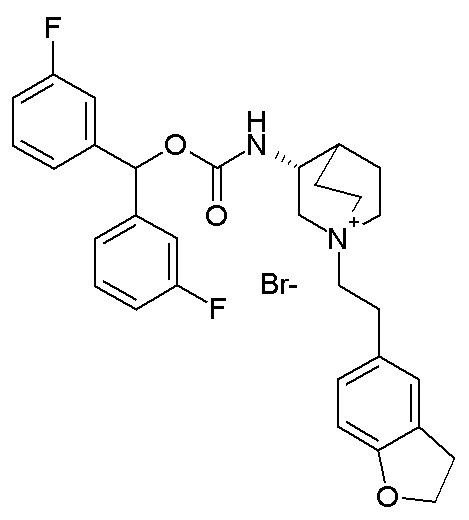
К раствору (R)-бис(3-фторфенил)метилхинуклидин-3-илкарбамата (53 мг, 0,14 ммоль, полученного как в примере 1) в этилацетате (2 мл) добавляли 5-(2-бромэтил)-2,3-дигидробензофуран (32,3 мг, 0,14 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 8 дней, и затем растворитель упаривали, и неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=95/5), собирая (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(2,3-дигидробензофуран-5-ил)этил)-1-азониабицикло[2.2.2]октанбромид (22,2 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,08 (д, 1H), 7,36-7,51 (м, 2H), 7,26 (д, 4H), 7,06-7,19 (м, 3H), 7,00 (д, 1H), 6,64-6,81 (м, 2H), 4,50 (т, 2H), 3,92-4,13 (м, 1H), 3,83 (т, 1H), 3,37-3,59 (м, 4H), 3,33 (д, 2H), 3,19-3,25 (м, 1H), 3,15 (т, 2H), 2,90 (м, 2H), 2,04-2,25 (м, 2H), 1,68-2,04 (м, 3H);
LC-MS (ESI POS): 519,30 (M+).
ПРИМЕР 14
Получение (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(4-фторфенэтил)-1-азониабицикло[2.2.2]октанбромида (соединение 33)
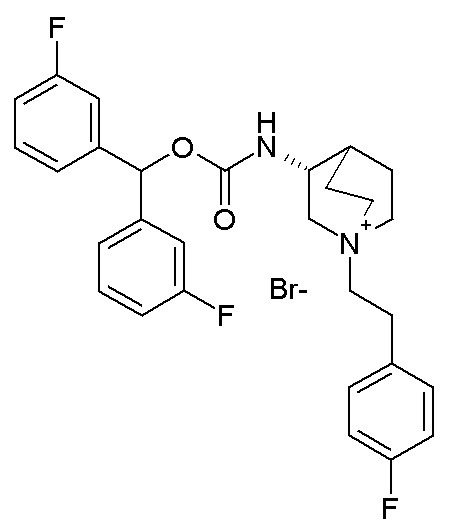
(R)-бис(3-Фторфенил)метилхинуклидин-3-илкарбамат (55 мг, 0,14 ммоль, полученный как в примере 1) растворяли в этилацетате (2 мл) и добавляли 1-(2-бромэтил)-4-фторбензол (21 мкл, 0,15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, затем снова добавляли 1-(2-бромэтил)-4-фторбензол (6,21 мкл, 0,04 ммоль). После перемешивания при комнатной температуре в течение 2 дней, реакционную смесь концентрировали в вакууме, и неочищенный остаток вначале очищали флэш-хроматографией (DCM/MeOH=9/1) и затем растирали с DCM/Et2O (1/1), получая (R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(4-фторфенэтил)-1-азониабицикло[2.2.2]октанбромид (45 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,09 (д, 1H), 7,39-7,48 (м, 2H), 7,35 (дд, 2H), 7,23-7,30 (м, 4H), 7,09-7,23 (м, 4H), 6,74 (с, 1H), 3,94-4,15 (м, 1H), 3,64-3,94 (м, 1H), 3,41-3,59 (м, 4H), 3,35 (д, 2H), 3,14-3,25 (м, 1H), 3,00 (дд, 2H), 2,04-2,25 (м, 2H), 1,69-2,04 (м, 3H);
LC-MS (ESI POS): 495,24 (M+).
ПРИМЕР 15
Получение (R)-3-(бензгидрилоксикарбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (соединение 34)
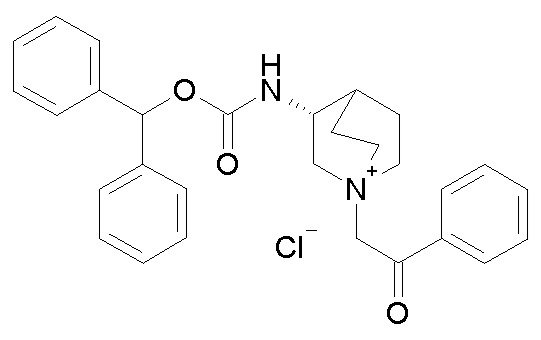
2-Хлор-1-фенилэтанон (55,0 мг, 0,36 ммоль) добавляли к раствору (R)-бензгидрилхинуклидин-3-илкарбамата (100 мг, 0,30 ммоль, полученного как в примере 2) в этилацетате (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Суспензию декантировали, раствор удаляли, и продукт упаривали досуха. Добавляли Et2O, суспензию обрабатывали ультразвуком. Осадок собирали фильтрованием с отсасыванием и сушили в вакууме при 40°C, получая (R)-3-(бензгидрилоксикарбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорид (110 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,15 (д, 1H), 7,94-8,05 (м, 2H), 7,69-7,81 (м, 1H), 7,52-7,66 (м, 2H), 7,11-7,45 (м, 10H), 6,71 (с, 1H), 5,15 (с, 2H), 3,92-4,19 (м, 2H), 3,44-3,86 (м, 5H), 1,96-2,32 (м, 4H), 1,73-1,96 (м, 1H);
LC-MS (ESI POS): 455,21 (M+).
ПРИМЕР 16
Получение (R)-3-((бис(4-фторфенил)метокси)карбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (соединение 35)
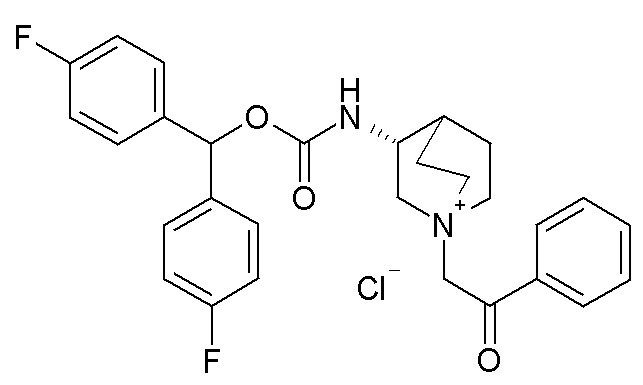
2-Хлор-1-фенилэтанон (13 мг, 0,09 ммоль) добавляли к раствору (R)-бис(4-фторфенил)метилхинуклидин-3-илкарбамата (32,0 мг, 0,09 ммоль, полученного как в примере 3) в этилацетате (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем к раствору добавляли другую порцию 2-хлор-1-фенилэтанона (2,7 мг, 0,02 ммоль), и реакционную смесь перемешивали в течение дополнительных 24 часов. Реакционную смесь упаривали досуха, и остаток растирали в Et2O. Затем добавляли изопропиловый эфир, и продукт обрабатывали ультразвуком, и осадок собирали фильтрованием с отсасыванием, получая (R)-3-((бис(4-фторфенил)метокси)карбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорид (32,4 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,09-8,22 (м, 1H), 7,91-8,05 (м, 2H), 7,69-7,81 (м, 1H), 7,55-7,69 (м, 2H), 7,34-7,48 (м, 4H), 7,09-7,31 (м, 4H), 6,73 (с, 1H), 5,14 (с, 2H), 3,86-4,20 (м, 2H), 3,47-3,87 (м, 5H), 1,61-2,41 (м, 5H);
LC-MS (ESI POS): 491,21 (M+).
ПРИМЕР 17
Получение (3R)-3-(((4-метоксифенил)(фенил)метокси)-карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 36)
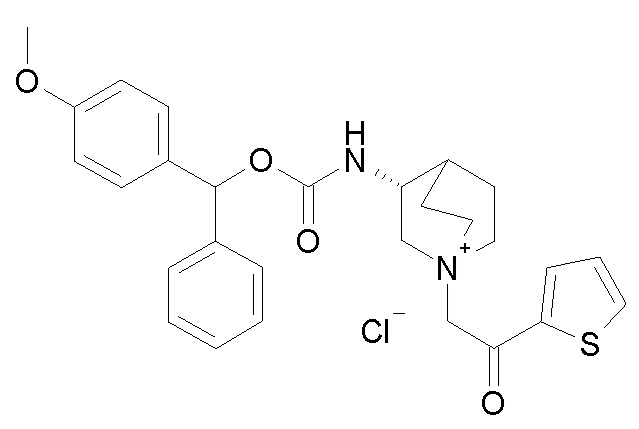
2-Хлор-1-(тиофен-2-ил)этанон (12,0 мг, 0,07 ммоль) добавляли к раствору (4-метоксифенил)(фенил)метил (R)-хинуклидин-3-илкарбамата (22,0 мг, 0,06 ммоль, полученного как в примере 3) в этилацетате (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение двух дней. Реакционную смесь упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=95/5), получая (3R)-3-(((4-метоксифенил)(фенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (23 мг, смесь диастереомеров).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,20 (д, 1H), 8,08 (д, 1H), 8,04-8,13 (м, 1H), 7,35 (м, 5H), 7,29 (м, 3H), 6,74-7,04 (м, 2H), 6,66 (с, 1H), 5,00 (с, 2H), 3,88-4,19 (м, 2H), 3,74 (с, 3H), 3,57-3,72 (м, 4H), 3,42-3,57 (м, 1H), 1,92-2,26 (м, 4H), 1,64-1,92 (м, 1H);
LC-MS (ESI POS): 491,21 (M+).
Следующие соединения получали, следуя способу, описанному в примере 17, применяя подходящие промежуточные соединения (пример 3, таблица 1) вместо (4-метоксифенил)(фенил)метил (R)-хинуклидин-3-илкарбамата. Данные соединения получали в виде смеси диастереомеров.
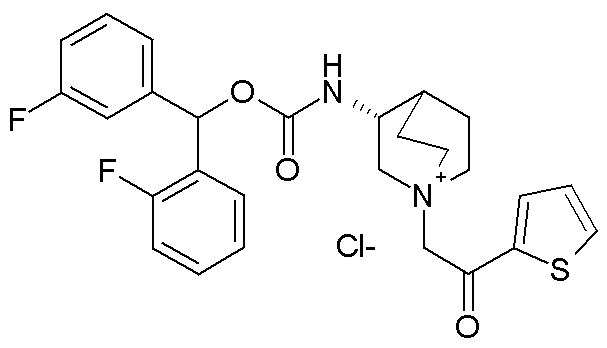
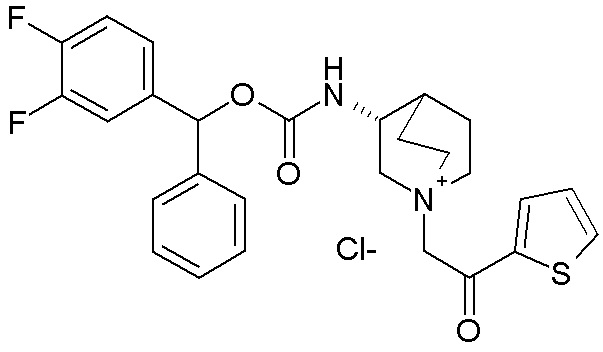
ПРИМЕР 18
Получение (3R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-((фенил(4-(трифторметил)фенил)метокси)карбониламино)-1-азониабицикло-[2.2.2]октанхлорида (соединение 41)
1- Получение фенил(4-(трифторметил)фенил)метил (R)-хинуклидин-3-илкарбамата (соединение 40)
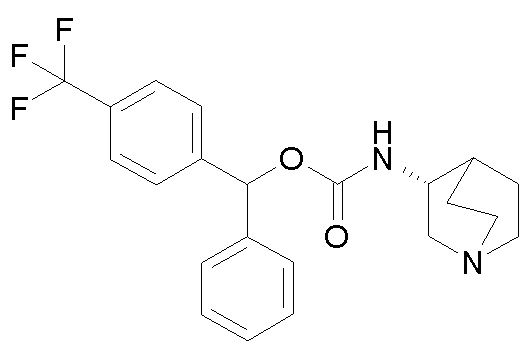
В первой колбе дигидрохлорид (R)-хинуклидин-3-амина (100 мг, 0,50 ммоль) растворяли в MeOH (5 мл) и воде (0,5 мл). Добавляли NaHCO3 (84,0 г, 1,00 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь упаривали, и остаток растворяли в сухом DMF (5 мл) и добавляли CDI (81,0 мг, 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи.
Во второй колбе фенил(4-(трифторметил)фенил)метанол (0,25 г, 1,00 ммоль) растворяли в сухом DMF (3 мл) и добавляли порциями при 0°C гидрид натрия (60% суспензия в минеральном масле, 24,0 мг, 1,00 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут и затем выливали в первую колбу.
Полученную в результате смесь перемешивали при комнатной температуре в течение ночи, затем ее распределяли между Et2O и водой. Органическую фазу промывали соляным раствором и сушили над сульфатом натрия. Растворитель удаляли в вакууме, и неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=95/5 - DCM/MeOH/TEA=75/25/0,5), получая фенил(4-(трифторметил)-фенил)метил (R)-хинуклидин-3-илкарбамат (56,0 мг, смесь диастереомеров).
2- Получение (3R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-((фенил(4-(трифторметил)фенил)метокси)карбониламино)-1-азониабицикло[2.2.2]октанхлорида (соединение 41)
Фенил(4-(трифторметил)фенил)метил (R)-хинуклидин-3-илкарбамат (56,0 мг, 0,14 ммоль) растворяли в этилацетате (1 мл) и ацетонитриле (0,5 мл). Полученный в результате раствор обрабатывали 2-хлор-1-(тиофен-2-ил)этаноном (24,0 мг, 0,15 ммоль) и перемешивали при комнатной температуре в течение ночи. Растворитель упаривали, и неочищенный остаток вначале очищали флэш-хроматографией (DCM/MeOH=95/5) и затем препаративной ВЭЖХ (CH3CN/H2O), получая (3R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-((фенил(4-(трифторметил)фенил)метокси)карбониламино)-1-азониабицикло[2.2.2]октанхлорид (30,0 мг, смесь диастереомеров).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,18-8,26 (м, 2H), 8,09 (д, 1H), 7,69-7,79 (м, 2H), 7,55-7,69 (м, 2H), 7,17-7,49 (м, 6H), 6,80 (с, 1H), 5,01 (с, 2H), 3,92-4,27 (м, 2H), 3,44-3,88 (м, 5H), 1,73-2,37 (м, 5H);
LC-MS (ESI POS): 529,13 (M+).
ПРИМЕР 19
Получение (3R)-3-(((2-хлорфенил)(4-хлорфенил)метокси)-карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло-[2.2.2]октанхлорида (соединение 43)
1- Получение (2-хлорфенил)(4-хлорфенил)метил (R)-хинуклидин-3-илкарбамата (соединение 42)

В первой колбе дигидрохлорид (R)-хинуклидин-3-амина (80,0 мг, 0,40 ммоль) растворяли в MeOH (8 мл) и воде (0,8 мл). Добавляли бикарбонат натрия (67,5 мг, 0,80 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь упаривали досуха, и твердое вещество растворяли в сухом DMF (8 мл). Добавляли CDI (65,1 мг, 0,40 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи.
Во второй колбе (2-хлорфенил)(4-хлорфенил)метанол (100 мг, 0,39 ммоль) растворяли в сухом DMF (3 мл) и обрабатывали гидридом натрия (60% суспензия в минеральном масле, 15,8 мг, 0,39 ммоль) при 0°C. Баню со льдом удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 20 минут и затем ее добавляли к первой реакционной смеси.
Полученную в результате смесь перемешивали при комнатной температуре в течение ночи и затем ее распределяли между Et2O и водой. Органическую фазу промывали соляным раствором, сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1 - DCM/MeOH=75/25), получая (2-хлорфенил)(4-хлорфенил)метил (R)-хинуклидин-3-илкарбамат (95 мг, смесь диастереомеров).
2- Получение (3R)-3-(((2-хлорфенил)(4-хлорфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 43)
К раствору (2-хлорфенил)(4-хлорфенил)метил (R)-хинуклидин-3-илкарбамата (95,0 мг, 0,23 ммоль) в этилацетате (3 мл) добавляли 2-хлор-1-(тиофен-2-ил)этанон (37,6 мг, 0,23 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме, и остаток брали в диэтиловом эфире и фильтровали. Продукт очищали флэш-хроматографией (DCM/MeOH=95/5), получая (3R)-3-(((2-хлорфенил)(4-хлорфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (45,0 мг, смесь диастереомеров).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,15-8,31 (м, 2H), 8,02-8,15 (м, 1H), 7,27-7,66 (м, 9H), 6,81-7,00 (м, 1H), 5,03 (с, 2H), 3,85-4,23 (м, 2H), 3,59-3,85 (м, 4H), 3,46-3,59 (м, 1H), 1,95-2,28 (м, 4H), 1,82-1,93 (м, 1H);
LC-MS (ESI POS): 529,09 (M+).
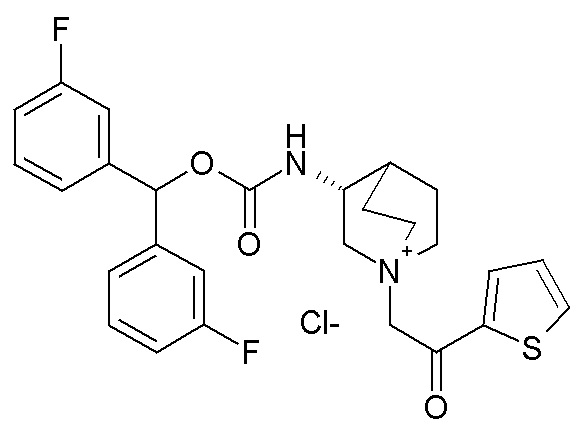
ПРИМЕР 20
Получение (R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-((тиофен-2-илметокси)карбониламино)-1-азониабицикло[2.2.2]октанхлорида (соединение 45)
1- Получение (R)-тиофен-2-илметилхинуклидин-3-илкарбамата (соединение 44)
В первой колбе дигидрохлорид (R)-хинуклидин-3-амина (100 мг, 0,50 ммоль) растворяли в MeOH (10 мл) и воде (1 мл). Добавляли бикарбонат натрия (84 мг, 1,00 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь упаривали досуха, и твердое вещество растворяли в сухом DMF (10 мл). Добавляли CDI (81 мг, 0,50 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи.
Во второй колбе тиофен-2-илметанол (114 мг, 1,00 ммоль) растворяли в сухом DMF (3 мл) и обрабатывали гидридом натрия (60% суспензия в минеральном масле, 40,0 мг, 1,00 ммоль) при 0°C. Баню со льдом удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 20 минут и затем ее выливали в первую колбу.
Полученную в результате смесь перемешивали при комнатной температуре в течение ночи, затем ее распределяли между Et2O и водой. Органическую фазу промывали соляным раствором, сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1-75/25), получая (R)-тиофен-2-илметилхинуклидин-3-илкарбамат (50 мг).
2- Получение (R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-((тиофен- 2-илметокси)карбониламино)-1-азониабицикло[2.2.2]октанхлорида (соединение 45)
2-Хлор-1-(тиофен-2-ил)этанон (30,2 мг, 0,19 ммоль) добавляли к раствору (R)-тиофен-2-илметилхинуклидин-3-илкарбамата (50,0 мг, 0,19 ммоль) в EtOAc (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и остаток растирали в Et2O, фильтровали и сушили, получая (R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-((тиофен-2-илметокси)карбониламино)-1-азониабицикло[2.2.2]октанхлорид (60 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,21 (дд, 1H), 8,12 (дд, 1H), 7,96 (ушир.с, 1H), 7,55 (дд, 1H), 7,35 (дд, 1H), 7,16 (дд, 1H), 7,03 (дд, 1H), 5,22 (с, 2H), 5,04 (с, 2H), 3,90-4,19 (м, 2H), 3,58-3,84 (м, 4H), 3,44-3,58 (м, 1H), 1,72-2,24 (м, 5H);
LC-MS (ESI POS): 391,23 (M+).
ПРИМЕР 21
Получение (R)-3-((бис(3-фторфенил)метокси)-карбонотиоиламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 47)
1- Получение (R)-O-бис(3-фторфенил)метилхинуклидин-3-илкарбамотиоата (соединение 46)
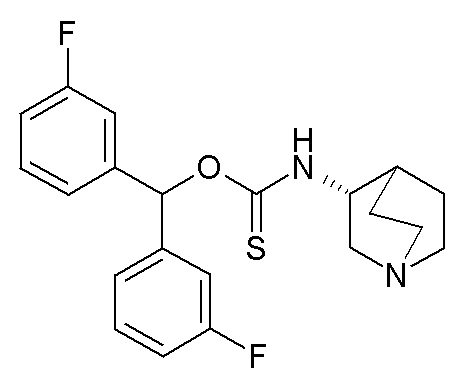
В первой колбе дигидрохлорид (R)-хинуклидин-3-амина (500 мг, 2,51 ммоль) растворяли в MeOH (25 мл) и воде (2,5 мл). Добавляли бикарбонат натрия (211 мг, 2,51 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель упаривали досуха, и остаток суспендировали в сухом DMF (25 мл) и обрабатывали ди(1H-имидазол-1-ил)метантионом (448 мг, 2,51 ммоль). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре.
Во второй колбе NaH (60% суспензия в минеральном масле, 187 мг, 4,68 ммоль) добавляли порциями к раствору бис(3-фторфенил)метанола (1,03 г, 4,68 ммоль) в сухом DMF (25 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и затем ее добавляли к первой колбе.
Полученную в результате смесь перемешивали при комнатной температуре в течение ночи и затем реакционную смесь распределяли между Et2O и водой. Органическую фазу сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1 и затем DCM/MeOH=75/25+0,5% TEA), получая (R)-O-бис(3-фторфенил)метилхинуклидин-3-илкарбамотиоат (60 мг).
2- Получение (R)-3-((бис(3-фторфенил)метокси)-карбонотиоиламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 47)
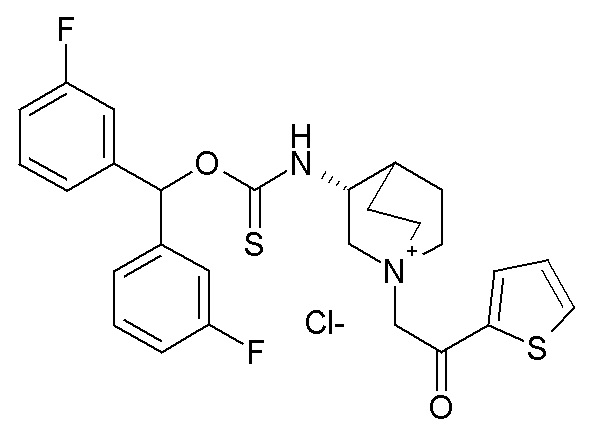
К раствору (R)-O-бис(3-фторфенил)метилхинуклидин-3-илкарбамотиоата (60 мг, 0,15 ммоль) в этилацетате (1 мл) добавляли 2-хлор-1-(тиофен-2-ил)этанон (27,3 мг, 0,17 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней. Растворитель упаривали, и остаток очищали флэш-хроматографией (DCM/MeOH=75/25), получая (R)-3-((бис(3-фторфенил)метокси)карбонотиоиламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (20 мг).
1H ЯМР (300 МГц, ДМСО-d6 353K) δ м.д. 9,74 (ушир.с, 1H), 8,16 (дд, 1H), 8,07 (дд, 1H), 7,38-7,52 (м, 3H), 7,33 (дд, 1H), 7,20-7,30 (м, 4H), 7,04-7,20 (м, 2H), 4,97 (с, 2H), 4,45-4,70 (м, 1H), 4,13-4,35 (м, 1H), 3,61-3,90 (м, 5H), 2,32-2,46 (м, 2H), 1,87-2,26 (м, 3H);
LC-MS (ESI POS): 513,25 (M+).
ПРИМЕР 22
Получение (3R)-1-бензил-3-((бис(3-фторфенил)метокси)-карбониламино)-1-метилпирролидиниййодида (соединение 49)
1- Получение (R)-бис(3-фторфенил)метил 1-бензилпирролидин-3-илкарбамата (соединение 48)
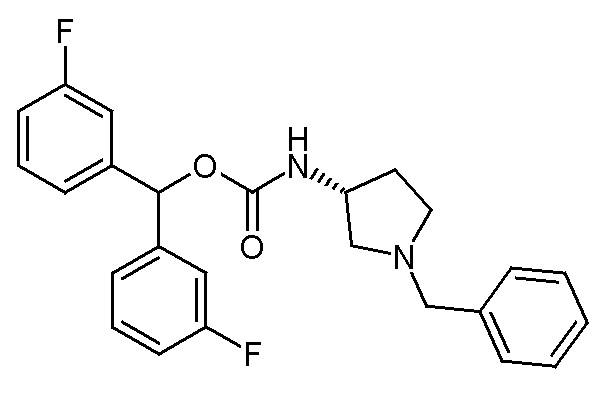
В первой колбе CDI (184 мг, 1,135 ммоль) добавляли к раствору (R)-1-бензилпирролидин-3-амина (200 мг, 1,13 ммоль) в сухом DMF (20 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи.
Во второй колбе бис(3-фторфенил)метанол (375 мг, 1,70 ммоль) растворяли в сухом DMF (9 мл) и обрабатывали гидридом натрия (60% суспензия в минеральном масле, 68,1 мг, 1,703 ммоль) при 0°C. Баню со льдом удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 20 минут и затем ее добавляли в первую колбу.
Полученную в результате смесь перемешивали при комнатной температуре в течение ночи и затем распределяли между Et2O и водой. Органическую фазу промывали соляным раствором, сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=98/2), получая (R)-бис(3-фторфенил)метил 1-бензилпирролидин-3-илкарбамат (300 мг).
2- Получение (3R)-1-бензил-3-((бис(3-фторфенил)метокси)-карбониламино)-1-метилпирролидиниййодида (соединение 49)
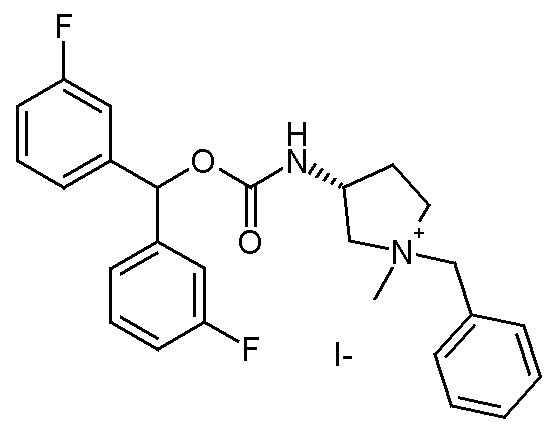
К раствору (R)-бис(3-фторфенил)метил 1-бензилпирролидин-3-илкарбамата (300 мг, 0,71 ммоль) в EtOAc (9 мл) добавляли йодметан (44,2 мкл, 0,71 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем растворитель удаляли в вакууме, и остаток растирали с диэтиловым эфиром. Продукт очищали флэш-хроматографией (DCM/MeOH=99/1) и затем препаративной ВЭЖХ, получая (3R)-1-бензил-3-((бис(3-фторфенил)метокси)карбониламино)-1-метилпирролидиниййодид (25 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,08 (д, 1H), 7,34-7,68 (м, 8H), 7,03-7,34 (м, 5H), 6,71 и 6,74 (с, 1H), 4,56 и 4,62 (с, 2H), 4,32-4,51 (м, 1H), 3,33-4,05 (м, 4H), 2,95 и 3,02 (с, 3H), 2,56-2,67 (м, 1H), 1,98-2,25 (м, 1H);
LC-MS (ESI POS): 437,24 (M+).
ПРИМЕР 23
Получение 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (соединение 50)
Бис(3-фторфенил)метилхинуклидин-3-карбоксилат (95 мг, 0,27 ммоль, полученный как в примере 4) и 2-бром-1-фенилэтанон (58,2 мг, 0,29 ммоль) растворяли в ацетонитриле (5 мл) и перемешивали при комнатной температуре в течение ночи. Растворитель упаривали, и полученный в результате остаток очищали флэш-хроматографией (DCM/MeOH=95/5-9/1), получая 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромид (110 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,92-8,06 (м, 2H), 7,71-7,82 (м, 1H), 7,56-7,69 (м, 2H), 7,30-7,52 (м, 6H), 7,10-7,22 (м, 2H), 6,93 (с, 1H), 5,24 (с, 2H), 4,03-4,21 (м, 1H), 3,91 (т, 1H), 3,59-3,82 (м, 4H), 3,45-3,59 (м, 1H), 2,64-2,71 (м, 1H), 2,02-2,25 (м, 2H), 1,83-2,02 (м, 1H), 1,40-1,64 (м, 1H);
LC-MS (ESI POS): 475,97 (M+).
Следующие соединения получали, следуя способу, описанному в примере 23, применяя подходящие алкилирующие агенты вместо 2-бром-1-фенилэтанона. Данные соединения получали в виде рацемической смеси.
ПРИМЕР 24
Получение 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-п-толилэтил)-1-азониабицикло[2.2.2]октанбромида (соединение 54)
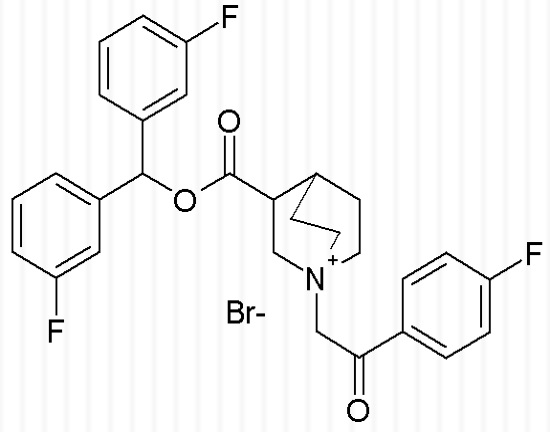
Бис(3-фторфенил)метилхинуклидин-3-карбоксилат (95 мг, 0,27 ммоль, полученный как в примере 4) и 2-бром-1-п-толилэтанон (62,3 мг, 0,29 ммоль) растворяли в ацетонитриле (10 мл) и перемешивали при комнатной температуре в течение ночи. Растворитель упаривали, и остаток растирали с Et2O/EtOAc (1/1) и выделяли фильтрованием с отсасыванием, получая 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-п-толилэтил)-1-азониабицикло[2.2.2]октанбромид (117,6 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,82-7,97 (м, 2H), 7,33-7,49 (м, 6H), 7,30-7,37 (м, 2H), 7,10-7,23 (м, 2H), 6,93 (с, 1H), 5,20 (с, 2H), 4,13 (дд, 1H), 3,83-3,97 (м, 1H), 3,59-3,78 (м, 4H), 3,48-3,59 (м, 1H), 2,61-2,71 (м, 1H), 2,42 (с, 3H), 2,01-2,23 (м, 2H), 1,85-2,01 (м, 1H), 1,43-1,65 (м, 1H);
LC-MS (ESI POS): 490,15 (M+).
Следующие соединения получали, следуя способу, описанному в примере 24, применяя подходящие алкилирующие агенты вместо 2-бром-1-п-толилэтанона. Данные соединения получали в виде рацемической смеси.
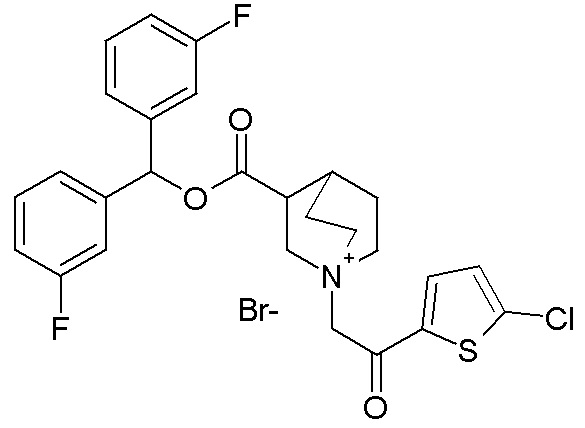
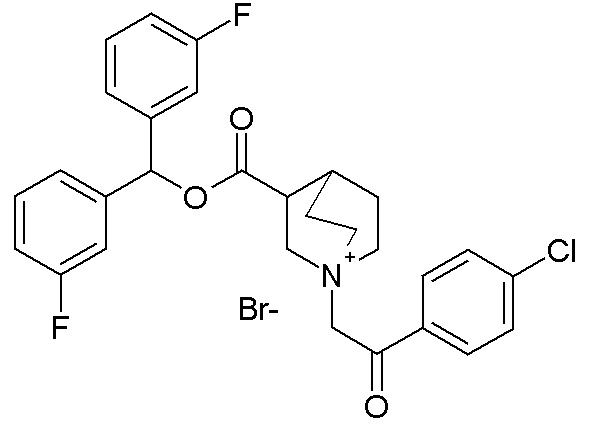
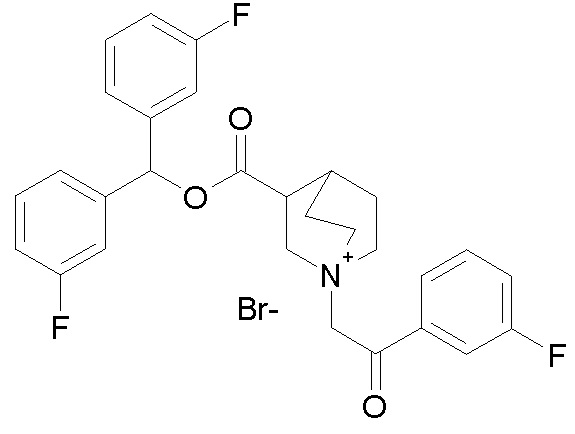
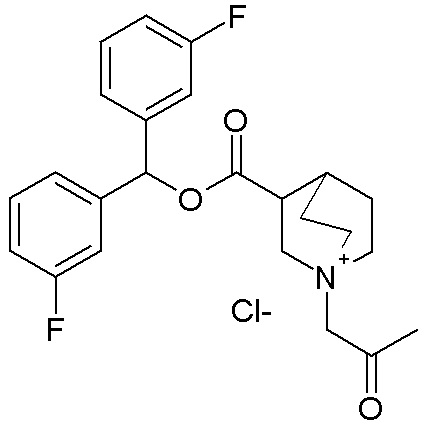
ПРИМЕР 25
Получение 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-трет-бутокси-2-оксоэтил)-1-азониабицикло[2.2.2]октанхлорида (соединение 59)
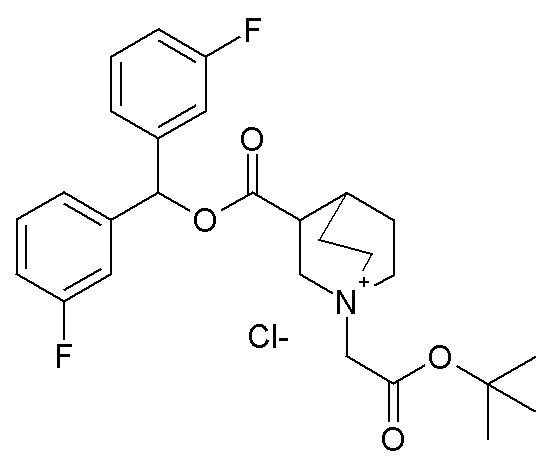
Бис(3-фторфенил)метилхинуклидин-3-карбоксилат (70 мг, 0,20 ммоль, полученный как в примере 4) и трет-бутил 2-хлорацетат (31 мкл, 0,21 ммоль) растворяли в ацетонитриле (3 мл) и перемешивали при комнатной температуре в течение ночи. Снова добавляли трет-бутил 2-хлорацетат (28 мкл, 0,20 ммоль), и смесь перемешивали в течение дополнительных 24 часов. Ацетонитрил упаривали, и полученное в результате неочищенное соединение растирали с Et2O и фильтровали, получая 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-трет-бутокси-2-оксоэтил)-1-азониабицикло[2.2.2]октанхлорид (42,4 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,26-7,56 (м, 6H), 7,09-7,23 (м, 2H), 6,91 (с, 1H), 4,31 (с, 2H), 4,03 (дд, 1H), 3,80 (т, 1H), 3,38-3,71 (м, 5H), 2,59-2,69 (м, 1H), 1,77-2,17 (м, 4H), 1,48 (с, 9H);
LC-MS (ESI POS): 472,19 (M+).
ПРИМЕР 26
Получение 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 60)
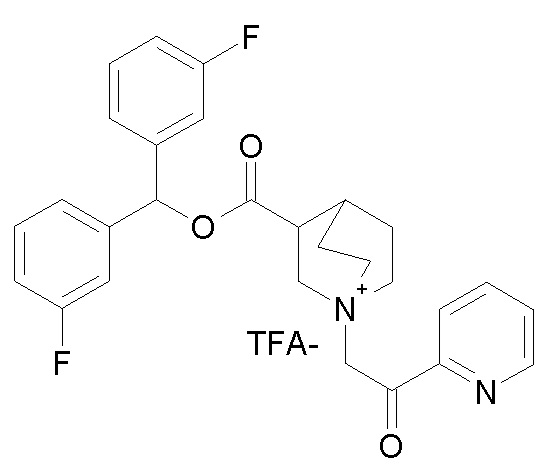
Бис(3-фторфенил)метилхинуклидин-3-карбоксилат (70 мг, 0,20 ммоль, полученный как в примере 4) и гидробромид 2-бром-1-(пиридин-2-ил)этанона (60,5 мг, 0,21 ммоль) суспендировали в ацетонитриле (5 мл) и перемешивали при комнатной температуре в течение 16 часов. Добавляли TEA (27 мкл, 0,20 ммоль), и полученный в результате чистый раствор перемешивали при комнатной температуре в течение ночи. Летучие компоненты упаривали, и остаток сначала очищали флэш-хроматографией (DCM/MeOH=9/1) и затем препаративной ВЭЖХ, получая 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетат (60,3 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,77 (м, 1H), 7,99-8,21 (м, 2H), 7,68-7,89 (м, 1H), 7,27-7,56 (м, 6H), 7,04-7,25 (м, 2H), 6,93 (с, 1H), 5,32 (с, 2H), 4,17 (м, 1H), 3,94 (т, 1H), 3,71 (м, 4H), 3,54 (т, 1H), 2,61-2,71 (м, 1H), 2,02-2,24 (м, 2H), 1,83-2,01 (м, 1H), 1,46-1,66 (м, 1H);
LC-MS (ESI POS): 477,08 (M+).
ПРИМЕР 27
Получение 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(3-(этоксикарбонил)изоксазол-5-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида (соединение 61)

Бис(3-фторфенил)метилхинуклидин-3-карбоксилат (86 мг, 0,24 ммоль, полученный как в примере 4) и этил 5-(2-бромацетил)изоксазол-3-карбоксилат (69,4 мг, 0,26 ммоль) растворяли в ацетонитриле (3 мл) и перемешивали при комнатной температуре в течение ночи. Осадок выделяли фильтрованием с отсасыванием, получая 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(3-(этоксикарбонил)изоксазол-5-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромид (26 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,90 (с, 1H), 7,23-7,52 (м, 6H), 7,16 (тд, 2H), 6,92 (с, 1H), 5,06 (с, 2H), 4,43 (кв, 2H), 4,00-4,16 (м, 1H), 3,81-3,97 (м, 1H), 3,48-3,80 (м, 5H), 2,65-2,71 (м, 1H), 2,01-2,24 (м, 2H), 1,78-2,01 (м, 1H), 1,45-1,65 (м, 1H), 1,35 (т, 3H);
LC-MS (ESI POS): 539,16 (M+).
ПРИМЕР 28
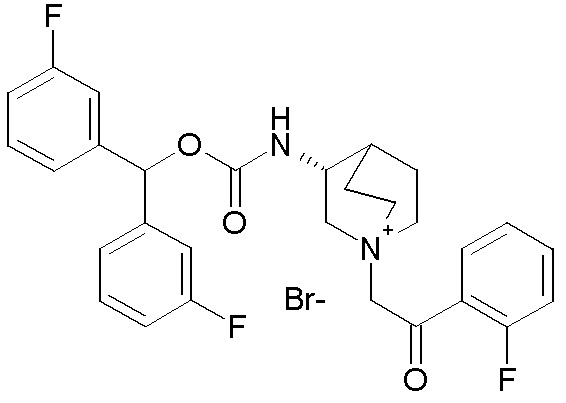
Получение 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(2-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида (соединение 62)
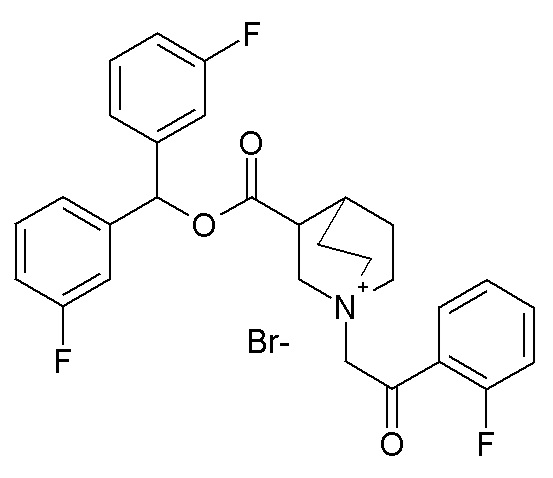
Бис(3-фторфенил)метилхинуклидин-3-карбоксилат (90 мг, 0,25 ммоль, полученный как в примере 4) и 2-бром-1-(2-фторфенил)этанон (65,6 мг, 0,30 ммоль) растворяли в ацетонитриле (4 мл) и перемешивали при комнатной температуре в течение ночи. Ацетонитрил упаривали, и остаток растирали с Et2O и фильтровали, получая 3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(2-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромид (51,3 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,97 (тд, 1H), 7,72-7,87 (м, 1H), 7,30-7,55 (м, 8H), 7,10-7,23 (м, 2H), 6,93 (с, 1H), 5,06 (д, 2H), 4,14 (дд, 1H), 3,92 (т, 1H), 3,60-3,82 (м, 4H), 3,49-3,59 (м, 1H), 2,64-2,71 (м, 1H), 2,01-2,19 (м, 2H), 1,82-2,01 (м, 1H), 1,41-1,70 (м, 1H);
LC-MS (ESI POS): 494,10 (M+).
Следующие соединения получали, следуя способу, описанному в примере 28, применяя подходящие алкилирующие агенты вместо 2-бром-1-(2-фторфенил)этанона. Данные соединения получали в виде рацемической смеси.
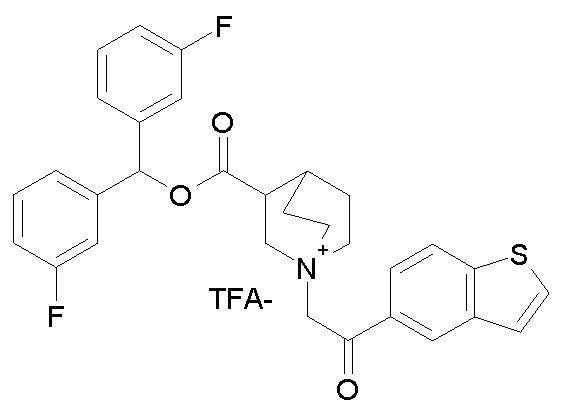
ПРИМЕР 29
Получение 1-(2-(бензо[b]тиофен-5-ил)-2-оксоэтил)-3-((бис(3-фторфенил)метокси)карбонил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 65)
Бис(3-фторфенил)метилхинуклидин-3-карбоксилат (86 мг, 0,24 ммоль, полученный как в примере 4) и 1-(бензо[b]тиофен-5-ил)-2-бромэтанон (67,5 мг, 0,26 ммоль) растворяли в ацетонитриле (3 мл) и перемешивали при комнатной температуре в течение ночи. Ацетонитрил упаривали, и неочищенное веество сначала растирали с Et2O и затем очищали препаративной ВЭЖХ, получая l-(2-(бензо[b]тиофен-5-ил)-2-оксоэтил)-3-((бис(3-фторфенил)-метокси)карбонил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетат (83 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,59 (д, 1H), 8,25 (д, 1H), 7,97 (д, 1H), 7,93 (дд, 1H), 7,66 (дд, 1H), 7,30-7,53 (м, 6H), 7,09-7,23 (м, 2H), 6,94 (с, 1H), 5,31 (с, 2H), 4,17 (дд, 1H), 3,94 (т, 1H), 3,62-3,84 (м, 4H), 3,47-3,62 (м, 1H), 2,66-2,73 (м, 1H), 2,02-2,24 (м, 2H), 1,88-2,04 (м, 1H), 1,40-1,69 (м, 1H);
LC-MS (ESI POS): 532,09 (M+).
ПРИМЕР 30
Получение 1-бензил-3-((бис(3-фторфенил)метокси)карбонил)-1-азониабицикло[2.2.2]октанбромида (соединение 66)
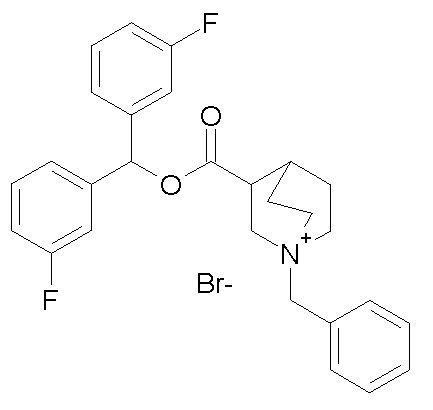
К раствору бис(3-фторфенил)метилхинуклидин-3-карбоксилата (90 мг, 0,25 ммоль, полученного как в примере 4) в EtOAc (3 мл) добавляли (бромметил)бензол (30,0 мкл, 0,25 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=95/5), получая 1-бензил-3-((бис(3-фторфенил)метокси)карбонил)-1-азониабицикло[2.2.2]октанбромид (50 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,52 (с, 5H), 7,42 (тд, 2H), 7,23-7,38 (м, 4H), 7,08-7,19 (м, 2H), 6,86 (с, 1H), 4,53 (с, 2H), 3,68-3,83 (м, 1H), 3,56-3,68 (м, 1H), 3,35-3,53 (м, 4H), 3,13-3,27 (м, 1H), 2,57-2,65 (м, 1H), 1,91-2,18 (м, 2H), 1,68-1,91 (м, 1H), 1,36-1,57 (м, 1H);
LC-MS (ESI POS): 448,23 (M+).
ПРИМЕР 31
Получение 3-((1,2-дифенилэтокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 67)
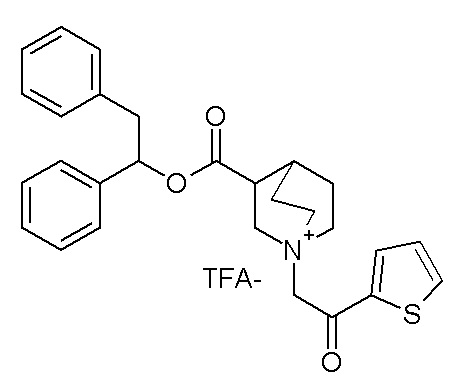
1,2-Дифенилэтилхинуклидин-3-карбоксилат (68 мг, 0,20 ммоль, полученный как в примере 5) и 2-хлор-1-(тиофен-2-ил)этанон (35,8 мг, 0,22 ммоль) растворяли в ацетонитриле (4 мл) и перемешивали при комнатной температуре в течение 64 часов. Ацетонитрил упаривали, и полученный в результате неочищенный остаток сначала очищали флэш-хроматографией (DCM/MeOH=9/1) и затем препаративной ВЭЖХ, получая 3-((1,2-дифенилэтокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетат (44 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,14-8,26 (м, 1H), 7,95-8,12 (м, 1H), 7,10-7,59 (м, 11H), 6,02 и 6,10 (дд, 1H), 5,03 и 5,05 (с, 2H), 3,90-4,06 (м, 1H), 3,59-3,89 (м, 5H), 2,98-3,33 (м, 3H), 1,45-2,45 (м, 4H), 0,83-1,31 (м, 1H);
LC-MS (ESI POS): 460,18 (M+).
ПРИМЕР 32
Получение 3-((бис(4-хлорфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 68)
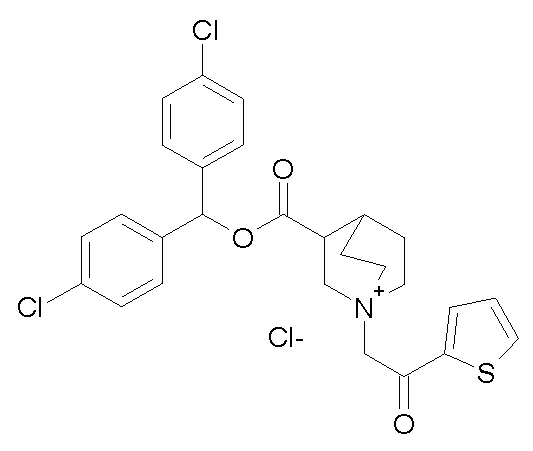
К раствору бис(4-хлорфенил)метилхинуклидин-3-карбоксилата (45 мг, 0,11 ммоль, полученного как в примере 6) в EtOAc (1,6 мл) добавляли 2-хлор-1-(тиофен-2-ил)этанон (20,0 мг, 0,13 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель удаляли, и неочищенный остаток очищали препаративной ВЭЖХ, получая 3-((бис(4-хлорфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (16 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,21 (дд, 1H), 8,09 (дд, 1H), 7,40-7,57 (м, 8H), 7,35 (дд, 1H), 6,92 (с, 1H), 5,09 (с, 2H), 4,01-4,16 (м, 1H), 3,80-4,00 (м, 1H), 3,54-3,78 (м, 3H), 3,47-3,54 (м, 2H), 2,59-2,69 (м, 1H), 2,01-2,21 (м, 2H), 1,82-2,01 (м, 1H), 1,41-1,64 (м, 1H);
LC-MS (ESI POS): 514,02 (M+).
ПРИМЕР 33
Получение 3-((бис(4-фторфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 70)
1- Получение бис(4-фторфенил)метилхинуклидин-3-карбоксилата (соединение 69)
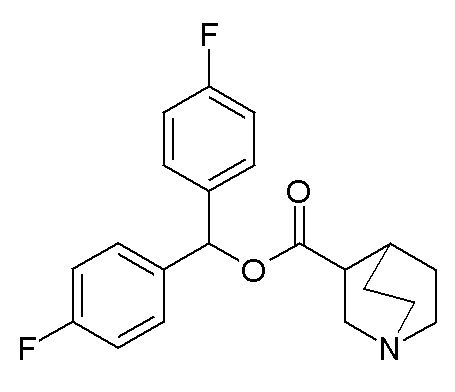
Гидрохлорид хинуклидин-3-карбоновой кислоты (96 мг, 0,50 ммоль), EDC (131 мг, 0,68 ммоль) и HOBT (104 мг, 0,68 ммоль) суспендировали в сухом DMF (5 мл). Добавляли бис(4-фторфенил)метанол (100 мг, 0,45 ммоль) и DIPEA (278 мкл, 1,59 ммоль), и смесь перемешивали при комнатной температуре в течение 24 часов.
Затем снова добавляли гидрохлорид хинуклидин-3-карбоновой кислоты (43,5 мг, 0,23 ммоль), EDC (43,5 мг, 0,23 ммоль), HOBT (34,8 мг, 0,23 ммоль) и DIPEA (79 мкл, 0,45 ммоль), и перемешивание продолжали в течение четырех дней.
Наконец, добавляли гидрохлорид хинуклидин-3-карбоновой кислоты (43,5 мг, 0,23 ммоль), EDC (43,5 мг, 0,23 ммоль) и DIPEA (79 мкл, 0,45 ммоль), и суспензию перемешивали в течение дополнительных 24 часов. Реакционную смесь распределяли между насыщенным NaHCO3 и Et2O, органический слой отделяли, снова промывали насыщенным NaHCO3 и затем соляным раствором. Органическую фазу сушили над Na2SO4, фильтровали и упаривали, получая бис(4-фторфенил)метилхинуклидин-3-карбоксилат (51 мг, рацемическая смесь). Соединение применяли как есть на следующей стадии.
2- Получение 3-((бис(4-фторфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 70)
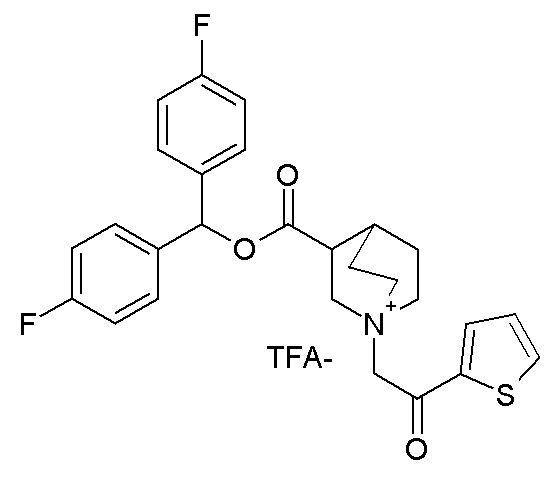
К раствору бис(4-фторфенил)метилхинуклидин-3-карбоксилата (51 мг, 0,14 ммоль) в ацетонитриле (5 мл) добавляли 2-бром-1-(тиофен-2-ил)этанон (32,2 мг, 0,16 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель упаривали, и полученный в результате неочищенный остаток очищали препаративной ВЭЖХ, получая 3-((бис(4-фторфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетат (73,4 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,21 (дд, 1H), 8,08 (дд, 1H), 7,43-7,60 (м, 4H), 7,35 (дд, 1H), 7,15-7,28 (м, 4H), 6,93 (с, 1H), 5,08 (с, 2H), 3,99-4,17 (м, 1H), 3,79-3,94 (м, 1H), 3,62-3,77 (м, 5H), 2,56-2,66 (м, 1H), 1,77-2,20 (м, 3H), 1,31-1,67 (м, 1H);
LC-MS (ESI POS): 482,17 (M+).
ПРИМЕР 34
Получение 3-(бензгидрилоксикарбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 72)
1- Получение бензгидрилхинуклидин-3-карбоксилата (соединение 71)
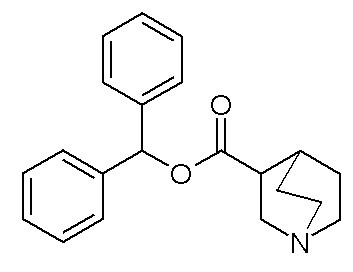
Гидрохлорид хинуклидин-3-карбоновой кислоты (114 мг, 0,60 ммоль), EDC (156 мг, 0,81 ммоль) и HOBT (125 мг, 0,81 ммоль) суспендировали в сухом DMF (5 мл). Добавляли дифенилметанол (100 мг, 0,54 ммоль) и DIPEA (332 мкл, 1,90 ммоль), и смесь перемешивали при комнатной температуре в течение 24 часов. Снова добавляли гидрохлорид хинуклидин-3-карбоновой кислоты (52,0 мг, 0,27 ммоль), EDC (52,0 мг, 0,27 ммоль) и HOBT (41,6 мг, 0,27 ммоль), с последующим добавлением DIPEA (95 мкл, 0,54 ммоль), и смесь перемешивали в течение 4 дней. Затем реакционную смесь распределяли между насыщенным NaHCO3 и Et2O. Органический слой отделяли, снова промывали насыщенным NaHCO3, водой и соляным раствором, сушили над Na2SO4, фильтровали и упаривали, получая бензгидрилхинуклидин-3-карбоксилат (54 мг, рацемическая смесь). Соединение применяли как есть на следующей стадии.
2- Получение 3-(бензгидрилоксикарбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 72)
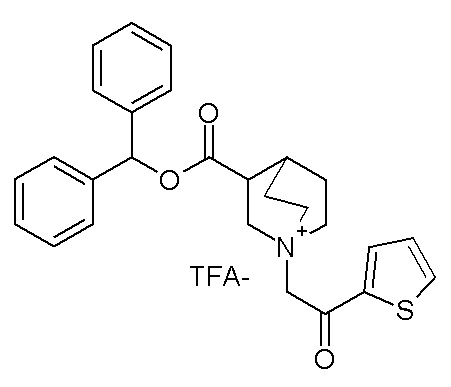
К раствору бензгидрилхинуклидин-3-карбоксилата (54 мг, 0,17 ммоль) в ацетонитриле (5 мл) добавляли 2-бром-1-(тиофен-2-ил)этанон (37,9 мг, 0,18 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель упаривали, и полученный в результате неочищенный остаток вначале очищали препаративной ВЭЖХ и затем флэш-хроматографией (DCM/MeOH=95/5-9/1), получая 3-(бензгидрилоксикарбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетат (53,8 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,20 (дд, 1H), 8,09 (дд, 1H), 7,23-7,52 (м, 11H), 6,90 (с, 1H), 5,09 (с, 2H), 4,04-4,16 (м, 1H), 3,79-3,95 (м, 1H), 3,56-3,79 (м, 4H), 3,33-3,56 (м, 1H), 2,57-2,70 (м, 1H), 1,99-2,24 (м, 2H), 1,76-1,99 (м, 1H), 1,40-1,68 (м, 1H);
LC-MS (ESI POS): 446,15 (M+).
ПРИМЕР 35
Получение 3-(((4-метоксифенил)(фенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 74)
1- Получение (4-метоксифенил)(фенил)метилхинуклидин-3-карбоксилата (соединение 73)
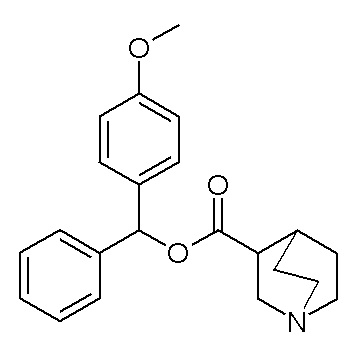
Гидрохлорид хинуклидин-3-карбоновой кислоты (150 мг, 0,78 ммоль), EDC (225 мг, 1,17 ммоль) и HOBT (180 мг, 1,17 ммоль) растворяли в сухом THF (7,5 мл). Добавляли (4-метоксифенил)(фенил)метанол (184 мг, 0,86 ммоль) и TEA (382 мкл, 2,74 ммоль), и полученную в результате реакционную смесь перемешивали при комнатной температуре в течение четырех дней. THF удаляли в вакууме, и неочищенный остаток распределяли между EtOAc и водой. Органическую фазу промывали насыщенным NaHCO3, сушили над Na2SO4, фильтровали и упаривали. Неочищенный остаток очищали флэш-хроматографией (EtOAc/MeOH=8/2 - EtOAc/MeOH=7:3 + 1% NH4OH), получая (4-метоксифенил)(фенил)метилхинуклидин-3-карбоксилат (37 мг, смесь диастереомеров). Соединение применяли как есть на следующей стадии.
2- Получение 3-(((4-метоксифенил)-(фенил)метокси)-карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 74)
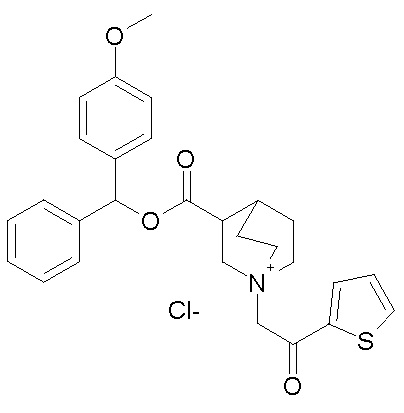
К раствору (4-метоксифенил)(фенил)метилхинуклидин-3-карбоксилата (37 мг, 0,10 ммоль) в EtOAc (1,5 мл) добавляли 2-хлор-1-(тиофен-2-ил)этанон (19 мг, 0,12 ммоль). Ацетонитрил добавляли до полного растворения, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Органические растворители удаляли в вакууме, и остаток очищали флэш-хроматографией (DCM/MeOH=9/1), получая 3-(((4-метоксифенил)(фенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (16 мг, смесь диастереомеров).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,21 (дд, 1H), 8,09 (дд, 1H), 7,24-7,51 (м, 8H), 6,89-6,98 (м, 2H), 6,86 (с, 1H), 5,09 (с, 2H), 4,02-4,20 (м, 1H), 3,80-3,94 (м, 1H), 3,74 (с, 3H), 3,36-3,73 (м, 5H), 2,56-2,66 (м, 1H), 1,79-2,23 (м, 3H), 1,36-1,67 (м, 1H);
LC-MS (ESI POS): 476,17 (M+).
ПРИМЕР 36
Получение 3-(((2-хлорфенил)(4-хлорфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 76)
1- Получение (2-хлорфенил)(4-хлорфенил)метилхинуклидин-3-карбоксилата (соединение 75)
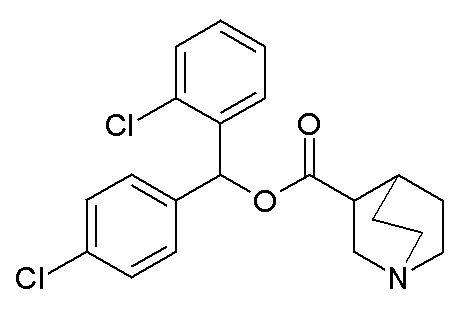
Гидрохлорид хинуклидин-3-карбоновой кислоты (0,1 г, 0,52 ммоль), EDC (150 мг, 0,78 ммоль) и HOBT (120 мг, 0,78 ммоль) растворяли в сухом THF (3 мл). (2-Хлорфенил)(4-хлорфенил)метанол (145 мг, 0,57 ммоль) растворяли в сухом THF (2 мл) и затем добавляли к реакционной смеси. Добавляли триэтиламин (253 мкл, 1,83 ммоль), и полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь упаривали, и неочищенный остаток брали в EtOAc и промывали водой и соляным раствором. Органическую фазу отделяли, сушили (сульфат натрия), фильтровали и упаривали в вакууме. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=95/5), получая (2-хлорфенил)(4-хлорфенил)метилхинуклидин-3-карбоксилат (40 мг, смесь диастереомеров). Соединение применяли как есть на следующей стадии.
2- Получение 3-(((2-хлорфенил)(4-хлорфенил)метокси)-карбонил)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 76)
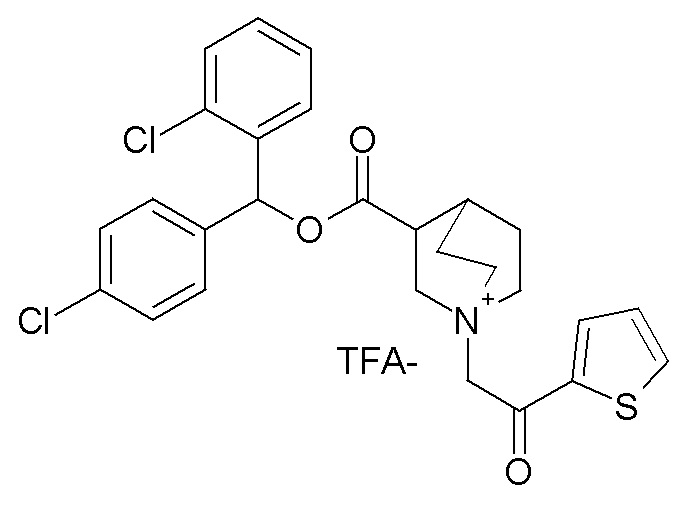
К раствору (2-хлорфенил)(4-хлорфенил)метилхинуклидин-3-карбоксилата (40 мг, 0,10 ммоль) в EtOAc (1,5 мл) добавляли 2-бром-1-(тиофен-3-ил)этанон (23 мг, 0,11 ммоль). Ацетонитрил добавляли до полного растворения, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Органические растворители упаривали в вакууме, и неочищенный остаток очищали препаративной ВЭЖХ, получая 3-(((2-хлорфенил)(4-хлорфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетат (28,6 мг, смесь диастереомеров).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,62 (дд, 1H), 7,74 (дд, 1H), 7,56 (дд, 1H), 7,35-7,66 (м, 8H), 7,12 (с, 1H), 5,05 (с, 2H), 4,09 (дд, 1H), 3,87 (т, 1H), 3,59-3,75 (м, 5H), 2,54-2,64 (м, 1H), 1,86-2,20 (м, 3H), 1,43-1,73 (м, 1H);
LC-MS (ESI POS): 514,08 (M+).
ПРИМЕР 37
Получение 3-(((3,4-дифторфенил)(фенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 78)
1- Получение (3,4-дифторфенил)(фенил)метилхинуклидин-3-карбоксилата (соединение 77)
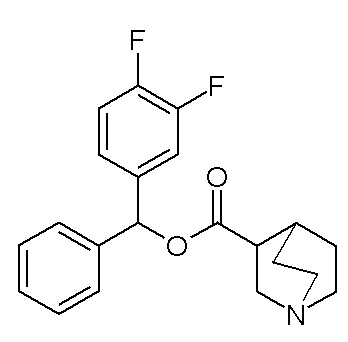
Гидрохлорид хинуклидин-3-карбоновой кислоты (80 мг, 0,42 ммоль), EDC (120 мг, 0,62 ммоль) и HOBT (96 мг, 0,62 ммоль) растворяли в сухом THF (3 мл). (3,4-Дифторфенил)(фенил)метанол (101 мг, 0,46 ммоль) растворяли в сухом THF (2 мл) и затем добавляли к реакционной смеси. Наконец, добавляли триэтиламин (203 мкл, 1,46 ммоль), и полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи. Летучие компоненты упаривали, и неочищенный остаток брали в EtOAc и промывали водой и затем насыщенным NaHCO3. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1), получая (3,4-дифторфенил)(фенил)метилхинуклидин-3-карбоксилат (65 мг, смесь диастереомеров).
2- Получение 3-(((3,4-дифторфенил)(фенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 78)
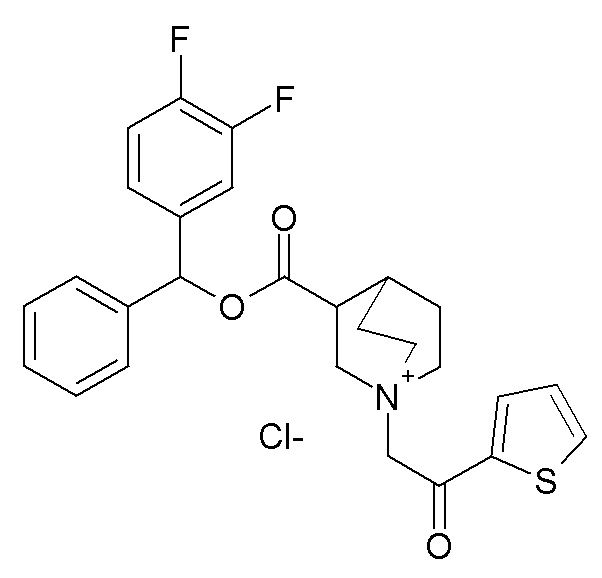
К раствору (3,4-дифторфенил)(фенил)метилхинуклидин-3-карбоксилата (65 мг, 0,18 ммоль) в этилацетате (3 мл) добавляли 2-хлор-1-(тиофен-2-ил)этанон (29,2 мг, 0,18 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме, и остаток растворяли с диэтиловым эфиром и фильтровали, получая 3-(((3,4-дифторфенил)(фенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (20 мг, смесь диастереомеров).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,21 (д, 1H), 8,11 (д, 1H), 7,52-7,71 (м, 1H), 7,18-7,52 (м, 8H), 6,90 (с, 1H), 5,14 (с, 2H), 3,98-4,19 (м, 1H), 3,82-3,98 (м, 1H), 3,41-3,82 (м, 5H), 2,59-2,69 (м, 1H), 1,40-2,21 (м, 4H);
LC-MS (ESI POS): 482,17 (M+).
ПРИМЕР 38
Получение 3-((3-фторбензилокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 80)
1- Получение 3-фторбензилхинуклидин-3-карбоксилата (соединение 79)
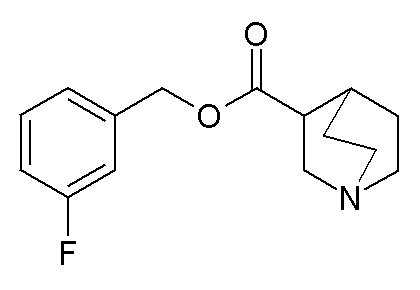
Гидрохлорид хинуклидин-3-карбоновой кислоты (95 мг, 0,50 ммоль), EDC (143 мг, 0,74 ммоль), HOBT (114 мг, 0,74 ммоль) и триэтиламин (242 мкл, 1,73 ммоль) растворяли в сухом THF (5 мл). (3-Фторфенил)метанол (68,8 мг, 0,54 ммоль) растворяли в сухом THF (2 мл) и добавляли к реакционной смеси. Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи. Летучие компоненты упаривали, и неочищенный остаток брали в EtOAc и промывали водой и затем насыщенным NaHCO3. Органическую фазу отделяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая 3-фторбензилхинуклидин-3-карбоксилат (120 мг, рацемическая смесь). Соединение применяли на следующей стадии без любой дополнительной очистки.
2- Получение 3-((3-фторбензилокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 80)
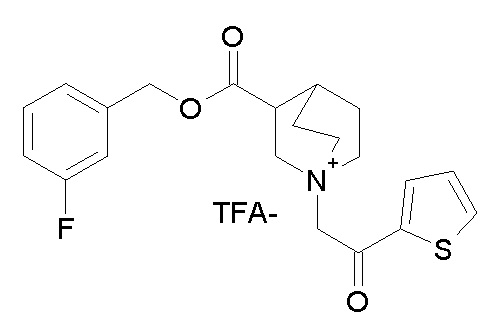
2-Хлор-1-(тиофен-2-ил)этанон (67 мг, 0,42 ммоль) добавляли к раствору 3-фторбензилхинуклидин-3-карбоксилата (100 мг, 0,38 ммоль) в смеси EtOAc (1,2 мл) с несколькими каплями DMF. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли вторую порцию 2-хлор-1-(тиофен-2-ил)этанона (67 мг, 0,42 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли, и остаток растворяли в ацетонитриле и нагревали при облучении микроволнами при 100°C в течение 2 часов. Ацетонитрил упаривали, и неочищенный остаток растирали с Et2O и затем очищали препаративной ВЭЖХ, получая 3-((3-фторбензилокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетат (70 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,21 (дд, 1H), 8,09 (дд, 1H), 7,45 (тд, 1H), 7,36 (дд, 1H), 7,24-7,30 (м, 2H), 7,11-7,23 (м, 1H), 5,26 (д, 1H), 5,20 (д, 1H), 5,10 (с, 2H), 4,10 (ддд, 1H), 3,81-3,93 (м, 1H), 3,57-3,76 (м, 4H), 3,31-3,49 (м, 1H), 2,54-2,61 (м, 1H), 1,85-2,18 (м, 3H), 1,63-1,85 (м, 1H);
LC-MS (ESI POS): 388,27 (M+).
ПРИМЕР 39
Получение 3-((9H-флуорен-9-илокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 82)
1- Получение 9H-флуорен-9-илхинуклидин-3-карбоксилата (соединение 81)
Гидрохлорид хинуклидин-3-карбоновой кислоты (95 мг, 0,50 ммоль), EDC (143 мг, 0,74 ммоль), триэтиламин (242 мкл, 1,73 ммоль) и HOBT (114 мг, 0,74 ммоль) растворяли в сухом THF (5 мл). 9H-флуорен-9-ол (99 мг, 0,54 ммоль) растворяли в сухом THF (2 мл) и затем добавляли к реакционной смеси. Полученную в результате суспензию перемешивали при комнатной температуре в течение ночи. Смесь упаривали, и неочищенный остаток брали в EtOAc и промывали последовательно водой и насыщенным NaHCO3. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1), получая 9H-флуорен-9-илхинуклидин-3-карбоксилат (102 мг, рацемическая смесь).
2- Получение 3-((9H-флуорен-9-илокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (соединение 82)
2-Хлор-1-(тиофен-2-ил)этанон (55 мг, 0,344 ммоль) добавляли к раствору 9H-флуорен-9-илхинуклидин-3-карбоксилата (100 мг, 0,31 ммоль) в ацетонитриле (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Снова добавляли 2-хлор-1-(тиофен-2-ил)этанон (55 мг, 0,34 ммоль), и смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли, и остаток повторно растворяли в ацетонитриле и нагревали при облучении микроволнами при 100°C в течение 3 часов. Ацетонитрил удаляли, и остаток растирали с Et2O и затем очищали препаративной ВЭЖХ, получая 3-((9H-флуорен-9-илокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетат (46 мг, рацемическая смесь).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,22 (дд, 1H), 8,11 (дд, 1H), 7,87 (д, 2H), 7,55-7,65 (м, 2H), 7,49 (т, 2H), 7,31-7,42 (м, 3H), 6,85 (с, 1H), 5,12 (с, 2H), 4,11-4,27 (м, 1H), 3,85-3,96 (м, 1H), 3,59-3,78 (м, 4H), 3,35-3,52 (м, 1H), 2,39-2,46 (м, 1H), 1,82-2,13 (м, 4H);
LC-MS (ESI POS): 444,30 (M+).
ПРИМЕР 40
Получение 4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиофен-2-ил)этил)пиперидинийхлорида (соединение 84)
1- Получение бис(3-фторфенил)метил 1-метилпиперидин-4-карбоксилата (соединение 83)
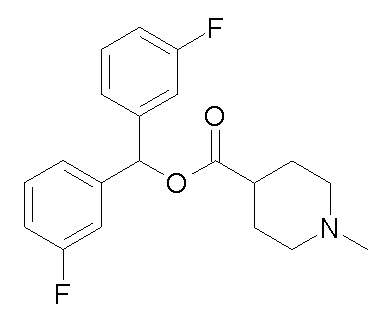
Гидрохлорид 1-метилпиперидин-4-карбоновой кислоты (250 мг, 1,39 ммоль), EDC (400 мг, 2,09 ммоль) и HOBT (320 мг, 2,09 ммоль) растворяли в сухом THF (10 мл). Бис(3-фторфенил)метанол (337 мг, 1,53 ммоль) растворяли в сухом THF (4 мл) и затем добавляли к реакционной смеси. Наконец, добавляли триэтиламин (679 мкл, 4,87 ммоль), и полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь упаривали, и остаток брали в EtOAc и промывали водой и насыщенным NaHCO3. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=9/1), получая бис(3-фторфенил)метил 1-метилпиперидин-4-карбоксилат (150 мг).
2- Получение 4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиофен-2-ил)этил)пиперидинийхлорида (соединение 84)
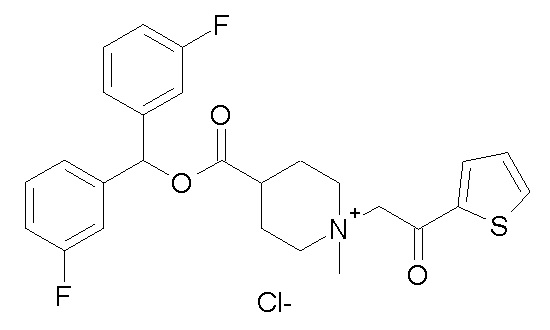
К раствору бис(3-фторфенил)метил 1-метилпиперидин-4-карбоксилата (150 мг, 0,43 ммоль) в этилацетате (5 мл) добавляли 2-хлор-1-(тиофен-2-ил)этанон (69,8 мг, 0,43 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме, и остаток брали в диэтиловом эфире, фильтровали и очищали флэш-хроматографией (DCM/MeOH=95/5), получая 4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиофен-2-ил)этил)пиперидинийхлорид (18 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,20 (д, 1H), 8,14 (д, 1H), 7,23-7,54 (м, 7H), 7,04-7,23 (м, 2H), 6,87 (с, 1H), 5,24 (ушир.с, 2H), 3,90-4,20 (м, 2H), 3,44-3,60 (м, 2H), 3,31 (ушир.с, 3H), 2,80-3,00 (м, 1H), 1,96-2,32 (м, 4H);
LC-MS (ESI POS): 470,18 (M+).
ПРИМЕР 41
Получение 4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиофен-3-ил)этил)пиперидинийбромида (соединение 85)
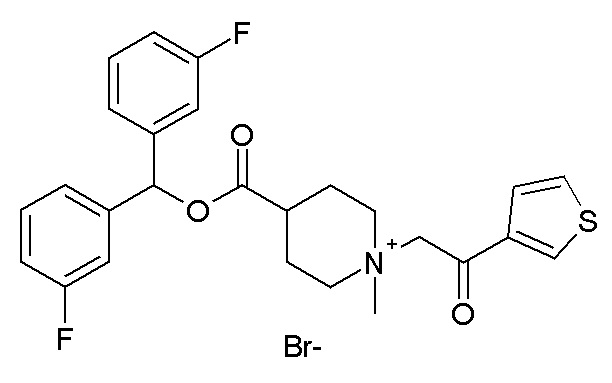
2-Бром-1-(тиофен-3-ил)этанон (23,7 мг, 0,12 ммоль) добавляли к раствору бис(3-фторфенил)метил 1-метилпиперидин-4-карбоксилата (40 мг, 0,12 ммоль, полученного как в примере 40) в EtOAc (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель упаривали, и неочищенный остаток очищали препаративной ВЭЖХ (элюент: CH3CN/H2O), получая 4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиофен-3-ил)этил)пиперидинийбромид (36,1 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,63-8,71 (м, 1H), 7,67-7,83 (м, 1H), 7,50-7,62 (м, 1H), 7,24-7,50 (м, 6H), 7,01-7,24 (м, 2H), 6,87 и 6,88 (с, 1H), 5,17 и 5,20 (с, 2H), 3,73-4,07 (м, 2H), 3,39-3,71 (м, 2H), 3,30 и 3,32 (с, 3H), 2,79-3,05 (м, 1H), 2,11-2,36 (м, 4H);
LC-MS (ESI POS): 470,24 (M+).
ПРИМЕР 42
Получение 4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиазол-2-ил)этил)пиперидинийбромида (соединение 86)
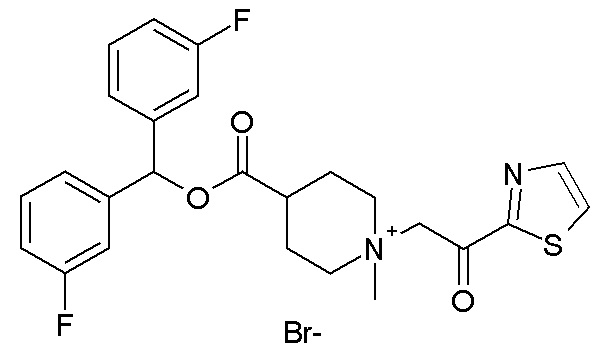
К раствору бис(3-фторфенил)метил 1-метилпиперидин-4-карбоксилата (106 мг, 0,31 ммоль, полученного как в примере 40) в EtOAc (3 мл) добавляли 2-бром-1-(тиазол-2-ил)этанон (63,2 мг, 0,31 ммоль). Смесь перемешивали при комнатной температуре в течение 27 часов. Растворитель упаривали, и неочищенный остаток растирали с Et2O и собирали фильтрацией. Соединение очищали флэш-хроматографией (DCM/MeOH=95/5-9/1), получая 4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиазол-2-ил)этил)пиперидинийбромид (120 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,40 (д, 1H), 8,26 (д, 1H), 7,25-7,55 (м, 6H), 7,02-7,25 (м, 2H), 6,87 (с, 1H), 5,32 и 5,34 (с, 1H), 3,44-4,26 (м, 4H), 3,32 (с, 3H), 2,77-3,05 (м, 1H), 2,00-2,43 (м, 4H);
LC-MS (ESI POS): 471,15 (M+).
ПРИМЕР 43
Получение (1R,4R)-4-((1,2-дифенилэтокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромида (соединение 88a) и (1S,4S)-4-((1,2-дифенилэтокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромида (соединение 88b)
Получение 1,2-дифенилэтил 1-метилпиперидин-4-карбоксилата (соединение 87)
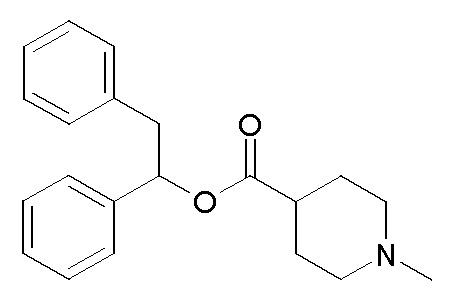
Оксалилдихлорид (141 мкл, 1,67 ммоль) добавляли по каплям к раствору гидрохлорида 1-метилпиперидин-4-карбоновой кислоты (300 мг, 1,67 ммоль) в DCM (20 мл) и нескольких капель DMF (каталитическое количество). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Летучие компоненты удаляли в вакууме, и неочищенный остаток брали в пиридине (20 мл). Добавляли 1,2-дифенилэтанол (331 мг, 1,67 ммоль), и полученную в результате суспензию нагревали при облучении микроволнами при 140°C в течение 1 часа. Пиридин удаляли в вакууме, и неочищенный остаток очищали флэш-хроматграфией (DCM/MeOH=97/3-94/6), получая 1,2-дифенилэтил 1-метилпиперидин-4-карбоксилат (200 мг).
2- Получение (1R,4R)-4-((1,2-дифенилэтокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромида (соединение 88a) и (1S,4S)-4-((1,2-дифенилэтокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромида (соединение 88b)
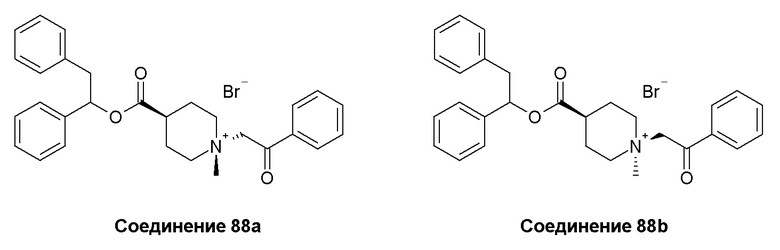
2-Бром-1-фенилэтанон (61,5 мг, 0,31 ммоль) добавляли к раствору 1,2-дифенилэтил l-метилпиперидин-4-карбоксилата (100 мг, 0,31 ммоль) в ацетонитриле (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель упаривали, и неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=98/2), собирая вначале (1R,4R)-4-((1,2-дифенилэтокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромид (82,3 мг, соединение 64a) и затем (1S,4S)-4-((1,2-дифенилэтокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромид (32,1 мг, соединение 64b).
Соединение 88a:
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,88-8,15 (м, 2H), 7,77 (тт, 1H), 7,50-7,69 (м, 2H), 7,15-7,48 (м, 10H), 5,98 (т, 1H), 5,30 (с, 2H), 3,44-3,84 (м, 4H), 3,27 (с, 3H), 3,10-3,20 (м, 2H), 2,57-2,71 (м, 1H), 1,89-2,18 (м, 4H);
LC-MS (ESI POS): 442,15 (M+).
Соединение 88b:
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,01 (м, 2H), 7,66-7,83 (м, 1H), 7,50-7,66 (м, 2H), 7,12-7,36 (м, 10H), 5,72-6,10 (м, 1H), 5,25 (с, 2H), 3,89-3,95 (м, 1H), 3,79-3,86 (м, 1H), 3,31-3,66 (м, 2H), 3,26 (с, 3H), 2,98-3,18 (м, 2H), 2,65-2,71 (м, 1H), 1,97-2,13 (м, 4H);
LC-MS (ESI POS): 442,2 (M+).
ПРИМЕР 44
Получение (1R,4R)-4-(((E)-1,2-дифенилвинилокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромида (соединение 90)
Получение (E)-1,2-дифенилвинил 1-метилпиперидин-4-карбоксилата (соединение 89)
Оксалилдихлорид (236 мкл, 2,78 ммоль) добавляли по каплям к раствору гидрохлорида 1-метилпиперидин-4-карбоновой кислоты (500 мг, 2,78 ммоль) в DCM (30 мл) и нескольких капель DMF (каталитическое количество). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Летучие компоненты удаляли в вакууме, и неочищенный остаток брали в пиридине (40 мл). Добавляли 1,2-дифенилэтанон (546 мг, 2,78 ммоль), и полученную в результате суспензию нагревали при облучении микроволнами при 140°C в течение 1 часа. Пиридин удаляли в вакууме, и неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=95/5), получая (E)-1,2-дифенилвинил 1-метилпиперидин-4-карбоксилат (62 мг).
Получение (1R,4R)-4-(((E)-1,2-дифенилвинилокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромида (соединение 90)
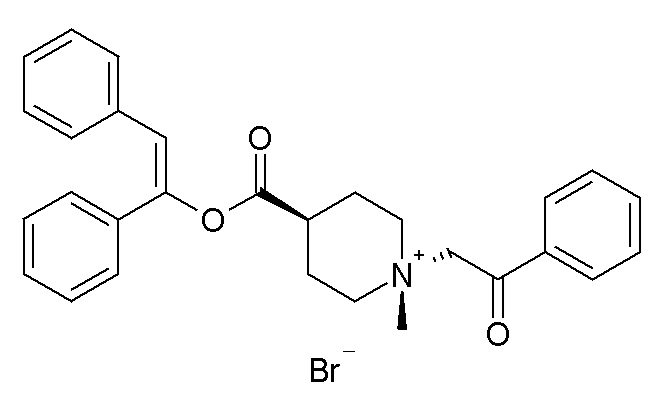
2-Бром-1-фенилэтанон (57,6 мг, 0,29 ммоль) добавляли к раствору (E)-1,2-дифенилвинил 1-метилпиперидин-4-карбоксилата (62 мг, 0,19 ммоль) в ацетонитриле (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем растворитель упаривали. Неочищенный остаток очищали флэш-хроматографией (DCM/MeOH=98/2) и затем флэш-хроматографией (DCM/MeOH=99/1), получая (1R,4R)-4-(((E)-1,2-дифенилвинилокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромид (18 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,96-8,08 (м, 2H), 7,73-7,84 (м, 1H), 7,60-7,70 (м, 4H), 7,52-7,60 (м, 2H), 7,38-7,50 (м, 5H), 7,28-7,36 (м, 1H), 7,06 (с, 1H), 5,36 (с, 2H), 3,77-3,95 (м, 2H), 3,60-3,76 (м, 2H), 3,36 (с, 3H), 3,12 (тт, 1H), 2,10-2,42 (м, 4H);
LC-MS (ESI POS): 440,23 (M+).
ПРИМЕР 45
Получение 4-((3-фторбензилокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 92)
Получение 3-фторбензилхинуклидин-4-карбоксилата (соединение 91)
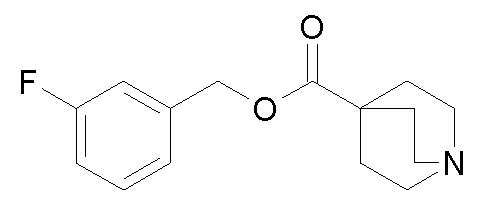
Смесь гидрохлорида хинуклидин-4-карбоновой кислоты (100 мг, 0,52 ммоль) и тионилхлорида (500 мкл, 6,85 ммоль) кипятили с обратным холодильником в течение 2 часов. Реакционную смесь охлаждали при комнатной температуре, и растворитель аккуратно удаляли. Остаток суспендировали в сухом DCM и обрабатывали (3-фторфенил)метанолом (65,8 мг, 0,52 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель упаривали, остаток растворяли в воде (1 мл), подщелачивали NaHCO3 и экстрагировали дважды EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали, получая 3-фторбензилхинуклидин-4-карбоксилат (41 мг, 29,8% выход), который применяли на следующей стадии без любой дополнительной очистки.
Получение 4-((3-фторбензилокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 92)
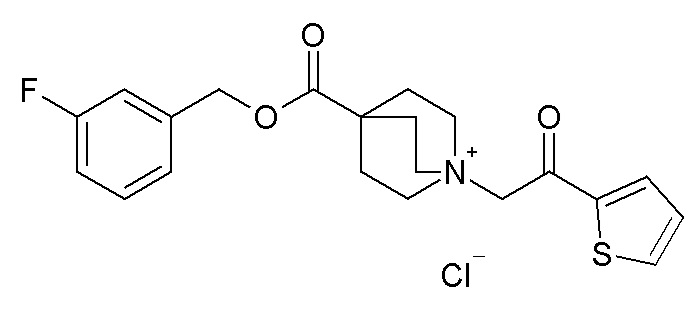
К раствору 3-фторбензилхинуклидин-4-карбоксилата (41 мг, 0,16 ммоль), растворенного в EtOAc (2 мл), добавляли 2-хлор-1-(тиофен-2-ил)этанон (20,0 мг, 0,12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение трех дней, затем осадок собирали фильтрованием с отсасыванием, получая 4-((3-фторбензилокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (30 мг, 45,4% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,21 (дд, 1H), 8,01-8,17 (м, 1H), 7,39-7,58 (м, 1H), 7,35 (дд, 1H), 7,06-7,28 (м, 3H), 5,18 (с, 2H), 4,97-5,13 (м, 2H), 3,68-3,87 (м, 6H), 2,04-2,30 (м, 6H);
LC-MS (ESI POS): 388,21 (M+).
ПРИМЕР 46
Получение 3-(бензилтиокарбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 94)
Получение S-бензилхинуклидин-3-карботиоата (соединение 93)
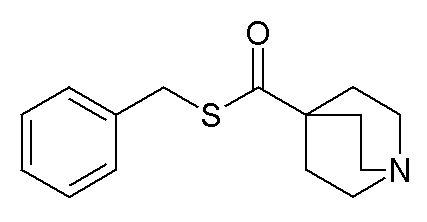
Гидрохлорид хинуклидин-3-карбоновой кислоты (55 мг, 0,29 ммоль), EDC (83 мг, 0,43 ммоль) и HOBT (65,9 мг, 0,43 ммоль) растворяли в сухом THF (5 мл) в атмосфере азота. Триэтиламин (140 мкл, 1,00 ммоль) и фенилметантиол (37,1 мкл, 0,32 ммоль) растворяли в сухом THF (2 мл) и затем добавляли к реакционной смеси. Полученную в результате суспензию перемешивали при комнатной температуре в течение ночи. Смесь упаривали, и неочищенный остаток распределяли между EtOAc и водой. Органическую фазу промывали насыщенным NaHCO3, сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая S-бензилхинуклидин-3-карботиоат (110 мг, неочищенный), который применяли на следующей стадии без любой дополнительной очистки.
2- Получение 3-(бензилтиокарбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (соединение 94)
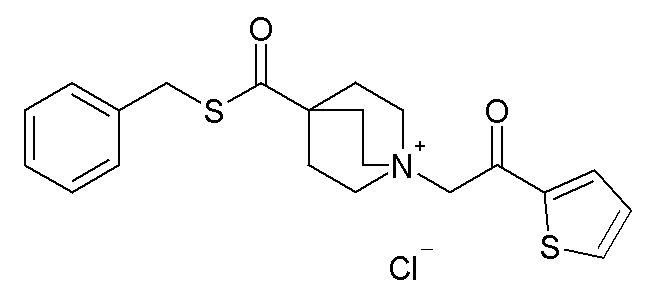
2-Хлор-1-(тиофен-2-ил)этанон (68,0 мг, 0,42 ммоль) добавляли к раствору S-бензилхинуклидин-3-карботиоата (100 мг, 0,38 ммоль) в ацетонитриле (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем добавляли вторую порцию 2-хлор-1-(тиофен-2-ил)этанона (68,0 мг, 0,42 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение дополнительных 3 часов. Растворитель удаляли в вакууме, и остаток растворяли в ацетонитриле и нагревали при облучении микроволнами при 100°C в течение 3 часов. Ацетонитрил удаляли, и остаток растирали с Et2O. Затем неочищенный остаток очищали препаративной ВЭЖХ (элюент: CH3CN/H2O), получая 3-(бензилтиокарбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорид (8,9 мг, 5,5% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,17 (д, 1H), 7,81 (д, 1H), 7,24-7,42 (м, 5H), 7,20 (дд, 1H), 5,23 (с, 2H), 4,24-4,33 (м, 2H), 4,25 (д, 1H), 4,19 (д, 1H), 3,94-4,14 (м, 3H), 3,71-3,87 (м, 1H), 3,38-3,52 (м, 1H), 2,52-2,67 (м, 1H), 2,07-2,36 (м, 2H), 1,81-2,07 (м, 2H);
LC-MS (ESI POS): 396,29 (M+).
Обозначения
* ЯМР
с = синглет
д = дублет
т = триплет
кв = квартет
дд = дублет дублетов
м = мультиплет
ушир. = уширенный
Получение биологических характеристик
Взаимодействие с M3 мускариновыми рецепторами можно оценить по результатам in vitro исследований, которые оценивают с помощью M3/M2 анализа связывания, эффективности исследуемых соединений и изменения ингибирующей активности, вызванного вымыванием антагонистов в отделенной трахее морской свинки, и in vivo продолжительности действия относительно бронхоспазма, индуцированного ацетилхолином, у морской свинки.
ПРИМЕР 47
Анализы на M3/M2 связывание
Клетки CHO-K1 клона, экспрессирующие человеческие M2 или M3-рецепторы (Swissprot P08172, P20309, соответственно) брали для анализа в фосфатно-буферном солевом растворе, не содержащем Ca++/Mg++, и повторно собирали центрифугированием при 1500 об/мин в течение 3 минут. Осадок после центрифугирования суспендировали в охлажденном на льду буфере A (15 мМ Tris-HCl pH 7,4, 2 мМ MgCl2, 0,3 мМ EDTA, 1 мМ EGTA) и гомогенизировали с помощью PBI politron (установленный на 5 в течение 15 секунд). Неочищенную мембранную фракцию собирали двумя последовательными стадиями центрифугирования при 40000 g в течение 20 минут при 4°C, разделенными стадией промывки в буфере A. Наконец, полученный осадок после центрифугирования повторно суспендировали в буфере B (75 мМ Tris HC1 pH 7,4, 12,5 мМ MgCl2, 0,3 мМ EDTA, 1 мМ EGTA, 250 мМ сахароза), и аликвоты хранили при -80°C.
В день эксперимента, замороженные мембраны повторно суспендировали в буфере C (50 мМ Tris-HCl pH 7,4, 2,5 мМ MgCl2, 1 мМ EDTA). Неселективный мускариновый лиганд, [3H]-N-метилскополамин (Mol. Pharmacol. 45:899-907), применяли для мечения M2 и M3 сайтов связывания. Эксперименты на связывание проводили в двух повторах (кривые концентраций по десяти точкам) в 96-луночных планшетах при концентрации радиолиганда 0,1-0,3 нМ. Неспецифическое связывание определяли в присутствии "холодного" N-метилскополамина 10 мкМ. Образцы (конечный объем 0,75 мл) выдерживали при комнатной температуре в течение 60 минут для M2 и 90 мин для M3 анализа связывания. Реакцию прекращали быстрой фильтрацией через GF/B Unifilter планшеты и двух промывок (0,75 мл) холодным буфером C, применяя Packard Filtermate Harvester. Радиоактивность на фильтрах измеряли сцинтилляционным счетчиком для микропланшетов TriCarb 2500 (PerkinElmer).
Значения ингибирующей M3 активности, полученные для соединений, находились в диапазоне 0,265-1514 нМ.
ПРИМЕР 48
In vitro взаимодействие с M3 рецепторами
Эффективность антагонистической активности в отделенной трахее морской свинки исследовали, следуя способу, описанному ранее Haddad EB et al. в Br J Pharmacol 127, 413-420, 1999, с небольшими изменениями.
Суммарную кривую зависимости эффекта от концентрации для исследуемых антагонистов строили на препаратах, у которых мышцы были предварительно сокращены карбахолом, до достижения полного ингибирования тонуса гладких мышц. Концентрацию антагониста, оказывающую 50% обращение тонического сокращения, индуцированного карбахолом (IC50), брали в качестве величины эффективности в данном биоанализе.
В экспериментах, нацеленных на оценку изменения ингибирующих эффектов, производимых исследуемыми соединениями, минимальную концентрацию исследуемых соединений, о которой известно, что она оказывает максимальный ингибирующий эффект, вводили в препараты, в которых мышцы были предварительно сокращены карбахолом. Как только тоническое сокращение полностью восстанавливалось, раствор для инкубатора органов обновляли, и препараты тщательно промывали свежим раствором Кребса. Карбахол (0,3 мкМ) вводили снова (с 30 минутным интервалом между удалением и следующим введением) в течение следующих 4 часов.
После введения карбахола ингибирующие эффекты соединений настоящего изобретения, введенных при концентрации 10 нМ, выражали в виде процентов восстановления реакции сокращения на карбахол. Процент восстановления через четыре часа после вымывания был меньшим, чем 50%.
ПРИМЕР 49
In vivo исследования
Испытания in vivo бронхоспазма, вызванного ацетилхолином у морской свинки, проводили согласно H. Konzett H и Rossler F Arch Exp Path Pharmacol 195, 71-74, 1940. Водные растворы исследуемых соединений вливали интратрахеально анестезированным морским свинкам при искусственном дыхании. Бронхиальный ответ на внутривенное введение ацетилхолина определяли перед и после введения лекарственного средства, и изменения легочной резистентности в несколько моментов времени выражали в виде процентов ингибирования бронхоспазма.
Бронхолитическая активность исследуемых соединений сохранялась неизменной вплоть до 24 часов после введения.
ПРИМЕР 50
Стабильность в легких
Во-первых, свежие легкие крыс (предварительно промытые соляным раствором) гомогенизировали в аммиачно-формиатном буфере 20 мМ. Для того чтобы показать, что соединения разрушаются, стабильность в гомогенате легких исследовали для соединения настоящего изобретения через 1 и 5 часов. Вкратце, 10 мкл исходного раствора 250 мкМ соединения в ацетонитриле добавляли к 1 мл гомогената легких, и образцы выдерживали при 37°C. Гомогенат легких (50 мкл) отбирали через 0,1 и 5 часов выдерживания и добавляли к 200 мкл ацетонитрила с добавлением варепамила в качестве внутреннего стандарта (250 нг/мл). Образцы анализировали ВЭЖХ-МС/МС анализом.
Стабильность в легких рассчитывали как процент, остающийся через 1 и 5 часов делением площади пика через 1 или 5 часов на площадь пика в момент времени 0.
Более чем 79% исследуемых соединений можно было все еще обнаружить после 1 часа выдерживания и более чем 57% через 5 часов, показывая, что данные соединения являются стабильными в присутствии гомогенизированного легкого.
Изобретение относится к соединениям общей формулы (IA) и их фармацевтически приемлемым солям, которые обладают действием антагониста М3 мускариновых рецепторов. В формуле (IA) R1 представляет собой группу формулы (а), где R3 и R4 являются одинаковыми или различными и представляют собой Н или арил, который может быть необязательно замещен одним или более заместителями, выбранными из группы, состоящей из атомов галогена, (C1-С6)алкокси и (C1-С6)галогеналкила, при условии, что R3 и R4 не являются одновременно Н; R2 представляет собой группу формулы (с), где А- представляет собой физиологически приемлемый анион, а R5 представляет собой группу формулы (е), где p равен 0, 1, 2 или 3, Р отсутствует или выбран из группы, состоящей из -O-, -С(О)- и -CON(H)-, q равен 0, 1 или 2, и W выбран из группы, состоящей из (C1-C6)алкила, (С2-С6)алкенила, арила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, ОН, CN, (C1-C6)алкила и (C1-C6)алкокси. Изобретение относится также к ряду конкретных соединений, фармацевтической композиции, применению указанных соединений для получения лекарственного средства для предотвращения и/или лечения бронхообструктивных или воспалительных заболеваний и устройству, содержащему фармацевтическую композицию. 5 н. и 2 з.п. ф-лы, 7 табл., 50 пр.
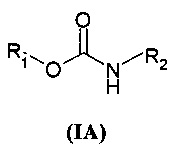
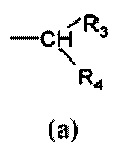
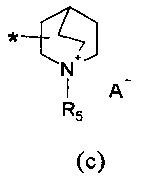
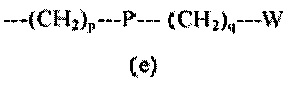
1. Соединение общей формулы (IA)
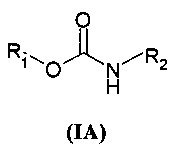 ,
,
где R1 представляет собой группу формулы (а)
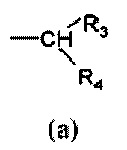 ,
,
где R3 и R4 являются одинаковыми или различными и представляют собой Н или арил, который может быть необязательно замещен одним или более заместителями, выбранными из группы, состоящей из атомов галогена, (C1-С6)алкокси и (C1-С6)галогеналкила, при условии, что R3 и R4 не являются одновременно Н, R2 представляет собой группу формулы (с)
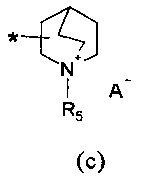 ,
,
где А- представляет собой физиологически приемлемый анион, а R5 представляет собой группу формулы (е)
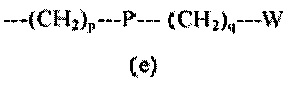 ,
,
где p равен 0, 1, 2 или 3, Р отсутствует или выбран из группы, состоящей из -O-, -С(О)- и -CON(H)-, q равен 0, 1 или 2, и W выбран из группы, состоящей из (C1-C6)алкила, (С2-С6)алкенила, арила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, ОН, CN, (C1-C6)алкила и (C1-C6)алкокси,
где указанный гетероарил представляет собой моно- или бициклическую кольцевую систему с 5-20 атомами в кольце, в которой по меньшей мере одно кольцо является ароматическим и в которой по меньшей мере один атом углерода заменен на гетероатом N, S или О,
и его фармацевтически приемлемые соли.
2. Соединение общей формулы (IA) по п.1, где R1 выбран из бис(3-фторфенил)метила, бензгидрила, бис(4-фторфенил)метила, (4-метоксифенил)(фенил)метила, (2-фторфенил)(4-фторфенил)метила, (3,4-дифторфенил)(фенил)метила, (4-(трифторметил)фенил)метила и (2-хлорфенил)(4-хлорфенил)метила, и R2 выбран из (2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октанила, (2-(4-хлорфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-п-толилэтил)-1-азониабицикло[2.2.2]октанила, (2-(4-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(3-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(2-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(4-метоксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(4-гидроксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-(5-хлортиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-(тиазол-2-ил)этил)-1-азониабицикло[2.2.2]октанила, (2-оксопропил)-1-азониабицикло[2.2.2]октанила, (3-метилбут-2-енил)-1-азониабицикло[2.2.2]октанила, бензил-1-азониабицикло[2.2.2]октанила, (3-феноксипропил)-1-азониабицикло[2.2.2]октанила, (2-(5-цианотиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октанила, (2-(изоксазол-3-иламино)-2-оксоэтил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанила, (2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанила, (2-феноксиэтил)-1-азониабицикло[2.2.2]октанила, ((2,3-дигидробензофуран-5-ил)этил)-1-азониабицикло[2.2.2]октанила и (4-фторфенэтил)-1-азониабицикло[2.2.2]октанила.
3. Соединение, выбранное из группы, состоящей из:
(R)-бис(3-фторфенил)метилхинуклидин-3-илкарбамата,
(R)-бензгидрилхинуклидин-3-илкарбамата,
(R)-бис(4-фторфенил)метилхинуклидин-3-илкарбамата,
(4-метоксифенил)(фенил)метил (R)-хинуклидин-3-илкарбамата,
(2-фторфенил)(4-фторфенил)метил (R)-хинуклидин-3-илкарбамата,
(2-фторфенил)(3-фторфенил)метил (R)-хинуклидин-3-илкарбамата,
(3,4-дифторфенил)(фенил)метил (R)-хинуклидин-3-илкарбамата,
бис(3-фторфенил)метилхинуклидин-3-карбоксилата,
1,2-дифенилэтилхинуклидин-3-карбоксилата,
бис(4-хлорфенил)метилхинуклидин-3-карбоксилата,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(4-хлорфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-п-толилэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(4-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(3-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(2-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(4-метоксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(4-гидроксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-1-(2-(бензо[b]тиофен-5-ил)-2-оксоэтил)-3-((бис(3-фторфенил)метокси)карбониламино)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(5-хлортиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(тиазол-2-ил)этил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксопропил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(3-метилбут-2-енил)-1-азониабицикло[2.2.2]октанбромида,
(R)-1-бензил-3-((бис(3-фторфенил)метокси)карбониламино)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(3-феноксипропил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(5-цианотиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(изоксазол-3-иламино)-2-оксоэтил)-1-азониабицикло[2.2.2]октанхлорида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-феноксиэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(2-(2,3-дигидробензофуран-5-ил)этил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-((бис(3-фторфенил)метокси)карбониламино)-1-(4-фторфенэтил)-1-азониабицикло[2.2.2]октанбромида,
(R)-3-(бензгидрилоксикарбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида,
(R)-3-((бис(4-фторфенил)метокси)карбониламино)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида,
(3R)-3-(((4-метоксифенил)(фенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
(3R)-3-(((2-фторфенил)(4-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
(3R)-3-(((2-фторфенил)(3-фторфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
(3R)-3-(((3,4-дифторфенил)(фенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
фенил(4-(трифторметил)фенил)метил (R)-хинуклидин-3-илкарбамата,
(3R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-((фенил(4-(трифторметил)фенил)метокси)карбониламино)-1-азониабицикло[2.2.2]октанхлорида,
(2-хлорфенил)(4-хлорфенил)метил (R)-хинуклидин-3-илкарбамата,
(3R)-3-(((2-хлорфенил)(4-хлорфенил)метокси)карбониламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
(R)-тиофен-2-илметилхинуклидин-3-илкарбамата,
(R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-((тиофен-2-илметокси)карбониламино)-1-азониабицикло[2.2.2]октанхлорида,
(R)-О-бис(3-фторфенил)метилхинуклидин-3-илкарбамотиоата,
(R)-3-((бис(3-фторфенил)метокси)карбонотиоиламино)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
(R)-бис(3-фторфенил)метил-1-бензилпирролидин-3-илкарбамата,
(3R)-1-бензил-3-((бис(3-фторфенил)метокси)карбониламино)-1-метилпирролидиниййодида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(4-метоксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(4-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-п-толилэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(5-хлортиофен-2-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(4-хлорфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(3-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксопропил)-1-азониабицикло[2.2.2]октанхлорида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-трет-бутокси-2-оксоэтил)-1-азониабицикло[2.2.2]октанхлорида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-(пиридин-2-ил)этил)-1-азониабицикло[2.2.2]октан-2,2,2-трифторацетата,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(3-(этоксикарбонил)изоксазол-5-ил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(2-фторфенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октанбромида,
3-((бис(3-фторфенил)метокси)карбонил)-1-(2-(4-гидроксифенил)-2-оксоэтил)-1-азониабицикло[2.2.2]октанбромида,
1-(2-(бензо[b]тиофен-5-ил)-2-оксоэтил)-3-((бис(3-фторфенил)метокси)карбонил)-1-азониабицикло[2.2.2]октан-2,2,2-трифторацетата,
1-бензил-3-((бис(3-фторфенил)метокси)карбонил)-1-азониабицикло[2.2.2]октанбромида,
3-((1,2-дифенилэтокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан-2,2,2-трифторацетата,
3-((бис(4-хлорфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
бис(4-фторфенил)метилхинуклидин-3-карбоксилата,
3-((бис(4-фторфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан-2,2,2-трифторацетата,
бензгидрилхинуклидин-3-карбоксилата,
3-(бензгидрилоксикарбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан-2,2,2-трифторацетата,
(4-метоксифенил)(фенил)метилхинуклидин-3-карбоксилата,
3-(((4-метоксифенил)(фенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
(2-хлорфенил)(4-хлорфенил)метилхинуклидин-3-карбоксилата,
3-(((2-хлорфенил)(4-хлорфенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азониабицикло[2.2.2]октан-2,2,2-трифторацетата,
(3,4-дифторфенил)(фенил)метилхинуклидин-3-карбоксилата,
3-(((3,4-дифторфенил)(фенил)метокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
3-фторбензилхинуклидин-3-карбоксилата,
3-((3-фторбензилокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан-2,2,2-трифторацетата,
9Н-флуорен-9-илхинуклидин-3-карбоксилата,
3-((9Н-флуорен-9-илокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октан-2,2,2-трифторацетата,
бис(3-фторфенил)метил 1-метилпиперидин-4-карбоксилата,
4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиофен-2-ил)этил)пиперидинийхлорида,
4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиофен-3-ил)этил)пиперидинийбромида,
4-((бис(3-фторфенил)метокси)карбонил)-1-метил-1-(2-оксо-2-(тиазол-2-ил)этил)пиперидинийбромида,
1,2-дифенилэтил-1-метилпиперидин-4-карбоксилата,
4-((1,2-дифенилэтокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромида,
(Е)-1,2-дифенилвинил-1-метилпиперидин-4-карбоксилата,
(1R,4R)-4-(((Е)-1,2-дифенилвинилокси)карбонил)-1-метил-1-(2-оксо-2-фенилэтил)пиперидинийбромида,
3-фторбензилхинуклидин-4-карбоксилата,
4-((3-фторбензилокси)карбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида,
S-бензилхинуклидин-3-карботиоата,
3-(бензилтиокарбонил)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида.
4. Фармацевтическая композиция, обладающая действием антагониста М3 мускариновых рецепторов, содержащая соединение по любому из пп.1-3 в эффективном количестве с одним или более фармацевтически приемлемыми носителями и/или эксципиентами.
5. Фармацевтическая композиция по п.4, которую вводят ингаляцией, в форме, выбранной из порошков для ингаляции, содержащих пропеллент дозированных аэрозолей или не содержащих пропелленты составов для ингаляции.
6. Применение соединения по любому из пп.1-3 для получения лекарственного средства для предотвращения и/или лечения бронхообструктивных или воспалительных заболеваний, выбранных из астмы, хронического бронхита и хронического обструктивного заболевания легких (COPD), опосредованных действием М3 мускариновых рецепторов.
7. Устройство, содержащее фармацевтическую композицию по п.5, которое представляет собой одно- или многодозовый порошковый ингалятор, ингалятор отмеренных доз и ингалятор мягкого тумана.
| УЗЕЛ КРЕПЛЕНИЯ ЗАЩИТНОГО ФАРТУКА К КАРНИЗНЫМ ПЛИТАМ И ПЛИТАМ ПОКРЫТИЙ | 1998 |
|
RU2154136C2 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| CN 101955457 A, 26.01.2011 | |||
| K | |||
| CHINNI MAHESH et al., A mild and efficient chemoselective protection of amines as N-benzyloxycarbonyl derivatives in the presence of La(NO 3 ) 3 6H 2 O under solvent-free conditions, TETRAHEDRON LETTERS, 2007, Vol | |||
| Приспособление для автоматической односторонней разгрузки железнодорожных платформ | 1921 |
|
SU48A1 |
| Устройство двукратного усилителя с катодными лампами | 1920 |
|
SU55A1 |
| K | |||
| FLINOIS et al., Enantioselective protonation and alkylation of non-covalent mixed aggregates of chiral 3-aminopyrrolidine lithium amides, TETRAHEDRON, 2002, Vol | |||
| Способ окисления боковых цепей ароматических углеводородов и их производных в кислоты и альдегиды | 1921 |
|
SU58A1 |
| Телефон | 1926 |
|
SU4707A1 |
| Способ изоляции водогазонасыщенных интервалов нефтяного пласта | 1988 |
|
SU1640364A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| S | |||
| BINIECKI et al., Syntezy R,S-estrow 2-, 3- i 4-pirydylometyloywch kwasow chinuklidyno-2- i chinuklidyno-3-karboksylowych, ACTA POLON | |||
| PHARM., 1981, Vol | |||
| Способ сужения чугунных изделий | 1922 |
|
SU38A1 |
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Устройство для дробления стружки | 1981 |
|
SU1014668A1 |
| R | |||
| B | |||
| BARLOW et al., A further search for selective antagonists at M 2 -muscarinic receptors, BRITISH J | |||
| PHARMAC., 1986, Vol | |||
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| Устройство катодов катодных ламп и катодных выпрямителей | 1924 |
|
SU837A1 |
| G | |||
| A | |||
| ROGERS et al., Inhibition of Acetylcholine Storage by Acetylcholine Analogs In Vitro, MOLECULAR PHARMACOLOGY, 1989, Vol | |||
| Коридорная многокамерная вагонеточная углевыжигательная печь | 1921 |
|
SU36A1 |
| Телефонная трансляция с катодными лампами | 1922 |
|
SU333A1 |
| S | |||
| BINIECKI et al., Synteza 1-hydroksy-4-(chinuklidyno-2-karboksy)-2-metylonaftalenu, ACTA POLON | |||
| PHARM., 1977, Vol | |||
| Нивелир для отсчетов без перемещения наблюдателя при нивелировании из средины | 1921 |
|
SU34A1 |
| Ребристый каток | 1922 |
|
SU121A1 |
| СТЕРИЛИЗОВАННОЕ МОЛОКО, ОБОГАЩЕННОЕ АНГИОГЕНИНОМ | 2000 |
|
RU2182791C2 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| J | |||
| S | |||
| BAJWA, Chemoselective Deprotection of Benzyl Esters in the Presence of Benzyl Ethers, Benzyloxymethyl Ethers and N-Benzyl Groups by Catalytic Transfer Hydrogenation, TETRAHEDRON LETTERS, 1992, Vol | |||
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |
| Телефонная трансляция | 1922 |
|
SU2299A1 |
| J | |||
| BLANCHET et al., A rapid and convenient synthesis of β -proline, TETRAHEDRON LETTERS, 2007, Vol | |||
| Приспособление для автоматической односторонней разгрузки железнодорожных платформ | 1921 |
|
SU48A1 |
| Приспособление для подачи сверла в ручных сверлильных приборах | 1926 |
|
SU5727A1 |
| Автооператор | 1984 |
|
SU1217654A1 |
| Отсадочная машина | 1983 |
|
SU1217476A1 |
| RN 1027980-30-8, 13.06.2008 | |||
| Устройство для уравновешивания подвижного узла | 1981 |
|
SU948551A1 |
| RN 913519-92-3, 17.11.2006 | |||
| RN 913517-08-5, 17.11.2006 | |||
| RN 913509-35-0, 17.11.2006 | |||
| RN 913490-74-1, 17.11.2006 | |||
| RN 913487-27-1, 17.11.2006 | |||
| Устройство для определения динамической термостойкости тиристоров | 1980 |
|
SU883808A1 |
| КАРБАМАТЫ ХИНУКЛИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2296762C2 |

