Изобретение относится к некоторым новым хроменоновым соединениям или их фармацевтически приемлемым солям, которые обладают противоопухолевой активностью и соответственно полезны в способах лечения организма человека или животного. Изобретение также относится к способам получения указанных хроменоновых соединений, содержащим их фармацевтическим композициям и их применению в терапевтических способах, например в изготовлении лекарственных средств для использования в профилактике или лечении злокачественных новообразований у теплокровного животного, такого как человек, включая использование в профилактике или лечении рака.
Настоящее изобретение также относится к хроменоновым соединениям, которые являются избирательными ингибиторами фосфоинозитид-3-киназы (PI3-киназы) β и полезны, например, для противоопухолевой терапии. Кроме того, настоящее изобретение также относится к применению хроменоновых соединений по изобретению, которые являются избирательными ингибиторами фосфоинозитид-3-киназы (PI3-киназы) β, в противоопухолевой терапии. Ингибиторы PI3-киназы β могут быть эффективными в лечении опухолей, которые имеют дефект в гене PTEN (фосфатаза и гомолог тензина с делецией на 10-й хромосоме), что составляет дополнительный аспект изобретения.
Настоящее изобретение также относится к хроменоновым соединениям, которые являются избирательными ингибиторами фосфоинозитид-3-киназы (PI3-киназы) δ, и полезны, например, для противоопухолевой терапии; а также к хроменоновым соединениям, которые являются избирательными ингибиторами как в отношении PI3-киназы β, так и в отношении PI3-киназы δ. Такие двойные ингибиторы PI3-киназы β/δ также полезны в лечении опухолей.
В области рака в последние годы было обнаружено, что клетки могут становиться злокачественными в результате трансформации части ее ДНК в онкоген, то есть ген, приводящий при его активации к образованию злокачественных опухолевых клеток (Bradshaw, Mutagenesis, 1986, 1, 91). Некоторые такие онкогены вызывают продуцирование пептидов, которые являются рецепторами факторов роста. Активация комплекса рецепторов факторов роста впоследствии приводит к увеличению пролиферации клеток. Известно, например, что некоторые онкогены кодируют ферменты тирозинкиназы и что некоторые рецепторы факторов роста также являются ферментами тирозинкиназами (Yarden etal., Ann. Rev. Biochem., 1988, 57, 443; Larsen et al., Ann. Reports in Med. Chem., 1989, Chpt. 13). Первая группа тирозинкиназ, подлежащая идентификации, возникла от таких вирусных онкогенов, как, например, тирозинкиназа pp60v-Src (также известная как v-Src), и соответствующих тирозинкиназ в нормальных клетках, например, тирозинкиназа pp60c-Src (также известная как c-Src).
Рецепторные тирозинкиназы являются важными в передаче биохимических сигналов, которые инициируют клеточную репликацию. Они представляют собой крупные ферменты, которые охватывают клеточную мембрану и имеют домен внеклеточного связывания для факторов роста, таких как эпидермальный фактор роста (EGF), и внутриклеточную область, которая функционирует как киназа для фосфорилирования тирозиновых аминокислот в белках и поэтому влияет на клеточную пролиферацию. Известны различные классы рецепторных тирозинкиназ (Wilks, Advances in Cancer Research, 1993, 60, 43-73), основанные на семействах факторов роста, которые связываются с разными рецепторными тирозинкиназами. Классификация включает рецепторные тирозинкиназы класса I, содержащие EGF семейство рецепторных тирозинкиназ, таких как рецепторы EGF, TGFα (трансформирующий фактор роста альфа), Neu и erbB.
Известно также, что некоторые тирозинкиназы относятся к классу нерецепторных тирозинкиназ, которые локализованы внутри клетки и вовлечены в передачу биохимических сигналов, таких как сигналы, влияющие на подвижность опухолевой клетки, распространение и инвазивность и впоследствии метастатический рост опухоли. Известны разные классы нерецепторных тирозинкиназ, включая Src семейство, такие как тирозинкиназы Src, Lyn, Fyn и Yes.
Кроме того, известно также, что некоторые тирозинкиназы относятся к классу серин/треонинкиназ, которые локализованы внутри клетки и по ходу транскрипции активации тирозинкиназы и вовлечены в передачу биохимических сигналов, таких как сигналы, влияющие на рост опухолевой клетки. Такие сериновые/треониновые пути передачи сигнала включают каскад Raf-MEK-ERK и те, которые расположены по ходу транскрипции PI3-киназы, например PDK-1, АКТ и mTOR (Blume-Jensen и Hunter, Nature, 2001, 411, 355).
Также известно, что некоторые другие киназы относятся к классу липидкиназ, которые локализованы внутри клетки и также вовлечены в передачу биохимических сигналов, таких как сигналы, влияющие на рост и инвазивность опухолевой клетки. Известны разные классы липидкиназ, включая вышеупомянутое семейство PI3-киназ, которое альтернативно известно как семейство фосфатидилинозитол-3-киназ.
В настоящее время хорошо понятно, что нарушение регуляции онкогенов и генов-супрессоров опухолевого роста вносит вклад в образование злокачественных опухолей, например посредством увеличенной клеточной пролиферации или повышенного клеточного выживания. Также в настоящее время известно, что пути передачи сигналов, опосредованные семейством PI3-киназ, имеют центральную роль в ряде клеточных процессов, включая пролиферацию и выживание, и разрегулирование этих путей является причинным фактором широкого спектра злокачественных новообразований и других заболеваний человека (Katso ef al., Annual Rev. Cell Dev. Biol., 2001, 17: 615-617 и Foster et al., J. Cell Science. 2003, 116: 3037-3040).
PI3-киназное семейство липидкиназ представляет собой группу ферментов, которые фосфорилируют третье положение инозитолового кольца фосфатидилинозитола (PI). Известны три основных группы ферментов PI3-киназ, которые классифицируют согласно их физиологической субстратной специфичности (Vanhaesebroeck et al., Trends in Biol. Sci., 1997, 22, 267). Ферменты PI3-киназы класса III фосфорилируют только PI. В противоположность этому, ферменты PI3-киназы класса II фосфорилируют и PI, и PI-4-фосфат [сокращенный ниже как PI(4)P]. Ферменты PI3-киназы класса I фосфорилируют PI, PI(4)P и PI-4,5-бисфосфат [сокращенный ниже как PI(4,5)P2], хотя, как полагают, только PI(4,5)P2 является физиологическим клеточным субстратом. Фосфорилирование PI(4,5)P2 продуцирует липидный вторичный мессенджер PI-3,4,5-трифосфат [сокращенный ниже как PI(3,4,5)P3]. Более отдаленные родственные члены этого суперсемейства представляют собой киназы класса IV, такие как mTOR и DNA-зависимая киназа, которые фосфорилируют остатки серина/треонина в белковых субстратах. Наиболее изученные и понятные из этих липидкиназ представляют собой ферменты PI3-киназы класса I.
PI3-киназа класса I представляет собой гетеродимер, состоящий из каталитической субъединицы p110 и регуляторной субъединицы, и это семейство дополнительно разделено на ферменты класса Ia и класса Ib на основе регуляторных партнеров и механизма регуляции. Ферменты класса Ia включают PI3-киназу β и состоят из трех отдельных каталитических субъединиц (р110α, р110β и р110δ), которые димеризуются с пятью отдельными регуляторными субъединицами (р85α, р55α, р50α, р85β и р55γ), причем все каталитические субъединицы способны взаимодействовать со всеми регуляторными субъединицами с образованием широкого ряда гетеродимеров. Ферменты PI3-киназы класса Ia в общем активируются в ответ на стимуляцию рецепторных тирозинкиназ фактором роста посредством взаимодействия SH2 доменов регуляторной субъединицы со специфическими фосфо-тирозиновыми остатками активированного рецептора или адаптерных белков, таких как IRS-1. И р110α, и р110β конститутивно экспрессируются во всех типах клеток, в то время как экспрессия р110δ более ограничена популяциями лейкоцитов и, некоторыми эпителиальными клетками. В противоположность этому, единственный фермент класса Ib состоит из каталитической субъединицы р110γ, которая взаимодействует с регуляторной субъединицей р101. Кроме того, ферменты класса Ib активируются в ответ на системы рецептора, сопряженного с G-белком (GPCR), а также посредством описанных выше механизмов.
В настоящее время имеются существенные свидетельства в пользу того, что ферменты PI3-киназы класса Ia, которые включают PI3-киназу β, вносят вклад в онкогенез в широком разнообразии человеческих раковых опухолей, либо непосредственно, либо опосредованно (Vivanco и Sawyers, Nature Reviews Cancer, 2002, 2, 489-501), например, субъединица р110α распространена при некоторых опухолях, таких как опухоли яичника (Shayesteh et al., Nature Genetics. 1999, 21: 99-102) и шейки матки (Ma et al., Oncogene. 2000, 19: 2739-2744). Активирующие мутации в пределах каталитического сайта р110α ассоциированы с различными другими опухолями, такими как опухоли колоректальной области, а также груди и легкого (Samuels et al., Science, 2004, 304, 554). Связанные с опухолями мутации в р85α также были идентифицированы при злокачественных новообразованиях, таких как рак яичника и толстой кишки (Philp et al., Cancer Research. 2001, 61, 7426-7429). PI3-киназа 5 играет решающую роль в B-клеточной функции, и было показано, что она является медиатором передачи сигнала выживания при разнообразных B-клеточных злокачественных новообразованиях. Это включает в себя хронический лимфоцитарный лейкоз (CLL), острый лимфобластный лейкоз (ALL), фолликулярную лимфому, диффузную крупноклеточную B-клеточную лимфому (DLBCL) и лимфому из клеток мантийной зоны (Ikeda et al., Blood, 2010, 116, 1460-1468; Herman et al., Blood, 2010, 116, 2078-2088; Lannutti et al., Blood, 2011, 117. 591-594; Hoellenriegel et al., Blood, 2011, 118, 3603-3612), но не ограничено ими. В дополнение к прямым эффектам, полагают, что активация PI3-киназы класса Ia вносит вклад в канцерогенные события, которые возникают в путях передачи сигнала против хода транскрипции, например, посредством лиганд-зависимой или лиганд-независимой активации рецепторных тирозинкиназ, GPCR-систем или интегринов (Vara et al., Cancer Treatment Reviews, 2004, 30, 193-204). Примеры таких путей передачи сигнала против хода транскрипции включают сверхэкспрессию рецепторной тирозинкиназы Erb2 при разных видах опухолей, приводящую к активации путей, опосредованных PI3-киназой (Harari et al., Oncogene, 2000, 19, 6102-6114), и сверхэкспрессию онкогена Ras (Kauffmann-Zeh et al., Nature, 1997, 385, 544-548). Кроме того, PI3-киназы класса Ia могут вносить вклад непосредственно в онкогенез, вызванный различными событиями в передаче сигнала по ходу транскрипции. Например, потеря эффекта подавляющей опухоль фосфатазы PTEN, которая катализирует превращение PI(3,4,5)P3 обратно в PI(4,5)P2, ассоциирована с чрезвычайно широким рядом опухолей посредством разрегулирования продуцирования PI(3,4,5)P3, опосредованного PI3-киназой (Simpson и Parsons, Exp. Cell Res., 2001, 264, 29-41). Более того, добавление эффектов от других событий в путях передачи сигнала, опосредованных PI3-киназой, как полагают, вносит вклад в разнообразие злокачественных новообразований, например посредством активации Akt (Nicholson и Anderson, Cellular Signalling, 2002, 14, 381-395).
Помимо роли в опосредовании передачи сигнала пролиферации и выживания в опухолевых клетках, также имеются свидетельства в пользу того, что ферменты PI3-киназы класса Ia вносят вклад в онкогенез посредством ее функции в ассоциированных с опухолями стромальных клетках. Например, известно, что передача сигнала PI3-киназы играет важную роль в опосредовании ангиогенных событий в эндотелиальных клетках в ответ на проангиогенные факторы, такие как VEGF (Abid et al., Arterioscler. Thromb. Vase. Biol., 2004, 24, 294-300). Поскольку ферменты PI3-киназы класса I также вовлечены в подвижность и миграцию (Sawyer, Expert Opinion Investig. Drugs, 2004, 13, 1-19), ингибиторы PI3-киназы обеспечат терапевтический благоприятный эффект посредством ингибирования инвазии и метастазов опухолевой клетки.
Кроме того, ферменты PI3-киназы класса I играют важную роль в регуляции иммунных клеток, причем PI3-киназная активность вносит вклад в проонкогенные эффекты воспалительных клеток (Coussens и Werb, Nature, 2002, 420, 860-867).
Эти обнаружения подтверждают, что фармакологические ингибиторы ферментов PI3-киназ класса I будут терапевтически ценными для лечения разных форм раковых заболеваний, включая солидные опухоли, такие как карциномы и саркомы, а также лейкозы и лимфоидные злокачественные новообразования. В частности, ингибиторы ферментов PI3-киназ класса I будут терапевтически ценными для лечения, например, рака груди, толстой и прямой кишки, легкого (включая мелкоклеточный рак легкого, немелкоклеточный рак легкого и бронхоальвеолярный рак) и предстательной железы, а также рака желчных протоков, кости, мочевого пузыря, головного мозга, головы и шеи, почки, печени, желудочно-кишечной ткани, пищевода, яичника, поджелудочной железы, кожи, семенников, щитовидной железы, матки, шейки матки и вульвы, а также лейкозов (включая ALL, CLL и CML [хронический миелоидный лейкоз]), множественной миеломы и лимфом (включая неходжкинские лимфомы, такие как диффузная крупноклеточная B-клеточная лимфома [DLBCL], фолликулярная лимфома и лимфома из клеток мантийной зоны).
В общем, исследователи изучали физиологические и патологические роли семейства ферментов PI3-киназ, используя вышеупомянутые ингибиторы PI3-киназ LY294002 и вортманнин. Хотя применение этих соединений может подтвердить роль PI3-киназы в клеточном событии, они являются недостаточно избирательными в пределах семейства PI3-киназ для того, чтобы обеспечить разделение на индивидуальные роли членов семейства. По этой причине более эффективные и избирательные фармацевтические ингибиторы PI3-киназ будут полезны для обеспечения более полного понимания функции PI3-киназы и для предложения полезных терапевтических агентов.
В дополнение к онкогенезу, имеются свидетельства в пользу того, что ферменты PI3-киназы класса I играют роль в других заболеваниях (Wymann et al, Trends in Pharmacological Science, 2003, 24, 366-376). Как ферменты PI3-киназы класса Ia, так и индивидуальный фермент класса Ib играют важные роли в клетках иммунной системы (Koyasu, Nature Immunology, 2003, 4, 313-319), и поэтому они являются терапевтическими мишенями для воспалительных и аллергических показаний. Ингибирование PI3-киназы также полезно, как описано ранее, для лечения сердечно-сосудистого заболевания посредством противовоспалительных воздействий или непосредственно путем воздействия на сердечные миоциты (Prasad et al., Trends in Cardiovascular Medicine, 2003, 13, 206-212). Игибирование PI3-киназы также полезно для лечения тромбоза. В WO 2004016607 предложен способ нарушения агрегации тромбоцитов и адгезии, возникающей в условиях высокого сдвига, и способ подавления активации тромбоцитов, вызванной сдвигом, где оба способа включают введение избирательного ингибитора PI3-киназы β. В WO 2004016607 также предложен антитромботический способ, включающий введение эффективного количества избирательного ингибитора PI3-киназы β. Согласно этому способу, специфическое ингибирование тромбоза может быть достигнуто без влияния на нормальный гемостаз благодаря направленному воздействию на PI3-киназу β, которая является важной для активации тромбоцитов, индуцируемой сдвигом. Указанный антитромботический способ, таким образом, не включает побочные эффекты, вызванные нарушением нормального гемостаза, например увеличение времени кровотечения.
Таким образом, ингибиторы ферментов PI3-киназ класса I, включая ингибиторы PI3-киназы β, как ожидают, будут ценными в предупреждении и лечении широкого ряда заболеваний помимо рака.
В настоящее время неожиданно обнаружено, что соединения, а именно хроменоновые соединения, по изобретению обладают сильной противоопухолевой активностью, являясь полезными в ингибировании неконтролируемой клеточной пролиферации, которая обусловлена злокачественным заболеванием. Не подразумевая, что соединения, раскрытые в настоящем изобретении, обладают фармакологической активностью только благодаря влиянию на единый биологический процесс, полагают, что соединения обеспечивают противоопухолевый эффект путем ингибирования ферментов PI3-киназ класса I, в частности путем ингибирования ферментов PI3-киназ класса Ia и/или фермента PI3-киназы класса Ib, более конкретно путем ингибирования ферментов PI3-киназ класса Ia, которые включают ингибирование PI3-киназы β.
Соединения по настоящему изобретению также полезны в ингибировании неконтролируемой клеточной пролиферации, обусловленной незлокачественными заболеваниями, такими как воспалительные заболевания (например ревматоидный артрит и воспалительное заболевание кишечника), фиброзные заболевания (например цирроз печени и фиброз легких), гломерулонефрит, рассеянный склероз, псориаз, доброкачественная гипертрофия простаты (ВРН), реакции гиперчувствительности кожи, заболевания кровяных сосудов (например атеросклероз и рестеноз), аллергическая астма, инсулинзависимый диабет, диабетическая ретинопатия и диабетическая нейропатия.
В общем, соединения по настоящему изобретению обладают сильной ингибирующей активностью в отношении ферментов PI3-киназ класса I, в частности в отношении ферментов PI3-киназ класса Ia, в том числе в отношении PI3-киназы β, в то же время обладая менее сильной ингибирующей активностью в отношении ферментов тирозинкиназ, таких как рецепторные тирозинкиназы, например тирозинкиназа EGF рецептора и/или тирозинкиназа VEGF рецептора, или в отношении нерецепторных тирозинкиназ, таких как Src. Более того, некоторые соединения по настоящему изобретению обладают существенно лучшей эффективностью в отношении ферментов PI3-киназ класса I, в частности в отношении ферментов PI3-киназ класса Ia, в том числе в отношении PI3-киназы β, чем в отношении тирозинкиназы EGF рецептора или тирозинкиназы VEGF рецептора или нерецепторной тирозинкиназы Src. Такие соединения обладают достаточной эффективностью в отношении ферментов PI3-киназ класса I, которые могут быть использованы в количестве, достаточном для ингибирования ферментов PI3-киназ класса I, в частности для ингибирования ферментов PI3-киназ класса Ia, включая PI3-киназу β, в то же время демонстрируя малую активность в отношении тирозинкиназы EGF рецептора или тирозинкиназы VEGF рецептора, или нерецепторной тирозинкиназы Src.
В дополнение, конкретные соединения по изобретению демонстрируют сильную ингибирующую активность в отношении как PI3-киназы β, так и PI3-киназы δ, в то же время обладая менее сильной ингибирующей активностью в отношении ферментов тирозинкиназ, таких как рецепторные тирозинкиназы, например тирозинкиназа EGF рецептора и/или тирозинкиназа VEGF рецептора, или в отношении нерецепторных тирозинкиназ, таких как Src. Более того, некоторые соединения по настоящему изобретению обладают существенно лучшей эффективностью в отношении как PI3-киназы β, так и PI3-киназы δ, чем в отношении тирозинкиназы EGF рецептора или тирозинкиназы VEGF рецептора, или нерецепторной тирозинкиназы Src. Такие соединения обладают достаточной эффективностью в отношении и PI3-киназы β, и PI3-киназы δ, которые могут быть использованы в количестве, достаточном для ингибирования PI3-киназы β и PI3-киназы δ, в то же время демонстрируя незначительную активность в отношении тирозинкиназы EGF рецептора или тирозинкиназы VEGF рецептора, или нерецепторной тирозинкиназы Src.
Согласно одному аспекту изобретения предложено производное хроменона формулы I:
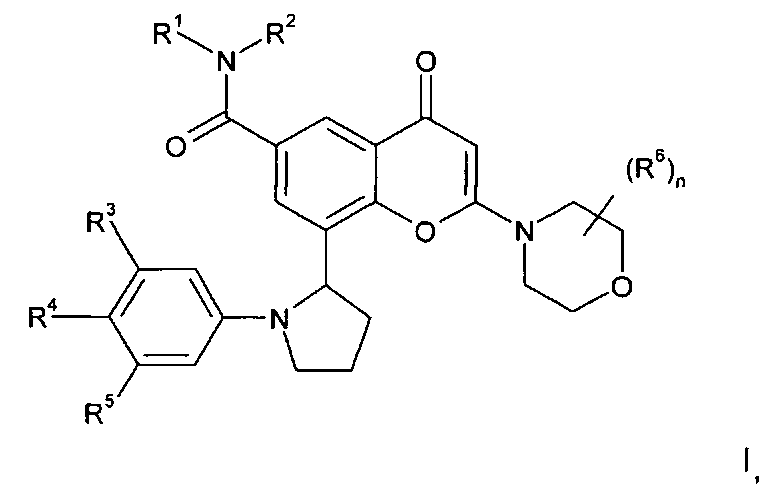
в которой:
R1 представляет собой С1-4алкил, возможно замещенный гидрокси;
R2 представляет собой Н или С1-4алкил; или
R1 и R2 вместе образуют 3-8-членную азотсодержащую гетероциклильную кольцевую систему, которая возможно содержит 1 или 2 дополнительных гетероатома, выбранных из кислорода, азота и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов), где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из H, галогена, C1-3алкокси и циано;
R4 представляет собой Н или фтор;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или его фармацевтически приемлемая соль.
В данном описании общий термин "C1-4алкил" включает как прямые, так и разветвленные алкильные группы, такие как пропил, изопропил и трет-бутил, а также C3-4циклоалкильные группы, такие как циклопропил и циклобутил, и также группы, такие как циклопропилметил. Однако, ссылки на индивидуальные алкильные группы, такие как "пропил", являются конкретным указанием только на вариант с прямой цепью, ссылки на индивидуальные разветвленные алкильные группы, такие как "изопропил", являются конкретным указанием только на вариант с разветвленной цепью, и ссылки на индивидуальные циклоалкильные группы, такие как "циклопропил", являются конкретным указанием только на 3-членное кольцо.
Специалисту в данной области техники понятно, что термины "C1-4алкил" и "C1-3алкил", как они использованы здесь, относятся к любой из алкильных групп, определенных выше, которые имеют от 1 до 4 и от 1 до 3 атомов углерода, соответственно. То же самое правило применимо в отношении других терминов, использованных здесь, таких как, например, "C1-3алкокси", "C1-10алкоксикарбонил" и "C1-10алканоил".
Следует понимать, что поскольку некоторые из соединений формулы I, определенной выше, могут существовать в оптически активных или рацемических формах благодаря одному или более асимметрическим атомам углерода, изобретение включает в его определении любую такую оптически активную или рацемическую форму, которая обладает ингибирующей активностью в отношении фосфоинозитид-3-киназы (PI3-киназы). Синтез оптически активных форм может быть осуществлен стандартными методиками органической химии, хорошо известными в данной области техники, например, синтезом из оптически активных исходных материалов или разделением рацемической формы. Подобным образом, вышеупомянутая активность может быть оценена с использованием стандартных лабораторных процедур.
Конкретный энантиомер описанных здесь соединений может быть более активным по сравнению с другими энантиомерами соединения. Например, (+)-энантиомер указанного в заголовке соединения Примера 1.03 (то есть, соединения Примера 1.03а, где (+) означает оптическое вращение, измеренное с использованием условий, описанных в Примере 1.03а) является энантиомером, активность которого слабее. Во избежание сомнений, рассматриваемый хиральный центр представляет собой атом углерода по положению 2 центрального пирролидинового кольца, которое присоединено к хроменоновому бициклическому кольцу.
Соответственно, в дополнительном аспекте изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, где хиральный центр по положению 2 центрального пирролидинового кольца, которое присоединено к хроменоновому бициклическому кольцу, находится в (R)-стереохимической конфигурации. В дополнительном аспекте изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, где хиральный центр по положению 2 центрального пирролидинового кольца, которое присоединено к хроменоновому бициклическому кольцу, находится в (S)-стереохимической конфигурации.
Согласно еще одному аспекту изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, которое представляет собой индивидуальный энантиомер, находящийся в энантиомерном избытке (%ee) не менее 95%, не менее 98% или не менее 99%. В одном воплощении этого аспекта изобретения хиральный центр по положению 2 центрального пирролидинового кольца, которое присоединено к хроменоновому бициклическому кольцу, находится в (R)-стереохимической конфигурации. В еще одном воплощении этого аспекта изобретения хиральный центр по положению 2 центрального пирролидинового кольца, которое присоединено к хроменоновому бициклическому кольцу, находится в (S)-стереохимической конфигурации.
Согласно еще одному аспекту изобретения предложена фармацевтическая композиция, содержащая производное хроменона формулы I, которое представляет собой индивидуальный энантиомер, находящийся в энантиомерном избытке (%ее) не менее 95%, не менее 98% или не менее 99%, или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем. Предпочтительно индивидуальный энантиомер присутствует в энантиомерном избытке (%ее) не менее 99%. В одном воплощении этого аспекта изобретения хиральный центр по положению 2 центрального пирролидинового кольца, которое присоединено к хроменоновому бициклическому кольцу, находится в (R)-стереохимической конфигурации. В еще одном воплощении этого аспекта изобретения хиральный центр по положению 2 центрального пирролидинового кольца, которое присоединено к хроменоновому бициклическому кольцу, находится в (S)-стереохимической конфигурации.
Некоторые соединения формулы I могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любую полиморфную форму или их смеси, которые обладают свойствами, полезными в ингибировании активности фосфоинозитид(PI)-3-киназы, причем в данной области хорошо известно как определить эффективность полиморфной формы в отношении ингибирования активности фосфоинозитид(PI)-3-киназы стандартными тестами, описанными ниже в данном описании.
В целом известно, что кристаллические вещества могут быть проанализированы с использованием традиционных методик, таких как анализ дифракции рентгеновских лучей на порошке (ниже XRPD), дифференциальная сканирующая калориметрия (ниже ДСК), термогравиметрический анализ (ниже ТГА), инфракрасная спектроскопия диффузного отражения с Фурье-преобразованием (DRIFT), коротковолновая инфракрасная спектроскопия (NIR), ядерная магнитно-резонансная спектроскопия жидкого и/или твердого состояния. Содержание воды таких кристаллических веществ может быть определено анализом по Карлу Фишеру.
В качестве примера, соединение Примера 1.03b проявляет полиморфизм, и были идентифицированы три кристаллические формы A, B и C. Конкретные кристаллические формы могут проявлять благоприятные свойства, такие как улучшенная стабильность, которая делает их особенно-подходящими для фармацевтических разработок.
Соответственно, в дополнительном аспекте изобретения предложена Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета примерно равном 4,8°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета примерно равном 8,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета примерно равных 4,8° и 8,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета примерно равных 4,8, 6,4, 8,1, 9,6, 15,8, 19,5, 20,3, 22,7, 23,4, 25,9°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на Фиг.1.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета примерно равном 4,8° плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета примерно равном 8,1° плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета примерно равных 4,8° и 8,1°, где указанные значения могут отклоняться на плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета примерно равных 4,8, 6,4, 8,1, 9,6, 15,8, 19,5, 20,3, 22,7, 23,4, 25,9°, где указанные значения могут отклоняться на плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета равном 4,8°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета равном 8,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета равных 4,8° и 8,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета равных 4,8, 6,4, 8,1, 9,6, 15,8, 19,5, 20,3, 22,7, 23,4, 25,9°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма А 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке как показано на Фиг.1.
В дополнительном аспекте изобретения предложена Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма B 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета примерно равном 11,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета примерно равных 6,9° и 11,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета примерно равных 6,9, 9,4, 9,8, 11,1, 12,7, 13,1, 13,7, 17,8, 18,7, 19,7°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на Фиг.2.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета примерно равном 11,1° плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета примерно равных 6,9 и 11,1°, где указанные значения могут отклоняться на плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета примерно равных 6,9, 9,4, 9,8, 11,1, 12,7, 13,1, 13,7, 17,8, 18,7, 19,7°, где указанные значения могут отклоняться на плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере одним характеристическим пиком при 2-тета равном 11,1°.
Согласно еще одному аспекту настоящего изобретения, предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета равных 6,9° и 11,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета равных 6,9, 9,4, 9,8, 11,1, 12,7, 13,1, 13,7, 17,8, 18,7, 19,7°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма В 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке как показано на Фиг.2.
В дополнительном аспекте изобретения предложена Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета примерно равных 5,9 и 12,2°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета примерно равных 5,9, 12,2, 11,8, 13,5, 15,2, 15,4, 17,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке по существу такую же, как картина дифракции рентгеновских лучей на порошке, показанная на Фиг.4.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета примерно равных 5,9 и 12,2°, где указанные значения могут отклоняться на плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета примерно равных 5,9, 12,2, 11,8, 13,5, 15,2, 15,4, 17,1°, где указанные значения могут отклоняться на плюс или минус 0,5° 2-тета.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с по меньшей мере двумя характеристическими пиками при 2-тета равных 5,9° и 12,2°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке с характеристическими пиками при 2-тета равных 5,9, 12,2, 11,8, 13,5, 15,2, 15,4, 17,1°.
Согласно еще одному аспекту настоящего изобретения предложена кристаллическая форма Форма С 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она, которая имеет картину дифракции рентгеновских лучей на порошке как показано на Фиг.4.
Следует понимать, что значения 2-тета на картинах дифракции рентгеновских лучей могут слегка отличаться в зависимости от оборудования или от образца, и поэтому зарегистрированные значения не следует воспринимать как абсолютные.
Известно, что может быть получена картина дифракции рентгеновских лучей на порошке, которая имеет одну или более ошибок измерений в зависимости от условий измерения (таких как используемое оборудование или аппаратура). В частности, известно в общем, что интенсивности на картине дифракции рентгеновских лучей на порошке могут колебаться в зависимости от условий измерения. Таким образом, следует понимать, что кристаллические формы по настоящему изобретению, описанные выше, если не указано иное, не ограничены кристаллами, которые дают картины дифракции рентгеновских лучей на порошке, идентичные картинам дифракции рентгеновских лучей на порошке, показанным на Фиг.1, 2 и 4, и любые кристаллы, дающие картины дифракции рентгеновских лучей на порошке по существу такие же, как показаны на Фиг.1, 2 и 4, входят в объем настоящего изобретения. Специалист в области рентгеновской порошковой дифракции способен оценить сходство по существу картин дифракции рентгеновских лучей на порошке.
Специалисты в области рентгеновской порошковой дифракции также понимают, что на относительную интенсивность пиков может влиять, например, размер кристаллических зерен выше 30 микрон и неунитарные соотношения размеров, которые могут влиять на анализ образцов. Специалисты также понимают, что на позицию отражений может влиять точная высота, на которую располагают образец в дифрактометре и калибровка нуля дифрактометра. Плоскостность поверхности образца также может оказывать небольшое влияние. Поэтому представленные данные картин дифракции не следует принимать за абсолютные значения (см. Jenkins, R & Snyder, R.L. ′Introduction to X-Ray Powder Diffractometry′ John Wiley & Sons 1996; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; Klug, H.P. & Alexander, L.E. (1974), X-Ray Diffraction Procedures).
В общем, ошибка измерения угла дифракции на рентгеновской порошковой дифрактограмме составляет приблизительно плюс или минус 0,5° 2-тета, и такую степень ошибки измерений следует принимать во внимание при рассмотрении данных рентгеновской порошковой дифракции. Кроме того, следует понимать, что интенсивности могут слегка колебаться в зависимости от экспериментальных условий и приготовления образца (предпочтительная ориентация).
Конкретные соединения по изобретению представляют собой каждое из соединений Примеров и их фармацевтически приемлемых солей, каждое(ая) из которых образуют дополнительный независимый аспект изобретения.
Согласно еще одному аспекту изобретения предложено производное хроменона формулы I, которое может быть получено согласно следующему любому из Примеров, как описано ниже.
Дополнительный аспект по изобретению представляет любой из определенных здесь объемов защиты, с оговоркой что конкретные соединения Примеров, таких как Пример 1.00, 1.01, 1.03, 1.03b, 1.05, 1.06, 1.07, 1.08, 2.00, 3.00, 3.02, 3.03 и так далее, индивидуально не заявляются.
Еще один аспект по изобретению представляет любой из определенных здесь объемов защиты, с оговоркой что одно конкретное соединение Примера, такого как Пример 1.00, 1.01, 1.03, 1.03b, 1.05, 1.06, 1.07, 1.08, 2.00, 3.00, 3.02, 3.03 и так далее, индивидуально не заявляется.
Соответственно, в одном из дополнительных аспектов изобретения предложено производное хроменона формулы I:
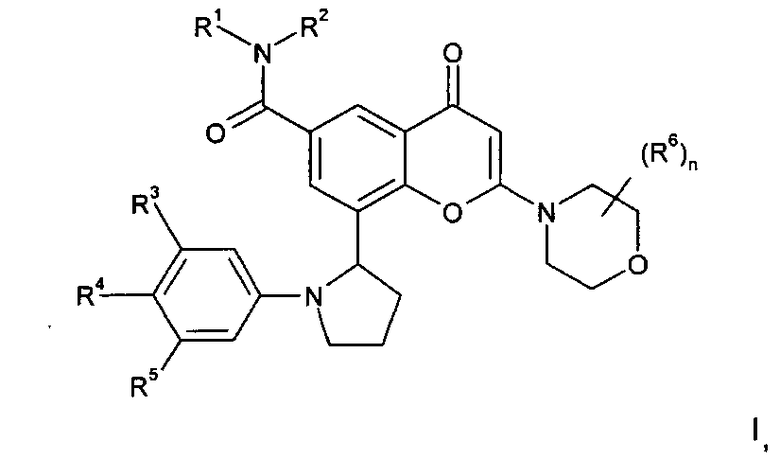
в которой:
R1 представляет собой C1-4алкил, возможно замещенный гидрокси;
R2 представляет собой Н или C1-4алкил; или
R1 и R2 вместе образуют 3-8-членную азотсодержащую гетероциклильную кольцевую систему, которая возможно содержит 1 или 2 дополнительных гетероатома, выбранных из кислорода, азота и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов), где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из Н, галогена, C1-3алкокси и циано;
R4 представляет собой H или фтор;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или его фармацевтически приемлемая соль; при условии, что соединение формулы I не представляет собой 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он.
Следует понимать, что некоторые соединения формулы I, определенной выше, могут проявлять явление таутомерии. Следует понимать, что настоящее изобретение включает в своем определении любую такую таутомерную форму или их смесь, обладающую ингибирующей активностью в отношении фосфоинозитид(PI)-3-киназы, и не должно быть ограничено лишь какой-либо одной таутомерной формой, использованной в изображениях структурных формул или названной в Примерах. В общем, лишь одна из любых таких таутомерных форм названа в Примерах, описанных ниже, или представлена в любом релевантном изображении формул, описанных ниже.
Подходящие значения для общих радикалов, на которые ссылаются выше, включают те, которые изложены ниже.
Подходящее значение для 3-8-членной азотсодержащей гетероциклильной системы, образованной R1 и R2 группами формулы I, представляет собой, например, азотсодержащее неароматическое насыщенное или частично насыщенное 3-8-членное кольцо, которое возможно, содержит 1 или 2 дополнительных гетероатома, выбранных из кислорода, азота и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов). Подходящие примеры включают азепанил, оксазепанил, азиридинил, азетидинил, пирролинил, пирролидинил, имидазолинил, имидазолидинил, пиразолинил, пиразолидинил, морфолинил, тиоморфолинил, тетрагидро-1,4-тиазинил, 1,1-диоксотетрагидро-1,4-тиазинил, пиперидинил, гомопиперидинил, пиперазинил, гомопиперазинил, дигидропиридинил, тетрагидропиридинил, дигидропиримидинил или тетрагидропиримидинил. В конкретной группе соединений конкретные примеры гетероциклильного кольца включают азетидинил, морфолинил, 1-оксотетрагидро-1,4-тиазинил и пиперидинил, и в частности, азетидин-1-ил, морфолин-4-ил, 1-оксотетрагидро-1,4-тиазин-4-ил и пиперидин-1-ил.
Подходящие значения для любой из ′R′ групп (от R1 до R6) включают, например:
для галогена: фтор, хлор, бром и йод;
для C1-4алкила: метил, этил, пропил, изопропил, трет-бутил, циклопропил и циклобутил;
для C1-3залкокси: метокси, этокси, пропокси и изопропокси.
Подходящая фармацевтически приемлемая соль соединения формулы I представляет собой, например, соль присоединения кислоты соединения формулы I, например, соль присоединения кислоты с неорганической или органической кислотой, такой как соляная, бромистоводородная, серная, трифторуксусная или лимонная кислота; или, например, соль соединения формулы I, которая является достаточно кислой, например, соль щелочного или щелочноземельного металла, такая как соль кальция или магния, или соль аммония, или соль с органическим основанием, таким как метиламин, диметиламин, триметиламин, пиперидин, морфолин или трис-(2-гидроксиэтил)амин. Дополнительная подходящая фармацевтически приемлемая соль соединения формулы I представляет собой, например, соль, образуемую в организме человека или животного после введения соединения формулы I.
Также следует понимать, что подходящий фармацевтически приемлемый сольват соединения формулы I также образует аспект настоящего изобретения. Подходящий фармацевтически приемлемый сольват представляет собой, например, гидрат, такой как полугидрат, моногидрат, дигидрат или тригидрат или его альтернативный эквивалент.
Также следует понимать, что подходящее фармацевтически приемлемое пролекарство соединения формулы I также образует аспект настоящего изобретения. Соответственно, соединения по изобретению могут быть введены в форме пролекарства, которое представляет собой соединение, распадающееся в организме человека или животного с высвобождением соединения по изобретению. Пролекарство может быть использовано для изменения физических свойств и/или фармакокинетических свойств соединения по изобретению. Пролекарство может быть образовано, когда соединение по изобретению содержит подходящие группу или заместитель, к которым может быть присоединена группа, модифицирующая свойства. Примеры пролекарств включают in vivo расщепляемые сложноэфирные производные, которые могут быть образованы по гидроксигруппе в соединении формулы I, и in vivo расщепляемые амидные производные, которые могут быть образованы по аминогруппе в соединении формулы I.
Соответственно, настоящее изобретение включает те соединения формулы I, как определено выше, которые могут быть доступны путем органического синтеза и могут быть доступны в организме человека или животного посредством расщепления его пролекарства. Соответственно, настоящее изобретение включает те соединения формулы I, которые получают средствами органического синтеза, а также такие соединения, которые образуются в организме человека или животного посредством метаболизма соединения-предшественника, то есть соединение формулы I может быть синтетически получаемым соединением или метаболически получаемым соединением.
Подходящее фармацевтически приемлемое пролекарство соединения формулы I представляет собой такое, которое основано на надлежащем медицинском заключении как подходящем для введения в организм человека или животного без проявления нежелательных фармакологических активностей и без чрезмерной токсичности.
Различные формы пролекарств описаны, например, в следующих документах:
a) Methods in Enzvmoloqy. Vol.42, p.309-396, edited by K. Widder, et al. (Academic Press, 1985);
b) Design of Pro-drugs, edited by H. Bundgaard, (Elsevier, 1985);
c) A Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H. Bundgaard, Chapter 5 "Design and Application of Pro-drugs", by H. Bundgaard p.113-191 (1991);
d) H. Bundgaard, Advanced Drug Delivery Reviews. 8, 1-38 (1992);
e) H. Bundgaard, et al., Journal of Pharmaceutical Sciences. 77, 285 (1988); t) N. Kakeya, et al., Chem. Pharm. Bull., 32, 692 (1984);
g) T. Higuchi and V. Stella, "Pro-Drugs as Novel Delivery Systems", A.C.S. Symposium Series, Volume 14; и
h) E. Roche (editor), "Bioreversible Carriers in Drug Design", Pergamon Press, 1987.
Подходящее фармацевтически приемлемое пролекарство соединения формулы I, которое имеет гидроксигруппу, представляет собой, например, его in vivo расщепляемый сложный или простой эфир. In vivo расщепляемый сложный или простой эфир соединения формулы I, содержащего гидроксигруппу, представляет собой, например, фармацевтически приемлемый сложный или простой эфир, который расщепляется в организме человека или животного с образованием исходного гидроксисоединения. Подходящие группы, образующие фармацевтически приемлемые сложные эфиры, для гидроксигруппы включают неорганические сложные эфиры, такие как фосфатные сложные эфиры (включая фосфорамидиновые циклические сложные эфиры). Дополнительные подходящие группы, образующие фармацевтически приемлемые сложные эфиры, для гидроксигруппы включают Смоалканоильные группы, такие как ацетил, бензоил, фенилацетил и замещенные бензоильные и фенилацетильные группы, C1-10алкоксикарбонильные группы, такие как группы этоксикарбонил, N,N-[ди-C1-4алкил]карбамоил, 2-диалкиламиноацетил и 2-карбоксиацетил. Примеры заместителей на кольце по фенилацетильным и бензоильным группам включают аминометил, N-алкиламинометил, N,N-диалкиламинометил, морфолинометил, пиперазин-1-ил метил и 4-С1-4алкилпиперазин-1-илметил. Подходящие группы, образующие фармацевтически приемлемые простые эфиры, для гидроксигруппы включают α-ацилоксиалкильные группы, такие как ацетоксиметильные и пивалоилоксиметильные группы.
Подходящее фармацевтически приемлемое пролекарство соединения формулы I, которое имеет аминогруппу, представляет собой, например, его in vivo расщепляемое амидное производное. Подходящие фармацевтически приемлемые амиды из аминогруппы включают, например, амид, образованный с C1-10алканоильными группами, такими как ацетильные, бензоильные, фенилацетильные и замещенные бензоильные и фенилацетильные группы. Примеры заместителей на кольце по фенилацетильным и бензоильным группам включают аминометил, N-алкиламинометил, N,N-диалкиламинометил, морфолинометил, пиперазин-1-илметил и 4-C1-4алкилпиперазин-1-илметил.
In vivo эффекты соединения формулы I могут проявляться отчасти благодаря одному или более метаболитам, которые образуются в организме человека или животного после введения соединения формулы I. Как изложено выше, in vivo эффекты соединения формулы I могут также проявляться посредством метаболизма соединения-предшественника (пролекарство).
Во избежание сомнений следует понимать, что когда в описании группа описана как "определенная здесь" или "определенная выше", указанная группа охватывает первое встречающееся и наиболее широкое определение, а также каждое и все конкретные определения для такой группы.
Конкретные новые соединения по изобретению включают, например, хроменоновые соединения формулы I или их фармацевтически приемлемые соли, где, если не указано иное, каждый из R1, R2, R3, R4, R5, n и R6 имеет любое из значений, определенных выше или в абзацах (a)-(u) ниже:
(a) R1 представляет собой C1-4алкил;
(b) R1 представляет собой метил, этил или 2-гидроксиэтил;
(c) R1 представляет собой метил или этил;
(d) R1 представляет собой метил;
(e) R2 представляет собой C1-4алкил;
(f) R2 представляет собой метил или этил;
(g) R2 представляет собой метил;
(h) R1 и R2 вместе образуют 4-6-членную азотсодержащую гетероциклильную кольцевую систему, которая возможно содержит 1 дополнительный гетероатом, выбранный из кислорода, азота и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов), где указанное кольцо возможно замещено гидрокси;
(i) R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидинила, морфолинила, 1-оксотетрагидро-1,4-тиазинила и пиперидинила, где указанное кольцо возможно замещено гидрокси;
(j) R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидин-1-ила, морфолин-4-ила, 1-оксотетрагидро-1,4-тиазин-4-ила и пиперидин-1-ила, где указанное кольцо возможно замещено гидрокси;
(k) R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидин-1-ила или морфолин-4-ила;
(I) R3 и R5 независимо выбраны из Н, фтора, метокси и циано;
(m) R3 и R5 независимо выбраны из Н и фтора;
(n) R3 и R5 представляют собой Н;
(o) R3 и R5 представляют собой фтор;
(р) R4 представляет собой Н;
(q) R3 и R4 представляют собой Н, и R5 представляет собой фтор;
(r) R3 и R5 представляют собой фтор, и R4 представляет собой H;
(s) n равен 0;
(t) n равен 1; и
(u) n равен 1; и R6 представляет собой метильную группу, находящуюся в положении 2 морфолинового кольца;
(v) R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, где указанное кольцо представляет собой морфолин-4-ил.
Конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I, определенной выше, где:
R1 представляет собой C1-4алкил, возможно замещенный гидрокси;
R2 представляет собой C1-4алкил; или
R1 и R2 вместе образуют 4-6-членную азотсодержащую гетероциклильную кольцевую систему, которая возможно содержит 1 дополнительный гетероатом, выбранный из кислорода, азота и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов), где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из Н или галогена, и R4 представляет собой Н;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 представляет собой C1-4алкил, возможно замещенный гидрокси;
R2 представляет собой C1-4алкил; или
R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидинила, морфолинила, 1-оксотетрагидро-1,4-тиазинила и пиперидинила, где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из H или галогена, и R4 представляет собой Н;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 представляет собой метил, этил или 2-гидроксиэтил;
R2 представляет собой метил или этил; или
R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидинила, морфолинила, 1-оксотетрагидро-1,4-тиазинила и пиперидинила, где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из Н или галогена, и R4 представляет собой Н;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 представляет собой метил или этил;
R2 представляет собой метил или этил; или
R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидин-1-ила, морфолин-4-ила, 1-оксотетрагидро-1,4-тиазин-4-ила и пиперидин-1-ила, где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из Н или фтора, и R4 представляет собой Н;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I, определенной выше, или их фармацевтически приемлемые соли, где: R1 и R2 являются предпочтительно такими, как определено в любом из подпунктов от (a) до (k) выше, в частности как определено в подпункте (d), (g) или от (i) до (k) выше; R3, R4 и R5 являются предпочтительно такими, как определено в любом из подпунктов от (l) до (r) выше, и в частности такими, как определено в подпункте (r) или (s) выше; и n и R6 являются предпочтительно такими, как определено в любом из подпунктов от (s) до (u) выше.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I, определенной выше, или их фармацевтически приемлемые соли, где: R1 и R2 являются предпочтительно такими, как определено в любом из подпунктов от (a) до (k) выше или (v) выше, в частности как определено в подпункте (d), (g), (v) или от (i) до (k) выше, и более конкретно, в подпункте (v) выше; R3, R4 и R5 являются предпочтительно такими, как определено в любом из подпунктов от (l) до (r) выше, и в частности такими, как определено в подпункте (r) или (s) выше; и n и R6 являются предпочтительно такими, как определено в любом из подпунктов от (s) до (u) выше.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 представляет собой метил или 2-гидроксиэтил;
R2 представляет собой метил; или
R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидин-1-ила, морфолин-4-ила, 1-оксотетрагидро-1,4-тиазин-4-ила, пиперидин-1-ила и 4-гидроксипиперидин-1-ила;
R3 и R5 независимо выбраны из Н, фтора, метокси и циано;
R4 представляет собой Н или фтор;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 представляет собой метил или 2-гидроксиэтил;
R2 представляет собой метил; или
R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидин-1-ила, морфолин-4-ила, 1-оксотетрагидро-1,4-тиазин-4-ила, пиперидин-1-ила и 4-гидроксипиперидин-1-ила;
R3 и R5 независимо выбраны из Н, фтора, метокси и циано;
R4 представляет собой Н или фтор;
n равен 0; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидин-1-ила, морфолин-4-ила, 1-оксотетрагидро-1,4-тиазин-4-ила, пиперидин-1-ила и 4-гидроксипиперидин-1-ила;
R3 и R5 независимо выбраны из Н, фтора, метокси и циано;
R4 представляет собой Н или фтор;
n равен 0; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 представляет собой метил;
R2 представляет собой метил; или
R1 и R2 вместе образуют морфолин-4-ильное кольцо;
R3 и R5 независимо выбраны из Н и фтора;
R4 представляет собой Н;
n равен 1, и группа R6 представляет собой метил; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 и R2 вместе образуют морфолин-4-ильное кольцо;
R3 и R5 независимо выбраны из Н и фтора;
R4 представляет собой Н;
n равен 1, и группа R6 представляет собой метил; или их фармацевтически приемлемые соли.
Дополнительная конкретная группа соединений по изобретению представляет собой хроменоновые соединения формулы I выше, где:
R1 и R2 вместе образуют морфолин-4-ильное кольцо;
R3 и R5 представляют собой фтор;
R4 представляет собой Н;
n равен 0; или их фармацевтически приемлемые соли.
Конкретные соединения по изобретению представляют собой, например, хроменоновые соединения формулы I, которые раскрыты в Примерах, изложенных здесь ниже. Во избежание сомнений, хотя соединения названы согласно номенклатуре ИЮПАК, может существовать много других правильных названий для конкретных Примеров. Например, соединение Примера 1.03b может быть названо как 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он или как 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он.
Например, конкретное соединение по изобретению представляет собой производное хроменона формулы I, выбранное из любого из следующих:
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он;
8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4Н-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-хромен-6-карбоксамид;
8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-хромен-6-карбоксамид;
8-(1-(3-фторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он;
8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-(1-(3-фторфенил)пирролидин-2-ил)-2-морфолино-4Н-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он; и
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксамид; или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой производное хроменона формулы I, выбранное из любого из следующих:
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он;
8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4Н-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-хромен-6-карбоксамид;
8-(1-(3-фторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он;
8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-(1-(3-фторфенил)пирролидин-2-ил)-2-морфолино-4Н-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он; и
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксамид; или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой производное хроменона формулы I, выбранное из любого из следующих:
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он;
8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4Н-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-хромен-6-карбоксамид;
8-(1-(3-фторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он;
8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-(1-(3-фторфенил)пиррол идин-2-ил)-2-морфолино-4Н-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он; и
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксамид; или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой соединение Примера 1.03 или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой соединение Примера 1.05 или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой соединение Примера 1.06 или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой соединение Примера 1.07 или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой соединение Примера 1.03b или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой (-)-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он или его фармацевтически приемлемую соль, где (-)- в химическом названии означает оптическое вращение, измеренное с использованием условий, описанных в Примере 1.03b.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой (-)-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он; где (-)- в химическом названии означает оптическое вращение, измеренное с использованием условий, описанных в Примере 1.03b.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль (-)-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она; где (-)- в химическом названии означает оптическое вращение, измеренное с использованием условий, описанных в Примере 1.03b.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль 8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-(1-(3-фторфенил)пирролидин-2-ил)-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-он или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-(1-(3-фторфенил)пирролидин-2-ил)-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-он.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль 8-(1-(3-фторфенил)пирролидин-2-ил)-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-она.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2R)-1-(3-фторфенил)пирролидин-2-ил)-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-он или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-он.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль 8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-она.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2S)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-он или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2S)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-он.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль 8-[(2S)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-она.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль 8-[1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2R)-1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2R)-1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль 8-[(2R)-1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2S)-1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он или его фармацевтически приемлемую соль.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой 8-[(2S)-1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он.
Согласно еще одному аспекту изобретения конкретное соединение по изобретению представляет собой фармацевтически приемлемую соль 8-[(2S)-1-(3-метоксифенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она.
В другом аспекте настоящего изобретения предложен способ получения соединения формулы I или его фармацевтически приемлемой соли. Соответствующий способ иллюстрируется следующими репрезентативными вариантами способа, в которых, если не указано иное, R1, R2, R3, R4, R5, n и R6 имеют любое из значений, определенных в данном описании выше. Необходимые исходные материалы могут быть получены стандартными процедурами органической химии. Получение таких исходных материалов описано в сочетании со следующими репрезентативными вариантами способа и в рамках сопровождающих Примеров. Альтернативно необходимые исходные материалы могут быть получены процедурами, аналогичными проиллюстрированным, которые находятся в рамках компетенции специалиста в области органической химии.
Подходящие варианты способа включают, например, следующие.
(а) Реакция перекрестного сочетания соединения формулы II:
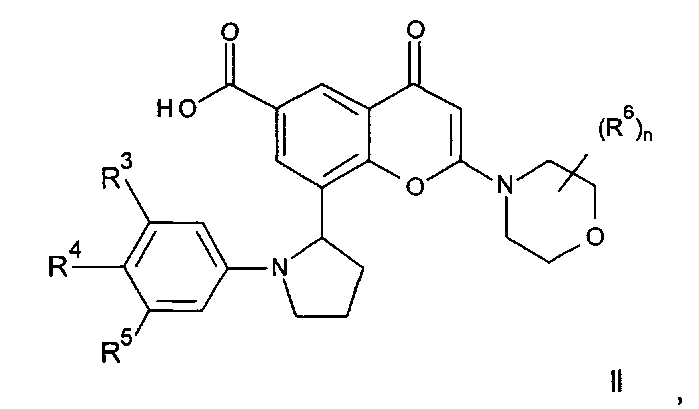
где R3, R4, R5, n и R6 имеют любое из значений, определенных в данном описании ранее, за исключением того, что любая присутствующая функциональная группа при необходимости защищена, с амином формулы III:
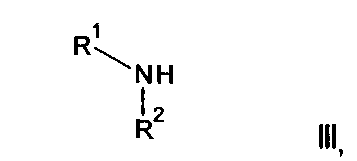
где R1 и R2 имеют любое из значений, определенных в данном описании ранее, за исключением того, что любая присутствующая функциональная группа при необходимости защищена. Взаимодействие может быть осуществлено в присутствии подходящего агента сочетания, такого как, например, TSTU (тетрафторборат 2-(2,5-диоксопирролидин-1-ил)-1,1,3,3-тетраметилизоурония), или TBTU (тетрафторборат 2-(1Н-бензо[d][1,2,3]триазол-1-ил)-1,1,3,3-тетраметилизоурония), или циклический тример ангидрида 1-пропилфосфоновой кислоты, с последующим удалением любой присутствующей защитной группы.
Реакцию удобно осуществлять в присутствии подходящего основания. Подходящее основание представляет собой, например, основание в виде органического амина, такого как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, N-метилморфолин, диазабицикло[5.4.0]ундец-7-ен, диизопропилэтиламин, или, например, карбонат или гидроксид щелочного или щелочно-земельного металла, например карбонат натрия, карбонат калия, карбонат кальция, гидроксид натрия или гидроксид калия.
Реакцию удобно осуществлять в присутствии подходящего инертного растворителя, такого как, например, N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилен, метанол, этанол, галогенированные растворители, такие как дихлорметан, хлороформ или тетрахлористый углерод, и при температуре в диапазоне, например, от -50°C до 100°C, предпочтительно в диапазоне от 0°C до 30°C.
Альтернативно, соединение формулы II, представляющее собой карбоновую кислоту, может быть превращено в активированную форму (такую как хлорангидрид, посредством, например, обработки оксалилхлоридом), которая затем может быть подвергнута взаимодействию с соединением формулы III в условиях, хорошо известных в данной области.
Соединения формулы II могут, например, быть получены посредством реакции омыления соединения формулы IIa:
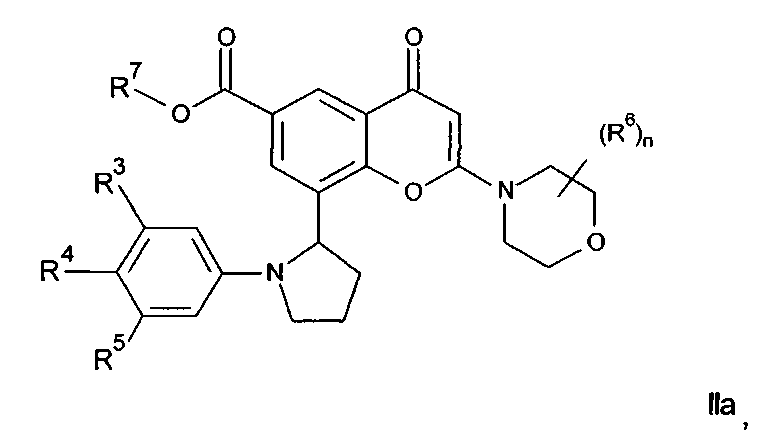
где R3, R4, R5, n и R6 имеют любое из значений, определенных в данном описании ранее, и R7 представляет собой C1-6алкил, обычно метил или этил.
Реакция омыления может быть осуществлена, например, посредством обработки соединения формулы IIa гидроксидом щелочного или щелочноземельного металла, таким как гидроксид лития, гидроксид калия или гидроксид натрия, в подходящем растворителе, таком как, например, смесь этанола и воды или воды и смешивающегося с водой растворителя, такого как, например, тетрагидрофуран или диоксан, при температуре в диапазоне, например, от 0°C до -100°C, предпочтительно в диапазоне 20-40°C.
Соединения формулы IIa могут, например, быть получены посредством взаимодействия, обычно в присутствии подходящего катализатора, соединения формулы IV:
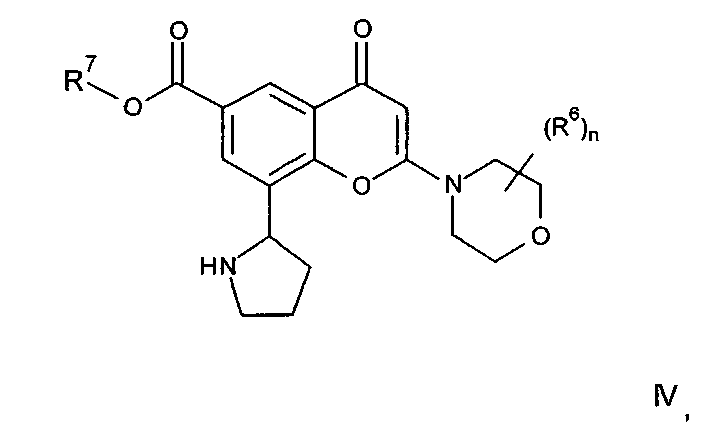
где n и R6 имеют любое из значений, определенных в данном описании ранее, и R7 представляет собой C1-6алкил, обычно метил или этил, с соединением формулы V
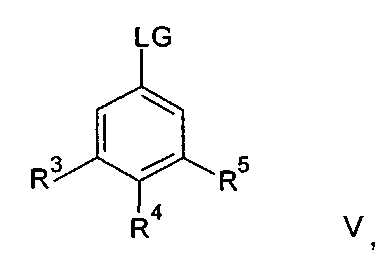
где R3, R4 и R5 имеют любое из значений, определенных в данном описании ранее, за исключением того, что любая функциональная группа при необходимости защищена, и LG представляет собой подходящую уходящую группу, такую как, например, галогеногруппа, такая как группа хлора, брома, йода (обычно бром или йод), с последующим удалением любой присутствующей защитной группы.
Походящий катализатор для этого взаимодействия включает, например, металлический катализатор, такой как палладий(0), например тетракис(трифенилфосфин)палладий(0), или катализатор, образуемый in-situ из соли палладия (II), например ацетата палладия(II), хлорида палладия(II), бромида палладия(II), хлорида бис(трифенилфосфин)палладия(II), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) или трис(дибензилиденацетон)дипалладия, и фосфинового лиганда, такого как, например, (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфин).
Взаимодействие обычно осуществляют в подходящем растворителе, таком как N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилен, и при температуре в диапазоне, например, от 20°C до 150°C, предпочтительно в диапазоне от 60°C до 120°C.
Взаимодействие также целесообразно проводить в присутствии основания, такого как, например, карбонат цезия, карбонат калия или карбонат натрия, обычно карбоната цезия.
Подходящие реакции этого типа описаны как катализируемые палладием реакции сочетания Бухвальда в ′Metal-Catalyzed Cross-Coupling Reactions′, Second Edition, Edited by Armin Meijere, Francois Diederich, Wiley-VCH, 2004, Volume 1, p 699.
Пример такой реакции описан на Стадии 4 получения исходного материала, 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты, в Примере 1.00.
Альтернативно соединения формулы На могут быть получены посредством реакций сочетания Чана-Лама, при которых соединение формулы IV подвергают взаимодействию с соединением формулы Va:
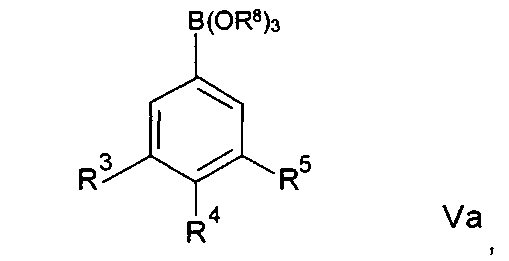
где R3, R4 и R5 имеют любое из значений, определенных в данном описании ранее, за исключением того, что любая функциональная группа при необходимости защищена, и R8 представляет собой C1-33алкил или Н.
Такое взаимодействие целесообразно осуществлять в присутствии источника меди, такого как, например, ацетат меди(II) в DCM, и проводят его посредством воздействия атмосферным кислородом при температуре окружающей среды (Tetpahedron Letters, 1998, 2933).
Соединения формулы IV могут быть получены гидрированием соединения формулы VI:
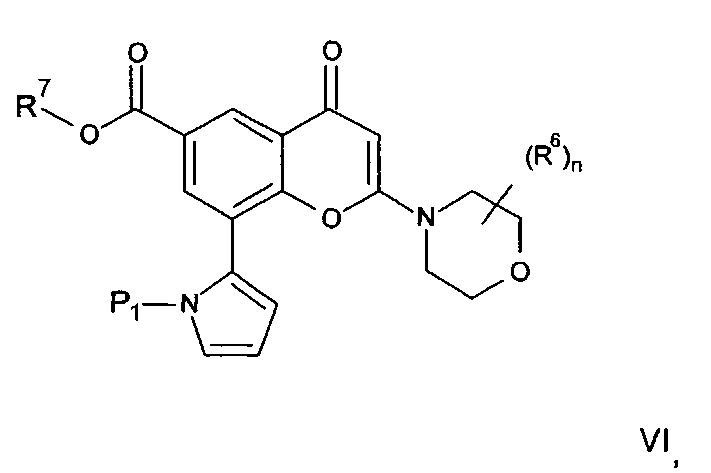
где n и R6 имеют любое из значений, определенных в данном описании ранее, R7 представляет собой C1-6алкил, обычно метил или этил, и P1 представляет собой защитную группу, такую как, например, карбамат, такой как трет-бутоксикарбонил, с последующим удалением P1.
Взаимодействие целесообразно осуществлять в присутствии подходящего растворителя, например спирта, такого как метанол, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилен; обычно в метаноле, и при температуре в диапазоне, например, от 20°C до 100°C, предпочтительно в диапазоне от 50°C до 80°C, под давлением водорода (1-100 атм (101,3-10130 кПа), предпочтительно около 5 атм (506,5 кПа), в присутствии катализатора, такого как палладий, родий, платина, предпочтительно 5% родия на окиси алюминия.
Если P1 представляет собой трет-бутоксикарбонил, то удаление P1 может быть удобным образом осуществлено с помощью хлористого водорода (растворенного, например, в диоксане) в растворителе, таком как DCM, при комнатной температуре.
Альтернативно соединения формулы IV могут быть получены посредством взаимодействия соединения формулы VII (где n и R6 имеют любое из значений, определенных в данном описании ранее, и R7 представляет собой (1-6C)алкил, обычно метил или этил), с соединением формулы VIa:

где P1 представляет собой защитную группу, предпочтительно алкоксикарбонильную группу, такую как трет-бутоксикарбонил, в условиях реакции сочетания Хека, с последующим снятием защиты в виде группы P1 и последующим восстановлением полученных изомерных дигидропирролов (или наоборот).
В отношении дополнительных подробностей условий реакции сочетания Хека смотри, например, в ′Metal-Catalyzed Cross-Coupling Reactions′, Edited by Francois Diederich and P.J. Stang, Wiley-VCH, 1998, p 99).
Подходящий катализатор для этого взаимодействия включает, например, металлический катализатор, такой как палладий(II), например дихлорбис(трифенилфосфин)палладий(II); или катализатор, образуемый in-situ из соли палладия (II), например ацетата палладия(II), хлорида палладия(II), бромида палладия(II), обычно в присутствии фосфинового лиганда, например трифенилфосфина.
Взаимодействие целесообразно проводить в подходящем растворителе, таком как N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилен, и при температуре в диапазоне, например, от 20°C до 150°C, предпочтительно в диапазоне от 60°C до 120°C.
Взаимодействие также целесообразно проводить в присутствии основания, такого как, например, карбонат цезия, карбонат калия или карбонат натрия, предпочтительно карбонат калия.
Если P1 представляет собой трет-бутоксикарбонил, то удаление P1 может быть удобным образом осуществлено с помощью хлористого водорода (растворенного, например, в диоксане), в растворителе, таком как DCM, при комнатной температуре.
Реакцию восстановления полученных изомерных дигидропирролов целесообразно проводить в присутствии подходящего растворителя, такого как, например, спирт, такой как метанол, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилен; обычно в метаноле, и при температуре в диапазоне, например, от 20°C до 100°C, предпочтительно в диапазоне от 20°C до 60°C, под давлением водорода (1-100 атм (101,3-10130 кПа), предпочтительно около 5 атм (506,5 кПа), в присутствии катализатора, такого как палладий, родий или платина, предпочтительно 5% палладия на угле.
Соединения формулы VI могут, например, быть получены взаимодействием соединения формулы VII:
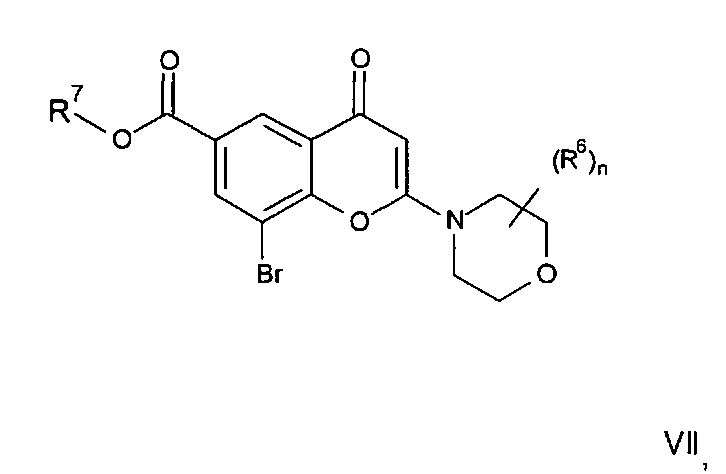
где n и R6 имеют любое из значений, определенных в данном описании ранее, и R7 представляет собой C1-6алкил, обычно метил или этил, с соединением формулы VIII:
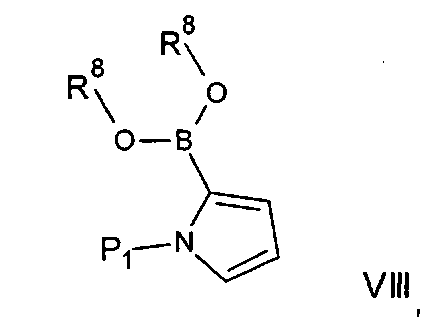
где P1 представляет собой защитную группу, такую как, например, карбамат, такой как, например, трет-бутоксикарбонил, и R8 представляет собой C1-3алкил или H, в условиях реакции сочетания Сузуки, с последующим удалением P1.
В отношении дополнительных подробностей условий реакции сочетания Сузуки смтори, например, в ′Metal-Catalyzed Cross-Coupling Reactions′, Edited by Francois Diederich and P.J. Stang, Wiley-VCH, 1998, p 49).
Походящий катализатор для этого взаимодействия включает, например, металлический катализатор, такой как палладий(0), например тетракис(трифенилфосфин)палладий(0); или катализатор, образуемый in-situ из соли палладия (II), например ацетата палладия(II), хлорида палладия(II), бромида палладия(II), хлорида бис(трифенилфосфин)палладия(II), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) или трис(дибензилиден-ацетон)дипалладия, обычно в присутствии фосфинового лиганда, такого как, например, (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфин).
Взаимодействие целесообразно осуществлять в подходящем растворителе, таком как N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилен, и при температуре в диапазоне, например, от 20°C до 150°C, предпочтительно в диапазоне от 60°C до 120°C.
Взаимодействие также удобно осуществлять в присутствии водного раствора основания, такого как, например, карбонат цезия, карбонат калия или карбонат натрия, предпочтительно карбоната натрия.
Соединения формулы VII могут быть получены в результате сочетания соединения формулы IX:
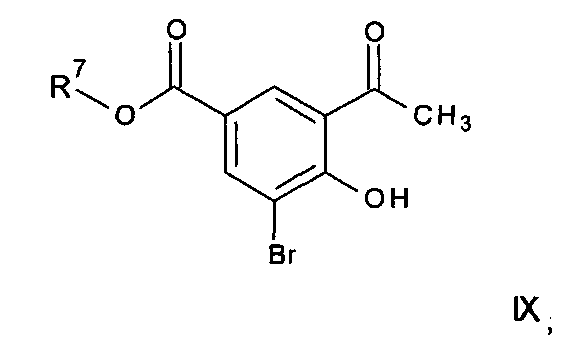
где R7 представляет собой C1-6алкил, обычно метил или этил, с соединением формулы X:
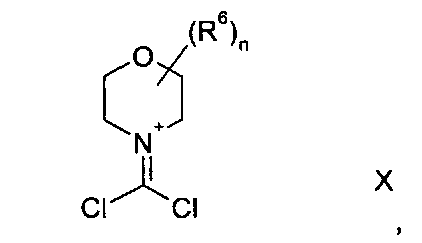
где n и R6 имеют любое из значений, определенных в данном описании ранее, в присутствии подходящего активирующего агента, такого как, например, кислота Льюиса, такая как комплекс диэтиловый эфират трифторида бора.
Взаимодействие соединений формулы IX с соединениями формулы X удобно проводить в присутствии подходящего растворителя, такого как, например, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилен, или галогенированные растворители, такие как дихлорметан, хлороформ или четыреххлористый углерод, и при температуре в диапазоне от 20°C до 150°C, предпочтительно в диапазоне от 60°C до 120°C.
Соединения формулы IX могут быть получены, например, как показано в синтезе метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата, используемого в качестве исходного материала в Примере 1.00. Соединения формулы IX также описаны в литературе (Ger. Often, DE 4318756, 1994 и Aust. J. Chem. 2003, 56, 1099), либо они могут быть получены посредством стандартных способов, известных из уровня техники.
В частности, соединения формулы VII могут быть получены посредством процедур в соответствии со следующей схемой:
метил-8-бром-2-морфолино-4Н-хромен-карбоксилат Соединения формулы VII альтернативно могут быть получены посредством процедур в соответствии со следующей схемой, которая более детально описана в Примере 1.00 в данном описании, где представлен способ получения метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата с использованием следующего пути:
где Br2 представляет собой бром, Pd(OAc)2 представляет собой ацетат палладия(II), DCM представляет собой дихлорметан, LiHMDS представляет собой бис(триметилсилил)амид лития, EtOH представляет собой этанол, Dppf представляет собой 1,1′-бис(дифенилфосфино)ферроцен, TEA представляет собой триэтиламин, THF представляет собой тетрагидрофуран, и TF2O представляет собой ангидрид трифторметансульфоновой кислоты.
Соединения формулы VII также могут быть получены посредством крупномасштабной процедуры в соответствии со следующей схемой, которая более подробно раскрыта в Примере 1.00:
Соединения формулы VII также могут быть получены посредством взаимодействия соединения формулы IX:
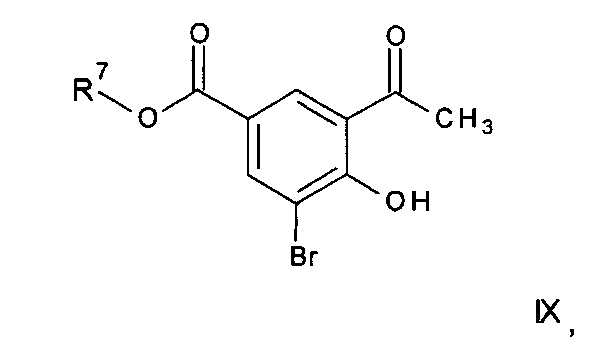
где R7 представляет собой C1-6алкил, обычно метил или этил, с соединением формулы Xa:
где n и R6 имеют любое из значений, определенных в данном описании ранее, в присутствии подходящего активирующего агента, такого как, например, сильное основание, такое как, например, бис(триметилсилиламид лития, с получением соединения формулы IXa:
после чего может быть осуществлена реакция замыкания кольца с образованием соединения формулы VII.
Взаимодействие соединений формулы IX с соединениями формулы Xa целесообразно проводить в присутствии подходящего растворителя или разбавителя, такого как, например, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилен, и при температуре в диапазоне, например, от -100°C до температуры окружающей среды, предпочтительно в диапазоне от -80°C до 20°C.
Реакция замыкания кольца с превращением соединения формулы IXa в соединение формулы VII может быть осуществлена, например, посредством обработки дегидратирующим агентом, таким как, например, ангидрид трифторметансульфоновой кислоты, в подходящем растворителе, таком как, например, дихлорэтан, при температуре в диапазоне, например, от 0°C до -100°C, обычно в диапазоне 20-60°C.
(б) Взаимодействие, обычно в присутствии подходящего катализатора, как определено ранее в данном описании в варианте способа (а), соединения формулы XI:
где R1, R2, n и R6 имеют любое из значений, определенных в данном описании ранее, за исключением того, что любая присутствующая функциональная группа при необходимости защищена, с соединением формулы V
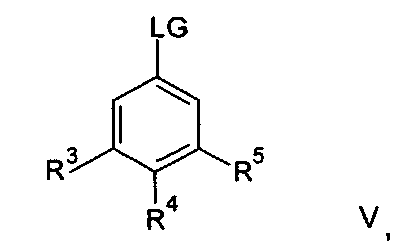
где R3, R4, R5 имеют любое из значений, определенных в данном описании ранее, и LG представляет собой подходящую уходящую группу, такую как, например, галогеногруппа, такая как группа хлора, брома, йода (обычно бром или йод), с последующим удалением любой присутствующей защитной группы.
Подходящие реакции этого типа описаны как катализируемые палладием реакции сочетания Бухвальда в ′Metal-Catalyzed Cross-Coupling Reactions′, Second Edition, Edited by Armin Meijere, Francois Diederich, Wiley-VCH, 2004, Volume 1, p699).
Подходящие условия для таких реакций описаны в варианте способа (а) в данном описании ранее.
Соединения формулы XI могут, например, быть получены посредством сначала омыления соединения формулы XII.
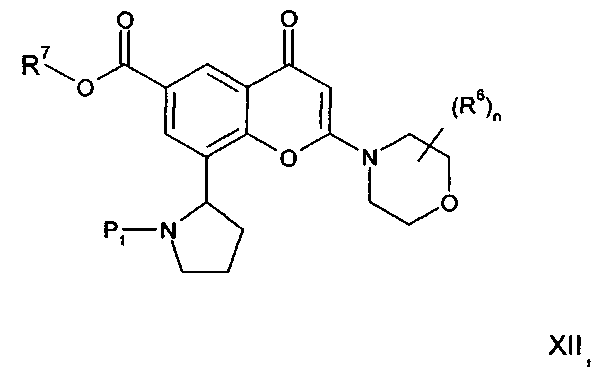
где n, R6 и P1 имеют любое из значений, определенных в данном описании ранее, и R7 представляет собой C1-6алкил, обычно метил или этил, и любая присутствующая функциональная группа при необходимости защищена, а затем образования амида посредством приведения полученной кислоты во взаимодействие, обычно в присутствии подходящего основания и агента сочетания, с амином формулы III:
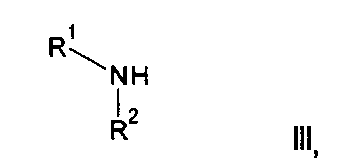
где R1 и R2 имеют любое из значений, определенных в данном описании ранее, за исключением того, что любая присутствующая функциональная группа при необходимости защищена, в присутствии подходящего агента сочетания, такого как, например, TSTU (тетрафторборат 2-(2,5-диоксопирролидин-1-ил)-1,1,3,3-тетраметилизоурония) или TBTU (тетрафторборат 2-(1H-бензо[d][1,2,3]триазол-1-ил)-1,1,3,3-тетраметил-изоурония), с последующим удалением P1 и любой другой присутствующей защитной группы.
Реакцию омыления можно осуществить, например, посредством обработки соединения формулы XII гидроксидом щелочного или щелочноземельного металла, таким как гидроксид лития, гидроксид калия или гидроксид натрия, в подходящем растворителе, таком как, например, смесь этанола и воды или воды и смешивающегося с водой растворителя, такого как, например, тетрагидрофуран или диоксан, при температуре в диапазоне, например, от 0°C до -100°C, предпочтительно в диапазоне 20-40°C.
Реакцию амидного сочетания целесообразно проводить в присутствии подходящего основания. Подходящим основанием является, например, основание в виде органического амина, такое как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, N-метилморфолин, диазабицикло[5.4.0]ундец-7-ен, диизопропилэтиламин, или, например, карбонат или гидроксид щелочного или щелочно-земельного металла, например карбонат натрия, карбонат калия, карбонат кальция, гидроксид натрия или гидроксид калия.
Взаимодействие удобно осуществлять в присутствии подходящего инертного растворителя или разбавителя, такого как, например, N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилен, метанол, этанол, галогенированные растворители, такие как дихлорметан, хлороформ или четыреххлористый углерод, и при температуре в диапазоне, например, от -50°C до 100°C, предпочтительно в диапазоне от 0°C до 30°C.
Альтернативно карбоновую кислоту для амидного сочетания можно трансформировать в активированную форму (такую как хлорангидрид, удобно посредством обработки оксалилхлоридом), которую затем подвергают взаимодействию с соединением формулы III, обычно в присутствии органического основания, такого как триэтиламин.
Если P1 представляет собой трет-бутоксикарбонил, то удаление P1 может удобным образом быть осуществлено с помощью хлористого водорода (растворенного, например, в диоксане), в растворителе, таком как DCM, при комнатной температуре.
Альтернативно соединение формулы XI может быть получено в результате взаимодействия соединения формулы XIII:
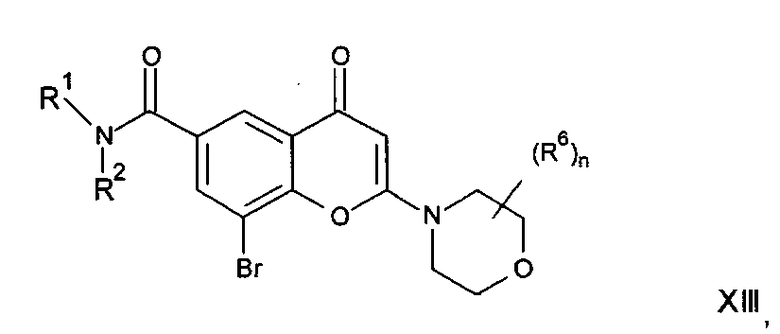
где R1, R2, n и R6 имеют любое из значений, определенных в данном описании ранее, за исключением того, что любая присутствующая функциональная группа при необходимости защищена, посредством реакции Сузуки с соединением формулы VIII:
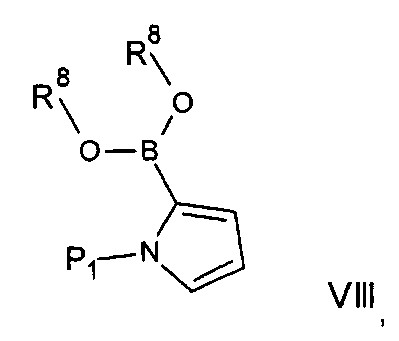
где P1 представляет собой защитную группу, такую как, например, карбамат, такой как, например, трет-бутоксикарбонил, и R8 представляет собой C1-3алкил или H, с последующим гидрированием и снятием защиты в виде P1.
Подходящим катализатором для реакции Сузуки является, например, металлический катализатор, такой как палладий(0), например тетракис(трифенилфосфин)палладий(0); или катализатор, образуемый in-situ из соли палладия (II), например ацетата палладия(И), хлорида палладия(М), бромида палладия(II), хлорида бис(трифенилфосфин)палладия(II), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II), трис(дибензилиден-ацетон)дипалладия, обычно в присутствии фосфинового лиганда, такого как, например, (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфин).
Взаимодействие удобно проводить в подходящем растворителе, таком как N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилен, и при температуре в диапазоне, например, от 20°C до 150°C, предпочтительно в диапазоне от 60°C до 120°C.
Взаимодействие также удобно проводить в присутствии водного раствора основания (обычно карбоната цезия, карбоната калия или карбоната натрия; предпочтительно карбоната натрия).
Если P1 представляет собой трет-бутоксикарбонил, то удаление P1 может удобным образом быть осуществлено с помощью хлористого водорода (растворенного, например, в диоксане) в растворителе, таком как DCM, при комнатной температуре.
Реакцию гидрирования удобно проводить в присутствии подходящего растворителя, такого как, например, спирты, такие как метанол; или N,N-диметилформамида, тетрагидрофурана, 1,4-диоксана, 1,2-диметоксиэтана, бензола, толуола или ксилена; предпочтительно метанола, и при температуре в диапазоне, например, от 20°C до 100°C, предпочтительно в диапазоне от 50°C до 80°C, под давлением водорода (1-100 атм (101,3-10130 кПа)), предпочтительно около 5 атм (506,5 кПа), в присутствии катализатора, такого как палладий, родий или платина, предпочтительно родия 5% на окиси алюминия.
Альтернативно соединения формулы XI могут быть получены посредством взаимодействия соединения XIII, где R1, R2, n и R6 имеют любое из значений, определенных в данном описании ранее, за исключением того, что любая присутствующая функциональная группа при необходимости защищена, с соединением формулы VIIIa
где P1 представляет собой защитную группу, предпочтительно алкоксикарбонильную группу, такую как трет-бутоксикарбонил, в условиях реакции сочетания Хека с последующим снятием защиты в виде группы P1 и последующим восстановлением полученных изомерных дигидропирролов (или наоборот).
В отношении дополнительных деталей условий реакции сочетания Хека смотри, например ′Metal-Catalyzed Cross-Coupling Reactions′, Edited by Francois Diederich and P.J. Stang, Wiley-VCH, 1998, p 99).
Подходящий катализатор для реакции включает, например, металлический катализатор, такой как палладий(II), например дихлорбис(трифенилфосфин)палладий(II); или катализатор, образуемый in-situ из соли палладия (II), например ацетата палладия(II), хлорида палладия(II), бромида палладия(II), обычно в присутствии фосфинового лиганда, например трифенилфосфина.
Взаимодействие удобно осуществлять в подходящем растворителе, таком как N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилен, и при температуре в диапазоне, например, от 20°C до 150°C, предпочтительно в диапазоне от 60°C до 120°C.
Взаимодействие также удобно осуществлять в присутствии основания, такого как, например, карбонат цезия, карбонат калия или карбонат натрия, обычно карбоната калия.
Если P1 представляет собой трет-бутоксикарбонил, то удаление P1 может быть удобным образом осуществлено с помощью хлористого водорода (растворенного, например, в диоксане) или трифторуксусной кислоты в растворителе, таком как DCM, при комнатной температуре.
Реакцию восстановления полученных изомерных дигидропирролов целесообразно проводить в присутствии подходящего растворителя, такого как, например, спирт, такой как метанол, N,N-диметилформамида, тетрагидрофурана, 1,4-диоксана, 1,2-диметоксиэтана, бензола, толуола или ксилена; обычно в метаноле, и при температуре в диапазоне, например, от 20°C до 100°C, предпочтительно в диапазоне от 20°C до 60°C, под давлением водорода (1-100 атм (101,3-101300 кПа)), предпочтительно около 5 атм (506,5 кПа), в присутствии катализатора, такого как палладий, родий, платина, такого как оксид платины(IV), предпочтительно 5% палладия на угле.
Пример схемы способа, который может быть использован для синтеза соединения формулы XIII, такого как, например. 8-бром-N,N-диметил-2-(морфолин-4-ил)-4-оксо-4Н-хромен-6-карбоксамид, является следующим:
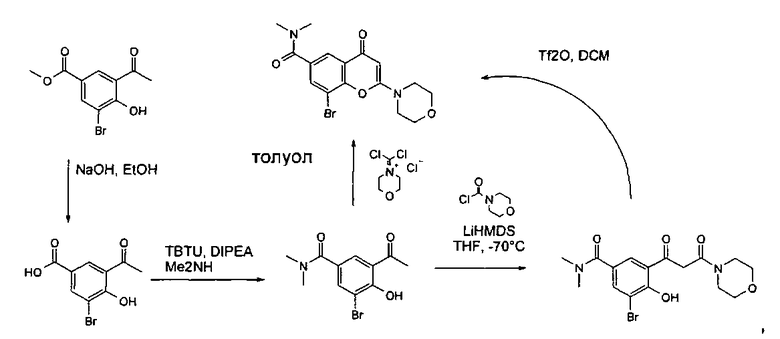
где NaOH представляет собой гидроксид натрия, Me2NH представляет собой диметиламин, DCM представляет собой дихлорметан, LiHMDS представляет собой бис(триметил силил)амид лития, EtOH представляет собой этанол, DIPEA представляет собой диизопропилэтиламин, THF представляет собой тетрагидрофуран, Tf2O представляет собой ангидрид трифторметансульфоновой кислоты, и TBTU представляет собой тетрафторборат 2-(1H-бензо[d][1,2,3]триазол-1-ил)-1,1,3,3-тетраметил изоурония.
Альтернативно соединение формулы XIII может быть получено посредством взаимодействия соединения формулы III
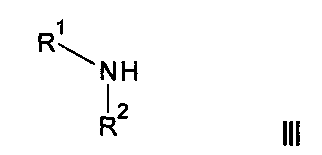
с соединением формулы XIV
в реакции амидного сочетания.
Соединения формулы XIV могут быть получены посредством процедур в соответствии со следующей схемой:

метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилат
Соединения формулы XIV могут, например, быть получены посредством омыления соединения формулы VII:
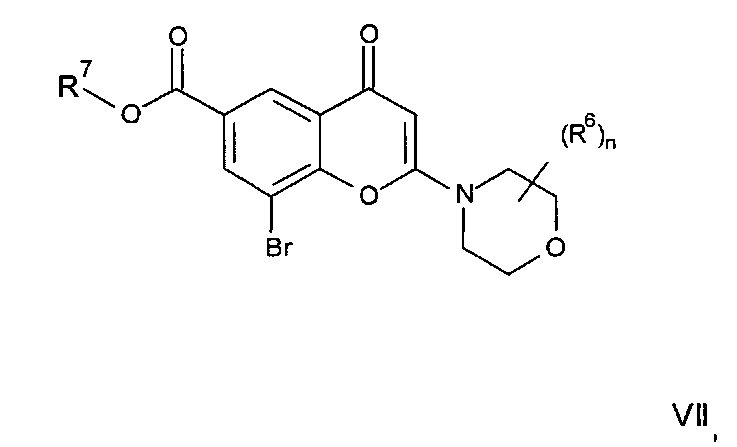
где n и R6 имеют любое из значений, определенных в данном описании ранее, R7 представляет собой C1-6алкил, обычно метил или этил.
Реакция омыления может быть осуществлена, например, посредством обработки соединения формулы VII гидроксидом щелочного или щелочноземельного металла, таким как гидроксид лития, гидроксид калия или гидроксид натрия, в подходящем растворителе, таком как, например, смесь этанола и воды или воды и смешивающегося с водой растворителя, такого как, например, тетрагидрофуран или диоксан, при температуре в диапазоне, например, от 0°C до -100°C, предпочтительно в диапазоне 20-40°C.
Следует понимать, что некоторые стадии могут быть объединены без выделения или очистки промежуточных соединений. Например, соединения формулы XIII могут быть непосредственно получены из соединений формулы VII, как проиллюстрировано в Примере 1.03b (крупномасштабная процедура), где соединение формулы XIV получают из соединения формулы VII. Здесь, карбоксилатную соль соединения формулы XIV (полученную в результате омыления VII без выделения) непосредственно подвергают взаимодействию с активирующим агентом (таким как 2-хлор-4,6-диметокси-1,3,5-триазин) с образованием активированной формы (′активированный сложный эфир′) с последующим взаимодействием с амином формулы III с получением соединения формулы XIII.
Следует понимать, что возможны также другие перестановки стадий способа в вариантах способа, описанных выше. Например, соединение формулы I может быть получено с использованием процедур, аналогичных описанным в вариантах способа (а)-(б), но где заключительной стадией в процедуре является введение группы морфолин-(R6)n.
Следует понимать, что любое соединение формулы I, полученное любым из описанных выше в данном описании способов, может быть при необходимости превращено в другое соединение формулы I.
Когда требуется фармацевтически приемлемая соль производного хроменона формулы I, например соль присоединения кислоты, она может быть получена посредством, например, взаимодействия указанного производного хроменона с подходящей кислотой.
Когда требуется фармацевтически приемлемое пролекарство производного хроменона формулы I, оно может быть получено с помощью обычной процедуры. Например, расщепляемый in vivo сложный эфир производного хроменона формулы I может быть получен, например, посредством взаимодействия соединения формулы I, содержащего гидроксигруппу, с фармацевтически приемлемой карбоновой кислотой. Например, расщепляемый in vivo амид производного хроменона формулы I может быть получен посредством, например, взаимодействия соединения формулы I, содержащего аминогруппу, с фармацевтически приемлемой карбоновой кислотой.
Специалисту в области органического синтеза будет очевидно, что некоторые заместители кольца в соединениях по настоящему изобретению могут быть введены посредством стандартных реакций ароматического замещения или получены посредством обычных модификаций функциональных групп либо до, либо немедленно сразу после упомянутых выше процедур, и как таковые включены в аспект, касающийся способа по изобретению. Такие реакции и модификации включают, например, введение заместителя посредством реакции ароматического замещения, восстановление заместителей, алкилирование заместителей, ацилирование заместителей, амидирование заместителей и окисление заместителей. Реагенты и реакционные условия для таких процедур хорошо известны в области химии. Частные примеры реакций ароматического замещения включают введение нитрогруппы с помощью концентрированной азотной кислоты, введение ацильной группы с использованием, например, ацилгалогенида и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фиделя-Крафтса; введение алкильной группы с помощью алкилгалогенида и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фиделя-Крафтса; и введение галогеногруппы. Частные примеры модификаций включают восстановление нитрогруппы до аминогруппы посредством, например, каталитического гидрирования с никелевым катализатором или обработки железом в присутствии соляной кислоты с нагреванием; окисление алкилтио до алкилсульфинила или алкилсульфонила.
Также следует понимать, что в ряде упомянутых выше реакций может быть необходимым или желательным защитить любые чувствительные группы в соединениях. Случаи, когда защита является необходимой или желательной, и подходящие способы защиты известны специалистам в данной области. Могут быть использованы обычные защитные группы в соответствии со стандартной практикой (в качестве иллюстрации смотри T.W. Green, Protective Groups in Organic Synthesis, John Wiley и Sons, 1991). Так, если взаимодействующие соединения включают такие группы, как амино, карбокси или гидрокси, может быть желательным защитить эти группы в ряде упомянутых выше реакций.
Подходящей защитной группой для амино- или алкиламиногруппы является, например, ацильная группа, например алканоильная группа, такая как ацетильная, алкоксикарбонильная группа, например метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группа, арилметоксикарбонильная группа, например бензилоксикарбонил, или ароильная группа, например бензоил. Условия снятия защиты для вышеуказанных защитных групп по необходимости варьируются в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа, или ароильная группа могут быть удалены посредством, например, гидролиза с помощью подходящего основания, такого как гидроксид щелочного металла, например гидроксида лития или натрия. Альтернативно ацильная группа, такая как трет-бутоксикарбонильная группа, может быть удалена, например, посредством обработки подходящей кислотой, такой как соляная, серная или фосфорная кислота либо трифторуксусная кислота, а арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть удалена, например, посредством гидрирования над катализатором, таким как палладий-на-углероде, или посредством обработки кислотой Льюиса, например трис(трифторацетатом) бора. Подходящей альтернативной защитной группы для первичной аминогруппы является, например, фталоильная группа, которая может быть удалена посредством обработки алкиламином, например диметиламинопропиламином, или гидразином.
Подходящей защитной группой для гидроксигруппы является, например, ацильная группа, например алканоильная группа, такая как ацетильная, ароильная группа, например бензоильная, или арилметильная группа, например бензильная. Условия снятия защиты для вышеуказанных защитных групп будут естественно варьировать в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа, могут быть удалены, например, посредством гидролиза с помощью подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно арилметильная группа, такая как бензильная группа, может быть удалена, например, посредством гидрирования над катализатором, таким как палладий-на-углероде.
Защитные группы могут быть удалены на любой подходящей стадии в синтезе, используя подходящие методики, хорошо известные в области химии.
Некоторые промежуточные соединения (например соединения формул II, На, IV, VI, VII, IXa, XI, XIII и XIV), определенные в данном описании, являются новыми и поэтому составляют дополнительный аспект изобретения. Например, соединения формулы VII (где n и R6 имеют любые из определенных в данном описании ранее значений) могут быть полезны в качестве промежуточных соединений в получении конкретных соединений по изобретению:

Биологические анализы
Следующие анализы могут быть использованы для измерения эффектов соединений по настоящему изобретению как ингибиторов ферментов PI3-киназ, как ингибиторов in vitro фосфо-АКТ (ser473) в клетках MDA-MB-468, как ингибиторов in vivo фосфо-АКТ (ser473) у бестимусных мышей Swiss nu/nu и как ингибиторов in vivo роста опухоли у бестимусных мышей Swiss nu/nu с трансплантированной клеточной линией человеческой аденокарциномы предстательной железы PC3.
(a) In vitro анализ ингибирования фермента
Ингибирование PI3Kβ, PI3Kα, PI3Kγ и PI3Kδ оценивали в анализе ферментативной активности на основе киназы Glo (Kinase Glo) с использованием человеческих рекомбинантных ферментов. В анализе измеряют снижение уровня АТР после инкубации с ферментом, PIP2 и АТР плюс соединение. АТР в конце реакции детектируют посредством добавления реагента Kinase Glo, в котором Ultra Glo™ люцифераза (Promega) использует АТР в качестве субстрата для катализа монооксигенации люциферина и генерирования света. Прямая зависимость существует между люминесценцией, измеренной с помощью реагента Kinase-Glo Plus, и количеством АТР, остающимся в завершенной киназной реакции, и люминесценция обратно пропорциональна активности киназы. Было протестировано двенадцать разных концентраций соединений, и необработанные данные по ингибированию PI3Kβ, PI3Kα, PI3Kγ и PI3Kδ наносили на график в зависимости от концентрации ингибитора.
Подробное описание методики:
Соединения в 100%-ном DMSO добавляли в планшеты для анализа посредством акустического распределения. PI3Kβ добавляли в трис-буфер (50 мМ трис с pH 7,4, 0,05% CHAPS (3-[(3-холамидопропил)диметиламмонио]-1-пропансульфонат), 2,1 мМ DTT (дитиотрейтол) и 10 мМ MgCl2) и оставляли для предварительной инкубации с соединением в течение 20 мин, а затем добавляли раствор субстрата, содержащий PIP2 и АТР. Ферментативную реакцию останавливали через 80 мин путем добавления раствора для детекции Kinase Glo. Планшеты оставляли на 30 мин при комнатной температуре, затем считывали на оборудовании Pherastar (программа Luminescence АТР 384), установленном в направлении максимальной лунки. Конечная концентрация DMSO, АТР и PIP2 в анализе составила 1%, 8 мкМ и 80 мкМ, соответственно.
Анализ данных:
Значения IC50 рассчитывали с использованием логарифмической кривой, подгоняемой к пакету программ нелинейной регрессии, нанося необработанные данные против концентрации ингибитора. Значение IC50 представляет собой концентрацию тестируемого соединения, которая ингибировала на 50% активность фермента.
(b) Протокол обнаружения фосфо-АКТ (ser473) в клетках MDA-MB-468
Клетки MDA-MB-468 (человеческая аденокарцинома груди АТСС НТВ 132) высевали на 384-луночные черные планшеты с плоским дном Greiner с помощью автоматического устройства для клеточных культур (Selec T). Клетки также можно поддерживать вручную и высевать в планшеты с использованием Multidrop или Wellmate. Клетки высевали в концентрации 1500 клеток/лунка в 40 мкл среды DMEN, содержащей 10% FCS (эмбриональная телячья сыворотка) и 1% глутамин. Планшеты с клетками инкубировали в течение 18 часов в инкубаторе при 37°C.
Соединения наносили на клетки с использованием акустического диспенсера Echo, который распределяет нанолитровые количества соединения или DMSO. Соединения наносили в диапазоне 12 разных концентраций, исходя из 30 мкМ высшей дозы, 28 соединений наносили на один планшет. Также было 17 лунок на планшет с одним только DMSO в качестве положительного контроля и 16 лунок отрицательного контроля, в которые вносили концентрацию референсного соединения, которое будет блокировать сигнал рАКТ.
Планшеты инкубировали при 37°C в течение 2 часов, клетки фиксировали добавлением 10 мкл 3,7% раствора формальдегида в вытяжном шкафу с использованием Wellmate. Через 30 мин для обеспечения фиксации, фиксатор и среды удаляли и планшеты промывали Proclin PBS/A, используя устройство отмывки планшетов Tecan PW384, в вытяжном шкафу. Клетки блокировали и пермеабилизировали добавлением 40 мкл PBS, содержащего 0,5% Twee20 и 1% Marvel, используя Wellmate, и инкубировали в течение 60 мин при комнатной температуре.
Буфер для пермеабилизации и блокирования удаляли, используя устройство отмывки планшетов Tecan PW384, затем добавляли раствор 20 мкл первичного антитела с использованием Wellmate. Раствор первичного антитела представляет собой разведение 1:500 кроличьего антитела против фосфо-АКТ Ser 473 (Cell signalling technologies, номер по каталогу #3787) в PBS/T, содержащем 1% marvel (сухой молочный порошок), и инкубировали в течение ночи при 4°C.
Планшеты промывали три раза, используя устройство отмывки планшетов Tecan PW384, забуференным фосфатом физиологическим раствором вместе с 0,05% (об./об.) Polysorbate20 и Proclin300 (Supelco). Затем в каждую лунку добавляли 20 мкл раствора вторичного антитела, используя Wellmate, и инкубировали в течение 1 часа при комнатной температуре. Раствор вторичного антитела представляет собой разведение 1:1000 антикроличьего антитела Alexa Fluor 488 (Molecular Probes, номер по каталогу А11008), разбавленного в забуференном фосфатом физиологическом растворе вместе с 0,05% (об./об.) Polysorbate20, содержащим 1% marvel. Планшеты промывали три раза как описано выше, затем в каждую лунку добавляли 20 мкл PBS и планшеты герметично закрывали с помощью приспособления для заклеивания планшетов.
Планшеты как можно быстрее считывали на устройстве для считывания планшетов Acumen. Используя эту систему, можно генерировать значения IC50 и характеристики планшетов, определенные по контрольным лункам.
Референсные соединения пропускали каждый раз для выполнения мониторного анализа.
(c) Протокол обнаружения ФосФо-АКТ (ser473) у бестимусных мышей Swiss пи/пи
Бестимусные мыши Swiss nu/nu могут быть трансплантированы подкожно клеточной линией человеческой аденокарциномы предстательной железы PC3 (АТСС CRL1435) для определения противоопухолевой активности ингибиторов PI3-киназы. На сутки 0, 106 клеток в 50% Matrigel™ (BD Biosciences #354234) инъецировали подкожно в левый бок животных. Животных рандомизировали на группы требуемой численности (обычно 5 на группу лечения), когда опухоли достигали объема примерно 400-600 мм3, и начинали лечение. Опухоли в конце извлекали и быстро замораживали в жидком азоте и хранили при -80°C до анализа. К каждой опухоли в пробирке Fastprep добавляли 1 мл буфера для лизиса плюс ингибиторы фосфатазы Sigma #Р2850, Sigma #Р5726 (разбавление 1:100) и ингибиторы протеазы Sigma #Р8340 (разбавление 1:200). Опухоли гомогенизировали в течение 1 минуты на аппарате Fastprep и затем оставляли на льду в течение 10 мин.
Образцы вращали в течение 10 мин при 13000 об/мин в охлаждаемой центрифуге. Осветленные лизаты затем отбирали в чистые пробирки и по 5 мкл использовали для анализа определения белка. Все образцы опухолей разбавляли до той же концентрации, так чтобы 15 мкг пропустить на одну дорожку на 4-15% NuPAGE Бис-Трис гелей (Invitrogen) в течение 90 мин при 140 Вольт. Образцы рандомизировали таким образом, чтобы минимизировать эффекты геля. После пропускания через нитроцеллюлозные мембраны их блокировали в течение одного часа, затем инкубировали в течение ночи с разведением 1:500 антитела против либо суммарной АКТ (CST #9272), либо фосфо-AKT-ser 473 (CST #9271). Блоты промывали три раза в PBST, затем инкубировали в течение одного часа при комнатной температуре с разведением 1:2000 вторичного антикроличьего антитела, связанного с пероксидазой хрена (HRP) (CST #7074). Буфер для блокирования и инкубации с антителом представляет собой 5% сухой молочный порошок в PBS с 0,05% Polysorbate.
Блоты промывали три раза в PBS/T, затем визуализировали с использованием набора Pierce West Dura ECL и ChemiGenius. Полосы подсчитывали и для каждого образца получали соотношение фосфо к общему сигналу. Контроли усредняли и каждый образец обработки приводили к среднему контрольному значению.
(d) Протокол обнаружения ингибирования роста опухоли в клеточной линии человеческой аденокарциномы предстательной железы РСЗ, трансплантированной бестимусным мышам Swiss nu/nu
Бестимусные мыши Swiss nu/nu могут быть трансплантированы подкожно клеточной линией человеческой аденокарциномы предстательной железы PC3 (АТСС CRL1435) для определения противоопухолевой активности ингибиторов PI3-киназы. На сутки 0, 106 клеток в 50% Matrigel™ (BDM) инъецировали подкожно в левый бок животных. Животных рандомизировали на группы по 10-15 животных, когда опухоли достигали объема примерно 200-300 мм3, и начинали лечение. Животным вводили в течение 2-4 недель пероральным, внутривенным или внутрибрюшинным путями соединение (и возможно сур-ингибитор, такой как 1-аминобензотриазол) в подходящем носителе в определенных дозах. Опухоли измеряли обычно дважды в неделю с помощью циркуля и объем опухолей рассчитывали с использованием формулы для объема эллипса (π/6×ширина×ширина×длина).
(e) Протокол обнаружения фосфо-АКТ (ser473) в клетках Jeko
Соединения в конечной концентрации x10 в 10 мкл 1% (об./об.) DMSO добавляли к лункам 96-луночного планшета Greiner с V-образным дном. Соединения вводили в диапазоне 10 концентраций, исходя из 1 мкМ высшей дозы, 8 соединений вводили на один планшет. Также было по 8 лунок на планшет с DMSO в качестве положительного контроля, в которые вносили носитель и анти-IgM, и 8 лунок отрицательного контроля, в которые вносили носитель и буфер для анализа. Конечная концентрация носителя равна 0,1% DMSO. Референсное PI3Kδ-избирательное соединение включали в каждый пробег. Клетки Jeko В (лимфома из человеческих клеток мантийной зоны, АТСС CRL-3006) высевали в 96-луночные планшеты Greiner с V-образным дном, содержащие соединения. Клетки высевали в концентрации 100000 клеток/лунка в 70 мкл среды RPMI, содержащей 1% глутамин.
Клеточные планшеты инкубируют в течение 1 часа в инкубаторе при 37°C. После этого предварительного времени инкубации анти-IgM (F(ab′)2 фрагмент козьего IgM против человека, Stratech 109-006-129) добавляют в планшеты в конечной концентрации х5 в 20 мкл буфера для анализа (среда RPMI, содержащая 1% глутамин). Конечная концентрация анти-IgM равна 0,06 мкг/мл или эквивалентной EC90 дозе. Планшеты инкубируют при 37°C в течение 10 мин, затем планшеты немедленно помещают на лед и центрифугируют при 12000 об/мин в течение 4 мин. На льду супернатанты осторожно удаляют с помощью ручной пипетки и добавляют 40 мкл буфера для лизиса. Планшеты инкубируют на льду в течение 5 мин и хранят при -80°C до анализа с помощью набора цельноклеточного лизата Phosphor(ser473)/Total Akt согласно инструкциям производителя (Mesoscale Diagnostics, K11100D-3).
Хотя фармакологические свойства соединений формулы I варьируют при структурных изменениях, как ожидают, в целом активность, которой обладают соединения формулы I, может быть продемонстрирована в следующих концентрациях или дозах в одном или более чем одном из вышеуказанных тестов (а) и (b):
Тест (a): IC50 в отношении PI3Kβ в диапазоне, например, от 1НМ до 25 мкМ;
Тест (a): IC50 в отношении PI3Kδ в диапазоне, например, от 1НМ до 25 мкМ;
Тест (b): IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-МВ-468, в диапазоне, например, от 1НМ до 25 мкМ;
Тест (е): IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках Jeko, в диапазоне, например, от 1НМ до 25 мкМ.
Предпочтительно, конкретные соединения по изобретению обладают активностью в следующих концентрациях или дозах в одном или более чем одном из вышеуказанных тестов (а) и (b):
Тест (a): IC50 в отношении PI3Kβ в диапазоне, например, от 1НМ до 10 мкМ;
Тест (а): IC50 в отношении PI3Kδ в диапазоне, например, от 1НМ до 10 мкМ;
Тест (b): IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468, в диапазоне, например, от 1НМ до 20 мкМ;
Тест (е): IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках Jeko, в диапазоне, например, от 1НМ до 20 мкМ.
Предпочтительно, конкретные соединения по изобретению обладают активностью в следующих концентрациях или дозах в одном или более чем одном из вышеуказанных тестов (а), (b), (с) и (d):
Тест (a): IC50 в отношении PI3Kβ в диапазоне, например, от 1НМ до 10 мкМ;
Тест (a): IC50 в отношении PI3Kδ в диапазоне, например, от 1НМ до 10 мкМ;
Тест (b): IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468, в диапазоне, например, от 1НМ до 20 мкМ;
Тест (с): более чем 50%-ное ингибирование in vivo фосфо-АКТ (ser473) в диапазоне, например, от 1 до 200 мг/кг/сутки;
Тест (d): активность ксенотрансплантата в диапазоне, например, от 1 до 200 мг/кг/сутки;
Тест (е): IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках Jeko, в диапазоне, например, от 1НМ до 20 мкМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.04, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 11НМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 24 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 5 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 2.00, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 9 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 12 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 2 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.03b, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 7 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 9 нМ; активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 1НМ; и активность в Тесте (е) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках Jeko приблизительно 7 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.03, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 12 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 22 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 2 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.03а, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 0,198 мкМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 0,282 мкМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 27 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.05, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 7 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 9 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 1НМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.06, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 7 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 7 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 1НМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.07, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 6 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 5 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 2 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.08, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 6 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 6 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 2 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.10, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 8 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 21НМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 4 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 1.11, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 9 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 15 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 8 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 3.00, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 11НМ; в Тесте (а) со значением IC50 в отношении PI3Kδ 18 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 3 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 3.02, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 14 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 10 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 3 нМ.
Например, хроменоновое соединение, раскрытое как соединение Примера 3.03, проявляет активность в Тесте (а) со значением IC50 в отношении PI3Kβ приблизительно 10 нМ; в Тесте (а) со значением IC50 в отношении PI3Kδ приблизительно 7 нМ; и активность в Тесте (b) со значением IC50 в отношении клеточной фосфо-АКТ (ser473) в клетках MDA-MB-468 приблизительно 1НМ.
Например, хроменоновые соединения, раскрытые в Примерах, проявляют активность в Тесте (а) в уровнях, проиллюстрированных в Таблице А.
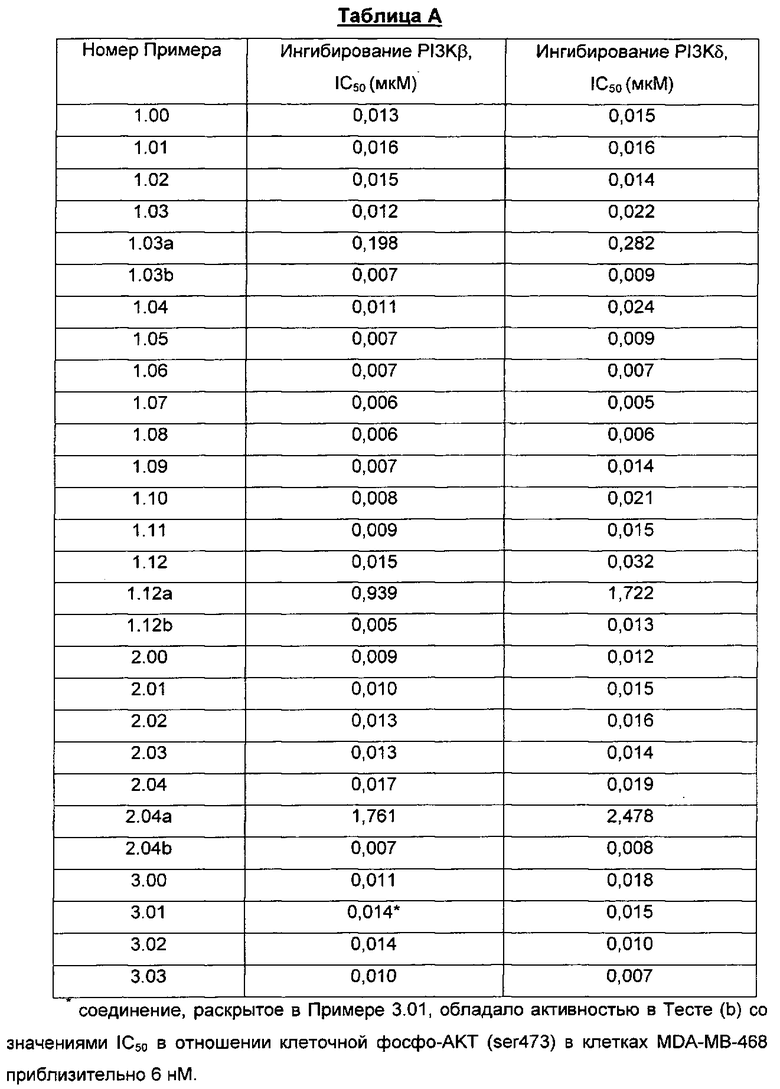
Согласно еще одному аспекту изобретения предложена фармацевтическая композиция, содержащая производное хроменона формулы I или его фармацевтически приемлемую соль, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем.
Композиции по изобретению могут быть представлены в форме, подходящей для перорального применения (например, в виде таблеток, лепешек, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде тонкоизмельченного порошка или жидкого аэрозоля), для введения путем инсуффляции (например, в виде тонкоизмельченного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного, внутрибрюшинного или внутримышечного введения) или в виде суппозиториев для ректального введения.
Композиции по изобретению могут быть получены традиционными методиками с использованием общепринятых фармацевтических эксципиентов, хорошо известных в данной области техники. Так, композиции, предназначенные для перорального применения, могут содержать, например, один или более красителей, подсластителей, корригентов и/или консервантов.
Количество активного ингредиента, которое объединяют с одним или более чем одним эксципиентом для получения единой лекарственной формы, как правило варьирует в зависимости от организма хозяина, подвергаемого лечению, и конкретного пути введения. Например, композиция, предназначенная для перорального введения людям, как правило содержит, например, от 1 мг до 1 г активного агента (более предпочтительно от 1 до 250 мг, например от 1 до 100 мг), объединенного с соответствующим и подходящим количеством эксципиентов, которое может варьировать от примерно 5 до примерно 98 процентов по массе от общей массы композиции.
Размер дозы соединения формулы I для терапевтических или профилактических целей, конечно, будет варьировать в зависимости от природы и тяжести болезненного состояния, возраста и пола животного или пациента, а также от пути введения, в соответствии с хорошо известными принципами медицины.
При использовании соединения формулы I для терапевтических или профилактических целей его обычно вводят в суточной дозе в интервале, например, от 1 мг/кг до 100 мг/кг массы тела, даваемой при необходимости в разделенных дозах. В целом, при парентеральном введении применяются более низкие дозы. Так, например, для внутривенного введения обычно применяют дозу в интервале, например, от 1 мг/кг до 25 мг/кг массы тела. Подобным образом, для введения путем ингаляции применяется доза в интервале, например, от 1 мг/кг до 25 мг/кг массы тела. Однако, пероральное введение является предпочтительным, в частности в форме таблетки. Обычно, стандартные лекарственные формы содержат приблизительно от 10 мг до 0,5 г соединения по изобретению.
Как указано в настоящей заявке, известно, что ферменты PI3-киназы принимают участие в образовании опухолей в результате одного или более эффектов опосредования пролиферации раковых и других клеток, опосредования ангиогенных событий и опосредования подвижности, миграции и инвазивности раковых клеток. Авторами было обнаружено, что хроменоновые соединения по настоящему изобретению обладают эффективным противоопухолевым действием, которое, как полагают, достигается путем ингибирования одного или более ферментов PI3-киназ класса I (таких как ферменты PI3-киназы класса Ia и/или фермент PI3-киназа класса Ib), которые вовлечены в стадии передачи сигнала, приводящие к пролиферации и выживанию опухолевых клеток, а также инвазивности и мигрирующей способности метастазирующих опухолевых клеток.
Соответственно, соединения по настоящему изобретению являются ценными в качестве противоопухолевых агентов, в частности в качестве избирательных ингибиторов пролиферации, выживания, подвижности, диссеминирования и инвазивности раковых клеток млекопитающих, приводя к ингибированию опухолевого роста и выживания и к ингибированию метастатического опухолевого роста. В частности, хроменоновые соединения по настоящему изобретению являются ценными в качестве антипролиферативных и антиинвазивных средств для сдерживания распространения и/или лечения солидного опухолевого заболевания. В частности, соединения по настоящему изобретению, как ожидают, являются полезными в предупреждении или лечении тех опухолей, которые чувствительны к ингибированию одного или более из множества ферментов PI3-киназ, таких как ферменты PI3-киназы класса Ia и фермент PI3-киназа класса Ib, которые вовлечены в стадии передачи сигнала, приводящие к пролиферации и выживанию опухолевых клеток и инвазивности метастазирующих опухолевых клеток. Кроме того, соединения по настоящему изобретению, как ожидают, являются полезными для предотвращения или лечения тех опухолей, которые опосредованы только или частично ингибированием ферментов PI3-киназ, таких как ферменты PI3-киназы класса Ia и фермент PI3-киназа класса Ib, то есть соединения могут быть использованы для получения ингибирующего действия в отношении фермента PI3-киназы у теплокровного животного, нуждающегося в таком лечении.
Как указано в настоящей заявке, ингибиторы ферментов PI3-киназ являются терапевтически ценными для лечения различных форм ракового заболевания, включая солидные опухоли, такие как карциномы и саркомы, и лейкозы и лимфолейкозы. В частности, ингибиторы ферментов PI3-киназ класса I являются терапевтически ценными для лечения, например, рака груди, ободочной и прямой кишки, легкого (включая мелкоклеточный рак легкого, немелкоклеточный рак легкого и бронхоальвеолярный рак) и предстательной железы, и рака желчных протоков, кости, мочевого пузыря, головы и шеи, почек, печени, желудочно-кишечной ткани, пищевода, яичника, поджелудочной железы, кожи, яичек, щитовидной железы, матки, шейки матки и наружных женских половых органов, и лейкозов (включая хронический лимфоидный лейкоз (CLL), острый лимфоидный лейкоз (ALL) и хронический миелоидный лейкоз (CML)), множественной миеломы и лимфом (включая неходжкинские лимфомы, такие как диффузная крупноклеточная B-клеточная лимфома [DLBCL], фолликулярная лимфома и лимфома из клеток мантийной зоны).
Согласно еще одному аспекту изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, как определено выше, для применения в качестве лекарственного средства у теплокровного животного, такого как человек.
Согласно еще одному аспекту изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, как определено выше, для применения в продуцировании антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно дополнительному воплощению этого аспекта изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, как определено выше, для применения у теплокровного животного, такого как человек, в качестве антиинвазивного агента для сдерживания распространения и/или лечения солидного опухолевого заболевания.
Согласно еще одному аспекту изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, для продуцирования антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно дополнительному воплощению этого аспекта изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в продуцировании антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно дополнительному воплощению этого аспекта изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования у теплокровного животного, такого как человек, в качестве антиинвазивного агента для сдерживания распространения и/или лечения солидного опухолевого заболевания.
Согласно дополнительному воплощению этого аспекта изобретения предложен способ продуцирования антипролиферативного эффекта у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Согласно дополнительному воплощению этого аспекта изобретения предложен способ продуцирования антиинвазивного эффекта путем сдерживания распространения и/или лечения солидного опухолевого заболевания у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества производного хроменона формулы I или его фармацевтически приемлемых соли, сольвата или пролекарства, как определено выше.
Согласно еще одному аспекту изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, как определено выше, для применения в предупреждении или лечении рака у теплокровного животного, такого как человек.
Согласно еще одному аспекту изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в предупреждении или лечении рака у теплокровного животного, такого как человек.
Согласно дополнительному воплощению этого аспекта изобретения предложен способ предупреждения или лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Согласно еще одному аспекту изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в предупреждении или лечении солидного опухолевого заболевания у теплокровного животного, такого как человек.
Согласно дополнительному воплощению этого аспекта изобретения предложен способ предупреждения или лечения солидного опухолевого заболевания у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Согласно еще одному аспекту изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, как определено выше, для применения в предупреждении или лечении тех опухолей, которые являются чувствительными к ингибированию ферментов PI3-киназ (таких как ферменты класса Ia и/или фермент PI3-киназа класса Ib), которые вовлечены в стадии передачи сигнала, приводящие к пролиферации, выживанию, инвазивности и мигрирующей способности опухолевых клеток.
Согласно дополнительному воплощению этого аспекта изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в предупреждении или лечении тех опухолей, которые являются чувствительными к ингибированию ферментов PI3-киназ (таких как ферменты класса Ia и/или фермент PI3-киназа класса Ib), которые вовлечены в стадии передачи сигнала, приводящие к пролиферации, выживанию, инвазивности и мигрирующей способности опухолевых клеток.
Согласно дополнительному воплощению этого аспекта изобретения предложен способ предупреждения или лечения тех опухолей, которые являются чувствительными к ингибированию ферментов PI3-киназ (таких как ферменты класса Ia и/или фермент PI3-киназа класса Ib), которые вовлечены в стадии передачи сигнала, приводящие к пролиферации, выживанию, инвазивности и мигрирующей способности опухолевых клеток, включающий введение указанному животному эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Согласно еще одному аспекту изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, как определено выше, для применения в обеспечении ингибирующего действия в отношении фермента PI3-киназы (такого как ингибирующее действие в отношении фермента PI3-киназы класса Ia или фермента PI3-киназы класса Ib).
Согласно дополнительному воплощению этого аспекта изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в обеспечении ингибирующего действия в отношении фермента PI3-киназы (такого как ингибирующее действие в отношении фермента PI3-киназы класса Ia или фермента PI3-киназы класса Ib).
Согласно еще одному аспекту изобретения также предложен способ обеспечения ингибирующего действия в отношении фермента PI3-киназы (такого как ингибирующее действие в отношении фермента PI3-киназы класса Ia или фермента PI3-киназы класса Ib), включающий введение эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Как изложено выше в данном описании, некоторые соединения по настоящему изобретению обладают значительно лучшей эффективностью в отношении ферментов PI3-киназ класса Ia, чем в отношении фермента PI3-киназы класса Ib или в отношении ферментов тирозинкиназы EGF рецептора, тирозинкиназы VEGF рецептора, или нерецепторной тирозинкиназы Src. Такие соединения обладают достаточной эффективностью в отношении ферментов PI3-киназ класса Ia, которые могут быть использованы в количестве, достаточном для ингибирования ферментов PI3-киназ класса Ia, в то же время демонстрируя незначительную активность в отношении фермента PI3-киназы класса Ib или в отношении тирозинкиназы EGF рецептора или тирозинкиназы VEGF рецептора, или нерецепторной тирозинкиназы Src. Такие соединения вероятно полезны для избирательного ингибирования ферментов PI3-киназ класса Ia и вероятно полезны для эффективного лечения, например, опухолей, управляемых ферментом PI3-киназой класса Ia.
Согласно этому аспекту изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, как определено выше, для применения в обеспечении ингибирующего действия в отношении фермента PI3-киназы класса Ia.
Согласно дополнительному воплощению этого аспекта изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в обеспечении ингибирующего действия в отношении фермента PI3-киназы класса Ia.
Согласно еще одному аспекту изобретения также предложен способ обеспечения ингибирующего действия в отношении фермента PI3-киназы класса Ia, включающий введение эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Под "ингибирующим действием в отношении фермента PI3-киназы класса Ia" понимают, что хроменоновые соединения формулы I являются более эффективными в отношении ферментов PI3-киназ класса Ia, чем в отношении других ферментов-киназ. В частности, некоторые соединения по изобретению являются более эффективными в отношении ферментов PI3-киназ класса Ia, чем в отношении других киназ, таких как рецепторные или нерецепторные тирозинкиназы или серин/треонинкиназы. Например, избирательный ингибитор фермента PI3-киназы класса Ia по изобретению по меньшей мере в 5 раз более эффективен, предпочтительно по меньшей мере в 10 раз более эффективен, более предпочтительно по меньшей мере в 100 раз более эффективен в отношении ферментов PI3-киназ класса Ia, чем в отношении других киназ.
Согласно дополнительному воплощению изобретения предложено производное хроменона формулы I или его фармацевтически приемлемая соль, как определено выше, для применения в лечении рака груди, толстой и прямой кишки, легкого (включая мелкоклеточный рак легкого, немелкоклеточный рак легкого и бронхоальвеолярный рак) и предстательной железы.
Согласно дополнительному воплощению этого аспекта изобретения предложено производное хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, для применения в лечении рака желчных протоков, кости, мочевого пузыря, головы и шеи, почки, печени, желудочно-кишечной ткани, пищевода, яичника, поджелудочной железы, кожи, семенников, щитовидной железы, матки, шейки матки и вульвы, а также лейкозов (включая ALL и CML), множественной миеломы и лимфом.
Согласно дополнительному воплощению этого аспекта изобретения предложено производное хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, для применения в лечении рака желчных протоков, кости, мочевого пузыря, головы и шеи, почки, печени, желудочно-кишечной ткани, пищевода, яичника, поджелудочной железы, кожи, семенников, щитовидной железы, матки, шейки матки и вульвы, а также лейкозов (включая ALL и CML), множественной миеломы и лимфом (включая неходжкинские лимфомы, такие какие диффузная крупноклеточная B-клеточная лимфома [DLBCL], фолликулярная лимфома и лимфома из клеток мантийной зоны).
Согласно дополнительному воплощению этого аспекта изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в лечении рака груди, толстой и прямой кишки, легкого (включая мелкоклеточный рак легкого, немелкоклеточный рак легкого и бронхоальвеолярный рак) и предстательной железы.
Согласно дополнительному воплощению этого аспекта изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в лечении рака желчных протоков, кости, мочевого пузыря, головы и шеи, почки, печени, желудочно-кишечной ткани, пищевода, яичника, поджелудочной железы, кожи, семенников, щитовидной железы, матки, шейки матки и вульвы, а также лейкозов (включая ALL и CML), множественной миеломы и лимфом.
Согласно дополнительному воплощению этого аспекта изобретения предложено применение производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для использования в лечении рака желчных протоков, кости, мочевого пузыря, головы и шеи, почки, печени, желудочно-кишечной ткани, пищевода, яичника, поджелудочной железы, кожи, семенников, щитовидной железы, матки, шейки матки и вульвы, а также лейкозов (включая ALL, CLL и CML), множественной миеломы и лимфом (включая неходжкинские лимфомы, такие какие диффузная крупноклеточная B-клеточная лимфома [DLBCL], фолликулярная лимфома и лимфома из клеток мантийной зоны).
Согласно дополнительному воплощению этого аспекта изобретения предложен способ лечения рака груди, толстой и прямой кишки, легкого (включая мелкоклеточный рак легкого, немелкоклеточный рак легкого и бронхоальвеолярный рак) и предстательной железы, у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Согласно дополнительному воплощению этого аспекта изобретения предложен способ лечения рака желчных протоков, кости, мочевого пузыря, головы и шеи, почки, печени, желудочно-кишечной ткани, пищевода, яичника, поджелудочной железы, кожи, семенников, щитовидной железы, матки, шейки матки и вульвы, а также лейкозов (включая ALL и CML), множественной миеломы и лимфом, у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Согласно дополнительному воплощению этого аспекта изобретения предложен способ лечения рака желчных протоков, кости, мочевого пузыря, головы и шеи, почки, печени, желудочно-кишечной ткани, пищевода, яичника, поджелудочной железы, кожи, семенников, щитовидной железы, матки, шейки матки и вульвы, а также лейкозов (включая ALL, CLL и CML), множественной миеломы и лимфом (включая неходжкинские лимфомы, такие какие диффузная крупноклеточная B-клеточная лимфома [DLBCL], фолликулярная лимфома и лимфома из клеток мантийной зоны), у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение эффективного количества производного хроменона формулы I или его фармацевтически приемлемой соли, как определено выше.
Как изложено выше в данном описании, in vivo эффекты соединения формулы I могут быть обусловлены отчасти одним или более метаболитами, которые образуются в организме человека или животного после введения соединения формулы I.
Как изложено выше в данном описании, конкретные соединения по изобретению обладают лучшей эффективностью в отношении определенных изоформ ферментов PI3-киназ по сравнению с другими, в частности соединения по изобретению обладают лучшей эффективностью в отношении PI3-киназы р и PI3-киназы 8, чем в отношении других изоформ PI3-киназ класса I, таких как α и γ.
Таким образом, в настоящем изобретении также предусмотрен способ ингибирования фосфоинозитид-3-киназы β у пациента, включающий введение пациенту количества соединения формулы I или его фармацевтически приемлемой соли, эффективного в ингибировании фосфоинозитид-3-киназы β у пациента.
Аналогично, в настоящем изобретении также предусмотрен способ ингибирования фосфоинозитид-3-киназы δ у пациента, включающий введение пациенту количества соединения формулы I или его фармацевтически приемлемой соли, эффективного в ингибировании фосфоинозитид-3-киназы δ у пациента.
Соединение формулы I или его фармацевтически приемлемая соль, являясь ингибитором PI3-киназы, также имеет потенциальные терапевтические применения при разных других болезненных состояниях. Например, PI3-киназа играет важную роль в стимулировании пролиферации гладких мышц в сосудистом дереве, а именно гладкомышечных клеток сосудов, Thyberg, 1998, European Journal of Cell Biology 76(1): 33-42, а также в легких (гладкомышечные клетки дыхательных путей), Krymskaya, V.P., BioDrugs, 2007. 21(2): 85-95. Избыточная пролиферация гладкомышечных клеток сосудов играет важную роль в образовании атеросклеротических бляшек и в развитии гиперплазии неоинтимы после инвазивных сосудистых процедур, Scwartz et al., 1984, Progress in Cardiovascular Disease 26: 355-372; Clowes et al., 1978, Laboratory Investigations 39: 141-150. Более того, избыточная пролиферация гладкомышечных клеток дыхательных путей приводит к развитию хронической обструктивной болезни легких (COPD) при установленных астме и хроническом бронхите. Ингибиторы активности PI3-киназы, таким образом, могут быть использованы для предупреждения сосудистого рестеноза, атеросклероза и COPD.
PI3-киназы также играют важную роль в регуляции опухолевых клеток и в склонности этих клеток подвергаться апоптозному росту (Sellers et al., 1999, The Journal of Clinical Investigation 104: 1655-1661). Кроме того, неконтролируемая регуляция PI3-киназных липидных продуктов PI(3,4,5)P3 и PI(3,4)P2 посредством липидной фосфатазы PTEN играет важную роль в прогрессировании ряда злокачественных опухолей у людей (Leevers et al., 1999, Current Opinion in Cell Biology 11: 219-225). Специфическая роль изоформы β фосфоинозитид-3-киназы (PI3Kβ) была описана при этих видах рака (Jia S et al., 2008, Nature 454 (7205): 776-9; Wee et al., 2008, PNAS 105(35): 13057-62). Таким образом, соединение формулы I или его фармацевтически приемлемая соль, являясь ингибитором PI3-киназы, может быть использовано для лечения новообразований у людей.
PI3-киназа также играет важную роль в функции лейкоцитов (Fuller et al., 1999, The Journal of Immunology 162(11): 6337-6340; Eder et al., 1998, The Journal of Biological Chemistry 273(43):28025-31) и функции лимфоцитов (Vicente-Manzanares et al., 1999, The Journal of Immunology 163(7): 4001-4012). Например, адгезия лейкоцитов к воспаленному эндотелию включает активацию эндогенных лейкоцитарных интегринов посредством зависимого от PI3-киназы процесса передачи сигнала. Кроме того, передача сигнала PI3-киназы, по-видимому, вовлечена в окислительный взрыв (Nishioka et al, 1998, FEBS Letters 441(1):63-66 и Condliffe, A.M., et al., Blood, 2005. 106(4): 1432-40) и перестройку цитоскелета (Kirsch et al., 1999, Proceedings National Academy of Sciences USA 96(11): 6211-6216) в нейтрофилах. Миграция и направленное движение нейтрофилов также зависит от активности PI3-киназы (Camps, М., et al., Nat Med, 2005. 11(9): p.936-43 и Sadhu, С., et al., J Immunol, 2003. 170(5): 2647-54). Таким образом, ингибиторы PI3-киназы могут быть полезны в снижении адгезии и активации лейкоцитов в участках воспаления и тем самым могут быть полезны для лечения острых и/или хронических воспалительных расстройств. PI3-киназа также играет важную роль в пролиферации и активации лимфоцитов, Fruman et al., 1999, Science 283 (5400): 393-397. Учитывая важную роль лимфоцитов в аутоиммунных заболеваниях, ингибиторы активности PI3-киназы могут быть использованы в лечении таких расстройств.
Противораковое лечение, описанное выше, можно применять в качестве единственной терапии, либо оно может включать, в дополнение к соединению по изобретению, традиционное хирургическое вмешательство, или радиотерапию, или химиотерапию. Такая химиотерапия может включать один или более агентов из следующих категорий противоопухолевых агентов:
(1) другие антипролиферативные/противоопухолевые лекарственные средства и их комбинации, применяемые в медицинской онкологии, такие как алкилирующие агенты (например цис-платина, оксалиплатин, карбоплатин, циклофосфамид, хлорметин, мелфалан, хлорамбуцил, бусульфан, бендамустин, темозоламид и нитрозомочевины); антиметаболиты (например гемцитабин и антифолаты, такие как фторпиримидины типа 5-фторурацила и тегафура, ралтитрексед, метотрексат, цитозинарабинозид и гидроксимочевина, а также аналоги пурина, такие как флударабин); противоопухолевые антибиотики (например антрациклины типа адриамицина, блеомицина, доксорубицина, дауномицина, эпирубицина, идарубицина, митомицина-С, дактиномицина и митрамицина); антимитотические агенты (например винкаалкалоиды типа винкристина, винбластина, виндезина и винорелбина, таксоиды типа таксола и таксотера и ингибиторы полокиназы); и ингибиторы топоизомеразы (например эпиподофиллотоксины типа этопозида и тенипозида, амсакрина, топотекана и камптотецина);
(2) цитостатические агенты, такие как антиэстрогены (например тамоксифен, фулвестрант, торемифен, ралоксифен, дролоксифен и йодоксифен), антиандрогены (например бикалутамид, флутамид, нилутамид и кипротерона ацетат), антагонисты рецепторов андрогенов MDV3100 или ARN-509, которые предотвращают ядерную транслокацию рецептора андрогена и его связывание с ДНК или белками-коактиваторами, ингибиторы CYP17A1, такие как абиратерон [ZYTIGA™], и смешанные ингибиторы функции рецепторов андрогенов и CYP17A1, такие как ТОК-001 (галетерон), антагонисты рилизинг-фактора лютеинизирующего гормона (LHRH) или агонисты LHRH (например гозерелин, лейпрорелин и бусерелин), прогестогены (например мегестрола ацетат), ингибиторы ароматазы (например анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы, такие как финастерид;
(3) антиинвазивные агенты (например ингибиторы семейства c-Src-киназ типа 4-(6-хлор-2,3-метилендиоксианилино)-7-[2-(4-метилпиперазин-1-ил)-этокси]-5-тетрагидропиран-4-илоксихиназолина (AZD0530; публикация международной заявки WO 01/94341], N-(2-хлор-6-метилфенил)-2-{6-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метилпиримидин-4-иламино}тиазол-5-карбоксамида (дасатиниб, BMS-354825; J. Med. Chem., 2004, 47, 6658-6661) и босутиниб (SKI-606), и ингибиторы металлопротеиназы типа маримастата, ингибиторы функции урокиназных рецепторов активаторов плазминогена или антитела к гепараназе);
(4) ингибиторы функции фактора роста: например, такие ингибиторы включают антитела к факторам роста и антитела к рецепторам фактора роста (например, анти-егЬВ2 антитело трастузумаб [Herceptin™], анти-EGFR антитело панитумумаб, анти-erbB1 антитело цетуксимаб [Erbitux, С225] и любые антитела к факторам роста и рецепторам фактора роста, раскрытые Stern et al. Critical reviews in oncology/haematology, 2005, Vol.54, pp 11-29); такие ингибиторы также включают ингибиторы тирозинкиназы, например ингибиторы семейства эпидермального фактора роста (например, ингибиторы тирозинкиназы семейства EGFR, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, ZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)-хиназолин-4-амин (Cl 1033), ингибиторы erB2 тирозинкиназы, такие как лапатиниб); ингибиторы семейства факторов роста гепатоцитов; ингибиторы инсулинового семейства факторов роста; ингибиторы семейства тромбоцитарных факторов роста, такие как иматиниб и/или нилотиниб (AMN107); ингибиторы серин/треонинкиназ (например, ингибиторы передачи сигнала Ras/Raf, такие как ингибиторы фарнезилтрансферазы, например сорафениб (BAY 43-9006), типифарниб (R115777) и лонафарниб (SCH66336)), ингибиторы передачи сигнала через MEK- и/или АКТ-киназы, ингибиторы c-kit, ингибиторы abl-киназы, ингибиторы PI3-киназы, ингибиторы PI3-киназы, ингибиторы CSF-IR-киназы, ингибиторы IGF рецепторной (инсулиноподобный фактор роста) киназы; ингибиторы aurora-киназы (например AZD1152, РН739358, VX-680, MLN8054, R763, МР235, МР529, VX-528 и АХ39459) и ингибиторы циклин-зависимой киназы, такие как CDK2 и/или CDK4 ингибиторы;
(5) антиангиогенные агенты, такие как агенты, которые ингибируют эффекты сосудистого эндотелиального фактора роста [например, антитело против сосудистого эндотелиального фактора роста бевацизумаб [Avastin™] и, например, ингибитор тирозинкиназы VEGF-рецептора, такие как вандетаниб (ZD6474), ваталаниб (РТК787), сунитиниб (SU 11248), акситиниб (AG-013736), пазопаниб (GW 786034) и 4-(4-фтор-2-метилиндол-5-илокси)-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолин (AZD2171; Пример 240 в заявке WO 00/47212), соединения, такие как раскрыты в публикациях международных заявок WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354), и соединения с другими механизмами действия (например, линомид, ингибиторы функции ανβ3 интегрина и ангиостатин);
(6) сосудоповреждающие агенты, такие как Комбретастатин A4 и соединения, раскрытые в публикациях международных заявок WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 и WO 02/08213;
(7) антагонист рецептора эндотелина, например зиботентан (ZD4054) или атрасентан;
(8) антисмысловые терапии, например такие, которые направлены на мишени, перечисленные выше, такие как ISIS 2503, антисмысловая anti-ras;
(9) подходы генной терапии, включая, например, подходы для замещения аберрантных генов, таких как аберрантный р53, или аберрантный BRCA1 или BRCA2, подходы GDEPT (ген-направленная фермент-пролекарственная терапия), такие как подходы, использующие цитозиндеаминазу, тимидинкиназу или бактериальный фермент нитроредуктазу, и подходы увеличения толерантности пациента к химиотерапии или радиотерапии, такие как терапия гена множественной лекарственной устойчивости; и
(10) иммуннотерапевтические подходы, включая, например, подходы ех-vivo и in-vivo для увеличения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4, или гранулоцитарно-макрофагеальный колониестимулирующий фактор, подходы для уменьшения анергии Т-клеток, подходы, использующие трансфицированные иммунные клетки, такие как цитокин-трансфицированные дендритные клетки, подходы, использующие цитокин-трансфицированные опухолевые клеточные линии, и подходы, использующие антиидиотипические антитела, подходы для усиления T-клеток, включая CTLA4 антитела и антитела против CD137, PD-1 или В7-Н1, агонисты toll-рецепторов; агонистические антитела к CD40, такие как SGN-40 (дацетузумаб), или к Tweak-рецептору, например PDL-192; агонистические антитела к FAS; подходы, использующие антитела к опухолеассоциированным антигенам, и антитела, которые истощают типовые клетки-мишени (например, неконъюгированные анти-CD20 антитела, такие как ритуксимаб, офатумумаб, обинутузумаб, анти-CD19 антитела, такие как MEDI-551, анти-CD52 антитела, такие как алемтузумаб, анти-CD37 антитела, такие как TRU-016, анти-CD22 антитела, такие как инотузумаб, меченные радиоактивным изотопом анти-CD20 антитела Bexxar и Zevalin, и анти-CD54 антитело Campath; иммунотоксины, такие как моксетумумаб пасудотокс), подходы, использующие антиидиотипические антитела, подходы, которые усиливают функцию клеток естественных киллеров, и подходы, которые используют конъюгаты антитело-токсин (например, анти-CD33 антитело Mylotarg), иммунные модификаторы, такие как ревлимид (леналидомид).
Согласно этому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы I, как определено выше, или его фармацевтически приемлемую соль и любой из противоопухолевых агентов, перечисленных выше в подпунктах (1)-(10).
Таким образом, в дополнительном аспекте изобретения предложено соединение формулы I или его фармацевтически приемлемая соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в подпунктах (1)-(10).
В дополнительном аспекте изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы I, как определено выше, или его фармацевтически приемлемую соль и любой из противоопухолевых агентов, перечисленных выше в подпункте (1).
В дополнительном аспекте изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы I, как определено выше, или его фармацевтически приемлемую соль и таксоид, такой как, например, таксол или таксотер, предпочтительно таксотер.
В данном описании, когда используют термин "комбинация", следует понимать, что он может относиться к одновременному, раздельному или последовательному введению. В одном аспекте изобретения "комбинация" относится к одновременному введению. В другом аспекте изобретения "комбинация" относится к раздельному введению. В еще одном аспекте изобретения "комбинация" относится к последовательному введению. Когда введение является последовательным или раздельным, время выдержки до введения второго компонента не должно быть таким, чтобы утрачивалось преимущество эффекта, возникающее в результате применения комбинации.
Согласно еще одному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в подпунктах (1)-(10), в сочетании с фармацевтически приемлемым разбавителем или носителем.
Согласно еще одному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в подпунктах (1)-(10), в сочетании с фармацевтически приемлемым разбавителем или носителем для применения в лечении рака.
Согласно другому аспекту изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в подпунктах (1)-(10), в изготовлении лекарственного средства для использования в лечении рака у теплокровного животного, такого как человек.
Таким образом, в дополнительном аспекте изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы I или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в подпунктах (1)-(10).
Согласно еще одному аспекту настоящего изобретения предложен набор, содержащий соединение формулы I или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из перечисленных выше в подпунктах (1)-(10).
Согласно еще одному аспекту настоящего изобретения предложен набор, содержащий:
a) соединение формулы I или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
b) противоопухолевый агент, выбранный из перечисленных выше в подпунктах (1)-(10), во второй стандартной лекарственной форме; и
c) контейнерные средства для содержания указанных первой и второй лекарственных форм.
Хотя соединения формулы I являются ценными главным образом в качестве терапевтических агентов для применения у теплокровных животных (включая человека), они также полезны в любых случаях, когда требуется ингибирование действий фермента PI3-киназы класса I, в частности ферментов PI3-киназ класса Ia и/или фермента PI3-киназы класса Ib, более конкретно ферментов PI3-киназ класса Ia, который включает PI3-киназу β. Так, они являются полезными фармакологическими стандартами для использования в разработке новых биологических тестов и при поиске новых фармакологических агентов.
Ссылки сделаны в данном описании на следующие фигуры:
Фиг.1: Картина дифракции рентгеновских лучей на порошке Формы A соединения Примера 1.03b;
Фиг.2: Картина дифракции рентгеновских лучей на порошке Формы B соединения Примера 1.03b;
Фиг.3: Термограмма дифференциальной сканирующей калориметрии (ДСК) Формы B соединения Примера 1.03b;
Фиг.4: Картина дифракции рентгеновских лучей на порошке Формы C соединения Примера 1.03b.
Изобретение будет проиллюстрировано в следующих далее Примерах, в которых, в общем:
1) операции проводили при температуре окружающей среды, то есть в диапазоне от 17 до 25°C, и в атмосфере инертного газа, такого как азот, если не указано иного;
2) выпаривание осуществляли с помощью роторного испарителя или с помощью оборудования Genevac в вакууме, а процедуры исследования проводили после удаления остаточных твердых веществ посредством фильтрования;
3) очистку посредством флэш-хроматографии выполняли на автоматизированном Armen Glider Flash: Spot II Ultimate (Armen Instrument, Saint-Ave, France), используя предварительно упакованные картриджи Merck с нормальной фазой Si60 диоксида кремния (гранулометрия: 15-40 или 40-63 мкм), полученные от Merck, Darmstad, Germany;
4) препаративную хроматографию выполняли на приборе Waters (600/2700 или 2525), снабженном ZMD или ZQ ESCi масс-спектрометрами и колонке с обращенной фазой Waters X-Terra, или Waters X-Bridge, или Waters SunFire (C-18, 5 микрон диоксид кремния, 19 мм диаметр, 100 мм длина, скорость потока 40 мл/минуту), используя в качестве элюента понижающие полярные смеси воды (содержащей 0,2% карбоната аммония) и ацетонитрила;
5) выходы, где указаны, не обязательно являются максимально достижимыми;
6) в общем, структуры конечных продуктов формулы I подтверждали с помощью спектроскопии ядерного магнитного резонанса (ЯМР); величины химических сдвигов ЯМР измеряли по дельта шкале [спектры протонного магнитного резонанса определяли с помощью прибора Bruker Avance 500 (500 МГц)]; измерения проводили при температуре окружающей среды, если не указано иного; были использованы следующие аббревиатуры: s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; dd, дублет дублетов; ddd, дублет дублета дублетов; dt, дублет триплетов; bs, широкий сигнал;
7) в общем, конечные продукты формулы I также характеризовали посредством масс-спектроскопиии с последующей жидкостной хроматографией (ЖХ-МС); ЖХ-МС проводили, используя Waters Alliance НТ (2790 & 2795), снабженный масс-спектрометром Waters ZQ ESCi или ZMD ESCi и колонкой X Bridge 5 мкм C-18 (2,1×50 мм), при скорости потока 2,4 мл/мин, используя систему растворителей от 95% A +5% C до 95% B +5% С в течение 4 мин, где A - вода, B - метанол, C - смесь 1:1 метанол:вода (содержащая 0,2% карбоната аммония);
8) промежуточные соединения в общем не были полностью охарактеризованы и чистоту оценивали с помощью анализов посредством тонкослойной хроматографии, масс-спектрального, ВЭЖХ и/или ЯМР анализа;
9) спектры дифракции рентгеновских лучей на порошке определяли с помощью прибора Bruker D4, помещая образец кристаллического материала на держатель Bruker, сделанный из монокристалла кремния (SSC), и распределяя данный образец до тонкого слоя с помощью предметного стекла микроскопа. Образец вращали со скоростью 30 оборотов в минуту (для улучшения статистики подсчета) и облучали рентгеновскими лучами, генерированными медной трубкой с длинным точным фокусом, работающей при 40 кВ и 40 мА при длине волны 1,5418 ангстрем. Коллимированный источник рентгеновских лучей пропускали через автоматическую переменную дивергентную щель, установленную на V20, и отраженное излучение направляли через 5,89 мм противорассеивающую щель и 9,55 мм детекторную щель. Образец подвергали воздействию в течение 0,03 секунд с приращением в 0,00570° 2-тета (непрерывный режим сканирования) в интервале от 2 градусов до 40 градусов 2-тета в режиме тета-тета. Время прогона составляло 3 минуты и 36 секунд. Данный прибор был оснащен сенсорным детектором положения (Lynxeye). Контроль и получение данных осуществляли посредством Dell Optiplex 686 NT 4.0 Workstation, работающей с программным обеспечением Diffract+. Специалистам в области дифракции рентгеновских лучей на порошке будет понятно, что на относительную интенсивность пиков могут влиять, например, зерна, размер которых больше 30 микрон, и неунитарные соотношения сторон, которые могут влиять на анализ образцов. Специалисту также будет понятно, что на положение отражений может влиять точная высота, на которой находится образец в дифрактометре, и калибровка нуля дифрактометра. Плоскостность поверхности образца также может оказывать небольшое влияние. Следовательно, представленные данные по картине дифракции не следует принимать как абсолютные значения;
10) дифференциальную сканирующую калориметрию проводили с помощью прибора ТА Instruments Q1000 DSC. Типично меньше чем 5 мг материала, содержащегося в стандартной алюминиевой чашке с подходящей по размеру крышкой, нагревали в интервале температур от 25°C до 300°C при постоянной скорости нагрева 10°C в минуту; в качестве продувочного газа использовали азот со скоростью потока 50 мл в минуту;
11) анализ дифракции рентгеновских лучей на одиночном кристалле выполняли при 200 K, используя диффрактометр Bruker APEX-II CCD с графитным монохроматическим MoKα излучением (λ=0,71073 Å). Структуру определяли прямыми методами и уточняли с F2 против всех отражений. Геометрия: длину связей, углы и т.д. рассчитывали, используя скругленные фракционные координаты. Все величины ′su′ (стандартная неопределенность) устанавливают из отклонений (полной) ковариационной матрицы. Величины ′esd′ (заданное стандартное отклонение) ячеек учитывают при установлении длин, углов и углов кручения. Уточнение: уточнение F2 против ВСЕХ отражений. Взвешенный R-фактор wR и критерий согласия S основаны на F2, конвенционные факторы R основаны на F, где F установлен равным нулю для отрицательного F2. Пороговое выражение F2>2σ(F2) используют только для рассчета R факторов (gt) и т.д., и оно не является релевантным для выбора отражений для уточнения. R-факторы, основанные на F2, статистически примерно в два раза больше, чем факторы, основанные на F, а R-факторы, основанные на ВСЕХ данных, даже еще больше. Компьютерные программы: сбор данных: Bruker АРЕХ2; уточнение ячейки: Bruker SAINT; восстановление данных: Bruker SAINT; программа(ы), используемая(ые) для установления структуры: SHELXS97 (Sheldrick, 2008); программа(ы), используемая(ые) для уточнения структуры: SHELXL97 (Sheldrick, 2008); молекулярная графика: ORTEPII (Johnson, 1976), PLATO N (Spek, 2007); программное обеспечение, используемое для подготовки материала для публикации: PLATON (Spek, 2007);
12) были использованы следующие аббревиатуры:
Пример 1.00
8-[1-(3,5-дифторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
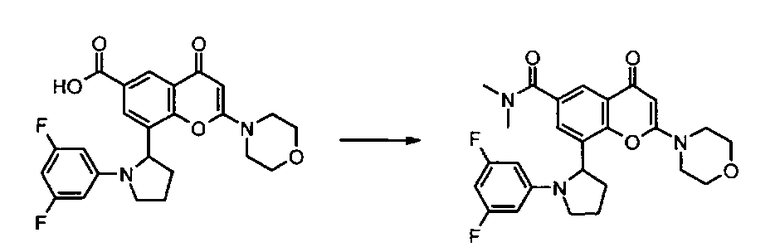
К суспензии 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (60 мг, 0,13 ммоль) в DMF (1 мл) при комнатной температуре под азотом добавляли DIPEA (0,046 мл, 0,26 ммоль), а затем TSTU (43,5 мг, 0,14 ммоль) и перемешивали в течение ночи. Затем добавляли диметиламин (2 н. в THF) (0,131 мл, 0,26 ммоль) и смесь перемешивали в течение еще одного часа. Реакционную смесь очищали препаративной ВЭЖХ. Фракции выпаривали досуха, остаток переносили в минимальное количество DCM, разбавляли петролейным эфиром, перемешивали в течение 3 ч, собирали фильтрованием и сушили с получением 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (43 мг, 68%) в виде белого твердого вещества.
Масс-спектр: m/z [M+H]+=484.
Спектр протонного ЯМР: (DMSO-d6) 1.75-1.88 (m, 1Н), 1.97-2.09 (m, 2Н), 2.45-2.57 (m частично скрыт DMSO-d5, 1Н), 2.70 (s, 3H), 2.92 (s, 3H), 3.33-3.41 (m, 1Н), 3.50-3.57 (m, 2Н), 3.37-3.64 (m, 2Н), 3.71-3.80 (m, 5Н), 5.25 (d, 1Н), 5.61 (s, 1Н), 6.13 (d, 2Н), 6.32 (t, 1Н), 7.11 (d, 1Н), 7.80 (d, 1Н).
8-(1-(3,5-Дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновую кислоту, используемую в качестве исходного материала, получали следующим образом.
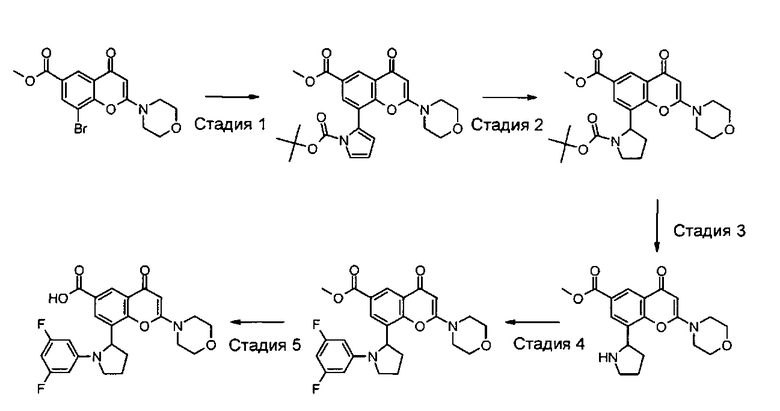
Стадия 1
К перемешиваемой суспензии метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (23 г, 62,47 ммоль) в DME (300 мл) и воде (30 мл) добавляли 1-(трет-бутоксикарбонил)-1Н-пиррол-2-илбороновую кислоту (14,50 г, 68,72 ммоль), Na2CO3 (19,87 г, 187,41 ммоль) и хлорид бис(трифенилфосфин)палладия(П) (0,877 г, 1,25 ммоль). Смесь дегазировали аргоном и нагревали до 80°C в течение 7 ч. Реакционную смесь охлаждали и концентрировали, остаток растворяли в 300 мл DCM и 300 мл воды. Органическую фазу декантировали, промывали рассолом, затем сушили над MgSO4, фильтровали и концентрировали с получением неочищенного соединения, которое очищали на диоксиде кремния, элюируя 80%-ным AcOEt в DCM. Растворители выпаривали с получением трет-бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-1Н-пиррол-1-карбоксилата (20 г, 70%) в виде белого порошка.
Масс-спектр: m/z [M+H]+=455.
Стадия 2
трет-Бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-1Н-пиррол-1-карбоксилат (17,1 г, 37,63 ммоль) и 5% родий на оксиси алюминия (50% влажности) (3,4 г, 0,80 ммоль) в MeOH (175 мл) перемешивали в атмосфере водорода при 5 бар (500 кПа) и 65°C в течение 7 ч. Катализатор удаляли из реакционной смеси фильтрованием через набивку целита и промывали MeOH. Целит и катализатор суспендировали в 500 мл 10%-ного MeOH в DCM и снова фильтровали. Органические растворы объединяли и выпаривали с получением трет-бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (14,4 г, 83%) в виде желто-коричневого твердого вещества.
Масс-спектр: m/z [M+H]+=459.
Стадия 3
К перемешиваемому раствору трет-бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (440 мг, 0,96 ммоль), растворенного в диоксане (3 мл) и DCM (3 мл) при комнатной температуре добавляли хлористый водород (4Mb диоксане) (2,4 мл, 9,6 ммоль) и реакционную смесь перемешивали в течение 4 ч. Растворители выпаривали и остаток дважды подвергали азеотропной перегонке с MeCN и сушили под вакуумом при 50°C. Оставшееся твердое вещество затем экстрагировали DCM. DCM сушили над MgSO4 и концентрировали с получением метил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилата (286 мг, 83%) в виде бледно-бежевого твердого вещества.
Масс-спектр: m/z [M+H]+=359.
Стадия 4
Палладиевый комплекс (33 мг, 0,04 ммоль, смотри ниже) добавляли к перемешиваемой смеси метил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилата (270 мг, 0,75 ммоль), 1-бром-3,5-дифторбензола (0,095 мл, 0,83 ммоль) и карбоната цезия (368 мг, 1,13 ммоль), растворенных в 1,4-диоксане (5 мл). Полученную суспензию дегазировали аргоном и затем перемешивали при 80°C в течение 23 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры, фильтровали и концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 6% MeOH в EtAc. Растворитель выпаривали досуха с получением метил-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (206 мг, 58%) в виде прозрачной желтой пены.
Масс-спектр: m/z [M+H]+=471.
Палладиевый комплекс, используемый в качестве реагента, получали следующим образом.
Раствор (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфина) (256 мг, 0,44 ммоль), трис(дибензилиденацетон)дипалладия (184 мг, 0,20 ммоль) и 1-бром-3,5-дифторбензола (0,213 мл, 1,85 ммоль) в бензоле (10 мл) перемешивали при комнатной температуре в течение 96 ч. Смесь затем фильтровали через набивку целита и концентрировали в вакууме. К остатку добавляли эфир (10 мл) и желтое кристаллическое твердое вещество оставляли образовываться при стоянии в течение 4 ч. Твердое вещество собирали фильтрованием, промывали эфиром и сушили под вакуумом с получением целевого палладиевого комплекса (139 мг, 39%).
Стадия 5
Водный 2 н. раствор NaOH (0,622 мл, 1,24 ммоль) добавляли к метил-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоксилату (195 мг, 0,41 ммоль) в MeOH (2 мл) и реакционную смесь перемешивали при 40°C в течение 6 ч. Смесь охлаждали до 0°C и к реакционной смеси по каплям добавляли водный 2 н. раствор HCl (0,684 мл, 1,37 ммоль) до тех пор, пока pH не становился равным приблизительно 5. Раствор частично концентрировали, и появлялся осадок. К суспензии добавляли воду и экстрагировали DCM. Органическую фазу промывали водой, сушили над MgSO4, фильтровали и концентрировали. Образовавшийся осадок растирали с эфиром, фильтровали, промывали эфиром, сушили под вакуумом при 50°C с получением 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (67 мг, 35%) в виде бледно-бежевого твердого вещества. Масс-спектр: m/z [M+H]+=457.
Метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилат, используемый в качестве исходного материала на Стадии 1 описанного выше способа получения 8-(1-(3,5-дифторфенил)-пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты, получали следующим образом.
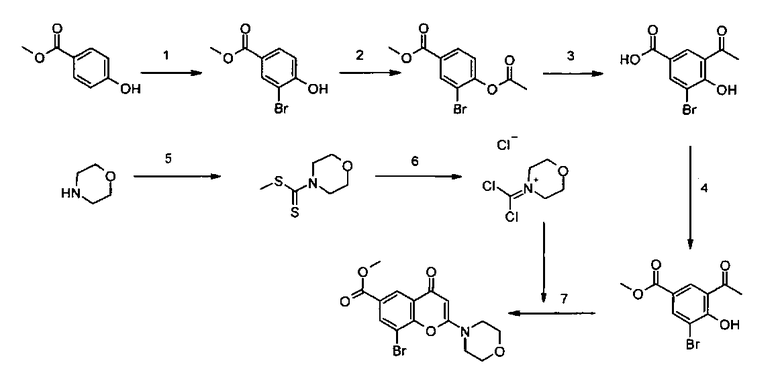
Стадия 1
К перемешиваемой суспензии метил-4-гидроксибензоата (180 г, 1183 ммоль) в DCM (3 л) под азотом и при 0°C по каплям добавляли бром (64 мл, 1242 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 36 ч. Затем добавляли раствор тиосульфата натрия (500 мл 10%-ного раствора), поддерживая температуру около 15°C, после чего добавляли MeOH (250 мл). Органический слой промывали водой, затем рассолом, сушили над MgSO4, фильтровали и концентрировали досуха с получением метил-3-бром-4-гидроксибензоата (290 г) в виде белого твердого вещества. Масс-спектр: m/z [M-H]+=229.
Стадия 2
К перемешиваемой суспензии метил-3-бром-4-гидроксибензоата (270 г, 1168 ммоль) в DCM (1,5 л) добавляли пиридин (150 мл). Затем по каплям при комнатной температуре и под азотом добавляли ацетилхлорид (87 мл, 1227 ммоль). Смесь оставляли перемешиваться в течение 2 ч при комнатной температуре. Затем добавляли воду (1 л) с последующим добавлением 2 н. HCl до pH 1. Органический слой затем промывали водой, рассолом, сушили над MgSO4, фильтровали и выпаривали досуха с получением метил-4-ацетокси-3-бромбензоата (300 г, 94%) в виде белого порошка.
Спектр протонного ЯМР: (DMSO-d6) 2.34 (s, 3H), 3.87 (s, 3H), 7.47 (d, 1Н), 8.01 (dd, 1Н), 8.20 (d, 1Н).
Стадия 3
К метил-4-ацетокси-3-бромбензоату (150 г, 549,3 ммоль) добавляли трихлорид алюминия (220 г, 1647,9 ммоль) и смесь нагревали при 140°C в отсутствие растворителя в течение 3 ч. При охлаждении до комнатной температуры твердое вещество измельчали и осторожно при перемешивании добавляли к воде (1,5 л). Затем добавляли HCI (250 мл 12 н.) и перемешивание продолжали в течение 30 минут. Полученное твердое вещество собирали фильтрованием, промывали водой (2×2 л) и сушили в течение ночи с получением 3-ацетил-5-бром-4-гидроксибензойной кислоты (120 г, 84%) в виде желтого порошка. Масс-спектр: m/z [M-H]+=258.
Стадия 4
К перемешиваемой суспензии 3-ацетил-5-бром-4-гидроксибензойной кислоты (240 г, 926 ммоль) в MeOH (2 л) по каплям под азотом добавляли дихлорид серы (68 мл, 926,5 ммоль) и смесь нагревали при 80°C в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали, разбавляли DCM. Органический слой промывали рассолом, сушили над MgSO4, фильтровали и концентрировали с получением неочищенного соединения, которое очищали на диоксиде кремния, элюируя 70%-ным DCM в петролейном эфире. Растворители выпаривали досуха с получением метил-3-ацетил-5-бром-4-гидроксибензоата (108 г, 42,7%) в виде белого порошка. Масс-спектр: m/z [M-H]+=229.
Стадия 5
К перемешиваемому раствору морфолина (201 мл, 2295 ммоль) в воде (2 л) под азотом добавляли дисульфид углерода (0,138 л, 2295,67 ммоль). Затем по каплям добавляли гидроксид натрия (96 г, 2410 ммоль в растворе в 1 л воды). Полученную смесь перемешивали при комнатной температуре в течение 1 ч, затем охлаждали до 5°C с помощью ледяной бани и по каплям добавляли диметилсульфат (217 мл, 2295 ммоль). Смесь перемешивали в течение 1 ч при комнатной температуре, полученное твердое вещество собирали фильтрованием, промывали водой (2×1 л) и сушили под вакуумом над пентоксидом фосфора при 50°C с получением метил-морфолин-4-карбодитиоата (360 г, 88%). Спектр протонного ЯМР: (CDCl3): 2.68 (s, 3H), 3.71-3.84 (m, 4Н), 4.02 (bs, 2Н), 4.30 (bs, 2Н).
Стадия 6
Газообразный хлор (455 г, 6417 ммоль) барботировали через раствор метил-морфолин-4-карбодитиоата (170 г, 959 ммоль) в DCM (1,5 л) в течение периода времени 2 ч, поддерживая температуру около 10-15°C. Как только добавление хлора было завершено, перемешивание продолжали в течение дополнительных 1,5 ч, в течение которых происходило образование осадка. Затем через смесь в течение 30 мин пропускали азот. Твердое вещество собирали фильтрованием под азотом, промывали DCM и хранили под азотом в холодильнике. Таким образом получали хлорид 4-(дихлорметилен)морфолин-4-ия (180 г, 92%) в виде белого гигроскопичного твердого вещества.
Стадия 7
К перемешиваемому раствору метил-3-ацетил-5-бром-4-гидроксибензоата (106 г, 388 ммоль) в толуоле (1 л) по каплям под азотом добавляли (диэтилоксонио)трифторборат (0,201 л, 1630 ммоль). Полученный раствор оставляли перемешиваться в течение ночи при комнатной температуре, затем добавляли хлорид 4-(дихлорметилен)морфолин-4-ия (143 г, 698 ммоль) и смесь нагревали при 90°C в течение 12 ч. После охлаждения до комнатной температуры добавляли эфир (1,5 л) и твердое вещество собирали фильтрованием. Это твердое вещество затем суспендировали в MeOH (1 л) и смесь нагревали при 50°C в течение 2 ч. После охлаждения до комнатной температуры твердое вещество собирали фильтрованием, затем солюбилизировали в DCM (1 л) и промывали водой и насыщенным раствором бикарбоната натрия. Органический слой сушили над MgSO4, фильтровали и выпаривали досуха с получением метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (68,0 г, 47,6%) в виде не совсем белого твердого вещества. Масс-спектр: m/z [M+H]+=368.
Альтернативным является следующий путь получения метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата.
Стадия 1
Дибром (0,185 л, 3614,92 ммоль) при 0°C под N2 добавляли по каплям к перемешиваемой суспензии метил-4-гидроксибензоата (500 г, 3286 ммоль) в DCM (4 л). Смесь оставляли перемешиваться в течение 24 ч при к.т. под N2. Затем добавляли раствор метабисульфита натрия (62,5 г, 329 ммоль) в 2 л воды при поддержании температуры около 15°C, после чего добавляли 500 мл MeOH. Органический слой промывали водой, рассолом, сушили над MgSO4, фильтровали и концентрировали досуха с получением метил-3-бром-4-гидроксибензоата (710 г, 94%) в виде белого твердого вещества. Спектр протонного ЯМР (CDCl3): 3.89 (s, 3H), 5.95 (s, 1Н), 7.05 (d, 1Н), 7.92 (dd, 1Н), 8.19 (d, 1Н).
Стадия 2
К дегазированному раствору метил-3-бром-4-гидроксибензоата (350 г, 1514,87 ммоль) в этаноле (3 л) под азотом добавляли триэтиламин (0,528 л, 3787,17 ммоль), 1-(винилокси)бутан (0,588 л, 4544,60 ммоль), 1,1′-бис(дифенилфосфино)ферроцен (33,1 г, 60,6 ммоль) и диацетоксипалладий (8,50 г, 37,9 ммоль). Смесь нагревали при 70°C в течение ночи. Реакционную смесь охлаждали, фильтровали и фильтрат концентрировали. Полученное твердое вещество солюбилизировали с помощью DCM (2 л) и при перемешивании добавляли 4 н. HCl (1,14 л, 4544 ммоль). Перемешивание продолжали в течение 2 ч, органическую фазу отделяли, сушили над MgSO4, фильтровали и концентрировали с получением твердого вещества, которое перемешивали в эфире (5 л) в течение 2 ч. Твердое вещество отфильтровывали и фильтрат концентрировали досуха с получением метил-3-ацетил-4-гидроксибензоата (240 г, 82%) в виде бежевого порошка. Масс-спектр: m/z [M-H]-=193.
Стадия 3
К перемешиваемому раствору метил-3-ацетил-4-гидроксибензоата (240 г, 1236 ммоль) в DCM (2 л) добавляли пиридин (0,400 л, 4944 ммоль), а затем при 0°C по каплям добавляли дибром (0,070 л, 1360 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем охлаждали до 5°C и по каплям добавляли 4 н. HCl (0,927 л, 3708 ммоль). Органическую фазу отделяли, сушили над MgSO4, фильтровали и концентрировали с получением коричневого твердого вещества, которое перемешивали в смеси эфир/петролейный эфир (1:1, 1 л) в течение 1 ч. Твердое вещество собирали фильтрованием и сушили с получением метил-3-ацетил-5-бром-4-гидроксибензоата (270 г, 80%) в виде бежевого порошка. Масс-спектр: m/z [M+H]+=273.
Стадия 4
К раствору бис(триметилсилил)амида лития (1,41 л, 1406 ммоль) при -65°C под азотом по каплям добавляли метил-3-ацетил-5-бром-4-гидроксибензоат (120 г, 439 ммоль) в THF (1,2 л). Раствор оставляли нагреваться до 0°C и выдерживали при этой температуре в течение 1 ч. Раствор снова охлаждали до -65°C и добавляли морфолин-4-карбонилхлорид (0,055 л, 483 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч, затем охлаждали до -30°C, добавляли DCM (1,5 л) и воду (1 л), после чего по каплям добавляли 6 н. HCl (500 мл), затем 2 н. HCl (300 мл) до pH 7, водный раствор экстрагировали DCM (3X). Объединенные экстракты сушили над MgSO4 и выпаривали. Неочищенный продукт растирали в МТВЕ с получением метил-3-бром-4-гидрокси-5-(3-морфолино-3-оксопропаноил)-бензоата (153 г, 90%) в виде бежевого твердого вещества. Масс-спектр: m/z [M+H]+=388.
Стадия 5
Ангидрид трифторметансульфоновой кислоты (0,755 л, 4487 ммоль) при комнатной температуре под азотом (экзотерма) добавляли к перемешиваемому раствору метил-3-бром-4-гидрокси-5-(3-морфолино-3-оксопропаноил)бензоата (433 г, 1122 ммоль, объединенный материал из нескольких партий), растворенного в 1,2-дихлорэтане (1 л). Полученный раствор перемешивали при 50°C в течение ночи. Смесь частично выпаривали и остаток при 0°C (экзотерма) разбавляли MeOH (1,6 л) и перемешивали в течение 1 ч при KT. Растворитель снова выпаривали и остаток разбавляли DCM, гасили насыщенным водным раствором карбоната натрия и экстрагировали DCM. Объединенные органические фазы промывали рассолом, сушили над MgSO4 и концентрировали с получением неочищенного продукта. Неочищенное вещество растирали под МТВЕ (2х), EtAc (1х) и МТВЕ (1х). Твердое вещество сушили с получением метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (208 г, 50%) в виде бежевого твердого вещества. Масс-спектр: m/z [M+H]+=370.
Такой же путь получения метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата осуществляли в более крупном масштабе следующим образом.
Стадия 1
В сосуд A, продуваемый азотом (10 л/мин), загружали дихлорметан (1060 кг) и метил-4-гидроксибензоат (100 кг). Реакционную смесь охлаждали до -2°C (исходная темп. 3,3°C, конечная темп. -1,7°C, время 1 час 45 мин). Используя давление азота, загружали бром (115 кг) и температуру поддерживали в диапазоне от -2 до +2°C (исходная темп. -1,7°C, конечная темп. -0,1°C, высшая темп. 1,2°C, время 4 ч). Реакционную смесь перемешивали в течение 24 ч, давая ей возможность нагреться вплоть до 20°C (исходная темп. -0,1°C, конечная темп. 18,2°C). Загружали раствор метабисульфита натрия (12,5 кг) в деминерализованной воде (400 кг) (исходная темп. 17,5°C, конечная темп. 11,0°C, высшая темп. 28,2°C, время добавления 3 ч 30 мин). Полученную суспензию фильтровали двумя приблизительно равными порциями. Партию 1 фильтровали и промывали деминерализованной водой (200 кг), а затем гептаном (272 кг), в результате чего получали влажный брикет партии 1 массой 204 кг. Партию 2 фильтровали и промывали деминерализованной водой (213 кг). Осадок на фильтре (162 кг) перезагружали в сосуд A и ресуспендировали в гептане (274 кг). Суспензию затем снова фильтровали, получая влажный брикет партии 2 массой 132 кг. Партию 1 сушили при 50°C в двухконусной ваккумной сушилке с получением сухой массы 67,2 кг.
В сосуд B, продуваемый азотом (10 л/мин), загружали партию 2 метил-3-бром-4-гидроксибензоата (132 кг) и воду (1200 кг). Реакционную смесь нагревали до 15-20°C (исходная температура 11,3°C, конечная температура 15,0°C, время 20 мин) и перемешивали в течение 75 мин. Полученную суспензию фильтровали на фильтре из нержавеющей стали. Полученный влажный брикет (194 кг) снова загружали в сосуд B, продуваемый азотом (10 л/мин), после чего добавляли воду (600 кг). Реакционную смесь нагревали до 15-20°C (исходная температура 11,7°C, конечная температура 15,0°C, время 20 мин) и перемешивали в течение 30 мин. Полученную суспензию фильтровали на фильтре из нержавеющей стали. Полученный влажный брикет (200 кг) сушили в тарельчатой сушилке для супензионных материалов при 50°C с получением сухой массы 50,2 кг. Получен метил-3-бром-4-гидроксибензоат (117,4 кг, 77,3%).
Эту процедуру выполняли четыре раза с небольшими различиями в реакционных условиях и процедурах выделения с получением всего 429,2 кг метил-3-бром-4-гидроксибензоата (выход варьировал от 62,3% до 77,3%).
Стадия 2
В продуваемый азотом (10 л/мин) сосуд A загружали метил-3-бром-4-гидроксибензоат (200 кг) и этанол (1250 кг). Сосуд дважды вакуумировали - заполняли азотом. Загружали триэтиламин (220 кг) с последующей этанольной промывкой (15 кг). Загружали винилбутиловый эфир (266 кг) с последующей этанольной промывкой (15 кг). Загружали 1,1′-бис(дифенилфосфино)ферроцен (18,9 кг), а затем диацетоксипалладий (4,96 кг). Реакционную смесь нагревали до 70°C (исходная температура 17,9°C, конечная температура 70,1°C, время 2 ч 30 мин). Реакционную смесь перемешивали в течение 13 ч. Реакционную смесь охлаждали до 50°C (исходная температура 68,7°C, конечная температура 53,5°C, время 40 мин). Реакционную смесь горячей фильтровали через футерованный стеклом фильтр из мягкой стали с 3-х картриджным фильтром на входе с последующей промывкой этанолом (120 кг). Этанольный растворитель затем отгоняли при максимальной температуре рубашки 50°C (мксимальная темп, основания составляла 50,7°C, время 7 ч 25 мин, дистиллят 1010 кг). При охлаждении до 20-25°C в сосуд A загружали разбавленную соляную кислоту (680 кг, получена из 90 кг 36%-ной HCI и 590 кг воды) (исходная темп.35°C, конечная температура 24,9°C, время 1 час). Реакционную смесь перемешивали в течение 2 ч 30 мин. Полученную суспензию фильтровали на фильтре в 2 загрузки и осадок на фильтре промывали деминерализованной водой (2500 кг). В сосуд A загружали метанол (2150 кг) и сырой осадок с фильтра (326 кг). Реакционную смесь нагревали до 60°C (исходная температура 10,7°C, конечная температура 59,3°C, время 3 ч 45 мин). Реакционную смесь горячей фильтровали из сосуда A в сосуд B посредством горячего пресс-фильтра и 3-х картриджных фильтров на входе в сосуд B, а затем разбавляли метанолом (120 кг). В сосуд B загружали деминерализованную воду (815 кг). Реакционную смесь охлаждали до 20°C и эту реакционную смесь перемешивали в течение 8 ч. Полученную суспензию фильтровали на фильтре и осадок на фильтре промывали смесью метанола (105 кг) и деминерализованной воды (60 кг). Влажный продукт (162 кг) сушили при 45°C с получением 116,5 кг метил-3-ацетил-4-гидроксибензоата (чистота 99,6% согласно ВЭЖХ, выход 69,3%).
Эту процедуру проводили три раза с небольшими различиями в реакционных условиях и процедурах выделения с получением всего 251,4 кг метил-3-ацетил-4-гидроксибензоата (выход варировал от 69,3% до 71,7%).
Стадия 3
В продуваемый азотом сосуд A (10 л/мин) загружали метил-3-ацетил-4-гидроксибензоат (80 кг) и дихлорметан (660 кг). Загружали пиридин (98 кг) с последующей промывкой дихлорметаном (13 кг). Реакционную смесь охлаждали до -2°C (исходная температура 7,6°C, конечная температура -4,0°C, время 2 ч). Загружали бром (73,5 кг), поддерживая температуру между -5 и 0°C (исходная температура -4,0°C, конечная температура -2,5°C, высшая темп. -2,0°C, время 5 ч 35 мин). Реакционную смесь перемешивали в течение 1 часа. Загружали раствор метабисульфита натрия (12,8 кг) в деминерализованной воде (168 кг), поддерживая температуру в диапазоне 0-5°C (исходная температура -7,2°C, конечная темп. -4,0°C, высшая темп. -4,0°C, время добавления 30 мин). Через 30 минут перемешивания слои разделяли и нижний органический слой выгружали (808 кг). В сосуд A загружали дихлорметан (111 кг) и через 30 минут перемешивания слои разделяли и нижний органический слой выгружали (126 кг). Верхнюю водную фазу выгружали в отходы (301 кг). Объединенные органические фазы (867 кг) загружали в сосуд A. Загружали 4 М соляную кислоту (336 кг) и спустя 30 минут перемешивания слои разделяли и нижний органический слой выгружали (856 кг). Загружали дихлорметан (111 кг) и спустя 30 минут перемешивания слои разделяли и нижний органический слой выгружали (106 кг). В сосуд A загружали дихлорметан (111 кг) и через 30 минут перемешивания слои разделяли и нижний органический слой выгружали (113 кг). Верхнюю водную фазу выгружали в отходы (404 кг). Три объединенных органических слоя (1075 кг) загружали в сосуд A. Загружали деминерализованную воду (168 кг) и через 30 минут перемешивания слои разделяли и нижний органический слой выгружали (1050 кг). Верхнюю водную фазу выгружали в отходы (195 кг). Органический слой загружали в сосуд B, сосуд оснащали для перегонки и растворитель отгоняли при атмосферном давлении (высшая темп, основания 47,8°C, дистиллят 500 кг). Загружали метанол (531 кг), сосуд оборудовали для перегонки и растворитель отгоняли при атмосферном давлении с максимальной темп, основания 65°C (высшая темп, основания 65,8°C, высшая темп, паров 62,9°C, дистиллят 685 кг). Реакционную смесь охлаждали до 20-25°C (исходная температура 65,8°C, конечная температура 23°C, время 2 ч). Загружали деминерализованную воду (504 кг) и полученную суспензию фильтровали на фильтр-прессе и осадок на фильтре промывали смесью вода:метанол 2:1 (84 кг). Влажный продукт (129 кг) сушили при 45°C с получением метил-3-ацетил-5-бром-4-гидроксибензоата (сухая масса: 99,7 кг, чистота согласно ВЭЖХ 98%, выход 88,6%).
Эту процедуру проводили три раза с небольшими различиями в реакционных условиях и процедурах выделения с получением всего 313 кг метил-3-ацетил-5-бром-4-гидроксибензоата (выход варьировал от 87,6% до 89,5%).
Стадия 4
В продуваемый азотом сосуд A (10 л/мин) загружали метил-3-ацетил-5-бром-4-гидроксибензоат (75,3 кг) и тетрагидрофуран (640 кг). Смесь перемешивали в течение 15 мин до достижения полного растворения. Раствор (714,5 кг) выгружали в продуваемые азотом барабаны вместе с тетрагидрофурановой промывкой (10 кг). В продуваемый азотом сосуд А (10 л/мин) загружали 20%-ный раствор бис(триметилсилил)амида лития в THF (726 кг) с последующей промывкой THF (10 кг). Содержимое сосуда охлаждали до температуры между -10 и -15°C (исходная температура 12,8°C, конечная температура -15,6°C, время 45 мин). Добавляли раствор метил-3-ацетил-5-бром-4-гидроксибензоата в THF (743 кг), поддерживая температуру между -10 и -15°C (исходная температура -15,6°C, конечная температура -12,9°C, высш. темп. -11,4°C, время 1 час 10 мин) с последующей промывкой тетрагидрофураном (10 кг). Реакционную смесь перемешивали в течение 1 часа 30 мин при температуре от -10 до -15°C (исходная температура -12,9°C, конечная температура -12,1°C). Загружали морфолин-4-карбонилхлорид (45,2 кг), поддерживая температуру от -5 до -10°C (исходная температура -11,7°C, конечная температура -5,6°C, высшая темп. -2,1°C, время 30 мин) с последующей промывкой тетрагидрофураном (5 кг). Реакционную смесь перемешивали в течение 9 ч при температуре от 5 до 10°C. Реакционную смесь перемешивали в течение 2 дополнительных часов при температуре от 5 до 10°C. Загружали деминерализованную воду (375 кг), поддерживая температуру ниже 10°C (исходная температура 7,5°C, конечная температура 6,4°C, высшая темп.6,1°C, время 34 мин). Загружали дихлорметан (700 кг), поддерживая температуру ниже 10°C (исходная температура 6,5°C, конечная температура 10,1°C, высшая темп.10,1°C, время 40 мин). Реакционную смесь переносили из сосуда A в сосуд В и промывали дихлорметаном (48 кг). Величину pH доводили до 0,5-2 посредством добавления 4 М раствора соляной кислоты (459 кг) и меняя ее на 1 М соляную кислоту (32,6 кг) близко к целевому pH (начальное pH 12,07, конечное pH 1,8, исходная температура 6,1°C, конечная температура 6,6°C, высшая темп.10,0°C, время добавления 4 ч). Слои разделяли и нижний водный слой (983 кг) выгружали и затем загружали в сосуд R103 для повторной экстракции. В сосуд C загружали дихлорметан (124 кг) и через 15 мин перемешивания слои разделяли. Нижний органический слой (435 кг) выгружали и затем загружали в сосуд D. Верхний водный слой (618 кг) выгружали в отходы. В течение 2 ч загружали гексан (1105 кг), поддерживая температуру при 5-10°C (исходная температура 6,4°C, конечная температура 5,8°C). Смесь охлаждали до -8°C (исходная температура 5,8°C, конечная температура -7,1°C), при -3°C загружали затравочные кристаллы. Полученную суспензию фильтровали на фильтре из нержавеющей стали двумя партиями, весь влажный осадок с фильтра промывали холодным гексаном (240 кг) (в партии 1 влажный продукт составлял 120 Кг, и в партии 2 влажный продукт составлял 312 Кг). Объединенные влажные продукты (432 кг) сушили в суспензионной вакуумной сушилке при 50°C в течение 9 суток (сутки 1 и 2: сушка под вакуумом; сутки 3 и 4: нагрев и отключение вакуума и выдерживание под азотом; сутки 5-7: сушка под вакуумом; сутки 7: продукт дробили посредством перемалывания и выгружали в сушилку) с получением метил-3-бром-4-гидрокси-5-[3-(морфолин-4-ил)-3-оксопропаноил]бензоата (сухая масса составляла 79,84 кг, чистота согласно ВЭЖХ 93,4%, выход 75%).
Эту процедуру повторяли пять раз в небольшими различиями в условиях реакций и процедурах выделения с получением 308,2 кг метил-3-бром-4-гидрокси-5-[3-(морфолин-4-ил)-3-оксопропаноил]бензоата (выход варьировал от 66% до 75%).
Стадия 5
В продуваемый азотом сосуд А (10 л/мин) загружали метил-3-бром-4-гидрокси-5-[3-(морфолин-4-ил)-3-оксопропаноил]бензоат (70 кг) и хлорбензол (770 кг). Загружали ангидрид трифторметансульфоновой кислоты (205 кг), поддерживая температуру ниже 25°C (исходная температура 11,7°C, конечная температура 19,8°C, высшая темп. 19,8°C, время 30 мин) с последующей промывкой хлорбензолом (6 кг). Реакционную смесь нагревали вплоть до 70°C (исходная температура 19,8°C, конечная температура 71,0°C, время 2 ч). Реакционную смесь перемешивали в течение 6 ч при 70°C. Реакционную смесь охлаждали до 5°C (исходная температура 71,0°C, конечная температура 5°C, время 3 ч). Загружали метанол (250 кг), поддерживая температуру ниже 15°C (исходная температура 5°C, конечная температура 13,9°C, высшая темп.14,1°C, время 2 ч 10 мин). Реакционную смесь перемешивали в течение 20 ч при 20-25°C.Загружали 20%-ный раствор карбоната натрия (403 кг) до pH 7,5, поддерживая температуру ниже 25°C (исходное pH менее 1, конечное pH 7,55, исходная температура 12,0°C, конечная температура 19,3°C, высшая темп.20,6°C, время добавления 2 часа 5 мин). Загружали дихлорметан (371 кг). Реакционную смесь перемешивали в течение 5 ч. Загружали деминерализованную воду (280 кг) и после перемешивания в течение 1 часа слои разделяли. Нижний органический слой (1259 кг) выгружали. В сосуд A загружали дихлорметан (371 кг). После перемешивания в течение 30 мин слои разделяли. Нижний органический слой (394 кг) выгружали и объединяли с предыдущими органическими слоями. Верхний водный слой (1031 кг) выгружали в отходы. В сосуд A загружали объединенные органические слои (1689 кг), а затем деминерализованную воду (280 кг). После перемешивания в течение 30 мин слои разделяли. Нижний органический слой (1538 кг) выгружали. Верхний водный слой (390 кг) выгружали в отходы. Органические слои (1538 Кг) снова загружали в сосуд A, сосуд оснащали для перегонки и растворитель отгоняли до 100 мбар и при 50°C до тех пор, пока содержание DCM в реакционной смеси не становилось ниже 3,5%. Полученную суспензию охлаждали до 0-5°C (исходная температура 54,4°C, конечная температура 5,0°C), перемешивали в течение 1 часа и затем фильтровали на фильтре из нержавеющей стали. Осадок на фильтре промывали этилацетатом (2×63 кг) и влажный осадок (38,2 кг) сушили в вакуумной печи при 50°C с получением метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (сухая масса равна 34,5 кг, чистота согласно ВЭЖХ 98,1%, выход 51,7%).
Эту процедуру повторяли несколько раз с небольшими различиями в реакционных условиях и процедурах выделения с получением всего 112,6 кг метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата.
Пример 1.01
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-N-(2-гидроксиэтил)-N-метил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
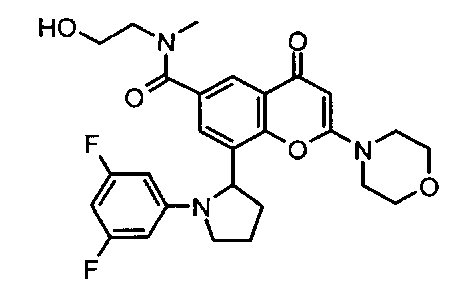
TBTU (102 мг, 0,32 ммоль) добавляли к перемешиваемому раствору 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (105 мг, 0,21 ммоль), 4-метилморфолина (0,070 мл, 0,63 ммоль) и 2-(метиламино)этанола (0,022 мл, 0,28 ммоль), растворенных в NMP (1,5 мл). Полученный раствор перемешивали при комнатной температуре в течение 16 ч, затем очищали препаративной ВЭЖХ. Фракции выпаривали досуха и растирали в диэтиловом эфире с получением твердого вещества. Этот материал собирали фильтрованием, промывали диэтиловым эфиром и сушили под вакуумом при 50°C с получением 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-N-(2-гидроксиэтил)-N-метил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (77 мг, 71%) в виде белого твердого вещества.
Масс-спектр: m/z [M+H]+=514.
Спектр протонного ЯМР: (DMSO-d6) 1.77-1.92 (m, 1Н), 1.97-2.09 (m, 2Н), 2.51-2.60 (m частично скрыт DMSO-d5, 1Н), 2.78 (s, 1.2Н), 2.93 (s, 1.8Н), 3.00-3.09 (m, 0.8Н), 3.13-3.23 (m, 0.8H), 3.33-3.49 (m частично скрыт H2O, 2.4Н), 3.50-3.66 (m, 5Н), 3.70-3.81 (m, 5Н), 4.77 (t, 0.6Н), 4.78 (t, 0.4H), 5.25 (d, 1H), 5.62 (s, 1H), 6.06-6.18 (m, 2H), 6.32 (t, 1H), 7.13 (s, 0.4H), 7.23 (s, 0.6H), 7.80 (s, 0.6H), 7.84 (s, 0.4H).
Пример 1.02
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(4-гидроксипиперидин-1-карбонил)-2-морфолино-4Н-хромен-4-он
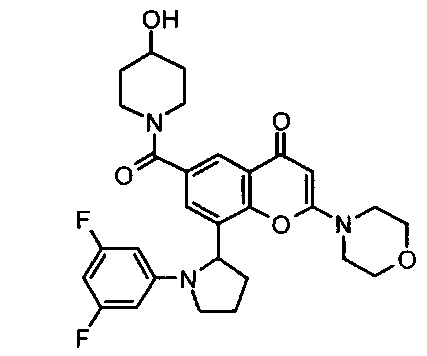
Указанное в заголовке соединение получали, используя процедуру, аналогичную описанной в Примере 1.01. 8-(1-(3,5-Дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновую кислоту (105 мг, 0,21 ммоль) подвергали взаимодействию с пиперидин-4-олом (22,48 мг, 0,22 ммоль) с получением 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(4-гидроксипиперидин-1-карбонил)-2-морфолино-4Н-хромен-4-она (75 мг, 66%) в виде белого твердого вещества.
Масс-спектр: m/z [M+H]+=540.
Спектр протонного ЯМР: (DMSO-d6) 1.02 (bs, 0.5Н), 1.19-1.50 (m, 2.5Н), 1.74 (bs, 1H), 1.79-1.89 (m, 1H), 1.99-2.13 (m, 2H), 2.52-2.61 (m частично скрыт DMSO-d5, 1Н), 2.89 (bs, 1Н), 2.94 (bs, 0.5Н), 3.26 (bs, 1H), 3.31-3.43 (m частично скрыт Н20, 1.5Н), 3.50-3.66 (m, 4Н), 3.67 (bs, 1Н), 3.71-3.86 (m, 5.5Н), 4.00 (bs, 0.5Н), 4.75 (d, 1H), 5.22-5.30 (m, 1H), 5.63 (s, 1H), 6.14 (d, 2H), 6.34 (t, 1H), 4.07 (d, 1H), 7.80 (d, 1H).
Пример 1.03
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он
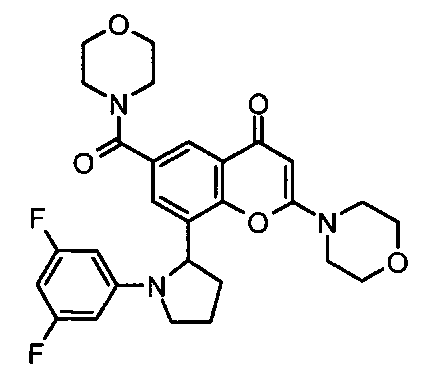
Гидрохлорид N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (71,4 мг, 0,37 ммоль) при комнатной температуре под азотом порциями добавляли к перемешиваемому раствору 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (85 мг, 0,19 ммоль), 2-гидроксипиридин-1-оксида (41,4 мг, 0,37 ммоль) и морфолина (0,033 мл, 0,37 ммоль) и перемешивали в течение 4 ч. Реакционную смесь разбавляли DCM, промывали водой, рассолом, сушили над MgSO4 и концентрировали. Реакционную смесь очищали препаративной ВЭЖХ. Фракции выпаривали досуха и остаток растворяли в DCM, сушили над MgSO4 и концентрировали. Оставшееся твердое вещество растирали с диэтиловым эфиром, фильтровали, промывали диэтиловым эфиром и сушили под вакуумом при 50°C с получением 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (58,0 мг, 59,3%) в виде бледно-бежевого твердого вещества.
Масс-спектр: m/z [M+H]+=526.
Спектр протонного ЯМР: (DMSO-d6) 1.74-1.89 (m, 1Н), 1.98-2.09 (m, 2Н), 2.50-2.57 (m частично скрыт DMSO-d5, 1Н), 3.07 (bs, 3H), 3.24 (bs, 1Н), 3.74-3.41 (m, 2Н), 3.45 (bs, 2Н), 3.49-3.67 (m, 5Н), 3.70-3.81 (m, 5Н), 5.22-5.29 (m, 1Н), 5.62 (s, 1Н), 6.13 (d, 2Н), 6.34 (t, 1Н), 7.07 (d, 1Н), 7.82 (d, 1Н).
Пример 1.03а
8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он
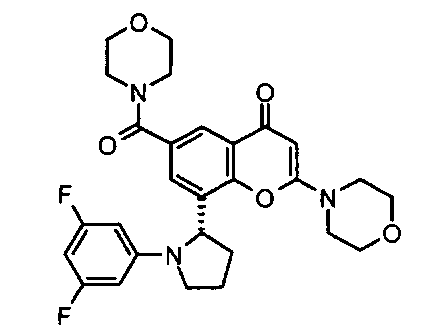
TSTU (75,0 мг, 0,23 ммоль) одной порцией при комнатной температуре добавляли к перемешиваемому раствору 8-[(2S)-(1-(3,5-дифторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (102 мг, 0,21 ммоль, энантиомерная чистота более 98%, получена из первого элюируемого энантиомера сложного эфира из хирального разделения трет-бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-пирролидин-1-карбоксилата - смотри ниже) и DIPEA (0,041 мл, 0,23 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1,5 ч. Затем к реакционной смеси добавляли морфолин (0,037 мл, 0,42 ммоль) и перемешивание продолжали в течение выходных. Реакционную смесь очищали препаративной ВЭЖХ. Фракции выпаривали досуха, остаток растворяли в DCM, сушили над MgSO4 и концентрировали. Оставшееся твердое вещество растирали с диэтиловым эфиром, фильтровали, промывали диэтиловым эфиром и сушили под вакуумом при 50°C с получением 8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (64 мг, 57%) в виде бледно-желтого твердого вещества.
Масс-спектр: m/z [M+H]+=526.
Оптическое вращение: 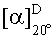
Спектр протонного ЯМР (CDCl3) 1.96-2.17 (m, 3H), 2.45-2.57 (m, 1Н), 3.07-3.83 (m, 14Н), 3.83-3.95 (m, 4Н), 5.07 (d, 1Н), 5.58 (s, 1Н), 5.92 (dd, 2Н), 6.11 (ddt, 1Н), 7.24 (d, 1Н), 8.15 (s, 1Н).
Пример 1.03b
8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он

TSTU (106 мг, 0,33 ммоль) при комнатной температуре добавляли одной порцией к перемешиваемому раствору 8-[(2R)-(1-(3,5-дифторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (144 мг, 0,30 ммоль, энантиомерная чистота более 98%, получена из второго энантиомера элюированного сложного эфира из хирального разделения трет-бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата - смотри ниже) и DIPEA (0,057 мл, 0,33 ммоль) и полученную смесь перемешивали в течение 1,5 ч. Затем к реакционной смеси добавляли морфолин (0,052 мл, 0,60 ммоль) и перемешивание продолжали в течение выходных. Реакционную смесь очищали препаративной ВЭЖХ. Фракции выпаривали досуха, остаток растворяли в DCM, сушили над MgSO4 и концентрировали. Оставшееся твердое вещество растирали с диэтиловым эфиром, фильтровали, промывали диэтиловым эфиром и сушили под вакуумом при 50°C с получением 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она (69 мг, 44%) в виде бледно-желтого твердого вещества.
Масс-спектр: m/z [M+H]+=526.
Оптическое вращение: 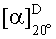
Спектр протонного ЯМР (CDCl3) 1.96-2.17 (m, 3H), 2.45-2.57 (m, 1Н), 3.07-3.83 (m, 14Н), 3.83-3.95 (m, 4Н), 5.07 (d, 1Н), 5.58 (s, 1Н), 5.92 (dd, 2Н), 6.11 (ddt, 1Н), 7.24 (d, 1Н), 8.15 (s, 1Н).
(R) и (S)-Энантиомеры 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты, используемой в качестве исходного материала, получали следующим образом.
Стадия 1
Условия хиральной препаративной ВЭЖХ:
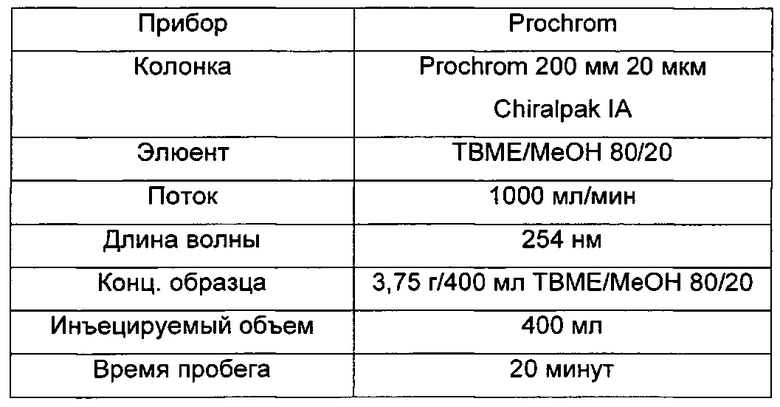
15 г трет-Бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (полученного, как описано в Примере 1.00 выше) разделяли в 5 инъекциях посредством хиральной ВЭЖХ. Первый элюируемый энантиомер: 6,32 г, показатель = 91%, αD = -72,7°; второй элюируемый энантиомер: 6,94 г, показатель = 89%, αD = +69,4°. Энантиомерная чистота более 98% для каждого энантиомера.
Условия альтернативной хиральной препаративной ВЭЖХ:
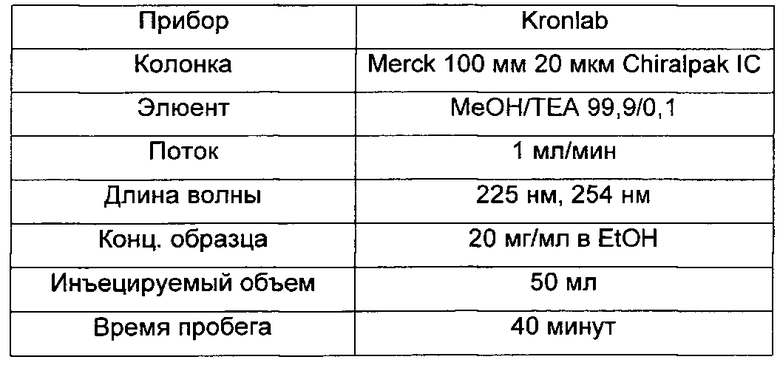
6,1 г трет-Бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата разделяли в 5 инъекциях. Первый элюируемый энантиомер: 1,88 г; второй элюируемый энантиомер: 1,98 г. Энантиомерная чистота более 98% для каждого энантиомера.
Абсолютную стереохимию двух энантиомеров трет-бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата, полученных после хиральной ВЭЖХ, устанавливали в соответствии со следующей процедурой.
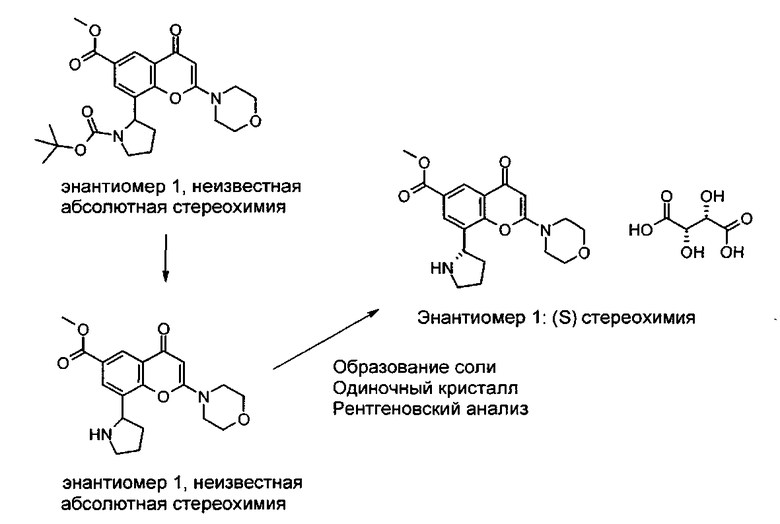
Метил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилат, энантиомер 1, получали из трет-бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата, энантиомера 1, посредством кислотного снятия защиты с помощью хлористого водорода (смотри стадию 1 получения 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она (то есть Пример 1.03b) ниже для аналогичной процедуры). Полученный метил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилат, энантиомер 1, затем смешивали с D-(S,S)-винной кислотой (0,5 экв.) с получением метил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилата соли (2S,3S)-2,3-дигидроксисукцината, которую подвергали кристаллизации из смеси метанол/изопропанол, используя метод паровой диффузии. Небольшое количество образца растворяли в метаноле во флаконе и этот флакон помещали внутрь большого флакона, содержавшего большой объем изопропанольного антирастворителя. Диффузия изопропанольного антирастворителя в метанольный раствор соли с течением времени вызывала кристаллизацию, и абсолютную конфигурацию полученного кристалла определяли посредством дифракции рентгеновских лучей на одиночном кристалле. В соответствии с правилами последовательности Кахна-Ингольда-Прелога (Cahn-lngold-Prelog), хиральный углеродный атом метил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилата, соли (2S,3S)-2,3-дигидроксисукцината, определяли как имеющий (S)-конфигурацию. Как результат, энантиомер 2 трет-бутил-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата имеет (R)-конфигурацию.
Следующая процедура описывает получение 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она (то есть Примера 1.03b) из второго элюируемого энантиомера сложного эфира выше (то есть трет-бутил-(2R)-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата). Аналогичную процедуру использовали для синтеза 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она (то есть Примера 1.03а) из первого элюируемого энантиомера сложного эфира выше на стадии 1 (то есть Апрет-бутил-(2S)-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата).
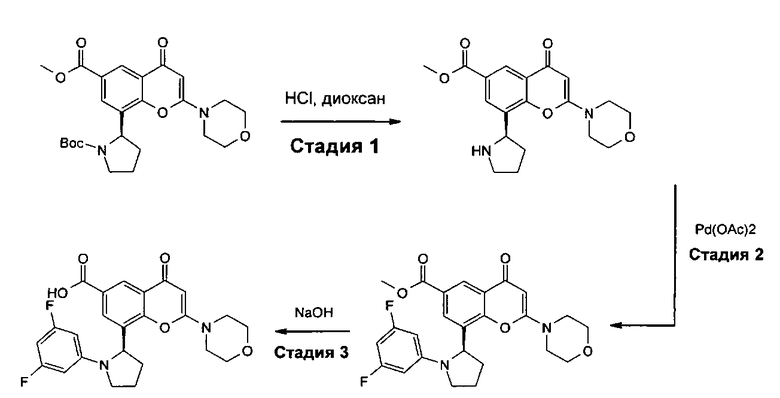
Стадия 1
Хлористый водород (4 M в диоксане) (8,81 мл, 35,22 ммоль) добавляли к перемешиваемому раствору трет-бутил-(2R)-2-(6-(метоксикарбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (1,90 г, 3,52 ммоль), растворенного в DCM (15 мл), и реакционную смесь перемешивали в течение 8 ч при комнатной температуре. После концентрирования добавляли 10%-ный метанольный аммиак (7 н.) в DCM, реакционную смесь адсорбировали на силикагеле и затем очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 8% метанольного аммиака (7 н.) в DCM. Растворители выпаривали досуха с получением после растирания с диэтиловым эфиром метил-2-морфолино-4-оксо-8-[(2R)-пирролидин-2-ил]хромен-6-карбоксилата (1,11 г, 88%) в виде светло-оранжевого кристаллического твердого вещества.
Масс-спектр: m/z [M+H]+=359.
Стадия 2
Диацетоксипалладий (0,028 г, 0,13 ммоль) добавляли к перемешиваемой смеси метил-2-морфолино-4-оксо-8-[(2R)-пирролидин-2-ил]хромен-6-карбоксилата (0,9 г, 2,51 ммоль), 1-бром-3,5-дифторбензола (0,361 мл, 3,14 ммоль), (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфина) (145 мг, 0,25 ммоль) и карбоната цезия (1,227 г, 3,77 ммоль), растворенных в 1,4-диоксане (16 мл). Полученную суспензию дегазировали аргоном и затем перемешивали при 100°C в течение 15 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры, концентрировали в присутствии силикагеля и очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 10% MeOH в EtAc. Растворитель выпаривали досуха с получением метил-8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-4-оксо-хромен-6-карбоксилата (0,792 г, 67%) в виде желтой пены. Масс-спектр: m/z [M+H]+=471. Стадия 3
Водный 2 н. раствор NaOH (2,55 мл, 5,10 ммоль) добавляли к метил-8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-4-оксо-хромен-6-карбоксилату (0,79 г, 1,68 ммоль) в смеси MeOH (9 мл) и DCM (6 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь охлаждали до 0°C и по каплям к реакционной смеси добавляли водный 2 н. раствор HCl (2,5 мл, 5,0 ммоль) до pH приблизительно 3. После разбавления водой (9 мл) реакционную смесь концентрировали до половины объема. Водную фазу экстрагировали DCM, органическую фазу концентрировали досуха с получением пены, которую растирали в EtAc, собирали фильтрованием, промывали EtAc, диэтиловым эфиром, сушили под вакуумом при 50°C с получением 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-4-оксо-хромен-6-карбоновой кислоты (0,638 г, 83%) в виде не совсем белого твердого вещества. Масс-спектр: m/z [M+H]+=457.
Соединения из примеров 1.03а и 1.03b также синтезировали в крупном масштабе в соответствии со следующей процедурой.
Стадия 1
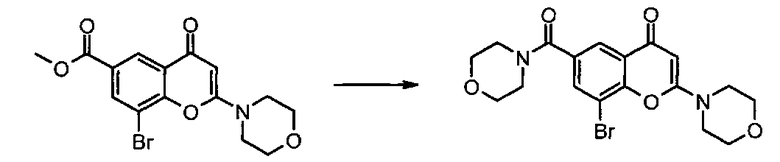
КОН (0,419 л, 4889 ммоль) при 20°C в течение периода времени 1 час добавляли порциями к метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилату (900 г, 2444 ммоль) в воде (5 л). Полученную суспензию перемешивали при 20°C в течение 4 часов до тех пор, пока не завершалось омыление. Реакционную смесь фильтровали для удаления частиц нерастворимых веществ и переносили в сосуд, содержащий воду (2 л) (сосуд 1). Добавляли морфолин (0,639 л, 7333 ммоль) и продолжали перемешивание. 2-Хлор-4,6-диметокси-1,3,5-триазин (1931 г, 11000 ммоль) и воду (8 л) загружали в сосуд 2 и температуру доводили до приблизительно 7°C, загружали 4-метилморфолин (134 мл, 1222 ммоль) с такой скоростью, чтобы поддерживать содержимое при 10°C, и содержимое перемешивали в течение 4 часов. Содержимое сосуда 2 переносили в сосуд 1 и перемешивали при комнатной температуре в течение ночи. Затем добавляли DCM (5 л) и смесь переносили в 30-литровый сепаратор, экстрагировали дополнительной порцией DCM и органические экстракты объединяли, сушили (Na2SO4) и выпаривали досуха. Твердое вещество перемешивали в этилацетате и фильтровали с получением 8-бром-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (520 г, 50,3%) в виде слегка серого твердого вещества. Масс-спектр: m/z [M+H]+=425.
Стадия 2
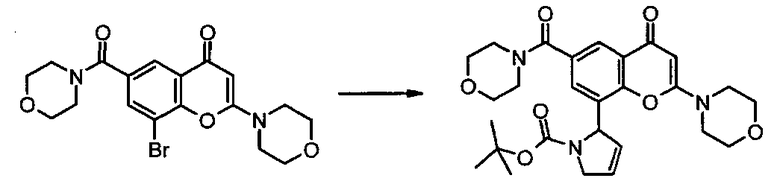
DMF (4 л) дегазировали посредством барботирования потоком азота в течение 15 минут. В 10-литровый сосуд с рубашкой загружали порцию DMF (600 мл), затем 8-бром-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (440 г, 1040 ммоль), трет-бутил-2,3-дигидро-1Н-пиррол-1-карбоксилата (246 г, 1455 ммоль), трифенилфосфина (27,3 г, 104 ммоль) и карбоната калия (431 г, 3119 ммоль). Добавляли оставшийся DMF (3 л), а затем диацетоксипалладий (11,67 г, 52 ммоль). Полученную суспензию перемешивали под азотом и нагревали при 100°C в течение 16 часов. Смесь охлаждали до комнатной температуры и разбавляли DCM (4 л). Смесь фильтровали через целит, фильтрат и промывки вливали в воду (38 л) и добавляли дополнительное количество DCM (2,5 л). Органический слой отделяли и водную фазу экстрагировали дополнительным количеством DCM (2,5 л). Органические фазы сушили с помощью сульфата магния, фильтровали и концентрировали с получением коричневой смолы. Смолу очищали посредством флэш-колоночной хроматографии на диоксиде кремния, элюируя DCM, затем 0-10% MeOH/DCM. Предфракции, содержавшие Ph3P, содержали значительное количество продукта. Их концентрировали с получением неочищенного материала. Чистые фракции давали 442 г чистого продукта. Предфракции повторно очищали как ранее (картридж 750 г диоксида кремния) с получением дополнительно 24 г чистого продукта. Обе партии объединяли с получением трет-бутил-2-(6-(морфолин-4-карбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-2,5-дигидро-1Н-пиррол-1-карбоксилата (466 г, 88%) в виде желтого твердого вещества. Масс-спектр: m/z [M+H]+=512.
Стадия 3
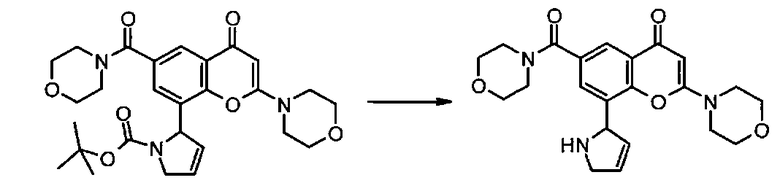
Трифторуксусную кислоту (1,6 л) добавляли к раствору трет-бутил-2-(6-(морфолин-4-карбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-2,5-дигидро-1Н-пиррол-1-карбоксилата (444 г, 867,92 ммоль) в DCM (3 л). Смесь перемешивали в течение 16 часов при комнатной температуре. Раствор концентрировали при пониженном давлении. Остаток разбавляли DCM (2,5 л) и добавляли к интенсивно перемешиваемой смеси DCM (1 л) и конц. водного аммиака (4 л). Водный слой промывали дополнительным количеством DCM (2 л). Объединенный органический раствор сушили с помощью сульфата магния, фильтровали и концентрировали при пониженном давлении с получением оранжевой сухой пленки, которую использовали на следующей стадии без дополнительной очистки.
Стадия 4
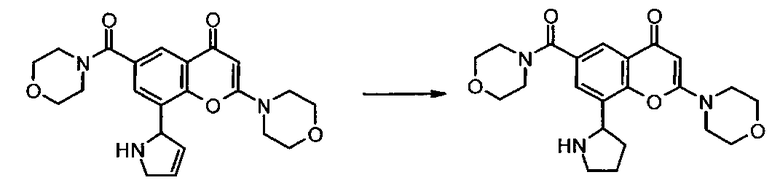
8-(2,5-Дигидро-1Н-пиррол-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он (353 г, 858 ммоль) и палладий на углероде 5% JM Type 87L (70 г, 16 ммоль) в MeOH (3500 мл) перемешивали в атмосфере водорода при 5 бар (50 кПа) и 45°C в течение 3 часов. Реакционную смесь фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали посредством флэш-колоночной хроматографии на диоксиде кремния, используя градиент элюирования (от 1% метанол/DCM до 20% метанол/DCM, содержавшие 1% конц. водного аммиака). Целевой продукт, 6-(морфолин-4-карбонил)-2-морфолино-8-(пирролидин-2-ил)-4Н-хромен-4-он (187 г, 53%), в результате выделяли в виде бесцветной сухой пленки. Большее количество материала получали повторением этого взаимодействия.
Стадия 5
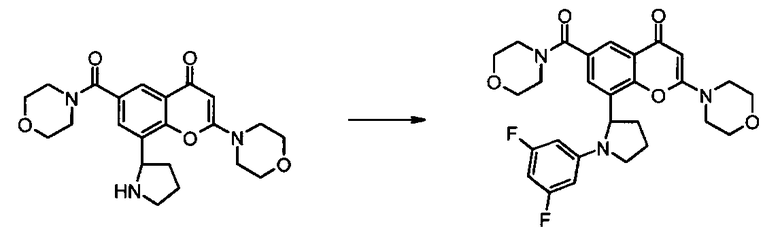
Через 6-(морфолин-4-карбонил)-2-морфолино-8-(пирролидин-2-ил)-4Н-хромен-4-он (296 г, 716 ммоль), 1-бром-3,5-дифторбензол (173 г, 895 ммоль) и карбонат цезия (700 г, 2148 ммоль), суспендированные в диоксане (3 л), барботировали азот в течение 10 минут. Добавляли диацетоксипалладий (8,04 г, 36 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфин) (41,4 г, 72 ммоль) и смесь барботировали азотом в течение 2 минут, затем нагревали при 100°C в течение 2 часов. После охлаждения до комнатной температуры смесь распределяли между DCM (500 мл) и водой (250 мл). Органическую фазу промывали рассолом и сушили с помощью сульфата магния, фильтровали и концентрировали при пониженном давлении с получением коричневой смолы. Смолу очищали посредством флэш-колоночной хроматографии на диоксиде кремния, используя градиент элюирования (от 0% метанол/DCM до 5% метанол/DCM). Целевой продукт, 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он (282 г, 75%), в результате выделяли в виде бледно-желтой сухой пленки. Масс-спектр: m/z [M+H]+=526. Большее количество материала получали повторением этого взаимодействия.
Стадия 6
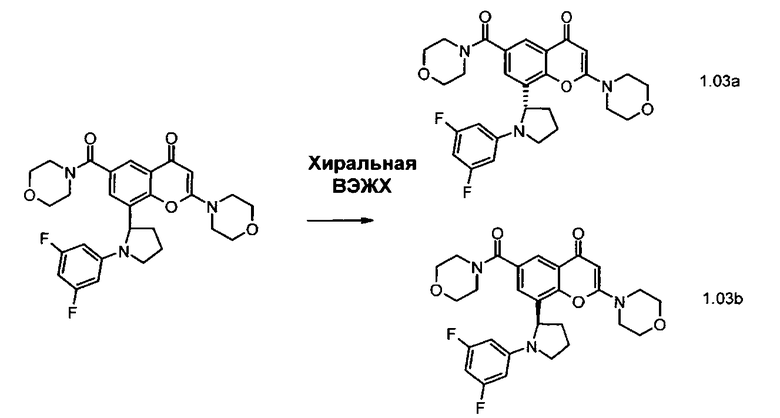
Каждый энантиомер выделяли посредством препаративной ВЭЖХ следующим образом.
Раствор рацемического 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (325 г) растворяли в 50% об./об. смеси метанола в этаноле. Аликвоты по 800 мл этой смеси, каждая содержавшая 12 г рацемата, инъецировали последовательно в колонку Prochrom 200 мм 20 мкм Chiralpak AD (5 кг CSP), используя ВЭЖХ прибор Prochrom. Энантиомеры элюировали при комнатной температуре, используя элюент, содержавший 25% гептан:37,5% метанол:37,5% этанол, со скоростью потока 1,2 л·мин-1 и временем прогона 25 минут. Первый элюированный энантиомер имел время удерживания 6,5 мин, а второй энантиомер начинал элюироваться после 10 минут. Первый элюируемый энантиомер, 143 г, 272 ммоль, 50,7%, получали в виде твердого вещества кремового цвета, а второй элюируемый энантиомер (59 г) выделяли в виде коричневой пленки.
Аналитические условия пост-хиральной очистки.
Колонка Chiralpak ID (внутренний диаметр) 4,6×250 мм 5 мкм. Элюировали смесью EtOH:MeOH (50:50) при 1 мл/мин. Время пробега 30 минут при температуре 25°C.
Показатели времени удерживания для энантиомера, которого элюировали первым из разделения на стадии 6 выше, составляло 17,73 минуты. Это сравнимо с аутентичным образцом из Примера 1.03b, полученным в соответствии с описанным выше способом, и идентифицировали его как энантиомер из Примера 1.03b, то есть 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он. Время удерживания второго элюируемого энантиомера составляло 13,52 минут, идентифицируя его как энантиомер из Примера 1.03а, то есть 8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он.
Хиральная хроматография установила уровень противоположного энантиомера в первом элюируемом энантиомере со стадии 6 выше (то есть энантиомера из Примера 1.03b, 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-она) как приблизительно 0,7%, что давало энантиомерную чистоту 99,3% и энантиомерный избыток 98,6%.
Кристаллическую форму (Форма А) соединения из Примера 1.03b также получали согласно следующему методу.
8-[(2R)-1-(3,5-Дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он (9 г) суспендировали в эфире (180 мл) в течение 72 часов. Полученный не совсем белый порошок выделяли фильтрованием и сушили. Его затем анализировали посредством дифракции рентгеновских лучей на порошке, и было показано, что он является кристаллическим (Фиг.1), имея следующие значения 29, измеренные с использованием CuKα излучения:
Другую кристаллическую форму (Форма В) соединения из Примера 1.ОЗЬ также получали согласно следующему методу.
8-[(2R)-1-(3,5-Дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он (1,77 г) суспендировали в эфире (40 мл) в течение 12 часов. Полученный не совсем белый порошок (720 мг) выделяли фильтрованием и сушили. Его анализировали посредством дифракции рентгеновских лучей на порошке (Фиг.2), и было показано, что он представляет собой другую кристаллическую форму, нежели Форма A, со следующими значениями 20, измеренными с использованием CuKα излучения:
Также выполняли ДСК анализ Формы B (Фиг.3), который показал точку плавления 180,2°C (начало).
Другую кристаллическую форму (Форма С) соединения из Примера 1.03b также получали посредством суспендирования материала Формы B в метаноле. Приблизительно 20 мг материала Формы B помещали во флакон с магнитной мешалкой и добавляли приблизительно 2 мл метанола. Флакон затем плотно закрывали крышкой и оставляли для перемешивания на столе магнитной мешалки. Через 3 суток образец удаляли со стола, крышку снимали и суспензию оставляли сушиться в условиях окружающей среды перед анализом посредством дифракции рентгеновских лучей на порошке (Фиг.4). Посредством дифракции рентгеновских лучей на порошке было определено, что эта форма (Форма C) является кристаллической и отлична от ранее наблюдавшихся форм со следующими значениями 20, измеренными с помощью CuKα излучения:
Пример 1.04
8-[1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-6-[(1-оксидотиоморфолин-4-ил)карбонил]-4Н-хромен-4-он
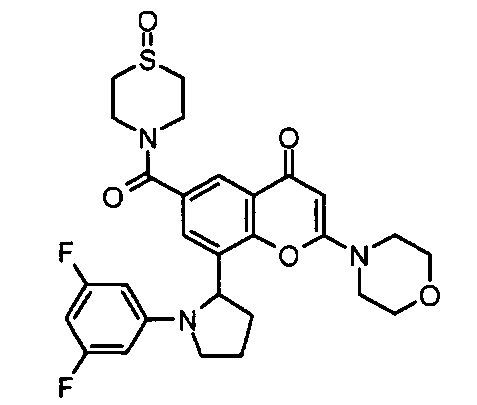
TBTU (176 мг, 0,55 ммоль) при комнатной температуре добавляли одной порцией к перемешиваемому раствору 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (125 мг, 0,27 ммоль) и DIPEA (0,286 мл, 1,64 ммоль) и перемешивали в течение 2,5 ч. К реакционной смеси добавляли гидрохлорид тиоморфолин-1-оксида (85 мг, 0,55 ммоль) и перемешивание продолжали в течение 3 ч. Реакционную смесь разбавляли DCM, промывали водой, рассолом, сушили над MgSO4 и концентрировали. Остаток очищали препаративной ВЭЖХ. Фракции выпаривали досуха, остаток растворяли в DCM, сушили над MgSO4 и концентрировали. Оставшееся твердое вещество растирали с диэтиловым эфиром, фильтровали, промывали диэтиловым эфиром и сушили под вакуумом при 50°C с получением 8-[1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-6-(1-оксо-1,4-тиазинан-4-карбонил)хромен-4-она (87 мг, 57%) в виде бледно-бежевого твердого вещества.
Масс-спектр: m/z [M+H]+=558.
Спектр протонного ЯМР: (DMSO-d6) 1.75-1.86 (m, 1Н), 1.98-2.08 (m, 2Н), 2.14 (bs, 0.5Н), 2.51-2.58 (m частично скрыт DMSO-d5, 1Н), 2.64 (bs, 1Н), 2.81 (bs, 1.5Н), 2.98 (bs, 1H), 3.27 (bs, 1H), 3.40 (bs, 1H), 3.49-3.65 (m, 5H), 3.64-3.83 (m, 6H), 4.21 (bs, 0.5H), 4.34 (bs, 0.5H), 5.25 (d, 1H), 5.62 (s, 1H), 6.13 (d, 2H), 6.34 (t, 1H), 7.18 (s, 1H), 7.87 (d, 1H).
Пример 1.05
6-(азетидин-1-карбонил)-8-[(2R)-(1-(3,5-дифторфенил)пирролидин-2-ил)]-2-морфолино-4Н-хромен-4-он
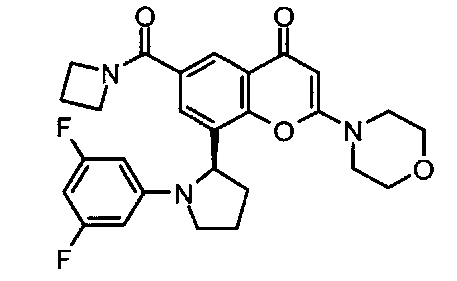
TBTU (211 мг, 0,66 ммоль) под азотом добавляли к перемешиваемому раствору 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-4-оксо-хромен-6-карбоновой кислоты (150 мг, 0,33 ммоль, энантиомерная чистота более 98%, смотри Пример 1.03b в отношении особенностей получения) и DIPEA (0,229 мл, 1,31 ммоль), растворенных в CHCl3 (2 мл). Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Добавляли гидрохлорид азетидина (61,5 мг, 0,66 ммоль) и реакционную смесь перемешивали при 50°C в течение 2-3 ч. Раствор охлаждали до комнатной температуры, гасили 10%-ным раствором водной лимонной кислоты и экстрагировали дихлорметаном. Объединенные органические фазы промывали насыщенным водным раствором гидрокарбоната натрия, рассолом, сушили над MgSO4 и концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 10% метанола в смеси EtAc/DCM (1/1). Растворитель выпаривали досуха, сухую пленку растирали в диэтиловом эфире и осадок собирали фильтрованием, промывали диэтиловым эфиром и сушили до постоянной массы с получением 6-(азетидин-1-карбонил)-8-[(2R)-(1-(3,5-дифторфенил)пирролидин-2-ил)]-2-морфолино-4Н-хромен-4-она (126 мг, 0,254 ммоль, 77%) в виде твердого вещества.
Масс-спектр: m/z [M+H]+=496.
Спектр протонного ЯМР: (DMSO-d6) 1.74-1.88 (m, 1Н), 1.97-2.08 (m, 2Н), 2.13-2.24 (m, 2Н), 2.43-2.51 (m частично скрыт DMSO-d5, 1Н), 3.33-3.39 (m частично скрыт H2O, 1Н), 3.49-3.66 (m, 4Н), 3.70-3.81 (m, 5Н), 3.93-4.09 (m, 4Н), 5.25 (d, 1Н), 5.62 (s, 1Н), 6.15 (d, 2Н), 6.33 (t, 1Н), 7.30 (d, 1Н), 8.04 (d, 1Н).
Пример 1.06
8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
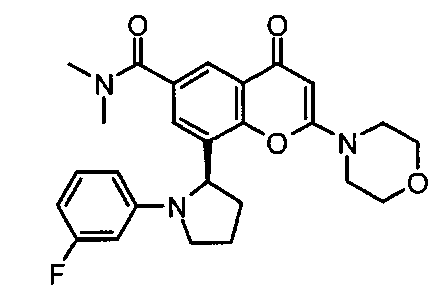
TSTU (429 мг, 1,34 ммоль) при комнатной температуре под азотом добавляли одной порцией к перемешиваемой суспензии 8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-2-морфолино-4-оксо-хромен-6-карбоновой кислоты (488 мг, 1,11 ммоль, энантиомерная чистота более 98%), гидрохлорида диметиламина (127 мг, 1,56 ммоль) и DIPEA (0,582 мл, 3,34 ммоль), растворенных в DCM (5 мл). Полученную суспензию перемешивали при комнатной температуре в течение 20 мин. Реакционную смесь гасили водой и экстрагировали DCM (1×40 мл). Органическую фазу сушили над MgSO4 и концентрировали с получением неочищенного продукта в виде желтой пены. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 10% MeOH в EtAc. Растворитель выпаривали досуха, пену растирали в диэтиловом эфире (10 мл), полученное твердое вещество отфильтровывали и сушили при пониженном давлении с получением 8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (276 мг, 53%) в виде желтого твердого вещества.
Масс-спектр: m/z [M+H]+=466.
Спектр протонного ЯМР: (DMSO-d6) 1.78-1.92 (m, 1Н), 1.96-2.09 (m, 2Н), 2.46-2.56 (m частично скрыт DMSO-d5, 1Н), 2.68 (s, 3H), 2.90 (s, 3H), 3.33-3.42 (m частично скрыт H2O, 1Н), 3.50-3.66 (m, 4Н), 3.70-3.81 (m, 5Н), 5.23 (d, 1Н), 5.61 (s, 1Н), 6.21-6.29 (m, 2Н), 6.36 (ddd, 1Н), 7.09 (ddd, 1Н), 7.14 (d, 1Н), 7.80 (d, 1Н).
8-[(2R)-(1-(3-Фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновую кислоту, использовавшуюся в качестве исходного материала, получали следующим образом.
Стадия 1
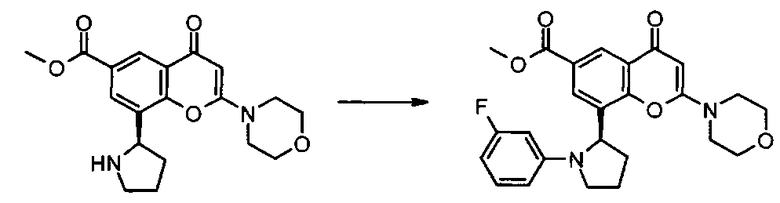
Диацетоксипалладий (3,3 мг, 0,01 ммоль) добавляли к перемешиваемой смеси метил-2-морфолино-4-оксо-8-[(2R)-пирролидин-2-ил]хромен-6-
карбоксилата (121 мг, 0,34 ммоль), (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфина) (16,61 мг, 0,03 ммоль), 1-бром-3-фторбензола (0,047 мл, 0,42 ммоль) и карбоната цезия (165 мг, 0,51 ммоль), суспендированных в 1,4-диоксане (3,3 мл). Полученную суспензию дегазировали аргоном и затем перемешивали при 100°C в течение 20 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и неочищенный продукт очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 7% пропанола в DCM. Растворитель выпаривали досуха с получением метил-8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (120 мг, 79%) в виде желтого масла, которое затвердевало при стоянии. Масс-спектр: m/z [M+H]+=453.
Стадия 4
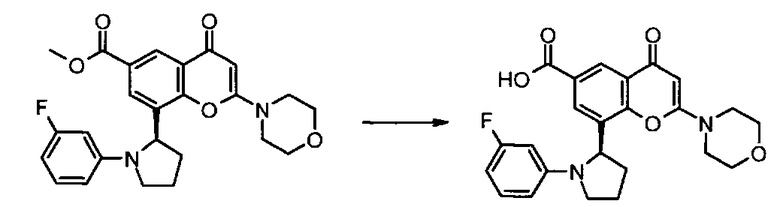
2 н. NaOH (0,398 мл, 0,80 ммоль) добавляли к метил-8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоксилату (120 мг, 0,27 ммоль) в смеси THF (2,4 мл) и MeOH (2,4 мл). Полученный раствор перемешивали при 25°C в течение выходных. Добавляли водн. HCl до pH приблизительно 2. Растворители удаляли под вакуумом и желтое твердое вещество собирали фильтрованием, промывали диэтиловым эфиром с получением 8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (100 мг, 86%). Масс-спектр: m/z [M+H]+=439.
Пример 1.07
8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он
TSTU (220 мг, 0,68 ммоль) при комнатной температуре под азотом добавляли одной порцией к перемешиваемой суспензии 8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (250 мг, 0,57 ммоль, энантиомерная чистота более 98%, смотри Пример 1.06 в отношении особенностей получения), морфолина (0,075 мл, 0,86 ммоль) и DIPEA (0,149 мл, 0,86 ммоль), растворенных в DCM (5 мл). Полученную суспензию перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь очищали препаративной ВЭЖХ. Фракции выпаривали досуха, полученную пену растворяли в DCM (0,5 мл) и добавляли диэтиловый эфир (1 мл). Полученное кристаллическое твердое вещество собирали фильтрованием и сушили под вакуумом с получением 8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (126 мг, 44%) в виде белого кристаллическое твердого вещества.
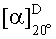
Масс-спектр: m/z [M+H]+=508.
Спектр протонного ЯМР: (DMSO-d6) 1.81-1.93 (m, 1Н), 1.97-2.10 (m, 2Н), 2.50-2.59 (m частично скрыт DMSO-d5, 1Н), 3.19-3.24 (m частично скрыт H2O, 1Н), 3.34-3.42 (m, 4Н), 3.44 (bs, 4Н), 3.50-3.64 (m, 4Н), 3.71-3.81 (m, 5Н), 5.23 (d, 1Н), 5.58 (s, 1Н), 6.21-6.28 (m, 2Н), 7.36 (ddd, 1Н), 7.08 (dd, 1Н), 7.10 (d, 1Н), 7.82 (d,1H).
Пример 1.08
6-(азетидин-1-карбонил)-8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-2-морфолино-4Н-хромен-4-он
HATU (368 мг, 0,97 ммоль) под азотом добавляли к перемешиваемому раствору 8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (212 мг, 0,48 ммоль, энантиомерная чистота более 98%, смотри Пример 1.06 в отношении особенностей получения) и DIPEA (0,674 мл, 3,87 ммоль), растворенных в DCM (1 мл). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Добавляли гидрохлорид азетидина (271 мг, 2,90 ммоль) и смесь перемешивали при комнатной температуре в течение 15 ч. Реакционную смесь очищали препаративной ВЭЖХ. Фракции выпаривали досуха с получением 6-(азетидин-1-карбонил)-8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-2-морфолино-4Н-хромен-4-она (28 мг, 12%) в виде бледно-желтой пены.
Масс-спектр: m/z [M+H]+=478.
Спектр протонного ЯМР (CDCl3) 1.97-2.14 (m, 3H), 2.18-2.33 (m, 2Н), 2.40-2.52 (m, 1Н), 3.37-3.46 (m, 1Н), 3.48-3.61 (m, 4Н), 3.75-3.82 (m, 1Н), 3.82-3-92 (m, 4Н), 4.02-4.10 (m, 1Н), 4.12-4.20 (m, 2Н), 4.21-4.30 (m, 1Н), 5.08 (d, 1Н), 5.57 (s, 1Н), 6.14 (ddd, 1Н), 6.21 (dd, 1Н), 6.37 (ddd, 1Н), 7.08 (dd, 1Н), 7.64 (d, 1Н), 8.26 (d, 1Н).
Пример 1.09
8-[(2R)-(1-(3,5-дифторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-он
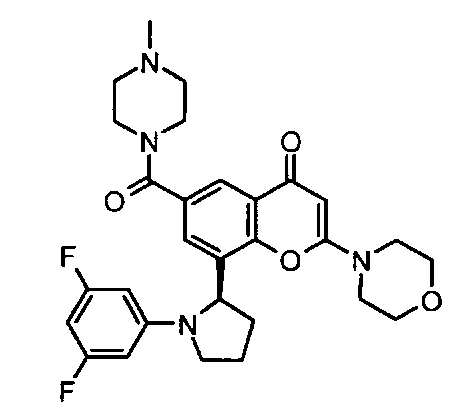
8-[(2R)-1-(3,5-Дифторфенил)пирролидин-2-ил]-2-морфолино-4-оксо-хромен-6-карбоновую кислоту (смотри Пример 1.03b в отношении особенностей получения, 97 мг, 0,21 ммоль), DIPEA (0,185 мл, 1,06 ммоль) и 1-метилпиперазин (0,047 мл, 0,43 ммоль) смешивали при комнатной температуре в DCM (3 мл). Затем добавляли ангидрид N-пропилфосфоновой кислоты, циклический тример (50 масс.% раствор в EtAc) (0,633 мл, 1,08 ммоль), и реакционную смесь перемешивали при 25°C в течение 1 часа. Реакционную смесь очищали препаративной ВЭЖХ с получением 8-[(2R)-(1-(3,5-дифторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-она (67 мг, 59%) в виде белой пены.
Масс-спектр: m/z [M+H]+=539.
Спектр протонного ЯМР: (DMSO-d6): 1.76-1.89 (m, 2Н), 1.98-2.07 (m, 2Н), 2.11 (s, 3Н), 2.13-2.22 (m, 2Н), 2.36 (br, 2Н), 2.93-3.13 (br, 2Н), 3.35-3.42 (m частично скрыт H2O, 2Н), 3.51-3.67 (m, 4H), 3.70-3.78 (m, 6 Н), 5.26 (d, 1Н), 5.62 (s, 1Н), 6.14 (d, 2Н), 6.33 (m, 1Н), 7.02 (d, 1Н), 7.78 (d, 1Н).
Пример 1.10
8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-он

8-[(2R)-(1-(3-Фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновую кислоту (смотри Пример 1.06 в отношении особенностей получения; 100 мг, 0,23 ммоль) подвергали взаимодействию с 1-метилпиперазином (0,076 мл, 0,68 ммоль), используя процедуру, аналогичную процедуре, описанной в примере 1.00, с получением 8-[(2R)-(1-(3-фторфенил)пирролидин-2-ил)]-6-(4-метилпиперазин-1-карбонил)-2-морфолино-4Н-хромен-4-она (39 мг, 33%).
Масс-спектр: m/z [M+H]+=521.
Спектр протонного ЯМР: (CDCl3): 1.97-2.13 (m, 4Н), 2.24 (br s, 3Н), 2.29-2.54 (m, 4Н), 3.00-3.28 (m, 2Н), 3.40-3.47 (m, 1Н), 3.49-3.61 (m, 4Н), 3.69-3.81 (m, 3Н), 3.81-3.94 (m, 4Н), 5.10 (d, 1Н), 5.57 (s, 1Н), 6.12 (d, 1Н), 6.20 (d, 1Н), 6.33-6.41 (m, 1Н), 7.06-7.11 (m, 1Н), 7.26 (s скрыт хлороформом, 1Н), 8.12 (d, 1Н).
Пример 1.11
8-[(2R)-(1-(3-метоксифенил)пирролидин-2-ил)]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он
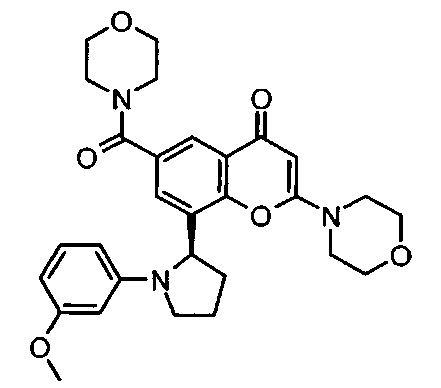
8-[(2R)-(1-(3-Метоксифенил)пирролидин-2-ил)-2-морфолино-4-оксо-4H-хромен-6-карбоновую кислоту (63 мг, 0,14 ммоль) подвергали взаимодействию с морфолином (0,12 мл, 0,14 ммоль), используя процедуру, аналогичную процедуре, описанной в примере 1.04, с получением 8-[(2R)-(1-(3-метоксифенил)пирролидин-2-ил)]-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (37 мг, 51%) в виде белой пены.
Масс-спектр: m/z [M+H]+=520.
Спектр протонного ЯМР: (DMSO-d6): 1.78-1.91 (m, 1Н), 1.95-2.06 (m, 2Н), 2.50-2.56 (m частично скрыт DMSO-d5, 1Н), 2.96-3.26 (m, 4Н), 3.35-3.42 (m, 2Н), 3.50-3.62 (m, 6 Н), 3.63 (s, 3Н), 3.71-3.79 (m, 6 Н), 5.20 (d, 1Н), 5.61 (s, 1Н), 5.94-5.98 (m, 1Н), 6.01 (dd, 1Н), 6.21 (dd, 1Н), 6.99 (dd, 1Н), 7.12 (d, 1Н), 7.81 (d, 1Н) ММА-04957-98-01-109269.
8-(1-(3-Метоксифенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновую кислоту, использовавшуюся в качестве исходного материала, получали, используя процедуры, аналогичные процедурам, описанным в примере 1.05, где 1-бром-3-метоксибензол использовали вместо 1-бром-3-фторбензола.
Стадия 1

Метил-2-морфолино-4-оксо-8-[(2R)-пирролидин-2-ил]хромен-6-карбоксилат (получение описано в Примере 1.03b, 125 мг, 0,35 ммоль) подвергали взаимодействию с 1-бром-3-метоксибензолом (0,049 мл, 0,38 ммоль), используя процедуру, аналогичную процедуре, описанной в примере Пример 1.0, с получением метил-8-[(2R)-(1-(3-метоксифенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (90 мг, 56%) в виде желтой смолы. Масс-спектр: m/z [M+H]+=465.
Стадия 2
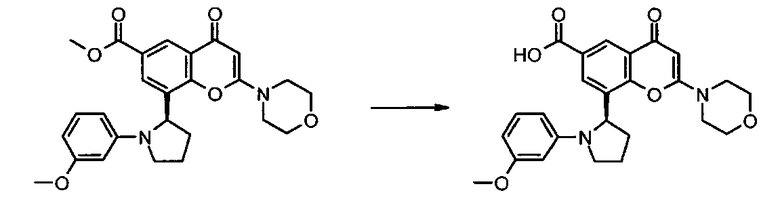
Метил-8-[(2R)-(1-(3-метоксифенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоксилат (90 мг, 0,19 ммоль) подвергали взаимодействию с гидроксидом натрия (38,7 мг, 0,97 ммоль) с получением 8-[(2R)-(1-(3-метоксифенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (64 мг, 73%) в виде бежевого твердого вещества. Масс-спектр: m/z [M+H]+=451.
Пример 1.12
8-(1-(4-фторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он
Диацетоксипалладий (6,79 мг, 0,03 ммоль) добавляли к перемешиваемой смеси 6-(морфолин-4-карбонил)-2-морфолино-8-(пирролидин-2-ил)-4Н-хромен-4-она (250 мг, 0,60 ммоль), бифенил-2-илдициклогексилфосфина (21 мг, 0,06 ммоль), 1-бром-4-фторбензола (0,083 мл, 0,76 ммоль) и карбоната цезия (296 мг, 0,91 ммоль), растворенных в 1,4-диоксане (5 мл). Полученную суспензию дегазировали аргоном и затем перемешивали при 100°C в течение 15 ч. Реакционную смесь очищали препаративной ВЭЖХ. Фракции, содержащие целевое соединение, выпаривали досуха с получением 8-(1-(4-фторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (140 мг, 46%) в виде смолы.
Масс-спектр: m/z [M+H]+=508.
Спектр протонного ЯМР (DMSO-d6): 1.79-1.92 (m, 1Н), 1.95-2.07 (m, 2Н), 2.52-2.57 (m частично скрыт DMSO-d6, 1Н), 2.92-3.28 (m, 4Н), 3.39-3.67 (m, 1Н), 3.48-3.67 (m, 6 Н), 3.70-3.80 (m, 6 Н), 5.17 (d, 1Н), 5.61 (s, 1Н), 6.41 (dd, 2Н), 6.95 (dd, 2Н), 7.13 (d, 1Н), 7.81 (d, 1Н).
Вышеуказанную рацемическую смесь очищали посредством хиральной ВЭЖХ.
Оба образца, полученные в виде прозрачной пленки, при растирании с диэтиловым эфиром давали кремообразные белые вещества. Эти материалы сушили в течение ночи в вакууме при 40°C.
1-ый элюируемый энантиомер: 30 мг (энантиомерная чистота 99%) пример 1.12а
2-ой элюируемый энантиомер: 26 мг (энантиомерная чистота 99%) пример 1.12b
Пример 1.12а
8-(1-(4-фторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он (энантиомер 1)
Условия анализа:
1-ый элюируемый энантиомер: пример 1.12а, время удерживания 2,92 мин, чистота 99,7%.
Пример 1.12b
8-(1-(4-фторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он (энантиомер 2)
Условия анализа:
2-ой элюируемый энантиомер: пример 1.12b, время удерживания 3,56 мин, чистота 99,6%.
6-(Морфолин-4-карбонил)-2-морфолино-8-(пирролидин-2-ил)-4Н-хромен-4-он, используемый в качестве исходного материала, получали следующим образом.
Стадия 1
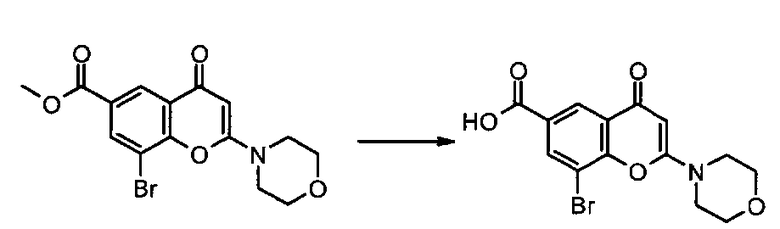
2 н. NaOH (40,7 мл, 81,48 ммоль) добавляли к метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилату (10 г, 27,2 ммоль) в смеси метанола (225 мл) и DCM (75 мл). Суспензию перемешивали при комнатной температуре в течение 16 ч. Растворители выпаривали, к суспензии добавляли воду (150 мл), раствор охлаждали до 0°C и по каплям к реакционной смеси добавляли 6 н. раствор HCl (15 мл, 89,6 ммоль) до pH приблизительно 3,4. Образовавшееся твердое вещество собирали фильтрованием, промывали водой (3×50 мл), затем толуолом (3×50 мл), затем диэтиловым эфиром (3×50 мл), сушили при 55°C под вакуумом над P2O5 с получением 8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (9,47 г, 98%) в виде бежевого твердого вещества. Неочищенный продукт использовали без дополнительной очистки. Масс-спектр: m/z [M+H]+=354.
Стадия 2
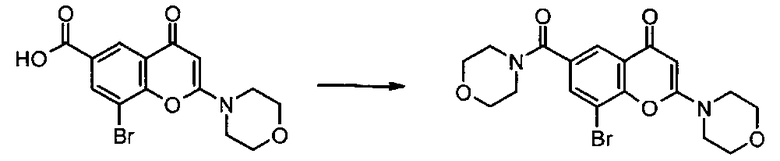
Ангидрид N-пропилфосфоновой кислоты, циклический тример (50 масс.% раствор в EtAc) (8,57 мл, 14,68 ммоль), добавляли при комнатной температуре одной порцией к перемешиваемому раствору 8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (2 г, 5,65 ммоль), DIPEA (4,9 мл, 28,2 ммоль) и морфолина (0,543 мл, 6,2 ммоль) в DCM (14 мл). Смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли воду (1 мл). Реакционную смесь перемешивали в течение 15 минут и экстрагировали DCM. Объединенные органические слои промывали водой; органический слой промывали рассолом, сушили над сульфатом магния и концентрировали с получением 8-бром-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (1,90 г, 79%) в виде бежевого твердого вещества. Масс-спектр: m/z [M+H]+=423.
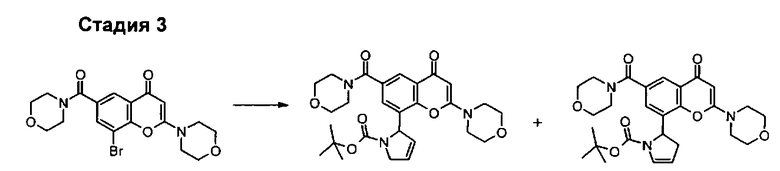
Диацетоксипалладий (0,095 г, 0,42 ммоль) под азотом добавляли к перемешиваемой суспензии 8-бром-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-она (1,79 г, 4,23 ммоль), трет-бутил-2,3-дигидро-1H-пиррол-1-к0арбоксилата (1,431 г, 8,46 ммоль), трифенилфосфина (0,222 г, 0,85 ммоль) и ацетата калия (1,245 г, 12,69 ммоль), растворенных в DMF (24,88 мл). Полученную суспензию дегазировали под азотом и перемешивали при 100°C в течение 16 ч. Смесь упаривали, адсорбировали на силикагеле и очищали посредством флэш-хроматографии на силикагеле, элюируя от 5 до 8% метанола в DCM. Растворитель выпаривали досуха с получением смеси трет-бутил-2-(6-(морфолин-4-карбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-2,5-дигидро-1H-пиррол-1-карбоксилата и трет-бутил-2-(6-(морфолин-4-карбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-2,3-дигидро-1Н-пиррол-1-карбоксилата (2,20 г, 102%) в виде оранжевого масла. Масс-спектр: m/z [M+H]+=512.
Стадия 4
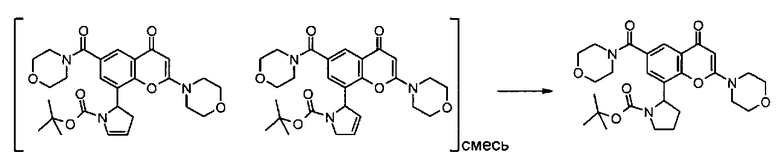
Вышеуказанную смесь трет-бутил-2-(6-(морфолин-4-карбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-2,3-дигидро-1Н-пиррол-1-карбоксилата и трет-бутил-2-(6-(морфолин-4-карбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-2,5-дигидро-1Н-пиррол-1-карбоксилата (1:1) (2,1 г, 2,05 ммоль) и оксида платины(IV) (0,093 г, 0,41 ммоль) в этаноле (30 мл) гидрировали под 1,2 атм (121,56 кПа) при комнатной температуре в течение 4 ч. Полученный раствор фильтровали и фильтрат концентрировали досуха с получением неочищенного трет-бутил-2-(6-(морфолин-4-карбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (1,60 г, 76%) в виде оранжевого твердого вещества. Масс-спектр: m/z [M+H]+=514.
Стадия 5
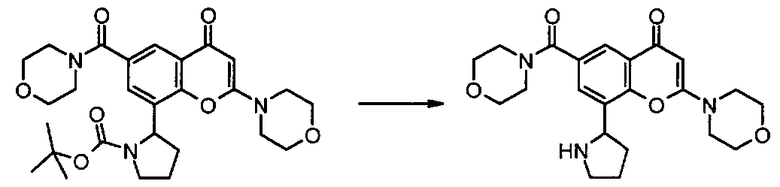
4 М HCl (9,74 мл, 39 ммоль) при к.т. добавляли к перемешиваемому раствору трет-бутил-2-(6-(морфолин-4-карбонил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (2 г, 3,89 ммоль), растворенного в DCM (15 мл), и перемешивали в течение выходных. После концентрирования добавляли DCM (15 мл) и МеОН (15 мл), затем раствор 10%-ного метанольного аммиака (7 н.) в DCM (10 мл). Неочищенный продукт адсорбировали на силикагеле и очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 10% метанола в DCM. Растворитель выпаривали досуха с получением твердого вещества и сушили под вакуумом с получением 6-(морфолин-4-карбонил)-2-морфолино-8-(пирролидин-2-ил)-4Н-хромен-4-она (1,0 г, 62%) в виде бежевого твердого вещества. Масс-спектр: m/z [M+H]+=414.
Пример 2.00
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
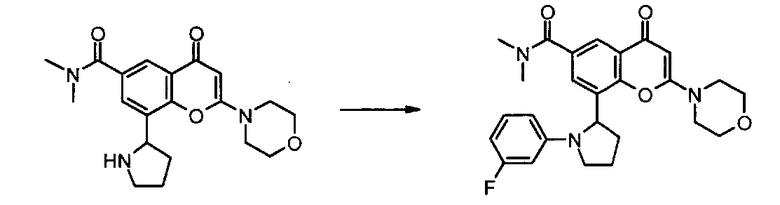
Диацетоксипалладий (3,9 мг, 0,02 ммоль) добавляли к перемешиваемой смеси N,N-диметил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксамида (130 мг, 0,35 ммоль), 1-бром-3-фторбензола (0,049 мл, 0,44 ммоль), (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфина) (20,25 мг, 0,03 ммоль) и карбоната цезия (171 мг, 0,52 ммоль), растворенных в 1,4-диоксане (4 мл). Полученную суспензию дегазировали аргоном и перемешивали при 100°C в течение 16 ч, затем охлаждали до комнатной температуры, фильтровали и концентрировали. Неочищенный продукт растворяли в DMA (2 мл) и очищали препаративной ВЭЖХ. Фракции, содержащие целевое соединение, выпаривали досуха с получением 8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (67 мг, 41%) в виде твердого вещества.
Масс-спектр: m/z [M+H]+=466.
Спектр протонного ЯМР: (DMSO-d6) 1.78-1.90 (m, 1Н), 1.97-2.08 (m, 2Н), 2.52-2.60 (m частично скрыт DMSO-d5, 1Н), 2.68 (s, 3H), 2.90 (s, 3H), 3.33-3.39 (m частично скрыт H2O, 1Н), 3.50-3.57 (m, 2Н), 3.57-3.64 (m, 2Н), 3.71-3.79 (m, 5Н), 5.23 (d, 1Н), 5.61 (s, 1Н), 6.22-6.29 (m, 2Н), 6.37 (ddd, 1Н), 7.09 (dd, 1Н), 7.14 (d, 1Н), 7.80 (d, 1Н).
N,N-Диметил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксамид, используемый в качестве исходного материала, получали следующим образом.
Стадия 1
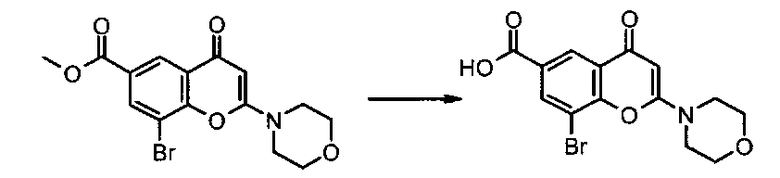
Гидроксид натрия (32,6 мл, 65,19 ммоль) при 0°C добавляли к перемешиваемой суспензии метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (8 г, 21,73 ммоль), растворенного в МеОН (42 мл) и THF (21 мл). Полученную суспензию перемешивали при комнатной температуре в течение 1 ч. Затем для облегчения перемешивания добавляли МеОН (60 мл), THF (10 мл) и воду (10 мл). По завершении реакционную смесь охлаждали до 0°C и к суспензии добавляли 2 н. HCl до pH 2. Твердое вещество собирали фильтрованием, промывали водой, AcOEt, диэтиловым эфиром и сушили с помощью Р205 под вакуумом при 50°C с получением 8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (7,0 г, 91%). Масс-спектр: m/z [M+H]+=354.
Стадия 2
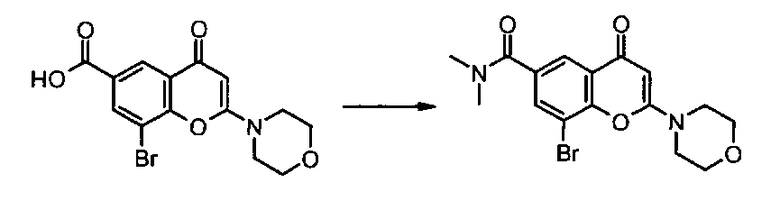
TSTU (6,55 г, 21,74 ммоль) под азотом добавляли к перемешиваемому раствору 8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (7 г, 19,77 ммоль) и DIPEA (3,79 мл, 21,74 ммоль), растворенных в DCM (100 мл). Полученную суспензию перемешивали при комнатной температуре в течение 4 ч. Добавляли дополнительное количество TSTU (6,55 г, 21,74 ммоль) и DIPEA (3,79 мл, 21,74 ммоль) и перемешивание продолжали в течение дополнительных 16 ч. Реакционную смесь гасили насыщенным водным раствором гидрокарбоната натрия и экстрагировали DCM и небольшим количеством МеОН. Объединенные органические фазы сушили над MgSO4, концентрировали и очищали посредством флэш-хроматографии на силикагеле, элюируя от 3 до 5% МеОН в DCM. Растворитель выпаривали досуха с получением 8-бром-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбокс-амида (7,70 г, 102%) в виде бежевого твердого вещества. Масс-спектр: m/z [M+H]+=381.
8-Бром-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид также синтезировали в крупном масштабе в соответствии со следующей процедурой. Гидроксид калия (39,8 мл, 518,37 ммоль) добавляли с постоянной скоростью в течение 30 мин к метил-8-бром-2-морфолино-4-оксо-4Н-хромен-6-карбоксилату (99,3 г, 259,18 ммоль) в воде (572 мл) при 22°C в 3-литровом сосуде с рубашкой, снабженном верхней магнитной мешалкой (сосуд 1). Полученную смесь перемешивали при 22°C в течение 4 ч. Одной порцией добавляли воду (286 мл) и смесь перемешивали при 22°C в течение 2 ч до завершения омыления. Реакционную смесь фильтровали для удаления частиц нерастворимых веществ, в сосуд загружали воду (95 мл) в качестве промывки сетчатого фильтра и объединенные фильтраты переносили в 3-литровый сосуд с рубашкой (сосуд 1). В сосуд 1 добавляли раствор гидрохлорида диметиламина (64,4 г, 777,55 ммоль) в воде (191 мл) и продолжали перемешивание.
В отдельный 2-литровый сосуд с рубашкой (сосуд 2) загружали 2-хлор-4,6-диметокси-1,3,5-триазин (208 г, 1,17 моль) и воду (978 мл) и температуру доводили до приблизительно 8°C. Загружали 4-метилморфолин (214 мл, 1,94 моль) с такой скоростью, чтобы поодерживать содержимое сосуда 2 при температуре ниже 12°C, и содержимое перемешивали в течение 2,5 ч при 12°C.
Содержимое сосуда 2 переносили с постоянной скоростью в течение 3,5 ч в сосуд 2 посредством капельной воронки и перемешивали при 22°C в течение 6 ч. Реакционную смесь затем подвергали следующему циклическому температурному режиму.
Реакционную смесь фильтровали и твердое вещество промывали водой (381 мл). Твердое вещество промывали водой (381 мл) и сушили в вакууме при 50°C с получением 8-бром-N,N-диметил-2-морфолино-4-оксо-хромен-6-карбоксамида (78 г, 78%) в виде бежевого твердого вещества.
Масс-спектр: m/z [М+Н] 381.
Спектр протонного ЯМР: (400 МГц, DMSO, 30°C) 2.93 (3H, br. s), 2.99 (3H, br. s), 3.57-3.60 (4Н, m), 3.74-3.76 (4Н, m), 5.61 (1Н, s), 7.87 (1Н, d, J=1,9 Гц), 7.99 (1Н, d, J=1,9 Гц).
Стадия 3
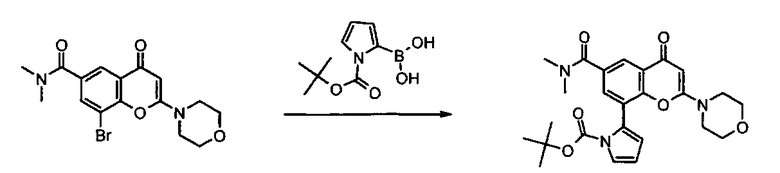
Хлорид бис(трифенилфосфин)палладия (II) (0,116 г, 0,17 ммоль) добавляли одной порцией к перемешиваемой суспензии 8-бром-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (3,15 г, 8,26 ммоль), 1-(трет-бутоксикарбонил)-1Н-пиррол-2-илбороновой кислоты (2,1 г, 9,92 ммоль) и карбоната натрия (2,63 г, 24,79 ммоль) в DME (50 мл) и воде (10 мл). Полученную смесь перемешивали при 80°C в течение 8 ч. После охлаждения к реакционной смеси добавляли воду и экстрагировали DCM. Объединенные органические фазы промывали водой и рассолом, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле, элюируя 5%-ным МеОН в DCM. Растворители выпаривали досуха с получением трет-бутил-2-(6-(диметилкарбамоил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-1Н-пиррол-1-карбоксилата (2,40 г, 62%). Масс-спектр: m/z [M+H]+=468.
Стадия 4
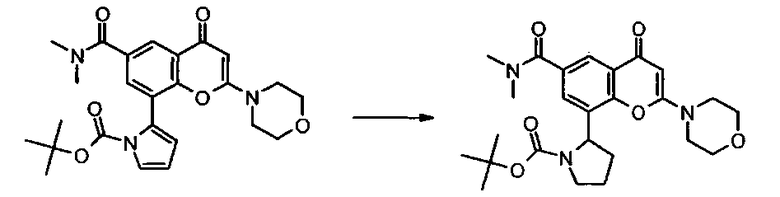
Раствор трет-бутил-2-(6-(диметилкарбамоил)-2-морфолино-4-оксо-4Н-хромен-8-ил)-1Н-пиррол-1-карбоксилата (0,6 г, 1,28 ммоль) в МеОН (10 мл) гидрировали над 5% Rh/Al2O3 (картридж NanoThales CatCart, продукт ID THS 02118) с помощью H-Cube (проточный аппарат для непрерывного гидрирования HC-2.SS от THALES Nanotechnology Inc. Budapest Н-1031; Zahony u.7.; Венгрия) при 10 бар (1000 кПа), 50°C и скорости потока 1 мл/мин. в течение 3,5 ч с непрерывным рециклингом. Картридж заменяли на картридж 10% Pd/c и гидрирование продолжали в течение 6 ч при 60 бар (6000 кПа) и 60°C. Смесь упаривали досуха с получением твердого вещества, которое растирали в диэтиловом эфире, фильтровали и сушили под вакуумом при 50°C с получением чистого трет-бутил-2-(6-(диметилкарбамоил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (0,30 г, 50%) в виде белого твердого вещества. Неочищенный продукт использовали как таковой для следующей стадии. Масс-спектр: m/z [M+H]+=472.
Стадия 5
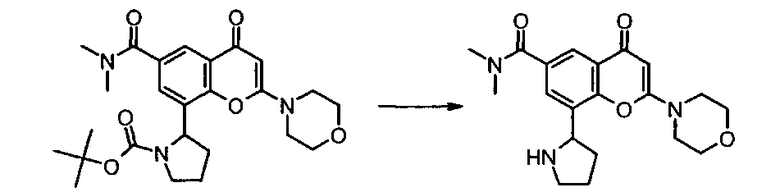
HCl (4,50 мл, 18 ммоль, 4 М раствор) при комнатной температуре добавляли к перемешиваемому раствору трет-бутил-2-(6-(диметилкарбамоил)-2-морфолино-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (850 мг, 1,80 ммоль), растворенного в DCM (10 мл), и перемешивали в течение 8 ч. После выпаривания летучих веществ добавляли DCM (5 мл) и МеОН (5 мл), а затем 10%-ный метанольный аммиак (7 н., 5 мл) в DCM. Твердое вещество отфильтровывали, промывали смесью 1:1 DCM и МеОН. Растворитель выпаривали досуха с получением после растирания с диэтиловым эфиром N,N-диметил-2-морфолино-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксамида (675 мг, 101%) в виде бледно-бежевого твердого вещества. Масс-спектр: m/z [M+H]+=372.
Пример 2.01
8-(1-(3-метоксифенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
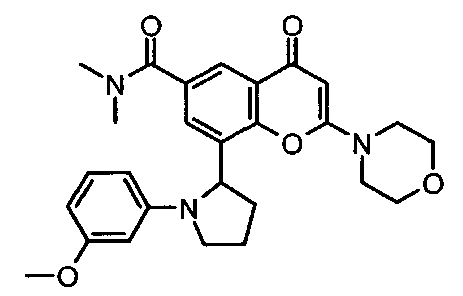
Указанное в заголовке соединение получали, используя процедуру, аналогичную описанной в Примере 2.00, за исключением того, что 1-бром-3-метоксибензол (0,055 мл, 0,44 ммоль) использовали вместо 1-бром-3-фторбензола, с получением 8-(1-(3-метоксифенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (46 мг, 28%) в виде твердого вещества.
Масс-спектр: m/z [M+H]+=478.
Спектр протонного ЯМР: (DMSO-d6) 1.78-1.90 (m, 1Н), 1.94-2.07 (m, 2Н), 2.46-2.55 (m частично скрыт DMSO-d5, 1Н), 2.70 (s, 3H), 2.90 (s, 3H), 3.33-3.39 (m частично скрыт H2O, 1Н), 3.49-3.57 (m, 2Н), 3.57-3.63 (m, 2Н), 3.64 (s, 3H), 3.69-3.79 (m, 5Н), 5.19 (d, 1Н), 5.61 (s, 1Н), 5.98 (s, 1Н), 6.02 (dd, 1Н), 6.20 (dd, 1Н), 6.98 (dd, 1Н), 7.17 (d, 1Н), 7.79 (d, 1Н).
Пример 2.02
8-(1-(3-циано-5-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
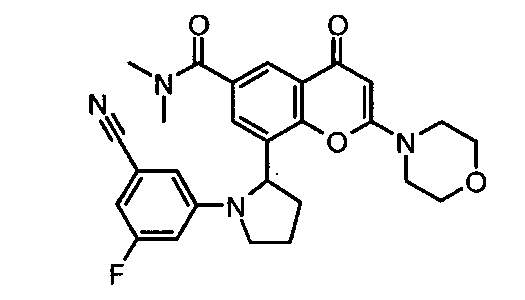
Указанное в заголовке соединение получали, используя процедуру, аналогичную описанной в Примере 2.00, за исключением того, что 3-бром-5-фторбензонитрил (88 мг, 0,44 ммоль) использовали вместо 1-бром-3-фторбензола, с получением 8-(1-(3-циано-5-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (90 мг, 52%) в виде твердого вещества.
Масс-спектр: m/z [M+H]+=491.
Спектр протонного ЯМР: (DMSO-d6) 1.74-1.88 (m, 1Н), 1.99-2.10 (m, 2Н), 2.45-2.56 (m частично скрыт DMSO-d5, 1Н), 2.68 (s, 3H), 2.90 (s, 3H), 3.35-3.43 (m частично скрыт H2O, 1Н), 3.49-3.58 (m, 2Н), 3.58-3.66 (m, 2Н), 3.71-3.77 (m, 4Н), 3.77-3.85 (m, 1Н), 5.31 (d, 1Н), 5.62 (s, 1Н), 6.61 (d, 1Н), 6.75 (s, 1Н), 6.92 (s, 1Н), 7.07 (d, 1Н), 7.80 (d, 1Н).
Пример 2.03
N,N-диметил-2-морфолино-4-оксо-8-(1-фенилпирролидин-2-ил)-4Н-хромен-6-карбоксамид
Указанное в заголовке соединение получали, используя процедуру, аналогичную описанной в Примере 2.00, за исключением того, что бромбензол (0,046 мл, 0,44 ммоль) использовали вместо 1-бром-3-фторбензола, с получением N,N-диметил-2-морфолино-4-оксо-8-(1-фенилпирролидин-2-ил)-4Н-хромен-6-карбоксамида (59 мг, 38%) в виде твердого вещества.
Масс-спектр: m/z [M+H]+=448.
Спектр протонного ЯМР: (DMSO-d6) 1.80-1.93 (m, 1Н), 1.95-2.10 (m, 2Н), 2.45-2.56 (m частично скрыт DMSO-d5, 1Н), 2.67 (s, 3H), 2.89 (s, 3H), 3.33-3.40 (m частично скрыт H2O, 1Н), 3.50-3.57 (m, 2Н), 3.57-3.65 (m, 2Н), 3.70-3.81 (m, 5Н), 5.20 (d, 1Н), 5.61 (s, 1Н), 6.44 (d, 2Н), 6.58 (t, 1Н), 7.10 (dd, 2Н), 7.17 (d, 1Н), 7.79 (d, 1Н).
Пример 2.04
8-(1-(4-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
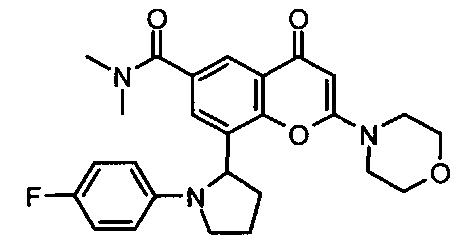
Указанное в заголовке соединение получали, используя процедуру, аналогичную описанной в Примере 2.00, за исключением того, что 1-бром-4-фторбензол (0,048 мл, 0,44 ммоль) использовали вместо 1-бром-3-фторбензола, с получением 8-(1-(4-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (31 мг, 19%) в виде твердого вещества.
Масс-спектр: m/z [M+H]+=466.
Спектр протонного ЯМР: (DMSO-d6) 1.79-1.91 (m, 1Н), 1.93-2.07 (m, 2Н), 2.45-2.56 (m частично скрыт DMSO-d5, 1Н), 2.68 (s, 3H), 2.90 (s, 3H), 3.32-3.42 (m частично скрыт H2O, 1Н), 3.49-3.57 (m, 2Н), 3.57-3.64 (m, 2Н), 3.70-3.80 (m, 5Н), 5.16 (d, 1Н), 5.61 (s, 1Н), 6.42 (dd, 2Н), 6.94 (dd, 2Н), 7.18 (d, 1Н), 7.79 (d, 1Н)
Рацемический 8-(1-(4-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид (200 мг, Пример 2.04) разделяли посредством хиральной ВЭЖХ с получением соединений из Примеров 2.04а и 2.04b.
Оба образца получали в виде тонких прозрачных пленок, которые при растирании с диэтиловым эфиром давали твердые вещества. Эти материалы сушили в течение ночи в вакууме при 40°C.
1-ый элюируемый энантиомер: 63 мг (энантиомерная чистота 99,7%) пример 2.04а
2-ой элюируемый энантиомер: 58 мг (энантиомерная чистота 99,0%) пример 2.04b
Пример 2.04а
8-[(2S)-1-(4-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
Масс-спектр: m/z [M+H]+=466.
Спектр протонного ЯМР (CDCl3): 1.95-2.12 (m, 3Н), 2.40-2.52 (m, 1Н), 2.78 (s, 3Н), 3.03 (s, 3Н), 3.30-3.43 (m, 1Н), 3.46-3.60 (m, 4Н), 3.71-3.80 (m, 1Н), 3.81-3.93 (m, 4Н), 5.04 (d, 1Н), 5.57 (s, 1Н), 6.36 (dd, 2Н), 6.86 (dd, 2Н), 7.40 (d, 1Н), 8.11 (d, 1Н).
Пример 2.04b
8-[(2R)-1-(4-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид
Масс-спектр: m/z [M+H]+=466.
Спектр протонного ЯМР (CDCl3): 1.95-2.12 (m, 3Н), 2.40-2.52 (m, 1Н), 2.78 (s, 3Н), 3.03 (s, 3Н), 3.30-3.43 (m, 1Н), 3.46-3.60 (m, 4Н), 3.71-3.80 (m, 1Н), 3.81-3.93 (m, 4Н), 5.04 (d, 1Н), 5.57 (s, 1Н), 6.36 (dd, 2Н), 6.86 (dd, 2Н), 7.40 (d, 1Н), 8.11 (d, 1Н).
Абсолютную стереохимию каждого из соединений из Примеров 2.04а и 2.04b определяли посредством энантиоселективного синтеза следующим образом. Соединение из Примера 2.04b получали, используя хиральный исходный материал с известной абсолютной химией (то есть метил-2-морфолино-4-оксо-8-[(2R)-пирролидин-2-ил]хромен-6-карбоксилат, получение которого описано в Примере 1.03b). По аналогии соединению из Примера 2.04а была присвоена (S)-конфигурация. Подробности энантиоселективного синтеза соединения из Примера 2.04b являются следующими.
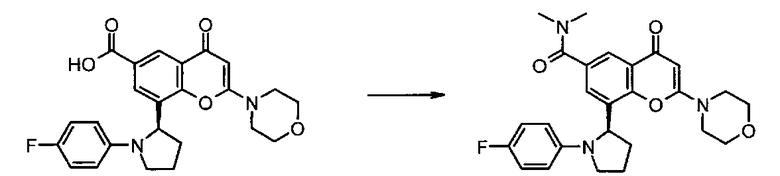
8-[(2R)-(1-(4-Фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновую кислоту (56 мг, 0,13 ммоль, для получения смотри Пример 1.03b) подвергали взаимодействию с гидрохлоридом диметиламина (52 мг, 0,64 ммоль), используя процедуру, аналогичную описанной в примере 1.04, с получением 8-(1-(4-фторфенил)пирролидин-2-ил)-1N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамида (9 мг, 15%), анализ данных как выше.
8-[(2R)-(1-(4-Фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоновую кислоту, использовавшуюся в качестве исходного материала, получали посредством процедур, аналогичных описанным в примере 1.05, с использованием 1-бром-4-фторбензола вместо 1-бром-3-фторбензола.
Стадия 1
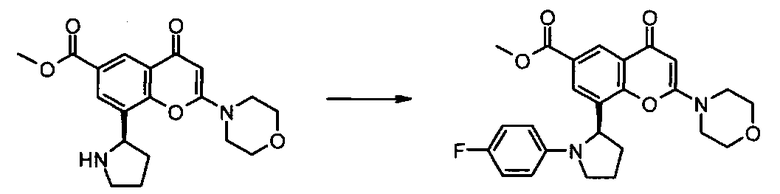
Метил-2-морфолино-4-оксо-8-[(2R)-пирролидин-2-ил]хромен-6-карбоксилат (130 мг, 0,36 ммоль) подвергали взаимодействию с 1-бром-4-фторбензолом (0,239 мл, 2,18 ммоль) с получением метил-8-[(2R)-(1-(4-фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоксилата (энантиомер 2, 24 мг, 15%) в виде бежевого твердого вещества. Масс-спектр: m/z [M+H]+=453.
Стадия 2
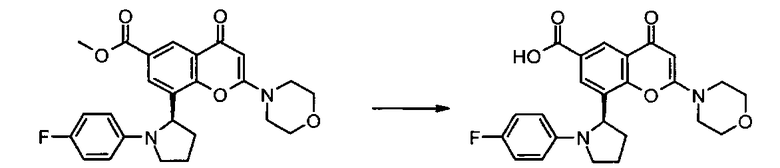
Метил-8-[(2R)-(1-(4-фторфенил)пирролидин-2-ил)]-2-морфолино-4-оксо-4Н-хромен-6-карбоксилат (119 мг, 0,26 ммоль из 2 разных партий) подвергали взаимодействию с гидроксидом натрия с получением 8-(1-(4-фторфенил)пирролидин-2-ил)-2-морфолино-4-оксо-4Н-хромен-6-карбоновой кислоты (энантиомер 2, 95 мг, 82%) в виде желтого твердого вещества. Масс-спектр: m/z [M+H]+=439.
Пример 3.00
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он
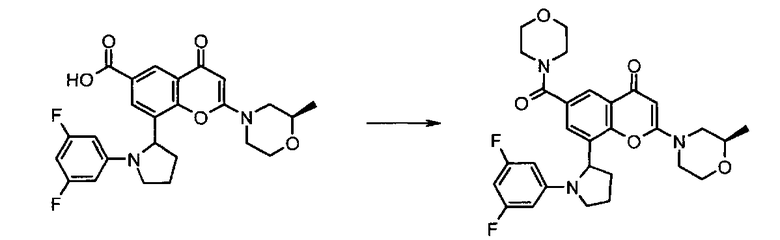
TBTU (154 мг, 0,48 ммоль) под азотом добавляли к перемешиваемому раствору 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоновой кислоты (90 мг, 0,19 ммоль), DIPEA (0,083 мл, 0,48 ммоль), растворенных в DMA (2 мл). Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Добавляли морфолин (0,050 мл, 0,57 ммоль) и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь очищали препаративной ВЭЖХ. Фракции, содержавшие целевое соединение, выпаривали досуха с получением 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-она (68 мг, 66%) в виде желтого твердого вещества.
Масс-спектр: m/z [M+H]+=540.
Спектр протонного ЯМР: (DMSO-d6) 1.17 (d, 3H), 1.75-1.88 (m, 1Н), 1.99-2.08 (m, 2Н), 2.45-2.57 (m, частично скрыт DMSO d6, 1Н), 2.77-2.86 (m, 1Н), 3.06 (bs, 2Н), 3.09-3.19 (m, 1Н), 3.23 (bs, 2Н), 3.33-3.41 (m частично скрыт H2O, 1Н), 3.44 (bs, 2Н), 3.55 (bs, 2Н), 3.58-3.71 (m, 2Н), 3.73-3.80 (m, 1Н), 3.85-4.06 (m, 3H), 5.26 (d, 1Н), 5.64 (s, 1Н), 6.13 (d, 2Н), 6.34 (t, 1Н), 7.06 (d, 0.5Н), 7.07 (d, 0.5Н), 7.82 (d, 1Н).
8-(1-(3,5-Дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоновую кислоту, используемую в качестве исходного материала, получали следующим образом.
Стадия 1

К суспензии метил-3-ацетил-5-бром-4-гидроксибензоата (70 г, 243,52 ммоль) в THF (700 мл) при -50°C под азотом (колба, снабженная ловушкой из хлорной извести) добавляли бис(триметилсилил)амид лития (828 мл, 828 ммоль). Темный раствор оставляли нагреваться до -5°C и перемешивали в течение 2 ч. К раствору при -20°C одной порцией добавляли дисульфид углерода (22 мл, 365 ммоль), затем смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду (700 мл), THF слои промывали водой (2x 350 мл). Водные фазы объединяли и охлаждали до 0°C и гасили H2SO4 (108 мл, 1948 ммоль) в сосуде, снабженном ловушкой из хлорной извести для нейтрализации образующегося H2S. Смесь перемешивали при комнатной температуре в течение 3 ч, добавляли DCM (700 мл) и смесь перемешивали при комнатной температуре в течение ночи. Водную фазу экстрагировали DCM (2х). Органические фазы объединяли, промывали рассолом, сушили над MgSO4, фильтровали и выпаривали с получением оранжевого твердого вещества. Это твердое вещество растирали в диэтиловом эфире, фильтровали и сушили с получением метил-8-бром-4-гидрокси-2-тиоксо-2Н-хромен-6-карбоксилата (48,7 г, 63%) в виде желтого/оранжевого твердого вещества. Масс-спектр: m/z [M+H]+=317.
Стадия 2

Йодэтан (37,1 мл, 463,51 ммоль) одной порцией добавляли к перемешиваемой суспензии метил-8-бром-4-гидрокси-2-тиоксо-2Н-хромен-6-карбоксилата (48,69 г, 154,5 ммоль) и карбоната калия (21,35 г, 154,50 ммоль) в ацетоне (490 мл). Полученную суспензию под азотом перемешивали при кипячении с обратным холодильником в течение 1 ч в колбе, снабженной ловушкой из хлорной извести. Реакционную смесь концентрировали, остаток разбавляли смесью DCM (980 мл)/вода (490 мл). Фазы разделяли и водный слой экстрагировали DCM (490 мл). Объединенные органические фазы промывали рассолом, сушили над MgSO4 и концентрировали. Остаток растирали с петролейным эфиром, фильтровали и сушили под вакуумом с получением метил-8-бром-2-(этилтио)-4-оксо-4Н-хромен-6-карбоксилата (52,9 г, 100%) в виде бледно-коричневого твердого вещества. Масс-спектр: m/z [M+H]+=345.
Стадия 3
3-Хлорпероксибензойную кислоту (76 г, 264,90 ммоль) порциями добавляли к перемешиваемому раствору метил-8-бром-2-(этилтио)-4-оксо-4Н-хромен-6-карбоксилата (47,35 г, 132,45 ммоль) в DCM (420 мл), охлажденному с помощью ледяной бани. Полученную смесь затем перемешивали под азотом при комнатной температуре в течение 2 ч. Суспензию охлаждали до -15°C и фильтровали, твердое вещество промывали холодным DCM, фильтрат затем промывали раствором пентагидрата сульфотиоата натрия (16,44 г, 66,23 ммоль) в воде (290 мл), затем насыщенным раствором NaHCO3 (2×300 мл). Органический слой декантировали, сушили над MgSO4 и выпаривали с получением красного порошка, который растирали с диэтиловым эфиром с получением твердого вещества, которое собирали фильтрованием и сушили с получением метил-8-бром-2-(этилсульфонил)-4-оксо-4Н-хромен-6-карбоксилата (44,6 г, 90%) в виде не совсем белого материала.
Масс-спектр: m/z [M+H]+=375.
Стадия 4
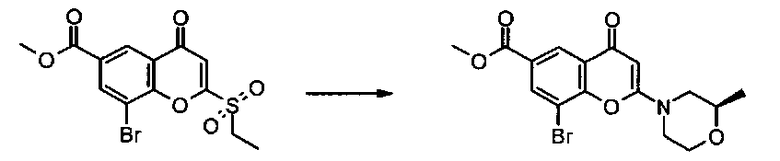
DIPEA (51,7 мл, 296,91 ммоль) при 5°C под азотом добавляли по каплям к перемешиваемому раствору метил-8-бром-2-(этилсульфонил)-4-оксо-4Н-хромен-6-карбоксилата (44,56 г, 118,77 ммоль) и гидрохлорида (R)-2-метилморфолина (22,88 г, 166,27 ммоль) в DCM (430 мл). Полученную суспензию перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли водой и медленно гасили 1 М HCl (119 мл, 118,77 ммоль) (pH 7). Фазы разделяли и органическую фазу промывали водой, рассолом, сушили над MgSO4 и концентрировали. Неочищенный продукт растирали в EtAc в течение 1 ч, твердое вещество собирали фильтрованием и сушили с получением метил-8-бром-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксилата (32,6 г, 67%) в виде белого твердого вещества. Масс-спектр: m/z [M+H]+=384.
Стадия 5

Хлорид бис(трифенилфосфин)палладия(И) (0,448 г, 0,63 ммоль) одной порцией добавляли к перемешиваемой суспензии (R)-метил-8-бром-2-(2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксилата (10,06 г, 25 ммоль), 1-(трет-бутоксикарбонил)-1Н-пиррол-2-илбороновой кислоты (6,46 г, 30,00 ммоль) и карбоната натрия (7,95 г, 75,00 ммоль) в DME (140 мл) и воде (28 мл). Смесь дегазировали аргоном в течение 15 минут, затем нагревали при 80°C в течение 6 ч. Растворитель выпаривали, к остатку добавляли воду и смесь экстрагировали DCM. Объединенные органические фазы промывали водой, рассолом, сушили над MgSO4 и концентрировали с получением неочищенного (R)-трет-бутил-2-(6-(метоксикарбонил)-2-(2-метилморфолино)-4-оксо-4Н-хромен-8-ил)-1Н-пиррол-1-карбоксилата, содержавшего приблизительно 20% исходного материала (13,98 г). Масс-спектр: m/z [M+H]+=469.
Стадия 6
(R)-трет-Бутил-2-(6-(метоксикарбонил)-2-(2-метилморфолино)-4-оксо-4Н-хромен-8-ил)-1Н-пиррол-1-карбоксилат (2 г, 4,27 ммоль) и 5% родий на окиси алюминия (влажность 50%) (0,4 г, 0,09 ммоль) в МеОН (50 мл) перемешивали в атмосфере водорода при 5 бар (500 кПа) и 65°C в течение 2 ч. Добавляли дополнительное количество катализатора и перемешивание продолжали в течение ночи. Затем добавляли 2 г 5% Rh/C с последующим добавлением через 1 ч такого же количества, затем спустя еще 2 ч. Катализатор отфильтровывали через целит и промывали смесью MeOH/DCM и растворитель выпаривали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле, элюируя от 10 до 15% МеОН в EtAc. Растворитель выпаривали досуха с получением трет-бутил-2-(6-(метоксикарбонил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-8-ил)-пирролидин-1-карбоксилата (1,18 г, 59%) в виде белого твердого вещества. Масс-спектр: m/z [M+H]+=473.
Стадия 7
4 н. HCl (6,24 мл, 24,97 ммоль) при комнатной температуре добавляли к перемешиваемому раствору трет-бутил-2-(6-(метоксикарбонил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-8-ил)пирролидин-1-карбоксилата (1,18 г, 2,50 ммоль) в DCM (10 мл) и перемешивали в течение 16 ч. Растворители выпаривали и добавляли DCM (5 мл) и МеОН (5 мл), а затем 10%-ный метанольный аммиак (7 н.) в дихлорметане (5 мл). Твердое вещество отфильтровывали и промывали смесью 1:1 DCM и МеОН. Растворитель выпаривали досуха и неочищенный продукт очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 10% МеОН в DCM. Растворитель выпаривали досуха, растирали с диэтиловым эфиром с получением твердого вещества, которое собирали фильтрованием и сушили под вакуумом с получением метил-2-((R)-2-метилморфолино)-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилата (0,760 г, 82%) в виде белого твердого вещества. Масс-спектр: m/z [M+H]+=373.
Стадия 8
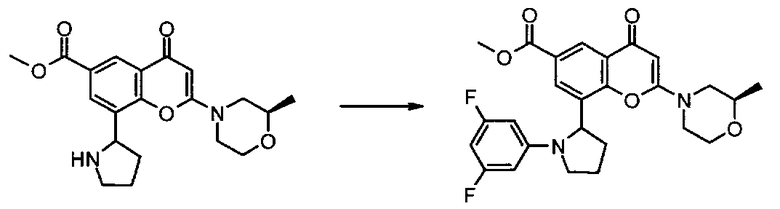
Диацетоксипалладий (4,58 мг, 0,02 ммоль) добавляли к перемешиваемой смеси метил-2-((R)-2-метилморфолино)-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилата (190 мг, 0,51 ммоль), (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфина) (25,09 мг, 0,04 ммоль), 1-бром-3,5-дифторбензола (73,4 мкл, 0,64 ммоль) и карбоната цезия (249 мг, 0,77 ммоль), суспендированных в 1,4-диоксане (5 мл). Полученную суспензию дегазировали аргоном и затем перемешивали при 100°C в течение 20 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры, нерастворимые вещества удаляли фильтрованием и фильтрат концентрировали. Неочищенный продукт адсорбировали на силикагеле и очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 6% МеОН в EtAc. Растворитель выпаривали досуха с получением метил-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксилата (150 мг, 61%) в виде бежевой пены.
Масс-спектр: m/z [M+H]+=485.
Стадия 9
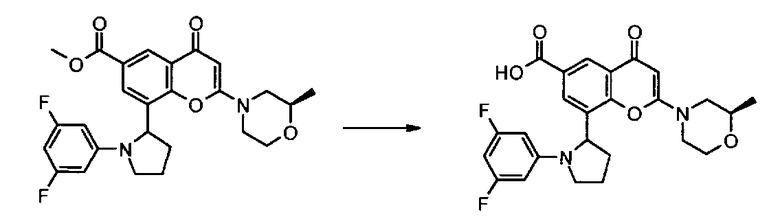
Гидроксид натрия (2 н. в воде) (0,351 мл, 0,70 ммоль) добавляли к перемешиваемой суспензии метил-2-((R)-2-метилморфолино)-4-оксо-8-(1-фенилпирролидин-2-ил)-4Н-хромен-6-карбоксилата (136 мг, 0,28 ммоль) в смеси МеОН (2 мл)/вода (2 мл). Полученную смесь перемешивали при комнатной температуре в течение 18 ч, затем при 50°C до завершения реакции. Реакционную смесь при 5°C подкисляли до pH 2-3 с помощью 2 н. HCl (0,379 мл, 0,76 ммоль). Полученный осадок собирали фильтрованием, промывали водой и диэтиловым эфиром и сушили до постоянной массы с получением 8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоновой кислоты (95 мг, 72%), которую использовали без дополнительной очистки. Масс-спектр: m/z [M+H]+=471.
Гидрохлорид (R)-2-метилморфолина, используемый в качестве исходного материала на стадии 4 выше, получали следующим образом.
Стадия 1
2-(Бензиламино)этанол (207 мл, 1454,97 ммоль) и (R)-2-метилоксиран (153 мл, 2182,46 ммоль) смешивали вместе и прокачивали при скорости потока 1 мл/мин через 10 мл петлю, нагретую при 150°C, в проточной химической системе. Регулятор обратного давления регулировали таким образом, чтобы достигалось давление внутри петли 250 фунт-сила на кв. дюйм (1723,7 кПа). Избыток пропиленоксида удаляли под вакуумом с получением (R)-1-(бензил(2-гидроксиэтил)амино)пропан-2-ола (300 г, 99%) в виде бесцветной жидкости. Масс-спектр: m/z [M+H]+=210.
Стадия 2
К перемешиваемому раствору (R)-1-(бензил(2-гидроксиэтил)амино)пропан-2-ола (110 г, 525,60 ммоль) в диоксане (500 мл) под азотом последовательно добавляли порошок гидроксида калия (88 г, 1576,80 ммоль) и трис(2-(2-метоксиэтокси)этил)амин (1,681 мл, 5,26 ммоль). Смесь охлаждали до 0°C и по каплям добавляли раствор 4-метилбензол-1-сульфонилхлорида (105 г, 551,88 ммоль) в диоксане (500 мл). Реакционную смесь перемешивали при 0°C в течение 45 мин, затем при комнатной температуре в течение дополнительных 4 ч. Смесь фильтровали и фильтрат выпаривали. Остаток растворяли в DCM и очищали на диоксиде кремния, элюируя 25% EtAc в DCM. Растворители выпаривали с получением масла, которое разбавляли EtAc (250 мл). Затем по каплям при перемешивании добавляли 4 н. раствор HCl в диоксане (0,066 л, 262,8 ммоль). Спустя 30 мин образовавшееся твердое вещество собирали фильтрованием, промывали диэтиловым эфиром и сушили с получением гидрохлорида (R)-4-бензил-2-метилморфолина (40 г, 33%) в виде белого твердого вещества. Спектр протонного ЯМР: (DMSO-d6): 1.09 (d, 3H), 2.67-2.77 (m, 1Н), 2.93-3.04 (m, 1Н), 3.14-3.23 (m, 2Н), 3.83-4.00 (m, 3H), 4.29-4.34 (m, 2Н), 7.43-7.49 (m, 3H), 7.60-7.68 (m, 2Н), 11.62 (bs, 1Н).
Стадия 3
Раствор гидрохлорида (R)-4-бензил-2-метилморфолина (40 г, 175,65 ммоль) в этаноле (1 л) гидрировали в присутствии палладия 10%/С (3 г, 28,19 ммоль) при комнатной температуре в течение ночи при давлении водорода 1 атм (101,3 кПа). Смесь фильтровали и фильтрат концентрировали досуха с получением гидрохлорида (R)-2-метилморфолина (23 г, 95%) в виде белого твердого вещества. Спектр протонного ЯМР: (DMSO-d6): 1.10 (d, 3H), 2.62 (dd, 1Н), 2.85-2.95 (m, 1Н), 3.08-3.20 (m, 2Н), 3.70-3.77 (m, 1Н), 3.78-3.83 (m, 1Н), 3.90 (dd, 1Н), 9.49 (bs, 2Н).
Пример 3.01
2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-8-(1-фенилпирролидин-2-ил)-4Н-хромен-4-он
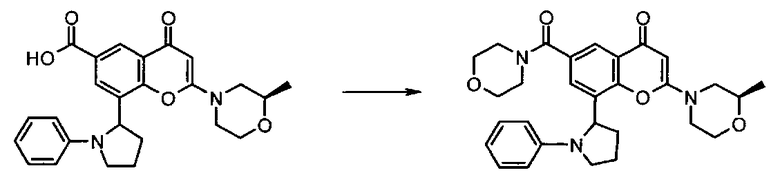
TBTU (148 мг, 0,46 ммоль) под азотом добавляли к перемешиваемому раствору 2-((R)-2-метилморфолино)-4-оксо-8-(1-фенилпирролидин-2-ил)-4Н-хромен-6-карбоновой кислоты (80 мг, 0,18 ммоль), DIPEA (0,080 мл, 0,46 ммоль), растворенных в DMA (2 мл). Полученный раствор перемешивали при комнатной температуре в течение 30 мин. К смеси добавляли морфолин (0,048 мл, 0,55 ммоль) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь очищали препаративной ВЭЖХ. Фракции, содержащие целевое соединение, выпаривали досуха с получением 2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-8-(1-фенилпирролидин-2-ил)-4Н-хромен-4-она (60 мг, 65%) в виде смолистого твердого вещества.
Масс-спектр: m/z [M+H]+=504.
Спектр протонного ЯМР: (DMSO-d6) 1.17 (d, 3H), 1.79-1.93 (m, 1Н), 1.98-2.09 (m, 2Н), 2.47-2.57 (m, частично скрыт DMSO d6, 1Н), 2.77-2.86 (m, 1Н), 3.02 (bs, 2Н), 3.09-3.18 (m, 1Н), 3.19 (bs, 2Н), 3.34-3.40 (m частично скрыт Н20, 1Н), 3.42 (bs, 2Н), 3.55 (bs, 2Н), 3.58-3.71 (m, 2Н), 3.72-3.81 (m, 1Н), 3.85-4.07 (m, 3H), 5.22 (d, 1Н), 5.64 (s, 1Н), 6.44 (d, 2Н), 6.53 (t, 1Н), 7.10 (t, 2Н), 7.12 (d, 1Н), 7.81 (d, 1Н).
2-((R)-2-Метилморфолино)-4-оксо-8-(1-фенилпирролидин-2-ил)-4Н-хромен-6-карбоновую кислоту, используемую в качестве исходного материала, получали следующим образом.
Стадия 1
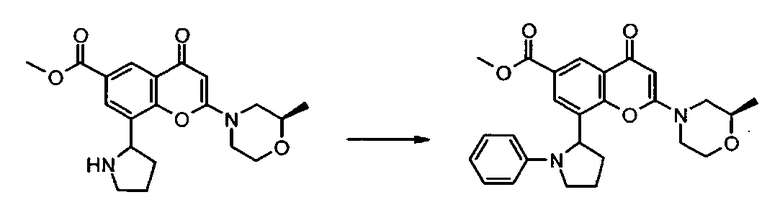
Диацетоксипалладий (4,58 мг, 0,02 ммоль) добавляли к перемешиваемой смеси метил-2-((R)-2-метилморфолино)-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилата (190 мг, 0,51 ммоль), (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфина) (25,09 мг, 0,04 ммоль), бромбензола (67 мкл, 0,64 ммоль) и карбоната цезия (249 мг, 0,77 ммоль), суспендированных в 1,4-диоксане (5 мл). Полученную суспензию дегазировали аргоном и перемешивали при 100°C в течение 20 ч. При охлаждении до комнатной температуры нерастворимые вещества удаляли фильтрованием и фильтрат концентрировали. Неочищенный продукт адсорбировали на силикагеле и очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 6% МеОН в EtAc. Растворитель выпаривали досуха с получением метил-2-((R)-2-метилморфолино)-4-оксо-8-(1-фенилпирролидин-2-ил)-4Н-хромен-6-карбоксилата (112 мг, 49%) в виде бежевой пены. Масс-спектр: m/z [M+H]+=449.
Стадия 2
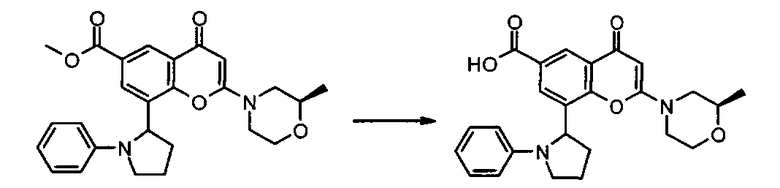
Гидроксид натрия (2 н. в воде) (0,293 мл, 0,59 ммоль) добавляли к перемешиваемой суспензии метил-2-((R)-2-метилморфолино)-4-оксо-8-(1-фенилпирролидин-2-ил)-4Н-хромен-6-карбоксилата (105 мг, 0,23 ммоль) в смеси МеОН (2 мл)/вода (2 мл), как описано в примере 3.00, с получением 2-((R)-2-метилморфолино)-4-оксо-8-(1-фенилпирролидин-2-ил)-4Н-хромен-6-карбоновой кислоты (87 мг, 86%), которую использовали без дополнительной очистки. Масс-спектр: m/z [M+H]+=435.
Пример 3.02
8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он
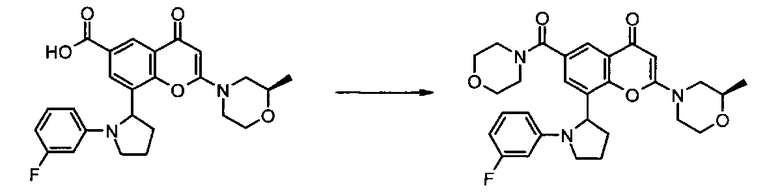
TBTU (138 мг, 0,43 ммоль) под азотом добавляли к перемешиваемому раствору 8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоновой кислоты (97 мг, 0,21 ммоль), DIPEA (0,075 мл, 0,43 ммоль), растворенных в DMF (1,5 мл). Полученный раствор перемешивали при комнатной температуре в течение 1,5 ч. Добавляли морфолин (0,028 мл, 0,32 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь очищали препаративной ВЭЖХ. Фракции выпаривали досуха, остаток растворяли в DCM, сушили над MgSO4 и выпаривали досуха с получением 8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-она (63 мг, 56%) в виде желтой пены.
Масс-спектр: m/z [M+H]+=533.
Спектр протонного ЯМР: (DMSO-d6) 1.17 (d, 3H), 1.77-1.90 (m, 1Н), 1.98-2.10 (m, 2Н), 2.47-2.57 (m, частично скрыт DMSO d6, 1Н), 2.78-2.86 (m, 1Н), 3.04 (bs, 2Н), 3.09-3.18 (m, 1Н), 3.22 (bs, 2Н), 3.34-3.41 (m частично скрыт H2O, 1Н), 3.42 (bs, 2Н), 3.54 (bs, 2Н), 3.58-3.70 (m, 2Н), 3.73-3.80 (m, 1Н), 3.85-4.06 (m, 3H), 5.25 (d, 1Н), 5.63 (s, 0.5Н), 3.64 (s, 0.5Н), 6.19-6.29 (m, 2Н), 6.38 (ddd, 1Н), 7.05-7.13 (m, 2Н), 7.81 (d, 1Н).
8-(1-(3-Фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоновую кислоту, используемую в качестве исходного материала, получали следующим образом.
Стадия 1
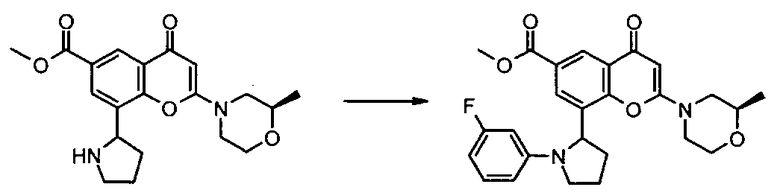
Диацетоксипалладий (8,44 мг, 0,04 ммоль) добавляли к перемешиваемой смеси метил-2-((К)-2-метилморфолино)-4-оксо-8-(пирролидин-2-ил)-4Н-хромен-6-карбоксилата (0,35 г, 0,94 ммоль), (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфина) (0,046 г, 0,08 ммоль), 1-бром-3-фторбензола (0,131 мл, 1,17 ммоль) и карбоната цезия (0,459 г, 1,41 ммоль), суспендированных в 1,4-диоксане (9 мл). Полученную суспензию дегазировали аргоном и затем перемешивали при 100°C в течение 20 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры, нерастворимые вещества удаляли фильтрованием и фильтрат концентрировали. Неочищенный продукт адсорбировали на силикагеле и очищали посредством флэш-хроматографии на силикагеле, элюируя от 0 до 10% МеОН в EtAc. Растворители выпаривали досуха с получением метил-8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксилата (0,279 г, 64%) в виде бежевой пены. Масс-спектр: m/z [M+H]+=467.
Стадия 2
Водный 2 н. раствор NaOH (0,804 мл, 1,61 ммоль) добавляли к метил-8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксилату (0,25 г, 0,54 ммоль), суспендированному в МеОН (4 мл). Раствор перемешивали при 40°C в течение ночи, затем охлаждали до 0°C и по каплям добавляли к реакционной смеси водный раствор 2 н. HCl (0,670 мл, 1,34 ммоль) до pH приблизительно 3. Полученный осадок собирали фильтрованием, промывали водой и сушили под вакуумом при 50°C в присутствии Р205 до постоянной массы с получением 8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоновой кислоты (201 мг, 83%) в виде бежевого твердого вещества.
Масс-спектр: m/z [M+H]+=453.
Пример 3.03
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-((R)-2-метил-морфолино)-4-оксо-4Н-хромен-6-карбоксамид
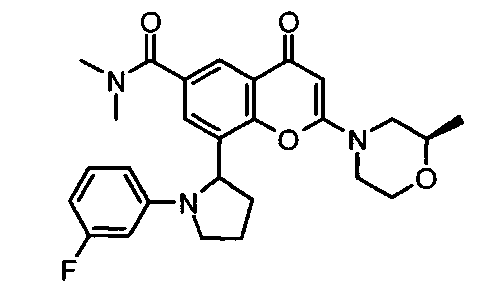
8-(1-(3-Фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоновую кислоту (102 мг, 0,23 ммоль) подвергали взаимодействию с диметиламином (2 н. в THF) (0,169 мл, 0,34 ммоль), как описано в Примере 3.02, с получением 8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксамида (51 мг, 47%) в виде бледно-желтой пены.
Масс-спектр: m/z [M+H]+=480.
Спектр протонного ЯМР: (DMSO-d6) 1.17 (d, 3H), 1.77-1.90 (m, 1Н), 1.98-2.08 (m, 2Н), 2.45-2.58 (m, частично скрыт DMSOd6, 1Н), 2.69 (s, 3H), 2.78-2.86 (m, 1Н), 2.90 (s, 3H), 3.09-3.18 (m, 1Н), 3.33-3.39 (m частично скрыт Н20, 1Н), 3.58-3.70 (m, 2Н), 3.72-7.79 (m, 1Н), 3.85-4.07 (m, 3H), 6.24 (d, 1Н), 5.63 (s, 0.5Н), 5.64 (s, 0.5Н), 6.21-6.29 (m, 2Н), 6.33-6.40 (m, 1Н), 7.04 (ddd, 1Н), 7.13 (d, 0.5Н), 7.14 (d, 0.5Н), 7.80 (d, 1Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ТЕТРАЗОЛОНА | 2014 |
|
RU2664644C2 |
| ПИРИМИДИЛЦИКЛОПЕНТАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ AKT-ПРОТЕИНКИНАЗЫ | 2008 |
|
RU2486178C2 |
| СОЕДИНЕНИЕ АМИНОПИРАЗОЛОПИРИМИДИНА, ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ТИРОЗИНКИНАЗНОГО РЕЦЕПТОРА НЕЙРОТРОФИЧЕСКОГО ФАКТОРА | 2017 |
|
RU2764523C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ИНГИБИТОРЫ ОКСАЗИНМОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ (MAGL) | 2019 |
|
RU2794334C2 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ, ПРОТИВОВИРУСНЫЙ АКТИВНЫЙ КОМПОНЕНТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2010 |
|
RU2452735C1 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2013 |
|
RU2627706C2 |
| Триазолпиримидиновые соединения и их применение в лечении рака | 2019 |
|
RU2793249C2 |
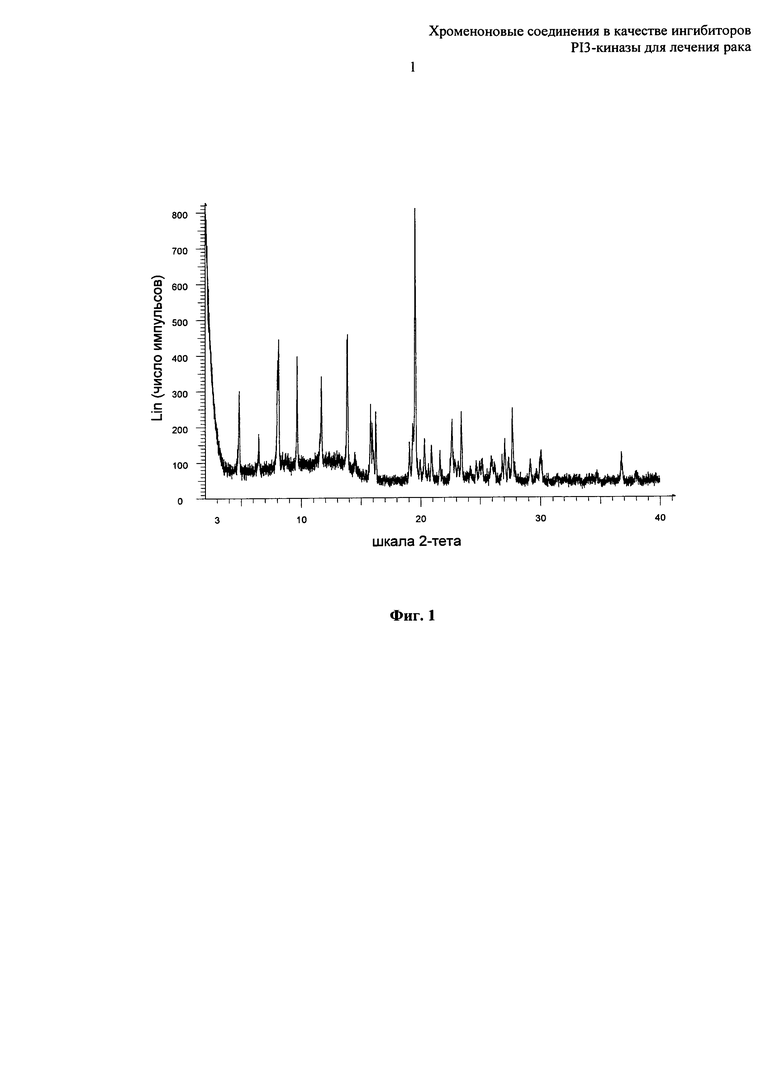
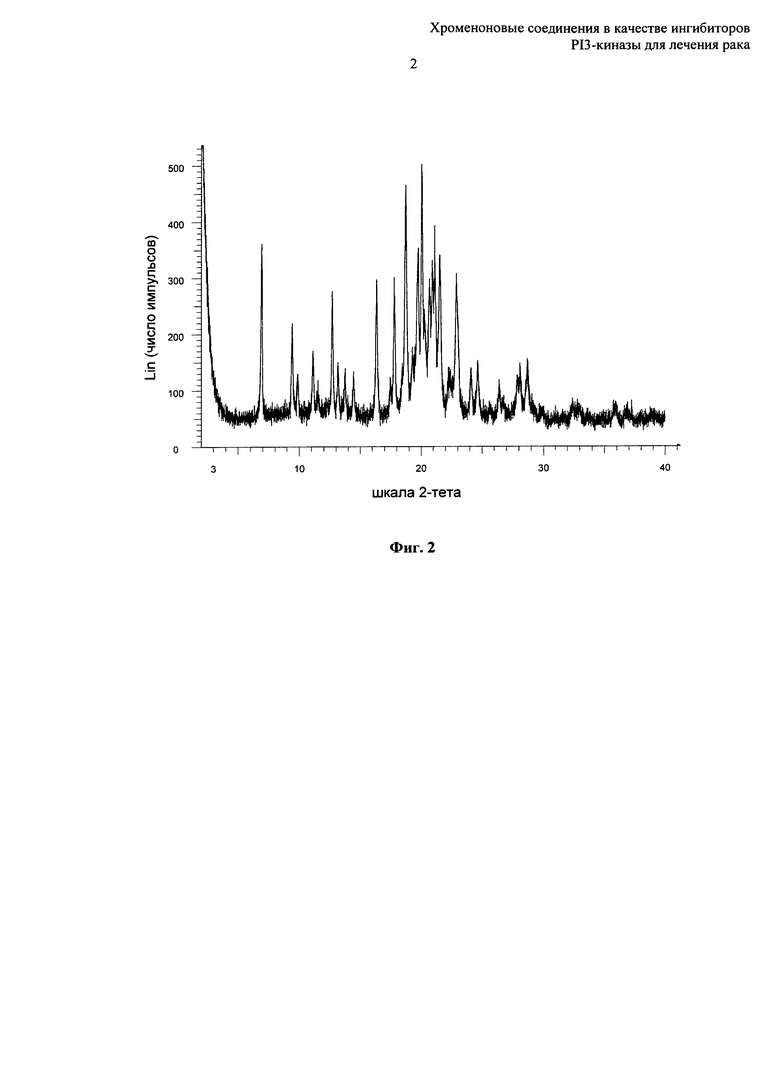

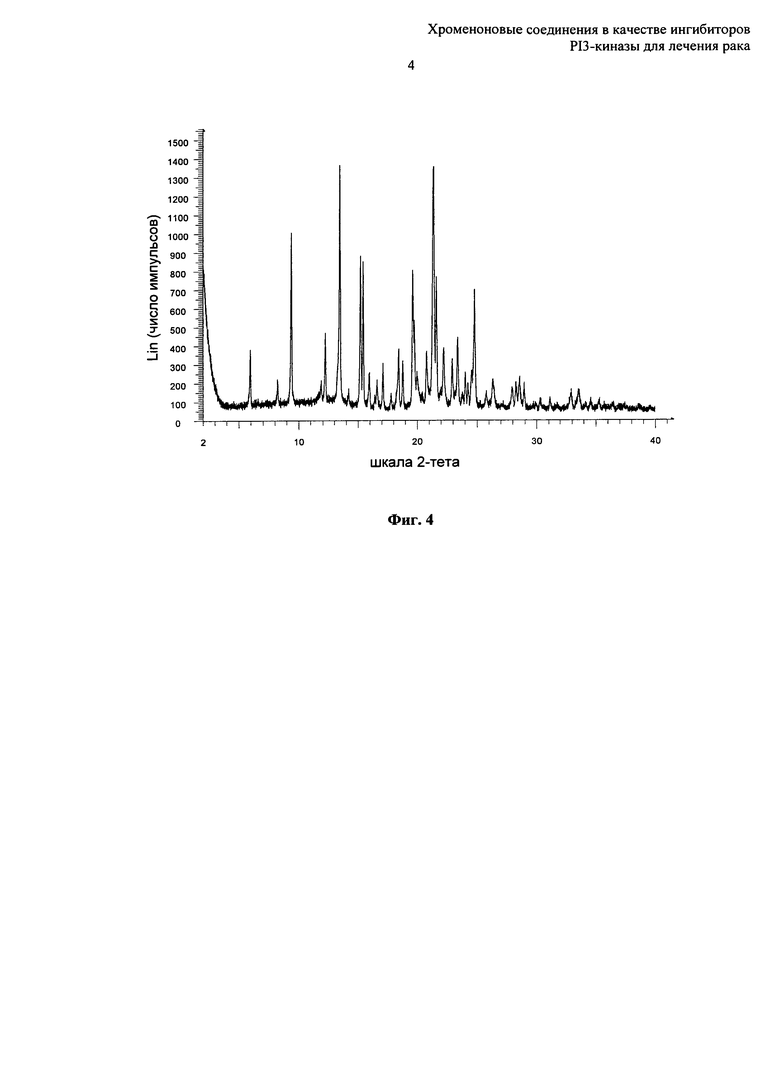
Изобретение относится к хроменоновым соединениям формулы I, обладающим свойствами ингибитора ферментов PI3-киназ, или его фармацевтически приемлемым солям, фармацевтическим композициям на их основе и способу лечения или профилактики опухолей, чувствительных к ингибированию ферментов PI3-киназ, с их использованием. В общей формуле I R1 представляет собой С1-4алкил, возможно замещенный гидрокси; R2 представляет собой С1-4алкил; или R1 и R2 вместе образуют 4-6-членную азотсодержащую гетероциклильную кольцевую систему, которая возможно содержит 1 дополнительный гетероатом, выбранный из кислорода и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов), где указанное кольцо возможно замещено гидрокси; R3 и R5 независимо выбраны из Н, галогена, C1-3алкокси и циано; R4 представляет собой Н или фтор; n равен 0 или 1. 4 н. и 11 з.п. ф-лы, 4 ил., 3 пр., 1 табл.
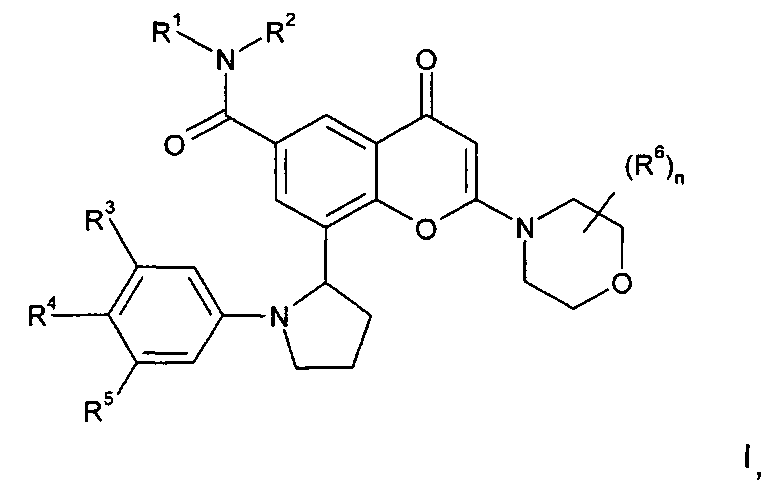
1. Соединение формулы I:
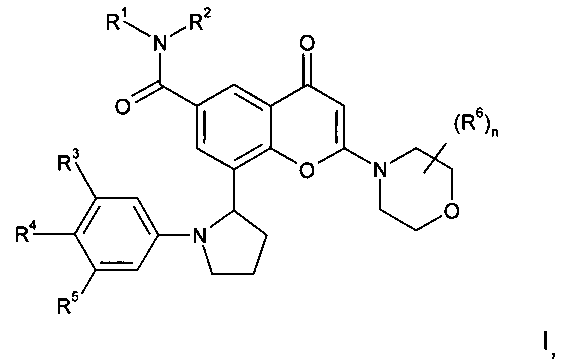
в которой:
R1 представляет собой С1-4алкил, возможно замещенный гидрокси;
R2 представляет собой С1-4алкил; или
R1 и R2 вместе образуют 4-6-членную азотсодержащую гетероциклильную кольцевую систему, которая возможно содержит 1 дополнительный гетероатом, выбранный из кислорода и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов), где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из Н, галогена, C1-3алкокси и циано;
R4 представляет собой Н или фтор;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или его фармацевтически приемлемая соль.
2. Соединение формулы I по п. 1, где:
R1 представляет собой метил или этил, возможно замещенный гидрокси;
R2 представляет собой метил или этил; или
R1 и R2 вместе образуют 4-6-членную азотсодержащую гетероциклильную кольцевую систему, которая возможно содержит 1 дополнительный гетероатом, выбранный из кислорода и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов), где указанное кольцо возможно замещено гидрокси; или его фармацевтически приемлемая соль.
3. Соединение формулы I по п. 1, где R3 и R5 независимо выбраны из Н, фтора, метокси и циано; или его фармацевтически приемлемая соль.
4. Соединение формулы I по п. 1, где R4 представляет собой Н; или его фармацевтически приемлемая соль.
5. Соединение формулы I по п. 1, где n равен 0; или его фармацевтически приемлемая соль.
6. Соединение формулы I по п. 1, где:
R1 представляет собой С1-4алкил, возможно замещенный гидрокси;
R2 представляет собой С1-4алкил; или
R1 и R2 вместе образуют 4-6-членную азотсодержащую гетероциклильную кольцевую систему, которая возможно содержит 1 дополнительный гетероатом, выбранный из кислорода и серы, где кольцевой атом серы возможно окислен с образованием S-оксида(ов), где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из Н или галогена;
R4 представляет собой Н;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или его фармацевтически приемлемая соль.
7. Соединение формулы I по п. 1, где:
R1 представляет собой метил, этил или 2-гидроксиэтил;
R2 представляет собой метил или этил; или
R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидинила, морфолинила, 1-оксотетрагидро-1,4-тиазинила и пиперидинила, где указанное кольцо возможно замещено гидрокси;
R3 и R5 независимо выбраны из Н или галогена;
R4 представляет собой Н;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или его фармацевтически приемлемая соль.
8. Соединение формулы I по п. 1, где:
R1 представляет собой метил или 2-гидроксиэтил;
R2 представляет собой метил; или
R1 и R2 вместе образуют азотсодержащую гетероциклильную кольцевую систему, выбранную из азетидин-1-ила, морфолин-4-ила, 1-оксотетрагидро-1,4-тиазин-4-ила, пиперидин-1-ила и 4-гидроксипиперидин-1-ила;
R3 и R5 независимо выбраны из Н, фтора, метокси и циано;
R4 представляет собой Н или фтор;
n равен 0 или 1, и когда n равен 1, группа R6 представляет собой метил; или его фармацевтически приемлемая соль.
9. Соединение формулы I по п. 1, выбранное из любого из следующих:
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он;
8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-морфолино-4Н-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2S)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид;
8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-хромен-6-карбоксамид;
8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-N,N-диметил-2-морфолино-4-оксо-хромен-6-карбоксамид;
8-(1-(3-фторфенил)пирролидин-2-ил)-6-(морфолин-4-карбонил)-2-морфолино-4Н-хромен-4-он;
8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-(1-(3-фторфенил)пирролидин-2-ил)-2-морфолино-4Н-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2S)-1-(3-фторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
6-(азетидин-1-карбонил)-8-[(2R)-1-(3-фторфенил)пирролидин-2-ил]-2-морфолино-хромен-4-он;
8-(1-(3,5-дифторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он;
8-(1-(3-фторфенил)пирролидин-2-ил)-2-((R)-2-метилморфолино)-6-(морфолин-4-карбонил)-4Н-хромен-4-он; и
8-(1-(3-фторфенил)пирролидин-2-ил)-N,N-диметил-2-((R)-2-метилморфолино)-4-оксо-4Н-хромен-6-карбоксамид; или его фармацевтически приемлемая соль.
10. Соединение формулы I, представляющее собой 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он; или его фармацевтически приемлемая соль.
11. Соединение формулы I по п. 10, представляющее собой 8-[(2R)-1-(3,5-дифторфенил)пирролидин-2-ил]-6-(морфолин-4-карбонил)-2-морфолино-хромен-4-он.
12. Фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль по любому из пп. 1-11 вместе с фармацевтически приемлемым разбавителем или носителем, для использования в предупреждении или лечении опухолей, которые чувствительны к ингибированию ферментов PI3-киназ.
13. Соединение формулы I или его фармацевтически приемлемая соль по любому из пп. 1-11 для применения в терапии в качестве ингибитора ферментов PI3-киназ.
14. Производное хроменона формулы I или его фармацевтически приемлемая соль по любому из пп. 1-11 для применения в предупреждении или лечении опухолей, которые чувствительны к ингибированию ферментов PI3-киназ.
15. Способ лечения или профилактики опухолей, которые чувствительны к ингибированию ферментов PI3-киназ, у теплокровного животного, включающий введение указанному животному эффективного количества соединения формулы I или его фармацевтически приемлемой соли по любому из пп. 1-11.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ИНГИБИТОРЫ ЦИКЛИН-ЗАВИСИМЫХ КИНАЗ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2334746C2 |

