Изобретение относится к области фармацевтической химии, а именно к химии пептидов и более конкретно к новому способу получения нонапептидэтиламида формулы
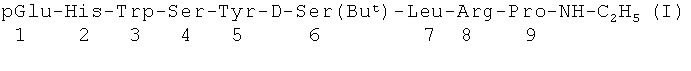
и промежуточным соединениям для его получения.
Нонапептидэтиламид I известен под названием бусерелин и представляет собой синтетический аналог природного гонадотропин-рилизинг гормона, обладающий сильной LH-RH/FSH-RH-активностью (Sandow J., Konig W. Studies with Fragments a Highly active Analogues of Luteinizing Hormone - Releasing Hormone, J. Endocrinol, 1979, 81(2), 175-182) [1].
Из RU №2086561, 10.08.1997 (смотри [2]), известен способ получения нонапептидэтиламида, обладающего сильной LH-RH/FSH-RH активностью, формулы

Нонапептидэтиламид получается конденсацией пептидных фрагментов 1-4 формулы

и 5-10 формулы
H-Tyr-D-Ser(But)-Leu-Arg-Pro-NH-C2H5
с последующей очисткой и выделением.
Способ осуществляется по следующей схеме 4+5, согласно которому тетрапептид формулы (II)

конденсируют с пентапептидом формулы (III)
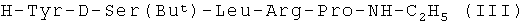 ,
,
полученным каталитическим гидрогенолизом защищенного пентапептида формулы (IV)
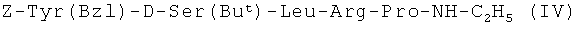 ,
,
где Z - карбобензоксигруппа;
Bzl - бензильная группа.
При конденсации тетрапептида (II) и пентапептида (III) могут быть использованы обычные в пептидном синтезе методы конденсации, например карбодиимидный метод.
Тетрапептид формулы (II) является известным и используется в синтезе полипептидов.
Известный способ позволяет получить конечный продукт с высоким выходом, но не обеспечивает высокий выход промежуточных продуктов на отдельных стадиях.
Наиболее близким по технической сущности и достигаемому результату к предложенному способу является способ получения нонапептидэтиламида формулы (I) конденсаций пептидных фрагментов по схеме 3+6 азидным методом, согласно которому трипептид формулы pGlu-His-Trp-NH-NH2 подвергают взаимодействию с гексапептидом формулы H-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NH-C2H5·2HCl в диметилформамиде при -30°С в присутствии трет-бутилнитрита и 6 н. HCl. Целевой продукт очищают и выделяют в виде моноацетатной соли (Konig W, Geiger R., Sandow J.K. Peptides having LH-RH/FSH-RH activity. Патент США №4024248 (1977) [3].
Недостатком известного способа получения бусерелина является низкий выход целевого продукта и необходимость использования азидного метода на последней стадии. Азидный метод предполагает использование высокотоксичных соединений, таких как трет-бутилнитрит. Кроме того, синтез пептидов азидным методом является длительными процессом, который проводят при строго контролируемом низком температурном режиме (см. Пептиды. Основные методы образования пептидных связей. М.: Мир, 1983 г.) [4].
Все это делает известный способ малопригодным для получения бусерелина в промышленном масштабе.
Технической задачей заявленного способа является упрощение процесса получения бусерелина и повышение выхода конечного продукта. Поставленная задача достигается заявленной группой изобретений.
Поставленная задача достигается описываемым способом, согласно которому синтез бусерелина осуществляют путем конденсации С-концевого тетрапептида формулы
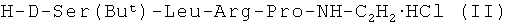
с дипептидом формулы Х-Ser-Tyr-OH,
где Х - защитная группа,
полученный N-защищенный гексапептидэтиламид формулы
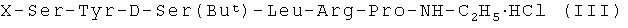
обрабатывают деблокирующими агентом для удаления N-защитной группы, а затем конденсируют с трипептидом формулы 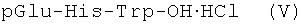 ,
,
конечный продукт очищают с помощью хроматографии и выделяют в виде моноацетата (или моноацетатной соли).
Изобретение относится также и к промежуточным соединениям, используемым для получения нонапептидэтиламида заявленным способом.
Итак, заявленное изобретение относится также к N-защищенному дипептиду формулы (IV)
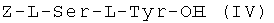 ,
,
где Z - бензилоксикарбонил,
в качестве промежуточного соединения для синтеза нонапептидэтиламида формулы (I)

В группу заявленного изобретения входит также и другое промежуточное соединение - трипептид формулы (V)
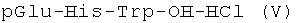
в качестве промежуточного соединения для синтеза нонапептидэтиламида формулы (I)
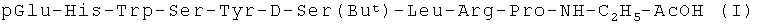
При этом Х в формуле дипептида (IV) представляет собой бензилоксикарбонил, в частности, это Z в формуле N-защищенного дипептида формулы (IV).
Деблокирование N-защищенного гексапептидэтиламида осуществляют путем каталитического гидрирования в присутствии 5% Pd/C (палладия на угле).
Список сокращений
Boc - трет-бутилоксикарбонил
Z - бензилоксикарбонил
DCC - N,N'-дициклогексилкарбодиимид
DCCU - N,N'-дициклогексилмочевина
ДМФА - N,N-диметилформамид
NMM - N-метилморфолин
HONB - N-гидрокси-5-нонборнен-2,3-дикарбоксиимид
ТГФ - тетрагидрофуран
HOBt - 1-гидроксибензотриазол
BSA - N,O-бис-триметилсилилацетамид
МеОН - метанол
ВЭЖХ - высокоэффективная жидкостная хроматография
ТСХ - тонкослойная хроматография
ПЭ - петролейный эфир
ЭА - этилацетат
Экспериментальная часть
В синтезе используют производные L-аминокислот фирмы Bachem (Швейцария), DCC, HONB, HOBt, BSA фирмы Fluka (Швейцария). ДМФА очищают перегонкой над нингидрином и окисью бария. Аналитическую ВЭЖХ проводят на хроматографе фирмы Gilson (Франция).
ТСХ проводят на стеклянных хроматографических пластинках Merck-Kiselgel (ФРГ).
1Н-ЯМР - спектры снимают на спектрометре WH-500 Bruker 500 МГц (ФРГ) в DMSO-d6 при 300 К. Химические сдвиги (δ, м.д.) измеряют относительно тетраметилсилана. Масс-спектры регистрировали на масс-спектрометре Analytical Compact MALDI 4 фирмы Kratos (Великобритания).
Изобретение иллюстрируется следующим примером.
Пример. 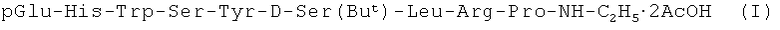 .
.
1. Nα-трет-Бутилоксикарбонил-L-лейцил-L-аргинил-L-пролинэтиламида гидрохлорид Boc-Leu-Arg-Pro-NHEt·HCl.
В трехгорлую колбу на 1 л, снабженную механической мешалкой, термометром и капельной воронкой, загружают 74,4 г (0,19 моль) Boc-Leu-Arg-OH, 35,7 г (0,2 моль) H-Pro-NHEt·HCl, 44,1 г (0,29 моль) HOBt·H2O, 300 мл ДМФА и перемешивают до полного растворения. Реакционную смесь охлаждают на ледяной бане до 0°С и при интенсивном перемешивании и температуре 0 - -5°С прикапывают раствор 39,6 г (0,19 моль) DCC в 50 мл, реакционную массу перемешивают 30 мин при t=0-5°С, оставляют на ночь, выпавшую DCCU отфильтровывают, промывают на фильтре 50 мл ДМФА. Фильтрат упаривают на роторном испарителе. Остаток растворяют в 400 мл воды и оставляют на ночь в холодильнике при t=5°С, выпавший осадок отфильтровывают, фильтрат экстрагируют эфиром (2×50 мл), упаривают на роторном испарителе и к остатку добавляют при перемешивании 100 г NaCl. Выпавшее масло экстрагируют хлороформом (2×200 мл) и экстракт промывают водой (3×40 мл). Хлороформ упаривают, остаток растворяют в 200 мл бутанола. Раствор упаривают на роторном испарителе, остаток затирают в 1 л эфира, продукт отфильтровывают, промывают на фильтре 100 мл эфира и сушат на воздухе.
В итоге получают 93,7 г (90%) Boc-Leu-Arg-Pro-NHEt·HCl. Продукт хроматографически однороден.
2. N-Оксинорборнен-2,3-дикарбоксиимидный эфир N-карбобензокси-О-трет-бутил-D-серина Z-D-Ser(But)-ONB.
В трехгорлую колбу на 1 л, снабженную механической мешалкой, термометром и капельной воронкой, загружают 51,7 г (0,17 моль) Z-D-Ser(But)-OH, 34,4 г (0,19 моль) HONB, 350 мл этилацетата и перемешивают до образования однородной суспензии. Реакционную смесь охлаждают на лед. бане до t=0°C и при интенсивном перемешивании прикапывают при температуре 0-5°С раствор 41,3 г (0,2 моль) DCC в 50 мл этилацетата. После добавления DCC реакционную массу перемешивают 30 мин при t=0-5°С и оставляют при перемешивании на ночь. Выпавший осадок DCCU отфильтровывают, промывают на фильтре 50 мл этилацетата, фильтрат упаривают досуха на роторном испарителе, к остатку добавляют 200 мл изопропанола и перемешивают при t=60°С до полного растворения (20 мин). Раствор охлаждают до t=0°С и перемешивают до выпадения обильного осадка, добавляют 200 мл петролейного эфира и помещают реакционную массу на ночь в морозильную камеру при температуре -20°С. Затем выпавший осадок отфильтровывают, промывают на фильтре 100 мл петролейного эфира и сушат на воздухе. В итоге получают 66,0 г (85%) Z-D-Ser(But)-ONB.
3. N-Карбобензокси-O-трет-бутил-D-серил-L-лейцил-L-аргинил-L-пролинэтиламид гидрохлорид Z-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl.
В одногорлую колбу емкостью 500 мл загружают 73,5 г (0,13 моль) Boc-Leu-Arg-Pro-NHEt·HCl, 150 мл диоксана, перемешивают до полного растворения, добавляют 150 мл 11% HCl в диоксане. Реакционную смесь выдерживают 1,5 ч, периодически перемешивая, выпавший осадок отфильтровывают, промывают на фильтре эфиром (2×100 мл) и сушат в вакууме. Полученный продукт загружают в двугорлую колбу емкостью 500 мл, снабженную механической мешалкой, добавляют 220 мл ДМФА и перемешивают до полного растворения. Добавляют 30,8 мл (0,28 моль) NММ, 63,9 г (0,14 моль) Z-D-Ser(But)-ONB, перемешивают 3 ч и помещают реакционную массу на ночь в холодильник при t=5°C. Отфильтровывают выпавший осадок NMM·HCl. Фильтрат упаривают на роторном испарителе до объема 70 мл, добавляют 70 мл воды, промывают эфиром (3×25 мл), (ТСХ 2) и упаривают на роторном испарителе. Остаток растворяют в 150 мл хлороформа, промывают водой (3×20 мл) и упаривают на роторном испарителе. Остаток затирают в эфире (500 мл), эфир декантируют, осадок сушат в вакууме. В итоге получают 91,4 г (96,3%) Z-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl. По данным ТСХ (хлороформ - метанол - уксусная кислота - вода 30:6:2:0,8) продукт хроматографически однороден.
4. О-трет-Бутил-D-серил-L-лейцил-L-аргинил-L-пролинэтиламида гидрохлорид (II).
H-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl,
где Et - C2H5.
В двугорлую колбу на 1 л, снабженную механической мешалкой, загружают 91,4 г (0,13 моль) Z-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl, добавляют 300 мл абс. этанола и перемешивают до полного растворения. Добавляют 0,1 г Pd(OH)2 и барботируют реакционную массу газообразным водородом при перемешивании в течение ночи. Затем катализатор отфильтровывают, фильтрат упаривают на роторном испарителе. Остаток затирают в эфире (400 мл), осадок отфильтровывают, промывают на фильтре 100 мл эфира и сушат на воздухе. В итоге получают 73,9 г (96,5%) H-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl (II).
По данным ТСХ (хлороформ - метанол - уксусная кислота - вода 30:6:2:0,8) продукт однороден.
5. N-Карбобензокси-L-серил-L-тирозина (IV) Z-Ser-Tyr-OH (Z-L-Ser-L-Tyr-OH).
В трехгорлую колбу на 1 л, снабженную обратным холодильником, загружают 29,3 г (0,16 моль) Н-Tyr-ОН, 109,8 г (0,54 моль) BSA, 250 мл хлористого метилена, полученную реакционную смесь кипятят 7-8 часов до полного растворения Н-Tyr-ОН. Реакционную смесь охлаждают на ледяной бане до 0°С и при интенсивном перемешивании присыпают при 0-5°С 70 г (0,16 моль) Z-Ser-OPfp. Затем реакционную массу перемешивают 30 мин при 0-5°С и оставляют при перемешивании и комнатной температуре на ночь. Контроль реакции ведут с помощью ТСХ в системе: ПЗ : ЭА 1:1. Упаривают реакционную массу на роторном испарителе (t≤40°C) и растворяют остаток в 150 мл ЭА, раствор помещают на ледяную баню, охлаждают до t=0°C и при интенсивном перемешивании прикапывают 50 мл 5% H2SO4. Через 10 минут охлаждение прекращают и перемешивают реакционную массу при 30-35°С до выпадения обильного однородного осадка и помещают на ночь в холодильник (5°С). Затем реакционную массу фильтруют, промывают на фильтре 2×50 мл смесью: водн. NaCl(насыщ): Н2О (1:1), 50 мл смеси: ПЭ : ЭА (3:2) и сушат на воздухе. В итоге получают 61,2 г (95%) Z-Ser-Tyr-OH в виде хроматографически однородного продукта. ТСХ в системе: ПЭ : ЭА 1:1.
6. N-Карбобензокси-L-серил-L-тирозил-О-трет-бутил-D-серил-L-лейцил-L-аргинил-L-пролинэтиламида гидрохлорид (III).
H-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl, где Et - C2H5.
В трехгорлую колбу на 1 л, снабженную механической мешалкой, термометром и капельной воронкой, загружают 73,9 г (0,12 моль) H-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl, 55,1 г (0,14 моль) Z-Ser-Tyr-OH, 27,3 г (0,18 моль) HOBt·Н2О, 500 мл ТГФ, нагревают до 45°С и перемешивают до полного растворения. Затем реакционную смесь охлаждают до комнатной температуры и при перемешивании прикапывают в течение 4-5 ч раствор 28,3 г (0,14 моль) DCC в 70 мл ТГФ. После добавления DCC реакционную массу перемешивают 1 ч и помещают на ночь в морозильную камеру при -20°С. Выпавший осадок отфильтровывают, промывают на фильтре 130 мл ТГФ и используют в дальнейшем без сушки.
7. L-Серил-L-тирозил-О-трет-бутил-D-серил-L-лейцил-L-аргинил-L-пролинэтиламида гидрохлорид.
H-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl, где Et - C2H5.
В двугорлую колбу на 1 л, снабженную механической мешалкой, загружают 105 г (0,12 моль) Z-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl, добавляют 350 мл абс. этанола, перемешивают до получения однородной суспензии и отфильтровывают DCCU. Промывают осадок на фильтре 70 мл абс. этанола, добавляют Pd(OH)2 и барботируют реакционную массу газообразным водородом при перемешивании в течение ночи. Затем катализатор отфильтровывают, фильтрат упаривают на роторном испарителе. Остаток затирают в этилацетате (650 мл), отфильтровывают, промывают на фильтре 150 мл ЭА и сушат на воздухе. В итоге получают 91 г (90%) H-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl, где Et - C2H5.
8. L-Гистидил-L-триптофана дигидрохлорид H-His-Trp-OH·2HCl.
В одногорлую колбу емкостью 1 л загружают 124,6 г (0,23 моль) (Вос)2-His-Trp-OH, 300 мл диоксана и перемешивают до полного растворения, раствор помещают на лед. баню и добавляют 300 мл 11% HCl в диоксане. Реакционную смесь выдерживают 1 ч на лед. бане, периодически встряхивая. Доводят температуру реакционной массы до комнатной, выдерживают 1,5 часа, выпавший осадок отфильтровывают, осадок, затирают в 500 мл эфира, отфильтровывают, промывают на фильтре эфиром (2×100 мл) и сушат в вакууме. В итоге получают 80 г (84%) H-His-Trp-OH·2HCl.
9. L-Пироглутамил-L-гистидил-L-триптофана гидрохлорид pGlu-His-Trp-OH·HCl (V).
В двугорлую колбу на 1 л, снабженную механической мешалкой, загружают 75,8 г (0,18 моль) H-His-Trp-OH·2HCl, добавляют 400 мл ДМФА и перемешивают до полного растворения. Колбу помещают на лед. баню, охлаждают до t=0°C, добавляют при перемешивании 40,6 мл (0,37 моль) NMM и 59,0 г (0,2 моль) Pyr-Glu-OPfp. Перемешивают 30 мин при t=0-5°С, затем 2 ч при комнатной температуре. Помещают реакционную массу на ночь в холодильник при t=5°С, отфильтровывают выпавший NMM·HCl и промывают осадок 100 мл ДМФА. ДМФА упаривают на роторном испарителе. Остаток затирают в ацетоне (600 мл), отфильтровывают, продукт на фильтре промывают 100 мл ацетона и сушат в вакууме. В итоге получают 70 г (80%) pGlu-His-Trp-ОН·HCl.
10. L-Пироглутамил-L-гистидил-L-триптофил-L-серил-L-тирозил-О-трет-бутил-D-серил-L-лейцил-L-аргинил-L-пролин-N-этиламид ацетат
pGlu-His-Trp-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NHEt·АсОН (бусерелин ацетат).
В двугорлую колбу на 1 л, снабженную механической мешалкой и капельной воронкой, загружают 53,8 г (0,11 моль) H-Pyr-His-Trp-OH·HCl, 84,1 г (0,1 моль) H-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NHEt·HCl, 21,4 г (1,4 моль) HOBt·Н2О, добавляют 400 мл ДМФА и перемешивают до полного растворения. Добавляют 11,1 г (0,11 моль) NMM и при перемешивании прикапывают в течение 6-8 ч раствор 35,1 г (0,17 моль) DCC в 50 мл ДМФА. Затем реакционную массу перемешивают при комнатной температуре 10 ч. Отфильтровывают выпавшую DCCU, промывают на фильтре 50 мл ДМФА, фильтрат упаривают на роторном испарителе. Остаток затирают в ТГФ (900 мл), осадок отфильтровывают, промывают 100 мл ТГФ и, не высушивая, подвергают хроматографической очистке.
Для очистки продукт растворяют в 0,05 М пиридин-ацетатном буфере, наносят на колонку 50×600 мм с SP-сефадексом и элюируют в градиенте 0,05-1 М пиридин-ацетатного буфера. Выделенные фракции упаривают, растворяют в 2%-ной уксусной кислоте и лиофилизуют.
После выделения продукта с помощью препаративной ВЭЖХ получено 70,3 г (50%) продукта с 98% чистотой.
Продукт хроматографически однороден (ТСХ, система Е), Rf=0,53 (1). Аминокислотный анализ: Glu 0,91 (1), His 0,93 (1), Trp 0,83 (1), Ser 1,90 (2), Tyr 0,93 (1), Leu 1,00 (1), Arg 1,06 (1), Pro 1,11 (1). [α]D 20=-45.3°(с=1; вода). ТСХ проводится на пластинках Merck Kieselgel-60 в системах: Д. Проявление производится обработкой пластинок хлором и затем раствором толидина.
Как видно из приведенного примера, предложенный способ позволяет значительно упростить процесс получения бусерелина за счет отказа от использования азидного метода на стадии получения конечного продукта и за счет отказа от использования бензильной защиты для боковой функции тирозина и повысить выход целевого продукта.
Дополнительными преимуществами заявленного способа являются:
- по целевой стадии - конденсация трипептида с гексапептидом с выходом целевого продукта 70% против 62% при конденсации тетрапептида с пентапептидом;
- выходы промежуточных продуктов возросли (с 60 до 70% и с 70 до 80% по однотипным реакциям), что сказалось на увеличении суммарного выхода целевого продукта;
- количество защитных групп существенно уменьшилось: с 6 до 4 (ранее применялись 1 Вос-защита и 5 Z-защит, в настоящем патенте соответственно 2 и 2), что снизило расход дорогостоящего катализатора Pd(OH)2 и повысило безопасность процесса благодаря снижению числа стадий каталитического гидрирования.
Источники информации
1. Sandow J., Konig W. Studies with Fragments a Highly active Analogues of Luteinizing Hormone - Releasing Hormone, J. Endocrinol, 1979, 81 (2), 175-182.
2. Патент Российской Федерации №2086561, опублик. 10.08.1997.
3. Konig W., Geiger R., Sandow J. K., Peptides having LH-RH/FSH-RH activity.
Патент США №4024248 (1977) (ближайший аналог).
4. Пептиды. Основные методы образования пептидных связей. - М.: Мир, 1983 г.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БУСЕРЕЛИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2010 |
|
RU2442791C1 |
| СПОСОБ СИНТЕЗА ДЕЗ-ГЛИ 10, /D-ЛЕЙ 6/ LH-RH-ЭТИЛАМИДА | 1994 |
|
RU2074191C1 |
| СПОСОБ ПОЛУЧЕНИЯ НОНАПЕПТИДЭТИЛАМИДА | 1995 |
|
RU2086561C1 |
| ПЕПТИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1994 |
|
RU2067000C1 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА (ADC) И КОНЪЮГАТЫ АНТИТЕЛА И ПРОЛЕКАРСТВА (APDC), СОДЕРЖАЩИЕ ФЕРМЕНТАТИВНО РАСЩЕПЛЯЕМЫЕ ГРУППЫ | 2016 |
|
RU2751512C2 |
| ЗАЩИЩЕННЫЕ ПЕПТИДЫ КАЛЬЦИТОНИНА | 2000 |
|
RU2193567C2 |
| МОДУЛЬНЫЙ МОЛЕКУЛЯРНЫЙ КОНЪЮГАТ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ ГЕНЕТИЧЕСКИХ КОНСТРУКЦИЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2529034C2 |
| Способ получения полипептидов или их солей | 1977 |
|
SU910116A3 |
| ДЕКАПЕПТИД, ОБЛАДАЮЩИЙ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2084458C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПЕПТИДАМИДОВ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ СОВМЕСТИМЫХ АЦЕТАТОВ ИЛИ ГИДРОХЛОРИДОВ | 1991 |
|
RU2036200C1 |
Назначение: изобретение относится к способам получения химическим синтезом нонапептидэтиламида, обладающего сильной LH-RH/FSH-RH активностью, формулы pGlu-His-Trp-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NH-С2Н5·2АсОН (I) и к промежуточным соединениям для его получения. Нонапептидэтиламид получают путем конденсации С-концевого тетрапептида формулы H-D-Ser(But)-Leu-Arg-Pro-NH-C2H5·HCl (II) с дипептидом формулы X-Ser-Tyr-OH (IV), где Х - защитная группа, полученный N-защищенный гексапептидэтиламид формулы X-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NH-C2H5·HCl (III) обрабатывают деблокирующим агентом для удаления N-защитной группы, а затем конденсируют с трипептидом формулы pGlu-His-Trp-OH-HCl (V), конечный продукт очищают с помощью хроматографии и выделяют в виде моноацетатной соли. 3 н. и 1 з.п. ф-лы.
1. Способ получения нонапептидэтиламида формулы:
,
заключающийся в том, что синтез осуществляют путем конденсации С-концевого тетрапептида формулы:
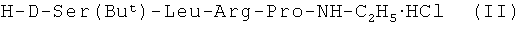
с дипептидом формулы:
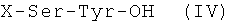 ,
,
где Х - защитная группа и полученный N-защищенный гексапептидэтиламид формулы:
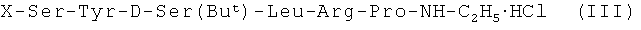
обрабатывают деблокирующим агентом для удаления N-защитной группы и полученный гексапептид формулы:
H-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NH-C2H5
конденсируют с трипептидом формулы:

и конечный продукт очищают с помощью хроматографии и выделяют в виде моноацетатной соли.
2. Способ по п.1, отличающийся тем, что Х представляет собой бензилоксикарбонил (Z), а деблокирование N-защищенного гексапептидэтиламида проводят путем каталитического гидрирования в присутствии 5% Pd/C.
3. Применение N-защищенного дипептида формулы (IV)
Z-L-Ser-L-Tyr-OH,
где Z - бензилоксикарбонил (IV), в качестве промежуточного соединения для синтеза нонапептидэтиламида формулы (I)
.
4. Трипептид формулы (V):
в качестве промежуточного соединения для синтеза нонапептидэтиламида формулы:
pGlu-His-Trp-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-NH-C2H5·2AcOH.
| Устройство для сборки гидравлической запрессовкой деталей типа вал-втулка | 1986 |
|
SU1407744A1 |
| US 4024248 A, 17.03.1977 | |||
| СПОСОБ ПОЛУЧЕНИЯ НОНАПЕПТИДЭТИЛАМИДА | 1995 |
|
RU2086561C1 |
| Способ определения октатиона | 1981 |
|
SU1008656A1 |

