Изобретение относится к усовершенствованному способу получения производных пептидамидов или их физиологически совместимых ацетатов или гидрохлоридов, который может найти применение при получении пептидов с С-концевым азидом азааминокислоты методом синтеза в твердой фазе.
Известны способы получения производных пептидов синтезом в твердой фазе [1,2] путем конденсации N-защищенной аминокислоты с аминокислотой или пептидом, связанными карбоксильной группой с твердым полимером-носителем.
Однако пептиды с С-терминальной азааминогруппой твердофазным синтезом получить до сих пор не удавалось.
Задача изобретения синтез пептидов, содержащих С-концевую терминальную азааминогруппу твердофазным способом при минимальной рацемизации.
Поставленная задача решается описываемым способом получения пептидов общей формулы
(X)n-A-NH2, (I)
в которой Х α -аминокислота из ряда Ac-Nal(2), Ac-D-Nal(2), p-Cl-Phe, p-Cl-D-Phe, Trp,D-Trp, Ser, D-Ser, Tyr, D-Tyr, Ser/ -L-Rhe-D-Ser/α -L-Rhe/, Leu, D-Leu, Arg, D-Arg, Pro, D-Pro, p_Glu, His, D-Ser(tBu);
n 3-10;
A азаглицин,
или их физиологически совместимых ацетатов или гидрохлоридов, заключающийся в том, что соединение общей формулы
R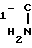 H
H Y3
Y3
(II) в которой у1, у2, у3, у4 и у5 водород, С1-4-алкокси или -(СН2)n-СООН, причем радикалы могут иметь одинаковое или различное значение и по меньшей мере один радикал представляет собой -(СН2)n-СООН; n целое число от 1 до 3,
R1 водород или 4-метоксифенил,
подвергают взаимодействию с силилирующим агентом, выбранным из ряда трет-бутилдиметил-силилхлорид, трет-бутилфенил-силилхлорид, триметилхлорсилан или бис-триметилсилилацетамид, в растворителе с последующей обработкой силилированного соединения производным хлормуравьиной кислоты с образованием соединения формулы Y3
Y3
(III) в которой R1, у1, у2, у3, у4 и у5 имеют вышеуказанные значения, а R2 фенил, замещенный нитрогруппой и галогеном, и взаимодействием полученных соединений формулы с гидразидом аминокислоты формулы
R3-X-CO-NH-NH2 HCl (IV)
в которой Х указанная выше α -аминокислота;
R3 Fmoc-, Bpoc- или Вос-защитные группы,
в растворителе с образованием соединений формулы Y3
Y3
(V) в которой R1, R3 и у1, у2, у3, у4 и у5 имеют указанное выше значение и в случае, если R3 Вос-защитная группа, ее удаляют гидрированием на Pd-катализаторе и перед дальнейшим взаимодействием преобразуют в Fmoc- или Врос-защитную группу, затем соединение формулы (V), где R1, у1, у2, у3, у4 и у5 имеют указанные выше значения, а R3 представляет собой Fmoc или Врос-уретановую защитную группу, копулируют с помощью обычного в химии пептидов реагента через -(CH2)n-COOH-группировку на смоле, защитную группу R3 отщепляют, затем постадийно присоединяют Fmoc- и Врос-защищенные α -аминокислоты, иногда в форме их активированных производных, и по окончании синтеза пептида формулы (I) обработкой кислотой средней силы освобождают от смолы, причем одновременно или с помощью подходящих приемов в заключение отщепляют временно введенные защитные группы боковой цепи.
В случае, если необходимо предотвращение побочных реакций или для синтеза специальных пептидов, функциональные группы в боковых ветвях амино-, азаамино или иминокислот дополнительно защитить подходящими защитными группами (см. T. W. Greene, "Protective Groups in Organic Synthesis", New York, John Wiley a Sons, 1981 Hubbuch, Kontakte (Merck) 1979, Nr 3 Seiten 14-23; Bullebach, Kontakte (merck) 1980, Nr.1, Seiten 23-35) используются в первую очередь Arg(Tos), Arg(Mts), Arg(Mtr), Arg(Pmc), Asp(OBzl), Asp(Otbu), Cys(4-MeBzl), Cys(Acm), Cys(StBu), Glu(OBzl), Glu(OtBu), His(Tos), His(Fmoc), His(Dnp), His(Trt), Lys(Cl-2), Lys(Boc), Met(O), Ser(BZl), Ser(tBu), Thr(Bzl), Thr(tBu).
В дальнейшем функциональные группы в боковых ветвях могут быть также гликозилированы, как, например, в ЕР-А 263521 (НОЕ 86) F 253.
Используемые в качестве носителя смолы имеются в продаже или могут быть изготовлены например алкоксибензилалко- гольные смолы, аминометилсмолы или бензгидриламиносмолы. Предпочтительны аминометил-, бензгидриламино- (ВНА) и метилбензгидраламин-смолы (МВНА). Загрузка определяется анализом аминокислоты и/или элементов.
Под подходящими спейсерами нужно понимать, например, описанные в Atherton, Sheppard in Perspek lives in Peptide Chemistry Seiten 101-117 (Karger, Basel 1981), EP-A 264 802 (HOE 86/F 259), EP-A 287 882 (HOE 87) F 101/ и ЕР-А 322 348 (НОЕ 87/F 386К), а также получаемые от них производные, как, например, те, у которых защитные группы отщеплены. Предпочти- тельны 4-карбоксилатопропокси-4'-метоксибензгидриламин и 5-карбоксилатоэтил-2,4-диметокси-4'-метоксибензгидриламин.
В качестве реагента соединения для спейсера формулы (V) и других производных аминокислоты могут быть использованы все возможные применяемые в синтезе пептидов активирующие реагенты, (см. Houben-Weyl). Методы органической химии, т. XV/2, Штутгарт 1974), но особенно карбодиимиды N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид или N-этил-N'-3- диметиламинопропилкарбодиимид. Соединение при этом может происходить непосредственно через присоединение производного аминокислоты с активирующим реагентом и в данном случае добавкой, подавляющей рацемизирование, как, например, 4-диметиламинопиридин, 1-гидроксибензотриазол (HObt) (W.Konig, R.Geiger, Chem. Ber. 103 (1970) 788-798), 3-гидрокси-4-оксо-3,4- дигидробензотриазин (HOOBt) (W.Konig, R. Geiger, Chem. Ber. 103(1970) 2034-2040)к смоле или предварительное активирование производного аминокислоты в качестве симметричного ангидрида или HOBt или HOOBt эфира может происходить отдельно и раствор активированного вещества в подходящем растворителе добавляться к способной к присоединению пептидной смоле.
Соединение или активирование спейсера формулы V и производных аминокислоты вышеуказанным активирующим реагентом может быть проведено в диметилформамиде или метиленхлориде, или в их смеси. Активированное производное аминокислоты обычно используется в 1,5 до 4-кратного избытке. В случаях, когда соединение неполное, реакция повторяется, без проведения деблокирования α -аминогрупп пептидной смолы, необходимой раньше для сцепления последующих аминокислот.
Окончание реакции соединения может быть проверено посредством нингидрин-реакции (см. E.Kaiser et al. (Anal. Biochem. 34(1970) 595).
Синтез может быть проведен также автоматизировано, например, Peptid-Synthesizer Model 430 A der Fa. Applied Biosystems, при этом могут быть использованы или программы синтеза, предусмотренные изготовителями прибора, или программы, разработанные пользователем. Последнее особенно применяется при использовании производных аминокислот, защищенных Fmoc-группами.
Отщепление пептидамида от смолы происходит путем обработки кислотой средней силы, обычно используемой в синтезе пептидов (например, трифторуксусной кислотой), при этом в качестве ловушки катионов добавляют фенол, крезол, тиокрезол, анизол, тиоанизол, этандитиол, диметилсульфид, этилметилсульфид или подобные, используемые обычно в синтезе твердых фаз, в отдельности или в смеси из двух или более. Трифторуксусная кислота при этом может быть использована разбавленной подходящим растворителем, например метиленхлоридом. При отщеплении спейсера от смолы происходит одновременно отщепление защитных групп боковой цепи.
Очистка полученных таким образом пептидов происходит посредством хроматографии на Sephadex ионообменных смолах или HPLC.
Предпочтительно использование метода синтеза твердых фаз для получения Ac-D-Nal(2)-p-Cl-D-Phe-D-Trp-Ser(α-L-Rha) -Leu -Arg-Pro- азалли-NH2 и pGlu-His-Trp-Ser-Tyr-D-Ser(tBu)-Leu-Arg-Pro-азагли- NH2(золадекс).
Использованные сокращения:
BSA двузамещенный триметилсилилацетамид
Cha циклогексилаланин
Chg циклогексилглицин
DСС дициклогексикарбодиимид
DIC диизопропилкарбодиимид
DMAP диметиламинопиридин
Fmoc 9-фторенилметоксикарбонил
HOBt 1-гидроксибензотриазол
HOOBt 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бенгзотриазин
Nal нафтилаланин
Npg неопентилглицин
Pmc 2,2,5,7,8-пентаметилхроман-6-сульфонил
Tbg третичный бутилглицин
THF тетрагидрофуран
Thia 2-тиенилаланин
П р и м е р 1. а) 5-карбоксилатэтил-2,4-диметокси-4'-метоксибензгидриламин.
17,5 г 5-Карбоксилатэтил-2,4-диметокси-4'-метоксибензофеноноксима растворяют в 450 мл (1:1) раствора из этанола и DMF и добавляют 2 мл концентрированного NH3. После добавления Pt/C-катализатора 5 дней гидрируют при нормальном давлении. По окончании реакции катализатор отсасывают, фильтрат сгущают и продукт осаждают эфиром. Продукт используют в таком виде без дальнейшей очистки.
b) N-(p-нитрофенилоксикарбонил)-5-карбоксилатоэтил-2,4-диметокси- 4'-метоксибензгидриламин.
10 г соединения, указанного в а, растворяют в 100 мл раствора (4:1) THF (DMF и смешивают при комнатной температуре с 2,1 эквив. двухзамещенного метилсилилацетамида (BSA). Суспензия отстаивалась за короткое время полностью и прозрачный раствор в течение 2 ч размешивают. Затем добавляют 3 г нитрофенилового эфира хлормуравьиной кислоты, смесь перемешивают еще 1 ч. По окончании реакции растворитель удаляют в высоком вакууме. Осадок переносят с 300 мл воды, появившееся масло экстрагируется с эфиром уксусной кислоты. Фазу эфира уксусной кислоты промывают раствором 1н. KHSO4 и водой. Органическую фазу сушат над MgSO4 и выпаривали до сухого. Осадок (12 г) характеризуется NMR, 1R и MS.
N-(p-нитрофенилоксикарбонил)-5-карб- оксилатэтил-2,4-диметокси-4'- метоксибензгидриламин непосредственно превращался в подходящий замещенный анкер с гидразидами аминокислоты.
с) Бензилоксикарбонил-4-пролилазаглицил-(5-карбоксилатэтил-2,4- диметокси-4'-метоксибензилгидрид)амид.
3,27 г бензилоксикарбонилпролилгидразида гидрохлорида и 6,94 г соединения, указанного в b растворяют в 40 мл диметилформамида (DMF) и смешивают с 3 эквив. N-этилморфолина и каталитическим количеством диметиламинопиридина (ДМАР). Оставляют на 16 ч реагировать. По окончании реакции выпаривают до сухого. Осадок помещают в эфир уксусной кислоты бутанол и органическую фазу промывают насыщенным раствором NaHCO3, 1н. KHSO4 и водой. Органическую фазу сушат над MgSO4, после фильтрации выпаривают до сухого. Осадок перекристаллизовывается из чистого эфира уксусной кислоты. Получают 6,6 г указанного соединения.
FAB-MS:641 (M+Li+)
IR:CO 1695 см-1
1H-NMR (ДMSO): δ= 3,7 S (6H, OCH3) ppm
d) 9-фторенилметоксикарбонил-L-пролилазаглицил(5-карбоксилатэтил- 2,4-диметокси-4'-метоксибензгидрил)амид.
26,5 г соединения, указанного в с растворяют в 300 мл метанола и обрабатывают 2 г Pd-катализатора. Через 1 ч гидрирование закончено. Катализатор отфильтровывают и фильтрат сгущают до сухого. Без дальнейшей очистки осадок переносят (17,5 г) в раствор, состоящий из 80 мл воды и 80 мл диоксана и обрабатывают 8 г гидрокарбоната Na и 17 г 9-фтор-енилметоксикарбонил-N- гидроксисукцинимида (Fmoc-ONSu). Оставляют на один день реагировать. По окончании реакции реакционную смесь фильтруют через осветлительный складчатый фильтр. Фильтрат подкисляют 2н. H2SO4 до рН 6 и выпаривают в вакууме до 80 мл. Разбавляют 100 мл воды и экстрагируют раствором эфира уксусной кислоты (n-бутанола (8,5: 1,5). Органическую фазу промывают с полунасыщенным раствором NaCl и затем выпаривают до сухого. Осадок фильтруют через 500 г силикагеля с уксусным эфиром. Получают 20 г указанного соединения.
FAB-MS: 729 (MFLi+)
IR: CO 1695 см-1
е) Сцепление соединения из примера 1, d с полистироловой смолой.
1,0 г аминометилполистиролсмолы (загрузка 1,07 ммоль) и 1,2 г соединения из примера 1,d) суспендируют в 10 мл диметилформамида и переносят с 216 мг 1-гидроксибензтриазола (HOBt) и 0,75 мл диизопропилкарбодиимида (DIC). Оставляют на ночь реагировать, пока нингидрин-тест не покажет полную реакцию. Смолу отфильтровывают и промывают диметилформамидом и метиленхлоридом и основательно высушивают в вакууме. Загрузка смолы с пролином составляет 0,51 ммоль/г
f) Ac-D-Nal(2)-p-Cl-D-Phe-D-Trp-Ser-Tyr-D-Ser-α -L-Rha/-Leu-Arg-Pro-Азагли-NH2
9-фторенилметоксикарбонил-N2-амино- защитные группы соединения из примера 1, е отщепляют 20%-ным раствором пиперидина/диметилформамида (2х3 мин 2х8 мл). Далее смолу окончательно промывают N-метилпирролидином, пептид синтезируется на смоле (785 мг смолы из примера 1,с)), при этом циклически проводят следующие шаги:
отщепление Fmoc-защитных групп 20%-ным пиперидином в DMF;
промывание смолы DMF/N-метилпирролидиноном;
присоединение Fmoc-аминокислоты при in situ-активировании HOBt-эфиром при использовании диизопропилкарбодиимида в качестве реагента активирования (1,5 ммоль аминокислоты, 2,25 ммоль HOBt, 1,6 ммоль диизопропилкарбодиимида).
Если присоединение было неполным (Кайзер-тест), цикл присоединения повторяют. В качестве последней аминокислоты используют Fmoc-D-Nal(2)-OH,N концевую ацетилгруппу вводят при помощи реакции с ацетгидридом.
По окончании синтеза твердых фаз смолу промывают (DMF, CH2Cl2) и высушивают. Получают 1,35 г замещенной смолы. Высушенную смолу суспендируют при комнатной температуре с 0,75 мл этандитиола. Через 15 мин добавляют 7,5 мл трифторуксусной кислоты и суспензию перемешивают 1,5 ч. Затем смолу отфильтровывают и основательно промывают 80%-ной трифторуксусной кислотой. Фильтрат выпаривают в вакууме и к нему добавляют 30 мл воды. Добавлением NaHCO3 устанавливают рН 6-7, пептид извлекают встряхиванием с n-пентанолом (4х30 мл). Фаза n-пентанола выпаривается и в 10 мл метанола/вода (9:1) впитывается и переносится с 0,5 г K2CO3. Перемешивают 30 мин, фильтруют, сгущают фильтрат. Осадок помещают в 100 мл n-пентанола и органическую фазу промывают водой. После сушки с MgSO4 и фильтрации органическую фазу выпаривают. Получают 740 мг продукта-сырца. После хроматографии на Sephadex G25 (1 М уксусная кислота) и на силикагеле получают 185 г чистого указанного соединения.
FAB-MS: 1531 (M+H+)
Соединения, приведенные в примерах 2-9, получают аналогично примеру 1.
П р и м е р 2. Ac-Nal(2)-p-Cl-D-Phe-D-Trp-Ser-Tyr-D-Ser(α-L-Rha)- Leu-Arg-Pro-Азагли-NH2, FAB-масс-спектр: 1531 (М+Н+).
П р и м е р 3. Ac-D-Nal(2)-p-Cl-Phe-D-Trp-Ser-Tyr-D-Ser(α -L-Rha)-Leu-Arg-Pro-Азагли- NH2, FAB-масс-спектр: 1531 (М+Н+).
П р и м е р 4. Ac-D-Nal(2)-p-Cl-D-Phe-Trp-Ser-Tyr-D-Ser(α -L-Rha)-Leu-Arg-Pro-Азагли- NH2, FAB-масс-спектр: 1531 (М+Н+).
П р и м е р 5. Ac-D-Na(2)-p-Cl-D-Phe-D-Trp-D-Ser-Tyr-D-Sor(α-L-Rha)-Leu-Arg-Pro- Азагли-NH2, FAB-масс-спектр: 1531 (М+Н+).
П р и м е р 6. Ac-D-Nal(2)-p-Cl-D-Phe-D-Trp-Ser-D-Tyr-D-Ser(α -L-Rha)-Leu-Arg-Pro-Аза- гли-NH2, FAB-масс-спектр: 1531 (М+Н+).
П р и м е р 7. Ac-D-Nal(2)-p-Cl-D-Phe-D-Trp-Ser-Tyr-Ser(α -L-Rha)- Leu-Arg-Pro-Азагли-NH2, FAB-масс-спектр: 1531 (М+Н+).
П р и м е р 8. Ac-D-Nal(2)-p-Cl-D-Phe-D-Trp-Ser-Tyr-D-Ser(α -L-Rha)-D-Leu-Arg-Азагли- -NH2, FAB-масс-спектр: 1531 (М+Н+).
П р и м е р 9. Ac-D-Nal(2)-p-Cl-D-Phe-D-Trp-Ser-Tyr-D-Ser(α-L-Rha)-Leu-D-Agr-Pro- Азагли-NH2, FAB-масс-спектр: 1531 (М+Н+).
П р и м е р 10. Ac-D-Nal(2)-p-Cl-D-Phe-D-Trp-Ser-Tyr-D-Ser(α -L-Rha)-Leu-Arg-D-Pro- Азагли-NH2.
а) Бензилоксикарбонил-4-D-пропилазаглицил(5-карбоксилатоэтокси- 2,4-диметокси-4'-метоксибензилгидрил)амид.
1,19 г бензилоксикарбонил-D-пролилгидразида гидрохлорида растворяют в 15 мл диметилформамида и разбавляют 1,84 г указанного в заголовке соединения из примера 1,b, 1,5 мл -этилморфолина, а также каталитическим количеством диметиламинопиридина.
Перемешивают в течение ночи при комнатной температуре. По окончании реакции диметилформамид отгоняют в вакууме и остаток растворяют в диметиленхлориде. Органическую фазу промывают 15%-ным раствором KHSO4/K2SO4, сушат над сульфатом магния и пропускают через ротационную печь. Получают 3,4 г масла. Затем обрабатывают на хроматографической колонке с силикагелем/градиент из чистого этилацетата (500 мл) до смеси этилацетат/метанол 50:2 (1000 мл).
Выход 2,1 г.
FAB-масс-спектр: 635 (М+Н+).
b) H-D-пролилазаглицил(5-карбоксилатэтокси-2,4-диметокси-4'- метоксибензгидрил)амид.
800 г указанного в заголовке соединения из примера 10,а растворяют в 20 мл метанола и смешивают с маленьким количеством Pd на угле (на кончике шпателя) (10%) в атмосфере азота.
Затем пропускают водород. Через 45 мин процесс гидрирования заканчивается. Реакционную смесь фильтруют через осветляющий фильтр и фильтрат отгоняют в ротационном испарителе под вакуумом. Получают 600 мг указанного в заголовке соединения.
с) 9-флуоренилметоксикарбонил-D-пролилазаглицил(5- карбоксилатоэтил-2,4-диметокси-4'-метоксибензгидрил)амид.
1 г указанного в заголовке соединения из примера 10,b растворяют в 15 мл смеси диоксан/вода (1: 1) и разбавляют 336 мг кислого карбоната натрия, а также порциями 674 мг 9-флуоренилметоксикарбонил-N-гидроксисукцинимида и выдерживают в течение ночи при комнатной температуре. По окончании реакции все, кроме воды, отгоняют в вакууме, а оставшуюся водную фазу подкисляют 1н. H2SO4. Продукт дважды экстрагируют этилацетатом, органическую фазу один раз промывают насыщенным раствором NaHCO3, сушат над сульфатом магния и растворитель отгоняют в вакууме.
Получают 1,4 г сырого продукта, который обрабатывают на хроматографической колонке с силикагелем, элюируя смесью метиленхлорид:метанол:вода:уксусная кислота (95:5:0,5-0,5).
Выход 870 мг.
FAB-масс-спектр: (M+Na+).
d) Связывание указанного в заголовке соединения из примера 10,с с полистироловой смолой.
1,0 г аминометилполистировой смолы (загрузка 1,07 ммоль) и 1,2 г указанного в заголовке соединения из примера 10,с суспендируют в 15 мл диметилформамида и смешивают 216 мг 12-гидроксибензотриазола и 0,75 мл диизопропилкарбодиимида.
Реакция протекает в течение ночи при комнатной температуре до полного окончания, определяемого в тесте нингидрином. Смолу отфильтровывают и промывают диметилформамидом, метиленхлоридом и метилтретбутиловым эфиром.
Выход смолы 1,5 г.
Количество D-пролина на смоле составляет 0,256 ммоль/г.
П р и м е р 11. p-Glu-His-Trp-Ser-Tyr-D-Ser(tBu)-Leu-Arg-Pro- Азагли-NH2.
Указанное соединение получают по аналогии с примером 1.
FAB-масс-спектр: 1398 (М+Н+).
П р и м е р 12. Общая методика получения солей.
100 мг соединения из примеров 1-11 (трифторацетатная форма) растворяют в 5-10 мл воды (рН 1,5-2) и смешивают взятым на кончике шпателя ионообменником (амберлит IRA 45) в форме ацетата или хлорида). Ионообменник перемешивают и измеряют рН смеси. В случае, если уксусной кислотой не достигнуто значение рН 3,5-4 или соляной кислотой рН 1,5-2, добавляют еще некоторое количество ионообменника. Затем ионообменник отсасывают и промывают водой. Пептидный раствор отгоняют под вакуумом или лиофилизируют.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДЫ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2083586C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ | 1992 |
|
RU2081880C1 |
| АГОНИСТЫ РЕЦЕПТОРОВ НЕЙРОМЕДИНА В И СОМАТОСТАТИНА | 2000 |
|
RU2263680C2 |
| ЦИКЛОГЕКСАПЕПТИДЫ, ИХ СМЕСИ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2163242C2 |
| ПТГ-СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ФРАГМЕНТ ДНК И СЛИТЫЙ БЕЛОК | 1993 |
|
RU2130945C1 |
| АНАЛОГИ ПЕПТИДА ЛГ-РФ, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1998 |
|
RU2212247C2 |
| СПОСОБ ТВЕРДОФАЗНОГО СИНТЕЗА ПЕПТИДОВ | 1991 |
|
RU2043362C1 |
| ПЕПТИДНЫЕ КОМПОЗИЦИИ | 2014 |
|
RU2725150C2 |
| ПРОИЗВОДНЫЕ МЕТАСТИНА И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2430107C2 |
| ПРОИЗВОДНЫЕ МЕТАСТИНА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2454425C2 |
Использование: в химии пептидов, в частности в способе получения производных пептидамидов. Сущность изобретения: способ получения производных пептидамидов ф-лы 1: (X)n-A-NH2, где X-Ac-L(или D)-Nal(2), p-Cl-L(илиD)-Phe, L(или D)-Ser, L(или D)-Tyr, L(или D)-Trp, L(или D)-Ser(α -L-Phe), L(или D)-Leu, L(или D)-Arg, L(или D)-Pro, p-Glu, His, D-ser(t-Bu); n = 3 - 10; а - азаглицин, или их физиологически совместимых ацетатов или гидрохлоридов. Реагент 1 - соединения ф-лы II  где Y1-Y5 - водород; C1-C4 -алкоксигруппа; -(CH2)m-C(O)-OH [радикалы одинаковы или различны, но по меньшей мере один из них (CH2)n-C(O)-OH ]; m = 1 - 3; R1 водород; 4-метоксифенил. Реагент 2: силилирующий агент из группы: третбутилдиметилсилилхлорид, третбутилфенилсилилхлорид, триметилхлорсилан, бис-триметилсилилацетамид. Условия реакции: процесс ведут в среде растворителя с последующей обработкой силилированного соединения производным хлормуравьиной кислоты. Получают соединения ф-лы III R2 -
где Y1-Y5 - водород; C1-C4 -алкоксигруппа; -(CH2)m-C(O)-OH [радикалы одинаковы или различны, но по меньшей мере один из них (CH2)n-C(O)-OH ]; m = 1 - 3; R1 водород; 4-метоксифенил. Реагент 2: силилирующий агент из группы: третбутилдиметилсилилхлорид, третбутилфенилсилилхлорид, триметилхлорсилан, бис-триметилсилилацетамид. Условия реакции: процесс ведут в среде растворителя с последующей обработкой силилированного соединения производным хлормуравьиной кислоты. Получают соединения ф-лы III R2 - 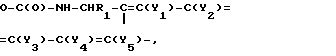 где Y1-Y5 и R1 указаны выше; R2 - галогено(или нитро)фенил. Эти соединения обрабатывают гидрохлоридом гидразида R3 -защищенной соответствующей α-аминокислоты (значения X, где R3 - Fmoc, Bpoc, ВОС-защитные группы, в среде растворителя с образованием соединения ф-лы IV
где Y1-Y5 и R1 указаны выше; R2 - галогено(или нитро)фенил. Эти соединения обрабатывают гидрохлоридом гидразида R3 -защищенной соответствующей α-аминокислоты (значения X, где R3 - Fmoc, Bpoc, ВОС-защитные группы, в среде растворителя с образованием соединения ф-лы IV  . Защитную группу -ВОС- удаляют гидрированием на Pd-катализаторе. Перед дальнейшей реакцией преобразуют защитную группу в Fmoc- или ВОС-защитную группу. Затем соединения ф-лы IV (со значением R3 - Fmoc, Bpoc-уретановая защитной группой) копулируют с помощью обычного для пептидов реагента (через -(CH)n-C(O)-OH - группировку на смоле), защитную группу - R3 - отщепляют, затем постадийно присоединяют Fmoc - или Bpoc-защищенные α -аминокислоты (иногда в форме активированных производных). По окончании синтеза пептида ф-лы I его освобождают от смолы обрабткой кислотой средней силы, причем одновременно или с помощью подходящих приемов отщепляют временно введенные защитные группы боковой цепи.
. Защитную группу -ВОС- удаляют гидрированием на Pd-катализаторе. Перед дальнейшей реакцией преобразуют защитную группу в Fmoc- или ВОС-защитную группу. Затем соединения ф-лы IV (со значением R3 - Fmoc, Bpoc-уретановая защитной группой) копулируют с помощью обычного для пептидов реагента (через -(CH)n-C(O)-OH - группировку на смоле), защитную группу - R3 - отщепляют, затем постадийно присоединяют Fmoc - или Bpoc-защищенные α -аминокислоты (иногда в форме активированных производных). По окончании синтеза пептида ф-лы I его освобождают от смолы обрабткой кислотой средней силы, причем одновременно или с помощью подходящих приемов отщепляют временно введенные защитные группы боковой цепи.
(X)n -A- NH2,
где X α -аминокислота из ряда Ac-Nal(2),Ac-D-Nal(2), p-Cl-Phe, p-Cl-D-Phe, Trp, D-Trp, Ser, D-Ser, Tyr, D-Tyr, Ser ( a -L-Phe D - Ser( a -L-Phe), Leu,D-Leu, Arg, D-Arg, Pro, D-Pro, p-GLu, His, D-Ser (tBu);
n целое число от 3 до 10,
А азаглицин,
или их физиологически совместимых ацетатов или гидрохлоридов, отличающийся тем, что соединение общей формулы II
где Y1, Y2, Y3, Y4 и Y5 водород, C1-C4 алкокси или -(СН2)m СООН, причем радикалы могут иметь одинаковое или различное значение и по меньшей мере один радикал представляет собой -(CH2)m COOH;
m целое число от 1 до 3;
R1 водород или 4-метоксифенил,
подвергают взаимодействию с силилирующим агентом, выбранным из ряда: трет-бутилдиметил-силилхлорид, трет-бутилфенил-силилхлорид, триметилхлорсилан или бис-триметилсилилацетамид, в растворителе с последующей обработкой силилированного соединения производным хлормуравьиной кислоты с образованием соединения общей формулы III
где R1, Y1, Y2, Y3, Y4 и Y5 имеют указанные значения,
R2 фенил, замещенный нитрогруппой и галогеном,
и взаимодействием полученных соединений с гидрозидом аминокислоты общей формулы IV
R3 X CO NH NH2 · HCl,
где X указанная α -аминокислота;
R3 Fmoc-, Bpoc- или Boc-защитные группы,
в растворителе с образованием соединений общей формулы V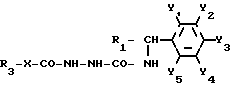
где R1, R3 и Y1, Y2, Y3, Y4 и Y5 имеют указанные значения,
и, в случае, если R3 Boc-защитная группа, ее удаляют гидрованием на Pd-катализаторе и перед дальнейшим взаимодействием преобразуют в Fmoc- или Bpoc-защитную группу, затем соединение формулы V, где R1, Y1, Y2, Y3, Y4 и Y5 имеют указанные значения, R3 - Fmoc или Bpoc-уретановая защитная группа, копулируют с помощью обычного в химии пептидов реагента через -(CH2)n COOH-группировку на смоле, защитную группу R3 отщепляют, затем постадийно присоединяют Fmoc- и Bpoc-защищенные α -аминокислоты, иногда в форме их активированных производных, и после окончания синтеза пептида формулы I обработкой кислотой средней силы освобождают от смолы, причем одновременно или с помощью подходящих приемов в заключение отщепляют временно введенные защитные группы боковой цепи.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ОСЬСОЮЗНАЯ mmm-jimriu'im | 0 |
|
SU384278A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
