Диамины
Изобретение относится к диаминам и в особенности к замещенным дифенилэтилендиаминам, а также получаемым из них катализаторам. Такие катализаторы пригодны для ускорения реакций асимметрического гидрирования, продукты которых полезны, например, в качестве химических промежуточных продуктов или реагентов для применения в получении высококачественных химических реактивов или фармацевтических промежуточных продуктов.
Каталитическое асимметрическое гидрирование включает активацию молекулярного водорода хиральными комплексами металлов. Однако органические молекулы также могут применяться в качестве донора водорода в присутствии подходящего хирального катализатора в процессе, известном как гидрирование с переносом водорода. Донор водорода, такой как изопропанол или муравьиная кислота, обычно используется с катализаторами типа [(сульфонилированный диамин)RuCl(арен)] для восстановления карбонильных групп. Эта технология обеспечивает мощное дополнение к каталитическому асимметрическому гидрированию. Гидрирование с переносом водорода, фактически, является особенно подходящим для асимметрического восстановления кетонов, которые представляют собой сложные субстраты для гидрирования, такие как ацетиленовые кетоны и циклические кетоны.
До этого времени сульфонилированный диаминовый компонент катализаторов гидрирования с переносом водорода был ограничен сульфонилированным дифенилэтилендиамином (Dpen) и циклоалкил-1,2-диаминами, такими как 1,2-циклогексан. Например, гидрирование с переносом водорода применяли, используя [(тозил-Dpen)RuCl(арен)]катализатор для фармацевтических продуктов, таких как 10-гидрокси-дигидро-дибенз[b,f]-азепины (см. WO 2004/031155).
Сульфонилированные диаминовые компоненты, используемые до этого времени, будучи полезными, не являются одинаково эффективными во всем диапазоне желаемых субстратов. Таким образом, существует потребность в расширении ряда диаминов, пригодных для использования в катализе гидрирования с переносом водорода, что обеспечит катализаторы с повышенной активностью, селективностью и стабильностью. Мы обнаружили, что стерические и электронные свойства диаминового компонента могут быть полезным образом перестроены путем введения одного или нескольких заместителей в фенольные кольца дифенилэтилендиаминов и путем варьирования свойств сульфонатов.
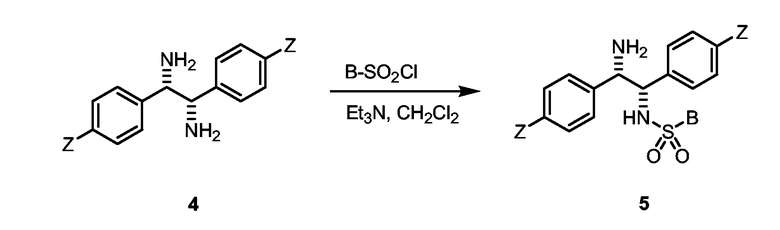
Соответственно, настоящее изобретение обеспечивает диамин формулы (I)
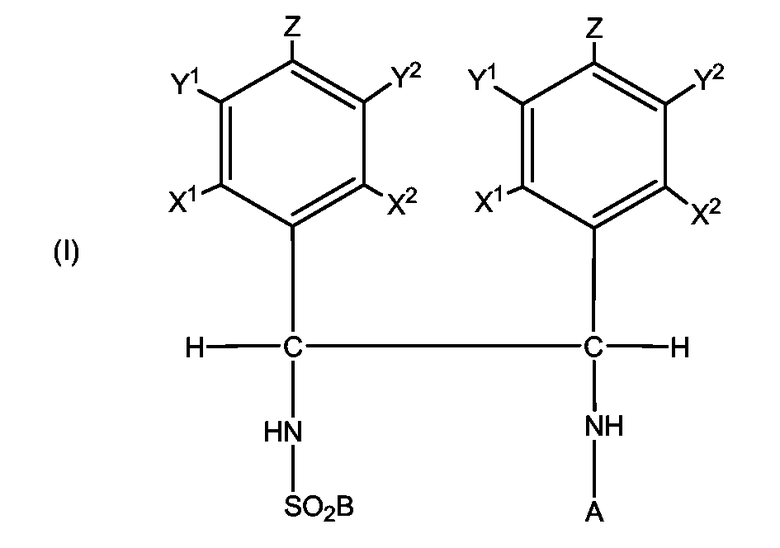
в которой А является водородом или насыщенной или ненасыщенной С1-С20 алкильной группой или арильной группой; В представляет собой замещенную или незамещенную С1-С20 алкильную, циклоалкильную, алкиларильную, арилалкильную или арильную группу или алкиламино группу и по меньшей мере один из X1, X2, Y1, Y2 или Z является С1-С10 алкильным, циклоалкильным, алкиларильным, арилалкильным или алкокси заместителем.
Изобретение, кроме того, обеспечивает способ получения диамина формулы (I), включающий стадии образования замещенного спироимидазола из замещенного дикетона формулы (II), где X1, X2, Y1, Y2 и Z являются такими, как описано выше, восстановления замещенного спироимидазола с образованием замещенного диамина, необязательного превращения замещенного диамина в энантиомерно обогащенную форму и сульфонилирования замещенного диамина.

Изобретение также обеспечивает катализатор, содержащий продукт реакции диамина формулы (I) и подходящего соединения каталитически активного металла.
В формуле (I) А является водородом или насыщенной или ненасыщенной С1-С20 алкильной группой или арильной группой. С1-С20 алкильная группа может быть разветвленной или линейной, например может быть метилом, этилом, н-пропилом, изопропилом, н-бутилом, изобутилом, втор-бутилом, трет-бутилом, пентилом, гексилом, циклогексилом, этил-гексилом, изооктилом, н-нонилом, н-децилом, изодецилом, тридецилом, октадецилом и изооктадецилом. Арильная група может быть незамещенным или замещенным фенилом, нафтилом или антрацилфенилом. Подходящими заместителями являются гидрокси группа, галогенид (например, F, Cl, Br, I), С1-С20 алкокси, амино, амидо, нитрильная и тиольная группы. Предпочтительно А представляет собой водород, метил, этил, пропил или фенил. Наиболее предпочтительно, когда А является водородом.
В формуле (I) В вводится при сульфонилировании необязательно энантиомерно обогащенного замещенного диамина. Для внесения изменения в свойства сульфонилированного диамина формулы (I) может быть использован широкий ряд сульфонилирующих соединений. Соответственно В может представлять собой замещенную или незамещенную С1-С20 алкильную, циклоалкильную, алкиларильную, арилалкильную или арильную группу, например, такую как описано выше или алкиламино группу. Под «алкиламино» мы подразумеваем, что В может иметь формулу -NR'2, где R' представляет собой, например, метил, циклогексил или изопропил или азот образует часть алкильной циклической структуры. Могут использоваться фторалкильная или фторарильная группы, например В может быть n-CF3-C6H4, C6H5 или CF2CF2CF2CF3 или CF3. Предпочтительно В является арильной группой. Арильная группа может быть незамещенным или замещенным фенилом, нафтилом или антрацилфенилом или арильным соединением с гетероатомом, таким как пиридил. Подходящими заместителями являются С1-С20 алкил, такой как описано выше, трифторметил, гидроксил, галогенид (например, F, Cl, Br, I), С1-С20 алкокси (особенно метокси), амино, амидо, нитрил, нитро и тиол. Таким образом, В может представлять собой, например, о-нитрофенил, п-нитрофенил, трихлорфенил, триметоксифенил, триизопропилфенил, о-аминофенил, бензил (-CH2C6H5), 2-фенилэтил (C2H4C6H5), фенил (C6H5), толил (p-CH3-C6H4), ксилил ((CH3)2C6H3, анизил (CH3O-C6H4), нафтил или данзил (5-диметиламино-1-нафтил). Предпочтительно В является толилом и сульфонилирование проводится тозилхлоридом (п-толуолсульфонилхлоридом).
Диамин настоящего изобретения имеет два хиральных центра, каждый содержит фенольное кольцо, имеющее, по меньшей мере, один заместитель X1, X2, Y1, Y2 или Z. Заместитель X1, X2, Y1, Y2 или Z является С1-С10 алкильной, циклоалкильной, алкиларильной, арилалкильной или алкокси группой. Следует понимать, что для того, чтобы удовлетворить валентности атомов углерода в фенольных кольцах, с которыми связан заместитель X1, X2, Y1, Y2 или Z, там где X1, X2, Y1, Y2 или Z не является С1-С10 алкильным, циклоалкильным, алкиларильным, арилалкильным или алкокси заместителем, X1, X2, Y1, Y2 или Z будет атомом водорода.
Таким образом, по меньшей мере, один из X1, X2, Y1, Y2 или Z может независимо являться С1-С10 алкильной группой, такой как метил, трифторметил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, циклопентил, гексил, циклогексил, этил-гексил, изооктил, н-нонил, н-децил или изодецил; алкиларильной группой, такой как бензил или этилфенил; арильной группой, такой как фенил, толил или ксилил; или С1-С10 алкокси группой, такой как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, циклопентокси, пентокси, гексокси, циклогексокси, этил-гексокси, изооктокси, н-нонокси, н-децокси или изодецокси.
Предпочтительно каждое фенильное кольцо имеет один или несколько заместителей. Фенильные кольца могут быть замещены в одном или нескольких положениях, то есть кольца могут быть моно-, ди-, три-, тетра- или пента- замещенными. Заместитель в фенильном кольце может находиться в орто (X1, X2), мета (Y1, Y2) или пара (Z) положении. Однако когда заместитель в фенильном кольце находится в мета-положении, то это минимизирует электронный эффект, оказываемый на аминогруппу, что может ускорять синтез получающегося в результате диамина. Таким образом в одном варианте осуществления замещенный диамин является 1,2-ди-(мета-замещенным фенил)этилендиамином. Когда присутствует более одного заместителя, они предпочтительно являются одинаковыми. Например, в одном варианте осуществления Y1, Y2 могут быть водородами, а X1, X2 и Z являются каждый предпочтительно алкильным, циклоалкильным, алкиларильным, арилалкильным или алкокси заместителем. В альтернативном варианте осуществления X1, X2 и Z могут быть водородами, а Y1, Y2 являются одинаковыми предпочтительно алкильными, циклоалкильными, алкиларильными, арилалкильными или алкокси заместителями. В предпочтительном варианте осуществления X1, X2, Y1, Y2 являются водородами, а Z представляет собой С1-С10 алкильный, циклоалкильный, алкиларильный, арилалкильный или алкокси заместитель. В особенно предпочтительном варианте осуществления X1, X2, Y1, Y2 являются водородами, а Z представляет собой метил. В другом особенно предпочтительном варианте осуществления X1, X2, Y1, Y2 являются водородами, а Z представляет собой метокси.
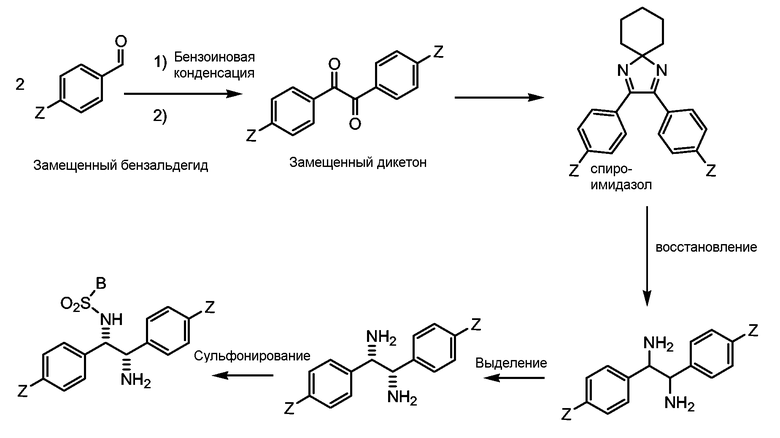
Замещенные диамины настоящего изобретения могут быть легко получены из замещенных дикетонов формулы (II), где X1, X2, Y1, Y2 и Z являются такими же, как описано выше, через спироимидазол, который затем восстанавливается до диамина и сульфонилируется.
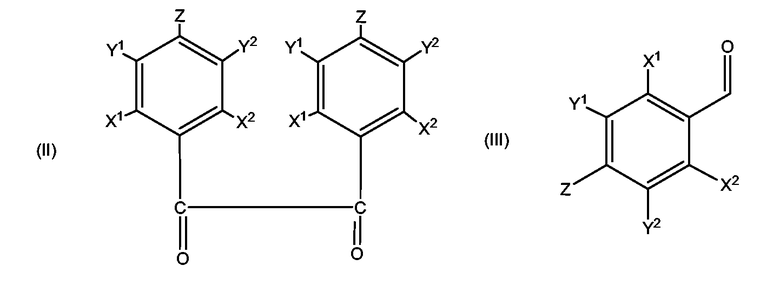
Замещенные дикетоны (бензилы) формулы (II) могут быть доступны из коммерческих источников или могут легко быть получены из замещенных бензальдегидов формулы (III), где X1, X2, Y1, Y2 и Z являются такими же, как описано выше, при помощи бензоиновой конденсации с последующим окислением получившегося в результате бензоина. Замещенные бензальдегиды могут быть получены из коммерческих источников или могут быть синтезированы, используя известные реакции замещения. Реакции бензоиновой конденсации являются хорошо известными и обычно выполняются путем реакции замещенного бензальдегида в подходящем растворителе в присутствии цианида натрия (см., например, Ide с соавт. Org. React. 1948, 4, 269-304). Окисление замещенного бензоина до дикетона может легко быть выполнено, используя ацетат меди и нитрат аммония (см., например, Weiss с соавт. J. Am. Chem. Soc. 1948, 3666).
Спироимидазол может быть получен обработкой замещенного дикетона формулы (II) уксусной кислотой, ацетатом аммония и циклогексаноном и кипячением с обратным холодильником. Восстановление получающегося в результате замещенного бензоина до замещенного амина может быть выполнено при смешивании раствора спироимидазола с литиевой проволокой и жидким аммонием при температуре ниже -60°С, обрабатывая смесь этанолом и хлоридом аммония и давая смеси нагреться до комнатной температуры. Замещенный диамин сульфонилируется, давая замещенные диамины настоящего изобретения.
Замещенный диамин может быть затем сульфонилирован при обработке замещенного диамина в подходящем растворителе желаемым сульфонил хлоридом, то есть Cl-SO2-B и основанием, таким как триэтиламин.
Атомы азота замещенного диамина связаны с хиральными центрами, и, таким образом, замещенный диамин является хиральным. Диамин может быть гомохиральным, то есть (R,R) или (S,S), или иметь один (R) и один (S) центр. Предпочтительно диамин является гомохиральным. Несмотря на то что диамин может быть использован в качестве рацемической смеси, амин предпочтительно является энантиомерно обогащенным. Разделение хирально замещенного диамина может быть выполнено, используя хиральную кислоту или любой другой способ, известный специалистам в данной области. Несмотря на то что разделение может быть проведено на стадии сульфонилированного диамина формулы (I), предпочтительнее проводить разделение на стадии замещенного диамина перед стадией сульфонилирования. Например, замещенный диамин может быть обработан хиральной карбоновой кислотой, такой как дитолуоилвинной кислотой или дибензоилвинной кислотой в подходящем растворителе. Разделенный замещенный диамин предпочтительно имеет энантиомерный избыток (ее %) >70%, более предпочтительно >90%.
Таким образом, этот путь обеспечивает эффективный и рентабельный способ получения энантиомерно обогащенных замещенных 1,2-дифенилэтилендиаминов. Путь описан ниже на предпочтительном примере, где А, X1, X2, Y1, Y2 являются водородами, В представляет собой, например, p-CH3-C6H5 и Z представляет собой С1-С10 алкильный, циклоалкильный, алкиларильный, арилалкильный или алкокси заместитель:
Катализаторы, пригодные для выполнения асимметрических реакций гидрирования с переносом водорода, могут быть получены по реакции замещенных сульфонилированных диаминов настоящего изобретения с подходящим соединением каталитически активного металла. Соединение металла предпочтительно является соединением металлов, выбираемых из списка, состоящего из Ru, Rh, Ir, Co, Ni, Fe, Pd или Pt. Предпочтительными соединениями являются соединения Ru, Rh и Ir, особенно Ru или Rh. Подходящие соединения Ru или Rh представляют собой [MX2(арен)]2, где М=Rh или Ru и Х=галоген, более предпочтительно [RuCl2(арен)]2. Арены представляют собой любые подходящие ароматические молекулы и включают бензолы и циклопентадиены, например бензол, пентаметилциклопентадиен и пара-цимен (4-изопропилтолуол). Особенно пригодные соединения металлов для получения катализаторов гидрирования включают [RhCp*Cl2]2 (где Cp* является CpMe5), [RuCl2(бензол)]2 и [RuCl2(п-цимен)]2.
Катализаторы могут быть получены простым смешиванием диамина и соединения металла в подходящем растворителе в мягких условиях (например, от 0 до 80°С при атмосферном давлении). Подходящие растворители включают углеводороды, ароматические углеводороды, хлорированные углеводороды, сложные эфиры, спирты, простые эфиры, ДМФ и тому подобное. По желанию реакция может проводиться отдельно, и получающийся в результате катализатор может выделяться, например, при удалении растворителя под вакуумом. Альтернативно катализатор может образовываться на месте, то есть в присутствии субстрата, который нужно гидрировать, и источника водорода, опять смешивая соединение металла и диамин в присутствии реагирующих веществ, которые могут быть разбавлены подходящим растворителем.
Хиральные катализаторы настоящего изобретения могут применяться для реакций гидрирования. Обычно карбонильное соединение или имин, источник водорода, основание и растворитель смешивают в присутствии катализатора, который может образовываться на месте. Предпочтительными источниками водорода являются изопропанол или муравьиная кислота (или форматы). Катализаторы могут использоваться для восстановления широкого ряда карбонильных соединений до соответствующих хиральных спиртов и иминов до соответствующих хиральных аминов. Реакции могут проводиться в обычных условиях гидрирования с переносом водорода и в разнообразных подходящих растворителях, известных специалистам в данной области. Например, реакция может выполняться в простом эфире, сложном эфире или диметилформамиде (ДМФ) при температуре 0-75°С. Может присутствовать вода. С муравьиной кислотой предпочтительно используются триэтиламин, DBU или другие третичные амины. С изопропанолом предпочтительными являются t-BuOK, KOH, iPrOK.
Изобретение иллюстрируется следующими примерами.
Пример 1: Получение диаминовых лигандов
(I) Образование спироимидазола (превращение 1 в 2)

а) Z=Метил (СН3): уксусную кислоту (70 мл) добавляли в колбу, содержащую коммерчески доступный дикетон 1а (диметилбензил 11,9 г, 50 ммоль) и ацетат аммония (27 г, 350 ммоль). Добавляли циклогексанон (5,3 мл, 51,5 ммоль) и реакционную смесь кипятили с обратным холодильником 1-4 часа. После охлаждения до комнатной температуры смесь выливали в воду и оставляли на ночь для кристаллизации. Кристаллы отфильтровывали и сушили при пониженном давлении. Перекристаллизацию проводили из этилацетата/гексана и получали 8,22 и 3,32 г для выхода 2а и 2. Общий выход составил 11,54 г, 73%.
b) Z=Метокси (СН3О): уксусную кислоту (100 мл) добавляли в колбу, содержащую коммерчески доступный дикетон 1b (диметоксибензил 18,9 г, 70 ммоль) и ацетат аммония (37,7 г, 490 ммоль). Добавляли циклогексанон (7,45 мл, 72,1 ммоль) и реакционную смесь кипятили с обратным холодильником 1-4 часа. После охлаждения до комнатной температуры смесь выливали в воду и оставляли на ночь для кристаллизации. Кристаллы отфильтровывали и сушили при пониженном давлении. Выход имидазола 2b составил 19,22 г, 79%. Дополнительной очистки можно достичь при перекристаллизации из этилацетата/гексана.
(II) Восстановление (превращение 2 в 3)

а) Z=Метил (СН3): газообразный аммиак медленно конденсировали в раствор спироимидазола 2а (6,95 г, 22 ммоль) в безводном ТГФ (50 мл) при -78°С под аргоном. Как только объем реакционной смеси становился больше приблизительно в два раза, поток газа останавливали. Литиевую проволоку (0,62 г, 88 ммоль) медленно добавляли, будучи уверенными, что температура не превышает -60°С. После перемешивания в течение 30-60 минут добавляли этанол (2,6 мл) и спустя 30-60 минут хлорид аммония (6,2 г). Смеси давали нагреться до комнатной температуры и добавляли воду (приблизительно 100 мл) и MTBE (приблизительно 100 мл). Слои разделяли и водный слой дважды экстрагировали примерно 100 мл MTBE. Объединенные органические слои промывали солевым раствором и выпаривали в вакууме. Получающееся в результате масло растворяли в MTBE и добавляли 10% HCl (2-3 эквив.). Двухфазную смесь перемешивали 30-90 минут и разбавляли водой. Слои разделяли и органический слой экстрагировали водой. Объединенные водные слои промывали дихлорметаном и затем нейтрализовали водным КОН до тех пор, пока рН не становился >10. Неочищенный диамин экстрагировали дихлорметаном (3 раза). Объединенные органические экстракты сушили (Na2SO4) и выпаривали до получения масла или твердого вещества. Выход рацемического диамина 3а как 19:1 смеси диастереомеров составил 4,96 г, 94%. Дополнительной очистки можно достичь при перекристаллизации из этилацетата/гексана.
b) Z=Метокси (СН3О): газообразный аммиак медленно конденсировали в раствор спироимидазола 2b (6,96 г, 20 ммоль) в безводном ТГФ (40 мл) при -78°С под аргоном. Как только объем реакционной смеси становился больше приблизительно в два раза, поток газа останавливали. Литиевую проволоку (0,56 г, 80 ммоль) медленно добавляли, будучи уверенными, что температура не превышает -60°С. После перемешивания в течение 30-60 минут добавляли этанол (2,4 мл) и спустя 30-60 минут хлорид аммония (2,8 г). Смеси давали нагреться до комнатной температуры и добавляли воду (приблизительно 100 мл) и MTBE (приблизительно 100 мл). Слои разделяли и водный слой экстрагировали дважды примерно 100 мл MTBE. Объединенные органические слои промывали солевым раствором и выпаривали в вакууме. Получающееся в результате масло растворяли в MTBE и добавляли 10% HCl (2-3 эквив.). Двухфазную смесь перемешивали 30-90 минут и разбавляли водой. Слои разделяли и органический слой экстрагировали водой. Объединенные водные слои промывали дихлорметаном и затем нейтрализовали водным КОН до тех пор, пока рН не становился >10. Неочищенный диамин экстрагировали дихлорметаном (3 раза). Объединенные органические экстракты сушили (Na2SO4) и выпаривали до получения масла или твердого вещества. Выход рацемического диамина 3b как 19:1 смеси диастереомеров составил 4,69 г, 86%. Дополнительной очистки можно достичь при перекристаллизации из этилацетата/гексана.
(III) Хиральное разделение диаминов
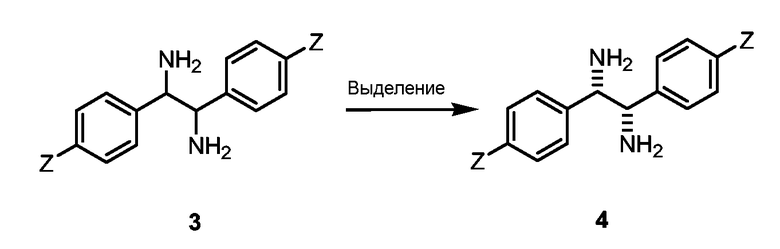
а) разделение диамина 3а. Образование соли с дитолуоилвинной кислотой в метаноле и перекристаллизация из метанола первоначально давала (R,R) 4a c 94% ee. Он мог быть повышен до >99% ее при еще одной дополнительной перекристаллизации.
b) разделение диамина 3b. Образование соли с дитолуоилвинной кислотой в метаноле и перекристаллизация из метанола первоначально давала (S,S) 4b c 74% ee. Возможно увеличить ее при дополнительной перекристаллизации. Образование соли с дибензоилвинной кислотой в метаноле и перекристаллизация из метанола первоначально давала 4b c 98% ee.
(IV) Синтез моносульфонилированных диаминов
а) Тозил-5а (Z=CH3, B=4-CH3-C6H4). Триэтиламин (210 мкл, 1,5 ммоль) добавляли к раствору диамина 4а (180 мг, 0,75 ммоль) в безводном дихлорметане (8 мл) и раствор охлаждали до 0°С. Медленно добавляли раствор тозил хлорида (п-толуолсульфонил хлорида, 148 мг, 0,77 ммоль) в безводном дихлорметане (4 мл). Смесь перемешивали при 0°С в течение 30-120 минут и давали нагреться до комнатной температуры за 1-24 часа. Добавляли воду и разделяли слои. Водный слой дважды экстрагировали дихлорметаном и объединенные органические слои промывали солевым раствором, сушили (Na2SO4) и выпаривали. Неочищенный моносульфонилированный диамин может быть очищен колоночной хроматографией. Очистка колоночной хроматографией дала 270 мг (91%) Ts-5a в виде белого твердого вещества.
b) Тозил-5b (Z=OCH3, B=4-CH3-C6H4). Триэтиламин (190 мкл, 1,3 ммоль) добавляли к раствору диамина 4b (175 мг, 0,65 ммоль) в безводном дихлорметане (8 мл) и раствор охлаждали до 0°С. Медленно добавляли раствор тозил хлорида (п-толуолсульфонил хлорида, 128 мг, 0,67 ммоль) в безводном дихлорметане (5 мл). Смесь перемешивали при 0°С в течение 30-120 минут и давали нагреться до комнатной температуры за 1-24 часа. Добавляли воду и разделяли слои. Водный слой дважды экстрагировали дихлорметаном и объединенные органические слои промывали солевым раствором, сушили (Na2SO4) и выпаривали. Неочищенный моносульфонилированный диамин может быть очищен колоночной хроматографией. Очистка колоночной хроматографией дала 270 мг (91%) Ts-5b в виде белого твердого вещества.
Пример 2: Получение катализатора гидрирования с переносом водорода
Смесь [Ru(п-цимен)Cl2]2 (0,5 эквив.), триэтиламина (2 эквив.), моно-сульфонилированного диамина (1 эквив.) 5b и безводного изопропанола нагревали при 70-90°С в течение 1-4 часов в инертной атмосфере. После охлаждения до комнатной температуры раствор концентрировали при пониженном давлении и оранжевое твердое вещество отделяли фильтрованием. Твердое вещество промывали дегазированной водой и небольшим количеством метанола, затем дополнительно сушили при пониженном давлении. Дальнейшая очистка может быть проведена при осаждении/перекристаллизации из горячего метанола.
Пример 3: Применение моно-сульфонилированных диаминов 5 для асимметрического гидрирования с переносом водорода
Тестирование катализаторов гидрирования с переносом водорода на смесях кетонов
Катализаторы гидрирования с переносом водорода, содержащие диамины 5, были получены на месте и тестированы на предварительно составленной смеси кетонов в ДМФ. [Ru(п-цимен)Cl2]2 (0,0025 ммоль) или [RhСр*Cl2]2 (0,0025 ммоль) и моносульфонилированный диамин (0,0055 ммоль) 5 в безводном ДМФ (2 мл) нагревали до 40°С в течение 10 минут в инертной атмосфере (аргон). Добавляли раствор, содержащий пять кетонов (0,5 мл, 1 ммоль в общем, по 0,2 ммоль каждого) в ДМФ (S/C 40 по отношению к каждому субстрату), после чего следовало 0,6 мл смеси муравьиной кислоты/триэтиламина в соотношении 1/1 и 1 мл ДМФ. Реакционную смесь нагревали в течение ночи (20 час) при 60°С и анализировали методом газовой хроматографии (ChiraDex CB колонка, 10 psi He, 100°С 12 мин, затем до 180°С при 1,5°С/мин, затем до 200°С при 5°С/мин).
Для справки: исследуемыми диаминами были следующие
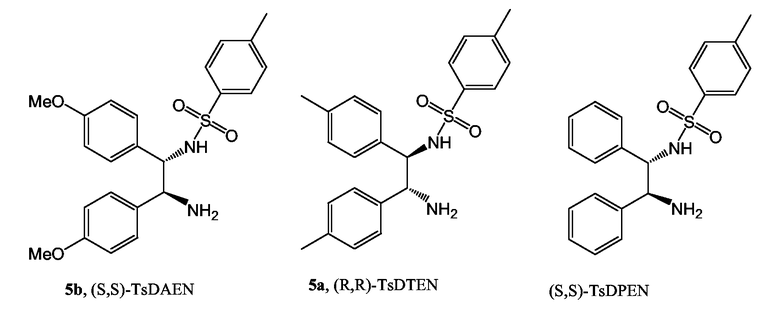


55% ее
40% ее
96% ее
29% ее
92% ее
57% ее
42% ее
98% ее
27% ее
94% ее
53% ее
35% ее
96% ее
28% ее
91% ее
72% ее
13% ее
98% ее
13% ее
90% ее
ее - энантиомерный избыток
Результаты показывают, что особенно высокие степени превращения и ее получаются для циклического кетона, то есть там, где кетон является частью кольцевой структуры, такой как α-тетралон.
Пример 4: Активность Ts-DAEN по отношению к Ts-DРEN
Активность (S,S)-TsDAEN относительно обычного (S,S)-TsDРEN в асимметрическом гидрировании с переносом водорода проверяли, используя α-тетралон в качестве субстрата. Реакцию проводили в масштабе 15 ммоль, при S/C 500/1, используя [RuCl2(п-цимен)]2 в качестве предшественника металла и ДМФ в качестве растворителя при 60°С. Один эквивалент триметиламмоний формата добавляли в начале реакции и дополнительное количество HCOOH добавляли в ходе реакции для поддержания рН при 8,2. Результаты, представленные ниже, показывают, что (S,S)-TsDAEN является более активным, чем (S,S)-TsDРEN.
5
22
71
90
2
3
4
6
22
25
31
36
45
70
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИФОСФИНЫ И МЕТАЛЛОКОМПЛЕКСЫ | 2006 |
|
RU2408600C2 |
| РУТЕНИЕВЫЕ КОМПЛЕКСЫ, СОДЕРЖАЩИЕ ПАРАЦИКЛОФАНОВЫЕ И КАРБОНИЛЬНЫЕ ЛИГАНДЫ, И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ КАТАЛИЗАТОРА | 2012 |
|
RU2614415C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ-3 ГЛИКОГЕНСИНТАЗЫ | 2011 |
|
RU2623427C2 |
| СПОСОБ РАЦЕМИЗАЦИИ ОПТИЧЕСКИ АКТИВНЫХ АЛЬФА-АМИНОАЦЕТАЛЕЙ | 2008 |
|
RU2470912C2 |
| СПОСОБ РАЦЕМОСЕЛЕКТИВНОГО СИНТЕЗА АНСА-МЕТАЛЛОЦЕНОВ | 2005 |
|
RU2391350C2 |
| ПРОИЗВОДНЫЕ 2-ОКСО-1-ПИРРОЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2001 |
|
RU2292336C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА И ЕГО АНАЛОГОВ | 2013 |
|
RU2619928C2 |
| СПОСОБ СТЕРЕОСЕЛЕКТИВНОГО ПОЛУЧЕНИЯ ЭНАНТИОМЕРА ГЕТЕРОБИЦИКЛИЧЕСКОГО СПИРТА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2185379C2 |
| СПОСОБ АСИММЕТРИЧЕСКОГО ГИДРИРОВАНИЯ ПРОИЗВОДНЫХ АКРИЛОВОЙ КИСЛОТЫ, КАТАЛИЗИРУЕМОГО ПЕРЕХОДНЫМИ МЕТАЛЛАМИ, И НОВАЯ КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ АСИММЕТРИЧЕСКОГО КАТАЛИЗА ПЕРЕХОДНЫМИ МЕТАЛЛАМИ | 2005 |
|
RU2415127C2 |
| ПРОИЗВОДНЫЕ 2-ОКСО-1-ПИРРОЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2355680C2 |
Изобретение относится к диамину формулы (I), в котором А представляет собой водород; В представляет собой С1-С6 алкильную группу, фенил-С1-С6 алкильную группу; или группу, выбранную из фенила, нафтила, необязательно замещенную заместителем, выбранным из С1-С6 алкила, C1-С6 алкокси, или галогена; X1, X2, Y1, Y2 представляют собой водород; Z представляет собой С1-С6 алкильную группу или С1-С6 алкокси группу. Также изобретение относится к способу получения диамина формулы (I). Также изобретение относится к катализатору асимметрического гидрирования, полученному в результате реакции диамина формулы (I) и соединения формулы [МХ2(арен)]2, где М выбирают из списка, состоящего из Ru, Rh, Ir, Co, Ni, Fe, Pd, Pt, и Х представляет собой галоген. Катализатор применяют для проведения реакции гидрирования с переносом водорода и реакцию гидрирования с переносом водорода проводят с циклическим кетоном. Технический результат - сульфонилированные дифенилэтилендиамины, используемые в катализе гидрирования с переносом водорода. 4 н. и 8 з.п. ф-лы, 2 табл., 4 пр.

1. Диамин формулы (I)
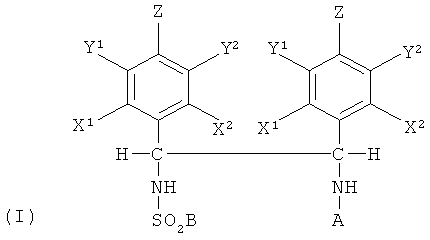
в которой
А представляет собой водород;
В представляет собой С1-С6 алкильную группу, фенил-С1-С6 алкильную группу или группу, выбранную из фенила, нафтила, необязательно замещенную заместителем, выбранным из С1-С6 алкила, C1-С6 алкокси или галогена;
X1, X2, Y1, Y2 представляют собой водород;
Z представляет собой С1-С6 алкильную группу или С1-С6 алкоксигруппу.
2. Диамин по п.1, в котором В является фенилом, нафтилом, необязательно замещенными заместителем, выбранным из С1-С6 алкила, C1-С6 алкокси, галогена.
3. Диамин по п.1, в котором Z представляет собой метил.
4. Диамин по п.1, в котором Z представляет собой метокси.
5. Диамин по п.1, в котором диамин является гомохиральным.
6. Диамин по п.2, в котором диамин является гомохиральным.
7. Диамин по пп.3 и 4, в котором диамин является гомохиральным.
8. Способ получения диамина формулы (I) по любому из пп.1-7, включающий стадии:
a) образования замещенного спироимидазола из замещенного дикетона формулы (II),
b) восстановление замещенного спироимидазола до образования замещенного диамина,
c) необязательное превращение замещенного диамина в энантиомерно обогащенную форму и
d) сульфонилирование замещенного диамина

9. Катализатор асимметрического гидрирования, полученный в результате реакции диамина формулы (I) по любому из пп.1-7 и соединения формулы [МХ2(арен)]2, где М выбирают из списка, состоящего из Ru, Rh, Ir, Co, Ni, Fe, Pd, Pt, и Х представляет собой галоген.
10. Катализатор по п.9, в котором соединением металла является [МХ2(арен)]2, где M - Rh или Ru и Х - галоген.
11. Применение катализатора по п.9 или 10 для проведения реакции гидрирования с переносом водорода.
12. Применение катализатора по п.11, в котором реакцию гидрирования с переносом водорода проводят с циклическим кетоном.
| CORVEY E.J | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| XIAOGUANG LI et al | |||
| Asymmetric transfer hydrogenation of ketones with a polymer-supported chiral diamine // TETRAHEDRON LETTERS, 2004, | |||

