Предпосылки создания изобретения
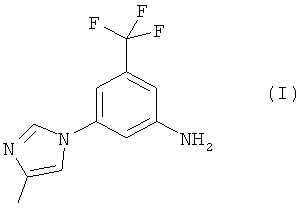
В настоящем изобретении предлагается эффективный, безопасный и коммерчески доступный способ получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина, имеющего формулу (I):
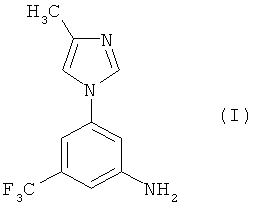
который является промежуточным соединением для получения замещенных пиримидиниламинобензамидов формулы (II):
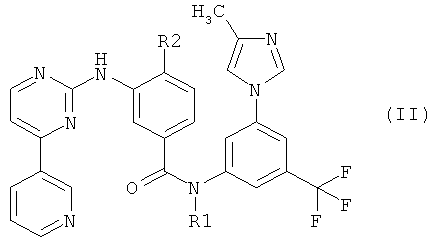
Соединения формулы (II) описаны в публикации W.Breitenstein et al. WO 04/005281 A1, которая включена в текст данной заявки посредством ссылки. Было установлено, что эти соединения ингибируют одну или более из тирозинкиназ, как, например, c-Abl, Bcr-Abl, рецепторы тирозинкиназ PDGF-R, Flt3, VEGF-R, EGF-R и c-Kit. Вследствие этого соединения формулы (II) могут быть использованы для лечения некоторых неопластических заболеваний таких, как лейкоз.
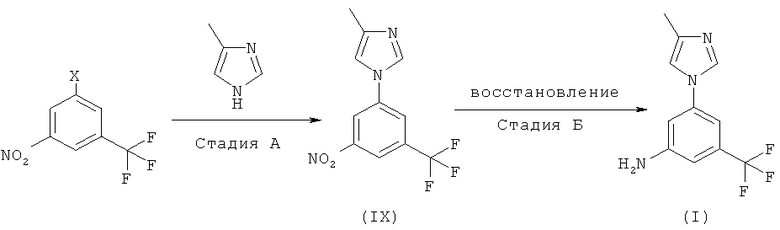
Известный ранее метод получения соединения (I) представляет собой четырехстадийный синтез, начинающийся с реакции ароматического замещения соединения (III) соединением (IV), который требует использование высокой температуры (150°С) (схема 1).
Схема 1
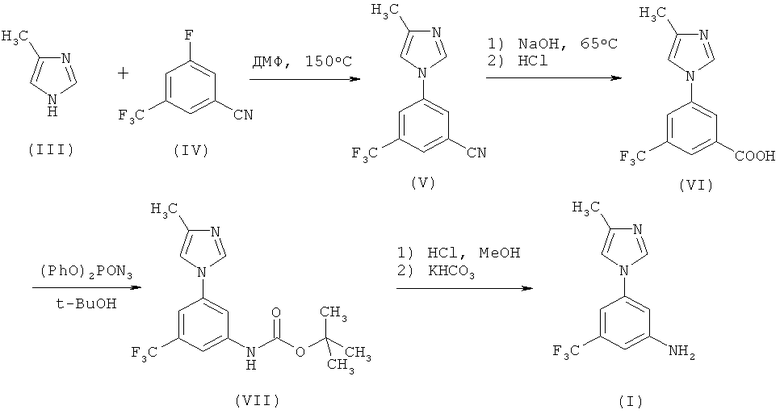
Кроме того, превращение соединения (VI) в соединение (VII) с помощью перегруппировки Курциуса проводится с использованием небезопасного реагента такого, как дифенилфосфорилазид. В результате этой реакции выходы и качественные характеристики продукта не являются постоянными, а удаление дифенилфосфорной кислоты, образующейся в качестве побочного продукта, вызывает трудности. Получаемый карбамат (VII) нуждается в хроматографической очистке, которая является дорогостоящей и длительной по времени процедуре.
Задачей настоящего изобретения является предоставление эффективных и альтернативных способов получения соединения формулы (I) с высоким выходом.
Следующим объектом по настоящему изобретению является получение соединения формулы (I) из более дешевых исходных веществ и реагентов.
Еще одним объектом по настоящему изобретению является способ получения соединения формулы (I) с использованием менее опасных реагентов.
Следующая цель - сокращение циклов нагрева и охлаждения, приводящее к уменьшению разложения и более чисто протекающей реакции, достигается с помощью использования микроволнового облучения, или дополнительной теплообменной способности обогревающих реакционные сосуды приборов, или посредством использования реакционного оборудования для непрерывного проведения реакции.
Настоящее изобретение преодолевает проблемы реакции, представленной выше на схеме 1.
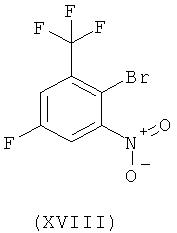
Настоящее изобретение включает также промежуточное соединение (XVIII) и его получение.
Краткое изложение сущности изобретения
Настоящее изобретение предоставляет новые синтетические способы получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина, имеющего формулу (I):
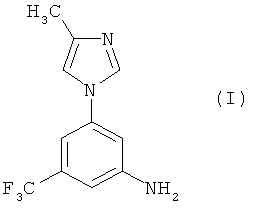
Соединение формулы (I) является промежуточным соединением для получения замещенных пиримидиниламинобензамидов формулы (II):
Соединения формулы (II) описаны в публикации W.Breitenstein et al. WO 04/005281, которая опубликована 15 января 2004 г. и включена в настоящее описание посредством ссылки. Предпочтительным соединением формулы (II) является 4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]-N-[5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)фенил]бензамид. Соединения формулы (II) могут быть использованы для лечения некоторых неопластических заболеваний таких, как лейкоз.
Более предпочтительно, настоящее изобретение предлагает общий способ получения соединения (I), представленный на следующей схеме:
Схема 2
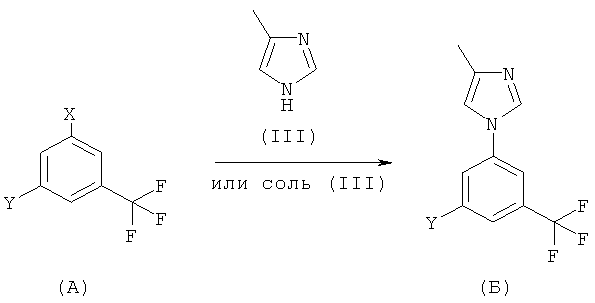
где
Х обозначает галоген, сульфонат или NO2; и
Y обозначает NH2, NO2, галоген или CN.
Общая реакционная схема включает реакцию соединения (А) с соединением (III) в соответствующих реакционных условиях с получением соединения (Б). Когда Y обозначает NH2, тогда (Б) обозначает соединение формулы (I). Когда Y обозначает NO2 или CN, или Х и Y оба являются галогенами, необходима дополнительная стадия процесса, как показано ниже.
Детальное описание изобретения
Общая реакционная схема по изобретению может быть проиллюстрирована посредством следующих вариантов осуществления изобретения.
Первый вариант представлен на реакционной схеме 3.
Схема 3
где Y в соединении А обозначает NO2:
Х может обозначать галоген, сульфонат или NO2.
Когда Х обозначает Br, стадия А включает использование переходного металла в качестве катализатора и основания (от среднего до сильного), а стадия Б включает реакцию восстановления с использованием переходного металла в качестве катализатора в подходящем полярном растворителе.
Когда Х обозначает водород, реакцию модифицируют в соответствии со схемой 4:
Схема 4

Этот процесс включает:
(i) обработку 1-нитро-3-трифторметилбензола (X) бромирующим агентом, предпочтительно 1,3-дибром-5,5-диметилгидантоином (1,3-дибром-5,5-диметилимидазолидин-2,4-дион), в присутствии сильной кислоты, предпочтительно концентрированной серной кислоты, в инертном растворителе, предпочтительно дихлорметане, при температуре в интервале от 25 до 40°С, предпочтительно при температуре 35°С, с получением 1-бром-3-нитро-5-трифторметилбензола (XI) в качестве основного продукта,
(ii) взаимодействие 1-бром-3-нитро-5-трифторметилбензола (XI) и 4-метил-1H-имидазола в присутствии переходного металла в качестве катализатора такого, как соединения меди, палладия или никеля, предпочтительно медной(I) соли, и основания (от умеренно сильного до среднего), предпочтительно карбоната, алканоата или гидрокарбонатной соли, и необязательно координирующей добавки такой, как 1,2-диамин, предпочтительно этилендиамин, в диполярном апротонном растворителе, предпочтительно N,N-диметилформамиде или 1-метил-2-пирролидиноне, при повышенной температуре, предпочтительно при температуре от 100 до 120°С, с получением 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (IX) в качестве основного продукта,
(iii) восстановление 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (IX), предпочтительно с использованием водорода в присутствии переходного металла в качестве катализатора, в полярном растворителе, предпочтительно метаноле или этаноле, и предпочтительно при повышенной температуре, с получением 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I). Исходные соединения 1-нитро-3-трифторметилбензол (X) и 4-метил-1Н-имидазол коммерчески доступны.
Когда Х обозначает йод на вышеприведенной схеме 3, стадия А включает использование переходного металла в качестве катализатора и от среднего до сильного основания, а стадия Б включает стадию восстановления с использованием переходного металла в качестве катализатора в соответствующем полярном растворителе, как представлено ниже на схеме 5:
Схема 5
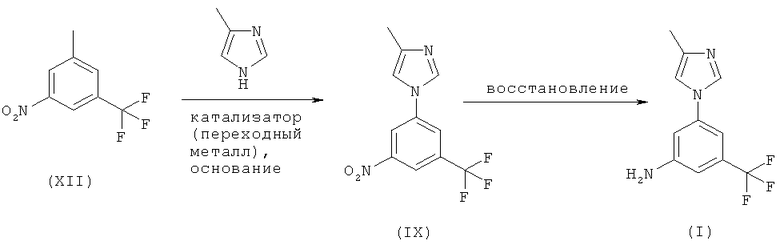
Соединение (I) может быть получено, исходя из 1-йод-3-нитро-5-трифторметилбензола (XII), с использованием методик стадий (ii) и (iii), описанных выше. Получение 1-йод-3-нитро-5-трифторметилбензола (XII) описано в J Med Chem, vol.44, p.4641 (2001).
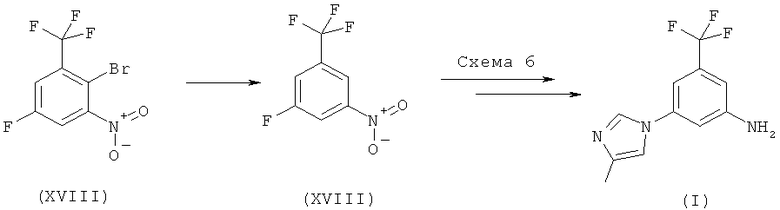
Когда X обозначает F на вышеприведенной схеме 3, стадия А включает использование от сильного до среднего основания в растворителе при повышенной температуре в интервале от 70 до 130°С, а стадия Б включает стадию восстановления с использованием переходного металла в качестве катализатора в соответствующем полярном растворителе, как указано ниже:
Схема 6
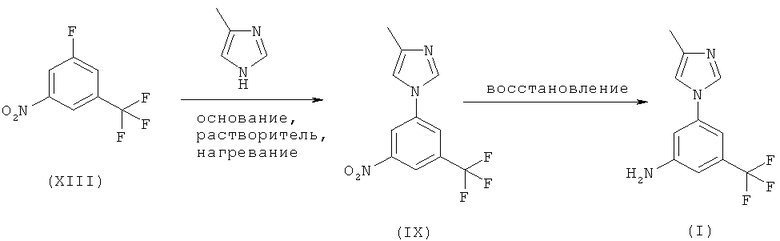
Этот процесс включает:
(i) реакцию смеси 1-фтор-3-нитро-5-трифторметилбензола (XIII) с 4-метил-1H-имидазолом в присутствии от умеренно сильного до среднего основания, предпочтительно, карбонатной или гидрокарбонатной соли, в соответствующем растворителе, предпочтительно N,N-диметилформамиде, N,N-диметилацетамиде или 1-метил-2-пирролидиноне, при температуре в интервале от 70 до 130°С, предпочтительно при 75-100°С, с получением 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (IX) в качестве основного продукта; и
(ii) восстановление 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (IX), предпочтительно с использованием водорода в присутствии переходного металла в качестве катализатора, в соответствующем полярном растворителе, предпочтительно метаноле или этаноле, и, предпочтительно, при повышенной температуре с получением 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I). Этот вариант может также представлять собой реакцию конденсации.
Кроме того, каждый из методов, описанных выше, может необязательно включать превращение соединения (IX) в соль формулы (XV), например, с целью очистки, как проиллюстрировано на следующей схеме:
Схема 7
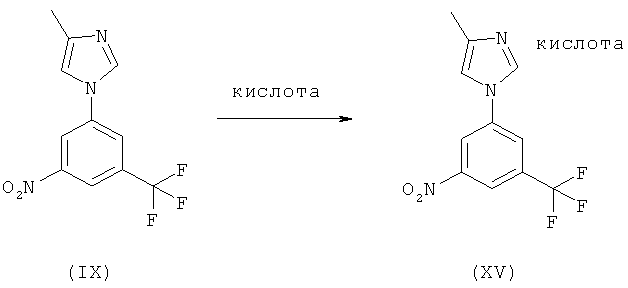
По этой схеме раствор соединения (IX) обрабатывают кислотой или раствором кислоты в воде или органическом растворителе с последующим выделением соли (XV), например, с помощью фильтрования. Соединение (IX) затем получают при обработке соли (XV) основанием, предпочтительно, водным раствором гидроксида натрия, и выделением свободного основания (IX) посредством экстракции или кристаллизации.
Для первого варианта основанием (от сильного до среднего) является предпочтительно карбонат, алконат или гидрокарбонат; более предпочтительно алкоксид калия, алкоксид натрия, алкоксид лития, гидрид калия, гидрид натрия или карбонаты лития, натрия, калия или цезия.
Вторым вариантом схемы 2 является вариант, когда Y обозначает NH2. При первом подварианте Х обозначает галоген. Когда Х обозначает Br, реакция описывается схемой 8:
Схема 8
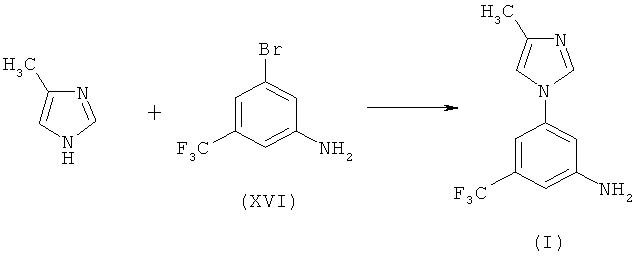
Эта реакция включает взаимодействие 3-бром-5-трифторметилфениламина (XVI) с 4-метил-1H-имидазолом в присутствии переходного металла в качестве катализатора такого, как соединение меди, палладия или никеля, предпочтительно медной(I) соли, и от сильного до среднего основания, предпочтительно карбоната, алканоата или гидрокарбонатной соли, и необязательно координирующей добавки такой, как, предпочтительно, циклогександиамин, в диполярном апротонном растворителе, предпочтительно диглиме, N,N-диметилформамиде или 1-метил-2-пирролидиноне, при повышенной температуре, предпочтительно в интервале от 100 до 150°С, с получением 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I) в качестве основного продукта.
Когда Х обозначает F, альтернативный синтез соединений (XIX) и (I) предусматривает применение недорогого исходного соединения 2-бром-5-фторбензотрифторида (XVII). Кроме того, соединение формулы (I) может быть синтезировано по следующей схеме:
Схема 9
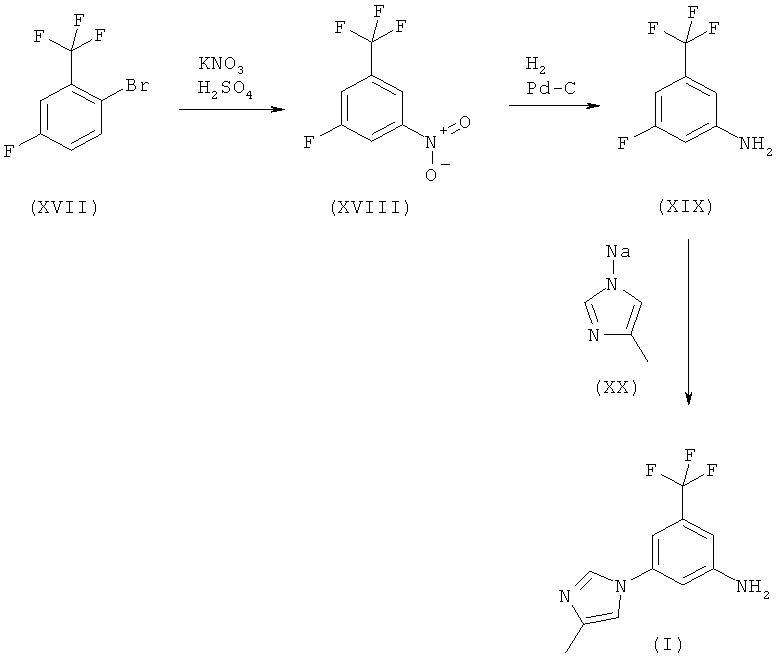
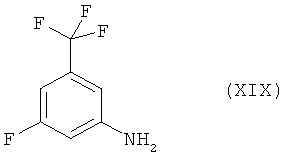
Нитрование коммерчески легко доступного 2-бром-5-фторбензотрифторида (XVII) нитратом калия и серной кислотой приводит к новому соединению 2-бром-5-фтор-1-нитро-3-трифторметилбензолу (XVIII). Восстановление соединения (XVIII) посредством гидрирования на катализаторе палладий/уголь дает 3-фтор-5-трифторметилфениламин [соединение (XIX)], который реагирует с натриевой солью 4-метилимидазола с получением соединения (I). Сырое соединение (I) включает требуемый продукт в качестве основного продукта и по крайней мере один изомер положения (regioisomer) в качестве побочного продукта. Сырой продукт (I) может быть перекристаллизован из толуола с получением чистого соединения (I) с >99,8%-ной степенью чистоты (по площади) по данным ВЭЖХ.
Следует отметить, что 3-фтор-5-трифторметилфениламин (XIX) также коммерчески доступен в небольших количествах, например, от фирмы ABCR. Синтетический путь, описанный в данной заявке, представляет собой новый способ получения соединения (XIX) из нового универсального соединения (XVIII). 3-Фтор-5-трифторметилфениламин (XIX), полученный этим методом, оказался идентичным коммерческому образцу, приобретенному у фирмы ABCR (ABCRF 01075).
Новое соединение (XVIII), описанное в данном тексте, является универсальным соединением и может быть использовано для синтеза целого ряда интересных трифторметилбензольных соединений, которые являются промежуточными соединениями для получения замещенных пиримидиниламинобензамидов (II), которые, как установлено, обладают антилейкозными свойствами (см. WO 04/005281).
Третьим вариантом схемы 2 является вариант, когда Х обозначает F и Y обозначает CN. Реакция (расщепление по Гофману) представлена на нижеприведенной схеме 10:
Схема 10
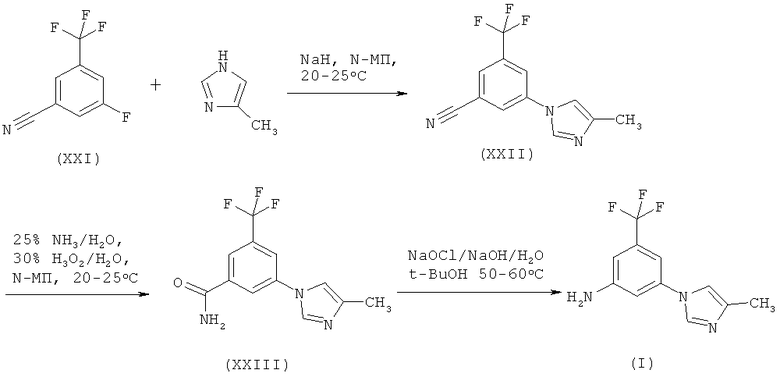
В четвертом варианте схемы 2 как X, так и Y оба являются галогенами. Эта реакция представлена на следующей схеме:
Схема 11

Согласно этому методу 3-бром-5-фтор-бензотрифторид (XXV) вводят в реакцию с 4-метилимидазолом (III) при температуре 25°С в присутствии сильного основания такого, как гидрид натрия, получая таким путем сырое соединение (XXVI) [содержащее 16% региоизомера]. Сырое соединение (XXVI) может быть перекристаллизовано из гептана и превращено в чистый бромарен (XXVI) без детектируемого количества региоизомера. Ариламинирование соединения (XXVI) дифениламином (XXVII) в присутствии палладиевого катализатора, фосфинового лиганда и основания такого, как комбинация Pd(ОАс)2/ксантфос/трет-бутоксид натрия или Pd(OAc)2/БИНАП/трет-бутоксид натрия, приводит к соединению (XXVIII). Содержание остатков палладия в соединении (XXVIII) может быть доведено до 3,4 м.д. после обработки на PICA угле. Гидролиз соединения (XXVIII) с помощью водного раствора хлористоводородной кислоты дает соединение (I) в форме его гидрохлорида. Эта соль может быть превращена затем в свободное основание [соединение (I)] обработкой бикарбонатом калия с получением таким образом чистого соединения (I) высокого качества: чистота по данным ВЭЖХ составляет >99%; содержание палладия составляет 0,5 м.д. Способ по настоящему изобретению является более безопасным, практичным и коммерчески приемлемым, по сравнению с применяемым ранее синтетическим методом (схема 1). Другой палладиевый катализатор, используемый в вышеприведенной реакции, включает тетракис(трифенил)фосфин палладий(0); трис(дибензилиденацетон)дипалладий(0) или хлорид палладия, а также другие катализаторы, известные специалистам в данной области техники. Другие лиганды, применяемые в приведенной выше реакции, включают трифенилфосфин или триалкилфосфин.
Следующие примеры служат исключительно для иллюстрации настоящего изобретения, никоим образом не ограничивая его объем.
Примеры
Пример 1: Синтез 1-[3-бром-5-(трифторметил)фенил]-4-метил-1H-имидазола (XXVI)
Схема 12
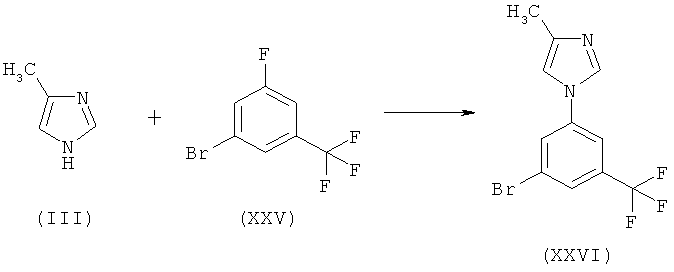
В двухлитровую, 4-горлую, круглодонную колбу, снабженную механической мешалкой, электронным термометром, нагревающей/охлаждающей емкостью, капельной воронкой и устройством для входа/выхода азота, помещают 1-метил-2-пирролидинон (113 г) и гидрид натрия (8,0 г, 60%-ный в масле) в атмосфере азота. Смесь перемешивают при температуре 20-25°С в течение 15 мин. Раствор 4-метилимидазола (17,6 г) и 1-метил-2-пирролидинона (181 г) медленно вносят в смесь в течение 30 мин, поддерживая температуру бани в интервале от 20 до 25°С. После добавления смесь перемешивают при температуре от 20 до 25°С в течение 2 ч. Затем к реакционной смеси медленно добавляют раствор 3-бром-5-фторбензотрифторида (XXV) (40 г) и 1-метил-2-пирролидинона (76 г) в течение 10 мин, поддерживая температуру бани в интервале от 20 до 25°С. Затем смесь перемешивают при температуре от 20 до 25°С в течение 16 ч.
Воду (720 г) медленно добавляют к смеси в течение 3 ч, поддерживая температуру бани в интервале от 20-25°С. После добавления смесь перемешивают при температуре 20-25°С в течение 1 ч. Твердое вещество выделяют фильтрованием, промывают раствором 1-метил-2-пирролидинона (41 г), а затем промывают водой (100 г). Твердое вещество высушивают на воздухе на воронке в течение 1 ч.
В двухлитровую, 4-горлую, круглодонную колбу в атмосфере азота загружают полученное твердое вещество (около 50 г) и этилацетат (361 г). Смесь перемешивают в течение 5 мин при температуре 20-25°С до растворения. Раствор промывают водой (дважды по 100 г). Органический слой дистиллируют при 100 мм Hg при температуре 40°С до тех пор, пока остаточный объем не составит 100 мл. Затем добавляют гептан (342 г) и смесь дистиллируют при 400 мм Hg при температуре 60°С до тех пор, пока остаточный объем не составит 300 мл. Эту операцию повторяют несколько раз. Остаток охлаждают с 55°С до 20°С в течение 5 ч и дополнительно перемешивают в течение 1 ч при температуре 20°С. Затем смесь охлаждают до 5°С в течение 1 ч и дополнительно перемешивают в течение 1 ч при температуре 5°С. Твердое вещество выделяют фильтрованием и промывают холодным (5°С) гептаном (68 г), затем высушивают при 5 мм Hg/20-25°C в течение 4 ч, получая соединение (XXVI) (24,3 г, 48% выход) в виде белого твердого вещества:
1Н ЯМР (300 МГц, ДМСО-d6): δ 8,45 (s, 1Н), 8,30 (s, 1H), 8,10 (s, 1H), 7,90 (s, 1H), 7,70 (s, 1H), 2,10 (s, 3H).
Пример 2: 5-(4-Метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамин (I)
Схема 13
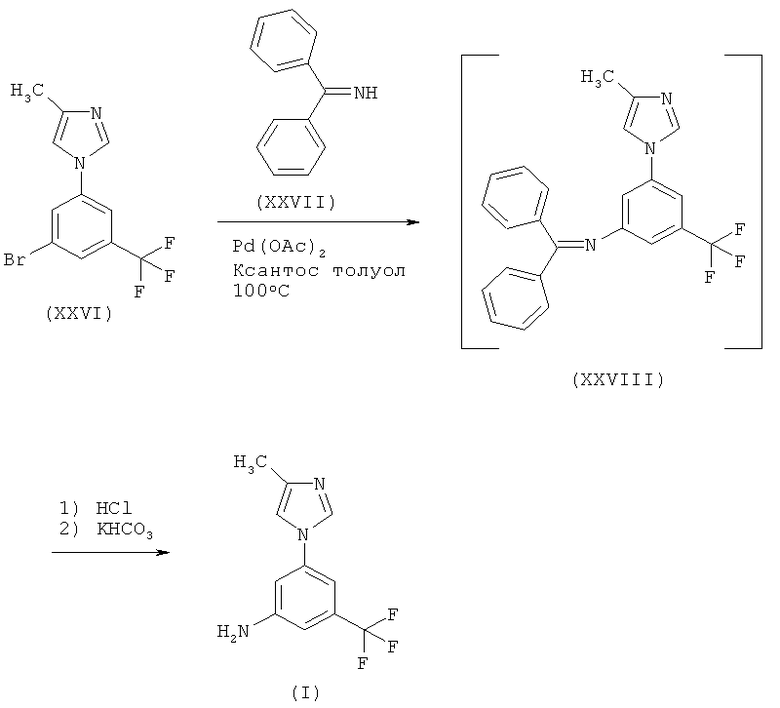
В 1-литровую, 4-горлую, круглодонную колбу, снабженную механической мешалкой, электронным термометром, нагревающей/охлаждающей емкостью, холодильником, капельной воронкой и устройством для входа/выхода азота, загружают толуол (400 мл) в атмосфере азота. Толуол нагревают до температуры 113°С, перемешивают при этой температуре дополнительно 1 ч, а затем охлаждают до температуры 20-25°С. В отдельную 1-литровую, 4-горлую, круглодонную колбу, снабженную механической мешалкой, электронным термометром, нагревающей/охлаждающей емкостью, холодильником, капельной воронкой и устройством для входа/выхода азота, загружают соединение (XXVI) (40 г) и дегазированный, как описано выше, толуол (240 мл). Суспензию перемешивают при температуре 20-25°С в течение 5 мин, получая при этом прозрачный раствор. Вносят в смесь трет-бутоксид, натрия (17,6 г), а затем смесь 4,5-бис(дифенилфосфино)-9,9-диметилксантена (1,5 г), ацетата палладия(II) (0,3 г) и дегазированного толуола (120 мл). После этого добавляют раствор бензофенонимина (XXVII) (26,4 г) и дегазированного толуола (40 мл). Смесь нагревают до температуры от 97 до 103°С и перемешивают при этой температуре дополнительно в течение 3 ч, а затем охлаждают до 60°С. Добавляют воду (200 мл), поддерживая в это время температуру в интервале от 20 до 40°С. Затем органический слой отделяют.
Суспензию PICA P1400 активированного угля (8 г) в толуоле (80 мл) вносят в органический слой. Образовавшуюся суспензию нагревают до температуры 80-85°С и перемешивают дополнительно в течение 5 ч. Смесь охлаждают до 20-25°С и перемешивают при этой температуре в течение 1 ч. Затем смесь фильтруют через фильтр Hyflo Super Celite (4 г) и промывают толуолом (160 мл). Те же операции, которые проводятся в предыдущей методике, повторяют несколько раз. Органический раствор концентрируют в вакууме до объема, составляющего 200 мл. К этому остатку добавляют ацетон (600 мл) и нагревают смесь до температуры 35±3°С. Затем добавляют концентрированную (37%-ную) хлористоводородную кислоту (14,2 г), поддерживая в это время температуру ниже 40°С. Смесь перемешивают при температуре 35-40°С в течение 2 ч, охлаждают до 20-25°С и дополнительно перемешивают в течение 1 ч. Твердое вещество отделяют фильтрованием, промывают ацетоном (40 мл) и высушивают при температуре 60°С/5 мм Hg в течение 8 ч, получая хлористоводородную соль соединения (I) (31,2 г) в виде белого твердого вещества. Твердое вещество растворяют в метаноле (312 мл) при температуре 40°С. Затем добавляют раствор бикарбоната калия (15,7 г) в воде (936 мл) в течение 2 ч, поддерживая в это время температуру бани при 30°С. После этого смесь охлаждают до 20°С и дополнительно перемешивают при температуре 20°С в течение 1 ч. Твердое вещество отделяют фильтрованием, промывают водой (80 г) и высушивают при температуре 60-75°С/5 мм Hg в течение 16 ч, получая соединение (I) (23,5 г, выход 74%) в виде белого твердого вещества:
1Н ЯМР (300 МГц, ДМСО-d6): δ 8,05 (s, 1Н), 7,40 (s, 1H), 7,00 (s, 1H), 6,95 (s, 1H), 6,85 (s, 1H), 5,90 (s, 2H), 2,15 (s, 3H).
Пример 3: Получение 2-бром-5-фтор-1-нитро-3-трифторметилбензола, соединение формулы (XVIII)
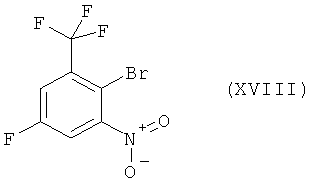
2-Бром-5-фторбензотрифторид (XVII) (50 г, получен от фирмы ABCR, F01421) растворяют в 750 мл дихлорметана. При перемешивании добавляют нитрат калия (60,54 г), а затем медленно добавляют серную кислоту (587,3 г 20% SO3, Riedel de Haen 30736). Температуру реакционной среды во время добавления серной кислоты поддерживают при 25-30°С посредством мягкого охлаждения. Реакционную смесь затем перемешивают дополнительно в течение 25 ч при комнатной температуре, после чего по данным IPS конверсия составляет >97%. Далее слои разделяют и кислый слой экстрагируют дихлорметаном при перемешивании (дважды по 300 мл). Дихлорметановые фазы объединяют и последовательно промывают 1000 мл насыщенного водного раствора NaHCO3, 1000 мл водного раствора сульфаминовой кислоты (5 масс.%), 1000 мл насыщенного водного раствора NaHCO3 и 1000 мл воды. Дихлорметановый раствор высушивают над безводным MgSO4 и растворитель удаляют в вакууме, получая 2-бром-5-фтор-1-нитро-3-трифторметилбензол (XVIII) в виде желтой жидкости. ГХ-МС: m/z: 287, 268, 257, 241, 229. Эти пики МС сопровождаются соответствующими изотопными пиками, характерными для бромсодержащих соединений. ИК (тонкий слой): 3101, 1618, 1591, 1554, 1454, 1423,1365, 1319, 1232, 1186,1153, 1113, 883 см-1.
1Н-ЯМР (400 МГц, ДМСО-d6): δ 8,13 (dd, J=8,5 и J=2,5 Гц), 8,42 (dd, J=7,6 и J=3,0 Гц).
Пример 4: 3-Фтор-5-трифторметилфениламин, соединение формулы (XIX)
2-Бром-5-фтор-1-нитро-3-трифторметилбензол (XVIII) (55,5 г) растворяют в 500 мл этанола. Добавляют в раствор триэтиламин (19,63 г) и палладий на угле (6 г, Pd/C 10%-ный, Engelhard 4505) и смесь гидрируют при температуре 20-25°С. Через 20 ч от начала реакции поглощение водорода прекращается. Подачу водорода останавливают и раствор отделяют от катализатора фильтрованием на Cellflock. Осадок на фильтре, содержащий катализатор, промывают этанолом (дважды по 100 мл). Фильтрат и промывные фракции объединяют и полученный таким образом раствор концентрируют при температуре 45°С при пониженном давлении до конечного объема около 400 мл. После добавления толуола (400 мл) образовавшийся раствор концентрируют до конечного объема около 250 мл, получая суспензию. Осадок отделяют фильтрованием и промывают его толуолом (дважды по 100 мл). Раствор снова концентрируют до конечного объема 200 мл и образовавшийся осадок снова отделяют фильтрованием. Осадок на фильтре промывают толуолом (трижды по 50 мл). Процесс разбавления толуолом, концентрирование и фильтрование повторяют до тех пор, пока в толуольном растворе не обнаруживается никаких существенных следов осадка. Наконец, растворитель выпаривают при температуре 45-50°С и пониженном давлении, а остаток высушивают в вакууме при температуре 45°С, получая 3-фтор-5-трифторметилфениламин в виде желтого масла. ГХ-МС: m/z: 179, 160, 151, 140, 132. Продукт идентифицирован (по данным ГХ и ВЭЖХ) с образцом 3-амино-5-фтор-бензотрифторида, полученным от фирмы ABCR (ABCR F01075). ЯМР-спектр также идентичен образцу, полученному от фирмы ABCR.
Пример 5: 3-(4-Метилимидазол-1-ил)-5-трифторметилфениламин (I)
Гидрид натрия (12,18 г, 55-65 мас.%, Fluka 71620) суспендируют в тетрагидрофуране (60 мл) и раствор 4-метилимидазола (24,5 г) в тетрагидрофуране (65 мл) медленно добавляют к перемешиваемой суспензии при температуре 20-25°С. Чтобы поддерживать температуру при 20-25°С во время добавления, необходимо применять мягкое охлаждение. После прибавления реакционную смесь перемешивают дополнительно 15 мин при 20-25°С, пока не прекратиться выделение газа. Раствор 3-фтор-5-трифторметилфениламина (XIX) (25 г) в 1-метил-2-пирролидоне (125 мл) медленно вносят в реакционную смесь и дополнительно продолжают перемешивание в течение 15 мин при температуре 20-25°С. Затем реакционную смесь нагревают на масляной бане при температуре 100°С для удаления летучего растворителя (тетрагидрофурана). Наконец, температуру повышают до 165°С (масляная баня) и реакционную смесь перемешивают в течение 22 ч при этой температуре. После этого реакционную смесь переносят в воду (500 мл) и водную фазу экстрагируют трет-бутилметиловым эфиром (дважды по 500 мл). Эфирные фазы объединяют и экстрагируют водой (дважды по 500 мл). Органический слой высушивают над безводным сульфатом магния (19 г) и растворитель выпаривают при температуре 45°С и пониженном давлении, получая сырой 3-(4-метил-имидазол-1-ил)-5-трифторметилфениламин в виде желтоватого твердого вещества. Сырой продукт, содержащий, по крайней мере, 1 региоизомер, растворяют в толуоле (93,4 г) при температуре 80-90°С и раствор оставляют самопроизвольно охлаждаться до комнатной температуры. Затем проводят кристаллизацию продукта при температуре около 35-40°С. Затем суспензию перемешивают дополнительно в течение 2 ч при комнатной температуре и продукт выделяют фильтрованием. Осадок на фильтре промывают охлажденным льдом толуолом (25 мл) и высушивают в вакууме при температуре 50°С, получая чистый 5-(4-метилимидазол-1-ил)-3-трифторметилфениламин (I). ГХ-МС: m/z 241, 222, 213, 200, 186, 172, 160.
1Н-ЯМР (400 МГц, ДМСО-d6): δ 2,15 (3H), 5,85 (2Н), 6,79 (1Н), 6,91 (1Н), 6,95 (1Н), 7,34 (1Н), 8,04 (1Н).
В частности, как описано выше, заместитель (бром) может быть селективно удален с получением 3-фтор-5-нитробензотрифторида (XIII). Синтез соединения (I) из соединения (XIII) представлен выше на схеме 6.
Схема 15
Пример 6: 5-(4-Метилимидазол-1-ил)-3-трифторметилбензонитрил (XXII)
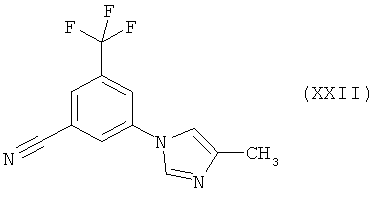
Раствор 4-метил-1Н-имидазола (1,98 г, 24,11 ммоля) в N-метилпирролидиноне (N-МП) (18 мл) прибавляют к раствору гидрида натрия (0,82 г, 60%-ный, 20,5 ммоля) в N-МП (18 мл) при температуре 20-25°С в атмосфере азота. Смесь перемешивают в течение 1 ч перед добавлением раствора 3-фтор-5-трифторметилбензонитрила (XXI) (3,2 г, 16,4 ммоля) в N-МП (8 мл). Реакционную смесь перемешивают в течение 2 ч при температуре 20-25°С, затем добавляют воду (120 мл) с 20-минутными интервалами и образовавшуюся суспензию перемешивают в течение 16 ч.
Осадок отфильтровывают, промывают водой (20 мл), растворяют в этилацетате (70 мл) и органический слой промывают водой (50 мл). Водную фазу экстрагируют этилацетатом (дважды по 40 мл) и объединенные органические слои выпаривают в вакууме до объема смеси 50 мл. После добавления гептана (68 мл) происходит кристаллизация продукта. Суспензию охлаждают до 0°С и перемешивают в течение 2 ч перед фильтрованием. Осадок на фильтре промывают холодным гептаном (дважды по 15 мл) и высушивают в вакууме, получая 3,1 г названного в заголовке соединения (выход 75,3%) в виде белых кристаллов (чистота продукта составляет 73,7% по данным ВЭЖХ).
Пример 7: 3-(4-Метилимидазол-1-ил)-5-трифторметилбензамид (XXIII)
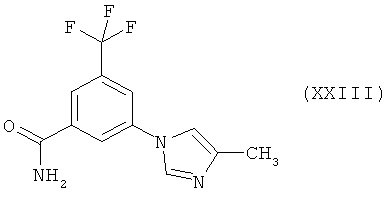
Раствор 5-(4-метилимидазол-1-ил)-3-трифторметилбензонитрила (3,5 г, 13,93 ммоля) в N-МП (28 мл) обрабатывают водным раствором аммиака (9,8 мл, 25%-ный) и водной перекисью водорода (3,5 мл, 30%-ная). Образовавшуюся смесь перемешивают в течение 1 ч при температуре 20-25°С, а затем переносят в охлажденную воду (420 мл). Образовавшуюся суспензию фильтруют, осадок на фильтре промывают водой (50 мл) и высушивают в вакууме при температуре 50°С, получая 3,2 г названного в заголовке соединения (XXIII) (выход 85,4%) в виде белых кристаллов (чистота составляет 98% по данным ВЭЖХ).
Пример 8: 5-(4-метил-1Н-имидазол-1-ил)-3-трифторметилфениламин (I)
Раствор 3-(4-метилимидазол-1-ил)-5-трифторметилбензамида (XXIII) (1 г, 3,71 ммоля) в трет-бутаноле (10 мл) и воде (3,8 мл) обрабатывают водными растворами гипохлорита натрия (3,7 мл, 9%-ный) и гидроксида натрия (1,5 мл, 30%-ный). Реакционную смесь перемешивают в течение 16 ч при температуре 60°С, после чего добавляют раствор гидросульфита натрия (2 мл, 10%-ный). Органические фазы отделяют и обрабатывают толуолом (5 мл) и водой (2,5 мл), а затем добавляют 2-молярный водный раствор НСl (5 мл). Образовавшуюся суспензию перемешивают в течение 1,5 ч, охлаждают до 0°С и фильтруют. Остаток на фильтре промывают толуолом (3 мл) и высушивают в вакууме, получая 0,39 г гидрохлорида названного в заголовке соединения (выход 43,2%) в виде оранжевых кристаллов (чистота 99,7% по данным ВЭЖХ). Для отделения анилина продукт обрабатывают водным раствором гидрокарбоната калия (2,2 мл, 5%-ный) в этаноле (1 мл) при температуре 45°С в течение 0,5 ч. Реакционную смесь затем охлаждают до 0°С в течение 1 ч и перемешивают в течение 2 ч. Продукт выделяют фильтрованием, промывают этанолом (дважды по 0,75 мл) и высушивают в вакууме при температуре 50°С, получая 0,27 г названного в заголовке соединения (I) (выход 32,8%) в виде почти белых кристаллов (чистота >99,9% по данным ВЭЖХ).
Пример 9: 5-(4-Метил-1Н-имидазол-1-ил)-3-трифторметилфениламин (I)
В одногорлую колбу, снабженную холодильником, добавляют CuI (89,5 мг, 0,47 ммоля), циклогексадиамин (107,3 мг, 0,94 ммоля) и диглим (10 мл). Смесь перемешивают в течение 10 мин при комнатной температуре. К гетерогенной смеси багрового цвета добавляют 3-бром-5-трифторметилфениламин (XVI) (1,13 г, 4,7 ммоля), 4-метил-1Н-имидазол (0,77 г, 9,4 ммоля) и Cs2CO3 (1,53 г, 4,7 ммоля). Смесь нагревают при температуре 150°С и дополнительно перемешивают в течение 24 ч. Затем смесь охлаждают до 25°С и очищают с помощью колоночной хроматографии (силикагель; EtOAc/MeOH в соотношении 95:5), получая соединение (I) в качестве основного продукта (выход 840 мг).
Пример 10: Получение 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (IX) (получение посредством каталитической конденсации)
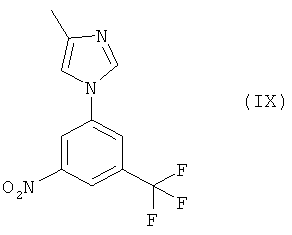
К перемешиваемой суспензии 1-бром-3-нитро-5-трифторметилбензола (4,05 г, 15 ммоля), 4-метил-1Н-имидазола (2,01 г, 24 ммоля, 98%-ный) и карбоната калия (3,73 г, 27 ммоля) в N,N-диметилформамиде (10 мл) добавляют диэтиламин (0,141 мл, 2,1 ммоля) и йодид меди(I) (0,204 г, 1,05 ммоля). Смесь при энергичном перемешивании нагревают до температуры 110°С в течение 23 ч. После этого большая часть 1-бром-3-нитро-5-трифторметилбензола успевает прореагировать, и суспензию оставляют самопроизвольно охлаждаться до комнатной температуры. Затем смесь разбавляют трет-бутилметиловым эфиром (30 мл) и добавляют 5%-ный водный раствор NaCl (30 мл) и изопропилацетат (15 мл). Водный слой отделяют и экстрагируют трет-бутилметиловым эфиром (10 мл) и изопропилацетатом (5 мл). Органические слои объединяют и фильтруют. Фильтрат промывают водой (10 мл), обрабатывают в течение 5 мин этилендиамином (0,303 мл), промывают водой (10 мл), 5%-ным водным раствором метабисульфита натрия (10 мл) и водой (10 мл), после чего обрабатывают углем (1,2 г) при комнатной температуре в течение 1 ч. Затем суспензию фильтруют с использованием ускорителя фильтрования и фильтрат выпаривают без досушивания при пониженном давлении, получая прозрачное, красно-коричневое масло, которое превращается в твердое вещество при стоянии при комнатной температуре. Полученное твердое вещество очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью (в соотношении 4:5) этилацетата и гексана (в присутствии 0,5 об.% триэтиламина) с получением в качестве основного вещества 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазол в виде бледно-желтого твердого вещества. Выход: 21,1% (чистота по данным ВЭЖХ: 96,7%). Т.пл.: 118-119°С.
Пример 11: 3-(4-Метилимидазол-1-ил)-5-трифторметилфениламин (I)
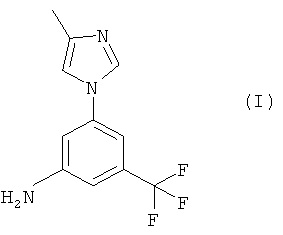
В автоклаве обрабатывают перед гидрированием суспензию 5%-ного палладия на активированном угле (0,6 г) в 94%-ном водном этаноле (200 мл). После этого добавляют 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазол (6,0 г, 22,1 ммоля) и смесь гидрируют при температуре 70°С и давлении 4 бар в течение 3 ч. За это время большая часть исходного вещества успевает прореагировать. Суспензию отфильтровывают с помощью ускорителя фильтрования. Фильтрат медленно переносят в воду (250 мл) при температуре 0-5°С. Образовавшуюся смесь концентрируют до массы 270 г, перемешивают, охлаждают до 0°С и снова перемешивают в течение почти 3 ч. Образовавшееся твердое вещество фильтруют, промывают водой (20 мл) и высушивают при температуре 50°С и пониженном давлении, получая 3-(4-метилимидазол-1-ил)-5-трифторметилфениламин в виде почти белого твердого вещества. Выход: 85,8% (чистота по ВЭЖХ составляет 94%), т.пл. 123-124°С.
Пример 12: Соль 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола с метансульфоновой кислотой (XXIX)
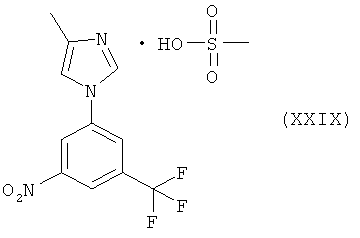
Сырой 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазол (IX) (1,85 г, 6 ммоля, чистота 88% по ВЭЖХ) растворяют в этилацетате (6 мл) при температуре около 50°С. К образовавшемуся черному раствору при перемешивании медленно прибавляют метансульфоновую кислоту (0,397 мл, 6 ммоля) при температуре около 50°С. В конце добавления начинается выпадение блестящего твердого вещества. Смесь оставляют самопроизвольно охлаждаться до комнатной температуры и продолжают перемешивание при температуре около 5°С в течение 75 мин. Образовавшееся твердое вещество отфильтровывают, промывают этилацетатом (4 мл) и высушивают при комнатной температуре и пониженном давлении. Суспензию получившегося вещества перемешивают в 2-пропаноле (5 мл) при температуре 50°С в течение 90 мин, оставляют самопроизвольно охлаждаться до комнатной температуры, а затем перемешивают в течение 1 ч и дополнительно при температуре 0-5°С в течение еще 1 ч. Образовавшееся твердое вещество отфильтровывают, промывают холодным 2-пропанолом (5 мл) и высушивают при комнатной температуре и пониженном давлении, получая соль метансульфоновой кислоты и 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола в виде бежевого твердого вещества. Выход: 54,1% (чистота по ВЭЖХ: 99,5%), т.пл. 208-213°С.
Пример 13: Получение 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (IX) (посредством ароматического замещения)
4-Метилимидазол (10,5 г, 125,5 ммоля) и карбонат калия (12,0 г, 119,6 ммоля) суспендируют в N,N-диметилформамиде (80 мл) и перемешивают при температуре 100°С в течение 1 ч, затем добавляют раствор 1-фтор-3-нитро-5-трифторметилбензола (12,5 г, 59,8 ммоля) в N,N-диметилформамиде (20 мл) в течение 10 мин. Смесь перемешивают при температуре реакционной смеси 108°С в течение 3 ч. ВЭЖХ анализ подтверждает полное вступление в реакцию исходного соединения. Смесь охлаждают приблизительно до температуры 20°С и добавляют воду (200 мл) в течение 1 ч. Образовавшуюся суспензию отфильтровывают, получая 17,5 г влажного твердого вещества (ВЭЖХ: 88,8% нужного изомера, 8,9% нежелательного изомера/побочного продукта).
Суспензию полученного продукта в воде (100 мл) перемешивают в течение 1 ч при комнатной температуре. Твердое вещество отфильтровывают, промывают водой (100 мл) и высушивают при температуре 50°С и пониженном давлении, получая сырой продукт по анализу ВЭЖХ, состоящий на 90% из требуемого продукта. Перекристаллизацию проводят путем обработки раствора вышеназванного сырого продукта (9,5 г) в этилацетате (50 мл) в течение 2 ч при температуре 70°С активированным углем (1 г) и ускорителем фильтрования (1 г), после чего фильтруют, а фильтрат выпаривают без досушивания, получая 11,1 г остатка, который растворяют в этилацетате (3,25 г) и гептане (50 мл) при нагревании с обратным холодильником. В раствор вносят затравку 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола при температуре 65°С и оставляют самопроизвольно охлаждаться до комнатной температуры в течение ночи, после чего перемешивают при 0°С в течение 3 ч. Образовавшееся твердое вещество отфильтровывают, промывают гептаном (20 мл) и высушивают при 50°С при пониженном давлении, получая 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазол в виде твердого вещества. Общий выход составляет 53,3% (чистота по ВЭЖХ 98,2%), т.пл. 117-118°С.
Пример 14: 1-Бром-3-нитро-5-трифторметилбензол (XI)
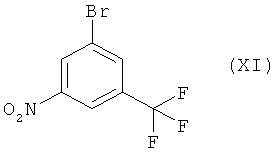
К раствору 1-нитро-3-трифторметилбензола (41,1 мл, 300 ммолей, 97%-ный, получен от фирмы Aldrich) в дихлорметане (240 мл) добавляют 98%-ную серную кислоту (45,7 мл, 840 ммоля) в течение 10 мин. При энергичном перемешивании двухфазную смесь нагревают до температуры 35°С и шестью одинаковыми порциями вносят 1,3-дибром-5,5-диметилимидазолидин-2,4-дион (в сумме 53,1 г, 180 ммоля) в течение 5 ч. Смесь дополнительно перемешивают при температуре 35°С в течение 19 ч. После этого более 97% исходного вещества вступает в реакцию согласно анализу ВЭЖХ. Реакционную смесь оставляют самопроизвольно охлаждаться до комнатной температуры, после чего в течение 20 мин при перемешивании добавляют 2-молярный водный раствор NaOH (210 мл) при охлаждении на водяной бане до 0-5°С. Внутренняя температуре поднимается временно до 35°С. Затем разделяют два слоя и водный слой экстрагируют гексаном (трижды по 200 мл). Объединенные органические слои промывают водой (200 мл), 5%-ным водным раствором метабисульфита натрия (дважды по 200 мл), 8%-ным водным раствором NaHCO3 (200 мл) и 10%-ным водным раствором NaCl (200 мл). После этого растворители выпаривают при пониженном давлении и температуре 45°С. Оставшуюся жидкость перегоняют при давлении 0,71 мбар и температуре бани 70-80°С, получая 1-бром-3-нитро-5-трифторметилбензол в виде бледно-желтой жидкости. Выход: 89,6% (чистота по 1Н-ЯМР около 95%).
1Н-ЯМР (400 МГц, CDCl3): 8,11 м.д. (m, 1Н), 8,45 м.д. (m, 1H), 8,58-8,59 м.д (m, 1Н). Т кип. 68°С при 0,71 мбар.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СРЕДСТВО С КВИСКВАЛАТ-АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ НА ИХ ОСНОВЕ | 1992 |
|
RU2117663C1 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ | 2000 |
|
RU2261862C2 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНДИОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1994 |
|
RU2140420C1 |
| СПОСОБ СИНТЕЗА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2404167C2 |
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ АНТАГОНИСТОВ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2327697C2 |
| ИНГИБИТОРЫ КИНАЗЫ, ИХ ПРОЛЕКАРСТВЕННЫЕ ФОРМЫ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2010 |
|
RU2568639C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ, И СПОСОБ ПОЛУЧЕНИЯ АНТИАНГИОГЕННОГО ЭФФЕКТА И/ИЛИ ЭФФЕКТА СНИЖЕНИЯ СОСУДИСТОЙ ПРОНИЦАЕМОСТИ С ИХ ПРИМЕНЕНИЕМ | 1996 |
|
RU2194701C2 |
| БЕНЗОИМИДАЗОЛЫ КАК ИНГИБИТОРЫ ПРОЛИЛГИДРОКСИЛАЗЫ | 2009 |
|
RU2531354C2 |
| ПРОИЗВОДНОЕ СКОНДЕНСИРОВАННОГО ПИРАЗИНА | 1991 |
|
RU2095352C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА | 2005 |
|
RU2414468C2 |
Настоящее изобретение относится к способу получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I), заключающийся в том, что 4-метил-1H-имидазол или его соль вводят в реакцию с соединением формулы
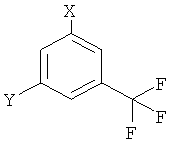
где Х обозначает галоген и Y обозначает NH2, в присутствии подходящего основания или соответствующего переходного металла в качестве катализатора или их комбинации в соответствующем растворителе. Также изобретение относится к другим вариантам способа получения соединения формулы (I) и используемым промежуточным соединениям. Технический результат: разработаны новые варианты способа получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I), который является промежуточным соединением для получения биологически активных соединений. 7 н. и 2 з.п. ф-лы, 14 пр.
1. Способ получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I), заключающийся в том, что 4-метил-1Н-имидазол или его соль вводят в реакцию с соединением формулы
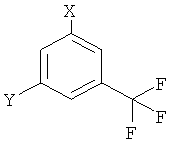
где Х обозначает галоген и
Y обозначает NH2,
в присутствии подходящего основания или соответствующего переходного металла в качестве катализатора или их комбинации в соответствующем растворителе.
2. Способ получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I), заключающийся в осуществлении следующих стадий:
а) нитрование 2-бром-5-фторбензотрифторида нитратом калия и серной кислотой с получением 2-бром-5-фтор-1-нитро-3-трифторметилбензола (XVIII);
б) восстановление 2-бром-5-фтор-1-нитро-3-трифторметилбензола посредством гидрирования на катализаторе палладий/уголь с получением 3-фтор-5-трифторметилфениламина и
с) взаимодействие 3-фтор-5-трифторметилфениламина с натриевой солью 4-метилимидазола с получением соединения (I).
3. Соединение формулы
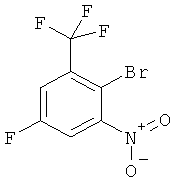 .
.
4. Способ получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I), заключающийся в осуществлении следующей реакции:
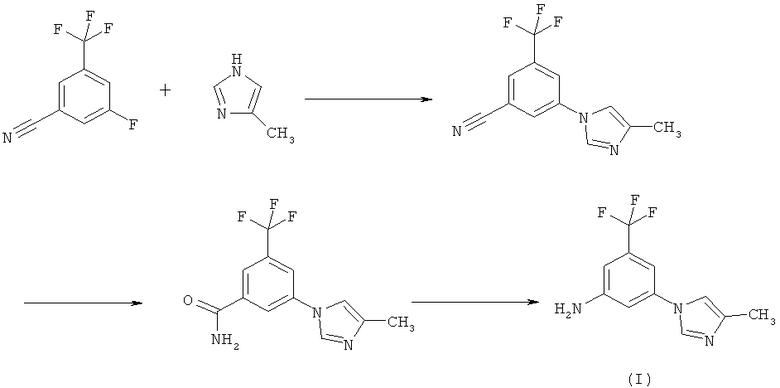
с использованием гидрида натрия в N-метилпирролидиноне (N-МП) на первой реакционной стадии и
с использованием водного раствора аммиака и водной перекиси водорода в N-метилпирролидиноне (N-МП) на второй реакционной стадии.
5. Способ получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I), заключающийся в осуществлении следующих стадий:
а) реакция 3-бром-5-фторбензотрифторида с 4-метилимидазолом в присутствии сильного основания;
б) перекристаллизация из гептана сырого соединения, полученного на стадии (а);
в) ариламинирование соединения, полученного на стадии (б), и дифенилимина в присутствии палладиевого катализатора, фосфинового лиганда и основания;
г) гидролиз продукта стадии (в) водным раствором хлористоводородной кислоты с получением 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I) в виде его хлористоводородной соли и
д) необязательное превращение соли 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I) в свободное основание.
6. Способ по п.5, где на стадии (в) катализатор выбирают из Pd(OAc)2;
тетракис(трифенил)фосфинпалладия(0);
трис(дибензилиденацетон)дипалладия(0) или хлорида палладия и где лиганды выбирают из Ксантфос, БИНАП, трифенилфосфина и триалкилфосфинов.
7. Способ получения 1-бром-3-нитро-5-трифторметилбензола, заключающийся в обработке 1-нитро-3-трифторметилбензола бромирующим агентом в присутствии сильной кислоты.
8. Способ по п.7, заключающийся в обработке 1-нитро-3-трифторметилбензола бромирующим агентом, выбранным из 1,3-дибром-5,5-диметилгидантоина (1,3-дибром-5,5-диметилимидазолидин-2,4-дион), в присутствии сильной кислоты в инертном растворителе при температуре от 25 до 40°С с получением 1-бром-3-нитро-5-трифторметилбензола.
9. Соединение 1-бром-3-нитро-5-трифторметилбензол формулы
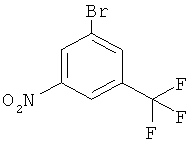 .
.
| ВЫСОКОЧАСТОТНЫЙ СПЕКТРОМЕТР | 0 |
|
SU166533A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| RU 96122865 A, 27.01.1999 | |||
| Способ получения производных имидазола | 1982 |
|
SU1051076A1 |

