Настоящее изобретение относится к 1H-хиназолин-2,4-дионам, их получению, их применению в качестве фармацевтических веществ и фармацевтическим композициям, содержащим их.
В частности, настоящее изобретение обеспечивает соединения формулы (I)
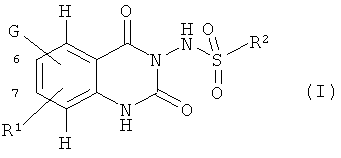
где
G означает NR3R4 или OR5
где
R3, R4 и R5 означают независимо водород, арил, аралкил, ацил или алкил, необязательно замещенный арилом, гетероциклилом, арилоксигруппой, аралкилоксигруппой или алкоксикарбониламиногруппой, или
R3 и R4 образуют вместе со смежным атомом азота гетероарильную или гетероциклильную группу, содержащую по меньшей мере один кольцевой атом азота и присоединенную посредством данного кольцевого атома азота, где гетероарильная и гетероциклильная группы необязательно замещены арилом, аралкилом, арилоксиалкилом, аминокарбонилалкилом, моно- или диалкиламинокарбонилалкилом или морфолинокарбонилалкилом,
R1 означает нитрогруппу или трифторметильную группу, и
R2 означает алкил, арил или аралкил,
и их соли.
Как указано выше, алкил, гетероарил и гетероциклил являются необязательно замещенными, предпочтительно незамещенными, моно-, ди- или тризамещенными упомянутыми заместителями, более предпочтительно незамещенными или монозамещенными упомянутыми заместителями.
Если не указано иначе, выражения, использованные в данном изобретении, имеют следующие значения.
Ацил означает алкилкарбонил, арилкарбонил или аралкилкарбонил.
Алкил означает линейный, разветвленный или циклический, насыщенный или ненасыщенный, предпочтительно насыщенный алкил, предпочтительно (С1-С8)алкил, более предпочтительно (С1-С6)алкил, наиболее предпочтительно (С1-С4)алкил. Алкил является необязательно замещенным одним или несколькими заместителями, предпочтительно от одного до трех заместителей. Заместителями предпочтительно являются галоид, гидроксил, циангруппа, (С1-С4)алкоксигруппа, (С1-С4)алкоксикарбонил, (С1-С4)алкиламиногруппа, ди(С1-С4)алкиламиногруппа, (С1-С4)алкоксикарбониламиногруппа или (С1-С4)алкилкарбониламиногруппа.
Алкан (например, в алкансульфониле) и алк (например, в алкокси) определяются аналогично алкилу, особенно в отношении линейности, насыщенности, предпочтительного размера и необязательного замещения.
Арил означает предпочтительно фенил, нафтил или 5-10-членный гетероарил, более предпочтительно фенил или 5-6-членный гетероарил. Арил является необязательно замещенным, предпочтительно незамещенным, моно-, ди- или тризамещенным. Заместителями предпочтительно являются галоид, более предпочтительно фтор или хлор, нитрогруппа, циангруппа, формил, карбоксамидогруппа, гидроксил, аминогруппа, (С1-С4)алкиламиногруппа, ди(С1-С4)алкиламиногруппа, (С1-С4)алкил, (С1-С4)алкоксигруппа, (C1-С4)алкоксикарбонил, (С1-С4)алкансульфонил, (С1-С4)алкилкарбонил, (С1-С4)алкоксикарбониламиногруппа или (С1-С4)алкилкарбониламиногруппа.
Аралкил означает алкил, замещенный арилом.
Галоид означает предпочтительно бром, хлор или фтор.
Гетероарил означает моно- или полициклический, предпочтительно моно- или бициклический, наиболее предпочтительно моноциклический ароматический остаток, содержащий один или несколько, предпочтительно от одного до трех, кольцевых гетероатомов, предпочтительно выбранных из атомов азота, кислорода и серы, наиболее предпочтительно азот. Гетероарил означает предпочтительно 5-10-членный гетероарил, более предпочтительно 5-6-членный гетероарил. Гетероарил является необязательно замещенным, предпочтительно незамещенным, моно-, ди- или тризамещенным. Заместителями предпочтительно являются галоид, более предпочтительно фтор или хлор, нитрогруппа, циангруппа, формил, карбоксамидогруппа, гидроксил, аминогруппа, (C1-С4)алкиламиногруппа, ди(С1-С4)алкиламиногруппа, (С1-С4)алкил, (С1-С4)алкоксигруппа, (С1-С4)алкоксикарбонил, (С1-С4)алкансульфонил, (С1-С4)алкилкарбонил, (С1-С4)алкоксикарбониламиногруппа или (C1-С4)алкилкарбониламиногруппа.
Гетероциклил означает моно- или полициклический, предпочтительно моно- или бициклический, наиболее предпочтительно моноциклический, насыщенный или частично ненасыщенный циклический остаток, содержащий три или несколько кольцевых атомов, предпочтительно от трех до десяти кольцевых атомов, из которых один или несколько, предпочтительно от одного до трех, являются гетероатомами, предпочтительно выбранными из атомов азота, кислорода и серы. Гетероциклил является необязательно замещенным, предпочтительно галоидом, циангруппой, карбоксамидогруппой, гидроксилом, аминогруппой, (С1-С4)алкиламиногруппой, ди(С1-С4)алкиламиногруппой, (С1-С4)алкилом, (С1-С4)алкоксигруппой, (С1-С4)алкоксикарбонилом, (C1-С4)алкилкарбонилом, (С1-С4)алкоксикарбониламиногруппой или (C1-С4)алкилкарбониламиногруппой.
Соли означают предпочтительно физиологически приемлемые соли, образованные, как соответствует, прибавлением кислоты или основания.
Упомянутые необязательные заместители, перечисленные в определениях для алкила, арила, гетероарила и гетероциклила, следует понимать как заместители в добавление к тем, которые перечислены в общих формулах, т.е., например, алкил может иметь заместители, перечисленные в общей формуле и/или определениях.
Таутомерные формы соединений формулы (I) также охватываются изобретением. В тех случаях, где имеется один или несколько асимметрических атомов, особенно углеродных атомов, соединения существуют в индивидуальной оптически активной изомерной форме или в качестве их смесей, например в качестве рацемических или диастереомерных форм. Настоящее изобретение охватывает индивидуальные оптически активные изомеры, а также их смеси, например рацемические или диастереомерные смеси.
Предпочтение отдается соединениям формулы (Ia)
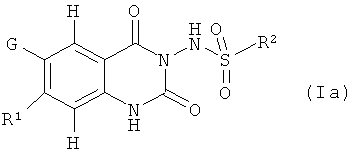
где R1, G и R2 имеют такие значения, которые определены в данном описании.
Дополнительное предпочтение отдается соединениям формулы (Ib)

где R2, R3 и R4 имеют такие значения, которые определены в данном описании.
Дальнейшее предпочтение отдается соединениям формулы (1с)

где R1, R2 и R5 имеют такие значения, которые определены в данном описании.
Дальнейшее предпочтение отдается соединениям формулы (Id)
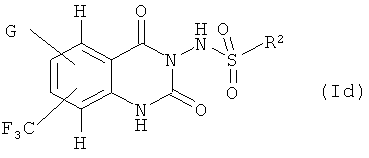
где G и R2 имеют такие значения, которые определены в данном описании.
Дальнейшее предпочтение отдается соединениям формулы (Ie)

где R1 и R2 имеют такие значения, которые определены в данном описании.
Дальнейшее предпочтение отдается соединениям формулы (Ih)

где G и R2 имеют такие значения, которые определены в данном описании.
Предпочтительно G означает R3R4N.
Предпочтительно R3 означает водород, арил, аралкил, ацил или алкил, необязательно замещенный гетероциклилом, арилоксигруппой, аралкилоксигруппой или алкоксикарбониламиногруппой, и R4 означает водород или алкил, или R3 и R4 образуют вместе со смежным атомом азота гетероарил или гетероциклил, содержащий по меньшей мере один кольцевой атом азота и присоединенный посредством этого кольцевого атома азота, где гетероарил и гетероциклил являются необязательно замещенными арилом, аралкилом, арилоксиалкилом, аминокарбонилалкилом или морфолинокарбонилалкилом. Предпочтительно R3 и R4 образуют вместе со смежным атомом азота 5-членный гетероарил, содержащий по меньшей мере один кольцевой атом азота и связанный посредством данного кольцевого атома азота, наиболее предпочтительно имидазол-1-ил.
Предпочтительно R1 представляет нитрогруппу.
Предпочтительно R2 представляет метил, этил, фенил, бензил, нитрофенил или пиридил, наиболее предпочтительно метил, этил или фенид.
В другом варианте воплощения изобретение обеспечивает соединения формулы (I),
где
G означает NR3R4 или OR5,
где
R3 означает водород, аралкил или алкил, необязательно замещенный арилом, гетероциклилом, арилоксигруппой, аралкилоксигруппой или алкоксикарбониламиногруппой,
R4 означает водород или алкил, или
R3 и R4 образуют вместе со смежным атомом азота гетероарил или
гетероциклил, содержащий по меньшей мере один кольцевой атом азота и связанный посредством данного кольцевого атома азота, где гетероарил и гетероциклил являются необязательно замещенными арилом, аминокарбонилалкилом, моно- или
диалкиламинокарбонилалкилом или морфолинокарбонилалкилом, и
R5 означает алкил,
R1 означает нитрогруппу или трифторметил, и
R2 означает алкил,
и их соли.
Особенно предпочтительными являются соединения формулы (Ia), где R1 представляет нитрогруппу и R2 представляет метил.
Особенно предпочтительными являются соединения формулы (Ia), где R1
представляет трифторметил и R2 представляет метил.
Особенно предпочтительными являются соединения формулы (Ia), где R1 представляет нитрогруппу и R2 представляет этил.
Особенно предпочтительными являются соединения формулы (Ia), где R1 представляет трифторметил и R2 представляет этил.
Особенно предпочтительными являются соединения формулы (Ia), где R1 представляет нитрогруппу и R2 представляет фенил.
Особенно предпочтительными являются соединения формулы (Ia), где R1 представляет трифторметил и R2 представляет фенил.
Особенно предпочтительными являются соединения формулы (I), где G представляет насыщенный незамещенный гетероцикл.
Особенно предпочтительными являются соединения формулы (I), где R3 представляет метил и R4 представляет гидроксиэтил.
Предпочтительные, особенно предпочтительные диапазоны и формулы могут быть сопоставлены по усмотрению. Определения применимы к соединениям формулы (I) и соответствующим исходным материалам и промежуточным веществам.
В дальнейшем аспекте настоящее изобретение обеспечивает способы получения соединений по изобретению.
Способ 1
Соединения формулы (Ib)
где R2, R3 и R4 имеют значения, указанные выше,
могут быть получены взаимодействием соединения формулы (II)

где R2 имеет значение, указанное выше,
с соединением формулы (III)
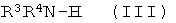 ,
,
где R3 и R4 имеют значения, указанные выше.
С этой целью смесь соединения формулы (II) с избытком соединения формулы (III), предпочтительно 1,5 до 30 эквивалентов, наиболее предпочтительно 2 до 10 эквивалентов, беспримесная или растворенная в соответствующем инертном растворителе, таком как 1,3-диметилимидазолидин-2-он, диметилсульфоксид, уксусная кислота или этанол, может быть нагрета в закрытом сосуде при высоких температурах, например 150°С, используя масляную баню или микроволновой реактор, в течение требуемого времени, например 5 мин до 1 ч, или альтернативно в соответствующем высококипящем инертном растворителе, как диметилсульфоксид, в открытой системе до более низкой температуры, например 120°С, в течение более длительного времени, например 16 ч. При необходимости защищенные части соединения, такие как гидроксил или аминогруппа, могут быть деблокированы в продукте реакции или продукт реакции может быть далее трансформирован, например, восстановлением или окислением.
Соединение формулы (II) может быть получено общепринятыми способами из амина (IV).
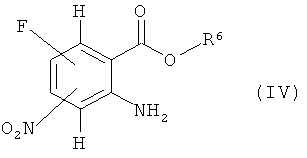
где R6 означает алкил,
конверсией в изоцианат (V)
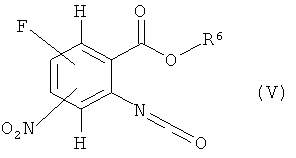
где R6 имеет значение, указанное выше,
например, реакцией с фосгеном и последующей циклоконденсацией с сульфонилгидразином (VI)
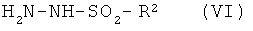
где R2 имеет значение, указанное выше,
в соответствующем инертном растворителе, таком как тетрагидрофуран, с последующим прибавлением основания, например водного раствора гидроксида натрия, или органического основания, такого как триэтиламин или основание Хюнига (N,N-диизопропилэтиламин). Соединения формул (IV) и (V) известны или могут быть получены по известным в литературе методикам или по аналогии с ними.
Следующая реакционная схема является иллюстративной для способа 1.
Схема 1

Способ 2
Соединения формулы (Ic)
где R1, R2 и R5 имеют значения, указанные выше, могут быть получены реакцией соединения формулы (VII)

где R1 и R5 имеют значения, указанные выше, и R7 означает алкил, с соединением формулы (VI) в соответствующем инертном растворителе, таком как тетрагидрофуран, необязательно в присутствии соответствующего основания, такого как водный раствор гидроксида натрия, или органического основания, такого как триэтиламин или основание Хюнига. Соответствующие температуры для данной реакции находятся в интервале 0-40°С, предпочтительно 22°С.
Изоцианат (VII) может быть получен из амина (VIII)

где R1 и R7 имеют значения, указанные выше,
реакцией с алкоголятом (IX)

где R5 имеет значение, указанное выше, и М+ означает металл, предпочтительно ион щелочного металла, в соответствующем инертном растворителе, например соответствующем спирте R5-ОН, с последующим гидролизом ацетамида, например с 98% серной кислотой, в амин (X).
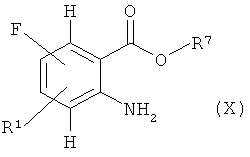
где R1 и R7 имеют значения, указанные выше,
и конверсией в изоцианат (VII), например, с фосгеном или трифосгеном. Соединения формулы (VIII) известны или могут быть получены по известным в литературе методикам или по аналогии с ними.
Следующая реакционная схема является иллюстративной для способа 2.
Схема 2

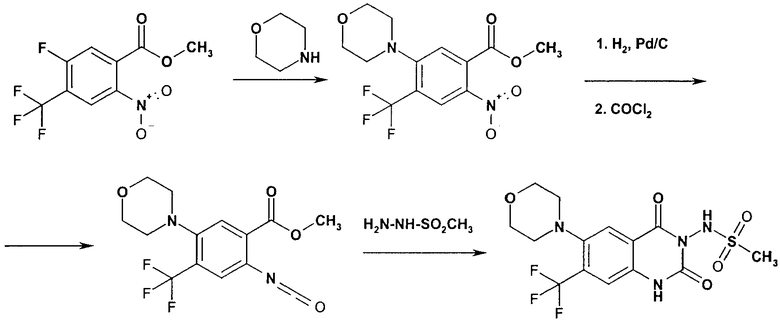
Способ 3
Соединения формулы (Id)
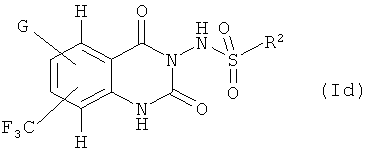
где G и R2 имеют значения, указанные выше,
могут быть получены реакцией соединения формулы (XI)

где G имеет значение, указанное выше,
с соединением формулы (VI).
Реакцию осуществляют в соответствующем инертном растворителе, таком как тетрагидрофуран, с последующим действием основания, например водного раствора гидроксида натрия, или органического основания, такого как триэтиламин или основание Хюнига.
Изоцианат (XI) может быть получен нуклеофильным замещением фторида (XII)

с амином (III) или алкоксидом (IX), давая нитросоединение (XIII),

где G имеет значение, указанное выше,
с последующим восстановлением в амин (XIV),

где G имеет значение, указанное выше,
с соответствующим восстанавливающим агентом, например, гидрированием с водородом, используя палладий на угле в качестве катализатора, и трансформацией в изоцианат (XI), например, с фосгеном или трифосгеном.
Соединения формулы (XII) известны или могут быть получены по известным в литературе методикам или по аналогии с ними.
Следующая реакционная схема является иллюстративной для способа 3.
Схема 3
Способ 4
Соединения формулы (Ie)
где
R1 и R2 имеют значения, указанные выше, и
G' означает R3'R4'N или R5'O,
где
R3', R4' и R5' имеют значения R3, R4 и R5 соответственно, как указано выше, при условии, что углеродные атомы R3', R4' и R5', смежные с азотом и кислородом
R3'R4'N и R5'O соответственно, являются первичными или вторичными углеродными атомами,
могут быть получены конденсацией, алкилированием или восстановительным алкилированием соединений формулы (If),
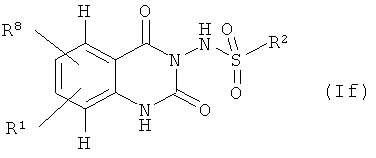
где
R1 и R2 имеют значения, указанные выше, и
R8 означает аминогруппу или гидроксил,
с соответствующими галоидными или альдегидными предшественниками R3', R4' или R5' в присутствии катализатора конденсации, основания или восстанавливающего агента, например натрийборциангидрида, соответственно.
Соединения формулы (If) могут быть получены деблокированием соединений формулы (Ig)
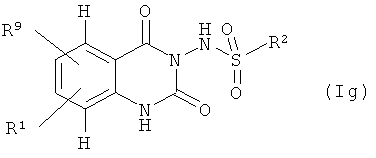
где
R1 и R2 имеют значения, указанные выше, и
R9 означает замещенные аминогруппу или гидроксил,
которое, в свою очередь, может быть получено одним из приведенных выше способов 1-3. Соответствующие галоидные или альдегидные предшественники R3', R4' или R5' известны или могут быть получены по известным в литературе методикам или по аналогии с ними.
Следующая реакционная схема является иллюстративной для способа 4.
Схема 4
Способ 5
Соединения формулы (Ih)

где G и R2 имеют значения, указанные выше, получаются реакцией соединения формулы (XVI)
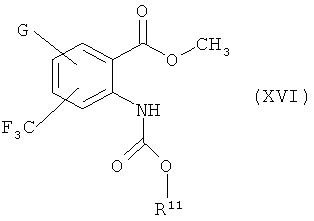
где
G имеет значение, указанное выше,
R11 представляет фенил, который необязательно замещен галоидом или (С1-С4)алкилом, с соединением формулы (VI).
Реакция предпочтительно осуществляется в соответствующем инертном растворителе, таком как тетрагидрофуран, с последующим прибавлением основания, например водного раствора гидроксида натрия, или органического основания, такого
как триэтиламин, этилдиизопропиламин или основание Хюнига.
Реакция особенно предпочтительна для соединений формулы (If), в которых G
находится в положении 6, и трифторметильная группа находится в положении 7.
Карбамат (XVI) может быть получен реакцией замещения соответствующего
аминосоединения формулы (XVII)

с хлорформиатом (XVIII)

где R11 имеет значения, указанные выше, в присутствии разбавителя, такого как диоксан, давая карбамат (XVI).
Соединения формул (XVIII) и (XVII) известны или могут быть получены по известным в литературе методикам или по аналогии с ними.
Следующая реакционная схема является иллюстративной для способа 5.
Схема 5

Следующие соображения могут применяться в зависимости от обстоятельств ко всем способам, описанным в контексте, а также к получению соответствующих исходных материалов и промежуточных веществ.
Одна или несколько функциональных групп, например карбоксигруппа, гидроксигруппа, аминогруппа или меркаптогруппа, могут нуждаться в блокировании в исходных соединениях с помощью защитных групп. Используемые защитные группы могут уже присутствовать в предшественниках и должны защищать реакционно-способные функциональные группы от нежелательных вторичных реакций, таких как реакции алкилирования, образования простых эфиров, образования сложных эфиров, сольволиз и подобные реакции. Отличительной чертой защитных групп является то, что они сами легко, т.е. без нежелательных вторичных реакций, подвергаются удалению, обычно сольволизом, восстановлением, фотолизом или также с помощью ферментативной активности, например, в условиях, аналогичных физиологическим условиям, и что они не присутствуют в конечных продуктах. Специалист знает или может легко установить, какие защитные группы соответствуют реакциям, упомянутым ранее и впредь.
Защита подобных функциональных групп такими защитными группами, сами защитные группы и реакции их удаления описаны, например, в стандартных справочных публикациях, таких как J.F.W.McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York, 1973, в Т.W.Greene, "Protective Groups in Organic Synthesis", Wiley, New York, 1981, в "The Peptides"; том 3 (редакторы: E.Gross и J.Meienhofer), Academic Press, London and New York, 1981, в "Methoden der organischen Chemie" (Methods of organic chemistry), Houben Weyl, 4-е издание, том 15/1, Georg Thieme Verlag, Stuttgart, 1974, в H.-D.Jakubke и H.Jescheit, "Aminosäuren, Peptide, Proteine" (Amino acids, peptides, proteins), Verlag Chemie, Weinheim, Deerfield Beach, and Basel, 1982 и в Jochen Lehmann, "Chemie der Kohlenhydrate: Monosaccharide und Derivate" (Chemistry of carbohydrates: monosaccharides and derivatives), Georg Thieme Verlag, Stuttgart, 1974.
Кислотно-аддитивные соли могут быть получены из свободных оснований известным образом и наоборот. Соединения формулы (I) в оптически чистой форме могут быть получены из соответствующих рацематов согласно хорошо общепринятым процедурам, например ВЭЖХ с хиральной матрицей. Альтернативно могут быть использованы оптически чистые исходные соединения.
Стереоизомерные смеси, например смеси диастереомеров, могут быть разделены на соответствующие изомеры общепринятым по сути образом посредством соответствующих способов разделения. Диастереомерные смеси, например, могут быть разделены на их индивидуальные диастереомеры посредством фракционной кристаллизации, хроматографии, распределением между растворителями и подобными процедурами. Данное разделение может происходить или на уровне исходного соединения, или на уровне самого соединения формулы (I). Энантиомеры могут быть разделены посредством образования диастереоизомерных солей, например солеобразованием с энантиомерно чистой хиральной кислотой, или посредством хроматографии, например ВЭЖХ, с использованием хроматографических субстратов с хиральными лигандами.
В дальнейшем аспекте соединения обеспечивают новые промежуточные соединения, как определено выше. Данные промежуточные соединения применимы для получения соединений формулы (I) и также обладают интересными фармацевтическими свойствами. Данные соединения также являются объектом по настоящему изобретению.
Соединения по изобретению обладают фармакологической активностью и, следовательно, пригодны в качестве фармацевтических средств. В частности, соединения являются мощными конкурентными антагонистами рецептора АМРА (α-амино-3-гидрокси-5-метилизоксазол-4-пропионовой кислоты) с некоторой активностью в отношении каинатных рецепторов.
Соединения по изобретению особенно эффективны в качестве фармацевтических средств в лечении эпилепсии, особенно частичных эпилептических припадков (простых, сложных и частичных, эволюционирующих во вторичные генерализованные эпилептические припадки) и генерализованных эпилептических припадков [малого эпилептического припадка (типичного и атипичного), миоклонического, клонического, тонического, тонического-клонического и атонического].
Соединения по изобретению особенно эффективны также в качестве фармацевтических средств в лечении психозов при шизофрении, при биполярном нарушении, болезни Паркинсона и при индуцированных препаратами психозах, а также в улучшении позитивных и негативных симптомов и эффективны при лечении пациентов, устойчивых к действию препаратов (сравни Н.О.Kalkman, E. GAD67: the link between GABA-deficit hypothesis and the dopaminergic- and glutamatergic theories of psychosis. J. Neural. Transm. 2003, 1110, c.c.803-812).
Кроме того, соединения по изобретению пригодны в качестве фармацевтических средств для лечения любой патологии, нарушения или клинического состояния, включающего опосредованное рецептором АМРА нейронное повреждение, например нейродегенеративные нарушения, такие как множественный склероз, амиотрофический латеральный склероз, болезнь Паркинсона, болезнь Хантингтона или болезнь Альцгеймера, шизофрения, особенно хроническая шизофрения, тревога, депрессия, биполярные нарушения настроения, нарушения сна, нарушения познавательной способности, рвота, шум в ушах, боль, неврональная боль, мигрень, рост опухоли, симптомы аутизма, ишемические и гипоксические состояния, такие как инсульт, субарахноидальная геморрагия, перинатальная гипоксия, травмы мозга и позвоночника, повреждение головы, высокое внутричерепное давление и любая хирургическая процедура, потенциально ассоциируемая с гипоксией центральной нервной системы, и состояния, вызываемые действиями окружающей среды, экзогенными нейротоксинами, включая те, которые вызваны инфекциями, а также те, которые вызваны метаболическими изменениями и гепатоэнцефалопатией, ассоциированной с печеночной недостаточностью.
Для всех этих показаний соответствующая доза будет, безусловно, меняться, завися, например, от используемого соединения по изобретению, пациента, способа введения и природы и тяжести подлежащих обработке состояний. Однако, в общем, удовлетворительные результаты у животных, как показано, получаются при ежедневных дозах от примерно 0,1 до примерно 30 мг/кг массы тела животного. У крупных млекопитающих, например у человека, указанная ежедневная доза находится в интервале от примерно 5 мг до примерно 2 г соединения по изобретению, удобно вводимых, например, в раздельных дозах до четырех раз в день.
Активные препараты по изобретению могут быть введены любым общепринятым путем, особенно энтерально, предпочтительно перорально, например, в форме таблеток или капсул, или парентерально, например, в форме инъекционных растворов или суспензий.
Согласно вышеупомянутому настоящее изобретение обеспечивает соединения для применения в качестве фармацевтических средств, в особенности для применения в лечении любой патологии, нарушения или клинического состояния, включающих этиологию рецепторов АМРА или включающих опосредованное рецепторами АМРА неврональное повреждение, и особенно для применения в любых специфических показаниях, перечисленных выше.
Настоящее изобретение обеспечивает также фармацевтическую композицию, включающую соединения в сочетании по меньшей мере с одним фармацевтическим носителем или разбавителем. Такие композиции могут быть получены обычным образом. Стандартные лекарственные формы содержат, например, от примерно 1 мг до примерно 400 мг активного агента согласно изобретению.
Кроме того, настоящее изобретение обеспечивает применение соединения по изобретению для получения лекарственного средства для лечения любой патологии, нарушения или клинического состояния, включающих этиологию рецепторов АМРА или включающих опосредованное рецепторами АМРА неврональное повреждение.
Более того, настоящее изобретение обеспечивает способ лечения любой патологии, нарушения или клинического состояния, включающих этиологию рецепторов АМРА или включающих опосредованное рецепторами АМРА неврональное повреждение у субъекта, нуждающегося в таком лечении, которое включает введение такому субъекту терапевтически эффективного количества соединения по изобретению.
Кроме того, соединения по изобретению могут быть скомбинированы с другими препаратами, пригодными для различных показаний, например, в случае эпилепсии с другими антиэпилептическими препаратами, как барбитураты и их производные, бензодиазепины, карбоксамиды, гидантоины, сукцинимиды, вальпроевая кислота и другие производные жирных кислот, другие антагонисты АМРА. Соединения по изобретению могут быть также скомбинированы с нейролептическими препаратами, выбранными из списка, состоящего из атипичных антипсихотических препаратов, таких как клозапин, оланзапин, рисперидон, и типичных антипсихотических препаратов, таких как галоперидол.
Данная заявка раскрывает в дополнительном аспекте применение соединений формулы (I) для лечения патологических состояний, которые реагируют на блокаду возбудительных аминокислотных рецепторов, таких как рецепторы АМРА, например нейродегенеративных нарушений, инсульта, эпилепсии, тревоги и боли.
В настоящее время неожиданно было найдено, что соединения полезны также в лечении невропатической боли.
Активность соединений в лечении нейропатической боли подтверждается, например, на следующей модели невропатической боли у крыс.
Крысам линии Wistar давали наркоз с энфлураном и производили небольшой разрез в верхней части середины одного бедра, чтобы выделить седалищный нерв. Нерв освобождали от соединительной ткани и шелковую шовную нить вводили в нерв, используя на 3/8 изогнутую хирургическую режущую мини-иглу, и плотно лигировали так, чтобы 1/3-1/2 тыльной толщины нерва удерживалась внутри лигатуры. Мышцу и кожу закрывали швами и скобками и рану посыпали порошком антибиотика. Данная процедура создает механическую гипералгезию, которая развивается в интервале 2-3 дней и поддерживается в течение по крайней мере 4 недель. Механическая гипералгезия оценивается измерением порогов подергивания лапки как на одной стороне (лигированной), так и на противоположной стороне (нелигированной) задней лапки в ответ на увеличивающееся воздействие раздражителя, нанесенного на лапку с использованием анальгезиметра (Ugo-Basile) с клинообразным зондом (площадь 1,75 мм2) и прерыванием порога в 250 г. Конечная точка принимается в качестве первого признака болевой реакции (сопротивление, звуковой сигнал или подергивание лапки). На гипералгезию указывает различие порогов подергивания на одной стороне и противоположной стороне. Обратимость установившейся гипералгезии с помощью введенных соединений измеряется 12-14 дней после хирургического вмешательства с использованием 6 животных на группу обработки. Пороги подергивания лапки измеряются до введения и затем до 6 ч после введения препарата или наполнителя. Статистический анализ осуществляется по показателям порога подергивания, используя дисперсионный анализ ANOVA, затем тест Тьюки правдоподобно достоверных различий (HSD), сравнивая животных, обработанных препаратом и согласованным по времени наполнителем.
На этой модели соединения в значительной степени обращали невропатическую механическую гипералгезию при 10 мг/кг перорально. С отобранными соединениями формулы (I) максимальное обращение невропатической механической гипералгезии 35% достигается через 3 ч после введения 10 мг/кг перорально.
Активность соединений формулы I в лечении невропатической боли может быть подтверждена в клинических испытаниях, например в следующем исследовании, проведенном с целью оценки эффективности соединения в лечении хронической боли у пациентов с диабетической невропатией.
Пациенты рандомизируются в соотношении 1:1, чтобы получать 2400 мг/день соединения или плацебо.
Исследование состоит из фазы, предшествующей рандомизации (1 неделя), и двойной слепой фазы (5 недель). Двойная слепая фаза состоит из трех периодов: однонедельный период титрования дозы, трехнедельный период поддерживающей терапии и однонедельный период последующего наблюдения.
В течение однонедельной фазы перед рандомизацией оценивается пригодность пациентов. Пациенты, отвечающие критериям включения в исследование/исключения из исследования, рандомизировались для получения или соединения, или плацебо в двойной слепой фазе исследования. В ходе 1 недели периода титрования дозы изучаемый препарат титровали от 800 мг/день (данных дважды в день) до 2400 мг/день (данных дважды в день). Пациенты, которые завершали однонедельный период титрования дозы, затем входили в трехнедельный период поддерживающей терапии. Пациенты, которые завершали трехнедельный период поддерживающей терапии или преждевременно прекращали обработку двойным слепым способом, затем входили в однонедельный период последующего наблюдения. Исследование назначения лекарственного средства полностью прекращалось или переходило в период последующего наблюдения. В ходе двойной слепой фазы получали оценки серийной эффективности и безопасности.
Амбулаторные больные, 120 мужчин и женщин, в возрасте 18-65 лет с клиническим диагнозом сахарного диабета (типа I или II) и историей боли, ассоциированной с диабетической невропатией в течение 6 месяцев до 3 лет перед вхождением в исследование, случайно распределяются в соотношении 1:1 для получения соединения или плацебо.
Общий балл краткой болевой анкеты МакГилла (SF-MPQ) в конце периода поддерживающей терапии используется в качестве параметра первичной эффективности. Средняя еженедельная оценка тяжести боли (ежедневный дневник боли пациента) от начала рандомизированной обработки до конца периода поддерживающей терапии, использование «спасательной» лекарственной терапии в период титрования дозы и поддерживающей терапии и средняя оценка тяжести боли в ходе периода последующего наблюдения (возвратная боль) используются в качестве вторичных параметров эффективности.
Общий болевой балл SF-MPQ в конце периода поддерживающей терапии анализируется с использованием анализа ковариантной модели, скорректированной для эффекта обработки по баллам после обработки с использованием базисной линии общего болевого балла SF-MPQ в качестве коварианта. Средняя еженедельная тяжесть боли анализируется с применением анализа ковариантной модели с повторяющимися измерениями с использованием недельной обработки и среднего показателя тяжести боли в ходе фазы, предшествующей рандомизации, в качестве коварианта. Использование «спасательной» лекарственной терапии в ходе двойной слепой фазы анализируется с использованием контроля с помощью критерия Кохрана-Мантеля-Гензеля для центра. Показатель средней тяжести боли в ходе периода последующего наблюдения (возвратная боль) анализируется с использованием анализа ковариантной модели, скорректированной для эффекта обработки по показателю средней тяжести боли периода последующего наблюдения с показателем средней тяжести боли в ходе периода, предшествующего рандомизации, в качестве коварианта.
В данном исследовании найдено, что соединения
N-{6-[(2-гидроксиэтил)метиламино]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид,
N-(6-[1,4]оксазепан-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид и
N-(6-морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид
наиболее специфично и статистически значимым образом снижают показатели тяжести боли относительно плацебо в ходе периодов поддерживающей терапии и последующего наблюдения.
Следовательно, соединения пригодны для лечения невропатической боли и ассоциированной гипералгезии, включая невралгию тройничного нерва и герпетическую, диабетическую невропатическую боль, мигрень, каузалгию и деафферентационные синдромы, такие как авульсия плечевого сплетения.
В дальнейшем аспекте по изобретению неожиданно было обнаружено, что соединения пригодны также для лечения эмоциональных расстройств и расстройств внимания.
Активность соединений в лечении эмоциональных расстройств, включая биполярные нарушения, например маниакально-депрессивные психозы, экстремальные психотические состояния, например манию, подтверждается, например, в следующих анализах, подходящих для определения препаратов, обращающих психомоторные стимулирующие эффекты.
Анализ 1: локомоция, индуцированная антагонистом N-метил-D-аспартата (NMDA)
Использовали самцов крыс линии Wistar Kyoto (Iffa Credo, Lion, France) с массой между 250 и 310 г. В принципе образовывали 4 группы обработки: 1) соединение (дозы 1, 3 или 10 мг/кг), затем конкурентный антагонист рецептора NMDA, (S)-2-амино-3-(2'-хлор-5-фосфонометилбифенил-3-ил)пропионовая кислота, впредь SDZ 220-581 (10 мг/кг), 2) предварительная обработка растворителем, затем SDZ 220-581 (10 мг/кг), 3) растворитель, сопровождавшийся растворителем, 4) соединение (1, 3, 10 мг/кг), затем растворитель. Крысы произвольно распределялись по этим группам предварительной обработки (n=10/дозовую группу). Препараты вводили подкожно за 15 мин до SDZ 220-581. Сразу после того, как животные получали SDZ 220-581, их помещали в монитор активности на период 60 мин. Локомоторную активность анализировали в течение первых 30 мин.
Локомоцию регистрировали с видеоследящей системой (VideoMot2, TSE Technical and Scientific Equipment GmbH, Bad Hombourg, Germany), используя цифровую видеокамеру с замкнутым круговым обращением (WV-BP.330/GE, Panasonic, Osaka, Japan). Видеосигнал от камеры переводился в цифровую форму и использовался для анализа данных. Животные находились при нормальном световом цикле 12 ч день/12 ч ночь, свет зажигали в 6 ч. Эксперименты проводили в тускло освещенной комнате между 7 ч и 15 ч. Животных помещали в круглую арену (диаметр 42 см, высота 32 см), сделанную из серого поливинилхлоридного пластика. Камера помещалась таким образом, что четыре животных (одно на арену) могли быть записаны одновременно.
В данном анализе соединения (1-10 мг/кг подкожно) значительно не изменяют локомоторную активность по сравнению с животными, обработанными наполнителем, в любое время в течение периода 30 мин. Однако конкурентный антагонист рецептора NMDA-SDZ 220-581 (10 мг/кг подкожно) индуцирует сильную локомоторную реакцию. Так, в то время когда контрольные животные проходят приблизительно 8-10 м в течение 30 мин, животные, обработанные SDZ 220-581, проходят приблизительно 30 м. Данная локомоторная реакция снижается дозозависимым образом с помощью соединений. С отобранными соединениями формулы (I) (например, при 10 мг/кг) эффект антагониста NMDA-SDZ 220-581 почти нормализуется.
Анализ 2: блокатор канала NMDA индуцирует качание и кружение головы
Использовали взрослых самцов крыс линии Wistar Kyoto (340-380 г; Iffa Credo. Lyon, France). Животных произвольно распределяли в следующие группы обработки (n=10): соединение (в дозе 0,3 или 10 мг/кг), затем фенциклидин (РСР; блокатор канала NMDA в дозе 0 или 10 мг/кг). Соединения (во время t=-15 мин) и РСР (во время t=0 мин) вводили подкожно в объеме 1 мл/кг. Видеозаписи поведения животных в течение периода 0-30 мин после РСР оценивались в баллах наблюдателем, не знающим о предварительной обработке животных. Качание головой (неоднократное раскачивание головой по меньшей мере на 2 см влево и вправо) и кружение (поворачивание вокруг с использованием передних лап, тогда как задние лапы оставались более или менее на исходной позиции) оценивалось в баллах как присутствие (1) или отсутствие (0) эффекта каждые 5 мин для продолжительности в 1 мин. Баллы для отдельных животных суммировали и групповые баллы использовали для статистического анализа (t-критерий с коррекцией Бонферрони).
В данном анализе РСР (10 мг/кг подкожно) индуцировал слабое покачивание головой и кружение. Предварительная обработка соединениями (3 и 10 мг/кг подкожно) значительно усиливала эти поведенческие реакции на РСР (p<0,05).
Индуцированные антагонистом NMDA локомоторные реакции отражают состояние, подобное мании. Блокада данной активности указывает на антиманиакальную активность. Кроме того, усиление качания головой и кружения предполагает поведенческое дезингибирование (анксиолитически-/антидепрессантноподобное) и социотропную активность. Следовательно, соединения пригодны в лечении эмоциональных нарушений, включая биполярные нарушения, например маниакально-депрессивные психозы, экстремальные психотические состояния, например мания и чрезмерные колебания настроения, где желательна поведенческая стабилизация. Кроме того, соединения показаны при нарушениях дефицита внимания и гиперреактивности (ADHD) и других нарушениях внимания, например аутизме, состояниях страха, генерализованного страха и агорафобии, а также при тех поведенческих состояниях, которые характеризуются социальным синдромом, например негативными симптомами.
В дальнейшем аспекте по настоящему изобретению неожиданно было обнаружено, что соединения также пригодны в лечении шизофрении и подобных психозам симптомов при других показаниях, например болезни Паркинсона.
Антишизофреническая активность соединений показана в стандартных анализах, например в анализе индуцированной амфетамином гиперлокомоции. Блокада индуцированной амфетамином гиперлокомоции хорошо известна в качестве скрининговой парадигмы для антишизофренической активности.
Использовали самцов крыс линии Wistar (Iffa Credo, Lion, France) с массой между 215 и 315 г. В принципе образовывали 4 группы обработки: 1) соединение (дозы 1, 3 или 10 мг/кг), затем амфетамин (1 мг/кг), 2) предварительная обработка растворителем, затем амфетамин (1 мг/кг), 3) растворитель, сопровождавшийся растворителем, 4) соединение (10 мг/кг), затем растворитель. Крысы произвольно распределялись по этим группам предварительной обработки (n=10/дозовую группу). Препараты вводили подкожно за 15 мин до амфетамина. Сразу после того, как животные получали амфетамин, их помещали в монитор активности на период 60 мин. Локомоторную активность анализировали в течение первых 30 мин.
Локомоцию регистрировали с видеоследящей системой (VideoMot2, TSE Technical and Scientific Equipment GmbH, Bad Hombourg, Germany), используя цифровую видеокамеру с замкнутым круговым обращением (WV-BP.330/GE, Panasonic, Osaka, Japan). Видеосигнал от камеры переводился в цифровую форму и использовался для анализа данных. Животные находились при нормальном световом цикле 12 ч день/12 ч ночь, свет зажигали в 6 ч. Эксперименты проводили в тускло освещенной комнате между 7 ч и 15 ч. Животных помещали в круглую арену (диаметр 42 см, высота 32 см), сделанную из серого поливинилхлоридного пластика. Камера помещалась таким образом, что четыре животных (одно на арену) могли быть записаны одновременно.
Амфетамин растворяли в физиологическом растворе в концентрации 1 мг/мл и вводили подкожно в объеме 1 мл/кг. Соединение растворяли в нескольких каплях раствора гидроксида натрия (0,1 н.) и затем разбавляли физиологическим раствором, как требовалось, чтобы получить растворы с концентрацией 10, 3 и 1 мг/мл. Их вводили подкожно в объеме 1 мл/кг.
Сравнение между группами проводили с использованием t-критерия Стьюдента, скорректированного для многократного анализа с использованием процедуры Бонферрони.
В данном анализе соединения формулы (I) снижали индуцированную амфетамином локомоцию в дозах от примерно 1 мг/кг до примерно 10 мг/кг подкожно.
Еще в одном дополнительном аспекте по изобретению было неожиданно обнаружено, что соединения пригодны также в лечении шума в ушах.
Воздействие соединений на активность шума в ушах показана в стандартных анализах, например, на модели шума в ушах, индуцированного салицилатом.
Было показано [С.A.Bauer и др., Hearing Research, 147 (2000) с.с.175-182], что хроническое воздействие салицилата вызывает в нижнем холмике (IC) крысы сверхрегуляцию экспрессии декарбоксилазы глутаминовой кислоты (GAD), ассоциированную с развитием шума в ушах. Более того, электрофизиологическая регистрация нейронов слухового нерва, использующая методику фиксации потенциала [D.Peruzzi и др., Neuroscience, 101 (2000) с.с.403-416, X.Lin и др., Journal of Neurophysiology, 79 (1998) c.c.2503-2512] и регистрации одного нейрона [J.J.Eggermont и М.Kenmochi, Hearing Research, 117 (1998) с.с.149-160], показала, что возбудимость нейронов изменяется после обработки салицилатом и хинином.
Введение салицилата или хинина вызывало увеличение доли работающих нейронов слухового нерва, измеренное с помощью методики внеклеточной электрофизиологической регистрации. Использование in vitro электрофизиологической регистрирующей методики переохлаждения с салицилатом повышает возбудимость регистрируемых нейронов. При введении соединений в концентрациях от примерно 1 нМ до 100 мкМ эффекты салицилата обращаются.
Для лечения упомянутых выше показаний соответствующие дозы будут, безусловно, меняться, например, в зависимости от используемого соединения, пациента, способа введения и природы и тяжести состояния, подлежащего лечению. Однако, в общем, удовлетворительные результаты у животных, как указывается, получаются при ежедневной дозе от примерно 1 мг/кг до примерно 50 мг/кг массы тела. У крупных млекопитающих, например у человека, показанная ежедневная доза находится в диапазоне от примерно 10 мг до примерно 1000 мг соединения по изобретению, удобно вводимых, например, в раздельных дозах до четырех раз в день.
В дальнейшем аспекте по настоящему изобретению неожиданно было обнаружено, что соединения пригодны также в лечении миопии и других нарушений зрения.
Такие нарушения включают, но без ограничения, связанную с возрастом макулярную дегенерацию, диабетическую ретинопатию, цистоидную макулярную эдему (СМЕ), патологическую миопию, наследственную зрительную невропатию Лебера, пигментозный ретинит и другие наследственные ретинальные дегенерации.
Активность соединений в отношении миопии показана в стандартных анализах, например, на модели согласно R.A.Stone и др. [Proc. Natl. Acad. Sci. (USA) 86, с.с.704-706 (1989)], где экспериментальная миопия осуществляется на цыплятах при введении от примерно 0,1 мг/кг до примерно 1 мг/кг в глазных каплях.
Эффективность при описанных зрительных нарушениях могла быть установлена на следующих животных моделях.
1) Спонтанное развитие вторичной формы глаукомы (например, пигментной дисперсии, закрытоугольной глаукомы или угловой дисгенезии) у мышей (например, но не исключительно, штаммы DBA/2J, DBA/2Nnia и мыши AKXD28/Ty, как описано у Anderson и др., ВМС Genetics, 2001; 2:1, Chang и др., Nature Genetics, 1999; 21:405-409, John и др., Invest. Ophthalmol. Vis. Sci., 1998; 39:951-962, Sheldon и др., Lab. Animal Sci., 1995; 15:508-518).
2) Генетическая животная модель для ретинальной дегенерации, например мышь
с ретинальной дегенерацией (rd) (как описано у Li и др., Invest. Ophthalmol. Vis. Sci., 2001; 42:2981-2989), Rpe65-дефицитная мышь (Van Hooser и др., PNAS, 2000; 97: 8623-8628), крыса RCS (Faktorovich и др., Nature, 1990; 347:83-86), мышь с медленной ретинальной дегенерацией (rds) (Ali и др., Nature Genetics, 2000, 25:306-310), собака с дегенерацией типа 1 ретинальных клеток (rcd1) (Suber и др., PNAS, 1993; 90:3968-3972).
3) Экспериментальная ретинальная дегенерация, индуцированная:
- световым воздействием на мышей (как описано у Wenzel и др., Invest. Ophthalmol. Vis. Sci., 2001; 42:1653-1659) или крыс (Faktorovich и др., J. Neurosci, 1992; 12:3554-3567),
- введением N-метил-N-нитрозомочевины (Kiuchi и др., Exp. Eye Res., 2002, 74:383-392) или йодата натрия (Sorsby и Harding, Vision Res., 1962, 2:139-148).
4) Экспериментальная модель повреждения зрительного нерва (ON):
- путем раздавливания зрительного нерва у мышей ((Levkovitch-Verbin и др., Invest. Ophthalmol. Vis. Sci., 2000, 41:4169-4174) и крыс (Yoles и Schwartz, Exp. Neurol., 1998, 153:1-7),
- путем рассечения зрительного нерва у крыс (как описано у Martin и др., Invest. Ophthalmol. Vis. Sci., 2002, 43:2236-2243, Solomon и др. J. Neurosci. Methods, 1996, 70:21-25),
- путем экспериментальной временной (острой) ретинальной ишемии у крыс после сшивания глазного сосуда (как описано у Lafuente и др.. Invest. Ophthalmol. Vis. Sci., 2001, 42:2074-2084) или канюлирования передней камеры (Buchi и др., Ophthalmologica, 1991, 203:138-147),
- путем внутриглазной инъекции эндотелина-1 у крыс (Stokely и др., Invest. Ophthalmol. Vis. Sci., 2002, 43:3223-3230) или кроликов (Takei и др., Graefes Arch. Clin. Exp.Ophthalmol., 1993, 231:476-481).
Для обработки миопии и других нарушений зрения соответствующая доза, безусловно, будет зависеть, например, от используемого соединения, пациента, способа введения и природы и тяжести миопии. Однако, в общем, показано, что удовлетворительные результаты у животных получаются при ежедневной дозе от примерно 0,01 мг/кг до примерно 1 мг/кг массы тела животного. У крупных млекопитающих, например у человека, показанная ежедневная доза находится в диапазоне от примерно 0,25 мг до примерно 10 мг соединения по изобретению, удобно введенного, например, в раздельных дозах до четырех раз в день.
Для упомянутых выше показаний соединения могут быть введены любым обычным способом, например перорально, например, в форме таблеток или капсул, или парентерально, например, в форме инъекционных растворов или суспензий.
Для лечения миопии и других нарушений зрения соединения могут быть введены местно в глаз или около глаза, например, в виде глазных капель, глазных суспензий или мазей, субконъюнктивальных, перибульбарных, ретробульбарных или интравитриальных инъекций, возможно с применением приспособлений медленного высвобождения, таких как конъюнктивальные вкладыши, микросферы или другие периокулярные или интраокулярные депо-приспособления.
Соединения предпочтительно наносятся в глаз приблизительно в виде 0,002% до приблизительно 0,02% офтальмологических растворов. Наполнитель глазного лекарства является таким, что соединение поддерживается в контакте с глазной поверхностью в течение достаточного периода времени, чтобы дать возможность соединению проникнуть в роговичную и внутреннюю области глаза. Фармацевтически приемлемыми наполнителями для глазного лекарства могут быть, например, мазь, растительное масло или инкапсулирующий материал.
Соответствующие соединения для лечения упомянутых выше состояний включают
N-{6-[(2-гидроксиэтил)метиламино]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид,
N-(6-[1,4]оксазепан-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид,
N-(6-морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид
и их фармацевтически приемлемые соли.
Настоящее изобретение обеспечивает также фармацевтические композиции, включающие соединение формулы (I) в сочетании по меньшей мере с одним фармацевтическим носителем или разбавителем, для применения в лечении невропатической боли, эмоциональных расстройств и расстройств внимания, шизофрении, шума в ушах, миопии и других нарушений зрения. Такие композиции могут быть получены общепринятым способом. Стандартные лекарственные формы для лечения невропатической боли, эмоциональных расстройств и расстройств внимания, шизофрении и шума в ушах могут содержать, например, от приблизительно 2,5 мг до приблизительно 500 мг соединения формулы (I). Стандартные лекарственные формы для лечения миопии и других нарушений зрения могут содержать, например, от приблизительно 0,05 мг до приблизительно 5 мг соединения формулы (I).
Изобретение, кроме того, обеспечивает применение соединения формулы (I) для получения фармацевтической композиции для предупреждения, лечения или задержки прогрессирования невропатической боли, эмоциональных расстройств и расстройств внимания, шизофрении, шума в ушах, миопии и других нарушений зрения.
Более того, изобретение обеспечивает способ для предупреждения, лечения или задержки прогрессирования невропатической боли, эмоциональных расстройств и нарушения внимания, шизофрении, шума в ушах, миопии и других нарушений зрения у субъекта, подлежащего такому лечении, способ, который включает введение упомянутому субъекту терапевтически эффективного количества соединения формулы (I).
В дальнейшем аспекте в настоящее время неожиданно было обнаружено, что соединения пригодны также в лечении множественного склероза и родственных заболеваний, связанных с демиелинизацией, например нейромиелита зрительного нерва.
Активность соединений в лечении множественного склероза подтверждается, например, на следующей модели экспериментального аутоиммунного энцефаломиелита (ЕАЕ), первичной животной модели для симптома множественного склероза (MS), аутоиммунного заболевания центральной нервной системы.
Самок крыс линии Dark Agouti (DA-крыс) содержат в помещении с контролируемым климатом при 12-часовом цикле свет/темнота размещенными в наполненных древесными опилками клетках по 4-5 крыс на клетку и получающими стандартный корм для грызунов и воду на усмотрение исполнителя. Сильно парализованным животным специально обеспечивается более легкий подход к корму и воде. Крысам дается по меньшей мере одна неделя, чтобы адаптироваться к окружающей обстановке, затем они произвольно разделяются на экспериментальные группы (10 крыс на группу) и получают индивидуальную цифровую метку на хвосте. Возраст крыс составляет 8-9 недель (примерно 135 г) во время иммунизации в день 0. Для оптимальной индукции экспериментального аутоиммунного энцефаломиелита (ЕАЕ) для иммунизационной процедуры используется свежевыделенный мозг и спинной мозг (соотношение 40:60) от взрослых DA-крыс в качестве источника сингенных энцефалитогенных нейроантигенов. Образцы ткани центральной нервной системы (ЦНС), называемые также как DA-b/sc, хранятся в пробирках Эппендорфа при -70°С, пока не понадобятся.
Крысы получают легкий наркоз с изофлураном и иммунизируются с помощью одной внутрикожной (i.d.) инъекции в тыльную часть хвоста с 200 мкл инокулята, содержащего 1 часть (объем:объем) ткани ЦНС, эмульгированной в соответствующем разбавителе, до 1 части неполного адъюванта Фрейнда (IFA), дополненного штаммом Mycobacterium tuberculosis H37RA (Difco, Detroit, MI). Смесь IFA-микобактерии ниже обозначается CFA (полный адъювант Фрейнда). Более конкретно, DA-b/sc эмульгируется в «шприце А», содержащем 0,9% NaCl, с использованием гомогенизатора Polytron PT 3100 (Kinematica, Lucerne, Switzerland) при 28000 об/мин в течение примерно 3 мин. Эмульсия антигена постепенно прибавляется к CFA в «шприце Б» по мере гомогенизации. Все растворы содержатся на льду, и им не дают перегреться с помощью высокоскоростного перемешивания.
Вспомогательных контрольных животных инъецировали одним полным адъювантом Фрейда (CFA) (1,6 мг М. tuberculosis на крысу) и обрабатывали наполнителем. Животных в других экспериментальных группах инъецировали с эмульсией нейроантиген-CFA (65 мг DA-b/sc на 1,6 мг микобактерий на крысу) и обрабатывали одним наполнителем или наполнителем, содержащим исследуемое соединение. Исследование обычно заканчивается в день 63, через девять недель после иммунизации в день 0.
Клиническая оценка в баллах экспериментального аутоиммунного энцефаломиелита (ЕАЕ)
Животных исследовали ежедневно на неврологические симптомы и изменение массы тела. Клиническая степень ЕАЕ оценивается с использованием шкалы заболевания от 0 до 4:
0 - отсутствие заболевания,
1 - полная потеря хвостового тонуса,
2 - слабость задней конечности (конечностей) или атаксия,
3 - полный паралич или обеих задних конечностей, или передних конечностей,
4 - летальное состояние с параличом как передних конечностей, так и задних конечностей; (смертность, связанная с заболеванием).
Баллы 3-4 часто сопровождались недержанием мочи. Смертность, связанная с ЕАЕ, регистрируется максимальным баллом 4. Другие регистрируемые данные включают день начала ЕАЕ и % случаев заболевания в группе.
Применение соединений: соединения принимаются в течение 14 до 21 дня, начиная со дня 0 (профилактическая обработка) или в день 12 после иммунизации (терапевтическая обработка). Соединения принимались дважды в день или трижды в день и вводились перорально, внутрибрюшинно или подкожно.
Статистический анализ: поскольку тяжесть заболевания и продолжительность - обе являются ключевыми параметрами для рассмотрения в исследовании препарата, клинические баллы анализируются как площадь под кривой (AUC) баллов во времени.
В данной модели ЕАЕ 10-20% иммунизированных антигеном контрольных животных могут погибать по причинам, связанным с заболеванием. Для объяснения этой важной информации в статистическом анализе была использована методика Гулда (A.L.Gould. A new approach to the analysis of clinical drug trials with withdrawals. Biometrics, 1980; 36:721-727). Животные в каждой группе классифицировались по их значению AUC (усилению тяжести заболевания). Погибшие крысы позиционировались в соответствии с временем их гибели, тем самым давая им более высокую оценку, чем выжившим. Оценки сравнивали, используя непараметрические критерии Вилкоксона и Манна-Уитни. Коэффициент достоверности р≤0,05 считается статистически значимым.
На данной модели соединения уменьшают тяжесть симптомов ЕАЕ. Для отобранных соединений формулы (I) эффект проявляется при 2×10 мг/кг внутрибрюшинно.
Соответствующие соединения для лечения множественного склероза:
N-{6-[(2-гидроксиэтил)метиламино]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид,
N-(6-[1,4]оксазепан-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид,
N-(6-морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид
и их фармацевтически приемлемые соли.
Настоящее изобретение обеспечивает также фармацевтические композиции, включающие соединение формулы (I) в сочетании по меньшей мере с одним фармацевтическим носителем или разбавителем для применения в лечении множественного склероза и родственных заболеваний, связанных с демиенилизацией, например нейромиелита зрительного нерва. Такие композиции могут быть получены общепринятым способом. Стандартные лекарственные формы для лечения множественного склероза и связанных с демиелинизацией заболеваний, например нейромиелита зрительного нерва, могут содержать, например, от примерно 2,5 мг до примерно 500 мг соединения формулы (I).
Изобретение, кроме того, обеспечивает применение соединения формулы (I) для получения фармацевтической композиции для предупреждения, лечения или задержки прогрессирования множественного склероза или связанных с демиелинизацией заболеваний, например нейромиелита зрительного нерва.
Более того, изобретение обеспечивает способ предупреждения, лечения или задержки прогрессирования множественного склероза и связанных с демиелинизацией заболеваний, например нейромиелита зрительного нерва, у пациента, нуждающегося в таком лечении, способ, который включает введение упомянутому пациенту терапевтически эффективного количества соединения формулы (I).
В дальнейшем аспекте настоящее изобретение относится к способам и соединениям для лечения деменции.
Неожиданно было найдено, что деменцию можно лечить введением антагониста рецептора α-амино-3-гидрокси-5-метилпропионовой кислоты (АМРА). Следовательно, настоящее изобретение относится к способу лечения и/или предупреждения деменции, включающему этап введения теплокровному животному, включая человека, нуждающемуся в этом, эффективного количества антагониста рецептора АМРА.
Термин «антагонист рецептора АМРА», как он использован в контексте, относится к соединениям формулы (I).
Особенно подходящими соединениями для лечения деменции являются:
N-{6-[(2-гидроксиэтил)метиламино]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид,
N-(6-[1,4]оксазепан-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид,
N-(6-морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид
и их фармацевтически приемлемые соли.
Термин «деменция», как он использован в контексте, включает, но без ограничения, деменцию Альцгеймера с психотическими симптомами или без них. В частности, способы и соединения, описанные в контексте, пригодны для лечения поведенческих расстройств, наблюдаемых при таких типах симптомов.
Будет понятно, что при обсуждении способов ссылки на активные ингредиенты подразумевают также включение фармацевтически активных солей. Если данные активные ингредиенты имеют, например, по меньшей мере один основный центр, то они могут образовать кислотно-аддитивные соли. Соответствующие кислотно-аддитивные соли могут быть также образованы, имея при необходимости дополнительно присутствующий основный центр. Активные ингредиенты, имеющие кислотную группу (например, СООН), могут образовать также соли с основаниями. Активный ингредиент или его фармацевтически приемлемая соль могут быть также использованы в форме гидрата или включать другие растворители, использованные для кристаллизации.
Фармакологическая активность соединения формулы (I) может быть, например, продемонстрирована в клиническом исследовании. Такие клинические исследования предпочтительно являются рандомизированными, проводимыми двойным слепым способом клиническими исследованиями пациентов с болезнью Альцгеймера. Благотворные воздействия на болезнь Альцгеймера могут быть определены непосредственно с помощью результатов данных исследований или с помощью изменений в планировании исследования, которые как таковые известны специалисту в данной области.
Одной целью данного изобретения является обеспечение фармацевтической композиции, включающей количество, которое терапевтически эффективно в отношении деменции, композиции, включающей по меньшей мере один антагонист рецептора АМРА и по меньшей мере один фармацевтически приемлемый носитель.
Фармацевтические композиции по изобретению могут быть получены общепринятым по сути способом и таковым, пригодным для энтерального введения, такого как пероральное или ректальное, и парентерального введения млекопитающим (теплокровным животным), включая человека, способом, включающим терапевтически эффективное количество по меньшей мере одного фармакологически активного ингредиента, одного или в комбинации с одним или несколькими фармацевтически приемлемыми носителями, особенно пригодными для энтерального или парентерального применения. Предпочтительным путем введения лекарственной формы по настоящему изобретению является пероральный.
Новые фармацевтические композиции содержат, например, от примерно 10% до примерно 100%, предпочтительно от примерно 20% до примерно 60% активных ингредиентов. Фармацевтическими препаратами для терапии для энтерального или парентерального введения являются, например, препараты в стандартных лекарственных формах, таких как таблетки с нанесенным сахарным покрытием, таблетки, капсулы или суппозитории и к тому же ампулы. Если не указано иначе, они получаются общепринятым по сути способом, например посредством обычных процессов смешивания, грануляции, нанесения сахарного покрытия, растворения и лиофилизации. Будет признано, что стандартная доля активного ингредиента или ингредиентов, содержащаяся в индивидуальной дозе каждой лекарственной формы сама по себе необязательно составляет эффективное количество, поскольку необходимое эффективное количество может быть достигнуто введением множества дозированных долей.
В приготовлении композиций для пероральной лекарственной формы может быть использована любая из обычных сред, такая как, например, вода, гликоли, масла или спирты; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие препараты, смазки, связующие препараты, дезинтегрирующие препараты и тому подобное в случае пероральных твердых препаратов, таких как, например, порошки, капсулы и таблетки. Из-за легкости введения таблетки и капсулы представляют наиболее благоприятную пероральную стандартную лекарственную форму, в каковом случае, безусловно, используются твердые фармацевтические носители.
Более того, настоящее изобретение относится к применению соединения формулы (I) для получения лекарственного средства для лечения деменции.
Дополнительно настоящее изобретение обеспечивает способ лечения теплокровных животных с деменцией, включающий введение животному соединения формулы (I) в количестве, которое объединено терапевтически эффективно в отношении деменции и в котором соединения могут также присутствовать в форме их фармацевтически приемлемых солей.
Эффективная доза каждого из активных ингредиентов, используемых в соединении формулы (I), может изменяться в зависимости от специфического соединения или используемой фармацевтической композиции, способа введения, тяжести состояния, подлежащего обработке. Так, дозовый режим соединения формулы (I) выбирается в соответствии с различными факторами, включающими путь введения и функции почек и печени пациента. Врач, клиницист или ветеринар обычной квалификации может легко определить и назначить эффективное количество одного активного ингредиента, требуемого для того, чтобы предупредить, противодействовать или остановить прогрессирование состояния. Оптимальная точность в достижении концентрации активных ингредиентов в диапазоне, который обеспечивает эффективность без токсичности, требует создания режима, основанного на кинетике доступности активных ингредиентов к сайтам-мишеням. Это включает рассмотрение распределения, равновесия и удаления активных ингредиентов из организма.
Изобретение иллюстрируется, но без ограничения, следующими примерами.
Рабочие примеры
Сокращения:
Пример 1
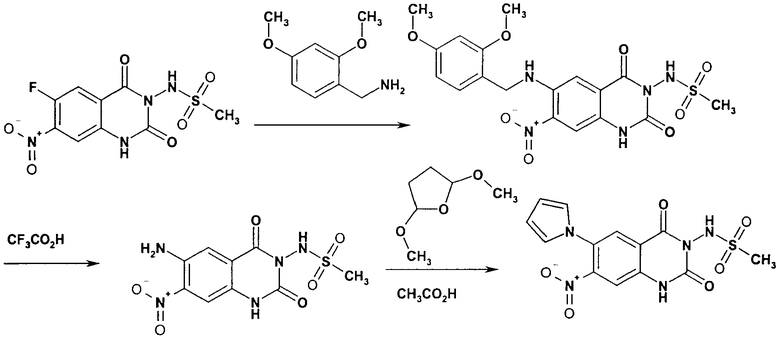
N-(6-Имидазол-1-ил-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
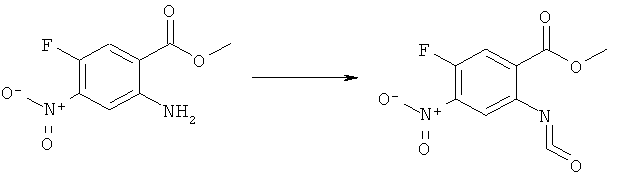
Раствор 15 мл фосгена в толуоле (20%) прибавляли по каплям к суспензии 1 г метилового эфира 2-амино-5-фтор-4-нитробензойной кислоты в 20 мл безводного толуола при -15°С. Вводили медленный ток фосгена и реакционную смесь нагревали до кипения. Через 45 мин аргоном продували желтый раствор и растворитель отгоняли, получая таким образом 1,1 г метилового эфира 5-фтор-2-изоцианато-4-нитробензойной кислоты в виде твердого вещества желтого цвета, достаточно чистого для использования на следующей стадии. ИК (CHCl3): 2240 см-1.
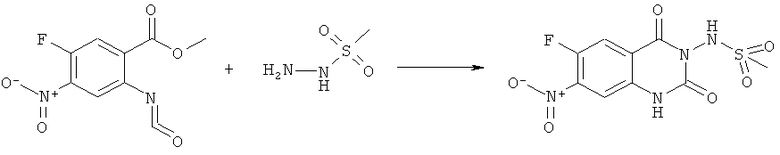
К раствору 1,1 г (4,58 ммоля) метилового эфира 5-фтор-2-изоцианато-4-нитробензойной кислоты в 20 мл безводного тетрагидрофурана прибавляли 0,504 г (4,58 ммоля) метансульфонилгидразида в 7 мл безводного тетрагидрофурана. Полученную в результате суспензию перемешивали в течение 1 ч, затем обрабатывали 4,58 мл 1 М раствора гидроксида натрия и перемешивали в течение 3,5 ч. После подкисления 5,7 мл 2 М раствора HCl смесь концентрировали до образования осадка, фильтровали и остаток промывали водой и перекристаллизовывали из тетрагидрофурана, получая 0,774 г указанного в заголовке соединения в виде порошка бежевого цвета, tпл 240-255°С (разл.).

Смесь 530 мг (1,665 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 567 мг (8,325 ммоля) имидазола и 7,7 мл безводного 1,3-имидазолидинона нагревали при 140°С (температура масляной бани) в течение 90 мин в закрытом сосуде. После охлаждения темный раствор выливали в 130 мл воды и доводили значение рН до ~5 добавлением 2 М уксусной кислоты. Образованный осадок отделяли фильтрованием и четыре раза перекристаллизовывали из смеси диметилсульфоксид/вода, получая 214 мг указанного в заголовке соединения в виде порошка красного цвета, tпл>270°С.
Пример 2
N-[6-(4-Гидроксиметилимидазол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

Смесь 100 мг (0,314 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 333 мг (1,57 ммоля) 4-(трет-бутилдиметил-силанилоксиметил)-1H-имидазола и 1 мл безводного 1,3-диметил-2-имидазолидинона нагревали при 140°С (температура масляной бани) в закрытом сосуде в течение 4 ч. После охлаждения оранжевый раствор разбавляли этилацетатом и органическую фазу промывали водой и насыщенным солевым раствором и сушили над сульфатом натрия. Упаривание растворителя дало масло, которое очищали с помощью хроматографии среднего давления на силикагеле со смесью этилацетат/уксусная кислота (98:2) в качестве элюента, получая 53 мг N-{6-[4-(трет-бутилдиметилсиланилоксиметил)-имидазол-1-ил]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид, tпл>270°С.
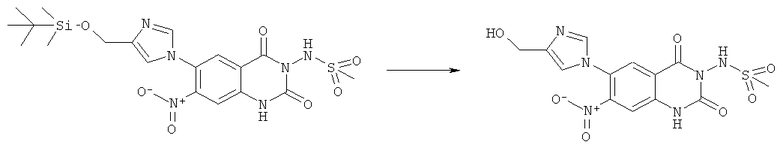
Смесь 1 г (1,96 ммоля) N-{6-[4-(трет-бутилдиметилсиланилоксиметил)-имидазол-1-ил]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамида, 0,738 г (2,34 ммоля) тригидрата тетрабутиламмонийфторида и 20 мл тетрагидрофурана перемешивали при 50°С в течение 18 ч. Реакционную смесь концентрировали досуха и остаток фракционировали хроматографией среднего давления на колонке с обращенной фазой RP-C18 (сорбент 20 мкм) со смесью ацетонитрил/вода (1:1) в качестве элюента. Остаток, полученный упариванием фракций 2-5, растворяли в воде и доводили значение рН до ~5 с разбавленным раствором гидроксида аммония. Продукт осаждали и отделяли фильтрацией. Остаток, полученный от фракций 6-13, фракционировали хроматографией среднего давления на колонке RP-C18 (сорбент 20 мкм) со смесью ацетонитрил/вода (1:2) в качестве элюента. В ходе концентрирования продукт осаждался и собирался фильтрацией. Кристаллизация всех объединенных фракций продукта из смеси диметилсульфоксид/вода дала 0,328 г N-[6-(4-гидроксиметилимидазол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида в виде порошка желтого цвета, tпл>300°С.
Пример 3
N-(6-Морфолин-4-ил-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:

Смесь 100 мг (0,314 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида и 0,274 мл (3,14 ммоля) морфолина нагревали при 140°С (температура масляной бани) в течение 1 ч в закрытом сосуде. После охлаждения остаток растворяли в воде, раствор подкисляли 2 М уксусной кислотой до рН~5 и оставляли при комнатной температуре. Образовывался оранжевый осадок, который отделяли фильтрованием и перекристаллизовывали из смеси диметилсульфоксид/вода, получая 81 мг указанного в заголовке соединения в виде порошка оранжевого цвета, tпл>260°C.
Пример 4
N-(6-Диметиламино-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:

Смесь 100 мг (0,314 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида и 0,185 мл (0,925 ммоля) диметиламина (33% в этаноле) в закрытом сосуде нагревали в микроволновом реакторе при 150°С в течение 15 мин. После охлаждения реакционную смесь концентрировали досуха и остаток кристаллизовали из смеси тетрагидрофуран/вода, получая 90 мг указанного в заголовке соединения в виде порошка оранжевого цвета, tпл 245-260°С (разл.).
Пример 5
N-[6-(2-Гидрокси-1-гидроксиметилэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
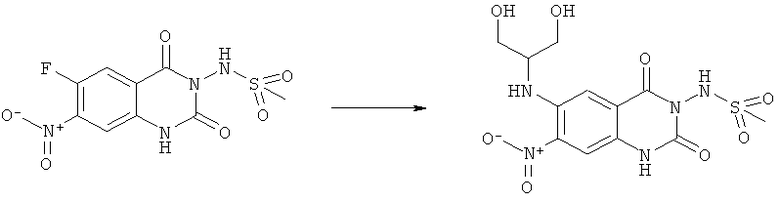
Смесь 100 мг (0,314 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида и 142 мг (1,55 ммоля) 2-амино-1,3-пропандиола в закрытом сосуде нагревали в микроволновом реакторе при 150°С в течение 20 мин. После охлаждения реакционную смесь концентрировали досуха и продукт очищали ВЭЖХ на колонке с обращенной фазой RP-C18, используя в качестве элюента градиент вода/ацетонитрил/0,1% трифторуксусная кислота. Получали 35 мг указанного в заголовке соединения в виде порошка красного цвета, tпл 126-145°С (разл.).
Способом, аналогичным описанному в предыдущем примере, получали следующее соединение.
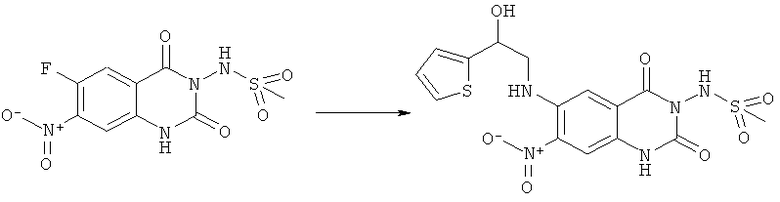
Пример 6
N-[6-(2-Гидрокси-2-тиофен-2-илэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
Порошок красного цвета, tпл 110°С (разл.).
Пример 7
N-[7-Нитро-2,4-диоксо-6-(2,2,2-трифторэтиламино)-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

Смесь 200 мг (0,628 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида и 1,08 мл (13,68 ммоля) 2,2,2-трифторэтиламина в закрытом сосуде нагревали в микроволновом реакторе при 140°С в течение 20 ч. После охлаждения реакционную смесь распределяли между этилацетатом и водой, органическую фазу сушили и концентрировали, получая осадок, который отделяли фильтрованием и подвергали очистке хроматографией среднего давления на колонке с обращенной фазой RP-C18 (сорбент 20 мкм) со смесью тетрагидрофуран/вода (4:3) в качестве элюента, содержащего 0,1% трифторуксусной кислоты. В ходе концентрации фракций продукт осаждался, его собирали фильтрацией, получая 63 мг порошка красного цвета, tпл 250°C (разл.).
Пример 8
N-(6-Амино-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:

К желтому раствору N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида (500 мг, 1,57 ммоля) в этаноле (0,5 мл) прибавляли 2,4-диметоксибензиламин (4,72 мл, 31,4 ммоля) в атмосфере аргона. Раствор нагревали при 150°С в закрытом сосуде в микроволновом реакторе в течение 4 мин. После удаления растворителя и 2,4-диметоксибензиламина упариванием в высоком вакууме на роторном испарителе оставшееся масло интенсивного пурпурного цвета обрабатывали диэтиловым эфиром, чтобы получить суспензию, которую фильтровали, осадок сушили, получая упомянутое в заголовке неочищенное соединение в виде твердого вещества интенсивного пурпурного цвета, tпл 232-237°С.

К раствору N-[6-(2,4-диметоксибензиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида (725 мг, 1,56 ммоля) в дихлорметане (4 мл) прибавляли трифторуксусную кислоту (0,5 мл) и смесь перемешивали при комнатной температуре в течение 30 мин. Затем растворитель удаляли упариванием на роторном испарителе, получая твердое вещество оранжевого цвета, которое суспендировали в диэтиловом эфире, фильтровали и остаток промывали три раза диэтиловым эфиром и два раза этилацетатом, получая указанное в заголовке соединение в виде чистого твердого вещества оранжевого цвета, tпл 326-335°С (разл.).
Пример 9
N-(7-Нитро-2,4-диоксо-6-пиррол-1-ил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид:

Раствор N-(6-амино-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида (90 мг, 0,285 ммоля) и 2,5-диметокситетрагидрофурана (0,038 мл, 0,291 ммоля) в уксусной кислоте (0,5 мл) нагревали при кипении в течение 80 мин. Образующуюся в результате суспензию фильтровали и осадок промывали этилацетатом. Фильтрат концентрировали в вакууме, получая твердое вещество коричневого цвета, которое суспендировали в диэтиловом эфире и фильтровали, получая твердое вещество коричневато-оранжевого цвета в качестве требуемого чистого продукта, tпл 243-250°С.
Пример 10
N-[6-(3-Формилпиррол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

Раствор N-(6-амино-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида (470 мг, 1,49 ммоля) и 2,5-диметокситетрагидрофуран-3-альдегида (796 мг, 4,47 ммоля) в уксусной кислоте (0,5 мл) нагревали при кипении в течение 5 ч. Растворитель удаляли упариванием на роторном испарителе, получая масло коричневого цвета, которое растворяли в этилацетате и промывали три раза водой. Органический слой сушили над сульфатом магния, фильтровали и растворитель удаляли упариванием на роторном испарителе, получая почти чистое соединение, указанное в заголовке, в виде твердого вещества коричневого цвета, tпл 230-245°С (разл.).
Пример 11
N-[6-(3-Гидроксиметилпиррол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

К раствору N-[6-(3-формилпиррол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида (580 мг, 1,47 ммоля, неочищенного, с предыдущей стадии) в метаноле (5 мл) прибавляли боргидрид натрия (112 мг, 2,95 ммоля) при 0°С и оставляли перемешиваться в течение 30 мин. Прибавляли небольшое количество уксусной кислоты, чтобы разрушить избыток боргидрида натрия. Реакционную смесь разбавляли этилацетатом и промывали два раза водой, и растворитель органического слоя удаляли упариванием на роторном испарителе, получая масло красно-коричневого цвета, которое очищали флэш-хроматографией со смесью дихлорметан/метанол от 98:2 до 95:5, получая указанное в заголовке соединение в виде твердого вещества оранжевого цвета, tпл 260-270°С (разл.).
Пример 12
N-[6-(2-Метокси-1-метилэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
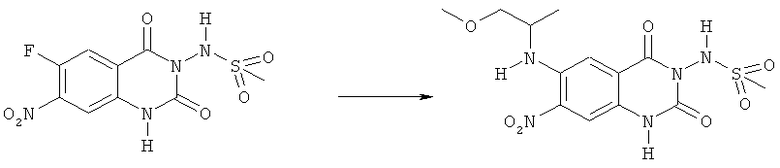
Смесь 200 мг (0,63 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 0,27 мл (2,51 ммоля) 2-метокси-1-метилэтиламина и 3 мл этанола нагревали при 150°С в закрытом сосуде в микроволновом реакторе в течение 15 мин. После охлаждения до комнатной температуры прибавляли 1 М водную хористоводородную кислоту до тех пор, пока не достигли значения рН 5-6. Полученную в результате суспензию фильтровали, осадок промывали водой и сушили при 60°С. Кристаллизация из смеси эфир-гексан дала 82 мг (34%) указанного в заголовке соединения в виде кристаллов красного цвета, tпл 103-111°С.
Пример 13
N-[6-(2-Бензилоксиэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
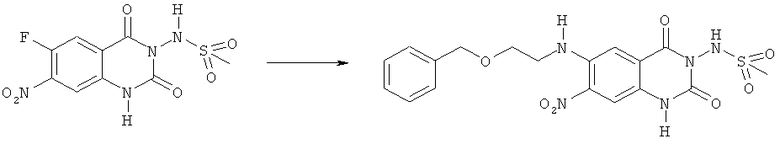
Смесь 318 мг (1 ммоль) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 605 мг (4,0 ммоля) 2-бензилоксиэтиламина и 3,2 мл этанола нагревали при 120°С в закрытом сосуде в микроволновом реакторе в течение 10 мин. После охлаждения до комнатной температуры прибавляли 1 М водную хлористоводородную кислоту до тех пор, пока значение рН не достигло ~3. Образующуюся в результате смесь экстрагировали этилацетатом. Органическую фазу отделяли, промывали водой и насыщенным солевым раствором, сушили над сульфатом магния и упаривали. Хроматография остатка на диоксиде кремния с элюированием смесью дихлорметан-метанол (93:7) и кристаллизация из смеси тетрагидрофуран-гексан дала 261 мг (58%) указанного в заголовке соединения в виде кристаллов красного цвета, tпл 204°С (разл.).
Пример 14
N-(6-Циклопентиламино-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:

Смесь 140 мг N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 0,17 мл (1,76 ммоля) циклопентиламина и 3 мл этанола нагревали при 150°С в закрытом сосуде в микроволновом реакторе в течение 5 мин. После охлаждения до комнатной температуры прибавляли воду и полученную в результате суспензию фильтровали. К фильтрату прибавляли концентрированную водную хлористоводородную кислоту до тех пор, пока значение рН не достигло 2-3, и образованную суспензию фильтровали. Остаток кристаллизовали из смеси эфир-гексан, получая 52 мг (31%) указанного в заголовке соединения в виде кристаллов красного цвета, tпл 146-155°C.
Пример 15
N-(6-Метокси-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:

Прибавляли 2,06 мл (4,12 ммоля) 2 М раствора диазометилтриметилсилана в гексане при слабом охлаждении при 20°С к раствору 1,0 г (4,13 ммоля) 2-ацетиламино-5-фтор-4-нитробензойной кислоты в 15 мл метанола и 35 мл бензола. Анализ с помощью ВЭЖХ через 15 мин показал, что еще присутствует около 50% исходного материала. Прибавляли дополнительные 2,4 мл (4,8 ммоля) диазометилтриметилсилана. После стояния в течение примерно 16 ч никакой исходный материал не мог быть определен с помощью ВЭЖХ. Прибавляли 0,5 мл ледяной уксусной кислоты и реакционную смесь упаривали досуха. Прибавляли толуол и смесь снова упаривали. Полученный в результате остаток растворяли в кипящем этилацетате, прибавляли гексан, образовавшиеся кристаллы отделяли фильтрованием, промывали гексаном и сушили примерно при 0,01 мм рт.ст. и 50°С, получая 912 мг (86%) метилового эфира 2-ацетиламино-5-фтор-4-нитробензойной кислоты, tпл 124-126°С.

Растворяли 256 мг (1,0 ммоль) метилового эфира 2-ацетиламино-5-фтор-4-нитробензойной кислоты в 5,1 мл кипящего абсолютного метанола и охлаждали до 23°С, таким образом некоторое количество вещества перекристаллизовывалось. Прибавляли 81 мг (1,5 ммоля) метилата натрия и смесь перемешивали в атмосфере аргона в течение 16 ч. Прибавляли уксусную кислоту до тех пор, пока значение рН не достигало 5, полученную в результате смесь распределяли между этилацетатом и водой, органическую фазу отделяли, промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на 100 г силикагеля (0,04-063 мм), элюируя смесью толуол-этилацетат (3:1), объем фракции 12 мл. Фракции 37-46 объединяли и упаривали, получая 77 мг (28%) метилового эфира 2-ацетиламино-5-метокси-4-нитробензойной кислоты в виде кристаллов желтоватого цвета, tпл 141-143°С.

Прибавляли по каплям 0,12 мл 98% серной кислоты при 0° (экзотермическая реакция) к суспензии 69 мг (0,257 ммоля) метилового эфира 2-ацетиламино-5-метокси-4-нитробензойной кислоты, 0,69 мл метанола и 0,12 мл воды. Затем смесь нагревали при 70°С в течение 30 мин, охлаждали до комнатной температуры, выливали в смесь льда, воды и этилацетата и доводили значение рН до 6 концентрированным раствором бикарбоната калия. Органическую фазу отделяли, промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали, получая 46 мг (79%) метилового эфира 2-амино-5-метокси-4-нитробензойной кислоты в виде кристаллов красно-коричневого цвета, tпл 127-129°С.
Прибавляли по каплям 0,5 мл раствора фосгена в толуоле (20%) к суспензии 35 мг (0,155 ммоля) метилового эфира 2-амино-5-метокси-4-нитробензойной кислоты в толуоле при -15°С. Смеси давали нагреться до комнатной температуры в течение 1 ч и затем нагревали при кипении. Через 45 мин аргон продували через кипящий желтый раствор в течение 30 мин и в течение 2 ч при комнатной температуре. Реакционную смесь упаривали и остаток сушили при комнатной температуре и давлении 9 мбар в течение 1 ч, что привело к 45 мг неочищенного метилового эфира 2-изоцианато-5-метокси-4-нитробензойной кислоты. Суспендировали 43 мг данного вещества в 0,91 мл абсолютного тетрагидрофурана, прибавляли 17 мг метансульфонилгидразида и суспензию перемешивали при комнатной температуре в течение 1 ч. Прибавляли 1,0 мл абсолютного тетрагидрофурана и 0,045 мл основания Хюнига и продолжали перемешивание при комнатной температуре в течение 18 ч. Реакционную смесь упаривали, остаток растворяли в смеси воды и этилацетата и водный слой доводили до значения рН~2 добавлением 1 М водной хлористоводородной кислоты. Органическую фазу отделяли, промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали. Полученное в результате вещество кристаллизовали из тетрагидрофурана, прибавляли 2 мл толуола и 0,02 мл основания Хюнига и смесь нагревали при кипении в течение 3 ч. Реакционную смесь упаривали, прибавляли 1 мл воды и значение рН доводили до 1 прибавлением 1 н. водной хлористоводородной кислоты. Суспензию перемешивали при комнатной температуре в течение 1 ч, затем фильтровали и остаток промывали водой. Остающиеся кристаллы растворяли в кипящем тетрагидрофуране, прибавляли пентан, образовавшиеся кристаллы отделяли фильтрованием и сушили в вакууме при 100°С, получая 7 мг (13%) N-(6-метокси-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в виде кристаллов светло-бежевого цвета, tпл>280°C.
Пример 16
N-[6-(4-Бромимидазол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]-метансульфонамид:

Смесь 20 мг (0,062 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 56 мг (0,372 ммоля) 4-бром-1H-имидазола и 0,1 мл диметилсульфоксида нагревали при 120°С в течение 16 ч. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом и водой, органическую фазу отделяли, промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, используя смесь дихлорметан-метанол (95:5), и фракции, содержащие требуемый продукт, объединяли и упаривали, получая 6 мг (21%) указанного в заголовке соединения. МС (ионизация электрораспылением (ES), m/e): 445 (М+).
Пример 17
N-[7-Нитро-2,4-диоксо-6-(4-фенилимидазол-1-ил)-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

Смесь 50 мг (0,156 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 139 мг (0,936 ммоля) 4-фенил-1H-имидазола и 0,25 мл диметилсульфоксида нагревали при 120°С в течение 3,5 ч. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом и водой, органическую фазу отделяли, промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, используя смесь тетрагидрофуран-гексан (80:20), и фракции, содержащие требуемый продукт, объединяли и упаривали, получая 53 мг (77%) указанного в заголовке соединения. MC(ES, m/e): 441 (М-1).
Пример 18
N-{6-[4-(4-Метоксифенил)имидазол-1-ил]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:

Смесь 75 мг (0,233 ммоля) N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 209 мг (1,165 ммоля) 4-(4-метоксифенил)-1H-имидазола и 0,3 мл диметилсульфоксида нагревали при 120°С в течение 3,5 ч. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом и водой, органическую фазу отделяли, промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, используя смесь дихлорметан-метанол (95:5), и фракции, содержащие требуемый продукт, объединяли и упаривали. Остаток кристаллизовали из дихлорметана, следов метанола и изопропилацетата, получая 56 мг (51%) указанного в заголовке соединения, tпл 268-271°С.
Общая методика А (ОМА)
К суспензии N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида (30 мг, 0,0943 ммоля) в этаноле прибавляли нуклеофил (0,943 ммоля, 10 экв) при комнатной температуре. Реакционная смесь быстро изменяла окраску до желто-оранжевой, и ее нагревали при 150°С в течение периода 6-40 мин, соответствующего исчезновению исходного материала (контроль с помощью ВЭЖХ). Растворитель упаривали, неочищенный материал растворяли в диметилсульфоксиде или адсорбировали на диатомовой земле Isolute® sorbent HM-N и очищали жидкостной хроматографией среднего давления на обращенной фазе С18.
Общая методика Б (ОМБ)
К раствору N-(6-фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида (30 мг, 0,0943 ммоля) в диметилсульфоксиде прибавляли нуклеофил (0,471 ммоля, 5 экв) при комнатной температуре. Реакционную смесь нагревали при 120°С в атмосфере азота в течение 16 ч. Неочищенный раствор очищали жидкостной хроматографией среднего давления на обращенной фазе С18.
Специфика ВЭЖХ: значения времен удерживания (Rt) получали с помощью системы Waters® Alliance HT HPLC с колонкой СС 70/4.6 Nucleosil 100-3 С18, используя градиент аода + 0,1% ТФК/ацетонитрил + 0,1 ТФК (5:95) до (95:5) в течение 8 мин, и током растворителя 1,4 мл/мин.
Пример 19
N-(6-Бензиламино-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (20 мг, 0,0628 ммоля) реагировал с бензиламином согласно ОМА, давая 8 мг (31%) порошка красного цвета. Rt=3,45 мин.
Пример 20
N-[6-(4-Фторбензиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 4-фторбензиламином согласно ОМА, давая 5 мг (12,5%) порошка красного цвета. Rt=5,00 мин.
Пример 21
N-[6-(2-Гидроксиэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-аминоэтанолом согласно ОМА, давая 20 мг (58%) порошка красного цвета. Rt=2,86 мин.
Пример 22
N-[6-(2-Морфолин-4-илэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-морфолин-4-илэтиламином согласно ОМА, давая 21 мг (54%) порошка красного цвета. Rt=2,33 мин.
Пример 23
N-[6-(4-Метоксибензиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 4-метоксибензиламином согласно ОМА, давая 24,7 мг (60%) порошка красного цвета. Rt=4,97 мин.
Пример 24
N-{6-[(Бензо[1,3]диоксол-5-илметил)амино]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с бензо[1,3]диоксол-5-илметиламином согласно ОМА, давая 9,1 мг (21,5%) порошка красного цвета. Rt=4,86 мин.
Пример 25
N-[7-Нитро-2,4-диоксо-6-(4-фенилпиперазин-1-ил)-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 1-фенилпиперазином согласно ОМА, давая 28,6 мг (66%) порошка оранжевого цвета. Rt=4,49 мин.
Пример 26
N-[6-(2-Метилимидазол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (25 мг, 0,0786 ммоля) реагировал с 2-метил-1H-имидазолом согласно ОМА, давая 14,5 мг (49%) порошка желтоватого цвета. Rt=1,98 мин.
Пример 27
N-[7-Нитро-2,4-диоксо-6-(4-фенилбутиламино)-1,4-дигидро-2H-хиназолин-3-ил]-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (20 мг, 0,0628 ммоля) реагировал с 4-фенилбутиламином согласно ОМА, но из-за низкой растворимости продукт реакции подвергался очистке препаративной тонкослойной хроматографией (элюент: гексан-этилацетат-уксусная кислота (3:7:0,1), давая 5,4 мг (19%) порошка красного цвета. Rt=5,93 мин.
Пример 28
N-{6-[4-(4-Ацетилфенил)пиперазин-1-ил]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (20 мг, 0,0628 ммоля) реагировал с 1-(4-пиперазин-1-илфенил)этаноном согласно ОМА, давая 14 мг (44%) порошка оранжевого цвета. Rt=3,85 мин.
Пример 29
N-(7-Нитро-2,4-диоксо-6-[1,2,4]триазол-1-ил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (20 мг, 0,0628 ммоля) реагировал с 1H-[1,2,4]триазолом согласно ОМБ, давая 19 мг (83%) порошка желтоватого цвета. Rt=2,87 мин.
Пример 30
N-[7-Нитро-2,4-диоксо-6-(2-феноксиэтиламино)-1,4-дигидро-2H-хиназолин-3-ил]-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (20 мг, 0,0628 ммоля) реагировал с 2-феноксиэтиламином согласно ОМА, давая 14 мг (52%) порошка красного цвета. Rt=5,25 мин.
Пример 31
N-[6-(2-Метоксиэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (20 мг, 0,0628 ммоля) реагировал с 2-метоксиэтиламином согласно ОМА, давая 8,5 мг (36%) порошка красного цвета. Rt=3,84 мин.
Пример 32
N-(7-Нитро-2,4-диоксо-6-пиразол-1-ил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 1H-пиразолом согласно ОМБ, давая 9,4 мг (27%) порошка красного цвета. Rt=3,64 мин.
Пример 33
N-[6-(4-Метилимидазол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (25 мг, 0,0786 ммоля) реагировал с 4-метил-1H-имидазолом согласно ОМБ, давая 12,6 мг (43%) порошка желтоватого цвета в виде смеси региоизомеров (5:1), Rt=2,11 мин.
Пример 34
N-{6-[2-(1H-Имидазол-4-ил)этиламино]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (20 мг, 0,0628 ммоля) реагировал с 2-(1H-имидазол-4-ил)этиламином согласно ОМА, давая 9,6 мг (37%) порошка красного цвета. Rt=2,65 мин.
Пример 35
N-(6-Циклопропиламино-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с циклопропиламином согласно ОМА, давая 10 мг (31%) порошка красного цвета. Rt=3,15 мин.
Пример 36
N-[6-(Циклопропилметиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (33 мг, 0,1037 ммоля) реагировал с циклопропилметиламином согласно ОМА, но с очисткой продукта реакции жидкостной хроматографией среднего давления (гексан-этилацетет-уксусная кислота (95:5:0,1) до (40:60:0,1), давая 11 мг (32%) порошка красного цвета. Rt=4,66 мин.
Пример 37
N-[6-(3-Гидроксипирролидин-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с пирролидин-3-олом согласно ОМА, давая 21 мг (58%) порошка красного цвета. Rt=4,22 мин.
Пример 38
N-[6-(2-Гидроксипропиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 1-аминопропан-2-олом согласно ОМА, давая 23,1 мг (65%) порошка красного цвета. Rt=3,07 мин.
Пример 39
N-(6-Изопропиламино-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с изопропиламином согласно ОМА, давая 19,6 мг (58%) порошка красного цвета. Rt=4,44 мин.
Пример 40
N-(7-Нитро-2,4-диоксо-6-пирролидин-1-ил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с пирролидином согласно ОМА, давая 12,2 мг (35%) порошка светло-оранжевого цвета. Rt=4,37 мин.
Пример 41
N-{6-[4-(2-Морфолин-4-ил-2-оксоэтил)имидазол-1-ил]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-(1H-имидазол-4-ил)-1-морфолин-4-илэтаноном согласно ОМБ, давая 18,1 мг (39%) порошка желтого цвета. Rt=4,37 мин.
Пример 42
N-{6-[(2-Гидроксиэтил)метиламино]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-метиламиноэтанолом согласно ОМА, давая 25 мг (71%) порошка желтого цвета. Rt=2,98 мин.
Пример 43
N-[6-(3-Гидроксипропиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 3-аминопропан-1-олом согласно ОМА, давая 24,1 мг (68,5%) порошка желтого цвета. Rt=3,09 мин.
Пример 44
N-[6-(2-Гидрокси-1-метилэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-аминопропан-1-олом согласно ОМА, давая 25 мг (71%) порошка красного цвета. Rt=3,21 мин.
Пример 45
N-[6-(2-Гидрокси-1,1-диметилэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-амино-2-метилпропан-1-олом согласно ОМА, давая 14,2 мг (39%) порошка красного цвета. Rt=3,55 мин.
Пример 46
N-[6-(4,5-Диметилимидазол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 4,5-диметил-1H-имидазолом согласно ОМБ, давая 18 мг (48%) порошка белого цвета. Rt=2,43 мин.
Пример 47
N-{6-[бис-(2-Гидроксиэтил)амино]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-(2-гидроксиэтиламино)-этанолом согласно ОМА, давая 21 мг (55%) порошка красного цвета. Rt=2,36 мин.
Пример 48
N-Аллил-2-[1-(3-метансульфониламино-7-нитро-2,4-диоксо-1,2,3,4-тетрагидро-хиназолин-6-ил)-1H-имидазол-4-ил]ацетамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с N-аллил-2-(1H-имидазол-4-ил)ацетамидом согласно ОМА, давая 28 мг (52%) порошка желтоватого цвета. Rt=2,36 мин.
Пример 49
трет-Бутиловый эфир [2-(3-метансульфониламино-7-нитро-2,4-диоксо-1,2,3,4-тетрагидрохиназолин-6-иламино)этил]карбаминовой кислоты:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с трет-бутиловым эфиром (2-аминоэтил)карбаминовой кислоты согласно ОМА, давая 27 мг (62%) порошка красного цвета. Rt=4,43 мин.
Пример 50
N-[6-(2-Аминоэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]-метансульфонамид:
К охлажденному льдом раствору трет-бутилового эфира [2-(3-метансульфониламино-7-нитро-2,4-диоксо-1,2,3,4-тетрагидрохиназолин-6-иламино)этил]карбаминовой кислоты (10 мг, 0,022 ммоля) в дихлорметане (0,5 мл) прибавляли трифторуксусную кислоту (0,25 мл). Реакционную смесь перемешивали при 0°С в течение 3 ч. Смесь фильтровали и полученное в результате твердое вещество красного цвета сушили в вакууме, получая 9 мг (87%) порошка красного цвета. Rt=2,13 мин.
Пример 51
N-[6-(1-Гидроксиметил-2-метилпропиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-амино-3-метилбутан-1-олом согласно ОМА, давая 17 мг (45%) порошка красного цвета. Rt=4,00 мин.
Пример 52
N-[6-(1-Гидроксиметилпропиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-аминобутан-1-олом согласно ОМА, давая 14,5 мг (40%) порошка красного цвета. Rt=3,60 мин.
Пример 53
N-[6-((S)-2-Гидрокси-1-метилэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (S)-2-аминопропан-1-олом согласно ОМА, давая 21,4 мг (61%) порошка красного цвета. Rt=3,20 мин.
Пример 54
N-[6-((R)-2-Гидрокси-1-фенилэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (R)-2-амино-2-фенилэтанолом согласно ОМА, давая 31,2 мг (75%) порошка красного цвета. Rt=4,11 мин.
Пример 55
N-[6-((S)-3-Гидроксипирролидин-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (S)-пирролидин-3-олом согласно ОМА, давая 26 мг (69%) порошка красного цвета. Rt=3,11 мин.
Пример 56
N-[6-((R)-3-Гидроксипирролидин-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (R)-пирролидин-3-олом согласно ОМА, давая 25,6 мг (60%) порошка красного цвета. Rt=3,09 мин.
Пример 57
N-(6-Азетидин-1-ил-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с азетидином согласно ОМА, давая 15 мг (45%) порошка красного цвета. Rt=3,92 мин.
Пример 58
N-{6-[(R)-1-(3,4-Диметоксифенил)-2-гидроксиэтиламино]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (R)-2-амино-2-(3,4-диметоксифенил)этанолом согласно ОМА, давая 24,8 мг (53%) порошка красного цвета. Rt=3,80 мин.
Пример 59
N-[6-(1-Гидроксиметилпентиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-аминогексан-1-олом согласно ОМА, давая 24 мг (61%) порошка красного цвета. Rt=4,5 мин.
Пример 60
N-(7-Нитро-2,4-диоксо-6-[1,2,3]триазол-1-ил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 1-H-[1,2,3]триазолом согласно ОМБ, давая 8,5 мг (25%) порошка белого цвета. Rt=4,5 мин.
Пример 61
N-{6-[(S)-2-Гидрокси-1-(1H-имидазол-4-илметил)этиламино]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (S)-2-амино-3-(1H-имидазол-4-ил)пропан-1-олом согласно ОМА, давая 24,1 мг (46%) порошка красного цвета. Rt=1,73 мин.
Пример 62
N-[6-((S)-2-Гидрокси-1-фенилэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (S)-2-амино-2-фенилэтанолом согласно ОМА, давая 25,8 мг (76%) порошка красного цвета. Rt=4,10 мин.
Пример 63
N-[6-((R)-1-Гидроксиметил-2-фенилэтиламино)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (R)-2-амино-3-фенилпропан-1-олом согласно ОМА, давая 22,2 мг (65%) порошка пурпурного цвета. Rt=4,40 мин.
Пример 64
N-{6-[2-Гидрокси-1-(1H-индол-3-илметил)этиламино]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с 2-амино-3-(1H-индол-3-ил)пропан-1-олом согласно ОМА, давая 18,8 мг (33%) порошка красного цвета. Rt=4,27 мин.
Пример 65
N-{6-[(R)-2-Гидрокси-1-(1H-имидазол-4-илметил)этиламино]-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (R)-2-амино-3-(1H-имидазол-4-ил)пропан-1-олом согласно ОМА, давая 22,7 мг (43%) порошка красного цвета. Rt=1,73 мин.
Пример 66
N-[6-(4-Цианометилимидазол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (30 мг, 0,0943 ммоля) реагировал с (1H-имидазол-4-ил)ацетонитрилом согласно ОМБ, давая 18,5 мг (47%) порошка желтого цвета. Rt=2,56 мин.
Пример 67
N-[6-(4-Метоксиметилимидазол-1-ил)-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
N-(6-Фтор-7-нитро-2,4-диоксо-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид (50 мг, 0,157 ммоля) реагировал с 4-метоксиметил-1H-имидазолом согласно ОМБ, давая 36 мг (44%) порошка желтого цвета. Rt=2,20 мин.
Пример 68
N-(6-Морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид:
Растворяли 70 мг (0,23 ммоля) метилового эфира 2-амино-5-морфолин-4-ил-4-трифторметилбензойной кислоты в 5 мл дихлорметана и обрабатывали 23 мг (0,076 ммоля) трифосгена. Через 15 мин к суспензии прибавляли 0,025 мл триэтиламина. Прозрачный раствор перемешивали в течение дополнительных 3 ч при комнатной температуре (КТ), после чего прибавляли с помощью шприца раствор 26 мг (0,23 ммоля) метансульфонилгидразида в 2,5 мл безводного тетрагидрофурана. Образующуюся в результате суспензию перемешивали в течение дополнительного часа при комнатной температуре, обрабатывали 0,3 мл 1 М водного раствора гидроксида натрия и перемешивали в течение ночи. После упаривания растворителя с использованием роторного испарителя остаток растворяли в дихлорметане и очищали препаративной тонкослойной хроматографией, используя смесь растворителей дихлорметан/метанол (9:1), получая N-(6-морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид с tпл 255-260°С.
Исходное вещество, метиловый эфир 2-амино-5-морфолин-4-ил-4-трифторметил-бензойной кислоты получали следующим образом.
Раствор 20 г (0,173 ммоля) трет-бутилата калия в 300 мл безводного тетрагидрофурана охлаждали до -78°С в атмосфере азота. Раствор 10 г (46,87 моля) 1-фтор-4-нитро-2-трифторметилбензола и 8,1 мл (0,1 моля) хлороформа в 100 мл безводного тетрагидрофурана медленно прибавляли так, чтобы температура держалась ниже -75°С. Темно-коричневый раствор обрабатывали последовательно дополнительными 4 мл хлороформа и раствором 10 г трет-бутилата калия в 100 мл тетрагидрофурана. Через 3 ч при -78°С раствор перемешивали, пока он не достиг комнатной температуры, и упаривали досуха. Прибавляли 100 мл смеси (1:1) уксусной кислоты и метанола. Остаток растворяли в 500 мл этилацетата и экстрагировали три раза 100 мл насыщенного водного раствора хлористого натрия. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. После хроматографии на 400 г силикагеля и элюирования смесью дихлорметан/метанол (9:1) получали чистый 1-фтор-4-нитро-5-трихлорвинил-2-трифторметилбензол в виде масла желтого цвета.
К 300 мл смеси растворителей тетрахлорметан, ацетонитрил и вода (10:10:15) прибавляли последовательно 8,5 г (25 ммолей) 1-фтор-4-нитро-5-трихлорвинил-2-трифторметилбензола, 1 г (8 моль-%) моногидрата трихлорида рутения и 23 г (0,1 моля) перйодата натрия. После перемешивания при 60°С в течение 16 ч прибавляли дополнительные 1 г моногидрата трихлорида рутения и 23 г перйодата натрия и перемешивание при 60°С продолжали в течение 4 ч. Охлажденную реакционную смесь фильтровали через слой целита и фильтрат упаривали досуха. Прибавляли 150 мл 1 М раствора гидроксида натрия и экстрагировали два раза 150 мл дихлорметана. Водную фазу подкисляли концентрированной хлористоводородной кислотой и экстрагировали четыре раза по 100 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия, фильтровали и упаривали. Остаток перекристаллизовывали из смеси ацетон/петролейный эфир, получая 5-фтор-2-нитро-4-трифторметилбензойную кислоту с tпл 142-145°С.
Раствор 2 г (24,5 ммоля) 5-фтор-2-нитро-4-трифторметилбензойной кислоты в 250 мл метанола обрабатывали 9,28 мл (73,5 ммоля) триметилхлорсилана и нагревали при кипении в течение 6 дней, в продолжении которых каждый вечер прибавляли дополнительные 9,28 мл триметилхлорсилана. Охлажденный раствор упаривали в вакууме и распределяли между дихлорметаном и водой. Объединенные органические фазы экстрагировали дважды по 50 мл 0,5 М раствора гидроксида натрия и 50 мл насыщенного солевого раствора. После высушивания над сульфатом натрия, фильтрования и упаривания кристаллический остаток перекристаллизовывали из смеси ацетон/петролейный эфир, получая метиловый эфир 5-фтор-2-нитро-4-трифторметил-бензойной кислоты с tпл 66-68°С.
Раствор 134 мг (0,5 ммоля) метилового эфира 5-фтор-2-нитро-4-трифторметил-бензойной кислоты в 5 мл безводного тетрагидрофурана обрабатывали 0,05 мл (0,55 ммоля) морфолина и нагревали при кипении в течение 90 мин. После упаривания растворителя получали метиловый эфир 5-морфолин-4-ил-2-нитро-4-трифторметилбензойной кислоты в виде масла желтого цвета.
Раствор 140 мл (0,42 ммоля) метилового эфира 5-морфолин-4-ил-2-нитро-4-трифторметилбензойной кислоты в 10 мл метанола обрабатывали 30 мг 10% палладия на угле и гидрировали при комнатной температуре под давлением в 5 бар в течение 23 ч. После отделения катализатора фильтрованием и упаривания растворителя получали чистый метиловый эфир 2-амино-5-морфолин-4-ил-4-трифторметилбензойной кислоты в виде масла желтого цвета.
Пример 69
N-(2,4-Диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид:
Раствор 65,0 г (300 ммолей) N-(2-метил-5-трифторметилфенил)ацетамида в 520 мл концентрированной серной кислоты обрабатывали по каплям раствором 152,5 г (1,5 ммоля) нитрата калия в 520 мл концентрированной серной кислоты в атмосфере азота и смесь держали при комнатной температуре в бане со льдом. Затем смесь перемешивали в течение еще 2 ч при комнатной температуре и затем выливали на лед. Суспензию фильтровали и отфильтрованный осадок растворяли в этилацетате, раствор сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали флэш-хроматографией (диоксид кремния, гексан/этилацетат (1:1)), получая продукт нитрования в виде смеси региоизомеров (62,44 г, 238 ммолей). Растворяли 51,0 г (195 ммолей) неочищенного продукта в 1000 мл воды и нагревали при 100°С и порциями обрабатывали смесью 184 г (1167 ммолей) перманганата калия и 70,4 г (585 ммолей) моногидрата сульфата магния. Смесь выдерживали при 100°С в течение дополнительных 2 ч и затем давали охладиться до комнатной температуры. Смесь затем фильтровали и фильтрат экстрагировали этилацетатом. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме, получая 19,3 г (27% на 2 стадии) 2-ацетиламино-5-нитро-4-трифторметилбензойной кислоты в виде кристаллов желтого цвета. 1Н-ЯМР (ДМСО-d6, 400 МГц): 9,13 (s, 1H), 8,67 (s, 1H), 1,98 (s, 3H).
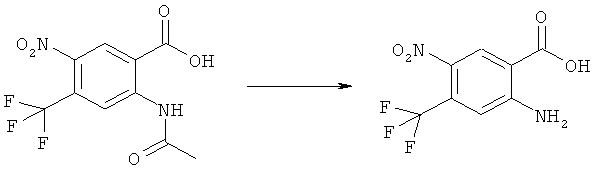
Раствор 49,5 г (169 ммолей) 2-ацетиламино-5-нитро-4-трифторметилбензойной кислоты в 600 мл метанола и 100 мл воды охлаждали до 0°С и прибавляли по каплям 71,1 мл (1,33 моля) концентрированной серной кислоты. По завершении прибавления смесь нагревали при кипении в течение 1 ч. Затем смесь охлаждали до 0°С, значение рН доводили до 10 с 30% водным раствором гидроксида натрия и перемешивали ее в течение 1 ч. Метанол отгоняли и оставшийся водный раствор разбавляли водой и экстрагировали трет-бутилметиловым эфиром. Водную фазу подкисляли концентрированным раствором HCl, образующуюся в результате желтую суспензию фильтровали и осадок промывали водой. Твердое вещество сушили в вакууме при 100°С, получая 28,3 г (67%) 2-амино-5-нитро-4-трифторметилбензойной кислоты, tпл 237°С.

Раствор 28 г (112 ммолей) 2-амино-5-нитро-4-трифторметилбензойной кислоты в 550 мл метанола охлаждали до 0°С в атмосфере азота и прибавляли по каплям концентрированную серную кислоту так, чтобы держать температуру около 20°С. По завершении прибавления смесь нагревали при кипении в течение 24 ч. Затем давали охладиться до комнатной температуры и смесь концентрировали в вакууме до объема примерно 50 мл. Данный остаток выливали на лед и экстрагировали этилацетатом. Органическую фазу промывали насыщенным водным раствором бикарбоната натрия насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт перекристаллизовывали из толуола, получая 26,7 г (90%) метилового эфира 2-амино-5-нитро-4-трифторметилбензойной кислоты, tпл 174-175°C.
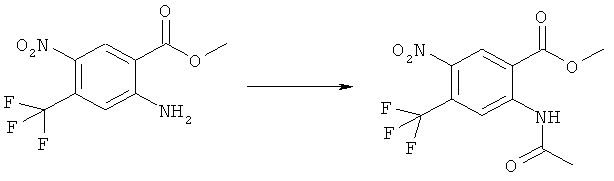
Раствор 2,0 г (7,57 ммоля) метилового эфира 2-амино-5-нитро-4-трифторметилбензойной кислоты в 20 мл уксусного ангидрида нагревали при кипении в течение 4 ч. Смеси давали охладиться до комнатной температуры и концентрировали в вакууме. Остаток забирали в этилацетат и промывали водой, водным насыщенным раствором бикарбоната натрия и насыщенным солевым раствором. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (диоксид кремния, гексан/этилацетат (3:1)) дала 1,79 г (77%) метилового эфира 2-ацетиламино-5-нитро-4-трифторметилбензойной кислоты в виде кристаллов желтого цвета, tпл 75-80°С.

Раствор 1,7 г (5,55 ммоля) метилового эфира 2-ацетиламино-5-нитро-4-трифторметилбензойной кислоты в 29 мл метанола обрабатывали 250 мг Pd/C (10%) и смесь перемешивали при давлении водорода 5 бар в течение 20 мин. Смесь фильтровали и фильтрат концентрировали в вакууме, получая 1,58 г (выход количественный) метилового эфира 2-ацетиламино-5-амино-4-трифторметилбензойной кислоты в виде кристаллов желтого цвета, tпл 143-152°С.

Раствор 1,55 г (5,6 ммоля) метилового эфира 2-ацетиламино-5-амино-4-трифторметилбензойной кислоты, 1,48 г (16,8 ммоля) 1,2-диформилгидразина и 5,5 мл (329,2 ммоля) триэтиламина в 40 мл пиридина обрабатывали по каплям 10,8 мл (85,3 ммоля) триметилсилилхлорида. Затем смесь нагревали при 100°С в течение 18 ч и давали охладиться до комнатной температуры. Затем смесь обрабатывали снова 1,48 г (16,8 ммоля) 1,2-диформилгидразина, 5,5 мл (39,2 ммоля) триэтиламина и 10,8 мл (85,3 ммоля) триметилсилилхлорида и нагревали при 100°С в течение 24 ч. Смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток забирали в воду и экстрагировали этилацетатом. Водную фазу подкисляли до рН 4-5 с 4 М водным раствором хлористоводородной кислоты и насыщали хлористым натрием. Данную водную фазу экстрагировали этилацетатом и эту органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт перекристаллизовывали из этилацетата, получая 1,34 г (76%) 2-ацетиламино-5-[1,2,4]триазол-4-ил-4-трифторметилбензойной кислоты. МС (ES, m/е): 315 (М+Н].
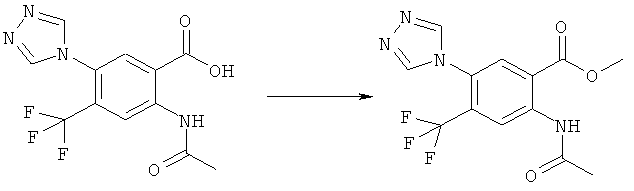
Раствор 1,3 г (4,14 ммоля) 2-ацетиламино-5-[1,2,4]триазол-4-ил-4-трифторметил-бензойной кислоты в 15 мл метанола обрабатывали по каплям 3,1 мл (6,21 ммоля) 2 М раствора триметилсилилдиазометана в гексане. Смесь перемешивали в течение 1 ч при комнатной температуре и снова прибавляли 1,5 мл (3,00 ммоля) раствора триметилсилилдиазометана в гексане (2 М). Через 1 ч реакцию прекращали добавлением ледяной уксусной кислоты до тех пор, пока более не наблюдалось выделение газа. Смесь концентрировали в вакууме и остаток забирали в этилацетат, раствор промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (диоксид кремния, градиент 100% этилацетата до этилацетат/метанол (9:1)) дала 456 мг (34%) метилового эфира 2-ацетиламино-5-[1,2,4]триазол-4-ил-4-трифторметилбензойной кислоты в виде кристаллов желтого цвета, tпл 180-184°С.
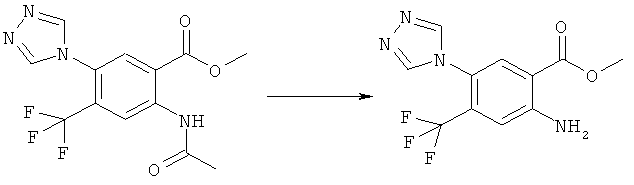
Раствор 450 мг (1,43 ммоля) метилового эфира 2-ацетиламино-5-[1,2,4]триазол-4-ил-4-трифторметилбензойной кислоты в 5 мл метанола и 1 мл воды охлаждали до 0°С и по каплям обрабатывали 0,6 мл (11,3 ммоля) концентрированной серной кислоты. Затем смесь нагревали при кипении в течение 30 мин и давали охладиться до комнатной температуры. Смесь выливали на лед и полученную в результате смесь экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт перекристаллизовывали из смеси этилацетат/гексан, получая 292 мг (71%) метилового эфира 2-амино-5-[1,2,4]триазол-4-ил-4-трифторметилбензойной кислоты, tпл 184-186°С.
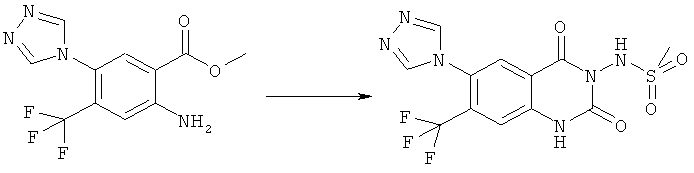
Раствор 270 мг (0,94 ммоля) метилового эфира 2-амино-5-[1,2,4]триазол-4-ил-4-трифторметилбензойной кислоты в 5 мл тетрагидрофурана при комнатной температуре в атмосфере азота обрабатывали 93 мг (0,31 ммоля) трифосгена. Смесь перемешивали в течение 10 мин, прибавляли 0,13 мл (0,94 ммоля) триэтиламина и перемешивание продолжали в течение 3 ч. Затем прибавляли 104 мг (0,94 ммоля) метансульфонилгидразида и смесь перемешивали в течение 1 ч при комнатной температуре. Затем смесь обрабатывали 2 мл 1 М водного раствора гидроксида натрия и перемешивали в течение дополнительных 2 ч. Значение рН смеси доводили до 4-5 с 4 М водным раствором HCl и смесь концентрировали в вакууме. Остаток забирали в этилацетат, раствор промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт перекристаллизовывали из этилацетата, получая 219 мг (56%) N-(2,4-диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в виде бесцветных кристаллов, tпл 231-237°С.
Пример 70
(2,4-Диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)амид этансульфоновой кислоты:
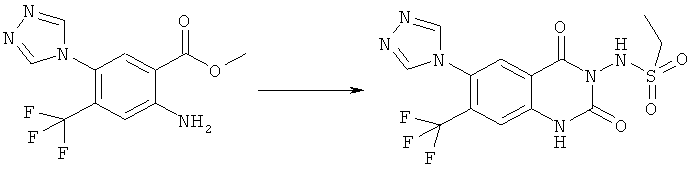
Раствор 650 мг (2,27 ммоля) метилового эфира 2-амино-5-[1,2,4]триазол-4-ил-4-трифторметилбензойной кислоты в 15 мл тетрагидрофурана при комнатной температуре в атмосфере азота обрабатывали 225 мг (0,76 ммоля) трифосгена. Смесь перемешивали в течение 20 мин, прибавляли 0,32 мл (2,27 ммоля) триэтиламина и продолжали перемешивание в течение 3 ч. Прибавляли 282 мг (2,27 ммоля) этансульфонилгидразида (полученного по аналогии со способом, описанным J.W.Powell и М.С.Whiting: The decomposition of sulphonylhydrazone salts - I, Tetrahedron, том 7 (1959) с.305) и смесь перемешивали в течение 1 ч при комнатной температуре. Затем смесь обрабатывали 5 мл 1 М водного раствора гидроксида натрия и перемешивали в течение 18 ч. Значение рН смеси доводили до 4-5 с 4 М водным раствором HCl и смесь концентрировали в вакууме. Неочищенный продукт очищали флэш-хроматографией (диоксид кремния, этилацетат/метанол (19:1)), дающей бесцветные кристаллы, которые перекристаллизовывали из смеси этилацетат/гексан, чтобы получить 462 мг (2,4-диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)амида этансульфоновой кислоты в виде бесцветных кристаллов, tпл 185-195°С.
Пример 71
Этиловый эфир 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-[1,2,4]триазол-3-карбоновой кислоты:

Раствор 613 мг (14,0 ммолей) гидрида натрия в 10 мл 1-метил-2-пирролидинона охлаждали до 0-5°С и по каплям обрабатывали раствором 1,74 г (12,4 ммоля) этилового эфира 1H-[1,2,4]триазол-3-карбоновой кислоты в 10 мл 1-метил-2-пирролидинона в течение 8 мин. Смесь перемешивали в течение 1 ч при 0-5°С и прибавляли раствор 3,00 г (11,2 ммоля) метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты в 10 мл 1-метил-2-пирролидинона. Смеси давали медленно нагреться до комнатной температуры и перемешивали в течение 18 ч в атмосфере азота. Смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу отделяли и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (диоксид кремния, гексан/этилацетат (1:1)) и смешанные фракции перекристаллизовывали из толуола, получая всего 3,12 г (71%) этилового эфира 1-(5-метоксикарбонил-4-нитро-2-трифторметилфенил)-1H-[1,2,4]триазол-3-карбоновой кислоты, tпл 190-192°С.

Раствор 3,00 г (7,73 ммоля) этилового эфира 1-(5-метоксикарбонил-4-нитро-2-трифторметилфенил)-1H-[1,2,4]триазол-3-карбоновой кислоты в 75 мл смеси метанол/тетрагидрофуран (1:1) обрабатывали 400 мг Pd/C при давлении водорода 5 бар в течение 30 мин. Затем смесь фильтровали и фильтрат концентрировали в вакууме. Неочищенный продукт перекристаллизовывали из изопропанола, получая 2,47 г (82%) этилового эфира 1-(4-амино-5-метоксикарбонил-2-трифторметилфенил)-1H-[1,2,4]триазол-3-карбоновой кислоты в виде кристаллов белого цвета, tпл 189-190°С.

Раствор 1,07 г (3,00 ммоля) этилового эфира 1-(4-амино-5-метоксикарбонил-2-трифторметилфенил)-1H-[1,2,4]триазол-3-карбоновой кислоты в 9 мл дихлорметана обрабатывали 296 мг (1,00 ммоль) трифосгена при комнатной температуре в атмосфере аргона. Смесь перемешивали в течение 15 мин и по каплям прибавляли 0,42 мл (3,00 ммоля) триэтиламина. Смесь перемешивали в течение 3 ч и прибавляли раствор 330 мг (3,00 ммоля) метансульфонилгидразида в 3,3 мл безводного тетрагидрофурана. Смесь перемешивали в течение 17 ч при комнатной температуре в атмосфере аргона. Суспензию фильтровали, осадок промывали дихлорметаном и водой и сушили в вакууме. Данные бесцветные кристаллы растворяли в 20 мл безводного диоксана и обрабатывали 0,83 мл (4,86 ммоля) этилдиизопропиламина при комнатной температуре в атмосфере аргона. Смесь перемешивали в течение 17 ч и затем концентрировали в вакууме. Остаток забирали в этилацетат и воду и значение рН доводили до 3-4 с 1 М водным раствором HCl. Органическую фазу отделяли, промывали дважды насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт перекристаллизовывали из смеси тетрагидрофуран/гексан, получая 855 мг (62%) этилового эфира 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-[1,2,4]триазол-3-карбоновой кислоты в виде бесцветных кристаллов, tпл 267-270°C.
Пример 72
Метиламид 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-[1,2,4]триазол-3-карбоновой кислоты:
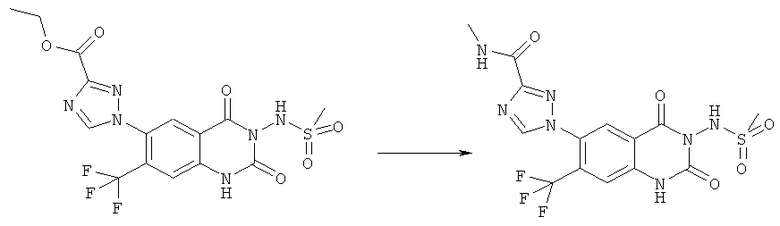
Раствор 150 мг (0,324 ммоля) этилового эфира 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-[1,2,4]триазол-3-карбоновой кислоты в 0,8 мл метанола обрабатывали 0,40 мл (3,24 ммоля) ~8 М раствора метиламина в этаноле. Смесь перемешивали в течение 22 ч при комнатной температуре и затем концентрировали в вакууме. Неочищенный продукт забирали в воду и значение рН раствора доводили до 3 с 1 М водным раствором HCl, смесь перемешивали в течение 3 ч при 0°С. Кристаллы отделяли фильтрованием, промывали холодной водой и сушили в вакууме, получая 124 мг (86%) метиламида 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-[1,2,4]триазол-3-карбоновой кислоты в виде кристаллов белого цвета, tпл 257-260°С.
Пример 73
N-(6-Имидазол-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид:

К суспензии 100 мг (0,379 ммоля) метилового эфира 2-амино-5-нитро-4-трифторметилбензойной кислоты в 1,5 мл безводного толуола прибавляли при -15°С 1,5 мл 20% раствора трифосгена в толуоле. После нагревания до комнатной температуры в суспензию вводили ток фосгена и одновременно начинали нагревание. При кипении ток фосгена поддерживали в течение 1 ч, затем заменяли током аргона в течение дополнительного часа. Толуол отгоняли, оставляя 110 мг (100%) метилового эфира 2-изоцианато-5-нитро-4-трифторметилбензойной кислоты в виде твердого вещества бежевого цвета. ИК (CHCl3, см-1): 2260 (s). 1H-ЯМР (CDCl3, 360 МГц): 4,05 (s, 3H); 7,55 (s, 1H); 8,65 (s, 1H).
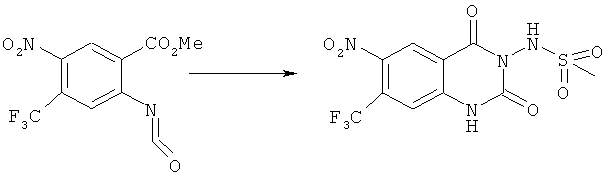
К раствору 110 мг (0,379 ммоля) метилового эфира 2-изоцианато-5-нитро-4-трифторметилбензойной кислоты в 1,7 мл безводного тетрагидрофурана прибавляли при комнатной температуре 41,7 г (0,379 ммоля) метансульфонилгидразида в 0,6 мл безводного тетрагидрофурана. Раствор превращался в белую суспензию, которую перемешивали в течение 1 ч, затем прибавляли 0,379 мл 1 М раствора гидроксида натрия и перемешивание прозрачного раствора продолжали в течение 4 ч. После прибавления 0,472 мл 2 М раствора HCl и упаривания тетрагидрофурана осадок отделяли фильтрованием и сушили при 50°С и 0,1 мм рт.ст., получая 114 мг (81%) N-(6-нитро-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в виде порошка светло-желтого цвета, tпл 220-232°С (разл.). 1Н-ЯМР (ДМСО-d6, 400 МГц): 3,15 (s, 3H); 7,66 (s, 1H); 8,62 (s, 1H); 10,50 (s, 1H); 12,41 (s, 1H).

Раствор 109 мг N-(6-нитро-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в 3 мл этанола и 3 мл уксусной кислоты гидрировали в присутствии 30 мг 10% палладия на угле. После исчезновения по данным ТСХ исходного материала реакционную смесь разбавляли этанолом и уксусной кислотой и слегка подогревали. Катализатор отделяли фильтрованием и фильтрат концентрировали досуха. Растирание остатка с этилацетатом дало 61 мг (61%) N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида в виде порошка желтого цвета, tпл 240°С (разл.). 1Н-ЯМР (ДМСО-d6, 400 МГц): 3,12 (s, 3Н); 5,66 (s, 2Н); 7,24 (s, 1H); 7,46 (s, 1H); 10,3 (уш. s, 1H); 11,4 (уш. s, 1H).

Смесь 500 мг (1,478 ммоля) N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 0,111 мл (1,478 ммоля) раствора формальдегида (37% в воде), 0,170 мл (1,478 ммоля) раствора глиоксаля (40% в воде) и 114 мг (1,478 ммоля) ацетата аммония в 3,7 мл уксусной кислоты нагревали в течение 26 ч в масляной бане при 70°С. Через 2 ч, 7 ч и 24 ч прибавляли половину эквивалента (0,739 ммоля) раствора формальдегида, раствора глиоксаля и ацетата аммония. Реакционную смесь концентрировали досуха и остаток фракционировали с помощью хроматографии среднего давления на колонке с обращенной фазой RP-18 (размер частиц 20 мкм) со смесью тетрагидрофуран/вода (3:4). После удаления тетрагидрофурана на роторном испарителе фракции лиофилизировали, пены собирали и кристаллизовали из смеси этанол/вода (3:1), получая 217 мг N-(6-имидазол-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в виде порошка светло-желтого цвета, tпл 282-301°С (разл.).
Пример 74
N-(2,4-Диоксо-6-тиоморфолин-4-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид:
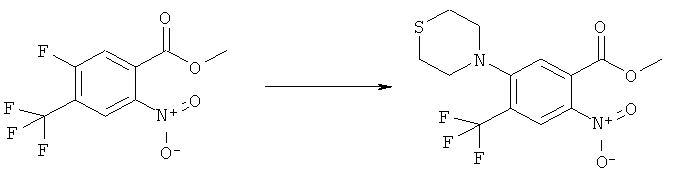
Раствор 240 мг (0,90 ммоля) метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты и 0,189 мл (2,00 ммоля) тиоморфолина в 2,5 мл тетрагидрофурана нагревали при кипении в течение 2 ч. После упаривания тетрагидрофурана остаток распределяли между водой и этилацетатом, органическую фазу отделяли и промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали, получая 310 мг метилового эфира 2-нитро-5-тиоморфолин-4-ил-4-трифторметилбензойной кислоты в виде порошка коричневого цвета, tпл 68-82°C.
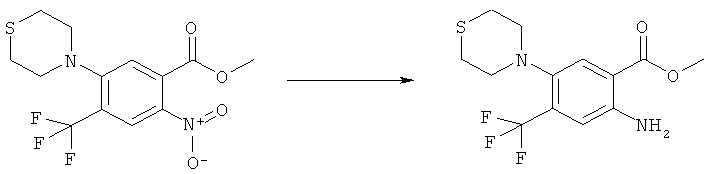
Гидрировали 300 мг метилового эфира 2-нитро-5-тиоморфолин-4-ил-4-трифторметилбензойной кислоты в 10 мл безводного этанола в присутствии 60 мг никеля Ренея. После отделения катализатора фильтрованием раствор упаривали досуха, получая 233 мг метилового эфира 2-амино-5-тиоморфолин-4-ил-4-трифторметилбензойной кислоты в виде порошка желтого цвета, tпл 85-117°С (разл.).

К суспензии 100 мг (0,312 ммоля) метилового эфира 2-амино-5-тиоморфолин-4-ил-4-трифторметилбензойной кислоты в 2 мл безводного толуола при 0°С прибавляли 1,5 мл 20% раствора фосгена в толуоле. После нагрева до комнатной температуры в суспензию вводили ток фосгена и одновременно начинали нагревание. При кипении ток фосгена поддерживали в течение 2 ч, затем в течение дополнительного 1 ч заменяли его током аргона. Толуол отгоняли, оставляя 126 мг (>100%) метилового эфира 2-изоцианато-5-тиоморфолин-4-ил-4-трифторметилбензойной кислоты в виде твердого вещества коричневого цвета, достаточно чистого для следующей стадии. 1Н-ЯМР (CDCl3, 360 МГц): 2,75-2,80 (m, 2H); 3,15-3,20 (m, 2H); 4,00 (s, 3H); 7,40 (s, 1H); 7,95 (s, 1H).
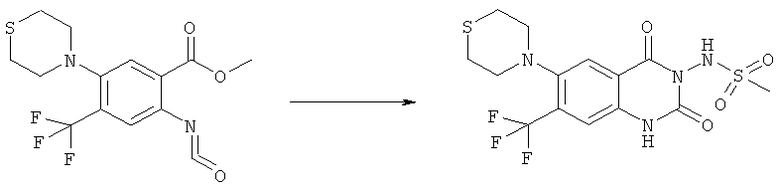
К раствору 120 мг (0,310 ммоля) метилового эфира 2-изоцианато-5-тиоморфолин-4-ил-4-трифторметилбензойной кислоты в 1,5 мл безводного тетрагидрофурана при комнатной температуре прибавляли 37,5 г (0,341 ммоля) метансульфонилгидразида в 0,5 мл безводного тетрагидрофурана. После перемешивания в течение 2,5 ч прибавляли 0,340 мл 1 М раствора гидроксида натрия и перемешивание продолжали в течение 1,5 ч с последующим прибавлением 0,412 мл 2 М раствора НС1. Тетрагидрофуран упаривали и водную фазу экстрагировали три раза этилацетатом. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали досуха. Остаток очищали хроматографией среднего давления на диоксиде кремния (размер частиц 20 мкм) со смесью этилацетат/циклогексан (2:1), получая N-(2,4-диоксо-6-тиоморфолин-4-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид в виде порошка светло-желтого цвета, tпл 245-260°С (разл.).
Той же самой последовательностью реакций, как в предыдущем примере, исходя из метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты и соответствующего амина, получали следующие соединения.
Пример 75
N-(6-[1,4]Оксазепан-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид
Порошок светло-желтого цвета, tпл 198-203°С.

Пример 76
N-[6-(4,4-Дифторпиперидин-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид
Порошок желтого цвета, tпл 249-261°С (разл.).

Пример 77
N-[6-(1,4-Диокса-8-азаспиро[4,5]дец-8-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид
Порошок желтого цвета, tпл 253-262°С.

Пример 78
N-[2,4-Диоксо-6-(4-оксопиперидин-1-ил)-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
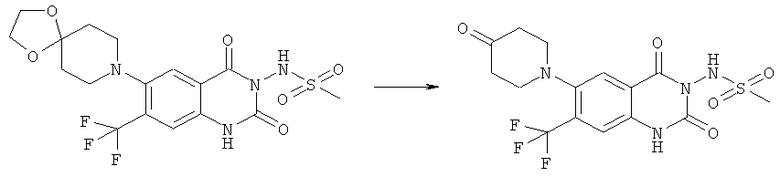
Обрабатывали 500 мг N-[6-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида 3,4 мл 6 М раствора HCl в 15 мл диоксана в течение 26 ч при комнатной температуре. После разбавления водой водную фазу экстрагировали этилацетатом, органическую фазу отделяли, промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали досуха, оставляя 490 мг порошка желтого цвета. Из данного порошка 280 мг фракционировали хроматографией среднего давления на колонке с обратимой фазой RP-18 (размер частиц 20 мкм) со смесью ацетонитрил/вода (1:2). Фракции, содержащие продукт, объединяли и экстрагировали этилацетатом, органическую фазу сушили над сульфатом натрия и концентрировали, получая N-[2,4-диоксо-6-(4-оксопиперидин-1-ил)-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид в виде порошка светло-желтого цвета, tпл 238-242°С.
Пример 79
N-(6-Азетидин-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:
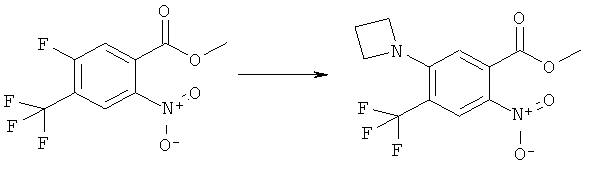
Раствор 1,07 г (4,0 ммоля) метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты и 0,461 мл (6,8 ммоля) азетидина в 10 мл тетрагидрофурана нагревали при кипении в течение 90 мин. После упаривания тетрагидрофурана остаток распределяли между водой и этилацетатом, органическую фазу отделяли и промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали, получая 1,19 г метилового эфира 5-азетидин-1-ил-2-нитро-4-трифторметилбензойной кислоты в виде порошка желтого цвета, tпл 127-136°С.
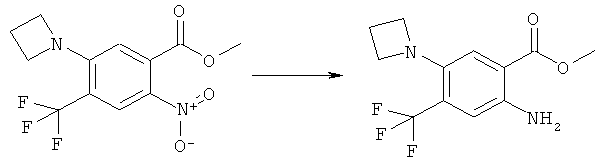
Гидрировали 1,10 г метилового эфира 5-азетидин-1-ил-2-нитро-4-трифторметилбензойной кислоты в 12 мл тетрагидрофурана в присутствии 200 г палладия на угле. После отделения катализатора фильтрованием раствор упаривали досуха, получая 0,99 г метилового эфира 2-амино-5-азетидин-1-ил-4-трифторметилбензойной кислоты в виде порошка желтого цвета, tпл 80-86°С.
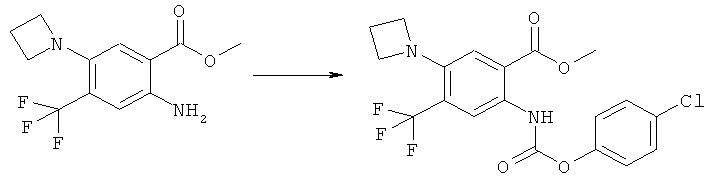
Смесь 129 мг (0,470 ммоля) метилового эфира 2-амино-5-азетидин-1-ил-4-трифторметилбензойной кислоты, 0,082 мл (0,470 ммоля) этилдиизопропиламина, 0,066 мл (0,470 ммоля) 4-хлорформилхлорформиата и 4 мл диоксана перемешивали в течение 45 мин и затем концентрировали досуха. Остаток распределяли между водой и этилацетатом, органическую фазу отделяли и промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали, оставляя вязкую массу, которую подвергали хроматографии среднего давления на диоксиде кремния со смесью этилацетат/циклогексан (1:4). Из первой фракции после упаривания получали 123 мг метилового эфира 5-азетидин-1-ил-2-(4-хлорфеноксикарбониламино)-4-трифторметилбензойной кислоты в виде пены желтого цвета. 1Н-ЯМР (ДМСО-d6+D2O): 7,20 d, J=10 Гц, 2H; 7,20 s, 1H; 7,15 s, 1H; 6,75 d, J=10 Гц, 2Н; 3,85 s, 3Н; 3,75 t, J=7 Гц, 4H; 2,15 pent, J=7 Гц, 2H.

Раствор 115 мг (0,268 ммоля) метилового эфира 5-азетидин-1-ил-2-(4-хлорфеноксикарбониламино)-4-трифторметилбензойной кислоты, 30 мг (0,268 ммоля) метансульфонилгидразида, 0,070 мл (0,402 ммоля) этилдиизопропиламина и 3 мл диоксана перемешивали в течение 24 ч при 70°С. После упаривания досуха остаток фракционировали хроматографией среднего давления на колонке с обращенной фазой RP-18 (размер частиц 20 мкм) со смесью ацетонитрил/вода (3:4). Фракции, содержащие продукт, объединяли и экстрагировали этилацетатом, органическую фазу сушили над сульфатом натрия и концентрировали, оставляя аморфный порошок, который кристаллизовали из смеси ацетонитрил/вода, получая 47 мг N-(6-азетидин-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в виде порошка желтого цвета, tпл 266-283°C.
Пример 80
N-[2,4-Диоксо-6-(пиридин-3-илокси)-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
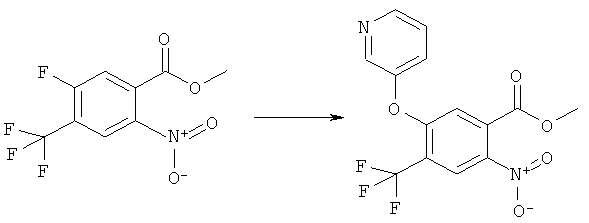
К суспензии 198 мг (4,53 ммоля) гидрида натрия (65% в масле, промыт пентаном) в 2,5 мл безводного тетрагидрофурана прикапывали раствор 356 мг (3,74 ммоля) 3-гидроксипиридина в 5 мл тетрагидрофурана и смесь перемешивали в течение 1 ч при комнатной температуре. После охлаждения до 0°С прибавляли 1,0 г (3,74 ммоля) метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты, растворенного в 10 мл тетрагидрофурана, и перемешивание продолжали в течение 7 ч. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия и упаривали, получая масло, которое фракционировали хроматографией среднего давления на силикагеле со смесью этилацетат/циклогексан (1:1). Фракции, содержащие продукт, объединяли и упаривали, получая 977 мг метилового эфира 2-нитро-5-(пиридин-3-илокси)-4-трифторметилбензойной кислоты в виде порошка белого цвета, tпл 98-100°C.

Гидрировали 919 мг метилового эфира 2-нитро-5-(пиридин-3-илокси)-4-трифторметилбензойной кислоты в 20 мл тетрагидрофурана в присутствии 227 мг палладия на угле. После отделения катализатора фильтрованием раствор упаривали досуха и остаток очищали хроматографией среднего давления на силикагеле со смесью этилацетат/циклогексан (1:2), получая 611 мг метилового эфира 2-амино-5-(пиридин-3-илокси)-4-трифторметилбензойной кислоты в виде порошка желтого цвета, tпл 132-134°С.
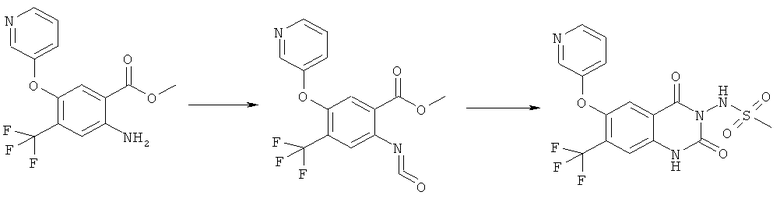
Обрабатывали 300 мг 2-амино-5-(пиридин-3-илокси)-4-трифторметилбензойной кислоты фосгеном в соответствии с методикой, описанной в примере 74, получая 291 мг метилового эфира 2-изоцианато-5-(пиридин-3-илокси)-4-трифторметилбензойной кислоты в виде масла коричневатого цвета. 1Н-ЯМР (CDCl3): 8,50 уш. s, 1Н; 8,40 s, 1H; 7,60 s, 1H; 7,50 s, 3H; 3,95 s, 3H.
Циклизовали 288 мг данного масла с метансульфонилгидразидом в соответствии с методикой, описанной в примере 74, получая 211 мг N-[2,4-диоксо-6-(пиридин-3-илокси)-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида в виде порошка белого цвета, tпл 194-196°С (ацетонитрил).
Пример 81
N-(6-Диметиламино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамид:
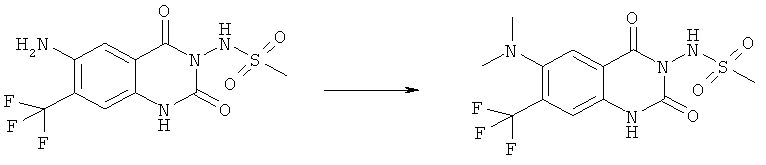
Смесь 370 мг (1,094 ммоля) N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида, 0,083 мл (1,02 ммоля) раствора формальдегида (37% в воде), 0,0033 мл уксусной кислоты, 21 мл тетрагидрофурана и 21 мл воды гидрировали в присутствии 100 мг палладия на угле в течение 10 дней. Через день 1, 3, 4, 5 и 6 прибавляли новую порцию формальдегида (0,083 мл) и уксусной кислоты (0,0033 мл). После фильтрования реакционной смеси тетрагидрофуран упаривали и остающуюся водную фазу экстрагировали этилацетатом. Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали досуха, оставляя порошок желтого цвета, который фракционировали хроматографией среднего давления на колонке с обращенной фазой RP-18 (размер частиц 20 мкм) со смесью ацетонитрил/вода (3:4). Фракции, содержащие продукт, объединяли и экстрагировали этилацетатом, органическую фазу сушили над сульфатом натрия и концентрировали, получая 277 мг N-(6-диметиламино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в виде порошка желтого цвета, tпл 254-272°C.
Аналогичным предыдущему примеру способом получали следующее соединение.
Пример 82
N-[6-(2-Гидроксиэтиламино)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид
Порошок желтого цвета, tпл 240-246°C.
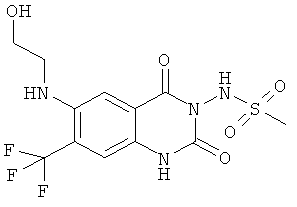
Пример 83
N-{6-[(2-Гидроксиэтил)метиламино]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:

Смесь 370 мг (0,968 ммоля) N-[6-(2-гидроксиэтиламино)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида, 0,074 мл (0,912 ммоля) раствора формальдегида (37% в воде), 4 мл уксусной кислоты, 15 мл тетрагидрофурана и 15 мл воды гидрировали в присутствии 115 мг палладия на угле в течение 3 дней. После дня 1 и 2 прибавляли новую порцию формальдегида (0,074 мл). После фильтрования реакционной смеси тетрагидрофуран упаривали и оставшуюся водную фазу экстрагировали этилацетатом. Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали досуха, оставляя масло коричневого цвета, которое фракционировали хроматографией среднего давления на обращенной фазе RP-18 (размер частиц 20 мкм) со смесью ацетонитрил/вода (1:2). Фракции, содержащие продукт, объединяли и экстрагировали этилацетатом, органическую фазу сушили над сульфатом натрия и концентрировали, получая 165 мг N-{6-[(2-гидроксиэтил)метиламино]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамида в виде порошка желтого цвета, tпл 220-224°С.
Пример 84
N-{6-[4-(4-Метоксифенил)имидазол-1-ил]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:

Смесь 1,200 г (4,49 ммоля) метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты и 0,861 г (4,94 ммоля) 4-(4-метоксифенил)-1H-имидазола в 15 мл тетрагидрофурана нагревали при кипении в течение 5 ч. После охлаждения реакционную смесь распределяли между этилацетатом и водой, органическую фазу отделяли и промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали досуха. Остаток очищали флэш-хроматографией на диоксиде кремния (размер частиц 20 мкм) со смесью этилацетат/гексан (2:3), получая 1,511 г метилового эфира 5-[4-(4-метоксифенил)имидазол-1-ил]-2-нитро-4-трифторметилбензойной кислоты в виде порошка желтого цвета. 1Н-ЯМР (ДМСО-d6, 400 МГц): 3,80 s, 3Н; 3,95 s, 3Н; 7,00 d, J=10,4 Гц, 2H; 7,78 d, J=10,4 Гц, 2H; 7,95 s, 1H; 8,00 s, 1H; 8,28 s, 1H; 8,72 s, 1H.
Гидрировали 1,470 г метилового эфира 5-[4-(4-метоксифенил)имидазол-1-ил]-2-нитро-4-трифторметилбензойной кислоты в 25 мл тетрагидрофурана в присутствии 200 мг палладия на угле. После отделения катализатора фильтрованием раствор упаривали досуха и остаток очищали флэш-хроматографией на диоксиде кремния (размер частиц 40-63 мкм) со смесью дихлорметан/метанол (98:2), получая 1,270 г метилового эфира 2-амино-5-[4-(4-метоксифенил)имидазол-1-ил]-4-трифторметилбензойной кислоты в виде твердого вещества бежевого цвета. 1Н-ЯМР (ДМСО-d6, 400 МГц): 3,80 s, 3Н; 3,95 s, 3Н; 6,95 d, J=10,4 Гц, 2H; 7,30 s, 2H; 7,40 s, 1H; 7,70 s, 1H; 7,75 s, 1H; 7,78 d, J=10,4 Гц, 2H; 7,80 s, 1H.
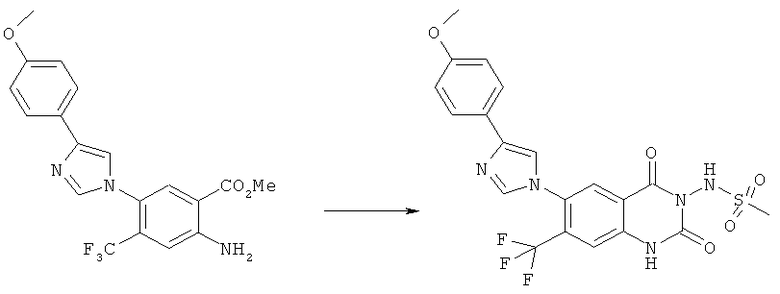
Смесь 0,923 г (2,31 ммоля) метилового эфира 2-амино-5-[4-(4-метоксифенил)-имидазол-1-ил]-4-трифторметилбензойной кислоты и 0,976 мл (6,93 ммоля) триэтиламина обрабатывали 1,04 г (3,47 ммоля) трифосгена и перемешивали при комнатной температуре в течение 90 мин. Прибавляли раствор 0,520 г (4,62 ммоля) метансульфонилгидразида в 20 мл диоксана и продолжали перемешивание при 80°С. После 2 и 3 ч прибавляли две дополнительные порции - 400 мг и 200 мг соответственно метансульфонилгидразида в диоксане. После 4 ч реакционную смесь концентрировали, разбавляли 80 мл смеси диоксан/вода (1:1) и обрабатывали 4,5 мл 1 М раствора гидроксида натрия при комнатной температуре в течение 1 ч. Значение рН реакционной смеси доводили до 5,5 с уксусной кислотой, смесь концентрировали и остаток забирали в этилацетат. Органическую фазу промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия и упаривали досуха. Остаток очищали флэш-хроматографией на диоксиде кремния (размер частиц 40-63 мкм) со смесью дихлорметан/метанол (95:5) и полученный продукт перекристаллизовывали из смеси метанол/дихлорметан и из смеси метанол/дихлорметан/диизопропиловый эфир, получая 0,725 г N-{6-[4-(4-метоксифенил)имидазол-1-ил]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамида в виде порошка белого цвета, tпл 293-294°С.
Пример 85
N-{6-[4-(4-Метоксиметилфенил)имидазол-1-ил]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамид:
К суспензии 3,00 г (7,71 ммоля) 4-бром-1-тритил-1H-имидазола в 30 мл диоксана прибавляли 1,58 г (9,23 ммоля) 4-метоксиметилбороновой кислоты, 3,47 г (10,5 ммоля) карбоната цезия и 0,121 г (0,131 ммоля) трис(дибензилиденацетон)дипалладия, затем 0,315 мл (0,308 ммоля) раствора 5 г три-трет-бутилфосфина в 25 мл диоксана. Смесь нагревали при 80°С и перемешивали в течение 6,5 ч. После охлаждения до комнатной температуры суспензию разбавляли дихлорметаном и фильтровали, осадок на фильтре промывали этилацетатом и фильтрат концентрировали досуха. Остаток очищали флэш-хроматографией на диоксиде кремния (размер частиц 40-63 мкм) со смесью гексан/этилацетат (7:3), получая 2,805 г 4-(4-метоксиметилфенил)-1-тритил-1H-имидазола, Rt=4,659 мин по данным ВЭЖХ на колонке Nucleosil C18HD с элюентом ацетонитрил + 0,05% ТФК/вода + 0,05% ТФК (20:80) до (100:0) в течение 6 мин, скорость потока растворителя 1,0 мл/мин. МС (ионизация при атмосферном давлении (API)-ES, положительная развертка, е/m): 431 (М+1).
Смесь 2,800 г 4-(4-метоксиметилфенил)-1-тритил-1H-имидазола в 50 мл трифторуксусной кислоты перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали, забирали в этилацетат и органическую фазу промывали насыщенным раствором бикарбоната натрия и насыщенным солевым раствором и сушили над сульфатом натрия. Упаривание растворителя дало остаток, который очищали флэш-хроматографией на диоксиде кремния (размер частиц 40-63 мкм) со смесью дихлорметан/метанол (93:7), получая 1,169 г 4-(4-метоксиметил-фенил)-1H-имидазола в виде порошка бежевого цвета. Rt=2,906 мин по данным ВЭЖХ на колонке Nucleosil C18HD с элюентом ацетонитрил + 0,05% ТФК/вода + 0,05% ТФК (20:80) до (100:0) в течение 6 мин, скорость потока растворителя 1,0 мл/мин. МС (API-ES, положительная развертка, е/m): 189 (М+1).

Смесь 1,00 г (3,74 ммоля) метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты и 0,775 г (4,12 ммоля) 4-(4-метоксиметил-фенил)-1H-имидазола в 10 мл тетрагидрофурана нагревали при кипении в течение 2 ч. После охлаждения реакционную смесь распределяли между этилацетатом и водой, органическую фазу отделяли и промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали досуха. Остаток очищали флэш-хроматографией на диоксиде кремния (размер частиц 40-63 мкм) со смесью этилацетат/гексан (2:3), получая 1,55 г метилового эфира 5-[4-(4-метоксиметилфенил)-имидазол-1-ил]-2-нитро-4-трифторметилбензойной кислоты в виде порошка желтого цвета, tпл 135-137°C. 1Н-ЯМР (ДМСО-d6, 400 МГц): 3,30 s, 3Н; 3,95 s, 3Н; 4,42 s, 3Н; 7,35 d, J=10,4 Гц, 2Н; 7,82 d, J=10,4 Гц, 2H; 8,02 s, 1H; 8,05 s, 1H; 8,30 s, 1H; 8,72 s, 1H.
Гидрировали 1,00 г метилового эфира 5-[4-(4-метоксиметилфенил)имидазол-1-ил]-2-нитро-4-трифторметилбензойной кислоты в 20 мл тетрагидрофурана в присутствии 200 мг платины на угле. После отделения катализатора фильтрованием раствор упаривали досуха, получая 0,661 г метилового эфира 2-гидроксиамино-5-[4-(4-метоксиметилфенил)имидазол-1-ил]-4-трифторметилбензойной кислоты в виде твердого вещества желтого цвета. 1Н-ЯМР (ДМСО-d6, 400 МГц): 3,30 s, 3Н; 3,82 s, 3Н; 4,40 s, 2H; 7,35 d, J=10,4 Гц, 2H; 7,62 s, 1H; 7,80 d, J=10,4 Гц, 2H; 7,85-7,90 m, 3Н. МС (API-ES, положительная развертка, е/m): 422 (М+1).
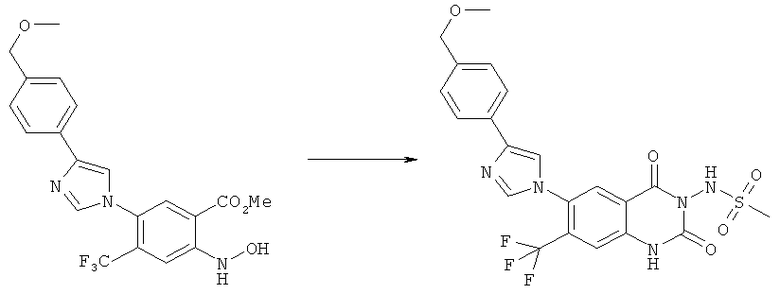
Смесь 0,519 г (1,23 ммоля) метилового эфира 2-гидроксиамино-5-[4-(4-метоксиметилфенил)имидазол-1-ил]-4-трифторметилбензойной кислоты и 0,530 мл (3,76 ммоля) триэтиламина обрабатывали 0,564 г (1,88 ммоля) трифосгена и перемешивали при комнатной температуре в течение 105 мин. Прибавляли раствор 0,282 г (2,51 ммоля) метансульфонилгидразида в 20 мл диоксана и продолжали перемешивание при 80°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли 25 мл воды и обрабатывали 2,5 мл 1 М раствора гидроксида натрия в течение 30 мин. После подкисления 1 М уксусной кислотой до рН 5,5 смесь концентрировали, остаток забирали в этилацетат и органическую фазу промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали досуха. Остаток очищали флэш-хроматографией на диоксиде кремния (размер частиц 40-63 мкм) со смесью дихлорметан/метанол (95:5), получая 0,196 г N-{6-[4-(4-метоксиметилфенил)имидазол-1-ил]-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил}метансульфонамида в виде порошка бежевого цвета, tпл 252-257°С.
Пример 86
N-[2,4-Диоксо-6-(2-оксо-2H-пиридин-1-ил)-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид
Анализы ВЭЖХ осуществляли, используя систему, включающую насосы Gilson 331, соединенные с детектором Gilson UV/VIS 152 и спектрометром Finnigan AQA (ионизация электрораспылением (ESI)), клапаном сброса в петле ввода 50 мкл и ВЭЖХ-колонкой Waters XTerra MS размером 4,6х50 мм со стационарной фазой С 18 и размером частиц 3,5 мкм и проточным градиентом от 5 до 90% ацетонитрила, содержащего 0,05% трифторуксусной кислоты. Времена удерживания (Rt) регистрировали для всех новых соединений.

К суспензии гидрида натрия (315 мг, 1,4 экв) в 50 мл тетрагидрофурана прибавляли по каплям раствор 2-гидроксипиридина (801 мг, 1,5 экв) в 5 мл тетрагидрофурана. Смесь перемешивали при комнатной температуре в течение 30 мин перед прибавлением метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты (1,5 г, 5,61 ммоля) в 10 мл тетрагидрофурана. Образующуюся в результате смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель удаляли в вакууме и неочищенное масло растворяли в этилацетате. Органическую фазу промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия и концентрировали в вакууме, получая неочищенное масло желтого цвета. Неочищенный продукт очищали флэш-хроматографией (этилацетат/гексан (0:100 до 100:0), чтобы получить метиловый эфир 2-нитро-5-(2-оксо-2H-пиридин-1-ил)-4-трифторметилбензойной кислоты в виде твердого вещества желтого цвета (1,3 г, выход 68%). МС (ES, m/z): 328 [М+Н+СН3СN]+, Rt 4,67 мин.
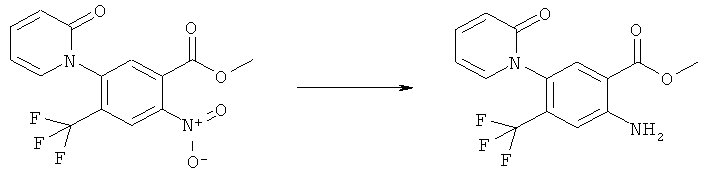
Метиловый эфир 2-нитро-5-(2-оксо-2H-пиридин-1-ил)-4-трифторметилбензойной кислоты (1,3 г, 3,8 ммоля) гидрировали над Ni-Ренея (400 мг) при давлении водорода 3 бара в течение 6 ч. Затем смесь фильтровали через слой целита и промывали его метанолом и диоксаном. Растворитель удаляли в вакууме, получая после высушивания в высоком вакууме метиловый эфир 2-амино-5-(2-оксо-2H-пиридин-1-ил)-4-трифторметилбензойной кислоты в виде твердого вещества белого цвета (1,2 г, 100%). МС (ES, m/z): 313 [М+Н]+, Rt 4,45 мин.
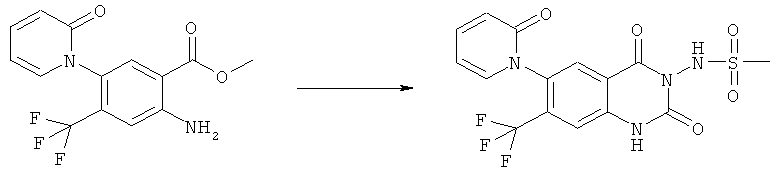
К раствору метилового эфира 2-амино-5-(2-оксо-2H-пиридин-1-ил)-4-трифторметилбензойной кислоты (500 мг, 1,6 ммоля) в 20 мл диоксана прибавляли 4-хлорфенилхлорформиат (0,273 мл, 1,25 экв). Образующуюся в результате смесь перемешивали при 100°С в течение 1 ч. Затем растворитель удаляли в вакууме. Неочищенное масло растворяли в 20 мл диоксана и прибавляли этилдиизопропиламин (0,550 мл, 2 экв) и метансульфонилгидразид (177 мг, 1 экв). Образующуюся в результате смесь перемешивали при 100°С в течение 2 ч. Растворитель удаляли в вакууме досуха и полученный в результате неочищенный остаток растворяли в 10 мл дихлорметана, раствор оставляли при комнатной температуре на 24 ч. Образовавшийся в итоге осадок отделяли фильтрованием, промывали дихлорметаном и сушили в высоком вакууме, получая N-[2,4-диоксо-6-(2-оксо-2H-пиридин-1-ил)-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид в виде твердого вещества белого цвета (100 мг, выход 15%). МС (ES, m/z): 458,3 [М+Н+СН3СN]+, Rt 3,73 мин. 1Н-ЯМР (ДМСО-d6, 400 МГц): 8,02 (s, 1Н), 7,66 (s, 1H), 7,49-7,50 (m, 2H), 6,46 (d, 1H, J=7,8 Гц), 6,31 (t, 1H, J=7,8 Гц), 3,15 (s, 3H).
Пример 87
N-(3-Метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидро-хиназолин-6-ил)-N-метилацетамид
Анализы ВЭЖХ осуществляли, используя систему, включающую насосы Gilson 331, соединенные с детектором Gilson UV/VIS 152 и спектрометром Finnigan AQA (ESI)), клапаном сброса в петле ввода 50 мкл и ВЭЖХ-колонкой Waters XTerra MS размером 4,6×50 мм со стационарной фазой С18 и размером частиц 3,5 мкм и проточным градиентом от 5 до 90% ацетонитрила, содержащего 0,05% трифторуксусной кислоты. Времена удерживания (Rt) регистрировали для всех новых соединений.

Метиловый эфир 5-фтор-2-нитро-4-трифторметилбензойной кислоты (300 мг, 1,12 ммоля) растворяли в диоксане и после прибавления метиламина (0,490 мл, 3,5 экв) желтую смесь перемешивали при комнатной температуре в течение 2 дней. Смесь упаривали досуха. Прибавляли дихлорметан и суспензию фильтровали. Упаривание фильтрата дало метиловый эфир 2-нитро-5-метиламино-4-трифторметилбензойной кислоты в виде твердого вещества желтого цвета (291,3 мг, выход 95,4%). Продукт использовали на следующей стадии без дальнейшей очистки.

Метиловый эфир 2-нитро-5-метиламино-4-трифторметилбензойной кислоты растворяли в смеси метанол/тетрагидрофуран и после прибавления 10% Pd/C смесь перемешивали в течение 45 мин при комнатной температуре в атмосфере водорода. Смесь фильтровали через слой целита, раствор упаривали при пониженном давлении, получая метиловый эфир 2-амино-5-метиламино-4-трифторметилбензойной кислоты в виде масла оранжевого цвета (240 мг, выход 103%). Продукт использовали на следующей стадии без дальнейшей очистки.
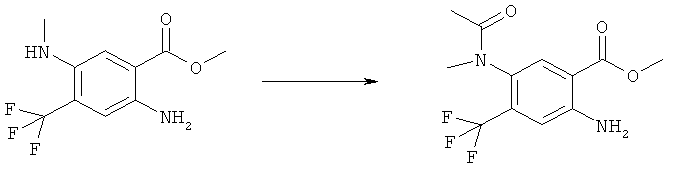
Растворяли метиловый эфир 2-амино-5-метиламино-4-трифторметилбензойной кислоты (26 мг, 0,96 ммоля) в диоксане и после прибавления ацетилхлорида (54 мкл, 1 экв) смесь перемешивали в течение 2 ч (контроль по ТСХ) при 80°С. Затем прибавляли ацетилхлорид (0,9 экв) и перемешивание продолжали при 80°С в течение 18 ч. Смесь упаривали досуха. Остаток растворяли в дихлорметане, раствор фильтровали, упаривали и остаток сушили в высоком вакууме. Неочищенный продукт очищали флэш-хроматографией (силикагель, градиент: гексан до этилацетата), получали метиловый эфир 5-(ацетилметиламино)-2-амино-4-трифторметилбензойной кислоты в виде твердого вещества желтого цвета (36 мг, выход 12,8%) (МН+ 291, Rt 5.0 мин). Выделяли также другое моноацетильное производное (50 мг) и диацетилпроизводное (92 мг).

К раствору метилового эфира 5-(ацетилметиламино)-2-амино-4-трифторметил-бензойной кислоты (36 мг, 0,12 ммоля) в диоксане (0,2 мл) медленно прибавляли 4-хлорфенилхлорформиат (26 мг, 1,1 экв). Раствор перемешивали при 80°С в течение 2 ч. Раствор упаривали, получая метиловый эфир 5-(ацетилметиламино)2-(4-хлорфеноксикарбониламино)-4-трифторметилбензойной кислоты после повторного растирания с гексаном. Использовали такой, какой есть, на следующей стадии.
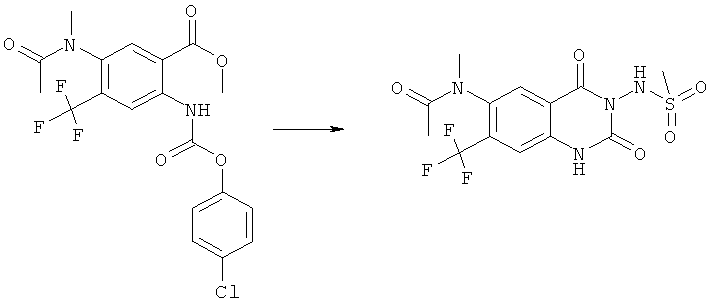
К раствору метилового эфира 5-(ацетилметиламино)2-(4-хлорфенокси-карбониламино)-4-трифторметилбензойной кислоты (55 мг, 0,12 ммоля) в диоксане (0,5 мл) прибавляли метансульфонилгидразид (15 мг, 1,1 экв) и этилдиизопропиламин (0,042 мл, 2 экв). Смесь перемешивали в течение 18 ч (контроль по ТСХ) при 85°С. Смесь стала мутной бело-желтой. Затем смесь упаривали досуха. Остаток растворяли в дихлорметане, твердое вещество отделяли фильтрованием и сушили в высоком вакууме (32,8 мг, выход 67,3%) (MH+ 395, Rt 3,58 мин).
Пример 88
Метиловый эфир 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты:
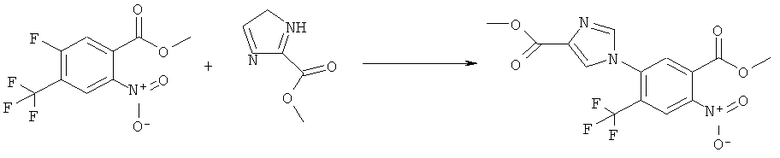
Раствор метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты (1 г, 3,74 ммоля) и метилового эфира 1H-имидазол-4-карбоновой кислоты (0,53 г, 4,12 ммоля) в 10 мл тетрагидрофурана и 2 мл диметилсульфоксида нагревали при кипении в течение 90 ч. Раствору давали охладиться до комнатной температуры и упаривали. Остаток кристаллизовали из дихлорметана и гексана, получая 1,12 г (3 ммоля, 80%) метилового эфира 1-(5-метоксикарбонил-4-нитро-2-трифторметилфенил)-1H-имидазол-4-карбоновой кислоты, tпл 136-138°С. МС (ES, m/z): 374 [М+Н]+.

Раствор 1,1 г (2,95 ммоля) метилового эфира 1-(5-метоксикарбонил-4-нитро-2-трифторметилфенил)-1H-имидазол-4-карбоновой кислоты в 400 мл метанола обрабатывали 163 мг 10% палладия на угле и гидрировали при комнатной температуре под давлением водорода 5 бар в течение 16 ч. После отделения катализатора фильтрованием и упаривания растворителя остаток хроматографировали на силикагеле, используя дихлорметан и увеличивающиеся до 15% количества метанола. Перекристаллизация остатка из дихорметана и гексана дала 932 мг (2,715 ммоля, 92%) метилового эфира 1-(4-амино-5-метоксикарбонил-2-трифторметилфенил)-1H-имидазол-4-карбоновой кислоты, tпл 206-208°С. МС (ES, m/z): 344 [M+H]+.
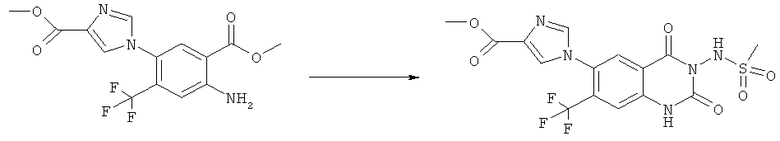
Раствор метилового эфира 1-(4-амино-5-метоксикарбонил-2-трифторметил-фенил)-1H-имидазол-4-карбоновой кислоты (932 мг, 2,715 ммоля) в диоксане (500 мл) обрабатывали трифосгеном (814 мг, 2,715 ммоля) и смесь перемешивали при 80°С в течение 3 ч. Прибавляли метансульфонилгидразид (302 мг, 2,715 ммоля) и перемешивание продолжали в течение 30 мин. После охлаждения до комнатной температуры и концентрации до объема 50 мл прибавляли 3,0 мл 1 М раствора гидроксида натрия, смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали в вакууме и остаток хроматографировали на силикагеле, используя градиент дихлорметана и метанола, получая после перекристаллизации из смеси дихлорметан/гексан 1,035 г (2,31 ммоля, 85%) метилового эфира 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты, tпл 287-288°С. МС (ES, m/z): 448 [M+H]+.
Пример 89
1-(3-Метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидро-хиназолин-6-ил)-1H-имидазол-4-карбоновая кислота:
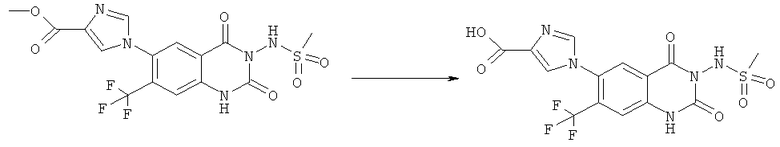
К раствору метилового эфира 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты (150 мг, 0,335 ммоля) в 10 мл диметилформамида прибавляли 1,7 мл 1 М водного раствора гидроксида натрия и перемешивали при комнатной температуре в течение 1 ч и при 50°С в течение 2 ч. После охлаждения до комнатной температуры растворитель упаривали в вакууме, остаток забирали в воду, подкисляли 5 мл 1 М хлористоводородной кислоты и экстрагировали три раза по 15 мл дихлорметана. Органический слой сушили над сульфатом натрия, фильтровали и упаривали, получая 126 мг (0,29 ммоля, 87%) аморфной 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты, МС (ES, m/z): 434 [М+Н]+.
Пример 90
Диметиламид 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты:

Раствор 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты (120 мг, 0,277 ммоля) в 10 мл диметилформамида обрабатывали гидрохлоридом диметиламина (46 мг, 0,554 ммоля), гидрохлоридом N-3-диметиламинопропил-N'-этилкарбодиимида (60 мг, 0,305 ммоля), N-гидроксибензотриазолом (11 мг, 0,08 ммоля) и триэтиламином (0,1 мл, 0,72 ммоля). Раствор нагревали при 100°С в течение 1 ч, охлаждали до комнатной температуры, упаривали в вакууме и остаток хроматографировали на силикагеле, получая 22 мг (0,048 ммоля, 17%) диметиламида 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты, tпл 284-286°С. МС (ES, m/z): 461 [М+Н]+.
Пример 91
Метиламид 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты:
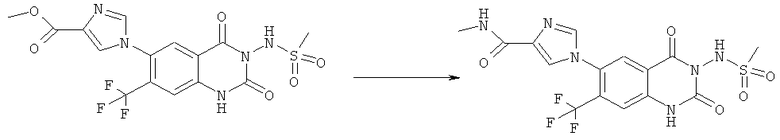
Раствор метилового эфира 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты (800 мг, 1,79 ммоля) и N-метилформамида (0,357 мл, 6 ммолей) в 5 мл диметилформамида нагревали до 120°С и при перемешивании прибавляли метилат натрия (100 мг, 1,79 ммоля). После 2 ч при 120°С прибавляли еще 100 мг метилата натрия и перемешивание при 120°С продолжали в течение дополнительных 2 ч. После охлаждения до комнатной температуры раствор упаривали в вакууме и остаток хроматографировали, получая 560 мг (1,25 ммоля, 70%) метиламида 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты, tпл 292-295°С. МС (ES, m/z): 447 [М+Н]+.
Пример 92
Амид 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидро-хиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты:

Раствор метилового эфира 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты (50 мг, 0,112 ммоля) и формамида (0,15 мл, 0,374 ммоля) в 5 мл диметилформамида нагревали до 120°С и при перемешивании прибавляли метилат натрия (6 мг, 0,112 ммоля). После 2 ч при 120°С раствор охлаждали до комнатной температуры, упаривали в вакууме и остаток хроматографировали, получая 30 мг (0,069 ммоля, 62%) аморфного амида 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты, МС (ES, m/z): 433 [М+Н]+.
Пример 93
N-[6-(4-Гидроксиметилимидазол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
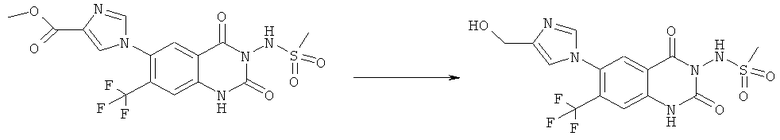
Раствор метилового эфира 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты (300 мг, 0,67 ммоля) в 5 мл смеси (1:1) диоксана и воды обрабатывали боргидридом натрия (40 мг, 1 ммоль) и перемешивали в течение ночи. После упаривания в вакууме остаток очищали препаративной ВЭЖХ, получая 20 мг (0,048 ммоля, 7%) N-[6-(4-гидроксиметилимидазол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида, tпл 225-230°С. МС (ES, m/z): 420 [М+Н]+.
Пример 94
N-[6-(4-Цианоимидазол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
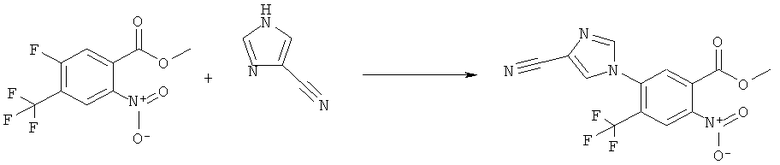
Раствор метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты (3,2 г, 11,98 ммоля), 1H-имидазол-4-карбонитрила (2,017 г, 14,37 ммоля) и этилдиизопропиламина (8,4 мл, 47,9 ммоля) в 10 мл диоксана нагревали при кипении в течение 24 ч. Раствору давали охладиться до комнатной температуры и упаривали. Остаток хроматографировали на силикагеле, используя градиент дихлорметана и метанола, получая 0,36 г (1,05 ммоля, 8,8%) аморфного метилового эфира 5-(4-цианоимидазол-1-ил)-2-нитро-4-трифторметилбензойной кислоты, МС (ES, m/z): 341 [М+Н]+.

Раствор 350 мг (1,03 ммоля) метилового эфира 5-(4-цианоимидазол-1-ил)-2-нитро-4-трифторметилбензойной кислоты в 100 мл метанола обрабатывали 22 мг 10% палладия на угле и гидрировали при комнатной температуре под давлением водорода 1 бар в течение 16 ч. После отделения катализатора фильтрованием остаток хроматографировали на силикагеле, используя дихлорметан и увеличивающиеся до 15% количества метанола, получая 35 мг (0,113 ммоля, 11%) аморфного метилового эфира 2-амино-5-(4-цианоимидазол-1-ил)-4-трифторметилбензойной кислоты, МС (ES, m/z): 311 [М+Н]+.

Раствор метилового эфира 2-амино-5-(4-цианоимидазол-1-ил)-4-трифторметил-бензойной кислоты (35 мг, 0,113 ммоля) в диоксане (25 мл) обрабатывали этилдиизопропиламином (0,5 мл, 2,86 ммоля) и трифосгеном (34 мг, 0,113 ммоля) и смесь перемешивали при 80°С в течение 1 ч. Прибавляли метансульфонилгидразид (13 мг, 0,113 ммоля) и перемешивание продолжали в течение 1 ч. После охлаждения до комнатной температуры раствор концентрировали в вакууме и остаток хроматографировали на силикагеле, получая 20 мг (0,048 ммоля, 42%) N-[6-(4-цианоимидазол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида, tпл 143-145°С. МС (ES, m/z): 415 [M+H]+.
Пример 95
N-[6-(4-Бромимидазол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

Подобно тому, как в предыдущем примере, раствор метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты (1 г, 3,73 ммоля), 4-бромимидазола (0,68 г, 4,5 ммоля) и N-этилдиизопропиламина (2,62 мл, 14,9 ммоля) в 10 мл диоксана нагревали при кипении в течение 16 ч. После соответствующей процедуры обработки получали 1,4 г (3,55 ммоля, 95%) метилового эфира 5-(4-бромимидазол-1-ил)-2-нитро-4-трифторметилбензойной кислоты, tпл 90°С. МС (ES, m/z): 395 [М+Н]+.
Подобным образом раствор метилового эфира 5-(4-бромимидазол-1-ил)-2-нитро-4-трифторметилбензойной кислоты (300 мг, 0,8 ммоля) в 50 мл метанола гидрировали над 43 мг 10% палладия на угле, чтобы получить после обычной процедуры обработки 160 мг (0,466 ммоля, 58%) метилового эфира 2-амино-5-(4-бромимидазол-1-ил)-4-трифторметилбензойной кислоты, tпл 163-165°С. МС (ES, m/z): 365 [М+Н]+.
Подобным образом раствор метилового эфира 2-амино-5-(4-бромимидазол-1-ил)-4-трифторметилбензойной кислоты (160 мг, 0,439 ммоля) и этилдиизопропиламина (0,52 мл, 3 ммоля) в 100 мл диоксана обрабатывали сначала трифосгеном (132 мг, 0,439 ммоля) и затем метансульфонилгидразидом (49 мг, 0,439 ммоля), получая после обычной обработки 95 мг (0,2 ммоля, 46%) N-[6-(4-бромимидазол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида, tпл 229-233°С.МС (ES, m/z): 469 [М+Н]+.
Пример 96
N-[6-(4-Трифторметилимидазол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

Подобным образом раствор метилового эфира 5-фтор-2-нитро-4-трифтометилбензойной кислоты (1,5 г, 5,62 ммоля), 4-трифторметилимидазола (0,945 г, 6,74 ммоля) и этилдиизопропиламина (3,94 мл, 22,5 ммоля) в 10 мл диоксана нагревали при кипении в течение 48 ч. После соответствующей процедуры обработки получали 1,4 г (3,65 ммоля, 65%) аморфного метилового эфира 5-(4-трифторметилимидазол-1-ил)-2-нитро-4-трифторметилбензойной кислоты, МС (ES, m/z): 384 [М+Н]+.
Подобным образом раствор метилового эфира 5-(4-трифторметилимидазол-1-ил)-2-нитро-4-трифторметилбензойной кислоты (1,0 г, 2,6 ммоля) в 100 мл метанола гидрировали над 55 мг 10% палладия на угле, чтобы получить после обычной процедуры обработки 840 мг (2,38 ммоля, 91%) аморфного метилового эфира 2-амино-5-(4-трифторметилимидазол-1-ил)-4-трифторметилбензойной кислоты, МС (ES, m/z): 354 [М+Н]+.
Подобным образом раствор метилового эфира 2-амино-5-(4-трифторметил-имидазол-1-ил)-4-трифторметилбензойной кислоты (840 мг, 2,38 ммоля) и этилдиизопропиламина (4 мл, ммоля) в 100 мл диоксана обрабатывали сначала трифосгеном (706 мг, 2,38 ммоля) и затем метансульфонилгидразидом (262 мг, 2,38 ммоля), получая после обычной обработки 470 мг (1,03 ммоля, 43%) N-[6-(4-трифторметилимидазол-1 -ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида, tпл 144-147°С.МС (ES, m/z): 458 [М+Н]+.
Пример 97
N-(2,4-Диоксо-6-пиррол-1-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамид:

Раствор 80 мг (0,236 ммоля) N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида и 0,092 мл (0,709 ммоля) 2,5-диметокситетра-гидрофурана в 1,5 мл уксусной кислоты нагревали при кипении в течение 2 ч. После упаривания растворителя на роторном испарителе остаток очищали хроматографией на обращенной фазе (С18) с градиентом ацетонитрил-вода и продукт лиофилизировали, получая 54 мг (59%) N-(2,4-диоксо-6-пиррол-1-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в виде твердого вещества коричневого цвета. МС (ES, m/e): 389 [М+Н]+.
Пример 98
N-[6-(3-Формилпиррол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
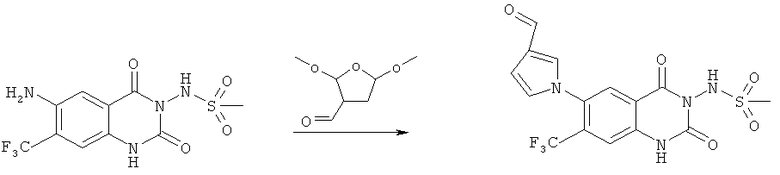
К раствору 100 мг (0,296 ммоля) N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в 2 мл уксусной кислоты прибавляли раствор 2,5-диметокситетрагидрофуран-3-карбальдегида в 1 мл уксусной кислоты и смесь нагревали при кипении в течение 3 ч. Растворитель удаляли упариванием на роторном испарителе и коричневое масло очищали хроматографией на обращенной фазе (С18) с градиентом ацетонитрил-вода, получая 80 мг (65%) N-[6-(3-формилпиррол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида в виде твердого вещества черного цвета. МС (ES, m/e):
417 [М+Н]+.
Пример 99
N-[6-(2-Гидрокси-1-фенилэтиламино)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

К раствору 0,101 мл (0,887 ммоля) оксида стирола в 1 мл ацетонитрила прибавляли 242 мг (1,77 ммоля) хлорида цинка и смесь перемешивали в течение 15 мин. К образующейся в результате белой суспензии прибавляли 50 мг (0,148 ммоля) N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида и смесь перемешивали при 70°С в течение 90 мин. Прибавляли дополнительно 0,017 мл (0,147 ммоля) оксида стирола и смесь перемешивали еще 30 мин при 70°С. Растворитель упаривали на роторном испарителе. Остаток растворяли в этилацетате и промывали водным раствором карбоната калия (1 М) и водным раствором лимонной кислоты (10%). Неочищенный продукт очищали хроматографией на обращенной фазе (С18), используя градиент ацетонитрил-вода. При лиофилизации получали 12 мг N-[6-(2-гидрокси-1-фенилэтиламино)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамида в виде твердого вещества желтоватого цвета. МС (ES, m/e): 459 [М+Н]+.
Пример 100
1-(3-Бензолсульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидро-хиназолин-6-ил)-1H-имидазол-4-карбоновая кислота:

Суспендировали 200 мг (0,58 ммоля) метилового эфира 1-(4-амино-5-метоксикарбонил-2-трифторметилфенил)-1Н-имидазол-4-карбоновой кислоты в 3 мл тетрагидрофурана и прибавляли 209 мг (0,71 ммоля) трифосгена. Десятью минутами позднее прибавляли к суспензии 0,112 мл диизопропилэтиламина. Прозрачный раствор перемешивали в течение дополнительных 2 ч при комнатной температуре и половину растворителя упаривали. Затем через шприц прибавляли раствор 121 мг (0,705 ммоля) бензолсульфонилгидразида в безводном тетрагидрофуране. Образующуюся в результате суспензию перемешивали в течение 20 мин при 60°С, потом обрабатывали 2 мл 1 М водного раствора гидроксида натрия и перемешивали в течение 6 ч при комнатной температуре, чтобы завершить омыление сложного эфира. После упаривания растворителей остаток растворяли в этилацетате, растворитель экстракта сушили, фильтровали и упаривали, получая 1-(3-бензолсульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновую кислоту. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 8,15 (s, 1H, имидазол); 8,10 (s, 1H, имидазол); 7,99 (s, 1H, ароматика); 7,71 (s, 1H, ароматика); ЖХ-МС: 494 [М-Н]-; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; отрицательные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Исходное соединение, метиловый эфир 1-(4-амино-5-метоксикарбонил-2-трифторметилфенил)-1H-имидазол-4-карбоновой кислоты, получали следующим образом:
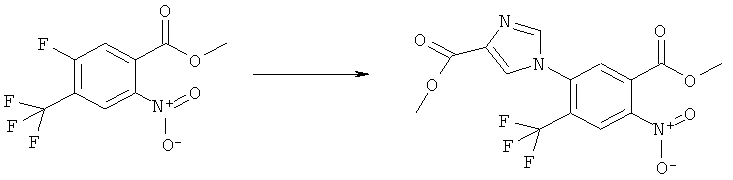
К раствору 8,00 г (29,95 ммоля) метилового эфира 5-фтор-2-нитро-4-трифторметилбензойной кислоты в 40 мл безводного тетрагидрофурана прибавляли 5,40 г (42,00 ммоля) метилового эфира 1H-имидазол-4-карбоновой кислоты. Реакционную смесь перемешивали при 70°С в течение 48 ч (после 16 ч добавление 0,3 эквивалента метилового эфира 1H-имидазол-4-карбоновой кислоты). Затем растворитель упаривали и остаток светло-коричневого цвета экстрагировали дихлорметаном. Объединенные органические экстракты сушили, растворитель упаривали, получая кристаллы светло-пурпурного цвета. Неочищенный продукт очищали флэш-хроматографией (дихлорметан/метанол (100:90/0:10), получая метиловый эфир 1-(5-метоксикарбонил-4-нитро-2-трифторметилфенил)-1H-имидазол-4-карбоновой кислоты. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 8,71 (s, 1H, ароматика); 8,31 (s, 1H, ароматика); 8,27 (s, 1H, имидазол); 8,09 (s, 1H, имидазол); 3,92 (s, 3Н, Ar-СООСН3); 3,80 (s, 3Н, СООСН3); ЖХ-МС: 374 [M+H]+; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; положительные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
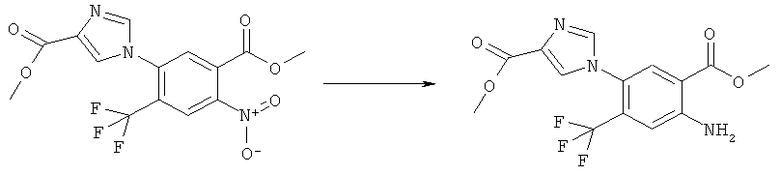
Раствор 4,4 г (11,79 ммоля) метилового эфира 1-(5-метоксикарбонил-4-нитро-2-трифторметилфенил)-1H-имидазол-4-карбоновой кислоты в 400 мл метанола обрабатывали 400 мг 10% палладия на угле и гидрировали при комнатной температуре под давлением водорода 60 фунтов/дюйм2 в течение 2 ч. После отделения катализатора фильтрованием через слой Hyflo SuperCel® и упаривания растворителя получали чистый метиловый эфир 1-(4-амино-5-метоксикарбонил-2-трифторметилфенил)-1H-имидазол-4-карбоновой кислоты в виде твердого вещества белого цвета. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 8,03 (s, 1Н, ароматика); 7,87 (s, 1H, ароматика); 7,78 (s, 1H, имидазол); 7,35 (s, 1H, имидазол); 3,82 (s, 3Н, Ar-СООСН3); 3,77 (s, 3H, СООСН3); ЖХ-МС: 344 [М+Н]+; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; положительные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Пример 101
Метиловый эфир (3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)карбаминовой кислоты:

К раствору 40 мг (0,12 ммоля) N-(2,5-диамино-4-трифторметилбензоил)-метансульфонгидразида в 1 мл тетрагидрофурана и 0,04 мл этилдиизопропиламина прибавляли 0,11 мл (0,20 ммоля) раствора фосгена в толуоле (0,94 г/мл). Реакционную смесь перемешивали при 110°С в микроволновом реакторе в течение 10 мин. Смесь охлаждали до комнатной температуры и выливали в метанол, растворители упаривали, давая твердое вещество желтого цвета. Неочищенный продукт экстрагировали этилацетатом и водой. Органические фракции объединяли, сушили и упаривали, получая метиловый эфир (3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)карбаминовой кислоты, tпл 232,4-240,2°C. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 7,95 (s, 1H, ароматика); 7,51 (s, 1H, ароматика); 3,64 (s, 3Н, СООСН3); 3,15 (s, 3H, SO2CH3). ЖХ-МС: 397 [М+Н]+; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; положительные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Исходное соединение, N-(2,5-диамино-4-трифторметилбензоил)метансульфон-гидразид, получали следующим образом.

К раствору 490 мг (1,85 ммоля) метилового эфира 2-амино-5-нитро-4-трифторметилбензойной кислоты в 5 мл метанола прибавляли 4,7 мл (9,27 ммоля) 2 М раствора гидроксида натрия. Желтый раствор перемешивали при 60°С в течение 12 ч. Затем прибавляли хлористоводородную кислоту (1 М), чтобы довести значение рН до 2. Объем растворителя уменьшали упариванием и остаток экстрагировали этилацетатом. Органические слои объединяли, сушили и растворитель упаривали, получая 2-амино-5-нитро-4-трифторметилбензойную кислоту. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 8,58 (s, 1H, ароматика); 8,10 (s (уш.), 2Н, NH2); 7,35 (s. 1H, ароматика).; ЖХ-МС: 249 [М-Н]-; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6х50 мм, размер частиц 3,5 мкм; отрицательные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.

К раствору 400 мг (1,60 ммоля) 2-амино-5-нитро-4-трифторметилбензойной кислоты в 1 мл диметилформамида прибавляли 363 мг (1,76 ммоля) N,N'-дициклогексилкарбодиимида и 0,21 мл (1,92 ммоля) N-метилморфолина и раствор оставляли перемешиваться при комнатной температуре в течение 10 мин. Затем прибавляли 73 мг (0,48 ммоля) моногидрата 1-гидроксибензотриазола и 705 мг (6,40 ммоля) метансульфонилгидразида и реакционную смесь перемешивали при 40°С в течение 12 ч. Растворители упаривали и желтый осадок экстрагировали этилацетатом и раствором 1 М хлористоводородной кислоты. Органические фракции объединяли, сушили, упаривали и очищали флэш-хроматографией (градиент 0-40% циклогексан/этилацетат), получая N-(2-амино-5-нитро-4-трифторметилбензоил)-метансульфонгидразид, tпл 221,2-226,8°С; 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 8,54 (s, 1H, ароматика); 7,89 (s (уш.), 2Н, NH2); 7,33 (s, 1H, ароматика); 3,02 (s, 3Н, SO2-СН3). ЖХ-МС: 341 [М-Н]-; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; отрицательные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.

К раствору 80 мг (0,23 ммоля) N-(2-амино-5-нитро-4-трифторметилбензоил)-метансульфонгидразида в 1,5 мл концентрированной хлористоводородной кислоты прибавляли 270 мг (1,17 ммоля) дигидрата хлорида олова(II) и смесь перемешивали при комнатной температуре в течение 20 мин. Смесь нейтрализовали при 0°С водным раствором аммиака. Продукт экстрагировали этилацетатом. Объединенные органические фракции сушили, упаривали и очищали флэш-хроматографией (градиент 0-70% циклогексан/этилацетат), получая N-(2,5-диамино-4-трифторметилбензоил)-метансульфонгидразид в виде гидрохлоридной соли, tпл 220,8-225,5°С; 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 6,98 (s, 1H, ароматика); 6,88 (s, 1H, ароматика); 2,98 (s, 3Н, SO2-СН3). ЖХ-МС: 313 [М+H]+; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; положительные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Пример 102
N-[6-(2-Метилпиррол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

К раствору 200 мг (0,59 ммоля) N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в 10 мл уксусной кислоты прибавляли 87 мг (0,60 ммоля) 2-метил-2,5-диметокситетрагидрофурана и реакционную смесь перемешивали при кипении в течение 5 ч. Затем растворитель упаривали и остаток сушили в течение 1 дня при 60°С и высоком вакууме, получая N-[6-(2-метилпиррол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 7,69 (s, 1Н, ароматика); 7,54 (s, 1H, ароматика); 6,6 (s, 1H, N-CH=СН в пирроле); 5,97 (t, 1H, СН=CH=СН в пирроле); 5,83 (m, 1H, СН=CH-С(СН3)-N в пирроле); 3,05 (s, 3Н, SO2-СН3); 1,82 (s, 3H, пиррол-1-CH 3). ЖХ-МС: 403 [М+Н]+; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; положительные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Пример 103
Метиловый эфир 1-(3-бензолсульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты:
Раствор 20 мг (0,04 ммоля) 1-(3-бензолсульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты в 50 мл 6 М раствора хлористоводородной кислоты в метаноле перемешивали при 70°С в течение 2 дней. Каждые полдня прибавляли дополнительные 50 мл 6 М раствора хлористоводородной кислоты в метаноле. Затем растворитель и хлористоводородную кислоту упаривали, получая метиловый эфир 1-(3-бензолсульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-имидазол-4-карбоновой кислоты; 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 8,16 (s, 1H, имидазол); 8,10 (s, 1H, ароматика); 7,98 (s, 1H, имидазол); 7,70 (s, 1H, ароматика); 3,79 (s, 3H, СООСН3). ЖХ-МС: 510 [М+Н]+; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; положительные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Пример 104
1-(3-Метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-пиррол-3-илметиловый эфир уксусной кислоты:

К раствору 130 мг (0,39 ммоля) N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в 5 мл уксусной кислоты прибавляли раствор 95 мг (0,39 ммоля) 2-(2,5-диметокситетрагидрофуран-3-илметокси)тетрагидро-пирана (полученного согласно Frydman, Benjamin; Ojea, Maria I. 1,4-Diaminobutanes from furans: a new synthetic approach to substituted putrescines. Tetrahedron Letters (1998), 39(27), с.с.4765-4768) и реакционную смесь перемешивали при кипении в течение 3 ч. Растворитель упаривали в высоком вакууме, получая 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-пиррол-3-илметиловый эфир уксусной кислоты. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 7,85 (s, 1H, ароматика); 7,62 (s, 1H, ароматика); 7,03 (s, 1H, N-CH=С(СН2) в пирроле); 6,91 (m, 1H, N-CH=CH в пирроле); 6,27 (m, 1H, СН=CH-С(СН2)-N в пирроле); 4,96 (s, 2Н, CH 2-ОСОСН3); 3,16 (s, 3H, SO2-CH3); 2,03 (s, 3H, СН2-OCOCH 3); ЖХ-МС: 459 [М-Н]-; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; отрицательные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Пример 105
N-[6-(3-Гидроксиметилпиррол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:
К раствору 100 мг (0,22 ммоля) 1-(3-метансульфониламино-2,4-диоксо-7-трифторметил-1,2,3,4-тетрагидрохиназолин-6-ил)-1H-пиррол-3-илметилового эфира уксусной кислоты в 0,5 мл метанола прибавляли 36,4 мг (0,26 ммоля) карбоната калия и реакционную смесь перемешивали при 55°С в течение 8 ч. Смесь охлаждали до комнатной температуры и перемешивали еще 12 ч. Затем к реакционной смеси для нейтрализации прибавляли раствор фосфатного буфера, рН 7 и растворители осторожно упаривали (опасность деградации). Неочищенный остаток очищали препаративной тонкослойной хроматографией (дихлорметан/метанол (8:2)), получая N-[6-(3-гидроксиметилпиррол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 7,82 (s, 1Н, ароматика); 7,64 (s, 1H, ароматика); 6,86 (m, 1H, N-СН=CH в пирроле); 6,84 (s, 1H, N-CH=С(СН2) в пирроле); 6,21 (m, 1H, СН=CH-С(СН2)-N в пирроле); 4,37 (d, 2Н, СН2-ОН); 3,16 (s, 3H, SO3-CH3); ЖХ-МС: 417 [М-Н]-; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6х50 мм, размер частиц 3,5 мкм; отрицательные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Пример 106
N-[6-(4-Метил-2-оксо-2,3-дигидропиррол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид:

К раствору 60 мг N-(6-амино-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида в 5 мл уксусной кислоты прибавляли 26 мг (0,18 ммоля) 2,5-диметокси-3-метилтетрагидрофурана (полученного согласно Markwell, Roger Edward; Hadley, Michael Stewart; Blaney, Frank Edward. Azabicycloalkane derivatives and medicaments containing them. Eur. Pat. Appl. (1983) EP 95262 A1). Реакционную смесь перемешивали при кипении в течение 10 ч. Затем растворители упаривали и неочищенный продукт очищали флэш-хроматографией (циклогексан/этилацетат от 100:0 до 20:80), получая N-[6-(4-метил-2-оксо-2,3-дигидропиррол-1-ил)-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил]метансульфонамид. 1Н-ЯМР (ДМСО-d6, 400 МГц, м.д.): 8,06 (s, 1Н, ароматика); 7,60 (s, 1H, ароматика); 7,13 (m, 1H, Н-CH=С(СН3)); 4,24 (s, 2H, СО-CH 2-С(СН3)); 3,16 (s, 3H, SO2-СН3); 1,84 (d, 3H, СН3); ЖХ-МС: 417 [М-Н]-; прибор Agilent LC/MSD серия 1100; ЖХ-МС: колонка SunFireC18, 4,6×50 мм, размер частиц 3,5 мкм; отрицательные ионы; вода/ацетонитрил (95:5 до 5:95 за 5 мин), скорость потока 1,5 мл/мин.
Биологические анализы
Связывание рецептора АМРА
Это может быть продемонстрировано в стандартных тестах, например в тесте связывания [3Н]-6-циано-7-нитрохиноксалин-2,3-диона ([3H]-CNQX) (Ноnоré и др., Biochem. Pharmacol. 1989, 38: с.с.3207-3212). Этот тест проводят следующим образом.
Мембраны мозга. Животных обезглавливают, мозг удаляют и гомогенизируют в 10 объемах охлажденной льдом 10% сахарозы в стеклянном/тефлоновом гомогенизаторе в положениях 5 в течение 30 с. Мембраны центрифугируют при 1000 g в течение 10 мин и супернатант центрифугируют при 20000 g в течение 15 мин. Полученный в итоге осадок вновь суспендируют в 10 объемах холодной воды с гомогенизатором тканей (Brinkman, Polytron) в положении 5 в течение 15 с и суспензию центрифугируют при 8000 g в течение 10 мин. Супернатант, включающий желтый слой, центрифугируют при 40000 g в течение 20 мин, осадок вновь суспендируют в 5 объемах воды и суспензию замораживают (20-30 мин в смеси сухой лед/метанол) и оттаивают (водяная баня при 37°С) дважды. Суспензию центрифугируют при 40000 g в течение 20 мин, осадок вновь суспендируют в 50 мМ HEPES/KOH, рН 7,5, и центрифугируют при 40000 g в течение 10 мин. Конечный осадок вновь суспендируют со стеклянным/тефлоновым гомогенизатором в 5 объемах буфера HEPES/KOH; аликвоты по 2 мл замораживают и хранят в жидком азоте.
Предварительная обработка мембран. Мембраны оттаивают при 35°С и один раз промывают 50 мМ HEPES/KOH путем центрифугирования при 39000 g в течение 10 мин. Конечный осадок вновь суспендируют со стеклянным/тефлоновым гомогенизатором в том же буфере.
Анализ связывания радиолиганда. Он осуществляется с использованием 96-луночных титрационных микропланшетов в объеме, содержащем 0,3 мл 50 мМ HEPES/KOH, рН7,2, 100 мкг мембранного белка, 5нМ [3H]-CNQX (NEN) и соединение, которое должно исследоваться. Инкубацию проводят при 4°С в течение 40 мин и реакцию останавливают центрифугированием (Sigma 4K10) при 3700 g в течение 30 мин. Осадок промывают один раз холодным буфером и затем растворяют в 0,02 мл тканевого солюбилизатора Soluene в течение 20 мин. Прибавляют двести мкл сцинтилляционной жидкости Microscint 20 (Packard) и радиоактивность подсчитывают в сцинтилляционном счетчике Packard Topcount при эффективности 40-45%. Неспецифическое связывание определяется с помощью 10 мкМ CNQX. Анализы проводятся троекратно. Например, в данном анализе соединение из примера 4 имело значение IC50 0,29 мкМ.
Функциональный тест для АМРА-рецепторной активности
Для определения функционального агонизма или антагонизма в отношении рецептора АМРА были осуществлены эксперименты на ооцитах Xenopus, как ранее подробно описано (Urwyler и др., Mol. Pharmacol. 2001, 60, с.с.963-971). Кратко, регистрацию данных двухэлектродного вольт-клампа осуществляли от ооцитов Xenopus laevis, экспрессирующих рецепторы GluR3 АМРА. Плазмиды для крысиной субъединицы GluR3-(flop) (Hollmann и др., Science 1991, 252, с.с.851-853) линеаризуются и транскрибируются в кэппированную кРНК с использованием набора синтеза РНК in vitro (Ambion, Texas) с полимеразой Т7. Исходные растворы содержатся в 70% этаноле. Перед использованием кРНК осаждается и вновь суспендируется в воде, обработанной диэтилпирокарбонатом (DEPC). Ооцитам вводят РНК, кодирующую крысиный рецептор GluR3-(flop) АМРА. Для регистрации ооциты помещают в перфузионную камеру с непрерывным самотеком лягушачьего раствора Рингера. Для регистрации данных от ооцитов, экспрессирующих рецепторы rGluR3-(flop), используется лягушачий раствор Рингера, содержащий Mg2+ (81 мМ NaCl; 2,5 мМ KCl; 1 мМ CaCl2; 1 мМ MgCl2, 2,5 мМ NaHCO3, 5 мМ HEPES, pH 7,4). Исследуемые соединения промываются с самотеком. Например, в данном анализе соединение из примера 4 является антагонистом в отношении рецептора rGluR3 AMPA со значением IC50 2,3 мкМ.
Аудиогенная модель припадков
Соединения по изобретению обладают, например, выраженными антиконвульсивными свойствами, которые определяют in vivo, например, у мышей, со ссылкой на их выраженное защитное действие в отношении конвульсий, вызываемых звуком, электрическим шоком или метразолом. Вызванные звуком припадки выявляются у мышей линии DBA/2 (R.L.Collins в: Experimental models of epilepsy, ред. Pupura, Penry Tower, Woodbury Walter; Raven Press, New York, 1972). Для исследования 20-дневных животных помещают в камеру с ослабленным звуком. После периода привыкания продолжительностью 60 с животных возбуждают, используя узкополосный шум (14-20 кГц, уровень давления звука (SPL) 118 дБ), продолжающийся самое большее 60 с. Мыши DBA/2 отвечают на акустические раздражители последовательностью неконтролируемого бега, клонических припадков, тонизирующих припадков и остановкой дыхания. Для анализа данных измеряется наличие, а также продолжительность различных поведенческих фаз. Вычисляются значения ED50 для различных поведенческих фаз. Значения ED50 после системного применения препаратов (внутрибрюшинного, подкожного, перорального) находятся в диапазоне между 0,5 и 100 мг/кг.
Кроме того, соединения по изобретению проявляют выраженное действие на хорошо разработанной мышиной модели электрического шока или мышиной модели конвульсий, вызванных метразолом, согласно Schmutz и др., Naunyn-Schmiedeberg's Arch. Pharmacol. 1990, 342, с.с.61-66. Значения ED50 находятся в диапазоне между 1 и 200 мг/кг.
Антишизофреническая активность соединений по изобретению может быть продемонстрирована, например, в тесте гиперлокомоции, вызванной амфетамином. Блокада гиперлокомоции, вызванной амфетамином, хорошо известна как разновидность скрининга на антишизофреническую активность.
Кроме того, соединения формулы (I) (упоминаемые также как антагонисты рецептора АМРА) могут быть объединены с другими активными ингредиентами («комбинированный препарат»).
Структура других активных ингредиентов, идентифицированная кодовыми номерами, родовым или торговым названиями, может быть взята из современного издания стандартного справочного издания "The Merck Index" или из баз данных, например, международные патенты (например, IMS World Publications). Соответствующее их содержание тем самым включено путем цитирования. Любой специалист в данной области вполне способен идентифицировать активные ингредиенты и на основании этих ссылок способен также получить их и исследовать фармацевтические показания и свойства на стандартных тест-моделях как in vitro, так и in vivo.
Термин «комбинированный препарат», как он использован в контексте, определяет главным образом «набор частей» в том смысле, что первый и второй активные ингредиенты, как определено выше, могут дозироваться независимо или путем применения различных заданных комбинаций с различающимися количествами ингредиентов, т.е. одновременно или в различные временные точки. Части набора частей затем могут, например, быть введены одновременно или хронологически дифференцировано, то есть в различные временные точки и с равными или различными интервалами времени для любой части набора частей. Весьма предпочтительно выбирать интервалы времени так, чтобы воздействие на подвергающееся лечению заболевание при комбинированном применении частей было больше, чем воздействие, которое могло быть получено при использовании только одного из активных ингредиентов. Отношение общих количеств активного ингредиента 1 к активному ингредиенту 2, которые должны быть введены в комбинированном препарате, может изменяться, например, для того, чтобы удовлетворить запросы субпопуляции пациентов, подвергающихся лечению, или одного пациента, чьи запросы могут отличаться вследствие возраста, пола, массы тела и т.д. Предпочтительно имеется по меньшей мере один благоприятный эффект, например совместное усиление воздействия первого и второго активных ингредиентов, в частности синергизм, например более чем аддитивный эффект, дополнительные благоприятные эффекты, меньше побочных эффектов, комбинированный терапевтический эффект в неэффективной дозе одного или обоих первого и второго активных ингредиентов, и особенно сильный синергизм действия первого и второго активных ингредиентов.
При обсуждении способов будет понятно, что ссылки на активные ингредиенты подразумевают также включение фармацевтически приемлемых солей. Если данные активные ингредиенты имеют, например, по меньшей мере один основный центр, они могут образовать кислотно-аддитивные соли. Соответствующие кислотно-аддитивные соли могут также образоваться, имея при необходимости дополнительно присутствующий основный центр. Активные ингредиенты, имеющие кислотную группу (например, СООН), могут также образовать соли с основаниями. Активный ингредиент или его фармацевтически приемлемая соль могут быть также применены в форме гидрата или могут включать другие растворители, использованные для кристаллизации.
Такие комбинированные препараты обладают благоприятными фармацевтическими эффектами, например, такие препараты проявляют синергизм действия. Комбинированные препараты могут быть использованы по показаниям, перечисленным в данном описании. Изобретение обеспечивает способ применения комбинированных препаратов для предупреждения, лечения, задержки прогрессирования нарушений и заболеваний, названных в данном описании.
Ниже приведены комбинации и применения особой значимости.
Таким образом, в дальнейшем аспекте изобретение относится к комбинации, которая включает по меньшей мере одно соединение формулы (I) («антагонист рецептора АМРА») и по меньшей мере один ноотроп. В такой комбинации активные ингредиенты присутствуют в каждом случае в свободной форме или в форме фармацевтически приемлемой соли, и необязательно присутствует по меньшей мере один фармацевтически приемлемый носитель; комбинация предназначена для одновременного, раздельного или последовательного применения.
Термин «ноотропы», как он использован в контексте, включает, но без ограничения, ноотропные растительные экстракты, антагонисты кальция, ингибиторы холинэстеразы, дигидроэрготоксин, ницерголин, пирацетам, производные пурина, пиритинол, винкамин и винпоцетин. В предпочтительном варианте воплощения изобретения компонентом комбинации является ингибитор холинэстеразы.
Термин «ноотропные растительные экстракты», как он использован в контексте, включает, но без ограничения, экстракты из листьев гинкго. Термин «антагонисты кальция», как он использован в контексте, включает, но без ограничения, циннаризин и нимодипин. Термин «ингибиторы холинэстеразы», как он использован в контексте, включает, но без ограничения, гидрохлорид донеперезила, ривастигмин и гидробромид галантамина. Термин «производные пурина», как он использован в контексте, включает, но без ограничения, пентифиллин.
Экстракты из листьев гинкго могут быть введены, например, в форме, которая реализуется на рынке, например, под торговой маркой гинкодилат™, согласно информации, предусмотренной во вкладыше упаковки. Циннаризин может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой циннаризин форте-ратиофарм™. Нимодипин может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой нимотоп™. Гидрохлорид донеперезила может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой арисепт™. Ривастигмин может быть получен, как раскрыто в патенте US 5602176. Он может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой экселон™. Гидробромид галантамина может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой реминил™. Дигидроэрготоксин может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой гидергин™. Ницерголин может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой сермион™. Пирацетам может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой цереброфорте™. Пентифиллин может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой косалдон™. Пиритинол может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой энцефабол™. Винпоцетин может быть введен, например, в форме, которая реализуется на рынке, например, под торговой маркой кавинтон™.
Структура активных ингредиентов, идентифицированная кодовыми номерами, родовым или торговым названиями, упомянутыми в контексте, может быть взята из современного издания стандартного справочного издания "The Merck Index" или из баз данных, например, международные патенты (например, IMS World Publications). Соответствующее их содержание тем самым включено путем цитирования. Любой специалист в данной области вполне способен идентифицировать активные ингредиенты и на основании этих ссылок способен также получить и исследовать фармацевтические показания и свойства на стандартных тест-моделях как in vitro, так и in vivo.
При обсуждении способов будет понятно, что ссылки на активные ингредиенты подразумевают также включение фармацевтически приемлемых солей. Если данные активные ингредиенты имеют, например, по меньшей мере один основный центр, они могут образовать кислотно-аддитивные соли. Соответствующие кислотно-аддитивные соли могут также образоваться, имея при необходимости дополнительно присутствующий основный центр. Активные ингредиенты, имеющие кислотную группу (например, СООН), могут также образовать соли с основаниями. Активный ингредиент или его фармацевтически приемлемая соль могут быть также применены в форме гидрата или могут включать другие растворители, использованные для кристаллизации.
Когда составные части комбинации, используемые в комбинации по изобретению, применяются в форме, в которой они реализуются на рынке в качестве отдельных препаратов, их дозирование и тип введения могут осуществляться согласно информации, предусмотренной на листке коробки соответствующего реализуемого на рынке препарата для того, чтобы привести в результате к благоприятному эффекту, описанному в контексте, если в нем не указано иначе. В частности, циннаризин может быть введен пациенту в общей ежедневной дозе между примерно 75 мг до примерно 150 мг. Нимодипин может быть введен пациенту в общей ежедневной дозе между примерно 60 мг до примерно 120 мг. Гидрохлорид донепезила может быть введен пациенту в общей ежедневной дозе между примерно 5 мг и 10 мг. Ривастигмин может быть введен пациенту в общей ежедневной дозе между примерно 6 мг до примерно 12 мг. Галантамин может быть введен пациенту в общей ежедневной дозе между примерно 12 мг и 24 мг, например 12 мг дважды в день. Дигидроэрготоксин может быть введен в форме его метансульфоната пациенту в общей ежедневной дозе между примерно 4 мг и 10 мг, например около 8 мг. Ницерголин может быть введен в форме его тартрата внутримышечной инъекцией пациенту в общей ежедневной дозе между примерно 4 мг и 8 мг. Пирацетам может быть введен пациенту в общей ежедневной дозе между примерно 1200 мг и 5000 мг, например 4800 мг/день. Пентифиллин может быть введен пациенту в общей ежедневной дозе между примерно 400 мг и 800 мг. Пиритинол может быть введен в форме его гидрохлорида пациенту в общей ежедневной дозе примерно 600 мг. Винпоцетин может быть введен пациенту в общей ежедневной дозе между примерно 10 мг и 15 мг.
В дальнейшем аспекте изобретение обеспечивает комбинацию, которая включает по меньшей мере один антагонист рецептора АМРА и по меньшей мере одно соединение, выбранное из группы, состоящей из (а) антиэпилептического препарата, выбранного из барбитуратов и их производных, бензодиазепинов, карбоксамидов, гидантоинов, сукцинимидов, валпроевой кислоты и других производных жирных кислот и других антиэпилептических препаратов, и/или (б) обычных антипсихотических агентов, и/или (в) атипичных антипсихотических агентов, эффект которых в комбинированном препарата более чем аддитивный. Более того, комбинации, раскрытые в контексте, могут быть применены для лечения шизофрении, которая устойчива к монотерапии, использующей только один из компонентов комбинации.
Термин «барбитураты и их производные», как он использован в контексте, включает, но без ограничения, фенобарбитал, пентобарбитал, мепобарбитал и примидон. Термин «бензодиазепины», как он использован в контексте, включает, но без ограничения, клоназепам, диазепам и лоразепам. Термин «карбоксамиды», как он использован в контексте, включает, но без ограничения, карбамазепин, окскарбазепин, 10-гидрокси-10,11-дигидрокарбамазепин и соединения формулы (II)
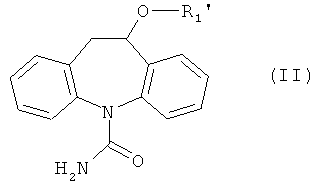
где R1' представляет (С1-С3)алкилкарбонил. Термин «гидантоины», как он использован в контексте, включает, но без ограничения, фенитоин. Термин «сукцинимиды», как он использован в контексте, включает, но без ограничения, этосуксимид, фенсуксимид и мезуксимид. Термин «валпроевая кислота и другие производные жирных кислот», как он использован в контексте, включает, но без ограничения, натриевую соль валпроевой кислоты, моногидрат гидрохлорида тиагабина и виграбатрин. Термин «другие антиэпилептические препараты», как он использован в контексте, включает, но без ограничения, леветирацетам, ламотригин, габапентин, султиам, фелбамат, 1,2,3-1H-триазолы, раскрытые в европейском патенте ЕР 114347, и 2-арил-8-оксодигидро-пурины, раскрытые в международной заявке WO 99/28320.
Термин «обычные антипсихотические препараты», как он использован в контексте, включает, но без ограничения, галоперидол, флуфеназин, тиотиксен и флупентиксол.
Термин «атипичные антипсихотические препараты», как он использован в контексте, относится к клозарилу, рисперидону, оланзапину, кветиапину, зипрасидону и арипипразолу.
Структура активных ингредиентов, идентифицированная кодовыми номерами, родовым или торговым названиями, и их получение могут быть взяты из современного издания стандартного справочного издания "The Merck Index" (например, M.J.O'Nell и др., ред., "The Merck Index", 13-e издание, Merck Research Laboratories, 2001) или из баз данных, например международные патенты (например, IMS World Publications). Соответствующее их содержание тем самым включено путем цитирования. Любой специалист в данной области вполне способен идентифицировать активные ингредиенты и на основании этих ссылок способен также получить их и исследовать фармацевтические показания и свойства на стандартных тест-моделях как in vitro, так и in vivo.
Еще в дальнейшем аспекте изобретение обеспечивает комбинацию, которая включает по меньшей мере один антагонист рецептора АМРА и по меньшей мере одно соединение, выбранное из группы, состоящей из препаратов, препятствующих тревоге, антидепрессантов, антигистаминов, антиконвульсантов, сосудорасширяющих агентов, цинковых солей и анестетиков, эффект которых в комбинированных препаратах более чем аддитивный. Более того, комбинации, раскрытые в контексте, могут применяться для лечения шума в ушах, который устойчив к монотерапии, использующей только один из компонентов комбинации.
Следовательно, изобретение относится к комбинации, такой как комбинированный препарат или фармацевтическая композиция, которая включает по меньшей мере один антагонист рецептора АМРА и по меньшей мере одно соединение, выбранное из группы, состоящей из препаратов, препятствующих тревоге, антидепрессантов, антигистаминов, антиконвульсантов, сосудорасширяющих агентов, цинковых солей и анестетиков, в которой активные ингредиенты в каждом случае присутствуют в свободной форме или в форме фармацевтически приемлемой соли, и включает необязательно по меньшей мере один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного введения.
Термин «антагонисты рецептора АМРА», как он использован в контексте, включает соединения формулы (I).
Термин «препарат, препятствующий тревоге», как он использован в контексте, включает, но без ограничения, алпразолам.
Термин «антидепрессанты», как он использован в контексте, включает, но без ограничения, нортриптилин (N-метил-3-(10,11-дигидро-5H-дибензо[а,d]циклогептен-5-илиден)пропиламин).
Термин «антиконвульсанты», как он использован в контексте, включает, но без ограничения, окскарбазепин.
Термин «анестетики», как он использован в контексте, включает, но без ограничения, лидокаин.
Термин «сосудорасширяющие препараты», как он использован в контексте, включает, но без ограничения, пентоксифиллин.
Термин «цинковые соли», как он использован в контексте, включает, но без ограничения, сульфат цинка.
Топирамат может быть введен, например, в форме, в которой он реализуется на рынке, например, под торговой маркой топамакс™. Соединения формулы (I), а также способы их получения и их композиции известны, например, из международной заявки WO 98/17672. Алпразолам может быть введен, например, в форме, в которой он реализуется на рынке, например, под торговой маркой ксанакс™. Нортриптилин может быть введен, например, в форме, в которой он реализуется на рынке, например, под торговой маркой трилептал™. Окскарбазепин может быть введен, например, в форме, в которой он реализуется на рынке, например, под торговой маркой трилептал™. Лидокаин может быть введен в форме его гидрохлорида, например в форме, в которой он реализуется на рынке в качестве инъекционного раствора, например, под торговой маркой хевенеурал™, сульфат цинка может быть введен, например, в форме, в которой он реализуется на рынке, например, под торговой маркой цинк-сандоз™. Пентоксифиллин может быть введен, например, в форме, в которой он реализуется на рынке, например, под торговой маркой трентал™.
Структура активных ингредиентов, идентифицированная кодовыми номерами, родовым или торговым названиями, может быть взята из современного издания стандартного справочного издания "The Merck Index" или из баз данных, например международные патенты (например, IMS World Publications). Соответствующее их содержание тем самым включено путем цитирования. Любой специалист в данной области вполне способен идентифицировать активные ингредиенты и на основании этих ссылок способен также получить их и исследовать фармацевтические показания и свойства на стандартных тест-моделях как in vitro, так и in vivo.
Еще в дальнейшем аспекте изобретение обеспечивает комбинацию, которая включает соединение формулы (I) и антиэпилептический препарат, выбранный из перечня, состоящего из барбитуратов и их производных, бензодиазепинов, карбоксамидов, гидантоинов, сукцинимидов, валпроевой кислоты и других производных жирных кислот и других антиэпилептических препаратов. Терапевтический эффект такой комбинации выше аддитивного эффекта одного отдельного препарата. Более того, комбинации, раскрытые в контексте, могут быть применены для лечения эпилепсии, которая устойчива к монотерапии, использующей только один из компонентов комбинации.
Следовательно, изобретение относится к комбинации, такой как комбинированный препарат или фармацевтическая композиция, которая включает два антиэпилептических агента, выбранных из перечня, состоящего из барбитуратов и их производных, бензодиазепинов, карбоксамидов, гидантоинов, сукцинимидов, валпроевой кислоты и других производных жирных кислот, антагонистов АМРА и других антиэпилептических препаратов, в которой активные ингредиенты в каждом случае присутствуют в свободной форме или в форме фармацевтически приемлемой соли, и необязательно включает по меньшей мере один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного применения.
Термин «барбитураты и их производные», как он использован в контексте, включает, но без ограничения, фенобарбитал, пентобарбитал, мепобарбитал и примидон. Термин «бензодиазепины», как он использован в контексте, включает, но без ограничения, клоназепам, диазепам и лоразепам. Термин «карбоксамиды», как он использован в контексте, включает, но без ограничения, карбамазепин, окскарбазепин, 10-гидрокси-10,11-дигидрокарбамазепин и соединения формулы (II).
Еще в дальнейшем аспекте изобретение обеспечивает комбинацию, которая включает по меньшей мере одно соединение формулы (I) («антагонист рецептора АМРА») и по меньшей мере одно соединение, выбранное из группы, состоящей из лития, натриевой соли валпроевой кислоты, обычных антипсихотических агентов, атипичных антипсихотических агентов, ламотригина, метилфенидата, антидепрессантов и антиэпилептических агентов, и которая сильнее, чем аддитивный эффект комбинированных препаратов.
Более того, такие комбинации могут быть применены для лечения эмоциональных нарушений и нарушений внимания, которые устойчивы к монотерапии, использующей только один из компонентов комбинации.
Следовательно, изобретение относится к комбинации, такой как комбинированный препарат или фармацевтическая композиция, которая включает по меньшей мере один антагонист рецептора АМРА и по меньшей мере одно соединение, выбранное из группы, состоящей из лития, натриевой соли валпроевой кислоты, обычных антипсихотических агентов, атипичных антипсихотических агентов, ламотригина, метилфенидата, антидепрессантов и антиэпилептических агентов, в которой активные ингредиенты в каждом случае присутствуют в свободной форме или в форме фармацевтически приемлемой соли, и необязательно включает по меньшей мере один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного применения.
Термин «эмоциональные нарушения и нарушения внимания», как он использован в контексте, включает, но без ограничения, биполярное нарушение, например маниакально-депрессивные психозы, манию с психотическими признаками или без них, нарушение внимания и гиперактивность (ADHD) и другие нарушения внимания, например аутизм, а также те поведенческие состояния, которые характеризуются социальной самоизоляцией, например негативные симптомы.
Термин «литий», как он использован в контексте, включает, но без ограничения, ацетат лития, карбонат лития, хлорид лития, цитрат лития и сульфат лития. Термин «обычный антипсихотический агент», как он использован в контексте, включает, но без ограничения, галоперидол и флуфеназин. Термин «атипичный антипсихотический агент», как он использован в контексте, включает, но без ограничения, оланзапин, хетиапин и рисперидон. Термин «антидепрессанты», как он использован в контексте, включает, но без ограничения, трициклические антидепрессанты, селективные ингибиторы повторного поглощения серотонина (SSRI's) или селективные ингибиторы повторного поглощения серотонина и норэпинефрина (SNRI's). Трициклический антидепрессант, пригодный по настоящему изобретению, выбирают главным образом из амитриптилина, бутриптилина, кломипрамина, дезипрамина, дибензепина, дотиепина, доксепина, имипрамина, нортриптилина, опипрамола, протриптилина, тримипрамина, мапротилина, миансерина и миртазепина. Пригодный по настоящему изобретению селективный ингибитор повторного поглощения серотонина выбирают главным образом из флуоксетина, флувоксамина, сертралина, пароксетина, циталопрама и эсциталопрама, а селективный ингибитор повторного поглощения серотонина и норэпинефрина выбирают из венлафаксина и дулоксетина.
Термин «антиэпилептические препараты», как он использован в контексте, включает, но без ограничения, барбитураты и их производные, бензодиазепины, карбоксамиды, гидантоины, сукцинимиды, валпроевую кислоту и другие производные жирных кислот, антагонисты АМРА и другие антиэпилептические препараты, в которых активные ингредиенты в каждом случае присутствуют в свободной форме или в форме фармацевтически приемлемой соли, и необязательно включает один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного применения.
Термин «барбитураты и их производные», как он использован в контексте, включает, но без ограничения, фенобарбитал, пентобарбитал, мепобарбитал и примидон. Термин «бензодиазепины», как он использован в контексте, включает, но без ограничения, клоназепам, диазепам и лоразепам. Термин «карбоксамиды», как он использован в контексте, включает, но без ограничения, карбамазепин, окскарбазепин, 10-гидрокси-10,11-дигидрокарбамазепин и соединения формулы (II).
где R1' представляет (С1-С3)алкилкарбонил. Термин «гидантоины», как он использован в контексте, включает, но без ограничения, фенитоин. Термин «сукцинимиды», как он использован в контексте, включает, но без ограничения, этосуксимид, фенсуксимид и мезуксимид. Термин «валпроевая кислота и другие производные жирных кислот», как он использован в контексте, включает, но без ограничения, натриевую соль валпроевой кислоты, моногидрат гидрохлорида тиагабина и виграбатрин. Термин «другие антиэпилептические препараты», как он использован в контексте, включает, но без ограничения, леветирацетам, ламотригин, габапентин, султиам, фелбамат, 1,2,3-1H-триазолы, раскрытые в европейском патенте ЕР 114347, особенно руфинамид, амид [1-(2,6-дифторбензил)-1H-[1,2,3]триазол-4-карбоновой кислоты, и 2-арил-8-оксодигидропурины, раскрытые в международной заявке WO 99/28320.
Еще в дальнейшем аспекте изобретение обеспечивает комбинацию, которая включает по меньшей мере одно соединение формулы (I) («антагонист рецептора АМРА») и по меньшей мере одно соединение, выбранное из группы, состоящей из бензодиазепинов, селективных ингибиторов повторного поглощения серотонина (SSRIs), селективных ингибиторов повторного поглощения серотонина и норэпинефрина (SNRIs), буспирона и прегабалина, которая сильнее, чем аддитивный эффект препаратов комбинации. Более того, комбинации, раскрытые в контексте, могут быть применены для лечения тревожных расстройств или других психиатрических расстройств с лежащей в основе тревожной симптоматикой, которые устойчивы к монотерапии, использующей только один компонент комбинации.
Следовательно, изобретение относится к комбинации, такой как комбинированный препарат или фармацевтическая композиция, которая включает по меньшей мере один антагонист рецептора АМРА и по меньшей мере одно соединение, выбранное из группы, состоящей из бензодиазепинов, селективных ингибиторов повторного поглощения серотонина (SSRIs), селективных ингибиторов повторного поглощения серотонина и норэпинефрина (SNRIs), буспирона и прегабалина, в которой активные ингредиенты в каждом случае присутствуют в свободной форме или в форме фармацевтически приемлемой соли, и необязательно включает по меньшей мере один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного применения.
Термин «тревожные или другие психиатрические расстройства с лежащей в основе тревожной симптоматикой», как он использован в контексте, включает, но без ограничения, тревожные расстройства, такие как общее тревожное расстройство, социальное тревожное расстройство, посттравматическое стрессовое расстройство, навязчивое компульсивное расстройство, паника и тревога, случающиеся после прекращения приема психостимуляторов, или прием других психотропных препаратов с возможностью неправильного употребления.
Селективный ингибитор повторного поглощения серотонина (SSRI), пригодный по настоящему изобретению, выбирают главным образом из флуоксетина, фувоксамина, сертралина, пароксетина, циталопрама и эсциталопрама.
Селективный ингибитор повторного поглощения серотонина и норэпинефрина (SNRI), пригодный по настоящему изобретению, выбирают главным образом из венлафаксина и дулоксетина.
Термин «бензодиазепины», как он использован в контексте, включает, но без ограничения, клоназепам, диазепам и лоразепам.
Еще в дальнейшем аспекте изобретение обеспечивает комбинацию, которая включает по меньшей мере одно соединение формулы (I) («антагонист рецептора АМРА») и по меньшей мере одно соединение, выбранное из группы, состоящей из пирензепина, телензепина, орто-метоксисилагексоциклиума, γ-аминомасляной кислоты (GABA) и агонистов GABA, комбинацию, которая сильнее, чем аддитивный эффект объединенных препаратов. Более того, комбинации, раскрытые в контексте, могут быть применены для лечения миопии, которая устойчива к монотерапии, использующей только один из компонентов комбинации.
Следовательно, изобретение относится к комбинации, такой как комбинированный препарат или фармацевтическая композиция, которая включает по меньшей мере один антагонист рецептора АМРА и по меньшей мере одно соединение, выбранное из группы, состоящей из пирензепина, телензепина, орто-метоксисила-гексоциклиума, γ-аминомасляной кислоты (GABA) и агонистов GABA, в которой активные ингредиенты в каждом случае присутствуют в свободной форме или в форме фармацевтически приемлемой соли, и необязательно включает по меньшей мере один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного применения.
Топирамат может быть введен, например, в форме, в которой он реализуется на рынке, например, под торговой маркой топамакс™. Соединения формулы (I), а также способ их получения и их фармацевтические композиции известны, например, из международной заявки WO 98/17672.
Пирензепин, телензепин и орто-метоксисилагексоциклиум могут быть применены, как описано в патенте US 5122522.
Термин «γ-аминомасляная кислота (GABA) и агонисты GABA», как он использован в контексте, включает, но без ограничения, соединения, раскрытые в международной заявке WO 03/032975.
Еще в дальнейшем аспекте изобретение обеспечивает комбинацию, которая включает по меньшей мере один антагонист рецептора АМРА и по меньшей мере один компонент комбинации, выбранный из группы, состоящей из ингибиторов циклооксигеназы, антагонистов ваниллоидного рецептора, опиоидов, трициклических антидепрессантов, антиконвульсантов, ингибиторов катепсина S и агонистов рецептора САВАВ, которая сильнее, чем аддитивный эффект объединенных препаратов. Более того, комбинации, раскрытые в контексте, могут быть применены для лечения боли, которая устойчива к монотерапи, использующей только один из компонентов комбинации.
Следовательно, изобретение относится к комбинации, такой как комбинированный препарат или фармацевтическая композиция, которая включает по меньшей мере один антагонист рецептора АМРА и по меньшей мере один компонент комбинации, выбранный из группы, состоящей из ингибиторов циклооксигеназы, антагонистов ваниллоидного рецептора, опиоидов, трициклических антидепрессантов, антиконвульсантов, ингибиторов катепсина S и агонистов рецептора GABAB, в которой активные ингредиенты в каждом случае присутствуют в свободной форме или в форме фармацевтически приемлемой соли, и необязательно включает по меньшей мере один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного применения.
Термин «боль» относится главным образом, но без ограничения, к нервнопатической боли.
Термин «ингибиторы циклооксигеназы», как он использован в контексте, включает, но без ограничения, специфические ингибиторы циклооксигеназы-2 (СОХ-2), например целекоксиб и рофекоксиб, и нестероидные противовоспалительные препараты (NSAIDs), например ацетилсалициловую кислоту и производные пропионовой кислоты.
Термин «трициклические антидепрессанты», как он использован в контексте, включает, но без ограничения, анафранил®, асендин®, авентил®, элавил®, эндеп®, норфранил®, норпрамин®, памелор®, синекван®, сурмонтил®, типрамин®, тофранил®, вивактил® и тофранил-РМ®.
Термин «антиконвульсанты», как он использован в контексте, включает, но без ограничения, окскарбазепин и габапентин. Термин «ингибиторы катепсина S», как он использован в контексте, включает, но без ограничения, соединения, раскрытые в международной заявке WO 03/020287. Термин «агонисты рецептора OABAB», как он использован в контексте, включает, но без ограничения, L-баклофен.
Термин «опиоид», как он использован в контексте, относится ко всем препаратам, как природным, так и синтетическим, с морфиноподобным действием. Опиоид, пригодный по настоящему изобретению, выбирают главным образом из группы, включающей алфентанил, аллилпродин, альфапродин, анилеридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, циклорфан, дезоморфин, декстроморамид, дезоцин, диампромид, дигидрокодеин, дигидроморфин, эптазоцин, этилморфин, фентанил, гидрокодон, гидроморфон, гидроксипетидин, левофенацилморфан, леворфанол, лофентанил, метилморфин, морфин, некоморфин, норметадон, норморфин, опиум, оксикодон, оксиморфон, фолкодин, профадол и суфентанил.
Биологические данные
Анализ
Связывание АМРА-рецептора соединениями по настоящему изобретению (примеры 69 и 73) было оценено, как описано на стр.109-111 описания настоящей заявки.
Результаты:
Биологические испытания in vivo
Анализ
Соединения по настоящему изобретению (примеры 69 и 73) были испытаны на животной модели эпилепсии, так называемый анализ максимального электрошока (тест MES, как описано на стр.111 (строки 24-28) описания настоящей заявки).
Вкратце, соединения по настоящему изобретению были испытаны на мышах OF1 с использованием теста максимального электрошока (тест MES), как подробно описано Schmutz и соавт., Naunyn-Schmiedeberg's Arch Pharmacol, 1990, т.342, стр.61-66. Вызванные клонические судороги задних конечностей индуцированы пропусканием электрического тока посредством временных электродов (%0 Гц, 18 А, 0,2 с). Мыши, обработанные носителем, показали среднюю продолжительность приступов 12-14 с. В качестве позитивного контроля использовали 30 мг/кг карбамазепина; мышей считали защищенными соединением, если продолжительность приступа составляла только 3 с или менее. Для каждых условий обработки использовали по 5 мышей, и в качестве считываемых данных принимался процент защищенных мышей (т.е. соединение могло давать 0%, 20%, 40%, 60%, 80% или 100% защиту).
Результаты:
Соединение по примеру 69 давали в дозе 50 мг/кг, перорально, за 1 час до вызывания судорог. Оно дало 20% защиту.
Соединение по примеру 73 давали дозированно, перорально, за 0,25 часа до вызывания судорог, Значения ED50 (ED - эффективная доза) рассчитывали с помощью GraphPad Prism, v 4,02. Величина ED50 составила 10,7 мг/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ ДИКАРБОНИЛЬНЫЕ ПРОИЗВОДНЫЕ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1995 |
|
RU2145606C1 |
| ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА И ЛЕКАРСТВЕННОЕ СРЕДСТВО, ИХ СОДЕРЖАЩЕЕ | 2000 |
|
RU2259360C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОБЕНЗО[b][1,4]ДИАЗЕПИН-2-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ I mGluR2 | 2002 |
|
RU2270197C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОБЕНЗО[b][1,4]ДИАЗЕПИН-2-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ MGLUR2 II | 2002 |
|
RU2263112C2 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1995 |
|
RU2135484C1 |
| ПРОИЗВОДНЫЕ АМИНОФОСФОНОВЫХ И -ФОСФИНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ | 1997 |
|
RU2181362C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ И КОСМЕТИКЕ | 2015 |
|
RU2712971C2 |
| АМИНОМЕТИЛХИНОЛОНЫ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2629111C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИН-2,4-ДИОНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ РАКА | 2010 |
|
RU2497812C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
Настоящее изобретение относится к новым антагонистам рецептора АМРА-производным 1H-хиназолин-2,4-диона, выбранных из группы: N-(6-имидазол-1-ил-7-нитро-2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида; N-(6-морфолин-4-ил-7-нитро-2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида; N-(7-нитро-2,4-диоксо-6-пиррол-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида; N-(7-нитро-2,4-диоксо-6-[1,2,4]триазол-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида; N-(7-нитро-2,4-диоксо-6-пиразол-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида; N-(7-нитро-2,4-диоксо-6-пирролидин-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида; N-(6-азетидин- 1-ил-7-нитро-2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида; N-(7-нитро-2,4-диоксо-6-[1,2,3]триазол-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида; N-(6-морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида; N-(2,4-диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида; (2,4-диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)амида этансульфоновой кислоты;
N-(6-имидазол-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида; N-(2,4-диоксо-6-тиоморфолин-4-ил-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида; N-(6-[1,4]оксазепан-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида и N-(6-азетидин-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида и их физиологически приемлемых солей. Соединения могут найти применение при лечении таких заболеваний, как эпилепсия или шизофрения. Изобретение также относится к фармацевтической композиции на основе указанных выше соединений. 4 н. и 5 з.п. ф-лы.
1. 1Н-хиназолин-2,4-дион, выбранный из группы, состоящей из
N-(6-Имидазол-1-ил-7-нитро-2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида,
N-(6-Морфолин-4-ил-7-нитро-2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида,
N-(7-Нитро-2,4-диоксо-6-пиррол-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида,
N-(7-Нитро-2,4-диоксо-6-[1,2,4]триазол-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида,
N-(7-Нитро-2,4-диоксо-6-пиразол-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида,
N-(7-Нитро-2,4-диоксо-6-пирролидин-1-ил-1,4-дигидро-2H-хиназолин-3-ил)-метансульфонамида,
N-(6-Азетидин-1-ил-7-нитро-2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида,
N-(7-Нитро-2,4-диоксо-6-[1,2,3]триазол-1-ил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида,
N-(6-Морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида,
N-(2,4-Диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида,
(2,4-Диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)амида этансульфоновой кислоты,
N-(6-Имидазол-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2H-хиназолин-3-ил)метансульфонамида,
N-(2,4-Диоксо-6-тиоморфолин-4-ил-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида.
N-(6-[1,4]Оксазепан-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамида и
N-(6-Азетидин-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамида
и их физиологически приемлемых солей.
2. 1Н-хиназолин-2,4-дион по п.1, который представляет собой N-(6-имидазол-1-ил-7-нитро-2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамид.
3. 1Н-хиназолин-2,4-дион по п.1, который представляет собой N-(6-морфолин-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамид.
4. 1Н-хиназолин-2,4-дион по п.1, который представляет собой N-(2,4-диоксо-6-[1,2,4]триазол-4-ил-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)-метансульфонамид.
5. 1Н-хиназолин-2,4-дион по п.1, который представляет собой N-(6-[1,4]оксазепан-4-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамид.
6. 1Н-хиназолин-2,4-дион по п.1, который представляет собой N-(6-имидазол-1-ил-2,4-диоксо-7-трифторметил-1,4-дигидро-2Н-хиназолин-3-ил)метансульфонамид.
7. Фармацевтическая композиция, обладающая антагонистической активностью в отношении рецептора АМРА, включающая соединение по любому из пп.1-6 в терапевтически эффективном количестве.
8. Применение соединения по любому из пп.1-6 для получения лекарственного средства для предупреждения, лечения или задержки прогрессирования состояния, опосредованного рецептором АМРА.
9. Применение соединения по любому из пп.1-6 для получения лекарственного средства для предупреждения, лечения или задержки прогрессирования эпилепсии или шизофрении.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| ХИНАЗОЛИНОВЫЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2135481C1 |

