Предпосылки создания изобретения
В настоящем изобретении предлагается эффективный, безопасный и коммерчески доступный способ получения 5-(4-метил-1H-имидазол-1-ил)-3-(трифторметил)бензамина следующей формулы (I):
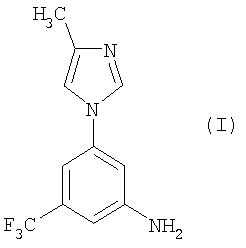 .
.
Соединение формулы (I) является промежуточным соединением для получения замещенных пиримидиниламинобензамидов формулы (II):
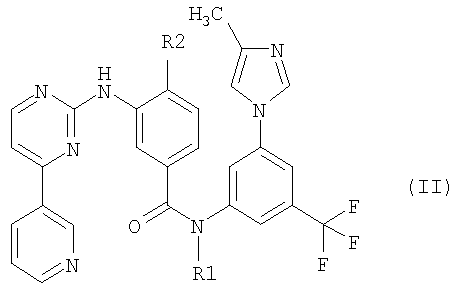
Соединения формулы (II) описаны в публикации W. Breitenstein et al., WO 04/005281 A1, которая включена в текст данной заявки посредством ссылки. Было установлено, что эти соединения ингибируют одну или более из тирозинкиназ, как, например, с-Ab1, Bcr-Ab1, рецепторы тирозинкиназ PDGF-R, Flt3, VEGF-R, EGF-R и c-Kit. Вследствие этого соединения формулы (II) могут быть использованы для лечения некоторых неопластических заболеваний, таких как лейкоз.
Ранее известный метод получения соединения (I) представляет собой четырехстадийный синтез, начиная с реакции ароматического замещения соединения (IIIa), 4-метил-1H-имидазола, соединением (IV), который требует использования высокой температуры (150°С) (схема 1).
Схема 1
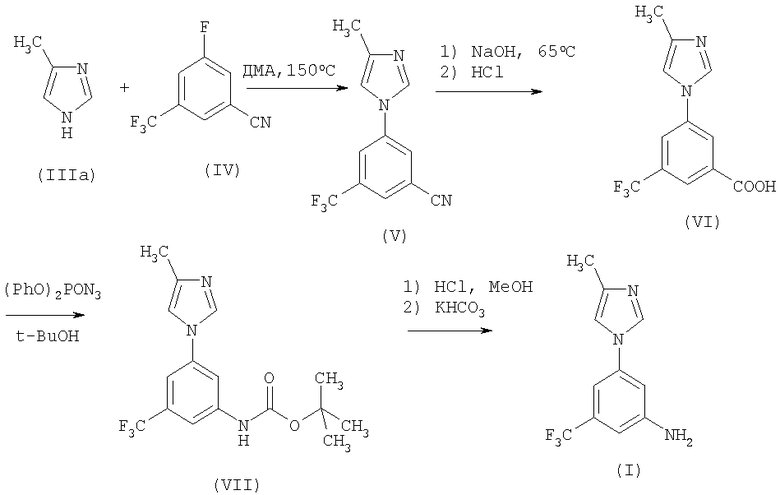
Кроме того, превращение соединения (VI) в соединение (VII) с помощью перегруппировки Курциуса проводится с использованием небезопасного реагента, такого как дифенилфосфорилазид. В результате этой реакции выходы и качественные характеристики продукта не являются постоянными, а удаление дифенилфосфорной кислоты, образующейся в качестве побочного продукта, вызывает трудности. Получаемый карбамат (VII) нуждается в хроматографической очистке, которая является дорогостоящей и длительной по времени процедурой.
Задача настоящего изобретения заключается в предоставлении альтернативных эффективных способов получения соединения формулы (I) с высоким выходом.
Следующим объектом по настоящему изобретению является получение соединения формулы (I) из более дешевых исходных веществ и реагентов.
Еще одним объектом по настоящему изобретению является способ получения соединения формулы (I) с использованием менее опасных реагентов.
Настоящее изобретение преодолевает проблемы синтеза, представленного выше на схеме 1.
Краткое изложение сущности изобретения
Настоящее изобретение предоставляет новые синтетические способы получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина, имеющего формулу (I)
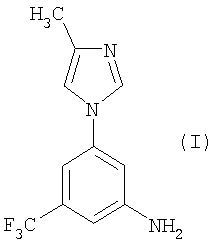
Соединение формулы (I) является промежуточным соединением для получения замещенных пиримидиниламинобензамидов формулы (II), описанных W. Breitenstein et al. в публикации WO 04/005281, опубликованной 15 января 2004 г., которая включена в настоящее описание посредством ссылки. Предпочтительным соединением формулы (II) является 4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]-N-[5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)фенил]бензамид.
Детальное описание изобретения
Общая реакционная схема по изобретению может быть проиллюстрирована посредством следующих вариантов.
В первом варианте осуществления настоящего изобретения предлагается общий способ получения соединения (I) по следующей схеме:
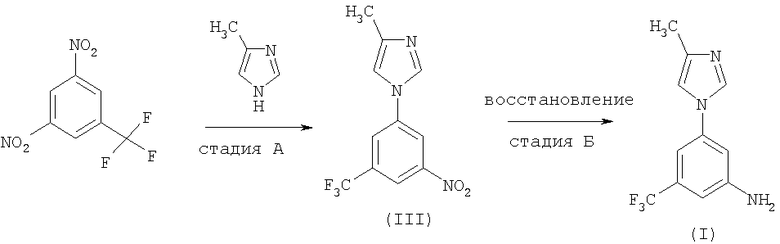
Стадия А включает синтез 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (III) посредством нуклеофильного ароматического замещения в присутствии основания. Стадия Б представляет собой реакцию восстановления, приводящую к соединению (I).
Основание может быть выбрано из группы, включающей алкоксид, гидрид, карбонат или фосфат. Предпочтительно основанием является алкоксид калия, алкоксид натрия, гидрид натрия, карбонат калия или фосфат калия. Растворитель, применяемый на стадии А, выбирают из N,N-диметилформамида (ДМФ), N,N-диметилацетамида (ДМА) или 1-метил-2-пирролидинона (N-МП) или их смесей.
Второй вариант осуществления изобретения включает конденсацию динитробензотрифторида с 4-метил-1Н-имидазолом с последующим гидрированием.
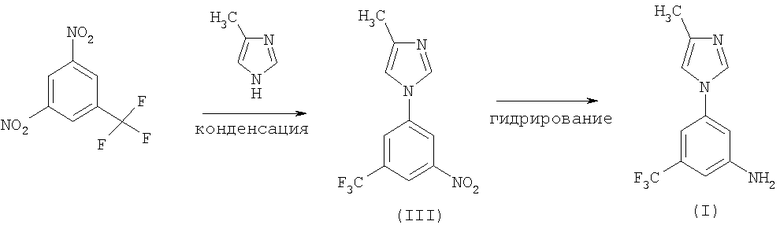
Кроме того, третий вариант включает дополнительную стадию для каждого процесса, описанного выше, необязательно с выделением соединения (III) в виде соли формулы (IV) с целью очистки, как представлено на следующей схеме:
Схема 2
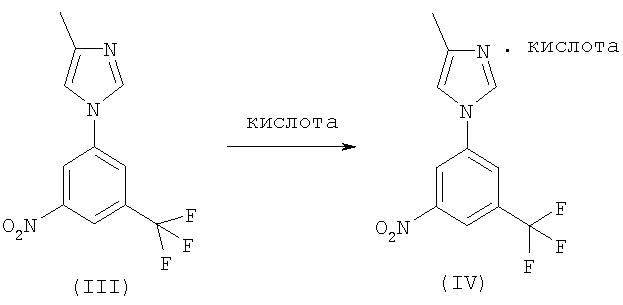
По этой схеме раствор соединения (III) обрабатывают кислотой или ее раствором в воде или органическом растворителе с последующим выделением соли (IV), например, посредством фильтрования.
Соединение (III) может быть затем получено обработкой соли (IV) основанием, предпочтительно водным раствором гидроксида натрия, и выделением свободного основания (III) посредством экстракции или кристаллизации.
Конденсацию проводят в нескольких стандартных растворителях, включая диметилсульфоксид (ДМСО), ДМФ, диглим, ТГФ, N-МП и ДМА.
При создании настоящего изобретения было установлено, что конденсация метилимидазола с динитробензотрифторидом лучше протекает в ДМА в качестве растворителя, в температурном интервале от 80 до 150°С, предпочтительно при температуре от 90 до 140°С. В случае, когда присутствует K2CO3 или другие основания, довольно быстро происходит разложение. Так как реакционная смесь нестабильна, температуру и время реакции снижают, насколько это возможно. Сокращение циклов нагрева и охлаждения, приводящее к уменьшению разложения и более чисто протекающей реакции, достигается с помощью использования микроволнового облучения или дополнительной теплообменной способности обогревающих реакционные сосуды приборов, или посредством использования реакционного оборудования для непрерывного проведения реакции.
K3PO4 проявляет сравнимый с K2CO3 эффект, но реакция во втором случае протекает быстрее. Сырой продукт с выходом >40% может быть получен согласно способу, описанному в данной заявке.
Восстановление нитроимидазольного интермедиата (III) может быть проведено с использованием газообразного водорода или переносящих водород агентов, таких как муравьиная кислота или формиат аммония, в присутствии обычно применяемых для этих целей переходных металлов VIII группы, таких как палладий, платина, никель или их комбинация. Металл наносится на подложку в количестве от 0,1 до 20 мас.% относительно общей массы металла и подложки. Могут быть использованы также комбинированные катализаторы. В пределах объема настоящего изобретения катализатор может также включать промоторы и сопромоторы. Предпочтительным методом восстановления является гидрирование, включающее использование газообразного водорода и палладиевого катализатора. Гидрирование обычно проводят при давлении водорода в интервале от 1 до 20 бар, предпочтительно от 5 до 10 бар. Сырой продукт может быть выделен также в виде хлористоводородной соли. Конечная очистка достигается с помощью кристаллизации с выделением соединения (I) в виде свободного основания.
Следующие примеры служат исключительно для иллюстрации настоящего изобретения, никоим образом не ограничивая его объема.
Пример 1
В сосуд емкостью 200 л загружают 9 кг динитробензотрифторида, 5,3 кг карбоната калия и 84,6 кг ДМА. Через 10 мин энергичного перемешивания до образования темно-красной смеси в нее загружают 3,8 кг 4-метил-1H-имидазола и смесь нагревают при перемешивании при температуре 95°С в течение 15-20 ч до тех пор, пока анализ не подтверждает отсутствие исходных веществ. Темную красно-коричневую смесь охлаждают до температуры 30°С, переносят в воду при энергичном перемешивании, фильтруют и промывают водой, получая около 5 кг сырого продукта в виде темно-коричневого влажного твердого вещества. Анализ указывает на наличие ненужного изомера в соотношении 1:9. Полученное твердое вещество обрабатывают циклогексаном и активированным углем при нагревании, затем смесь осветляют и осадок на фильтре промывают горячим циклогексаном. Объединенные фильтраты охлаждают до комнатной температуры, получая бежевое твердое вещество. Предполагаемый выход 2,6-3,6 кг (25-35%).
Пример 2: гидрирование с использованием Pd/C катализатора
Промежуточное нитросоединение (III) (34,4 г), полученное согласно примеру 1, 1,72 г 5%-ного Pd/C и 217 мл метанола загружают в емкость для гидрирования. Гидрирование проводят при температуре от 70 до 75°С и давлении 4,2-7,5 бар в течение 2 ч. После завершения реакции (согласно анализу с помощью газожидкостной хроматографии) катализатор отфильтровывают и промывают метанолом. Фильтраты объединяют и большую часть растворителя удаляют в вакууме. К твердому остатку прибавляют 174 мл метанола и 526 мл ацетона. После прибавления 17 г безводной хлористоводородной кислоты в осадок выпадает хлористоводородная соль. Суспензию охлаждают до температуры от -10 до -5°С и перемешивают в течение 30 мин. Затем соль отфильтровывают, промывают 58 мл ацетона, к влажной хлористоводородной соли добавляют 319 мл метанола и суспензию нагревают до температуры от 58 до 62°С. После добавления 18 г бикарбоната натрия и 756 г воды раствор фильтруют и охлаждают до температуры 3-7°С. Соединение (I) в виде кристаллического продукта фильтруют, промывают водой и высушивают в вакууме при температуре от 60 до 75°С (выход 19,1 г, 62% от теоретического, чистота >99%).
Пример 3
Следующий процесс включает гидрирование с использованием никеля Ренея в качестве катализатора. Промежуточное нитросоединение (III) (7,5 кг), никель Ренея (0,375 кг) и метанол (32,5 кг) загружают в реакционную емкость. Несколько раз повторяют поочередное продувание азотом и вакуумирование, а затем трижды повторяют поочередное продувание водородом и вакуумирование. После этого давление поднимают до 4 бар и нагревают реакционную смесь до температуры 70°С. Поддерживают давление при 4 бар до прекращения поглощения водорода с последующим дополнительным перемешиванием при этой температуре в течение 2 ч. Сбрасывание давления и отбор пробы осуществляют с помощью нижнего клапана. Если по данным анализа реакция не закончилась, повторяют нагревание при 70°С под давлением газообразного водорода (4 бар) и перемешивают еще в течение часа. После завершения реакции осветляют реакционную смесь, пропуская ее через фильтрационный картридж. Растворитель удаляют посредством вакуумной перегонки (максимально 60°С) и к остатку добавляют толуол (44 кг) и ацетон (121 кг). Затем к полученной смеси добавляют по каплям хлористоводородную кислоту (3,7 кг). Белый твердый остаток центрифугируют и промывают ацетоном. Твердое вещество растворяют в метаноле (55 кг), при температуре 60°С к этому раствору прибавляют раствор бикарбоната натрия (3,95 кг) в воде (165 кг) при температуре ниже 60°С, затем добавляют 0,7 кг угля и перемешивают смесь при температуре 60°С в течение 1 ч. Затем смесь осветляют и охлаждают до температуры 15-20°С. После перемешивания в течение 1 ч при этой температуре смесь центрифугируют и дважды промывают водой. Твердое вещество высушивают до содержания воды ниже 0,5%. Ожидаемое количество составляет 5,5 кг (выход 82,5%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 5-(4-МЕТИЛ-1Н-ИМИДАЗОЛ-1-ИЛ)-3-(ТРИФТОРМЕТИЛ)БЕНЗАМИНА (ВАРИАНТЫ) | 2006 |
|
RU2446162C2 |
| СПОСОБ СИНТЕЗА 5-(МЕТИЛ-1Н-ИМИДАЗОЛ-1-ИЛ)-3-(ТРИФТОРМЕТИЛ)БЕНЗАМИДА | 2006 |
|
RU2444520C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ 2-АМИНОТИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2008 |
|
RU2456285C2 |
| ПРОИЗВОДНЫЕ БЕТА-КАРБОЛИНА, ОБЛАДАЮЩИЕ ДЕЙСТВИЕМ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), СПОСОБ ЕЕ ПОЛУЧЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ ДЕЙСТВИЯ ФОСФОДИЭСТЕРАЗЫ (ВАРИАНТЫ) И СПОСОБ УВЕЛИЧЕНИЯ КОНЦЕНТРАЦИИ ЦГМФ | 2001 |
|
RU2271358C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗ | 2008 |
|
RU2445309C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1993 |
|
RU2130453C1 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ПИРИМИДИНАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ (ЛЕЧЕНИЯ) ОПУХОЛИ | 1994 |
|
RU2135491C1 |
| N-АРИЛАМИДЫ АНТРАНИЛОВОЙ КИСЛОТЫ И ТИОАНТРАНИЛОВОЙ КИСЛОТЫ | 1999 |
|
RU2286338C2 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНДИОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1994 |
|
RU2140420C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА | 2005 |
|
RU2414468C2 |
Изобретение относится к новому способу получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I), заключающийся в том, что:
а) соединение формулы
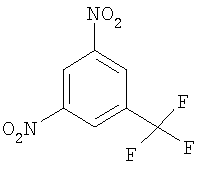
вводят в реакцию конденсации с 4-метил-1Н-имидазолом (IIIa) с получением 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (III) и
б) восстанавливают образовавшийся 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазол до соединения формулы (I).
Изобретение также относится к способу получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I). Технический результат : предлагается эффективный, безопасный и коммерчески доступный способ получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина, являющегося ключевым промежуточным соединением для получения пиримидиниламинобензамидов 2 н. и 8 з.п. ф-лы.
1. Способ получения 5-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)бензамина (I), заключающийся в том, что:
а) соединение формулы
вводят в реакцию конденсации с 4-метил-1H-имидазолом (IIIa) с получением 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (III) и
б) восстанавливают образовавшийся 4-метил- 1-(3-нитро-5-трифторметилфенил)-1Н-имидазол до соединения формулы (I).
2. Способ по п.1, где стадию а) осуществляют в температурном интервале 80 - 150°С.
3. Способ по п.1, где стадию а) осуществляют в температурном интервале 90 - 140°С.
4. Способ по п.1, где реакция восстановления на стадии б) включает применение металла VIII группы в качестве катализатора, газообразного водорода или агента-переносчика водорода.
5. Способ по п.4, где катализатором является палладий, платина или никель Ренея или их комбинации.
6. Способ получения 5-(4-метил-1H-имидазол-1-ил)-3-(трифторметил)бензамина (I), заключающийся в том, что
а) соединение формулы
в присутствии подходящего основания и соответствующего растворителя вводят в реакцию с 4-метил-1H-имидазолом с получением 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазола (III) и
б) образовавшийся 4-метил-1-(3-нитро-5-трифторметилфенил)-1Н-имидазол (III) гидрируют газообразным водородом в присутствии подходящего катализатора в соответствующем растворителе с получением соединения формулы (I).
7. Способ по п.6, где основанием является алкоксид, гидрид, карбонат или фосфат.
8. Способ по п.1 или 6, где стадию а) проводят в полярном апротонном растворителе, выбранном из диметилсульфоксида (ДМСО), диметилформамида (ДМФ), диглима, ТГФ, N-метилпирролидона (N-МП) и диметилацетамида (ДМА).
9. Способ по п.1, где применяют микроволновое излучение.
10. Способ по п.1, где сокращение циклов нагрева и охлаждения для достижения более высокой селективности осуществляют путем использования дополнительной теплообменной способности обогревающих реакционные сосуды приборов или посредством использования реакционного оборудования для непрерывного проведения реакции.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ рафинирования олова от сурьмы и мышьяка | 1976 |
|
SU588762A1 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ПИРИМИДИНАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ (ЛЕЧЕНИЯ) ОПУХОЛИ | 1994 |
|
RU2135491C1 |

