Настоящее изобретение относится к ряду соединений, которые, как было установлено, модулируют пути обмена, определяющие активность циклооксигеназы и простагландинсинтаз, а также синтез простагландинов, и, таким образом, могут быть использованы для лечения и профилактики различных заболеваний и нарушений, которые оказались невосприимчивыми к предшествующим схемам лечения.
Простагландины (PG), определяемые также как простаноиды, представляют собой широко распространенную группу оксигенированных липидов, которые модулируют функции клеток в физиологическом и патологическом отношении. Биосинтез PG происходит в результате высвобождения из клеточных мембран арахидоновой кислоты (АА), главной жирной кислоты, катализируемого фосфолипазой А2 (PLA2). Высвобожденная арахидоновая кислота (АА) под воздействием циклооксигеназы (СОХ) превращается в неустойчивый оксигенированный промежуточный простагландин (PGH2). Образованный промежуточный простагландин PGH2 может быть превращен в разные простагландины, такие как простагландин Е2 (PGE2), простациклин (PGI2), простациклин F2α (PGF2α) или простагландин D2 (PGD2), под воздействием специфических ферментов, именуемых простагландинсинтазами (например, РGE-синтаза или PGE-S, PGD-синтаза или PGD-S и т.д.).
Циклооксигеназа (СОХ) присутствует в трех изоформах: СОХ-1, СОХ-2 и СОХ-3, которая является сплайсированным вариантом СОХ-1. СОХ-1 в основном экспрессирована в большинстве тканей, в то время как индукция СОХ-2 обычно происходит под воздействием провоспалительных цитокинов и стресса. Полученные результаты неоспоримо доказали, что СОХ-2 отвечает за вредное провоспалительное действие простаноидов. Поэтому ингибирование СОХ-2 считается главной мишенью при создании лекарственных средств для лечения воспалительных заболеваний. Однако исследования показали, что СОХ-2 играет также важную роль в гомеостазе основных органов и тканей.
Недавно выполненные клинические исследования показали, что продолжительное лечение ингибиторами СОХ-2 повышает вероятность инсульта и инфаркта миокарда, которые являются осложнениями, возникающими вследствие блокирования вазозащитного действия PGI2, образуемого под воздействием СОХ-2. Однако блокирование ферментов СОХ также уменьшает продуцирование PGD2. В периферических тканях PGD2 стимулирует вазодилатацию и ингибирует агрегацию тромбоцитов. PGD2 является наиболее распространенным простагландином в головном мозге, и недавно выполненное исследование показало, что РGD2 опосредует нейрозащитное действие в нейронах гиппокампа. Поэтому блокирование СОХ-2-образуемого PGD2 может способствовать повышению вероятности инсульта и инфаркта миокарда, наблюдаемому при проведении клинических испытаний с использованием ингибиторов СОХ-2.
PGD2 существует в течение очень короткого периода времени и подвергается дегидратации in vivo и in vitro с образованием биологически активных простагландинов серии J2, включающих 15-дезоксипростагландин J2 (15d-PGJ2). Данный простагландин является природным химически устойчивым противовоспалительным производным РGD2. 15d-PGJ2 является высокоаффинным лигандом рецептора, активируемого пролифератором пероксисомы, (PPAR) подтипа PPARγ, который представляет собой лигандзависимый ядерный фактор транскрипции, участвующий в целом ряде функций клеток, включая противовоспалительное действие. 15d-PGJ2 подавляет несколько воспалительных генов, таких как гены синтазы оксида азота, простагландин Е-синтазы (PGES) и α-фактора некроза опухолей (TNFα), при помощи PPARγ-зависимых и PPARγ-независимых механизмов. В хондроцитах крыс 15d-PGJ2 почти полностью прекращает стимулируемый цитокинами синтез PGE2 и экспрессию PGES, из чего следует, что 15d-PGJ2 является противовоспалительным мессенджером, который выключает продуцирование провоспалительного простагландина PGE2 в указанных клетках. Независимо от действующего механизма 15d-PGJ2 присутствует in vivo в фазе разрешения воспалительного процесса, что также позволяет предположить его функционирование в качестве регулятора обратной связи воспалительной реакции. Кроме того, установлено, что введение 15d-PGJ2 уменьшает развитие экспериментально вызванного колита у крыс, являющегося моделью воспалительного заболевания кишечника. Поэтому следует ожидать, что соединения и условия, которые смещают баланс в сторону продуцирования простагландинов PGD2 и 15d-PGJ2, должны оказывать противовоспалительное действие.
В центральной нервной системе противовоспалительные действия 15d-PGJ2 могут быть благоприятными при лечении нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона и расеянный склероз, а также инсульта, травмы спинного мозга и черепно-мозговой травмы, когда воспаление вызывает поражение головного мозга и гибель клеток. Во всех указанных случаях поражение головного мозга обусловлено чрезмерной активацией микроглии. Микроглия, природные резидентные иммунокомпетентные клетки, играет важную родь в воспалительных процессах, происходящих в центральной нервной системе. Неконтролируемая активация микроглии может оказывать токсическое воздействие на клетки головного мозга вследствие высвобождения разных веществ, таких как воспалительные цитокины (IL-1β, TNF-α, IL-6), NO, PGE2 и супероксид. В результате выполнения нескольких исследований было установлено, что 15d-PGJ2 может подавлять продуцирование воспалительных цитокинов и NO активированными микроглиоцитами, и астроцитами, из чего следует, что простагландины могут играть важную роль в предотвращении поражения головного мозга, обусловленного чрезмерной активацией глиоцитов. Факты, свидетельствующие о том, что введение 15d-PGJ2 до и во время экспериментально вызванного аутоиммунного энцефалита (ЕАЕ), животной модели рассеянного склероза, значительно уменьшает тяжесть ЕАЕ, позволяют далее предположить, что 15d-PGJ2 может эффективно предотвращать поражение головного мозга в случае воспалительных нейродегенеративных заболеваний.
Недавно выполненные исследования показали, что 15d-PGJ2 уменьшает воспаление тканей головного мозга, нарушение поведения и гибель нейронов после внутримозгового кровоизлияния у крыс и защищает головной мозг от реперфузии вследствие ишемии в экспериментальной модели удара. Интравентрикулярное введение 15d-PGJ2 уменьшает объем инфаркта головного мозга, ингибирует апоптоз головного мозга и нейронов, подавляет активацию NF-κВ и повышает уровень гем-оксигеназы-1 (НО-1), сильного эндогенного антиоксиданта, в зависимости от PPARγ. Вышеуказанные результаты, свидетельствующие о нейрозащитном действии 15d-PGJ2 в случае ишемического удара, подтверждены недавно выполненным исследованием, показывающим, что у субъектов, страдающих острым ишемическим ударом, наблюдаются более высокие уровни 15d-PGJ2 в плазме, чем у нормальных субъектов, и что повышенное содержание 15d-PGJ2 в плазме ассоциировано с лучшим неврологическим состоянием на ранней и поздней стадиях заболевания. Повышенное содержание 15d-PGJ2 в плазме было также ассоциировано с меньшим объемом инфаркта, и данный эффект не зависел от других важных прогностических показателей. Поэтому можно предположить, что соединения и условия, смещающие баланс продуцирования простагландинов в сторону 15d-PGJ2, должны оказывать благоприятное воздействие при лечении как хронических воспалительных нейродегенеративных заболеваний, таких как болезнь Альцгеймера (AD), болезнь Паркинсона (PD) и рассеянный склероз, так и острых заболеваний, таких как удар, травма спинного мозга и черепно-мозговая травма.
Болезнь Альцгеймера (AD) является прогрессирующим и смертельным нейродегенеративным заболеванием, характеризующимся отложением внеклеточных бляшек Аβ (β-амилоида) и образованием внутриклеточных сплетений в головном мозге. Бляшки Аβ состоят главным образом из β-амилоида и пептидов Аβ. Накопление Аβ вызывает воспалительную реакцию, которая, как предполагается, способствует патогенезу AD и увеличению поражения нейронов. Кроме того, считается, что повышенные уровни растворимых пептидов Аβ в головном мозге вызывают дисфункцию нейронов и нарушение познавательной способности до отложения бляшек. Поэтому образование пептидов Аβ в головном мозге является главной мишенью при патогенезе AD.
Образование Аβ вызывает протеаза, которая расщепляет более крупный белок-предшественник β-амилоида (βАРР) со стороны N-конца пептида Аβ. Указанная протеаза, именуемая также β-секретазой или АРР-расщепляющим ферментом β-сайта (ВАСЕ1), является трансмембранной аспартилпротеазой. Уровни белка ВАСЕ1 и β-секретазы (С-концевой β-фрагмент) повышены в головном мозге субъектов, страдающих спорадической болезнью Альцгеймера. Поэтому уменьшение продуцирования Аβ в головном мозге субъектов, страдающих болезнью Альцгеймера, является главной терапевтической целью.
В научной литературе сообщалось, что уровни мРНК и белка ВАСЕ1 повышаются под воздействием провоспалительных медиаторов и снижаются лекарственными средствами, которые являются агонистами γ-рецептора, активируемого пролифератором пероксисомы (PPARγ). Недавно было установлено, что уменьшение PPARγ вызывает повышение уровней мРНК β-секретазы в результате увеличения активности промотора гена ВАСЕ1, в то время как сверхэкспрессия PPARγ, а также активаторов PPARγ специфически модулирует транскрипцию ВАСЕ1 путем подавления активности промотора гена ВАСЕ1, из чего следует, что PPARγ может быть ингибитором ВАСЕ1. Кроме того, лечение трансгенных hAPP-мышей агонистами PPARγ снижало уровни как мРНК ВАСЕ1, так и внутриклеточного β-амилоида в нейронах коры головного мозга.
Кроме того, установлено, что сверхэкспрессия PPARγ уменьшает секрецию Аβ в культивируемых клетках в результате активации повсеместного и опосредуемого протеасомой разрушения белка-предшественника βАРР и активация PPARγ непосредственно влияет на устойчивость Аβ, добавляемого в культуру клеток. Такое снижение устойчивости позволяет предположить, что активация PPARγ может вызывать включение механизма быстрого клиренса клеток для амилоидного пептида.
Полученные данные свидетельствуют о значительной роли PPARγ в модуляции образования β-амилоида и наличии защитного механизма, благодаря которому активация PPARγ уменьшает продуцирование пептидов β-амилоида в головном мозге.
15d-PGJ2, высокоаффинный лиганд рецептора, активируемого пролифератором пероксисомы (PPAR) подтипа PPARγ, подавляет несколько воспалительных генов при помощи PPARγ-зависимых и PPARγ-независимых механизмов. Поэтому можно предположить, что условия, при которых увеличивается эндогенное продуцирование 15d-PGJ2, должны подавлять воспалительные процессы в головном мозге и уменьшать образование амилоидного пептида в головном мозге субъектов, страдающих болезнью Альцгеймера. Следовательно, соединения и условия, повышающие эндогенное продуцирование 15d-PGJ2, должны оказывать благоприятное воздействие при лечении болезни Альцгеймера.
Простагландины серии J2 являются сильнодействующими индукторами фактора роста нервов (NGF) и образования фактора роста нервов, выделенного из головного мозга (BDNF), и стимулируют разрастание аксонов под воздействием NGF в культуре клеток. Способность 15d-PGJ2 стимулировать аксоны не связана с PPARγ, так как синтетический агонист и антагонист РPAR-γ не изменяет стимулирующее аксоны действие 15-дезокси-PGJ2. В экспериментах на животных было показано, что введение NGF в желудочки головного мозга защищает холинергические нейроны, стимулирует рост аксонов и улучшает холинергическую функцию. Аналогичным образом, введение NGF в желудочки головного мозга уменьшает гибель нейронов в гиппокампе песчанок, страдающих ишемией головного мозга. Было установлено, что BDNF стимулирует выживание и рост развивающихся нейронов in vitro и улучшает функции двигательных нейронов в животных моделях. У животных, у которых была вызвана ишемия переднего мозга, BDNF уменьшал ишемическое поражение нейронов. К сожалению, из-за плохого проникновения указанных нейротрофинов через гематоэнцефалический барьер клинические испытания не позволили получить значительного эффекта. Однако следует ожидать, что условия или лечение, позволяющие увеличить эндогенные уровни 15d-PGJ2, должны улучшить местное продуцирование указанных факторов роста и стимулировать рост нейронов и, следовательно, облегчить восстановление нейронов в спинном и головном мозге.
Повышенные уровни воспалительных маркеров ассоциированы с ишемией сосудов, и воспаление опосредует патогенез острых синдромов коронарных артерий. Воспаление играет значительную роль в возникновении и развитии атеросклероза, а также может играть главную роль в развитии тромбоза в результате активации процесса коагуляции. Поэтому условия и соединения, уменьшающие воспаление сосудов, могут оказывать благоприятное воздействие на сердечно-сосудистые заболевания, в которых воспаление опосредует гибель или поражение клеток, такие как заболевание коронарной артерии сердца.
мРНК простагландин D-синтазы (PGDS) экспрессирована в сердце. Таким образом, локально продуцируемый PGD2 может вызвать образование 15d-PGJ2 в миоцитах или окружающих клетках. PPARγ также присутствует и функционирует в миоцитах сердца. Недавно выполненное исследование показало, что 15d-PGJ2, природный метаболит PGD2, оказывает противовоспалительное действие в миоцитах сердца, модулируя IL-1β-стимулируемые СОХ-2, РGE-S и iNOS в зависимости от PPAR. 15d-PGJ2 блокировал стимулируемое IL-1β продуцирование PGE2, но не модифицировал стимуляцию IL-1β простагландинов PGI2 или PGF2α, из чего следует, что воздействие лиганда PPARγ является специфическим для PGE-S. Блокирование IL-1β-индуцированного PGE-S под воздействием 15d-PGJ2 предположительно уменьшает продуцирование провоспалительного простагландина PGE2 в ткани сердца. Кроме того, установлено, что 15d-PGJ2 повышает экспрессию гем-оксигеназы-1 (НО-1) в миоцитах сердца и уменьшает величину инфаркта миокарда в животной модели инфаркта миокарда у крыс, вызванного реперфузией при регионарной ишемии. Среди исследованных различных лигандов PPARγ 15d-PGJ2 вызывал наиболее выраженное уменьшение величины инфаркта. Хотя данное действие опосредовано PPARγ, повышенная экспрессия антиоксиданта и цитозащитного белка НО-1 не зависит от PPARγ. В целом полученные результаты позволяют предположить, что условия и соединения, повышающие эндогенные уровни 15d-PGJ2, могут уменьшать воспаление сосудов и, таким образом, оказывать благоприятное воздействие при лечении сердечно-сосудистых заболеваний.
Все больше данных свидетельствуют о том, что жировая ткань является главным источником циркулирующих воспалительных факторов, особенно в случае ожирения. Жир продуцирует провоспалительные адипоцитокины, включающие TNF-α, лептин, PAI-1, IL-6 и ангиотензиноген. TNF-α является главным активатором NFκB. TNF-α ингибирует также передачу сигнала инсулина, вызывая, таким образом, устойчивость к инсулину. Уровни PAI-1 позволяют прогнозировать CAD и диабет и являются основным фактором тромбообразования в случае ожирения. IL-6 стимулирует продуцирование в печени С-реактивного белка (CRP) и способствует повышению уровней высокочувствительного (hs)CRP в сыворотке субъектов, страдающих ожирением. HsCRP позволяет прогнозировать инфаркт миокарда, инсульт, заболевание периферических артерий и внезапную смерть. Ангиотензиноген является предшественником Ang II, который, как хорошо известно, активирует многочисленные механизмы поражения сосудов. Как правило, уровни всех указанных адипокинов повышены у инсулинустойчивых субъектов, страдающих висцеральным ожирением, создающим провоспалительную среду. Поэтому сахарный диабет типа 2 (T2D) и ожирение являются воспалительными состояниями.
Жировая ткань экспрессирует самые высокие уровни PPARγ по сравнению с другими тканями. Лиганды PPARγ стимулируют дифференцировку жировых клеток и поглощение свободных жирных кислот жировой тканью. Указанные лиганды способствуют также ослаблению провоспалительной среды путем уменьшения экспрессии TNF-α, PAI-1 и IL-6 и увеличения экспрессии адипонектина в жире. Таким образом, активация PPARγ непосредственно подавляет воспаление в клетках сосудов и оказывает косвенное воздействие, регулируя экспрессию генов в жировой ткани.
T2D и синдром обмена веществ характеризуются устойчивостью к воздействию инсулина в периферических тканях, включая скелетные мышцы, печень и жировую ткань. Активация PPARγ определенными синтетическими лигандами, такими как тиазолидиндионы, улучшает восприимчивость к инсулину и снижает в крови уровни глюкозы, триглицеридов и свободных жирных кислот, не стимулируя секрецию инсулина, в животных моделях T2D у грызунов. Агонисты PPARγ также снижают устойчивость к инсулину периферических тканей у человека и были эффективно использованы для лечения субъектов, страдающих сахарным диабетом типа 2 (T2D).
15-Дезоксипростагландин J2 (15d-PGJ2) является природным химически устойчивым противовоспалительным простагландином, который, по-видимому, является предполагаемым эндогенным высокоаффинным лигандом рецептора, активируемого пролифератором пероксисомы, подтипа PPARγ. Соединения и условия, которые повышают эндогенное продуцирование 15d-PGJ2, предположительно должны подавлять воспаление сосудов и улучшать восприимчивость к инсулину в случае диабета типа 2.
Провоспалительные цитокины, такие как альфа-фактор некроза опухолей (TNF-α), сверхэкспрессированы в случае псориаза и атопического дерматита. TNF-α играет важную роль в возникновении и развитии воспаления, и недавно полученные экспериментальные данные показывают, что возникновение поражений в экспериментальной модели псориаза опосредовано TNF-α. Полученные данные, позволяющие предположить роль TNF-α в патогенезе псориаза, подтверждены результатами недавно выполненных клинических испытаний, показывающими, что введение моноклонального антитела (mAb) против TNF-α (инфликсимаба) или растворимого гибридного белка рецептора TNF (этанерцепта) вызывает улучшение состояния у больных псориазом.
15d-PGJ2, химически устойчивый метаболит простагландина РGD2, является высокоаффинным лигандом рецептора γ, активируемого пролифератором пероксисомы (РPARγ).
15d-PGJ2 подавляет несколько провоспалительных генов в активированных макрофагах, микроглиоцитах и астроцитах человека, включая гены индуцибельной NO-синтазы (iNOS) и α-фактора некроза опухолей (TNF-α), и указанное подавление по меньшей мере частично зависит от экспрессии PPARγ. Кроме того, установлено, что синтетические лиганды PPARγ, такие как инсулинсенсибилизирующие тиазолидиндионы, улучшают состояние больных псориазом. В целом полученные результаты позволяют предположить, что соединения и условия, повышающие эндогенное продуцирование 15d-PGJ2, могут оказывать эффективное воздействие при лечении псориаза.
Недавно полученные данные показывают, что определенные синтетические агонисты PPARγ обладают умеренной антипролиферативной активностью в отношении многих линий раковых клеток, выделенных из эпителия человека. Кроме того, недавно полученные данные свидетельствуют о том, что нормальные эпителиальные клетки предстательной железы и Т-лимфоциты более устойчивы к апоптозу под воздействием указанных лигандов PPARγ. С учетом вышеуказанного специфического действия в отношении рака возможное использование агонистов PPARγ в качестве химиотерапевтических средств заслуживает серьезного внимания.
Кроме того, установлено, что 15d-PGJ2, природный лиганд PPARγ, обладает противоопухолевым действием. Например, 15d-PGJ2 существенно ингибирует рост клеток и вызывает апоптоз раковых клеток нескольких типов, включая раковые клетки ободочной и прямой кишки, желудка, молочной железы и печени. Механистические исследования позволяют предположить, что указанное ингибирующее рост действие опосредовано механизмами, не зависящими от PPARγ. Однако независимо от действующего механизма соединения и условия, повышающие эндогенные уровни 15d-PGJ2, предположительно должны ингибировать рост и развитие опухоли.
Повышенные уровни простагландина Е2 (PGE2) были обнаружены в различных злокачественных новообразованиях. Различные данные, помимо обнаружения повышенных уровней PGE2 в опухолях, позволяют предположить, что PGE2 играет определенную роль в возникновении и развитии рака. Например, PGE2 может стимулировать пролиферацию и подвижность клеток, ингибируя апоптоз и иммунный надзор. Важно отметить, что PGE2 также может, по меньшей мере частично, индуцировать образование кровеносных сосудов, повышая продуцирование проангиогенных факторов, включая эндотелиальный фактор роста сосудов. С вышеуказанными результатами согласуется тот факт, что более высокие уровни PGE2 в образцах раковой ткани в значительной степени соответствуют возникновению метастазов и повышенной васкуляризации опухоли.
Недавно выполненные исследования с использованием экспериментальных животных также позволяют предположить, что PGE2 может стимулировать канцерогенез. Установлено, что генетическое разрушение рецептора ЕР2 простагландина PGE2 уменьшает число и размер экспериментальных опухолей, при этом другие исследования показали, что лечение моноклональным антителом против PGE2 ингибирует рост трансплантируемых опухолей. Исходя из вышеизложенного, можно ожидать, что соединения и условия, ингибирующие ферментативные пути, вызывающие повышение содержания PGE2 в опухоли, также должны ингибировать рост опухоли.
Синтез PGE2 из арахидоновой кислоты требует двух ферментов, действующих последовательно, циклооксигеназы (СОХ) и простагландин Е-синтазы (PGES). Повышенная экспрессия PGES была обнаружена в нескольких злокачественных новообразованиях человека, из чего с большой вероятностью следует, что аберрантная экспрессия PGES вызывает повышенное продуцирование РGE2, что способствует пролиферации клеток и росту опухоли. Как сообщалось в научной литературе, 15d-PGJ2 почти полностьью ингибирует индуцируемый цитокином синтез PGE2 и экспрессию мембранной PGE-синтазы. Следовательно, соединения и условия, повышающие эндогенные уровни 15d-PGJ2, предположительно должны уменьшать синтез PGE2, а также пролиферацию клеток и рост опухоли и оказывать благоприятное воздействие при лечении рака.
Авторы настоящего изобретения ранее показали, что 7β-гидроксиэпиандростерон (7β-ОН-EPIA), эндогенный 7-гидроксистероид, оба, оказывают нейрозащитное и кардиозащитное действие (WO 02/00224, WO 02/00225 и WO 03/015791). Указанный стероид защищает нервные клетки от гибели in vitro (органотипические культуры среза гиппокампа (OTHSC) и клетки РС12) и головной мозг от поражения в многочисленных экспериментальных моделях in vivo и обеспечивает эффективную защиту от индуцированного регионарной ишемией инфаркта миокарда в перфузированных сердцах крыс. В целом полученные результаты позволяют предположить, что 7β-ОН-EPIA может оказывать благоприятное воздействие в процессе предотвращения и лечения нейродегенеративных заболеваний, таких как удар, травма спинного мозга, черепно-мозговая травма и болезнь Альцгеймера, и сердечно-сосудистых заболеваний, таких как инфаркт миокарда (MI).
Авторы настоящего изобретения недавно показали, что инкубация клеток РС12 с индометацином, ингибитором циклооксигеназы (СОХ), полностью отменяла обеспечиваемую 7β-ОН-EPIA защиту от ишемии. Полученные результаты позволяют предположить, что активность СОХ необходима для нейрозащитного действия 7β-ОН-EPIA.
Авторы настоящего изобретения показали, что инкубация моноцитов клеток крови человека (hMBC) с наномолярными концентрациями 7β-ОН-EPIA вызывает почти 10-кратное увеличение продуцирования простагландина 15-дезокси-Δ12,14-J2 (15d-PGJ2). Подобное действие 7-гидроксистероида, по-видимому, является специфическим для 15d-PGJ2, так как указанный стероид существенно не изменяет продуцирование простагландина Е2 (PGE2) в указанных клетках. В отличие от этого инкубация hMBC с α-фактором некроза опухолей (TNFα) провоспалительного цитокина повышает продуцирование обоих простагландинов примерно в 3 раза. Кроме того, совместная инкубация с TNF-α и 7β-ОН-EPIA вызывает увеличение 15d-PGJ2 аналогично увеличению, наблюдаемому при использовании только 7β-ОН-EPIA, в то время как добавление наномолярных концентраций 7β-ОН-EPIA полностью устраняло увеличение РGE2 под воздействием TNFα.
В научной литературе сообщалось, что 15d-PGJ2 почти полностью ингибирует индуцируемый цитокином синтез PGE2 и экспрессию мембранной PGE-синтазы (mPGES) в хондроцитах крыс, из чего следует, что 15d-PGJ2 является противовоспалительным мессенджером, который выключает продуцирование провоспалительного простагландина PGE2 в указанных клетках. Поэтому полученные результаты могут свидетельствовать о том, что ингибирование TNFα-индуцированного продуцирования провоспалительного простагландина PGE2 под воздействием 7β-ОН-EPIA опосредовано повышенным высвобождением 15d-PGJ2.
15d-PGJ2 является высокоаффинным лигандом рецептора, активируемого пролифератором пероксисомы, (PPAR) подтипа PPARγ, лигандзависимым ядерным фактором транскрипции, участвующим в целом ряде функций клетки, включая противовоспалительное, нейрозащитное, кардиозащитное, метаболическое и противоопухолевое действие. Авторы настоящего изобретения обнаружили, что соединения, такие как 7β-ОН-EPIA, способны избирательно облегчать продуцирование 15d-PGJ2 и, таким образом, могут оказывать благоприятное воздействие в случае воспаления и кожных заболеваний, таких как воспалительное заболевание кишечника и псориаз, в случае неврологических, сердечно-сосудистых заболеваний и нарушений обмена веществ, таких как удар, травма спинного мозга, черепно-мозговая травма, болезнь Альцгеймера, болезнь Паркинсона, заболевание коронарной артерии сердца и диабет типа 2, в которых воспаление вызывает дисфункцию и гибель клеток, и в случае различных типов рака, в которых повышенное продуцирование РGE2 способствует пролиферации клеток и росту опухоли.
Таким образом, одним объектом настоящего изобретения является применение вещества, усиливающего продуцирование 15-дезоксипростагландина J2, для приготовления лекарственного средства, предназначенного для лечения или профилактики состояний, опосредованных повышенными уровнями простагландина Е2 или других метаболитов циклооксигеназы и простагландинсинтазы либо для лечения или профилактики состояний, ухудшение которых обусловлено пониженным уровнем или пониженной доступностью 15-дезоксипростагландина J2.
Другим объектом настоящего изобретения является применение вещества, облегчающего продуцирование 15-дезоксипростагландина J2 и избирательно ингибирующего продуцирование простагландина Е2 в присутствии вызывающего воспаление агента, для приготовления лекарственного средства, предназначенного для лечения или профилактики состояний, опосредованных повышенными уровнями простагландина Е2 или других метаболитов циклооксигеназы-2, либо для лечения или профилактики состояний, ухудшение которых обусловлено пониженным уровнем или пониженной доступностью 15-дезоксипростагландина J2.
Другим объектом настоящего изобретения является применение вещества, усиливающего продуцирование 15-дезоксипростагландина J2, для приготовления лекарственного средства, предназначенного для стимуляции разрастания аксонов или для лечения периферической невропатии.
Периферическая невропатия может возникнуть вследствие лечения химиотерапевтическими средствами, такими как цисплатин, или вследствие других причин, таких как диабетическая невропатия.
Еще одним объектом настоящего изобретения является применение вещества, усиливающего продуцирование 15-дезоксипростагландина J2 и активирующего PPAR-гамма, для приготовления лекарственного средства, предназначенного для лечения состояния, требующего активации PPAR-гамма.
Другим объектом настоящего изобретения является применение соединения, усиливающего продуцирование 15-дезоксипростагландина J2, для приготовления лекарственного средства, предназначенного для лечения или профилактики рака.
Еще одним объектом настоящего изобретения является применение соединения, усиливающего продуцирование 15-дезоксипростагландина J2, для приготовления лекарственного средства, предназначенного для ингибирования пролиферации раковых клеток, индукции апоптоза раковых клеток или торможения роста и развития опухоли.
Раковые клетки, к которым в наибольшей степени относится данный объект настоящего изобретения, включают раковые клетки ободочной и прямой кишки, желудка, молочной железы, печени, предстательной железы, мочевого пузыря, щитовидной железы и пищевода.
Кроме того, соединения по настоящему изобретению могут быть использованы для лечения и профилактики:
боли, ассоциированной с воспалением;
заболеваний периферических артерий (включая нарушения, препятствующие притоку крови к конечностям или вызванные периферическим атеросклерозом) и их осложнений, таких как критическая ишемия конечностей;
заболевания коронарной артерии и его осложнений, таких как ишемическая болезнь сердца (например, стабильная и нестабильная стенокардия) и инфаркт миокарда (MI);
сердечно-сосудистых заболеваний и их осложнений, таких как удар и преходящее ишемическое нарушение мозгового кровообращения (TIA);
ишемии печени и почки, например, атеросклеротического стеноза почечной артерии;
заболеваний обмена веществ, таких как диабет типа 2 и его осложения, такие как заболевания периферических артерий, заболевание коронарной артерии, сосудистые заболевания почки и диабетическая невропатия;
ожирения и его осложнений, таких как диабет типа 2, заболевания периферических артерий и заболевание коронарной артерии;
воспалительных заболеваний дыхательных путей, таких как астма и хроническое обструктивное заболевание легких (COPD), например, хронический бронхит;
хронических нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона, рассеянный склероз и периферические невропатии;
острых неврологических дегенеративных состояний, таких как черепно-мозговая травма и травма спинного мозга;
воспалительного заболевания кишечника;
воспалительных заболеваний, характеризующихся дегенерацией суставного хряща, таких как ревматоидный артрит, первичный и вторичный остеоартрит, и их осложений;
ран; и
токсичности или периферических невропатий, вызванных химиотерапевтическими средствами.
Указанные вещества могут быть предпочтительно использованы для лечения сахарного диабета и его осложнений; ишемических заболеваний сосудов; боли, ассоциированной с воспалением; воспалительных заболеваний кожи; травмы спинного мозга; периферической невропатии; рассеянного склероза; воспалительного заболевания кишечника; ревматоидного артрита; синдрома обмена веществ Х, ожирения, акромегалии и заживления ран.
Такие вещества могут быть также пригодны для лечения или профилактики таких состояний, как воспалительные заболевания периферических органов, например, печени и почек, вызванных, например, ишемией печени и почки.
Другие воспалительные состояния, которые можно лечить указанными веществами, включают воспалительные заболевания дыхательных путей, такие как астма, ринит, бронхит и хроническое обструктивное заболевание легких (COPD).
Настоящее изобретение иллюстрировано прилагаемыми чертежами, на которых 7β-гидроксиэпиандростерон определяется как 7β-ОН-EPIA.
На фиг.1 представлены объединенные данные среднего процентного значения гибели клеток, полученные в результате выполнения 4 отдельных экспериментов в примере 1, которые выражены в виде среднего±стандартная ошибка среднего.
На фиг.2 показано среднее процентное значение гибели клеток в отдельных экспериментах, описанных в примере 1.
На фиг.3 показано среднее процентное значение гибели клеток в отдельных экспериментах, описанных в примере 1.
На фиг.4 показано среднее процентное значение гибели клеток в отдельных экспериментах, описанных в примере 1.
На фиг.5(а) и 5(b) показано воздействие повышения концентраций 7β-ОН-EPIA на уровни простагландина PGD2, обнаруженного в супернатантах мононуклеарных клеток периферической крови, инкубированных с 7β-ОН-EPIA, в присутствии и в отсутствие TNF-α, как описано в примере 2.
На фиг.6(а) и 6(b) показано воздействие повышения концентраций 7β-ОН-EPIA на уровни простагландина PGE2, обнаруженного в супернатантах мононуклеарных клеток периферической крови, инкубированных с 7β-ОН-EPIA, в присутствии и в отсутствие TNF-α, как описано в примере 2.
На фиг.7(а) и 7(b) показано воздействие повышения концентраций 7β-ОН-EPIA на уровни простагландина 15d-PGJ2, обнаруженного в супернатантах мононуклеарных клеток периферической крови, инкубированных с 7β-ОН-EPIA, в присутствии и в отсутствие TNF-α, как описано в примере 2.
На фиг.8 показано воздействие введения 7β-гидрокси-EPIA на (А) миелопероксидазу (МРО) и маркеры окислительного стресса, а именно (В) Prot CO, (C) T-образные складки и (D) маркер антиоксидантной защиты GSH в ткани ободочной кишки, как более подробно описано в примере 4.
На фиг.9 показан уровень 15d-PGJ2 в ободочной кишке (А) и количественное определение относительной экспрессии мРНК СОХ-2, mPGES-1 и Н-PGDS (В) в разные периоды времени введения 7β-гидрокси-EPIA в примере 4.
На фиг.10 показано воздействие 7β-гидрокси-EPIA на синтез в ободочной кишке простагландина Е2, D2 и 15d-PGJ2 во время введения DSS в примере 4.
На фиг.11 показана экспрессия в ободочной кишке мРНК СОХ-2, mPGES-1 и H-PGDS во время индукции колита в примере 4.
Соединения, которые могут быть использованы в настоящем изобретении, включают соединения формулы (I):

где
пунктирная окружность показывает, что содержащее ее кольцо может быть полностью насыщенным или может иметь одну, две или три углерод-углеродные двойные связи;
пунктирная линия показывает, что указанная связь может быть углерод-углеродной простой или двойной связью;
R1 означает атом водорода или метильную группу;
R2, R3 и R4 имеют одинаковые или разные значения и означают оксогруппу, гидроксильную группу, меркаптогруппу, атом водорода, атом галогена, алкоксильную группу, арилоксигруппу или ацильную группу;
их фармацевтически приемлемые соли и сложные эфиры.
Цифры (3) и (7) в вышеуказанной формуле приведены только для справки и служат для определения использованной системы нумерации. Конечно, когда R2 означает оксогруппу, пунктирная окружность может означать только полностью насыщенное кольцо (в смысле отсутствия двойных связей в кольце) либо одну или две двойные связи.
Из указанных соединений предпочтительными соединениями являются соединения формулы (II):

(где R1, R2, R3 и R4 имеют указанные выше значения) и их сложные эфиры.
Другим предпочтительным классом указанных соединений являются соединения формулы (III):

(где R2а означает оксогруппу, гидроксильную группу, меркаптогруппу или атом галогена; и R1, R3 и R4 имеют указанные выше значения) и их сложные эфиры.
Еще одним предпочтительным классом указанных соединений являются соединения формулы (IV):

(где R1, R2, R3 и R4 имеют указанные выше значения) и их сложные эфиры.
Еще одним предпочтительным классом указанных соединений являются соединения формулы (V):

где R2, R3 и R4 имеют указанные выше значения.
Примеры соединений формулы (II) включают 7-гидрокситестостерон, имеющий формулу (IIa):

и его сложные эфиры. 7-Гидроксильная группа в данном соединении может находиться в альфа- или бета-конфигурации, или может быть использована смесь двух изомеров.
Примеры соединений формулы (III) включают 7α-гидроксидегидроэпиандростерон (7α-гидрокси-DHEA), имеющий формулу (IIIa):

и его сложные эфиры, 7β-аналог, имеющий формулу (IIIb):

и его сложные эфиры, 7β-гидроксипрегненолон, имеющий формулу (IIIc):

и его сложные эфиры, 7α-гидроксильный аналог и его сложные эфиры.
Примеры соединений формулы (IV) включают 7-гидроксиэпиандростерон, который может иметь форму 7α- или 7β-изомеров, и его сложные эфиры. 7β-гидроксиэпиандростерон, далее именуемый 7β-ОН-EPIA, имеет формулу (IVa):

и 7α-гидроксиэпиандростерон, далее именуемый 7α-ОН-EPIA, имеет формулу (IVb):

Примерами соединений формулы (V) являются 7β-гидрокси-17β-эстрадиол, имеющий формулу (Va):

и его сложные эфиры, 7α-аналоги и их сложные эфиры, и 7β-гидроксиэстрон, имеющий формулу (Vb):

и его сложные эфиры, 7α-аналоги и их сложные эфиры.
Когда R2, R2а, R3 или R4 в соединениях по настоящему изобретению означают атом галогена, такой атом может быть атомом фтора, хлора, брома или йода, предпочтительно атомом хлора.
Когда R2, R2а, R3 или R4 означают алкоксильную группу, такая группа может быть группой с прямой или разветвленной цепью, предпочтительно содержащей 1-6 атомов углерода. Примеры таких групп включают метоксильную, этоксильную, пропоксильную, изопропоксильную, бутоксильную, втор-бутоксильную, изобутоксильную, трет-бутоксильную, пентилокси и гексилокси группы.
Когда R2, R2а, R3 или R4 означают арилоксигруппу, такая группа предпочтительно является феноксильной или нафтилоксигруппой.
Когда R2, R2а, R3 или R4 означают ацильную группу, такая группа может быть, например, алифатической ацильной группой или ароматической ацильной группой. Примеры алифатических ацильных групп включают группы, содержащие 1-6 атомов углерода, такие как формильная, ацетильная, пропионильная, бутирильная, валерильная, изовалерильная, пивалоильная и гексаноильная группы. Примеры ароматических ацильных групп включают бензоильную, нафтоильную и толуоильную группы.
Следует отметить, что когда соединение содержит группу формулы -OR, где R означает любые из вышеуказанных групп и атомов, приведенных для R2 и т.д., активным типом, по-видимому, является соединение, содержащее свободную гидроксильную группу. Таким образом, вместо гидроксильной группы может быть использована любая группа, которая может быть превращена in vivo в гидроксильную группу.
Указанные соединения могут быть получены разными хорошо известными способами при использовании исходных стероидов. Например, такие соединения могут быть получены способами, описанными в европейском патенте ЕР 1294382.
Соединения формулы (I), описанные в европейском патенте ЕР 1294382, публикациях WO 2002/000224 и WO 2002/000225, предназначены для лечения или профилактики хронических и острых нейродегенеративных заболеваний или состояний, и соединения, описанные в публикации WO 2002/015791, предназначены для лечения или профилактики хронических и острых кардиодегенеративных заболеваний или состояний, и такое лечение или профилактика, несомненно, исключены из настоящей формулы изобретения только в отношении соединений формулы (I).
К другому классу соединений, которые могут быть использованы в настоящем изобретении, относятся соединения формулы (VI):

где
Х означает группу формулы >CR5R6 или в том случае, когда R10 не является атомом водорода, группу формулы >SO2;
Y означает группу формулы >NH или >CR5R6;
Z означает группу формулы >C=O, группу формулы >CH2 или прямую связь;
R5 означает атом водорода и R6 означает атом водорода, карбоксильную группу или гидроксильную группу; либо
R5 и R6 вместе означают оксогруппу, метилендиоксигруппу или гидроксииминогруппу;
R7 означает атом водорода или низшую алкильную группу;
R8 означает два атома водорода, или оксогруппу, или гидроксииминогруппу;
R9 означает атом водорода, низшую алкильную группу или атом галогена;
R10 означает атом водорода, низшую алкоксильную группу или карбоксильную группу;
R11 и R12 имеют одинаковые или разные значения и означают атом водорода, низшую алкильную группу или атом галогена;
их соли и сложные эфиры, когда указанные соединения содержат карбоксильную группу.
В соединениях формулы (VI) Z может означать прямую связь, и в данном случае Z может быть частью 5-членного кольца, конденсированного с 5-членным азотсодержащим гетероциклическим кольцом, или может быть группой формулы >CН2 или >C=O, и в данном случае Z является частью 6-членного кольца.
Когда R7, R9, R11 или R12 означает низшую алкильную группу, такая группа может быть алкильной группой с прямой или разветвленной цепью, содержащей 1-10, предпочтительно 1-6 атомов углерода. Примеры таких групп включают метильную, этильную, пропильную, изопропильную, бутильную, втор-бутильную, трет-бутильную, пентильную, изопентильную, неопентильную, 2-метилбутильную, 1-этилпропильную, 4-метилпентильную, 3-метилпентильную, 2-метилпентильную, 1-метилпентильную, 3,3-диметилбутильную, 2,2-диметилбутильную, 1,1-диметилбутильную, 1,2-диметилбутильную, 1,3-диметилбутильную, 2,3-диметилбутильную, 2-этилбутильную, гексильную, изогексильную, гептильную, октильную, нонильную и децильную группы, из которых предпочтительными являются метильная, этильная, пропильная, бутильная и гексильная группы, более предпочтительными являются метильная и этильная группы и наиболее предпочтительной является метильная группа.
Когда R9, R11 или R12 означает атом галогена, такой атом может быть атомом фтора, хлора, брома или йода, из которых предпочтительными являются атомы фтора и хлора.
Когда R10 означает низшую алкоксильную группу, такая группа может быть алкоксильной группой с прямой или разветвленной цепью, содержащей 1-10, предпочтительно 1-6 атомов углерода. Примеры таких групп включают метоксильную, этоксильную, пропоксильную, изопропоксильную, бутоксильную, втор-бутоксильную, трет-бутоксильную, пентилокси, изопентилокси, неопентилокси, 2-метилбутоксильную, 1-этилпропоксильную, 4-метилпентилокси, 3-метилпентилокси, 2-метилпентилокси, 1-метилпентилокси, 3,3-диметилбутоксильную, 2,2-диметилбутоксильную, 1,1-диметилбутоксильную, 1,2-диметилбутоксильную, 1,3-диметилбутоксильную, 2,3-диметилбутоксильную, 2-этилбутоксильную, гексилокси, изогексилокси, гептилокси, октилокси, нонилокси и децилокси группы, из которых предпочтительными являются метоксильная, этоксильная, пропоксильная, бутоксильная и гексилокси группы, более предпочтительными являются метоксильная и этоксильная группы и наиболее предпочтительной является метоксильная группа.
Из соединений по настоящему изобретению особенно предпочтительными являются соединения, в которых
Х означает группу формулы >CR5R6, где R5 означает атом водорода и R6 означает атом водорода, гидроксильную группу или карбоксильную группу, либо R5 и R6 вместе означают оксогруппу или метилендиоксигруппу;
Y означает группу формулы >CR5R6, где R5 означает атом водорода и R6 означает атом водорода или карбоксильную группу;
R7 означает атом водорода;
R8 означает два атома водорода или оксогруппу;
R9 означает атом водорода;
R10 означает атом водорода, С1-С4 алкоксильную группу или карбоксильную группу; и R11 и R12 имеют одинаковые или разные значения и означают атом водорода или С1-С4 алкильную группу;
их соли и сложные эфиры.
В приведенной ниже таблице 1 представлены конкретные примеры соединений по настоящему изобретению.

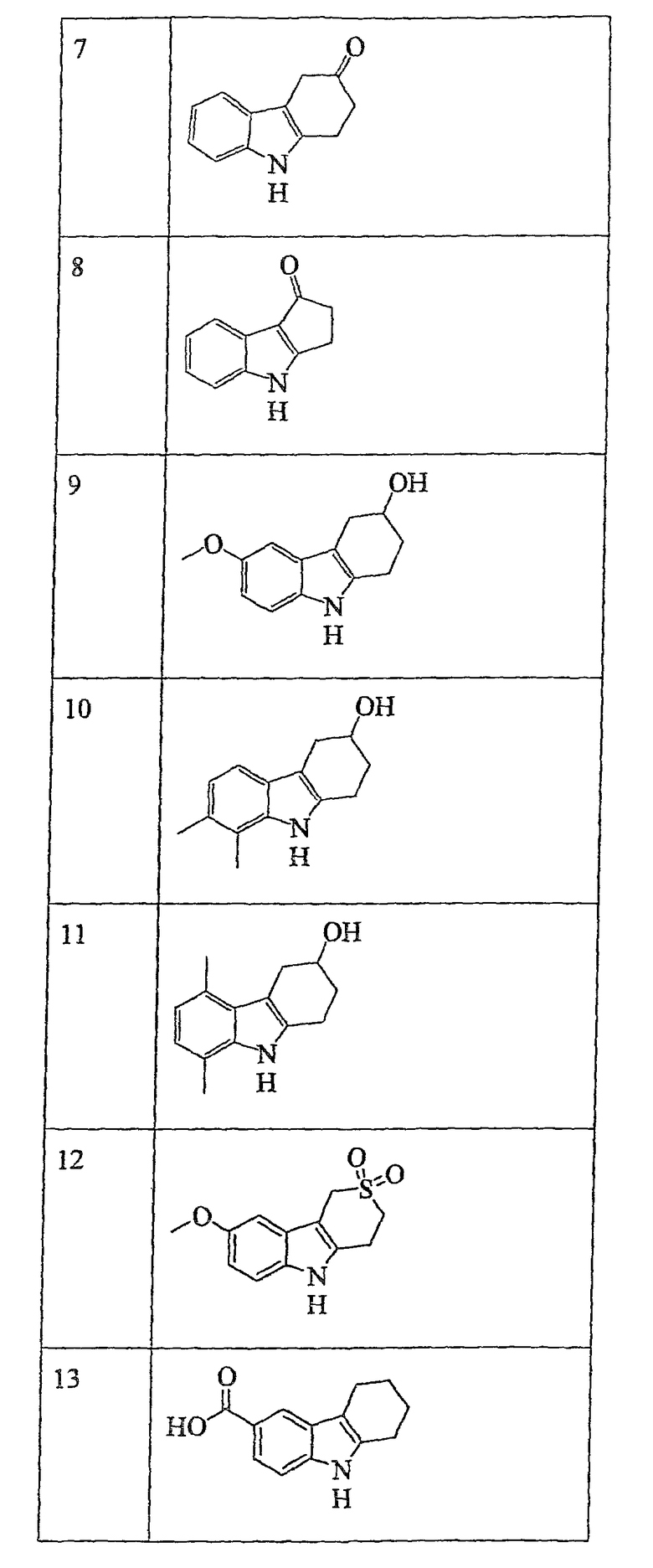


Наиболее предпочтительными соединениями формулы (VI) являются соединения, указанные в приведенной выше таблице под номерами 4, 6, 7, 8, 10, 14, 15, 16, 17, 20, 21, 22 и 23.
Соединения по настоящему изобретению, содержащие карбоксильную группу, например когда R5 или R10 означает карбоксильную группу, могут образовывать сложные эфиры, которые могут быть получены стандартными методами этерификации. Не существует каких-либо конкретных ограничений относительно природы сложного эфира при условии, что в случае медицинского применения полученного соединения такое соединение должно быть фармацевтически приемлемым, то есть не должно быть менее активным или неприемлемо менее активным, более токсичным или неприемлемо более токсичным, чем исходное соединение. Однако, если данное соединение предназначено для использования в немедицинских целях, например, в качестве промежуточного продукта при получении других соединений, вышеуказанное ограничение является недействительным, при этом отсутствуют какие-либо ограничения относительно природы сложных эфиров, которые могут быть получены.
Примеры сложноэфирных групп включают
алкильные группы, содержащие 1-20 атомов углерода, более предпочтительно 1-10 атомов углерода, приведенные для R7, R9, R11 или R12, и высшие алкильные группы, хорошо известные в данной области, такие как додецильная, тридецильная, пентадецильная, октадецильная, нонадецильная и икозильная группы;
циклоалкильные группы, содержащие 3-7 атомов углерода, такие как, например, циклопропильная, циклобутильная, циклопентильная, циклогексильная и циклогептильная группы;
аралкильные группы, в которых алкильная часть содержит 1-3 атома углерода и арильная часть является карбоциклической ароматической группой, содержащей 6-14 атомов углерода, которая может быть замещенной или незамещенной; примеры таких аралкильных групп включают бензильную, фенетильную, 1-фенилэтильную, 3-фенилпропильную, 2-фенилпропильную, 1-нафтилметильную, 2-нафтилметильную, 2-(1-нафтил)этильную, 2-(2-нафтил)этильную, бензгидрильную (то есть дифенилметильную), трифенилметильную, бис(о-нитрофенил)метильную, 9-антрилметильную, 2,4,6-триметилбензильную, 4-бромбензильную, 2-нитробензильную, 4-нитробензильную, 3-нитробензильную, 4-метоксибензильную и пиперонильную группы;
алкенильные группы, содержащие 2-6 атомов углерода, такие как винильная, аллильная, 2-метилаллильная, 1-пропенильная и изопропенильная группы;
галогензамещенные алкильные группы, содержащие 1-6, предпочтительно 1-4 атома углерода, такие как 2,2,2-трихлорэтильная, 2-галогенэтильная (например, 2-хлорэтильная, 2-фторэтильная, 2-бромэтильная или 2-йодэтильная), 2,2-дибромэтильная и 2,2,2-трибромэтильная группы;
замещенные силилалкильные группы, например, 2-три(С1-С4)-алкилсилилэтильные группы, в частности, 2-триметилсилилэтильную группу;
замещенные и незамещенные фенильные группы, такие как, например, фенильная, толильная и бензамидофенильная группы;
замещенные и незамещенные фенацильные группы, такие как, например, фенацильная группа или п-бромфенацильная группа;
циклические и ациклические терпенильные группы, такие как, например, геранильная, нерильная, линалильная, фитильная, ментильная (в частности, м- и п-метильная), туйильная, карильная, пинанильная, борнильная, норкарильная, норпинанильная, норборнильная, ментенильная, камфенильная и норборненильная группы;
алкоксиметильные группы, в которых алкоксильная часть содержит 1-6, предпочтительно 1-4 атома углерода и может быть замещена одной незамещенной алкоксильной группой, такие как метоксиметильная, этоксиметильная, пропоксиметильная, изопропоксиметильная, бутоксиметильная и метоксиэтоксиметильная группы;
алифатические ацилоксиалкильные группы, в которых ацильная группа предпочтительно является алканоильной группой и более предпочтительно алканоильной группой, содержащей 2-6 атомов углерода, и алкильная часть содержит 1-6, предпочтительно 1-4 атома углерода, такие как ацетоксиметильная, пропионилоксиметильная, бутирилоксиметильная, изобутирилоксиметильная, пивалоилоксиметильная, 1-пивалоилоксиэтильная, 1-ацетоксиэтильная, 1-изобутирилоксиэтильная, 1-пивалоилоксипропильная, 2-метил-1-пивалоилоксипропильная, 2-пивалоилоксипропильная, 1-изобутирилоксиэтильная, 1-изобутирилоксипропильная, 1-ацетоксипропильная, 1-ацетокси-2-метилпропильная, 1-пропионилоксиэтильная, 1-пропионилоксипропильная, 2-ацетоксипропильная и 1-бутирилоксиэтильная группы;
циклоалкил-замещенные алифатические ацилоксиалкильные группы, в которых ацильная группа предпочтительно является алканоильной группой и более предпочтительно алканоильной группой, содержащей 2-6 атомов углерода, циклоалкильный заместитель содержит 3-7 атомов углерода и алкильная часть содержит 1-6, предпочтительно 1-4 атома углерода, такие как циклогексилацетоксиметильная, 1-(циклогексилацетокси)этильная, 1-(циклогексилацетокси)пропильная, 2-метил-1-(циклогексилацетокси)пропильная, циклопентилацетоксиметильная, 1-(циклопентилацетокси)этильная, 1-(циклопентилацетокси)пропильная и 2-метил-1-(циклопентилацетокси)пропильная группы;
алкоксикарбонилоксиалкильные группы, в частности, 1-(алкоксикарбонилокси)этильные группы, такие как 1-метоксикарбонилоксиэтильная, 1-этоксикарбонилоксиэтильная, 1-пропоксикарбонилоксиэтильная, 1-изопропоксикарбонилоксиэтильная, 1-бутоксикарбонилоксиэтильная, 1-изобутоксикарбонилоксиэтильная, 1-втор-бутоксикарбонилоксиэтильная, 1-трет-бутоксикарбонилоксиэтильная, 1-(1-этилпропоксикарбонилокси)этильная и 1-(1,1-дипропилбутоксикарбонилокси)этильная группы, и другие алкоксикарбонилалкильные группы, в которых как алкоксильные, так и алкильные группы содержат 1-6, предпочтительно 1-4 атома углерода, такие как 2-метил-1-(изопропоксикарбонилокси)пропильная, 2-(изопропоксикарбонилокси)пропильная, изопропоксикарбонилоксиметильная, трет-бутоксикарбонилоксиметильная, метоксикарбонилоксиметильная и этоксикарбонилоксиметильная группы;
циклоалкилкарбонилоксиалкильные и циклоалкилоксикарбонилоксиалкильные группы, такие как, например, 1-метилциклогексилкарбонилоксиметильная, 1-метилциклогексилоксикарбонилоксиметильная, циклопентилоксикарбонилоксиметильная, циклопентилкарбонилоксиметильная, 1-(циклогексилоксикарбонилокси)этильная, 1-(циклогексилкарбонилокси)этильная, 1-(циклопентилоксикарбонилокси)этильная, 1-(циклопентилкарбонилокси)этильная, 1-(циклогептилоксикарбонилокси)этильная, 1-(циклогептилкарбонилокси)этильная, 1-метилциклопентилкарбонилоксиметильная, 1-метилциклопентилоксикарбонилоксиметильная, 2-метил-1-(1-метилциклогексилкарбонилокси)пропильная, 1-(1-метилциклогексилкарбонилокси)пропильная, 2-(1-метилциклогексилкарбонилокси)пропильная, 1-(циклогексилкарбонилокси)пропильная, 2-(циклогексилкарбонилокси)пропильная, 2-метил-1-(1-метилциклопентилкарбонилокси)пропильная, 1-(1-метилциклопентилкарбонилокси)пропильная, 2-(1-метилциклопентилкарбонилокси)пропильная, 1-(циклопентилкарбонилокси)пропильная, 2-(циклопентилкарбонилокси)пропильная, 1-(1-метилциклопентилкарбонилокси)этильная, 1-(1-метилциклопентилкарбонилокси)пропильная, адамантилоксикарбонилоксиметильная, адамантилкарбонилоксиметильная, 1-адамантилоксикарбонилоксиэтильная и 1-адамантилкарбонилоксиэтильная группы;
циклоалкилалкоксикарбонилоксиалкильные группы, такие как, например, циклопропилметоксикарбонилоксиметильная, циклобутилметоксикарбонилоксиметильная, циклопентилметоксикарбонилоксиметильная, циклогексилметоксикарбонилоксиметильная, 1-(циклопропилметоксикарбонилокси)этильная, 1-(циклобутилметоксикарбонилокси)этильная, 1-(циклопентилметоксикарбонилокси)этильная и 1-(циклогексилметоксикарбонилокси)этильная группы;
терпенилкарбонилоксиалкильные и терпенилоксикарбонилоксиалкильные группы, такие как, например, 1-(ментилоксикарбонилокси)этильная, 1-(ментилкарбонилокси)этильная, ментилоксикарбонилоксиметильная, ментилкарбонилоксиметильная, 1-(3-пинанилоксикарбонилокси)этильная, 1-(3-пинанилкарбонилокси)этильная, 3-пинанилоксикарбонилоксиметильная и 3-пинанилкарбонилоксиметильная группы;
5-алкил- или 5-фенил-(2-оксо-1,3-диоксолен-4-ил)алкильные группы, такие как, например, (5-метил-2-оксо-1,3-диоксолен-4-ил)метильная, (5-фенил-2-оксо-1,3-диоксолен-4-ил)метильная, (5-изопропил-2-оксо-1,3-диоксолен-4-ил)метильная, (5-трет-бутил-2-оксо-1,3-диоксолен-4-ил)метильная и 1-(5-метил-2-оксо-1,3-диоксолен-4-ил)этильная группы; и
другие группы, в частности группы, которые легко удаляются in vivo, такие как фталидильная, инданильная и 2-оксо-4,5,6,7-тетрагидро-1,3-бензодиоксолен-4-ильная группы.
Кроме того, соединения по настоящему изобретению, содержащие карбоксильную группу, могут быть превращены стандартными методами в соли с основанием. Не существует каких-либо конкретных ограничений относительно природы таких солей при условии, что соединения, предназначенные для медицинского применения, должны быть фармацевтически приемлемыми. Однако если данное соединение должно быть использовано для немедицинских целей, например, в качестве промежуточного продукта при получении других соединений, вышеуказанное ограничение является недействительным, при этом отсутствуют какие-либо ограничения относительно природы солей, которые могут быть получены. Примеры таких солей включают соли с щелочным металлом, таким как натрий, калий или литий; соли с щелочноземельным металлом, таким как барий или кальций; соли с другим металлом, таким как магний или алюминий; соли аммония; соли с органическими основаниями, такие как соль с метиламином, диметиламином, триэтиламином, диизопропиламином, циклогексиламином или дициклогексиламином; и соли с основной аминокислотой, такой как лизин или аргинин. Предпочтительными являются фармацевтически приемлемые соли.
Соединения по настоящему изобретению могут быть также превращены стандартными методами в соли с кислотами. Не существует каких-либо конкретных ограничений относительно природы таких солей при условии, что соединения, предназначенные для медицинского применения, должны быть фармацевтически приемлемыми. Однако если данное соединение должно быть использовано для немедицинских целей, например, в качестве промежуточного продукта при получении других соединений, вышеуказанное ограничение является недействительным, при этом отсутствуют какие-либо ограничения относительно природы солей, которые могут быть получены. Примеры таких солей включают соли с минеральными кислотами, в частности, с галогенводородными кислотами (такими как фтористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота или хлористоводородная кислота), азотной кислотой, перхлорной кислотой, угольной кислотой, серной кислотой или фосфорной кислотой; соли с низшими алкилсульфоновыми кислотами, такими как метансульфоновая кислота, трифторметансульфоновая кислота или этансульфоновая кислота; соли с арилсульфоновыми кислотами, такими как бензолсульфоновая кислота или п-толуолсульфоновая кислота; соли с органическими карбоновыми кислотами, такими как уксусная кислота, фумаровая кислота, винная кислота, щавелевая кислота, малеиновая кислота, яблочная кислота, янтарная кислота, бензойная кислота, миндальная кислота, аскорбиновая кислота, молочная кислота, глюконовая кислота или лимонная кислота; и соли с аминокислотами, такими как глутаминовая кислота или аспарагиновая кислота. Предпочтительными являются фармацевтически приемлемые соли.
Соединения формулы (VI), описанные в публикации WO 2006/082409, предназначены для лечения или профилактики хронических и острых нейродегенеративных заболеваний или состояний, и такое лечение или профилактика, несомненно, исключены из настоящей формулы изобретения только в отношении соединений формулы (VI).
Соединения по настоящему изобретению могут быть использованы для лечения или профилактики различных хронических и острых заболеваний или состояний, и для достижения указанных целей могут быть получены в виде стандартных фармацевтических препаратов, хорошо известных в данной области. Таким образом, указанные соединения можно вводить перорально, например, в виде таблеток, капсул, гранул, порошков, сиропов, распыляемых растворов и других хорошо известных форм, или парентерально, например, в виде инъекций, распыляемых растворов, глазных капель, лейкопластырей или суппозиториев и т.д.
Указанные фармацевтические препараты могут быть получены стандартными методами и могут содержать известные адъюванты, обычно используемые в данной области, например, наполнители, связывающие вещества, дезинтеграторы, смазывающие вещества, стабилизаторы, корригенты и т.д., в зависимости от предполагаемого применения и формы препарата. Величина дозы зависит от состояния, возраста и массы тела субъекта, а также от характера и тяжести подлежащего лечению заболевания, но в случае перорального введения взрослому человеку общая суточная доза обычно составляет от 0,01 до 50 мг/кг массы тела (более предпочтительно от 0,05 до 20 мг/кг массы тела) и может быть введена в виде однократной дозы или разделенных доз, вводимых, например, один-три раза в сутки.
Как правило, соединения по настоящему изобретению могут быть использованы для лечения или профилактики различных воспалительных состояний или состояний, обусловленных путями обмена веществ, вызывающими воспаление. Примеры применения указанных соединений включают стимуляцию роста аксонов; лечение или профилактику сахарного диабета (в частности, сахарного диабета типа 2) и его осложнений; лечение и профилактику ишемических заболеваний сосудов; лечение боли, ассоциированной с воспалением; лечение и профилактику воспалительных кожных заболеваний, включающих псориаз и заживление ран. Другие воспалительные состояния, которые могут быть подвергнуты лечению или профилактике либо тяжесть которых может быть ослаблена при использовании соединений по настоящему изобретению, включают травму спинного мозга, периферическую невропатию, рассеянный склероз, воспалительное заболевание кишечника, ревматоидный артрит, периферическую невропатию или токсичность, вызванную химиотерапевтическими средствами, такими как цисплатин, или другими причинами, такими как диабетическая невропатия. И наконец, состояния, которые могут быть подвергнуты лечению или профилактике либо тяжесть которых может быть ослаблена при использовании соединений по настоящему изобретению, включают также разные типы рака.
Настоящее изобретение далее проиллюстрировано приведенными ниже примерами, не ограничивающими объем изобретения.
Пример 1
Защитное действие 7β-гидроксиэпиандростерона от ишемии в клетках РС-12: ингибирование индометацином, ингибитором циклооксигеназы (СОХ)
Целью данного эксперимента было исследование способности 7β-ОН-EPIA сохранять нейрозащитную эффективность в модели гипоксии при ослаблении синтеза простагландинов индометацином. Главной использованной экспериментальной системой была вызванная ишемией цитотоксичность в клетках РС12. Конечной точкой эксперимента была гибель клеток.
Вызванная ишемией гибель клеток РС-12 была значительно уменьшена 7β-ОН-EPIA. Индометацин (10 мкМ), блокирующий синтез простагландинов, не оказывал прямого воздействия на вызванную ишемией гибель клеток, но полностью блокировал нейрозащитное действие 1 мкМ и 10 мкМ 7β-ОН-EPIA, подтверждая гипотезу, что синтез простагландинов необходим для нейрозащитного действия 7β-гидрокси-EPIA.
Методика выполнения эксперимента
Культура клеток РС-12
Клетки РС-12 помещали в колбы, сенсибилизированные коллагеном типа 1, в среды для РС-12 следующего состава: RPMI 1640 без L-глутамина, с 2 мМ L-глутамина, 10 мМ HEPES, 1 мМ пирувата натрия, дополнительное количество глюкозы до достижения конечной концентрации, равной 4,5 г/л (RPMI обычно содержит 2 г/л), 10% инактивированной нагреванием лошадиной сыворотки, 5% фетальной телячьей сыворотки и 50 единиц пенициллина/стрептомицина. Среды меняли через каждые 2 дня.
Анализы клеток РС-12
Конфлюентные культуры клеток РС-12 дифференцировали, культивируя в течение 7 дней в среде для РС-12 без сыворотки, но с 50 нг/мл NGF (среда NGF для РС-12). Клетки собирали, промывали и подсчитывали, 1×105 клеток РС-12/лунка культивировали в течение ночи на титрационном микропланшете в среде NGF для РС-12 без глюкозы. Затем указанную среду заменяли средой NGF для РС-12 без глюкозы, содержащей испытуемые соединения, и планшеты выдерживали в нормальных условиях в течение 30 минут.
На данной стадии среды и испытуемые вещества, предназначенные для нахождения в бескислородных условиях, помещали в камеру и дезоксигенировали 95% N2/5% CO2. Среды на планшетах заменяли бескислородными средами и планшеты помещали в анаэробную камеру, в которую подавали 95% N2/5% CO2 в течение 10 минут, затем герметично закрывали и инкубировали при 37°С в течение ночи (18 часов). В контрольных экспериментах клетки обрабатывали так же, как в условиях ишемии за исключением того, что все инкубации выполняли в атмосфере 5% СО2/95% воздуха.
Жизнеспособность клеток определяли методом исключения при помощи трипанового синего.
Результаты
Клетки РС-12 были относительно устойчивы к гипоксии. В связи с этим были созданы более суровые условия объединенного снижения содержания кислорода-глюкозы (ишемия) для инициации токсичности (фиг.1). 7β-ОН-EPIA оказывал в данном анализе цитозащитное действие в зависимости от дозы, при этом значительная нейрозащита наблюдалась при дозе 1 мкМ (26% уменьшение гибели клеток) и 10 мкМ (53% уменьшение гибели клеток). На фиг.1 представлены объединенные данные 4 экспериментов, в которых был обнаружен вышеуказанный эффект. На фиг.1 показано среднее процентное значение гибели клеток, полученное на основании объединенных данных 4 отдельных экспериментов. Данные выражены в виде среднего±стандартная ошибка среднего. В случае ишемии гибель клеток была равна 76%±2,2%. Статистически значимое уменьшение гибели клеток наблюдалось при использовании 1 мкМ 7β-ОН-EPIA (26% уменьшение гибели клеток, р<0,001), 10 мкМ 7β-ОН-EPIA (53% уменьшение гибели клеток, р<0,001), *** = р<0,001 по сравнению с полностью ишемическими условиями.
Антагонист рецептора NMDA МК-801 также уменьшал вызванную ишемией токсичность клеток РС-12 (фиг.2). Ингибитор циклооксигеназы индометацин оказывал среднее защитное действие при дозе 100 мкМ (29% уменьшение гибели клеток), которое отсутствовало при более низких концентрациях (фиг.2). На фиг.2 показано среднее процентное значение гибели клеток. Данные выражены в виде среднего±стандартная ошибка среднего. В полностью ишемических условиях гибель клеток была равна 79%±5,3%. Статистически значимое уменьшение гибели клеток наблюдалось при использовании 100 мкМ индометацина (IM) (29% уменьшение гибели клеток, р<0,05), 10 мкМ МК801 (62% уменьшение гибели клеток, р<0,001), ** = р<0,01, *** = р<0,001 по сравнению с полностью ишемическими условиями.
Нейрозащитное действие 7β-ОН-EPIA было полностью блокировано 10-100 мкМ индометацина (IM) (фиг.3), при этом токсичность в указанных культурах не отличалась от культур, находящихся в полностью ишемических условиях. На фиг.3 показано среднее процентное значение гибели клеток. Данные выражены в виде среднего±стандартная ошибка среднего. В полностью ишемических условиях гибель клеток была равна 76%±4,2%. Статистически значимое уменьшение гибели клеток наблюдалось при использовании 10 мкМ 7β-ОН-EPIA (50% уменьшение гибели клеток, р<0,001), 10 мкМ МК801 (42% уменьшение гибели клеток, р<0,001), *** = р<0,001 по сравнению с полностью ишемическими условиями.
Как показано на фиг.4, 10 мкМ индометацина полностью блокировали защитное действие 1 мкМ и 10 мкМ 7β-ОН-EPIA. На фиг.4 показано среднее процентное значение гибели клеток. Данные выражены в виде среднего±стандартная ошибка среднего. В полностью ишемических условиях гибель клеток была равна 76%±4,8%. Статистически значимое уменьшение гибели клеток наблюдалось при использовании 1 мкМ 7β-ОН-EPIA (25% уменьшение гибели клеток, р<0,05), 10 мкМ 7β-ОН-EPIA (50% уменьшение гибели клеток, р<0,001), * = р<0,05, *** = р<0,001 по сравнению с полностью ишемическими условиями.
Заключение
Вызванная ишемией гибель клеток РС-12 была значительно уменьшена 7β-ОН-EPIA. Индометацин, который блокирует синтез простаглиндинов, не оказывал прямого влияния на гибель клеток РС-12 при концентрациях до 10 мкМ. Однако при дальнейшем увеличении концентрации до 100 мкМ наблюдалось среднее нейрозащитное действие.
Индометацин (10 мкМ) не оказывал прямого воздействия на вызванную ишемией гибель клеток, но значительно ослаблял нейрозащитное действие 7β-ОН-EPIA, подтверждая гипотезу о том, что синтез простагландинов необходим для нейрозащитного действия 7β-ОН-EPIA.
Пример 2
Воздействие 7β-гидроксиэпиандростерона на продуцирование простагландинов D2, E2 и 15-дезокси-Δ 12,14 -J2 мононуклеарными клетками человека
Целью данного эксперимента была оценка способности 7β-ОН-EPIA индуцировать биосинтез специфических метаболитов арахидоновой кислоты в моноцитах человека, а именно простагландина D2 (PGD2), простагландина Е2 (PGE2) и 15-дезокси-Δ12,14-простагландина J2 (15d-PGJ2).
Моноциты крови подвергали воздействию разных концентраций 7β-ОН-EPIA в отсутствие или в присутствии фактора некроза опухолей (TNF-α), провоспалительного фактора, и при помощи иммуноферментного анализа (EIA) измеряли количество продуцированного PGD2, PGE2 и 15d-PGJ2.
7β-ОН-EPIA (0,1 нМ - 100 нМ) вызывал в зависимости от концентрации увеличение продуцирования PGD2 из нормальных мононуклеарных клеток периферической крови человека. TNF-α увеличивал продуцирование PGD2 по сравнению с контрольными клетками, и подобное увеличение происходило при самой высокой концентрации 7β-ОН-EPIA. 7β-ОН-EPIA (1 нМ - 1000 нМ), по-видимому, не оказывает существенного влияния на биосинтез PGE2 в нормальных мононуклеарных клетках периферической крови человека, но полностью подавляет TNF-α-индуцированное увеличение продуцирования РGE2. Было установлено, что 7β-ОН-EPIA (0,1 нМ - 1000 нМ) увеличивает продуцирование 15d-PGJ2 в нормальных мононуклеарных клетках периферической крови человека примерно в 9-12 раз при отсутствии TNF-α и в 2-2,5 раза в присутствии TNF-α по сравнению с соответствующими контрольными клетками.
Методика выполнения эксперимента
Мононуклеарные клетки периферической крови
Мононуклеарные клетки (моноциты и лимфоциты) были получены путем центрифугирования в градиенте плотности 48 мл цельной крови человека с фиколл-гипаком. Кровь помещали поверх фиколла (плотность = 1,077 г/мл) и центрифугировали с ускорением 400 g в течение 1 часа (22°С), после чего клетки в интерфазе удаляли пипеткой и переносили в чистые пробирки. Пробирки заполняли культуральной средой RPMI 1640 (2 объема), тщательно смешивали и центрифугировали с ускорением 400 g в течение 5 минут (22°С). Супернатант удаляли, клеточный дебрис ресуспендировали в культуральной среде RPMI 1640 и объем доводили до получения соответствующего числа клеток в одной инкубации, как указано в приведенном ниже разделе ”Результаты”. Клетки инкубировали в стерильных пластиковых 1,5-мл пробирках в конечном объеме 1 мл среды RPMI 1640 в течение 18 часов при 37°С в воздушной атмосфере с 5% СО2 и 100% влажности. Затем добавляли 7β-ОН-EPIA и клетки инкубировали еще один час при 37°С, после чего добавляли рекомбинантный α-фактор некроза опухолей человека (TNF-α) и продолжали инкубировать клетки еще 3 часа. 7β-ОН-EPIA был получен в диметилсульфоксиде (ДМСО), все остальные агенты были получены в среде RPMI 1640. Необходимые контрольные образцы содержали среду или ДМСО в такой же концентрации, которая всегда была <0,1% об./об. Инкубацию прекращали, центрифугируя пробирки с ускорением 11000 g в течение 30 секунд при 22°С, и переносили супернатанты в чистые 1,5-мл пробирки. Образцы сразу же обрабатывали для определения PGD2 нижеописанным способом или хранили при -20°С до измерения PGE2 или 15d-PGJ2.
Иммуноферментные анализы (EIA) простагландинов
Продуцирование простагландинов моноцитами человека под воздействием 7β-ОН-EPIA без стимуляции или при стимуляции TNF-α определяли, измеряя внеклеточные уровни эйкозаноида при помощи коммерчески доступных наборов для EIA.
Иммуноферментный анализ PGD2
Анализ PGD2 нельзя выполнить непосредственно из супернатантов клеток, так как данный простагландин является химически неустойчивым и быстро разлагается на ряд простагландинов серии J, включающих PGJ2, Δ12-PGJ2 и 15-дезокси-Δ12,14-PGJ2. Для устранения указанной проблемы неустойчивый простагландин PGD2 подвергали химической обработке с образованием устойчивого производного, в данном случае простагландина D2-метоксамина (PGD2-MOX), который хранили для последующего выполнения анализа. Сразу же после окончания инкубации в 1,5-мл пробирки, содержащие 100 мкл реагента для оксимирования метила (гидрохлорид метоксиламина (МОХ-HCl) и ацетат натрия, растворенный в растворе этанола/воды с соотношением 10:90 об./об., добавляли 100 мкл супернатанта образца. Пробирки помещали на водяную баню и осуществляли взаимодействие в течение 30 минут при 60°С. По окончании указанного периода образцы хранили при -80°С. Уровни PGD2 определяли при помощи набора для иммуноферментного анализа простагландина D2-MOX компании Cayman Chemical (№ по каталогу 512011).
Иммуноферментный анализ PGE2
Внеклеточные уровни PGE2 определяли при помощи набора для иммуноферментного анализа PGE2 Parameter компании R&D Systems в соответствии с инструкциями изготовителя для высокочувствительной методики выполнения эксперимента.
Иммуноферментный анализ 15d-PG2
Внеклеточные уровни 15d-PGJ2 определяли при помощи набора для иммуноферментного анализа 15-дезокси-Δ12,14-простагландина J2 Correlate-EIA Prostaglandin компании Assay Designs (№ по каталогу 900-023) в соответствии с инструкциями изготовителя.
Статистический анализ
Результаты выражены в виде среднего±стандартное отклонение для n = 3 инкубаций. Вероятность (Р) того, что две совокупности данных отличаются друг от друга, определяли при помощи непарного t-критерия Стьюдента. Разница считалась значимой при Р<0,05. Статистические вычисления производили в компьютере Apple Macintosh при помощи программного обеспечения Statview компании Abacus Concepts Inc.
Результаты
Воздействие 7β-ОН-EPIA на продуцирование PGD2 мононуклеарными клетками человека
На фиг.5(а) показано воздействие возрастающих концентраций 7β-ОН-EPIA (0,1 нМ - 1000 нМ) на уровни PGD2, обнаруженные в супернатантах 1×107 мононуклеароных клеток периферической крови/мл, инкубированных в течение 4 часов с 7β-ОН-EPIA. 7β-ОН-EPIA, по-видимому, вызывает увеличение продуцирования PGD2 в зависимости от концентрации, которое достигает максимального значения при 100 нМ 7β-ОН-EPIA (102±24 пг/мл PGD2), что значительно выше, чем у контрольного образца (40±13 пг/мл PGD2; Р=0,01).
На фиг.5(b) показано воздействие возрастающих концентраций 7β-ОН-EPIA (0,1 нМ - 1000 нМ) на уровни PGD2, обнаруженные в супернатантах мононуклеарных клеток, инкубированных в присутствии TNF-α (0,5 мкг/мл). TNG-α вызывал 2,3-кратное увеличение PGD2 по сравнению с контрольным наполнителем ДМСО (Р<0,05). При концентрациях 0,1 нМ - 100 нМ 7β-ОН-EPIA, по-видимому, не влияет на TNF-α-индуцированный биосинтез PGD2. При самой высокой концентрации 7β-ОН-EPIA, использованной в данном эксперименте (1000 нМ), уровни РGD2 увеличивались до 164±31 пг/мл (Р=0,03 по сравнению с использованием только TNF-α).
Воздействие 7β-ОН-EPIA на продуцирование РGE2 мононуклеарными клетками человека
На фиг.6(а) показано воздействие возрастающих концентраций 7β-ОН-EPIA (1 нМ - 1000 нМ) на уровни PGE2, обнаруженные в супернатантах 6×105 мононуклеарных клеток периферической крови/мл, инкубированных в течение 4 часов с 7β-OH-EPIA. 7β-ОН-EPIA, по-видимому, повышает уровни PGE2 по сравнению с контрольным ДМСО, однако такие увеличения не были статистически значимыми.
На фиг.6(b) показано воздействие возрастающих концентраций 7β-ОН-EPIA (1 нМ - 1000 нМ) на уровни PGE2, обнаруженные в супернатантах мононуклеарных клеток, инкубированных в присутствии TNF-α (10 нг/мл). TNF-α стимулировал значительное 1,97-кратное увеличение PGE2 по сравнению с конторольным ДМСО (Р=0,001). При концентрациях 1 нМ - 100 нМ 7β-ОН-EPIA значительно подавлял TNF-α-индуцированный биосинтез PGE2 со 167±6 пг/мл под воздействием только TNF-α до 75±25 пг/мл, 82±23 пг/мл и 74±12 пг/мл соответственно для 1 нМ, 10 нМ и 100 нМ 7β-ОН-EPIA (все значения Р<0,02 по сравнению с использованием только TNF-α). При самой высокой концентрации 7β-ОН-EPIA, использованной в данном эксперименте (1000 нМ), не было обнаружено воздействия на TNF-α-индуцированное продуцирование PGE2.
Воздействие 7β-ОН-EPIA на продуцирование 15d-PGJ2 мононуклеарными клетками человека
На фиг.7(а) показано воздействие возрастающих концентраций 7β-ОН-EPIA (0,1 нМ - 1000 нМ) на уровни 15d-PGJ2, обнаруженные в супернатантах мононуклеарных клеток, инкубированных в течение 4 часов с 7β-ОН-EPIA. 7β-ОН-EPIA значительно повышал уровни 15d-PGJ2 примерно в 9-12 раз при всех использованных концентрациях. Уровни увеличивались с 51±6 пг/мл для контрольного ДМСО до 506±101 пг/мл, 539±51 пг/мл, 520±45 пг/мл, 450±133 пг/мл и 590±84 пг/мл соответственно для 0,1 нМ, 1 нМ, 10 нМ, 100 нМ и 1000 нМ 7β-OH-EPIA (все значения Р<0,05 по сравнению с контрольным ДМСО).
На фиг.7(b) показано воздействие возрастающих концентраций 7β-ОН-EPIA (0,1 нМ - 1000 нМ) на уровни 15d-PGJ2, обнаруженные в супернатантах мононуклеарных клеток, инкубированных с TNF-α (10 нг/мл). При концентрации цитокина, использованной в данном эксперименте, TNF-α, по-видимому, стимулирует незначительное увеличение 15d-PGJ2, однако такое увеличение существенно не отличалось от контрольного ДМСО. При всех использованных концентрациях 7β-ОН-EPIA повышал уровни 15d-PGJ2 в присутствии TNF-α. Уровни 15d-PGJ2 были увеличены со 157±39 пг/мл в присутствии только TNF-α до 348±48 пг/мл, 334±24 пг/мл, 356±85 пг/мл, 406±30 пг/мл и 318±100 пг/мл в присутствии TNF-α вместе с соответственно 0,1 нМ, 1 нМ, 10 нМ, 100 нМ и 1000 нМ 7β-ОН-EPIA (все значения Р<0,05 по сравнению с использованием только TNF-α).
Пример 3
7α-Гидрокси-DHEA, 7β-гидрокси-DHEA и 7β-гидрокси-EPIA являются природными метаболитами дегидроэпиандростерона (DHEA) и эпиандростерона (EPIA). Так как в научной литературе отмечено, что многие стероиды препятствуют воспалительным и иммунным процессам, целью настоящего изобретения было исследование воздействия указанных гидроксистероидов на продуцирование PG и экспрессию генов родственных ферментов. Моноциты периферической крови человека (PBMC) культивировали в течение 4 часов и 24 часов в присутствии каждого из указанных стероидов (1-100 нМ) с добавлением и без добавления провоспалительного цитокина TNF-α (10 нг/мл). Уровни PGE2, PGD2 и 15-дезокси-Δ12,14-PGJ2 (15d-PGJ2) измеряли в инкубационной среде, и содержание в клетках мРНК циклооксигеназы (СОХ-2) и PGE-синтазы (m-PGES1) определяли количественным методом ПЦР с обратной транскриптазой (RT-PCR). Добавление TNF-α вызывало повышенное продуцирование PG и увеличение уровней мРНК СОХ-2 и m-PGES1. Из трех исследованных стероидов только 7β-гидрокси-EPIA уменьшал экспрессию СОХ-2 и m-PGES1 при значительном уменьшении продуцирования РGE2 и увеличении продуцирования 15d-PGJ2. Полученные результаты показывают, что 7β-гидрокси-EPIA оказывает противовоспалительное действие.
1.1. Получение и культивирование моноцитов периферической крови человека (РВМС)
Цельную кровь, взятую у доноров, собирали в мешки, содержащие EDTA, в Etablissement Francais du Sang (Brest, France). РВМС выделяли из цельной крови в стерильных условиях в течение 36 часов после сбора крови. Центрифугирование в градиенте плотности выполняли на фиколле (Eurobio). Клетки промывали в среде RPMI 1640 (Eurobio) и суспендировали в среде RPMI 1640, содержащей 10% инактивированной нагреванием фетальной телячьей сыворотки (Eurobio), 2 мМ глутамина (D. Dutscher), 100 единиц пенициллина/мл (D. Dutscher) и 100 мкг стрептомицина/мл (D. Dutscher). Моноциты отбирали путем адгезии к пластику в течение 1 часа и примерно 107 клеток культивировали на 6-луночных культуральных планшетах для ткани (3 мл среды/лунка). Все инкубации выполняли во влажной камере при 37°С и 5% СО2. Клетки выделяли и диспергировали в 2 мл свежей инкубационной среды, содержащей или 7β-гидрокси-EPIA, или 7β-гидрокси-DHEA, или 7α-гидрокси-DHEA (в 20 мкл этанола) в присутствии или в отсутствие 0,01 мкг/мл TNF-α (Sigma-Аldrich). В контрольных инкубациях использовали 20 мкл этанола, но без стероида. Супернатанты собирали после инкубации в течение 4 часов и 24 часов для измерения содержания РG и клетки использовали для последующего выделения РНК. В результате выполнения метода одностадийной экстракции с использованием реагента тризола (Invitrogen, Cergy-Pontoise, France) была получена общая РНК.
1.2. Полимеразная цепная реакция с обратной транскриптазой в реальном времени
кДНК синтезировали из РНК, обработанной ДНКазой I TURBO (Ambion, Huntingdon, UK), при помощи набора для системы синтеза первой цепи Superscript (Invitrogen). Смеси для амплификации методом RT-PCR (50 мкл) содержали раствор 2,5-кратного количества смеси RealMaster/20-кратного количества SYBR (11,25 мкл) (Eppendorf, Le Pecq, France) и 200 нМ верхних и нижних затравок. Реакции выполняли в термоблоке для проведения реакций RealPlex ep gradient S (Eppendorf). Циклизацию выполняли в следующих условиях: 10 минут при 95°С и 45 циклов при 95°С, 55°С и 68°С в течение соответственно 15 секунд, 30 секунд и 30 секунд. В результате каждого анализа была построена стандартная кривая для четырех последовательных точек разведения контрольной кДНК. Для количественного анализа был использован обязательный ген HPRT1. Все олигонуклеотидные затравки (таблица 2) были синтезированы при помощи Genecust/Distribio (Evry, France). Специфичность амплифицированного продукта контролировали при помощи кривой плавления продукта и подтверждали, выполняя анализ методом электрофореза в агарозном геле.
1.3. Измерения простагландинов (PG)
Для определения уровней РGE2 (Oxford Biomedical Research, UK) и 15d-PGJ2 (Assay designs, Euromedex, France) в культуральной среде были использованы коммерчески доступные наборы для EIA. Уровни PGD2 были измерены при помощи набора для иммуноферментного анализа простагландина D2-MOX (Cayman Chemical, Euromedex). В данном случае до выполнения анализа только что полученные образцы сразу же обрабатывали реагентом МОХ-HCl, который превращал PGD2 в PGD2-MOX, предотвращая таким образом любое дальнейшее химическое разложение.
1.4. Статистический анализ данных
Все анализы были выполнены трижды и результаты были представлены в виде кривой среднего±стандартная ошибка среднего. Дисперсионный анализ по одному признаку выполняли на основании многократных тестов Дункана для сравнения различий между группами. Различия считались статистически значимыми при р<0,05.
2.1. Воздействие 7α-гидрокси-DHEA
РВМС человека культивировали с добавлением и без добавления TNF-α в течение 4 или 24 часов. Уровни PGE2, PGD2 и 15d-PGJ2 измеряли в культуральной среде, и продуцирование мРНК родственных генов (СОХ-2, m-PGES1) измеряли в клетке. Полученные данные приведены в таблице 2. При отсутствии TNF-α и добавлении 7α-гидрокси-DНEA в трех разных концентрациях не происходило существенного изменения уровней PGE2, PGD2 и 15d-PGJ2 в культурах в течение 4 часов. Только уровень 15d-PGJ2 умеренно повысился после культивирования в течение 24 часов. Инкубация с 7α-гидрокси-DHEA значительно повышала уровни мРНК m-PGES1 в культурах через 24 часа.
Присутствие TNF-α вызывало предполагаемое повышение уровней всех простагландинов через 24 часа. Инкубация с 7α-гидрокси-DHEA в течение 4 часов не изменяла уровни PG при всех испытанных концентрациях. В отличие от этого уровни PGE2 и двух PGD2-15d-PGJ2 были соответственно значительно повышены и понижены после инкубации с 7α-гидрокси-DHEA и TNF-α в течение 24 часов по сравнению с использованием только TNF-α. Кроме того, инкубация с TNF-α (то есть воспалительная активация) вызывала заметное увеличение продуцирования мРНК СОХ-2 и m-PGES1. Инкубация с 7α-гидрокси-DHEA не вызывала значительных изменений уровней мРНК по сравнению с использованием только TNF-α.
2.2. Воздействие 7β-гидрокси-DНEA
РВМС человека культивировали с добавлением и без добавления TNF-α в течение 4 или 24 часов. Уровни PGE2, PGD2 и 15d-PGJ2 измеряли в культуральной среде, и продуцирование мРНК родственных генов (СОХ-2, m-PGES1) измеряли в клетке. Полученные данные приведены в таблице 3. Инкубация с 7β-гидрокси-DHEA в трех концентрациях в течение 4 часов при отсутствии TNF-α не вызывала существенных изменений уровней PGE2, PGD2 и 15d-PGJ2 в культуральной среде. Однако уровни PGD2 и 15d-PGJ2, а также мРНК m-PGES1 увеличились после 24 часов инкубации с 7β-гидрокси-DHEA в культуре.
Присутствие TNF-α вызывало предполагаемое повышение содержания всех простагландинов (РG) через 24 часа. Инкубация с 7β-гидрокси-DHEA в течение 4 часов не вызывала дальнейшего изменения уровней PG при любых испытанных концентрациях. В отличие от этого уровни PGD2 и 15d-PGJ2 значительно снизились после инкубации с 7β-гидрокси-DHEA и TNF-α в течение 24 часов по сравнению с использованием только TNF-α. Кроме того, инкубация с ТNF-α (то есть воспалительная активация) вызывала значительное увеличение продуцирования мРНК СОХ-2 и m-PGES1. Инкубация с 7β-гидрокси-DHEA не вызывала сопоставимых изменений уровней мРНК по сравнению с использованием только TNF-α.
2.3. Воздействие 7β-гидрокси-EPIA
РВМС человека культивировали с добавлением и без добавления TNF-α в течение 4 или 24 часов. Уровни PGE2, PGD2 и 15d-PGJ2 измеряли в культуральной среде, и продуцирование мРНК родственных генов (СОХ-2, m-PGES1) измеряли в клетке. Полученные данные приведены в таблице 4. Инкубация с 7β-гидрокси-EPIA в трех концентрациях в течение 4 часов при отсутствии TNF-α не вызывала существенных изменений уровней PGE2, PGD2 и 15d-PGJ2 в культуральной среде. Однако содержание РGD2 и 15d-PGJ2 заметно увеличивалось при инкубации культур клеток с 7β-гидрокси-EPIA в течение 24 часов. Экспрессия СОХ-2 не была значительно изменена указанным стероидом через 4 или 24 часа, но уровни мРНК m-PGES1 значительно снизились и затем увеличились соответственно через 4 часа и 24 часа.
Присутствие TNF-α вызывало предполагаемое увеличение содержания всех простагландинов (PG) через 24 часа. Инкубация с 7β-гидрокси-EPIA в течение 4 часов не вызывала дальнейшего изменения уровней PG при любых испытанных концентрациях. В отличие от этого через 24 часа две более низкие дозы 7β-гидрокси-EPIA снижали уровень PGD2 и повышали уровень 15d-PGJ2 по сравнению с использованием только TNF-α. Инкубация с TNF-α (то есть воспалительная активация) также вызывала заметное увеличение мРНК СОХ-2 и m-PGES1. Увеличение продуцирования мРНК СОХ-2 и m-PGES1 под воздействием TNF-α замедлялось при инкубации культур клеток с 10 и 100 нМ 7β-гидрокси-EPIA в течение 24 часов.
В целом полученные результаты ясно показывают, что 7β-гидрокси-EPIA оказывает значительное противовоспалительное действие.



Пример 4
Воздействие 7β-ОН-EPIA на экспериментальную модель колита
Введение крысам сульфата натрия декстрана (DDS) в питьевой воде в течение 6 последовательных дней вызывало воспаление ободочной кишки (колит), характеризующееся уменьшением длины ободочной кишки, повышенной активностью МРО, отсутствием слизи в бокаловидных клетках и повышенной экспрессией СОХ-2 и mPGES-1-синтазы и продуцированием простагландина Е2 (PGE2). Введение DSS также увеличивало содержание маркеров окислительного стресса, таких как протеинкарбонил (Prot CO) и Т-образные складки в кишечнике. Авторы настоящего изобретения приводят данные о том, что введение 7β-гидрокси-EPIA один раз в сутки в течение 7 дней до введения DSS позволяет предотвратить развитие DSS-индуцированного колита. Введение 0,01 мг/кг 7β-гидрокси-EPIA полностью предотвращало поражение колитом и воспаление ткани, и указанное воздействие 7β-гидрокси-EPIA было ассоциировано с заметным уменьшением маркеров окислительного стресса и продуцирования PGE2, что обусловлено начальным, но временным увеличением экспрессии СОХ-2 и стабильным увеличением продуцирования противовоспалительного простагландина 15d-PGJ2. Полученные результаты показывают, что 7β-гидрокси-EPIA оказывает значительное противовоспалительное действие при очень низких дозах в данной общепринятой экспериментальной модели воспалительного заболевания кишечника (IBD).
Методика выполнения эксперимента
Животные
Все методики и способы выполнения эксперимента находились в соответствии с директивой 86/609/CEE Евросоюза об использовании лабораторных животных. Самцы крыс Wistar (180-200 г), приобретенные в компании Charles River (L'Arbresle, France), получали лабораторный корм для грызунов и воду по потребности.
Введение лекарственного средства и индукция колита
После 7-дневного периода адаптации животных распределяли в две контрольные группы (контрольная группа животных, получавших псевдолечение, и контрольная группа животных, которым вводили 7β-гидрокси-EPIA), и две группы животных, страдающих колитом (колит, не подвергаемый лечению, и колит, подвергаемый лечению 7β-гидрокси-EPIA). 7β-гидрокси-EPIA (0,01, 0,1 и 1 мг/кг массы тела, растворенный в ДМСО) или только ДМСО (наполнитель) вводили внутрибрюшинно один раз в сутки в течение 7 дней, с 0-го по 7-й день. Колит индуцировали с 7-го по 14-й день, добавляя DSS (молекулярная масса 36-50 кДа; MP Biomedicals, France) в питьевую воду. Две контрольные группы получали только водопроводную воду.
Макроскопическая оценка поражения ободочной кишки
На 9-й, 11-й, 13-й, 14-й день регистрировали массу тела, длину ободочной кишки, консистенцию стула и путем визуального осмотра оценивали свежее ректальное кровотечение. Тяжесть поражения ободочной кишки оценивали слепым методом в соответствии с описанием, приведенным в публикации Mabley et al., 2001, Inflamm. Res.; 50: 561-569) (0: хорошо сформированные гранулы и отсутствие поражения ободочной кишки; 1: ободочная кишка с небольшим количеством крови, смешанной с фекалиями; 2: ободочная кишка с большим количеством крови в фекалиях; 3: ободочная кишка, заполненная кровью при отсутствии фекалий).
Гистологическое исследование
Часть проксимального отдела ободочной кишки (1 см) фиксировали в 4% формальдегиде (Labonord, Templemars, France) и заливали парафином. Готовили срезы ткани (5 мкм, которые затем очищали, гидратировали и окрашивали гематоксилином/эозином или альциановым синим в соответствии со стандартными методиками гистологического исследования соответственно поражения ободочной кишки и содержания слизи в бокаловидных клетках.
Получение гомогенатов ткани
Ободочную кишку раскрывали по границе брыжеечной части и эпителиальные клетки соскабливали тупым краем предметного стекла, взвешивали, промывали и центрифугировали. Клеточный дебрис гомогенизировали в 9 объемах буфера ТКЕ (10 нМ трис-буфера с HCl; 150 мМ KCl; 1 мМ EDTA; 0,25 мМ PМSF, рН 7,4) и замораживали при -80°С до последующего использования. Содержание белка в гомогенатах измеряли в соответствии с описанием, приведенным в публикации Lowry et al. (J. Biol. Chem. 1951; 193: 265-74).
Активность МРО
Активность МРО определяли в гомогенатах методом на основе о-дианизидина, описанным в публикации Krawisz et al. (Gastroenterology 1984; 87(6): 1344-50), который был модифицирован в соответствии с описанием, приведенным в публикации Pelissier et al. (Steroids 2006; 71(3): 240-8). Активность МРО была выражена в виде количества фермента, необходимого для изменения в минуту оптической плотности при 460 нм с использованием коэффициента экстинкции для окисленного о-дианизидина (1,13×104 моль-1 см-1).
Биохимическое определение окислительного стресса
Перокисление липидов (Т-образные складки) измеряли методом, описанным в публикации Ohkawa et al. (Anal. Biochem. 1979; 95: 315-58), который был модифицирован в соответствии с описанием, приведенным в публикации Albrecht et al., (1992, Toxicol. Lett; 63: 91-96). Оптическую плотность аддукта малондиальдегида и тиобарбитуровой кислоты с соотношением 1:2 (Sigma-Aldrich, St. Quentin-Fallavier, France) измеряли при 532 нм (использованный коэфициент экстинкции: 0,156 мкмоль-1см-1). Содержание карбонила в окисленных белках (Prot CО) определяли методом, описанным в публикации Levine et al. (Methods Enzymol. 1990; 186: 464-78). Содержание небелковых сульфгидрильных групп (главным образом GSH), принятое в качестве маркера антиоксидантной защиты в гомогенатах, определяли методом, описанным в публикации Sedlak and Lindsay (Anal. Biochem. 1968; 25(1): 192-205).
Иммуноанализы простагландинов
Для определения уровней PGE2 (Oxford Biomedical Research, UK) и уровней 15d-PGJ2 (Assay designs, Euromedex, France) в супернатантах ободочной кишки, полученных из только что приготовленных гомогенатов, использовали коммерчески доступные наборы для EIA. Уровни PGD2 измеряли при помощи набора для EIA простагландина D2-MOX (Cayman Chemicdal, Euromedex). В данном случае до выполнения анализа только что приготовленные образцы сразу же обрабатывали реагентом МОХ-HCl, который превращал PGD2 в PGD2-MOX, предотвращая таким образом любое дальнейшее химическое разложение.
ПЦР с обратной транскриптазой в реальном времени
Общую РНК из только что приготовленных образцов ободочной кишки (300 мг) экстрагировали, используя реагент тризол (Invitrogen, Cergy-Pontoise, France). Поли(А)-РНК очищали от общей РНК при помощи набора MicroPoly(A)Purist (Amvion, Huntingdon, UK). кДНК синтезировали, используя набор для системы синтеза первой цепи Superscript (Invitrogen). ПЦР в реальном времени выполняли, используя смеси (25 мкл), содержащие раствор 2,5-кратного количества смеси RealMaster/20-кратного количества SYBR (11,25 мкл) (Eppendorf, Le Pecq, France) и 300 нМ верхних и нижних затравок. Реакции выполняли в термоблоке для проведения реакций RealPlex ep gradient S (Eppendorf). Циклизацию выполняли в следующих условиях: 10 минут при 95°С и 40 циклов при 95°С, 55°С и 68°С в течение соответственно 15 секунд, 30 секунд и 30 секунд. В результате каждого анализа была построена стандартная кривая для четырех последовательных точек разведения контрольной кДНК. Олигонуклеотидные затравки были синтезированы при помощи Genecust/Distribio (Evry, France). Специфичность амплифицированного продукта контролировали при помощи кривой плавления продукта и подтверждали, выполняя анализ методом электрофореза в агарозном геле. Уровни мРНК были выражены относительно контрольного ДМСО после нормализации до HPRT1.
Статистический анализ
Все анализы были выполнены трижды для каждого животного и результаты были представлены в виде графика среднего±стандартная ошибка среднего. Дисперсионный анализ по одному признаку выполняли на основании многократных тестов Дункана для сравнения различий между группами. Различия считались статистически значимыми при р<0,05.
Результаты
Исследование индукции колита во времени
Введение крысам 5% DSS в питьевой воде в течение 7 дней (D7-D14) вызывало клинические и гистологические признаки колита без гибели животных. У всех крыс обычно появлялась сильная диарея через 5 дней после индукции колита (12-й день) и на следующий день наблюдалось ректальное кровотечение. У всех крыс, которые получали DSS, на 11-й день было зарегистрировано уменьшение массы тела, массы эпидидимальной жировой ткани и массы печени по сравнению с контрольными животными, подвергаемыми псевдолечению (соответственно -5%, -17%, -8%; р<0,05). Снижение вышеуказанных показателей было также отмечено на 14-й день (таблица 5). Изменение массы селезенки отмечено не было. По сравнению с контрольной группой животных, подвергаемых псевдолечению, крысы, получавшие DSS, также характеризовались значительным укорачиванием ободочной кишки с 11-го дня (-14%, р<0,05) по 14-й день (-26%; р<0,05) (cм. таблицу 5), обусловленным утолщением ткани ободочной кишки и отсутствием стула. Активность МРО в слизистой оболочке ободочной кишки измеряли в виде показателя инфильтрации нейтрофилов. На 13-й и 14-й день в группе животных, страдающих колитом, было обнаружено 9-кратное и 7-кратное увеличение активности МРО в слизистой оболочке по сравнению с контрольными животными, подвергаемыми псевдолечению (р<0,05) (фиг.8,А). До 13-го дня не было отмечено изменение активности МРО. Полученные результаты сопоставимы с результатами гистологического анализа. Действительно основные признаки воспаления ободочной кишки, а именно скрытая деформация, инфильтрация нейтрофилов в ткань слизистой оболочки и утрата бокаловидных клеток, содержащих меньше муцина, появились в группе животных, страдающих колитом, на 13-й день и стали более выраженными на 14-й день по сравнению с контрольной группой животных, подвергаемых псевдолечению (данные не показаны).
Как показано на фиг.8, два маркера окислительного стресса, а именно Prot CO, Т-образные складки, и уровни GSH антиоксидантной защиты, были значительно повышены по сравнению с контрольными уровнями на 13-й и 14-й день в слизистой оболочке ободочной кишки животных, страдающих колитом.
Защитное воздействие 7β -гидрокси-EPIA в случае колита
Две низкие дозы 7β-гидрокси-EPIA (0,01, 0,1 мг/кг) предотвращали DSS-индуцированное поражение ободочной кишки, о чем свидетельствовало прекращение диареи и ректального кровотечения на 14-й день. Внутрибрюшинная инъекция 0,01 мг/кг 7β-гидрокси-EPIA один раз в сутки в течение 7 дней до введения DSS восстанавливала массу тела до контрольных уровней без изменения массы эпидидимиса (таблица 5). В период между 9-м и 14-м днями уменьшение длины ободочной кишки было менее выраженным в группах, которым вводили две более низкие дозы стероида (0,01, 0,1 мг/кг) по сравнению с группой животных, страдающих колитом (данные не показаны для 9-го дня, таблица 5). 7β-гидрокси-EPIA предотвращал уменьшение слизи в бокаловидных клетках на 14-й день и гистологические изменения, такие как патологические крипты и инфильтрация нейтрофилов (данные не показаны), и значительно снижал активность МРО при введение всех доз, 0,01, 0,1 и 1 мг/кг (фиг.8,А). Параметры окислительного стресса (Prot CО, Т-образные складки) и уровни GSH были одинаковыми на 9-й день (не показано) и 11-й день во всех группах, а именно в контрольной группе животных, подвергаемых псевдолечению, в группе животных, страдающих колитом, которых не подвергали лечению, и в группе животных, страдающих колитом, которым вводили 7β-гидрокси-EPIA (0,01, 0,1 и 1 мг/кг) (фиг.8, В, С и D). Все маркеры окислительного стресса и маркер антиоксидантной защиты не изменялись у животных, которым вводили 7β-гидрокси-EPIA (дни D0-D7), в то время как указанные параметры были значительно увеличены в группе животных, страдающих колитом (р<0,05). Полученные результаты показывают, что 7β-гидрокси-EPIA оказывает заметное противовоспалительное действие при очень низких дозах в указанной экспериментальной модели воспалительного заболевания кишечника (IBD).
Продуцирование простагландинов (PG) в ткани ободочной кишки: воздействие β-гидрокси-EPIA
Введение 7β-гидрокси-EPIA не изменяло уровни PGE2 и РGD2 в ткани ободочной кишки у контрольных крыс, не страдающих колитом (данные не показаны). В отличие от этого введение 7β-гидрокси-EPIA с 0-го дня по 7-й день вызывало значительное увеличение уровней 15d-PGJ2 со 2-го дня по 14-й день по сравнению с исходными уровнями. Введение 0,1 мг/кг 7β-гидрокси-EPIA вызывало 51-кратное увеличение уровня на 2-й день, который постепенно снижался с 4-го дня по 14-й день (уменьшение с 44-кратного до 5-кратного значения) (фиг.9,А, фиг.10,С).
Введение воспалительного агента DSS с 7-го дня по 14-й день вызывало заметное увеличение продуцирования провоспалительного простагландина PGE2 на 14-й день, при этом уровень противовоспалительного простагландина 15d-PGJ2 значительно снизился (фиг.10).
Введение 0,01 мг/кг 7β-гидрокси-EPIA с 0-го дня по 7-й день полностью предотвращало DSS-индуцированное продуцирование PGE2 в ободочной кишке и при этом значительно увеличивало уровень 15d-PGJ2 (фиг.10).
Экспрессия СОХ-2 и PG-синтазы в группах псевдобольных животных и животных, страдающих колитом: воздействие предварительно вводимого 7β-гидрокси-EPIA
В связи со значительными изменениями уровней простагландинов при введении DSS и 7β-гидрокси-EPIA авторы настоящего изобретения исследовали изменение экспрессии СОХ-2, mPGES-1 и H-PGDS в результате предварительного введения стероида путем количественного определения специфической мРНК при помощи ПЦР с обратной транскриптазой (RT-PCR) в реальном времени. Исследовали транскрипцию указанных генов и сравнивали с транскрипцией обязательного гена HPRT1. У контрольных крыс, не страдающих колитом, введение 0,1 мг/кг 7β-гидрокси-EPIA вызывало значительное 1,5-кратное увеличение уровней мРНК СОХ-2 через 15 часов при значительном уменьшении на 2-й день и 4-й день с последующим возвратом к исходным значениям (фиг.9,В). Экспрессия мРНК mPGES-1 временно повышалась в период между 6-ю и 15-ю часами и возвращалась к исходному уровню на 2-й день. Синтез мРНК H-PGDS не изменялся в ходе эксперимента.
После индукции колита под воздействием DSS на 13-й и 14-й день наблюдалось 2,5-кратное увеличение экспрессии мРНК СОХ-2 (фиг.11,А), в то время как количество мРНК mPGES-1 значительно увеличивалось только на 13-й день (фиг.11,В). При введении DSS количество H-PGDS не изменялось.
Предварительное введение 7β-гидрокси-EPIA (с 0-го дня по 7-й день) подавляло увеличение синтеза мРНК как СОХ-2, так и mPGES-1 под воздействием DSS (фиг.11).
Полученные результаты показывают, что противовоспалительное действие 7β-гидрокси-EPIA обусловлено одновременным уменьшением продуцирования РGE2 и увеличением продуцирования 15d-PGJ2. Длительное воздействие 7β-гидрокси-EPIA на 15d-PGJ2, имевшее место в данном исследовании, позволяет предположить, что 7β-гидрокси-EPIA вызывает продолжительное изменение экспрессии генов, участвующих в воспалении и его разрешении.
На фиг.8 показано воздействие 7β-гидрокси-EPIA на (А) миелопероксидазу (МРО) и маркеры окислительного стресса, а именно (В) Prot CO, (C) Т-обратные складки и (D) маркер антиоксидантной защиты GSH в ткани ободочной кишки. Уровни биомаркеров показывают, что крысы, получавшие DSS, существенно отличались от контрольных животных на 13-й и 14-й день (р<0,05). 7β-Гидрокси-EPIA значительно снижал активность МРО на 13-й и 14-й день по сравнению с группой крыс, страдающих колитом (р<0,05), и восстанавливал параметры окислительного стресса (В, С и D) до контрольных уровней на 13-й и 14-й день при всех вводимых дозах. Значения выражены в виде среднего±стандартная ошибка среднего (А) и процентного значения для группы псевдобольных крыс (В, С и D) при наличии 3-23 крыс в группе. На фиг.8,А-8,D черные столбцы = контрольная группа псевдобольных крыс; белые столбцы = группа крыс, страдающих DSS-индуцированным колитом; темно-серые столбцы = DSS + 0,01 мг/кг 7β-гидрокси-EPIA; умеренно серые столбцы = DSS + 0,1 мг/кг 7β-гидрокси-EPIA и светло-серые столбцы = DSS + 1,0 мг/кг 7β-гидрокси-EPIA.
Группа крыс, страдающих DSS-индуцированным колитом, по сравнению с контрольной группой всевдобольных крыс (р<0,05) группы крыс, которым вводили 7β-гидрокси-EPIA, по сравнению с группой крыс, страдающих DSS-индуцированным колитом (р<0,05).
На фиг.9 показан уровень 15d-PGJ2 в ободочной кишке (А) и количественное определение относительной экспрессии мРНК СОХ-2, mPGES-1 и H-PGDS (В) в разные периоды введения 7β-гидрокси-EPIA. Значения выражены в виде среднего±стандартная ошибка среднего (n=3-15) (А) и величины мРНК представлены в сравнении с величинами мРНК в контрольной группе псевдобольных животных после нормализации до HPRT1 (В). На фиг.9,А черный столбец = контрольная группа псевдобольных крыс; темно-серый столбец = 0,01 мг/кг 7β-гидрокси-EPIA; умеренно серый столбец = 0,1 мг/кг 7β-гидрокси-EPIA и светло-серый столбец = 1,0 мг/кг 7β-гидрокси-EPIA. На фиг.9,В темно-серый столбец с белыми точками = СОХ-2; светло-серый столбец с черными точками = mPGES-1 и белый столбец с черными точками = Н-PGDS.
На фиг.10 показано воздействие 7β-гидрокси-EPIA на синтез в ободочной кишке простагландинов E2, D2 и 15d-PGJ2 во время введения DSS. Введение DSS значительно увеличивало синтез PGE2 (A), D2 (B) и 15d-PGJ2 (C)(р<0,05) на 13-й и 14-й день (р<0,05). Введение 7β-гидрокси-EPIA в течение 7 дней до введения DSS уменьшало синтез PGE2 на 13-й и 14-й день (А) при значительном увеличении продуцирования 15d-PGJ2 при всех вводимых дозах (С). Данные выражены в виде среднего±стандартная ошибка среднего при значении n=3-23. На фиг.10,А-10,С черный столбец = контррольная группа псевдобольных животных; белый столбец = группа животных, страдающих DSS-индуцированным колитом; темно-серый столбец с белыми точками = DSS + 0,01 мг/кг 7β-гидрокси-EPIA; светло-серый столбец с черными точками = DSS + 0,1 мг/кг 7β-гидрокси-EPIA; и белый столбец с черными точками = DSS + 1,0 мг/кг 7β-гидрокси-EPIA.
На фиг.5 показана экспрессия в ободочной кишке мРНК СОХ-2, mPGES-1 и H-PGDS во время индукции колита. Колит вызывал значительное увеличение синтеза мРНК СОХ-2 на 13-й и 14-й день (А), при этом синтез mPGES-1 возрастал только на 13-й день (В). Возврат к исходным уровням экспрессии генов наблюдался при всех дозах 7β-гидрокси-EPIA. На фиг.10,А-10,С белый столбец = группа животных, страдающих DSS-индуцированным колитом; темно-серый столбец = DSS + 0,01 мг/кг 7β-гидрокси-EPIA; умеренно серый столбец = DSS + 0,1 мг/кг 7β-гидрокси-EPIA; и светло-серый столбец = DSS + 1,0 мг/кг 7β-гидрокси-EPIA.
Пример 5
1. Воздействие 7β-ОН-EPIA на индуцированный коллагеном артрит
При выполнении данного эксперимента была исследована эффективность контролирования 7β-ОН-EPIA воспалительных и патологических изменений, возникших в модели индуцированного коллагеном артрита. Животная модель с использованием мышей демонстрирует клиническую реминисценцию артрита человека.
Самцам мышей DBA/1 (в возрасте 10-12 недель) вводили 100 мкг куриного коллагена типа II, эмульгированного в полном адъюванте Фрейнда (CII/CFA), в 0-й день путем инъекции в основание хвоста. Введение проводили ежедневно с 20-го по 50-й день путем подкожной инъекции (группы 2, 3 и 4) и с 0-го по 30-й день путем подкожной инъекции (группа 5) следующим образом:
Экспериментальные группы (n=10/группа)
В конце эксперимента задние конечности удаляли, коленные гомогенаты собирали для измерения простагландинов (только группы 1 и 4) и лапы фиксировали для исследования патологии стандартными методами. Каждую лапу наполовину надрезали в продольном направлении, рассекали и декальцинировали для изготовления срезов. Декальцинированные образцы подвергали стандартной обработке, делали срезы и изготавливали один окрашенный срез для исследования. Срезы получали из обеих половин каждого образца. Каждую лапу оценивали по стандартной системе оценок. Образцы оценивали слепым методом, не имея представления о методике эксперимента или принадлежности к группе.
Клиническое опухание суставов оценивали два раза в неделю с 21-го по 50-й день. Каждой из четырех конечностей присваивали число баллов в соответствии с нижеследующей системой оценок (объединенная клиническая оценка заболевания):
Результаты приведены в таблице 6. Из указанной таблицы видно, что введение 7β-ОН-EPIA вызывало заметное уменьшение опухания сустава, измеренное вышеуказанным методом клинической оценки заболевания, по сравнению с контрольными группами (контрольные группы псевдобольных животных 1 и 6 были объединены). Данный результат был подтвержден данными патологического исследования, свидетельствующими о меньшем опухании лап (таблица 7). Из таблицы 7 видно, что самая низкая оценка патологии, в соответствии с которой у 9 из 10 животных полностью отсутствовало гистопатологическое изменение в суставах лап, имела место в том случае, когда 7β-гидрокси-EPIA начинали вводить одновременно с введением коллагена, индуцирующего артрит (0-й день), и продолжали лечение до 30-го дня. Таким образом, лечение 7β-гидрокси-EPIA таких заболеваний, как ревматоидный артрит, может оказывать благоприятное воздействие.
2. Измерение простагландинов в коленных гомогенатах
Уровни 15d-PGJ2 и PGE2 в коленных гомогенатах, полученных выше, измеряли стандартными методами. Как видно из таблицы 8, уровни PGE2 были ниже в группе, которой вводили 0,1 мг/кг 7β-ОН-EPIA, по сравнению с контрольной группой, не подвергаемой лечению. В группе, которой вводили 0,1 мг/кг 7β-ОН-EPIA, уровни 15d-PGJ2 были выше по сравнению с контрольной группой, не подвергаемой лечению.
Заключение
7β-Гидрокси-EPIA оказывал четко выраженное противовоспалительное действие. Лечение во время индукции заболевания оказывало наибольшее воздействие. Такой же результат был получен для других испытуемых соединений, включая стероиды, испытанных в данной модели, и отражает относительную легкость предотвращения поражения суставов по сравнению с восстановлением существующих изменений. Состояние животных, которым вводили более высокие дозы 7β-гидрокси-EPIA, было таким же, когда лечение начинали с 20-го дня. Такая степень защиты от заболевания является достаточно высокой для схемы лечения. Самая низкая доза 7β-гидрокси-EPIA, по-видимому, оказывает меньшее воздействие на заболевание, хотя необходим статистический анализ для выявления того, что полученный результат является следствием зависимости доза-эффект.
Анализ уровней простагландинов Е2 и 15d-J2 показал наличие ясно выраженного эффекта лечения. Уровни PGE2 были ниже у мышей, подвергаемых лечению, по сравнению с контрольными животными, не подвергаемыми лечению. В отличие от этого уровни 15d-PGJ2 повысились после лечения.
Клиническое наблюдение за течением болезни и данные гистопатологии являются убедительным свидетельством противовоспалительного действия 7β-гидрокси-EPIА в рассмотренной модели артрита. Лечение до возникновения заболевания было наиболее эффективным, что находит почти неизменное подтверждение в данной модели. Однако то, что 7β-гидрокси-EPIA может подавлять развитие заболевания при введении во время его возникновения, выделяет данное соединение из многих конкурентных препаратов и является весьма обнадеживающим.
На основании приведенных результатов можно сделать вывод о том, что 7β-гидрокси-EPIA, вводимый в низких дозах, таких как 1,0 мкг/кг, может уменьшать поражение суставов.
На основании приведенных результатов можно сделать вывод о том, что 7β-гидрокси-EPIA, вводимый в дозе 10,0 мкг/кг, почти полностью предотвращает развитие заболевания, то есть поражение суставов.
На основании приведенных результатов можно сделать вывод о том, что 7β-гидрокси-EPIA ослабляет тяжесть клинических симптомов индуцированного коллагеном артрита и изменяет продуцирование простагландинов в тканях в пользу разрешения воспаления, защиты клеток и восстановления тканей.
Пример 6
Действие 7β-ОН-EPIA в модели индуцированной цисплатином периферической невропатии
Цисплатин является антимитотическим соединением, используемым для лечения разных типов рака. Однако его применение ограничено из-за нескольких вредных эффектов, среди которых особенно неприятными являются периферические невропатии. Индуцируемые цисплатином невропатии являются главным образом сенсорными невропатиями. У субъектов пропадает чувствительность в дистальных отделах конечностей с последующим возникновением сенсорной атаксии. Гистологические исследования позволили выявить дегенерацию аксонов. Воздействие цисплатинов на сенсорные нейроны в культурах клеток вызывало уменьшение плотности сети аксонов и последующую дегенерацию клеточного тельца. Фактор роста нервов (NGF), специфический фактор роста сенсорных нейронов, защищает нейроны от указанной интоксикации. Таким образом, интоксикация сенсорных нейронов в культуре цисплатином является приемлемой моделью для исследования нейрозащитного действия соединений в случае периферических невропатий.
Нейрозащитное действие 7β-ОН-EPIA оценивали в отношении сенсорных нейронов крыс в модели периферической невропатии.
Первичные культуры сенсорных нейронов диссоциированного ганглия задних корешков спинного мозга инкубировали с 3 мкг/мл цисплатина в присутствии и в отсутствие 7β-ОН-EPIA в течение 48 и 72 часов и оценивали следующие параметры:
- клеточные тельца нейронов, окрашенные антителом против МАР2 (ассоциированный с микроканальцами белок 2);
- плотность сети аксонов, окрашенных антителом против β-тубулина.
Культуры нейронов
Сенсорные нейроны крыс были получены методом, описанным в публикации Холла и др., 1997 [Hall et al. (J. Neurosci. 1997 Apr. 15; 17(8); 2775-84]. Самок крыс (15-дневная беременность) умерщвляли путем смещения шейного отдела позвоночника (Rats Wistar; Janvier, Le Genest-St-Isle, France) и удаляли из матки плоды. Удаляли спинной мозг с ганглием задних корешков (DRG) и помещали в охлаждаемую льдом среду Лейбовица (L15, Fisher 11415-049), содержащую пенициллин 50 ед./мл - стрептомицин 50 мкг/мл (PS, 1%) и бычий сывороточный альбумин (BSA 1%, Sigma A6003). Выделяли DRG и диссоциировали, обрабатывая трипсином, в течение 20 минут при 37°С (трипсин EDTA, 10-кратное количество, 10%, Fisher 15400054), разведенным в PBS без кальция и магния (Fisher 2007-03). Реакцию прекращали, добавляя модифицированную по способу Дульбекко среду Игла (DMEM, Fisher 21969-035), содержащую ДНКазу I сорта II (0,1 мг/мл, Roche diagnostic 104159) и фетальную телячью сыворотку (FBS 10%, Fisher 10270-098). Взвесь клеток растирали 10 мл пипеткой и центрифугировали с ускорением 350×g в течение 10 минут при комнатной температуре. Дебрис диссоциированных клеток ресуспендировали в культуральной среде определенного состава.
Жизнеспособные клетки подсчитывали в цитометре Neubauer при помощи теста на исключение с окрашиванием трипановым синим (Sigma) и высевали с плотностью 30000 клеток/лунка на 96-луночные планшеты (Nunc). Лунки предварительно сенсибилизировали поли-L-лизином (10 мкг/мл, Sigma P2636) в ультрачистой стерильной воде (Merck Eurolab 60759.01).
Клетки оставляли для адгезии в течение 2 часов и выдерживали во влажной камере при 37°С в атмосфере 5% СО2/95% воздуха.
Инкубация культур нейронов с 7β-ОН-EPIA
После культивирования в течение 5 дней культуральную среду заменяли культуральной средой определенного состава в соответствии с нижеуказанными разными условиями:
- наполнитель (ДМСО 0,1%)
- наполнитель (ДМСО 0,1%) + цисплатин (3 мкг/мл, Sigma ref: p4394)
- испытуемое соединение 7β-ОН-EPIA (1 нМ, 10 нМ и 100 нМ) + цисплатин (3 мкг/мл)
- эталонное соединение NGF (10 нг/мл) + цисплатин (3 мкг/мл)
Выживание нейронов оценивали, используя шесть лунок для каждого условия. После инкубации в течение 48 часов и 72 часов нейроны фиксировали в течение 5 минут в растворе этанола/уксусной кислоты (95%/5%) при -20°С и трижды промывали в PBS.
Нейротрофическое действие соединений контролировали, инкубируя NGF (10 нг/мл) и 7β-ОН-EPIA (1 нМ, 10 нМ и 100 нМ) в течение 48 часов и 72 часов. В конце инкубации клетки фиксировали в течение 5 минут в растворе этанола/уксусной кислоты (95%/5%) при -20°С и трижды промывали PBS.
Анализ числа клеточных телец и сети аксонов в поле зрения
Клеточные тельца сенсорных нейронов метили моноклональным антителом против МАР-2 (Sigma M4403) и аксоны сенсорных нейронов метили моноклональным антителом против β-тубулина (Sigma T8660). Указанные антитела разводили в отношении 1:400 в растворе для инкубации (PBS с 5% FCS и 0,1% сапонина, Sigma S-7900). Указанные антитела специфически метят клеточные тельца нейронов и аксоны.
Клетки инкубировали в течение 2 часов, промывали в PBS и инкубировали с IgG козы против мыши Alexa Fluor 488 (Molecular Probes A11001), разведенным в отношении 1:300 в растворе для инкубации, для выявления антител против МАР-2 и β-тубулина. Ядра клеток окрашивали флуоресцентным маркером (окрашивающий раствор Hoechst, SIGMA H6024, 1 мкг/мл в растворе для инкубации в течение 1 часа).
Для каждого условия было сделано 2 снимка каждой лунки (12 снимков для каждого условия) в анализаторе In Cell Analyzer 1000 (Amersham Biosciences) с использованием компьютерного программного обеспечения для In Cell Analyzer 1000 3.2. Для мечения МАР-2 было использовано 10-кратное увеличение, для мечения β-тубулина было использовано 20-кратное увеличение. Все снимки были сделаны для каждого мечения в одинаковых условиях.
Анализ числа клеточных телец, меченных антителами против МАР-2, и общей длины аксонов, меченных антителами против β-тубулина, был выполнен при помощи программного обеспечения для рабочей станции In Cell Analyser 1000 3.2. Результаты были выражены в процентах по сравнению с наполнителем. Каждую группу сравнивали при помощи непарного t-критерия.
Результаты
Защита, обеспечиваемая 7β-ОН-EPIA, от индуцируемой цисплатином утраты плотности аксонов
Инкубация с 3 мкг/мл цисплатина в течение 48 часов
Плотность аксонов была в среднем равна 5459 мкм от общей длины аксонов в поле зрения после инкубации сенсорных нейронов в среде (“наполнитель”), то есть без цисплатина, в течение 48 часов. Инкубация с цисплатином в течение 48 часов уменьшала общую длину аксонов в поле зрения примерно до 4585 мкм. Такое уменьшение плотности сети аксонов цисплатином было статистически значимым (-16%, р<0,001) по сравнению с наполнителем.
Инкубация с 10 нг/мл NGF в течение 48 часов предотвращала индуцированную цисплатином утрату аксонов и вызывала значительное увеличение длины аксонов по сравнению с культурами, инкубируемыми только в среде.
Инкубация с 1 нМ и 10 нМ 7β-ОН-EPIA защищала сенсорные нейроны от индуцированной цисплатином утраты аксонов через 48 часов. Указанный эффект был статистически значимым. Общая длина аксонов была равна 5378 мкм и 5549 мкм после инкубации в течение 48 часов соответственно с 1 нМ и 10 нМ 7β-ОН-EPIA, что соответствует уменьшению индуцированной цисплатином токсичности соответственно на 90,7% и 101%.
Инкубация с 3 мкг/мл цисплатина в течение 72 часов
Сенсорные нейроны в среде “наполнитель” без цисплатина были в среднем равны 5320 мкм от общей длины аксонов в поле зрения. Инкубация с цисплатином в течение 72 часов уменьшала общую длину аксонов в поле зрения примерно до 4046 мкм. Такое уменьшение плотности сети аксонов цисплатином было статистически значимым (-24%, р<0,001) по сравнению с наполнителем.
Инкубация с 10 нг/мл NGF в течение 72 часов предотвращала индуцированную цисплатином утрату аксонов и вызывала значительное увеличение длины аксонов по сравнению с культурами, инкубируемыми только в среде.
Инкубация с 1 нМ 7β-ОН-EPIA защищала сенсорные нейроны от индуцированной цисплатином утраты аксонов через 72 часа. Указанный эффект был статистически значимым. Общая длина аксонов была равна 4948 мкм после инкубации в течение 72 часов с 1 нМ 7β-ОН-EPIA, что соответствует уменьшению индуцированной цисплатином токсичности на 70,8%.
10 нМ
1 нМ
5549,41
5377,89
146
106
12
12
102
99
p<0,001
p<0,001
10 нМ
1 нМ
4275,04
4947,99
125,14
130,81
12
12
80
93
p>0,05
p<0,001
Защита, обеспечиваемая 7β-ОН-EPIA от индуцируемой цисплатином гибели нейронов (утрата клеточных телец нейронов)
Инкубация с 3 мкг/мл цисплатина в течение 48 часов
После инкубации в среде с “наполнителем” без цисплатина в течение 48 часов в среднем был обнаружен 61 сенсорный нейрон в поле зрения. Инкубация с 3 мкг/мл цисплатина уменьшала число нейронов в среднем до 41 сенсорного нейрона в поле зрения. Утрата клеточных телец нейронов под воздействием цисплатина была статистически значимой (-33%, р<0,001) по сравнению с числом клеточных телец, обнаруженных после инкубации в течение 48 часов без цисплатина, то есть в среде, содержащей только наполнитель.
Инкубация с 10 нг/мл NGF в течение 48 часов почти полностью предотвращала индуцируемую цисплатином утрату клеточных телец.
Инкубация с 1 нМ, 10 нМ и 100 нМ 7β-ОН-EPIA защищала сенсорные нейроны от индуцируемой цисплатином гибели клеток через 48 часов. Указанный эффект был статистически значимым. Общее число сенсорных нейронов в поле зрения было равно 51, 45 и 50 после инкубации в течение 48 часов соответственно с 1 нМ, 10 нМ и 100 нМ 7β-ОН-EPIA, что соответствует уменьшению индуцируемой цисплатином гибели клеток соответственно на 53,1%, 18,7% и 43,8%.
Инкубация с 3 мкг/мл цисплатина в течение 72 часов
После инкубации в среде с “наполнителем” без цисплатина в течение 72 часов в среднем было обнаружено 54 сенсорных нейрона в поле зрения. Инкубация с 3 мкг/мл цисплатина уменьшала число нейронов в среднем до 33 сенсорного нейронов в поле зрения. Утрата клеточных телец нейронов под воздействием цисплатина была статистически значимой (-39%, р<0,001) по сравнению с числом клеточных телец, обнаруженных после инкубации в течение 72 часов без цисплатина, то есть в среде, содержащей только наполнитель.
Инкубация с 10 нг/мл NGF в течение 48 часов почти полностью предотвращала индуцируемую цисплатином утрату клеточных телец.
Инкубация с 1 нМ, 10 нМ и 100 нМ 7β-ОН-EPIA почти полностью защищала сенсорные нейроны от индуцируемой цисплатином гибели клеток через 72 часа. Указанный эффект был статистически значимым. Общее число сенсорных нейронов в поле зрения было равно 52, 55 и 52 после инкубации в течение 72 часов соответственно с 1 нМ, 10 нМ и 100 нМ 7β-ОН-EPIA, что соответствует уменьшению индуцируемой цисплатином гибели клеток соответственно на 93%, 104% и 93%.
10 нМ
1 нМ
45,50
51,42
1,26
2,11
12
12
75
85
p<0,05
p<0,001
10 нМ
1 нМ
54,58
52,42
1,37
1,65
12
12
1,2
98
p<0,001
p<0,001
Пример 7
В настоящем исследовании оценивали стимулирующее воздействие 7β-ОН-EPIA на рост аксонов в диссоциированных двигательных и сенсорных нейронах спинного мозга в первичных культурах. Нейроны окрашивали антителом против β-тубулина и определяли общую длину нейронов в анализаторе “In Cell Analyzer”. Культуры нейронов инкубировали с 10-9 М - 10-4 М 7β-ОН-EPIA и определяли плотность сети нейронов после инкубации в течение 6 часов, 12 часов и 24 часов. В качестве эталонных соединений использовали выделенный из головного мозга фактор роста нервов (BDNF), специфический фактор роста для двигательных нейронов и фактор роста нервов (NGF), специфический фактор роста для сенсорных нейронов. Полученные данные показывают, что 7β-ОН-EPIA оказывает значительное нейротрофическое действие как на двигательные нейроны, так и на сенсорные нейроны. Указанные воздействия, которые были сравнимы с воздействиями BDNF и NGF, были особенно выраженными при наномолярных концентрациях гидроксистероида. В целом полученные результаты подтверждают возможность использования 7-гидроксистероидов для стимуляции разрастания аксонов и лечения периферической невропатии.
1. Материалы и методы
1.1. Получение культур двигательных нейронов спинного мозга крыс
Двигательные нейроны спинного мозга крыс были получены методом, описанным в публикации Мартиноу и др., 1992 (Martinou JC, Martinou I, Kato AC, Cholinergic differentiation factor (CDF/LIF) promotes survival of isolated rat embryonic motoneurons in vitro. Neuron. 1992 Apr.; 8(4): 737-44). Беременных самок крыс Wistar с 15-дневным сроком беременности умерщвляли путем смещения шейного отдела позвоночника и удаляли из матки плоды. Спинной мозг удаляли и помещали в охлаждаемую льдом среду Лейбовица (L15, Invitrogen, 11415-049), содержащую 1% бычьего сывороточного альбумина (BSA без жирной кислоты, Eurobio, Les Ulis, France, GXXBSA01-65). Мозговые оболочки тщательно удаляли.
Двигательные нейроны спинного мозга диссоциировали, обрабатывая трипсином, в течение 20 минут при 37°С (трипсин EDTA, 10-кратное количество, Invitrogen 15400054), разводили в PBS без кальция и магния (Invitrogen 2007-03). Реакцию прекращали, добавляя модифицированную по способу Дульбекко среду Игла (DMEM, Invitrogen 21969-035), содержащую ДНКазу I сорта II (0,1 мг/мл, Roche diagnostic 104159) и 10% фетальной телячьей сыворотки (FСS, Invitrogen 10270098). Взвесь клеток механически диссоциировали путем 3-кратного пропускания через 10-мл пипетку. Затем клетки центрифугировали с ускорением 580 g в течение 10 минут при комнатной температуре. Дебрис диссоциированных клеток ресуспендировали в среде L15 и полученную взвесь обогащали двигательными нейронами, осуществляя центрифугирование с ускорением 180×g в течение 10 минут при комнатной температуре на слое раствора BSA (3,5%) в среде L15. Супернатант удаляли и дебрис ресуспендировали в среде L15, содержащей ДНКазу I (1%). Затем взвесь помещали на градиент оптипрепа (плотность: 1,06 г/мл; Abcys, Paris, France; 1030061) и центрифугировали с ускорением 335×g в течение 15 минут при комнатной температуре. Верхнюю фазу, содержащую очищенные двигательные нейроны, собирали, ресуспендировали в среде L15 и центрифугировали с ускорением 800×g в течение 10 минут при комнатной температуре. Затем дебрис ресуспендировали в культуральной среде определенного состава, состоящей из нейробазальной среды, содержащей 2% В27, 2 мМ L-глутамина и пенициллин 50 ед./мл - стрептомицин 50 мкг/мл. Жизнеспособные клетки подсчитывали в цитометре Neubauer при помощи теста на исключение с окрашиванием трипановым синим (Sigma Т8154), затем высевали с плотностью 30000 клеток/лунка на 96-луночные планшеты (предварительно сенсибилизированные поли-L-лизином; Nunclon, Invitrogen P5899) и культивировали при 37°С во влажной камере в атмосфере, состоящей из воздуха (95%) и СО2 (5%).
1.2. Получение культур сенсорных нейронов крыс
Сенсорные нейроны крыс были получены методом, описанным в публикации Холла и др. 1997 (Hall AK, Ai X, Hickman GE, MacPhedran SE, Nduaguba CO, Robertson CP. The generation of neuronal heterogeneity in a rat sensory ganglion. J. Neurosci. 1997, Apr 15; 17(8): 2775-84). Самок крыс (15-дневная беременность) умерщвляли путем смещения шейного отдела позвоночника (Rats Wistar; Janvier, Le Genest-St-Isle, France) и удаляли из матки плоды. Удаляли спинной мозг с ганглием задних корешков (DRG) и помещали в охлаждаемую льдом среду Лейбовица (L15, Fisher 11415-049), содержащую пенициллин 50 ед./мл - стрептомицин 50 мкг/мл (PS, 1%) и бычий сывороточный альбумин (BSA 1%, Sigma A6003). DRG выделяли и диссоциировали, обрабатывая трипсином, в течение 20 минут при 37°С (трипсин EDTA, 10-кратное количество, 10%, Fisher 15400054), разводили в PBS без кальция и магния (Fisher 2007-03). Реакцию прекращали, добавляя модифицированную по способу Дульбекко среду Игла (DMEM, Fisher 21969-035), содержащую ДНКазу I сорта II (0,1 мг/мл, Roche diagnostic 104159) и фетальную телячью сыворотку (FBS 10%, Fisher 10270-098). Взвесь клеток растирали 10-мл пипеткой и центрифугировали с ускорением 350×g в течение 10 минут при комнатной температуре. Дебрис диссоциированных клеток ресуспендировали в культуральной среде определенного состава.
Жизнеспособные клетки подсчитывали в цитометре Neubauer при помощи теста на исключение с окрашиванием трипановым синим (Sigma) и высевали с плотностью 25000 клеток/лунка на 96-луночные планшеты (Nunc). Лунки предварительно сенсибилизировали поли-L-лизином (10 мкг/мл, Sigma P2636) в ультрачистой стерильной воде (Merck Eurolab 60759.01).
Клетки оставляли для адгезии в течение 2 часов и выдерживали во влажной камере при 37°С в атмосфере 5% СО2/95% воздуха.
1.3. Инкубация культур двигательных нейронов спинного мозга с активными соединениями
Культуральную среду заменяли культуральной средой определенного состава после инкубации в течение 12 часов в соответствии с описанными ниже условиями:
контрольная культура (наполнитель, 0,1% ДМСО);
7β-ОН-EPIA 10-4 М, 10-5 М, 10-6 М, 10-7 М, 10-8 М и 10-9 М (в 0,1% ДМСО);
BDNF 50 нг/мл и 10 нг/мл (в 0,1% ДМСО).
После инкубации в течение 6 часов, 12 часов и 24 часов клетки фиксировали в течение 5 минут в растворе этанола/уксусной кислоты (95%/5%) при -20°С и трижды промывали в PBS.
1.4. Инкубация культур сенсорных нейронов с активными соединениями
Культуральную среду заменяли культуральной средой определенного состава после инкубации в течение 12 часов в соответствии с описанными ниже условиями:
контрольная культура (наполнитель, 0,1% ДМСО);
7β-ОН-EPIA 10-4 М, 10-5 М, 10-6 М, 10-7 М, 10-8 М и 10-9 М (в 0,1% ДМСО);
NGF 50 нг/мл и 10 нг/мл (в 0,1% ДМСО).
После инкубации с 7β-ОН-EPIA в течение 6 часов, 12 часов и 24 часов клетки фиксировали в течение 5 минут в растворе этанола/уксусной кислоты (95%/5%) при
-20°С и трижды промывали в PBS.
1.5. Анализ роста аксонов
Двигательные и сенсорные нейроны метили моноклональным антителом против β-тубулина (Sigma T8660), разведенным в отношении 1:400 в растворе для инкубации (PBS с 5% FCS и 0,1% самонина, Sigma S-7900). Указанное антитело специфически метит клеточные тельца и аксоны нейронов.
После инкубации в течение 2 часов клетки промывали в PBS и инкубировали с IgG козы против мыши Alexa Fluor 488 (Molecular Probes A11001), разведенным в отношении 1:300 в растворе для инкубации для выявления антител против β-тубулина III.
Анализ общей длины нейронов, меченных антителами против β-тубулина (нейроны), был выполнен при помощи программного обеспечения для рабочей станции In Cell Abalyser 1000 3.2.
Результаты выражены в процентнах по сравнению с наполнителем. Группы сравнивали при помощи непарного t-критерия Стьюдента.
2. Результаты
2.1. Воздействие введения лекарственного средства на длину аксонов двигательных нейронов
Анализ общей величины удлинения клеток свидетельствует о нейротрофических свойствах испытуемых соединений.
а) После инкубации в течение 6 часов
Результаты приведены в таблице 13. Введение BDNF в количестве 50 нг/мл и 10 нг/мл значительно увеличивало плотность сети аксонов, образованной двигательными нейронами, соответственно на 180% и 193% по сравнению с наполнителем (р<0,001).
Инкубация с 10-8 М и 10-9 М 7β-ОН-EPIA в течение 6 часов значительно увеличивала плотность сети аксонов двигательных нейронов соответственно на 150% и 188% по сравнению с наполнителем (р<0,001).
b) После инкубации в течение 12 часов
Результаты приведены в таблице 14. Введение 50 нг/мл и 10 нг/мл BDNF значительно увеличивало плотность сети аксонов, образованной двигательными нейронами, соответственно на 152% и 173% по сравнению с наполнителем (р<0,001).
Инкубация с 10-4 М - 10-9 М 7β-ОН-EPIA в течение 12 часов знчительно увеличивала плотность сети аксонов на 226%-137% по сравнению с наполнителем.
с) После инкубации в течение 24 часов
Результаты приведены в таблице 15. Инкубация с 10 нг/мл BDNF в течение 24 часов значительно увеличивала плотность сети аксонов по сравнению с наполнителем (170%, р<0,01). Однако инкубация с 50 нг/мл BDNF не вызывала значительного изменения плотности сети аксонов.
Инкубация с 10-8 М 7β-ОН-EPIA в течение 24 часов увеличивала плотность сети аксонов на 174% по сравнению с наполнителем (р<0,005).
2.2. Воздействие введения лекарственного средства на длину аксонов сенсорных нейронов
а) После инкубации в течение 6 часов
Результаты приведены в таблице 16. Введение 50 нг/мл и 10 нг/мл NGF в течение 6 часов значительно увеличивало плотность сети аксонов, образованной сенсорными нейронами, соответственно на 273% (р<0,001) и 191% (р<0,01).
Инкубация с 10-7 М, 10-8 М и 10-9 М 7β-ОН-EPIA в течение 6 часов увеличивала плотность сети аксонов сенсорных нейронов соответственно на 171% (р<0,001), 176% (р<0,005) и 149% (р<0,05) по сравнению с контрольной культурой клеток.
b) После инкубации в течение 12 часов
Результаты приведены в таблице 17. Инкубация с 50 нг/мл и 10 нг/мл NGF значительно увеличивала плотность сети аксонов, образованной сенсорными нейронами, по сравнению с наполнителем соответственно на 216% (р<0,001) и 128% (р<0,05).
Инкубация с 10-9 М и 10-7 М 7β-ОН-EPIA значительно увеличивала плотность сети аксонов по сравнению с наполнителем соответственно на 145% (р<0,001) и 134% (р<0,05).
с) После инкубации в течение 24 часов
Результаты приведены в таблице 18. После инкубации в течение 24 часов с 50 нг/мл и 10 нг/мл NGF значительно увеличивалась плотность сети аксонов, образованной сенсорными нейронами, соответственно на 309% и 371% по сравнению с наполнителем (р<0,001).
После инкубации с 10-7 М, 10-8 М и 10-9 М 7β-ОН-EPIA в течение 24 часов плотность сети аксонов увеличивалась по сравнению с наполнителем соответственно на 173% (р<0,005), 174% (р<0,01) и 147% (р<0,05).
10 нг/мл
489,5
19,0
12
193
р<0,001
10-5 М
10-6 М
10-7 М
10-8 М
10-9 М
293,3
354,5
350,5
382,4
479,3
22,8
33,7
34,3
25,8
40,8
12
12
12
12
12
115
139
138
150
188
р<0,05
p<0,05
p<0,05
p<0,001
p<0,001
10 нг/мл
731,3
34,8
12
173
р<0,001
10-5 М
10-6 М
10-7 М
10-8 М
10-9 М
680,9
580,3
956,7
759,0
764,9
30,8
55,5
54,6
61,6
59,9
12
12
12
12
12
161
137
137
179
180
р<0,001
p<0,05
p<0,001
p<0,001
p<0,001
10 нг/мл
366,1
29,6
12
170
р<0,001
10-5 М
10-6 М
10-7 М
10-8 М
10-9 М
200,9
224,8
271,7
374,6
265,9
20,7
36,7
34,0
22,3
26,2
12
12
12
12
12
93
104
126
174
123
р<0,05
p<0,05
p<0,05
p<0,005
p<0,05
10 нг/мл
195,2
28,8
12
191
р<0,01
10-5 М
10-6 М
10-7 М
10-8 М
10-9 М
114,8
97,7
151,8
179,3
174,2
8,2
11,4
11,7
16,0
9,1
12
12
12
12
12
112
96
149
176
171
р<0,05
p<0,05
p<0,05
p<0,005
p<0,001
10 нг/мл
381,3
31,4
12
128
р<0,05
10-5 М
10-6 М
10-7 М
10-8 М
10-9 М
321,0
287,0
398,4
368,3
431,8
18,4
24,3
35,8
32,2
24,2
12
12
12
12
12
108
96
134
123
145
р<0,05
p<0,05
p<0,05
p<0,05
p<0,001
10 нг/мл
819,9
61,5
12
371
р<0,001
10-5 М
10-6 М
10-7 М
10-8 М
10-9 М
244,8
189,9
380,8
384,3
324,9
28,2
24,4
45,8
52,2
45,1
12
12
12
12
12
111
86
173
174
147
р<0,05
p<0,05
p<0,005
p<0,01
p<0,05
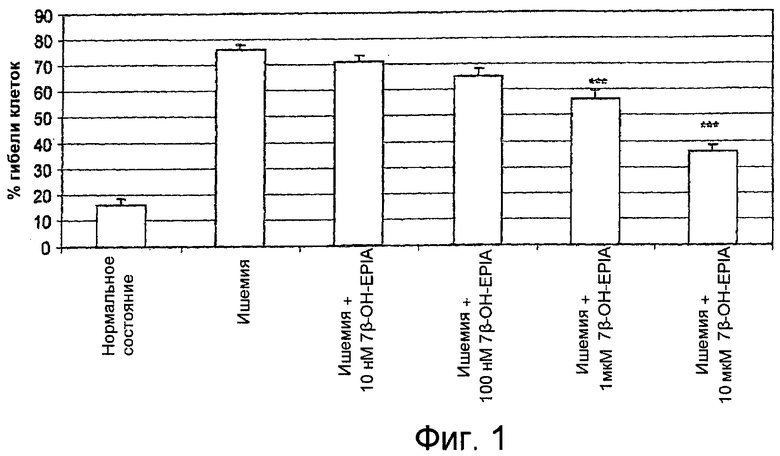


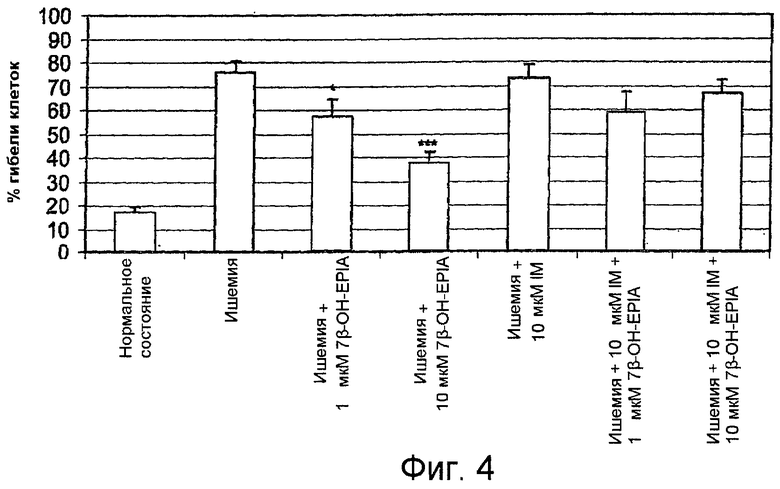

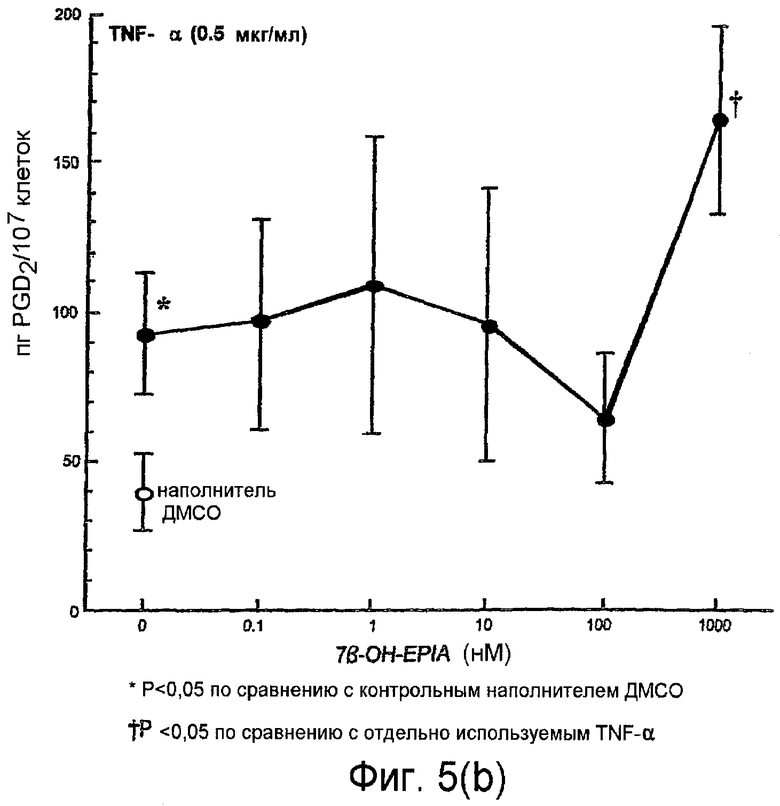

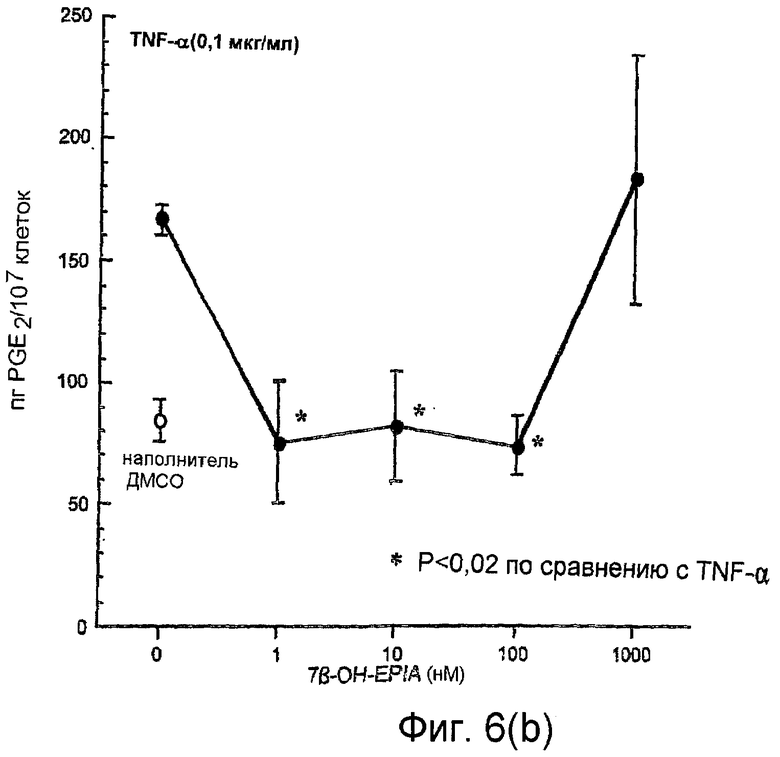
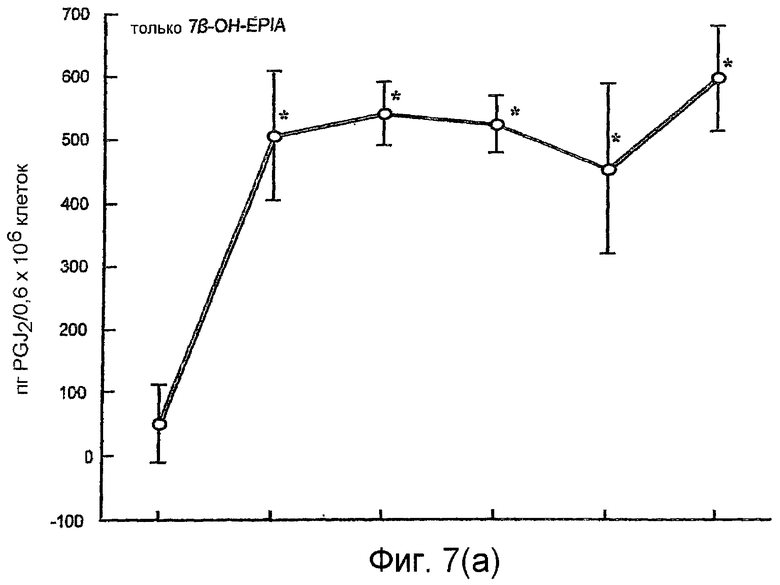





Настоящее изобретение относится к области фармацевтики и медицины и касается применения 7α-гидроксиэпиандростерона или 7β-гидроксиэпиандростерона для приготовления лекарственного средства для лечения воспалительного состояния, выбранного из воспаления, воспалительного заболевания периферических органов, выбранных из печени или почек, воспалительного заболевания дыхательных путей, сахарного диабета, ассоциированной с воспалением боли, воспалительных заболеваний кожи, рассеянного склероза, ревматоидного артрита, остеоартрита и их осложнений. 3 з.п. ф-лы, 14 ил., 18 табл., 7 пр.
1. Применение соединения, представляющего собой 7α-гидроксиэпиандростерон, или 7β-гидроксиэпиандростерон, или его фармацевтически приемлемую соль, или сложный эфир, для приготовления лекарственного средства, предназначенного для лечения или профилактики воспалительного состояния, выбранного из группы, состоящей из: воспаления, воспалительного заболевания периферических органов, выбранных из печени или почек, воспалительного заболевания дыхательных путей, сахарного диабета и его осложнений, боли, ассоциированной с воспалением, воспалительных заболеваний кожи, рассеянного склероза, заболеваний периферических артерий и их осложнений, заболевания коронарной артерии и его осложнений, атеросклеротического стеноза почечной артерии, ревматоидного артрита, первичного и вторичного остеоартрита и их осложнений, воспалительных заболеваний, характеризующихся дегенерацией суставного хряща, акромегалии и заживления ран.
2. Применение по п.1 для приготовления лекарственного средства, предназначенного для лечения или профилактики боли, ассоциированной с воспалением, заболеваний периферических артерий и их осложнений, заболевания коронарной артерии и его осложнений, воспалительного заболевания дыхательных путей, воспалительных заболеваний, характеризующихся дегенерацией суставного хряща.
3. Применение по п.1 или 2, где воспалительное заболевание дыхательных путей включает астму, ринит, бронхит или хроническое обструктивное заболевание легких.
4. Применение по п.1 для приготовления лекарственного средства, предназначенного для лечения или профилактики боли, ассоциированной с воспалением, атеросклеротического стеноза почечной артерии, диабета типа 2 и его осложнений, астмы и хронического обструктивного заболевания легких, ревматоидного артрита, первичного и вторичного остеоартрита и их осложнений.
5. Применение по п.1 или 2, в котором указанное соединение представляет собой 7α- или 7β-гидроксиэпиандростерон или его сложный эфир.
| НЕЙРОПРОТЕКТОРНЫЕ 7-БЕТА-ГИДРОКСИСТЕРОИДЫ | 2001 |
|
RU2258511C2 |
| ПРОФИЛАКТИЧЕСКОЕ И ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ ГИДРОКСИСТЕРОИДОВ | 2002 |
|
RU2329049C2 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Харкевич Д.А | |||
| Фармакология | |||
| - М.: Медицина, 1987, с.47, 48. | |||

