Изобретение относится к способу получения адамантилалкиловых и адамантилоксиалкиловых эфиров тозилоксиметилфосфоновой кислоты. Алкиловые и алкоксиалкиловые эфиры тозилоксиметилфосфоновой кислоты используются для синтеза фармацевтических препаратов, обладающих противовирусной активностью (цидофовир, адефовир, тенофовир и их аналоги).
CHIMERIX INC [US]; UNIV CALIFORNIA [US] Patent WO 2007/130783 A2, Metabolically stable alkoxyalkyl esters of antiviral or antiproliferative phosphonates, nucleoside phosphonates and nucleoside phosphates, 15.11.2007.
Beadle J.R., Wan W.B., Ciesia S.L. et al. // J. Med. Chem. 2006. V.49. N 6. Р.2010-2015.
Данным изобретением решена задача создания липофильных модификаторов синтетических аналогов нуклеозидов, которые, за счет наличия липофильного адамантанового фрагмента, обеспечат высокую биодоступность нуклеозидфосфоната, обладающих способностью проникать внутрь клетки и постепенно высвобождать активный нуклеозид. Это позволит поддерживать внутриклеточную концентрацию препарата, достаточную для терапевтического действия в течение длительного времени, и, таким образом, снизить разовую дозу препарата и частоту приема.
Известен метод синтеза диметилового эфира тозилоксиметилфосфоновой кислоты (Holy А. // Coll. Czech. Chem. Commun. 1982. V.47. N 12. P.3447-3463), диэтилового эфира тозилоксиметилфосфоновой кислоты (Barral К., Balzarini J., Neyts J. et al. // J. Med. Chem. 2006. V.49. N 1. P.43-50. Farrington G.K., Kumar A., Wedler F.C. // J. Med. Chem. 1985. V.28. N 11. P.1668-1673), диизопропилового эфира тозилоксиметилфосфоновой кислоты (Hammerschmidt F., Kaehlig H., Mueller N. // J. Chem. Soc., Perkin Trans. 1991. N 2. P.365-369, Holy A. // Coll. Czech. Chem. Commun. 1993. V.58. N 3. P.649-674, Kelley J.L., Linn J.A, McLean Ed.W, Tuttle J.V. // J. Med. Chem. 1993. V.36. N 22. P.3455-3463) взаимодействием соответствующих алкиловых эфиров гидроксиметилфосфоновой кислоты с тозилхлоридом в присутствии органического основания. Однако полученные эфиры не имеют в своем составе липофильных фрагментов, поэтому сами по себе не могут быть использованы в качестве липофильных модификаторов нуклеозидов.
Наиболее близким по технической сущности к заявляемому способу является способ получения натриевых солей O-алкоксиалкил(тозилоксиметил)фосфонатов из O,O-диэтил(тозилоксиметил)фосфоната обработкой последнего бромтриметилсиланом в растворе хлористого метилена с последующим взаимодействием образующегося бистриметилсилилового эфира с оксалилхлоридом в присутствии каталитических количеств N,N-диметилформамида и реакцией полученного дихлорфосфоната с алкоксиалканолами (12-метилтридецилоксипропан-1-олом, 13-метилтетрадецилоксипропан-1-олом, 14-метилпентадецилоксипропан-1-олом, 15-метилгексадецилоксипропан-1-олом, 17-метилоктадецилоксиэтан-1-олом и 15-метилгексадецилоксиэтан-1-олом), взятыми в соотношении 0.73 моль на 1 моль исходного O,O-диэтил(тозилоксиметил)фосфоната в присутствии пиридина.
CHIMERIX INC [US]; UNIV CALIFORNIA [US] Patent EP 2012799 (A2) Metabolically stable alkoxyalkyl esters of antiviral or antiproliferative phosphonates, nucleoside phosphonates and nucleoside phosphates, 14.01.2009.
CHIMERIX INC [US]; UNIV CALIFORNIA [US] Patent WO 2007/130783 (A2) Metabolically stable alkoxyalkyl esters of antiviral or antiproliferative phosphonates, nucleoside phosphonates and nucleoside phosphates, 15.11.2007.
Способ заключается в том, что к раствору 29.5 ммоль O,O-диэтил(тозилоксиметил)фосфоната в 150 мл хлористого метилена добавляли 175 ммоль бромтриметилсилана и реакционную смесь перемешивали при комнатной температуре 18 ч. Затем хлористый метилен и избыток бромтриметилсилана удаляли в вакууме, нелетучий остаток растворяли в 150 мл хлористого метилена и охлаждали полученный раствор до 0°С; к нему добавляли 0.5 мл ДМФА и затем добавляли по каплям раствор 175 ммоль оксалилхлорида в 50 мл хлористого метилена в течение 30 мин. После прибавления всего оксалилхлорида перемешивание продолжали в течение 5 ч. Хлористый метилен отгоняли и оставшееся масло растворяли в 100 мл диэтилового эфира. К полученному эфирному раствору добавляли при перемешивании раствор 21.5 ммоль соответствующего алкоксиалканола и 10 мл пиридина в 50 мл диэтилового эфира и перемешивали 3 ч. После этого реакционную смесь выливали в холодный насыщенный раствор гидрокарбоната натрия и перемешивали 1 ч. Органический слой отделяли, сушили сульфатом магния и эфир удаляли в вакууме. Полученный таким образом сырой продукт очищали хроматографией с использованием элюента хлористый метилен - 15% этанола.
Указанный способ обладает целым рядом существенных недостатков:
1. Используется большой избыток оксалилхлорида (5.9 моль на 1 моль исходного O,O-диэтил(тозилоксиметил)фосфоната), регенерирование которого из отгона с хлористым метиленом затруднительно, что приводит к усложнению технологического процесса и неоправдано с экономической точки зрения.
2. Реакция тозилоксиметилдихлорфосфоната с 0.73 эквивалентами спиртов проходит недостаточно селективно, поскольку, помимо целевого O-алкоксиалкил(тозилоксиметил)хлорфосфоната, на этой стадии образуются полные эфиры тозилоксиметилфосфоновой кислоты и остается исходный дихлорфосфонат, что приводит к снижению выходов целевых продуктов. Поэтому более целесообразным представляется синтез полных эфиров тозилоксиметилфосфоновой кислоты в качестве липофильных модификаторов нуклеозидов как более селективный процесс.
3. Применяемые в качестве липофильных модификаторов алкоксиалканолы разветвленного строения весьма труднодоступны и для их получения требуется многостадийный синтез.
Технический результат - селективный способ получения адамантилалкиловых и адамантилоксиалкиловых эфиров тозилоксиметилфосфоновой кислоты общей формулы I
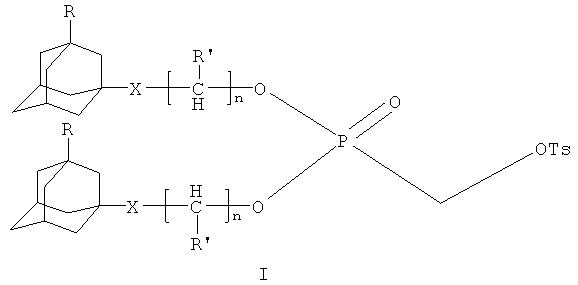
где Х - отсутствует, R=Н, R'=Н, n=1 (Ia); X - отсутствует, R=Н, R'=Н, n=2 (Iб); X - отсутствует, R=Н, R'=CH3, n=1 (Iв); Х=О, R=Н, R'=Н, n=2 (Iг); Х=О, R=C2H5, R'=Н, n=2 (Iд); Ts - тозил (n-толуолсульфонил),
как более доступных липофильных модификаторов нуклеозидов; распространение метода на синтез полных эфиров тозилоксиметилфосфоновой кислоты.
Технический результат достигается тем, что в способе получения эфиров тозилоксиметилфосфоновой кислоты на стадии синтеза дихлорфосфоната используется меньший избыток оксалилхлорида (3 моль на 1 моль исходного О,О-диэтил(тозилоксиметил)фосфоната), а этерификация дихлорфосфоната проводится двумя эквивалентами спиртов адамантанового ряда общей формулы II
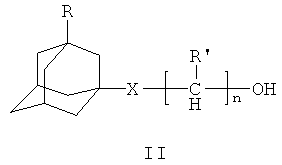
где Х - отсутствует, R=Н, R'=Н, n=1 (IIa), Х - отсутствует, R=Н, R'=Н, n=2 (IIб), X - отсутствует, R=Н, R'=CH3, n=1 (IIв), Х=О, R=Н, R'=Н, n=2 (IIг), Х=О, R=C2H5, R'=Н, n=2 (IIд).
Отличительные признаки:
1. Добавление оксалилхлорида из расчета 3 моль оксалилхлорида на 1 моль исходного O,O-диэтил(тозилоксиметил)фосфоната.
2. Использование спиртов адамантанового ряда для синтеза эфиров тозилоксиметилфосфоновой кислоты.
3. Синтез полных эфиров тозилоксиметилфосфоновой кислоты путем добавления 2 эквивалентов спиртов адамантанового ряда общей формулы II к образующемуся после реакции с оксалилхлоридом дихлорфосфонату.
4. Использование в качестве органического основания триэтиламина на стадии реакции дихлорфосфоната со спиртами.
5. Использование в качестве элюента системы петролейный эфир - этилацетат на стадии хроматографического выделения соединений Ia-д.
6. Использование перекристаллизации для конечной очистки соединений Ia-в.
Заявляемое изобретение имеет следующие преимущества:
Распространение метода, указанного в прототипе, на синтез полных эфиров тозилоксиметилфосфоновой кислоты, образующихся с более высокой селективностью.
Использование в качестве липофильных модификаторов спиртов адамантанового ряда, более доступных и обладающих противовирусной активностью.
Уменьшение количества оксалилхлорида.
Примеры выполнения способа
Анализ был проведен на хромато-масс-спектрометре Finnigan Trace DCQ с использованием капиллярной колонки ВРХ-5 30□0.32 компании SGE при энергии ионизирующих электронов 70 эВ.
Спектры ЯМР регистрировали на приборах Brucker AC200 и Brucker AM300 с использованием растворителя CDCl3 (200.13, 300.13 (1Н), 50.32, 75.47 (13С) и 121.49 МГц (31Р)). Измерения проводили без использования дополнительных эталонов с привязкой частоты к сигналу дейтерированного растворителя.
Пример 1
O,O-Бис(адамант-1-илмстил)(тозилоксиметил)фосфонат (Ia)
Все операции проводили в атмосфере аргона. К 10.6 г (33.0 ммоль) O,O-диэтил(тозилоксиметил)фосфоната в 215 мл хлористого метилена добавили 28.5 г (186 ммоль) триметилбромсилана и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель и избыток триметилбромсилана удаляли в вакууме. Остаток растворяли в 200 мл хлористого метилена, охлаждали до 0°С, добавляли 0.25 мл ДМФА и по каплям при перемешивании 12.7 г (100 ммоль) оксалилхлорида в 15 мл хлористого метилена, поддерживая температуру 0-5°С. После добавления всего необходимого количества оксалилхлорида реакционную смесь перемешивали при той же температуре в течение 6 ч. Растворитель удаляли в вакууме. Остаток растворяли в 60 мл абсолютного диэтилового эфира, фильтровали. К полученному эфирному раствору дихлорфосфоната добавляли по каплям при перемешивании и 0°С раствор 7.0 г (69.3 ммоль) триэтиламина и 11.0 г (66.0 ммоль) адамант-1-илметанола (IIа) в 120 мл эфира. Смесь выдерживали при 0°С в течение 12 ч, выливали в 500 мл холодной воды. Эфирный слой отделяли, промывали насыщенным водным раствором гидрокарбоната натрия (2×150 мл), сушили сульфатом натрия. Растворитель отгоняли. Остаток хроматографировали на силикагеле с диаметром зерен 60-200 мкм с использованием элюента петролейный эфир (40-70°С) - этилацетат с постепенным увеличением полярности (0-20% этилацетата). Продукт перекристаллизовывали из 100 мл смеси петролейного эфира и этилацетата. Выход: 7.28 г (39.2%). Т. пл. 132°С. Масс-спектр m/z (%): 563 [М]+, 407 (13), 150 (16), 148 (100), 135 (32), 107 (14), 93 (22), 91 (25), 79 (21), 67 (14), 55 (6). ЯМР 1Н, δ, м.д.: 1.50 с. (12Н, CH2, Ad), 1.65 кв. (12Н, СН2, Ad), 2.00 с. (6Н, СН, Ad), 2.45 с. (3Н, СН3, р-Tol), 3.60 м. (4Н, AdCH 2O), 4.20 д. (2Н, СН2О, 2JHP 7.0 Гц), 7.35 д., 7.8 д. (4Н, р-Tol, AB-система); δC 21.71 с. (СН3, p-Tol), 27.98 с. (СН, Ad), 33.93 д. (С, Ad, 3JCP 6.0 Гц). 36.91 с. (СН2, Ad), 38.77 с. (СН2, Ad), 60.99 д. (CH2O, 1JCP 169.8 Гц), 63.97 д. (AdCH2O, 2JCP 32.5 Гц), 128.25 с. (м-СН, р-Tol), 130.03 с. (o-СН, p-Tol), 131.95 с. (i-С, p-Tol), 145.47 с. (n-C, p-Tol); δP 16.05 м.д.
Пример 2
O,O-Бис(2-(адамант-1-ил)этил)(тозилоксиметил)фосфонат (Iб)
К 14.8 г (46.0 ммоль) 0,0-диэтил(тозилоксиметил)фосфоната в 300 мл хлористого метилена добавили 40.6 г (265 ммоль) триметилбромсилана и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель и избыток триметилбромсилана удаляли в вакууме. Остаток растворяли в 270 мл хлористого метилена, охлаждали до 0°С, добавляли 0.34 мл ДМФА и по каплям при перемешивании 17.7 г (139 ммоль) оксалилхлорида в 20 мл хлористого метилена, поддерживая температуру 0-5°С. После добавления всего необходимого количества оксалилхлорида реакционную смесь перемешивали при той же температуре в течение 6 ч. Растворитель удаляли в вакууме. Остаток растворяли в 80 мл абсолютного диэтилового эфира, фильтровали. К полученному эфирному раствору дихлорфосфоната добавляли по каплям при перемешивании и 0°С раствор 9.76 г (96.6 ммоль) триэтиламина и 16.6 г (92.0 ммоль) 2-(адамант-1-ил)этанола (IIб) в 170 мл эфира. Смесь выдерживали при 0°С в течение 12 ч, выливали в 700 мл холодной воды. Эфирный слой отделяли, промывали насыщенным водным раствором гидрокарбоната натрия (2×210 мл), сушили сульфатом натрия. Растворитель отгоняли. Остаток хроматографировали на силикагеле с диаметром зерен 60-200 мкм с использованием элюента петролейный эфир (40-70°С) - этилацетат с постепенным увеличением полярности (0-20% этилацетата). Продукт перекристаллизовывали из 140 мл смеси петролейного эфира и этилацетата. Выход: 9.53 г (35.0%). Т. пл. 67°С. Масс-спектр m/z (%): 591 [М]+, 435 (12), 273 (12), 267 (33), 163 (100), 135 (74), 105 (31), 91 (37), 79 (36), 67 (16), 55 (11). ЯМР 1Н, δ, м.д.: 1.45 т. (4Н, AdCH 2CH2O), 1.49 с. (12Н, CH2, Ad), 1.65 кв. (12Н, CH2, Ad), 1.95 с. (6Н, СН, Ad), 2.45 с. (3Н, СН3, p-Tol), 4.12 м. (4Н, CH2O), 4.19 д. (2Н, CH2O, 2JHP 8.2 Гц), 7.35 д., 7.8 д. (4Н, р-Tol, AB-система); δC 21.77 с. (CH3, p-Tol), 28.60 с. (СН, Ad), 31.84 с. (С, Ad), 37.01 с. (CH2, Ad), 42.54 с. (СН2, Ad), 44.28 д. (AdCH 2CH2O, 3JCP 6.03 Гц), 61.20 д. (CH2O, 1JCP 167.5 Гц), 63.97 д. (AdCH2 CH 2O, 2JCP 6.8 Гц), 128.32 с. (м-СН, p-Tol), 130.08 с. (о-CH, p-Tol), 131.97 с. (i-С, р-Tol), 145.52 с. (n-С, р-Tol); δP 18.01 м.д.
Пример 3
O,O-Бис(1-(адамант-1-ил)этил)(тозилоксиметил)фосфонат (Iв)
К 14.8 г (46.0 ммоль) O,O-диэтил(тозилоксиметил)фосфоната в 300 мл хлористого метилена добавили 40.6 г (265 ммоль) триметилбромсилана и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель и избыток триметилбромсилана удаляли в вакууме. Остаток растворяли в 270 мл хлористого метилена, охлаждали до 0°С, добавляли 0.34 мл ДМФА и по каплям при перемешивании 17.7 г (139 ммоль) оксалилхлорида в 20 мл хлористого метилена, поддерживая температуру 0-5°С. После добавления всего необходимого количества оксалилхлорида реакционную смесь перемешивали при той же температуре в течение 6 ч. Растворитель удаляли в вакууме. Остаток растворяли в 80 мл абсолютного диэтилового эфира, фильтровали. К полученному эфирному раствору дихлорфосфоната добавляли по каплям при перемешивании и 0°С раствор 9.76 г (96.6 ммоль) триэтиламина и 16.6 г (92.0 ммоль) 1-(адамант-1-ил)этанола (IIв) в 170 мл эфира. Смесь выдерживали при 0°С в течение 12 ч, выливали в 700 мл холодной воды. Эфирный слой отделяли, промывали насыщенным водным раствором гидрокарбоната натрия (2×210 мл), сушили сульфатом натрия. Растворитель отгоняли. Остаток хроматографировали на силикагеле с диаметром зерен 60 - 200 мкм с использованием элюента петролейный эфир (40-70°С) - этилацетат с постепенным увеличением полярности (0-20% этилацетата). Продукт перекристаллизовывали из 140 мл смеси петролейного эфира и этилацетата. Выход: 1.35 г (4.97%). Т. пл. 165°С. Масс-спектр m/z (%): 591 [М]+, 273 (25), 267 (84), 162 (89), 135 (100), 107 (30), 93 (44), 79 (37), 67 (17), 55 (12). ЯМР 1Н, δ, м.д.: 1.20 д. (6Н, СН3), 1.40-1.75 м. (24Н, CH2, Ad), 2.0 с. (6Н, СН, Ad), 2.45 с. (СН3, p-Tol), 4.22 м. (2Н, СН2О, 2Н, СНО), 7.35 д., 7.8 д. (4Н, р-Tol, AB-система); δC 15.0 с. (СН3), 21.50 с. (СН3, р-Tol), 28.18 с. (СН, Ad), 36.68 с. (С, Ad), 37.01 с. (СН2, Ad), 37.70 с. (СН3, Ad), 62.54 д. (CH2O, 1JCP 170.0 Гц), 83.00 д. (AdCHCH3)O, 128.27 с. (м-СН, р-Tol), 129.98 с. (о-СН, p-Tol), 131.97 с. (i-С, p-Tol), 145.39 с. (n-С, р-Tol); δP 14.16 м.д.
Пример 4
O,O-Бис(2-(адамант-1-илокси)этил)(тозилоксиметил)фосфонат (Iг).
К 6.98 г (21.7 ммоль) O,O-диэтил(тозилоксиметил)фосфоната в 140 мл хлористого метилена добавили 19.3 г (126 ммоль) триметилбромсилана и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель и избыток триметилбромсилана удаляли в вакууме. Остаток растворяли в 130 мл хлористого метилена, охлаждали до 0°С, добавляли 0.17 мл ДМФА и по каплям при перемешивании 8.34 г (65.7 ммоль) оксалилхлорида в 10 мл хлористого метилена, поддерживая температуру 0-5°С. После добавления всего необходимого количества оксалилхлорида реакционную смесь перемешивали при той же температуре в течение 6 ч. Растворитель удаляли в вакууме. Остаток растворяли в 40 мл абсолютного диэтилового эфира, фильтровали. К полученному эфирному раствору дихлорфосфоната добавляли по каплям при перемешивании и 0°С раствор 4.60 г (45.6 ммоль) триэтиламина и 8.50 г (43.4 ммоль) 2-(адамант-1-илокси)этанола (IIг) в 80 мл эфира. Смесь выдерживали при 0°С в течение 12 ч, выливали в 210 мл холодной воды. Эфирный слой отделяли, промывали насыщенным водным раствором гидрокарбоната натрия (2×100 мл), сушили сульфатом натрия. Растворитель отгоняли. Остаток хроматографировали на силикагеле с диаметром зерен 60-200 мкм с использованием элюента петролейный эфир (40-70°С) - этилацетат с постепенным увеличением полярности (0-50% этилацетата). Продукт перекристаллизовывали из 70 мл смеси петролейного эфира и этилацетата. Выход: 5.23 г (38.7%). Т. пл. 53-55°С. Масс-спектр m/z (%): 624 [М-Н]+, 579 (5), 293 (5), 268 (5), 155 (7), 135 (100), 121 (7), 107 (12), 93 (22), 91 (19), 79 (17), 67 (7). ЯМР 1Н, δ, м.д.: 1.52 кв. (12H, Ad), 1.70 с. (12Н, Ad), 2.12 с. (6Н, Ad), 2.45 с. (3Н, СН3, p-Tol), 3.58 м. (4Н, AdOCH 2CH2O), 4.15 м. (4Н, AdOCH2 CH 2O), 4.30 д. (2Н, CH2O, 2JHP 8.2 Гц), 7.35 д., 7.80 д. (4Н, p-Tol, AB-система); δC 21.66 с. (СН3, р-Tol), 30.47 с. (СН, Ad), 36.36 с., (CH2, Ad), 41.39 с. (СН2, Ad), 59.09 с. (AdOCH 2CH2O), 61.74 д. (CH2O, 1JCP 169.8 Гц), 67.02 д. (AdOCH2 CH 2O, 2JCP 7.55 Гц), 72.62 с. (С, Ad), 128.26 с. (м-СН, р-Tol), 129.92 с. (о-СН, p-Tol), 132.01 с. (i-C, p-Tol), 145.24 с. (n-С, р-Tol); δP 18.15 м.д.
Пример 5
O,O-Бис(2-(3-этиладамант-1-ил)этил)(тозилоксиметил)фосфонат (Iд)
К 10.6 г (33.0 ммоль) O,O-диэтил(тозилоксиметил)фосфоната в 215 мл хлористого метилена добавили 28.5 г (186 ммоль) триметилбромсилана и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель и избыток триметилбромсилана удаляли в вакууме. Остаток растворяли в 200 мл хлористого метилена, охлаждали до 0°С, добавляли 0.25 мл ДМФА и по каплям при перемешивании 12.7 г (100 ммоль) оксалилхлорида в 15 мл хлористого метилена, поддерживая температуру 0-5°С. После добавления всего необходимого количества оксалилхлорида реакционную смесь перемешивали при той же температуре в течение 6 ч. Растворитель удаляли в вакууме. Остаток растворяли в 60 мл абсолютного диэтилового эфира, фильтровали. К полученному эфирному раствору дихлорфосфоната добавляли по каплям при перемешивании и 0°С раствор 7.0 г (69.3 ммоль) триэтиламина и 14.8 г (66.0 ммоль) 2-(3-этиладамант-1-илокси)этанола (IIд) в 120 мл эфира. Смесь выдерживали при 0°С в течение 12 ч, выливали в 500 мл холодной воды. Эфирный слой отделяли, промывали насыщенным водным раствором гидрокарбоната натрия (2×150 мл), сушили сульфатом натрия. Растворитель отгоняли. Остаток хроматографировали на силикагеле с диаметром зерен 60-200 мкм с использованием элюента петролейный эфир (40-70°С) - этилацетат с постепенным увеличением полярности (0-20% этилацетата). Выход: 8.70 г (38.8%). Масс-спектр m/z (%): 679 [М]+, 636 (12), 500 (7), 473 (62), 381 (7), 355 (12), 337 (22), 311 (48), 293 (20), 267 (11), 177 (11), 162 (100), 155 (25), 149 (13), 121 (42), 107 (56), 93 (44), 91 (35), 79 (32), 55 (12). ЯМР 1Н, δ, м.д.: 0.80 т. (6Н, СН3, Et), 1.20 кв. (4Н, СН2, Et), 1.35 с., 1.40 с., 1.63 кв., 2.18 с. (28Н, Ad), 2.45 с. (СН3, p-Tol), 3.58 т. (4Н, AdOCH 2CH2O), 4.15 м. (4Н, AdOCH2 CH 2O), 4.30 д. (2Н, CH2O, 2JHP 8.2 Гц), 7.35 д., 7.8 д. (4Н, р-Tol, АВ-система); δC 7.18 с. (СН3, Et), 21.73 с. (СН3, р-Tol), 30.57 с. (СН, Ad), 35.89 с. (CH2, Et), 40.99 с. (CH2, Ad), 45.78 с. (СН2, Ad), 59.41 с. (AdOCH 2CH2O), 61.66 д. (CH2O, 1JCP 187.0 Гц), 67.09 д. (AdOCH2 CH 2O, 2JCP 6.8 Гц), 73.69 с. (С, Ad), 128.30 с. (м-СН, р-Tol), 129.99 с. (о-СН, р-Tol), 132.06 с. (i-С, p-Tol), 145.31 с. (n-С, р-Tol); δP 17.18 м.д.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, СОЕДИНЕНИЯ, КОМПЛЕКСНЫЙ МЕТАЛЛООРГАНИЧЕСКИЙ КАТАЛИЗАТОР | 2012 |
|
RU2652807C2 |
| ФОТОХРОМНЫЕ ПРОИЗВОДНЫЕ 5'-ВИНИЛ-6-НИТРО-СПИРОБЕНЗОПИРАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2458927C1 |
| N @ -(фосфонато)-диазен-N-оксиды | 1990 |
|
SU1747450A1 |
| N,N-бис(2-(диалкиламино)этил)карбоксамиды и их дигидрохлориды, проявляющие антиаритмическую активность, и фармацевтические композиции на их основе | 2017 |
|
RU2645080C1 |
| ЛУПАНОВЫЕ А-СЕКОТРИТЕРПЕНОИДЫ, ПРОЯВЛЯЮЩИЕ ПРОТИВОВИРУСНУЮ АКТИВНОСТЬ | 2011 |
|
RU2470003C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2,3,5,6-ТЕТРАОКСАБИЦИКЛО[2.2.1]ГЕПТАНОВ | 2012 |
|
RU2494102C1 |
| Четвертичные аммониевые соли на основе производных витамина В6 | 2015 |
|
RU2607522C1 |
| СПОСОБ ПОЛУЧЕНИЯ (4E, 6Z)-ГЕКСАДЕКА-4,6-ДИЕН-1-ОЛА | 2015 |
|
RU2582619C1 |
| ЗАМЕЩЁННЫЕ ИЗОКСАЗОЛЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2733945C2 |
| α-Гидроксифосфонаты с алкильными заместителями у атома фосфора, обладающие антимикробной активностью | 2024 |
|
RU2834476C1 |
Изобретение относится к способу получения адамантилалкиловых и адамантилоксиалкиловых эфиров тозилоксиметилфосфоновой кислоты формулы I, используемых для синтеза фармацевтических препаратов с противовирусной активностью, из соединений формулы II
где Х - отсутствует, R=H, R'=H, n=1; или X - отсутствует, R=H, R'=H, n=2; или X - отсутствует, R=H, R'=CH3, n=1; или Х=O, R=H, R'=H, n=2; или Х=O, R=C2H5, R'=H, n=2; Ts - тозил (n-толуолсульфонил). Способ включает обработку раствора O,O-диэтил(тозилоксиметил)фосфоната в хлористом метилене бромтриметилсиланом с последующим взаимодействием образующегося бистриметилсилилового эфира с оксалилхлоридом в присутствии каталитических количеств N,N-диметилформамида при соотношении 3 моль оксалилхлорида на 1 моль O,O-диэтил(тозилоксиметил)фосфоната, затем образующийся дихлорфосфонат обрабатывают в присутствии Et3N двумя эквивалентами спиртов формулы II. Способ с улучшенной селективностью реакции и использованием доступных реагентов упрощает и удешевляет производство. 5 пр.
Способ получения адамантилалкиловых и адамантилоксиалкиловых эфиров тозилоксиметилфосфоновой кислоты общей формулы I
где Х отсутствует, R=H, R'=H, n=1; или X отсутствует, R=H, R'=H, n=2; или X отсутствует, R=H, R'=CН3, n=1; или Х=O, R=H, R'=H, n=2; или Х=O, R=C2H5, R'=H, n=2;
Ts - тозил (n-толуолсульфонил), при котором раствор O,O-диэтил(тозилоксиметил)фосфоната в хлористом метилене обрабатывают бромтриметилсиланом с последующим взаимодействием образующегося бистриметилсилилового эфира с оксалилхлоридом в присутствии каталитических количеств N,N-диметилформамида при соотношении 3 моль оксалилхлорида на 1 моль O,O-диэтил(тозилоксиметил)фосфоната, после чего образующийся дихлорфосфонат обрабатывают в присутствии триэтиламина двумя эквивалентами спиртов формулы II
,
где X отсутствует, R=H, R'=H, n=1; или X отсутствует, R=H, R'=H, n=2; или X отсутствует, R=H, R'=CН3, n=1; или Х=O, R=H, R'=H, n=2; или Х=O, R=C2H5, R'=H, n=2.
| НУКЛЕОЗИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ПРОТИВ ВИРУСА ГЕПАТИТА В | 1999 |
|
RU2237479C2 |
| JAMES R | |||
| BEADLE ET ALL, «Synthesis and Antiviral Evaluation of Alkoxyalkyl Derivatives of 9-(S)-(3-Hydroxy-2-phosphonomethoxypropyl)adenine against Cytomegalovirus and Orthopoxviruses», J | |||
| Med | |||
| Chem., 2006, 49 (6), 2010-2015 | |||
| FARRINGTON G.K | |||
| ET ALL, «Design and synthesis of new transition-state analog | |||

