Область техники
Изобретение относится к новому тиопроизводному пиридина, представленному общей формулой (I), которое будет описано далее, или его фармакологически приемлемой соли и к фармацевтической композиции, содержащей тиопроизводное пиридина или его соль.
Уровень техники
Гастрит, язвы желудка и язвы двенадцатиперстной кишки являются болезнями, развивающимися при усложненном участии таких факторов, как стрессы, генетические факторы и образ жизни. В последние годы внимание сфокусировано на Helicobacter pylori (Н. pylori) как одном из таких факторов. Интенсивные исследования были проведены в отношении связей между Helicobacter pylori и гастритом, язвами желудка, язвами двенадцатиперстной кишки и раком желудка с тех пор, как Warren и Marshall достигли успеха в изолированной культуре спиралевидной бактерии из биопсии желудочного образца в 1983 г. В результате представлен доклад, касающийся зараженности Helicobacter pylori, в которой реальная доля Helicobacter pylori составляет около 4% для здорового желудка, тогда как реальные доли Helicobacter pylori являются настолько высокими как около 83% при хроническом гастрите, около 69% при язве желудка, около 92% при язве двенадцатиперстной кишки и около 51% при синдромах неязвенной диспепсии (см. непатентный документ 1). Кроме того, заражение Helicobacter pylori также имеет стойкую связь с частотой желудочных раковых заболеваний, и International Agency for Research on Cancer of the World Health Organization (WHO) сделан вывод в 1994, что Helicobacter pylori был канцерогеном, играющим причинную роль в развитии желудочных раковых заболеваний.
Традиционные методы лечения гастрита, язвы желудка, язвы двенадцатиперстной кишки и тому подобного осуществляют, главным образом, путем симптоматического лечения с применением лекарств, таких как Н2 блокатор и ингибитор протонного насоса (PPI), которые подавляют секрецию желудочной кислоты, защитные агенты слизистой оболочки или тому подобное. Однако, даже если множество пациентов временно исцеляются этими лекарствами, указано, что около 80% этих пациентов будут иметь рецидив в течение одного года, если лечение прекращается (см. непатентный документ 1). Обнаружено также, что если Helicobacter pylori (Н. pylori) уничтожают, одногодичная частота рецидивов язвы двенадцатиперстной кишки находится в пределах 10%, а частота рецидивов язвы желудка явно ниже (см. непатентный документ 2). Это приводит к методу, при котором в дополнение к ингибитору протонного насоса (PPI) назначают большую дозу антибактериальных агентов, таких как амоксициллин, кларитромицин и метронидазол, в то же самое время в течение одной недели или более. Однако введение большого количества антибактериальных агентов имеет результатом уничтожение эффективных бактерий. Как результат, есть опасение в отношении возможности промотирования проявления побочных эффектов, таких как жидкий стул, диаррея, извращение вкуса, воспаление языка, стоматит, ухудшение функции печени, аномальная функция печени и геморрагический энтерит, и дополнительно возможность реализации метициллин-резистентного Staphylococcus aureus (MRSA).
Были предприняты попытки разработать фармацевтические средства, которые проявляют удовлетворительную антибактериальную активность против Helicobacter pylori и имеют высокую безопасность в регулярной дозе с такими исходными данными (см. патентные документы 1-5).
Чтобы создать такой же эффект, как антибиотики, в отношении уничтожения Helicobacter pylori в клинической практике, эти фармацевтические средства должны проявлять антибактериальную активность, равную или более высокую чем активность против Helicobacter pylori, например, антибиотиков, проявляющих антибактериальную активность против Helicobacter pylori в клинической практике. Иными словами, желательно, чтобы эти фармацевтические средства имели активность при концентрации более высокой, чем минимальная ингибирующая концентрация (MIC) 0,3 мкг/мл.
Кроме того, некоторые соединения из карбоксилатных производных гуанидинометилциклогексана, описанных в патентном документе 6, каждое, имеют MIC менее чем 1 мкг/мл, как активность против Helicobacter pylori. Однако в действительности каждое из этих соединений разлагается очень быстро ферментом разложения в тонком кишечнике или крови. Этот характер разработанных соединений, основанный на таких метаболических характеристиках, что соединения разлагаются в тонком кишечнике или крови, рассматривается как доказательство селективности в антибактериальной активности против Helicobacter pylori, исходя из описанных в патентном документе 7 следующих соображений: "Когда антибиотики или синтетические антимикробные агенты вводят, они метаболически разносятся, включая, например, те, которые проходят пищеварительный тракт и абсорбируются из кишечника, чтобы поступить в кровь, и те, которые выделяются с фекалиями, с тем результатом, что большое число бактерий, живущих в кишечнике, уничтожаются фармацевтическим средством, проходящим через кишечник, что приводит к нарушению баланса в кишечной флоре, и поэтому необходимо избегать введения в течение длительного времени". Однако метаболические ферменты в кишечнике и крови и кишечные бактерии, как известно, варьируются в зависимости от индивидуальных особенностей и потребляемой пищи, и поэтому мало вероятно, что пациенты с различными исходными данными обеспечат такие метаболические характеристики стабильно.
Между тем, существуют производные пиридина (см. патентный документ 8), известные как противоязвенные агенты, производные пиридина (см. патентный документ 2), проявляющие антибактериальную активность против Helicobacter pylori, и производные пиридина (см. патентный документ 9), используемые в качестве ингибиторов секреции желудочной кислоты. Как производные пиридина, 2-[{4-(2-гидроксиэтокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол, 2-[{4-(3-гидроксипропокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол и тому подобные являются соединениями, применимыми в качестве противоязвенных агентов, как указано в примерах 26 и 34 в патентном документе 5. Однако нет описания, касающегося экспериментальных данных относительно противоязвенной активности каждого из указанных соединений, и, кроме того, нет ни описания, ни какого-либо намека в отношении активностей указанных соединений против Helicobacter pylori.
Более того, авторы данного изобретения обнаружили производное пиридилметилтио-1Н-бензимидазола, имеющее чрезвычайно высокую активность против Helicobacter pylori (см. патентный документ 10).
В этой ситуации были проведены исследования в отношении противоязвенных агентов, ингибиторов секреции желудочной кислоты или производных пиридина, имеющих активность против Helicobacter pylori. Никакое пригодное соединение не было разработано до сих пор, несмотря на эти исследования и попытки.
Между тем, эти 1Н-бензимидазол-2-тиопроизводные могут быть получены легко при использовании в качестве исходного материала 2-меркаптометил-1Н-бензимидазолов, которые легко могут быть получены, используя, например, тиомочевину и тому подобное в качестве исходного материала. Однако необходимо, чтобы эти производные 2-меркаптометил-1Н-бензимидазола могли быть получены совсем разными методами синтеза, и поэтому исследования с использованием указанных производных время от времени продолжались до настоящего времени.
Когда, например, соединение, имеющее структуру пиридин-2-ил-тиометил-1Н-бензимидазола, было исследовано путем CAS в режиме онлайн, число удачных попыток соединений находилось в пределах 10. Восемь соединений из этих десяти соединений были соединениями, в которых атом азота бензимидазола является бензолсульфонилированным и атом серы тиогруппы является сульфинированным. Одно из оставшихся двух соединений является соединением, в котором атом хлора находится в 4 положении пиридинового кольца (см. непатентный документ 3). Другое является соединением, которое содержит метоксигруппу и цианогруппу в 3 и 4 положениях пиридинового кольца, соответственно, и его используют в качестве исходного продукта для синтеза инденофлуоренольного кольца путем циклизации цианогруппы (см. непатентный документ 4).
Удачные 10 соединений описаны в двух документах, которые указаны как непатентные документы 3 и 4.
СПИСОК ЦИТИРУЕМЫХ ИСТОЧНИКОВ
ПАТЕНТНЫЙ ДОКУМЕНТ
Патентный документ 1: Японская выложенная патентная заявка (JP-A) No. 2-209809
Патентный документ 2: JP-A No. 3-173817
Патентный документ 3: JP-A No. 3-48680
Патентный документ 4: JP-A No. 7-69888
Патентный документ 5: JP-A No. 5-247035
Патентный документ 6: WO 96/06825
Патентный документ 7: WO 97/23207
Патентный документ 8: JP-A No. 61-50979
Патентный документ 9: JP-A No. 58-39622
Патентный документ 10: WO 2008/075462
НЕПАТЕНТНЫЙ ДОКУМЕНТ
Непатентный документ 1: Martin J. Blaser, Clin. Infectious Disease, 15: 386-393, 1992
Непатентный документ 2: Graham D.Y., et al., Ann. Intern. Med., 116; 705-708, 1992
Непатентный документ 3: Ismail, M. M., et al., Chem Papers (2004), 58(2), 117-125 (Chem. Abstr. 141: 366111)
Непатентный документ 4: Kaigorodoves, E. A., et al., Chem. Heter. Compd. (1997), 33(6), 752-753 (Chem. Abstr. 128: 127976)
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ПРОБЛЕМЫ, КОТОРЫЕ ДОЛЖНЫ БЫТЬ РАЗРЕШЕНЫ ИЗОБРЕТЕНИЕМ
Задачей данного изобретения является создание нового типа соединения, имеющего высокую и селективную активность против Helicobacter pylori. Иными словами, данное изобретение относится к новому производному пиридина и фармацевтической композиции с использованием производного пиридина и, особенно, к фармацевтической композиции, имеющей селективную антибактериальную активность против Helicobacter pylori.
СРЕДСТВА ДЛЯ РАЗРЕШЕНИЯ УКАЗАННЫХ ПРОБЛЕМ
Авторы данного изобретения обратили внимание на сшитую часть между пиридиновым кольцом и 1H-бензимидазольным кольцом и провели исследования, касающиеся корреляции между сшитой структурой и активностью против Helicobacter pylori и в результате неожиданно обнаружили, что соединение пиридина, имеющее структуру, в которой метиленовая группа и тиогруппа заменяются одна другой, имеют очень высокую и селективную активность против Helicobacter pylori.
Т.е. данное изобретение относится к тиопроизводному пиридина, представленному следующей общей формулой (I), или к фармакологически приемлемой его соли:

где R1 и R2, соответственно, представляют атом водорода, C1-8алкиловую группу, C1-8алкоксигруппу или атом галогена; R3 и R4, соответственно, представляют атом водорода, C1-8алкиловую группу или C1-8алкоксигруппу; X представляет -O-, -S- или -NH-; Y представляет C1-12алкиловую группу, которая может быть замещена заместителем, выбранным из группы, состоящей из гидроксильной группы, C1-8алкоксигруппы и атома галогена, или -(R5-O)n-R6 (где R5 представляет C1-5алкиленовую группу, R6 представляет C1-8алкиловую группу, которая может быть замещена заместителем, выбранным из группы, состоящей из гидроксильной группы, C1-8алкоксигруппы и атома галогена, и n означает целое число от 1 до 10); и Z представляет атом водорода или С1-8алкиловую группу.
Кроме того, данное изобретение относится к фармацевтической композиции, содержащей тиопроизводное пиридина, представленное указанной общей формулой (I), или его фармакологически приемлемую соль и фармацевтически приемлемый носитель, и, особенно, к фармацевтической композиции, которая предотвращает или лечит инфекции Helicobacter pylori (H. pylori) или болезни, в которых участвует Helicobacter pylori (H. pylori).
Данное изобретение также относится к тиопроизводному пиридина, представленному указанной общей формулой (I), или его фармакологически приемлемой соли в качестве эффективного компонента, который предотвращает или лечит инфекции Helicobacter pylori (H. pylori) или болезни, в которых участвует Helicobacter pylori (H. pylori).
Дополнительно данное изобретение относится к способу профилактики или лечения болезней, в которых участвует Helicobacter pylori (H. pylori), включающему введение фармацевтической композиции, содержащей тиопроизводное пиридина, представленное указанной общей формулой (I), или его фармакологически приемлемую соль, в эффективном количестве пациенту, инфицированному Helicobacter pylori (H. pylori).
Кроме того, данное изобретение относится к применению тиопроизводного пиридина, представленного указанной общей формулой (I), или его фармакологически приемлемой соли для получения фармацевтической композиции, которая предотвращает или лечит инфекции Helicobacter pylori или болезни, в которых участвует Helicobacter pylori.
Следующие описания даны для более подробного пояснения данного изобретения.
(1) Тиопроизводное пиридина, представленное следующей общей формулой (I), или его фармакологически приемлемая соль:
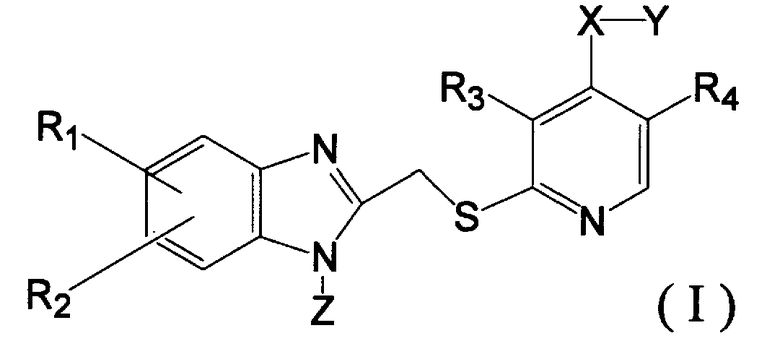
где R1 и R2, соответственно, представляют атом водорода, C1-8алкиловую группу, C1-8алкоксигруппу или атом галогена; R3 и R4, соответственно, представляют атом водорода, C1-8алкиловую группу или C1-8алкоксигруппу; X представляет -O-, -S- или -NH-; Y представляет C1-12алкиловую группу, которая может быть замещена заместителем, выбранным из группы, состоящей из гидроксильной группы, C1-8алкоксигруппы и атома галогена, или -(R5-O)n-R6 (где R5 представляет C1-5алкиленовую группу, R6 представляет C1-8алкиловую группу, которая может быть замещена заместителем, выбранным из группы, состоящей из гидроксильной группы, C1-8алкоксигруппы и атома галогена, и n означает целое число от 1 до 10) и Z представляет атом водорода или C1-8алкиловую группу.
(2) Тиопроизводное пиридина или его фармакологически приемлемая соль по п.(1), где Y в общей формуле (I) означает C3-10алкиловую группу, которая может быть замещена заместителем, выбранным из группы, состоящей из гидроксильной группы или C1-8алкоксигруппы.
(3) Тиопроизводное пиридина или его фармакологически приемлемая соль по п.(1), где Y в общей формуле (I) представляет -(R5-O)n-R6 (где R5 представляет C1-5алкиленовую группу, R6 представляет C1-8алкиловую группу, которая может быть замещенной заместителем, выбранным из группы, состоящей из гидроксильной группы, C1-8алкоксигруппы и атома галогена, и n означает целое число от 1 до 10), где R5 означает этиленовую группу и R6 означает C1-8алкиловую группу, которая может быть замещенной атомом фтора.
(4) Тиопроизводное пиридина или его фармакологически приемлемая соль по любому из предшествующих пп.(1)-(3), где X в общей формуле (I) означает -O- или -S-.
(5) Тиопроизводное пиридина или его фармакологически приемлемая соль по любому из предшествующих пп.(1)-(4), где R3 и R4 в общей формуле (I), соответственно, представляют атом водорода, метиловую группу или метоксигруппу.
(6) Фармацевтическая композиция, содержащая тиопроизводное пиридина или его фармакологически приемлемую соль по любому из предшествующих пп.(1)-(5) и фармацевтически приемлемый носитель.
(7) Фармацевтическая композиция по п.(6), применяемая для профилактики или лечения инфекции Helicobacter pylori (H. pylori).
(8) Фармацевтическая композиция по п.(6), применяемая для профилактики или лечения болезней, в которых участвует Helicobacter pylori (H. pylori).
(9) Фармацевтическая композиция по п.(8), где болезнями, в которых принимает участие Helicobacter pylori (H. pylori), являются гастрит, язва желудка, язва двенадцатиперстной кишки, синдромы неязвенной диспепсии, MALT лимфома желудка, гиперплазированные желудочные полипы, желудочные злокачественные заболевания, желудочно-кишечные злокачественные заболевания, панкреатит или воспалительные кишечные болезни.
(10) Способ профилактики или лечения болезней, в которых участвует Helicobacter pylori (H. pylori), включающий введение фармацевтической композиции, содержащей тиопроизводное пиридина или его фармакологически приемлемую соль по любому из предшествующих пп.(1)-(5), в эффективном количестве пациенту, инфицированному Helicobacter pylori (H. pylori).
(11) Способ по п.(10), где болезнями, в которых участвует Helicobacter pylori, являются гастрит, язва желудка, язва двенадцатиперстной кишки, синдромы неязвенной диспепсии, желудочная MALT лимфома, гиперплазированные желудочные полипы, желудочные злокачественные заболевания, желудочно-кишечные злокачественные заболевания, панкреатит или воспалительные кишечные болезни.
(12) Применение тиопроизводного пиридина или его фармакологически приемлемой соли по любому из предшествующих пп.(1)-(5) для получения фармацевтической композиции для профилактики или лечения инфекций Helicobacter pylori или болезней, в которых участвует Helicobacter pylori.
(13) Применение по п.(12), где болезнями, в которых участвует Helicobacter pylori, являются гастрит, язва желудка, язва двенадцатиперстной кишки, синдромы неязвенной диспепсии, желудочная MALT лимфома, гиперплазированные желудочные полипы, желудочные злокачественные заболевания, желудочно-кишечные злокачественные заболевания, панкреатит или воспалительные кишечные болезни.
(14) Тиопроизводное пиридина или его фармакологически приемлемая соль по любому из пп.(1)-(5), применяемое в качестве эффективного компонента для профилактики или лечения инфекций Helicobacter pylori или болезней, в которых участвует Helicobacter pylori.
(15) Тиопроизводное пиридина или его фармакологически приемлемая соль по п.(14), где болезнями, в которых участвует Helicobacter pylori, являются гастрит, язва желудка, язва двенадцатиперстной кишки, синдромы неязвенной диспепсии, желудочная MALT лимфома, гиперплазированные желудочные полипы, желудочные злокачественные заболевания, желудочно-кишечные злокачественные заболевания, панкреатит или воспалительные кишечные болезни.
ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Новое тиопроизводное пиридина или его фармакологически приемлемая соль по данному изобретению очень полезно в качестве антибактериального агента против Helicobacter pylori, который проявляет селективную антибактериальную активность против Helicobacter pylori, не затрагивая бактерии, и его можно вводить в течение продолжительного периода времени. Дополнительно, соединение по данному изобретению имеет величину MIC в пределах 0,003 мкг/мл как активность против Helicobacter pylori, показывающую, что соединение имеет очень высокую антибактериальную активность, и, следовательно, применимо в качестве профилактических или лечебных агентов против различных болезней, в которых участвует Helicobacter pylori.
Кроме того, антибактериальный агент против Helicobacter pylori по данному изобретению может быть использован в сочетании с ингибитором секреции желудочной кислоты, таким как H2 блокатор или ингибитор протонного насоса (PPI), и может быть применен в качестве профилактических или лечебных агентов против различных болезней, в которых участвует Helicobacter pylori, таких как гастрит, язва желудка, язва двенадцатиперстной кишки, синдромы неязвенной диспепсии, желудочная MALT лимфома, гиперплазированные желудочные полипы, желудочные злокачественные заболевания, желудочно-кишечные злокачественные заболевания, панкреатит или воспалительные кишечные болезни.
СПОСОБЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Данное изобретение будет пояснено подробно.
В данном изобретении, во-первых, было обнаружено, что тиопроизводное пиридина проявляет селективную и высокую антибактериальную активность против Helicobacter pylori, когда атом углерода во 2 положении бензимидазольного кольца замещен 4-замещенной-пиридилтиогруппой, даже если 2 положение бензимидазольного кольца не занято гетероатомом, таким как атом кислорода или атом серы, но занято атомом углерода. Следовательно, атом водорода, присоединенный в том же положении, например, пиридинового кольца, бензимидазольного кольца и тому подобного в производном 2-(4'-замещенный-пиридилтиометил)бензимидазола по данному изобретению, может быть замещенным различными заместителями в соответствии с необходимостью. Важно для соединения по данному изобретению, что второе положение бензимидазольного кольца является замещенным 4-замещенной-пиридилтиометилгруппой.
Примеры C1-8алкиловой группы по данному изобретению включают линейные или разветвленные алкиловые группы, имеющие 1-8, предпочтительно 1-5 и более, предпочтительно 1-3 атома углерода, например, метиловую группу, этиловую группу, н-пропиловую группу, изопропиловую группу, н-бутиловую группу, изобутиловую группу, втор-бутиловую группу, трет-бутиловую группу, н-пентиловую группу и тому подобное.
Примеры С1-12алкиловой группы, представленные Y в общей формуле (I) по данному изобретению, включают линейные или разветвленные алкиловые группы, имеющие 1-12, предпочтительно 1-10 или 3-12, и более предпочтительно 3-10 атомов углерода, например, метиловую группу, н-пропиловую группу, н-бутиловую группу, изобутиловую группу, втор-бутиловую группу, н-пентиловую группу, н-гексиловую группу, н-гептиловую группу, н-октиловую группу, н-нониловую группу и тому подобное. Предпочтительные примеры C1-12алкиловой группы по данному изобретению включают линейные алкиловые группы, имеющие от 1 до 12, предпочтительно от 1 до 10 и более предпочтительно 3-10 атомов углерода, например, н-пропиловую группу, н-бутиловую группу, н-гексиловую группу, н-гептиловую группу, н-октиловую группу, н-нониловую группу и тому подобное.
Примеры С1-8алкоксигруппы по данному изобретению включают линейные или разветвленные алкиловые группы, имеющие 1-8, предпочтительно 1-5, и более, предпочтительно 1-3 атома углерода, например, метоксигруппу, этоксигруппу, н-пропилоксигруппу, изопропилоксигруппу, н-бутилоксигруппу, изобутилоксигруппу, втор-бутилоксигруппу, трет-бутилоксигруппу, н-пентилоксигруппу и тому подобное.
Примеры атома галогена по данному изобретению включают такие атомы галогена, как атом фтора, атом хлора, атом брома, атом иода и тому подобное.
Примеры С1-5алкиленовой группы по данному изобретению включают линейные или разветвленные алкиленовые группы, имеющие от 1 до 5, предпочтительно от 2 до 5 и более предпочтительно 2-4 атома углерода, например, метиленовую группу, этиленовую группу, н-пропиленовую группу и тому подобное.
В данном изобретении n представляет число повторений оксиалкиленовой группы, и примеры n включают целые числа от 1 до 10, предпочтительно от 2 до 10 и более предпочтительно от 2 до 5. Хотя целые числа n могут быть необязательно фиксированной величиной, например величины n являются смесью 2 и 3 или смесью от 2 до 10, предпочтительно величины n являются фиксированной величиной.
Алкиловая группа, алкоксигруппа и тому подобное по данному изобретению могут быть замещенными каким-либо заместителем. Предпочтительные примеры заместителя включают гидроксильную группу, указанные С1-8алкоксигруппы, указанные атомы галогенов и тому подобное.
Предпочтительные примеры R1 и R2 в общей формуле (I) по данному изобретению включают атом водорода или С1-8алкиловые группы, и атом водорода является более предпочтительным.
Предпочтительные примеры R3 и R4 в общей формуле (I) по данному изобретению включают атом водорода или С1-8алкиловые группы. Более предпочтительные примеры R3 и R4 включают атом водорода, метиловую группу и метоксигруппу.
Предпочтительные примеры X в общей формуле (I) по данному изобретению включают атом кислорода или атом серы.
Примеры одного из Y в общей формуле (I) по данному изобретению включают, предпочтительно, C1-12алкиловые группы, более предпочтительно линейные C1-12алкиловые группы и еще более предпочтительно линейные C3-12алкиловые группы, которые могут быть замещенными гидроксильной группой или C1-8алкоксигруппой. Предпочтительные примеры С1-8алкоксигруппы в качестве заместителя включают С1-3алкоксигруппы, такие как метоксигруппа и этоксигруппа. Гидроксильная группа С1-8алкоксигруппа предпочтительно присоединена к атому углерода на конце С1-12алкиловой группы, хотя она может быть присоединена к атому углерода в любом положении C1-12алкиловой группы. Соответственно, предпочтительные примеры Y включают C1-12алкиловые группы, ω-гидрокси-C1-12алкиловые группы и ω-C1-8алкокси-C1-12алкиловые группы.
Другие предпочтительные примеры Y в общей формуле (I) по данному изобретению включают группы, представленные формулой -(R5-O)n-R6 (в формуле R5 представляет С1-5алкиленовую группу, R6 представляет С1-8алкиловую группу, которая может быть замещена гидроксильной группой или атомом галогена, и n означает целое число от 1 до 10). Более предпочтительные примеры Y включают группы, представленные формулой -(CH2CH2-O)n-R6 (в формуле R6 представляет С1-8алкиловую группу, которая может быть замещена гидроксильной группой или атомом галогена, и n означает целое число от 2 до 5). Предпочтительные примеры атома галогена в этом случае включают атом фтора, и число атомов фтора равно от 1 до 6 и предпочтительно от 1 до 3. Предпочтительные примеры положения замещения атома галогена включают концы алкиловой группы, хотя атом галогена может быть присоединен к атому углерода в любом положении С1-8алкиловой группы. Соответственно, предпочтительные примеры R6 включают С1-8алкиловые группы, ω-моно- или поли-фтор-С1-8алкиловые группы и ω-гидрокси-С1-8алкиловые группы.
Предпочтительные примеры Z в общей формуле (I) по данному изобретению включают атом водорода.
Конкретные примеры фармакологически приемлемой соли тиопроизводного пиридина, представленного общей формулой (I), по данному изобретению включают кислотные аддитивные соли тиопроизводных пиридина и неорганических кислот (например, таких как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное) или органических кислот (например, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, аспарагиновая кислота, глутаминовая кислота и тому подобное) и тому подобное.
Кроме того, тиопроизводное пиридина или его фармакологически приемлемая соль может образовывать сольват с водой или органическим растворителем. Предпочтительные примеры сольвата включают гидрат и различные сольваты (например, сольваты со спиртами, такими как этанол).
В качестве конкретных примеров соединения по данному изобретению соединения, представленные общей формулой (I), в которой R1, R2, R4 и Z означают, соответственно, атом водорода и R3 означает метиловую группу, или их фармакологически приемлемые соли показаны в таблице 1.
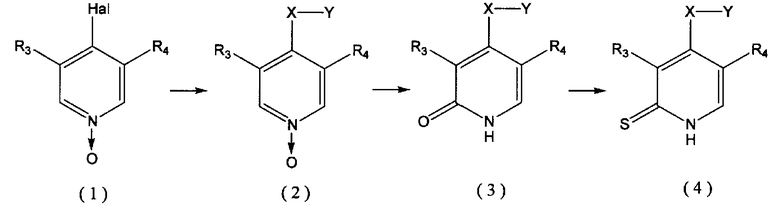
Соединение, представленное общей формулой (I), по данному изобретению или его фармакологически приемлемая соль могут быть получены согласно способу, описанному в примерах, которые будут приведены далее. Эти соединения могут быть получены путем образования производного 1,2-дигидропиридин-N-оксид-2-тиона (4) из производного пиридин-N-оксида (1) способом, показанным ниже.
(в формуле R3, R4, X и Y имеют указанные выше значения и Hal представляет атом галогена), и затем путем взаимодействия производного 1,2-дигидропиридин-N-оксид-2-тиона (4) с производным 2-замещенного метилбензимидазола (5) по следующему способу:
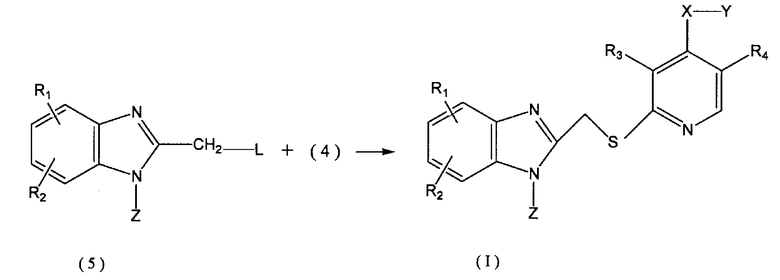
(в формуле, R1, R2, R3, R4, X, Y и Z имеют указанные выше значения и L представляет уходящую группу).
Во-первых, производное пиридина превращают в N-оксид (I) и затем Y-XH подвергают взаимодействию с N-оксидом (1) с получением производного пиридина (2), в котором группа -X-Y присоединена к атому углерода в 4-м положении пиридинового кольца. В этом случае, когда X означает атом кислорода, Y-XH может быть превращена в присутствии основания, такого как гидроксид натрия, в 4-X-Y замещенную структуру (2) в одну стадию. Однако когда X представляет атом серы, предпочтительно, чтобы производное пиридина было превращено в 4-меркапто структуру при использовании, например, бисульфида натрия и тому подобного, и затем 4-меркапто структуру превращают в 4-S-Y замещенную структуру при использовании галогенированного соединения, такого как Y-Hal (где Hal представляет атом галогена). В качестве атома галогена в формуле (1) и Y-Hal предпочтителен атом хлора.
Затем полученную 4-X-Y замещенную структуру (2) превращают в структуру дигидропиридин-2-она (3) в присутствии ангидрида уксусной кислоты и тому подобного, которую затем превращают в сульфид при использовании реагента Лавессона, чтобы тем самым получить структуру дигидропиридин-2-тиона (4).
Полученная структура дигидропиридин-2-тиона (4) может быть подвергнута взаимодействию с производным бензимидазола (5) с получением целевого соединения, представленного общей формулой (I). В качестве уходящей группы L в производном бензимидазола (5) предпочтителен атом галогена и, особенно, атом хлора.
Когда активная функциональная группа имеется в указанных реакциях, предпочтительно защищать функциональную группу с помощью защитной группы при необходимости, чтобы провести реакцию.
Соединение, представленное общей формулой (I), по данному изобретению или его соль могут уничтожать или оказывать бактериостатическое действие на Helicobacter pylori в организмах животных, принадлежащих к млекопитающим, таких как человек, и поэтому эффективны в качестве агента против Helicobacter pylori.
Фармацевтический агент, содержащий соединение, представленное общей формулой (I), или его соль по данному изобретению, является эффективным для профилактики или лечения болезней, в которых участвует Helicobacter pylori. Выражение "болезни, в которых участвует Helicobacter pylori," в данном описании изобретения означает болезни, вызываемые или обостряемые инфекцией, наличием или пролиферацией Helicobacter pylori в живых организмах. Иными словами, "болезнями, в которых участвует Helicobacter pylori," являются болезни, симптомы которых могут быть облегчены путем устранения Helicobacter pylori. Примеры таких болезней включают гастрит, язву желудка, язву двенадцатиперстной кишки, синдромы неязвенной диспепсии, желудочную MALT лимфому, гиперплазированные желудочные полипы и желудочные злокачественные заболевания (особенно, желудочные злокачественные заболевания, которые повторяются после эндоскопического удаления начальных желудочных злокачественных новообразований). Другие примеры "болезней, в которых участвует Helicobacter pylori," включают желудочно-кишечные злокачественные заболевания и панкреатит, вызываемые Helicobacter pylori. Соединение или его соль по данному изобретению может задерживать или подавлять развитие желудочно-кишечных злокачественных заболеваний, связанных с Helicobacter pylori. Дополнительные примеры "болезней, в которых участвует Helicobacter pylori," включают воспалительные кишечные болезни, вызываемые Helicobacter pylori.
Профилактические или лечебные агенты против болезней, в которых участвует Helicobacter pylori, являются композициями, содержащими в качестве эффективных компонентов производное пиридина, представленное общей формулой (I), или его фармакологически приемлемую соль по данному изобретению, и могут быть использованы как фармацевтические композиции. В этом случае соединение по данному изобретению, фармакологически приемлемый носитель и/или добавки обычно вводят в состав и используют как фармацевтическую композицию, хотя соединение по данному изобретению может быть использовано как таковое.
В фармацевтической композиции по данному изобретению различные дозированные формы могут быть приспособлены для соответствующих профилактических и терапевтических целей, и примеры дозированных форм включают порошки, тонкие гранулы, гранулы, таблетки, капсулы, сухие сиропы, сиропы, препараты для инъекций и тому подобное. Эти дозированные формы могут быть введены перорально или парентерально.
Когда готовят твердый состав для перорального введения, наполнитель и, если необходимо, связующее, дезинтегрирующая добавка, смазка, краситель, корректирующий/нейтрализующий агент и тому подобное могут быть добавлены к соединению по данному изобретению, и затем полученная композиция может быть приготовлена в виде таблеток, глазированных таблеток, гранул, порошков, капсул или тому подобного обычным способом. Добавки могут быть такими, которые обычно используют в соответствующих областях. Например, кукурузный крахмал, лактоза, сахароза, хлорид натрия, маннит, сорбит, глюкоза, крахмал, карбонат кальция, каолин, микрокристаллическая целлюлоза, кремниевая кислота или тому подобное могут быть использованы в качестве наполнителя. Вода, этанол, гуммиарабик, трагакант, пропанол, чистый сироп, раствор глюкозы, раствор крахмала, раствор желатина, карбоксиметилцеллюлоза, гидроксипропилцеллюлоза, желатин, гидроксипропилкрахмал, метилцеллюлоза, этилцеллюлоза, шеллак, фосфат кальция, поливиниловый спирт, поливиниловый простой эфир, поливинилпирролидон или тому подобное могут быть использованы в качестве связующего. Желатиновые порошки, кристаллическая целлюлоза, сухой крахмал, альгинат натрия, пектин, порошки агара, карбоксиметилцеллюлоза, бикарбонат натрия, карбонат кальция, цитрат кальция, лаурилсульфат натрия, моноглицерид стеариновой кислоты, лактоза или тому подобное могут быть использованы в качестве дезинтегрирующей добавки. Диоксид кремния, очищенный тальк, стеарат, бура, полиэтиленгликоль или тому подобное могут быть использованы в качестве смазки. В качестве красителя могут быть использованы такие красители, как оксид титана и оксид железа, которые разрешено добавлять. В качестве корректирующего/нейтрализующего агента могут быть использованы сахароза, цедра горького апельсина, лимонная кислота, винная кислота или тому подобное.
В случае приготовления жидкого состава для перорального введения корректирующий агент, буфер, стабилизатор, нейтрализующий агент и тому подобное могут быть добавлены к соединению по данному изобретению, чтобы получить жидкий препарат для перорального введения, сироп, эликсир и тому подобное обычным способом. В этом случае корректирующим/нейтрализующим агентом могут быть указанные выше. Примеры буфера включают натрия цитрат и тому подобное. Примеры стабилизатора включают трагакант, гуммиарабик, желатин и тому подобное.
В случае приготовления препарата для инъекций регулятор рН, буфер, стабилизатор, изотонический агент, местное обезболивающее и тому подобное могут быть добавлены к соединению по данному изобретению, чтобы получить препарат для подкожной, внутримышечной или внутривенной инъекции обычным способом. Примеры регулятора pH и буфера, в этом случае, включают цитрат натрия, ацетат натрия, фосфат натрия и тому подобное. Примеры стабилизатора включают пиросульфит натрия, EDTA, тиогликолевую кислоту, тиомолочную кислоту и тому подобное. Примеры местного обезболивающего включают прокаин гидрохлорид, лидокаин гидрохлорид и тому подобное. Примеры изотонического агента включают хлорид натрия, глюкозу и тому подобное.
Фармацевтическая композиция и профилактические и лечебные агенты по данному изобретению могут дополнительно содержать один или два вида или более видов декстринов в добавление к тиопроизводному пиридина, представленному общей формулой (I), или его фармакологически приемлемой соли. Примеры указанного декстрина, применимого по данному изобретению, включают, но без ограничения перечисленным, α-декстрин, β-декстрин, γ-декстрин, α-циклодекстрин, β-циклодекстрин, γ-циклодекстрин и тому подобное.
Фармацевтическая композиция и профилактические и лечебные агенты по данному изобретению могут дополнительно содержать один или два вида или более видов медицинских компонентов, подавляющих секрецию желудочной кислоты, в дополнение к тиопроизводному пиридина, представленному общей формулой (I), или его фармакологически приемлемой соли, или могут быть использованы в сочетании с указанными медицинскими компонентами. Примеры медицинских компонентов, подавляющих секрецию желудочной кислоты, включают H2 блокатор, ингибитор протонного насоса (PPI) и тому подобное.
Примеры H2 блокаторов, которые могут быть использованы по данному изобретению, включают, но без ограничения перечисленным, фамотидин, ранитидин и тому подобное. Дополнительно примеры ингибитора протонного насоса, которые могут быть использованы по данному изобретению, включают, но без ограничения перечисленным, ланзопразол, рабепразол, пантопразол и тому подобное.
Согласно указанной выше комбинированной рецептуре эффект данного изобретения ожидается более усовершенствованным.
Хотя количество соединения по данному изобретению, которое может быть приготовлено в виде каждой указанной унифицированной дозированной формы, зависит, например от симптома у пациента, которому соединение вводят, от дозированной формы и тому подобного, и поэтому не является фиксированным, количество на унифицированную дозированную форму желательно от около 1 - 1200 мг в случае медикамента для перорального введения и от около 0,1 до 500 мг в случае инъекции. Кроме того, хотя суточная доза медикамента, имеющего указанную дозированную форму, различается в зависимости от симптома, массы, возраста, пола и тому подобного пациента и не является определенной безоговорочно, обычно она от около 0,1 до 5000 мг и предпочтительно от 1 до 1200 мг в сутки для взрослых, и медикамент предпочтительно вводят за раз или раздельно от двух до четырех раз в сутки.
Данное изобретение будет пояснено подробно на примерах, которые не предназначаются для ограничения данного изобретения.
Сравнительный пример 1
Получение 4-меркапто-3-метилпиридин-1-оксида
130,6 г (0,910 моль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида и 330,0 г (3,788 моль, 4,2 эквивалента) тригидрата гидросульфида натрия добавляли к 2,17 л этанола и смесь подвергали реакции при кипении с обратным холодильником с перемешиванием в течение 5,5 ч. Реакционный раствор охлаждали и перегоняли при пониженном давлении, чтобы удалить растворитель, чтобы таким образом получить остаток, к которому добавляли 1,31 л воды, чтобы растворить остаток. 379,2 г (3,64 моль, 4,0 эквивалента) 35% хлористоводородной кислоты постепенно добавляли к полученному раствору с перемешиванием, чтобы осадить кристаллы. Полученные кристаллы собирали фильтрованием с последующей сушкой при пониженном давлении, получая 124,4 г 4-меркапто-3-метилпиридин-1-оксида (HPLC: 97,7 % по площади, выход: 96,9%).
1H-ЯМР (400 МГц, CDCl3) δ:
0,94 (3H, т, J=7 Гц), 1,35-1,45 (4H, м), 1,76-1,81 (2H, м), 2,13 (3H, c), 3,97 (2H, т, J=6 Гц), 4,71 (1H, д, J=14 Гц), 4,82 (1H, д, J=14 Гц), 6,69 (1H, д, J=6 Гц), 7,33-7,28 (2H, м), 7,63 (2H, м), 8,30 (1H, д, J=6 Гц)
MC m/z: 357 (М+)
Пример 1
Получение гидрохлорида 2-[[[4-[(4-метоксибутил]тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
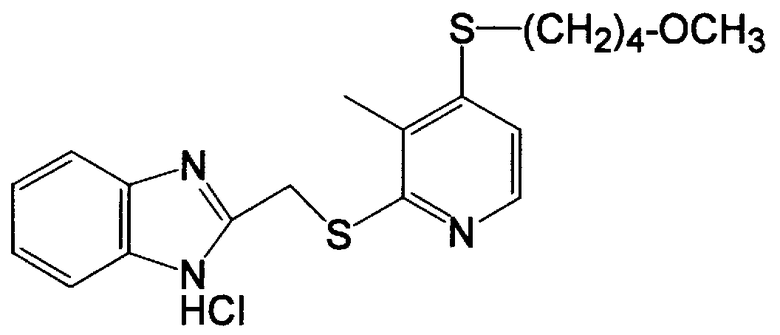
(1) Получение 4-[(4-метоксибутил)тио-3-метилпиридин-1-оксида
173,3 г (1,5 эквивалента) водного 30% раствора гидроксида натрия, 106,7 г (1,0 эквивалент) 1-хлор-4-метоксибутана и 1,85 л этанола добавляли к 122,8 г (0,870 моль, 1,0 эквивалент) 4-меркапто-3-метилпиридин-1-оксида, полученного в сравнительном примере 1, и смесь кипятили с обратным холодильником с перемешиванием в течение 7 ч и затем реакцию прекращали. Затем реакционную смесь охлаждали, перегоняли ее при 40°C или ниже при пониженном давлении, чтобы удалить растворитель, с получением остатка, и к полученному остатку добавляли 0,93 л воды и затем смесь экстрагировали 1,5 л этилацетата и затем 1,0 л (×2) дихлорметана. Полученные органические фазы объединяли вместе и затем сушили ангидридом сульфата натрия. Сухую органическую фазу перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая таким образом 183,2 г 4-[(4-метоксибутил)тио-3-метилпиридин-1-оксида.
(2) Получение 4-[(4-метоксибутил)тио-2-кето-3-метилпиридина
1796,8 г (17,60 моль, 22,1 эквивалента) ангидрида уксусной кислоты добавляли к 181,0 г (0,796 моль, 1,0 эквивалент) (4-[(4-метоксибутил)тио-3-метилпиридин-1-оксида, чтобы осуществить реакцию при нагревании/кипячении с обратным холодильником в течение 10 ч с перемешиванием. Реакционный раствор концентрировали при 40°C или менее при пониженном давлении, затем 1,5 л этилацетата и 0,24 л метанола добавляли к концентрированной смеси, которую дополнительно перемешивали при нагревании/кипячении с обратным холодильником в течение 6 ч и 20 мин. Реакционный раствор концентрировали при 40°C или менее при пониженном давлении и полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 100,6 г 4-[(4-метоксибутил)тио-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[(4-метоксибутил)тио]-3-метилпиридин-2-тиона
96,3 г (0,238 моль, 1,1 эквивалента) реагента Лавессона и 0,76 л толуола добавляли к 100,0 г (0,440 моль, 1,0 эквивалент) 4-[(4-метоксибутил)тио-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 14 ч. Затем реакционную смесь охлаждали, осажденное твердое вещество промывали дихлорметаном, получая таким образом 17,1 г 4-[(4-метоксибутил)тио]-3-метилпиридин-2-тиона.
(4) Получение гидрохлорида 2-[[[4-[(4-метоксибутил)тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
10,02 г (41,2 ммоль, 1,0 эквивалент) 4-[(4-метоксибутил)тио]-3-метилпиридин-2-тиона и 7,50 г (45,0 ммоль, 1,1 эквивалента) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 6,0 г водного 30% раствора гидроксида натрия и 315 мл этанола и смесь перемешивали при 50°C в течение 6 ч. Чтобы завершить реакцию, 1,40 г (8,4 ммоль, 0,2 эквивалента) 2-(хлорметил)бензимидазола и 1,1 г водного 30% раствора гидроксида натрия дополнительно добавляли к реакционному раствору, который дополнительно перемешивали при 50°С в течение 4 ч, чтобы завершить реакцию. Затем реакционный раствор концентрировали при пониженном давлении, 240 мл воды добавляли к реакционному раствору и реакционный раствор экстрагировали дихлорметаном три раза (470 мл, 200 мл, 200 мл). Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, кристаллизовали из этанольного раствора хлористоводородной кислоты, получая 9,23 г бесцветных кристаллов целевого гидрохлорида 2-[[[4-[(4-метоксибутил)тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола.
Общий выход был 4,7% и чистота, измеренная путем HPLC, была 99,2%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
1,62-1,66 (4H, м), 2,21 (3H, c), 3,04-3,07 (2H, м), 3,21 (3H, c) 3,31-3,34 (2H, м), 4,94 (3H, c), 7,09 (1H, д, J=5,6 Гц), 7,50-7,55 (2H, м), 7,74-7,79 (2H, м), 8,14 (1H, д, J=5,6 Гц)
MC m/z: 373 (М+; свободное тело)
Пример 2
Получение 2-[[[4-[(8-гидроксиоктил)тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола

(1) Получение 4-[(8-гидроксиоктил)тио]-3-метилпиридин-1-оксида
88,7 г (1,6 эквивалента) водного 30% раствора гидроксида натрия, 88,7 г (1,0 эквивалент) 8-бром-1-октанола и 0,9 л этанола добавляли к 59,85 г (0,424 моль, 1,0 эквивалент) 4-меркапто-3-метилпиридин-1-оксида, полученного в сравнительном примере 1, и смесь кипятили с обратным холодильником с перемешиванием в течение 5 ч и затем реакцию прекращали. Потом реакционную смесь охлаждали, перегоняли ее при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая остаток, к полученному остатку добавляли 0,76 л воды и затем 1,5 л этилацетата. Осадок собирали фильтрованием и сушили при пониженном давлении, получая таким образом 103,6 г 4-[(8-гидроксиоктил)тио]-3-метилпиридин-1-оксида.
(2) Получение 4-[(8-ацетоксиоктил)тио]-2-кето-3-метилпиридина
55,0 г (15,2 эквивалента) ангидрида уксусной кислоты добавляли к 97,43 г (0,362 моль, 1,0 эквивалент) 4-[(8-гидроксиоктил)тио]-3-метилпиридин-1-оксида, чтобы кипятить с обратным холодильником с перемешиванием в течение 7 ч. Потом реакционный раствор перегоняли, чтобы удалить ангидрид уксусной кислоты, 520 мл этилацетата и 69 мл метанола добавляли к реакционному раствору, полученный реакционный раствор дополнительно кипятили с обратным холодильником с перемешиванием в течение 5 ч и 30 мин. Реакционный раствор концентрировали при пониженном давлении и полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 46,24 г 4-[(8-ацетоксиоктил)тио]-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[(8-ацетоксиоктил)тио]-3-метилпиридин-2-тиона
32,5 г (0,080 моль, 1,1 эквивалента) реагента Лавессона и 255 мл толуола добавляли к 46,13 г (0,148 моль, 1,0 эквивалент) 4-[(8-ацетоксиоктил)тио]-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 12 ч. Потом реакционную смесь охлаждали, перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель. 600 мл воды затем добавляли к реакционному раствору, который экстрагировали 6000 мл дихлорметана. Полученные органические фазы объединяли вместе, промывали три раза 600 мл воды и затем сушили ангидридом сульфата натрия. Сухую органическую фазу затем перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая таким образом 8,40 г 4-[(8-ацетоксиоктил)тио]-3-метилпиридин-2-тиона.
(4) Получение 2-[[[4-[(8-гидроксиоктил)тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
8,40 г (25,6 ммоль, 1,0 экв.) 4-[(8-ацетоксиоктил)тио]-3-метилпиридин-2-тиона и 4,32 г (25,9 ммоль, 1,0 экв.) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 3,20 г водного 30% раствора гидроксида натрия и 192 мл этанола и смесь перемешивали при 50°C в течение 2 ч и 30 мин. Так как реакция не завершалась, 0,76 г (4,6 ммоль, 0,2 экв.) 2-(хлорметил)бензимидазола и 0,56 г водного 30% раствора гидроксида натрия добавляли к реакционному раствору, который дополнительно подвергали реакции при 50°С в течение 1 ч. Чтобы завершить реакцию, 1,53 г (9,2 ммоль, 0,4 экв.) 2-(хлорметил)бензимидазола и 1,15 г водного 30% раствора гидроксида натрия снова добавляли к реакционному раствору, который дополнительно перемешивали при 20°C в течение 13 ч. 3,21 г водного 30% раствора гидроксида натрия дополнительно добавляли к реакционному раствору, который затем еще перемешивали при 20°С в течение 10 ч. Осадок подвергали перекристаллизации из водного ацетона, получая 6,45 г бесцветных кристаллов целевого 2-[[[4-[(8-гидроксиоктил)тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола.
Общий выход был 3,9% и чистота, измеренная путем HPLC, была 98,4%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
1,41-1,26 (10H, м), 1,63 (2H, м), 2,17 (3H, c), 3,04 (2H, т, J=7,2Гц), 3,37-3,34 (2H, м), 4,34 (1H, ушир.с), 4,66 (2H, c), 7,08 (1H, д, J=5,6 Гц), 7,12-7,15 (2H, м), 7,48 (2H, ушир.c), 8,25 (1H, д, J=5,6 Гц), 12,25 (1H, ушир.с)
MC m/z : 415 (М+)
Пример 3
Получение гидрохлорида 2-[[[4-[(8-метокси-1-октил]тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
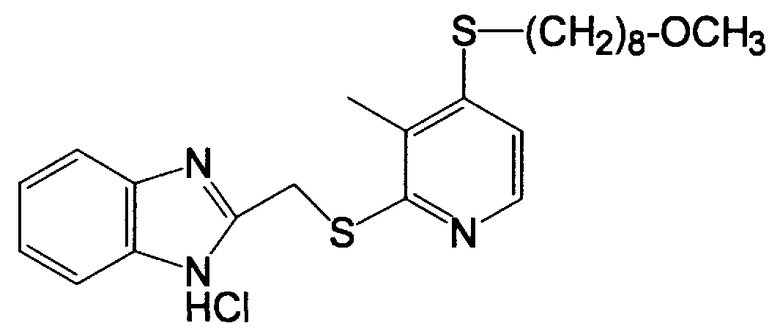
(1) Получение 4-[(8-метоксиоктил)тио-3-метилпиридин-1-оксида
2,25 г (1,5 эквивалента) водного 30% раствора гидроксида натрия, 1,93 г (1,0 эквивалент) 8-хлор-1-метоксиоктана и 30 мл этанола добавляли к 1,6 г (11,3 ммоль, 1,0 эквивалент) 4-меркапто-3-метилпиридин-1-оксида, полученного в сравнительном примере 1, смесь кипятили с обратным холодильником с перемешиванием в течение 19 ч и затем реакцию прекращали. Потом реакционную смесь охлаждали, концентрировали ее при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая остаток, к полученному остатку добавляли 20 мл воды и затем смесь экстрагировали дважды 400 мл этилацетата. Полученные органические фазы объединяли вместе и затем сушили ангидридом сульфата натрия. Сухую органическую фазу затем перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая таким образом 2,36 г 4-[(8-метоксиоктил)тио-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-[(8-метоксиоктил)тио]-2-кето-3-метилпиридина
19,4 г (22,9 эквивалента) ангидрида уксусной кислоты добавляли к 2,36 г (8,3 ммоль, 1,0 эквивалент) 4-[(8-метоксиоктил)тио-3-метилпиридин-1-оксида для кипячения с обратным холодильником в течение 28 ч с перемешиванием. Реакционный раствор перегоняли, чтобы удалить ангидрид уксусной кислоты, затем 18 мл этилацетата и 2,5 мл метанола добавляли к реакционному раствору и реакционный раствор дополнительно кипятили с обратным холодильником с перемешиванием в течение 11 ч. Реакционный раствор концентрировали при пониженном давлении и полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 1,41 г 4-[(8-метоксиоктил)тио]-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[(8-метоксиоктил)тио]-3-метилпиридин-2-тиона
1,10 г (2,7 ммоль, 1,1 эквивалент) реагента Лавессона и 10 мл толуола добавляли к 1,40 г (4,9 ммоль, 1,0 эквивалент) 4-[(8-метоксиоктил)тио-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 4 ч. Потом реакционную смесь охлаждали, перегоняли ее при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая таким образом 0,18 г 4-[(8-метоксиоктил)тио]-3-метилпиридин-2-тиона.
(4) Получение гидрохлорида 2-[[[4-[(8-метокси-1-октил)тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
111,6 мг (0,37 ммоль, 1,0 эквивалент) 4-[(8-метоксиоктил)тио]-3-метилпиридин-2-тиона и 84,1 мг (0,5 ммоль, 1,0 эквивалент) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 0,068 мл водного 30% раствора гидроксида натрия и 3 мл этанола и смесь перемешивали при 50°C в течение 5 ч. Так как реакция не была завершена, 13,6 мг (0,08 ммоль, 0,2 эквивалента) 2-(хлорметил)бензимидазола и 0,011 мл водного 30% раствора гидроксида натрия дополнительно добавляли к реакционному раствору, который дополнительно подвергали реакции при 50°C в течение 14 ч и 30 мин. 6 мл воды добавляли к реакционному раствору и реакционный раствор экстрагировали 18 мл дихлорметана три раза. Потом органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольного раствора хлористоводородной кислоты, получая 66,5 мг бесцветных кристаллов целевого гидрохлорида 2-[[[4-[(8-метоксиоктил)тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола.
Общий выход был 2,3% и чистота, измеренная путем HPLC, была 97,3%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
1,42-1,22 (10H, м), 1,61 (2H, м, J=6,8 Гц), 2,21 (3H, c), 3,02 (2H, т, J=7,2 Гц), 3,19 (3H, c), 3,27 (2H, т, J=6,8 Гц), 4,92 (2H, c), 7,08 (1H, д, J=5,6 Гц), 7,50-7,54 (2H, м), 7,73-7,78 (2H, м), 8,12 (1H, д, J=5,2 Гц)
MC m/z : 429 (М+: свободное тело)
Пример 4
Получение гидрохлорида 2-[[(4-н-пропокси-3-метил-2-пиридил)тио]метил]-1H-бензимидазола

(1) Получение 4-пропокси-3-метилпиридин-1-оксида
8,32 г (208 ммоль, 2,0 эквивалента) 60% гидрированного натрия добавляли к смешанному раствору 12,5 г (208 ммоль, 1,0 эквивалент) 1-пропанола и 200 мл ДМСО в течение 1 ч в струе азота при перемешивании смешанного раствора при нагревании при 50°C. После завершения добавления и после перемешивания смешанного раствора при нагревании при 53-57°C к смеси добавляли 15,0 г (104,5 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида. Смесь дополнительно перемешивали при нагревании при 44-46°C в течение 3 ч, добавляли 15 мл воды и затем перегоняли при пониженном давлении, чтобы удалить растворитель. Потом 400 мл воды добавляли к концентрированному остатку, раствор экстрагировали 500 мл дихлорметана и затем 400 мл дихлорметана. Экстракт сушили ангидридом сульфата магния и затем концентрировали при пониженном давлении досуха, получая таким образом 24,1 г 4-пропокси-3-метилпиридин-1-оксида.
(2) Получение 4-пропокси-2-кето-3-метилпиридина
233,6 г (2,29 моль, 16 эквивалентов) ангидрида уксусной кислоты добавляли к 24,0 г (143,5 ммоль, 1,0 эквивалент) 4-пропокси-3-метилпиридин-1-оксида для проведения реакции при 110-112°С в течение 8 ч. Реакционный раствор перегоняли, чтобы удалить ангидрид уксусной кислоты, затем 244 мл этилацетата и 31 мл метанола добавляли к полученному концентрированному остатку, реакционный раствор дополнительно кипятили с обратным холодильником с перемешиванием в течение 2 ч. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 9,67 г 4-пропокси-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-пропокси-3-метилпиридин-2-тиона
11,8 г (29,2 ммоль, 1,1 экв.) реагента Лавессона и 93 мл толуола добавляли к 9,0 г (53,8 ммоль, 1,0 эквивалент) 4-пропокси-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 9 ч. 1,3 г (3,2 ммоль, 0,1 экв.) реагента Лавессона дополнительно добавляли к реакционному раствору, который подвергали реакции при кипении с обратным холодильником в течение 10 ч. Потом реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая таким образом 0,39 г 4-пропокси-3-метилпиридин-2-тиона.
(4) Получение гидрохлорида 2-[[(4-н-пропокси-3-метил-2-пиридил)тио]метил]-1H-бензимидазола
370 мг (2,0 ммоль, 1,0 эквивалент) 4-пропокси-3-метилпиридин-2-тиона и 370 мг (2,2 ммоль, 1,1 эквивалент) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 297 мг водного 30% раствора гидроксида натрия и 16 мл этанола и смесь перемешивали при 50°С в течение 2 ч. Затем реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 26 мл воды добавляли к раствору, который затем экстрагировали 112 мл дихлорметана. Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольного раствора хлористоводородной кислоты, получая 431 мг бесцветных кристаллов целевого гидрохлорида 2-[[(4-пропокси-3-метил-2-пиридил)тио]метил]-1H-бензимидазола.
Общий выход был 0,14% и чистота, измеренная путем HPLC, была 98,7%.
1H-ЯМР (400 МГц, CDCl3) δ:
1,09 (3H, т, J=7,2 Гц), 1,90-1,99 (2H, м), 2,40 (3H, c), 4,22 (2H, т, J=6,4 Гц), 5,46 (2H, c), 7,11 (1H, д, J=6,8 Гц), 7,48-7,53 (2H, м), 7,79-7,83 (2H, м), 8,55 (1H, д, J=6,8 Гц)
MC m/z: 313 (М+: свободное тело)
Пример 5
Получение гидрохлорида 2-[[(4-тиометил-3-метил-2-пиридил)тио]метил]-1H-бензимидазола

(1) Получение 4-метилтио-3-метилпиридин-1-оксида
Смешанный раствор, содержащий 5,0 г (34,8 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида, 2,4 г (34,2 ммоль, 1,0 эквивалент) тиометоксида натрия и 74 мл этанола, перемешивали при нагревании/кипячении с обратным холодильником в течение 5 ч. 1,2 г (17 ммоль, 0,5 эквивалента) тиометоксида натрия дополнительно добавляли к раствору, который затем дополнительно подвергали реакции при нагревании/кипячении с обратным холодильником в течение 1 ч и затем охлаждали до температуры окружающей среды. Потом реакционный раствор концентрировали при 40°C при пониженном давлении и добавляли 20 мл воды, реакционный раствор экстрагировали дважды 37 мл этилацетата, дополнительно дважды 40 мл дихлорметана и затем дважды 50 мл дихлорметана. Объединенные экстрагированные слои сушили ангидридом сульфата магния и затем концентрировали при пониженном давлении досуха, получая 4,9 г 4-метилтио-3-метилпиридин-1-оксида в твердой форме.
(2) Получение 4-метилтио-2-кето-3-метилпиридина
68,0 г (666 ммоль, 22 эквивалента) ангидрида уксусной кислоты добавляли к 4,7 г (30,3 ммоль, 1,0 эквивалент) 4-метилтио-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 107-115°С в течение 20 ч. 63 мл этилацетата и 8,4 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, остаток дополнительно кипятили с обратным холодильником с перемешиванием в течение 3 ч. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 3,62 г 4-метилтио-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-метилтио-3-метилпиридин-2-тиона
4,8 г (11,9 ммоль, 1,1 эквивалента) реагента Лавессона и 38 мл толуола добавляли к 3,4 г (21,9 ммоль, 1,0 эквивалент) 4-метилтио-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 11 ч. Затем реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель, полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая таким образом 1,33 г 4-метилтио-3-метилпиридин-2-тиона в виде твердого вещества.
(4) Получение гидрохлорида 2-[[(4-тиометил-3-метил-2-пиридил)тио]метил]-1H-бензимидазола
1,29 г (7,5 ммоль, 1,0 эквивалент) 4-метилтио-3-метилпиридин-2-тиона и 1,38 г (8,3 ммоль, 1,1 эквивалента) 2- (хлорметил)бензимидазола добавляли в смешанный раствор 1,10 г водного 30% раствора гидроксида натрия и 100 мл этанола и смесь перемешивали при 50°C в течение 2 ч. Так как реакция не была завершена, 250 мг (1,5 ммоль, 0,20 эквивалента) 2-(хлорметил)бензимидазола дополнительно добавляли к реакционному раствору, который затем перемешивали при 50°C в течение 1 ч. 0,20 г водного 30% раствора гидроксида натрия дополнительно добавляли к реакционному раствору, который дополнительно подвергали реакции при 50°С в течение 30 мин. Затем реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 94 мл воды добавляли к раствору, который затем экстрагировали 300 мл дихлорметана и затем 100 мл дихлорметана. Потом органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольного раствора хлористоводородной кислоты, получая 62 мг бесцветных кристаллов целевого гидрохлорида 2-[[(4-тиометил-3-метил-2-пиридил)тио]метил]-1H-бензимидазола.
Общий выход был 0,62% и чистота, измеренная путем HPLC, была 97,8%.
1H-ЯМР (400 МГц, CDCl3) δ:
2,22 (3H, c), 4,91 (2H, c), 7,04 (1H, д, J=5,6 Гц), 7,50-7,54 (2H, м), 7,72-7,77 (2H, м), 8,14 (1H, д, J=5,6 Гц)
MC m/z: 301 (М+: свободное тело)
Пример 6
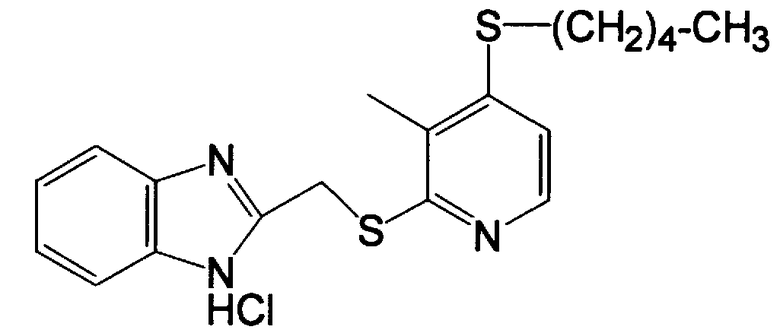
Получение гидрохлорида 2-[[(4-тиопропил-3-метил-2-пиридил)тио]метил]-1H-бензимидазола

(1) Получение 4-пропилтио-3-метилпиридин-1-оксида
5,0 г (34,8 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида, 7,0 г (52,5 ммоль, 1,5 эквивалента) водного 30% раствора гидроксида натрия и 2,7 г (35,5 ммоль, 1,0 эквивалент) 1-пропантиола добавляли к 74 мл этанола и смесь подвергали реакции при 72-73°C при нагревании/перемешивании в течение 1 ч и 30 мин. Так как реакция не была завершена, 0,5 г (6,6 ммоль, 0,2 эквивалента) 1-пропантиола и 1,4 г (10,5 ммоль, 0,3 эквивалента) водного 30% раствора гидроксида натрия дополнительно добавляли к реакционному раствору, который затем дополнительно подвергали реакции при 2-73°C при нагревании/перемешивании в течение 1 ч и 30 мин. После охлаждения до 30°C реакционный раствор перегоняли при пониженном давлении, чтобы удалить растворитель. Затем 20 мл воды добавляли к концентрированному остатку, остаток экстрагировали четыре раза в целом, т.е. дважды 37 мл этилацетата и дважды 30 мл этилацетата. Экстракт сушили ангидридом сульфата магния и затем концентрировали при пониженном давлении досуха, получая таким образом 6,25 г 4-пропилтио-3-метилпиридин-1-оксида в виде твердого вещества.
(2) Получение 4-пропилтио-2-кето-3-метилпиридина
40,8 г (730 ммоль, 22 эквивалента) ангидрида уксусной кислоты добавляли к 6,05 г (33,0 ммоль, 1,0 эквивалент) 4-пропилтио-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 108-110°С в течение 18 ч. 69 мл этилацетата и 9,5 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, который дополнительно кипятили с обратным холодильником с перемешиванием в течение 2 ч. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 3,5 г 4-пропилтио-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-пропилтио-3-метилпиридин-2-тион
0,83 г (11,9 ммоль, 1,1 эквивалента) реагента Лавессона и 6,6 мл толуола добавляли к 0,7 г (3,8 ммоль, 1,0 эквивалент) 4-пропилтио-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 11 ч. Затем реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и полученный концентрированный остаток очищали колоночной хроматографией на силикагеле. К очищенному остатку затем добавляли 2 мл 1-гексана, чтобы промыть остаток три раза, получая 0,081 г 4-пропилтио-3-метилпиридин-2-тиона в виде твердого вещества.
Целевой материал получали в большем количестве так же, как указано выше.
3,3 г (8,2 ммоль, 1,1 эквивалента) реагента Лавессона и 26 мл толуола добавляли к 2,8 г (15,3 ммоль, 1,0 эквивалент) 4-пропилтио-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 7 ч и 30 мин. Затем реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и 50 мл дихлорметана добавляли к полученному остатку, чтобы удалить нерастворимое вещество фильтрованием. Концентрированный остаток, полученный концентрированием фильтрата при пониженном давлении, очищали колоночной хроматографией на силикагеле, получая 0,29 г 4-пропилтио-3-метилпиридин-2-тиона в виде твердого вещества.
(4) Получение гидрохлорида 2-[[(4-тиопропил-3-метил-2-пиридил)тио]метил]-1H-бензимидазола
370 мг (1,86 ммоль, 1,0 эквивалент) 4-тиопропил-3-метилпиридин-2-тиона и 340 мг (2,0 ммоль, 1,1 эквивалента) 2- (хлорметил)бензимидазола добавляли в смешанный раствор 320 мг водного 30% раствора гидроксида натрия и 24 мл этанола и смесь перемешивали при 50°C в течение 2 ч. Так как реакция не была завершена, 62 мг (0,37 ммоль, 0,5 эквивалента) 2-(хлорметил)бензимидазола и 50 мг водного 30% раствора гидроксида натрия дополнительно добавляли к реакционному раствору, который дополнительно подвергали реакции при 50°C в течение 3 ч. Потом реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 39 мл воды добавляли к раствору, который затем экстрагировали 100 мл дихлорметана и затем 68 мл дихлорметана. Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольного раствора хлористоводородной кислоты, получая 93 мг бесцветных кристаллов гидрохлорида 2-[[(4-пропокси-3-метил-2-пиридил)тио]метил]-1H-бензимидазола.
Общий выход был 0,79% и чистота, измеренная путем HPLC, была 99,5%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
0,99 (3H, т, J=7,2 Гц), 1,61-1,66 (2H, м), 2,22 (3H, c), 3,02 (2H, т, J=7,2 Гц), 4,92 (2H, c), 7,09 (1H, д, J=5,6 Гц), 7,50-7,55 (2H, м), 7,73-7,78 (2H, м), 8,12 (1H, д, J=5,6 Гц)
MC m/z: 329 (М+: свободное тело)
Пример 7
Получение 2-[[(4-тиопентил-3-метил-2-пиридил)тио]метил]-1H-бензимидазола
(1) Получение 4-пентилтио-3-метилпиридин-1-оксида
Смесь 3,63 г (34,8 ммоль, 1,0 эквивалент) 1-пентантиола, 74 мл этанола и 7,0 г (52,5 ммоль, 1,5 эквивалента) водного 30% раствора гидроксида натрия перемешивали при нагревании/кипячении с обратным холодильником в течение 1 ч. Затем реакционную смесь охлаждали до 30°C, 5,0 г (34,8 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида добавляли к реакционному раствору, который снова перемешивали при нагревании в течение 1 ч. После охлаждения до температуры окружающей среды реакционный раствор перегоняли при пониженном давлении, чтобы удалить растворитель. Затем 20 мл воды добавляли к концентрированному остатку, остаток экстрагировали дважды в целом, т.е. один раз 20 мл этилацетата и один раз 37 мл этилацетата. Экстракт сушили ангидридом сульфата магния и затем концентрировали при пониженном давлении досуха, получая таким образом 6,0 г 4-пентилтио-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-пентилтио-2-кето-3-метилпиридина
78,2 г (766 ммоль, 21 эквивалент) ангидрида уксусной кислоты добавляли к 7,8 г (36,9 ммоль, 1,0 эквивалент) 4-пентилтио-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 108-111°С в течение 15 ч. 78 мл этилацетата и 10,4 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, его дополнительно кипятили с обратным холодильником с перемешиванием в течение 3 ч. Реакционный раствор охлаждали и затем перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 2,61 г 4-пентилтио-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-пентилтио-3-метилпиридин-2-тиона
0,56 г (1,4 ммоль, 1,1 эквивалента) реагента Лавессона и 5 мл толуола добавляли к 0,54 г (2,6 ммоль, 1,0 эквивалент) 4-пентилтио-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 9 ч. Потом реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая таким образом 0,343 г 4-пентилтио-3-метилпиридин-2-тиона в виде порошкообразного твердого вещества.
(4) Получение 2-[[(4-тиопентил-3-метил-2-пиридил)тио]метил]-1H-бензимидазола
330 мг (1,7 ммоль, 1,0 эквивалент) 4-тиопентил-3-метилпиридин-2-тиона и 304 мг (1,8 ммоль, 1,1 эквивалента) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 243 мг водного 30% раствора гидроксида натрия и 13 мл этанола и смесь перемешивали при 50°C в течение 2 ч. Затем реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 21 мл воды добавляли к раствору, который затем экстрагировали 91 мл дихлорметана. Потом органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, концентрировали и очищали, получая 222 мг бесцветных кристаллов целевого 2-[[(4-тиопентил-3-метил-2-пиридил)тио]метил]-1H-бензимидазола.
Общий выход был 6,8% и чистота, измеренная путем HPLC, была 99,6%.
1H-ЯМР (400 МГц, CDCl3) δ:
0,93 (3H, т, J=7,2 Гц), 1,35-1,51 (4H, м), 1,71-1,78 (2H, м), 2,25 (3H, c), 2,97 (2H, т, J=7,6 Гц), 4,57 (2H, c), 6,96 (1H, д, J=5,6 Гц), 7,18-7,23 (2H, м), 7,35 (1H, ушир.с), 7,72 (1H, ушир.с), 8,33 (1H, д, J=5,6 Гц), 10,90 (1H, ушир.с)
MC m/z: 357 (М+: свободное тело)
Пример 8
Получение гидрохлорида 2-[[4-[[2-[2-(2,2,2-трифторэтокси)этокси]этил]тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола

(1) Получение 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-3-метилпиридин-1-оксида
8,71 г (217,8 ммоль, 1,5 эквивалента) гидрохлорида натрия, 8,71 мл воды, 31,18 г (150,9 ммоль, 1,1 эквивалента) 1-хлор-2-[2-(2,2,2-трифторэтокси)этокси]этана и 300 мл этанола добавляли к 20,02 г (141,8 ммоль, 1,0 эквивалент) 4-меркапто-3-метилпиридин-1-оксида, полученного в сравнительном примере 1, смесь кипятили с обратным холодильником с перемешиванием в течение 8 ч и затем реакцию прекращали. Потом реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и 80 мл воды добавляли к полученному остатку, который затем экстрагировали 300 мл этилацетата. Полученную органическую фазу сушили ангидридом сульфата магния и затем сухую органическую фазу перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая таким образом 46,01 г 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-2-кето-3-метилпиридина
324 г (3,2 моль, 22 эквивалента) ангидрида уксусной кислоты добавляли к 45,73 г (146,9 ммоль, 1,0 эквивалент) 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 100°С в течение 15 ч. 300 мл этилацетата и 40 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, и полученную смесь дополнительно кипятили с обратным холодильником с перемешиванием в течение 2 ч. Потом реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 18,50 г 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-3-метилпиридин-2-тиона
12,57 г (31,1 ммоль, 1,1 эквивалента) реагента Лавессона и 100 мл толуола добавляли к 18,03 г (57,9 ммоль, 1,0 эквивалент) 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 8 ч. Реакционную смесь охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Концентрированный остаток очищали хроматографией на силикагеле, получая 4,90 г 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-3-метилпиридин-2-тиона в виде бурого порошка.
(4) Получение гидрохлорида 2-[[4-[[2-[2-(2,2,2-трифторэтокси)этокси]этил]тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
4,08 г (12,5 моль, 1,0 эквивалент) 4-[2-(2-(2,2,2-трифторэтокси)этокси)этил]тио-3-метилпиридин-2-тиона и 2,20 г (13,2 ммоль, 1,1 эквивалента) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 570 мг водного 30% раствора гидроксида натрия и 50 мл этанола и смесь перемешивали при 50°C в течение 3 ч. Затем реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 100 мл воды добавляли к раствору, который затем экстрагировали 300 мл дихлорметана. Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольного раствора хлористоводородной кислоты, получая 4,63 г бесцветных кристаллов целевого гидрохлорида 2-[[4-[[2-[2-(2,2,2-трифторэтокси)этокси]этил]тио]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола.
Общий выход был 11,0% и чистота, измеренная путем HPLC, была 99,1%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
2,22 (3H, c), 3,26 (2H, т, J=6,2 Гц), 3,50-3,60 (2H, м), 3,66 (2H, т, J=6,3 Гц), 3,69-3,71 (2H, м), 4,06 (2H, кв, J=9,3 Гц), 4,94 (2H, c), 7,14 (1H, д, J=5,1 Гц), 7,50-7,55 (2H, м), 7,74-7,80 (2H, м), 8,14 (1H, д, J=5,2 Гц), 15,2 (1H, ушир.c)
MC m/z: 457 (М+: свободное тело)
Пример 9
Получение гидрохлорида 2-[[4-[2-[2-(2,2,2-трифторэтокси)этокси]этокси]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
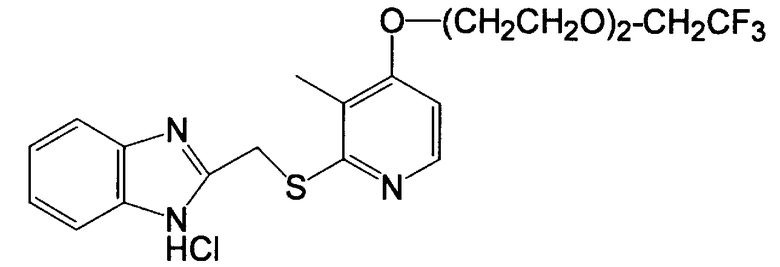
(1) Получение 4-[2-(2-(2,2,2-трифторэтокси)этокси)этокси]-3-метилпиридин-1-оксида
17,01 г (118,5 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида и 9,77 г (244 ммоль, 2,1 эквивалента) гидроксида натрия добавляли к 44,58 г (237 ммоль, 2,0 эквивалента) 2-[2-(2,2,2-трифторэтокси)этокси]этанола и 80 мл толуола и смесь кипятили с обратным холодильником с перемешиванием в течение 6 ч и затем реакцию прекращали. Потом реакционный раствор охлаждали, 140 мл воды и 11,84 г концентрированной хлористоводородной кислоты добавляли к реакционному раствору, который затем экстрагировали 280 мл этилацетата. Органическую фазу сушили ангидридом сульфата магния и затем сухую органическую фазу сушили при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая таким образом 48,90 г 4-[2-(2-(2,2,2-трифторэтокси)этокси)этокси]-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-[2-(2-(2,2,2-трифторэтокси)этокси)этокси]-2-кето-3-метилпиридина
324 г (3,2 моль, 19 эквивалентов) ангидрида уксусной кислоты добавляли к 48,69 г (164,9 ммоль, 1,0 эквивалент) 4-[2-(2-(2,2,2-трифторэтокси)этокси)этокси]-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 100°C в течение 11 ч. 300 мл этилацетата и 40 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, и полученную смесь кипятили с обратным холодильником с перемешиванием в течение 30 мин. Затем реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 8,70 г 4-[2-(2-(2,2,2-трифторэтокси)этокси)этокси]-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[2-(2-(2,2,2-трифторэтокси)этокси)этокси]тио-3-метилпиридин-2-тиона
6,25 г (16,8 ммоль, 1,2 эквивалента) реагента Лавессона и 50 мл толуола добавляли к 8,60 г (29,1 ммоль, 1,0 эквивалент) 4-[2-(2-(2,2,2-трифторэтокси)этокси)этокси]-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 11 ч. Потом реакционную смесь охлаждали до 30°C, 6,15 г (15,2 ммоль, 1,0 эквивалент) реагента Лавессона добавляли к реакционному раствору, который дополнительно подвергали реакции при нагревании/кипячении с обратным холодильником в течение 11 ч. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 1,40 г 4-[2-(2-(2,2,2-трифторэтокси)этокси)этокси]тио-3-метилпиридин-2-тиона в виде порошка.
(4) Получение гидрохлорида 2-[[4-[2-[2-(2,2,2-трифторэтокси)этокси]этокси]-3-метил-2-пиридил-тио]метил]-1H-бензимидазола
4,08 г (12,5 моль, 1,0 эквивалент) 4-[2-[2-(2,2,2-трифторэтокси)этокси]этокси]-3-метилпиридин-2-тиона и 2,20 г (13,2 ммоль, 1,1 эквивалента) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 570 мг водного 30% раствора гидроксида натрия и 50 мл этанола и смесь перемешивали при 50°C в течение 3 ч. Затем реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 100 мл воды добавляли к раствору, который затем экстрагировали 300 мл дихлорметана. Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольного раствора хлористоводородной кислоты, получая 4,63 г бесцветных кристаллов целевого гидрохлорида 2-[[4-[2-[2-(2,2,2-трифторэтокси)этокси]этокси]-3-метил-2-пиридил-тио]метил]-1H-бензимидазола.
Общий выход был 3,0% и чистота, измеренная путем HPLC, была 99,1%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
2,12 (3H, c), 3,62-3,66 (2H, м) 3,70-3,74 (2H, м), 3,76-3,80 (2H, м), 4,07 (2H, кв, J=9,3 Гц), 4,19-4,23 (2H, м), 4,94 (2H, c), 6,92 (1H, д, J=5,9 Гц), 7,50-7,56 (2H, м), 7,74-7,79 (2H, м), 8,19 (1H, д, J=5,9 Гц), 15,2 (1H, ушир.с)
MC m/z: 457 (М+: свободное тело)
Пример 10
Получение гидрохлорида 2-[[(4-пентилокси-3-метил-2-пиридил)тио]метил]-1H-бензимидазола

(1) Получение 4-пентилокси-3-метилпиридин-1-оксида
12,04 г (83,9 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида и 6,79 г (170 ммоль, 2,0 эквивалента) гидроксида натрия добавляли к 14,76 г (167 ммоль, 2,0 эквивалента) 1-пентанола и 58 мл толуола и смесь кипятили с обратным холодильником с перемешиванием в течение 5 ч и затем реакцию прекращали. Потом реакционный раствор охлаждали, 97 мл воды и 7,24 г концентрированной хлористоводородной кислоты добавляли к реакционному раствору, который затем экстрагировали 98 мл этилацетата. Органическую фазу сушили ангидридом сульфата магния и затем сухую органическую фазу сушили при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая таким образом 17,56 г 4-пентилокси-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-пентилокси-2-кето-3-метилпиридина
248 г (2,43 моль, 27 эквивалентов) ангидрида уксусной кислоты добавляли к 17,56 г (89,9 ммоль, 1,0 эквивалент) 4-пентилокси-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 105°C-110°С в течение 9 ч. 230 мл этилацетата и 34,5 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, и полученную смесь кипятили с обратным холодильником с перемешиванием в течение 8 ч. Затем реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 8,70 г 4-пентилокси-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-пентилокси-3-метилпиридин-2-тиона
6,18 г (15,3 ммоль, 1,0 эквивалент) реагента Лавессона и 52 мл толуола добавляли к 5,8 г (29,7 ммоль, 1,0 эквивалент) 4-пентилокси-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 11 ч и 30 мин. Потом реакционную смесь охлаждали до 30°C, 6,18 г (15,3 ммоль, 1,0 эквивалент) реагента Лавессона добавляли к реакционному раствору, который дополнительно подвергали реакции при нагревании/кипячении с обратным холодильником в течение 16 ч и 30 мин. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 0,338 г 4-пентилокси-3-метилпиридин-2-тиона в виде порошка.
(4) Получение гидрохлорида 2-[[(4-пентилокси-3-метил-2-пиридил)тио]метил]-1H-бензимидазола
338 мг (1,6 ммоль, 1,0 эквивалент) 4-пентилокси-3-метилпиридин-2-тиона и 362 мг (2,2 ммоль, 1,1 эквивалента) 2- (хлорметил)бензимидазола добавляли в смешанный раствор 0,290 мл водного 30% раствора гидроксида натрия и 12 мл этанола и смесь перемешивали при 50°С в течение 4 ч и 30 мин. 19 мл воды добавляли к реакционному раствору, который затем экстрагировали 84 мл дихлорметана и затем 20 мл дихлорметана. Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольного раствора хлористоводородной кислоты, получая 134,8 мг бесцветных кристаллов целевого гидрохлорида 2-[[(4-пентилокси-3-метил-2-пиридил)тио]метил]-1H-бензимидазола.
Общий выход был 0,64% и чистота, измеренная путем HPLC, была 97,4%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
0,89 (3H, т, J=7,6 Гц), 1,29-1,42 (4H, м), 1,73 (2H, м J=6,8 Гц), 2,11 (3H, c), 4,06 (2H, т, J=6,4 Гц), 4,93 (2H, c), 6,89 (1H, д, J=6,0 Гц), 7,50-7,55 (2H, м), 7,74-7,79 (2H, м), 8,17 (1H, д, J=5,6 Гц)
MC m/z: 341 (М+: свободное тело)
Пример 11
Получение 2-[[4-(3-этокси-1-пропокси)-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
(1) Получение 4-[(3-этокси-1-пропил)окси]-3-метилпиридин-1-оксида
3,52 г (24,5 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида и 2,0 г (50 ммоль, 2,0 эквивалента) гидроксида натрия добавляли к 4,80 г (46,1 ммоль, 1,9 эквивалента) 3-этокси-1-пропанола и 17 мл толуола и смесь кипятили с обратным холодильником с перемешиванием в течение 4 ч и затем реакцию прекращали. Потом реакционный раствор охлаждали, 30 мл воды и 1,7 г концентрированной хлористоводородной кислоты добавляли к реакционному раствору, который затем экстрагировали дважды 50 мл этилацетата и дважды 30 мл дихлорметана. Органическую фазу сушили ангидридом сульфата магния и затем сухую органическую фазу сушили при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая таким образом 5,6 г 4-[(3-этокси-1-пропил)окси]-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-[(3-этокси-1-пропил)окси]-2-кето-3-метилпиридина
64,9 г (636 ммоль, 24 эквивалента) ангидрида уксусной кислоты добавляли к 5,5 г (26,0 ммоль, 1,0 эквивалент) 4-[(3-этокси-1-пропил)окси]-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 118°C в течение 4 ч. 60 мл этилацетата и 9 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, и полученную смесь дополнительно кипятили с обратным холодильником с перемешиванием в течение 2 ч. Потом реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 1,74 г 4-[(3-этокси-1-пропил)окси]-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[(3-этокси-1-пропил)окси]-3-метилпиридин-2-тиона
1,60 г (4,0 ммоль, 1,0 эквивалент) реагента Лавессона и 13 мл толуола добавляли к 1,67 г (7,9 ммоль, 1,0 эквивалент) 4-[(3-этокси-1-пропил)окси]-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 14 ч и 30 мин. Затем реакционную смесь охлаждали до 30°C, 1,54 г (3,8 ммоль, 1,0 эквивалент) реагента Лавессона добавляли к реакционному раствору, который дополнительно подвергали реакции при нагревании/кипячении с обратным холодильником в течение 10 ч. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 0,44 г 4-[(3-этокси-1-пропил)окси]-3-метилпиридин-2-тиона в виде порошка.
(4) Получение 2-[[4-(3-этокси-1-пропокси)-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
420 мг (1,8 ммоль, 1,0 эквивалент) 4-[(3-этокси-1-пропил)окси]-3-метилпиридин-2-тиона и 350 мг (2,1 ммоль, 1,1 эквивалента) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 276 мг водного 30% раствора гидроксида натрия и 14 мл этанола и смесь перемешивали при 50°C в течение 3 ч. Затем реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 23 мл воды добавляли к реакционному раствору, который затем экстрагировали дихлорметаном (100 мл, 30 мл × 2) последовательно. Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле, получая таким образом 220 мг целевого 2-[[4-(3-этокси-1-пропокси)-3-метил-2-пиридил]тио]метил]-1H-бензимидазола.
Общий выход был 2,9% и чистота, измеренная путем HPLC, была 98,6%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
1,10 (3H, т, J=7,2 Гц), 1,93-1,99 (2H, м), 2,07 (3H, c), 3,42 (2H, кв, J=7,2 Гц), 3,51 (2H, т, J=6,4 Гц), 4,20 (2H, т, J=6,0 Гц), 4,66 (2H, c), 6,88 (1H, д, J=6,0 Гц), 7,12-7,15 (2H, м), 7,42-7,54 (2H, м), 8,27 (1H, д, J=6,0 Гц)
MC m/z : 357 (М+)
Пример 12
Получение гидрохлорида 2-[[[4-[2-(2-метоксиэтокси)этокси]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
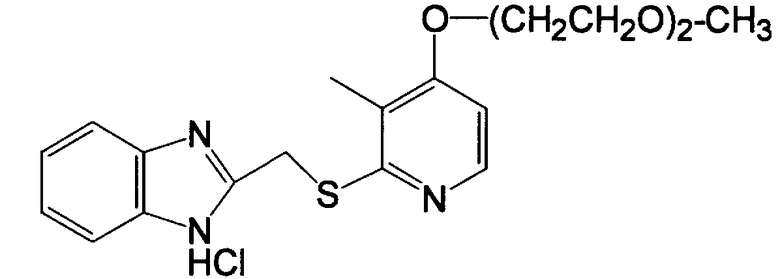
(1) Получение 4-[2-(2-метоксиэтокси)этокси]-3-метилпиридин-1-оксида
3,50 г (24,4 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида и 1,97 г (49,3 ммоль, 2,0 эквивалента) гидроксида натрия добавляли к 5,86 г (48,8 ммоль, 2,0 эквивалента) 2-(2-метоксиэтокси)этанола и 16 мл толуола и смесь кипятили с обратным холодильником с перемешиванием в течение 5 ч и затем реакцию прекращали. Потом реакционный раствор охлаждали, 28 мл воды и 0,90 г концентрированной хлористоводородной кислоты добавляли в реакционный раствор, который затем экстрагировали 31 мл этилацетата и дополнительно 60 мл (×2) дихлорметана. Органическую фазу сушили ангидридом сульфата магния и затем сухую органическую фазу сушили при 40°C или менее при пониженном давлении, получая таким образом 5,91 г 4-[2-(2-метоксиэтокси)этокси]-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-[2-(2-метоксиэтокси)этокси]-2-кето-3-метилпиридина
75,6 г (741 ммоль, 168 эквивалентов) ангидрида уксусной кислоты добавляли к 5,84 г (25,7 ммоль, 1,0 эквивалент) 4-[2-(2-метоксиэтокси)этокси]-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 100°С-113°C в течение 5 ч. 70 мл этилацетата и 15 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, и полученную смесь дополнительно кипятили с обратным холодильником с перемешиванием в течение 3 ч. Потом реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 1,35 г 4-[2-(2-метоксиэтокси)этокси]-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[2-(2-метоксиэтокси)этокси]-3-метилпиридин-2-тиона
1,30 г (3,2 ммоль, 1,1 эквивалента) реагента Лавессона и 11 мл толуола добавляли к 1,33 г (5,9 ммоль, 1,0 эквивалент) 4-[2-(2-метоксиэтокси)этокси]-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 12 ч. Затем реакционную смесь охлаждали до 30°C, 1,32 г (3,3 ммоль, 1,1 эквивалента) реагента Лавессона добавляли к реакционному раствору, который дополнительно подвергали реакции при нагревании/кипячении с обратным холодильником в течение 10 ч. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 87,1 мг 4-[2-(2-метоксиэтокси)этокси]-3-метилпиридин-2-тиона в виде порошка.
(4) Получение гидрохлорида 2-[[[4-[2-(2-метоксиэтокси)этокси]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
80,9 мг (0,33 ммоль, 1,0 эквивалент) 4-[2-(2-метоксиэтокси)этокси]-3-метилпиридин-2-тиона и 63,5 мг (0,38 ммоль, 1,2 эквивалента) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 0,051 мл водного 30% раствора гидроксида натрия и 3 мл этанола и смесь перемешивали при 50°С в течение 3 ч и 30 мин. 6 мл воды добавляли к реакционному раствору, который затем экстрагировали дихлорметаном (один раз 30 мл его и дважды 12 мл его) последовательно. Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольного раствора хлористоводородной кислоты, получая 77,5 мг бесцветных кристаллов целевого гидрохлорида 2-[[[4-[2-(2-метоксиэтокси)этокси]-3-метил-2-пиридил)тио]метил]-1H-бензимидазола.
Общий выход был 0,87% и чистота, измеренная путем HPLC, была 98,2%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
2,12 (3H, c), 3,44 (2H, т, J=4,8 Гц), 3,59 (2H, т, J=4,8 Гц), 3,75 (2H, т, J=4,4 Гц), 4,18 (2H, т, J=4,4 Гц), 4,92 (2H, c), 6,89 (1H, д, J=6,0 Гц), 7,51-7,55 (2H, м), 7,74-7,78 (2H, м), 8,15 (1H, д, J=5,6 Гц)
MC m/z: 373 (М+ свободное тело)
Пример 13
Получение 2-[[[4-(8-гидрокси-1-октилокси)-3-метил-2-пиридил]тио]метил]-1H-бензимидазола

(1) Получение 4-[(8-гидрокси-1-октил)окси]-3-метилпиридин-1-оксида
10,00 г (69,7 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида и 5,60 г (140 ммоль, 2,0 эквивалента) гидроксида натрия добавляли к 20,5 г (140 ммоль, 2,0 эквивалента) 1,8-октандиола и 46 мл толуола и смесь кипятили с обратным холодильником с перемешиванием в течение 5 ч и 30 мин и затем реакцию прекращали. Потом реакционный раствор охлаждали, 80 мл воды и 5,0 г концентрированной хлористоводородной кислоты добавляли к реакционному раствору и затем 1,8 г водного 30% раствора гидроксида натрия добавляли к реакционному раствору, который затем экстрагировали три раза 100 мл этилацетата. Органическую фазу сушили ангидридом сульфата магния и затем сухую органическую фазу сушили при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая таким образом 20,8 г 4-[(8-гидрокси-1-октил)окси]-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-[(8-ацетокси-1-октил)окси]-2-кето-3-метилпиридина
189,4 г (1,855 моль, 27 эквивалентов) ангидрида уксусной кислоты добавляли к 20,4 г (69,1 ммоль, 1,0 эквивалент) 4-[(8-гидрокси-1-октил)окси]-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 117°C в течение 7 ч. 175 мл этилацетата и 26 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, и полученную смесь дополнительно кипятили с обратным холодильником с перемешиванием в течение 5 ч. Потом реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 1,63 г 4-[(8-ацетокси-1-октил)окси]-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[(8-ацетокси-1-октил)окси]-3-метилпиридин-2-тиона
1,10 г (2,7 ммоль, 1,0 эквивалент) реагента Лавессона и 9,4 мл толуола добавляли к 1,60 г (5,4 ммоль, 1,0 эквивалент) 4-[(8-ацетокси-1-октил)окси]-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 14 ч и 30 мин. Так как реакция не была завершена, реагент Лавессона добавляли последовательно три раза в целом, отслеживая протекание реакции. При первом добавлении реагент Лавессона добавляли следующим образом: количество реагента: 1,10 г (2,7 ммоль, 1,0 эквивалент), реакция при нагревании/кипячении с обратным холодильником в течение 8 ч. При втором добавлении реагент Лавессона добавляли следующим образом: количество реагента: 0,44 г (1,1 ммоль, 0,4 эквивалент), реакция при нагревании/кипячении с обратным холодильником в течение 3 ч и 30 мин. При третьем добавлении реагент Лавессона добавляли следующим образом: количество реагента: 0,44 г (1,1 ммоль, 0,4 эквивалент), реакция при нагревании/кипячении с обратным холодильником в течение 15 ч. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 0,185 г 4-[(8-ацетокси-1-октил)окси]-3-метилпиридин-2-тиона в виде порошка.
(4) Получение 2-[[4-(8-гидрокси-1-октилокси)-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
180 мг (0,58 ммоль, 1,0 эквивалент) 4-[(8-ацетокси-1-октил)окси]-3-метилпиридин-2-тиона и 96,2 мг (0,58 ммоль, 1,0 эквивалент) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 0,051 мл водного раствора 2 моль/л гидроксида натрия и 4,4 мл этанола и смесь перемешивали при 50°C в течение 3 ч. Так как реакция не была завершена, 0,289 мл водного раствора 2 моль/л гидроксида натрия добавляли к реакционной смеси, которую дополнительно подвергали реакции при 20°C в течение 14 ч. Затем реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 7 мл воды добавляли к реакционному раствору, который затем экстрагировали дихлорметаном (30 мл, 12 мл × 2) последовательно. Потом органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанола, н-гексана и трет-бутил метилового простого эфира, получая 75,5 мг бесцветных кристаллов целевого 2-[[4-(8-гидрокси-1-октилокси)-3-метил-2-пиридил]тио]метил]-1H-бензимидазола.
Общий выход был 0,34% и чистота, измеренная путем HPLC, была 99,4%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
1,28-1,42 (10H, м), 1,69-1,76 (2H, м), 2,07 (3H, c), 3,31-3,38 (2H, м), 4,06 (2H, т, J=6,4 Гц), 4, 66 (2H, c), 6,87 (1H, д, J=6,0 Гц), 7,11-7,15 (2H, м), 7,48 (2H, ушир.с), 8,26 (1H, д, J=5,6 Гц), 12,23 (1H, ушир.с)
MC m/z: 399 (М+)
Пример 14
Получение гидрохлорида 2-[[[4-(8-метокси-1-октилокси]-3-метил-2-пиридил]тио]метил]-1H-бензимидазола

(1) Получение 4-[(8-метокси-1-октил)окси]-3-метилпиридин-1-оксида
8,6 г (59,9 ммоль, 1,0 эквивалент) 4-хлор-3-метилпиридин-1-оксида и 4,8 г (120 ммоль, 2,0 эквивалента) гидроксида натрия добавляли к 33,0 г (205,9 ммоль, 3,4 эквивалента) 8-метокси-1-октанола и 60 мл толуола и смесь кипятили с обратным холодильником с перемешиванием в течение 6 ч и затем реакцию прекращали. Потом реакционный раствор охлаждали, 100 мл воды и 7,35 г концентрированной хлористоводородной кислоты добавляли к реакционному раствору и затем добавляли 0,14 г водного 30% раствора гидроксида натрия. Реакционный раствор затем экстрагировали три раза этилацетатом (один раз 150 мл его и дважды 100 мл его). Органическую фазу сушили ангидридом сульфата магния и затем сухую органическую фазу сушили при 40°C или менее при пониженном давлении, чтобы удалить растворитель, получая таким образом 26,02 г 4-[(8-метокси-1-октил)окси]-3-метилпиридин-1-оксида в виде маслянистого вещества.
(2) Получение 4-[(8-метокси-1-октил)окси]-2-кето-3-метилпиридина
161,78 г (1,585 моль, 16 эквивалентов) ангидрида уксусной кислоты добавляли к 26,02 г (97,3 ммоль, 1,0 эквивалент) 4-[(8-метокси-1-октил)окси]-3-метилпиридин-1-оксида, чтобы осуществить реакцию при 110°С в течение 6 ч. 162 мл этилацетата и 30 мл метанола добавляли к концентрированному остатку, полученному после удаления ангидрида уксусной кислоты перегонкой, и полученную смесь дополнительно кипятили с обратным холодильником с перемешиванием в течение 3 ч. Потом реакционный раствор охлаждали, перегоняли его при 40°C или менее при пониженном давлении, чтобы удалить растворитель. Полученный концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 2,18 г 4-[(8-метокси-1-октил)окси]-2-кето-3-метилпиридина в виде маслянистого вещества.
(3) Получение 4-[(8-метокси-1-октил)окси]-3-метилпиридин-2-тиона
1,59 г (3,9 ммоль, 1,0 эквивалент) реагента Лавессона и 14 мл толуола добавляли к 2,1 г (7,9 ммоль, 1,0 эквивалент) 4-[(8-метокси-1-октил)окси]-2-кето-3-метилпиридина и смесь перемешивали при нагревании/кипячении с обратным холодильником в течение 6 ч. Так как реакция не была завершена, реагент Лавессона добавляли последовательно два раза в целом, отслеживая прохождение реакции. При первом добавлении реагент Лавессона добавляли следующим образом: количество реагента: 0,95 г (2,3 ммоль, 0,6 эквивалента), реакция при нагревании/кипячении с обратным холодильником в течение 1 ч. При втором добавлении реагент Лавессона добавляли следующим образом: количество реагента: 0,95 г (2,3 ммоль, 0,6 эквивалента), реакция при нагревании/кипячении с обратным холодильником в течение 3 ч и 30 мин. Реакционный раствор охлаждали и перегоняли при 40°C или менее при пониженном давлении, чтобы удалить растворитель, и концентрированный остаток очищали колоночной хроматографией на силикагеле, получая 0,565 г 4-[(8-метокси-1-октил)окси]-3-метилпиридин-2-тиона в виде порошка.
(4) Получение 2-[[4-(8-метокси-1-октилокси)-3-метил-2-пиридил]тио]метил]-1H-бензимидазола
565 мг (2,0 ммоль, 1,0 эквивалент) 4-[(8-метокси-1-октил)окси]-3-метилпиридин-2-тиона и 363 мг (2,2 ммоль, 1,1 эквивалента) 2-(хлорметил)бензимидазола добавляли в смешанный раствор 1,09 мл водного раствора 2 моль/л гидроксида натрия и 15,2 мл этанола и смесь перемешивали при 50°C в течение 3 ч. Потом реакционный раствор концентрировали при 40°C или менее при пониженном давлении, 24 мл воды добавляли к реакционному раствору, чтобы экстрагировать раствор 106 мл дихлорметана и затем 53 мл дихлорметана последовательно. Затем органическую фазу сушили ангидридом сульфата магния, растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле. Остаток, полученный концентрированием целевой фракции после хроматографии, подвергали кристаллизации из этанольной хлористоводородной кислоты, получая 168 мг кристаллов целевого гидрохлорида 2-[[4-(8-метокси-1-октилокси)-3-метил-2-пиридил]тио]метил]-1H-бензимидазола.
Общий выход был 0,65% и чистота, измеренная путем HPLC, была 94,7%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
1,29-1,47 (10H, м), 1,71-1,72 (2H, м), 2,11 (3H, c), 3,20 (3H, c), 4,05-4,06 (2H, м), 4,90 (2H, c), 6,85-6,86 (1H, м), 7,52-7,53 (2H, м), 7,74-7,76 (2H, м), 8,13-14 (1H, м)
MC m/z: 413 (М+: свободное тело)
Фармакологическое исследование 1
Антибактериальное исследование
В отношении соединений, полученных в примерах 1-14, было проведено исследование антибактериальной активности каждого соединения против Helicobacter pylori (H. Pylori).
Стандартный бактериальный штамм ATCC43504 использовали как Helicobacter pylori (H. pylori) для проведения исследования in vitro в колумбийской агаровой среде. Бактерию культивировали при 37°C и pH 7,0 в течение трех дней. Минимальную ингибирующую концентрацию (MIC, мкг/мл) определяли на четвертый день. Каждый образец растворяли в 1% ДМСО для применения. В качестве контрольных лекарств для антибактериального агента применяли ампициллин (AMPC), кларитромицин (CAM) и гентамицин (GEM). В качестве бактерии для сравнения использовали грамотрицательную бактерию, например E. coli (ATCC 10536 ATCC 25922), Klebsiella pneumonia (ATCC 10031), Proteus vulgaris (ATCC 13315), Pseudomonas aeruginosa (ATCC 9027) и Salmonella typhimurium (ATCC 13311), и грамположительную бактерию, например Staphylococcus aureus, MRSA (ATCC33591), Staphylococcus epidermidis (ATCC 12228), Streptococcus pneumonia (ATCC 6301), Mycobacterium ranae (ATCC 110) и Enterococcus faecalis (VRE, ATCC 51575).
Результаты исследования показаны в таблицах 2 и 3.
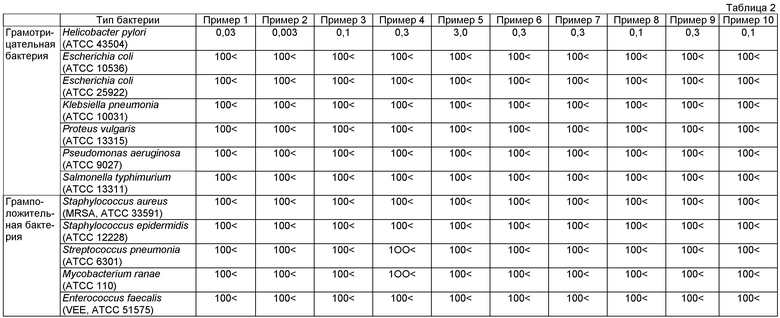
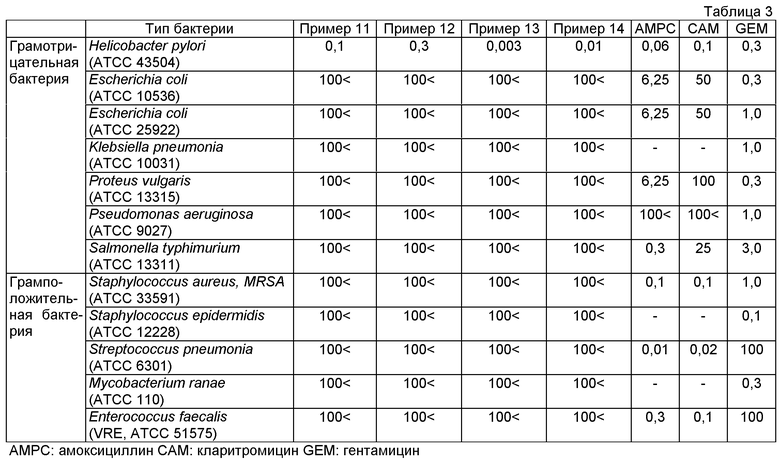
Величины в каждой таблице показывают минимальные ингибирующие концентрации (MIC, мкг/мл). "100<" означает, что никакая антибактериальная активность не проявлялась даже при дозе 100 мкг/мл.
Как видно из таблиц 2 и 3, новое тиопроизводное пиридина по данному изобретению обнаружило высокую антибактериальную способность против Helicobacter pylori (H. pylori). MIC каждого из соединений примеров 1, 2, 13 и 14 была 0,03 мкг/мл или менее, и особенно соединения примеров 2 и 13 имели удивительно высокую антибактериальную способность, и MIC каждого соединения была 0,003 мкг.
Все указанные соединения по данному изобретению не обнаруживали антибактериальной активности с MIC 100 мкг/мл или более против таких бактерий, как грамотрицательная бактерия или грамположительная бактерия, и, следовательно, установлено, что соединение по данному изобретению проявляло селективную и специфическую активность против Helicobacter pylori (H. pylori).
С другой стороны, гентамицин (GEM), используемый в качестве контрольного лекарства, имел MIC 0,3 мкг/мл против Helicobacter pylori. Однако GEM имел MIC 0,1-1,0 мкг/мл против грамотрицательной или грамположительной бактерии. Дополнительно каждый результат ампициллина (AMPC) и кларитромицина (CAM) был подобен результату GEM.
Указанные ингредиенты введены в состав в указанных соотношениях согласно обычному способу, чтобы получить таблетку с 145,2 мг/таблетка.
Указанные ингредиенты введены в состав в указанных соотношениях согласно обычному способу, чтобы получить гранулу с 1000 мг/упаковка.
ПРОМЫШЛЕННОЕ ПРИМЕНЕНИЕ
Тиопроизводное пиридина или его фармакологически приемлемая соль по данному изобретению проявляют селективную антибактериальную активность против Helicobacter pylori (H. pylori), не влияя на нормальные бактерии, и, следовательно, очень полезны в качестве антибактериального агента для лечения и профилактики различных болезней, в которых участвует Helicobacter pylori.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ БЕНЗИМИДАЗОЛА | 2006 |
|
RU2409573C2 |
| ПРЕПАРАТ ПРОТИВ Helicobacter pylori, ИНГИБИРУЮЩИЙ СЕКРЕЦИЮ ЖЕЛУДОЧНОГО СОКА | 2007 |
|
RU2451682C2 |
| ЗАМЕЩЕННЫЕ АРИЛАЛКИЛТИОАЛКИЛТИОПИРИДИНЫ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2139286C1 |
| ТИОПИРИДИНЫ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBAKTER | 1995 |
|
RU2142459C1 |
| НОВЫЙ ИНГИБИТОР КИСЛОТНОЙ СЕКРЕЦИИ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2815697C1 |
| ЗАМЕЩЕННЫЕ АРИЛТИОАЛКИЛТИОПИРИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА. | 1994 |
|
RU2154062C2 |
| ПИРИДИЛДИМЕТИЛСУЛЬФОНОВОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2404968C2 |
| ПРОИЗВОДНЫЕ ТИОФЕНА | 2003 |
|
RU2296758C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА | 1992 |
|
RU2035461C1 |
| ПИРИДИЛТИОСОЕДИНЕНИЯ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2149872C1 |
Изобретение относится к тиопроизводному пиридина, представленному общей формулой (I), или к его фармакологически приемлемой соли:

где R1 и R2, соответственно, представляют атом водорода; R3 и R4, соответственно, представляют атом водорода или С1-8алкиловую группу; Х представляет -О- или -S-; Y представляет С1-12алкиловую группу, которая может быть замещена заместителем, выбранным из группы, состоящей из гидроксильной группы, C1-8алкоксигруппы или -(R5-O)n-R6 (где R5 представляет С1-5алкиленовую группу, R6 представляет С1-8-алкиловую группу, которая может быть замещена атомом галогена, и n означает целое число от 1 до 2) и Z представляет атом водорода. Изобретение также относится к фармацевтическим композициям, обладающим антибактериальной активностью в отношении Helicobacter pyroli, на основе указанных соединений. Технический результат - получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для профилактики или лечения болезней, в которых участвует Helicobacter pylori (H.pylori), а именно гастрита, язвы желудка, язвы двенадцатиперстной кишки, MALT лимфомы желудка, гиперплазированных желудочных полипов или желудочно-кишечных злокачественных заболеваний. 3 н. и 7 з.п. ф-лы, 3 табл., 14 пр.
1. Тиопроизводное пиридина, представленное следующей общей формулой (I), или его фармакологически приемлемая соль:
где R1 и R2 соответственно представляют атом водорода; R3 и R4 соответственно представляют атом водорода или C1-8алкиловую группу; Х представляет -О- или -S-; Y представляет С1-12алкиловую группу, которая может быть замещена заместителем, выбранным из группы, состоящей из гидроксильной группы, С1-8алкоксигруппы или -(R5-O)n-R6 (где R5 представляет С1-5алкиленовую группу, R6 представляет С1-8-алкиловую группу, которая может быть замещена атомом галогена, и n означает целое число от 1 до 2 и Z представляет атом водорода.
2. Тиопроизводное пиридина или его фармакологически приемлемая соль по п.1, где Y в общей формуле (I) означает С3-10алкиловую группу, которая может быть замещена заместителем, выбранным группы, состоящей из гидроксильной группы или C1-8алкоксигруппы.
3. Тиопроизводное пиридина или его фармакологически приемлемая соль по п.1, где Y в общей формуле (I) представляет -(R5-O)n-R6 (где R5 представляет С1-5алкиленовую группу, R6 представляет C1-8алкиловую группу, которая может быть замещена атомом галогена, и n означает целое число от 1 до 2, где R5 означает этиленовую группу и R6 означает C1-8алкиловую группу, которая может быть замещенной атомом фтора.
4. Тиопроизводное пиридина или его фармакологически приемлемая соль по любому из пп.1-3, где R3 и R4 в общей формуле (I) соответственно представляют атом водорода, метиловую группу.
5. Фармацевтическая композиция, обладающая антибактериальной активностью в отношении Helicobacter pyroli, содержащая тиопроизводное пиридина или его фармакологически приемлемую соль по пп.1-3 и фармацевтически приемлемый носитель.
6. Фармацевтическая композиция, обладающая антибактериальной активностью в отношении Helicobacter pyroli, содержащая тиопроизводное пиридина или его фармакологически приемлемую соль по п.4 и фармацевтически приемлемый носитель.
7. Фармацевтическая композиция по п.5, применяемая для профилактики или лечения инфекций Helicobacter pylori (H.pylori).
8. Фармацевтическая композиция по п.6, применяемая для профилактики или лечения болезней, в которых участвует Helicobacter pylori (H.pylori).
9. Фармацевтическая композиция по п.7, где болезнями, в которых принимает участие Helicobacter pylori (H.pylori), являются гастрит, язва желудка, язва двенадцатиперстной кишки, MALT лимфома желудка, гиперплазированные желудочные полипы или желудочно-кишечные злокачественные заболевания.
10. Фармацевтическая композиция по п.8, где болезнями, в которых принимает участие Helicobacter pylori (H.pylori), являются гастрит, язва желудка, язва двенадцатиперстной кишки, MALT-лимфома желудка, гиперплазированные желудочные полипы или желудочно-кишечные злокачественные заболевания.
| WO 2008075462 A1, 26.06.2008 | |||
| RU 971000643 A, 20.02.1999 | |||
| US 5013743 A, 07.05.1991 | |||
| US 4727150 A, 23.02.1988 | |||
| US 4746667 A, 24.05.1988 | |||
| US 5045552 A, 03.09.1991. |

