Область технического применения
Данное изобретение относится к новым производным пиридина, обладающим превосходным анти-Helicobacter pylori действием, способу получения соединения и фармацевтической композиции, содержащей соединение.
Уровень техники
Гастрит, язва желудка и язва двенадцатиперстной кишки являются заболеваниями, вызванными сложной комбинацией факторов, такими как стрессы, генетическая предрасположенность и особенности образа жизни. В последние годы к Helicobacter pylori (H. pylori) как к одной из причин заболевания было привлечено всеобщее внимание. После того как в 1983 году Warren и Marshall успешно выделили и вырастили спиралевидные бактерии из образцов биопсии желудков, были проведены интенсивные исследования взаимосвязи между гастритом, язвой желудка, язвой двенадцатиперстной кишки и раком желудка и указанной бактерией. В результате зараженность H. pylori была таковой, что число положительных тестов было около 4% в здоровых желудках, тогда как число позитивных тестов было до 83% при хронических гастритах, около 69% при язве желудка, около 92% при язве двенадцатиперстной кишки и около 51% при синдроме неязвенной диспепсии (Martin J. Blaser.: Clin. Infectious Disease, 15; 386-393, 1992). Кроме того, инфекция H. pylori весьма характерна при заболеваемости раком желудка, и поэтому Международное агентство по изучению рака и Всемирная организация здравоохранения (ВОЗ) постановили в 1994 году, что H. pylori является сильным онкогенным фактором, являющимся причиной болезни.
Для лечения гастрита, язвы желудка, язвы двенадцатиперстной кишки и подобных симптоматическая терапия, в которой используются Н2-блокаторы, подавляющие секрецию кислоты желудочного сока, лекарства, подавляющие секрецию кислоты желудочного сока, такие как ингибиторы протонного насоса, и средства, защищающие слизистую, и подобные, составляют основное направление лечения. Однако известно, что, несмотря на то что эти лекарства временно излечивают патологические изменения, когда лечение приостанавливают, происходит рецидив приблизительно в 80% случаев в течение одного года (Martin J. Blaser.: Clin. Infectious Disease, 15; 386-393, 1992). С другой стороны, сообщается, что когда Helicobacter pylori (H. pylory) уничтожена, частота рецидивов в год была в пределах 10% для язвы двенадцатиперстной кишки, и также очевидно ниже был процент для язвы желудка (Graham D.Y., и др.: Ann. Intern. Med., 116; 705-708, 1992). Таким образом, стал общепринятым метод одновременного введения антибактериальных препаратов, таких как амоксицилин или кларитромицин или метронидазол с ингибиторами протонного насоса (PPI) в течение недели или больше, в больших количествах. Однако введение антибактериальных средств в больших количествах также убивает полезные бактерии в кишечнике. В результате, предполагается, что существует возможность возникновения жидкого стула, диареи и дизгевзии; побочные эффекты, такие как воспаление языка, стоматит, нарушение функции печени, печеночная дисфункция и геморрагический энтерит и образование резистентных к метицилину Staphylococcus aureus (MRSA).
При таких обстоятельствах были предприняты попытки разработать высокобезопасный медикамент, проявляющий удовлетворительное антибактериальное действие против H. Pylory при обычно используемых дозах. Например, Патентные Документы 1-4 и 9 предлагают такие лекарственные средства.
В клинической практике, для того чтобы вещество проявляло уничтожающий H. pylori эффект, который эквивалентен эффекту антибиотического вещества, необходимо, чтобы вещество показало активность, эквивалентную или лучше чем анти-H. pylori активность этих антибиотических веществ, показывающих клиническую эффективность против H. pylori. Значит желательно, чтобы активность была бы сильнее, чем активность при минимальной концентрации ингибирования (МКИ) - 0,3 мкг/мл.
Кроме того, некоторые соединения - производные эфира гуанидинметилциклогексанкарбоновой кислоты, описанные в Патентном Документе 5, проявляют анти-H. Pylory активность, как представлено при МКИ менее чем 1 мкг/мл. Несмотря на это эти соединения имеют свойство быстро разлагаться под действием расщепляющих энзимов в тонком кишечнике или в крови. Это свойство связано с соединениями, которые были разработаны, чтобы обладать селективностью по отношению к H. pylori, в соответствии с метаболитическим свойством разрушаться в тонком кишечнике или в крови, на основе идеи, что «антибиотические вещества или синтетические антибактериальные средства метаболитически распределяются при применении таким образом, чтобы эти вещества адсорбировались из тонкого кишечника через желудочно-кишечный тракт и всасывались в кровь, или выделялись вместе с испражнениями, и, таким образом, большинство бактерий, которые обитают в кишечнике, оказываются уничтоженными лекарствами, проходящими желудочно-кишечный тракт, благодаря чему разрушается бактериальный баланс кишечной флоры. Таким образом, введение в течение длительного периода времени следует избегать, как описано в Патентном Документе 6. Однако известно, что метаболитические энзимы в желудочно-кишечном тракте или крови и кишечные бактерии имеют индивидуальные вариации или флуктуации, получающиеся в результате диеты. Вследствие того маловероятно, что такие метаболитические характеристики стабилизированы и гарантированы у пациентов, имеющих различный анамнез.
Между тем, как известны производные пиридина, которые используются как противоязвенные препараты (см. Патентный документ 7), производные пиридина, проявляющие антибактериальное действие против Helicobacter pylori (см. Патентный документ 2), и производные пиридина, используемые для подавления секреции кислоты желудочного сока (см. Патентный Документ 8). Кроме того, соединения Сравнительных Примеров, а именно 2-[{4-(2-гидроксиэтокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол (Сравнительное Соединение 1) и 2-[{4-(3-гидроксипропокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол (Сравнительное соединение 2) - это соединения, описанные как используемые в качестве антиязвенных соединений в Примере 26 и Примере 24 в Патентном Документе 9. Однако не представлено описания экспериментальных данных, связанных с антиязвенной активностью, и нет описания или предположения об их активности против Helicobacter pylori.
Патентный Документ 1: JP-A № 2-209809
Патентный Документ 2: JP-A № 3-173817
Патентный Документ 3: JP-A № 3-48680
Патентный Документ 4: JP-A № 7-69888
Патентный Документ 5: WO 96/06825
Патентный Документ 6: WO 97/23207
Патентный Документ 7: JP-A № 61-50979
Патентный Документ 8: JP-A № 58-39622
Патентный Документ 9: JP-A № 5-247035
Описание изобретения
Проблемы, решаемые изобретением
При таких обстоятельствах изобретатели данного изобретения добились успеха в поиске соединения, которое обладает сильной анти-H. pylori активностью, как представлено при МКИ, меньше чем 0,3 мкг/мл, не оказывает действия на бактерии, постоянно живущие в человеческом организме, и проявляющего специфическое антибактериальное действие против H. pylori, и также открыли вещество, проявляющее эффективное действие против тех бактерий, которые резистентны к таким антибиотикам, как рокситромицин и офлоксацин, таким образом, дополняющие данное изобретение.
Таким образом, целью данного изобретения является соединение, проявляющее превосходную антибактериальную активность против H. pylory, и фармацевтическую композицию, содержащую соединение.
Средства для решения проблем
Данное изобретение предусматривает следующие изобретения от (1) до (12).
(1) Новое производное пиридина формулы (I):
(Химическая формула 1)

где R представляет гидроксиалкильную группу с нормальной или разветвленной цепью, имеющую от 5 до 10 атомов углерода, или его фармацевтически приемлемую соль.
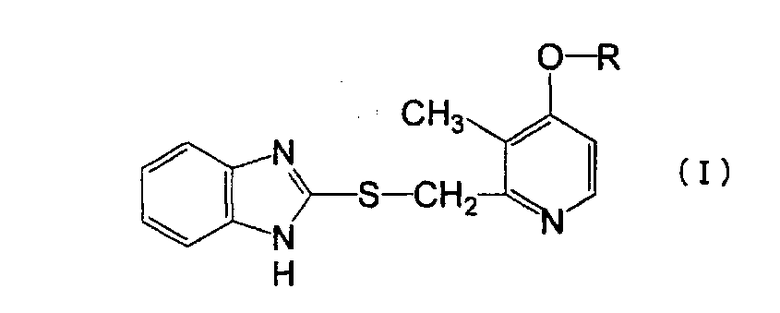
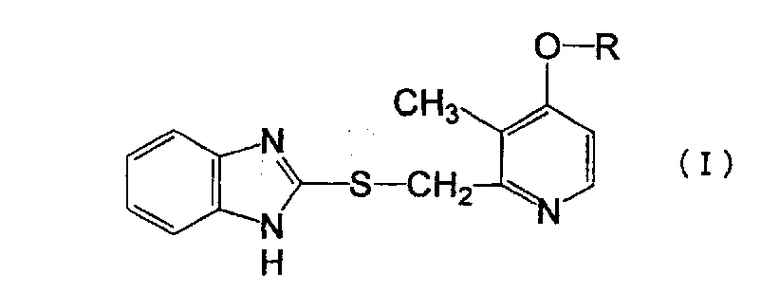
(2) Способ получения нового производного пиридина, представленного формулой (I):
(Химическая формула 4)

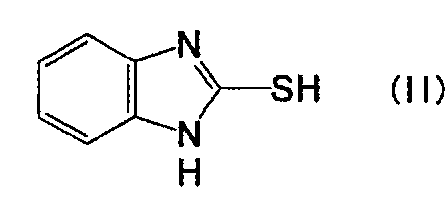
где R представляет гидроксиалкильную группу с нормальной или разветвленной цепью, имеющую от 5 до 10 атомов углерода, или его фармацевтически приемлемую соль. Способ, включающий реакцию соединения, представленного формулой (II):
(Химическая Формула 2)

с соединением, представленным формулой (III):
(Химическая Формула 3)

где R имеет то же значение, как определено выше, и Х представляет атом галогена или сульфонилоксигруппу.
(3) Фармацевтическая композиция, содержащая новое производное пиридина по пункту 1, или его фармацевтически приемлемую соль.
(4) Анти-Helicobacter pylori средство, содержащее новое производное пиридина по пункту 1, или его фармацевтически приемлемую соль.
(5) Анти-Helicobacter pylori средство по пункту 4, где Helicobacter pylori, подлежащая лечению - это бактерия, резистентная к антибиотикам на основе макролидов или новым антибиотикам на основе хинолона.
(6) Анти-Helicobacter pylori средство по пункту 4, кроме того, содержащее один или два или более декстринов.
(7) Анти-Helicobacter pylori средство по пункту 4, кроме того, содержащее одно или два или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
(8) Профилактическое или терапевтическое средство для лечения заболевания, связанного с Helicobacter pylori, средство, содержащее новое производное пиридина по пункту 1 или его фармацевтически приемлемую соль в качестве активного ингредиента.
(9) Профилактическое или терапевтическое средство по пункту 8, где заболеванием является гастрит, язва желудка, язва двенадцатиперстной кишки, неязвенный диспепсический синдром, MALT-лимформа желудка, гиперпластический полип желудка, рак желудка, рак пищеварительной системы, панкреатит или воспалительное заболевание кишечника.
(10) Профилактическое или терапевтическое средство по пункту 9, где рак желудка - это рак желудка, развившийся после эндоскопической резекции раннего рака желудка.
(11) Профилактическое или терапевтическое средство по любому из пунктов 8-10, кроме того, содержащее один или два или более декстринов.
(12) Профилактическое или терапевтическое средство по любому из пунктов 8-10, кроме того, содержащее одно или два или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
(13) Способ предупреждения или лечения заболевания у млекопитающих, включающий введение нового производного пиридина по пункту 1 или его фармацевтически приемлемой соли.
(14) Способ по пункту 13, дополнительно включающий введение одного или двух или более декстринов, или одного или двух или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
(15) Способ уничтожения или контролирования Helicobacter pylori у млекопитающих, включающий введение млекопитающему, нуждающемуся в этом, нового производного пиридина по пункту 1 или фармацевтически приемлемой соли в количестве, эффективном для уничтожения или контролирования Helicobacter pylori.
(16) Способ по пункту 15, дополнительно включающий один или два или более декстринов, или одно или два или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
(17) Способ предупреждения или лечения заболевания, связанного с Helicobacter pylori у млекопитающего, включающий введение млекопитающему, нуждающемуся в этом, нового производного пиридина по пункту 1 или его фармацевтически приемлемой соли в количестве, эффективном для предупреждения или лечения заболевания, связанного с Helicobacter pylori.
(18) Способ по пункту 17, дополнительно включающий введение одного или двух или более декстринов, или одного или двух или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
(19) Применение нового производного пиридина по пункту 1 или его фармацевтически приемлемой соли такового для изготовления лекарственного средства.
(20) Применение по пункту 19, где производное пиридина по пункту 1 или его фармацевтически приемлемая соль используется в комбинации с одним или двумя или более декстринами, или с одним или двумя или более лекарственными средствами, подавляющими секрецию кислоты желудочного сока.
(21) Применение по пунктам 19 или 20, где лекарственное средство представляет собой лекарственное средство для предупреждения или лечения заболевания, связанного с Helicobacter pylori.
(22) Применение нового производного пиридина по пункту 1 или фармацевтически приемлемой соли для изготовления анти-Helicobacter pylori средств.
(23) Применение по пункту 22, в котором новое производное пиридина по пункту 1 или его фармацевтически приемлемую соль используют в комбинации с одним или двумя или более декстринами, или с одним или двумя или более лекарственными средствами, подавляющими секрецию желудочного сока.
(24) Продукт, содержащий новое производное пиридина по пункту 1 или его фармацевически приемлемую соль и инструкцию или упаковочный контейнер, описывающие, что это соединение используют для уничтожения или контролирования Helicobacter pylori.
(25) Продукт, содержащий новое производное пиридина по пункту 1 или его фармацевически приемлемую соль и инструкцию или упаковочный контейнер, описывающие, что это соединение используют для предупреждения или лечения заболевания, связанного с Helicobacter pylori.
Эффект изобретения
Новое производное пиридина настоящего изобретения и его фармацевически приемлемая соль проявляют отличное антибактериальное действие против Helicobacter pylori (H. Pylori). Новое производное пиридина данного изобретения и его фармацевически приемлемая соль имеют большие преимущества как лекарственные средства, чем соединения, не проявляющие действия на бактерии, постоянно живущие в человеческом организме, но проявляют специфическое действие против H. Pylori и также имеют преимущество перед соединениями, проявляющими превосходную активность против H. Pylori, которые резистентны к антибиотикам на основе макролидов и новым антибиотикам на основе хинолонов. Кроме того, применение нового производного пиридина, согласно изобретению, и его фармацевически приемлемая соль позволяет уничтожить H. pylori у млекопитающего (в частности, у человека).
Данное описание изобретения включает объект изобретения, описанный в описании изобретения и/или чертежах Японской Заявки на Патент № 2006-66431, которая является основой для испрашивания приоритета по данной заявке.
Лучший вариант осуществления изобретения
R в формуле (I) или (III) представляет гидроксиалкильную группу с нормальной неразветвленной или разветвленной цепью, имеющую от 5 до 10 атомов углерода. Группа с нормальной неразветвленной или разветвленной цепью, имеющая от 5 до 10 атомов углерода, в соответствии с данным изобретением - это группа, в которой, по меньшей мере, один водород алкильной группы с нормальной или разветвленной цепью, имеющей от 5 до 10 атомов углерода, замещен на гидроксильную группу. Число гидроксильных групп в гидроксиалкильной группе не ограничивается, но предпочтительно от 1 до 3, более предпочтительно от 1 до 2 и более предпочтительно одна гидроксильная группа. Положение гидроксильной группы в гидроксиалкильной группе не ограничивается, и гидроксильная группа может располагаться в любом положении углеродной цепи. Однако предпочтительно, что одна гидроксильная группа (если есть множество гидроксильных групп, то, по крайней мере, из числа тех) располагается на атоме углерода у одного из концевых углеродов цепи. То есть, по меньшей мере, одна гидроксильная группа в гидроксиалкильной группе предпочтительно является группой, образованной путем замещения водорода в -СН3-группе у одного из концов алкильной группы с нормальной неразветвленной или разветвленной цепью, имеющей от 5 до 10 атомов углерода. Число атомов углерода в группе R может быть соответствующим любому числу в пределах значений от 5 до 10, но, в частности, предпочтительно число атомов углерода, равное 8. Число атомов углерода в группе R предпочтительно в диапазоне от 5 до 10, более предпочтительно в диапазоне от 6 до 9 и особенно предпочтительно 8 с точки зрения интенсивности анти-Helicobacter pylori активности. Группа R может быть любой - с неразветвленной цепью или разветвленной цепью, но предпочтительно, чтобы группа была с неразветвленной цепью. В частности, предпочтительные примеры группы R включают -(СН2)5ОН, -(СН2)6ОН, -(СН2)7ОН, -(СН2)8ОН, -(СН2)9ОН и -(СН2)10ОН.
В формуле (III) Х представляет атом галогена или сульфонилокси группу. Атом галогена означает фтор, хлор, бром и йод. Что касается сулфонилокси группы, могут быть использованы различные сульфонилокси группы и, как правило, могут быть использованы алкилсульфонилокси группа, которая может быть замещена, или арилсульфонилокси группа, которая может быть замещена. В частности, алкилсульфонилокси группа может быть представлена низшей алкилсульфонилокси группой, такой как метансульфонилокси группа или этансульфонилокси группа. Алкил, содержащийся в алкилсульфонилокси группе, более того может быть замещен с помощью таких заместителей как галоген. Арилсульфонилокси группа может быть представлена примером бензолсульфонилокси группы или подобными. Арил, содержащийся в арилсульфонилокси группе, более того, может быть замещен заместителем.
Производное пиридина, которое является целевым соединением данного изобретения, может быть получено при помощи исходных материалов соединений (II) и (III), способных вступать в реакцию. Предпочтительно, чтобы данная реакция проводилась в присутствии основания. Примеры основания включают гидриды щелочных металлов, такие как гидрид натрия и гидрид калия; алкоголяты, такие как трет-бутоксид калия и пропоксид натрия, этоксид натрия и метоксид натрия; карбонаты щелочных металлов, такие как карбонат калия и карбонат натрия; органические амины, такие как триэтиламин; и подобные. Как растворитель, используемый в реакции, можно отметить, например, спирты, такие как метанол и этанол, диметилсульфоксид и подобные. Количество основания, используемого в реакции, - это обычно небольшой избыток относительно одного эквивалентна, но больший избыток основания также может быть использован. То есть количество от 1 до 10 эквивалентов и предпочтительно от 1 до 4 эквивалентов. Температура реакции, как правило, от -40°С до температуры около точки кипения растворителя и предпочтительно 0-60°С. Время реакции - около 0,2-24 часа и предпочтительно 0,5-2 часа.
Целевое соединение (I), полученное в реакции, может быть выделено и очищено при помощи обычно используемых методов, таких как перекристаллизация и хроматография.
Соединение (I) настоящего изобретения может быть превращено в его фармацевтически приемлемую соль путем обычно используемых методов. Примеры таких солей включают гидрохлорид, гидробромид, гидройодид, фосфат, нитрат, сульфат, ацетат, цитрат и подобные.
Соединение, в соответствии с данным изобретением, или его соль может существовать в форме гидрата или сольвата с низшими спиртами или подобными. Объем соединений, в соответствии с данным изобретением, или его соль также включает их гидраты и сольваты.
Способ получения исходных материалов - соединения (III) - описан ниже.
Способ получения 1
(Химическая Формула 5)

Хлорпроизводное (VI) получают реакцией нитросоединения, формула (IV) с концентрированной соляной кислотой. Когда хлорпроизводное (IV) реагирует с производным спирта ROH (V) в присутствии основания, можно получить алкоксипроизводное формулы (VII). Основание включает, например, щелочные металлы, такие как литий, натрий и калий; гидриды щелочных металлов, такие как гидрид натрия и гидрид калия; алкооляты, такие как трет-бутоксид калия, пропоксид натрия, этоксид натрия и метоксид натрия; карбонаты или гидрокарбонаты щелочных металлов, такие как карбонат калия, карбонат лития, карбонат натрия, гидрокарбонат калия и гидрокарбонат натрия; щелочные металлы, такие как калий, натрий и литий; гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия, и т.п. Как растворитель, используемый в реакции в добавлении к низшим спиртам, представленным ROH, можно отметить простые эфиры, такие как тетрагидрофуран, диоксан и трет-бутилметиловый эфир; кетоны, такие как ацетон и метилэтилкетон; и ароматические углеводороды, такие как бензол, толуол, ксилол и триметилбензол. Примеры подходящих растворителей включают ацетонитрил, диметилформамид, диметилсульфоксид, N-метилпирролидон и подобные. Что касается температуры реакции, подходящая температура выбирается от -70°С до температуры около точки кипения растворителя. Время реакции около 1-48 часов.
Когда полученное таким образом соединение (VII) нагревают (около 80-120°С) в присутствии уксусного ангидрида или ацетата щелочного металла, такого как ацетат натрия или ацетат калия, или в присутствии минеральной кислоты, такой как серная кислота или перхлорная кислота, получают производное 2-ацетоксиметилпиридина, представленное формулой (VIII). Время реакции около 0,1-10 часов.
Производное 2-гидроксиметилпиридина, представленное формулой (IX), можно получать щелочным гидролизом соединения (VII). Примеры щелочей включают гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия и подобные. Как используемый растворитель можно отметить, например, метанол, этанол, воду. Время реакции приблизительно 0,1-2 часа.
Затем можно получить производное 2-галогенметилпиридина, представленное формулой (III), путем галогенирования соединения (IX) хлорующими агентами, такими как тионилхлорид, бромирующими агентами или йодирующими агентами. В качестве альтернативы, может быть получено производное 2-сульфонилметилпиридина, представленное формулой (III), путем проведения сульфонилоксилирования с различными сульфонилоксилирующими агентами, например алкилсульфонилоксилирующим агентом, таким как метансульфонилхлорид или этансулфонилхлорид, или ароматическим сульфонилоксилирующими агентами, такими как бензолсульфонилхлорид. Как используемый растворитель, можно отметить, например, хлороформ, дихлорметан, тетрахлорэтан, бензол, толуол, тетрагидрофуран, диоксан, метил-трет-бутиловый эфир, диоксан, диметилформамид, диметилсульфоксид, N-метилпирролидон и подобные. Что касается температуры реакции, подходящая температура выбирается от -70°С до температуры около точки кипения растворителя. Время реакции приблизительно 0,1-2 часа.
Соединение данного изобретения или его соль может уничтожать или контролировать Helicobacter pylori в организме млекопитающих (как правило, в его человеке). То есть соединение данного изобретения или соль - это эффективное средство противо-Helicobacter pylori.
Данное изобретение также предусматривает способ уничтожения или контролирования Helicobacter pylori у млекопитающих, способ, включающий введение эффективного количества соединения данного изобретения или его соли млекопитающим, нуждающимся в этом.
Данное изобретение также предусматривает применение соединения данного изобретения или его соли для изготовления средств против Helicobacter pylori.
Лекарственное средство, содержащее соединение данного изобретения или его соль, эффективно для предупреждения или лечения заболеваний, связанных с Helicobacter pylori. «Заболевания связаны с Helicobacter pylori», в соответствии с данным изобретением, относится к заболеванию, которое вызвано или обострено инфекцией, выживанием или распространением Helicobacter pylori в живом организме. Другими словами, «заболевание, связанное с Helicobacter pylori» - это заболевание при котором симптомы могут быть обличены при удалении Helicobacter pylori. Примеры таких заболеваний включают гастрит, язву желудка, язву двенадцатиперстной кишки, неязвенный диспепсический синдром, MALT-лимфому желудка, гиперпластический полип желудка, рак желудка (в частности, рак желудка, развившийся после эндоскопической резекции раннего рака желудка) и подобные. Другие примеры «заболевания, связанного с Helicobacter pylori» включают рак органов пищеварительной системы и панкреатиты, обусловленные Helicobacter pylori. Соединение данного изобретения или его соль способно замедлять или препятствовать развитию рака органов пищеварительной системы, обусловленному Helicobacter pylori. В качестве другого примера «заболевания, связанного с Helicobacter pylori» можно отметить воспалительную болезнь кишечника.
В рамках использования соединения данного изобретения в качестве лекарственного средства можно использовать различные лекарственные формы с целью профилактики или лечения, и примеры вводимых форм включают порошки, мелкие гранулы, гранулы, таблетки, капсулы, сухие сиропы, сиропы, инъекции и подобные. Фармацевтические композиции соединения данного изобретения могут включать фармацевтически приемлемый носитель или наполнитель или добавки.
В случае приготовления твердого лекарственного препарата для перорального приема наполнитель и, если требуется, связывающее вещество, дезинтегрант, вещества облегчающие скольжение, краситель, вкусовая или ароматизирующая добавка и т.п. могут быть добавлены к соединению данного изобретения и к таблеткам, таблеткам в оболочке, гранулам, порошкам, капсулам и т.п., и могут быть получены с помощью общепринятых методов. Что касается таких добавок, могут быть использованы общеиспользуемые в смежных областях техники. Например, как наполнитель, могут быть использованы кукурузный крахмал, лактоза, сахароза, хлорид натрия, маннит, сорбит, глюкоза, крахмал, карбонат кальция, каолин, микрокристаллическая целлюлоза, кремниевая кислота и подобные. Как связывающее вещество могут быть использованы вода, этанол, гуммиарабик, трагакант, пропанол, простой сироп, раствор глюкозы, раствор крахмала, раствор желатина, карбоксиметилцеллюлоза, гидроксипропилцеллюлоза, желатин, гидроксипропилкрахмал, метилцеллюлоза, этилцеллюлоза, шеллак, фосфат кальция, поливиниловый спирт, поливиниловый эфир, поливинилпирролидон и подобные. Как дезинтегранты могут быть использованы порошкообразный желатин, кристаллическая целлюлоза, сухой крахмал, альгинат натрия, пектин, порошкообразный агар, карбоксиметилцеллюлоза, гидрокарбонат натрия, карбонат кальция, цитрат кальция, лаурилсульфат натрия, моноглицерид стеариновой кислоты, лактоза и подобные. Как красители, одобренные для добавления, могут быть использованы диоксид титана и оксид железа. Как вкусовые или ароматизирующие добавки могут быть использованы сахароза, апельсиновая цедра, лимонная кислота, винная кислота и подобные.
В случае приготовления жидких лекарственных форм для перорального применения вкусовая добавка, буферный агент, стабилизатор, ароматизирующая добавка и подобные могут быть добавлены к соединению данного изобретения, и такая жидкая лекарственная форма для внутреннего применения - сиропы, эликсиры и т.п. можно приготовить при помощи общепринятых методов. В этом случае вкусовая и ароматизирующая добавки, отмеченные выше, могут быть использованы. Как буферные агенты можно отметить цитрат натрия и подобные. Как стабилизаторы можно отметить трагакант, гуммиарабик, желатин и подобные.
В случае приготовления лекарственных форм для инъекционного применения рН-контролирующий агент, буферный агент, изотоническое вещество, местный анестетик и т.п. можно добавить к соединению данного изобретения, и такие лекарственные формы для инъекционного применения для подкожного, внутримышечного, внутривенного введения можно приготовить при помощи общепринятых методов. Как рН-контролирующий агент и буферный агент в этом случае можно отметить цитрат натрия, ацетат натрия, фосфат натрия и подобные. Как стабилизатор могут быть отмечены пиросульфит натрия, ЭДТА, тиогликолевая кислота, тиомолочная кислота и подобные. Как местный анестетик могут быть отмечены прокаин гидрохорид, лидокаин гидрохлорид и т.п. Как изотоническое вещество можно отметить, например, хлорид натрия, глюкозу и подобные.
Фармацевтическая композиция и профилактическое или терапевтическое средство данного изобретения, описанное в формуле изобретения, может, кроме того, содержать один или два или более декстринов, в добавлении к новому производному пиридина, представленного формулой (I) или его фармацевтически приемлемой соли. Примеры декстринов, которые можно использовать в данном изобретении, включают α-декстрин, β-декстрин, γ-декстрин, α-циклодекстрин, β-циклодекстрин, γ-циклодекстрин и подобные, но не ограничиваясь этими.
Фармацевтическая композиция и профилактическое или терапевтическое средство данного изобретения, описанное в формуле изобретения, может, кроме того, содержать одно или два или более лекарственных средств, подавляющих секрецию кислоты желудочного сока, в дополнении к новому производному пиридина, представленному формулой (I) или его фармацевтически приемлемой соли. Как лекарства, подавляющие секрецию кислоты желудочного сока, можно отметить Н2-блокаторы, ингибиторы протонного насоса и подобные. Примеры Н2-блокаторов, которые можно использовать в данном изобретении, включают фамотидин, ранитидин и подобные; наряду с тем примеры ингибиторов протонного насоса, которые можно использовать в данном изобретении, включают ланзопразол, омепрозол, рабепразол и подобные, но примеры не ограничиваются этими.
При использовании комбинированных препаратов, как описано выше, ожидается расширение эффекта данного изобретения в будущем.
Количество соединения данного изобретения, включенное в каждую стандартную лекарственную форму, может изменяться в зависимости от симптомов пациента, нуждающегося в применении соединения, или от технологии приготовления лекарственного средства, но, в общем, желательно регулировать количество каждой стандартной лекарственной формы приблизительно 1 до 1200 мг для лекарственного средства для перорального применения, и приблизительно 0,1 до 500 мг для лекарственных средств для инъекционного применения. Кроме того, суточное количество лекарственного средства в виде описанных выше лекарственных форм варьируется в зависимости от симптомов, веса тела, возраста, риска и т.п. пациента, и его невозможно определить как фиксированное значение; однако количество, как правило, может быть приблизительно 0,1-5000 мг, и предпочтительно 1-1200 мг в день для взрослых, и предпочтительно, чтобы это количество назначалось один или около 2-4-разовыми порциями в день.
В последующем соединения исходных материалов, используемые в данном изобретении, сравнительные соединения и способ получения соединения данного изобретения будут описаны детально со ссылками на Примеры Синтеза и Примеры. В добавление, ВЭЖХ анализ проводили при следующих условиях:
Колонка: Interstil ODS-3 150 мм×4,6 мм внутр. диаметр.
Элюент: 0,05 М KH2PO4/ацетонитрил=50/50 (об.)
Скорость потока: 1,0 мл/мин
Температура колонки: 40°С
Количество инжектируемой пробы: 2 мкл
Длина волны детектирования: 254 нм
Пример Синтеза 1:
4-(5-гидроксипентилокси)-2,3-диметилпиридин-N-оксид
В токе азота и в бане из силиконового масла вводили 140 мл 1,5-пентандиола и при перемешивании добавляли 4,6 мг (0,2 моль, 2 экв.) металлического натрия. Затем нагревали баню из силиконового масла, и смесь реагировала при 100°С в течение 1 часа. К полученной реакционной жидкости добавляли 15,8 г (0,1 моль, 1 экв.) 4-хлор-2,3-диметилпиридин-N-оксида, и затем смесь реагировала при повышении температуры до 120°С в течение 2 часов. Реакционную жидкость охлаждали и концентрировали при уменьшенном давлении и упаривали досуха, получая 53,3 г концентрированного остатка, и сконцентрированный остаток очищали, используя колонку с силикагелем, получая 25,0 4-(5-гидроксипентилокси)-2,3-диметилпиридин-N-оксида.
Пример Синтеза 2:
4-(5-гидроксипентилокси)-2-ацетоксиметил-3-метилпиридин
К 24,5 г (0,1 моль, 1,0 экв.) 4-(5-гидроксипентилокси)-2,3-диметилпиридин-N-оксида добавляли 153,1 г (1,5 моль, 15 экв.) уксусного ангидрида, и смесь реагировала при 100°С в течение 5 часов. Уксусный ангидрит отгоняли, и затем полученный концентрированный остаток очищали, используя колонку с силикагелем, получая 12,1 г 4-(5-гидроксипентилокси)-2-ацетоксиметил-3-метилпиридина (выход 39,2%).
Пример Синтеза 3:
4-(5-гидроксипентилокси)-2-гидроксиметил-3-метилпиридин 11,8 г (0,038 моль, 1 экв.) 4-(5-гидроксипентилокси)-2-ацетоксиметил-3-метилпиридина добавляли по каплям к 30,4 г (0,152 моль, 4,0 экв.) 20% водного раствора гидроксида натрия, и смесь реагировала при комнатной температуре в течение часа. Затем реакционную смесь экстрагировали 150 мл хлороформа, сушили над сульфатом магния и затем концентрировали и упаривали досуха, получая 6,8 г 4-(5-гидроксипентилокси)-2-гидроксиметил-3-метилпиридина в виде бледно-желтых кристаллов (выход 79,1%).
Пример Синтеза 4:
4-(6-гидроксигексилокси)-2,3-диметилпиридин-N-оксид
В токе азота и на бане из силиконового масла помещали (в колбу) 47,3 г (0,4 моль, 4 экв.) 1,6-гександиола и 100 мл толуола, смесь растворялась при 55°С. Затем маленькими порциями в течение 2 часов добавляли 4,6 г (0,2 моль, 2 экв.) металлического натрия. Температура поднималась от 79°С до 91°С. Затем нагревали баню из силиконового масла, и смесь реагировала при 100°С в течение 1 часа. К полученной реакционной жидкости добавляли 15,8 г (0,1 моль, 1 экв.) 4-хлор-2,3-диметилпиридин-N-оксида, затем температуру повышали до 110°С и позволяли прореагировать смеси в течение 2 часов. Реакционную жидкость охлаждали, оставляя на ночь, и затем отделившийся толуольный слой отделяли декантацией. К остатку добавляли 100 мл метанола и смесь перемешивали. Нерастворимую часть отделяли фильтрованием и затем полученный фильтр концентрировали при уменьшенном давлении и упаривали досуха, получая 69,2 г 4-(6-гидроксикесилокси)-2,3-диметилпиридин-N-оксида.
Пример Синтеза 5:
4-(6-гидроксигексилокси)-2-ацетоксиметил-3-метилпиридин
К 68,2 г (0,1 моль, 1,0 экв.) 4-(6-гидроксигексилокси)-2,3-диметилпиридин-N-оксида добавляли 153,1 г (1,5 моль, 15 экв.) уксусного ангидрида, и смесь реагировала при 100°С в течение 4 часов. Уксусный ангидрид отгоняли, и затем 96,3 г образовавшегося концентрированного остатка очищали, используя колонку с силикагелем (хлороформ:метанол=40:1), получая 30,7 г 4-(6-гидроксигексилокси)-2-ацетоксиметил-3-метилпиридина в виде оранжевого маслянистого вещества (выход 95,0%).
Пример Синтеза 6:
4-(6-гидроксигексилокси)-2-гидроксиметил-3-метилпиридин
29,7 г (0,092 моль, 1,0 экв.) 4-(6-гидроксигексилокси)-2-ацетоксиметил-3-метилпиридина добавляли по каплям к 147 г (0,736 моль, 8,0 экв.) 20% водного раствора гидроксида натрия, и смесь реагировала при комнатной температуре в течение 1 часа. Затем реакционную смесь экстрагировали 200 мл хлороформа, сушили над сульфатом магния и затем концентрировали и упаривали досуха, получая 22,7 г оранжево-коричневого маслянистого вещества. Поскольку было обнаружено, что немного исходного вещества не подверглось гидролизу, продукт в дальнейшем реагировал в течение 3 часов при комнатной температуре в 20% водном растворе гидроксида натрия (12 экв.), и затем реакционную жидкость экстрагировали 150 мл хлороформа и концентрировали при уменьшенном давлении. Затем 19,0 г образовавшегося коричневого маслянистого вещества очищали, используют колонку с силикагелем (хлороформ), получая 7,1 г 4-(6-гидроксигексилокси)-2-гидроксиметил-3-метипиридина в виде масла (выход 58,2%).
Пример Синтеза 7:
4-(7-гидроксигептилокси)-2,3-диметилпиридин-N-оксид
В токе азота и в бане силиконового масла 24,8 г (0,188 моль, 2,2 экв.) 1,7 гептандиола и 100 мл толуола помещали (в колбу) при перемешивании, смесь растворялась при 60°С. Затем добавляли 3,1 г (0,136 моль, 1,6 экв.) металлического натрия. Далее силиконовую баню нагревали, и смесь реагировала при 100°С в течение 1 часа. К образовавшейся реакционной жидкости добавляли 13,4 г (0,085 моль, 1,0 экв.) 4-хлор-2,3-диметилпиридин-N-оксида, затем температуру повышали до 100°С, и смесь реагировала в течение 2 часов. Реакционную жидкость охлаждали, оставляя на ночь, и отделившийся толуольный слой отделяли декантацией. К образовавшемуся остатку добавляли 100 мл метанола и смесь перемешивали. Нерастворимую часть удаляли фильтрацией, и затем полученный фильтрат концентрировали при уменьшенном давлении и упаривали досуха, получая 45,5 г 4-(7-гидроксигептилокси)-2,3-диметилпиридин-N-оксида.
Пример Синтеза 8:
4-(7-гидроксигептилокси)-2-ацетоксиметил-3-метилпиридин
К 44,5 г (0,085 моль, 1,0 экв.) 4-(7-гидроксигептилокси)-2,3-диметилпиридин-N-оксида добавляли 130,2 г (1,275 моль, 15 экв.) уксусного ангидрида, и смесь реагировала при 100°С в течение 4 часов. Уксусный ангидрид отгоняли и затем 61,7 г образовавшегося концентрированного остатка очищали, используя колонку с силикагелем (хлорофрм:метанол=40:1), получая 26,3 г 4-(7-гидроксигептилокси)-2-ацетоксиметил-3-метилпиридина в виде масла (выход 91,6%).
Пример Синтеза 9:
4-(7-гидроксигептилокси)-2-гидроксиметил-3-метилпиридин
25,3 г (0,075 моль, 1,0 экв.) 4-(7-гидроксигеплтилокси)-2-ацетоксиметил-3-метилпиридина добавляли по каплям к 120 г (0,6 моль, 8,0 экв.) 20% раствора гидроксида натрия, и смесь реагировала при комнатной температуре в течение 1,5 часов. Затем реакционную жидкость экстрагировали 200 мл хлороформа, сушили сульфатом магния и затем концентрировали и упаривали досуха, получая 14,6 г 4-(7-гидроксигептилокси)-2-гидроксиметил-3-метилпиридина в виде светло-коричневого масла (выход 76,8%).
Пример Синтеза 10:
4-(8-гидроксиоктилокси)-2,3-диметилпиридин-N-оксид
В токе азота и на бане из силиконового масла 47,4 г (0,324 моль, 3,6 экв.) 1,8-октандиола и 100 мл толуола помещали (в колбу), и при перемешивании смесь растворялась при 60°С. Затем добавляли 4,1 г (0,18 моль, 2 экв.) металлического натрия. Далее баню из силиконового масла нагревали, и смесь реагировала при 100°С в течение 1 часа. К полученной реакционной жидкости добавляли 4-хлор-2,3-диметилпиридин-N-оксида и затем температуру повышали до 110°С, и смесь реагировала в течение 2 часов. Реакционную жидкость охлаждали, оставляя на ночь, и отделившийся толуольный слой отделяли декантацией. К образовавшемуся остатку добавляли 150 мл метанола, и смесь перемешивали. Нерастворимую часть отделяли фильтрованием и затем полученный фильтрат концентрировали при уменьшенном давлении и упаривали досуха, получая 68,1 г 4-(8-гидроксиоктилокси)-2,3-диметилпиридин-N-оксида.
Пример Синтеза 11:
4-(8-гидроксиоктилокси)-2-ацетоксиметил-3-метилпиридин
К 67,1 г (0,09 моль, 1,0 экв.) 4-(8-гидроксиоктилокси)-2,3-диметилпиридин-N-оксида добавляли 137,8 г (1,35 моль, 15 экв.) уксусного ангидрида, и смесь реагировала при 100°С в течение 4 часов. Уксусный ангидрид упаривали, и затем 98,5 г образовавшийся концентрированный остаток очищали, используя колонку с силикагелем (хлороформ:метанол=40:1), получая 41,6 г 4-(8-гидроксиоктилокси)-2-ацетоксиметил-3-метилпиридина в виде масла.
Пример Синтеза 12:
4-(8-гидроксиоктилокси)-2-гидроксиметил-3-метилпиридин
40,6 г (0,09 моль, 1 экв.) 4-(8-гидроксиоктилокси)-2-ацетоксиметил-3-метилпиридина добавляли по каплям к 144 г (0,72 моль, 8 экв.) 20% водного раствора гидроксида натрия, и смесь реагировала при комнатной температуре в течение 1 часа. Затем реакционную жидкость экстрагировали 200 мл хлороформа, сушили над сульфатом магния, и затем концентрировали и упаривали досуха, получая 28,0 г 4-(8-гидроксиоктилокси)-2-гидроксиметил-3-метилпиридина в виде оранжево-коричневого масла.
Пример Синтеза 13:
4-(9-гидроксинонилокси)-2,3-диметилпиридин-N-оксид
В токе азота и на бане из силиконового масла 50 г (0,312 моль, 4,0 экв.) 1,9-нонадиола и 86,7 мл толуола помещали (в колбу) и при перемешивании смесь в токе азота нагревали до 67°С. Затем добавляли 3,6 г (0,156 моль, 2 экв.) металлического натрия. Далее баню из силиконового масла нагревали, и смесь реагировала при 100°С в течение 1 часа. К полученной реакционной жидкости добавляли 4-хлор-2,3-диметилпиридин-N-оксида, и затем температуру повышали до 109°С, и смесь реагировала в течение 5 часов. Реакционную жидкость охлаждали, оставляя на ночь и отделившийся толуольный слой отделяли декантацией. К образовавшемуся остатку добавляли 130 мл метанола, и смесь перемешивали. Нерастворимую часть отделяли фильтрованием, и затем полученный фильтрат концентрировали при уменьшенном давлении и упаривали досуха, получая 56,4 г 4-(9-гидроксинонилокси)-2,3-диметилпиридин-N-оксида в виде масла.
Пример Синтеза 14:
4-(9-гидроксинонилокси)-2-ацетоксиметил-3-метилпиридин
К 56,4 г (0,078 моль, 1,0 экв.) 4-(9-гидроксинонилокси)-2,3-диметилпиридин-N-оксида добавляли 119,3 г (1,169 моль, 15 экв.) уксусного ангидрида, и смесь реагировала при 100°С в течение 5 часов. Уксусный ангидрид упаривали и затем 91,7 г образовавшегося концентрированного остатка очищали, используя колонку с силикагелем (хлороформ:метанол=40:1), получая 34,7 г 4-(9-гидроксинонилокси)-2-ацетоксиметил-3-метилпиридина в виде масла.
Пример Синтеза 15:
4-(9-гидроксинонилокси)-2-гидроксиметил3-диметилпиридин
34,7 г (0,078 моль, 1 экв.) 4-(9-гидроксинонилокси)-2-ацетоксиметил-3-метилпиридина добавляли по каплям к 125 г (0,625 моль, 8 экв.) 20% водного раствора гидроксида натрия, и смесь реагировала при комнатной температуре в течение 1 часа. Затем реакционную жидкость экстрагировали 173 мл хлороформа, сушили над сульфатом магния и затем концентрировали и упаривали досуха, получая 28,4 г 4-(9-гидроксинонилокси)-2-гидроксиметил-3-метилпиридина в виде коричневого масла.
Пример Синтеза 16:
4-(10-гидроксидецилокси)-2,3-диметилпиридин-N-оксид
В токе азота и на бане из силиконового масла 50 г (0,287 моль, 4,0 экв.) 1,10-декандиола и 79,7 мл толуола помещали (в колбу) и при перемешивании смесь в токе азота нагревали до 67°С. Затем добавляли 3,3 г (0,143 моль, 2 экв.) металлического натрия. Далее баню из силиконового масла нагревали, и смесь реагировала при 100°С в течение 1 часа. К полученной реакционной жидкости добавляли 11,3 г (0,072 моль, 1 экв.) 4-хлор-2,3-диметилпиридин-N-оксида и затем температуру повышали до 109°С, и смесь реагировала в течение 3 часов. Реакционную жидкость охлаждали, оставляя на ночь, и отделившийся толуольный слой отделяли декантацией. К образовавшемуся остатку добавляли 120 мл метанола, и смесь перемешивали. Нерастворимую часть отделяли фильтрованием, и затем полученный фильтрат концентрировали при уменьшенном давлении и упаривали досуха, получая 72,9 г 4-(10-гидроксидецилокси)-2,3-диметилпиридин-N-оксида в виде коричневого масла.
Пример Синтеза 17:
4-(10-гидроксидецилокси)-2-ацетоксиметил-3-метилпиридин
К 72,9 г (0,072 моль, 1,0 экв.) 4-(10-гидроксидецилокси)-2,3-диметилпиридин-N-оксида добавляли 109,7 г (1,075 моль, 15 экв.) уксусного ангидрида, и смесь реагировала при 100°С в течение 4 часов. Уксусный ангидрид упаривали, и затем 93,5 г образовавшегося концентрированного остатка очищали, используя колонку с силикагелем (хлороформ:метанол=40:1), получая 18,5 г 4-(10-гидроксидецилокси)-2-ацетоксиметил-3-метилпиридина в виде смеси, содержащей непрореагировавшее исходное вещество в виде оранжево-коричневого масла. К 18,5 г оранжево-коричневого масла добавляли 100 г (1,021 моль, 15 экв.) уксусного ангидрида, и смесь реагировала при 100°С в течение 3 часов. Реакционную жидкость концентрировали при уменьшенном давлении и упаривали досуха, получая 20,4 г 4-(10-гидроксидецилокси)-2-ацетоксиметил-3-метилпиридина.
Пример Синтеза 18:
4-(10-гидроксидецилокси)-2-гидроксиметил-3-диметилпиридин
14,0 г (0,078 моль, 1 экв.) 4-(10-гидроксидецилокси)-2-ацетоксиметил-3-метилпиридина добавляли по каплям к 59 г (0,296 моль, 8 экв.) 20% водного раствора гидроксида натрия, и смесь реагировала при комнатной температуре в течение ночи. Затем реакционную жидкость экстрагировали 100 мл хлороформа, сушили над сульфатом магния и затем концентрировали и упаривали досуха, получая 12,0 г 4-(10-гидроксидецилокси)-2-гидроксиметил-3-метилпиридина в виде оранжево-коричневого масла.
Пример 1:
2-[{4-(5-гидроксипентилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол
В атмосфере азота 6,5 г (0,029 моль, 1 экв.) 4-(5-гидроксипентилокси)-2-гидроксиметил-3-метилпиридина растворяли в 100 мл хлороформа при охлаждении льдом и солью, раствор добавляли к раствору, приготовленному при растворении 10,4 г (0,087 моль, 3 экв.) тионилхлорида в 90 мл хлороформа. Смесь реагировала при -10°С в течение 3 часов. Реакционную жидкость подщелачивали до рН 9, используя насыщенный водный раствор карбоната натрия, и затем образовавшуюся смесь экстрагировали 90 мл хлороформа, сушили сульфатом магния, концентрировали и упаривали досуха, получая 7,8 г 4-(5-гидроксипентилокси)-2-хлорметил-3-метилпиридина. К раствору, приготовленному при добавлении 7,5 г 4-(5-гидроксипентилокси)-2-хлорметил-3-метилпиридина к 2-меркаптобензимидазолу при охлаждении ледяной воды и перемешивании, добавляли NaOH в 60 мл этанола в течение 1,5 часов. Далее температуру поднимали до 50°С, и смесь реагировала в течение 15 минут, и затем концентрировали при уменьшенном давлении, получая 12,8 г оранжеватого масла. Концентрированный остаток очищали, используя колонку с силикагелем (хлороформ:метанол=40:1), получая 4,8 г оранжеватого масла. Масло перекристаллизовывали из смеси этилацетат:метанол=20:1 (21 об.), получая 2,8 г окрашенных кристаллов 2-[{4-(5-гидроксипентилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазола (ВЭЖХ: 99,4% площади, выход 26,9%)
1Н-ЯМР (400 МГц, CDCl3) δ: 1,47-1,73 (7Н, м), 2,25 (3Н, с), 3,70 (2Н, т J=6 Гц), 4,03 (2Н, т J=6 Гц), 4,37 (2Н, с), 6,72 (1Н, д J=6 Гц), 7,08-7,24 (2Н, м), 7,33-7,85 (2Н, м), 8,33 (1Н, д J=6 Гц), 12,52-13,43 (1Н, у.с.)
MS m/z: 357 (М+)
Пример 2:
2-[{4-(6-гидроксигексилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол
В атмосфере азота 6,9 г (0,029 моль, 1 экв.) 4-(6-гидроксигексилокси)-2-гидроксиметил-3-метилпиридина растворяли в 120 мл дихлорметана и к этому раствору добавляли раствор, приготовленный при растворении 10,4 г (0,087 моль, 3 экв.) тионилхлорида в 60 мл дихлорметана. Смесь реагировала при -10°С в течение 1,5 часов. Реакционную жидкость подщелачивали до рН 8, используя насыщенный водный раствор карбоната натрия, и затем дихлорметановый слой сушили сульфатом магния, концентрировали и упаривали досуха, получая 6,7 г 4-(6-гидроксигексилокси)-2-хлорметил-3-метилпиридина. К раствору, приготовленному при добавлении 6,5 г (0,025 моль, 1,0 экв.) 4-(6-гидроксигексилокси)-2-хлорметил-3-метилпиридина и 3,5 г (0,023 моль, 0,8 экв.) 2-меркаптобензимидазола при охлаждении ледяной водой и перемешивании, добавляли NaOH в 60 мл этанола в течение 1,5 часов. Далее температуру поднимали до 50°С, и смесь реагировала в течение 15 минут и затем концентрировали при уменьшенном давлении, получая 12,8 г оранжеватого масла. Концентрированный остаток очищали, используя колонку с силикагелем (хлороформ:метанол=40:1), получая 4,8 г оранжеватого масла. Масло перекристаллизовывали из смеси этилацетат:метанол=20:1 (21 об.), получая 2,8 г окрашенных кристаллов 2-[{4-(6-гидроксигексилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазола (ВЭЖХ: 99,4% площади, выход 26,9%)
1Н-ЯМР (400 МГц, CDCl3) δ: 1,34-1,99 (9Н, м), 2,26 (3Н, с), 3,67 (2Н, т J=6 Гц), 4,03 (2Н, т J=6 Гц), 4,37 (2Н, с), 6,73 (1Н, д J=6 Гц), 7,11-7,24 (2Н, м), 7,33-7,77 (2Н, м), 8,34 (1Н, д J=6 Гц), 12,69-13,34 (1Н, у.с.)
MS m/z: 371 (М+)
Пример 3:
2-[{4-(7-гидроксигептилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол
8,1 г (0,032 моль, 1 экв.) 4-(7-гидроксигептилокси)-2-гидроксиметил-3-метилпиридина растворяли в 120 мл дихлорметана и добавляли 11,4 г (0,096 моль, 3 экв.) тионилхлорида. Смесь реагировала при -10°С в течение 2 часов. Реакционную жидкость подщелачивали до рН 8, используя насыщенный водный раствор карбоната натрия и дихлорметановый слой, сушили над сульфатом магния, затем концентрировали и упаривали досуха, получая 8,7 г 4-(7-гидроксигептилокси)-2-хлорметил-3-метилпиридин в виде оранжево-коричневого масла.
К раствору, приготовленному при растворении и перемешивании 3,6 г (0,024 моль, 0,8 экв.) 2-меркаптоимидазола и 6,9 г (0,036 моль, 1,2 экв.) 28% метоксида натрия в 100 мл метанола, добавляли раствор 8,2 г 4-(7-гидроксигептилокси)-2-хлорметил-3-метилпиридина в 100 мл метанола при 27°С. Далее смесь нагревали до кипения в течение 30 минут, охлаждали и концентрировали при уменьшенном давлении, получая 9,0 г оранжеватого масла. 200 мл этилацетата и 12 г метанола добавляли, чтобы растворить концентрированный остаток, затем добавляли 150 мл воды и органический слой промывали водой. Органический слой экстрагировали 50 мл этилацетата и затем объединяли органические слои. К объединенному органическому слою добавляли 27 г силикагеля, смесь перемешивали 30 минут и затем отделяли силикагели фильтрацией. Фильтрат концентрировали при уменьшенном давлении, получая 8,4 г оранжево-белых кристаллов. Кристаллы суспендировали в 126 г (15 об.) этилацетата и суспензию нагревали до 57°С. Затем добавляли 2,0 г метанола до завершения растворения кристаллов. Кристаллы отделяли после того, как раствор охладится. После выдержки при 20°С в течение 30 минут кристаллы отделяли фильтрованием и затем кристаллы сушили при уменьшенном давлении, получая 4,0 г окрашенных кристаллов 2-[{4-(7-гидроксигептилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазола. (чистота по ВЭЖХ: 98,2% площади, выход 34,5%).
ЯМР-1Н (400 МГц, CDCl3) δ: 1,30-1,99 (11Н, м), 2,25 (3Н, с), 3,66 (2Н, т J=6 Гц), 4,02 (2Н, т J=6 Гц), 4,37 (2Н, с), 6,72 (1Н, д J=6 Гц), 7,10-7,25 (2Н, м), 7,31-7,81 (2Н, м), 8,33 (1Н, д J=6 Гц), 12,61-13,45 (1Н, у.с.)
MS m/z: 385 (М+)
Пример 4:
2-[{4-(8-гидроксиоктилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол
27,0 г (0,09 моль, 1,0 экв.) 4-(8-гидроксиоктилокси)-2-гидроксиметил-3-метилпиридина растворяли в 140 мл дихлорметана и добавляли 21,4 г (0,18 моль, 2,0 экв.) тионилхлорида. Смесь реагировала при -10°С в течение 3,5 часов. Реакционную жидкость подщелачивали до рН 8, используя 300 г насыщенного водного раствора карбоната натрия, и затем дихлорметановый слой сушили над сульфатом магния, затем концентрировали и упаривали досуха, получая 25,2 г 4-(8-гидроксиоктилокси)-2-хлорметил-3-метилпиридина в виде оранжево-коричневого масла (выход 98,1%).
К раствору, приготовленному при растворении и перемешивании 4,8 г (0,032 моль, 0,8 экв.) 2-меркаптоимидазола, 28% метоксида натрия в 100 мл метанола, прибавляли раствор, приготовленный при растворении 11,4 г 4-(8-гидроксиоктилокси)-2-хлорметил-3-метилпиридина в 60 мл метанола при 27°С. Затем смесь нагревали до кипения в течение 30 мин, охлаждали и затем концентрировали при уменьшенном давлении, получая 22,8 г оранжеватого масла. Концентрированный остаток растворяли в 250 мл этилацетата, и затем органический слой промывали 200 мл воды. Органический слой оставляли стоять на ночь и затем отделяли кристаллы. Соответственно, добавляли 20 мл метанола, и смесь нагревали до 42°С до растворения. Затем добавляли 27,6 г силикагеля, смесь перемешивали в течение 30 минут и затем отделяли силикагель фильтрованием. Фильтрат концентрировали при уменьшенном давлении, получая 9,0 г оранжево-белых кристаллов. Кристаллы суспендировали в 135 г (15 об.) этилацетата, суспензию нагревали до 57°С, и затем добавляли 12,8 г метанола для завершения растворения кристаллов. Кристаллы отделяли после того, как раствор охладился. После стояния при 20°С в течение 1 часа кристаллы отделяли фильтрованием. Кристаллы сушили при уменьшенном давлении, получая 3,7 г окрашенных кристаллов 2-[{4-(8-гидроксиоктилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазола (чистота по ВЭЖХ: 98,4% площади, выход 23,1%).
ЯМР-1Н (400 МГц, CDCl3) δ: 1,10-1,95 (13Н, м), 2,26 (3Н, с), 3,65 (2Н, т J=6 Гц), 4,02 (2Н, т J=6 Гц), 4,37 (2Н, с), 6,73 (1Н, д J=6 Гц), 7,11-7,24 (2Н, м), 7,44-7,64 (2Н, м), 8,33 (1Н, д J=6 Гц), 12,26-13,84 (1Н, у.с.)
MS m/z: 399 (М+)
Пример 5:
2-[{4-(9-гидроксинонилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол
28,4 г (0,078 моль, 1,0 экв.) 4-(9-гидроксинонилокси)-2-гидроксиметил-3-метилпиридина растворяли в 121 мл дихлорметана и добавляли раствор 18,5 г (0,155 моль, 2,0 экв.) тионилхлорида, растворенного в 69 мл дихлорметана. Смесь реагировала при -17°С в течение 3 часов. Реакционную жидкость подщелачивали до рН 9, используя насыщенный водный раствор карбоната натрия, и затем экстрагировали, дихлорметановый слой сушили сульфатом магния, затем концентрировали и упаривали досуха, получая 17,2 г 4-(9-гидроксинонилокси)-2-хлорметил-3-метилпиридина (выход 74,1%).
К раствору, приготовленному при растворении и перемешивании 7,2 г (0,048 моль, 0,6 экв.) 2-меркаптоимидазола и 14,0 г (0,073 моль, 0,93 экв.) 28% метоксида натрия в 181 мл метанола, добавляли 17,2 г (0,057 моль, 1,0 экв.) 4-(9-гидроксинонилокси)-2-хлорметил-3-метилпиридина при 27°С. Затем смесь нагревали до кипения в течение 30 минут, охлаждали и затем концентрировали при уменьшенном давлении, получая оранжеватое масло. Концентрированный остаток растворяли в 377 мл этилацетата, и затем органический слой промывали 302 мл воды. После того как из органического слоя выпали в осадок кристаллы, добавляли 30 мл метанола и смесь нагревали до 35°С до растворения. Затем добавляли 41,6 г силикагеля, и смесь перемешивали 30 минут, и затем силикагель отделяли фильтрованием. Фильтрат концентрировали при уменьшенном давлении, получая 13,9 г оранжевого масла. Масло нагревали в 208,9 г (15 об.) этилацетата до растворения, и затем выпали в осадок кристаллы при охлаждении раствора. После стояния при 20°С в течение 1 часа кристаллы отделяли фильтрованием. Кристаллы сушили при уменьшенном давлении, получая 4,3 г 2-[{4-(9-гидроксинонилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазола (чистота по ВЭЖХ: 96,4% площади, выход 17,7%).
1Н-ЯМР (400 МГц, CDCl3) δ: 1,19-1,90 (15Н, м), 2,26 (3Н, с), 3,64 (2Н, т J=6 Гц), 4,02 (2Н, т J=6 Гц), 4,37 (2Н, с), 6,73 (1Н, д J=6 Гц), 7,11-7,24 (2Н, м), 7,36-7,66 (2Н, м), 8,33 (1Н, д J=6 Гц), 12,40-13,60 (1Н, у.с.)
MS m/z: 413 (М+)
Пример 6:
2-[{4-(10-гидроксидецилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол
11,5 г (0,035 моль, 1 экв.) 4-(10-гидроксидецилокси)-2-гидроксиметил-3-метилпиридина растворяли в 120 мл дихлорметана и добавляли раствор 12,5 г (0,105 моль, 3,0 экв.) тионилхлорида, растворенного в 60 мл дихлорметана. Смесь реагировала при -17°С в течение 3 часов. Реакционную жидкость подщелачивали до рН 8, используя насыщенный водный раствор карбоната натрия, и затем экстрагировали, дихлорметановый слой сушили над сульфатом магния, затем концентрировали и упаривали досуха, получая 10,6 г 4-(10-гидроксидецилокси)-2-хлорметил-3-метилпиридина (выход 96,4%).
К раствору, приготовленному при растворении 4,3 г (0,029 моль, 0,9 экв.) 2-меркаптобензимидазола и 4,3 г (0,029 моль, 0,9 экв.) 4-(10-гидроксидецилокси)-2-хлорметил-3-метилпиридина и 200 мл метанола добавляли 6,8 г (0,035 моль, 1 экв.) 28% метоксида натрия при 22°С. Далее смесь нагревали до кипения в течение 30 минут, охлаждали и затем концентрировали при уменьшенном давлении, получая 18,0 г оранжевого масла. Концентрированный остаток растворяли в жидкой смеси 250 мл этилацетата и 2 мл метанола, и затем органический слой промывали 200 мл воды. К органическому слою добавляли 18,0 г силикагеля, смесь перемешивали в течение 30 минут и затем силикагель отделяли фильтрованием. Фильтрат концентрировали при уменьшенном давлении, получая 13,4 желто-белых кристаллов. Кристаллы суспендировали в 201 г (15 об.) этилацетата, суспензию нагревали до 55°С, и затем добавляли 17,5 г метанола до завершения растворения кристаллов. Кристаллы выпадали в осадок при охлаждении раствора. После стояния при от 10 до 15°С в течение 30 минут отделяли 6,5 г кристаллов при фильтровании. К кристаллам добавляли 280 г (43-кратное количество) метанола, и смесь нагревали при перемешивании. Нерастворимые примеси отделяли фильтрованием, и затем фильтрат концентрировали при уменьшенном давлении и сушили, получая 6,7 г бледно-желтых кристаллов. Кристаллы растворяли в 134 мл (20-кратное количество) этилацетата и 6,7 г метанола при нагревании до 60°С и затем раствор охлаждали при стоянии. После стояния в течение ночи при 20°С кристаллы отделяли фильтрованием и сушили при уменьшенном давлении, получая 5,0 г окрашенных кристаллов 2-[{4-(10-гидроксидецилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазола (чистота по ВЭЖХ: 96,3% площади, выход 36,5%).
1Н-ЯМР (400 МГц, CDCl3) δ: 1,10-1,95 (17Н, м), 2,26 (3Н, с), 3,64 (2Н, т J=6 Гц), 4,02 (2Н, т J=6 Гц), 4,37 (2Н, с), 6,73 (1Н, д J=6 Гц), 7,11-7,24 (2Н, м), 7,44-7,64 (2Н, м), 8,33 (1Н, д J=6 Гц), 12,30-13,68 (1Н, у.с.)
MS m/z: 427 (М+)
Фармакологический Тест. Пример 1:
Испытание Антибактериального действия
(Метод)
In vitro тест выполняли в среде Колумбийского агара, используя стандартный штамм H. pylori, ATCC 43504. Штамм культивировали в среде Колумбийского агара при 37°С и рН 7 в течение 3 дней и на 4-й день определяли минимальную концентрацию ингибирования (MIC, мкг/мл). Каждый тестируемый образец растворяли в 1% растворе ДМСО. Кроме того, как антибиотическое контрольное лекарство использовали гентамицин из аминогликозидного ряда (контрольное лекарство 2), тетрациклин из тетрациклинового ряда (контрольное лекарство 3) и офлоксацин из ряда новых хинолонов (контрольное лекарство 4).
(Результаты)
Результаты в виде активности in vitro против Helicobacter pylori представлены в Таблице 1.
In vitro активность против Helicobacter pylori новых производных пиридина
средство 2 (Гентамицин)
(Тетрациклин)
(Офлоксацин)
В качестве результата данного теста соединения данного изобретения (Примеры 1-6) признали обладающими сильным антибактериальным эффектом против H. pylori, так же как та же самая активность различных антибактериальных средств (контрольные лекарства 1-4).
Что касается противо-Helicobacter pylori активности (MIC мкг/мл), было найдено, что соединения данного изобретения показали активность явно в 10 раз или более сильно, чем активность, присущая двум подобным соединениям (сравнительные соединения 1 и 2). Кроме того, соединении Сравнительного Примера - это вышеупомянутые 2-[{4-(2-гидроксиэтилокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол (сравнительное соединение 1) и 2-[{4-(3-гидроксипропокси)-3-метилпиридин-2-ил}метилтио]-1Н-бензимидазол (сравнительное соединение 2).
Фармакологический Тест. Пример 2
(Метод)
In vitro тест выполняли в среде Колумбийского агара, используя стандартный штамм H. pylori, NCTC 11637 и 11916, клинические штаммы РТ#1045482, PT#1045483 и PT#1045484 и резистентные к офлоксацину и рокситромицину клинические штаммы TY2,4 и 5. Каждый из тестируемых объектов был растворен в виде 1% раствора в ДМСО. Более того, как антибиотические контролирующие лекарства, использовались рокситромицин из ряда макролидов и офлоксамицин из ряда новых хинолонов. Штаммы культивировали при 37°С и рН 7,0 в течение 3 дней и на 4-й день определяли минимальную концентрацию ингибирования (MIC, мкг/мл).
(Результаты)
Результаты в виде действия (MIC, мкг/мл) на резистентные бактерии Helicobacter pylori in vitro представлены в Таблице 2.
Действие новых производных пиридина на резистентные бактерии Helicobacter pylori
LX
В таблице 2 различные численные значения представляют минимальную концентрацию ингибирования (MIC, мкг/мл) различных тестируемых объектов против штаммов бактерий, RMX представляют рокситромицин и OFLX представляют офлоксацин.
Исходя из результатов данного теста, соединения данного изобретения (Примеры 1-6) показали тождественную или более сильную антибактериальную активность по сравнению с рокситомицином или офлоксацином к стандартным штаммам и клиническим штаммам. Более того, соединения так же проявили сильную антибактериальную активность против рокситомицин- или офлоксацин-резистентных клинических штаммов. То есть соединения признали обладающими сильной активностью даже в отношении штаммов, которые резистентны к рокситромицину из ряда макролидов и офлоксацину из ряда новых хинолонов.
Следующая Таблица 3 показывает минимальную концентрацию ингибирования (MIC, мкг/мл) против стандартных штаммов NCTC11637 и NCTC11916 соединений ((а)-(f)), имеющих структуру, аналогичную структуре соединения данного изобретения. Эти данные описаны в Таблице 6 JP No. 7-69888.
Действие, которое проявляют производные пиридина на Helicobacter pylori
В таблице 3 (а) - это 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-ил)метилсульфенил-1Н-бензимидазол, (b) - это 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-ил)метилтио-1Н-бензимидазол, (с) - это 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-ил]метилсульфенил-1Н-бензимидазол, (е) - это 2-[4-(3-метоксипропокси)-3-метилпиридин-2-ил]метилсульфенил-1Н-бензимидазола натриевая соль, (f) - это 2-[4-(3-метоксипропрокси)-3-метилпиридин-2-ил]метилтио-1Н-бензимидазол, XM - это рокситромицин. Ряд численных значений представляет минимальную концентрацию ингибирования (MIC, мкг/мл) ряда тестируемых объектов против различных штаммов бактерий.
При сравнении данных Таблицы 2 и Таблицы 3 видно, что соединения данного изобретения (Примеры 1-6) обладают против стандартного штамма NCTC11637 приблизительно 2,7-1,667-кратной активностью и против штамма NCNC11916 приблизительно 5,2-1,667-кратной активностью по сравнению с активностью соединений аналогичных классов ((а)-(f)). В частности, Пример 4 показал то, что он имеет наибольшую активность среди соединений аналогичных классов и имеет активность сильнее в 26,6 раз против стандартного штамма NCTC11637 и в 55 раз против стандартного штамма NCTC11916 в сравнении с активностью соединения (е) и (f).
Фармакологический Тест. Пример 3
(Метод)
Проводили in vitro антибактериальный тест для соединений Примеров 1-6 против разных бактерий. В качестве грамотрицательных бактерий использовали Escherichia coli (ATCC 10536, ATCC 25992), Klebsiella pneumonia (ATCC 10031), Proteus vulgaris (ATCC 13315), Pseudomonas aeruginosa (ATCC 9027), Salmonella typhimurin (ATCC 13311), и в качестве грамположительных бактерий использовали Staphylococcus aureus, MRSA (ATCC 33591), Staphylococcus epidermidis (ATCC 12228), Staphylococcus pneumonia (ATCC 6301), Mycobacterium ranae (ATCC 110) и Enterococcus feacalis (VRE, ATCC 51575). Различные бактерии культивировали при 37°С в течение 20-48 часов при помощи стандартного метода и определяли минимальную концентрацию ингибирования (MIC, мкг/мл). Каждый тестируемый образец был растворен в виде 1% раствора в ДМСО. Кроме того, в качестве антибиотического контрольного лекарства использовали гентамицин (GEM) из ряда аминогликозидов.
(Результаты)
Результаты в виде эффекта (MIC, мкг/мл) соединений на различные бактерии in vitro представлены в Таблице 4.
Эффект (мкг/мл) новых производных пиридина на грамотрицательные и грамположительные бактерии
coccus epidermidis (ATCC 12228)
Как результат, ни одно из соединений не было признанным обладающим антибактериальным действием против различных грамотрицательных и грамположительных бактерий. С другой стороны, гентомицин из ряда аминогликозидов проявляет сильное антибактериальное действие против различных грамотрицательных и грамположительных бактерий. Исходя их этого, было предположено, что соединения данного изобретения не обладают воздействием на все кишечные бактерии.
Пример приготовления 1. Таблетки
Таблетки, каждая весом 145,2 мг, изготовили смешением в вышеописанных пропорциях при помощи стандартного метода
Пример приготовления 2. Гранулы
Гранулы весом 1000 мг на пачку изготовили смешением в вышеописанных пропорциях в соответствии со стандартным методом.
Пример приготовления 3. Капсулы
Капсулы, каждая весом 96,5 мг, приготовили смешением в вышеописанных пропорциях в соответствии со стандартным методом.
Пример приготовления 4. Инъекции
Препарат для инъекций изготовили смешением в вышеописанных пропорциях в соответствии со стандартным методом.
Пример приготовления 5. Сироп
Препарат в виде сиропа изготовили смешением в вышеописанных пропорциях в соответствии со стандартным методом.
Пример приготовления 6. Таблетки
Таблетки, каждая весом 110 мг, приготовили смешением в вышеуказанных пропорциях в соответствии со стандартным методом.
Применяемость в производственных условиях
Новое производное пиридина данного изобретения или его фармацевтически приемлемая соль является клинически весьма перспективным лекарственным средством, поскольку соединение не обладает действием на симбиотические бактерии человека, проявляет специфическое антибактериальное действие против H. pylori и также проявляет высокую антибактериальную активность против штаммов, которые резистентны к антибактериальным средствам.
Все публикации, патенты и заявки на патенты, цитируемые в данном описании, были полностью включены в данное описание как ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРЕПАРАТ ПРОТИВ Helicobacter pylori, ИНГИБИРУЮЩИЙ СЕКРЕЦИЮ ЖЕЛУДОЧНОГО СОКА | 2007 |
|
RU2451682C2 |
| ТИОПРОИЗВОДНОЕ ПИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, КОТОРАЯ ЕГО СОДЕРЖИТ И ИМЕЕТ СПОСОБНОСТЬ ДЕЙСТВОВАТЬ ПРОТИВ HELICOBACTER PYLORI | 2010 |
|
RU2484089C1 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОЛЕКАРСТВ ИНГИБИТОРОВ ПРОТОННОГО НАСОСА | 2003 |
|
RU2292342C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА | 1992 |
|
RU2035461C1 |
| НОВЫЙ ИНГИБИТОР КИСЛОТНОЙ СЕКРЕЦИИ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2815697C1 |
| ПИРРОЛОПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ | 2001 |
|
RU2254335C2 |
| Способ получения производных пиридина или их фармацевтически приемлемых солей | 1987 |
|
SU1780535A3 |
| ТИОПИРИДИНЫ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBAKTER | 1995 |
|
RU2142459C1 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ АЗОЛОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1994 |
|
RU2128659C1 |
Настоящее изобретение относится к новым производным пиридина, общей формулы I, где R представляет гидроксиалкильную группу с нормальной неразветвленной цепью, имеющую 5-10 атомов углерода или его фармацевтически приемлемую соль. Также изобретение относится к способу получения соединения формулы I, фармацевтической композиции на основе соединения формулы I, а также к профилактическому и терапевтическому анти-Helicobacter pylori средству на основе соединения формулы I. Технический результат: получено новое производное пиридина, обладающее анти-Helicobacter pylori активностью. 8 н. и 16 з.п. ф-лы, 4 табл.
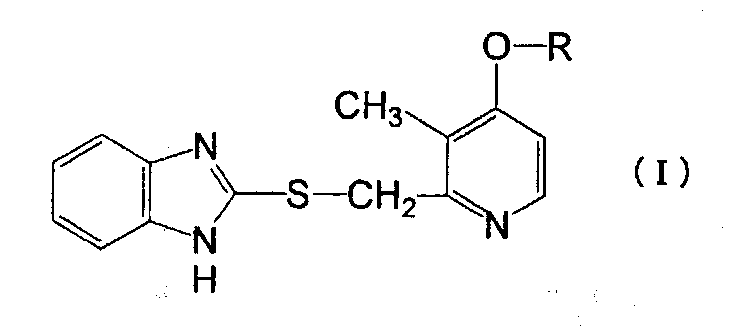
1. Производное пиридина, представленное формулой (I):
где R представляет гидроксиалкильную группу с нормальной неразветвленной цепью, имеющую 5-10 атомов углерода или его фармацевтически приемлемую соль.
2. Способ получения нового производного пиридина, представленного формулой (I)
где R представляет собой гидроксиалкильную группу с нормальной неразветвленной цепью, имеющую 5-10 атомов углерода или его фармацевтически приемлемой соли, включающий реакцию соединения, представленного формулой (II):
с соединением, представленным формулой (III):
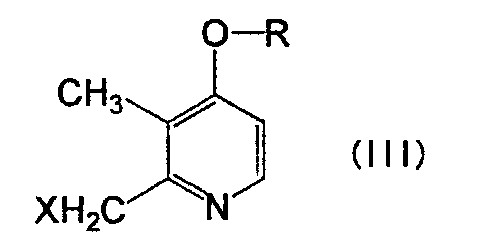
где R имеет такое же значение, как определено выше; и X представляет атом галогена или сульфонилоксигруппу.
3. Фармацевтическая композиция, обладающая анти-Helicobacter pylori активностью, содержащая производное пиридина по п.1 или его фармацевтически приемлемую соль в эффективном количестве.
4. Анти-Helicobacter pylori средство, содержащее производное пиридина по п.1 или фармацевтически приемлемую соль.
5. Анти-Helicobacter pylori средство по п.4, где Helicobacter pylori представляет собой бактерию, которая резистентна к антибиотикам на основе макролида или новым антибиотикам на основе хинолона.
6. Анти-Helicobacter pylori средство по п.4, дополнительно включающее один, или два, или более декстринов.
7. Анти-Helicobacter pylori средство по п.4, дополнительно включающее одно, или два, или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
8. Профилактическое или терапевтическое средство от заболеваний, связанных с Helicobacter pylori, включающее производное пиридина по п.1 или его фармацевтически приемлемую соль в качестве активного ингредиента.
9. Профилактическое или терапевтическое средство по п.8, где заболевание представляет собой гастрит, язву желудка, язву двенадцатиперстной кишки, неязвенный диспепсический синдром, MALT-лимфома желудка, гиперпластический полип желудка, рак желудка, рак органов пищеварения, панкреатит или воспалительная болезнь желудка.
10. Профилактическое или терапевтическое средство по п.9, где рак желудка - это рак желудка, развивающийся после эндоскопической резекции раннего рака желудка.
11. Профилактическое или терапевтическое средство по любому из пп.8-10, дополнительно содержащее один, или два, или более декстринов.
12. Профилактическое или терапевтическое средство по любому из пп.8-10, дополнительно содержащее одно, или два, или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
13. Производное пиридина или его фармацевтически приемлемая соль по п.1, где R представляет гидроксиалкильную группу с нормальной неразветвленной цепью, имеющую 5-10 атомов углерода, которая замещена гидроксильной группой у одного концевого углерода цепи.
14. Способ по п.2, где R представляет гидроксиалкильную группу с нормальной неразветвленной цепью, имеющую 5-10 атомов углерода, которая замещена гидроксильной группой у одного концевого углерода цепи.
15. Фармацевтическая композиция, обладающая анти-Helicobacter pyroli активностью, содержащая производное пиридина по п.13 или его фармацевтически приемлемую соль в эффективном количестве.
16. Анти-Helicobacter pylori средство, содержащее производное пиридина по п.13 или его фармацевтически приемлемую соль.
17. Анти-Helicobacter pylori средство по п.16, где Helicobacter pylori является бактерией, которая резистентна к антибиотикам на основе макролидов или новым антибиотикам на основе хинолона.
18. Анти-Helicobacter pylori средство по п.16, дополнительно содержащее один, или два, или более декстринов.
19. Анти-Helicobacter pylori средство по п.16, дополнительно содержащее одно, или два, или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
20. Профилактическое или терапевтическое средство от заболевания, связанного с Helicobacter pylori, содержащее производное пиридина по п.13 или его фармацевтически приемлемую соль в качестве активного ингредиента.
21. Профилактическое или терапевтическое средство по п.20, где заболеванием является гастрит, язва желудка, язва двенадцатиперстной кишки, неязвенный диспепсический синдром, MALT-лимфома желудка, гиперпластический полип желудка, рак желудка, рак органов пищеварения, панкреатит или воспалительная болезнь желудка.
22. Профилактическое или терапевтическое средство по п.21, где раком желудка является рак желудка, развивающийся после эндоскопической резекции раннего рака желудка.
23. Профилактическое или терапевтическое средство по любому из пп.20-22, дополнительно содержащее один, или два, или более декстринов.
24. Профилактическое или терапевтическое средство по любому из пп.20-22, дополнительно содержащее одно, или два, или более лекарственных средств, подавляющих секрецию кислоты желудочного сока.
| Ьм1ШМЕШ'?КНАВ| | 0 |
|
SU382489A1 |
| КОМПЛЕКТ ПРИСПОСОБЛЕНИЙ ДЛЯ СБОРКИр — п | 0 |
|
SU174726A1 |
| ПИРИДИЛТИОСОЕДИНЕНИЯ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2149872C1 |
| Способ получения производныхбЕНзиМидАзОлА или иХ СОлЕй | 1975 |
|
SU795476A3 |
| Способ получения производных имидазола или их солей с кислотами | 1986 |
|
SU1396965A3 |

