Настоящее изобретение относится к лекарственным средствам, предназначенным для лечения и/или профилактики болезней, прежде всего тромбоэмболических заболеваний, более конкретно к пролекарствам, являющимся производными 5-хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамида.
Пролекарства являются производными действующего вещества, которые перед высвобождением собственно действующего вещества претерпевают протекающую in vivo одностадийную или многостадийную биотрансформацию ферментативного и/или химического характера. Комплекс свойств того или иного действующего вещества как правило оптимизируют путем варьирования структуры остатка пролекарства, лежащего в основе этого действующего вещества [Р.Ettmayer et al., J. Med. Chem. 47, 2393 (2004)]. При этом для обеспечения оптимального лечебного эффекта структуру остатка Пролекарства и требуемый механизм высвобождения действующего вещества следует приводить в чрезвычайно точное соответствие с индивидуальным действующим веществом, его показанием, местом действия и способом применения. Множество лекарственных средств применяют в виде пролекарств, которые по сравнению с лежащим в их основе действующим веществом обладают более высокой биодоступностью, что достигается, например, путем оптимизации физико-химических характеристик, в частности, растворимости подобных пролекарств, а также их активных и пассивных абсорбционных свойств или специфического распределения в тканях. Из обширного перечня посвященной пролекарствам литературы в качестве примера следует упомянуть Н.Bundgaard (издатель), Design of Prodrugs: Bioreversible derivatives for various functional groups and chemical entities, Elsevier Science Publishers B.V., 1985.
В международной заявке WO 2005/028473 описаны используемые для повышения оральной биодоступности ацилоксиметилкарбаматные пролекарства оксазолидинонов. Из международной заявки WO 01/00622 известно об ацильных пролекарствах карбаматных ингибиторов инозин-5'-монофосфат-дегидрогеназы. Иной тип амидных пролекарств оксазолидинонов, которые высвобождают лежащее в их основе действующее вещество путем многостадийного механизма активирования, приведен в международной заявке WO 03/006440
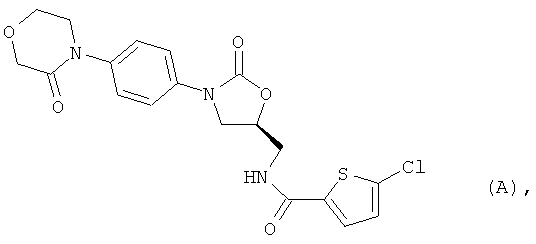
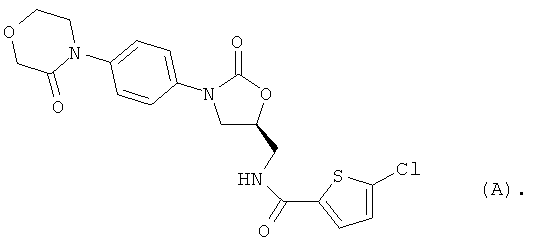
5-Хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамид [BAY 59-7939, соединение (А)] является орально действующим прямым ингибитором фактора Ха серин-протеазы, которая выполняет незаменимую функцию при регулировании свертывания крови. В настоящее время указанное соединение находится на стадии углубленного клинического испытания в качестве нового действующего вещества для потенциально возможного предупреждения и лечения тромбоэмболических заболеваний [S.Roehrig et al., J. Med. Chem. 48, 5900 (2005)].
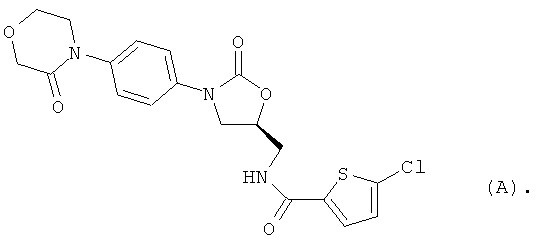
Однако соединение (А) обладает ограниченной растворимостью в воде и физиологических средах, что осложняет, например, внутривенное применение этого действующего вещества.
В связи с вышеизложенным в основу настоящего изобретения была положена задача найти производные или пролекарства соединения (А), которые обладают повышенной растворимостью в указанных средах и одновременно допускают возможность контролируемого высвобождения действующего вещества (А) в организме пациента после применения.
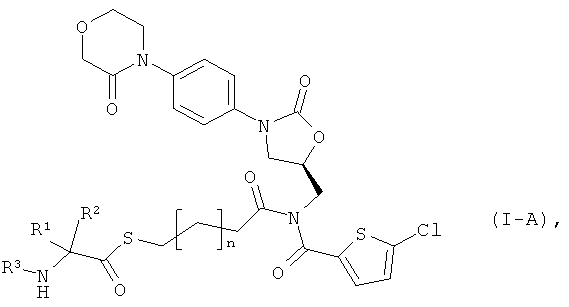
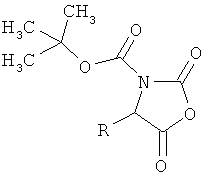
Поставленная задача решается соединениями общей формулы (I)
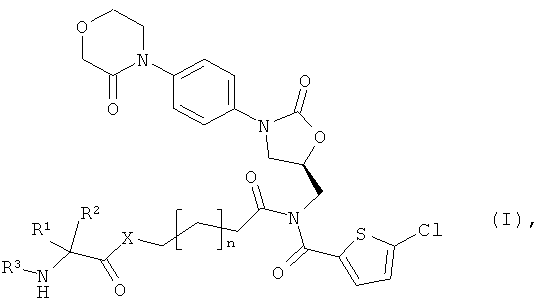
в которой
n означает 1 или 2,
Х означает атом кислорода, атом серы или NH,
R1 означает боковую группу природной α-аминокислоты или ее гомологов, или изомеров, выбранную из водорода, метила, пропан-2-ила, пропан-1-ила, 2-метил-пропан-1-ила, имидазол-4-илметила, гидроксиметила, 1-гидрокси-этила, карбоксиметила, 2-карбоксиэтила, карбамоил-метила, 2-карбамоилэтила, 4-аминобутан-1-ила, 3-аминопропан-1-ила, 3-гуанидинопропан-1-ила, бензила или 4-гидроксибензила,
R2 означает водород или метил,
R3 означает водород,
или
R1 и R3 соединены между собой посредством группы (СН2)3- или (СН2)4- и совместно с атомами азота и углерода, к которым они присоединены, образуют пятичленное или шестичленное кольцо,
а также их солями, сольватами и сольватами солей.
Предпочтительны соединения вышеприведенной формулы (I), в которой
n означает 1 или 2,
Х означает NH,
R1 означает боковую группу природной α-аминокислоты или ее гомологов или изомеров, выбранную из водорода, метила, пропан-2-ила, 2-метил-пропан-1-ила, имидазол-4-илметила, гидроксиметила, 1-гидроксиэтила, карбоксиметила, 2-карбоксиэтила, карбамоилметила, 2-карбамоилэтила, 4-аминобутан-1-ила, бензила или 4-гидроксибензила,
R2 означает водород,
R3 означает водород,
а также их соли, сольваты и сольваты солей.
Предлагаемые соединения формулы (I) в зависимости от их структуры могут находиться в форме стереоизомеров (энантиомеров, диастереомеров). В связи с этим настоящее изобретение относится также к энантиомерам или диастереомерам и их смесям. Индивидуальные стереоизомеры могут быть выделены из подобных смесей энантиомеров и/или диастереомеров известными методами.
В случае если предлагаемые соединения формулы (I) находятся в виде таутомеров, изобретение относится ко всем без исключения формам таутомеров.
В соответствии с настоящим изобретением предпочтительными солями являются физиологически приемлемые соли предлагаемых соединений формулы (I). Под подобными солями подразумевают также соли, которые не пригодны для фармацевтического применения, однако могут быть использованы, например, для выделения или очистки предлагаемых соединений формулы (I).
Физиологически приемлемыми солями предлагаемых соединений формулы (I) являются соли, образующиеся по реакции присоединения минеральных кислот, карбоновых кислот и сульфокислот, например, соли соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфокислоты, этансульфокислоты, толуолсульфокислоты, бензолсульфокислоты, нафталиндисульфокислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты и бензойной кислоты.
В соответствии с настоящим изобретением под сольватами подразумевают такие формы предлагаемых соединений формулы (I), которые являются комплексами, образуемыми этими находящимися в твердом или жидком состоянии соединениями путем координации с молекулами растворителя. Гидраты являются особой формой сольватов и образуются вследствие координации указанных соединений с молекулами воды. В соответствии с настоящим изобретением предпочтительными сольватами являются гидраты.
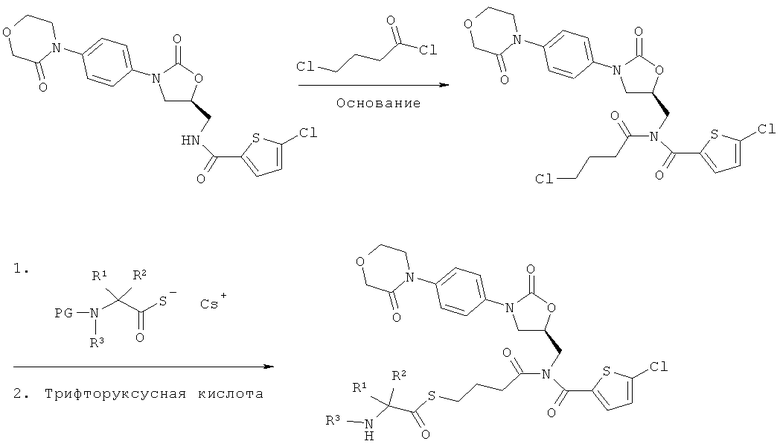
Соединения вышеприведенной формулы (I) можно получать за счет того, что
[А] сначала соединение формулы:
в инертном растворителе в присутствии основания
подвергают взаимодействию с соединением формулы:
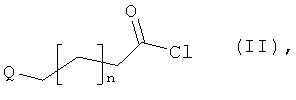
в которой
n такой, как указано выше, и
Q означает удаляемую группу, например, такую как хлор, бром или йод,
получая соединение формулы:
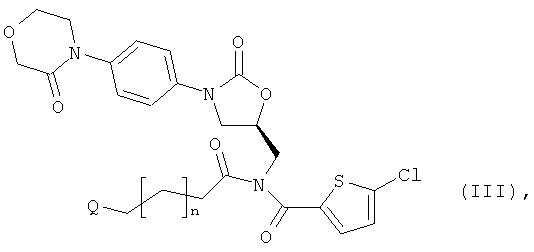
в которой n и Q такие, как указано выше,
а затем
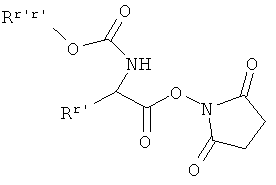
[А1] соединение формулы (III) в инертном растворителе подвергают взаимодействию с цезиевой солью α-аминокарбоновой кислоты или α-аминотиокарбоновой кислотой формулы:
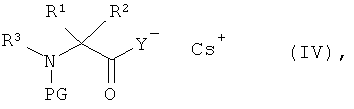
в которой
R1, R2 и R3 такие, как указано выше,
PG означает аминозащитную группу, например, такую как трет-бутоксикарбонил (Boc) или бензилоксикарбонил (Z), и
Y означает кислород или серу,
получая соединение формулы:
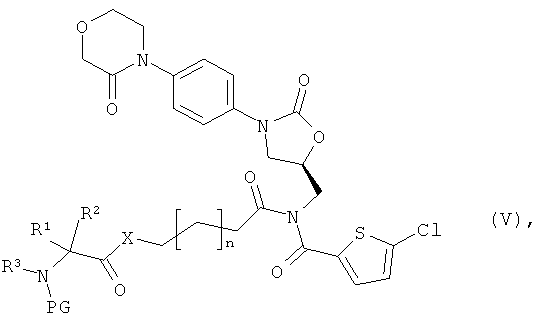
в которой
n, R1, R2, R3 и PG такие, как указано выше, и
Х означает кислород или серу,
после чего защитную группу PG удаляют обычными методами, получая соединение формулы:
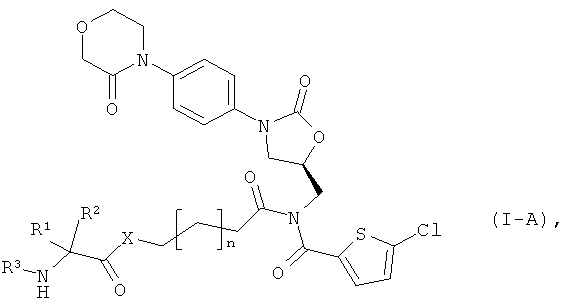
в которой
n, R1, R2 и R3 такие, как указано выше, и
Х означает кислород или серу,
или
[А2] соединение формулы (III) в инертном растворителе в присутствии основания подвергают взаимодействию с α-аминотиокарбоновой кислотой формулы:
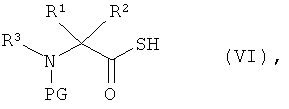
в которой
R1, R2 и R3 такие, как указано выше, и
PG означает аминозащитную группу, например, такую как трет-бутоксикарбонил (Boc) или бензилоксикарбонил (Z),
получая соединение формулы:
в которой n, R1, R2, R3 и PG такие, как указано выше,
после чего защитную группу PG удаляют обычными методами,
получая соединение формулы:
в которой n, R1, R2 и R3 такие, как указано выше,
или
[В] соединение (А) в инертном растворителе в присутствии основания подвергают взаимодействию с соединением формулы:
в которой n такой, как указано выше,
получая соединение формулы:
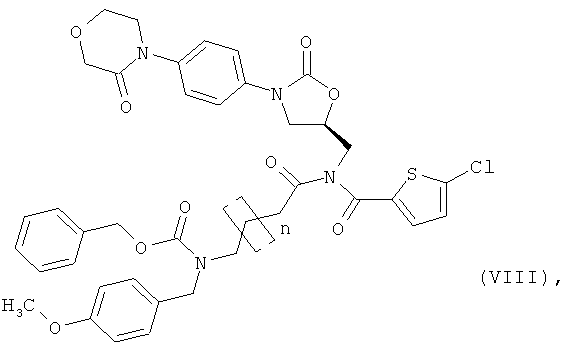
в которой n такой, как указано выше,
затем обычными методами удаляют защитные группы, получая соединение формулы:

в которой n такой, как указано выше,
и соединение формулы (IX) в присутствии основания
подвергают взаимодействию с соединением формулы:
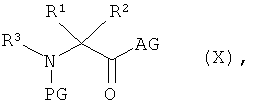
в которой
R1, R2 и R3 такие, как указано выше,
AG означает гидрокси или галоген, предпочтительно хлор или бром, или совместно с карбонильной группой образует активный сложный эфир, предпочтительно N-гидроксисукцинимидоэфир, или смешанный ангидрид, предпочтительно алкиловый эфир угольной кислоты, особенно предпочтительно этиловый эфир угольной кислоты, и
PG означает аминозащитную группу, например, такую как трет-бутоксикарбонил (Boc) или бензилоксикарбонил (Z),
получая соединение формулы:
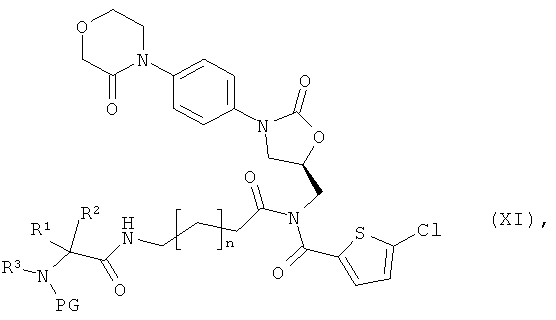
в которой n, R1, R2, R3 и PG такие, как указано выше,
после чего защитную группу PG удаляют обычными методами, получая соединение формулы:
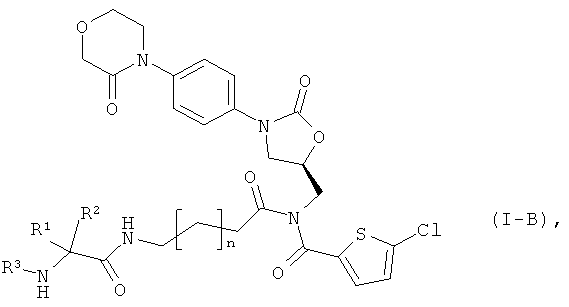
в которой n, R1, R2 и R3 такие, как указано выше,
и каждое из конечных соединений формул (I-A) или (I-B), посредством соответствующих (i) растворителей и/или (ii) кислот при необходимости переводят в их сольваты, соли и/или сольваты солей.
Соединения формул (I-А), (I-В) и (IX) могут находиться также в форме соответствующих солей. Подобные соли при необходимости можно перевести в свободные основания посредством соответствующих (i) растворителей и/или (ii) оснований.
При осуществлении приведенных выше последовательностей реакций при необходимости присутствующие в остатке R1 функциональные группы в случае целесообразности или необходимости могут находиться также во временно защищенной форме. При этом введение и удаление соответствующих защитных групп, а также аминозащитной группы PG выполняют обычными известными из химии пептидов методами [смотри, например, T.W.Greene, P.G.M.Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 1999; M.Bodanszky, A.Bodanszky, The Practice of Peptide Synthesis, Springer, Berlin, 1984].
Указанные, при необходимости присутствующие в R1 защитные группы можно удалять одновременно с отщеплением аминозащитной группы PG или на отдельной реакционной стадии, реализуемой до или после отщепления группы PG.
В соответствии с рассмотренным выше способом в качестве аминозащитной группы PG предпочтительно используют трет-бутоксикарбонил (Boc) или бензилоксикарбонил (Z). Отщепление подобных защитных групп, а также отщепление защитных групп на реакционной стадии (VIII)→(X) осуществляют обычными методами, предпочтительно путем взаимодействия с сильной кислотой, такой как хлороводородная, бромоводородная или трифторуксусная кислота, в инертном растворителе, таком как диоксан, дихлорметан или уксусная кислота.
В качестве инертного растворителя на реакционных стадиях (А)+(II)→(III) и (А)+(VII)→(VIII) предпочтительно используют тетрагидрофуран, N,N-диметилформамид или диметилсульфоксид, причем особенно предпочтительным растворителем является N,N-диметилформамид. Основанием, пригодным для выполнения указанных взаимодействий, прежде всего является гидрид натрия. Указанные взаимодействия в общем случае выполняют в температурном интервале от 0 до +40°С при нормальном давлении.
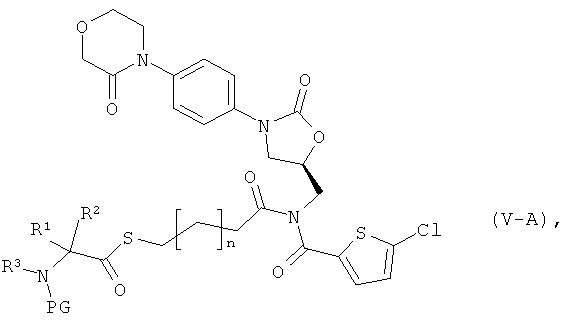
В качестве инертного растворителя на реакционных стадиях (III)+(VI)→(V-A) и (IX)+(X)→(XI) предпочтительно используют тетрагидрофуран, N,N-диметилформамид или диметилсульфоксид, причем особенно предпочтительным растворителем является N,N-диметилформамид.
Основанием, пригодным для выполнения указанных взаимодействий, прежде всего является этилдиизопропиламин. Указанные взаимодействия в общем случае выполняют в температурном интервале от 0 до +40°С при нормальном давлении.
Реакционную стадию (III)+(IV)→(V) предпочтительно выполняют в N,N-диметилформамиде в качестве растворителя. Реакцию в общем случае осуществляют при нормальном давлении в температурном интервале от 0 до +50°С, предпочтительно от +20 до +50°С. Реакцию благоприятно осуществлять также в условиях воздействия ультразвука.
Соединения формул (II), (IV), (VI), (VII) и (X) являются коммерчески доступными или известными из литературы продуктами, или могут быть получены известными из литературы методами. Получение соединения (А) описано в S.Roehrir et al., J. Med. Chem. 48, 5900 (2005).
Получение предлагаемых соединений формулы (I) можно представить в виде приведенной ниже реакционной схемы.
Реакционная схема
Предлагаемые соединения формулы (I) и их соли являются полезными пролекарствами действующего вещества - соединения (А). С одной стороны, они обладают хорошей стабильностью, например, при показателе pH 4, а, с другой стороны, эффективно превращаются в действующее вещество - соединение (A) in vivo. Кроме того, предлагаемые соединения формулы (I) обладают хорошей растворимостью в воде и других физиологически совместимых средах, в связи с чем они пригодны для лечения, реализуемого прежде всего путем внутривенного применения.
Предлагаемые соединения формулы (I) можно применять для лечения и/или профилактики заболеваний, предпочтительно тромбоэмболических заболеваний и/или тромбоэмболических осложнений.
К тромбоэмболическим заболеваниям в соответствии с настоящим изобретением прежде всего относятся такие заболевания, как инфаркт миокарда с повышением и без повышения сегмента ST, стабильная стенокардия, нестабильная стенокардия, повторные закупорки и рестенозы после вмешательств в венечные сосуды сердца, таких как ангиопластика или аортокоронарное шунтирование, периферические облитерирующие эндартерииты, эмболии легочной артерии, глубокие венозные тромбозы и тромбозы почечной вены, временные приступы ишемии, а также тромботическое и тромбоэмболическое кровоизлияние в мозг.
Таким образом, предлагаемые соединения формулы (I) пригодны также для предупреждения и лечения тромбоэмболии сердечного происхождения, например, таких как мозговые ишемии, инсульт и систематические тромбоэмболии и ишемии, возникающие у пациентов с острой, периодически возникающей или постоянной аритмией сердца, например, такой как мерцание предсердий, у подвергаемых кардиоверсии пациентов, а также у пациентов, страдающих заболеваниями сердечных клапанов или с искусственными сердечными клапанами. Кроме того, предлагаемые соединения формулы (I) пригодны для лечения пациентов с распространенным внутрисосудистым свертыванием крови.
Тромбоэмболические осложнения возникают также при микроангиопатических гемолитических анемиях, экстракорпоральных кровообращениях, таких как гемодиализ, а также при наличии у пациентов искусственных клапанов сердца.
Кроме того, предлагаемые соединения формулы (I) пригодны для профилактики и/или лечения атеросклеротических заболеваний сосудов и воспалительных заболеваний, таких как ревматические заболевания двигательного аппарата, а также для профилактики и/или лечения болезни Альцгеймера. Наряду с этим предлагаемые соединения формулы (I) можно использовать для торможения опухолевого роста и метастазирования, при микроангиопатии, старческой дегенерации макулы, диабетической ретинопатии, диабетической нефропатии и других микрососудистых заболеваниях, а также для предупреждения и лечения тромбоэмболических осложнений, например, таких как венозные тромбоэмболии, возникающих у онкологических пациентов, прежде всего подвергнутых обширному хирургическому вмешательству, а также химиотерапии или лучевой терапии.
Предлагаемые соединения формулы (I) можно переводить в лекарственные средства, которые содержат еще одно или несколько других действующих веществ, которые прежде всего предназначены для лечения и/или профилактики указанных выше заболеваний. Примерами предпочтительных действующих веществ, пригодных для использования в соответствующих комбинациях, являются:
- вещества, снижающие уровень липидов, прежде всего ингибиторы HMG-CoA-(3-гидрокси-3-метилглутарил-коэнзим А)-редуктазы;
- вещества, используемые для коронарной терапии/вазодилататоры, прежде всего ингибиторы ферментов превращения ангиотензина (АСЕ-ингибиторы); антагонисты рецептора AII (ангиотензина II); антагонисты β-адреноблокатора; антагонисты альфа-1-адреноблокатора; диуретики; блокаторы кальциевого канала; вещества, способствующие повышению уровня циклического гуанозинмонофосфата (cGMP), например, такие как стимуляторы растворимой гуантилатциклазы;
- активаторы плазминогена (тромболитики/фибринолитики) и соединения, повышающие тромболиз/ фибринолиз, такие как ингибиторы ингибитора активатора плазминогена (PAI-ингибиторы) или ингибиторы активированного тромбином ингибитора фибринолиза (TAFI-ингибиторы);
- вещества с противосвертывающим действием (антикоагулянты);
- вещества, подавляющие агрегацию тромбоцитов (ингибиторы агрегации тромбоцитов);
- антагонисты рецептора фибриногена (антагонисты гликопротеида-IIb/IIIa);
- антиаритмические средства.
Кроме того, предлагаемые соединения формулы (I) можно переводить в лекарственные средства, которые содержат еще одно или несколько инертных, нетоксичных, фармацевтически пригодных вспомогательных веществ.
Предлагаемые соединения формулы (I) могут обладать систематическим и/или местным действием. В соответствии с этим они пригодны, например, для орального, парентерального, пульмонального или назального применения. Предлагаемые соединения формулы (I) можно применять в виде форм, соответствующих указанным выше способам применения.
Для орального применения пригодны функционирующие согласно уровню техники лекарственные формы, которые содержат предлагаемые соединения формулы (I) в кристаллическом, аморфизованном и/или растворенном состоянии и способны быстро и/или модифицированно их высвобождать:
Под подобными формами применения подразумевают, например, таблетки (без покрытия или с покрытием, которое, например, обладает устойчивостью к воздействию желудочного сока, замедленной растворимостью или отсутствием растворимости и обеспечивает контролируемое высвобождение предлагаемого в изобретении соединения), таблетки, способные к быстрому распадаться в полости рта или пленки/облатки, пленки/лиофилизаты, капсулы (например, капсулы из жесткого или мягкого желатина), драже, гранулы, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное применение (например, внутривенное, внутриартериальное, интракардиальное, интраспинальное или интралюмбальное) позволяет исключить стадию резорбции или включает стадию резорбции (например, внутримышечное, подкожное, внутрикожное, чрезкожное или внутрибрюшинное применение). Для парентерального применения, в частности, пригодны такие лекарственные формы, как, например, инъекционные и инфузионные препараты в виде растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для прочих способов применения пригодны, например, ингаляционные лекарственные формы (в частности, порошковые ингаляторы, распылители), или назально применяемые лекарственные формы, такие как капли, растворы или распыляемые растворы.
Предпочтительным является парентеральное, прежде всего внутривенное применение.
Предлагаемые соединения формулы (I) могут быть переведены в указанные выше лекарственные формы. Соответствующий процесс может быть реализован известным образом благодаря смешиванию предлагаемых соединений с инертными, нетоксичными, фармацевтически пригодными вспомогательными веществами. К подобным вспомогательным веществам относятся, в частности, основы (например, микрокристаллическая целлюлоза, лактоза, маннитол), растворители (например, жидкие полиэтиленгликоли), эмульгаторы, диспергаторы, смачивающие агенты (например, додецилсульфат натрия, полиоксисорбитанолеат), связующие вещества (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (в частности, антиоксиданты, например, такие как аскорбиновая кислота), красители (в частности, неорганические пигменты, например, такие как оксиды железа) и средства для корректировки вкуса и/или запаха.
Предпочтительная дозировка действующего вещества, обеспечивающая достижение эффективного результата при парентеральном применении, в общем случае составляет примерно от 0,001 до 1 мг, предпочтительно от 0,01 до 0,5 мг на кг массы тела. В случае орального применения оптимальной дозировке соответствует примерный интервал от 0,01 до 100 мг, предпочтительно от 0,01 до 20 мг и еще более предпочтительно от 0,1 до 10 мг на кг массы тела.
Несмотря на это в некоторых случаях может возникать необходимость в корректировке указанных дозировок, которая определяется массой тела, способом применения действующего вещества, индивидуальным отношением пациентов к нему, а также типом препарата и моментом времени, соответственно периодичностью его применения. Так, например, в определенных случаях могут оказаться достаточными дозировки, более низкие по сравнению с указанными выше нижними предельными значениями, тогда как в иных случаях могут потребоваться дозировки, превышающие указанные выше максимальные значения. В последнем случае рекомендуется распределять дозировку на несколько применяемых в течение суток доз.
Приведенные ниже примеры предназначены для пояснения изобретения и не ограничивают его объема.
В отсутствие особых указаний процентные данные в нижеследующих опытах и примерах приведены в массовых процентах, части в массовых частях. Количественные соотношения между растворителями, степени разбавления и концентрации растворов жидких компонентов в жидкостях указаны в соответствующих объемных единицах.
А. Примеры
Сокращения и аббревиатуры:
Методы ЖХ-МС и ВЭЖХ
Метод 1a (препаративная ВЭЖХ). Колонка VP 250/21 Nukleodur 100-5 С18 ec, Macherey & Nagel Nr. 762002; элюент А: вода/0,01% трифторуксусной кислоты, элюент В: ацетонитрил/0,01% трифторуксусной кислоты; градиент: 0 мин 0% В → 20 мин 20% В → 40 мин 20% В → 60 мин 30 % В → 80 мин 30% В → 90 мин 100% В → 132 мин 100% В; расход 5 мл/мин; комнатная температура; УФ-детектирование 210 нм.
Метод 1b (препаративная ВЭЖХ). Колонка SymmetryPrep™ С18 7µМ; 19×300 мм, Waters; элюент А: вода/0,01% трифторуксусной кислоты, элюент В: ацетонитрил/0,01% трифторуксусной кислоты; градиент: 0 мин 0% В → 20 мин 20% В → 40 мин 20% В → 60 мин 30% В → 80 мин 30% В → 90 мин 100% В → 132 мин 100% В; расход 5 мл/мин; комнатная температура; УФ-детектирование при 210 нм.
Метод 2 (аналитическая ВЭЖХ). Колонка XTerra 3,9×150 WAT 186000478; элюент А: 10 мл 70%-ной перхлорной кислоты в 2,5 л воды, элюент В: ацетонитрил; градиент: 0,0 мин 20% В → 1 мин 20% В → 4 мин 90% В → 9 мин 90% В; комнатная температура; расход 1 мл/мин. В соответствии с вариантом 2а элюирование выполняют при температуре колонки 40°С.
Метод 3 (ЖХ-МС). Прибор Micromass ZQ; тип прибора ВЭЖХ: HP серии 1100; UV DAD; колонка: Phenomenex Gemini 3µ 30 мм × 3,00 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 2,5 мин 30% А → 3,0 мин 5% А → 4,5 мин 5% А; расход: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура 50°С; УФ-детектирование при 210 нм.
Метод 4 (ЖХ-МС). Прибор Micromass ZQ с ВЭЖХ HP серии 1100; UV DAD; колонка Phenomenex Synergi 2µ Hydro-RP Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 2,5 мин 30% А → 3,0 мин 5% А → 4,5 мин 5% А; расход: 0,0 мин 1 мл/мин → 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура 50°С; УФ-детектирование при 210 нм.
Метод 5 (ЖХ-МС). Прибор Micromass Quattro LCZ с ВЭЖХ Agilent серии 1100; колонка Phenomenex Synergi, 2 мкм, Hydro-RP Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 2,5 мин 30% А → 3,0 мин 5% А → 4,5 мин 5% А; расход: 0,0 мин 1 мл/мин → 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура 50°С; УФ-детектирование в диапазоне 208-400 нм.
Метод 6 (ЖХ-МС). Масс-спектрометр типа Micromass ZQ; ВЭЖХ: Waters Alliance 2795; колонка Phenomenex Synergi, 2 мкм, Hydro-RP Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 2,5 мин 30% А → 3,0 мин 5% А → 4,5 мин 5% А; расход: 0,0 мин 1 мл/мин → 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура 50°С; УФ-детектирование при 210 нм.
Метод 7 (хиральная аналитическая ВЭЖХ). Хиральная силикагелевая фаза (250 мм × 4,6 мм) на основе поли(N-метакрилоил-L-лейциндициклопропилметиламида); элюент: изогексан/этилацетат 35:65 (об/об.); температура 24°С; расход 2 мл/мин; УФ-детектирование при 270 нм.
Метод 8 (хиральная аналитическая ВЭЖХ). Хиральная силикагелевая фаза (250 мм × 4,6 мм) на основе поли(N-метакрилоил-L-лейцин-трет-бутиламида); элюент: изогексан/этилацетат 35:65 (об/об.); температура 24°С; расход 2 мл/мин; УФ-детектирование при 270 нм.
Метод 9 (хиральная аналитическая ВЭЖХ). Хиральная силикагелевая фаза (250 мм × 4,6 мм) на основе поли(N-метакрилоил-L-лейцин-трет-бутиламида); элюент: изогексан/этилацетат 65:35 (об/об.); температура 24°С; расход 2 мл/мин; УФ-детектирование при 270 нм.
Метод 10 (хиральная препаративная ВЭЖХ). Хиральная силикагелевая фаза (670 мм × 40 мм) на основе поли(N-метакрилоил-L-лейцин-дицикло-пропилметиламида); элюент: изогексан/этилацетат 25:75 (об/об.); температура 24°С; расход 80 мл/мин; УФ-детектирование при 270 нм.
Метод 11 (хиральная препаративная ВЭЖХ). Хиральная силикагелевая фаза (670 мм × 40 мм) на основе поли(Л/-метакрилоил-1-лейцин-трет-бутиламида); элюент: изогексан/этилацетат 65:35 (об/об.); температура 24°С; расход 50 мл/мин; УФ-детектирование при 260 нм.
Метод 12 (ЖХ-МС). Прибор: Micromass Quattro LCZ с ВЭЖХ Agilent серии 1100; колонка: Phenomenex Onyx Monoolithic C18, 100 мм × 3 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 2 мин 65% А → 4,5 мин 5% А → 6 мин 5% А; расход 2 мл/мин; температура термостата 40°С; УФ-детектирование в диапазоне 208-400 нм.
Метод 13 (ЖХ-МС). Прибор: Micromass Platform LCZ с ВЭЖХ Agilent серии 1100; колонка: Thermo Hypersil GOLD 3µ 20 мм × 4 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 100% А → 0,2 мин 100% А → 2,9 мин 30% А → 3,1 мин 10% А → 5,5 мин 10% А; температура термостата 50°С; расход 0,8 мл/мин; УФ-детектирование при 210 нм.
Метод 14 (ЖХ-МС). Тип масс-спектрометра: Micromass ZQ; тип прибора ВЭЖХ: Waters Alliance 2795; колонка: Merck Chromolith SpeedROD RP-18e, 100 мм × 4,6 мм; элюент A: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты; элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 10% В → 7,0 мин 95% В → 9,0 мин 95% В; температура термостата 35°С; расход 0,0 мин 1,0 мл/мин → 7,0 мин 2,0 мл/мин → 9,0 мин 2,0 мл/мин; УФ-детектирование при 210 нм.
ЯМР-спектроскопия
ЯМР-измерения осуществляли при частоте 400,13 или 500,13 МГц. Образцы обычно растворяли в ДМСО-d6; температура измерения 302К.
Исходные соединения
В качестве исходного продукта используют 5-хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамид [соединение (А)], получение которого описано в публикации S.Roehrig et al,. J. Med. Chem. 48, 5900 (2005):
Пример 1А
5-Хлор-N-(4-хлорбутаноил)-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамид
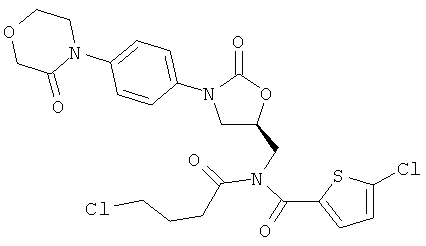
1 г (2,3 ммоль) 5-хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамида [соединения (А)] растворяют под аргоном в 100 мл абсолютного диметилформамида. Добавляют 110 мг (4,6 ммоль) гидрида натрия (98%-ного), и полученный раствор в течение 20 минут перемешивают при комнатной температуре. Затем добавляют 4,37 г (30,97 ммоль) хлорбутаноилхлорида, причем реакционную температуру поддерживают на уровне комнатной. Раствор перемешивают при комнатной температуре в течение 16 ч и затем медленно при охлаждении добавляют 25 мл воды. После этого добавляют 300 мл этилацетата и еще 50 мл воды. Осуществляют разделение фаз, причем фазу этилацетата концентрируют в вакууме. Остаток смешивают с этилацетатом и фильтруют. Маточный раствор концентрируют и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь толуола с этанолом в соотношении 5:1. Фракции, содержащие целевое соединение, а также фракции, которые содержат образующееся в результате енолизации бисацилированное соединение, объединяют и удаляют растворитель. Остаток смешивают с насыщенным раствором хлористого водорода в дихлорметане и в течение ночи перемешивают при комнатной температуре. Затем смесь концентрируют в вакууме и остаток вновь подвергают очистке флэш-хроматографией на силикагеле используемой в качестве элюента смесью толуол/этанол (6:1). Соответствующие фракции концентрируют, и остаток лиофилизируют из диоксана. При этом получают 94 мг целевого соединения (выход 7,5% от теоретического).
ВЭЖХ (метод 2): Rt=5,23 мин;
ЖХ-МС (метод 6): Rt=2,13 мин; m/z=540 (М+Н)+.
Пример 2А
5-Хлор-N-(4-хлорпентаноил)-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамид
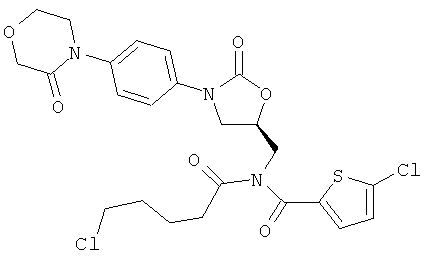
Синтез осуществляют аналогично примеру 1А исходя из 3 г (6,88 ммоль) соединения (А) и 5-хлорпентаноилхлорида. Получают 1008 мг целевого соединения (выход 26% от теоретического).
ВЭЖХ (метод 2): Rt=5,35 мин;
ЖХ-МС (метод 6): Rt=2,22 мин; m/z=554 (М+Н)+.
Пример 3А
N-(4-аминобутаноил)-5-хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамид гидрохлорид
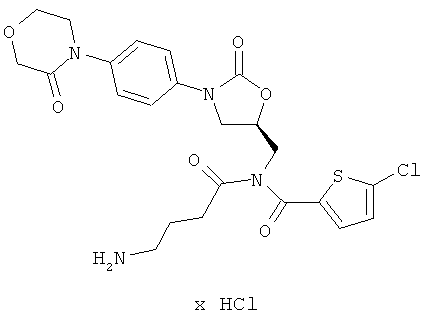
Стадия а)
1,33 г (3,06 ммоль) соединения (А) растворяют в 75 мл абсолютного диметилформамида, полученный раствор смешивают с 220 мг (9,2 ммоль) гидрида натрия (98%-ного) и перемешивают в течение 30 минут при комнатной температуре. Затем добавляют 11,5 г (30,6 ммоль) растворенного в 10 мл абсолютного диметилформамида соединения из примера 5А. Смесь перемешивают еще в течение 15 минут при комнатной температуре и затем добавляют 20 мл воды. Смесь концентрируют, и остаток смешивают с 300 мл этилацетата. Выполняют трехкратное встряхивание с 300 мл 10-процентного раствора карбоната натрия. Органическую фазу отделяют, концентрируют и смешивают с 50 мл дихлорметана. Затем добавляют 25 мл диэтилового эфира. После кратковременного перемешивания нерастворенные остатки отделяют фильтрованием, и фазу дихлорметана концентрируют. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь этилацетат/толуол (5:1). Соответствующие фракции, которые содержат бисацилированный побочный продукт массой М=1113, образовавшийся в результате енолизации моноацильного соединения, концентрируют. Затем остаток в течение 2 ч перемешивают с 10 мл насыщенного раствора хлористого водорода в дихлорметане, при этом происходит расщепление первоначально образовавшегося эфира енола. После этого осуществляют концентрирование, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь этилацетат/толуол (5:1). Соответствующие фракции концентрируют и получают 151 мг соединения с полной защитой аминогрупп (выход 7% от теоретического).
ВЭЖХ (метод 2): Rt=5,83 мин;
ЖХ-МС (метод 6): Rt=2,61 мин; m/z=775 (М+Н)+.
Стадия b)
151 мг (0,2 ммоль) полученного защищенного промежуточного соединения перемешивают в течение ночи при комнатной температуре в 8 мл безводной трифторуксусной кислоты. Смесь концентрируют в высоком вакууме, поддерживая температуру около 20°С. Остаток смешивают с 100 мл соляной кислоты с показателем pH 3 и встряхивают с 75 мл дихлорметана, а затем двукратно с этилацетатом. Водную фазу концентрируют, и остаток лиофилизируют из соляной кислоты с показателем pH 3. Получают 70 мг целевого соединения (выход 64% от теоретического).
ВЭЖХ (метод 2): Rt=4,13 мин;
ЖХ-МС (метод 5): Rt=1,38 мин; m/z=521 (М+Н)+.
Пример 4А
N-(5-аминопентаноил)-5-хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамид гидрохлорид
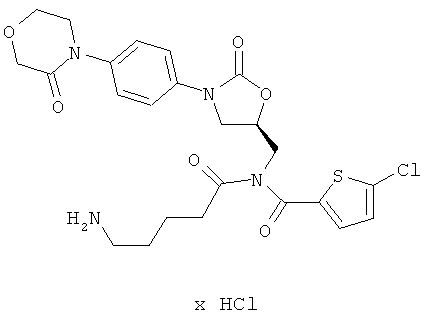
Стадия а)
2.83 г (6.5 ммоль) соединения (А) под аргоном растворяют в 100 мл абсолютного диметилформамида, полученный раствор смешивают с 468 мг (19,5 ммоль) гидрида натрия, и смесь в течение 30 минут перемешивают при комнатной температуре. После этого добавляют 7,6 г (19,5 ммоль) растворенного в 10 мл диметилформамида соединения из примера 10А. Смесь перемешивают еще в течение 15 минут при комнатной температуре, а затем медленно смешивают с 20 мл воды. Смесь концентрируют, и остаток в течение 1 ч перемешивают со 150 мл насыщенного раствора хлористого водорода в дихлорметане, причем перемешивание сопровождается расщеплением первоначально образовавшегося в результате енолизации бисацильного соединения массой М=1141. Затем смесь концентрируют, и остаток смешивают с 700 мл этилацетата. Осуществляют двукратное встряхивание, используя по 200 мл 10%-ного раствора карбоната натрия. Органическую фазу отделяют, концентрируют, смешивают с 30 мл этилацетата и затем с 30 мл диэтилового эфира. После кратковременного перемешивания нерастворенные остатки отделяют фильтрованием, органическую фазу концентрируют. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь этилацетат/толуол (4:1). Соответствующие фракции концентрируют, остаток смешивают с 10 мл этилацетата. Добавляют 100 мл холодного диэтилового эфира, и смесь в течение 30 минут выдерживают при 0°С. Осуществляют фильтрование и повторную обработку остатка 100 мл диэтилового эфира. После вторичного фильтрования собирают и сушат фильтровальный остаток. Получают 1 г соединения с полной защитой аминогрупп (выход 20% от теоретического).
ВЭЖХ (метод 2): Rt=5,92 мин;
ЖХ-МС (метод 6): Rt=2,68 мин; m/z=789 (М+Н)+.
Стадия b)
1 г (1,3 ммоль) полученного выше защищенного промежуточного соединения в 70 мл безводной трифторуксусной кислоты в течение 6 ч обрабатывают в ультразвуковой ванне. Смесь концентрируют в высоком вакууме, поддерживая температуру на уровне 20°С. Остаток смешивают с 350 мл соляной кислоты с показателем pH 3 и после 15-минутного перемешивания при комнатной температуре встряхивают со 100 мл дихлорметана. Затем остаток встряхивают со 100 мл этилацетата. Водную фазу отделяют и осуществляют кратковременную дистилляцию в высоком вакууме с целью удаления остаточного этилацетата, а затем лиофилизацию. Получают 586 мг целевого соединения (выход 81% от теоретического).
ВЭЖХ (метод 2): Rt=4,2 мин;
ЖХ-МС (метод 6): Rt=1,17 мин; m/z=535 (М+Н)+.
Пример 5А
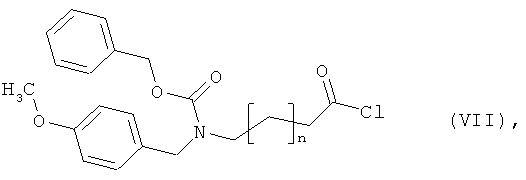
Бензил-(4-хлор-4-оксобутил)(4-метоксибензил)карбамат
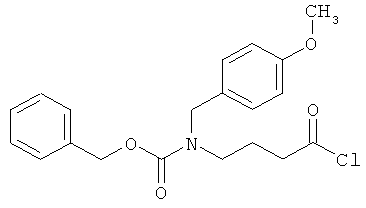
Синтез этого соединения выполняют аналогично примеру 10А исходя из 4-аминомасляной кислоты.
Пример 6А
(2S)-2-[(трет-бутоксикарбонил)амино]-3-метилбутантио-S-кислота
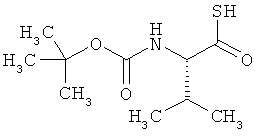
Соединение получают из Boc-валина в соответствии с известной из литературы методикой [R.Michelot et al., Bioorg. Med. Chem., 1996, 4, 2201).
Пример 7А
[(трет-Бутоксикарбонил)амино]этантио-S-кислота
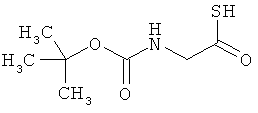
Соединение получают из Boc-глицина в соответствии с известной из литературы методикой [R.Michelot et al., Bioorg. Med. Chem., 1996, 4, 2201).
Пример 8А
(2S)-2,6-бис[(трет-бутоксикарбонил)амино]гексантио-S-кислота
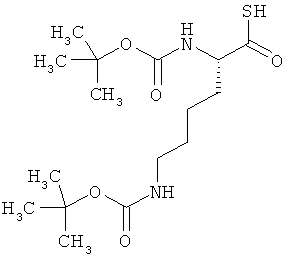
Соединение получают из бис-Boc-лизина в соответствии с известной из литературы методикой [R.Michelot et al., Bioorg. Med. Chem., 1996, 4, 2201).
Пример 9А
(2S)-2-[(трет-бутоксикарбонил)амино]пропантио-S-кислота
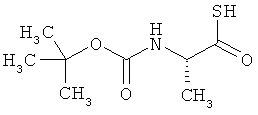
Соединение получают из Boc-аланина в соответствии с известной из литературы методикой [R.Michelot et al., Bioorg. Med. Chem., 1996, 4, 2201).
Пример 10А
Бензил(5-хлор-5-оксопентил)(4-метоксибензил)карбамат
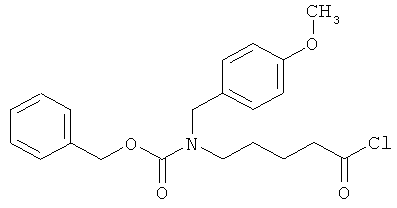
10 г (85,4 ммоль) 5-аминовалерьяновой кислоты, 17,4 г (128 ммоль) п-анисового альдегида и 10,3 г (85,4 ммоль) сульфата магния смешивают с 330 мл этанола и полученную смесь в течение 1 часа нагревают с обратным холодильником. Осуществляют фильтрование и промывку смеси этанолом, после чего раствор смешивают с 1,94 г (51,2 ммоль) боргидрида натрия, который добавляют порциями в течение 15 минут. Добавляют 10 мл воды, а затем 128 мл 2М раствора едкого натра. Через 5 минут смесь разбавляют 300 мл воды и последовательно встряхивают с тремя порциями этилацетата объемом по 200 мл. Показатель pH водной фазы посредством 4М соляной кислоты устанавливают на уровне 2, и водную фазу концентрируют в вакууме. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь ацетонитрил/вода/уксусная кислота (5:1:0,1). Соответствующие фракции концентрируют и смешивают с этилацетатом и диэтиловым эфиром. Остаток фильтруют и сушат в высоком вакууме. Получают 9,1 г защищенной п-метоксибензилом аминокислоты (выход 45% от теоретического).
Полученный продукт смешивают с 1,6 л смеси диоксан/вода (1:1), показатель pH посредством раствора едкого натра устанавливают на уровне 10, и осуществляют смешивание с 12,97 г (76 ммоль) добавляемого по каплям бензилхлоркарбоната. После 15-минутного перемешивания при комнатной температуре диоксан отгоняют в вакууме, и показатель pH оставшегося раствора посредством 2М соляной кислоты устанавливают на уровне 2. Осуществляют экстракцию этилацетатом и последующую двукратную промывку органической фазы водой. Затем органическую фазу концентрируют, остаток сушат в высоком вакууме. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента ацетонитрил. Соответствующие фракции концентрируют, остаток сушат в высоком вакууме. Получают 5,6 г защищенной аминокислоты (выход 38% от теоретического).
ЖХ-МС (метод 3): Rt=2,47 мин; m/z=372 (М+Н)+.
5,6 г (15 ммоль) 5-{[(бензилокси)карбонил](4-метоксибензил)амино}-валерьяновой кислоты растворяют в 60 мл дихлорметана, и полученный раствор смешивают с 2,2 мл тионилхлорида. Смесь в течение 30 минут нагревают с обратным холодильником. Затем ее концентрируют в вакууме, остаток вновь смешивают с дихлорметаном и концентрируют. Остается вязкое масло, которое подвергают сушке в высоком вакууме. Получают 5,7 г целевого соединения (выход 98% от теоретического), которое подвергают дальнейшему превращению без дополнительной очистки и определения характеристик.
Примеры осуществления изобретения
Общая методика 1 получения цезиевых солей карбоновых кислот или пригодных защищенных производных аминокислоты
1 ммоль соответствующей карбоновой кислоты растворяют в смеси 10 мл диоксана с 10 мл воды, и полученный раствор смешивают с 0,5 ммоль карбоната цезия. После этого выполняют лиофилизацию.
Общая методика 2 получения уретанзащищенных N-карбоксиангидридов пригодных защищенных производных аминокислоты
Уретанзащищенные N-карбоксиангидриды производных аминокислоты являются коммерчески доступными продуктами или могут быть получены известными из литературы методами (М. Johnston et al., J. Org. Chem., 1985, 50, 2200; W.D. Fuller et al., J.Am. Chem. Soc., 1990, 112, 7414; S. Mobasheri et al., J.Org. Chem., 1992, 57, 2755).
Общая методика 3 получения N-гидроксисукцинимидоэфиров пригодных защищенных производных аминокислоты
N-гидроксисукцинимидоэфиры производных аминокислоты являются коммерчески доступными продуктами или могут быть получены стандартными методами химии пептидов.
Пример 1
2-[[(5-Хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино]-4-оксобутилглицинат гидрохлорид
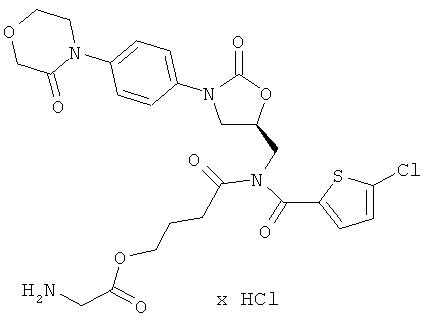
Стадия а)
14 мг (26 мкмоль) соединения из примера 1А и 9,5 мг (31 мкмоль) цезиевой соли Boc-глицина, полученной из Boc-глицина в соответствии с общей методикой 1, растворяют в 5 мл диметилформамида. После 16-часового перемешивания при 50°С раствор концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 8 мг защищенного промежуточного соединения (выход 45% от теоретического).
ВЭЖХ (метод 2): Rt=5,18 мин;
ЖХ-МС (метод 3): Rt=2,38 мин; m/z=679 (М+Н)+.
Стадия b)
7 мг (11 мкмоль) полученного на стадии а) защищенного промежуточного соединения, которое еще содержит примеси, смешивают с 1 мл 22%-ного раствора хлористого водорода в диоксане. Через 30 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и лиофилизируют из диоксана. Получают 0,6 мг целевого соединения (выход 8% от теоретического).
ВЭЖХ (метод 2): Rt=4,2 мин;
ЖХ-МС (метод 3): Rt=1,33 мин; m/z=579 (M+H)+.
Пример 2
2-[[(5-Хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино]-5-оксопентилглицинат гидрохлорид
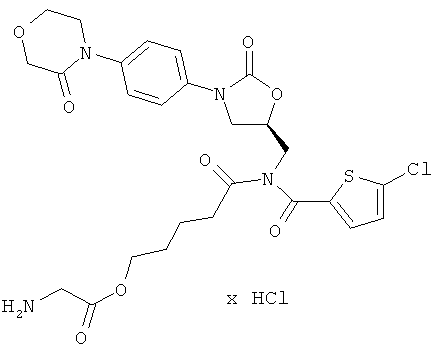
Стадия а)
59 мг (106 мкмоль) соединения из примера 2А и 43 мг (138,4 мкмоль) цезиевой соли Boc-глицина, полученной из Boc-глицина в соответствии с общей методикой 1, растворяют в 10 мл диметилформамида. После 16-часового перемешивания раствор концентрируют при 50°С, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 26 мг защищенного промежуточного соединения (выход 35% от теоретического).
ВЭЖХ (метод 2): Rt=5,27 мин;
ЖХ-МС (метод 6): Rt=2,23 мин; m/z=693 (М+Н)+.
Стадия b)
12 мг (17 мкмоль) полученного на стадии а) защищенного промежуточного соединения смешивают с 3 мл 22%-ного раствора хлористого водорода в диоксане. Спустя 30 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и затем лиофилизируют соляной кислотой с pH 4. Получают 7,2 мг целевого соединения (выход 66% от теоретического).
ВЭЖХ (метод 2): Rt=4,32 мин;
ЖХ-МС (метод 5): Rt=1,48 мин; m/z=593 (M+H)+.
Пример 3
2-[[(5-Хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино]-5-оксопентил-L-валинат гидрохлорид
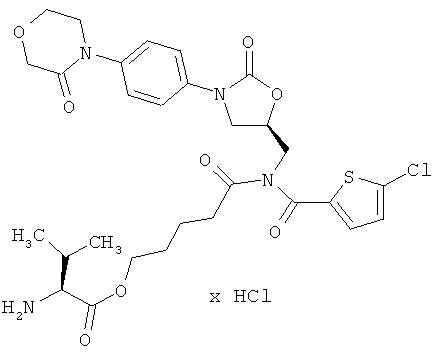
Стадия а)
50 мг (90 мкмоль) соединения из примера 2А и 41 мг (117 мкмоль) цезиевой соли Boc-валина, полученного из Boc-валина в соответствии с общей методикой 1, растворяют в 10 мл диметилформамида. После 42-часового перемешивания при 50°С раствор концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 26 мг защищенного промежуточного соединения (выход 39% от теоретического).
ВЭЖХ (метод 2): Rt=5,71 мин;
ЖХ-МС (метод 6): Rt=2,56 мин; m/z=733 (M-Н)+.
Стадия b)
26 мг (35 мкмоль) полученного на стадии а) защищенного промежуточного соединения вводят в 5 мл дихлорметана и смешивают с 2 мл безводной трифторуксусной кислоты. Спустя 30 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток перемешивают с ацетонитрилом, после чего растворитель удаляют. Остаток лиофилизируют из соляной кислоты с pH 3. Получают 24 мг целевого соединения (количественный выход).
ВЭЖХ (метод 2): Rt=4,5 мин;
ЖХ-МС (метод 3): Rt=1,52 мин; m/z=635 (М+Н)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=0,95 (2d, 6Н), 1,65 (m, 4H), 2,15 (m, 1H), 2,6 (m, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,9 (d, 1H), 3,95 (t, 2H), 4,1-4,3 (m, 7H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,3 (m, 3Н).
Пример 4
S-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)(2S)-2-амино-3-метилбутантиоат гидрохлорид
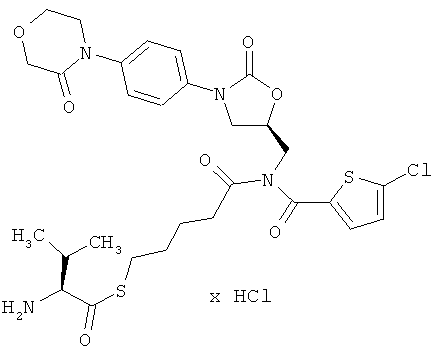
Стадия а)
50 мг (90 мкмоль) соединения из примера 2А и 42 мг (180 мкмоль) соединения из примера 6А растворяют в 10 мл диметилформамида. Добавляют 16 мкл этилдиизопропиламина и раствор перемешивают в течение 16 ч при 60°С. Затем раствор концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1b). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 17 мг защищенного промежуточного соединения (выход 25% от теоретического).
ВЭЖХ (метод 2): Rt=5,56 мин.
Стадия b)
17 мг (23 мкмоль) полученного на стадии а) защищенного промежуточного соединения смешивают с 3 мл безводной трифторуксусной кислоты. Спустя 15 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток смешивают с соляной кислотой с pH 3 и двукратно встряхивают с небольшими количествами дихлорметана и этилацетата. Водную фазу концентрируют и лиофилизируют из соляной кислоты с pH 3. Получают 7 мг целевого соединения (выход 45% от теоретического).
ВЭЖХ (метод 2): Rt=4,65 мин;
ЖХ-МС (метод 6): Rt=1,5 мин; m/z=651 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=0,95 и 1,0 (2d, 6H), 1,5-1,7 (m, 4H), 2,15 (m, 1H), 2,55 (t, 2H), 3,0 (m, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (t, 2H), 4,1-4,3 (m, 6H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,3 (m, 3H).
Пример 5
S-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)аминоэтан-тиоат гидрохлорид
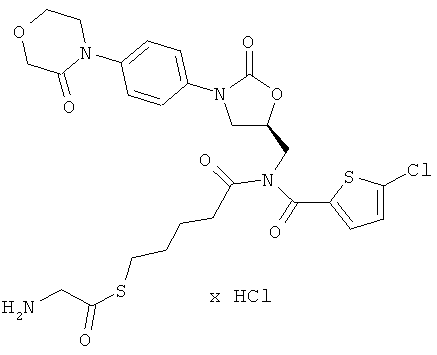
Стадия а)
50 мг (90 мкмоль) соединения из примера 2А и 52 мг (271 мкмоль) соединения из примера 7А растворяют в 15 мл диметилформамида. Добавляют 16 мкл этилдиизопропиламина, и раствор в течение 40 ч перемешивают при 60°С. В течение указанного времени еще пять раз добавляют по 52 мг соединения из примера 7А. Затем реакционный раствор концентрируют. Остаток смешивают с этилацетатом и двукратно встряхивают с 10%-ным раствором карбоната натрия. Органическую фазу концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции наряду с целевым соединением еще содержат исходный продукт. Из них в вакууме удаляют растворитель и используют на следующей стадии. Получают 38 мг защищенного промежуточного соединения (выход сырого продукта 59% от теоретического).
ВЭЖХ (метод 2): Rt=5,43 мин.
Стадия b)
37 мг (52 мкмоль) полученного на стадии а) защищенного промежуточного соединения смешивают с 3 мл безводной трифторуксусной кислоты. Спустя 15 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток подвергают очистке препаративной ВЭЖХ (метод 1а). При этом выделяют остаточный исходный продукт. Соответствующие фракции концентрируют и затем лиофилизируют из соляной кислоты с pH 3. Получают 8 мг целевого соединения (выход 24% от теоретического).
ВЭЖХ (метод 2): Rt=4,4 мин;
ЖХ-МС (метод 12): Rt=2,1 мин; m/z=609 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=1,5-1,7 (m, 4Н), 2,55 (m, 2H), 3,0 (t, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (t, 2H), 4,05-4,25 (m, 7H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,3 (m, 3Н).
Пример 6
S-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)(2S)-2,6-диаминогексантиоат дигидрохлорид
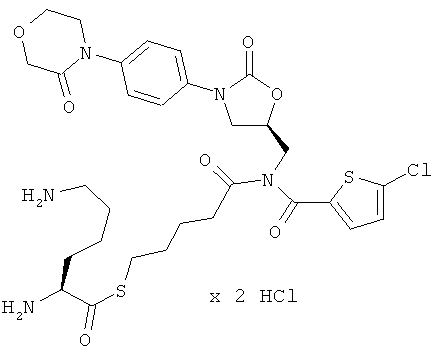
Стадия а)
50 мг (90 мкмоль) соединения из примера 2А и 98 мг (271 мкмоль) соединения из примера 8А растворяют в 15 мл диметилформамида. Добавляют 16 мкл этилдиизопропиламина, и раствор в течение 40 ч перемешивают при 60°С. В течение указанного времени еще пять раз добавляют по 98 мг соединения из примера 8А. Затем реакционный раствор концентрируют. Остаток смешивают с этилацетатом и двукратно встряхивают с 10%-ным раствором карбоната натрия. Органическую фазу концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции целевого соединения концентрируют и подвергают двукратной очистке препаративной ВЭЖХ (метод 1а). Фракции, содержащие чистое целевое соединение, объединяют и концентрируют. Получают 26 мг защищенного промежуточного соединения (выход 33% от теоретического).
ВЭЖХ (метод 2): Rt=5,85 мин.
Стадия b)
25 мг (28 мкмоль) полученного на стадии а) защищенного промежуточного соединения смешивают с 5 мл безводной трифторуксусной кислоты. Спустя 5 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и лиофилизируют из соляной кислоты с pH 3. Получают 10 мг целевого соединения (выход 49% от теоретического).
ВЭЖХ (метод 2): Rt=4,2 мин;
ЖХ-МС (метод 13): Rt=2,6 мин; m/z=680 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=1,3-1,5 (m, 2H), 1,5-1,7 (m, 6H), 1,7-1,9 (m, 2H), 2,55 (m, 2H), 2,75 (m, 2H), 2,95 (t, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (t, 2H), 4,1-4,3 (m, 6H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1 H), 7,85 (m, 3H), 8,5 (m, 3H).
Пример 7
S-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)(2S)-2-аминопропантиоат гидрохлорид
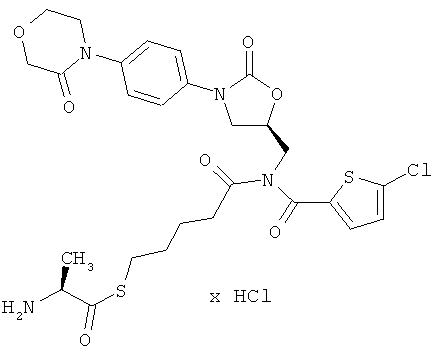
Стадия а)
50 мг (90 мкмоль) соединения из примера 2А и 55 мг (270 мкмоль) соединения из примера 9А растворяют в 15 мл диметилформамида. Добавляют 16 мкл этилдиизопропиламина, и раствор в течение 40 ч перемешивают при 60°С. В течение указанного времени еще пять раз добавляют по 55 мг соединения из примера 9А. Затем реакционную смесь концентрируют. Остаток смешивают с этилацетатом и двукратно встряхивают с 10%-ным раствором карбоната натрия. Органическую фазу концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции объединяют и удаляют растворитель. Получают 28 мг защищенного промежуточного соединения (выход 43% от теоретического).
Стадия b)
28 г (19 мкмоль) полученного на стадии а) защищенного промежуточного соединения смешивают с 3 мл безводной трифторуксусной кислоты. Спустя 15 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и лиофилизируют из соляной кислоты с pH 3. Получают 10 мг целевого соединения (выход 81% от теоретического).
ВЭЖХ (метод 2): Rt=4,46 мин;
ЖХ-МС (метод 14): Rt=3,37 мин; m/z=623 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=1,45 (d, 3H), 1,5-1,7 (m, 4H), 2,55 (t, 2H), 2,95 (t, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (t, 2H), 4,1-4,25 (m, 5H), 4,3 (q, 1H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,4 (m, 3H).
Пример 8
S-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-4-оксобутил)-(2S)-2-амино-3-метилбутантиоат гидрохлорид
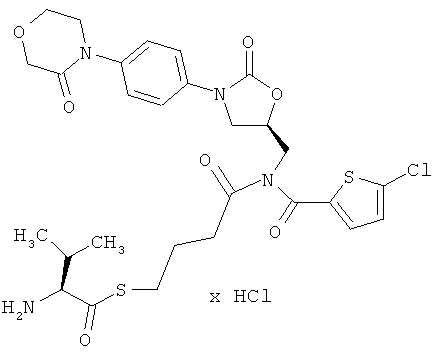
Стадия а)
48 мг (89 мкмоль) соединения из примера 1А и 62 мг (266 мкмоль) соединения из примера 6А растворяют в 15 мл диметилформамида. Добавляют 16 мкл этилдиизопропиламина, и раствор в течение 40 ч перемешивают при 60°С. В течение указанного времени еще пять раз добавляют по 62 мг соединения из примера 6А. Затем реакционную смесь концентрируют. Остаток смешивают с этилацетатом и двукратно встряхивают с 10%-ным раствором карбоната натрия. Органическую фазу концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 22 мг защищенного промежуточного соединения (выход 34% от теоретического).
ВЭЖХ (метод 2): Rt=5,76 мин.
Стадия b)
22 мг (30 мкмоль) полученного на стадии а) защищенного промежуточного соединения смешивают с 3 мл безводной трифторуксусной кислоты. Спустя 5 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и лиофилизируют из соляной кислоты с pH 3. Получают 11 мг целевого соединения (выход 52% от теоретического).
ВЭЖХ (метод 2): Rt=4,55 мин;
ЖХ-МС (метод 6): Rt=1,43 мин; m/z=637 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=0,95 и 1,0 (2d, 6H), 1,8-1,9 (m, 2H), 2,2 (m, 1H), 2,65 (m, 2H), 3,0 (m, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (t, 2H), 4,1-4,3 (m, 6H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,65 (d, 1H), 8,4 (m, 3Н).
Пример 9
5-Хлор-N-[4-(глициламино)бутаноил]-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамид гидрохлорид
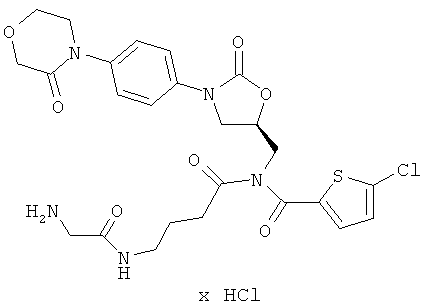
Стадия а)
40 мг (72 мкмоль) соединения из примера 3А и 17 мг (86 мкмоль) трет-бутил-2,5-диоксо-1,3-оксазолидин-3-карбоксилата растворяют в 5 мл диметилформамида. Порциями добавляют 13 мкл этилдиизопропиламина, и раствор в течение 10 минут перемешивают при комнатной температуре. Затем раствор концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 22 мг защищенного промежуточного соединения (выход 44% от теоретического).
ВЭЖХ (метод 2): Rt=4,74 мин;
ЖХ-МС (метод 5): Rt=2,07 мин; m/z=678 (М+Н)+.
Стадия b)
22 мг (32 мкмоль) полученного на стадии а) защищенного промежуточного соединения вводят в 10 мл насыщенного раствора хлористого водорода в диоксане. Добавляют 1 мл воды, и раствор в течение 5 минут перемешивают при комнатной температуре. Спустя 5 минут смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток подвергают очистке препаративной ВЭЖХ (метод 1b). Соответствующие фракции концентрируют и лиофилизируют из соляной кислоты с pH 3. Получают 4 мг целевого соединения (выход 21% от теоретического).
ВЭЖХ (метод 2): Rt=4,14 мин;
ЖХ-МС (метод 3): Rt=1,26 мин; m/z=578 (М+Н)+.
Пример 10
5-Хлор-N-[4-(глициламино)пентаноил]-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамид гидрохлорид
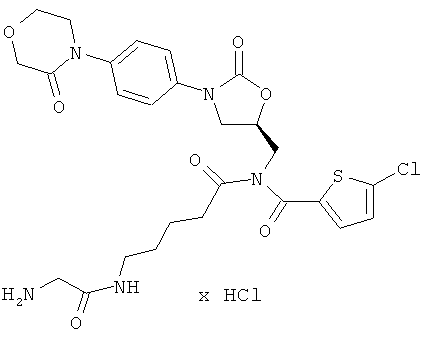
Стадия а)
35 мг (61 мкмоль) соединения из примера 4А и 37 мг (184 мкмоль) трет-бутил-2,5-диоксо-1,3-оксазолидин-3-карбоксилата растворяют в 5 мл диметилформамида. Порциями добавляют 12 мкл этилдиизопропиламина, и раствор в течение 10 минут перемешивают при комнатной температуре. Затем его концентрируют, остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 15 мг защищенного промежуточного соединения (выход 36% от теоретического).
ВЭЖХ (метод 2): Rt=4,85 мин;
ЖХ-МС (метод 6): Rt=1,95 мин; m/z=692 (M+H)+.
Стадия b)
15 мг (22 мкмоль) полученного на стадии а) защищенного промежуточного соединения смешивают с 3 мл насыщенного раствора хлористого водорода в диоксане и с одной каплей вода. После 10-минутного перемешивания при комнатной температуре реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре. Остаток смешивают с 30 мл водной соляной кислоты (pH 3), после чего двукратно встряхивают с дихлорметаном и двукратно с этилацетатом. Водную фазу концентрируют и лиофилизируют из соляной кислоты с pH 3. Получают 8 мг целевого соединения (выход 58% от теоретического).
ВЭЖХ (метод 2): Rt=4,24 мин;
ЖХ-МС (метод 3): Rt=1,36 мин; m/z=592 (М+Н)+.
Пример 11
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-пролинамид гидрохлорид
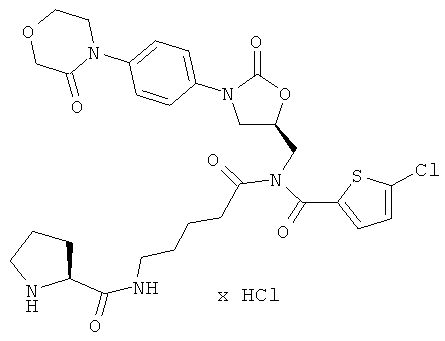
Стадия а)
50 мг (87 мкмоль) соединения из примера 4А и 47 мг (175 мкмоль) бензил-(2S)-2-(хлоркарбонил)пирролидин-1-карбоксилата вводят в 80 мл дихлорметана. В течение 3 минут тремя порциями добавляют 263 мкмоль 0,1М раствора этилдиизопропиламина, растворенного в диметилформамиде, и реакционную смесь в течение 10 минут перемешивают при комнатной температуре. Затем ее подкисляют уксусной кислотой и концентрируют. Остаток смешивают с 2 мл диметилформамида и подвергают очистке препаративной ВЭЖХ (метод 1b). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 40 мг защищенного промежуточного соединения (выход 60% от теоретического).
ВЭЖХ (метод 2): Rt=5,05 мин.
Стадия b)
40 мг (52 мкмоль) полученного на стадии а) защищенного промежуточного соединения переводят в 40 мл безводной трифторуксусной кислоты. После 16-часового перемешивания при комнатной температуре реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, и остаток подвергают очистке препаративной ВЭЖХ (метод 1b). Соответствующие фракции концентрируют и лиофилизируют из соляной кислоты с pH 3. Получают 16 мг целевого соединения (выход 46% от теоретического).
ВЭЖХ (метод 2): Rt=4,28 мин;
ЖХ-МС (метод 3): Rt=1,39 мин; m/z=632 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=1,4 (m, 2Н), 1,55 (m, 2H), 1,9 m (2H), 1,8 и 2,25 (2m, 2H), 2,55 (m, 2H), 3,0-3,3 (m, 4H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (t, 2H), 4,05-4,25 (m, 6H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,5 (m, 2H).
Пример 12
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-гистидинамид гидрохлорид
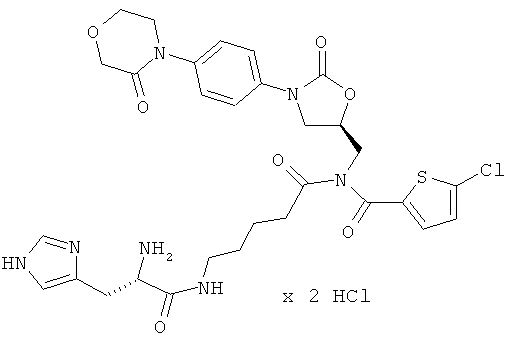
Стадия а)
199 мг (441 мкмоль) 2,5-диоксопирролидин-1-ил-N,1-бис(трет-бутоксикарбонил)-1-гистидината совместно с 661 мкл 0,1М раствора этилдиизопропиламина в диметилформамиде вводят в 1 мл диметилформамида. В течение 30 минут шприцем по каплям добавляют 42 мг (73 мкмоль) растворенного в 2,5 мл диметилформамида соединения из примера 4А. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента сначала ацетонитрил, а затем смесь ацетонитрил/вода (10:1). Соответствующие фракции целевого соединения, которые еще содержат примеси, объединяют и концентрируют в вакууме. Остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции, которые содержат смесь бис-Boc-защищенного и моно-Boc-защищенного продукта, концентрируют и сушат в высоком вакууме. Получают 18 мг защищенного промежуточного соединения (выход 28% от теоретического).
ВЭЖХ (метод 2): Rt=4,48 мин; 4,92 мин;
ЖХ-МС (метод 3): Rt=1,60 мин; m/z=772 (М+Н)+;
Rt=2,58 мин; m/z=872 (M+H)+.
Стадия b)
18 мг смеси бис-Boc-защищенного и моно-Boc-защищенного промежуточного соединения переводят в 4 мл безводной трифторуксусной кислоты и в течение 20 минут перемешивают при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, и остаток двукратно лиофилизируют из соляной кислоты с pH 3. Получают 15 мг целевого соединения (выход 98% от теоретического).
ВЭЖХ (метод 2): Rt=4,12 мин;
ЖХ-МС (метод 3): Rt=1,09 мин; m/z=672 (М+Н)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=1,35 (m, 2H), 1,5 (m, 2H), 2,55 (m, 2H), 3,0-3,3 (m, 4H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (t, 2H), 4,1-4,3 (m, 6H), 4,95 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,45 (s, 1H), 7,5 (d, 2H), 7,65 (d, 1H), 8,5 (m, 3H), 8,7 (t, 1H), 9,0 (s, 1H).
Пример 13
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-валинамид гидрохлорид
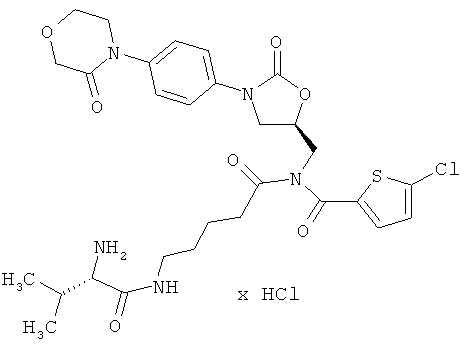
Стадия а)
50 мг (87 мкмоль) соединения из примера 4А совместно с 64 мг (262 мкмоль) трет-бутил-(43)-4-изопропил-2,5-диоксо-1,3-оксазолидин-3-карбоксилата вводят в 20 мл дихлорметана. Порциями добавляют 874 мкл 0,1М раствора этилдиизопропиламина в диметилформамиде, и реакционную смесь перемешивают в течение 10 минут при комнатной температуре. Затем ее разбавляют дихлорметаном и двукратно встряхивают с водой. Органическую фазу концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1b). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 4,5 мг защищенного промежуточного соединения (выход 7% от теоретического).
ВЭЖХ (метод 2): Rt=5,14 мин.
Стадия b)
4.5 мг (6 мкмоль) защищенного промежуточного соединения смешивают с 2 мл безводной трифторуксусной кислоты и в течение 15 мин перемешивают при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, остаток смешивают с 20 мл разбавленной соляной кислоты (pH 3) и двукратно встряхивают с дихлорметаном и однократно с этилацетатом. Водную фазу лиофилизируют из соляной кислоты с pH 3. Получают 3 мг целевого соединения (выход 73% от теоретического).
ВЭЖХ (метод 2): Rt=4,36 мин;
ЖХ-МС (метод 3): Rt=1,46 мин; m/z=634 (М+Н)+.
Пример 14
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-1-лизинамид гидрохлорид
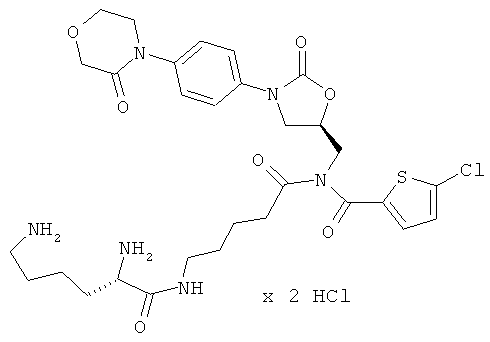
Стадия а)
39 мг (87 мкмоль) 2,5-диоксопирролидин-1-ил-N2,N6-бис(трет-бутоксикарбонил)-L-лизината совместно с 25 мг (44 мкмоль) соединения из примера 4А растворяют в 40 мл диметилформамида, и раствор порциями смешивают с 350 мкл 0,1М раствора этилдиизопропиламина в диметилформамида. После 10-минутного перемешивания при комнатной температуре реакционную смесь концентрируют. Остаток смешивают с этилацетатом и двукратно встряхивают с 10%-ным раствором карбоната натрия. Органическую фазу концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента сначала этилацетат, а затем смесь толуол/этанол (1:1). Соответствующие фракции целевого соединения, еще содержащие примеси, объединяют и концентрируют в вакууме. Затем остаток вновь подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 5 мг защищенного промежуточного соединения (выход 11% от теоретического).
ВЭЖХ (метод 2): Rt=5,27 мин;
ЖХ-МС (метод 3): Rt=2,59 мин; m/z=863 (М+Н)+.
Стадия b)
5 мг защищенного промежуточного соединения переводят в 2,5 мл безводной трифторуксусной кислоты и в течение 20 минут перемешивают при комнатной температуре. Реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, остаток смешивают с соляной кислотой (pH 3) и двукратно встряхивают с дихлорметаном. Затем отделяют и лиофилизируют водную фазу. Получают 3,8 мг целевого соединения (выход 89% от теоретического).
ВЭЖХ (метод 2): Rt=4,12 мин;
ЖХ-МС (метод 3): Rt=1,02 мин; m/z=663 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=1,3 (m, 2Н), 1,4 (m, 2H), 1,5-1,6 (m, 4H), 1,7 (m, 2H), 2,55 (m, 2H), 2,75 (m, 2H), 3,1 (m, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (t, 2H), 4,1-4,3 (m, 6H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 7,85 (m, 3Н), 8,15 (m, 3H), 8,45 (t, 1H).
Пример 15
5-Хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)-N-[5-(L-треониламино)пентаноил]тиофен-2-карбоксамид гидрохлорид

Стадия а)
277 мг (875 мкмоль) 2,5-диоксопирролидин-1-ил-N-(трет-бутоксикарбонил)-L-треонината совместно 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 1 ч по каплям добавляют 50 мг (87 мкмоль) растворенного в 5 мл диметилформамида соединения из примера 4А. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента сначала этилацетат, а затем смесь толуол/этанол (1:1). Соответствующие фракции целевого соединения, еще содержащие примеси, объединяют и концентрируют в вакууме. Затем остаток вновь подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 22 мг защищенного промежуточного соединения (выход 34% от теоретического).
ВЭЖХ (метод 2а): Rt=4,8 мин;
ЖХ-МС (метод 12): Rt=3,13 мин; m/z=736 (M+H)+.
Стадия b)
22 мг (30 мкмоль) полученного защищенного промежуточного соединения переводят в 5 мл безводной трифторуксусной кислоты и в течение 20 минут перемешивают при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, остаток смешивают с 30 мл соляной кислоты (pH 3) и двукратно встряхивают с 30 мл дихлорметана и однократно с 30 мл этилацетата. После этого отделяют и лиофилизируют водную фазу. Получают 15 мг целевого соединения (выход 75% от теоретического).
ВЭЖХ (метод 2): Rt=4,2 мин;
ЖХ-МС (метод 3): Rt=1,39 мин; m/z=636 (M+H)+.
1H-ЯМР (500 МГц, ДMCO-d6): δ=1,1 (d, 3Н), 1,4 (m, 2H), 1,6 (m, 2H), 2,6 (t, 2H), 3,0 и 3,15 (2m, 2H), 3,4 (m, 1H), 3,7 (t, 2H), 3,8 (m, 2H), 3,95 (t, 2H), 4,1-4,3 (m, 5H), 4,95 (m, 1H), 5,5 (d, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,05 (m, 3H), 8,4 (t, 1H).
Пример 16
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-тирозин-амид гидрохлорид
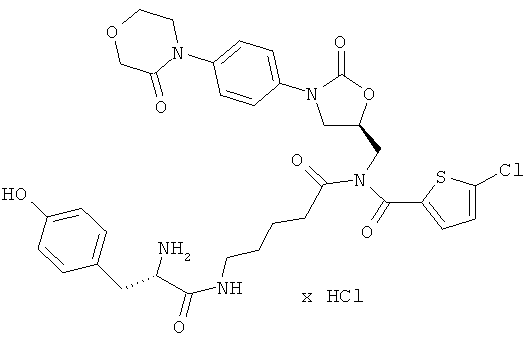
Стадия а)
331 мг (875 мкмоль) 2,5-диоксопирролидин-1-ил-N-(трет-бутоксикарбонил)-L-тирозината совместно с 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 1 ч по каплям добавляют 50 мг (87 мкмоль) растворенного в 5 мл диметилформамида соединения из примера 4А. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента сначала этилацетат, а затем смесь толуол/этанол (1:1). Соответствующие фракции целевого соединения, еще содержащие примеси, объединяют и концентрируют в вакууме. Затем остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 26 мг защищенного промежуточного соединения (выход 37% от теоретического).
ВЭЖХ (метод 2а): Rt=5,0 мин;
ЖХ-МС (метод 3): Rt=2,38 мин; m/z=798 (М+Н)+.
Стадия b)
26 мг (33 мкмоль) полученного защищенного промежуточного соединения смешивают с 5 мл безводной трифторуксусной кислоты и в течение 10 минут перемешивают при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, и остаток смешивают с 60 мл соляной кислоты (pH 3). Нерастворенные компоненты отделяют фильтрованием. Затем лиофилизируют водную фазу. Получают 23 мг целевого соединения (выход 96% от теоретического).
ВЭЖХ (метод 2): Rt=4,4 мин;
ЖХ-МС (метод 12); Rt=2,09 мин; m/z=698 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=1,3 (m, 2H), 1,5 (m, 2H), 2,8-3,2 (m, 4H), 3,7 (t, 2H), 3,8 (m, 2H), 3,95 (t, 2H), 4,1-4,3 (m, 5H), 4,9 (m, 1H), 6,7 (d, 2H), 7,0 (d, 2H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,65 (d, 1H), 8,1 (m, 3H), 8,3 (t, 1H), 9,4 (s, 1H).
Пример 17
N1-(5-{[5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-аспартамид гидрохлорид
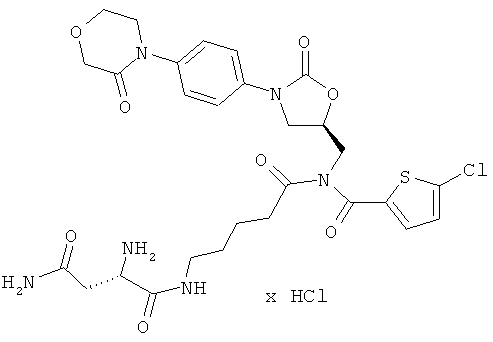
Стадия а)
288 мг (875 мкмоль) 2,5-диоксопирролидин-1-ил-N2-(трет-бутоксикарбонил)-L-аспарагината совместно с 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 30 минут по каплям добавляют 50 мг (87 мкмоль) растворенного в 5 мл диметилформамида соединения из примера 4А. После дополнительного 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют, и остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции, еще содержащие небольшое количество соединения (А), концентрируют и сушат в высоком вакууме. Получают 29 мг защищенного промежуточного соединения в виде сырого продукта, который без дополнительной очистки используют для превращения на следующей стадии.
ВЭЖХ (метод 2): Rt=4,5 мин;
ЖХ-МС (метод 3): Rt=2,07 мин; m/z=749 (М+Н)+.
Стадия b)
26 мг полученного на стадии а) защищенного сырого продукта смешивают с 5 мл безводной трифторуксусной кислоты и в течение 10 минут перемешивают при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, и остаток смешивают с 50 мл соляной кислоты (pH 3). Нерастворенные компоненты отделяют фильтрованием и концентрируют водную фазу. После этого остаток подвергают очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Остаток лиофилизируют из соляной кислоты с показателем pH, установленным на уровне 3. Получают 14 мг целевого соединения (выход 53% от теоретического).
ВЭЖХ (метод 2а): Rt=4,1 мин;
ЖХ-МС (метод 12): Rt=1,84 мин; m/z=649 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ=1,4 (m, 2Н), 1,55 (m, 2H), 2,55 (m, 2H), 2,65 (m, 2H), 3,0-3,1 (m, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 3,95 (m, 3H), 4,1-4,3 (m, 5H), 4,9 (m, 1H), 7,2 (s, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (m, 2H), 8,0 (m, 3H), 8,3 (t, 1H).
Пример 18
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-фенилаланин-амид гидрохлорид
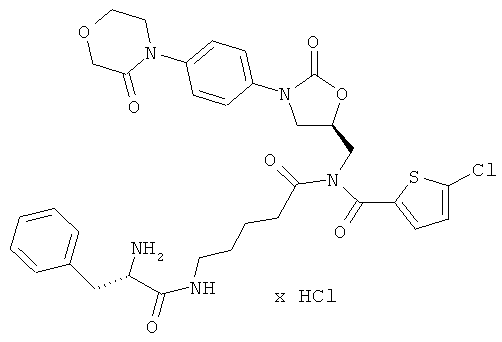
Стадия а)
317 мг (875 мкмоль) 2,5-диоксопирролидин-1-ил-N-(трет-бутоксикарбонил)-L-фенилаланината совместно с 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 30 минут по каплям добавляют 50 мг (87 мкмоль) растворенного в 5 мл диметилформамида соединения из примера 4А. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют. Остаток подвергают очистке флэш-хроматографией, сначала используя в качестве элюента смеси дихлорметан/этилацетат с соотношением компонентов соответственно 3:1, 2:1 и 1:1. Затем для элюирования используют чистый этилацетат, а в заключение - этанол. Соответствующие фракции целевого соединения, которые еще содержат примеси, объединяют и концентрируют в вакууме. После этого остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 34 мг защищенного промежуточного соединения (выход 50% от теоретического).
ВЭЖХ (метод 2а): Rt=5,34 мин;
ЖХ-МС (метод 12): Rt=3,47 мин; m/z=782 (M+H)+.
Стадия b)
33 мг (42 мкмоль) полученного защищенного промежуточного соединения растворяют в дихлорметане, раствор совмещают с 1,5 мл безводной трифторуксусной кислоты и в течение 10 минут перемешивают при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, и остаток смешивают с 5 мл соляной кислоты (pH 3). После этого лиофилизируют водную фазу. Получают 28 мг целевого соединения (выход 93% от теоретического).
ВЭЖХ (метод 2): Rt=4,5 мин;
ЖХ-МС (метод 12): Rt=2,08 мин; m/z=682 (M+H)+,
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,25 (m, 2H), 1,5 (m, 2H), 2,9-3,2 (m, 4H), 3,7 (m, 2H), 3,8 (t, 2H), 3,9 (m, 1H), 4,0 (t, 2H), 4,1-4,3 (m, 5H), 4,9 (m, 1H), 7,2 (d, 2H), 7,2-7,35 (m, 4H), 7,4 (d, 2H), 7,5 (d, 2H), 7,65 (d, 1H), 8,2 (m, 2H), 8,3 (t, 1H).
Пример 19
N1-(5-{[5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-глутамамид гидрохлорид
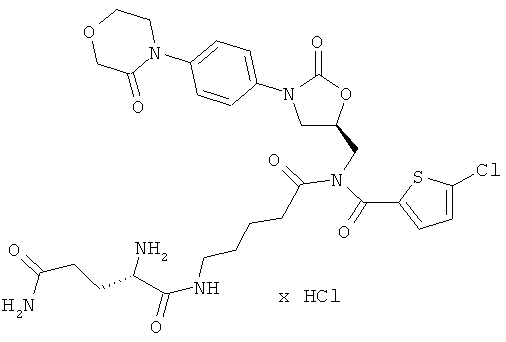
Стадия а)
300 мг (875 мкмоль) 2,5-диоксопирролидин-1-ил-N2-(трет-бутоксикарбонил)-L-глутамината совместно с 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 30 минут по каплям добавляют 50 мг (87 мкмоль) соединения из примера 4А, растворенного в 5 мл диметилформамида. Реакционную смесь после 30-минутного перемешивания при комнатной температуре концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента смеси дихлорметан/этилацетат/метанол с соотношением компонентов сначала 150:50:5, затем 150:50:10 и наконец 150:50:20. Соответствующие фракции целевого соединения, которые еще содержат примеси, объединяют и концентрируют в вакууме. После этого остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 24 мг защищенного промежуточного соединения (выход 34% от теоретического).
ВЭЖХ (метод 2а): Rt=4,57 мин;
ЖХ-МС (метод 12): Rt=2,97 мин; m/z=763 (M+H)+.
Стадия b)
24 мг (35 мкмоль) полученного защищенного промежуточного соединения растворяют в дихлорметане, раствор смешивают с 2 мл безводной трифторуксусной кислоты, и компоненты перемешивают в течение 10 минут при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, и остаток смешивают с 15 мл соляной кислоты (pH 3). Смесь встряхивают сначала двукратно с дихлорметаном, а затем однократно с этилацетатом. После этого лиофилизируют водную фазу. Получают 14 мг целевого соединения (выход 59% от теоретического).
ЖХ-МС (метод 12): Rt=1,60 мин; m/z=663 (M+H)+.
1H-ЯМР (400МГц, AMCO-d6): δ=1,4 (m, 2H), 1,6 (m, 2H), 1,9 (q, 2H), 2,15 (m, 2H), 2,55 (m, 2H), 3,1 (m, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 4,0 (t, 2H), 4,1-4,3 (m, 5H), 4,9 (m, 1H), 6,9 (s, 1H), 7,3 (d, 1H), 7,4 (m, 3Н), 7,5 (d, 2H), 7,65 (d, 1H), 8,1 (m, 3H), 8,4 (t, 1H).
Пример 20
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-альфа-глутамин гидрохлорид
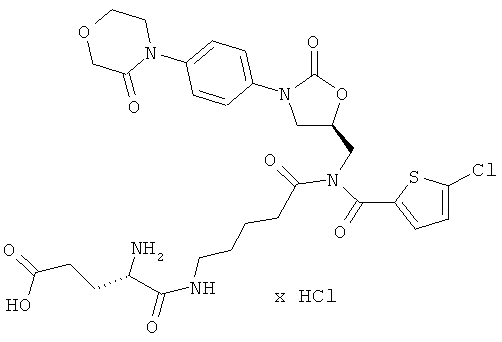
Стадия а)
350 мг (875 мкмоль) 5-трет-бутил-1-(2,5-диоксопирролидин-1-ил)-N-(трет-бутоксикарбонил)-L-глутамата совместно с 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 30 минут по каплям добавляют 50 мг (87 мкмоль) соединения из примера 4А, растворенного в 5 мл диметилформамида. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента смеси дихлорметан/этилацетат/метанол с соотношением компонентов сначала 150:50:5, затем 150:50:10 и наконец 150:50:20. Соответствующие фракции целевого соединения, которые еще содержат примеси, объединяют и концентрируют в вакууме. После этого остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 35 мг защищенного промежуточного соединения (выход 49% от теоретического).
ВЭЖХ (метод 2а): Rt=5,4 мин;
ЖХ-МС (метод 3): Rt=2,63 мин; m/z=820 (M+H)+.
Стадия b)
35 мг (43 мкмоль) полученного защищенного промежуточного соединения растворяют в дихлорметане, раствор смешивают с 1,5 мл безводной трифторуксусной кислоты, и компоненты перемешивают в течение 2 ч при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, остаток смешивают с 10 мл соляной кислоты (pH 3) и лиофилизируют. Получают 29 мг целевого соединения (выход 97% от теоретического).
ЖХ-МС (метод 12): Rt=1,72 мин; m/z=664 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,4 (m, 2Н), 1,6 (m, 2H), 1,9 (q, 2H), 2,3 (m, 2H), 2,55 (m, 2H), 3,1 (m, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 4,0 (t, 2H), 4,1-4,3 (m, 5H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,1 (m, 3H), 8,45 (t, 1H).
Пример 21
5-Хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)-N-[5-(L-сериламино)пентаноил]тиофен-2-карбоксамид гидрохлорид
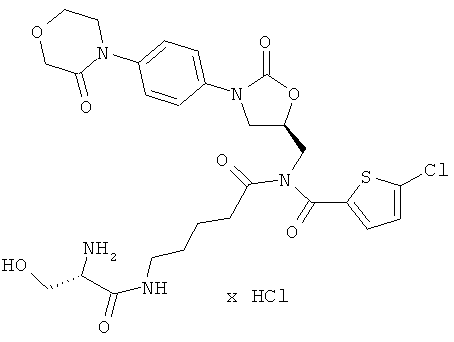
Стадия а)
350 мг (875 мкмоль) 2,5-диоксопирролидин-1-ил N-(трет-бутоксикарбонил)-L-серината совместно с 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 30 минут по каплям добавляют 50 мг (87 мкмоль) соединения из примера 4А, растворенного в 5 мл диметилформамида. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента сначала смесь дихлорметан/этилацетат (3:1), а затем смеси дихлорметан/этилацетат/метанол с соотношением компонентов сначала 150:50:5, затем 150:50:10 и наконец 150:50:20. Соответствующие фракции целевого соединения, которые еще содержат примеси, объединяют и концентрируют в вакууме. После этого остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 33 мг защищенного промежуточного соединения (выход 52% от теоретического).
ЖХ-МС (метод 12): Rt=2,87 мин; m/z=722 (M+H)+.
Стадия b)
33 мг (46 мкмоль) полученного защищенного промежуточного соединения растворяют в дихлорметане, раствор смешивают с 1,6 мл безводной трифторуксусной кислоты, и компоненты в течение 10 мин перемешивают при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, остаток смешивают с 5 мл соляной кислоты (pH 3) и лиофилизируют. Получают 24 мг целевого соединения (выход 80% от теоретического).
ЖХ-МС (метод 12): Rt=1,81 мин; m/z=622 (M+H)+;
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,4 (m, 2H), 1,6 (m, 2H), 2,55 (m, 2H), 3,1 (dt, 2H), 3,6-3,8 (m, 5H), 3,85 (dd, 1H), 4,1-4,3 (m, 5H), 4,95 (m, 1H), 5,4 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,1 (m, 3Н), 8,4 (t, 1H).
Пример 22
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-лейцинамид гидрохлорид
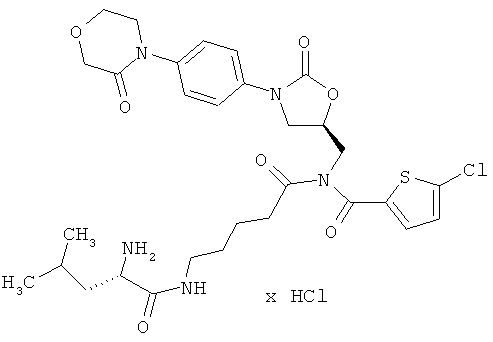
Стадия а)
287 мг (875 мкмоль) 2,5-диоксопирролидин-1-ил-N-(трет-бутоксикарбонил)-L-лейцината совместно с 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 30 минут по каплям добавляют 50 мг (87 мкмоль) соединения из примера 4А, растворенного в 5 мл диметилформамида. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента сначала смесь дихлорметан/этилацетат (3:1), а затем смеси дихлорметан/этилацетат/метанол с соотношением компонентов сначала 150:50:5, затем 150:50:10 и наконец 150:50:20. Соответствующие фракции целевого соединения, которые еще содержат примеси, объединяют и концентрируют в вакууме. После этого остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 10 мг защищенного промежуточного соединения (выход 15% от теоретического).
ЖХ-МС (метод 12): Rt=3,44 мин; m/z=748 (M+H)+.
Стадия b)
10,2 мг (14 мкмоль) защищенного промежуточного соединения растворяют в дихлорметане, полученный раствор смешивают с 0,5 мл безводной трифторуксусной кислоты, и компоненты перемешивают в течение 15 минут при комнатной температуре. Затем реакционный раствор концентрируют в вакууме при 25°С или более низкой температуре, остаток смешивают с 5 мл разбавленной соляной кислоты (pH 3) и лиофилизируют. Получают 7 мг целевого соединения (выход 73% от теоретического).
ЖХ-МС (метод 12): Rt=2,25 мин; m/z=648 (M+H)+.
Пример 23
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксобутил)-L-гистидинамид гидрохлорид
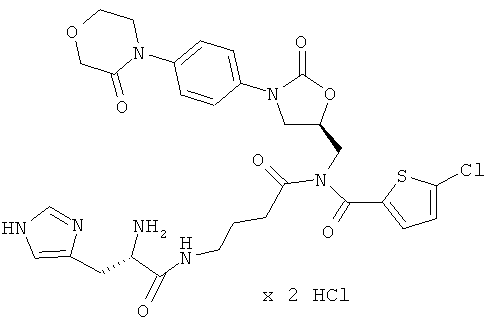
Стадия а)
195 мг (431 мкмоль) 2,5-диоксопирролидин-1-ил-N,1-бис(трет-бутоксикаронил)-L-гистидината совместно с 645 мкл 0,1М раствора этилдиизопропилмина в диметилформамида вводят в 3 мл диметилформамида. Затем в течение 1 ч по каплям добавляют 40 мг (72 мкмоль) соединения из примера 3А, растворенного в 17 мл диметилформамида. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента сначала ацетонитрил, а затем смесь ацетонитрил/вода (10:1). Соответствующие фракции целевого соединения, которые еще содержат примеси, объединяют и концентрируют в вакууме. Затем остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции, которые содержат смешанное бис-Boc-защищенное и моно-Boc-защищенное соединение, концентрируют и сушат в высоком вакууме. Получают 30 мг смешанного моно- и бис-Boc-защищенного промежуточного соединения (выход 55% от теоретического).
ВЭЖХ (метод 2): Rt=4,47 мин; 4.91 мин;
ЖХ-МС (метод 3): Rt=1,53 мин; m/z=758 (М+Н)+. Rt=2,45 мин; m/z=858 (M+H)+.
Стадия b)
30 мг полученного смешанного бис-Boc-защищенного и моно-Boc-защищенного промежуточного соединения смешивают с 2 мл безводной трифторксусной кислоты и перемешивают в течение 10 минут при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, и остаток двукратно лиофилизируют из соляной кислоты с pH 3. Получают 24 мг целевого соединения (выход 83% от теоретического).
ВЭЖХ (метод 2): Rt=4,07 мин;
ЖХ-МС (метод 13): Rt=2,41 мин; m/z=658 (M+H)+.
Пример 24
N-(5-{[(5-хлор-2-тиенил)карбонил]({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)-фенил]-1,3-оксазолидин-5-ил}метил)амино}-5-оксопентил)-L-альфа-аспарагин гидрохлорид
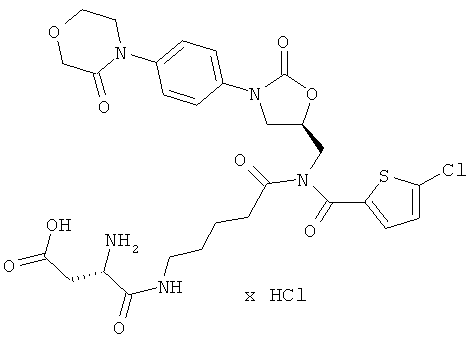
Стадия а)
338 мг (875 мкмоль) 5-трет-бутил-1-(2,5-диоксопирролидин-1-ил)-N-(трет-бутоксикарбонил)-L-аспартата совместно с 13,7 мкл этилдиизопропиламина вводят в 1 мл диметилформамида. Затем в течение 30 минут по каплям добавляют 50 мг (87 мкмоль) соединения из примера 4А, растворенного в 5 мл диметилформамида. После 30-минутного перемешивания при комнатной температуре реакционную смесь концентрируют, и остаток подвергают очистке флэш-хроматографией, используя в качестве элюента сначала смесь дихлорметан/этилацетат (3:1), а затем смеси дихлорметан/этилацетат/метанол с соотношением компонентов сначала 150:50:5, затем 150:50:10 и, наконец, 150:50:20. Соответствующие фракции целевого соединения, которые еще содержат примеси, объединяют и концентрируют в вакууме. После этого остаток подвергают повторной очистке препаративной ВЭЖХ (метод 1а). Соответствующие фракции концентрируют и сушат в высоком вакууме. Получают 36 мг защищенного промежуточного соединения (выход 51% от теоретического).
ВЭЖХ (метод 2а): Rt=5,4 мин;
ЖХ-МС (метод 12): Rt=3,52 мин; m/z=806 (M+H)+.
Стадия b)
36 мг (45 мкмоль) полученного защищенного промежуточного соединения растворяют в дихлорметане, раствор смешивают с 1,5 мл безводной трифторуксусной кислоты и перемешивают в течение 2 часов при комнатной температуре. Затем реакционную смесь концентрируют в вакууме при 25°С или более низкой температуре, остаток смешивают с 5 мл соляной кислоты (pH 3) и лиофилизируют. Получают 28 мг целевого соединения (выход 91% от теоретического).
1H-ЯМР (400МГц, ДМСО-d6): δ=1,4 (m, 2H), 1,6 (m, 2H), 2,7-2,9 (m, 4H), 3,1 (m, 2H), 3,7 (t, 2H), 3,8 (dd, 1H), 4,0 (t, 2H), 4,1-4,3 (m, 5H), 4,9 (m, 1H), 7,3 (d, 1H), 7,4 (d, 2H), 7,5 (d, 2H), 7,6 (d, 1H), 8,2 (m, 3H), 8,4 (t, 1H), 13,0 (m, 1H).
В. Определение растворимости, стабильности и способности действующего вещества к высвобождению
а) Определение растворимости
Испытуемое вещество суспендируют в воде или разбавленной соляной кислоте с показателем pH, равным 4. Полученную суспензию в течение 24 ч встряхивают при комнатной температуре. После 30-минутного ультрацентрифугирования с ускорением 224000g надосадочную жидкость разбавляют диметилсульфоксидом и анализируют методом ВЭЖХ. Количественное определение осуществляют по двухпозиционной калибровочной кривой испытуемого соединения в диметилсульфоксиде.
ВЭЖХ
Прибор Agilent 1100 с УФ-детектором DAD (G1315A), четырехходовым насосом (G1311A), автоматической пипеткой СТС HTS PAL, дегазатором (G1322A) и термостатом колонки (G1316A); колонка Kromasil C18, 60×2,1 мм, 3,5 мкм; температура 30°С; элюент А: вода+5 мл перхлорной кислоты в литре, элюент В: ацетонитрил; расход: 0,75 мл/мин; градиент: 0-0,5 мин 98% А, 2% В; линейно 0,5-4,5 мин 10% А, 90% В; 4,5-6 мин 10% А, 90% В; линейно 6,5-6,7 мин 98% А, 2% В; 6,7-7,5 мин 98% А, 2% В.
В таблице 1 приведена растворимость соединений, полученных в наиболее типичных примерах осуществления изобретения, в разбавленной соляной кислоте (pH 4).
Деструкция соединений в указанных растворах не наблюдалась.
Определенная при этом тестировании растворимость лежащего в основе биологически активной субстанции [соединение (А)] в разбавленной соляной кислоте (pH 4) составляет 8,1 мг/л.
b) Стабильность в буферных растворах с различными значениями показателя pH
Навеску испытуемого вещества (0,3 мг) помещают в пробирку для ВЭЖХ емкостью 2 мл и смешивают с 0,5 мл ацетонитрила. Для растворения испытуемого образца пробирку около 10 секунд подвергают воздействию ультразвука. Затем добавляют 0,5 мл соответствующего буферного раствора, и образец вновь обрабатывают ультразвуком.
Использовали следующие буферные растворы:
pH 4,0: 1 л подвергнутой ультратонкой фильтрации воды с показателем pH, установленным посредством 1н. соляной кислотой, 4,0,
pH 7,4: к 90 г хлорида натрия, 13,61 г гидрофосфата калия и 83,35 г 1М раствора едкого натра добавляли подвергнутую ультратонкой фильтрации воду до объема 1 л, а затем разбавляли в соотношении 1:10.
Образцы раствора объемом 5 мкл в течение 24 часов при 25°С ежечасно анализируют методом ВЭЖХ на содержание неизменившегося испытуемого вещества. Количественную оценку выполняют по выраженным в процентах площадям соответствующих пиков.
ВЭЖХ-метод:
Прибор Agilent 1100 с УФ-детектором DAD (G1312A), бинарным насосом (G1312A), автосамплером (G1329A), печью (G1316A), термостатом (G1330A); колонка Kromasil 100 С18, 60 мм × 2,1 мм, 3,5 мкм; температура колонки 30°С; элюент А: вода + 5 мл перхлорной кислоты в литре, элюент В: ацетонитрил; градиент: 0-1,0 мин 98% А, 2% В; 1,0-9,0 мин 2% А, 98% В; 9,0-13,0 мин 2% А, 98% В; 13,0-13,5 мин 98% А, 2% В; 13,5-15,0 98% А, 2% В; расход 0,75 мл/мин; УФ-детектирование при 210 нм.
В таблице 2 приведены отношения площадей пиков (F) в соответствующие моменты времени к площадям пиков в начальный момент времени для наиболее типичных примеров осуществления изобретения.
В результате этого испытания установлено, что при показателе pH 7,4 одновременно со снижением содержания испытуемого вещества происходит увеличение содержания соединения (А) (действующего вещества).
c) Стабильность in vitro в плазме крысы и человека*)
Определенный объем (например, 2,0 мл) помещенной в закрытую пробирку плазмы В нагревают на водяной бане до температуры 37°С. По достижении заданной температуры добавляют определенное количество испытуемого вещества в виде раствора (объем растворителя не превышает 2% от объема плазмы). Плазму встряхивают и тут же отбирают первый образец объемом от 50 до 100 мкл. Затем в течение двух часов инкубации в разные моменты времени отбирают еще от 4 до 6 аликвотных образца.
Для осаждения белка образцы плазмы смешивают с ацетонитрилом. После центрифугирования соответствующим методом ЖХ/МС-МС выполняют количественное определение испытуемого вещества и в некоторых случаях известных продуктов его расщепления в надосадочной жидкости.
*) Стабильность соединений в гепаринизованной крови крысы или человека определяют аналогично ее определению в плазме
d) Определение метаболической стабильности в гепатоцитах
Для обеспечения максимально линейных экспериментальных кинетических условий метаболическую стабильность испытуемых новых соединений по отношению к гепатоцитам определяют инкубированием соединений при пониженных концентрациях (предпочтительно менее 1 мкмоль) и незначительных количествах клеток (предпочтительно 1·106 в мл). С целью определения периода полувыделения (то есть деструкции соединений) методом ЖХ-МС в определенный промежуток времени отбирают семь образцов инкубационного раствора. На основании указанных полупериодов рассчитывают различные параметры очищения крови (CL) и значения Fmax (смотри ниже).
Показатели CL и Fmax представляют собой меру "фазы 1" и "фазы 2" метаболизма соединения в гепатоцитах. С целью максимально возможного устранения воздействия органического растворителя на ферменты в инкубационных составах его концентрацию в общем случае ограничивают 1% для ацетонитрила или 0,1% для диметилсульфоксида.
Для всех видов и пород животных число гепатоцитов в печени принимают равным 1,1·108 в расчете на грамм печени. Параметры CL, расчет которых основан на значениях периода полувыделения, выходящих за пределы инкубационного периода, который обычно составляет 90 минут, можно рассматривать лишь в виде грубого приближения.
Ниже приведены расчетные параметры их значения.
(QH означает специфичный для разных видов печеночный кровоток)

е) Исследование фармакокинетики на крысах Wistar (внутривенные инъекции)
В день дачи испытуемого вещества в яремную вену подопытных животных (мужских особей крыс Wistar с массой тела 200-250 г) под наркозом (Isofluran®) имплантируют катетер для отбора крови.
В день выполнения экспериментов в хвостовую вену стеклянным шприцем Hamilton® вводят определенную дозу испытуемого вещества в виде раствора (дача пилюли, длительность применения менее 10 секунд). В течение 24 часов после инъекции через катетер от 8 до 12 раз последовательно отбирают образцы крови. Для выделения плазмы образцы центрифугируют в гепаринизованных трубках. Для осаждения белка в соответствующий момент времени определенный объем плазмы смешивают с ацетонитрилом. После центрифугирования соответствующим методом ЖХ/МС-МС выполняют количественное определение испытуемого вещества и в некоторых случаях известных продуктов его расщепления в надосадочной жидкости.
На основании измеренных концентраций в плазме выполняют расчет фармакокинетических характеристик испытуемого вещества, соответственно высвобождающегося из него в качестве действующего вещества соединения (А), таких как AUC, Cmax, T1/2 (период полувыделения) и CL (коэффициент очищения крови).
f) Определение антитромботического действия на модели артериовенозного шунта для крыс
Мужским особям крыс (штамм HSD CPB:WU) натощак дают наркоз путем внутрибрюшинной инъекции раствора Ромпун/Кетавет (12 мг/кг/ 50 мг/кг). Тромбообразование в артериовенозном шунте провоцируют методом, описанным в публикации Р.С.Wong et al., Thrombosis Research, 83 (2), 117-126 (1996). С этой целью осуществляют открытое препарирование левой яремной вены и правой сонной артерии. В артерию вводят полиэтиленовый катетер длиной 8 см (РЕ60, фирма Becton-Dickinson), являющийся продолжением шланга из полимера Tygon длиной 6 см (R-3606, внутренний диаметр 3,2 мм, фирма Kronlab), который с целью формирования тромбогенной поверхности снабжен ворсованной и уложенной в виде двойной петли нейлоновой нитью (60×0,26 мм, фирма Berkley Trilene). В яремную вену вводят полиэтиленовый катетер длиной 2 см (РЕ60, фирма Becton-Dickinson), который соединяют полиэтиленовым катетером длиной 6 см (РЕ 160, фирма Becton-Dickinson) со шлангом из полимера Tygon. Шланги заполняют физиологическим раствором натрия хлорида. В течение 15 минут обеспечивают экстракорпоральную циркуляцию. Затем шунт удаляют и сразу же взвешивают тромбированную нейлоновую нить. Перед экспериментом определяют массу исходной нейлоновой нити. До начала экстракорпоральной циркуляции выполняют инъекцию испытуемого вещества (в виде раствора в физиологическом растворе поваренной соли с показателем pH, установленным 0,1Н соляной кислотой на уровне 4).
С. Примеры приготовления фармацевтических композиций
Предлагаемые в изобретении соединения могут быть преобразованы в следующие фармацевтические композиции:
Раствор для внутривенных инъекций
Предлагаемое в изобретении соединение растворяют в физиологически совместимом растворителе (например, изотоническом растворе хлорида натрия, 5% растворе глюкозы и/или 30% растворе полиэтиленгликоля PEG 400, показатель pH каждого из которых устанавливают в интервале от 3 до 5) в концентрации ниже точки насыщения. Раствор при необходимости подвергают фильтрованию в стерильных условиях и/или расфасовывают в стерилизованные и освобожденные от пирогенных примесей сосуды для инъекций.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИНОАЦИЛЬНЫЕ ПРОИЗВОДНЫЕ ПРОЛЕКАРСТВА И ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ТРОМБОЭМБОЛИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2435768C2 |
| ГЕТЕРОЦИКЛИЛАМИДОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 2006 |
|
RU2415851C2 |
| СОЛИ ГЕТЕРОЦИКЛИЛАМИДЗАМЕЩЕННЫХ ИМИДАЗОЛОВ С СУЛЬФОНОВОЙ КИСЛОТОЙ | 2012 |
|
RU2606639C2 |
| ЗАМЕЩЕННЫЕ АРИЛСУЛЬФОНАМИДЫ КАК ПРОТИВОВИРУСНЫЕ СРЕДСТВА | 2007 |
|
RU2423352C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ ДЛЯ ЛЕЧЕНИЯ КАРДИОВАСКУЛЯРНЫХ И ГЕМАТОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ, ИХ ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ | 2012 |
|
RU2611012C2 |
| АЦИЛИРОВАННЫЕ НОНАДЕПСИПЕПТИДЫ В КАЧЕСТВЕ ПРОИЗВОДНЫХ ЛИЗОБАКТИНА | 2005 |
|
RU2414477C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТА[B]БЕНЗОФУРАНА И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2415847C9 |
| ТИЕНОПИРАЗОЛЫ | 2004 |
|
RU2358978C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЛАМИДОМ ТИАЗОЛЫ, ПИРРОЛЫ И ТИОФЕНЫ | 2006 |
|
RU2425829C2 |
| ДИПЕПТИДНОЕ ПРОЛЕКАРСТВО, ЕГО ПРИМЕНЕНИЕ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2013 |
|
RU2629558C2 |
Изобретение относится к соединению формулы (I):
в которой n означает 1 или 2, Х означает атом кислорода, атом серы или NH, R1 означает боковую группу природной α-аминокислоты или ее гомологов или изомеров, выбранную из водорода, метила, пропан-2-ила, пропан-1-ила, 2-метил-пропан-1-ила, имидазол-4-илметила, гидроксиметила, 1-гидроксиэтила, карбоксиметила, 2-карбоксиэтила, карбамоилметила, 2-карбамоилэтила, 4-аминобутан-1-ила, 3-аминопропан-1-ила, 3-гуанидинопропан-1-ила, бензила или 4-гидроксибензила, R2 означает водород или метил, R3 означает водород, или R1 и R3 соединены между собой посредством группы (СН2)3- или (СН2)4- и совместно с атомами азота и углерода, к которым они присоединены, образуют пятичленное или шестичленное кольцо, а также их солям, сольватам и сольватам солей, которые предназначены для лечения и/или профилактики болезней, прежде всего тромбоэмболических заболеваний. 1 з.п. ф-лы, 2 табл., 24 пр.
1. Соединение формулы (I)
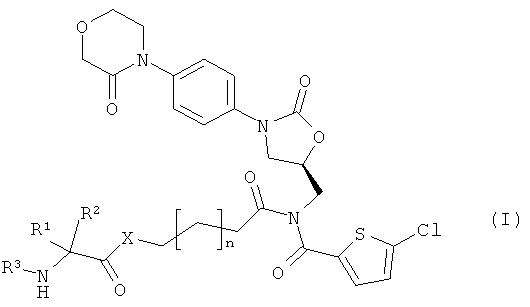
в которой n означает 1 или 2,
Х означает атом кислорода, атом серы или NH,
R1 означает боковую группу природной α-аминокислоты или ее гомологов или изомеров, выбранную из водорода, метила, пропан-2-ила, пропан-1-ила, 2-метил-пропан-1-ила, имидазол-4-илметила, гидроксиметила, 1-гидроксиэтила, карбоксиметила, 2-карбоксиэтила, карбамоилметила, 2-карбамоилэтила, 4-аминобутан-1-ила, 3-аминопропан-1-ила, 3-гуанидинопропан-1-ила, бензила или 4-гидроксибензила,
R2 означает водород или метил,
R3 означает водород,
или
R1 и R3 соединены между собой посредством группы (СН2)3- или (СН2)4- и совместно с атомами азота и углерода, к которым они присоединены, образуют пятичленное или шестичленное кольцо,
а также их соли, сольваты и сольваты солей.
2. Соединение формулы (I) по п.1, в которой
n означает 1 или 2,
Х означает NH,
R1 означает боковую группу природной α-аминокислоты или ее гомологов или изомеров, выбранную из водорода, метила, пропан-2-ила, 2-метил-пропан-1-ила, имидазол-4-илметила, гидроксиметила, 1-гидроксиэтила, карбоксиметила, 2-карбоксиэтила, карбамоилметила, 2-карбамоилэтила, 4-аминобутан-1-ила, бензила или 4-гидроксибензила,
R2 означает водород,
R3 означает водород,
а также их соли, сольваты и сольваты солей.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА АМИДНОГО ТИПА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ | 2001 |
|
RU2232761C2 |

