Данное изобретение относится к новым замещенным производным дигидропиразолонов для лечения и/или профилактики заболеваний, их применению для лечения и/или профилактики заболеваний кровообращения, сердечной недостаточности, анемий, хронических заболеваний почек и почечной недостаточности, а также к лекарственному средству, содержащему производное дигидропиразолона, и способу лечения и/или профилактики заболеваний сердечного кровообращения, сердечной недостаточности, анемий, хронических заболеваний почек и почечной недостаточности у человека и животных.
Недостаточное обеспечение организма человека или его части кислородом, которое либо неблагоприятно отражается на функционировании организма или его части вследствие продолжительности и/или масштаба этого процесса, либо полностью парализует его функции, называют гипоксией. Гипоксия может быть вызвана снижением содержания кислорода во вдыхаемом воздухе (например, при нахождении на большой высоте), нарушениями внешнего дыхания (например, вследствие нарушения функции легких или закупорки дыхательных путей), снижением минутного объема сердца (например, при сердечной недостаточности, острой перегрузке правой половины сердца при легочной эмболии), слишком низкой пропускной способностью кислорода в крови (например, вследствие малокровия (анемии) или интоксикации, например, монооксидом углерода), локальным снижением кровоснабжения вследствие тромбоза сосуда (типичные ишемические состояния, например, сердца, нижних конечностей или головного мозга, диабетические макро- и микроангиопатии) или также повышенной потребностью тканей в кислороде (например, вследствие усиленной работе мускулов или местными воспалениями) [Eder, Gedigk (авторы), Allgemeine Pathologie und pathologische Anatomie, 33-е издание, изд-во Springer, Berlin, 1990].
Организм человека условно способен приспосабливаться к ситуациям острого или хронического уменьшения снабжения кислородом. Наряду с немедленным откликом, который за счет контрольного механизма вегетативной нервной системы среди прочих включает увеличение минутного объема сердца и дыхания, а также локальное расширение кровеносных сосудов, гипоксия влечет за собой изменение транскрипции многочисленных генов. Функция продуктов, вырабатываемых генами, служит при этом компенсацией недостатка кислорода. Так многими энзимами усиленно экспримируются гликолиз и транспорт глюкозы 1, за счет чего усиливается анаэробное получение АТФ, что позволяет пережить недостаток кислорода [Schmidt, Thews (авторы), Physiologie des Menschen, 27-е издание, изд-во Springer, Berlin, 1997; Löffler, Petridea (авторы), Biochemie und Pathobiochemie, 7-е издание, изд-во Springer, Berlin, 2003].
Далее, гипоксия приводит к усиленной экспрессии васкулярного фактора роста эндотелиальных клеток, вследствие чего в тканях с кислородным голоданием стимулируется новообразование кровеносных сосудов (ангиогенез). Таким образом улучшается долговременное кровоснабжение ишемической ткани. При различных заболеваниях сердечного кровообращения и тромбозе сосудов эта противорегуляция очевидно происходит в недостаточной степени [Обзор у: Simons и Ware, Therapeutic angiogenesis in cardiovascular disease, Nat. Rev. Drug. Discov. 2 (11), 863-71 (2003)].
Далее, при систематической гипоксии усиленно экспримируется образующийся преимущественно в интерстициальных фибропластах почек пептидный гормон эритропоэтин. Этим стимулируется образование красных клеток крови в костном мозге и тем самым повышается пропускная способность кислорода в крови. Этот эффект необходим спортсменам-разрядникам при так называемых высотных тренировках. Снижение пропускной способности кислорода в крови, например, вследствие малокровия обычно вызывает уменьшение продуцирования эритропоэтин в почках. При некоторых формах анемии этот механизм регулирования может быть нарушен или уровень его номинального значения может устанавливаться ниже. Так, например, у пациентов, которые страдают почечной недостаточностью, хотя и продуцируется эритропоэтин в паренхиме почки, но в расчете на пропускную способность кислорода в крови в заметно меньших количествах, что имеет следствием так называемую ренальную (почечную) анемию. В частности, ренальная анемия, а также анемии, вызванные опухолями и ВИЧ-инфекцией, обычно лечатся путем парентерального применения рекомбинантного человеческого эритропоэтина (rhEPO). К настоящему времени этой дорогостоящей терапии нет альтернативы с орально применяемыми лекарственными средствами [Обзоры у: Eckardt, The potentiell of erythropoietin and related strategies to stimulate erythropoiesis, Curr. Opin. Investig. Drugs 2(8), 1081-5 (2001); Berns, Should the target hemoglobin for patients with chronic kidney disease treated with erythropoietic replacement therapy be changed?, Semin. Dial. 18 (1), 22-9 (2005)]. Последние исследования показали, что эритропоэтин наряду с его усиливающим эритропоэз действием оказывает также независимое от этого защитное (анти-апоптическое) действие на ткани, страдающие от кислородного голодания, в частности, на сердце и головной мозг. Далее, терапия эритропоэтином, согласно новым исследованиям, у пациентов с сердечной недостаточностью снижает тяжесть заболевания. [Обзоры у: Caiola и Cheng, Use of erythropoietin in heart failure management, Ann. Pharmacotber. 38 (12), 2145-9 (2004); Katz, Mechanisms and treatment of anemia in chronic heart failure, Congest. Heart. Fail. 10 (5), 243-7 (2004)].
Для описанных выше индуцированных гипоксией генов является общим, что наращивание их экспрессии при гипоксии вызывается так называемым танскрипционным фактором (HIF), индуцируемым гипоксией. В случае HIF речь идет о гетеродимерном танскрипционном факторе (HIF), который состоит из альфа- и бета-субъединиц. Описаны три HIF-альфа-изоформы, из которых HIF-1-альфа и HIF-2-альфа высоко гомологичны и значимы для индуцированной гипоксией экспрессии генов. В то время как обозначенная как ARNT (арилуглеводородный ядерный транслокатор рецепторов) бета-субъединица (описана в 2 изоформах) экспримирована конститутивно, экспрессия альфа-субъединицы зависит от содержания кислорода в клетке. При нормоксемии HIF-альфа-протеин поли-убиквитирует и затем протеасомально деградирует. При гипоксии эта деградация замедляется, так что HIF-альфа димеризуется с ARNT и его целевые гены могут активироваться. При этом HIF-димер соединен с так называемым отвечающим за гипоксию элементом (HRE) в регуляторной последовательности его целевых генов. Эти HRE определены с помощью консенсусной последовательности. Функциональные HRE доказаны в регуляторных элементах многочисленных индуцированных гипоксией генов [Обзоры у: Semenza, Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology, Trends Mol. Med. 7 (8), 345-50 (2001); Wenger и Gassmann, Oxygen(es) and the hypoxia-inducible factor-1, Biol. Chem. 378 (7), 609-16 (1997)].
Молекулярный механизм, лежащий в основе этого регулирования HIF-альфа, был разъяснен работами нескольких независимых друг от друга групп исследователей. Механизм сохранен с охватом видов: HIF-альфа гидролизуется с помощью обозначенных как PHD или EGLN подклассов кислородозависимых пролил-4-гидроксилаз на два отдельных пролильных остатка (Р402 и Р564 субъединицы человеческого HIF-1-альфа). В случае HIF-пролил-4-гидроксилаз речь идет о железозависимых, преобразованных с помощью 2-оксоглутарата диоксигеназах [Epstein et al., С. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107 (1), 43-54 (2001); Bruick и McKiught, A conserved family of prolyl-4-hydroxylases that modify HIF, Science 294 (5545), 1337-40 (2001); Ivan et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor, Proc. Natl. Acad. Sei. U.S.A. 99 (21), 13459-64 (2002)]. Энзимы впервые в 2001 г. аннотированы как пролил-гидроксилазы [Aravind и Koonin, The DNA-repair protein AIkB, EGL-9, and leprecan define new f amities of 2-oxoglutarate- and iron- dependent dioxygenases, Genome Biol. 2 (3), research0007.1-0007.8, Epub 2001 Feb 19].
К пролил-гидролизованной HIF-альфа-субъединице прикрепляется pVHL белок, подавляющий опухоли (Tumor Suppressor Protein), который вместе с элонгином В и С образует так называемый VBC-комплекс, который адаптирует HIF-альфа-субъединицу к Е3 убиквитин-лигазе. Так как пролил-4-гидроксилирование HIF-альфа-субъединицы и ее последующая деградация происходит в зависимости от внутриклеточной концентрации кислорода, HIF-пролил-4-гидроксилазы назвали также клеточным сенсором кислорода. Идентифицированы три изоморфы этого энзима: EGLN1/PHD2, EGLN2/PHD1 и EGLN3/PHD3. Два этих энзима (EGLN2/PHD1 и EGLN3/PHD3) сами транскрипционно индуцируются при гипоксии и, возможно, являются ответственными за наблюдаемое при хронической гипоксии снижение уровня HIF-alpha [Обзор у: Schofield и Ratcliffe, Oxygen sensing by HIF hydroxylases, Nat. Rev. Mol. Cell. Biol. 5 (5), 343-54 (2004)].
Избирательное фармакологическое торможение HIF-пролил-4-гидроксилаз влечет за собой усиление экспрессии HIF-зависящих целевых генов и, поэтому, является полезным для терапии многочисленных болезней. В частности, для заболеваний системы сердечного кровообращения следует ожидать улучшения в течении болезни от индукции новых кровеносных сосудов и изменения ситуации с обменом веществ в ишемическом органе от аэробного к анаэробному синтезу АТФ. Улучшение васкуляризации хронических ран способствует процессу заживления, особенно для трудно поддающегося лечению Ulcera cruris (хронической венозной недостаточности, сопровождающейся трофическими нарушениями) и других хронических кожных ран. Индукция аутогенного эритропоэтина при некоторых формах заболеваний, в частности у пациентов с почечной анемией, также представляет собой преследуемую терапевтическую цель.
Описанные к настоящему времени в научной литературе ингибиторы HIF-пролил-4-гидроксилазы не выполняют требований, предъявляемых к лекарственным средствам. При этом речь идет о конкурентных аналогах оксоглутарата (таких как, например, N-оксалилглицин), которые характеризуются очень слабым воздействия и поэтому в моделях in viva до сих пор не смогли проявить активности в смысле индукции целевых HIF-генов. Или речь идет о комплексообразователях с железом (хелатирующих агентах), таких как десферроксамин, которые действуют как неспецифические ингибиторы железосодержащих диоксигеназ и, хотя они вызывают индукцию целевых генов, как например эритропоэтина in vivo, за счет связывания в комплекс имеющегося железа явно противодействуют эритропоэзу.
Задача данного изобретения заключается в предоставлении новых соединений, которые могут использоваться для лечения заболеваний, в особенности кардиоваскулярных и гематологических заболеваний.
В рамках данного изобретения описываются соединения, которые действуют как специфические ингибиторы HIF-пролил-4-гидроксилаз и вследствие их специфического механизма действия in vivo после парентерального или орального применения приводят к индукции целевых HIF-генов, как, например, эритропоэтин, и вызванных ими биологических процессов, такого как, например, эритропоэз.
2-Гетероарил-4-арил-1,2-дигидропиразолоны с бактерицидным и/или фунгицидным действием описаны в ЕР 165448 и ЕР 212281. Применение 2-гетероарил-4-арил-1,2-дигидропиразолонов в качестве ингибиторов липоксигеназы для лечения болезней дыхательных путей, сердечного кровообращения и воспалительных заболеваний показано в ЕР 183159. В DE 2651008 описаны 2,4-дифенил-1,2-дигидропиразолоны с гербицидной активностью. О получении и фармакологических свойствах некоторых 2-пиридил-1,2-дигидропиразолонов сообщается в HeIv. Chim. Acta 49 (1), 272-280 (1966). В WO 96/12706, WO 00/51989 и WO 03/074550 описаны соединения с частично дигидропиразолоновой структурой для лечения различных заболеваний и в WO 2006/101903 опубликованы бипиразолы с гидрокси- или алкокси-заместителями для лечения нервно-психиатрических заболеваний. Далее, в WO 03/051833 и WO 2004/089303 описаны производные пиразола с гетероарильными заместителями для лечения боли и различных заболеваний ЦНС. В WO 2006/114213 описаны 2,4-дипиридил-1,2-дигидропиразолоны как ингибиторы HIF-пролил-4-гидроксилаз.
О рентгеновской кристаллической структуре соединения 3-метил-1-(пиридин-2-ил)-4-(1-пиридин-2-ил-3-метил-1H-пиразол-5-ил)-2H-3-пиразолин-5(1H)-он (другое название: 5,5'-диметил-2,2'-дипиридин-2-ил-1',2'-дигидро-2H,3'H-3,4'-бипиразол-3'-он) сообщалось в Acta Crystallogr., Section E: Structure Reports Online E57 (11), ol 126-ol 127 (2001) [Chem. Abstr. 2001:796190]. Синтез некоторых производных 3',5-диметил-2-фенил-1’-(1,3-тиазол-2-ил)- 1'Н,2Н-3,4'-бипиразол-5’-ола описан в Indian J. Heterocyclic Chem. 3 (1), 5-8 (1993) [Chem. Abstr. 1994: 323362]. О синтезе и таутомерии некоторых производных 4-(пиразол-5-ил)-пиразолин-5-она сообщается в J. Heterocyclic Chem. 27 (4), 865-870 (1990) [Chem. Abstr. 1991:428557]. Терапевтическое применение соединений, названных в этих публикациях, не описано. В WO 2007/008541 сообщается о соединении 2-трет.-бутил-1'-[4-(4-хлорфенил)-1,3-тиазол-2-ил]-3',5-диметил-1'H,2H-3,4'-бипиразол-5'-ол в качестве экспериментального примера.
Предметом данного изобретения являются соединения общей формулы (I)
 ,
,
в которой
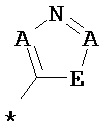
R1 означает гетероарильную группу формул
 ,
,  ,
,  или
или  ,
,
в которых
* означает место соединения с дигидропиразолоновым циклом,
А означает для каждого отдельного представителя C-R4 или N, при этом максимум два члена цикла А одновременно означают N, и
Е означает О, S или N-R5,
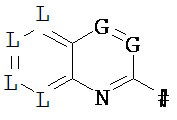
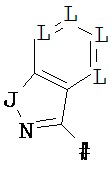
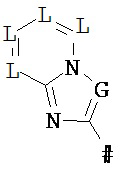
R2 означает гетероарильную группу формул
 ,
,  ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,
 ,
,  или
или  ,
,
в которых
# означает место соединения с дигидропиразолоновым циклом,
G означает дня каждого отдельного появления C-R6 или N, при этом максимум два члена цикла G одновременно означают N, и
J означает О, S иди N-R7, и
L означает для каждого отдельного появления C-R8 или N, при этом максимум два члена цикла L одновременно означают N, где
R4, R6 и R8 являются одинаковыми или разными и в каждом отдельном случае независимыми друг от друга и означают водород или заместители, выбранные из группы, включающей галоген, циано, нитро, (С1-С6)-алкил, (С3-С7)-циклоалкил, 4-10-членный гетероциклоалкил, фенил, 5-6-членный гетероарил, -C(=O)-R9, C(=O)-OR10, -C(=O)-NR11R12, -O-C(=O)-R13, -O-C(=O)-NR14R15, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -NR20-C(=O)-NR21R22, -NR23-SO2-R24, -SO2-R25, -SO2-NR26R27, -OR28, -SR29 и -NR30R31, где
(i) (С1-С6)-алкил, в свою очередь, может иметь от одного до трех заместителей, одинаковых или разных, выбранных из группы, включающей галоген, циано, оксо, (С3-С7)-циклоалкил, 4-10-членный гетероциклоалкил, фенил, 5-6-члснный гетероарил, -C(=O)-R9, C(=O)-OR10, -C(=O)-NR11R12, -O-C(=O)-R13, -O-C(=O)-NR14R15, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -NR20-C(=O)-NR21R22, -NR23-SO2-R24, -SO2-R25, -SO2-NR26R27, -OR28, -SR29 и -NR30R31,
при этом названные последними циклоалкильный, гетероциклоалкильный, фенильный и гетероарильный остатки, в свою очередь, могут соответственно иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей, галоген, циано, (С1-С4)алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
(ii) (С3-С7)-циклоалкил, 4-10-членный гетероциклоалкил, фенил и 5-6-членный гетероарил, в свою очередь, могут соответственно иметь от одного до трех заместителей, одинаковых иди разных, из группы, включающей (С1-С6)-алкил, галоген, циано, оксо, -C(=O)-R9, C(=O)-OR10, -C(=O)-NR11R12, -O-C(=O)-R13, -O-C(=O)-NR14R15, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -NR20-C(=O)-NR21R22, -NR23-SO2-R24, -SO2-R25, -SO2-NR26R27, -OR28, -SR29 и -NR30R31,
при этом названный последним алкильный остаток в свою очередь может иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей галоген, пиано, гидрокси, трифторметокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил, (С3-С7}-циклоалкил, 4-7-членный гетероциклоалкил, фенил и/или 5- или 6-членный гетероарил,
(iii) R9, R10, R11, R13, R14, R17, R19, R21, R24, R25, R26, R28, R29 и R30 независимо друг от друга при каждом отдельном появлении означают остаток, выбранный из группы, включающей водород, (С1-С6)-алкил, (С3-С7)циклоалкил, 4-10-членный гетероциклоалкил, фенил и 5- иди 6-членный гетероарил, причем
(С3-С7)-циклоалкил, 4-10-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, в свою очередь, могут соответственно иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей галоген, пиано, (С1-С4)алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (C1-C4)-алкоксикарбонил,
(С1-С6)-алкил может иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей галоген, пиано, гидрокси, трифторметокси, (C1-C4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил, (С3-С7)-циклоалкил, 4-7-членный гетероциклоалкил, фенил и/иди 5- или 6-членный гетероарил,
(iv) R12, R15, R16, R18, R20, R22, R23, R27 и R31 независимо друг от друга при каждом отдельном появлении означают остаток, выбранный из группы, включающей водород и (С1-С6)-алкил,
причем (С1-С6)-алкил может иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей галоген, пиано, гидрокси, трифторметокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил,
и/или где
(v) R11 и R12, R14 и R15, R16 и R17, R18 и R19, R20 и R21, R21 и R22, R23 и R24, R26 и R27 а также R30 и R31 соответственно попарно вместе с атомами, с которыми они связаны, могут образовывать 5- или 6-членное гетероциклоалкильное кольцо, которое может тлеть от одного до трех заместителей, одинаковых иди разных, выбранных из группы, включающей галоген, циано, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
и
R5 и R7 одинаковы иди различны и независимо друг от друга означают водород или выбраны из группы, включающей (С1-С6)-алкил, (С3-С7)циклоалкил, 4-7-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, где
(i) (С1-С6)-алкил, в свою очередь, может иметь от одного до трех заместителей, одинаковых или разных, выбранных из группы, включающей галоген, циано, оксо, (С3-С7)-циклоалкил, 4-7-членный гетероциклоалкил, фенил, 5- или 6-членный гетероарил, -C(=O)-R9, C(=O)-OR10, -C(=O)-NR11R12, -O-C(=O)-R13, -O-C(=O)-NR14R15, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -NR20-C(=O)-NR21R22, -NR23-SO2-R24, -SO2-R25, -SO2-NR26R27, -OR28, -SR29 и -NR30R31,
при этом названные последними циклоалкильный, гетероциклоалкильный, фенильный и гетероарильный остатки, в свою очередь, могут соответственно иметь до трех заместителей, одинаковых или разных, из группы, включающей, галоген, циано, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил, и
(ii) (С3-С7)циклоалкил, 4-7-членный гетероциклоалкил, фенил и 5- иди 6-членный гетероарил, в свою очередь, могут соответственно иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей (С1-С6)-алкил, галоген, циано, оксо, -C(=O)-R9, C(=O)-OR10, -C(=O)-NR11R12, -O-C(=O)-R13, -O-C(=O)-NR14R15, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -NR20-C(=O)-NR21R22, -NR23-SO2-R24, -SO2-R25, -SO2-NR26R27, -OR28, -SR29 и -NR30R31,
при этом названный последним алкильный остаток, в свою очередь, может иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей галоген, циано, гидрокси, трифторметокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил, (С3-С7)-циклоалкил, 4-7-членный гетероциклоалкил, фенил и/или 5- или 6-членный гетероарил,
где
(a) R9, R10, R11, R13, R14, R17, R19, R21, R24, R25, R26, R28, R29 и R30 независимо друг от друга при каждом отдельном появлении означают остаток, выбранный из группы, включающей водород, (С1-С6)-алкил, (С3-С7)циклоалкил, 4-7-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, причем
(С3-С7)циклоалкил, 4-7-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, в свою очередь, могут соответственно иметь до трех заместителей, одинаковых или разных, из группы, включающей галоген, пиано, (С1-С4)алкил, трифторметил, гидрокси, (С1-С4)алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил
(С1-С6)-алкил может иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей галоген, пиано, гидрокси, трифторметокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил, (С3-С7)циклоалкил, 4-7-членный гетероциклоалкил, фенил и/или 5- или 6-членный гетероарил,
(b) R12, R15, R16, R18, R20, R22, R23, R27 и R31 независимо друг от друга при каждом отдельном появлении означают остаток, выбранный из группы, включающей водород и (С1-С6)-алкил,
причем (С1-С6)-алкил может иметь от одного до трех заместителей, одинаковых или разных, выбранных из группы, включающей галоген, пиано, гидрокси, трифторметокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
и/или
(с) R11 и R12, R14 и R15, R16 и R17, R18 и R19, R20 и R21, R21 и R22, R23 и R24, R26 и R27 а также R30 и R31 соответственно попарно вместе с атомами, с которыми они связаны, могут образовывать 5- или 6-членное гетероциклоалкильное кольцо, которое может иметь от одного до трех заместителей, одинаковых или разных, выбранных из группы, включающей галоген, циано, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
и
R2 означает водород, (С1-С6)-алкил или (С3-С7)-циклоалкил,
а также их соли, сольваты и сольваты солей,
за исключением соединений
3-метил-1-(пиридин-2-ил)-4-(1-пиридин-2-ил-3-метил-1H-пиразол-5-ил)-2H-3-пиразолин-5(1H)-ол,
3',5-диметил-2-фенил-1'-(4-фенил-1,3-тиазол-2-ил)-1'H,2Н-3,4'-бипиразол-5'-ол,
3',5-диметил-2-фенил-1'-(4-тиофен-2-ил-1,3-тиазол-2-ил)-1'H,2H-3,4'-бипиразол-5'-ол,
3',5-диметил-1'-(4-метил-1,3-тиазол-2-ил)-2-фенил-1'H,2H-3,4'-бипиразол-5'-ол,
2-(4-хлорфенил)-3',5-диметил-1'-(4-фенил-1,3-тиазол-2-ил)-1'H,2H-3,4'-бипиразол-5'-ол
и
2-трет.-бутил-1'-[4-(4-хлорфенил)-1,3-тиазол-2-ил]-3',5-диметил-1'H,2H-3,4'-бипиразол-5'-ол.
Соединениями согласно изобретению являются соединения формулы (I) и их соли, сольваты и сольваты солей, охватываемые формулой (I) соединения приведенных далее формул и их соли, сольваты и сольваты солей, а также охватываемые формулой (I), названные далее в качестве примеров осуществления соединения и их соли, сольваты и сольваты солей, в общем и целом в случае названных ниже соединений, охватываемых формулой (I), речь не идет еще о солях, сольватах и сольватах солей.
Соединения согласно изобретению в зависимости от их строения могут существовать в стереоизомерной форме (энантиомеры, диастереомеры). Поэтому изобретение включает в себя энантиомеры или диастереомеры и их соответствующие смеси. Из таких смесей энантиомеров и/или диастереомеров можно известными методами выделить стереоизомерно однородные компоненты.
Поскольку соединения согласно изобретению могут встречаться в таутомерных формах, данное изобретение включает все таутомерные формы.
В качестве солей в рамках данного изобретения предпочтительными являются соединения, физиологически не вызывающие опасений. Включены также соли, которые сами не пригодны для фармацевтического использования, но, например, могут применяться для выделения или очистки соединений согласно изобретению.
Физиологически не вызывающие опасений соли соединений согласно изобретению включают соли, полученные присоединением минеральных кислот, карбоновых кислот и сульфокислот, например, соли хлористоводородной, бромистоводородной, серной, фосфорной кислот, метансульфокислоты, этансульфокислоты, толуолсульфокислоты, бензолсульфокислоты, нафталиндисульфокислоты, уксусной, трифторуксусной, пролионовой, молочной, винной, яблочной, лимонной, фумаровой, малеиновой и бензойной кислот.
Физиологически не вызывающие опасений соли соединений согласно изобретению включают также соли обычных оснований, такие как, например и предпочтительно, соли щелочных металлов (например, натриевые и калийные соли), соли щелочноземельных металлов (например, кальциевые и магниевые соли) и соли аммония, ведущие свое происхождение от аммиака или органических аминов с числом углеродных атомов от 1 до 16, таких как, например и предпочтительно, этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метилморфолин, аргинин, лизин, этилендиамин и N-метилпиперидин.
Сольватами в рамках изобретения называют такие формы соединений согласно изобретению, которые в твердом или жидком состоянии образуют комплекс с молекулами растворителя за счет координации. Гидраты представляют собой специальную форму сольватов, у которых координация происходит с водой. В качестве сольватов в рамках данного изобретения предпочтительными являются гидраты.
Кроме того, данное изобретение включает также пролекарства соединений согласно данному изобретению. Понятие «пролекарство» охватывает соединения, которые сами могут быть биологически активными или неактивными, но во время нахождения в организме превращаются в соединения согласно изобретению (например, путем метаболизма или гидролитически).
В рамках данного изобретения заместители, если ничего другого не оговорено, имеют следующие значения:
(C1-С6)-Алкил и (С1-С4)-алкил в рамках данного изобретения означают неразветвленный или разветвленный алкильный остаток с числом углеродных атомов от 1 до 6 или от 1 до 4.
Предпочтительным является неразветвленный или разветвленный алкильный остаток с числом углеродных атомов от 1 до 4. Для примера и предпочтительно следует назвать: метил, этил, н-пропил, изопропил, н-бутил, изо-бутил, втор.-бутил, трет.-бутил, 1-этилпропил, н-пентил и н-гексил.
(C1-С6)-Алкокси и (С1-С4)-алкокси в рамках данного изобретения означают неразветвленный или разветвленный алкоксильный остаток с числом углеродных атомов от 1 до 6 или от 1 до 4. Предпочтительным является неразветвленный или разветвленный алкоксильный остаток с числом углеродных атомов от 1 до 4. Для примера и предпочтительно следует назвать: метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет.-бутокси, н-пентокси и н-гексокси.
Моно-(С1-С6)-алкиламино и моно-(С1-С4)-алкиламино в рамках данного изобретения означают аминогруппу с одним неразветвленным или разветвленным алкильным заместителем, имеющим 1-6 или 1-4 атома углерода. Предпочтительным является неразветвленный или разветвленный остаток моноалкиламина с числом углеродных атомов от 1 до 4. Для примера и предпочтительно следует назвать: метиламино, этиламино, н-пропиламино, изопропиламино, н-бутиламино, трет.-бутиламино, н-пентиламино и н-гексиламино.
Ди-(С1-С6)-алкиламино и ди-(С1-С4)-алкиламино в рамках данного изобретения означают аминогруппу с двумя неразветвленными или разветвленными алкильными заместителями, имеющими 1-6 иди 1-4 атома углерода. Предпочтительными являются неразветвленные или разветвленные диалкиламино-остатки с числом углеродных атомов от 1 до 4. Для примера и предпочтительно следует назвать: N,N-диметиламино, N,N-диэтиламино, N-этил-N-метиламино, N-метил-N-н-пропиламино, N-изопропил-N-н-пропиламино, N,N-диизопропиламино, N-н-бутил-N-метиламино, N-трет.-бутил-N-метиламино, N-метил-N-н-пентиламино и N-н-гексил-N-метиламино.
(С1-С6)-Алкоксикарбонил и (C1-C4)-алкоксикарбонил в рамках данного изобретения означают неразветвленный иди разветвленный алкоксильный остаток с числом углеродных атомов от 1 до 6 или от 1 до 4, который соединен через карбонильную группу. Предпочтительным является неразветвленный или разветвленный алкоксикарбонильный остаток с числом углеродных атомов от 1 до 4 в алкокси-группе. Для примера и предпочтительно следует назвать: метоксикарбонил, этоксикарбонил, н-пропоксикарбонил, изопропоксикарбонил, н-бутоксикарбонил и трет.-бутоксикарбонил.
(С3-С7)-Циклоалкил и (С3-С6)-циклоалкил в рамках данного изобретения означают моноциклический, насыщенный карбоцикл с числом углеродных атомов в цикле от 3 до 7 или от 3 до 6. Для примера и предпочтительно следует назвать: циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
4-10-Членный гетероциклоалкил в рамках данного изобретения означает моно- или при необходимости бициклический, насыщенный и имеющий одну двойную связь гетероцикл с общим числом атомов в цикле от 4 до 10, который содержит два атома в цикле из ряда N, О и/или S и присоединен по атому углерода из цикла иди при необходимости по атому азота из цикла. Для примера следует назвать: азетидинил, оксетанил, тиетанил, пирролидинил, пирролинил, пиразолидинил, дигидропиразолил, тетрагидрофуранил, тиоланил, 1,3-оксазолидинил, 1,3-тиазолидинил, пиперидинил, тетрагидропиридил, пиперазинил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, 1,4-диоксанил, 1,4-диоксанил, морфолинил, тиоморфолинил, гексагидроазепинил, гексагидро-1,4-диазепинил, октагидроазолинил, октагидропирроло[3,4-b]пирролил, октагидроизоиндолил, октагидропирроло[3,4-b]пиридил, гексагидропирроло[3,4-с]пиридил, октагидропирроло[1,2-а]пирацинил, декагидроизохинолинил, октагидропиридо[1,2-а]пиразинил, 7-азабицикло[2.2.1]гептанил, 3-азабицикло[3.2.0]гептанил, 3-азабицикло[3.2.1]октанил, 8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил. Предпочтительным в рамках изобретения является моноциклический, насыщенный 4-7-членный гетероциклоалкильный остаток с общим числом атомов в цикле от 4 до 7, который имеет один или два гетероатома в цикле из ряда N, О и/или S и присоединен по атому углерода из цикла или при необходимости по атому азота из цикла. Для примера следует назвать: азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, тетрагидрофуранил, тиоланил, 1,3-оксазолидинил, пиперидинил, пиперазинил, тетрагидропиранил, тетрагидротиопиранил, 1,3-диоксанил, 1,4-диоксанил, морфолинил, тиоморфолинил, гексагидроазепинил, гексагидро-1,4-диазепинил. Особенно предпочтительным является 4-6-членный гетероциклоалкильный остаток с общим числом атомов в цикле от 4 до 6, который имеет один или два гетероатома в цикле из ряда N и/или О, такой как, например, пирролидинил, тетрагидрофуранил, пиперидинил, пиперазинил, тетрагидропиранил и морфолинил.
5- или 6-Членный гетероарил в рамках изобретения означает ароматический гетероцикл (гетероароматическое соединение) с общим числом атомов в цикле 5 или 6, который содержит до четырех одинаковых или разных атомов в цикле из ряда N, О и/или S и присоединен по атому углерода из цикла или при необходимости по атому азота из цикла. Для примера следует назвать: фурил, пирролил, тиснил, пиразолил, имидазолил, тиазолил, оксазолил, изоксазолил, изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридил, пиримидинил, пиридазинил, пиразинид, триазинил.
Предпочтительными являются 5- иди 6-членные гетероарильные остатки, содержащие до трех гетероатомов в цикле из ряда N, О и/или S, такие как, например, фурил, тиснил, тиазолил, оксазолил, изотиазолил, изоксазолил, пиразолил, имидазолил, триазолил, оксадиазолил, тиадиазолил, пиридил, пиримидинил, пиридазинил, пиразинил.
Галоген в рамках данного изобретения включает фтор, хлор, бром и йод. Предпочтительными являются фтор, хлор и бром, особенно предпочтительны фтор и хлор.
Если в соединениях согласно изобретению остатки являются замещенными, то остатки, если не определено ничего другого, могут иметь один или два заместителя. В рамках данного изобретения действительно то, что для всех остатков, которые встречаются многократно, их значения не зависят друг от друга. Предпочтительным является замещение одним, двумя или тремя одинаковыми или разными заместителями. Особенно предпочтительным является замещение одним или двумя одинаковыми или разными заместителями.
В рамках данного изобретения предпочтительными являются соединения формулы (I), в которой
R1 означает гетероарильную группу формулы
или
где
* означает место соединения с дигидропиразолоновым циклом,
А означает при каждом появлении C-R4 или N, при этом максимум два члена А в цикле равны N, где
R4 в каждом отдельном случае, независимо друг от друга, означает водород или заместитель, выбранный из группы, включающей фтор, хлор, бром, циано, нитро, (С1-С4)-алкил, гидрокси, (C1-C4)-алкокси, трифторметокси, амино, моно-(С1-С6)-алкиламино, ди-(С1-С6)-алкиламино, гидроксикарбонил и (С1-С6)-алкоксикарбонил,
при этом названный (С1-С6)-алкильный остаток, в свою очередь, может иметь до трех одинаковых или разных заместителей, выбранных из группы, включающей фтор, хлор, бром, циано, гидрокси, трифторметокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и (С1-С4)-алкоксикарбонил, и
Е означает О, S или N-R5, где
R5 означает водород или (С1-С6)-алкил,
R2 означает гетероарильную группу формулы
 или
или
где
# означает место соединения с дигидропиразолоновым циклом,
G соответственно означает C-R6 или N, при этом не более одного из двух членов цикла G означают N, в котором
R6 в каждом отдельном случае, независимо друг от друга, означает водород или заместитель, выбранный из группы, включающей фтор, хлор, бром, циано, (С1-С6)-алкил, (С1-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил, 5- или 6-членный гетероарил, -C(=O)-OR10, -C(=O)-NR11R12, -O-С(=O)-R13, -O-C(=O)-NR14R15, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -NR20-C(=O)-NR21R22, -NR23-SO2-R24, -OR28 и -NR30R31, где
(i) (С1-С6)-алкил, в свою очередь, может иметь от одного до трех заместителей, одинаковых или разных, выбранных из группы, включающей фтор. хлор, бром, циано, (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил, 5- или 6-членный гетероарил, -C(=O)-OR10, -C(=O)-NR11R12, -O-С(=O)-R13, -O-C(=O)-NR14R15, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -NR20-C(=O)-NR21R22, -NR23-SO2-R24, -OR28 и -NR30R31,
при этом названные последними циклоалкильный. гетероциклоалкильный, фенильный и гетероарильный остатки, в свою очередь, могут соответственно иметь до двух, одинаковых или разных, заместителей, выбранных из группы, включающей фтор, хлор, бром, циано, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
(ii) (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, в свою очередь, могут соответственно иметь один или два одинаковых или разных заместителя, выбранных из группы, включающей фтор, хлор, бром, циано, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
(iii) R10, R11, R13, R14, R17, R19, R21, R24, R28 и R30 независимо друг от друга при каждом отдельном появлении означают остаток, выбранный из группы, включающей водород, (С1-С6)-алкил, (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, причем
(С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, в свою очередь, могут соответственно иметь до трех заместителей, одинаковых или разных, из группы, включающей фтор, хлор, бром, циано, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (C1-C4)-алкоксикарбонил
(С1-С6)-алкил может иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей фтор, хлор, бром, циано, гидрокси, трифторметокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил, (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил и/или 5- или 6-членный гетероарил,
(iv) R12, R15, R16, R18, R20, R22, R23 и R31 независимо друг от друга при каждом отдельном появлении означают остаток, выбранный из группы, включающей водород и (С1-С6)-алкил,
причем (С1-С6)-алкил может иметь один или два заместителя, одинаковых или разных, выбранных из группы, включающей фтор, хлор, бром, циано, гидрокси, трифторметокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
и/или где
(v) R11 и R12, R14 и R15, R16 и R17, R18 и R19, R20 и R21, R21 и R22, R23 и R24, а также R30 и R31 соответственно попарно вместе с атомами, с которыми они связаны, могут образовывать 5- или 6-членное гетероциклоалкильное кольцо, которое может иметь один или два заместителя, одинаковых или разных, выбранных из группы, включающей, фтор, хлор, бром, циано, (С1-С4)алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)алкоксикарбонил,
и
J означает О или S,
и
R3 означает водород или метил,
а также их соли, сольваты и сольваты солей.
Предпочтительными в рамках данного соединения являются также соединения формулы (I), в которой
R1 означает гетероарильную группу формулы
или
где
* означает место соединения с дигидропиразолоновым циклом,
А означает при каждом появлении C-R4 или N, при этом максимум один из членов цикла А равен N, где
R4 в каждом отдельном случае, независимо друг от друга, означает водород или заместитель, выбранный из группы, включающей фтор, хлор, бром, циано, нитро, (С1-С6)-алкил, гидрокси, (С1-С6)-алкокси, трифторметокси, амино, моно-(С1-С6)-алкиламино, ди-(С1-С6)-алкиламино, гидроксикарбонил и (С1-С6)-алкоксикарбонил,
при этом названный (С1-С6)-алкильный остаток, в свою очередь, может иметь до трех, одинаковых или разных, заместителей, выбранных из группы, включающей фтор, гидрокси, трифторметокси, (С1-С4)алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)алкиламино, гидроксикарбонил и/или (С1-С4)алкоксикарбонил,
и
Е означает О или S,
R2 означает гетероарильную группу формулы
или
где
# означает место соединения с дигидропиразолоновым циклом,
G соответственно означает C-R6 или N, при этом не более одного из двух членов цикла G означают N, где
R6 в каждом отдельном случае, независимо друг от друга, означает водород или заместитель, выбранный из группы, включающей фтор, хлор, бром, циано, (С1-С6)-алкил, (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил, 5- или 6-членный гетероарил, -C(=O)-OR10, -C(=O)-NR11R12, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -OR28 и -NR30R31, где
(i) (С1-С6)-алкил, в свою очередь, может иметь от одного до трех заместителей, одинаковых или разных, выбранных из группы, включающей фтор, (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, 5- или 6-членный гетероарил, -C(=O)-OR10, -C(=O)-NR11R12, -NR16-C(=O)-R17, -NR18-C(=O)-OR19, -OR28 и -NR30R31,
при этом названные последними циклоалкильный, гетероциклоалкильный и гетероарильный остатки, в свою очередь, могут соответственно иметь до двух одинаковых или разных заместителей, выбранных из группы, включающей фтор, хлор, бром, циано, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
(ii) (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, в свою очередь, могут соответственно иметь один или два одинаковых или разных заместителя, выбранных из группы, включающей фтор, хлор, бром, циано, (С1-С6)-алкил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (C1-C4)-алкоксикарбонил,
при этом (С1-С6)-алкил, в свою очередь, может иметь до трех заместителей, одинаковых или разных, выбранных из группы, включающей фтор, гидрокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил, (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил и/или 5-или 6-членный гетероарил,
(iii) R10, R11, R17, R19, R28 и R30 независимо друг от друга при каждом отдельном появлении означают остаток, выбранный из группы, включающей водород, (C1-C6)-алкил, (С3-С6)-циклоалкил и 4-6-членный гетероциклоалкил, причем
(С3-С6)-циклоалкил и 4-6-членный гетероциклоалкил, в свою очередь, могут соответственно иметь до трех заместителей, одинаковых или разных, из группы, включающей фтор, (С1-С4)алкил, трифторметил, гидрокси, (С1-С4)-алкокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил
(С1-С6)-алкил может иметь от одного до трех заместителей, одинаковых или разных, из группы, включающей фтор, гидрокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил, (С3-С6)-циклоалкил, 4-6-членный гетероциклоалкил, фенил и/или 5- или 6-членный гетероарил,
(iv) R12, R16, R18 и R31 независимо друг от друга при каждом отдельном появлении означают остаток, выбранный из группы, включающей водород и (С1-С6)-алкил, причем (С1-С6)-алкил может иметь один или два заместителя, одинаковых или разных, выбранных из группы, включающей фтор, гидрокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
и/или где
(v) R11 и R12, R16 и R17, R18 и R19, а также R30 и R31 соответственно попарно вместе с атомами, с которыми они связаны, могут образовывать 5- или 6-членное гетероциклоалкильное кольцо, которое может иметь один или два заместителя, одинаковых или разных, выбранных из группы, включающей, фтор, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4-алкокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
и
J означает О или S,
и
R3 означает водород,
а также их соли, сольваты и сольваты солей.
Особенно предпочтительными в рамках данного соединения являются соединения формулы (I), в которой
R1 означает гетероарильную группу формулы
 или
или 
где
* означает место соединения с дигидропиразолоновым циклом,
и
R4 означает водород, фтор, хлор, бром, циано, (С1-С4)-алкил, трифторметил, гидроксиметил, (С1-С4)алкокси, трифторметокси, гидроксикарбонил и (С1-С4)-алкоксикарбонил,
R2 означает гетероарильную группу формулы
 или
или 
где
# означает место соединения с дигидропиразолоновым циклом,
и
R6, R6A и R6B являются одинаковыми или разными и независимо друг от друга означают водород или заместитель, выбранный из группы, включающей фтор, хлор, бром, циано, (C1-C6)-алкил, трифторметил, гидрокси, (С1-С6)-алкокси, трифторметокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил, 4-6-членный гетероциклоалкил, фенил и 5- или 6-членный гетероарил, причем
(С1-С6)-алкил, в свою очередь, может иметь заместитель, такой как гидрокси, (С1-С4)-алкокси или амино
и
4-6-членный гетероциклоалкил, фенил и 5- или 6-чденный гетероарил, в свою очередь, может соответственно иметь один или два заместителя, одинаковых иди разных, таких как фтор, хлор, бром, циано, (С1-С4)алкил, трифторметил, гидрокси, (С1-С4)-алкокси, трифторметокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
и
R3 означает водород,
а также их соли, сольваты и сольваты солей.
Также наиболее предпочтительными в рамках данного изобретения являются соединения формулы (I), в которой
R1 означает гетероарильную группу формулы

где
* означает место соединения с дигидропиразолоновым циклом,
и
R4 означает водород фтор, хлор, бром, циано, (С1-С4)-алкил, трифторметил, гидроксиметил, (С1-С4)-алкокси, трифторметокси, гидроксикарбонил или (С1-С4)алкоксикарбонил,
R2 означает гетероарильную группу формул
или
где
# означает место соединения с дигидропиразолоновым циклом,
и
R6, R6A и R6B являются одинаковыми или разными и независимо друг от друга означают водород или заместитель, выбранный из группы, включающей фтор, хлор, бром, циано, (C1-C6)-алкил, трифторметил, гидрокси, (С1-С6)-алкокси, трифторметокси, амино, моно(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил, (С1-С4)-алкоксикарбонил и 4-6-члснный гетероциклоалкил, причем
(С1-С6)-алкил, в свою очередь, может иметь заместитель, такой как гидрокси, (C1-C4)-алкокси или амино
и
4-6-членный гетероциклоалкил, в свою очередь, может соответственно иметь один или два заместителя, одинаковых или разных, таких как фтор, циано, (С1-С4)-алкил, трифторметил, гидрокси, (С1-С4)-алкокси, оксо, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино, гидроксикарбонил и/или (С1-С4)-алкоксикарбонил,
и
R3 означает водород,
а также их соли, сольваты и сольваты солей.
Подробные определения остатков, приведенные в соответствующих комбинациях и предпочтительных комбинациях остатков, заменяются независимо от соответствующих указанных комбинаций остатков любыми определениями остатков из других комбинаций.
Наиболее предпочтительными являются комбинации двух или более из выше упомянутой предпочтительной области.
Производные 1,2-дигидролиразол-3-онов формулы (I) также могут существовать в таутомерной форме 1H-пиразол-5-олов (I’) (см. нижеследующую схему 1); обе таутомерные формы включаются в данное изобретение.
Схема 1

Следующим предметом изобретения является способ получения соединений формулы (I) согласно изобретению, отличающийся тем, что соединение формулы (II)
 ,
,
в которой R1 и R3 имеют указанные выше значения и
Z1 означает метил или этил,
взаимодействует в инертном растворителе при необходимости в присутствии кислоты с соединением формулы (III)
 ,
,
в которой R2 имеет указанное выше значение,
с образованием соединений формулы (IV)

в которой Z1, R1, R2 и R3 имеют указанные выше значения,
которые уже при этих условиях проведения реакции или в последующей стадии реакции под влиянием оснований циклизуются с образованием соединений формулы (I),
и соединения формулы (I) при необходимости с помощью соответствующих (i) растворителей и/или (ii) оснований или кислот переводятся в их сольваты, соли и/или сольваты солей.
Соединения формулы (I) согласно изобретению, в которых R3 означает водород, могут также быть получены следующим образом: сначала соединение формулы (V)
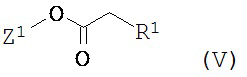 ,
,
в которой Z1 и R1 имеют указанные выше значения,
конденсируется с соединением формулы (VI)
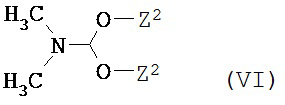 ,
,
в которой
Z2 означает метил или этил,
с образованием соединения формулы (VII)
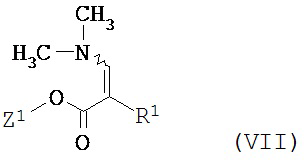 ,
,
в которой Z1 и R1 имеют указанные выше значения,
и затем в присутствии кислоты взаимодействует с соединением формулы (III) с образованием соединений формулы (IV-A)
 ,
,
в которой Z1, R1 и R2 имеют указанные выше значения,
которые уже при этих условиях проведения реакции или в последующей стадии реакции под влиянием оснований циклизуются с образованием соединений формулы (I), в которых R3 означает водород.
Другие соединения согласно изобретению при необходимости могут быть получены путем превращений функциональных групп некоторых заместителей, в частности обозначенных как R1 и R2, исходя из полученных указанными выше способами соединений формулы (I). Эти превращения осуществляются обычными, известными специалисту методами и включают, например, такие реакции, как нуклеофильное или электрофильное замещение, окисление, восстановление, гидрирование, катализируемые переходными металлами реакции сочетания, алкилирование, ацилирование, аминирование, этерификация, расщепление сложного эфира, образование простого эфира, расщепление простого эфира, образование карбонамидов, сульфонамидов, карбаматов и мочевин, а также введение и удаление временных защитных групп.
В качестве инертных растворителей для стадий способа (II)+(III)→(IV), (IV)→(I), (VII)+(III)→(IV-A) и (IV-A)→(I) годятся, в частности, простые эфиры, такие как, диэтиловый эфир, метил-трет.-бутиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран и диоксан, или спирты, такие как метанол, этанол, н-пропанол, изо-пропанол, н-бутанол и трет.-бутанол. Предпочтительно используются метанол, этанол, тетрагидрофуран или смеси этих растворителей.
Стадия способа (V)+(VI)→(VII) предпочтительно осуществляется в диметилформамиде в качестве растворителя или также в присутствии избытка (VI) без другого растворителя. При необходимости реакцию можно проводить также при микроволновом облучении. В общем случае взаимодействие происходит в области температур от +20°С до +150°С, предпочтительно при температуре от +80°С до +120°С [ср. также J.P. Bazureau et al., Synthesis 1998, 967; там же 2001 (4), 581].
Стадии способа (II)+(III)→(IV) и (VII)+(III)→(IV-A) могут осуществляться при необходимости при добавлении кислот. Для этого годятся обычные неорганические или органические кислоты, такие как, например, хлористый водород, уксусная кислота, трифторуксусная кислота, метансульфокислота, п-толуолсульфокислота или 10-камфор-сульфокислота. Предпочтительно используются уксусная кислота или, особенно, 10-камфорсульфокислота иди п-толуолсульфокислота.
Взаимодействие (II)+(III)→(IV) в общем случае происходит при температурах от 0°С до +100°С, предпочтительно при температуре от +10°С до +50°С. Реакция (VII)+(III)→(IV-A) в общем случае происходит при температурах от +20°С до +120°С, предпочтительно при температуре от +50°С до +100°С.
Последовательности способа (II)+(III)→(IV)→(I) и (VII)+(III)→(IV-A)→(I) могут осуществляться в две стадии иди также как реакция в одной колбе без выделения промежуточных продуктов (IV) и (IV-A). Для последнего варианта годится, в частности, взаимодействие компонентов при микроволновом облучении; при этом реакция в общем случае происходит при температуре от +50°С до +200°С, предпочтительно при температуре от +100°С до +180°С.
Отчасти замыкание кольца с образованием соединений (I) происходит уже при получении (IV) и (IV-A); тогда циклизацию при необходимости можно усовершенствовать путем обработки реакционной смеси in situ основанием.
В качестве основания для такой отдельной стадии циклизации (IV)→(I) и (IV-A)→(I) годятся обычные неорганические и органические основания. К ним относятся гидроксиды щелочных металлов, такие как, например, гидроксиды натрия или калия, карбонаты щелочных и щелочноземельных металлов, такие как карбонаты натрия, калия, кальция или цезия, алкоголяты щелочных металлов, такие как метанолаты натрия или калия, этанолаты натрия или калия, трет.-бутилаты натрия или калия или гидриды щелочных металлов, такие как гидрид натрия. Предпочтительно применяются метанолат натрия или этанолат натрия.
Индуцированные основаниями реакции (IV)→(I) и (IV-A)→(I) в общем случае происходят при температуре от 0°С до +60°С, предпочтительно при температуре от 0°С до +30°С.
Все стадии способа могут осуществляться при нормальном, повышенном или при пониженном давлении (например, от 0.5 до 5 бар). В общем случае работают при нормальном давлении.
Соединения формулы (II) можно получать обычными, известными из литературы методами С-ацилирования сложных эфиров карбоновых кислот из соединений формулы (V). Соединения формул (III), (V) и (VI) являются коммерчески доступными, известными из литературы или могут быть получены по аналогии со способами, описанными в литературе.
Получение соединений согласно изобретению можно пояснить следующей схемой 2:
Схема 2:
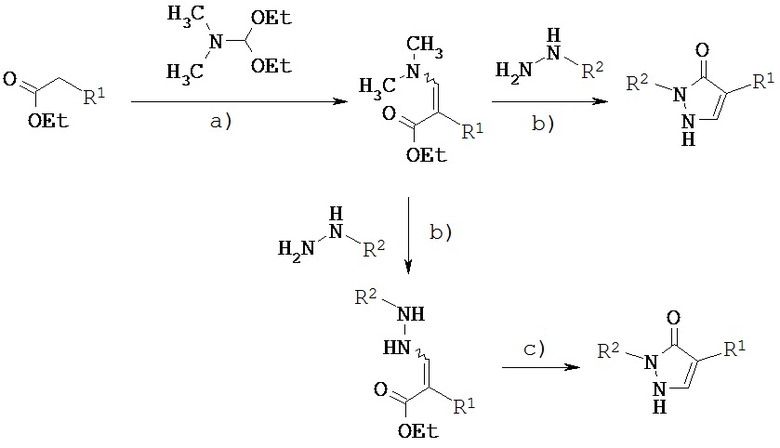
[а): диметилформамид, 16 ч, +100°C; b) этанол, катализатор 10-камфорсульфокислота, +78°С; с): NaOEt, этанол, 1 ч, комнатная температура].
Соединения согласно изобретению проявляют непредсказуемый, весьма ценный фармакологический спектр активности. Поэтому они годятся для применения в качестве лекарственных средств для лечения и/или профилактики болезней у человека и животных.
Соединения согласно изобретению выделяются как специфические ингибиторы HIF-пролил-4-гидроксилаз.
Соединения согласно изобретению из-за своих фармакологических свойств могут применяться для лечения и/или профилактики кардиоваскулярных заболеваний, в особенности сердечной недостаточности, коронарных болезней сердца, Angina pectoris, инфаркта миокарда, аполлексии, артериосклероза, эссенциальной, легочной и злокачественной гипертонии, а также периферических облитерирующих заболеваний.
Далее, соединения согласно изобретению годятся для лечения и/или профилактики расстройств кроветворения, таких как, например, идиопатические анемии, почечные анемии и анемии в сопровождении опухолевых заболеваний (особенно анемии, индуцированные химиотерапией), инфекций (особенно ВИЧ-инфекции) или других воспалительных заболеваний, таких как, например, ревматоидный артрит. Кроме того, соединения согласно изобретению пригодны для поддерживающего лечения анемий вследствие потери крови, железодефицитной анемии, витаминодефицитной анемии (например, из-за недостатка витамина В12 или недостатка фолиевой кислоты), гипопластической и апластической анемий, гемолитической анемии или для поддерживающего лечения анемий вызванных нарушением усвоения железа (железорефрактерная анемия) или анемий, как следствия других эндокринных нарушений (например, гипотиреоз).
Далее, соединения годятся для усиления гематокрита перед операцией с целью получения крови для последующего переливания пациенту собственной крови.
Кроме того, соединения согласно изобретению могут применяться для лечения и/или профилактики вызванных операциями ишемических состояний и их последствий после хирургических вмешательств, в особенности операций на сердце при использовании аппарата сердце-легкие (например, операции по шунтированию, имплантации сердечных клапанов), операции на сонной артерии, операции на аорте и операции с инструментальным вскрытием крыши черепа иди проникновением под нее. Далее, соединения годятся для общего лечения и/или профилактики при хирургических операциях с целью ускорения заживления ран и сокращения времени реконвалесценции.
Кроме того, соединения годятся для лечения и профилактики последствий острых и затяжных ишемических состояний головного мозга (например, апоплексия, асфиксия новорожденных).
Далее, соединения могут использоваться для лечения и/или профилактики рака и для лечения и/или профилактики появляющегося в ходе лечения рака нанесения вреда состоянию здоровья, в частности после терапии цитостатиками, антибиотиками и облучения.
Далее, соединения годятся для лечения и профилактики заболеваний ревматического характера и других форм болезней, причисляемых к аутоиммунным заболеваниям, и особенно для лечения и/или профилактики появляющегося в ходе медикаментозного лечения таких заболеваний нанесения вреда состоянию здоровья.
Далее, соединения согласно изобретению используются для лечения и/или профилактики болезней глаз (например, глаукомы), головного мозга (например, болезни Паркинсона, болезни Альцгеймера, деменций, хронической чувствительности к боли), хронических болезней почек, почечной недостаточности и острой почечной недостаточности, а также для способствования заживлению ран.
Далее, соединения годятся для лечения и/или профилактики общей телесной слабости, вплоть до кахексии, особенно часто встречающейся в преклонном возрасте.
Далее, годятся соединения для лечения и/или профилактики сексуальных дисфункций.
Кроме того, соединения годятся для лечения и/или профилактики сахарного диабета (Diabetes mellitus) и заболеваний, являющихся его последствием, таких как, например, диабетическая макро- и микроангиопатия, диабетическая нефропатия и невропатия.
Далее, соединения согласно изобретению пригодны для лечения и/иди профилактики фиброзных заболеваний, например, сердца, легких и печени.
Особенно годятся соединения согласно изобретению для профилактики и лечения ретинопатии у недоношенных детей (Retinopathia praematurorum).
Следующим предметом данного изобретения является применение соединений согласно изобретению для лечения и/или предупреждения заболеваний, особенно выше названных заболеваний.
Следующим предметом данного изобретения является применение соединений согласно изобретению для приготовления лекарственного средства для лечения и/иди предупреждения заболеваний, особенно названных выше заболеваний.
Следующим предметом данного изобретения является способ лечения и/или профилактики заболеваний, в особенности вышеназванных заболеваний, с применением эффективных количеств по меньшей мере одного из соединений согласно изобретению.
Соединения согласно изобретению могут использоваться одни или при необходимости в комбинации с другими биологически активными веществами. Следующим предметом данного изобретения являются лекарственные средства, содержащие по меньшей мере одно из соединений согласно изобретению и одно или несколько других биологически активных веществ, особенно для лечения и/или предупреждения названных выше заболеваний. В качестве подходящих для комбинирования биологически активных веществ для примера и предпочтительно следует назвать: ингибиторы ангиотензинпревращающего фермента, антагонисты рецепторов ангиотензина II, бета-адреноблокаторы, антагонисты ионов кальция, ингибиторы фосфодиэстеразы, антагонисты рецепторов минералокортикоидов, диуретики, аспирин, добавки, содержащие железо, добавки, содержащие витамин В12 и фолиевую кислоту, статины, производные дигиталиса (дигоксина), противоопухолевые химиотерапевтические средства, а также антибиотики.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с ингибиторами ангиотензинпревращающего фермента, такими как, например и предпочтительно эналаприл, каптоприл, лизиноприл, рамиприл, делаприл, фозиноприл, квиноприл, периндоприл или трандоприл.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с антагонистами рецепторов ангиотензина AII, такими как, например и предпочтительно лозартан, кандесартан, вальсартан, телмисартан или эмбусартан.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с бета-адреноблокаторами, такими как, например и предпочтительно: пропранолол, атенолол, тимолол, пиндолол, алпренолол, окспренолол, пенбутолол, бупранолол, метипранолол, надолол, мепиндолол, каразолол, зоталол, метопролол, бетаксолол, целипролол, бисопролол, картеолол, эсмолол, лабеталол, карведилол, адапролол, ландиолол, небиволол, эпанолол или буциндолол.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с антагонистами ионов кальция, такими как, например и предпочтительно: нифедилин, амлодилин, верапамил или дилтиазем.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с ингибиторами фосфодиэстеразы, такими как, например и предпочтительно: милринон, амринон, пимобендан, цилостазол, силденафил, варденафил или тадалафил.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с антагонистами рецепторов минералокортикоидов, такими как, например и предпочтительно: спиронолактон, эплеренон, канренон или калия канреноат.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с диуретиками, такими как, например и предпочтительно: фуросемид, буметанид, торсемид, бендрофлуметиазид, хлортиазид, гидрохлортиазид, гидрофлуметиазид, метиклотиазид, политиазид, трихлорметиазид, хлорталидон, индапамид, метолазон, квинетазон, ацетазоламид, дихлорфенамид, метазоламид, глицерин, изосорбид, маннитол, амилорид или триамтерен.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с ингибиторами ГМГ-КоА-редуктазы из класса статинов, такими как, например и предпочтительно: ловастатин, симвастатин, правастатин, флувастатин, аторвастатин, розувастатин, церивастатин или литавастатин.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с противоопухолевыми химиотерапевтическими средствами из группы препаратов платины, такими как, например, дисплатин и карбоплатин, алкилирующими средствами, такими как, например, циклофосфамид (циклофосфан) и хлорамбуцил (хлорбутин), антиметаболитами, такими как, например, 5-фторурацил и метотрексат, ингибиторами толоизомеразы, такими как, например, этолозид и камптотецин, антибиотиками, такими как, например, зорафениб и зунитиниб.
В случае предпочтительного варианта осуществления изобретения соединения согласно изобретению даются в комбинации с антибиотиками, например и предпочтительно из группы пенициллинов, цефалоспоринов иди хинолонов, такими как, например, ципрофлоксацин и моксифлоксацин.
Следующим предметом данного изобретения являются лекарственные средства, которые содержат по меньшей мере одно соединение согласно изобретению, обычно вместе с одним или несколькими инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами, а также их применение для указанных выше цепей.
Соединения согласно изобретению могут воздействовать системно и/или местно. Для этого они могут аплицироваться подходящим образом, как, например, орально, парентерально, пульмонально (легочным путем), назально, подъязычно (сублингвально), на язык (лингвально), щечно, ректально, дермально, трансдермально, конъюктивально, в ухо или как имплантант и эндопротез сосуда.
Для указанных способов приема лекарств соединения согласно изобретению могут отпускаться в подходящей форме применения.
Для орального применения согласно современному уровню техники подходят действующие формы применения, которые быстро и/или модифицировано отдают соединения согласно изобретению и которые содержат соединения согласно изобретению в кристаллической и/или аморфизированной форме и/или в растворенном виде, такие как, например, таблетки (таблетки с покрытием или без покрытия, например, с устойчивой к желудочному соку оболочкой или с покрытием, растворимым или нерастворимым, которым контролируется высвобождение соединения согласно изобретению), быстро распадающиеся в полости рта таблетки или пластинки/облатки, пластинки/лиолизаты, капсулы (например, твердые или мягкие желатиновые капсулы), драже, вещество в гранулах, гранулы, порошки, эмульсии, суспензии, аэрозоли иди растворы.
Парентеральное применение может происходить в обход стадии ресорбции (например, внутривенно, внутриартериально, интракардиально (внутрисердечно), интраспинально (внутрь спинного мозга) или интралюмбально) или при включении ресорбции (например, внутримышечно, подкожно, внутрикожно, чрезкожно или внутрибрюшинно). Для парентерального применения в качестве форм применения годятся среди прочего препараты для инъекций и вливаний в форме растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для других способов применения годятся, например, ингаляционные лекарственные формы (порошковые ингаляторы, распылители), капли в нос, растворы или спреи для носа, таблетки, применяемые на язык, под язык или за щеку, пластинки/облатки или капсулы, суппозитории, препараты для ушей или глаз, вагинальные капсулы, водные суспензии (лосьоны, микстуры «болтушки»), лилофильные суспензии, мази, кремы, трансдермальные терапевтические системы (например, пластыри), молочко, пасты, пены, присыпки, имплантанты и эндопротезы сосудов.
Предпочтительными являются оральное и парентеральное применение, особенно оральное и внутривенное применение.
Соединения согласно изобретению могут переводиться в указанные формы применения. Это может происходить известными сами по себе способами путем смешивания с инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами. К вспомогательным веществам относятся, в частности, вещества-носители (например, микрокристаллическая целлюлоза, лактоза, маннитол), растворители (например, жидкие полиэтиленгликоли), эмульгаторы и диспергаторы или смачиватели (например, натрийдодецилсульфат, полиоксисорбитанолеат), связующие (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (например, антиоксиданты, такие как, например, аскорбиновая кислота), красители (например, неорганические пигменты, такие как, например, оксиды железа) и добавки, корректирующие вкус и/или запах.
В общем случае оказалось выгодным давать при парентеральном применении количества от примерно 0.001 до 1 мг/кг, предпочтительно от 0.01 до 0.5 мг/кг веса тела для достижения эффективного результата. При оральном применении дозировка составляет около 0.01 - 100 мг/кг, предпочтительно 0.01 - 20 мг/кг и наиболее предпочтительно 0.1 - 10 мг/кг веса тела.
Однако при необходимости можно отклониться от названных количеств, а именно в зависимости от веса тела, способа дачи лекарства, индивидуальных особенностей по отношению к биологически активному веществу, типа композиции и времени и интервала, в котором происходит применение. Так, в некоторых случаях достаточно обходиться меньшими, чем было указано минимальными количествами, в то время как в других случаях нужно превысить названные верхние границы. В случае применения больших количеств можно порекомендовать разделить их на несколько отдельных приемов в течение дня.
Нижеследующие примеры исполнения поясняют изобретение. Изобретение не ограничивается этими примерами.
Если ничего не оговорено другого, процентные данные в следующем далее тексте и примерах являются весовыми процентами; части являются весовыми частями. Соотношения растворителей, соотношения при разбавлении и концентрационные данные растворов жидкость/жидкость соответственно относятся к объемам.
А. Примеры Сокращения и аббревиатуры
ЖХ-МС-, ГХ-МС- и ВЖХ-методы:
Метод 1 (ЖХ-МС):
Прибор: Micromass Platform LCZ с ВЭЖХ Agilent Serie 1100; Колонка: Thenno Hypersil GOLD 3 μ, 20 мм × 4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 100% А→0.2 мин 100% А→2.9 мин 30% А→3.1 мин 10% А→5.5 мин 10% А; печь: 50°С; поток: 0.8 мл/мин; УФ-детектирование: 210 нм.
Метод 2 (ЖХ-МС)
Тип прибора МС: Micromass ZQ; Тип прибора ВЭЖХ: серия HP 1100; УФ DAD; колонка: Phenomenex Gemini 3μ 30 мм × 3.00 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А→2.5 мин 30% А→3.0 мин 5% А→4.5 мин 5% А; поток: 0.0 мин 1 мл/мин → 2.5 мин/3.0 мин/4.5 мин 2 мл/мин; печь: 50°С; УФ-детектирование: 210 нм.
Метод 3 (ЖХ-МС):
Прибор: Micromass Quattro LCZ с ВЭЖХ Agilent Serie 1100; колонка: Phenomenex Syncrgi 2μ Hydro-RP Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А→2.5 мин 30% А→3.0 мин 5% А→4.5 мин 5% А; поток: 0.0 мин 1 мл/мин → 2.5 мин/3.0 мин/4.5 мин 2 мл/мин; печь: 50°С; УФ-детектирование: 208-400 нм.
Метод 4 (ЖХ-МС):
Тип прибора МС: Micromass ZQ; Тип прибора ВЭЖХ: Waters Alliance 2795; колонка: Phenomenex Synergi 2μ Hydro-RP Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А→2.5 мин 30% А→3.0 мин 5% А→4.5 мин 5% А; поток: 0.0 мин 1 мл/мин → 2.5 мин/3.0 мин/4.5 мин 2 мл/мин; печь: 50°С; УФ-детектирование: 210 нм.
Метод 5 (ЖХ-МС):
Тип прибора МС: Micromass ZQ; Тип прибора ВЭЖХ: HP 1100 Scries; УФ DAD; колонка: Phenomenex Synergi 2μ Hydro-RP Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А→2.5 мин 30% А→3.0 мин 5% А→4.5 мин 5% А; поток: 0.0 мин 1 мл/мин → 2.5 мин/3.0 мин/4.5 мин 2 мл/мин; печь: 50°С; УФ-детектирование: 210 нм.
Метод 6 (ЖХ-МС):
Тип прибора МС: Waters ZQ; Тип прибора ВЭЖХ: Agilent 1100 Scries; УФ DAD; колонка: Thermo Hypersil GOLD 3μ 20 мм × 4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 100% А→3.0 мин 10% А→4.0 мин 10% А→4.1 мин 100% А; поток: 2.5 мл/мин; печь: 55°С; УФ-детектирование: 210 нм.
Метод 7 (ЖХ-МС):
Тип прибора МС: Micromass ZQ; Тип прибора ВЭЖХ: Waters Alliance 2795; колонка: Phenomenex Syncrgi 2.5μ MAX-RP 100A Mercury 20 мм x 4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А→0.1 мин 90% А→3.0 мин 5% А→4.0 мин 5% А→4.01 мин 90% А; поток: 2 мл/мин; печь: 50°С; УФ-детектирование: 210 нм.
Метод 8 (ЖХ-МС):
Прибор: Micromass Quanro Micro MS с ВЭЖХ Agilent Serie 1100; колонка: Thermo Hypersil GOLD 3μ 20 мм × 4 мм; элюент A: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 100% А→3.0 мин 10% А→4.0 мин 10% А→4.01 мин 100% А (поток 2.5 мл/мин) → 5.00 мин 100% А; печь: 50°С; поток: 2 мл/мин; УФ-детектирование: 210 нм.
Метод 9 (ЖХ-МС):
Прибор: Micromass Quattro LCZ с ВЭЖХ Agilent Serie 1100; колонка: Phenomenex Syncrgi 2.5μ MAX-RP 100A Mercury 20 мм × 4 мм; элюент А: 1 л вода + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А→0.1 мин 90% А→3.0 мин 5% А→4.0 мин 5% А→4.1 мин 90% А; поток: 2 мл/мин; печь: 50°С; УФ-детектирование: 208-400 нм.
Метод 10 (ЖХ-МС):
Прибор: Micromass QuattroPremier с Waters UPLC Acquity; колонка: Thermo Hypersil GOLD 1.9μ 50 мм × 1 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А→0.1 мин 90% А→1.5 мин 10% А→2.2 мин 10% А; поток: 0.33 мл/мин; печь: 50°С; УФ-детектирование: 210 нм.
Метод 11 (ЖХ-МС):
Прибор: HP 1100 с DAD-детектированием; колонка: Kiomasil 100 RP-18,60 мм × 2.1 мм, 3.5 мкм; элюент А: 5 мл HClO4 (70%-ная)/литр воды, элюент В: ацетонитрил; градиент: 0 мин 2% В→0.5 мин 2% В→4.5 мин 90% В→6.5 мин 90% В→6.7 мин 2% В→7.5 мин 2% В; поток: 0.75 мл/мин; температура колонки: 30°С; УФ-детектирование: 210 нм.
Метод 12 (ВЭЖХ):
Колонка: Kromasil 100 С18 5 мкм, 250 мм × 20 мм; элюент А: 0.2%-ная трифторуксусная кислота, элюент В: ацетонитрил; градиент: 0.0 мин 95% А→10 мин 5% А→15 мин 5% А→15.1 мин 95% А→20 мин 95% А; печь: 30°С; поток: 25 мл/мин; УФ-детектирование: 240 нм.
Метод 13 (ЖХ-МС):
Прибор: Micromass Quattro LCZ с ВЭЖХ Agilent Scrie 1100; колонка: Phenomenex Onyx Monolithic C18, 100 мм × 3 мм; элюент A: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А→2 мин 65% А→4.5 мин 5% А→6 мин 5% А; поток: 2 мл/мин; печь: 40°С; УФ-детектирование: 208-400 нм.
Метод 14 (ГХ-MC):
Прибор: Micromass GCT, GC6890; колонка: Restek RTX-35,15 м × 200 мкм × 0.33 мкм; постоянный поток с гелием: 0.88 мл/мин; печь: 70°С; ввод (Inlet): 250°С; градиент: 70°С, 30°С/мин → 310°С (3 мин выдерживать)
Метод 15 (препаративная ВЭЖХ):
Колонка: Chromatorex C18 5 мкм, 250 мм × 20 мм; элюент А: водный 0.1%-ный раствор диизопропилэтиламина, элюент В: ацетонитрил; градиент 0.0 мин 60% А→4 мин 60% А; печь: 30°С; поток: 25 мл/мин; УФ-детектирование: 260 нм.
Метод 16 (препаративная ЖХ-МС):
Прибор МС: Waters ZQ 2000; Прибор ВЭЖХ: Agilent 1100, соединение двух колонок; автоматический пробоотборник: НТС PAL; колонка: YMC-ODS-AQ, 50 мм × 4.6 мм, 3.0 мкм; элюент А: вода + 0.1% муравьиная кислота, элюент В: ацетонитрил+0.1% муравьиная кислота; градиент: 0.0 мин 100% А→0.2 мин 95% А→1.8 мин 25% А→1.9 мин 10% А→2.0 мин 5% А→3.2 мин 5% А→3.21 мин 100% А→3.35 мин 100% А; печь: 40°С; поток: 3.0 мл/мин; УФ-детектирование: 210 нм.
Метод 17 (препаративная ВЭЖХ):
Колонка: Kfomasil 100 C18 5 мкм, 250 мм × 20 мм; элюент А: водный 0.1%-ный раствор диизопропилэтиламина, элюент В: ацетонитрил; градиент: 0.0 мин 95% А→10 мин 65% А→10.1 мин 95% А→15 мин 95% А; печь: 40°С; поток: 25 мл/мин; УФ-детектирование: 210 нм.
Исходные соединения и промежуточные вещества:
Пример 1А
2-Гидразино-4-метилпиридин
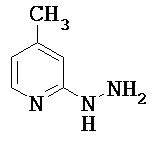
Растворяли 3.33 г (30 ммоль) 2-фтор-4-метилпиридина в 40 мл 2-этоксиэтанола, раствор смешивали с 14,6 мл (15.0 г, 300 ммоль) гидразингидрата и смесь перемешивали при температуре кипения (температура бани 150°С) в течение 16 ч. После этого реакционный раствор упаривали на ротационном испарителе, остаток смешивали со 100 мл воды и экстрагировали этиловым эфиром уксусной кислоты (трижды по 100 мл). Объединенные органические фазы сушили над сульфатом натрия, фильтровали и упаривали. Полученный остаток сушили в вакууме.
Выход: 1.90 г (51% от теоретического)
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.83 (d, 1H), 7.22 (s, 1H), 6.51 (s, 1H), 6.38 (d, 1H), 4.04 (s, 2H), 2.17 (s, 3H).
ЖХ-МС (Метод 1): Rt=0.80 мин; MC (ESIpos): m/z=124 [M+H]+.
Пример 2А
Метиловый эфир 3-(диметиламино)-2-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]акриловой кислоты
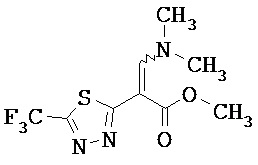
3.05 г (13.5 ммоль) метилового эфира 5-(трифторметил)-1,3,4-тиадиазол-2-илуксусной кислоты [получение смотри DE 4240168-А1] нагревали в 6.9 мл (40.5 ммоль) диметилформамид-диэтилацеталя в течение ночи при 100°С. После охлаждения смесь упаривали и очищали остаток с помощью препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент).
Выход: 2.8 г (74% от теоретического)
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.28 (s, 1H), 3.74 (s, 3H), 3.32 (s, 6H).
ЖХ-МС (Метод 4): Rt=1.88 мин; МС (ESIpos): m/z=282 [М+Н]+.
Пример 3А
Этиловый эфир 3-(диметиламино)-2-(1H-1,2,3-триазол-1-ил)акриловой кислоты

Получение названного соединения происходит аналогично примеру 2А, исходя из 1.00 г (6.45 ммоль) этилового эфира 2-(1H-1,2,3-триазол-1-ил)уксусной кислоты.
Выход: 1.4 г (100% от теоретического)
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.10 (d, 1H), 7.78 (d, 1H), 7.65 (s, 1H), 4.03 (q, 2H), 3.06 (уш.:, 3H), 2.10 (уш. s, 3H), 1.12 (t, 3H).
ЖХ-МС (Метод 5): Rt=1.40 мин: МС (ESIpos): m/z=211 [М+Н]+.
Пример 4А
(6-Гидразинопиридин-3-ил)метанол

220.0 г (1.5 моля) (6-хлорпиридин-3-ил)метанола [Evans et al., Jrganic Letters 2001, 19, 3009-3012] помещали в 746 мл (767.1 г, 15.3 моля) гидразингидрата и перемешивали в течение 5 ч при температуре бани 150°С. Концентрировали реакционную смесь в вакууме, смешивали остаток с 500 мл воды и добавляли 86.0 г (1.5 моля) гидроксида калия. Перемешивали 15 мин, затем удаляли воду на ротационном испарителе почти полностью, а остающуюся воду за несколько раз отгоняли в виде азеотрола с толуолом. Маслянистый остаток смешивали с этанолом, охлаждали до примерно 10°С, отфильтровывали выпавший осадок хлорида калия, фильтрат упаривали и остаток смешивали с диэтиловым эфиром. После чего продукт отфильтровывали, остаток на фильтре промывали диэтиловым эфиром и кристаллы сушили в вакууме.
Выход: 149.0 г (68% от теоретического)
ЖХ-МС (Метод 1): Rt=0.46 мин; МС (ESIpos): m/z=140 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.91 (d, 1H), 7.40 (dd, 1H), 7.29 (s, 1H), 6.66 (d, 1H), 4.94 (уш. s, 1H), 4.34-4.28 (m, 2H), 4.04 (уш. s, 2Н).
Пример 5A
1-(6-Гидразинопиридин-3-ил)-N-метилметанамин

1.0 г (6.4 ммоль) 1-(б-хлорпиридин-3-ил)-N-метилметанамина [получение смотри ЕР 0556684-А1] помещали в 1.5 мл (1.6 г, 31.9 ммоль) гидразингидрата и перемешивали 12 ч при кипении (при температуре бани 150°С). Охлажденный реакционный раствор упаривали и остаток сушили в вакууме. Получали 1.1 г названного соединения, которое использовали без последующей очистки.
ЖХ-МС (Метод 1): Rt=0.52 мин; МС (ESIpos): m/z=153 [М+Н]+.
Пример 6А
6-Гидразиноникотиновая кислота

5.0 г (31.7 ммоль) 6-хлорникотиновой кислоты и 30.9 мл (31.8 г, 634.7 ммоль) гидразингидрата помещали в 10 мл этанола и перемешивали при кипении в течение 16 ч при температуре бани 100°С. Растворитель и избыток гидразингидрата отгоняли на ротационном испарители, остаток помещали в воду, затем смешивали с 1.8 г (31.7 ммоль) гидроксида калия и перемешивали 15 мин. Растворитель полностью удаляли на ротационном испарителе, остаток сушили в вакууме и получали 7.5 г продукта, который подвергали дальнейшему превращению.
ЖХ-МС (Метод 1): Rt=0.48 мин; MS (ESIpos): m/z=154 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.49 (d, 1H), 7.89 (уш. s, IH), 7.84 (dd, 1H), 6.63 (d, 1H), 5.37 (уш. s, 2H).
Пример 7А
4-Хлор-6-гидразинолиримидин
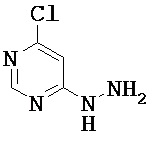
В раствор 20.0 г (134.3 ммоль) 4,6-дихлорпиримидина в 300 мл этанола прибавляли по каплям 11.8 мл (12.1 г, 241.6 ммоль) гидразингидрата при комнатной температуре. Если при добавлении гидразингидрата происходило помутнение раствора, добавляли еще этанол (около 400 мл). Реакционный раствор перемешивали затем 12 ч при комнатной температуре. Затем отфильтровывали выпавший осадок, остаток на фильтре промывали дважды по 150 мл воды и дважды по 100 мл диэтилового эфира и продукт сушили в вакууме. Из сконцентрированного маточного раствора получали дополнительные количества кристаллического продукта.
Выход: 16.% г (87% от теоретического)
ЖХ-МС (Метод I): Rt=1.17 мин; МС (ESIpos): m/z=145 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.81 (s, 1H), 8.17 (уш. s, 1Н), 6.75 (s, 1H), 4.48 (уш. s, 2H).
Пример 8А
4-Гидразино-6-пиперидин-1-илпиримидин

Стадия а): 4-Хлор-6-пиперидин-1-илпиримидин

Смесь из 10.0 г (67.1 ммоль) 4,6-дихлорпиримидина и 5.7 г (67.1 ммоль) пиперидина в 100 мл воды перемешивали 16 ч при температуре бани 115°С. После охлаждения до комнатной температуры осадок отфильтровывали, промывали водой и сушили в вакууме.
Выход: 6.4 г (47% от теоретического)
ЖХ-МС (Метод 4): Rt=2.16 мин; MS (ESIpos): m/z=198 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.29 (s, 1H), 6.92 (s, 1H), 3.65-3.58 (m, 4Н), 1.66-1.62 (m, 2Н), 1.60-1.48 (m, 4Н).
Стадия b): 4-Гидразино-6-пиперидин-1-илпиримидин
В раствор 6.0 г (30.4 ммоль) 4-хлор-6-пиперидин-1-илпиримидина в 50 мл этанола добавляли по каплям при перемешивании 17.7 мл (18.2 г, 364.2 ммоль) гидразингидрата при комнатной температуре. Реакционный раствор перемешивали еще 16 ч при 80°С. Затем концентрировали реакционную смесь в вакууме, остаток смешивали с водой, выпавшее твердое вещество отфильтровывали, остаток на фильтре промывали дважды водой по 150 мл и дважды диэтиловым эфиром по 100 мл и продукт сушили в вакууме.
Выход: 4.0 г (69% от теоретического)
ЖХ-МС (Метод 1): Rt=2.06 мин; MS (ESIpos): m/z=194 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.91 (s, 1H), 7.54 (уш. s, 1H), 5.89 (s, 1H), 4.11 (уш. s, 2Н), 3.50-3.47 (m, 4Н), 1.61-1.58 (m, 2Н), 1.51-1.46 (m, 4Н).
Пример 9А
2-Гидразино-5-(метилсульфонил)пиридин

Смешивали 2.0 г (8.5 ммоль) 2,5-бис(метилсульфонил)пиридина [Wooda et al., J. Heterocycl. Chem. 1984, 27, 97-101] в 15 мл этанола с 1.7 мл (1.7 г, 34.0 ммоль) гидразингидрата и перемешивали 4 ч с обратным холодильником. После чего реакционный раствор охлаждали до 15°С, выпавшее твердое вещество отфильтровывали, остаток на фильтре промывали этанолом и диэтиловым эфиром и продукт сушили в вакууме.
Выход: 1.4 г (89% от теоретического)
ЖХ-МС (Метод 1): Rt=0.51 мин; МС (ESIpos): m/z=188 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.56 (s, 1Н), 8.38 (d, 1H), 7.81 (dd, 1H), 6.79 (d, 1H), 4.42 (s, 2H), 3.11 (s, 3H).
Пример 10А
5-Бром-2-гидразинопиридин
Раствор 1.8 г (9.5 ммоль) 5-бром-2-хлорпиридина в 25 мл этанола смешивали при перемешивании при комнатной температуре с 9.3 мл (9.5 г, 190.2 ммоль) гидразингидрата и затем перемешивали еще 46 ч при 90°С. После концентрирования реакционной смеси в вакууме остаток смешивали с водой, твердое вещество отфильтровывали, промывали водой и диэтиловым эфиром и сушили в вакууме.
Выход: 0.8 г (44% от теоретического)
ЖХ-МС (Метод 8): Rt=0.50 мин; МС (ESIpos): m/z=188 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.02 (d, 1H), 7.66 (s, 1H), 7.58 (dd, 1H), 6.69 (d, 1H), 4.16 (s, 2H).
Пример 11А
1-(6-Гидразинолиримидин-4-ил)азетидин-3-ол

Стадия а): 1-(6-Хлорпиримидин-4-ил)азетидин-3-ол

Помещали 7.2 г (48.7 ммоль) 2,4-дихлорпиримидина в 140 мл воды. Добавляли 5.3 г (48.7 ммоль) 3-гидроксиазетидина-гидрохлорида и 48.7 мл 1 N раствора едкого натра и нагревали 72 ч при 90°С (при этом 2,4-дихлорпиримидин оказался летучим и осаждался в виде кристаллов на холодильнике). Удаляли растворитель в вакууме, сушили остаток и получали 10.4 г сырого продукта, который использовали для дальнейших превращений без дополнительной очистки.
ЖХ-МС (Метод 10): Rt=0.36 мин; МС (ESIpos): m/z=186 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.29 (s, 1H), 6.50 (s, 1H), 4.63-4.57 (m, 1H), 4.26 (t, 2H), 3.82-3.75 (m, 2H).
Стадия b): 1-(6-Гидразинолиримидин-4-ил)азетидин-3-ол
Помещали 960 мг (5.2 ммоль) 1-(б-хлорпиримидин-4-ил)азетидин-3-ола и 2.5 мл (51.7 ммоль) гидразингидрата в смесь из 10 мл этанола и 10 мл ТГФ. Реакция происходила в течение 1 ч при 130°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). Концентрировали смесь примерно до 20-50% первоначального объема жидкости и оставляли при комнатной температуре на 48 ч. Жидкость декантировали с образовавшегося осадка и твердое вещество трижды промывали 1.5 мл холодного этанола. Сушили в высоком вакууме.
Выход: 300 мг (32% от теоретического)
ЖХ-МС (Метод 8): Rt=0.23 мин; МС (ESIpos): m/z=182 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.88 (s, 1H), 7.63 (s, 1H), 5.68 (уш. s, 1H), 5.52 (s, 1H), 4.55 (уш. s, 1H), 4.18-4.06 (m, 4H), 3.65-3.60 (m, 2H).
Пример 12А
5-(2,2-Диметилпропокси)-2-гидразинопиридин

Стадия а): 2-Хлор-5-(2,2-диметилпропокси)пиридин

Разделили 5.2 г (40.0 ммоль) 6-хлорпиридин-3-ола, 11.9 г (60.0 ммоль) 1-йод-2,2-диметилпролана, 19.6 г (60.0 ммоль) карбоната цезия и 120 мл диметилового эфира диэтиленгликоля на пять равных по величине порций и по порциям заставляли взаимодействовать при 160° в течение 4 ч при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). После чего объединили полученные пять реакционных смесей, отфильтровывали твердое вещество, промывали его диметиловым эфиром диэтиленгликоля и объединяли фильтрат и промывные растворы. Удаляли большую часть растворителя и сконцентрированный раствор (около 50 мл) смешивали с 300 мл воды. Перемешивали 30 мин, отфильтровывали полученное твердое вещество, промывали один раз водой и сушили в высоком вакууме.
Выход: 7.0 г (88% от теоретического)
ЖХ-МС (Метод 8): Rt=2.47 мин; МС (ESIpos): m/z=200 [М+Н]+;
1Н-ЯМР (400 МГц, CDCl3): δ=8.05 (d, 1Н), 7.25-7.15 (m, 2H), 3.61 (s, 2H), 1.03 (s, 9H).
Стадия b): 5-(2,2-Диметилпропокси)-2-гидразинопиридин
Разделили 6.2 г (30.8 ммоль) 2-хлор-5-(2,2-диметилпропокси)пиридина вместе с 60 мл (1.2 моль) гидразингидрата соответственно на четыре равных порции и смешали соответственно каждую с 10 мл этанола. Каждую порцию заставляли взаимодействовать в течение 12 ч при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer) при 170° (200 ватт). После чего объединяли четыре смеси и удаляли растворитель. Остаток смешивали с этиловым эфиром уксусной кислоты и промывали по одному разу насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия. Сушили над сульфатом магния и удаляли растворитель в вакууме.
Выход: 6.0 г (76% от теоретического)
ЖХ-МС (Метод 8): Rt=1.28 мин; МС (ESIpos): m/z=196 [M+H]+;
1Н-ЯМР (400 МГц, CDCl3): δ=7.84 (s, 1H), 7.17 (dd, 1H), 6.68 (d, 1H), 5.54 (уш. s, 1H), 3.80 (уш. s, 2H), 3.56 (s, 2H), 1.02 (s, 9H).
Пример 13А
2-Хлор-5-(метоксиметил)пиридин

Растворяли 2.6 г (23.0 ммоль) трет.-бутилата калия в 50 мл ТГФ. Добавляли туда 3.0 г (20.9 ммоль) (6-хлорпиридин-3-ил)метанола и перемешивали 15 мин при комнатной температуре. Затем добавляли 4.4 г (31.3 ммоль) йодметана и перемешивали около 30 мин до тех пор, пока не затихала слабо экзотермическая реакция. Удаляли растворитель, смешивали остаток с дихлорметаном и промывали дважды водой. Сушили над сульфатом магния, упаривали и очищали остаток колоночной хроматографией на силикагеле (Biotage-хроматография, растворитель: циклогексан/этиловый эфир уксусной кислоты 85:15).
Выход: 2.2 г (68% от теоретического)
ЖХ-МС (Метод 1): Rt=2.62 мин; МС (ESIpos): m/z=158 [М+Н]+;
1Н-ЯМР (400 МГц, CDCl3): δ=8.34 (d, 1H), 7.65 (dd, 1H), 7.32 (d, 1H), 4.45 (s, 2H), 3.41 (s, 3H).
Пример 14A
2-Бром-4,5-диметилпиридин
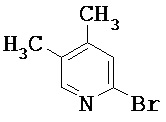
Помещали 71.3 г (0.8 моль) 2-(диметиламино)-этанола в 500 мл н-гексана и охлаждали до 0°С. Медленно добавляли 1.0 литр (1.6 моль) раствора н-бутиллития (1.6 М в н-гексане) и перемешивали 15 мин при 0°С. Затем добавляли по каплям раствор 17.9 г (166.7 ммоль) 3,4-лутидина в 500 мл н-гексана и перемешивали 1 ч при 0°С. После чего охлаждали до -78°С и смешивали с раствором 331.7 г (1.0 моль) тетрабромметана в 1.0 литре ТГФ. Перемешивали 1 ч при -78°С и затем поднимали температуру реакционной смеси до комнатной. Снова охлаждали до 0°С и медленно по каплям добавляли 1.5 литра воды. Отделяли органическую фазу, промывали ее водой, сушили над сульфатом магния и концентрировали в вакууме. Остаток подвергали сначала предварительной очистке на ~ 1 кг силикагеля (растворитель: циклогексан/этиловый эфир уксусной кислоты 9:1, затем 7:3). Фракции, содержащие продукт, объединяли и упаривали в вакууме. Остаток затем еще раз очищали на силикагеле (растворитель: циклогексан/этиловый эфир уксусной кислоты 9:1). Полученный таким образом продукт содержит примерно 10% региоизомерного 2-бром-3,4-диметилпиридина.
Выход: 6.7 г (20% от теоретического)
ГХ-МС (Метод 14): Rt=4.24 мин; MS (ESIpos): m/z=187 [M+H]+;
1Н-ЯМР (400 МГц, CDCl3): δ=8.07 (s, 1H), 7.25 (s, 1H), 2.24 (s, 3H), 2.18 (s, 3H).
Пример 15A
трет.-Бутил[(6-хлорпиридин-3-ил)метил]карбамат

Помещали 25.0 г (175.3 ммоль) 5-аминометил-2-хлорпиридина в 175 мл дихлорметана. Добавляли 175 мл 10%-ного раствора едкого натра и по каплям прибавляли раствор 383 г (175.3 ммоль) ди-трет.-бутилдикарбоната в 175 мл дихлорметана. Перемешивали 16 ч при комнатной температуре. Разбавляли затем 175 мл дихлорметана, отделяли органическую фазу, а водную фазу экстрагировали 175 мл дихлорметана. Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Продукт сушили высоком вакууме.
Выход: 42.0 г (99% от теоретического)
ЖХ-МС (Метод 7): Rt=1.58 мин; МС (ESIpos): m/z=243 [М+Н]+;
1Н-ЯМР (400 МГц, CDCl3): δ=8.31 (d, 1H), 7.61 (dd, 1H), 730 (d, 1H), 4.99 (уш. s, 1H), 4.30 (d, 2H), 1.46 (s, 9Н).
Пример 16А
4-(6-Гидразинолиримидин-4-ил)морфолин

Стадия а): 4-(6-Хлорпиримидин-4-ил)морфолин
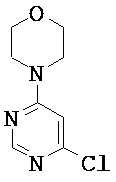
Помещали 45.0 г (302.1 ммоль) 4,6-дихлорпиримидина в 450 мл воды. Добавляли 26.3 г (302.1 ммоль) морфолина и перемешивали 16 ч при 90°С. После чего охлаждали до 0°С и отфильтровывали образовавшийся осадок. Промывали осадок один раз 50 мл воды и сушили на воздухе.
Выход: 51.0 г (85% от теоретического)
ЖХ-МС (Метод 4): Rt=1.09 мин; МС (ESIpos): m/z=200 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.35 (s, 1Н), 6.95 (s, 1H), 3.62 (s, 8H).
Стадия b): 4-(6-Гидразинолиримидин-4-ил)морфолин
Помещали 53.0 г (2.7 моль) 4-(6-хлорпиримидин-4-ил)морфолина в 260 мл этанола. Добавляли 132.9 г (2.7 моль) гидразингидрата и перемешивали при нагревании 16 ч с обратным холодильником. После чего охлаждали до комнатной температуры и удаляли примерно половину растворителя отгонкой. Охлаждали до 0°С и отфильтровывали образовавшийся осадок. Промывали его холодным этанолом и сушили сначала на воздухе, а затем в вакууме.
Выход: 35.0 г (68% от теоретического)
ЖХ-МС (Метод 1): Rt=0.17 мин; МС (ESIpos): m/z=196 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.94 (s, 1H), 7.70 (s, 1H), 5.91 (s, 1H), 4.15 (s, 2H), 3.66-3.60 (m, 4H), 3.45-3.37 (m, 4H).
Пример 17А
2-Гидразинолиразин

Прибавляли по каплям 20.0 г (174.6 ммоль) 2-хлорпиразина к 60.0 мл (61.7 г, 1.2 моль) гидразингидрата. Перемешивали 45 мин при температуре бани 120°С. Затем оставляли охлажденную реакционную смесь на 12 ч при 2°С, отфильтровывали выпавшие кристаллы и промывали осадок на фильтре дважды петролейным эфиром. После чего перекристаллизовывали осадок из толуола.
Выход: 6.5 г (34% от теоретического)
ЖХ-МС (Метод 1): Rt=0.49 мин; МС (ESIpos): m/z=111 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.11 (s, 1H), 7.94 (s, 1H), 7.90 (s, 1H), 7.70 (d, 1H), 4.29 (ш. s, 2H).
Пример 18А
5-(трет.-Бутоксиметил)-2-гидразинопиридин

Стадия а): 5-(трет.-Бутоксиметил)-2-хлорпиридин

Помещали 7.2 г (50.0 ммоль) (6-хлорпиридин-3-ил)метанола в 50 мл дихлорметана. Добавляли 25.1 г (115.0 ммоль) ди-трет.-бутилдикарбоната и 1.2 г (5.0 ммоль) перхлората магния и перемешивали 24 ч при 40°С. Затем охлаждали до комнатной температуры, добавляли еще 12.5 г (87.1 ммоль) ди-трет.-бутилдикарбоната и 600 мг (2.7 ммоль) перхлората магния и снова перемешивали 2.5 ч с обратным холодильником. Снова добавляли 12.5 г (87.1 ммоль) ди-трет.-бутилдикарбоната и перемешивали еще 3 ч обратным холодильником. Затем разбавляли дихлорметаном и промывали один раз водой и один раз раствором хлорида натрия. Сушили над сульфатом магния, концентрировали и очищали остаток хроматографией на колонке с силикагелем (растворитель: циклогексан/этиловый эфир уксусной кислоты 85:15).
Выход: 7.9 г (79% от теоретического)
ЖХ-МС (метод 10): Rt=1.12 мин; МС (ESIpos): m/z=200 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.36 (d, 1Н), 7.78 (dd, 1H), 6.98 (d, 1H), 4.45 (s, 2H), 1.222 (s, 9H).
Стадия b): 5-(трет.-Бутоксиметил)-2-гидразинопиридин

7.9 г (39.6 ммоль) 5-(трет.-Бутоксиметил)-2-хлорпиридина вместе с 19.8 г (395.6 ммоль) гидразингидрата разделили соответственно на три равных порции и каждую смешали соответственно с 15 мл этанола. Каждую порцию заставляли взаимодействовать в течение 4 ч при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer) при 170° (200 ватт). После чего объединяли все три смеси и удаляли растворитель. Остаток соединяли с этиловым эфиром уксусной кислоты и промывали один раз насыщенным раствором гидрокарбоната натрия. Водную фазу экстрагировали один раз этиловым эфиром уксусной кислоты. Обе фазы с этиловым эфиром уксусной кислоты объединяли и промывали один раз насыщенным раствором хлорида натрия. Сушили над сульфатом магния и удаляли растворитель. Полученный остаток смешивали с петролейным эфиром и отфильтровывали твердое вещество.
Выход: 1.6 г (21% от теоретического)
ЖХ-МС (Метод 10): Rt=0.77 мин; МС (ESIpos): m/z=196 [M+H]+;
1Н-ЯМР (400 МГц, CDCl3): δ=8.08 (s, 1H), 7.50 (dd, 1H), 6.68 (d, 1H), 5.77 (уш. s, 1H), 4.32 (s, 2H), 3.80 (уш. 8,2Н), 1.28 (s, 9Н).
Пример 19А
6-Гидразинопиридин-3-карбонитрил

Перемешивали 2.0 г (14.4 ммоль) нитрила 6-хлорникотиновой кислоты в течение 15 мин и 7.0 мл (7.3 г, 144.4 ммоль) гидразингидрата при температуре бани 100°С. Охлажденную до комнатной температуры реакционную смесь разбавляли водой и перемешивали при комнатной температуре 30 мин. Отфильтровывали выпавший осадок, промывали осадок на фильтре водой, сушили кристаллы в течение ночи на воздухе и перекристаллизовывали из этилового эфира уксусной кислоты.
Выход: 1.5 г (80% от теоретического)
ЖХ-МС (Метод 1): Rt=0.51 мин; МС (ESIpos): m/z=135 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.56 (s, 1H), 8.35 (s, 1H), 7.73 (d, 1H), 6.75 (m, 1H), 4.42 (s, 1H).
Пример 20А
2-Гидразино-5-метилпиридин
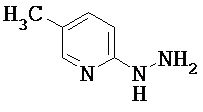
Перемешивали 1.0 г (7.8 ммоль) 2-хлор-5-метилпиридина и 5.7 мл (5.9 г, 117.6 ммоль) гидразингидрата 12 ч с обратным холодильником. Охлажденную реакционную смесь смешивали с 10 мл моноэтилового эфира этиленгликоля и затем растворитель полностью удаляли на ротационном испарителе. Повторяли эту операцию дважды, после чего смешивали остаток с дихлорметаном, отфильтровывали осадок, концентрировали фильтрат в вакууме и сушили остаток в вакууме.
Выход: 644 мг (67% от теоретического)
ЖХ-МС (Метод 8): Rt=0.35 мин; МС (ESIpos): m/z=124 [М+Н]+.
Пример 21A
4-Циклопропил-6-гидразинопиримидин
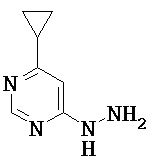
Перемешивали 1.6 г (6.9 ммоль) 4-хлор-6-циклопропилпиримидина [FR 1519069 (1966); Chem. Abctr. 71, 49965y, 1969] и 3.4 мл (3.5 г, 69.0 ммоль) гидразингидрата перемешивали 16 ч при температуре бани 90°С. Охлажденную реакционную смесь смешивали с моноэтиловым эфиром этиленгликоля и концентрировали в вакууме. Этот процесс повторяли еще раз. Затем хроматографировали остаток на силикагеле 60 (растворитель: ацетонитрил/вода 8:2).
Выход: 0.8 г (69% от теоретического)
ГХ-МС (метод 14): Rt=5.72 мин; (ESIpos): m/z=151 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.33 (уш. s, IH), 8.19 (s, 1H), 6.59 (s, 1H), 4.85 (уш. s, 2Н), 1.90-1.87 (m, 1H), 0.96-0.85 (m, 4H).
Аналогичным образом получали соединения, приведенные в нижеследующей таблице 1, из указанных эдуктов по следующей прописи:
Использовали гидразингидрат или 1 М раствор гидразина в ТГФ. Взаимодействие может осуществляться также при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer или Emrys Optimizer) обычно в этаноле или ТГФ при 150°С в течение периода времени от 15 мин до 4 ч. Очистка происходила так, как описано в примере 9А, или аналогичным образом, например, путем промывания выпавшего осадка водой и перекристаллизацией из этилового эфира уксусной кислоты или путем смешивания с этиловым эфиром уксусной кислоты или с петролейным эфиром. Избыток гидразингидрата может быть удален путем растворения сырого продукта, например, в этиловом эфире уксусной кислоты и промыванием насыщенным раствором гидрокарбоната натрия.
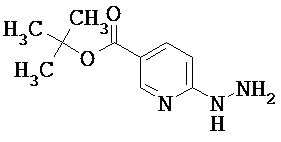



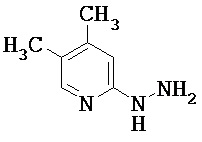
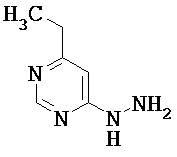

Пример 30А
4-Гидразино-6-{4-пирролидин-1-илпиперидин-1-ил)пиримидин
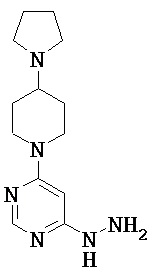
Смесь из 2.0 г (13.8 ммоль) соединения из примера 7А и 4.3 г (27.7 ммоль) 4-пирролидин-1-илпиперидина в 20 мл воды перемешивали при 100°C в течение 16 ч. Выпавший осадок отфильтровывали, промывали сначала этанолом, затем диэтиловым эфиром и сушили в вакууме.
Выход: 3.0 г (82% от теоретического)
ЖХ-МС (метод 8): Rt=0.21 мин; МС (ESIpos): m/z=263 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.92 (s, 1H), 7.58 (s, 1H), 5.91 (s, 1H), 4.17-4.07 (m, 4H), 2.95-2.85 (m, 2H), 2.54 (s, 2H), 2.52-2.46 (m, 2H), 2.24-2.15 (m, 1H), 1.90-1.80 (m, 2H), 1.71-1.61 (m, 4H), 1.39-1.24 (m, 2H).
Пример 31А
4-Гидразино-6-[4-(2-метоксиэтил)пиперазин-1-ил]пиримидин

Смесь из 1.0 г (6.9 ммоль) соединения из примера 7А и 1.1 г (7.6 ммоль) 1-(2-метоксиэтил)пиперазина в 10 мл воды перемешивали 2 ч при 100°C. Добавляли еще 0.9 г (6.2 ммоль) 1-(2-метоксиэтил)пиперазина и реакционную смесь перемешивали еще 16 ч при 100°C. После концентрирования в вакууме остаток смешивали с ацетонитрилом. Выпавший осадок отфильтровывали, промывали сначала этанолом, затем диэтиловым эфиром и сушили в вакууме.
Выход: 0.8 г (42% от теоретического)
ЖХ-МС (Метод 8): Rt=0.22 мин; МС (ESIpos): m/z=253 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.93 (s, 1H), 7.64 (s, 1H), 5.91 (s, 1H), 4.14 (s, 2H), 3.50-3.40 (m, 6H), 3.24 (s, 3Н), 2.47-2.39 (m, 4H).
Пример 32А
Этиловый эфир [4-(трифторметил)-1H-имидазол-1-ил]уксусной кислоты
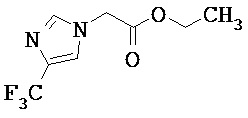
Помещали 2.0 г (14.7 ммоль) 4-(трифторметил)-1H-имидазола в 5.5 мл (4.7 г, 14.7 ммоль) 21%-ного раствора этилата натрия в этаноле и смешивали с 1.8 мл (2.7 г, 16.2 ммоль) этилового эфира бромуксусной кислоты. Перемешивали реакционную смесь 16 ч при комнатной температуре. Затем отфильтровывали выпавший осадок, осадок на фильтре промыли этанолом и фильтрат концентрировали в вакууме. Остаток смешали с диизопропиловым эфиром, еще раз фильтровали, фильтрат снова упаривали на ротационном испарителе и остаток сушили в вакууме. Продукт получается в виде смеси 9:1 обоих региоизомеров и в таком виде используется для дальнейших превращений.
Выход: 3.3 г (95% от теоретического)
ЖХ-МС (Метод 1): Rt=1.75 мин + 1.80 мин; МС (ESIpos): m/z=223 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.93 (s, 1H), 7.82 (s, 1H), 5.04 (s, 2H), [5.07 (s, 2H)], 4.18 (q, 2H) [4.12 (q, 2H)], 1.22 (t, 3Н), [1.19 (t, 3Н)].
Пример 33А
Этиловый эфир [4-циано-1H-имидазол-1-ил]уксусной кислоты

Помещали 3.3 г (35.3 ммоль) 1H-имидазол-4-карбонитрила [Matthews et al., J. Org. Chem. 1986, 51, 3228-3231] в 13.2 мл (11.5 г, 35.3 ммоль) 21%-ного раствора этилата натрия в этаноле и смешивали с 4.3 мл (6.5 г, 38.9 ммоль) этилового эфира бромуксусной кислоты. Перемешивали реакционную смесь 16 ч при комнатной температуре. Затем отфильтровывали выпавший осадок, осадок на фильтре промыли этанолом и фильтрат концентрировали в вакууме. Остаток смешали с диизопропиловым эфиром, еще раз фильтровали, фильтрат снова упаривали на ротационном испарителе и остаток сушили в вакууме.
Выход: 3.8 г (60% от теоретического)
ЖХ-МС (Метод 1): Rt=1.17 мин; МС (ESIpos): m/z=180 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.12 (s, 1H), 7.88 (s, 1H), 5.06 (s, 2H), 4.18 (q, 2H), 1.22 (t, 3H).
Пример 34А
Этиловый эфир (4-метил-1H-имидазол-1-ил)уксусной кислоты

Помещали 4.4 г (53.6 ммоль) 4-метил-1H-имидазола в 20.0 мл (17.4 г, 53.6 ммоль) 21%-ного раствора этилата натрия в этаноле и смешивали с 6.5 мл (9.8 г, 58.9 ммоль) этилового эфира бромуксусной кислоты. Перемешивали реакционную смесь 16 ч при комнатной температуре. Затем отфильтровывали выпавший осадок, осадок на фильтре промывали этанолом и фильтрат концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (растворитель: ацетонитрил/вода 9:1). Продукт получается в виде смеси 3:2 обоих региоизомеров и в таком виде используется для дальнейших превращений.
Выход: 1.8 г (20% от теоретического)
ЖХ-МС (Метод 1): Rt=1.02 мин; МС (ESIpos): m/z=169 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.48 (s, 1H), [7.52 (s, 1H)], 6.82 (s, 1H), [6.64 (s, 1H)], 4.86 (s, 2H), [4.88 (s, 2H)], 4.22-4.11 (m, 2H), 2.07 (s, 3H), [2.06 (s, 3H)], 1.25-1.19 (m, 3H).
Пример 35А
Этиловый эфир (2-метил-1H-имидазол-1-ил)уксусной кислоты
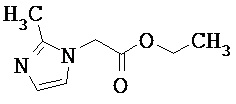
Помещали 2.0 г (24.4 ммоль) 2-метил-1H-имидазола в 9.1 мл (7.9 г, 24.4 ммоль) 21%-ного раствора этилата натрия в этаноле и смешивали с 2.9 мл (4.5 г, 26.8 ммоль) этилового эфира бромуксусной кислоты. Перемешивали реакционную смесь 16 ч при комнатной температуре. Затем отфильтровывали выпавший осадок, осадок на фильтре промыли этанолом и фильтрат концентрировали в вакууме. Остаток смешали с диизопропиловым эфиром, еще раз фильтровали и фильтрат снова упарили в вакууме.
Выход: 2.9 г (70% от теоретического)
ЖХ-МС (Метод 1): Rt=0.49 мин; МС (ESIpo:): m/z=169 [М+Н]+;
Пример 36А
Этиловый эфир (4-метил-1H-1,2,3-триазол-1-ил)уксусной кислоты

Растворяли 2.0 г (14J ммоль) (4-метил-1H-1,2,3-триазол-1-ил)уксусной кислоты в 30 мл этанола и смешивали с 10 каплями концентрированной серной кислоты. Перемешивали реакционную смесь 16 ч при комнатной температуре. Смесь концентрировали в вакууме, остаток смешивали с этиловым эфиром уксусной кислоты и суспензию промывали полуконцентрированным раствором гидрокарбоната натрия. Органическую фазу сушили над сульфатом магния, растворитель полностью удаляли на ротационном испарителе и твердое вещество сушили в вакууме.
Выход: 1.5 г (61% от теоретического)
ЖХ-МС (Метод 1): Rt=2.33 мин; МС (ESIpos): m/z=170 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.82 (s, 1Н), 5.31 (s, 2H), 4.17 (q, 2H), 2.25 (s, 3Н), 1.21 (t, 3H).
Приведенные в таблице 2 соединения получены из соответствующих эдуктов аналогично примеру 36А:
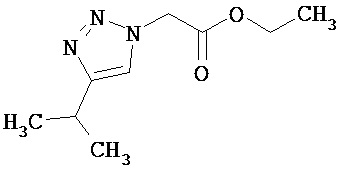
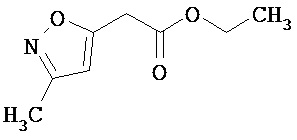
80%
2Н), 2.21 (s, 3Н), 1.21 (t, 3H)
Пример 39А
Этиловый эфир 2-(1H-1,2,3-триазол-1-ил)уксусной кислоты

Медленно прибавляли 129.2 г (5.6 моль) натрия к 4.0 литрам этанола. После чего добавляли 400.0 г (5.6 моль) 1,2,3-1H-триазола и по каплям прибавляли 623 мл (938.2 г, 5.6 моль) этилового эфира бромуксусной кислоты при температуре реакционной смеси 20-25°С. Перемешивали при комнатной температуре 48 ч. Выпавший осадок отфильтровывали, этанол удаляли в вакууме и снова фильтровали. Остаток помещали в этиловый эфир уксусной кислоты, фильтровали, снова концентрировали в вакууме и очищали перегонкой с колонкой длиной 30 см Продукт получали при температуре бани 140°С, температуре верха колонны 60-115°С и давлении 1 мбар.
Выход: 440.0 г (50% от теоретического)
ВЭЖХ (Метод 11): Rt=1.58 мин;
ЖХ-МС (Метод 1): Rt=0.71 мин; МС (ESIpos): m/z=156 [M+H]+.
Пример 40А
Этиловый эфир 1H-имидазол-1-илуксусной кислоты
Медленно прибавляли 118.2 г (5.1 моль) натрия к 2.5 литрам этанола. После чего добавляли 350.0 г (5.1 моль) имидазола и по каплям прибавляли 570 мл (858.6 г, 5.1 моль) этилового эфира бромуксусной кислоты при температуре реакционной смеси 20-25°С. Перемешивали при комнатной температуре 24 ч. Выпавший осадок отфильтровывали, этанол удаляли в вакууме и снова фильтровали. Остаток очищали хроматографией на колонке с силикагелем (растворитель: этиловый эфир уксусной кислоты)
Выход: 639.0 г (81% от теоретического)
ГХ-МС (Метод 14): Rt=4.55 mm; МС (ESIpos): m/z=155 [M+H]+.
Пример 41А
Этиловый эфир (4-циано-1H-1,2,3-триазол-1-ил)уксусной кислоты
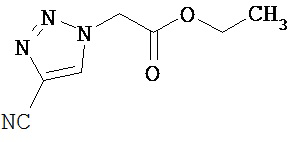
Перемешивали 4.1 г (31.9 ммоль) этилового эфира азидоуксусной кислоты и 2.8 г (31.9 ммоль) нитрила 2-хлоракриловой кислоты в 32 мл воды 16 ч при температуре бани 80°С. После охлаждения до комнатной температуры раствор подкисляли 1 N соляной кислотой и экстрагировали этиловым эфиром уксусной кислоты. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток смешивали с 50 мл этанола и 10 каплями концентрированной серной кислоты и перемешивали 16 ч с обратным холодильником. Затем реакционную смесь концентрировали в вакууме, остаток смешивали с этиловым эфиром уксусной кислоты, суспензию промывали полуконцентрированным раствором гидрокарбоната натрия и сушили органическую фазу над сульфатом натрия. Растворитель полностью удаляли на ротационном испарителе и твердое вещество сушили в вакууме.
Выход: 1.5 г (25% от теоретического)
ЖХ-МС (Метод 7): Rt=0.96 мин; МС (ESIpos): m/z=181 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.06 (s, 1H), 5.57 (s, 2H), 4.19 (q, 2H), 1.22 (t, 3Н).
Пример 42А
Этиловый эфир 3-(диметиламино)-2-(1H-имидазол-1-ил)акриловой кислоты

Перемешивали 38.0 г (244.9 ммоль) соединения из примера 39А с 126 мл (108.1 г, 734.7 ммоль) диметилформамид-диэтилацеталя 16 ч при температуре бани 90°С. После охлаждения концентрировали в вакууме, смешивали с диизопропиловым эфиром, отфильтровывали твердое вещество, промывали его затем диизопропиловым эфиром.
Выход: 49.0 г (95% от теоретического)
ЖХ-МС (Метод 4): Rt=2.42 мин; МС (ESIpos): m/z=211 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.52 (s, 1H), 7.49 (s, 1H), 7.05 (s, 1H), 6.91 (s, 1H), 4.02 (q, 2H), 2.63 (уш. s, 6H), 1.12 (t, 3H).
Пример 43А
Этиловый эфир 3-(диметиламино)-2-(1H-1,2,4-триазол-1-ил)-акриловой кислоты

Перемешивали 1.9 г (9.8 ммоль) этилового эфира [1,2,4]триазол-1-илуксусной кислоты [Ainsworth et al., J. Am. Chem. Soc. 1955, 77, 621-623] и 3.6 мл (2.9 г, 19.6 ммоль) диметилформамид-диэтилацеталя 12 ч при температуре бани 100°C. Затем охлажденный реакционный раствор упаривали на ротационном испарителе и остаток сушили в вакууме.
Выход: 2.3 г (90%-ной чистоты, 100% от теоретического)
ЖХ-МС (Метод 1): Rt=2.32 мин; МС (ESIpos): m/z=211 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.48 (s, 1H), 8.04 (s, 1H), 7.61 (s, 1H), 4.03 (q, 2H), 3.04 (уш. s, 3H), 2.25 (уш. s, 3H), 1.12 (t, 3H).
Пример 44А
Этиловый эфир 3-(диметиламино)-2-[4-(трифторметил)-1H-имидазол-1-ил]акриловой кислоты

Перемешивали 38.0 г (170.8 ммоль) соединения из примера 32А и 58.5 мл (50.3 г, 341.6 ммоль) диметилформамид-диэтилацеталя 16 ч при температуре бани 100°C. Затем охлажденный реакционный раствор упаривали на ротационном испарителе и остаток сушили в вакууме.
Выход: 49.5 г (97% от теоретического)
ЖХ-МС (Метод 8): Rt=1.68 мин; МС (ESIpos): m/z=278 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.81 (s, 1H), 7.27 (s, 1H), 7.58 (s, 1H), 4.03 (q, 2H), 2.68 (ш. s, 6Н), 1.13 (t, 3H).
Пример 45А
Этиловый эфир 3-(диметиламино)-2-[4-циано-1H-имидазол-1-ил]акриловой кислоты

Перемешивали 3.8 г (21.4 ммоль) соединения из примера 33А и 7.4 мл (6.3 г, 42.8 ммоль) диметилформамид-диэтилацеталя 16 ч при температуре бани 100°С. Затем охлажденный реакционный раствор упаривали на ротационном испарителе и остаток сушили в вакууме.
Выход: 5.0 г (73%-ной чистоты, 73% от теоретического)
ЖХ-МС (Метод 1): Rt=2.69 мин; МС (ESIpos): m/z=235 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.13 (s, 1H), 7.85 (s, 1H), 7.58 (s, 1H), 4.03 (q, 2H), 2.69 (уш. s, 6H), 1.12 (t, 3H).
Пример 45A
Этиловый эфир 3-(диметиламино)-2-[4-метил-1H-имидазол-1-ил]акриловой кислоты

Перемешивали 310 мг (1.5 ммоль. 80% -ной чистоты) соединения из примера 34А и 0.5 мл (434 мг, 3.0 ммоль) диметилформамид-диэтилацеталя 16 ч при температуре бани 100°С. Затем охлажденный реакционный раствор упаривали на ротационном испарителе и остаток сушили в вакууме. Продукт получался в виде смеси 3:2 обоих региоизомеров и в таком виде использовался для дальнейших превращений.
Выход: 329 мг (99% от теоретического)
ЖХ-МС (Метод 1): Rt=2.05 мин; МС (ESIpos): m/z=224 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.50 (s, 1H), [7.58 (s, 1H)], 7.32 (d, 1H), [7.38 (d, 1H)], 6.73 (s, 1H), [6.66 (s, 1H)], 4.04-3.98 (m, 2H), 2.64 (уш. s, 6H), 2.08 (s, 3H), [1.97 (s, 3H)], 1.12 (t, 3H).
Приведенные в таблице 3 соединения получены из соответствующих эдуктов и диметилформамид-диэтилацеталя аналогично примеру 43А:


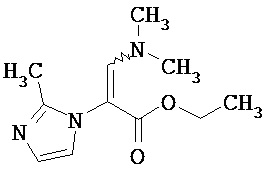


Пример 52А
2-(6-Хлорпиримидин-4-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Помещали 10.0 г (47.7 ммоль) соединения из примера 3А и 8.3 г (57.1 ммоль) соединения из примера 7А в 100 мл этанола и смешивали с 1.5 мл (2.2 г, 19.0 ммоль) трифторуксусной кислоты. Перемешивали с обратным холодильником 12 ч. Затем к охлажденной реакционной смеси добавили избыток 4 М раствора хлористого водорода в диоксане, перемешивали примерно 1 ч, отфильтровывали выпавшие кристаллы и промывали осадок на фильтре диоксаном и этанолом. Полученный таким образом промежуточный продукт растворяли в 150 мл этанола, смешивали с 50 мл 25%-ного метанольного раствора метилата натрия и перемешивали при комнатной температуре 2 ч. После этого доводили рН реакционной смеси с помощью 1 N соляной кислоты до 5, перемешивали еще 2 ч при комнатной температуре, твердое вещество отфильтровывали, осадок на фильтре промывали этанолом и продукт сушили в вакууме.
Выход: 7.0 г (49% от теоретического)
ЖХ-МС (Метод 10): Rt=1.20 мин; МС (ESIpos): m/z=264 [M+H]+.
Пример 53А
2-(6-Хлорпиримидин-4-ил)-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Помещали 10.0 г (47.8 ммоль) соединения из примера 42А и 8.3 г (57.3 ммоль) соединения из примера 7А в 100 мл этанола и смешивали с 1.5 мл (2.2 г, 19.0 ммоль) трифторуксусной кислоты. Перемешивали с обратным холодильником 12 ч. Отфильтровывали выпавшие кристаллы, промывали осадок на фильтре этанолом и сушили промежуточный продукт в течение ночи в вакууме. Затем этот продукт суспендировали в 20 мл метанола и смешивали с 100 мл 4 М раствора хлористого водорода в диоксане и перемешивали при комнатной температуре еще 1 ч. Твердое вещество отфильтровывали, осадок на фильтре промывали диоксаном, этиловым эфиром уксусной кислоты и диизопропиловым эфиром и продукт сушили в вакууме.
Выход: 4.6 г (32% от теоретического)
ВЭЖХ (Метод 11): Rt=2.81 мин; МС (ESIpos): m/z=263 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.46 (s, 1H), 8.96 (s, 1H), 8.56 (s, 1H), 8.51 (d, 1H), 8.07-8.04 (m, 1H), 7.85-7.82 (m, 1H).
Пример 54А
2-(6-Морфолин-4-илпиримидин-4-ил)-3-оксо-2,3-дигидро-1H-пиразол-4-этиловый эфир

Растворяли 1.4 г (10.0 ммоль) карбоната калия в 50 мл воды. Добавляли к раствору 2.0 г (10.0 ммоль) соединения из примера 16А и затем 2.2 г (10.0 ммоль) диэтилового эфира этоксиметиленмалоновой кислоты и перемешивали после этого 2 ч при 100°С. Охлаждали до комнатной температуры, отфильтровывали твердое вещество, промывали дважды водой и сушили в высоком вакууме.
Выход: 2.4 г (75% от теоретического)
ЖХ-МС (Метод 8): Rt=1.31 мин; МС (ESIpos): m/z=320 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.30 (s, 1H), 7.97 (s, 1H), 7.48 (s, 1H), 4.00 (q, 2H), 3.75- 3.61 (m, 4H), 3.55-3.45 (m, 4H), 1.18 (t, 3H).
Примеры осуществления:
Пример 1
2-Пиридин-2-ил-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он

Растворяли 250 мг (1.19 ммоль) соединения из примера 3А, 108 мг (0.99 ммоль) 2-гидразинопиридина и 23 мг (99 мкмоль) 10-камфорсульфокислоты в 5 мл безводного этанола и кипятили с обратным холодильником в течение ночи. Снова добавляли в общей сложности четырежды по 108 мг (0.99 ммоль) 2-гидразинопиридина и кипятили снова с обратным холодильником до тех пор, пока соединение из примера 3А полностью не вступило в реакцию. После охлаждения реакционную смесь очищали многократной препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент при добавлении 0.1% концентрированной соляной кислоты) и получали 6 мг (2% от теоретического) названного соединения.
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.52 (d, 1H), 8.44 (s, 1H), 8.37 (s, 1H), 8.29-8.23 (m, 1H), 8.11-8.06 (m, 1H), 7.90 (s, 1H), 7.43-7.38 (m, 1H).
ЖХ-МС (Метод 2): Rt=1.17 мин; MC (ESIpos): m/z=229 [M+H]+.
Пример 2
2-(4-Метилпиридин-2-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
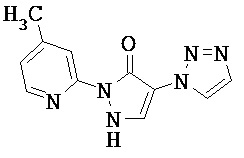
Растворяли 250 мг (1.19 ммоль) соединения из примера 3А, 122 мг (0.99 ммоль) соединения из примера 1А и 23 мг (99 мкмоль) 10-камфорсульфокислоты в 5 мл безводного этанола и кипятили с обратным холодильником в течение ночи. После чего охлаждали и предварительно очищали реакционную смесь с помощью препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент) и последующей Flash-хроматографией на силикагеле (растворитель: дихлорметан/метанол-градиент). Повторная очистка с помощью препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент) давала в конце концов 21 мг (9% от теоретического) названного соединения.
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1H), 8.38 (d, 1H), 8.29 (s, 1H), 8.09 (s, 1H), 7.90 (s, 1H), 7.27 (d, 1H), 2.46 (s, 3H).
ЖХ-МС (Метод 4): Rt=1.05 мин; MC (ESIpos): m/z=243 [M+H]+.
Пример 3
2-(6-Морфолин-4-ил-пиримидин-4-ил)-4-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]-1,2-дигидро-3H-пиразол-3-он
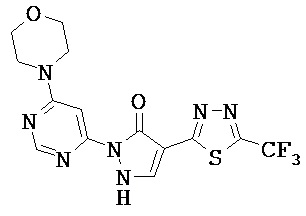
Растворяли 2.43 г (8.62 ммоль) соединения из примера 2А, 1.53 г соединения из примера 16А и 182 мг (784 мкмоль) 10-камфорсульфокислоты в 35 мл безводного метанола и кипятили с обратным холодильником в течение ночи. После охлаждения упаривали, снова растворяли остаток в 325 мл метанола, добавляли 0.5 мл (8.62 ммоль) 25%-ного метанольного раствора метилата натрия и снова кипятили с обратным холодильником в течение ночи. После охлаждения отфильтровывали выделившийся осадок, промывали его диэтиловым эфиром, суспендировали в небольшом количестве воды, смешивали с 1 М соляной кислотой и суспензию снова упаривали. Остаток промывали водой и диэтиловым эфиром. Получали 1.34 г (43% от теоретического) названного соединения.
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.55 (s, 1H), 8.26 (s, 1H), 7.49 (s, 1H), 3.84-3.70 (m, 8H).
ЖХ-МС (Метод 4): Rt=1.68 мин; MC (ESIpos): m/z=400 [M+H]+.
Пример 4
2-(6-Пиперидин-1-ил-пиримидин-4-ил)-4-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]-1,2-дигидро-3H-пиразол-3-он

Растворяли 240 мг (854 мкмоль) соединения из примера 2А, 150 мг (776 мкмоль) соединения из примера 8А и 18 мг (78 мкмоль) 10-камфорсульфокислоты в 3 мл безводного этанола и кипятили с обратным холодильником в течение ночи. После охлаждения добавляли 64 мг (931 мкмоль) этилата натрия и снова перемешивали с обратным холодильником в течение ночи при комнатной температуре. Образовавшийся остаток отфильтровывали, промывали этанолом и диэтиловым эфиром и сушили. Фильтрат упаривали и остаток очищали с помощью препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент). Обе полученных таким образом фракции промежуточного продукта вместе суспендировали в 5 мл метанола, смешивали с 15 мг (278 мкмоль) метилата натрия и кипятили с обратным холодильником в течение ночи. После охлаждения очищали реакционную смесь с помощью препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент) и получали 26 мг (8% от теоретического) названного соединения.
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.33 (s, 1H), 7.87 (s, 1H), 7.76 (s, 1H), 3.63-3.58 (m, 4H), 1.68-1.51 (m, 6H).
ЖХ-МС (Метод 3): Rt=2.23 мин; MC (ESIpos): m/z=398 [M+H]+.
Соединения, указанные в таблице 4, получали из соответствующих эдуктов аналогично примеру 4:


Пример 7
2-(4-Метилпиридин-2-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Растворяли 3.7 г (17.6 ммоль) соединения из примера 3А и 2.6 г (21.1 ммоль) соединения из примера 1А в 100 мл безводного этанола и смешивали с 0.7 мл (1.0 г, 8.8 ммоль) трифторуксусной кислоты. Перемешивали 16 ч при температуре бани 100°С. После чего охлаждали до комнатной температуры и упаривали в вакууме. Остаток растворяли в 35 мл ацетонитрила и смешивали при комнатной температуре с 9 мл (35.2 ммоль) 4 N раствора хлористого водорода в диоксане. Осадок отделяли и промывали 35 мл ацетонитрила. Промывали твердое вещество диизопропиловым эфиром, затем нагревали в 10 мл метанола до 40°С, смешивали с 10 мл этилового эфира уксусной кислоты, фильтровали и промывали 5 мл смеси 1:1 из метанола и этилового эфира уксусной кислоты и 2 мл этилового эфира уксусной кислоты.
Выход: 2.0 г (40% от теоретического)
ЖХ-МС (Метод 2): Rt=1.05 мин; МС (ESIpos): m/z=243 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.42-8.40 (m, 1Н), 8.37 (d, 1H), 8.29 (s, 1H), 8.09 (s, 1H), 7.89 (d, 1H), 7.27 (d, 1H), 2.46 (s, 3H).
Пример 8
Гидрохлорид этилового эфира 6-[5-Оксо-4-(1H-1,2,3-триазол-1-ил)-2,5-дигидро-1H-пиразол-1-ил]никотиновой кислоты
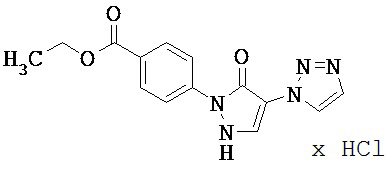
Перемешивали 227 мг (1.1 ммоль) соединения из примера 3А, 245 мг (1.1 ммоль) этилового эфира 6-гидразиноникотиновой кислоты [получение см. в WO 2006/114213] и 50 мг (0.2 ммоль) 10-камфорсульфокислоты в 6 мл этанола 3 ч с обратным холодильником. Охлажденную реакционную смесь после этого смешивали еще с 490 мг (2.2 ммоль) этилового эфира 6-гидразиноникотиновой кислоты и 37 мг (0.2 ммоль) п-толуолсульфокислоты и перемешивали 1 день при температуре кипения. Затем реакционную смесь концентрировали в вакууме и остаток хроматографировали с помощью препаративной ВЭЖХ (метод 12). Полученную из разделения методом ВЭЖХ лиофилизированную трифторацетатную соль смешивали с 4 мл 4 N раствора хлористого водорода в диоксане, суспензию частично упаривали, твердое вещество отфильтровывали, осадок промывали на фильтре диэтиловым эфиром и продукт сушили в вакууме.
Выход: 139 мг (38% от теоретического)
ЖХ-МС (Метод 1):): Rt=2.88 мин; МС (ESIpos): m/z=301 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.00 (s, 1H), 8.60-8.57 (m, 1H), 8.53-8.46 (m, 2H), 8.45 (s, 1H), 7.91 (s, 1H), 4.38 (q, 2H), 1.5 (t, 3H).
Пример 9
2-(6-Пиперидин-1-илпиримидин-4-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
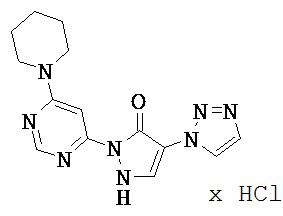
Помещали 400 мг (1.8 ммоль) соединения из примера 3А, 338 мг (1.8 ммоль) соединения из примера 8А и 60 мг (0.4 ммоль) п-толуолсульфокислоты в смесь из 2 мл ТГФ и 2 мл этанола и осуществляли реакцию в течение 1 ч при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer) при температуре 140°С. Охлажденную реакционную смесь концентрировали в вакууме и остаток смешивали с 2 мл 4 N раствора хлористого водорода в диоксане, диэтиловом эфире и ацетонитриле. Выпавший осадок отфильтровывали и хроматографировали осадок с помощью препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент при добавлении 0.1% муравьиной кислоты в воде). Полученную таким образом лиофилизированную формиатную соль смешивали с 4 мл 4 N раствора хлористого водорода в диоксане, суспензию частично упаривали, твердое вещество отфильтровывали, осадок на фильтре промывали диэтиловым эфиром и продукт сушили в вакууме.
Выход: 75 мг (12% от теоретического)
ЖХ-МС (Метод 1): Rt=2.75 мин; МС (ESIpos): m/z=313 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.51 (s, 1Н), 8.38 (s, 1H), 8.21 (s, 1H), 7.85 (s, 1H), 7.38 (s, 1H), 3.76-3.72 (m, 4H), 1.74-1.63 (m, 2H), 1.59 (s, 9H).
Пример 10
4-(1H-Имидазол-1-ил)-2-(6-пиперидин-1-илпиримидин-4-ил)-1,2-дигидро-3H-пиразол-3-он

Растворяли 5.5 г (26.3 ммоль) соединения из примера 42А и 6.1 г (31.5 ммоль) соединения из примера 8А в 55 мл этилового эфира уксусной кислоты и смешивали с 1.2 г (10.5 ммоль) п-толуолсульфокислоты. Смесь кипятили с обратным холодильником и перемешивали при этой температуре 16 ч. После охлаждения до комнатной температуры осадок отфильтровывали и промывали этиловым эфиром уксусной кислоты. Твердое вещество растворяли в 50 мл воды и устанавливали величину рН раствора на 7 с помощью 1 N соляной кислоты. Осадок отфильтровывали, промывали водой и диизопропиловым эфиром и затем сушили над пятиокисью фосфора.
Выход: 7.8 г (95% от теоретического)
ЖХ-МС (Метод 1): Rt=2.32 мин; МС (ESIpos): m/z=312 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.42 (s, 1Н), 8.53 (s, 1H), 8.28 (s, 1H), 8.04 (s, 1H), 7.82 (s, 1H), 7.45 (s, 1H), 3.76-3.72 (m, 4H), 1.74-1.52 (m, 6H).
Пример 11
4-(1H-Имидазол-1-ил)-2-(4-метилпиридин-2-ил)-1,2-дигидро-3H-пиразол-3-он

Растворяли 200 мг (1.0 ммоль) соединения из примера 42А и 118 мг (1.0 ммоль) соединения из примера 1А в 2 мл этанола и смешивали с 44 мг (0.2 ммоль) 10-камфорсульфокислоты. Смесь кипятили с обратным холодильником 16 ч, затем концентрировали и остаток очищали с помощью препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты в воде).
Выход: 4 мг (2% от теоретического)
ЖХ-МС (Метод 1): Rt=1.94 мин; МС (ESIpos): m/z=242 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.34 (d, 1H), 8.18 (s, 1H), 8.12-8.09 (m, 2H), 7.53 (s, 1H), 7.19 (d, 1H), 7.09 (s, 1H), 2.42 (s, 3H).
Пример 12
2-[5-(Гидроксиметил)пиридин-2-ил]-4-[4-{трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он
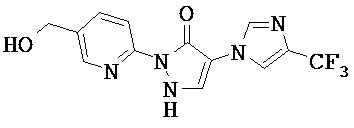
Растворяли 1.0 г (3.6 ммоль) соединения из примера 44А и 502 мг (3.6 ммоль) соединения из примера 4А в 1 мл ледяной уксусной кислоты и перемешивали при комнатной температуре 16 ч. Затем снова добавляли 200 мг (1.4 ммоль) соединения из примера 4А и перемешивали при комнатной температуре еще 20 ч. Смешивали реакционную смесь с 5 мл этилового эфира уксусной кислоты и с помощью разбавленного водного раствора гидрокарбоната натрия устанавливали величину рН 7. Водную фазу упаривали в вакууме, остаток смешивали с 1.5 мл (4.0 ммоль) 21%-ного этанольного раствора этилата натрия и перемешивали при комнатной температуре 4 ч. Осадок отфильтровывали и промывали этанолом.
Выход: 111 мг (9% от теоретического)
ЖХ-МС (Метод 4): Rt=1.70 мин; МС (ESIpos): m/z=326 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.44 (s, 1H), 8.39-8.21 (m, 2H), 8.19 (s, 1H), 8.14 (s, 1H), 7.98 (dd, 1H), 5.41 (t, 1H), 4.58 (d, 2H).
Пример 13
2-[5-(Гидроксиметил)пиридин-2-ил]-4-[4-(трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Растворяли 7.5 г (27.0 ммоль) соединения из примера 44А и 3.9 г (27.0 ммоль) соединения из примера 4А в 50 мл этанола, смешивали с 1.3 г (5.4 ммоль) 10-камфорсульфокислоты и перемешивали 16 ч при комнатной температуре. Далее, растворяли 1.5 г (5.4 ммоль) соединения из примера 44А и 0.8 г (5.4 ммоль) соединения из примера 4А в 10 мл этанола, смешивали с 0.2 г (1.1 ммоль) п-толуолсульфокислоты и перемешивали 16 ч при комнатной температуре. После чего объединяли обе смеси, упаривали в вакууме и очищали хроматографированием на колонке с силикагелем (растворитель: ацетонитрил/вода 4:1). После удаления растворителя твердое вещество смешивали с 2 мл 4 N раствора хлористого водорода в диоксане, упаривали в вакууме и остаток сушили.
Выход: 1.5 г (14% от теоретического)
ВЭЖХ (Метод 11): Rt=3.38 мин; МС (ESIpos): m/z=326 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.43 (s, 1H), 8.39-8.21 (m, 2H), 8.19 (s, 1H), 8.15 (s, 1H), 7.98 (dd, 1H), 5.48-5.38 (m, 1H), 4.57 (d, 2H).
Пример 14
2-(4-Метилпиридин-2-ил)-4-[4-(трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

4.1 г (13.4 ммоль) соединения из примера 44А, 1.9 г (15.7 ммоль) соединения из примера 1А и 0.5 г (2.7 ммоль) п-толуолсульфокислоты в смеси из 8 мл ТГФ и 12 мл этанола взаимодействовали 1 ч при микроволновом облучении (single-mode-Mikrowelle) (Emrys Optimizer) при 160°С. После удаления всех летучих компонентов в вакууме остаток смешивали с этиловым эфиром уксусной кислоты и водой. Отделенную органическую фазу промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия и отфильтровывали осушитель. Фильтрат концентрировали в вакууме и сырой продукт хроматографировали методом ВЭЖХ (метод 15). Полученный лиофилизат смешивали с 5 мл 4 N раствора хлористого водорода в диоксане, частично упаривали в вакууме, суспензию смешивали с ацетонитрилом и диэтиловым эфиром, кристаллы отфильтровывали, осадок на фильтре промывали н-пентаном и продукт сушили в вакууме.
Выход: 0.4 г (9% от теоретического)
ЖХ-МС (Метод 1): Rt=3.11 мин; МС (ESIpos): m/z=310 [М+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.37 (d, 1Н), 8.31 (s, 1H), 8.19 (s, 1H), 8.19 (s, 1H), 8.16 (s, 1H), 8.12 (s, 1H), 7.23 (d, 1H), 2.44 (s, 3H).
Пример 15
2-(6-Пиперидин-1-илпиримидин-4-ил)-4-[4-(трифторметил)-1Н-имидазол-1-ил]-1,2-дигидро-3Н-пиразол-3-он-гидрохлорид

200 мг (0.7 ммоль) Соединения из примера 44А, 139 мг (0.7 моль) соединения из примера 8А и 24 мг (0.1 ммоль) п-толуолсульфокислоты в 3 мл ТГФ взаимодействовали 1 ч при 160°С при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). После удаления всех летучих компонентов в вакууме остаток хроматографировали с помощью препаративной ВЭЖХ (колонка RP18; элюент: ацетонитрил/вода-градиент при добавлении в воду 0.1% муравьиной кислоты). Сконцентрированную фракцию с продуктом смешивали с 2 мл 4 N раствора хлористого водорода в диоксане и диэтиловом эфире, выпавшие кристаллы отфильтровывали, осадок на фильтре промывали диэтиловым эфиром и продукт сушили в вакууме.
Выход: 72 мг (24% от теоретического)
ЖХ-МС (Метод 1): Rt=3.29 мин; МС (ESIpos): m/z=380 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.49 (s, 1H), 8.25 (s, 1H), 8.15 (s, 1H), 8.11 (s, 1H), 7.43 (s, 1H), 3.69 (s, 4H), 1.69-1.66 (m, 2H), 1.58 (s, 9H).
Пример 16
1-[3-Оксо-2-(6-пиперидин-1-илпиримидин-4-ил)-2,3-дигидро-1Н-пиразол-4-ил]-1Н-имидазол-4-карбонитрил-гидрохлорид

Растворяли 200 мг (0.9 ммоль) соединения из примера 45А и 165 мг (0.9 ммоль) соединения из примера 8А в 2 мл этанола и смешивали с 29 мг (0.2 ммоль) п-толуолсульфокислоты. Перемешивали 48 ч при 90°С. После охлаждения до комнатной температуры упаривали и остаток очищали методом препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты). Сконцентрированную фракцию с продуктом смешивали с избытком 4 N раствора хлористого водорода в диоксане и диэтиловом эфире, выпавшие кристаллы отфильтровывали, осадок на фильтре промывали диэтиловым эфиром и продукт сушили в вакууме.
Выход: 18 мг (6% от теоретического)
ВЭЖХ (Метод 11): Rt=3.64 мин; МС (ESIpos): m/z=337 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.49 (s, 1H), 8.40 (s, 1H), 8.21 (s, 1H), 8.18 (s, 1H), 7.42 (s, 1H), 3.75-3.65 (m, 4H), 1.73-1.51 (m, 6H).
Пример 17
2-(6-Циклопропилпиримидин-4-ил)-4-(1Н-1,2,3-триазол-1-ил)-1,2-дигидро-3Н-пиразол-3-он-гидрохлорид

Растворяли 280 мг (1.3 ммоль) соединения из примера 3А и 200 мг (1.3 ммоль) соединения из примера 37А в 2 мл этанола и смешивали с 21 мкл (30 мг, 0.3 ммоль) трифторуксусной кислоты. Перемешивали 12 ч с обратным холодильником. После охлаждения до комнатной температуры реакционный раствор сразу хроматографировали с помощью препаративной ВЭЖХ (колонка RPI8; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты). При этом во фракции с продуктом названное соединение отчасти выпадает в осадок (в виде свободного основания). Отфильтровывали осадок, промывали его на фильтре диэтиловым эфиром и фильтрат смешивали с избытком 4 N раствора хлористого водорода в диоксане. Перемешивали при комнатной температуре, выпавший осадок отфильтровывали, осадок на фильтре промывали диэтиловым эфиром и продукт сушили в вакууме.
Выход: 16 мг (4% от теоретического)
ВЭЖХ (Метод 11): Rt=3.11 мин;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.90 (s, 1H), 8.59 (m, 1H), 8.44 (s, 1H), 8.29 (s, 1H), 7.91 (s, 1H), 2.26-2.24 (m, 1H), 1.17-1.05 (m, 4Н).
Пример 18
2-(6-Циклопропилпиримидин-4-ил)-4-(1Н-1,2,3-триазол-1-ил-1,2-дигидро-3Н-пиразол-3-он

Растворяли 280 мг (13 ммоль) соединения из примера 3А и 200 мг (1.3 ммоль) соединения из примера 37А в 2 мл этанола и смешивали с 21 мкл (30 мг, 0.3 ммоль) трифторуксусной кислоты. Перемешивали 12 ч с обратным холодильником. После охлаждения до комнатной температуры реакционный раствор сразу хроматографировали с помощью препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты). При этом во фракции с продуктом названное соединение отчасти выпадает в осадок. Отфильтровывали осадок и сушили в вакууме.
Выход: 5 мг (1.3% от теоретического)
ВЭЖХ (Метод 11): Rt=3.10 мин;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.89 (s, 1H), 8.59-8.55 (m, 1H), 8.44 (s, 1H), 8.29 (s, 1H), 7.91 (s, 1H), 2.25-2.23 (m, 1H), 1.17-1.05 (m, 4H).
Пример 19
2-[5-(Гидроксиметил)пиридин-2-ил]-4-(1Н-1,2,3-триазол-1-ил)-1,2-дигидро-3Н-пиразол-3-он

Растворяли 15.1 г (71.9 ммоль) соединения из примера 3А и 10.0 г (71.9 ммоль) соединения из примера 4А в 375 мл этанола и смешивали с 1.7 г (7.2 ммоль) 10-камфор-сульфокислоты. Кипятили 16 ч с обратным холодильником. После охлаждения до комнатной температуры реакционную смесь упаривали и остаток чистили хроматографированием на колонке с силикагелем (растворитель: дихлорметан/метанол 9:1, затем 1:1). Фракцию с продуктом концентрировали в вакууме, остаток смешивали с диизопропиловым эфиром, отфильтровывали и сушили.
Выход: 1.0 г (5% от теоретического)
ЖХ-МС (Метод 1): Rt=2.14 мин; МС (ESIpos): m/z=259 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.46 (d, 1H), 8.43 (s, 1H), 8.34 (s, 1H), 8.23 (d, 1H), 8.01 (dd, 1H), 7.90 (s, 1H), 5.43 (уш. s, 1H), 4.58 (s, 2H).
Пример 20
1-[3-Оксо-2-(6-пиперидин-1-илпиримидин-4-ил)-2,3-дигидро-1Н-пиразол-4-ил]-1Н-1,2,3-триазол-4-карбонитрил-гидрохлорид
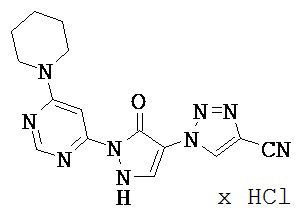
Растворяли 400 мг (1.7 ммоль) соединения из примера 51A и 328 мг (1.7 ммоль) соединения из примера 8А в 4 мл этанола и смешивали с 59 мг (0.3 ммоль) п-толуолсульфокислоты. Взаимодействие осуществляли при 120°С в течение 1 ч при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). После охлаждения до комнатной температуры реакционную смесь концентрировали и остаток очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты). Выпавшую при этом в осадок соль муравьиной кислоты переводили в гидрохлорид добавлением 0.2 мл 4 N раствора хлористого водорода в диоксане.
Выход: 4 мг (1% от теоретического)
ЖХ-МС (Метод 8): Rt=1.73 мин; МС (ESIpos): m/z=338 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.35 (s, 1H), 8.47 (s, 1H), 8.47 (s, 1H), 8.33 (s, 1H), 7.05 (s, 1H), 3.75-3.65 (m, 4H), 1.67-1.64 (m, 2H), 1.57-1.55 (m, 4H).
Пример 21
2-[6-(Диметиламино)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3Н-пиразол-3-он

3.7 г (16.5 ммоль) соединения из примера 3А, 2.4 г соединения из примера 7А и 0.6 г (3.3 ммоль) п-толуолсульфокислоты в смеси из 10 мл этанола и 5 мл ТГФ взаимодействовали в течение 1 ч при 140°С при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). Выпавший при этом осадок отфильтровывали, осадок промывали на фильтре смесью этанола и диэтилового эфира и продукт сушили в вакууме.
Выход: 0.8 г (17% от теоретического)
ЖХ-МС (Метод 10): Rt=0.47 мин; МС (ESIpos): m/z=273 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.53 (s, 1H), 8.38 (s, 1H), 8.20 (s, 1H), 7.85 (s, 1H), 7.22 (s, 1H), 3.21 (s, 6H).
Пример 22
2-(5-Бромпиридин-2-ил)-4-(1Н-1,2,3-триазол-1-ил)-1,2-дигидро-3Н-пиразол-3-он-гидрохлорид

Смесь из 280 мг (1.3 ммоль) соединения из примера 3А, 250 мг (1.3 ммоль) соединения из примера 10A и 46 мг (0.3 ммоль) п-толуолсульфокислоты в 5 мл ТГФ взаимодействовала в течение 30 мин при 170°С при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). После добавления 2 мл муравьиной кислоты к реакционному раствору выпавший осадок отфильтровывали, смешивали с 3 мл 4 N раствора хлористого водорода в диоксане, снова отфильтровывали осадок, промывали его диэтиловым эфиром и сушили в вакууме.
Выход: 181 мг (40% от теоретического)
ЖХ-МС (Метод 8): Rt=1.50 мин; МС (ESIpos): m/z=306 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.66 (s, 1H), 8.48 (s, 1H), 8.44 (s, 1H), 8.33-8.25 (m, 2H), 7.90 (s, 1H).
Пример 23
2-(5-Бромпиридин-2-ил)-4-(1Н-имидазол-1-ил)-1,2-дигидро-3Н-пиразол-3-он-гидрохлорид

Смесь из 278 мг (1.3 ммоль) соединения из примера 42А, 250 мг (1.3 ммоль) соединения из примера 8А и 46 мг (0.3 ммоль) п-толуолсульфокислоты в 5 мл ТГФ взаимодействовала 30 мин при 170°С при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). После охлаждения до комнатной температуры реакционную смесь упаривали и остаток очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты в воду). Выпавшую при этом в осадок соль муравьиной кислоты переводили в гидрохлорид добавлением 2 мл 4 N раствора хлористого водорода в диоксане. Продукт промывали диэтиловым эфиром и сушили в вакууме.
Выход: 60 мг (13% от теоретического)
ЖХ-МС (Метод 8): Rt=1.04 мин; МС (ESIpos): m/z=307 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.49 (s, 1H), 8.67 (s, 1H), 8.60 (s, 1H), 8.39-8.25 (m, 2H), 8.06 (s, 1H), 7.85 (s, 1H).
Пример 24
2-(6-Хлорпиримидин-4-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Смесь из 7.3 г (34.6 ммоль) соединения из примера 3А и 6.0 г (41.5 ммоль) соединения из примера 7А в 70 мл этанола смешивали с 1.1 мл (1.6 г, 13.8 ммоль) трифторуксусной кислоты и перемешивали 20 ч при 100°С. Выпавший осадок отфильтровывали и фильтрат упаривали в вакууме. Остаток суспендировали в 100 мл этанола, смешивали с 30 мл 30%-ного раствора метилата натрия в метаноле и перемешивали 1.5 ч при комнатной температуре. После добавления 42 мл 4 N раствора хлористого водорода в диоксане (рН=5-6) и 30 мин перемешивания твердое вещество отфильтровывали, промывали сначала этанолом, затем диэтиловым эфиром и сушили в вакууме. Последующая очистка происходила путем многократного смешивания с этанолом и ацетонитрилом. После чего добавляли 10 мл 4 N раствора хлористого водорода в диоксане и перемешивали 16 ч при комнатной температуре. Продукт отфильтровывали, промывали сначала ацетонитрилом, затем диэтиловым эфиром и сушили в вакууме.
Выход: 7.9 г (76% от теоретического)
ЖХ-МС (Метод 8): Rt=1.20 мин; МС (ESIpos): m/z=264 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.97 (s, 1H), 8.59 (s, 1H), 8.47 (s, 1H), 8.43 (s, 1H), 7.89 (s, 1H).
Пример 25
2-[6-(4-Пирролидин-1-илпиперидин-1-ил)пиримидин-4-ил]-4-[4-(трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Смесь из 683 мг (2.5 ммоль) соединения из примера 44А и 711 мг (2.7 ммоль) соединения из примера 30А в 10 мл этанола смешивали с 76 мкл (112 мг, 1.0 ммоль) трифторуксусной кислоты и перемешивали 16 ч при 100°С. После добавления 10 мл муравьиной кислоты реакционный раствор очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты в воду). Выпавший осадок соли муравьиной кислоты переводили в гидрохлорид путем добавления 2 мл 4 N раствора хлористого водорода в диоксане. Гидрохлорид промывали диэтиловым эфиром и сушили в вакууме.
Выход: 315 мг (25% от теоретического)
ЖХ-МС (Метод 10): Rt=0.65 мин; МС (ESIpos): m/z=449 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=11.42 (уш.:, 1H), 8.54 (s, 1H), 8.35 (s, 1H), 8.19 (a, 1H), 8.14 (s, 1H), 7.56 (s, 1H), 4.51 (уш. 9,2H), 3.53-3.37 (m, 4H), 3.13-2.97 (m, 4H), 2.24-2.14 (m, 2H), 2.03-1.68 (m, 5H).
Пример 26
2-(5-Бромпиридин-2-ил)-4-[4-(трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Смесь из 369 мг (1.3 ммоль) соединения из примера 44А, 250 мг (1.3 ммоль) соединения из примера 10А и 46 мг (0.3 ммоль) п-толуолсульфокислоты в 5 мл ТГФ взаимодействовала в течение 30 мин при 170°С при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). После добавления к реакционному раствору 2 мл муравьиной кислоты выпавший осадок отфильтровывали, смешивали с 3 мл 4 N раствора хлористого водорода в диоксане, снова отфильтровывали, промывали сначала ацетонитрилом, затем диэтиловым эфиром и сушили в вакууме.
Выход: 163 мг (30% от теоретического)
ЖХ-МС (Метод 10): Rt=1.06 мин; МС (ESIpos): m/z=374 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.65 (s, 1H), 8.45 (s, 1H), 8.38-8.23 (m, 2H), 8.19 (s, 1H), 8.15 (s, 1H).
Пример 27
2-[5-(Гидроксиметил)пиридин-2-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
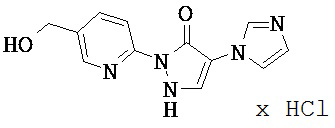
Растворяли 200 мг (1.0 ммоль) соединения из примера 42А и 133 мг (1.0 ммоль) соединения из примера 4А в 2 мл этанола и смешивали с 44 мг (0.2 ммоль) 10-камфор-сульфокислоты. Перемешивали с обратным холодильником 12 ч. Охлажденную реакционную смесь концентрировали в вакууме. После двукратной очистки методом препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты в воду) фракцию с продуктом смешивали с 1 мл 4 N раствора хлористого водорода в диоксане, перемешивали 1 ч, растворитель полностью удаляли на роторном испарителе и остаток сушили в вакууме.
Выход: 86 мг (31% от теоретического)
ЖХ-МС (Метод 1): Rt=1.77 мин; МС (ESIpos): m/z=258 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.50 (s, 1H), 8.51 (s, 1H), 8.47 (s, 1H), 8.29 (d, 1H), 8.09-8.01 (m, 2H), 7.87 (s, 1H), 3.58 (s, 2H).
Пример 28
2-[6-(Диметиламино)пиримидин-4-ил]-4-[4-(трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он

8.3 г (27.7 ммоль) соединения из примера 44А, 4.0 г (27.7 ммоль) соединения из примера 7А и 1.0 г (5.5 ммоль) п-толуолсульфокислоты в смеси из 10 мл этанола и 5 мл ТГФ взаимодействовали в течение 1 ч при 140°С при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). Выпавший осадок отфильтровывали, осадок промывали на фильтре смесью из диэтилового эфира и этанола и продукт сушили в вакууме.
Выход: 1.3 мг (14% от теоретического)
ЖХ-МС (Метод 10): Rt=0.84 мин; МС (ESIpos): m/z=340 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.50 (s, 1H), 8.24 (s, 1H), 8.15 (s, 1H), 8.11 (s, 1H), 7.28 (s, 1H), 2.54 (s, 6H).
Соединения, указанные в таблице 5, получали из соответствующих эдуктов аналогично указанным примерам:

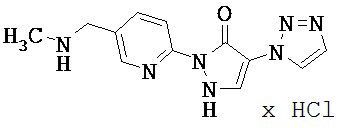

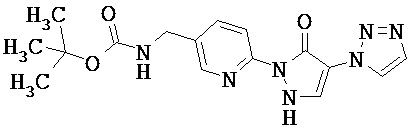




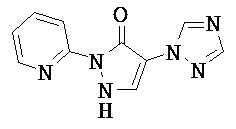






[пример]; выход (% от теоретического)
(метод)

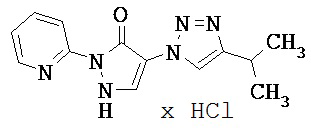











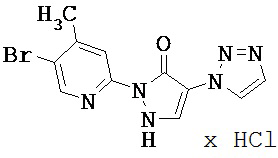


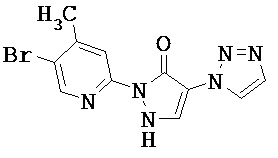
Пример 62
Гидрохлорид трет.-бутилового эфира 6-[5-Оксо-4-(1H-1,2,3-триазол-1-ил)-2,5-дигидро-1H-пиразол-1-ил]пиридин-3-карбоновой кислоты

Помещали 3.2 г (15.0 ммоль) соединения из примера 3А в 100 мл этанола. Добавляли 3.1 г (15.0 ммоль) соединения из примера 22А и 571 мг (3.0 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали с обратным холодильником 16 ч. Затем смесь упаривали и остаток очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, удаляли большую часть растворителя и отфильтровывали образовавшийся осадок. Последний сушили в высоком вакууме и затем смешивали с 4 N раствором хлористого водорода в диоксане. Перемешивали 1 ч при комнатной температуре, отфильтровывали осадок и сушили в высоком вакууме.
Выход: 1.6 г (28% от теоретического)
ЖХ-МС (Метод 1): Rt=3.32 мин; МС (ESIpo:): m/z=329 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.94 (s, 1Н), 8.53 (s, 1H), 8.50-8.40 (m, 3H), 7.91 (s, 1H), 1.58 (s, 9H).
Пример 63
Гидрохлорид трет.-бутилового эфира 6-[4-(1H-имидазол-1-ил)-5-оксо-2,5-дигидро-1H-пиразол-1-ил]пиридин-3-карбоновой кислоты
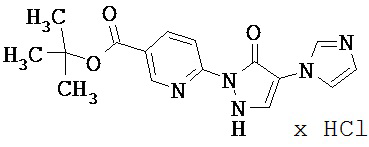
Помещали 3.1 г (15.0 ммоль) соединения из примера 42А в 100 мл этанола. Добавляли 3.1 г (15.0 ммоль) соединения из примера 22А и 571 мг (3.0 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали сначала 16 ч при комнатной температуре. После чего смесь перемешивали с обратным холодильником еще 24 ч и после этого удаляли растворитель. Остаток очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, удаляли большую часть ацетонитрила. Оставшийся раствор лиофилизировали. Смешивали лиофилизат с 4 N раствором хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Осадок отфильтровывали и сушили в высоком вакууме.
Выход: 1.3 г (23% от теоретического)
ЖХ-МС (Метод 7): Rt=0.99 мин; МС (ESIpos): m/z=328 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.55 (s, 1H), 8.94 (s, 1H), 8.70 (s, 1H), 8.53-8.42 (m, 2H), 8.10 (s, 1H), 7.88 (s, 1H), 1.58 (s, 9H).
Пример 64
Гидрохлорид трет.-бутилового эфира 6-{5-оксо-4-[4-(трифторметил)-1H-имидазол-1-ил]-2,5-дигидро-1H-пиразол-1-ил}пиридин-3-карбоновой кислоты

Помещали 4.2 г (15.0 ммоль) соединения из примера 44А в 100 мл этанола. Добавляли 3.1 г (15.0 ммоль) соединения из примера 22А и 571 мг (3.0 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 16 ч при комнатной температуре. Затем удаляли растворитель и остаток очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли и удаляли большую часть содержащегося ацетонитрила и часть воды. Отфильтровывали образовавшийся осадок и сушили его на воздухе. После чего смешивали с 20 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Осадок отфильтровывали и снова очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, смешивали с 1 N соляной кислотой и лиофилизировали.
Выход: 750 мг (12% от теоретического)
ЖХ-МС (Метод 7): Rt=2.10 мин; МС (ESIpos): m/z=396 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.93 (s, 1H), 8.59-8.38 (m, 3H), 8.19 (s, 1H), 8.14 (s, 1H), 1.59 (s, 9H).
Пример 65
6-[5-Оксо-4-(1H-1,2,3-триазол-1-ил)-2,5-дигидро-1H-пиразол-1-ил]пиридин-3-карбоновой кислоты гидрохлорид

Растворяли 50 мг (0.1 ммоль) соединения из примера 62 в 1 мл смеси 1:1 из дихлорметана и трифторуксусной кислоты и перемешивали при комнатной температуре 1 ч. Реакционный раствор концентрировали в вакууме, остаток суспендировали в 2 мл 1 N соляной кислоты и затем лиофилизировали.
Выход: 42 мг (99% от теоретического)
ЖХ-МС (Метод 9): Rt=0.82 мин; МС (ESIpos): m/z=273 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.99 (s, 1H), 8.53 (s, 1H), 8.51-8.41 (m, 3Н), 7.91 (s, 1Н).
Синтез приведенных в таблице 6 соединений осуществляется аналогично получению соединения из примера 65:


Пример 68
6-[5-Оксо-4-(1H-1,2,3-триазол-1-ил)-2,5-дигидро-1H-пиразол-1-ил]пиридин-3-карбоновая кислота

Перемешивали 491 мг (1.8 ммоль, с чистотой 80%) соединения из примера 3А и 289 мг (1.6 ммоль) этилового эфира 6-гидразиноникотиновой кислоты [получение см. в WO 2006/114213] в 10 мл уксусной кислоты 12 ч при комнатной температуре. После чего к реакционной смеси добавили еще 120 мг (0.7 ммоль) этилового эфира 6-гидразиноникотиновой кислоты и снова перемешивали 13 ч при комнатной температуре. После выдерживания в течение последующих двух суток при комнатной температуре реакционный раствор концентрировали в вакууме, остаток смешивали с этиловым эфиром уксусной кислоты, промывали насыщенным раствором гидрокарбоната натрия до нейтральной реакции, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали на роторном испарителе. Полученный таким образом промежуточный продукт растворяли в 10 мл этанола, смешивали с 0.3 мл (1.8 ммоль) 30%-ного раствора метилата натрия в метаноле и перемешивали 17 ч при комнатной температуре. Доводили рН реакционного раствора с помощью 1 N соляной кислоты до 5 и перемешивали еще 2 ч при комнатной температуре. Упаривали в вакууме, остаток смешивали с ацетонитрилом, выпавший осадок отфильтровывали, осадок промывали на фильтре диэтиловым эфиром и сушили в вакууме. Полученный таким образом сложный эфир смешивали с 9.4 мл 0.1 М этанольного раствора гидроокиси калия и перемешивали 16 ч при комнатной температуре. С помощью 1 N соляной кислоты устанавливали рН=2, растворитель удаляли на роторном испарителе, остаток смешивали с водой и экстрагировали этиловым эфиром уксусной кислоты. Отфильтровывали выпавшие кристаллы из водной фазы, осадок промывали на фильтре диэтиловым эфиром и продукт сушили в вакууме.
Выход: 32 мг (7% от теоретического)
ЖХ-МС (Метод 1): Rt=2.31 мин: MS (ESIpos): m/z=273 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=13.49 (уш. s, 1Н), 9.00 (s, 1H), 8.71-8.33 (m, 4H), 8.41 (s, 1H).
Пример 69
Этиловый эфир 6-[5-оксо-4-(1H-1,2,3-триазол-1-ил)-2,5-дигидро-1H-пиразол-1-ил]никотиновой кислоты
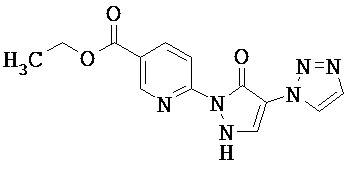
Перемешивали 2.7 г (13.1 ммоль) соединения из примера 3А, 2.0 г (13.1 ммоль) соединения из примера 6А и 1.1 г (6.5 ммоль) п-толуолсульфокислоты в 50 мл этанола 16 ч с обратным холодильником. К охлажденной реакционной смеси добавляли 50 мл диметилформамида и после этого перемешивали еще 10 ч при температуре бани 130°С. Реакционный раствор концентрировали в вакууме и остаток смешивали с 10 мл этанола и 1 мл концентрированной серной кислоты. Перемешивали при кипячении 12 h. К охлажденной реакционной смеси добавляли воду, отфильтровывали осадок, промывали осадок на фильтре этанолом и сушили продукт в вакууме.
Выход: 0.14 г (3% от теоретического)
ЖХ-МС (Метод 9): Rt=2.89 мин; МС (ESIpos): m/z=300 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.00 (s, 11H), 8.57-8.54 (m, 11H), 8.53-8.46 (m, 2H), 8.45 (s, 11H), 7.91 (s, 11H), 4.38 (q, 2H), 1.35 (t, 3H).
Пример 70
2-[5-(Аминометил)пиридин-2-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3Н-пиразол-3-он-гидрохлорид

Перемешивали 83 мг (0.2 ммоль) соединения из примера 32 в 5 мл 4 М раствора хлористого водорода в диоксане 2 ч при комнатной температуре. После чего концентрировали в вакууме и хроматографировали остаток с помощью препаративной ВЭЖХ (колонка RP18; градиент: ацетонитрил/вода с добавкой 0.1% трифторуксусной кислоты). Объединенные фракции с продуктом концентрировали и остаток смешивали с 2 мл 1 N соляной кислоты. Полученную суспензию сушили вымораживанием. Лиофилизат смешивали с этанолом, осадок отфильтровывали и кристаллы сушили в вакууме.
Выход: 10 мг (14% от теоретического)
ЖХ-МС (Метод 8): Rt=0.76 мин; МС (ESIpos): m/z=258 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.64 (s, 1H), 8.53 (s, 3H), 8.47 (s, 2H), 8.31 (s, 1H), 8.18 (s, 1H), 7.91 (s, 1H), 4.12-4.10 (m, 2H).
Пример 71
2-(6-Морфолин-4-илпиримидин-4-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он

Помещали 1.9 г (8.8 ммоль) соединения из примера 3А и 1.9 г (9.7 ммоль) соединения из примера 16А в 25 мл этилового эфира уксусной кислоты и при комнатной температуре смешивали с 504 мг (4.4 ммоль) трифторуксусной кислоты. Перемешивали 16 ч с обратным холодильником, затем охлаждали до 5°С и перемешивали еще 2 ч. Отфильтровывали образовавшийся осадок, промывали этиловым эфиром уксусной кислоты и сушили сначала на воздухе, а затем в высоком вакууме. Получили 1.7 г продукта. Маточный раствор объединяли с промывным раствором и растворитель удаляли. Остаток (2.4 г), по данным ЖХ-МС еще содержит промежуточное соединение этиловый эфир 3-[2-(6-морфолин-4-илпиримидин-4-ил)-гидразино]-2-(1H-1,2,3-триазол-1-ил)проп-2-еновой кислоты (промежуточная ступень циклизации), который применяется непосредственно для получения в примере 72 (см. там).
Выход: 1.7 г (61% от теоретического)
ЖХ-МС (Метод 9): Rt=0.90 мин; МС (ESIpos): m/z=315 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1Н), 8.38 (s, 1H), 8.01 (s, 1H), 7.73 (s, 1H), 7.70 (s, 1H), 3.71-3.65 (m, 4H), 3.57-3.51 (m, 4Н).
Пример 72
2-(6-Морфолин-4-илпиримидин-4-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Партия 1: 1.7 г (5.4 ммоль) соединения из примера 71 смешивали с 7.5 мл 4 N раствора хлористого водорода в диоксане. Перемешивали при комнатной температуре, смешивали с 5 мл диоксана и перемешивали при комнатной температуре 16 ч. Отфильтровывали осадок и промывали 5 мл диоксана. Сушили 16 ч в высоком вакууме, затем смешивали с 10 мл метанола и перемешивали 1 ч при комнатной температуре. Осадок отфильтровывали, промывали 4 мл метанола и сушили в высоком вакууме. Получили 1.6 г названного соединения.
Партия 2: Другие количества названного соединения получены следующим образом: Растворяли осадок, полученный при синтезе соединения из примера 71 из маточника (2.4 г), который содержит промежуточную стадию циклизации с открытым циклом - этиловый эфир 3-[2-(6-морфолин-4-илпиримидин-4-ил)гидразино]-2-(1H-1,2,3-триазол-1-ил)проп-2-еновой кислоты, в 12 мл этанола и смешивали при перемешивании при комнатной температуре с 1.5 мл 30%-ного раствора метилата натрия в метаноле. Перемешивали 45 мин при комнатной температуре, затем устанавливали с помощью 2 N соляной кислоты величину рН 5 и перемешивали еще 16 ч при комнатной температуре. Охлаждали до 10°С, отфильтровывали осадок и промывали 3.5 мл диоксана. Сушили 16 ч в высоком вакууме, затем смешивали с 5 мл метанола и перемешивали еще 1 ч при комнатной температуре. Отфильтровывали осадок, промывали 2 мл метанола, сушили в высоком вакууме и получали таким образом еще 997 мг названного соединения.
Выход: общий 2.6 г (83% от теоретического)
ЖХ-МС (Метод б): Rt=0.89 мин; МС (ESIpos): m/z=315 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.54 (a, 1H), 8.39 (s, 1H), 8.28 (s, 1H), 7.88 (s, 1H), 7.42 (s, 1H), 3.71 (s, 8H).
Пример 73
4-(1H-Имидазол-1-ил)-2-(6-морфолин-4-илпиримидин-4-ил)-1,2-дигидро-3Н-пиразол-3-он-трифтор-ацетат
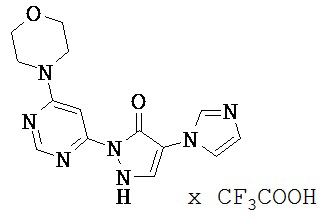
Растворяли 209 мг (1.0 ммоль) соединения из примера 42А в 7 мл ТГФ. Добавляли 195 мг (1.0 ммоль) соединения из примера 16А и 38 мг (0.2 ммоль) п-толуолсульфокислоты-моногидрата при комнатной температуре и затем осуществляли взаимодействие в течение 1 ч при 150°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). Затем полученную смесь сразу же очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты).
Выход: 71 мг (17% от теоретического)
ЖХ-МС (Метод 1): Rt=2.03 мин; МС (ESIpos): m/z=314 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.38 (s, 1H), 8.57 (s, 1H), 8.31 (s, 1H), 8.02 (s, 1H), 7.79 (s, 1H), 7.47 (s, 1H), 3.72 (s, 8H).
Пример 74
4-(1H-Имидазол-1-ил)-2-(6-морфолин-4-илпиримидин-4-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Смешивали 60 мг (0.1 ммоль) соединения из примера 73 с 0.5 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Отфильтровывали осадок, промывали дважды по 0.5 мл диоксана и затем сушили в вакууме.
Выход: 46 мг (94% от теоретического)
ЖХ-МС (Метод 1): Rt=0.91 мин; МС (ESIpos): m/z=314 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.46 (s, 1H), 8.57 (s, 1H), 8.35 (s, 1H), 8.06 (s, 1H), 7.82 (s, 1H), 7.46 (s, 1H), 3.73 (s, 8H).
Пример 75
2-(6-Морфолин-4-илпиримидин-4-ил)-4-[4-(трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Растворяли 309 мг (1.0 ммоль) соединения из примера 44А в 7 мл ТГФ. Добавляли при комнатной температуре 195 мг (1.0 ммоль) соединения из примера 16A и 38 мг (0.2 ммоль) п-толуолсульфокислоты-моногидрата и затем осуществляли реакцию в течение 1 ч при 150°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). После чего полученную смесь сразу же очищали методом препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Объединяли полученные фракции с продуктом и растворитель удаляли. Остаток смешивали с 4 N раствором хлористого водорода в диоксане. Перемешивали 1 ч при комнатной температуре, отфильтровывали осадок и сушили в высоком вакууме.
Выход: 77 мг (19% от теоретического)
ЖХ-МС (Метод 7): Rt=1.31 мин; МС (ESIpos): m/z=382 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.53 (s, 1H), 8.33 (s, 1H), 8.18 (s, 1H), 8.13 (s, 1H), 7.49 (s, 1H), 3.67 (s, 8H).
Пример 76
1-[2-(6-Морфолин-4-илпиримидин-4-ил)-3-оксо-2,3-дигидро-1H-пиразол-4-ил]-1H-имидазол-4-карбонитрил-трифторацетат

Помещали 976 мг (с чистотой 72%, 3.0 ммоль) соединения из примера 45А в 20 мл ТГФ. Добавляли 586 мг (3.0 ммоль) соединения из примера 16A и 114 мг (0.6 ммоль) п-толуолсульфокислоты-моногидрата и затем осуществляли реакцию в течение 1 ч при 150°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). Реакционную смесь очищали хроматографически сначала на силикагеле (Biotage-хроматография; растворитель: дихлорметан/метанол 10:1 с добавкой небольшого количества водного раствора аммиака). Полученный таким образом сырой продукт очищали затем двукратной препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Объединяли фракции с продуктом после ВЭЖ-хроматографии, смешивали с таким же объемом воды и лиофилизировали.
Выход: 11 мг(1% от теоретического)
ЖХ-МС (Метод 4): Rt=1.54 мин; МС (ESIpos): m/z=339 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.53 (s, 1H), 8.42 (s, 1H), 8.29 (s, 1H), 8.19 (s, 1H), 7.46 (s, 1H), 3.70-3.64 (m, 8H).
Пример 77
1-[2-(6-Морфолин-4-илпиримидин-4-ил)-3-оксо-2,3-дигидро-1H-пиразол-4-ил]-1H-имидазол-4-карбоксамид-гидрохлорид

Помещали 976 мг (с чистотой 72%, 3.0 ммоль) соединения из примера 45А в 20 мл ТГФ. Добавляли 586 мг (3.0 ммоль) соединения из примера 16А и 114 мг (0.6 ммоль) п-толуолсульфокислоты-моногидрата и затем осуществляли реакцию в течение 1 ч при 150°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). Реакционную смесь очищали хроматографически сначала на силикагеле (Biotage-хроматография; растворитель: дихлорметан/метанол 10:1 с добавкой небольшого количества водного раствора аммиака). Полученный таким образом сырой продукт очищали затем путем двукратной препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Объединяли фракции с продуктом после ВЭЖ-хроматографии, упаривали в вакууме, остаток помещали в 1 М соляную кислоту и раствор лиофилизировали.
Выход: 14 мг (1.2% от теоретического)
ЖХ-МС (Метод 10): Rt=0.47 мин; МС (ESIpos): m/z=357 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.20 (s, 1Н), 8.57 (s, 1H), 8.53 (s, 1H), 8.29 (s, 1H), 8.23 (уш. s, 1H), 7.80 (уш. s, 1H), 7.49 (s, 1H), 3.72 (s, 8H).
Пример 78
Трет.-бутилового эфира 6-[4-(4-циано-1H-имидазол-1-ил)-5-оксо-2,5-дигидро-1H-пиразол-1-ил]пиридин-3-карбоновой кислоты гидрохлорид

Помещали 500 мг (с чистотой 72%, 1.5 ммоль) соединения из примера 45А в 11 мл ТГФ. Добавляли 322 мг (1.5 ммоль) соединения из примера 22А и 58 мг (0.3 ммоль) п-толуолсульфокислоты-моногидрата и затем осуществляли реакцию в течение 3 ч при 150°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). После чего очищали смесь сразу же препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, упаривали, смешивали с 1 N раствором хлористого водорода в диоксане и перемешивали при комнатной температуре 1 ч. Отфильтровывали осадок, промывали его дважды по 0.5 мл диоксана и сушили в высоком вакууме.
Выход: 144 мг (24% от теоретического)
ЖХ-МС (Метод 8): Rt=2.10 мин; МС (ESIpos): m/z=353 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.94 (s, 1H), 8.54-8.42 (m, 4H), 8.20 (s, 1H), 1.58 (s, 9H).
Соединения, представленные в таблице 7, получены из соответствующих эдуктов аналогично указанным примерам. Вместо 10-камфорсульфокислоты может также использоваться п-толуолсульфокислоты моногидрат.


2.27 (s,3H)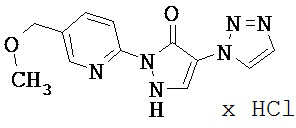
Пример 82
2-[5-(трет.-Бутоксиметил)пиридин-2-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Помещали 631 мг (3.0 ммоль) соединения из примера ЗА и 586 мг (3.0 ммоль) соединения из примера 18A в 10 мл этанола. Добавляли 114 мг (0.6 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 16 ч при комнатной температуре. Затем удаляли растворитель и очищали остаток препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, упаривали и остаток сушили в высоком вакууме. Добавляли 10 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Отфильтровывали осадок, промывали его трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 260 мг (25% от теоретического)
ЖХ-МС (Метод 8): Rt=1.77 мин; МС (ESIpos): m/z=315 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.47.8.42 (m, 2Н), 8.35 (s, 1Н), 8.22 (d, 1H), 7.99 (dd, 1H), 7.90 (s, 1H), 4.50 (s, 2H), 1.25 (s, 9H).
Пример 83
4-(1H-1,2,3-Триазол-1-ил)-2-[4-(трифторметил)пиридин-2-ил]-1,2-дигидро-3H-пиразол-3он-гидрохлорид

Помещали 631 мг (3.0 ммоль) соединения из примера 3А и 531 мг (3.0 ммоль) соединения из примера 25А в 10 мл этанола. Добавляли 114 мг (0.6 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 16 ч с обратным холодильником. Затем концентрировали и очищали остаток препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, растворитель удаляли и остаток сушили в высоком вакууме. Смешивали с 20 мл 4 N раствора хлористого водорода в диоксане и перемешивали при комнатной температуре 1 ч. Отфильтровывали осадок, дважды промывали его трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 231 мг (23% от теоретического)
ЖХ-МС (Метод 8): Rt=1.65 мин; МС (ESIpos): m/z=297 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.81 (d, 1H), 8.70 (s, 1H), 8.59 (s, 1H), 8.47 (s, 1H), 7.92 (s, 1H), 7.77 (d, 1H).
Пример 84
2-[5-(2,2-Диметилпропокси)пиридин-2-ил]-4-{1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он

Помещали 171 мг (0.8 ммоль) соединения из примера 3А и 455 мг (чистота 35%, 0.8 ммоль) соединения из примера 12А в 7 мл этанола. Добавляли 31 мг (0.2 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 64 ч с обратным холодильником. Затем концентрировали и очищали остаток препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, растворитель удаляли и остаток сушили в высоком вакууме.
Выход: 28 мг (11% от теоретического)
ЖХ-МС (Метод 10): Rt=1.17 мин; МС (ESIpos): m/z=315 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1H), 8.29 (s, 1H), 8.23 (s, 1H), 8.18-8.08 (m, 1H), 7.90 (s, 1H), 7.69 (dd, 1H), 3.78 (s, 2H), 1.02 (s, 9H).
Пример 85
2-[5-(2,2-Диметилпропокси)пиридин-2-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Смешивали 22 мг (0.1 ммоль) соединения из примера 84 с 2 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Осадок отфильтровывали и сушили в высоком вакууме.
Выход: 17 мг (97% от теоретического)
ЖХ-МС (Метод 10): Rt=1.17 мин; МС (ESIpos): m/z=315 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1H), 8.29 (s, 1H), 8.23 (s, 1H), 8.18-8.08 (m, 1H), 7.90 (s, 1H), 7.69 (dd, 1H), 3.78 (s, 2H), 1.02 (s, 9H).
Пример 86
1-[2-(6-Морфолин-4-илпиримидин-4-ил)-3-оксо-2,3-дигидро-1H-пиразол-4-ил]-1H-1,2,3-триазол-4-карбонитрил-трифторацетат

Помещали 250 мг (1.1 ммоль) соединения из примера 41А, 207 мг (1.1 ммоль) соединения из примера 16 A и 40 мг (0.2 ммоль) п-толуолсульфокислоты-моногидрата в 10 мл ТГФ. Реакцию проводили при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer) сначала 3.5 ч при 120°С, затем 4 ч при 130°С. Охлаждали до комнатной температуры, смешивали реакционную смесь с 5 мл ацетонитрила и оставляли на 24 ч. Декантировали жидкость и осадок промывали один раз ацетонитрилом Промывной раствор и декантированную жидкость объединяли и упаривали. Полученный таким образом остаток очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли и упаривали. Твердый остаток дважды смешивали со смесью из небольшого количества трет.-бутилметилового эфира и нескольких капель ацетонитрила. Жидкость декантировали с осадка и твердое вещество затем сушили в высоком вакууме.
Выход: 15 мг(3% от теоретического)
ЖХ-МС (Метод 4): Rt=1.57 мин; МС (ESIpos): m/z=340 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.58 (s, 1H), 8.24 (s, 1H), 7.42 (s, 1H), 3.80-3.63 (m, 8H).
Пример 87
1-[2-(6-Морфолин-4-илпиримидин-4-ил)-3-оксо-2,3-дигидро-1H-пиразол-4-ил]-1H-1,2,3-триазол-4-карбонитрил-гидрохлорид
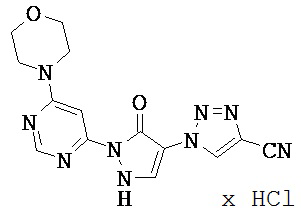
Смешивали 18 мг (0.04 ммоль) соединения из примера 86 с 3 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Отфильтровывали осадок, промывали трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 15 мг (97% от теоретического)
ЖХ.МС (Метод 8): Rt=1.38 мин; МС (ESIpos): m/z=340 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.58 (s, 1H), 8.25 (s, 1H), 7.41 (s, 1H), 3.82-3.57 (m, 8H).
Пример 88
2-[6-(3-Гидроксиазетидин-1-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он

Помещали 348 мг (1.7 ммоль) соединения из примера ЗА и 300 мг (1.7 ммоль) соединения из примера НА в смесь из 6 мл ТГФ и 6 мл этанола. Добавляли 63 мг (0.3 ммоль) п-толуолсульфокислоты-моногидрата и реакцию осуществляли при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer) в течение 1.5 ч при 130°С. Охлаждали до комнатной температуры, добавляли еще 50 мг соединения из примера 3А и на кончике шпателя п-толуолсульфокислоты-моногидрат и нагревали 1 ч до 150°С. Охлаждали до комнатной температуры, декантировали жидкость с осадка и промывали осадок многократно ТГФ (осадок, партия 1). Декантированную жидкость и промывные растворы объединяли, упаривали и полученный остаток смешивали с 5 мл этанола и 5 мл ТГФ и с п-толуолсульфокислотой-моногидратом (на кончике шпателя). Вновь проводили реакцию при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer) в течение 2 ч при 150°С. Охлаждали до комнатной температуры и отфильтровывали образовавшийся осадок (осадок, партия 2). Фильтрат упаривали, смешивали с 3 мл DMSO и очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли и упаривали. Растворяли остаток в этаноле, смешивали с небольшим количеством ТГФ и концентрировали смесь с образованием осадка. Твердое вещество отфильтровывали и промывали ТГФ (осадок, партия 3). Объединяли все три партии полученных осадков и сушили в высоком вакууме.
Выход: 108 мг (22% от теоретического)
ЖХ-МС (Метод 8): Rt=1.00 мин; МС (ESIpos): m/z=301 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.47 (s, 1H), 8.36 (s, 1H), 8.18 (s, 1H), 7.84 (s, 1H), 6.97 (s, 1H), 5.92 (d, 1H), 4.69-4.49 (m, 1H), 4.41-4.33 (m, 2H), 3.95-3.86 (m, 2H).
Пример 89
2-[6-(3-Гидроксиазетидин-1-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
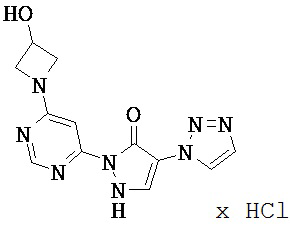
Смешивали 105 мг (0.4 ммоль) соединения из примера 88 с 3 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Отфильтровывали осадок, промывали его трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 117 мг (99% от теоретического)
ЖХ-МС (Метод 8): Rt=0.99 мин; МС (ESIpos): m/z=301 [М+Н]+:
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.50 (s, 1H), 8.38 (s, 1H), 8.22 (s, 1H), 7.87 (s, 1H), 6.95 (s, 1H), 4.68-4.60 (m, 1H), 4.43-4.35 (m, 2H), 3.97-3.88 (m, 2H).
Пример 90
2-(4,5-Диметилпиридин-2-ил)-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он

Помещали 262 мг (1.3 ммоль) соединения из примера 42А и 172 мг (1.3 ммоль) соединения из примера 26А в 5 мл этанола. Добавляли 48 мг (0.3 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 24 ч с обратным холодильником. Охлаждали до комнатной температуры, при этом продукт выкристаллизовывался. Осадок отфильтровывали и промывали по одному разу этанолом и петролейным эфиром. После этого сушили в высоком вакууме.
Выход: 65 мг (20% от теоретического)
ЖХ-МС (Метод 8): Rt=0.92 мин; МС (ESIpos): m/z=256 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.23 (s, 1H), 8.15 (s, 1H), 8.07 (s, 2H), 7.52 (s, 1H), 7.08 (s, 1H), 2.36 (s, 3H), 2.26 (s, 3H).
Пример 91
2-(4,5-Диметилпиридин-2-ил)-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
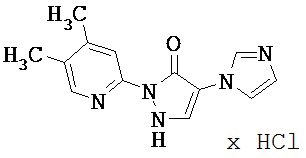
Смешивали 200 мг (0.5 ммоль) соединения из примера 90 с 5 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Затем отфильтровывали, промывали по одному разу диоксаном и трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 117 мг (74% от теоретического)
ЖХ-МС (Метод 10): Rt=0.28 мин; МС (ESIpos): m/z=256 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.50 (s, 1Н), 8.40 (s, 1H), 8,26 (s, 1H), 8.13 (s, 1H), 8.08 (s, 1H), 7.87 (s, 1H), 2.40 (s, 3H), 2.28 (s, 3H).
Пример 92
2-[5-(2,2-Диметилпропокси)пиридин-2-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он

Помещали 233 мг (1.0 ммоль, чистота 90%) соединения из примера 42А в 4 мл этанола. Добавляли 260 мг (1.0 ммоль, чистота 75%) соединения из примера 12А и 38 мг (0.2 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали с обратным холодильником 16 ч. Охлаждали затем до комнатной температуры, отфильтровывали выпавший осадок, промывали небольшим количеством этанола и сушили осадок в высоком вакууме. Другие количества названного выше соединения получали, когда очищали маточный раствор после фильтрования методом препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты).
Выход: 264 мг (78% от теоретического)
ЖХ-МС (Метод 10): Rt=0.88 мин; МС (ESIpos): m/z=314 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.22-8.08 (m, 4H), 7.67-7.62 (dd, 1Н), 7.52 (s, 1H), 7.10 (s, 1H), 3.75 (s, 2H), 1.02 (s, 9H).
Пример 93
2-[5-(2,2-Диметилпропокси)пиридин-2-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Смешивали 200 мг (0.638 ммоль) соединения из примера 92 с 5 мл 4 N раствора хлористого водорода в диоксане и перемешивали 30 мин при комнатной температуре. Затем отфильтровывали, промывали по одному разу диоксаном и трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 129 мг (64% от теоретического)
ЖХ-МС (Метод 10): Rt=0.87 мин; МС (ESIpos): m/z=314 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.52 (s, 1H), 8.46 (s, 1H), 8.22 (d, 1H), 8.18-8.15 (d, 1H), 8.07 (s, 1H), 7.87 (s, 1H), 7.71-7.68 (dd, 1H), 3.79 (s, 2H), 1.02 (s, 9H).
Пример 94
2-(4,5-Диметилпиридин-2-ил)-4-[4-(трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он-трифторацетат
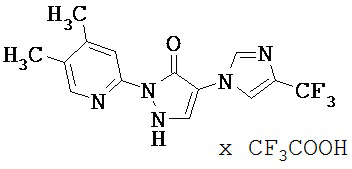
Помещали 347 мг (1.3 ммоль) соединения из примера 44А и 171 мг (1.3 ммоль) соединения из примера 26А в 5 мл этанола. Добавляли 48 мг (03 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 16 ч с обратным холодильником. Реакционный раствор сразу же очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Содержащие продукт фракции объединяли и частично упаривали. Отфильтровывали осадок, промывали водой и сушили в высоком вакууме.
Выход: 84 мг (15% от теоретического)
ЖХ-МС (Метод 8): Rt=1.88 мин: МС (ESIpos): m/z=324 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.27 (s, 1H), 8.25 (s, 1H), 8.18 (s, 1H), 8.15 (s, 1H), 8.08 (s, 1H), 2.38 (s, 3H), 2.27 (s, 3H).
Пример 95
2-(4,5-Диметилпиридин-2-ил)-4-[4-(трифторметил)-1H-имидазол-1-ил]-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
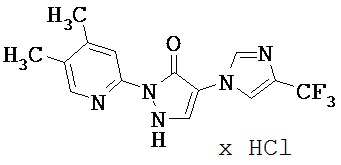
Смешивали 80 мг (0.2 ммоль) соединения из примера 94 с 2.5 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Осадок отфильтровывали, промывали по одному разу диоксаном и трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 65 мг (99% от теоретического)
ЖХ-МС (Метод 10): Rt=1.03 мин: МС (ESIpos): m/z=324 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.27 (s, 1H), 8.25 (s, 1H), 8.18 (s, 1H), 8.13 (s, 1H), 8.07 (s, 1H), 2.37 (s, 3H), 2.27 (s, 3H).
Пример 96
2-[5-(2,2-Диметилпропокси)пиридин-2-ил]-4-[4-(трифторметил)-1H-имидазол-1-ил]-1H-дигидро-3H-пиразол-3-он-гидрохлорид

Помещали 308 мг (1.0 ммоль, чистота 90%) соединения из примера 44А в 4 мл этанола. Добавляли 260 мг (1.0 ммоль, чистота 75%) соединения из примера 12А и 38 мг (0.2 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 16 ч с обратным холодильником. После этого упаривали и очищали остаток препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли и растворитель удаляли. Смешивали остаток с 5 мл 4 N раствора хлористого водорода в диоксане и перемешивали 30 мин. Осадок отфильтровывали и сушили в высоком вакууме.
Выход: 140 мг (34% от теоретического)
ЖХ-МС (Метод 10): Rt=1.38 мин; МС (ESIpos): m/z=382 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.29 (s, 1H), 8.21-8.12 (m, 4H), 7.71-7.68 (dd, 1H), 3.76 (s, 2H), 1.02 (s, 9H).
Пример 97
2-[6-(Азетидин-3-илокси)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
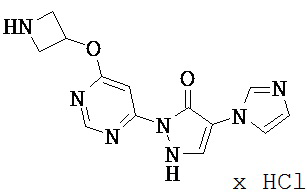
Стадия а):
3-({6-[5-Оксо-4-(1Н-1,2,3-триазол-1-ил)-2,5-дигидро-1Н-пиразол-1-ил]пиримидин-4-ил}окси)азетидин-1-трет.-бутиловый сложный эфир - трифторацетат

Помещали 345 мг (2.0 ммоль) 3-гидроксиазетидин-1-трет.-бутилового сложного эфира в 15 мл диоксана. Затем добавляли туда при комнатной температуре 1 мл (2.0 ммоль) 2 М раствора фосфазен-основания (Phosphazcn-Base) Р2-трет.-бутил в ТГФ. Перемешивали 15 мин при комнатной температуре и добавляли 350 мг (1.3 ммоль) соединения из примера 52А. Реакцию проводили в течение 1 ч при 120°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). Осадок отделяли фильтрованием, реакционный раствор упаривали и осадок затем очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли и частично упаривали. Отфильтровывали осадок, промывали водой и сушили в высоком вакууме.
Выход: 82 мг (12% от теоретического)
ЖХ-МС (Метод 10): Rt=0.98 мин; МС (ESIpos): m/z=401 [M+H]+:
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.77 (s, 1H), 8.61 (s, 1H), 8.43 (s, 1H), 7.90 (s, 1H), 7.78 (s, 1H), 5.47-5.38 (m, 1H), 4.32-4.22 (m, 2H), 3.96-3.86 (m, 2H), 1.40 (s, 9H).
Стадия b):
2-[6-(Азетидин-3-илокси)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Суспендировали 60 мг (0.2 ммоль) 3-({6-[5-оксо-4-(1H-1,2,3-триазол-1-ил)-2,5-дигидро-1H-пиразол-1-ил]пиримидин-4-ил}окси)азетидин-1-трет.-бутилового сложного эфира в 2 мл дихлорметана. Добавляли 1 мл ТГФ и перемешивали 1 ч при комнатной температуре. После чего упаривали и сушили остаток в высоком вакууме. Остаток смешивали затем с 3 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Затем фильтровали, промывали по одному разу диоксаном и трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 54 мг (97% от теоретического)
ЖХ-МС (Метод 8): Rt=0.80 мин; МС (ESIpos): m/z=301 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.50 (уш. s, 1H), 9.37 (уш. s, 1H), 8.80 (s, 1H), 8.63 (s, 1H), 8.44 (s, 1H), 7.90 (s, 1H), 7.80 (s, 1H), 5.57-5.48 (m, 1H), 4.43-432 (m, 2H), 4.18-4.07 (m, 2H).
Пример 98
1-{2-[5-(2,2-Диметилпропокси)пиридин-2-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-1,2,3-триазол-4-карбонитрил-гидрохлорид

Помещали 150 мг (0.6 ммоль) соединения из примера 41 А в 2.5 мл этанола. Добавляли 166 мг (0.6 ммоль, чистота 75%) соединения из примера 12А и 24 мг (0.1 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 16 ч с обратным холодильником. Реакционный раствор сразу очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли и упарили. Полученный остаток смешивали с 1 мл 4 N раствора хлористого водорода в диоксане и перемешивали 1 ч при комнатной температуре. Упаривали и сушили остаток в высоком вакууме.
Выход: 3 мг (1% от теоретического)
ЖХ-МС (Метод 8): Rt=2.42 мин; МС (ESIpos): m/z=340 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.39 (s, 1H), 8.41 (s, 1H), 8.24 (d, 1H), 8.12 (d, 1H), 7.70 (dd, 1H), 3.78 (s, 2H), 1.03 (s, 9H).
Пример 99
6-[4-(4-Циано-1H-1,2,3-триазол-1-ил)-5-оксо-2,5-дигидро-1H-пиразол-1-ил]пиридин-3-трет.-бутиловый сложный эфир - гидрохлорид

Помещали 150 мг (0.6 ммоль) соединения из примера 41 А в 2.5 мл этанола. Добавляли 178 мг (0.6 ммоль) соединения из примера 22А и 24 мг (0.1 ммоль) п-толуолсульфокислоты-моногидрата и перемешивали 16 ч с обратным холодильником. Реакционный раствор сразу очищали препаративной ВЭЖХ (колонка RPI8; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, упаривали и сушили в высоком вакууме. Полученный остаток смешивали с 5 мл 4 N раствора хлористого водорода в диоксане и перемешивали 30 мин при комнатной температуре. Осадок отфильтровывали, промывали трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 21 мг (8% от теоретического)
ЖХ-МС (Метод 7): Rt=1.96 мин; МС (ESIpos): m/z=354 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.39 (s, 1H), 8.94 (s, 1H), 8.63 (s, 1H), 8.46 (s, 2H), 1.58 (s, 9H).
Пример 100
2-[6-(3-Гидроксиазетидин-1-ил)пиримидин-4-ил]-4-(1H-имидазол-1-ил)-1H-дигидро-3H-пиразол-3-он-трифторацетат

Суспендировали 400 мг (1.3 ммоль) соединения из примера 53А, 518 мг (4.0 ммоль) N,N-диизопропиламина и 293 мг (2.6 ммоль) азетидин-3-ол-гидрохлорида в 8 мл этанола. Реакцию проводили в течение 30 мин при 120°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). После фильтрования реакционный раствор очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли и упаривали, остаток смешивали со смесью, состоящей из 5 мл трет.-бутилметилового эфира и 10 мл метанола, и затем отсасывали. После чего снова очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, упаривали и остаток сушили в высоком вакууме.
Выход: 115 мг(21% от теоретического)
ЖХ-МС (Метод 8): Rt=0.80 мин; МС (ESIpos): m/z=300 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.38 (s, 1H), 8.50 (s, 1H), 8.23 (s, 1H), 8.02 (t, 1H), 7.80 (t, 1H), 6.97 (s, 1H), 4.70-4.61 (m, 1H), 4.47-4.38 (m, 2H), 3.98-3.90 (m, 2H).
Пример 101
2-[6-(3-Гидроксиазетидин-1-ил)пиримидин-4-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3он-гидрохлорид

Перемешивали 100 мг (0.2 ммоль) соединения из примера 100 с 3 мл 4 N раствора хлористого водорода в диоксане. Отфильтровывали осадок, промывали дважды трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 78 мг (96% от теоретического)
ЖХ-МС (Метод 8): Rt=0.80 min; МС (ESIpos): m/z=300 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.42 (s, 1H), 8.50 (s, 1H), 8.25 (s, 1H), 8.03 (t, 1H), 7.82 (t, 1H), 6.97 (s, 1H), 4.69-4.60 (m, 1H), 4.47-4.37 (m, 2H), 3.99-3.90 (m, 2H).
Пример 102
2-[6-(Этилсульфанил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он

Суспендировали 200 мг (0.7 ммоль) соединения из примера 52А под аргоном в 1.1 мл диметилформамида. Добавили 61 мг (1.0 ммоль) этантиола. При охлаждении льдом осторожно добавили 35 мг (0.8 ммоль, 60%-ный в минеральном масле) гидрида натрия. Перемешивали 2.5 ч при комнатной температуре. Затем добавили по каплям 1 мл воды и перемешивали еще 15 мин при комнатной температуре. Полученный прозрачный раствор сразу очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли, упаривали и остаток сушили в высоком вакууме.
Выход: 39 мг (18% от теоретического)
ЖХ-МС (Метод 10): Rt=0.81 мин; МС (ESIpos): m/z=290 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.90 (s, 1H), 8.60 (s, 1H), 8.44 (s, 1H), 8.27 (s, 1H), 7.90 (s, 1H), 3.20 (q, 2H), 1.35 (t, 3H).
Пример 103
2-[6-(Этилсульфанил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1H-дигидро-3H-пиразол-3-он-гидрохлорид

Смешивали 35 мг (0.1 ммоль) соединения из примера 102 с 1.5 мл 4 N раствора хлористого водорода в диоксане. Отфильтровывали осадок, промывали дважды трет.-бутилметиловым эфиром и сушили в высоком вакууме.
Выход: 36 мг (90% от теоретического)
ЖХ-МС (Метод 10): Rt=0.80 мин; МС (ESIpos): m/z=290 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.90 (s, 1H), 8.62 (s, 1H), 8.44 (s, 1H), 8.26 (s, 1H), 7.91 (s, 1H), 3.21 (q, 2H), 1.35 (t, 3H).
Пример 104
2-(6-{[2-(Диэтиламино)этил]сульфанил}пиримидин-4-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-трифторацетат

Растворяли 200 мг (0.7 ммоль) соединения из примера 52А под аргоном в 2 мл диметилформамида. Прибавляли по каплям 131 мг (1.0 ммоль) 2-{диэтиламино)этантиола и затем охлаждали до 0°С. Прибавляли 35 мг (0.8 ммоль, 60%-ный в минеральном масле) гидрида натрия, поднимали температуру до комнатной и перемешивали при комнатной температуре 2.5 ч. После чего медленно прибавляли 3 мл воды и перемешивали 15 мин. Осадок отфильтровывали и фильтрат упаривали. Полученный таким образом остаток очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты).
Выход: 129 мг (41% от теоретического)
ЖХ-МС (Метод 10): Rt=0.29 мин; МС (ESIpo:): m/z=361 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.42 (уш. s, 1Н), 8.91 (s, 1H), 8.49 (s, 1H), 8.40 (s, 1H), 8.37 (s, 1H), 7.88 (s, 1H), 3.58-3.51 (m, 2H), 3.42-3.33 (m, 2H), 3.28-3.20 (m, 4H), 1.30-1.20 (m, 6H).
Пример 105
2-{6-[4-(2-Метоксиэтил)пипсразин-1-ил]пиримидин-4-ил}-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Помещали 500 мг (2.4 ммоль) соединения из примера 3А, 600 мг (2.4 ммоль) соединения из примера 31A и 82 мг (0.5 ммоль) п-толуолсульфокислоты в 8 мл ТГФ и осуществляли взаимодействие в течение 30 мин при 170°С при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). После охлаждения до комнатной температуры остаток сразу очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% муравьиной кислоты в воду). Образовавшуюся соль муравьиной кислоты переводили в гидрохлорид добавлением 4 мл 4 N раствора хлористого водорода в диоксане. Продукт промывали диэтиловым эфиром и сушили в вакууме.
Выход: 212 мг (20% от теоретического)
ЖХ-МС (Метод 11): Rt=2.79 мин; МС (ESIpos): m/z=372 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=11.28 (уш. s, 1H), 8.61 (s, 1H), 8.41 (s, 1H), 8.38 (s, 1H), 7.90 (s, 1H), 7.56 (s, 1H), 4.64-4.43 (m, 2H), 3.76 (t, 2H), 3.65-3.51 (m, 4H), 3.36-3.30 (m, 5H), 3.23- 3.08 (m, 2H).
Пример 106
2-[6-(3,5-Диметилпиперидин-1-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-
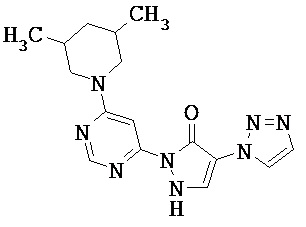
Растворяли 2.9 г (9.6 ммоль) соединения из примера 52А в 40 мл диметилформамида и получали таким образом концентрированный базовый раствор. Помещали 23 мг (0.1 ммоль) 3,5-диметилпиперидина в 200 мкл диметилформамида и смешивали последовательно с 400 мкл (0.1 ммоль) концентрированного базового раствора соединения из примера 52А и 35 мг (0.3 ммоль) карбоната калия. Перемешивали реакционную смесь 16 ч при 100°С. Затем суспензию фильтровали и фильтрат хроматографировали методом препаративной ЖХ-МС (метод 16). Фракции с продуктом концентрировали в вакууме и остаток сушили.
Выход: 3 мг (10% от теоретического)
ЖХ-МС (Метод 16): Rt=1.90 мин; МС (ESIpos): m/z=341 [M+H]+.
Аналогично рабочей прописи примера 106 получены приведенные в таблице 8 соединения из 0.1 ммоль соединения из примера 52А и 0.1 ммоль соответствующего вторичного амина:

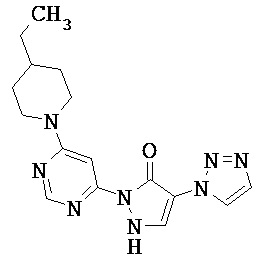
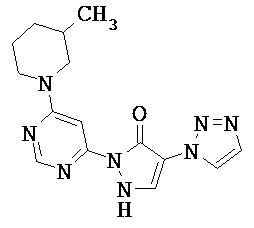

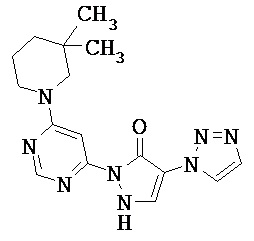
MC: Rt (метод 16)



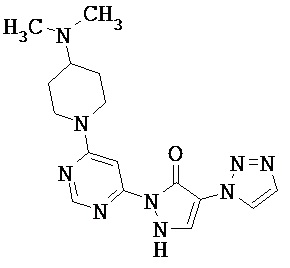




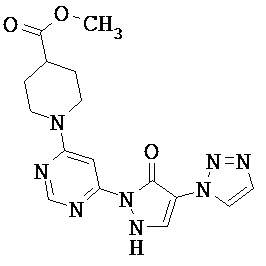
№
теоретического)
MC: Rt (метод 16)
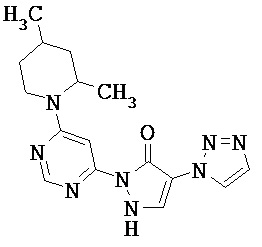



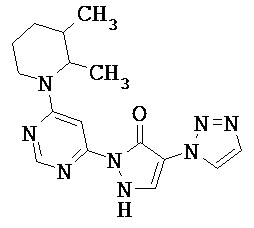
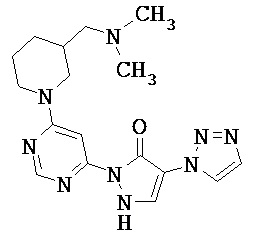






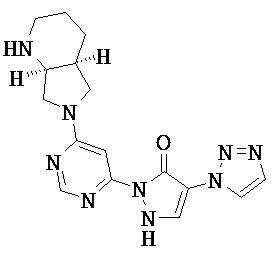

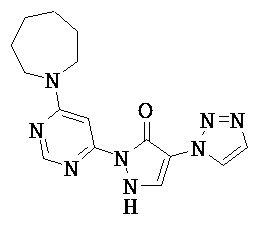
Пример 138
2-[6-(2,5-Диметил-2,5-дигидро-1H-пиррол-1-ил)пиримидин-4-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он

Растворяли 2.9 г (9.6 ммоль) соединения из примера 53А в 40 мл диметилформамида и получали таким образом концентрированный базовый раствор.
Помещали 19 мг (0.1 ммоль) 2,5-диметил-2,5-дигидро-1H-пиррола в 200 мкл диметилформамида и смешивали последовательно с 400 мкл (0.1 ммоль) концентрированного базового раствора соединения из примера 53А и 35 мг (0.3 ммоль) карбоната калия. Перемешивали реакционную смесь 16 ч при 100°С.
Затем суспензию фильтровали и фильтрат хроматографировали методом препаративной ЖХ-МС (метод 16). Фракции с продуктом концентрировали в вакууме и остаток сушили.
Выход: 5 мг (16% от теоретического)
ЖХ-МС (Метод 16): Rt=1.42 мин; МС (ESIpos): m/z=324 [M+H]+.
Аналогично рабочей прописи примера 140 получены приведенные в таблице 9 соединения из 0.1 ммоль соединения из примера 53А и 0.1 ммоль соответствующего вторичного амина:
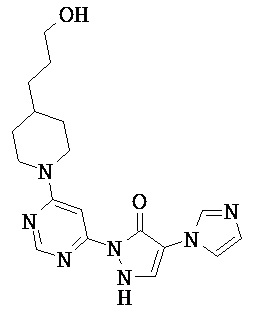
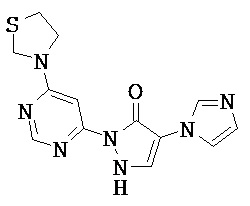


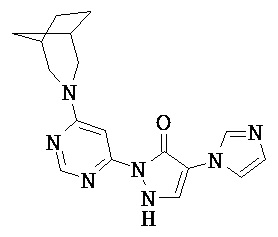





№
теоретического)
Rt (метод 16)

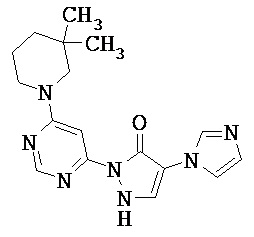




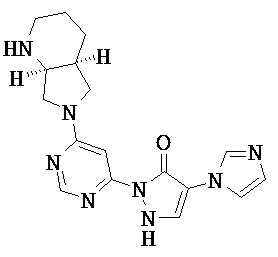


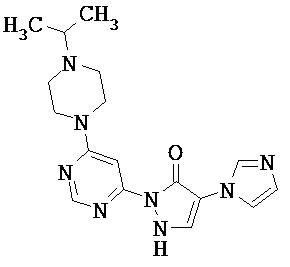

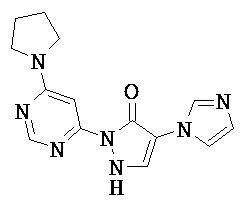
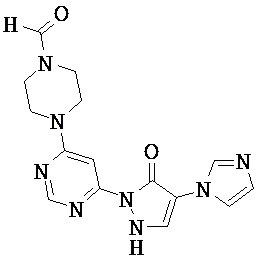

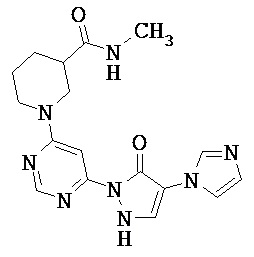

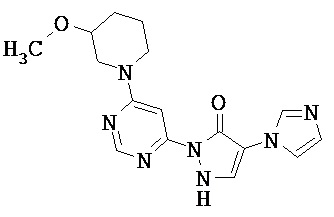



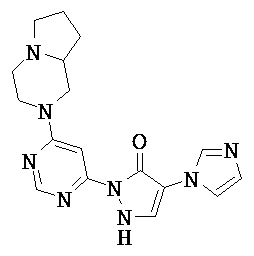


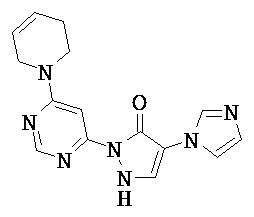



Пример 177
4-(3-Метил-1,2,4-оксадиазол-5-ил)-2-(6-морфолин-4-илпиримидин-4-ил)-1,2-дигидро-3H-пиразол-3-он-трифторацетат

Суспендировали 22 мг (0.5 ммоль, 60%-ный в минеральном масле) гидрида натрия под аргоном в 1 мл диметилформамида. Добавляли 38 мг (0.5 ммоль) 1-N'-гидроксиэтанимидамида ("ацетамидоксим") и нагревали при перемешивании до 50°С. Через час добавили 50 мг (0.2 ммоль) соединения из примера 54А. Перемешивали реакционную смесь 2 ч при 80°С. Затем снова добавляли друг за другом дважды смесь из 22 мг (0.5 ммоль, 60%-ный в минеральном масле) гидрида натрия и 38 мг (0.5 ммоль) 1-N’-гидроксиэтанимидамида в 1 мл диметилформамида, которая перед этим нагревалась в течение 30 минут при перемешивании до 50°С, и реакционную смесь перемешивали после каждого добавления еще 30 мин при 80°С. После чего состав охлаждали и упаривали. Остаток растворяли в смеси, содержащей по 2 мл воды, метанола и 1 N соляной кислоты, и очищали препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты).
Выход: 12 мг (17% от теоретического)
ЖХ-МС (Метод 4): Rt=1.35 мин; МС (ESIpos): m/z=330 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.58 (s, 1H), 8.04 (s, 1H), 7.29 (s, 1H), 3.85-3.65 (m, 8H), 2.23 (s, 3H).
Пример 170
4-(3-Метил-1,2,4-оксадиазол-5-ил)-2-(6-морфолин-4-илпиримидин-4-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид

Перемешивали 10 мг (0.02 ммоль) соединения из примера 177 2 ч при комнатной температуре с 4 N раствором хлористого водорода в диоксане. Растворитель декантировали, трижды последовательно смешивали с трет.-бутилметиловым эфиром и декантировали растворитель. Оставшийся осадок сушили в высоком вакууме.
Выход: 7 мг (86% от теоретического)
ЖХ-МС (Метод 8): Rt=1.24 мин; МС (ESIpos): m/z=330 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.58 (s, 1H), 8.08 (s, 1H), 7.29 (s, 1H), 4.00-3.50 (m, 8H), 2.23 (s, 3H).
Пример 179
2-(6-Метоксипиримидин-4-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3Р-пиразол-3-он-гидрохлорид
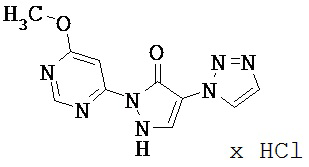
Помещали 0.3 мл (6.6 ммоль) метанола в 15 мл диоксана. При перемешивании медленно добавляли 1.3 мл (2.7 ммоль) 2 М раствора фосфазен-основания Р2-трет.-бутил (Phosphazen-Base P2-tert.-Butyl) в ТГФ и перемешивали 15 мин при комнатной температуре. Затем добавляли 350 мг (1.3 ммоль) соединения из примера 52А и проводили реакцию 2 ч при 150°С при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). Затем добавляли еще 2 мл (49.2 ммоль) метанола и проводили реакцию снова 2 h при тех же условиях при микроволновом облучении (single mode-Mikrowelle) (СЕМ Explorer). После охлаждения реакционную смесь упаривали и очищали остаток препаративной ВЭЖХ (колонка RP18; растворитель: ацетонитрил/вода-градиент с добавкой 0.1% трифторуксусной кислоты). Фракции с продуктом объединяли и упаривали на роторном испарителе. Остаток сушили в высоком вакууме и после этого смешивали с 3 мл 4 N раствора хлористого водорода в диоксане. Перемешивали 30 мин при комнатной температуре, затем отфильтровывали осадок и сушили в высоком вакууме.
Выход: 60 мг (15% от теоретического)
ЖХ-МС (Метод 7): Rt=0.82 мин; МС (ESIpos): m/z=260 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.80 (s, 1H), 8.59 (s, 1H), 8.43 (s, 1H), 7.90 (s, 1H), 7.70 (s, 1H), 4.00 (s, 3H).
Пример 180
2-[6-(4-Пирролидин-1-илпиперидин-1-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
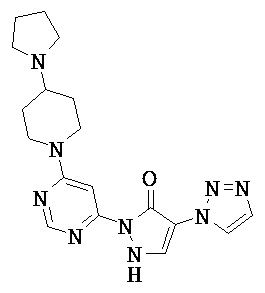
Помещали 481 мг (2.3 ммоль) соединения из примера 3А, 600 мг (2.3 ммоль) соединения из примера ЗОА и 79 мг (0.5 ммоль) п-толуолсульфокислоты в 8 мл ТГФ и осуществляли взаимодействие в течение 30 мин при 170°С при микроволновом облучении (single mode-Mikrowelle) (Emrys Optimizer). Затем реакционную смесь концентрировали в вакууме и остаток хроматографировали методом препаративной ВЭЖХ (метод 17). Фракции с продуктом лиофилизировали и после этого смешивали со смесью вода/ацетонитрил (5:1). Раствор нагревали, обрабатывали ультразвуком и фильтровали через микропористый фильтр (0.45 мкМ). Фильтрат упаривали в вакууме досуха.
Выход: 17 мг (2% от теоретического)
ЖХ-МС (Метод 8): Rt=0.94 мин; МС (ESIpos): m/z=382 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.41 (s, 1H), 8.35 (s, 1H), 8.07 (s, 1H), 7.70 (s, 1H), 7.68 (s, 1H), 4.43-4.23 (m, 2H), 3.18-2.88 (m, 6H), 2.14-2.00 (m, 2H), 1.92-1.76 (m, 4H), 1.56-1.40 (m, 2H), 1.16 (t, 1H).
В. Оценка фармакологической активности
Фармакологические свойства соединений согласно изобретению могут быть показаны в следующих отчетах об исследовании:
Сокращении:
DMEM Dulbecco's Modified Eagle Medium (модифицированная Eagle-среда фирмы Далбекко)
FCS Fetal Calf Serum (эмбриональная телячья сыворотка)
ТМВ 3,3',5,5'-Tetramethylbenzidin = 3,3',5,5'-тетраметилбензидин
Tris Tris(hydroxymethyl)-aminomethan = трис(гидроксиметил)-аминометан
1. Опыты in vitro для определения активности и селективности ингибиторов HIF-пролил-4-гидроксилазы
1.a) Ингибирование активности HIF-пролилгидроксилазы:
Гидроксилированный HIF особым образом привязывается к комплексу протеин-элонгин В-элонгин С (Protein-Elongin B-Elongin C-Komplex) (VBC-комплексу) Хиппеля-Линдау. Это взаимодействие происходит только тогда, когда HIF гидролизован на законсервированном пролильном остатке. Это является основой для биохимического определения активности HIF-пролилгидроксилазы. Тест осуществляется, как описано в [Oehme F., Jongbaus W., Narouz-Ott L., Huetter J., Flamme I., Anal. Biochem. 330 (1), 74-80 (2004)]:
Прозрачный микротитровальный планшет (фирмы Pierce) с 96 лунками, покрытый NeutrAvidin HBC, инкубировали в течение 30 минут блокер-казеином. После чего планшет трижды промыли по 200 мкл буферного раствора для промывки (50 мМ Tris, рН 7.5, 100 мМ NaCl, 10% (v/v) Blocker-Casein, 0.05% (v/v) Tween 20) для каждой лунки. Добавили пептид биотин-DLDLEMLAPYIPMDDDFQL (фирмы Eurogentec, 4102 Seraing, Belgien) с концентрацией 400 нМ в 100 мкл буферного раствора для промывки. Этот пептид служит субстратом для пролилгидроксилирования и присоединяется к микротитровальному планшету. Спустя 60 минут инкубирования планшет трижды промывали буферным раствором для промывки, инкубировали 30 минут 1 мМ биотина в блокер-казеине и после этого снова трижды промывали буферным раствором для промывки.
Для осуществления реакции пролилгидроксилазы пептидный субстрат, соединенный с планшетом, инкубировали 1-60 минут клеточным лизатом, содержащим пролилгидроксилазу. Реакция идет при комнатной температуре в 100 мкл буферного раствора для реакции (20 мМ Tris, pH 7.5, 5 мМ KCl, 1.5 мМ MgCl2, 1 мкМ-1 мМ 2-оксоглутарата, 10 мкМ FeSO4, 2 мМ аскорбата). Кроме того, реакционная смесь содержит исследуемый ингибитор пролилгидроксилазы в различных концентрациях. Изучаемые вещества используются предпочтительно, но не обязательно с концентрациями от 1 нМ до 100 мкМ. За счет трехкратного промывания планшета буферным раствором для промывания реакция останавливается.
Для количественного определения пролилгидроксилирования добавляется смешанный протеин, который содержит как тиоредоксин из Е. Coli, так и VBC-комплекс, в 80 мкл связующего буфера (50 мМ Tris, pH 7.5, 120 мМ NaCl). Через 15 минут добавляли 10 мкл раствора политональных анти-тиоредоксиновых антител кроликов в связующем буфере. Спустя еще 30 минут добавляли 10 мкл раствора анти-иммуноглобулина кролика в соединении с пероксидазой хрена в связующем буфере. Через 30 минут инкубирования при комнатной температуре трижды промывали буферным раствором для промывки, чтобы удалить несвязанный VBC-комплекс и антитела. Для определения количества связанного VBC-комплекса инкубировали 15 минут с ТМВ. Цветная реакция заканчивается добавлением 100 мкл 1М раствора серной кислоты. Количества связанного VBC-комплекса определяли измерением оптической плотности при 450 нм. Они являются пропорциональными количеству гидроксилированного пролина в пептидном субстрате.
Для детектирования пролингидроксилирования можно использовать альтернативно VBC-комплекс, связанный с европием (фирма Perkin Elmer). В этом случае количества связанного VBC-комплекса определяются путем флуоресценции. Кроме того, можно применять маркированный [35S]-метионином VBC-комплекс. Для этого VBC-комплекс с радиоактивной меткой можно получить путем транскрипции-трансляции in vitro в лизате ретикулоцитов.
Примеры осуществления замедляют активность HIF-пролилгидроксилазы в этом тесте с величины IC50≤30 мкМ. В нижеследующей таблице приведены репрезентативные величины IC50 в примерах осуществления.
1.b) Клеточный тест, действующий in vitro:
Количественное определение активности соединений согласно изобретению происходит с помощью рекомбинантной линии клеток. Клетки изначально происходят от линии клеток карциномы легких человека (А549, АТСС: American Type Culture Collection, Manassas, VA 20108, USA). Тестовая линия клеток стабильно трансфизируется с помощью вектора, который содержит ген-репортер Photinus pyralis-луциферазы (далее называемый луциферазой) под контролем искусственного минимального промотора. Минимальный промотор состоит из двух отвечающих за гипоксию элементов выше ТАТА-бокса [Oehme F., Ellinghaus P., Kolkhof P., Smith TJ., Ramakrishnan S., Hütter J., Schramm M., Flamme I., Biochem. Biophys. Res. Commun. 296 (2), 343-9 (2002)]. Под воздействием гипоксии (например, культивирование в присутствии 1% кислорода в течение 24 часов) иди под воздействием неселективных ингибиторов диоксигеназы (например, десферроксамин с концентрацией 100 мкМ, хлорид кобальта с концентрацией 100 мкМ или диэтиловый эфир N-оксалилглицина с концентрацией 1 mM) тестовая линия клеток продуцирует луциферазу, которая может детектироваться и количественно оцениваться помощью подходящих реагентов, обладающих биолюминесценцией (например, Steady-Glo® Luciferase Assay System, Promega Corporation, Madison, WI 53711, USA), и подходящего прибора для измерения люминесценции.
Последовательность осуществления теста: Клетки за сутки перед тестом наносили в точно измеренном количестве культуральной среды (DMEM, 10% FCS, 2 мМ глутамин) на микротитровальный планшет (фирмы Pierce) с 384 или 1536 лунками и помещали в клеточный инкубатор (влажность воздуха 96%, 5% v/v СО2, 37°C). В день проведения теста в культуральную среду добавляли тестируемые вещества в ступенчатой концентрации. В композиции, служащие отрицательным контролем, к клеткам не добавляли никаких тестируемых веществ. В качестве положительного контроля для определения чувствительности клеток к ингибиторам добавляли, например, десферроксамин в конечной концентрации 100 мкМ. Спустя 6-24 ч после переноса тестируемых веществ в лунки микротитровального планшета измеряли полученный световой сигнал в приборе для измерения люминесценции. Пользуясь измеренными значениями, устанавливали корреляции между действующими дозами, которые служили основой для определения полумаксимальной активной концентрации (обозначенной как ЕС50).
1.c) Клеточный тест, действующий in vitro для изменения экспрессии генов:
Чтобы изучить изменение экспрессии специфических иРНК в линии клеток человека после обработки тестируемыми веществами, культивировали следующие линии клеток на планшетах с 6- или 24 лунками: клетки печени человека (HUH, JCRB Cell Bank, Japan), эмбриональные почечные фибробласты человека (НЕК/293, АТСС, Manassas, VA 20108, USA), клетки рака шейки матки человека (HeLa, ATCC, Manassas, VA 20108, USA), эндотелиальные клетки пупочной вены человека (HUVEC, Cambrex, East Rutherford, New Jersey 07073, USA). Спустя 24 часа после добавления тестируемых веществ промывали клетки солевым фосфатным буферным раствором и получали из них общую - РНК с применением подходящих методов (например, Trizol®-реагент, Invitrogen GmbH, 7613I Karlsruhe, Deutschland).
Для типичного эксперимента по анализу переваривали по 1 мкг полученной таким образом общей РНК с помощью ДНКазы и с применением подходящей реакции реверсной транскриптазы (ImProm-II Reverse Transcription System, Promega Corporation, Madison, WI 5371 1, USA) переводили в кДНК. Использовали 2.5% полученного таким образом состава кДНК соответственно для цепной реакции полимеразы. Уровень экспрессии иРНК исследуемого гена изучали с помощью количественной цепной реакции полимеразы в режиме реального времени (real time quantitative Polymerase chain reaction) [TaqMan-PCR; Held CA., Stevens J., Livak KJ., William: P.M., Genome Res. 6 (10), 986-94 (1996)] с применением ABI Prism 7700 Sequenz-Detektionsinstruments (устройства детектирования последовательностей типа ABI Prism 7700) (фирмы Applied Biosystems, Inc.). Используемые при этом комбинации праймер-зонд генерировали с помощью программного обеспечения Primer Express 1.5 (фирмы Applied Biosystems, Inc.). В частности, исследовали иРНК эритропоэтина, карбоангидразы IX, лактатдегидрогеназы А и васкулярный фактор роста клеток эндотелия.
Вещества согласно данному изобретению приводили к заметному, зависящему от дозы, росту индуцированных гипоксией генов в иРНК в клетках человека.
2. Тест in vivo для доказательства активности (действия) в кардиоваскулярной системе
2.а) Тест in vivo для изменения экспрессии генов:
Мышам и крысам давали исследуемые соединения, растворенные в подходящих растворителях, либо орально через желудочный зонд, либо внутрибрюшинно, либо внутривенно. Типичными дозировками были 0.1, 0.5, 1, 5, 10, 20, 50, 100 и 300 мг вещества на кг веса тела и на прием. Контрольные животные получали только растворитель. Через 4, 8 иди 24 часа после дачи испытуемого вещества животных умерщвляли с помощью сверхдозы изофурана и последующего перелома шейного отдела позвоночника и извлекали исследуемые органы. Части органов подвергали глубокому замораживанию в жидком азоте. Из частей органов получали описанную в В.1.а) общую РНК и ее переводили в кДНК. Уровень экспрессии иРНК изучаемых генов исследовали с помощью количественной цепной реакции полимеразы в режиме реального времени (real time quantitative Polymerase chain reaction) [TaqMan-PCR; Heid CA., Stevens J., Livak KJ., Williams P.M., Genome Res. 6 (10), 986-94 (1996)] с применением устройства детектирования последовательностей типа ABI Prism 7700 (ABI Prism 7700 Sequenz-Detektionsinstruments) (фирмы Applied Biosystems, Inc.).
Вещества согласно изобретению по сравнению с плацебо-контролем через оральное иди парентеральное применение приводили к выраженному, зависящему от дозы, росту иРНК эритропоэтина в почках.
2.b) Определение уровня эритропоэтина в сыворотке:
Мышам и крысам давали исследуемые соединения, растворенные в подходящих растворителях, либо внутрибрюшинно, либо орально один или два раза в день. Типичными дозировками были 0.1, 0.5, 1, 5, 10, 20, 50, 100 и 300 мг вещества на кг веса тела и на прием. Плацебо-контрольные животные получали только растворитель. Перед аппликацией и через четыре часа после последней дачи веществ у животных отбирали из ретроорбитального венозного сплетения или из хвостовой вены 50 мкл крови под наркозом короткого действия. Добавкой гепарин-лития кровь делали несвертывающейся. Центрифугированием получали плазму крови. В плазме крови с помощью эритропоэтин-ELISA (Quantikine® mouse Epo Immunoassay, R&D Systems, Inc., Minneapolis, USA) определяли содержание эритропоэтина в соответствии с руководством фирмы-изготовителя. Измеренные значения пересчитывали в пг/мл (pg/ml), пользуясь заявленными для эритропоэтина мышей сравнительным измерениями.
Вещества согласно изобретению по сравнению с плацебо-контролем после орального иди парентерального применения приводили к выраженному, зависящему от дозы, росту эритопоэтина в плазме по сравнению с исходными значениями и плацебо-контролем.
2.с) Определение клеточного состава периферийной крови:
Мышам и крысам давали исследуемые соединения, растворенные в подходящих растворителях, либо внутрибрюшинно, либо орально один или два раза в день в течение нескольких дней. Типичными дозировками были 0.1, 0.5, 1, 5, 10, 20, 50, 100 и 300 мг вещества на кг веса тепа и на прием. Контрольные животные получали только растворитель. В конце опыта отбирали у животных под наркозом короткого действия кровь из сплетения вен в уголке глаза или из хвостовой вены и добавкой цитрата натрия делали кровь несвертывающейся. С помощью подходящего электронного измерительного прибора определяли концентрацию эритроцитов, лейкоцитов и тромбоцитов. Концентрацию ретикулоцитов определяли при помощи мазков крови, которые окрашивали подходящим для этого красящим раствором (фирма КАВЕ Labortechnik, Nümbrecht), путем тщательного осмотра под микроскопом каждых 1000 эритроцитов. Для определения гематокрита кровь отбирали из ретроорбитального венозного сплетения с помощью гематокритного капилляра и величину гематокрита после центрифугирования капилляров в подходящей для этого центрифуге считывали вручную.
Вещества согласно данному изобретению после орального или парентерального применения приводили к выраженному, зависящему от дозы, росту гематокрита, увеличению числа эритроцитов и ретикулоцитов по сравнению с исходными значениями и плацебо-контролем.
3. Определение растворимости
Приготовление исходного раствора:
Точно отвешивали не менее 1.5 мг тестируемого соединения в Wide Mouth 10 mm Screw V-Vial (фирмы Glastechnik Gräfenroda GmbH, Art.-Nr. 8004-WM-H/V15μ) с подходящим навинчивающимся колпачком и перегородкой, смешивали с DMSO до концентрации 50 мг/мл и встряхивали 30 минут с помощью Vortexcrs.
Приготовление калиброванных растворов:
Необходимые операции пипетирования осуществляли с помощью обращающегося с жидкостями робота (Liquid-Handling-Roboter) в микротитровальный планшет с 96 лунками объемом 1.2 мл [96er Deep Well Plate (DWP)]. В качестве растворителя использовали смесь ацетонитрил/вода 8:2.
Приготовление исходного раствора для калибровочных растворов (концентрированного раствора): 10 мкл Исходного раствора смешивали с 833 мкл смеси растворителей (концентрация=600 мкг/мл) и гомогенизировали. Для каждого тестируемого соединения в отдельном микроимировальном планшете (DWP) готовили разбавление 1:100 и снова гомогенизировали.
Калибровочный раствор 5 (600 нг/мл): 30 мкл концентрированного раствора смешивали с 270 мкл смеси растворителей и гомогенизировали.
Калибровочный раствор 4 (60 нг/мл): 30 мкл калибровочного раствора 5 смешивали с 270 мкл смеси растворителей и гомогенизировали.
Калибровочный раствор 3 (12 нг/мл): 100 мкл калибровочного раствора 4 смешивали с 400 мкл смеси растворителей и гомогенизировали.
Калибровочный раствор 2 (1.2 нг/мл): 30 мкл калибровочного раствора 3 смешивали с 270 мкл смеси растворителей и гомогенизировали.
Калибровочный раствор 1 (0.6 нг/мл): 150 мкл калибровочного раствора 2 смешивали с 150 мкл смеси растворителей и гомогенизировали.
Приготовление растворов проб:
Необходимые операции пипетирования осуществляли с помощью обращающегося с жидкостями робота в микротитровальный планшет с 96 лунками объемом 1.2 мл [96er Deep Well Plate (DWP)]. Смешивали 10.1 мкл концентрированного раствора с 1000 мкл PBS-буферного раствора с рН 6.5 (PBS-буферный раствор с рН 6.5: 61.86 г хлорида натрия, 39.54 г дигидрофосфата натрия и 83.35 г 1 N раствора едкого натра помещали в мерную колбу емкостью 1 литр, добавляли воду и перемешивали около 1 часа. Отбирали 500 мл этого раствора в мерную колбу емкостью 5 литров и добавляли воду. Устанавливали величину рН 6.5 с помощью 1 N раствора едкого натра).
Осуществление:
Необходимые операции пилотирования осуществляли с помощью обращающегося с жидкостями робота в микротитровальный планшет с 96 лунками объемом 1.2 мл [96er Deep Well Plate (DWP)]. Приготовленные таким образом растворы проб встряхивали 24 часа на термостатированном устройстве для встряхивания со скоростью 1400 об/мин при 20°С. От этих растворов отбирали соответственно по 180 мкл и переносили в пробирку из полиалломера центрифуги Бекмана (Beckman Polyallomer Centrifuge Tubes). Центрифугировали эти растворы 1 час с примерно 223.000×g. Отбирали от каждого раствора по 100 мкл раствора сверху и разбавляли PBS-буферным раствором с рН 6.5 в соотношении 1:10 и 1:1000.
Анализ:
Образцы анализировали с помощью ВЭЖХ/МС-МС. Количественную оценку давали по калибровочной кривой тестируемого соединения из пяти точек. Растворимость выражали в мг/л. Последовательность анализа: 1) холостой опыт (смесь растворителей); 2) калибровочный раствор 0.6 нг/мл; 3) калибровочный раствор 1.2 нг/мл; 4) калибровочный раствор 12 нг/мл; 5) калибровочный раствор 60 нг/мл; 6) калибровочный раствор 600 нг/мл; 7) холостой опыт (смесь растворителей); 8) раствор пробы 1:1000; 7) раствор пробы 1:10.
Метод ВЭЖХ/МС-МС:
ВЭЖХ: Agilent I100, квадратичный насос (G1311A), автоматический пробоотборник (Autosampier) CTC HTS PAL, дегазатор (Degaser) (G1 1322A) и термостат для колонки (Saulenthermostat) (01316A); колонка: Oasis HLB 20 мм × 2.1 мм, 25 μ; температура: 40°С; элюент А: вода + 0.5 мл муравьиной кислоты/л; элюент В: ацетонитрил + 0.5 мл муравьиной кислоты/л; скорость потока: 2.5 мл/мин; время останова (Stoptime) 1.5 мин; градиент: 0 мин 95% А, 5% В; Rampe: 0-0.5 мин 5% А, 95% В; 0.5-0.84 мин 5% А, 95% В; Rampe: 0.84-0.85 мин 95% А, 5% В; 0.85-1.5 мин 95% А, 5% В.
MS/MS: WATERS Quattro Micro Tandem MS/MS; Z-Spray API-Interface; ВЭЖХ-МС-прибор (HPLC-MS-Eingangssplitter) 1:20; измерение в режиме ESI.
С. Примеры осуществления для фармацевтических составов
Соединения согласно данному изобретению могут переводиться следующим образом в фармацевтические составы:
Таблетки:
Состав:
100 мг соединения согласно изобретению, 50 мг лактозы (моногидрата), 50 мг кукурузного крахмала (натурального), 10 мг поливинилпирролидона (PVP 25) (фирмы BASF, Людвигсхафен, Германия) и 2 мг стеарата магния.
Вес таблетки 212 мг. Диаметр 8 мм, радиус свода 12 мм.
Получение:
Смесь соединения согласно изобретению, лактозы и крахмала гранулировали с 5%-ным раствором (м/м) поливинилпирролидона в воде. После высушивания гранулят смешивали в течение 5 минут со стеаратом магния. Эту смесь с помощью обычного пресса для таблеток прессовали (формат таблеток смотри выше). В качестве контрольной цифры для прессования использовали сипу прессования 15 кН.
Суспензия для орального применения:
Состав:
1000 мг соединения согласно изобретению, 1000 мг этанола (96%), 400 мг Rhodigel® (ксантановая смола фирмы FMC, Пенсильвания, США) и 99 г воды.
Разовой дозе 100 мг соединения согласно изобретению соответствует 10 мл суспензии для орального применения.
Получение:
Суспендировали Rhodigel в этаноле, добавляли в суспензию соединение согласно изобретению. При перемешивании добавляли воду. До завершения набухания Rhodigel перемешивали около 6 часов.
Раствор для орального применения:
500 мг соединения согласно изобретению, 2.5 г полисорбата и 97 г полиэтиленгликоля 400. Разовой дозе 100 мг соединения согласно изобретению соответствует 20 г раствора для орального применения.
Получение:
Суспендировали соединение согласно изобретению в смеси из полиэтиленгликоля и полисорбата (Polysorbat) при перемешивании. Процесс перемешивания продолжали до полного растворения соединения согласно изобретению.
Раствор для внутривенного вливания:
Растворяли соединение согласно изобретению в концентрации, равной половине растворимости насыщения, в физиологически совместимом растворителе (например, изотоническом растворе поваренной соли, 5%-ном растворе глюкозы и/или 30%-ном растворе PEG-400). Раствор стерильно фильтровали и заполняли стерильные и апирогенные инъекционные емкости.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИПЕРИДИНИЛПИРАЗОЛОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2713937C2 |
| ГЕТЕРОЦИКЛИЛАМИДОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 2006 |
|
RU2415851C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТА[B]БЕНЗОФУРАНА И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2415847C9 |
| АЦИЛИРОВАННЫЕ НОНАДЕПСИПЕПТИДЫ В КАЧЕСТВЕ ПРОИЗВОДНЫХ ЛИЗОБАКТИНА | 2005 |
|
RU2414477C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HIF-ПРОЛИЛ-4-ГИДРОКСИЛАЗЫ | 2009 |
|
RU2509080C9 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| НОВЫЕ АНТИГЕЛЬМИНТНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2828007C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
| Селективный ингибитор киназы JAK1 | 2020 |
|
RU2818002C2 |
Данное изобретение относится к применению производных дигидропиразолонов формулы (I), в которой радикалы и символы определены в п.1 формулы изобретения, для изготовления лекарственного средства для лечения и/или профилактики заболеваний сердечного кровообращения, сердечной недостаточности, анемий, хронических заболеваний почек и почечной недостаточности, а также к лекарственному средству, содержащему указанное производное дигидропиразолона, и к способу лечения и/или профилактики указанных выше заболеваний у человека и животных. 3 н. и 1 з.п. ф-лы, 10 табл., 180 пр.

1. Применение соединений формулы (I)

в которой
R1 означает гетероарильную группу формулы
 ,
, 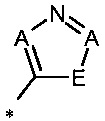 или
или 
в которых
* означает место соединения с дигидропиразоловым циклом,
А означает для каждого отдельного представителя C-R4 или N, при этом максимум два члена цикла А одновременно означают N, и
R4 в каждом отдельном случае независимо друг от друга означают водород или заместители, выбранные из группы циано, (С1-С6)-алкил, C(=O)NH2, где
(С1-С6)-алкил, в свою очередь, может иметь до трех атомов галогена;
Е означает О или S,
R2 означает гетероарильную группу формулы
 или
или  ,
,
в которых
# означает место соединения с дигидропиразолоновым циклом,
G соответственно означает C-R6 или N, при этом не более одного из двух членов цикла G означают N,
R6 в каждом отдельном случае, независимо друг от друга означают водород или заместитель, выбранный из галогена, циано, (С1-С6)-алкила, (С3-С6)-циклоалкила, 4-10-членного гетероциклоалкила, содержащего 1 или 2 гетероатома, выбранного из N, О и S, C(=O)-OR10, -SO2R25, -OR28, -SR29 и -NR30R31, где
(i) (С1-С6)-алкил, в свою очередь, может иметь от одного до трех заместителей, одинаковых или разных, выбранных из группы, включающей фтор, хлор, бром, -NR18-C(=O)-OR19, -OR28 и -NR30R31,
(ii) 4-10-членный гетероциклоалкил, в свою очередь, может соответственно иметь один или два одинаковых или разных заместителя, выбранных из группы (С1-С6)-алкил, -C(=O)-R9, -C(=O)-OR10, CONHR11R12, гидрокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино, ди-(С1-С4)-алкиламино и пирролидинила,
при этом (С1-С6)-алкил, в свою очередь, может быть замещен заместителем, выбранным из циано, гидрокси, (С1-С4)-алкокси, амино, моно-(С1-С4)-алкиламино и ди-(С1-С4)-алкиламино;
(iii) R9, R10, R11, R19, R25, R29 и R30 независимо друг от друга при каждом отдельном проявлении означают остаток, выбранный из группы, включающей водород и (С1-С6)-алкил,
R28 при каждом отдельном проявлении означают остаток, выбранный из водорода, (С1-С6)-алкила и азетидинила, причем (С1-С6)-алкил, в свою очередь, может быть замещен амино, моно-(С1-С4)-алкиамино и ди-(С1-С4)-алкиламино, а азетидинил, в свою очередь, может быть замещен (С1-С4)-алкоксикарбонилом;
(iv) R12, R18 и R31 независимо друг от друга при каждом отдельном проявлении означают остаток, выбранный из водорода и (С1-С6)-алкила; и
J означает S и
R3 означает водород,
или их фармацевтически приемлемых солей,
для изготовления лекарственного средства для лечения и/или профилактики заболеваний сердечного кровообращения, сердечной недостаточности, анемий, хронических заболеваний почек и почечной недостаточности.
2. Лекарственное средство, ингибирующее HIF-пролилгидроксилазу, содержащее соединение формулы (I), которое определено в п. 1, в комбинации с инертным, не токсичным, фармацевтически пригодным вспомогательным веществом.
3. Лекарственное средство по п. 2 для лечения и/или профилактики заболеваний сердечного кровообращения, сердечной недостаточности, анемий, хронических заболеваний почек и почечной недостаточности.
4. Способ лечения и/или профилактики заболеваний сердечного кровообращения, сердечной недостаточности, анемий, хронических заболеваний почек и почечной недостаточности у человека и животных с применением активного количества по меньшей мере одного соединения формулы (I), которое определено в п. 1, или лекарственного средства по п. 2 или 3.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| US 6878729 B2, 12.04.2005 | |||
| RU 2005141499 A? 10.05.2006. | |||

