Предпосылки создания изобретения
Область изобретения
Изобретение относится к кристаллическим солевым формам соединения 8-азабицикло[3.2.1]октана, которые можно использовать в качестве антагонистов мю-опиоидного рецептора. Изобретение также относится к фармацевтическим композициям, содержащим такие кристаллические соединения, к способам получения таких соединений для лечения или облегчения медицинских состояний, опосредованных активностью мю-опиоидного рецептора, и к способам получения таких соединений.
Предшествующий уровень техники
В находящихся одновременно на рассмотрении предварительных заявках на патент США N 60/777962, поданной 1 марта 2006 г., и N 60/841028, поданной 30 августа 2006, и заявке на патент США сер. No. 11/711961 описываются соединения 8-азабицикло[3.2.1]октана, которые являются антагонистами мю-опиоидного рецептора и которые, как ожидается, могут найти применение для лечения или облечения медицинских состояний, опосредованных активностью мю-опиоидного рецептора. В частности, в данных заявках конкретно описано соединение - сульфат 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида, как продемонстрировавшее антагонистическую активность в отношении мю-опиоидного рецептора.
Химическая структура 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (далее упоминаемого как соединение 1) показана ниже:

Для эффективного применения данного соединения в качестве терапевтического агента было бы желательно иметь его твердую солевую форму, которую легко было бы получать, и которая обладала бы приемлемой химической и физической стабильностью. Например, было бы весьма желательно иметь солевую форму, которая является термически стабильной, например, при температурах, превышающих примерно 175°С или примерно 180°C, и не является ни гигроскопичной, ни расплывающейся во влаге воздуха, облегчая тем самым обработку и хранение вещества. Кристаллические твердые вещества обычно являются более предпочтительными по сравнению с аморфными формами с позиций повышения чистоты и стабильности получаемого продукта.
Ранее кристаллические солевые формы соединения 1 известны не были. Соответственно, существует потребность в стабильной кристаллической солевой форме соединения 1, которая не является гигроскопичной, не расплывается во влаге воздуха и проявляет подходящую термическую стабильность.
Краткое изложение сущности изобретения
Настоящее изобретение относится к кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида или ее сольвату. В одном аспекте, кристаллическая солевая форма по изобретению представляет собой кристаллическую сульфатную соль соединения 1. В другом аспекте, кристаллическая солевая форма по изобретению представляет собой кристаллический гидрат сульфатной соли соединения 1.
Неожиданно было установлено, что кристаллическая сульфатная соль по изобретению не проявляет существенных термических процессов ниже температуры плавления в диапазоне от примерно 190 до примерно 205°C и теряет массу менее чем примерно 0,3% при воздействии относительной влажности между примерно 2% и примерно 90% при комнатной температуре. Кроме того, ни кристаллическая сульфатная соль по изобретению, ни ее гидрат не расплываются в поглощенной из воздуха влаге при воздействии до примерно 90% относительной влажности при комнатной температуре.
Наряду с другими применениями, ожидается, что кристаллические солевые формы по изобретению можно будет использовать для получения фармацевтических композиций для лечения или облегчения медицинских состояний, опосредованных активностью мю-опиоидного рецептора. Соответственно, в другом составляющем его аспекте изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и кристаллическую сульфатную соль 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида или ее сольват.
Изобретение также относится к способу лечения или облегчения заболевания или состояния, облечение при котором достигается при лечении с использованием антагониста мю-опиоидного рецептора, например, нарушения, связанного с пониженной моторикой желудочно-кишечного тракта, причем данный способ включает введение млекопитающему терапевтически эффективного количества кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида или ее сольвата.
Изобретение дополнительно относится к способу лечения вызванной опиоидами дисфункции кишечника или послеоперационной кишечной непроходимости, причем данный способ включает введение млекопитающему терапевтически эффективного количества кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида или ее сольвата.
В другом аспекте способа, изобретение относится к способу получения кристаллической сульфатной соли по изобретению, который включает контактирование 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида с серной кислотой с образованием реакционной смеси и выделение кристаллической сульфатной соли из реакционной смеси.
Изобретение относится к дополнительному способу получения кристаллической сульфатной соли по изобретению, который включает диспергирование кристаллического гидрата сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида в разбавителе, включающем метанол, с образованием реакционной смеси и выделение кристаллической сульфатной соли из реакционной смеси.
Еще в одном аспекте способа, изобретение относится к способу получения кристаллической сульфатной соли соединения 1, где данный способ включает: (а) контактирование защищенного предшественника соединения 1, в котором гидроксильные группы являются защищенными, с серной кислотой с образованием первой реакционной смеси; (b) выделение твердой сульфатной соли соединения 1 промежуточного качества из первой реакционной смеси; с) диспергирование твердой сульфатной соли промежуточного качества в разбавителе, включающем метанол, с образованием второй реакционной смеси и (d) выделение кристаллической сульфатной соли из второй реакционной смеси.
В родственном составляющем аспекте, изобретение относится к бисульфитному аддукту бензилового эфира N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты, который можно использовать для получения вышеуказанного защищенного предшественника соединения 1.
Изобретение также относится к кристаллической сульфатной соли по изобретению, как описано в данном описании, для применения при терапевтическом лечении или в качестве лекарственного средства, а также к применению кристаллической сульфатной соли по изобретению для получения лекарственного средства, в особенности для получения лекарственного средства для лечения нарушения, связанного с активностью мю-опиоидного рецептора у млекопитающего.
Краткое описание чертежей
Различные аспекты настоящего изобретения иллюстрируются со ссылкой на прилагаемые чертежи.
На фиг.1 показана порошковая рентгенограмма (XRPD) кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по изобретению.
На фиг.2 показан график, полученный методом дифференциальной сканирующей калориметрии (ДСК) (правая сторона вертикальной оси), и график, полученный при термогравиметрическом анализе (ТГА) (левая сторона вертикальной оси), кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по изобретению.
На фиг.3 показан график, полученный при определении динамической сорбции влаги (ДСВ), для кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по изобретению.
На фиг.4 показана порошковая рентгенограмма (XRPD) кристаллического гидрата сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по изобретению.
На фиг.5 показан график, полученный методом дифференциальной сканирующей калориметрии (ДСК) (правая сторона вертикальной оси), и график, полученный при термогравиметрическом анализе (ТГА) (левая сторона вертикальной оси), кристаллического гидрата сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по изобретению.
На фиг.6 показан график, полученный при определении динамической сорбции влаги (ДСВ), для кристаллического гидрата сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по изобретению.
Подробное описание изобретения
Изобретение относится к кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида или ее сольвату.
Определения
При описании соединений, композиций и способов по изобретению следующие термины имеют следующие значения, если не указано другого.
Термин «терапевтически эффективное количество» означает количество, достаточное для проведения лечения при введении пациенту, нуждающемуся в лечении.
Термин «лечение», как он использован в данном описании, означает лечение заболевания, нарушения или медицинского состояния у пациента, такого как млекопитающее (в частности, человек), которое включает:
(а) предотвращение появления заболевания, нарушения или медицинского состояния, т.е. профилактическое лечение пациента;
(b) облегчение течения заболевания, нарушения или медицинского состояния, т.е. устранение или индуцирование регрессии заболевания, нарушения или медицинского состояния у пациента, включая нейтрализацию действия других терапевтических агентов;
(с) подавление заболевания, нарушения или медицинского состояния, т.е. замедление или остановка развития заболевания, нарушения или медицинского состояния у пациента, или
(d) смягчение симптомов заболевания, нарушения или медицинского состояния у пациента.
Термин «сольват» означает комплекс или агрегат, образованный одной или несколькими молекулами растворенного вещества, т.е. соединения по изобретению или его фармацевтически приемлемой соли, и одной или несколькими молекулами растворителя. Такие сольваты обычно представляют собой кристаллические твердые вещества, имеющие по существу фиксированное мольное соотношение растворенного вещества и растворителя. Иллюстративные растворители включают, в качестве примера, воду, метанол, этанол, изопропанол, уксусную кислоту и тому подобное. Когда растворителем является вода, образованный сольват конкретно определяют термином «гидрат».
Термин «кристаллическая сульфатная соль» или, альтернативно, «кристаллическая сульфатная соль (безводная форма)» или «безводная сульфатная соль», как они использованы в данном описании, означают кристаллическое твердое вещество, которое не включает в существенном количестве фиксированной молярной доли молекул растворителя в кристаллической решетке, т.е. которое не является ни сольватом, ни гидратом. Сольваты, или более конкретно гидраты, по изобретению однозначно определены.
Следует отметить, что, как использовано в описании и прилагаемой формуле изобретения, указания на единственное число могут включать ссылку на множественное число, если контекст четко не указывает на другое.
Активный агент
Активный агент в настоящих солевых формах, т.е. соединение 1, обозначает 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид. Альтернативно, с использованием правил ИЮПАК, как осуществляется программным обеспечением AutoNom, (MDL Information Systems, GmbH, Франкфурт, Германия), соединение обозначается 3-((1R,3R,5S)-8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид. Использованное в данном описании название, следовательно, соответствует записи по правилам ИЮПАК с эндо-ориентацией замещенной фенильной группы относительно ясно определенной группы 8-азабицикло[3.2.1]октана. Еще в одной общепринятой номенклатуре, часть "((S)-2,3-дигидрокси-пропионил)амино" молекулы обозначается по-разному как ((S)-2,3-дигидрокси-1-оксопропил)амино или ((S)-2,3-дигидроксипропанамидо).
Солевые формы по изобретению
В одном аспекте изобретение относится к кристаллическому сульфату 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида.
В одном аспекте, кристаллическая сульфатная соль по настоящему изобретению характеризуется порошковой рентгенограммой (XRPD), имеющей два или более пика дифракции, включая три или более, и четыре или более пика дифракции при значениях 2θ, выбранных из 6,58±0,20, 7,52±0,20, 9,35±0,20, 14,69±0,20, 16,01±0,20, 17,45±0,20, 17,99±0,20, 18,62±0,20, 19,76±0,20, 21,11±0,20, 22,07±0,20, 23,18±0,20, 23,74±0,20, 24,56±0,20, 25,63±0,20, 26,45±0,20, 27,86±0,20, 28,31±0,20, 29,54±0,20, 30,59±0,20, 31,58±0,20, 33,89±0,20 и 36,02±0,20. В частности, в данном аспекте кристаллическая форма характеризуется порошковой рентгенограммой, имеющей два или более пиков дифракции, включая три или более, и четыре или более пиков дифракции при значениях 2θ, выбранных из 14,69±0,20, 16,01±0,20, 21,11±0,20, 22,07±0,20 и 23,18±0,20.
Как хорошо известно в области рентгеновской порошковой дифрактометрии, положения пиков в спектре XRPD относительно менее чувствительны к экспериментальным деталям, таким как подробности получения образца и геометрия прибора, чем относительная высота пиков. Таким образом, в одном аспекте кристаллическая сульфатная соль соединения 1 характеризуется порошковой рентгенограммой, на которой положение пиков по существу соответствует тем, которые показаны на фиг.1.
Кристаллическая структура сульфата была дополнительно охарактеризована рентгенодифракционным анализом монокристалла, с помощью которого были определены следующие параметры кристаллической пространственной решетки: элементарная ячейка орторомбическая с размерами a = 6,8239 Å, b = 16,2275 Å, c = 24,2021 Å, α=β=γ=90°; объем элементарной ячейки (V) 2680,0 Å3; рассчитанная плотность составляет 1,38 г/см3; пространственная группа P212121(#19). Полученная молекулярная структура подтверждает, что химический состав сульфатной соли соединения 1 соответствует молярному соотношению 1:1 сульфатного противоиона и соединения 1, и что асимметрическая элементарная ячейка не содержит воды или молекул других растворителей. Пики на порошковой рентгенограмме, предсказанные из полученных положений атомов, находятся в превосходном соответствии с наблюдаемым характером распределения пиков на XRPD рентгенограмме.
В другом аспекте кристаллическая сульфатная соль по настоящему изобретению характеризуется своим поведением при воздействии высокой температуры. Как продемонстрировано на фиг.2, график, зарегистрированный методом дифференциальной сканирующей калориметрии (ДСК), для высококристаллического образца имеет пик эндотермического теплового потока, идентифицированный как переход плавления в диапазоне примерно от 190°C до примерно 205°C. На графике, полученном методом термогравиметрического анализа (ТГФ), не наблюдается значительной потери веса при температуре ниже температуры плавления. Термическое разложение происходит приблизительно после плавления.
Еще в одном аспекте кристаллическая сульфатная соль характеризуется спектром инфракрасного поглощения, в котором наблюдаются значимые полосы поглощения при примерно 430, 590, 639, 705, 867, 1036, 1053, 1105, 1171, 1231, 1277, 1375, 1391, 1452, 1476, 1553, 1596, 1639, 1664, 2852, 2907, 2928, 2967, 3168 и 3357 см-1.
Было продемонстрировано, что кристаллическая сульфатная соль соединения 1 обладает обратимым профилем сорбции/десорбции с исключительно низким уровнем гигроскопичности (т.е. менее чем примерно 0,3% прибавления веса в диапазоне влажности от 2% относительной влажности до 90% относительной влажности при комнатной температуре), как показано на фиг.3.
Дополнительно было установлено, что кристаллическая сульфатная соль соединения 1 является стабильной при воздействии повышенной температуры и влажности. При хранении в течение 4 недель при 40°C и 75% относительной влажности анализ, проведенный методом ВЭЖХ, показал отсутствие химического разложения и при этом не обнаружено изменений в результатах ДСК, ТГА или XRPD.
В другом аспекте изобретение относится к кристаллическому гидрату сульфатной соли соединения 1.
В одном аспекте кристаллический гидрат сульфатной соли по настоящему изобретению характеризуется порошковой рентгенограммой (XRPD), имеющей два или более пика дифракции, включая три или более, и четыре или более пика при значениях 2θ, выбранных из 9,41±0,20, 9,98±0,20, 15,17±0,20, 16,70±0,20, 18,59±0,20, 19,46±0,20, 19,91±0,20, 20,63±0,20, 21,35±0,20, 21,89±0,20, 23,00±0,20, 24,20±0,20, 25,40±0,20, 26,03±0,20, 27,44±0,20, 28,46±0,20, 29,45±0,20, 31,22±0,20, 31,82±0,20, 33,17±0,20, 33,56±0,20 и 36,89±0,20. В частности, в данном аспекте кристаллическая форма характеризуется порошковой рентгенограммой, имеющей два или более пика дифракции, включая три или более, и четыре или более пика при значениях 2θ, выбранных из 16,70±0,20, 18,59±0,20, 19,46±0,20, 19,91±0,20, 23,00±0,20 и 24,20±0,20.
В другом аспекте кристаллический гидрат сульфатной соли соединения 1 характеризуется порошковой рентгенограммой, на которой положение пиков по существу соответствует тем, которые показаны на фиг.4.
Кристаллический гидрат сульфатной соли по настоящему изобретению также характеризуется графиком, полученным методом диференциально сканирующей калориметрии (ДСК), на котором проявляется два эндотермических процесса: первый пик в эндотермическом потоке тепла в области от примерно 125 до примерно 133°C, и второй пик в диапазоне от примерно 178 до примерно 183°C при проведении анализа при скорости нагрева 10°C в минуту, как проиллюстрировано на фиг.5. На графике, полученном методом термогравиметрического анализа (ТГА), наблюдается первый термический процесс при температуре между примерно 60 и примерно 140°C и второй термический процесс в диапазоне между примерно 140 и примерно 190°C. Анализ методом ТГА в сочетании с ИК спектроскопией испаренного вещества в первом термическом процессе согласуется с составом гидрата, где имеется примерно один моль воды на моль сульфата соединения 1.
Неожиданно кристаллический гидрат сульфатной соли соединения 1 продемонстрировал низкую гигроскопичность. Как показано на фиг.6, кристаллический гидрат обладает обратимым профилем сорбции/десорбции при комнатной температуре во всем диапазоне от примерно 2% до примерно 90% относительной влажности с приростом веса менее примерно 0,3% во всем диапазоне относительной влажности.
Данные свойства солевых форм по данному изобретению дополнительно проиллюстрированы ниже в примерах.
Способы синтеза
Активный агент, 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид, может быть получен из легко доступных исходных веществ с использованием способов, описанных далее в примерах, или способов, описанных в заявках на патент США, находящихся одновременно на рассмотрении и перечисленных в разделе «Предпосылки создания изобретения» настоящей заявки.
В одном способе получения, кристаллическую сульфатную соль по изобретению получают путем контактирования 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида с серной кислотой в количестве примерно от 0,5 до примерно 1,5 мольных эквивалентов, включая примерно 1 мольный эквивалент. Обычно данную реакцию проводят в инертном разбавителе при температуре в диапазоне от примерно 0°C до примерно 65°C, включая диапазон от примерно 60 до примерно 65°C. Подходящие инертные разбавители включают, например, метанол, толуол, дихлорметан и их комбинации, такие как толуол и ацетонитрил, дихлорметан и ацетонитрил и толуол, ацетонитрил и вода в дополнение к комбинации метанола и воды, включающей примерно 10% воды. С использованием данных разбавителей получают реакционную смесь с концентрацией от примерно 5 до примерно 400 мг/мл, включая диапазон от примерно 50 до примерно 100 мг/мл, и выдерживают ее в течение от примерно 2 до примерно 24 часов при необязательном перемешивании. Смесь может быть охлаждена до температуры от примерно 5 до примерно 20°C во время периода выдерживания.
По завершении реакции кристаллическую соль по изобретению выделяют из реакционной смеси с помощью любых обычных способов, таких как фильтрование, концентрирование, центрифугирование и тому подобные.
Альтернативно, кристаллическую сульфатную соль по изобретению получают путем перекристаллизации гидратной формы. Кристаллический гидрат диспергируют в инертном разбавителе, как описано выше, в концентрации от примерно 5 до примерно 400 мг/мл. Особенно полезными разбавителями для данной реакции являются метанол или сочетание метанол:вода, в соотношении от примерно 3:1 до примерно 9:1. Реакционную смесь поддерживают при температуре в диапазоне от примерно 0 до примерно 65°C, обычно при перемешивании, в течение от примерно 1 до примерно 24 часов, включая от примерно 1 до примерно 6 часов. Обычно, во время периода выдерживания реакционную смесь охлаждают от температуры примерно 65°C до температуры от примерно 5 до примерно 20°C. Для улучшения выхода объем раствора может быть уменьшен примерно на 50% перед выдерживанием реакционной смеси в течение промежутка времени от примерно 1 до примерно 24 часами, включая промежуток от примерно 1 до примерно 6 часов, при температуре от примерно 5 до примерно 20°C. Полученные кристаллы выделяют обычным образом.
Как кристаллическую сульфатную соль, так и кристаллический гидрат сульфатной соли соединения 1 преимущественно получают из защищенного предшественника соединения 1. Как описано далее в примерах, для получения активного агента защищенный альдегид 2, бензиловый эфир N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты, регенерированный из его бисульфитного аддукта 3, конденсируют с гидрохлоридом 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида 4 для получения защищенного промежуточного соединения 5, из которого удаляют защитную группу для получения 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида 6.
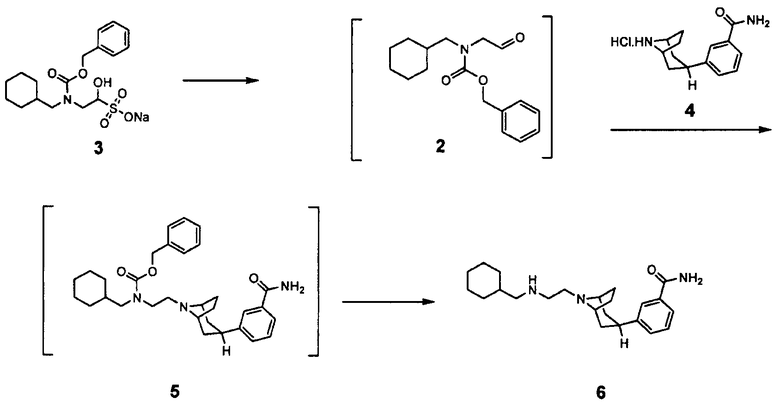
Реакция промежуточного соединения 6 с (4S)-2,2-диметил-1,3-диоксолан-4-карбоксилатом лития 7 приводит к защищенному промежуточному соединению - {2-[3-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметиламиду (S)-2,2-диметил-[1,3]диоксолан-4-карбоновой кислоты 8.

Защищенное промежуточное соединение 8 приводят в контакт с примерно от 0,8 до примерно 1,3 эквивалентами, обычно от примерно 1 до примерно 1,2 эквивалентами, водной серной кислоты в инертном растворителе, таком как этилацетат или изопропилацетат, при температуре между примерно 20 и примерно 30°C. Обычно в реакционную смесь включают второй разбавитель, который смешивается с реакционной смесью, и в котором продукт менее растворим. В качестве второго разбавителя можно использовать ацетонитрил. Реакционную смесь обычно перемешивают в течение от примерно 2 до примерно 72 часов, что приводит к удалению защитной группы из соединения 8 и образованию сульфатной соли соединения 1 промежуточного качества, которая обычно главным образом представляет собой кристаллический гидрат сульфатной соли соединения 1. Продукт промежуточного качества может быть выделен обычным образом, например, путем фильтрования.
Гидратная форма может быть получена перекристаллизацией сульфатного продукта промежуточного качества, например, путем суспендирования продукта промежуточного качества в ацетонитриле при нагревании, добавления воды для ускорения растворения, охлаждения до температуры окружающей среды и выделения перекристаллизованной гидратной формы, как описано ниже в примере 2.
Кристаллическая сульфатная соль соединения 1 может быть получена из твердого продукта промежуточного качества, полученного на описанной выше стадии удаления защитной группы. Продукт промежуточного качества диспергируют в инертном разбавителе, включающем метанол, в концентрации между примерно 5 и 400 мг/мл, включая от примерно 50 до примерно 200 мг/мл. Неожиданно было установлено, что комбинация метанола и воды, содержащая до 25% воды, включая диапазон между примерно 0 и примерно 15% воды и между примерно 5 и примерно 15% воды, представляет собой растворитель, который можно использовать для получения безводной кристаллической соли. В частности, комбинацию метанола и воды, содержащую примерно 10% воды можно использовать для перекристаллизации продукта промежуточного качества, получая безводную сульфатную соль согласно настоящему изобретению.
В обычном способе перекристаллизации, реакционную смесь нагревают до тех пор, пока не получают полное растворение, например, реакционную смесь нагревают до примерно 65°C и затем охлаждают до температуры от примерно 5 до примерно 22°C в течение промежутка времени от примерно 2 до примерно 24 часов. Необязательно, когда реакционная смесь находится при температуре ниже температуры растворения, могут быть добавлены кристаллы безводной сульфатной соли в качестве затравки для кристаллизации. Полученные кристаллы выделяют обычным образом, например, путем фильтрования.
В соответствии с еще одним способом гидратная форма может быть получена из кристаллической сульфатной (безводной) формы. Обычно, кристаллический сульфат первоначально преобразуют в более растворимую аморфную форму, например, путем лиофилизации или быстрого упаривания раствора, полученного из кристаллического сульфата. Затем аморфный твердый сульфат диспергируют в водной системе растворителей, например, содержащей 25% воды и 75% ацетонитрила, и необязательно перемешивают в течение промежутка времени, превышающего примерно 12 часов или более чем примерно 24 часа, при температуре в диапазоне примерно от 0 до примерно 65°C. Обычно температуру первоначально поднимают до примерно 65°C, а затем снижают до температуры в диапазоне от примерно 5 до примерно 20°C. Полученную кристаллическую гидратную форму затем выделяют обычным образом.
В соответствии с аспектом способа, среди других способов изобретение относится к способу получения кристаллической сульфатной соли соединения 1, данный способ включает а) контактирование защищенного предшественника соединения 1, в котором гидроксильные группы являются защищенными, с серной кислотой с образованием первой реакционной смеси; (b) выделение твердой сульфатной соли соединения 1 промежуточного качества из первой реакционной смеси; с) диспергирование твердой сульфатной соли промежуточного качества в разбавителе, включающем метанол, с образованием второй реакционной смеси и (d) выделение кристаллической сульфатной соли из второй реакционной смеси.
Дополнительно, в составляющем его аспекте, изобретение относится к бисульфитному аддукту 3 бензилового эфира N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты, который можно использовать для получения соединения 1. Как описано в получении 1, бисульфитный аддукт 3 может быть получен путем восстановительного аминирования циклогексанкарбоксальдегида 2,2-диэтоксиэтиламином с использованием триацетоксиборгидрида натрия с последующим присоединением защитной группы аминогруппы, удалением альдегидной функциональной группы и преобразованием в бисульфитный аддукт путем взаимодействия с бисульфитом натрия. Альтернативно, первоначальное восстановительное аминирование может быть осуществлено посредством каталитического гидрирования. Подходящие катализаторы гидрирования включают, но не ограничиваются указанным, палладиевые, платиновые катализаторы и никель Ренея.
Фармацевтические композиции
Кристаллические сульфатные солевые формы по изобретению обычно вводят пациенту в виде фармацевтической композиции или препарата. Такие фармацевтические композиции можно вводить пациенту с помощью любого приемлемого пути введения, включая, но не ограничиваясь указанным, пероральный, ректальный, влагалищный, назальный, путем ингаляции, наружный (включая чрезкожный) и парентеральный пути введения.
Соответственно, в одном из составляющих его аспектов, изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель или эксципиент и терапевтически эффективное количество кристаллической сульфатной соли соединения 1 или ее сольвата. Необязательно такие фармацевтические композиции могут содержать, при желании, другой терапевтический агент и/или агенты для получения препаратов. При обсуждении композиций, следует понимать, что термин «соль по изобретению» включает кристаллическую сульфатную соль соединения 1, а также ее сольваты, в частности ее гидрат.
Фармацевтические композиции по изобретению обычно содержат терапевтически эффективное количество активного агента, присутствующего в виде соли по изобретению. Обычно такие фармацевтические композиции будут содержать от примерно 0,1 до примерно 95% по весу активного агента, предпочтительно от примерно 5 до примерно 70% по весу и более предпочтительно от примерно 10 до примерно 60% по весу активного агента.
В фармацевтических композициях по изобретению можно использовать любой общепринятый носитель или эксципиент. Выбор конкретного носителя или эксципиента, или комбинации носителей или эксципиентов будет зависеть от способа введения, используемого для лечения конкретного пациента или от типа медицинского состояния или заболевания. В данном отношении, получение подходящей фармацевтической композиции для определенного пути введения хорошо известно в рамках знаний специалистов в области фармацевтики. Дополнительно носители или эксципиенты, используемые в фармацевтических композициях по данному изобретению, являются коммерчески доступными. В качестве иллюстрации обычные методы получения рецептур описаны в Remington: The Science и Practice of Pharmacy, 20е издание, Lippincott Williams & White, Baltimore, Maryland (2000); и H.C. Ansel et al., Pharmaceutical Dosage Forms и Drug Delivery Systems, 7е издание, Lippincott Williams & White, Baltimore, Maryland (1999).
Иллюстративные примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничиваются указанным, следующие: сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу, такую как микрокристаллическая целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; порошкообразную трагакантовую камедь; солод; желатин; тальк; эксципиенты, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этиолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; не содержащую пирогена воду; изотонический физиологический раствор; раствор Рингера; этиловый спирт; фосфатные буферные растворы и другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции обычно получают путем тщательного и однородного смешивания или перемешивания активного агента с фармацевтически приемлемым носителем и одним или несколькими необязательными ингредиентами. Полученная однородная смесь затем может быть сформована или введена в таблетки, капсулы, пилюли и тому подобное с использованием общепринятых способов и оборудования.
Фармацевтические композиции по изобретению предпочтительно упаковывают в виде единичной дозированной формы. Термин «единичная дозированная форма» относится к физически дискретной единице, подходящей для введения дозировки пациенту, т.е. каждая единица содержит предварительно определенное количество активного агента, рассчитанного для оказания желаемого терапевтического действия либо самого по себе, либо в сочетании с одной или несколькими дополнительными единицами. Например, такие единичные дозированные формы могут представлять собой капсулы, таблетки, пилюли и тому подобное или единичные упаковки, подходящие для парентерального введения.
В одном варианте осуществления фармацевтические композиции по изобретению являются подходящими для перорального введения. Подходящие фармацевтические композиции для перорального введения могут находиться в виде капсул, таблеток, пилюль, пастилок, облаток, драже, порошков, гранул или в виде раствора или суспензии в водной или неводной жидкости; или в виде эмульсии масло-в-воде или вода-в-масле; или в виде эликсира или сиропа и тому подобного; где каждое из них содержит предварительно определенное количество соединения по настоящему изобретению в виде активного ингредиента.
В том случае, если они предназначены для перорального введения в виде твердой дозированной формы (т.е. в виде капсул, таблеток, пилюль и тому подобного), фармацевтические композиции по изобретению обычно будут включать активный агент и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия или дикальций фосфат. Необязательно или альтернативно, такие твердые дозированные формы могут также включать: наполнители или заполнители, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или аравийская камедь; увлажнители, такие как глицерин; дезинтегрирующие агенты, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и/или карбонат натрия; замедляющие растворение агенты, такие как парафин; ускорители абсорбции, такие как четвертичные аммониевые соединения; увлажнители, такие как цетиловый спирт и/или глицерин моностеарат; адсорбенты, такие как каолин и/или бентонитовая глина; лубриканты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; красители и буферные агенты.
Высвобождающие агенты, смачивающие агенты, агенты для получения оболочки, подсластители, вкусовые агенты и ароматизаторы, консерванты и антиоксиданты также могут присутствовать в фармацевтических композициях по изобретению. Примеры фармацевтически приемлемых антиоксидантов включают: водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и тому подобные; масло-растворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, лецитин, пропилгаллат, альфа-токоферол и тому подобные; и металлохелатирующие агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота, сорбит, винная кислота, фосфорная кислота и тому подобные. Агенты для получения покрытий для таблеток, капсул, пилюль и тому подобного, включая те, которые используются для получения покрытий, растворимых в кишечнике, такие как ацетатфталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы, сополимеры метакриловой кислоты/сложного эфира метакриловой кислоты, ацетаттримеллитат целлюлозы, карбоксиметилэтилцеллюлоза, ацетат-сукцинат гидроксипропилметилцеллюлозы и тому подобные.
Фармацевтические композиции по изобретению также могут быть получены таким образом, чтобы обеспечить медленное или контролируемое высвобождение активного агента с использованием, в качестве примера, гидроксипропилметилцеллюлозы в различных пропорциях или других полимерных матриц, липосом и/или микросфер. Кроме того, фармацевтические композиции по изобретению могут необязательно содержать заглушающие агенты и могут быть получены таким образом, что они будут высвобождать активный ингредиент только или предпочтительно в определенной части желудочно-кишечного тракта, необязательно замедленным образом. Примеры герметизирующих композиций, которые можно использовать, включают полимерные вещества и воски. Активный агент также может существовать в микроинкапсулированном виде, если это является подходящим, с одним или несколькими из вышеуказанных эксципиентов.
Подходящие жидкие дозированные формы для перорального введения включают, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие дозированные формы обычно включают активный агент и инертный разбавитель, такой как, например, вода и другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (главным образом, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли, и сложные эфиры жирных кислот сорбита и их смеси. Суспензии, в дополнение к активному ингредиенту, могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбита, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакантовая камедь и их смеси.
Соли по данному изобретению также можно вводить парентерально (например, путем внутривенной, подкожной, внутримышечной или внутрибрюшинной инъекции). Для парентерального введения активный агент обычно смешивают с подходящим носителем для парентерального введения, включая, например, стерильные водные растворы, физиологический раствор, низкомолекулярные спирты, такие как пропиленгликоль, полиэтиленгликоль, растительные масла, желатин, сложные эфиры жирных кислот, такие как этилолеат и тому подобные. Парентеральные препараты могут также содержать один или несколько антиоксидантов, солюбилизаторов, стабилизаторов, консервантов, смачивающих агентов, эмульгаторов или диспергирующих агентов. Данные препараты можно сделать стерильными посредством использования стерильной среды для инъекций, стерилизующего агента, фильтрации, облучения или нагревания.
Альтернативно, фармацевтические композиции по изобретению можно составить для введения путем ингаляции. Подходящие фармацевтические композиции для введения путем ингаляции будут обычно представлены в виде аэрозоля или порошка. Такие композиции обычно вводят с использованием хорошо известных устройств доставки, таких как дозированный ингалятор, порошковый ингалятор, небулайзер или аналогичное устройство доставки.
При введении посредством ингаляции с использованием контейнера под давлением, фармацевтические композиции по изобретению обычно будут включать активный ингредиент и подходящий пропеллент, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другие подходящие газы. Кроме того, фармацевтическая композиция может быть в виде капсулы или картриджа (изготовленного, например, из желатина), содержащего соединение по изобретению и порошок, подходящий для использования в порошковом ингаляторе. Подходящие порошковые основы включают, в качестве примера, лактозу или крахмал.
Соли по изобретению также можно вводить чрезкожно с использованием известных чрезкожных систем доставки и эксципиентов. Например, активный агент может быть смешан с усилителями проницаемости, такими как пропиленгликоль, полиэтиленгликоль монолаурат, азациклоалкан-2-оны и тому подобные и введен в пластырь или аналогичную систему доставки. При желании в таких чрезкожных композициях можно использовать дополнительные эксципиенты, включая желирующие агенты, эмульгаторы и буферы.
При желании, соли по данному изобретению можно вводить в комбинации с одним или несколькими другими терапевтическими агентами. В данном варианте осуществления соль по данному изобретению либо физически смешивают с другим терапевтическим агентом с образованием композиции, содержащей оба агента; либо каждый агент присутствует в отдельной и индивидуальной композиции, которые вводят пациенту одновременно или последовательно.
Например, соль по изобретению может быть объединена со вторым терапевтическим агентом с использованием обычных способов и оборудования с образованием композиции, включающей соединение 1 и второй терапевтический агент. Дополнительно, терапевтические агенты могут быть объединены с фармацевтически приемлемым носителем с образованием фармацевтической композиции, включающей соль по изобретению, второй терапевтический агент и фармацевтически приемлемый носитель. В данном варианте осуществления компоненты композиции обычно смешивают или перемешивают для создания физической смеси. Физическую смесь затем вводят в терапевтически эффективном количестве с использование одного из описанных в данном описании путей. Альтернативно, терапевтические агенты могут оставаться по отдельности и индивидуально перед введением пациенту. В данном варианте осуществления агенты не смешивают вместе физически перед введением, вводят одновременно или через определенные промежутки времени в виде отдельных композиций. Такие композиции могут быть запакованы по отдельности или могут быть упакованы вместе в виде набора. Два терапевтических агента в наборе можно вводить с использованием одного и того же пути введения или с использованием различных путей введения.
В качестве второго терапевтического агента можно использовать любой терапевтический агент, совместимый с настоящим активным агентом. В частности, в сочетании с настоящими соединениями можно использовать прокинетические агенты, действующие по механизмам, отличающимся от антагонизма мю-опиоидного рецептора. Например, в качестве второго терапевтического агента можно использовать агонисты 5-HT4 рецептора, такие как тегазерод, рензаприд, мозаприд, прукалоприд, {(1S,3R,5R)-8-[2-(4-ацетилпиперазин-1-ил)этил]-8-азабицикло[3.2.1]окт-3-ил}амид, 1-изопропил-1H-индазол-3-карбоновой кислоты, {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид, 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты или метиловый эфир 4-(4-{[(2-изопропил-1H-бензимидазол-4-карбонил)амино]метил}-пиперидин-1-илметил)пиперидин-1-карбоновой кислоты.
Дополнительные полезные прокинетические агенты включают, но не ограничиваются указанным, агонисты рецептора, 5-HT3 (например, пумосетраг) антагонисты рецептора 5-HT1A (например, AGI 001), альфа-2-дельта лиганды (например, PD-217014), открыватели хлоридных каналов (например, лубипростон), антагонисты допамина (например, итоприд, метаклопрамид, домперидон), агонисты GABA-B (например, баклофен, AGI 006), каппа-опиоидные агонисты (например, азимадолин), мускариновые M1 и M2 антагонисты (например, акотиамид), агонисты мотилина (например, митемцинал), активаторы гуанилатциклазы (например, MD-1100) и агонисты грелина (например, Tzp 101, RC 1139).
Кроме того, соли по изобретению могут быть объединены с опиоидными терапевтическими агентами. Такие опиоидные агенты включают, но не ограничиваются указанным, морфин, петидин, кодеин, дигидрокодеин, оксиконтин, оксикодон, гидрокодон, суфентанил, фентанил, ремифентанил, бупренорфин, метадон и героин.
Многочисленные дополнительные примеры таких терапевтических агентов известны в данной области и любые такие известные терапевтические агенты можно использовать в сочетании с соединениями по данному изобретению. Вторичный(ые) агент(ы), когда они включены в состав, присутствуют в терапевтически эффективном количестве, т.е. в любом количестве, которое оказывает терапевтически благоприятное действие при совместном введении вместе с соединением по изобретению. Подходящие дозировки для других терапевтических агентов, вводимых в сочетании с соединением по изобретению, обычно колеблются от примерно 0,05 мкг/день до примерно 100 мг/день.
Соответственно, фармацевтические композиции по изобретению необязательно включают второй терапевтический агент, как описано выше.
Следующие примеры иллюстрируют показательные фармацевтические композиции по настоящему изобретению.
Пример рецептуры A: твердые желатиновые капсулы для перорального введения
Соль по изобретению (50 г), высушенную распылением лактозу (200 г) и стеарат магния (10 г) тщательно перемешивают. Полученную композицию загружают в твердую желатиновую капсулу (260 мг композиции на капсулу).
Пример рецептуры B: твердые желатиновые капсулы для перорального введения
Соль по изобретению (20 мг), крахмал (89 мг), микрокристаллическую целлюлозу (89 мг) и стеарат магния (2 мг) тщательно смешивают и затем пропускают через сито U.S. No. 45 меш. Полученную композицию загружают в твердую желатиновую капсулу (200 мг композиции на капсулу).
Пример рецептуры C: желатиновые капсулы для перорального введения
Соль по изобретению (10 мг), полиоксиэтиленсорбит моноолеат (50 мг) и порошок крахмала (250 мг) тщательно перемешивают и затем загружают в желатиновую капсулу (310 мг композиции на капсулу)
Пример рецептуры D: таблетки для перорального введения
Соль по изобретению (5 мг), крахмал (50 мг) и микрокристаллическую целлюлозу (35 мг) пропускают через сито U.S. No. 45 меш и тщательно перемешивают. Раствор поливинилпирролидона (10 вес.% в воде, 4 мг) смешивают с полученными порошками и данную смесь затем пропускают через сито U.S. No. 14 меш. Полученные таким образом гранулы сушат при 50-60°C и пропускают через сито U.S. No. 18 меш. Затем к гранулам добавляют карбоксиметилкрахмал натрия (4,5 мг), стеарат магния (0,5 мг) и тальк (1 мг), которые предварительно были пропущены через сито U.S. No. 60 меш. После перемешивания смесь прессуют на таблетирующей машине, получая таблетки весом 100 мг.
Пример рецептуры E: таблетки для перорального введения
Соль по изобретению (25 мг), микрокристаллическую целлюлозу (400 мг), испаренный диоксид кремния (10 мг) и стеариновую кислоту (5 мг) тщательно перемешивают и затем прессуют с получением таблеток (440 мг композиции на таблетку).
Пример рецептуры F: таблетки с одной насечкой для перорального введения
Соль по изобретению (15 мг), кукурузный крахмал (50 мг), кросскармелозу натрия (25 мг), лактозу (120 мг) и стеарат магния (5 мг) тщательно перемешивают и затем прессуют с получением таблеток с одной насечкой (215 мг композиции на таблетку).
Пример рецептуры G: суспензия для перорального введения
Следующие ингредиенты тщательно смешивают с получением суспензии для перорального введения, содержащей 100 мг активного ингредиента на 10 мл суспензии.
(Vanderbilt Co.)
Пример рецептуры H: сухая порошкообразная композиция
Тонкоизмельченную соль по изобретению (1 мг) смешивают с лактозой (25мг) и затем загружают в ингаляционный картридж из желатина. Содержимое картрижда вводят с использованием порошкового ингалятора.
Пример рецептуры J: препараты для инъекций
Соль по изобретению (0,1 г) смешивают с 0,1 M буферным раствором цитрата натрия (15 мл). pH полученного раствора доводят до pH 6 с использованием 1 н. водной соляной кислоты или 1 н. водного раствора гидроксида натрия. В цитратный буфер затем добавляют стерильный обычный физиологический раствор для достижения общего объема 20 мл.
Следует понимать, что в обсуждавшихся выше фармацевтических композициях можно использовать любую форму соли по изобретению (т.е. кристаллическую соль или сольват), которая является подходящей для определенного способа введения.
Применение
Настоящий активный агент, сульфат 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида, представляет собой антагонист мю-опиоидного рецептора, и, следовательно, ожидается, что соли по изобретению можно будет использовать для лечения медицинских состояний, опосредованных мю-опиоидными рецепторами или связанных с активностью мю-опиоидного рецептора. В частности, ожидается, что соли по изобретению можно будет использовать для лечения неблагоприятных действий, связанных с применением опиоидных анальгетиков, т.е. таких симптомов как запор, пониженное опустошение желудка, боль в брюшной области, вздутие, тошнота и гастроэзофагиальный рефлюкс, определяемые совместно как индуцированная опиоидами дисфункция кишечника. Также ожидается, что настоящие солевые формы можно будет использовать для лечения послеоперационной непроходимости кишечника, нарушения пониженной моторики желудочно-кишечного тракта, которое происходит после абдоминального или другого хирургического вмешательства. Кроме того, было предположено, что соединения-антагонисты мю-опиоидного рецептора, такие как соединение 1, можно использовать для изменения состояния при вызванной опиоидами тошноте и рвоте.
Поскольку было показано, что соединение 1 повышает моторику желудочно-кишечного (ЖК) тракта на животных моделях, ожидается, что соли по изобретению можно будет использовать для лечения нарушений ЖК-тракта, вызванных пониженной моторикой, у млекопитающих, включая людей. Такие нарушения ЖК моторики включают, в качестве иллюстрации, хронический запор, синдром воспаленного кишечника с доминированием запора (C-IBS), диабетические и идиопатические парезы желудка и функциональную диспепсию.
Соответственно, в одном аспекте изобретение относится к способу повышения моторики желудочно-кишечного тракта у млекопитающего, причем данный способ включает введение млекопитающему терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соль по изобретению.
При использовании для лечения нарушений, связанных с пониженной моторикой ЖК-тракта или других состояний, опосредованных мю-опиоидными рецепторами, соли по изобретению обычно будут вводить перорально в виде однократной дневной дозы или множества дозировок в день, хотя можно использовать и другие формы введения. Например, в особенности при использовании для лечения послеоперационной непроходимости кишечника, соединения по изобретению можно вводить парентерально. Количество активного агента, вводимого из расчета на дозу или общее количество, вводимое в течение дня, обычно будет определяться лечащим врачом, с точки зрения значимых обстоятельств, включая состояние, подвергаемое лечению, выбранный путь введения, конкретное вводимое соединение и его относительная активность, возраст, вес и ответная реакция индивидуального пациента, тяжесть симптомов у пациента и тому подобное.
Подходящие дозы для лечения нарушений, связанных с пониженной моторикой ЖК-тракта, или других нарушений, опосредованных мю-опиоидными рецепторами, будут колебаться примерно от 0,0007 до примерно 20 мг/кг/день активного агента, включая примерно от 0,0007 до примерно 1,4 мг/кг/день. Для среднего человека весом 70 кг, это будет составлять количество примерно от 0,05 до примерно 100 мг в день активного агента.
В одном аспекте изобретения соединения по изобретению используют для лечения вызванной опиоидами дисфункции кишечника. При использовании для лечения вызванной опиоидами дисфункции кишечника соединения по изобретению обычно будут вводить перорально в виде единичной дневной дозировки или в виде множества доз в день. Предпочтительно, дозировка для лечения вызванной опиоидами дисфункции кишечника будет колебаться примерно от 0,05 до примерно 100 мг в день.
В другом аспекте изобретения соединения по изобретению используют для лечения послеоперационной кишечной непроходимости. При использовании для лечения послеоперационной кишечной непроходимости соединения по изобретению обычно будут вводить перорально или внутривенно в виде единичной дневной дозировки или в виде множества доз в день. Предпочтительно, дозировка для лечения послеоперационной кишечной непроходимости будет колебаться примерно от 0,05 до примерно 100 мг в день.
Изобретение также относится к способу лечения млекопитающего, имеющего заболевание или состояние, связанное с активностью мю-опиоидного рецептора, где способ включает введение млекопитающему терапевтически эффективного количества соединения по изобретению или фармацевтической композиции, содержащей соединение по изобретению.
Настоящие активные агенты необязательно вводят в сочетании с другим терапевтическим агентом или агентами, в частности, в сочетании с прокинетическими агентами, действующими посредством не мю-опиоидных механизмов. Соответственно, в другом аспекте способы и композиции по изобретению дополнительно включают терапевтически эффективное количество другого прокинетического агента.
Как описано выше, соли по изобретению являются антагонистами мю-опиоидного рецептора. Изобретение, соответственно, дополнительно относится к способу антагонизации мю-опиоидного рецептора у млекопитающего, где способ включает введение млекопитающему соли по изобретению.
Среди других свойств было установлено, что настоящие активные агенты в виде свободного основания и сульфатной соли проявляют эффективное связывание с мю-опиоидными рецепторами и не проявляют или проявляют в незначительной степени агонизм в мю-рецепторных функциональных анализах. Следовательно, соли по изобретению являются эффективными антагонистами мю-опиоидного рецептора. Кроме того, активный агент проявляет главным образом периферическую активность по сравнению с активностью центральной нервной системы на модели животных. Таким образом, можно ожидать, что соли по изобретению будут восстанавливать вызванное опиоидами снижение моторики ЖК-тракта, не мешая благоприятным центральным анальгетическим эффектам. Данные свойства, так же как и применение соединений по изобретению, могут быть продемонстрированы с использованием различных анализов in vitro и in vivo, хорошо известных специалистам в данной области. Иллюстративные анализы дополнительно подробно описаны в следующих примерах.
ПРИМЕРЫ
Следующие синтетические и биологические примеры предназначены для иллюстрации изобретения, и их не следует рассматривать как ограничивающие каким-либо образом объем изобретения. В приведенных ниже примерах следующие сокращения имеют следующие значения, если не указано другого. Сокращения, не определенные ниже, имеют общепринятые значения.
DIPEA = N,N-диизопропилэтиламин
ДМФ = N,N-диметилформамид
EtOAc = этилацетат
EtOH = этанол
МеТГФ = 2-метилтетрагидрофуран
MTBE = трет-бутил-метиловый простой эфир
NaHMDS = бис(триметилсилил)амид натрия
PyBop = гексафторфосфат бензотриазол-1-илоксипирролидинофосфония
фт/кв.дюйм = фунты на квадратный дюйм
Rt = время удерживания
ТГФ = тетрагидрофуран
Реагенты и растворители были получены из коммерческих источников (Aldrich, Fluka, Sigma, и т.д.), и использованы без дополнительной очистки. Реакции проводили в атмосфере азота, если не отмечено другого. Мониторинг хода протекания реакции проводили с помощью тонкослойной хроматографии (ТСХ), аналитической высокоэффективной жидкостной хроматографии (анал.ВЭЖХ) и масс-спектрометрии. Соотношения эндо/экзо определяли с помощью ВЭЖХ анализа с использованием описанных ниже протоколов. Реакционные смеси обрабатывали, как конкретно описано для каждой реакции; обычно их очищали путем экстракции или другими методами очистки, такими как кристаллизация и осаждение, зависимые от температуры и растворителя. Получение характеристик продуктов реакций, как положено, проводили с помощью масс- и 1Н-ЯМР спектроскопии. Для измерения ЯМР образцы растворяли в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d 6), и 1H-ЯМР спектр регистрировали с использованием прибора Varian Gemini 2000 (300 МГц) в стандартных условиях наблюдения. Масс-спектрометрическую идентификацию соединений проводили с использованием метода ионизации электрораспылением (ESMS) на приборе Applied Biosystems (Foster City, CA), модель API 150 EX, или приборе Agilent (Palo Alto, CA), модель1100 LC/MSD.
Общие условия для ВЭЖХ
B = вода/ACN (10:90) + 0,1% ТФУК,
ВЭЖХ - Метод 1
Неочищенные соединения растворяли в смеси вода/ACN (50:50) в концентрации примерно 1 мг/мл и анализировали с использованием следующего градиента в течение 20 минут (время (мин)/% B): 0/10, 2,5/20, 9/75, 15/90, 17/90, 18/10, 20/10.
ВЭЖХ - Метод 2
Соединения растворяли в смеси вода/ACN (90:10) в концентрации примерно 1 мг/мл и анализировали с использованием следующего градиента в течение 30 минут (время (мин)/% B): 0/10, 13/10, 23/65, 28/90, 29/90, 30/10.
ВЭЖХ - Метод 3
Соединения растворяли в смеси вода/ACN (90:10) в концентрации примерно 1 мг/мл и анализировали с использованием следующего градиента в течение 55 минут (время (мин)/% B): 0/10, 10/20, 46/75, 47/90, 50/10, 55/10.
Получение 1: Синтез бисульфитного аддукта бензилового эфира N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты
a. Получение N-циклогексилметил-(2,2-диэтоксиэтил)амина
К смеси 2,2-диэтоксиэтиламина (209 мл, 1,43 моль) и МеТГФ (1050 л) добавляли циклогексанкарбальдегид (107 мл, 0,89 моль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре и охлаждали до 0°C. В течение 40 минут добавляли триацетоксиборгидрид натрия (378 г, 1,79 моль) и реакционную смесь перемешивали в течение 2 часов и охлаждали до 0°C. Добавляли 1 M NaOH (1 л). Органический слой промывали насыщенным раствором соли в воде (1:1, 2 × 1 л) и объем уменьшали до ~20%. Добавляли МеТГФ (1 л) и объем уменьшали до ~20%. Раствор неочищенного указанного в заголовке промежуточного соединения использовали непосредственно на следующей стадии.
b. Получение бензилового эфира N-циклогексилметил-(2,2-диэтоксиэтил)карбаминовой кислоты
К продукту предшествующей стадии (~213 г, ~0,9 моль) добавляли МеТГФ (2 л) и DIPEA (233 мл, 1,34 моль). Реакционную смесь охлаждали до 0°C и добавляли по каплям бензилхлорформиат (140 мл, 0,98 моль). Реакционную смесь перемешивали в течение 30 минут при 0°C, в течение 2 часов при подъеме температуры от 0°C до комнатной температуры, а затем в течение 1 часа при комнатной температуре. Добавляли воду (1,6 л) и реакционную смесь перемешивали в течение 10 минут. Фазы отделяли и органический слой промывали бикарбонатом натрия (1,6 л) и водой (1,6 л). Слои отделяли и объем органического слоя уменьшали примерно до 20%. Добавляли МеТГФ (1 л) и объем уменьшали до ~20%. Раствор неочищенного указанного в заголовке промежуточного соединения использовали непосредственно на следующей стадии.
c. Синтез бисульфитного аддукта бензилового эфира N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты
К продукту предшествующей стадии (~302 г, ~0,62 моль) и ацетонитрилу (2 л) добавляли 1 M HCl (2 л) и реакционную смесь перемешивали при 30°C в течение 7 часов. Добавляли этилацетат (2 л) и реакционную смесь перемешивали в течение 10 минут. Фазы отделяли, органический слой промывали 1 M HCl (1,5 л), фазы опять отделяли и органический слой промывали 0,5 M HCl (1 л). Добавляли бисульфит натрия (71,4 г, 0,69 моль), реакционную смесь перемешивали в течение ночи и затем фильтровали. Реактор и осадок на фильтре промывали этилацетатом (1 л). Полученный раствор сушили на воздухе в течение 2 часов и в вакууме в течение ночи, получая указанное в заголовке соединение в виде белого твердого вещества (199 г, >99% чистота по площади пика по данным ВЭЖХ). Фильтрат обрабатывали в соответствии с такой же методикой, получая вторую порцию указанного в заголовке соединение (30 г).
Получение 2: Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
a. Получение 8-бензил-3-экзо-(3-метоксифенил)-8-азабицикло[3.2.1]октан-3-ола
В колбу емкостью 3 л добавляли порошок хлорида церия (194 г, 0,79 моль). Колбу продували азотом и добавляли ТГФ (800 мл). Реакционную смесь перемешивали при 25°C в течение 1 часа. К смеси добавляли по каплям ~1M 3-метоксифенилмагний бромид в ТГФ (800 мл, 0,87 моль). Полученную суспензию перемешивали при 3°C в течение 1,5 часов. Затем добавляли по каплям раствор 8-бензил-8-азабицикло[3.2.1]октан-3-она (120,4 г, 0,56 моль) в ТГФ (200 мл), поддерживая внутреннюю температуру при -5°C. Полученный раствор перемешивали в течение 15 минут. Реакционную смесь добавляли в колбу, содержащую 6 н. HCl (800 мл), поддерживая температуру при 10°C. После удаления растворителя с использованием роторного испарителя реакционную смесь перемешивали при комнатной температуре в течение ночи. Твердые вещества выделяли фильтрованием, промывали 6 н. HCl (70 мл) и ацетонитрилом (3 × 70 мл) и сушили, получая HCl соль указанного в заголовке промежуточного соединения в виде не совсем белого твердого вещества (161 г).
b. Получение 8-бензил-3-(3-метоксифенил)-8-азабицикло[3.2.1]окт-2-ена
В колбу емкостью 3 л добавляли гидрохлорид 8-бензил-3-экзо-(3-метоксифенил)-8-азабицикло[3.2.1]октан-3-ола (383,9 г, 1,06 моль), 6 M HCl (800 мл) и МеТГФ (200 мл). Полученную суспензию нагревали при 70°C в течение 2,5 часов в атмосфере азота. Реакционную смесь переносили в 12 л реактор и охлаждали до 10°C. Реакционную колбу промывали МеТГФ (1 л), который добавляли в 12 л реактор. Добавляли NaOH (50 вес.% в воде, 200 мл) и дополнительно добавляли порциями NaOH (50 вес.%, 150 мл) до достижения pH ~13. Фазы отделяли, водный слой экстрагировали МеТГФ (1 л) и объединенные МеТГФ слои промывали насыщенным раствором соли (1 л). Растворительно удаляли путем упаривания на роторе при 30-40°C, получая указанное в заголовке промежуточное соединение (360 г) в виде густого масла. Добавляли EtOH (1,5 л) и объем уменьшали до ~500 мл и затем доводили до 1,8 л.
c. Получение 3-эндо-(3-метоксифенил)-8-азабицикло[3.2.1]октана
К 8-бензил-3-(3-метоксифенил)-8-азабицикло[3.2.1]окт-2-ену (в 95% EtOH, 400 мл, 0,20 моль), полученному на предшествующей стадии, добавляли 6 M HCl (45 мл) и затем МеТГФ (50 мл). Реакционную смесь продували азотом, нагревали до 40°C и добавляли палладий-на-угле (10 вес.%, 8 г). В реакторе поддерживали повышенное давление водорода (3 × 20 фт/кв.дюйм) и затем гидрировали при давлении 20 фт/кв.дюйм при 40°C в течение 18 часов. Реакционную смесь фильтровали через целит, концентрировали, промывали МеТГФ (2 × 100 мл), фильтровали через крупный стеклянный фильтр, промывали МеТГФ (10 мл) и сушили на фильтре, получая HCl соль указанного в заголовке промежуточного соединения в виде белого твердого вещества (31 г, один изомер, (экзо-изомер, не определяемый посредством ВЭЖХ)). Дополнительно 5,2 г продукта выделяли из маточного раствора.
d. Получение 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
В колбу емкостью 500 мл добавляли гидрохлорид 3-эндо-(3-метоксифенил)-8-азабицикло[3.2.1]октана (115 г, 0,45 моль) и бромистоводородную кислоту (48 вес.% в воде, 100 мл, 0,88 моль). Смесь нагревали до 120°C и выдерживали при этой температуре в течение 24 часов при перемешивании. Дополнительно добавляли раствор бромистоводородной кислоты (25 мл), реакционную смесь нагревали при перемешивании в течение 6 часов и затем охлаждали до 70°C. Добавляли ацетонитрил (200 мл), полученную суспензию охлаждали до 10°C и затем фильтровали, а осадок на фильтре промывали ацетонитрилом (50 мл), получая HBr соль указанного в заголовке промежуточного соединения (99 г, >99% чистота) в виде белого гранулированного твердого вещества.
e. Получение 2,2,2-трифтор-1-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этанона
К раствору гидробромида 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола (54,4 г, 0,19 моль), толуола (210 мл) и триэтиламина (40 мл, 0,29 моль) добавляли трифторуксусный ангидрид (54 мл, 0,38 моль) в течение 20 минут. Реакционную смесь перемешивали при 40°C в течение 2 часов. Добавляли этилацетат (370 мл) и насыщенный раствор соли в воде (1:1, 265 мл). Реакционную смесь перемешивали в течение 15 минут, фазы отделяли. К органическому слою добавляли насыщенный раствор бикарбоната натрия (300 мл) и смесь интенсивно перемешивали в течение ночи. Фазы отделяли, и органический слой промывали насыщенным раствором соли в воде (1:1, 265 мл), сушили над сульфатом натрия и большую часть растворителя удаляли с использованием роторного растворителя. Добавляли толуол (100 мл) и растворитель удаляли с использованием роторного растворителя, получая неочищенное указанное в заголовке промежуточное соединение.
f. Получение 3-эндо-[8-(2,2,2-трифторацетил)-8-азабицикло[3.2.1]окт-3-ил]фенилового эфира трифторметансульфоновой кислоты
В колбу емкостью 500 мл добавляли раствор в этилацетате (220 мл) промежуточного соединения, полученного на предшествующей стадии (32,8 г, 0,11 моль) и триэтиламин (23 мл, 0,17 моль). Раствор охлаждали до 5°C и добавляли по каплям трифторметансульфонилхлорид (14 мл, 0,13 моль). Смесь оставляли нагреваться до 25°C и перемешивали при данной температуре в течение 1 часа. Добавляли насыщенный раствор бикарбоната натрия (200 мл), слои отделяли, к органическому слою добавляли насыщенный раствор соли (150 мл), слои опять отделяли и из органического слоя удаляли растворитель, получая неочищенное указанное в заголовке промежуточное соединение.
г. Получение 3-эндо-[8-(2,2,2-трифторацетил)-8-азабицикло[3.2.1]окт-3-ил]бензонитрила
В колбу емкостью 100 мл добавляли 3-эндо-[8-(2,2,2-трифторацетил)-8-азабицикло[3.2.1]окт-3-ил]фениловый эфир трифторметансульфоновой кислоты (25,3 г, 58,7 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,81 г, 0,9 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (1,01 г, 1,8 ммоль) и цианид цинка (4,2 г, 35,8 ммоль). Три раза колбу продували азотом в течение 5 минут и затем помещали в вакуум лабораторного насоса на 5 минут. В колбу добавляли ДМФ (150 мл) и дистиллированную воду (2,5 мл). Раствор продували азотом при перемешивании в течение 10 минут, нагревали до 120°C и перемешивали при 120°C в атмосфере азота в течение 4 часов. Когда реакция завершалась, добавляли 20 г продукта из предыдущей партии, полученного таким же способом и перемешивали в течение 20 минут.
Большую часть растворителя удаляли путем отгонки и раствор охлаждали до 22°C. К раствору добавляли этилацетат (445 мл) и полученный раствор фильтровали через целит. Добавляли бикарбонат натрия (450 мл) и раствор перемешивали в течение 15 минут. Слои отделяли и органический слой промывали разбавленным насыщенным раствором соли (2 × 95 мл) и фильтровали через сульфат натрия. Объем раствора уменьшали примерно до 50 мл путем удаления этилацетата. Добавляли изопропиловый спирт (150 мл) и раствор перемешивали при 22°C в течение 1 часа. Твердые вещества выделяли путем фильтрования и промывали изопропиловым спиртом (2 × 25 мл), получая указанное в заголовке промежуточное соединение (33,5 г, 100% чистота по данным ВЭЖХ) в виде не совсем белого/светло-коричневого твердого вещества. Вторую порцию продукта (6,3 г, >98% чистота по данным ВЭЖХ) выделяли из фильтрата.
h. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
Раствор 3-эндо-[8-(2,2,2-трифторацетил)-8-азабицикло[3.2.1]окт-3-ил]бензонитрила (10 г, 32 ммоль) в серной кислоте (96%, 12 мл) нагревали до 50°C при перемешивании и выдерживали при данной температуре при перемешивании в течение 2 часов. Реакционную смесь охлаждали до 22°C и медленно добавляли в колбу емкостью 500 мл, содержащую 5 н. NaOH (90 мл) и метанол (100 мл), которую охлаждали до 10°C. Выпавшую в осадок соль отфильтровывали и фильтрат перемешивали при 22°C в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли МеТГФ (150 мл) и реакционную смесь перемешивали при 22°C в течение 5 минут. Слои отделяли и к водному слою добавляли МеТГФ (100 мл). Слои отделяли и к объединенным органическим слоям добавляли насыщенный раствор соли (150 мл). Слои отделяли, органический слой сушили над карбонатом калия и фильтровали и растворитель удаляли. К остатку при перемешивании добавляли смесь EtOH (25 мл) и концентрированной HCl (2,6 мл), затем добавляли MTBE (25 мл) и раствор перемешивали при 22°C. Выпавшие в осадок твердые вещества отфильтровывали и сушили на воздухе, получая HCl соль указанного в заголовке соединения (8 г, 97% чистота по данным ВЭЖХ) в виде белого твердого вещества.
Получение 3: Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
a. Получение 8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-илового эфира трифторметансульфоновой кислоты
В колбу емкостью 500 мл добавляли гидрохлорид 8-бензил-8-азабицикло[3.2.1]октан-3-она (50,4 г, 200 ммоль), EtOAc (160 мл) и 4 н NaOH (50 мл). Реакционную смесь нагревали до 30°C и перемешивали при данной температуре в течение 1 часа. Слои отделяли и водный слой отбрасывали. Объем органического слоя уменьшали до ~40 мл с использованием роторного испарителя и добавляли ТГФ (270 мл).
Полученный раствор добавляли в колбу емкостью 1 л и охлаждали до -20°C. В колбу в течение 15 минут добавляли раствор NaHMDS (1 M в ТГФ, 230 мл, 230 ммоль). Реакционную смесь перемешивали при -20±5°C в течение 1 часа. К реакционной смеси в течение 5 минут порциями добавляли N-фенил-бис(трифторметансульфонимид) (82,2 г, 230 ммоль) и смесь перемешивали при температуре от -20°C до -10°C в течение 1 часа. К реакционной смеси добавляли 1 н. NaOH (200 мл) и смесь оставляли нагреваться до 22°C при перемешивании. Растворитель частично удаляли с использованием роторного испарителя при 30°C, получая объем, равный 450 мл. К оставшейся реакционной смеси добавляли EtOAc (300 мл) и гептан (150 мл). Смесь перемешивали при 22°C в течение 5 минут. Слои отделяли и водный слой отбрасывали. Органический слой промывали 1 н. NaOH (3 × 450 мл). Водный слой отбрасывали. Органический слой концентрировали с использованием роторного испарителя, получая указанное в заголовке промежуточное соединение (77 г, >96% чистота по данным ВЭЖХ, метод 1).
1H ЯМР (d 6-ДМСО, 400 МГц): δ (м.д.) 7,25-7,35 (м, 5H), 6,05 (д, J=5,2, 1H), 3,64 (кв, J=13,2, 2H), 3,40-3,44 (м, 2H), 2,77 (д, J=16,4, 1H), 1,79-2,09 (м, 5H), 1,52-1,59 (м, 1H).
b. Получение 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида
К неочищенному продукту предшествующей стадии добавляли ТГФ (420 мл) и раствор продували азотом в течение 5 минут. В колбу емкостью 2 л добавляли 3-карбамоилфенилбороновую кислоту (98%, 33,0 г, 200 ммоль), ацетат палладия (II) (98%, 0,46 г, 2 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (97%, 1,1 г, 2 ммоль) и фторид калия (34,9 г, 600 ммоль) с последующим добавлением раствора в ТГФ 8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-илового эфира трифторметансульфоновой кислоты. Полученную смесь продували азотом в течение 5 минут, нагревали при кипении с обратным холодильником (67°C) в атмосфере азота и перемешивали в течение 2 часов. Реакционную смесь охлаждали до 30°C, затем добавляли EtOAc (500 мл) и 1 н. NaOH (500 мл) и смесь перемешивали при 22°C в течение 10 минут. Слои отделяли и водный слой отбрасывали. Органический слой промывали смесью насыщенного раствора соли (250 мл) и воды (250 мл) и перемешивали в течение 5 минут. Слои отделяли и водный слой отбрасывали. Органический слой недолго сушили над Na2SO4, фильтровали и растворитель частично удаляли. Продукт выпадал в осадок в виде светло-желтого твердого вещества в процессе удаления растворителя. Полученную суспензию (примерно 200 мл) фильтровали и твердые вещества промывали холодным EtOAc (0°C, 100 мл) и сушили в высоком вакууме при 25°C, получая указанное в заголовке промежуточное соединение (42,5 г) в виде светло-желтого твердого вещества.
Маточный раствор и вышеуказанные растворители после промывания объединяли и концентрировали и полученную суспензию (примерно 100 мл) перемешивали при 5°C в течение 30 минут и фильтровали. Отфильтрованные твердые вещества промывали холодным EtOAc (0°C, 30 мл) и сушили в высоком вакууме, получая вторую порцию указанного в заголовке промежуточного соединения (7 г, объединенный выход 78%, >98,5% чистота по данным ВЭЖХ метод 1).
(m/z): [M+H]+ вычислено для C21H22N2O, 319,18; найдено 319,4. 1H ЯМР (CDCl3, 400 МГц): δ (м.д.) 7,9 (с, 1H), 7,63 (д, J=6,4, 1H), 7,57 (d, J=6,4, 1H), 7,21-7,42 (м, 6H), 6,38 (д, J=4,4, 1H), 6,13 (шир.с, 1H), 5,83 (шир.с, 1H), 3,68-3,76 (м, 2H), 3,46-3,51 (м, 2H), 2,92 (д, J=17,2, 1H), 2,18-2,26 (м, 1H), 2,04-2,12 (м, 2H), 1,86-1,92 (м, 1H), 1,58-1,65 (м, 1H).
c. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
В сосуд для гидрирования емкостью 1 л добавляли 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамид (40 г, 125 ммоль), EtOH (800 мл), 6 M HCl (42 мл) и воду (80 мл) и смесь перемешивали при 22°C до тех пор, пока не наблюдалось полное растворение. Реакционную смесь продували азотом в течение 5 минут при нагревали при 30°C в течение 5 минут. К смеси добавляли 10 вес.% Pd/C (50% в воде, 4 г). Смесь продували при атмосферном давлении водородом в течение 5-10 минут при нагревании. Смесь перемешивали при 50°C в токе водорода при давлении <5 фт/кв.дюйм (<0,34 атмосферы) в течение 5 часов, получая >99% конверсию реагентов в соответствии с результатами анализа методом ВЭЖХ. Раствор охлаждали до 30°C и фильтровали через целит, получая раствор неочищенной HCl соли указанного в заголовке соединения с соотношением эндо/экзо ~93:7 по данным ВЭЖХ, метод 2. эндо Rt=10,97, экзо Rt=12,67. (m/z): [M+H]+ вычислено для C14H18N2O, 231,15; найдено 231,2.
Воду удаляли из неочищенного продукта путем азеотропной отгонки при 30°C в EtOH (~80 мл), получая суспензию, которую нагревали до 60°C до полного растворения. Раствор охлаждали до 35°C и добавляли кристаллы продукта (0,05 г) для затравки. (Кристаллы для затравки получали в соответствии со способом, описанным в получении 2). Полученную суспензию перемешивали при 22°C в течение 30 минут, медленно добавляли MTBE (120 мл) и суспензию перемешивали при 22°C в течение 4 часов и затем при 0°C в течение 1 часа. Полученные твердые вещества отфильтровывали, промывали холодным EtOH и сушили в высоком вакууме, получая HCl соль указанного в заголовке соединение (24,5 г) в виде белого порошка (75% выход, >98,5% чистота, < 0,4% экзо изомера по данным ВЭЖХ метод 3, эндо Rt=8,67, экзо Rt=9,43).
(m/z): [M+H]+ вычислено для C14H18N2O 231,15; найдено 231,2. 1H ЯМР (d 6-ДМСО, 400 МГц): δ (м.д.) 9,13 (шир.с, 1H), 9,03 (шир.с, 1H), 8,05 (с, 1H), 7,93 (с, 1H), 7,73 (д, J=7,6, 1H), 7,58 (д, J=7,6, 1H), 7,40 (т, J=7,6, 2H), 3,97 (с, 2H), 3,17-3,23 (м, 1H), 2,39-2,46 (м, 2H), 2,19-2,24 (м, 2H), 1,86-1,89 (м, 2H), 1,59-1,63 (м, 2H).
Пример 1A: Синтез кристаллического сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
a. Получение бензилового эфира N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты
В колбу емкостью 100 мл добавляли бисульфитный аддукт бензилового эфира N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты (3,94 г, 1 ммоль) и МеТГФ (35 мл), с последующим добавлением воды (25 мл). Полученную суспензию перемешивали при комнатной температуре в течение 5 минут и добавляли 1 M NaOH (8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 45 минут. Слои отделяли и объем органического слоя уменьшали до ~8 мл, получая неочищенное указанное в заголовке промежуточное соединение.
b. Получение бензилового эфира 2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]-этил}циклогексилметилкарбаминовой кислоты
К продукту предшествующей стадии добавляли ДМФ (15 мл), с последующим добавлением гидрохлорида 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)-бензамида (2,67 г, 1 ммоль) и затем ДМФ (10 мл). Смесь перемешивали при комнатной температуре в течение 30 минут, охлаждали до 10°C и затем добавляли триацетоксиборгидрид натрия (4,25 г, 2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 90 минут и затем охлаждали до 10°C. Добавляли изопропилацетат (100 мл), затем 1 M NaOH (50 мл). Смесь перемешивали в течение 15 минут и фазы отделяли. Органический слой промывали насыщенным раствором соли в воде (1:1, 2 × 50 мл) и объем органического слоя уменьшали до ~10 мл, получая неочищенное указанное в заголовке промежуточное соединение.
c. Получение 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
К продукту предыдущей стадии добавляли EtOH (30 мл) и концентрированную HCl (1,5 мл). Раствор продували азотом, добавляли 10% палладий-на-угле (470 мг) и смесь продували азотом в течение 5 минут и затем гидрировали при давлении 30 фт/кв.дюйм в течение ночи. После продувания азотом в течение 2 минут раствор фильтровали через целит и растворитель удаляли до объема ~10 мл. Добавляли изопропилацетат (40 мл) и 1 M NaOH (20 мл). Слои отделяли и органический слой промывали насыщенным раствором соли (20 мл), фазы отделяли и органический растворитель удаляли до объема 5-10 мл. Добавляли изопропилацетат (20 мл) и объем уменьшали до ~8 мл, к данному раствору добавляли изопропилацетат (20 мл). Полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Продукт выделяли путем фильтрования, реакционную колбу и осадок на фильтре промывали изопропилацетатом (10 мл), получая указанное в заголовке промежуточное соединение (2,4 г, 98% чистота) в виде не совсем белого твердого вещества.
d. Получение сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (гидратная форма)
В колбу емкостью 500 мл добавляли 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид (31 г, 83,9 ммоль) и ДМФ (150 мл). Смесь перемешивали в течение 10 минут, затем добавляли гексафторфосфат бензотриазол-1-илокситрис(пирролидино)фосфония (56,8 г, 109 ммоль) и (4S)-2,2-диметил-1,3-диоксолан-4-карбоксилат лития (15,6 г, 92,3 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли этилацетат (600 мл) и 0,5 M NaOH (300 мл) и фазы отделяли. Органический слой содержал неочищенный ({2-[3-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметиламид (S)-2,2-диметил-[1,3]диоксолан-4-карбоновой кислоты (~84 ммоль), который не выделяли в индивидуальном состоянии.
Органический слой промывали насыщенным раствором соли в воде (1:1, 2 × 300 мл) и фазы отделяли. К органическому слою добавляли 2 M H2SO4 (42 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли ацетонитрил (300 мл) и полученную суспензию перемешивали в течение 2 часов. Продукт выделяли фильтрованием, осадок на фильтре промывали ацетонитрилом (200 мл), сушили на воздухе в течение 2 часов и затем в вакууме при комнатной температуре в течение 20 часов, получая указанное в заголовке соединение (40 г, 97% чистота по данным ВЭЖХ) в виде белого порошка.
e. Синтез кристаллического сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
В колбу емкостью 100 мл добавляли гидратную форму сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидрокси-пропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (2 г) и MeOH (40 мл). Полученную суспензию нагревали до 65°C в атмосфере азота в течение 20 минут, получая полное растворение вещества. Раствор охлаждали до комнатной температуры при перемешивании. Примерно 20 мл растворителя удаляли при слегка пониженном давлении и полученную суспензию перемешивали при комнатной температуре в течение ночи. Продукт выделяли путем фильтрования и колбу и осадок на фильтре промывали ацетонитрилом (2 × 5 мл). Осадок на фильтре сушили на воздухе в течение 2 часов и затем в вакууме при комнатной температуре в течение ночи, получая указанное в заголовке соединение (1,71 г, >99% чистота по данным ВЭЖХ, ~85% выход) в виде белого порошка.
Образец, полученный в соответствии с вышеуказанным способом, характеризовали с помощью 1H ЯМР (400 МГц, ДМСО d 6): δ (м.д.) 9,08 и 8,94 (два набора ушир.с, 1H), 7,99-8,04 (м, 2H), 7,74-7,76 (м, 1H), 7,68-7,70 (м, 1H), 7,41-7,45 (м, 2H), 4,81, 5,00 и 5,30 (три набора ушир.с, 2H), 4,34 (деформированный м, 1H), 4,00 и 4,05 (деформированный м, 2H), 3,01-3,25 и 3,47-3,55 и 3,75-3,82 (три набора м, 10H), 2,50-2,55 (м, 2H), 1,99 (деформированный м, 2H), 1,56-1,70 (м, 8H), 1,15-1,19 (м, 3H), 0,89-0,99 (м, 2H).
Пример 1B: Синтез кристаллического сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
a. Получение сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидрокси-пропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Смесь 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (100 г, 270,6 ммоль) и ДМФ (480 мл) перемешивали в течение 10 минут и затем охлаждали до 0°C. Добавляли гексафторфосфат безотриазол-1-илокситрис(пирролидино)фосфония (183 г, 352 ммоль) и (4S)-2,2-диметил-1,3-диоксолан-4-карбоксилат лития (49,3 г, 324 ммоль) в виде одной порции при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли изопропилацетат (2,0 л) и 1 M NaOH (1,0 л), реакционную смесь перемешивали в течение 15 минут и фазы отделяли. Органический слой промывали насыщенным раствором соли в воде (1:1, 2 × 1,0 л) и фазы отделяли. Объем органического слоя уменьшали до четверти первоначального объема (~500 мл), добавляли ацетонитрил (500 мл) и реакционную смесь перемешивали до гомогенизации, получая раствор промежуточного 2-[3-эндо-(3-карбамоил-фенил)-8-азабицикло[3.2.1]окт-8-ил]этил-циклогексилметил-амида (S)-2,2-диметил-1,3-диоксолан-4-карбоновой кислоты в изопропилацетате и ацетонитриле.
Аликвоту вышеуказанного раствора промежуточного соединения (3,03 г, 6,09 ммоль) в смеси изопропилацетат/ацетонитрил (22,5 мл) объединяли с 2,0 М серной кислотой в воде (3,68 мл) и выдерживали при 25°C в течение 20 часов и затем выдерживали при 10°C при перемешивании в течение 5 часов. Реакционный раствор фильтровали и осадок на фильтре промывали ацетонитрилом (25 мл) и сушили, получая сульфатную соль указанного в заголовке соединения промежуточного качества (2,91 г, 99,4% чистота по данным ВЭЖХ) в виде белого твердого вещества, преимущественно в виде кристаллического гидрата.
b. Синтез кристаллического сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Смесь сульфата 3-эндо-(8-2-[циклогексилметил-((S)-2,3-дигидроксипропионил)-амино]-этил-8-азабицикло[3.2.1]окт-3-ил)бензамида промежуточного качества (154,0 г, 277,1 ммоль), полученного на предыдущей стадии, с метанола/10% воды (616 мл) нагревали при 65°C в течение 45 минут при перемешивании. Реакционную смесь охлаждали до 55°C, добавляли кристаллы указанного в заголовке продукта (120 мг) для затравки, реакционную смесь перемешивали при 55°C в течение 1 часа, температуру снижали до 20°C при скорости 10°C/час и затем выдерживали в течение 8 часов. Реакционную смесь охлаждали до 5°C, выдерживали в течение 30 минут и фильтровали. Осадок на фильтре промывали метанолом (2 × 25 мл) и сушили в течение ночи в высоком вакууме, получая указанное в заголовке соединение (126,3 г, 99,9% чистота).
Пример 2: Перекристаллизация сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (гидратная форма)
Сульфат соединения 1 (гидратная форма) (920 мг) суспендировали в ацетонитриле (5 мл) и нагревали до 65°C. Затем по каплям добавляли воду (2,4 мл) до достижения полного растворения. Полученный раствор охлаждали до температуры окружающей среды в течение 20 минут. Появление зародышей кристаллов наблюдали примерно при 35°C. Твердые вещества выделяли фильтрованием в вакууме, промывали ацетонитрилом (5 мл) и сушили, получая указанное в заголовке соединение.
Пример 3: Кристаллизация сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Сульфат соединения 1 (гидратная форма) (50 мг) диспергировали в смеси растворителей (0,83 мл), состоящей из воды (10%) и метанола (90%), и нагревали до 60°C при перемешивании. Полученный раствор оставляли охлаждаться до температуры окружающей среды в течение 2 часов. Полученные твердые вещества выделяли фильтрованием в вакууме, получая указанное в заголовке соединение (8 мг).
Пример 4: Кристаллизация сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Сульфат соединения 1 (гидратная форма) (42 мг) диспергировали в смеси растворителей (0,42 мл), состоящей из воды (25%) и метанола (75%), и нагревали до 60°C при перемешивании. Полученный раствор оставляли охлаждаться до температуры окружающей среды. Объем раствор уменьшали на 50% путем упаривания с использованием роторного испарителя и раствор оставляли при температуре окружающей среды на ночь. Полученные твердые вещества выделяли фильтрованием в вакууме, получая указанное в заголовке соединение (8 мг).
Пример 5: Кристаллизация сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Соединение 1 (11 мг) растворяли в смеси растворителей (0,2 мл), состоящей из толуола (22%) и ацетонитрила (78%). Добавляли ацетонитрил (0,15 мл), а затем 0,04 M серную кислоту в ацетонитриле (0,59 мл). При добавлении кислоты образовывался твердый осадок. Реакционную смесь оставляли при температуре окружающей среды на 12 часов. Полученное твердое вещество выделяли фильтрованием, получая указанное в заголовке соединение.
Пример 6: Кристаллизация сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Соединение 1 (38 мг) растворяли в дихлорметане (0,5 мл). К раствору добавляли 0,04 M серную кислоту в ацетонитриле (1,91 мл). При добавлении кислоты образовывался твердый осадок. Реакционную смесь оставляли при температуре окружающей среды на 12 часов. Полученное твердое вещество выделяли фильтрованием, получая указанное в заголовке соединение.
Пример 7: Кристаллизация сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Соединение 1 (22 мг) растворяли в смеси растворителей (0,41 мл), состоящей из толуола (23%) и ацетонитрила (77%). К раствору добавляли 0,04 M серную кислоту в ацетонитриле (1,20 мл). При добавлении кислоты образовывался твердый осадок. К реакционной смеси добавляли воду (0,16 мл) для растворения осадка. Появление зародышей кристаллов наблюдали через 2 часа. Полученное твердое вещество выделяли фильтрованием в вакууме, получая указанное в заголовке соединение.
Пример 8: Кристаллизация сульфата 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (гидратная форма)
Кристаллический сульфат соединения 1 (7,1 г) растворяли в смеси растворителей, состоящей из воды (42 мл) и ацетонитрила (25 мл). Раствор подвергали лиофилизации, получая аморфную сульфатную соль. Аморфную соль (6,6 г) диспергировали в смеси растворителей (34,6 мл), состоящей из ацетонитрила (75%) и воды (25%), и нагревали до 65°C в течение 10 минут при перемешивании и оставляли охлаждаться при перемешивании до достижения температуры окружающей среды. Через 12 часов полученное твердое вещество выделяли фильтрованием в вакууме, получая указанное в заголовке соединение (5,4 г).
Примеры 9-17: Свойства солевых форм по изобретению
Образцы кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (соединение 1), полученной как описано в примере 1A, и кристаллического гидрата сульфатной соли соединения 1, полученного, как описано в примере 2, анализировали с помощью метода рентгеновской дифракции порошка (XRPD), диференциальной сканирующей калориметрии (ДСК), термогравиметрического анализа (ТГА), инфракрасной спектроскопии (ИК) и ионной хроматорафии.
Пример 10: Рентгеновская дифракция порошка
Порошковые рентгенограммы, представленные на фиг.1 и фиг.4, получены на дифрактометре Rigaku с использованием Cu Kα (30,0 Кв, 15,0 мА) облучения. Анализ проводили с использованием гониометра, функционирующего в режиме непрерывного сканирования 3° в минуту с размером шага 0,03° в диапазоне от 2 до 40°. Образцы получали на кварцевых держателях образца в виде тонкого слоя порошкообразного вещества. Прибор был откалиброван по кремниевому стандарту.
Пример 11: Термический анализ
Эксперимент методом дифференциальной сканирующей калориметрии (ДСК) проводили с использованием модуля TA Instruments модель Q-100. Данные получали и анализировали с использованием программного обеспечения TA Instruments Thermal Advantage для Q Series™. Образец примерно в 1-10 мг точно взвешивали в алюминиевом тигле с крышкой. Оценку образца проводили, используя линейный уровень нагрева в 10°C/мин от 5°C обычно до 265°C. Ячейку ДСК продували сухим азотом во время использования. Иллюстративные графики ДСК для образцов кристаллической сульфатной соли и кристаллического гидрата сульфатной соли соединения 1 показаны на фиг.2 и 5 соответственно.
Термогравиметрический анализ (ТГА) проводили с использованием модуля TA Instruments модель Q-500. Данные получали и анализировали с использованием программного обеспечения TA Instruments Thermal Advantage для Q Series™. Образец весом примерно 1-5 мг помещали в алюминиевый тигель или на платиновый лоток и сканировали при температуре от температуры окружающей среды до 300°C при скорости линейного нагрева 10°C/мин. Балансировочную и печную ячейки продували азотом во время использования. Иллюстративные графики, полученные при ТГА образцов кристаллической сульфатной соли и кристаллического гидрата сульфатной соли соединения 1, также показаны на фиг.2 и 5 соответственно.
Пример 12: Оценка динамической сорбции влаги
Оценку динамической сорбции влаги (ДСВ) проводили при 25°C с использованием атмосферного микробаланса VTI, системы SGA-100 (VTI Corp., Hialeah, FL 33016). Использовали образец размером приблизительно в 5-10 мг и влажность устанавливали при значении окружающей среды в начале анализа. Обычный ДСВ анализ состоял из трех сканирований: от влажности окружающей среды до 2% относительной влажности (ОВ), от 2% ОВ до 90% ОВ, от 90% ОВ до 5% ОВ при скорости сканирования 5% ОВ/шаг. Массу измеряли каждые две минуты и ОВ изменяли на следующее значение (±5% ОВ), когда масса образца была стабильной в пределах 0,02% для 5 последовательных точек. Иллюстративные графики ДСВ для образцов кристаллической сульфатной соли и кристаллического гидрата сульфатной соли соединения 1 показаны на фиг.3 и 6 соответственно.
Пример 13: Инфракрасный анализ
Спектр инфракрасного поглощения (ИК) регистрировали в частотном диапазоне от 4000 до 400 см-1 с использованием спектрометра Avatar 360 FT-IR, оборудованного диффузным отражающим инфракрасным спектроскопическим модулем с Фурье-преобразованием (DRIFTS). Иллюстративный спектр ИК поглощения для образца кристаллической сульфатной соли по изобретению имел значимые полосы поглощения при 430±1, 590±1, 639±1, 705±1, 867±1, 1036±1, 1053±1, 1105±1, 1171±1, 1231±1, 1277±1, 1375±1, 1391±1, 1452±1, 1476±1, 1553±1, 1596±1, 1639±1, 1664±1, 2852±1, 2907±1, 2928±1, 2967±1, 3168±1 и 3357±1 см-1.
Пример 14: Рентгеноструктурный анализ кристаллической структуры
Крупный образец кристалла сульфатной соли соединения 1, имеющий размеры 0,43 × 0,05 × 0,031 мм устанавливали на стекловолокно. Структурные данные для рентгеновской дифракции получали с использованием Bruker SMART 6K CCD детектора рентгеновской площади с диаметром окна 13,5 см, контролируемого программным обеспечением SMART, версия 5,630 (Bruker, 2003) с использованием Cu Kα излучения. Расстояние от образца до детектора составляло 5,039 см. Данные получали при температуре -153±1°C и анализировали с использованием программного обеспечения SHELXS, версия 6.14 (Bruker, 2003). Установлены следующие параметры кристаллической решетки: единичная ячейка орторомбическая с размерами a = 6,8239 Å, b = 16,2275 Å, c = 24,2021 Å, α=β=γ=90°; объем ячейки (V) 2680,0 Å3; вычисленная плотность составляет 1,38 г/см3; пространственная группа P212121(#19). Пики на порошковой рентгенограмме, предсказанные из определенных положений атомов в соответствии с программным обеспечением Mercury 1.4, при визуальном сравнении находились в превосходном соответствии с экспериментальными результатами, показанными на фиг.1.
Пример 15: Анализ стабильности твердого состояния
Образцы сульфатной соли по изобретению хранили во множестве открытых стеклянных ампул при 20°C и 60% относительной влажности (ОВ) и при 40°C и 75% ОВ. Через определенные промежутки времени содержимое репрезентативных ампул удаляли и анализировали методами ДСК, ТГА, PXRD и методом ВЭЖХ для оценки химической чистоты. После 4 недель хранения обнаружимых изменений не имелось ни на термограммах ДСК или ТГА, ни на порошковых рентгенограммах XRPD образцов, хранившихся при любых из двух указанных условий. Химическая чистота хранившихся образцов по данным ВЭЖХ была неизменной при 99,7%.
Пример 16: Определение содержания противоиона
Образец сульфатной соли по изобретению анализировали методом хроматографии сульфатного иона с использованием системы ионной хроматографии Dionex ICS-2000, оборудованной анионным саморегенерирующим супрессором, детектором проводимости, аналитической анионообменной колонкой, IonPac AS11-HC и защитной колонкой IonPac AG11-HC. Определенное содержание сульфата в образце составляло 17,1%, что можно сравнить с теоретическим содержанием сульфата в 17,6% для одного мольного эквивалента сульфатного иона на моль исходного соединения.
Пример 17: Определение содержания воды в гидрате
Образец гидрата по изобретению анализировали методом ТГА в сочетании с ИК анализом вещества, испаренного во время первоначальной потери веса. Зарегистрированный методом ТГА график и оказывает потерю веса 3,2% при температуре ниже 100°С, что можно сравнить с теоретической потерей веса 3,1% для моногидрата сульфата соединения 1. ИК спектр испаренного вещества соответствовал известному ИК спектру воды.
Анализ 1: радиолигандный анализ связывания для опиоидных рецепторов мю человека, дельта человека и каппа морской свинки.
a. Получение мембран
Клетки CHO-K1 (яичники китайского хомячка) стабильно трансфицированные кДНК мю-опиоидного рецептора человека или каппа рецептора морской свинки выращивали в среде, состоящей из среды Ham's-F12, дополненной 10% FBS, 100 единиц/мл пенициллина - 100 мкг/мл стрептомицина и 800 мкг/мл генетицина в 5% CO2, увлажненном инкубаторе при 37°C. Уровни экспрессии рецептора (Bmax ~2,0 и ~0,414 пмоль/мг белка соответственно) определяли с использованием [3H]-дипренорфина (удельная активность ~50-55 Ки/ммоль) в мембранном анализе радиолигандного связывания.
Клетки выращивали до 80-95% конфлюенции (<25 пассирований субкультуры). Для пассирования клеточной линии монослой клеток инкубировали в течение 5 минут при комнатной температуре и собирали путем механического встряхивания в 10 мл PBS, дополненного 5 мМ EDTA. После повторного суспендирования клетки переносили в 40 мл свежей среды для роста для центрифугирования в течение 5 минут при 1000 об/мин и повторно суспендировали в свежей среде для роста при подходящем соотношении разделения.
Для получения мембран клетки собирали путем осторожного механического перемешивания с 5 мМ EDTA в PBS с последующим центрифугированием (2500 g в течение 5 минут). Осадок повторно суспендировали в буфере для анализа (50 мМ 4-(2-гидроксиэтил)пиперазин-1-этансульфоновой кислоты, N-(2-гидроксиэтил)пиперазин-N'-(2-этансульфоновая кислота) (HEPES)), pH 7,4 и гомогенизирвоали с использованием политронного разрушителя на льду. Полученные гомогенаты центрифугировали (1200 g в течение 5 минут), осадок отбрасывали и супернатант центрифугировали (40000 g в течение 20 минут). Осадок промывали один раз путем повторного суспендирования в буфере для анализа с последующим дополнительным центрифугированием (40000 g в течение 20 минут). Полученный осадок повторно суспендировали в буфере для анализа (эквивалент 1 колба T-225 /1 мл буфера для анализа). Концентрацию белка определяли с использованием набора для анализа белка Bio-Rad Bradford и мембраны хранили в виде замороженных аликвот при -80°C до тех пор, пока не потребуется.
Мембраны дельта-опиоидного рецептора человека (hDOP) получали от Perkin Elmer. Сообщавшиеся значения Kd и Bmax для данных мембран, определенные анализом насыщения в анализах радиолигандного связывания [3H]-натриндола составляли 0,14 нМ (pKd = 9,85) и 2,2 пмоль/мг белка соответственно. Концентрацию белка определяли с использованием набора для анализа белка Bio-Rad Bradford. Мембраны хранили в виде замороженных аликвот при -80°C до тех пор, пока не потребуется.
b. Анализы радиолигандного связывания
Анализы радиолигандного связывания проводили в 96-луночных полипропиленовых планшетах для анализа Axygen с глубокими 1,1 мл лунками при общем анализируемом объеме в 200 мкл, содержащем подходящее количество мембранного белка (~3, ~2 и ~20 мкг для мю, дельта и каппа рецепторов соответственно) в буфере для анализа, дополненном 0,025% бычьего сывороточного альбумина (БСА). Исследования насыщения связывания для определения значений Kd радиоактивного лиганда проводили с использованием [3H]-дипренорфина при 8-12 различных концентрациях, колеблющихся от 0,001 нМ до 5 нМ. Анализ замещения для определения значений pKi для соединений проводили с использованием [3H]-дипренорфина в концентрации 0,5, 1,2 и 0,7 нМ для мю, дельта и каппа рецепторов соответственно и одиннадцати концентрациях соединения, колеблющихся от 10 пМ до 100 мкМ.
Данные связывания анализировали с помощью нелинейного регрессионного анализа с использованием программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) м 3-параметровой модели для односайтовой конкуренции. Минимум на кривой фиксировали как значение, полученное для неспецифического связывания, определенное в присутствии 10 мкМ налоксона. Значения Ki для тестируемых соединений рассчитывали в программе Prism из наиболее хорошо соответствующих значений IC50 и значения Kd радиоактивного лиганда с использованием уравнения Ченга-Прусова (Ki= IC50/(1+([L]/Kd)), где [L] = концентрация [3H]-дипренорфина. Результаты выражают в качестве отрицательного десятичного логарифма значений Ki, pKi.
Тестируемые соединения, имеющие более высокое значение pKi в данном анализе, обладают более высокой аффинностью связывания в отношении мю, дельта или каппа опиоидного рецептора. Сульфатная соль соединения 1 имела значение pKi, равное 9,9 для мю-опиоидного рецептора человека.
Анализ 2: Агонист-опосредованная активация мю-опиоидного рецептора в мембранах, полученных из клеток CHO-K1, экспрессирующих мю-опиоидный рецептор человека
В данном анализе определяли эффективность и значения собственной активности тестируемых соединений путем измерения количества связанного GTP-Eu, присутствующего после активации рецептора в мембранах, полученных из клеток CHO-K1, экспрессирующих мю-опиоидный рецептор человека.
a. Получение мю-опиоидных рецепторных мембран:
мю-Опиоидные рецепторные мембраны человека (hMOP) получали, как описано выше или закупали в компании Perkin Elmer. Сообщалось, что значения pKd и Bmax для закупленных мембран, определенные анализом насыщения в анализах связывания [3H]-дипренорфинового радиоактивного лиганда, составляли 10,06 и 2,4 пмоль/мг белка соответственно. Концентрацию белка определяли с использованием набора для анализа белка Bio-Rad Bradford. Мембраны хранили в виде замороженных аликвот при -80°C до тех пор, пока не потребуется. Лиофилизованные GTP-Eu и GDP, разведенные до 10 мкМ и 2 мМ соответственно, в дважды перегнанной H2O затем перемешивали и давали возможность постоять при комнатной температуре на 30 минут перед перенесением в индивидуальные аликвоты образцов для хранения при -20°C.
b.Анализ нуклеотидного обмена мю-GTP-Eu человека
Анализ нуклеотидного обмена GTP-Eu проводили с использованием набора для GTP-связывания DELPHIA (Perkin/Elmer) в 96-луночных фильтрующихся планшетах AcroWell 96 в соответствии с инструкциями производителя. Мембраны получали, как описано выше, и перед началом анализа аликвоты разводили до концентрации 200 мкг/мл в буфере для анализа (50 мМ HEPES, pH 7,4 при 25°C), затем подвергали гомогенизации в течение 10 секунд с использованием гомогенизатора Polytron. Тестируемые соединения получали в виде исходных растворов в ДМСО с концентраций 10 мМ, разводили до 400 мкМ в буфере для анализа, содержащем 0,1% БСА и затем делали серийные разведения (1:5) для получения десяти концентраций соединения в диапазоне от 40 пМ - 80 мкМ; GDP и GTP-Eu разводили до концентрации 4 мкМ и 40 нМ соответственно в буфере для анализа. Анализ проводили в общем объеме 100 мкл, содержащем 5 мкг мембранного белка, концентрация тестируемого соединения колебалась от 10 пМ до 20 мкМ), 1 мкМ GDP и 10 нМ GTP-Eu, разведенного в 10 мМ MgCl2, 50 мМ NaCl и 0,0125% БСА (конечные концентрации в анализе). В каждый планшет включали кривую концентрация-ответная реакция для DAMGO (Tyr-D-Ala-Gly-(метил)Phe-Gly-ол) (в диапазоне концентрации 12,8 пМ - 1 мкМ).
Планшеты для анализа получали непосредственно перед анализом после добавления 25 мкл буфера для анализа, 25 мкл тестируемого соединения и 25 мкл GDP и GTP-Eu. Анализ начинали посредством добавления 25 мкл мембранного белка и оставляли инкубироваться в течение 30 мину. Планшеты для анализа затем фильтровали с использованием вакуумного коллектора Waters, соединенного с лабораторным вакуумным насосом, отрегулированным на 10-12 дюймов рт.ст. (250-300 мм рт.ст), и промывали раствором для промывки GTP комнатной температуры (2 × 300 мл). Дно планшетов блоттировали для удаления избытка жидкости. Планшеты сразу же считывали для определения количества связанного GTP-Eu путем измерения флуоресценции с временным разрешением (TRF) на планшетном ридере Packard Fusion. Носитель: конечная концентрация ДМСО в анализе не превышала 1%.
Количество связанного GTP-Eu пропорционально степени активации мю-опиоидных рецепторов тестируемыми соединениями. Собственная активность (IA), выраженная в процентах, была определена как соотношение количества связанного GTP-Eu, наблюдаемого для активации с помощью тестируемого соединения, к количеству, наблюдавшемуся для активации с помощью DAMGO, который считается полным агонистом (IA=100). Сульфатная соль соединения 1 в данном анализе продемонстрировала собственную активность, составляющую -5. Таким образом, было показано, что настоящая сульфатная соль является антагонистом.
Анализ 3: Крысиная модель эффективности In Vivo
В данном анализе эффективность тестируемых соединений оценивали на модели желудочно-кишечного транзита, который оценивает периферическую активность. Данное исследование одобрено учрежденным комитетом по уходу и применению животных (Institutional Animal Care и Use Committee) при Theravance, Inc. и соответствует руководству по уходу и применению лабораторных животных (Guide for the Care и Use of Laboratory Animals), опубликованному национальной Академией наук (©1996).
a. Анализ опустошения желудка крысы
Тестируемые соединения оценивали в анализе опустошения желудка крысы для определения их способности обращать индуцированное лоперамидом опустошение желудка. Крыс не кормили в течение ночи перед введением тестируемого соединения или носителя посредством внутривенного, подкожного, внутримышечного или перорального пути введения в дозах, изменяющихся в диапазоне от 0,001 до примерно 30 миллиграмм/килограмм (мг/кг). После введения тестируемого соединения проводили подкожное введение лоперамида в дозе 1 мг/кг или носителя. Через пять минут после введения лоперамида или носителя с помощью зонда в ротовую полость вводили непищевой, не поглощаемый активированным углем тонкоизмельченный материал и животным оставляли свободный доступ к воде в течение 60 минут продолжительности эксперимента. Затем проводили эвтаназию животных посредством удушения диоксидом углерода с последующей торакотомией и желудок осторожно вырезали. Желудок перевязывали у нижнего пищеводного сфинктера и пилорического сфинктера, чтобы воспрепятствовать дополнительному опустошению во время удаления ткани. Затем, после удаления лигатуры, определяли вес желудка.
b. Данные анализа и результаты
Данные были проанализированы с использованием программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA). Кривые процента обратимости строили с помощью нелинейного регрессионного анализа с использованием сигмоидальной модели доза-ответная реакция (переменный наклон) и рассчитывали наиболее подходящие значения ID50. Минимумы и максимумы на кривой фиксировали по контрольным значениям для лоперамида (означающим 0% обратимости) и контрольным значениям носителя (означающим 100% обратимости) соответственно. Результаты выражены в виде ID50, дозы, требуемой для 50% обратимости действия лоперамида, в миллиграммах на килограмм. Для сульфатной соли соединения 1 при пероральном введении значение ID50 составляло 0,26 мг/кг в модели опустошения желудка крысы.
В то время как настоящее изобретение было описано со ссылкой на его определенные варианты осуществления, специалистам в данной области следует понимать, что могут быть сделаны различные изменения, и эквиваленты могут быть заменены, не отступая от истинной сути и объема изобретения. Дополнительно, могут быть выполнены многочисленные модификации, чтобы адаптировать конкретную ситуацию, материал, композицию вещества, способ, стадию или стадии способа к предмету, сути и объему настоящего изобретения. Подразумевается, что все такие модификации входят к объем прилагаемой формулы изобретения. Дополнительно, все процитированные выше публикации, патенты и патентные документы включены в данное описание путем ссылки во всей своей полноте, как если бы индивидуально включены путем ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ 8-АЗАБИЦИКЛО[3.2.1]ОКТАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ МЮ-ОПИОИДОВ | 2007 |
|
RU2423362C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО ПРОДУКТА ДЛЯ СИНТЕЗА АНТАГОНИСТОВ мю-РЕЦЕПТОРА ОПИОИДОВ | 2008 |
|
RU2469035C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ 3-КАРБОКСИПРОПИЛ-АМИНОТЕТРАЛИНА | 2009 |
|
RU2512390C2 |
| ПРОИЗВОДНОЕ АМИНОБЕНЗОЙНОЙ КИСЛОТЫ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1993 |
|
RU2067979C1 |
| ХИНОЛИНОНКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ 5-HT РЕЦЕПТОРОВ | 2005 |
|
RU2394033C2 |
| ПРОИЗВОДНОЕ 8-АЗАБИЦИКЛО [3.2.1]ОКТ-2-ЕНА, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2186780C2 |
| ИНДАЗОЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2005 |
|
RU2404179C2 |
| ДИГИДРОБЕНЗОФУРАНОВЫЕ КАРБОКСАМИДЫ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2043991C1 |
| АЗАБИЦИКЛИЧЕСКИЕ АЛКАНОВЫЕ ПРОИЗВОДНЫЕ, ЗАМЕЩЕННЫЕ КОНДЕНСИРОВАННЫМ БИЦИКЛОГЕТЕРОЦИКЛОМ | 2007 |
|
RU2437884C2 |
| ПРОИЗВОДНЫЕ 1,2,5-ТИАДИАЗОЛА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2158262C2 |


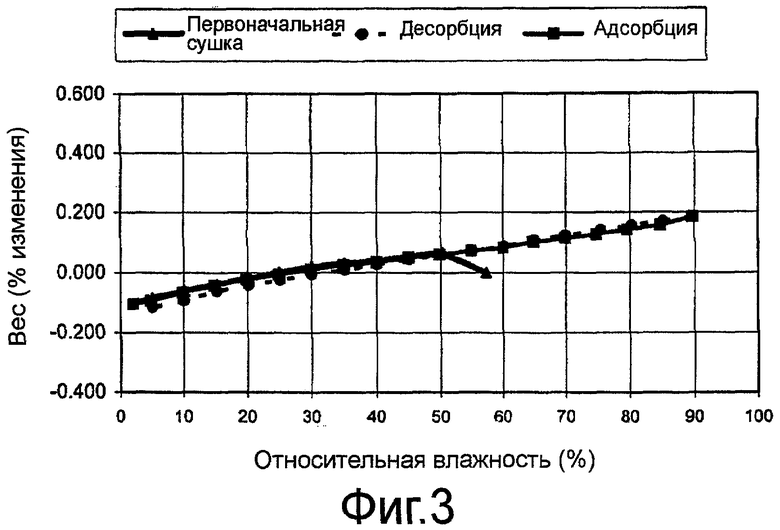
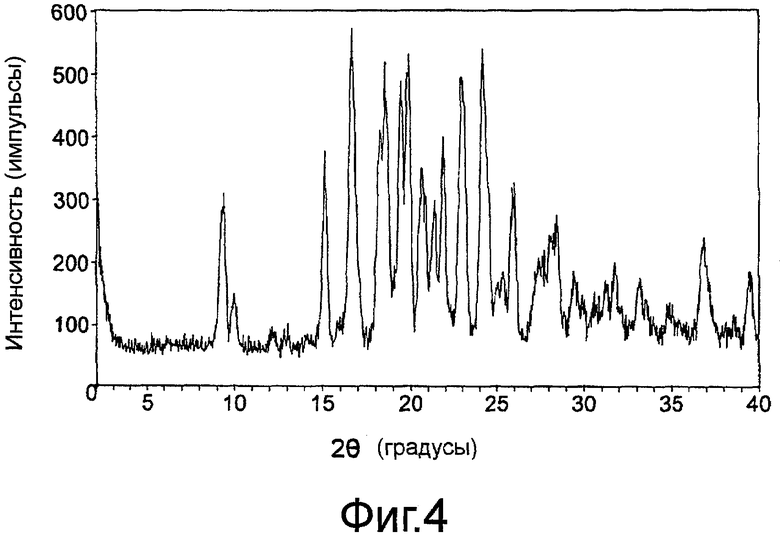
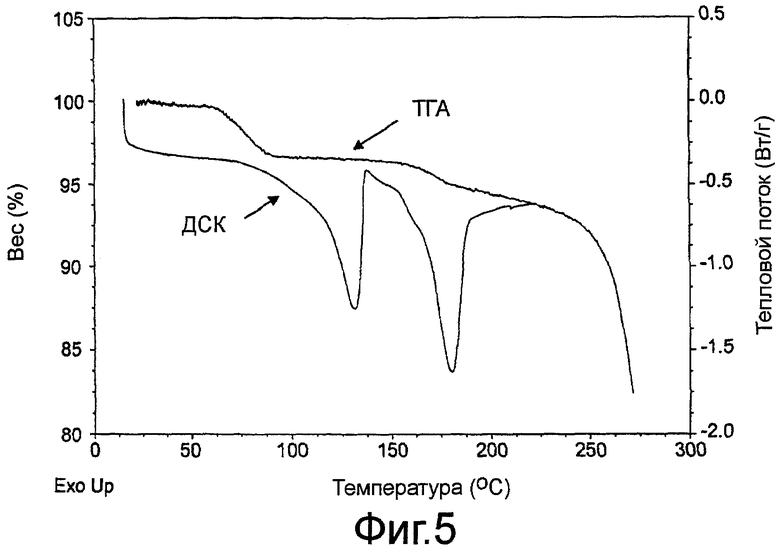

Настоящее изобретение относится к кристаллической солевой форме, представляющей собой сульфатную соль 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида или ее гидрат, которые используются в качестве антагонистов мю-опиоидного рецептора, к фармацевтической композиции на их основе, к их применению для получения лекарственного средства, а также к способам лечения, повышения моторики желудочно-кишечного тракта и антагонизации мю-опиоидного рецептора. Также изобретение относится к вариантам способа получения кристаллической сульфатной соли 3-эндо(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида и к бисульфитному аддукту бензилового эфира N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты. 9 н. и 17 з.п. ф-лы, 26 пр., 6 ил.
1. Кристаллическая солевая форма, представляющая собой сульфатную соль 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида или ее гидрат.
2. Кристаллическая солевая форма по п.1, где кристаллическая солевая форма представляет собой кристаллическую сульфатную соль.
3. Кристаллическая солевая форма по п.2, где кристаллическая солевая форма характеризуется порошковой рентгенограммой, имеющей два или более пиков дифракции при значениях θ, выбранных из 6,58±0,20, 7,52±0,20, 9,35±0,20, 14,69±0,20, 16,01±0,20, 17,45±0,20, 17,99±0,20, 18,62±0,20, 19,76±0,20, 21,11±0,20, 22,07±0,20, 23,18±0,20, 23,74±0,20, 24,56±0,20, 25,63±0,20, 26,45±0,20, 27,86±0,20, 28,31±0,20, 29,54±0,20, 30,59±0,20, 31,58±0,20, 33,89±0,20 и 36,02±0,20.
4. Кристаллическая солевая форма по п.3, где порошковая рентгенограмма включает два или более пиков дифракции при значениях 2θ, выбранных из 14,69±0,20, 16,01±0,20, 21,11±0,20, 22,07±0,20 и 23,18±0,20.
5. Кристаллическая солевая форма по п.2, где кристаллическая солевая форма характеризуется порошковой рентгенограммой, на которой положение пиков по существу соответствует положению пиков на рентгенограмме, приведенной на фиг.1.
6. Кристаллическая солевая форма по п.2, где кристаллическая солевая форма характеризуется графиком, полученным методом дифференциальной сканирующей калориметрии, зарегистрированным при скорости нагрева 10°С в минуту, который имеет максимум в эндотермическом тепловом потоке при температуре между примерно 190°С и примерно 205°С.
7. Кристаллическая солевая форма по п.2, где кристаллическая солевая форма характеризуется графиком, полученным методом дифференциальной сканирующей калориметрии, по существу соответствующим графику, приведенному на фиг.2.
8. Кристаллическая солевая форма по п.1, где кристаллическая солевая форма представляет собой гидрат сульфатной соли.
9. Кристаллическая солевая форма по п.8, где кристаллическая солевая форма характеризуется порошковой рентгенограммой, имеющей два или более пиков дифракции при значениях 2θ, выбранных из 9,41±0,20, 9,98±0,20, 15,17±0,20, 16,70±0,20, 18,59±0,20, 19,46±0,20, 19,91±0,20, 20,63±0,20, 21,35±0,20, 21,89±0,20, 23,00±0,20, 24,20±0,20, 25,40±0,20, 26,03±0,20, 27,44±0,20, 28,46±0,20, 29,45±0,20, 31,22±0,20, 31,82±0,20, 33,17±0,20, 33,56±0,20 и 36,89±0,20.
10. Кристаллическая солевая форма по п.9, где порошковая рентгенограмма включает два или более пиков при значениях 2θ, выбранных из 16,70±0,20, 18,59±0,20, 19,46±0,20, 19,91±0,20, 23,00±0,20 и 24,20±0,20.
11. Кристаллическая солевая форма по п.8, где кристаллическая солевая форма характеризуется порошковой рентгенограммой, на которой положение пиков по существу соответствует положению пиков на рентгенограмме, приведенной на фиг.4.
12. Кристаллическая солевая форма по п.8, где кристаллическая солевая форма характеризуется графиком, полученным методом дифференциальной сканирующей калориметрии, по существу соответствующим графику, приведенному на фиг.5.
13. Фармацевтическая композиция, обладающая антагонистической активностью в отношении мю-опиоидного рецептора, содержащая фармацевтически приемлемый носитель и эффективное количество кристаллической солевой формы по любому из пп.1-12.
14. Способ получения кристаллической сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по п.3, включающий:
(a) контактирование защищенного предшественника 3-энгдо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида, в котором гидроксильные группы являются защищенными, с серной кислотой с образованием первой реакционной смеси, где защищенный предшественник представляет собой {2-[3-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметиламид (S)-2,2-диметил-[1,3]диоксолан-4-карбоновой кислоты;
(b) выделение твердой сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида, преимущественно в виде кристаллического гидрата, из первой реакционной смеси;
(c) диспергирование твердой сульфатной соли, полученной на стадии (b), в разбавителе, включающем метанол, с образованием второй реакционной смеси; и
(d) выделение кристаллической сульфатной соли из второй реакционной смеси.
15. Способ по п.14, где разбавитель включает метанол, дополнительно содержащий до 25% воды.
16. Способ по п.14, где разбавитель включает метанол, дополнительно содержащий от примерно 5% до примерно 15% воды.
17. Способ получения кристаллической сульфатной соли 2-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по п.3, включающий:
(a) диспергирование кристаллического гидрата сульфатной соли 3-эндо-(8-{2-[циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по п.9 в разбавителе, содержащем метанол с образованием реакционной смеси; и
(b) выделение кристаллической сульфатной соли из реакционной смеси.
18. Способ по п.17, где разбавитель включает метанол, дополнительно содержащий до 25% воды.
19. Бисульфитный аддукт бензилового эфира N-циклогексилметил-(2-оксоэтил)карбаминовой кислоты.
20. Кристаллическая солевая форма по любому из пп.1-12 для применения в лечении заболевания или медицинского состояния у млекопитающего, которое улучшается при лечении с использованием антагониста мю-опиоидного рецептора.
21. Применение кристаллической солевой формы по любому из пп.1-12 для получения лекарственного средства для лечения заболевания или медицинского состояния у млекопитающего, которое улучшается при лечении с использованием антагониста мю-опиоидного рецептора.
22. Применение по п.21, где заболевание или состояние представляет собой вызванную опиоидами дисфункцию кишечника или послеоперационную кишечную непроходимость.
23. Способ лечения млекопитающего, имеющего медицинское состояние, облегчение при котором наступает при лечении с использованием антагониста мю-опиоидного рецептора, включающий введение млекопитающему фармацевтической композиции, содержащей фармацевтически приемлемый носитель и эффективное количество кристаллической солевой формы по любому из пп.1-12.
24. Способ по п.23, где медицинское состояние представляет собой вызванную опиоидами дисфункцию кишечника или послеоперационную кишечную непроходимость.
25. Способ повышения моторики желудочно-кишечного тракта у млекопитающего, включающий введение млекопитающему фармацевтической композиции, содержащей фармацевтически приемлемый носитель и эффективное количество кристаллической солевой формой по любому из пп.1-12.
26. Способ антагонизации мю-опиоидного рецептора у млекопитающего, включающий введение млекопитающему фармацевтической композиции, содержащей фармацевтически приемлемый носитель и эффективное количество кристаллической солевой формы по любому из пп.1-12.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Батарея гальванических элементов | 1927 |
|
SU7580A1 |

