Область техники, к которой относится изобретение
Изобретение относится к способу получения производного арил-8-азабицикло[3.2.1]октана, которое используется в качестве промежуточного продукта для получения лекарственных средств. В частности, изобретение относится к получению промежуточных продуктов для синтеза агентов-антагонистов мю-рецептора опиоидов.
Уровень техники
Предполагается, что соединения, которые демонстрируют антагонизм в отношении мю-рецепторов опиоидов, можно использовать для лечения или уменьшения интенсивности симптомов медицинских состояний, опосредуемых активностью мю-рецептора опиоидов, таких как расстройства, связанные с пониженной моторикой желудочно-кишечного тракта. Например, предполагается, что такие соединения можно использовать для лечения индуцированной опиоидами дисфункции кишечника или послеоперационной непроходимости кишечника. В заявке на патент США № 11/711961 недавно описан ряд производных 3-эндо(8-азабицикло[3.2.1]окт-3-ил)-(замещенный)фенила, которые проявляют активность в качестве антагонистов мю-рецептора опиоидов. Особенно представляющими интерес в данном ряду являются соединения формулы (I)
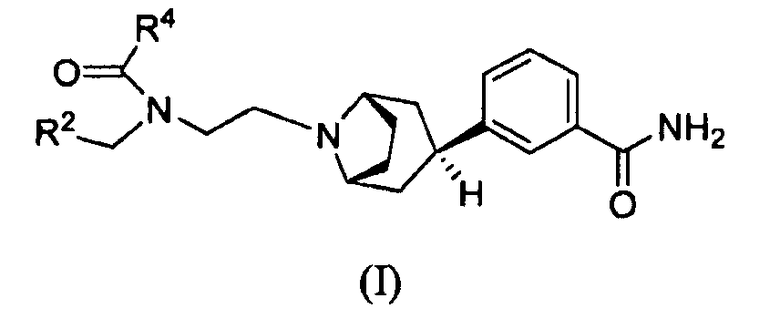
где, например, R2 представляет собой циклогексил или 4,4-дифторциклогексил и R4 представляет собой С1-4алкил, замещенный одним или двумя гидроксильными заместителями. Как описано в указанной заявке, получение таких соединений зависит от ключевого промежуточного продукта, соединения 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида
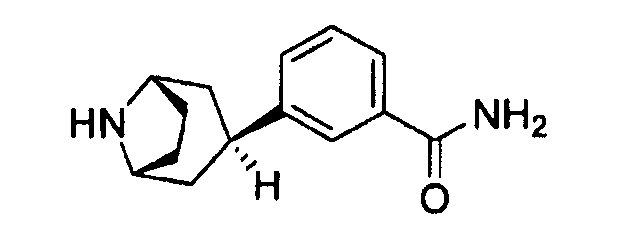
имеющего эндоориентацию бензамидной части относительно 8-азабицикло[3.2.1]октильной группы конечного продукта.
Для использования указанного выше класса антагонистов мю-рецептора опиоидов в качестве лекарственных средств желательно иметь эффективный способ получения 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида с высокой стереоспецифичностью. Поскольку эффективность синтеза органических соединений резко уменьшается с числом стадий способа, для синтетического способа желательно иметь минимальное число стадий способа.
Сущность изобретения
Настоящее изобретение относится к эффективному способу получения 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида. Обнаружено, что гидрирование промежуточного продукта, 3-(8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида, дает продукт с хорошей селективностью для требуемой эндоконфигурации. Далее было обнаружено, что в подходящих условиях гидрирования продукт можно получить в одну стадию с хорошим выходом и стереоселективностью из промежуточного продукта, арил-8-азабицикло[3.2.1]окт-2-ена, защищенного аминозащитной группой, удаляемой каталитическим гидрированием.
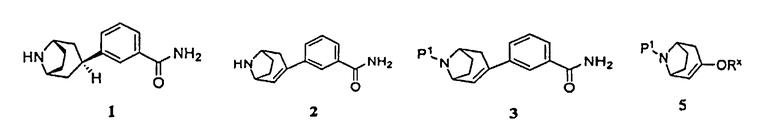
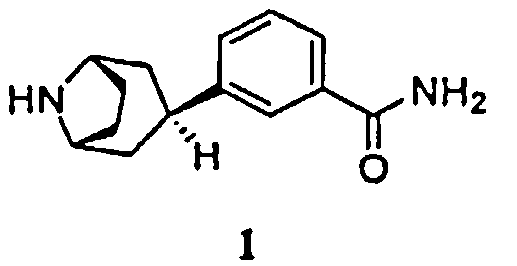
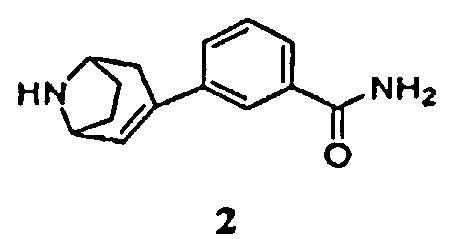
Соответственно, в одном аспекте изобретение относится к способу получения соединения формулы 1, имеющего химическое название 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамид, или его соли
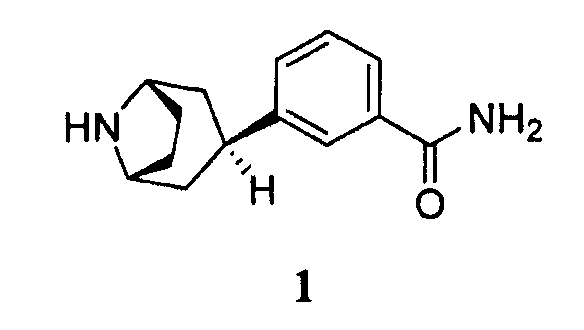
включающему гидрирование соединения формулы 2, имеющего химическое название 3-(8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамид:
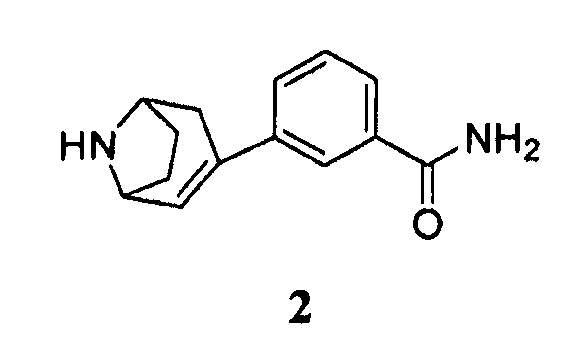
или его соли с кислотой в присутствии палладиевого металлического катализатора и, когда соединение формулы 2 находится в форме свободного основания, в присутствии кислоты, с получением соединения формулы 1 или его соли.
В другом аспекте изобретение относится к способу получения соединения формулы 1 или его соли, включающему
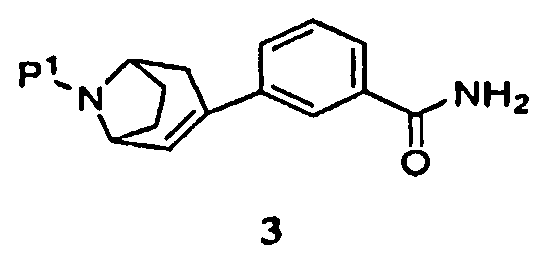
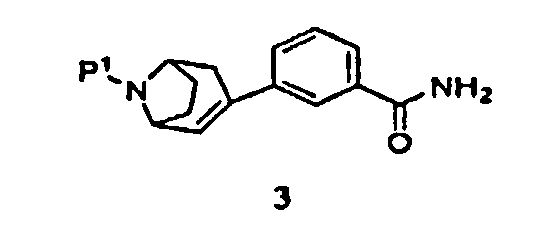
(а) пропускание через реакционный сосуд, содержащий соединение формулы 3
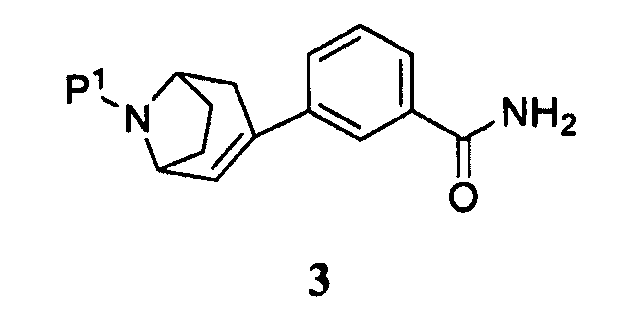
и кислоту, нереакционноспособного газа, где Р1 представляет собой аминозащитную группу, удаляемую каталитическим гидрированием;
(b) добавление палладиевого металлического катализатора в реакционный сосуд;
(с) пропускание водорода через реакционный сосуд и
(d) подачу в реакционный сосуд газообразного водорода при давлении менее чем приблизительно пол атмосферы, таким образом, чтобы общее давление в реакционном сосуде составляло менее чем приблизительно полторы атмосферы, с получением соединения формулы 1 или его соли.
В одном аспекте в данном способе используют промежуточный продукт формулы 3, где Р1 представляет собой бензил.
В одном аспекте кислотой является хлористоводородная кислота.
В другом аспекте способ дополнительно включает преобразование продукта стадии (d) в кристаллическую форму.
Далее было установлено, что промежуточный продукт формулы 3 эффективно получают в одну стадию взаимодействием промежуточного продукта, защищенного 8-азабицикло[3.2.1]окт-2-ена, с замещенной фенилбороновой кислотой.
Поэтому в другом аспекте изобретение относится к способу получения соединения формулы 3
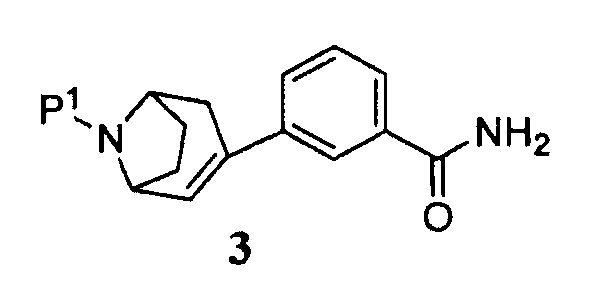
где Р1 представляет собой аминозащитную группу, включающую
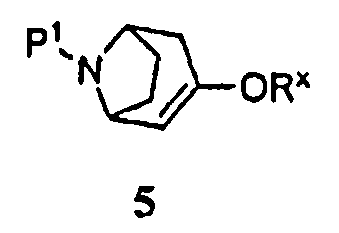
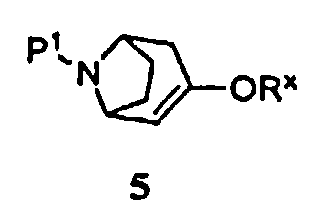
(а) приведение соединения формулы 5
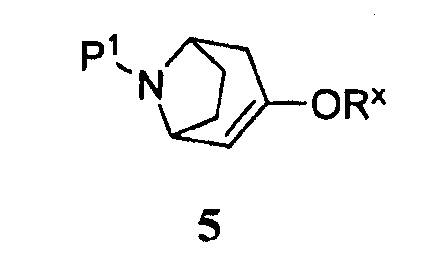
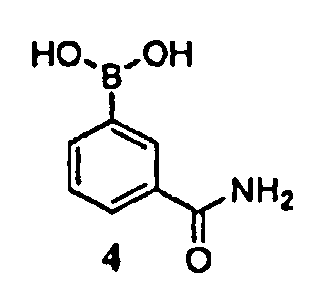
где -ORx представляет собой сульфонатную уходящую группу, в контакт с соединением формулы 4
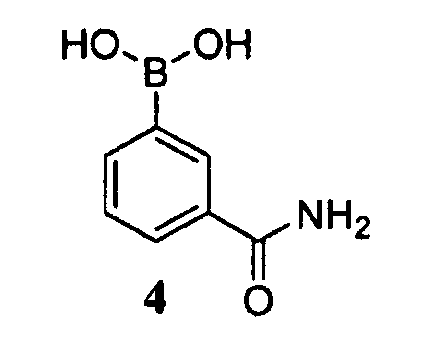
в присутствии палладиевого катализатора и фосфинового лиганда, с получением соединения формулы 3.
С учетом указанного выше еще в одном следующем аспекте изобретение относится к способу получения соединения формулы 1 или его соли, включающему
(а) приведение соединения формулы 5
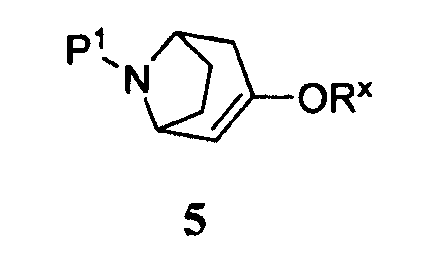
где Р1 представляет собой аминозащитную группу, удаляемую каталитическим гидрированием, и -ORx представляет собой сульфонатную уходящую группу, с соединением формулы 4
в присутствии палладиевого катализатора и фосфинового лиганда, с получением соединения формулы 3
(b) пропускание через реакционный сосуд, содержащий соединение формулы 3 и кислоту, нереакционноспособного газа;
(с) добавление палладиевого металлического катализатора в реакционный сосуд;
(d) пропускание водорода через реакционный сосуд и
(е) подачу в реакционный сосуд газообразного водорода при давлении менее чем приблизительно пол атмосферы, таким образом, чтобы общее давление в реакционном сосуде составляло менее чем приблизительно полторы атмосферы, с получением соединения формулы 1 или его соли.
Некоторые промежуточные продукты в описанных выше реакциях являются новыми. Поэтому в аспекте конкретных соединений изобретение далее относится к соединению формулы 5, где Р1 представляет собой бензил, и -ORx представляет собой сульфонатную уходящую группу, соединению формулы 3, где Р1 представляет собой аминозащитную группу, и соединению формулы 2.
Подробное описание изобретения
Общий способ получения 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида (1) с требуемой стереоспецифичностью для эндоориентации иллюстрируется на следующей схеме:
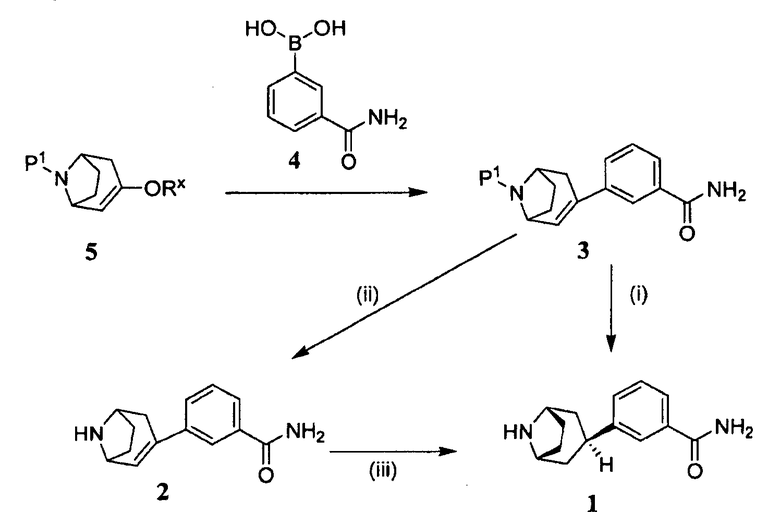
Защищенный (8-азабицикло[3.2.1]окт-2-ен (5), где Р1 представляет собой аминозащитную группу, и -ORx представляет собой сульфонатную уходящую группу, является полезным исходным соединением. Бициклооктеновый промежуточный продукт 5 подвергают взаимодействию с замещенной фенилбороновой кислотой 4, получая при этом промежуточный продукт 3. Когда аминозащитную группу, удаляемую каталитическим гидрированием, используют в качестве Р1, промежуточный продукт 3 может быть подвергнут как удалению защитной группы, так и восстановлению каталитическим гидрированием в регулируемых условиях в одну стадию, как показано на стадии (i), с получением продукта 1. Когда в качестве Р1 используют аминозащитную группу, удаляемую другими методами, промежуточный продукт 3 сначала подвергают удалению защитной группы, получая при этом соединение 2 (стадия (ii)), и затем гидрируют (стадия (iii)), с получением продукта 1.
Аминозащитные группы, удаляемые каталитическим гидрированием, обычно воздействием газообразного водорода в присутствии палладиевого металлического катализатора, включают, но не ограничиваются перечисленным, арилметильные группы, такие как бензил (Bn), 4-метоксибензил и трифенилметил (Tr), и некоторые карбонильные группы, такие как бензилоксикарбонил (Cbz), п-нитробензилоксикарбонил (PNZ), 2,4-дихлорбензилоксикарбонил и 5-бензизоксазолилметоксикарбонил. Аминозащитные группы, удаляемые другими методами, например обработкой кислотой, включают, но не ограничиваются перечисленным, трет-бутоксикарбонил (Вос) и п-метоксибензилоксикарбонил (Moz). Многочисленные защитные группы и их введение и удаление описано в публикации T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, и цитированных там ссылках.
Символ -ORx представляет собой сульфонатную уходящую группу. Сульфонатные уходящие группы, используемые в настоящем способе, включают трифторметансульфонат (обычно называемый трифлатом), п-толуолсульфонат (обычно называемый тозилатом) и метилсульфонат (обычно называемый мезилатом). Промежуточные продукты формулы 5 получают преимущественно из защищенного 8-азабицикло[3.2.1]октанона 6:
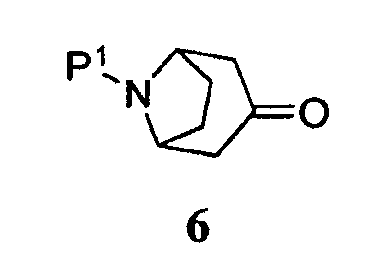
где Р1 представляет собой бензил. Например, как описано подробно ниже в разделе примеров, для получения бициклооктенового промежуточного продукта 5, где Р1 представляет собой бензил, и -ORx представляет собой трифлат, бензилзащищенный промежуточный продукт 6, 8-бензил-8-азабицикло[3.2.1]октан-3-он, приводят в контакт с приблизительно от 1 до приблизительно 1,5 эквивалентами N-фенилбис(трифторметансульфонимида) и приблизительно от 1 до приблизительно 1,5 эквивалентами основания, такого как бис(триметилсилил)амид натрия. Реакцию обычно проводят при температуре приблизительно от -20 до приблизительно 10°С в течение приблизительно от получаса до приблизительно двух часов или до тех пор, пока реакция по существу не завершится.
Для получения бициклооктенового промежуточного продукта 5, где Р1 представляет собой Вос, и -ORx представляет собой трифлат, Вос-защищенный промежуточный продукт 6 подвергают взаимодействию с N-фенилбис(трифторметансульфонимидом) и основанием, как описано выше. Взаимодействие Вос-промежуточного продукта, однако, обычно проводят при более низкой температуре, ниже приблизительно -70°С, в течение приблизительно от 2 до приблизительно 5 часов или до тех пор, пока реакция по существу не завершится. Вос-защищенный промежуточный продукт 6 является коммерчески доступным или его получают взаимодействием бензилзащищенной формы с ди-трет-бутилдикарбонатом (обычно обозначаемым Вос2О) и каталитическим гидрированием.
Бициклооктеновый промежуточный продукт 5, где Р1 представляет собой бензил, и -ORx представляет собой тозилат, можно получить из бензилзащищенного промежуточного продукта 6 способом, аналогичным способу получения бензилтрифлатного промежуточного продукта, с использованием ангидрида п-толуолсульфоновой кислоты (обычно обозначаемого Tos2O) в качестве реагента.
Бензилзащищенный 8-азабицикло[3.2.1]октанон 6 обычно получают из коммерческих источников и его можно получить взаимодействием 2,5-диметокситетрагидрофурана с бензиламином и 1,3-ацетондикарбоновой кислотой в кислотном водном растворе в присутствии буферного агента, как описано в US 2005/0228014 (см. также US 5753673).
В настоящем способе бициклооктеновый промежуточный продукт 5 подвергают взаимодействию с арилбороновой кислотой 4, с получением промежуточного продукта 3. Бициклооктен 5 необязательно можно использовать в качестве неочищенного продукта, описанного выше, без полного выделения и очистки. Реакцию обычно проводят путем контактирования соединения 5 в количестве приблизительно от 1 до приблизительно 1,2 эквивалента 4 в присутствии каталитического количества палладиевого катализатора и фосфинового лиганда (приблизительно от 0,005 до приблизительно 0,1 эквивалента) и приблизительно от 2 до приблизительно 4 эквивалентами основания.
Подходящие палладиевые катализаторы включают трис(дибензилиденацетон)дипалладий(0) (Pd2dba3), ацетат палладия (II) (Pd(OAc)2), дихлор(1,1'-бис(дифенилфосфино)ферроцен)дипалладий(II) (Pd(dppf)Cl2), дихлорбис(дифенилфосфино)палладий(II) (Pd(PPh3)2Cl2) и тому подобное, где в скобках указаны их обычные аббревиатуры. Фосфиновые лиганды, используемые в настоящей реакции, включают трициклогексилфосфин (PCy3), трициклогексилфосфинтетрафторборат (PCy3HBF4), 1,1'-бис(дифенилфосфино)ферроцен (dppf), 1,1'-бис(ди-трет-бутилфосфино)ферроцен, три(2-фурил)фосфин, 1,3-бис(дифенилфосфино)пропан (dppp), 1,5-бис(дифенилфосфино)пентан (dpppe), три-трет-бутилфосфин (P(t-Bu)3) и 9,9-диметил-4,5-бис(дифенилфосфино)ксантен (ксантфос). Нижеследующие системы являются примерами каталитических систем для реакции сочетания с бороновой кислотой: 0,01 эквивалента Pd(OAc2)/0,01 эквивалента dppf, 0,04 эквивалента Pd2dba3/0,08 эквивалента PCy3HBF4, 0,03 эквивалента Pd2dba3/0,06 эквивалента dpppe.
Конкретные основания для реакции сочетания включают фторид калия и фторид цезия. Альтернативно, в качестве основания можно использовать карбонат цезия, карбонат натрия, карбонат калия, ацетат натрия, трет-бутоксид натрия, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) или 1,4-диазабицикло[2.2.2]октан (DABCO). Реакцию обычно проводят в инертном растворителе, таком как тетрагидрофуран, N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон. Подходящие системы смешанных растворителей включают тетрагидрофуран и воду, тетрагидрофуран и N,N-диметилформамид, тетрагидрофуран и N-метилпирролидон, ацетон и воду, этанол и воду и изопропанол и воду. Реакцию обычно проводят при температуре приблизительно от 40 до приблизительно 80°С в течение приблизительно от 1 до приблизительно 4 часов или до тех пор, пока реакция по существу не завершится. Продукт 3 выделяют в виде твердого вещества общепринятыми методами. Продукт 3 необязательно можно дополнительно очистить выделением в виде соли кислоты, обычно гидрохлорида.
Затем, когда Р1 является аминозащитной группой, удаляемой каталитическим гидрированием, промежуточный продукт 3, защищенный 3-(8-азабицикло[3.2.1]окт-3-ен-3-ил)бензамид, подвергают удалению защитной группы и восстанавливают на одной стадии гидрирования, как показано схематично на пути (i), получая при этом продукт 1. В реакции гидрирования в реакционную смесь промежуточного продукта 3 в спиртовом разбавителе, например, этаноле и воде, и в присутствии приблизительно от 1,5 до приблизительно 2,5 эквивалента кислоты, например хлористоводородной кислоты, сначала пропускают нереакционноспособный газ для уменьшения количества кислорода в реакционной смеси. Обычно в качестве пропускаемого газа используют азот, хотя альтернативно можно использовать аргон или другой инертный газ. Газ обычно пропускают через трубку, вставленную в реакционную смесь, которая находится в сосуде, открытом для атмосферы. Отмечено, что благоприятным является ограничение продолжительности времени, в течение которого реакционная смесь подвергается воздействию пропускаемого газа. Например, для реакционной смеси при масштабе 40 граммов при использовании сосуда для гидрирования объемом 1 литр хорошая степень преобразования в продукт достигается, когда через реакционную смесь сначала пропускают азот в течение приблизительно от 5 до приблизительно 10 минут. Если первоначальное пропускание азота продолжительнее чем приблизительно получаса, предполагается, что это неблагоприятно влияет на степень преобразования смеси в продукт.
Затем добавляют палладиевый металлический катализатор, обычно палладий на углероде, и через реакционную смесь пропускают водород, как описано выше, что имеет эффект введения водорода в реакционную смесь для замены по меньшей мере некоторого количества пропускаемого газа (азота). Продолжительность времени, требуемого для введения водорода в реакционную смесь, зависит от масштаба реакции. Например, для реакции при масштабе 40 граммов благоприятным является пропускание водорода в течение времени приблизительно от 5 до приблизительно 10 минут, но обнаружено, что более продолжительный период является нежелательным. Предполагается, что для реакции в масштабе килограмма приемлемым является пропускание через реакционную смесь водорода в течение времени приблизительно от 20 до приблизительно 30 минут. Затем в реакционную смесь подают поток газообразного водорода при давлении менее чем приблизительно пол-атмосферы, обычно в закрытый сосуд, таким образом, чтобы общее давление в сосуде составляло менее чем приблизительно полторы атмосферы. Реакцию обычно проводят при температуре приблизительно от 45 до приблизительно 55°С в течение приблизительно от 3 до приблизительно 6 часов или до тех пор, пока реакция по существу не завершится. Включение кислоты в реакционную смесь дает продукт в виде соли кислоты.
Было отмечено, что результаты реакции гидрирования зависят от давления, при котором подают газообразный водород. Ограничение давления подаваемого водорода до уровня ниже приблизительно пол-атмосферы способствует как селективности образования требуемой эндоориентации продукта, так и степени преобразования реакционной смеси. При применении регулируемых условий гидрирования, описанных в выше в описании, процент экзосоединения в продукте не выше приблизительно 10%, обычно приблизительно от 6 до приблизительно 10%. Например, как описано ниже, реакция бензилзащищенного промежуточного продукта 3, при которой давление подаваемого газообразного водорода поддерживали ниже 34473,8 Па (5 фунтов на квадратный дюйм (psi), 0,34 атмосферы), давала после 5 часов >99% преобразование в гидрохлоридную соль продукта 1 с отношением изомеров эндо:экзо приблизительно 93:7.
В противоположность этому воздействие потока газообразного водорода выше атмосферного давления, 137895,2 Па (20 psi, 1,36 атмосферы), давало ~95% преобразование в продукт 1 с отношением изомеров эндо:экзо приблизительно 85:15.
Следовательно, в одном аспекте изобретение относится к способу получения соединения формулы 1 или его соли, где стадией подачи газообразного водорода является подача газообразного водорода при давлении, регулируемом таким образом, чтобы процент экзоизомера в продукте 1 составлял менее чем приблизительно 10%.
Когда Р1 представляет собой аминозащитную группу, которая не удаляется каталитическим гидрированием, сначала защитную группу удаляют подходящим общепринятым методом, с получением промежуточного продукта 2, который затем гидрируют, получая при этом продукт 1. Например, когда Р1 представляет собой Вос, промежуточный продукт 3 приводят в контакт с трифторуксусной кислотой, получая при этом 2 в виде трифторацетатной соли. Когда промежуточный продукт 2 получают в виде соли с кислотой, необходимо на стадии гидрирования добавить дополнительную кислоту. Гидрирование промежуточного продукта с удаленной защитной группой 2 дает продукт 1 с приемлемой стереоселективностью и, как оказалось, стереоселективность не является очень чувствительной к условиям гидрирования. Кроме того, в данном способе процент экзоизомера в продукте обычно составляет не более приблизительно 10%, обычно приблизительно от 6 до приблизительно 10%. Например, обработка 2 водородом из баллона (приблизительно одна атмосфера) дает продукт 1 с отношением изомеров эндо:экзо приблизительно 93:7. Это наблюдение согласуется с гипотезой, что ограничение давления водорода при реакции гидрирования промежуточного продукта 3, когда Р1 является удаляемым каталитическим гидрированием, как описано выше, способствует снятию защиты у соединения 3 по сравнению с восстановлением двойной связи.
Наконец, стереоселективность продукта реакции гидрирования можно дополнительно повысить кристаллизацией в виде соли кислоты, например в виде гидрохлорида. Неочищенный продукт реакции гидрирования кристаллизуют общепринятыми методами. Например, для кристаллизации продукта в виде гидрохлоридной соли смесь неочищенного продукта, приблизительно от 1,5 до приблизительно 2,5 эквивалентов хлористоводородной кислоты, если она не присутствует в реакции гидрирования, и спирта, предпочтительно спирта, используемого в качестве разбавителя на предыдущей стадии, нагревают приблизительно до 60°С до тех пор, пока не будет достигнуто полное растворение любых твердых веществ. Реакционную смесь для кристаллизации охлаждают при перемешивании приблизительно до 35°С, затем приблизительно до комнатной температуры и перемешивают приблизительно при 0°С в течение приблизительно от одного часа до приблизительно 6 часов или до тех пор, пока кристаллизация по существу не завершится.
Для инициации кристаллизации необязательно добавляют затравочные кристаллы продукта, когда смесь имеет температуру приблизительно 35°С. Добавление антирастворителя, такого как простой метил-трет-бутиловый эфир (МТВЕ), когда смесь находится при комнатной температуре, является благоприятным для повышения выхода. При включении в способ процедуры кристаллизации гидрохлоридную соль соединения 1, например, получали с процентом нежелательного экзоизомера менее приблизительно 0,5%.
В случае, когда продукт гидрирования имеет неприемлемо высокое содержание экзокомпонента, например, стадию гидрирования проводят без применения регулируемого давления газообразного водорода, для улучшения стереоспецифичности до приблизительного уровня, описанного выше, можно использовать модифицированную методику кристаллизации. В модифицированной методике используют более продолжительный период кристаллизации. Смесь неочищенного продукта и спирта нагревают приблизительно до 60°С до тех пор, пока не наблюдают полное растворение, и затем охлаждают приблизительно до 30°С, при этой температуре добавляют затравочные кристаллы продукта. Используемое отношение затравочных кристаллов к неочищенному продукту составляет приблизительно от 1:800 до приблизительно 1:400 (масса:масса). Образовавшуюся суспензию охлаждают до комнатной температуры и перемешивают в течение приблизительно от 8 до приблизительно 24 часов до тех пор, пока не наблюдают кристаллический осадок, и затем добавляют антирастворитель, в частности 2-метилтетрагидрофуран. Суспензию обычно перемешивают в течение еще одного периода приблизительно от 3 до приблизительно 6 часов или по существу до завершения осаждения. Кристаллы выделяют общепринятыми способами. С применением данной методики гидрохлоридную соль соединения 1 с экзокомпонентом менее приблизительно 2%, в том числе менее приблизительно 1%, и менее приблизительно 0,5%, можно получить из неочищенного продукта с содержанием экзокомпонента приблизительно 15%.
ПРИМЕРЫ
Следующие примеры синтеза предоставлены для иллюстрации изобретения и не должны истолковываться никоим образом как ограничение объема изобретения. Ниже в примерах следующие аббревиатуры имеют следующие значения, если не оговорено особо. Аббревиатуры, не указанные ниже, имеют их обычно используемые значения.
ACN=ацетонитрил
ДМФА=N,N-диметилформамид
EtOAc=этилацетат
EtOH=этанол
NaHMDS=бис(триметилсилил)амид натрия
MeTHF=2-метилтетрагидрофуран
MTBE=трет-бутилметиловый эфир
psi=фунты на квадратный дюйм
Rt=время удерживания
Реагенты и растворители покупали у коммерческих поставщиков (Aldrich, Strem Chemicals, Inc., etc.) и использовали без дополнительной очистки. Наблюдение за протеканием реакции проводили с помощью аналитической высокоэффективной жидкостной хроматографии и масс-спектрометрии. Эндо/экзо отношения продуктов определяли анализом ВЭЖХ с применением описанных ниже протоколов. Реакционные смеси обрабатывали, как описано конкретно в каждой реакции; обычно их очищали экстракцией и другими методами очистки, такими как кристаллизация, в зависимости от температуры и растворителя, и осаждение. Характеризацию продуктов реакции общепринятым образом проводили с помощью масс- и 1Н-ЯМР-спектрометрии. Для ЯМР измерения образцы растворяли в дейтерированном растворителе (ДМСО-d6 или CDCl3) и 1Н-ЯМР-спектры регистрировали инструментом Varian Gemini 2000 (400 МГц) в стандартных условиях измерения. Масс-спектрометрическую идентификацию соединений проводили с использованием инструмента Agilent (Palo Alto, CA) model 1100 LC/MSD.
Общие условия ВЭЖХ
Колонка: Zorbax SB-Aq, 5 мкм, 4,6×250 мм
Температура колонки: 40°С
Скорость потока: 1,0 мл/мин
Подвижные фазы: А=вода/ACN (98:2)+0,1% ТФУК
B=вода/ACN (10:90)+0,1% ТФУК
Объем впрыскивания: 10 мкл
Длина волны детектора: 214 нм.
Способ 1 ВЭЖХ
Неочищенные соединения растворяли в смеси вода/ACN (50:50) в количестве приблизительно 1 мг/мл и анализировали с применением следующего градиента в течение 20 мин (время (мин)/% В): 0/10, 2,5/20, 9/75, 15/90, 17/90, 18/10, 20/10.
Способ 2 ВЭЖХ
Соединения растворяли в смеси вода/ACN (90:10) в количестве приблизительно 1 мг/мл и анализировали с применением следующего градиента в течение 30 мин (время (мин)/% В): 0/10, 13/10, 23/65, 28/90, 29/90, 30/10.
Способ 3 ВЭЖХ
Соединения растворяли в смеси вода/ACN (90:10) в количестве приблизительно 1 мг/мл и анализировали с применением следующего градиента в течение 55 мин (время (мин)/% В): 0/10, 10/20, 46/75, 47/90, 50/10, 55/10.
Пример 1: синтез гидрохлорида 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида
a. Получение 8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-илового эфира трифторметансульфоновой кислоты
В колбу на 500 мл добавляли гидрохлорид 8-бензил-8-азабицикло[3.2.1]октан-3-она (50,4 г, 200 ммоль), EtOAc (160 мл) и 4н. NaOH (50 мл). Реакционную смесь нагревали до 30°С и перемешивали при такой температуре в течение 1 час. Слои разделяли и водный слой удаляли. Объем органического слоя уменьшали до ~40 мл на роторном испарителе и добавляли ТГФ (270 мл).
Образовавшийся раствор добавляли в колбу на 1 л и охлаждали до -20°С. В колбу в течение 15 мин добавляли раствор NaHMDS (1М в ТГФ, 230 мл, 230 ммоль). Реакционную смесь перемешивали при -20±5°С в течение 1 часа. К реакционной смеси порциями в течение 5 мин добавляли N-фенилбис(трифторметансульфонимид) (82,2 г, 230 ммоль) и смесь перемешивали при температуре от -20°С до -10°С в течение 1 час. К реакционной смеси добавляли 1н. NaOH (200 мл) и смеси давали возможность нагреться до 22°С при перемешивании. Растворитель частично удаляли на роторном испарителе при 30°С до объема 450 мл. К оставшейся реакционной смеси добавляли EtOAc (300 мл) и гептан (150 мл). Смесь перемешивали при 22°С в течение 5 мин. Слои разделяли и водный слой удаляли. Органический слой промывали 1н. NaOH (3×450 мл). Водные слои удаляли. Органический слой концентрировали на роторном испарителе, получая при этом указанный в заголовке промежуточный продукт (77 г, чистота, определенная способом 1 ВЭЖХ, >96%).
1Н ЯМР (d6-ДМСО, 400 МГц): δ (м.д.) 7,25-7,35 (м, 5H), 6,05 (д, J=5,2, 1Н), 3,64 (кв, J=13,2, 2H), 3,40-3,44 (м, 2H), 2,77 (д, J=16,4, 1Н), 1,79-2,09 (м, 5H), 1,52-1,59 (м, 1Н).
b. Получение 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида
К неочищенному продукту предыдущей стадии добавляли ТГФ (420 мл) и раствор продували азотом в течение 5 мин. В колбу на 2 л добавляли 3-карбамоилфенилбороновую кислоту (98%, 33,0 г, 200 ммоль), ацетат палладия (II) (98%, 0,46 г, 2 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (97%, 1,1 г, 2 ммоль) и фторид калия (34,9 г, 600 ммоль), с последующим добавлением раствора 8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-илового эфира трифторметансульфоновой кислоты в ТГФ. Образовавшуюся смесь продували азотом в течение 5 мин, кипятили с обратным холодильником (67°С) в атмосфере азота и перемешивали в течение 2 час. Реакционную смесь охлаждали до 30°С, затем добавляли EtOAc (500 мл) и 1н. NaOH (500 мл) и смесь перемешивали при 22°С в течение 10 мин. Слои разделяли и водный слой удаляли. Органический слой промывали смесью насыщенного раствора соли (250 мл) и воды (250 мл) и перемешивали в течение 5 мин. Слои разделяли и водный слой удаляли. Органический слой быстро сушили над Na2SO4, фильтровали и растворитель частично удаляли. Во время удаления растворителя продукт осаждался в виде бледно-желтого твердого вещества. Образовавшуюся суспензию (приблизительно 200 мл) фильтровали, твердое вещество промывали холодным EtOAc (0°С, 100 мл) и сушили в высоком вакууме при 25°С, получая при этом указанный в заголовке промежуточный продукт (42,5 г) в виде бледно-желтого твердого вещества.
Маточный раствор и указанные выше промывочные жидкости объединяли, концентрировали, образовавшуюся суспензию (приблизительно 100 мл) перемешивали при 5°С в течение 30 мин и фильтровали. Полученное фильтрованием твердое вещество промывали холодным EtOAc (0°С, 30 мл) и сушили в высоком вакууме, получая при этом вторую порцию указанного в заголовке промежуточного продукта (7 г, объединенный выход 78%, чистота, определенная способом 1 ВЭЖХ >98,5%).
(m/z): [M+H]+ вычислено для C21H22N2O, 319,18; найдено 319,4. 1H ЯМР (CDCl3, 400 МГц): δ (м.д.) 7,9 (с, 1Н), 7,63 (д, J=6,4, 1Н), 7,57 (д, J=6,4, 1Н), 7,21-7,42 (м, 6H), 6,38 (д, J=4,4, 1H), 6,13 (с, ушир., 1Н), 5,83 (с, ушир., 1Н), 3,68-3,76 (м, 2H), 3,46-3,51 (м, 2H), 2,92 (д, J=17,2, 1Н), 2,18-2,26 (м, 1Н), 2,04-2,12 (м, 2H), 1,86-1,92 (м, 1Н), 1,58-1,65 (м, 1Н).
c. Синтез гидрохлорида 3-эндо-8-азабицикло[3.2.1]окт-3-ил)бензамида
В сосуд для гидрирования на 1 л добавляли 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамид (40 г, 125 ммоль), EtOH (800 мл), 6М HCl (42 мл) и воду (80 мл) и смесь перемешивали при 22°С до тех пор, пока не наблюдали полное растворение. Реакционную смесь продували азотом в течение 5 мин при нагревании до 30°С в течение 5 мин. К смеси добавляли 10 мас.% Pd/C (50% воды, 4 г). Смесь продували при атмосферном давлении водородом в течение 5-10 мин при нагревании. Смесь перемешивали при 50°С в токе водорода при <34473,8 Па (<5 фунт/кв. дюйм, <0,34 атмосферы) в течение 5 час, что приводило к >99% превращению реагентов, согласно анализу ВЭЖХ. Раствор охлаждали до 30°С и фильтровали через целит, получая при этом раствор неочищенного указанного в заголовке соединения с эндо:экзо отношением ~93:7, определенным способом 2 ВЭЖХ, Rt эндоизомера=10,97, Rt экзоизомера=12,67. (m/z): [M+H]+ вычислено для C14H18N2O 231,15; найдено 231,2.
Воду удаляли из неочищенного продукта азеотропной перегонкой при 30°С в EtOH (~80 мл), получая при этом суспензию, которую нагревали до 60°С до полного растворения. Раствор охлаждали до 35°С и добавляли затравочные кристаллы продукта (0,05 г). (Затравочные кристаллы получали согласно способу, описанному в заявке на патент США № 12/072534.) Образовавшуюся суспензию перемешивали при 22°С в течение 30 мин, медленно добавляли МТВЕ (120 мл), суспензию перемешивали при 22°С в течение 4 час и затем при 0°С в течение 1 час. Образовавшееся твердое вещество отделяли фильтрованием, промывали холодным EtOH и сушили в высоком вакууме, получая при этом указанное в заголовке соединение (24,5 г) белого порошка (выход 75%, чистота >98,5%, <0,4% экзоизомера по данным способа 3 ВЭЖХ, Rt эндоизомера 8,67, Rt экзоизомера=9,43).
(m/z): [M+H]+ вычислено для C14H18N2O 231,15; найдено 231,2. 1H ЯМР (d6-ДМСО, 400 МГц): δ (м.д.) 9,13 (с, ушир., 1Н), 9,03 (с, ушир., 1Н), 8,05 (с, 1Н), 7,93 (с, 1Н), 7,73 (д, J=7,6, 1Н), 7,58 (д, J=7,6, 1Н), 7,40 (т, J=7,6, 2H), 3,97 (с, 2H), 3,17-3,23 (м, 1Н), 2,39-2,46 (м, 2H), 2,19-2,24 (м, 2H), 1,86-1,89 (м, 2H), 1,59-1,63 (м, 2H).
Пример 2: синтез гидрохлорида 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение трет-бутилового эфира 3-оксо-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
В сосуд для гидрирования на 1 л добавляли 8-бензил-8-азабицикло[3.2.1]октан-3-он (86,1 г, 400 ммоль), ди-трет-бутилдикарбонат (98,2 г, 450 ммоль), 10 мас.% Pd/C (24 г, 11 ммоль) и EtOAc (400 мл). Суспензию перемешивали и продували азотом в течение 10 мин. Реакционную смесь перемешивали при давлении водорода 344738 Па (50 фунт/кв. дюйм, 3,4 атмосферы) и при 20°С в течение 28 час. Реакционную смесь затем фильтровали через целит. Неочищенный твердый осадок промывали EtOAc (100 мл). Фильтрат и промывочные жидкости объединяли и добавляли смесь насыщенный NaHCO3/насыщенный раствор соли (смесь 1:1, 400 мл). Раствор в EtOAc отделяли, сушили над Na2SO4 и концентрировали, с получением липкого бледно-желтого масла, которое отверждалось при стоянии, с образованием указанного в заголовке промежуточного продукта (93 г, количественный выход).
1H ЯМР (CDCl3, 400 МГц): δ (м.д.) 4,47 (с, ушир., 2H), 2,63 (с, ушир., 2H), 2,32 (д, J=16,4, 2H), 2,08 (м, 2H), 1,65 (т, J=8, 2H), 1,49 (с, 9H).
b. Получение трет-бутилового эфира 3-трифторметансульфонилокси-8-азабицикло[3.2.1]окт-2-ен-8-карбоновой кислоты
В колбу на 1 л добавляли продукт предыдущей стадии (93 г, 400 ммоль) и ТГФ (400 мл). Смесь перемешивали при комнатной температуре до тех пор, пока не наблюдали полное растворение, и затем охлаждали до -74°С. В реакционный сосуд медленно, в течение 3 час (темп. <-70°С) добавляли раствор NaHMDS (1М в ТГФ, 460 мл, 460 ммоль). К реакционной смеси порциями добавляли N-фенилбис(трифторметансульфонимид) (164 г, 460 ммоль) при поддержании внутренней температуры <-70°С. Добавляли дополнительный ТГФ (200 мл) и смесь перемешивали при -74°С в течение 90 мин. Смесь нагревали до комнатной температуры и слегка концентрировали для удаления приблизительно 200 мл растворителя. К оставшемуся раствору добавляли 1н. NaOH (600 мл), гексаны (200 мл) и EtOAc (400 мл). Слои разделяли и органический слой промывали 1н. NaOH (3×200 мл) и насыщенным раствором соли (200 мл), сушили над Na2SO4 и затем концентрировали. Остаток сушили и медленно отверждали, получая при этом указанный в заголовке промежуточный продукт в виде желтоватого твердого вещества (117,8 г, выход 82%).
1H ЯМР (d6-ДМСО, 400 МГц): δ (м.д.) 6,32 (д, J=6, 1Н), 4,30-4,40 (м, 2H), 2,90 (д, J=16,4, 2H), 2,18-2,23 (м, 2H), 1,91-1,98 (м, 2H), 1,65-1,72 (м, 2H), 1,39 (с, 9H).
c. Получение трет-бутилового эфира 3-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-2-ен-8-карбоновой кислоты
В колбу на 250 мл добавляли трет-бутиловый эфир 3-трифторметансульфонилокси-8-азабицикло[3.2.1]окт-2-ен-8-карбоновой кислоты (20 г, 56 ммоль), 3-карбамоилфенилбороновую кислоту (10,2 г, 61 ммоль), трис(дибензилиденацетон)дипалладий(0) (Pd2dba3) (2 г, 2,2 ммоль), трициклогексилфосфинтетрафторборат (PCy3HBF4) (1,65 г, 4,4 ммоль) и KF (9,8 г, 168 ммоль). Реагенты продували азотом в течение 5 мин и затем добавляли ТГФ (120 мл) и ДМФА (30 мл). Суспензию перемешивали и продували азотом в течение еще 5 мин, затем нагревали до 70°С. После нагревания 2 час при 70°С смесь охлаждали до комнатной температуры и фильтровали через целит. Фильтрат концентрировали и остаток распределяли между EtOAc (350 мл) и смесью 0,5н. NaOH (400 мл)/насыщенный раствор соли (50 мл). Органический слой отделяли и сушили Na2SO4. Четверть раствора удаляли. Оставшуюся часть раствора концентрировали до ~100 мл, к остатку добавляли гексаны (50 мл). Наблюдали осаждение твердого вещества. Объем раствора уменьшали на 10 мл, добавляли гексаны (10 мл) и суспензию перемешивали при 0°С в течение 1 час. Твердое вещество отделяли фильтрованием и сушили, получая при этом указанный в заголовке промежуточный продукт (8,4 г) в виде бледно-желтого твердого вещества. Маточный раствор далее концентрировали, с образованием при этом дополнительного твердого осадка, который отделяли фильтрованием, получая при этом дополнительный продукт (0,5 г) (объединенный выход 66%).
1H ЯМР (d6-ДМСО, 400 МГц): δ (м.д.) 8,01 (с, 1Н), 7,89 (т, J=1,6, 1Н), 7,73-7,76 (м, 1Н), 7,55 (д, J=8, 1Н), 7,36-7,42 (м, 2H), 6,62 (д, J=5,2, 1Н), 4,35-4,41 (м, 2H), 2,95-2,99 (м, 1Н), 1,65-2,33 (м, 5H), 1,38 (с, 9H).
d. Получение 3-(8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида
В колбу на 50 мл добавляли трет-бутиловый эфир 3-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-2-ен-8-карбоновой кислоты (4 г, 12 ммоль) и ТФУК (10 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. Раствор концентрировали и остаток сушили в вакууме. В реакционной смеси наблюдали образование кристаллического твердого вещества, смесь затем перемешивали в ТГФ (~10 мл) при 0°С в течение 1 час. Твердое вещество отделяли фильтрованием, получая при этом соль ТФУК указанного в заголовке промежуточного продукта (2,4 г) в виде бледно-желтого твердого вещества. Дополнительный продукт (0,32 г) выделяли из маточного раствора (объединенный выход 66%).
1H ЯМР (d6-ДМСО, 400 МГц): δ (м.д.) 8,96 (с, ушир., 2H), 8,05 (с, 1Н), 7,96 (с, 1Н), 7,82 (д, J=7,6, 1Н), 7,62 (д, J=1,6, 1Н), 7,42-7,48 (м, 2H), 6,54 (д, J=6, 1Н), 4,29-4,38 (м, 2H), 3,03-3,08 (м, 2H), 2,65 (д, J=18, 1Н), 2,05-2,22 (м, 3H), 1,83-1,89 (м, 1Н).
e. Синтез гидрохлорида 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида
В колбу на 25 мл добавляли трифторацетат 3-(8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида (0,25 г, 0,7 ммоль), EtOH (5 мл) и воду (0,5 мл). Смесь перемешивали при комнатной температуре и продували азотом. К раствору добавляли 10 мас.% Pd/C (0,025 г) и смесь продували водородом в течение 5 мин. Затем смесь нагревали до 50°С и перемешивали в атмосфере водорода из баллона в течение 16 час. Смесь охлаждали до комнатной температуры и фильтровали через целит. Осадок на фильтре промывали EtOH (10 мл), растворы объединяли и концентрировали приблизительно до 1 мл, получая при этом указанный в заголовке неочищенный промежуточный продукт с отношением изомеров эндо/экзо 93:7, определенным способом 3 ВЭЖХ, Rt эндоизомера=8,67, Rt экзоизомера=9,43. (m/z): [M+H]+ вычислено для C14H18N2O 231,15; найдено 231,4.
К неочищенному продукту добавляли концентрированный HCl (~0,1 мл) и реакционную смесь нагревали до 60°С до полного растворения. Раствор охлаждали до 35°С и добавляли затравочные кристаллы (приблизительно 2 мг). (Затравочные кристаллы получали, как описано в примере 1.) Суспензию дополнительно охлаждали до комнатной температуры и добавляли МТВЕ (2 мл). Суспензию перемешивали при комнатной температуре в течение ~2 час, затем при 0°С в течение 30 мин. Твердое вещество отделяли фильтрованием, получая при этом указанное в заголовке соединение (~0,13 г, <0,4 экзоизомера по данным способа 3 ВЭЖХ, Rt эндоизомера=8,67, Rt экзоизомера=9,43). (m/z): [M+H]+ вычислено для C14H18N2O 231,15; найдено 231,4.
Пример 3: Синтез 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида
a. Получение 8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-илового эфира толуол-4-сульфоновой кислоты
В колбу на 1 л добавляли 8-бензил-8-азабицикло[3.2.1]октан-3-он (12,9 г, 60 ммоль) и ТГФ (100 мл). Смесь перемешивали при комнатной температуре до тех пор, пока не наблюдали полное растворение, и затем охлаждали до -30°С. К реакционному раствору в течение 30 мин добавляли раствор бис(триметилсилил)амида лития (LiHMDS) (1М раствор в ТГФ, 69 мл, 69 ммоль). Раствор перемешивали при -25±5°С в течение 1 час. К реакционной смеси при температуре -7±3°С добавляли раствор ангидрида п-толуолсульфоновой кислоты (22,5 г, 69 ммоль) в ТГФ (150 мл). Образовавшийся раствор перемешивали при температуре от -10°С до 10°С в течение 1 час. К раствору добавляли EtOAc (250 мл) и 1н. NaOH (200 мл) и раствор перемешивали при комнатной температуре в течение 5 мин. Органический слой отделяли и промывали 0,5н. NaOH (200 мл), с последующим промыванием смесью 1:1 насыщенный раствор соли/вода (200 мл). Органический слой сушили над Na2SO4 и концентрировали, получая при этом желтый твердый остаток. Твердое вещество суспендировали в смеси гексан (40 мл)/следовое количество EtOAc (<5 мл) и суспензию фильтровали, с получением указанного в заголовке промежуточного продукта (19,1 г, выход 86%).
(m/z): [M+H]+ вычислено для C21H23NO3S 370,15; найдено 370,2. 1H ЯМР (CDCl3, 400 МГц): δ (м.д.) 7,84-7,86 (м, 2H), 7,21-7,37 (м, 7H), 5,4 (дт, J=5,6, 1,6, 1Н), 3,58 (д, J=1,2, 2H), 3,24-3,32 (м, 2H), 2,58-2,63 (м, 1Н), 2,46 (с, 3H), 2,05-2,13 (м, 1Н), 1,89-1,95 (м, 1Н), 1,70-1,77 (м, 2H), 1,41-1,49 (м, 1Н).
b. Синтез 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида
В круглодонную колбу на 100 мл добавляли 8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-иловый эфир толуол-4-сульфоновой кислоты (0,75 г, 2 ммоль), 3-карбамоилфенилбороновую кислоту (0,36 г, 2,2 ммоль) Pd2dba3 (0,055 г, 0,06 ммоль), 1,5-бис(дифенилфосфино)пентан (0,053 г, 0,12 ммоль) и CsF (0,91 г, 6 ммоль). Реакционную смесь продували азотом в течение 5 мин и добавляли ТГФ (8 мл) и 1,5 мл ДМФА (1,5 мл). Суспензию перемешивали при комнатной температуре и продували азотом в течение еще 5 мин, затем нагревали до 68°С. После нагревания 21 час при 68°С реакционную смесь охлаждали до комнатной температуры и распределяли между EtOAc (20 мл) и 1н. NaOH (20 мл). Органический слой промывали смесью 1:1 насыщенный раствор соли:вода (2×20 мл) и сушили над Na2SO4. Органический слой концентрировали до 7-8 мл, что приводило к образованию суспензии, которую фильтровали, получая при этом указанное в заголовке соединение (0,41 г, выход 64%) в виде не совсем белого твердого вещества. (m/z): [M+H]+ вычислено для C21H22N2O 319,18; найдено 319,4.
Пример 4: синтез гидрохлорида 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида
a. Синтез гидрохлорида 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида
В сосуд для гидрирования добавляли 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамид (5 г, 15,7 ммоль), EtOH (100 мл) и 6н. HCl (7,5 мл). Смесь перемешивали при комнатной температуре и продували азотом в течение 5 мин. К раствору добавляли 10 мас.% Pd/C (0,5 г) и смесь продували водородом в течение 5 мин. Смесь нагревали до 50°С и перемешивали при такой температуре в токе водорода при 1378952,0 Па (20 фунт/кв. дюйм, 1,36 атмосферы) в течение 40 час, что приводило приблизительно к 95% преобразованию в указанное в заголовке соединение с отношением изомеров эндо/экзо 85:15 согласно способу 2 ВЭЖХ (Rt эндоизомера=10,97, Rt экзоизомера=12,67). Смесь охлаждали до комнатной температуры и фильтровали через целит. Маточный раствор концентрировали, получая при этом неочищенный, указанный в заголовке промежуточный продукт.
b. Кристаллизация гидрохлорида 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида
К неочищенному продукту предыдущей стадии добавляли EtOH (20 мл) и реакционную смесь нагревали до 60°С до завершения растворения. Раствор охлаждали до 30°С и в это время добавляли затравочные кристаллы указанного в заголовке соединения (приблизительно 10 мг). (Затравочные кристаллы получали, как описано в примере 1.) Суспензию охлаждали до комнатной температуры при перемешивании и перемешивали в течение ночи при комнатной температуре. Наблюдали осаждение кристаллов. К суспензии добавляли МеТГФ (15 мл), суспензию перемешивали при комнатной температуре в течение еще 4 час и фильтровали, получая при этом указанное в заголовке соединение (2,1 г, выход 50%, 0,3% экзоизомера по данным способа 2 ВЭЖХ (Rt эндоизомера=10,97, Rt экзоизомера 12,67)).
Хотя настоящее изобретение было описано с обращением к его определенным вариантам осуществления, специалисту в данной области будет понятно, что могут быть сделаны различные изменения и могут быть заменены эквиваленты, не выходящие за пределы подлинного существа и объема изобретения. Кроме того, могут быть сделаны многие модификации для адаптации конкретной ситуации, вещества, рассматриваемой композиции, способа, стадии или стадий способа для задачи, существа и объема настоящего изобретения. Предполагается, что все такие модификации входят в объем прилагаемой формулы изобретения. Кроме того, все публикации, патенты и патентные документы, цитированные выше в данном контексте, включены в данное описание во всей своей полноте посредством ссылки, как если бы каждый из них был включен по отдельности посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ 8-АЗАБИЦИКЛО[3.2.1]ОКТАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ МЮ-ОПИОИДОВ | 2007 |
|
RU2423362C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ 8-АЗАБИЦИКЛО[3.2.1]ОКТАНА | 2008 |
|
RU2458061C2 |
| ХИНОЛИНОНКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ 5-HT РЕЦЕПТОРОВ | 2005 |
|
RU2394033C2 |
| ИНДАЗОЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2005 |
|
RU2404179C2 |
| ПРОИЗВОДНЫЕ 2-ПИПЕРИДИНО-1-АЛКАНОЛА | 1991 |
|
RU2029769C1 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛООКТАНА, ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, СОЕДИНЕНИЯ | 2000 |
|
RU2262505C2 |
| АЗАБИЦИКЛИЧЕСКИЕ АЛКАНОВЫЕ ПРОИЗВОДНЫЕ, ЗАМЕЩЕННЫЕ КОНДЕНСИРОВАННЫМ БИЦИКЛОГЕТЕРОЦИКЛОМ | 2007 |
|
RU2437884C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МУСКАРИНОВЫЕ АГОНИСТЫ И КОМПОЗИЦИИ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ЛЕЧЕНИЯ | 2002 |
|
RU2292346C2 |
| ПРОИЗВОДНОЕ АМИНОБЕНЗОЙНОЙ КИСЛОТЫ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1993 |
|
RU2067979C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН- ИЛИ БЕНЗОДИАЗЕПИНКАРБОНОВОЙ КИСЛОТЫ И ИХ СОЛИ | 1992 |
|
RU2040524C1 |
Изобретение относится к способам получения 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида формулы 1. Способы осуществляют путем гидрирования в присутствии хлористоводородной или трифторуксусной кислоты промежуточного продукта, аминозащищенного 3-(8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида формулы 3, где аминозащитная группа является удаляемой каталитическим гидрированием. Также изобретение относится к промежуточным соединениям 2, 3 и 5, к способу получения соединения формулы 3 и к способу увеличения стереоспецифичности для эндоориентации образца 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида. Технический результат - усовершенствованные способы получения 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамида. 8 н. и 6 з.п. ф-лы, 1 сx., 4 пр.
1. Способ получения соединения формулы 1, имеющего химическое название 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамид, или его соли,
включающий
(а) пропускание через реакционный сосуд, содержащий соединение формулы 3
и кислоту, представляющую собой хлористоводородную кислоту или трифторуксусную кислоту, азота, где Р1 представляет собой аминозащитную группу, удаляемую каталитическим гидрированием,
(b) добавление в реакционный сосуд катализатора - палладия на углероде;
(c) пропускание водорода через реакционный сосуд и
(d) подачу в реакционный сосуд газообразного водорода при давлении, менее чем приблизительно половина атмосферы, таким образом, чтобы общее давление в реакционном сосуде составляло менее чем приблизительно полторы атмосферы, с получением соединения формулы 1 или его соли.
2. Способ по п.1, где Р1 представляет собой бензил.
3. Способ по п.2, где кислотой является хлористоводородная кислота.
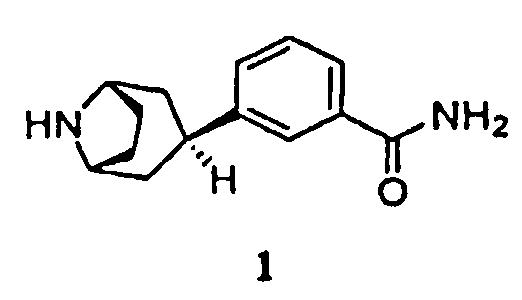
4. Способ получения соединения формулы 1, имеющего химическое название 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамид, или его соли
включающий
(а) приведение соединения формулы 5
где Р1 представляет собой аминозащитную группу, удаляемую каталитическим гидрированием, и -ORx представляет собой сульфонатную уходящую группу, в контакт с соединением формулы 4
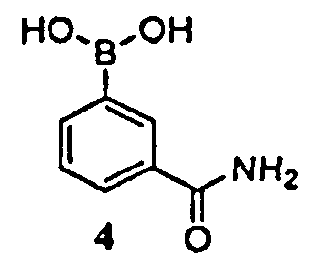
в присутствии палладиевого катализатора, представляющего собой ацетат палладия (II) или трис(дибензилиденацетон)дипалладий(0), и фосфинового лиганда, представляющего собой 1,1'-бис(дифенилфосфино)ферроцен, трициклогексилфосфинтетрафторборат или 1,5-бис(дифенилфосфино)пентан, с получением соединения формулы 3
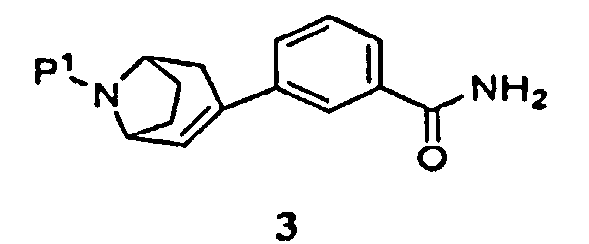
(b) пропускание через реакционный сосуд, содержащий соединение формулы 3 и кислоту, представляющую собой хлористоводородную кислоту или трифторуксусную кислоту, азота;
(c) добавление в реакционный сосуд катализатора - палладия на углероде;
(d) пропускание водорода через реакционный сосуд и
(е) подачу в реакционный сосуд газообразного водорода при давлении, менее чем приблизительно половина атмосферы, таким образом, чтобы общее давление в реакционном сосуде составляло менее чем приблизительно полторы атмосферы, с получением соединения формулы 1 или его соли.
5. Способ по п.4, где Р1 представляет собой бензил и -ORx представляет собой трифторметансульфонат.
6. Способ получения соединения формулы 1, имеющего химическое название 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамид, или его соли
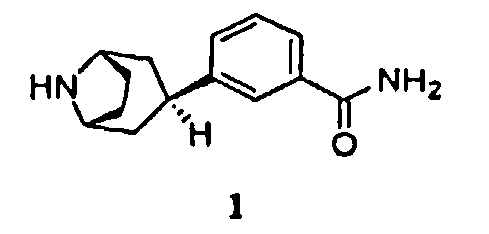
включающий гидрирование соединения формулы 2, имеющего химическое название 3-(8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамид,
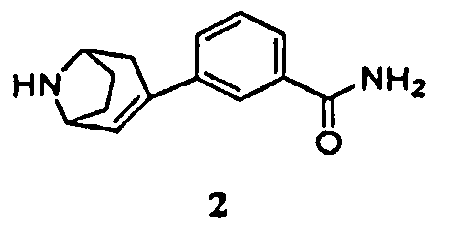
или его соли в присутствии катализатора - палладия на углероде и, когда соединение формулы 2 находится в форме свободного основания, в присутствии кислоты, представляющей собой хлористоводородную кислоту или трифторуксусную кислоту, с получением соединения формулы 1 или его соли.
7. Способ по п.6, где процесс проводят в присутствии кислоты, представляющей собой хлористоводородную кислоту или трифторуксусную кислоту, и продуктом является соль соединения формулы 1.
8. Способ по п.7, где кислотой является хлористоводородная кислота и продуктом является гидрохлоридная соль соединения формулы 1.
9. Способ получения соединения формулы 3
где Р1 представляет собой аминозащитную группу, включающий (а) приведение соединения формулы 5
где -ORx представляет собой сульфонатную уходящую группу, в контакт с соединением формулы 4
в присутствии палладиевого катализатора, представляющего собой ацетат палладия (II) или трис(дибензилиденацетон)дипалладий(0), и фосфинового лиганда, представляющего собой 1,1'-бис(дифенилфосфино)ферроцен, трициклогексилфосфинтетрафторборат или 1,5-бис(дифенилфосфино)пентан, с получением соединения формулы 3.
10. Способ по п.9, где Р1 представляет собой бензил и -ORx представляет собой трифторметансульфонат.
11. Соединение формулы 2
имеющее химическое название 3-(8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамид.
12. Соединение формулы 3
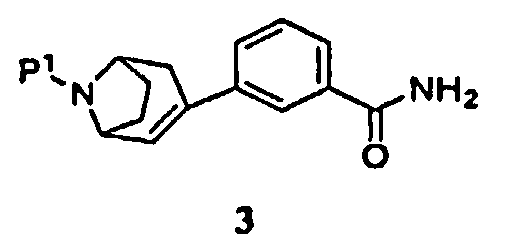
где Р1 представляет собой трет-бутоксикарбонил.
13. Соединение формулы 5
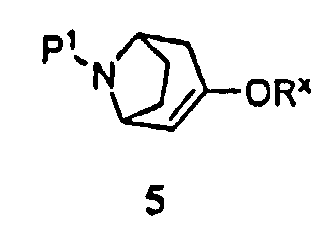
где Р1 представляет собой бензил или трет-бутоксикарбонил и -ORx представляет собой п-толуолсульфонат.
14. Способ увеличения стереоспецифичности для эндоориентации образца 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида, причем образец содержит более чем приблизительно 10% 3-экзо-(8-азабицикло[3.2.1]окт-3-ил)бензамида, где 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамид и 3-экзо-(8-азабицикло[3.2.1]окт-3-ил)бензамид находятся в форме соли, включающий
(a) нагревание смеси образца 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида и спирта до тех пор, пока не будет наблюдаться полное растворение, с получением реакционного раствора;
(b) охлаждение реакционного раствора, добавление затравочных кристаллов 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида и перемешивание смеси до тех пор, пока не будет наблюдаться образование осадка, и
(c) добавление 2-метилтетрагидрофурана к реакционному раствору и перемешивание, по существу, до полного осаждения с получением 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида, содержащего менее чем приблизительно 2% 3-экзо-(8-азабицикло[3.2.1]окт-3-ил)бензамида.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| KEVERLINE et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |
| FORBES | |||
| Highly stereoselective synthesis of exo and endo indolotropanes // Tetrahedron letters | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |

