Область техники, к которой относится изобретение
Настоящее изобретение относится к средствам, проявляющим сродство к ГАМКА-рецепторам, конкретно - к соединениям пиразоло[1,5-a]пиримидина, более конкретно - N-{2-заместитель-5-[3-заместитель-пиразоло[1,5-a]-пиримидин-7-ил]-фенил}-N-метилацетамидам и N-{2-заместитель-5-[3-заместитель-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метан-сульфонамидам.
Уровень техники
ГАМКА-рецептор (γ-аминомасляная кислота A) является пентамерным белком, который формирует мембранный ионный канал. ГАМКА-рецептор участвует в регуляции седативного эффекта, тревожности, мышечного тонуса, активности, приводящей к эпилепсии, и функционировании памяти. Эти эффекты обусловлены различными субъединицами ГАМКА-рецептора, особенно α1- и α2-субъединицами.
Седативный эффект модулируется α1-субъединицей. Золпидем характеризуется высокой аффинностью к α1-рецепторам, и его седативный и снотворный эффекты обусловлены этими рецепторами in vivo. Сходным образом снотворное действие залеплона также опосредовано α1-рецепторами.
Нейролептическое действие диазепама опосредовано усилением ГАМК-эргического переноса в популяции нейронов, экспрессирующих α2-рецепторы. Это указывает на то, что α2-рецепторы являются высокоспецифичными мишенями при лечении тревожности.
Миорелаксация при приеме диазепама в основном опосредована α2-рецепторами, поскольку эти рецепторы высокоэффективно экспрессируются в спинном мозге.
Противосудорожный эффект диазепама частично опосредован α1-рецепторами. При приеме диазепама компонент, снижающий память, антероградная амнезия опосредована α1-рецепторами.
ГАМКА-рецептор и его α1- и α2-субъединицы описаны Меллером и коллегами (J. Pharmacol. Exp. Ther., 300, 2-8, 2002; Curr. Opin. Pharmacol., 1, 22-25, 2001); Рудольфом и коллегами (Nature, 401, 769-800, 1999) и Нутом и коллегами (Br. J. Psychiatry, 179, 390-396, 2001).
Диазепам и другие классические бензодиазепины широко используются в качестве седативных агентов, снотворных агентов, противосудорожных агентов и миорелаксантов. Их побочными эффектами являются антероградная амнезия, угнетение двигательной активности и усиление действия алкоголя.
В данном контексте соединения по настоящему изобретению являются лигандами α1- и α2-ГАМКА-рецепторов для клинического применения при расстройствах сна, предпочтительно инсомнии, тревожности и эпилепсии.
Инсомния является широко распространенным заболеванием. Хронической его формой страдают 10% населения и 30% - кратковременной формой заболевания. Инсомния характеризуется проблемами при засыпании или пробуждении и связана с проявлением эффектов на следующий день, таких как усталость, слабость, низкая концентрация внимания и раздражительность. Важен социальный вклад этого заболевания и его вклад в здоровье, что приводит к очевидным социально-экономическим последствиям.
Фармакологическая терапия при лечении инсомнии в первую очередь включает в себя барбитураты и хлоралгидрат, но эти препараты вызывают многочисленные побочные эффекты, например, отравление при передозировке, повышение уровня метаболизма, а также развитие зависимости и повышение устойчивости. Кроме того, они влияют на структуру сна посредством сокращения его общей продолжительности и количества стадий быстрого сна. Также бензодиазепины имеют важное терапевтическое преимущество за счет своей низкой токсичности, но до сих пор не решена серьезная проблема привыкания, миорелаксации, амнезии и возобновления симптомов инсомнии после прекращения приема препарата.
Новейший известный терапевтический подход заключается в применении снотворных бензодиазепинового ряда, таких как пирроло[3,4-b]пиразины (зопиклон), имидазо[1,2-a]пиридины (золпидем) и пиразоло[1,5-a]пиримидины (залеплон). Позже начали разработку двух новых пиразоло[1,5-a]пиримидинов, индиплона и оцинаплона, последний с выраженным седативным действием. Все эти соединения вызывают быстрое засыпание и имеют менее выраженные отрицательные эффекты на следующий день, меньший риск злоупотребления препаратом и пониженный риск возобновления симптомов инсомнии по сравнению с бензодиазепинами. Механизм действия этих соединений заключается в аллостерической активации ГАМКА-рецепторов за счет присоединения к сайту связывания бензодиазепина (C.F.P.George, The Lancet, 358, 1623-1626, 2001). Тогда как бензодиазепины являются неспецифическими лигандами сайта связывания ГАМКА-рецептора, золпидем и залеплон проявляют большую селективность к α1-субъединице. Несмотря на это данные препараты все же влияют на структуру сна и могут вызывать зависимость при долгосрочном лечении.
Ряд родственных пиразоло[1,5-a]пиримидинов были описаны в патентных публикациях US 178449, US 4281000, US 4521422 (2-пиридинил-[7-(4-пиридинил)пиразоло[1,5-а]пиримидин-3-ил]метанон, оцинаплон), US 4576943, US 4626538 (N-{3-[3-(цианопиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-этил-ацетамид, залеплон), US 4654347, US 6399621 (N-{3-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил }-N-метил-ацетамид, индиплон), WO 2005014596, WO 2005014597 и в WO 2006136530.
Поиск новых активных соединений для лечения инсомнии является необходимым для здравоохранения, поскольку даже недавно выпущенные снотворные препараты тем не менее изменяют структуру сна и могут вызывать зависимость при долгосрочном лечении.
Тем не менее, желательно сфокусироваться на разработке новых снотворных препаратов с меньшим количеством побочных эффектов.
Раскрытие изобретения
Авторы настоящего изобретения открыли новые пиразоло[1,5-а]-пиримидины, которые обладают активностью в отношении ГАМКА и особенно против ее α1- и α2-субъединиц. Следовательно, соединения по настоящему изобретению эффективны для лечения и профилактики заболеваний, опосредованных и α2-субъединицами ГАМКА-рецептора. Примерами, не ограничивающими данное изобретение, являются расстройства сна, особенно инсомния, тревожность и эпилепсия. Не ограничивающими данное изобретение примерами относительных показаний к применению соединений, описанных здесь, являются заболевания и состояния, такие как инсомния или анестезия, при которых требуется индукция сна, индукция седативного эффекта или индукция миорелаксации.
Структура залеплона, основного соединения в ряду пиразоло[1,5-a] пиримидинов, является сходной со структурой соединений по настоящему изобретению. Тем не менее, залеплон вызывает интенсивную биотрансформацию за счет альдегидоксидазы (B.G.Lake et al., Metabolism of zaleplon by human liver: evidence for involvement of aldehyde oxidase, Xenobiotica, 2002, Oct; 32 (10): 835-47; and K.Kawashima et al., Aldehyde oxidase-dependent marked species difference in hepatic metabolism of the sedative-hypnotic zaleplon, between monkeys and rats, Drug Metab Dispos. 1999 Mar; 27 (3): 422-8). Хотя в литературе описан другой пиразоло[1,5-a]пиримидин, более метаболически стабильный по сравнению с залеплоном, это соединение имеет недостаток - его токсический эффект выше, что было показано в экпериментах на жизнеспособность клеток.
Чувствительность соединений к биотрансформации связана с их метаболической стабильностью, т.е. периодом полужизни препарата в организме и возможности образования метаболитов. Эти параметры являются важными для оценки биодоступности, токсичности и возможности дозирования для взаимодействия с другими препаратами, что в свою очередь является важными параметрами при определении их возможного применения для лечения людей. С этой точки зрения соединения с максимальной метаболической стабильностью минимизируют вероятные взаимодействия с другими препаратами и требуют более редкого приема доз препарата.
Соединения по настоящему изобретению меньше подвержены спонтанным биотрансформациям, т.е. их метаболическая стабильность выше по сравнению с другими известными родственными пиразоло[1,5-a]-пиримидинами, что улучшает их фармакокинетический профиль, помогая сохранять фармакологический эффект, и позволяет поддерживать сон в течение всей ночи при различных показаниях. Это свойство связано с замещением фенольного кольца, т.е. заместителей R3 и R4. Особенно эффективными являются соединения, которые несут электрон, полученный от удаленных заместителей в фенольном кольце.
Кроме того, соединения по настоящему изобретению обладают сильным седативным/снотворным действием, как показано в примерах, и низким токсическим эффектом, что было показано в эксперименте на жизнеспособность клеток.
Таким образом, настоящее изобретение относится к соединениям, охватываемым общей формулой (I):
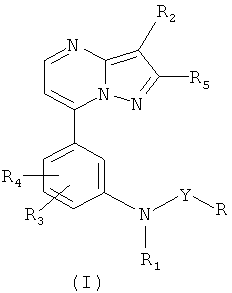
и их фармацевтически приемлемым солям и гидратам, которые являются лигандами ГАМКА-рецептора, где R и R1 представляют собой алкил(C1-C6), R2 выбран из группы, включающей циано-, нитрогруппу и тиофен-2-карбонил, R3 выбран из группы, включающей водород и галоген, R4 выбран из группы, включающей водород, галоген, алкил(C1-C6), R5 выбран из группы, включающей водород и алкил(C1-C6) и Y выбиается из группы, состоящей из -CO- и -SO2-; и его фармацевтически приемлемых солей и гидратов.
Другим объектом настоящего изобретения являются новые способы лечения или профилактики тревожности, эпилепсии и расстройств сна, в том числе инсомнии, и индукции седативно-гипнотического, анестезирующего, снотворного эффектов и миорелаксации путем введения терапевтически эффективных количеств указанных соединений или их фармацевтически приемлемых солей или гидратов.
Осуществление изобретения
Как указано выше, настоящее изобретение относится к соединениям общей формулой (I):
и их фармацевтически приемлемым солям и гидратам, где R, R1, R2, R3, R4, R5 и Y являются такими, как указано выше.
Используемый здесь термин "фармацевтически приемлемая соль" означает любую соль, образованную органической или неорганической кислотой, такой как бромоводородная, соляная, фосфорная, азотная, серная, уксусная, адипиновая, аспарагиновая, бензенсульфоновая, бензойная, лимонная, этансульфоновая, муравьиная, фумаровая, глутаминовая, молочная, малеиновая, яблочная, малоновая, миндальная, метансульфоновая, 1,5-нафталенсульфоновая, щавелевая, пиваловая, пропионовая, p-толуолсульфоновая, янтарная, виннокаменная кислоты и другие.
Конкретные соединения по формуле (1) выбираются из группы, включающей:
N-{2-фтор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамида,
N-{2-фтор-5-[3-циано-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамида,
N-{2-хлор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамида,
N-{2-хлор-5-[3-циано-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамида,
N-{2-фтор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамида,
N-{2-фтор-5-[3-циано-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамида,
N-{2-хлор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамида,
N-{2-хлор-5-[3-циано-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамида,
N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамида,
N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамида,
N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-метансульфонамида,
N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-метансульфонамида,
N-{2-метил-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида,
N-{2-метокси-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида,
N-{2,4-дифтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида и
N-{5-фтор-2-метокси-3-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида.
Нижеследующие схемы реакций иллюстрируют получение соединений по настоящему изобретению.
Схема 1
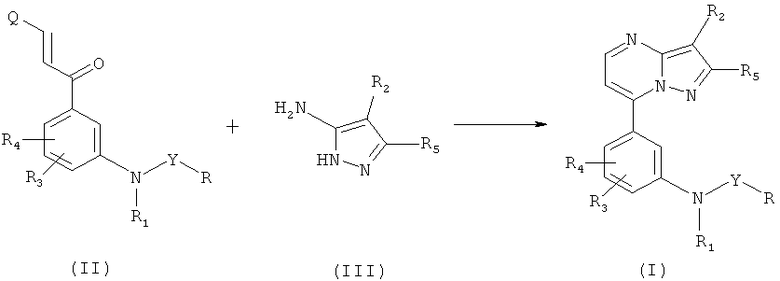
где R, R1, R2, R3, R4, R5 и Y являются такими, как указано выше, a Q является соответствующей уходящей группой, выбранной из группы, включающей N(диалкил(C1-C6)), алкилтио(C1-C6)- и алкокси(C1-C6)-группу. Предпочтительно, Q выбрана из группы, включающей диметиламино-, метилтио- и метоксигруппу.
Реакция аминопиразола общей формулы (III) с соединением, соответствующим образом замещенным 1-арил-2-пропен-1-группой, проводится в инертном полярном растворителе, содержащем или не содержащем протоны, таком как ледяная уксусная кислота, этанол, метанол, диметилформамид или диметилсульфоксид при температуре от 50 до 130°C. По истечении нескольких часов (реакционное время) растворитель удаляется и полученный остаток разделяется и помещается в водный раствор бикарбоната натрия и в дихлорметан. Грубый осадок после выпаривания органического слоя досуха может быть очищен при помощи одного из следующих методов: (a) хроматография на силикатном геле в присутствии этилацетата или дихлорметана/метанола в качестве элюента или (b) кристаллизации в подходящем растворителе (этилацетате, этаноле, метаноле и т.д.).
Промежуточное соединение формулы (II), если Q является диметиламиногруппой [промежуточное соединение (VI)] может быть получено в следующей последовательности реакций, представленных на Схеме 2:
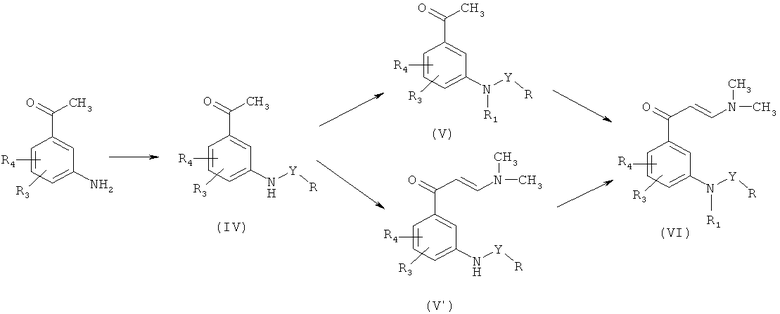
где R, R1, R2, R3, R4, R5 и Y являются такими, как указано выше. Промежуточные соединения формулы (IV), где Y является сульфонильной группой, получают в соответствии с методом, описанным R.H.Uloth et al. (J. Med. Chem. 9, 88-96, 1966).
Алкилирование промежуточных продуктов формулы (IV), приводящее к образованию промежуточных продуктов по формуле (V), проводят в соответствии с методами, хорошо известными специалистам в области органической химии, через образование аниона и последующего взаимодействия с галогенпроизводным алкана.
Енаминоны формул (V′) и (VI) получают в соответствии с традиционными методиками синтеза енаминов, описанных у J.М.Domagala et al. (J. Heterocyclic Chem., 26 (4), 1147-58, 1989) и K.Sawada et al. (Chem. Pharm. Bull., 49 (7), 799-813, 2001) путем взаимодействия ацетофенона с N,N-диметилформамид диметилацетатом (ДМФДМА) или реагентом Бредерека (трет-бутоксибис(диметиламино)метан).
Промежуточные продукты формулы (II), где Q является диметиламиногруппой, Y - сульфонилом, a R1 - метилом (VII), могут быть получены в соответствии с альтернативной Схемой 3:
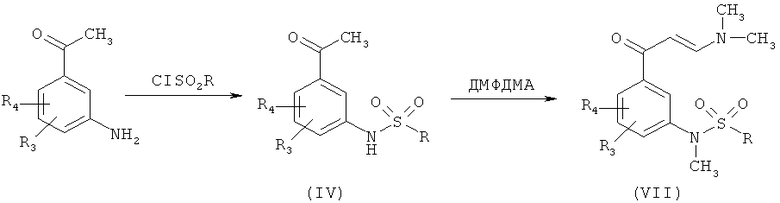
где R, R3 и R4 являются такими, как указано выше.
Превращение (IV) в (VII) приводит к образованию енаминона и одновременно к образованию N-метилсульфонамида как результат использования свойств N,N-диметилформамида диметилацетата в качестве метилирующего агента.
Промежуточные продукты формулы (II), где Q является диметиламиногруппой, R1 является метилом (X), также могут быть получены в соответствии со Схемой 4:
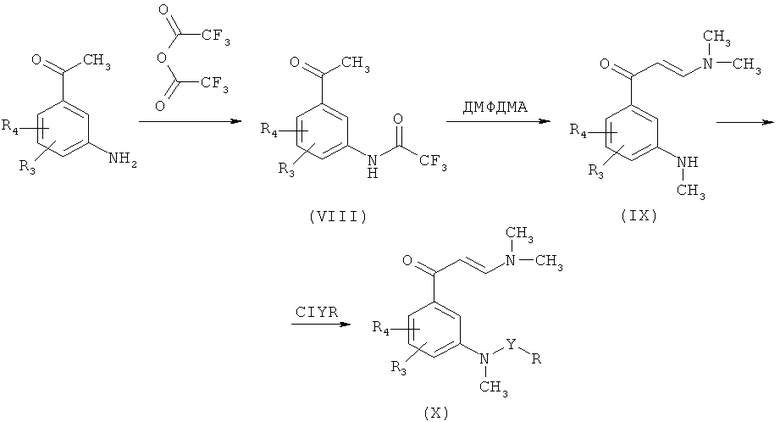
где R, R3, R4 и Y являются такими, как указано выше.
Преимущество этого способа заключается в том, что образование сульфонамида или карбоксамида происходит на последней стадии процесса. В результате общее количество этапов способа уменьшается при получении больших количеств продукта. Более того, как показано на схеме, превращение (VIII) в (IX) приводит к трем последующим реакциям, которые протекают одновременно: (a) образование енаминона, (b) метилирование трифторацетамида и (c) деацилирование, в результате которого образуется N-метилированный амин. Последующая реакция (IX) с соответствующим хлоридом сульфоновой кислоты или хлоридом карбоновой кислоты приводит к получению промежуточных продуктов (X).
Соединения по настоящему изобретению или их фармацевтически приемлемые соли или гидраты могут применяться для приготовления лекарственного средства для лечения и профилактики заболеваний, связанных с модуляцией ГАМКА-рецепторов у человека и других млекопитающих. Более конкретно, для лечения заболеваний, связанных с модуляцией ГАМКА-рецепторов и представляющих собой заболевания, связанные с модуляцией α1-ГАМКА-рецептора и/или α2-ГАМКА-рецептора. Перечень подобных заболеваний не ограничивается только тревожностью, эпилепсией, нарушениями сна, в том числе инсомнией, и им подобными.
В соответствии с другим вариантом осуществления изобретения предлагается применение соединений по настоящему изобретению или их фармацевтически приемлемых солей или гидратов для приготовления лекарственного средства для лечения и профилактики тревожности у человека и других млекопитающих.
В соответствии с другим вариантом осуществления изобретения предлагается применение соединений по настоящему изобретению или их фармацевтически приемлемых солей или гидратов для приготовления лекарственного средства для лечения и профилактики эпилепсии у человека и других млекопитающих, нуждающихся в этом.
В соответствии с другим вариантом осуществления изобретения предлагается применение соединений по настоящему изобретению или их фармацевтически приемлемых солей или гидратов для приготовления лекарственного средства для лечения и профилактики нарушений сна у человека и других млекопитающих, нуждающихся в этом.
В соответствии с другим вариантом осуществления изобретения предлагается применение соединений по настоящему изобретению или их фармацевтически приемлемых солей или гидратов для приготовления лекарственного средства для лечения и профилактики инсомнии у человека и других млекопитающих, нуждающихся в этом.
В соответствии с другим вариантом осуществления изобретения предлагается применение соединений по настоящему изобретению или их фармацевтически приемлемых солей или гидратов для приготовления лекарственного средства для индукции седативно-гипнотического эффекта у человека и других млекопитающих, нуждающихся в этом.
В соответствии с другим вариантом осуществления изобретения предлагается применение соединений по настоящему изобретению или их фармацевтически приемлемых солей или гидратов для приготовления лекарственного средства для индукции анестезии у человека и других млекопитающих, нуждающихся в этом.
В соответствии с другим вариантом осуществления изобретения предлагается применение соединений по настоящему изобретению или их фармацевтически приемлемых солей или гидратов для приготовления лекарственного средства для модулирования времени, необходимого для индукции сна и его продолжительности у человека и других млекопитающих, нуждающихся в этом.
В соответствии с другим вариантом осуществления изобретения предлагается применение соединений по настоящему изобретению или их фармацевтически приемлемых солей или гидратов для приготовления лекарственного средства для индукции миорелаксации у человека и других млекопитающих, нуждающихся в этом.
Настоящее изобретение также относится к способу лечения или профилактики заболеваний, связанных с модуляцией ГАМКА-рецепторов человека и других млекопитающих, который заключается во введении указанному человеку или другому млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли или гидрата вместе с фармацевтически приемлемыми растворителями или носителями. Более конкретно, заболевания, ассоциированные с модуляцией ГАМКА-рецепторов, включают заболевания, связанные с изменениями α1-ГАМКА-рецептора и/или α2-ГАМКА-рецептора. Список подобных заболеваний не ограничивается только тревожностью, эпилепсией, расстройствами сна, в том числе инсомнией, и им подобных.
Термин "млекопитающее" обозначает здесь представителей класса Млекопитающие высших позвоночных. Термин "млекопитающие" включает в себя человека, но не этим не ограничивается.
В соответствии с другим вариантом осуществления изобретения предлагается фармацевтическая композиция, включающая соединение по настоящему изобретению или его фармацевтически приемлемые соли или гидраты совместно с терапевтически инертными носителями.
Композиции по изобретению могут включать носители, пригодные для перорального, ректального или парентерального (в том числе подкожного, внутримышечного и внутривенного) введения, хотя наиболее подходящий способ введения зависит от природы и тяжести состояния пациента, которое подвергается лечению. Наиболее предпочтительным способом введения соединений по настоящему изобретению является пероральный способ введения. Композиции могут быть легко представлены в виде дозированных лекарственных форм и приготовлены любым из известных в фармацевтической литературе методов.
Активное соединение можно комбинировать с фармацевтическим носителем в соответствии с традиционными фармацевтическими методиками приготовления лекарственных препаратов. Носитель может принимать ряд форм, зависящих от формы лекарственного препарата, необходимой для введения, например, пероральной или парентеральной (в том числе для внутривенных инъекций или инфузий). При приготовлении композиций для перорального введения можно применять любую из обычных фармацевтических сред. Традиционные фармацевтические среды включают, например, воду, гликоли, масла, спирты, вкусовые добавки, консерванты, красители и т.п., в случае жидкой пероральной композиции (такой как, например, суспензии, раствора, эмульсии или эликсира), аэрозолей, или носители, такие как крахмал, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие вещества, скользящие вещества, связывающие вещества, дезинтегрирующие вещества и другие, в случае твердых пероральных препаратов (таких как, например, порошки, капсулы и таблетки), причем твердые препараты более предпочтительны по сравнению с жидкими.
Благодаря простоте введения пациенту таблетки и капсулы представляют собой наиболее предпочтительную лекарственную дозированную форму для введения препарата, в этом случае применяют твердые фармацевтические носители. В случае необходимости таблетки могут быть покрыты оболочкой в соответствии с водной или безводной методиками.
Подходящий интервал дозировки для применения составляет от примерно 0,01 мг до примерно 100,00 мг ежедневно, эта доза дается один раз в день или делится на субдозы, если требуется.
Соединения по настоящему изобретению имеют высокое сродство к α1- и α2-ГАМКА-рецепторам. Результаты, полученные in vitro, согласуются с результатами, полученными in vivo, в седативно-гипнотических исследованиях.
Фармакологическую активность соединений по настоящему изобретению определяли, как будет описано ниже.
а) Анализ связывания лигандов. Определение аффинности анализируемых соединений к α1- и α2-ГАМКА-рецептору.
В эксперименте использовались самцы крыс линии Sprague-Dawley массой 200-250 г. После декапитации животного удаляли мозжечок (ткань, содержащая преимущественно α1-ГАМКА-рецепторы) и спинной мозг (ткань, содержащая преимущественно α2-ГАМКА-рецепторы). Мембраны готовили в соответствии с методикой, предложенной J.Lameh et al. (Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 24, 979-991, 2000) и H.Noguchi et al. (Eur. J. Pharm., 434, 21-28, 2002) с небольшими изменениями. Взвешенные ткани суспендировали в 50 мМ Трис-HCl (рН 7,4), 1:40 (об./об.) или в сахарозе 0,32 М в случае спинного мозга, гомогенизировали и затем центрифугировали при 20000 g в течение 10 минут при 7°C. Образовавшийся осадок ресуспендировали в тех же условиях и снова центрифугировали. Наконец, осадок ресуспендировали в минимальном объеме и хранили при -80°C с течение ночи. На следующий день процедуру повторяли, осадок ресуспендировали при соотношении 1:10 (об./об.) в случае процедуры с мозжечком и при соотношении 1:5 (об./об.) в случае со спинным мозгом.
Аффинность определяли при помощи конкурентного анализа, используя флумазенил с радиоактивной меткой в качестве лиганда. Исследования проводили в соответствии с методиками, описанными S.Arbilla et al. (Eur. J. Pharmacol., 130, 257-263, 1986) и Y.Wu et al. (Eur. J. Pharmacol., 278, 125-132, 1995) на 96-луночных микропланшетах. Инкубировали мембраны, содержащие исследуемые рецепторы, флумазенил (конечная концентрация радиоактивной метки 1 нМ) и повышающиеся концентрации исследуемых соединений (в общем объеме 230 мкл в 50 мМ [pH 7,4] Трис-HCl). Одновременно инкубировали одни только мембраны с флумазенилом с радиоактивной меткой (общее связывание 100%) и в присутствии повышающихся концентраций флумазенила без радиоактивной метки (неспецифическое связывание, % оценка лиганда с радиоактивной меткой). Реакции начинали добавлением лиганда с радиоактивной меткой и инкубировали в течение 60 минут при 4°C. В конце периода инкубации 200 мкл реакционной смеси переносили на планшет для микротитрования (Millipore) и фильтровали при помощи вакуумного насоса и затем промывали трижды холодным буфером для исследования. Планшеты для микротитрования снабжали фильтром GF/B, после фильтрования оставались мембраны, содержащие рецепторы и лиганд с радиоактивной меткой, который связался с рецепторами. После промывания планшетам давали высохнуть. На следующий день осуществляли подсчет при помощи сцинтилляционного счетчика Perkin-Elmer Microbeta.
Для анализа результатов вычисляли процент специфического связывания для каждой концентрации анализируемого соединения по формуле:
% специфического связывания=(X-N/T-N)·100,
где
X - количество связавшегося лиганда для каждой концентрации соединения,
T - общее связывание, максимальное количество, связавшееся с лигандом, содержащим радиоактивную метку,
N - неспецифическое связывание, количество лиганда с радиоактивной меткой, связавшееся неспецифически, вне зависимости от того, какой был использован рецептор.
Каждая концентрация исследуемых соединений анализировалась трижды, средние значения использовались для определения экспериментальных значений процента специфического связывания, в зависимости от концентрации соединения. Данные по аффинности выражали в % ингибирования при концентрациях 10-5 M и 10-7 M для α1-субъединицы и 10-5 М для α2-субъединицы. Результаты этих исследований представлены в Таблицах 1 и 2 соответственно.
б) Определение прогностической седативно-гипнотической активности in vivo.
Активность этих соединений in vivo оценивали при помощи прогностического седативно-гипнотического исследования на мышах (D.J.Sanger et al., Eur. J. Pharmacol., 313, 35-42, 1996 и G.Griebel et al., Psychopharmacology, 146, 205-213, 1999).
Для исследования использовали группы по 5-8 самцов мышей линии CD1 весом 22-26 г. Исследуемые соединения вводили внутрибрюшинно одной эквимолекулярной дозой, растворенной в 0,25% агаре с каплей Tween 80 в объеме 10 мл/кг. Для каждого способа введения анализировали две дозы. Контрольные животные получали только носитель. При помощи Smart System (Panlab, S.L., Spain) записывали пройденное расстояние в см для каждой мыши с пятиминутными интервалами в течение 30 минут после внутрибрюшинной инъекции. Вычисляли процент ингибирования на пройденном расстоянии у животных, получавших лечение, против контроля (первые 5 минут отбрасывали) и значения ED50. Результаты этого анализа представлены в Таблицах 3 и 4.
По сравнению с другими представителями пиразоло[1,5-a]-пиримидинов, описанных в литературных источниках, соединение из примера 11 настоящего изобретения обладает значительно более низким ED50. Это означает, что соединение из примера 11 является более активным in vivo, поскольку для индукции терапевтического эффекта требуется более низкая доза.
в) Определение метаболической стабильности in vitro в цитозольной фракции гепатоцитов человека
Соединения растворяли в диметилсульфоксиде так, чтобы начальная концентрация составляла 10 мМ. Этот стоковый раствор затем разводили растворителем и буфером, чтобы получить конечную концентрацию для анализа 5 мкМ. Соединения анализировали при одной концентрации 5 мкМ дважды, инкубируя с 1,0 мг/мл пулированного цитозоля человека (полученного из Xenotech plc) при 37°C. Метаболизм оценивали в присутствии или отсутствии кофакторов и измеряли как расход исходного соединения при помощи анализа ЖХ/МС во временных точках 0, 60 и 120 минут. Затем вычисляли процентное содержание оставшегося исходного соединения. Результаты представлены в Таблице 5. Использовали общий ЖХ метод:
Подвижная фаза: А=0,1% муравьиная кислота в воде
В=0,1% муравьиная кислота в ацетонитриле
Колонка для ВЭЖХ: Higgins Clipius C18 5 мкм, 50×3 мм
Скорость потока: 2 мл.мин-1.
Неожиданно для соединений по экспериментальным примерам 6 и 11 было замечено, что процентные соотношения (10-20%) оставшегося исходного соединения по сравнению с залеплоном и соединением по WO 200514596 оказались выше после инкубации в течение 60 и 120 минут. С другой стороны, для залеплона было показано, что процентные соотношения оставшегося соединения в любой временной точке ниже, а биотрансформация между 60 и 120 минутами выше.
г) Определение токсичности для клеток in vitro в клетках HepG2, СНО-K1 и HeLa в течение 24 часов.
HepG2 (клетки гепатоцеллюлярной карциномы человека) и CHO-K1 (клетки яичников китайского хомяка), полученные из коллекции American Туре Culture Collection (АТСС). HepG2 культивировали в минимальной необходимой среде (MEM), содержащей солевой раствор Эрла с 1,87 мМ Glutamax® и дополненной 1 мМ пирувата натрия, 0,1 мМ заменимых аминокислот, 100000 Ед/л пенициллина, 10000 Ед/л стрептомицина и 10% фетальной бычьей сыворотки. СКО-K1 поддерживали в среде Хама F-12, содержащей 1 мМ Glutamax® и дополненной 1 мМ L-глутамина, 10000 Ед/л пенициллина, 10000 мкг/л стрептомицина и 10% фетальной бычьей сыворотки. Система анализа Promega CellTiter 96® Aqueous One Solution Cell Viability содержит соль тетразолия (MTS), которая под действием ферментов с дегидрогеназной активностью в метаболически активных клетках превращается в растворимый продукт формазан. Количество формазана пропорционально количеству живых клеток в культуре.
Соединения растворяли в ДМСО так, чтобы начальная концентрация составляла 100 мМ. Из этого раствора готовили серию разведений в ДМСО так, чтобы интервал концентраций составлял от 50 до 0,25 мМ. Стоковый раствор и серию разведений затем разводили 1:100 соответствующей культуральной средой. В случае СНО-K1 клеток готовили концентрации 1000, 500, 250, 100, 50, 25, 10, 5 и 2,5 мкМ, чтобы оценить IC50, тогда как в случае с клетками HepG2 анализировали конечные концентрации 1000, 100, 10 и 1 мкМ, чтобы вычислить процентное соотношение клеток. Конечная концентрация ДМСО во всех лунках составляла 1% (об./об.). Обе линии клеток инкубировали с исследуемыми соединениями в течение 24 часов. Относительную жизнеспособность клеток определяли спектрофотометрически при 490 нм после добавления красителя MTS и последующей инкубации в течение 1 часа. В качестве положительного контроля использовали тамоксифен.
Аналогичный протокол использовали для определения токсичности для клеток в эксперименте с клетками HeLa в течение 24 часов. Результаты представлены в Таблице 6.
Эти результаты показывают, что соединение из примера 11 по настоящему изобретению менее токсично, чем контрольное соединение индиплон, поскольку выживаемость клеток для соединения из примера 11 выше по сравнению с индиплоном (84,5% против 70%) в культуре клеток HepG2. Эти результаты также подтверждаются двумя другими проанализированными линиями клеток. Экспериментальный пример 1:
N-{2-фтор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид
Смесь 0,048 г (0,38 мМ) 4-нитро-2Н-пиразол-3-иламина и 0,1 г (0,38 мМ) N-[5-(3-диметиламино-акролеил)-2-фторфенил]-N-метилацетамидав 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, которое подвергали хроматографии (на силикагеле) с использованием ацетат-дихлорметана в качестве элюента, таким образом получая 61 мг (выход 49%) твердого N-{2-фтор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида.
1Н ЯМР (400 МГц, CDCl3): δ 1,97 (3H, s), 3,29 (3H, s), 7,29 (1H, d, J=4,4 Гц), 7,45 (1H, t, J=8,4 Гц), 7,89-8,02 (1Н, m), 8,07-8,09 (1Н, m), 8,83 (1H, s), 9,0 (1Н, d, J=4,4 Гц).
MS (ES) m/z=330 (MH+).
ВЭЖХ=95,7%.
Экспериментальный пример 2:
N-{2-фтор-5-[3-циано-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамид
Смесь 0,041 г (0,38 мМ) 5-амино-1H-пиразол-4-карбонитрила и 0,1 г (0,38 мМ) N-[5-(3-диметиламино-акролеил)-2-фторфенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 95 мг N-{2-фтор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамида (выход 81%).
1Н ЯМР (400 МГц, CDCl3): δ 1,96 (3H, s), 3,28 (3H, s), 7,18 (1H, d, J=4,4 Гц), 7,42 (1H, t, J=8,8 Гц), 7,99-8,02 (1Н, m), 8,09-8,12 (1H, m), 8,42 (1H, s), 8,79 (1Н, d, J=4,4 Гц).
MS (ES) m/z=310 (MH+).
ВЭЖХ=97,8%.
Экспериментальный пример 3:
N-{2-хлор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамид
Смесь 0,054 г (0,43 мМ) 4-нитро-2Н-4-пиразол-3-иламина и 0,120 г (0,43 мМ) N-[5-(3-диметиламино-акролеил)-2-фторфенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха чтобы получить масло, которое подвергали хроматографии (на силикагеле) с использованием ацетат-дихлорметана в качестве элюента, таким образом, получая 35 мг (выход 24%) твердого N-{2-хлор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида.
1Н ЯМР (400 МГц, CDCl3): δ 1,90 (3H, s), 3,26 (3H, s), 7,30 (1H, d, J=4,4 Гц), 7,77 (1Н, t, J=8 Гц), 7,93 (1H, dd, J=2,4 и 8,4 Гц), 8,08 (1H, d, J=2 Гц), 8,83 (1H, s), 9,01 (1H, d, J=4,8 Гц).
MS (ES) m/z=346 (MH+).
ВЭЖХ=91%.
Экспериментальный пример 4:
N-{2-хлор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид
Смесь 0,046 г (0,43 мМ) 5-амино-1H-пиразол-4-карбонитрила и 0,120 г (0,43 мМ) N-[5-(3-диметиламино-акролеил)-2-хлорфенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 108 мг N-{2-фтор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамида (выход 77%).
1Н ЯМР (400 МГц, CDCl3): δ 1,90 (3H, s), 3,25 (3H, s), 7,20 (1H, d, J=4,4 Гц), 7,74 (1Н, t, J=8,8 Гц), 7,94 (1H, dd, J=2,4 и 8,4 Гц), 8,10 (1H, d, J=2 Гц), 8,43 (1H, s), 8,80 (1H, d, J=4,8 Гц).
MS (ES) m/z=326 (MH+).
ВЭЖХ=97,7%.
Экспериментальный пример 5:
N-{2-фтор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамид
Смесь 0,043 г (0,33 мМ) 4-нитро-2Н-4-пиразол-3-иламина и 0,1 г (0,33 мМ) N-[5-(3-диметиламино-акролеил)-2-фторфенил]-N-метилметансульфонамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана.
Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, которое подвергали хроматографии (на силикагеле) с использованием ацетат-дихлорметана в качестве элюента, таким образом, получая 58 мг (выход 48%) твердого N-{2-фтор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамида.
1Н ЯМР (400 МГц, CDCl3): δ 3,02 (3H, s), 3,39 (3H, s), 7,29 (1H, d, J=4,4 Гц), 7,38-7,42 (1H, m), 8,05-8,13 (2H, m), 8,83 (1H, s), 8,98 (1H, d, J=4,4 Гц).
MS (ES) m/z=366 (MH+).
ВЭЖХ=97,6%.
Экспериментальный пример 6:
N-{2-фтор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид
Смесь 0,036 г (0,33 мМ) 5-амино-1Н-пиразол-4-карбонитрила и 0,1 г (0,33 мМ) N-[5-(3-диметиламино-акролеил)-2-фторфенил]-N-метилметансульфонамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана.
Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 81 мг N-{2-фтор-5-[3-циано-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамида (выход 70%).
1Н ЯМР (400 МГц, CDCl3): δ 3,01 (3H, s), 3,38 (3H, s), 7,29 (1H, d, J=4,4 Гц), 7,36-7,41 (1H, m), 8,08-8,15 (2H, m), 8,42 (1H, s), 8,77 (1H, d, J=4,4 Гц).
MS (ES) m/z=346 (MH+).
ВЭЖХ=99,1%.
Экспериментальный пример 7:
N-{2-хлор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамид
Смесь 0,050 г (0,39 мМ) 5-нитро-2Н-пиразол-3-иламина и 0,124 г (0,39 мМ) N-[5-(3-диметиламино-акролеил)-2-хлорфенил]-N-метилметансульфонамида в 12 мл ледяной уксусной кислоты нагревали в течение 1,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 56 мг N-{2-хлор-5-[3-нитро-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамида (выход 77%).
1Н ЯМР (400 МГц, CDCl3): δ 3,08 (3H, s), 3,38 (3H, s), 7,30 (1H, d, J=4,4 Гц), 7,71 (1H, d, J=8,4 Гц), 8,04 (1H, dd, J=2 и 8,4 Гц), 8,14 (1H, d, J=2,4 Гц), 8,83 (1H, s), 8,99 (1H, d, J=4,4 Гц).
MS (ES) m/z=382 (MH+).
ВЭЖХ=98,5%.
Экспериментальный пример 8:
N-{2-хлор-5-[3-циано-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилметансульфонамид
Смесь 0,042 г (0,39 мМ) 5-амино-1H-пиразол-3-карбонитрила и 0,124 г (0,39 мМ) N-[5-(3-диметиламино-акролеил)-2-хлорфенил]-N-метилметансульфонамида в 12 мл ледяной уксусной кислоты нагревали в течение 1,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 99 мг N-{2-хлор-5-[3-циано-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-метансульфонамида (выход 70%).
1Н ЯМР (400 МГц, CDCl3): δ 3,08 (3H, s), 3,37 (3H, s), 7,20 (1H, d, J=4,4 Гц), 7,69 (1H, d, J=8,8 Гц), 8,05 (1Н, dd, J=2,4 и 8,8 Гц), 8,16 (1H, d, J=1,6 Гц), 8,42 (1H, s), 8,78 (1H, d, J=4,4 Гц).
MS (ES) m/z=362 (MH+).
ВЭЖХ=93,7%.
Экспериментальный пример 9:
N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамид
Смесь 0,046 г (0,39 мМ) 5-амино-3-метил-1H-пиразол-4-карбонитрила и 0,1 г (0,38 мМ) N-[5-(3-диметиламино-акролеил)-2-фторфенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 92 мг N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамида (выход 75%).
1Н ЯМР (400 МГц, CDCl3): δ 1,98 (3H, s), 2,61 (3H, s), 3,3 (3H, s), 7,09 (1H, d, J=4 Гц), 7,39-7,44 (1H, m), 7,89-8,02 (1H, m), 8,08-8,11 (1H, m), 8,70 (1H, d, J=4,4 Гц).
MS (ES) m/z=324 (MH+).
ВЭЖХ=98,4%.
Экспериментальный пример 10:
N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил ацетамид
Смесь 0,055 г (0,43 мМ) 5-амино-3-метил-1H-пиразол-4-карбонитрила и 0,120 г (0,43 мМ) N-[5-(3-диметиламино-акролеил)-2-хлорфенил]-N-метилацетамида в 12 мл ледяной уксусной кислоты нагревали в течение 1,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 106 мг N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метилацетамида (выход 73%).
1Н ЯМР (400 МГц, CDC3): δ 1,91 (3H, s), 2,61 (1H, s), 3,25 (3H, s), 7,10 (1H, d, J=4,8 Гц), 7,73 (1Н, d, J=8,4 Гц), 7,97 (1H, dd, J=2 и J=8 Гц), 8,08 (1H, d, J=2,4 Гц), 8,71 (1Н, d, J=4,4 Гц).
MS (ES) m/z=340 (MH+).
ВЭЖХ=99,6%.
Экспериментальный пример 11:
N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-а]пиршмидин-7-ил]-фенил}-N-метил-метансульфонамид
Смесь 0,041 г (0,33 мМ) 5-амино-3-метил-1H-пиразол-4-карбонитрила и 0,1 г (0,33 мМ) N-[5-(3-диметиламино-акролеил)-2-фторфенил]-N-метилметансульфонамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 66 мг N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил }-N-метил-метансульфонамида (выход 55%).
1Н ЯМР (400 МГц, CDCl3): δ 2,78 (3H, s), 3,17 (3H, s), 3,54 (3H, s), 7,24 (1H, d, J=4,4 Гц), 7,51-7,56 (1H, m), 8,25-8,31 (2H, m), 8,84 (1H, d, J=4,4 Гц).
MS (ES) m/z=360 (MH+).
ВЭЖХ=98,9%.
Экспериментальный пример 12:
N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид
Смесь 0,048 г (0,39 мМ) 5-амино-3-метил-1H-пиразол-4-карбонитрила и 0,124 г (0,39 мМ) N-[5-(3-диметиламино-акролеил)-2-хлорфенил]-N-метилметансульфонамида в 12 мл ледяной уксусной кислоты нагревали в течение 1,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 89 мг N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-метансульфонамида (выход 60,5%).
1Н ЯМР (400 МГц, CDCl3): δ 2,61 (3H, s), 3,08 (1H, s), 3,66 (3H, s), 7,10 (1H, d, J=4,8 Гц), 7,68 (1H, d, J=8,8 Гц), 8,04 (1Н, dd, J=2,4 и J=8,8 Гц), 8,15 (1H, d, J=2,4 Гц), 8,70 (1H, d, J=4,4 Гц).
MS (ES) m/z=376 (MH+).
ВЭЖХ=98,1%.
Экспериментальный пример 13:
N-{2-метил-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамид
Смесь 0,074 г (0,38 мМ) (5-амино-1H-пиразол-4-ил)-тиофен-2-ил-метанона и 0,1 г (0,38 мМ) N-[5-(3-диметиламино-акролеил)-2-метилфенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 132 мг N-{2-метил-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида (выход 88%).
1Н ЯМР (400 МГц, CDCl3): δ 1,87 (3H, s), 2,37 (3H, s), 3,25 (3H, s), 7,13 (1H, d, J=4 Гц), 7,18-7,20 (1H, m), 7,54 (1H, D, J=7,6 Гц), 7,70 (1H, d, J=5,2 Гц), 7,94-7,98 (2H, m), 8,08 (1H, d, J=2,8 Гц), 8,71 (1H, s), 8,81 (1H, d, J=4 Гц).
MS (ES) m/z=391 (МН+).
ВЭЖХ=98,3%.
Экспериментальный пример 14:
N-{2-метокси-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамид
Смесь 0,070 г (0,36 мМ) (5-амино-1Н-пиразол-4-ил)-тиофен-2-ил-метанона и 0,1 г (0,38 мМ) N-[5-(3-диметиламино-акролеил)-2-метокси-фенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 135 мг N-{2-метокси-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида (выход 92%).
1Н ЯМР (400 МГц, CDCl3): δ 1,90 (3H, s), 3,23 (3H, s), 3,97 (3H, s), 7,13 (1H, d, J=4,8 Гц), 7,17-7,21 (2H, m), 7,70 (1H, D, J=4,4 Гц), 8,02 (1H, S), 8,09 (1Н, d, J=4 Гц), 8,15 (1Н, d, J=8,8 Гц), 8,71 (1H, s), 8,79 (1H, d, J=4,4 Гц).
MS (ES) m/z=407 (MH+).
ВЭЖХ=100%.
Экспериментальный пример 15:
N-{2,4-дифтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримид ин-7-ил]-фенил}-N-метил-ацетамид
Смесь 0,217 г (1,12 мМ) (5-амино-1Н-пиразол-4-ил)-тиофен-2-ил-метанона и 0,3 г (1,12 мМ) N-[5-(3-диметиламино-акролеил)-2,4-дифторфенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 320 мг N-{2,4-дифтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида (выход 69%).
1Н ЯМР (250 МГц, CDCl3): δ 1,82 (3H, s), 3,11 (3H, s), 6,96-7,06 (3H, m), 7,55 (1H, d, J=4,9 Гц), 7,76 (1H, t, J=8,2 Гц), 7,91 (1H, dd, J=1 и 3,6 Гц), 8,52 (1Н, s), 8,68 (1H, d, J=4,1 Гц).
MS (ES) m/z=413 (MH+).
ВЭЖХ=99,0%.
Экспериментальный пример 16:
N-{5-фтор-2-метокси-3-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]-пиримидин-7-ил]-фенил}-N-метил-ацетамид
Смесь 0,180 г (0,93 мМ) (5-амино-1Н-пиразол-4-ил)-тиофен-2-ил-метанона и 0,275 г (0,93 мМ) N-[3-(3-диметиламино-акролеил)-5-фтор-2-метоксифенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревали в течение 2,5 часов, затем растворитель удаляли при помощи выпаривания при пониженном давлении. К образовавшемуся осадку добавляли 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделялись, водный слой промывали дважды 10 мл дихлорметана. Органические слои промывали 10 мл воды и высушивали над сульфатом магния. Слой, содержащий дихлорметан, выпаривали досуха, чтобы получить масло, из которого в присутствии этилацетата образовывалось 160 мг N-{5-фтор-2-метокси-3-[3-(тиофен-2-карбонил)-пиразоло[1,5-a]пиримидин-7-ил]-фенил}-N-метил-ацетамида (выход 40%).
1Н ЯМР (250 МГц, CDCl3): δ 2,04 (3H, s), 3,32 (3H, s), 3,56 (3H, s), 7,09-7,25 (3H, m), 7,35 (1H, dd, J=2,2 и J=7,1 Гц), 7,72 (1H, d, J=4,9 Гц), 8,12 (1H, d, J=3,8), 8,68 (1H, s), 8,85 (1H, d, J=4 Гц).
MS (ES) m/z=425 (MH+).
ВЭЖХ=98,4%.
Пример композиции 1: таблетки 5 мг
Пример композиции 2: капсулы 10 мг
Пример композиции 3: пероральные капли
Пример композиции 4: таблетки 2,5 мг
Пример композиции 5: капсулы 5 мг
Пример композиции 6: пероральные капли
| название | год | авторы | номер документа |
|---|---|---|---|
| ГАЛОГЕНИЗИРОВАННЫЕ ПИРАЗОЛО[1,5-а]ПИРИМИДИНЫ (ВАРИАНТЫ), СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2006 |
|
RU2478101C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРОВ ГАМК | 2011 |
|
RU2582337C2 |
| НОВЫЕ ЛИГАНДЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2492164C2 |
| СОЕДИНЕНИЕ АМИНОПИРАЗОЛОПИРИМИДИНА, ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ТИРОЗИНКИНАЗНОГО РЕЦЕПТОРА НЕЙРОТРОФИЧЕСКОГО ФАКТОРА | 2017 |
|
RU2764523C2 |
| ТИАЗОЛОПИРИМИДИНОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРОВ NMDA | 2014 |
|
RU2703273C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ БЛОКИРОВАНИЯ ОБРАТНОГО ЗАХВАТА НОРЭПИНЕФРИНА, ДОПАМИНА И СЕРОТОНИНА | 2005 |
|
RU2388751C2 |
| НОВЫЕ 1,2-БИС-СУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ КАК МОДУЛЯТОРЫ ХЕМОКИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2654213C9 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2826628C1 |
Настоящее изобретение относится к новым соединениям пиразоло[1,5-а]пиримидина, их фармацевтически приемлемым солям и гидратам, обладающим способностью ингибировать ГАМКА-рецепторы, и пригодным для лечения и профилактики тревожности, эпилепсии и расстройств сна, в том числе инсомнии, а также для индуцирования седативно-гипнотического, обезболивающего и снотворного эффектов и миорелаксации. Соединениям пиразоло[1,5-а]-пиримидина согласно изобретению выбираются из группы, включающей: N-{2-фтор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид, N-{2-фтор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид, N-{2-хлор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид, N-{2-хлор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид, N-{2-фтор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид, N-{2-фтор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид, N-{2-хлор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид, N-{2-хлор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид, N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид, N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид, N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид, N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид, N-{2-метил-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид, N-{2-метокси-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид, N-{2,4-дифтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид и N-{5-фтор-2-метокси-3-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид. 11 н. и 3 з.п. ф-лы, 6 табл., 4 схемы, 22 пр.
1. Соединения пиразоло[1,5-а]пиримидина, выбранные из группы, включающей:
N-{2-фтор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид,
N-{2-фтор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид,
N-{2-хлор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид,
N-{2-хлор-5-[3-циано-пиразоло[l,5-a]пиримидин-7-ил]-фенил}-N-метилацетамид,
N-{2-фтор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид,
N-{2-фтор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид,
N-{2-хлор-5-[3-нитро-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид,
N-{2-хлор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид,
N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид,
N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид,
N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид,
N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид,
N-{2-метил-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид,
N-{2-метокси-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид,
N-{2,4-дифтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид и
N-{5-фтор-2-метокси-3-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид,
и их фармацевтически приемлемые соли и гидраты.
2. Соединения по п.1, выбранные из группы, включающей:
N-{2-фтор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид,
N-{2-хлор-5-[3-циано-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид,
N-{2-фтор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид,
N-{2-хлор-5-[3-циано-2-метил-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид,
и их фармацевтически приемлемые соли и гидраты.
3. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для лечения или профилактики заболеваний, связанных с ингибированием ГАМКА-рецепторов у нуждающегося в таком лечении человека или другого млекопитающего.
4. Применение по п.3, в котором ГАМКА-рецептор представляет собой α1-ГАМКА-рецептор.
5. Применение по п.3, в котором ГАМКА-рецептор представляет собой α2-ГАМКА-рецептор.
6. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для лечения или профилактики тревожности, опосредованной активностью ГАМКА рецепторов, у нуждающегося в этом человека или других млекопитающих.
7. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для лечения или профилактики эпилепсии, опосредованной активностью ГАМКА-рецепторов, у нуждающегося в этом человека или других млекопитающих.
8. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для лечения или профилактики нарушений сна, опосредованной активностью ГАМКА-рецепторов, у нуждающегося в этом человека или других млекопитающих.
9. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для лечения или профилактики инсомнии, опосредованной активностью ГАМКА-рецепторов, у нуждающегося в этом человека или других млекопитающих.
10. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для индукции седативно-гипнотического эффекта, опосредованного активностью ГАМКА-рецепторов, у нуждающегося в этом человека или других млекопитающих.
11. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для индукции анестезии, опосредованной активностью ГАМКА-рецепторов, у нуждающегося в этом человека или других млекопитающих.
12. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для модулирования времени, необходимого для индукции сна и его продолжительности, опосредованной активностью ГАМКА-рецепторов, у нуждающегося в этом человека или других млекопитающих.
13. Применение соединений по п.1 для приготовления лекарственного средства, предназначенного для индукции миорелаксации, опосредованной активностью ГАМКА-рецепторов, у нуждающегося в этом человека или других млекопитающих.
14. Фармацевтическая композиция, обладающая способностью ингибировать ГАМКА-рецепторы, и включающая терапевтически эффективное количество соединения по п.1 и приемлемое количество фармацевтических наполнителей или носителей.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| US 6399621 В1, 04.06.2002 | |||
| US 4626538 А, 02.12.1986 | |||
| С.F.P.GEORGE "Pyrazolopyrimidines"; THE LANGET, 2001, v.358, p.1623-1626 | |||
| ПИРАЗОЛОПИРИМИДИНИЛ- И ПИРИМИДИНИЛАЛКИЛИДЕНБИСФОСФОНОВЫЕ КИСЛОТЫ, ИХ ЭФИРЫ ИЛИ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2079506C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-{5-[3-(ТИОФЕН-2-КАРБОНИЛ)-ПИРАЗОЛО[1,5-а]ПИРИМИДИН-7-ИЛ]-2-ФТОР-ФЕНИЛ}-N-МЕТИЛ-АЦЕТАМИДА В ПОЛИМОРФНОЙ МОДИФИКАЦИИ В | 2007 |
|
RU2404984C1 |

