Область техники, к которой относится изобретение
Данное изобретение относится к агентам, обладающим аффинностью к рецептору ГАМКА, в частности к галогенизированным пиразоло-[1,5-а]пиримидинам, и более конкретно к [7-(3-амино-4-галогенил)-пиразоло[1,5-а]пиримидин-3-ил]-тиофен-2-ил-метанонацильным и сульфонильным соединениям.
Уровень техники
Рецептор ГАМКА (γ-аминомасляной кислотыА) представляет собой пентамерный белок, который формирует мембранный ионный канал. Рецептор ГАМКА участвует в регуляции седативного эффекта, тревоги, мышечного тонуса, эпилептогенной активности и функций памяти. Данные действия обусловлены определенными субъединицами рецептора ГАМКА, в частности α1- и α2-субъединицами.
Седативный эффект модулируется α1-субъединицей. Золпидем характеризуется высокой аффинностью в отношении α1-рецепторов, и его седативное и снотворное действие опосредуется данными рецепторами in vivo. Аналогично снотворное действие залеплона также опосредуется α1-рецепторами.
Анксиолитическое действие диазепама опосредуется путем усиления ГАМКергической передачи в популяции нейронов, экспрессирующих α2-рецепторы. Это показывает, что α2-рецепторы являются высокоспецифическими мишенями для лечения тревоги.
Мышечная релаксация при использовании диазепама в основном опосредована α2-рецепторами, поскольку данные рецепторы демонстрируют высокоспецифическую экспрессию в спинном мозге. Противосудорожный эффект диазепама отчасти обусловлен α1-рецепторами. При использовании диазепама, соединения ослабляющего память, антероградная амнезия опосредована α1-рецепторами.
Широкий обзор по рецептору ГАМКА и его α1- и α2-субъединицам приведен в статьях Н.Mohler et al. (J. Pharmacol. Exp. Ther., 300, 2-8, 2002); H.Mohler et al. (Curr. Opin. Pharmacol., 1, 22-25, 2001); U. Rudolph et al. (Nature, 401, 796-800, 1999) и D.J.Nutt et al. (Br. J. Psychiatry, 179, 390-396, 2001).
Диазепам и другие классические бензодиазепины широко используют в качестве анксиолитических агентов, снотворных агентов, противосудорожных препаратов и мышечных релаксантов. Данные побочные эффекты включают антероградную амнезию, снижение двигательной активности и потенцирование эффектов этанола.
В данном контексте соединения, соответствующие данному изобретению, представляют собой лиганды α1- и α2-рецептора ГАМКА, предназначенные для клинического применения при нарушениях сна, предпочтительно при бессоннице, тревоге и эпилепсии.
Бессонница является широко распространенным заболеванием. Ее хроническая форма поражает 10% населения и 30%, когда учитывают также временную бессонницу. Бессонница характеризуется трудностями, связанными с засыпанием или сном, и связана с неприятными эффектами на следующий день, такими как усталость, отсутствие энергии, низкая сосредоточенность и раздражительность. Воздействие на социальные аспекты и состояние здоровье данного заболевания важно и приводит в результате к заметным социоэкономическим последствиям.
Фармакотерапия в лечении бессонницы в основном включает барбитураты и хлоралгидрат, но данные лекарственные препараты вызывают многочисленные известные неблагоприятные эффекты, например токсичность передозировки, метаболическая индукция и повышенная зависимость и толерантность. Кроме того, они воздействуют на структуру сна посредством снижения относительно всех остальных продолжительности и числа REM-стадий сна (стадий быстрого сна). Далее бензодиазепины имеют важное терапевтическое преимущество вследствие своей пониженной токсичности, но они еще характеризуются серьезными проблемами зависимости, мышечной релаксации, амнезии и реактивной бессонницы после прекращения приема препарата.
Последний известный терапевтический подход представляет собой введение небензодиазепиновых снотворных, таких как пирроло[3,4-b]-пиразины (зопиклон), имидазо[1,2-а]пиридины (золпидем) и, наконец, пиразоло[1,5-а]пиримидины (залеплон). Позднее была начата разработка двух новых пиразоло[1,5-а] пиримидинов, индиплона и окинаплона, последний со значительным анксиолитическим действием. Все данные соединения проявляют быстрое индуцирование сна и имеют пониженные неприятные эффекты на следующий день, более низкий потенциал привыкания и пониженный риск реактивной бессонницы, чем бензодиазепины. Механизм действия данных соединений представляет собой аллостерическую активацию рецептора ГАМКА посредством его связывания с центром связывания бензодиазепина (см. статью С.F.P.George, The Lancet, 358, 1623-1626, 2001). Хотя бензодиазепины является неспецифическими лигандами центра связывания рецептора ГАМКА, золпидем и залеплон проявляют повышенную избирательность в отношении α1-субъединицы. Тем не менее, данные лекарственные препараты еще воздействуют на структуру сна и могут вызывать зависимость при длительном лечении.
Настоящее изобретение структурно связано, но обладает самостоятельной патентоспособностью относительно соединения N-{3-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамида, индиплона, которое описано в US 6399621, и соединений N-{3-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамида и N-{3-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]-пиримидин-7-ил]-фенил}-N-проп-2-инилметансульфонамида, которые описаны в WO 2005014597, примеры 3 и 16 соответственно, вследствие своих усовершенствованных признаков, как показано в разделе "Детальное описание изобретения". Аналогичные индиплону соединения описаны ранее в US 4521422.
Исследование новых активных соединений при лечении бессонницы отвечает основополагающим требованиям здравоохранения, поскольку даже недавно введенные снотворные препараты еще воздействуют на структуру сна и могут вызывать зависимость при длительном применении.
Вследствие этого необходимо сосредоточиться на разработке новых снотворных агентов с пониженным риском побочных эффектов.
Таким образом, настоящее изобретение направлено на новые галогенизированные пиразоло[1,5-а] пиримидины, которые активны в отношении ГАМКА и, в частности, в отношении ее α1- и α2-субъединиц. Следовательно, соединения, соответствующие данному изобретению, используют для лечения и предупреждения всех данных заболеваний, опосредованных α1- и α2-субъединицами рецептора ГАМКА.
Неограничивающими примерами данных заболеваний являются нарушения сна, предпочтительно бессонница, тревога и эпилепсия. Неограничивающими примерами соответствующих показаний для соединения, соответствующих данному изобретению, являются все данные заболевания или состояния, такие как бессонница или анестезия, при которых требуется вызвать сон, вызвать седативный эффект или вызвать мышечную релаксацию.
Раскрытие изобретения
В настоящем изобретении описан новый класс соединений, представленных формулой (I):
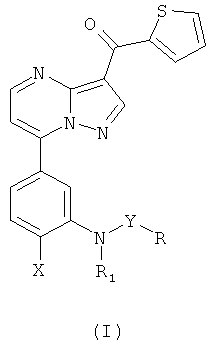
где R, R1, X и Y определены ниже, которые являются лигандами рецептора ГАМКА, и их фармацевтически приемлемые соли.
В отношении другого объекта настоящее изобретение направлено на разработку новых способов лечения или предупреждения тревоги, эпилепсии и нарушений сна, включая бессонницу, индуцирование седативно-снотворного эффекта, анестезии, сна и мышечной релаксации путем введения терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Синтетические способы получения указанных соединений и ряда промежуточных продуктов также входят в объем изобретения. Сами соответствующие промежуточные продукты также составляют другой объект изобретения.
Осуществление изобретения
Настоящее изобретение относится к галогенизированным пиразоло-[1,5-а]пиримидинам, в частности к новым [7-(3-амино-4-галогенил)-пиразоло[1,5-а]пиримидин-3-ил]-тиофен-2-ил-метанонацильным и сульфонильным соединениям формулы (I):
где
R представляет собой алкил(C1-C6);
R1 выбран из группы, состоящей из алкил(C1-C6)и алкинил(C1-C6);
Х представляет собой атом галогена и
Y выбран из группы, состоящей из -СО- и -SO2-;
и их фармацевтически приемлемым солям.
Предпочтительно, когда R означает метил, R1 выбран из группы, включающей метил и проп-2-инил, а X выбран из группы, включающей фтор и хлор.
Термин "фармацевтически приемлемая соль", используемый в данном контексте, охватывает любую соль, образованную из органических и неорганических кислот, таких как бромистоводородная, хлористоводородная, фосфорная, азотная, серная, уксусная, адипиновая, аспарагиновая, бензолсульфоновая, бензойная, лимонная, этансульфоновая, муравьиная, фумаровая, глутаминовая, молочная, малеиновая, яблочная, малоновая, миндальная, метансульфоновая, 1,5-нафталиндисульфоновая, щавелевая, пивалиновая, пропионовая, п-толуолсульфоновая, янтарная, винная кислоты и т.п.
Настоящее изобретение включает соединения:
N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид;
N-{2-хлор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид;
N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид;
N-{2-хлор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид и
N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-проп-2-инил-метансульфонамид.
Другой вариант осуществления настоящего изобретения состоит в представлении способа получения соединений формулы (I) и их фармацевтически приемлемых солей.
Соединения, соответствующие настоящему изобретению, могут быть использованы для лечения или предупреждения заболеваний, ассоциированных с модуляцией рецептора ГАМКА у млекопитающих, которое заключается во введении указанному нуждающемуся в этом млекопитающему эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Более конкретно, заболевания, ассоциированные с модуляцией рецептора ГАМКА, включают заболевания, ассоциированные с модуляцией рецептора α1-ГАМКА и/или модуляцией рецептора α2-ГАМКА. Неограничивающий перечень данных заболеваний включает тревогу, эпилепсию, нарушения сна, в том числе бессонницу, и т.п.
Другой вариант осуществления настоящего изобретения относится к применению соединения формулы (I) для лечения или предупреждения тревоги у нуждающегося в этом млекопитающего, которое включает введение указанному млекопитающему эффективного количества указанного соединения или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к применению соединения формулы (I) для лечения или предупреждения эпилепсии у нуждающегося в этом млекопитающего, которое включает введение указанному млекопитающему эффективного количества указанного соединения или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к применению соединения формулы (I) для лечения или предупреждения нарушений сна у нуждающегося в этом млекопитающего, которое включает введение указанному млекопитающему эффективного количества указанного соединения или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к применению соединения формулы (I) для лечения или предупреждения бессонницы у нуждающегося в этом млекопитающего, которое включает введение указанному млекопитающему эффективного количества указанного соединения или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к применению соединения формулы (I) для индуцирования седативно-снотворного эффекта у нуждающегося в этом млекопитающего, которое включает введение указанному млекопитающему эффективного количества указанного соединения или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к применению соединения формулы (I) для индуцирования анестезии у нуждающегося в этом млекопитающего, которое включает введение указанному млекопитающему эффективного количества указанного соединения или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к применению соединения формулы (I) для модуляции времени, необходимого для индуцирования сна и его продолжительности у нуждающегося в этом млекопитающего, которое включает введение указанному млекопитающему эффективного количества указанного соединения или его фармацевтически приемлемой соли.
Другой вариант осуществления настоящего изобретения относится к применению соединения формулы (I) для индуцирования мышечной релаксации у нуждающегося в этом млекопитающего, которое включает введение указанному млекопитающему эффективного количества указанного соединения или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к способу лечения или предупреждения у млекопитающего, страдающего от заболеваний, ассоциированных с модуляцией рецептора ГАМКА у млекопитающих, который заключается во введении указанному нуждающемуся в этом млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли совместно с фармацевтически приемлемыми разбавителями или носителями. Более подробно, заболевания, ассоциированные с модуляцией рецептора ГАМКА, включают заболевания, ассоциированные с модуляцией рецептора α1-ГАМКА и/или с модуляцией рецептора α2-ГАМКА. Неограничивающий перечень таких заболеваний включает тревогу, эпилепсию, нарушения сна, в том числе бессонницу, и т.п.
В данном контексте термин "млекопитающее" будет относиться к классу Mammalia высших позвоночных животных. Термин "млекопитающее" включает, без ограничения только этим, человека.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с терапевтически инертными носителями.
Другой вариант осуществления настоящего изобретения относится к способам получения промежуточных соединений формулы (VI):
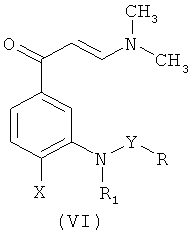
где R, R1, X и Y такие, как описано выше.
Конкретные промежуточные соединения формулы (VI), а именно:
N-[5-(3-диметиламино-акрилоил)-2-фтор-фенил]-N-метил-ацетамид;
N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метил-ацетамид;
N-[5-(3-диметиламино-акрилоил)-2-фтор-фенил]-N-метил-метан-сульфонамид;
N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метил-метан-сульфонамид и
N-[5-(3-диметиламино-акрилоил)-2-фтор-фенил]-N-проп-2-инил-метансульфонамид, составляют другой вариант осуществления настоящего изобретения.
Композиции включают подходящие для перорального, ректального и парентерального (в том числе подкожного, внутримышечного и внутривенного) введения, хотя наиболее подходящий путь будет зависеть от природы и тяжести состояния, которое лечат. Наиболее предпочтительный способ, соответствующий настоящему изобретению, представляет собой пероральный способ. Композиции могут быть удобно представлены в единичной лекарственной форме и получены любым из способов, хорошо известных в области фармации.
Активное соединение можно скомбинировать с фармацевтическим носителем в соответствии с принятыми методами составления фармацевтических смесей. Носитель может принимать широкий ряд форм в зависимости от формы препарата, требующегося для применения, например пероральной или парентеральной (включая внутривенные инъекции или инфузии). При изготовлении композиций для пероральной лекарственной формы можно использовать любую из обычных фармацевтических сред. Обычные фармацевтические среды включают, например, воду, гликоли, масла, спирты, вкусовые добавки, консерванты, красители и т.п. в случае пероральных жидких препаратов (таких как, например, суспензии, растворы, эмульсии и эликсиры); аэрозоли или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, скользящие вещества, связующие агенты, разрыхлители и т.п., в случае пероральных твердых препаратов (таких как, например, порошки, капсулы и таблетки), причем пероральные твердые препараты предпочтительны относительно пероральных жидких препаратов.
Вследствие простоты применения таблетки и капсулы представляют собой наиболее удобную пероральную единичную лекарственную форму, в данном случае используют твердые фармацевтические носители. При необходимости таблетки могут быть покрыты с помощью стандартных водных или неводных методов.
Подходящий интервал доз для применения составляет общую суточную дозу от приблизительно 0,01 мг до приблизительно 100,00 мг, которую принимают один раз в сутки или, при необходимости, в виде разделенных доз.
Соединения общей формулы (I) могут быть получены согласно реакции, показанной на Схеме 1.
Схема 1
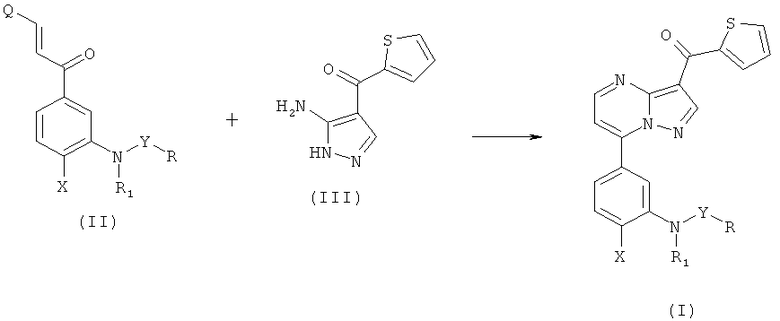
В промежуточных продуктах формулы (II), R, R1, X и Y такие, как определено для формулы (I) и Q представляет собой подходящую уходящую группу, выбранную из группы, включающей N(диалкил(C1-C6)), алкилтио(C1-C6) и алкокси(C1-C6). Предпочтительно, когда Q выбран из группы, включающей диметиламино-, метилтио- и метоксигруппу. Обработка полученных в результате соединений в форме свободного основания кислотой позволяет получить их соответствующие соли.
Реакцию аминопиразола (III) с соответствующим образом замещенным 1-арил-2-пропен-1-оном (II) проводят в инертном полярном протонном или непротонном растворителе, таком как ледяная уксусная кислота, этанол, метанол, диметилформамид или диметилсульфоксид, при температуре, находящейся в интервале от 50° до 130°C. Через несколько часов (время реакции) раствор удаляют и полученный остаток разделяют между водным раствором бикарбоната натрия и дихлорметаном. Сырец, полученный в результате выпаривания органического слоя досуха, можно очистить одним из следующих методов: (а) хроматографией на силикагеле с использованием этилацетата или дихлорметан/метанола в качестве элюента или (b) кристаллизацией в подходящем растворителе (этилацетате, этаноле, метаноле и т.п.).
Промежуточное соединение формулы (II), в котором Q означает диметиламиногруппу [промежуточное соединение (VI)] можно получить, следуя последовательности реакций, показанной на Схеме 2.
Схема 2
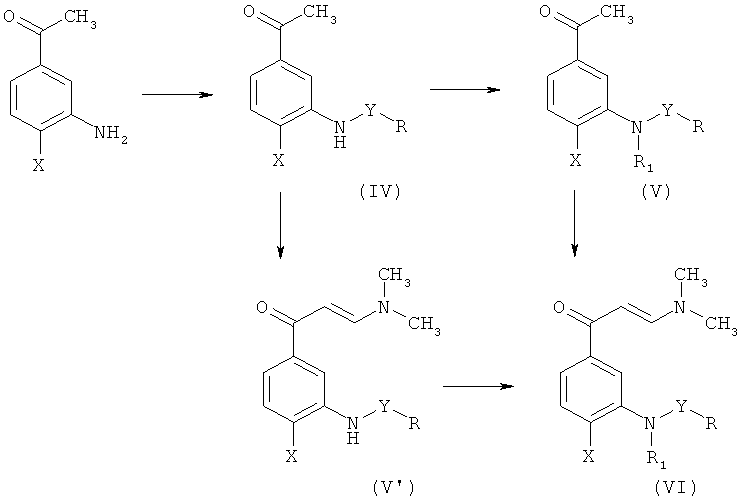
где R, R1, X и Y такие, как описано выше.
Промежуточные соединения формулы (IV), в которых Y представляет собой сульфонильную группу [промежуточные соединения (IV')] получают согласно способу, описанному в статье R.H.Uloth et al. (J. Med. Chem. 9, 88-96, 1966).
Алкилирование промежуточных соединений (IV), приводящее к промежуточным соединениям формулы (V), осуществляют посредством образования аниона и последующей реакции с алкилгалогенидом.
Энаминоны формулы (V') и (VI) получают посредством реакции соответствующих ацетофенонов (IV) и (V) соответственно с N,N-диметилформамиддиметилацеталем (DMFDMA) или реагентом Бредерека (трет-бутоксибис(диметиламино)метаном).
Промежуточные соединения формулы (II), в которой Q представляет собой диметиламиногруппу, Y означает сульфонил и R1 представляет собой метил [промежуточные соединения (VII)], могут быть альтернативно получены согласно Схеме 3.
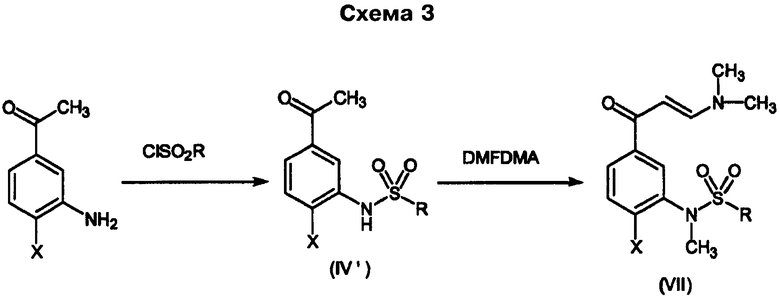
Превращение соединения (IV') в (VII) приводит к образованию энаминона и, одновременно, образованию N-метилсульфонамида как результата использования свойств N,N-диметилформамиддиметилацеталя в качестве метилирующего агента.
Промежуточные соединения формулы (II), в которой Q представляет собой диметиламиногруппу и R1 представляет собой метил (X), могут быть получены согласно Схеме 4.
Схема 4
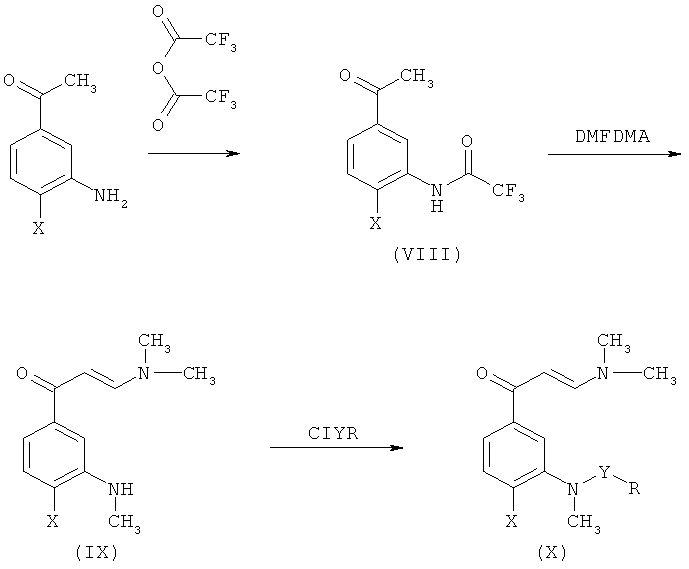
Преимущество данного способа основано на том факте, что образование сульфонамида или карбоксамида имеет место на последней стадии способа. В результате общее число стадий реакции уменьшают при получении больших партий продуктов. Более того, как показано на схеме, превращение соединения (VIII) в (IX) приводит к трем следующим реакциям в процессе, проводимом в одной реакционной емкости: (а) образование энаминона; (b) метилирование трифторацетамида и (с) дезацилирование с образованием N-метилированного амина. Последующая реакция соединения (IX) с соответствующим хлоридом сульфоновой кислоты или карбоновой кислоты приводит к получению промежуточных соединений (X).
Соединения, соответствующие настоящему изобретению, обладают высокой аффинностью к α1- и α2-рецепторам ГАМКА. Данные полученные in vitro результаты согласуются с результатами, полученными in vivo в тестах на седативно-снотворный эффект.
В соответствии с полученными результатами соединения, представленные в настоящем изобретении, проявили фармакологическую активность как in vitro, так и in vivo, которая аналогична или превышает активность соединений, соответствующих предшествующему уровню техники. Все данные результаты поддерживают их применение при заболеваниях или состояниях, модулируемых α1- и α2-рецепторами ГАМКА, таких как бессонница или анестезия, при которых необходимо индуцирование сна, индуцирование седативного эффекта или индуцирование мышечной релаксации. Более того, обнаружено, что при введении соединений, соответствующих настоящему изобретению, в низких дозах обнаруживают неожиданное повышение седативно-снотворной активности относительно достигаемой при использовании соединений, соответствующих предшествующему уровню техники (т.е. индиплона и залеплона и примеров 3 и 16 из WO 200501497), как продемонстрировано ниже.
Как будет показано ниже, установлены фармакологическая и цитотоксическая активности, метаболическая стабильность и фармакокинетический профиль соединений, соответствующих настоящему изобретению.
а) Фармакологические активности
1-Лиганд-связывающие анализы. Определение аффинности тест-соединений для α1- и α2-рецептора ГАМКА
Во время эксперимента используют самцов мышей Sprague-Dawley массой 200-250 г. После декапитации удаляют мозжечок (ткань, которая в основном содержит α1-рецептор ГАМКА) и спинной мозг (ткань, которая в основном содержит α2-рецептор ГАМКА). Мембраны получают согласно методу J.Lameh et al. (см. Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 24, 979-991, 2000) и Н.Noguchi et al. (см. Eur J Pharm, 434, 21-28, 2002) с незначительными модификациями. После взвешивания тканей их суспендируют в 50 мМ Трис·HCl (pH 7,4), 1:40 (об./об.) или сахарозе 0,32 М в случае спинного мозга, гомогенизируют и затем центрифугируют при 20000 g в течение 10 мин при 7°C. Полученную в результате пеллету ресуспендируют в тех же условиях и снова центрифугируют. Пеллету в заключение ресуспендируют в минимальном объеме и хранят при -80°C в течение ночи. На следующий день процесс повторяют до тех пор, пока конечную пеллету не ресуспендируют в соотношении 1:10 (об./об.) в случае мозжечка и в соотношении 1:5 (об./об.) в случае спинного мозга.
Аффинность определяют с помощью конкурентных тестов, используя флумазенил с радиоактивной меткой в качестве лиганда. Тесты проводят согласно методам, описанным в статье S.Arbilla et al. (см. Eur. J. Pharmacol., 130, 257-263, 1986) и в статье Y.Wu et al. (см. Eur. J. Pharmacol., 278, 125-132, 1995) с использованием 96-луночных планшетов для микротитрования. Инкубируют мембраны, содержащие исследуемые рецепторы, флумазенил (с радиоактивной меткой в конечной концентрации 1 нМ) и повышающиеся концентрации тест-соединений (в общем объеме 230 мкл в 50 мМ [pH 7,4] буфере Трис·HCl). Одновременно мембраны инкубируют только с флумазенилом с радиоактивной меткой (общее связывание 100%) и в присутствии повышенной концентрации флумазенила без радиоактивной метки (неспецифическое связывание, оценка в % лиганда с радиоактивной меткой). Реакции начинают с добавления лиганда с радиоактивной меткой с последующим инкубированием в течение 60 минут при 4°C. В конце периода инкубирования 200 мкл реакции переносят в планшет для мультискрининга (Millipore) и фильтруют с использованием вакуумного насоса, а затем трижды промывают холодным тест-буфером. Планшеты для мультискрининга снабжены фильтром GF/B, который удерживает мембраны, содержащие рецепторы и лиганд с радиоактивной меткой, который связан с рецепторами. После промывания планшеты оставляют до тех пор, пока они не высохнут. После высыхания добавляют сцинтилляционную жидкость и оставляют при перемешивании в течение ночи. На следующий день планшеты подсчитывают, используя сцинтилляционный счетчик Perkin-Elmer Microbeta.
Для анализа результатов процент специфического связывания для каждой концентрации тест-соединения подсчитывают следующим образом:
% специфического связывания = (X-N/T-N)×100
где,
X: количество связанного лиганда для каждой концентрации соединения.
Т: общее связывание, максимальное количество, связанное с лигандом с радиоактивной меткой.
N: неспецифическое связывание, количество лиганда с радиоактивной меткой, связанного неспецифическим путем независимо от используемого рецептора.
Каждую концентрацию каждого соединения тестируют в трех повторностях и их средние значения используют для определения экспериментальных значений % специфического связывания относительно концентрации соединения. Данные по аффинности выражают как % ингибирования при концентрациях 10-5 М и 10-7 М и получают Ki для ряда соединений, в котором рассчитывают соотношения между аффиностями α1 и α2. Результаты данных тестов приводят в таблицах 1 и 2. Преимущественно некоторые соединения, соответствующие настоящему изобретению, демонстрируют повышенную избирательность в качестве седативно-снотворных агентов относительно активности мышечной релаксации, что доказывают с помощью повышенного соотношения α2/α1 по сравнению с соединениями предшествующего уровня техники.
В данном контексте соотношение избирательности α2/α1 для соединения и примера получения 2 составляет 9,6 в отличие от 7,7 для индиплона и 5,0 для соединения из примера 3 в WO 2005014597, что, таким образом, приводит в результате к повышению избирательности на 25% и 92% соответственно. Следовательно, у настоящих соединений ожидают пониженные побочные эффекты.
2. Определение in vivo предсказанного седативно-снотворного действия
Эффекты данных соединений in vivo оценивают в тесте предсказанного седативно-снотворного эффекта на мышах (см. статьи D.J.Sanger et al., Eur. J. Pharmacol., 313, 35-42, 1996 и G.Griebel et al., Psychopharmacology, 146, 205-213, 1999).
Используют группы по 5-8 самцов мышей CD1 массой 22-26 г на момент тестирования. Тест-соединения вводят в однократных эквимолекулярных внутрибрюшинных дозах, суспендированные в 0,25% агара с одной каплей Твин 80 в объеме 10 мл/кг. В каждом способе тестируют две дозы. Контрольные животные получают только носитель. Используя Smart System (Panlab, S.L., Spain) регистрируют пройденное расстояние в см для каждой мыши через 5-минутные интервалы в течение 30 минут после внутрибрюшинного (ip) дозирования и 60 минут после перорального (po) дозирования. Рассчитывают процент сокращения пройденного расстояния у леченых животных относительно контрольных животных (первые 5 минут отбрасывают). Результаты данного теста приводятся в таблице 3.
Неожиданно, соответствующие соединения в настоящем изобретении показывают повышенную седативно-снотворную активность по сравнению с соединениями, соответствующими предшествующему уровню техники.
В частности, соединения, соответствующие настоящему изобретению, в низких дозах обусловливают более сильное повышение седативно-снотворной активности относительно достигаемой при использовании соединений, соответствующих предшествующему уровню техники (т.е. индиплон, залеплон и Примеры 3 и 16 из WO 2005014597). Это очень важно, поскольку возможно получить требуемый терапевтический эффект (т.е. седативно-снотворный) при использовании более низкой дозы с дополнительным преимуществом, состоящим в том, что могут быть сведены к минимуму близкие побочные эффекты.
Сравнение между соединениями, представленными в настоящем изобретении, и соответствующими соединениями, представленными в предшествующем уровне техники, показывает, что присутствие атома галогена в структуре, представленной формулой (I), дает возможность повышения седативно-снотворной активности, особенно в низких дозах. Так, например, при сравнении активности соединения, соответствующего Примеру 10 настоящего изобретения, с активностью, полученной при использовании соединения, соответствующего Примеру 16 из WO 2005014597, достигают повышения более чем 20% при использовании низкой дозы независимо от пути введения.
b) Цитотоксическая активность
Определение in vitro клеточной токсичности на HepG2 в течение 24 часов
Клетки HepG2 (гепатоцеллюларной карциномы человека) получают из Американской коллекции типовых культур (АТСС) и культивируют в минимальной основной среде Игла (Игл) со сбалансированным солевым раствором Игла, подведенным так, чтобы он содержал 1,87 мМ Glutamax™ I, 0,1 мМ ненезаменимых аминокислот, 1,0 мМ пирувата натрия, 100000 ед./л пенициллина, 10000 мкг/л стрептомицина, 90% сыворотку бычьих эмбрионов, 10%. Водный нерадиоактивный анализ выживаемости клеток Promega CellTiter 96® включает соединение тетразолия [3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий, внутреннюю соль (MTS). Превращение MTS в водный растворимый продукт формазана осуществляют с помощью ферментов дегидрогеназ, обнаруженных в метаболически активных клетках. Количество продукта формазана прямо пропорционально числу живых клеток в культуре.
Соединения растворяют в ДМСО до достижения исходной концентрации 100 мМ. Делают серийные разведения из данного маточного раствора в ДМСО для достижения концентраций 10, 1, 0,1 и 0,01 мМ. Маточный раствор и серийные разведения затем разводят 1:100 средой для культивирования клеток, чтобы получить шесть конечных концентраций для анализа 1000, 100, 10, 1, 0,1 и 0,01 мкМ. Конечная концентрация ДМСО во всех лунках составляет 1% об./об. Клетки HepG2 инкубируют с тест-соединениями в течение 24 часов. Относительную выживаемость клеток определяют спектрофотометрически при длине волны 490 нм после добавления красителя MTS и последующего инкубирования в течение одного часа. В качестве положительного контроля используют тамоксифен.
Процент поглощения образцов, обработанных тест-продуктом, сравнивают с необработанным образцом, чтобы рассчитать процент контроля. Результаты данного теста приводят в таблице 4.
Соответственно, соединения, полученные в примерах получения 2, 4, 6 и 8, неожиданно проявляют более низкую цитотоксичность, чем соединения, соответствующие предшествующему уровню техники, улучшая, таким образом, профиль безопасности соединений, соответствующих настоящему изобретению.
с) Метаболическая стабильность
Определение in vitro метаболической стабильности в цитозольной фракции гепатоцитов человека
Соединения растворяют в ДМСО, чтобы получить исходную концентрацию 10 мМ. Затем данный маточный раствор разводят растворителем и буфером, чтобы получить конечную концентрацию для анализа 5 мкМ. Соединения тестируют в одной концентрации 5 мкМ в двух повторностях, инкубируя с 1,0 мг/мл объединенного цитозоля человека (полученного из фирмы Xenotech ple) при 37°C. Метаболизм оценивают в присутствии или в отсутствие кофакторов и измеряют как уменьшение исходного соединение с помощью анализа ЖХ/МС (жидкостная хроматография/масс-спектрометрия) в точках времени 0, 60 и 120 минут. Затем рассчитывают процент остающегося исходного соединения. Используют общий метод ЖХ:
Результаты данного теста приводятся в таблице 5.
Неожиданно, некоторые соединения, соответствующие настоящему изобретению, проявляют повышенную метаболическую стабильность по сравнению с соединениями, соответствующими предшествующему уровню техники, предсказывая, таким образом, улучшенный фармакокинетический профиль для данных соединений.
d) Фармакокинетический профиль
Определение in vivo фармакокинетического профиля после введения однократной дозы
Соединение, полученное в примере получения 2, тестируют на фармакокинетический профиль после внутривенного введения. Индиплон используют как эталонное соединение. Для каждого соединения используют трех самцов крыс Sprague-Dawley массой 250-300 г. Отбор образцов осуществляют посредством пункции ретроорбитального синуса в следующих точках времени 2,5, 5, 30, 60, 120, 180, 300 и 420 мин после введения. Образцы хранят в ледяной бане до отделения плазмы. Животных анестезируют ингаляцией изофлурана при каждом отборе образца. Плазму отделяют центрифугированием (10 мин, 4°C, 4500 об/мин) и хранят при температуре ниже -70°C до проведения анализа.
Используют аналитический способ, основанный на экстракции каждого соединения путем экстракции типа жидкость-твердое вещество с последующим определением посредством ЖХ/МС или ЖХ/МС/МС с использованием внутреннего стандарта (IS).
Осуществляют расчет фармакокинетических параметров (AUC0-t = площадь под кривой от нуля до последней точки времени экстракции, Cl = клиренс, t1/2 = полупериод существования и Vd = объем распространения) согласно анализу без компартментализации. Результаты представлены в таблице 6.
Экспериментальные результаты демонстрируют совершенно иной фармакокинетический профиль для соединения, представленного в примере 2, по сравнению с соединением, соответствующим предшествующему уровню техники - индиплоном. Действительно, площадь под кривой на 57% больше у соединения, представленного в примере получения 2, указывая тем самым на повышенный уровень воздействия продукта; клиренс снижен на 20%, тогда как его полупериод существования увеличен на 76%, показывая тем самым замедленную скорость выведения, и конечный объем распространения выше на 212%, позволяя предположить широкое распространение в глубоких неводных отделах (т.е. головном мозге) по сравнению с индиплоном. Фармакокинетические параметры коррелируют с рядом фактов, обнаруженных в фармакологии животных. Например, в тесте in vivo седативно-снотворной активности на мышах (3 мкмоль/кг) процент ингибирования снижается с 74% (5 минут) до 67% (60 минут) для индиплона, напротив, указанный параметр остается постоянным на уровне 84% для соединения, полученного в примере получения 2. Указанные неожиданные фармакокинетические свойства показывают, что соединение, соответствующее настоящему изобретению, обеспечивает улучшенное качество сна, избегая, таким образом, ночных пробуждений и гарантируя здоровый и продолжительный сон.
Следующие неограничивающие примеры иллюстрируют объем настоящего изобретения.
Пример получения 1: N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-метилацетамид
3,3 г (16,9 ммоль) N-(5-ацетил-2-фторфенил)-ацетамида растворяют в 8,36 мл (7,49 г) (62,89 ммоль) N,N-диметилформамиддиметилацеталя и полученный в результате раствор нагревают с обратным холодильником в течение 6,5 часов. Избыток летучего реагента удаляют отгонкой при пониженном давлении, получая сырец, который кристаллизуют из этилацетата. Получают 3,32 г N-[5-(3-диметиламино-акрилоил)-2-фтор-фенил]-ацетамида в виде твердого вещества желтовато-белого цвета (выход 78,6%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 2,21 (3H, s), 2,89 (3H, s), 3,11 (3H, s), 5,65 (1H, d, J=12,8 Гц), 7,05-7,1 (1H, m), 7,62-7,68 (2H, m), 7,77 (1H, d, J=12,4 Гц), 8,71-8,73 (1H, m).
MC (ES) m/z=251 (MH+).
ВЭЖХ=99,8%.
1,5 г (5,99 ммоль) N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-ацетамида растворяют в 15 мл сухого N,N-диметилформамида. К образовавшемуся раствору при 0°C и в инертной атмосфере добавляют 0,29 г (7,31 ммоль) гидрида натрия. После перемешивания в течение 30 минут добавляют раствор 0,94 г (6,59 ммоль) йодистого метила в 5 мл сухого N,N-диметилформамида и продолжают перемешивание при комнатной температуре в течение 5 часов. Растворитель удаляют отгонкой при пониженном давлении. К полученному в результате остатку добавляют 30 мл дихлорметана и 10 мл воды. Разделяют два слоя и водный слой промывают 30 мл дихлорметана. Органические слои промывают 40 мл воды и сушат над сульфатом магния. Слой дихлорметана выпаривают досуха, получая масло, которое при кристаллизации из этилацетата дает 804 мг N-[5-(3-диметиламино-акрилоил)-2-фтор-фенил]-N-метил-ацетамида в виде желтовато-белого твердого вещества (выход 50,8%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 1,85 (3H, s), 2,94 (3H, s), 3,17 (3H, s), 3,22 (3H, s), 5,62 (1H, d, J=12,4 Гц), 7,16-7,25 (1H, m), 7,78-7,89 (3H, m).
MC (ES) m/z=265 (MH+).
ВЭЖХ=94,9%.
Пример получения 2: N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло-[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид
Смесь 0,073 г (0,38 ммоль) (5-амино-1Н-пиразол-4-ил)-тиофен-2-ил-метанона и 0,1 г (0,38 ммоль) N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-метилацетамида в 10 мл ледяной уксусной кислоты нагревают с обратным холодильником в течение 2,5 часов и затем растворитель удаляют отгонкой при пониженном давлении. К полученному в результате остатку добавляют 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделяют и водный слой промывают 10 мл дихлорметана. Органические слои промывают 10 мл воды и сушат над сульфатом магния. Слой дихлорметана упаривают досуха, получая масло, которое в присутствии этилацетата дает 112 мг N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамида в виде твердого вещества (выход 75%).
Данные 1H ЯМР-спектроскопии(400 МГц, CDCl3): δ 1,98 (3H, s,), 3,3 (3H, s), 7,13 (1H, d, J=4 Гц), 7,18-7,20 (1H, m), 7,42 (1H, t, J=8,8 Гц), 7,71 (1H, d, J=5,2 Гц), 8,02-8,08 (2Н, m), 8,12 (1H, dd, J=2,4 и 7,6 Гц), 8,71 (1H, s), 8,82 (1H, d, J=4 Гц).
MC (ES) m/z=395 (MH+).
ВЭЖХ=99,2%
Т.п. (температура плавления) = 165-167°C
Пример получения 3: N-[2-Хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метилацетамид
4,46 г (21,1 ммоль) N-(5-ацетил-2-хлорфенил)-ацетамида растворяют в 10,4 мл (9,34 г) (78,39 ммоль) N,N-диметилформамиддиметилацеталя и полученный в результате раствор нагревают с обратным холодильником в течение 6,5 часов. Избыток летучего реагента удаляют отгонкой при пониженном давлении, получая сырец, который кристаллизуют из этилацетата. Получают 4,53 г N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-ацетамида в виде твердого вещества желтовато-белого цвета (выход 80,5%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 2,24 (3H, s), 2,90 (3H, s), 3,12 (3H, s), 5,66 (1H, d, J=12,4 Гц), 7,38 (1H, d, J=8,8 Гц), 7,62 (1H, d, J=8,8 Гц), 7,69 (1H, s), 7,77 (1H, d, J=12,4 Гц), 8,7 (1H, s).
MC (ES) m/z=267 (MH+).
ВЭЖХ=98,3%.
1,0 г (3,75 ммоль) N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-ацетамида растворяют в 10 мл сухого N,N-диметилформамида. К образованному раствору при 0°C и в инертной атмосфере добавляют 0,18 г (4,57 ммоль) гидрида натрия. После перемешивания в течение 30 минут добавляют раствор 0,59 г (4,12 ммоль) йодистого метила в 3 мл сухого N,N-диметилформамида и продолжают перемешивание при комнатной температуре в течение 5 часов. Растворитель удаляют отгонкой при пониженном давлении. К полученному в результате остатку добавляют 30 мл дихлорметана и 10 мл воды. Разделяют два слоя и водный слой промывают 30 мл дихлорметана. Органические слои промывают 40 мл воды и сушат над сульфатом магния. Слой дихлорметана выпаривают досуха, получая масло, при кристаллизации которого из этилацетата-гексана получают 928 мг N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метилацетамида в виде твердого вещества желтовато-белого цвета (выход 88,16%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 1,79 (3H, s), 2,94 (3H, s), 3,17 (3H, s), 3,19 (3H, s), 5,61 (1H, d, J=12,4 Гц), 7,50 (1H, d, J=8,4 Гц), 7,79-7,85 (3H, m).
MC (ES) m/z=281 (MH+).
ВЭЖХ=100%.
Пример получения 4: N-{2-Хлор-5-[3-(тиофен-2-карбонил)-пиразоло-[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид
Смесь 0,083 г (0,43 ммоль) (5-амино-1Н-пиразол-4-ил)-тиофен-2-ил-метанона и 0,12 г (0,43 ммоль) N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метилацетамида в 12 мл ледяной уксусной кислоты нагревают с обратным холодильником в течение 1,5 часов и затем растворитель удаляют отгонкой при пониженном давлении. К полученному в результате остатку добавляют 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Два слоя разделяют и водный слой промывают 10 мл дихлорметана. Органические слои промывают 10 мл воды и сушат над сульфатом магния. Слой дихлорметана упаривают досуха, получая масло, которое в присутствии этилацетата дает 139 мг N-{2-хлор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метилацетамид в виде твердого вещества (выход 79%).
Данные 1H ЯМР-спектроскопии(400 МГц, CDCl3): δ 1,92 (3H, s,), 3,27 (3H, s), 7,15 (1H, d, J=4,8 Гц), 7,19-7,21 (1H, m), 7,70-7,71 (1H, m), 7,73 (1H, d, J=8,8 Гц), 8,02 (1H, dd, J=2,4 и 7,6 Гц), 8,06-8,07 (1H, m), 8,12 8 (1H, d, J=2 Гц), 8,71 (1H, s), 8,83 (1H, d, J=4 Гц).
MC (ES) m/z=411 (MH+).
ВЭЖХ=99,6%.
Т.п.=191-193°C.
Пример получения 5: N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-метилметан-сульфонамид
1,66 г (6,77 ммоль) N-(5-ацетил-2-фторфенил)-N-метилметан-сульфонамида растворяют в 3,35 мл (3,0 г) (25,18 ммоль) N,N-диметилформамиддиметилацеталя и полученный в результате раствор нагревают с обратным холодильником в течение 2,5 часов. Смесь охлаждают до комнатной температуры. К образованному твердому веществу добавляют 20 мл н-гексана и фильтруют, получая твердое вещество, которое кристаллизуют из этилацетата. Получают 1,37 г N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-метилметансульфонамида в виде твердого вещества желтовато-белого цвета (выход 67,4%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 2,92 (3H, s), 2,96 (3H, s), 3,15 (3H, s), 3,31 (3H, s), 5,61 (1H, d, J=12,8 Гц), 7,13-7,18 (1H, m), 7,78 (1H, d, J=12,8 Гц), 7,88-7,93 (2Н, m).
МС (ES) m/z=301 (MH+).
ВЭЖХ=97,99%.
Пример получения 6: N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид
Смесь 0,064 г (0,33 ммоль) (5-амино-1Н-пиразол-4-ил)-тиофен-2-ил-метанона и 0,1 г (0,33 ммоль) N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-метилметансульфонамида в 10 мл ледяной уксусной кислоты нагревают с обратным холодильником в течение 2,5 часов и затем растворитель удаляют отгонкой при пониженном давлении. К полученному в результате остатку добавляют 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Разделяют два слоя и водный слой промывают 10 мл дихлорметана. Органические слои промывают 10 мл воды и сушат над сульфатом магния. Слой дихлорметана выпаривают досуха, получая масло, которое в присутствии этилацетата дает 111 мг N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфон амида в виде твердого вещества (выход 77%).
Данные 1H ЯМР-спектроскопии(400 МГц, CDCl3): δ 3,01 (3H, s,), 3,39 (3H, s), 7,13 (1H, d, J=4,4 Гц), 7,18-7,20 (1H, m), 7,36-7,41 (1H, m), 7,70 (1H, dd, J=1,2 и 5,2 Гц), 8,07-8,09 (1H, m), 8,11-8,17 (2Н, m), 8,7 (1H, s), 8,80 (1H, d, J=4,8 Гц).
MC (ES) m/z=431 (MH+).
ВЭЖХ=98,6%.
Т.п.=194-196°C
Пример получения 7: N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метилметансульфонамид
1,0 г (4,04 ммоль) N-(5-ацетил-2-хлор-фенил)-метансульфонамида растворяют в 10 мл сухого N,N-диметилформамида и 2,69 мл (2,41 г) (20,19 ммоль) N,N-диметилформамид диметилацеталя. Полученный в результате раствор нагревают с обратным холодильником в течение 2 часов. Растворитель и избыток летучего реагента удаляют отгонкой при пониженном давлении, получая масло, которое в присутствии этилацетата дает 1,04 г сырца. Его хроматографируют (силикагель), используя этилацетат/2-пропанол в качестве растворителя. Получают 0,51 г
N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метил-метансульфонамида в виде твердого вещества желтовато-белого цвета (выход 40%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 2,9 (3H, s), 3,04 (3H, s), 3,15 (3H, s), 3,3 (3H, s), 5,61 (1H, d, J=12,4 Гц), 7,48 (1H, d, J=8,4 Гц), 7,78 (1H, d, J=12,8 Гц), 7,83 (1H, dd, J=8,8-1,6 Гц), 7,93 (1H, d, J=1,6 Гц).
MC (ES) m/z=317 (MH+).
ВЭЖХ=87,58%.
Пример получения 8: N-{2-хлор-5-[3-(тиофен-2-карбонил)-пиразоло-[1,5-а]пиримидин-7-ил]-фенил}-N-метилметансульфонамид
Смесь 0,076 г (0,39 ммоль) (5-амино-1Н-пиразол-4-ил)-тиофен-2-ил-метанона и 0,124 г (0,39 ммоль) (N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метилметансульфонамида в 10 мл ледяной уксусной кислоты нагревают с обратным холодильником в течение 1,5 часов и затем растворитель удаляют отгонкой при пониженном давлении. К полученному в результате остатку добавляют 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Разделяют два слоя и водный слой промывают 10 мл дихлорметана. Органические слои промывают 10 мл воды и сушат над сульфатом магния. Слой дихлорметана выпаривают досуха, получая масло, которое в присутствии этилацетата дает 128 мг
N-{2-хлор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамида в виде твердого вещества (выход 73%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 3,09 (3H, s,), 3,38 (3H, s), 7,15 (1H, d, J=4,8 Гц), 7,19-7,20 (1H, m), 7,68-7,71 (2H, m), 8,07-8,09 (2H, m), 8,19 (1H, d, J=2 Гц), 8,71 (1H, s), 8,82 (1H, d, J=4,4 Гц).
MC (ES) m/z=447 (MH+).
ВЭЖХ=98,1%.
Т.п.=241-243°C.
Пример получения 9: N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-проп-2-инилметансульфонамид
1,2 г (4,46 ммоль) N-(5-ацетил-2-фторфенил)-N-проп-2-инил-метансульфонамида растворяют в 3 мл (2,7 г) (22,58 ммоль) N,N-диметилформамиддиметилацеталя и нагревают с обратным холодильником в течение 2,5 часов. Смесь охлаждают при комнатной температуре и добавляют 20 мл н-гексана. Полученное масло хроматографируют (силикагель), используя этилацетат/2-пропанол в качестве растворителя. Получают 0,46 г твердого вещества желтовато-белого цвета. Данное твердое вещество кристаллизуют в этилацетате и получают 0,213 г N-[5-(3-диметиламино-акрилоил)-2-фтор-фенил]-N-проп-2-инил-метансульфонамида (выход 14,7%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 2,35 (1H, m), 2,92 (3H, s), 3,11 (3H, s), 3,15 (3H, s), 4,43 (2H, m), 5,61 (1H, d, J=12,8 Гц), 7,16-7,21 (1H, m), 7,79 (1H, d, J=12,8 Гц), 7,91-7,94 (1H, m), 8,01-8,04 (1H, m).
MC (ES) m/z=325 (MH+).
ВЭЖХ=91,63%.
Пример получения 10: N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло-[1,5-а]пиримидин-7-ил]-фенил}-N-проп-2-инил-метансульфонамид
Смесь 0,108 г (0,56 ммоль) (5-амино-1Н-пиразол-4-ил)-тиофен-2-ил-метанона и 0,198 г (0,61 ммоль) N-[5-(3-диметиламино-акрилоил)-2-фтор-фенил]-N-проп-2-инил-метансульфонамида в 10 мл ледяной уксусной кислоты нагревают с обратным холодильником в течение 2 часов и затем растворитель удаляют отгонкой при пониженном давлении. К полученному в результате остатку добавляют 15 мл дихлорметана и 10 мл насыщенного раствора бикарбоната натрия. Разделяют два слоя и водный слой промывают 10 мл дихлорметана. Органические слои промывают 10 мл воды и сушат над сульфатом магния. Слой дихлорметана выпаривают досуха, получая масло, которое в присутствии этилацетата дает 156 мг
N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-проп-2-инил-метансульфонамида в виде твердого вещества (выход 61%).
Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): δ 2,39 (1H, s), 3,16 (3H, s,), 4,50 (2H, s), 7,14 (1H, d, J=4,4 Гц), 7,18-7,20 (1H, m), 7,40-7,44 (1H, m), 7,70 (1H, m), 8,07-8,09 (1H, m), 8,18-8,21 (1H, m), 8,24-8,26 (1H, m), 8,7 (1H, s), 8,80 (1H, d, J=4,8 Гц).
MC (ES) m/z=455 (MH+).
ВЭЖХ=94,9%.
Т.п.=149-153°C.
Изобретение относится к новым галогенизированным пиразоло[1,5-а]-пиримидинам общей формулы (I) и их фармацевтически приемлемым солям, обладающим аффинностью в отношении α1-, α2 субъединиц рецептора ГАМКА. В формуле 
R представляет собой алкил(C1-C6); R1 выбран из группы, включающей алкил(C1-C6) и алкинил(C1-C6); Х представляет собой атом галогена и Y выбран из группы, включающей -CO- и -SO2-. Изобретение относится также к способу получения соединений формулы (I), фармацевтической композиции на их основе, к применению указанных соединений для получения лекарственного средства для лечения или предупреждения тревоги, эпилепсии, нарушений сна, в том числе бессонницы, а также для индуцирования седативно-снотворного эффекта, анестезии и мышечной релаксации. Кроме того, изобретение относится к промежуточным енаминоновым соединениям и способам их получения. 16 н. и 7 з.п. ф-лы, 6 табл., 10 пр.
1. Галогенизированные пиразоло[1,5-а]пиримидины, охватываемые общей структурной формулой (I)
где R представляет собой алкил(C1-C6);
R1 выбран из группы, включающей алкил(C1-C6) и алкинил(C1-C6);
X представляет собой атом галогена и
Y выбран из группы, включающей -СО- и -SO2-,
и их фармацевтически приемлемые соли.
2. Пиразоло[1,5-а]пиримидины по п.1, в котором R означает метил.
3. Пиразоло[1,5-а]пиримидины по п.1, в котором R1 означает метил.
4. Пиразоло[1,5-а]пиримидины по п.1, в котором R1 означает проп-2-инил.
5. Пиразоло[1,5-а]пиримидины по п.1, в котором Х означает фтор.
6. Пиразоло[1,5-а]пиримидины по п.1, в котором Х означает хлор.
7. Галогенизированные пиразоло[1,5-а]пиримидины, выбранные из группы, включающей:
N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид;
N-{2-хлор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамид;
N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид;
N-{2-хлор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-метансульфонамид и
N-{2-фтор-5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-проп-2-инил-метансульфонамид;
и его фармацевтически приемлемую соль.
8. Способ получения пиразоло[1,5-а]пиримидинов общей формулы (I) по п.1, в котором осуществляют взаимодействие промежуточного соединения формулы (II)

где R, R1, Х и Y такие, как определено для формулы (I), a Q означает соответствующую уходящую группу, выбранную из группы, включающей ди(алкилC1-C6)аминогруппу, алкилтио(C1-C6)группу и алкокси(C1-C6)группу, с промежуточным соединением формулы (III)
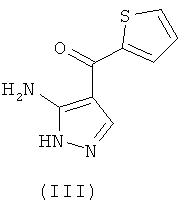
и альтернативно осуществляют взаимодействие образующихся в ходе реакции соединений в форме свободного основания с кислотой для получения солей указанных соединений.
9. Способ по п.8, в котором используют промежуточные соединения формулы (II), где Q выбран из группы, включающей диметиламиногруппу, метилтиогруппу и метоксигруппу.
10. Фармацевтическая композиция, обладающая аффинностью в отношении α1-, α2 субъединиц рецептора ГАМКА, включающая терапевтически эффективное количество пиразоло[1,5-а]пиримидинов по п.1, совместно с адекватными количествами фармацевтических наполнителей или носителей.
11. Применение пиразоло[1,5-а]пиримидинов по п.1 для получения лекарственного средства для лечения или предупреждения тревоги у млекопитающего.
12. Применение пиразоло[1,5-а]пиримидинов по п.1 для получения лекарственного средства для лечения или предупреждения эпилепсии у млекопитающего.
13. Применение пиразоло[1,5-а]пиримидинов по п.1 для получения лекарственного средства для лечения или предупреждения нарушений сна у млекопитающего.
14. Применение пиразоло[1,5-а]пиримидинов по п.1 для получения лекарственного средства для лечения или предупреждения бессонницы у млекопитающего.
15. Применение пиразоло[1,5-а]пиримидинов по п.1 для получения лекарственного средства для индуцирования седативно-снотворного эффекта у млекопитающего.
16. Применение пиразоло[1,5-а]пиримидинов по п.1 для получения лекарственного средства для анестезии у млекопитающего.
17. Применение пиразоло[1,5-а]пиримидинов по п.1 для получения лекарственного средства для модуляции времени, необходимого для индуцирования сна и его продолжительности у млекопитающего.
18. Применение пиразоло[1,5-а]пиримидинов по п.1 для получения лекарственного средства для индуцирования мышечной релаксации у млекопитающего.
19. Промежуточные енаминоновые соединения, выбранные из группы, включающей:
N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-метилацетамид;
N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метилацетамид;
N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-метил-метансульфонамид;
N-[2-хлор-5-(3-диметиламино-акрилоил)-фенил]-N-метилметан-сульфонамид и
N-[5-(3-диметиламино-акрилоил)-2-фторфенил]-N-проп-2-инил-метансульфонамид.
20. Способ получения енаминонового промежуточного соединения формулы (VI)

где R, R1, X и Y такие, как определено выше, в котором осуществляют взаимодействие соответствующего ацетофенона формулы (IV)
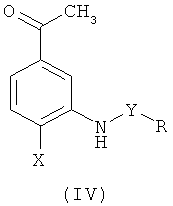
где R, Х и Y такие, как определено выше, с N,N-диметилформамид-диметилацеталем или трет-бутоксибис(диметиламино)метаном, а затем проводят алкилирование полученного в ходе вышеуказанной реакции енаминона формулы (V')
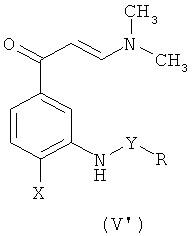
где R, Х и Y такие, как определено выше, через образование аниона с гидридным соединением, таким как NaH, после чего осуществляют взаимодействие полученного продукта реакции с алкилгалогенидом формулы ZR1, в которой Z означает атом галогена, а R1 означает алкил(C1-C6).
21. Способ по п.20, в котором гидридное соединение представляет собой гидрид натрия, а Z означает иод.
22. Способ получения енаминонового промежуточного соединения формулы (VI), в котором осуществляют взаимодействие соответствующего ацетофенона формулы (V)

где R, R1, X и Y такие, как определено выше, с N,N-диметилформамид-диметилацеталем или трет-бутоксибис(диметиламино)метаном.
23. Способ получения енаминонового промежуточного соединения формулы (VII)
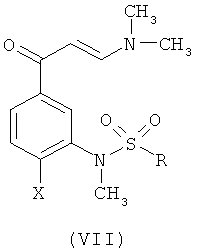
где R и Х такие, как определено выше, в котором осуществляют взаимодействие ацетофенона формулы (IV')
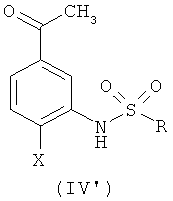
с N,N-диметилформамид-диметилацеталем.
| US 6399621 B1, 04.06.2002 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| US 4521422, 04.06.1985 | |||
| RU 2003129060 A, 10.04.2005. | |||

