Изобретение относится к получению новых постметаллоценовых катализаторов (каталитических систем) циглеровского типа.
К постметаллоценовым каталитическим системам циглеровского типа относится смесь соединений, содержащая не менее двух компонентов, способная после формирования каталитически активных центров вести процесс полимеризации олефиновых углеводородов. Основой системы служит комплексное соединение переходного металла, второй компонент - активатор или сокатализатор (в общем случае эту роль исполняет металлорганическое соединение непереходного металла, например метилалюмоксан, или слабокоординирующееся основание типа фенилборатов).
Данный тип катализаторов с наибольшей эффективностью может быть использован для получения как известных, так и новых полимерных материалов с необычными свойствами, к примеру термоэластопластичного полипропилена или сополимеров низших олефинов с высшими. Образующиеся полиолефины могут иметь степень изотактичности между 15-70%, определяемую по содержанию пентад [mmmm], высокий молекулярный вес от 150 000 до 4 000 000 г/моль, молекулярно-массовое распределение от 1,2 до 10.
Пост-металлоценовый катализ с участием комплексных соединений переходных металлов и сокатализаторов в виде алюминийорганических соединений, алкилалюмоксанов или перфторфенилборатов, является одним из активно развивающихся направлений в химии координационных и высокомолекулярных соединений. Это обусловлено, с одной стороны, необходимостью создания нового поколения дешевых, высокоэффективных и стереоспецифичных катализаторов полимеризации олефинов, сочетающих в себе достоинства гомогенных и классических гетерогенных катализаторов Циглера-Натта, а с другой, требованиями рынка к новым типам высокомолекулярных соединений с заданным комплексом полезных свойств.
В 1998 году независимо группами Брукхарта [Small, В.L.; Brookhart, M.J. Am. Chem. Soc. 1998, 720, 7143] и Гибсона [Britovsek, G.J.P.; Gibson, V.С.; Kimberley, В.S.; Maddox, P.J.; McTavish, S.J.; Solan, G.A.; White, A.J.P.; Williams, D.J. Chem. Commun. 1998, 849] были опубликованы первые работы по исследованию катализаторов полимеризации этилена на основе комплексов железа (II) и кобальта (II) с бис(имино)пиридильными лигандами (прекатализатор 1 на схеме 1) и полиметилалюмоксана (МАО) в качестве сокатализатора.
Схема 1
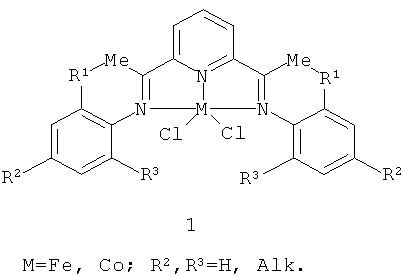
Такие системы показали очень высокую активность - до 11000
(кг ПЭ)·моль-1·ч-1·бар-1 (это выше активности металлоценовых комплексов металлов четвертой группы), - чем привлекли внимание исследователей и специалистов, работающих в химической промышленности. С использованием катализаторов данного типа получают линейный полиэтилен с широким разбросом молекулярных масс: от олигомеров до высокомолекулярного полиэтилена. Наличие и объем заместителей в фенильных кольцах при иминных атомах азота сильно влияют на активность катализатора и молекулярную массу полимера. К существенным недостаткам катализаторов данного типа относится отсутствие каталитической активности в реакции полимеризации пропилена и высших олефинов.
В 2002 г. появились сообщения [G.W.Coates, P.D.Hustad, S.Reinartz. Angew. Chem. Int. Ed., 2002, 41, 2237, Makio H, Kashiwa N, Fujita T. Adv. Syth. Catal. 2002. 344, 1, 1] о синтезе новых высокоактивных катализаторов полимеризации олефинов на основе комплексов металлов четвертой группы с бис(феноксииминовыми) лигандами (катализатор 2 на схеме 2).
Схема 2
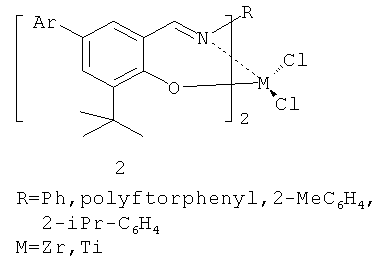
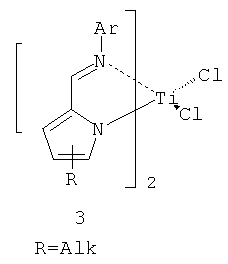
В зависимости от заместителей активность представленных катализаторов, названных постметаллоценовыми катализаторами FI, достигала и даже превышала активность металлоценовых систем. Позже появились высокоэффективные бис(пирролилиминовые) прекатализаторы 3 (схема 2).
Наиболее перспективные прекатализаторы данного типа в присутствии МАО характеризуются следующими значениями активностей: 3300 (кг ПЭ)·моль-1·ч-1·бар-1 для титанового комплекса; 6500 (кг ПЭ)·моль-1·ч-1·бар-1 для гафниевого и 51900 (кг ПЭ)·моль-1·ч-1·бар-1 для циркониевого комплекса (при температуре 25°C и времени полимеризации 5 минут). Максимум активности в присутствии МАО достигается при температуре 40°С (58700 (кг ПЭ)·моль-1·ч-1·бар-1); при этом получен полиэтилен с молекулярной массой Mw~104. Отличительной чертой катализаторов группы FI является возможность варьировать их электронные и стерические характеристики путем изменения состава и структуры лигандов, что позволяет изменять каталитические свойства в очень широких пределах («настройка» на синтез полиолефина с определенными характеристиками). Необходимо, однако, отметить, что высокая активность этих катализаторов наблюдается лишь в течение первых 5-10 мин, а максимальное время их работы не превышает 30 минут.
Другим типом постметаллоценовых прекатализаторов являются соединения металлов 4 группы с тридентатным ONO лигандом [Z.Ziniuk, I.Goldberg, M.Kol Inorganic Chemistry Communications, V 2, N11, 1999, Р.549-551], общей формулы LMCl2 (формула 4, схема 3).
Схема 3
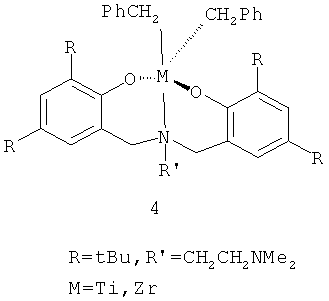
Данный тип веществ характеризуется высокой активностью в полимеризации этилена, пропилена и гексена-1. В полимеризации гексена-1 активность составляет 50 кг/моль Zr·ч. Как видно из структурной формулы, эти вещества содержат металлорганический фрагмент со связью М-С, что, с одной стороны, облегчает процесс превращения прекатализатора в истинную каталитическую частицу в процессе формирования каталитической системы, но с другой, усложняет и удорожает их синтез.
Наиболее близкой каталитической системой (прототипом) к предложенной является система, в которой в качестве соединения переходного металла используется титановый хлоридный комплекс с хиральными лигандами типа (4R,5R)-2,2-диметил-α,α,α',α'-тетрафенил-1,3-диоксолан-4,5-диметанола 5 общей формулы LMCl2 (схема 4), а в качестве сокатализатора - МАО [Изв. АН. Сер. хим. 2005, Т.54. №10. С.2348, Белоконь Ю.Н., Гагиева С.Ч., Сухова Т.А., Дмитриев А.В., Лысенко К.А., Бравая Н.М., Булычев Б.М., Зеебах Д.].
Схема 4
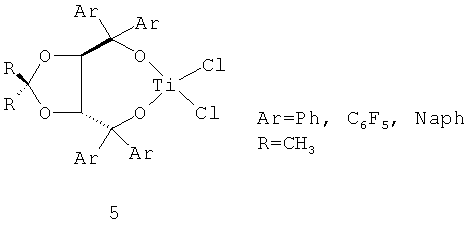
Данный тип комплексов, после активации метилалюмоксаном, образует систему, которая катализирует реакцию полимеризации этилена с активностью до 530 кг ПЭ(моль Ti·ч·атм)-1.
Дешевизна и простота синтеза комплексов LMCl2 делает эти соединения перспективными для разработки новых гомогенных каталитических систем для полимеризации олефинов практически всех типов.
Однако гомогенные каталитические системы, используемые для проведения полимеризации в газовой или суспензионной фазе, обладают рядом существенных недостатков. В частности, для активации всех металлоценовых и постметаллоценовых прекатализаторов необходимо использовать большой (от 1:1000 до 1:10 000) избыток МАО по отношению к соединению переходного металла. Стоимость МАО на несколько порядков выше стоимости обычных алюминийорганических соединений, используемых в классическом катализе Циглера-Натта и в существенно меньших количествах, что заметно ухудшает экономику всего каталитического процесса. Далее, активные центры, образующиеся в каталитических системах LMCl2+MAO, характеризуются низкой стабильностью, что приводит к их быстрой дезактивации в процессе полимеризации (5-20 мин). Этот недостаток в наибольшей степени присущ гомогенным системам, поэтому все реальные технологии ориентированы на использование гетерогенных систем и отсюда на иммобилизацию гомогенных катализаторов на носители различной природы несмотря на заметное снижение активности таких катализаторов. Использование гетерогенных катализаторов позволяет получать полиолефины с определенным гранулометрическим составом.
Задачей изобретения является устранение всех присущих известному техническому решению недостатков, что достигается получением гетерогенной постметаллоценовой каталитической системы, способной вести полимеризацию этилена, пропилена и высших альфа-олефинов, а также сополимеризацию вышеперечисленных мономеров в любой комбинации.
Поставленная задача решается получением каталитической системы для синтеза полиолефинов, содержащей галоидный комплекс металла 4 группы периодической системы с полидентантным лигандом (из числа производных (4R,5R)-1,1-R1,R2-α,α,α',α'-тетра(C6R3R4R5R6R7)-1,3-диоксолан-4,5-диметанола или 2-(2-гидрокси-2,2-дифенилэтиламино)-1,1-дифенилэтанола и алюминийорганическое соединение, отличающейся тем, что она дополнительно содержит, по меньшей мере, один галогенид металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий и алюминий. Молярное соотношение между галоидным комплексом металла 4 группы, галогенидом металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий и алюминий, и алюминийорганическим соединением находится в диапазоне 1:0,1-500:10-5000, предпочтительно 1:1-300:100-1000.
Лиганд в качестве заместителей содержит R1 и R2 группы, одинаковые или различные, которые выбирают из группы, включающей замещенный или незамещенный C1-C20 линейный или разветвленный алкил, С3-С20 циклоалкил, С6-С20 арил и С10-С20 конденсированные ароматические группы.
В частных воплощениях изобретения поставленная задача решается тем, что лиганд в качестве заместителей R3-R7 содержит, по меньшей мере, один заместитель, выбранный из группы, включающей алкил (С1-С5), арил, фтор, трифторметил.
Первоначально переходный металл связан с атомами галогена, которые впоследствии замещаются на алкильные (арильные или бензильные) группы под действием активатора. Предлагаемая каталитическая система в качестве такого активатора содержит, по меньшей мере, одно алюминийорганическое соединение, выбранное из группы, включающей AlR3 (например, триметилалюминий, триэтилалюминий, три-н-пропилалюминий, три-н-бутилалюминий, триизобутилалюминий), R2AlHal (например, диэтилалюминийхлорид) и (Al(R)O)n, где R - линейная или разветвленная алкильная труппа, или любую их комбинацию. В Европейских патентах EU 1178057 A2 и WO 0246247 A2 в качестве активатора также используются разветвленные или циклические алюминоксаны (формулы 6 и 7):
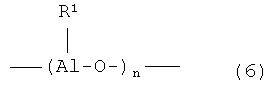
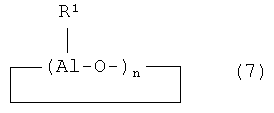
где R1 включает замещенный или незамещенный С1-С20 линейный или разветвленный алкил, С3-С20 циклоалкил.
Кроме того, эти активаторы могут быть комбинированы со специфичными соединениями, представленными формулами 8, 9, 10, которые могут быть использованы в сочетании с активаторами 6 и 7.
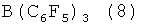

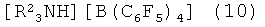
В формулах 9, 10 R2 представляет собой C1-C4 алкильные или арильные группы.
Поставленная задача также решается способом получения каталитической системы, в соответствии с которым последовательно осуществляют следующие стадии: (а) добавление к полидентантному лиганду из числа производных ТАДДОЛа или иминодиэтанола металлоорганического производного металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий или алюминий с получением полупродукта, (б) последующее смешение полученного в соответствии со стадией (а) полупродукта с галогенидом металла из 4 группы с получением смеси комплекса галогенида металла 4 группы с одним из вышеперечисленных лигандов и галогенида металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий или алюминий, (в) добавление к полученной смеси активатора - алюминийорганического соединения.
В систему после осуществления стадии (б) желательно дополнительно ввести тонкодисперсный галогенид металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий или алюминий, как минимум в эквимолярном по отношению к хлоридному комплексу металла 4 группы количестве, после чего осуществляют стадию (в).
Введение в состав каталитической системы высокодисперсного галогенида металла 1, 2, 3, 12 или 13 группы позволяет значительно повысить каталитическую активность и снизить количество дорогостоящего сокатализатора - метилалюмоксана.
Самоиммобилизованное соединение образуется при последовательном действии на полидентатные органические лиганды, содержащие донорные атомы кислорода, азота, металлорганического соединения 1, 2, 3, 12 или 13 групп, и затем добавлением к смеси галогенида металла 4 группы периодической системы. Каталитическая система образуется при действии на самоиммобилизованное на галогениды металла соединение алюминийорганического соединения, используемого в качестве активатора. Для создания большего числа активных центров на поверхности носителя в систему может вводиться дополнительное количество мелкодисперсного галогенида металла 1, 2, 3, 12 или 13 групп или индивидуальный галогенид добавляется к готовой форме постметаллоценового комплекса.
Поставленная задача также решается полиолефином, получаемым полимеризацией этилена или другого альфа-олефина с использованием вышеописанной каталитической системы.
Синтезы комплексов проводили в атмосфере аргона. Тетрагидрофуран, дихлорметан, толуол, изопропанол, гексан и этилацетат марок «х.ч.»; TiCl4 фирмы Fluka дополнительно перегоняли в атмосфере аргона, SiO2, дибензофуран, использовали фирмы Fluka. Галогениды металла (LiCl, MgCl2, AlCl3, ScCl3, ZnCl2) фирмы Aldrich дополнительно подвергали механическому воздействию в шаровой мельнице в течение 1 часа в атмосфере аргона.
Спектры ЯМР растворов лигандов в CDCl3 записывали на приборах "Bruker WP-300" и "Bruker АМХ-400", ИК спектры - на спектрофотометре "Magna-IR 750". Оптическое вращение измеряли на поляриметре "Perkin-Elmer 241". Элементный анализ проводился на приборах "Carlo Erba-1106" и "Carlo Erba-1108".
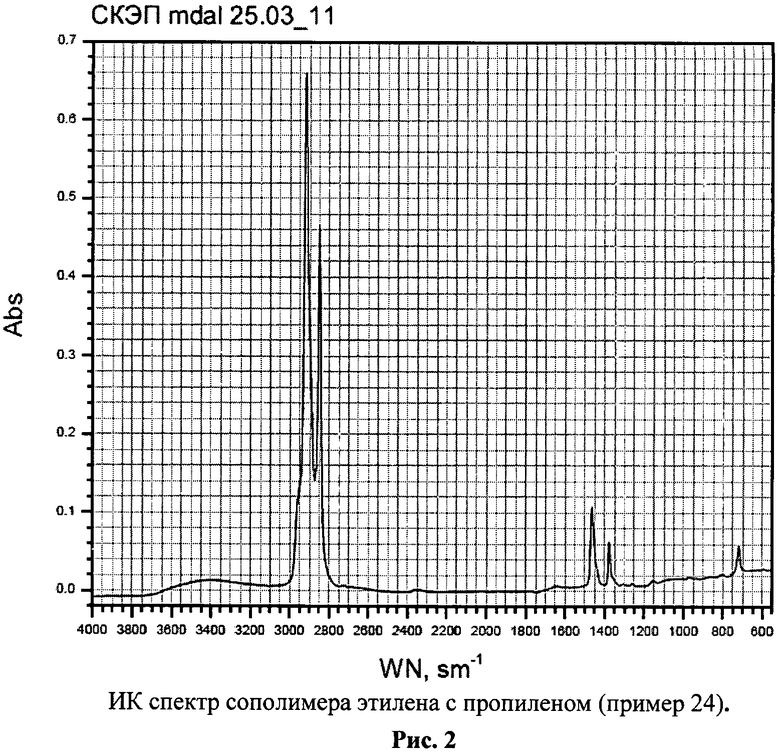
Содержание этилена в сополимерах находили с помощью соотношения оптических плотностей полос поглощения D1170 и D720 в ИК спектрах (D1170/D720) [Kissin Yu.V., Advances in Polym. Sci. 1974. V.15. P.92], а также из спектров ЯМР 13С, согласно [Soga K., Shiono T., Doi Yo., Polym. Bul., 1983. №10. P.168., Kakugo M., Naito Yu., Mizunumo K., Miyatake Т., Macromolecules. 1982. V.15. №4. P.1150].
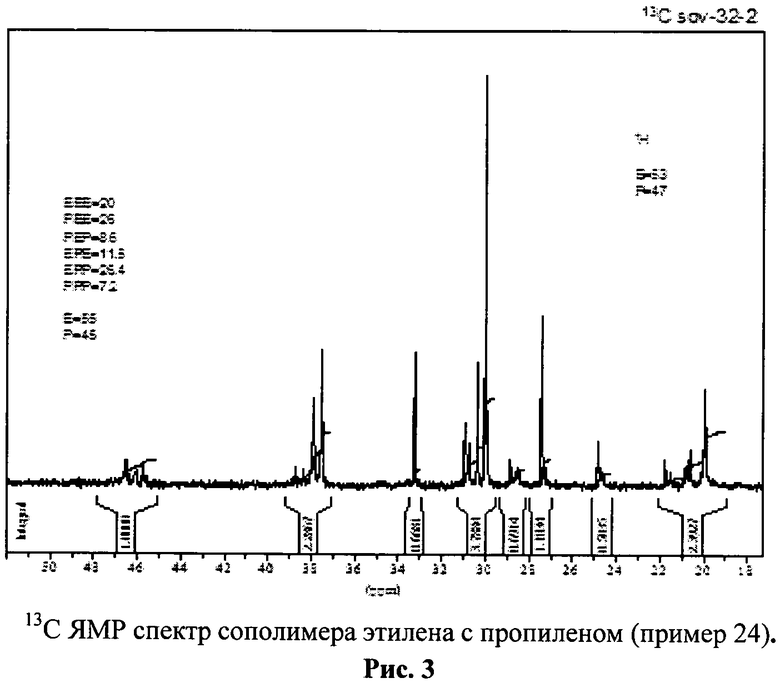
Из спектров ЯМР 13С полимерных образцов определяли также содержание стерических пентад и константы сополимеризации. Спектры 13С ЯМР 5%-ных растворов полимеров в о-дихлорбензоле записывали на приборе "Bruker AVANCE-400" (частота 100.613 МГц) при 90°C.
Теплофизические характеристики (температуру плавления Тпл, теплоту плавления ΔНпл, температуру кристаллизации Ткр и теплоту кристаллизации ΔНкр) полимеров определяли методом ДСК на анализаторе DSC-7 "Perkm-Elmer" для образцов массой 3-8 мг при скорости сканирования 10 град/мин.
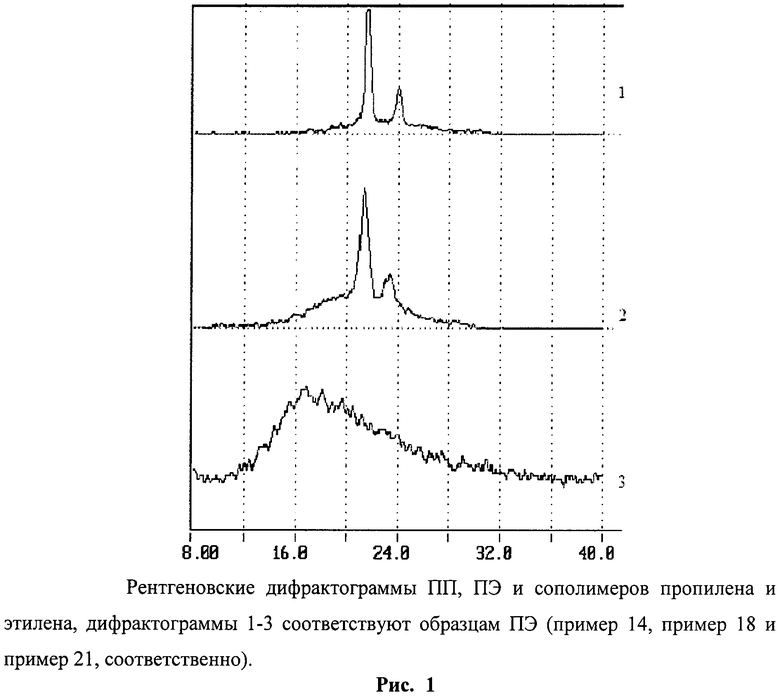
Рентгеновские спектры насцентных полимеров записывали на дифрактометре "ДРОН-2" (CuKα-излучение, Ni-фильтр, скорость сканирования 1 град (2θ)/мин). Степень кристалличности χ образцов находили по соотношению интегральной интенсивности кристаллической составляющей и общей интенсивности, α-модификацию ПП идентифицировали по рефлексу (130)α при 2θ=18,6° [Turner-Jones A., Aizewood J.M., Beckert D.R., Macromol. Chem. 1964. B.75. №1. S.134, Turner-Jones A., Polymer. 1971. V.12. №8. Р.487]. Отнесение рефлексов ПЭ осуществляли согласно [Мартынов М.А., Вылегжанина К.А. Рентгенография полимеров. Л: Химия, 1972].
Гель-хроматограммы образцов полимеров получали на гель-хроматографе "Waters 150-С" с использованием µ-styragel НТ колонки в 1,2,4-трихлорбензоле при 130°C. Средние ММ рассчитывали по универсальной калибровочной кривой с использованием ПС-стандартов.
Образцы для испытания деформационно-прочностных свойств готовили прессованием полимеров при 190°C и давлении 10 МПа при скорости охлаждения расплава 20 град/мин. В качестве стабилизатора использовали антиоксидант «Ирганокс» в количестве 0.5 мас.%. Испытания на растяжение проводили при 20°C на машине "Instron 1122" на образцах сечением 0.75×5 мм, длина базы 35 мм. Режим испытаний: растяжение со скоростью 500 мм/мин до 100% деформации, обратный ход траверсы с той же скоростью до нулевой величины растягивающего усилия, повторное растяжение образца со скоростью 500 м/мин до разрыва.
Для характеристики эластомерных свойств материала использовали величину остаточного удлинения ε300=(L1-L0)×100/L300 (%), где L1 - длина образца после снятия нагрузки при удлинении на 300%, L0 - длина исходного образца, L300 - деформация растяжения.
Синтез прекатализаторов и катализаторов
Пример 1. Синтез (4R,5R)-2,2-диметил-α,α,α',α'-тетра(перфторфенил)-1,3-диоксолан-4,5-диметанола.
В двугорлую колбу, высушенную и заполненную аргоном, поместили 1.25 мл перфторбензола (0.01 моль), растворенного в 30 мл эфира, при температуре -78°C и при перемешивании прибавили 1 мл (0,00892 моль) бутиллития (10 М раствора в гексане). Перемешивание продолжали 1 час, затем прибавили 0,62 мл диэтилового эфира (R,R) винной кислоты (0,0025 моль). Температуру постепенно довели до комнатной и реакционную смесь перемешивали 12 часов. Нейтрализовали насыщенным раствором хлорида аммония, органический слой отделяли, упарили, продукт выделяли хроматографированием на колонке 2 см × 15 см (SiO2, элюент эфир:гексан = 1:5), перекристаллизовали из метанола. Выход целевого продукта 0.7 г (24%), Т.пл.=106°C; [α]D 25=-18.4 (с=1, CHCl3). Спектр ЯМР 1Н (d-CDCl3) (δ, м.д.): 5,61 (с, 2Н-ОН), 4,36 (с, 2Н), 1,6 (с, 6Н). Найдено: С 45,05; Н 1,37%. Для C31H10O4F20 вычислено: C 45,20; H 1,18%.
Пример 2. Синтез [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорида.
В двугорлую колбу, снабженную магнитной мешалкой, в атмосфере аргона помещали (4R,5R)-2,2-диметил-α,α,α',α'-тетра(перфторфенил)-1,3-диоксолан-4,5-диметанол (0.16 г, 0.0002 моля), 10 мл толуола, затем при -78°С прибавляли по каплям 0.042 мл (0.00042 моля) 10 М раствора бутиллития в н-гексане. После этого температуру реакционной среды медленно доводили до комнатной, перемешивали 4 ч, охлаждали до -40°C, прибавляли TiCl4 (0.02 мл, 0.00020 моля) и смесь отфильтровывали, органический слой упаривали, продукт перекристаллизовали из толуола. Выход - 0.16 г, 87%. Найдено, %: С 39,29; Н 0,92; Ti 4,95; Cl, 7.29; для C31H8TiF20Cl2 вычислено, %: С 39.48; Н 0.85; Ti 5.08; Cl, 7.52. Т. пл.=294°С; [α]D RT=+18.40 (с=1, толуол). Спектр ЯМР 1Н (d8-толуол), (δ, м.д.): 4.69 (с, 2Н, СН3); 1.43 (с, 6Н, СН3).
Пример 3. Синтез [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол] титан (IV) дихлорида, иммобилизованного на хлорид лития.
В двухгорлую колбу, снабженную магнитной мешалкой, в атмосфере аргона помещали (4R,5R)-2,2-диметил-α,α,α',α'-тетра(перфторфенил)-1,3-диоксолан-4,5-диметанол (0.16 г, 0.00020 моля), 10 мл толуола, затем при -78°C прибавляли по каплям 0.042 мл (0.00042 моля) 10 М раствора бутиллития в н-гексане. После этого температуру реакционной среды медленно доводили до комнатной, перемешивали 4 ч, охлаждали до -40°C, прибавляли TiCl4 (0.02 мл, 0.00020 моля), медленно доводили до комнатной температуры, органический слой упаривали. Выход - 0.25 г, 88%. Найдено, %: С, 37.12; Н, 0.84 Ti, 4.47; Cl, 13.45; для C31H8O4Li2TiF20Cl4 вычислено, %: C, 37.20; H, 0.77; Ti, 4.56; Cl, 13.77. Т.пл.=294°C; [α]D RT=+18.40 (с=1, толуол). Спектр ЯМР 1Н (d8-толуол), (δ, м.д.): 4.69 (с, 2Н, СН3); 1.43 (с, 6Н, СН3).
Пример 4. Синтез титанового комплекса [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорид, иммобилизованного на хлорид магния.
В двугорлую колбу, снабженную магнитной мешалкой, в атмосфере аргона помещали (4R,5R)-2,2-диметил-α,α,α',α'-тетра(перфторфенил)-1,3-диоксолан-4,5-диметанол (0.20 г, 0.00024 моля), 10 мл толуола, затем при -78°C прибавляли по каплям 0.43 мл (0.00070 моля) 1,9 М раствора бутилмагнийхлорида в диэтиловом эфире. После этого температуру реакционной среды медленно доводили до комнатной температуры, упаривали эфир в противотоке аргона. Реакционную смесь охлаждали до -40°C, прибавляли TiCl4 (0.03 мл, 0.00024 моля), медленно доводили до комнатной температуры, упаривали органический слой. Выход - 0.25 г, 76%. Найдено, %: C, 35.69; H, 0.72; Ti, 4.55; Cl, 13.39; для C31H8O4MgTiF20Cl4 вычислено, %: С, 35.91; Н, 0.77; Ti, 4.54; Cl, 13.71. Т.пл.=249°C. Спектр ЯМР 1Н (d8-толуол), (δ, м.д.): 4.69 (с, 2Н, СН3); 1.43 (с, 6Н, СН3). Полученный in-situ комплекс, без дополнительной очистки, был использован в качестве прекатализатора.
Пример 5. Синтез титанового комплекса [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорид, иммобилизованного на хлорид алюминия.
В двугорлую колбу, снабженную магнитной мешалкой, в атмосфере аргона помещали (4R,5R)-2,2-диметил-α,α,α',α'-тетра(перфторфенил)-1,3-диоксолан-4,5-диметанол (0.20 г, 0.00024 моля), 10 мл толуола, затем при -78°C прибавляли по каплям 0.72 мл (0.00072 моля) 1,0 М раствора диэтилалюминийхлорида в гексане. После этого температуру реакционной среды медленно доводили до комнатной температуры, упаривали эфир в противотоке аргона. Реакционную смесь охлаждали до -40°C, прибавляли TiCl4 (0.03 мл, 0.00024 моля), медленно доводили до комнатной температуры, упаривали органический слой. Выход - 0.16 г, 76%. Найдено, %: C, 34.47; H, 0.71; Ti, 4.31; Cl, 16.22; для C31H8O4AlTiF20Cl5 вычислено, %: С, 34.60; Н, 0.74; Ti, 4.37; Cl, 16.51. Т.пл.=269°С. Спектр ЯМР 1Н (d8-толуол), (δ, м.д.): 4.69 (с, 2Н, СН3); 1.43 (с, 6Н, СН3). Полученный in-situ комплекс, без дополнительной очистки, был использован в качестве катализатора.
Пример 6. Синтез титанового комплекса [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорид, иммобилизованного на хлорид цинка.
В двугорлую колбу, снабженную магнитной мешалкой, в атмосфере аргона помещали (4R,5R)-2,2-диметил-α,α,α',α'-тетра(перфторфенил)-1,3-диоксолан-4,5-диметанол (0.20 г, 0.00024 моля), 10 мл толуола, затем при -78°C прибавляли по каплям 0.37 мл (0.00070 моля) 1,9 М раствора диэтилцинка в диэтиловом эфире. После этого температуру реакционной среды медленно доводили до комнатной температуры, упаривали эфир в противотоке аргона. Реакционную смесь охлаждали до -40°C, прибавляли TiCl4 (0.03 мл, 0.00024 моля), медленно доводили до комнатной температуры, упаривали органический слой. Выход - 0.16 г, 76%. Найдено, %: С, 32.55; Н, 0.72; Ti, 4.15; Cl, 12.37; для C31H8O4ZnTiF20Cl4 вычислено, %: С, 32.60; Н, 0.70; Ti, 4.12; Cl, 12.44. Т.пл.=270°С. Полученный in-situ комплекс, без дополнительной очистки, был использован в качестве прекатализатора.
Пример 7. Синтез 2-(2-гидрокси-2,2-дипентафторфенилэтиламино)-1,1-дипентафторфенилэтанола.
В двугорлую колбу, высушенную и заполненную аргоном, поместили 1.25 мл перфторбензола (0.01 моль), растворенного в 30 мл эфира при температуре -78°С, и при перемешивании прибавили 1.14 мл (0.0102 моль) бутиллития (10 М раствор в гексане). Перемешивание продолжали 1 час, затем прибавили 0,48 г диэтилового эфира аминодиуксусной кислоты (0.0025 моль). Температуру постепенно довели до комнатной и реакционную смесь перемешивали 12 часов. Нейтрализовали насыщенным раствором хлорида аммония, органический слой отделяли, упарили, продукт выделяли хроматографированием на колонке 2 см × 25 см (SiO2, элюент эфир:гексан = 1:5), перекристаллизовали из метанола. Выход 0.84 г, 43.5%, Т.пл.=186°C. Спектр ЯМР 1Н (d-CDCl3) (δ, м.д.): 3.70 (с, СН2-4Н), 4.60 (с, 2Н-ОН). Найдено: С 43.45; Н 0.87 N 1.77%. Для C31H10O4F20 вычислено: С 43.71; Н 0.92, N 1.82%.
Пример 8. Синтез титанового комплекса [2-(2-гидрокси-2,2-дипентафторфенилэтиламино)-1,1-дипентафторфенилэтанола]титан (IV) дихлорид.
В двугорлую колбу, снабженную магнитной мешалкой, в атмосфере аргона помещали 2-(2-гидрокси-2,2-дипентафторфенилэтиламино)-1,1-дипентафторфенилэтанол (0.15 г, 0.0002 моля), 10 мл толуола, затем при -78°С прибавляли по каплям 0.042 мл (0.00042 моля) 10 М раствора бутиллития в н-гексане. После этого температуру реакционной среды медленно доводили до комнатной, перемешивали 4 ч, охлаждали до -78°С, прибавляли TiCl4 (0.02 мл, 0.0002 моля) и смесь отфильтровывали, органический слой упаривали, продукт перекристаллизовали из толуола. Выход - 0.16 г, 87%. Найдено, %: С, 39.29; Н, 0.92; Ti, 4.95; Cl, 7.29; для C31H8O4TiF20Cl2 вычислено, %: С, 39.48; Н, 0.85; Ti, 5.08; Cl, 7.52. Т.пл.=294°С. Спектр ЯМР 1Н (d8-толуол), (δ, м.д.): 3.58 (с, СН2-4Н).
Пример 9. Синтез титанового комплекса [2-(2-гидрокси-2,2-дипентафторфенилэтиламино)-1,1-дипентафторфенилэтанола] титан (IV) дихлорид, иммобилизованного на хлорид лития.
В двухгорлую колбу, снабженную магнитной мешалкой, в атмосфере аргона помещали 2-(2-гидрокси-2,2-дипентафторфенилэтиламино)-1,1-дипентафторфенилэтанол (0.15 г, 0.0002 моля), 10 мл толуола, затем при -78°С прибавляли по каплям 0.042 мл (0.00042 моля) 10 М раствора бутиллития в н-гексане. После этого температуру реакционной среды медленно доводили до комнатной, перемешивали 4 ч, охлаждали до -78°С, прибавляли TiCl4 (0.02 мл, 0.0002 моля), доволили до комнатной температуры, органический слой упаривали. Выход - 0.16 г, 87%. Найдено, %: С, 39.29; Н, 0.92; Ti, 4.95; Cl, 7.29; для C31H8O4Ti30Сl4 вычислено, %: С, 39.48; Н, 0.85; Ti, 5.08; Cl, 7.52. Т.пл.=294°С; [α]D RT=+18.40 (с=1, толуол). Спектр ЯМР 1Н (d8-толуол), (δ, м.д.): 3.60 (с, СН2-4Н). Полученный in-situ комплекс, без дополнительной очистки, был использован в качестве катализатора.
Пример 10. Синтез титанового комплекса [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорид, иммобилизованного на хлорид магния.
Комплекс, полученный в примере 4, представляющий собой титан дихлорид (4R,5R)-2,2-диметил-α,α,α',α'-тетра(перфторфенил)-1,3-диоксолан-4,5-диметанол (0.16 г, 0.0002 моля), растворяли в 10 мл толуола, добавляли хлорид магния (0.02 г, 0.0002 моля), который предварительно был подвергнут механическому воздействию в шаровой мельнице в течение 1 часа в атмосфере аргона. Полученную суспензию, представляющую собой иммобилизованный на хлорид магния комплекс 4 (C31H8O4MgTiF20Cl4), без дополнительной очистки, использовали в качестве компонента каталитической системы.
Пример 11. Синтез титанового комплекса [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорид, иммобилизованного на хлорид магния.
Комплекс, полученный в примере 4, представляющий собой титан дихлорид (4R,5R)-2,2-диметил-α,α,α',α'-тетра(перфторфенил)-1,3-диоксолан-4,5-диметанол (0.16 г, 0.0002 моля), растворяли в 10 мл толуола, добавляли хлорид магния (9.5 г, 0.1 моля), который предварительно был подвергнут механическому воздействию в шаровой мельнице в течение 1 часа в атмосфере аргона. Полученную суспензию, представляющую собой иммобилизованный на хлорид магния комплекс 4 (C31H8O4MgTiF20Cl4), без дополнительной очистки, использовали в качестве компонента каталитической системы.
Пример 12. Синтез титанового комплекса [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорид, иммобилизованного на хлорид алюминия.
Комплекс, полученный в примере 5 (0.16 г, 0.0002 моля), растворяли в 10 мл толуола, добавляли хлорид алюминия (0.03 г, 0.0002 моля), который предварительно был подвергнут механическому воздействию в шаровой мельнице в течение 1 часа в атмосфере аргона. Полученную суспензию, представляющую собой иммобилизованный на хлорид алюминия комплекс 5 (C31H8O4AlTiF20Cl5), без дополнительной очистки, использовали в качестве компонента каталитической системы.
Пример 13. Синтез титанового комплекса [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорид, иммобилизованного на хлорид цинка.
Комплекс, полученный в примере 6 (0.16 г, 0.0002 моля), растворяли в 10 мл толуола, добавляли хлорид цинка (0.03 г, 0.0002 моля), который предварительно был подвергнут механическому воздействию в шаровой мельнице в течение 1 часа в атмосфере аргона. Полученную суспензию, представляющую собой иммобилизованный на хлорид магния комплекс 4 (C31H8O4ZnTiF20Cl4), без дополнительной очистки, использовали в качестве компонента каталитической системы.
Пример 14. Синтез титанового комплекса [(4R,5R)-2,2-диметил-α,α,α',α'-тетра-(перфторфенил)-1,3-диоксолан-4,5-диметанол]титан (IV) дихлорид, иммобилизованного на хлорид скандия.
Комплекс, полученный в примере 3 (0.16 г, 0.0002 моля), растворяли в 10 мл толуола, добавляли хлорид скандия (0.03 г, 0.0002 моля), который предварительно был подвергнут механическому воздействию в шаровой мельнице в течение 1 часа в атмосфере аргона. Полученную суспензию, представляющую собой иммобилизованный на хлорид магния комплекс 4 (C31H8O4ZnTiF20Cl4), без дополнительной очистки, использовали в качестве компонента каталитической системы.
Полимеризация этилена
Пример 15
При изучении каталитических реакций полимеризации этилена применяли толуол и гептан марки "осч". Перед использованием растворители очищали от возможных примесей по стандартной для данного процесса методике. Полиметилалюмоксан фирмы Witco использовали в виде 10%-ного раствора в толуоле, молекулярная масса - 58 г/моль. Аргон и этилен ("осч") сушили пропусканием газов через колонку, заполненную молекулярными ситами 5 Å.
Полимеризацию этилена осуществляли в толуоле в стальном реакторе объемом 200 см3 при давлении этилена 1-11 атм. Предварительно реактор откачивали 1 ч при 90°C, затем охлаждали до нужной температуры, добавляли при перемешивании 100 мл толуола, 3,5 мл (6·10-3 моль) полиметилалюмоксана (МАО) и этилен (1 атм). Реакцию полимеризацию начинали через 15-20 минут после окончания подготовительных операций, разбивая в реакторе стеклянную ампулу, содержащую 0,05 г катализатора (6·10-5 моль Ti), полученного согласно примеру 2 (соотношение Al/Ti=100). Полимеризацию прекращали введением в содержимое реактора 10%-ного раствора НСl в этиловом спирте. Полимерный продукт отфильтровывали, промывали спиртом и водой и высушивали в вакууме при 50-60°С до постоянного веса. Выход 0,1 г. Температура плавления 139°С. ΔНпл, 180 Дж/г. Степень кристалличности 27%.
Таким образом, полученная каталитическая система содержит:
Пример 16
Полимеризацию этилена осуществляли в толуоле в стальном реакторе объемом 200 см3 при давлении этилена 1 атм. Предварительно реактор откачивали 1 ч при 90°С, затем охлаждали до нужной температуры, добавляли при перемешивании 100 мл толуола, 3,5 мл (6·10-3 моль) полиметилалюмоксана (МАО) и этилен. Реакцию полимеризацию начинали через 15-20 минут после окончания подготовительных операций, разбивая в реакторе стеклянную ампулу, содержащую 0,06 г катализатора (6·10-5 моль Ti), полученного согласно примеру 3 (соотношение Al/Ti=100). Полимеризацию прекращали введением в содержимое реактора 10%-ного раствора НСl в этиловом спирте. Полимерный продукт отфильтровывали, промывали спиртом и водой и высушивали в вакууме при 50-60°С до постоянного веса. Выход 6,7 г. Температура плавления 140.45°С. ΔHпл, 185,3 Дж/г. Степень кристалличности 32% (рис.1).
Таким образом, полученная каталитическая система содержит:
Пример 17
Полимеризацию этилена осуществляли в гептане в стальном реакторе объемом 200 см3 при давлении этилена 1 атм. Предварительно реактор откачивали 1 ч при 90°С, затем охлаждали до нужной температуры, добавляли при перемешивании 100 мл гептана, 3,5 мл (6·10-3 моль) полиметилалюмоксана (МАО) и этилен. Реакцию полимеризацию начинали через 15-20 минут после окончания подготовительных операций, разбивая в реакторе стеклянную ампулу, содержащую 0,06 г катализатора (6·10-5 моль Ti), полученного согласно примеру 4 (соотношение Al/Ti=100). Полимеризацию прекращали введением в содержимое реактора 10%-ного раствора НСl в этиловом спирте. Полимерный продукт отфильтровывали, промывали спиртом и водой и высушивали в вакууме при 50-60°С до постоянного веса. Выход 12 г. Температура плавления 128°С, ΔHпл, 171 Дж/г. Mw=240000, Mw/Mn=2,1. Степень кристалличности 44%.
Таким образом, полученная каталитическая система содержит:
Пример 18
Полимеризацию этилена осуществляли в гептане в стальном реакторе объемом 200 см3 при давлении этилена 1 атм. Предварительно реактор откачивали 1 ч при 90°С, затем охлаждали до нужной температуры, добавляли при перемешивании 100 мл гептана, 6,5 15 мл (0.01 моль) полиметилалюмоксана (МАО) и этилен. Реакцию полимеризацию начинали через 15-20 минут после окончания подготовительных операций, разбивая в реакторе стеклянную ампулу, содержащую 0,002 г катализатора (2·10-6 моль Ti), полученного согласно примеру 3 (соотношение Al/Ti=5000). Полимеризацию прекращали введением в содержимое реактора 10%-ного раствора НСl в этиловом спирте. Полимерный продукт отфильтровывали, промывали спиртом и водой и высушивали в вакууме при 50-60°С до постоянного веса. Выход 17 г. Температура плавления 132°С, ΔHпл, 165 Дж/г. Mw=1200000, Mw/Mn=2,6. Степень кристалличности 43%.
Таким образом, полученная каталитическая система содержит:
Пример 19
Полимеризацию этилена осуществляли в гептане в стальном реакторе объемом 200 см3 при давлении этилена 1 атм. Предварительно реактор откачивали 1 ч при 90°С, затем охлаждали до нужной температуры, добавляли при перемешивании 100 мл гептана, 3,5 мл (6·10-3 моль) полиметилалюмоксана (МАО) и этилен. Реакцию полимеризацию начинали через 15-20 минут после окончания подготовительных операций, разбивая в реакторе стеклянную ампулу, содержащую 0,07 г катализатора (6·10-5 моль Ti) в 1 мл толуола, полученного согласно примеру 10 (соотношение Al/Ti=100). Полимеризацию прекращали введением в содержимое реактора 10%-ного раствора НСl в этиловом спирте. Полимерный продукт отфильтровывали, промывали спиртом и водой и высушивали в вакууме при 50-60°С до постоянного веса. Выход 14 г. Температура плавления 131°С, ΔHпл, 163 Дж/г. Mw=256000, Mw/Mn=2,5. Степень кристалличности 34%.
Таким образом, полученная каталитическая система содержит:
Пример 20
Полимеризацию этилена осуществляли в гептане в стальном реакторе объемом 200 см3 при давлении этилена 1 атм. Предварительно реактор откачивали 1 ч при 90°С, затем охлаждали до нужной температуры, добавляли при перемешивании 100 мл гептана, 6,5 мл (0.01 моль) полиметилалюмоксана (МАО) и этилен. Реакцию полимеризацию начинали через 15-20 минут после окончания подготовительных операций, разбивая в реакторе стеклянную ампулу, содержащую 2,8 г катализатора (6·10-5 моль Ti), полученного согласно примеру 11 (соотношение Al/Ti=5000). Полимеризацию прекращали введением в содержимое реактора 10%-ного раствора НСl в этиловом спирте. Полимерный продукт отфильтровывали, промывали спиртом и водой и высушивали в вакууме при 50-60°C до постоянного веса. Выход 18 г. Температура плавления 129°С, ΔHпл, 175 Дж/г. Mw=1150000, Mw/Mn=2,9. Степень кристалличности 40%.
Таким образом, полученная каталитическая система содержит:
Полимеризация пропилена
Пример 21. Полимеризация пропилена «в массе»
Полимеризацию пропилена проводили в интервале температур 40-70°C в стальном реакторе с мешалкой в режиме полного заполнения реактора жидким мономером при давлении, превышающем насыщенную упругость паров пропилена, соответствующем данной температуре. Перед проведением эксперимента стальной реактор (объем 200 мл) вакуумировали в течение 1 ч при температуре опыта, несколько раз промывали пропиленом, затем понижали температуру до 10°С и заполняли жидким пропиленом (10.89 моль/л). После достижения рабочей температуры (50°С) включали мешалку, вводили 3,1 мл (5,3·10-3 моль) МАО и разбивали стеклянную ампулу стеклянную ампулу, содержащую 0,045 г катализатора (4,8·10-5 моль Ti), полученного согласно примеру 3 (соотношение Al/Ti=110), размещенную в нижней части реактора. Скорость полимеризации определяли по количеству пропилена, который вводили градуированным шприцем для поддержания постоянного давления в реакторе в ходе опыта. После завершения времени полимеризации (1 ч) давление в аппарате стравливали до атмосферного, реактор открывали, в реакционную смесь добавляли этиловый спирт, содержащий 10% раствор НСl. Высадившийся полимер отфильтровывали, промывали водой, сушили при комнатной температуре, затем - в вакуумном сушильном шкафу при температуре 80°С до постоянной массы.
Выход 8 г. 13С NMR (mmmm 25.5%, mmmr 8.9%, rmmr 3.4%, mmrr 9.6%, mmrm+rmmr 14.2%, rmrm 5.0%, rrrr 14.4%, mrrr 9.7, mrrm 9.2%), Mw=550900 г/моль, Mw/Mn=10.1. Механические свойства: модуль упругости 2.9 МПа, εр, % 1800, σт, МПа 6.4%. Температура плавления 150°С, ΔHпл, 22 Дж/г. Mw=1180000, Mw/Mn=4,8. Степень кристалличности 17% (рис.1).
Таким образом, полученная каталитическая система содержит:
Пример 22. Полимеризация пропилена в гептане
При изучении каталитических реакций полимеризации пропилена применяли толуол марки «осч», гептан «осч». Растворители очищали от возможных примесей перегонкой над натриевой пылью дважды. Аргон и пропилен («ос.ч.») сушили пропусканием газов через колонку, заполненную молекулярными ситами 5Å. Все операции по сборке аппаратуры, порядку подготовки и способам введения в реактор тестируемых комплексов и газообразного пропилена, а также измерению кинетики полимеризации аналогичны описанным ранее в примере 14. После полного растворения пропилена в гептане и достижения рабочей температуры (50°С) в реактор последовательно вводили 4,5 мл (5,3·10-3 моль) МАО и 0,07 г катализатора (7,8·10-5 моль Ti), полученного согласно примеру 3 (соотношение Al/Ti=68). После прекращения времени полимеризации (1 ч) реакцию останавливали обработкой содержимого 10%-ным раствором НСl в этаноле. Полимерный продукт отфильтровывали, промывали этанолом и водой и сушили в вакууме при 50-60°С до постоянной массы. Выход 1,3 г.
13С NMR (mmmm 27.1%, mmmr 3.6%, rmmr 9.6%, mmrr 9.9%, mmrm+rmmr 15.6%, rmrm 6.2%, rrrr 14.0%, mrrr 10.0, mrrm 7.1%). Температура плавления 150°С, ΔHпл, 22 Дж/г. Mw=311000, Mw/Mn=13,2. Степень кристалличности 40%.
Таким образом, полученная каталитическая система содержит:
Пример 23. Полимеризация пропилена в толуоле
Все операции по сборке аппаратуры, порядку подготовки и способам введения в реактор тестируемых комплексов и газообразного пропилена, а также измерению кинетики полимеризации аналогичны описанным ранее в примере 14. После полного растворения пропилена в толуоле и достижения рабочей температуры (50°С) в реактор последовательно вводили 6,3 мл (1,1·10-2 моль) МАО и 0,07 г катализатора (7,8·10-5 моль Ti), полученного согласно примеру 3. После прекращения времени полимеризации (1 ч) реакцию останавливали обработкой содержимого 10%-ным раствором НСl в этаноле. Полимерный продукт отфильтровывали, промывали этанолом и водой и сушили в вакууме при 50-60°С до постоянной массы. Выход 1,1 г.
13С NMR (mmmm 28.9%, mmmr 9.4%, rmmr 3.2%, mmrr 10.0%, mmrm+rmmr 12.6%, rmrm 5.0%, rrrr 16.2%, mrrr 10.1, mrrm 5.6%). Mw=143432, Mw/Mn=9.4. Степень кристалличности 23%.
Таким образом, полученная каталитическая система содержит:
Пример 24. Сополимеризация пропилена и этилена
Сополимеризацию пропилена и этилена также проводили в режиме полного заполнения реактора жидким пропиленом. Концентрацию этилена поддерживали постоянной в ходе опыта (соотношение этилен-пропилен = 60-40). Все операции по сборке аппаратуры, порядку подготовки и способам введения в реактор тестируемых комплексов и газообразного пропилена, а также измерению кинетики полимеризации аналогичны описанным ранее в примере 14. После полного растворения смеси этилена и пропилена в толуоле и достижения рабочей температуры (50°С) в реактор последовательно вводили 6,3 мл (1,1·10-2 моль) МАО и 0,07 г катализатора (7,8·10-5 моль Ti), полученного согласно примеру 3. Полимерный продукт отфильтровывали, промывали этанолом и водой и сушили в вакууме при 50-60°С до постоянной массы. Выход 13,5 г.
Mw=560000, Mw/Mn=2.8. Степень кристалличности 8% (рис.1-3).
Таким образом, полученная каталитическая система содержит:
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОНЕНТ КАТАЛИЗАТОРА ДЛЯ ПОЛИМЕРИЗАЦИИ ЭТИЛЕНА, ЕГО ПОЛУЧЕНИЕ И КАТАЛИЗАТОР, СОДЕРЖАЩИЙ ЭТОТ КОМПОНЕНТ | 2006 |
|
RU2375378C1 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ, ПРЕКАТАЛИЗАТОР ДЛЯ КАТАЛИТИЧЕСКОЙ СИСТЕМЫ И СПОСОБ ПОЛУЧЕНИЯ КАТАЛИТИЧЕСКОЙ СИСТЕМЫ | 2017 |
|
RU2676764C1 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ПОЛИМЕРИЗАЦИИ ЭТИЛЕНА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ ПОЛИМЕРИЗАЦИИ ЭТИЛЕНА | 1995 |
|
RU2142472C1 |
| ГОМОГЕННАЯ КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ СИНТЕЗА НИЗКОМОЛЕКУЛЯРНОГО РАЗВЕТВЛЕННОГО ПОЛИЭТИЛЕНА И СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО РАЗВЕТВЛЕННОГО ПОЛИЭТИЛЕНА | 1999 |
|
RU2161628C1 |
| СПОСОБ РЕГУЛИРОВАНИЯ СТРУКТУРЫ ЦЕПИ СОПОЛИМЕРА | 2011 |
|
RU2542992C2 |
| МЕТАЛЛООРГАНИЧЕСКОЕ СОЕДИНЕНИЕ В ТВЕРДОЙ ФОРМЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2013 |
|
RU2615128C2 |
| КОМПОНЕНТ КАТАЛИЗАТОРА ДЛЯ ПОЛИМЕРИЗАЦИИ ЭТИЛЕНА, ПРИГОТОВЛЕНИЕ ТАКОВОГО И КАТАЛИЗАТОР, ВКЛЮЧАЮЩИЙ КОМПОНЕНТ КАТАЛИЗАТОРА | 2010 |
|
RU2567391C2 |
| ПРЕКУРСОР ДЛЯ КАТАЛИЗАТОРА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2013 |
|
RU2623228C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРА | 2010 |
|
RU2500691C2 |
| КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 2018 |
|
RU2783537C2 |
Изобретение относится к каталитической системе для получения полиолефинов. Каталитическая система содержит галоидный комплекс металла 4 группы периодической системы с полидентантным органическим лигандом из числа производных (4R,5R)-2,2-R1,R2-α,α,α',α'-тетра(C6R3R4R5R6R7)-1,3-диоксолан-4,5-диметанола, где R1 и R2 группы, одинаковые или различные, которые выбирают из группы, включающей замещенный или незамещенный С1-С20 линейный или разветвленный алкил, С3-С20 циклоалкил, С6-С20 арил и С10-С20 конденсированные ароматические группы; R3-R7-алкил (С1-С5), арил, фтор, трифторметил; или 2-(2-гидрокси-2,2-дифенилэтиламино)-1,1-дифенилэтанола и алюминийорганическое соединение. При этом система дополнительно содержит, по меньшей мере, один галогенид металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий и алюминий. Молярное соотношение между галоидным комплексом металла 4 группы, галогенидом металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий и алюминий, и алюминийорганическим соединением находится в диапазоне 1:0,1-500:10-5000, предпочтительно 1:1-300:100-1000. Также предложены способ получения каталитической системы и полиолефин. Изобретение позволяет повысить каталитическую активность и снизить количество дорогостоящего сокатализатора - метилалюмоксана. 3 н. и 2 з.п. ф-лы, 3 ил., 24 пр.
1. Каталитическая система для получения полиолефинов, содержащая галоидный комплекс металла 4 группы Периодической системы с полидентантным органическим лигандом из числа производных (4R,5R)-2,2-R1,R2-α,α,α',α'-тетра(C6R3R4R5R6R7)-1,3-диоксолан-4,5-диметанола, где R1 и R2 группы, одинаковые или различные, которые выбирают из группы, включающей замещенный или незамещенный С1-С20 линейный или разветвленный алкил, С3-С20 циклоалкил, С6-С20 арил и С10-С20 конденсированные ароматические группы; R3-R7-алкил (С1-С5), арил, фтор, трифторметил; или 2-(2-гидрокси-2,2-дифенилэтиламино)-1,1-дифенилэтанола и алюминийорганическое соединение, отличающаяся тем, что она дополнительно содержит, по меньшей мере, один галогенид металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий и алюминий, молярное соотношение между галоидным комплексом металла 4 группы, галогенидом металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий и алюминий, и алюминийорганическим соединением находится в диапазоне 1:0,1-500:10-5000, предпочтительно, 1:1-300:100-1000.
2. Система по п.1, отличающаяся тем, что в качестве алюминийорганического соединения содержит соединение, выбранное из группы, включающей AlR3 (например, триметилалюминий, триэтилалюминий, три-н-пропилалюминий, три-н-бутилалюминий, триизобутилалюминий), R2AlHal (например, диэтилалюминийхлорид).
3. Способ получения каталитической системы в соответствии с любым из предшествующих пунктов формулы, отличающийся тем, что последовательно осуществляют следующие стадии: (а) добавление к одному из вышеперечисленных лигандов металлоорганического производного металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий или алюминий с получением полупродукта, (б) последующее смешение полученного в соответствии со стадией (а) полупродукта с галогенидом металла из 4 группы с получением смеси комплекса галогенида металла 4 группы с одним из вышеперечисленных лигандов и галогенида металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий или алюминий, (в) добавление к полученной смеси активатора алюминийорганического соединения.
4. Способ по п.2, отличающийся тем, что в систему после осуществления стадии (б) дополнительно вводят тонкодисперсный галогенид металла, выбранного из группы, включающей литий, натрий, магний, цинк, скандий или алюминий, как минимум, в эквимолярном по отношению к хлоридному комплексу металла 4 группы количестве, после чего осуществляют стадию (в).
5. Полиолефин, отличающийся тем, что он получен полимеризацией этилена или другого альфа-олефина с использованием каталитической системы в соответствии с любым из пп.1 и 2 формулы.
| БЕЛОКОНЬ Ю.Н | |||
| и др | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Известия Академии наук | |||
| Серия химическая, 2005, №10, с.2275-2280 | |||
| JP 56151707 A, 24.11.1981 | |||
| КАТАЛИЗАТОР ДЛЯ ПОЛИМЕРИЗАЦИИ ИЛИ СОПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1970 |
|
SU414770A3 |
| КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ПРОПИЛЕНА | 1989 |
|
RU2081883C1 |

