Эта заявка опубликована как частичное продолжение заявки PCT/CA2006/000484, опубликованной 31 марта 2006, по которой был испрошен приоритет на основании заявки PCT/CA2005/000819, опубликованной 27 мая 2005. Далее, эта заявка представляет собой частичное продолжение заявки на патент США 11/759154, которая представляет собой частичное продолжение заявки PCT/CA2006/000484 и по которой был испрошен приоритет на основании предварительной заявки на патент США №60/804067, опубликованной 6 июня 2006. Далее, по этой заявке испрошен приоритет на основании предварительных заявок на патент США №№ 60/807639, опубликованной 18 июля 2006, и 60/887188, опубликованной 30 января 2007.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к высвобождающим сероводород (H2S) производным лекарственных веществ, имеющим улучшенную активность и/или уменьшенные побочные эффекты. В частности, настоящее изобретение относится к производным лекарственных веществ, включающим H2S-высвобождающий фрагмент 4-гидрокситиобензамида, либо ковалентно связанный с лекарственным веществом, либо образующий соль с лекарственным веществом.
УРОВЕНЬ ТЕХНИКИ
Оксид азота (NO) и монооксид углерода (CO), синтезируемые, соответственно, из L-аргинина под действием NO-синтетазы и из гема под действием оксигеназы гема, представляют собой, соответственно, хорошо известные нейромедиаторы и также участвуют в регуляции сосудистого тонуса. Недавние исследования допускают, что сероводород (H2S) представляет собой третий газообразный медиатор млекопитающих. H2S синтезируется из L-цистеина или под действием цистатионин бета-синтетазы (CBS) или цистатионин гамма-лиазы (CSE), оба фермента используют пиридоксаль 5'-фосфат (витамин B6) в качестве кофактора.
Предполагается, что H2S возбуждает АТФ-чувствительные калиевые каналы (KATP) В гладких мышечных клетках сосудов, нейронах, кардиомиоцитах и панкреатических бета-клетках. В добавление, H2S может реагировать с реакционноспособными кислородными и/или азотными молекулами, ограничивая их токсичные эффекты и также смягчая их физиологические функции, действуя аналогично оксиду азота.
Недавние исследования показали, что H2S участвует в регуляции сосудистого тонуса, сокращениях миокарда, нейротрансмиссии и секреции инсулина. Недостаток H2S наблюдается в различных животных моделях артериальной и легочной гипертензии, болезни Альцгеймера, повреждения слизистой оболочки желудка и цирроза печени. Предполагается, что экзогенный H2S улучшает состояние при миокардиальной дисфункции, связанной с ишемией/реперфузионным повреждением, и уменьшает повреждение слизистой желудка, обусловленное противовоспалительными лекарственными веществами.
Более конкретно, недавно наблюдали, что H2S приводит в действие противовоспалительную и аналгезирующую активности. H2S представляет собой эндогенное соединение, продуцируемое в многочисленных тканях и влияющее на многочисленные функции (Wang, Two's company, three's a crowd: can H 2 S be the third endogenous gaseous transmitter? FASEB J. 2002; 16: 1792-1798). Также, как было показано, он представляет собой вазодилатор и может подавлять адгезию лейкоцитов к эндотелию сосудов (Wang, 2002; Fiorucci et al., Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005; 129: 1210-1224). Далее, Fiorucci et al. (2005) было продемонстрировано, что предварительная обработка донором H2S может уменьшать тяжесть обусловленного NSAID желудочного повреждения на крысах.
Предполагается, что продукция эндогенного H2S изменяется при многих болезнях. Кроме того, уровни H2S могут зависеть от применяемых в настоящее время лекарственных веществ. Например, ацетилсалициловая кислота и нестероидные противовоспалительные лекарственные вещества (NSAID), как было показано, имеют подавляющий эффект на CSE-H2S путь в слизистой оболочке желудочно-кишечного тракта (Fiorucci, S. et al.). Этот эффект может вносить вклад в повреждение слизистой оболочки желудка, обусловленное этими лекарственными веществами. Таким образом, фармакологическая модуляция уровней H2S может иметь значительный терапевтический потенциал.
Также предполагается, что H2S может играть роль при сердечно-сосудистой патологии и, как таковой, его уровень должен исследоваться у пациентов с различным факторами риска атеросклероза, такими как артериальная гипертензия, гиперлипидемия, сахарный диабет и т.д. Показано, что H2S гасится реакционноспособными частицами кислорода (ROS) (Whiteman, M. et al., The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrate 'scavenger'?, J Neurochem. 2004; 90: 765-768), и обсуждается важная роль окислительного стресса при многочисленных болезнях, таких как атеросклероз, артериальная гипертензия, болезнь Альцгеймера и т.д., предполагается, что избыточная продукция ROS может вызывать дефицит H2S.
Бета-блокаторы, которые применяются для лечения ангины, гипертонии и сердечной аритмии, обнаруживают респираторные побочные эффекты, такие как одышка, бронхостеноз и т.д., и, следовательно, могут вызывать проблемы у пациентов, подверженных астме, бронхиту и подобному. Следовательно, бета-блокаторы дополнительно ухудшают состояние при респираторных болезнях, таких как астма. Однако у астматических пациентов должны применяться уменьшенные дозы упомянутых лекарственных веществ для того, чтобы не подвергать дополнительной опасности функцию дыхания. Таким образом, эффективность бета-блокаторов является уменьшенной.
Антитромботические средства, такие как, например, дипиридамол, аспирин и т.д., применяемые для профилактики тромбозных явлений, имеют ряд побочных эффектов, таких как боль в желудке, тошнота и другие осложнения в отношении желудочно-кишечного тракта. У пациентов, имеющих патологии, связанные с окислительным стрессом, терапевтическое действие или переносимость этих лекарственных веществ, как в случае аспирина, значительно уменьшены.
Бронходилататоры, например салбутамол и т.д., применяются при лечении астмы и бронхита и лекарственные вещества, активные в отношении холинергической системы, применяются при патологиях, таких как недержание мочи. Их введение может вызывать побочные эффекты, повреждающие сердечно-сосудистую систему пациента, вызывая проблемы как у сердечно-сосудистых, так и гипертензивных пациентов.
Отхаркивающие и муколитические средства, которые применяются в терапии воспалительных состояний органов дыхания, могут приводить к увеличению изжоги и раздражению желудка, особенно у пациентов пожилого возраста.
Ингибиторы резорбции кости, такие как дифосфонаты (например, алендронат и т.д.) представляют собой лекарственные вещества, имеющие высокую желудочно-кишечную токсичность.
Ингибиторы фосфодиэстеразы, такие как, например, силденафил, запринаст, используемые при лечении сердечно-сосудистых и респираторных заболеваний, характеризуются аналогичными проблемами, такими как переносимость и/или эффективность, в частности, в патологических состояниях при окислительном стрессе.
Противоаллергические лекарственные средства, например цетиризин, монтелукаст, и т.д., имеют подобные проблемы в упомянутых патологических состояниях, особенно в отношении их эффективности.
Антиангиотензиновые средства, такие как ACE-ингибиторы, например эналаприл, каптоприл, и т.д., и ингибиторы рецепторов, например лозартан и т.д., применяются при лечении сердечно-сосудистых заболеваний. Эти лекарственные вещества могут вызывать респираторные побочные эффекты (то есть кашель и т.д.), в частности, в патологических состояниях при окислительном стрессе.
Антидиабетические лекарственные вещества, как инсулин-сенсибилизирующие, так и вызывающие гипогликемию, такие как, например, сульфонилмочевины, толбутамид, глипирид, гликлазид, глибурид, никотинамид и т.д., неэффективны при профилактике осложнений при диабете. Их введение может давать побочные эффекты, такие как, например, повреждения желудка. Эти явления становятся более интенсивными при патологических состояниях окислительного стресса.
Антибиотики, например ампициллин, кларитромицин и т.д., и противовирусные средства, например ацикловир и т.д., обнаруживают проблемы в отношении их переносимости, например они вызывают раздражение желудочно-кишечного тракта.
Противоопухолевые лекарственные вещества, например доксорубицин, даунорубицин, цисплатин и т.д., имеют высокую токсичность, в ряде органов, среди которых желудок и кишечник. Упомянутая токсичность дополнительно ухудшает состояние при вышеупомянутых патологиях при окислительном стрессе.
Лекарственные вещества против деменции, например никотин и холиномиметические препараты, характеризуются слабой переносимостью, особенно при патологических состояниях при окислительном стрессе.
Таким образом, существует необходимость в создании доступных лекарственных средств, имеющих улучшенные терапевтические характеристики, то есть имеющих пониженную токсичность и/или более высокую эффективность, с тем чтобы что они могли вводиться пациентам при болезненных состояниях при окислительном стрессе и/или эндотелиальных дисфункциях, избегая серьезных недостатков лекарственных веществ предшествующего уровня техники.
Неожиданно, авторы настоящего изобретения впервые обнаружили, что 4-гидрокситиобензамид (также обозначаемый здесь как 4-HTB или TBZ) является эффективным высвобождающим H2S фрагментом в тканях, когда он либо ковалентно связан с лекарственным веществом, либо образует соль с лекарственным веществом, причем полученные производные лекарственных веществ имеют пониженные побочные эффекты. Например, производные лекарственные вещества по настоящему изобретению создают значительно меньшие желудочно-кишечные и/или сердечно-сосудистые побочные эффекты.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте настоящего изобретения предоставлены производные лекарственных веществ, где упомянутые производные включают H2S-высвобождающий фрагмент 4-гидрокситиобензамида (также обозначаемый здесь как 4-HTB или TBZ), который либо связан ковалентно с лекарственным веществом, либо образует соль с лекарственным веществом. Соединения по настоящему изобретению проявляют неожиданно сильную активность по сравнению с лекарственным веществом, взятым отдельно, 4-гидрокситиобензамидом, взятым отдельно, и сочетанием лекарственного вещества и 4-гидрокситиобензамида, вводимых раздельно, но последовательно, обнаруживают уменьшенные побочные эффекты, или и то, и другое.
Соединения по настоящему изобретению создают умеренный, кратковременный прирост концентраций H2S в плазме. Без необходимости быть связанным с любой теорией кратковременный прирост концентраций H2S в плазме, который остается в пределах физиологического диапазона, может вносить вклад в усиление активности лекарственных веществ, уменьшенное повреждение желудочно-кишечного тракта и/или уменьшенную сердечно-сосудистую токсичность.
Далее, соединения по настоящему изобретению неожиданно вызывают значительно меньший прирост в систолическом давлении крови при введении гипертензивным крысам, что наблюдалось при введении лекарственного вещества как такового. Уменьшенная склонность повышать давление крови может уменьшать сердечно-сосудистые побочные эффекты, часто наблюдаемые при продолжительном применении некоторых из лекарственных веществ.
Согласно настоящему изобретению предоставлены соединения, имеющие следующую общую формулу:
где A представляет собой остаток лекарственного вещества, Y является выбранным из группы, состоящей из -C(O)O-, -C(O)NH-, -C(O)OC(O)-, -C(O)NHCH2C(O)-, O, S, N, 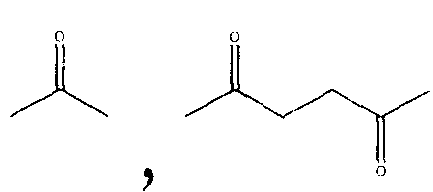 или группа отсутствует, и X представляет собой
или группа отсутствует, и X представляет собой 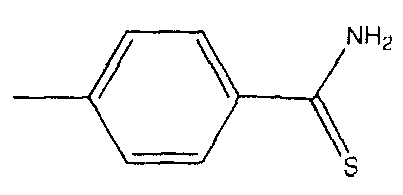 или
или 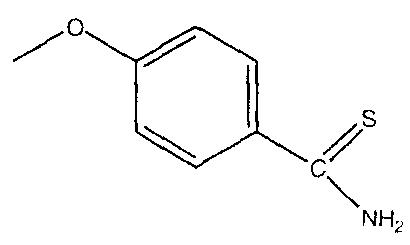 , и фармацевтически приемлемые соли соединений, в которых если Y отсутствует, то производное лекарственного вещества может представлять собой соль A и X. В предпочтительном варианте осуществления A и X связаны через эфирный мостик, ангидридный мостик, тиоэфирный мостик, амидный мостик или азо-мостик. В одном варианте осуществления, соли образуются с остатком лекарственных веществ, с использованием тиокарбамоилбензоата вместо 4-гидрокситиобензамида.
, и фармацевтически приемлемые соли соединений, в которых если Y отсутствует, то производное лекарственного вещества может представлять собой соль A и X. В предпочтительном варианте осуществления A и X связаны через эфирный мостик, ангидридный мостик, тиоэфирный мостик, амидный мостик или азо-мостик. В одном варианте осуществления, соли образуются с остатком лекарственных веществ, с использованием тиокарбамоилбензоата вместо 4-гидрокситиобензамида.
Лекарственное вещество может быть выбрано из множества известных классов лекарственных веществ, включая, например, белки, пептиды, нуклеотиды, лекарственные вещества против ожирения, пищевые продукты с увеличенной питательной ценностью, кортикостероиды, ингибиторы эластазы, аналгетики, противогрибковые препараты, противораковые средства, противорвотные средства, аналгетики, сердечно-сосудистые средства, противовоспалительные препараты, антигельминтные средства, антиаритмические средства, антибиотики (включая пенициллины), антикоагулянты, антидепрессанты, антидиабетические агенты, антиэпилептические средства, антигистамины, антигипертензивные средства, антимускариновые средства, антимикобактериальные агенты, антинеопластические агенты, иммуносупрессанты, средства, направленные на щитовидную железу, противовирусные агенты, анксиолитические успокаивающие (снотворные средства и нейролептики), вяжущие средства, бета-андреноблокаторы, сердечные ионотропные агенты, кортикостероиды, противокашлевые средства (отхаркивающее и муколитические средства), диуретики, допаминергические средства (лекарственные вещества против синдрома Паркинсона), гематостатические средства, иммунологические средства, препараты, регулирующие липидный обмен, мышечные релаксанты, парасимпатомиметические средства, тиреокальцитонин и бисфосфонаты паращитовидной железы, простагландины, половые гормоны, антиаллергические агенты, стимуляторы, анорексические средства, симпатомиметические средства, средства, активные в отношении щитовидной железы, вазодилаторы и ксантины.
Следующие лекарственные вещества являются особенно применимыми по настоящему изобретению:
нестероидные противовоспалительные лекарственные вещества (NSAID): ацетилсалициловая кислота (ASA), диклофенак, напроксен, индометацин, флурбипрофен, сулиндак, ибупрофен, ацеклофенак, ацеметацин, беноксапрофен, бензофенак, бромфенак, буклоксовая кислота, бутибуфен, карпрофен, целекоксиб, циклопрофен, цинметацин, клиденак, клопирак, дифлусинал, этодолак, эторикоксиб, фенбуфен, фенклофенак, фенхлорак, фенопрофен, фентиазак, флуноксапрофен, фурапрофен, фуробуфен, фурафенак, ибуфенак, индопрофен, изоксепак, кетопрофен, кеторолак, локсопрофен, лоназолак, лумиракоксиб, метиазиновая и мефенамовая кислоты, меклофенамовая кислота, мелоксикам, набуметон, пиромидовая кислота, салсалат, миропрофен, оксапрозин, оксепинак, паракоксиб, фенилбутазон, пирпрофен, пироксикам, пирозолак, протизиновая кислота, рофекоксиб, салицилат натрия, супрофен, тиапрофеновая кислота, толметин, валдекоксиб, зомепирак, и подобные;
аналгезирующие лекарственные вещества: ацетаминофен, ацетаминосалол, аминохлортеноксазин, ацетилсалициловая 2-амино-4-пиколиновая кислота, ацетилсалицилсалициловая кислота, анилеридин, беноксапрофен бензилморфина, ацетат 5-бромсалициловой кислоты, буцетин, бупренорфин, буторфанол, капсаицин, цинцгофен, цирамадол, клометацин, клониксин, кодеин, дезоморфин, дезоцин, дигидрокодеин, дигидроморфин, димепгептанол, дипироцетил, эптазоцин, этоксазен, этилморфин, эугенол, флоктафенин, фосфосал, глафенин, гидрокодон, гидроморфон, гидроксипетидин, ибуфенак, пара-лактофенетид, леворфанол, мептазинол, метазоцин, метопон, морфин, налбуфин, никоморфин, норлеворфанол, норморофин, оксикодон, оксиморфон, пентазоцин, феназоцин, феноколл, феноперидин, фенилбутазон, фенилсалицилат, фенилрамидол, салицин, салициламид, тиорфан, трамадол, диацереин, актарит, и подобные;
лекарственные вещества против воспаления толстой кишки: 4- или 5-аминосалициловая кислота, тримебутин, и подобные;
респираторные и урогенитальные средства (бронходилаторы и лекарственные вещества, активные в отношении холинергической системы, отхаркивающие средства/муколитики, антиастматические/антиаллергические антигистаминовые лекарственные вещества): бронходилаторы и лекарственные вещества, активные в отношении холинергической системы: ацефилин, албутерол, бамбутерол, бамифиллин, метилсульфат бевониума, битолтерол, карбутерол, кленбутерол, хлорпреналин, диоксетдрин, дифиллин, эфедрин, эпинефрин, эпроксинол, этафредин, этилнорепинефрин, этофиллин, фенотерол, бромид флутоприума, гексопреналин, бромид ипратропиума, изоэтарин, изопротенерол, мабутерол, метапротеренол, оксибутинин, бромид окситропиума, пирбутерол, прокатерол, протокилол, проксифиллин, репротерол, римитерол, салметерол, зотеренол, тербуталин, 1-теобромуксусная кислота, бромид тиотропиума, третохинол, тулобутерол, запринаст, циклодрин, NS-21, 2-гидрокси-2,2-дифенил-N-(1,2,3,6-тетрагидропиридин-4-илметил)ацетамид и подобные;
отхаркивающие/муколитические средства: амброксол, бромгексин, домиодол, эрдостеин, гуаиакол, гуаифенесин, иодированный глицерин, летостеин, месна, зобрерол, степронин, терпин, тиопронин, и подобные;
антиастматические/антиаллергические антигистаминные лекарственные вещества: акривастин, аллокламид, амлексанокс, цетиризин, клобензепам, хромогликат, хромолин, эпинастин, фексофенадин, формотерол, гистамин, гидроксизин, левокабастин, лодоксамид, мабутерол, монтелукаст, недокромил, репиринаст, сератродаст, суплатаст, тозилат, терфенадин, тиарамид, урусгиол, бромгексин, и подобные;
ACE-ингибиторы: алацеприл, беназеприл, каптоприл, церонаприл, цилазаприл; делаприл, эналаприл, эналаприлат, фосиноприл, имидаприл, лисиноприл, лосартан, мовелтиприл, нафтопидил, периндоприл, хинаприл, рамиприл, спираприл, темокаприл, трандолаприл, урапидил, и подобные;
бета-блокаторы: ацебутолол, алпренолол, амосулалол, аротинолол, атенолол, бетаксолол, бевантолол, букомол, буфетолол, буфуралол, бунитролол, бупранолол, бутолфилол, каразолол, картеолол, карведилол, целипролол, цетамолол, дилевалол, эпанолол, эсмолол, инденолол, лабеталол, мепиндолол, метипранолол, метопролол, мопролол, надолол, надоксолол, небиволол, нифеналол, нипридалол, окспренолол, пенбутолол, пиндолол, практолол, пронеталол, пропранолол, зоталол, сульфиналол, талинолол, тертатолол, тилизолол, тимолол, толипролол, ксибенолол, и подобные;
антитромботические средства и вазодилаторы: ацеторфан, ацетилсалициловая кислота, аргатробан, баметан, гемисукцинат бенфуродила, бензиодарон, бетагистин, бромвинцамин, буфениод, цитиколин, клобенфурол, клопидогрел, цикланделат, далтепарин, дипиридамол, дропениламин, эноксапарин, фендилин, ифенпродил, илопрост, индобуфен, исбогрел, изоксисуприн, гепарин, ламифибан, мидродин, надропарин, никотиноильный спирт, нилидрин, озагрел, пергексилин, фенилпропаноламин, прениламин, папверолин, натриевая соль ревипарина, ридогрел, сулоктидил, тинэфедрин, тинзапарин, трифлусал, ниацинат ксантинола, и подобные;
антидиабетические средства: акарбос, карбутамид, глиборнурид глибутиазол, миглитол, репаглинид, троглитазон, 1-бутил-3-метанил-мочевина, толрестат, никотинамид, и подобные;
противоопухолевые лекарственные вещества: анцитабин, антрамицин, азацитидин, азасерин, 6-азауридин, бикалутамид, карубицин, карзинофилин, хлорамбуцил, хлорзотоцин, цитарабин, даунорубицин, дефосфамид, демеколцин, деноптерин, 6-диазо-5-оксо-1-норлейцин, доцетаксел, доксифлуридин, доксорубицин, дролоксифен, эдатрексат, эфлорнитин, эноцитабин, эпирубицин, эпитиостанол, этанидазол, этопозид, фенретинид, флударабин, фторурацил, гемцитабин, гексестрол, идарубицин, лонидамин, манномустин, мелфалан, меногарил, 6-меркаптопурин, метотрексат, митобронитол, митолактол, митомицины, митоксантрон, мопидамол, микофеноловая кислота, ниноптерин, ногаламицин, паклитаксель, пентостатин, пирарубицин, пиритрексим, пликамицин, подофилиловая кислота, порфимер-натрий, порфиромицин, пропагерманий, пуромицин, ранимустин, ретиноевая кислота, роквинимекс, стрептонигрин, стрептозоцин, тенипозид, тенуазоновая кислота, тиамиприн, тиогуанин, томудекс, топотекан, триметрексат, туберцидин, убенимекс, винбластин, винкристин, виндезин, винорелбин, зорубицин, и подобные;
противоязвенные лекарственные вещества: ε-ацетмаидокапровая кислота, арбапростил, цетраксат, циметидин, экабет, энпростил, эсапразол, ирзогладин, мизопростол, омепразол, орнопростил, пантопразол, плаунотол, риопростил, росапростол, ротраксат, зофалкон, тримопростил, и подобные;
противогиперлипидемические лекарственные вещества (статины): аторвастатин, циластатин, дермостатин, флувастатин, ловастатин, мевастатин, нистатин, пентостатин, пепстатин, привастатин-натрий, симвастатин, и подобные;
антибиотики: амдиноциллин, амоксициллин, ампициллин, апалциллин, апициклин, аспоксициллин, азидамфеникол, азидоциллин, азлоциллин, азтреонам, бензоилпас, бензил пенициллиновая кислота, биапенем, бикозамицин, капреомицин, карбенициллин, кариндациллин, карумонан, цефаклор, цефадроксил, цефамандол, цетиризин, цефазедон, цефазолин, цефбуперазон, цефклидин, цефдинир, цефдиторен, цефепим, цефетамет, цефиксим, цефменоксим, цефметазол, цефминокс, цефодизин, цефоницид, цефоперазон, цефоранид, цефотаксим, цефотертан, цефотиам, цефокситин, цефозопран, цефпимизол, цефпирамид, цефпиром, цефпрозил, цефроксадин, цефсулодин, цефтазидим, цефтерам, цефтезол, цефтибутен, цефтиофур, цефтизоксим, цефтриаксон, цефуроксим, цефузонам, цефацетрил натрий, цефалексин, цефалоглицин, цефалоридин, цефалоспорин C, цефалотин, цефапирин-натрий, цефрадин, хлорамфеникол, хлортетрациклин, циноксацин, клавулановая кислота, клометоциллин, клоксациллин, циклациллин, циклосерин, демеклоциклин, диклоксациллин, эпициллин, фенбециллин, фломоксеф, флоксациллин, этациллин, имипенем, ленампициллин, лоракарбеф, лимециклин, мафенид, меклоциклин, меропенем, метампициллин, метациклин, метициллин-натрий, мезлоциллин, миноциклин, моксалактам, мупироцин, миксин, негамицин, новобиоцин, оксациллин, панипенем, калиевая соль пенициллина G, пенициллин N, пенициллин O, пенициллин V, калиевая соль фенетициллина, пипациклин, пиперациллин, пирлимицин, порфиромицин, пропициллин, хинациллин, ритипенем, ролитертрациклин, санциклин, седекамицин, спектиномицин, сулбактам, сулбенициллин, темоциллин, тетрациклин, тикарциллин, тигемонам, туберцидин, азитромицин, кларитромицин, диртромицин, энвиомицин, эритромицин, джосамицин, мидекамицин, миокамицин, олеандомицин, рифабутин, рифамид, фиамицин, рифаксимин, рокитамицин, спирамицин, тролеандромицин, виомицин, виргиниамицин; амикацин, апрамицин, арбекацин, дибекацин, дигидрострептомицин, фортимицины, гентамицин, микрономицин, неомицин, нетилмицин, паромомицин, рибостамицин, сизомицин, спектиномицин, стрептомицин, тобрамицин, троспектромицин; бакампициллин, цефкапен-пивоксил, цефподоксим-проксетил, панипенем, пивампициллин, пивцефалексин, султамициллин, талампициллин; карбомицин, клиндамицин, линкомицин, микамицин, росарамицин, ципрофлоксацин, клинафлоксацин, дифлоксацин, эноксацин, энрофлоксацин, флероксацин, флумеквин, грепафлоксацин, ломефлоксацин, надифлоксацин, налидиксовая кислота, норфлоксацин, офлоксацин, пазуфлоксазин, пефлоксацин, пипемидовая кислота, пиромидовая кислота, руфлоксацин, сарфлоксацин, тосульфоксаин, тровафлоксацин, кломоциклин, гуамециклин, окситетрациклин, нифурпиринол, нифурпразин; пара-аминосалициловая кислота, гидразид пара-аминосалициловой кислоты, клофазимин, дезоксидигидрострептомицин, этамубтол, гликониазид, изониазид, опиниазид, фениламиносалициклат, рифампин, рифапентин, салиназид, 4-4'-сульфинилдианилин, ацедиасульфон, дапзон, сукцисульфон, пара-сульфанилилбензиламин, тиазолсульфон, ацетил сульфаметоксипиразин, мафенид, 4'-(метилсульфамоил)сульфаниланилид, салазосульфадимидин, сульфабензамид, сульфацетамид, сульфахлорпиридазин, сульфахризоидин, сульфацитин, сульфадиазин, сульфадикрамид, сульфадиметоксин, сульфадоксин, сульфаэтидол, сульфагуанидин, сульфагуанол, сульфален, сульфамеразин, сульфаметер, сульфаметазин, сульфаметизол, сульфаметомидин, сульфаметоксазол, сульфаметоксипиридазин, сульфаметилтиазол, сульфаметрол, сульфамидохризоидин, сульфамоксол, сульфаниламид, 2-пара-сульфанилиланилиноэтанол, N,4-сульфанилилсульфаниламид, сульфанилилмочевина, N-сульфанилил-3,4-ксиламид, сульфаперин, сульфафеназол, сульфапроксилин, сульфапиразин, сульфапиридин, сульфазомизол, сульфасимазин, сульфатиазол, сульфатиомочевина, сульфизомидин, сульфизоксазол, 4-сульфаниламидо салициловая кислота; негамицин, карумонан, клоксихин, нитроксолин, аргинин, метронидазол, и подобные;
противовирусные лекарственные вещества: ацикловир, амантадин, цидофовир, цитарабин, диданозин, дидезоксиаденозин, эдоксудин, фамцикловир, флоксуридин, ганцикловир, идоксуридин, инданавир, кетоксал, ламивудин, MADU, пенцикловир, подофиллотоксин, рибавирин, римантадин, сахинавир, зоривудин, ставудин, трифлуридин, валацикловир, видарабин, ксеназоевая кислота, залцитабин, зидовудин; и подобные;
ингибиторы резорбции кости (бисфосфонаты): алендроновая кислота, бутедроновая кислота, этидроновая кислота, оксидроновая кислота, памидроновая кислота, риседроновая кислота, и подобные;
лекарственные вещества против деменции: амиридин, лазабемид, мофегилин, салбелузол, оксирацетам, ипидакрин, небрацетам, такрин, велнакрин, и подобные.
Вышеупомянутые предшественники лекарственных веществ приготовляются согласно способам, известным в предшествующем уровне техники. См., например, The Merck Index, 13 th Edition (2001), Merck & Co., Whitehouse Station, N.J., которая включена в данное описание посредством ссылки. Если доступны, могут быть применены соответствующие изомеры, включая оптические изомеры.
Фармацевтически приемлемые соли соединений по настоящему изобретению, такие как, например, соли с щелочными металлами и щелочноземельными металлами, нетоксичными аминами и аминокислотами также представляют собой часть настоящего изобретения. Предпочтительные соли соединений по настоящему изобретению представляют собой соли с аргинином и агматином. Также включенными являются фармацевтически приемлемые соли присоединения кислоты.
Производные по изобретению могут быть применены согласно терапевтическим показаниям для лекарства-предшественника, делая возможным достижение преимуществ, приводимых далее для этих лекарственных веществ.
Производные NSAID по настоящему изобретению являются очень хорошо переносимыми и эффективными, даже когда организм ослаблен и находится в состоянии окислительного стресса. Производные NSAID могут быть применены при тех патологиях, в которых воспаление играет значительную патогенетическую роль, как, например, без ограничения, в случае рака, астмы, инфаркта миокарда.
Более конкретно, производные NSAID по настоящему изобретению могут быть применены для, без ограничения, лечения воспаления у индивидуума, и для лечения других расстройств, ассоциированных с воспалением, например, в качестве аналгетика в лечении боли и головных болей, или как жаропонижающее для лечения лихорадки. Например, соединения по изобретению могут быть применены для лечения артрита, включая, без ограничения, ревматоидный артрит, спондилоатропатии, подагрический артрит, остеоартрит, системную красную волчанку и ювенильный артрит. Такие соединения по изобретению могут быть применены для лечения астмы, бронхита, менструальных колик, тендинита, бурсита, болезненных состояний кожи, таких как псориаз, экзема, ожоги и дерматит, и постоперационного воспаления, включая глазное хирургическое вмешательство, такое как операция на катаракте и хирургическое вмешательство, относящееся к рефракции. Соединения по изобретению также могут быть применены для лечения желудочно-кишечных состояний, таких как воспалительное заболевание кишечника, болезнь Крона, гастрит, спастический колит и язвенный колит, и для предотвращения или лечения рака, такого как колоректальный рак. Соединения по изобретению могут быть применены для лечения воспаления при таких болезнях, как сосудистые болезни, головные боли при мигрени, узелковый периартериит, тиреоидит, апластическая анемия, болезнь Ходжкина, склеродома, ревматическая атака, диабет I типа, заболевания нейромышечного соединения, включая миастения gravis, заболевание белого вещества мозга, включая множественный склероз, саркоидоз, невротический синдром, синдром Бехчета, полимиозит, гингивиты, нефрит, гиперчувствительность, опухание, имеющее место после повреждения, миокардиальная ишемия, и подобное. Соединения также могут быть применены при лечении офтальмических заболеваний, таких как ретинит, ретинопатии, увеит, светобоязнь, и острое повреждение глазной ткани. Соединения также могут быть применены при лечении воспаления органов дыхания, таких как воспаления, которые ассоциированы с вирусными инфекциями и фиброзно-кистозной дегенерацией. Соединения также могут быть применены для лечения определенных расстройств центральной нервной системы, таких как кортикальные деменции, включая болезнь Альцгеймера. Соединения по изобретению могут быть применены в качестве противовоспалительных агентов, например при лечении артрита, с дополнительным преимуществом значительно меньших вредных побочных эффектов. Эти соединения также могут быть применены при лечении аллергического ринита, синдрома респираторного дистресса, синдрома эндотоксического шока, атеросклероза и повреждения центральной нервной системы, полученного при инсульте, ишемии и травме. Соединения также могут быть применены при лечении боли, без ограничения, послеоперационной боли, зубной боли, мышечной боли, и боли, имеющей место при раке. Кроме возможности быть примененными для лечения человека, эти соединения также могут быть применены для лечения млекопитающих, включая лошадей, собак, кошек, крыс, мышей, овец, свиней и т.д.
Производные противоколитного лекарственного вещества по настоящему изобретению, например производные 4- или 5-аминосалициловой кислоты, производные тримебутина, и подобные, могут быть применены для профилактики или лечения различных заболеваний, особенно воспалительных состояний желудочно-кишечного тракта, включая, без ограничения, воспалительные состояния рта, такие как воспаление слизистой оболочки, инфекционные заболевания (например, вирусные, бактериальные и грибковые болезни), и болезнь Крона; воспалительные состояния пищевода, такие как эзофагит, состояния, полученные при химическом повреждении (например, проглатывании щелочи), желудочно-пищеводный рефлюкс, билиарный рефлюкс, эзофагит Баррета, болезнь Крона и доброкачественная стриктура пищевода; воспалительные состояния, такие как гастрит (например, вызываемый Helicobacter pylori, нарушение желудочной кислотности и атрофический гастрит), глютеиновая болезнь, язва желудка и двенадцатиперстной кишки, предраковые повреждения желудка, неязвенная диспепсия и болезнь Крона; воспалительные состояния желудка, такие как болезнь Крона, чрезмерное развитие микрофлоры, пептическая язва и трещина кишки; воспалительные состояния толстой кишки, такие как болезнь Крона, язвенный колит, спастический колит, инфекционный колит (например, псевдомембранный колит, такой как колит, вызываемый бактерией Clostridium difficile, сальмунелезный энтерит, шигелловые инфекции, иерсиниозы, криптоспоридиоз, микроспридиальные инфекции, и вирусные инфекции), обусловленный радиацией колит, колит у пациентов со сниженной иммунной реакцией (например, тифлит), предраковые состояния толстой кишки (например, дисплазия, воспалительные состояния кишки и полипы толстой кишки), проктит, воспаление, ассоциированное с геммороем, спастическая прокталгия и ректальные трещины; печеночные, относящиеся к желчному пузырю и/или желчным протокам состояния, такие как холангит, первичный склерозирующий холангит, первичный желчный цирроз печени и холецистит; и гнойное воспаление кишечника.
Статины применяются для профилактики и лечения атеросклероза, который вызывает боль в груди, сердечные приступы, инсульты и перемежающуюся хромоту у индивидуумов, которые находятся или могут находиться в группе риска по атеросклерозу. Факторы риска для атеросклероза включают ненормально повышенные уровни холестерина, наследственность при сердечных приступах (особенно в молодости), пожилой возраст и диабет. Большинству пациентов статины назначают в связи с высокими уровнями холестерина. Несмотря на то что уменьшение уровней холестерина является важным, болезнь сердца является сложной и могут играть роль другие факторы, такие как воспаление. Известно, однако, что статины проявляют неблагоприятные эффекты, такие как, например, заболевание печени, возможно имеют канцерогенный потенциал, создают мышечные побочные эффекты и миопатию.
Производные статина по настоящему изобретению могут уменьшать побочные эффекты, ассоциированные со статинами, и/или иметь улучшенную фармакологическую активность. Неожиданно, производное симвастатина, сложный 4-тиокарбамоилфениловый эфир-2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8a-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-иловый эфир янтарной кислоты, значительно уменьшает агрегацию тромбоцитов при концентрациях 3, 10 и 30 мкМ по сравнению с соответствующим статином, вводимым отдельно. Далее, производное симвастатина по настоящему изобретению вызывает значительное увеличение тромбоцитарного cAMP по сравнению с теми же концентрациями симвастатина, вводимого отдельно.
Производные адренергических блокаторов, или α- или β-блокаторов, по настоящему изобретению могут быть применены для профилактики или лечения гипертензии, стенокардии, выпадения митрального клапана, застойной сердечной недостаточности, инфаркта миокарда, глаукомы, головных болей при мигрени, тахикардии и тремора с уменьшенными побочными эффектами.
Производные антитромботических лекарственных средств по настоящему изобретению, например производные аспирина, потенциируют антитромбоцитную активность с улучшенной желудочной переносимостью. Основное предназначение антитромботических лекарственных веществ состоит в профилактике и лечении венозной тромбоэмболии (VTE), предотвращении инсульта у пациентов с фибрилляцией предсердия и профилактике и лечении острого ишемического синдрома (ACS).
Производные бронходилаторов и производные лекарственных веществ, активных в отношении холинергической системы, могут быть применены для облегчения симптомов астмы путем расслабления напряжения мышц, которые сужают просвет воздушных путей. В краткодействующих формах производные бронходилаторов смягчают или останавливают симптомы астмы и являются очень полезными во время приступа астмы. В продолжительно действующих формах производные бронходилаторов помогают контролировать симптомы астмы и предотвращают приступы астмы. Производные по настоящему изобретению уменьшают побочные эффекты путем влияния на сердечно-сосудистую систему, такие как тахикардия, гипертония и т.д.
Производные отхаркивающих и муколитических средств по настоящему изобретению могут быть применены для разрыхления и удаления слизи и мокроты из дыхательных путей. Желудочно-кишечная переносимость отхаркивающих и муколитических средств может быть улучшена в случае модифицированных 4-гидрокситиобензамидом производных, как впервые описано в настоящем изобретении.
Производные бисфосфонатов по настоящему изобретению могут быть применены при лечении или профилактике нарушений кальциевого метаболизма или болезни, например остеопорозе, болезни Бехтерева, метастазах кости, мочекаменной болезни, гетеротропных окостенениях, ревматоидном артрите, остеоартрите или дегенеративном артрозе. Токсичность в отношении желудочно-кишечного тракта у производных по настоящему изобретению может быть уменьшена.
Терапевтическая эффективность производных ингибиторов фосфодиэстеразы (PDE) (бронходилататоров) по настоящему изобретению является улучшенной и побочные эффекты уменьшены. PDE ингибиторы имеют доказанный потенциал в качестве противовоспалительных лекарственных веществ, особенно при дыхательных заболеваниях. Они подавляют высвобождение воспалительных сигналов, например, цитокинов, и ингибируют продукцию реакционноспособных кислородных молекул. PDE-ингибиторы имеют высокий терапевтический и коммерческий потенциал как нестероидные противовоспалительные средства при воспалительных дыхательных заболеваниях, таких как астма, COPD и риниты.
Более высокая эффективность и/или меньшие побочные эффекты могут также наблюдаться для производных антилейкотриеновых лекарственных веществ, ACE-ингибиторов, антидиабетических лекарственных веществ, антибиотика, противовирусных и противоопухолевых лекарственных веществ.
Соединения по настоящему изобретению могут быть получены по следующим схемам:
Схема 1, представленная ниже, использует в качестве примера синтез производного NSAID, 4-тиокарбамоилфенилового эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты. В этой схеме реагент Лавессона применяется для введения серы в высвобождающий сероводород фрагмент после того, как группа ковалентно связывается с остатком NSAID.
Схема 1

Диклофенак (1), который имеет свободную карбоксильную группу, сначала растворяли в диметилформамиде и добавляли гидроксибензотриазол (HOBt) и 1,3-дициклогексилкарбодиимид (DCC). К этой смеси добавляли 4-гидроксибензамид в условиях, пригодных для образования промежуточного соединения по настоящему изобретению (например, 4-карбамоилфенил 2-(2-(2,6-дихлорфениламино)фенил)ацетата (2)); в указанном промежуточном соединении отсутствует сера. Пригодное соединение, которое может вводить серу, такое как реагент Лавессона, добавляли для получения соединения по настоящему изобретению (например, 4-тиокарбамоилфениловый эфир [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (3).
В зависимости от специфического состояния или болезненного состояния, которые излечиваются, индивидуумам могут вводиться соединения по настоящему изобретению при любой приемлемой терапевтически эффективной и безопасной дозировке, которая может быть легко определена специалистом в данной области. Эти соединения, наиболее желательно, могут вводиться в дозировках, варьирующих от около 1 до около 2000 мг в день, в виде отдельной или разделенных доз, хотя неизбежно будут иметь место варьирования в зависимости от веса и состояния индивидуума, который излечивается, и выбранного конкретного пути введения. Понятно, что дозировки будут зависеть от применяемого конкретного лекарственного вещества, образующего соединения по настоящему изобретению. Однако уровень дозировок, который находится в диапазоне от около 0,1 до приблизительно 100 мг/кг, предпочтительно между приблизительно 5 и 90 мг/кг, и более предпочтительно между приблизительно 5 и 50 мг/кг, является наиболее желательным. Варьирования могут, тем не менее, иметь место в зависимости от массы и состояния пациентов, которые излечиваются, и их индивидуальной реакции на упомянутый медикамент, также как от типа выбранной фармацевтической композиции, периода и интервала времени, во время которого такое введение проводится. В некоторых случаях уровни дозировки ниже нижнего предела упомянутого выше диапазона могут являться более чем достаточными, в то время как в других случаях гораздо большие дозы могут применяться, не вызывая каких-либо нежелательных побочных эффектов, при условии, что такие большие дозы сначала разделяются на несколько небольших доз для введения в течение дня.
Соединения по настоящему изобретению могут быть введены в любой фармацевтической форме, природа которой будет зависеть от пути введения. Эти фармацевтические композиции могут быть приготовлены традиционными способами, с применением совместимых, фармацевтически приемлемых наполнителей или носителей. Примеры таких композиций включают капсулы, таблетки, чрескожные пластыри, ромбы, пастилки, спреи, сиропы, порошки, гранулы, гели, эликсиры, суппозитории, и подобное, препараты для приготовления растворов для немедленного приема, инъецируемые препараты, ректальные, назальные, глазные, вагинальные и т.д. препараты. Предпочтительным путем введения являются пероральный и ректальный пути.
Таблетки, содержащие различные наполнители, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, фосфат дикальция и глицин, могут употребляться для перорального введения в соответствии с различными разрыхляющими агентами, такими как крахмал (предпочтительно кукурузный, картофельный крахмал или крахмал из тапиоки), алгиновая кислота и определенные комплексные силикаты, совместно с гранулирующими связующими, подобными поливинилпирролидону, сахарозе, желатину и гуммиарабику. Кроме того, смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк, могут применяться для целей таблетирования. Твердые композиции подобного типа также могут употребляться в качестве наполнителей в желатиновых капсулах; предпочтительные вещества для этой цели также включают лактозу или молочный сахар, также как высокомолекулярные полиэтиленгликоли. Если водные суспензии и/или эликсиры желательны для перорального введения активного компонента, они могут быть объединены с подсластителями или ароматизирующими веществами, окрашивающим агентом и, если так необходимо, эмульсифицирующими и/или суспендирующими агентами, совместно с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и их различные сочетания.
Лекарственная форма может быть составлена для непосредственного высвобождения, контролируемого высвобождения, продолжительного высвобождения, отсроченного высвобождения или направленного отсроченного высвобождения. Определения этих терминов известны для специалистов в данной области. Кроме того, профиль высвобождения дозированной формы может управляться полимерной смесевой композицией, композицией с покрытой матрицей, композицией в форме отдельных частиц, композицией в форме отдельных покрытых частиц, композицией, основанной на ионообменной смоле, основанной на осмосе композицией, или поддающейся биологическому разложению полимерной композицией. Без необходимости быть связанным с любой теорией, предполагается, что высвобождение может управляться посредством благоприятной диффузии, растворением, размывом покрытия, ионным обменом, осмосом или их сочетаниями.
Для парентерального введения может применяться раствор активного соединения либо в кунжутном, либо в арахисовом масле или в водном пропиленгликоле. Водные растворы должны быть соответствующим образом забуферены {предпочтительно до pH более 8), если необходимо, и жидкий разбавитель сначала приводится к изотонии. Водные растворы пригодны для целей внутривенной инъекции. Приготовление всех этих растворов в стерильных условиях легко достигается стандартными фармацевтическими методиками, хорошо известными специалистам в данной области.
Следующие примеры далее описывают и делают возможным для специалиста в данной области техники осуществлять и применять изобретение. Однако, будет понятно, что эти варианты осуществления предоставлены с целью иллюстрации изобретения, и не должны обсуждаться как ограничивающие объем изобретения, который определен в формуле изобретения.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фигуре 1 представлен индекс активности заболевания на мышах, имеющих TNBS-обусловленный колит после обработки 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислотой (соединение XXVII), месаламином отдельно, 4-гидрокситиобензамидом (4-HTB) отдельно и смесью месаламина и 4-HTB.
На фигуре 2 показана активность миелопероксидазы (MPO) мыши, имеющей TNBS-обусловленный колит после обработки 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислотой (соединение XXVII), месаламином отдельно, 4-HTB отдельно и смесью месаламина и 4-гидрокситиобензамида (4-HTB).
На фигуре 3 показаны величины восприятия боли для месаламина и 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты (соединение XXVII) в присутствии или в отсутствие глибенкламида.
На фигуре 4 представлены индексы восприятия боли для 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты (соединение XXVII), месаламина и 4-гидрокситиобензамида (4-HBT).
На фигуре 5 представлена диаграмма адгезии лейкоцитов на отрезке времени 60-65 минут для 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты (соединение XXVII) в присутствии аспирина или аспирина вместе с глибенкламидом.
На фигуре 6 представлена диаграмма, показывающая генерацию H2S из цистеина, 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты (соединение XXVII) и 4-гидрокситиобензамида (4-HBT).
На фигуре 7a показан индекс абдоминального рефлекса отдергивания (величина AWR) в модели на крысах восприятия висцеральной боли с применением носителя, малеата тримебутина и тиокарбамоилбензоата тримебутина (соединение III).
На фигуре 7b показан индекс абдоминального рефлекса отдергивания (величина AWR) в модели восприятия висцеральной боли на крысах с применением носителя и тиокарбамоилбензоата отдельно.
На фигуре 8 показан индекс желудочного повреждения, измеренный у крыс, обработанных носителем, диклофенаком, 4-гидрокситиобензамидом (TBZ) и 4-тиокарбамоилфениловым эфиром [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (соединение XVII).
На фигуре 9 представлено содержание простагландина E2 (PGE2) в желудке, полученного на крысах, обработанных носителем, диклофенаком, 4-гидрокситиобензамидом (TBZ) и 4-тиокарбамоилфениловым эфиром [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (соединение XVII).
На фигуре 10 представлен индекс желудочного повреждения, измеренный у крыс, обработанных носителем, напроксеном, и 4-тиокарбамилфениловым эфиром 2-(6-метоксинафталин-2-ил)пропионовой кислоты (соединение XX).
На фигуре 11 представлен уровень синтеза тромбоксана B2 в крови крыс фигуры 10.
На фигуре 12 показано количество экссудируемого PGE2, полученного в подкожном кармане крысы с применением исследования воздушного кармана на крысах, при обработке носителем, диклофенаком и 4-тиокарбамоилфениловым эфиром [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (соединение XVII).
На фигуре 13 показано содержание всего тромбоксана B2 (TXB2) в крови крыс по фигуре 12.
На фигуре 14 показано ингибирование увеличения объема лапы крыс, обработанных носителем, диклофенаком и 4-тиокарбамоилфениловым эфиром [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (соединение XVII).
На фигуре 15 показано количество экссудируемого PGE2, полученное в подкожном кармане крысы с применением исследования кармана на крысах при введении носителя, напроксена, и 4-тиокарбамилфенилового эфира 2-(6-метоксинафталин-2-ил)пропионовой кислоты (соединение XX).
На фигуре 16 представлен синтез тромбоксана (нг/мл) в крови человека (in vitro) в виде функции концентрации индометацина и 4-тиокарбамоилфенилового эфира [1-(4-хлорбензоил)-5-метокси-2-метил-1-H-индол-3-ил]уксусной кислоты (соединение XIX).
На фигуре 17 представлена площадь поверхности, в мм2, желудочных язв на крысах после дневного лечения в течение одной недели носителем, диклофенаком, 4-тиокарбамилфениловым эфиром 2-(6-метоксинафталин-2-ил)пропионовой кислоты (соединение XVII), напроксеном и 4-тиокарбамилфениловым эфиром 2-(6-метоксинафталин-2-ил)пропионовой кислоты (соединение XX).
На фигуре 18 показан прирост систолического давления крови (мм Hg) на крысах, обработанных носителем, напроксеном и 4-тиокарбамилфениловым эфиром 2-(6-метоксинафталин-2-ил)пропионовой кислоты (соединение XX).
На фигуре 19 показано количество сероводорода, образуемого из 4-гидрокситиобензамида (TBZ) и 4-тиокарбамоилфенилового эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (соединение XVII) при инкубировании в буфере и в гомогенате печени.
На фигуре 20 показано действие симвастатина и смешанного 4-тиокарбамоилфенилового эфира 2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8a-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-илового эфира янтарной кислоты (соединение I) на ADP-обусловленную агрегацию тромбоцитов человека.
На фигуре 21 показано действие симвастатина и 4-тиокарбамоилфенилового эфира 2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8a-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-илового эфира янтарной кислоты (соединение I) на концентрации cAMP тромбоцитов человека.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Получение соединений
Тонкослойную хроматографию проводили на силикагелевых пластинках Macherey-Nagel 50 с флуоресцентным индикатором и пластинки осматривали в УФ-свете (254 нм). Для колоночной хроматографии использовали Kieselgel 60. Все синтетические реагенты покупались в химической компании Aldrich-Sigma и использовались без очистки. Растворители были аналитического класса чистоты или высшей чистоты и использовались в том виде, как они поставлялись. Роторный испаритель Buchi R-114 использовали для удаления растворителей в вакууме. Структуры устанавливались спектроскопическими методами 1H-ЯМР на протонах и 13C-ЯМР. Спектры измеряли на приборе Varian Mercury Plus 400. Химические сдвиги измеряли относительно Me4Si в качестве внутреннего стандарта. Масс-спектры синтезированных продуктов получали на масс-спектрометрометре Applied BioSystem API 2000. Точки плавления измеряли на приборе Buchi B-540. Чистоту конечного соединения определяли методом RP-HPLC. Колонка была присоединена к инжектору модели Rheodyne 7725, система для HPLC Waters 600, перестраиваемым детектором оптической плотности Waters 486, установленным на длину волны 215 или 235 нм, и записывающему устройству Waters 746. Синтезированные соединения имели удовлетворительные элементные анализы; анализы проводились только по ключевым элементам, результаты находились в пределах ±0,4% от теоретических величин.
ПРИМЕР 1. Синтез 4-тиокарбамоилфенилового эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (также обозначается как Соединение XVII)

Синтез 4-карбамоилфенил 2-[2-(2,6-дихлорфениламино)фенил]ацетата (5)
К раствору 1 (диклофенак, 890 мг, 3,0 ммоль) в 50 мл N,N-диметилформамида, добавляли гидроксибензотриазол (445 мг, 3,3 ммоль) и DCC (680 мг, 3,3 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси добавляли 4-гидроксибензамид (4, 616 мг, 4,5 ммоль) и перемешивали в течение 1 часа при 0°C и 3 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении, масляный остаток, полученный таким образом, растворяли в хлороформе; органический слой промывали насыщенным солевым раствором, высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9/1), c которой получали 4-карбамоилфенил-2-(2-(2,6-дихлорфениламино)фенил)ацетат (5) (212 мг, 17% выход).
Синтез 4-тиокарбамоилфенилового эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (6)
4-Карбамоилфенил 2-(2-(2,6-дихлорфениламино)фенил)ацетат (5, 480 мг, 1,14 ммоль) и реагент Лавессона (460 мг, 1,14 ммоль) растворяли в 20 мл безводного бензола. Реакционную смесь отогревали до 50°C и перемешивали в течение 6 часов. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5/0,5) для получения чистого соединения 6 (446 мг, 91% выход).
1H-ЯМР (CDCl3): δ 4,07 (с, 2H), 6,59 (д, 1H), 6,67 (с, 1H), 6,98 (т, 1H), 7,14 (т, 1H), 7,19 (д, 1H), 7,28 (т, 1H), 7,33 (д, 2H), 7,63 (с, 1H), 7,97 (д, 2H);
13C-ЯМР (ДМСО-d6): δ 38,8, 118,8, 121,8, 122,6, 123,7, 124,4, 128,7, 129,1, 129,6, 131,2, 137,2, 137,8, 142,9, 153,5, 170,5, 193,2, 201,7.
MS (EI), m/e 431 (M+);
т.пл. 170-172°C.
ПРИМЕР 2. Синтез 4-тиокарбамоилфенил-2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (также обозначается как Соединение XVIII)

Синтез 4-карбамоилфенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (5)
К раствору 1 (лумиракоксиб, 223 мг, 0,75 ммоль) в 15 мл диметилформамида, добавляли гидроксибензотриазол (111 мг, 0,825 ммоль) и DCC (170 мг, 0,825 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси добавляли 4-гидроксибензамид (4, 154 мг, 1,125 ммоль) и перемешивали в течение 1 часа при 0°C и 3 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении для удаления растворителя. Масляный остаток, полученный таким образом, растворяли в хлороформе; органический слой промывали насыщенным солевым раствором, высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9/1), после колонки получали 4-карбамоилфенил-2-(2-(2-хлор-6-фторфениламино)-5-метилфенил) ацетат (5) (111 мг, 35% выход).
Синтез 4-тиокарбамоилфенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (6)
4-Карбамоилфенил-2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетат, 5 (110 мг, 0,27 ммоль) и реагент Лавессона (109 мг, 0,27 ммоль) растворяли в 15 мл безводного бензола. Реакционную смесь отогревали до 60°C и перемешивали в течение 3 часов. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5:0,5) для получения чистого соединения 6 (59 мг, выход 51%).
1H-ЯМР (CDCl3): δ 2,32 (с, 3H), 4,01 (с, 2H), 6,46 (с, 1H), 6,70 (д, 1H), 6,92 (т, 1H), 7,01 (д, 2H), 7,11 (д, 2H), 7,19 (д, 1H), 7,62 (с, NH), 7,84 (д, 2H);
13C-ЯМР (ДМСО-d6): δ 20,8, 30,7, 115,1, 119,2, 122,0, 122,3, 124,1, 124,9, 126,1, 128,2, 129,2, 132,3, 134,8, 138,6, 140,9, 153,7, 154,6, 156,2, 170,4, 201,7
MS (EI), m/e 429 (M+); т.пл.: 120-122°C.
ПРИМЕР 3. Синтез 4-тиокарбамоилфенилового эфира 2-ацетоксибензойной кислоты (также обозначается как Соединение XVI)

Синтез 4-карбамоилфенил 2-ацетоксибензоата (5)
К раствору 1 (ацетилсалициловая кислота, 500 мг, 2,77 ммоль) в 15 мл диметилформамида, добавляли гидроксибензотриазол (412 мг, 3,05 ммоль) и DCC (628 мг, 3,05 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси добавляли 4-гидроксибензамид (4, 418 мг, 3,05 ммоль) и перемешивали в течение 1 часа при 0°C и 3 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении для удаления растворителя. Масляный остаток, полученный таким образом, растворяли в хлороформе; органический слой промывали насыщенным солевым раствором, высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9/1), с колонки получали 4-карбамоилфенил 2-ацетоксибензоат (5) (410 мг, 47% выход).
Синтез 4-тиокарбамоилфенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (6)
4-Карбамоилфенил 2-ацетоксибензоат, 5 (410 мг, 1,37 ммоль) и реагент Лавессона (554 мг, 1,37 ммоль) растворяли в 35 мл безводного бензола. Реакционную смесь отогревали до 60°C и перемешивали в течение 3 часов. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5:0,5) для получения 470 мг неочищенного соединения 6. Полученное соединение очищали методом препаративной HPLC, проводимой в системах двух растворителей: A: 100% ацетонитрил с 0,1% TFA, B: 100% H2O с 0,1% TFA (линейный градиент от 10% A до 60% A в течение 35 мин, УФ-детектирование при 254 нм, скорость потока 30 мл/мин), что дало чистое соединение 6 (324 мг, выход 71%).
1H-ЯМР (CDCl3): δ 2,30 (с, 3H), 7,17 (д, 1H), 7,21 (д, 2H), 7,40 (т, 1H), 7,66 (т, 1H), 7,94 (д, 2H), 8,2 (д, 1H).
13C-ЯМР (ДМСО-d6): δ 21,2, 121,9, 122,4, 124,3, 126,4, 128,7, 132,4, 135,1, 137,3, 151,5, 153,7, 162,7, 169,8, 201,8.
MS (EI), m/e 316(M+); т.пл.: 154-156°C.
ПРИМЕР 4. Синтез 4-тиокарбамоилфенилового эфира [1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-ил]уксусной кислоты (также обозначается как Соединение XIX)
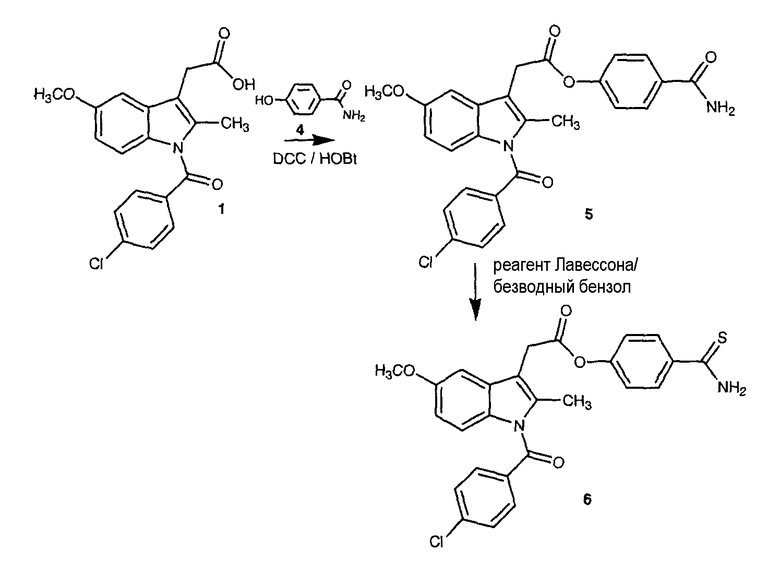
Синтез 4-карбамоилфенил 2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетата (5)
К раствору 1 (индометацин, 3 г, 8,38 ммоль) в 60 мл диметилформамида добавляли гидроксибензотриазол (1,25 г, 9,22 ммоль) и DCC (1,9 г, 9,22 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси 4-гидроксибензамида (4, 1,72 г, 12,6 ммоль) добавляли и перемешивали в течение 1 часа при 0°C и 2 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении для удаления растворителя. Масляный остаток, полученный таким образом, растворяли в этилацетате; органический слой промывали насыщенным солевым раствором, 5% раствором NaHCO3, 10% лимонной кислотой и затем высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), с колонки получали 4-карбамоилфенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетат (5) (479 мг, 12% выход).
Синтез 4-тиокарбамоилфенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетата (6)
Растворяли 4-карбамоилфенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетат 5 (340 мг, 0,71 ммоль) и реагент Лавессона (287 мг, 0,71 ммоль) в 15 мл безводного бензола. Реакционную смесь отогревали до 60°C и перемешивали в течение 4 часов. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5:0,5) для получения 178 мг неочищенного соединения 6. Полученное соединение очищали методом препаративной HPLC, проводимой в системах с двумя растворителями: A: 100% ацетонитрил с 0,1% TFA, B: 100% H2O 0,1% TFA (линейный градиент от 10% A до 80% A в течение 30 мин, УФ-детектирование при 254 нм, скорость потока 30 мл/мин), что дало чистое соединение 6 (56 мг, 16% выход).
1H-ЯМР (CDCl3): δ 2,45 (с, 3H), 3,83 (с, 3H, OCH3), 3,91 (с, 2H), 6,70 (д, 1H), 6,88 (д, 1H), 7,04 (с, 1H), 7,11 (д, 2H), 7,47 (д, 2H), 7,67 (д, 2H), 7,88 (д, 2H).
13C-ЯМР (ДМСО-d6): δ 13,6, 30,8, 56,0, 101,5, 111,9, 112,0, 115,3, 121,7, 128,6, 129,4, 130,8, 131,2, 131,4, 134,0, 136,8, 137,1, 139,7, 156,2, 157,9, 167,6, 169,8, 201,8.
MS (EI), m/e 493 (M+); т.пл.: 224-226°C.
ПРИМЕР 5. Синтез 4-тиокарбамилфенилового эфира 2-(6-метоксинафталин-2-ил)пропионовой кислоты (также обозначается как Соединение XX)
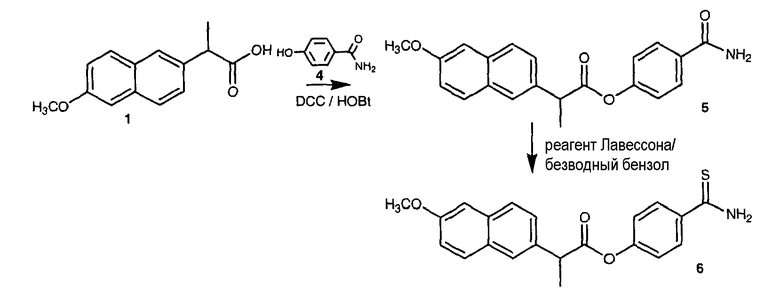
Синтез 4-карбамоилфенил 2-(2-метоксинафталин-6-ил)пропаноата (5).
К раствору 1 (напроксен, 4 г, 17,4 ммоль) в 80 мл диметилформамида добавляли гидроксибензотриазол (2,59 г, 19,14 ммоль) и DCC (2,59 г, 19,14 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси добавляли 4-гидроксибензамид (4, 3,58 г, 26,1 ммоль) и перемешивали в течение 1 часа при 0°C и 2 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении для удаления растворителя. Масляный остаток, полученный таким образом, растворяли в этилацетате; органический слой промывали насыщенным солевым раствором, 5% раствором NaHCO3, 10% лимонной кислотой и затем высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), с колонки получали 4-карбамоилфенил-2-(2-метоксинафталин-6-ил)пропаноат (5) (1,91 г, 32% выход).
Синтез 4-тиокарбамоилфенил 2-(2-метоксинафталин-6-ил)пропаноат (6)
4-Карбамоилфенил 2-(2-метоксинафталин-6-ил)пропаноат 5 (1,80 г, 4,34 ммоль) и реагент Лавессона (1,75 г, 4,34 ммоль) растворяли в 130 мл безводного бензола. Реакционную смесь отогревали до 60°C и перемешивали в течение 4 часов. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,75:0,25) для получения 2,9 г неочищенного соединения 6. Полученное соединение очищали на открытой колонке с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5)), что дало чистое соединение 6 (970 мг, выход 61%).
1H-ЯМР (ДМСО-d6): δ 1,59 (д, 3H), 3,86 (с, 3H, OCH3), 4,24 (дд, 1H), 7,06 (д, 2H), 7,18 (д, 1H), 7,31 (с, 1H), 7,50 (д, 1H), 7,84 (с, 1H) 7,85 (д, 1H), 7,86 (с, 1H), 7,89 (д, 2H), 9,47 и 9,84 (с, 2H, NH2).
13C-ЯМР (ДМСО-d6): δ 19,1, 45,2, 55,9, 106,5, 119,6, 121,6, 126,6, 126,9, 128,0, 129,4, 129,9, 134,2, 135,6, 137,8, 153,4, 158,1, 173,3, 199,7. MS (EI), m/e 366 (M+);
т.пл.: 196-198°С.
ПРИМЕР 6. Синтез 4-тиокарбамоилфенил 2-(4-изобутилфенил)пропаноата
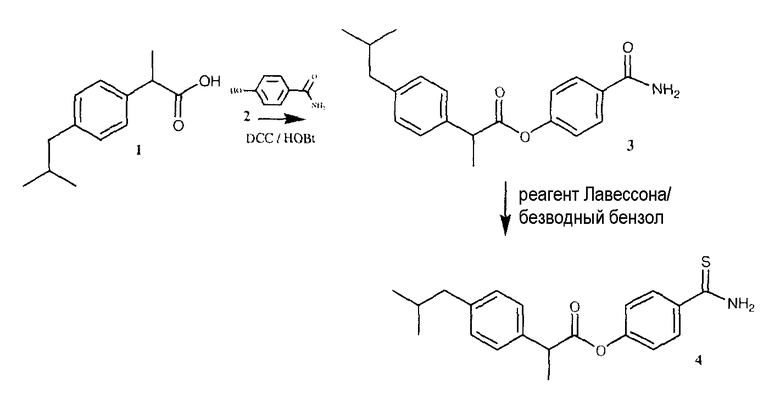
К раствору 1 (ибупрофен, 3,87 г, 18,8 ммоль) в 80 мл диметилформамида, добавляли гидроксибензотриазол (2,8 г, 20,7 ммоль) и DCC (4,27 г, 20,7 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси добавляли 4-гидроксибензамид (2, 3,9 г, 28 ммоль) и перемешивали в течение 1 часа при 0°C и 2 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении для удаления растворителя. Масляный остаток, полученный таким образом, растворяли в этилацетате; органический слой промывали насыщенным солевым раствором, 5% раствором NaHCO3, 10% лимонной кислотой и затем высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенный продукт 3 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), с колонки получали 4-карбамоилфенил 2-(4-изобутилфенил)пропаноат (3) (2,48 г, выход 40%).
Синтез 4-тиокарбамоилфенил 2-{4-изобутилфенил)пропаноата (4)
4-карбамоилфенил 2-{4-изобутилфенил)пропаноат, 3 (2,48 г, 7,62 ммоль) и реагент Лавессона (3,1 г, 7,62 ммоль) растворяли в 130 мл безводного бензола. Реакционную смесь отогревали до 60°C и перемешивали в течение 4 часов. Растворитель удаляли при пониженном давлении. Полученное соединение очищали на открытой колонке с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), что дало чистое соединение 4 (1,45 г, 55% выход).
1H-ЯМР (ДМСО-d6): δ 0,84 (д, 6H), 1,48 (д, 3H), 1,79-1,82 (м, 1H), 2,42 (д, 2H), 4,05 (дд, 1H), 7,05 (д, 2H), 7,15 (д, 2H), 7,28 (д, 2H) 7,88 (д, 2H), 9,49 и 9,87(с, 2H, NH2).
13C-ЯМР (ДМСО-d6): δ 19,2, 22,9, 30,3, 44,9, 121,6, 127,9, 129,5, 130,0, 137,8, 138,0, 140,8, 153,3, 173,3, 199,6.
MS (EI), m/e 341 (M+); Т.пл.: 121-123°C.
ПРИМЕР 7. Синтез 4-тиокарбамоилфенил-2-(4-оксофенил)фенилпропаноата
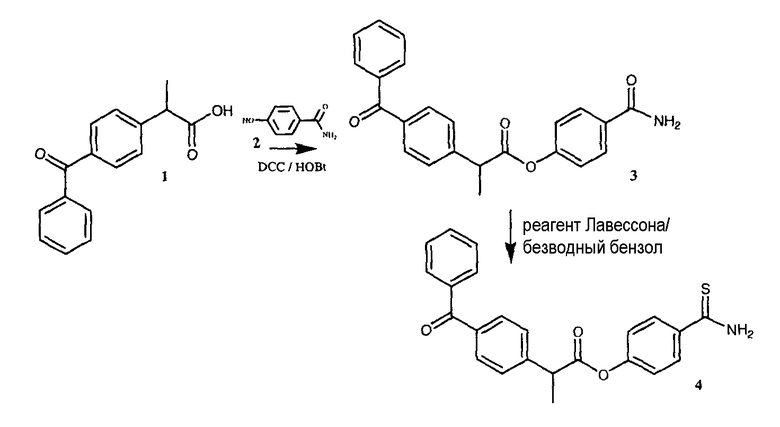
Синтез 4-карбамоилфенил-2-(4-оксофенил)фенилпропаноата(3)
К раствору 1 (кетопрофен, 3 г, 11,8 ммоль) в 80 мл диметилформамида добавляли гидроксибензотриазол (1,76 г, 13 ммоль) и DCC (2,68 г, 13 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси добавляли 4-гидроксибензамид (2, 2,43 г, 17,7 ммоль) и перемешивали в течение 1 часа при 0°C и 2 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении для удаления растворителя. Масляный остаток, полученный таким образом, растворяли в этилацетате; органический слой промывали насыщенным солевым раствором, 5% раствором NaHCO3, 10% лимонной кислотой и затем высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенный продукт 3 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), из которой получали 4-карбамоилфенил-2-(4-оксофенил)фенилпропаноат (3) (1,84 г, 42% выход).
Синтез 4-тиокарбамоилфенил-2-(4-оксофенил)фенилпропаноата (4)
4-карбамоилфенил-2-(4-оксофенил)фенилпропаноат (3) (1,84 г, 4,93 ммоль) и реагент Лавессона (2 г, 4,93 ммоль) растворяли в 100 мл безводного бензола. Реакционную смесь отогревали до 60°C и перемешивали в течение 4 часов. Растворитель удаляли при пониженном давлении. Полученное соединение очищали на открытой колонке с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), что дало чистое соединение 4 (0,45 г, выход 23%).
1H-ЯМР (ДМСО-d6): δ 1,53 (д, 3H), 4,25 (дд, 1H), 7,08 (д, 2H), 7,54-7,73 (м, 9H), 7,90 (д,2H), 9,51 и 9,88 (с, 2H, NH2).
13C-ЯМР (ДМСО-d6): δ 19,2, 44,9, 121,6, 129,3, 129,5, 129,8, 130,3, 132,6, 133,5, 137,6, 137,9, 138,1, 141,2, 153,3, 154,5, 156,1, 163,8, 172,9, 199,6.
MS (EI), m/e 390 (M+); Т.пл.: 114-116°C.
ПРИМЕР 8. Синтез 4-тиокарбамоилфенил-2-(3-фтор-4-фенил)фенилпропаноата
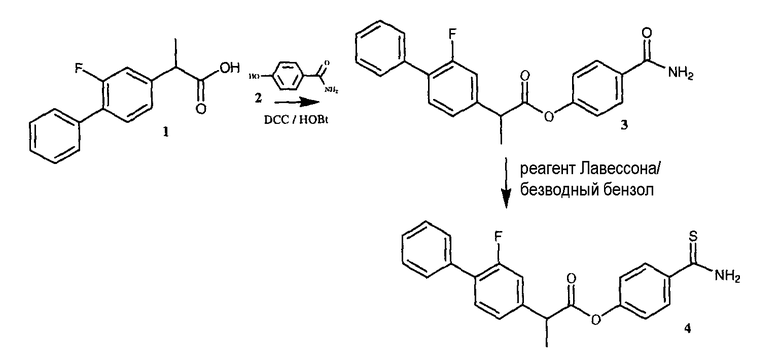
Синтез 4-карбамоилфенил-2-(3-фтор-4-фенил)фенилпропаноата (3)
К раствору 1 (флурбипрофен, 2 г, 8,2 ммоль) в 80 мл диметилформамида добавляли гидроксибензотриазол (1,22 г, 9,02 ммоль) и DCC (1,86 г, 9,02 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси добавляли 4-гидроксибензамид (2, 1,7 г, 12,2 ммоль) и перемешивали в течение 1 часа при 0°C и 2 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении для удаления растворителя. Масляный остаток, полученный таким образом, растворяли в этилацетате; органический слой промывали насыщенным солевым раствором, 5% раствором NaHCO3, 10% лимонной кислотой и затем высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенный продукт 3 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), из которой 4-карбамоилфенил-2-(3-фтор-4-фенил)фенилпропаноат (3) получали (1,09 г, 37% выход).
Синтез 4-тиокарбамоилфенил-2-(3-фтор-4-фенил)фенилпропаноата (4)
4-карбамоилфенил-2-(3-фтор-4-фенил)фенилпропаноат 3 (1,09 г, 3 ммоль) и реагент Лавессона (1,21 г, 3 ммоль) растворяли в 70 мл безводного бензола. Реакционную смесь отогревали до 60°C и перемешивали в течение 4 часов. Растворитель удаляли при пониженном давлении. Полученное соединение очищали на открытой колонке с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), что дало чистое соединение 4 (0,35 г, 31% выход).
1H-ЯМР (ДМСО-d6): δ 1,55 (д, 3H), 4,21 (дд, 1H), 7,32-7,55 (м, 8H), 7,90 (д, 2H), 9,51 и 9,88 (с, 2H, NH2).
13C-ЯМР (ДМСО-d6): δ 19,1, 44,7, 115,9, 116,2, 121,7, 124,8, 128,6, 129,3, 129,4, 129,5, 131,7, 135,8, 137,7, 142,6, 153,7, 158,3, 163,5, 173,1, 199,6.
MS (EI), m/e 380 (M+); Т.пл.: 142-144°C.
ПРИМЕР 9. Общая схема синтеза для 4-тиокарбамоилфениловых эфиров 4- или 5-амино-2-гидроксибензойной кислоты (8) (также обозначается как Соединение XXVII)
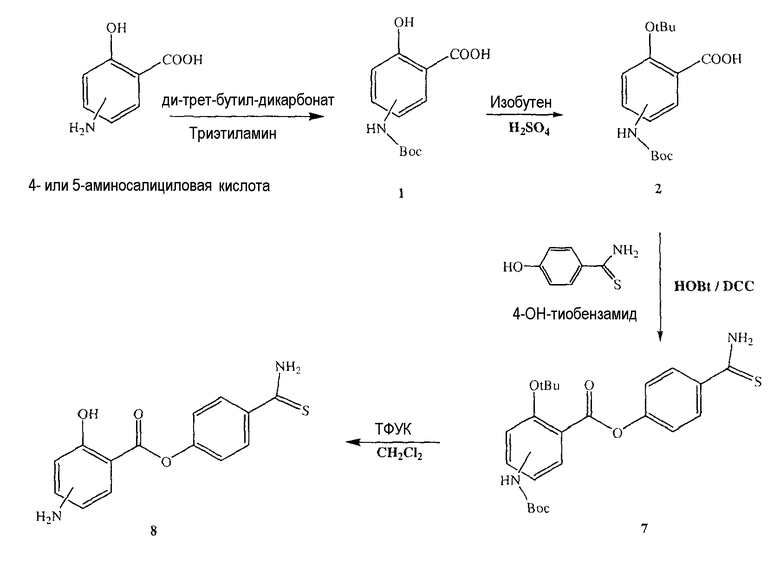
Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 4- или 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды, добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль) при перемешивании при 0°C в течение 1/2 часа. Реакционную смесь перемешивали механически в течение 24 часов при комнатной температуре. После упаривания растворителя к остатку по каплям добавляли 3М HCl (15 мл). Осадок фильтровали, промывали водой и высушивали. Остаток загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9/1), с колонки получали 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2)
Смесь соединения (1) (12,0 ммоль), концентрированной H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере изобутиленового газа (5 psi) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл), насыщенным солевым раствором (100 мл), высушивали (Na2SO4) и упаривали. Остаток растворяли в смеси 1:1 MeOH/CCl4 (400 мл), промывали водой (300 мл), затем экстрагировали смесью 1:1 MeOH/вода (2×200 мл). Экстракт сушили (Na2SO4) и упаривали до получения белого твердого вещества (2), которое перекристаллизовывали из смеси DCM/гексан (выход 83%).
Синтез 4-тиокарбамоилфенилового эфира 4- или 5-амино-2-гидроксибензойной кислоты (8)
К раствору 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (2) (3,0 ммоль) в 50 мл диметилформамида, добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль) при перемешивании при 0°C в течение 1 часа. К реакционной смеси добавляли 4-гидрокситиобензамид (3,0 ммоль) и перемешивали механически в течение 3 часов при 0°C и 72 часа при комнатной температуре. После фильтрования фильтрат упаривали при пониженном давлении для удаления растворителя. Масляный остаток, полученный таким образом, растворяли в этилацетате; органический слой промывали насыщенным солевым раствором, высушивали над безводным MgSO4, фильтровали и растворитель упаривали. Неочищенное промежуточное соединение (7) обрабатывали 40% TFA раствором в CH2Cl2. Через 2 часа растворитель удаляли, что приводило к соединению (8) в виде неочищенного остатка. Остаток загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (8/2), с колонки получали 4-тиокарбамоилфениловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (8), соединение формулы XXVII (выход 48%).
ПРИМЕР 10. Синтез тримебутинтиокарбамоилбензоата
Получение 4-тиокарбамоилбензоата 2-(диметиламино)-2-фенилбутилового эфира 3,4,5-триметоксибензойной кислоты (тримебутинтиокарбамоилбензоат)
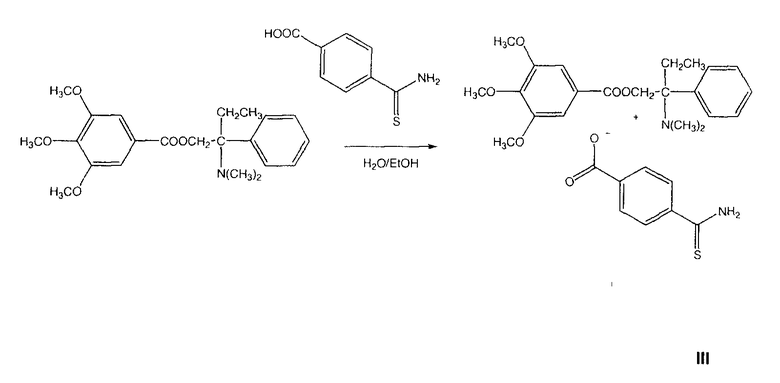
К смеси 4-(тиокарбамоил)бензойной кислоты (0,1 моль) и тримебутина (0,1 моль) добавляли смесь воды (200 мл) и этилового спирта (20 мл) и полученную суспензию перемешивали при комнатной температуре до тех пор, пока она не становилась прозрачной. Затем раствор замораживали и лиофилизировали для получения желаемой соли (выход количественный).
1H-ЯМР (400 МГц, ДМСО-d6): δ 0,60 (т, 3H), 1,45-1,75 (м, 4H), 1,80-1,90 (м, 2H), 2,28 (с, 6H), 2,90-3,40 (м, 2H), 3,69 (с, 9H), 3,95 (м, 1H), 4,73 (дд, 2H), 7,01 (с, 2H), 7,22 (т, 1H), 7,35 (т, 2H), 7,46 (д, 2H) 7,93 (дд, 4H), 9,65 (ушир.с, 1H, NH), 10,05 (ушир.с, 1H, NH).
13C-ЯМР (400 МГц, ДМСО-d6): δ 9,07, 28,9, 56,5, 60,8, 64,5, 65,7, 107,1, 125,3, 127,4, 128,1, 128,6, 129,5, 129,7, 132,3, 141,8, 142,5, 148,5, 153,4, 154,8, 165,9, 169,4, 172,5, 188,6.
Т.пл. 66-68°C (разл).
Синтез 4-(тиокарбамоил)бензойной кислоты
Соединение синтезировали по методике, ранее описанной в литературе (Fairfull, E.S., Lowe J.L, Peak D.A. J. Chem. Soc. 1952, 742), которая включена в данное описание посредством ссылки.
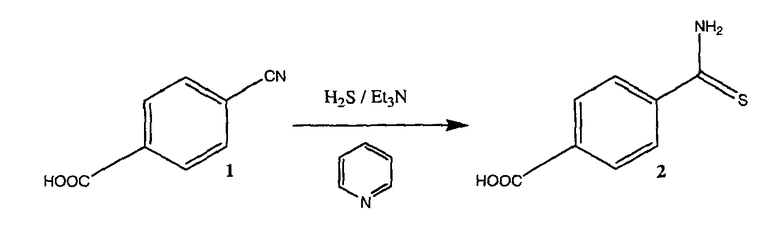
4-{Тиокарбамоил)бензойная кислота (2)
3 г 4-цианобензойной кислоты 1 (20,4 ммоль) растворяли в 40 мл пиридина и добавляли 2,1 мл триэтиламина (20,4 ммоль). Сухой сероводород пропускали через раствор постоянным потоком в течение 4 часов. Затем смесь выливали в воду и твердое вещество собирали фильтрованием, перекристаллизация из петролейного эфира приводила к 2,51 г чистого соединения 2 (выход 68%).
MS (ESI), m/e 182,2 (M+).
1H-ЯМР (ДМСО-d6): δ 7,92 (дд, 4H), 9,68 (с, 1H, NH), 10,12 (с, 1H, NH), 13,25 (с, 1H, OH).
13C-ЯМР (ДМСО-d6): δ 127,3, 129,6, 132,0, 148,5, 169,4, 188,6. Т. пл. 296-298°C (разл.)
ПРИМЕР 11. Синтез смешанного 4-тиокарбамоилфенилового эфира - 2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8a-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-ильного эфира янтарной кислоты (3) (также обозначается как Соединение I)

Синтез смешанного 4-карбамоилфенилового эфира - 2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8a-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-илового эфира янтарной кислоты (2)
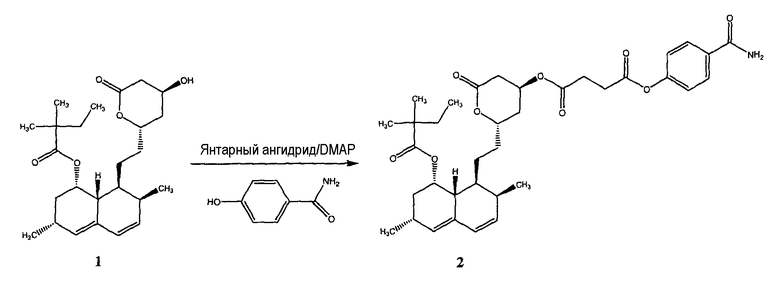
Раствор 420 мг (0,001 моль) симвастатина (1) в 3 мл дихлорметана обрабатывали 110 мг янтарного ангидрида и 10 мг DMAP. Через 36 часов при перемешивании добавляли 210 мг (0,001 моль) EDCI и 170 мг (0,0012 моль) 4-гидроксибензамида.
Через 1 час растворитель удаляли при пониженном давлении и неочищенный остаток очищали на колонке с силикагелем, при элюировании смесью дихлорметан/метиловый спирт (9,5/0,5) с получением соединения 2 в виде белого твердого вещества (350 мг; выход 55%).
MS (EI), m/e 638 (M+);
1H-ЯМР (ДМСО-d6) δ 0,831 (м, 6H, 2-Me), 1,075 (м, 9H, 3-Me), 1,53 (м, 6H), 1,97 (м, 2H), 2,27 (м, 5H), 2,52 (д, 2H), 2,62 (д, 2H), 3,68 (м, 1H), 4,07 (м, 1H), 5,52 (м, 1H), 5,50 (ушир.т, 1H), 5,77 (дд, 1H), 5,96 (д, 1H); 7,08 (д, 2H), 7,87 (д, 2H), 7,94 (ушир.с, 2H).
Синтез смешанного 4-тиокарбамоилфенилового эфира - 2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8a-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-илового эфира янтарной кислоты (3)
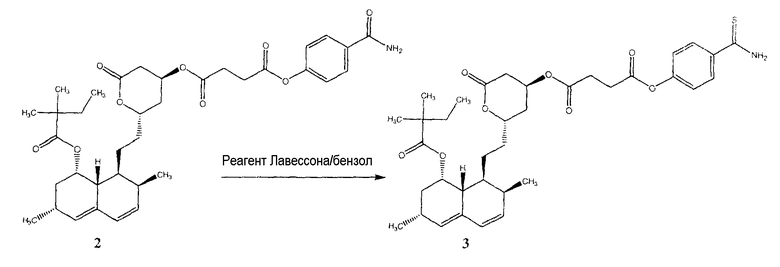
Смешанный 4-карбамоилфениловый эфир - 2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8a-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-иловый эфир янтарной кислоты (2) (0,35 г, 0,000548 моль) и реагент Лавессона (0,221 г, 0,000548 моль) растворяли в 30 мл безводного бензола. Реакционную смесь отогревали до 50°C и перемешивали в течение 6 часов. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5:0,5) для получения 35 мг чистого соединения 3 (выход 10%).
MS (EI), m/e 654 (M+);
1H-ЯМР (ДМСО) δ 0,831 (м, 6H, 2-Me), 1,075 (м, 9H, 3-Me), 1,53 (м, 6H), 1,97 (м, 2H), 2,27 (м, 5H), 2,52 (д, 2H), 2,62 (д, 2H), 3,68 (м, 1H), 4,07 (м, 1H), 5,52 (м, 1H), 5,50 (ушир.т, 1H), 5,77 (дд, 1H), 5,96 (д, 1H); 7,11 (д, 2H), 7,9 (д, 2H), 9,48 (с, 1H), 9,86 (с, 1H).
ТЕСТИРОВАНИЕ СОЕДИНЕНИЙ
ПРИМЕР 12. Сравнение индексов активности заболевания и миелопероксидазной (MPO)-активности 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты
Стандартная экспериментальная животная модель колита, вызванного введением в толстую кишку мыши 2,4,6-тринитробензолсульфоновой кислоты (TNBS) используется в следующем примере. Подробное описание этой модели опубликовано (Santucci et al. (2003) Gastroenterology 124:1381-94), работа включена в данное описание в качестве справочного материала. Кратко, Balb/c-мыши 6-8-недельного возраста получали TNBS введением в толстую кишку при дозе 1,5 мг в 0,1 мл 30% этанола. Мышей рандомизовали для различных групп обработки (n=6 на группу). Начиная на один час позже и продолжая каждые 12 часов в течение 5 дней, мышей обрабатывали перорально носителем (1% карбоксиметилцеллюлозой (CMC)), отдельно 5-ASA (месаламин) (100 мг/кг), 4-гидрокситиобензамидом (обозначается на фигурах как 4-HTB) (100 мг/кг), 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислотой (100 мг/кг) (в дальнейшем обозначается как Соединение XXVII) и эквимолярными дозами месаламина (50 мг/кг) и (4-HTB) (50 мг/кг). *p<0,05 приведено по отношению к группе, обработанной носителем. Каждая группа состояла из по меньшей мере 5 животных.
Мышей исследовали (слепым методом) в день окончания эксперимента на присутствие диареи, тестировали на скрытую кровь и измеряли их массы тел. "Коэффициент развития болезни" вычисляли, основываясь на этих данных (по шкале от 0 до 4, как описано в работе, цитированной выше). После умерщвления вырезали образец толстой кишки для измерения активности миелопероксидазы (MPO) как маркера просачивания гранулоцитов. Все результаты приводили по сравнению с результатами, полученными на здоровых мышах аналогичным образом.
На фигуре 1 показано, что соединение XXVII является почти в три раза более эффективным по сравнению или с месаламином отдельно, 4-HTB отдельно или смесью месаламина и 4-HTB в отношении ослабления симптомов болезни. Далее, Фигура 2 показывает, что соединение XXVII значительно уменьшает воспаление, что показано уменьшением просачивания гранулоцитов (уменьшенная MPO-активность).
ПРИМЕР 13. Сравнение действия 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты и месаламина в модели восприятия висцеральной боли на крысах
В следующем примере применяли модель восприятия висцеральной боли на крысах, преклиническую модель спастического колита. Крысы (самец, Вистар, 200-250 г, получены от Charles River, Monza, Italy), выдерживались в пластиковых клетках и в контролируемых условиях с 12-часовым циклом день-ночь, с включением света в 7:00 утра. Водопроводная вода и стандартное лабораторное питание были неограниченно доступными. До экспериментов крысы индивидуально тренировались, находясь 2-3 часа в день в клетке из оргстекла в течение 2-3 дней. Это позволяло им приспосабливаться к среде движение-ограничение. Корм был недоступен в течение 12 часов до проведения регистрации колоректального растяжения (CRD). Эксперименты проводили с пробужденными крысами и проводили слепым методом, в котором наблюдатель не был проинформирован о природе лекарственного вещества, вводимого каждому животному.
В день тестирования крыс усыпляли эфирной ингаляцией и 2-см резиновый баллон вставляли ректально на 2 см от края анального отверстия и фиксировали при основании хвоста. Баллон был связан через двухканальный катетер с датчиком давления для постоянного контроля ректального давления компьютером (PowerLab PC, A.D. Instruments, Milford, MA, USA) и со шприцом для накачивания/спускания воздуха из баллона. Затем крысы помещались в небольшие клетки (20×8×8 см) на повышенной платформе Plexiglas™, им позволяли проснуться и приспособиться в течение 1 часа. После восстановления от усыпления с животными проводили методику CRD и тестировали их поведенческие отклики. За ночь до экспериментов баллоны накачивали и оставляли в течение ночи, так что латекс растягивался и баллоны становились податливыми.
20-секундный CRD, проводимый каждые 5 минут, применяли с приращением в 0,4 мл от исходных 0,4 мл до 1,6 мл воды. Для достижения точного измерения характеристик толстой кишки и восприятия растяжения повторяли дважды для каждой интенсивности и данные для каждого животного усредняли для анализа. Каждое животное проходило двойной набор CRD. Через двадцать минут после первой последовательности CRD (0,4 мл - 1,6 мл воды), лекарственные вещества вводили внутрибрюшинно (i.p.) и проводили вторую последовательность CRD. Поведенческие отклики в течение первой и второй последовательности CRD регистрировали и сравнивали.
Поведенческий отклик на CRD оценивали путем измерения абдоминального рефлекса отдергивания (AWR) с применением полуколичественного измерения (1). AWR представляет собой неконтролируемый двигательный рефлекс, аналогичный висцеромоторному рефлексу, но он имеет большое преимущество в том, что, в противоположность последнему, не требует абдоминального хирургического вмешательства для имплантирования пишущих электродов и наличия проволок в стенке абдоминальной мышцы, которые могут вызывать добавочное повышение чувствительности (Ness, T.J. and Gebhart, G.F. (1990) Pain 41:167-234, которая включена в данное описание посредством ссылки).
Измерение AWR, состоящее в визуальном наблюдении отклика животного CRD различных уровней наблюдателем в слепом методе и соотнесении величины AWR согласно поведенческой шкале, которая ранее описана в Al-Chaer, E.D. et al. (2000) Gastroenterology 19: 1276-85, работа включена в данное описание посредством ссылки, в которой величине 0 соответствует отсутствие поведенческого отклика на CRD, величине 1 соответствует краткое движение головой при начале действия раздражителя, затем наблюдается неподвижность, величине 2 соответствует мягкое сокращение абдоминальных мускулов, хотя крысы не поднимают живот с платформы, величине 3 соответствует сильное сокращение абдоминальных мускулов с подъемом живота с платформы и величине 4 соответствует жесткое сжатие абдоминальной мышцы, которое подтверждается изгибанием тела дугой и подъемом живота, тазовых структур и мошонки.
Модель восприятия висцеральной боли на крысах, как описано выше, применяли для сравнения величин реакции на 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойную кислоту (соединение XXVII) или с, или в отсутствие глибенкламида, ингибитора АТФ-чувствительных K+ (KATP) каналов.
Фигура 3 показывает единицы восприятия боли как отклик на 0,8 мл колоректального растяжения в группах крыс (по меньшей мере 5 на группу), получавших носитель, месаламин (100 мг/кг), и соединение XXVII (100 мг/кг). Соединение XXVII значительно уменьшает восприятие боли (*p<0,05 по отношению к группе, обработанной носителем), в то время как месаламин не имеет значительного эффекта. Уменьшение восприятия боли соединением XXVII обращалось путем предварительной обработки глибенкламидом (10 мг/кг i.p. за 30 мин до), в то время как предварительная обработка глибенкламидом не влияет на восприятие боли в группах, получавших носитель или месаламин, в предположении, что антиноцицептивная активность соединения XXVII может быть опосредована АТФ-чувствительными K+ (KATP) каналами. Фигура 4 показывает, что 4-гидрокситиобензамид (4-HTB) отдельно (100 мг/кг) не имеет значительного эффекта на восприятие боли.
ПРИМЕР 14. Влияние 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты на адгезию лейкоцитов к эндотелию сосудов in vivo.
Адгезию лейкоцитов исследовали с применением прижизненной микроскопия, как описано в деталях ранее (Wallace et al., (1993) Am. J. Physiol. 265: 993-998, которая включена в данное описание посредством ссылки). Крыс анестезировали действием пентобарбитала натрия (60 мг/кг i.p.) и производили рассечения прижиганием вдоль абдоминальной области. Трахеотомию проводили для облегчения дыхания. Крыс помещали в положение на спине, и сегмент брыжейки временно выводили на поверхность через абдоминальный разрез. Брыжейку аккуратно помещали над оптически прозрачным основанием для осмотра, который позволял просвечивание 2-см2 сегмента ткани. Всю незащищенную ткань покрывали марлей, смоченной в физиологическом растворе для минимизации обезвоживания. Температуру основания удерживали при 37°C и брыжейку поливали подогретым физиологическим раствором, забуференным бикарбонатом (pH 7,4). Прижизненный микроскоп (Nikon L25/0,35) и окуляр ×10 использовали для наблюдения за мезентериальным капиллярным кровообращением. Для исследования выбирались посткапиллярные венулы с диаметрами, варьирующими от 20 до 40 мкм. Видеокамера, установленная на микроскопе (Panasonic™, цифровой 5000), передавала изображение на монитор, и изображения записывали для анализа при воспроизведении с применением кассетного видеомагнитофона. Изображения брыжеечного капиллярного кровообращения записывали за 5 минут перед введением аспирина (фоновый уровень), в момент введения аспирина (время 0-5) и каждые 15 минут в течение 60 минут. Адгезию лейкоцитов измеряли количественно слепым методом из видеоизображений сосудов, произведенных в течение 5-минутных периодов, как число лейкоцитов, которые остаются неподвижными вдоль стенки сосуда в течение 30 с или более (выражали на 100 мкм длины венулы). Группы крыс (по меньшей мере 5 в каждой группе) предварительно обрабатывали 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислотой (соединение XXVII) (100 мг/кг), месаламином (50 мг/кг) или носителем за 60 мин перед введением аспирина (или носителя). Эти лекарственные вещества вводились внутрижелудочно. В некоторых экспериментах крыс обрабатывали глибенкламидом (10 мг/кг i.p.) или носителем за 30 мин перед введением этих соединений.
Фигура 5 показывает адгезию лейкоцитов на конечный момент времени эксперимента (минуты 60-65). Эта фигура иллюстрирует способность соединения XXVII подавлять обусловленную аспирином адгезию лейкоцитов и способность предварительной обработки глибенкламидом обращать этот эффект ингибирования адгезии лейкоцитов.
ПРИМЕР 15. Генерация H 2 S из 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты
5-Амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойную кислоту (соединение XXVII) тестировали на генерацию H2S в трех различных условиях. Измерялись концентрации H2S, которые генерировались в пределах 1 часа при 1 нМ концентрациях L-цистеина, 4-HBT (4-гидрокситиобензамид) и 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты. Высвобождение H2S тестировали в трех условиях: (i) когда соединение находилось в буфере, (ii) когда соединение находилось в гомогенате печени, и (iii) когда соединение находилось в гомогенате печени совместно с ингибитором цистатионин γ-лиазы (PAG = DL-пропаргилглицин; 2 нМ). Результаты представлены на Фигуре 6. *p<0,05 по сравнению с высвобождением в группе, обработанной носителем. *p>0,05 по отношению к соответствующей группе 'гомогенатов печени'. Энзиматическую емкость продукции H2S определяли с применением того же реактора, который описан ранее (Khan et al. (1980) Microchem J. 25, 388-395, которая включена в данное описание посредством ссылки). 2-мл реакционную смесь для исследования вводили в реактор. Смесь содержала 1 нМ L-цистеина (или соединения), 2 нМ пиридоксаль 5'-фосфата и 100 нМ буфера с фосфатом калия (pH=7,4). Постоянный поток азота пропускали через смесь через газоподводящий капилляр. Реакции инициировали путем переноса пробирок из ледяной бани в баню с водой при 37°C. Поток азота переносил сероводородную кислоту во второй реактор, содержащий 4 мл неокисляющего сероводород буферного раствора (SAOB), состоящего из 2 М KOH, 1 М салициловой кислоты и 0,22 М аскорбиновой кислоты при pH 12,8[5]. После инкубирования при 37°C
в течение 90 минут добавляли 1 мл 10% раствора трихлоруксусной кислоты к смеси, с тем чтобы останавить реакцию. Остаток H2S в смеси переводили в потоке азота в течение еще 60 минут инкубации при 37°C. Концентрацию сульфида в SAOB-растворе измеряли посредством сульфидчувствительного электрода (Модель 9616 S2-/Ag+ электрод, Orion Research, Beverly, MA, USA). Для исследований, в которых тестовые соединения инкубировали в гомогенате печени, 100-150 мг выделенных печени крыс гомогенизировали в 1 мл экстракторе белка T-PER, охлаждаемого льдом. Гомогенаты добавляли к реакционной смеси при концентрации 10% (мас./об.). 2 нМ DL-пропаргилглицина инкубировали с гомогенатом печени в течение 5 мин при 37°C перед энзиматической реакцией. Khan, S.U. Morris, G.F. и Hidiroglou, M. (1980). Быстрая оценка концентрации сульфида в рубце и крови с сульфидспецифическим ионным электродом описана в Microchem. J. 25:388-395, которая включена в данное описание посредством ссылки.
Результаты, представленные на фигуре 6, допускают, что соединение XXVII имеет следующие отличительные признаки:
1. Соединение XXVII высвобождает H2S самопроизвольно (в буфере), что желательно для местного эффекта в пищеварительном тракте. 4-HTB и L-цистеин не высвобождают значимые количества H2S, если инкубируются только в буфере;
2. Высвобождение H2S больше в присутствии ткани;
3. Высвобождение H2S из соединения XXVII происходит независимо от активности двух главных энзимов для эндогенного синтеза H2S (цистатионин β-синтетаза и цистатионин-γ-лиаза). Это было продемонстрировано путем отсутствия эффекта ингибитора указанных энзимов (PAG; DL-пропаргилглицин), на генерацию H2S из соединения XXVII. В противоположность этому, высвобождение H2S из L-цистеина значительно ингибируется PAG;
4. Концентрация H2S, полученного из соединения XXVII находится в диапазоне 10-20 мкМ, если используется 1 нМ соединения. Концентрации до 5 нМ месаламина могут быть измерены в просвете толстой кишки после того, как пациенты приняли обычные дозы этого лекарственного вещества (Dig. Dis. Sci. 1989; 34: 573-578). Эндогенные концентрации H2S могут быть достаточно велики, порядка 160 мкМ (Antioxid. Redox Signal. 2003; 5, 493-501). Соединение XXVII высвобождает H2S с концентрациями в пределах физиологического диапазона, таким образом минимизируя вероятность токсичности, обусловленной H2S.
ПРИМЕР 16. Сравнение действия тиокарбамоилбензоата тримебутина с тримебутином отдельно и с тиокарбамоилбензоатом отдельно на модели восприятия висцеральной боли на крысах
Эксперименты проводили, как описано в Примере 13, за исключением того, что каждая группа из 5 крыс получала носитель, малеат тримебутина (10 мг/кг), или с эквимолярными дозами тиокарбамоилбензоата тримебутина (соединение III), или отдельно тиокарбамоилбензоат.
Фигуры 7(a) и 7(b) показывают, что тиокарбамоилбензоат тримебутина является более эффективным, чем или малеат тримебутина, или тиокарбамоилбензоат, в отношении уменьшения висцеральной боли как отклик на колоректальное растяжение.
Таким образом, тиокарбамоилбензоат тримебутина может быть применен для лечения абдоминальной боли, ассоциированной с различными воспалительными состояниями пищеварительного тракта, также как функциональными желудочно-кишечными расстройствами, такими как спастический колит, диспепсия и т.д., которые характеризуются повышенной висцеральной ноцицепцией (с или без сопровождающего воспаления).
ПРИМЕР 17. Желудочно-кишечная безопасность NSAID-соединений по настоящему изобретению
Производное диклофенака по настоящему изобретению, 4-тиокарбамоилфенилового эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты, также обозначаемого здесь как соединение XVII, исследовали на его желудочно-кишечную безопасность на крысах. В частности, измеряли желудочное повреждение, желудочный синтез PGE2, изъязвление тонкой кишки и гематокрит.
Самцы крыс Вистар с массой 175-200 г не получали пищу в течение 18 часов перед пероральным введением 1% карбоксиметилцеллюлозы (носитель; 0,2 мл) отдельно, или одного из следующих растворов в этом носителе: диклофенак (20 мг/кг), 4-тиокарбамоилфениловый эфир [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (соединение XVII) (27,3 мг/кг), 4-гидрокситиобензамид (TBZ) (7,3 мг/кг) или диклофенак совместно с TBZ. Доза соединения XVII была эквимолярна дозе диклофенака 20 мг/кг. Ананлогично, доза TBZ была эквимолярна дозе соединения XVII.
В каждой группе было по 5 крыс. Через три часа после введения тестируемых соединений крыс усыпляли и величину желудочного геморрагического повреждения измеряли слепым методом (в мм). "Коэффициент желудочного повреждения" получали путем суммирования длин всех повреждений в желудке. Как можно видеть из Фигуры 8, не наблюдалось повреждения желудка в группах "носителя" или "соединения XVII". Соединение XVII вызывает значительно меньшее повреждение желудка, чем диклофенак. Кроме того, эффект неповреждения желудка не наблюдается, если фрагмент NSAID (диклофенака) и TBZ вводится раздельно, но одновременно.
Эти наблюдения были подтверждены последующим гистологическим исследованием слепым методом. Образцы (100-200) желудочной ткани вырезали для измерения синтеза простагландина Е2 (PGEa), как описано в деталях ранее (Wallace et al., Cyclooxygenase 1 contributes to inflammatory responses in rats and mice: implications for gastrointestinal toxicity. Gastroenterology 1998; 115; 101-109, которая включена в данное описание посредством ссылки). Кратко, образцы ткани крошили с помощью ножниц в течение 30 мин, затем помещали в 1 мл буфера фосфата натрия (pH 7,4) и помещали во взбалтываемую водяную баню (37°C) на 20 мин. Затем образцы немедленно центрифугировали в течение 1 мин при 9000g и супернатант немедленно замораживали до -80°C в течение последующего измерения концентрации PGE2 с применением специфического теста ELISA (Wallace et al., 1998).
Как можно видеть из Фигуры 9, диклофенак (с, или без, сопутствующим введением TBZ) и соединение XVII значительно уменьшают величину синтеза PGE2 в желудке, что указывает на ингибирование COX-1 и/или COX-2. TBZ отдельно не уменьшает синтеза PGE2 в желудке по сравнению с носителем. Таким образом, отсутствие желудочного повреждения у крыс, обработанных соединением XVII, как представлено на фигуре 1, не относится к изменению способности этих лекарственных веществ подавлять синтез простагландина в желудке. Подавление синтеза PGE2 в желудке является практически равным в случае этих лекарственных веществ и в случае эквимолярной дозы диклофенака.
Фигура 10 показывает, что производное напроксена по настоящему изобретению, 4-тиокарбамилфениловый эфир 2-(6-метоксинафталин-2-ил)пропионовой кислоты (соединение XX), вызывает значительно меньшее повреждение, чем напроксен отдельно. Этот эксперимент проводили точно таким же образом, как те, что представлены на фигуре 8. Напроксен и соединение XX, каждый, вводили перорально при дозе 60 мкмоль/кг, и 3 часа спустя повреждение желудка исследовали слепым методом. Повреждение желудка не поддавалось обнаружению у любой из крыс, обработанных соединением XX. Каждая группа состояла из 5 крыс. Эти наблюдения были подтверждены последующим гистологическим исследованием слепым методом.
Ингибирование COX-1 также измеряли с использованием таких же крыс. Сразу после сбора экссудатов из кармана, 1 мл крови отбирали из нижней половой вены каждой крысы и помещали в стеклянную пробирку и позволяли сворачиваться в течение 45 мин, как описано ранее (Wallace et al., Gastroenterology 1998). Образцы затем центрифугировали в течение 3 мин при 9000g и супернатант замораживали при -80°C в течение последующего измерения концентраций тромбоксана B2 с применением специфического теста ELISA. Как представлено на фигуре 11, напроксен и соединение XX, каждый, значительно (*p<0,05) ингибируют активность COX-1 по сравнению с группой, обработанной носителем.
ПРИМЕР 18. Ингибирование циклооксигеназы-2 (COX-2) и циклооксигеназы-1 (COX-1) действием 4-тиокарбамоилфенилового эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты.
Ингибирование COX-2 in vivo измеряли с применением модифицированного варианта ранее опубликованной модели (Wallace et al., Limited anti-inflammatory efficacy of cyclo-oxygenase-2 inhibition in carrageenan-airpouch inflammation. Br. J. Pharmacol. 1999; 126:1200-1204, которая включена в данное описание посредством ссылки). Кратко, подкожный "карман" создавали путем повторяющихся инъекций воздуха в течение нескольких дней. Один раз возникнув, воспаление в кармане может быть вызвано путем инъекции 1 мл 1% зимосана. Это вызывает большой прирост простагландина E2 (PGE2) в пределах кармана, что, как было показано, достигается почти исключительно действием COX-2. Каждую группу из 5 крыс перорально обрабатывали, за 30 мин до инъекции каррагенана, носителем (1% карбоксиметилцеллюлоза), диклофенаком (3 мг/кг) или 4-тиокарбамоилфениловым эфиром [2-(2,6-дихлорфениламино)фенил]уксусной кислоты, соединением XVII (4,1 мг/кг). Другая группа из 5 крыс получала носитель, но также инъекцию 0,9% стерильного физиологического раствора в карман, в количестве, которое поступает с зимосаном.
Как можно видеть из Фигуры 12, предварительная обработка или диклофенаком, или соединением XVII значительно уменьшала концентрации PGE2 в пределах кармана, которые возникали как отклик на инъекцию зимосана. *p<0,05 по отношению к группе, получавшей носитель + зимосан. Эти результаты показывают, что оба соединения значительно ингибируются COX-2. В противоположность этому, TBZ один не влияет значительно на активность COX-2.
Ингибирование COX-1 также измеряли с применением тех же крыс, с применением того же способа, как описано для Фигуры 11. Как представлено на фигуре 13, диклофенак и соединение XVII, каждый, ингибируют полный синтез тромбоксана в крови, который проходит под действием COX-1 более чем на 80%. В противоположность этому, TBZ не влияет значительно на активность COX-1.
ПРИМЕР 19. Влияние производных NSAID на повреждение желудка, активность COX-1 и COX-2 in vivo
Противовоспалительные эффекты (ингибирование COX-2 и COX-1) и желудочную безопасность ряда соединений сравнивали с применением исследований, описанных выше. Результаты обобщены в таблице 1. Все исходные NSAID вызывают значительное желудочное повреждение. Однако производные TBZ по настоящему изобретению показывают улучшенную желудочную безопасность по сравнению с исходными лекарственными веществами. Также можно видеть из таблицы 1, что производные TBZ или сохраняют, или, как ни странно, увеличивают их способность ингибировать COX-1 и/или COX-2 по сравнению с исходным лекарственным веществом.
Влияние производных NSAID на повреждение желудка и активность COX-1 и COX-2 in vivo
↑: статистически значимый прирост по отношению к исходному лекарственному веществу (p<0,05)
↓: статистически значимое уменьшение по отношению к исходному лекарственному веществу (p<0,05)
↔: нет значимого изменения по отношению к исходному лекарственному веществу
ПРИМЕР 20. Влияние производных NSAID на воспаление
Противовоспалительные эффекты 4-тиокарбамоилфенилового эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (соединение XVII) с тем же диклофенаком исследовали с применением модели каррагенанового отека задней лапы, как ранее описано у Wallace et al., Gastroenterology 1998. Самцы крыс Вистара с массой 175-200 г получали тестируемые соединения перорально за 30 мин перед подподошвенной инъекцией 100 мкл 1% лямбда-каррагенана. Объем лапы измеряли с применением гидроплетизмометра Ugo Basile перед инъекцией каррагенана и затем через 1-часовые интервалы в течение 5 часов. Каждую группу, которая состояла из 5 крыс, обрабатывали диклофенаком при дозах 1, 3 или 10 мг/кг или с соединением XVII при дозе, эквимолярной диклофенаку 3 мг/кг.
Как представлено на фигуре 14, диклофенак зависимым от дозы образом уменьшал отек лапы, обусловленный подподошвенной инъекцией каррагенана. Соединение XVII, вводимое при дозе, эквимолярной диклофенаку 3 мг/кг, уменьшало отек лапы в большей степени. Действительно, эффект соединения XVII на отек лапы являлся сравнимым с эффектом диклофенака при дозе 10 мг/кг.
Так как соединение XVII подавляет синтез простагландина в той же степени, как диклофенак, увеличенная активность новых соединений по изобретению в модели отека лапы наиболее вероятно относится к другому свойству соединения. Ранее было продемонстрировано, что доноры сероводорода могут значительно уменьшать обусловленный каррагенаном отек лапы у крыс (Zanardo et al., Hydrogen sulphide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J 2006; 20: 2118-2120, которая включена в данное описание посредством ссылки), таким образом, без необходимости быть связанным с любой теорией, предполагается, что высвобождение H2S из соединения XVII является фактором, усиливающим противовоспалительные характеристики по сравнению с диклофенаком.
Без необходимости быть связанным с любой теорией, также предполагается, что некоторая часть дополнительной активности соединений по этому изобретению в моделях воспаления может быть приписана усиленному ингибированию активности COX-2. Эффекты носителя, напроксена и 4-тиокарбамилфенилового эфира 2-(6-метоксинафталин-2-ил)пропионовой кислоты (соединение XX) были сравнены с моделью воздушного кармана на крысах (как описано для Фигуры 12). Каждая группа состояла из 5 крыс. Напроксен и соединение XX, каждый, вводились при дозе 60 мкмоль/кг. Как представлено на фигуре 15, как напроксен, так и соединение XX значительно подавляют активность COX-2 по сравнению с группой, получавшей носитель (*p<0,05. **p<0,01).
Также, без необходимости быть связанным с любой теорией, возможно, что некоторая часть дополнительной активности производных NSAID по этому изобретению в моделях воспаления может быть приписана усиленному ингибированию активности COX-1. Эффекты носителя, индометацина и 4-тиокарбамоилфенилового эфира [1-(4-хлорбензоил)-5-метокси-2-метил-1-H-индол-3-ил]уксусной кислоты (соединение XIX), были сравнены с их эффектами на полное количество тромбоксана В2 в крови человека in vitro. Аликвоты (0,5 мл) крови от здоровых добровольцев добавляли в стеклянные пробирки, содержащие 10 мкл метанола отдельно, или одно из тестируемых лекарственных веществ, приготовленных так, что конечная концентрация должна составлять 0,1, 0,3, 1 или 3 мкМ. Пробирки помещали в водяную баню (37°C) с мягким потряхиванием на 45 мин, после чего их центрифугировали (1000g) в течение 10 минут. Концентрацию тромбоксана B2 в каждом образце затем определяли с применением специфического теста ELISA, как в исследованиях, представленных на фигуре 11. Как представлено на фигуре 16, как индометацин, так и соединение XIX создавали зависимое от концентрации ингибирование активности COX-1 по сравнению с группой, обработанной носителем. Однако, при концентрациях 1 и 3 мкМ, соединение XIX создавало значительно большее (*p<0,05) ингибирование активности COX-1, чем то, что получали действием индометацина.
ПРИМЕР 21. Адгезия лейкоцитов к сосудистому эндотелию; производные NSAID по настоящему изобретению
Адгезия лейкоцитов к эндотелию сосудов представляет собой первое событие в воспалительных реакциях и вносит вклад в образование тромбов. Доноры сероводорода, как было показано, уменьшают адгезию лейкоцитов, обусловленную аспирином или провоспалительным трипептидом, fMLP (Zanardo et al., FASEB J. 2006; 20: 2118-2120). Эффекты нескольких производных NSAID по настоящему изобретению на адгезию лейкоцитов исследовали с применением прижизненной микроскопии на крысах, как описано в деталях Zanardo et al. FASEB J 2006; 20: 2118-2120.
Кратко, посткапиллярные брыжеечные венулы в анестезированных крысах исследовали под оптическим микроскопом. После записи фонового уровня на отрезке в 5 мин одно из тестовых соединений, перечисленных в таблице 2 ниже, вводили внутрижелудочно при дозе 30 мкмоль/кг, за исключением напроксена и 4-тиокарбамилфенилового эфира 2-(6-метоксинафталин-2-ил)пропионовой кислоты (соединение XX), которые вводили при дозе 60 мкмоль/кг. Все тестируемые соединения приготовляли в среде 1% карбоксиметилцеллюлозы. Изменения адгезии лейкоцитов в пределах венулы записывали на видеокамеру, присоединенную к микроскопу, и количественное определение числа прикрепленных лейкоцитов проводили слепым методом путем оценки записанных на видеопленку изображений. Каждая группа состояла из 5 самцов крыс Вистар, с массой 150-175 г. Лейкоцит считался "адгезированным", если он оставался неподвижным в течение 30 секунд или более (результаты ниже выражали как значение ± SEM). В конце эксперимента желудок открывали и исследовали на присутствие желудочного повреждения под рассекающим микроскопом.
Адгезия лейкоцитов к эндотелию сосудов
Из таблицы 2 можно видеть, что TBZ-производное аспирина по настоящему изобретению, соединение XVI, значительно уменьшает число прикрепленных лейкоцитов на 100 мкм длины сосуда по сравнению с аспирином отдельно. В добавление, соединение XVI значительно уменьшает процент желудочного повреждения по сравнению с аспирином отдельно. Аналогично, далее таблица 2 показывает, что TBZ-производное диклофенака по настоящему изобретению, соединение XVII, значительно уменьшает число прикрепленных лейкоциов на 100 мкм длины сосуда и значительно уменьшает процент желудочного повреждения по сравнению с диклофенаком отдельно. Аналогичным образом, далее таблица 2 показывает, что TBZ-производное напроксена по настоящему изобретению, соединение XX, значительно уменьшает число прикрепленных лейкоцитов на 100 мкм длины сосуда и значительно уменьшает процент желудочного повреждения по сравнению с напроксеном отдельно.
Интересно, что TBZ-производное лумиракоксиба, COX-2 селективный ингибитор, имеющий уменьшенный желудочный побочный эффект, соединение XVIII, по-прежнему не показывает случаев желудочного повреждения, но число прикрепленных лейкоцитов на 100 мкм длины сосуда значительно уменьшено по сравнению с лумиракоксибом отдельно. Таким образом, ковалентное связывание TBZ с селективными в отношении COX-2 NSAID также может уменьшать сердечно-сосудистые побочные эффекты этих ингибиторов COX-2.
Таким образом, производные NSAID по настоящему изобретению могут приводить к уменьшению сердечно-сосудистых побочных эффектов NSAID путем уменьшения адгезии лейкоцитов.
ПРИМЕР 22. Влияние производных NSAID по настоящему изобретению на излечение язвы желудка
NSAID, включая те, что являются селективными в отношении COX-2, часто ингибируют выздоровление ранее возникших желудочных язв (Stadler et al., Diclofenac delays healing of gastroduodenal mucosal lesions. Double-blind, placebo-controlled endoscopic study in healthy volunteers. Digestive Diseases and Sciences 1991; 36: 594-600). Для определения эффектов двух соединений по настоящему изобретению на заживление язвы (соединение XVII и соединение XX), по сравнению с диклофенаком и напроксеном, соответственно, крыс обрабатывали этими лекарственными веществами после того, как язвы были созданы в их желудках. Желудочные язвы созданы путем серозного нанесения уксусной кислоты, как описано Elliott et al., A nitric oxide-releasing nonsteroidal anti-inflammatory drug accelerates gastric ulcer healing in rats. Gastroenterology 1995; 109: 524-530. Через три дня после начала, группами из 5 крыс, каждую группу обрабатывали дважды в день, перорально, носителем, диклофенаком, (30 мкмоль/кг), соединением XVII (30 мкмоль/кг), напроксеном (60 мкмоль/кг) или соединением XX (60 мкмоль/кг). Через 4 дня такого лечения крыс усыпляли и желудок вырезали и фотографировали. Размер язвы (в мм2) определяли путем планиметрической индивидуальной обработки слепым методом, использованной для крыс. В подгруппе из 5 крыс, усыпленных через 3 дня после создания желудочных язв (то есть перед началом лекарственного лечения), средняя площадь поверхности язв составляла 24±2 мм2. Как показано на фигуре 17, крысы, обработанные носителем, диклофенаком или напроксеном, проявляют аналогичные степени выздоровления. Однако крысы, обработанные соединением XVII или соединением XX, проявляют значительно большее выздоровление (*p<0,05 по сравнению с диклофенаком и напроксеном, соответственно). Обработка TBZ отдельно не влияет значительно на выздоровление желудочных язв по сравнению с группой, обработанной носителем.
ПРИМЕР 23. Влияния производных NSAID по настоящему изобретению на давление крови
NSAID, включая те, что проявляют селективность в отношении COX-2, могут обострять гипертензию, существовавшую до этого, и мешать эффективности некоторых антигипертензивных средств (Whelton, A. Nephrotoxicity of nonsteroidal antiinflammatory drugs: physiologic foundations and clinical implications. Am. J. Med. 1999; 106 (5B): 13S-24S). Крысы получали эти лекарственные вещества внутрибрюшинно после создания гипертензии, для определения эффектов производных напроксена по настоящему изобретению соединение XX сравнивалось по давлению крови с напроксеном, взятым отдельно. Крысам предоставляли питьевую воду, включающую метиловый эфир Nω-нитро-L-аргинина (400 мг/л) в течение 7 дней перед экспериментом, как описано ранее Ribeiro et al. (Chronic inhibition of nitric oxide synthesis: A new model or arterial hypertension. Hypertension 1992; 20: 298-303). Крыс (от 5 до 8 на группу) анестезировали галотаном и в сонную артерию вставляли катетер для измерения давления крови, который вел запись постоянно на диаграммный самописец. После измерения давления крови, стабильного в течение по меньшей мере 15 минут, или напроксен, или соединение XX вводили внутрибрюшинно в виде болюсной инъекции с концентрацией 60 ммоль/кг. Изменения в давлении крови регистрировали в течение 60 минут после инъекции. Средний уровень давления крови составлял 150±6 мм Hg. Фигура 18 показывает, что напроксен вызывает значимый прирост систолического давления крови. В противоположность этому, соединение XX не увеличивает систолическое давление крови по сравнению с группой, обработанной носителем, и изменение в давлении крови является значительно меньшим, чем те давления, что обусловлены диклофенаком и напроксеном, соответственно.
ПРИМЕР 24. Измерение генерации H 2 S из 4-тиокарбамоилфенилового эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты
Для сравнения высвобождения H2S in vitro, обусловленного 4-тиокарбамоилфениловым эфиром [2-(2,6-дихлорфениламино)фенил]уксусной кислоты, соединение XVII, и TBZ, 100-150 мг выделенных образцов печени гомогенизировали в 1-мл охлаждаемом льдом белковом экстракторе T-PER. 2-мл реакционную смесь для исследование вводили в смеси с охлаждаемым льдом 250 мкл 0,1N NaOH в запаянный 3-горлый реактор. Смесь, содержащая 1 мМ соединение XVII, или 1 мМ TBZ, растворяли в PEG и 100 мМ буфера с фосфатом калия (pH=7,4). Инкубации проводили в присутствии, или без 10% (мас./об.) гомогената печени и 2 мМ пиридоксаль 5'-фосфата. Постоянный поток азота пропускали через смесь через подводящий газ капилляр. Реактор удерживали при 37°C и экстракцию H2S начинали введением 1 мл 10% раствора трихлоруксусной кислоты. Поток азота переводил сероводородную кислоту в другой реактор через охлажденный коннектор и пробулькиванием в 2 мл сульфидного неокисляющего буферного раствора (SAOB), состоящего из 2 M KOH, 1 M салициловой кислоты и 0,22 M аскорбиновой кислоты при pH 12,8. Через 30 минут SAOB-раствор удаляли и концентрацию сульфида измеряли посредством сульфидчувствительного электрода (Модель 9616 S2-/Ag*-электрод, Orion Research, Beverly, MA, USA) и выражали в виде концентрации H2S (Ubuka, 2002; Khan et al., 1980). Реакции инициировали перенесением пробирки из ледяной бани в водную баню при 37°C. Поток азота переводил сероводородную кислоту во второй реактор, содержащий 2 мл SAOB, описанный ранее. После инкубирования при 37°C в течение 90 минут 1 мл 50% раствора трихлоруксусной кислоты добавляли к смеси для остановки реакции. Остающийся в смеси H2S переводили с током азота в течение еще 30 минут инкубации при 37°C. Концентрацию сульфида в SAOB-растворе измеряли сульфидчувствительным электродом, как описано выше (Ubuka, 2002; Khan et al., 1980).
Как представлено на фигуре 19, инкубация соединения XVII в буфере приводит к значительно большему высвобождению H2S, чем в случае эквивалентного количества TBZ. Аналогично, имеется большее высвобождение H2S из соединения XVII, чем из TBZ при инкубировании с гомогенатом печени.
ПРИМЕР 25
Действие смешанного 4-тиокарбамоилфенилового 2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8a-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-иловый эфира янтарной кислоты (соединение I) и симвастатина на агрегацию тромбоцитов человека (in vitro).
Обогащенную тромбоцитами плазму (PRP) получали, как описано в деталях ранее (Ma L, Elliott SN, Cirino G, Buret A, Ignarro LJ, Wallace JL. Platelets modulate gastric ulcer healing through release of endostatin and VEGF. Proc Natl Acad Sci USA 98: 6470-6475, которая включена в данное описание посредством ссылки). Концентрацию тромбоцитов в PRP приводили к 1×108 на мл путем разбавления буфером Тирода (Tyrode) (pH 7,4). Аликвоты (400 мкл) тромбоцитов помещали в стеклянную кювету и помещали в агрегометр тромбоцитов ChronoLog. Агрегацию как ответ на добавление в кювету аденозиндифосфата (ADP) контролировали в течение периода в 5 мин. Сначала строили кривую концентрация-отклик на ADP,
затем концентрацию ADP, создающую 70-80% максимальной агрегации, применяли для всех последующих исследований. Суспензии PRP предварительно инкубировали в течение 10 мин при 37°C
с различными концентрациями (3-30 мкМ) симвастатина или соединения I, или с носителем (метанол). Затем оценивали агрегационный отклик на ADP. Эксперименты повторяли 4-6 раз для каждой концентрации каждого лекарственного вещества.
На фигуре 20 показаны эффекты симвастатина и соединения I на ADP-обусловленную агрегацию тромбоцитов человека. Только симвастатин уменьшает агрегацию тромбоцитов при концентрации 30 пМ, в то время как соединение I значительно уменьшает агрегацию тромбоцитов при концентрациях 3, 10 и 30 мкМ (звездочки показывают значительное уменьшение агрегации тромбоцитов по сравнению с соответствующей группой, обработанной носителем; p<0,05).
ПРИМЕР 26. Действие соединения I и симвастатина на cAMP тромбоцитов человека (in vitro)
Обогащенную тромбоцитами плазму (PRP) получали, как описано выше. Аликвоты 400 мкл PRP помещали в стеклянные пробирки, которые содержали IBMX (изобутил-1-метилксантин; 0,5 мМ), неселективный ингибитор фосфодиэстеразы. Через две минуты в пробирки добавляли носитель (метанол) или различные концентрации (3-100 мкМ) симвастатина или соединения I. В качестве положительного контроля некоторые аликвоты тромбоцитов обрабатывали форсколином (10 мкМ), известным возбудителем аденилатциклазы. Через десять минут образцы PRP центрифугировали при 9000g в течение 2 мин и супернатант отбрасывали. Гранулы повторно суспендировали в буфере, облучали ультразвуком в течение 2 мин, затем концентрации cAMP определяли с применением специфического энзимсвязанного иммуносорбентного исследования (Cayman Chemical Co., Ann Arbor, MI, USA). Эксперименты повторяли 4-6 раз для каждой концентрации каждого лекарственного вещества.
На фигуре 21 показаны эффекты симвастатина и соединения I на концентрации cAMP тромбоцитов человека. Пунктирная линия показывает уровни cAMP в тромбоцитах, обработанных форсколином (10 мкМ). Только симвастатин значительно увеличивает cAMP тромбоцитов при наивысшей концентрации (100 мкМ), в то время как соединение I вызывает значительный прирост в cAMP тромбоцитов при концентрациях 10, 30 и 100 мкМ). (Звездочки показывают значительное уменьшение агрегации тромбоцитов по сравнению с соответствующей группой, обработанной носителем; p<0,05).
| название | год | авторы | номер документа |
|---|---|---|---|
| СЕРОВОДОРОДНЫЕ ПРОИЗВОДНЫЕ НЕСТЕРОИДНЫХ ПРОТИВОВОСПАЛИТЕЛЬНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2007 |
|
RU2468019C2 |
| ПРОИЗВОДНЫЕ 4- ИЛИ 5-АМИНОСАЛИЦИЛОВОЙ КИСЛОТЫ | 2006 |
|
RU2414476C2 |
| ПРОИЗВОДНОЕ ГИАЛУРОНОВОЙ КИСЛОТЫ И СОДЕРЖАЩЕЕ ЕГО ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2005 |
|
RU2390529C2 |
| НИТРОЭФИРЫ, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ И/ИЛИ АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1994 |
|
RU2136653C1 |
| СОКРИСТАЛЛИЧЕСКИЕ ФОРМЫ ТРАМАДОЛА И NSAID | 2009 |
|
RU2599717C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |
| АНАЛЬГЕЗИРУЮЩИЕ КОМПОЗИЦИИ | 2019 |
|
RU2820449C2 |
| ФЕНИЛКАРБАМАТЫ И ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА ГИДРОЛАЗЫ АМИДОВ ЖИРНЫХ КИСЛОТ (FAAH) И МОДУЛЯТОРОВ D3 ДОПАМИНОВОГО РЕЦЕПТОРА (D3DR) | 2014 |
|
RU2693457C2 |
| Способ ингибирования бактериальной цистатионин-γ-лиазы | 2023 |
|
RU2841071C1 |
| ИНГИБИТОРЫ ЦИСТАТИОНИН-Г-ЛИАЗЫ (CSE) | 2013 |
|
RU2640418C2 |
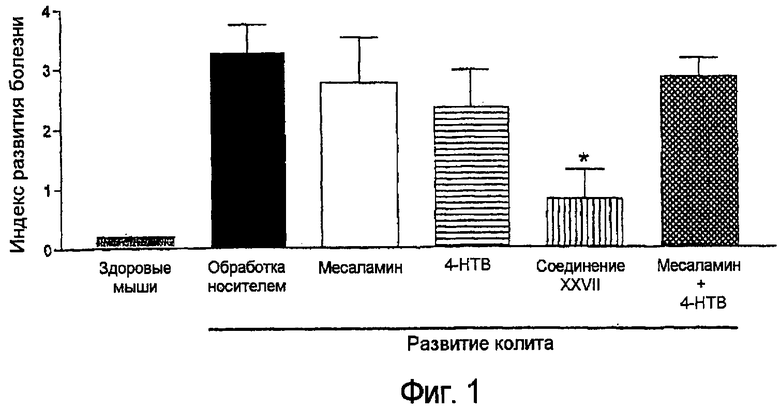

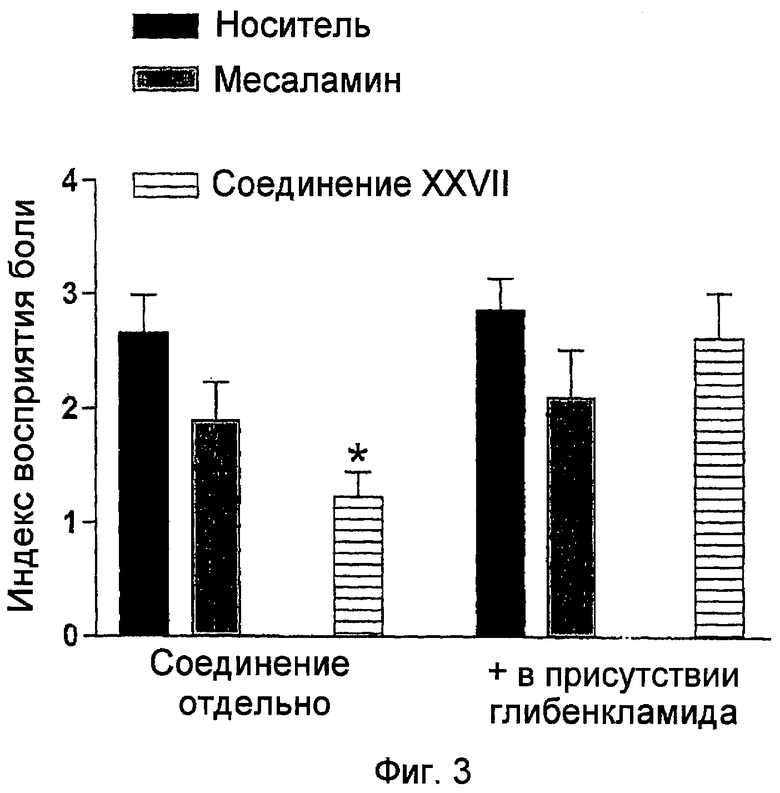
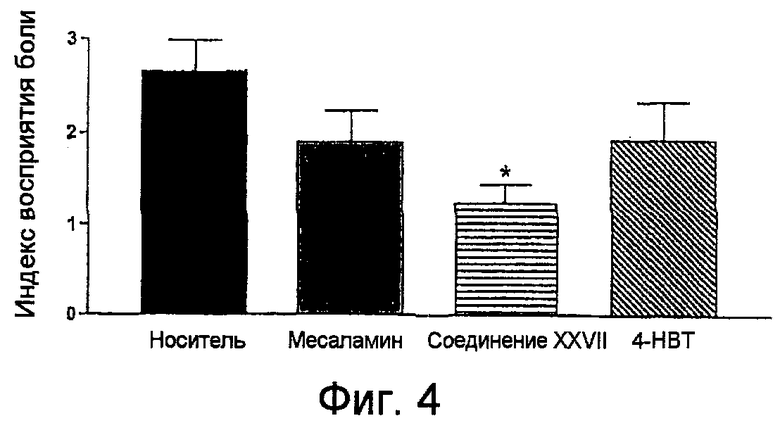
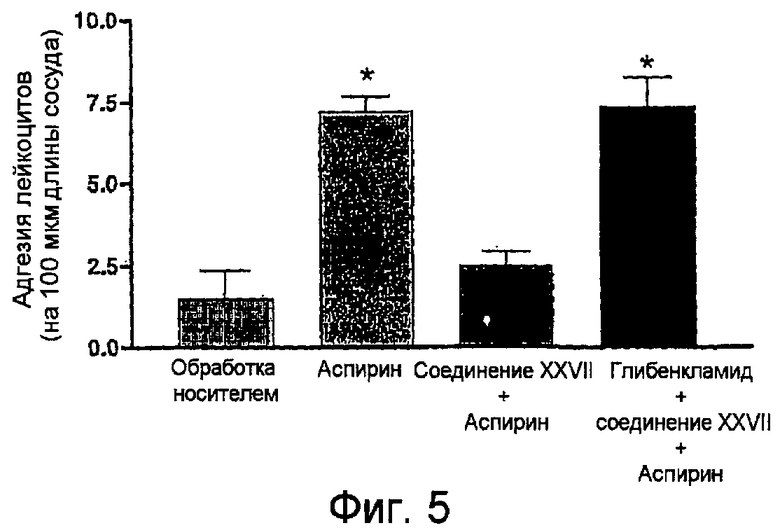


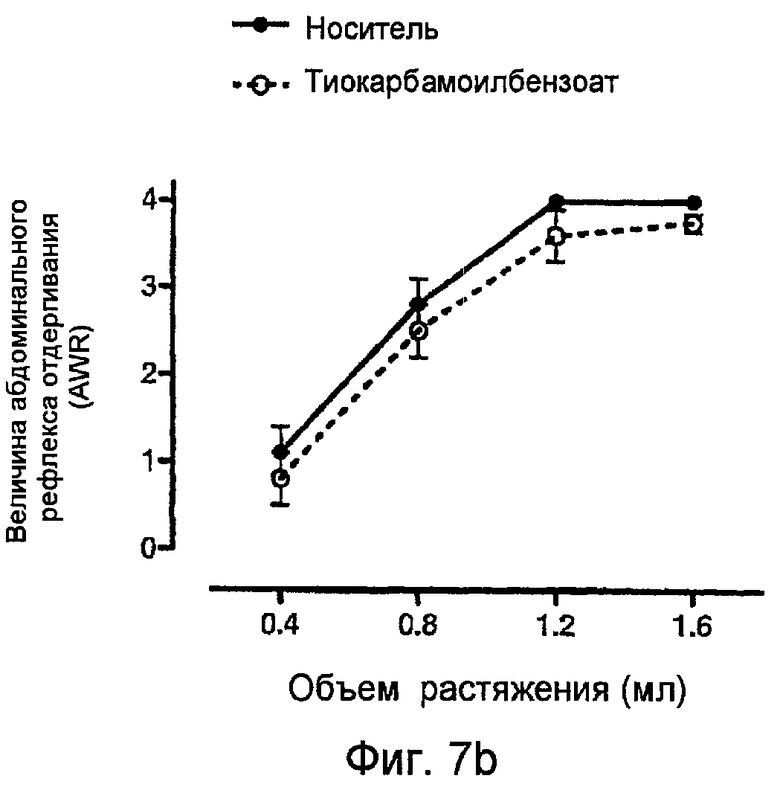
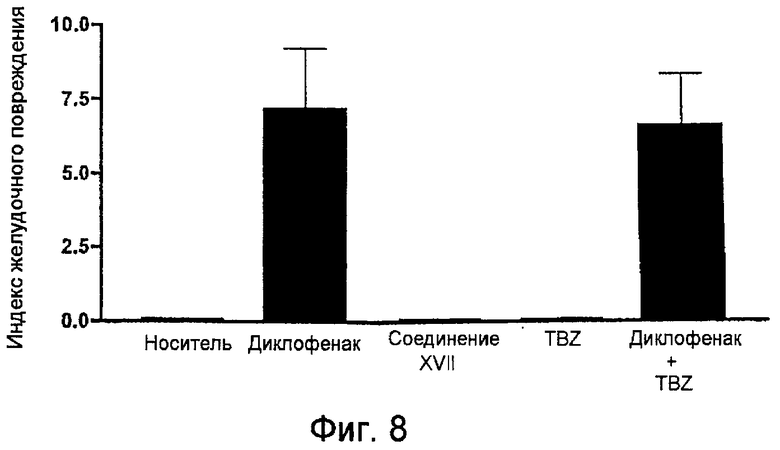
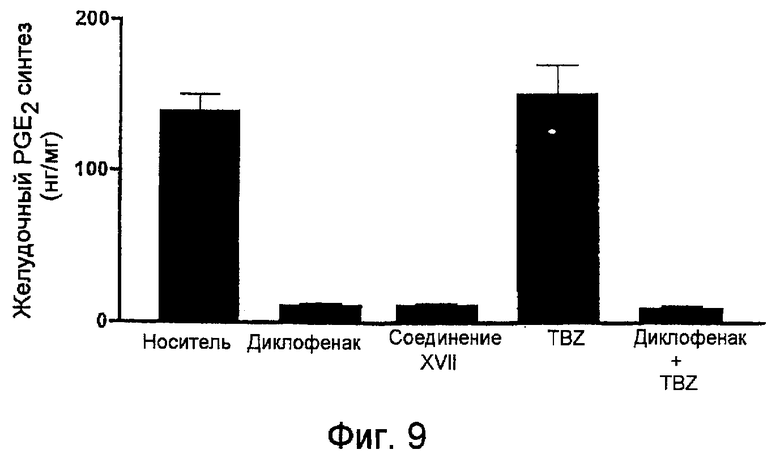
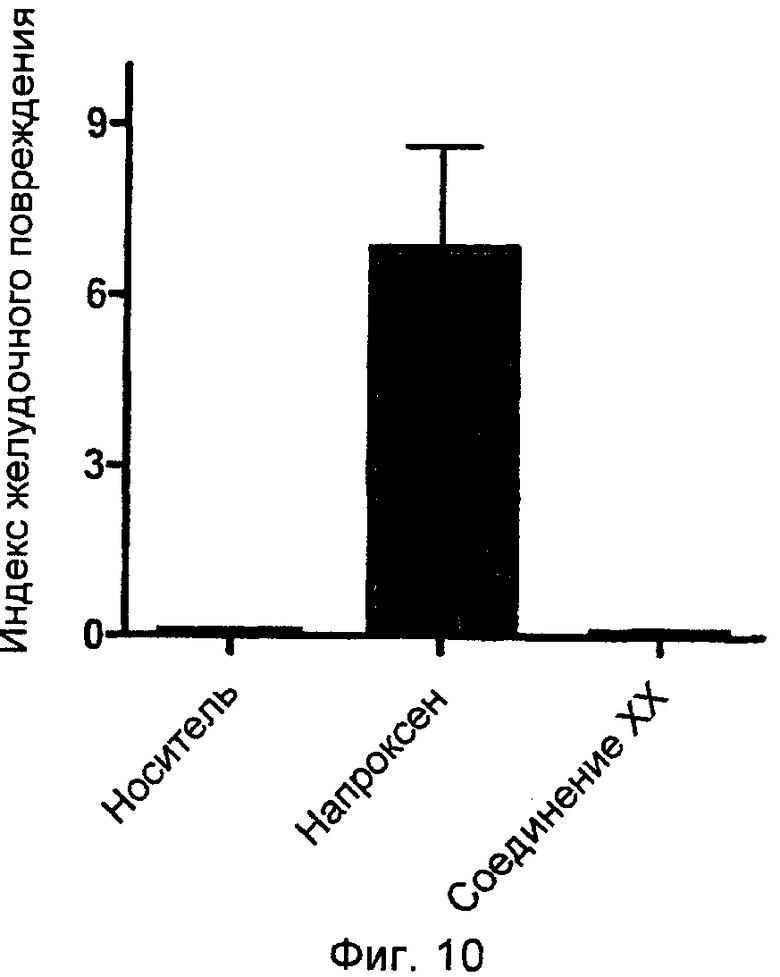
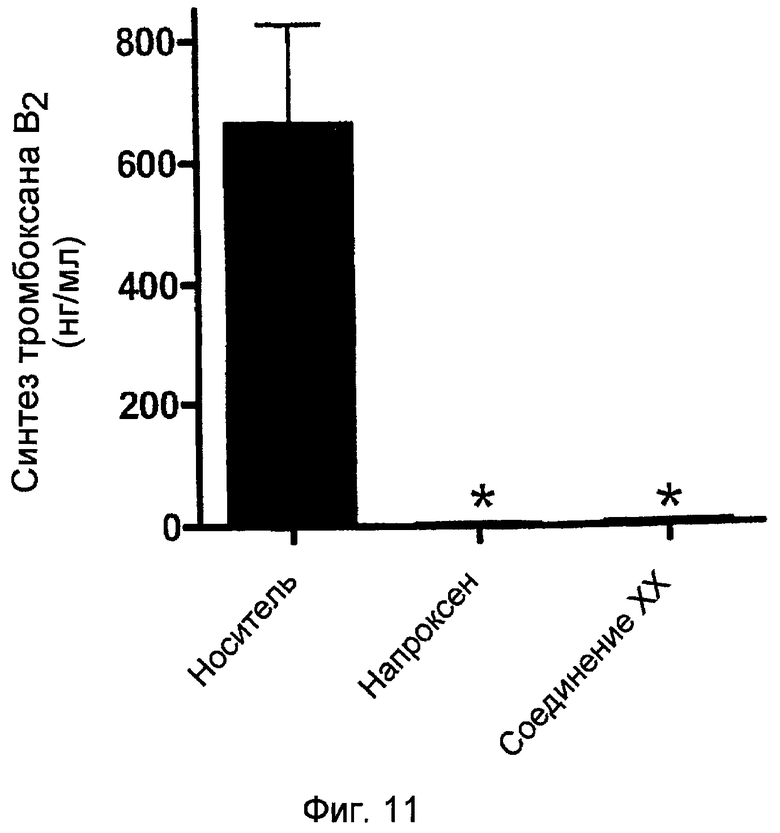
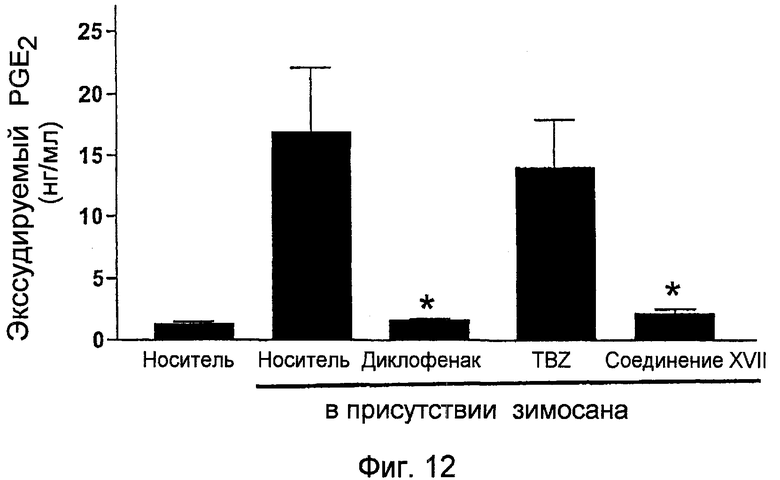
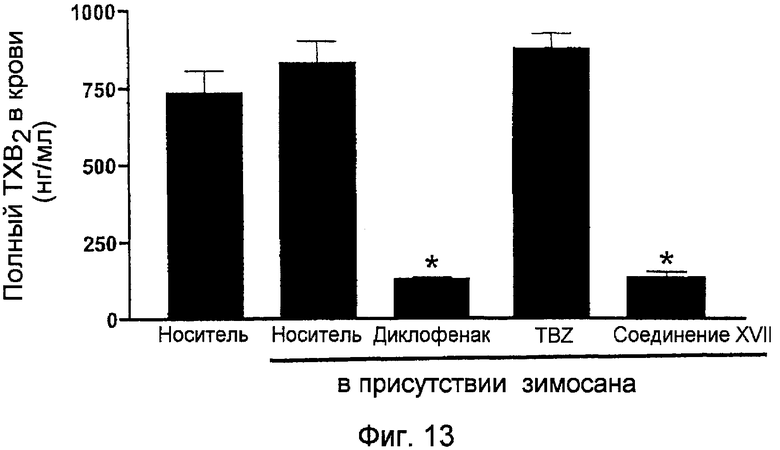
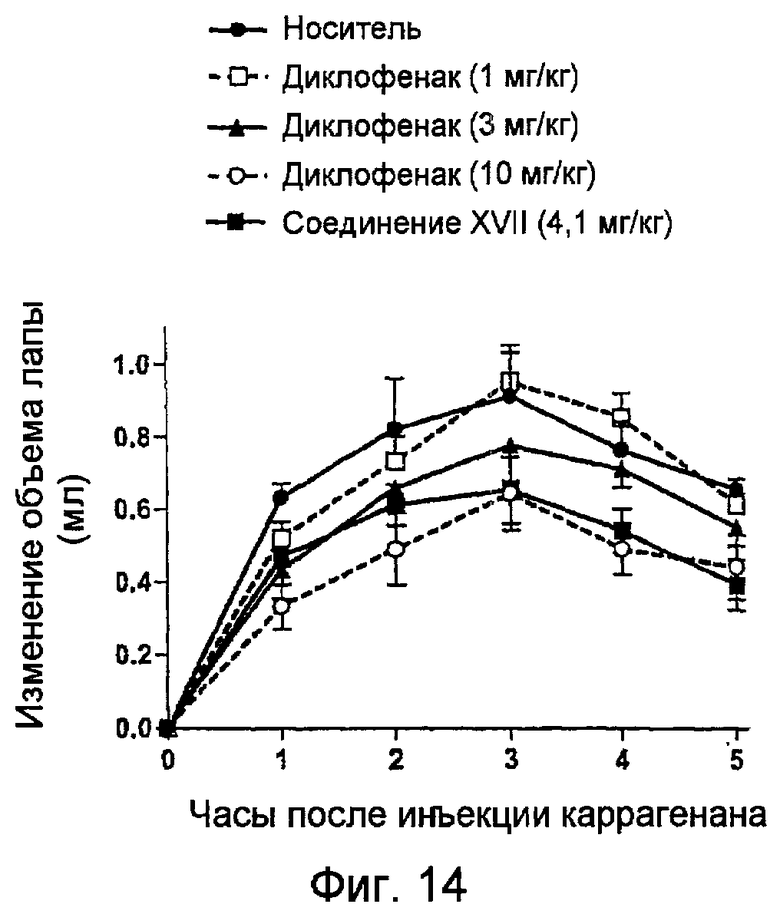
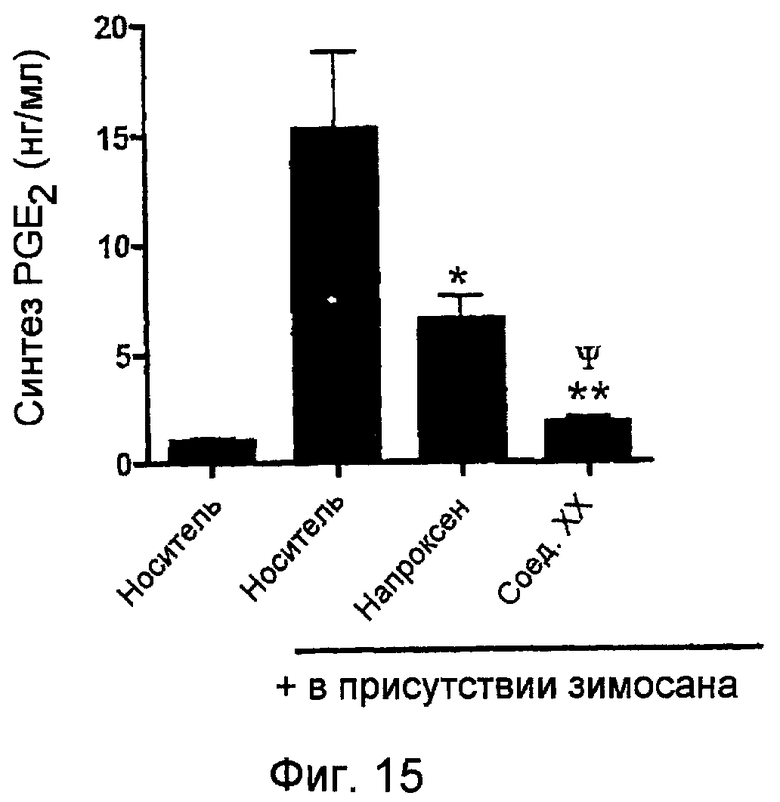

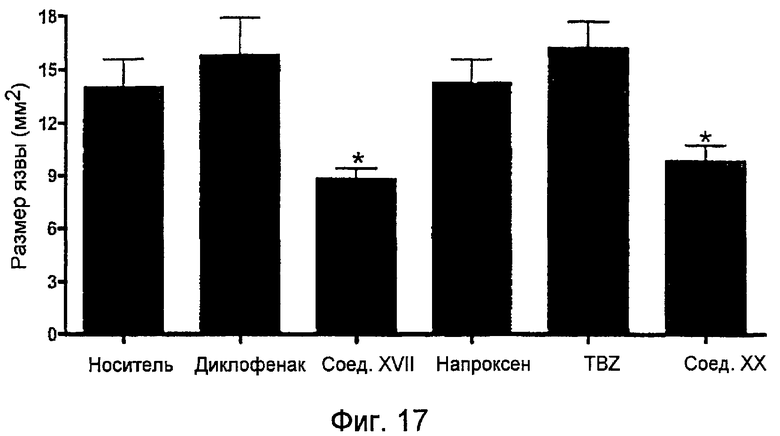



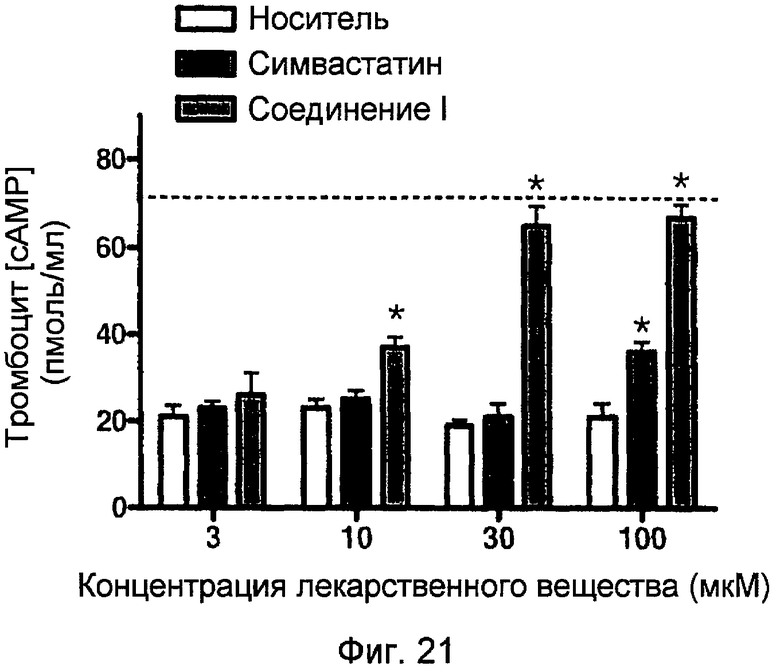
Предоставлены производные лекарственных веществ, где упомянутые производные включают H2S-высвобождающий фрагмент 4-гидрокситиобензамида, который или ковалентно связан с лекарственным веществом, или образует фармацевтически приемлемую соль с противолипидемическим лекарственным веществом. Соединения по настоящему изобретению проявляют повышенную активность, или уменьшенные побочные эффекты, или и то, и другое. 4 з.п. ф-лы, 26 пр., 21 ил., 1 сх.
1. Соединение или его фармацевтически приемлемая соль общей формулы:

где А представляет собой противогиперлипидемическое лекарственное вещество, Y выбран из группы, состоящей из  или отсутствует, и Х выбран из группы, состоящей из
или отсутствует, и Х выбран из группы, состоящей из 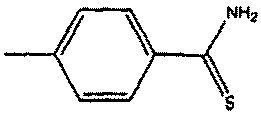 или
или  причем если Y отсутствует, соединение может представлять собой соль А и X.
причем если Y отсутствует, соединение может представлять собой соль А и X.
2. Соединение по п.1, где Y представляет собой группу  и Х
и Х
представляет собой 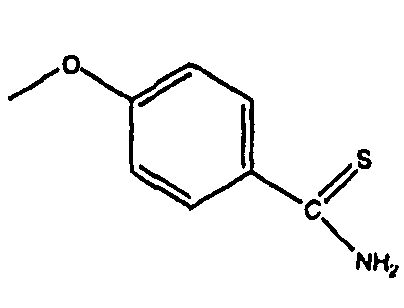
3. Соединение по п.1 или 2, в котором антигиперлипидемическое лекарственное вещество представляет собой статин.
4. Соединение по п.3, в котором статин выбран из группы, состоящей из аторвастатина, циластатина, дермостатина, флувастатина, ловастатина, мевастатина, правастатина натрия и симвастатина.
5. Соединение по п.4, в котором соединение представляет собой смешанный 4-тиокарбамоилфениловый эфир - 2-{2-[8-(2,2-диметилбутирилокси)-2,6-диметил-1,2,6,7,8,8а-гексагидронафталин-1-ил]этил}-6-оксотетрагидропиран-4-иловый эфир янтарной кислоты.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| RU 2009105491 A, 27.08.2010 | |||
| Leffler et al, «Carbamate antimalarials», J | |||
| of the American Chemical Society, 1948, vol.70, no.10, p.p.3439-3442. | |||

