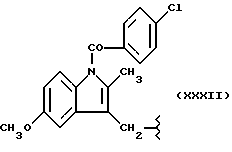
Настоящее изобретение относится к нитроэфирам производным пропионовой кислоты, 1-(п-хлорбензоил)-5-метокси-2-метил-3-индолилуксусной кислоты, 5-бензоил-1,2-дигидро-3H-пирроло[1,2-a] пиррол-1-карбоновой кислоты, 6-метокси-2-нафтилуксусной кислоты, их фармацевтическому использованию и способу их получения. Настоящее изобретение также относится к фармацевтическим композициям, содержащим по крайней мере один из вышеуказанных нитроэфиров в качестве активного ингредиента.
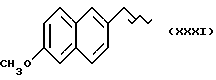
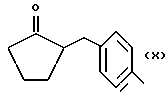
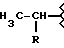
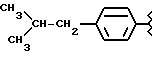
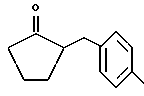
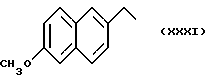
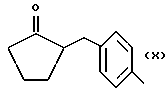
Некоторые производные пропионовой кислоты, такие как например, 2-(6-метокси-2-нафтил) пропионовая кислота, 2-(4-изобутилфенил)пропионовая кислота или альфа-метил-4-[(2-оксоциклопентил)метил] бензолуксусная кислота, длительное время использовали как фармацевтические препараты с противовоспалительной активностью и рекламировали на различных международных рынках много лет. Способ получения 2-(6-метокси-2-нафтил)пропионовой кислоты был описан в Южноафриканском Патенте N 6707, 597, в Германском Патенте N 1 934,460, соответствующем US Патенту N 3,637,767, а также в С.А. 71,91162j (1969); HARRISON et. al. J. Med. Chem. 13, 203 (1970); способ получения 2-(4-изобутилфенил)пропионовой кислоты был описан в патенте GB N 971,700, US N 3,228,831 и US N 3,385,886, а также в T.SHIORI, N. KAWAJ. J. Org. Chem. 43, 2936 (1978); J. T. PINHEY, B.A. ROWE, Tetragedron Letters 21, 965 (1980); в то же время способ получения альфа-метил-4-[(2-оксоциклопентил)метил] бензолуксусной кислоты был описан в Германском Патенте N 2,814,556 и в US Патенте N 4,161,538.
В случае 2-(6-метокси-2-нафтил)пропионовой кислоты, фармакологические данные описаны в ROSZNOWSKI et.al., J. Pharmacol. Exp. Ther. 179,114 (1971), в то же время фармакологические данные 2-(4-изобутилфенил)пропионовой кислоты описаны в ADAMS et. al. Arch. Pharmacodyn. Ther. 178,115 (1969).
Использование этих производных пропионовой кислоты в качестве противовоспалительных агентов, как известно, сопровождается сильно неблагоприятными реакциями, например, в желудочно-кишечной системе, также возможны повреждения в печени и в почках.
Другие, особенно токсичные продукты, например, 5-бензоил-2-дигидро-3H-пирроло [1,2-а]пиррол-1-карбоновая кислота или Кеторолак [W.H. ROOKS et.al. Agents Actions 12,684 (1982)] и 1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-уксусная кислота или Индометацин [C. D. KLAASSEN, Toxicol. Appl. Prarmacol, 28, 127 (1976)]. В частности, в некоторых странах Кеторолак был изъят с рынка из-за желудочно-кишечной токсичности, в то же время Индометацин - один из лекарственных препаратов, который был причиной высокого коэффициента смертности за годы его применения на рынке. В сравнении с другими известными противовоспалительными и/или анальгетическими лекарственными препаратами Кеторолак и Индоментацин являются причиной - из-за уже описанных неблагоприятных реакций - очень экстенсивных повреждений и, в частности, в отношении желудочно-кишечной токсичности, и, как установлено, являлись причиной смерти даже у детей.
Поэтому очевидна необходимость в лекарственных препаратах, обладающих хорошей противовоспалительной и/или анальгетической активностью, но не обладающих в целом токсичностью.
Целью настоящего изобретения является получение продуктов, которые по крайней мере обеспечивают высокий уровень фармакологической активности, которая характерна для известных противовоспалительных и/или анальгетических агентов, но при этом способны подавлять неблагоприятные реакции, которые вызываются действием описанных выше средств и имеют хорошую толерантность.
Другой целью настоящего изобретения является реализация способа получения производных пропионовой кислоты, 1-(п-хлорбензоил)-5-метокси-2-метил-1-индолилуксусной кислоты, 5-бензоил-1,2-дигидро-3H-пирроло [1,2-a]пиррол-1-карбоновой кислоты, 6-метокси-2-нафтилуксусной кислоты, обладающих противовоспалительной и/или анальгетической активностью, с хорошей толерантностью и свободных от неблагоприятных реакций, которые типичны для противовоспалительных и анальгетических агентов.
Еще одной целью настоящего изобретения является получение фармацевтических композиций, обладающих противовоспалительной и/или анальгетической активностью и хорошей толерантностью.
Эти и еще другие цели и преимущества, которые будут показаны из последующего описания, достигнуты производными пропионовой кислоты, 1-(п-хлорбензоил)-5-метокси-2-метил-3-индолилуксусной кислоты, 5-бензоил-1,2-дигидро-3H-пирроло[1,2-a]пиррол-1-карбоновой кислоты, 6-метокси-2-нафтилуксусной кислоты, которые согласно настоящему изобретению имеют следующую общую формулу: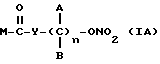
где A и B выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила, M выбран из: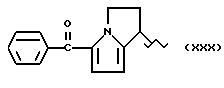
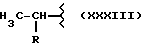
где R выбран из: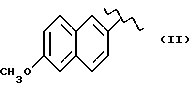
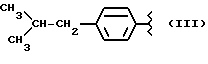
Y выбран из кислорода, NH, NR1, где R1 означает линейную или разветвленную алкильную группу, и n равно от 1 до 10.
Наиболее предпочтителен фрагмент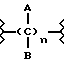
с линейными, разветвленными или циклическими алкиленовыми группами C2-C10.
Фактически установлено, что введение группы, такой как концевая нитроэфирная группа, в производные формулы (IA) приводит к увеличению фармакологической активности, которая характерна для противовоспалительных нестероидных и/или анальгетических агентов, приводит к продуктам с хорошей толерантностью, и в то же время уничтожает неблагоприятные реакции, возникающие при лечении такими лекарственными препаратами. Кроме того, введение концевой нитроэфирной группы в производные пропионовой кислоты приводит к усилению противовоспалительного эффекта по сравнению с хорошо известными нестероидными противовоспалительными лекарственными средствами; такое увеличение достигается концевой нитроэфирной группой, которая может рассматриваться как источник окиси азота и которая может дополнительно усиливать противовоспалительные эффекты.
Также было замечено, что производные (IA) пригодны для лечения различных тяжелых состояний, например, тяжелых состояний, требующих одновременно применения противовоспалительных и анальгетических лекарственных препаратов, или ревматических заболеваний общего характера, иммунологических расстройств, а также они могут облегчать болевые состояния средней тяжести любого характера.
Кроме того, производные (IA), являющиеся предметом настоящего изобретения, можно использовать при лечении заболеваний сердечно-сосудистой системы и центральной нервной системы, особенно при лечении миокарда и ишемии головного мозга, а также в случае артериальных тромбозов и в случае старческого слабоумия.
В соответствии с этим изобретением особенно полезным является нитроэфир (IA), где водород выбран как A и B, M выбран как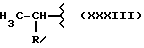
где R выбран как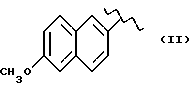
NH выбран как Y, и n равно четырем, согласно следующей формуле: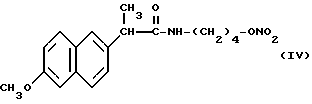
Также особенно полезен согласно настоящему изобретению нитроэфир (IA), где водород выбран как A и B, M выбран как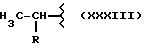
где R выбран как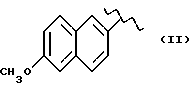
кислород выбран как Y, и n равно четырем, согласно следующей формуле: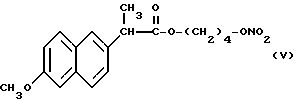
Также особенно полезны согласно настоящему изобретению нитроэфиры производные 2-(4-изобутилфенил)пропионовой кислоты, имеющие следующую формулу: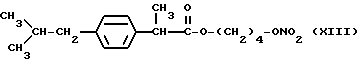
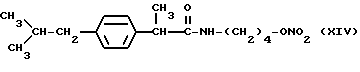
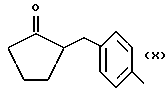
Также особенно полезны согласно настоящему изобретению нитроэфиры (IA), имеющие следующую формулу;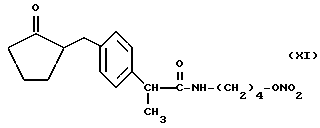
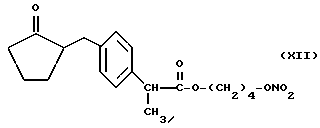
Также согласно настоящему изобретению нитроэфиры (IA), где M выбран как
кислород выбран как Y, водород выбран как A и B и n равно четырем согласно следующей формуле: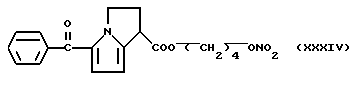
проявляют очень хорошую толерантность.
Для получения нитроэфиров (IA), которые являются предметом настоящего изобретения, особенно важен первый способ, который согласно настоящему изобретению, включает следующие стадии.
Получение натриевой соли производных, имеющих следующую общую формулу: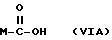
где M выбран из (XXX), (XXXI), (XXXII),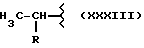
где R выбран из следующих структур: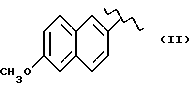
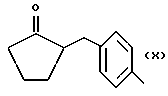
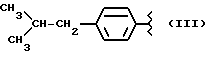
или получение производных (VIA), замещенных по карбоксильной группе, таких как хлорангидриды, ангидриды или подобные.
Реакцию натриевой соли вышеуказанных производных (VIA) или вышеуказанных производных (VIA), замещенных по карбоксильной группе, с соединением, имеющим следующую общую формулу: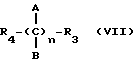
где R4 выбран из хлора, брома, NHR5 с R5 - водород, линейный или разветвленный алкил, A и B выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила, R3 выбран из хлора, брома, иода, и n равно от 1 до 10, с получением таким путем соответствующих эфиров или соответствующих амидов.
Реакцию вышеуказанных эфиров или вышеуказанных амидов с нитрующим агентом, таким как AgNO3 или подобным, с получением в результате нитроэфиров (IA).
Второй способ, являющийся также очень важным, согласно настоящему изобретению, включает следующие стадии.
Получение натриевой соли производных, имеющих следующую общую формулу: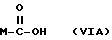
где M выбран из (XXX), (XXXI), (XXXII),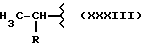
где R выбран среди следующих структур: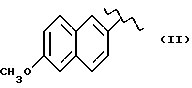
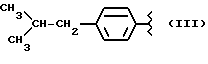
при получении производных (VIA), замещенных по карбоксильной группе, таких как хлорангидриды, ангидриды или подобные.
Реакцию натриевой соли вышеуказанных производных (VIA) или вышеуказанных производных (VIA), замещенных по карбоксильной группе, с соединением, имеющим следующую общую формулу.
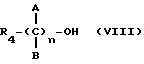
где R4 выбран из хлора, брома, NHR5 с R5 - водород, линейный или разветвленный алкил, A и B выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила, и n равно от 1 до 10, с получением таким путем соответствующих эфиров или соответствующих амидов.
Реакцию вышеуказанных эфиров или вышеуказанных амидов галоидирующим соединением, таким как PBr3 или подобным, с получением продуктов вышеуказанных эфиров или вышеуказанных амидов, характеризующихся наличием концевой галоидной группой.
Реакцию вышеуказанных эфиров или вышеуказанных амидов, характеризующихся наличием концевой галоидной группы, с нитрующим агентом, таким как AgNO3 или подобным, с получением нитроэфиров производных (IA).
Растворители, которые используют в способах, являющихся предметов настоящего изобретения, преимущественно выбирают из хлороформа, хлористого метилена, ацетонитрила, диметилформамида, тетрагидрофурана, 1,4-диоксана, и подобных.
Такие способы для получения производных (IA), являющихся предметом настоящего изобретения состоят из ограниченного числа стадий и позволяют получать продукты этих процессов на короткое время с хорошими выходами и в больших количествах даже в промышленном плане.
Согласно способам, которые являются предметом настоящего изобретения, получение нитроэфира производного пропионовой кислоты, имеющего следующую формулу: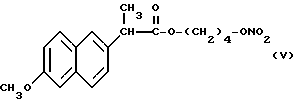
особенно полезно и описано в следующем примере, иллюстрирующем, но не ограничивающем данное изобретение.
Пример 1
а) 0,59 г EtONa, растворенного в 10 мл этилового спирта, добавили, медленно прикапывая, к раствору 2 г 2-(6-метокси-2-нафтил) пропионовой кислоты, растворенной в 20 мл этилового спирта. Реакционную смесь перемешивали 5 минут при комнатной температуре, затем упарили при пониженном давлении, получая 2,1 г натриевой соли 2-(6-метокси-2-нафтил)пропионовой кислоты.
2,1 г натриевой соли 2-(6-метокси-2-нафтил)пропионовой кислоты, полученной таким образом, диспергировали в 40 мл диметилформамида и 1,5 г 1-Br-4-Cl-бутана, растворенного в 30 мл диметилформамида, добавили при перемешивании к этой дисперсии. Реакционную смесь перемешивали 12 часов при комнатной температуре, затем разбавили водой и экстрагировали хлористым метиленом. Органическую фазу, проэкстрагированную таким образом, высушили сульфатом натрия и упарили растворитель при пониженном давлении до тех пор, пока получили 2 г сухого остатка.
Остаток очистили хроматографией на силикагеле, используя в качестве элюента смесь гексан/эфир 7/3 (по объему).
Головные фракции собрали, растворитель упарили при пониженном давлении получили 1 г 2-(6-метокси-2-нафтил)пропионата 4-хлорбутила (IX).
ИК (см-1): C=O, 1669
1H-ЯМР (300 МГц) (CDCl3): 1,6 м.д. (d, 3H); 1,75 м.д. (m, 4H); 3,45 м.д. (m, 2H); 3,88 м.д. (q, 1H); 3,91 м.д. (l, 3H); 4,1 м.д. (m, 2H); 7,1 - 7,7 м.д. (m, ароматические).
Масс-спектрометрия: M+ • 320.
b) 0,79 г AgNO3 растворенного в 1,3 мл ацетонитрила прикапывали к 1 г (IX), полученного, как описано в a), растворенному в 4,5 мл ацетонитрила. Реакционную смесь перемешивали 12 часов при температуре 85oC и затем отфильтровали.
Из полученного раствора был упарен растворитель при пониженном давлении и в полученный остаток добавили 10 мл хлористого метилена. Смесь, полученную таким образом, отфильтровали снова, органическую фазу промыли водой и затем высушили над сульфатом натрия. Растворитель упарили при пониженном давлении и получили 1,8 г сухого остатка, который очистили хроматографией на силикагеле, используя в качестве элюента смесь гексан/эфир 7/3 (по объему). Фракции, содержащие продукт, были собраны, растворитель упарили при пониженном давлении и получили 1,5 г нитроэфира 2-(6-метокси-2-нафтил) пропионата 4-гидроксибутила (V).
ИК (см-1): C=O, 1733; ONO2, 1637.
1H-ЯМР (300 МГц) (CDCl3): 1,6 м.д. (d, 3H); 1,65 м.д. (m, 4H); 3,8 м.д. (q, 1H); 3,9 м.д. (s, 3H); 4,1 м.д. (m, 2H); 4,3 м.д. (m, 2H); 7,1 - 7,7 м. д. (m, ароматические).
Масс-спектрометрия: M+ • 347.
Согласно способу, который является предметом настоящего изобретения, также получают нитроэфир производный пропионовой кислоты, который является особенно полезным и имеет следующую формулу: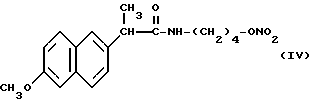
который получили, как описано в следующем примере, и который дан для иллюстрации данного изобретения, но не ограничивает его объема.
Пример 2
a) 23,9 г фталимида калия, диспергированного в 200 мл безводного диметилформамида добавили к раствору 55,7 г 1,4-ди-бромбутана, растворенного в 300 мл безводного диметилформамида. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре, затем разбавили водой и экстрагировали хлористым метиленом. Хлористый метилен упарили из полученной таким образом органической фазы при пониженном давлении и затем отогнали диметилформамид при давлении 10 мм Hg.
Остаток промывали водой и проэкстрагировали хлористым метиленом. Органическая фаза, полученная таким образом, была высушена и растворитель отогнали при пониженном давлении, получив 14,8 г 1-фталимид-4-бромбутана, который обработали изопропиловым эфиром и затем выделили. Т. пл. = 77oC.
b) 32 мл иодистоводородной кислоты осторожно добавили к 8,25 г 1-фталимидо-4-бромдутана; затем смесь нагрели и выдерживали при бурном кипении 24 часа.
После охлаждения смесь разбавили водой и после фильтрования упарили растворитель при пониженном давлении, полученный остаток перекристаллизовали один раз из этилового эфира и получили 6 г 4-иодбутиламмонийиодида.
Т. пл. = 103oC.
c) 7 мл тионилхлорида осторожно добавили к раствору 2,3 г 2-(6-метокси-2-нафтил) пропионовой кислоты в 15 мл безводного хлороформа. Реакционную смесь перемешивали 40 минут при комнатной температуре, а затем упарили растворитель при пониженном давлении, получив 2,23 г 2-(6-метокси-2-нафтил) пропионилхлорида.
2,3 г 2-(6-метокси-2-нафтил) пропионилхлорида растворили в пиридине и раствор охладили до температуры 0oC.
3,27 г 4-иодбутиламмонийиодида прибавили к этому раствору, и смесь, полученную таким образом, перемешивали 1 час при 0oC и затем разбавили водой и проэкстрагировали хлористым метиленом.
Органическую фазу, полученную таким образом, сначала промыли 10% раствором соляной кислоты, а затем насыщенным раствором бикарбоната натрия, затем упарили растворитель при пониженном давлении, получив 3,2 г сухого остатка. Остаток очищали хроматографией на силикагеле, используя в качестве элюента хлористый метилен.
Промежуточные фракции собрали, упарили растворитель при пониженном давлении и получили 1,6 г 2-(6-метокси-2-нафтил)-4-иодбутилпропионамида (XX).
ИК (см-1): NH, 3294; C=O, 1651.
1H-ЯМР (300 МГц) (CDCl3): 1,1 - 1,75 м.д. (m, 4H); 1,6 м.д. (d, 3H); 3,1 м. д. (t, 2H); 3,2 м.д. (q, 2H); 3,7 м.д. (q, 1H); 3,9 м.д. (s, 3H); 5,35 м. д. (m, NH); 7,1 - 7,7 м.д. (m, ароматические).
d) Суспензию 1,6 г 2-(6-метокси-2-нафтил)-4-иодбутилпропионамида в 20 мл ацетонитрила нагрели до температуры около 40oC и перемешивали, пока 1,0 г AgNO3 был добавлен.
Смесь перемешивали 1 час при комнатной температуре, затем отфильтровали и упарили при пониженном давлении. Полученный остаток смешали с хлористым метиленом, образовавшуюся смесь отфильтровали и упарили растворитель при пониженном давлении, и получили 0,8 г сухого остатка, который очищали хроматографией на силикагеле, используя в качестве элюента смесь хлористый метилен/этилацетат 9/1 (по объему).
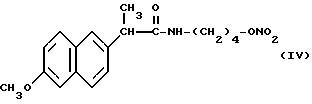
Головные фракции собрали, растворитель упарили при пониженном давлении и получили 0,75 г нитроэфира 2-(6-метокси-2-нафтил)-4- гидроксибутилпропионамида (IV).
ИК (см-1): C=O, 1672; NH, 3294; ONO2, 1637.
Масс-спектрометрия: M+ • 346.
1H-ЯМР (80 МГц) (CDCl3): 1,3 - 1,6 м.д. (m, 4H); 1,7 м.д. (d, 3H); 3,1 м.д. (q, 2H); 3,7 м.д. (q, 1H); 3,9 м.д. (s, 3H); 4,3 м.д. (m, 2H); 5,6 м.д. (m, NH); 7,05 - 7,8 м.д. (m, ароматические).
Согласно настоящему изобретению нитроэфир, имеющий следующую формулу: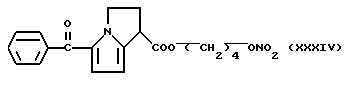
являющийся особенно полезным, был получен, как описано в приведенном ниже примере, который дан для иллюстрации данного изобретения, но не ограничивает его объема.
Пример 3
Получение соединения, имеющего формулу: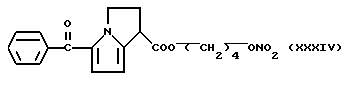
a) В суспензию 80% гидрида натрия (0,16 г) в ДМФ (15 мл) осторожно при перемешивании, прикапали 1,15 г Кеторолака, растворенного в 20 мл ДМФ.
Реакционную смесь выдерживали при перемешивании при 40oC 15 минут, затем добавили 1 мл 1,4-дибромбутана и смесь перемешивали при комнатной температуре всю ночь.
Затем упарили растворитель при пониженном давлении и остаток обработали водой и хлористым метиленом. Органическую фазу отделили, высушили над сульфатом натрия и удалили растворитель при пониженном давлении, полученный остаток очищали хроматографией на силикагеле, используя в качестве элюента смесь петролейный эфир/эфир 4/6 (по объему). Головные фракции собрали, растворитель упарили при пониженном давлении и получили 0,75 г продукта, имеющего формулу: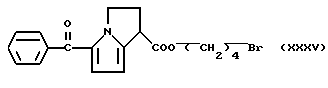
1H-ЯМР (80 МГц) (CDCl3): 1,83 (6H, m); 2,81 (2H, m); 3,38 (2H, t); 4,12 (2H, t); 4,48 (1H, m); 6,03 (1H, d); 6,78 (1H, d); 7,41 (3H, m); 7,73 (2H, m).
b) Раствор AgNO3 (0,5 г) в 5 мл ацетонитрила добавили к раствору (XXXV) в 20 мл ацетонитрила. Реакционную смесь выдерживали при перемешивании при комнатной температуре 48 часов. Растворитель удалили при пониженном давлении и остаток обработали водой и хлористым метиленом. Органическую фазу отделили, высушили над сульфатом натрия и удалили растворитель при пониженном давлении. Остаток очищали хроматографией на силикагале, используя в качестве элюента смесь петролейный эфир /эфир 4/ 6 (по объему). Головные фракции собрали, растворитель упарили при пониженном давлении и получили 0.35 г (XXXIV).
1H-ЯМР (80 МГц) (CDCl3) (м.д.): 1.78 (6H, m); 2.82 (2H, m); 4.14 (2H, m); 4.47 (3H, m); 6.03 (1H, d); 6,79 (1Н, d); 7,46 (3Н,m); 7.77 (2H, m).
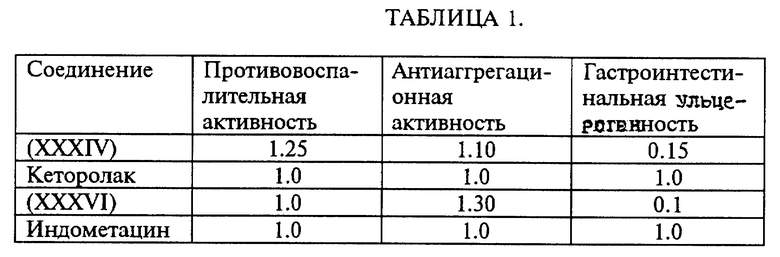
По биологическим тестам противовоспалительная и анальгетическая активность была определена, например, для нитроэфиров (LA), имеющих следующую формулу: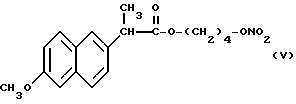
Противовоспалительная активность указанных выше нитроэфиров производных пропионовой кислоты была изучена на крысах вида Вистар с использованием метода воздействия на индуцированный каррагином отек, как описано в C.F. WINTER, E. RISLEY, G. W. NUSS, Proc. Soc. Exp. Biol. Med. 111, 544 - 547 (1962), в то время как анальгетическая активность указанных выше производных была изучена на мышах породы Свисс, как описано L.C.HENDERSHOT, J.FORSAITH, J.Pharmacol. Exp. Ter. 125, 237 - 249 (1959).
Противовоспалительная и анальгетическая активность вышеуказанных производных получена в сравнении с 2-(6-метокси-2-нафтил)пропионовой кислотой, взятой как эталон.
Антитромбоцитарная активность указанных выше производных была изучена на человеческих тромбоцитах. Тромобоциты были инкубированы с соединениями на 10 минут при 37oC до стимуляции тромбином. Антитромбоцитраная активность указанных выше производных получена в сравнении с 2-(6-метокси-2-нафтил) пропионовой кислотой, взятой за эталон.
Затем острая токсичность вышеупомянутых производных (IV) и (V) была оценена при оральном применении отдельных доз каждого соединения (IV) и (V), используя для каждого производного группы из 10 мышей породы Свисс.
Частота летальности и начала симптомов интоксикации были описаны для периода 14 дней.
Даже после применения дозы 750 мг/кг соединения (IV) или соединения (V) не установлено симптомов их токсичности у обрабатываемых животных.
Дальнейшие биологические исследования были проведены в отношении фармакотоксикологического профиля изучаемых соединений, в частности, соединения (V), в сравнении с 2-(6-метокси-2-нафтил) пропионовой кислотой, взятой за эталон.
А.Фармакодинамическая активность.
Острые модели.
Воздействие на индуцированный каррагином отек у крыс. Исходя из предварительных экспериментов, соединение (V) и 2-(6-метокси-2-нафтил) пропионовая кислота показывают сравнимую эффективность; эффективная доза находится в диапазоне от 1 до 10 мг/кг.
Подострые модели.
Адъювант артрита крыс. Животные, обработанные в течение 19 дней (с 3 по 20 день после инъекции) соединением (V) или 2-(6-метокси-2-нафтил) пропионовой кислотой, и тем и другим в дозе 3 мг/кг, показали значительное снижение симптомов артрита по сравнению с контролем.
В.Гастроинтестинальная толерантность.
Повреждение слизистой оболочки крыс. Соединение (V) было изучено в сравнении с 2-(6-метокси-2-нафтил) пропионовой кислотой, взятой в качестве эталона, каждый в дозе в диапазоне от 3 до 30 мг/кг; соединение (V) показало значительно лучшую толерантность, чем 2-(6-метокси-2-нафтил) пропионовая кислота. 2-(6-метокси-2-нафтил) пропионовая кислота уже при 3 мг/кг вызывала повреждение слизистой, и этот эффект зависел от дозы, в то время как соединение (V) проявляло хорошую толерантность даже при дозах 30 мг/кг.
C.Общая фармакология.
Вторичная формакологическая экспертиза соединения (V) была выполнена при сравнении с 2-(6-метокси-нафтил) пропионовой кислотой. Не было обнаружено значительных побочных эффектов, что касается фармакологической активности при действии на центральную нервную систему, на вегетативную нервную систему, на сердечно-сосудистую, респираторную и гастроинтестинальную системы.
D.Токсикология.
Острая токсикология у грызунов. Предварительные исследования были проведены на мышах с использованием двух способов введения. Никаких явных симптомов токсичности не было обнаружено у обработанных животных при оральном или интраперитонеальном введении при дозе 300 мг/кг.
Максимальная переносимая доза у грызунов. Предварительные исследования показали, что соединение (V) хорошо переносится собакой, теми видами животных, которые, как известно, особенно чувствительны к ульцерогенной активности противовоспалительных препаратов. Животные, получая оральную дозу соединения (V) 30 мг/кг, не проявляли вышеупомянутых симптомов. В сравнении, 2-(6-метокси-2-нафтил) пропионовая кислота в дозе 10 мг/кг вызывала смерть животных.
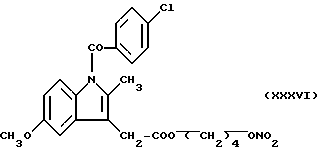
Кроме того, были проведены биологические исследования, относящиеся к нитроэфирам (IA), имеющим следующую формулу: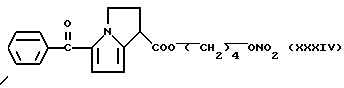
Затем была определена противовоспалительная активность, гастроинтестинальная толерантность и тромбоцитарная антиагрегационная активность вышеуказанных соединений.
Противовоспалительная активность была определена на крысах методом воздействия на индуцированный каррагином отек, как описано в C.A. WINTER и др. (1962) Proc. Aoc. Exp. Biol. Med. III, 544. Гастроинтестинальная толерантность определялась оральным введением на крысах. Тромбоцитарная антиагрегационная активность определялась на человеческих тромбоцитах, стимулированных арахидоновой кислотой согласно методу, описанному в V.BERTELE и др. (1983) Science 220, 517.
Результаты, приведенные в табл. 1 оценки, относящихся к противовоспалительной, антиагрегационной активности и гастроинтестинальной толерантности ниже исследованных соединений, показывают относительно высокую степень по сравнению с продуктом, взятым в качестве стандарта.
Острая токсичность указанных соединений была оценена при оральном применении отдельных доз для каждого производного в группе из 10 мышей. Доказательство смерти и начала симптомов интоксикации было описано для периода 14 дней. Даже после применения дозы 100 мг/кг каждого соединения, животные не показали симптомов явной токсичности.
Пример 4.
Синтез (S)6-метокси -α- метил-2-нафталинуксусной кислоты 1-нитрокси-2-метил-2-пропил эфира /HCT3017/: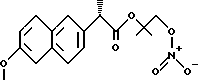
(S)-6-метоки -α- метил-2-нафталинуксусная кислота 1-хлор-2-метил-2-пропил эфир.
К раствору (S)-6-метоки -α- метил-2-нафталинуксусной кислоты (4 г, 17 молей) в хлороформе (45 мл) добавляют при комнатной температуре 1-хлор-2-метил-2-пропанол (1,89 г, 17 ммолей, N,N'-дициклогексилкарбодиимида (3,59 г, 17 молей) и 4-диметиламинопиридина (0,11 г, 0,09 ммолей). Смесь перемешивают при комнатной температуре в течение 24 часов. Твердую массу фильтруют и раствор хлороформа промывают водой. Органический слой отделяют, просушивают с сульфатом натрия и выпаривают в вакууме. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 8-2). Выделяют промежуточный продукт в виде белой твердой массы (3,34 г, точка плавления 48-51oC) и используют для последующей реакции.
(S)-6-метокси -α- метил-2-нафталинуксусная кислота 1-нитроокси-2-метил-2-пропил эфир.
Смесь (S)-6-метоки -α- метил-2-нафталинуксусная кислота 1-хлор-2-метил-2-пропил эфира (3,2 г, 10 ммолей) и нитрата серебра (2,39 г, 14 ммолей) в ацетонитриле (30 мл) орошают в темноте в течение 2 дней. После охлаждения твердую массу фильтруют и растворитель выпаривают. Осадок очищают в хроматографической колонне (элюент гексан-этил-ацетат 8-2). Конечное соединение выделяют в чистом виде как светло-желтое масло (1,5 г).
1H-NMR(CDCl3, ppm): 7,84-7,73 (3H, m), 7,50 (1H,dd), 7,21-7,18 (2H,m), 4,15 (2H,s), 3,90 (4H,m), 1,48 (3H,d), 1,29 (6H,s).
Пример 5.
Синтез (S)-6-метокси -α- метил-2-нафталинуксусной кислоты 6-(нитроокси)гексил эфира [HCT3018].
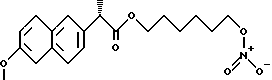
(S)-6-метокси -α- метил-2-нафталинуксусная кислота 6-хлоргексил эфир.
Раствор (S)-6-метокси -α- метил-2-нафталинуксусной кислоты (6 г, 24 ммоли) в хлороформе (50 мл) охлаждают до температуры 0oC и обрабатывают каплями триэтиламина (4,88 г, 48 ммолей). К полученной охлажденной смеси добавляют каплями раствор 6-хлор-1-гексанола (4,94 г, 36 молей) в хлороформе (15 мл). Смесь перемешивают при комнатной температуре в течение 2 часов и затем промывают водой. Органический слой отделяют, просушивают с сульфатом натрия и выпаривают в вакууме. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 8-2). Выделяют промежуточный продукт в виде светлого желтого масла (5,54 г) и используют для следующей реакции.
(S)-6-метокси -α- метил-2-нафталинуксусная кислота 6-(нитроокси)гексил эфир.
Смесь (S)-6-метокси -α- метил-2-нафталинуксусной кислоты 6-хлоргексил эфира (5,36 г, 15 ммолей) и нитрата серебра (5,48 г, 32 ммолей) в ацетонитриле (50 мл) орошают в темноте в течение 7 дней. После охлаждения твердую массу фильтруют и растворитель выпаривают. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 9-1). Конечное соединение выделяют в чистом виде как светло-желтое масло (5,2 г).
1H-NMR(CDCl3, ppm): 7,74-7,70 (3H,m), 7,44 (1H,dd), 7,20-7,13 (2H,m), 4,26 (2H, t), 4,10 (2H,m), 3,90 (3H,s), 3,88 (1H,q), 1,61 (3H,d), 1,58-1,47 (4H,m), 1,28-1,15 (4H,m).
Производные бруфена.
Пример 6.
Синтез -α- -метил-4-(2-метилпропил) бензенуксусной кислоты 3-(нитроокиси)пропил эфира [NCX 322]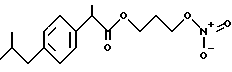
α- -метил-4-(2-метилпропил) бензенуксусная кислота 3-хлорпропил эфир.
К раствору α- -метил-4-(2-метилпропил) бензенуксусной кислоты (4,1 г, 20 ммолей) в хлороформе (45 мл) добавляют при комнатной температуре 1-хлор-3-пропанол (1,89 г, 20 ммолей), N,N'-дициклогексил карбодиимида (4,1 г, 20 ммолей) и 4 диметиламинопиридина (0,11 г, 0,9 ммолей). Смесь перемешивают при комнатной температуре в течение 4 часов. Твердую массу фильтруют и раствор хлороформа промывают водой. Органический слой отделяют, просушивают с сульфатом натрия и выпаривают в вакууме. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 1-1). Выделяют промежуточный продукт в виде светло-желтого масла (4,8 г) и используют для следующей реакции.
α- -метил-4-(2-метилпропил) бензенуксусная кислота 3-(нитроокси) пропил эфир.
Смесь α- -метил-4-(2-метилпропил) бензенуксусной кислоты 3-хлорпропил эфира (4,47 г, 15,8 ммолей) и нитрата серебра (5,3 г, 31,7 ммолей) в ацетонитриле (20 мл) орошают в темноте в течение 36 часов. После охлаждения твердую массу фильтруют и растворитель выпаривают. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 18-1). Конечное вещество выделяют в чистом виде как светло-желтое масло (3,7 г).
1H-NMR(CDCl3, ppm): 7,19 (4H,dd), 4,39 (2H2t), 4,21 (2H,t), 3,73 (1H,q), 2,50 (2H,d), 2,04 (2H,t), 1,90 (1H,m), 1,54 (3H,d), 0,94 (6H,d).
Пример 7.
Синтез α- метил-4-(2-метилпропил) бензенуксусной кислоты 6-(нитроокси) гексил эфира [NCX 321]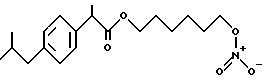
хлор α -метил-4-(2-метилпропил) бензенуксусной кислоты.
Раствор α -метил-4-(2-метилпропил) в бензенуксусной кислоте (6,97 г, 33,8 ммолей) в толуоле (20 мл) и диметилформамиде (2 мл) охлаждают до 0oC и обрабатывают каплями оксалил хлорида (8,6 г, 67,8 ммолей). Смесь перемешивают в течение 24 часов при комнатной температуре и затем выпаривают в вакууме. Твердую массу используют для последующей реакции.
α -метил-4-(2-метилпропил) бензенуксусная кислота 6-хлоргексил эфир.
Раствор массы хлорида α -метил-4-(2-метилпропил) бензенуксусной кислоты (33,8 ммолей) в хлороформе (35 мл) обрабатывают при комнатной температуре триэтиламином (6,87 г, 67,9 ммолей). Смесь охлаждают до температуры 0oC и обрабатывают каплями раствора 6-хлор-1-гексанол (6,95 г, 50,9 молей). Смесь перемешивают при комнатной температуре в течение 5 часов и промывают водой. Органический слой просушивают с сульфатом натрия и выпаривают в вакууме. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 7-3). Выделяют промежуточный продукт в виде светло-желтого масла (9,9 г) и используют для последующей реакции.
α -метил-4-(2-метилпропил) бензенуксусная кислота 6-(нитроокси) гексил эфира.
Смесь α -метил-4-(2-метилпропил) бензенуксусной кислоты 6-хлоргексил эфира (5,58 г, 17,2 ммолей) и нитрата серебра (4,09 г, 24,1 ммолей) в ацетонитриле (20 мл). орошают в темноте в течение 40 часов. После охлаждения массу фильтруют и растворитель выпаривают в вакууме. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 27-1). Конечный продукт выделяют в чистом виде как светло-желтое масло (2,4 г).
1H-MNR (CDCl3, ppm): 7,18 (2H, d), 7,07 (2H, d), 4,36 (2H, t), 4,05 (2H, m), 3,67 (1H, q), 2,43 (2H, d), 1,83 (1H, m), 1,60 (4H, m), 1,46 (3H, d), 1,36 - 1,23 (4H, m), 0,88 (6H, d).
Производные локсопрофена
Пример 8.
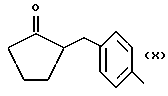
Синтез α метил-4-[(2-оксоциклопентил)метил] бензенуксусной кислоты 4-(нитроокси)бутил эфира [NCX 628]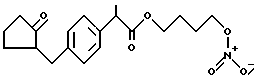
α метил-4-[(2-оксоциклопентил)метил]бензеновая кислота 4-бромбутил эфир.
К раствору 1,4-дибромбутана (7,3 г, 33,79 ммолей) в диметилформамиде (30 мл) добавляют каплями при комнатной температуре раствор соли натрия ∞ метил-4-[(2-оксоциклопентил)метил]бензенуксусной кислоты (3,03 г, 11,3 ммолей) в диметилформамиде (60 мл). Смесь перемешивают при комнатной температуре в течение 15 часов, раствор выпаривают в вакууме и осадок промывают водой и диэтил эфиром. Органическую фазу выделяют, промывают водой, просушивают сульфатом натрия и выпаривают в вакууме. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 8 - 2). Промежуточный продукт выделяют в виде светло-желтого масла (3,9 г) и используют для последующей реакции.
α метил-4-[(2-оксоциклопентил)метил] бензенуксусная кислота 4-(нитроокси)бутил эфир.
Смесь α метил-4-[(2-оксоциклопентил)метил] бензенуксусной кислоты 4-бромбутил эфира (3,0 г, 10,2 ммолей) и нитрата серебра (4,69 г, 27,6 ммолей) в ацетонитриле (20 мл) орошают в темноте в течение 18 часов. Затем охлаждают, массу фильтруют и раствор выпаривают в вакууме. Осадок очищают в хроматографической колонне (элюент гексан-этил ацетат 8 - 2). Конечный продукт выделяют в чистом виде как светло-желтое масло (2,84 г).
1H-NMR (CDCl3, ppm): 7,19 - 7,06 (4H, m), 4,34 (2H, t), 4,06 (2H, е), 3,65 (1H, q), 3,07 (1H, dd), 2,47 (1H, dd), 2,42 - 1,43 (11H, m), 1,23 (3H, d).
Индометацин.
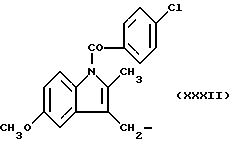
Синтез 1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-уксусной кислоты 4-(нитроокси)бутил эфира (NCX 534).
1-(4-хлорбензоил-5-метокси-2-метил-1H-индол-3-уксусная кислота 4-хлорбутил эфир.
К раствору 1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол- 3-уксусной кислоты (5,04 г, 14 ммолей) в хлороформе (50 мл) добавляют при комнатной температуре 1-хлор-4-бутанол (1,4 мл, 14 ммолей), N,N1-дициклоэтилкарбодиимид (2,87 г, 14 ммолей) и 4-диметиламинопиридин (0,11 г, 0,09 ммолей).
Смесь перемешивают при комнатной температуре в течение 6 часов. Массу фильтруют и раствор хлороформа промывают водой. Органический слой выделяют, просушивают в вакууме.
Осадок очищают в хроматографической колонне (элюент n-гексан/этил ацетат 9/1). Промежуточный продукт выделяют в виде желтого масла (5,2 г) и используют для последующей реакции.
1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-уксусная кислота 4-(нитроокси)бутил эфир.
Смесь 1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-уксусной кислоты 4-хлорбутил эфира (5 г, 11 ммолей) и нитрата серебра (3,8 г, 22 ммолей) в ацетонитриле (25 мл) орошают в темноте в течение 48 часов.
После охлаждения массу фильтруют и растворитель выпаривают в вакууме. Осадок очищают в хроматографической колонне (элюент n-гексан/этил ацетат 9/1). Конечный продукт выделяют в виде масла (4,2 г).
1H-NMR (CDCl3, ppm): 7,65 (2H, m), 7,45 (2H, m), 6,95 (1H, d), 6,84 (1H, d), 6,66 (1H, dd), 4,10 (2H, t), 3,82 (3H, s), 3,65 (2H, s), 3,35 (2H, t), 2,39 (3H, s), 1,80 (4H, m).
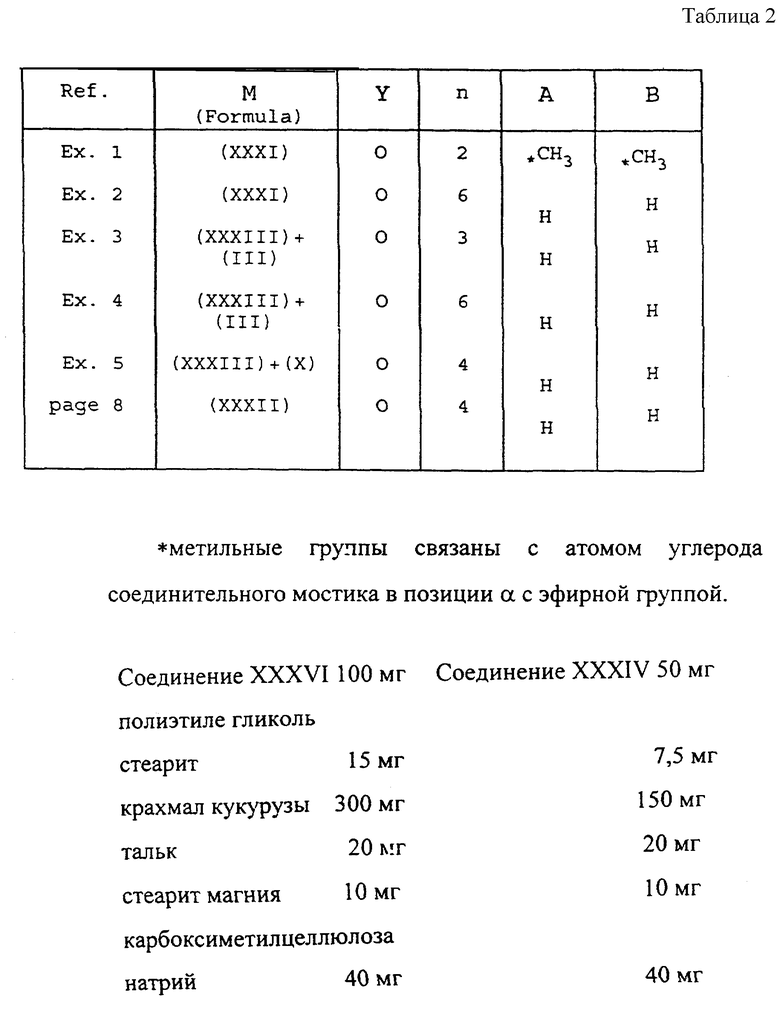
Полученные вещества приведены в следующей табл. 2, в которой делается ссылка на формулу (1A) пункта 1 формулы изобретения: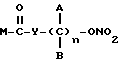
Указанные грануляты были приготовлены в лаборатории методом влажной грануляции. Ниже описывается приготовление гранулята, включающего соединение XXXVI.
В растворе смешивали 10 г соединения XXXVI с 3 гр крахмала, 1,5 г полиэтилен гликоль стеарита, 2 г талька и 1 г стеарита магния. Порошок обрабатывали в растворе до тех пор, пока не появилась очень тонкая консистенция. Раствор из 4 г карбоксиметилцеллюлозы натрия в 50 мл воды добавляли порциями в порошок в растворе, перемешивая, при промежуточном просушивании при температуре 60oC в вакууме опять размалывая и просушивая. Полученные гранулы продавливали через сито 6-меш.
п. Животные модели воспалений.
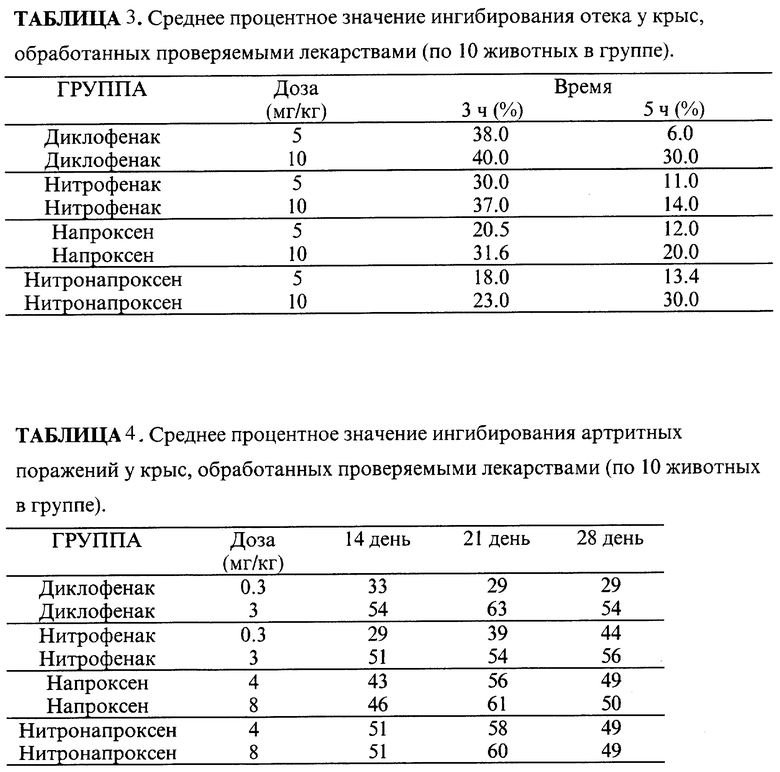
Для проверки эффективности этих новых соединений мы использовали две модели с острым (отек, вызванный каррагинаном) и хроническим (адъювантный артрит) воспалением у крыс.
Отек вызывали инъекцией 0,1 мл 1% каррагинана, суспендированного в стерильном физиологическом растворе (растворитель) в плантарное пространство правой задней ступни каждой крысы. Объем ступни измеряли через 3 и 5 часов после инъекции каррагинана и анализировали отек с точки зрения увеличения объема ступни по отношению к объему ступни до раздражающей инъекции. Измерения проводили водным плетизмометром (mod. 7150, Ugo Basile, Milano, Italy). Диклофенак, нитрофенак, напроксен и нитронапроксен (нитронапроксен соответствует нитроксибутиловому эфиру напроксена) дозой 5 и 10 мг/кг, суспендированные в 0,5% карбоксиметилцеллюлозе, орально вводили за час до инъекции каррагинана. Среднее значение увеличения ступни в группах обработанных животных сравнивали со значением у животных, обработанных растворителем. Адъювантный артрит вызывали инъекцией в основание хвоста 0,6 мл убитых нагреванием клеток Mycobacterium butyricum, суспендированных в 0,1 мл минерального масла. Через 14, 21 и 28 дней развитие артрита оценивалось одним и тем же наблюдателем. Первичные и вторичные артритные поражения оценивались по условной шкале следующим образом: левая и правая задние ступни каждая 0 - 7, левая и правая передние ступни каждая 0 - 4,5, хвост 0 - 5, уши 0 - 2, нос и глаза каждый 0 - 1. Дифлофенак вводили в дозах 0,3 и 3 мг/кг веса тела, напроксен и нитронапроксен давали в дозах 4 и 8 мг/кг веса тела соответственно; лекарства вводили перорально ежедневно с 3 по 21 день после вызывания артрита, следуя профилактическому протоколу. Для 14, 21 и 28 дней противовоспалительную активность лекарств выражали как процент ингибирования значения оценки артрита по шкале против контрольной группы.
Как исходные лекарства, так и нитропроизводные в обеих дозах вызывали значительное уменьшение отека лапки через три часа, тогда как через пять часов только диклофенак и нитронапроксен при более высоких дозах обеспечивали значительное уменьшение, вероятно благодаря своим фармакокинетическим свойствам. В каждом случае активности лекарств, по-видимому, сравнимы (7,8) с похожим средним процентным значением ингибирования отека: среднее значение около 38,5% через три часа для диклофенака и нитрофенака при дозе 10 мг/кг, среднее значение около 27% через три часа для напроксена и нитронапроксена при дозе 10 мг/кг (табл. 3).
Диклофенак и нитрофенак при высоких дозах (3 мг/кг) и напроксен и нитронапроксен при обеих дозах (4 и 8 мг/кг) значительно ингибировали артрит.
Воздействие высвобождающего оксид азота производного напроксена на гипертензию и поражение желудка, вызванные хроническим ингибированием оксида азота у крыс. Mаrcelo N. Muscara, Webb McKnight, Piero Del Soldato & John L. Wallace Department of Pharmacology and Therapeutics, University of Calgary, Alberta, Canada; NicOx S.A., Paris, France. (Submtter December 4, 1997; accepted January 21, 1998; received in final Form Janyary 22, 1998)
Резюме: NSAIDs могут повышать кровяное давление посредством таких механизмов, как почечное сужение кровеносных сосудов и ретенция натрия. Такое воздействие проявляется особенно очевидно на гипертензивных особях. Как было показано, высвобождающие оксид кислорода NSAID производные обладают значительно пониженной токсичностью в желудочно-кишечном тракте и в почках. Кроме того, мы оценили воздействие 4х-недельной обработки или напроксеном, или его высвобождающим оксид азота и производным (NO-напроксен) на общее кровяное артериальное давление и на поражение желудка у крыс, в которых гипертензия была вызвана L-NAME. Крысы получали L-NAME или растворенным в питьевой воде (400 мг/л), или в водопроводной воде (контроль). Растворитель, напроксен (10 мг/кг) или эквимолярная доза NO-напроксена (14.5 мг/кг) вводились орально ежедневно. Через 4 недели измеряли кровяное давление, брали образцы крови для измерения синтеза тромбоксана и оценивали поражение желудка посредством слепой макроскопической оценки. И напроксен, и NO-напроксен ингибировали общую циклооксигеназную активность на > 90%. У крыс, обработанных NO-напроксеном, не проявлялись значительные повреждения желудка. Поражение желудка, вызванное одним L-NAME, усиливалось напроксеном, но предотвращалось NO-напроксеном. Лечение L-NAME значительно повышало кровяное давление. В отсутствие L-NAME группа, получившая напроксен, имела значительно более высокое кровяное давление, чем в контрольной группе и в группе, получившей NO-напроксен. Для крыс, получивших L-NAME, применимы те же результаты, но сопутствующее введение NO-напроксена приводило к значительному уменьшению кровяного давления по сравнению с введением только L-NAME. Основываясь на этих результатах, мы делаем вывод, что NO-напроксен может представлять собой более безопасную альтернативу по сравнению со стандартными NSAIDs при лечении воспалительных состояний у гипертензивных пациентов.
Ключевые слова: нестероидное противовоспалительное лекарство, гипертензия, оксид азота, циклооксигеназа.
Введение.
Токсическое воздействие нестероидных противовоспалительных лекарств (NSAIDs) на почечную функцию происходит путем ингибирования циклооксигеназы, в результате чего уменьшается продукция сосудорасширяющих простаноидов (в основном PGE2 и PGI2), которые вместе с другими медиаторами (такими как ангиотензин II, предсердный натриуретический пептид и аргинин-вазопрессин) вовлечены непосредственно в контроль почечного гомеостаза (1,2). Экскреция натрия, секреция ренина и симпатический тонус косвенно контролируются простагландинами (3,4), ингибирование которых приводит к увеличению общего кровяного давления (КД). Однако у человека воздействие NSAID на кровяное давление (КД) нельзя четко оценить, при этом расхождения возникают из-за различий в возрасте, этническом происхождении и/или состоянии здоровья среди изучаемых субъектов (5). Более пожилые, чернокожие и с пониженным ренином гипертензивные пациенты описаны как особенно чувствительные к гипертензии, вызванной NSAID (6). Индометацин и напроксен являются NSAIDs, которые вызывают у гипертензивного населения не только значительное повышение КД, но также проявляют значительный антагонизм к положительным воздействиям антигипертензивных лекарств, таких как триазиды и β- блокаторы (6).
Образование язвы желудка, вызванное приемом NSAID, по-видимому, связано со снижением тока крови в желудке и с увеличением слипания лейкоцитов в микроциркуляции желудка, что является побочным эффектом снижения синтеза простагландинов (см. обзор в ссылке 7). Предполагают, что такие воздействия NSAIDs являются основными в их ульцерогенном действии (7). Недавно была описана новая группа NSAID производных со значительно сниженным вредным воздействием на слизистую оболочку желудка. Такие производные называют "NO-NSAIDs", поскольку они обладают способностью высвобождать оксид азота (NO), хорошо известный вазодилатор (8,9,10) с ингибирующим воздействием на слипание тромбоцитов (11,12) и лейкоцитов (13). Добавление высвобождающей оксид азота части к NSAIDs не привело к потере способности этих лекарств ингибировать циклооксигеназу. Среди NO-NSAIDs, которые недавно охарактеризованы таким образом, находятся NO-фторбипрофен и NO-кетопрофен (14), NO-диклофенак (15), NO-напроксен (16) и NO-аспирин (17). Когда NO-NSAIDs давали крысам с нормальным КД, они не оказывали значительного влияния на общее кровяное артериальное давление (14, 17). Однако, исследования таких соединений на гипертензивных животных не проводились.
В этом исследовании мы изучали вероятность того, что NO-NSAIDs могут оказывать меньше вазопрессорного воздействия на гипертензивных крыс, чем стандартные NSAID. Таким образом, мы тестировали воздействие лечения напроксеном или NO-напроксеном за период в размере четырех недель на КД и поражение желудка у крыс, в которых гипертензия была вызвана хроническим введением L-NAME, ингибитора синтеза NO.
Методы.
Лечение. Самцов крыс Wistar, весом примерно 250 г, получили от Charles River Breeding Farms (Montreal, Quebec). Их содержали в полипропиленовых клетках группами по два или три животных в клетке, и они получали лабораторную еду ad libitum. Группы крыс (n=10 в каждой) получали ежедневно оральную дозу растворителя (1 мл/кг), напроксена (10 мг/кг) или эквимолярную дозу NO-напроксена (14.5 мг/кг) в течение четырех недель. В каждой группе половина животных получала Nω- нитро-L-аргинин метил эфир (L-NAME; Sigma Chemical Co. , 400 мг/л питьевой воды) ad libium в течение четырех недель исследуемого периода, тогда как оставшиеся крысы получали водопроводную воду. Исходя из индивидуального ежедневного потребления жидкости, такая концентрация L-NAME привела к дозе приблизительно 45 мг/кг/день. Растворитель содержал 0.5% карбоксиметилцеллюлозы/диметилсульфоксида (95/5, об/об). Напроксен (Sigma) и NO-напроксен (NicOx) растворяли в таком растворе до 10 мг/мл и 14.5 мг/мл соответственно.
Измерение кровяного давления. Через недели лечения животные имели систолическое артериальное давление, измеренное на хвосте посредством манжеты для измерения артериального давления (аппарат Гарварда), как описано ранее (18). Измерения учитывались только тогда, когда три последовательных показания не отличалось более чем на 5 мм Hg, и записывали среднее значение. Человек, проводящий измерения, не знал о воздействии, которому подверглись крысы.
Синтез тромбоксана. В конце четвертой недели периода лечения и приблизительно через 20 часов после последней дозы растворителя, напроксена и NO-напроксена крыс анастезировали натрий пентобарбиталом и определяли синтез тромбоксана B2 в цельной крови, как описано ранее (17). Кратко, образцы крови отбирали из нисходящей брюшной аорты, и аликвоту 1 мл немедленно переносили в стеклянную пробирку и инкубировали при 37oC в течение 45 минут. В конце инкубационного периода добавляли 10 мг индометацина и пробирки центрифугировали (2,000 г, 10 мин). Сыворотку отделяли и хранили при -20oC, затем анализировали концентрацию тромбоксана B2, используя коммерческий набор для твердофазного иммуноферментного анализа (Cayman Chemical Company, Ann Arbor, MI). В отдельном эксперименте три группы крыс (n=6 в каждой) обрабатывали ежедневно в течение пяти дней растворителем, напроксеном или NO-напроксеном, как в исследовании, описанном выше. Через два часа после последней дозы тестируемых лекарств брали цельную кровь для измерения синтеза тромбоксана B2, как описано выше.
Поражение слизистой оболочки желудка. После измерения КД и сбора образцов крови крыс умерщвляли и вырезали желудок. Макроскопически видимое поражение слизистой оболочки оценивалось наблюдателем, не знавшем о лечении, как описано ранее (19).
Статистический анализ. Все данные представлены как среднее значение ± SEM. Различия между группами были проанализированы, используя односторонний анализ дисперсии, за которым последовал тест Student-Neuman-Keuls для множественных сравнений. Значения с вероятностью менее чем 5% (p < 0.05) принимались значимыми.
Синтез тромбоксана: Табл. 5 демонстрирует, что различия в синтезе TXB2 не обнаружены во всех группах, когда образцы крови отбирали через 20 ч после последней дозы NSAID (или растворителя). Однако, в образцах крови, отобранных через 2 часа после последней дозы NSAID, и напроксен, и NO-напроксен ингибировали синтез тромбоксана примерно на > 90% (p < 0.001 против группы, обработанной растворителем). Супрессия тромбоксанового синтеза, обнаруженная для напроксена, была более значительна, чем супрессия, обнаруженная для NO-напроксена (p < 0.05).
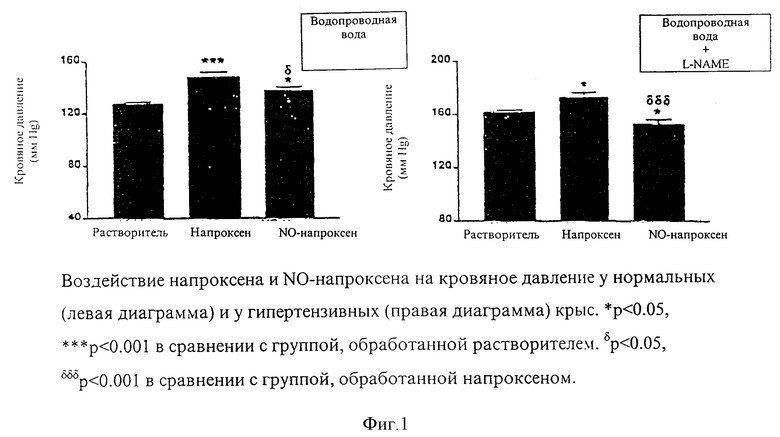
Кровяное давление: Как показано на фиг. 1, обработка L-NAME вызвала значительное увеличение значений КД у животных, получавших растворитель (127±2 против 162±2, p < 0.001), напроксен (148±4 против 173±4 p < 0.001) или NO-напроксен (137±3 против 152±4, p < 0.01). В отсутствие L-NAME, и напроксен и NO-напроксен вызывали значительное повышение значений КД по сравнению со значениями у животных, обработанных растворителем (p < 0.001 и p < 0.05 соответственно), однако вазопрессорный эффект напроксена проявлялся более значительно, чем вызванный NO-напроксеном (p < 0.05). В то время как вазопрессорный эффект L-NAME значительно усиливался при сопутствующем введении напроксена (p < 0.05), NO-напроксен способствовал значительному уменьшению гипертензии, вызванной L-NAME (p < 0.05).
Повреждение желудка: Повреждение слизистой желудка не было обнаружено в крысах, обработанных или контролем, или NO-напроксеном, тогда как у всех крыс, обработанных напроксеном, были обнаружены повреждения (фиг. 2). Введение L-NAME также привело к развитию геморрагических поражений в желудке. Введение напроксена крысам, получавшим L-NAME в питьевой воде, привело к значительному увеличению тяжести поражения слизистой желудка. Напротив, введение NO-напроксена препятствовало повреждению слизистой желудка, связанному с введением L-NAME в питьевой воде.
Обсуждение.
Хроническое ингибирование синтеза NO посредством введения L-аргининовых аналогов обеспечивает полезную модель гипертензии (20, 21). Эта модель демонстрирует некоторое сходство с клиническими патологиями, связанными с эндотелиальной дисфункцией, такими как атеросклероз (22), гиперлипидемия (23) и ишемия-реперфузия (24). Некоторые экспериментальные протоколы демонстрировали, что гипертензивное воздействие L-аргининовых аналогов может быть обратимо или ослаблено введением большого количества L-аргинина, а также, что почки являются одними из органов-мишеней, ответственных за гемодинамическое воздействие в результате ингибирования синтеза NO (20, 21, 25-27). Сосудорасширяющие простагландины представляют собой другую группу медиаторов фундаментальной важности при поддержании почечного гомеостаза (1, 2). Ингибирование почечной циклооксигеназы посредством NSAIDs приводит к нарушению почечного контроля КД, особенно у гипертензивных пациентов (6).
Наши результаты показывают, что и напроксен, и его NO-высвобождающее производное значительно увеличивают КД, причем воздействие NO-напроксена более дискретно, чем воздействие, вызванное исходным соединением. Мы также показали, что напроксен усиливает гипертензию, вызванную введением L-NAME, таким образом мимикрируя клиническую ситуацию (6). С другой стороны, NO-напроксен не только не усиливает, но даже частично ингибирует гипертензию, вызванную L-NAME. Как было показано, фторбипрофен или NO-фторбипрофен не проявляли вазопрессорного воздействия у нормальных крыс в течение периода, равного одному часу после внутрижелудочного введения (14). Более того, аспирин и NO-аспирин не изменяли общего КД у нормальных крыс после внутривенного введения (17). Таким образом, нам следует предположить, что вазопрессорное воздействие NSAIDs проявляется только после пролонгированного периода лечения. Более того, эти результаты подтверждают, что NO и простагландины действуют синергитически в контроле КД, и что в отсутствие эндогенного NO наблюдается значительное возрастание чувствительности к NO-напроксену, высвобождающему NO, что проявляется в различных воздействиях NO-напроксена в отсутствие и присутствии сопутствующей обработки L-NAME.
Введение напроксена или L-NAME вызывало заметное повреждение желудка, которое значительно усиливалось, когда эти два соединения вводились вместе. Эти наблюдения находятся в соответствии с описанными защитными воздействиями NO и простагландинов на слизистую оболочку кишечника (29). Несмотря на тот факт, что NO-напроксен уменьшал общую циклооксигеназную активность более чем на 90%, он не вызывал повреждений желудка, когда вводился нормальным или гипертензивным крысам. В последнем случае NO-напроксен защищал желудок от повреждений, обычно вызываемых хроническим введением L-NAME. Способность некоторых NO-NSAIDs защищать желудок от повреждения, несмотря на такую же эффективную супрессию синтеза простагландинов в желудке как и посредством исходных лекарств, хорошо документирована (14, 15). Следует подчеркнуть, что предыдущие исследования, проверяющие желудочно-кишечную безопасность NO-NSAIDs, включали их введение на короткие периоды времени. Таким образом, настоящие данные об уменьшенных повреждениях желудка в течение 4 недель ежедневной терапии усиливают заявление об относительной безопасности этой новой группы лекарств по сравнению с исходными лекарствами.
Из табл. 5 можно явно понять, что и напроксен, и NO-напроксен вызывали значительное ингибирование тромбоксановой генерации в цельной крови, когда образцы крови отбирали через 2 ч после последней дозы. Однако, синтез TXB2 был значительно ниже в группе, обработанной напроксеном, чем в группе, обработанной NO-напроксеном. Несмотря на эти различия, NO-напроксен ингибировал синтез тромбоксана на 92%; следовательно, защитное воздействие NO-напроксена или при увеличении КД, или при повреждении слизистой желудка не может быть описано чистым ингибированием COX. В предыдущем исследовании NO-напроксен не вызывал значительного повреждения желудка при более высокой дозе, чем использовалась в настоящем исследовании.
Следует также заметить, что, как это было предварительно продемонстрировано, NO-напроксен проявляет противовоспалительную активность, сравнимую по силе с напроксеном, и анальгетическую активность, превосходящую напроксен (16).
Приблизительно 20 миллионов человек в США подвергаются одновременно противогипертензивной и NSAID терапии (6), таким образом подвергаясь сердечно-почечному и желудочному риску, обсужденному выше. Результаты, представленные в этой статье, подтверждают, что NO-напроксен может представлять более безопасную альтернативу стандартным NSAIDs для лечения воспалительных заболеваний у гипертензивных пациентов.
Эта работа поддерживалась грантом от Heart and Stroke Foundation of Canada, Dr. Wallace является представителем Medical Research Council of Canada Senior Scientist and an Alberta Heritage Foundation for Medical Research Scientist. Dr. Muscara поддерживается Merck Pharmacology Fellowship.
Сравнение NO-напроксена с напроксеном: ульцерогенное, анальгетическое и противовоспалительное воздействия. N. M. Davies, A.G. Roseth*, C.B. Appleyard, W. McKnight, P. Del Soldato , A. Calignano , Cirino & J.L. Wallace Intestinal Disease Research Unit, Faculty of Medicine, University of Calgary, Calgari Alberta, Canada; *Department of Medicine, Aker University Hospital, Oslo, Norway; Nicox SA. Paris, France; and Department of Experimental Pharmacology? Univercity of Naples, Naples, Italy
Принято к печати 3 сентября 1996
Резюме
Предпосылки: Недавно был описан новый класс высвобождающего оксид азота нестероидного противовоспалительного лекарственного (NO-NSAID) производного, которое проявляет противовоспалительную активность, но вызывает явно меньше желудочно-кишечных повреждений, чем исходное NSAID, из которого его получают. Настоящие исследования были проведены, чтобы определить, является ли нитроксибутил-эфирное производное напроксена менее ульцерогенным по отношению к желудочно-кишечному тракту, чем исходное NSAID, и проявляет ли оно анальгетические и противовоспалительные свойства исходного NSAID.
Методы: Оба лекарственных средства сравнивали на модели с острым повреждением желудка, на модели с антральной язвой, и после ежедневного введения по два раза в день в течение 18 дней (модель с небольшим кишечным повреждением). Противовоспалительную активность проверяли на модели с вызванным каррагином отеком лапы, тогда как анальгетическая активность проверялась на модели с вызванной уксусной кислотой болевыми корчами. Фармакокинетические профили напроксена против NO-напроксена сравнивали ВЭЖХ анализом.
Результаты: Было обнаружено, что NO-накроксен вызывает значительно меньшее повреждение желудка, хотя вызывает увеличение TNFa в плазме подобно напроксену. При хроническом введении NO-напроксена небольшое кишечное повреждение было заметно меньше, чем при введении исходного NSAID. Однако, NO-напроксен проявлял повышенное анальгетическое и сравнимое противовоспалительное воздействие по сравнению с напроксеном.
NO-напроксен полностью не превращался в напроксен, но пониженный уровень последнего в крови не являлся основной причиной пониженной желудочно-кишечной токсичности NO-напроксена.
Вывод: NO-напроксен является новым щадящим желудочно-кишечный тракт производным NSAID с повышенными анальгетическими и сравнимыми противовоспалительными свойствами по сравнению с напроксеном.
Введение.
NSAIDs имеют широко распространенное клиническое использование благодаря своим противовоспалительным, противогипертермическим, анальгетическим и антитромботическим свойствам. Однако, использование этих лекарств проводится ограниченно из-за их способности вызывать повреждение слизистой на всем протяжении желудочно-кишечного тракта. NSAID-связанные желудочно-кишечные побочные эффекты объясняют более чем 70000 госпитализаций и 7000 смертей ежегодно в США. Напроксен является стереохимически чистой 2-арилпропионовой кислотой NSAID и, как было показано, вызывает повреждение слизистой на всем протяжении желудочно-кишечного тракта. В США напроксен недавно продавался как over-the-counter лекарственное средство. При увеличении использования over-the-counter NSAIDs существует определенная возможность увеличения распространения вредных воздействий, вызываемых этими лекарствами.
Мы недавно описали новый класс высвобождающего оксид азота нестероидного противовоспалительного лекарственного препарата (NO-NSAID), который вызывает заметно пониженные желудочно-кишечные реакции.

Нитроэфиры формулы M-CO-Y-(C)nAB-ONO2, где А и В - водород или метил; М выбирают из соединений формулы I-IV
R - остаток формулы V-VII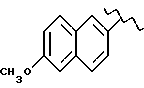
Y - О, NH, NR1; R1 - алкил, n = 1-6, пригодны для лечения тяжелых состояний, требующих одновременно применения противовоспалительных и анальгетических лекарственных препаратов, или ревматических заболеваний общего характера. 5 с. и 12 з.п. ф-лы, 2 ил., 5 табл.
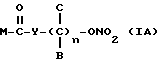
где А и В выбраны из водорода, или метила, М выбран из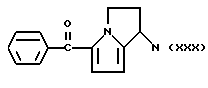
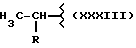
где R выбран из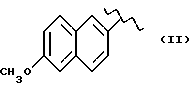
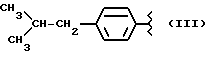
Y выбран из кислорода, NH, NR1, где R1 означает линейную или разветвленную алкильную группу, и n равно от 1 до 6.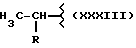
где R означает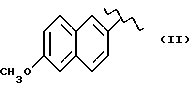
А и В равны водороду, Y равен кислороду и n равно четырем.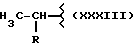
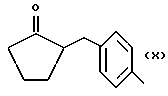
где R равен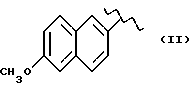
А и В равны водороду, Y равен NH и n равно четырем.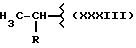
где R равен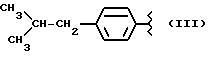
Y равен кислороду, А и В равны водороду, и n равно четырем.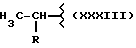
где R равен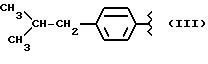
Y равен NH, А и В равны водороду и n равно четырем.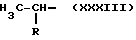
где R равен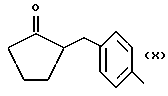
А и В равны водороду, Y равен кислороду и n равно четырем.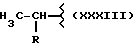
где R равен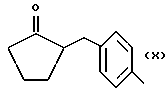
А и В равны водороду, Y равен NH и n равно четырем.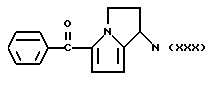
А и В равны водороду, Y равен кислороду и n равно четырем.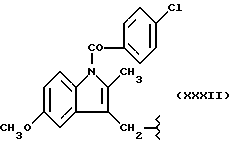
А и В равны водороду, Y равен кислороду и n равно четырем.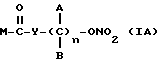
где А и В выбраны из водорода или метила, М выбран из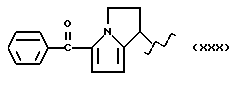
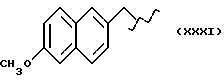
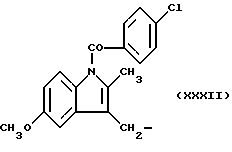
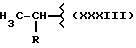
где R выбран из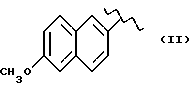
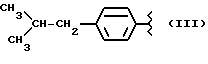
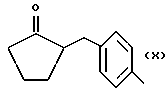
Y выбран из кислорода, NH, NR1, где R1 означает линейный или разветвленный алкил, и n равно от 1 до 10, отличающийся тем, что включает следующие стадии: - получение натриевой соли производных, имеющих следующую общую формулу: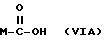
где М выбран из (XXX), (XXXI), (XXXII),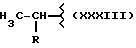
где R выбран из следующих структур: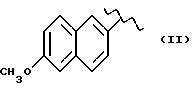
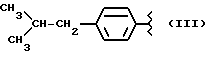
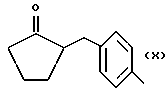
или получение производных (VIA), замещенных по карбоксильной группе, таких как хлорангидриды, ангидриды или подобные; реакцию натриевой соли вышеуказанных производных (VIA) или вышеуказанных производных (VIA), замещенных по карбоксильной группе, с соединением, имеющим следующую общую формулу: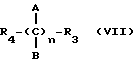
где R4 выбран из хлора, брома, NHR5, где R5 - водород, линейный или разветвленный алкил, А и В выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила, R3 выбран из хлора, брома, иода и n равно от 1 до 10, с получением таким путем соответствующих эфиров или соответствующих амидов; реакцию вышеуказанных эфиров или вышеуказанных амидов с нитрующим агентом, таким, как AgNO3 или подобным, с получением в результате нитроэфиров (IA).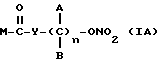
где А и В выбраны из водорода или метила, М выбран из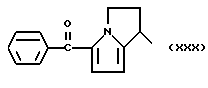
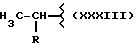
где R выбран из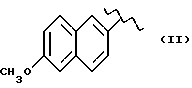
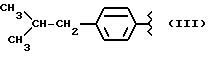
Y выбран из кислорода, NH, NR1, где R1 означает линейную или разветвленную алкильную группу, и n равно от 1 до 10, отличающийся тем, что он включает следующие стадии: получение натриевой соли производных имеющих следующую общую формулу: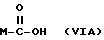
где М выбран из (XXX), (XXXI), (XXXII),
где R выбран из следующих структур: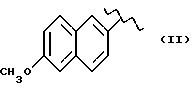
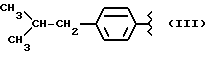
или получение производных (VIA), замещенных по карбоксильной группе, таких как хлорангидриды, ангидриды или подобные; реакцию натриевой соли вышеуказанных производных (VIA) или вышеуказанных производных (VIA), замещенных по карбоксильной группе, с соединением, имеющим следующую общую формулу: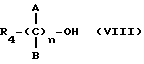
где R4 выбран из хлора, брома, NHR5, где R5 - водород, линейный или разветвленный алкил, А и В выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила, и n равно от 1 до 10, с получением таким образом соответствующих эфиров или соответствующих амидов; реакцию вышеуказанных эфиров или вышеуказанных амидов с галоидирующим соединением, таким как PBr3 или подобным, с получением таким образом продуктов вышеуказанных эфиров или вышеуказанных амидов, характеризующихся наличием концевой галоидной группы; реакцию вышеуказанных эфиров или вышеуказанных амидов, характеризующихся наличием концевой галоидной группы, с нитрирующим агентом, таким как AgNO3 или подобным, с получением таким образом нитроэфиров (IA).
Приоритет по признакам:
06.10.93, где М представляет собой 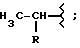
10.05.94 по всем другим значениям радикалов.
| Способ получения производных 5-ароил1,2-дигидро-3н-пиррол(1,2-а)-пиррол-1карбоновой кислоты или их солей | 1977 |
|
SU695558A3 |
| Тест-система для выявления ДНК возбудителя пастереллеза (Pasteurella multocida) в биологическом материале животных и кормах с помощью полимеразной цепной реакции в режиме реального времени | 2023 |
|
RU2814556C1 |
| Ротационный рабочий орган почвообрабатывающего орудия | 1991 |
|
SU1793828A3 |
| DE 1443429 A, 1968 | |||
| Экономайзер | 0 |
|
SU94A1 |
| Экономайзер | 0 |
|
SU94A1 |

