Область техники
Настоящее изобретение относится к производным гиалуроновой кислоты, к которым нестероидное противовоспалительное лекарственное средство или облегчающее заболевание противоревматическое лекарственное средство присоединено посредством спейсера, который способен к биодеградации, и к способам их получения.
Уровень техники
Раствор гиалуроната натрия используют в качестве терапевтического средства для лечения таких артритов, как остеоартрит колена (ОА) или ревматоидный артрит колена (RA). Раствор гиалуроната натрия, в основном, используют в качестве инъекций посредством прямого введения в пораженный коленный сустав и плечевой сустав и часто используют с целью улучшения состояния при функциональных нарушениях и подавления боли, вызванной артритом.
Нестероидные противовоспалительные лекарственные средства (здесь и далее обозначаемые как «NSAIDs» или «NSAID») и облегчающие заболевание противоревматические лекарственные средства (здесь и далее обозначаемые как «DMARD»), которые улучшают такие болезненные состояния, как суставной ревматизм, также используют в качестве средств для подавления или смягчения болей, вызванных такими артритами. В основном, такие NSAID во многих случаях вводят перорально, и существуют также частые случаи, при которых применение инъекции описанного выше раствора гиалуроната натрия сопровождается пероральным введением NSAID. В случае перорального введения таких NSAID существует проблема в том, что большая часть NSAID метаболизируется во время циркуляции в кровотоке до того, как они достигнут поврежденного участка. Для того чтобы обойти данную проблему, необходимы высокие дозы NSAID, чтобы поддержать эффективную концентрацию в крови для того, чтобы доставить NSAID к пораженной части. Тем не менее такие высокие дозы NSAID при пероральном введении вызывают серьезные желудочно-кишечные побочные эффекты.
В дополнение, средства для иммунотерапии (иммуномодуляторы и иммуносупрессанты) используют в качестве DMARD для контроля иммунного нарушения или тому подобное, что, как полагают, вызывает воспаление.
С другой стороны, гиалуроновая кислота представляет собой полисахарид, состоящий из повторяющейся структуры с дисахаридной единицей N-ацетил-D-глюкозамина и D-глюкуроновой кислоты в качестве основной центральной структуры, и, как известно, является сильно гидрофильной из-за карбоксильной группы и большого количества гидроксильных групп в дисахаридной единице. В качестве примера того, что гиалуроновая кислота обладает гидрофильными свойствами, а именно высокой степенью гидратации молекулой воды, гиалуроновая кислота может удерживать количество воды приблизительно в 1000 раз большее, чем ее собственная масса. Тем не менее, когда сильно гидрофобные средства, такие как NSAID, присоединяют к гиалуроновой кислоте, обладающей такими высоко гидрофильными свойствами, общеизвестно, что гидрофобное свойство самой молекулы гиалуроновой кислоты возрастает так, что образуется полунерастворимый в воде гель или нерастворимое вещество. Следовательно, такой полунерастворимый в воде гель или нерастворимое вещество не подходят для использования в виде инъекций. Более того, с увеличением степени замещения лекарственного средства с целью более длительного замедленного высвобождения нерастворимость также возрастает так, что средство принимает форму, неподходящую для инъекций.
В качестве примера, в котором не только NSAID, но и другие лекарственные средства присоединяли к гиалуроновой кислоте, существует сообщение о конъюгате, в котором ингибитор матриксной металлопротеиназы (ингибитор ММР) в качестве средства для лечения артрита и гиалуроновая кислота были связаны друг с другом через спейсер или не через спейсер (ссылка на патент 1). Тем не менее в качестве подходящего способа связывания ингибитора ММР с гиалуроновой кислотой в сообщении приведен пример более сильной ковалентной связи, и это предполагает, что можно было ожидать синергического медикаментозного эффекта действия ингибитора ММР и эффекта гиалуроновой кислоты, исходя из того, что конъюгат не диссоциирует и не разрушается до ингибитора ММР и гиалуроновой кислоты в участке введения. В дополнение, в качестве примера приведена карбоксильная группа как участок связывания с гиалуроновой кислотой, однако, степень замещения лекарственным средством карбоксильной группы существенно невелика, или лечение в отношении выполнения подходящего варианта осуществления (раствора) в виде инъекции не проводили.
В качестве других примеров существуют случаи, в которых гиалуроновая кислота активирована растворимым в воде карбодиимидом, и нуклеофильные реагенты способны вступать с ним в реакцию (ссылка на патент 2), но данные лекарственные средства не являлись NSAID, и конечная дозированная форма представляла собой нерастворимую пленку. В дополнение, существуют случаи, в которых различные лекарственные средства присоединяли к гиалуроновой кислоте, используя в качестве конденсирующего агента галогенизированный низший диалкилфосфинотиоил (Rpt-X) (ссылка на патент 3), но дозированные формы полученных производных не описаны, и лечение, предоставляющее возможность их использования в качестве раствора в препарате, не охвачено в этом способе.
Ссылка на патент 1: WO 99/59603.
Ссылка на патент 2: JP-T-3-502704.
Ссылка на патент 1: JP-A-9-188705.
Описание изобретения
Проблемы, которые решаются изобретением
Можно рассматривать способ, в котором нерешенный вопрос, такой как побочные эффекты на желудочно-кишечный тракт, вызванные пероральным введением NSAID, обходят непосредственной инъекцией NSAID в пораженный участок. Даже теоретически, но, например, когда NSAID непосредственно вводят в полость коленного сустава, период времени для продолжающегося эффекта NSAID является коротким вследствие быстрой абсорбции, так что такой способ не подходит. В дополнение, так как NSAID сами по себе предназначены смягчать или подавлять боль, такой способ не становится основным средством лечения артритов.
Соответственно, настоящее изобретение предназначено предложить фармацевтическое средство, которое может внести большой вклад в смягчение или подавление боли, сопровождающей артриты, и основное средство лечения артритов, путем получения нового производного, в котором одно из NSAID или DMARD химически присоединено к средству для лечения артрита, гиалуронату натрия, и введения его в пораженный участок, и предложить фармацевтическое средство, которое проявляет свой продолжительный эффект посредством контролируемого высвобождения NSAID или DMARD.
Способы решения проблем
Принимая во внимание описанные выше проблемы, авторы настоящего изобретения провели интенсивные исследования с целью получения производных гиалуроновой кислоты, содержащих NSAID, и производных гиалуроновой кислоты, содержащих DMARD, которые можно использовать в виде инъекций в пораженный участок у пациентов с артритом, а также они обладают сильным эффектом не только при радикальном лечении артритов, но также и для смягчения или подавления боли и воспаления.
В результате было обнаружено, что такие производные, в которых NSAID и DMARD присоединены к гиалуроновой кислоте посредством спейсера, содержащего биодеградируемый участок, подходят для описанных выше целей, и далее предпочтительно, чтобы растворимые производные гиалуроновой кислоты, содержащие NSAID, и растворимые производные гиалуроновой кислоты, содержащие DMARD, которые можно использовать в качестве пригодных для инъекций растворов в виде инфузий (инъекций), можно было бы получить посредством увеличения растворимости, добавляя щелочную обработку в процесс получения, тем самым выполняя настоящее изобретение.
Таким образом, настоящее изобретение относится к тому, что перечислено ниже:
(1) производные гиалуроновой кислоты, в которых противовоспалительные лекарственные средства ковалентно связаны посредством спейсера, содержащего биодеградируемый участок.
(2) производные гиалуроновой кислоты по описанному выше пункту (1), где противовоспалительные лекарственные средства выбраны из нестероидных противовоспалительных лекарственных средств и облегчающих заболевание противоревматических лекарственных средств.
(3) производные гиалуроновой кислоты по описанным выше пунктам (1) или (2), где противовоспалительные лекарственные средства содержат карбоксильную группу.
(4) производные гиалуроновой кислоты по описанному выше пункту (3), где противовоспалительное лекарственное средство представляет собой остаток соединения, выбранного из группы, состоящей из салициловой кислоты, аспирина, мефенамовой кислоты, толфенамовой кислоты, флуфенамовой кислоты, диклофенака, сулиндака, фенбуфена, индометацина, ацеметацина, амфенака, этодолака, фелбинака, ибупрофена, флурбипрофена, кетопрофена, напроксена, пранопрофена, фенопрофена, тиапрофеновой кислоты, оксапрозина, локсопрофена, алминопрофена, залтопрофена, пироксикама, теноксикама, лорноксикама, мелоксикама, тиарамида, толметина, дифлюнизала, ацетаминофена, флоктафенина, тиноридина и актарита.
(5) производные гиалуроновой кислоты по любому из описанных выше пунктов (1)-(4), где спейсер представляет собой соединение, содержащее, по крайней мере, одну функциональную группу, которая связана с гиалуроновой кислотой, и одну функциональную группу, которая связана с противовоспалительным лекарственным средством.
(6) производные гиалуроновой кислоты по любому из описанных выше пунктов (1)-(5), где спейсер выбран из диаминоалкана, содержащего от 2 до 18 атомов углерода, аминоалкилового спирта, содержащего от 2 до 12 атомов углерода, которая может иметь заместитель(заместители), и аминокислоты.
(7) производные гиалуроновой кислоты по любому из описанных выше пунктов (1)-(6), где гиалуроновая кислота имеет средневзвешенную молекулярную массу от 500000 до 3000000.
(8) Производные гиалуроновой кислоты по любому из описанных выше пунктов (1)-(7), где противовоспалительное лекарственное средство введено в соотношении от 5 до 50% мол. на повторяющуюся дисахаридную единицу гиалуроновой кислоты.
(9) Производные гиалуроновой кислоты, в которых нестероидное противовоспалительное лекарственное средство связано с гиалуроновой кислотой посредством ковалентной связи, которая содержит частичную структуру дисахаридной единицы гиалуроновой кислоты, к которой присоединено противовоспалительное лекарственное средство, как показано нижеследующей формулой (1):
Y-CO-NH-R 1 -(O-R 2 ) n (1)
где Y-CO- представляет собой один остаток дисахаридной единицы гиалуроновой кислоты;
R2 представляет собой остаток нестероидного противовоспалительного лекарственного средства, представленного группой Z-CO- или атомом водорода, при условии, что все R2 не являются атомом водорода;
-NH-R1-(O)n представляет собой спейсерный остаток в соединении-спейсере, представленном формулой H2N-R1-(OH)n, имеющем гидроксильные группы в количестве n;
R1 представляет собой линейную или разветвленную углеводородную группу, содержащую от 2 до 12 атомов углерода, которая может иметь заместитель;
-CO-NH- представляет собой амидную связь карбоксильной группы гиалуроновой кислоты в качестве составляющей сахарид гиалуроновой кислоты с аминогруппой соединения-спейсера;
-О-СО- представляет собой сложноэфирную связь гидроксильной группы соединения-спейсера с карбоксильной группой в остатке нестероидного противовоспалительного лекарственного средства и
n равно целому числу от 1 до 3,
где производное гиалуроновой кислоты имеет степень замещения нестероидным противовоспалительным лекарственным средством от 5 до 50% мол. на повторяющуюся единицу гиалуроновой кислоты и
карбоксильная группа в остатке гиалуроновой кислоты, составляющем производное гиалуроновой кислоты, присутствует в качестве участвующей в образовании амидной связи при связывании со связывающим спейсер остатком нестероидного противовоспалительного лекарственного средства или в качестве не принимающей в этом участие свободной карбоксильной группы, согласно степени замещения остатка нестероидного противовоспалительного лекарственного средства.
(10) Производные гиалуроновой кислоты по описанному выше пункту (9), где нестероидное противовоспалительное лекарственное средство представляет собой соединение, представленное следующей ниже формулой (2):

где R3 представляет собой заместитель, выбранный из низшей алкильной группы и низшей алкоксильной группы или атома водорода;
R4, R5 и R6 каждый независимо представляет собой заместитель, выбранный из группы, состоящей из низшей алкильной группы, низшей алкоксильной группы и гидроксильной группы, атома галогена или атома водорода, и
Х являются одинаковыми или различными и каждый представляет собой заместитель, выбранный из низшей алкильной группы и трифторметильной группы или атома галогена и, по крайней мере, один из Х представляет собой атом галогена.
(11) Производные гиалуроновой кислоты по описанному выше пункту (10), где нестероидное противовоспалительное лекарственное средство представляет собой диклофенак или его производные.
(12) Производные гиалуроновой кислоты по любому из описанных выше пунктов (9)-(11), где R1 в формуле (1) представляет собой этиленовую группу, триметиленовую группу или пропиленовую группу, которые могут иметь заместитель(заместители).
(13) Производные гиалуроновой кислоты по любому из описанных выше пунктов (1)-(12), которые можно получить способом, включающим взаимодействие гиалуроновой кислоты со связанным со спейсером нестероидным противовоспалительным лекарственным средством или взаимодействие связанной со спейсером гиалуроновой кислоты с нестероидным противовоспалительным лекарственным средством и доведение реакционного раствора до щелочной среды.
(14) Производные гиалуроновой кислоты по любому из описанных выше пунктов (1)-(13), где раствор, получаемый растворением производного гиалуроновой кислоты в водной среде до концентрации 1,0% масс, способен проходить через пористый фильтр, имеющий размер пор 0,45 мкм и диаметр 25 мм со скоростью 2 мл в минуту или более при температуре 24°С под давлением 5,0 кг/см2.
(15) Производные гиалуроновой кислоты по любому из описанных выше пунктов (1)-(13), где раствор, получаемый растворением производного гиалуроновой кислоты в водной среде до концентрации 1,0% масс, способен проходить через пористый фильтр, имеющий размер пор 0,22 мкм и диаметр 25 мм со скоростью 2 мл в минуту или более при температуре 24°С под давлением 5,0 кг/см2.
(16) Раствор производного гиалуроновой кислоты, который можно пропускать через инжектор и который содержит производное гиалуроновой кислоты по любому из описанных выше пунктов (1)-(15), растворенное в водной среде.
(17) Раствор производного гиалуроновой кислоты по описанному выше пункту (16), в котором водная среда представляет собой водную среду, выбранную из забуференного фосфатом физиологического раствора, физиологического раствора и воды для инъекций.
(18) Раствор производного гиалуроновой кислоты по описанному выше пункту (17), который простерилизован через фильтр.
(19) Фармацевтическое средство, которое содержит производное гиалуроновой кислоты по любому из описанных выше пунктов (1)-(15) в качестве активного ингредиента.
(20) Фармацевтическое средство по описанному выше пункту (19), которое представляет собой средство для лечения артрита, противовоспалительное лекарственное средство или болеутоляющее средство.
(21) Фармацевтическое средство по описанному выше пункту (19) или (20), которое применимо для парентерального введения.
(22) Фармацевтическое средство по описанному выше пункту (21), которое представляет собой инъекцию, применимую для местного введения.
(23) Фармацевтическое средство по описанному выше пункту (21) или (22), которое представляет собой инъекцию, применимую для внутрисуставного введения.
(24) Фармацевтическое средство, которое можно пропускать через инжектор и которое содержит раствор, в котором производное гиалуроновой кислоты по любому из описанных выше пунктов (1)-(15) в качестве активного ингредиента растворено в водной среде.
(25) Набор для инъекции производного гиалуроновой кислоты, который включает раствор производного гиалуроновой кислоты по любому из описанных выше пунктов (16)-(18), которым заполнен инжектор, способный пропустить раствор.
(26) Набор по описанному выше пункту (25), в котором раствор для заполнения представляет собой фармацевтическое средство по любому из описанных выше пунктов (19)-(24).
(27) Медицинский набор для инъекции, который герметично запакован с плунжером для вытеснения лекарственного средства таким образом, что он может плавно двигаться, и который включает шприц, заполненный раствором, в котором производное гиалуроновой кислоты по любому из описанных выше пунктов (1)-(15) растворено в фармацевтически приемлемом забуференном фосфатом физиологическом растворе, физиологическом растворе или воде для инъекций.
(28) Производное, в котором спейсер, содержащий биодеградируемый участок, связан с противовоспалительным лекарственным средством через ковалентную связь.
(29) Производное по описанному выше пункту (28), где спейсер, содержащий биодеградируемый участок, представляет собой остаток диаминоалкана, аминоалкилового спирта или аминокислоты.
(30) Производное по любому из описанных выше пунктов (28) или (29), где спейсер, содержащий биодеградируемый участок, представляет собой остаток соединения, способный связывать два или более противовоспалительных лекарственных средства на один моль спейсера.
(31) Производное по любому из описанных выше пунктов (28)-(30), где противовоспалительное лекарственное средство представляет собой остаток соединения, выбранного из группы, состоящей из салициловой кислоты, аспирина, мефенамовой кислоты, толфенамовой кислоты, флуфенамовой кислоты, диклофенака, сулиндака, фенбуфена, индометацина, ацеметацина, амфенака, этодолака, фелбинака, ибупрофена, флурбипрофена, кетопрофена, напроксена, пранопрофена, фенопрофена, тиапрофеновой кислоты, оксапрозина, локсопрофена, алминопрофена, залтопрофена, пироксикама, теноксикама, лорноксикама, мелоксикама, тиарамида, толметина, дифлюнизала, ацетаминофена, флоктафенина, тиноридина и актарита.
(32) Производное по любому из описанных выше пунктов (28)-(31), где ковалентная связь представляет собой сложноэфирную связь или амидную связь.
(33) Производное по описанному выше пункту (32), которое представлено следующей ниже формулой (3):
H 2 N-R 1 -(O-R 2 ) n (3)
где R2 представляет собой атом водорода или остаток нестероидного противовоспалительного лекарственного средства, представленного формулой Z-CO-, при условии, что все R2 не являются атомом водорода;
H2N-R1-(O-)n представляет собой остаток спейсера в соединении-спейсере, представленном формулой H2N-R1-(OH)n, содержащем гидроксильные группы в количестве n;
R1 представляет собой линейную или разветвленную углеводородную группу, содержащую от 2 до 12 атомов углерода, которая может иметь заместители;
-О-СО- представляет собой сложноэфирную связь, состоящую из гидроксильной группы соединения-спейсера и карбоксильной группы в остатке нестероидного противовоспалительного лекарственного средства, и
n равно целому числу от 1 до 3.
(34) Способ получения производного гиалуроновой кислоты, которое содержит гиалуроновую кислоту, связанную с противовоспалительным лекарственным средством посредством ковалентной связи через спейсер, содержащий биодеградируемый участок, причем указанный способ включает:
взаимодействие гиалуроновой кислоты со связанным со спейсером противовоспалительным лекарственным средством или
взаимодействие связанной со спейсером гиалуроновой кислоты с противовоспалительным лекарственным средством.
(35) Способ получения производного гиалуроновой кислоты по описанному выше пункту (34), который включает обработку раствора продукта реакции гиалуроновой кислоты со связанным со спейсером противовоспалительным лекарственным средством или раствора продукта реакции связанной со спейсером гиалуроновой кислоты с противовоспалительным лекарственным средством в щелочных условиях.
Преимущество изобретения
По настоящему изобретению предлагаются производные гиалуроновой кислоты, в которых противовоспалительное лекарственное средство связано с гиалуроновой кислотой посредством ковалентной связи через спейсер, содержащий биодеградируемый участок, в частности, производное гиалуроновой кислоты, содержащее нестероидное противовоспалительное лекарственное средство, в котором нестероидное противовоспалительное лекарственное средство связано посредством ковалентной связи (здесь и далее обозначаемое как «вещество 1 по настоящему изобретению»), также производное гиалуроновой кислоты, содержащее облегчающее заболевание противоревматическое средство, в котором облегчающее заболевание противоревматическое средство связано посредством ковалентной связи (здесь и далее обозначаемое как «вещество 2 по настоящему изобретению», в этой связи вещество 1 по настоящему изобретению и вещество 2 по настоящему изобретению также в целом называют «вещество по настоящему изобретению») и фармацевтическое средство, которое включает одно из данных производных в качестве активного ингредиента (здесь и далее обозначаемое как «фармацевтическое средство по настоящему изобретению»). Поскольку вещество по настоящему изобретению в достаточной степени растворяется в буфере, физиологическом растворе, воде для инъекций или тому подобное, которое используется в качестве растворителя при инъекциях или тому подобное, его можно использовать в качестве инъекции, которую можно вводить непосредственно в пораженный участок. В дополнение, фармацевтическое средство по настоящему изобретению можно использовать при лечении артритов, подавлении воспаления и подавлении боли, и также возможно его парентеральное или местное введение в качестве инъекций (например, внутрисуставное введение).
Краткое описание чертежей
Фиг.1 представляет собой график, показывающий показатели боли на модели, вызванной брадикинином боли у крысы.
Фиг.2 представляет собой график, показывающий показатели боли на модели вызванной 1% раствором нитрата серебра боли у крысы.
Фиг.3 представляет собой график, показывающий степень весовой нагрузки (%) на модели боли, вызванной 1% раствором нитрата серебра у крысы.
Фиг.4 представляет собой график, показывающий остаточное соотношение в коленном суставе кролика со времени введения гиалуроната натрия, содержащего аминопропанолкетопрофен (КР-НА), смеси кетопрофена и НА и кетопрофена в коленный сустав кролика.
Фиг.5 представляет собой график, показывающий эффект гиалуроната натрия, содержащего аминопропанолдиклофенак, имеющего различную степень замещения (DS) на модели боли, вызванной 1% раствором нитрата серебра у крысы.
Фиг.6 представляет собой график, показывающий эффект перорального введения диклофенака натрия на модели боли, вызванной 1% раствором нитрата серебра у крысы.
Фиг.7 представляет собой график, показывающий эффект одного лекарственного средства диклофенака и гиалуроновой кислоты на модели боли, вызванной 1% раствором нитрата серебра у крысы.
Фиг.8 представляет собой график, показывающий сравнение эффектов гиалуроната натрия, содержащего аминопропанолдиклофенак (65 кДа), гиалуроната натрия, содержащего диаминопропандиклофенак, и гиалуроната натрия, содержащего аминоэтанолдиклофенак, на модели боли, вызванной 1% раствором нитрата серебра у крысы.
Фиг.9(а) представляет собой график, показывающий эффекты (in vitro) одного лекарственного средства диклофенака натрия и производных гиалуроновой кислоты, содержащего диклофенак, в СОХ-2.
Фиг.9(b) представляет собой график, показывающий эффекты (in vitro) одного лекарственного средства гиалуроната натрия и производных гиалуроновой кислоты, содержащих диклофенак, в СОХ-2.
Фиг.10(а) представляет собой график, показывающий эффект введения производного гиалуроновой кислоты, содержащего диклофенак, в лапу с предварительной инъекцией адъюванта на модели вызванного адъювантом артрита (AIA) у крысы.
Фиг.10(b) представляет собой график, показывающий эффект введения производного гиалуроновой кислоты, содержащего диклофенак, в лапу без предварительной инъекции адъюванта на модели вызванного адъювантом артрита (AIA) у крысы.
Наилучший способ осуществления изобретения
Настоящее изобретение описано ниже, основываясь на вариантах осуществления настоящего изобретения.
Вещество по настоящему изобретению представляет собой производное гиалуроновой кислоты, в котором противовоспалительное лекарственное средство связано с гиалуроновой кислотой посредством ковалентной связи через спейсер, содержащий биодеградируемый участок. По настоящему изобретению противовоспалительное лекарственное средство выбрано из нестероидных противовоспалительных лекарственных средств (NSAID или NSAIDs) и ослабляющих заболевание противоревматических лекарственных средств (DMARD).
В этой связи терминология «NSAIDs» в общем случае обозначает более чем одно нестероидное противовоспалительное лекарственное средство, к которому относятся два или более лекарственных средств, и «NSAID» обозначает каждое нестероидное противовоспалительное лекарственное средство в некоторых случаях, но данные термины строго не различаются в данном описании.
Вещество 1 по настоящему изобретению представляет собой производное гиалуроновой кислоты, в котором нестероидное противовоспалительное лекарственное средство связано через ковалентную связь, и его структура по существу представлена следующей ниже формулой (4):
HA-SP-NSAID (4)
где НА представляет собой цепь гиалуроновой кислоты; SP представляет собой остаток спейсера; NSAID представляет собой остаток нестероидного противовоспалительного лекарственного средства и «-» представляет собой ковалентную связь.
В дополнение, вещество 2 по настоящему изобретению представляет собой производное гиалуроновой кислоты, в котором ослабляющее заболевание противоревматическое лекарственное средство связано через ковалентную связь, и его структура по существу представлена следующей ниже формулой (5):
HA-SP-DMARD (5)
где НА представляет собой цепь гиалуроновой кислоты; SP представляет собой остаток спейсера; DMARD представляет собой остаток ослабляющего заболевание противоревматического лекарственного средства и «-» представляет собой ковалентную связь.
Вещество по настоящему изобретению можно растворить в водном растворителе, и он представляет собой вязкий раствор.
Используемый в настоящем описании термин «водный растворитель» означает воду, буферный раствор, содержащий воду, и водный раствор или буферный раствор, содержащий фармацевтически приемлемую соль металла, средство доведения рН или тому подобное. Конкретные примеры включают воду для инъекций, забуференный фосфатом физиологический раствор, физиологический раствор и тому подобное.
Гиалуроновая кислота, используемая в веществе по настоящему изобретению, особенно не ограничена при условии, что она представляет собой гликозаминогликан, который состоит из дисахаридной единицы, состоящей из N-ацетил-D-глюкозамина и D-глюкуроновой кислоты, связанной посредством β1,3 связи, в качестве основной центральной структуры и создана повторением β1,4 связи в дисахаридной единице, а именно обычно используемой гиалуроновой кислотой. В дополнение, возможно использовать гиалуроновые кислоты, которые получают из животных или микроорганизмов или химическим синтезом.
Средневзвешенная молекулярная масса гиалуроновой кислоты особенно не ограничена, но в качестве примера можно привести массу от 10000 до 5000000. В качестве примера можно привести массу предпочтительно от 500000 до 3000000 и более предпочтительно от 600000 до 1500000 и от 1500000 до 3000000 в качестве стандартов, используемых в средствах для лечения артритов.
В этой связи гиалуроновая кислота, используемая по настоящему изобретению, может находиться либо в не образующей соли свободной форме, либо в виде фармацевтически приемлемой соли. Фармацевтически приемлемая соль гиалуроновой кислоты включает соли ионов щелочных металлов, такие как соль натрия, соль калия, и соли ионов щелочноземельных металлов, такие как соль магния и соль кальция. Если производное гиалуроновой кислоты используют в фармацевтических препаратах или тому подобное для применения на живом организме, то соль гиалуроновой кислоты, которую можно использовать, предпочтительно представляет собой соль с ионом щелочного металла, особенно соль с ионом натрия, вследствие его высокой аффинности к живому организму.
NSAID в качестве одного из противовоспалительных лекарственных средств в отношении настоящего изобретения в общем случае обозначает целое соединение, которое обычно называют нестероидным противовоспалительным лекарственным средством и которые особо не ограничены, но лекарственные средства, которые применяются для лечения артритов особенно предпочтительны. По общепринятому способу классификации NSAID существует классификация, основанная на различии их центральной структуры в химической структуре. Если NSAID, которые будут применять по настоящему изобретению, привести в пример на основе такой классификации, NSAID типа салициловой кислоты включают салициловую кислоту, аспирин и тому подобное; NSAID типа фенамовой кислоты включают мефенамовую кислоту, толфенамовую кислоту, флуфенамовую кислоту и тому подобное; NSAID типа арилацетата включают диклофенак, сулиндак, фенбуфен, индометацин, ацеметацин, амфенак, этодолак, фелбинак и тому подобное; NSAID типа пропионовой кислоты включают ибупрофен, флурбипрофен, кетопрофен, напроксен, пранопрофен, фенопрофен, тиапрофеновую кислоту, оксапрозин, локсопрофен, алминопрофен, залтопрофен и тому подобное; NSAID типа оксикама включают пироксикам, теноксикам, лорноксикам, мелоксикам и тому подобное и другие NSAID включают тиарамид, толметин, дифлюнизал, ацетаминофен, флоктафенин, тиноридин и тому подобное.
В качестве NSAID, которые будут применять по настоящему изобретению, предпочтительны NSAID, которые содержат такую функциональную группу, как карбоксильная группа, гидроксильная группа или аминогруппа в химической структуре. Поскольку можно выбрать функциональную группу спейсера согласно функциональным группам таких NSAID, вещество 1 по настоящему изобретению особо не ограничено, но наиболее предпочтительно используют NSAID, которые, по крайней мере, содержат карбоксильную группу.
Среди них более предпочтительно используют соединения, которые содержат центральную структуру, представленную следующей ниже формулой (6):
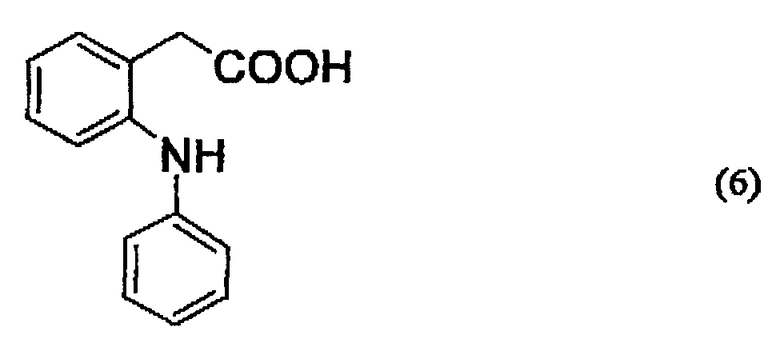
Более того, особенно предпочтительно используют соединения, представленные следующей ниже формулой (2):
R3 представляет собой заместитель, выбранный из низших алкильных групп и низших алкоксильных групп или атома водорода; каждый R4, R5 и R6 независимо представляет собой заместитель, выбранный из низшей алкильной группы, низшей алкоксильной группы и гидроксильной группы, атома галогена или атома водорода, и Х одинаковые или отличаются друг от друга и каждый независимо представляет собой заместитель, выбранный из низшей алкильной группы и трифторметильной группы или атома галогена, где, по крайней мере, один из Х представляет собой атом галогена. В дополнение, описанные выше низшая алкильная группа и низшая алкоксильная группа предпочтительно представляют собой низшую алкильную группу и низшую алкоксильную группу, содержащую от 1 до 12 атомов углерода, которые могут быть разветвленными, и более предпочтительно представляют собой низшую алкильную группу и низшую алкоксильную группу, содержащую от 1 до 6 атомов углерода, которые могут быть разветвленными.
Также, если карбоксиметильная группа и остаток амина расположены в положении 1 и положении 2 бензольного кольца соответственно, с которым связан R3, то R3 предпочтительно присоединен в положении 5.
В качестве соединений, представленных описанной выше формулой (2), например, можно привести соединения, описанные в WO 99/11605, и описанная в нем информация включена в данное описание посредством ссылки. Карбоксильная группа в NSAID не ограничена свободной формой, но также может быть и в виде соли.
DMARD в качестве другого из противовоспалительных лекарственных средств в отношении настоящего изобретения в общем случае означает целые фармацевтические препараты, обычно используемые в качестве противоревматических лекарственных средств и особо не ограничены, но предпочтительны DMARD, которые содержат такую функциональную группу, как карбоксильная группа, гидроксильная группа, аминогруппа или меркаптогруппа в химической структуре. DMARD включает актарит, метотрекскат, салазосульфапиридин, буцилламин и тому подобное.
В связи с этим можно привести описанные выше NSAID и DMARD в качестве противовоспалитеных лекарственных средств по настоящему изобретению, но особенно предпочтительны соединения, которые содержат карбоксильную группу.
В связи с этим возможно введение таких функциональных групп в гиалуроновую кислоту через требуемый способ связывания путем выбора функциональной группы фрагмента спейсера в зависимости от функциональных групп, содержащихся в описанных выше NSAID и DMARD. В дополнение, не всегда необходимо, чтобы один вид NSAID или DMARD присоединяли к веществу по настоящему изобретению, и производные гиалуроновой кислоты, к которым присоединено два или более видов NSAID и DMARD, также охватываются настоящим изобретением.
Описанный выше спейсер, представленный SP, является спейсером, который содержит участок, который способен к биодеградации, и представляет собой остаток соединения, содержащего, по крайней мере, одну функциональную группу, которая связана с гиалуроновой кислотой, и одну функциональную группу, которая связана с NSAID или DMARD (здесь и далее обозначаемое как «соединение-спейсер»). Участок спейсера, который способен к биодеградации особо не ограничен, при условии, что NSAID или DMARD, высвобождаемые из производных гиалуроновой кислоты, обладают эффектом, но предпочтительно, что участок расщепляется в участке связывания NSAID или DMARD со спейсером.
Соответствующие функциональные группы соединения-спейсера можно необязательно выбрать в зависимости от способов связывания с гиалуроновой кислотой и NSAID или DMARD. Например, если молекула спейсера присоединена к карбоксильной группе гиалуроновой кислоты посредством амидной связи, можно выбрать соединение-спейсер с аминогруппой, и в случае сложноэфирной связи с карбоксильной группой гиалуроновой кислоты можно выбрать спейсер с гидроксильной группой. Если спейсер присоединен к гидроксильной группе гиалуроновой кислоты посредством сложноэфирной связи, можно выбрать спейсер с карбоксильной группой. В данном случае с позиции краткости, для введения молекулы спейсера в гиалуроновую кислоту и стабильности в живом организме, соединение спейсер, содержащее аминогруппу, которую можно соединить с карбоксильной группой гиалуроновой кислоты посредством амидной связи, можно привести в качестве одного из предпочтительных вариантов осуществления.
Таким же образом функциональную группу соединения спейсера, которая связана с NSAID или DMARD, также можно выбрать, основываясь на функциональной группе, принадлежащей NSAID или DMARD. Например, в случае NSAID или DMARD, содержащего гидроксильную группу, оно может связываться посредством сложноэфирной связи, если выбрано соединение-сейсер, содержащее карбоксильную группу, в случае NSAID или DMARD, содержащего карбоксильную группу, оно может связываться посредством амидной связи, если выбрано соединение-спейсер, содержащее аминогруппу, и в случае NSAID или DMARD, содержащего меркаптогруппу, оно может связываться посредством тиоэфирной связи, если выбрано соединение-сейсер, содержащее карбоксильную группу.
В данном случае, если принять во внимание способность к биодеградации, предпочтительно соединение-спейсер, содержащее функциональную группу, которая может связываться посредством сложноэфирной связи с карбоксильной группой NSAID или DMARD, и особенно предпочтительно, чтобы карбоксильная группа NSAID или DMARD и гидроксильная группа соединения спейсера связывались посредством сложноэфирной связи.
Как описано выше, можно выбрать соединение-спейсер необязательно согласно характеристикам гиалуроновой кислоты и NSAID или DMARD, но, например, диаминоалкан, содержащий от 2 до 18 атомов углерода, аминоалкиловый спирт, содержащий от 2 до 12 атомов углерода, которые могут иметь заместитель(заместители), аминокислоту и тому подобное, можно привести в качестве примера. Аминокислота может представлять собой природную или неприродную аминокислоту, и она особо не ограничена, но предпочтительно в качестве примера можно привести глицин, β-аланин и γ-аминомасляную кислоту.
Как описано выше, если принять во внимание способ связывания гиалуроновой кислоты с NSAID, аминоалкиловый спирт, содержащий от 2 до 12 атомов углерода, который может иметь заместитель, можно привести в качестве предпочтительного примера соединения-спейсера.
В дополнение, он может представлять собой соединение-спейсер, которое содержит две или более таких функциональных групп, способных связываться с NSAID или DMARD, в одной молекуле (здесь и далее обозначаемое как «мультивалентное соединение-спейсер»).
Если выбрано мультивалентное соединение-спейсер, два или более NSAID или DMARD может одновременно связываться с одним спейсером. Соответственно, два или более NSAID или DMARD может одновременно присоединяться к функциональной группе, например к одной карбоксильной группе гиалуроновой кислоты, к которой присоединены NSAID или DMARD. Примеры таких мультивалентных соединений-спейсеров включают серинол и его производное, производное серина, производное треонина, 2-амино-1,5-пентандиол и его производное, 3-амино-1,2-пропандиол и его производное, трис(гидроксиметил)аминометан и его производное, бисгомотрис и его производные, и тому подобное.
Преимущество использования такого мультивалентного соединения-спейсера состоит в том, что большее количество NSAID или DMARD можно присоединить, не предоставляя большое количество карбоксильных групп и гидроксильных групп, приводящих к гидрофильным свойствам гиалуроновой кислоты в реакции замещения, так что гидрофильные свойства, а именно растворимость в водной среде, может сохраняться, несмотря на то, что большое количество NSAID или DMARD присоединено к молекуле.
Способ синтеза вещества по настоящему изобретению особо не ограничен, при условии, что это способ, согласно которому можно получить растворимое вещество по настоящему изобретению, как описано выше.
В этой связи в случае производного гиалуроновой кислоты, в котором соединение присоединено к гиалуроновой кислоте, карбоксильная группа и гидроксильная группа, принадлежащая гиалуроновой кислоте, главным образом, принимает участие в связывании с соединением таким образом, что гидрофильные свойства производного гиалуроновой кислоты уменьшаются, тогда как степень замещения вещества увеличивается.
Пример способа для синтеза вещества 1 по настоящему изобретению включает способ, который включает проведение щелочной обработки после присоединения NSAID к гиалуроновой кислоте через спейсер, содержащий участок, способный к биодеградации.
Описанный выше способ щелочной обработки после реакции присоединения для того, чтобы сделать щелочным реакционный раствор, особо не ограничен, при условии, что это обработка, при которой раствор становится щелочным. Более подробно, в качестве примера можно привести способ, в котором либо органическое основание, либо неорганическое основание добавляют в раствор, но предпочтительно неорганическое основание, если принять во внимание дальнейшую обработку и тому подобное. В дополнение, даже среди неорганических оснований более слабое основание, такое как гидрокарбонат натрия или карбонат натрия, более предпочтительно, чем более сильное основание, такое как гидроксид натрия, вследствие более слабого влияния на гиалуроновую кислоту и NSAID. В качестве условий рН щелочной обработки в данном случае в качестве примера можно привести значения от 7,2 до 11, предпочтительно от 7,5 до 10.
Время воздействия щелочной обработки особо не ограничено, при условии, что не оказано влияние на уменьшение молекулярной массы гиалуроновой кислоты, но можно указать от 2 до 12 часов, предпочтительно от 2 до 6 часов, и что можно получить растворимое производное гиалуроновой кислоты, не оказывая влияния на гиалуроновую кислоту, если проводится обработка в течение данного периода времени.
В качестве конкретного примера заданное растворимое производное гиалуроновой кислоты можно получить, давая возможность производному NSAID, связанному со спейсером, вступать в реакцию с гиалуроновой кислотой, добавляя в реакционный раствор слабую щелочь, такую как гидрокарбонат натрия, с последующим перемешиванием в течение нескольких часов и затем проводя последующие обработки, такие как нейтрализация, осаждение этанолом и сушка.
Описанный выше способ можно также применять для синтеза вещества 2 по настоящему изобретению таким образом, что можно получить растворимое вещество 2 по настоящему изобретению.
В связи с этим способ присоединения спейсера и NSAID или DMARD к гиалуроновой кислоте может представлять собой либо способ, в котором спейсер присоединяют к гиалуроновой кислоте и затем NSAID или DMARD присоединяют к связанной со спейсером гиалуроновой кислоте, либо способ, в котором сначала присоединяют спейсер к NSAID или DMARD и затем связанный со спейсером NSAID или связанный со спейсером DMARD присоединяют к гиалуроновой кислоте, но последний способ является предпочтительным.
Способ для соответственного связывания NSAID или DMARD, гиалуроновой кислоты и спейсера особо не ограничен, но можно использовать обычно используемый общепринятый способ в качестве средства проведения реакции связывания, при условии, что это способ, который может привести к образованию сложноэфирной связи, образованию амидной связи, образованию тиоэфирной связи и тому подобное. Специалист в данной области может несомненно оценить и выбрать условия реакции.
В связи с этим конденсацию гиалуроновой кислоты со связанным со спейсером NSAID или со связанным со спейсером DMARD или с соединением-спейсером можно осуществить, используя либо карбоксильную группу, либо гидроксильную группу гиалуроновой кислоты. Но с карбоксильной группой можно более легко провести конденсацию вследствие более высокой реакционной способности, которой обладает эта функциональная группа. Способ осуществления такой конденсации, например, включает способ, в котором используют растворимый в воде конденсирующий агент, такой как растворимый в воде карбодиимид (например, гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (EDCI·HCl), йодметилат 1-этил-3-(3-диметиламинопропил)карбодиимида и т.д.), способ, в котором используют добавочное средство для конденсации, такое как N-гидроксисукцинимид (HOSu) или N-гидроксибензотриазол (HOBt) и описанный выше конденсирующий агент, способ активирующих эфиров, способ ангидридов кислот и тому подобное. Среди них способ, в котором используют растворимый в воде конденсирующий агент, или способ, в котором используют добавочное средство для конденсации и растворимый в воде конденсирующий агент, как реакция в присутствии водного растворителя, является предпочтительным, и способ, в котором используют добавочное средство для конденсации и растворимый в воде конденсирующий агент, является особенно предпочтительным с точки зрения ингибирования побочных реакций. Предпочтительно, чтобы карбоксильная группа гиалуроновой кислоты связывалась со связанным со спейсером NSAID или со связанным со спейсером DMARD или с соединением-спейсером посредством сложноэфирной связи или амидной связи, более предпочтительно посредством амидной связи.
Можно подбирать степень замещения NSAID или DMARD гиалуроновой кислоты в отношении вещества по настоящему изобретению, изменяя количество конденсирующего агента, добавочного средства для конденсации, связанного со спейсером NSAID или связанного со спейсером DMARD во время процедуры синтеза вещества по настоящему изобретению. В связи с этим степень замещения можно определить, измеряя поглощение, или способом, который использует ВЭЖХ, ЯМР и тому подобное.
По настоящему изобретению степень замещения NSAID или DMARD особо не ограничена при условии, что сохраняется растворимость производного в водной среде, но предпочтительна степень от 0,1 до 80% мол. и более предпочтительно от 5 до 50% мол., в расчете на повторяющуюся дисахаридную единицу гиалуроновой кислоты. В дополнение, если вещество по настоящему изобретению используют в качестве активного ингредиента фармацевтического препарата, то оптимальную степень замещения определяют, принимая во внимание эффективную концентрацию или эффективность замедленного высвобождения NSAID или DMARD в участке, затронутом при конденсации.
Как описано выше, связанный со спейсером NSAID или связанное со спейсером DMARD присоединяют к карбоксильной группе гиалуроновой кислоты, при этом карбоксильная группа образует амидную связь или сложноэфирную связь с уменьшением или потерей ее гидрофильных свойств.
В качестве одного из средств для решения данной проблемы становится возможным присоединение большого количества NSAID или DMARD, в то время как сохраняются гидрофильные свойства, при использовании мультивалентного соединения-спейсера. Например, если производное аминотриола, содержащее 3 гидроксильные группы и 1 аминогруппу, используют в качестве соединения-спейсера, присоединение NSAID ко всем 3 гидроксильным группам приводит к присоединению 3 молекул NSAID к одной молекуле спейсера. Если такие связанные с аминотриолом NSAID присоединяют к карбоксильной группе гиалуроновой кислоты, например, при степени замещения (степень замещения в расчете на гиалуроновую дисахаридную единицу) 20%, то это означает, что степень замещения NSAID составляет 60%, эквивалентной в 3 раза большей степени замещения связанных с аминотриолом NSAID.
В дополнение, как описано выше, растворимость производного гиалуроновой кислоты в водной среде сохраняется, если применяют способ, в котором проводят щелочную обработку после реакции присоединения для синтеза производного гиалуроновой кислоты с присоединенным противовоспалительным лекарственным средством, которое приводилось в качестве примера способа синтеза вещества по настоящему изобретению. Такой эффект сохранения растворимости явно полезен, поскольку нет необходимости учитывать ни тип соединения-спейсера, ни степень замещения лекарственного средства и тому подобное, и обработка является общепринятой.
Обобщая описанные выше объяснения, можно, например, привести в качестве особо предпочтительного варианта осуществления вещества 1 по настоящему изобретению производное гиалуроновой кислоты, содержащее дисахаридную единицу, состоящую из гиалуроновой кислоты, представленной формулой (1). В связи с этим следующая ниже формула (1) демонстрирует частичную структуру через дисахаридную единицу, состоящую из гиалуроновой кислоты, где N-ацетил-D-глюкозамин с присоединенным противовоспалительным лекарственным средством и D-глюкуроновая кислота связаны через связь β-1,3.
Y-CO-NH-R 1 -(O-R 2 ) n (1)
где Y-CO- представляет собой один остаток дисахаридной единицы, составляющей гиалуроновую кислоту; R2 представляет собой остаток NSAID, представленный группой Z-CO- или атомом водорода, при условии, что в нем не все R2 являются атомом водорода; -HN-R1-(O-)n представляет собой спейсерный остаток в соединении-спейсере, представленном формулой H2N-R1-(OH)n, содержащем гидроксильные группы в количестве n; R1 представляет собой линейную или разветвленную углеводородную группу, содержащую от 2 до 12 атомов углерода, которая может иметь заместитель(заместители); -CO-NH- представляет собой амидную связь карбоксильной группы гиалуроновой кислоты в качестве составляющей сахарид гиалуроновой кислоты с аминогруппой соединения-спейсера; -О-СО- представляет собой сложноэфирную связь гидроксильной группы соединения-спейсера с карбоксильной группой, принадлежащей NSAID, и n равно целому числу от 1 до 3. В связи с этим карбонильная группа в остатке гиалуроновой кислоты, составляющей производное гиалуроновой кислоты, присутствует в качестве амидной связи, вовлеченной в связывание с остатком связанного со спейсером противовоспалительного лекарственного средства, или в качестве свободной карбоксильной группы не вовлеченного в него согласно степени замещения остатком NSAID.
Заместитель в группе R1 включает алкильную группу, алкенильную группу, арильную группу, алкоксильную группу, ацильную группу, карбоксильную группу, галоген и тому подобное, где количество атомов водорода в алкильной группе, алкенильной группе, алкоксильной группе и ацильной группе предпочтительно составляет от 1 до 11, более предпочтительно от 1 до 4, и фенильная группа предпочтительна в качестве арильной группы. Например, в качестве примера можно привести серин в качестве соединения-спейсера, содержащего карбоксильную группу в качестве заместителя, и треонин в качестве соединения-спейсера, содержащего карбоксильную группу и метильную группу.
В связи с этим согласно описанной выше формуле (1) Y-COOH представляет собой одну дисахаридную единицу, составляющую гиалуроновую кислоту до реакции; H2N-R1-(OH)n представляет собой соединение-спейсер до реакции и HOOC-Z представляет собой NSAID до реакции.
В качестве наиболее предпочтительного способа для синтеза дисахаридной единицы, составляющей гиалуроновую кислоту по описанной выше формуле (1), в качестве примера можно привести способ, в котором связывают соединение-спейсер и NSAID и затем проводят реакцию с гиалуроновой кислотой. Концептуальное выражение данной реакции представлено ниже:

R7 представляет собой защитную группу для аминогруппы, где защитная группа особо не ограничена, поскольку можно использовать защитные группы, обычно используемые в качестве защитных групп для аминогруппы, и примеры включают защитную группу уретанового типа, такую как трет-бутоксикарбонильная группа, бензилоксикарбонильная группа и 9-флуоренилметилоксикарбонильная группа, и защитную группу ацильного типа, такую как формильная группа и фталоильная группа, и защитная группа уретанового типа предпочтительна. В связи с этим R1, R2 и Z представляют собой группы, как описано выше.
Тем не менее приведенное выше описание представляет собой концептуальное объяснение пути реакции, и схема и тому подобное для эффективного осуществления реакции, которую специалисты в данной области могут вывести, в настоящее описание не включены.
В описанной выше формуле (1) R1 более предпочтительно представляет собой линейную или разветвленную углеводородную группу, содержащую от 2 до 5 атомов углерода, которая может иметь заместитель (заместители), особенно предпочтительно содержащую 2 или 3 атома углерода, и примеры включают этиленовую группу, триметиленовую группу и пропиленовую группу.
Также в качестве NSAID, используемого в описанной выше формуле (1), возможно выбрать из описанных выше NSAID. В дополнение, соединения, представленные следующей ниже формулой (7), предпочтительно можно привести в качестве примера.
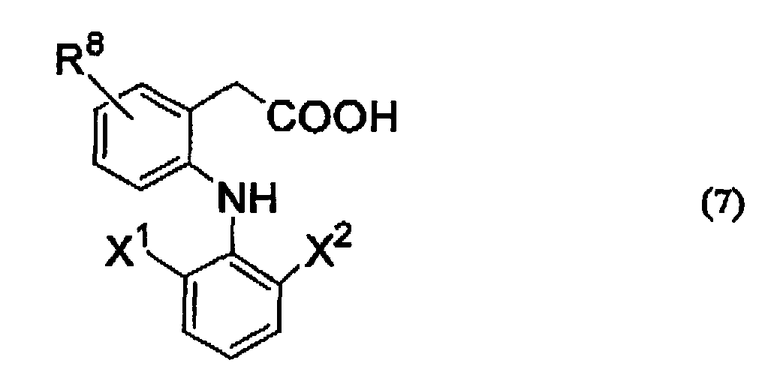
R8 представляет собой заместитель, выбранный из низшей алкильной группы и низшей алкоксильной группы или атома водорода, и более предпочтительно представляет собой низшую алкильную группу, содержащую от 1 до 12 атомов углерода, которые могут быть разветвленными, или атом водорода, и особенно предпочтительно, низшую алкильную группу, содержащую от 1 до 4 атомов углерода, или атом водорода.
Каждый X1 и X2 независимо представляет собой заместитель, выбранный из низшей алкильной группы и трифторметильной группы или атома галогена, где, по крайней мере, один из них представляет собой атом галогена. X1 и X2 предпочтительно представляют собой атомы галогена, которые являются одинаковыми или различными, и более предпочтительно выбраны из атома фтора и атома хлора.
В дополнение, предпочтительно, чтобы R8 связывался в положении 5 бензольного кольца, с которым связан R8, если карбоксиметильная группа и аминогруппа расположены в положении 1 и положении 2 бензольного кольца соответственно.
Конкретные примеры соединений, представленных описанной выше формулой (7), включают соединения, представленные следующими ниже формулами (8) и (9):
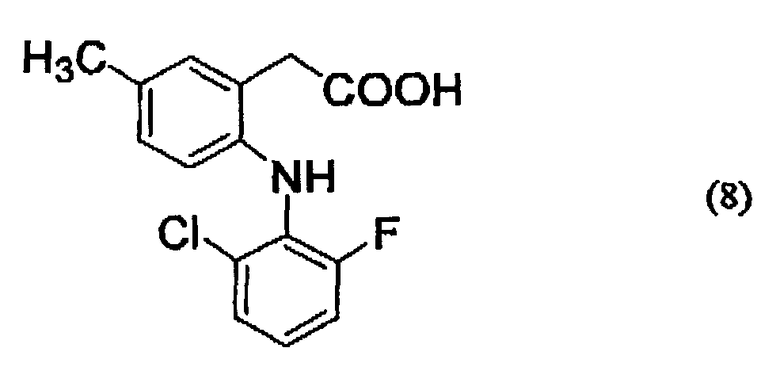
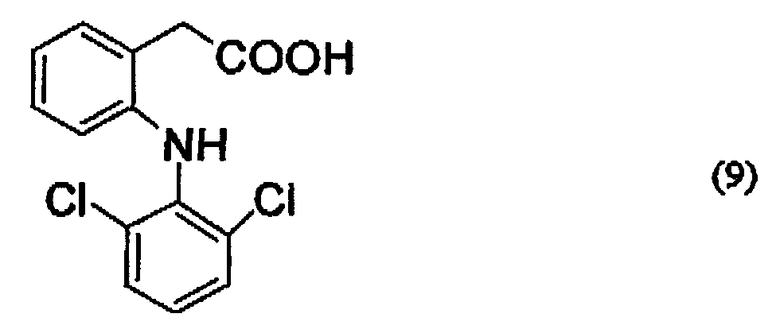
Например, если производное гиалуроновой кислоты, содержащее диклофенак, синтезируют, используя диклофенак, представленный формулой (9), то группа -CO-Z в описанной выше формуле (1) представлена следующей ниже формулой (10):
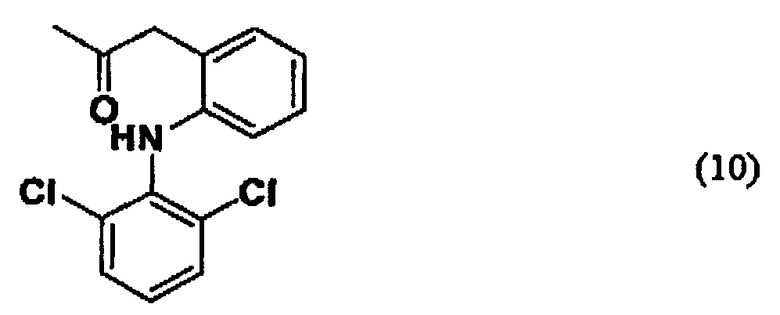
В связи с этим производные гиалуроновой кислоты, содержащие диклофенак, обладают очень сильным обезболивающим действием и противовоспалительным действием.
В качестве гиалуроновой кислоты, которую можно использовать в веществе по настоящему изобретению, содержащем дисахаридную единицу, составляющую гиалуроновую кислоту, представленную описанной выше формулой (1), предпочтительно выбрана гиалуроновая кислота, имеющая средневзвешенную молекулярную массу от 50000 до 3000000, более предпочтительна гиалуроновая кислота, имеющая средневзвешенную молекулярную массу от 50000 до 2000000.
Степень замещения (DS) NSAID в веществе по настоящему изобретению, содержащем дисахаридную единицу, составляющую гиалуроновую кислоту, представленную описанной выше формулой (1), предпочтительно составляет от 5 до 50% мол., более предпочтительно от 10 до 50% мол. в расчете на повторяющуюся дисахаридную единицу гиалуроновой кислоты.
В качестве значительной характеристики вещества по настоящему изобретению можно привести такую особенность, что вещество по настоящему изобретению можно растворять в водной среде, а именно оно легко растворимо в воде, так что если добавляют водный растворитель к веществу по настоящему изобретению, оно растворяется без проведения нагревания, солюбилизационной обработки и тому подобного. В связи с этим даже если степень замещения высока, а именно 5% или более, или более того 10% или более, оно может раствориться. Таким образом, раствор, приготовленный растворением вещества по настоящему изобретению в водной среде, представляет собой пригодную для инъекций жидкость и обладает способностью проходить через фильтр для фильтрации. Кстати, как описано выше, известно, что если лекарственное средство, обладающее сильными гидрофобными свойствами, такое как NSAID или DMARD, присоединяют к гиалуроновой кислоте, обладающей сильными гидрофильными свойствами, то продукт становится полурастворимым в воде гелем, обладающим сильной вязкоупругой или нерастворимой природой, поскольку увеличиваются гидрофобные свойства самой молекулы гиалуроновой кислоты, так что продукт не подходит для инъекций, который вводят инжектором.
Тем не менее поскольку растворимость производного гиалуроновой кислоты сохраняется, например, при проведении щелочной обработки во время способа ее получения, как описано выше, вещество по настоящему изобретению может представлять собой прозрачный раствор, обладающий способностью проходить через фильтр для фильтрации.
Таким образом, раствор вещества по настоящему изобретению можно подвергать фильтрации таким образом, что возможно удаление пыли, удаление микроорганизмов и стерилизация от микроорганизмов при фильтрации. То есть удаление пыли и микроорганизмов может быть эффективно при прохождении через фильтр размером 5 мкм или 0,45 мкм и более предпочтительно стерилизация также становится возможна при прохождении через фильтр размером 0,22 мкм.
Более конкретно, предпочтительно, чтобы раствор, приготовленный растворением вещества по настоящему изобретению в водной среде с концентрацией 1,0% масс, был способен проходить через пористый фильтр (размер пор 0,45 мкм, диаметр 25 мм) при скорости 2 мл в минуту или более при температуре 24°С под давлением 5,0 кг/см2.
Также более предпочтительно, чтобы раствор, приготовленный растворением вещества по настоящему изобретению в водной среде с концентрацией 1,0% масс, был способен проходить через пористый фильтр (размер пор 0,22 мкм, диаметр 25 мм) при скорости 2 мл в минуту или более в тех же самых описанных выше условиях.
Как описано ниже, если вещество по настоящему изобретению используют в качестве лекарственного средства, которое будут применять на живых организмах (млекопитающие, особенно предпочтительно человек), то удаление пыли и удаление микроорганизмов и стерилизация от микроорганизмов становится существенным элементом, так что такая характеристика вещества по настоящему изобретению весьма полезна. В дополнение, в случае стерилизации нагреванием, облучением ультрафиолетом и тому подобное существует возможность вызвать деградацию, уменьшение молекулярной массы и тому подобное у производного гиалуроновой кислоты, но такой проблемы можно избежать в случае фильтрационной стерилизации.
Фармацевтическое средство по настоящему изобретению представляет собой фармацевтическое средство, содержащее производное гиалуроновой кислоты в качестве вещества по настоящему изобретению, в качестве активного ингредиента. Путем использования отмеченных выше характеристик вещества по настоящему изобретению фармацевтическое средство по настоящему изобретению может относиться к варианту осуществления, в котором вещество можно вытеснять из инжектора и тому подобное, и его также используют в качестве раствора вещества по настоящему изобретению, растворенного в водной среде. Например, можно привести раствор, в котором физиологический раствор, забуференный фосфатом физиологический раствор или вода для инъекций, пригодные для введения в живой организм, используют в качестве растворителя и который содержит вещество по настоящему изобретению в концентрации от 0,1% масс до 10% масс. Предпочтительно, чтобы такой раствор не был мутным, а был прозрачным.
Как описано выше, фармацевтическое средство по настоящему изобретению применимо для удаления пыли, удаления микроорганизмов и стерилизации от микроорганизмов при фильтрации через фильтр. Удаление пыли и микроорганизмов становится возможным при прохождении через фильтр размером 5 мкм или 0,45 мкм, и стерилизация также становится возможной при прохождении через фильтр размером 0,22 мкм. В дополнение, также возможно использовать фармацевтическое средство по настоящему изобретению совместно с веществом по настоящему изобретению и фармацевтически приемлемым носителем в таком диапазоне, что преимущество, присущее фармацевтическому средству по настоящему изобретению, а именно свойство стерилизации фильтрацией, не утрачивается.
Предпочтительно, чтобы фармацевтическое средство по настоящему изобретению, приготовленное таким образом, можно было подвергнуть фильтрационной стерилизации, а также в таком состоянии, чтобы оно обладало определенной степенью вязкоупругости.
Возможно использовать фармацевтическое средство по настоящему изобретению в качестве лекарственного средства для использования при парентеральном введении или лекарственного средства для использования при местном введении. В качестве варианта использования его при парентеральном введении и местном введении предпочтителен раствор, приготовленный растворением описанного выше вещества по настоящему изобретению в водной среде, и такие способы введения, как инъекция и инфузия, можно предпочтительно привести в качестве примера (согласно данному описанию, «инфузия» иногда включает «инъекцию»). Осуществляя местное введение инфузией, можно избежать появления побочных эффектов в системе органов пищеварения. В дополнение, поскольку можно избежать метаболизма в системе органов пищеварения, можно уменьшить дозу по сравнению со случаем перорального введения и, кроме того, также можно избежать проблемы системной токсичности, вызываемой большой дозой при пероральном введении.
Относительно устройства для вытеснения, используемого при инъекции, инфузии и тому подобное, возможно использование инструментов, обычно используемых с целью введения фасованных лекарственных средств вытеснением, таких как инжектор и устройство для инфузии.
В связи с этим можно также предложить набор, в котором раствором фармацевтического средства по настоящему изобретению или веществом по настоящему изобретению заполняют пригодное для вытеснения устройство для инфузии, оборудованное плунжером для вытеснения лекарственного средства или тому подобное. В дополнение, можно создать набор в виде набора для медицинских инъекций, в котором раствором, приготовленным растворением вещества по настоящему изобретению в фармацевтически приемлемом забуференном фосфатом физиологическом растворе, физиологическом растворе или в воде для инъекций, заполнен шприц и герметично упакован со скользящим плунжером для вытеснения таким образом лекарственного средства. В связи с этим возможно использование обычно применяемого устройства в качестве плунжера для вытеснения лекарственного средства, который сделан из эластичной основы, такой как резина или синтетический каучук, и вставлен в шприц в состоянии плотного контакта таким образом, чтобы он мог скользить. В дополнение, стержень плунжера для вытеснения лекарственного средства путем вытеснения плунжера, также можно включить в набор.
Хотя заболевание, которое необходимо лечить, и способ введения фармацевтического средства по настоящему изобретению особо не ограничены, можно использовать его в качестве терапевтического средства для лечения артритов, подавления воспаления, подавления боли и тому подобное (здесь и далее обозначаемого как «терапевтическое средство по настоящему изобретению»), которое является предпочтительным. В связи с этим согласно данному описанию «терапевтическое средство» охватывает не только «терапевтическое средство», но также и лекарственное средство, которое используют с целью предупреждения заболевания или смягчения симптомов.
Терапевтическое средство по настоящему изобретению не только обладает действием при замедленном высвобождении противовоспалительного лекарственного средства, такого как NSAID, и действием в системе доставки лекарственного средства, как описано ниже, но также одновременно можно ожидать и эффекта фармацевтических препаратов гиалуроновой кислоты, используемых в настоящее время в клинической области лечения артритов, в дополнение к терапевтическому эффекту противовоспалительного лекарственного средства при лечении артритов.
Кроме того, доза терапевтического средства по настоящему изобретению особо не ограничена, поскольку представляет собой вопрос, который следует решать индивидуально согласно способу введения, форме введения, целей использования и определенных симптомов, возраста, массы тела и состояния животного, которое будут лечить, таким образом, что его терапевтический эффект проявляется наиболее соответствующим образом. Например, в случае инъекций для применения человеку можно указать приблизительно от 1 мг до 1000 мг, предпочтительно приблизительно от 5 мг до 500 мг, более предпочтительно приблизительно от 10 мг до 100 мг на взрослого человека однократно, в расчете на производное гиалуроновой кислоты. Тем не менее полагают, что сила действия лекарственного средства, присущая NSAID или DMARD, используемым в веществе по настоящему изобретению в качестве активного ингредиента, имеет большое влияние на терапевтическое средство по настоящему изобретению, так что описанный выше диапазон не всегда подходит, и необходимо его устанавливать, принимая во внимание дозу, преобразованную в стандартный отдельный препарат NSAID или DMARD. В дополнение, как показано в примерах, описанных ниже, в отличие от случая, в котором вводят один препарат NSAID, фармацевтическое средство по настоящему изобретению присутствует в участке введения стабильно и продолжительно, так что необходимо устанавливать дозу, также принимая во внимание этот момент.
Участок, в котором необходимо использовать терапевтическое средство по настоящему изобретению, особо не ограничен, при условии, что он представляет собой участок применения при парентеральном введении, и среди частей тела предпочтительны суставы, коленный сустав, плечевой сустав, бедренный сустав, челюстной сустав и тому подобное особенно предпочтительны. В частности, применение при остеоартрите колена (ОА) и ревматоидном артрите колена (RA) предпочтительно.
В этой связи, если терапевтическое средство по настоящему изобретению используют в качестве терапевтического средства для лечения артрита, подходящую концентрацию инфузий (инъекций) в сустав можно необязательно выбрать, как описано выше, и концентрация раствора предпочтительно составляет от 0,3 до 3,0% масс, более предпочтительно от 0,5 до 1,5% масс.
В качестве одного из наиболее предпочтительных вариантов фармацевтического средства по настоящему изобретению можно привести следующую ниже конструкцию.
NSAID:
Соединение, представленное описанной выше формулой (2)
Спейсер и способ связывания:
аминоалкиловый спирт связан с NSAID через сложноэфирную связь и связан с гиалуроновой кислотой через амидную связь.
Молекулярная масса гиалуроновой кислоты:
средневзвешенная молекулярная масса от 500000 до 3000000.
Степень замещения NSAID:
от 5 до 50% мол. на дисахаридную единицу гиалуроновой кислоты.
Концентрация и растворитель:
забуференный фосфатом физиологический раствор, имеющий концентрацию от 0,3 до 3,0% масс.
Условие предоставления:
заполненный шприц в стерильном состоянии.
В дополнение, в качестве NSAID более предпочтительно соединение, представленное описанной выше формулой (7), и дополнительно предпочтительны соединения, представленные описанной выше формулой (8) и описанной выше формулой (9), диклофенак или его производное особенно предпочтителен. В качестве спейсера более предпочтительно, если он выбран из аминопропилового спирта или аминоэтилового спирта.
В качестве степени замещения предпочтительно замещение от 10 до 50% мол. на дисахаридную единицу гиалуроновой кислоты.
Также наиболее предпочтительно, если возможна фильтрация через фильтр размером 5 мкм или 0,45 мкм и дополнительно если возможна фильтрация через фильтр размером 0,22 мкм.
Как показано в примерах, которые описаны ниже, особенно подходит для использования фармацевтическое средство по настоящему изобретению в качестве терапевтического средства для артритов, особенно в качестве инфузий в суставы для лечения артритов. Например, если низкомолекулярные соединения, такие как NSAID, непосредственно вводить в полость сустава, то такие соединения немедленно удаляются в кровоток через синовиальную оболочку, так что невозможно ожидать большого эффекта.
С одной стороны, если раствор производного гиалуроновой кислоты, содержащего NSAID, к которому NSAID в качестве вещества по настоящему изобретению присоединен посредством ковалентной связи, вводят в полость сустава, NSAID продолжительно присутствует в синовиальной ткани, как показано далее в примерах, в то время как низкомолекулярное соединение само по себе быстро метаболизируется в синовиальной оболочке. Как общеизвестно, гиалуроновая кислота обладает аффинностью к синовиальной оболочке. По этой причине полагают, что фармацевтическое средство по настоящему изобретению задерживается до определенной степени в синовиальной оболочке в состоянии, когда гиалуроновая кислота и NSAID связаны, и после постепенного проникновения в ткани или клетки NSAID высвобождаются из гиалуроновой кислоты и оказывают действие. То есть в случае введения фармацевтического средства по настоящему изобретению NSAID не удаляются сразу в кровоток, а NSAID постоянно присутствуют в суставной жидкости и синовиальной оболочке так, что проявляет постоянный эффект.
На основании этого предпочтительно, чтобы связывание гиалуроновой кислоты с соединением-спейсером в фармацевтическом средстве по настоящему изобретению проявляло устойчивость к его биодеградации по сравнению со связыванием NSAID с соединением-спейсером. В дополнение, предпочтителен вариант осуществления, в котором участок связывания NSAID с соединением-спейсером не разрушается в полости сустава, а разрушается в синовиальной ткани после прохождения в синовиальную оболочку. Изменяя способы связывания NSAID с соединением-спейсером и гиалуроновой кислоты с соединением-спейсером, можно изменять устойчивость к биодеградации, тем самым давая возможность контролировать способность к высвобождению и скорость высвобождения. Например, если рассматривать гидролиз, происходящий в живом организме, то сложноэфирная связь более восприимчива к разрушению, чем амидная связь. Таким образом, при выборе спейсера, который связывается с гиалуроновой кислотой через амидную связь и с NSAID через сложноэфирную связь, сложноэфирные связи восприимчивы к гидролизу, и NSAID высвобождаются из вещества по настоящему изобретению, которое гидролизуется, чтобы стать активным. Фармацевтический препарат для использования с замедленным высвобождением также возможно получить с помощью фармацевтического средства по настоящему изобретению.
В связи с этим в примерах, которые описаны ниже, если соответственно вводили 2 типа веществ по настоящему изобретению, в которых гиалуроновая кислота была связана с соединением-спейсером через амидную связь и NSAID был связан с соединением-спейсером через амидную связь или сложноэфирную связь, вещество по настоящему изобретению, в котором NSAID и соединение-спейсер были связаны через сложноэфирную связь, проявляли более значительный подавляющий боль эффект.
Известно, что ингибирование продукции простагландина посредством ингибирующей активности в отношении циклооксигеназы (СОХ) в клетках-мишенях играет роль в механизме действия NSAID, которые подавляют воспаление и боль, сопутствующие артриту. Оценку вещества по настоящему изобретению проводили, используя набор Chemiluminescent COX Inhibitor Screening Assay (производимый Cayman), который представляет собой способ, в общем случае используемый для оценки ингибирующей активности в отношении СОХ-2. Как результат, ингибирующей активности в отношении СОХ-2 не обнаружили для вещества 1 по настоящему изобретению, используя дозу, при которой NSAID в качестве одного препарата явно показывала ингибирующую активность в отношении СОХ-2, а также при дозе вещества 1 по настоящему изобретению в количестве NSAID, которое соответствует дозе одного препарата, преобразованного из NSAID.
Такие результаты in vitro не могут быть перенесены на живые организмы, которые согласуются различными условиями и состояниями, однако легко предположить, что высвобождение NSAID в область действия фармацевтическим средством по настоящему изобретению является предпочтительным.
В дополнение, вещество по настоящему изобретению также применимо в качестве основного вещества в системе доставки лекарственного средства (DDS) NSAID или DMARD, которое, будучи низкомолекулярным соединением, как известно, затруднительно для эффективной доставки к участку-мишени (клетки) при введении одного лекарственного средства вследствие быстрого метаболизма в живом организме. Для этого с получением эффективных результатов путем уменьшения влияния метаболизма весьма важно доставлять NSAID или DMARD к клетке-мишени в виде производного гиалуроновой кислоты, содержащего NSAID или DMARD, по настоящему изобретению и для дальнейшего прохождения в клетки в том же самом виде для эффективного постоянного присутствия в участке-мишени.
Количество лекарственного средства, эффективное для лечения в области введения, можно эффективно поддерживать посредством использования вещества по настоящему изобретению по сравнению с однократным введением лекарственного средства, так что гораздо более сильный терапевтический эффект можно ожидать при гораздо меньшей дозе при пероральном введении. В дополнение, поскольку можно улучшить способность к замедленному высвобождению и постоянство эффекта, также можно ожидать уменьшения количества введений и тому подобное в клинической практике.
Примеры
Настоящее изобретение описано ниже более подробно, основываясь на примерах. Тем не менее нет намерения ограничить тем самым технический объем настоящего изобретения.
В связи с этим все гиалуроновые кислоты и гиалуронаты натрия, используемые в следующих ниже примерах, приобретены в Seikagaku Corporation.
Здесь и далее в качестве забуференного фосфатом физиологического раствора (PBS) в следующих ниже примерах использовали 5 мМ PBS, если не указано иначе.
Тестовый пример
Тест на прохождение через фильтр
Приготавливали PBS, в котором каждое вещество, которое необходимо было испытать, растворяли до концентрации 1,0% масс. При температуре 24°С под давлением 5,0 кг/см2 каждый раствор веществ, которые необходимо было испытать, приготовленный в следующих ниже примерах, пропускали через пористый фильтр (25 мм в диаметре) и измеряли прошедшее количество (мл) за 1 минуту. Случай, в котором проходило 2 мл или более, показан буквой «А», и случай, в котором проходило менее чем 2 мл, показан буквой «В», и случай, в котором не проходило ничего, показан буквой «С».
Примеры получения
Справочный пример 1
Синтез трет-бутоксикарбониламинопропанола (Boc-NH(CH2)3OH) (Boc-аминопропанола)
В 10 мл дихлорметана растворяли 1,542 г (20,5 ммоль) аминопропанола и медленно добавляли 10 мл 4,484 г (20,5 ммоль) раствора ди-трет-бутилдикарбоната (Вос2О) в дихлорметане по каплям при охлаждении льдом. После этого температуру реакционного раствора доводили обратно до комнатной температуры и перемешивали в течение 2 часов и 40 минут, исчезновение исходных продуктов подтверждали тонкослойной хроматографией (ТСХ) и затем дихлорметан выпаривали при пониженном давлении. Реакция протекала количественно, и маслянистое вещество получали с выходом 3,92 г. Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,46 (9H, с, Boc), 1,66 (2H, квант, -NHCH2 CH 2CH2O-), 3,27 (3H, м, -NHCH 2CH2CH2O-), 3,66 (2H, м, -NHCH2CH2 CH 2O-), 4,91 (1H, ушир., CH2 ОH).
Пример 1
Синтез гидрохлорида аминопропанолкетопрофена
1) Синтез Boc-аминопропанолкетопрофена
В 14 мл дихлорметана растворяли 2,371 г (13,5 ммоль) Boc-аминопропанола и 3,441 г (13,5 ммоль) кетопрофена (производимого Tokyo Kasei Kogyo) и добавляли 323 мг (2,6 ммоль) 4-диметиламинопиридина (DMAP) и 2,833 г (14,8 ммоль) растворимого в воде гидрохлорида карбодиимида (WSCI·HCl)/14 мл дихлорметана в указанном порядке при охлаждении льдом. После доведения температуры реакционной смеси обратно до комнатной температуры и перемешивания в течение ночи дихлорметан выпаривали при пониженном давлении и добавляли этилацетат с последующим выделением вещества промыванием дважды 5% лимонной кислотой, водой, дважды 5% гидрокарбонатом натрия, водой и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия, этилацетат выпаривали при пониженном давлении с получением 5,430 г указанного в заголовке соединения (выход 98%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,43 (9H, с, Boc), 1,54 (3H, д, -OCOCH(CH 3)-), 1,77 (2H, квант, -NHCH2 CH 2CH2O-), 3,09 (2H, м, -NHCH 2CH2CH2-), 3,82 (1H, кв, -OCOCH(CH3)-), 4,15 (2H, м, -NHCH2CH2 CH 2O-), 4,69 (1H, ушир., -NHCH2-), 7,42-7,83 (9H, м, ароматический H).
2) Синтез гидрохлорида аминопропанолкетопрофена
При охлаждении льдом 20 мл 4М раствора хлористый водород/этилацетат добавляли к 5,330 г (12,95 ммоль) полученного выше Boc-аминопропанолкетопрофена с последующим перемешиванием при охлаждении льдом в течение 15 минут и в течение 2 часов при комнатной температуре. После подтверждения исчезновения Boc-аминопропанолкетопрофена при помощи ТСХ растворитель выпаривали при пониженном давлении и осадок дважды подвергали декантации диэтиловым эфиром. После этого осадок сушили при пониженном давлении с количественным получением указанного в заголовке соединения с выходом 4,569 г. Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,50 (5H, д, -OCOCH(CH 3)-), 2,08 (2H, м, -NHCH2 CH 2CH2O-), 3,04 (2H, ушир., -NH CH2CH2CH2O-), 3,82 (1H, кв, -OCOCH(CH3)-), 4,16 (2H, м, -NHCH2CH2 CH 2O-), 7,36-7,80 (9H, м, ароматический H), 8,20 (ушир., H 3 N +CH2-).
Пример 2
Синтез гиалуроната натрия, содержащего аминопропанолкетопрофен
В смеси 22,5 мл воды/22,5 мл диоксана растворяли 200 мг (0,5 ммоль/дисахаридная единица) гиалуроната натрия, имеющего средневзвешенную молекулярную массу 900000, и затем добавляли 0,25 мл 2М водного раствора гидроксисукцинимида (HOSu), 0,25 мл водного раствора WSCI·HCl в концентрации 1 моль/л и 0,5 мл 0,5М водного раствора гидрохлорида аминопропанолкетопрофена, полученного в примере 1, в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 3 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 3 часов и 20 минут. После нейтрализации реакционного раствора добавлением 86 мкл 50% уксусной кислоты добавляли 800 мг хлорида натрия с последующим перемешиванием. Смесь осаждали, добавляя 200 мл этанола, осадок дважды промывали 80% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением порции из 198 мг белого твердого вещества. Степень замещения кетопрофеном составляла 15,5% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 3
Синтез гиалуроната натрия, содержащего аминопропанолкетопрофен
В смеси 45 мл воды/45 мл диоксана растворяли 400 мг (1,0 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 900000, и затем добавляли HOSu в количестве 1,66 ммоль/1 мл воды, WSCI·HCl в количестве 0,83 ммоль/1 мл воды и полученный в примере 1 гидрохлорид аминопропанолкетопрофена в количестве 0,83 ммоль/4 мл воды в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли гидрокарбонат натрия в количестве 300 мг/1 мл с последующим перемешиванием в течение 3 часов и 10 минут. После нейтрализации реакционного раствора добавлением 86 мкл уксусной кислоты добавляли 400 мг хлорида натрия с последующим перемешиванием. Смесь осаждали, добавляя 300 мл этанола, осадок дважды промывали 80% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 246 мг белого твердого вещества. Степень замещения кетопрофеном составляла 26,3% согласно анализу ВЭЖХ. Для приготовления раствора, полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 4
Синтез гидрохлорида аминопропанолнапроксена
1) Синтез Boc-аминопропанолнапроксена
В 2 мл дихлорметана растворяли 350 мг (2 ммоль) Boc-аминопропанола и 462 г (2 ммоль) напроксена (производимого Wako Pure Chemical Industries) и добавляли 48 мг (0,4 ммоль) DMAP и 422 г WSCI·HCl (2,2 ммоль)/2 мл дихлорметана в указанном порядке при охлаждении льдом. После доведения температуры реакционной смеси обратно до комнатной температуры и перемешивания в течение 4 часов и 50 минут дихлорметан выпаривали при пониженном давлении и добавляли этилацетат с последующим выделением вещества промыванием дважды 5% лимонной кислотой, водой, дважды 5% гидрокарбонатом натрия, водой и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия этилацетат выпаривали при пониженном давлении с получением 720 мг белых кристаллов указанного в заголовке соединения (выход 93%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,42 (9H, c, Boc), 1,58 (3H, д, -OCOCH(CH3)-), 1,75 (2H, квант, -NHCH2 CH 2CH2O-), 3,07 (2H, м, -NHCH 2CH2CH2O-), 3,85 (1H, кв, -OCOCH(CH3)-), 3,91 (3H, с, -OCH 3), 4,13 (2H, м, -NHCH2CH2 CH 2O-), 4,63 (1H, ушир., -NHCH2-), 7,09-7,75 (6H, м, ароматический H).
2) Синтез гидрохлорида аминопропанолнапроксена
В 1 мл дихлорметана растворяли 684 мг (1,76 ммоль) полученного выше Boc-аминопропанолнапроксена и добавляли 2 мл 4М раствора хлористый водород/этилацетат при охлаждении льдом с последующим перемешиванием при охлаждении льдом в течение 20 минут и при комнатной температуре в течение 1 часа. После подтверждения исчезновения Boc-аминопропанолнапроксена при помощи ТСХ добавляли диэтиловый эфир с последующей декантацией три раза. После этого осадок сушили при пониженном давлении с количественным получением указанного в заголовке соединения с выходом 564 мг. Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3+CD3OD) δ (м.д.)=1,57 (3H, д, -OCOCH(CH 3)-), 2,02 (2H, квант, -NHCH2 CH 2CH2O-), 2,88 (2H, м, -NHCH 2CH2CH2O-), 3,87 (1H, кв, -OCOCH(CH3)-), 3,90 (3H, с, -OCH 3), 4,17 (2H, м, -NHCH2CH2 CH 2O-), 7,08-7,73 (6H, м, ароматический H), 8,10 (ушир., H 3 N +CH2-).
Пример 5
Синтез гиалуроната натрия, содержащего аминопропанолнапроксен
В смеси 11,5 мл воды/11,5 мл диоксана растворяли 100 мг (0,25 ммоль/дисахаридная единица) гиалуроната натрия, имеющего средневзвешенную молекулярную массу 900000, и затем добавляли HOSu (0,2 ммоль)/0,1 мл воды, WSCI·HCl (0,1 ммоль)/0,1 мл воды и гидрохлорид аминопропанолнапроксена (0,1 ммоль)/0,3 мл воды, полученный в примере 4, в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 1,5 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 3 часов и 35 минут. После нейтрализации реакционного раствора добавлением 43 мкл 50% уксусной кислоты добавляли 500 мг хлорида натрия с последующим перемешиванием. Смесь осаждали добавлением 50 мл этанола, осадок дважды промывали 80% этанолом, дважды этанолом и диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 95 мг белого твердого вещества. Степень замещения напроксеном составляла 13,1% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 6
Синтез гидрохлорида аминопропанолибупрофена
1) Синтез Boc-аминопропанолибупрофена
В 2 мл дихлорметана растворяли 352 мг (2 ммоль) Boc-аминопропанола и 412 г (2 ммоль) ибупрофена (производимого Wako Pure Chemical Industries) и добавляли 48 мг (0,4 ммоль) DMAP и 423 г (2,2 ммоль) WSCI·HCl/2 мл дихлорметана в указанном порядке при охлаждении льдом. После доведения температуры реакционной смеси обратно до комнатной температуры и перемешивания в течение ночи добавляли этилацетат с последующим выделением вещества промыванием дважды 5% лимонной кислотой, водой, дважды 5% гидрокарбонатом натрия, водой и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия этилацетат выпаривали при пониженном давлении с получением 665 мг указанного в заголовке соединения (выход 91%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=0,88 (6H, д, -CH(CH 3)2), 1,44 (9H, с, Boc), 1,49 (3H, д, -OCOCH(CH 3)-), 1,75 (2H, м, -NHCH2 CH 2CH2O-), 1,85 (1H, м, -CH2 CH(CH3)2), 2,45 (2H, д, -CH 2CH(CH3)2), 3,05 (2H, м, -NHCH 2CH2CH2O-), 3,69 (1H, кв, -OCOCH(CH3)-), 4,13 (2H, т, -NHCH2CH2 CH 2O-), 4,63 (1H, ушир., -NHCH2-), 7,07-7,21 (4H, м, ароматический H).
2) Синтез гидрохлорида аминопропанолибупрофена
В 1 мл дихлорметана растворяли 636 мг (1,75 ммоль) полученного выше Boc-аминопропанолибупрофена и добавляли 4 мл 4М раствора хлористый водород/этилацетат при охлаждении льдом с последующим перемешиванием при охлаждении льдом в течение 10 минут и при комнатной температуре в течение 3 часов. После подтверждения исчезновения Boc-аминопропанолибупрофена при помощи ТСХ добавляли диэтиловый эфир с последующей декантацией три раза. После этого осадок сушили при пониженном давлении с получением указанного в заголовке соединения с выходом 406 мг (77%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=0,89 (6H, д, -CH(CH 3)2), 147 (3H, д, -OCOCH(CH 3)-), 1,83 (1H, м, -CH2 CH(CH3)2), 2,08 (2H, квант, -NHCH2 CH 2CH2O-), 2,44 (2H, д, -CH 2CH(CH3)2), 3,01 (2H, т, -NHCH 2CH2CH2O-), 3,71 (1H, кв, -OCOCH(CH3)-), 4,11-4,27 (2H, м, -NHCH2CH2 CH 2O-), 7,06-7,20 (4H, м, ароматический H), 8,25 (ушир., H 3 N +CH2-).
Пример 7
Синтез гиалуроната натрия, содержащего аминопропанолибупрофен
В смеси 11,5 мл воды/11,5 мл диоксана растворяли 100 мг (0,25 ммоль/дисахаридная единица) гиалуроната натрия, имеющего средневзвешенную молекулярную массу 900000, и затем добавляли HOSu (0,2 ммоль)/0,1 мл воды, WSCI·HCl (0,1 ммоль)/0,1 мл воды и гидрохлорид аминопропанолибупрофена (0,1 ммоль)/0,3 мл воды, полученный в примере 6, в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 1,5 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 3 часов и 35 минут. После нейтрализации реакционного раствора добавлением 43 мкл 50% уксусной кислоты добавляли 500 мг хлорида натрия с последующим перемешиванием. Смесь осаждали добавлением 50 мл этанола, осадок дважды промывали 80% этанолом, дважды этанолом и диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 93 мг белого твердого вещества. Степень замещения ибупрофеном составляла 16,4% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 8
Синтез гидрохлорида аминопропанолфлурбипрофена
1) Синтез Boc-аминопропанолфлурбипрофена
В 2 мл дихлорметана растворяли 352 мг (2 ммоль) Boc-аминопропанола и 489 г (2 ммоль) флурбипрофена (производимого Wako Pure Chemical Industries) и добавляли 48 мг (0,4 ммоль) DMAP и 423 г (2,2 ммоль) WSCI·HCl/2 мл дихлорметана в указанном порядке при охлаждении льдом. После доведения температуры обратно до комнатной температуры и перемешивания в течение ночи добавляли этилацетат с последующим выделением вещества промыванием дважды 5% лимонной кислотой, водой, дважды 5% гидрокарбонатом натрия, водой и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия этилацетат выпаривали при пониженном давлении с получением 753 мг указанного в заголовке соединения (выход 94%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,26 (9H, с, Boc), 1,54 (3H, д, -OCOCH(CH 3)-), 1,80 (2H, квант, -NHCH2 CH 2CH2O-), 3,13 (2H, м, -NHCH 2CH2CH2O-), 3,76 (1H, кв, -OCOCH(CH3)-), 4,15 (2H, м, -NHCH2CH2 CH 2O-), 4,66 (1H, ушир., -NHCH2-), 7,10-7,55 (9H, м, ароматический H).
2) Синтез гидрохлорида аминопропанолфлурбипрофена
В 1 мл дихлорметана растворяли 720 мг (1,79 ммоль) полученного выше Boc-аминопропанолфлурбипрофена и добавляли 4 мл 4М раствора хлористый водород/этилацетат при охлаждении льдом с последующим перемешиванием при охлаждении льдом в течение 3 минут и при комнатной температуре в течение 3 часов и 10 минут. После подтверждения исчезновения Boc-аминопропанолфлурбипрофена при помощи ТСХ добавляли диэтиловый эфир с последующей декантацией два раза. После этого осадок сушили при пониженном давлении с получением указанного в заголовке соединения с выходом 352 мг (94%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,51 (3H, д, -OCOCH(CH 3)-), 2,10 (2H, квант, -NHCH2 CH 2CH2O-), 3,05 (2H, т, -NHCH 2CH2CH2O-), 3,76 (1H, кв, -OCOCH(CH3)-), 4,13-4,29 (2H, м, -NHCH2CH2 CH 2O-), 7,07-7,53 (9H, м, ароматический H), 8,27 (ушир., H 3 N +CH2-).
Пример 9
Синтез гиалуроната натрия, содержащего аминопропанолфлурбипрофен
В смеси 11,5 мл воды/11,5 мл диоксана растворяли 100 мг (0,25 ммоль/дисахаридная единица) гиалуроната натрия, имеющего средневзвешенную молекулярную массу 900000, и затем добавляли HOSu (0,2 ммоль)/0,1 мл воды, WSCI·HCl (0,1 ммоль)/0,1 мл воды и гидрохлорид аминопропанолфлурбипрофена (0,1 ммоль)/0,3 мл воды, полученный в примере 8, в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 1,5 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 3 часов и 35 минут. После нейтрализации реакционного раствора добавлением 43 мкл 50% уксусной кислоты добавляли 500 мг хлорида натрия с последующим перемешиванием. Смесь осаждали добавлением 50 мл этанола, осадок дважды промывали 80% этанолом, дважды этанолом и диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 94 мг белого твердого вещества. Степень замещения флурбипрофеном составляла 21,1% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 10
Синтез гидрохлорида аминопропанолацетилсалициловой кислоты
1) Синтез Boc-аминопропанолацетилсалициловой кислоты
Boc-аминопропанол (2,11 ммоль), ацетилсалициловую кислоту (2,11 ммоль) (производимую Wako Pure Chemical Industries) и DMAP (0,42 ммоль) растворяли в дихлорметане и диоксане (2:1, 6 мл) и добавляли WSCI·HCl (2,35 ммоль) при охлаждении льдом. После доведения температуры обратно до комнатной температуры и перемешивания в течение ночи добавляли этилацетат с последующим выделением вещества промыванием 5% лимонной кислотой, 5% гидрокарбонатом натрия и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия этилацетат выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=3:1, 0,5% триэтиламин) с получением указанного в заголовке соединения (298,0 мг, выход 48%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,44 (9H, с, Boc), l,90-1,96 (2H, м, -BocHNCH2 CH 2CH2O-), 2,35 (3H, с, -COCH 3), 3,24-3,28 (2H, м, BocHNCH 2CH2CH2O-), 4,35 (2H, т, BocHNCH2CH2 CH 2O-), 4,78 (1H, с, NH), 7,11 (1H, дд, ароматический), 7,32 (1H, тд, ароматический), 7,55-7,59 (1H, м, ароматический), 8,01 (1H, дд, ароматический).
2) Синтез гидрохлорида аминопропанолацетилсалициловой кислоты
Полученную выше Boc-аминопропанолацетилсалициловую кислоту растворяли в дихлорметане (1 мл) и добавляли 4н. раствор хлористый водород/этилацетат (3 мл) при охлаждении льдом с последующим перемешиванием в течение 2 часов. После подтверждения исчезновения Boc-аминопропанолацетилсалициловой кислоты при помощи ТСХ добавляли диэтиловый эфир. Образовавшийся таким образом осадок центрифугировали и надосадочную жидкость подвергали декантации. Полученный таким образом осадок сушили при пониженном давлении с получением 213,9 мг (выход 96%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=2,22 (2H, т, -H2NCH2 CH 2CH2O-), 2,35 (3H, с, -COCH 3), 3,13 (2H, т, -H2NCH 2CH2CH2O-), 4,41 (2H, т, -H2NCH2CH2 CH 2O-), 7,09 (1H, дд, ароматический), 7,31 (1H, дт, ароматический), 7,56 (1H, дт, ароматический), 7,99 (1H, дд, ароматический).
Пример 11
Синтез гиалуроната натрия, содержащего аминопропанолацетилсалициловую кислоту
Гиалуровую кислоту (100 мг) 0,25 ммоль/дисахаридная единица, имеющую средневзвешенную молекулярную массу 900000, растворяли в смеси вода-диоксан (1:1) и добавляли HOSu в концентрации 2 моль/л (0,1 мл), WSCI·HCl в концентрации 1 моль/л (0,1 мл) и полученный выше в примере 10 раствор гидрохлорида аминопропанолацетилсалициловой кислоты в смеси вода-диоксан (1:1) (2 мл) в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 5% водный раствор гидрокарбоната натрия (1,5 мл) с последующим перемешиванием в течение 3 часов. После нейтрализации реакционного раствора добавлением 50% уксусной кислоты (43 мкл) добавляли хлорид натрия (0,4 г) с последующим перемешиванием. Смесь осаждали добавлением этанола (100 мл), осадок промывали 80% этанолом, этанолом и диэтиловым эфиром, два раза для каждого вещества, последовательно. После этого осадок сушили при пониженном давлении с получением указанного в заголовке соединения (97,7 мг). Степень замещения ацетилсалициловой кислотой составляла 13,5% при определении абсорбциометрическим способом. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 12
Синтез гидрохлорида аминопропанолфелбинака
1) Синтез Boc-аминопропанолфелбинака
Boc-аминопропанол (2,04 ммоль), фелбинак (2,04 ммоль) (производимый Aldrich Chem. Co.) и DMAP (0,41 ммоль) растворяли в диоксане (7 мл) и затем добавляли раствор WSCI·HCl (2,35 ммоль) в смеси диоксан-дихлорметан (3:4) при охлаждении льдом. Реакционный раствор осветляли добавлением диметилформамида (ДМФА) (3 мл) и затем доводили его температуру обратно до комнатной температуры с последующим перемешиванием в течение ночи. Добавляли этилацетат с последующим выделением вещества промыванием 5% водным раствором лимонной кислоты, 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия растворитель выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=3:1, 0,5% триэтиламин), с получением указанного в заголовке соединения (623,0 мг, выход 83%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,44 (9H, с, Boc), 1,80-1,85 (2H, м, BocHNCH2 CH 2CH2O-), 3,15-3,19 (2H, м, BocHNCH 2CH2CH2O-), 3,67 (2H, с, PhCH 2), 4,18 (2H, т, BocHNCH2CH2 CH 2O-), 4,67 (1H, с, NH), 7,34-7,59 (9H, м, ароматический).
2) Синтез гидрохлорида аминопропанолфелбинака
Полученный выше Boc-аминопропанолфелбинак (1,69 ммоль) растворяли в дихлорметане (1 мл) и добавляли 4н. раствор хлористый водород/этилацетат (3 мл) при охлаждении льдом. Температуру смеси доводили обратно до комнатной температуры с последующим перемешиванием в течение 2 часов. После подтверждения исчезновения Boc-аминопропанолфелбинака при помощи ТСХ добавляли диэтиловый эфир и образовавшийся таким образом осадок центрифугировали. Полученный таким образом осадок три раза подвергали декантации диэтиловым эфиром и затем сушили при пониженном давлении с получением указанного в заголовке соединения (511,7 мг, выход 99%). Структуру определяли 1Н-ЯМР (CDCl3: CD3OD=1:1).
1H-ЯМР (500 МГц, CDCl3:CD3OD=1:1) δ (м.д.)=1,98-2,04 (2H, м, -H2NCH2 CH 2CH2O-), 2,95 (2H, т, -H2NCH 2CH2CH2O-), 3,73 (2H, с, -PhCH 2-), 4,23 (2H, т, -H2NCH2CH2 CH 2O-), 7,33-7,59 (9H, м, ароматический).
Пример 13
Синтез гиалуроновой кислоты, содержащей аминопропанолфелбинак
Гиалуроновую кислоту (200 мг) 0,5 ммоль/дисахаридная единица, имеющую средневзвешенную молекулярную массу 900000, растворяли в смеси вода-диоксан (1:1, 45 мл) и добавляли HOSu в концентрации 2 моль/л (0,1 мл), WSCI·HCl в концентрации 1 моль/л ((0,1 мл) и 0,5М водный раствор гидрохлорида пропаноламинфелбинака, полученный в примере 12, (0,5 мл) в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 5% водный раствор гидрокарбоната натрия (3 мл) с последующим перемешиванием в течение 3 часов. После нейтрализации реакционного раствора добавлением 50% водного раствора уксусной кислоты (86 мкл) добавляли хлорид натрия (0,8 г), с последующим перемешиванием. Добавляли этанол (200 мл) с последующим перемешиванием. Образовавшийся таким образом осадок центрифугировали и полученный осадок промывали 80% водным раствором этанола, этанолом и диэтиловым эфиром, по два раза для каждого вещества, последовательно. Осадок сушили при комнатной температуре при пониженном давлении с получением указанного в заголовке соединения (205,1 мг). Степень замещения фелбинаком составляла 27,8% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 14
Синтез гидрохлорида аминопропанолфенбуфена
1) Синтез Boc-аминопропанолфенбуфена
Boc-аминопропанол (2,18 ммоль), фенбуфен (2,18 ммоль) (производимый ICN Biochemical Inc.) и DMAP (0,44 ммоль) растворяли в смеси ДМФА-дихлорметан (5:3, 8 мл) и затем добавляли дихлорметановый раствор (5 мл) WSCI·HCl (2,48 ммоль) при охлаждении льдом. После постепенного доведения температуры реакционной смеси обратно до комнатной температуры смесь перемешивали в течение ночи. Добавляли этилацетат с последующим выделением вещества промыванием 5% водным раствором лимонной кислоты, 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия растворитель выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем (хлороформ:этилацетат=40:1, 0,5% триэтиламин) с получением указанного в заголовке соединения (747,8 мг, выход 83%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,44 (9H, с, Boc), 1,82-1,87 (2H, м, BocHNCH2 CH 2CH2O-), 2,79 (2H, т, -COC 2 H 4CO-), 3,20-3,24 (2H, м, BocHNCH 2CH2CH2O-), 3,36 (2H, т, -COC 2 H 4CO-) 4,19 (2H, т, BocHNCH2CH2 CH 2O-), 4,76 (1H, с, NH), 7,39-7,64 (5H, м, ароматический), 7,70 (2H, тд, ароматический), 8,06 (2H, тд, ароматический).
2) Синтез гидрохлорида аминопропанолфенбуфена
Полученный выше Boc-аминопропанолфенбуфен (1,82 ммоль) растворяли в дихлорметане (4 мл), при охлаждении льдом добавляли 4н. раствор хлористый водород/этилацетат (4 мл), затем температуру смеси постепенно доводили обратно до комнатной температуры и перемешивали в течение 90 минут. Выпадение белого осадка наблюдали сразу же после начала реакции. После подтверждения исчезновения Boc-аминопропанолфенбуфена при помощи ТСХ к реакционному раствору добавляли диэтиловый эфир и полученный белый осадок центрифугировали. Осадок три раза промывали диэтиловым эфиром и затем сушили при пониженном давлении с получением указанного в заголовке соединения (621,4 мг, выход 98%). Структуру определяли 1Н-ЯМР (CDCl3:CD3OD=1:1).
1H-ЯМР (500 МГц, CDCl3:CD3OD=1:1) δ (м.д.)=2,01-2,07 (2H, м, -H2NCH2 CH 2CH2O-), 2,79 (2H, т, -COC 2 H 4CO-), 3,05 (2H, т, -H2NCH 2CH2CH2O-), 3,44 (2H, т, -COC 2 H 4CO-), 4,26 (2H, т, -H2NCH2CH2 CH 2O-), 7,41-7,50 (3H, м, ароматический), 7,66 (дд, 2H, ароматический), 7,75 (d, 2H, ароматический), 8,08 (d, 2H, ароматический).
Пример 15
Синтез гиалуроновой кислоты, содержащей аминопропанолфенбуфен
Гиалуроновую кислоту (200 мг) 0,5 ммоль/дисахаридная единица, имеющую средневзвешенную молекулярную массу 900000, растворяли в смеси вода-диоксан (1:1, 45 мл) и затем добавляли HOSu в концентрации 2 моль/л (0,25 мл), WSCI·HCl в концентрации 1 моль/л (0,25 мл) и раствор (0,66 мл) гидрохлорида аминопропанолфенбуфена, полученного в примере 14, в смеси вода-диоксан (25:8) с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 5% водный раствор гидрокарбоната натрия (3 мл) с последующим перемешиванием в течение 3 часов. После нейтрализации реакционного раствора добавлением 50% водного раствора уксусной кислоты (86 мкл) добавляли хлорид натрия (0,8 г) с последующим перемешиванием. Добавляли этанол (200 мл) с последующим перемешиванием. Образовавшийся таким образом осадок центрифугировали и полученный таким образом осадок промывали 80% водным раствором этанола, этанолом и диэтиловым эфиром, по два раза каждым. Осадок сушили при пониженном давлении с получением указанного в заголовке соединения (214,1 мг). Степень замещения фенбуфеном составляла 23,8% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 16
Синтез гидрохлорида аминопропанолмефенамовой кислоты
1) Синтез Boc-аминопропанолмефенамовой кислоты
Boc-аминопропанол (0,616 ммоль), мефенамовую кислоту (0,620 ммоль) (производимую Wako Pure Chemical Industries) и DMAP (0,126 ммоль) растворяли в дихлорметане (3 мл) и добавляли дихлорметановый раствор (1,5 мл) WSCI·HCl (0,758 ммоль) при охлаждении льдом. После постепенного доведения температуры реакционной смеси обратно до комнатной температуры смесь перемешивали в течение ночи. Реакционный раствор снова охлаждали на льду и добавляли дихлорметановый раствор (1 мл) WSCI·HCl (0,207 ммоль) с последующим перемешиванием в течение 5 часов до тех пор, пока температура постепенно не становилась обратно комнатной. В реакционный раствор добавляли этилацетат и смесь промывали 5% водным раствором лимонной кислоты, 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия растворитель выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=6:1, 0,5% триэтиламин) с получением указанного в заголовке соединения (190,4 мг, выход 78%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,45 (9H, с, Boc), 1,96-2,01 (2H, м, BocHNCH2 CH 2CH2O-), 2,18 (3H, с, PhCH 3), 2,33 (3H, с, PhCH 3), 3,31-3,32 (2H, м, BocHNCH 2CH2CH2O-), 4,38 (2H, т, BocHNCH2CH2 CH 2O-), 4,78 (1H, с, NH), 6,64-6,67 (1H, м, ароматический), 6,74 (1H, дд, ароматический), 7,02-7,26 (4H, м, ароматический), 7,94 (1H, дд, ароматический), 9,24 (1H, с, -PhNHPh-).
2) Синтез гидрохлорида аминопропанолмефенамовой кислоты
Полученную выше Boc-аминопропанолмефенамовую кислоту (0,462 ммоль) растворяли в дихлорметане (0,5 мл) и при охлаждении льдом добавляли 4н. раствор хлористый водород/этилацетат (1,5 мл) с последующим перемешиванием в течение 3 часов. После подтверждения исчезновения Boc-аминопропанолмефенамовой кислоты при помощи ТСХ к реакционному раствору добавляли диэтиловый эфир и образовавшийся таким образом осадок центрифугировали. Полученный таким образом осадок промывали диэтиловым эфиром и затем сушили при пониженном давлении с получением указанного в заголовке соединения (154,4 мг, количественный). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=2,16 (3H, с, PhCH 3), 2,25-2,30 (2H, м, -H2NCH2 CH 2CH2O-), 2,31 (3H, с, PhCH 3), 3,20 (2H, т, -H2NCH 2CH2CH2O-), 4,44 (2H, т, -H2NCH2CH2 CH 2O-), 6,63-6,66 (1H, м, ароматический), 6,70-6,72 (1H, дд, ароматический), 7,02 (1H, д, ароматический), 7,09 (1H, т, ароматический), 7,14 (1H, д, ароматический), 7,22-7,25 (1H, м, ароматический), 7,92 1H, дд, ароматический), 9,17 (1H, с, ароматический).
Пример 17
Синтез гиалуроновой кислоты, содержащей аминопропанолмефенамовую кислоту
Гиалуроновую кислоту (100 мг) 0,25 ммоль/дисахаридная единица, имеющую средневзвешенную молекулярную массу 900000, растворяли в смеси вода-диоксан (1:1, 22,5 мл) и затем добавляли HOSu в концентрации 2 моль/л (0,25 мл), WSCI·HCl в концентрации 1 моль/л (0,25 мл) и раствор (2 мл) гидрохлорида аминопропанолмефенамовой кислоты (0,10 ммоль), полученного в примере 16, в смеси вода-диоксан (1:1) в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 5% водный раствор гидрокарбоната натрия (1,5 мл) с последующим перемешиванием в течение 4 часов. После нейтрализации добавлением 50% водного раствора уксусной кислоты (43 мкл) добавляли хлорид натрия (0,4 г) с последующим перемешиванием. Этанол (100 мл) добавляли туда с последующим перемешиванием и образовавшийся таким образом осадок центрифугировали. Полученный таким образом осадок промывали 80% водным раствором этанола, этанолом и диэтиловым эфиром, по два раза каждым, последовательно. Осадок сушили при пониженном давлении с получением указанного в заголовке соединения (101,7 мг). Степень замещения мефенамовой кислотой составляла 17,5% при измерении абсорбциометрическим способом. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 18
Синтез гидрохлорида аминопропанолдиклофенака
1) Синтез Boc-аминопропанолдиклофенака
В 1 мл дихлорметана растворяли 135,8 мг (0,775 ммоль) Boc-аминопропанола, добавляли 4 мл дихлорметанового раствора 229,6 мг (0,775 ммоль) диклофенака (производимого Wako Pure Chemical Industries), 1 мл дихлорметанового раствора 18,9 мг (0,155 ммоль) DMAP и 0,5 мл ДМФА в указанном порядке и добавляли 2 мл дихлорметанового раствора 191,4 мг (0,998 ммоль) WSCI·HCl при охлаждении льдом с последующим перемешиванием в течение 7 часов, в то время как температура постепенно доходила обратно до комнатной температуры. Реакционный раствор снова охлаждали на льду и в качестве дополнительной операции добавляли 1 мл дихлорметанового раствора 91,9 мг (0,310 ммоль) диклофенака, 7,5 мг (0,061 ммоль) DMAP и 1 мл дихлорметанового раствора 70,9 мг (0,370 ммоль) WSCI·HCl в указанном порядке с последующим перемешиванием, в то время как температура постепенно доходила обратно до комнатной температуры. Данную дополнительную операцию проводили 5 раз. Добавляли этилацетат с последующим выделением вещества промыванием дважды 5% водным раствором лимонной кислоты, дважды 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно. После обезвоживания сульфатом натрия этилацетат выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем с получением 282,2 мг (80%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,44 (9H, с, Boc), 1,85 (2H, квант, -NHCH2CH2CH2O-), 3,16 (2H, кв, -NHCH 2CH2CH2O-), 3,82 (2H, с, Ph-CH 2-CO), 4,22 (2H, т, -NHCH2CH2 CH 2O-), 4,68 (1H, с, NH), 6,54-7,35 (8H, м, ароматический H, NH).
2) Синтез гидрохлорида аминопропанолдиклофенака
В 2 мл дихлорметана растворяли 1019 мг полученного выше Boc-аминопропанолдиклофенака и при охлаждении льдом добавляли 4М раствор хлористый водород/этилацетат в количестве 8 мл с последующим перемешиванием в течение 3 часов. После добавления 150 мл диэтилового эфира для осаждения осадок сушили при пониженном давлении. Указанное в заголовке соединение получали в количестве 791 мг (90%). Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=2,13 (2H, квант, -NHCH2 CH 2CH2O-), 3,08 (2H, т, -NHCH 2CH2CH2O-), 3,84 (2H, с, Ph-CH 2-CO), 4,25 (2H, т, -NHCH2CH2 CH 2O-), 6,52-7,33 (8H, м, ароматический H, NH).
Пример 19
Синтез гиалуроната натрия, содержащего аминопропанолдиклофенак
В смеси 56,3 мл воды/56,3 мл диоксана растворяли 500 мг (1,25 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 800000, и затем добавляли HOSu (1 ммоль)/0,5 мл воды, WSCI·HCl (0,5 ммоль)/0,5 мл воды и 0,5 ммоль/(вода:диоксан=1:1, 5 мл) гидрохлорида аминопропанолдиклофенака, полученного в примере 18, туда в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 7,5 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 3 часов и 40 минут. После нейтрализации реакционного раствора добавлением 215 мкл 50% водного раствора уксусной кислоты добавляли 2,5 г хлорида натрия с последующим перемешиванием. Смесь осаждали добавлением 400 мл этанола, осадок промывали дважды 85% водным раствором этанола, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 541 мг белого твердого вещества. Степень замещения диклофенаком составляла 18,2% при измерении при помощи спектрофотометра.
Пример 20
Синтез гидрохлорида аминопропанолэтодолака
1) Синтез Boc-аминопропанолэтодолака
В 4 мл дихлорметана растворяли 178,8 мг (1,02 ммоль) Boc-аминопропанола, 293,8 (1,02 ммоль) этодолака (производимого Wako Pure Chemical Industries) и 23,8 мг (0,20 ммоль) DMAP и добавляли 2 мл дихлорметанового раствора 233,8 мг (1,22 ммоль) WSCI·HCl при охлаждении льдом с последующим перемешиванием в течение ночи, в то время как температура постепенно повышалась до комнатной температуры. Далее при охлаждении на льду добавляли 2 мл дихлорметанового раствора 68,8 мг (0,36 ммоль) WSCI·HCl с последующим перемешиванием в течение 80 минут, в то время как температура постепенно повышалась обратно до комнатной температуры. Добавляли этилацетат с последующим выделением вещества промыванием дважды 5% водным раствором лимонной кислоты, дважды 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия, этилацетат выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем с получением 436,3 мг (96%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=0,83 (3H, т, -CH2 CH 3), 137 (3H, т, -CH2 CH 3), 1,43 (9H, с, Boc), 1,79 (2H, квант, -NHCH2 CH 2CH2O-), 3,14 (2H, кв, -NHCH 2CH2CH2O-), 4,10-4,22 (2H, м, -NHCH2CH2 CH 2O-), 4,63 (1H, с, NH), 7,00-7,37 (3H, м, ароматический H), 8,97 (1H, с, NH).
2) Синтез гидрохлорида аминопропанолэтодолака
В 1 мл дихлорметана растворяли 421,5 мг (0,948 ммоль) полученного выше Boc-аминопропанолэтодолака и при охлаждении льдом добавляли 4М раствор хлористый водород/этилацетат в количестве 3 мл с последующим перемешиванием в течение 3 часов. Диэтиловый эфир и гексан добавляли для осаждения и осадок сушили при пониженном давлении. Осадок очищали хроматографией на колонке с силикагелем с получением 197,6 мг (55%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=0,81 (3H, т, -CH2 CH 3), 1,35 (3H, т, -CH2 CH 3), 1,92-2,17 (4H, м, -CH2 CH 3, -NHCH2 CH 2CH2O-), 4,12 (1H, квант, -NHCH2CH2 CH 2O-), 4,20 (1H, квант, -NHCH2CH2 CH 2O-), 6,99-7,35 (3H, м, ароматический H), 8,99 (1H, с, NH).
Пример 21
Синтез гиалуроната натрия, содержащего аминопропанолэтодолак
В смеси 12,8 мл воды/12,8 мл диоксана растворяли 114 мг (0,285 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 800000, и затем добавляли 0,228 ммоль HOSu/0,1 мл воды, 0,114 ммоль WSCI·HCl/0,1 мл воды и 0,114 ммоль/(2 мл вода:диоксан=1:1) гидрохлорида аминопропанолэтодолака, полученного в примере 20, в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 1,71 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 4,5 часов. После нейтрализации реакционного раствора добавлением 49 мкл 50% уксусной кислоты добавляли 456 мг хлорида натрия с последующим перемешиванием. Смесь осаждали добавлением 110 мл этанола, осадок промывали дважды 80% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 111 мг белого твердого вещества. Степень замещения этодолаком составляла 14,4% согласно анализу ВЭЖХ.
Пример 22
Синтез гидрохлорида аминопропанолактарита
1) Синтез Boc-аминопропанолактарита
В 2 мл дихлорметана растворяли 123,1 мг (0,703 ммоль) Boc-аминопропанола, полученного в справочном примере 1, затем добавляли раствор в ДМФА (1 мл) 136,0 мг (0,704 ммоль) актарита и добавляли 17,1 мг (0,140 ммоль) DMAP и 175,4 мг (0,915 ммоль) WSCI·HCl в указанном порядке при охлаждении льдом с последующим перемешиванием в течение ночи, в то время как температура постепенно повышалась обратно до комнатной температуры. Добавляли этилацетат с последующим выделением вещества промыванием 5% водным раствором лимонной кислоты, 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия этилацетат выпаривали при пониженном давлении. Осадок очищали хроматографией на колонке с силикагелем с получением 203,1 мг (83%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,44 (9H, с, Boc), 1,80 (2H, квант, -NHCH2 CH 2CH2O-), 2,18 (3H, с, NAc), 3,14 (2H, кв, -NHCH 2CH2CH2O-), 3,59 (2H, с, Ph-CH 2-CO), 4,15 (2H, т, -NHCH2CH2 CH 2O-), 4,66 (1H, с, NH), 7,13 (1H, с, NH), 7,23 (2H, д, ароматический H), 7,46 (2H, д, ароматический H).
В связи с этим актарит получали следующим способом синтеза.
п-Аминофенилуксусную кислоту (1,02 ммоль) (производимую Wako Pure Chemical Industries) растворяли в смеси дихлорметан:метанол:вода (1:3:1, 50 мл) и добавляли уксусный ангидрид при охлаждении на льду с последующим перемешиванием в течение ночи, в то время как температура постепенно повышалась обратно до комнатной температуры. Растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (196,4 мг, выход 99%). Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CD3OD) δ (м.д.)=2,11 (3H, с, Ac), 3,55 (2H, с, Ph-CH 2-), 7,21-7,49 (4H, м, ароматический H).
2) Синтез гидрохлорида аминопропанолактарита
В 2 мл дихлорметана растворяли 201,3 мг (0,574 ммоль) полученного выше Boc-аминопропанолактарита и при охлаждении льдом добавляли 3 мл 4М раствора хлористый водород/этилацетат с последующим перемешиванием в течение 3 часов. Диэтиловый эфир добавляли для осаждения, осадок промывали дважды диэтиловым эфиром и затем сушили при пониженном давлении с получением 161,3 мг (98%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CD3OD) δ (м.д.)=1,94-1,99 (2H, м, -NHCH2 CH 2CH2O-), 2,11 (3H, с, NAc), 2,94 (2H, т, -NHCH 2CH2CH2O-), 3,63 (2H, с, Ph-CH 2-CO), 4,19 (2H, т, -NHCH2CH2 CH 2O-), 7,22-7,51 (4H, м, ароматический H).
Пример 23
Синтез гиалуроната натрия, содержащего аминопропанолактарит
В смеси 11,25 мл воды/11,25 мл диоксана растворяли 110 мг (0,25 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 800000, и затем добавляли HOSu (0,2 ммоль)/0,1 мл воды, WSCI·HCl (0,1 ммоль)/0,1 мл воды и гидрохлорида аминопропанолактарита, полученного в примере 22 (0,1 ммоль)/(вода:диоксан=1:1, 2 мл), в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 1,5 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 3 часов. После нейтрализации реакционного раствора добавлением 43 мкл 50% уксусной кислоты добавляли 400 мг хлорида натрия с последующим перемешиванием. Смесь осаждали добавлением 100 мл этанола, осадок промывали дважды 80% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 97 мг белого твердого вещества. Степень замещения актаритом составляла 13,2% согласно анализу ВЭЖХ.
Пример 24
Синтез гиалуроната натрия, содержащего аминопропанолкетопрофен
В смеси 43 мл воды/43 мл диоксана растворяли 200 мг (0,5 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 900000, и затем добавляли водный раствор HOSu в количестве 0,3 ммоль/2 мл воды, водный раствор WSCI·HCl в количестве 0,15 ммоль/2 мл воды и водный раствор гидрохлорида аминопропанолкетопрофена, полученного в примере 1, в количестве 1,5 ммоль/2 мл воды в указанном порядке с последующим перемешиванием в течение ночи. После того как собирали 11,5 мл реакционного раствора, добавляли 100 мг гидрокарбоната натрия с последующим перемешиванием. Смесь осаждали, добавляя 50 мл этанола, осадок дважды промывали 80% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 35 мг белого порошка. Степень замещения кетопрофеном составляла 7,2% согласно анализу ВЭЖХ.
Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «С».
Справочный пример 2
Синтез Вос-серинола
Серинол (10,1 ммоль) (производимый Aldrich Chem. Co.) растворяли в смеси вода-диоксан (1:1, 20 мл) и затем добавляли диоксановый раствор (15 мл) Вос2О (10,8 ммоль) при охлаждении льдом с последующим перемешиванием в течение ночи, в то время как температура постепенно повышалась обратно до комнатной температуры. Растворитель выпаривали при пониженном давлении. Осадок промывали гексаном и затем сушили при пониженном давлении с получением указанного в заголовке соединения (1847 мг, выход 95%). Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CD3OD) δ (м.д.)=1,44 (9H, с, Boc), 3,57-3,58 (5H, м, серинол).
Пример 25
Синтез гидрохлорида серинолкетопрофена
1) Синтез Вос-серинолкетопрофена
Кетопрофен (1,11 ммоль) (производимый Tokyo Kasei Kogyo) растворяли в дихлорметане (3 мл) и добавляли триэтиламин (1,11 ммоль) и дихлорметановый раствор (2 мл) диметилфосфинотиоилхлорида (Mpt-Cl) (1,11 ммоль) в указанном порядке с последующим перемешиванием в течение 25 минут. Дополнительно добавляли триэтиламин (0,36 ммоль) с последующим перемешиванием в течение 20 минут. Реакционный раствор охлаждали на льду и добавляли триэтиламин (1,11 ммоль), DMAP (0,19 ммоль) и Вос-серинол (0,50 ммоль), полученный в справочном примере 2, в указанном порядке с последующим перемешиванием в течение ночи при комнатной температуре. Реакционный раствор основа охлаждали на льду и добавляли 25% водный аммиак (2 мл) и диоксан (10 мл) в указанном порядке с последующим перемешиванием в течение 20 минут. Реакционный раствор концентрировали до 5 мл и добавляли этилацетат. Вещества выделяли промыванием водой, 5% водным раствором лимонной кислоты, 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно и после обезвоживания сушкой над сульфатом натрия растворитель выпаривали при пониженном давлении. Осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=2:1, 0,5% триэтиламин) с получением указанного в заголовке соединения (287,3 мг, выход 87%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,38-1,40 (9H, м, Boc), 1,51-1,53 (6H, м, -OCOCH(CH 3)-), 3,76-3,81 (2H, м, -OCOCH(CH 3)-), 3,96-4,11 (4H, м, -CH2CH(NHBoc)CH 2-), 4,61 (1H, ушир.тд, -CH2 CH(NHBoc)CH2-), 7,40-7,80 (18H, м, ароматический).
2) Синтез гидрохлорида серинолкетопрофена
Boc-серинолкетопрофен (0,428 ммоль) растворяли в дихлорметане (1 мл) и при охлаждении льдом добавляли 4н. раствор хлористый водород/этилацетат (4 мл) с последующим перемешиванием в течение 2 часов, в то время как температура постепенно повышалась обратно до комнатной температуры. После подтверждения исчезновения Boc-серинолкетопрофена при помощи ТСХ диэтиловый эфир, добавляли гексан и полученный осадок центрифугировали. Полученный таким образом осадок сушили при пониженном давлении с получением указанного в заголовке соединения (243,6 мг, количественный). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,49 (6H, т, -OCOCH(CH 3)-), 3,79 (1H, м, -CH2 CH(NHBoc)CH2-), 4,00-4,53 (6H, м, серинол, кетопрофен), 7,31-7,80 (18H, м, ароматический).
Пример 26
Синтез гиалуроновой кислоты, содержащей серинолкетопрофен
В смеси вода-диоксан (1:1, 22,5 мл) растворяли 0,25 ммоль/дисахаридная единица гиалуроновой кислоты (100 мг), имеющей средневзвешенную молекулярную массу 900000, и затем добавляли HOSu в концентрации 2 моль/л (0,1 мл), WSCI·HCl в концентрации 1 моль/л (0,1 мл) и диоксановый раствор (2 мл) гидрохлорида серинолкетопрофена (0,10 ммоль), полученного в примере 25, с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 5% водный раствор гидрокарбоната натрия (1,5 мл) с последующим перемешиванием в течение 4 часов. Смесь нейтрализовали, добавляя 50% водного раствора уксусной кислоты (43 мкл), и добавляли хлорид натрия (0,4 г) с последующим перемешиванием. Добавляли этанол (100 мл) с последующим перемешиванием и полученный таким образом осадок центрифугировали. Полученный таким образом осадок промывали 80% водным раствором этанола, этанолом и диэтиловым эфиром, последовательно по два раза каждым. Осадок сушили при пониженном давлении с получением указанного в заголовке соединения (92,3 мг). Степень замещения кетопрофеном составляла 11,2% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 27
Синтез гидрохлорида 2-амино-1,5-пентандиолкетопрофена
1) Синтез Вос-амино-1,5-пентандиолкетопрофена
Вос-амино-1,5-пентандиол (Вос-NHCH(CH2OH)CH2CH2CH2OH, производимый Aldrich Chem. Co.) (1,98 ммоль) растворяли в дихлорметане (2 мл) и добавляли дихлорметановый раствор (4 мл) кетопрофена (производимого Tokyo Kasei Kogyo) и дихлорметановый раствор (1 мл) DMAP (0,791 ммоль) в указанном порядке с последующим перемешиванием. Реакционный раствор охлаждали на льду и добавляли дихлорметановый раствор (5 мл) WSCI·HCl (4,93 ммоль) с последующим перемешиванием в течение ночи, в то время как температура повышалась до комнатной температуры. Реакционный раствор разбавляли этилацетатом, промывали 5% водным раствором лимонной кислоты, 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно с последующим обезвоживанием сушкой над сульфатом натрия и затем растворитель выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=5:2, 0,5% триэтиламин), с получением указанного в заголовке соединения (1,361 г, выход 99%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,39-1,40 (9H, м, Boc), 1,51-1,55 (6H, м, -OCOCH(CH 3)-), 3,75-4,55 (8H, м, кетопрофен, 2-амино-l,5-пентандиол), 7,40-7,80 (18H, м, ароматический).
2) Синтез гидрохлорида 2-амино-1,5-пентандиолкетопрофена
Полученный выше Boc-амино-1,5-пентандиолкетопрофен (1,95 ммоль) растворяли в дихлорметане (1 мл) и при охлаждении льдом добавляли 4н. раствор хлористый водород/этилацетат (4 мл) с последующим перемешиванием в течение 3 часов, в то время как температура постепенно повышалась до комнатной температуры. К реакционному раствору добавляли гексан и полученный таким образом осадок центрифугировали. Полученный таким образом осадок сушили при пониженном давлении с получением указанного в заголовке соединения (1,20 г, выход 98%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,50 (3H, д, -OCOCH(CH 3)-), 1,51 (3H, д, -OCOCH(CH 3)-), 3,47 (1H, ушир.д, 2-амино-l,5-пентандиол), 3,44-4,48 (6H, м, кетопрофен, 2-амино-l,5-пентандиол), 7,33-7,84 (18H, м, ароматический).
Пример 28
Синтез гиалуроновой кислоты, содержащей 2-амино-1,5-пентандиолкетопрофен
В смеси вода-диоксан (1:1, 30,8 мл) растворяли 0,34 ммоль/дисахаридная единица гиалуроновой кислоты (137 мг), имеющей средневзвешенную молекулярную массу 900000, и добавляли HOSu в концентрации 2 моль/л (0,137 мл), WSCI·HCl в концентрации 1 моль/л (0,137 мл) и водно-диоксановый (1:1) раствор (4 мл) гидрохлорида 2-амино-1,5-пентандиолкетопрофена (0,137 ммоль), полученного в примере 27, в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 5% водный раствор гидрокарбоната натрия (2,1 мл) с последующим перемешиванием в течение 5 часов. После проведения нейтрализации добавлением 50% водного раствора уксусной кислоты (59 мкл) добавляли хлорид натрия (0,548 г) с последующим перемешиванием. Добавляли этанол (140 мл) с последующим перемешиванием и полученный таким образом осадок центрифугировали.
Полученный таким образом осадок промывали 80% водным раствором этанола, этанолом и диэтиловым эфиром. Осадок сушили при пониженном давлении с получением указанного в заголовке соединения (135,1 мг). Степень замещения кетопрофеном составляла 18,5% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Пример 29
Синтез гидрохлорида 3-амино-1,2-пропандиолкетопрофена
1) Синтез Вос-амино-1,2-пропандиолкетопрофена
Вос-амино-1,2-пропандиол (Вос-NHCH2СН(OH)CH2OH, производимый Aldrich Chem. Co.) (2,05 ммоль) растворяли в дихлорметане (2 мл) и добавляли дихлорметановый раствор (4 мл) кетопрофена (4,11 ммоль) (производимого Tokyo Kasei Kogyo) и дихлорметановый раствор (1 мл) DMAP (0,803 ммоль) в указанном порядке с последующим перемешиванием. Реакционный раствор охлаждали на льду и добавляли дихлорметановый раствор (5 мл) WSCI·HCl (4,94 ммоль) с последующим перемешиванием в течение ночи, в то время как температура повышалась до комнатной температуры. Реакционный раствор охлаждали на льду и добавляли дихлорметановый раствор (1 мл) WSCI·HCl (1,24 ммоль) с последующим перемешиванием при комнатной температуре в течение 1 часа и затем при 35°С в течение 2 часов. Добавляли этилацетат с последующим промыванием 5% водным раствором лимонной кислоты, 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно. Проводили обезвоживание сушкой над сульфатом натрия и затем растворитель выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=2:1, 0,5% триэтиламин) с получением указанного в заголовке соединения (1,175 г, выход 87%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,36-1,40 (9H, м, Boc), 1,42-1,53 (6H, м, -OCOCH(CH 3)-), 3,10-3,30 (2H, м, BocNHCH 2-), 3,65-3,82 (2H, м, -OCOCH(CH3)-), 3,99-4,36 (2H, м, BocNHCH2(CHO-)CH 2O-), 4,49-4,76 (1H, м, BocNH-), 5,04-5,09 (1H, м, BocNHCH2(CHO-)CH2O-), 7,38-7,80 (18H, м, ароматический).
2) Синтез гидрохлорида 3-амино-1,2-пропандиолкетопрофена
Полученный выше Boc-амино-1,2-пропандиолкетопрофен (1,76 ммоль) растворяли в дихлорметане (1 мл) и при охлаждении льдом добавляли 4н. раствор хлористый водород/этилацетат (4 мл) с последующим перемешиванием в течение 3 часов. К реакционному раствору добавляли гексан и полученный таким образом осадок центрифугировали. Полученный таким образом белый осадок сушили при пониженном давлении с получением указанного в заголовке соединения (1,029 г, выход 97%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,33-1,49 (6H, м, -OCOCH(CH 3)-), 3,02-3,20 (м, 2H, H2NCH 2(CHO-)CH2O-), 3,56-3,82 (1H, м, -OCOCH(CH3)-), 3,90-4,15 (2H, м, H2NCH2(CHO-)CH 2O-), -OCOCH(CH3)-), 4,18-4,50 (1H, м, H2NCH2CH(O-)CH 2O-), 5,35-5,37 (1H, м, H2NCH2 CH(O-)CH2O-), 7,30-7,8Q (18H, м, ароматический).
Пример 30
Синтез гиалуроновой кислоты, содержащей 3-амино-1,2-пропандиолкетопрофен
В смеси вода-диоксан (1:1, 45 мл) растворяли 0,5 ммоль/дисахаридная единица гиалуроновой кислоты (200 мг), имеющей средневзвешенную молекулярную массу 900000, и добавляли HOSu в концентрации 2 моль/л (0,25 мл), WSCI·HCl в концентрации 1 моль/л (0,25 мл) и водно-диоксановый (1:1) раствор (4 мл) гидрохлорида 3-амино-1,2-пропандиолкетопрофена (0,20 ммоль), полученного в примере 29, в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 5% водный раствор гидрокарбоната натрия (3 мл) с последующим перемешиванием в течение 4 часов. Смесь нейтрализовали добавлением 50% водного раствора уксусной кислоты (86 мкл), добавляли хлорид натрия (0,8 г) с последующим перемешиванием. Добавляли этанол (200 мл) с последующим перемешиванием и полученный таким образом осадок центрифугировали. Полученный таким образом осадок промывали 80% водным раствором этанола, этанолом и диэтиловым эфиром. Осадок сушили при пониженном давлении с получением указанного в заголовке соединения (217,4 мг). Степень замещения кетопрофеном составляла 40,3% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Справочный пример 3
Синтез Вос-трис(гидроксиметил)аминометана
Трис(гидроксиметил)аминометан (10,1 ммоль) растворяли в смеси вода и добавляли диоксан (1:2, 30 мл) и водно-диоксановый раствор (1:9, 10 мл) Вос2О (10,8 ммоль) с последующим перемешиванием при комнатной температуре в течение 45 минут и затем при 40°С в течение 70 минут. Добавляли диоксановый раствор (3 мл) Вос2О (5,41 ммоль) с последующим перемешиванием в течение ночи, в то время как температура постепенно повышалась до комнатной температуры. Растворитель выпаривали при пониженном давлении. Осадок промывали гексаном и затем сушили при пониженном давлении с получением указанного в заголовке соединения (2,21 г, выход 99%). Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CD3OD) δ (м.д.)=1,44 (9H, с, Boc), 3,68 (6H, с, -C(CH 2OH)3).
Пример 31
Синтез гидрохлорида трис(гидроксиметил)аминометанкетопрофена
1) Синтез Вос-трис(гидроксиметил)аминометанкетопрофена
В 3 мл дихлорметана растворяли 419 мг (1,65 ммоль) кетопрофена (производимого Tokyo Kasei Kogyo) и добавляли 230 мкл (1,65 ммоль) триэтиламина и 213 мг Mpt-Cl (1,65 ммоль)/2 мл дихлорметана в указанном порядке с последующим перемешиванием в течение 10 минут. Далее, 230 мкл (1,65 ммоль) триэтиламина, 33 мг (0,27 ммоль) DMAP и 110 мг (0,5 ммоль) Вос-трис(гидроксиметил)аминометана (Вос-NHC(CH2OH)3), полученного в справочном примере 3, добавляли в указанном порядке с последующим перемешиванием в течение ночи, в то время как температура повышалась до комнатной температуры. После добавления 2 мл водного аммиака добавляли диоксан до тех пор, пока дихлорметан и водный аммиак не становились равномерно распределенными, и смесь перемешивали в течение 40 минут. Дихлорметан выпаривали при пониженном давлении, к осадку добавляли этилацетат с последующим выделением вещества промыванием дважды 5% лимонной кислотой, водой, дважды 5% гидрокарбонатом натрия, водой и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия этилацетат выпаривали при пониженном давлении и осадок очищали хроматографией на колонке с силикагелем (дихлорметан:метанол=100:1→75:1). Указанное в заголовке соединение получали количественно с выходом 467 мг. Структуру определяли 1Н-ЯМР (CDCl3) для подтверждения того, что 3 молекулы кетопрофена присоединено к 1 молекуле Вос-трис(гидроксиметил)аминометана.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,29 (9H, с, Boc), 1,44-1,54 (3H×3, м, -OCOCH(CH 3)-), 3,76 (1H×3, кв, -OCOCH(CH3)-), 4,04-4,27 (6H, м, -NHC(CH 2O-KP)3), 4,81 (1H, ушир., -NH-), 7,37-7,85 (9H×3, м, ароматический H).
2) Синтез гидрохлорида трис(гидроксиметил)аминометанкетопрофена
В 1 мл дихлорметана растворяли 453 мг (0,49 ммоль) полученного выше Boc-трис(гидроксиметил)аминометанкетопрофена и при охлаждении льдом добавляли 3 мл 4М раствора хлористый водород/этилацетат с последующим перемешиванием в течение 30 минут при охлаждении льдом и затем при комнатной температуре в течение 1 часа и 30 минут. После подтверждения исчезновения Boc-трис(гидроксиметил)аминометанкетопрофена при помощи ТСХ диэтиловый эфир и гексан добавляли для декантации. После этого осадок сушили при пониженном давлении с получением указанного в заголовке соединения с выходом 411 мг (97%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,39-1,50 (3H×3, м, -OCOCH(CH 3)-), 3,96 (1H×3, кв, -OCOCH(CH3)-), 4,09-4,46 (6H, м, -NHC(CH 2O-KP)3), 7,25-7,80 (9H×3, м, ароматический), 9,31 (ушир., H 3 N +CH2-).
Пример 32
Синтез гидрохлорида глицинтрис(гидроксиметил)аминометанкетопрофена
1) Синтез Вос-глицинтрис(гидроксиметил)аминометанкетопрофена
В 1 мл хлороформа растворяли 133 мг (0,76 ммоль) Вос-глицина и добавляли 106 мкл (0,76 ммоль) триэтиламина и 98 мг Mpt-Cl (0,76 ммоль)/1 мл хлороформа при охлаждении льдом с последующим перемешиванием в течение 10 минут. После этого постепенно добавляли 433 мг (0,5 ммоль) полученного в примере 31 гидрохлорида трис(гидроксиметил)аминометанкетопрофена/70 мкл (0,5 ммоль) триэтиламина/2 мл хлороформа и 106 мкл (0,76 ммоль) триэтиламина, разделив на 4 части. После перемешивания при комнатной температуре в течение 1 часа дополнительно добавляли 106 мкл (0,76 ммоль) триэтиламина при охлаждении льдом и добавляли смешанный ангидрид кислоты Вос-глицина, активированный растворением 131 мг (0,75 ммоль) Вос-глицина в 1 мл хлороформа и добавлением при охлаждении льдом 105 мкл (0,75 ммоль) триэтиламина и 95 мг (0,75 ммоль) Mpt-Cl/1 мл хлороформа, и смесь перемешивали при комнатной температуре в течение ночи. Добавляли этилацетат с последующим выделением вещества промыванием дважды 5% лимонной кислотой, водой, дважды 5% гидрокарбоната натрия, водой и насыщенным раствором соли последовательно. После обезвоживания сушкой над сульфатом натрия этилацетат выпаривали при пониженном давлении и осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=3:2) с получением 411 мг указанного в заголовке соединения (выход 55%). Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,43 (9H, с, Boc), 1,45-1,52 (3H×3, м, -OCOCH(CH 3)-), 3,56 (2H, ушир., -NHCH 2CO-), 3,76 (1H×3, кв, -OCOCH(CH3)-), 3,98-4,28 (6H, м, -NHC(СH 2O-KP)3), 5,51 (1H, ушир., -NHCH2CO-), 6,63 (1H, ушир., -NHC(CH2O-KP)3), 7,34-7,83 (9H×3, м, ароматический H).
2) Синтез гидрохлорида глицинтрис(гидроксиметил)аминометанкетопрофена
К 361 мг (0,37 ммоль) полученного выше Boc-трис(гидроксиметил)аминометанкетопрофена добавляли 2 мл 4М раствора хлористый водород/этилацетат при охлаждении льдом и перемешивали при комнатной температуре в течение 2 часов. После подтверждения исчезновения Boc-глицин-трис(гидроксиметил)аминометанкетопрофена при помощи ТСХ, добавляли диэтиловый эфир и гексан для декантации. После этого осадок сушили при пониженном давлении с количественным получением указанного в заголовке соединение с выходом 336 мг. Структуру определяли 1Н-ЯМР (CDCl3).
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,40 (3H×3, м, -OCOCH(CH 3)-), 3,68-4,24 (11H, м, 2H; -NHCH 2CO-, 1H×3; -OCOCH(CH3)-, 6H; -NHC(CH 2O-KP)3), 7,27-7,82 (9H×3, м, ароматический H), 8,31 (ушир., H 3 N +CH2-).
Пример 33
Синтез гиалуроновой кислоты, содержащей глицин-трис(гидроксиметил)аминометанкетопрофен
В смеси 11,5 мл воды/11,5 мл диоксана растворяли 100 мг (0,25 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 900000, и добавляли 2 моль/л HOSu/0,1 мл воды, 1 моль/л WSCI·HCl/0,1 мл воды и 93 мг (0,1 ммоль)/3 мл диоксана гидрохлорида глицин-трис(гидроксиметил)аминометанкетопрофена, полученного в примере 32, в указанном порядке с последующим перемешиванием в течение ночи. К реакционному раствору добавляли 5% водный раствор гидрокарбоната натрия с последующим перемешиванием в течение 4 часов и 45 минут. После нейтрализации реакционного раствора добавлением 43 мкл 50% уксусной кислоты добавляли 400 мг хлорида натрия с последующим перемешиванием. Смесь осаждали добавлением 100 мл этанола, осадок промывали дважды 80% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 95 мг белого твердого вещества. Степень замещения кетопрофеном составляла 39% согласно анализу ВЭЖХ. Для приготовления раствора полученное таким образом вещество растворяли в PBS до концентрации 1,0% масс. Раствор представлял собой бесцветную и прозрачную жидкость, и результат при проверке его прохождения через фильтр представлял собой «А».
Справочный пример 4
Синтез Вос-аминопропилбромида
В 20 мл дихлорметана растворяли 1,222 г (5,58 ммоль) гидробромида 3-бромпропиламина, добавляли 0,778 мл (5,58 ммоль) триэтиламина при охлаждении льдом и дополнительно добавляли 50 мл дихлорметанового раствора 1,214 г (5,56 ммоль) Вос2О по каплям через 10 минут с последующим перемешиванием. После перемешивания при комнатной температуре в течение 50 минут добавляли этилацетат с последующим выделением вещества промыванием 5% водным раствором лимонной кислоты, водой и насыщенным раствором соли последовательно. После обезвоживания сульфатом натрия растворитель выпаривали при пониженном давлении с получением 1,304 указанного в заголовке соединения (98%). Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,44 (9H, с, Boc), 2,05 (2H, квант, -NHCH2 CH 2CH2Br), 3,28 (2H, кв, -NHCH 2CH2CH2Br), 3,44 (2H, т, -NHCH2CH2 CH 2Br), 4,64 (1H, с, NH).
Пример 34
Синтез гидрохлорида аминопропанолдиклофенака
1) Синтез Boc-аминопропанолдиклофенака
В 3 мл ДМФА растворяли 1,476 г (4,64 ммоль) диклофенака натрия и добавляли 7 мл раствора в ДМФА 1,105 г (4,64 ммоль) Boc-аминопропилбромида, полученного в справочном примере 4, по каплям при охлаждении льдом с последующим перемешиванием в течение ночи при комнатной температуре, затем перемешиванием при 60°С в течение 10 часов. Смесь перемешивали в течение ночи при комнатной температуре, затем перемешивали при 60°С в течение 9 часов и далее перемешивали при комнатной температуре в течение 3 дней. Добавляли этилацетат с последующим выделением вещества промыванием дважды 5% водным раствором гидрокарбоната натрия, водой и насыщенным раствором соли последовательно. После обезвоживания сульфатом натрия этилацетат выпаривали при пониженном давлении. Полученный таким образом осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=7:1, 0,5% триэтиламин) с получением 1,702 г (81%) указанного в заголовке соединения.
2) Синтез гидрохлорида аминопропанолдиклофенака
В 2 мл дихлорметана растворяли 1019 мг (2,25 ммоль) полученного выше Boc-аминопропанолдиклофенака и при охлаждении льдом добавляли 8 мл 4М раствора хлористый водород/этилацетат с последующим перемешиванием в течение 3 часов. 150 мл диэтилового эфира добавляли для осаждения и осадок сушили при пониженном давлении с получением 791 мг (90%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=2,13 (2H, квант, -NHCH2 CH 2CH2O-), 3,08 (2H, т, -NHCH 2CH2CH2O-), 3,84 (2H, с, Ph-CH 2-CO), 4,25 (2H, т, -NHCH2CH2 CH 2O-), 6,52-7,33 (8H, м, ароматический H, NH).
Пример 35
Синтез гиалуроната натрия, содержащего аминопропанолдиклофенак (DS 4,3%)
В смеси 57,5 мл воды/57,5 мл диоксана растворяли 500 мг (1,25 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 800000, и затем добавляли 0,33М HOSu/0,75 мл воды, 0,16М WSCI·HCl/0,75 мл воды и 0,16М полученного выше в примере 34 гидрохлорида аминопропанолдиклофенака/0,75 мл воды в указанном порядке с последующим перемешиванием в течение ночи. Затем добавляли 375 мг гидрокарбоната натрия/3 мл воды к реакционному раствору с последующим перемешиванием в течение 4 часов. После нейтрализации реакционного раствора добавлением 108 мкл уксусной кислоты добавляли 3,0 г хлорида натрия с последующим перемешиванием. Смесь осаждали, добавляя 200 мл этанола, осадок дважды промывали 80% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 505 мг белого твердого вещества. Степень замещения диклофенаком составляла 4,3% при измерении с использованием спектрофотометра.
Пример 36
Синтез гиалуроната натрия, содержащего аминопропанолдиклофенак (DS 9,7%)
В смеси 57,5 мл воды/57,5 мл диоксана растворяли 500 мг (1,25 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 800000, и затем добавляли 0,5М HOSu/1,0 мл (вода:диоксан=1:1), 0,25М WSCI·HCl/1,0 мл (вода:диоксан=1:1) и 0,25М полученного выше в примере 34 гидрохлорида аминопропанолдиклофенака/1,0 мл (вода:диоксан=1:1) в указанном порядке с последующим перемешиванием в течение ночи. Затем 380 мг гидрокарбоната натрия/5 мл воды добавляли к реакционному раствору с последующим перемешиванием в течение 4 часов. После нейтрализации реакционного раствора добавлением 108 мкл уксусной кислоты добавляли 3,0 г хлорида натрия с последующим перемешиванием. Смесь осаждали, добавляя 200 мл этанола, осадок промывали три раза 80% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 503 мг белого твердого вещества. Степень замещения диклофенаком составляла 9,7% при измерении с использованием спектрофотометра.
Пример 37
Синтез гиалуроната натрия (65 кДа), содержащего аминопропанолдиклофенак (DS 17,1%), используя гиалуроновую кислоту, имеющую среднюю молекулярную массу 65 кДа
В смеси 22,5 мл воды/22,5 мл диоксана растворяли 200,8 мг (0,50 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей среднюю молекулярную массу 65 кДа, и затем добавляли 1М HOSu в количестве 0,4 мл, 0,5М WSCI·HCl в количестве 0,4 мл и 0,1М/2,0 мл (вода:диоксан=1:1) полученного выше в примере 34 гидрохлорида аминопропанолдиклофенака в указанном порядке с последующим перемешиванием в течение ночи. Затем 3 мл 5% водного раствора гидрокарбоната натрия добавляли к реакционному раствору с последующим перемешиванием в течение 3 часов. После нейтрализации реакционного раствора добавлением 86 мкл 50% уксусной кислоты добавляли 1,0 г хлорида натрия с последующим перемешиванием. Смесь осаждали, добавляя 200 мл этанола, осадок промывали дважды 85% этанолом, дважды этанолом и диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 190,5 мг белого твердого вещества. Степень замещения диклофенаком составляла 17,1% при измерении с использованием спектрофотометра.
Справочный пример 5
Синтез Вос-аминоэтилбромида
В 20 мл дихлорметана растворяли 2,155 г (10,5 ммоль) гидробромида 3-бромэтиламина, 1,463 мл (10,5 ммоль) триэтиламина добавляли при охлаждении льдом и далее добавляли 5 мл дихлорметанового раствора 2,299 г (10,5 ммоль) Вос2О с последующим перемешиванием. После перемешивания при комнатной температуре в течение 90 минут добавляли этилацетат с последующим выделением вещества промыванием 5% водным раствором лимонной кислоты, водой и насыщенным раствором соли последовательно. После обезвоживания сульфатом натрия растворитель выпаривали при пониженном давлении с получением 2,287 г указанного в заголовке соединения (97%). Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,45 (9H, с, Boc), 3,45-3,55 (4H, м, -NHCH 2 CH 2Br), 4,93 (1H, с, NH).
Пример 38
Синтез гидрохлорида аминоэтанолдиклофенака
1) Синтез Boc-аминоэтанолдиклофенака
После охлаждения льдом 5 мл раствора ДМФА, содержащего 2,287 г (10,2 ммоль) Boc-аминоэтилбромида, полученного в справочном примере 5, добавляли 6 мл раствора ДМФА, содержащего 3,255 г (10,2 ммоль) диклофенака натрия с последующим перемешиванием при комнатной температуре в течение ночи. Смесь перемешивали при 60°С в течение 11 часов и затем перемешивали при комнатной температуре в течение ночи. Добавляли этилацетат и выделяли вещества промыванием 5% водным раствором гидрокарбоната натрия, водой и насыщенным раствором соли последовательно. После обезвоживания сульфатом натрия этилацетат выпаривали при пониженном давлении. Полученный осадок очищали хроматографией на колонке с силикагелем (толуол:этилацетат=20:1, 0,5% триэтиламин) с получением 2,675 г (60%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,42 (9H, с, Boc), 3,41 (2H, д, -NHCH 2CH2O-), 3,83 (2H, с, Ph-CH 2-CO), 4,21 (2H, т, -NHCH2 CH 2O-), 4,72 (1H, с, NH), 6,54-7,47 (8H, м, ароматический H, NH).
2) Синтез гидрохлорида аминоэтанолдиклофенака
В 5 мл дихлорметана растворяли 2,108 г (4,80 ммоль) полученного выше Boc-аминоэтанолдиклофенака и при охлаждении льдом добавляли 20 мл 4М раствора хлористый водород/этилацетат с последующим перемешиванием в течение 2,5 часов. Добавляли диэтиловый эфир и гексан для осаждения и осадок сушили при пониженном давлении с получением 1,775 г (98%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=3,18 (2H, т, NH2 CH 2CH2O-), 3,94 (2H, с, Ph-CH 2-CO), 4,37 (2H, т, NH2CH2 CH 2O-), 6,47-7,31 (8H, м, ароматический H, NH).
Пример 39
Синтез гиалуроната натрия, содержащего аминоэтанолдиклофенак (DS 14,7%)
В 57,5 мл воды/57,5 мл диоксана растворяли 500 мг (1,25 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 800000, и затем добавляли 2М HOSu в количестве 0,5 мл, 1М WSCI·HCl в количестве 0,5 мл и 3 мл раствора (вода:диоксан=1:1) 188,6 мг (0,5 М) полученного в примере 38 гидрохлорида аминоэтанолдиклофенака в указанном порядке с последующим перемешиванием в течение ночи. Затем добавляли 7,5 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 4 часов. После нейтрализации реакционного раствора добавлением 215 мкл 50% уксусной кислоты добавляли 2,5 г хлорида натрия с последующим перемешиванием. Смесь осаждали, добавляя 500 мл этанола, осадок промывали дважды 85% этанолом, дважды этанолом и дважды диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 473,7 мг белого твердого вещества. Степень замещения диклофенаком составляла 14,7% при измерении с использованием спектрофотометра.
Пример 40
Синтез гидрохлорида диаминопропандиклофенака
1) Boc-пропиламиддиклофенак
В 3 мл дихлорметана растворяли 338,4 мг (1,94 ммоль) трет-бутил-N-(2-аминопропил)карбаминовой кислоты (производимой Tokyo Kasei Kogyo) и 694,4 мг (2,34 ммоль) диклофенака и при охлаждении льдом добавляли 59,0 мг (0,483 ммоль) DMAP и 505,3 мг (2,64 ммоль) WSCI·HCl с последующим перемешиванием в течение 70 минут, затем перемешивали при комнатной температуре в течение 90 минут. Добавляли этилацетат с последующим выделением вещества промыванием 5% водным раствором лимонной кислоты, 5% водным раствором гидрокарбоната натрия и насыщенным раствором соли последовательно. После обезвоживания сульфатом натрия этилацетат выпаривали при пониженном давлении. Полученный осадок очищали хроматографией на колонке с силикагелем (гексан:этилацетат=2:1) с получением 835,5 г (95%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,45 (9H, с, Boc), 1,60 (2H; квант, -NHCH2 CH 2CH2NHBoc), 3,14 (2H, кв, -NHCH2CH2 CH 2NHBOC), 3,31 (2H, кв, -NHCH 2CH2CH2NHBoc), 3,69 (2H, с, Ph-CH 2-CO), 4,93 (1H, с, NH), 6,50-7,60 (9H, м, ароматический H, NH).
2) Гидрохлорид диаминопропандиклофенака
При охлаждении льдом 20 мл 4М раствора хлористый водород/этилацетат добавляли к 1 мл дихлорметанового раствора 825,0 мг (1,82 ммоль) полученного выше Boc-пропиламиддиклофенака с последующим перемешиванием в течение 2 часов. Диэтиловый эфир добавляли для осаждения и осадок сушили при пониженном давлении с получением 714,5 мг (101%) указанного в заголовке соединения. Структуру определяли 1Н-ЯМР.
1H-ЯМР (500 МГц, CDCl3) δ (м.д.)=1,90 (2H, т, -NHCH2 CH 2CH2NH2), -2,99 (2H, т, -NHCH2CH2 CH 2NH2), 3,26 (2H, д, -NHCH 2CH2CH2NH2), 3,71 (2H, с, Ph-CH 2-CO), 6,40-7,49 (8Н, м, ароматический Н).
Пример 41
Синтез гиалуроната натрия, содержащего диаминопропандиклофенак (DS 18,1%)
В смеси 22,5 мл воды/22,5 мл диоксана растворяли 200 мг (0,5 ммоль/дисахаридная единица) гиалуроновой кислоты, имеющей средневзвешенную молекулярную массу 800000, и затем добавляли 2М HOSu в количестве 0,2 мл, 1М WSCI·HCl в количестве 0,2 мл и 1 мл раствора (вода:диоксан=1:1) 78,4 мг (0,2 ммоль) полученного в примере 40 гидрохлорида диаминопропандиклофенака в указанном порядке с последующим перемешиванием в течение ночи. Затем добавляли 3 мл 5% водного раствора гидрокарбоната натрия с последующим перемешиванием в течение 4 часов. После нейтрализации реакционного раствора добавлением 86 мкл 50% уксусной кислоты добавляли 1 г хлорида натрия с последующим перемешиванием. Смесь осаждали, добавляя 200 мл этанола, осадок промывали дважды 85% этанолом, дважды этанолом и диэтиловым эфиром и сушили при комнатной температуре в течение ночи при пониженном давлении с получением 206,2 мг белого твердого вещества. Степень замещения диклофенаком составляла 18,1% при измерении с использованием спектрофотометра.
Пример 42
Получение 1% раствора гиалуроната натрия, содержащего аминопропанолкетопрофен, для использования в проведении тестов
К 22 мг гиалуроната натрия, содержащего аминопропанолкетопрофен (степень замещения 26,3%), полученного в примере 3, добавляли 5 мМ забуференного фосфатом физиологического раствора, с получением общего количества 2,19 г, с последующим перемешиванием в течение ночи, с получением 1% раствора. Раствор пропускали через фильтр, размером 0,45 мкм и использовали в качестве указанного в заголовке раствора. Когда измеряли содержание эндотоксина в данном растворе при помощи метода тестирования эндотоксина (колориметрический метод), который представляет собой способ тестирования, описанный в фармакопее Японии, количество эндотоксина составляло 0,0073 EU/MI.
Тесты введения
Пример 43
Эффект гиалуроната натрия, содержащего аминопропанолкетопрофен на модель вызванной брадикинином боли у крыс
1) Введение тестовых веществ
В качестве общей анестезии применяли ингаляцию изофлурана (Foran, зарегистрированный товарный знак, Dainippon Pharmaceutical Co., Ltd., концентрация 3,0%, скорость потока 2,0 литров/мин), которым заполняли малое устройство для анестезии животных (ТК-4, произведенный BioMachinery Co., Ltd.).
В качестве тестовых веществ использовали PBS, 1% раствор гиалуроната натрия (НА), раствор кетопрофена натрия в концентрации 3,7 мг/мл (КР), полученный растворением кетопрофена в PBS, и 1% раствор в PBS гиалуроната натрия, содержащего аминопропанолкетопрофен (КР-НА), полученный в примере 42.
Крыс (Crj:SD(SPF), самцы, возраст 7 недель) фиксировали в положении лежа на спине под эфирной анестезией и широкую область вокруг коленного сустава левой задней лапы выбривали с использованием электрической машинки для стрижки. После дезинфекции участка вокруг сустава распылением 70% спирта описанные выше тестовые вещества вводили в полость коленного сустава левой задней лапы в дозе 20 мкл/сустав, используя шприц с иглой 29G для использования инсулина (производимого Terumo Corp.). Данную процедуру проводили, используя 5 случаев (n=5) для каждой группы исследуемых веществ.
2) Введение вызывающих боль веществ (раствор ВК+PGE2)
Через 1 день после введения каждого тестового вещества крыс фиксировали в положении лежа на спине без анестезии. После дезинфекции участка вокруг сустава распылением 70% спирта смешанный раствор брадикинина (ВК) и простагландина Е2 (PGE2) в качестве вызывающих боль веществ вводили в полость коленного сустава левой задней лапы в дозе 50 мкл/сустав, используя шприц с иглой 29G для использования инсулина (производимого Terumo Corp., толщина иглы составляет 0,33 мм). Дополнительно данный раствор вызывающих боль веществ получали таким образом, чтобы конечная концентрация ВК и PGE2 составляла 4 мкг/мл и 2 мкг/мл соответственно. Реакции на боль наблюдали визуально сразу после введения вызывающего боль средства.
3) Наблюдение боли
В течение приблизительно 2 минут после введения вызывающих боль веществ наблюдали визуально и оценивали поведенческие проявления при ходьбе, такие как «поднимание лапы», «ходьба на трех лапах» и «хромота». Оценки боли обозначали так, что поднимание лапы: добавление 1 балла и хромота или ходьба на трех лапах: добавление 1 балла, и оценивали с шагом от 0 до 2 баллов. В дополнение, оценку проводили в условиях слепого способа. График, на котором оценены реакции на боль соответствующих индивидуумов, показан на фиг.1.
На фиг.1 результаты показаны в средних значениях показателей боли±стандартное отклонение.
Соответственно, эффекты подавления боли наблюдали в порядке КР-НА>КР>НА по сравнению с получавшей PBS группой.
Пример 44
Эффект внутрисуставного введения гиалуроната натрия, содержащего аминопропанолкетопрофен, на модель вызванной 1% нитратом серебра боли у крыс
1) Ведение вызывающих боль веществ
В качестве общей анестезии применяли ингаляцию изофлурана (Foran, зарегистрированный товарный знак, Dainippon Pharmaceutical Co., Ltd., концентрация 3,0%, скорость потока 2,0 литров/мин), которым заполняли малое устройство для анестезии животных (ТК-4, произведенный BioMachinery Co., Ltd.).
Крыс (Crj:SD(SPF), самцы, возраст 6 недель) фиксировали в положении лежа на спине под эфирной анестезией и широкую область вокруг коленного сустава левой задней лапы выбривали с использованием электрической машинки для стрижки. После дезинфекции участка вокруг сустава распылением 70% спирта, 1% раствор нитрата серебра вводили в полость коленного сустава левой задней лапы в дозе 50 мкл/сустав, используя шприц с иглой 29G для использования инсулина (производимого Terumo Corp.).
2) Введение тестовых веществ
В качестве тестовых веществ готовили 1% раствор гиалуроната натрия (НА) и 1% раствор гиалуроната натрия, содержащего аминопропанолкетопрофен (КР-НА), полученного в примере 42, каждый с использованием PBS в качестве растворителя. Крыс разделяли на 2 группы, каждая включающая по 5 животных, и каждое тестовое вещество вводили соответствующей группе через 24 часа после введения 1% раствора нитрата серебра. Что касается способа введения, как и в случае веществ, вызывающих боль, участок вокруг сустава дезинфицировали распылением 70% спирта под ингаляционной анестезией изофлураном и каждое тестовое вещество вводили в полость коленного сустава левой задней лапы в дозе 40 мкл/сустав, используя шприц с иглой 29G для использования инсулина (n=5).
3) Способ оценки
За ходьбой в каждой группе наблюдали визуально и оценивали ее, используя следующую таблицу показателей боли, которая обозначает цифрами ходьбу в показателях в условиях слепого способа. Результаты показаны на фиг.2.
На фиг.2 результаты показаны в средних значениях показателей боли ± стандартное отклонение.
Показатель:
0: норма (включая сюда почти нормальное поведение)
1: средняя хромота
2: сильная хромота
3: ходьба на трех лапах
В дополнение, весовую нагрузку на лапу, подвергшуюся инъекции нитрата серебра (левая задняя лапа), измеряли при помощи измерителя взвешенного обезболивающего действия (произведенного Tokken Inc.), и степень весовой нагрузки рассчитывали, деля измеренное значение на массу тела. (В этой связи степень весовой нагрузки составляла приблизительно 32% у нормальных животных). Измерение проводили один раз в день до 2 дней после введения каждого тестового вещества. Результаты показаны на фиг.3.
Как показано на фиг.2, показатель боли постепенно уменьшался как в группе введения НА, так и в группе введения КР-НА, но степень подавления боли (степень восстановления после боли) была более быстрой в группе введения КР-НА, чем в группе введения НА. В дополнение, степень весовой нагрузки, главным образом, становится выше, по мере прогрессирования восстановления после боли при измерении степени, но, как показано на фиг.3, степень весовой нагрузки становилась значительно выше за короткий период времени у животных, получавших КР-НА, по сравнению с животными, получавшими НА. Взаимоотношение между группой КР-НА и группой НА по результатам на фиг.2 и фиг.3 было одинаковым.
Пример 45
Проверка свойства замедленного высвобождения гиалуроновой кислоты, содержащей NSAID, в коленном суставе кролика
1) Способ введения тестовых веществ
1% раствор гиалуроната натрия, содержащего аминопропанолкетопрофен (КР-НА), полученного в примере 42, раствор кетопрофена (КР), в котором 1,42 мг кетопрофена растворяли в 1 мл PBS, и смешанный раствор кетопрофена и НА (КР+НА), в котором 1,41 мг кетопрофена растворяли в 1 мл 1% раствора НА, использовали в качестве тестовых веществ.
Использовали по пять кроликов для каждого тестового вещества, кроликов фиксировали в положении лежа на спине под общей анестезией кетамином (1 мл/голова, в/в) и широкую область вокруг коленного сустава левой задней лапы выбривали с использованием электрической машинки для стрижки. Каждое из описанных выше веществ вводили в полость сустава с внешней стороны колена кролика в дозе 300 мкл, используя шприц на 1 мл с иглой 25G (производимый Terumo Corp., толщина иглы составляет 0,5 мм).
Вскрытие проводили через 6, 12, 24 часа и через 2, 4 дня после введения тестовых веществ.
2) Способ измерения количества КР свободного типа и КР связанного типа в синовиальной жидкости
Кроликов умерщвляли обескровливанием под общей анестезией кетамином. После того как собирали всю синовиальную жидкость, полость сустава промывали 2 раза 2 мл физиологического раствора в полости сустава препарированного колена, используя иглу 25G. Промывочную жидкость также собирали. Количество КР и КР-НА в синовиальной жидкости, объединенной с извлеченными промывочными жидкостями, измеряли следующим ниже способом.
Добавлением 1н. HCl (0,2 мл) в синовиальную жидкость (4 мл об.) подтверждали кислотность соляной кислоты и затем добавляли этилацетат в том же самом объеме раствора и энергично перемешивали и извлекали верхний органический слой. Данную операцию экстракции проводили в общем 3 раза. Раствор ацетонитрила добавляли к извлеченному органическому слою для получения его раствора в ацетонитриле, и количество свободного КР измеряли, используя ВЭЖХ (количество КР свободного типа в синовиальной жидкости).
Затем водный слой, полученный при описанной выше операции экстракции, доводили до сильно щелочного состояния добавлением 1н. NaOH и перемешивали при комнатной температуре в течение 1 часа. Затем водный слой охлаждали на льду, доводили до солянокислого состояния, медленно добавляя 4н. HCl при перемешивании, и затем энергично перемешивали, добавляя этилацетат в том же самом объеме раствора к извлеченному верхнему органическому слою. Данную операцию экстракции проводили в общем 3 раза. Раствор ацетонитрила добавляли к извлеченному органическому слою для получения его раствора в ацетонитриле, и количество НА-КР (количество КР связанного типа) измеряли, используя ВЭЖХ (количество связанного НА-КР в синовиальной жидкости).
3) Способ измерения количества КР в расщепленной жидкости синовиальной оболочки
Синовиальную оболочку отделяли и собирали из коленного сустава после описанного выше извлечения синовиальной жидкости (2). Собранную синовиальную оболочку тщательно промывали 100 мл физиологического раствора до полного удаления связанной синовиальной жидкости. После удаления коленной чашечки синовиальную оболочку помещали в пробирку, в нее добавляли 5 мл протеиназы К (лот № 102К8633, производимый SIGMA), приготовленной в концентрации 2 мг/мл с использованием 10 мМ раствора ацетата натрия (рН 7,5), и ферментативное расщепление проводили при 55°С в течение 41 часа при необязательном перемешивании с использованием Vortex. После расщепления фермент инактивировали инкубацией при 100°С в течение 5 минут и количество КР в полученной таким образом расщепленной жидкости измеряли по следующей ниже методике.
1/4 объема 4н. NaOH добавляли к полученной таким образом расщепленной жидкости и перемешивали при комнатной температуре в течение 1 часа. Затем раствор охлаждали на льду, доводили до солянокислого состояния, медленно добавляя 4н.HCl, и затем энергично перемешивали, добавляя диэтиловый эфир в том же самом объеме раствора к извлеченному верхнему органическому слою. Данную операцию обезжиривания проводили в общем 3 раза. При охлаждении на льду 4н. HCl добавляли к раствору после операции обезжиривания и перемешивали, для укрепления солянокислого состояния, и затем энергично перемешивали, добавляя этилацетат в том же самом объеме раствора к извлеченному верхнему органическому слою. Раствор ацетонитрила добавляли к извлеченному органическому слою, для получения его раствора в ацетонитриле, и количество свободного КР измеряли, используя ВЭЖХ.
Количество КР свободного типа в расщепленной жидкости синовиальной оболочки
Остаточные соотношения КР и НА-КР в синовиальной жидкости и в расщепленной жидкости синовиальной оболочки вычисляли в разное время (см. таблицу, фиг.4).
Когда в качестве тестовых веществ вводили КР (одно лекарственное средство) или КР-НА (смесь), то КР исчезало из синовиальной жидкости и синовиальной оболочки через 6 часов и 12 часов. Тем не менее, когда вводили КР-НА (конъюгат) в качестве вещества по настоящему изобретению, присутствие КР подтверждали как в синовиальной жидкости, так и в синовиальной оболочке даже через 4 дня, так что считается, что КР проявляет свой продолжительный эффект, постоянно присутствуя в участке введения.
Полагают, что боль в суставе возникает не через хрящ, который представляет собой безнервную ткань, а через синовиальную оболочку, и в дополнение предполагают, что когда NSAID вводят в полость сустава, NSAID быстро проникает в синовиальную оболочку и оказывает эффект, перемещаясь в синовиальной оболочке. Таким образом, предполагают, что сохранение концентрации NSAID в синовиальной оболочке в значительной степени имеет отношение к продолжительному обезболивающему действию и эффекту замедленного высвобождения. Как показано в таблице, когда вводили КР (одно лекарственное средство), он исчезал из синовиальной оболочки через 6 часов путем прохождения через синовиальную оболочку и метаболизма. Тем не менее, когда вводили КР-НА (конъюгат), КР постоянно сохранялся также в синовиальной оболочке, указывая таким образом на то, что он более эффективен в качестве препаратов с замедленным высвобождением NSAID.
Пример 46
Эффекты внутрисуставных инъекций гиалуроната натрия, содержащего аминопропанолдиклофенак, имеющего различные степени замещения (DS), на модели вызванной 1% нитратом серебра боли у крыс
Определение эффекта внутрисуставной инъекции следующих ниже веществ проводили согласно способу в описанном выше примере 44.
Тестовые вещества:
(i) раствор в PBS 1% гиалуроната натрия, содержащего аминопропанолдиклофенак (DS 18,2%), полученного в примере 19;
(ii) раствор в PBS 1% гиалуроната натрия, содержащего аминопропанолдиклофенак (DS 9,7%), полученного в примере 36;
(iii) раствор в PBS 1% гиалуроната натрия, содержащего аминопропанолдиклофенак (DS 4,3%), полученного в примере 35;
(iv) PBS.
По методике, аналогично описанной в примере 44, ходьбу каждой группы наблюдали визуально и оценивали, используя таблицу показателей боли, которая обозначает цифрами ходьбу в показателях в условиях слепого способа. Результаты показаны на фиг.5. Результаты показаны в средних значениях показателей боли ± стандартное отклонение, и DS 18%, DS 10% и DS 4% относятся к DS 18,2%, DS 9,7% и DS 4,3 соответственно. В дополнение, на фиг.5 * указывает на то, что существует значимое различие между PBS и уровнем значимости 0,01<р<0,05, и ** указывают на то, что существует значимое различие между PBS и уровнем значимости р<0,01.
Следовательно, все производные гиалуроната натрия, содержащие аминопропанолдиклофенак с DS 18,2%, DS 9,7% и DS 4,3% в качестве тестовых веществ, показали обезболивающий эффект. В частности, тестовые вещества с DS 18,2% и DS 9,7% показали значительный обезболивающий эффект по сравнению с PBS.
В дополнение, обезболивающий эффект улучшался в зависимости от степени замещения (DS) диклофенаком.
Справочный пример 6
Эффект перорального введения диклофенака натрия на модели вызванной 1% нитратом серебра боли у крыс
Исследование проводили согласно методике, описанной выше в примере 44, и следующие ниже тестовые вещества вводили перорально и оценивали. Тестовые вещества вводили перорально с использованием зонда для перорального введения крысам (производимые Fuchigami Kikai) в дозе 1 мл/голова.
Тестовые вещества:
(i) 1% суспензия диклофенака натрия (10% гуммиарабик);
(ii) 0,02% суспензия диклофенака натрия (10% гуммиарабик).
Дополнительно (i) (группа высокой дозы) соответствует введению 50 мкг/кг диклофенака натрия и (ii) (группа низкой дозы) соответствует введению 1 мкг/кг диклофенака натрия, которое является почти таким же количеством, что и количество клинической дозы.
По методике, аналогичной описанной в примере 46, ходьбу каждой группы наблюдали визуально и оценивали, используя таблицу показателей боли, которая обозначает цифрами ходьбу в показателях в условиях слепого способа. Результаты показаны на фиг.6. На фиг.6 «диклофенак Na (р.о.) 50 мг/кг» соответствует описанной выше (i) (группе высокой дозы) и «диклофенак Na (р.о.) 1 мг/кг» соответствует описанной выше (ii) (группе низкой дозы). Дополнительно результаты показателей боли при внутрисуставном введении производного гиалуроновой кислоты, содержащей диклофенак (DS 18,2%) и PBS, измеренные выше в примере 46, также показаны для сравнения на фиг.6 как «диклофенак-НА» и «PBS» соответственно.
Следовательно, пероральное введение высокой дозы (50 мг/кг) диклофенака натрия показывало обезболивающий эффект, однако на следующий день наблюдали положительную реакцию пробы на скрытую кровь в кале, подобный желтухе симптом и потерю массы тела. Доза в группе высокой дозы является во много раз большей, чем клиническая доза и представляет собой неиспользуемую на практике дозу из-за побочных эффектов и токсичности.
В группе низкой дозы (1 мг/кг) при пероральном введении, которая является почти клинической дозой, эффект не обнаружили по сравнению с группой, получавшей внутрисуставную инъекцию PBS.
C другой стороны, внутрисуставное введение производного гиалуроновой кислоты, содержащей диклофенак, показывало обезболивающий эффект после введения. Дополнительно побочного эффекта, вызванного при пероральном введении высокой дозы (50 мг/кг) диклофенака натрия, не наблюдали, так что подтвердили его высокую степень пригодности.
Справочный пример 7
Эффект внутрисуставного введения единичного лекарственного средства диклофенака на модели вызванной 1% нитратом серебра боли у крыс
Исследование проводили согласно методике, описанной выше в примере 44, и следующие ниже тестовые вещества вводили внутрь сустава и оценивали.
Тестовые вещества:
(i) 0,1% раствор диклофенака;
(ii) смешанный раствор 0,1% диклофенака/1% гиалуроновой кислоты;
(iii) PBS.
По методике, аналогично описанной в примере 46, ходьбу каждой группы (n=9) наблюдали визуально и оценивали, используя таблицу показателей боли, которая обозначает цифрами ходьбу в показателях в условиях слепого способа. Результаты показаны на фиг.7. На фиг.7 результаты показаны в средних значениях показателей боли ± стандартное отклонение и «0,1% диклофенак Na» соответствует вышеописанному (i) 0,1% раствору диклофенака, и «0,1% Дикло+НА» соответствует описанному выше смешанному раствору 0,1% диклофенака/1% гиалуроновой кислоты. Дополнительно результаты для производного гиалуроновой кислоты, содержащей диклофенак (DS 18,2%), измеренные в описанном выше примере 46, также показаны для сравнения на фиг.7 в качестве «Дикc-HA(конъюгат)».
Следовательно, одно лекарственное средство диклофенак и смесь диклофенака и гиалуроновой кислоты не проявили значительного эффекта по сравнению с PBS в качестве контрольной группы.
Пример 47
Эффект внутрисуставного введения гиалуроната натрия (65 кДа), содержащего аминопропанолдиклофенак, гиалуроната натрия, содержащего диаминопропандиклофенак, и гиалуроната натрия, содержащего аминоэтанолдиклофенак на модели вызванной 1% нитратом серебра боли у крыс
Исследование проводили согласно методике, описанной выше в примере 44, и следующие ниже тестовые вещества вводили внутрь сустава и оценивали.
Тестовые вещества:
(i) раствор в PBS 1% гиалуроната натрия (65 кДа), содержащего аминопропанолдиклофенак, полученного в примере 37;
(ii) раствор в PBS 1% гиалуроната натрия, содержащего диаминопропандиклофенак, полученного в примере 41;
(iii) раствор в PBS 1% гиалуроната натрия, содержащего аминоэтанолдиклофенак, полученного в примере 39;
(iv) PBS.
По методике, аналогично описанной в примере 44, ходьбу каждой группы (n=7) наблюдали визуально и оценивали, используя таблицу показателей боли, которая обозначает цифрами ходьбу в показателях в условиях слепого способа. Результаты показаны на фиг.8. На фиг.8 результаты показаны в средних значениях показателей боли и «65 кДа» соответствует описанному выше (i) раствору в PBS 1% гиалуроната натрия (65 кДа), содержащего аминопропанолдиклофенак, «диамид» соответствует описанному выше (ii) раствору в PBS 1% гиалуроната натрия, содержащего диаминопропандиклофенак и «С2» соответствует описанному выше (iii) раствору в PBS 1% гиалуроната натрия, содержащего аминоэтанолдиклофенак. В дополнение, на фиг.8 Δ указывает на то, что существует значимое различие между PBS и уровнем значимости 0,05<р<0,1, * указывает на то, что существует значимое различие между PBS и уровнем значимости 0,01<р<0,05, и ** указывает на то, что существует значимое различие между PBS и уровнем значимости р<0,01.
Следовательно, раствор гиалуроната натрия, содержащего аминоэтанолдиклофенак, в котором использован аминоэтанол, где количество спейсерных углеродов меньше, чем в аминопропаноле на фактор 1, также показывает значительный обезболивающий эффект. Тем не менее раствор гиалуроната натрия, содержащего диаминопропандиклофенак, в котором диклофенак присоединен посредством амидной связи, не оказал эффекта на тестовую систему данного примера. В дополнение, производное гиалуроновой кислоты, содержащее диклофенак, где используется гиалуроновая кислота, имеющая молекулярную массу 65 кДа, показало уменьшенный обезболивающий эффект по сравнению со случаем с молекулярной массой 800000.
Данные результаты показывают, что обезболивающий эффект зависит от типа связывания диклофенака или молекулярной массы гиалуроновой кислоты.
Пример 48
Эффект одного лекарственного средства диклофенака натрия и производного гиалуроновой кислоты на СОХ-2 (in vitro)
Ингибирующую активность в отношении СОХ-2 следующих тестовых соединений оценивали, используя набор Chemiluminescent COX Inhibitor Screening Assay (производимый Cayman) (набор для проверки ингибиторов, использующий пероксидазную активность полученной из овцы СОХ-2 в качестве индекса).
Тестовые вещества:
(i) водный раствор гиалуроната натрия, содержащего аминопропанолдиклофенак, полученного в примере 19 (Дик-C3-HA) (соответствует 200 мкг/мл НА, соответствует 80 мкМ диклофенака);
(ii) водный раствор гиалуроната натрия, содержащего аминоэтанолдиклофенак, полученного в примере 39 (Дик-C2-HA, DS 18,5%) (соответствует 200 мкг/мл НА, соответствует 80 мкМ диклофенака);
(iii) водный раствор 80 мкМ диклофенака натрия;
(iv) водный раствор 200 мкг/мл гиалуроната натрия (НА).
Получали исходные растворы различных описанных выше тестовых веществ и данные тестовые вещества далее разбавляли в 10 раз, 100 раз и 1000 раз дистиллированной водой, их ингибирующую активность в отношении СОХ-2 измеряли, используя набор COX Inhibitor Screening Assay (группа, не получавшая тестовых веществ n=6, группа, получавшая тестовое веществе n=3). Величину активности фермента для каждой группы, получавшей тестовое вещество, выражали в качестве относительного % при помощи следующей ниже формулы, на основании ферментативной активности СОХ-2 группы, не получавшей тестовое вещество, которую определили как 100%. Результаты показаны на фиг.9(а) и фиг.9(b). На фиг.9(а) «диклофенак Na» соответствует описанному выше водному раствору диклофенака натрия, и «Дик-C3-HA» соответствует описанному выше водному раствору гиалуроната натрия, содержащего аминопропанолдиклофенак, и «Дик-C2-HA» соответствует описанному выше водному раствору гиалуроната натрия, содержащего аминоэтанолдиклофенак. На фиг.9(b) «НА» соответствует описанному выше водному раствору гиалуроната натрия.
Величина ферментативной активности (%)=(группа, получавшая тестовое вещество)/группа, не получавшая тестовое вещество×100
Следовательно, производные гиалуроновой кислоты, содержащие диклофенак (Дик-C3-HA и Дик-C2-HA), в концентрации, соответствующей концентрации, при которой одно лекарственное средство диклофенак натрия показывал явную ингибирующую активность в отношении СОХ-2, не показывали ингибирующей активности в отношении СОХ-2. Одно лекарственное средство НА также не показало ингибирующей активности в отношении СОХ-2. Данные результаты предполагают, что эффект производных гиалуроновой кислоты, содержащих диклофенак, in vitro не является действием их самих, а может передаваться через высвобождение диклофенака из НА.
В качестве одной из причин, почему эффект производных гиалуроновой кислоты, содержащих диклофенак in vivo (примеры 46 и 47) сильнее, чем эффект одного лекарственного средства диклофенака, считают, что НА несет большее количество диклофенака в клетки-мишени в СОХ-2 вследствие ее большей аффинности к клеткам.
Пример 49
Эффект производного гиалуроновой кислоты, содержащего диклофенак, на модель вызванного адъювантом артрита (AIA) у крыс
1) Введение адъюванта
После нагревания 6 мг/мл Mycobacterium butyricum (лот № 2115687, Difco) при 121°С в автоклаве его вводили в подушечки правой задней лапы в дозе 50 мкл/сустав шприцем с иглой 29G для использования инсулина (Terumo).
2) Введение тестовых веществ
Тестовые вещества:
(i) раствор в PBS 1% гиалуроната натрия, содержащего аминопропанолдиклофенак (DS 18%), полученного в примере 19;
(ii) PB.
Как и в случае введения адъюванта, описанные выше тестовые вещества вводили без анестезии в полости тибиотарзальных суставов обеих лап в дозе 50 мкл/сустав шприцем с иглой 29G для использования инсулина (Terumo), сразу же после инъекции адъюванта и через 1, 2 и 3 недели после инъекции (всего 4 раза) (n=14).
4) Оценка
Оценку проводили перед инъекцией адъюванта и на 3, 5, 7, 10, 12, 14, 21 и 28 день после инъекции.
* масса тела
* объем обеих задних лап (устройство для измерения объема отека задних лап у крыс и мышей (ТК-101СМР), производимое Unicom).
5) Результаты
Степень отечности лап, подвергшихся инъекции адъюванта и не подвергавшихся инъекции адъюванта, определяли по следующей ниже формуле. Результаты степени отечности лап, подвергшихся инъекции адъюванта, показаны на фиг.10(а) и результаты степени отечности лап, не подвергшихся инъекции адъюванта, показаны на фиг.10(b). На фиг.10 «диклофенак-НА» обозначает описанный выше (i) раствор в PBS 1% гиалуроната натрия, содержащего аминопропанолдиклофенак (DS 18%) и «норма» обозначает группы без введения адъюванта и тестового вещества. На фиг.10 Δ указывает на то, что существует значимое различие между PBS и уровнем значимости 0,05<р<0,1, * указывает на то, что существует значимое различие между PBS и уровнем значимости 0,01<р<0,05, и ** указывает на то, что существует значимое различие между PBS и уровнем значимости р<0,01.
Степень отечности (%)=(объем лапы на день измерения-объем лапы перед введением адъюванта)/объем лапы перед введением адъюванта×100
Отек наблюдали в лапах, подвергшихся инъекции (R), c 3 дня после введения адъюванта, и диклофенак-НА показывал эффект подавления объема отека на 3 день, 5 день, 21 день после введения статистически значимой по сравнению с группой, получавшей PBS, и также на 7 день, 26 день и 28 день, хотя и незначительно. Также в другие моменты времени группа, получавшая диклофенак-НА, показала низкие значения в каждый момент времени, хотя и незначительные.
Явную отечность (вторичное воспаление) наблюдали на лапах, не подвергавшихся инъекции (L), c 14 дня после введения. Диклофенак-НА статистически значимо подавлял данный отек на 14 день и 26 день после введения. Более того, он проявлял тенденцию подавлять отек на 21 день и 28 день, хотя и незначительную.
В дополнение, поскольку модель вызванного адъювантом артрита (AIA) у крысы в общем случае используют в качестве модели ревматоидного артрита, который представляет собой артрит, вызванный аутоиммунным заболеванием, предполагают, что вещество по настоящему изобретению оказывает эффект на ревматоидный артрит.
Хотя изобретение описано подробно и со ссылками на определенные его осуществления, специалисту в данной области будет очевидно, что различные изменения и модификации можно провести, не отступая от его сущности и объема.
Данная заявка основана на заявке на патент Японии № 2004-2478, поданной 7 января 2004, полное содержание которой включено в данное описание посредством ссылки. Все ссылки, приведенные в настоящем описании, включены в описание во всей своей полноте.
Промышленная применимость
По настоящему изобретению предлагаются производные гиалуроновой кислоты, содержащие NSAID, в которых NSAID связаны с гиалуроновой кислотой посредством ковалентной связи через спейсер, содержащий биодеградируемый участок, производные гиалуроновой кислоты, содержащие DMARD, в которых DMARD связаны с ней посредством ковалентной связи таким же образом, и фармацевтическое средство, которое содержит данные производные в качестве активного ингредиента. Поскольку производные гиалуроновой кислоты, содержащие NSAID, и производные гиалуроновой кислоты, содержащие DMARD, достаточно растворяются в буферах, которые используют в качестве растворителей в продуктах для инъекций и тому подобное, их можно использовать в качестве продуктов для инъекций, которые можно вводить непосредственно в пораженный участок. Более того, фармацевтическое средство по настоящему изобретению можно использовать для лечения артритов, подавления воспаления и подавления боли, и также возможно его парентеральное введение или местное введение (например, внутрисуставное введение) в качестве наполнителей.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕЛЬ, ПОЛУЧЕННЫЙ ИЗ ФОТОСШИТОЙ ГИАЛУРОНОВОЙ КИСЛОТЫ С ВВЕДЕННЫМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ | 2006 |
|
RU2429018C2 |
| 4-ГИДРОКСИТИОБЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ | 2007 |
|
RU2465271C2 |
| СЕРОВОДОРОДНЫЕ ПРОИЗВОДНЫЕ НЕСТЕРОИДНЫХ ПРОТИВОВОСПАЛИТЕЛЬНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2007 |
|
RU2468019C2 |
| С-С-АЦИЛИРОВАННОЕ ПРОИЗВОДНОЕ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, НАНОМИЦЕЛЛЯРНАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ СТАБИЛИЗИРОВАННОЙ НАНОМИЦЕЛЛЯРНОЙ КОМПОЗИЦИИ И ЕЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2640287C2 |
| КРИСТАЛЛИЧЕСКИЙ D-ИЗОГЛУТАМИЛ-D-ТРИПТОФАН И МОНОАММОНИЙНАЯ СОЛЬ D-ИЗОГЛУТАМИЛ-D-ТРИПТОФАНА | 2007 |
|
RU2483077C2 |
| КОНЪЮГАТЫ: ТЕРАПЕВТИЧЕСКОЕ СОЕДИНЕНИЕ - ЖИРНАЯ КИСЛОТА | 1994 |
|
RU2137755C1 |
| СОДЕРЖАЩИЕ АЗОТ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ИХ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2008 |
|
RU2465276C2 |
| НИТРОЭФИРЫ, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ И/ИЛИ АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1994 |
|
RU2136653C1 |
| ДВОЙНЫЕ АНТИТРОМБОТИЧЕСКИЕ ИНГИБИТОРЫ, СОДЕРЖАЩИЕ ОСТАТОК БИОТИНА | 2005 |
|
RU2403259C2 |
| СИСТЕМЫ ДОСТАВКИ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩИЕ БЕССЛЕДНЫЕ ЛИНКЕРЫ | 2018 |
|
RU2772690C2 |
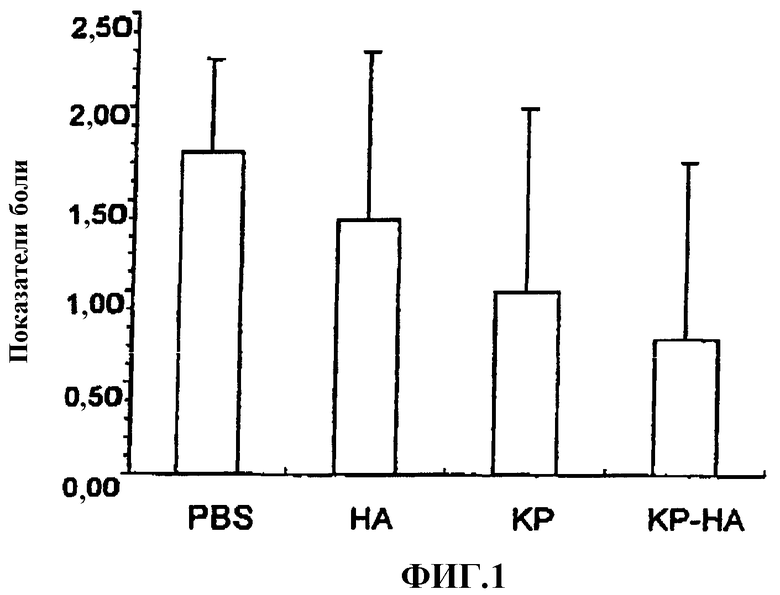
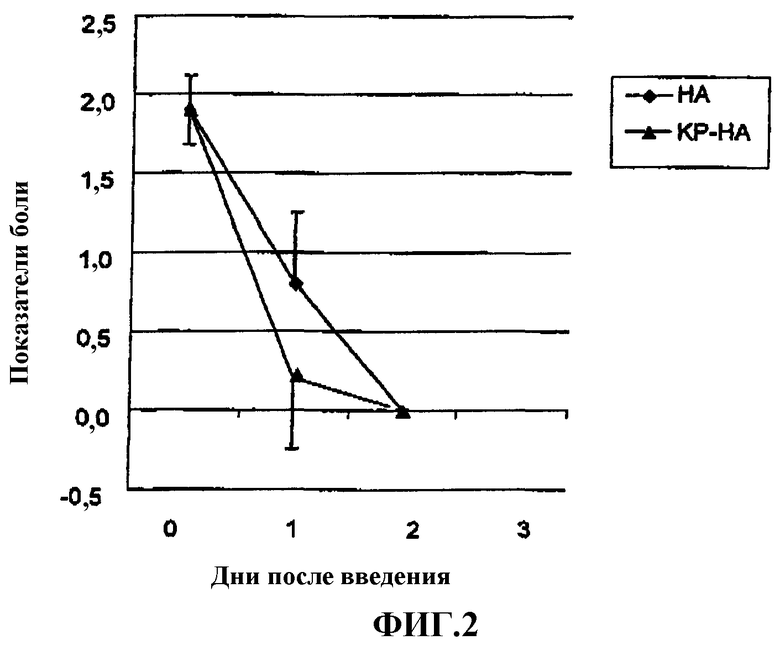
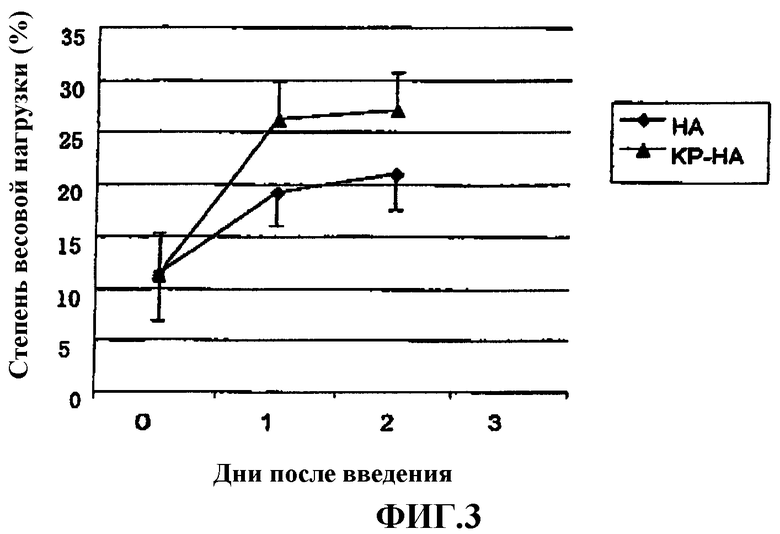

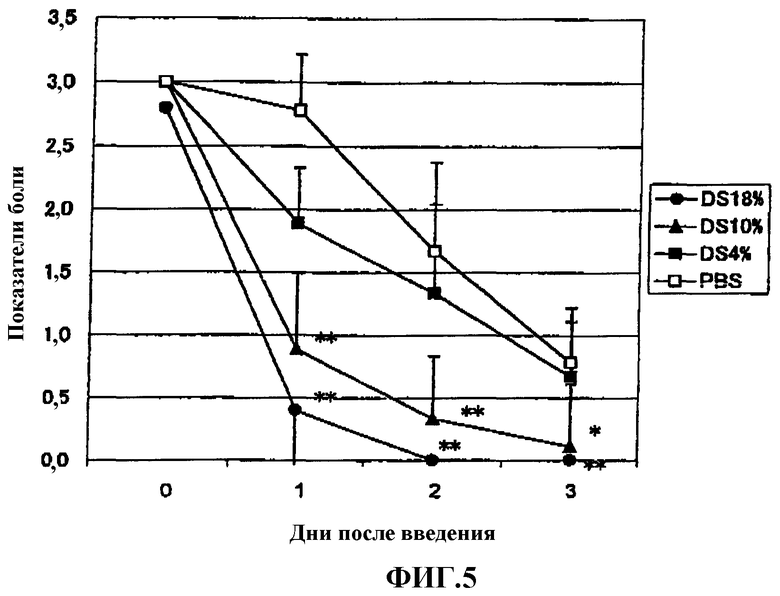
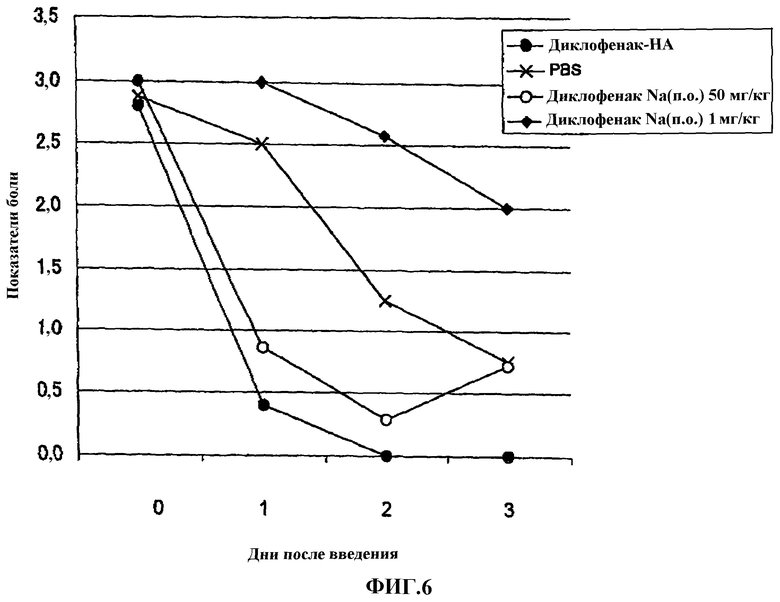
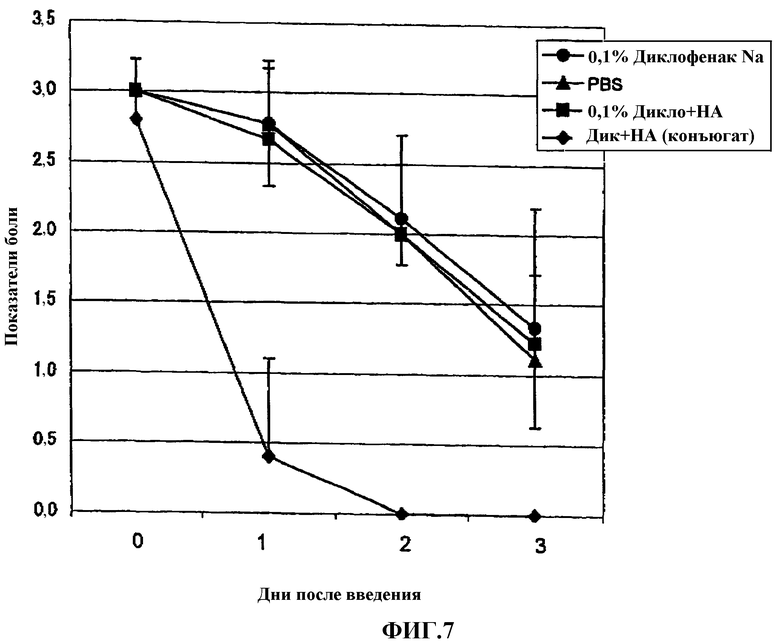
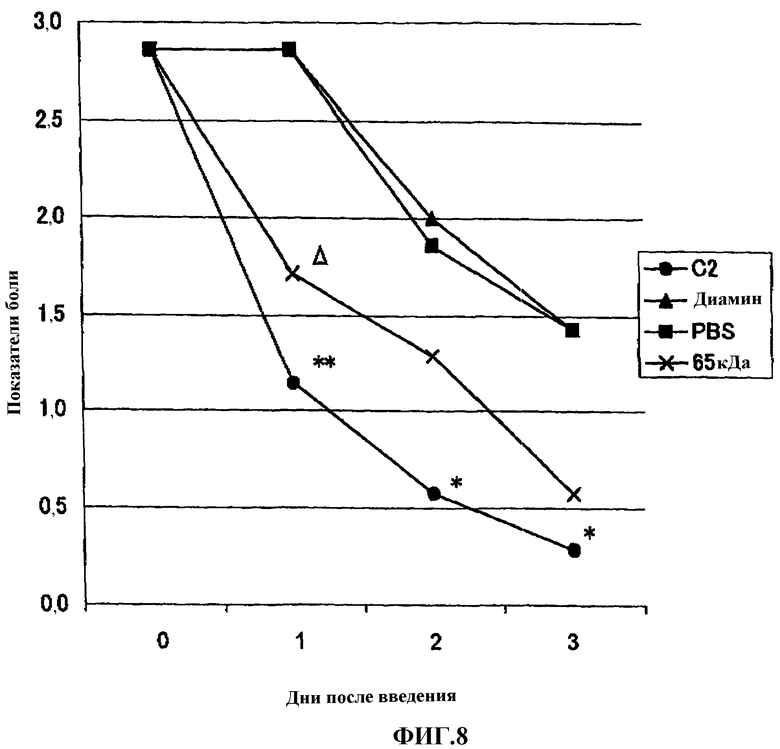
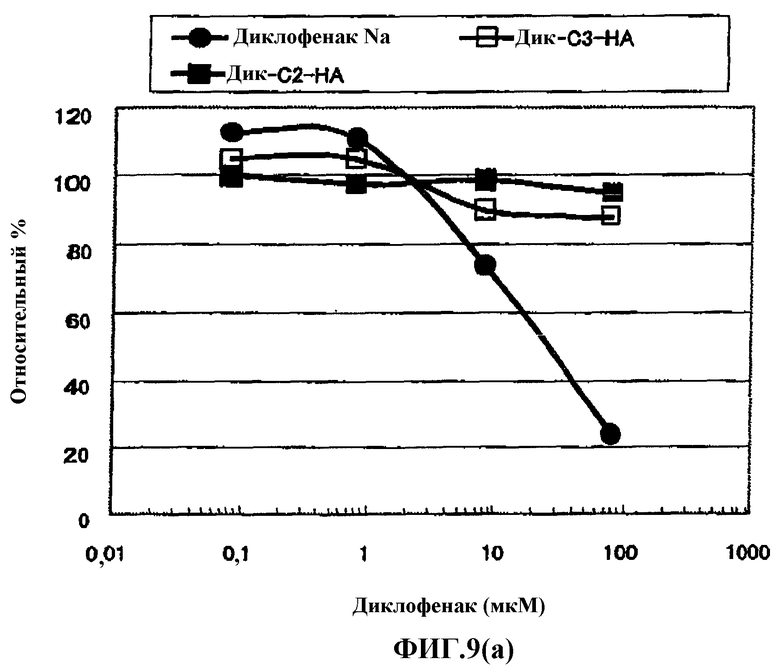
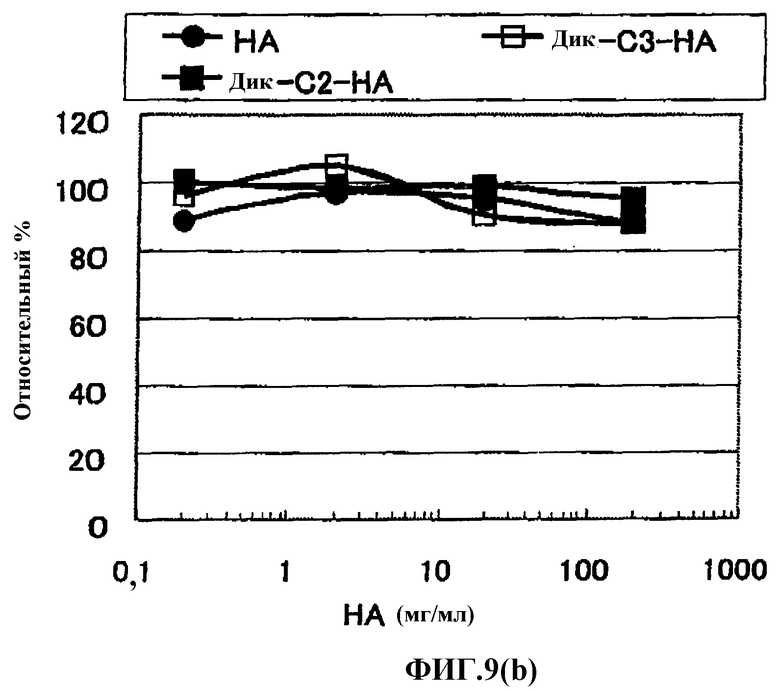
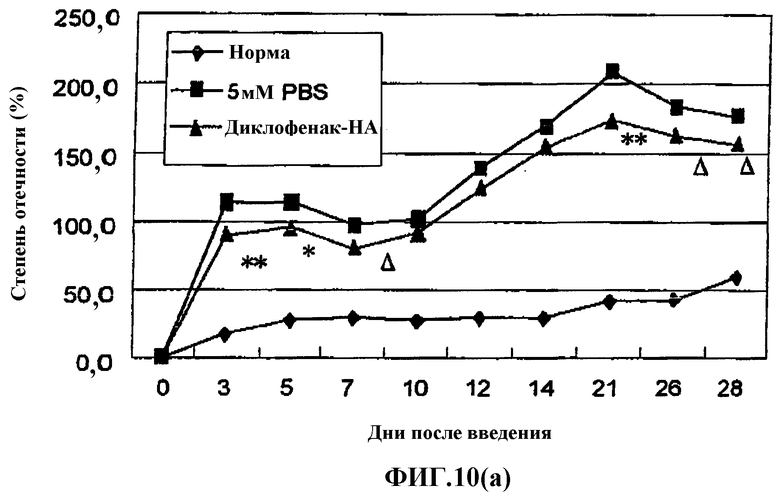
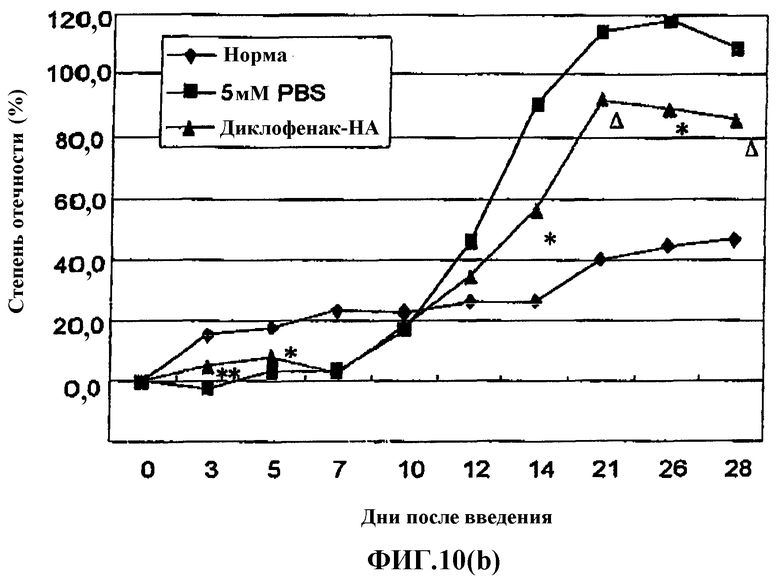
Изобретение относится к производному гиалуроновой кислоты, где нестероидное противовоспалительное лекарственное средство связано с гиалуроновой кислотой посредством ковалентной связи, которое содержит частичную структуру дисахаридной единицы гиалуроновой кислоты, к которой присоединено противовоспалительное лекарственное средство, представленное следующей ниже формулой (I):
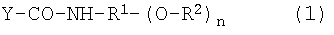
где Y-CO- представляет собой один остаток дисахаридной единицы гиалуроновой кислоты; R2 представляет собой остаток нестероидного противовоспалительного лекарственного средства, представленного группой Z-CO- или атомом водорода, при условии, что все R2 не являются атомом водорода; -NH-R1-(-O-)n представляет собой спейсерный остаток в соединении-спейсере, представленном формулой H2N-R1-(OH)n, имеющем гидроксильные группы в количестве n; R1 представляет собой линейную или разветвленную углеводородную группу, содержащую от 2 до 12 атомов углерода, которая может иметь заместитель; -СО-NН- представляет собой амидную связь карбоксильной группы гиалуроновой кислоты в качестве составляющей сахарид гиалуроновой кислоты с аминогруппой соединения-спейсера; -O-СО- представляет собой сложноэфирную связь гидроксильной группы соединения-спейсера с карбоксильной группой в остатке нестероидного противовоспалительного лекарственного средства и n равно целому числу от 1 до 3, где производное гиалуроновой кислоты имеет степень замещения нестероидным противовоспалительным лекарственным средством от 5 до 50% мол. на повторяющуюся единицу гиалуроновой кислоты и карбонильная группа в остатке гиалуроновой кислоты, составляющем производное гиалуроновой кислоты, присутствует в качестве участвующей в образовании амидной связи при связывании со связывающим спейсер остатком нестероидного противовоспалительного лекарственного средства или в качестве не принимающей в этом участие свободной карбоксильной группы, согласно степени замещения остатка нестероидного противовоспалительного лекарственного средства. Также изобретение относится к раствору и фармацевтическому средству для подавления боли и/или подавления воспаления и/или лечения артрита производного гиалуроновой кислоты, набору и медицинскому набору для инъекции, включающему раствор производного гиалуроновой кислоты. 7 н. и 19 з.п. ф-лы, 12 ил., 1 табл.
1. Производное гиалуроновой кислоты, где нестероидное противовоспалительное лекарственное средство связано с гиалуроновой кислотой посредством ковалентной связи, которое содержит частичную структуру дисахаридной единицы гиалуроновой кислоты, к которой присоединено противовоспалительное лекарственное средство, представленное следующей формулой (1)
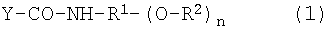
где Y-CO- представляет собой один остаток дисахаридной единицы гиалуроновой кислоты;
R2 представляет собой остаток нестероидного противовоспалительного лекарственного средства, представленного группой Z-CO- или атомом водорода, при условии, что все R2 не являются атомом водорода;
-NH-R1-(O-)n представляет собой спейсерный остаток в соединении-спейсере, представленном формулой H2N-R1-(OH)n, имеющем гидроксильные группы в количестве n;
R1 представляет собой линейную или разветвленную углеводородную группу, содержащую от 2 до 12 атомов углерода, которая может иметь заместитель;
-CO-NH- представляет собой амидную связь карбоксильной группы гиалуроновой кислоты в качестве составляющей сахарид гиалуроновой кислоты с аминогруппой соединения-спейсера;
-O-СО- представляет собой сложноэфирную связь гидроксильной группы соединения-спейсера с карбоксильной группой в остатке нестероидного противовоспалительного лекарственного средства и
n равно целому числу от 1 до 3,
где производное гиалуроновой кислоты имеет степень замещения нестероидным противовоспалительным лекарственным средством от 5 до 50 мол.% на повторяющуюся единицу гиалуроновой кислоты и
карбонильная группа в остатке гиалуроновой кислоты, составляющем производное гиалуроновой кислоты, присутствует в качестве участвующей в образовании амидной связи при связывании со связывающим спейсер остатком нестероидного противовоспалительного лекарственного средства или в качестве не принимающей в этом участие свободной карбоксильной группы, согласно степени замещения остатка нестероидного противовоспалительного лекарственного средства.
2. Производное гиалуроновой кислоты по п.1, где указанное нестероидное противовоспалительное лекарственное средство выбирают из группы, включающей кетопрофен, напроксен, ибупрофен, флурбипрофен, ацетилсалициловую кислоту, фелбинак, мефенамовую кислоту, диклофенак и этодолак.
3. Производное гиалуроновой кислоты по п.1, где нестероидное противовоспалительное лекарственное средство представляет собой соединение, представленное следующей формулой (2)
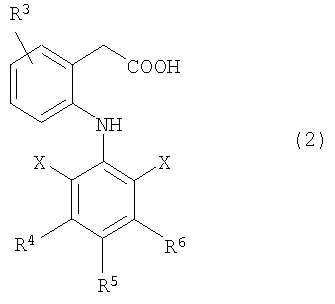
где R3 представляет собой заместитель, выбранный из низшей алкильной группы и низшей алкоксильной группы или атома водорода;
R4, R5 и R6 каждый независимо представляет собой заместитель, выбранный из группы, состоящей из низшей алкильной группы, низшей алкоксильной группы и гидроксильной группы, атома галогена или атома водорода, и
Х являются одинаковыми или различными и каждый представляет собой заместитель, выбранный из низшей алкильной группы и трифторметильной группы или атома галогена и, по крайней мере, один из Х представляет собой атом галогена.
4. Производное гиалуроновой кислоты по п.3, где нестероидное противовоспалительное лекарственное средство представляет собой соединение, представленное следующей формулой (7):
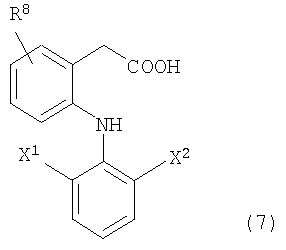
где R8 представляет собой заместитель, выбранный из низшей алкильной группы и низшей алкоксильной группы или атома водорода, и
каждый X1 и X2 независимо представляет собой заместитель, выбранный из низшей алкильной группы и трифторметильной группы или атома галогена, при условии, что по крайней мере один из X1 и X2 представляет собой атом галогена.
5. Производное гиалуроновой кислоты по п.1, где нестероидное противовоспалительное лекарственное средство, представленное группой Z-CO- является остатком, представленным следующей формулой (10)
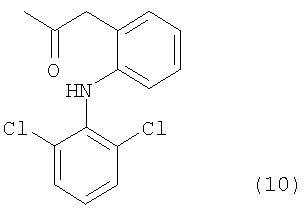
6. Производное гиалуроновой кислоты по любому из пп.1-5, где гиалуроновая кислота имеет средневзвешенную молекулярную массу от 500000 до 3000000.
7. Производное гиалуроновой кислоты по любому из пп.1-5, где R1 в формуле (1) представляет собой этиленовую группу, триметиленовую группу или пропиленовую группу, которая может иметь заместитель(заместители).
8. Производное гиалуроновой кислоты по любому из пп.1-6, полученное способом, включающим взаимодействие гиалуроновой кислоты со связанным со спейсером противовоспалительным лекарственным средством или взаимодействие связанной со спейсером гиалуроновой кислоты с противовоспалительным лекарственным средством и доведение реакционного раствора до щелочной среды.
9. Производное гиалуроновой кислоты по любому из пп.1-5, где раствор, получаемый растворением производного гиалуроновой кислоты в водной среде до концентрации 1,0 мас.%, способен проходить через пористый фильтр, имеющий размер пор 0,45 мкм и диаметр 25 мм со скоростью 2 мл в минуту или более при температуре 24°С под давлением 5,0 кг/см2.
10. Производное гиалуроновой кислоты по любому из пп.1-5, где раствор, получаемый растворением производного гиалуроновой кислоты в водной среде до концентрации 1,0 мас.%, способен проходить через пористый фильтр, имеющий размер пор 0,22 мкм и диаметр 25 мм со скоростью 2 мл в минуту или более при температуре 24°С под давлением 5,0 кг/см2.
11. Раствор производного гиалуроновой кислоты для подавления боли и/или подавления воспаления и/или лечения артрита, который можно пропускать через инжектор и который содержит производное гиалуроновой кислоты по любому из пп.1-10, растворенное в водной среде.
12. Раствор производного гиалуроновой кислоты по п.11, в котором водная среда представляет собой водную среду, выбранную из забуференного фосфатом физиологического раствора, физиологического раствора и воды для инъекций.
13. Раствор производного гиалуроновой кислоты по п.12, который простерилизован через фильтр.
14. Фармацевтическое средство для подавления боли и/или подавления воспаления и/или лечения артрита, которое содержит производное гиалуроновой кислоты по любому из пп.1-10 в качестве активного ингредиента,
15. Фармацевтическое средство по п.14, которое представляет собой средство для лечения артрита, противовоспалительное лекарственное средство или болеутоляющее средство.
16. Фармацевтическое средство по п.14 или 15, которое применимо для парентерального введения.
17. Фармацевтическое средство по п.16, которое представляет собой инъекцию, применимую для местного введения.
18. Фармацевтическое средство по п.16, которое представляет собой инъекцию, применимую для внутрисуставного введения.
19. Фармацевтическое средство для подавления боли и/или подавления воспаления и/или лечения артрита, которое можно пропускать через инжектор и которое содержит раствор, в котором производное гиалуроновой кислоты по любому из пп.1-10 в качестве активного ингредиента растворено в водной среде.
20. Набор для инъекции производного гиалуроновой кислоты, который включает раствор производного гиалуроновой кислоты по любому из пп.11-13, которым заполняют инжектор, способный пропустить раствор.
21. Набор по п.20, в котором раствор для заполнения представляет собой фармацевтическое средство по любому из пп.14-19.
22. Медицинский набор для инъекции, который герметично запакован с плунжером для вытеснения лекарственного средства таким образом, что оно может плавно двигаться, и который включает шприц, заполненный раствором, в котором производное гиалуроновой кислоты по любому из пп.1-10 растворено в фармацевтически приемлемом забуференном фосфатом физиологическом растворе, физиологическом растворе или воде для инъекций.
23. Производное, представленное следующей формулой (3)
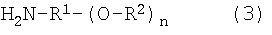
где R2 представляет собой атом водорода или остаток нестероидного противовоспалительного лекарственного средства, представленного формулой Z-CO- при условии, что все R2 не являются атомом водорода;
H2N-R1-(O-)n представляет собой остаток спейсера в соединении-спейсере, представленном формулой H2N-R1-(OH)n, содержащем гидроксильные группы в количестве n;
R1 представляет собой линейную или разветвленную углеводородную группу, содержащую от 2 до 12 атомов углерода, которая может иметь заместители;
-O-СО- представляет собой сложноэфирную связь, состоящую из гидроксильной группы соединения-спейсера и карбоксильной группы в остатке нестероидного противовоспалительного лекарственного средства и n равно целому числу от 1 до 3.
24. Производное по п.23, где нестероидное противовоспалительное лекарственное средство выбирают из группы, включающей кетопрофен, напроксен, ибупрофен, флурбипрофен, ацетилсалициловую кислоту, фелбинак, мефенамовую кислоту, диклофенак и этодолак.
25. Производное по п.23, где противовоспалительное лекарственное средство представляет собой соединение, представленное вышеуказанной формулой (2).
26. Производное по любому из пп.23-25, где R1 в вышеуказанной формуле (3) представляет собой этиленовую группу, триметиленовую группу или пропиленовую группу, которая может иметь заместитель(заместители).
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| Способ крашения тканей | 1922 |
|
SU62A1 |
| Способ приготовления сернистого красителя защитного цвета | 1915 |
|
SU63A1 |
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| СПОСОБ ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОЙ ГИАЛУРОНОВОЙ КИСЛОТЫ | 2000 |
|
RU2191782C2 |

