ПРАВИТЕЛЬСТВЕННАЯ ПОДДЕРЖКА
Настоящее изобретение было сделано при правительственной поддержке Национальных институтов здравоохранения США, грант R44-AA-009930. Правительство США обладает определенными правами на данное изобретение.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к анальгезирующим композициям, содержащим аминохинолиновые соединения вместе с опиоидами, ингибиторами обратного захвата норэпинефрина/серотонина и/или нестероидными противовоспалительными средствами (НПВС).
УРОВЕНЬ ТЕХНИКИ
Хотя острая боль в ответ на травму является важным механизмом уменьшения степени повреждения у человека, нервная система может подвергаться адаптивному изменению, которое приводит к боли, которая ощущается намного дольше после заживления травмы (хроническая боль) (Costigan et al., 2009). Эта хроническая боль может быть вызвана раздражителем, который обычно ее не вызывает (аллодиния), или реакция на опасный раздражитель может быть многократно усилена (гипералгезия). По некоторым оценкам, хроническая боль поражает не меньше 100 миллионов взрослых в США и может отрицательно влиять на качество жизни (Institute of Medicine, 2011). Медикаментозное лечение нейропатической (вызванной повреждением нерва) или другой хронической боли в значительной степени основано на применении опиатов или их производных (Reuben et al., 2015). Эти препараты обладают множеством побочных эффектов, включая толерантность/гипералгезию, что приводит к повышению дозы и развитию опиатной зависимости (Chou et al., 2015). Дополнительные побочные эффекты при длительном приеме опиатов в высоких дозах включают запор, нарушение дыхания во сне, переломы, дисрегуляцию гипоталамо-гипофизарно-надпочечниковой оси и передозировку, а также эффекты со стороны сердечно-сосудистой и иммунной системы (Baldini et al., 2012). Другие препараты, включая нестероидные противовоспалительные средства (НПВС), противосудорожные средства, миорелаксанты и антидепрессанты, а также средства, воздействующие на потенциалзависимые кальциевые каналы (габапентин и прегабалин), применяют для лечения хронической боли, однако эти методы лечения дают ограниченное облегчение (Lunn et al., 2014; Moore et al., 2014; Schreiber et al., 2015; Smith et al., 2012; Sofat et al., 2017; Lozada, et al., 2008). Кроме того, длительное введение высоких доз НПВС, следующих по популярности после опиоидов средств для лечения хронической боли, ассоциировано с множеством побочных эффектов, включая проблемы с желудком (такие как кровотечение, язва и расстройство желудка), почечную недостаточность, повышенное артериальное давление или нарушения работы сердца, задержку жидкости, сыпь или другие аллергические реакции (Marcum and Hani, 2010). Третья категория средств для лечения хронической боли - это недавно представленные блокаторы систем обратного захвата 5-ГТ и НЭ (Smith et al., 2012; Sofat et al., 2017). Ингибиторы обратного захвата 5-ГТ/НЭ также вызывают ряд побочных эффектов, включая тошноту, нарушения со стороны ЖКТ, утомляемость, но при этом также нарушение сна. Более опасными являются последствия быстрого прекращения применения этих лекарственных средств, если они не снимают хроническую боль. Эти эффекты "отмены" включают резкие перепады настроения, возбуждение, агрессию, ночные кошмары, спутанность сознания и ощущения наподобие электрошока в голове и других частях тела (Fava et al., 2018; Carvalho et al., 2016). Во всех случаях, будь то опиаты/опиоиды, НПВС или ингибиторы обратного захвата 5-ГТ/НЭ, повышение дозы для терапевтического успеха при лечении хронической боли приводит к появлению тяжелых побочных эффектов и более тяжелых симптомов отмены, если лекарственную терапию резко прекращают.
Большинство лиц, страдающих хронической болью, у которых боль не удается контролировать другими классами лекарственных средств, продолжают применять назначенные опиаты/опиоиды в течение длительных периодов времени и в высоких дозах. Из-за растущей обеспокоенности по поводу значительного увеличения применения опиатов для лечения хронической боли и сопутствующих проблем, связанных с передозировкой, ненадлежащим применением и передачей третьим лицам, называемых "опиоидным кризисом", были разработаны рекомендации по ограничению применения назначаемых опиоидов до наименьшей эффективной дозы и в течение минимально коротких сроков эффективного применения, и, что важно, по разработке новых неопиоидных лекарственных средств на основе научной информации об этиологии хронической боли (Volkow and McLellan, 2016; Taneja et al., 2017; Kirkpatrick et al., 2016).
Что касается боли, можно сосредоточиться на системах, которые передают сенсорную информацию от периферических рецепторов, и на системах, которые передают информацию внутри и между сенсорными нейронами. Одним из наиболее изученных молекулярных механизмов, приводящих к развитию синдромов хронической нейропатической боли, является повышенная активность периферических потенциалзависимых натриевых каналов (ПЗНК) (Wood et al., 2004; Lai, et al., 2004; Black et al., 2004; Coggeshall et al., 2004; Dib-Hajj et al., 2007). Тетродотоксин-чувствительный канал Nav1.7 расположен вдоль отростков и тел медленно проводящих ноцицептивных нейронов, и его роль, как при острой, так и при хронической боли, была четко продемонстрирована с помощью генетических манипуляций у животных и при природных генетических мутациях у людей (Black et al., 2004; Wang et al., 2011; Lawrence, 2012). В частности, канал Nav1.7 был связан с болью, ассоциированной с воспалением, при этом его активация способствует увеличению генерации и проводимости потенциалов действия при хронических болевых синдромах (Eijkelkamp et al., 2012). Кроме того, активность канала Nav1.7 может усиливать генераторные потенциалы и способствовать активации ПЗНК других сенсорных нейронов, включая устойчивый к тетродотоксину канал Nav1.8 (Dib-Hajj et al., 2007; Choi & Waxman, 2011). Канал Nav1.8 был связан с развитием как воспалительных, так и нейропатических болевых состояний. В целом, усиление активности каналов Nav1.7 и Nav1.8 в периферических сенсорных нейронах составляет общий компонент индукции и поддержания хронических болевых синдромов (Wang et al., 2011; Theile & Cummins, 2011; Laedermann et al., 2015).
Роль возбуждающей аминокислоты, глутамата, в физиологии нормального ощущения и передачи боли в случае хронической боли также хорошо известна (Davies & Lodge, 1987; Dickenson & Sullivan, 1987; Childers & Baudy, 2007). Активация или повреждение сенсорных нейронов вызывает усиленное высвобождение глутамата как периферическими, так и центральными нейронами, при этом высвобождаемый глутамат может действовать на соседние рецепторы глутамата (NMDA), способствуя периферической сенсибилизации (Fernandez-Montoya et al., 2017; Jang et al., 2004). Взаимодействие глутамата с NMDA-рецепторами в ганглиях задних корешков (DRG) также участвует в усилении сенсорных сигналов (Ferrari et al., 2014; Rozanski et al., 2013). Таким образом, NMDA-рецепторы участвуют как в инициации, так и в усилении ощущения боли и его передаче в ЦНС. Апрегуляция NMDA-рецепторов наблюдается как в периферических нейронах, так и в спинном мозге после повреждения сенсорных нервов, причем считается, что такая апрегуляция способствует хронической нейропатической боли (Petrenko et al., 2003). В частности, количество NMDA-рецепторов, содержащих субъединицу GluN2B (NR2B), играет наиболее важную роль в развитии и поддержании хронических болевых синдромов (Karlsson et al., 2002; Iwata et al., 2007; Gaunitz et al., 2002; Wilson et al., 2005).
На основании этих доводов, лекарственное средство, которое может одновременно ингибировать активность каналов Nav1.7 и Nav1.8, а также ингибировать активность NMDA-рецепторов (особенно NMDA-рецепторов, которые содержат субъединицу GluN2B), может быть полезным как для предотвращения развития хронической боли путем ингибирования апрегуляции рецепторов/каналов, так и для уменьшения боли даже после развития хронического болевого синдрома. Такое лекарственное средство не должно действовать в центральной нервной системе, однако должно быть способным предотвращать периферическую сенсибилизацию, которая приводит к развитию центральной сенсибилизации и хронической боли, и/или ослаблять инициацию и передачу сигналов хронической боли в головной мозг.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
N-замещенные-4-уреидо-5,7-дихлор-2-карбокси (или карбоксиэфир) хинолины в комбинации с опиоидами, ингибиторами обратного захвата НЭ/5-ГТ или НПВС являются эффективными при лечении и предупреждении хронической нейропатической боли у человека.
Анальгезирующие композиции, в которых реализовано настоящее изобретение, содержат аминохинолиновое соединение вместе с опиоидом, ингибитором обратного захвата НЭ/5-ГТ, нестероидным противовоспалительным средством (НПВС) или их комбинацией. Аминохинолиновое соединение потенцирует биоактивность опиоидов, средств, которые блокируют обратный захват (5-ГТ) серотонина и норэпинефрина (НЭ), и НПВС. В результате совместное введение аминохинолинового соединения позволяет использовать более низкую дозу опиоида, блокатора обратного захвата НЭ или 5ГТ или НПВС для достижения требуемого анальгезирующего (антигипералгезического) действия. Кроме того, аминохинолиновое соединение, в случае раннего введения в ходе развития хронической боли, может предотвращать развитие хронической боли и/или обострение хронического болевого синдрома.
В аминохинолиновых соединениях, представленных Формулой (I):
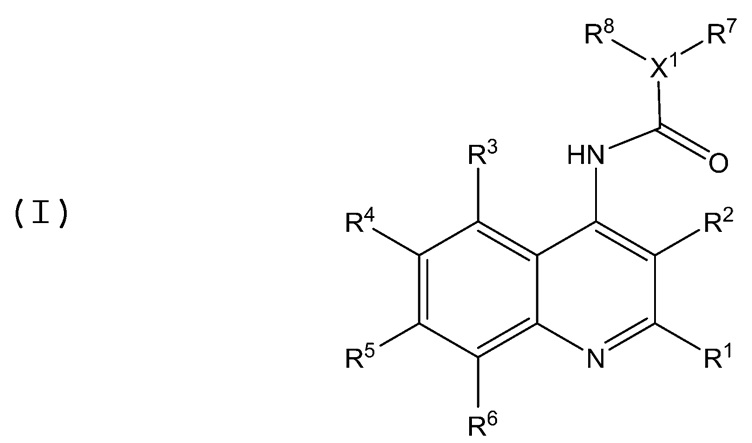
заместители определены следующим образом: R1 представляет собой H, C2-C4 алкил, C2-C4 алкенил, галоген, Z1R9 или N(R10)(R11). R2 представляет собой H, C1-C4 алкил, C2-C4 алкенил, галоген, Z2R12, N(R13)(R14), или C1-C4 алкил, замещенный одной или более группами, выбранными из группы, состоящей из C1-C4 алкила, C2-C4 алкенила, галогена, Z3R15, N(R16)(R17); каждый R3, R4, R5 и R6 независимо представляют собой H, C1-C4 алкил, C2-C4 алкенил, галоген, Z3R18 или N(R19)(R20); X1 представляет собой N или CH; каждый R7 и R8 независимо представляют собой H, C1-С6 алкил, C2-C4 алкенил, C2-C4 алкинил, арил или C1-С6 алкил, замещенный одной или более группами, выбранными из группы, состоящей из C1-C4 алкила, C2-C4 алкенила, нитро, галогена, Z4R21 и N(R22)(R23); или R7 и R8 вместе с X1 образуют 5-8-членную насыщенную, ненасыщенную или ароматическую органическую циклическую или гетероциклическую группу; каждый R9, R10, R11, R12, R13, R14, R15, R16, R17, R18, R19, R20, R21, R22 и R23 независимо представляет собой H, C1-C4 алкил или C1-C4 алкил, замещенный одной или более группами, выбранными из группы, состоящей из C1-C4 алкила, C2-C4 алкенила, C2-C4 алкинила, галогена, гетероарила, Z5R24 и N(R25)(R26). Каждый из Z1, Z2, Z3, Z4 и Z5 независимо представляет собой O, S, NH, C(=O)O, O-C(=O), C(=O) или C(=O)NH. Каждый R24, R25 и R26 независимо представляет собой C1-C4 алкил при условии, что в случае, когда R1 представляет собой Z1R9, Z1 представляет собой C(=O)O, R9 представляет собой H или C1-C2 алкил, каждый из R3 и R5 представляет собой галоген, X1 представляет собой N, и каждый из R4 и R6 представляет собой H, то тогда по меньшей мере один из R7 и R8 не является фенилом, алкокси-замещенным фенилом или C1-С6 алкильной группой.
В аминохинолиновых соединениях Формулы (II):
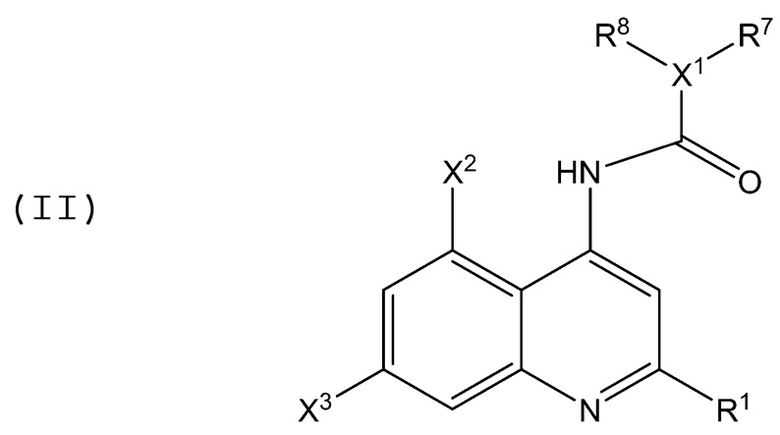
X1, R1, R7 и R8 имеют значение, определенное в Формуле (I) выше, и каждый X2 и X3 независимо представляет собой электроноакцепторную группу, такую как галоген, нитро и т.п., при условии, что в случае, когда R1 представляет собой Z1R9, Z1 представляет собой C(=O)O или C(=O), R9 представляет собой H или C1-C4 алкил, и X1 представляет собой N, то тогда по меньшей мере один из R7 и R8 не является фенилом или алкокси-замещенной группой.
В аминохинолиновых соединениях Формулы (III):
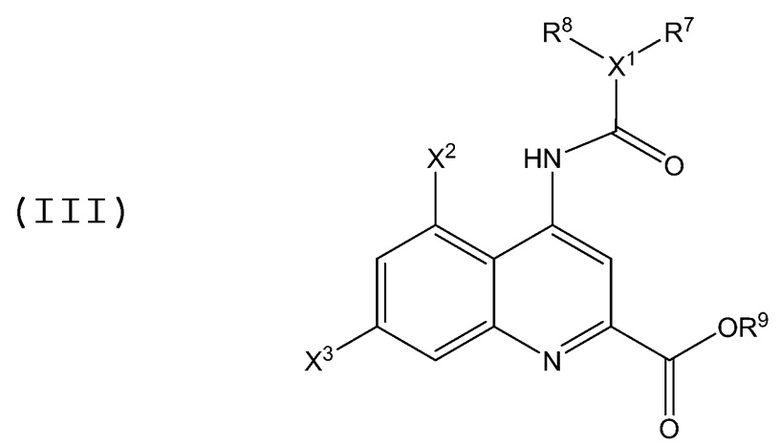
каждый X2 и X3 независимо является галогеном, и каждый из X1, R1, R7, R8 и R9 имеет значение, определенное в Формулах (I) и (II), описанных выше, при условии, что в случае, когда R9 представляет собой H или C1-C2 алкил, и X1 представляет собой N, то тогда по меньшей мере один из R7 и R8 не является фенилом или алкокси-замещенной фенильной группой.
В аминохинолиновых соединениях Формулы (IV):
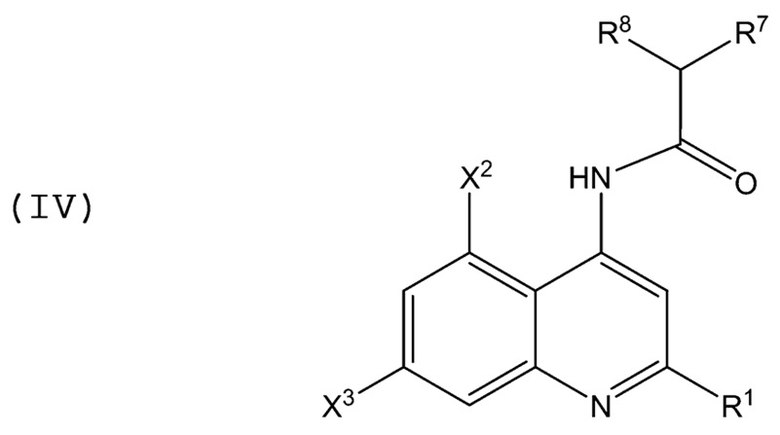
каждый из X2, X3, R1, R7 и R8 имеет значение, определенное в Формулах (I) и (II) выше.
В аминохинолиновых соединениях Формулы (V):
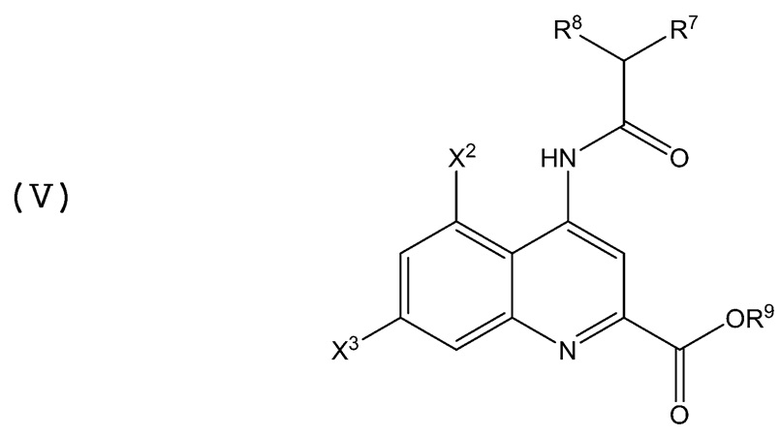
каждый из X2, X3, R7, R8 и R9 имеет значение, определенное в Формулах (I) и (II) выше.
В аминохинолиновых соединениях Формулы (VI):
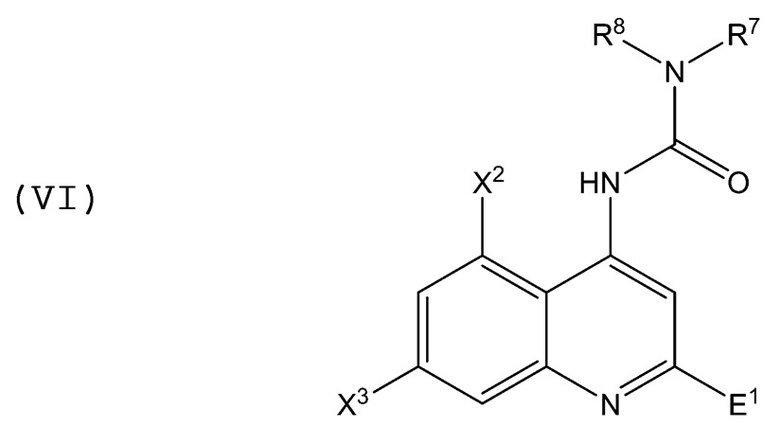
R7 представляет собой алкил, циклоалкил, аминоалкил или фенил; R8 представляет собой H, алкил, циклоалкил, аминоалкил или фенил; E1 представляет собой -C(=O)OR9, -C(=O)R9, -C(=O)N(R9)2 и -[C(R9)2]n-OR9; "n" равно 1, 2, 3 или 4; каждый R9 независимо представляет собой H, C1-C4 алкил или C1-C4 алкил, замещенный одной или более группами, выбранными из группы, состоящей из C1-C4 алкила, C2-C4 алкенила, C2-C4 алкинила, галогена, гетероарила, Z5R24 и N(R25)(R26); Z5 представляет собой O, S, C(=O)O или O-C(=O); каждый R24, R25 и R26 независимо представляет собой C1-C4 алкил; каждый X2 и X3 независимо представляет собой электроноакцепторную группу (предпочтительно галоген или нитро); алкильные, циклоалкильные, аминоалкильные и фенильные группы могут быть незамещенными или могут быть замещены один или более раз алкильной (1-3 углерода) группой или алкилоксигруппой (например, алкильной или алкоксигруппой, содержащей 1-3 углерода); и в случае присутствия кислотных или основных функциональных групп соединение может находиться в форме свободной кислоты, свободного основания или может быть фармакологически приемлемой солью присоединения. Когда E1 представляет собой C(=O)OR9, по меньшей мере один из R7 и R8 не является фенилом.
Особенно предпочтительными для применения в настоящих анальгезирующих композициях являются соединения Формулы (VI) в форме свободной кислоты, свободного основания или фармакологически приемлемой соли присоединения, где:
R7 представляет собой алкил (предпочтительно алкил, содержащий 3-6 атомов углерода) или фенил;
R8 представляет собой алкил (предпочтительно алкил, содержащий 3-6 атомов углерода) или фенил;
E1 представляет собой -C(=O)R9, или E1 представляет собой -C(=O)OR9;
каждый R9 представляет собой H или C1-C4 алкил; и
каждый X2 и X3 независимо представляет собой электроноакцепторную группу (предпочтительно галоген или нитро).
Введение анальгезирующих композиций может осуществляться пероральным, подкожным, внутривенным, внутримышечным, внутрибрюшинным, трансдермальным или трансбуккальным путями.
Неограничивающими примерами соединений общей Формулы (VI) являются производные 2-карбокси-хинолинов, например, (N, N-дибутил)-4-уреидо-5,7-дихлор-2-карбокси-хинолин (BCUKA), (N, N-дифенил)-4-уреидо-5,7-дихлор-2-карбокси-хинолин (DCUKA) и т.п.
Дизамещенные 4-уреидо-5,7-дихлор-2-карбокси-хинолиновые соединения Формулы (VI) обладают сродством к некоторым или всем из следующего: Nav1.7, Nav1.8 и NMDA-рецепторы. Эти соединения обладают полезной активностью при лечении хронических болевых синдромов, возникающих вследствие боли при дегенеративных заболеваниях суставов (например, остеоартрозе) и воспалительного и механического повреждения периферических нервов, эффективно снижая тяжесть механической или термической аллодинии/гипералгезии.
Соединения Формулы (VI) могут быть получены путем амидирования промежуточного 5,7-дихлорхинолон-2-карбоксилата (например, который может быть получен при присоединении по Михаэлю 3,5-дихлоранилина к диметилацетилендикарбоксилату с последующей термической циклизацией полученного арилмалеата) с хлорсульфонилизоцианатом, с получением сложного этилового эфира (4-амино)-5,7-дихлор-2-карбокси-хинолина (ключевого промежуточного соединения), который может быть функционализирован посредством реакций с соответствующими электрофилами. Для получения монозамещенных мочевин реакционноспособное промежуточное соединение мочевины получают в реакции первичного амина с карбонилдиимидазолом. Реакция получаемой имидазолмочевины со сложным этиловым эфиром амином-5,7-дихлор-2-карбокси-хинолина в присутствии гидроксида натрия дает целевую монозамещенную мочевину в 4-ом положении хинолина с сопутствующим гидролизом сложного эфира. Удаление защитной группы, такой как трет-бутоксикарбонильная (BOC) защитная группа, если такая используется в синтезе реакционноспособного промежуточного соединения мочевины, может быть выполнено при использовании трифторуксусной кислоты (ТФУ) с получением требуемой соли ТФУ.
Для получения дизамещенных производных мочевины метиловый или этиловый сложный эфир (4-амино)-5,7-дихлор-2-карбокси-хинолина ацетилируют по 4-аминоположению двузамещенным карбамоилхлоридом с образованием (N, N-дизамещенного) 4-уреидо-5,7-дихлор-2-карбокси-хинолинового сложного эфира. Необязательно (N, N-дизамещенный) 4-уреидо-5,7-дихлор-2-карбокси-хинолиновый сложный эфир может быть гидролизован до (N, N-дизамещенного) 4-уреидо-5,7-дихлор-2-карбокси-хинолина.
DCUKA и DCUK-OEt демонстрируют сродство к Nav1.7 и Nav1.8, причем DCUKA также демонстрирует сродство к NMDA-рецептору. DCUK-OEt также может действовать как пролекарство для DCUKA с гидролизом сложного эфира при воздействии карбоксилэстеразы 1, который происходит после введения in vivo.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 графически иллюстрирует, что DCUK-OEt может действовать как пролекарство для DCUKA. Данные, полученные при пероральном введении DCUK-OEt крысам в суспензии высушеного распылением дисперсионного состава, показывают, что уровни DCUKA в крови зависели от дозы DCUK-OEt, которую вводили. Пиковый уровень DCUKA составил ~3 мкМ после дозы DCUK-OEt 50 мг/кг и ~11 мкМ после дозы DCUK-OEt 100 мг/кг.
Фиг. 2 графически иллюстрирует лечение индуцированной цисплатином нейропатической боли DCUKA (50 мг/кг). У крыс, обработанных противоопухолевым химиотерапевтическим средством, цисплатином, лечение цисплатином снижает болевой порог при механическом воздействии, а лечение DCUKA устраняет этот эффект и увеличивает болевой порог при механическом воздействии до контрольных уровней. Данные показывают отношение порога механической боли после введения цисплатина или цисплатина плюс DCUKA к порогу механической боли до введения цисплатина.
Фиг. 3 графически иллюстрирует сравнение эффектов эквимолярных доз DCUKA, BCUKA и габапентина, требуемых для устранения индуцированной цисплатином нейропатической боли, измеряемой по изменениям порога механической боли.
Фиг. 4 графически иллюстрирует купирование с помощью DCUKA (50 мг/кг) нейропатической боли, вызванной при введении крысам полного адъюванта Фрейнда (CFA). Введение CFA в лапу крысам вызывает воспаление и снижает порог механический боли. Лечение DCUKA устраняет снижение порога механической боли у получивших CFA крыс и возвращает порог к исходному уровню. Данные показывают отношение порога механической боли после введения CFA или CFA плюс DCUKA к порогу механической боли до введения CFA.
Фиг. 5 иллюстрирует сравнение эффекта DCUKA (50 мг/кг) и BCUKA (50 мг/кг) при купировании нейропатической боли, вызванной введением CFA крысам. Данные показывают отношение порога механической боли после введения CFA или CFA плюс DCUKA или BCUKA к исходному порогу механической боли (до введения CFA).
На Фиг. 6 показаны результаты мета-анализа экспериментов по определению дозозависимого эффекта DCUKA при купировании CFA-индуцированной нейропатической боли.
Фиг. 7 графически иллюстрирует лечение боли, вызванной диабетической нейропатией, с применением DCUKA (50 мг/кг). Крысам вводили стрептозотоцин (STZ) для индукции диабета, который снижал порог механической боли по сравнению с исходным уровнем (до введения STZ). Лечение DCUKA возвращало порог механической боли к исходному уровню. Данные показывают отношение порога механической боли после введения STZ или STZ плюс DCUKA к порогу механической боли до введения STZ.
На Фиг. 8 показаны результаты мета-анализа экспериментов по определению дозозависимого эффекта DCUKA при купировании STZ-индуцированной нейропатической боли.
Фиг. 9 графически иллюстрирует лечение остеоартрозной боли с применением DCUKA. Крысам вводили монойодуксусную кислоту (MIA), которая вызывает воспалительную реакцию. Повреждение и разрушение хряща приводит к хронической нейропатической боли, которая выражается в сниженном пороге механической боли по сравнению с исходным уровнем, измеренным у животных, не получавших MIA или лекарственное средство. DCUKA устранял снижение порога механической боли дозозависимым образом. Данные показывают отношение порога механической боли после введения MIA или MIA плюс DCUKA к порогу механической боли у животных, не получавших MIA или лекарственное средство.
На Фиг. 10A графически показано, что введение DCUKA улучшает способность морфина купировать CFA-индуцированную нейропатическую боль.
На Фиг. 10B графически показано, что введение DCUK-OEt улучшает способность морфина купировать CFA-индуцированную нейропатическую боль.
На Фиг. 10C показаны результаты изоболографического анализа, которые демонстрируют, что DCUK-OEt значительно снижал полумаксимальную эффективную дозу морфина, требуемую для купирования боли.
Фиг. 11 иллюстрирует, что DCUKA потенцирует способность оксикодона купировать CFA-индуцированную нейропатическую боль.
Фиг. 12 иллюстрирует, что DCUKA потенцирует способность метадона купировать CFA-индуцированную нейропатическую боль.
Фиг. 13 иллюстрирует, что DCUKA потенцирует способность трамадола купировать MIA-индуцированную остеоартрозную (нейропатическую) боль.
Фиг. 14 иллюстрирует, что DCUKA потенцирует способность аспирина купировать STZ-индуцированную нейропатическую боль (диабетическая нейропатия).
Фиг. 15 иллюстрирует, что DCUKA потенцирует способность диклофенака купировать CFA-индуцированную нейропатическую боль.
Фиг. 16 графически иллюстрирует, что введение DCUKA после инъекции CFA предотвращает развитие CFA-индуцированной нейропатической боли, измеряемой по изменениям порога механической боли.
Фиг. 17 графически иллюстрирует, что введение DCUKA одновременно с противоопухолевым химиотерапевтическим средством цисплатином предотвращает развитие индуцированной цисплатином нейропатической боли, измеряемой по изменениям порога механической боли.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Анальгезирующие композиции, описанные в настоящем документе, хорошо подходят для лечения хронического (нейропатического) болевого синдрома.
Способы, описанные в настоящем документе, включают лечение субъекта, нуждающегося в облегчении боли (например, пациента-человека или животного), с применением содержащих аминохинолин композиций, которые также включают опиоид, ингибитор обратного захвата НЭ/5-ГТ и/или нестероидное противовоспалительное средство (НПВС).
Опиоиды, подходящие для применения в анальгезирующих композициях, включают опиаты, т.е. природные растительные алкалоиды, такие как морфин, кодеин, папаверин, тебаин и т.п.; полусинтетические опиоиды, такие как оксикодон, диаморфин, дигидрокодеин и т.п.; а также синтетические опиоиды, такие как производные фенилпиридина, например, метиламид 6-амино-5-(2,3,5-трихлорфенил)-пиридин-2-карбоновой кислоты и т.п.; производные фенилпиперидина, например, фентанил, сульфентанил, алфентанил и т.п.; производные морфинана, например, леворфанол, буторфанол и т.п.; производные дифенилгептана, например, метадон, пропоксифен и т.п.; производные бензоморфана, например, пентазоцин, феназоцин и т.п.
Мультитаргетные лекарственные средства, подходящие для применения в анальгезирующих композициях, включают средства, которые воздействуют на опиатный рецептор и/или переносчики обратного захвата моноаминов, т.е. трамадол и т.п.
НПВС, подходящие для применения в анальгезирующих композициях, включают аспирин, производные уксусной кислоты, такие как индометацин, сулиндак, этодолак, толметин, кеторолак, набуметон, диклофенак и т.п., производные пропионовой кислоты, такие как ибупрофен, напроксен, фенопрофен, кетопрофен, флурбипрофен, оксапрозин и т.п., производные еноловой кислоты, такие как пироксикам, мелоксикам, теноксикам и т.п., производные фенамовой кислоты, такие как мефенамовую кислоту, меклофенамовую кислоту, флуфенамовую кислоту и т.п., а также фармацевтически приемлемые соли вышеуказанного.
Иллюстративные соли НПВС, подходящие для применения в настоящих композициях, представляют собой фармацевтически приемлемые соли вышеуказанных производных уксусной кислоты, например, соли индометацина, такие как индометацин натрия, индометацин меглумин и т.п., соли толметина, такие как толметин натрия и т.п., соли кеторолака, такие как кеторолака трометамин и т.п., соли диклофенака, такие как диклофенак натрия, диклофенак диэтиламин, диклофенак эполамин и т.п., а также фармацевтически приемлемые соли вышеуказанных производных пропионовой кислоты, например, соли ибупрофена, такие как ибупрофен-лизин, ибупрофен метилглюкамин и т.п., соли напроксена, такие как напроксен пиперазин, напроксен натрия и т.п., соли фенопрофена, такие как фенопрофен кальция и т.п.
Аминохинолиновые соединения Формулы (I), (II), (III), (IV), (V) и (VI) могут быть получены любым удобным способом, известным специалистам в данной области. Например, в патенте США 6,962,930 (Tabakoff et al.) и в патенте США 7,923,458 (Tabakoff), которые включены в настоящий документ посредством отсылки во всей своей полноте, описано получение некоторых соединений хинолина, аналогичных соединениям настоящего изобретения, которое с легкостью может быть адаптировано для получения требуемых аминохинолиновых соединений. На Схеме 1 представлена общая схема получения аминохинолинового соединения Формулы (I) и структурно близких или аналогичных соединений из 4-аминозамещенного хинолина, Соединения (A), в котором заместители R совпадают с заместителями в Формуле (I). Аминогруппу Соединения (A) подвергают реакции с активированным ацилирующим Соединением (B), включающим уходящую группу (LG), которая способна реагировать с ароматическими аминогруппами с образованием соединения Формулы (I). Замещенные хинолиновые соединения, имеющие аминогруппу в 4-ом положении кольцевой структуры хинолина, такие как Соединение (A), имеющие различные профили замещения на кольцевой системе хинолина, а также их получение, известны средним специалистам в области химических наук. Защитные группы, такие как раскрытые в публикации Protective Groups in Organic Synthesis, 3rd Ed., Green and Wuts, Eds., John Wiley & Sons, Inc. (1999), которая включена в настоящий документ посредством отсылки, могут использоваться при получении Соединения (A), Соединения (B) и/или при сочетании Соединения (A) и Соединения (B), в случае необходимости или потребности в облегчении получения и/или выделения соединений Формулы (I).
Схема 1
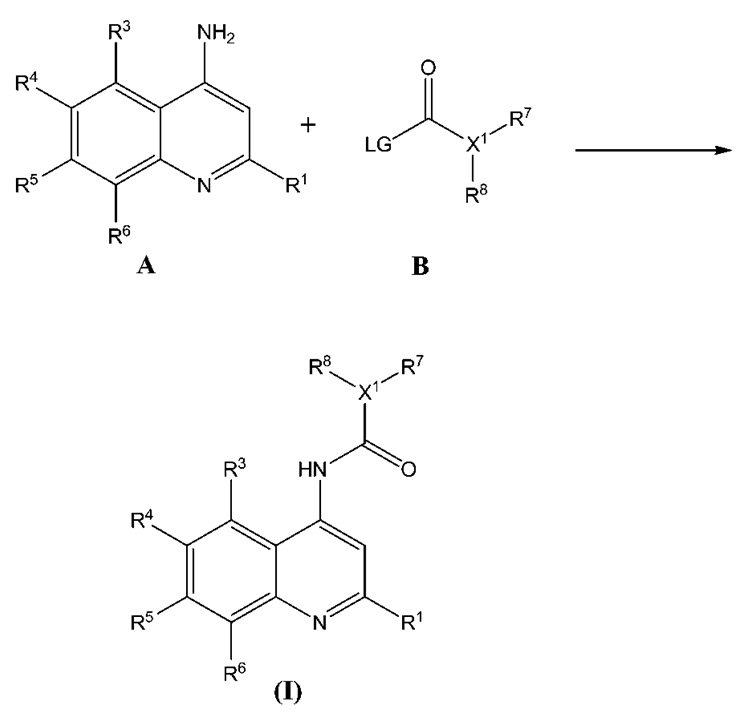
При использовании в настоящем документе термин "аминохинолиновое соединение" относится к соединениям, представленным в Формулах (I), (II), (III), (IV), (V) и (VI), как описано в настоящем документе. Аминохинолиновые соединения могут применяться при хронической боли и множестве других состояний.
Термин "алкил" при использовании в настоящем документе направлен на насыщенную углеводородную группу (обозначенную формулой CnH2n+1), которая является нормальной, разветвленной или циклизованной ("циклоалкил") и которая является незамещенной или замещенной, т.е. в такой группе один или более атомов водорода заменены другим атомом или молекулой.
"Арил" обозначает либо бензольное кольцо из 6 углеродов, либо конденсированные 6-углеродные кольца других ароматических производных (см., например, Hawley's Condensed Chemical Dictionary (13 ed.), R. J. Lewis, ed., J. Wiley & Sons, Inc., New York (1997)). Арильные группы включают, без ограничения, фенил и нафтил.
"Гетероарильные" кольца являются ароматическими кольцами, включающими по меньшей мере один атом углерода в кольце, при этом один или более, как правило от 1-4, атомов, формирующих кольцо, не являются атомами углерода, т.е. являются гетероатомом (как правило, O, N или S). Гетероарил включает, без ограничения: морфолинил, пиперазинил, пиперидинил, пиридил, пирролидинил, пиримидинил, триазинил, фуранил, хинолинил, изохинолинил, тиенил, имидазолил, тиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензотиазолил, бензоксазолил, изоксазолил, триазолил, тетразолил, индазолил, индолинил, индолил-4,7-дион, 1,2-диалкил-индолил, 1,2-диметил-индолил и 1,2-диалкил-индолил-4,7-дион.
"Алкокси" означает -OR, где R представляет собой алкил, как определено выше, например, метокси, этокси, пропокси, 2-пропокси и т.п.
"Алкенил" означает линейный моновалентный углеводородный радикал из двух - шести атомов углерода или разветвленный моновалентный углеводородный радикал из трех - шести атомов углерода, содержащий по меньшей мере одну двойную связь, например, винил, пропенил и т.п.
"Алкинил" означает линейный моновалентный углеводородный радикал из двух - шести атомов углерода или разветвленный двухвалентный углеводородный радикал из трех - шести атомов углерода, содержащий по меньшей мере одну тройную связь, например, этинил, пропинил и т.п.
"Галогенид" и "галоген" относятся к атому галогена, включающему фтор, хлор, бром и иод.
Группы заместителей, например, C1-6 алкил, известны и, как указано в настоящем описании, включают каждый из индивидуальных заместителей в группе, например, C1 алкил, C2 алкил, C3 алкил и C4 алкил.
"Замещенный" означает, что один или более атомов водорода на обозначенном атоме заменены заместителем, выбранным из указанной группы, при условии, что обычная валентность обозначенного атома не превышена, и что замещение приводит к образованию стабильного соединения.
"Незамещенные" атомы несут все возможные атомы водорода в соответствии со своей валентностью. Когда заместитель представляет собой, например, "кето", то тогда на атоме будут заменены два водорода. Комбинации заместителей и/или переменных допустимы только в том случае, если такие комбинации приводят к образованию стабильных соединений; под "стабильным соединением" или "стабильной структурой" подразумевается соединение, которое является достаточно устойчивым, чтобы перенести выделение до пригодной степени чистоты из реакционной смеси и изготовление композиции эффективного терапевтического средства.
"Фармацевтически приемлемый" при использовании в отношении солей или носителей относится к материалам, которые обычно считаются подходящими для введения или контакта с телом или частями тела человека. Фармацевтически приемлемые соли являются материалами, в которых исходное соединение (например, аминохинолиновое соединение Формулы (I)) или какое-либо другое терапевтическое средство или вспомогательно вещество модифицированы путем получения их солей с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, без ограничения перечисленными, соли неорганических или органических кислот с основными остатками, такими как амины, или соли щелочных металлов или органических оснований с кислотными остатками, такими как карбоновые кислоты. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Такие обычные нетоксичные соли включают соли, полученные из таких неорганических кислот, как хлороводородная, бромоводородная, серная, сульфаминовая, фосфорная, азотная и т.п.; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изетионовая и т.п. Фармацевтически приемлемые соли представляют собой такие формы соединений, которые подходят для применения в контакте с тканями человека и животных, не вызывая чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соизмеримо с разумным отношением выгоды/риска.
Фармацевтически приемлемые формы солей аминохинолиновых соединений, предложенных в настоящем документе, синтезируют из исходного соединения, которое содержит основную или кислотную группу, с помощью стандартных химических методов. Обычно такие соли получают, например, при взаимодействии форм свободных кислот или оснований этих соединений со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе, или в их смеси. Как правило, предпочтительны неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечень подходящих солей можно найти в справочнике Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418, содержание которого включено в настоящий документ посредством отсылки.
"Пролекарства" представляют собой любые ковалентно связанные носители, которые высвобождают активное исходное лекарственное средство аминохинолиновых соединений in vivo, когда такое пролекарство вводят млекопитающему. Пролекарства аминохинолиновых соединений согласно настоящему изобретению получают путем модификации функциональных групп, присутствующих в соединениях, таким образом, что модификации расщепляются, либо в результате стандартных манипуляций, либо in vivo, включая ферментативное превращение, с образованием исходных соединений. Пролекарства включают соединения, в которых гидрокси, амино или сульфгидрильные группы связаны с любой группой, которая при введении млекопитающему отщепляется с образованием свободной гидроксильной, амино или сульфгидрильной группы соответственно. Примеры пролекарств включают, без ограничения перечисленными, ацетатные, формиатные и бензоатные производные функциональных спиртовых и аминогрупп в аминохинолиновых соединениях согласно настоящему изобретению и т.п. Соединения, которые эффективно действуют в качестве пролекарств аминохинолиновых соединений согласно настоящему изобретению, могут быть идентифицированы с использованием стандартных методик, известных в данной области. Примеры таких производных пролекарств см., например, в следующих публикациях: (a) Design of Prodrugs, edited by H. Bundgaard, (Elsevier, 1985) and Methods in Enzymology, Vol. 42, p. 309-396, edited by K. Widder et al. (Academic Press, 1985); (b) А Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H. Bundgaard, Chapter 5 "Design and Application of Prodrugs", by H. Bundgaard p. 113-191 (1991); (с) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992); (d) H. Bundgaard et al., Journal of Pharmaceutical Sciences, 77:285 (1988); и (e) N. Kakeya et al., Chem. Pharm. Bull, 32: 692 (1984), каждая из которых прямо включена в настоящий документ посредством отсылки.
Кроме того, изобретение также включает сольваты, метаболиты и фармацевтически приемлемые соли аминохинолиновых соединений.
Термин "сольват" относится к агрегату молекулы с одной или более молекулами растворителя. "Метаболит" является фармакологически активным продуктом, образующимся in vivo в процессе метаболизма указанного соединения или его соли в организме. Такие продукты могут образовываться, например, в результате окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, деэтерификации, ферментативного расщепления и т.п. вводимого соединения. Таким образом, изобретение включает метаболиты аминохинолиновых соединений, в том числе соединения, получаемые в процессе, включающем контакт соединения настоящего изобретения с млекопитающим в течение периода времени, достаточного для образования его соответствующего продукта метаболизма.
Фармацевтические композиции и схемы лечения
В одном аспекте настоящего изобретения предложены фармацевтические композиции, которые содержат фармацевтически эффективное количество аминохинолинового соединения вместе с опиоидом или НПВС в фармацевтически приемлемом носителе (например, разбавителе, комплексообразующем веществе, добавке, вспомогательном веществе, адъюванте и т.п.). Аминохинолиновые композиции могут быть представлены, например, в форме соли, микрокристаллической форме, нанокристаллической форме, сокристаллической форме, в форме наночастиц, в форме микрочастиц и/или аморфной форме. Носитель может быть органическим или неорганическим носителем, который подходит для наружного, энтерального или парентерального применения. Аминохинолиновые композиции согласно настоящему изобретению могут быть изготовлены, например, с применением обычных нетоксичных, фармацевтически приемлемых носителей для таблеток, гранул, капсул, липосом, суппозиториев, интраназальных спреев, растворов, эмульсий, суспензий, аэрозолей, систем направленной химической доставки и любой другой формы, подходящей для такого применения, которые известны в области фармацевтики. Неограничивающие примеры носителей, которые могут применяться, включают воду, глюкозу, лактозу, гуммиарабик, желатин, маннит, крахмальную пасту, трисиликат магния, тальк, кукурузный крахмал, кератин, коллоидный диоксид кремния, картофельный крахмал, мочевину и другие носители, подходящие для применения в производстве, в твердой, полутвердой, жидкой или аэрозольной форме. Кроме того, могут использоваться вспомогательные, стабилизирующие, загущающие и окрашивающие вещества и отдушки.
Фармацевтические композиции включают по меньшей мере одно аминохинолиновое соединение, как описано в настоящем документе, в комбинации с опиоидом, ингибитором захвата НЭ или 5ГТ и/или НПВС и фармацевтически приемлемый носитель, растворитель или разбавитель, такой как водный буфер с физиологически приемлемым pH (например, pH 7-8,5), носитель в форме наночастиц на основе полимера, липосому и т.п. Фармацевтические композиции могут быть доставлены в любой подходящей лекарственной форме, такой как лекарственная форма в виде жидкости, геля, твердого вещества, крема или пасты. В одном варианте осуществления композиции могут обеспечивать замедленное высвобождение аминохинолинового соединения.
В некоторых вариантах осуществления фармацевтические композиции включают, без ограничения перечисленными, такие формы, которые подходят для перорального, ректального, назального, наружного (включая трансбуккальное и подъязычное), трансдермального, вагинального, парентерального (включая внутримышечное, внутрибрюшинное, подкожное и внутривенное), спинального (эпидурального, интратекального) и центрального (интрацеребровентрикулярного) введения. Композиции в случае необходимости могут быть представлены в виде дискретных единиц дозы. Фармацевтические композиции согласно изобретению могут быть изготовлены любым из способов, хорошо известных в области фармацевтики. Некоторые предпочтительные способы введения включают внутривенное (в/в), наружное, подкожное, пероральное и спинальное введение. В случае системного введения аминохинолиновое соединение обычно вводят субъекту в дозе в пределах от приблизительно 1 миллиграмма аминохинолинового соединения на килограмм массы тела (мг/кг) до приблизительно 200 мг/кг. Как правило, вводимая доза должна быть достаточной для обеспечения концентрации аминохинолинового соединения в организме субъекта порядка от приблизительно 100 наномолей (нМ) до приблизительно 100 микромолей (мкМ).
Фармацевтические композиции, подходящие для перорального введения, включают капсулы, облатки или таблетки, каждая из которых содержит заданное количество одного или нескольких аминохинолиновых соединений в виде порошка или гранул. В другом варианте осуществления композиция для перорального введения является раствором, суспензией или эмульсией. В альтернативе анальгезирующие композиции, содержащие аминохинолиновые соединения, могут быть представлены в виде болюса, электуария или пасты. Таблетки и капсулы для перорального введения могут содержать стандартные вспомогательные вещества, такие как связующие вещества, наполнители, смазывающие вещества, разрыхлители, красители, ароматизаторы, консерванты или смачивающие вещества. При необходимости таблетки могут быть покрыты оболочкой в соответствии со способами, хорошо известными в данной области. Жидкие препараты для перорального введения включают, например, водные или масляные суспензии, растворы, эмульсии, сиропы или настойки. В альтернативе композиции могут быть представлены в виде сухого продукта для смешивания с водой или другим подходящим растворителем перед применением. Такие жидкие препараты могут содержать обычные добавки, такие как суспендирующие вещества, эмульгирующие вещества, неводные растворители (которые могут включать пищевые масла), консерванты и т.п. Добавки, вспомогательные вещества и т.п. обычно включают в состав композиций для перорального введения в диапазоне концентраций, подходящих для их предполагаемого применения или функции в композиции, и которые хорошо известны в области фармацевтики.
Фармацевтические композиции для парентерального, спинального или центрального введения (например, путем болюсной инъекции или непрерывной инфузии) могут быть представлены в форме единичных доз в ампулах, предварительно заполненных шприцах, инфузиях малого объема или в мультидозовых контейнерах, и предпочтительно включают добавленный консервант. Композиции для парентерального введения могут быть суспензиями, растворами или эмульсиями, и могут содержать вспомогательные вещества, такие как суспендирующие вещества, стабилизирующие вещества и диспергирующие вещества. Добавки, вспомогательные вещества и подобные компоненты, как правило, включают в композиции для парентерального введения в диапазоне концентраций, подходящих для их надлежащего применения или функции в композиции, и которые известны в области производства фармацевтических композиций. Аминохинолиновые соединения включают в композиции в диапазоне терапевтически полезных и эффективных концентраций, определяемых стандартными методами, которые известны в области медицины и фармацевтики.
Фармацевтические композиции для наружного применения на эпидермис (слизистые оболочки или поверхность кожи) могут быть изготовлены в форме мазей, кремов, лосьонов, гелей или в виде трансдермального пластыря. Такие трансдермальные пластыри могут содержать усилители проникновения через кожу, такие как линалоол, карвакрол, тимол, цитраль, ментол, транс-анетол и т.п. Мази и кремы могут, например, включать водную или масляную основу с добавлением подходящих загущающих веществ, гелеобразующих веществ, красителей и т.п. Лосьоны и кремы могут включать водную или масляную основу и, как правило, также содержат одно или более эмульгирующих веществ, стабилизирующих веществ, диспергирующих веществ, суспендирующих веществ, загущающих веществ, окрашивающих веществ и т.п. Гели предпочтительно включают водный носитель-основу и включают гелеобразующее вещество, такое как поперечно сшитый полимер полиакриловой кислоты, дериватизированный полисахарид (например, карбоксиметилцеллюлозу) и т.п. Добавки, вспомогательные вещества и т.п., как правило, включают в композиции для наружного применения на эпидермис в диапазоне концентраций, подходящих для их надлежащего применения или функции в композиции, и которые известны в области производства фармацевтических составов.
Лекарственные формы фармацевтических композиций, подходящие для трансбуккального или подъязычного введения, включают таблетки для рассасывания, включающие анальгезирующие вещества в содержащей вкусовую добавку основе, такой как сахароза, гуммиарабик или трагакант; пастилки, включающие аминохинолиновое соединение в инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик; и ополаскиватели для полости рта, включающие действующее вещество в подходящем жидком носителе. Лекарственные формы фармацевтических композиций для наружного применения при необходимости могут включать вещества, улучшающие проникновение. Добавки, вспомогательные вещества и т.п., как правило, включают в композиции для наружного применения в полости рта в диапазоне концентраций, подходящих для их надлежащего применения или функции в композиции, и которые известны в области производства фармацевтических составов. Анальгезирующие средства присутствуют в композициях в диапазоне терапевтически полезных и эффективных концентраций, определяемых стандартными методами, которые известны в области медицины и фармацевтики.
Для ректального введения анальгезирующие средства представлены в твердом или полутвердом (например, крем или паста) носителе или растворителе. Например, такие композиции для ректального введения могут быть представлены в виде суппозиториев единичной дозы. Подходящие носители или растворители включают масло какао и другие материалы, обычно используемые в данной области. Добавки, вспомогательные вещества и т.п., как правило, включают в композиции для ректального введения в диапазоне концентраций, подходящих для их надлежащего применения или функции в композиции, и которые известны в области производства фармацевтических составов.
Анальгезирующие композиции настоящего изобретения, подходящие для вагинального введения, представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев, содержащих аминохинолин согласно изобретению в комбинации с носителями, как известно в данной области. В альтернативе композиции, подходящие для вагинального введения, могут доставлять в жидкой или твердой лекарственной форме. Добавки, вспомогательные вещества и т.п., как правило, включают в композиции для вагинального введения в диапазоне концентраций, подходящих для их надлежащего применения или функции в композиции, и которые известны в области производства фармацевтических составов.
Анальгезирующие композиции, подходящие для интраназального введения, также охвачены настоящим изобретением. Такие интраназальные композиции включают, в дополнение к анальгезирующим средствам, носитель для доставки и подходящее устройство для доставки жидкого спрея, диспергируемого порошка или капель. Капли могут быть приготовлены на водной или неводной основе, также содержащей одно или более диспергирующих веществ, солюбилизирующих веществ или суспендирующих веществ. Жидкие спреи удобно доставлять из упаковки под давлением, инсуффлятора, небулайзера или других удобных средств доставки аэрозоля, включающего аминохинолин. Упаковки под давлением включают подходящий пропеллент, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другой подходящий газ, хорошо известный в данной области. Дозы аэрозоля можно контролировать, обеспечив клапан для доставки дозированного количества аминохинолина. В альтернативе фармацевтические композиции для введения путем ингаляции или инсуффляции могут быть представлены в форме композиции сухого порошка, например, порошковой смеси анальгезирующих средств и подходящей порошковой основы, такой как лактоза или крахмал. Такая порошковая композиция может быть представлена в виде стандартной лекарственной формы, например, в капсулах, картриджах, желатиновых упаковках или блистерных упаковках, из которых порошок можно вводить с помощью ингалятора или инсуффлятора. Добавки, вспомогательные вещества и т.п., как правило, включают в композиции для интраназального введения в диапазоне концентраций, подходящих для их надлежащего применения или функции в композиции, и которые хорошо известны в области производства фармацевтических составов.
Способы облегчения хронической боли (например, нейропатической боли) включают введение пациенту, страдающему одним из вышеуказанных состояний, эффективного количества аминохинолинового соединения вместе с опиоидом и/или НПВС, и/или ингибитором захвата 5-ГТ/НЭ. Предпочтительно анальгетическую композицию вводят парентерально или энтерально. Доза, содержащая эффективное количество аминохинолиновых соединений, может изменяться в зависимости от возраста и состояния каждого отдельного пациента, подлежащего лечению. Подходящие дозы аминохинолинового соединения обычно составляют в пределах от приблизительно 1 мг/кг до приблизительно 200 мг/кг, при этом аминохинолиновое соединение можно вводить вместе с опиоидом, НПВС и/или ингибитором захвата 5-ГТ/НЭ в количестве от одной десятой до полной рекомендованной дозы конкретного соединения.
Такие дозы могут вводить один или более раз в день, один или более раз в неделю, один или более раз в месяц и т.п.
При использовании в настоящем документе термины "снижение", "ингибирование", "блокирование", "предупреждение", "облегчение", "ослабление" и "антагонист" в отношении композиции означают, что соединение снижает частоту, тяжесть, величину, уровень или ассоциированные симптомы состояния, события или активности по меньшей мере приблизительно на 7,5%, 10%, 12,5%, 15%, 17,5%, 20%, 22,5%, 25%, 27,5%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 90% или 100% по сравнению с тем, в каком качестве состояние, событие или активность обычно существовали бы без применения композиции, включающей такое соединение. Термины "увеличение", "подъем", "повышение", "апрегуляция", "улучшение", "активация" и "агонист" в отношении соединения означают, что соединение увеличивает возникновение или активность состояния, события или активности по меньшей мере приблизительно на 7,5%, 10%, 12,5%, 15%, 17,5%, 20%, 22,5%, 25%, 27,5%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 400%, 500%, 750% или 1000% по сравнению с тем, в каком качестве состояние, событие или активность обычно существовали бы без применения композиции.
Следующие примеры включены для демонстрации некоторых аспектов изобретения. Специалистам в данной области техники должно быть понятно, что методики, раскрытые в примерах, которые представляют собой методики, которые, как известно, хорошо работают при практическом осуществлении изобретения, могут рассматриваться как составляющие предпочтительные варианты его практического осуществления. Однако специалисты в данной области техники, с учетом настоящего описания, должны понимать, что в конкретные раскрытые варианты осуществления могут быть внесены многочисленные изменения, и при этом все же будет получен аналогичный или подобный результат, без отступления от сущности и объема изобретения. Примеры приведены лишь для иллюстрации и не должны являться ограничением.
Способы выявления потенцирования/синергического действия
Хотя определение потенцирования/синергического действия при взаимодействии лекарственных средств является относительно простым, подходы к демонстрации синергического действия не всегда были согласованными. Обзор методики, представленный в публикации Foucquier и Gued (2015), сосредоточен на аналитических подходах к измерению эффективности комбинаций лекарственных средств и представляет собой краткое описание методов, применяемых в настоящее время. Методы подразделяются на "Стратегию на основе эффектов", которая состоит из четырех подходов: (1) Определение подпороговой комбинации; (2) Метод отдельного средства в наибольшей дозе; (3) Аддитивность ответа и (4) Модель независимости действия по Блиссу; и "Стратегию на основе зависимости дозы-эффекта", которая была впервые описана Леве (1926) и в настоящее время называется изоболографическим анализом (Tallarida 2001). В рамках этой "Стратегии на основе эффекта" все четыре подхода были использованы для тестирования потенцирования/синергического действия с описанными комбинациями лекарственных средств. Все четыре подхода дали совпадающие результаты.
Применение DCUKA для повышения активности опиоидов/НПВС/ингибиторов обратного захвата 5-ГТ и НЭ
Представленные ниже данные демонстрируют, что доза DCUKA, эквивалентная пороговой дозе для уменьшения хронической боли в модели хронической воспалительной боли на крысах (Модель с адъювантом Фрейнда) или модели остеоартроза на крысах (MIA), при введении с низкой дозой опиоида, НПВС или соединения, которое уменьшает боль путем ингибирования синаптического захвата норэпинефрина и/или серотонина (например, трамадол), увеличивает активность опиоида, НПВС и/или ингибиторов обратного захвата серотонина/норэпинефрина при уменьшении хронической боли. Что касается опиоидов кроме морфина, стандарт "Показателя эквивалентной дозы морфина" или "Эквивалентов в миллиграммах морфина" (MME) можно использовать для определения количества другого анальгезирующего средства, эквивалентного по активности конкретной суточной дозе морфина. Введение DCUKA или DCUK-OEt (который действует как пролекарство для DCUKA) вместе с морфином увеличивает активность морфина в 4-5 раз. При использовании MME для вычисления дозы другого анальгезирующего средства для ежедневного введения (в зависимости от дозы и количества введений этой дозы рассматриваемого анальгезирующего средства в день), включение DCUKA или DCUK-OEt в схему лечения боли позволяет снизить дозу анальгезирующего средства так же, как снизить дозу морфина при введении морфина вместе с DCUKA/DCUK-OEt.
Вычисление дозы вводимого анальгезирующего средства при использовании DCUKA или DCUK-OEt для потенцирования обезболивающего действия
Доза другого анальгезирующего средства в случае введения вместе с DCUKA может быть вычислена согласно следующей формуле:
Доза опиоидного анальгетика=суточная доза морфина, используемая в аналогичной ситуации ÷ MME ÷ коэффициент, основанный на повышении активности опиоида в результате добавления DCUKA или DCUK-OEt.
В качестве примера вычисления дозы оксикодона используют следующую информацию: (1) MME интересующего опиоида (в данном случае оксикодона) составляет 1,5 (Von Korff et al., Clin. J. Pain 24(6): 521-527 (2008)); (2) суточная доза морфина, требуемая для уменьшения боли, о котором сообщает пациент. В этом отношении важно использовать шкалы оценки (Schneider et al., 2003). При умеренной или тяжелой боли разовая доза морфина (внутрь) может изменяться в пределах 10-30 мг, причем такие дозы принимают 6 раз в день (т.е. 60-180 мг/сутки); (3) коэффициент, на который можно уменьшить дозу опиоида при добавлении DCUKA в схему применения при лечении боли.
Для человека доза DCUKA может варьировать от 150 до 450 мг при введении 2-3 раза в день. Если DCUKA вводят в таком качестве, дозу используемого опиоида можно снизить в 4-5 раз (этот коэффициент определяют на основе данных, представленных на Фигуре 11 и 12). Специалист по лечению боли должен вести внимательное наблюдение за пациентом, чтобы скорректировать дозу до величины, необходимой для комфорта пациента. Это может быть достигнуто путем увеличения дозы опиоидов или DCUKA в границах рекомендуемых диапазонов.
Примеры фактических вычислений дозы
Начальная доза оксикодона/день = (60 мг [начальная ежедневная доза морфина])÷(1,5 [MME])÷(5=коэффициент снижения дозы опиоида) = 8 мг/день.
Начальная доза метадона/день = (60)÷(4)÷(5) = 3 мг/день.
Дозу метадона, применяемого для контроля боли, обычно повышают в течение шестинедельного периода, при этом более низкую дозу, вводимую вместе с DCUKA, можно изменять соответственно.
Аналогичные вычисления можно выполнить для других опиоидов, одобренных для медицинского применения при лечении хронических болевых синдромов. Специалистам в данной области должно быть понятно, что между пациентами существуют значительные различия в степени ощущаемой боли, и что ощущаемая боль также зависит от причины боли и степени повреждения. Приведенные примеры являются иллюстрацией того, что можно рассматривать в качестве способа поддержания уровня анальгезии/антигипералгезии при снижении доз опиоидов в сочетании с введением DCUKA. При необходимости, для дополнительного контроля боли, суточную дозу DCUKA и/или опиоида можно постепенно повышать (повышение может составлять 25-50% от суточной дозы DCUKA или опиоида).
В случае применения НПВС вместе с DCUKA применяются те же принципы, за исключением того, что коэффициент, на который можно уменьшить дозу НПВС при приеме вместе с DCUKA, находится в диапазоне от приблизительно 5 до приблизительно 6.
Суточная доза диклофенака = (60 мг [ежедневная доза морфина])÷(0,10)÷(6) = 100 мг. Для пациентов, принимающих диклофенак натрия, рекомендовано не превышать дозу 225 мг/день.
Таким образом, на основании открытия, что DCUKA и его пролекарство (DCUK-OEt) могут повышать эффективность опиоидов и НПВС, корректировка суточных доз опиоидов и НПВС в сторону снижения приводит к уменьшению побочных эффектов при сохранении купирования боли.
В отсутствие опубликованных значений MEE для ингибиторов обратного захвата НЭ/5-ГТ можно использовать данные, представленные авторами изобретения, по эффектам трамадола в сочетании с DCUKA при лечении остеоартроза (Фигура 9). Трамадол, как было показано, является слабым опиоидом, но обладает выраженным действием в качестве ингибитора обратного захвата НЭ/5-ГТ (Barber, 2011). Другими ингибиторами обратного захвата НЭ/5-ГТ являются дулоксетин, венлафаксин, милнаципран и т.п. Данные показывают, что дозу ингибитора обратного захвата НЭ/5-ГТ можно снизить в два раза при введении пациентам вместе с 150-450 мг DCUKA по две или три дозы/сутки.
ПРИМЕР 1. Получение соединений формулы (VI)
Производные кинуреновой кислоты, содержащей третичную уреидогруппу, включая 5,7-дихлор-4-(3,3-дифенилуреидо)хинолин-2-карбоновую кислоту (DCUKA, 7a), можно синтезировать, как описано ранее (Snell et al., 2000), при использовании реакционноспособного промежуточного карбамоилхлорида (6a-b). Однако этот синтез можно улучшить благодаря сопутствующему гидролизу сложного эфира в ходе заключительной реакции ацилирования. Один вариант соединения, 5,7-дихлор-4-(3,3-дибутилуреидо)хинолин-2-карбоновую кислоту (BCUKA, 7b), синтезировали согласно этому способу в фазах синтеза I-IV, как объяснено и проиллюстрировано на Схеме 2 {Реагенты и условия (I): MeOH, нагрев с обратным холодильником, 16 ч. (II): Ph2O, 250°C, 2 ч. (III): (a) ClSO2NCO, MeCN, нагрев с обратным холодильником, 2 ч. (b) HC1, MeOH, КТ, 30 мин (IV): NaH, ДМФА, от 0°C до КТ, 16 ч.
Схема 2
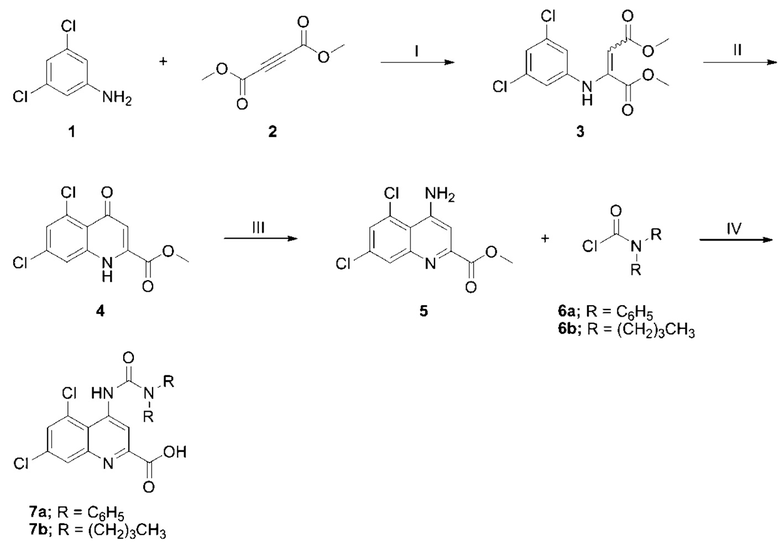
Фаза синтеза I
3,5-Дихлоранилин (1, 5,00 г, 30,9 ммоль) и диметил-ацетилендикарбоксилат (2, 3,80 мл, 30,9 ммоль) объединяли в безводном MeOH (60 мл) под азотом и нагревали с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и выпаривали досуха. Полученное в результате желтое твердое вещество перекристаллизовывали из MeOH (два раза) с получением смеси цис и транс-изомеров целевого диметиланилиномалеата (3) в виде тонких желтых кристаллов (5,23 г, 17,2 ммоль). Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в дейтерированном ДМСО, составили: 3,57 и 3,67 (3H, с), 3,72 и 3,80 (3H, с), 5,35 и 5,58 (1H, с), 6,98 и 7,12 (2H, скаж), 7,23 и 7,31 (1H, скаж), 9,52 и 9,64 (1H, шс).
Фаза синтеза II
Диметиланилиномалеат (3, 3,50 г, 11,5 ммоль) порциями добавляли к дифениловому эфиру (70 мл) при 250°C. Температуру полученного раствора поддерживали при 250°C в течение 2 часов, после чего охлаждали до комнатной температуры и разбавляли смесью гексанов (100 мл). Образовавшийся осадок удаляли путем фильтрации, промывали смесью гексанов (50 мл) и суспендировали в этаноле, нагреваемом под обратным холодильником, после чего фильтровали для удаления растворимых примесей. Твердый остаток на фильтре сушили в вакууме с получением требуемого хинолон-карбоксилата (4) в виде почти белого твердого вещества (3,10 г, 11,4 ммоль). Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в дейтерированном ДМСО, составили: 3,96 (3H, с), 6,59 (1H, с), 7,42 (1H, с), 7,97 (1H, с), 12,05 (1H, ш). Этот процесс может быть адаптирован для применения с аппаратом непрерывного потока (Cao, 2017) для снижения высоких температур в присутствии дифенилового эфира.
Фаза синтеза III
Хлорсульфонилизоцианат (1,20 мл, 13,8 ммоль) добавляли к суспензии хинолинкарбоксилата (4, 2,50 г, 9,19 ммоль) в безводном MeCN (35 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 1,5 часов, после чего нагревание прекращали и добавляли 1,0 М раствор HC1 в безводном MeOH (20 мл). Реакционной смеси позволяли охладиться до комнатной температуры при перемешивании, в результате чего через 1 час образовывался осадок. Осадок удаляли с помощью фильтрации, промывали MeCN и сушили на воздухе. Осадок с фильтра суспендировалии в воде (50 мл), в которую добавляли насыщенный раствор карбоната натрия (~5 мл) до pH 10, вызвав загущение суспензии. Полученное твердое вещество собирали с помощью фильтрации, промывали холодной водой и сушили в вакууме (40°C) с получением целевого аминохинолина (5) в виде почти белого твердого вещества (1,82 г, 6,71 ммоль). Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в дейтерированном ДМСО, составили: 4,05 (3H, с), 6,04 (2H, с), 7,33 (1H, с), 7,47 (1H, д, J=1,9 Гц), 8,10 (1H, d, J=1,9 Гц).
Фаза синтеза IV
Ацилирование аминохинолина (5) с сопутствующим гидролизом сложного эфира, с получением 5,7-дихлор-4-(3,3-дибутилуреидо)-хинолин-2-карбоновой кислоты (BCUKA, 7b) проводили следующим образом; N, N-дибутилкарбамоилхлорид (6b, 96 мг, 0,50 ммоль) и аминохинолин (5, 113 мг, 0,42 ммоль) растворяли в безводном ДМФА (2 мл) и охлаждали до 0°C. Добавляли дисперсию гидрида натрия в минеральном масле (60%, 35 мг, 0,83 ммоль), позволяли смеси нагреться до комнатной температуры и перемешивали в течение 16 часов. Реакцию останавливали путем добавления в насыщенный раствор NH4Cl (1 мл) с последующим доведением до pH 3 с помощью водной 1,0 М HCl. Экстракция EtOAc (2×10 мл) с последующей промывкой насыщенным солевым раствором (5 мл) и сушкой (Na2SO4) давала неочищенный продукт в виде бледно-желтого масла. Очистка соединения с помощью хроматографии на силикагеле (9:1 ДХМ:MeOH) давала 5,7-дихлор-4-(3,3-дибутилуреидо)хинолин-2-карбоновую кислоту (DBCUKA, 7b) в виде бледно-желтого твердого вещества (82 мг, 0,20 ммоль). Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в CDCl3, составили: 1,00 (6H, т, J=7,4 Гц), 1,36-1,45 (4H, м), 1,64-1,72 (4H, м), 3,39-3,45 (4H, м), 5,17 (1H, с), 7,69 (1H, с), 8,30 (1H, с), 9,16 (1H, с).
Карбамоилхлориды имеют ограниченную коммерческую доступность и, кроме того, отличаются высокой реакционной способностью, особенно в отношении гидролиза, и, как результат, низкой стабильностью. Это особенно очевидно в случае моно-н-замещенных карбамоилхлоридов. Таким образом, для получения моно-н-замещенных аналогов кинуреновой кислоты было предпочтительно использовать альтернативные эквиваленты карбамоильных катионов с ослабленной реакционной способностью. Карбамоилимидазолы (например, 9a-d), как было показано, оказались подходящими реакционноспособными соединениями для синтеза различных функциональных групп, включая мочевину, тиомочевины, карбаматы, тиокарбаматы и амиды (Grzyb et al., 2005) Производные кинуреновой кислоты, содержащие вторичную уреидогруппу, были получены при использовании этого подхода в фазах синтеза V-VII, как объяснено и проиллюстрировано на Схеме 3 (Реагенты и условия: (V): CDI, ДХМ, 0°C→КТ, 16 ч. (VI): 5, NaH, ДМФА, 0°C→КТ, 16 ч. (VII) ТФУ, ДХМ, КТ, 16 ч).
Схема 3
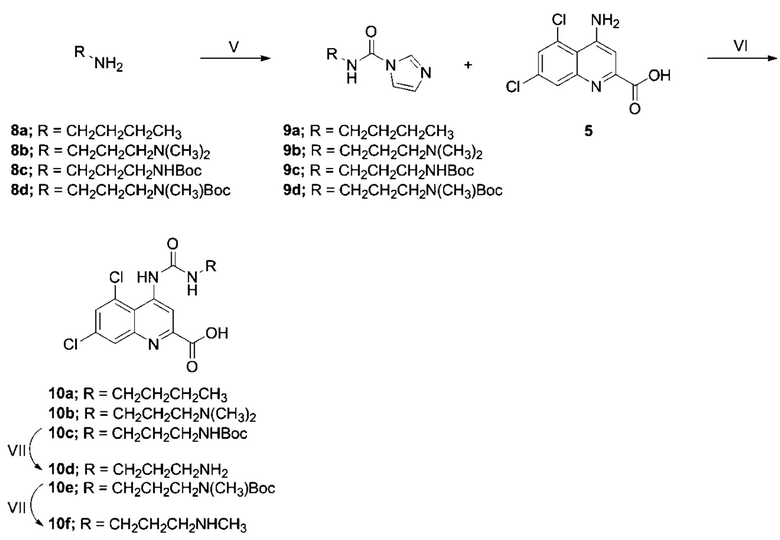
Общий пример фазы синтеза V
н-Бутиламин (8a, 100 мкл, 74 мг, 1,01 ммоль) в ДХМ (1 мл) добавляли к раствору CDI (0,197 г, 1,21 ммоль) в ДХМ (5 мл) при 0°C, после чего реакционной смеси позволяли нагреться до КТ и перемешивали в течение ночи. Раствор разбавляли ДХМ (10 мл), промывали водой (2×10 мл) и солевым раствором (10 мл), сушили (Na2SO4) и выпаривали досуха с получением целевого карбамоилимидазола (9a) в виде бесцветного масла (115 мг, 0,69 ммоль). Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в CDCl3, составили: 0,97 (3H, т, J=7,4 Гц), 1,42 (2H, к, J=7,6, 7,3 Гц), 1,63 (2H, тт, J=7,3, 7,0 Гц), 3,44 (2H, дт, J=7,0, 6,7 Гц), 6,75 (1H, ш), 7,07 (1H, с), 7,42 (1H, с), 8,17 (1H, с).
N-(3-(Диметиламино)пропил)-1H-имидазол-1-карбоксамид (9b) получали из 3-диметиламинопропиламина (8b), как описано в фазе синтеза V. Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в CDCl3, составили: 1,77 (2H, тт, J=5,7, 5,5 Гц), 2,32 (6H, с), 2,56 (2H, т, J=5,5 Гц), 3,54 (2H, дт, J=5,9, 5,5 Гц), 7,07 (1H, с), 7,27 (1H, с), 8,04 (1H, с), 9,34 (1H, ш).
трет-Бутил(3-(1H-имидазол-1-карбоксамидо)пропил)карбамат (9c) получали из трет-бутил(3-аминопропил)карбамат (8c), как описано в фазе синтеза V. Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в CDCl3, составили: 1,49 (9H, с), 1,73 (2H, тт, J=5,8, 5,7 Гц), 3,30 (2H, дт, J=6,4, 5,7 Гц), 3,48 (2H, дт, J=6,0, 5,8 Гц), 4,91 (1H, ш), 7,11 (1H, с), 7,52 (1H, с), 7,92 (1H, ш), 8,25 (1H, с).
трет-Бутил(3-(1H-имидазол-1-карбоксамидо)пропил)(метил)-карбамат (9d) получали из трет-бутилового эфира N-(3-аминопропил)-N-метилкарбаминовой кислоты (8d), как описано в фазе синтеза V. Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в CDCl3: 1,49 (9H, с), 1,74-1,80 (2H, ш), 2,87 (3H, с), 3,35-3,43 (4H, м), 7,09 (1H, с), 7,53 (1H, с), 8,11 (1H, ш), 8,25 (1H, с).
Общий пример фазы синтеза VI
Можно было использовать карбамоилимидазолы (9a-d) аналогично карбамоилхлоридам в фазе синтеза IV, что позволило провести одностадийное ацилирование аминохинолина (5) и одновременный гидролиз сложного эфира. Этот подход использовали для синтеза 4-(3-бутилуреидо)-5,7-дихлорхинолин-2-карбоновой кислоты (10a) следующим образом: N-бутил-7-имидазол-1-карбоксамид (9a, 125 мг, 0,95 ммоль) и аминохинолин (5, 215 мг, 0,79 ммоль) растворяли в безводном ДМФА (4 мл) и охлаждали до 0°C. Добавляли дисперсию гидрида натрия в минеральном масле (60%, 63 мг, 1,58 ммоль), позволяли смеси нагреться до комнатной температуры и перемешивали в течение 16 часов. Реакцию останавливали путем добавления в насыщенный раствор NH4Cl (3 мл) с последующим доведением до pH 3 с использованием водной 1,0 М HCl. Экстракция EtOAc (2×20 мл) с последующей промывкой насыщенным солевым раствором (10 мл) и сушкой (Na2SO4) давала неочищенный продукт в виде бледно-оранжевого остатка. Очистка соединения с помощью обращенно-фазовой хроматографии на силикагеле (C18) (H2O:MeCN, 1:1) давала 4-(3-бутилуреидо)-5,7-дихлорхинолин-2-карбоновую кислоту (10a) в виде бежевого твердого вещества (142 мг, 0,39 ммоль). Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в дейтерированном ДМСО, составили: 0,92 (3H, т, J=7,3 Гц), 1,31-1,38 (2H, м), 1,44-1,52 (2H, м), 3,16 (2H, дт, J=6,6, 6,0 Гц), 7,47 (1H, ш), 7,87 (1H, д, J=2,2 Гц), 8,11 (1H, д, J=2,2 Гц), 8,68 (1H, с), 9,12 (1Н, ш).
5,7-Дихлор-4-(3-(3-(диметиламино)пропил)уреидо)хинолин-2-карбоновую кислоту (10b) получали из N-(3-(диметиламино)-пропил)-1H-имидазол-1-карбоксамида (9b), как описано в фазе синтеза VI. Из-за цвиттер-ионной природы целевого соединения подкисление до pH 1/2 выполняли с использованием ТФУ перед проведением обращенно-фазовой (C18) хроматографии, с получением продукта в форме соли ТФУ. Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в D2O, составили: 1,87-2,00 (2H, м), 2,87 (6H, с), 3,12-3,20 (2H, м), 3,21-3,31 (2H, м), 7,13 (1H, с), 7,48 (1H, с), 8,06 (1H, с). Значение пика поглощения (в м.д.), обнаруженное в спектре 19F-ЯМР, полученном в D2O, составило -75,6.
4-(3-(3-((трет-Бутоксикарбонил)амин)пропил)уреидо)-5,7-дихлорхинолин-2-карбоновую кислоту (10c) получали из трет-бутил-(3-(1H-имидазол-1-карбоксамидо)пропил)карбамата (9c), как описано в фазе синтеза VI. Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в дейтерированном ДМСО, составили: 1,39 (9H, с), 1,60 (2H, тт, J=6,8, 6,6 Гц), 2,95-3,02 (2H, м), 3,12-3,18 (2H, м), 6,83 (1H, ш), 7,45 (1H, ш), 7,85 (1H, с), 8,10 (1H, с), 8,65 (1H, с), 9,15 (1H, ш).
4-(3-(3-((трет-Бутоксикарбонил)(метил)амино)пропил)уреидо)-5,7-дихлорхинолин-2-карбоновую кислоту (10е) получали из трет-бутил(3-(1H-имидазол-1-карбоксамидо)пропил)метилкарбамата (9d), как описано в фазе синтеза VI. Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в дейтерированном ДМСО, составили: 1,39 (9H, с), 1,63-1,71 (2H, м), 2,79 (3H, с), 3,08-3,16 (2H, м), 3,19-3,26 (2H, м), 7,27 (1H, ш), 7,68 (1H, д, J=1,8 Гц), 8,29 (1H, д, J=1,8 Гц), 8,40 (1H, ш), 8,97 (1H, ш).
Общий пример фазы синтеза VII
ТФУ (173 мкл, 2,25 ммоль) добавляли к раствору Boc-защищенного амина (10c, 103 мг, 0,23 ммоль) в ДХМ (4 мл). После перемешивания при комнатной температуре в течение 16 часов, растворитель удаляли при пониженном давлении, а остаток очищали непосредственно с помощью обращенно-фазовой хроматографии (C18, 1:1 H2O:MeCN) с получением 4-(3-(3-аминопропил)уреидо)-5,7-дихлорхинолин-2-карбоновой кислоты (10d) в форме соли ТФУ в виде белого твердого вещества (64 мг, 0,14 ммоль). Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в D2O (1,0% ТФУ), составили: 1,61 (2H, тт, J=6,9, 7,1 Гц), 2,72 (2H, т, J=7,1 Гц), 3,04 (2H, т, J=6,9 Гц), 7,62 (1H, с), 7,89 (1H, с), 8,71 (1H, с).
5,7-Дихлор-4-(3-(3-(метиламино)пропил)уреидо)хинолин-2-карбоновую кислоту (10f) получали из 4-(3-(3-((трет-бутоксикарбонил)(метил)амино)пропил)уреидо)-5,7-дихлорхинолин-2-карбоновой кислоты (10е), как описано в фазе синтеза VI. Значения пиков поглощения (в м.д.), обнаруженные в спектре 1H-ЯМР, полученном в D2O (1,0% ТФУ), составили: 1,81 (2H, тт, J=6,8, 7,7 Гц), 2,55 (3H, с), 2,94 (2H, т, J=7,8 Гц), 3,22 (2H, т, J=6,8 Гц), 7,78 (1H, д, J=1,9 Гц), 8,01 (1H, д, J=1,9 Гц), 8,77 (1H, с). Значение пика поглощения (в м.д.), обнаруженного в спектре 19F-ЯМР, полученном в D2O, составило -73,4. Структуры соединений показаны на Схеме 4.
Схема 4
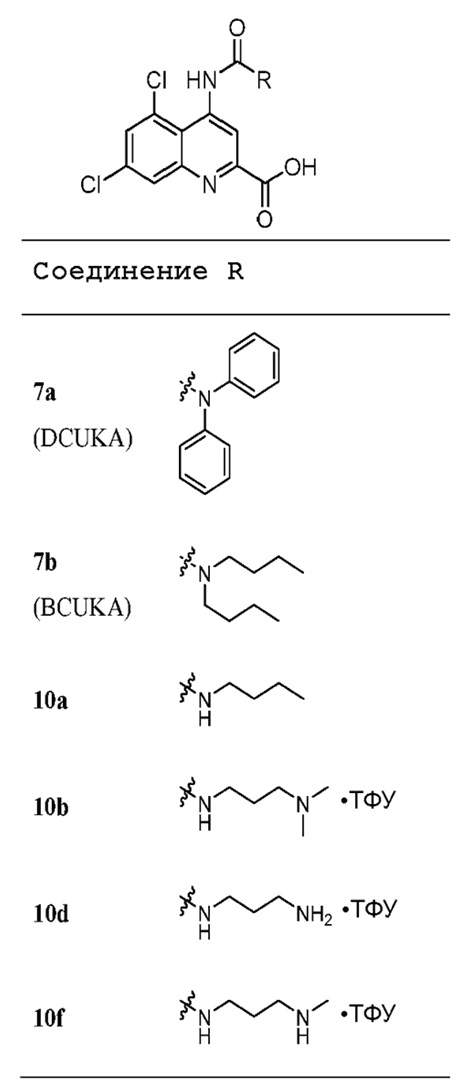
Синтез 3-(2-бутирил-5,7-дихлорхинолин-4-ил)-1,1-дифенилмочевины
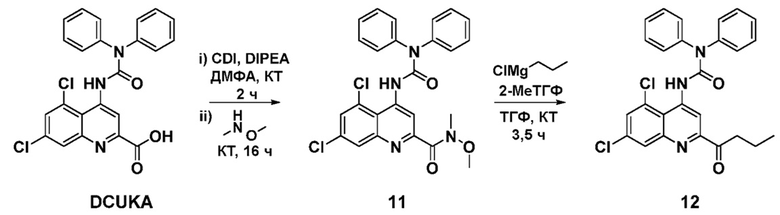
A. 5,7-Дихлор-4-(3,3-дифенилуреидо)-N-метокси-N-метилхинолин-2-карбоксамид (11)
Карбонилдиимидазол (72 мг, 0,44 ммоль) и диизопропилэтиламин (115 мкл, 0,66 ммоль) добавляли к раствору 5,7-дихлор-4-(3,3-дифенилуреидо)хинолин-2-карбоновой кислоты (DCUKA; 100 мг, 0,22 ммоль) в сухом N, N-диметилформамиде (15 мл). Реакционную смесь перемешивали при комнатной температуре под азотом в течение 2 часов, после чего добавляли гидрохлорид N, O-диметилгидроксиламина (86 мг, 0,88 ммоль). Полученный бледно-желтый раствор перемешивали при комнатной температуре еще в течение 16 часов, после чего растворитель удаляли при пониженном давлении, а остаток растворяли в этилацетате (20 мл) и промывали насыщенным раствором гидрокарбоната натрия (2×15 мл) и 0,1M HCl (2×15 мл), затем водой (15 мл) и солевым раствором (10 мл). Органическую фазу сушили (MgSO4) и выпаривали досуха. Целевое соединение получали после очистки с помощью хроматографии на силикагеле (смесь гексанов:EtOAc 1:1) в виде белого твердого вещества (81 мг, 0,16 ммоль, 73%). Rf 0,33 (смесь гексанов:EtOAc 1:1); Тп 207-210°C; 1H-ЯМР (400 МГц, CDCl3) 3,40 (3H, с), 3,75 (3H, ш), 7,28 (1H, с), 7,34-7,37 (2H, м), 7,41-7,49 (8H, м), 8,03 (1H, д, J=2,0 Гц), 8,87 (1H, с), 9,39 (1H, с).
B. 3-(2-Бутирил-5,7-дихлорхинолин-4-ил)-1,1-дифенилмочевина (12)
Раствор н-пропилмагнийхлорида в 2-метилтетрагидрофуране (1,0M, 1,12 мл, 1,12 ммоль) по каплям добавляли к раствору 5,7-дихлор-4-(3,3-дифенилуреидо)-N-метокси-N-метилхинолин-2-карбоксамида (11, 70 мг, 0,14 ммоль) в сухом тетрагидрофуране (10 мл) при -10°C, под азотом. После добавления реакционуую смесь перемешивали при -10°C в течение 30 мин, после чего позволяли нагреться до комнатной температуры и перемешивали еще в течение 3 ч. Реакцию останавливали насыщенным раствором хлорида аммония (10 мл) и продукт, кетон 12, экстрагировали этилацетатом (3×15 мл). Органический экстракт промывали солевым раствором (10 мл) и сушили (MgSO4), а затем выпаривали досуха. Остаток очищали с помощью хроматографии на силикагеле (смесь гексанов:EtOAc 4:1) с получением целевого соединения в виде бледно-желтого твердого вещества (32 мг, 0,07 ммоль, 47%). Rf 0,45 (смесь гексанов:EtOAc 4:1); Тп 161-164°C; 1H-ЯМР (400 МГц, CDCl3) 1,03 (3H, т, J=7,4 Гц), 1,80 (2H, к, J=7,3, 7,4 Гц), 3,24 (2H, т, J=7,3 Гц), 7,28 (1H, с), 7,34-7,38 (2H, м), 7,42-7,49 (8H, м), 8,09 (1H, д, J=2,1 Гц), 9,15 (1H, с), 9,31 (1H, с).
Синтез 5,7-дихлор-4-(3,3-дифенилуреидо)-N-этилхинолин-2-карбоксамида (13)
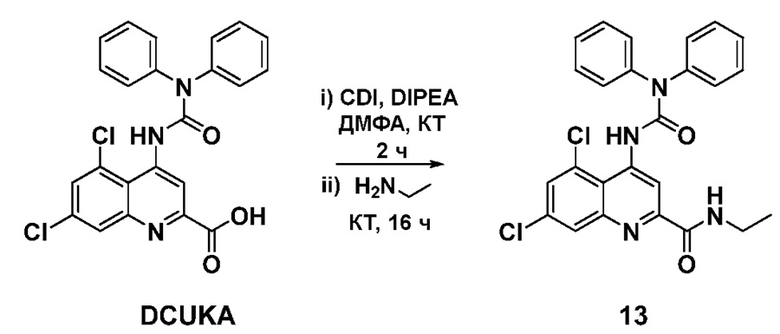
Карбонилдиимидазол (143 мг, 0,88 ммоль) и диизопропилэтиламин (230 мкл, 1,32 ммоль) добавляли к раствору 5,7-дихлор-4-(3,3-дифенилуреидо)хинолин-2-карбоновой кислоты (DCUKA; 200 мг, 0,44 ммоль) в сухом N, N-диметилформамиде (25 мл). Реакционную смесь перемешивали при комнатной температуре под азотом в течение 2 часов, после чего добавляли этиламин в ТГФ (2,0M, 0,66 мл, 1,32 ммоль). Полученный бледно-желтый раствор перемешивали при комнатной температуре еще в течение 16 часов, по истечении которых реакция проходила полностью. Растворитель удаляли при пониженном давлении, а остаток растворяли в этилацетате (50 мл) и промывали насыщенным раствором гтдрокарбоната натрия (2×30 мл) и 0,1M HC1 (2×30 мл), затем водой (25 мл) и солевым раствором (25 мл). Органическую фазу сушили (MgSO4) и выпаривали досуха. Целевой этиламид 13 получали после хроматографии на силикагеле (смесь гексанов:EtOAc 1:1) в виде почти белого твердого вещества (148 мг, 0,31 ммоль, 71%). Rf 0,43 (смесь гексанов:EtOAc 1:1); Тп 202-205°C; 1H-ЯМР (400 МГц, CDCl3) 1,31 (3H, т, J=7,2 Гц), 3,56 (2H, к, J=7,2 Гц), 7,28 (1H, с), 7,32-7,37 (2H, м), 7,41-7,49 (8H, м), 7,99 (1H, с), 8,01 (1H, ш), 9,28 (1H, с), 9,32 (1H, с).
Синтез 5,7-дихлор-4-(3,3-дифенилуреидо)-N-изопропилхинолин-2-карбоксамида (14)
Карбонилдиимидазол (146 мг, 0,88 ммоль) и диизопропилэтиламин (230 мкл, 1,32 ммоль) добавляли к раствору 5,7-дихлор-4-(3,3-дифенилуреидо)хинолин-2-карбоновой кислоты (DCUKA; 200 мг, 0,44 ммоль) в сухом N, N-диметилформамиде (25 мл). Реакционную смесь перемешивали при комнатной температуре под азотом в течение 2 часов, после чего добавляли изопропиламин (110 мкл, 1,32 ммоль). Полученный бледно-желтый раствор перемешивали при комнатной температуре еще в течение 16 часов, после чего растворитель удаляли при пониженном давлении, а остаток растворяли в этилацетате (50 мл) и промывали насыщенным раствором гидрокарбоната натрия (2×30 мл) и 0,1M HC1 (2×30 мл), затем водой (25 мл) и солевым раствором (25 мл). Органическую фазу сушили (MgSO4) и выпаривали досуха. Целевой изопропиламид 14 получали с помощью хроматографии на силикагеле (смесь гексанов:EtOAc 1:1) в виде белого твердого вещества (182 мг, 0,37 ммоль, 84%). Rf 0,52 (смесь гексанов:EtOAc 1:1); Тп 197-199°C; 1H-ЯМР (400 МГц, ДМСО-d6) 1,23 (6H, д, J=6,8 Гц), 4,15 (1H, м), 7,34-7,39 (2H, м), 7,46-7,55 (8H, м), 7,76 (1H, д, J=1,9 Гц), 8,11 (1H, д, J=1,9 Гц), 8,55 (1H, д, J=8,2 Гц), 9,00 (1H, с), 9,21 (1H, с).
ПРИМЕР 2. Применение DCUK-OEt в качестве пролекарства DCUKA in vivo
В данном примере показано, что DCUKA быстро образуется in vivo при гидролизе сложного эфира после перорального введения DCUK-OEt крысам. Это исследование проводили в соответствии с Руководством NIH по содержанию и применению лабораторных животных. Высушенная распылением дисперсия (SDD) DCUK-OEt с полимером ГПМЦАС-MG (HPMCAS-MG SDD) была получена в Catalent Pharma. Эта SDD содержала 15 мг DCUK-OEt и 85 мг полимера на 100 мг SDD. Препарат 0,5% гидроксипропилметилцеллюлозы (ГПМЦ) получали при нагревании 100 мл воды до 60-75°С. Отдельную аликвоту 100 мл воды охлаждали до 5°C. 500 мг ГПМЦ добавляли в 50 мл горячей воды при перемешивании, а затем добавляли 50 мл холодной воды. Перемешивание продолжали до образования прозрачного раствора. Для получения суспензии SDD, аликвоты 15 мл 0,5% ГПМЦ добавляли во флакон, содержащий 1 г SDD. Смесь растирали до образования однородной суспензии, содержащей 10 мг/мл DCUK-OEt. Четырем крысам в группе вводили 50 мг/кг или 100 мг/кг DCUK-OEt в этой суспензии через желудочный зонд. Образцы крови забирали из яремной вены за 0 минут до введения дозы и через 30 минут, 60 минут, 90 минут, 120 минут и 180 минут после введения дозы. Образцы крови собирали в пробирки, содержащие NaF, чтобы минимизировать гидролиз DCUK-OEt in vitro до DCUKA под действием ферментов, присутствующих в крови крыс.
Уровни DCUK-OEt и DCUKA измеряли в образцах цельной крови с помощью валидированного метода ЖХ-МС/МС. Внутренние стандарты DCUKA-d10 и DCUKA-OEt-d10 получали путем синтеза (Wempe laboratory, UC Denver School of Pharmacy, Med. Chem. Core Facility). Стоковые 10,0 мМ растворы DCUKA, DCUKA-OEt, DCUKA-d10 и DCUKA-OEt-d10 в ДМСО приготавливали для стандартных кривых и внутренних стандартов, и стандарты и образцы разбавляли в растворах 4:1 (метанол:ацетонитрил, 1:1):Вода (10 мМ NH4OAc, 0,1% муравьиная кислота), которые использовали для непосредственного введения в масс-спектрометр.
Использовали Applied Biosystems Sciex 4000 (Applied Biosystems; Foster City, CA), оборудованный ВЭЖХ Shimadzu (Shimadzu Scientific Instruments, Inc; Columbia, MD) и автосэмплером Leap (LEAP Technologies; Carrboro, NC). Для жидкостной хроматографии использовали колонку Agilent Technologies, Zorbax extended-C18 250×4,6 мм, 5 микрон, снабженную защитной колонкой, при 40°C и скорость потока 0,6 мл/мин. Подвижная фаза состояла из A: 10 мМ (NH4OAc), 0,1% муравьиной кислоты в H2O, и B: 50:50 ACN:MeOH. Использовали следующий метод хроматографии: 95% A в течение 2,0 мин; повышение до 95% B за 7,0 мин и удерживание в течение 9,0 мин, наконец, возврат к 95% A через 18,0 мин и удерживание в течение 2,0 мин (общее время работы 20,0 мин). Соединения контролировали при использовании режима детектирования положительных ионов при ионизации электрораспылением (ЭРИ+) с использованием следующих условий: i) напряжение ионораспыления 5500 В; ii) температура 450°C; iii) газом завесы (CUR; значение 10) и активированной столкновениями диссоциации (CAD; значение 12) являлся азот; iv) газ источника ионов один (GS1) и два (GS2); v) входной потенциал был установлен на 10 В; vi) квадруполь один (Q1) и (Q3) были установлены на единичное разрешение; vii) время измерения было установлено на 200 мсек; и viii) потенциал декластеризации (DP), энергия столкновения (CE) и потенциал на выходе ячейки столкновений (CXP) являются напряжениями (V). Образцы (10 мкл) анализировали с помощью ЖХ/МС-МС при использовании следующих фрагментов для количественного определения: DCUKA, 452→168 m/z, tR=5,3 мин; DCUKA-OEt: 480→168 m/z, tR=5,6 мин; внутренний стандарт DCUKA-d10: 462→178 m/z; и внутренний стандарт DCUKA-OEt-d10: 490→178 m/z.
На Фигуре 1 графически показано, что DCUK-OEt может служить в качестве пролекарства для DCUKA после введения in vivo. Данные представлены как средние значения±стандартное отклонение для 3-4 крыс на группу (данные для одного выброса через 60 мин после введения дозы 100 мг/кг не включены). 50 мг/кг или 100 мг/кг DCUK-OEt, приготовленного в виде высушенной распылением дисперсионной композиции с полимером ГПМЦАС-MG, вводили в виде суспензии ГПМЦ через желудочный зонд 4 крысам в группе. Кровь забирали из яремной вены в указанные точки времени, и уровни DCUKA определяли с помощью анализа ЖХ-МС/МС. Результаты показывают уровни DCUKA в крови, полученные после введения DCUK-OEt. DCUK-OEt обнаруживали только после введения дозы 100 мг/кг. Наиболее высокие уровни DCUK-OEt составляли 0,49 мкМ и 0,24 мкМ через 60 минут после введения DCUK-OEt и 0,08 мкМ через 90 минут после введения DCUK-OEt. Уровни DCUK-OEt были ниже предела обнаружения у четвертой крысы в группе. Напротив, как показано на Фигуре 1, уровни DCUKA достигали примерно 3 мкМ после введения 50 мг/кг DCUK-OEt и примерно 11 мкМ после введения 100 мг/кг DCUK-OEt.
ПРИМЕР 3. Лечение нейропатической боли с применением DCUKA, BCUKA и DCUK-OEt
Данный пример иллюстрирует способность DCUKA, BCUKA и DCUK-OEt купировать нейропатическую боль, измеряемую как механическую или термическую боль, индуцированную цисплатином (противоопухолевая химиотерапия), полным адъювантом Фрейнда (CFA) (воспалительная боль) или диабетом (стрептозотоцин-индуцированная боль), или моноиодацетатом (MIA) (остеоартрозная боль).
Все исследования проводили в соответствии с руководством NIH по содержанию и применению лабораторных животных.
Лекарственные средства
Для in vivo исследований индуцированной цисплатином, CFA и STZ боли DCUKA или BCUKA или DCUK-OEt, или габапентин, приготавливали в эмульсии 50% желатина/50% масла канолы (эту эмульсию использовали в качестве растворителя). Желатин подготавливали путем добавления 0,8 г желатина Knox, Kraft Foods North America, Tarrytown NY) и 0,06 г винной кислоты (McCormick and Co., Inc., Hunt Valley, MD) в 30 мл очищенной воды. Раствор нагревали при 98°C в течение 20 минут, затем охлаждали до 50°C. Шесть мл 95% спирта и воды добавляли с получением 50 мл желатина. Различные количества DCUKA или BCUKA или DCUK-OEt, или габапентина добавляли в 5 мл масла канолы (Safeway Inc., Pleasanton, CA) при перемешивании и обработке ультразвуком (VWR BIOSONIK IV, 70%) в течение 5 минут, а затем суспензии лекарственного средства добавляли к 5 мл желатина при перемешивании и обработке ультразвуком. Эмульсии разбавляли растворителем при необходимости и нагревали до 37°C для перорального введения животным. Непосредственно перед введением внутрь через желудочный зонд эмульсию перемешивали на вортексе.
Для in vivo исследований MIA-индуцированной боли, DCUKA объединяли с D-α-токоферол полиэтиленгликоль 1000 сукцинатом (TPGS 1000). DCUK-OEt (2,5 г) взвешивали в чистом стеклянном стакане и медленно добавляли TPGS 1000 (47,5 мл). Смесь гомогенизировали в течение 2-3 минут с получением кремово-белой водной суспензии. Эту суспензию инкапсулировали в капсулах Torpac (Torpac, Fairfield, NJ) и вводили перорально крысам с помощью шприца для капсул Torpac (Wempe et al., 2012).
Для индукции нейропатической боли использовали четыре различных средства. Цисплатин (Sigma-Aldrich, St. Louis, MO) растворяли в 0,9% растворе хлорида натрия. Стрептозотоцин (Sigma-Aldrich) растворяли в 20 мМ натрий-цитратном буфере, pH=4,5. Полный адъювант Фрейнда (CFA) получали в Sigma-Aldrich. Моноиодацетат натрия получали в Sigma-Aldrich и растворяли в солевом растворе.
Измерение механической гипералгезии
Эти исследования проводили на самцах крыс Спрег-Доули (Taconic, Germantown PA или Harlan, Indianapolis IN) возрастом примерно восемь недель. Крыс содержали в аккредитованном по стандарту AAALAC виварии с регулируемым освещением, температурой и влажностью. Боль исследовали с помощью электронного анестезиометра фон Фрэя (IITC Life Science, Woodland Hills, CA). Крыс помещали в подвешенные клетки с полом из металлической сетки и позволяли привыкнуть к обстановке в течение приблизительно 20 минут. Механические раздражители прикладывали к середине подошвенной поверхности задней лапы (лап). Использовали два разных метода. В первом методе использовали набор нитей фон Фрея с разной жесткостью (г) и прикладывали к лапе нити возрастающей жесткости. Сила, создаваемая при каждом приложении нити, отображалась на электронном сенсоре. Каждую нить прикладывали пять раз, пока не возникала реакция отдергивания лапы ("вздрагивание" после приложения нити). Когда нить вызывала отдергивание лапы в четырех из пяти тестов или достигали максимального раздражения (10% от массы тела), тестирование прекращали. Среднее из четырех значений использовали для вычисления порога отдергивания лапы в г. Во втором методе полугибкую нить прикладывали к середине подошвенной поверхности лапы с возрастающим давлением, пока не наблюдали отдергивание лапы. Давление (силу в граммах) при отдергивании лапы (порог отдергивания) регистрировали с помощью электронного сенсора. Для каждого теста производили пять измерений и вычисляли среднее значение. В обоих методах, чтобы избежать сенсибилизации, между измерениями устанавливали трехминутный перерыв.
Кратковременное действие DCUKA, BCUKA, DCUK-OEt и габапентина при купировании нейропатической боли (механической гипералгезии), индуцированной цисплатином, полным адъювантом Фрейнда (CFA), стрептозотоцином (STZ) или моноиодацетатом (MIA)
Кратковременное действие DCUKA при индуцированной цисплатином боли: Для введения цисплатина использовали два метода. 1) Цисплатин растворяли в 0,9% солевом растворе (1 мг/мл) и вводили в хвостовую вену в объеме 1,5 или 2,5 мл/кг массы тела. После внутривенной инъекции цисплатина делали инъекцию такого же количества солевого раствора (Joseph and Levine, 2009). Дозы цисплатина составляли 1,5 или 2,5 мг/кг в отдельных экспериментах. 2) Цисплатин растворяли в 0,9% солевом растворе и вводили путем внутрибрюшинной инъекции в дни 1, 4, 8 и 12. Дозы цисплатина составляли 2 мг/кг, 1 мг/кг, 2 мг/кг и 2 мг/кг, соответственно, с суммарной дозой 7 мг/кг. Новый раствор цисплатина приготавливали каждый день перед инъекцией, и 0,9% солевой раствор (2 мл) вводили подкожно после инъекции цисплатина (чтобы избежать нефротоксичности). Во всех экспериментах крыс тестировали на исходную болевую чувствительность (порог механической боли) перед началом введения цисплатина. Схемы эксперимента для исследований с использованием внутривенных инъекций были следующими: (1) Начиная через один час после инъекции цисплатина крысам перорально вводили растворитель (масло канолы/желатин) (через внутрижелудочный зонд) два раза в день (каждые 12 часов) в течение трех дней (эти крысы являются контролями для исследования предупреждения цисплатин-индуцированной боли, см. ниже). В четвертый день у крыс снова исследовали порог механической боли. В пятый день крысам вводили 50 мг/кг DCUKA или растворитель и через час определяли порог механической боли. 2) В 4 день после введения цисплатина крысам вводили различные дозы DCUKA (12,5, 25, 50 или 75 мг/кг) или растворитель через желудочный зонд, и через час определяли порог механической боли. 3) В 6 день после введения цисплатина измеряли порог механической боли и давали крысам 50 мг/кг DCUKA или растворителя перорально. Порог механической боли тестировали через час. 4) В 6 день после введения цисплатина крысам давали различные дозы DCUKA (25, 50 или 75 мг/кг) или растворителя, а через час измеряли порог механической боли. Схема экспериментов, в которых цисплатин вводили внутрибрюшинно, являлась следующей: в 14 день после введения цисплатина (дни 1, 4, 8, 12) исследовали порог механической боли. Затем крысам перорально вводили 50 мг/кг DCUKA или растворителя и через час измеряли порог механической боли. Данные представлены как отношение порога механической боли, измеренного после введения DCUKA, к исходному порогу механической боли до введения цисплатина (измеренному на той же лапе).
Анализ данных для экспериментов с цисплатином: Кратковременное действие DCUKA и мета-анализ: В зависимости от схемы эксперимента анализ состоял либо из однофакторного дисперсионного анализа с повторными измерениями, либо из двухфакторного дисперсионного анализа с повторными измерениями (Proc Mixed, SAS v9.3, Cary, NC). Лечение и время являются основными фиксированными независимыми исследуемыми эффектами. В одном эксперименте оценивали лечение, время и взаимодействие между ними. Идентификационный номер животного использовали в качестве повторяющегося параметра, так как на одном животном производили несколько измерений, в том числе до и после обработки, а также на левой и правой лапе. Все модели тестировали на равные дисперсии между группами лечения (тест Барлетта на однородность дисперсий) и нормальность (критерий согласия Колмогорова-Смирнова). Если данные не соответствовали этим условиям, их соответствующим образом корректировали в смешанной модели. В некоторых анализах использовали апостериорные LSD-критерии Фишера для сравнения статистической значимости (p-значение<0,05) между различными лечебными дозами, и во всех анализах использовали апостериорные LSD-критерии Фишера для сравнения статистической значимости между экспериментальной группой и исходным значением.
Эксперименты включали в мета-анализ, если они соответствовали следующим требованиям: 1. Механическую боль измеряли с помощью теста фон Фрея, 2. Введение цисплатина успешно индуцировало боль (снижение порога механической боли на 25%) и 3. Механическую боль измеряли в течение 90 минут после введения DCUKA. Смешанная модель с использованием дозы DCUKA в качестве фиксированной независимой переменной с 5 различными уровнями классов, идентификацию исследования в качестве как случайной, так и повторяющейся оценки и идентификацию крыс в качестве случайного эффекта использовали для определения общей эффективности DCUKA при нейропатической боли (Proc Mixed, SAS v9.3, Cory, NC). Для парного сравнения доз DCUKA использовали апостериорные LSD-критерии Фишера.
Сравнение действия DCUKA, BCUKA и габапентина при индуцированной цисплатином боли
Цисплатин вводили внутрибрюшинно в дни 1 (2 мг/кг), 4 (1 мг/кг), 8 (2 мг/кг) и 12 (2 мг/кг) (суммарная доза 7 мг/кг). Цисплатин приготавливали ежедневно, и после каждой инъекции цисплатина подкожно вводили 2 мл 0,9% раствора хлорида натрия. В 14 день измеряли порог механической боли, и крысы получали растворитель (масло канолы/желатин), DCUKA (50 мг/кг), BCUKA (50 мг/кг) или габапентин (30 мг/кг, доза, эквимолярная дозе DCUKA и BCUKA). Через один и два часа снова проверяли порог механической боли. Данные представлены как отношение порога механической боли, измеренного после введения DCUKA, BCUKA или габапентина, к исходному порогу механической боли (измеренному на той же лапе). Статистический анализ представлял собой двухфакторный дисперсионный анализ с повторными измерениями (Proc Mixed, SAS v9.3). Лечение и время являлись основными фиксированными независимыми эффектами, и также тестировали взаимодействие. Идентификационный номер животного использовали в качестве повторяющегося параметра. Апостериорные LSD-критерии Фишера использовали для сравнения значимости (p<0,05) между разными точками времени в группах лечения.
Кратковременное действие DCUKA при нейропатической боли, индуцированной полным адъювантом Фрейнда (CFA)
После измерения исходного порога отдергавания лапы, CFA (0,1 мл) вводили подкожно в подошвенную поверхность левой задней лапы крысы под легкой анестезией изофлураном (5% для вводного наркоза и 2% для поддержания). Крыс оставляли в своей домашней клетке на 48 часов. Бумажную подстилку использовали, чтобы избежать компрессионных нейропатий, вызванных жесткой подстилкой. Через 48 или 60 часов после инъекции CFA крысам перорально давали растворитель (масло канолы/желатин) или разные дозы DCUKA перорально, через внутрижелудочный зонд, и через один час определяли порог механической боли. Данные представлены как отношение порога механической боли, измеренного после введения DCUKA, к исходному порогу механической боли.
Анализ данных для экспериментов с CFA: Кратковременное действие 50 мг/кг DCUKA и мета-анализ зависимости эффекта от дозы DCUKA
Каждый эксперимент анализировали с использованием 1-факторного дисперсионного анализа (Proc Glm или Proc Mixed, SAS v9.3, Cary, NC). Тестируемым фиксированным независимым эффектом являлась группа лечения. Все модели тестировали на равные дисперсии между группами лечения (критерий Барлетта на однородность дисперсий) и нормальность (критерий согласия Колмогорова-Смирнова). Если данные не соответствовали этим условиям, использовали смешанную модель. Во всех анализах использовали апостериорные LSD-критерии Фишера для сравнения статистической значимости (p-значение<0,05) между различными группами лечения.
Сравнение действия DCUKA и BCUKA при CFA-индуцированной нейропатической боли
Исходный порог механической боли определяли и вводили животным CFA, как описано выше. Через 48 часов после введения CFA крысам давали растворитель (масло канолы/желатин) (n=17), 50 мг/кг DCUKA (n=17) или 50 мг/кг BCUKA (n=6). Порог механической боли тестировали через час, и данные представлены как отношение болевого порога после введения растворителя, DCUKA или BCUKA, к исходному болевому порогу, измеренному на той же лапе. Для определения статистической значимости использовали однофакторный дисперсионный анализ с последующим апостериорным LSD-критерием Фишера (p<0,05).
Кратковременное действие DCUKA при индуцированной стрептозотоцином (STZ) нейропатической боли (Модель диабетической нейропатической боли)
После измерения исходного порога отдергивания лапы определяли исходную массу тела и концентрацию глюкозы в крови (уровень глюкозы в крови измеряли в крови из хвоста при использовании системы мониторинга уровня глюкозы в крови ASCENSIA CONTOUR, Bayer, Pittsburgh, PA). Крыс не кормили в течение ночи и внутрибрюшинно вводили растворитель (20 мМ цитрат натрия, pH 4,5, Sigma-Aldrich) или 50 мг/кг STZ в растворителе. Раствор STZ приготавливали каждый день и использовали в течение 10 минут. Корм крысам давали через 30 минут после введения STZ. Через три дня после введения STZ снова измеряли уровни глюкозы в крови, и крыс с уровнем глюкозы в крови выше 350 мг/дл считали "больными диабетом". Если уровень глюкозы в крови был ниже 350 мг/дл, крысе вводили вторую дозу STZ (45 мг/кг), используя ту же процедуру. Через четырнадцать дней после первого введения STZ крысам давали растворитель (масло канолы/желатин) или различные дозы DCUKA перорально, и порог механической боли проверяли в разное время после этих обработок. Данные представлены как отношение порога механической боли после введения DCUKA к исходному порогу механической боли (измеренному на той же лапе).
Индивидуальный экспериментальный анализ
Точки данных считали выбросами и исключали из набора данных, если отношение порога механической боли к исходному уровню выходило за пределы соответствующего среднего значения в группе лечения ±2 стандартных отклонения. Каждый эксперимент анализировали с использованием однофакторного дисперсионного анализа (Proc Glm или Proc Mixed, SAS v9.3, Cary, NC). Тестируемым фиксированным независимым эффектом являлась группа лечения. Все модели тестировали на равные дисперсии между группами лечения (тест Барлетта на однородность дисперсий) и нормальность (критерий согласия Колмогорова-Смирнова). Если данные не соответствовали этим условиям, использовали смешанную модель. Во всех анализах использовали апостериорные LSD-критерии Фишера для сравнения статистической значимости (p-значение<0,05) между различными способами лечения.
Кратковременное действие DCUKA при индуцированной моноиодацетатом (MIA) нейропатической боли (модель остеоартрозной нейропатической боли)
Крысам делали анестезию изофлураном, брили правые колени и вводили растворитель или MIA для индукции болезни. Животных взвешивали еженедельно до измерения боли в день 20. Исходные измерения отдергивания лапы проводили у животных, не получавших MIA. В день 21 группам животных перорально вводили растворитель или DCUKA (1 или 2 капсулы перорально; дозы примерно 50 мг/кг или 100 мг/кг). Порог механической боли проверяли через 90 минут после введения лекарственного средства. В конце тестирования из хвостовой вены забирали кровь для определения уровней DCUKA с помощью анализа ЖХ-МС/МС. Данные представлены как отношение порога механической боли после введения DCUKA к порогу механической боли, определенному у животных без введения лекарственных средств или MIA.
Анализ данных для экспериментов с MIA
Эксперимент анализировали с использованием однофакторного дисперсионного анализа (SigmaPlot 12) и апостериорные критерии Холма-Шидака использовали для всех парных сравнений. P-значения <0,05 считали статистически значимыми. С учетом того, что введение 1 капсулы DCUKA не оказывало значимого влияния на порог механической боли (Фигура 8), после этого лечения оценивали уровни Киндолора в крови. Было определено, что уровни Киндолора в крови <500 нг/мл (<1,1 мкМ) были получены после введения одной капсулы, и этот уровень в крови был признан неэффективным при лечении нейропатической боли, индуцированной MIA.
На Фигуре 2 показано, что лечение DCUKA (50 мг/кг) купирует нейропатическую боль, вызванную введением крысым химиотерапевтического противоопухолевого средства цисплатина. Показаны объединенные результаты из шести экспериментов. На графике представлены средние значения ±1 SEM. *P-значение <0,0001 в сравнении с контролем (0 мг/кг DCUKA). Во всех экспериментах крыс проверяли на исходный порог механической боли до введения цисплатина. После введения цисплатина, описанного ранее, крысам вводили DCUKA, а через час снова определяли порог механической боли. Результаты показывают отношение порога механической боли, измеренного через один час после введения растворителя или DCUKA, в сравнении с исходным (до введения цисплатина) порогом механической боли.
На Фигуре 3 графически сравнивается действие DCUKA (50 мг/кг), BCUKA (50 мг/кг) и габапентина (Нейронтин, 30 мг/кг) при нейропатической боли, индуцированной химиотерапевтическим средством цисплатином. Средний порог механической боли ±1 стандартная ошибка нанесен на график для каждой группы лечения и точки времени. *P<0,05 в сравнении с соответствующей группой до лечения. Перед введением цисплатина у крыс определяли исходный порог механической боли и вводили цисплатин, как описано ранее. После введения цисплатина измеряли порог механической боли и крысам перорально вводили дозы растворителя, DCUKA, BCUKA или габапентина. Порог механической боли снова измеряли через 1 и 2 часа после этих процедур. Данные представляют собой отношение порога механической боли, измеренного до введения DCUKA, BCUKA или габапентина, а также через 1 и 2 часа. Отдельное введение только цисплатина ("предварительная обработка") значимо снижало порог механической боли по сравнению с исходным уровнем, а DCUKA и BCUKA в значительной степени устраняли снижение порога механической боли. Габапентин в дозе, эквимолярной дозе DCUKA и BCUKA, не оказывал значимого влияния на снижение порога механической боли, вызванное цисплатином.
Фигура 4 графически иллюстрирует, что DCUKA лечит нейропатическую боль, вызванную введением крысам полного адъюванта Фрейнда (CFA) с целью вызвать воспалительную реакцию. Данные объединены из трех экспериментов. В каждом эксперименте исходный порог механической боли (сила в граммах, вызывающая отдергивание лапы) сначала измеряли с помощью электронного анестезиометра фон Фрея. Затем крысам вводили 0,1 мл полного адъюванта Фрейнда (CFA) в подошвенную поверхность левой задней лапы. Через 48-60 часов, когда развивалась вызванная CFA боль, крысам перорально вводили растворитель (эмульсию желатина/масла канолы) или 50 мг/кг DCUKA. Через час снова измеряли порог механической боли. Результаты показывают отношение порога механической боли, измеренного через час после введения растворителя или DCUKA, к исходному порогу механической боли. Апостериорный LSD t-критерий Фишера для парных сравнений показал значимое (*) различие между 50 мг/кг DCUKA и 0 мг/кг DCUKA (p-значение <0,0001). Введение CFA снижало порог механической боли примерно на 60%, а лечение DCUKA устраняло этот эффект и повышало порог механической боли в лапе, обработанной CFA, до уровня, незначительно отличающегося от исходного уровня.
Фигура 5 графически иллюстрирует сравнение действия DCUKA (50 мг/кг) и BCUKA (50 мг/кг) при лечении нейропатической боли, вызванной введением CFA. Порог механической боли для каждого животного представлен отношением к исходному уровню только для лапы, в которую делали инъекцию. Средний порог механической боли ±1 стандартная ошибка нанесен на график для каждой группы лечения. *P<0,05 в сравнении с растворителем. Исходный порог механической боли измеряли и вводили крысам CFA, как описано ранее. Через 48 часов крысам вводили растворитель (масло канолы/желатин), DCUKA или BCUKA, а через час измеряли порог механической боли. Введение CFA снижало порог механической боли приблизительно до 40% от исходного уровня, а лечение DCUKA (n=17) или BCUKA (n=6) возвращало порог механической боли к уровню, незначительно отличающемуся от исходного уровня.
Дозозависимое действие DCUKA при нейропатической боли, индуцированной CFA, определяли с использованием подхода мета-анализа. Результаты показаны на Фигуре 6. Порог механической боли для каждого животного представлен отношением к исходному уровню только для лапы, в которую делали инъекцию. Средний порог механической боли ±1 стандартная ошибка наносили на график для каждой группы лечения. Оно основано на средних значениях при лечении в 5 исследованиях, включенных в мета-анализ. *P<0,05 по сравнению с группой лечения носителем (0 мг/кг). Во всех экспериментах с CFA исходный порог механической боли измеряли с помощью электронного анестезиометра фон Фрея. CFA вводили путем инъекции в подошвенную поверхность левой задней лапы, и через 48 часов после инъекции крысам перорально вводили растворитель (желатин/масло канолы) или DCUKA. Порог механической боли снова измеряли через 60 мин после введения носителя или DCUKA. Для включения эксперимента в мета-анализ присутствовали следующие требования: 1) введение CFA вызывало снижение порога механической боли не меньше чем на 25%; 2) болевой порог измеряли через 60 минут после введения DCUKA или растворителя. Этим требованиям соответствовали пять экспериментов, в которых тестировали разные дозы DCUKA. Порог механической боли в виде отношения к исходному порогу механической боли показан как среднее значение ±SEM. Наблюдали значимый общий эффект DCUKA (F(5125)=7,71, P<0,0001). Введение CFA снижало порог механической боли примерно на 60%, и этот эффект был в значимой степени купирован дозами DCUKA 30 мг/кг и выше, то есть болевой порог возвращался к исходному уровню.
На Фигуре 7 показано, что DCUKA купирует нейропатическую боль, которая сопутствует диабету. Диабет у крыс индуцировали инъекцией стрептозотоцина (STZ), как описано ранее. Апостериорный LSD t-критерий Фишера для парных сравнений показал значимое (*) различие между 50 мг/кг DCUKA и 0 мг/кг DCUKA (различие=0,60, p-значение<0,0001). Показаны объединенные данные из трех экспериментов. В каждом эксперименте исходный порог механической боли определяли с помощью электронного анестезиометра фон Фрея. Через четырнадцать дней после введения STZ, перорально вводили растворитель (желатин/масло канолы) или 40 или 50 мг/кг DCUKA (эти дозы не оказывали значимо различного действия), и через 90 минут оценивали механическую боль. Результаты показывают отношение порога механической боли после введения растворителя/DCUKA к исходному порогу механической боли. STZ снижал порог механической боли примерно на 40%, и этот эффект был купирован до исходного уровня при лечении DCUKA.
Дозозависимое действие DCUKA при STZ-индуцированной нейропатической боли определяли с помощью метода мета-анализа. Требования для включения эксперимента в мета-анализ являлись следующими: 1) введение STZ вызывало нейропатическую боль, измеряемую снижением порога механической боли по меньшей мере на 25%; 2) боль измеряли через 90 минут после введения растворителя или DCUKA. В мета-анализ включали четыре эксперимента, соответствовавшие этим критериям.
На Фигуре 8 показано дозозависимое действие DCUKA при лечении нейропатической боли, индуцированной STZ. Данные представлены в виде отношения порога механической боли после введения растворителя или DCUKA к исходному порогу боли, измеренному на той же лапе. *P<0,05 в сравнении с группой, получавшей растворитель. Наблюдали общий значимый эффект DCUKA в отношении порога механической боли (F(7115=8,48, p<0,0001). Введение STZ вызывало снижение порога боли примерно на 40%, а дозы DCUKA 30 мг/кг и выше купировали эффект STZ и повышали болевой порог до исходного уровня.
На Фигуре 9 показана дозозависимость DCUKA при лечении нейропатической боли, индуцированной MIA. Данные представлены в виде отношения среднего ±SEM порога механической боли после введения растворителя (0 капсул) или DCUKA (1 или 2 капсулы) к исходному болевому порогу, измеренному у животных, не получавших MIA или лекарственных средств. *P<0,05 в сравнении с группой введения растворителя (дисперсионный анализ и апостериорные сравнения). Наблюдали общий значимый эффект DCUKA в отношении порога механической боли (F(226)=4,07, p=0,029). Введение MIA приводило к снижению болевого порога приблизительно на 60%, и доза DCUKA (2 капсулы) приблизительно 100 мг/кг обеспечивала значимое купирование эффекта MIA.
ПРИМЕР 4 DCUKA и DCUK-OEt улучшает эффект морфина при CFA-индуцированной нейропатической боли.
Данные ниже демонстрируют, что низкие дозы DCUKA вместе с морфином приводят к синергическому эффекту при снижении механической аллодинической и термической гипералгезической боли, вызванной воспалительным агентом.
Введение CFA и измерение порога механической боли описаны в Примере 3. В этом эксперименте порог механической боли тестировали до начала исследования. Через 48 часов после введения CFA растворитель, DCUKA или морфин, или комбинацию DCUKA и морфина вводили за 30 минут до измерения порога механической боли.
Тест на термическую гиперчувствительность (тест на отдергивание лапы при воздействии теплового излучения)
Крыс помещали в прозрачные пластиковые камеры на поверхность стекла и позволяли привыкнуть к обстановке в течение 15 минут до начала тестирования. Тетмическую чувствительность измеряли по задержке отдергивания лапы при воздействии теплового излучения в качестве раздражителя. Источник теплового излучения (то есть инфракрасного) включали по таймеру и направляли на подошвенную поверхность левой задней лапы. Детектор движения, который останавливал и лампу, и таймер при отдергивании лапы, определял задержку отдергивания лапы. Время задержки измеряли до и после введения лекарственного средства или носителя. Максимальное время до выключения 33 секунды использовали, чтобы предотвратить повреждение тканей. В этом эксперименте крысам вводили DCUK-OEt, после чего сразу вводили морфин в возрастающих отношениях доз и тестировали через 30 минут.
Фигура 10А демонстрирует, что комбинация таких доз DCUKA и морфина, которые по отдельности неэффективны для обеспечения обезболивания, приводит к полному купированию хронической боли, вызванной воспалением. Эти данные согласуются с выводом, что комбинация DCUKA и морфина оказывает действие, превышающие аддитивные эффекты каждого лекарственного средства по отдельности, как определено согласно каждому из подходов "Стратегии на основе эффекта", обсуждаемых в публикации Foucquier & Guedj (2015). Эта фигура иллюстрирует подход "Подпороговой комбинации", когда комбинация неэффективных доз лекарственных средств производит значимый эффект. Фигуры 10B и 10C демонстрируют, что комбинация DCUK-OEt и морфина обеспечивает супрааддитивный эффект по сравнению с любым из этих средств, вводимым отдельно. В этом случае была протестирована термическая гипералгезия, вызванная введением CFA. Значение ED50 для антигипералгезического ответа на морфин значительно снижалась при введении DCUK-OEt при отношении дозы к морфину >30:1. Например, доза морфина 0,2 мг/кг вызывает примерно 20% антигипералгезического ответа. В комбинации с 6,4 мг DCUK-OEt ответ повышался до 40%. DCUK-OEt снижал ED50 для антигипералгезического действия морфина при отношении доз DCUK-OEt/морфина 18:1 (†P<0,7) и 32:1 (*P<0,05) по сравнению с одним морфином согласо однофакторному дисперсионному анализу с апостериорным критерием Даннета.
ПРИМЕР 5 DCUKA улучшает эффект оксикодона и метадона при лечении FA-индуцированной нейропатической боли.
Данные ниже демонстрируют, что низкие дозы DCUKA потенцируют эффект низких доз оксикодона или метадона, уменьшая механическую аллодинию, вызванную воспалительным агентом. Данные представлены в виде отношения средних значений ±SEM порога механической боли в обработанной лапе после введения лекарственного средства или растворителя (VEH) к порогу механической боли до введения адъюванта Фрейнда (FA). *P<0,05 в сравнении с группой, получавшей растворитель (n=8/группа, дисперсионный анализ и апостериорные сравнения).
После тестирования механической боли в исходном состоянии крысам в правую заднюю лапу вводили смесь неполного адъюванта Фрейнда (FA) и Mycobacterium butyricum (аналогично полному адъюванту Фрейнда). Через 72 часа тестирование механической боли повторяли с использованием модифицированной версии процедуры теста Фон Фрэя, описанного в Примере 3. Крыс тестировали с помощью набора нитей фон Фрэя в пределах от 3,16 до 5,18 абсолютного порога. Каждую нить применяли три раза, чтобы определить, на нить какой жесткости крыса будет отвечать 100% времени. Данные анализировали при использовании программы Psychofit. В день 4, для определения ответов на отдельные лекарственные средства, группам крыс давали растворитель или дозы DCUKA (одну или две капсулы, п/о, как описано в Примере 3) или дозы оксикодона (0,1, 0,25 или 1 мг/кг в/б), или дозы метадона (1,5, 2,5 или 3,5 мг/кг в/б), и через 75-90 минут после введения доз проводили тест фон Фрэя. В день 8 оценивали эффект комбинации низких доз (одна капсула) DCUKA с низкими дозами оксикодона или метадона. Крысам давали DCUKA плюс растворитель, оксикодон плюс растворитель, метадон плюс растворитель или DCUKA плюс оксикодон, или DCUKA плюс метадон, с использованием доз, определенных в начальной фазе исследования. На Фигуре 11 продемонстрировано, что комбинация доз DCUKA (1 капсула) и оксикодона (0,1 мг/кг), которые по отдельности неэффективны для обеспечения антигипералгезии, приводит к уменьшению вызванной воспалением хронической боли. Эти данные согласуются с выводом, что комбинация DCUKA и оксикодона обладает действием, превосходящим аддитивные эффекты каждого лекарственного средства в отдельности, как определено согласно всем четырем подходам "Стратегии на основе эффекта", обсуждаемым в публикации Foucquier и Guedj (2015). Эта фигура иллюстрирует подход "Подпороговой комбинации", т.е. когда комбинация неэффективных доз лекарственных средств производит значимый эффект. Сплошная линия указывает исходное отношение порога механической боли у животных, получавших адъювант Фрейнда и растворитель. Повышение выше этой линии указывает на (незначимые) эффекты DCUKA или оксикодона по отдельности. Пунктирная линия указывает ожидаемый аддитивный эффект комбинации лекарственных средств. Увеличенный эффект комбинации лекарственных средств (т.е. DCUKA плюс оксикодон 0,1 мг/кг) выше пунктирной линии указывает на "положительный эффект комбинации" (Foucquier and Guedj, 2015).
На Фигуре 12 показано, что DCUKA (1 капсула) усиливает эффект метадона (1,5 или 3,5 мг/кг) при уменьшении хронической боли, вызванной воспалением. Данные представлены в виде отношения среднего значения ±SEM порога механической боли в обработанной лапе после введения лекарственного средства или растворителя (VEH) к порогу механической боли до введения адъюванта Фрейнда (FA). *P<0,05 в сравнении с DCUKA/VEH или только 1,5 мг/кг метадона; **P<0,05 в сравнении с DCUKA/VEH или только 3,5 мг/кг метадона; +P<0,05 в сравнении с VEH (n=8/группа, дисперсионный анализ и апостериорные сравнения).
Эти данные согласуются с выводом, что комбинация DCUKA и метадона обладает действием, превышающим аддитивные эффекты каждого лекарственного средства по отдельности, как определено согласно каждому из подходов "Стратегии на основе эффекта", обсуждаемых в Foucquier and Guedj (2015). В случае 1,5 мг/кг метадона эта фигура иллюстрирует подход "Подпороговой комбинации", в котором комбинация неэффективных доз лекарственных средств производит значимый эффект. Сплошная черная линия указывает исходное отношение пороговых значений механической боли у животных, получавших адъювант Фрейнда и растворитель. Повышение выше этой линии отражает (незначительное) воздействие DCUKA или метадона (1,5 мг/кг) по отдельности. Серая линия указывает ожидаемый аддитивный эффект комбинации DCUKA и метадона 1,5 мг/кг. Повышенный эффект комбинации DCUKA и метадона 1,5 мг/кг отражает "положительный эффект комбинации" (Foucquier and Guedj, 2015). В случае 3,5 мг/кг метадона диаграмма иллюстрирует подход "Одного средства в наибольшей дозе", который отражает то, что эффект комбинации лекарственных средств превышает эффекты, производимые ее отдельными компонентами. Пунктирная линия указывает ожидаемый аддитивный эффект 3,5 мг/кг метадона и DCUKA. Повышенный эффект комбинации лекарственных средств выше этой линии отражает "положительный эффект комбинации" (Foucquier and Guedj, 2015).
ПРИМЕР 6 DCUKA улучшает эффект трамадола при лечении MIA-индуцированной нейропатической боли.
Данные ниже демонстрируют, что в другой модели хронической боли, модели MIA-индуцированного остеоартроза, низкие дозы DCUKA могут синергически взаимодействовать с низкой дозой другого наркотического анальгетика (трамадола), уменьшая гипералгезию. Трамадол воздействует на µ-опиатный рецептор, а также является ингибитором обратного захвата серотонина и норэпинефрина.
После введения MIA, как описано в Примере 3, на крысах первоначально исследовали кратковременный эффект различных доз DCUKA в день 21 после введения MIA, результаты показаны на Фигуре 13. Введение одной капсулы DCUKA (приблизительно 50 мг/кг) не оказывало значительного влияния на MIA-индуцированную боль. Уровни DCUKA в крови через 2-2,5 часа после введения дозы в этот момент составили <500 нг/мл (<1,1 мкМ). Такой уровень DCUKA в крови считался неэффективным для уменьшения MIA-индуцированной нейропатической боли. У крыс снова тестировали порог механической боли через 29 дней после введения MIA (болевой порог повторно оценивали в день 28, чтобы определить, что не присутствовало значимое отличие от первоначального теста в день 20). Крысам давали растворитель, 1 капсулу (низкая доза) DCUKA п/о, 5 мг/кг (низкая доза) п/о, трамадол или 1 капсулу DCUKA плюс 5 мг/кг трамадола, за 90 минут до измерения порога механической боли, как описано в Примере 3, и забирали кровь из яремной вены после тестирования для оценки уровней DCUKA с помощью методов ЖХ-МС/МС, описанных в Примере 2.
Анализ данных
Данные представлены в виде отношения порога механической боли после лечения лекарственным средством к исходному порогу механической боли до введения MIA и без лечения лекарственным средством. Данные двух выбросов в группе DCUKA плюс трамадол исключали из анализа. Считалось, что животные с уровнем DCUKA в крови <500 нг/мл, т.е. уровнем, который согласно предыдущим оценкам был неэффективным для купирования MIA-индуцированной нейропатической боли, получали "низкую дозу" DCUKA. Данные этих животных использовали для анализа различий между группами с использованием однофакторного дисперсионного анализа и критерия Холма-Шидака для всех парных сравнений.
Фигура 13 демонстрирует, что дозы DCUKA (1 капсула, когда уровень DCUKA в крови был <500 нг/мл) и трамадола (5 мг/кг), которые по отдельности не оказывают значимого влияния на порог механической боли по сравнению с растворителем, в комбинации приводят к значимому устранению снижения болевого порога, вызванного введением MIA. Данные представлены в виде среднего значения ±SEM отношения порога механической боли после лечения низкой дозой DCUKA, низкой дозой (5 мг/кг) трамадола или комбинацией низкой дозы DCUKA и низкой дозы трамадола к порогу механической боли до введения MIA. *P<0,05 в сравнении со всеми остальными группами (дисперсионный анализ и апостериорные сравнения).
Эти данные согласуются с выводом, что комбинация DCUKA и трамадола обладает действием, превышающим аддитивные эффекты каждого лекарственного средства по отдельности, как определено согласно каждому из подходов "Стратегии на основе эффекта", обсуждаемых в Foucquier and Guedj (2015). Эта фигура иллюстрирует подход "Одного средства в наибольшей дозе", который отражает тот факт, что действие комбинации лекарственных средств превышает действие, оказываемое ее отдельными компонентами. Сплошная линия указывает исходное отношение порога механической боли у животных, получавших МИА и растворитель. (Незначимый) эффект трамадола обозначен повышением по сравнению с исходным уровнем, это повышение указано пунктирной линией. Эффект комбинации лекарственных средств выше пунктирной линии отражает "положительный эффект комбинации" (Foucquier and Guedj (2015).
ПРИМЕР 7 DCUKA улучшает действие аспирина при лечении STZ-индуцированной нейропатической боли.
Данные ниже демонстрируют, что в другой модели хронической боли, модели STZ-индуцированной диабетической нейропатии, DCUKA может синергически взаимодействовать с другим анальгетиком (аспирином), уменьшая аллодинию.
После введения стрептозотоцина (STZ), как описано в Примере 3, крыс тестировали на аллодинию с использованием аппарата фон Фрэя. За шестьдесят минут до тестирования крыс делили на четыре группы. Группа 1 получала растворитель; Группа 2 получала DCUKA; Группа 3 получала аспирин; и Группа 4 получала комбинацию DCUKA и аспирина.
На Фигуре 14 показано, что дозы DCUKA (12,5 мг/кг) или аспирина (25 мг/кг), которые по отдельности не оказывают значимого эффекта при аллодинии, в комбинации полностью купируют гиперчувствительность, т.е. возвращают болевой порог к исходному уровню. Эти данные согласуются с выводом, что комбинация DCUKA и аспирина обладает действием, превышающим аддитивное действие каждого лекарственного средства по отдельности, что определяется согласно каждому из подходов "Стратегии на основе эффекта", обсуждаемых в Foucquier & Guedj (2015). Эта Фигура иллюстрирует подход "Подпороговой комбинации", когда комбинация неэффективных доз лекарственных средств производит значимый эффект.
ПРИМЕР 8 DCUKA потенцирует эффект диклофенака при лечении FA-индуцированной нейропатической боли.
Данные ниже демонстрируют, что низкая доза DCUKA (одна капсула, как описано в Примере 1) потенцирует эффект низкой дозы диклофенака, уменьшая механическую аллодинию, вызванную воспалительным агентом. Данные представлены в виде отношения среднего значения ±SEM порога механической боли в обработанной лапе после введения лекарственного средства или растворителя (VEH) к порогу механической боли до введения адъюванта Фрейнда (FA). *P<0,05 в сравнении с DCUKA или 1,5 мг/кг диклофенака по отдельности; **P<0,05 в сравнении с DCUKA или 10 мг/кг диклофенака по отдельности (дисперсионный анализ и апостериорные сравнения).
Крысам вводили адъювант Фрейнда и тестировали на механическую боль, как описано в Примерах 2 и 5. Группам крыс давали разные дозы диклофенака в день 4 для определения подходящих доз для эксперимента по тестированию комбинированных эффектов DCUKA и диклофенака. Различные группы крыс тестированы в день 4 после введения одной капсулы DCUKA или низких доз диклофенака отдельно и в комбинации. На Фигуре 15 показано, что DCUKA потенцирует действие диклофенака при уменьшении хронической боли, вызванной воспалением. Эти данные согласуются с выводом, что комбинация DCUKA и диклофенака обладает действием, превышающим аддитивное действие каждого лекарственного средства по отдельности, как определено согласно каждому из подходов "Стратегии на основе эффекта", обсуждаемых в Foucquier and Guedj (2015). В случае 1,5 мг/кг диклофенака эта фигура иллюстрирует подход "Подпороговой комбинации", в котором комбинация неэффективных доз лекарственных средств оказывает значимое действие. Сплошная черная линия указывает исходное отношение порогов механической боли у животных, получавших адъювант Фрейнда и растворитель. Повышение выше этой линии указывает на (незначимый) эффект DCUKA или 1,5 мг/кг диклофенака по отдельности. Серая линия указывает на ожидаемый аддитивный эффект DCUKA и 1,5 мг/кг диклофенака. Повышенный эффект комбинации DCUKA и 1,5 мг/кг диклофенака выше серой линии указывает на "положительный эффект комбинации". В случае 10 мг/кг диклофенака фигура иллюстрирует подход "Одного средства в наибольшей дозе", который отражает тот факт, что действие комбинации лекарственных средств превышает действие отдельных компонентов. Пунктирная линия указывает ожидаемые аддитивные эффекты DCUKA и 10 мг/кг диклофенака. Повышенный эффект комбинации лекарств выше пунктирной линии указывает на "положительный эффект комбинации" (Foucquier and Guedj, 2015).
ПРИМЕР 9 Предотвращение индуцированной цисплатином и CFA нейропатической боли с помощью DCUKA.
Данные ниже показывают, что введение животным DCUKA после повреждения, но до развития нейропатической боли, может предотвратить развитие боли. Крысам вводили цисплатин или CFA и измеряли порог механической боли, как описано в Примере 3.
Предотвращение CFA-индуцированной нейропатической боли с применением DCUKA
После измерения исходного порога механической боли крысам вводили CFA, как описано ранее. После инъекции CFA крысам давали растворитель (масло канолы/желатин) (n=7) или 50 мг/кг DCUKA (n=7) перорально через внутрижелудочный зонд. Затем крысы получали еще три введения растворителя или DCUKA с интервалами по 12 часов. Через 60 часов после введения CFA порог механической боли снова тестировали.
Предотвращение цисплатин-индуцированной нейропатической боли с применением DCUKA. Через один день после измерения исходного порога механической боли крысам в/б вводили цисплатин. Инъекции цисплатина снова делали в дни 4, 8 и 12. Через один час после первой инъекции цисплатина крысам начинали давать 50 мг/кг DCUKA или растворитель через внутрижелудочный зонд два раза в день в течение 14 дней. После последнего лечения (день 15) порог механической боли снова тестировали. Затем болевой порог тестировали один раз в неделю в течение 4 недель без дополнительного лечения.
На Фигуре 16 показано, что DCUKA может предотвратить развитие нейропатической боли, вызванной CFA. Данные показывают отношение порога механической боли через 60 часов после введения CFA к исходному порогу механической боли до введения CFA. Введение CFA значительно уменьшало болевой порог приблизительно на 70%. Однако, когда животным ежедневно давали DCUKA до тестирования, порог не уменьшался (P=0,16), а оставался близко к исходному болевому порогу, т.е. DCUKA обладает способностью предотвращать развитие воспалительной боли при введении после воспалительного агента. Для сравнения болевого порога между группами использовали t-критерий Стьюдента, а парный t-критерий использовали для сравнения болевого порога в группе.
На Фигуре 17 показано, что многократное введение DCUKA в период между введением цисплатина и тестированием болевого порого предотвращало развитие боли в модели цисплатин-индуцированной нейропатической боли на крысах. Данные представлены как среднее значение ±SEM порога механической боли (порога отдергивания лапы). Инъекция цисплатина приводила к значительному снижению порога механической боли в течение нескольких дней у крыс, длительно получавших растворитель. При многократном введении DCUKA статистически значимого снижения болевого порога по сравнению с исходным уровнем до введения цисплатина не наблюдали. Результаты показывают, что DCUKA обладает способностью предотвращать развитие нейропатической боли, вызванной химиотерапией, при введении после химиотерапевтического средства.
Двухфакторный дисперсионный анализ с повторными измерениями выявил статистически значимое различие (P=0,023) между двумя лечениями, DCUKA и растворителем. Индивидуальные P-значения, показанные на графике, относятся к DCUKA в сравнении с растворителем и вычислены методом множественного сравнения. Также обнаружены значимые различия в болевом пороге в группе цисплатина и растворителя (дни 15, 19, 25, 32) по сравнению с днем 0, до введения цисплатина, однако отсутствовали значимые различия в болевом пороге между группой цисплатина и DCUKA по сравнению со значениями в День 0 до инъекции цисплатина.
Выше были описаны предпочтительные варианты осуществления настоящего изобретения, включая лучший из известных авторам изобретения способ осуществления изобретения. Вариации этих предпочтительных вариантов осуществления в рамках объема настоящего изобретения будут очевидны специалистам в данной области техники после прочтения предыдущего описания. Таким образом, настоящее изобретение включает в себя все модификации и эквиваленты заявленного изобретения, определяемого прилагаемой формулой изобретения. Более того, любая комбинация вышеописанных элементов во всех их возможных вариантах охвачена изобретением, если иное не указано в настоящем документе или иным образом в явной форме не противоречит контексту.
Использованные источники
Каждый документ, перечисленный ниже, полностью включен в настоящий документ посредством отсылки.
Baldini A., Von Korff M., Lin E.H. (2012) A Review of Potential Adverse Effects of Long-Term Opioid Therapy: A Practitioner's Guide. Prim Care Companion CNS Disord 14.
Barber J. (2011) Examining the use of tramadol hydrochloride as an antidepressant. Exp Clin Psychopharmacol 19:123-130.
Basbaum A.I., Bautista D.M., Scherrer G., Julius D. (2009) Cellular and molecular mechanisms of pain. Cell 139:267-284.
Belkouch M., Dansereau M.A., Tetreault P., Biet M., Beaudet N., Dumaine R., Chraibi A., Melik-Parsadaniantz S., Sarret P. (2014) Functional up-regulation of Nav1.8 sodium channel in Abeta afferent fibers subjected to chronic peripheral inflammation. Journal of neuroinflammation 11:45.
Black J.A., Liu S., Tanaka M., Cummins T.R., Waxman S.G. (2004) Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108:237-247.
Bodnar R.J. (2000) Supraspinal circuitry mediating opioid antinociception: antagonist and synergy studies in multiple sites. J Biomed Sci 7:181-194.
Bozic I., Reiter J.G., Allen B., Antal T., Chatterjee K., Shah P., Moon Y.S., Yaqubie A., Kelly N., Le D.T., Lipson E.J., Chapman P.B., Diaz L.A., Jr., Vogelstein B., Nowak M.A. (2013) Evolutionary dynamics of cancer in response to targeted combination therapy. Elife 2:e00747.
Cao C. (2017) Flow Chemistry: pathway for continuous API manufacturing. Pharmaceutical Manufacturing, https://www.pharmasalmanac.com/articles/flow-chemistry-pathway-for-continuous-api-manufacturing.
Carvalho A.F., Sharma M.S., Brunoni A.R., Vieta E., Fava G.A. (2016) The Safety, Tolerability and Risks Associated with the Use of Newer Generation Antidepressant Drugs: A Critical Review of the Literature. Psychother Psychosom 85:270-288.
Cashman J.N. (1996) The mechanism of action of NSAIDs in analgesia. Drugs 52 Suppl 5:13-23.
Childers W.E., Jr., Baudy R.B. (2007) N-methyl-D-aspartate antagonists and neuropathic pain: the search for relief. J Med Chem 50:2557-2562.
Choi J.S., Waxman S.G. (2011) Physiological interactions between Na(v)1.7 and Na(v)1.8 sodium channels: a computer simulation study. J. Neurophysiol 106:3173-3184.
Chou R., Turner J.A., Devine E.B., Hansen R.N., Sullivan S.D., Blazina I., Dana T., Bougatsos C., Deyo R.A. (2015) The effectiveness and risks of long-term opioid therapy for chronic pain: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med 162:276-286.
Chou T.C. (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70:440-446.
Coggeshall R.E., Tate S., Carlton S.M. (2004) Differential expression of tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 in normal and inflamed rats. Neurosci Lett 355:45-48.
Costigan M., Scholz J., Woolf C.J. (2009) Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 32:1-32.
Davies S.N., Lodge D. (1987) Evidence for involvement of N-methylaspartate receptors in 'wind-up' of class 2 neurones in the dorsal horn of the rat. Brain Res 424:402-406.
Dib-Hajj S.D., Cummins T.R., Black J.A., Waxman S.G. (2007) From genes to pain: Na v 1.7 and human pain disorders. Trends Neurosci 30:555-563.
Dickenson A.H., Sullivan A.F. (1987) Evidence for a role of the NMDA receptor in the frequency dependent potentiation of deep rat dorsal horn nociceptive neurones following C fibre stimulation. Neuropharmacology 26:1235-1238.
Eijkelkamp N., Linley J.E., Baker M.D., Minett M.S., Cregg R., Werdehausen R., Rugiero F., Wood J.N. (2012) Neurological perspectives on voltage-gated sodium channels. Brain 135:2585-2612.
Fava G.A., Benasi G., Lucente M., Offidani E., Cosci F., Guidi J. (2018) Withdrawal Symptoms after Serotonin-Noradrenaline Reuptake Inhibitor Discontinuation: Systematic Review. Psychother Psychosom 87:195-203.
Fernandez-Montoya J., Avendano C., Negredo P. (2017) The Glutamatergic System in Primary Somatosensory Neurons and Its Involvement in Sensory Input-Dependent Plasticity. Int J Mol Sci 19.
Ferrari L.F., Lotufo C.M., Araldi D., Rodrigues M.A., Macedo L.P., Ferreira S.H., Parada C.A. (2014) Inflammatory sensitization of nociceptors depends on activation of NMDA receptors in DRG satellite cells. Proc Natl Acad Sci U S A 111:18363-18368.
Foucquier J., Guedj M (2015) Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect 3:e00149.
Galizzi J.P., Lockhart B.P., Bril A. (2013) Applying systems biology in drug discovery and development. Drug Metabol Drug Interact 28:67-78.
Gaunitz C., Schuttler A., Gillen C., Allgaier C. (2002) Formalin-induced changes of NMDA receptor subunit expression in the spinal cord of the rat. Amino Acids 23:177-182.
Institute of Medicine (2011), Relieving pain in America: A Blueprint for Transforming Prevention, Care, Education and Research, National Academics Press, Washington, D.C.
Iwata H., Takasusuki T., Yamaguchi S., Hori Y. (2007) NMDA receptor 2B subunit-mediated synaptic transmission in the superficial dorsal horn of peripheral nerve-injured neuropathic mice. Brain Res 1135:92-101.
Jang J.H., Kim D.W., Sang Nam T., Se Paik K., Leem J.W. (2004) Peripheral glutamate receptors contribute to mechanical hyperalgesia in a neuropathic pain model of the rat. Neuroscience 128:169-176.
Karlsson U., Sjodin J., Angeby Moller K., Johansson S., Wikstrom L., Nasstrom J. (2002) Glutamate-induced currents reveal three functionally distinct NMDA receptor populations in rat dorsal horn - effects of peripheral nerve lesion and inflammation. Neuroscience 112:861-868.
Kirkpatrick D.R., McEntire D.M., Smith T.A., Dueck N.P., Kerfeld M.J., Hambsch Z.J., Nelson T.J., Reisbig M.D., Agrawal D.K. (2016) Transmission pathways and mediators as the basis for clinical pharmacology of pain. Expert Rev Clin Pharmacol:1-25.
Laedermann C.J., Abriel H., Decosterd I. (2015) Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front Pharmacol 6:263.
Lai J., Porreca F., Hunter J.C., Gold M.S. (2004) Voltage-gated sodium channels and hyperalgesia. Annu Rev Pharmacol Toxicol 44:371-397.
Lawrence J. (2012) Nav1.7: a new channel for pain treatment. Pharmaceutical Journal.
Leung E.L., Cao Z.W., Jiang Z.H., Zhou H., Liu L. (2013) Network-based drug discovery by integrating systems biology and computational technologies. Brief Bioinform 14:491-505.
Li P., Huang C., Fu Y., Wang J., Wu Z., Ru J., Zheng C., Guo Z., Chen X., Zhou W., Zhang W, Li Y, Chen J, Lu A, Wang Y (2015) Large-scale exploration and analysis of drug combinations. Bioinformatics 31:2007-2016.
Liu Y., Hu B., Fu C., Chen X. (2010) DCDB: drug combination database. Bioinformatics 26:587-588.
Loewe SMH (1926) über Kombinations swirkungen. Arch für Exp Pathol 114:313-326.
Lozada C.J. and Diamond H.S. (2008) Osteoarthritis medication: Analgesics, other, nonsteroidal anti-inflammatory. Medscape.
Lunn M.P., Hughes R.A., Wiffen P.J. (2014) Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev:CD007115.
Marcum Z.A., Hanlon J.T. (2010) Recognizing the Risks of Chronic Nonsteroidal Anti-Inflammatory Drug Use in Older Adults. Ann Longterm Care 18:24-27.
Millan M.J. (2014) On 'polypharmacy' and multi-target agents, complementary strategies for improving the treatment of depression: a comparative appraisal. Int J Neuropsychopharmacol 17:1009-1037.
Minami K., Ogata J., Uezono Y. (2015) What is the main mechanism of tramadol? Naunyn Schmiedebergs Arch Pharmacol 388:999-1007.
Moore R.A., Wiffen P.J., Derry S., Toelle T., Rice A.S. (2014) Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev:CD007938.
Petrenko A.B., Yamakura T., Baba H., Shimoji K. (2003) The role of N-methyl-D-aspartate (NMDA) receptors in pain: a review. Anesth Analg 97:1108-1116.
Ramsay R.R., Popovic-Nikolic M.R., Nikolic K., Uliassi E., Bolognesi M.L. (2018) A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med 7:3.
Reuben D.B., Alvanzo A.A., Ashikaga T., Bogat G.A., Callahan C.M., Ruffing V., Steffens D.C. (2015) National Institutes of Health Pathways to Prevention Workshop: the role of opioids in the treatment of chronic pain. Ann Intern Med 162:295-300.
Rozanski G.M., Li Q., Stanley E.F. (2013) Transglial transmission at the dorsal root ganglion sandwich synapse: glial cell to postsynaptic neuron communication. Eur J Neurosci 37:1221-1228.
Schneider C., Yale, S.H. and Larson M. (2003) Principles of pain management. Clinical Medicine & Research 1:337-340
Schreiber A.K., Nones C.F., Reis R.C., Chichorro J.G., Cunha J.M. (2015) Diabetic neuropathic pain: Physiopathology and treatment. World J Diabetes 6:432-444.
Smith H.S., Smith E.J., Smith B.R. (2012) Duloxetine in the management of chronic musculoskeletal pain. Ther Clin Risk Manag 8:267-277.
Sofat N., Harrison A., Russell M.D., Ayis S., Kiely P.D., Baker E.H., Barrick T.R., Howe F.A. (2017) The effect of pregabalin or duloxetine on arthritis pain: a clinical and mechanistic study in people with hand osteoarthritis. J Pain Res 10:2437-2449.
Tabakoff B., Ren W., Vanderlinden L., Snell L.D., Matheson C.J., Wang Z.J., Levinson R., Smothers C.T., Woodward J.J., Honse Y., Lovinger D., Rush A.M., Sather W.A., Gustafson D.L., Hoffman P.L. (2016) A novel substituted aminoquinoline selectively targets voltage-sensitive sodium channel isoforms and NMDA receptor subtypes and alleviates chronic inflammatory and neuropathic pain. Eur J Pharmacol 784:1-14.
Talevi A. (2015) Multi-target pharmacology: possibilities and limitations of the "skeleton key approach" from a medicinal chemist perspective. Front Pharmacol 6:205.
Tallarida R.J. (2001) Drug synergism: its detection and applications. J Pharmacol Exp Ther 298:865-872.
Taneja A., Della Pasqua O., Danhof M. (2017) Challenges in translational drug research in neuropathic and inflammatory pain: the prerequisites for a new paradigm. Eur J Clin Pharmacol 73:1219-1236.
Theile J.W., Cummins T.R. (2011) Recent developments regarding voltage-gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front Pharmacol 2:54.
Volkow N.D., McLellan A.T. (2016) Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. N Engl J Med 374:1253-1263.
Wang W., Gu J., Li Y.Q., Tao Y.X. (2011) Are voltage-gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol Pain 7:16.
Wempe M.F., Lightner J.W., Miller B., Iwen T.J., Rice P.J., Wakui S., Anzai N., Jutabha P., Endou H. (2012) Potent human uric acid transporter 1 inhibitors: in vitro and in vivo metabolism and pharmacokinetic studies. Drug Des Devel Ther 6:323-339.
Wilson J.A., Garry E.M., Anderson H.A., Rosie R., Colvin L.A., Mitchell R., Fleetwood-Walker S.M. (2005) NMDA receptor antagonist treatment at the time of nerve injury prevents injury-induced changes in spinal NR1 and NR2B subunit expression and increases the sensitivity of residual pain behaviours to subsequently administered NMDA receptor antagonists. Pain 117:421-432.
Wood J.N., Boorman J.P., Okuse K., Baker M.D. (2004) Voltage-gated sodium channels and pain pathways. J Neurobiol 61:55-71.
Worley S.L. (2016) New Directions in the Treatment of Chronic Pain: National Pain Strategy Will Guide Prevention, Management, and Research. P T 41:107-114.
Yekkirala A.S., Roberson D.P., Bean B.P., Woolf C.J. (2017) Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov 16:810.
Zimmermann G.R., Lehar J., Keith C.T. (2007) Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today 12:34-42.
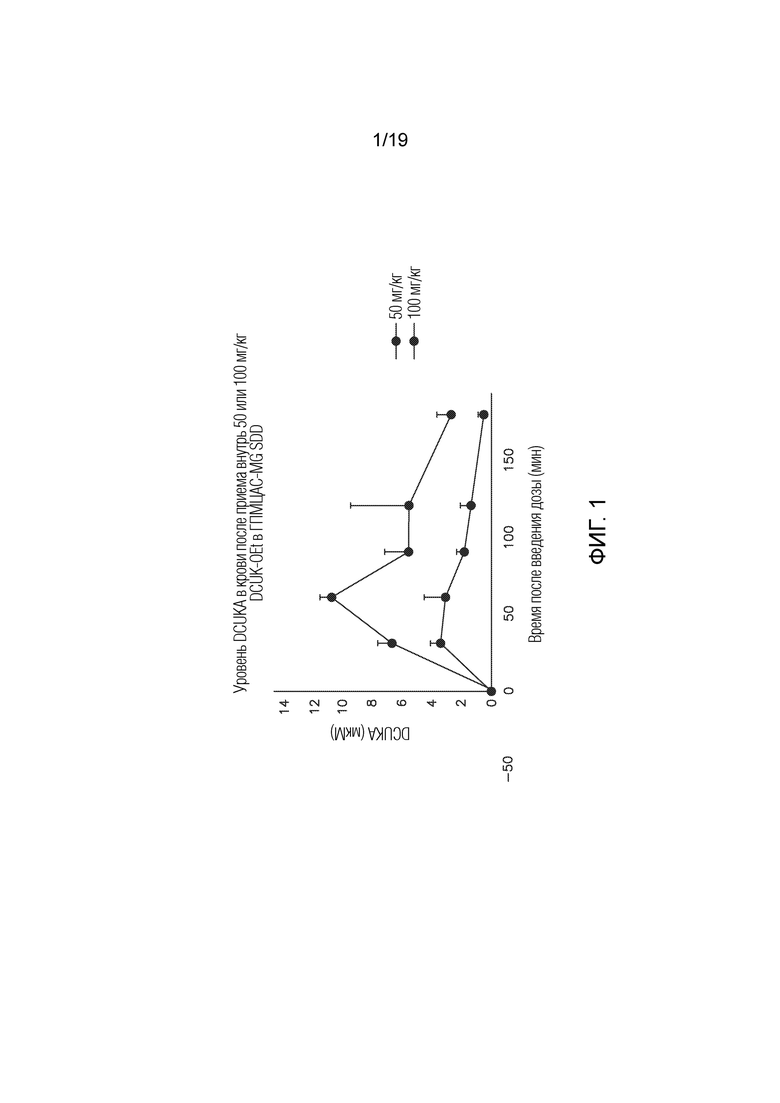
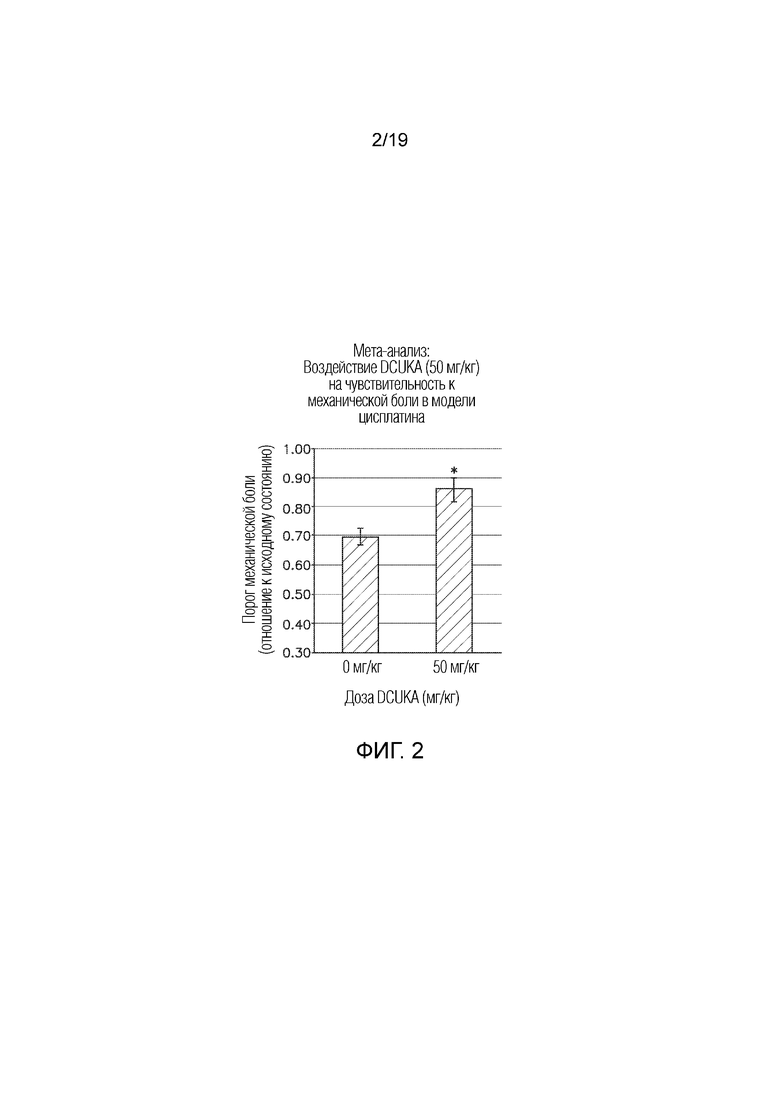
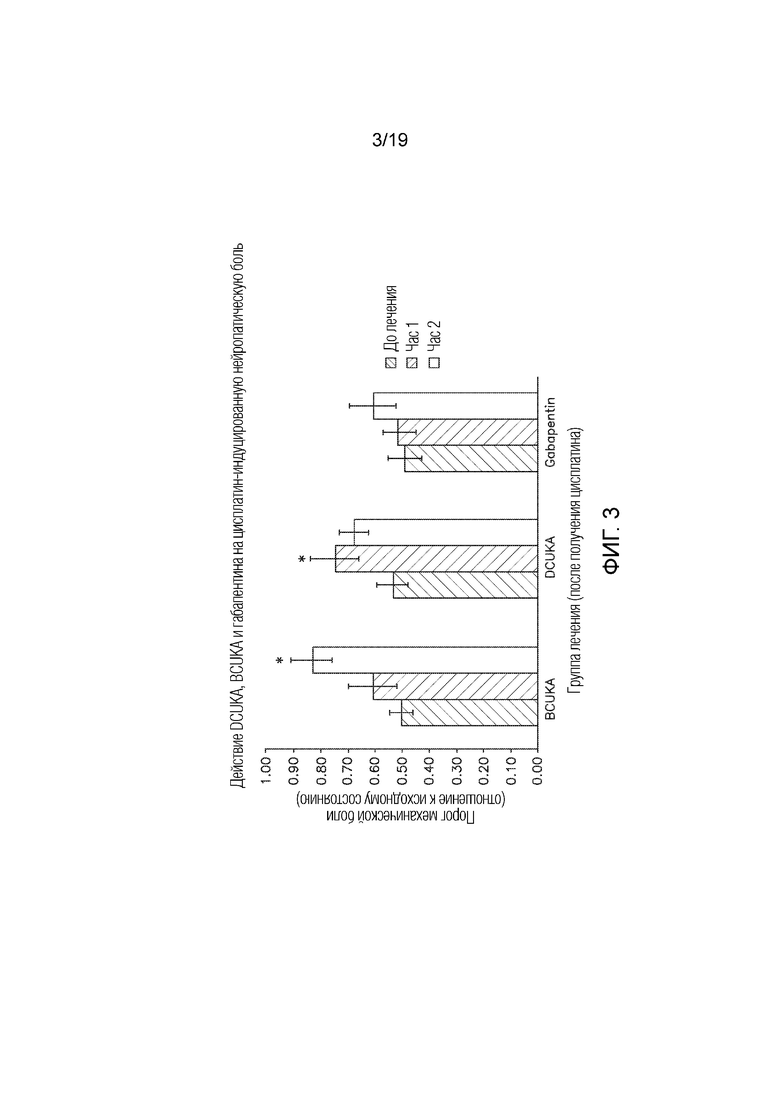
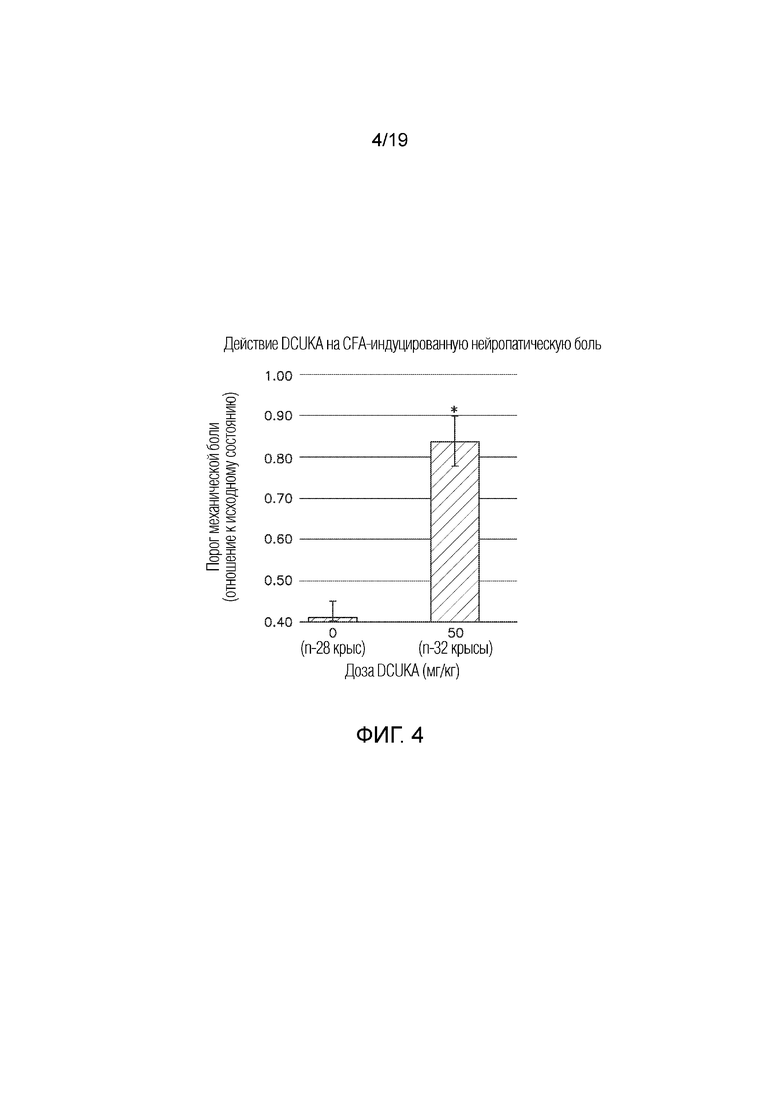
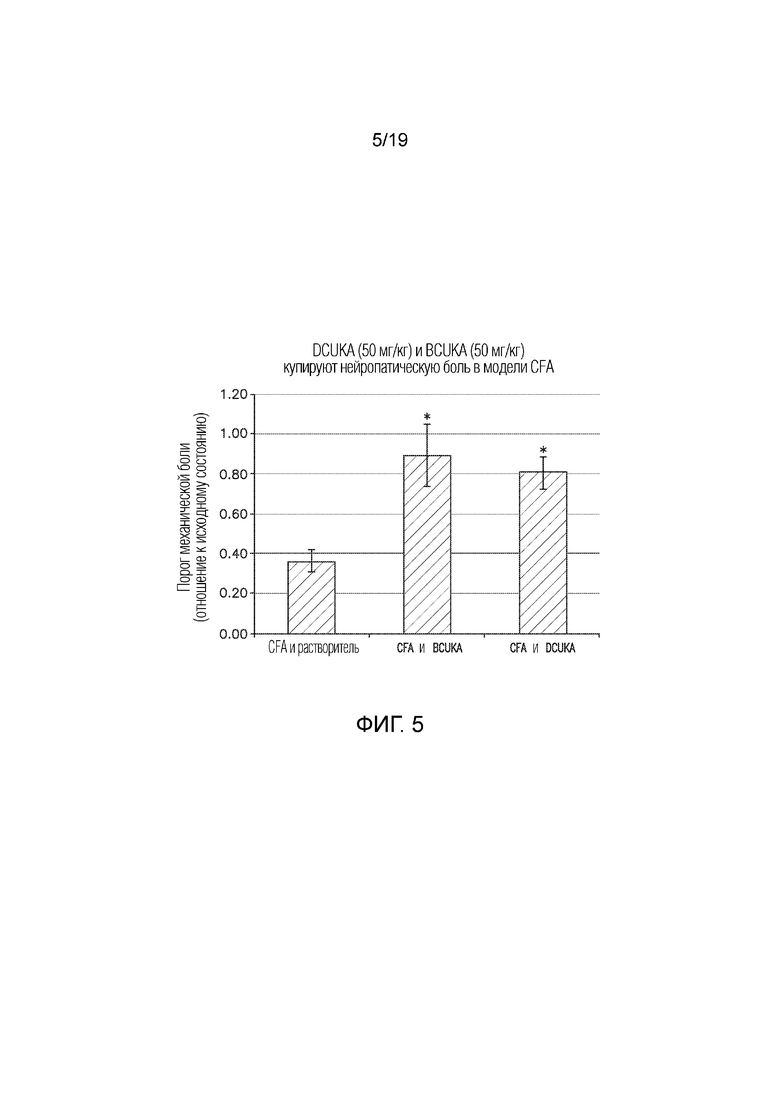

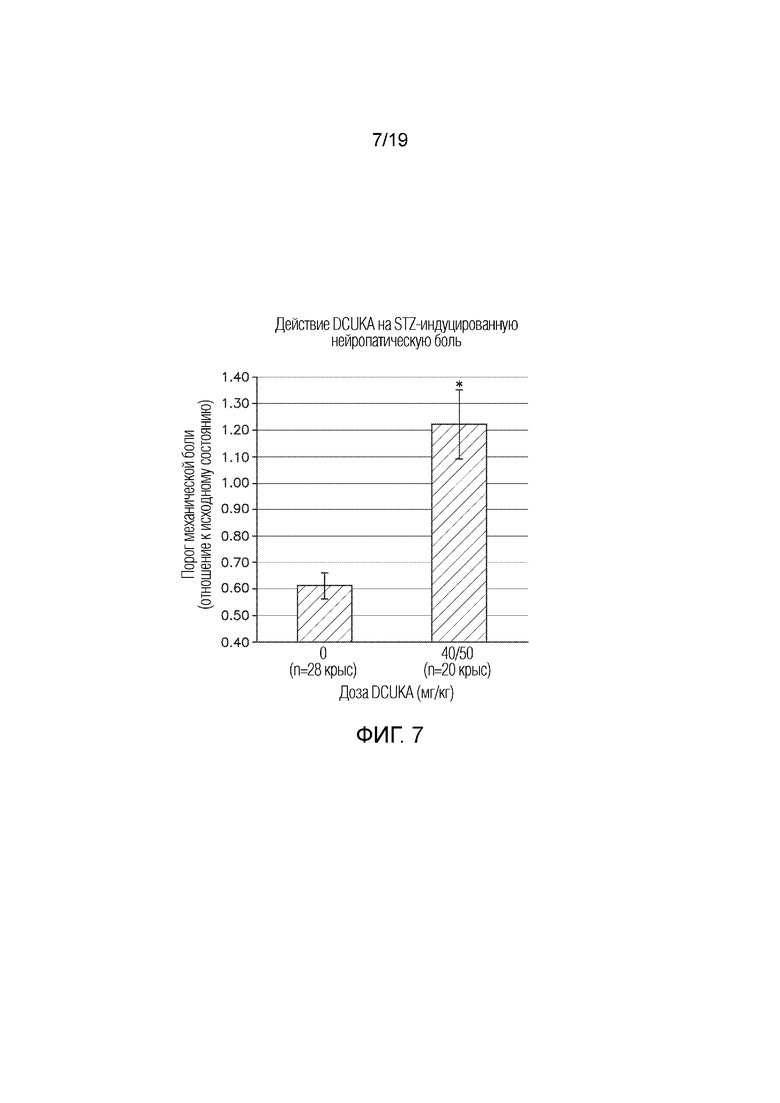
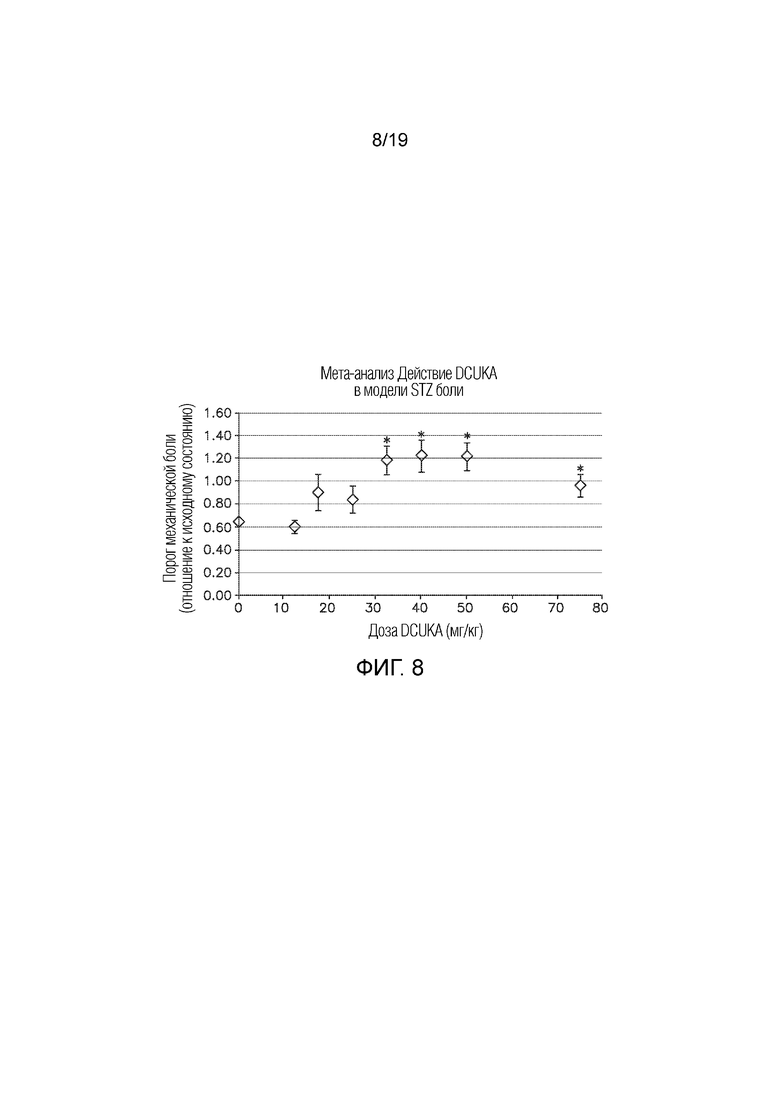
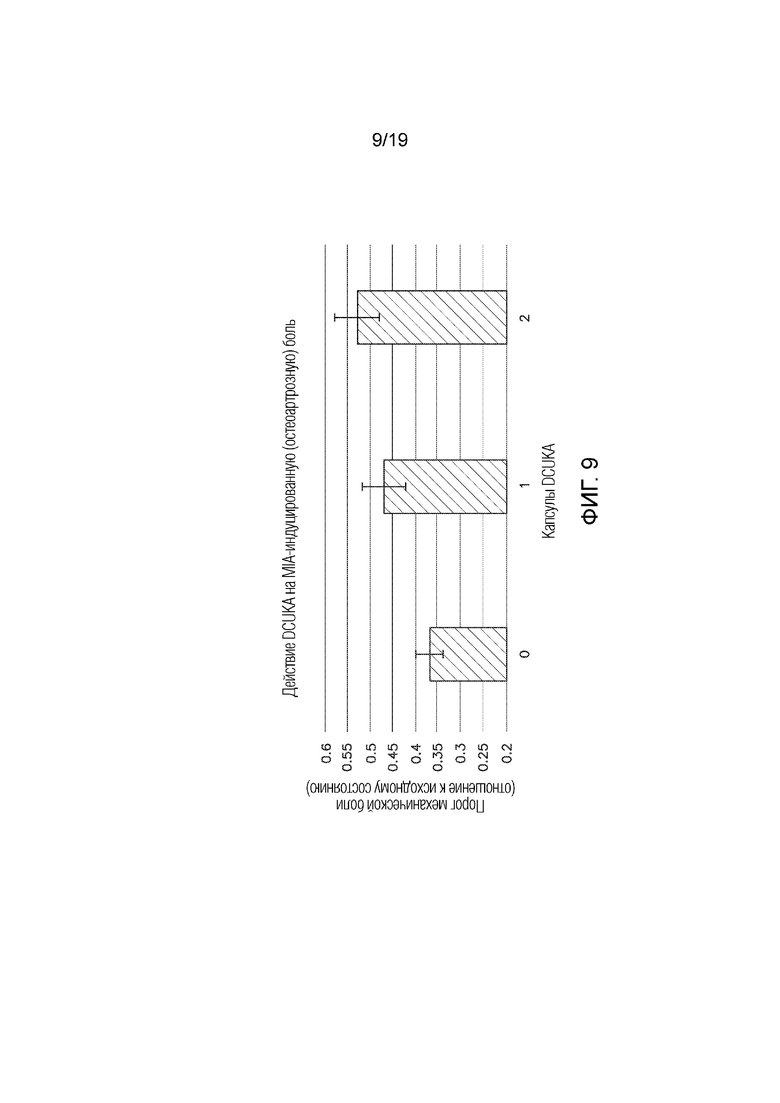
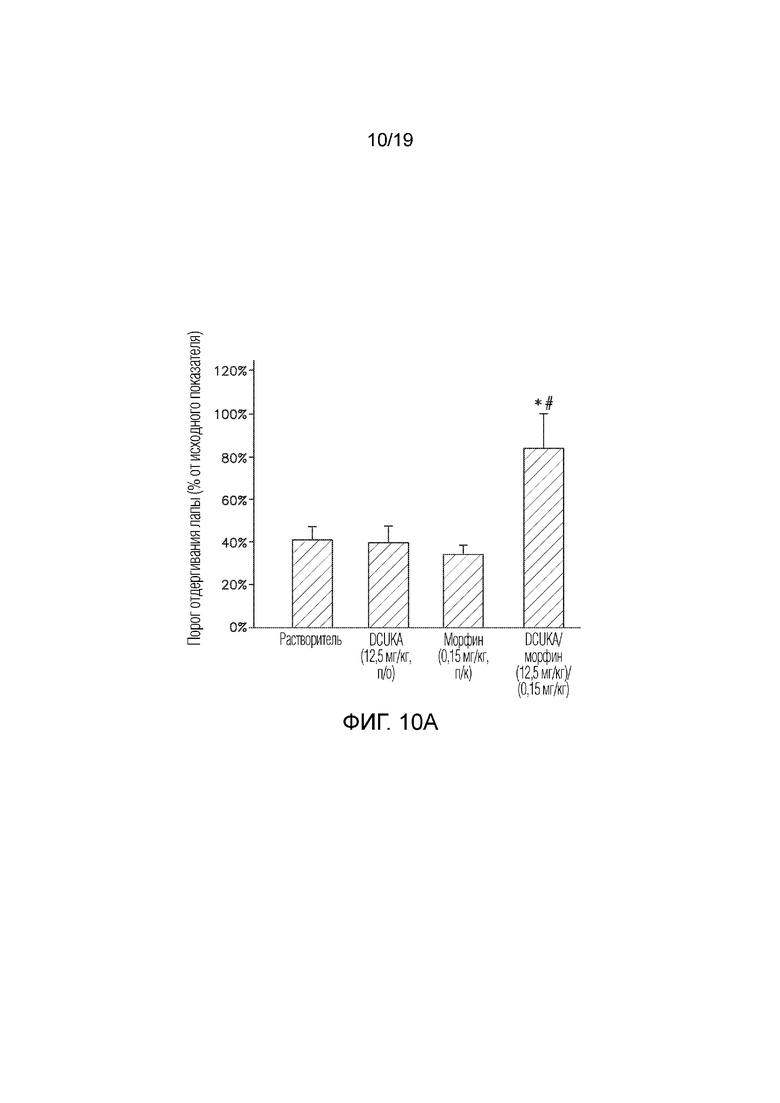
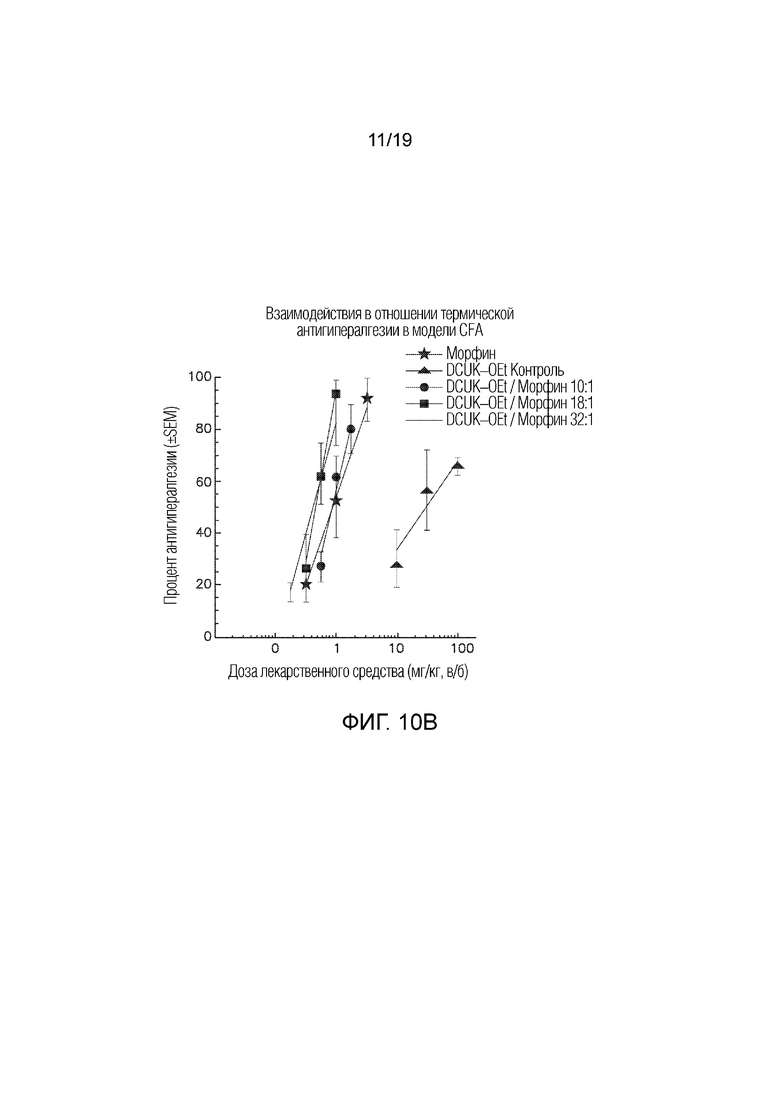
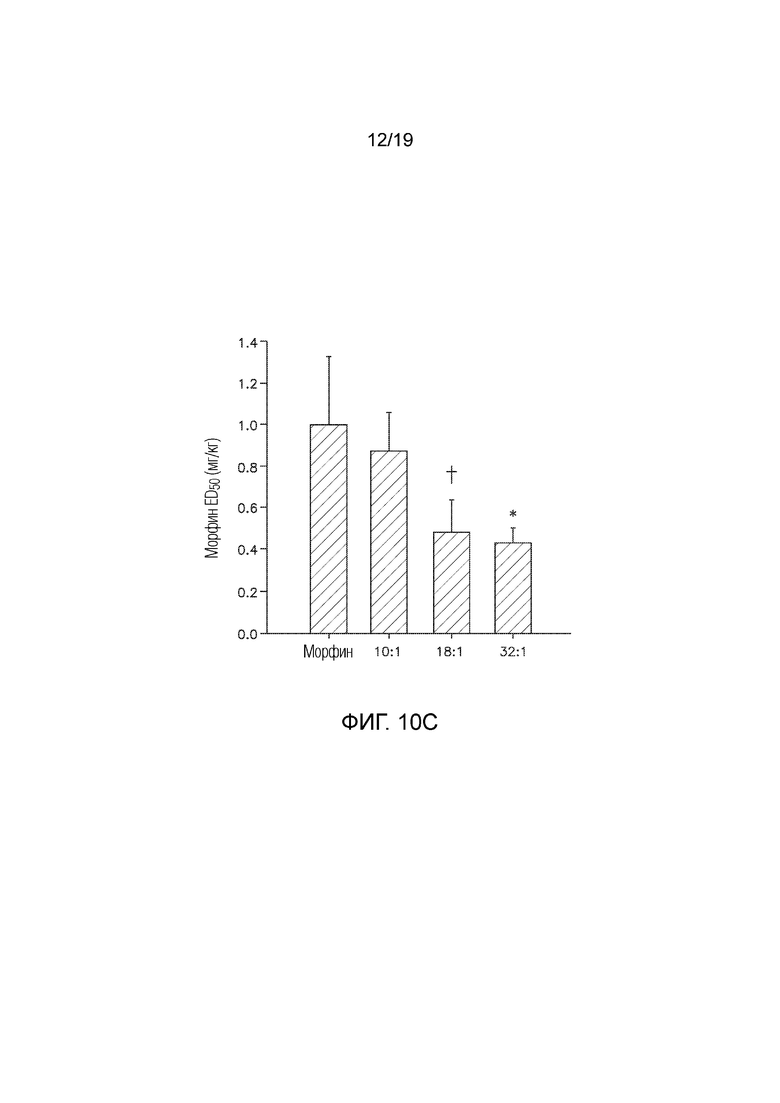
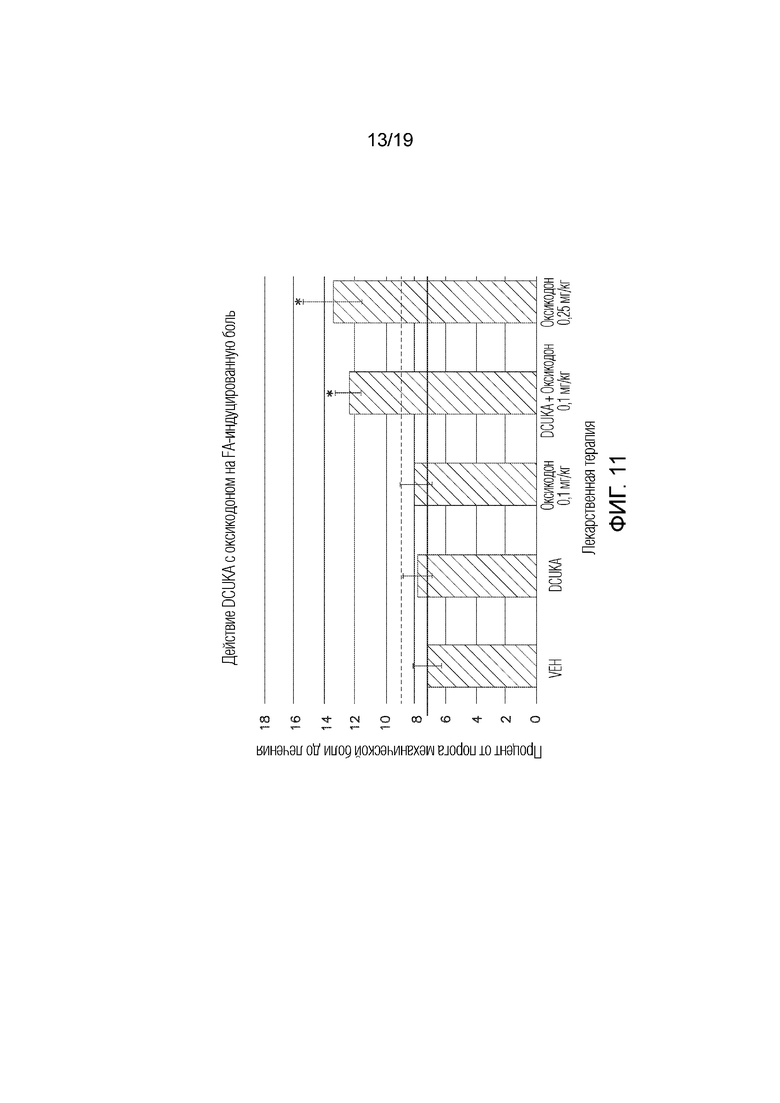
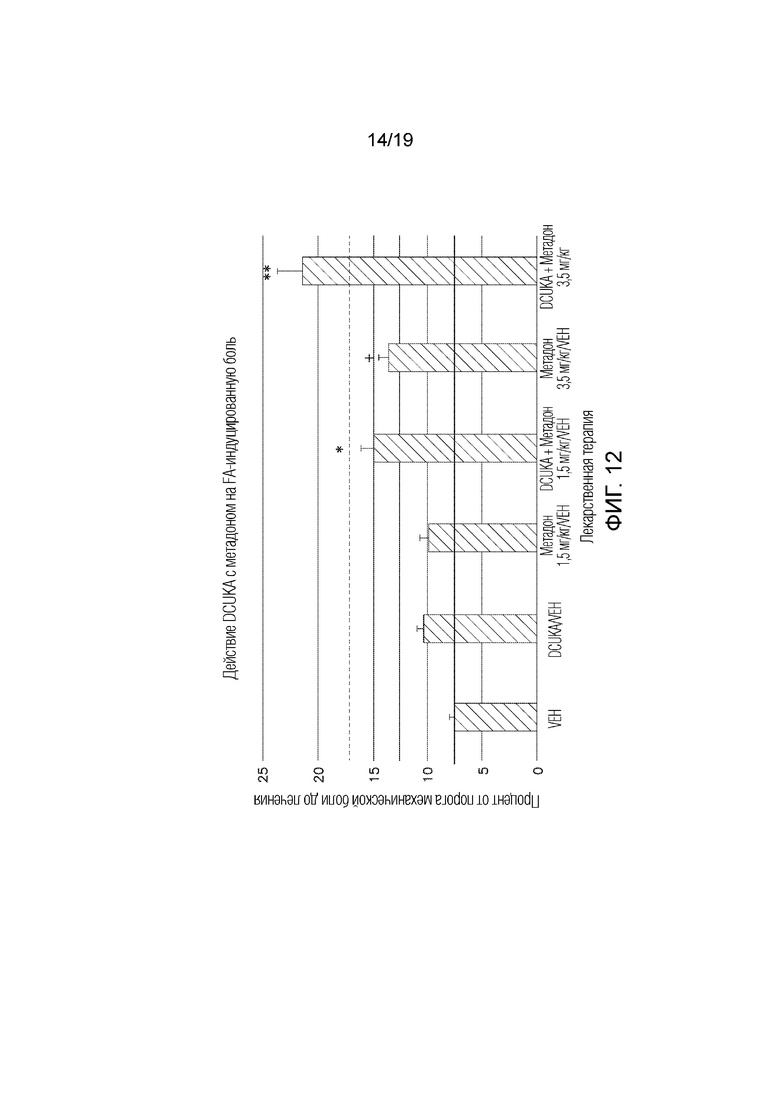
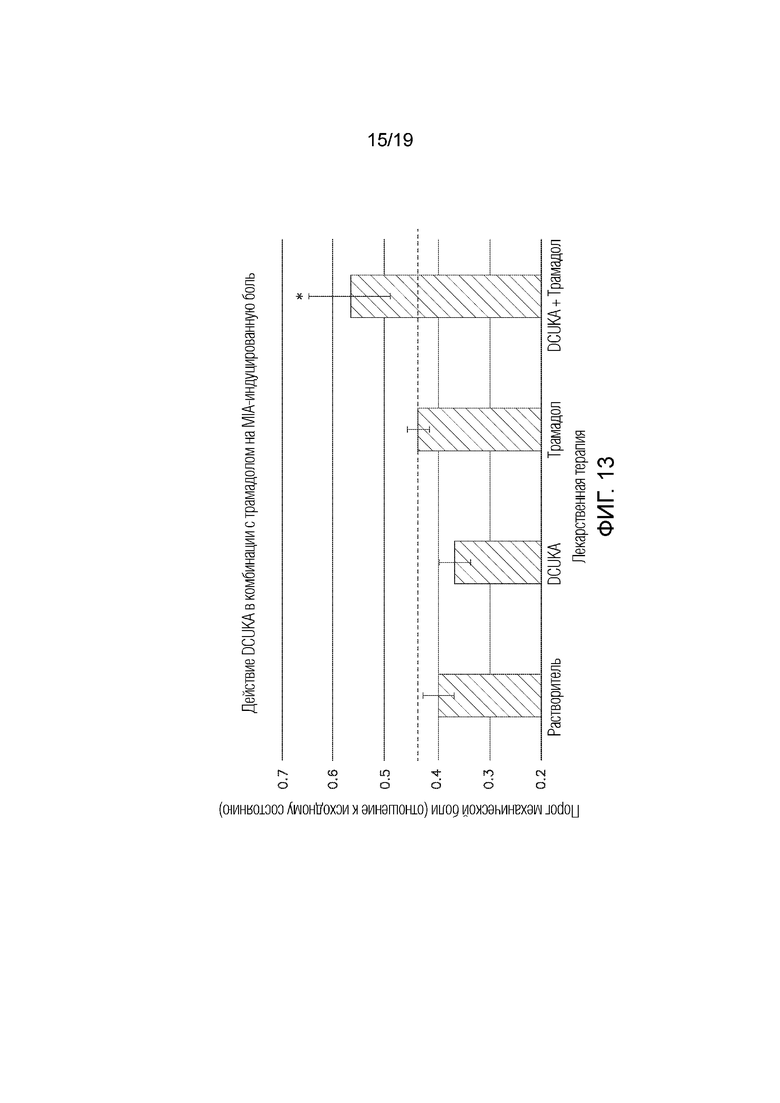
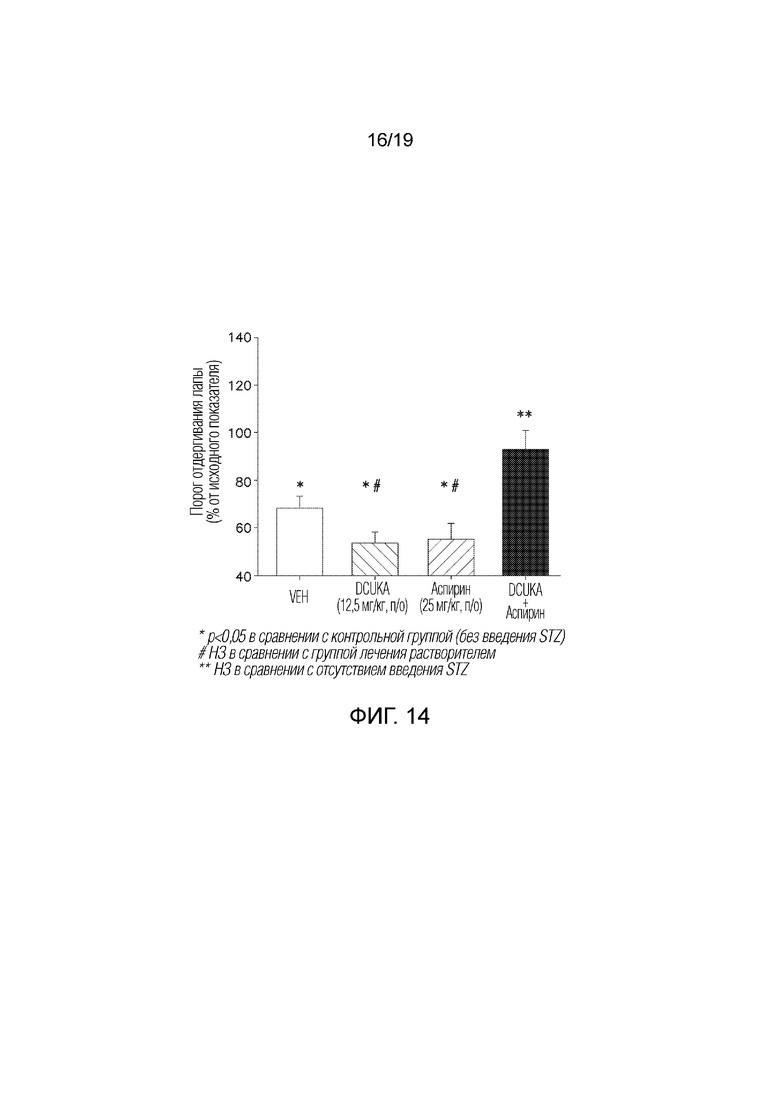
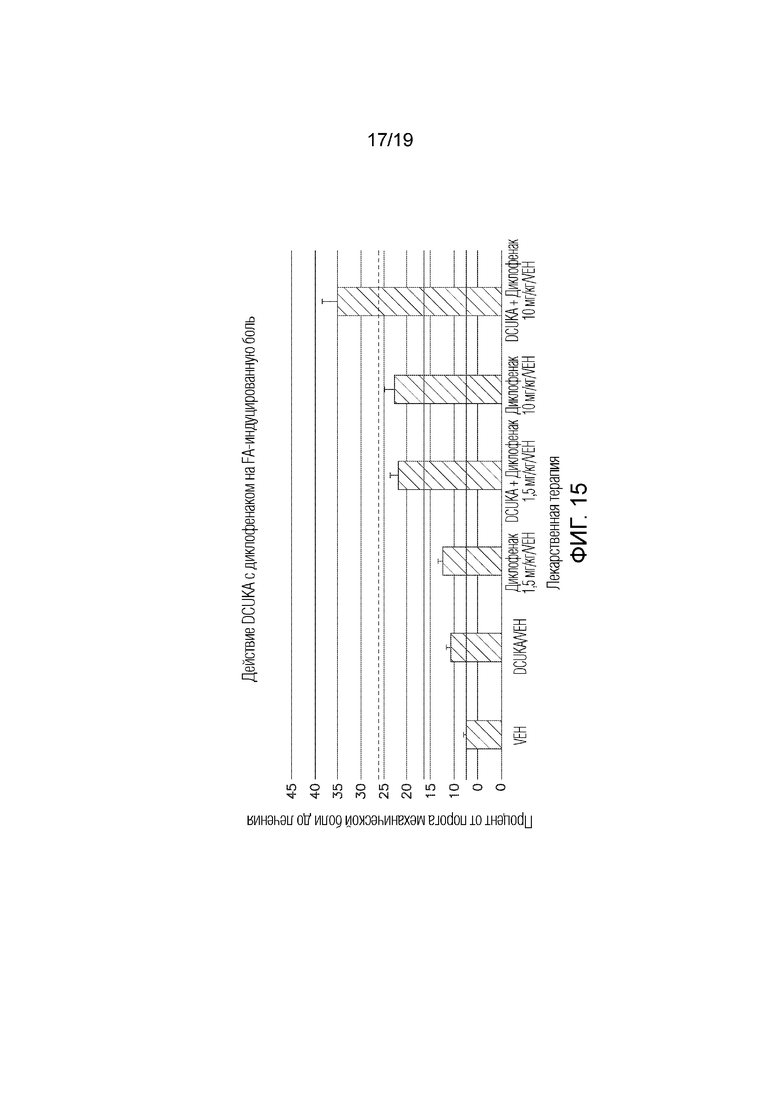
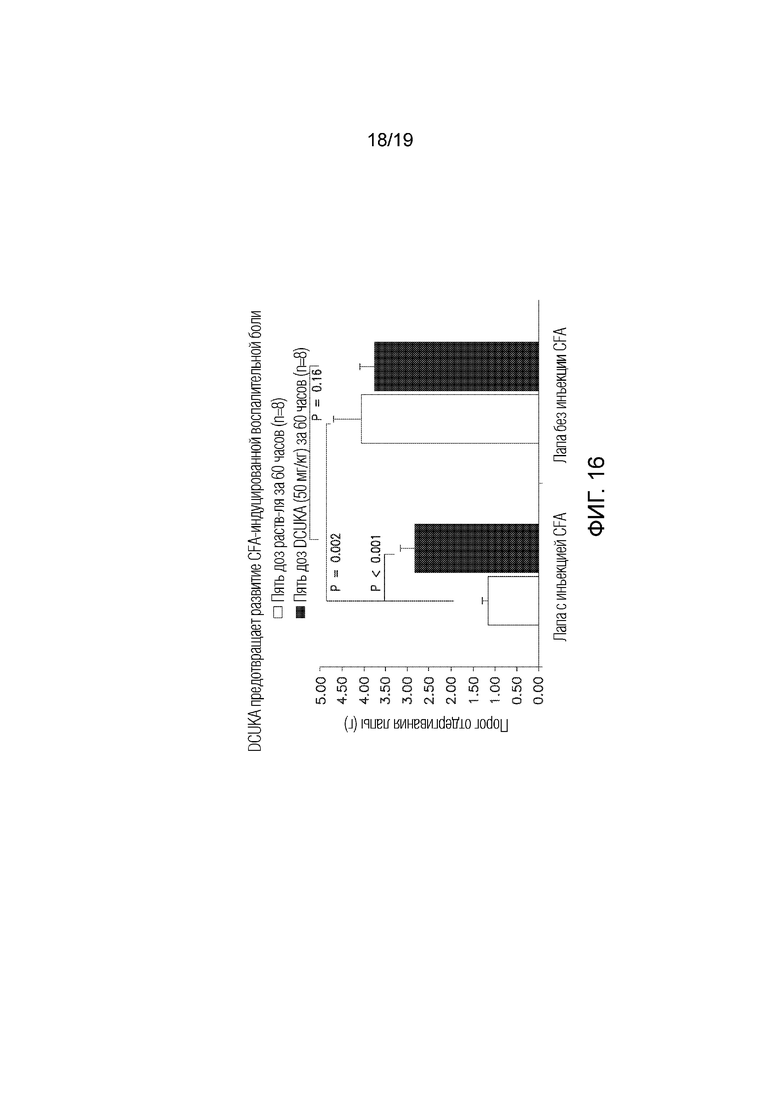
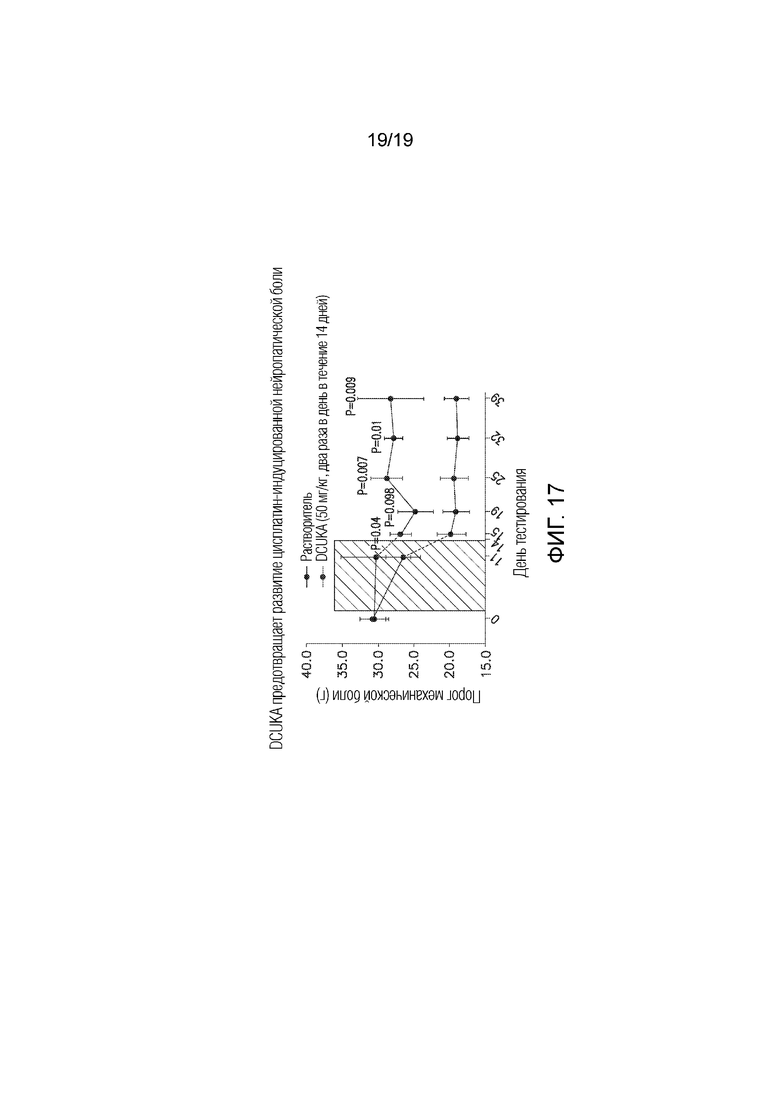
Группа изобретений относится к медицине и фармацевтике, а именно к фармацевтическим композициям для лечения хронической боли у субъекта и для потенцирования действия анальгезирующего соединения у пациента, страдающего хронической болью, включающим а) фармацевтически эффективное количество аминохинолинового соединения, которое представляет собой 5,7-дихлор-4-(3,3-дифенилуреидо)хинолон-2-карбоновую кислоту (DCUKA) в форме свободной кислоты, форме свободного основания или фармакологически приемлемой соли присоединения вместе с b) анальгезирующим средством, отличным от аминохинолинового соединения и выбранным из группы, состоящей из метадона, трамадола и их смесей. Использование группы изобретений позволяет эффективно лечить хроническую боль. 2 н.п. ф-лы, 17 ил., 9 пр.
1. Фармацевтическая композиция для лечения хронической боли у субъекта, включающая а) фармацевтически эффективное количество аминохинолинового соединения, которое представляет собой 5,7-дихлор-4-(3,3-дифенилуреидо)хинолон-2-карбоновую кислоту (DCUKA) в форме свободной кислоты, форме свободного основания или фармакологически приемлемой соли присоединения;
вместе с b) анальгезирующим средством, отличным от аминохинолинового соединения и выбранным из группы, состоящей из метадона, трамадола и их смесей.
2. Фармацевтическая композиция для потенцирования действия анальгезирующего соединения у пациента, страдающего хронической болью, включающая а) фармацевтически эффективное количество аминохинолинового соединения, которое представляет собой 5,7-дихлор-4-(3,3-дифенилуреидо)хинолон-2-карбоновую кислоту (DCUKA) в форме свободной кислоты, форме свободного основания или фармакологически приемлемой соли присоединения;
вместе с b) анальгезирующим средством, отличным от аминохинолинового соединения и выбранным из группы, состоящей из метадона, трамадола и их смесей.
| US 20170204064 A1, 20.07.2017 | |||
| US 20070087977 A1, 19.04.2007 | |||
| US 20170216212 A1, 03.08.2017 | |||
| Приспособление при токарном станке для обработки фрезером внутренней стороны ложек | 1929 |
|
SU21415A1 |
| Детекторный радиоприемник | 1929 |
|
SU13905A1 |

