Область техники, к которой относится изобретение
Изобретение относится к препарату, содержащему полимерную матрицу с пролонгированным высвобождением, который можно непосредственно прессовать в таблетки, и который содержит ниацин, вещество, замедляющее высвобождение, а также другие наполнители. Таблетки по изобретению демонстрируютулучшенные технологические характеристики, благоприятные характеристики высвобождения и снижение тяжести, продолжительности и количества случаев кожной гиперемии, обычно связанной с лечением ниацином.
Уровень техники
Известно, что ниацин (никотиновая кислота, называемая также 3-пиридинкарбоновой кислотой, химическая формула C6H5NO2) может с успехом использоваться для лечения гиперхолестеринемии, поскольку повышает содержание липопротеинов высокой плотности (HDL) и понижает общее содержание липопротеинов низкой плотности (LDL) и триглицеридов в холестерине сыворотки крови.
Хотя известно, что ниацин оказывает благоприятное влияние на липиды крови, за исключением препарата Ниаспан (NIASPAN®, Kos Pharmaceuticals, Inc., Cranbury, NJ), его повсеместное использование ограничено частыми случаями гиперемии, возникающими обычно при введении высоких доз ниацина, необходимых для эффективного лечения. Термин "гиперемия" широко используется для описания индуцированной ниацином вазодилятации. В результате у пациента после введения ниацина может развиться неприятное чувство жжения, а также иногда заметное покраснение. Хотя для предупреждения кожной гиперемии предлагались различные средства и/или препараты (см. US Pat. Nos. 4956252, 5023245 и 5126145), вышеуказанные нежелательные побочные эффекты по-прежнему остаются серьезной проблемой, ограничивающей широкое использование продуктов на основе ниацина.
Кроме того, качество веществ, замедляющих высвобождение (также часто называемых "агентами набухания") в коммерческих препаратах NIASPAN® неодинаково, в результате чего для достижения соответствия описанию спецификации продукта возникает необходимость в организации специального серийного производства.
Таким образом, в настоящее время в области фармацевтики существует потребность в препаратах с пролонгированным высвобождением на основе никотиновой кислоты, обеспечивающих более низкую частоту кожной гиперемии, по сравнению с существующими препаратами на основе ниацина, и легко получаемыми промышенными способами, а также характеризующимися улучшенными физическими, химическими и механическими свойствами.
Раскрытие изобретения
Настоящее изобретение относится к ниацин-содержащей фармацевтической композиции и ниацин-содержащему таблеточному препарату с пролонгированным (длительным) высвобождением (ПВ), которые включают ниацин и вещество, замедляющее высвобождение. В соответствии с одним аспектом, изобретение относится к таблеточному препарату типа ПВ (с пролонгированным высвобождением), содержащему 1000 мг ниацина и обладающему улучшенной текучестью, сжимаемостью, уплотняемостью и твердостью, по сравнению с известными лекарственными препаратами, содержащими 1000 мг ниацина. Помимо этого таблетки, содержащие 1000 мг ниацина с пролонгированным высвобождением (ПВ) по настоящему изобретению демонстрируют способность усиливать в два раза скорость высвобождения и/или поглощения коммерчески доступных 500 мг таблеток NIASPAN® без снижения технологической устойчивости (устойчивым процессом называется такой процесс, который позволяет воспроизводить целевую конечную точку при различных обстоятельствах или условиях, таких как небольшие изменения в исходных материалах или производственных процессах) или коммерческой желательности (например, размера). Поскольку полагают, что две 500 мг таблетки NIASPAN® характеризуются меньшей гиперемией, чем одна таблетка 1000 мг, одной из целей настоящего изобретения является создание таблеток с пролонгированным высвобождением, содержащих 1000 мг ниацина, биоэквивалентных двум 500 мг таблеткам NIASPAN®.
В частности, настоящее изобретение относится к ниацин-содержащей фармацевтической композиции, включающей:
(a) приблизительно от 70% до 92% ниацина по весу;
(b) приблизительно от 7% до 25% по весу вещества, замедляющего высвобождение;
(c) приблизительно от 0,1% до 4,3% по весу связывающего вещества, а также
(а) приблизительно от 0,5% до 1,5% по весу скользящего компонента.
В другом варианте осуществления, изобретение относится к ниацин-содержащей фармацевтической композиции, которая включает 1000 мг ниацина, что составляет приблизительно от 78% до 82% по весу от общего веса композиции; приблизительно от 14% до 18% по весу гидроксипропилметилцеллюлозы с метоксильной степенью замены около 1,39-1,41 и гидроксипропоксильной молярной заменой около 0,20-0,22; приблизительно от 2,5% до 3,0% по весу поливинилпирролидона, а также приблизительно от 0,95% до 1,05% по весу стеариновой кислоты.
В другом варианте осуществления, изобретение относится к ниацин-содержащей фармацевтической композиции, с уровнем гиперемии при введении больному, меньшем, чем с уровнем гиперемии от соответствующей дозы таблеток NIASPAN®, которая включает приблизительно от 70% до 92% по весу ниацина; приблизительно от 7% до 25% по весу вещества, замедляющего высвобождение; приблизительно от 0,1% до 4,3% по весу связывающего вещества, а также приблизительно от 0,5% до 1,5% по весу скользящего компонента.
В другом варианте осуществления, композиция по изобретению может быть выполнена в виде таблетки, содержащей 1000 мг ниацина с пролонгированным высвобождением.
В соответствии с другим вариантом осуществления, фармацевтическая таблетка по изобретению представляет собой таблетку, полученную методом прямой компрессии.
В другом варианте осуществления, изобретение относится к фармацевтической композиции, которая при дозировке больному один раз в день является эффективно снижающей содержание липидов в сыворотке и исключает явления гепатотоксичности и/или повышения содержания мочевой кислоты или глюкозы или того и другого, что может привести к необходимости прерывания в лечении.
В предпочтительном варианте, композиция по изобретению вводится пациенту один раз в день по вечерам или ночью.
В наиболее предпочтительном варианте, изобретение относится к фармацевтической композиции, в которой высвобождения ниацина замедлено.
В другом варианте осуществления, композиция по изобретению может дополнительно содержать ингибирующий гиперемию агент немедленного высвобождения.
В предпочтительном варианте осуществления, ингибирующий гиперемию агент может представлять собой простагландиновый D2 рецептор.
В наиболее предпочтительном варианте осуществления, простагландиновый D2 рецептор может представлять собой МК-0524.
В другом предпочтительном варианте осуществления ингибирующий гиперемию агент может представлять собой нестероидное противовоспалительное лекарственное средство (NSAID).
В другом варианте осуществления, вещество, замедляющее высвобождение, входящее в состав композиции, может быть выбрано из группы, включающей гидроксипропилцеллюлозу (НРС), гидроксипропилметилцеллюлозу (НРМС или гипромеллозу), метилцеллюлозу (МС), гидроксиэтилцеллюлозу (НЕС), поливинилпирролидон (PVP), ксантановую камедь и их смеси.
В предпочтительном варианте осуществления, вещество, замедляющее высвобождение может представлять собой гидроксипропилметилцеллюлозу.
В наиболее предпочтительном варианте осуществления, гидроксипропилметилцеллюлоза может иметь метоксильную степень замещения приблизительно от 1,2 до 2,0 и гидроксипропоксильное молярное замещение приблизительно от 0,1 до 0,3.
В еще более предпочтительном варианте осуществления, гидроксипропилметилцеллюлоза может иметь метоксильную степень замещения приблизительно от 1,4 до 1,9 и гидроксипропоксильное молярное замещение приблизительно от 0,19 до 0,24.
В другом еще более предпочтительном варианте осуществления, гидроксипропилметилцеллюлоза может иметь метоксильную степень замещения приблизительно 1,4 и гидроксипропоксильное молярное замещение приблизительно 0,21.
В другом предпочтительном варианте осуществления, гидроксипропилметилцеллюлоза может характеризоваться вязкостью приблизительно от 11000 до 22000 мПа·с.
В более предпочтительном варианте осуществления гидроксипропилметилцеллюлоза может характеризоваться вязкостью приблизительно от 13000 до 18000 мПа·с.
В другом варианте осуществления, фармацевтическая композиция по изобретению может включать связывающее вещество, которое может быть выбрано из группы, включающей поливинилпирролидон, гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу, этилцеллюлозу, полиметакрилат, воски и их смеси.
В предпочтительном варианте осуществления, связывающее вещество может представлять собой поливинилпирролидон.
В другом варианте осуществления, композиция по изобретению включает скользящий компонент, который может быть выбран из группы, включающей тальк, стеарат магния, стеарат кальция, стеариновую кислоту, гидрогенированные растительные масла и их смеси.
В предпочтительном варианте осуществления, скользящий компонент может представлять собой стеариновою кислоту.
В другом варианте осуществления, композиция по изоретению может дополнительно включать покрытие.
В предпочтительном варианте осуществления, покрытие композиции может представлять собой цветное покрытие, увеличивающее вес композиции приблизительно на 1,5-8,0%.
В более предпочтительном варианте, покрытие композиции может представлять собой цветное покрытие, увеличивающее вес композиции приблизительно на 1,75-5,0%.
В другом варианте осуществления, изобретение относится к фармацевтической композиции, которая может включать приблизительно от 76% до 88% по весу ниацина; приблизительно от 11,0% до 20,0% по весу вещества, замедляющего высвобождение; приблизительно от 0,2% до 3,25% по весу связывающего вещества, а также приблизительно от 0,75% до 1,25% по весу скользящего компонента.
В другом варианте осуществления, изобретение относится к фармацевтической композиции, которая может включать приблизительно, от 76% до 82% по весу ниацина; приблизительно, от 14% до 18% по весу вещества, замедляющего высвобождение; приблизительно, от 2,5% до 3,0% по весу связывающего вещества, а также приблизительно, от 0,85% до 1,05% по весу скользящего компонента.
В предпочтительном варианте, композиция может содержать приблизительно от 0,95 мас.% до 1,05 мас.% скользящего компонента.
В другом варианте осуществления, фармацевтическая композиция по изобретению может дополнительно включать антилипидемический агент.
В более предпочтительном варианте осуществления, антилипидемический агент в композиции может представлять собой ингибитор HMG-CoA редуктазы.
В другом варианте осуществления, композиция по по изобретению может дополнительно включать ингибирующий гиперемию агент.
В другом варианте осуществления, композиция по изобретению может дополнительно включать ингибирующий гиперемию агент.
В предпочтительном варианте, ингибирующий гиперемию агент может представлять собой нестероидное противовоспалительное лекарственное средство (NSAID).
В более предпочтительном варианте, ингибирующий гиперемию агент может представлять собой аспирин (ASA).
В другом более предпочтительном варианте, ингибирующий гиперемию агент может представлять собой антагонист D2 простамандиновых рецепторов.
В наиболее предпочтительном варианте, антагонист D2 простагландиновых рецепторов может представлять собой МК-0524.
В другом варианте осуществления, настоящее изобретение относится к ниацин-содержащей фармацевтической композиции с пролонгированным высвобождением, которая может включать ниацин в количестве 1000 мг и может обеспечивать in vivo при однократной дозе больному двух 1000 мг таблеток профиль в плазме с 90% доверительных интервалов натурального логарифма отношения в интервале от 80% до 125%, по меньшей мере для одного из параметров биодоступности, выбранных из нижеследующих:
максимальная наблюдавшаяся концентрация никотин-мочевой кислоты - 2601,8 нг/мл, общее восстановление ниацина в моче - 60,5%; максимальная наблюдавшаяся концентрация ниацина - 4958,9 нг/мл и площадь под фармакинетической кривой "концентрация/время" (показатель полноты всасывания - AUC) ниацина - 12414,5 нг/мл.
В предпочтительном варианте, вышеуказанный натуральный логарифм отношения может находиться в интервале от 90% до 115%.
В более предпочтительном варианте, натуральный логарифм отношения может находиться в интервале от 95% до 110%.
В другом предпочтительном варианте осуществления, изобретение относится к композиции, которая может дополнительно включать по меньшей мере один дополнительный терапевтический агент, который можно выбрать из группы, включающей ингибирующий гиперемию агент и антилипидемический агент.
В другом предпочтительном варианте осуществления, изобретение относится к фармацевтической композиции, которая при дозировке больному один раз в день является эффективно снижающей содержание липидов в сыворотке, и исключает явления гепатотоксичности и/или повышения содержания мочевой кислоты или глюкозы или того и другого, что приводит к необходимости прерывания в лечении.
Один вариант осуществления настоящего изобретения относится к ниацин-содержащей фармацевтической композиции с переработанным составом и пролонгированным высвобождением, включающей ниацина в количестве 1000 мг и обеспечивающей при введении субъектам в экспериментах по биоэквивалентности, в которых сравнивается однократная доза из четырех 500 мг таблеток NIASPAN® с однократной дозой указанных содержащих 1000 мг ниацина композиций замедленного высвобождения, 90% доверительных интервалов натурального логарифма отношения соответствующих параметров биодоступности в интервале от 80% до 125%.
В предпочтительном варианте осуществления, изобретение относится к композиции, в которой указанные параметры биодоступности представляют собой максимальную наблюдавшуюся концентрацию никотин-мочевой кислоты (нг/мл), общую восстанавливаемость ниацина в моче (%%), максимально наблюдавшуюся концентрацию ниацина (нг/мл) и площадь под фармакинетической кривой "концентрация/время" (показатель полноты всасывания - AUC) ниацина.
В другом варианте осуществления, изобретение относится к ниацин-содержащей фармацевтической композиции с пролонгированным высвобождением, которая включает ниацин в количестве 1000 мг и обеспечивает in vivo профиль в плазме при однократной дозе больному двух 1000 мг таблеток с 90% доверительных интервалов натурального логарифма отношения в интервале от 80% до 125%, по меньшей мере для одного из параметров биодоступности, выбранных из нижеследующих:
максимально наблюдавшаяся концентрация никотин-мочевой кислоты -приблизительно от 2111,0 нг/мл до 3253 нг/мл, общее восстановление ниацина в моче - приблизительно от 49,24% до 70,23%; максимально наблюдавшаяся концентрация ниацина - приблизительно от 3096 нг/мл до 6750 нг/мл и площадь подфармакинетической кривой "концентрация/время" (показатель полноты всасывания - AUC) ниацина - приблизительно от 6723 нг/мл до 18643 нг/мл.
В другом варианте осуществления, композиция при дозировке больному один раз в день является эффективно снижающей содержание липидов в сыворотке, исключающей явления гепатотоксичности и/или повышения содержания мочевой кислоты или глюкозы или того и другого, требующего перерыва в лечении.
В другом варианте осуществления, настоящее изобретение относится к ниацин-содержащему таблеточному препарату с пролонгированным высвобождением, который может быть получен методом прямой компрессии и включает 500 мг ниацина, что составляет приблизительно от 68% до 75% по весу от общего веса препарата; приблизительно от 24% до 29% по весу вещества, замедляющего высвобождение; приблизительно от 2,25% до 2,75% по весу связывающего вещества, а также приблизительно от 0,95% до 1,05% по весу скользящего компонента.
В другом варианте осуществления, изобретение относится к препарату, который может включать приблизительно от 68% до 75% по весу ниацина; приблизительно от 24% до 29% по весу вещества, замедляющего высвобождение; приблизительно от 2,25% до 2,75% по весу связывающего вещества, а также приблизительно от 0,95% до 1,05% по весу скользящего компонента.
В одном предпочтительном варианте, препарат по изобретению может дополнительно включать покрытие, обеспечивающее прибавку в весе препарата приблизительно на 1,5-8,0%.
В другом варианте осуществления, изобретение относится к ниацин-содержащему таблеточному препарату с пролонгированным высвобождением, который может быть получен методом прямой компрессии и включает 750 мг ниацина, что составляет приблизительно от 74% до 80% по весу от общего веса препарата; приблизительно от 16% до 22% по весу вещества, замедляющего высвобождение; приблизительно от 2,5% до 2,75% по весу связывающего вещества, а также приблизительно от 0,75% до 1,25% по весу скользящего компонента.
В более предпочтительном варианте осуществления, препарат по изобретению может включать приблизительно от 76% до 79% по весу ниацина; приблизительно от 18% до 21% по весу вещества, замедляющего высвобождение; приблизительно от 2,5% до 2,7% по весу связывающего вещества, а также приблизительно от 0,95% до 1,05% по весу скользящего компонента.
В одном варианте осуществления, препарат по изобретению может дополнительно включать покрытие, обеспечивающее прибавку в весе препарата приблизительно на 1,5-8,0%.
Фармацевтическая композиция по настоящему изобретению может также включать ингибирующий гиперемию компонент немедленного высвобождения и ниацин-содержащий компонент замедленного высвобождения, в котором ниацин высвобождается замедленно (то есть, по истечении времени задержки). В соответствии с предпочтительным аспектом, ниацин высвобождается, по меньшей мере, приблизительно, через 30 или 40 минут после высвобождения ингибирующего гиперемию агента.
Изобретение относится также к способу снижения у пациента гиперемии, связанной с лечением ниацином, причем указанный способ включает введение ниацин-содержащих таблеточных лекарственных форм с пролонгиированным высвобождением пациенту, нуждающемуся в терапии ниацином.
В соответствии с предпочтительным вариантом осуществления, ниацин-содержащую таблетку с пролонгированным высвобождением получают путем смешивания гранулированного ниацина.
В соответствии с настоящим изобретением, гиперемию можно еще сильнее снизить, путем введения ниацин-содержащего препарата с пролонгированным высвобождением по настоящему изобретению совместно с нестероидным противовоспалительным лекарственным средством (nonsteroidal anti-inflammatory drug (NSAID)). В соответствии с предпочтительным аспектом, NSAID представляет собой аспирин.
В соответствии с предпочтительным аспектом, ниацин-содержащий препарат по настоящему изобретению вводят один раз в день вечером или ночью.
В другом варианте осуществления, настоящее изобретение относится к способу снижения гиперемии, связанной с терапевтическим использованием ниацина, который включает введение пациенту один раз в день фармацевтической дозированной лекарственной формы, включающей приблизительно от 70 мас.% до 92 мас.% ниацина; приблизительно от 7 мас.% до 25 мас.% вещества, замедляющего высвобождение; приблизительно от 0,1 мас.% до 4,3 мас.% связывающего вещества, и приблизительно от 0,5 мас.% до 1,5 мас.% скользящего компонента.
В другом варианте осуществления, в вышеуказанном способе фармацевтическая дозированная лекарственная форма может включать две 1000 мг таблетки.
В другом варианте осуществления, в вышеуказанном способе по изобретению указанная фармацевтическая дозированная лекарственная форма может представлять собой 1000 мг таблетку, включающую приблизительно от 76 мас.% до 88 мас.% ниацина; приблизительно от 11,0 мас.% до 20,0 мас.% вещества, замедляющего высвобождение; приблизительно от 0,2 мас.% до 3,25 мас.% связывающего вещества, и приблизительно от 0,75 мас.% до 1,25 мас.% скользящего компонента.
В предпочтительном варианте осуществления, в вышеуказанном способе по изобретению указанная лекарственная форма может включать приблизительно от 78 мас.% до 82 мас.% ниацина; приблизительно от 14 мас.% до 18 мас.% вещества, замедляющего высвобождение; приблизительно от 2,5 мас.% до 3,0 мас.% связывающего вещества, и приблизительно от 0,85 мас.% до 1,05 мас.% скользящего компонента.
В другом варианте осуществления в вышеуказанном способе по изобретению лекарственная форма может включать приблизительно от 0,95 мас.% до 1,05 мас.% скользящего компонента.
В другом варианте осуществления, в вышеуказанном способе по изобретению указанное вещество, замедляющее высвобождение, может быть выбрано из группы, включающей гидроксипропилцеллюлозу (НРС), гидроксипропилметилцеллюлозу (НРМС или гипромеллозу), метилцеллюлозу (МС), гидроксиэтилцеллюлозу (НЕС), поливинилпирролидон (PVP), сополимеры метакрилата и триметиламмонийэтилметакрилата, ксантановую камедь и смеси указанных соединений.
В предпочтительном варианте осуществления, в вышеуказанном способе по изобретению вещество, замедляющее высвобождение, может представлять собой гидроксипропилметилцеллюлозу, которая может характеризоваться метоксильной степенью замещения приблизительно от 1,2 до 2,0, и гидроксипропоксильным молярным замещением приблизительно от 0,1 до 0,3.
В другом предпочтительном варианте осуществления, в вышеуказанном способе по изобретению гидроксипропилметилцеллюлоза может характеризоваться метоксильной степенью замещения приблизительно от 1,4 до 1,9, и гидроксипропоксильным молярным замещением приблизительно от 0,19 до 0,24.
В другом варианте осуществления, в вышеуказанном способе по изобретению гидроксипропилметилцеллюлоза может характеризоваться метоксильной степенью замещения приблизительно 1,4 и гидроксипропоксильным молярным замещением приблизительно 0,21.
В предпочтительном варианте осуществления, в вышеуказанном способе гидроксипропилметилцеллюлоза может характеризоваться вязкостью в интервале приблизительно от 11000 до 22000 мПа·с.
В другом предпочтительном варианте осуществления, гидроксипропилметилцеллюлоза может характеризоваться вязкостью приблизительно от 13000 до 18000 мПа·с.
В другом варианте осуществления, в вышеуказанном способе по изобретению фармацевтическая дозированная лекарственная форма может дополнительно содержать покрытие.
В предпочтительном варианте осуществления, вышеуказанное покрытие может представлять собой цветное покрытие, увеличивающее вес фармацевтической дозированной лекарственной формы приблизительно на 1,5-8,0%.
В предпочтительном варианте осуществления, вышеуказанное покрытие может представлять собой цветное покрытие, увеличивающее вес фармацевтической лекарственной формы приблизительно на 1,75-5,0%.
Также настоящее изобретение относится к способам изготовления ниациновых таблеток с пролонгированным высвобождением методом прямой компрессии, включающим следующие этапы:
(a) приготовление смеси, включающей приблизительно от 70% до 92% ниацина по весу, от 7% до 25% по весу вещества, замедляющего высвобождение, от 0,1% до 4,3% по весу связывающего вещества от 0,5% до 1,5% по весу скользящего компонента; а затем
(b) прессование полученной на этапе (а) смеси в таблетку.
В предпочтительном варианте осуществления, в вышеуказанном способе содержащая ниацин таблетка может представлять собой 1000 мг дозированный лекарственный препарат.
В другом предпочтительном варианте осуществления, в вышеуказанном способе на таблетку могут дополнительно наноситься покрытия.
В более предпочтительном варианте осуществления, на таблетку может дополнительно наноситься цветное покрытие, обеспечивающее прибавку в весе таблетки приблизительно на 1,5-8,0%.
В другом более предпочтительном варианте, в вышеуказанном способе по изобретению указанное цветное покрытие может обеспечивать прибавку в весе таблетки приблизительно на 1,75-5,0%.
В другом предпочтительном варианте осуществления, в вышеуказанном способе по изобретению, вещество, замедляющее высвобождение, может быть выбрано из группы, включающей гидроксипропилцеллюлозу (НРС), гидроксипропилметилцеллюлозу (НРМС или гипромеллозу), метилцеллюлозу (МС), гидроксиэтилцеллюлозу (НЕС), поливинилпирролидон (PVP), сополимеры метакрилата и триметиламмонийэтилметакрилата, ксантановую камедь и их смеси.
В другом варианте осуществления изобретения, указанное связывающее вещество может быть выбрано из группы, включающей поливинилпирролидон, гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу, этилцеллюлозу, полиметакрилат, воски и их смеси.
В другом предпочтительном варианте осуществления, в вышеуказанном способе скользящий компонент может быть выбран из группы, включающей тальк, стеарат магния, стеарат кальция, стеариновую кислоту, гидрогенированные растительные масла и их смеси.
В другом предочтительном варианте осуществления, в вышеуказанном способе по изобретению, таблетка может включать приблизительно от 76 мас.% до 88 мас.% ниацина; приблизительно от 11 мас.% до 20 мас.% вещества, замедляющего высвобождение; приблизительно от 0,2 мас.% до 3,25 мас.% связывающего вещества; и приблизительно от 0,75 мас.% до 1,25 мас.% скользящего компонента.
В более предпочтительном варианте, вышеуказанная таблетка может включать приблизительно от 78 мас.% до 82 мас.% ниацина;
приблизительно от 14 мас.% до 18 мас.% вещества, замедляющего высвобождение; приблизительно от 2,5 мас.% до 3,0 мас.% связывающего вещества; и приблизительно от 0,95 мас.% до 1,05 мас.% скользящего компонента.
В другом предпочтительном варианте, в вышеуказанном способе по изобретению, указанное вещество, замедляющее высвобождение может представлять собой гидроксипропилметилцеллюлозу, указанное связывающее вещество может быть представлено поливинилпирролидоном, указанный скользящий компонент - стеариновой кислотой, а гидроксипропилметилцеллюлоза может характеризоваться метоксильной степенью замещения приблизительно от 1,2 до 2,0 и гидроксипропоксильным молярным замещением приблизительно от 0,1 до 0,3.
В другом предпочтительном варианте осуществления, в способах, композициях и препаратах по настоящему изобретению, в качестве ниацина может применяться гранулированый ниацин.
В более предпочтительном варианте осуществления изобретения, размер частиц гранулированого ниацина соответствует не менее чем 85 мас.% для просеиваемой фракции 100-425 мкм и не более чем 10 мас.% для пыли размером менее 100 мкм.
Краткое описание чертежей
Фиг.1 представляет собой график, показывающий среднее растворение ниацина из содержащей 1000 мг ниацина таблетки с пролонгированным (замедленным) высвобождением, содержащей разные уровни METHOCEL® K-15M Premium.
Фиг.2 представляет собой график, показывающий влияние вязкости METHOCEL® K-15МР CR на растворение ниацина из содержащей 1000 мг ниацина таблетки с пролонгированным высвобождением (общий вес 1240 мг).
Фиг.3 представляет собой график, показывающий профили растворения ниацина из содержащей 1000 мг ниацина таблетки с пролонгированным высвобождением с использованием нерасфасованного лекарственного средства и сита PVP K-90 с ячейкой 40 меш.
Фиг.4 представляет собой график, показывающий профили растворения ниацина из содержащих 1000 мг ниацина таблеток с пролонгированным высвобождением (общий вес 1240 мг), полученных при различных этапах смешивания.
Фиг.5 представляет собой блок-схему, на которой показан процесс производства, представляющий собой прямую компрессию.
Фиг.6 представляет собой блок-схему клинического исследования, раскрытого в Примере 3.
Фиг.7 представляет собой гистограмму, показывающую частоту случаев гиперемии после введения двух покрытых пленкой содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест) и двух непокрытых 1000 мг таблеток NIASPAN® (Сравнение).
Фиг.8 представляет собой гистограмму, показывающую среднее значение интенсивности первого случая гиперемии после введения двух покрытых пленкой содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест) и двух непокрытых 1000 мг таблеток NIASPAN® (Сравнение).
Фиг.9 представляет собой гистограмму, показывающую среднее значение продолжительности первого случая гиперемии после введения двух покрытых пленкой содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест) и двух непокрытых 1000 мг таблеток NIASPAN® (Сравнение).
Фиг.10 представляет собой гистограмму, показывающую наличие отдельных симптомов гиперемии при первом случае гиперемии после введения двух покрытых пленкой содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест) и двух непокрытых 1000 мг таблеток NIASPAN® (Сравнение).
Фиг.11 представляет собой график, показывающий среднюю концентрацию ниацина в плазме после введения двух содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест или Измененный препарат) и двух 1000 мг таблеток NIASPAN® (Сравнение).
Фиг.12 представляет собой график, показывающий среднюю концентрацию NUA в плазме после введения двух содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест или Измененный препарат) и двух 1000 мг таблеток NIASPAN® (Сравнение).
Фиг.13 представляет собой гистограмму, показывающую среднее содержание в моче ниацина и его метаболитов (процент от дозы ниацина) через 96 часов после введения двух содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест или Измененный препарат) и двух 1000 мг таблеток NIASPAN® (Сравнение).
Фиг.14 представляет собой график, показывающий линейный профиль среднего содержания ниацина в плазме для трех тестовых ниацин-содержащих препаратов с пролонгированным высвобождением (ERN-1, ERN-2,ERN-3) и ниацин-содержащего препарата с пролонгированным высвобождением сравнения (NSP).
Фиг.15 представляет собой график, показывающий полулогарифмический профиль среднего содержания ниацина в плазме для трех тестовых препаратов и одного препарата сравнения.
Фиг.16 представляет собой график, показывающий линейный профиль среднего содержания NUA в плазме для трех тестовых ниацин-содержащих препаратов с пролонгированным высвобождением (ERN-1, ERN-2, ERN-3) и ниацин-содержащего препарата сравнения с пролонгированным высвобождением (NSP).
Фиг.17 представляет собой график, показывающий полулогарифмический профиль среднего содержания NUA в плазме для трех тестовых препаратов и одного препарата сравнения.
Фиг.18 представляет собой гистограмму, показывающую среднее содержание в моче ниацина и его метаболитов (процент от дозы ниацина) для трех тестовых ниацин-содержащих препаратов с пролонгированным высвобождением (ERN-1, ERN-2, ERN-3) и ниацин-содержащего препарата сравнения с пролонгированным высвобождением (NSP).
Фиг.19 представляет собой график, показывающий линейный профиль среднего содержания ниацина в плазме для двух покрытых пленкой содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест) и двух непокрытых содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Сравнение); фигура 20 представляет собой график, показывающий логарифмический профиль среднего содержания ниацина в плазме для тестовых препаратов и препаратов сравнения.
Фиг.21 представляет собой график, показывающий линейный профиль среднего содержания NUA в плазме для двух покрытых пленкой содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест) и двух непокрытых содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Сравнение); фиг.22 представляет собой график, показывающий логарифмический профиль среднего содержания NUA в плазме для тестовых препаратов и препаратов сравнения.
Фиг.23 представляет собой график, показывающий среднее содержание ниацина и его метаболитов в моче через 96 часов после введения двух покрытых пленкой содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Тест) и двух непокрытых содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению (Сравнение).
Фиг.24 представляет собой блок-схему дизайна эксперимента из Примера 6.
Фиг.25 представляет собой гистограмму, показывающую наличие отдельных симптомов гиперемии при первом случае гиперемии после введения двух содержащих 1000 мг ниацина препаратов с пролонгированным высвобождением по настоящему изобретению ("NIASPAN® CF"), если: (1) субъектам предварительно давали аспирин (ASA), (2) ASA вводили совместно с препаратами ниацина, и (3) давали только ниацин-содержащие препараты.
Фиг.26 представляет собой гистограмму, показывающую случаи гиперемии для Примеров 3 и 8.
Фиг.27 представляет собой гистограмму, иллюстрирующую интенсивность случаев гиперемии для Примеров 3 и 8.
Осуществление изобретения
Препараты для матричных таблеток с пролонгированным высвобождением по настоящему изобретению включают (1) ниацин в качестве активного ингредиента и (2) гидрофильную полимерную матрицу, обеспечивающую пролонгированное высвобождение активного ингредиента, то есть вещество, замедляюще высвобождение. В настоящей заявке термин "препарат с пролонгированным высвобождением" означает препарат, обеспечивающий эффективное лечение дислипидемии у пациента при введении препарата только раз в день.
Ниацин-содержащие препараты с пролонгированным высвобождением по настоящему изобретению могут приводить к улучшению липидного профиля у пациента. Например, введение пациенту ниацин-содержащего препарата с пролонгированным высвобождением по настоящему изобретению может понизить общее содержание холестерина, липопротеинов низкой плотности (LDL), триглицеридов и липопротеина A (Lp(a)), а также повысить содержание липопротеинов высокой плотности (HDL) в кровотоке пациента. Состояние, требующее снижения общего содержания холестерина, LDL, триглицеридов и/или липопротеина A (Lp(a)); и/или повышения HDL в кровотоке пациента, здесь будет называться "дислипидемией". Соответственно, настоящее изобретение охватывает лечение дислипидемий путем введения ниацин-содержащего препарата с пролонгированным высвобождением по настоящему изобретению пациенту, нуждающемуся в таком лечении.
Биоэквивалентность представляет собой отсутствие значимых различий в скорости и степени, с которыми активный ингредиент или активная группа в фармацевтических эквивалентах или фармацевтических альтернативах становится доступной в области действия лекарственного средства при введении в той же молярной дозе и в таких же условиях в правильно спроектированном клиническом исследовании. Как правило, чтобы сделать вывод о биоэквивалентности двух препаратов достаточно показать, что с 90% доверительным интервалом отношения натуральных логарифмов для Сmax и AUC или других соответствующих характеристик биоэквивалентности для Теста и Сравнения укладываются в интервал от 80% до 125%.
Препараты по настоящему изобретению считаются биоэквивалентными препаратам по изобретению, если 90% Cl для отношений натуральных логарифмов параметров биодоступности при лечении тестируемым / сравнительным препаратом укладываются в интервал от 80% до 125% (см., например, руководства Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products-General Considerations, U.S. Department of Health & Human Services, Food and Drug Administration, CDER, March 2003; Guidance for Industry Food-Effect Bioavailability and Fed Bioequivalence Studies, December 2002; содержание обеих публикаций включено сюда по ссылке). Как известно специалистам, такие препараты сравнивают с препаратами сравнения (такими, как описаны здесь или в описанных здесь вариантах осуществления изобретения) при одинаковых условиях (например, аналитические и технические условия) с использованием подходящих параметров биоэквивалентности, причем препарат сравнения используется в качестве контроля.
Ниацин
Ниацин, водорастворимое лекарственное средство, доступно коммерчески в виде мелких белых кристаллов, гранул или белого кристаллического порошка. Для получения фармацевтических композиций по настоящему изобретению можно использовать кристаллы, гранулы или порошок ниацина. В соответствии с предпочтительным вариантом осуществления, фармацевтические композиции получают из гранулированного ниацина, обладающего большей текучестью, по сравнению с порошковым ниацином. Текучесть является чрезвычайно важной характеристикой сжимаемости при изготовлении таблеток. Использование в настоящем изобретении гранулированого ниацина повышает текучесть и делает возможным осуществить прямое прессование ниацина в таблетки в промышленном масштабе. Для получения ниацин-содержащих таблеток по настоящему изобретению подходящим является любой размер частиц гранулированого ниацина. Предпочтительным размером частиц является не менее чем 85% (вес/вес) для просеиваемой фракции, так что размер гранул находится в интервале 100-425 мкм и не более чем 10%(вес/вес) для пыли с размером меньше 100 мкм. Текучесть ниацинового порошка можно повысить с помощью процесса сухого или влажного гранулирования.
Как правило, концентрация ниацина в таблетках по настоящему изобретению находится в интервале, приблизительно, от 70% до 95% по весу, предпочтительно, приблизительно, от 76% до 90% по весу, более предпочтительно, приблизительно, от 78% до 82% по весу. Ниацин в препарате по настоящему изобретению с пролонгированным высвобождением может присутствовать в количестве, приблизительно, от 100 до 3000 мг. В соответствии с некоторыми аспектами, в препарат по настоящему изобретению входит около 500 мг, около 750 мг или около 1000 мг ниацина. Предпочтительная суточная доза ниацина составляет около 1000 мг, около 1500 мг или около 2000 мг. Так, например, суточную дозу ниацина можно ввести пациенту, дав ему две 1000 мг таблетки раз в день.
Вещество, замедляющее высвобождение
Пролонгированное (замедленное) высвобождение из матричной системы, как правило, включает увлажнение полимера, гидратацию полимера, образование геля, набухание и растворение полимера. Что касается растворимых лекарственых средств, то они увлажняются, растворяются и диффундируют из гелевого слоя, образованного полимерной матрицей. Хотя механизмы высвобождения растворимых лекарственных средств из матричной таблетки зависят от различных факторов, общий принцип заключается в том, что присутствующий в таблетке водорастворимый полимер гидратируется на внешней поверхности таблетки и образует гелевый слой. По мере проникновения воды внутрь таблетки, толщина гелевого слоя увеличивается, и растворенное лекарственное средство диффундирует через него. Скорость высвобождения лекарственного средства из проглоченной таблетки на протяжении времени ее жизни определяется диффузией растворенного лекарства через гель и скоростью эрозии таблетки.
В качестве вещества, замедляющего высвобождение по настоящему изобретению может использоваться любое известное специалистам вещество, проявляюще благоприятные свойства набухания и гелеобразования. Примеры подходящих веществ, замедляющих высвобождение, включают, но этим не ограничиваются, гидроксипропил-целлюлозу (НРС), гидроксипропилметилцеллюлозу (обычно называемую НРМС или гипромеллозой), метилцеллюлозу (МС), гидроксиэтилцеллюлозу (НЕС) и поливинилпирролидон (PVP), ксантановую камедь, а также сополимеры метакрилата с триметиламмонийэтилметакрилатом (EUDRAGIT RS®), EUDRAGIT RL®)), а также смеси этих веществ, замедляющих высвобождение. В соответствии с одним вариантом осуществления изобретения, вещество, замедляющее высвобождение, представляет собой гидрофильный водорастворимый полимер. Предпочтительными гидрофильными полимерами являются гидроксипропилметилцеллюлоза средней вязкости и поливиниловый спирт средней вязкости.
Задерживающий высвобождение агент, как правило, представлен в таблетках настоящего изобретения в количестве, приблизительно, от 7,0% до 25,0% по весу (весовой процент по отношению к общему весу композиции), предпочтительно, приблизительно, от 11,0% до 20,0% по весу, более предпочтительно, приблизительно, от 14% до 18% по весу.
В соответствии с одним вариантом осуществления, веществом, замедляющим высвобождение, является гидроксипропилметилцеллюлоза. НРМС содержит полимерный каркас из целлюлозы, природного углевода, основной повторяющейся структурой которого является ангидроглюкоза. Растворимость (например, скорость гидратации) и толщина гелевого слоя, формируемого НРМС, определяется соотношением содержания двух химических заместителей, гидроксипропоксила (иногда называемого гидроксипропилом) и метоксила (иногда называемого метилом), связанных с целлюлозным каркасом НРМС (целлюлоза является природным углеводом, основной повторяющейся структурой которого является ангидроглюкоза). Гидроксипропоксильный заместитель относительно гидрофилен по своей природе и вносит значительный вклад в скорость гидратации, а метоксильный заместитель по своей природе относительно гидрофобен. Количество групп заместителей на ангидроглюкозном каркасе целлюлозы можно выразить через среднее количество групп заместителей, связанных с одним циклом ангидроглюкозы, эта концепция известна специалистам под названием "степень замещения". См. книги METHOCEL®) Cellulose Ethers Technical Handbook, Dow Chemical Company (Published Sept. 2002, Form No. 192-01062-0902 AMS); а также Using METHOCEL®) Cellulose Eithers for Controlled Release of Drugs in Hydrophilic Matrix Systems (Published July 2002, Form No. 198-02075-0702 AMS). В соответствии с одним вариантом осуществления изобретения, степень замещения метоксилом в веществе, замедляющем высвобождение НРМС колеблется, приблизительно, от 1,2 до 2,0, а степень замещения гидроксипропоксилом, приблизительно, от 0,1 до 0,3; предпочтительная метоксильная степень замещения колеблется, приблизительно, от 1,4 до 1,9, а гидроксипропоксильная молярная степень замещения - приблизительно, от 0,19 до 0,24, более предпочтительная метоксильная степень замещения колеблется, приблизительно, от 1,39 до 1,41, гидроксипропоксильная молярная степеньзамещения - приблизительно, от 0,2 до 0,22, более предпочтительно, если метоксильная степень замещения составляет около 1,4, а гидроксипропоксильная молярная степень замещения - около 0,21. Предпочтительным задерживающим высвобождение агентом является METHOCEL® K-15M (производства Dow Chemical Company, включая специфические суббренды, такие как K-15M premium и K-15M premium CR).
Кроме того, коммерчески доступны полимеры гидроксипропилметил-целлюлозы различных степеней вязкости. Например, вязкость METHOCEL® К может составлять 4000 и 15000 мПа·с (1 сантипуаз (cps)=1 мПа·с (миллипаскаль в секудну)). Это METHOCEL® K4M и METHOCEL® K15M производства Dow Chemical Co, USA; вязкость препарата Metalose 90 SH составляет 4000, 15000 и 39000 мПа·с, он доступен в компании Shin Etsu Ltd, Japan. В соответствии с вариантом осуществления настоящего изобретения, вязкость НРМС (определенная при концентрации 2% в воде при 20°С, например, ASTM D2363) составляет, приблизительно, от 11000 до 22000 мПа·с, предпочтительно, приблизительно, от 13000 до 18000 мПа·с.
Чтобы определить специфические характеристики, необходимые для замещения подходящих полимеров, отличающихся от НРМС, компетентный специалист может изменять степень замещения полимера (например, гидроксипропилцеллюлозы) и определить таким способом степень замещения, соответствующую профилю растворения препарата, в котором используется НРМС в соответствии с изобретением (например, препарата, соответствующего Примерам 1 или 2).
Наполнители
Таблетки по настоящему изобретению могут также содержать связывающее вещество. Это может быть любое традиционно используемое фармацевтически приемлемое связывающее вещество, такое как поливинилпирролидон (называемый также PVP, повидон, поливидон), гидроксипропилцеллюлоза, гидроксиэтилцеллюлоза, этилцеллюлоза, полиметакрилат, воски и подобные им. Могут применяться также смеси упомянутых выше связывающих веществ. В соответствии с вариантом осуществления изобретения, связывающее вещество составляет приблизительно от 0,1% до 4,3% по весу от общего веса таблетки, предпочтительно, приблизительно от 0,2% до 3,25% по весу, более предпочтительно, приблизительно от 2,5% до 3,0% по весу.
Помимо вышесказанного, таблетки по настоящему изобретению содержат скользящее вещество (смазку). Оно может быть гидрофобным или гидрофильным и включает скользящие компоненты, хорошо известные специалистам в соответствующей области, такие например как, но не ограничиваясь только ими, тальк, стеарат магния, стеарат кальция, стеариновая кислота, гидрированные растительные масла и подобные им. Предпочтительным скользящим компонентом является стеариновая кислота. Введение скользящего компонента в препарат снижает трение между стенкой матрицы и компонентами таблетки при ее прессовании, помогает движению порошка (потоку порошка, то есть, движению перемешанного препарата в приемное устройство и матрицу), а также позволяет избежать адгезии вещества таблетки на оборудование. В соответствии с одним вариантом осуществления, в препарат таблетки по изобретению входит приблизительно от 0,5% до 1,5% скользящего компонента по весу, предпочтительно, приблизительно от 0,75% до 1,25% по весу, более предпочтительно, приблизительно от 0,85% до 1,15% по весу, еще более предпочтительно, приблизительно от 0,95% to 1,05% по весу.
Покрытия
Таблеточные препараты по настоящему изобретению с пролонгированным высвобождением могут также содержать покрытия, известные в области фармацевтики твердых дозированных лекарственных форм. Покрытия обеспечивают цвет таблетки, улучшают ее визуальные характеристики, функционируют как барьер для влаги и запахов, защищают от разрушения под действием таких факторов окружающей среды, как солнечный свет, колебания температуры, а также маскируют вкус таблетки. Подобные покрытия, как хорошо известно специалистам, могут содержать полимер, пластификатор и/или пигмент-краситель. Примеры покрытий включают покрытия OPADRY®. Покрытия могут наноситься из раствора (например, водного) или суспензии любыми известными способами, такими как приспособление для нанесения покрытия в кипящем слое (например, покрытия Wurster) или приспособление для нанесения покрытия методом кристаллизации. В соответствии с одним вариантом осуществления изобретения, покрытие является окрашенным, конкретно, представляетсобой покрытие OPADRY®. В соответствии с другим аспектом, окрашенное покрытие наносится на таблетку в количестве, приблизительно, от 1,5 до 8,0% по весу, предпочтительно, приблизительно, от 1,75 до 5,0% по весу.
Эквивалентность 500 мг таблеткам NIASPAN®
Обзор проведенных клинических исследований показал, что две 1000 мг таблетки NIASPAN® (вес таблетки 1203,6 мг) не являются биоэквивалентом четырем 500 мг таблеткам NIASPAN®), и что из 1000 мг таблеток NIASPAN® ниацин выделяется быстрее, чем из 500 мг таблеток. Дополнительные исследования показали, что содержащие 1000 мг ниацина ER таблетки (вес таблетки 1419,0 мг), содержание компонентов в которой в два раза превышает соответствующее количество из 500 мг таблеток NIASPAN®, также не являются биоэквивалентными. В последнем случае, из содержащих 1000 мг ниацина таблеток высвобождается медленнее, чем из 500 мг таблеток NIASPAN® in vitro, и из содержащих 1000 мг ниацина таблеток ER абсорбируется медленнее, чем из продукта сравнения (500 мг) in vivo. В соответствии с данными еще одного исследования, содержащие 1000 мг ниацина ER таблетки весом 1300,0 мг и 1280,0 мг с переработанным составом также не являются биоэквивалентами 500 мг таблеткам NIASPAN® в связи с их более медленной скоростью высвобождения.
Для того, чтобы приготовить содержащие 1000 мг ниацина таблетки ER, биоэквивалентные двум 500 мг таблеткам NIASPAN®, авторы изобретения приготовили и протестировали in vitro несколько содержащих ниацин 1000 мг ниацина ER препаратов. Это позволило предсказать высвобождение in vivo и характеристики поглощения. Затем в тестовых таблетках ER, содержащих 1000 мг ниацина еще раз изменили препарат, используя полученную информацию о том, что растворение уменьшается с повышением содержания полимера (контролирующего высвобождение) в таблетке (по весу). Таким образом, в исследование вошли новые ингредиенты (такие как различные типы полимеров) и анализ альтернативных технологий производства (например, прямая компрессия или уплотнение валиком roller compaction)). В таблице 1 приведены различные тестовые формулы содержащих 1000 мг ниацина таблеток с различным весом.
ние
После первичной оценки нескольких параметров для дальнейшего исследования было выбрано четыре описанных ниже препарата. Различные препараты были проанализированы на основании профиля растворения, с использованием 500 мг NIASPAN® для сравнения и аппарата USP Type 3 в 250 мл жидкости, имитирующей желудочный сок, при рН 1,2, 37°С, в течение 60 минут, после чего эксперимент продолжали в 250 мл жидкости, имитирующей желудочный сок, при рН 6,8, 37°С для всех временных точек.
(i) METHOCEL® Е10М, полученный методом влажного гранулирования (WG)
Гранулированный ниацин, METHOCEL® E10М, повидон K90 и стеариновую кислоту взвесили в соответствии с формулами для препаратов весом 1240 мг, 1260 мг, 1280 мг и 1300 мг, а затем гранулировали в высокоскоростном грануляторе и использованием деионизированной воды в качестве раствора гранулирования. Влажные гранулы высушили, измельчили, после чего смешали с экстрагранулированым METHOCEL® E10M и стеариновой кислотой.
Готовую хорошо перемешанную смесь спрессовали в таблетки с помощью пресса BWI Manesty Beta Press (Thomas Eng, Hoffman Estate, IL) на скорости 500 таблеток в минуту, твердость получаемых таблеток колебалась от 16 до 18 Кр (килофунтов).
(ii) METHOCEL® E10M. полученный методом прямой компрессии (DC)
Гранулированный ниацин, METHOCEL® E10M, повидон K90 и стеариновую кислоту взвесили в соответствии с формулами, представленными в Таблице 1, после чего поместили в смеситель (блендер), рассчитанный на в 8 раз большее количество смеси (LB-9322, Petterson Kelly, East Stroudsburg, PA) и перемешивали 10 минут. Тщательно перемешанную смесь спрессовали в таблетки с помощью пресса BWI Manesty Beta Press (Thomas Eng, Hoffman Estate, IL) на скорости 500 таблеток в минуту, твердость получаемых таблеток колебалась от 16 до 18 Кр.
(iii) METHOCEL® K15M. полученный методом WG
Ниацин USP, METHOCEL® K15M и повидон K90 взвесили в соответствии с формулами, представленными в Таблице 1, а затем гранулировали в высокоскоростном грануляторе и использованием деионизированной воды в качестве раствора гранулирования. Влажные гранулы высушили, измельчили, после чего смешали с экстрагранулированым METHOCEL® E15M и стеариновой кислотой. Готовую хорошо перемешанную смесь спрессовали в таблетки с помощью пресса BWI Manesty Beta Press (Thomas Eng, Hoffman Estate, IL) на скорости 500 таблеток в минуту, твердость получаемых таблеток колебалась от 16 до 18 Кр.
(iv) METHOCEL® K15M. полученный методом DC
Гранулированный ниацин, METHOCEL® K15M, повидон K90 и стеариновую кислоту взвесили в соответствии с формулами, представленными в Таблице 1, после чего поместили в смеситель (блендер), рассчитанный на в 8 раз большее количество смеси (LB-9322, Petterson Kelly, East Stroudsburg, PA) и перемешивали 10 минут.Тщательно перемешанную смесь спрессовали в таблетки с помощью пресса BWI Manesty Beta Press (Thomas Eng, Hoffman Estate, IL) на скорости 500 таблеток в минуту, твердость получаемых таблеток колебалась от 16 до 18 Кр.
Анализ включал изменения параметров процесса машинной обработки; вариацию в содержании полимера (см. Таблицу 1); взаимообмен методов влажного гранулирования, прямой компрессии и роллерной компрессии; вариации содержания PVP; изменения твердости таблеток; вариации веса (±5%); воспроизводимость; вариации в скорости таблетирования и стабильности таблеток(скорость высвобождения послехранения, поглощение влаги и т.д.). Профиль высвобождения целевого вещества был достигнут для следующих трех препаратов: (i) METHOCEL® Е-10М методом влажного гранулирования; (iii) METHOCEL® K-15M методом влажного гранулирования, и (iv) METHOCEL® K15M методом прямой компрессии. Кроме того, эти три препарата продемонстрировали приемлемые результаты стабильности в трехмесячном исследовании на стабильность.
Таблетки прямой компрессии с использованием METHOCEL® K-15M были выбраны в качестве предпочтительного аспекта для дальнейшего анализа по экономическим соображениям и из-за более высокой стабильности, что удалось определить в ходе анализа, описанного выше. Соответственно, по отношению к содержащим 1000 мг ниацина таблеткам DC с переработанным составом, была проведена дополнительная оценка с целью определить влияние размера гранулирования; распределения размера частиц каждого компонента, общей плотности и плотности утряски каждого компонента; различных партий каждого компонента; однородности содержимого; срезом Хаузера и Кара (Hauser and Carr indices); текучести; уплотняемости и хрупкости. В Таблице 2 перечислены специфические первичные материалы, используемые в различных экспериментальных препаратах. DMF означает Drug Master File (основной файл для лекарства).
ный ниацин, USP
Таблица 3 иллюстрирует тестовые 1000 мг DC таблетки с переработанным составом, содержащие различные количества наполнителей и обладающие связанными с ними физическими свойствами. Эти препараты были приготовлены, как описано выше, причем весовой процент каждого компонента приведен в Таблице 3.
Фиг.1 представляется пример сравнения профилей растворимости для препаратов, проиллюстрированных в Таблице 3.
Таблица 4 иллюстрирует данные по растворимости и биодоступности нескольких экспериментальных препаратов 1000 мг ниацина по сравнению с 500 мг Niaspan® в клинических исследованиях. Для расчета растворения использовали аппарат USP Apparatus 1 с 900 мл деионизированной воды при 100 об/мин (метод "корзины", basket method) при 37°С.
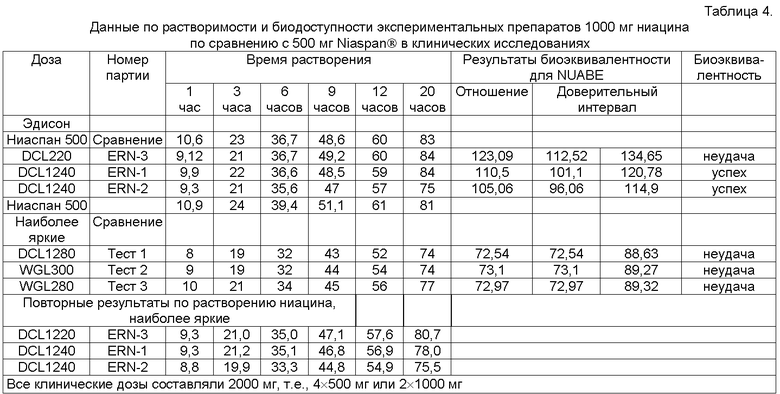
При исследовании воспроизводимости 1000 мг ниациновых ER таблеток с переработанным составом, полученных прямой компрессией, использовались следующие параметры:
Параметры препарата:
Вязкость и содержание гидроксипропоксила в METHOCEL® K-15МР CR
Размер частиц METHOCEL® K-15M
Размер частиц гранулированого ниацина
Содержание стеариновой кислоты
Просеивание PVP K-90
Параметры обработки
Последовательность и время перемешивания
Твердость таблетки
Скорость таблетирования
В следующей ниже Таблице 5 и на Фиг.2-4 проиллюстрированы данные, полученные в ходе исследований воспроизводимости, описанных выше:
По завершении анализа описанных выше переменных авторы не нашли значимых различий в растворении ниацина из таблеток, при изготовлении которых менялись значения следующих параметров: вязкость и содержание гидроксипропоксила в METHOCEL® K-15 premium CR, размеры частиц гранулированого ниацина, просеивание PVP K-90 через экраны с ячейками 40 mesh, содержание стеариновой кислоты в диапазоне от 0,5% до 2,0%, этапы смешивания, а также время перемешивания. Больший размер частиц METHOCEL® K-15M premium (CR) и твердость таблеток (в особенности, меньше 8 Kp) увеличивали растворение ниацина. Меньший размер частиц гранулированого ниацина и METHOCEL® K-15M premium CR показывали более высокую сжимаемость. Усилие выталкивания значительно уменьшалось при повышении в препарате количества стеариновой кислоты. Большая твердость таблеток достигалась при большем усилии компрессии и усилии выталкивания; для получения целевых таблеток твердостью 18 Кр при повышении скорости таблетирования также требовалось большее усилие компрессии.
В соответствии с вышеизложенным, настоящее изобретение охватывает содержащие 1000 мг ниацина таблетки с пролонгированным высвобождением (ER), полученные методом влажного гранулирования или прямой компрессии и включающие:
(a) приблизительно от 70% до 92% ниацина по весу;
(b) приблизительно от 7% до 25% по весу вещества, замедляющего высвобождение, метоксильная степень замещения которого составляет, приблизительно, от 1,2 до 2,0 и гидроксипропоксильное молярное замещение которого составляет приблизительно от 0,1 до 0,3;
(c) приблизительно от 0,1% до 4,3% по весу связывающего вещества, и
(d) приблизительно от 0,5% до 1,5% по весу скользящего компонента. В соответствии с предпочтительным вариантом осуществления, препарат готовят методом прямой компрессии.
Так как содержащий 1000 мг ниацина препарат с пролонгированным высвобождением по настоящему изобретению биоэквивалентен двум 500 мг таблеткам NIASPAN®, следует ожидать, что у них будет одинаковая эффективность и профиль токсичности. Таким образом, введение содержащего 1000 мг ниацина препарата с пролонгированным высвобождением по настоящему изобретению позволит получить те же преимущества при лечении, что и при использовании двух 500 мг таблеток NIASPAN®, но при этом избежать ограничивающей лечение гепатотоксичности или повышения содержания мочевой кислоты или глюкозы до такой степени, что лечение приходится прекращать. Специалистам хорошо известны проблемы токсичности, связанные с пролонгированным выделением ниацин-содержащих препаратов. См., например, "A comparison of the Efficacy and Toxic Effects of Sustained - v. Immediate-Release Niacin Hypercholesterolemic Patients", McKenneyet al., JAMA, vol.271, No.9, Mar.2, 1994; а также "Hepatic Toxicity of Unmodified and Time-Release Preparations of Niacin", Rader, et al., The Am. Jour. Of Med., vol.92, Jan. 1992, page 77.
Таким образом, в одном варианте настоящего изобретения введение фармацевтической композиции (препарата) по изобретению для лечения нуждающегося в этом пациента, причем указанное лечение может уменьшить содержание липидов в сыворотке крови, не вызывая ограничивающей лечение (i) гепатотоксичности и (ii) повышения содержания мочевой кислоты или глюкозы после введения препарата указанным пациентам, которое приводит к необходимости прервать лечение, в случае если пациент принимает указанную композицию один раз в день. В соответствии с другим аспектом, введение производят раз в день, по вечерам или по ночам (например, после обеда или перед сном).
Комбинированное лечение
Ежедневное введение ниацин-содержащих препаратов по настоящему изобретению можно сочетать с ингибитором редуктазы HMG-CoA. Термины "комбинированная терапия" и "комбинированное лечение" в настоящей заявке включают введение ниацин-содержащего препарата по настоящему изобретению и по меньшей мере одного дополнительного активного вещества в препарате той же или другой фармацевтической дозированной лекарственной формы. Комбинированная терапия здесь включает одновременное или последовательное введение активных веществ, являющееся частью режима лечения.
Примеры ингибиторов HMG-CoA редуктазы включают, но не ограничиваются, ловастатин и родственные соединения, описанные в патенте US Pat. No. 4231938, правастатин и родственные соединения, описанные в патентах US Pat. Nos. 4346227 и 4448979, мевастатин и родственные соединения, описанные в патенте US Pat. No. 3983140, велостатин и симвастатин и родственные соединения, описанные в патентах US Pat. Nos. 4448784 и 4450171, флувастатин, аторвастатин, ривастатин и флуиндостатин (SandozXU-62-320). Другие ингибиторы HMG-CoA редуктазы включают, но не ограничиваются, пиразоловые аналоги производных мевалонолактона, описанные в патенте US Pat. No. 4613610, другие аналоги мевалонолактона, описанные в заявке РСТ WO 86/03488, 6-[2-(замещенные-пиррол-1-ил)алкил]пиран-2-оны и его производные, описанные в патенте US Pat. No. 4647576, Searle's SC45355 (3-замещенное производное пентандионовой кислоты) дихлороацетат, имидазоловые аналоги, описанные в заявке РСТ WO 86/07054, производные 3-карбокси-2-гидрокси-пропанфосфорной кислоты, описанные во французском патенте No. 2596393, 2,3-ди-замещенные пиррольные, фурановые и тиофеновые производные, описанные в европейской патентной заявке No. 0221025 А 14, нафтильные аналоги мевалонолактона, описанные в патенте US Pat. No. 4686237, октагидронафталины, такие как описаны в патенте US Pat. No. 4499289, кето-аналогиловастина, описанные в европейской патентной заявке No. 0142146 А2, а также и другие известные ингибиторы HMG-СоАредуктазы, такие как описаны в британских патентах GB Patent No. 2205837 и 2205838 и в американских патентах US Pat. No. 5217992; 5196440; 5189180; 5166364; 5157134; 5110940; 5106992; 5099035; 5081136; 5049696; 5049577; 5025017; 5011947; 5010105; 4970221; 4940800; 4866058; 4686237.
Фармацевтические препараты по настоящему изобретению можно также вводить совместно с другими антилипидемическими агентами. Конкретные примеры антилипидемических агентов включают, но не ограничиваются, секвестранты желчной кислоты, например, холестирамин, колестепол DEAESephadex (Secholex.RTM. и Polidexide.RTM.), пробукол и родственные соединения, описанные в патенте US Pat. No. 3674836, липостабил-®hone-Poulanc), Eisai E5050 (N-замещенное производное этаноламина), иманиксил (НОЕ-402), тетрагидролипстатин (THL), иситигмастанил-фосфорилхолин (SPC Roche), аминоциклодекстрин (Tanabe Seiyoku), Ajinomoto A J-814 (производное азулена), мелинамид (Sumitomo), Sandoz 58-035, американский цианимид CL-277,082 и CL-283,546 (двузамещенные производные мочевины), неомицин, пара-аминосалициловая кислота, аспирин, четвертичный амин поли(диаллилдиметиламмония хлорид) и ионены, такие как описаны в патенте US Pat. No. 4759923, омега-3-жирные кислоты, содержащиеся в разнообразных продуктах из рыбьего жира, производные фибриновой кислоты, например гемфиброзил, хлофибрат, безафибрат, фенофибрат, ципрофибрат и клинофибрат, а также другие известные агенты, понижающие содержание холестерина в сыворотке, например, описанные в патенте US Pat. No. 5200424; европейской патентной заявке No. 0065835 A1, европейском патенте No. 164-698-A, GB Patent No. 1586152 и GB Patent Application No. 2162-179-A.
Кроме того, фармацевтический препарат по настоящему изобретению можно вводить совместно с агентом, ингибирующим гиперемию. Такие агенты включают, но не ограничиваются, нестероидные противовоспалительные вещества, такие как аспирин и соли салицилата; пропионовые кислоты, такие как ибупрофен, флурбипрофен, фенопрофен, кетопрофен, напроксен, напроксен натрия, карпрофен и супрофен; производные индолуксусной кислоты, такие как индометацин, этодолак и сулиндак; бензолуксусные кислоты, такие как ахлофенак, дихлофенак и фенхлофенак; пирролуксусные кислоты, такие как зомепирак и толмектин; пиразолы, такие какфенилбутазон и оксифенилбутазон; оксикамы, такие как пироксикам; а также антраниловые кислоты, такие как мехлофенамат и мефенамовая кислота.
В качестве агента, ингибирующего гиперемию, может также использоваться антагонист простагландиновых рецепторов D2, включая, но не ограничиваясь, соединения из патента US patent Publication Nos. 2004/0229844 и 2005/0154044. Предпочтительным антагонистом простагландиновых Е2 рецепторов является МК-0524 (Merck & Co.).
Замедленное высвобождение
Настоящее изобретение включает лекарственные формы с замедленным высвобождением. Термин "замедленное высвобождение" означает здесь, что в течение некоторого времени после введения пациенту происходит только незначительное высвобождение препарата, или его, вообще, не происходит (то есть, наблюдается период задержки, лаг-период). Содержащий ниацин препарат по настоящему изобретению можно приготовить в форме с замедленным высвобождением в качестве единственного активного агента фармацевтической композиции или в качестве одного из нескольких активных агентов фармацевтической лекарственной формы (другие активные агенты могут быть или не быть в форме для замедленного высвобождения). Так, например, фармацевтическая композиция может содержать ингибирующий гиперемию компонент немедленного высвобождения и ниацин-содержащий компонент с пролонгированным высвобождением. Например, при введении пациенту фармацевтической композиции по настоящему изобретению, ингибирующий гиперемию агент немедленного высвобождения поступает в кровь сразу же, а содержащий ниацин компонент - через некоторое время (то есть, по меньшей мере через 30 или 40 минут).
Замедленное высвобождение осуществляют с использованием веществ и методов, хорошо известных в уровне техники. Эти вещества и методы включают следующие. Цельная капсульная система доставки лекарственного средства, содержащая нерастворимую капсулу с лекарством и затычку. По истечении заранее определенного времени задержки затычка удаляется путем ее разбухания, эрозии или растворения. Примером такой системы является Pulsincap® (Scherer DDS, Ltd), в которой отверстие в капсуле закрыто набухаемой гидрогелевой пробкой. При контакте со средой растворения или с желудочно-кишечными жидкостями затычка набухает и по истечении периода задержки выталкивается из капсулы. После этого происходит быстрое выделение лекарственного средства. Для управления периодом задержки можно изменять размер и положение затычки. См., например, WO 90/09168; Wilding et al., Pharm Res. 1992; 9:654-657. В качестве материала затычки можно использовать нерастворимые, но проницаемые и набухаемые полимеры (например, полиметакрилаты). (см. Krogel I, Bodmeier R, Pharm Res. 1998; 15(3):474-481; Krogel I, Bodmeier R, Pharm Res. 1999; 16(9):1424-1429), разрушаемые сжимаемые полимеры (например, гидроксипропилметил-целлюлозу, поливиниловый спирт, полиэтиленоксид), расплавленные и застывшие полимеры (например, насыщенные полигликолированные глицериды, глицерилмоноолеат) и разрушаемые полимеры, деградация которых контролируется ферментативно (например, пектин). Потенциальную проблему различного времени удержания в желудке можно решить, нанеся на систему энтерическое покрытие, так чтобы растворение протекало только при более высоких значениях рН, характерных для тонкого кишечника Saeger Н, Virley P. Pulsincap& Mac226: Pulsed-Release Dosage Form. Product information from Scherer DDS, Ltd; 2004.
Система Port® System (Port Systems, LLC) представляет собой капсульную систему, основанную на явлении осмоса. Система состоит из желатиновой капсулы, покрытой полупроницаемой мембраной (например, из ацетата целлюлозы) и содержащей нерастворимую затычку (например, липидную) и осмотически активный агент, а также само лекарственное средство. Crison et al., Proceed Intern Symp Control Rel Bioact Mater. 1995; 22:278-279. При контакте с водной средой вода диффундирует через полупроницаемую мембрану, что приводит к повышению внутреннего давления и, вследствие этого, к выдавливанию затычки по истечении периода задержки. Контролируют время задержки, меняя толщину покрытия.
Если лекарственное средство надо ввести в жидкой форме, можно использовать осмотические капсульные системы, в которых жидкое лекарственное средство абсорбировано на высокопористых частицах, высвобождающих лекарственное средство через отверстие полупроницаемой капсулы под действием расширяющегося осмотического слоя после растворения барьерного слоя. См. US Patent No. 5318558. Капсульная система доставляет лекарственное средство в организм путем осмотической инфузии влаги. Стенка капсулы изготовлена из эластичного материала и содержит отверстие. При осмосе давление в капсуле возрастает и стенка растягивается. Отверстие настолько мало, что в нерастянутом состоянии стенки поток лекарственного средства через него практически останавливается, но если стенка растягивается выше порогового значения, отверстие сильно увеличивается в размерах, что обеспечивает выход из него лекарственного средства с требуемой скоростью. Могут использоваться эластомеры, например стиролбутадиеновые сополимеры. См. US Patent No. 5221278; US Patent No. 5209746.
Система Time Clock® system (West Pharmaceutical Services Drug Delivery & Clinical Research Centre) представляет собой твердую форму дозировки, покрытую липидными барьерами, которые содержат воск carnuba и пчелиный воск, а также поверхностно-активные вещества, такие как полиоксиэтилена сорбит моноолеат.Wilding et al., Int J Pharm. 1994; 111:99-102; Niwa et al., J Drug Target. 1995; 3:83-89. Это покрытие разрушается или эмульсифицируется в водной среде за время, пропорциональное толщине пленки, и открывает ядро таблетки для диспергирования. Клинические испытания на добровольцах показали, что период задержки не зависит от времени пребывания в желудке, а также что на редисперсию гидрофобной пленки не оказывают влияние ни ферменты кишечника, ни механическое воздействие желудка, ни рН в желудке или кишечнике. Gazzaniga et al., Int J Pharm. 1994; 2(108):77-83. Период задержки увеличивается с повышением толщины покрытия.
Система Chronotropic® представляет собой содержащее лекарственное средство ядро, покрытое гидрофильной набухающей гидроксипропил метил-целлюлозой (НРМС), отвечающей за период задержки до начала высвобождения лекарства. Gazzaniga et al., Eur J Biopharm. 1994; 40(4):246-250; Gazzaniga et al., Proceed Intern Symp Control Rel Bioact Mater. 1995:22:242-243; EP 0572942. Использование внешней энтерической пленки, предохраняющей от действия желудочного сока, позволяет решить проблемы вариабельности времени растворения в желудке. Sangalli et al., J Contr Rel. 2001:73:103-110. Период задержки определяется толщиной и степенью вязкости НРМС. Система приемлема как для таблеток, так и для капсул. Conte et al., Drug Dev Ind Pharm. 1989; 15(14-16):2583-2596.
Можно приготовить также многослойную таблетку, содержащую два активных агента. Так, трехслойная таблетка включает два содержащих активные агенты слоя, разделенные лишенным лекарственного средства гелеобразным полимерным барьерным слоем. US Patent No. 4865849; Conte et al., Eur J Pharm. 1992;38(6):209-212; Krogel I, Bodmeier R, Int J Pharm. 1999; 187:175-184. Эта трехслойная таблетка с трех сторон покрыта непроницаемой этилцеллюлозой, а верхняя часть таблетки не закрыта. При взаимодействии со средой растворения доза из верхнего слоя быстро выделяется через непокрытую поверхность. Вторая доза выделяется из нижнего слоя после разрушения и растворения желеобразного барьерного слоя НРМС. Скорость желеобразования и/или растворения барьерного слоя определяет доступность второй дозы. Желеобразующие полимеры могут включать производные целлюлозы, такие как НРМС, метилцеллюлоза или полимерные спирты различных молекулярных весов, а материалы покрытия включают этилцеллюлозу, целлюлозу-ацетат-пропионат, метакриловые полимеры, акриловые и метакриловые сополимеры и полиспирты.
В пульсирующих системах с разрушающимся покрытием высвобождение лекарственного средства происходит при распаде покрытия. Давление, необходимое для разрушения этого покрытия, создают шипучими наполнителями, набухающими агентами или осмотическим давлением. Например, можно сделать шипучую смесь лимонной кислоты и бикарбоната натрия и включить ее в ядро таблетки, покрытой этилцеллюлозой. Образующаяся после проникновения воды в ядро двуокись углерода разрушает покрытие и дает возможность лекарству выйти наружу. Bussemer Т, Bodmeier R, AAPS Pharm Sci. 1999; 1(4 suppl):434 (1999). Период задержки возрастает с увеличением толщины покрытия и твердости ядра таблетки.
Для изготовления капсул можно использовать хорошо разбухающие агенты, называемые также супердезинтегрантами. Такие капсулы содержат лекарство, набухающий агент и разрушаемый полимерный слой. US Patent No. 5229131. Примеры супердезинтегрантов включают кросскармелозу, натрия крахмала гликолят и низкозамещенную гидроксипропил-целлюлозу. Набухание этих веществ приводит к полному разрушению пленки с выходом лекарственного средства. Период задержки зависит от препарата внешнего полимерного слоя. Введение гидрофильного полимера, такого как НРМС, уменьшает период задержки. Система может использоваться для введения как твердых так и жидких лекарственных препаратов.
Для обеспечения пролонгированного высвобождения одного активного агента и пролонгированного или иного (например, немедленного) высвобождения другого активного агента могут использоваться системы доставки в форме отдельных частиц (например, шариков или катышей). См., например, US Patent No. 4871549.
Система Time-Controlled Explosion System (контролируемая по времени взрывная система) (Fujisawa Pharmaceutical Co., Ltd.) представляет собой систему из множества частей, лекарственное средство в которой нанесено на зерна из сахара типа non-pareil sugar seeds, затем на него наносится набухаемый слой и нерастворимый верхний слой. Ueda et al., J Drug Targeting. 1994:2:35-44; Ueda et al., Chem Pharm Bull. 1994; 42(2):359-363; Ueda et al., Chem Pharm Bull. 1994; 42(2):364-367; Hata et al., Int J Pharm. 1994; 110:1-7. Набухающие агенты могут включать супердезинтегранты, такие как карбоксиметилцеллюлозу натрия, натрия крахмала гликолят, L-гидроксипропилцеллюлозу, полимеры, подобные поливинилацетату, полиакриловой кислоте, полиэтиленгликолю и т.д. Альтернативно, можно использовать также "шипучую" систему, включающую смесь винной кислоты и бикарбоната натрия. При попадании воды набухающий слой расширяется, пленка из-за этого разрушается и лекарственное средство быстро выходит наружу. Высвобождение происходит независимо от факторов окружающей среды, таких как рН или растворимость лекарственного средства. Период задержки меняют, изменяя толщину покрытия или добавляя больше липофильного пластификатора во внешний слой. US Patent No. 5508040. Контролирующая проницаемость система основана на комбинации осмотического эффекта и эффекта набухания. Ядро содержит лекарственное средство, твердое и/или жидкое липидное соединение низкой общей плотности (например, минеральное масло) и дезинтегрант.Это ядро затем покрывают слоем ацетата целлюлозы. При погружении в водную среду вода проникает в ядро и вытесняет липидное соединение. После его удаления внутреннее давление начинает возрастать до достижения критического напряжения с последующим разрушением капсулы. U.S. Patent No. 5229131.
Еще одна система основана на капсулах или таблетках, состоящих из большого количества шариков, каждый из которых состоит из двух или больше более мелких шариков или частей (то есть, популяция). Schultz Р, Kleinebudde P. J Contr Rel. 1997:47:181-189. У каждого шарика имеется ядро, содержащее терапевтическое лекарственное средство и водорастворимый осмотический агент. Ядро окружено водопроницаемой, но нерастворимой в воде полимерной пленкой. В препарат полимерной пленки включен гидрофобный нерастворимый агент, влияющий на проницаемость (например, жирная кислота, воск или соль жирной кислоты). Скорость проникновения воды и высвобождения лекарственного средства обусловлена толщиной пленки вокруг каждой группы, которая отличается от толщины покрытия других шариков в форме дозировки. Осмотические агенты растворяются в воде, заставляя шарики набухать, и это регулирует скорость диффузии вещества. Поскольку каждая группа шариков высвобождает лекарственное средство в свое время, единичная дозированная лекарственная форма приводит к серии выбросов препарата. Толщину покрытия различных шариков можно менять.
Осмотически активные агенты, которые не подвергаются набуханию, также можно использовать для пролонгированного высвобождения. Schultz et al., J Contr Rel. 1997; 47:191-199; US Patent No. 5260069. Ядра шариков содержат лекарственное средство и хлорид натрия. Эти ядра покрыты полупроницаемым полимером ацетатом целлюлозы. Последний селективно пропускает воду, но не пропускает лекарственное средство. Период задержки увеличивается при повышении толщины покрытия и при увеличении содержания в нем талька или липофильного пластификатора. Хлорид натрия облегчает быстрое высвобождение лекарственного средства. В отсутствие хлорида происходит более медленное высвобождение по истечении периода задержки, поскольку ядро разбухает меньше и в оболочке образуются маленькие трещинки.
Для обеспечения пролонгированного (замедленного) высвобождения можно использовать систему, содержащую ядро из лекарственного средства и осмотически активного агента (хлорида натрия), покрытое нерастворимой проницаемой мембраной. US Patent No. 5260068. Покрытие состоит из различных типов поли(акрилат-метакрилат) сополимеров и стеарата магния, который снижает водную проницаемость мембраны и, тем самым, позволяет применять более тонкие пленки. Толстых пленок следует избегать, так как они могут не разрушиться полностью. При использовании в качестве покрытия этилцеллюлозы удается влиять на период задержки энтерического полимера и обеспечивать разрушение пленки в заранее определенное время. Bodmeier et al., Pharm Res. 1996; 13(1):52-56.
На проницаемость и поглощение воды акриловыми полимерами с четвертичными аммонийными группами можно повлиять, вводя различные противоионы в среду. Beckert et al., Proceed Int'l Symp Control Rel Bioact Mater. 1999; 26:533-534. Были разработаны различные основанные на этом ионном обмене системы доставки. Предпочтительным для этой цели полимером является Eudragit RS 30D, так как содержит положительные поляризованные четвертичные аммонийные группы в боковой цепи полимера, а также отрицательные противоионы гидрохлорида. Аммонийная группа гидрофильна и облегчает взаимодействие полимера с водой, что меняет его проницаемость и дает воде возможность попасть в активное ядро контролируемым образом. Шарики таблетки можно покрыть полимером EUDRAGIT RS30D® (10% - 40% увеличение веса), создав слои четырех различных видов, различающихся по толщине. Период задержки коррелирует с толщиной пленки. Проницаемость лекарственного средства через пленку EUDRAGIT зависит от количества ацетата натрия в ядре. По истечении периода задержки взаимодействие между ацетатом и полимером приводит к увеличению проницаемости покрытия, так что вся активная доза высвобождается за несколько минут. Guo X. Physicochemical and Mechanical Properties Influencing the Drug Release From Coated Dosage Forms. Doctoral Thesis. The University of Texas at Austin; 1996.
Система сигмоидального высвобождения включает ядра шариков, содержащие лекарственное средство и янтарную кислоту и покрытые аммонийно-метакрилатным сополимером USP/NF типа В. Narisawa et al., Pharm Res. 1994; 11(1):111-116. Период задержки определяется скоростью проникновения воды через полимерную мембрану. Вода растворяет янтарную кислоту и лекарственное средство в ядре. В свою очередь, раствор кислоты повышает проницаемость гидратированной пленки полимера. Помимо янтарной кислоты можно использовать уксусную, глутаровую, винную, яблочную или лимонную кислоты. Повышение проницаемости можно объяснить гидратацией пленки, что увеличивает свободный объем. На основании этих находок удалось спроектировать покрытую оболочкой систему доставки, ядро в которой содержит кислоту. Narisawa et al., Pharm Res. 1994; 11(1):111-116; Narisawa et al., J Contr Rel. 1995; 33:253-260. Период задержки, полученный в исследованиях in-vitro, хорошо коррелировал сданными in-vivo исследований с участием гончих собак. Narisawa et al., J Contr Rel. 1995; 33:253-260.
Настоящее изобретение относится к фармацевтической композиции, охватывающей ниациновый препарат по настоящему изобретению в форме пролонгированного высвобождения, а также ингибирующий гиперемию агент. Ниацин, высвобождающийся пролонгированным образом, и агент, ингибирующий гиперемию, можно включать в одну или в различные лекарственные формы. Так, например, фармацевтическая композиция может охватывать твердую лекарственную форму, имеющую снаружи агент, обеспечивающий немедленное высвобождение и ингибирующий гиперемию, и внутри, содержащий ниацин компонент с пролонгированным высвобождением. В соответствии с предпочтительным вариантом осуществления, ингибирующий гиперемию агент выделяется от 30 до 40 минут до высвобождения ниацина.
Приведенные ниже примеры предназначены для более лучшей иллюстрации, но не ограничивают различные аспекты и варианты осуществления настоящего изобретения.
Пример 1
В этом примере использовался следующий препарат:
Предпочтительно, если используется METHOCEL® K-15M Premium, то размер его частиц должен обеспечивать прохождение, минимум, 90% частиц через сито с ячейкой 100 меш (в соответствии с американским стандартом). При использовании METHOCEL® K-15M Premium CR, предпочтительно, чтобы, минимум, 99% частиц проходило через сито с ячейкой 40 меш (в соответствии с американским стандартом) и, минимум, 90% частиц - через сито с ячейкой 100 меш.
Чтобы получить порцию весом 20 кг, измельченный гранулированый ниацин и наполнители взвесили в соответствии с приведенной выше формулой, поместили в смеситель на 8 кварт (8,808 л) и перемешивали 10 минут со скоростью 24 об/мин. В частности, для измельчения METHOCEL® K-15М и стеариновой кислоты был выбран фильтре размером ячейки 12 меш (1,68 мм), а для измельчения гранулированого ниацина и Повидона K-90 (возможно, просеянного, размолотого или то и другое) - фильтр с размером ячейки 16 меш (1,19 мм). Полученную гранулированую композицию непосредственно спрессовали в таблетки с помощью пресса BWI Manesty Beta Press с 19 мм овальным инструментарием при 30 кН. Твердость таблеток (то есть, уплотняющую силу таблетки, измеренную стандартными методами компрессии, известными специалистам) контролировали и поддерживали в диапазоне от 16 до 22 кР (килофунтов) с помощью стандартного тестера твердости таблеток. Необязательно, в альтернативных вариантах осуществления, стеариновую кислоту или повидон можно просеять также через фильтр, такой как фильтр 40 мкш, и можно варьировать этапы перемешивания (один или два) и время перемешивания (10, 15 или 20).
Полученные таблетки были покрыты 2% по весу гранулированным цветным покрытием OPADRY® Orange 03 B 93199. Условия нанесения покрытия были следующие:
Обнаружено, что покрытые оболочкой содержащие 1000 мг ниацина таблетки, полученные методом прямой компрессии, были стабильны в течение трех месяцев при температуре и относительной влажности(РН) 40°С/75% и 25°С/60% RH. Это исследование проводили, сравнивая результаты анализа на ниацин, растворение ниацина, влажность таблеток и их физический вид до и после эксперимента по стабильности. Фиг.5 иллюстрирует блок-схему производственного процесса прямой компрессии при получении препарата для таблетки в соответствии с вариантом осуществления наятоящего изобретения.
Если не указано иначе, содержащие 1000 мг ниацина препараты с пролонгированным высвобождением по настоящему изобретению были приготовлены в соответствии с Примером 1.
Пример 2
Используя описанные здесь процессы, методом прямой компрессии можно изготовить содержащие 500 мг и 750 мг ниацина таблетки с пролонгированным высвобождением (покрытые или непокрытые оболочкой). Их содержание проиллюстрировано в приведенных ниже Таблице 8 и Таблице 9.
Чтобы получить 500 мг или 750 мг таблетки, измельченный гранулированный ниацин и наполнители взвесили в соответствии с проиллюстрированными в Таблицах 8 и 9 концентрациями компонентов и затем перемешали в подходящем смесителе или миксере в течение достаточного времени, чтобы адекватно перемешать компоненты. Полученные гранулированные композиции можно непосредственно спрессовать в таблетки с помощью подходящего пресса, такого как BWI Manesty Beta Press, получив 500 мг или 750 мг таблетки требуемой твердости. При желании эти таблетки можно также покрыть оболочкой, такой как цветное покрытие, методами, известными в уровне техники.
Пример 3
Сравнение числа случаев гиперемии при использовании покрытых оболочкой содержащих 1000 мг ниацина матричных таблеток прямой компрессии с пролонгированным высвобождением и 1000 мг NIASPAN®
Метод
Проводилось рандомизированное исследование, представляющее собой двойной слепой тест с плацебо и с трехсторонним кроссовером. Под контролем двух наблюдателей в едином центре проводилось исследование способности препаратов вызывать гиперемию. В ходе эксперимента субъекты не принимали аспирин или NSAIDs.
В эксперименте принимали участие здоровые, некурящие мужчины-добровольцы в возрасте от 18 до 70 лет с индексом массы тела (BMI) от 22 до 31. Субъекты признавались здоровыми после полного физического обследования, изучения истории болезни, снятия электрокардиограммы, а также клинического лабораторного исследования, проводившегося в ходе предварительного скринингового визита или при визите на первом этапе исследования. Субъектов исключали, если у них обнаруживали аллергию или гиперчувствительность к ниацину или родственным производным; если они принимали вызывающие зависимость препараты в течение последних трех лет; если у них были мигрени, диабет, камни в желчном пузыре, заболевания печени, тяжелая гипертония или гипотония, нарушения в работе сердца, заболевания почек или вызванная приемом лекарственных средств миопатия. Субъекты не должны были принимать никакие лекарственные средства по рецепту в течение 21 дня до начала исследования, а безрецептурные лекарства, витамины и растительные препараты в течение 10 дней до начала исследования.
Скрининговые процедуры завершились за 21 день до приема лекарственного средства в Периоде Испытания 1 (Фиг.6). В ходе каждого из трех периодов испытания субъекты изолировали, приблизительно, с 7 утра первого дня до завершения всех процедур утром второго дня (между 7 и 10 утра). В ходе каждого периода испытаний препарат еды и время приема пищи оставались одинаковыми. Пища соответствовала специфическому меню, в котором контролировалось содержание ниацина и жиров. В ходе исследования не допускался прием никаких сопутствующих лекарственных средств, витаминов или растительных препаратов.
Экспериментальное лечение в ходе испытания
Препараты, вводившиеся в ходе трех периодов клинических испытаний, проиллюстрированы на Фиг.6. Исследуемое лечение включало две покрытые оболочкой содержащие 1000 мг ниацина таблетки по изобретению (см. Пример 1) (Тест - ниацин-содержащие ER таблетки с переработанным составом), в качестве сравнения использовали две непокрытые оболочкой содержащие 1000 мг ниацина коммерческие ER-таблетки (Сравнение - NIASPAN®)). Для контроля давали две непокрытые таблетки плацебо (Контроль). Поскольку эксперимент основывался на сообщениях субъектов о гиперемии, было важно оставить пациентов и персонал в полном неведении о препаратах, назначаемых им в ходе лечения. Этого достигали несколькими способами. В процессе каждого активного лечения совместно с активными таблетками давали две покрытые оболочкой или две непокрытые таблетки плацебо, так что все пациенты получали две покрытые оболочкой и две непокрытые таблетки, независимо от лечения. Кроме того, препараты давали субъектам из непрозрачных дозировочных чашек, и субъектам завязывали глаза при приеме препарата. Чтобы скорректировать данные по гиперемии на непредсказуемый плацебо-ответ, в эксперимент включали плацебо (плацебо-контролируемое лечение).
В первый день каждого периода исследования субъектам давали одну дозу лекарственного средства, приблизительно, в 23.00. Это делалось перекрестным методом, в соответствии со схемой рандомизации. Между каждым периодом лечения был установлен минимальный период отмывки в 7 дней. Проводившие испытание люди и персонал медицинского центра не знали о назначенной схеме лечения, а персоналу центра, принимавшему участие в организации и администрировании лечения, запретили собирать или проверять возникающие при лечении нежелательные эффекты.
Каждую дозу вводили перорально вместе с 240 мл воды после обезжиренной легкой закуски. Эту закуску полностью съедали в течение 15 минут перед приемом лекарственного средства. Таблетки давали либо вместе, либо одну за другой, и от каждого субъекта требовали принять их все в течение не более одной минуты. Запрещали жевать или кусать таблетки. Если, чтобы запить таблетки, субъектам требовалась дополнительная вода, ее давали порциями по 120 мл. Чтобы убедиться в проматывании дозы, по завершении приема препарата рот каждого субъекта осматривали.
Характеристики гиперемии
Первичной характеристикой гиперемии было признано сообщение субъекта о случае или эпизоде гиперемии. Такой случай или эпизод был описан как один или более следующих одновременных симптомов гиперемии: краснота, тепло, покалывание, зуд. В течение каждого периода испытания субъектам каждый час предлагали сообщать о наличии или отсутствии симптомов гиперемии в течение 8 часов после введения препарата. Им предлагалось регистрировать начало и прекращение симптомов и оценивать интенсивность (тяжесть) каждого симптома, нанеся вертикальную линию на горизонтальную 10-см визуальную аналоговую шкалу (visual analog scale (VAS)), помеченную отметками, начиная от "отсутствуют" (0) слева и до "непереносимо" (100) справа. Информацию отмечали в электронном дневнике гиперемии.
Вторичные характеристики гиперемии включали количество эпизодов гиперемии, интенсивность и продолжительность гиперемии, как применительно к случаям гиперемии в целом, так и к индивидуальным симптомам (краснота, тепло, покалывание, зуд). Каждый субъект оценивал общую тяжесть первого случая или эпизода гиперемии, причем время начало первого эпизода было определено как начало первого или одного из одновременно возникших первых симптомов за время исследования. Время завершения эпизода гиперемии определено как время прекращения одного или нескольких симптомов гиперемии в эпизоде, за которым следовал бессимптомный период продолжительностью не менее 30 минут.
Статистический анализ
Установлено, что для того, чтобы показать статистически значимые различия случаев гиперемии между видами лечения при альфа (альфа) 5% по тесту Мак Немара (McNemar's test (nQuery Advisor®), version 5.0)), необходимо использовать выборку 144 субъекта. Для того чтобы обеспечить адекватное количество завершивших испытание субъектов и получить значимые данные, по меньшей мере, для двух видов лечения, субъектов, которые вышли из испытания значительно раньше срока, заменяли.
Первичную оценку эффективности (случаи гиперемии) сравнили между группами лечения методом равенства парных пропорций Мак Немара. Первичное сравнение проводили между исследуемыми и референсными препаратами ниацина ER среди субъектов, принимавших по меньшей мере одну дозу исследуемого препарата в течение по меньшей мере двух периодов исследования. Выполняли также сравнение между ниацином и плацебо. Вторичную оценку проводили с использованием как теста Мак Немара (для категоричных переменных) или соответствующий парный t-тест. Все сравнения были двусторонними и проводились при альфа (α)=0,05.
Результаты
В этом исследовании принимали участие 156 субъектов, каждый из них получил по меньшей мере одну дозу исследуемого препарата. Их средний возраст составлял 33,5 лет, а средний BMI был равен 26,2. Краткое описание демографии субъектов представлено ниже в Таблице 10.
Все 156 субъектов получали исследуемые лекарственные препараты в Период 1, 143 субъекта (92%) получали лекарственные препараты в Период 2, и 131 субъект (84%) - в Период 3. Всего 130 субъектов (83%) завершили все три периода. Преждевременно из исследования было исключено 26 человек (17%): 8 (5%) отозвали свое согласие, 3 (2%) не удалось проследить, у 2 (1%) наблюдались побочные эффекты, у 2 (1%) были нарушения в протоколе, у 1 (1%) был положительный результат теста на наркотики, и остальные 10 (6%) были исключены по "иным" причинам. 11 из вышедших из исследования субъектов заменили, чтобы обеспечить достаточную статистическую значимость результатов.
Гиперемия
Как и предполагалось, удалось спровоцировать гиперемию, так как у субъектов, получавших активное лечение, гиперемия наблюдалась в четыре раза чаще, чем у принимавших контрольное лечение. В Таблице 11 отображено количество, интенсивность и продолжительность первого случая гиперемии у группы, предназначенной для лечения (intent-to-treat (ITT)), определяемой как субъекты, получившие, по меньшей мере, одну дозу исследуемого препарата, и завершившие, по меньшей мере, один экспериментальный период, но не включающей замененных субъектов.
Обнаруженный в этом эксперименте плацебо-ответ представляет собой типичный общий плацебо-ответ.
Процент, общая интенсивность и продолжительность первого случая гиперемии у субъектов, получивших хотя бы одну дозу препарата при экспериментальном лечении по меньшей мере в двух периодах исследования, для Тест (ниацин-содержащий препарат с переработанным составом ER) против Сравнения (Коммерческий ниацин-содержащий препарат ЕР)(Сравнение): А) Вероятность первого случая гиперемии (р=0,0027); В) Интенсивность первого случая гиперемии на основании VAS; показаны значения медианы [среднее было 35,6±22,78 (min, max: 0,0, 99,0) для Теста и 52,8±23,86 (min, max: 0,0; 95,0) для сравнения, р<0,001]; С) Продолжительность первого события гиперемии (мин); показаны значения медианы [среднее было 130,3±95,01 (min, max: 9,0; 473,0) для Теста и 195,7±136,32 (min, max: 5,0; 984,0) для сравнения, р<0,0001].
Как показано на Фиг.7, для первичного анализа эффективности, среди субъектов, принимавших по меньшей мере одну дозу исследуемого препарата в течение, по меньшей мере, двух периодов исследования, 118 (89%) испытывали гиперемию в течении лечения тестовым препаратом и 130 (98%) - во время лечения препаратом сравнения (референсным препаратом). Разница была статистически значима, р=0,0027.
На Фиг.8 и 9 показана медиана интенсивности и продолжительности первого случая гиперемии. Сравнение средних значений интенсивности и продолжительности с соответствующими медианами позволяет предположить, что распределение данных отличается от нормального. Тестовое лечение приводило к 42% уменьшению медианы интенсивности гиперемии (33% снижение средней интенсивности гиперемии) и 43% снижение медианы продолжительности гиперемии (33% средней продолжительности гиперемии), по сравнению с референсным лечением. Парный t-тест продемонстрировал статистически значимые улучшения в случае тестового лечения, как в терминах средней интенсивности (р<0,0001), так и средней продолжительности (р<0,0001) первого случая гиперемии.
Тест, по сравнению с референсным лечением, приводил к меньшему проценту каждого из четырех симптомов гиперемии (краснота, тепло, покалывание и зуд) (Фиг.10). Различия между действиями двух препаратов было значимым, причем различие было в пользу тестового препарата для каждого из четырех индивидуальных симптомов гиперемии первого события гиперемии, при этом использовался тест Мак Немара. Покраснение наблюдалось в 71% случаев для тестового препарата и 86% для референсного препарата (р=0,0016); ощущение тепла - в 68% случаев для тестового препарата и 80% для референсного препарата (р=0,0163); покалывание - в 47% случаев для тестового препарата и 62% для референсного препарата (р=0,0039); зуд - в 48% случаев для тестового препарата и 65% для референсного препарата (р=0,0015).
Полученные данные показывают, что препараты по изобретению снижают количество случаев, интенсивность (тяжесть) и продолжительность гиперемии, по сравнению с коммерчески доступным препаратом. В целом, можно сказать, что у препаратов по изобретению наблюдалось значимое 9% снижение вероятности гиперемии (89%), по сравнению с коммерчески доступным ниацин-содержищим ER препаратом NIASPAN® (98%), несмотря на то, что лечение было так спроектировано, чтобы вызвать гиперемию введением однократной большой (2000 мг) дозы препарата субъектам, которых ранее ниацином не лечили (ниацин-наивные субъекты). Введение препаратов по изобретению в этом провоцирующем гиперемию исследовании приводило также к статистически высокозначимому снижению интенсивности и продолжительности гиперемии. Медиана интенсивности и продолжительности гиперемии уменьшалась на 42% и 43%, соответственно, по сравнению с лечением коммерческим ниацин-содержащим ER-препаратом. Кроме того, продолжительность первого события гиперемии в случае препаратов по изобретению была короче более чем на 1 час.
Пример 4
Целью настоящего исследования было определение биоэквивалентности (BE) содержащих 1000 мг ниацина таблеток с пролонгированным высвобождением по изобретению (называемых здесь таблетками с переработанным составом) (Тест), по сравнению с коммерчески доступными 1000 мг таблетками NIASPAN®) ®EF) при введении в виде однократной дозы 2000 мг.
Схема исследования
Проводилось рандомизированное исследование из одного центра с открытой меткой, с введением однократной дозы и двусторонним перекрестным исследованием. В эксперименте принимали участие 44 здоровых некурящих добровольцев мужчин и женщин в возрасте от 40 до 70 лет, включительно. Исключенных из эксперимента не заменяли. Каждый субъект получал два ниацин-содержащих препарата, Тест и Ref, в виде одной и той же однократной дозы 2000 мг, которые давали им в два разных сеанса. Период промывки между дозами составлял, по меньшей мере, 10 дней. Тестовый продукт представлял собой содержащие 1000 мг ниацина таблетки замедленного высвобождения с переработанным составом, а референсный продукт (продукт сравнения) ®EF) представлял собой 1000 мг таблетки NIASPAN®. Каждую дозу запивали 240 мл воды и давали после обедненной жирами легкой закуски, приблизительно, в 22:00 в День 1 каждого периода. В течение каждого периода исследования (5 дней для периода 1 и 6 дней для периода 2) субъекты находились в центре, где проводилось испытание, их пища соответствовала предоставленным спонсорами меню. На протяжении эксперимента запрещался прием других медикаментов, витаминов, растительных или пищевых добавок.
Последовательные образцы крови собирали в течение промежутка времени, начиная от 30 минут до введения дозы и в течение 24 часов после ее введения с интервалами: -30 мин (пре-доза), 1, 2, 3, 4, 4.5, 5, 6, 7, 8, 10, 12, 14, 16 и 24 часа (пост-дозы). Мочу собирали в течение промежутка времени, начиная от 24 часа до введения дозы и в течение 96 часов после ее введения за интервалы: от -24 до -18, от -18 до -12, от -12 до -6 и от -6 до 0 часов (пре-доза); от 0 до 6, от 6 до 12, от 12 до 18, от 18 до 24, от 24 до 48, от 48 до 72 и от 72 до 96 часов (пост-доза). Плазму анализировали на содержание ниацина и никотин-мочевой кислоты (nicotinuric acid (NUA)). Мочу анализировали на содержание ниацина и его метаболитов: NUA, N-метилникотинамида (MNA) и 2-PY (N-метил-2-пиридон-5-карбоксамид). Ниацин активно метаболизируется, и его концентрации в плазме демонстрируют значительно большую вариабельность, по сравнению с NUA, одним из его основных метаболитов.
Таким образом, для определения скорости абсорбции ниацина использовали максимальную концентрацию NUA в плазме (Сmax). Как показано в NIASPAN® NDA, общее восстановление в моче является более точной характеристикой степени абсорбции, чем AUC, поскольку AUC более подвержен нелинейной фармакокинетике. Таким образом, в качестве меры степени поглощения ниацина использовали общее его количество, экскретируемое в виде самого ниацина и трех его метаболитов NUA, MNA и 2PY в моче. Таким образом, первичными переменными, определенными в протоколе и используемыми для оценки биоэквивалентности NUA, являются Сmax для NUA и общее восстановление в моче ниацина и трех его метаболитов (NUA, MNA и 2PY).
Тестовые лекарственные препараты представляли собой две 1000 мг таблетки (т.е. содержащие 1000 мг ниацина) с пролонгированным высвобождением с переработанным составом по настоящему изобретению. Лекарственные препараты сравнения представляли собой две 1000 мг таблетки NIASPAN®. Лечение проводилось с интервалом по меньшей мере 10 дней.
Субъекты получали пищу в одно и то же время каждый день, в который они проводили в клинике в течение каждого периода. Для каждого периода еда была одинакова, и требовалось, чтобы субъекты съедали свои порции целиком. Завтрак, ланч, обед и вечерняя легкая закуска начинались ежедневно, приблизительно, в 07:00, 12:00, 18:00 и 21:45, соответственно. Фактическое время еды или приема легкой закуски для каждого субъекта было спланировано в соответствии с фактическим режимом введения препаратов. От субъектов требовали выпивать, минимум, 720 мл воды в День 1 и 1440 мл воды в День 1-5, помимо 240 мл воды, выпитой при приеме препарата в День 1.
В День 1 были поданы обед и вечерняя закуска. В Дни 1-5 подавали завтрак, ланч, обед и вечернюю закуску. Последняя съедалась за 15 минут непосредственно перед введением препарата в День 1 каждого периода. В День 6 второго периода еды не подавалось, поскольку субъекты выписывались из клиники после завершения всех клинических процедур.
Оценка фармакокинетики
а. Сбор и анализ плазмы
Последовательные образцы крови были собраны для каждого периода в промежуток времени, начиная от 30 минут до введения препарата, и заканчивая 24 часами после этого (15 образцов не лечение). Каждый образец брали в 10 мл пробирку VACUTAINER с гепарином натрия и охлаждали на водяной бане с размолотым льдом не менее 5 минут после сбора. Затем образцы центрифугировали при 4 градусов и, приблизительно, 3000 об/мин в течение 15 минут, чтобы отделить плазму. Каждый образец плазмы разделили на две аликвоты, А и В, и перенесли в две предварительно охлажденные, соответственно помеченные полипропиленовые пробирки. В этих пробирках образцы замораживали и хранили при -20°С.
Концентрации ниацина и NUA анализировали с помощью валидированной комбинации методов жидкостной хроматографии и масс-спектрометрии (LC/MS/MS). Данные об указанных концентрациях получали для одной и той же инъекции. Нижний предел количественного определения (lower limit of quantitation (LLQ)) для ниацина и NUA составлял 2 нг/мл в плазме. В каждом анализе оценивались также образцы, используемые для контроля качества.
b. Сбор и анализ мочи
Мочу собирали в следующие интервалы времени: от -24 до -18, от -18 до -12, от -12 до -6 и от -6 до 0 часов (до введения дозы); от 0 до 6, от 6 до 12, от 12 до 18, от 18 до 24, от 24 до 48, от 48 до 72 и от 72 до 96 часов после введения дозы (всего было 11 сборов).
Собранную мочу переносили в пластиковые контейнеры с плотно пригнанными крышками. На протяжении периода сборки мочу охлаждали в холодильнике или на бане с ледяной водой. Коллекторы для сборки были помечены с указанием номера и инициалов субъекта, интервала сборки и номера протокола. Пустые контейнеры взвешивали, округляя до ближайшей десятой доли грамма (например, до 100,1 г), эти данные записывали на контейнере и документировали в лабораторных журналах. В конце каждого интервала общий вес контейнера и собранной мочи измеряли и округляли также до ближайшей десятой доли грамма. Вес мочи определяли, вычитая вес пустого контейнера из общего веса контейнера с мочей. В некоторых случаях объем мочи за период сборки превышал вместимость контейнера, в таких случаях для завершения сбора мочи требовался второй контейнер. Регистрировали также дату (даты) и время начала и прекращения сбора мочи за каждый интервал. Две аликвоты (приблизительно 2,5 мл каждая) из каждого интервала сборки переносили в две соответственно помеченные полипропиленовые пробирки. Если для конкретного интервала сборки требовалось более одного контейнера, перед взятием аликвот мочу из обоих контейнеров смешивали. В этих пробирках образцы замораживали и хранили при -20°С до анализа.
Концентрации ниацина, NUA, MNA и 2-PY анализировали с помощью валидированной комбинации методов жидкостной хроматографии и масс-спектрометрии (LC/MS/MS). Данные о концентрациях ниацина и NUA получали для одной и той же инъекции, а данные о концентрациях MNA и 2-PY получали для другой инъекции. В моче значения LLQ составляли 20 нг/мл для ниацина и 200 нг/мл для NUA. LLQ для MNA и 2-PY составляли 500 нг/мл и 2500 нг/мл, соответственно. В каждом анализе оценивались также образцы, используемые для контроля качества.
с.Фармакокинетические параметры в плазме и восстановление в моче
В фармакокинетический (РК) анализ включали данные от субъектов, у которых удалось получить достаточно информации для расчета РК параметров, по меньшей мере, для одного лечения. После начала каждого лечения для каждого субъекта рассчитывали следующие РК параметры:
* Cmax: максимальная наблюдавшаяся концентрация
* Тmax: время достижения максимальной наблюдавшейся концентрации
* AUClast: площадь под кривой концентрация-время от момента времени О до момента получения последней измеримой (ненулевой) концентрации, определенная по линейной формуле трапеций.
* AUCinf: площадь под кривой концентрация-время от момента времени О до бесконечности; определенная как сумма AUClast и отношения Сt к λ, где Сt представляет собой последнюю наблюдавшуюся концентрацию, а λ -конечная константа скорости элиминации, полученная из графика зависимости натурального логарифма концентрации от времени.
* Т1/2: кажущийся конечный период полужизни, рассчитанный как отношение 0.693 к λ.
На основании данных анализа мочи на содержание ниацина и его метаболитов (NUA, MNA и 2-PY) были рассчитаны следующие параметры:
* CumXu: общее количество каждого метаболита, начиная от 0 до 96 часов после введения препарата.
* %Fe: доля каждого метаболита, экскретируемая в моче, по отношению к дозе ниацина после коррекции на базовое восстановление и молекулярный вес, рассчитанная за 96 часов после введения препарата.
* Общий %Fe: общая доля четырех метаболитов за 96 часов после введения.
%Fe для каждого аналита в моче рассчитывался по следующей формуле:
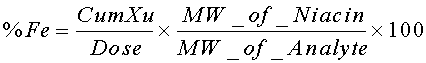
Концентрации ниже предела количественного определения принимались за ноль. Для расчета РК параметров при анализе плазмы использовали фактические времена сбора образцов. Количество ниацина и его метаболитов в моче определяли, умножая концентрацию каждого метаболита на объем мочи, собранной за каждый интервал. Общее количество в моче за 24 часа после введения препарата корректировали на базовую линию, вычитая количество, обнаруженное в моче за 24 часовой интервал до введения. Если результаты каких-либо измерений после введения препарата были меньше базовой линии, то соответствующее количество принимали за ноль. Молекулярные веса ниацина и его метаболитов составляли 123.1, 180.2, 137.1 и 153.1 для ниацина, NUA, MNA и 2-PY, соответственно. Сумма %Fe для четырех аналитов в моче принималась за общий %Fe.
Параметры биоэквивалентности (как описано выше) были рассчитаны с использованием программного обеспечения WinNonlin Linear Mixed Effects Modeling/bioequivalence, Version 5.0.1 (26 июля 2005). Статистический анализ
Статистический анализ рассчитанных выше параметров биодоступности проводили с помощью программы SAS® System for Windows™, версия 8.2. Фармакокинетические параметры в плазме (Сmax, Тmax, Т1/2, AUQigst и AUCinf), их натуральные логарифмы (кроме Тmax и Т1/2) и общая статистика (n, среднее, std, медиана, min, max, CV%) были определены за лечение и за период. Концентрации в плазме ниацина и NUA также суммированы за время и лечение.
При РК анализе ниацина и NUA предполагается, что натуральные логарифмы Сmax и AUQlast соответствуют нормальному распределению и независимы для двух лечений. Данные были обработаны на модели ANOVA со смешанными эффектами с использованием SAS PROC MIXED, причем лечение, период и последовательность принимались за фиксированные эффекты, а субъект в последовательности был случайным эффектом. Отношения Test/REF для Сmax и AUQlast и их соответствующие 90% доверительные интервалы определяли на основании этой модели. Среднее восстановление ниацина и его метаболитов из мочи было рассчитано и просуммировано по лечению и по интервалу. Значения CumXu и %Fe индивидуальных компонентов и общее за 96 часов также было рассчитано и просуммировано по лечению и по интервалу.
90% доверительные интервалы (Cls) для средних отношений Test/REF для общего %Fe были рассчитаны по такой же модели ANOVA, что и при анализе РК в плазме.
Демографические переменные субъектов (возраст, пол, раса, вес, рост и ширина локтя) были просуммированы по полу. Были рассчитаны также среднее, стандартное отклонение (SD), медиана, минимум и максимум для непрерывных демографических переменных.
Результаты
Диспозиция субъектов приведена в Таблице 12. После проверки на соответствие критериям включения и исключения по протоколу в исследовании приняли участие 44 субъекта. Все субъекты приняли, по меньшей мере, одну дозу исследуемого препарата, и 41 из них завершил исследование. 44 субъекта получили медикаменты в периоде 1, в соответствии с рандомизированным лечением по протоколу; 41 субъект получил медикаменты в периоде 2. Прервали испытание 3 субъекта. Субъекты 0012 и 0039 были исключены в периоде 2. Субъект 0038 отозвал свое согласие также в периоде 2. Количество прервавших испытание субъектов укладывается в допустимое 10% выпадение, и было принято решение, что оно не влияет на результаты или выводы из этого испытания.
Из 44 субъектов, принявших участие в испытаниях, было 25 мужчин и 19 женщин. Средний возраст составлял 54.5 лет; средний вес был 169.8 фунтов; средний рост 68.0 дюймов; средняя ширина локтя 2.6 дюймов. 37 человек были белые, 6 черные, один был американским индейцем. Подробная демография приведена в Таблице 13.

а. Анализ биоэквивалентности
При анализе мочи для конвертации веса мочи в объем использовалась плотность 1 г/мл. Это предположение основано на предыдущем исследовании с использованием NIASPAN®, где средняя плотность для 962 образцов составляла 1.009 г/мл, а максимальная плотность, определенная для 962 образцов, была 1.025 г/мл.
Графики средней концентрации ниацина и NUA в плазме, в зависимости от лечения, показаны на Фиг.11 и 12, соответственно. Среднее восстановление в моче представлено на Фиг.13.
b. Содержание NUA в плазме и общее количество, экскретируемое в моче.
В Таблице 14 показаны средние значения (SD) и статистические результаты для двух первичных переменных (Сmax для NUA и общее восстановление в моче ниацина и трех метаболитов) и для NUA AUClast В таблице приводятся результаты анализа BE с и без референсного лечения для субъектов 0001, 0003 и 0014, у которых после введения препарата наблюдались эпизоды рвоты.
У субъекта 0001 началась рвота через 7 часов 20 минут после введения продукта REF в период 2. У субъекта 0003 рвота была через 8 часов 34 минуты и через 9 часов 20 минут после введения продукта REF в период 2, соответственно. У субъекта 0014 рвота была через 11 часов 20 минут после введения продукта REF в период 1. Минимальное время начало рвоты для всех трех субъектов составляло, по меньшей мере, 7 часов 20 минут после введения препарата. Тmax для NUA и ниацина укладывалось в 6 часов после дозировки. Таким образом, было признано, что рвота не влияет на параметры РК этих субъектов.
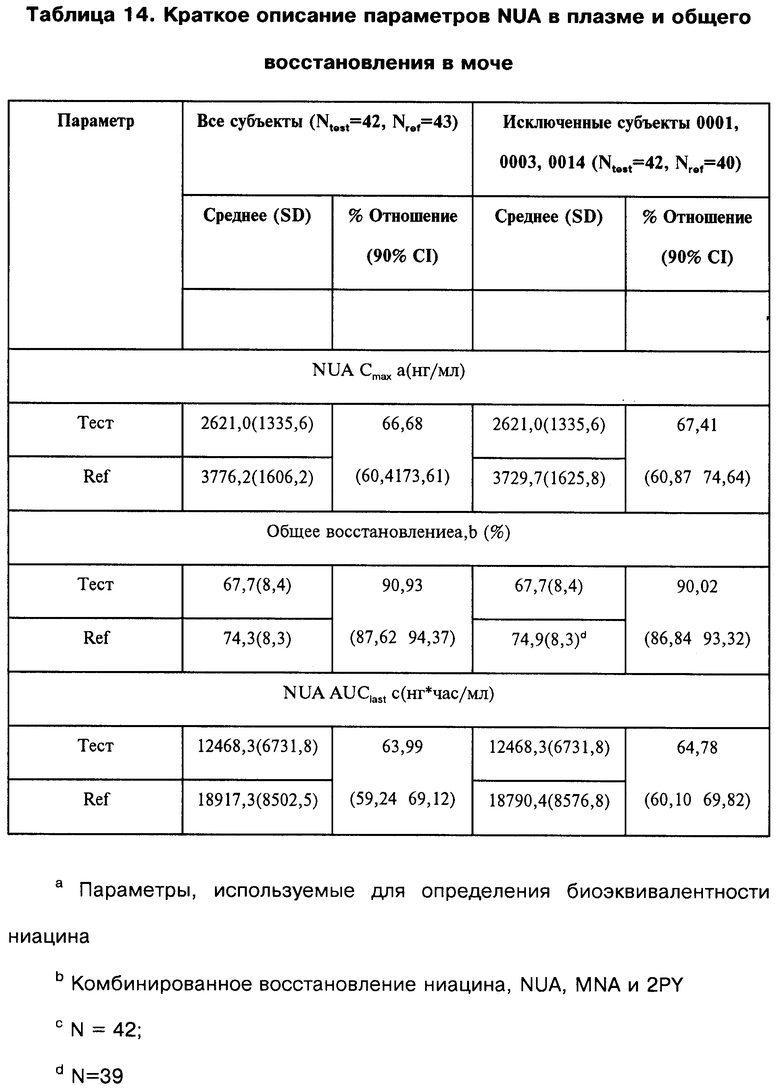
Как видно из приведенной выше таблицы, 90% Cl для среднего отношения Тест/REF для NUA Сmax было вне диапазона биоэквивалентности 80-125%, но 90% Cl для среднего отношения Тест/REF для ниацина и метаболитов, восстановленных в моче, укладывалось в диапазон 80-125%.
Для субъектов 0001, 0003 и 0014 результаты были одинаковы с и без референсного лечения.
Терминальная скорость элиминирования была рассчитана для каждого субъекта по лечению. Среднее NUA T1/2 составляло 3.16 и 3.47 часа, среднее NUA Тmax было 5.55 и 5.80 часов, а среднее NUA AUCinf было 12510.8 и 18980.8 нг·час/мл для Теста и Ref, соответственно.
с. Ниацин в плазме
Средние параметры РК для содержания ниацина в плазме, а также данные их статистического анализа, представлены в Таблице 15. Там приведены также данные анализа BE с и без референсного лечения для субъектов 0001, 0003 и 0014. Средние отношения Тест/REF для Сmax и AUClast ниацина были менее 100%. Соответствующие 90% Cl для отношений не укладывались в 80-125% интервал из-за высокой вариабельности. Для субъектов 0001, 0003 и 0014 результаты были одинаковы с и без референсного лечения.
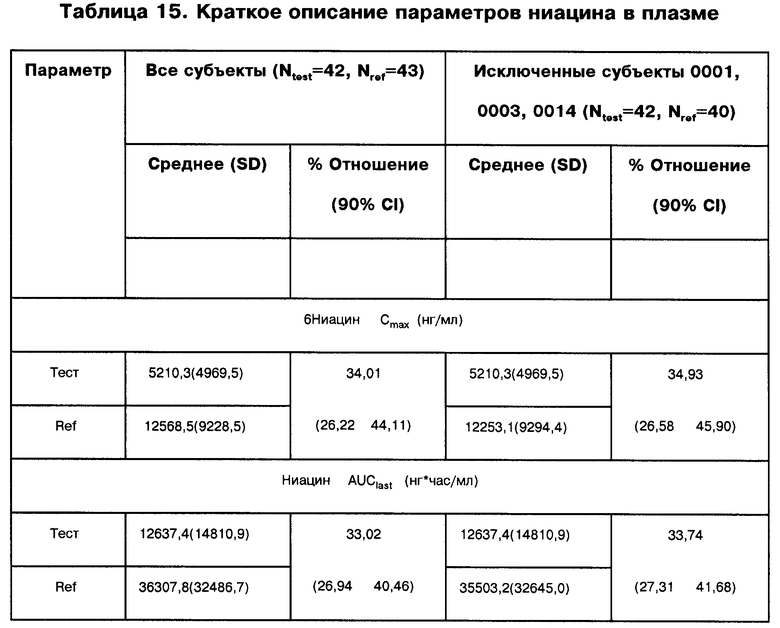
Среднее значение Т1/2 для ниацина было 5.46 и 4.42 часа, среднее Тmax соответствовало 5.56 и 5.55 часов, а среднее AUC,nf было 13987.8 и 35296.6 нг*час/мл для Теста и REF, соответственно.
d. Восстановление в моче индивидуальных аналитов
Средние значения восстановления в моче индивидуальных аналитов приведены в Таблице 16.
Как показано в приведенной выше таблице, среднее восстановление в моче было наибольшим для 2PY, за ним в порядке убывания следовали MNA, NUA и ниацин.
е. Заключение по данным анализа биоэквивалентности
Биоэквивалентность была оценена на основании 90% Cl для средних отношений Тест/Ref NUA Сmax и восстановления в моче ниацина и его метаболитов (Общий %Fe). 90% С1 для среднего отношения Тест/Ref для Общего %Fe укладывалось в требуемый диапазон BE 80-125%, но для NUA Сmax выходило из диапазона биоэквивалентности. 90% Cl для средних отношений Тест/Ref данных поддерживающих измерений, включая NUA AUQlast также выпадают из диапазона 80-125%. Таким образом, содержащие 1000 мг ниацина таблетки ER с переработанным составом (Тест) показывают меньшую скорость абсорбции и сравнимую степень абсорбции, по сравнению с 1000 мг таблетками NIASPAN® ®EF). Тестовое лечение не является биоэквивалентом референсному лечению.
Пример 5
Целью настоящего исследования было определение биоэквивалентности трех препаратов для содержащих 1000 мг ниацина таблеток с пролонгированным высвобождением по изобретению (называемыми здесь таблетками с переработанным препаратом), по сравнению с коммерчески доступными 500 мг таблетками NIASPAN® при введении ниацина в виде однократной дозы 2000 мг.
Схема исследования
Проводилось рандомизированное исследование из одного центра с открытой меткой, с введением однократной дозы и четырехсторонним кроссовером. В эксперименте принимали участие 44 здоровых некурящих добровольцев мужчин и женщин возраста от 40 до 70 лет, включительно. Исключенных из эксперимента не заменяли. Каждый субъект получал одинаковую дозу перорального препарата, содержащего 2000 мг ниацина, четыре раза. Период промывки между дозами составлял, по меньшей мере, 10 дней. Каждый субъект получал две таблетки ER препарата, содержащие 1000 мг ниацина (ERN-1, ERN-2, ERN-3) и четыре 500 мг таблетки NIASPAN®. Каждую дозу запивали 300 мл воды и давали после обедненной жирами легкой закуски, приблизительно, в 22:00 в День 1 каждого периода. Пища субъектов соответствовала предоставленным спонсорами меню. На протяжении эксперимента запрещался прием других медикаментов, витаминов, растительных или пищевых добавок. Образцы крови брали перед введением дозы и с частыми интервалами в течение 24 часов после этого;
мочу собирали в течение 24 часов до и 96 часов после введения дозы. Плазму анализировали на содержание NUA и ниацина. Мочу анализировали на содержание ниацина и его метаболитов: NUA, MNA и 2PY. Субъекты находились в клинике в течение 5-дневного периода каждого исследования. Пищу, которую получали субъекты на протяжении лечения, контролировали на предмет содержания в ней ниацина (завтрак, ланч, обед и вечерняя легкая закуска).
Лекарственные препараты сравнения представляли собой доступные 500 мг препараты NIASPAN® (NSP), содержащие высокоактивные гранулы (ниацин, повидон и гидроксипропилметилцеллюлоза [НРМС]) которые последовательно смешивали со стеариновой кислотой и дополнительным НРМС, после чего прессовали в таблетки.
Тестовые лекарственные препараты представляли собой три различные 1000 мг препарата NIASPAN® (ERN-1, ERN-2 и ERN-3) с переработанным составом, приготовленные в соответствии с приведенной ниже Таблицей 17.
Гранулированный ниацин, METROCEL® K-15M, повидон К90 и стеариновую кислоту взвешивали в соответствии с формулами из Таблицы 17 и помещали в 8-пинтовый смеситель (LB-9322, Petterson Kelly, East Stroudsburg, PA), после чего перемешивали 10 минут. Хорошо перемешанную смесь прессовали в таблетки с помощью пресса BWI Manesty Beta Press (Thomas Eng, Hoffman Estate, IL), работающего со скоростью 500 таблеток в минуту, причем целевая твердость таблеток составляла от 16 до 18 Кр.
Вся еда и напитки не содержали алкоголя и ксантина. Поскольку ниацин может присутствовать и в нормальной диете, пищу, которую получали испытуемые, контролировали на содержание ниацина, так чтобы его поступление с пищей составляло, приблизительно, 25 мг в день, пока субъекты находились в клинике. Каждую дозу давали, приблизительно, в 22:00 сразу после обезжиренной легкой закуски.
Во все дни в течение каждого периода субъекты начинали прием пищи в одно и то же время и находились в клинике на все время исследования. Для каждого периода еда была одинакова, и требовалось, чтобы субъекты съедали свои порции целиком. Завтрак, ланч, обед и вечерняя легкая закуска начинались ежедневно, приблизительно, в 07:00, 12:00, 17:00 и 21:45, соответственно. Фактическое время еды или приема легкой закуски для каждого субъекта было спланировано в соответствии с фактическим режимом введения препаратов. От субъектов требовали выпивать, минимум, 720 мл воды в День 1 и 1440 мл воды в День 1, 2, 3, 4 и 5, помимо 300 мл воды, выпитой при приеме препарата в День 1.
В День 1 были поданы обед и вечерняя закуска. В Дни 1-5 подавали завтрак, ланч, обед и вечернюю закуску. Последняя съедалась за 15 минут непосредственно перед введением препарата в День 1 каждого периода. В День 6 каждого указанного периода еда не подавалась, поскольку субъекты выписывались из клиники после завершения всех клинических процедур.
Оценка фармакокинетики
а. Сбор и анализ плазмы
Образцы крови получали за 30 минут до введения препарата (то есть, пре-доза) и через 1, 2, 3, 4, 4.5, 5, 6, 7, 8, 9, 10, 11, 12, 14, 16 и 24 часа после введения в течение каждого периода. Образцы брали в зоне тестирования, субъекты в это время сидели, выпрямившись в кресле. Кровь собирали в 7 мл пробирки VACUTAINER с гепарином натрия и охлаждали на водяной бане с размолотым льдом не менее 5 минут после сбора. Затем образцы центрифугировали при 4 градусов и, приблизительно, 3000 об/мин в течение 15 минут, чтобы отделить плазму. Фракцию плазмы переносили в две охлаждаемые предварительно помеченные полипропиленовые пробирки. До анализа образцы хранили при температуре, приблизительно, -70°С. Биоанализ содержания ниацина и NUA в плазме проводили методом хроматографии HPLC с детекцией MS/MS.
Данные об указанных концентрациях получали для одной и той же инъекции. Нижний предел количественного определения (lower limit of quantitation (LLQ)) для ниацина и NUA составлял 2 нг/мл в плазме. В каждом анализе оценивались также образцы, используемые для контроля качества.
b. Сбор и анализ мочи
Мочу собирали за следующие интервалы: от -24 до -18, от -18 до -12, от -12 до -6 и от -6 до 0 часов (до введения дозы); от 0 до 6, от 6 до 12, от 12 до 18, от 18 до 24, от 24 до 48, от 48 до 72 и от 72 до 96 часов после введения дозы (всего было 11 сборов).
Собранную мочу переносили в пластиковые контейнеры с плотно пригнанными крышками. На протяжении периода сборки мочу охлаждали в холодильнике или на бане с ледяной водой. Коллекторы для сборки были помечены с указанием номера и инициалов субъекта, интервала сборки и номера протокола. Регистрировали общий вес мочи, собранной в течение каждого интервала, округляя до ближайшей десятой доли грамма (например, до 100.1 г). Регистрировали также дату (даты) и время начала и прекращения сбора мочи за каждый интервал. Две аликвоты (приблизительно, 2.5 мл каждая) из каждого интервала сборки переносили в две соответственно помеченные полипропиленовые пробирки. Образцы для анализа помечали, чтобы идентифицировать Kos, номер протокола, номер субъекта, дату, интервал сборки, день исследования и период. Аликвоты хранили, приблизительно, при -20°С до готовности к отправке. Для определения плотности в клинике брали еще одну аликвоту.
Образцы мочи анализировали на содержание ниацина, NUA, MNA и 2-PY. Биоанализ концентраций ниацина, NUA, MNA и 2-PY в моче проводили методом хроматографии HPLC с MS/MS детекцией. Данные о концентрациях ниацина и NUA получали для одной и той же инъекции. В моче значения LLQ составляли 20 нг/мл для ниацина и 200 нг/мл для NUA. LLQ для MNA и 2-PY составляли 500 нг/мл и 2500 нг/мл, соответственно. В каждом анализе оценивались также образцы, используемые для контроля качества.
с.Фармакокинетические параметры в плазме и восстановление в моче
В фармакокинетический (РК) анализ включали данные от субъектов, у которых удалось получить достаточно информации для расчета РК параметров, по меньшей мере, для одного лечения. После начала каждого лечения для каждого субъекта рассчитывали следующие РК параметры:
- Сmax: максимальная наблюдавшаяся концентрация
- Тmax: время достижения максимальной наблюдавшейся концентрации
- AUC0-last: площадь под кривой концентрация-время от момента времени 0 до момента получения последней измеримой концентрации, определенная по линейной формуле трапеций.
Для NUA были получены дополнительные РК параметры:
- AUC0-inf: площадь под кривой концентрация-время от момента времени 0 до бесконечности; определенная как сумма AUC0-last и отношения Сt к Kcl, где Сt представляет собой последнюю наблюдавшуюся концентрацию, а Kcl - конечная константа скорости элиминации.
- Т1/2: конечный период полужизни, рассчитанный как отношение 0.693 к Kcl.
На основании данных анализа мочи на содержание ниацина, NUA, MNA и 2-PY были рассчитаны следующие параметры:
- CumXu: индивидуальное восстановление в моче каждого аналита (то есть, количество каждого аналита, восстановленное в моче).
- Fe: фракция, экскретируемая в моче и рассчитанная по формуле:
- Общий %Fe: общее восстановление ниацина, NUA, MNA и 2-PY.
Концентрации ниже LLQ принимались за ноль, и в ходе анализа использовали фактические времена сбора образцов. Индивидуальные графики концентрации ниацина и NUA в плазме сгенерировали с помощью программного обеспечения WinNonlin Professional Network Edition, Version 4.1. Графики среднего содержания ниацина и NUA в плазме и данные по восстановлению в моче были получены программами WinNonlin 4.1 и Microsoft®) Excel 2000. Фармакокинетические параметры в плазме для каждого профиля определяли программой WinNonlin. Наклон конечной фазы и кажущийся период полужизни для ниацина в плазме вычислен не был из-за слишком малого количества оцененных концентраций ниацина в каждом профиле плазмы и отсутствия ясно выраженной конечной фазы.
Все фармакокинетические параметры в моче определяли с помощью программ WinNonlin 4.1 и Excel 2000. Количество каждого аналита в моче определяли, умножая концентрацию аналита на объем мочи, собранной для каждого интервала; восстановленное в моче количество за 24-часовой интервал после введения препарата затем было скорректировано на базовое восстановление путем вычитания количества, содержащегося в 24-часовом интервале предварительной дозировки.
Молекулярные веса аналитов были следующие: ниацин, 123.1, NUA, 180.2; MNA, 137.1; 2PY, 153.1. Общий процент восстановленной дозы получали, просуммировав проценты восстановления индивидуальных аналитов.
Статистический анализ
Статистический анализ демографии проводили с помощью SAS System for Windows™, version 8.02. Для непрерывных демографических были рассчитаны среднее значение, стандартное отклонение (3D), медиана, минимум и максимум переменных.
Параметры биоэквивалентности определяли с помощью мастера биоэквивалентности, входящего в препарат программы WinNonlin 4.1 и натуральных логарифмов данных. Модель включает последовательность, субъекты в последовательности, период и лечение.
При анализе биоэквивалентности использовались классические 90% доверительные интервалы (Cl) отношений теста к сравнению (ERN-1/NSP, ERN-2/NSP, ERN-3/NSP) для средних наименьших квадратов, при этом данные были переведены в соответствующие натуральные логарифмы. Различные лечения считались биоэквивалентными, если 90% Cl укладывался в интервал от 80 до 125%. При определении биоэквивалентности использовали параметры Сmax для NUA в плазме и общее количество ниацина и метаболитов, экскретируемых в моче. Доверительные интервалы были определены также для ниацина в плазме (Сmax и AUC0-last) и индивидуальных %Fe (ниацин, NUA, MNA, 2PY) для мочи.
Результаты
В исследовании приняли участие 44 здоровых, некурящих мужчин и женщин, возраста от 40 до 70 лет, включительно, и прошедших проверку на соответствие критериям включения и исключения по протоколу. Субъекты были отобраны на основании неиспользования табака, по меньшей мере, 120 дней до получения первой дозы и отсутствия клинически значимых данных в их медицинской истории, а также физического экзамена, электрокардиограммы (ECG) и данных клинического лабораторного анализа.
Из 44 отобранных субъектов 41 завершил испытание. Мужчин было 28 человек, а женщин 16. Средний возраст составлял 51 год, средний вес был 171 фунт, средний рост 68 дюймов. 36 субъектов были белыми, шесть черными, а два латиноамериканцами. Подробная демография приведена в Таблице 18.
а. Анализ биоэквивалентности
43 субъекта предоставили плазму и мочу для референсного лечения (NSP). 42 субъекта предоставили плазму и мочу для тестового лечения ERN-1 и ERN-2, а 41 - для тестового лечения ERN-3. Для построения таблиц средних, графиков средних, индивидуальных графиков и листингов концентраций использовали номинальные значения времени. В РК-анализе были использованы следующие правила:
Для времени взятия образцов от 1 до 10 часов (включительно): в случае отклонения от 5 минут и более использовались фактические значения времени. При отклонении менее 5 минут использовали номинальные значения времени.
Для времени взятия образцов более 10 часов: в случае отклонения от 10 минут и более использовались фактические значения времени. При отклонении менее 10 минут использовали номинальные значения времени.
Статистическое описание фармакокинетических параметров ниацина и NUA в плазме показано в Таблице 18а.
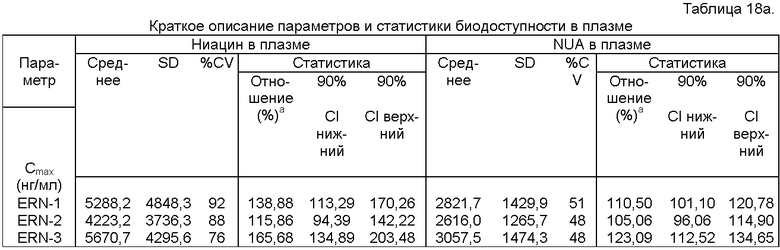
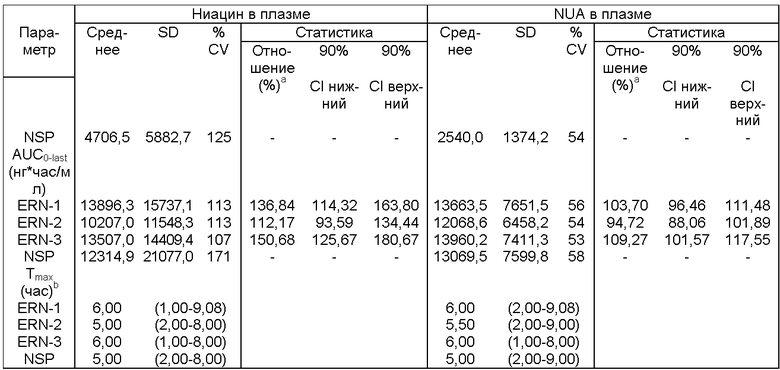
Каждое лечение включало 2000 мг ниацина, N=42 для ERN-1 и ERN-2, 41 для ERN-3 и 43 для NSP. NSP означает референсное лечение.
а Отношение среднего наименьших квадратов для натуральных логарифмов от Сmax и AUCo-last для ниацина и NUA.
b Медиана и среднее представлены в для Тmax
Средние профили содержания ниацина и NUA в плазме показаны на Фиг.14-17.
b. Данные для плазмы
Ниацин в плазме
Значения пре-дозы для всех субъектов было ниже LLQ. Через 4.5 - 12 часов после введения дозы препарата в каждом лечении у всех субъектов наблюдалось измеряемое содержание ниацина в плазме.
Среднее Сmax для ниацина было 5288, 4223, 5671 и 4707 нг/мл для ERN-1, ERN-2, ERN-3 и NSP, соответственно. Среднее AUC0-last было 13896, 10207, 13507 и 12315 нг*час/мл для ERN-1, ERN-2, ERN-3 и NSP, соответственно. Медиана Тmax для ниацина составляла 6,0 часов для ERN-1 и ERN-3 и 5 часов для ERN-2 и NSP.
Отношения натуральных логарифмов Сmax и AUC0-last было более 100% для всех трехтестовых лечений, по сравнению с NSP. Отношения для Сmax ниацина составляли 139%, 116% и 166% для ERN-1, ERN-2 и ERN-3, соответственно. Отношения для AUC0-last для ниацина составляли 137%, 112% и 151% для ERN-1, ERN-2 и ERN-3, соответственно. 90% Cl для натурального логарифма от Сmax составляли 113-170%, 94-142% и 135-203% для ERN-1, ERN-2 и ERN-3, соответственно. 90% Cl для натурального логарифма от AUC0-last составляли 114-164%, 94-134% и 126-181% для ERN-1, ERN-2 и ERN-3, соответственно. 90% Cl для Сmax и AUC0-last находились вне диапазона эквивалентности 80-125%.
Данные для ниацина были высоковариабельными, CV колебалось от 76 до 171% для Сmax и AUCo-last во всех четырех лечениях.
Содержание никотинмочевой кислоты в плазме
У трех субъектов обнаруживались положительные концентрации NUA на уровне пре-дозы. Это были субъект 0028 (Период 2, ERN-1, концентрация 4.47 нг/мл), субъект 30 (Период 2, ERN-3, концентрация 2,75 нг/мл) и субъект 33 (Период 2, ERN-2, концентрация 3,26 нг/мл). На эти профили в плазме коррекции не делалось, поскольку эти предварительные концентрации составляли всего около 0,24%, 0,06% и 0,53% от значения Сmax для субъектов 0028, 0030 и 0033, соответственно. У всех субъектов измеримые концентрации NUA обнаруживались через 3-16 часов после каждого лечения. Средние значения Сmax составляли 2822, 2616, 3058 и 2540 нг/мл для ERN-1, ERN-2, ERN-3 и NSP, соответственно. Средние значения NUA AUC0-last составляли 13664, 12069, 13960 и 13070 нг*час/мл для ERN-1, ERN-2, ERN-3 и NSP, соответственно. Медиана NUA Тmax составляла 6 часов для ERN-1 и ERN-3, 5,5 часов для ERN-2 и 5,0 часов для NSP.
Конечная скорость элиминирования была рассчитана для каждого субъекта и лечения, когда только это было возможно. Средняя t1/2 составляла 3,4 часов для ERN-1, ERN-2 и ERN-3, соответственно, и 3,1 часа для NSP. Среднее значение AUCinf составляло 13602, 11913, 14136 и 13009 нг*час/мл для ERN-1, ERN-2, ERN-3 и NSP, соответственно.
Отношения натуральных логарифмов Сmax и AUClast были более 100% для лечений ERN-1 и ERN-3, по сравнению с NSP. Однако, для ERN-2 отношение Стад было больше 100%, a AUC0-last меньше 100%. Отношения NUA Сmax составляли 111%, 105% и 123% для ERN-1, ERN-2 и ERN-3, соответственно. Отношения для NUA AUC0-last составляли 104, 95% и 109% для ERN-1, ERN-2 и ERN-3, соответственно. 90% Cl для натуральных логарифмов NUA Сmах составляли 101-121%, 96-115% и 113-135% для ERN-1, ERN-2 и ERN-3, соответственно. 90% Сl для натуральных логарифмов NUA AUC0-last составляли 96-111%, 88-102% и 102-118% для ERN-1, ERN-2 и ERN-3, соответственно. 90% Cl для Сmax и AUC0-last укладывались в интервал биоэквивалентности 80-125% для ERN-1 и ERN-2. В случае ERN-3 90% Cl для Сmax был вне диапазона 80-125%, но этот параметр для AUC0-last укладывался в указанный интервал.
Данные для NUA были высоковариабельны, значения CV составляли 48-58% для Сmax и AUC0-last Для всех четырех видов лечения.
с. Восстановление ниацина и метаболитов в моче
Для преобразования веса мочи в объем использовалось значение плотности 1. Оно основано на данных предыдущих исследований с участием NIASPAN®, когда средняя плотность, измеренная для 962 образцов, составляла 1,009 г/мл, а максимальная плотность составляла 1,025 г/мл.
Среднее восстановление в моче показано в Таблице 19 и отображено на Фиг.18.
Каждое лечение включало 2000 мг ниацина, N=42 для ERN-1, ERN-2 и NSP, и 41 для ERN-3.
NSP представляет собой референсное лечение.
а Отношение средних наименьших квадратов натуральных логарифмов восстановления ниацина, NUA, MNA, 2РY и общего восстановления.
b Комбинированное восстановление ниацина, NUA, MNA и 2PY.
с Предполагает биоэквивалентность (то есть, 90% Cl укладывается в 80-125% для натуральных логарифмов от восстановления MNA, 2PY и общего восстановления). Общее восстановление
Общее восстановление ниацина в моче, выраженное как сумма восстановлений ниацина, NUA, MNA и 2PY, составляло 67,14% для NSP и 63,91%, 63,44% и 66,16% для ERN-1, ERN-2 и ERN-3, соответственно. Отношение средних наименьших квадратов логарифмов %Fe для общего восстановления составляло 95%, 94% и 98%, соответственно, для ERN-1, ERN-2 и ERN-3. 90% Cl для отношений составлял 91-99%, 90-98% и 94-102%, соответственно, для ERN-1, ERN-2 и ERN-3, показывая, что общее количество, экскретируемое в моче тремя тестовыми препаратами, было эквивалентно NSP при условии доверительного интервала 80-125%.
d. Заключения по анализу биоэквивалентности
Фармакокинетический анализ данных для NUA показывает, что пиковая экспозиция, измеренная по С^, была выше для всех трех тестовых препаратов (ERN-1, ERN-2, ERN-3), по сравнению с NSP, на 5-23%. 90% Cl для отношений средних наименьших квадратов для натуральных логарифмов Сmax укладывалось в интервал 80-125% для ERN-1 и ERN-2, означая, что эти препараты биоэквивалентны NSP по отношению к NUA. В случае ERN-3 90% Cl был вне диапазона 80-125% для Сmax, показывая, что препарат ERN-3 не биоэквивалентен NSP.
Среднее общее количество ниацина и метаболитов, экскретируемых в моче, составляло 63-66% для трех тестовых препаратов и 67% для NSP. Экскретируемая фракция была минимальна для родительского соединения ниацин, а за ним по возрастанию следовали NUA, MNA и 2PY (37,2-40,0%). Общее измеренное восстановление было на 2-6% меньше для тестовых препаратов, по сравнению с NSP. 90% Cl для отношений средних наименьших квадратов для натуральныхлогарифмов общего восстановления укладывалось в интервал 80-125% и, таким образом, для всех трех тестовых препаратов было эквивалентно, по сравнению с NSP.
Соответственно, один аспект изобретения включает содержащие 1000 мг ниацина фармацевтические композиции с пролонгированным высвобождением и переработанным препаратом, которые при введении субъектам в исследовании биоэквивалентности, в которых сравниваются однократная доза из четырех 500 мг таблеток NIASPAN® и одна доза указанной переработанной, содержащие 1000 мг ниацина фармацевтической композиции с пролонгированным высвобождением, дают 90% Сl натурального логарифма отношения соответствующих параметров биодоступности, который укладывается в интервал 80%-125%.
В соответствии с предпочтительным аспектом, параметры биодоступности соответствуют NUA Сmax (нг/мл) и Общему восстановлению или Сmax ниацина (нг/мл) и AUC ниацина.
Пример 6
Целью настоящего исследования было определение биоэквивалентности (BE) покрытых и непокрытых содержащих 1000 мг ниацина таблеток с пролонгированным высвобождением по изобретению (называемыми здесь таблетками с переработанным препаратом), при введении в виде однократной дозы 2000 мг.
Схема исследования
Проводилось рандомизированное исследование из одного центра с открытой меткой, с введением однократной дозы и двусторонним кроссовером. В эксперименте принимали участие 44 здоровых некурящих добровольца мужчин и женщин возраста от 40 до 70 лет, включительно. Исключенных из эксперимента не заменяли. Каждый субъект получал два ниацин-содержащих препарата, Тест и Ref, в виде одной и той же однократной дозы 2000 мг, которые давали им в два разных сеанса. Период промывки между дозами составлял, по меньшей мере, 10 дней. Тестовый продукт представлял собой две таблетки покрытого оболочкой 1000 мг переработанного ниацина с пролонгированным высвобождением, а референсный продукт (продукт сравнения) ®EF) представлял собой две таблетки непокрытого 1000 мг ниацина с пролонгированным высвобождением. Каждую дозу запивали 240 мл воды и давали после обедненной жирами легкой закуски, приблизительно, в 22:00 в День 1 каждого периода. В течение 6-дневного периода исследования (день 1 - день 5) субъекты находились в центре, где проводилось испытание, их пицца соответствовала предоставленным спонсорами меню. На протяжении эксперимента запрещался прием других лекарственных препаратов, витаминов, растительных или пищевых добавок.
Последовательные образцы крови собирали в течение промежутка времени, начиная от 30 минут до введения дозы и в течение 24 часов после ее введения с интервалами: -30 мин (пре-доза), 1, 2, 3, 4, 4.5, 5, 6, 7, 8, 10, 12, 14, 16 и 24 часа (пост-дозы). Мочу собирали в течение промежутка времени, начиная от 24 часа до введения дозы и в течение 96 часов после ее введения за интервалы: от -24 до -18, от -18 до -12, от -12 до -6 и от -6 до О часов (пре-доза); от 0 до 6, от 6 до 12, от 12 до 18, от 18 до 24, от 24 до 48, от 48 до 72 и от 72 до 96 часов (пост-доза). Плазму анализировали на содержание ниацина и никотин-мочевой кислоты (NUA). Мочу анализировали на содержание ниацина и его метаболитов: NUA, MNA и 2-PY.
Субъекты получали пищу в одно и то же время каждый день, которые они проводили в клинике в течение каждого периода. Для каждого периода еда была одинакова, и требовалось, чтобы субъекты съедали свои порции целиком. Завтрак, ланч, обед и вечерняя легкая закуска начинались ежедневно, приблизительно, в 07:00, 12:00, 17:00 и 21:45, соответственно. Фактическое время еды или приема легкой закуски для каждого субъекта было спланировано в соответствии с фактическим режимом введения препаратов. От субъектов требовали выпивать, минимум, 720 мл воды в День 1 и 1440 мл воды в День 1-5, помимо 240 мл воды, выпитой при приеме препарата в День 1.
В День 1 были поданы обед и вечерняя закуска. В Дни 1 - 5 подавали завтрак, ланч, обед и вечернюю закуску. Последняя съедалась за 15 минут непосредственно перед введением препарата в День 1 каждого периода.
В День 6 второго периода еды не подавалось, поскольку субъекты выписывались из клиники после завершения всех клинических процедур.
Оценка фармакокинетики
а. Сбор и анализ плазмы
Последовательные образцы крови были собраны для каждого периода в промежуток времени, начиная от 30 минут до введения препарата, и заканчивая 24 часами после этого (15 образцов не лечение). Каждый образец брали в одну 17 мл пробирку VACUTAINER с гепарином натрия и охлаждали на водяной бане с измельченным льдом не менее 5 минут после сбора. Затем образцы центрифугировали при 4 градусов и, приблизительно, 3000 об/мин в течение 15 минут, чтобы отделить плазму. Каждый образец плазмы разделили на две аликвоты, А и В, и перенесли в две предварительно охлажденные, соответственно помеченные полипропиленовые пробирки. В этих пробирках образцы замораживали и хранили при -20°С.
Концентрации ниацина и NUA анализировали с помощью валидированной комбинации методов жидкостной хроматографии и масс-спектрометрии (LC/MS/MS). Данные об указанных концентрациях получали для одной и той же инъекции. Нижний предел количественного определения для ниацина и NUA составлял 2 нг/мл в плазме. В каждом анализе оценивались также образцы, используемые для контроля качества.
b. Сбор и анализ мочи
Мочу собирали за следующие интервалы: от -24 до -18, от -18 до -12, от -12 до -6 и от -6 до 0 часов (до введения дозы); от 0 до 6, от 6 до 12, от 12 до 18, от 18 до 24, от 24 до 48, от 48 до 72 и от 72 до 96 часов после введения дозы (всего было 11 сборов).
Собранную мочу переносили в пластиковые контейнеры с плотно пригнанными крышками. На протяжении периода сборки мочу охлаждали в холодильнике или на бане с ледяной водой. Коллекторы для сборки были помечены с указанием номера и инициалов субъекта, интервала сборки и номера протокола. Пустые контейнеры взвешивали, округляя до ближайшей десятой доли грамма (например, до 100,1 г), эти данные записывали на контейнере и документировали в лабораторных журналах. В конце каждого интервала общий вес контейнера и собранной мочи измеряли и округляли также до ближайшей десятой доли грамма. Вес мочи определяли, вычитая вес пустого контейнера из общего веса контейнера плюс мочи. В некоторых случаях объем мочи за период сборки превышал вместимость контейнера, в таких случаях для завершения сбора мочи требовался второй контейнер. Регистрировали также дату (даты) и время начала и прекращения сбора мочи за каждый интервал. Две аликвоты (приблизительно, 2,5 мл каждая) из каждого интервала сборки переносили в две соответственно помеченные полипропиленовые пробирки. Если для конкретного интервала сборки требовалось более одного контейнера, перед взятием аликвот мочу из обоих контейнеров смешивали. Образцы затем замораживали и хранили при -20°С до анализа.
Концентрации в моче ниацина, NUA, MNA и 2-PY анализировали с помощью валидированной комбинации методов жидкостной хроматографии и масс-спектрометрии (LC/MS/MS). Данные о концентрациях ниацина и NUA получали для одной и той же инъекции, а данные о концентрациях MNA и 2-PY получали для другой инъекции. В моче значения LLQ составляли 20 нг/мл для ниацина и 200 нг/мл для NUA. LLQ для MNA и 2-PY составляли 500 нг/мл и 2500 нг/мл, соответственно. В каждом анализе оценивались также образцы, используемые для контроля качества.
с. Фармакокинетические параметры в плазме и восстановление в моче
В фармакокинетический (РК) анализ включали данные от субъектов, у которых удалось получить достаточно информации для расчета РК параметров, по меньшей мере, для одного лечения. После начала каждого лечения для каждого субъекта рассчитывали следующие РК параметры для ниацина и NUA в плазме:
* Сmax: максимальная наблюдавшаяся концентрация
* Тmax: время достижения максимальной наблюдавшейся концентрации
* AUClast: площадь под кривой концентрация-время от момента времени
0 до момента получения последней измеримой (ненулевой) концентрации, определенная по линейной формуле трапеций.
* AUCinf: площадь под кривой концентрация-время от момента времени 0 до бесконечности, определенная как сумма AUClast, и Сt за λz, где Сt представляет собой последнюю наблюдавшуюся концентрацию, а λz - конечная константа скорости элиминации, полученная из графика зависимости натурального логарифма концентрации от времени.
* Т1/2: кажущийся конечный период полужизни, рассчитанный как отношение 0,693 к λz.
На основании данных анализа мочи на содержание ниацина и его метаболитов (NUA, MNA и 2-PY) были рассчитаны следующие параметры:
* CumXu: общее количество каждого метаболита, начиная от 0 до 96 часов после введения препарата.
* %Fe: доля каждого метаболита, экскретируемая в моче, по отношению к дозе ниацина после коррекции на базовое восстановление и молекулярный вес, рассчитанная за 96 часов после введения препарата.
* Общий %Fe: общая доля четырех метаболитов за 96 часов после введения.
%Fe для каждого аналита в моче рассчитывался по следующей формуле:
Концентрации ниже предела количественного определения принимались за ноль. Количество ниацина и его метаболитов в моче определяли, умножая концентрацию каждого метаболита на объем мочи, собранной за каждый интервал. Общее количество в моче за 24 часа после введения препарата корректировали на базовую линию, вычитая количество, обнаруженное в моче за 24-часовой интервал до введения. Если результаты каких-либо измерений после введения препарата были меньше базовой линии, то соответствующее количество принимали за ноль. Молекулярные массы ниацина и его метаболитов составляли 123,1, 180,2, 137,1 и 153,1 для ниацина, NUA, MNA и 2-PY, соответственно. Сумма %Fe для четырех аналитов в моче принималась за общий %Fe.
Параметры биодоступности (как описано выше) были рассчитаны с использованием программного обеспечения WinNonlin Linear Mixed Effects Modeling/bioequivalence, Version 5.0.1 (26 июля 2005).
Статистический анализ
Статистический анализ рассчитанных выше параметров биодоступности проводили с помощью программы SAS® System for Windows™, версия 8.2.
Фармакокинетические параметры в плазме (Сmax, Тmax, Т1/2, AUQlast и AUCinf), их натуральные логарифмы (кроме Тmax) и общая статистика (n, среднее, std, медиана, min, max, CV%) были определены за лечение и за период. Концентрации в плазме ниацина и NUA также суммированы за время и лечение.
При РК анализе ниацина и NUA предполагалось, что натуральные логарифмы Сmax и AUQlast соответствуют нормальному распределению и независимы для двух лечений. Данные были обработаны на модели ANOVA со смешанными эффектами с использованием SAS PROC MIXED, причем лечение, период и последовательность принимались за фиксированные эффекты, а субъект в последовательности был случайным эффектом. Отношения Test/REF для Сmax и AUQlast и их соответствующие 90% доверительные интервалы определяли на основании этой модели.
Среднее восстановление ниацина и его метаболитов из мочи было рассчитано и просуммировано по лечению и по интервалу. Значения CumXu и %Fe индивидуальных компонентов и общее за 96 часов после введения также было рассчитано и просуммировано по лечению.
90% доверительные интервалы (Cl) для средних отношений Test/REF для общего %Fe были рассчитаны по такой же модели ANOVA, что и при анализе РК в плазме.
Демографические переменные субъектов (возраст, пол, раса, вес, рост, размер и ширина локтя, а также ВМI) были просуммированы по полу. Были рассчитаны также среднее, стандартное отклонение (SD), медиана, минимум и максимум для непрерывных демографических переменных.
Результаты
Распределение субъектов приведено в Таблице 20. После проверки на соответствие критериям включения и исключения по протоколу в исследовании приняли участие 44 субъекта. Все субъекты приняли, по меньшей мере, одну дозу исследуемого препарата, и 42 из них завершил исследование. 44 субъекта получили лекарственные препараты в периоде 1, в соответствии с рандомизированным лечением по протоколу; 42 субъекта получили лекарственные препараты в периоде 2. Прервали испытание 2 субъекта. Количество прервавших испытание субъектов укладывается в допустимое 10% выпадение, и было принято решение, что оно не влияет на результаты или выводы из этого испытания.
Из 44 субъектов, принявших участие в испытаниях, было 20 мужчин и 24 женщины. Средний возраст составлял 53,1 лет; средний вес был 161,5 фунтов; средний рост 65,6 дюймов; средняя ширина локтя 2,7 дюйма, средний BMI составлял 26,3 кг/м2. Размер ворота определяли как маленький, средний и большой. У девяти человек был маленький размер, у 20 человек - средний, и у 15 субъектов - большой. 38 человек были латиноамериканцы, 4 белые, два черные. Подробные демографические данные приведены в Таблице 21.
ность
а. Анализ биоэквивалентности
Данные от 42 субъектов в тестовом лечении и 44 субъектов в лечении REF были проанализированы с целью определения биоэквивалентности. Во всех анализах использованы фактические времена относительно времени дозировки.
При анализе мочи для конвертации веса мочи в объем использовалась плотность 1 г/мл. Это предположение основано на предыдущем исследовании с использованием NIASPAN®, где средняя плотность для 962 образцов составляла 1,009 г/мл, а максимальная плотность, определенная для 962 образцов, была 1,025 г/мл.
Графики средней концентрации ниацина и NUA в плазме, в зависимости от лечения, показаны на Фиг.19-22, соответственно. Среднее восстановление в моче представлено на Фиг.23.
b. Содержание NUA в плазме и общее количество, экскретируемое в моче.
Первичные переменные, используемые для оценки биоэквивалентности ниацина, были определены как Сmax для NUA и общее восстановление в моче ниацина и трех метаболитов (NUA, MNA и 2PY).
В Таблице 22 показаны средние значения (SD) и статистические результаты для этих двух переменных. Приведены также средние значения (SD) и статистические результаты для NUA AUClast
Как видно из приведенной выше таблицы, 90% Cl для натурального логарифма отношений Теста к сравнению для первичных переменных BE, NUA Сmax и общего восстановления ниацина и метаболитов укладывалось в диапазон 80-125%. Отношение Теста к сравнению для натурального логарифма значения параметра NUA AUGlast также было в интервале 80-125%.
Конечная скорость элиминирования была рассчитана для каждого субъекта по лечению. Среднее NUA T1/2 составляло 3,16 и 3,04 часа, среднее NUA Tmax было 4,90 и 4,80 часа, а среднее NUA AUCint было 10914,7 и 11770,6 нг*час/мл для Теста и Ref, соответственно.
с. Ниацин в плазме
Средние параметры РК для содержания ниацина в плазме, а также данные их статистического анализа, представлены в Таблице 23. Отношения Тест/REF для натуральных логарифмов Сmax и AUClast ниацина были менее 100%. 90% Cl для отношений натурального логарифма Сmax и AUGlast ниацина не укладывались в интервал 80-125% из-за высокой вариабельности.
Среднее значение T1/2 для ниацина было 4,73 и 2,94 часа, среднее Тmax соответствовало 4,68 и 4,64 часов, а среднее AUCinf было 11553,1 и 16134,3 нг*час/мл для Теста и REF, соответственно.
а. Восстановление в моче индивидуальных аналитов
Средние значения восстановления в моче индивидуальных аналитов приведены в Таблице 24.
Среднее восстановление в моче было наибольшим для 2PY, за ним в порядке убывания следовали MNA, NUA и ниацин.
е. Заключение по данным анализа биоэквивалентности
Биоэквивалентность была оценена на основании 90% Cl для средних отношений Тест/Ref NUA Сmax и восстановления в моче ниацина и его метаболитов (Общий %Fe). 90% Cl для средних отношений Тест/Ref для натуральных логарифмов скорости (NUA Сmax) и степени (общий %Fe в моче) поглощения ниацина укладывались в требуемый диапазон BE 80-125% и показывают, что тестовый препарат и препарат сравнения биоэквивалентны. 90% Cl для NUA AUQlast также укладываются в интервал 80-125%, поддерживая заключение по BE.
Для Cmax и AUQlast ниацина верхние пределы 90% Cl средних отношений Test/REF укладываются в интервал биоэквивалентности, и нижние пределы находятся очень близко к нижнему пределу интервала биоэквивалентности, 80%.
Пример 7
Для 1000 мг препаратов изобретения, проанализированных в Примерах 4, 5 и 6, среднее значение Сmax для NUA (нг/мл), общее восстановление в моче (%), Сmax для ниацина (нг/мл) и AUC для ниацина представлены в Таблице 25 ниже (исключая ERN-3).
Соответственно, один аспект настоящего изобретения относится к содержащим 1000 мг ниацина фармацевтическим композициям с пролонгированным высвобождением, которые при введении нуждающемуся в этом пациенту в виде однократной дозы, состоящей из двух таблеток по 1000 мг, обеспечивают профиль в плазме in vivo с 90% Cl для натурального логарифма отношения, укладывающегося в интервал от 80% до 125%, по меньшей мере, для следующих параметров биодоступности:
(a) NUA Cmax 2601,8 нг/мл;
(b) общее восстановление ниацина в моче 60,5%;
(c) Сmax ниацина 4958,9 нг/мл; и
(d) AUC ниацина 12414,5 нг/мл.
Таблица 25а далее иллюстрирует верхний и нижний пределы отобранных параметров биодоступности из Таблицы 25 с учетом стандартной ошибки (показана в скобках). В частности, для расчета нижнего предела было найдено нижнее среднее значение из Примеров 4, 5 и 6 выше для каждого параметра, идентифицированного выше в Таблице 25 и затем из этого среднего значения вычли две стандартные ошибки. Для расчета стандартной ошибки стандартное отклонение делят на квадратный корень из размера выборки (например, 1430/V44=326). Аналогично, верхний предел представляет наибольшее среднее из примеров 4, 5 и 6 для каждого параметра плюс две стандартные ошибки.
Соответственно, один вариант осуществления настоящего изобретения относится к содержащим 1000 мг ниацина фармацевтическим композициям с пролонгированным высвобождением, которые при введении нуждающемуся в этом пациенту в виде однократной дозы, состоящей из двух таблеток, содержащих по 1000 мг ниацина, обеспечивают профиль в плазме in vivo с 90% Cl для натурального логарифма отношения, укладывающегося в интервал от 80% до 125%, по меньшей мере, для следующих параметров биодоступности:
(a) NUA Сmax, приблизительно, от 2111,0 нг/мл до 3253 нг/мл;
(b) общее восстановление ниацина в моче, приблизительно, от 49,24% до 70,23%;
(c) Сmax ниацина, приблизительно, от 3096 нг/мл до 6750 нг/мл; и
(d) AUC ниацина, приблизительно, от 6723 нг/мл до 18643 нг/мл.
Пример 8
Сравнительная вероятность случаев гиперемии, индуцированной 2000 мг дозой ниацина замедленного высвобождения при условии предварительной обработки или совместного введения с аспирином
Проводилось рандомизированное двойное слепое исследование под контролем двух наблюдателей, с введением однократной дозы и трехсторонним перекрестным исследованием, исследование проводилось в едином центре и было предназначено для того, чтобы изучить эффект предварительного лечения аспирином и совместного введения аспирина на реакции гиперемии, вызванные пероральным введением содержащих ниацин таблетокс пролонгированным высвобождением по настоящему изобретению. Схема эксперимента и схемы лечения показаны на Фиг.24. В течение всего времени исследования субъектам не разрешалось использовать аспирин помимо исследования или другие NSAIDS. Исследование было одобрено комиссией клиники, каждый субъект дал письменное информированное разрешение до своего участия.
Исследование включало здоровых взрослых мужчин от 19 до 70 лет с индексом массы тела (BMI) 22-31 кг/м2. Женщин исключили из эксперимента, чтобы избежать путаницы из-за наложения на индуцированную ниацином гиперемию пре-менопаузальной гиперемии. Субъекты признавались здоровыми после полного физического обследования, изучения медицинской истории, электрокардиограммы и результатов клинического лабораторного тестирования, проведенного при скрининговом визите и по завершении первого периода исследования. Субъектов исключали, если они использовали табак или никотин-содержащий продукт в течение 4 месяцев до начала исследования; если у них была аллергия или гиперчувствительность на ниацин, аспирин или родственные производные; если они злоупотребляли химическими веществами или у них была зависимость от них, по меньшей мере, в течение последних трех лет; если у них была история мигренозных головных болей, диабет, желчекаменная болезнь, заболевания печени, тяжелая гипер- или гипотония, нарушения в работе сердца, заболевания почек или индуцированная лекарствами миопатия. Субъектам не разрешали принимать любые лекарственные препараты по рецепту в течение 21 дня до начала исследования, а безрецептурные лекарственные препараты, витамины или растительные препараты в течение 10 дней до начала и во время исследования.
Скрининговые процедуры были завершены в течение 21 дня до клинического приема в Период 1. В каждом из трех исследуемых периодов субъектов изолировали, приблизительно, на 24 часа, перерыв между лечениями составлял не менее 7 дней, кроме того, субъекты получали еду в соответствии со специфическим меню с контролируемым содержанием ниацина и жиров. Препарат еды и время начала приема пищи было одинаковым для каждого периода исследования.
Экспериментальное лечение в ходе испытания
Исследуемые лекарственные препараты вводились перорально перекрестным способом, в соответствии со схемой рандомизации. Хотя дозировки аспирина (ASA) и плацебо были свои для каждого периода, дозы покрытых оболочкой ниацин-содержащих таблеток по изобретению (называемых также ниацин-содержащими ER-таблетками с переработанным препаратом или "rNER") - две 1000 мг таблетки - были одинаковы. В одном периоде субъекты получали две 325 мг таблетки аспирина за 30 минут до ниацин-содержащих ER 2000 с переработанным препаратом совместно с двумя таблетками плацебо ("предварительное лечение ASA"). В другом периоде субъекты получали две таблетки плацебо за 30 минут до ниацин-содержащих ER 2000 с переработанным препаратом совместно с двумя 325 мг таблетками аспирина ("параллельное введение ASA"). В третьем периоде субъекты получали контрольное лечение, состоящее из двух таблеток плацебо за 30 минут до ниацин-содержащих ER 2000 с переработанным препаратом совместно с двумя таблетками плацебо ("R-ниацин только ER").
Поскольку оценка случаев гиперемии является субъективной, пациентов и персонал оставляли в полном неведении о препарате назначаемых препаратов. Этого достигали несколькими способами. В процессе каждого периода субъекты получали одно и то же количество таблеток для каждой дозы (см. Фиг.25). Хотя таблетки плацебо и аспирина похожи по виду, они не идентичны; поэтому препараты давали субъектам из непрозрачных дозировочных чашек, и субъектам завязывали глаза при приеме препарата. Контрольное лечение, R-ниацин только ER, включали в исследование, чтобы определить реакции гиперемии в отсутствие аспирина. Только спонсор исследования и персонал медицинского центра, готовящий дозы для каждого периода, знал схему рандомизации лечения в ходе исследования. Исследователи, рабочий персонал и контролер исследования не знали схему рандомизации лечения, и персоналу, принимавшему участие в организации и администрировании лечения, запретили собирать или проверять возникающие при лечении нежелательные эффекты.
Каждый субъект получал лекарственные препараты предобработки и легкую закуску до приема ниацин-содержащего ER с переработанным препаратом. Субъекты принимали назначенное им лечение предобработки (аспирин или плацебо) перорально совместно с 180 мл воды, приблизительно, в 21:30, а обедненную жирами легкую закуску, приблизительно, в 21:45. Закуску съедали целиком, после чего субъект получал оставшуюся часть назначенного лечения. Это происходило, приблизительно, в 22:00, и таблетки запивались 240 мл воды. Каждая доза лекарственных препаратов включала несколько таблеток и съедалась в течение минуты, так как таблетки съедали либо все вместе, либо одну сразу же за другой. Если требовалось, то субъектам давали дополнительную воду порциями по 120 мл; жевать или кусать таблетки было запрещено. Чтобы убедиться в проглатывании дозы, по завершении приема препарата рот каждого субъекта осматривали.
Анализ гиперемии
Случай гиперемии был определен как сообщение субъекта об одном или более следующих симптомов гиперемии: покраснение, тепло, покалывание и зуд; эти симптомы могли возникать по отдельности или одновременно. В течение каждого периода испытания субъектам каждый час предлагали сообщать о наличии или отсутствии симптомов гиперемии каждый час в течение 8 часов после введения переработанного ниацин-содержащего ER. Им предлагалось регистрировать начало и прекращение симптомов и оценивать интенсивность (тяжесть) каждого симптома, делая запись об этом в электронном дневнике.
Каждый субъект ранжировал свое ощущение интенсивности симптомов как непрерывными, так и категориальными средствами. Субъекты отмечали интенсивность вертикальной линией на электронной визуальной аналоговой шкале (visual analog scale (VAS)), помеченной отметками, начиная от "отсутствуют" слева и до "непереносимо" справа, а также ранжировали их как мягкие, умеренные или тяжелые. Легко переносимые симптомы, не ограничивающие активность, были определена как мягкие; симптомы, вызывающие трудность в осуществлении активности, были тяжелыми. Каждый субъект сходным образом ранжировал первое общее событие гиперемии, определенное как первый один или несколько параллельных симптомов гиперемии, возникший (возникшие) после приема ниацина ER. Начало первого симптома было также началом первого общего события гиперемии, а общее событие завершалось, когда прекращался последний симптом в этом событии, и проходило не менее 30 минут без дополнительных симптомов гиперемии.
Статистический анализ
Планировалось, что в эксперименте примут участие 164 субъекта, что гарантирует, что хотя бы 144 субъекта завершать все три курса лечения. Субъекты, рано прервавшие исследование, не заменялись.
Все сравнения проводились как двусторонние с альфа (α)=0,05. Первичная конечная точка представляла собой количество субъектов, наблюдавших, по меньшей мере, одно событие гиперемии в исследовании. Случаи гиперемии сравнивали между "предварительным лечением ASA" и контрольным лечением, "R-ниацин только ER", используя для этого тест Мак Немара. Чтобы тест был включен в сравнение, требовалось, чтобы субъекты реагировали (в данном случае, чтобы у них начиналась гиперемия) после обоих видов лечения. Сравнение числа случаев гиперемии проводилось одинаково между лечениями типа "Параллельное введение ASA" и "R-ниацин только ER", а также между "Предварительное лечение ASA" и "Параллельное введение ASA".
Вторичные конечные точки включали количество событий гиперемии, интенсивность, время начала и продолжительность первых общих событий гиперемии, а также и отдельных ее симптомов. Количество событий было просуммировано с помощью счетчика частоты, и этот параметр сравнили тестом Мак Немара. Данные анализа интенсивности VAS преобразовали из графической формы в цифровые данные, выражая вертикальную отметку, сделанную субъектом, в виде расстояния от левого края VAS-линии (стандартизированного на 100 мм). Интенсивности, измеренные с помощью VAS и продолжительности, сравнили между различными лечениями с помощью парных t-тестов для средних и знаково-ранговых критериев Вилкоксона для медиан; интенсивность по категориальной шкале сравнивали с помощью теста симметрии Боукера, генерализации теста Мак Немара, который также требует, чтобы субъекты предоставили данные для обоих видов лечения в сравнении. Сравнение между лечениями для вторичных конечных точек было выполнено для тех же пар, что и для первичных конечных точек. Побочные эффекты (исключая гиперемию) были кодированы с использованием медицинского словаря регуляторной деятельности (Medical Dictionary for Regulatory Activities (MedDRA, Version 7.0)). Побочные эффекты разных видов лечения не сравнивали.
Результаты
В этом исследовании принимали участие 164 мужчины, средний возраст препаратил 29 лет и BMI 26,5 кг/м2. Все они приняли, по меньшей мере, одну дозу исследуемого препарата. Краткое описание демографических данных субъектов представлено ниже в Таблице 26. Из этих 164 субъектов 148 (90%) получили все три лечения и были оценены на гиперемические ответы. Шестнадцать субъектов (10%) завершили испытание рано: 4 (2%) отозвали согласие, 4 (2%) не удалось проследить, у 1 (1%) наблюдался побочный эффект, у 3 (2%) были нарушения в протоколе, у 3 (2%) был положительный результат теста на наркотики, и 1 (1%) был удален из-за ошибки в дозировке.
ность
Гиперемия
Среди 148 субъектов, получивших все три лечения, количество случаев гиперемии было значительно выше после лечения типа "R-ниацин только ER" (77%), чем после "Параллельное введение ASA" (61%, р<0,001) или "Предварительное лечение ASA" (53%, р<0,001; Таблица 27).
Ни одно аспирин-содержащее лечение сильно не отличалось от других по отношению к числу случаев гиперемии. Как показано на Фиг.25, количество индивидуальных симптомов (покраснение, теплота, покалывание, зуд) в первом общем случае гиперемии было снижено на 30%-50% после "Предварительного лечения ASA" по сравнению с "R-ниацин только ER". Наименьшее количество субъектов сообщило о наличии всех четырех симптомов после "Предварительное лечение ASA", а наибольшее количество субъектов - после "R-ниацин только ER". Индивидуальные симптомы между лечениями на сравнивали. Количество случаев гиперемии следовало такому же тренду, что и вероятность гиперемии, причем наибольшее количество было после лечения "R-ниацин только ER", а наименьшее количество - после "Предварительного лечения ASA" (данные не показаны).
У субъектов с гиперемией как после "Предварительного лечения ASA", так и "R-ниацин только ER" (Таблица 28 ниже), "Предварительное лечение ASA" значимо снижало интенсивность первых общих событий гиперемии, измеренных либо категориально, либо с помощью VAS (каждый р<0,001).
В случае обоих типов лечения, большая часть случаев гиперемии было ранжировано как мягкие, и только одно (после "R-ниацин только ER") было тяжелым. Число субъектов с мягкими случаями было на 36% больше после "Предварительное лечение ASA", чем после "R-ниацин только ER"; соответственно, количество субъектов с умеренной или тяжелой гиперемией было на 62% меньше. Рейтинги VAS были более чем на 30% ниже после "Предварительное лечение ASA", чем после "R-ниацин только ER". При анализе продолжительности первого общего случая гиперемии средние значения и медианы были несогласованны, предполагая ненормальное распределение. Медиана продолжительности для "Предварительное лечение ASA" была на 43% меньше, чем для для "R-ниацин только ER" (р=0,008). Индивидуальные симптомы, краснота, тепло и покалывание, были значительно менее интенсивными после "Предварительное лечение ASA" (р<=0,025, данные не показаны); значимых различий по продолжительности симптомов между лечениями замечено не было.
У субъектов, наблюдавших гиперемию после лечений типа "Параллельное введение ASA" и "R-ниацин только ER" (Таблица 29 ниже) интенсивность первого общего события гиперемии значимо различалась для категориальных данных (р=0,028), но не для данных VAS.
Здесь количество субъектов с мягкими случаями гиперемии после "Параллельное введение ASA" было на 22% выше, чем после "R-ниацин только ER", а умеренных или тяжелых случаев было на 38% меньше. Различие в продолжительности первых общих событий гиперемии было незначимо. Что касается индивидуальных симптомов, интенсивность покраснения и теплоты после "Параллельное введение ASA" была значимо меньше (р<0,024, данные не показаны); значимых различий в продолжительности любого симптома отмечено не было.
У субъектов с гиперемией после видов лечения "Предварительное лечение ASA" и "Параллельное введение ASA" (см. Таблицу 30 ниже) различия в интенсивности первых общих событий гиперемии были незначимы для категориальных характеристик, но ранг VAS был на 20% ниже для "Предварительное лечение ASA", и это различие было уже статистически значимым.
ность (мин)3
Продолжительность первых общих случаев гиперемии между этими видами лечения различалась незначимо. Для индивидуальных симптомов ни интенсивность, ни продолжительность, значимо также не различались между двумя видами лечения.
Приведенные выше результаты показывают, что 650 мг (2×325 мг) таблетки аспирина, принятые за 30 минут до таблеток с пролонгированным высвобождением по изобретению, значимо снижают количество случаев, интенсивность и продолжительность гиперемии (по сообщениям субъектов), по сравнению с применением только таблеток по изобретению. Параллельное введение 650 мг аспирина и таблеток по изобретению уменьшали количество случаев гиперемии, интенсивность и продолжительность в меньшей степени. Данные по количеству случаев гиперемии и интенсивности из Примеров 3 и 8 суммированы и проиллюстрированы на Фиг.26 и 27. Эти фигуры показывают, что фармацевтические композиции с пролонгированным высвобождением по настоящему изобретению снижают интенсивность и продолжительность (-40%) гиперемии, по сравнению с оригинальными 1000 мг таблетками (Nispan®) - см. Пример 3, хотя наблюдалось только незначительное уменьшение в количестве случаев гиперемии. Пример 8 показывает, что аспирин, принятый за 30 минут до или совместно с фармацевтическими композициями с пролонгированным высвобождением по настоящему изобретению, может уменьшить количество случаев гиперемии, а также снижает ее интенсивность и продолжительность. В Примере 3 почти все пациенты (98%) отмечали гиперемию (случаи), если принимали одну 2000 мг дозу оригинальных 1000 мг таблеток (доза из двух таблеток). В Примере 8 только 50-60% субъектов отмечали гиперемию, они принимали одну 2000 мг дозу таблеток замедленного высвобождения изобретения (доза из двух таблеток) плюс аспирин. В предыдущем исследовании показано, что медиана интенсивности при использовании оригинальных 1000 мг таблеток составляла 54 мм для VAS. В текущем исследовании при применении таблеток замедленного высвобождения изобретения плюс аспирина медиана интенсивности составляла только 19-23 мм, и абсолютное большинство испытуемых (около 80% или больше) называли гиперемию "мягкой".
Хотя выше изобретение было описано со ссылкой на специфические варианты осуществления, очевидно, что в него можно внести множество изменений, модификаций и вариаций, не отступая от изложенной здесь изменений, модификаций и вариаций, не отступая от изложенной здесь изобретательской концепции. Соответственно, предполагается, что изобретение охватывает все такие изменения, модификации и вариации, которые соответствуют идее и охвачены границами приведенной ниже формулой изобретения. Все процитированные здесь патентные заявки, патенты и другие публикации включены сюда по ссылке во всей своей полноте.
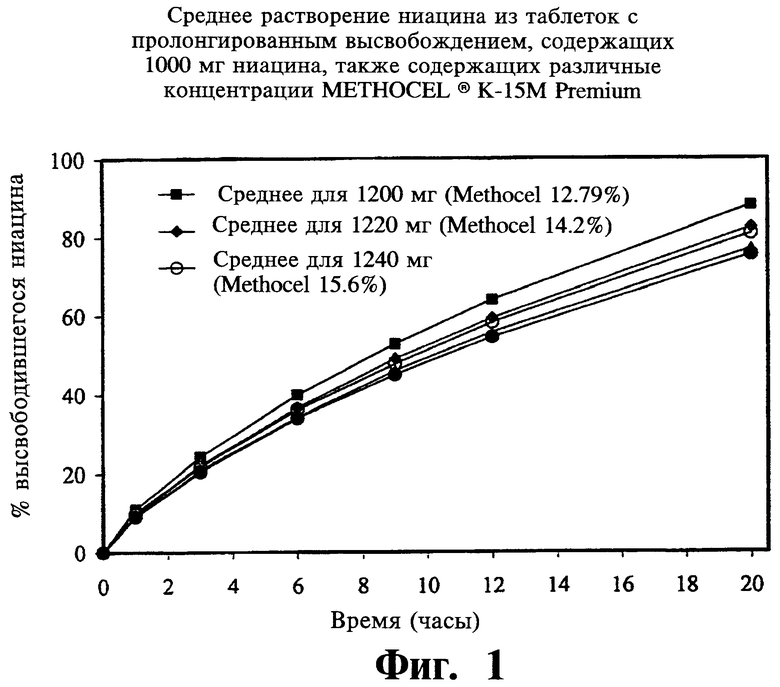
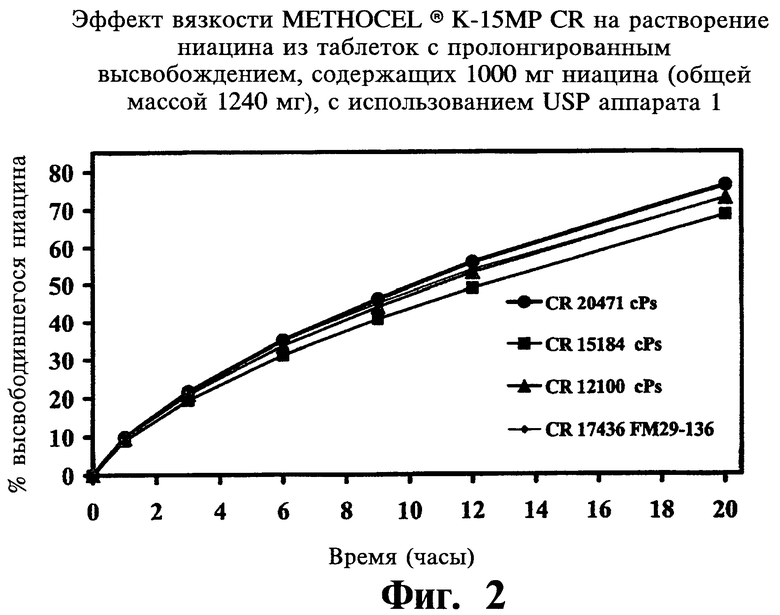
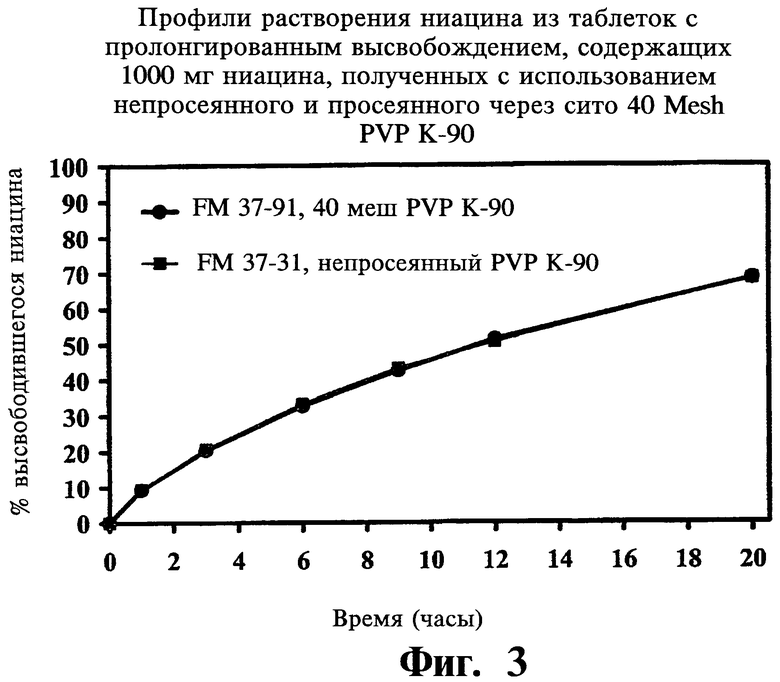
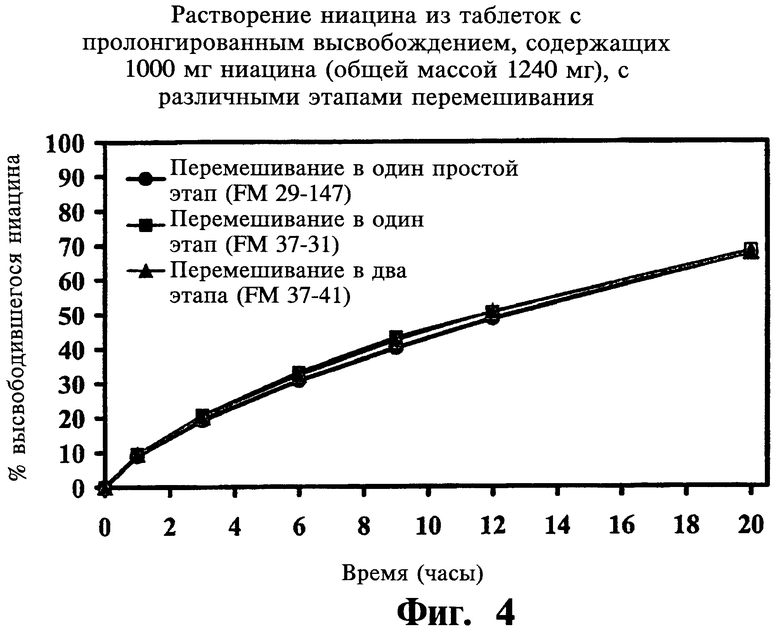
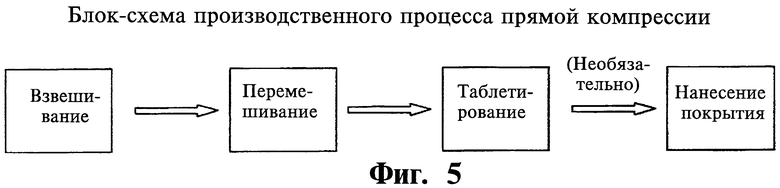

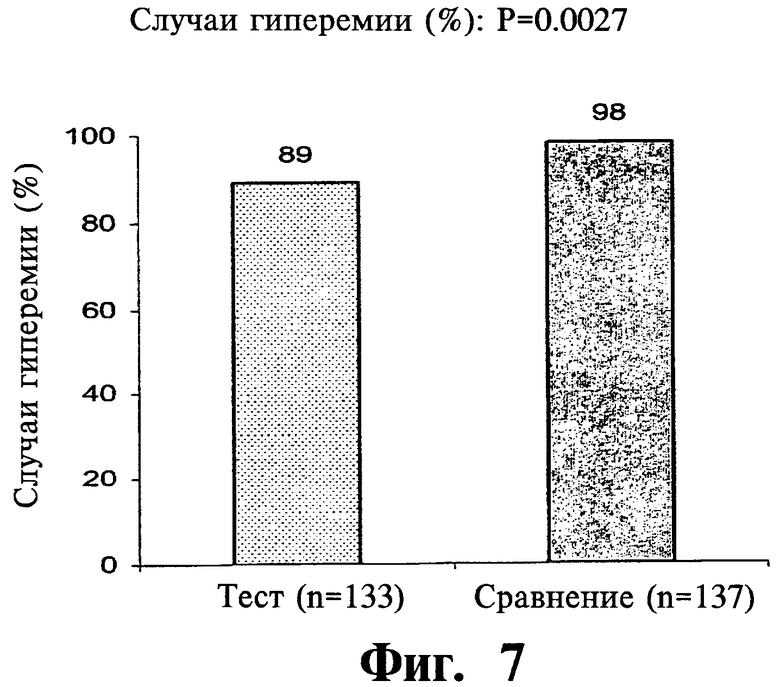
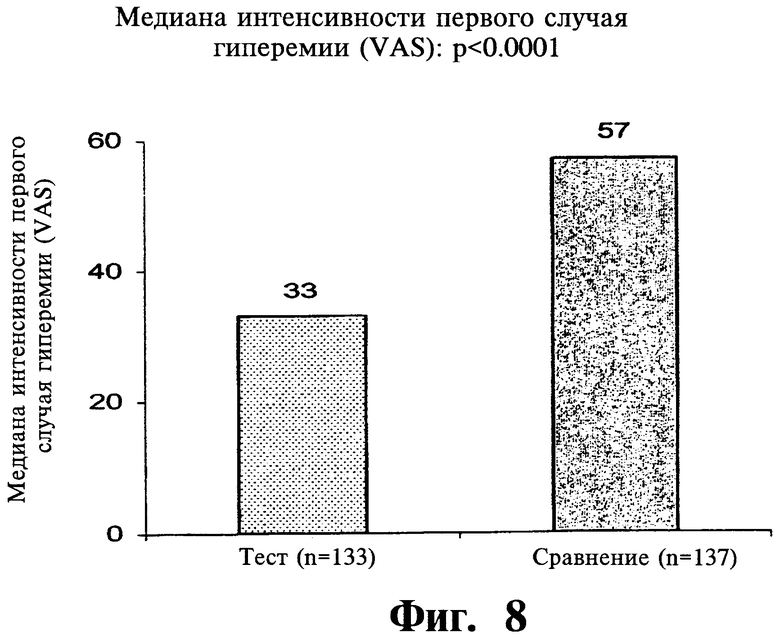
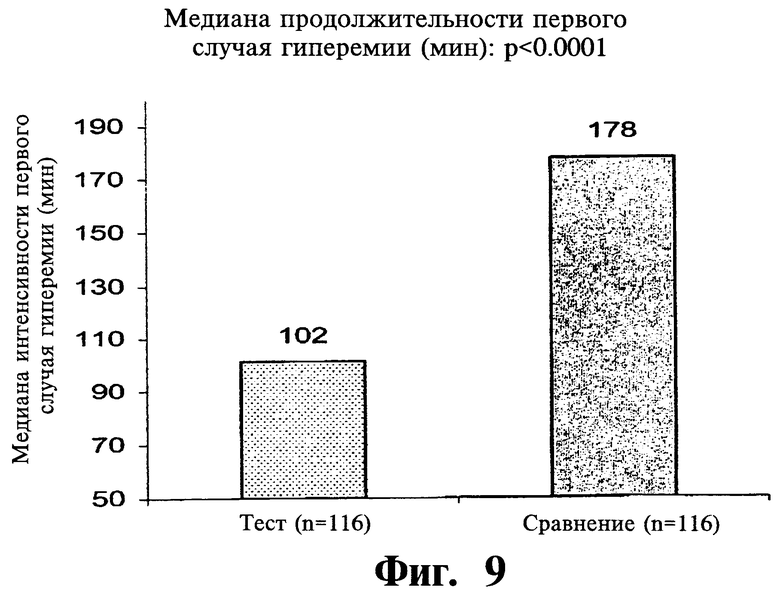
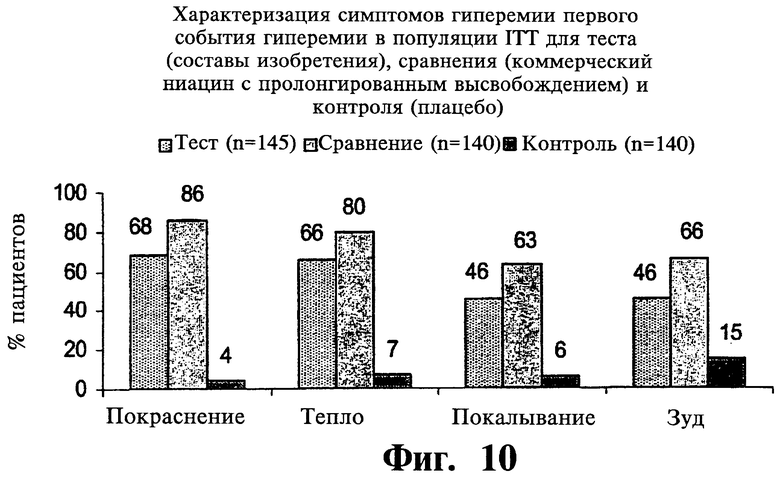
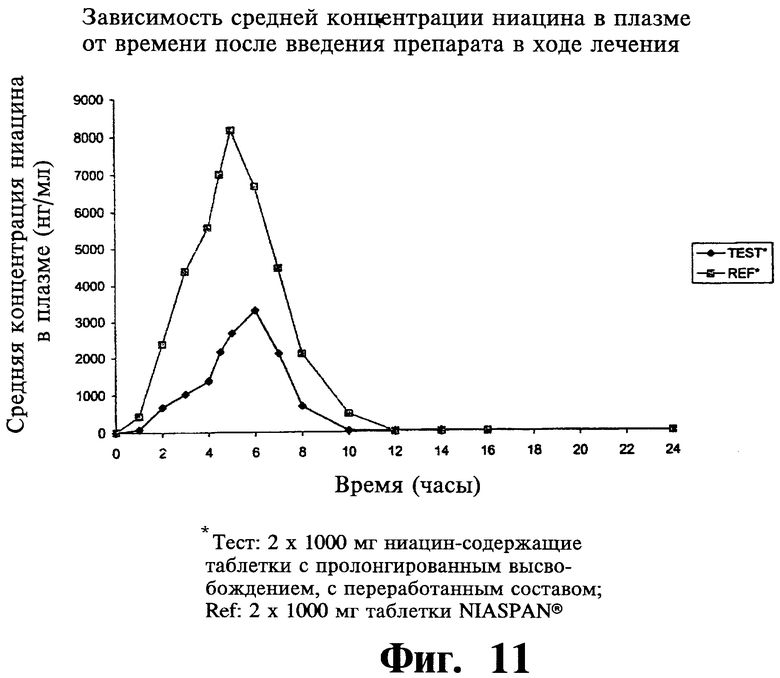
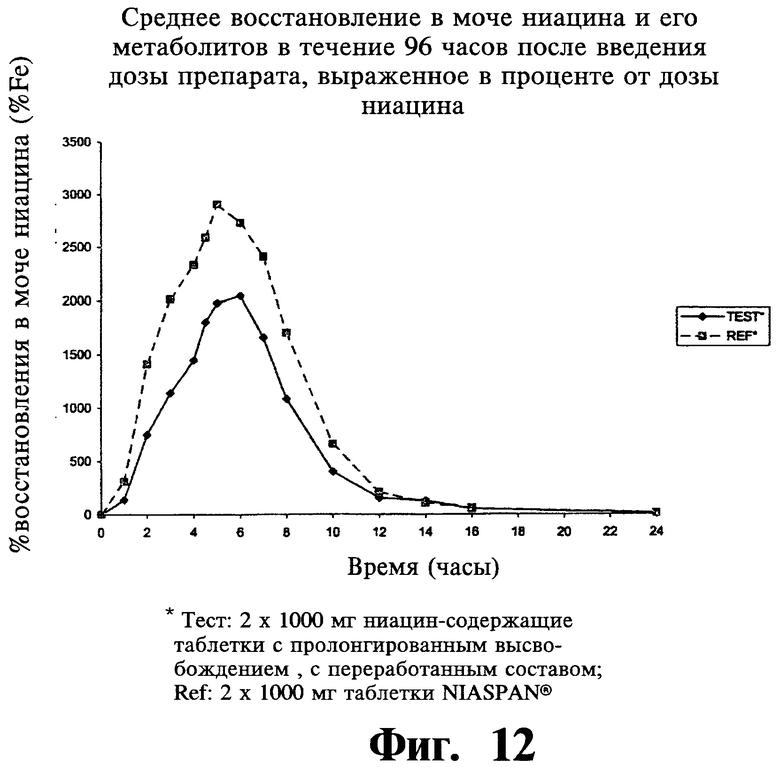
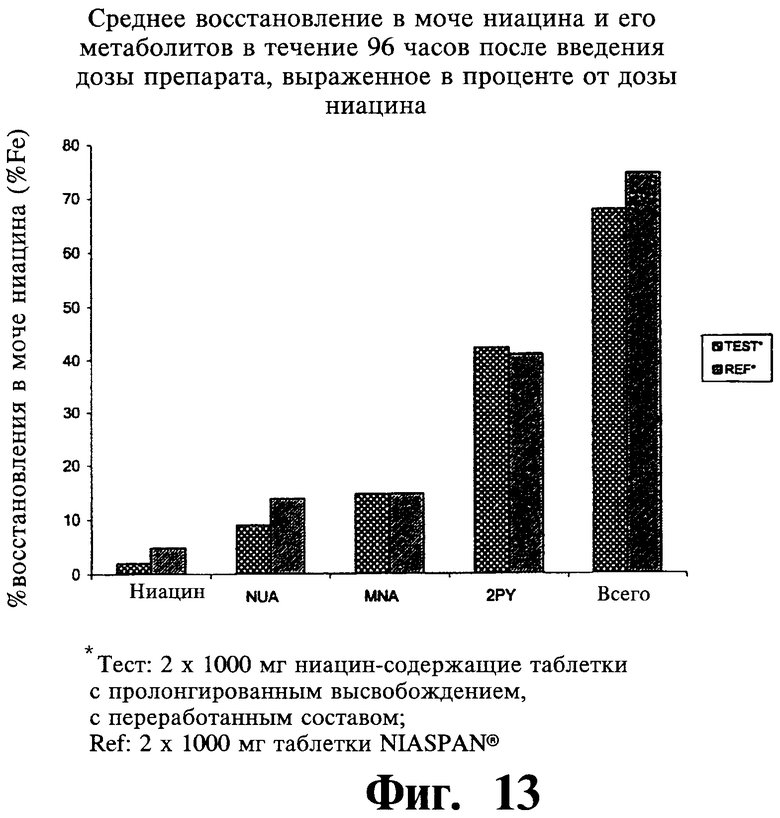
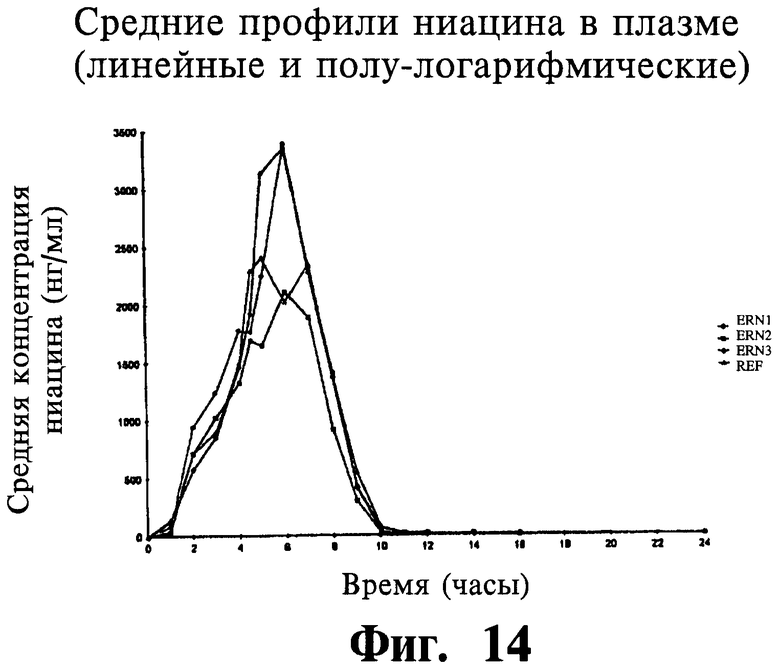
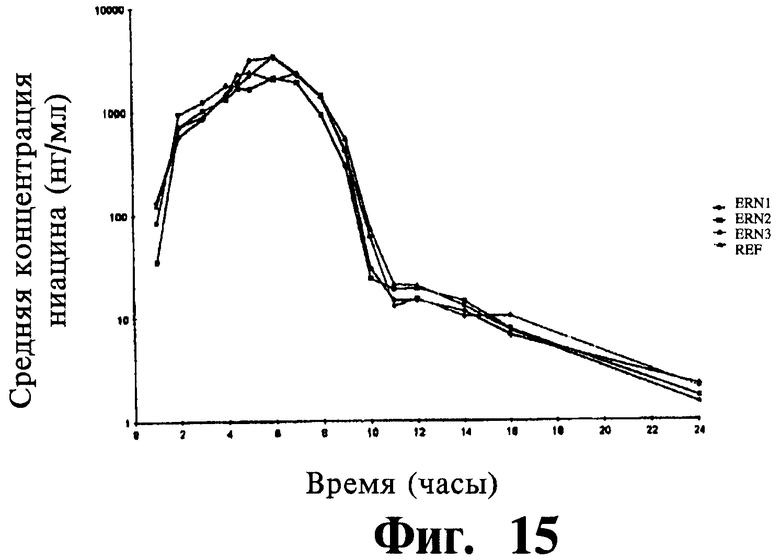
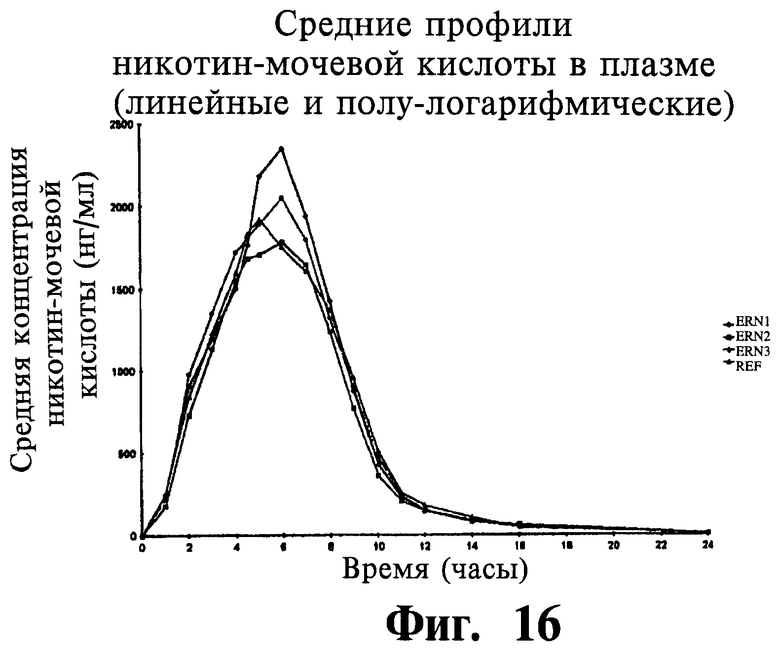
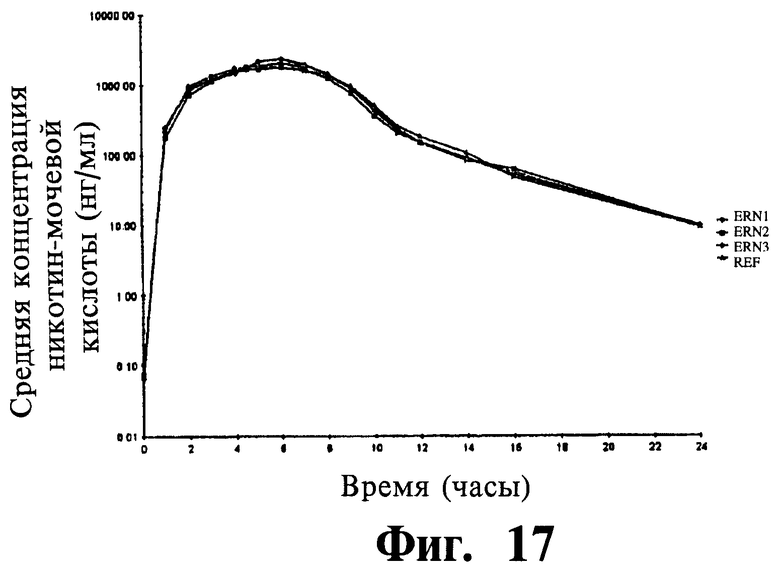
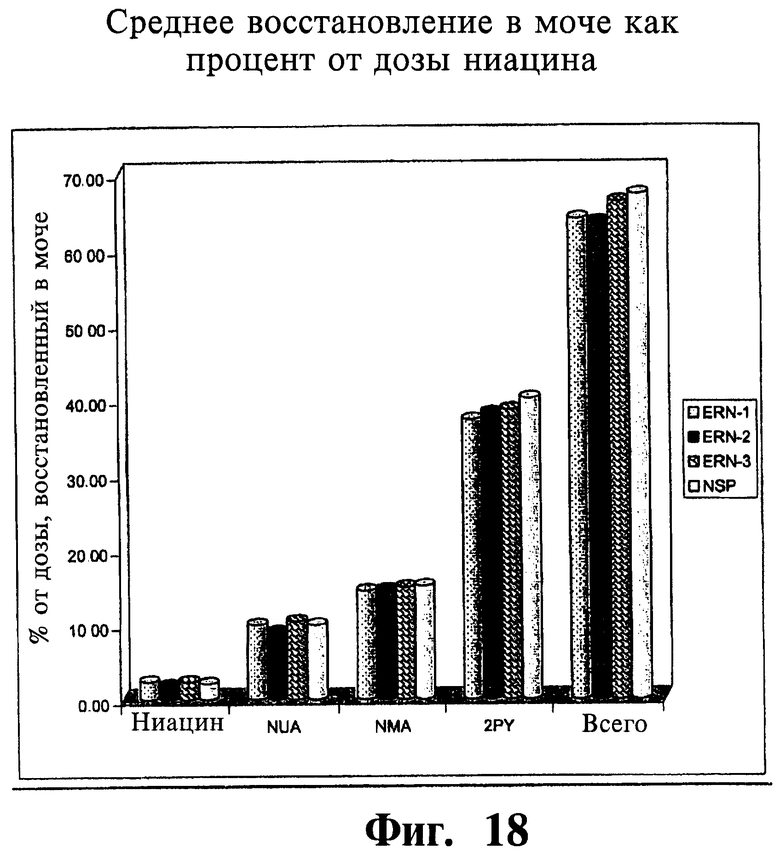
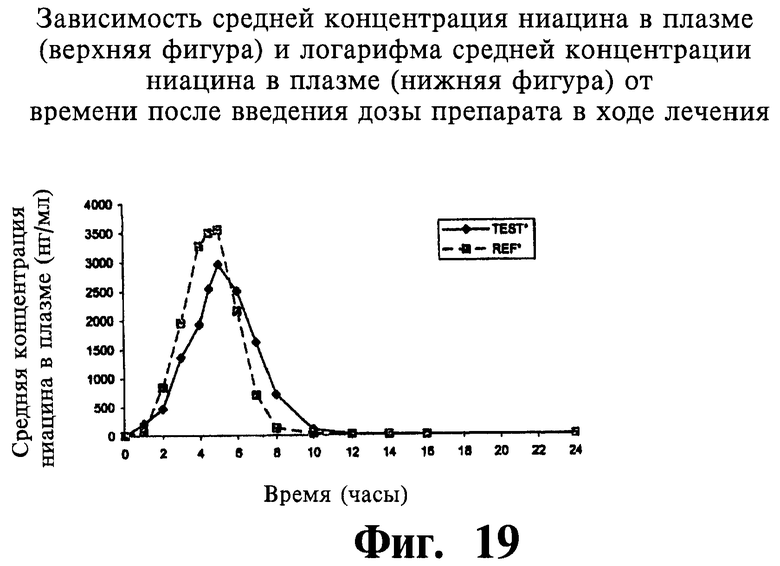
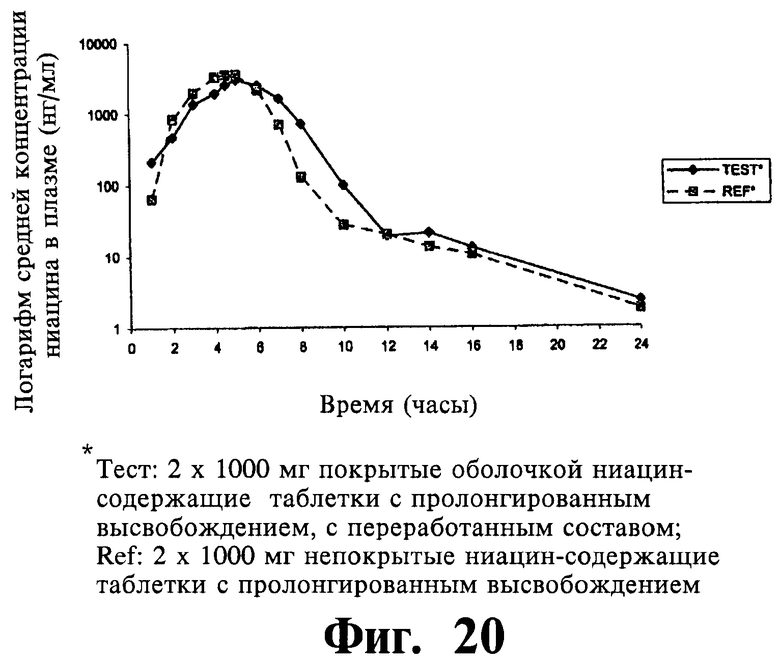
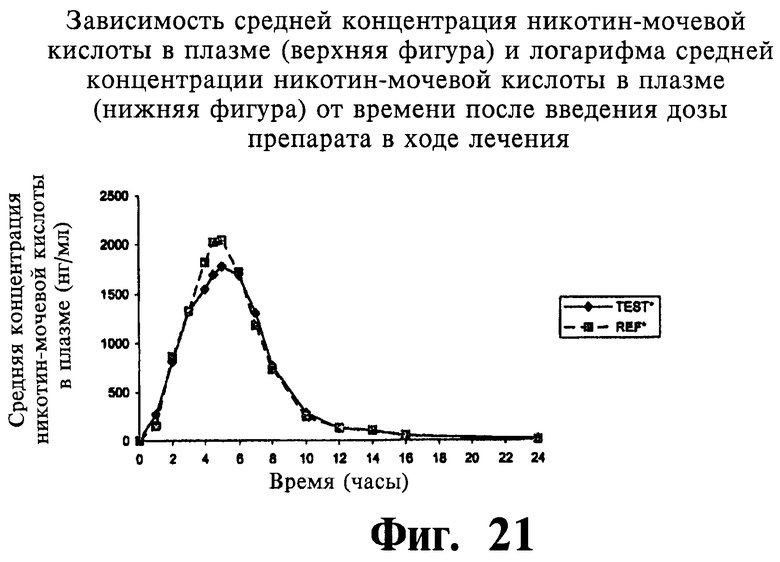
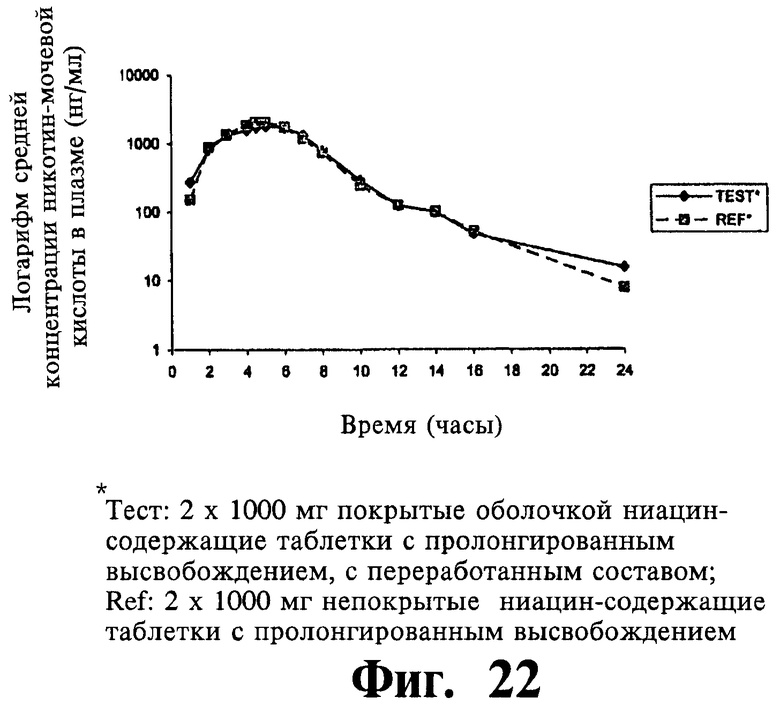
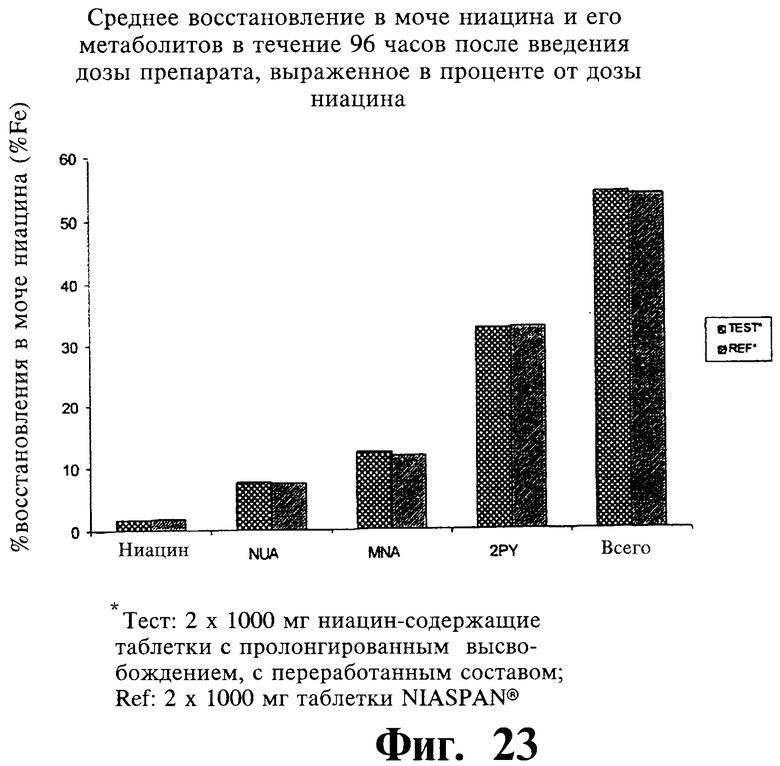
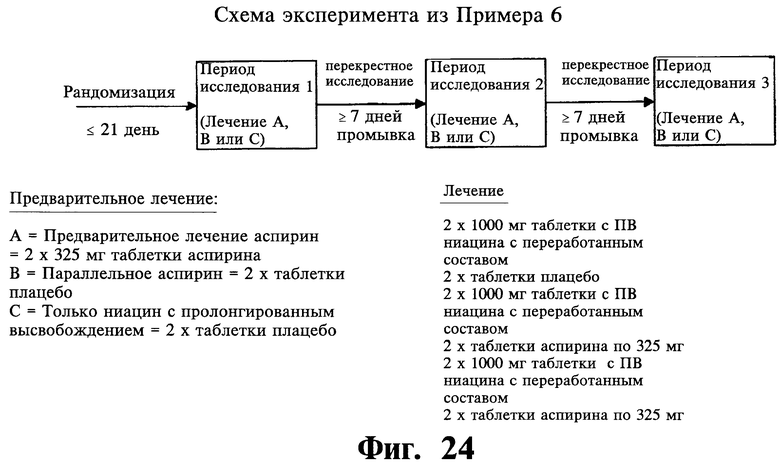
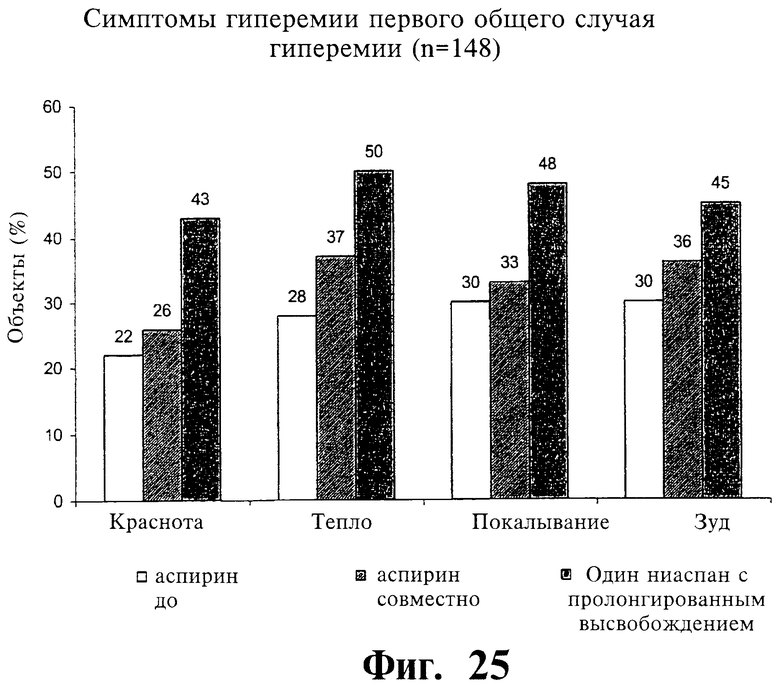
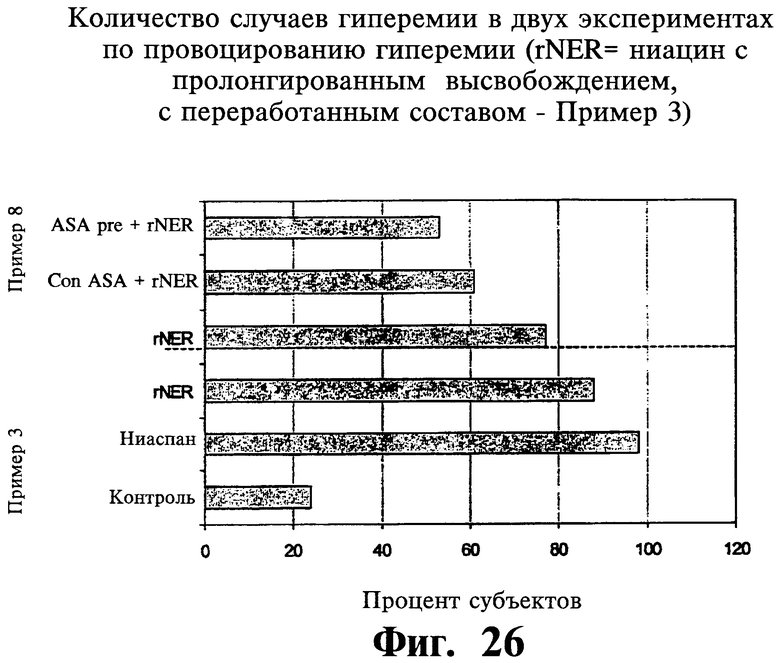
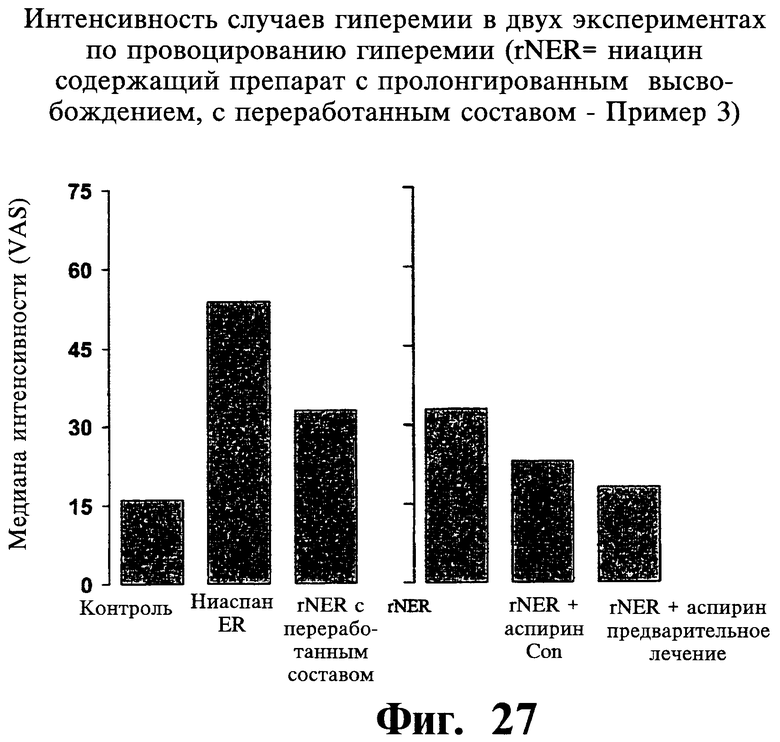
Изобретение относится к фармацевтической композиции, содержащей 1000 мг ниацина. Композиция включает от 78 до 82 мас.% гранулированного ниацина, при этом не менее 85 мас.% гранулированного ниацина имеет размер частиц в диапазоне от 100 до 425 микрометров и не более 10 мас.% гранулированного ниацина имеет размер частиц менее 100 микрометров. Также композиция содержит от 14 до 18 мас.% гидроксипропилметилцеллюлозы с метоксильной степенью замещения от 1,39 до 1,41 и гидроксипропоксильным молярным замещением от 0,20 до 0,22, от 2,5 до 3,0 мас.% поливинилпирролидона и от 0,5 до 1,5 мас.% стеариновой кислоты. Также описан способ получения указанной таблетки ниацина и способ снижения гиперемии. Таблетки по настоящему изобретению демонстрируют благоприятные характеристики высвобождения, а также снижение тяжести, продолжительности и количества случаев кожной гиперемии, обычно связанной с лечением ниацином. 3 н. и 21 з.п. ф-лы, 27 ил., 30 табл., 8 пр.
1. Фармацевтическая композиция, содержащая 1000 мг ниацина, включающая
(a) приблизительно от 78 мас.% до приблизительно 82 мас.% гранулированного ниацина, при этом не менее чем 85 мас.% указанного ниацина имеет размер частиц в диапазоне от приблизительно 100 мкм до приблизительно 425 мкм, и не более 10 мас.% указанного гранулированного ниацина имеет размер частиц менее 100 мкм;
(b) приблизительно от 14 мас.% до приблизительно 18 мас.% гидроксипропилметилцеллюлозы с метоксильной степенью замещения приблизительно от 1,39 до приблизительно 1,41 и гидроксипропоксильным молярным замещением от приблизительно 0,20 до приблизительно 0,22;
(c) приблизительно от 2,5 мас.% до приблизительно 3,0 мас.% поливинилпирролидона; и
(d) приблизительно от 0,5 мас.% до приблизительно 1,5 мас.% стеариновой кислоты;
(e) причем указанная композиция является биоэквивалентной двум 500 мг таблеткам NIASPAN®.
2. Композиция по п.1, отличающаяся тем, что указанная композиция является эффективно снижающей содержание липидов в сыворотке без оказания ограничивающих лечение (i) гепатотоксичности и (ii) повышения содержания мочевой кислоты, или глюкозы, или того и другого, при введении пациенту, требующем перерыва в таком лечении после приема указанной композиции указанным пациентом один раз в день.
3. Композиция по п.2, введение которой указанному пациенту осуществляется один раз в день вечером или ночью.
4. Композиция по п.1, дополнительно содержащая ингибирующий гиперемию агент.
5. Композиция по п.4, отличающаяся тем, что указанный ингибирующий гиперемию агент представляет собой простагландиновый D2 рецептор.
6. Композиция по п.5, отличающаяся тем, что простагландиновый D2 рецептор представляет собой МК-0524.
7. Композиция по п.4, отличающаяся тем, что указанный ингибирующий гиперемию агент представляет собой нестероидное противовоспалительное лекарственное средство (NSAID).
8. Фармацевтическая композиция по п.1, отличающаяся тем, что содержит приблизительно от 16 мас.% до приблизительно 18 мас.% гидроксипропилметилцеллюлозы.
9. Композиция по п.1 или 8, отличающаяся тем, что гидроксипропилметилцеллюлоза имеет метоксильную степень замещения приблизительно 1,4 и гидроксипропоксильное молярное замещение приблизительно 0,21.
10. Композиция по п.1 или 8, отличающаяся тем, что гидроксипропилметилцеллюлоза характеризуется вязкостью приблизительно от 11000 мПа·с до 22000 мПа·с.
11. Композиция по п.10, отличающаяся тем, что гидроксипропилметилцеллюлоза характеризуется вязкостью приблизительно от 13000 мПа·с до 18000 мПа·с.
12. Композиция по п.1, дополнительно включающая покрытие.
13. Композиция по п.12, отличающаяся тем, что указанное покрытие представляет собой цветное покрытие, увеличивающее массу композиции приблизительно на 1,5-8,0 мас.%.
14. Композиция по п.13, отличающаяся тем, что указанное покрытие представляет собой цветное покрытие, применяемое для обеспечения прибавки в массе таблетки приблизительно на 1,75-5,0 мас.%.
15. Композиция по п.1, дополнительно содержащая антилипидемический агент.
16. Композиция по п.15, отличающаяся тем, что указанный антилипидемический агент представляет собой ингибитор HMG-CoA редуктазы.
17. Композиция по п.4, отличающаяся тем, что ингибирующий гиперемию агент представляет собой аспирин (ASA).
18. Фармацевтическая композиция по п.1, отличающаяся тем, что указанная композиция представляет собой 1000 мг таблетку, содержащую ниацин с замедленным высвобождением, полученную методом прямой компрессии.
19. Способ снижения гиперемии, связанной с терапевтическим использованием ниацина, включающий введение пациенту фармацевтической композиции по п.1.
20. Способ получения содержащей ниацин таблетки методом прямой компрессии, включающий:
(а) получение смеси приблизительно от 70 мас.% до приблизительно 92 мас.% гранулированного ниацина, при этом не менее 85 мас.% указанного гранулированного ниацина имеет размер частиц в диапазоне приблизительно от 100 мкм до приблизительно 425 мкм, и не более чем 10 мас.% указанного гранулированного ниацина имеет размер частиц менее чем 100 мкм, приблизительно от 14 мас.% до приблизительно 18 мас.% гидроксипропилметилцеллюлозы с метоксильной степенью замещения приблизительно от 1,39 приблизительно до 1,41 и гидроксипропоксильным молярным замещением приблизительно от 0,20 до приблизительно 0,22, приблизительно от 2,5 мас.% до приблизительно 3 мас.% поливинилпирролидона и приблизительно от 0,5 мас.% до приблизительно 1,5 мас.% стеариновой кислоты; и
(b) прессование смеси, полученной на стадии (а), в таблетку.
21. Способ по п.20, отличающийся тем, что указанная содержащая ниацин таблетка представляет собой 1000 мг ниациновую дозированную лекарственную форму.
22. Способ по п.21, дополнительно включающий нанесение на таблетку покрытия.
23. Способ по п.22, дополнительно включающий нанесение на таблетку цветного покрытия для обеспечения прибавки в массе таблетки приблизительно на 1,5-8,0 мас.%.
24. Способ по п.23, отличающийся тем, что указанное цветное покрытие обеспечивает прибавку в массе приблизительно на 1,75-5,0 мас.%.
| WO 00/33818 A1, 15.06.2000 | |||
| Способ моделирования освещения объекта | 1976 |
|
SU577504A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 3065143 A, 20.11.1962. | |||

