Область техники, к которой относится изобретение
Настоящее изобретение относится к новому производному оксазолидинона. Более конкретно, настоящее изобретение относится к новому производному оксазолидинона, имеющему циклическую амидоксимную или циклическую амидразоновую группу. Кроме того, настоящее изобретение относится к фармацевтической композиции, обладающей активностью антибиотика, которая включает, в качестве активного ингредиента, производное оксазолидинона, его пролекарство, его гидрат, его сольват, его изомер или его фармацевтически приемлемую соль.
Предпосылки создания изобретения
С тех пор как о линезолиде, который является оксазолидиноновым антибиотиком, впервые было сообщено в 1984 г. (см. Публикацию Европейского Патента №127902), о множестве производных оксазолидинона сообщалось различными фармацевтическими фирмами. Однако, лекарственные средства, во время разработки, не обладают превосходящими свойствами перед линезолидом (название продукта: Zyvox) с точки зрения токсичности и эффективности. Вследствие таких проблем линезолид еще привлекает внимание в качестве наилучшей альтернативы ванкомицину при лечении заболеваний, вызванных метициллин резистентным staphylococcus aureus (MRSA). Если резистентные к линезолиду бактерии, о которых недавно сообщалось, продолжают распространяться, возникнут очень серьезные проблемы, то есть, отсутствие лечения заболеваний, вызванных резистентными к линезолиду бактериями.
По этой причине существует очень срочная необходимость в создании лекарственных средств, обладающих превосходящими свойствами перед линезолидом с точки зрения эффектов или токсичности и проявляющих эффективность против резистентных к линезолиду бактерий. В заявке на патент Кореи №10-2008-0093712, зарегистрированной авторами настоящего изобретения 24 сентября 2008 г., раскрывается, что оксазолидиноновый антибиотик, имеющий циклическую амидразоновую или циклическую амидоксимную группу, обладает превосходящими свойствами перед линезолидом с точки зрения эффективности и токсичности и оксазолидиноновый антибиотик имеет многие преимущества за счет введения циклической амидразоновой группы.
В частности, циклическая амидразоновая группа является слабо основной и, таким образом, образует соль. Когда циклическая амидразоновая группа образует гидрохлорид, гидрохлорид имеет кислотность, подобную уксусной кислоте, то есть, рКа составляет около 5. Вследствие такой слабой кислотности, антибактериальные эффекты не ухудшаются, и растворимость гидрохлорида в воде может быть значительно увеличена.
Однако, оксазолидиноновый антибиотик, раскрытый в вышеуказанной заявке на патент, также обладает незначительным воздействием на резистентные к линезолиду бактерии и, таким образом, не может быть использован для эффективного лечения инфекций из-за резистентных к линезолиду бактерий при данных обстоятельствах, что бактерии продолжают распространяться.
Техническая проблема
Как результат ряда обширных и интенсивных исследований и экспериментов для решения проблем, как описано выше, авторы настоящего изобретения обнаружили, что, как описано ниже, новые производные оксазолидинона, представленные нижеприводимой формулой (1), в частности, новые производные оксазолидинона, имеющие циклическую амидоксимную или циклическую амидразоновую группу, оказывают превосходное воздействие на резистентные к линезолиду бактерии, обладают более высокой антибактериальной активностью, чем обычные антибиотики, и имеют высокую растворимость, что дает возможность производным оксазолидинона быть легко переработаны в пероральные и инъецируемые лекарственные средства, таким образом, завершая настоящее изобретение, базирующееся на данном раскрытии.
В частности, настоящее изобретение относится к соединению, представленному нижеприводимой формулой (1).
Настоящее изобретение также относится к пролекарству соединения, сольвату соединения, изомеру соединения или фармацевтически приемлемой соли соединения.
Настоящее изобретение также относится к фармацевтической композиции, включающей соединение, и способу лечения антибиотиком, используя эффективное количество соединения.
Техническое решение
В соответствии с одним аспектом настоящего изобретения, оно относится к новому производному оксазолидинона, представленному нижеприводимой формулой (1), в частности, к новому производному оксазолидинона, имеющему циклическую амидоксимную или циклическую амидразоновую группу. Кроме того, настоящее изобретение также относится к новому производному оксазолидинона, представленному нижеприводимой формулой (1), его пролекарству, его гидрату, его сольвату, его изомеру и его фармацевтически приемлемой соли:
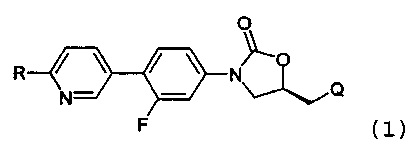
где
R означает гетероциклическую группу, выбираемую из следующих групп:
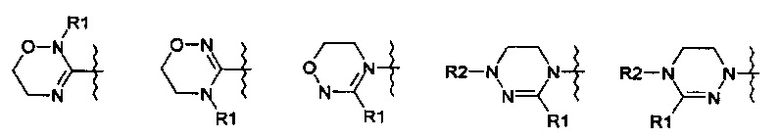
R1 означает водород, C1-С6-алкил или С3-С6-циклоалкил;
R2 означает водород, C1-С6-алкил или (СН2)mC(=O)R21, где R21 означает водород, (СН2)nNHR211, где R211 означает водород или C1-С6-алкил, CH2OH или СН(ОН)СН2ОН, и m и n, каждый, независимо означают целое число от 0 до 3; и
Q означает OR3, NHR3 или  где R3 означает водород, C1-С6-алкил, -C(=O)R31, где R31 означает водород, C1-С6-алкил, С3-С6-циклоалкил или О-(C1-С6)-алкил или гетероароматическую циклическую группу, выбираемую из следующих групп:
где R3 означает водород, C1-С6-алкил, -C(=O)R31, где R31 означает водород, C1-С6-алкил, С3-С6-циклоалкил или О-(C1-С6)-алкил или гетероароматическую циклическую группу, выбираемую из следующих групп:
Соединение представляет собой новое соединение, химическую структуру которого редко исследовали. Так, за счет введения циклической амидоксимной или циклической амидразоновой группы в оксазолидиноновый антибиотик, может быть значительно улучшена абсорбционная способность и может быть значительно увеличена растворимость соединения в воде, так как циклическая амидоксимная или циклическая амидразоновая группа обладает подходящей основностью и, таким образом, образует соль. Вследствие увеличения растворимости в воде соединение может быть получено в форме, пригодной для инъекции, без получения пролекарственной формы, и соединение имеет незначительную токсичность.
Производное оксазолидинона проявляет антибактериальную способность в отношении грамположительных бактерий, как например Staphylococcus aureus, Enterococcus faecalis и т.п., и грамотрицательных бактерий, как например Haemophilus influenza, Moraxella catarrhalis и т.п., которые резистентны к существующим антибиотикам, при гораздо более низкой концентрации, чем коммерчески доступный линезолид. В особенности, производное оксазолидинона обладает превосходной антибактериальной способностью против резистентной к линезолиду Enterococcus faecium.
Термин «алкил», как используется в данном контексте, включает линейные или разветвленные структуры. Например, C1-С6-алкил включает все возможные изомеры положения и геометрические изомеры, как например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, гексил и т.п.
Термин «С3-С6-циклоалкил» включает все изомеры положения циклического типа и геометрические изомеры, как например циклопропил, циклобутил, циклопентил, циклогексил, циклопропилметил и т.п.
Предпочтительно, производное оксазолидинона формулы (1) может представлять собой соединение, соответствующее соединению, выбираемому из соединений формул (2)-(4):
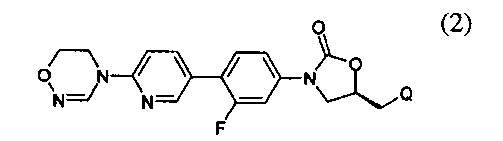
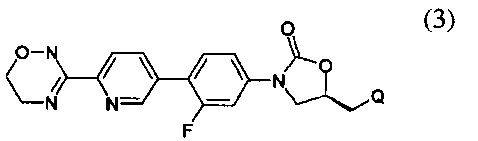
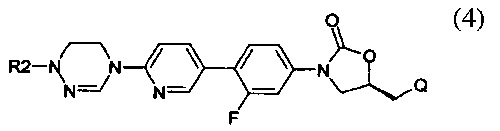
где R2 и Q имеют такие же значения, как указано выше, что касается формулы (1).
Производное оксазолидинона согласно настоящему изобретению может представлять собой одно из следующих соединений, но не исчерпывающим образом:
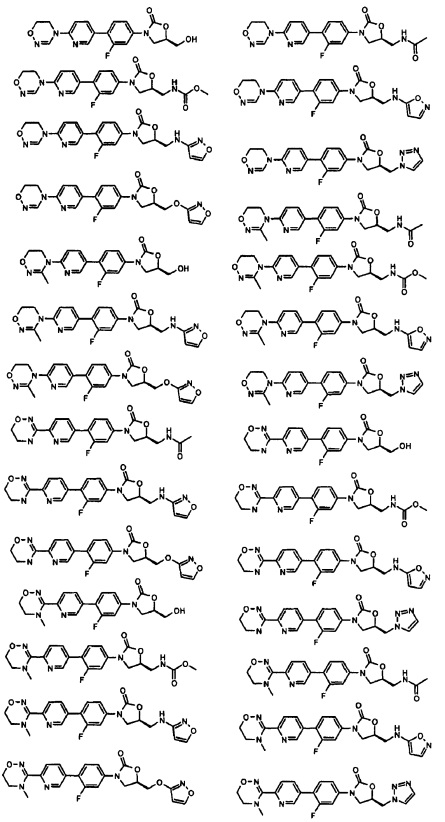
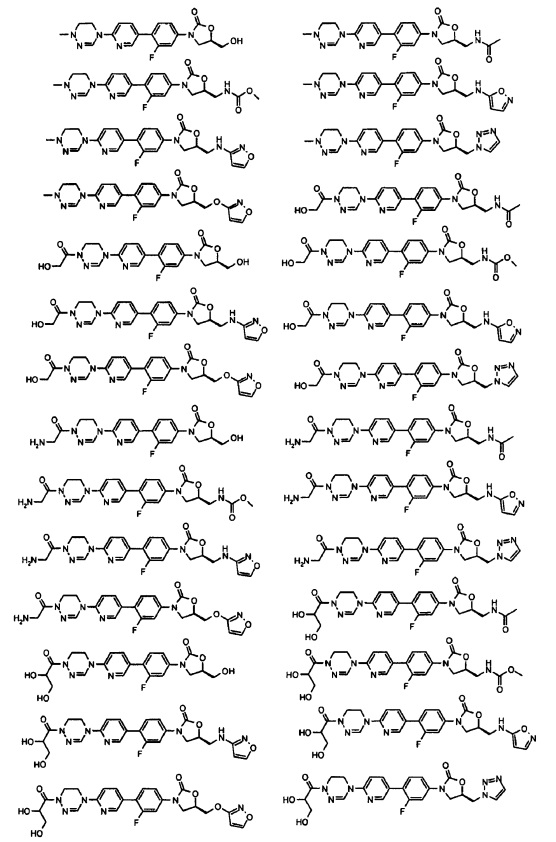
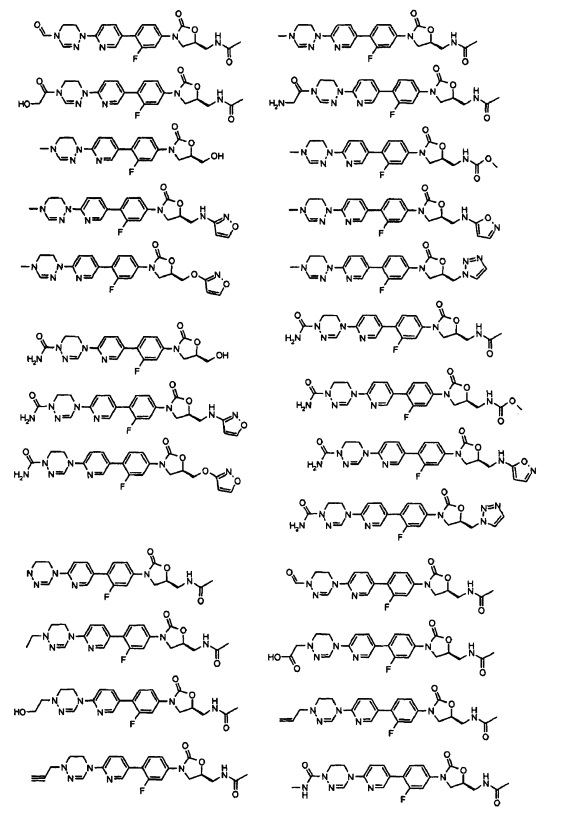
Производное оксазолидинона согласно настоящему изобретению может быть получено в виде пролекарства, гидрата, сольвата, изомера или фармацевтически приемлемой соли для усиления биодоступности или растворимости. Таким образом, пролекарство, гидрат, сольват, изомер и фармацевтически приемлемая соль производного оксазолидинона также входят в рамки настоящего изобретения.
В дальнейшем должны быть кратко описаны термины, как используемые в данном контексте.
Термин «фармацевтически приемлемая соль» относится к составам соединения, которые не вызывают тяжелого раздражения в организме, в который введено соединение, и не ухудшают биологической активности и физических свойств соединения. Термины «гидрат», «сольват», «изомер» и «пролекарство» также имеют значения такие же, как указано выше. Фармацевтически приемлемые соли включают фармацевтически приемлемые, содержащие анион, нетоксичные аддитивные соли, образуемые с кислотами, например, с неорганическими кислотами, как например хлороводородная (соляная) кислота, серная кислота, азотная кислота, фосфорная кислота, бромоводородная кислота, иодоводородная кислота и т.п., с органическими кислотами, как например винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота, малеиновая кислота, салициловая кислота, и т.п., и с сульфоновыми кислотами, как например метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и т.п. Примеры фармацевтически приемлемых солей карбоновых кислот включают соли (щелочных) металлов или соли щелочноземельных металлов лития, натрия, калия, кальция, магния и т.п., соли аминокислот, как например лизин, аргинин, гуанидин и т.п., и органические соли, как например дициклогексиламин, N-метил-D-глюкамин, трис(гидроксиметил)-метиламин, диэтаноламин, холин, триэтиламин и т.п. Соединение формулы (1) может быть превращено в соль, используя обычный способ.
Термин «гидрат» относится к соединению или его соли согласно настоящему изобретению, которое(которая) содержит стехиометрические или нестехиометрические количества воды, связанной с ним(ней) за счет нековалентной межмолекулярной силы.
Термин «сольват» относится к соединению или его соли согласно настоящему изобретению, которое(которая) содержит стехиометрические или нестехиометрические количества растворителя, связанного с ним(ней) за счет нековалентной межмолекулярной силы. В этом отношении, предпочтительными растворителями могут быть летучие растворители, нетоксичные растворители и/или растворители, подходящие для введения людям.
Термин «изомеры» относится к соединениям или их солям согласно настоящему изобретению, которые имеют одну и ту же химическую или молекулярную формулу, но различные структурные формулы. Такие изомеры включают структурные изомеры, такие как таутомеры и т.п., R- или S-изомеры, имеющие асимметрический углеродный центр, и стереоизомеры, такие как геометрические изомеры (транс-, цис-) и т.п. Все изомеры и их смеси также входят в рамки настоящего изобретения.
Термин «пролекарство» относится к агенту, который in vivo превращается в исходное лекарственное вещество. В некоторых случаях пролекарства часто используют из-за более легкого введения, чем исходные лекарственные вещества. Например, пролекарства обладают биодоступностью, при введении перорально, тогда как исходные лекарственные вещества могут не обладать биодоступностью. Кроме того, пролекарство может обладать улучшенной растворимостью в случае фармацевтической композиции по сравнению с исходным лекарственным веществом. Например, пролекарство может представлять собой in vivo гидролизуемый сложный эфир соединения согласно настоящему изобретению или его фармацевтически приемлемую соль. Кроме того, пролекарство может быть короткоцепочечным пептидом (полиаминокислота) со связанным с ним кислотным радикалом, который метаболизируется, так, что пептид открывает активный сайт.
Другие термины, которые используются в данном контексте, могут быть интерпретированы как обычно понимаемые в области, к которой имеет отношение настоящее изобретение.
В уровне техники известны различные типы пролекарств, и не являющиеся исчерпывающими примеры цитируемых ссылок включают:
a) Desin of Prodrugs, под ред. H. Bundgaard (Elsevier, 1985) и Methods in Enzymology, том 42, cc. 309-396, под ред. K. Widder и др. (Academic press, 1985);
b) A Textbook of Drug Design and Development, под ред. Krogsgaard-Larsen и H. Bundgaard, глава 5 «Design and Application of Prodrugs», H. Bundgaard, cc. 113-191 (1991);
c) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992);
d) H. Bundgaard и др., Journal of Pharmaceutical Sciences, 77, 285 (1988); и
e) N. Kakeya и др., Chem. Pharm. Bull., 32, 692 (1984).
Например, пролекарство согласно настоящему изобретению может представлять собой одно из следующих соединений:
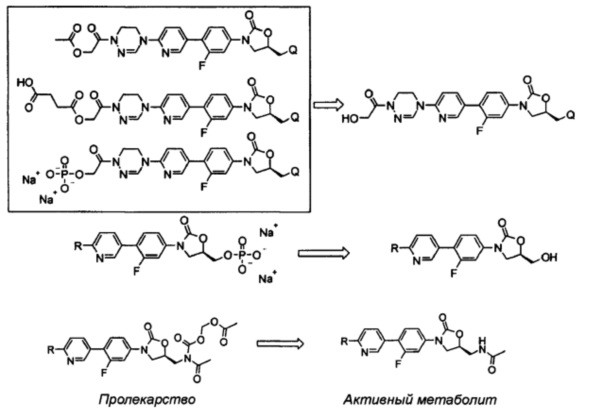
или представлять собой соединение, выбираемое из соединений формул (5)-(7):
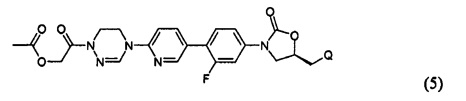
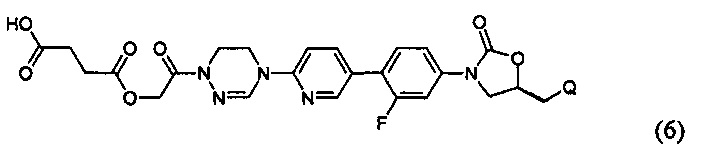
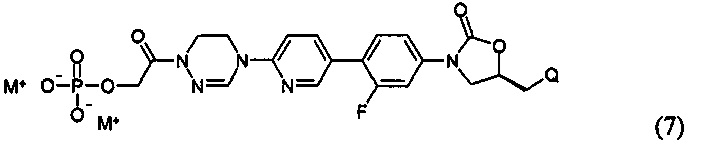
где Q означает OR3, NHR3 или  где R3 означает водород, C1-С6-алкил, -C(=O)R31, или гетероароматическую циклическую группу, выбираемую из следующих групп:
где R3 означает водород, C1-С6-алкил, -C(=O)R31, или гетероароматическую циклическую группу, выбираемую из следующих групп:
где R31 означает водород, C1-С6-алкил, С3-С6-циклоалкил или O-(C1-С6)алкил; и М+ представляет собой ион щелочного металла, такой как Na+ или K+, или ион аммония.
Более предпочтительно, производное оксазолидинона формулы (1) может представлять собой соединение вышеприведенной формулы (2), (3) или (4), где Q означает NHC(=O)CH3, NHC(=O)OCH3, 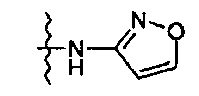 или
или 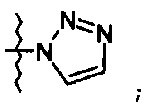 и R2 означает метил, С(=O)CH2OH, C(=O)CH2NH2 или С(=O)СН(ОН)CH2OH.
и R2 означает метил, С(=O)CH2OH, C(=O)CH2NH2 или С(=O)СН(ОН)CH2OH.
Как проиллюстрировано в предшествующих примерах, фосфонатная или ацетильная группа может быть присоединена по гидроксильной группе, так что пролекарство после введения превращается в активный метаболит. Согласно другому воплощению, может быть присоединена аминокислота или может быть использован способ получения в карбонатной форме. Такое пролекарство главным образом используют тогда, когда растворимость является относительно низкой или абсорбционная способность является низкой. Использование пролекарств может приводить к улучшению абсорбции, распределения, метаболизма и экскрекции (ADME) и профиля PK, в дополнение, к повышению растворимости и абсорбционной способности.
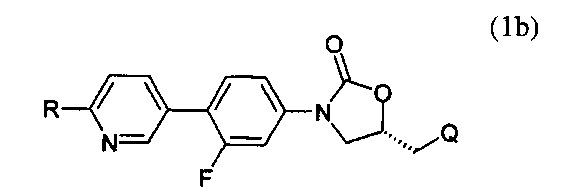
Соединение согласно настоящему изобретению имеет хиральный центр в положении С-5 оксазолидинонового цикла. Предпочтительный диастереоизомер производного оксазолидинона согласно настоящему изобретению представлен формулой (1), приведенной выше, и, по сравнению с эпимером, представленным формулой (1b), приводимой ниже, диастереоизомер проявляет превосходное действие.
Когда используют смесь эпимеров относительно формы хирального центра оксазолидинона, количество используемой смеси может быть установлено принимая во внимание доли диастереоизомеров в целях достижения такого же фармацевтического эффекта, как когда используют один зеркальный изомер.
Соединение формулы (1) или его соль может быть таутомеризовано(на) и, таким образом, хотя химические формулы или реакционные схемы, как используемые согласно данному контексту, представляют только один возможный таутомер, настоящее изобретение не ограничено одним таутомером, представленным химическими формулами или реакционными схемами, и произвольные таутомерные формы, обладающие антибактериальной активностью, также входят в объем настоящего изобретения.
Кроме того, соединение согласно настоящему изобретению может проявлять полиморфизм и, таким образом, все полиморфные соединения, обладающие антибактериальной активностью, также входят в объем настоящего изобретения.
Новое производное оксазолидинона согласно настоящему изобретению может быть получено, используя различные известные способы в зависимости от его заместителей. Например, производное оксазолидинона может быть получено, используя один из способов, проиллюстрированных на нижеприводимых реакционных схемах. Способы получения, представленные на нижеприводимых реакционных схемах, предусмотрены только для иллюстративных целей и ясно, что способы получения могут быть легко изменены квалифицированным специалистом в данной области в соответствии с конкретными заместителями. Таким образом, способ получения производного оксазолидинона согласно настоящему изобретению не ограничивается способами получения, проиллюстрированными на нижеприводимых реакционных схемах. В дополнение, если не указано иным образом, определение заместителей на нижеприводимых реакционных схемах является таким же, как указано в случае вышеприведенной формулы (1).
Получение производных оксазолидинона формулы (1) представлено на нижеприводимой реакционной схеме 1:
Реакционная схема 1
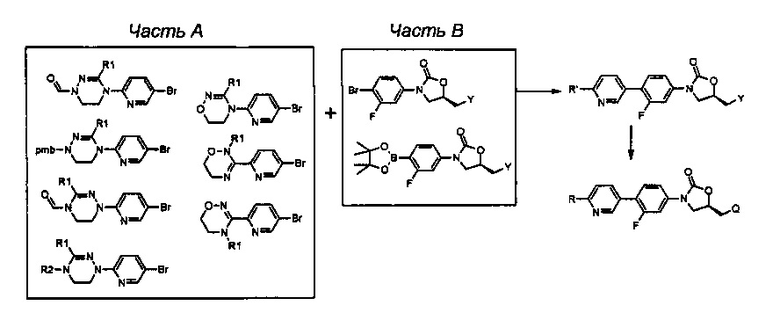
Как проиллюстрировано на вышеприведенной реакционной схеме 1, соединение формулы (1) синтезируют посредством реакции связывания между бромпиридиновой частью, имеющей циклическую амидоксимную или циклическую амидразоновую группу, то есть, частью А, и частью В, имеющей оксазолидиноновую группу. После реакции связывания, добавляют различные производные по отношению к сайту R' для превращения группы R' в группу R и группу Y превращают в группу Q за счет реакции У с различными производными, таким образом, завершая синтез соединения формулы (1). В дополнение, такая же группа, как Q, указанная в вышеприведенной формуле (1), может быть введена в сайт Y и Y может быть выбран из различных промежуточных реакционных групп, включая Q. Подобным образом, R' также может быть выбран из различных промежуточных реакционных групп, включая группу R, указанную в вышеприведенной формуле (1).
Сначала, бромпиридиновые производные, каждое, имеющее циклическую амидразоновую группу, согласно части А реакционной схемы 1, могут быть синтезированы согласно нижеприводимой реакционной схеме 2.
Реакционная схема 2
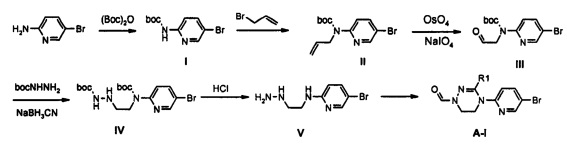
Как представлено на реакционной схеме 2, соединение I синтезируют путем введения во взаимодействие 2-амино-5-бромпиридина с (Вос)2O и соединение II синтезируют путем введения во взаимодействие соединения I с аллилбромидом. Соединение II превращают в альдегид III, используя OsO4 и NaIO4, и затем подвергают реакции с BocNHNH2, получая соединение IV. Синтезированное соединение IV обрабатывают кислотой с получением соединения V и затем подвергают реакции со сложным ортоэфиром, получая соединение A-I.
Однако способ согласно реакционной схеме 2 включает использование OsO4, который является относительно дорогостоящим и является очень токсичным, и, таким образом, может быть предложен другой способ, согласно которому не используют OsO4, и который осуществляют в соответствии с нижеприводимой реакционной схемой 3.
Реакционная схема 3
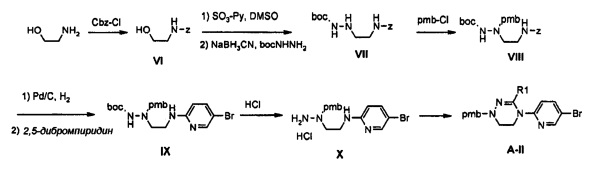
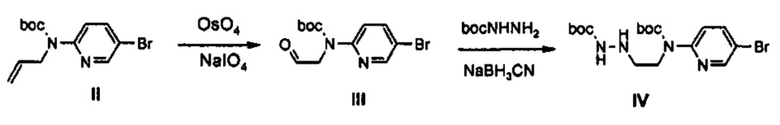
Как представлено на реакционной схеме 3, этаноламин подвергают реакции с Cbz-Cl и затем окисляют с помощью SO3-Py, получая альдегид, и альдегид подвергают реакции с BocNHNH2 для синтеза соединения VII. Затем соединение VII подвергают реакции с pmb-Cl для синтеза соединения VIII и Cbz-группу удаляют из полученного соединения с помощью Pd/C в присутствии водорода и подвергают реакции с дибромпиридином, получая соединение IX. После этого соединение IX обрабатывают соляной кислотой, получая соединение X, и полученное соединение X подвергают реакции со сложным ортоэфиром для синтеза соединения A-II.
Соединение, местоположение амина в котором отличается от такового циклического амидразона, синтезируют согласно нижеприводимой реакционной схеме 4.
Реакционная схема 4
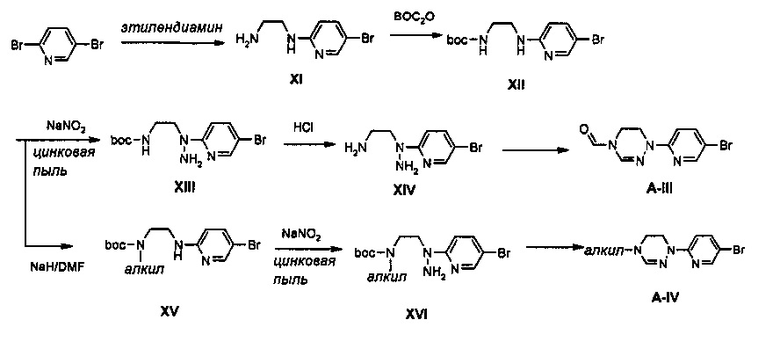
Boc-группу присоединяют к соединению IX, полученному путем введения во взаимодействие дибромпиридина с этилендиамином, получая соединение XII. Соединение XII подвергают аминированию с помощью NaNO2 и Zn, обработанного кислотой, и затем подвергают реакции со сложным ортоэфиром, получая соединение А-III. В качестве другого способа, синтез производного, имеющего введенную в него алкильную группу, осуществляют таким образом, что соединение XII подвергают алкилированию, получая соединение XV, имеющее введенную в него алкильную группу, и затем подвергают аминированию и циклизации с помощью сложного ортоэфира, синтезируя соединение А-IV, имеющее введенную в него алкильную группу.
Способы синтеза бромпиридинов, каждого, имеющих циклическую амидоксимную группу, представлены на нижеприводимой реакционной схеме 5.
Реакционная схема 5
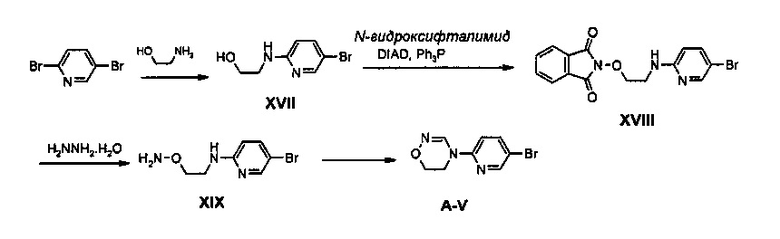
Сначала синтезируют соединение XVII путем введения во взаимодействие дибромпиридина с этаноламином и затем путем осуществления реакции Mitsunobu с гидроксифталимидом, получая соединение XVIII, из него удаляют фталимидную группу при использовании гидразина и после этого полученное в результате соединение подвергают циклизации с помощью сложного ортоэфира, получая соединение A-V.
Способ синтеза производного бромпиридина, имеющего другой тип циклической амидоксимной группы, представлен на реакционной схеме 6.
Реакционная схема 6
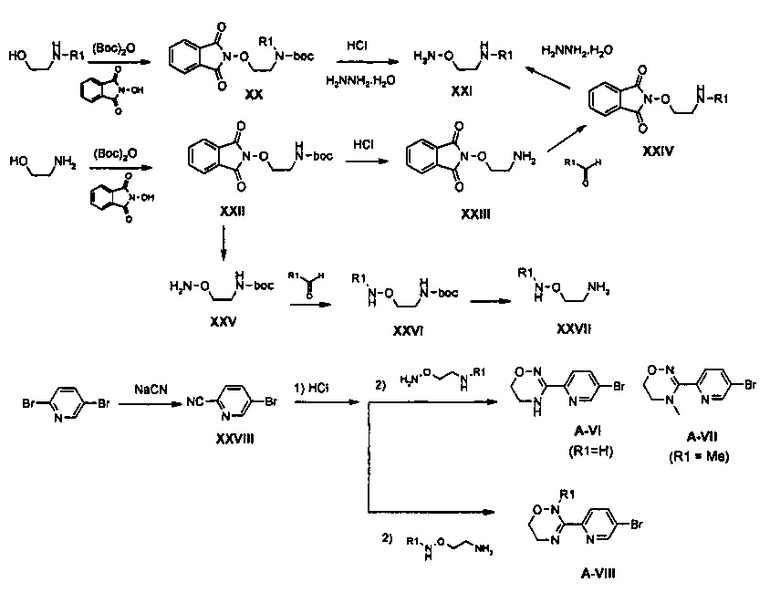
Соединение XX получают путем введения во взаимодействие N-алкилэтаноламина с Boc2O и затем путем осуществления реакции Mitsunobu с гидроксифталимидом, полученное в результате соединение обрабатывают соляной кислотой для удаления Boc-группы и фтал-имидную группу удаляют из него при использовании гидразина, получая соединение XXI, в которое введена алкильная группа R1. Кроме того, в качестве способа введения алкильной группы R1 позже, сначала соединение XXII может быть получено из этаноламина посредством реакции Mitsunobu и затем может быть обработано соляной кислотой, получая соединение XXIII, соединение XXIII может быть подвергнуто реакции с альдегидом, получая соединение XXIV, в которое введена алкильная группа R1, и затем фталимидная группа может быть удалена из соединения XXIV при использовании гидразина, получая соединение XXI.
Кроме того, в качестве способов синтеза других соединений, местоположение R1 в которых является отличным от местоположения в вышеуказанном соединении, сначала из соединения XXII удаляют фталимидную группу при использовании гидразина и затем подвергают реакции с алкилальдегидом, получая соединение XXVI, в которое введена алкильная группа, и полученное в результате соединение обрабатывают соляной кислотой, получая соединение XXVII, из которого удаляют Boc-группу. Полученные соединения XXI и XXVII, каждое, подвергают реакции с цианобромпиридином XXVIII для синтеза соединений A-VI, A-VII и A-VIII. Между тем, соединения формулы (1), где R1 означает водород, можно не подвергать реакции с алкилальдегидом для получения каждого результирующего соединения.
На вышеприведенной реакционной схеме 1, способы синтеза соединений В-части, имеющих оксазолидиноновую группу, представлены на нижеприводимой реакционной схеме 7.
Реакционная схема 7
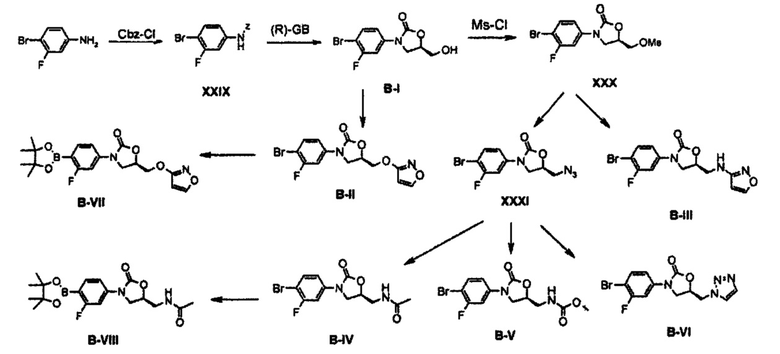
Соединение XXIX получают путем введения во взаимодействие 3-фтор-4-броманилина с Cbz-Cl и затем его вводят во взаимодействие с (R)-глицидилбутиратом для синтеза хирального соединения B-I. Спиртовую группу соединения B-I превращают в различные типы производных соединений Y, синтезируя соединения В-II, В-III, В-IV, B-V и B-VI, с последующим связыванием с А-частью, или бром-группу соединения B-I превращают в пинаколборановую группу, получая соединение B-VII, и затем подвергают связыванию, таким образом завершая синтез соединения формулы (1).
Настоящее изобретение также относится к фармацевтической композиции антибиотика, которая включает: (а) терапевтически эффективное количество нового производного оксазолидинона формулы (1), его пролекарства, его гидрата, его сольвата, его изомера или его фармацевтически приемлемой соли; и (b) фармацевтически приемлемый носитель, разбавитель, эксципиент или их комбинацию.
Термин «фармацевтическая композиция», как используемый в данном контексте, означает смесь соединения согласно настоящему изобретению и других химических компонентов, как например разбавитель или носитель. Фармацевтическая композиция облегчает введение соединения в организм. Введение соединения может быть осуществлено, используя различные способы. Примеры различных способов введения включают, но не исчерпывающим образом, пероральное введение, инъекцию, аэрозольное введение, парентеральное введение и локальное введение. Фармацевтическая композиция может быть получена через посредство реакции с кислотой, как например соляная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, метансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота или т.п.
Как используется в данном контексте, термин «терапевтически эффективное количество» означает эффективное количество активного ингредиента фармацевтической композиции для облегчения или уменьшения одного или более симптомов нарушений, подвергаемых лечению композицией, или для замедления стимулирования клинических маркеров или симптомов заболеваний, необходимых для предотвращения. Таким образом, термин «терапевтически эффективное количество» означает количество, которое обладает эффектом: (1) реверсирования скорости прогрессирования заболеваний, (2) до некоторой степени ингибирования дальнейшего прогрессирования заболеваний и/или (3) до некоторой степени ослабления (предпочтительно, устранения) одного или более связанных с заболеваниями симптомов. Терапевтически эффективное количество может быть экспериментально определено путем испытания соединения на известной in vivo и in vitro модельной системе для заболеваний, нуждающихся в лечении.
Термин «носитель» определяют как соединение, которое облегчает доставку соединения в клетки или ткани. Например, диметилсульфоксид (ДМСО) представляет собой обычно используемый носитель, который облегчает введение многих органических соединений в клетки или ткани организма.
Термин «разбавитель» определяют как соединение, которое стабилизирует биологически активную форму соединения-мишени и разводится в воде, используемой для растворения соединения. Соли, растворенные в буферных растворах, используют в качестве разбавителей в уровне техники. Обычно используемым буферным раствором является забуференный фосфатом солевой раствор, так как он обладает подобной соленостью человеческому организму. Буферные соли могут регулировать значение pH раствора при низких концентрациях и, таким образом, забуференный разбавитель редко модифицирует биологическую активность соединения.
Используемое соединение может быть введено пациенту индивидуально или может быть введено пациенту в виде фармацевтической композиции, получаемой путем смешивания соединения с другими активными ингредиентами или с соответствующим носителем или эксципиентом, как в случае комбинированной терапии. Способы получения готовой лекарственной формы и введения соединения согласно настоящей заявке можно найти в «Remington's Pharmaceutical Sciences», Mack Publishing Co, Easton, PA, восемнадцатое издание, 1990.
Фармацевтическая композиция согласно настоящему изобретению может быть получена известным образом посредством способов, как например обычное смешивание, растворение, грануляция, дражирование, растирание в порошок, эмульгирование, инкапсуляция, улавливание или лиофилизация.
Таким образом, фармацевтические композиции для применения согласно настоящему изобретению могут быть получены обычным образом, используя один или более фармацевтически приемлемых носителей, включая эксципиенты или вспомогательные агенты, которые облегчают обработку активных соединений до готовых лекарственных форм для фармацевтического применения. Подходящая готовая лекарственная форма зависит от выбранного пути введения. Любые подходящие хорошо известные способы, носители и эксципиенты могут быть использованы, как подразумеваемые в уровне техники, например, согласно указанному выше руководству «Remingston's Pharmaceutical Sciences». Соединение формулы (1) согласно настоящему изобретению может быть использовано для получения готовой лекарственной формы для инъекции, перорального введения или т.п. в соответствии с предназначенным применением.
Для инъекции, композиция согласно настоящему изобретению может быть получена в виде водного раствора, предпочтительно, физиологически приемлемого буфера, как, например, раствор Хэнкса, раствор Рингера или физиологический солевой буфер. Для трансмукозального введения, в композиции используют неинвазивные агенты, пригодные для барьера, через который проходит композиция. Такие неинвазивные агенты обычно известны в уровне техники.
Для перорального введения, соединения могут быть использованы для получения композиции путем комбинирования активных соединений с фармацевтически приемлемыми носителями, известными в уровне техники. Такие носители дают возможность использования соединений согласно настоящему изобретению для получения готовых лекарственных форм в виде таблеток, пилюль, порошков, гранул, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий, и т.п. Предпочтительными являются капсулы, таблетки, пилюли, порошки и гранулы и, в особенности, могут быть использованы капсулы и таблетки. Таблетки и пилюли могут быть получены с энтеросолюбильными покрытиями. Фармацевтические композиции для перорального использования могут быть получены путем смешивания одного или более твердых эксципиентов с одним или более соединениями согласно изобретению, необязательно измельчая полученную в результате смесь, и обработки смеси до гранул, после добавления вспомогательных агентов, если желательно, для получения таблеток или ядер драже. Подходящие эксципиенты включают, в особенности, наполнители, как например лактоза, сахароза, маннит или сорбит; вещества на основе целлюлозы, как например кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрийкарбоксиметилцеллюлоза и/или поливинилпирролидон (PVP). Если желательно, могут быть добавлены дезинтегрирующие агенты, как например сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, как например, альгинат натрия, смазочные вещества, как например стеарат магния, и носители, как например связующие вещества, и т.п.
Фармацевтические композиции, которые могут быть введены перорально, включают твердые капсулы, сделанные из желатина, а также мягкие, герметизированные капсулы, сделанные из желатина, и пластификатор, как например глицерин или сорбит. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, как например лактоза, связующим веществом, как например крахмал, и/или смазочным веществом, как например тальк или стеарат магния. В случае мягких капсул, активные соединения могут быть растворены или суспендированы в подходящих жидкостях, как например жировые масла, жидкий парафин или жидкий полиэтиленгликоль. В дополнение, могут быть добавлены стабилизаторы. Все готовые лекарственные формы для перорального введения должны быть в дозировках, подходящих для такого введения.
Соединения могут быть использованы для получения готовой лекарственной формы для парентерального введения путем инъекции, например, путем инъекции ударной дозы вещества или непрерывной инфузии. Композиции для инъекции могут быть предусмотрены в виде стандартной лекарственной формы, например, в ампулах или многодозовых контейнерах, с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных средах, и могут содержать агенты для приготовления лекарственной формы, как например суспендирующие агенты, стабилизаторы и/или диспергаторы.
Кроме того, соединения могут быть в форме сухого порошка, которую используют после растворения в стерильной непирогенной воде.
Соединения могут быть использованы для получения готовой лекарственной формы в виде суппозиториев, включая обычные вещества для суппозитория, как например масло какао или другие глицериды, или в виде композиций для ректального введения, как например удерживающая клизма.
Фармацевтические композиции, подходящие для использования согласно настоящему изобретению, включают композиции, где активные ингредиенты содержатся в количестве, эффективном для достижения предназначенной им цели. Более конкретно, термин «терапевтически эффективное количество» означает количество соединения, эффективное для продления продолжительности существования субъекта, подвергаемого лечению, или для предотвращения, ослабления или уменьшения интенсивности симптомов заболеваний. Определение терапевтически эффективного количества может быть в пределах компетенции квалифицированного специалиста в данной области, в особенности, с точки зрения предусмотренного в данном контексте подробного описания.
Когда получают готовую лекарственную форму в виде стандартной лекарственной формы, фармацевтическая композиция может включать соединение формулы (1) в качестве активного ингредиента в стандартной дозе приблизительно от 0,1 мг до 1500 мг. Подходящую дозу соединения формулы (1) вводят в соответствии с предписанием докторов в зависимости от факторов, как, например, масса тела и возраст пациентов и конкретных свойств и тяжести заболеваний. Однако включенное в готовую лекарственную форму соединение может быть введено от одного до трех раз в сутки для лечения взрослых, согласно частоте и интенсивности введения, и его доза обычно находится в диапазоне от примерно 1 мг до примерно 1500 мг. Когда вводят взрослому внутримышечно или внутривенно, фармацевтическая композиция может быть введена от одного до трех раз в сутки и доза соединения обычно может составлять от примерно 1 мг до примерно 1500 мг. Для некоторых пациентов, однако, может быть использована более высокая доза.
Фармацевтическая композиция согласно настоящему изобретению может быть составлена вместе, по меньшей мере, с одним известным лекарственным средством, выбираемым из клинически пригодных антибактериальных агентов (например, β-лактам, макролид, хинолон и аминогликозид) и противовоспалительных агентов (например, противогрибковый триазол или амфотерицин), или может быть совместно введена вместе с одним или более известных лекарственных средств. Соединение согласно настоящему изобретению может быть использовано для получения готовой лекарственной формы вместе с, или совместно вводимым, повышающим бактерицидность/проницаемость белком (BPI) или ингибитором насоса оттока, для увеличения активности против грамотрицательных бактерий и антибиотика, резистентного к бактериям.
Соединение согласно настоящему изобретению может быть использовано для получения готовой лекарственной формы вместе с витаминами или может быть введено вместе с витаминами, например, витамином В2, витамином В6 или витамином В12, и фолиевой кислотой. Соединение согласно настоящему изобретению может быть использовано для получения готовой лекарственной формы вместе с ингибитором циклооксигеназы (СОХ) или может быть введено вместе с ингибитором циклооксигеназы, в особенности, с ингибитором СОХ-2.
Настоящее изобретение также относится к способу лечения антибиотиком, осуществляемому при использовании эффективного количества нового производного оксазолидинона, представленного вышеприведенной формулой (1), его пролекарства, его гидрата, его сольвата, его изомера или его фармацевтически приемлемой соли.
Наилучший вариант осуществления настоящего изобретения
В дальнейшем, настоящее изобретение должно быть описано более подробно со ссылкой на нижеследующие примеры. Эти примеры предусмотрены только для иллюстрации настоящего изобретения и не должны быть истолкованы как ограничивающие объем и сущность настоящего изобретения.
Для синтеза соединения формулы (1), сначала осуществляют синтез части А и части B в соответствии с нижеследующими примерами получения.
Пример получения 1
Получение соединения I
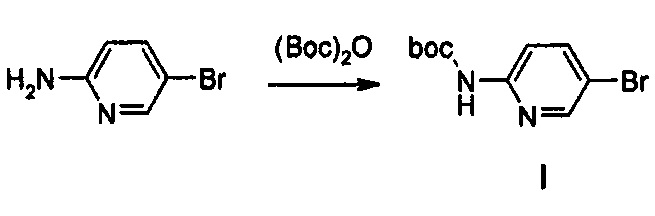
10 г (57,80 ммоль) 2-Амино-5-бромпиридина, 17,4 мл (124,47 ммоль) триэтиламина, 16,3 г (74,75 ммоль) ди-трет-бутилдикарбоната и 0,5 г (4,05 ммоль) диметиламинопиридина добавляют к 270 мл дихлорметана, при температуре 0°C, и полученный раствор перемешивают в течение 3 часов.
Реакционную смесь растворяют в 300 мл дихлорметана и затем промывают 200 мл водного насыщенного раствора бикарбоната натрия и полученный раствор обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении и использованием колоночной хроматографии, получая 9,8 г (36,21 ммоль) соединения I в виде твердого вещества желтого цвета (выход: 63%).
1H ЯМР (600 МГц, CDCl3) δ 8,32 (д, J=2,4 Гц, 1Н), 7,97 (с, 1Н), 7,90 (д, J=9,0 Гц, 1Н), 7,75 (дд, J=9,0 Гц, J2=2,4 Гц, 1Н), 1,55 (с, 9Н).
Пример получения 2
Получение соединения II
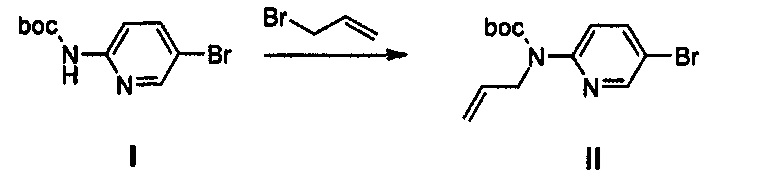
9,89 г (36,21 ммоль) Соединения I, 20 г (43,45 ммоль) карбоната цезия и 3,7 6 мл (43,45 ммоль) аллилбромида добавляют к 200 мл диметилформамида, при комнатной температуре, и полученный раствор перемешивают при температуре 70°C в течение 2,5 часов.
Реакционную смесь охлаждают до температуры 0°C, медленно добавляют 200 мл дистиллированной воды и полученный раствор разбавляют с помощью 600 мл этилацетата и затем последовательно промывают 500 мл дистиллированной воды, 250 мл 0,5 н. HCl и 200 мл водного насыщенного раствора хлорида натрия.
После этого, полученный раствор обезвоживают, используя безводный сульфат натрия, и концентрируют при пониженном давлении, получая 11 г (35,12 ммоль) соединения II в виде масла желтого цвета (выход: 97%).
1H ЯМР (600 МГц, CDCl3) δ 8,38 (д, J=1,8 Гц, 1Н), 7,70 (дд, J1=8,4 5 Гц, J2=2,4 Гц, 1Н), 7,66 (д, J=8,4 Гц, 1Н), 5,93 (м, 1Н), 5,12 (м, 2Н), 4,53 (м, 2Н) 1,50 (с, 9Н).
Пример получения 3
Получение соединения IV
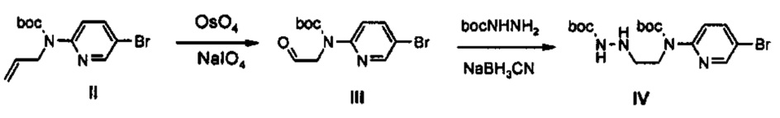
11 г (35,12 ммоль) Соединения II, 11 мл (0,88 ммоль) OsO4 и 30 г (140,48 ммоль) NaIO4 добавляют в указанной последовательности, при температуре 0°C, полученный раствор перемешивают при температуре 0°C в течение 5 часов, затем добавляют 7 г (32,73 ммоль) NaIO4 и полученный раствор перемешивают в течение 1 часа. Реакционную смесь отфильтровывают и промывают с помощью 300 мл этилацетата. Органический слой промывают с помощью 300 мл дистиллированной воды, обезвоживают безводным сульфатом натрия и потом концентрируют при пониженном давлении, получая 14,19 г соединения III в виде масла коричневого цвета.
14,19 г Полученного соединения III, 12,87 г (97,50 ммоль) BocNHNH2, 3,34 г (53,16 ммоль) цианоборгидрида натрия и 2,1 мл (35,44 ммоль) уксусной кислоты, добавляют в указанной последовательности, при температуре 0°C, и полученный раствор перемешивают при комнатной температуре в течение 3 часов. После этого, к реакционной смеси добавляют 150 мл дистиллированной воды и полученный раствор перемешивают при комнатной температуре в течение 20 минут и затем экстрагируют с помощью 500 мл этилацетата и 300 мл водного раствора бикарбоната натрия. Затем, водный слой промывают с помощью 300 мл этилацетата, обезвоживают безводным сульфатом натрия и потом концентрируют при пониженном давлении. Концентрат подвергают колоночной хроматографии, получая 10,92 г (25,32 ммоль) соединения IV в виде масла (выход: 72%).
1H ЯМР (600 МГц, CDCl3) δ 8,39 (д, J=2,4 Гц, 1Н), 7,71 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 7,60 (д, J=9,0 Гц, 1Н), 6,52 (с, 1Н), 4,01 (м, 1Н), 4,03 (т, J=6,0 Гц, 2Н), 3,07 (т, J=6,0 Гц, 2Н) 1,52 (с, 9Н), 1,46 (с, 9Н).
Пример получения 4
Получение соединения A-I
10,92 г (25,32 ммоль) Соединения VI добавляют к 7 0 мл метанола, добавляют 120 мл 4 М HCl, полученный раствор перемешивают при комнатной температуре в течение 12 часов и раствор после перемешивания концентрируют при пониженном давлении. 7,5 г полученного соединения V, 40 мл триметилортоформиата и 3,91 г (47,68 ммоль) ацетата натрия добавляют к 40 мл уксусной кислоты, и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 4 часов. Смесь охлаждают до комнатной температуры и затем концентрируют при пониженном давлении, добавляют 300 мл дихлорметана и полученный раствор дважды промывают с помощью 300 мл водного насыщенного раствора бикарбоната натрия, обезвоживают безводным сульфатом натрия и затем концентрируют при пониженном давлении, получая 5,9 г (22,03 ммоль) соединения A-I в виде твердого вещества желтого цвета (выход: 87%).
1H ЯМР (600 МГц, CDCl3) δ 8,59 (с, 1Н), 8,37 (д, J=2,4 Гц, 1Н), 7,82 (с, 1Н), 7,78 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 6,80 (д, J=9,0 Гц, 1Н), 4,00 (т, J=5,4 Гц, 2Н), 3,85 (т, J=5,4 Гц, 2Н).
Пример получения 5
Получение соединения VI
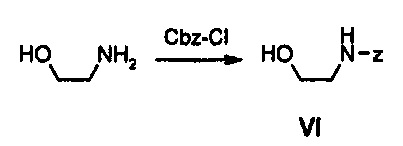
32 г (527,5 ммоль) 2-Аминоэтанола растворяют в 250 мл дихлорметана, добавляют 300 мл 1 н. водного раствора NaOH и медленно, по каплям, добавляют 60 г (351,7 ммоль) Cbz-Cl (бензилхлорформиат), при перемешивании получаемого раствора. Полученный раствор перемешивают при комнатной температуре в течение 2 часов, органический слой отделяют и дважды промывают водой, и органический слой после промывки обезвоживают, используя безводный сульфат натрия, и концентрируют при пониженном давлении, получая 62 г (317,6 ммоль) соединения VI в виде твердого вещества белого цвета (выход: 90%).
1H ЯМР (600 МГц, CDCl3) δ 7,36 (м, 5Н), 5,15 (с, 1Н), 5,11 (с, 2Н), 3,73 (м, 2Н), 3,37 (м, 2Н), 2,08 (с, 1Н).
Пример получения 6
Получение соединения VII
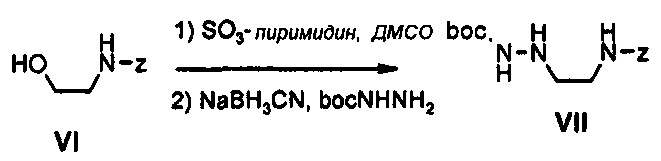
30 г (153,7 ммоль) Соединения VI, 49 г (307,4 ммоль) сульфонтриоксида, 86 мл (614,4 ммоль) триэтиламина и 120 мл ДМСО добавляют к 250 мл дихлорметана, при температуре 0°C, и полученный раствор перемешивают при комнатной температуре в течение 2 часов. После этого, к реакционной смеси добавляют 1000 мл диэтилового эфира и полученный раствор последовательно промывают 500 мл дистиллированной воды, 800 мл 0,5 н. HCl и 500 мл дистиллированной воды, обезвоживают безводным сульфатом натрия и затем концентрируют при пониженном давлении.
Полученный альдегид растворяют в 300 мл метанола, добавляют в указанной последовательности 22 г (169,0 ммоль) трет-бутилшабазита, 11,6 г (184,4 ммоль) цианоборгидрида натрия и 11 мл (184,4 ммоль) уксусной кислоты, при температуре 0°C, и затем полученный раствор перемешивают при комнатной температуре в течение 12 часов. После этого к реакционной смеси добавляют 11 мл (184,4 ммоль) уксусной кислоты и полученный раствор концентрируют при пониженном давлении, экстрагируют, используя 800 мл этилацетата и 500 мл водного насыщенного раствора бикарбоната натрия, и затем подвергают колоночной хроматографии, получая 25 г (80,8 ммоль) соединения VII в виде масла коричневого цвета (выход: 53%).
1H ЯМР (600 МГц, CDCl3) δ 7,32 (м, 5Н), 6,17 (с, 1Н), 5,47 (с, 1Н), 5,14 (с, 1Н), 5,10 (с, 2Н), 3,29 (м, 2Н), 2,90 (м, 2Н), 1,45 (с, 9Н).
Пример получения 7
Получение соединения VIII
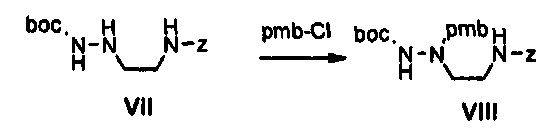
25 г (80,8 ммоль) Соединения VII, 32 г (200 ммоль) параметоксибензилхлорида и 43 мл (243 ммоль) диизопропилэтиламина добавляют к 50 мл диметилформамида, и полученный раствор перемешивают при температуре 80°C в течение 5 часов. Затем к реакционной смеси добавляют 500 мл этилацетата, полученный раствор последовательно промывают с помощью 500 мл дистиллированной воды и 500 мл водного насыщенного раствора бикарбоната натрия, и раствор после промывки обезвоживают, используя безводный сульфат натрия, концентрируют при пониженном давлении и затем подвергают колоночной хроматографии, получая 19 г (44,0 ммоль) соединения VIII в виде масла коричневого цвета.
1H ЯМР (600 МГц, CDCl3) δ 7,35 (м, 5Н), 7,25 (д, J=8,4 Гц, 2Н), 6,87 (д, J=8,4 Гц, 2Н), 5,99 (с, 1Н), 5,38 (с, 1Н), 3,92 (с, 2Н), 3,81 (с, 3Н), 3,29 (м, 2Н), 2,79 (м, 2Н), 1,40 (с, 9Н).
Пример получения 8
Получение соединения IX
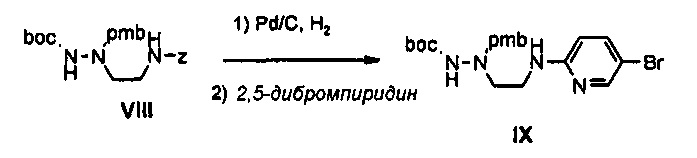
12 г (27,9 ммоль) Соединения VIII и 1,2 г Pd/C добавляют к 200 мл этанола, полученный раствор перемешивают в атмосфере водорода в течение 1 часа, раствор после перемешивания отфильтровывают через целит, и затем концентрируют при пониженном давлении, добавляют 13,3 г (55,87 ммоль) 2,5-дибромпиридина и полученный раствор перемешивают при температуре 140°C в течение 1 часа. Реакционную смесь растворяют в 200 мл дихлорметана и затем промывают 50 мл насыщенного раствора бикарбоната натрия. После этого, раствор после промывки обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении, и концентрат подвергают колоночной хроматографии, получая 4,2 г (9,3 ммоль) соединения IX в виде масла коричневого цвета (выход: 33%).
1H ЯМР (600 МГц, CDCl3) δ 8,07 (с, 1Н), 7,43 (д, J=7,8 Гц, 1Н), 7,23 (д, J=8,4 Гц, 2Н), 6,85 (д, J=8,4 Гц, 2Н), 6,42 (д, J=7,8 Гц, 1Н), 5,72 (с, 1Н), 5,39 (с, 1Н), 3,92 (с, 2Н), 3,82 (с, 3Н), 3,40 (м, 2Н), 2,88 (м, 2Н), 1,40 (с, 9Н).
Пример получения 9
Получение соединения А-II
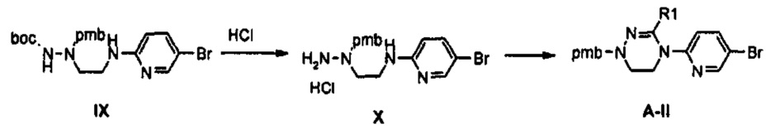
3 г (6,6 ммоль) Соединения IX и 15 мл 4 М HCl добавляют к 15 мл дихлорметана, полученный раствор перемешивают при комнатной температуре в течение 2 часов, и раствор после перемешивания концентрируют при пониженном давлении, получая соединение X в виде гидрохлорида, из которого удалена Boc-группа.
Затем, к полученному соединению X добавляют 10 мл триметоксиортоформиата и 20 мл уксусной кислоты, и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении и затем экстрагируют, используя 150 мл этилацетата и 50 мл водного насыщенного раствора бикарбоната натрия. После этого, экстракт обезвоживают, используя безводный сульфат натрия, и затем подвергают колоночной хроматографии, получая 1 г (2,8 ммоль) соединения А-II в виде твердого вещества цвета слоновой кости (выход: 42%).
1H ЯМР (600 МГц, CDCl3) δ 8,29 (д, J=3,0 Гц, 1Н), 7,87 (с, 1Н), 7,67 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 7,32 (д, J=8,4 Гц, 2Н), 6,89 (д, J=8,4 Гц, 2Н), 6,65 (д, J=9,0 Гц, 1Н), 4,09 (с, 2Н), 3,81 (с, 3Н), 3,79 (т, J=5,4 Гц, 2Н), 2,92 (т, J=5,4 Гц, 2Н).
Пример получения 10
Получение соединения XI

1 г (4,22 ммоль) 2,5-дибромпиридина добавляют к 10 мл этилендиамина и полученный раствор перемешивают при температуре 100°C в течение 15 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении для удаления этилендиамина. После этого, добавляют 50 мл дихлорметана для разбавления концентрата, разбавленный раствор промывают 30 мл дистиллированной воды и затем к собранной дистиллированной воде добавляют 50 мл дихлорметана для экстракции от органического слоя, эти процессы повторяют более двух раз. Объединенные органические слои обезвоживают, используя безводный сульфат натрия, затем концентрируют при пониженном давлении, получая 0,8 9 г (4,12 ммоль) соединения XI в виде жидкости светло-желтого цвета (выход: 98%).
1H ЯМР (400 МГц, CDCl3) δ 8,10 (д, J=1,6 Гц, 1Н), 7,45 (дд, J=8,8 Гц, J2=2,4 Гц, 1Н), 6,33 (д, J=8,8 Гц), 4,95 (уш.с, 1Н), 3,34 (кв, J=6 Гц, 2Н), 2,94 (т, J=6 Гц, 2Н).
Пример получения 11
Получение соединения XII
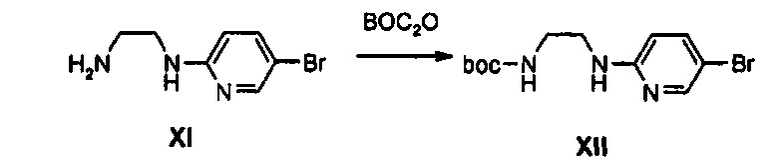
0,89 г (4,12 ммоль) Соединения XI растворяют в 10 мл 1,4-диоксана, полученный раствор охлаждают до температуры 0°C, добавляют 0,44 г (4,12 мл) карбоната натрия, растворенного в 20 мл дистиллированной воды, и медленно, по каплям, к полученному раствору добавляют 0,99 мл (4,32 ммоль) ди-трет-бутилдикарбоната, растворенного в 5 мл 1,4-диоксана. После этого, температуру реакционной смеси повышают до комнатной температуры, реакционную смесь перемешивают в течение 15 часов и затем концентрируют при пониженном давлении для удаления 1,4-диоксана, медленно добавляют 2 н. водный раствор хлороводорода, при температуре 0°C, для снижения значения pH до 4, и полученный раствор экстрагируют, используя 250 мл этилацетата, для получения органического слоя. Органический слой обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении, получая твердый продукт. Твердый продукт промывают 30 мл н-гексана, получая 1,28 г (4,05 ммоль) соединения XII в виде твердого вещества светло-желтого цвета (выход: 98%).
1H ЯМР (400 МГц, CDCl3) δ 8,09 (д, J=1,6 Гц, 1Н), 7,44 (дд, J=8,8 Гц, J2=2,4 Гц, 1Н), 6,33 (д, J=8,8 Гц, 1Н), 4,94 (уш.с, 1Н), 3,44 (м, 2Н), 3,35 (м, 2Н), 1,44 (с, 9Н).
Пример получения 12
Получение соединения XIII
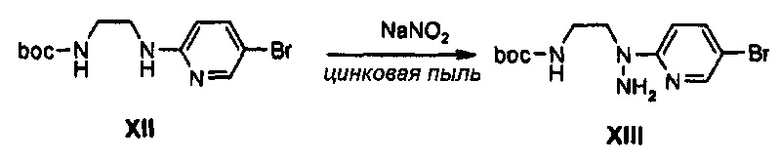
0,5 г (1,58 ммоль) соединения XII растворяют в 6 мл уксусной кислоты, полученный раствор охлаждают до температуры 0°C и медленно, по каплям, добавляют 0,11 г (1,66 ммоль) нитрита натрия, растворенного в 2 мл дистиллированной воды. Реакционную температуру повышают от 0°C до комнатной температуры, полученный раствор перемешивают в течение 30 минут, температуру снова понижают до температуры 0°C, и к полученному раствору добавляют 0,21 г (3,16 ммоль) цинка, и затем перемешивают еще в течение 1 часа. Раствор после перемешивания нейтрализуют при температуре 0°C, используя 50 мл дистиллированной воды и насыщенный раствор бикарбоната натрия, добавляют 0,4 мл 4 М раствора хлороводорода в 1,4-диоксане, полученный раствор экстрагируют с помощью 50 мл этилацетата и экстракт обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении, получая 0,41 г (1,23 ммоль) соединения XIII в виде твердого вещества красновато-коричневого цвета (выход: 79%).
1Н ЯМР (600 МГц, CDCl3) δ 8,18 (м, 1Н), 7,65 (м, 2Н), 7,15 (м, 1Н), 5,80 (уш.с, 1Н), 4,04 (м, 2Н), 3,57 (м, 2Н), 1,32 (с, 9Н).
Пример получения 13
Получение соединения XIV
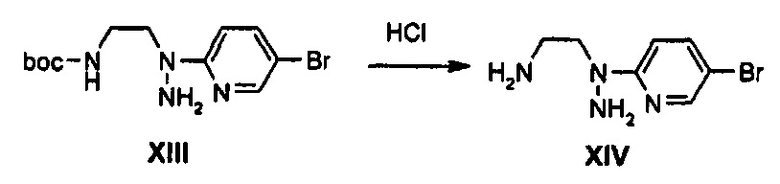
0,4 г (1,20 ммоль) Соединения XIII растворяют в 3 мл дихлорметана, полученный раствор охлаждают до температуры 0°C и, по каплям, добавляют 0,4 мл 4 М раствора хлороводорода в 1,4-диоксане, в атмосфере азота. После этого, полученный раствор перемешивают в течение 15 часов и затем концентрируют при пониженном давлении, получая твердый продукт. Твердый продукт промывают с помощью 20 мл диэтилового эфира, получая 0,28 г (0,97 ммоль) соединения XIV в виде твердого вещества светло-желтого цвета (выход: 80%).
1H ЯМР (600 МГц, CD3OD) δ 8,20 (д, J=2,4 Гц, 1Н), 8,12 (дд, J1=9,6 Гц, J2=2,4 Гц, 1Н), 7,30 (д, J=9,6 Гц, 1Н), 4,06 (т, J=6,6 Гц, 2Н), 3,40 (т, J=6,6 Гц, 2Н).
Пример получения 14
Получение соединения А-III
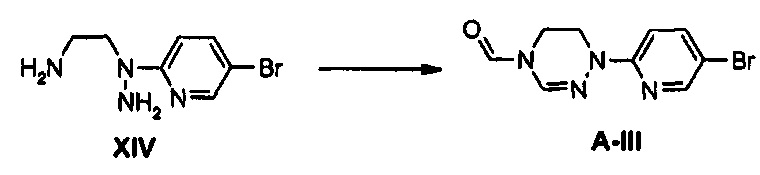
710 мг (2,45 ммоль) Соединения XIV и 2 мл триметилортоформиата добавляют к 8 мл уксусной кислоты и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 5 часов. Затем, реакционную смесь охлаждают до комнатной температуры и после этого концентрируют при пониженном давлении для удаления растворителя, добавляют 10 мл дистиллированной воды и полученный концентрат экстрагируют, используя 20 мл дихлорметана. После этого, к дистиллированной воде после экстракции добавляют 20 мл дихлорметана и повторно экстрагируют для получения органического слоя. Органический слой обезвоживают, используя безводный сульфат натрия, концентрируют при пониженном давлении и затем подвергают колоночной хроматографии, получая 290 мг (1,07 ммоль) соединения A-III в виде твердого вещества светло-желтого цвета (выход: 44%).
1H ЯМР (400 МГц, CDCl3) δ 8,49 (с, 1Н), 8,20 (с, 1Н), 7,65 (дд, J1=9,2 Гц, J2=2,4 Гц, 1Н), 7,33 (д, J=9,0 Гц, 1Н), 7,24 (с, 1Н), 4,07 (т, J=5,2 Гц, 2Н), 3,90 (т, J=5,2 Гц, 2Н).
Пример получения 15
Получение соединения XV
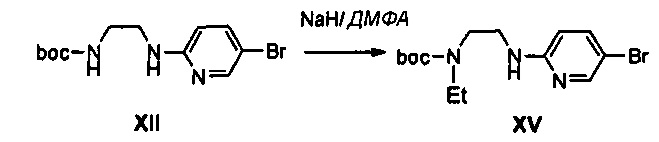
1 г (3,16 ммоль) соединения XII растворяют в 10 мл N,N-диметилформамида, к полученному раствору добавляют 0,21 г (74 ммоль) 55%-ного гидрида натрия, при температуре 0°C, полученный раствор перемешивают в течение 5 минут, и медленно, по каплям, к перемешиваемому раствору добавляют 0,3 мл (3,79 ммоль) иодэтана. После этого, температуру полученного раствора медленно повышают до комнатной температуры, и полученный раствор перемешивают в течение 3 часов. Температуру реакционной смеси снова понижают до температуры 0°C, медленно добавляют 10 мл дистиллированной воды, полученный раствор перемешивают в течение 5 минут и добавляют 30 мл этилацетата и 20 мл насыщенного раствора хлорида аммония, для экстракции органического слоя. Органический слой промывают с помощью 30 мл водного раствора тиосульфата натрия и обезвоживают, используя безводный сульфат натрия. Обезвоженный органический слой концентрируют при пониженном давлении и затем подвергают колоночной хроматографии, получая 0,48 г (1,39 ммоль) соединения XV в виде жидкости светло-желтого цвета (выход: 44%).
1H ЯМР (600 МГц, CDCl3) δ 8,09 (с, 1Н), 7,43 (м, 1Н), 6,31 (д, J=8,4 Гц, 1Н), 5,26 (уш.с, 1Н), 3,43 (м, 4Н), 3,22 (м, 2Н), 1,45 (с, 9Н), 1,10 (т, J=6,6 Гц, 1Н).
Пример получения 16
Получение соединения XVI
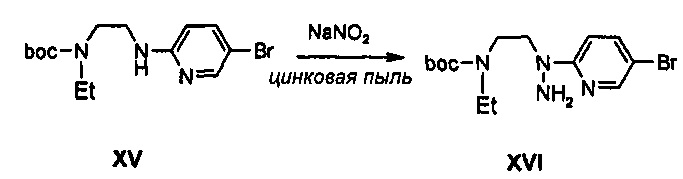
0,4 8 г (1,39 ммоль) соединения XV растворяют в 6 мл уксусной кислоты, полученный раствор охлаждают до температуры 0°C и медленно, по каплям, добавляют 0,11 г (1,66 ммоль) нитрита натрия, растворенного в 2 мл дистиллированной воды. Реакционную температуру повышают от 0°C до комнатной температуры, полученный раствор перемешивают в течение 30 минут, температуру снова понижают до температуры 0°C и к полученному раствору добавляют 0,27 г (4,18 ммоль) цинка, и затем перемешивают еще в течение 1 часа. Перемешиваемый раствор нейтрализуют при температуре 0°C, используя 50 мл дистиллированной воды и насыщенный раствор бикарбоната натрия, добавляют 0,4 мл 4 М раствора хлороводорода в 1,4-диоксане, полученный раствор экстрагируют 50 мл этилацетата и экстракт обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении, получая 0,51 г (1,29 ммоль) соединения XVI в виде твердого вещества светло-желтого цвета (выход: 93%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,24 (с, 1Н), 7,96 (с, 1Н), 7,09 (д, J=9 Гц, 1Н), 3,85 (м, 2Н), 3,40 (м, 2Н), 3,09 (м, 2Н), 1,39-1,30 (м, 9Н), 1,01 (т, J=6,6 Гц, 1Н).
Пример получения 17
Получение соединения А-IV
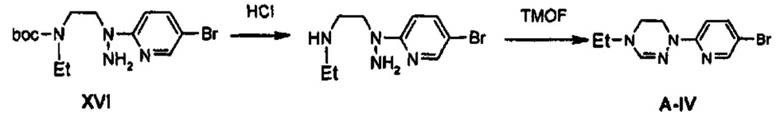
0,51 г (1,29 ммоль) соединения XVI растворяют в 20 мл дихлорметана, полученный раствор охлаждают до температуры 0°C и, по каплям, добавляют 20 мл 4 М раствора хлороводорода в 1,4-диоксане. После этого, полученный раствор перемешивают в течение 1 часа и затем концентрируют при пониженном давлении, получая твердый продукт. Твердый продукт промывают 20 мл диэтилового эфира, получая 0,42 г (1,26 ммоль) твердого соединения светло-желтого цвета, из которого удалена Boc-группа (выход: 98%).
1H ЯМР (400 МГц, ДМСО d-6) δ 9,20 (уш.с, 1Н), 8,29 (д, J=2 Гц, 1Н), 7,98 (дд, J1=9,2 Гц, J2=2,4 Гц, 1Н), 7,32 (д, J=9,2 Гц, 1Н), 4,04 (т, J=6,4 Гц, 1Н), 3,27 (т, J=6,4 Гц, 1Н), 2,97 (кв., J=7,2 Гц, 1Н), 1,21 (т, J=7,2 Гц, 1Н).
Затем, 0,4 г (1,20 ммоль) полученного соединения и 3 мл триметилортоформиата добавляют к 6 мл уксусной кислоты, и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и затем концентрируют при пониженном давлении, добавляют 10 мл дистиллированной воды и полученный раствор экстрагируют, используя 20 мл дихлорметана, для получения органического слоя. Органический слой обезвоживают безводным сульфатом натрия, концентрируют при пониженном давлении и затем подвергают колоночной хроматографии, получая 171 мг (0,63 ммоль) соединения А-IV в виде твердого вещества светло-коричневого цвета (выход: 53%).
1Н ЯМР (600 МГц, CDCl3) δ 8,11 (д, J=1,8 Гц, 1Н), 7,55 (дд, J1=9 Гц, J2=2,4 Гц, 1Н), 7,19 (д, J=9 Гц, 1Н), 6,72 (с, 1Н), 3,98 (т, J=4,8 Гц, 2Н), 3,39 (т, J=4,8 Гц, 2Н), 3,17 (кв, J=7,2 Гц, 2Н), 1,18 (т, J=7,2 Гц, 2Н).
Пример получения 18
Получение соединения XVII
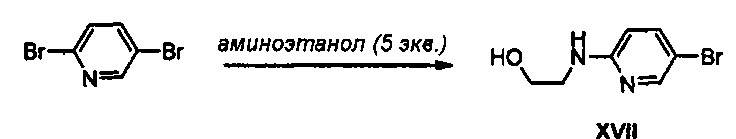
10 г (42,20 ммоль) 2,5-Дибромпиридина добавляют к 13 мл 2-аминоэтанола и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и растворяют в этилацетате, полученный раствор промывают водным насыщенным раствором бикарбоната натрия и затем обезвоживают безводным сульфатом натрия, потом концентрируют при пониженном давлении, получая 9,05 г (41,46 ммоль) соединения XVII в виде твердого вещества белого цвета (выход: 98%).
1H ЯМР (400 МГц, CDCl3) δ 8,08 (д, J=1,6 Гц, 1Н), 7,46 (дд, J1=8,8 Гц, J2=1,6 Гц, 1Н), 6,37 (д, J=8,8 Гц), 4,87 (с, 1Н), 3,81 (т, J=4,4 Гц, 2Н), 3,65 (с, 1Н), 3,45 (м, 2Н).
Пример получения 19
Получение соединения XVIII
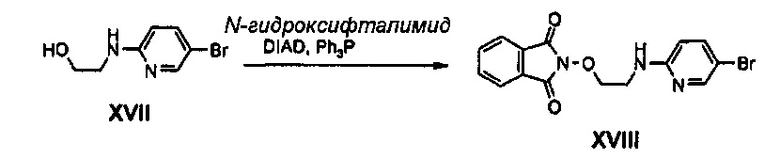
9 г (41,46 ммоль) Соединения XVII, 7,44 г (45,61 ммоль) N-гидроксифталимида и 14,14 г (53,90 ммоль) трифенилфосфина добавляют к 150 мл тетрагидрофурана, и полученный раствор перемешивают в атмосфере аргона. Затем, к перемешиваемому раствору медленно, по каплям, добавляют 10,61 мл (53,90 ммоль) диизопропилазодикарбоксилата, при температуре -5°C. После этого, полученное твердое вещество, спустя 1 час, отфильтровывают и фильтрат концентрируют при пониженном давлении, и затем подвергают колоночной хроматографии, получая 8,1 г соединения XVIII в виде твердого вещества белого цвета (выход: 54%).
1Н ЯМР (600 МГц, CDCl3) δ 8,10 (д, J=2,4 Гц, 1Н), 7,85 (м, 2Н), 7,78 (м, 2Н), 7,46 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 6,47 (дд, J1=8,4 Гц, J2=0,6 Гц, 1Н), 5,62 (м, 1Н), 4,37 (т, J=4,8 Гц, 2Н), 3,70 (м, 2Н).
Пример получения 20
Получение соединения XIX
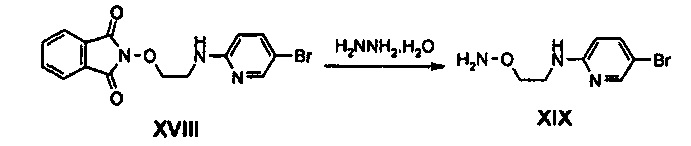
8,1 г (22,36 ммоль) Соединения XVIII добавляют к 100 мл этанола, по каплям добавляют 2,24 мл (44,73 ммоль) гидразинмоногидрата и полученный раствор нагревают до температуры 70°C и затем перемешивают в течение 2 часов. Полученное твердое вещество промывают дихлорметаном и диэтиловым эфиром и фильтрат концентрируют при пониженном давлении, получая 4,51 г (19,4 ммоль) соединения XIX в виде твердого вещества коричневого цвета (выход: 87%).
1H ЯМР (400 МГц, CDCl3) δ 8,10 (дд, J1=2,8 Гц, J2=0,8 Гц, 1Н), 7,46 (дд, J1=8,8 Гц, J2=2,4 Гц, 1Н), 6,33 (дд, J1=8,8 Гц, J2=0,4 Гц, 1Н), 3,86 (т, J=4,8 Гц, 2Н), 3,52 (м, 2Н).
Пример получения 21
Получение соединения A-V
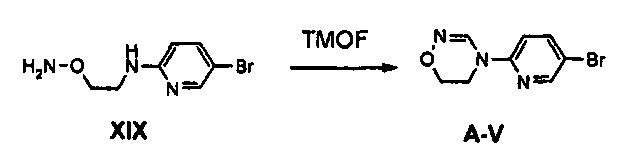
2,5 г (10,77 ммоль) Соединения XIX добавляют к 40 мл уксусной кислоты и 20 мл триметилортоформиата и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 1,5 часов. Полученную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении, добавляют 100 мл этилацетата, полученный раствор дважды промывают с помощью 80 мл водного насыщенного раствора бикарбоната натрия и затем обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 2,03 г (8,42 ммоль) соединения A-V в виде твердого вещества белого цвета (выход: 78%).
1H ЯМР (600 МГц, CDCl3) δ 8,39 (с, 1Н), 8,35 (дд, J1=2,4 Гц, J2=0,6 Гц, 1Н), 7,78 (дд, J1=13,8 Гц, J2=2,4 Гц, 1Н), 6,73 (дд, J1=9,0 Гц, J2=0,6 Гц, 1H), 4,21 (т, J=4,8 Гц, 2H), 3,83 (т, J=4,8 Гц, 2Н).
Пример получения 22
Получение соединения XX
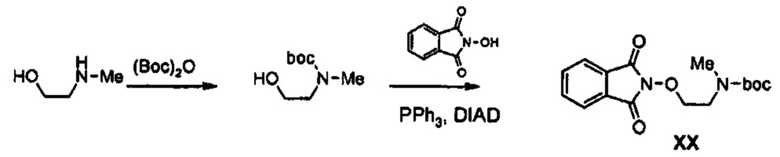
90,1 г (1,2 моль) 2-(Метиламино)этанола растворяют в 1,2 л метиленхлорида, медленно добавляют 218 г (1 моль) Boc2O, в время как полученный раствор перемешивают при температуре 0°C, и полученный раствор перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь последовательно промывают с помощью 700 мл водного насыщенного раствора хлорида аммония и 300 мл воды, обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении, получая 175 г (1 моль) бесцветного масла с защитной Boc-группой (выход: 100%).
1H ЯМР (600 МГц, CDCl3) δ 7,84 (уш.с, 2Н), 7,76 (уш.с, 2Н), 4,34 (д, J=15,0 Гц, 2Н), 3,63 (уш.с, 2Н), 3,04 (д, J=15,0 Гц, 3Н), 1,46 (д, J=16,2 Гц, 9Н).
90 г (0,514 моль) Полученного соединения растворяют в 1,5 л тетрагидрофурана, добавляют 88,0 г (539 моль) N-гидроксифталимида и 141 г (0,539 моль) трифенилфосфина, медленно добавляют 106 мл (0,539 моль) диизопропилазодикарбоксилата, в то время как получающийся в результате раствор перемешивают при температуре 0°С, и полученный раствор перемешивают в течение 3 часов, в то время как его температура повышается до комнатной температуры. После концентрирования реакционной смеси при пониженном давлении, добавляют 600 мл диизопропилового эфира, полученный раствор перемешивают при температуре 0°С в течение 1 часа и трифенилфосфиноксид типа твердого вещества белого цвета отфильтровывают. Твердое вещество промывают 200 мл диизопропилового эфира, охлажденного до температуры 0°С, и объединяют с первым фильтратом, и полученный в результате фильтрат концентрируют при пониженном давлении, получая 198 г смеси соединения XX и диизопропилгидразодикарбоксилата при отношении концентраций компонентов смеси от 10% до 15% (выход: 120%).
1H ЯМР (600 МГц, CDCl3) δ 7,84 (уш.с, 2Н), 7,76 (уш.с, 2Н), 4,34 (д, J=15,0 Гц, 2Н), 3,63 (уш.с, 2Н), 3,04 (д, J=15,0 Гц, 3Н), 1,46 (д, J=16,2 Гц, 9Н).
Пример получения 23
Получение соединения XXI
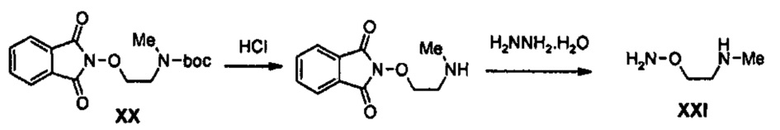
198 г (514 ммоль) Соединения XX растворяют в 260 мл 1,4-диоксана, медленно добавляют 385 мл (1,54 моль) 4 М раствора хлороводорода в 1,4-диоксане, в то время как перемешивают получающийся раствор при температуре 0°С, и полученный раствор перемешивают при комнатной температуре в течение 4 часов и далее перемешивают при температуре 0°С в течение 1 часа. Твердое вещество белого цвета, полученное после реакции между вышеуказанными веществами, отфильтровывают и затем промывают 200 мл 1,4-диоксана, охлажденного до температуры 0°С, получая 116 г (514 ммоль) твердого соединения белого цвета из которого удалена Boc-группа.
1H ЯМР (600 МГц, ДМСО-d6) δ 9,31 (уш.с, 1Н), 7,88-7,92 (м, 4Н), 4,46 (т, J=5,4 Гц, 2Н), 3,30 (т, J=5,4 Гц, 2Н), 2,66 (с, 3Н).
53 г (210 ммоль) Полученного соединения растворяют в 1,5 л этанола, добавляют 25,1 мл (0,518 моль) гидразинмоногидрата, в то время как получаемый раствор перемешивают при комнатной температуре, используя механическую мешалку, и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 4 часов. Реакционную смесь охлаждают до температуры 0°С и перемешивают в течение 1 часа. Полученное твердое вещество (то есть, фталгидразид) отфильтровывают и промывают 100 мл этанола, охлажденного до температуры 0°С, и фильтрат концентрируют при пониженном давлении. Концентрат далее концентрируют при пониженном давлении после добавления 250 мл дихлорметана и 500 мл толуола, добавляют 250 мл толуола и концентрирование при пониженном давлении повторяют дважды, для удаления избытка гидразина, получая 25,1 г (202 ммоль) соединения XXI в виде твердого вещества белого цвета (выход: 96%).
1H ЯМР (600 МГц, ДМСО-d6) δ 3,74 (т, J=4,8 Гц, 2Н), 3,08 (т, J=4,8 Гц, 2Н), 2,54 (с, 3Н).
Пример получения 24
Получение соединения XXVIII
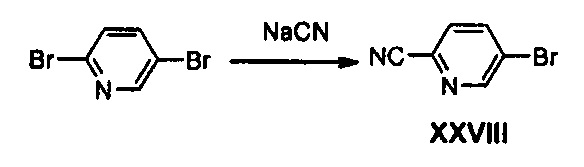
30 г (126, 63 ммоль) 2,5-дибромпиридина, 9,87 г (108,90 ммоль) цианида меди и 5,3 г (108,90 ммоль) цианида натрия добавляют к 300 мл N,N-диметилформамида и полученный раствор нагревают до температуры 150°С и перемешивают в течение 5 часов. Смесь охлаждают до комнатной температуры, добавляют 400 мл этилацетата и полученную смесь три раза промывают 300 мл воды. Полученный органический слой промывают 200 мл насыщенного раствора хлорида натрия и обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 12,17 г соединения XXVIII в виде твердого вещества белого цвета (выход: 53%).
1H ЯМР (600 МГц, CDCl3) δ 8,80 (дд, J=2,4 Гц, J2=0,6 Гц, 1Н), 8,00 (дд, J=8,4 Гц, J2=1,8 Гц, 1Н), 7,60 (дд, J,=8,4 Гц, J2=1,2 Гц, 1Н).
Пример получения 25
Получение соединения A-VI
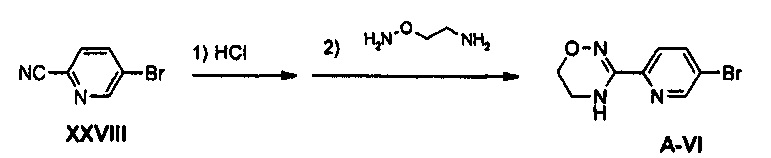
2 г (10,93 ммоль) Соединения XXVIII добавляют к 1,91 мл этанола, добавляют 13,7 мл 4 М раствора хлороводорода в диоксане, полученный раствор перемешивают в атмосфере аргона, при комнатной температуре, в течение 18 часов и затем концентрируют при пониженном давлении, и концентрат растворяют в 30 мл метанола. Затем, 2,46 г (21,86 ммоль) диамина, синтезированного из этаноламина, используя такую же методику, как используемая в примерах получения 22 и 23, добавляют к 100 мл метанола, добавляют 3,02 г (21,86 ммоль) карбоната калия, в то время как получаемый раствор перемешивают, полученный в результате раствор перемешивают при комнатной температуре в течение 30 минут и потом отфильтровывают, фильтрат концентрируют при пониженном давлении, добавляют предварительно полученный метанольный раствор и полученный в результате фильтрат перемешивают при комнатной температуре в течение 12 часов. Смесь концентрируют при пониженном давлении, добавляют 30 мл уксусной кислоты и полученную смесь кипятят с обратным холодильником, при перемешивании, в течение 4 часов. Полученную смесь охлаждают до комнатной температуры и затем концентрируют при пониженном давлении, добавляют 100 мл дихлорметана и полученный раствор промывают 100 мл насыщенного раствора бикарбоната натрия и обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 1,14 г (4,71 ммоль) соединения A-VI (выход: 43%).
1H ЯМР (600 МГц, CDCl3) δ 8,57 (д, J=2,4 Гц), 7,94 (дд, J=8,4 Гц, J2=0,6 Гц, 1Н), 7,85 (дд, J=8,4 Гц, J2=2,4 Гц, 1Н), 6,43 (м, 1Н), 4,06 (т, J=4,8 Гц, 2Н), 3,83 (м, 2Н).
Пример получения 26
Получение соединения A-VII
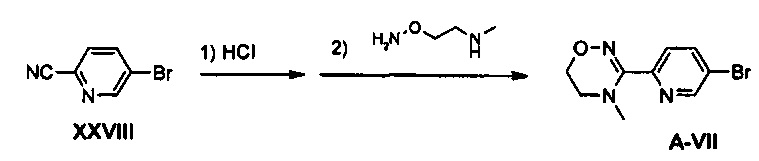
1,15 г (4,49 ммоль) соединения A-VII, в виде твердого вещества белого цвета, (выход: 41%) получают из 2 г (10,93 ммоль) соединения XXVIII путем введения во взаимодействие диамина XXI, полученного в соответствии с примером получения 23, таким же образом, как в примере 25.
1H ЯМР (600 МГц, CDCl3) δ 8,70 (д, J=2,4 Гц), 7,89 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,56 (д, J=8,4 Гц, 1Н), 4,14 (т, J=4,8 Гц, 2Н), 3,48 (т, J=4,8 Гц, 2Н), 2,88 (с, 3Н).
Пример получения 27 Получение соединения XXIX
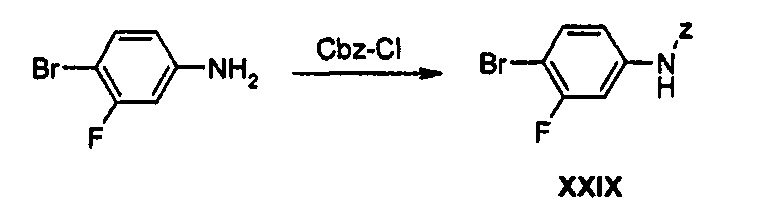
100 г (528 ммоль) 3-Фтор-4-броманилина растворяют в 500 мл дихлорметана, добавляют 800 мл 1 н. водного раствора NaOH и медленно, по каплям, добавляют 82 мл (580 ммоль) Cbz-Cl (бензилхлорформиат), в то время как получаемый раствор перемешивают. Полученный раствор перемешивают при комнатной температуре в течение 1 часа для отделения от него органического слоя. Органический слой дважды промывают водой, обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении, получая 173 г (528 ммоль) соединения XXIX в виде твердого вещества белого цвета.
1H ЯМР (600 МГц, CDCl3) δ 7,40 (м, 7Н), 6,93 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 6,71 (с, 1Н), 5,20 (с, 2Н).
Пример получения 28
Получение соединения B-I
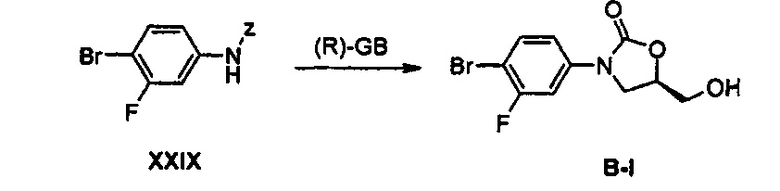
119 г (367 ммоль) Соединения XXIX растворяют в смеси из 300 мл тетрагидрофурана и 150 мл диметилформамида, медленно, по каплям, добавляют 38,19 г (477 ммоль) трет-бутоксида лития, при температуре 0°С, полученный раствор перемешивают в течение 10 минут, добавляют 63 мл (440 ммоль) (R)-глицидилбутирата и 21 мл (550 ммоль) метанола, и полученный раствор перемешивают при комнатной температуре в течение 3 часов. Затем, значение pH реакционной смеси доводят приблизительно до 6, используя водный раствор хлорида аммония, и после этого реакционную смесь концентрируют при пониженном давлении. Концентрат растворяют в 1000 мл смеси 80% этилацетат/гексан, последовательно промывают водой и водным насыщенным раствором хлорида натрия (насыщенный солевой раствор), и потом обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 93 г (320 ммоль) соединения B-I в виде твердого вещества белого цвета (выход: 87%).
1H ЯМР (600 МГц, CDCl3) δ 7,53 (м, 2Н), 7,15 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 4,77 (м, 1Н), 4,00 (м, 3Н), 3,77 (м, 1Н), 2,10 (т, J=6,0 Гц, 1Н).
Пример получения 29
Получение соединения В-II
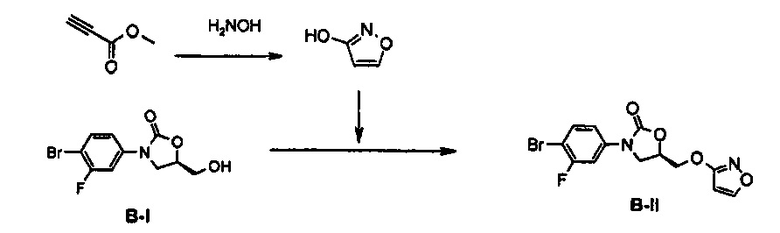
58 г (1,45 моль) Гидроксида натрия добавляют к 580 мл воды, добавляют 35 г (0,5 моль) гидроксиламингидрохлорида, в то время как полученный раствор перемешивают, и добавляют раствор, полученный путем разбавления 38 мл (0,43 моль) метилпропиолята в 600 мл метанола. Полученный в результате раствор перемешивают при комнатной температуре в течение 6 суток, значение pH доводят до 2, используя концентрированную соляную кислоту, и этот раствор насыщают хлоридом натрия и затем восьмикратно экстрагируют с помощью 500 мл дихлорметана. Экстракт обезвоживают, используя безводный сульфат натрия, и потом концентрируют при пониженном давлении, получая твердое вещество. Полученное твердое вещество три раза промывают 200 мл горячего гексана, получая 11,53 г (140 ммоль) гидроксиизоксазола в виде твердого вещества цвета слоновой кости (выход: 32%).
1H ЯМР (400 МГц, CDCl3) δ 11,25 (уш., 1Н), 8,52 (д, J=2,0 Гц, 1Н), 6,07 (д, J=2,0 Гц, 1Н).
5 г (17,24 ммоль) Полученного гидроксиизоксазола, 1,8 г
(20,68 ммоль) соединения B-I и 5,9 г (22,41 ммоль) трифенилфосфина добавляют к 90 мл тетрагидрофурана и медленно, по каплям, добавляют 4,4 мл (22,41 ммоль) диизопропилазодикарбоксилата, при температуре 0°C. Полученный раствор перемешивают при комнатной температуре в течение 1,5 часов, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 4,58 г (12,8 ммоль) соединения В-II в виде твердого вещества белого цвета (выход: 74%).
1H ЯМР (400 МГц, CDCl3) δ 8,16 (д, J=2,0 Гц, 1Н), 7,55 (м, 2Н), 7,18 (м, 1Н), 6,01 (д, J=2,0 Гц, 1Н), 5,03 (м, 1Н), 4,59 (дд, J1=11,6 Гц, J2=4,0 Гц, 1Н), 4,51 (дд, J1=11,6 Гц, J2=4,4 Гц, 1Н), 4,16 (т, J=8,8 Гц, 1Н), 3,97 (дд, J1=8,8 Гц, J2=6,0 Гц, 1Н).
Пример получения 30
Получение соединения XXX
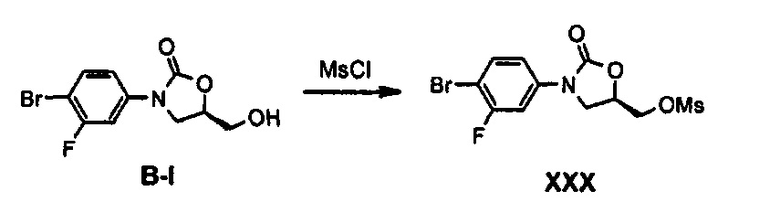
30 г (103 ммоль) Соединения B-I и 23 мл (134 ммоль) диизопропилэтиламина растворяют в 350 мл дихлорметана, медленно, по каплям, добавляют 9,6 мл (124 ммоль) метансульфонилхлорида (MsCl), при температуре 0°C, и полученный раствор перемешивают в течение 20 минут и затем перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь растворяют в 300 мл дихлорметана и затем последовательно промывают 200 мл 0,5 н. водного раствора хлороводорода, 100 мл водного насыщенного раствора бикарбоната натрия и 100 мл водного насыщенного раствора хлорида натрия, потом обезвоживают безводным сульфатом натрия и концентрируют при пониженном давлении, получая 38 г (103 моль) соединения XXX в виде твердого вещества коричневого цвета (выход: 99%).
1H ЯМР (400 МГц, CDCl3) δ 7,53 (м, 2Н), 7,14 (дд, J1=8,8 Гц, J2=2,4 Гц, 1Н), 4,95 (м, 1Н), 4,46 (м, 2Н), 4,13 (дд, J,=9,2 Гц, J2=9,2 Гц, 1Н), 3,94 (дд, J1=9,2 Гц, J2=6,4 Гц, 1Н), 3,10 (с, 3Н).
Пример получения 31 Получение соединения В-III
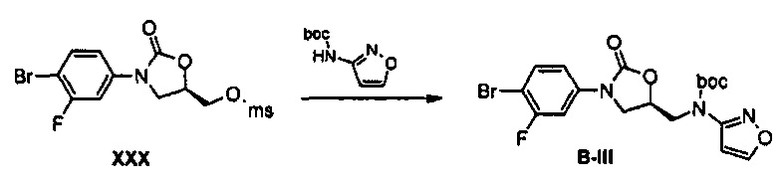
2,5 г (6,87 ммоль) Соединения XXX и 1,26 г (6,87 ммоль) Boc-аминоизоксазола растворяют в 7 мл диметилформамида, добавляют 0,33 г (7,56 ммоль) NaH, при температуре 0°C, и полученный раствор перемешивают при температуре 75°C в течение 2,5 часов. Реакционную смесь экстрагируют, используя этилацетат и дистиллированную воду, для получения органического слоя. Органический слой обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 2,61 г (5,72 ммоль) соединения B-III в виде твердого вещества белого цвета (выход: 83%).
1H ЯМР (600 МГц, CDCl3) δ 8,26 (д, J=1,8 Гц, 1Н), 7,53 (м, 2Н), 7,14 (дд, J=9,0 Гц, J=3,0 Гц, 1Н), 6,01 (уш., 1Н), 5,92 (м, 1Н), 4,37 (дд, J=7,8 Гц, 1Н), 4,12 (м, 2Н), 3,81 (дд, J=8,4 Гц, J=4 Гц, 1Н), 1,56 (с, 9Н).
Пример получения 32
Получение соединения XXXI
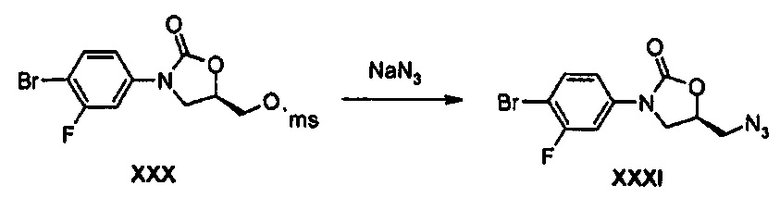
38 г (103 ммоль) Соединения XXX и 16,8 г (258 ммоль) азида натрия добавляют к 90 мл диметилформамида и полученный раствор перемешивают при температуре 90°C в течение 3 часов. Реакционную смесь растворяют в 500 мл этилацетата и затем промывают дистиллированной водой, потом обезвоживают безводным сульфатом натрия и концентрируют при пониженном давлении, получая 33 г (103 моль) соединения XXXI в виде твердого вещества светло-коричневого цвета (выход: 99%).
1Н ЯМР (600 МГц, CDCl3) δ 7,53 (м, 2Н), 7,15 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 4,80 (м, 1Н), 4,06 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 3,84 (дд, J1=9,0 Гц, J2=6,0 Гц, 1Н), 3,73 (дд, J1=13,2 Гц, J2=4,8 Гц, 1Н), 3,61 (дд, J1=13,2 Гц, J2=4,8 Гц, 1Н)
Пример получения 33
Получение соединения В-IV
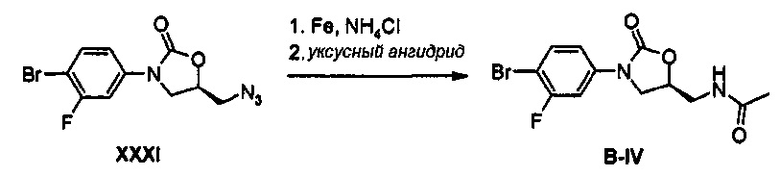
4,2 г (13,3 ммоль) Соединения XXXI, 2,2 г (40,0 ммоль) порошкообразного железа, 7,1 г (133,3 ммоль) хлорида аммония и 10 мл дистиллированной воды добавляют к 40 мл этанола, и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 12 часов. Реакционную смесь охлаждают до комнатной температуры, отфильтровывают через целит, концентрируют при пониженном давлении, экстрагируют дихлорметаном и водным раствором бикарбоната натрия, обезвоживают, используя безводный сульфат натрия, и затем отфильтровывают, добавляют 1,4 мл (14,0 ммоль) уксусного ангидрида и полученный раствор перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь экстрагируют дихлорметаном и водным раствором бикарбоната натрия, обезвоживают, используя безводный сульфат натрия, и затем отфильтровывают, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 3,5 г (10,6 моль) соединения B-IV в виде твердого вещества светло-коричневого цвета (выход: 7 9%).
1H ЯМР (600 МГц, ДМСО) δ 8,26 (т, J=6,0 Гц, 1Н), 7,72 (т, J=8,4 Гц, 1Н), 7,65 (дд, J1=12,0 Гц, J2=2,4 Гц, 1Н), 7,32 (дд, J1=8,4 Гц, J2=3,6 Гц, 1Н), 4,74 (м, 1Н), 4,12 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 3,73 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,42 (т, J=5,4 Гц, 2Н), 1,83 (с, 3Н).
Пример получения 34
Получение соединения B-V
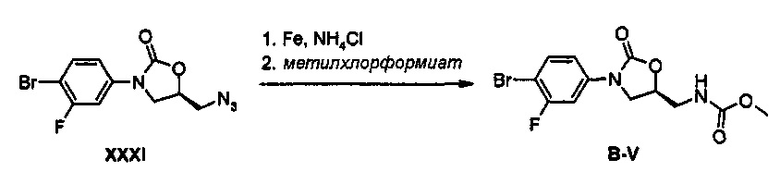
19,7 г (56,7 ммоль) Соединения B-V получают, в виде твердого вещества белого цвета, используя метилхлорформиат, по такому же способу, как в примере получения 33.
1H ЯМР (600 МГц, CDCl3) δ 7,53 (м, 2Н), 7,11 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 5,11 (с, 1Н), 4,78 (м, 1Н), 4,04 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 3,80 (м, 1Н), 3,68 (с, 3Н), 3,61 (м, 2Н).
Пример получения 35
Получение соединения B-VI
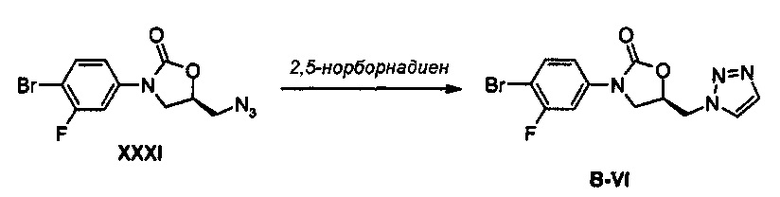
2,92 г (9,27 ммоль) Соединения XXXI и 9,4 мл (92,7 ммоль) 2,5-норборнадиена (бицикло[2,2,1]гепта-2,5-диен) добавляют к 50 мл 1,4-диоксана и полученный раствор кипятят с обратным холодильником, при перемешивании, в течение 2,5 часов. Реакционную смесь концентрируют при пониженном давлении и затем экстрагируют, используя 150 мл дихлорметана и 100 мл дистиллированной воды. Экстрагированный органический слой обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 2 г (5,8 ммоль) соединения В-VI (выход: 63%).
1H ЯМР (600 МГц, CDCl3) δ 7,78 (д, J=1,2 Гц, 1Н), 7,75 (д, J=1,2 Гц, 1Н), 7,49 (дд, J1=8,4 Гц, J2=7,8 Гц, 1Н), 7,42 (дд, J1=10,8 Гц, J2=2,4 Гц, 1Н), 7,00 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 5,08 (м, 1Н), 4,80 (дд, J1=4,2 Гц, J2=1,2 Гц, 2Н), 4,15 (дд, J1=9,6 Гц, J2=9,6 Гц, 1Н), 3,94 (дд, J1=9,6 Гц, J2=6,6 Гц, 1Н).
Пример получения 36
Получение соединения B-VII

К 20 мл N,N-диметилформамида последовательно добавляют 2 г (5,60 ммоль) соединения В-II, 2,1 г (8,40 ммоль) биспинаколятодиборана, 274 мг (0,34 ммоль) PdCl2(dppf) и 1,65 г (16,80 ммоль) ацетата калия, и полученный раствор перемешивают в атмосфере азота, при температуре 90°C, в течение 15 часов. Реакционную смесь охлаждают до комнатной температуры и затем экстрагируют 80 мл дистиллированной воды и 100 мл этилацетата. Экстрагированный органический слой обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении и осуществлением колоночной хроматографии, получая 1,11 г (2,75 ммоль) соединения B-VII (выход: 49%).
1H ЯМР (600 МГц, CDCl3) δ 8,16 (д, J=1,8 Гц, 1Н), 7,44 (дд, J1=8,4 Гц, J2=1,2 Гц, 1Н), 7,41 (дд, J1=11,4 Гц, J2=1,8 Гц, 1Н), 7,28 (дд, J1=7,8 Гц, J2=1,8 Гц, 1Н), 6,00 (д, J=1,8 Гц, 1Н), 5,04 (м, 1Н), 4,59 (м, 1Н), 4,05 (м, 1Н), 4,18 (т, J=9,0 Гц, 1Н), 3,99 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 1,36 (с, 12Н).
Пример получения 37
Получение соединения B-VIII

К 5 мл N,N-диметилформамида последовательно добавляют 0,3 г (0,90 ммоль) соединения B-IV, 0,3 г (1,18 ммоль) биспинаколятодиборана, 22 мг (0,03 ммоль) PdCl2(dppf) и 0,27 г (2,71 ммоль) ацетата калия, и полученный раствор перемешивают в атмосфере азота, при температуре 90°C, в течение 15 часов. Реакционную смесь охлаждают до комнатной температуры и затем экстрагируют 40 мл дистиллированной воды и 50 мл этилацетата. Экстрагированный органический слой обезвоживают, используя безводный сульфат натрия, с последующим концентрированием при пониженном давлении, получая 350 мг соединения В-VIII в виде смеси. Полученное соединение используют в последующем процессе без очистки.
Пример получения 38
Получение соединения В-IX

0,75 г (1,95 ммоль) Соединения B-IX получают (выход: 55%) из 1,2 г (3,52 ммоль) соединения B-VI, по такому же способу, как в примере получения 36.
1H ЯМР (600 МГц, CDCl3) δ 7,78 (д, J=1,2 Гц, 1Н), 7,45 (д, J=1,2 Гц, 1Н), 7,70 (дд, J1=8,4 Гц, J2=7,2 Гц, 1Н), 7,31 (дд, J1=11,4 Гц, J2=1,8 Гц, 1Н), 7,12 (дд, J=8,4 Гц, J2=1,8 Гц, 1Н), 5,07 (м, 1Н), 4,79 (м, 2Н), 4,18 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 3,93 (дд, J1=9,0 Гц, J2=5,4 Гц, 1Н), 1,32 (с, 12Н).
Пример получения 39
Получение соединения В-Х
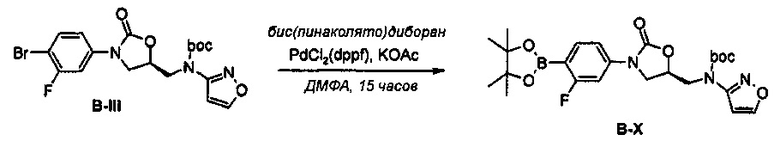
0,8 г (1,59 ммоль) Соединения В-Х получают (выход: 98%) из 0,74 г (1,62 ммоль) соединения В-III, по такому же способу, как в примере получения 36.
1H ЯМР (600 МГц, CDCl3) δ 8,26 (д, J=0,6 Гц, 1Н), 7,23 (дд, J1=7,8 Гц, J2=7,8 Гц, 1Н), 7,39 (дд, J1=11,4 Гц, J2=1,2 Гц, 1Н), 7,25 (дд, J1=8,4 Гц, J2=1,2 Гц, 1Н), 6,92 (уш., 1Н), 5,09 (м, 1Н), 4,38 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 4,12 (дд, J1=8,4 Гц, 2Н), 3,82 (дд, J1=8,4 Гц, J2=5,4 Гц, 1Н), 1,36 (с, 9Н), 1,26 (с, 12Н).
Пример получения 40
Получение соединения B-XI

0,88 г (2,24 ммоль) Соединения В-XI получают (выход: 78%) из 1 г (2,88 ммоль) соединения B-V, по такому же способу, как в примере получения 36.
1H ЯМР (600 МГц, CDCl3) δ 7,24 (дд, J=8,4 Гц, J2=8,4 Гц, 1Н), 7,39 (дд, J1=11,4 Гц, J2=1,2 Гц, 1Н), 7,23 (дд, J=8,4 Гц, J2=1,8 Гц, 1Н), 5,14 (м, 1Н), 4,78 (м, 1Н), 4,07 (дд, J=9,0 Гц, J2=9,0 Гц, 1Н), 3,81 (м, 1Н), 3,68 (с, 3Н), 3,62 (м, 1Н), 3,54 (м, 1Н) 1,36 (с, 12Н).
Пример получения 41
Получение соединения B-XII
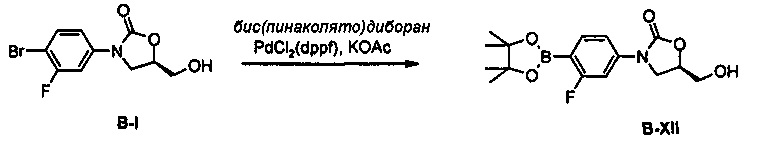
1,03 г (3,06 ммоль) Соединения B-XII получают (выход: 88%) из 1 г (3,45 ммоль) соединения B-I, по такому же способу, как в примере получения 36.
1H ЯМР (600 МГц, CDCl3) δ 7,45 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 7,43 (дд, J1=10,8 Гц, J2=1,8 Гц, 1Н), 7,29 (дд, J1=7,8 Гц, J2=1,8 Гц, 1Н), 4,77 (м, 1Н), 4,01 (м, 3Н), 3,78 (м, 1Н), 2,10 (т, J=5,4 Гц, 1Н), 1,37 (с, 12Н).
ПРИМЕРЫ
Пример 1
Получение соединения 1
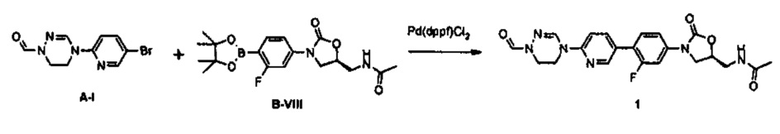
0,8 г (2,99 ммоль) Соединения A-I, синтезированного согласно вышеприведенному примеру получения, 1,3 г (3,30 ммоль) соединения B-VIII, PdCl2(dppf) и 2 М водный раствор карбоната натрия добавляют к 20 мл диметилформамида и полученный раствор перемешивают при температуре 90°C в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и затем экстрагируют с помощью 150 мл дихлорметана и 300 мл дистиллированной воды, затем обезвоживают, используя безводный сульфат натрия, и концентрируют при пониженном давлении.
Концентрат очищают колоночной хроматографией, получая 1,04 г (2,28 ммоль) соединения 1 в виде твердого вещества серого цвета (выход: 76%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,52 (с, 1Н), 8,50 (с, 1Н), 8,28 (т, J=6,0 Гц, 1Н), 8,06 (с, 1Н), 8,10 (д, J=9,0 Гц), 7,63 (м, 2Н), 7,43 (дд, J1=8,4 Гц, J2=2,4 Гц), 7,36 (д, J=8,4 Гц, 1Н), 4,76 (м, 1Н), 4,17 (т, J=9,0 Гц, 1Н), 4,18 (т, J=7,8 Гц, 2Н), 3,92 (м, 4Н), 3,78 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,44 (т, J=6,0 Гц, 2Н), 1,84 (с, 3Н).
LCMS: 441 (М+Н+) для C21H21FN6O4.
Пример 2
Получение соединения 2
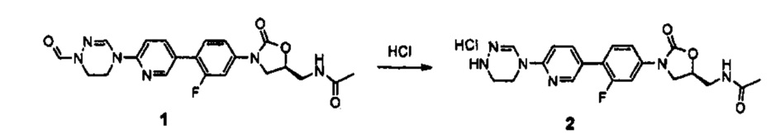
0,6 г (1,36 ммоль) Соединения 1, синтезированного в соответствии с примером 1, растворяют в 4,5 мл метанола и 4,5 мл дихлорметана, добавляют 1 мл 4 М HCl, при температуре 0°C, и полученный раствор перемешивают в течение 30 минут. Реакционную смесь концентрируют при пониженном давлении, получая 0,8 г (1,36 ммоль) соединения 2 в виде пены желтого цвета.
1H ЯМР (600 МГц, ДМСО d-6) δ 9,00 (с, 1Н), 8,62 (с, 1Н), 8,30 (т, J=6,0 Гц, 1Н), 8,14 (д, J=9,0 Гц, 1Н), 7,68 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 7,65 (дд, J1=7,8 Гц, J2=2,4 Гц, 1Н), 7,57 (д, J=8,4 Гц, 1Н), 7,46 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 4,78 (м, 1Н), 4,19 (т, J=9,0 Гц, 1Н), 4,03 (т, J=4,8 Гц, 2Н), 3,79 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,44 (м, 4Н), 1,84 (с, 3Н).
LCMS: 413 (М+Н+) для C20H21FN6O3.
Пример 3
Получение соединения 3
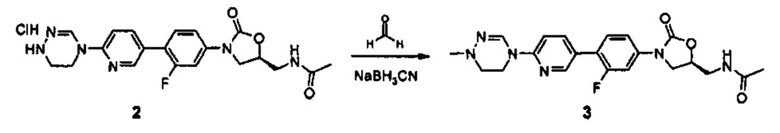
0,23 г (0,50 ммоль) Соединения 2, синтезированного в соответствии с примером 2, 0,06 мл (0,75 ммоль) формальдегида, 0,09 мл (0,50 ммоль) диизопропилэтиламина, 47 мг (0,75 ммоль) цианоборгидрида натрия и 0,03 мл (0,5 ммоль) уксусной кислоты последовательно добавляют к 2 мл метанола, при температуре 0°С, и полученный раствор перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь отфильтровывают и промывают 3 мл метанола, получая 78 мг (0,17 ммоль) соединения 3 (выход: 34%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,44 (с, 1Н), 8,25 (т, J=6,0 Гц, 1Н), 7,95 (с, 1Н), 7,92 (д, J=9,6 Гц, 1Н), 7,61 (м, 2Н), 7,41 (дд, J1=9,0 Гц, J2=2,4 5 Гц, 1Н), 7,16 (д, J=8,4 Гц, 1Н), 4,76 (м, 1Н), 4,17 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 3,84 (т, J=5,4 Гц, 2Н), 3,79 (дд, J1=9,0 Гц, J2=6,0 Гц, 1Н), 3,43 (т, J=5,4 Гц, 2Н), 2,95 (т, J=5,4 Гц, 2Н), 2,68 (с, 3Н), 1,84 (с, 3Н).
LCMS: 427 (М+Н+) для C21H23FN6O3.
Пример 4
Получение соединения 4
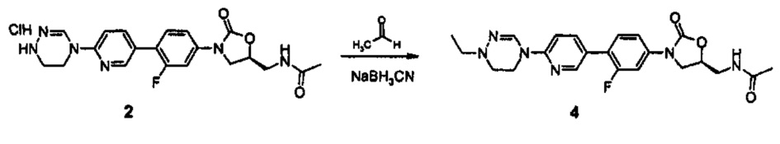
230 мг (0,52 ммоль) Соединения 4 получают (выход: 63%), используя ацетальдегид, по такому же способу, как в примере 3.
1H ЯМР (400 МГц, CDCl3) δ 8,45 (с, 1Н), 8,00 (с, 1Н), 7,81 (д, J=8,8 Гц, 1Н), 7,56 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,43 (дд, J1=8,8 Гц, J2=8,8 Гц, 1Н), 7,32 (дд, J1=9,2 Гц, J2=2,4 Гц, 1Н), 6,87 (д, J=8,8 Гц, 1Н), 6,09 (т, J=5,6 Гц, 1Н), 4,84 (м, 1Н), 4,11 (дд, J1=8,8 Гц, J2=8,8 Гц, 1Н), 3,97 (т, J=5,2 Гц, 2Н), 3,84 (дд, J1=9,2 Гц, J2=5,6 Гц, 1Н), 3,69 (м, 2Н), 3,08 (т, J=5, 6 Гц, 2Н), 3,04 (м, 2Н), 2,06 (с, 3Н), 1,28 (т, J=7,2 Гц, 3Н).
LCMS: 441 (М+Н+) для C22H25FN6O3.
Пример 5
Получение соединения 5
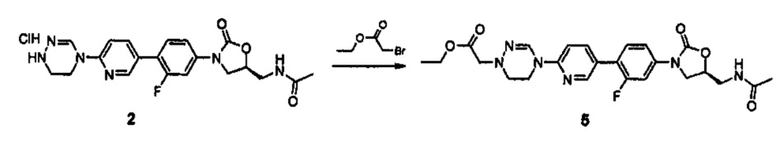
0,20 г (0,38 ммоль) Соединения 2, синтезированного в соответствии с примером 2, и 0,13 мл (0,76 ммоль) диизопропилэтиламина последовательно добавляют к 5 мл диметилформамида, медленно добавляют 0,05 мл (0,46 ммоль) этилбромацетата и полученный раствор перемешивают при температуре 80°C. Реакционную смесь охлаждают до комнатной температуры и затем дважды промывают 50 мл этилацетата и 60 мл дистиллированной воды, затем обезвоживают, используя безводный сульфат натрия, и концентрируют при пониженном давлении. Концентрат разделяют с помощью колоночной хроматографии, получая 110 мг (0,22 ммоль) соединения 5 в виде твердого вещества коричневого цвета.
1H ЯМР (600 МГц, CDCl3) δ 8,45 (с, 1Н), 7,98 (с, 1Н), 7,81 (д, J=9,0 Гц, 1Н), 7,56 (дд, J1=12,6 Гц, J2=2,4 Гц, 1Н), 7,42 (т, J=8,4 Гц, 1Н), 7,29 (м, 1Н), 6,87 (д, J=9,0 Гц, 1Н), 6,08 (т, J=6,6 Гц, 1Н), 4,83 (м, 1Н), 4,25 (кв, J=7,2 Гц, 2Н), 4,10 (м, 1Н), 3,99 (т, J=5,4 Гц, 2Н), 3,84 (с, 2Н), 3,75 (м, 1Н), 3,72 (м, 1Н), 3,66 (м, 1Н), 3,03 (т, J=5,4 Гц, 2Н), 2,05 (с, 3Н), 1,32 (т, J=12 Гц, 3Н).
LCMS: 4 99 (М+Н+) для C24H27FN6O5.
Пример 6
Получение соединения 6
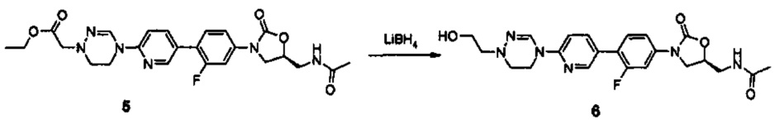
0,11 г (0,22 ммоль) соединения 5, синтезированного в соответствии с примером 5, растворяют в 10 мл тетрагидрофурана и медленно, по каплям, добавляют 0,2 мл 2 М раствора боргидрида лития в тетрагидрофуране. Полученный раствор перемешивают при комнатной температуре в течение 1 часа и, снова по каплям, добавляют 0,2 мл 2 М раствора боргидрида лития в тетрагидрофуране. Затем, к полученному раствору добавляют 0,5 мл водного насыщенного раствора хлорида аммония, и полученный раствор разбавляют 30 мл дихлорметана, затем обезвоживают безводным сульфатом натрия и концентрируют при пониженном давлении. Концентрат разделяют колоночной хроматографией, получая 24 мг (0,05 ммоль) соединения 6 в виде твердого вещества желтого цвета (выход: 24%).
1Н ЯМР (600 МГц, CDCl3) δ 8,44 (с, 1Н), 8,02 (с, 1Н), 7,82 (д, J=8,4 Гц, 1Н), 7,56 (дд, J1=12,6 Гц, J2=1,8 Гц, 1Н), 7,41 (т, J=8,4 Гц, 1Н), 7,29 (м, 1Н), 6,87 (д, J=9,0 Гц, 1Н), 6,17 (т, J=6,6 Гц), 4,83 (м, 1Н), 3,99 (м, 2Н), 3,97 (т, J=5,4 Гц, 1Н), 3,84 (м, 1Н), 3,73 (м, 1Н), 3,69 (м, 1Н), 3,50 (м, 2Н), 3,12 (м, 2Н), 3,06 (м, 2Н), 2,05 (с, 3Н).
LCMS: 457 (М+Н+) для C22H25FN6O4.
Пример 7
Получение соединения 7
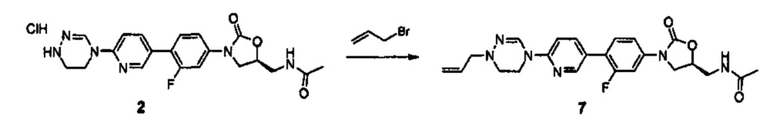
0,10 г (0,19 ммоль) Соединения 2 примера 2 и 0,06 мл (0,38 ммоль) диизопропилэтиламина последовательно добавляют к 3 мл диметилформамида, медленно добавляют 0,02 мл (0,23 ммоль) аллилбромида и полученный раствор перемешивают при комнатной температуре в течение 20 часов. Реакционную смесь дважды промывают 50 мл дихлорметана и 50 мл дистиллированной воды, затем обезвоживают безводным сульфатом натрия и концентрируют при пониженном давлении. Концентрат разделяют колоночной хроматографии, получая 25 мг (0,06 ммоль) соединения 7 в виде твердого вещества коричневого цвета (выход: 29%).
1H ЯМР (600 МГц, CDCl3) δ 8,43 (с, 1Н), 7,99 (с, 1Н), 7,79 (д, J=8,4 Гц, 1Н), 7,54 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,38 (т, J=9,0 Гц, 1Н), 7,31 (м, 1Н), 6,85 (д, J=9,0 Гц, 1Н), 6,30 (т, J=6,0 Гц, 1Н), 6,02 (м, 1Н), 5,31 (д, J=16,8 Гц, 1Н), 5,25 (д, J=9,6 Гц, 1Н), 4,83 (м, 1Н), 4,10 (м, 1Н), 3,94 (т, J=4,8 Гц, 2Н), 3,84 (м, 1Н), 3,73 (м, 1Н), 3,72 (м, 1Н), 3,65 (м, 2Н), 3,12 (кв, J=7,2 Гц, 2Н), 3,04 (т, J=5,4 Гц, 2Н), 2,04 (с, 3Н).
LCMS: 453 (М+Н+) для C23H25FN6O3.
Пример 8
Получение соединения 8
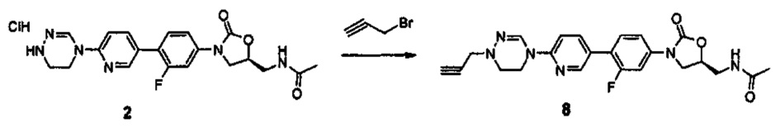
0,10 г (0,19 ммоль) Соединения 2 примера 2 и 0,06 мл (0,38 ммоль) диизопропилэтиламина последовательно добавляют к 3 мл диметилформамида, медленно добавляют 0,06 мл (0,38 ммоль) пропаргилбромида (80%-ный раствор в толуоле) и полученный раствор перемешивают при комнатной температуре в течение 20 часов. Реакционную смесь дважды промывают 50 мл дихлорметана и 50 мл дистиллированной воды, обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении. Концентрат разделяют с помощью колоночной хроматографии, получая 40 мг (0,09 ммоль) соединения 8 в виде твердого вещества коричневого цвета (выход: 47%).
1H ЯМР (600 МГц, CDCl3) δ 8,45 (с, 1Н), 8,04 (с, 1Н), 7,89 (д, J=9,6 Гц, 1Н), 7,55 (д, J=12,6 Гц, 1Н), 7,42 (т, J=8,9 Гц, 1Н), 7,29 (д, J=8,4 Гц, 1Н), 6,88 (д, J=9,0 Гц, 1Н), 5,97 (м, 1Н), 4,82 (м, 1Н), 4,09 (т, J=8,7 Гц, 1Н), 3,98 (м, 2Н), 3,88 (с, 2Н), 3,81 (т, J=7,8 Гц, 1Н), 3,67 (м, 1Н), 3,48 (м, 2Н), 3,18 (м, 2Н), 2,04 (с, 3Н)
LCMS: 451 (М+Н+) для C23H23FN6O3.
Пример 9
Получение соединения 9
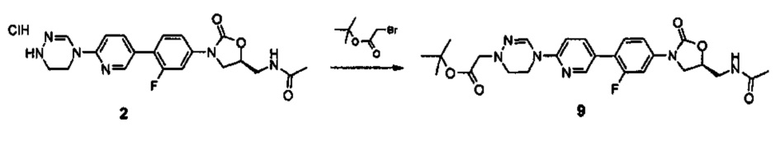
0,10 г (0,19 ммоль) Соединения 2 примера 2 и 0,06 мл (0,38 ммоль) диизопропилэтиламина последовательно добавляют к 2 мл диметилформамида, медленно добавляют 0,06 мл (0,38 ммоль) трет-бутилбромацетата и полученный раствор перемешивают при комнатной температуре в течение 20 часов. Реакционную смесь три раза промывают 50 мл этилацетата и 50 мл дистиллированной воды, обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении. Концентрат разделяют колоночной хроматографии, получая 47 мг (0,07 ммоль) соединения 9 в виде твердого вещества коричневого цвета (выход: 37%).
1H ЯМР (600 МГц, CDCl3) δ 8,43 (С, 1Н), 7,95 (с, 1Н), 7,80 (д, J=8,4 Гц, 1Н), 7,55 (дд, J1=12,6 Гц, J2=1,8 Гц, 1Н), 7,41 (т, J=8,7 Гц, 1Н), 7,28 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 6,86 (д, J=9,0 Гц, 1Н), 6,08 (т, J=6,3 Гц, 1Н), 4,82 (м, 1Н), 4,09 (т, J=9,0 Гц, 1Н), 3,98 (т, J=4,8 Гц, 2Н), 3,82 (дд, J1=9,6 Гц, J2=7,2 Гц, 1Н), 3,76 (с, 2Н), 3,72 (м, 2Н), 3,65 (м, 2Н), 3,30 (т, J=4,8 Гц, 2Н), 2,04 (с, 9Н), 1,50 (с, 9Н).
LCMS: 527 (М+Н+) для C26H31FN6O5.
Пример 10
Получение соединения 10
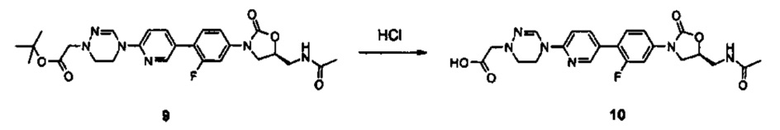
47 мг (0,09 ммоль) Соединения 9, синтезированного в соответствии с примером 9, добавляют к 2 мл дихлорметана, добавляют 5 мл 4 М раствора хлороводорода в диоксане, и полученный раствор перемешивают при комнатной температуре в течение 2 часов, затем концентрируют при пониженном давлении, получая 26 мг (0,06 ммоль) соединения 10 в виде твердого вещества желтого цвета (выход: 62%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,52 (с, 1Н), 8,37 (уш.с, 1Н), 8,30 (т, J=5,4 Гц, 1Н), 8,02 (д, J=7,8 Гц, 1Н), 7,65 (м, 2Н), 7,44 (д, J=8,4 Гц, 1Н), 7,34 (д, J=8,4 Гц, 1Н), 4,78 (м, 1Н), 4,18 (т, J=8,4 Гц, 1Н), 3,96 (м, 2Н), 3,81 (с, 2Н), 3,79 (м, 2Н), 3,44 (м, 2Н), 3,34 (м, 1Н), 1,84 (с, 3Н).
LCMS: 471 (М+Н+) для C22H23FN6O5.
Пример 11
Получение соединения 11
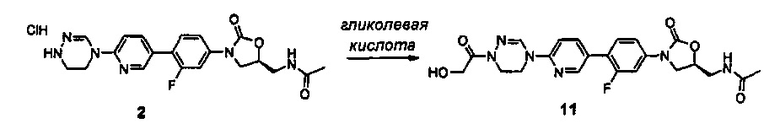
0,5 г (1,11 ммоль) соединения 2 примера 2, 0,1 г (1,13 ммоль) гликолевой кислоты, 0,39 мл (2,22 ммоль) диизопропил-этиламина и 0,7 г (1,33 ммоль) PyBoP добавляют к 3 мл диметилформамида, при температуре 0°C, и полученный раствор перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь разбавляют 30 мл дихлорметана, промывают 30 мл дистиллированной воды и затем обезвоживают, используя безводный сульфат натрия, затем концентрируют при пониженном давлении. Концентрат разделяют колоночной хроматографией, получая 70 мг (0,15 ммоль) соединения 11 в виде твердого вещества белого цвета (выход: 14%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,51 (с, 1Н), 8,27 (т, J=6,0 Гц, 1Н), 8,02 (с, 1Н), 8,00 (д, J=9,0 Гц, 1Н), 7,64 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 7,62 (дд, J1=12,0 Гц, J2=1,8 Гц, 1Н), 7,43 (дд, J1=9,0 Гц, J2=1,8 Гц, 1Н), 7,35 (д, J=8,4 Гц, 1Н), 4,78 (м, 1Н), 4,63 (т, J=6,0 Гц, 1Н), 4,37 (д, J=6,0 Гц, 1Н), 4,17 (т, J=9,0 Гц, 1Н), 3,93 (с, 4Н), 3,78 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,44 (т, J=6,6 Гц, 1Н), 1,84 (с, 3Н).
LCMS: 471 (М+Н+) для C22H23FN6O5.
Пример 12
Получение соединения 12
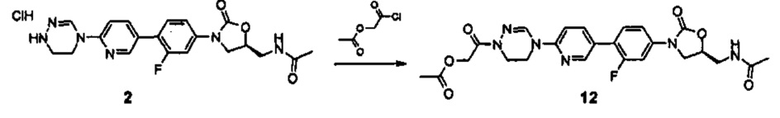
100 мг (0,19 ммоль) Соединения 2 примера 2 и 0,06 мл (0,38 ммоль) диизопропилэтиламина последовательно добавляют к 2 мл дихлорметана, медленно добавляют 0,02 мл (0,19 ммоль) ацетоксиацетилхлорида и полученный раствор перемешивают при комнатной температуре в течение 1 часа, потом концентрируют при пониженном давлении. Концентрат разделяют колоночной хроматографией, получая 38 мг (0,08 ммоль) соединения 12 в виде твердого вещества желтого цвета (выход: 39%).
1H ЯМР (600 МГц, CDCl3) δ 8,49 (д, J=0,6 Гц, 1Н), 7,91 (с, 1Н), 7,88 (дд, J1=8,4 Гц, J2=1,2 Гц, 1Н), 7,58 (д, J=12,6 Гц, 1Н), 7,42 (т, J=9,0 Гц, 1Н), 7,31 (д, J=8,4 Гц, 1Н), 6,95 (д, J=9,0 Гц, 1Н), 6,10 (т, J=6,3 Гц, 1Н), 5,13 (с, 2Н), 4,83 (м, 1Н), 4,10 (т, J=8,4 Гц, 1Н), 4,06 (т, J=4,8 Гц, 2Н), 3,93 (т, J=5,l Гц, 2Н), 3,84 (м, 1Н), 3,72 (м, 1Н), 3,67 (м, 1Н), 2,22 (с, 3Н), 2,04 (с, 3Н).
LCMS: 513 (М+Н+) для C24H25FN6O6.
Пример 13
Получение соединения 13
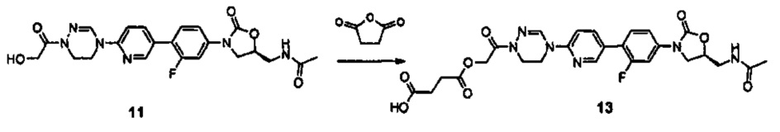
30 мг (0,06 ммоль) соединения 11, синтезированного в соответствии с примером 11, 70 мг (0,07 ммоль) ангидрида янтарной кислоты и 7,8 мг (0,01 ммоль) диметиламинопиридина последовательно добавляют к 2 мл тетрагидрофурана и полученный раствор перемешивают при комнатной температуре в течение 5 часов, и затем концентрируют при пониженном давлении. Концентрат разделяют колоночной хроматографией, получая 30 мг (0,05 ммоль) соединения 13 в виде твердого вещества цвета слоновой кости (выход: 83%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,52 (с, 1Н), 8,28 (т, J=6,0 Гц, 1Н), 8,08 (д, J=5,4 Гц, 1Н), 8,01 (д, J=9,0 Гц, 1Н), 7,64 (м, 2Н), 7,43 (д, J=8,4 Гц, 1Н), 7,40 (д, J=9,0 Гц, 1Н), 5,04 (с, 1Н), 4,81 (с, 1Н), 4,77 (м, 1Н), 4,18 (т, J=9,0 Гц, 1Н), 3,94 (м, 4Н), 3,79 (т, J=8,4 Гц, 1Н), 3,44 (т, J=5,4 Гц, 2Н), 3,38 (м, 2Н), 2,62 (м, 2Н), 1,84 (с, 3Н).
LCMS: 571 (М+Н+) для C26H27FN6O8.
Пример 14
Получение соединения 14
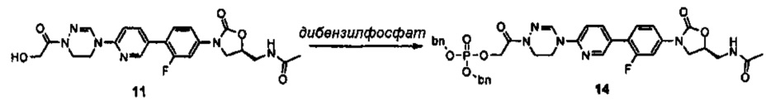
35 мг (0,07 ммоль) соединения 11 примера 11, 29 мг (0,11 ммоль) трифенилфосфина, 23 мкл (0,11 ммоль) DIAD и 31 мг (0,11 ммоль) дибензилфосфата добавляют к 1 мл тетрагидрофурана, при комнатной температуре, и полученный раствор перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении и затем разделяют колоночной хроматографией, получая 40 мг (0,05 ммоль) соединения 14 в виде твердого вещества белого цвета (выход: 71%).
1H ЯМР (600 МГц, CDCl3) δ 8,49 (с, 1Н), 7,88 (д, J=8,4 Гц, 1Н), 7,86 (с, 1Н), 7,58 (дд, J1=12,6 Гц, J2=2,4 Гц, 1Н), 7,43 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 7,35 (м, 11Н), 6,94 (д, J=7,8 Гц, 1Н), 6,00 (т, J=6,0 Гц, 1Н), 5,18 (м, 4Н), 5,11 (д, J1=11,4 Гц, 1Н), 4,82 (м, 1Н), 4,11 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 4,06 (т, J=5,4 Гц, 2Н), 3,90 (с, 2Н), 3,82 (дд, J1=9,0 Гц, J2=7,2 Гц, 1Н), 3,74 (м, 1Н), 3,66 (м, 1Н), 2,04 (с, 3Н).
LCMS: 741 (М+Н+) для C36H36FN6O8P.
Пример 15
Получение соединения 15
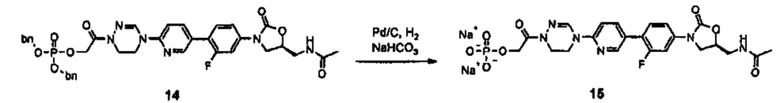
40 мг (0,05 ммоль) соединения 14, синтезированного в соответствии с примером 14, 20 мг Pd/C и 8 мг (0,1 ммоль) бикарбоната натрия добавляют к смеси тетрагидрофурана (1 мл) и дистиллированной воды (2 мл), и полученный раствор перемешивают в атмосфере водорода в течение 3 часов. Реакционную смесь отфильтровывают, используя целит, концентрируют при пониженном давлении и затем растворяют в 1 мл дистиллированной воды. После этого, к полученному раствору добавляют 3 мл этанола и полученный раствор отверждают и отфильтровывают, получая 15 мг (0,25 ммоль) соединения 15 в виде твердого вещества светло-серого цвета (выход: 50%).
1H ЯМР (600 МГц, D2O) δ 8,18 (с, 1Н), 7,78 (д, J=8,4 Гц, 1Н), 7,61 (с, 1Н), 7,29 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 7,23 (д, J=12,4 Гц, 1Н), 7,09 (д, J=8,4 Гц, 1Н), 6,97 (д, J=9,0 Гц, 1Н), 4,58 (м, 3Н), 4,00 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 3,75 (с, 2Н), 3,64 (м, 3Н), 3,43 (м, 1Н), 3,34 (дд, J1=15,0 Гц, J2=5,4 Гц, 1Н), 1,79 (С, 3Н).
LCMS: 551 (М+Н+) для C22H24FN6O8P.
Пример 16
Получение соединения 16
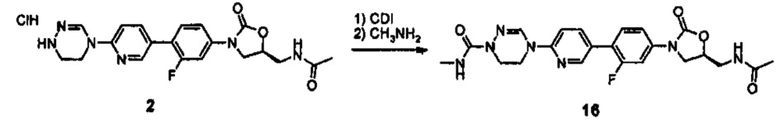
0,25 г (0,55 ммоль) Соединения 2 примера 2, 0,18 г (1,10 ммоль) карбонилдиимидазола и 0,19 мл (1,09 ммоль) диизопропил-этиламина добавляют к 20 мл дихлорметана и полученный раствор перемешивают в течение 1 часа. Реакционную смесь концентрируют при пониженном давлении до объема приблизительно 5 мл, добавляют 0,5 мл 33%-ного метиламина и полученную смесь перемешивают при температуре 50°C в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры, и выпавший осадок затем отфильтровывают, получая 0,17 г (0,36 ммоль) соединения 16 в виде твердого вещества белого цвета (выход: 65%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,49 (с, 1Н), 8,23 (т, J=6,0 Гц, 1Н), 7,97 (м, 2Н), 7,61 (м, 2Н), 7,42 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,29 (д, J=8,4 Гц, 1Н), 6,97 (кв, J=4,8 Гц, 1Н), 4,76 (м, 1Н), 4,17 (дд, J1=9,6 Гц, J2=9,6 Гц, 1Н), 3,90 (т, J=5,4 Гц, 2Н), 3,79 (м, 3Н), 3,44 (т, J=5,4 Гц, 1Н), 2,67 (д, J=4,2 Гц, 3Н), 1,84 (с, 3Н).
LCMS: 47 0 (М+Н+) для C22H24FN7O4.
Пример 17
Получение соединения 17
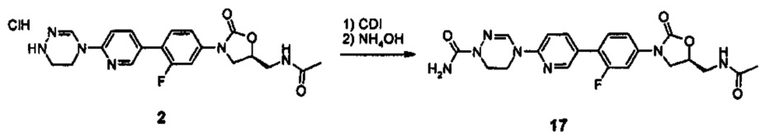
0,14 г (0,31 ммоль) Соединения 17, в виде твердого вещества белого цвета, получают (выход: 56%) по такому способу, как в примере 16, за исключением того, что используют водный раствор аммиака вместо метиламина.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,50 (с, 1Н), 8,27 (т, J=6,0 Гц, 1Н), 7,98 (д, J=6,0 Гц, 1Н), 7,96 (с, 1Н), 7,62 (м, 2Н), 7,43 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,30 (д, J=9,0 Гц, 1Н), 6,44 (с, 2Н), 4,77 (м, 1Н), 4,17 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 3,90 (т, J=5,4 Гц, 2Н), 3,80 (м, 3Н), 3,44 (т, J=5,4 Гц, 1Н), 1,84 (с, 3Н).
LCMS: 456 (М+Н+) для C21H22FN7O4.
Пример 18
Получение соединения 18
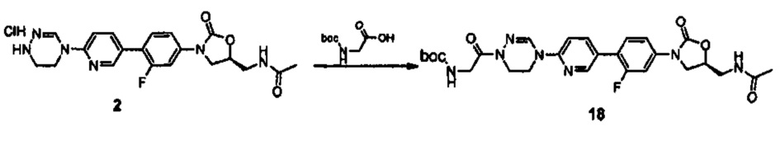
50 мг (0,09 ммоль) Соединения 18 получают (выход: 41%), используя Boc-глицин, по такому же способу, как в примере 11.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,52 (с, 1Н), 8,28 (т, J=6,0 Гц, 1Н), 8,05 (с, 1Н), 8,00 (дд, J1=7,2 Гц, J2=2,4 Гц, 1Н), 7,65 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 7,62 (дд, J1=13,8 Гц, J2=2,4 Гц, 1Н), 7,43 (дд, J1=8,4 Гц, J2=2,4 Гц, 1H), 7,35 (д, J=8,4 Гц, 1H), 6,48 (т, 6,0 Гц, 1Н), 4,77 (м, 1Н), 4,17 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 4,05 (д, J=6,0 Гц, 2Н), 3,93 (с, 4Н), 3,79 (дд, J1=9,0 Гц, J2=6,0 Гц, 1Н), 3,44 (т, J=6,0 Гц, 2Н) 1,84 (с, 3Н), 1,38 (с, 9Н).
LCMS: 57 0 (М+Н+) для C27H32FN7O6.
Пример 19
Получение соединения 19
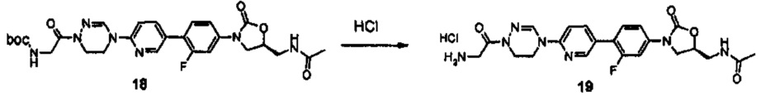
50 мг (0,09 ммоль) Соединения 18, синтезированного в соответствии с примером 18, растворяют в 10 мл дихлорметана, добавляют 1 мл 4 М HCl и полученный раствор перемешивают в течение 1 часа. Реакционную смесь концентрируют при пониженном давлении, получая 46 мг (0,09 ммоль) соединения 19 в виде твердого вещества желтого цвета (выход: 99%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,53 (с, 1Н), 8,29 (т, J=6,0 Гц, 1Н), 8,18 (м, 4Н), 8,02 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,63 (м, 2Н), 7,44 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,40 (д, J=7,2 Гц, 1Н), 6,48 (т, 6,0 Гц, 1Н), 4,77 (м, 1Н), 4,17 (дд, J1=7,8 Гц, J2=7,8 Гц, 1Н), 4,00 (м, 4Н), 3,80 (м, 1Н), 3,45 (м, 4Н), 1,84 (с, 3Н).
LCMS: 470 (М+Н+) для C22H24FN7O4.
Пример 20
Получение соединения 20
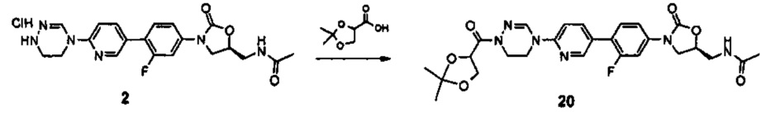
0,12 г (0,22 ммоль) соединения 20 получают (выход: 50%), в виде твердого вещества белого цвета, используя глицериновую кислоту, по такому же способу, как в примере 11.
1Н ЯМР (600 МГц, ДМСО d-6) δ 8,47 (с, 1Н), 8,23 (т, J=6,0 Гц, 1Н), 7,98 (с, 1Н), 7,96 (д, J=8,4 Гц, 1Н), 7,60 (м, 2Н), 7,39 (дд, J1=9,0 Гц, J2=1,8 Гц, 1Н), 7,32 (д, J=8,8 Гц, 1Н), 5,18 (т, 6,6 Гц, 1Н), 4,73 (м, 1Н), 4,26 (дд, J1=7,8 Гц, J2=7,8 Гц, 1Н), 4,13 (т, J=9,0 Гц, 1Н), 4,92 (м, 3Н), 3,85 (м, 2Н), 3,74 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,40 (т, J=5,4 Гц, 2Н), 1,80 (с, 3Н), 1,36 (с, 3Н), 1,30 (с, 3Н).
LCMS: 541 (М+Н+) для C26H29FN6O6.
Пример 21
Получение соединения 21
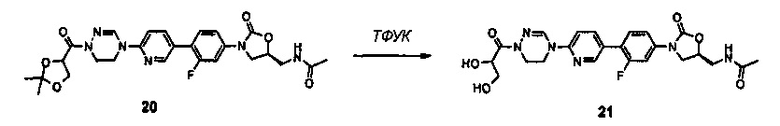
0,12 г (0,22 ммоль) соединения 20, полученного в соответствии с примером 20, растворяют в 2 мл тетрагидрофурана, добавляют 2 мл ТФУК и полученный раствор перемешивают при комнатной температуре в течение 1 часа. Затем, к реакционной смеси добавляют 15 мл диэтилового эфира и полученную смесь отфильтровывают, получая 65 мг (0,13 ммоль) соединения 21 в виде твердого вещества белого цвета (выход: 59%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,47 (с, 1Н), 8,25 (т, J=5,4 Гц, 1Н), 8,00 (с, 1Н), 7,96 (д, J=9,0 Гц, 1Н), 7,59 (м, 2Н), 7,39 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 7,32 (д, J1=9,0 Гц, 1Н), 4,81 (м, 1Н), 4,72 (м, 1Н), 4,65 (м, 2Н), 4,13 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 4,02 (м, 1Н), 3,95 (м, 1Н), 3,80 (м, 3Н), 3,60 (м, 2Н), 3,40 (т, J=5,4 Гц, 2Н), 1,80 (с, 3Н).
LCMS: 501 (М+Н+) для C22H25FN6O6.
Пример 22
Получение соединения 22
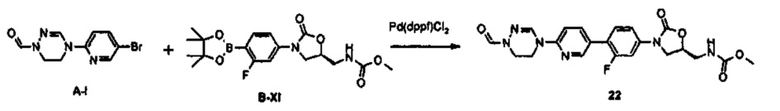
1,04 г (2,28 ммоль) соединения 22 получают (выход: 76%), в виде твердого вещества серого цвета, путем введения во взаимодействие соединения A-I с соединением B-XI, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,51 (м, 2Н), 8,06 (с, 1Н), 8,00 (д, J=8,4 Гц, 1Н), 7,63 (м, 2Н), 7,56 (т, J=5,4 Гц, 1Н), 7,43 (дд, J1=8,4 Гц, J2=1,8 Гц, 1Н), 7,35 (д, J=9,0 Гц, 1Н), 4,76 (м, 1Н), 4,18 (т, J=8,4 Гц, 1Н), 3,93 (т, J=4,8 Гц, 2Н), 3,88 (т, J=4,8 Гц, 2Н), 3,82 (дд, J1=9,0 Гц, J2=6,0 Гц, 1Н), 3,54 (с, 3Н), 3,38 (м, 2Н).
LCMS: 457 (М+Н+) для C21H21FN6O5.
Пример 23
Получение соединения 23
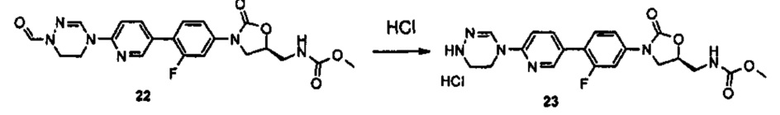
1 г (2,19 ммоль) Соединения 23, в виде твердого вещества коричневого цвета, получают с количественным выходом путем обработки соединения 22 соляной кислотой, по такому же способу, как в примере 2.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,51 (м, 2Н), 8,06 (с, 1Н), 8,00 (д, J=8,4 Гц, 1Н), 7,63 (м, 2Н), 7,56 (т, J=5,4 Гц, 1Н), 7,43 (дд, J1=8,4 Гц, J2=1,8 Гц, 1Н), 7,35 (д, J=9,0 Гц, 1Н),4,76 (м, 1Н), 4,18 (т, J=8,4 Гц, 1Н), 3,93 (т, J=4,8 Гц, 2Н), 3,88 (т, J=4,8 Гц, 2Н), 3,82 (дд, J1=9,0 Гц, J2=6,0 Гц, 1Н), 3,54 (с, 3Н), 3,38 (м, 2Н).
LCMS: 4 57 (М+Н+) для C21H21FN6O5.
Пример 24
Получение соединения 24
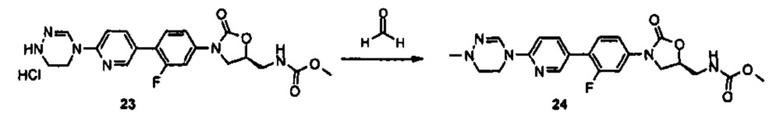
153 мг (0,35 ммоль) соединения 24, в виде твердого вещества желтого цвета, получают (выход: 81%) из соединения 23 по такому же способу, как в примере 3.
1Н ЯМР (600 МГц, CDCl3) δ 8,45 (с, 1Н), 7,98 (с, 1Н), 7,81 (м, 1Н), 7,54 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,41 (т, J=8,4 Гц, 1Н), 7,31 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 6,86 (д, J=8,4 Гц, 1Н), 5,14 (м, 1Н), 4,81 (м, 1Н), 4,09 (т, J=8,4 Гц, 1Н), 3,94 (т, J=4,8 Гц, 2H), 3,85 (т, J=8,4 Гц, 1Н), 3,69 (с, 3Н), 3,64 (м, 1Н), 3,58 (м, 1Н), 3,03 (т, J=4,8 Гц, 2Н), 2,84 (с, 3Н).
LCMS: 4 43 (М+Н+) для C21H23FN6O4.
Пример 25
Получение соединения 25
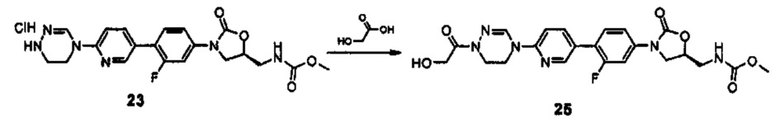
55 мг (0,11 ммоль) соединения 25 получают (выход: 18%), в виде твердого вещества белого цвета, из соединения 23, по такому же способу, как в примере 11.
1H ЯМР (600 МГц, CDCl3) δ 8,05 (с, 1Н), 7,93 (с, 1Н), 7,89 (д, J=8,4 Гц, 1Н), 7,71 (дд, J1=12,6 Гц, J2=2,4 Гц, 1Н), 7,43 (т, J=8,4 Гц, 1Н), 7,34 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 6,96 (д, J=7,8 Гц, 1Н), 5,12 (м, 1Н), 4,82 (м, 1Н), 4,55 (д, J=4,8 Гц, 2Н), 4,11 (т, J=5,4 Гц, 2Н), 4,06 (м, 1Н), 3,95 (т, J=5,4 Гц, 2Н), 3,87 (т, J=7,8 Гц, 1Н), 3,70 (с, 3Н), 3,65 (м, 1Н), 3,57 (м, 1Н).
LCMS: 487 (М+Н+) для C22H23FN6O6.
Пример 26
Получение соединения 26
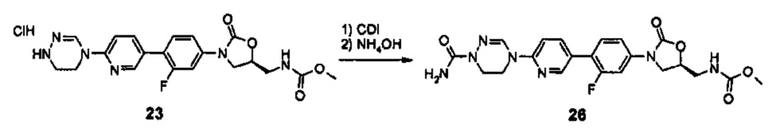
35 мг (0,07 ммоль) соединения 26 получают (выход: 32%), в виде твердого вещества белого цвета, из соединения 23, с помощью подобной методики, как в примере 17.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,50 (с, 1Н), 7,98 (д, J=9,0 Гц, 1Н), 7,95 (с, 1Н), 7,63 (м, 2Н), 7,56 (м, 1Н), 7,43 (д, J=9,0 Гц, 1Н), 7,30 (д, J=9,0 Гц, 1Н), 6,44 (с, 2Н), 4,76 (м, 1Н), 4,18 (т, J=9,6 Гц, 1Н), 3,90 (т, J=4,8 Гц, 2Н), 3,81 (м, 2Н), 3,54 (с, 3Н), 3,36 (м, 3Н).
LCMS: 472 (М+Н+) для C21H22FN7O5.
Пример 27
Получение соединения 27
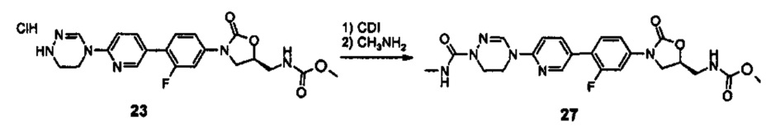
56 мг (0,12 ммоль) соединения 27 получают (выход: 46%), в виде твердого вещества белого цвета, из соединения 23, по такому же способу, как в примере 16.
1H ЯМР (600 МГц, CDCl3) δ 8,48 (с, 1Н), 7,90 (с, 1Н), 7,86 (д, J=9,0 Гц, 1Н), 7,56 (д, J=12,6 Гц, 1Н), 7,42 (т, J=9,0 Гц, 1Н), 7,33 (д, J=6,0 Гц, 1Н), 6,91 (д, J=8,4 Гц, 1Н), 6,34 (д, J=4,8 Гц, 1Н), 5,14 (м, 1Н), 4,81 (м, 1Н), 4,10 (т, J=8,4 Гц, 1Н), 4,01 (т, J=4,8 Гц, 2Н), 3,90 (т, J=4,8 Гц, 2Н), 3,86 (т, J=7,8 Гц, 1Н), 3,69 (с, 3Н), 3,64 (м, 1Н), 3,58 (м, 1Н), 2,91 (д, J=4,8 Гц, 3Н).
LCMS: 48 6 (М+Н+) для C22H24FN7O5.
Пример 28
Получение соединения 28
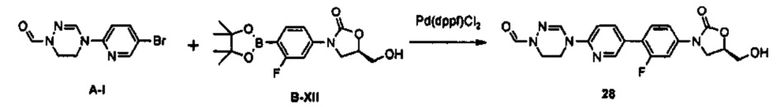
200 мг (0,50 ммоль) соединения 28 получают (выход: 36%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-I и соединения B-XII, по такому же способу, как в примере 1.
1Н ЯМР (400 МГц, ДМСО d-6) δ 8,50 (с, 1Н), 8,49 (с, 1Н), 8,05 (с, 1Н), 7,99 (д, J=8,4 Гц, 1Н), 7,63 (м, 2Н), 7,45 (дд, J1=8,4 Гц, J2=1,8 Гц, 1Н), 7,33 (д, J=9,6 Гц, 1Н), 5,24 (т, J=5,4 Гц, 1Н), 4,73 (м, 1Н), 4,18 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 3,89 (м, 5Н), 3,67 (м, 1Н), 3,58 (м, 1Н).
LCMS: 400 (М+Н+) для C19H18FN5O4.
Пример 29
Получение соединения 29
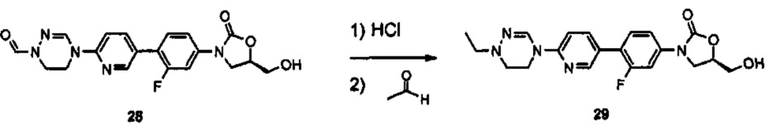
55 мг (0,12 ммоль) соединения 29 получают (выход: 28%), в виде твердого вещества белого цвета, из соединения 28, таким же образом, как в примерах 2 и 4.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,43 (с, 1Н), 7,94 (с, 1Н), 7,90 (д, J=9,0 Гц, 1Н), 7,62 (м, 2Н), 7,42 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 7,17 (д, J=8,4 Гц, 1Н), 4,75 (м, 1Н), 4,14 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 3,87 (дд, J1=9,0 Гц, J2=6,0 Гц, 1Н), 3,73 (м, 1Н), 3,58 (м, 1Н), 3,43 (т, J=5,4 Гц, 2Н), 2,97 (м, 4Н), 1,31 (т, J=5,4 Гц, 3Н).
LCMS: 400 (М+Н+) для C20H22FN5O3.
Пример 30
Получение соединения 30
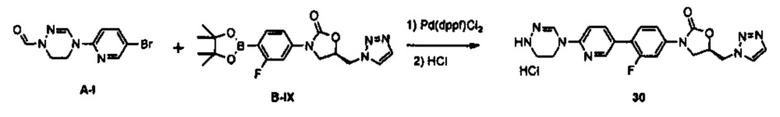
900 мг (1,96 ммоль) соединения 30 получают (выход: 68%), в виде твердого вещества коричневого цвета, путем введения во взаимодействие соединения A-I и соединения В-IX, таким же образом, как в примерах 1 и 2.
1H ЯМР (600 МГц, ДМСО d-6) δ 9,00 (с, 1Н), 8,61 (с, 1Н), 8,20 (с, 1Н), 8,14 (д, J=8,4 Гц, 1Н), 7,78 (с, 1Н), 7,66 (т, 9,0 Гц, 1Н), 7,62 (м, 1Н), 7,56 (с, 1Н), 7,40 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 5,19 (м, 1Н), 4,87 (д, J=4,8 Гц, 2Н), 4,30 (т, J=9,0 Гц, 1Н), 4,03 (т, J=4,8 Гц, 2Н), 3,96 (м, 2Н), 3,66 (м, 1Н), 3,46 (м, 2Н).
LCMS: 423 (М+Н+) для C20H19FN8O2.
Пример 31
Получение соединения 31
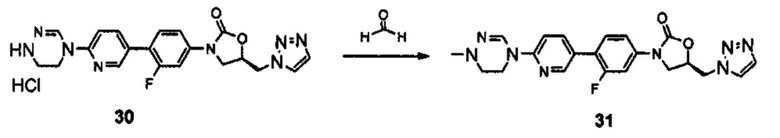
68 мг (0,15 ммоль) соединения 31 получают (выход: 63%), в виде твердого вещества коричневого цвета, из соединения 30, по такому же способу, как в примере 3.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,44 (с, 1Н), 8,19 (с, 1Н), 7,95 (с, 1Н), 7,91 (д, J=9,0 Гц, 1Н), 7,77 (с, 1Н), 7,60 (т, J=8,4 Гц, 1Н), 7,54 (дд, J1=13,8 Гц, J2=1,8 Гц, 1Н), 7,36 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,16 (д, J=9,0 Гц, 1Н), 5,18 (м, 1Н), 4,86 (м, 1Н), 4,29 (т, J=9,0 Гц, 1Н), 3,95 (м, 1Н), 3,84 (т, J=4,8 Гц, 2Н), 2,95 (т, 7=4,8 Гц, 2Н), 2,68 (с, 3Н).
LCMS: 437 (М+Н+) для C21H21FN8O2.
Пример 32
Получение соединения 32
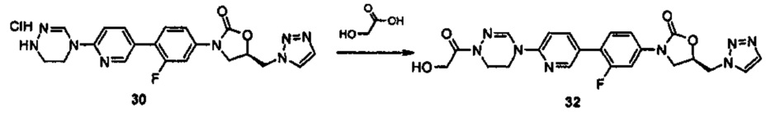
84 мг (0,17 ммоль) соединения 32 получают (выход: 35%), в виде твердого вещества желтого цвета, из соединения 30, по такому же способу, как в примере 11.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,51 (с, 1Н), 8,19 (с, 1Н), 8,02 (с, 1Н), 7,99 (д, J=8,4 Гц, 1Н), 7,78 (с, 1Н), 7,62 (т, J=8,4 Гц, 1Н), 7,55 (д, J=13,2 Гц, 1Н), 7,38 (д, J=8,4 Гц, 1Н), 7,35 (д, J=9,0 Гц, 1Н), 5,19 (м, 1Н), 4,86 (д, J=4,2 Гц, 2Н), 4,37 (с, 2Н), 4,29 (т, J=9,0 Гц, 1Н), 3,95 (м, 1Н), 3,93 (м, 4Н).
LCMS: 481 (М+Н+) для C21H22FN8O4.
Пример 33
Получение соединения 33
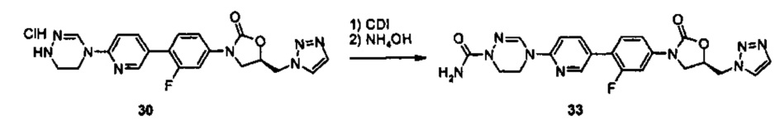
225 мг (0,48 ммоль) соединения 33 получают (выход: 71%), в виде твердого вещества коричневого цвета, из соединения 30, по такому же способу, как в примере 17.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,49 (с, 1Н), 8,19 (с, 1Н), 7,97 (д, J=9,0 Гц, 1Н), 7,95 (с, 1Н), 7,78 (с, 1Н), 7,62 (т, J=9,0 Гц, 1Н), 7,55 (д, J=13,2 Гц, 1Н), 7,38 (д, J=8,4 Гц, 1Н), 7,30 (д, J=9,0 Гц, 1Н), 6,44 (с, 2Н), 5,18 (м, 1Н), 4,86 (д, J=4,8 Гц, 2Н), 4,29 (т, J=9,0 Гц, 1Н), 3,96 (м, 1Н), 3,90 (т, J=4,8 Гц, 2Н), 3,82 (т, J=4,8 Гц, 2Н).
LCMS: 4 66 (М+Н+) для C21H20FN9O3.
Пример 34
Получение соединения 34
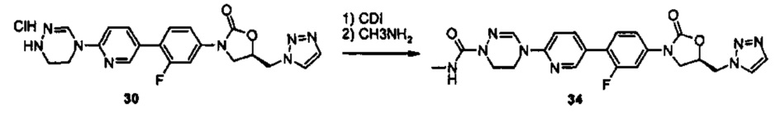
188 мг (0,39 ммоль) Соединения 34 получают (выход: 57%), в виде твердого вещества белого цвета, из соединения 30, по такому же способу, как в примере 16.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,49 (с, 1Н), 8,19 (с, 1Н), 7,96 (м, 2Н), 7,78 (с, 1Н), 7,62 (т, J=9,0 Гц, 1Н), 7,55 (м, 1Н), 7,38 (м, 1Н), 7,29 (д, J=9,0 Гц, 1Н), 6,97 (м, 1Н), 5,19 (м, 1Н), 4,86 (д, J=4,8 Гц, 2Н), 4,29 (т, J=9,0 Гц, 1Н), 3,95 (м, 1Н), 3,90 (т, J=4,8 Гц, 2Н), 3,81 (т, J=4,8 Гц, 2Н), 2,67 (д, J=4,2 Гц, 3Н).
LCMS: 480 (М+Н+) для C22H22FN9O3.
Пример 35
Получение соединения 35
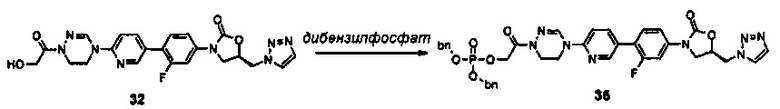
90 мг (0,12 ммоль) Соединения 35 получают (выход: 68%), в виде твердого вещества белого цвета, из соединения 32, по такому же способу, как в примере 14.
1H ЯМР (400 МГц, CDCl3) δ 8,48 (с, 1Н), 7,87 (м, 2Н), 7,81 (д, J=0,8 Гц, 1Н), 7,77 (дд, J=0,8 Гц, 1Н), 7,45 (дд, J1=8,8 Гц, J2=2,4 Гц, 1Н), 7,35 (м, 11Н), 7,20 (дд, J1=8,8 Гц, J2=2,4 Гц, 1Н), 6,94 (д, J=8,8 Гц, 1Н), 5,12 (м, 4Н), 5,14 (м, 3Н) 4,83 (д, J=4,0 Гц, 2Н), 4,15 (дд, J1=9,6 Гц, J2=9,6 Гц, 1Н), 4,03 (м, 3Н), 3,91 (т, J=5,2 Гц, 2Н).
LCMS: 741 (М+Н+) для C36H34FN8O7P.
Пример 36
Получение соединения 36
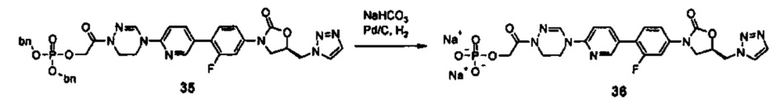
50 мг (0,08 ммоль) Соединения 36 получают (выход: 74%), в виде твердого вещества белого цвета, из соединения 35, по такому же способу, как в примере 15.
1H ЯМР (600 МГц, D2O) δ 8,17 (с, 1Н), 7,90 (с, 1Н), 7,77 (д, J=7,8 Гц, 1Н), 7,62 (с, 1Н), 7,58 (с, 1Н), 7,26 (дд, J1=8,4 Гц, J2=8,4 Гц, 1H), 7,05 (д, J=12,6 Гц, 1H), 6,97 (д, J=7,8 Гц, 1H), 6,93 (Д, J=8,4 Гц, 1H), 5,04 (м, 1H), 4,73 (м, 2H), 4,60 (м, 2Н), 4,13 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 3,75 (м, 3Н), 3,37 (м, 2Н).
LCMS: 561 (М+Н+) для C22H22FN8O7P.
Пример 37
Получение соединения 37
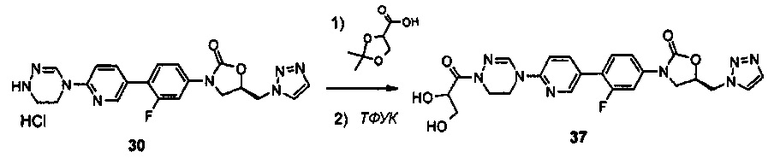
17 мг (0,03 ммоль) Соединения 37 получают (выход: 30%), в виде твердого вещества желтого цвета, из соединения 30, таким же образом, как в примерах 20 и 21.
1H ЯМР (600 МГц, ДМСО) δ 8,51 (с, 1Н), 8,18 (с, 1Н), 8,04 (с, 1Н), 7,99 (д, J=9,0 Гц, 1Н), 7,77 (с, 1Н), 7,62 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 7,55 (д, J=13,8 Гц, 1Н), 7,36 (м, 2Н), 5,17 (м, 1Н), 4,86 (д, J=4,2 Гц, 2Н), 4,29 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 4,04 (м, 1Н), 3,95 (м, 3Н), 3,83 (м, 2Н), 3,64 (м, 1Н), 3,55 (м, 1Н).
LCMS: 511 (М+Н+) для C23H23FN8O5.
Пример 38
Получение соединения 38
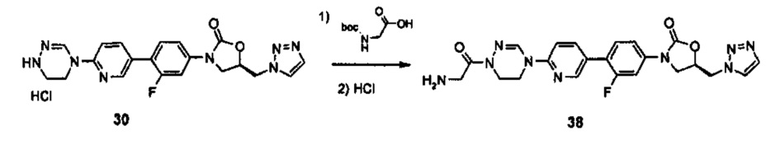
25 мг (0,05 ммоль) Соединения 38 получают (выход: 70%), в виде твердого вещества светло-желтого цвета, из соединения 30, таким же образом, как в примерах 18 и 19.
1H ЯМР (400 МГц, ДМСО) δ 8,53 (с, 1Н), 8,20 (д, J=1,2 Гц, 1Н), 8,13 (с, 1Н), 8,00 (д, J=7,2 Гц, 1Н), 7,77 (д, J=1,2 Гц, 1Н), 7,63 (дд, J1=8,8 Гц, J2=8,8 Гц, 1Н), 7,55 (дд, J1=13,2 Гц, J2=2,0 Гц, 1Н), 7,40 (м, 2Н), 5,19 (м, 1Н), 4,87 (д, J=5,4 Гц, 2Н), 4,29 (дд, J1=9,2 Гц, J2=9,2 Гц, 1Н), 4,04 (м, 3Н), 3,96 (м, 4Н).
LCMS: 480 (М+Н+) для C22H22FN9O3.
Пример 39
Получение соединения 39
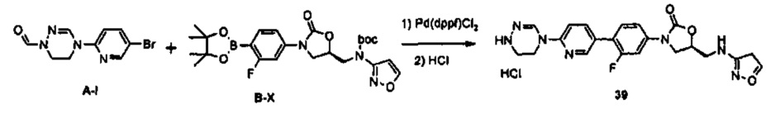
1,1 г (2,53 ммоль) Соединения 39 получают (выход: 45%), в виде твердого вещества желтого цвета, путем введения во взаимодействие соединения A-I и соединения В-Х, таким же образом, как в примерах 1 и 2.
1H ЯМР (600 МГц, ДМСО-6) δ 8,98 (с, 1Н), 8,62 (с, 1Н), 8,40 (д, J=1,8 Гц, 1Н), 8,14 (д, J=9,0 Гц, 1Н), 7,67 (м, 2Н), 7,58 (д, J=9 Гц, 1Н), 7,46 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 6,02 (д, J=1,8 Гц, 1Н), 4,93 (м, 1Н), 4,22 (дд, J1=9,0 Гц, J2=9,0 Гц 1Н), 4,12 (т, J=5,4 Гц, 2Н), 3,88 (дд, J=6,6 Гц, 1Н), 3,46 (м, 4Н).
LCMS: 439 (М+Н+) для C21H21FN7O3.
Пример 40
Получение соединения 40
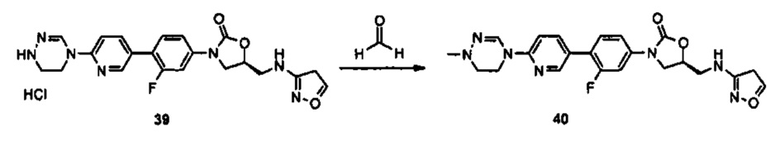
45 мг (0,10 ммоль) Соединения 40 получают (выход: 23%), в виде твердого вещества белого цвета, из соединения 39, по такому же способу, как в примере 3.
1H ЯМР (600 МГц, ДМСО) δ 8,94 (с, 1Н), 8,60 (с, 1Н), 8,39 (д, J=1,8 Гц, 1Н), 8,13 (д, J=9,0 Гц, 1Н), 7,66 (м, 2Н), 7,57 (д, J=9,0 Гц, 1Н), 7,46 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 6,01 (д, J=1,8 Гц, 1Н), 4,92 (м, 1Н), 4,21 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 4,12 (т, J=5,4 Гц, 2Н), 3,88 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,45 (м, 4Н), 2,92 (с, 2Н)
LCMS: 452 (М+Н+) для C22H22FN7O3.
Пример 41
Получение соединения 41
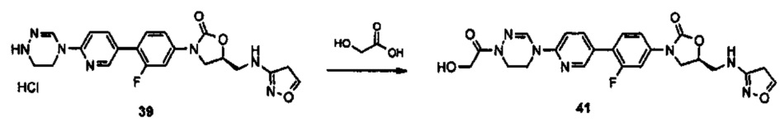
28 мг (0,06 ммоль) Соединения 41 получают (выход: 15%), в виде твердого вещества белого цвета, из соединения 39, по такому же способу, как в примере 11.
1H ЯМР (600 МГц, ДМСО) δ 8,05 (с, 1Н), 8,40 (с, 1Н), 8,00 (м, 2Н), 7,63 (м, 2Н), 7,44 (д, J=7,8 Гц, 1Н), 7,34 (д, J=8,4 Гц, 1Н), 6,60 (с, 1Н), 6,01 (с, 1Н), 4,92 (м, 1Н), 4,66 (с, 1Н), 4,36 (м, 2Н), 4,20 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 3,93 (с, 4Н), 3,86 (м, 1Н), 3,46 (с, 2Н).
LCMS: 4 96 (М+Н+) для C23H22FN7O5.
Пример 42
Получение соединения 42
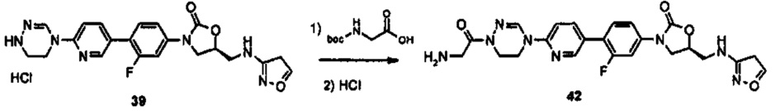
20 мг (0,04 ммоль) Соединения 42 получают (выход: 40%), в виде твердого вещества белого цвета, из соединения 39, таким же образом, как в примерах 18 и 19.
1H ЯМР (600 МГц, ДМСО) δ 8,55 (с, 1Н), 8,41 (д, J=0,6 Гц, 1Н), 8,24 (с, 2Н), 8,14 (с, 1Н), 8,03 (д, J=8,4 Гц, 1Н), 7,65 (м, 2Н), 7,45 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 7,40 (д, J=8,4 Гц, 1Н), 6,01 (д, J=1,2 Гц, 1Н), 4,93 (м, 1Н), 4,21 (дд, J1=9,0 Гц, J2=9,0 Гц, 1Н), 4,00 (м, 4Н), 3,87 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,46 (м, 2Н), 1,29 (м, 2Н).
LCMS: 495 (М+Н+) для C23H23FN8O4.
Пример 43
Получение соединения 43
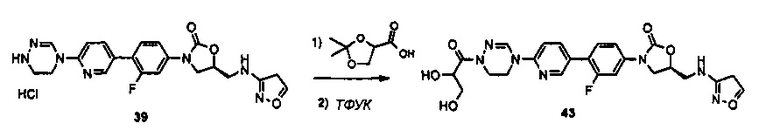
62 мг (0,12 ммоль) Соединения 43, в виде твердого вещества белого цвета, получают (выход: 41%) из соединения 39, таким же образом, как в примерах 20 и 21.
1H ЯМР (600 МГц, ДМСО) δ 8,52 (с, 1Н), 8,41 (д, J=1,2 Гц, 1Н), 8,05 (с, 1H), 8,00 (д, J=9,0 Гц, 1H), 7,64 (м, 2H), 7,44 (д, J=7,2 Гц, 1Н), 7,36 (д, J=8,4 Гц, 1Н), 6,61 (с, 1Н), 6,01 (д, 7=1,2 Гц, 1Н), 4,92 (м, 1Н), 4,86 (м, 1Н), 4,21 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 4,03 (м, 2Н), 3,84 (м, 5Н), 3,47 (м, 2Н).
LCMS: 52 6 (М+Н+) для C24H24FN7O6.
Пример 44
Получение соединения 44
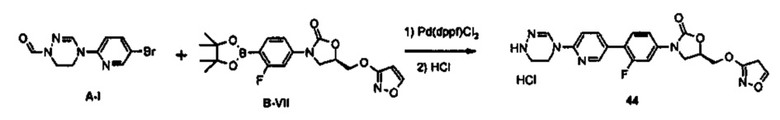
887 мг (1,84 ммоль) Соединения 44 получают (выход: 35%), в виде твердого вещества желтого цвета, путем введения во взаимодействие соединения A-I и соединения B-VII, таким же образом, как в примерах 1 и 2.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,95 (м, 1Н), 8,72 (д, J=1,8 Гц, 1Н), 8,61 (с, 1Н), 8,14 (д, J=7,8 Гц, 1Н), 7,69 (м, 2Н), 7,55 (д, J=8,4 Гц, 1Н), 7,50 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 6,41 (д, J=1,8 Гц, 1Н), 5,14 (м, 1Н), 4,51 (м, 2Н), 4,27 (т, J=9,0 Гц, 1Н), 3,99 (м, 3Н), 3,39 (м, 2Н)
LCMS: 440 (М+Н+) для C21H20FN6O4.
Пример 45
Получение соединения 45
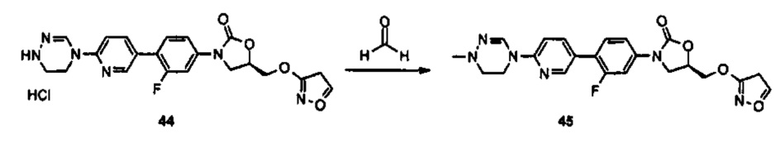
54 мг (0,12 ммоль) Соединения 45 получают (выход: 57%), в виде твердого вещества белого цвета, из соединения 44, по такому же способу, как в примере 3.
1H ЯМР (600 МГц, CDCl3) δ 8,46 (с, 1Н), 8,17 (д, J=1,8 Гц, 1Н), 7,99 (с, 1Н), 7,82 (м, 1Н), 7,57 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,43 (т, J=9,0 Гц, 1Н), 7,36 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 6,87 (д, J=8,4 Гц, 1Н), 6,02 (д, J=1,8 Гц, 1Н), 5,06 (м, 1Н), 4,61 (дд, J1=11,4 Гц, J2=3,6 Гц, 1Н), 4,53 (дд, J1=12,0 Гц, J2=4,8 Гц, 1Н), 4,21 (т, J=9,0 Гц, 1Н), 4,02 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,94 (т, J=5,4 Гц, 2Н), 3,03 (т, J=4,8 Гц, 2Н), 2,85 (с, 3Н)
LCMS: 454 (М+Н+) для C22H22FN6O4.
Пример 46
Получение соединения 46
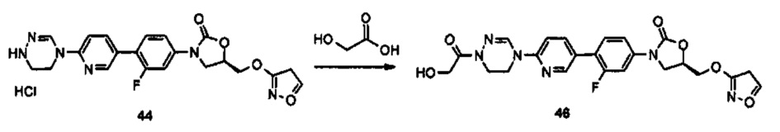
88 мг (0,18 ммоль) Соединения 46 получают (выход: 42%), в виде твердого вещества коричневого цвета, из соединения 44, по такому же способу, как в примере 11.
1H ЯМР (400 МГц, ДМСО d-6) δ 8,72 (д, J=1,6 Гц, 1Н), 8,52 (с, 1Н), 8,01 (м, 1Н), 7,64 (м, 2Н), 7,47 (дд, J1=8,4 Гц, J2=2,0 Гц, 1Н), 7,35 (д, J=8,8 Гц, 1Н), 6,41 (д, J=1,6 Гц, 1Н), 5,13 (м, 1Н), 4,64 (т, J=6,0 Гц, 1Н), 4,51 (м, 2Н), 4,37 (д, J=6,4 Гц, 2Н), 4,26 (т, J=9,2 Гц, 1Н), 3,99 (дд, J1=9,2 Гц, J2=6,0 Гц, 1Н), 3,93 (м, 4Н).
LCMS: 4 98 (М+Н+) для C23H22FN6O6.
Пример 47
Получение соединения 47
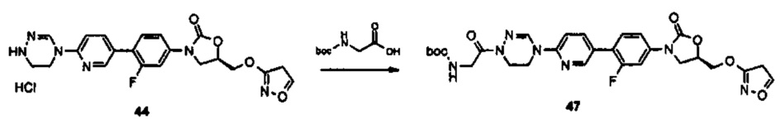
100 мг (0,17 ммоль) Соединения 47 получают (выход: 80%), в виде твердого вещества желтого цвета, из соединения 44, по такому же способу, как в примере 18.
1H ЯМР (400 МГц, ДМСО d-6) δ 8,72 (д, J=1,6 Гц, 1Н), 8,52 (с, 1Н), 8,01 (м, 1Н), 7,64 (м, 2Н), 7,47 (дд, J1=8,4 Гц, J2=2,0 Гц, 1Н), 7,35 (д, J=8,8 Гц, 1Н), 6,41 (д, J=1,6 Гц, 1Н), 5,13 (м, 1Н), 4,64 (т, J=6,0 Гц, 1Н), 4,51 (м, 2Н), 4,37 (д, J=6,4 Гц, 2Н), 4,26 (т, J=9,2 Гц, 1Н), 3,99 (дд, J1=9,2 Гц, J2=6,0 Гц, 1Н), 3,93 (м, 4Н).
LCMS: 498 (М+Н+) для C23H22FN6O6.
Пример 48
Получение соединения 48
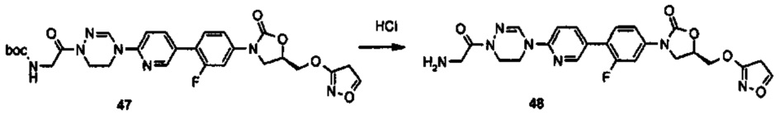
70 мг (0,14 ммоль) Соединения 48 получают (выход: 88%), в виде твердого вещества желтого цвета, из соединения 47, по такому же способу, как в примере 19.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,72 (д, J=1,8 Гц, 1Н), 8,54 (с, 1Н), 8,18 (т, J=5,4 Гц, 2Н), 8,14 (с, 1Н), 8,04 (д, J=9,0 Гц, 1Н), 7,65 (м, 2Н), 7,48 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 7,41 (д, J=9,0 Гц, 1Н), 6,41 (д, J=1,8 Гц, 1Н), 5,13 (м, 1Н), 4,53 (дд, J1=11,4 Гц, J2=3,0 Гц, 1Н), 4,49 (дд, J1=11,4 Гц, J2=6,0 Гц, 1Н), 4,26 (т, J=9,0 Гц, 1Н), 4,03 (м, 2Н), 3,98 (м, 5Н).
LCMS: 497 (М+Н+) для C23H23FN7O5.
Пример 49
Получение соединения 4 9
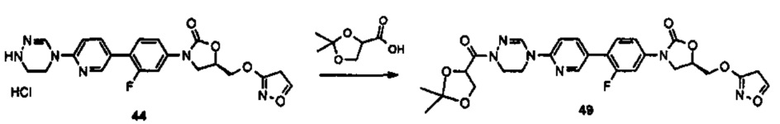
234 мг (0,41 ммоль) Соединения 49 получают (выход: 79%), в виде твердого вещества белого цвета, из соединения 44, по такому же способу, как в примере 20.
1H ЯМР (600 МГц, CDCl3) δ 8,50 (с, 1Н), 8,18 (д, J=1,8 Гц, 1Н), 7,90 (с, 2Н), 7,89 (м, 1Н), 7,60 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,44 (т, J=8,4 Гц, 1Н), 7,34 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 6,97 (д, J=9,0 Гц, 1Н), 6,02 (д, J=1,8 Гц, 1Н), 5,33 (т, J=6,6 Гц, 1Н), 5,07 (м, 1Н), 4,61 (дд, J1=11,4 Гц, J2=3,6 Гц, 1Н), 4,53 (дд, J1=12,4 Гц, J2=4,8 Гц, 1Н), 4,22 (т, J=9,0 Гц, 1Н), 4,12 (м, 1Н), 4,01 (м, 3Н), 3,94 (м, 1Н), 3,89 (м, 1Н), 1,56 (с, 3Н), 1,48 (с, 3Н).
LCMS: 568 (М+Н+) для C27H28FN6O7.
Пример 50
Получение соединения 50
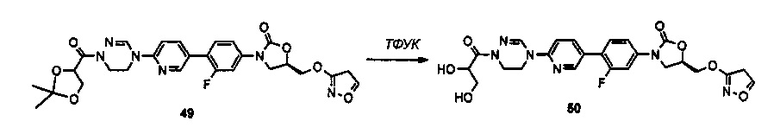
88 мг (0,17 ммоль) Соединения 50 получают (выход: 47%), в виде твердого вещества белого цвета, из соединения 49, по такому же способу, как в примере 21.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,72 (д, J=1,8 Гц, 1Н), 8,53 (с, 1Н), 8,05 (с, 1Н), 8,01 (м, 1Н), 7,66 (м, 2Н), 7,48 (м, 1Н), 7,37 (д, J=9,6 Гц, 1Н), 6,41 (д, J=1,8 Гц, 1Н), 5,12 (м, 1Н), 4,86 (м, 1Н), 4,67 (м, 2Н), 4,51 (м, 2Н), 4,26 (т, J=9,0 Гц, 1Н), 4,06 (м, 1Н), 4,00 (м, 2Н), 3,84 (м, 2Н), 3,65 (м, 1Н), 3,56 (м, 1Н).
LCMS: 528 (М+Н+) для C24H24FN6O7.
Пример 51
Получение соединения 51
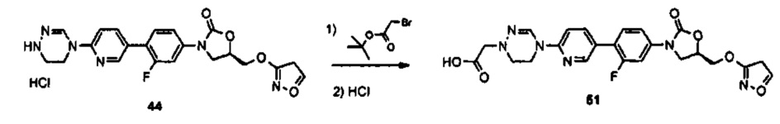
40 мг (0,08 ммоль) Соединения 51 получают (выход: 34%), в виде твердого вещества белого цвета, из соединения 44, таким же образом, как в примерах 9 и 10.
1H ЯМР (600 МГц, ДМСО d-6) δ 12,43 (с, 1Н), 8,72 (д, J=1,8 Гц, 1Н), 8,46 (с, 1Н), 7,94 (м, 2Н), 7,65 (м, 2Н), 7,46 (м, 1Н), 7,19 (д, J=9,0 Гц, 1Н), 6,42 (д, 1,8 Гц, 1Н), 5,13 (м, 1Н), 4,51 (дд, J1=11,4 Гц, J2=8,4 Гц, 1Н), 4,48 (т, J=6,0 Гц, 1Н), 4,26 (т, J=8,4 Гц, 1Н), 3,99 (м, 1Н), 3,87 (т, J=4,8 Гц, 2Н), 3,70 (с, 3Н), 3,23 (т, J=4,8 Гц, 2Н).
LCMS: 4 98 (М+Н+) для C23H22FN6O6.
Пример 52
Получение соединения 52
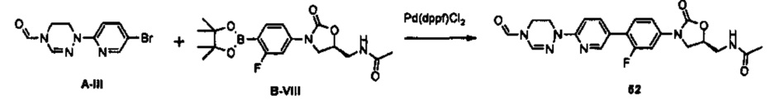
0,3 г (0,90 ммоль) Соединения А-III, синтезированного в соответствии с примером получения 14, растворяют в 5 мл N,N-диметилформамида, добавляют 350 мг соединения B-VIII, синтезированного в соответствии с примером получения 37, 22 мг (0,03 ммоль) PdCl2(dppf) и 0,9 мл 2 М водного раствора карбоната натрия, и полученный раствор перемешивают в атмосфере азота, при температуре 90°C, в течение 5 часов. Реакционную смесь охлаждают до комнатной температуры и отфильтровывают через целит, для удаления осадка. Полученную смесь экстрагируют с помощью 50 мл этилацетата и 40 мл насыщенного раствора хлорида аммония, для получения органического слоя. Органический слой концентрируют при пониженном давлении. Полученное твердое вещество промывают 20 мл этилацетата и 20 мл дихлорметана и затем высушивают, получая 75 мг (0,17 ммоль) соединения 52 в виде твердого вещества серого цвета (выход: 19%).
1H ЯМР (600 МГц, CD3OD) δ 8,57 (с, 1Н), 8,32 (с, 1Н), 7,85-7,81 (м, 2Н), 7,61 (м, 1Н), 7,50 (м, 2Н), 7,35 (м, 1Н), 4,18 (м, 2Н), 4,11 (м, 2Н), 3,91 (м, 2Н), 3,87 (м, 1Н), 3,57 (м, 2Н), 1,97 (с, 3Н).
LCMS: 441 (М+Н+) для C21H21FN6O4.
Пример 53
Получение соединения 53
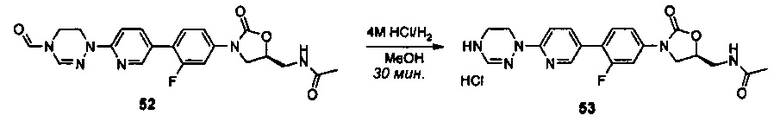
44 мг (0,1 ммоль) Соединения 52, синтезированного в соответствии с примером 52, растворяют в 7 мл метанола, последовательно добавляют 10 мг 10%-ного Pd/C и 1 мл 4 М раствора хлороводорода в 1,4-диоксане, при температуре 0°C, и полученный раствор перемешивают в атмосфере водорода в течение 30 минут. Реакционную смесь отфильтровывают через целит, используя 30 мл метанола, и затем фильтрат концентрируют при пониженном давлении, получая 44 мг (0,10 ммоль) соединения 53 (выход: 98%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,24 (с, 1Н), 7,71 (д, J=9,0 Гц, 1Н), 7,55 (м, 1Н), 7,42 (м, 1Н), 7,37 (д, J=9,0 Гц, 1Н), 7,26 (д, J=9,0 Гц, 1Н), 6,89 (с, 1Н), 6,81 (с, 1Н), 4,76 (м, 1Н), 4,16 (м, 2Н), 3,77 (м, 2Н), 3,44-3,37 (м, 4Н), 1,84 (с, 3Н).
LCMS: 413 (М+Н+) для C20H21FN6O3.
Пример 54
Получение соединения 54
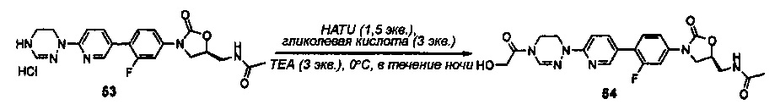
58,1 мг (0,14 ммоль) Соединения 53, синтезированного в соответствии с примером 53, 80,2 мг (0,21 ммоль) HATU и 32,2 мг (0,42 ммоль) гликолевой кислоты растворяют в 1,5 мл N,N-диметилформамида, медленно добавляют 59 мкл (0,42 ммоль) триэтиламина, при температуре 0°C, и полученный раствор перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь экстрагируют 30 мл этилацетата и 30 мл насыщенного раствора хлорида аммония, для получения органического слоя. Органический слой обезвоживают, используя безводный сульфат натрия, и затем концентрируют при пониженном давлении до получения твердого продукта. Твердый продукт промывают смешанным раствором из 5 мл этилацетата и 5 мл н-гексана, получая 20 мг (0,04 ммоль) соединения 54 в виде твердого вещества красновато-коричневого цвета (выход: 30%).
1H ЯМР (600 МГц, ДМСО d-6) δ 8,35 (с, 1Н), 8,26 (т, J=6,0 Гц, 1Н), 7,85 (д, J=8,4 Гц, 1Н), 7,61-7,57 (м, 2Н), 7,44-7,39 (м, 2Н), 5,32 (уш.с, 1Н), 4,76 (м, 1Н), 4,41 (уш.с, 1Н), 4,18 (м, 1Н), 4,06 (т, J=5,4 Гц, 1Н), 3,84-3,76 (м, 2Н), 3,42 (т, J=5,4 Гц, 1Н), 3,35 (м, 2Н), 1,84 (с, 3Н).
LCMS: 471 (М+Н+) для C22H23FN6O5.
Пример 55
Получение соединения 55
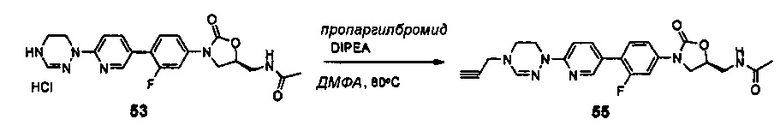
110 мг (0,24 ммоль) Соединения 53, синтезированного в соответствии с примером 53, растворяют в 2 мл N,N-диметилформамида, добавляют 0,21 мл (1,22 ммоль) диизопропилэтиламина, по каплям, добавляют 54 мкл (0,36 ммоль) пропаргилбромида (80%-ный раствор в толуоле), и полученный раствор перемешивают при температуре 80°C в течение 5 часов. Реакционную смесь охлаждают до комнатной температуры и экстрагируют с помощью 20 мл дистиллированной воды и 30 мл этилацетата, для получения органического слоя. Органический слой обезвоживают, используя безводный сульфат натрия, концентрируют при пониженном давлении и затем разделяют колоночной хроматографией, получая 14 мг (0,03 ммоль) соединения 55 в виде твердого вещества серого цвета (выход: 13%).
1H ЯМР (600 МГц, CDCl3) δ 8,43 (м, 2Н), 7,71 (д, J=9,0 Гц, 1Н), 7,54 (дд, J1=12,6 Гц, J2=2,4 Гц, 1Н), 7,39 (т, J=8,4 Гц, 1Н), 7,28 (дд, J1=9 Гц, J2=2,4 Гц, 1Н), 6,70 (д, J=9,0 Гц, 1Н), 6,00 (т, J=6,0 Гц, 1Н), 4,82 (м, 1Н), 4,66 (дд, J1=17,4 Гц, J2=2,4 Гц, 1Н), 4,55 (м, 1Н), 4,13 (дд, J1=17,4 Гц, J2=2,4 Гц, 1Н), 4,09 (т, J=9,0 Гц, 1Н), 3,81 (м, 1Н), 3,77-3,71 (м, 2Н), 3,63 (м, 1Н), 2,98 (м, 1Н), 2,90 (м, 1Н), 2,26 (т, J=2,4 Гц, 1Н), 2,04 (с, 3Н).
LCMS: 451 (М+Н+) для C23H23FN6O3.
Пример 56
Получение соединения 56
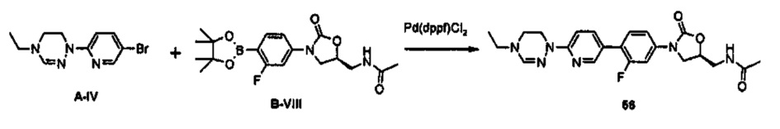
171 мг (0,63 ммоль) Соединения A-IV, синтезированного в соответствии с примером получения 17, растворяют в 4 мл N,N-диметилформамида, добавляют 180 мг соединения B-VIII, синтезированного в соответствии с примером получения 37, 16 мг (0,02 ммоль) PdCl2(dppf) и 0,95 мл (1,91 ммоль) 2 М водного раствора карбоната натрия, и полученный раствор перемешивают в атмосфере азота при температуре 85°C в течение 15 часов. Реакционную смесь охлаждают до комнатной температуры и отфильтровывают, используя целит, для удаления осадка. Полученную смесь экстрагируют 50 мл этилацетата и 40 мл насыщенного раствора хлорида аммония, для получения органического слоя. Органический слой обезвоживают, используя безводный сульфат натрия, концентрируют при пониженном давлении и затем разделяют колоночной хроматографией, получая 19 мг (0,04 ммоль) соединения 56 в виде твердого вещества желтого цвета (выход: 7,2%).
1H ЯМР (600 МГц, CDCl3) δ 8,30 (с, 1Н), 7,71 (д, J=9,0 Гц, 1Н), 7,50 (д, J=12,6 Гц, 1Н), 7,40 (т, J=8,4 Гц, 1Н), 7,35 (д, J=8,4 Гц, 1Н), 7,25 (м, 1Н), 6,73 (с, 1Н), 4,80 (м, 1Н), 4,11-4,06 (м, 3Н), 3,80 (м, 1Н), 3,73 (м, 1Н), 3,62 (м, 1Н), 3,44 (т, J=4,8 Гц, 2Н), 3,20 (кв, J=7,2 Гц, 2Н), 1,21 (т, J=12 Гц, 2Н).
LCMS: 441 (М+Н+) для C22H25FN6O3.
Пример 57
Получение соединения 57

113 мг (0,27 ммоль) Соединения 57 получают (выход: 41%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-V и соединения B-VIII, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,53 (с, 1Н), 8,50 (с, 1Н), 8,28 (т, J=5,4 Гц, 1Н), 8,00 (д, J=7,8 Гц, 1Н), 7,63 (м, 2Н), 7,43 (д, J=9,0 Гц, 1Н), 7,29 (д, J=9,0 Гц, 1Н), 4,76 (м, 1Н), 4,18 (т, J=9,0 Гц, 1Н), 4,13 (т, J=4,8 Гц, 2Н), 3,88 (т, J=4,8 Гц, 2Н), 3,79 (м, 1Н), 3,44 (т, J=4,8 Гц, 2Н), 1,84 (с, 3Н).
LCMS: 414 (М+Н+) для C20H20FN5O4.
Пример 58
Получение соединения 58
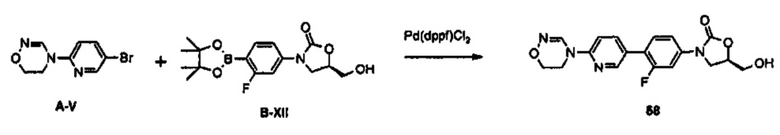
130 мг (0,35 ммоль) Соединения 58 получают (выход: 20%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-V и соединения B-XII, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,52 (с, 1Н), 7,99 (т, J=9,6 Гц, 1Н), 7,64 (м, 2Н), 7,47 (м, 2Н), 7,29 (д, J=9,0 Гц, 1Н), 5,28 (т, J=6,0 Гц, 1Н), 4,75 (м, 1Н), 4,22 (м, 1Н), 4,14 (м, 3Н), 3,89 (м, 2Н), 3,70 (м, 1Н), 3,57 (м, 1Н).
LCMS: 373 (М+Н+) для C18H17FN4O4.
Пример 59
Получение соединения 59
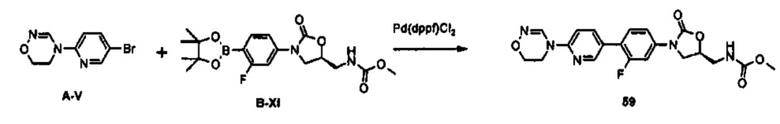
115 мг (0,27 ммоль) Соединения 59 получают (выход: 19%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-V и соединения B-XI, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,52 (с, 1Н), 8,50 (с, 1Н), 8,00 (дд, J1=9,0 Гц, J2=1,8 Гц, 1Н), 7,64 (т, J=9,0 Гц, 1Н), 7,62 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,55 (т, J=5,4 Гц, 1Н), 7,43 (дд, J1=8,4 Гц, J2=1,8 Гц, 1Н), 7,29 (д, J=8,4 Гц, 1Н), 4,77 (м, 1Н), 4,18 (т, J=9,0 Гц, 1Н), 4,11 (т, J=4,2 Гц, 2Н), 3,88 (м, 2Н), 3,81 (м, 1Н), 3,54 (с, 3Н), 3,33 (м, 2Н).
LCMS: 430 (М+Н+) для C20H20FN5O5.
Пример 60
Получение соединения 60
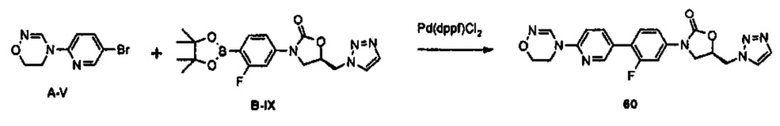
36 мг (0,08 ммоль) Соединения 60 получают (выход: 35%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-V и соединения В-IX, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,52 (с, 1Н), 8,49 (с, 1Н), 8,19 (с, 1Н), 7,99 (д, J=8,4 Гц, 1Н), 7,78 (с, 1Н), 7,62 (т, J=8,4 Гц, 1Н), 7,55 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,38 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,29 (д, J=8,4 Гц, 1Н), 5,18 (м, 1Н), 4,86 (м, 1Н), 4,29 (т, J=9,0 Гц, 1Н), 4,11 (т, J=4,8 Гц, 2Н), 3,95 (м, 1Н), 3,88 (т, J=4,8 Гц, 2Н).
LCMS: 424 (М+Н+) для C20H18FN7O3.
Пример 61
Получение соединения 61
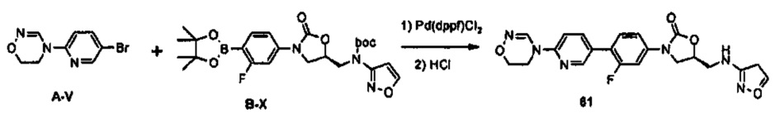
81 мг (0,18 ммоль) Соединения 61 получают (выход: 75%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-V и соединения В-Х, таким же образом, как в примерах 1 и 2.
1H ЯМР (600 МГц, ДМСО) δ 8,55 (с, 1Н), 8,50 (с, 1Н), 8,39 (д, J=1,8 Гц, 1Н), 8,00 (д, J=10,2 Гц, 1Н), 7,63 (м, 2Н), 7,44 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 7,29 (д, J=8,4 Гц, 1Н), 6,01 (д, J=1,8 Гц, 1Н), 4,92 (м, 1Н), 4,21 (дд, J1=20 8,4 Гц, J2=8,4 Гц, 1Н), 4,12 (т, J=7,2 Гц, 2Н), 3,88 (м, 3Н), 3,47 (м, 2Н).
LCMS: 439 (М+Н+) для C21H19FN6O4.
Пример 62
Получение соединения 62
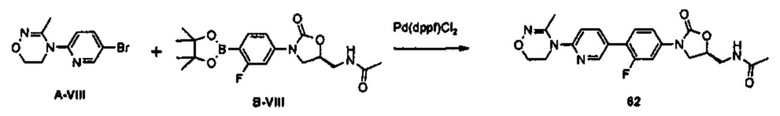
30 мг (0,07 ммоль) Соединения 62 получают (выход: 68%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-VIII и соединения B-VTII, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, CDCl3) δ 8,72 (м, 1Н), 7,88 (дд, J1=8,4 Гц, J2=3,6 Гц, 1Н), 7,59 (дд, J1=12,6 Гц, J2=2,4 Гц, 1Н), 7,44 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 7,33 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,08 (д, J=8,4 Гц, 1Н), 6,12 (т, J=6 Гц, 1Н), 4,83 (м, 1Н), 4,21 (т, J=4,8 Гц, 2Н), 4,10 (дд, J=9,0 Гц, 1Н), 3,93 (т, J=3,6 Гц, 2Н), 3,85 (дд, J=6,6 Гц, 1Н), 3,75-3,65 (м, J=6,6 Гц, 2Н).
LCMS: 428 (М+Н+) для C21H22FN5O4.
Пример 63
Получение соединения 63
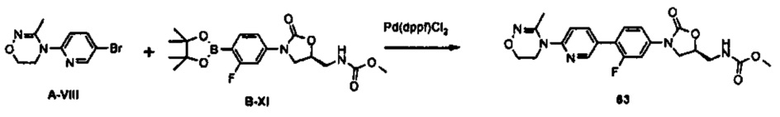
326 мг (0,73 ммоль) Соединения 63 получают (выход: 51%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-VIII и соединения B-XI, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,55 (с, 1Н), 7,86 (м, 1Н), 7,56 (дд, J1=12,6 Гц, J2=1,8 Гц, 1Н), 7,43 (т, J=8,4 Гц, 1Н), 7,33 (дд, J1=8,4 Гц, J2=1,8 Гц, 1Н), 7,05 (д, J=8,4 Гц, 1Н), 5,13 (м, 1Н), 4,79 (м, 1Н), 4,18 (т, J=4,8 Гц, 2Н), 4,08 (т, J=9,0 Гц, 1Н), 3,90 (т, J=4,8 Гц, 2Н), 3,85 (т, J=7,8 Гц, 1Н), 3,67 (с, 3Н), 3,63 (м, 1Н), 3,55 (м, 1Н), 2,10 (с, 3Н).
LCMS: 444 (М+Н+) для C21H22FN5O5.
Пример 64
Получение соединения 64
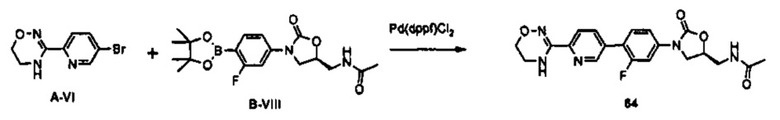
168 мг (0,41 ммоль) Соединения 64 получают (выход: 56%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-VI и соединения B-VIII, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,77 (с, 1Н), 8,27 (т, J=6,0 Гц, 1Н), 8,04 (дд, J1=7,2 Гц, J2=1,8 Гц, 1Н), 7,96 (д, J=9,0 Гц, 1Н), 7,71 (т, J=8,4 Гц, 1Н), 7,66 (дд, J1=13,2 Гц, J2=2,4 Гц, 1Н), 7,47 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,40 (т, J=3,6 Гц, 1Н), 4,78 (м, 1Н), 4,19 (т, J=8,4 Гц, 1Н), 3,88 (т, J=4,8 Гц, 2Н), 3,80 (дд, J1=9,0 Гц, J2=6,0 Гц, 1Н), 3,47 (дд, J1=8,4 Гц, J2=3,6 Гц, 2Н), 3,44 (т, J=4,8 Гц, 2Н), 1,84 (с, 3Н).
LCMS: 414 (М+Н+) для C20H20FN5O4.
Пример 65
Получение соединения 65
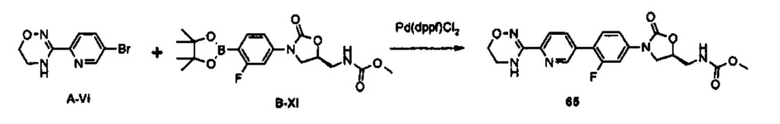
391 мг (0,91 ммоль) Соединения 65 получают (выход: 78%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-VI и соединения B-XI, по такому же способу, как в примере 1.
1H ЯМР (400 МГц, ДМСО d-6) δ 8,77 (с, 1Н), 8,05 (д, J=8,4 Гц, 1Н), 7,96 (д, J=8,0 Гц, 1Н), 7,72 (т, J=8,8 Гц, 1Н), 7,66 (дд, J1=13,6 Гц, J2=2,0 Гц, 1Н), 7,56 (т, J=5,6 Гц, 1Н), 7,47 (дд, J1=8,4 Гц, J2=2,0 Гц, 1Н), 7,40 (м, 1Н), 4,78 (м, 1Н), 4,19 (т, J=9,0 Гц, 1Н), 3,88 (т, J=4,6 Гц, 2Н), 3,84 (дд, J1=8,8 Гц, J2=6,0 Гц, 1Н), 3,55 (с, 3Н), 3,48 (м, 4Н).
LCMS: 430 (М+Н+) для C20H20FN5O5.
Пример 66
Получение соединения 66
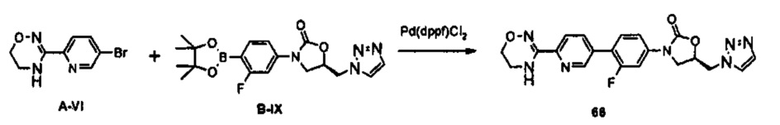
78 мг (0,18 ммоль) Соединения 66 получают (выход: 75%), в виде твердого вещества бледно-розового цвета, путем введения во взаимодействие соединения A-VI и соединения В-IX, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,77 (с, 1Н), 8,19 (с, 1Н), 8,04 (д, J=8,4 Гц, 1Н), 7,95 (д, J=8,4 Гц, 1Н), 7,78 (с, 1Н), 7,70 (т, J=9,0 Гц, 1Н), 7,60 (дд, J1=13,8 Гц, J2=2,4 Гц, 1Н), 7,42 (дд, J1=8,4 Гц, J2=2,4 Гц, 1Н), 7,40 (т, J=2,4 Гц, 1Н), 5,19 (м, 1Н), 4,87 (м, 1Н), 4,31 (т, J=9,0 Гц, 1Н), 3,97 (м, 1Н), 3,88 (т, J=4,8 Гц, 2Н), 3,47 (м, 2Н).
LCMS: 424 (М+Н+) для C20H18FN7O3.
Пример 67
Получение соединения 67
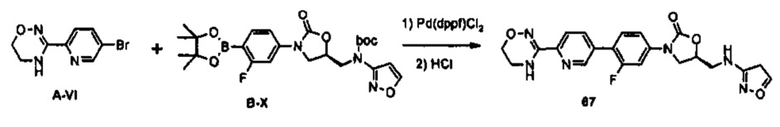
88 мг (0,20 ммоль) Соединения 67 получают (выход: 68%), в виде твердого вещества бледно-желтого цвета, путем введения во взаимодействие соединения A-VI и соединения В-Х, таким же образом, как в примерах 1 и 2.
1H ЯМР (600 МГц, ДМСО) δ 9,08 (с, 1Н), 8,89 (с, 1Н), 8,40 (д, J=1,8 Гц, 1Н), 8,20 (дд, J1=8,4 Гц, J2=1,2 Гц, 1Н), 8,13 (д, J=8,4 Гц, 1Н), 7,75 (дд, J1=8,4 Гц, J2=8,4 Гц, 1Н), 7,69 (дд, J1=13,8 Гц, J2=2,4 Гц, 1H), 7,50 (дд, J1=8,4 Гц, J2=1,8 Гц, 1H), 6,01 (д, J=1,8 Гц, 1H), 4,94 (м, 1H), 4,23 (дд, J1=9,0 Гц, J2=9,0 Гц, 1H), 4,13 (т, J=4,2 Гц, 2H), 3,90 (дд, J1=9,0 Гц, J2=6,6 Гц, 1Н), 3,60 (т, J=4,2 Гц, 2Н), 3,47 (м, 2Н).
LCMS: 439 (М+Н+) для C21H19FN6O4.
Пример 68
Получение соединения 68
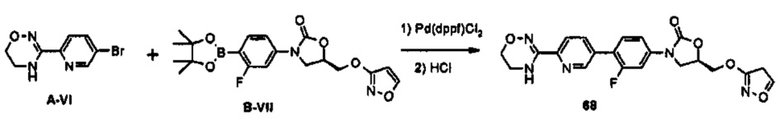
111 мг (0,25 ммоль) Соединения 68 получают (выход: 62%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-VI и соединения B-VII, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, ДМСО d-6) δ 8,78 (с, 1Н), 8,72 (д, J=1,8 Гц, 1Н), 8,05 (д, J=8,4 Гц, 1Н), 7,74 (д, J=8,4 Гц, 1Н), 7,70 (дд, J1=15,6 Гц, J2=2,4 Гц, 1Н), 7,52 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 7,43 (м, 1Н), 6,42 (д, J=1,8 Гц, 1Н), 5,14 (м, 1Н), 4,50 (м, 2Н), 4,27 (т, J=9,0 Гц, 1Н), 4,00 (дд, J1=9,6 Гц, J2=6,0 Гц, 1Н), 3,88 (т, J=4,8 Гц, 2Н), 3,47 (м, 2Н).
LCMS: 441 (М+Н+) для C21H19FN5O5.
Пример 69
Получение соединения 69
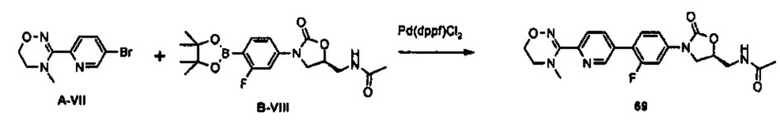
120 мг (0,28 ммоль) Соединения 69 получают (выход: 53%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-VII и соединения B-VIII, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, CDCl3) δ 8,80 (с, 1Н), 7,93 (д, J=8,4 Гц, 1Н), 7,72 (д, J=7,8 Гц, 1Н), 7,61 (дд, J1=12,6 Гц, J2=2,4 Гц, 1Н), 7,47 (т, J=9,0 Гц, 1Н), 7,33 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 6,03 (м, 1Н), 4,83 (м, 1Н), 4,18 (т, J=9,0 Гц, 1Н), 4,10 (т, J=9,0 Гц, 1Н), 3,84 (дд, J1=8,4 Гц, J2=6,6 Гц, 1Н), 3,74 (м, 1Н), 3,68 (м, 3Н), 3,52 (т, J=4,8 Гц, 2Н), 2,94 (с, 3Н), 2,04 (с, 3Н).
LCMS: 428 (М+Н+) для C21H22FN5O4.
Пример 70
Получение соединения 70
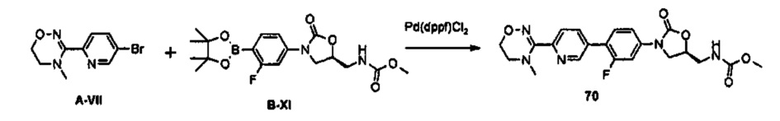
437 мг (0,98 ммоль) Соединения 70 получают (выход: 84%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-VII и соединения B-XI, по такому же способу, как в примере 1.
1H ЯМР (600 МГц, CDCl3) δ 8,81 (с, 1Н), 7,93 (д, J=7,8 Гц, 1Н), 7,72 (д, J=7,8 Гц, 1Н), 7,60 (дд, J1=13,2 Гц, J2=1,8 Гц, 1Н), 7,47 (т, J=8,4 Гц, 1Н), 7,35 (дд, J1=9,0 Гц, J2=2,4 Гц, 1Н), 5,13 (м, 1Н), 4,82 (м, 1Н), 4,17 (т, J=4,8 Гц, 2Н), 4,11 (т, J=9,0 Гц, 1Н), 3,87 (т, J=7,8 Гц, 1Н), 3,70 (с, 3Н), 3,65 (м, 1Н), 5 3,60 (м, 1Н), 3,51 (т, J=7,8 Гц, 2Н), 2,94 (с, 3Н).
LCMS: 444 (M+H+) для C21H22FN5O5.
Пример 71
Получение соединения 71
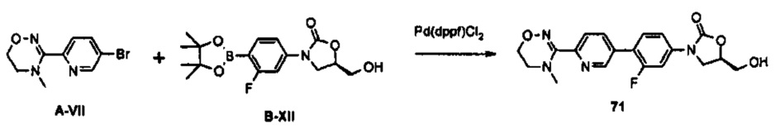
71 мг (0,16 ммоль) Соединения 71 получают (выход: 20%), в виде твердого вещества белого цвета, путем введения во взаимодействие соединения A-VII и соединения B-XII, по такому же способу, как в примере 1.
1H ЯМР (400 МГц, ДМСО d-6) δ 8,82 (с, 1Н), 8,06 (д, J=8,4 Гц, 1Н), 7,69 (м, 1Н), 7,63 (д, J=8,4 Гц, 1Н), 7,51 (м, 2Н), 5,27 (т, J=6,0 Гц, 1Н), 4,76 (м, 1Н), 4,15 (т, J=9,2 Гц, 1Н), 4,00 (т, J=4,8 Гц, 2Н), 3,90 (дд, J1=8,8 Гц, J2=6,1 Гц, 1Н), 3,70 (м, 1Н), 3,59 (м, 1Н), 3,42 (т, 4,4 Гц, 2Н), 2,80 (с, 1Н).
LCMS: 44 4 (М+Н+) для C19H19FN4O4.
Экспериментальный пример 1
Измерение антибактериальной активности in vitro
Для оценки антибактериальных активностей производных оксазолидинона, синтезированных в соответствии с примерами 1-71, осуществляли тест на активность in vitro, используя следующий метод.
Антибактериальные активности in vitro производных оксазолидинона согласно примерам 1-71 оценивали путем измерения ингибирующей минимально на 90% концентрации (MIC90, мкг/мл), представляющей собой минимальную концентрацию антибиотика, которая ингибирует рост бактерий вплоть до 90%, по сравнению с ростом бактерий в случае необработанного контроля, которую измеряли с помощью фотоспектроскопии. MIC90 измеряли по методу микроразведения бульона, базирующемуся на CLSI-стандартах [ссылка: Clinical and Laboratory Standards Institute Document. (2000) Methods for Dilution Antimicrobial Susceptibility Test for Bacteria that Grow Aerobically-Fifth Edition: M7-A5. CLSI, Villanova, PA].
1) Тест-штаммы
Измеряли активности 12 штаммов, включая Staphylococcus aureus, Methicillin Resistant Staphylococcus aureus, Staphylococcus epidermidis, Methicillin Resistant Staphylococcus epidermidis, Enterococcus faecalis, Vancomycin Resistant Enterococcus faecalis, Linezolid и Vancomycin Resistant Enterococcus faecalis, Enterococcus faecium, Vancomycin Resistant Enterococcus faecium, Linezolid и Vancomycin Resistant Enterococcus faecium и Moraxella catarrhalis, и результаты представлены в нижеприводимой таблице 1.
2) Метод получения тестируемого вещества
10240 мкг/мл Тестируемых веществ (то есть, производных оксазолидинона согласно примерам 1-71), каждое, растворяли в ДМСО, и каждый полученный раствор подвергали двухкратному серийному разведению и затем разводили в 20 раз с помощью стерильной трижды дистиллированной воды. Конечная концентрация тестируемого вещества в случае антибактериального теста составляла от 0,063 мкг/мл (минимум) до 128 мкг/мл (максимум), и конечная концентрация ДМСО, используемого в качестве эксципиента, составляла 2,5% (об./об.). Линезолид, представленный нижеприводимой формулой В (выпускается фирмой Pfizer), использовали в качестве контроля и его бактериальную активность сравнивали с таковой тестируемых веществ. Результаты представлены в нижеприводимой таблице 1.
Формула В
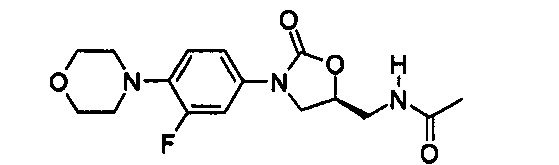
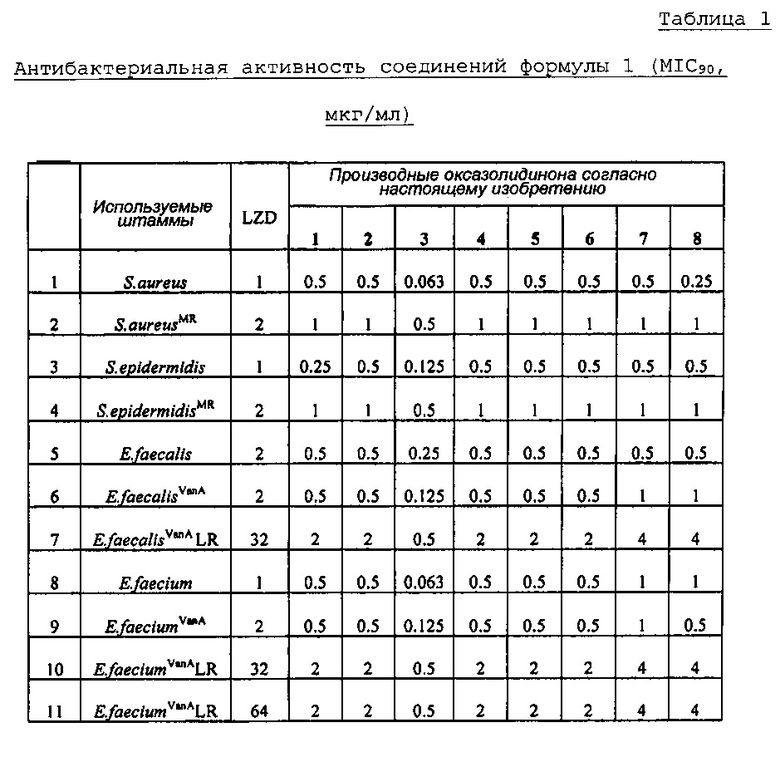

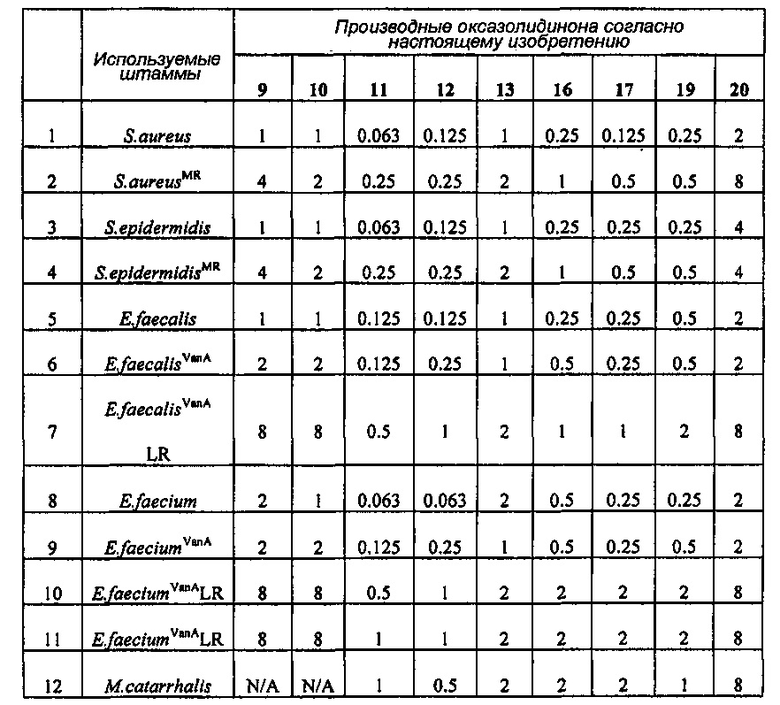
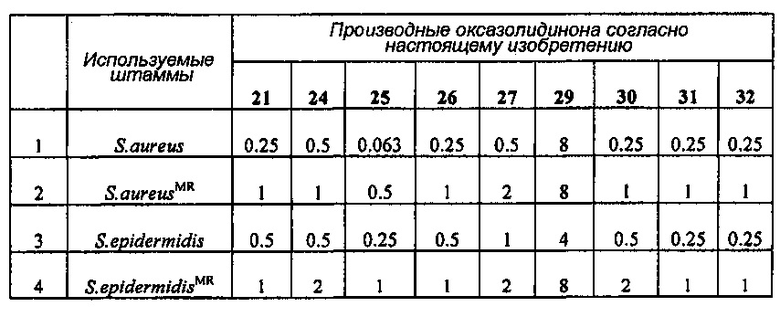
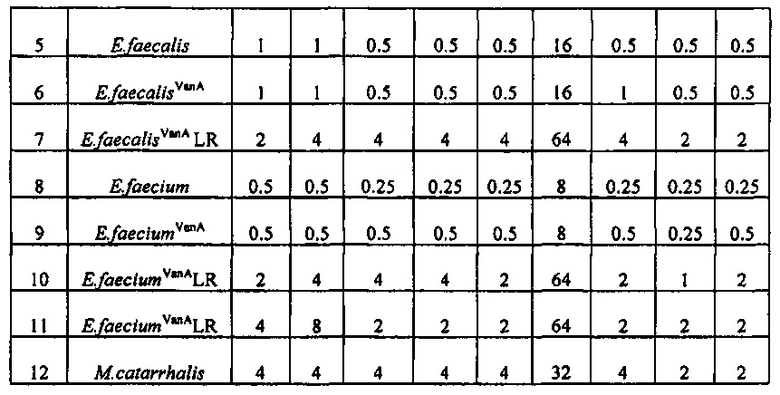
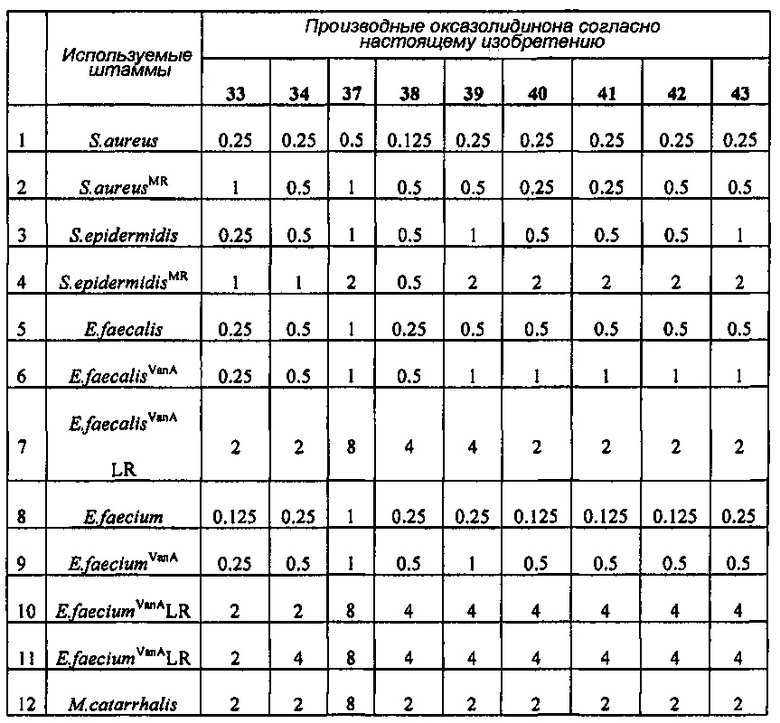
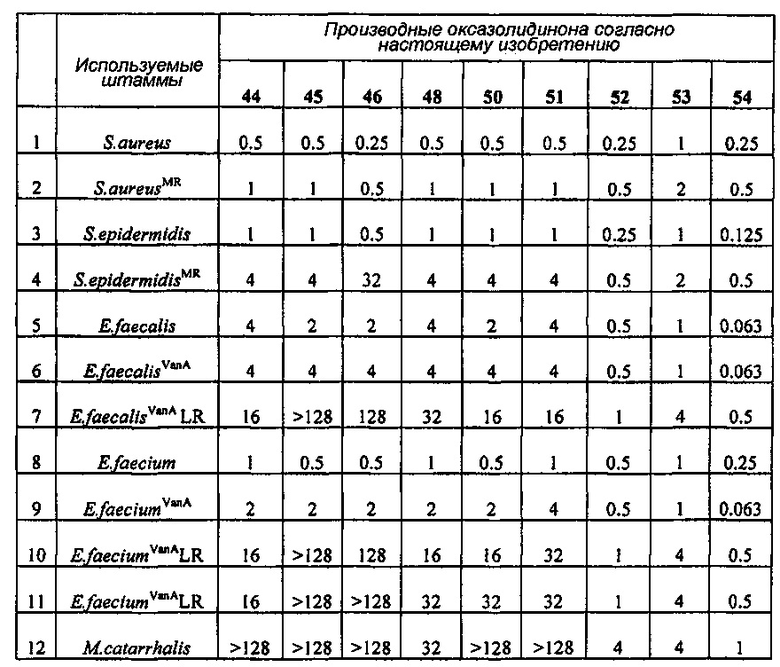
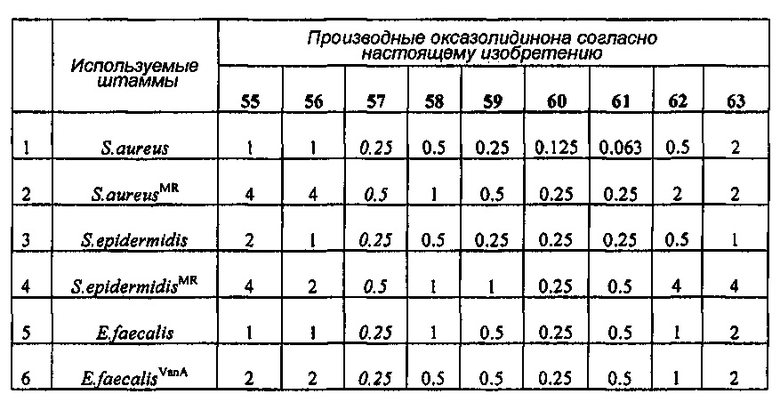
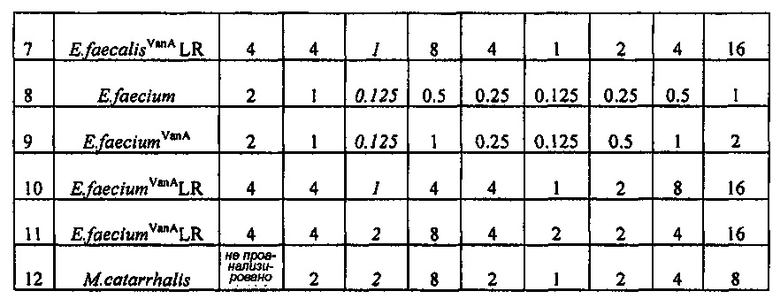
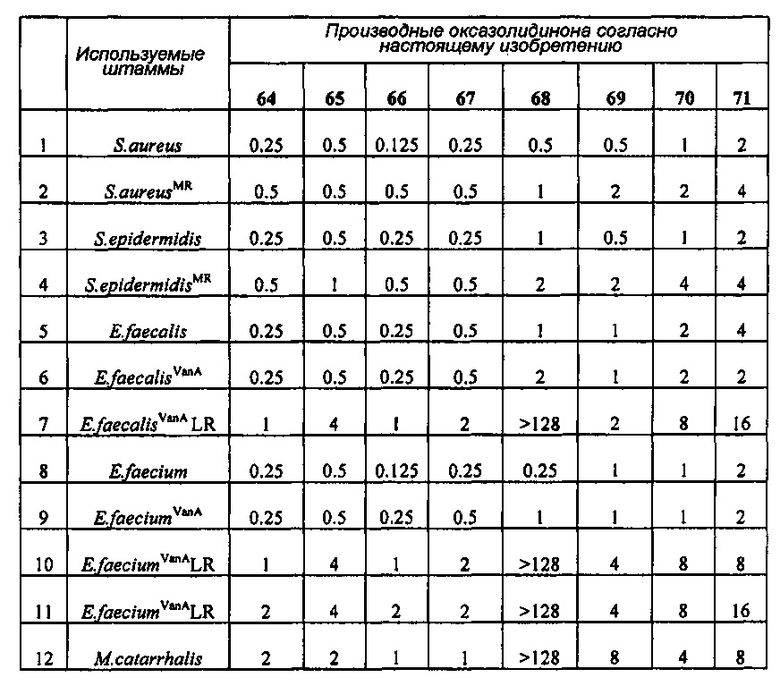
1. Staphylococcus aureus
2. Methicillin Resistant Staphylococcus aureus
3. Staphylococcus epidermidis
4. Methicillin Resistant Staphylococcus epidermidis
5. Enterococcus faecalis
6. Vancomycin Resistant Enterococcus faecalis
7. Linezolid и Vancomycin Resistant Enterococcus faecalis
8. Enterococcus faecium
9. Vancomycin Resistant Enterococcus faecium
10. 11. Linezolid и Vancomycin Resistant Enterococcus faecium
12. Moraxella catarrhalis
Как видно из таблицы 1, можнет быть подтверждено, что производные оксазолидинона согласно настоящему изобретению являются более эффективными против грамположительных бактерий (например, MRSA, VRE и т.п.), резистентных к существующим антибиотикам, при намного более низкой концентрации, по сравнению с линезолидом в качестве контроля, и, в особенности, производные оксазолидинона являются очень эффективными против резистентных к линезолиду бактерий. В частности, соединения 3, 11 и 54 обладают очень высокой антибактериальной активностью против Enterococcus faecalis и Enterococcus faecium, резистентных к линезолиду, что указывает на то, что соединения могут быть эффективно использованы против резистентных к линезолиду бактерий, которые в последнее время появились и могут вызывать большие проблемы в будущем.
Следовательно, производные оксазолидинона согласно настоящему изобретению могут быть эффективно использованы в качестве антибиотиков, имеющих широкий спектр активности против грамположительных бактерий, и в качестве терапевтических агентов для лечения инфекции за счет резистентных штаммов, таких как MRSA и VRE, в частности, резистентных к линезолиду бактерий.
Несмотря на то, что предпочтительные воплощения настоящего изобретения раскрыты для иллюстративных целей, квалифицированный специалист в данной области должно быть понятно, что возможны различные модификации, дополнения и замены, не отступая от объема и сущности данного изобретения, как раскрыто в сопровождающей формуле изобретения.
Промышленная применимость
Как описано выше, новые производные оксазолидинона согласно настоящему изобретению имеют широкий спектр антибактериальной активности против резистентных бактерий, включая MRSA и VRE. В особенности, производные оксазолидинона обладают высокой активностью против резистентных к линезолиду бактерий и, таким образом, могут быть эффективно использованы в качестве антибиотиков второго поколения на основе оксазолидинона. В дополнение, соединения согласно настоящему изобретению имеют циклическую амидоксимную или циклическую амидразоновую группу и, таким образом, могут образовывать соль. Соответственно, соединения обладают более высокой растворимостью по отношению к воде, чем существующие соединения, и, таким образом, соединения могут быть легко использованы для получения готовой лекарственной формы, как пероральная готовая лекарственная форма или инъекция.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА С ЦИКЛИЧЕСКИМ АМИДОКСИМОМ ИЛИ ЦИКЛИЧЕСКИМ АМИДРАЗОНОМ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2468023C1 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2822624C1 |
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| КОНЪЮГАТЫ БЕЛОК-АКТИВНОЕ ВЕЩЕСТВО И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2613906C2 |
| 1, 2, 4-ТРИАЗОЛЬНОЕ ПРОИЗВОДНОЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2003 |
|
RU2288223C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА, ПОДАВЛЯЮЩЕЕ РОСТ РАКОВОЙ КЛЕТКИ, И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2792849C1 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛПИРИМИДИНОНА, СПОСОБ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2263676C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
Изобретение относится к производному оксазолидинона, представленному формулой (1), где R означает гетероциклическую группу, выбираемую из следующих групп, представленных формулой (1а), R1 означает водород, C1-С6-алкил или С3-С6-циклоалкил; R2 означает водород, C1-С6-алкил, -СН2-СН=СН2, -СН2-С≡СН, -С(O)СН(ОН)СН2ОН, -СН2-С(О)ОС2Н5, -СН2СН2ОН, -СН2С(O)ОН, -СН2С(O)ОС(СН3)3, -С(O)СН2ОН, -С(O)СН2ОС(O)СН3, или (СН2)mC(=O)R21, где R21 означает водород, (СН2)nNHR211, где R211 означает водород или C1-С6 алкил, или СН(ОН)СН2ОН, и m и n, каждый, независимо означают целое число от 0 до 3; и Q означает OR3, NHR3 или 1,2,3-триазол, где R3 означает водород, C1-С6-алкил, -C(=O)R31 или гетероароматическую циклическую группу, выбираемую из изоксазол-3-ила или изоксазол-5-ила, R31 означает водород, C1-С6-алкил, С3-С6-циклоалкил или О-(C1-С6)алкил. Изобретение также относится к пролекарству оксазолидинона, представленному соединением, выбираемым из соединений формул (5)-(7), где Q означает те же заместители, которые указаны для соединения формулы (1), и М+ представляет собой ион щелочного металла, такой как Na+ или K+, или ион аммония. Соединения по изобретению также относятся к конкретным производным оксазолидинона, приведенным в формуле изобретения. Соединения по изобретению предназначены для изготовления фармацевтической композиции для антибиотика, включающей (а) терапевтически эффективное количество производного оксазолидинона и (b) фармацевтически приемлемый носитель, разбавитель, эксципиент или их комбинацию. Технический результат – производные оксазолидинона, обладающие активностью антибиотика. 4 н. и 2 з.п. ф-лы, 1 табл., 113 пр.
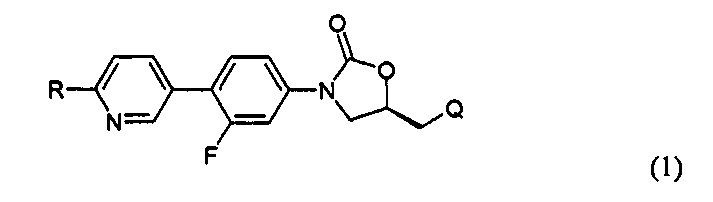 ,
, 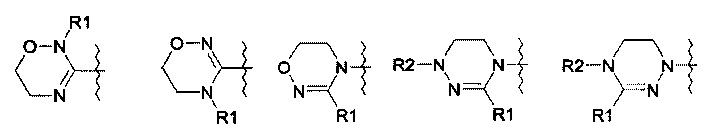 (1а),
(1а),
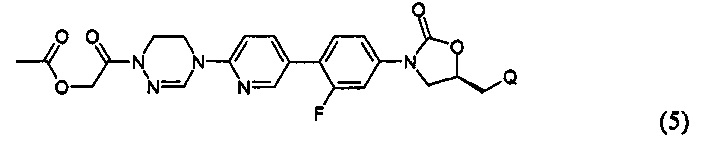 ,
, 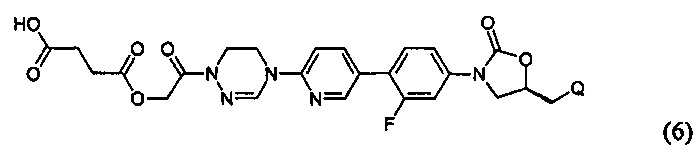 ,
, 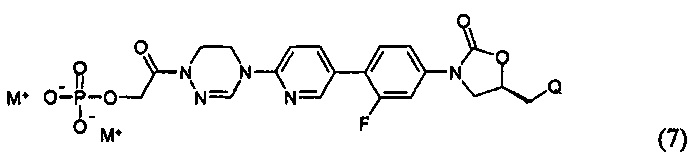
1. Новое производное оксазолидинона, представленное формулой (1):
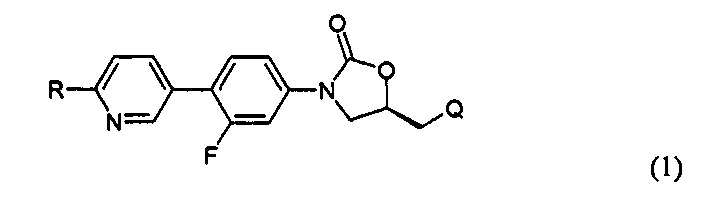
где R означает гетероциклическую группу, выбираемую из следующих групп:
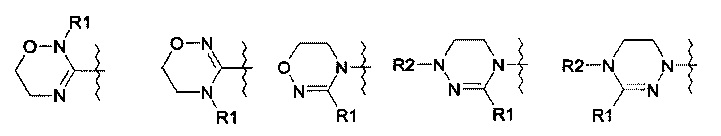
R1 означает водород, C1-С6-алкил или С3-С6-циклоалкил;
R2 означает водород, C1-С6-алкил, -СН2-СН=СН2, -СН2-С≡СН, -С(O)СН(ОН)СН2ОН, -СН2-С(О)ОС2Н5, -СН2СН2ОН, -СН2С(O)ОН, -СН2С(O)ОС(СН3)3, -С(O)СН2ОН, -С(O)СН2ОС(O)СН3, или (СН2)mC(=O)R21, где R21 означает водород, (СН2)nNHR211, где R211 означает водород или C1-С6 алкил, или СН(ОН)СН2ОН, и m и n, каждый, независимо означают целое число от 0 до 3; и
Q означает OR3, NHR3 или 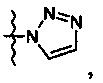 где R3 означает водород, C1-С6-алкил, -C(=O)R31, или гетероароматическую циклическую группу, выбираемую из следующих групп:
где R3 означает водород, C1-С6-алкил, -C(=O)R31, или гетероароматическую циклическую группу, выбираемую из следующих групп:
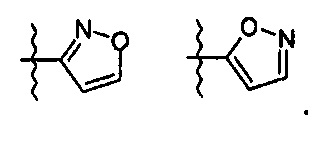
где R31 означает водород, C1-С6-алкил, С3-С6-циклоалкил или О-(C1-С6)алкил.
2. Новое производное оксазолидинона по п.1, представленное соединением, выбираемым из соединений формул (2)-(4):
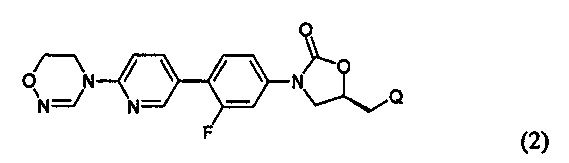
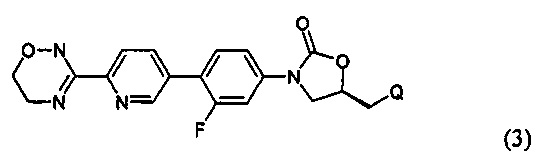
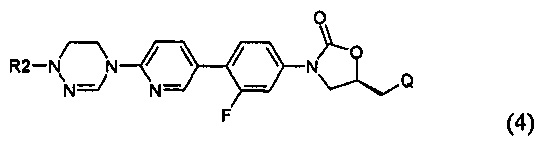
где R2 и Q имеют такие же значения, как указано в п. 1.
3. Новое производное оксазолидинона по п.2, где Q означает NHC(=O)CH3, NHC(=O)OCH3,  или
или  и R2 означает метил, С(=O)СН2ОН, C(=O)CH2NH2 или С(=O)СН(ОН)СН2ОН.
и R2 означает метил, С(=O)СН2ОН, C(=O)CH2NH2 или С(=O)СН(ОН)СН2ОН.
4. Новое пролекарство оксазолидинона, представленное соединением, выбираемым из соединений формул (5)-(7):
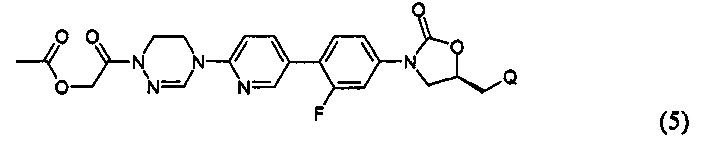
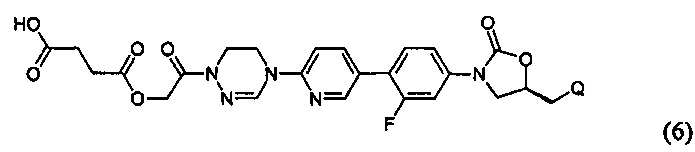
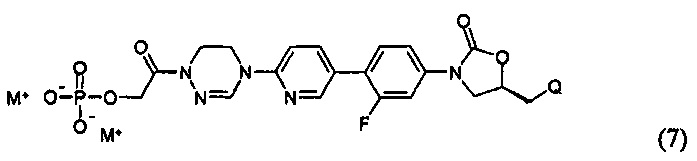
где Q означает OR3, NHR3 или  где R3 означает водород, C1-С6-алкил, -C(=O)R31, или гетероароматическую циклическую группу, выбираемую из следующих групп:
где R3 означает водород, C1-С6-алкил, -C(=O)R31, или гетероароматическую циклическую группу, выбираемую из следующих групп:
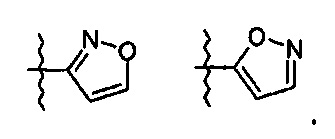
где R31 означает водород, C1-С6-алкил, С3-С6-циклоалкил или O-(C1-С6)алкил; и М+ представляет собой ион щелочного металла, такой как Na+ или K+, или ион аммония.
5. Новое производное оксазолидинона, выбираемое из следующих соединений:

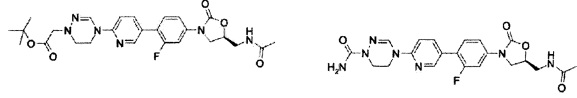
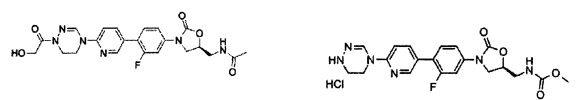
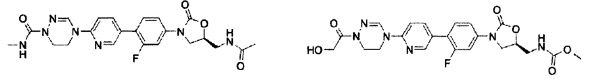
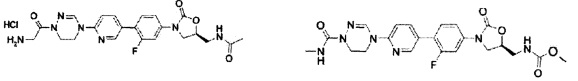
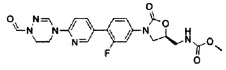
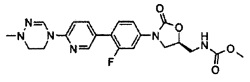
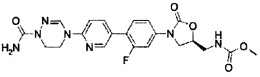


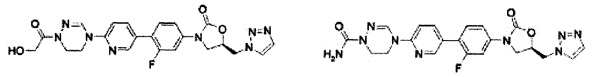
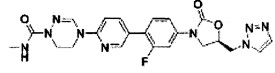
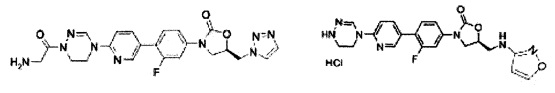

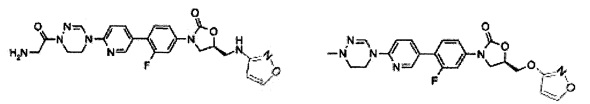

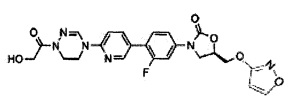
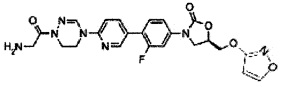
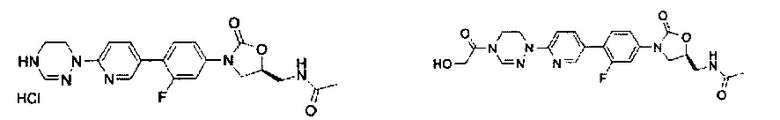







6. Фармацевтическая композиция для антибиотика, причем фармацевтическая композиция включает: (а) терапевтически эффективное количество нового производного оксазолидинона по любому одному из пп. 1-5 и (b) фармацевтически приемлемый носитель, разбавитель, эксципиент или их комбинацию.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА | 2004 |
|
RU2414469C2 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |

