Настоящее изобретение относится к способу получения производных 3-дигалогенметил-пиразол-4-карбоновой кислоты формулы (I) взаимодействием α-фтораминов формулы (III) в присутствии кислот Льюиса с производными акриловой кислоты формулы (II) с получением винамидиниевых солей формулы (IV) и их последующим взаимодействием с гидразинами, и к самим винамидиниевым солям формулы (IV).
Сложные эфиры, амиды и нитрилы 2-дифторметил-пиразол-4-карбоновой кислоты представляют собой важные промежуточные соединения для получения биологически активных веществ, применяемых в агрономии, особенно для получения пиразолилкарбоксанилидных фунгицидов.
В международной заявке WO-A-05042468 описан способ получения сложных эфиров 2-дигалогенацил-3-аминоакриловой кислоты путем реакции галогенангидридов кислот со сложными эфирами диалкиламиноакриловой кислоты, а также их взаимодействия с производными гидразина с получением сложных эфиров 3-дигалогенметил-пиразол-4-карбоновых кислот.
В международной заявке WO-A-03051820 описан способ получения сложных эфиров 2-галогенацил-3-аминоакриловой кислоты путем реакции N-замещенных сложных эфиров 3-аминоакриловой кислоты с ангидридами галогеналкилкарбоновых кислот и их последующего взаимодействия с производными гидразина с получением сложных эфиров 3-галогеналкил-пиразол-4-карбоновой кислоты. Реакция получения сложных эфиров 3-галогеналкил-пиразол-4-карбоновой кислоты при комнатной температуре проходит неселективно, и поэтому ее нужно проводить при низких температурах (-80°C).
В международной заявке WO-A-06005612 описан способ получения этилового эфира 4,4-дифтор-3-оксомасляной кислоты путем реакции 2,2-дифтор-N-диалкилацетамида со сложными эфирами уксусной кислоты в присутствии основания. После этого этиловый эфир 4,4-дифтор-3-оксомасляной кислоты, как описано в JACS, 73, 3684 (1951), подвергают взаимодействию с триметил-орто-формиатом и уксусным ангидридом в этил-(2-этоксиметилен)-4,4-дифторметилацетоацетат, который, согласно патентной заявке США US-A-5489624, реакцией с метилгидразином можно превратить в этиловый эфир 3-дифторметил-1-метил-4-пиразолкарбоновой кислоты. Описанный синтетический путь, во-первых, включает множество стадий, и, во-вторых, использующийся 2,2-дифтор-N-диалкилацетамид недоступен коммерчески и может быть получен лишь с невысоким выходом (около 70%) фторированием 2,2-дихлор-N-диалкилацетамида.
Описанные в предшествующем уровне техники способы имеют недостаток, состоящий в том, что используемые галогенангидриды карбоновых кислот, ангидриды галогеналкилкарбоновых кислот и галогенакриловые сложные эфиры являются дорогими, вызывают проблемы с коррозией и/или требуют больших технических затрат при очистке.
Поэтому в основе настоящего изобретения лежит задача - разработать более простой и экономичный способ получения производных 2-галоген-ацил-3-аминоакриловой кислоты, особенно сложных эфиров, нитрилов и амидов.
Описанная выше задача, согласно настоящему изобретению, решена с помощью способа получения производных 3-дигалогенметил-пиразол-4-карбоновой кислоты формулы (I)
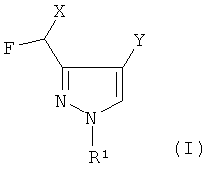
в которых
R1 выбирают из водорода, алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или арилалкила с 7-19 атомами углерода,
Y выбирают из (C=O)OR6, CN и (C=O)NR7R8, где R6 R7 и R8 независимо друг от друга выбирают из алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или арилалкила с 7-19 атомами углерода; и
X представляет собой F, Cl или CF3
взаимодействием α-фтораминов формулы (III)
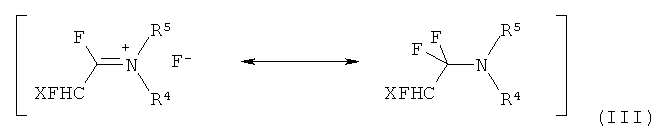
в которой
R4 выбирают из алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или арилалкила с 7-19 атомами углерода,
R5 независимо от R4 выбирают из C1-12-алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или арилалкила с 7-19 атомами углерода, и
X представляет собой F, Cl или CF3,
в присутствии кислот Льюиса (Z) с производными акриловой кислоты формулы (II)
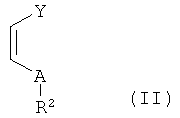
в которой
A выбирают из O, S и NR3,
Y выбирают из (C=O)OR6, CN и (C=O)NR7R8, где R6, R7 и R8 независимо друг от друга выбирают из алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или арилалкила с 7-19 атомами углерода; и
R2 и R3 независимо друг от друга выбирают из алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или арилалкила с 7-19 атомами углерода, -OR', -SR', -NR'2, где R' может представлять собой алкил с 1-5 атомами углерода,
или R2 и R3 вместе с атомом N, к которому они присоединены, могут образовывать пяти- или шестичленный цикл,
и последующим взаимодействием с гидразинами формулы (V)
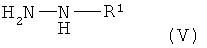
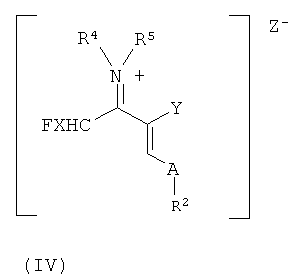
Другим объектом настоящего изобретения является образующаяся согласно настоящему изобретению в качестве промежуточного продукта винамидиниевая соль формулы (IV)
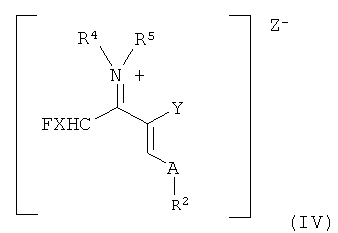
в которой
все заместители имеют указанные выше значения.
Другие варианты выполнения настоящего изобретения можно выбирать из зависимых пунктов формулы изобретения и описания.
Способ по настоящему изобретению можно проиллюстрировать с помощью следующей схемы (I):

Общие определения
В контексте настоящего изобретения, термин "галоген" (X) охватывает, если не указано иное, элементы, выбранные из группы, состоящей из фтора, хлора, брома и иода, где фтор, хлор и бром являются предпочтительными, и фтор и хлор особенно предпочтительными.
В данном случае, замещенные группы могут быть однократно замещенными или многократно замещенными, при этом в случае многократного замещения заместители могут быть одинаковыми или разными.
В контексте настоящего изобретения, группа -X означает атом галогена, выбранный из фтора, хлора, брома и иода, предпочтительно фтора, хлора и иода, особенно предпочтительно из фтора и хлора.
Алкильные группы, замещенные одним или более атомами галогена (-Х), выбирают, например, из трифторметила (CF3), дифторметила (CHF2), CF3CH2, ClCH2, CF3CCl2.
Алкил с 1-12 атомами углерода представляет собой линейные или разветвленные углеводородные группы. В частности, данное определение охватывает, например, метил, этил, н-, изо-пропил, н-, изо-, втор- и трет-бутил, н-пентил, н-гексил, 1,3-диметилбутил, 3,3-диметилбутил, н-гептил, н-нонил, н-децил, н-ундецил, н-додецил.
Соединения по настоящему изобретению могут, при необходимости, существовать в виде смесей различных возможных изомерных форм, в частности, стереоизомеров, таких как, например, Е- и Z-, трео- и эритро-, а также оптических изомеров, а при определенных условиях также таутомеров. В настоящем изобретении раскрываются и заявляется приоритет как на E-, так и на Z-изомеры, а также на трео- и эритро-, а также на оптические изомеры, любые смеси указанных изомеров, а также на возможные таутомерные формы.
Производные акриловой кислоты
В контексте настоящего изобретения, используемые производные акриловой кислоты представляют собой соединения, соответствующие общей формуле (II).
В указанной формуле A выбирают из O, S и NR3, и остатки R2 и R3 независимо друг от друга выбирают из алкила с 1-12 атомами углерода, C5-18-арила с 5-18 атомами углерода, C7-19-арилалкила, алкоксигрупп (-OR'), меркапто-групп (-SR'), амино-групп (-NR'2), где R' может представлять собой алкил с 1-5 атомами углерода.
Альтернативно, R2 и R3 могут вместе с N-атомом, к которому они присоединены, образовывать пяти- или шестичленный цикл.
Предпочтительно, остатки R2 и R3 независимо друг от друга выбирают из алкила с 2-8 атомами углерода, O-(С2-6-алкил), S-(C2-6-алкил), N(C2-6-алкил)2.
Особенно предпочтительно, остатки R2 и R3 независимо друг от друга выбирают из алкила с 3-6 атомами углерода, O-(C3-4-алкил), S-(C3-4-алкил), N(C3-4-алкил)2.
Предпочтительные, согласно настоящему изобретению, производные диалкиламиноакриловой кислоты изображены следующими формулами от (II-a) до (II-e).
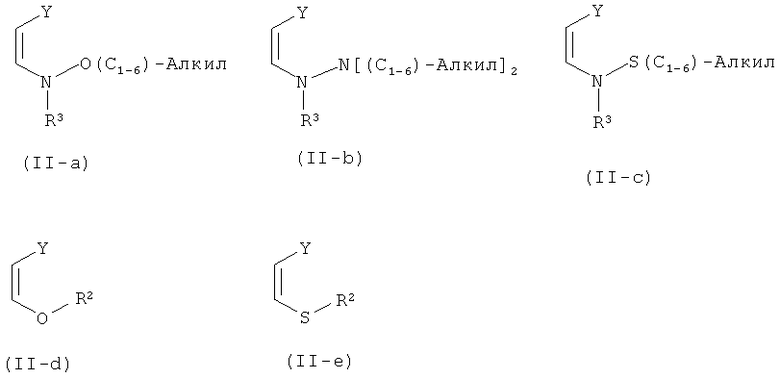
Группу Y выбирают из сложноэфирных групп ((C=O)OR6), нитрильной группы (CN) и амидных групп ((C=O)NR7R8), где R6 R7 и R8 независимо друг от друга выбирают из алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или C7-19-арилалкила; предпочтительно из алкила с 2-8 атомами углерода, особенно предпочтительно из алкила с 3-6 атомами углерода.
Примеры пригодных, согласно настоящему изобретению, сложных эфиров акриловой кислоты представляют собой сложный эфир метоксиакриловой кислоты, сложный эфир алкилтиоакриловой кислоты, метиловый эфир 3-(N,N-диметиламино)-акриловой кислоты, этиловый эфир 3-(N,N-диметиламино)-акриловой кислоты, этиловый эфир 3-(N,N-диэтиламино)-акриловой кислоты, 3-(N,N-диметиламино)-акрилонитрил, диметиламид 3-(N,N-диметиламино)-акриловой кислоты и диэтиламид 3-(N,N-диметиламино)-акриловой кислоты, из которых особенно предпочтительным является этиловый эфир 3-(N,N-диэтиламино)-акриловой кислоты.
Способ получения сложных эфиров диалкиламиноакриловой кислоты описан в предшествующем уровне техники, например, в европеской заявке на патент EP-A-0608725.
Способ получения диалкиламиноакрилонитрилов описан в предшествующем уровне техники, например, Rene и др. в Synthesis (1986), (5), 419-420.
Производные акриловой кислоты, в случае необходимости, можно очищать, например, перегонкой. Однако в целом это не является необходимым в случае реакции по настоящему изобретению.
α-Фторамины
В контексте настоящего изобретения, используемые α-фторамины представляют собой соединения, соответствующие общей формуле (III).
в которой
R4 выбирают из алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или C7-19-арилалкила, предпочтительно из алкила с 2-8 атомами углерода, особенно предпочтительно из алкила с 3-6 атомами углерода.
R5 независимо от R4 выбирают из алкила с 1-12 атомами углерода, арила с 5-18 атомами углерода или C7-19-арилалкила, предпочтительно из алкила с 2-8 атомами углерода, особенно предпочтительно из алкила с 3-6 атомами углерода.
X представляет собой CF3, F или Cl.
Указанные соединения доступны согласно статье Petrov и др. в Journal of Fluorine Chemistry 109 (2001) 25-31 и Dmowski и др. в Chemistry of Organic Fluorine Compounds II, A Critical Review, ACS, Washington DC (1995) 263 по реакции фторированных/галогенированных алкенов с вторичными аминами, и могут быть приобретены у, например, DuPont.
Согласно настоящему изобретению, упомянутые ранее α-фторамины выбирают, например, из группы, состоящей из 1,1,2,2-тетрафторэтил-N,N-диметиламина, 1,1,2,2-тетрафторэтил-N,N-диэтиламина, 1,1,2-трифтор-2-(трифторметил)этил-N,N-диметиламина, 1,1,2-трифтор-2-(трифторметил)-этил-N,N-диэтиламина (реагент Ишикавы), 1,1,2-трифтор-2-хлорэтил-N,N-диметиламина и 1,1,2-трифтор-2-хлорэтил-N,N-диэтиламина (реактив Яровенко), из которых предпочтительными являются 1,1,2,2-тетрафторэтил-N,N-диметиламин и 1,1,2,2-тетрафторэтил-N,N-диэтиламин, и 1,1,2,2-тетрафторэтил-N,N-диметиламин особенно предпочтителен.
Кислоты Льюиса
Описанные ранее α-фторамины реагируют в присутствии кислот Льюиса (Z) с образованием иммониевых солей, как описано Wakselman и др. в J.C.S. Chem. Comm. 565 (1975) 956.
Реакция α-фтораминов с кислотами Льюиса протекает предпочтительно при температуре от -80 до 50°C, предпочтительно от -40 до 40°C, особенно предпочтительно от 0 до 30°C.
При необходимости, кислоту Льюиса можно не добавлять.
Реакцию можно проводить в веществе или в растворителе. Преимущественно реакцию проводят в растворителе. Подходящие растворители выбирают, например, из группы, состоящей из алифатических и ароматических углеводородов, таких как, например, н-гексан, бензол или толуол, которые могут быть замещены атомами фтора и хлора, как в случае метиленхлорида, дихлорметана, трихлорметана, четыреххлористого углерода, фторбензола, хлорбензола или дихлорбензола; простых эфиров, таких как, например, диэтиловый эфир, дифениловый эфир, метил-трет-бутиловый эфир, изопропилэтиловый эфир, диоксан, диглим, диметилгликоль или ТГФ; нитрилов, таких как метилнитрил, бутилнитрил или фенилнитрил, из которых особенно предпочтительными являются дихлорметан и ацетонитрил.
В качестве кислот Льюиса подходящими являются, например, соединения, выбранные из группы, состоящей из BF3, AlCl3, AlF3, ZnCl2, PF5, SbF5, SnCl4, BiCl3, GaCl3, SiCl4.
Кислоту Льюиса и α-фторамин предпочтительно используют в эквимолярных количествах. Альтернативно, можно также использовать избыток кислоты Льюиса. Соотношение кислота Льюиса : α-фторамин, по настоящему изобретению, находится между 1:1 и 10:1, предпочтительно между 1:1 и 5:1, особенно предпочтительно между 1:1 и 1:1.3.
В предпочтительном варианте выполнения способа по настоящему изобретению, α-фторамин используют в виде вещества или растворяют в подходящем растворителе, и затем добавляют кислоту Льюиса.
Вследствие подверженности α-фтораминов гидролизу, реакцию α-фтораминов с кислотой Льюиса проводят в безводных условиях в атмосфере инертного газа.
Образующиеся винамидиниевые соли формулы (IV) не являются ни гигроскопичными, ни подверженными гидролизу, и с ними можно работать и хранить на воздухе.
Последующее взаимодействие иммониевых солей со сложными эфирами диалкиламиноакриловой кислоты формулы (II) осуществляют, предпочтительно, без выделения иммониевых солей. В другом варианте выполнения настоящего изобретения, иммониевые соли можно выделить и использовать по мере надобности.
Реакцию иммониевых солей с производными акриловой кислоты формулы (II) с получением винамидиниевых солей формулы (IV)
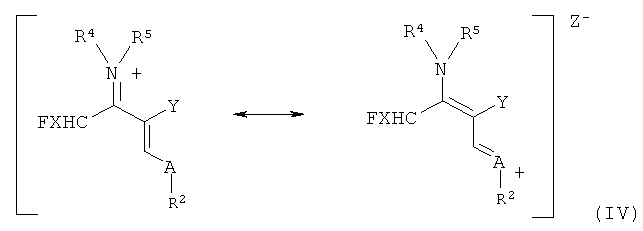
где анион Z- выбран, например, из группы, состоящей из [BF4]-, [AlCl3F]-, [AlF4]-, [ZnCl2F]-, [PF6]-, [SbF6]-, [SnCl4F]- [BiCl3F]-, [GaCl3F]-, [ZnCl2F]-, [SnCl4F]-, [BiCl3F]-, [GaCl3F]-, [SiCl4F]-, можно осуществлять при температурах от -40 до 60°C, предпочтительно от -20 до 40°C, особенно предпочтительно от 0 до 50°C.
Иммониевые соли и производные акриловой кислоты предпочтительно используют в эквимолярных количествах. Альтернативно, можно использовать избыток иммониевой соли или производного акриловой кислоты. Соотношение иммониевая соль : производное акриловой кислоты, по настоящему изобретению, находится между 1:10 и 10:1, предпочтительно между 1:5 и 5:1, особенно предпочтительно между 1.3:1 и 1:1.3.
В качестве растворителя предпочтительно используют те же растворители, которые использовались ранее также для синтеза иммониевых солей.
В предпочтительном варианте выполнения способа по настоящему изобретению, имониевую соль используют в виде чистого вещества или растворяют в подходящем растворителе, и затем добавляют производное акриловой кислоты.
В другом варианте выполнения способа по настоящему изобретению, производные акриловой кислоты (II) и α-фторамины (III), при необходимости, растворяют в растворителе и затем добавляют кислоту Льюиса. После этого добавляют гидразин формулы (V).
Иммониевые соли формулы (IV) можо выделять простым удалением растворителя.
Однако, предпочтительно, иммониевые соли формулы (IV) без предварительного выделения вводят в реакцию с гидразинами общей формулы (V), предпочтительно с метил гидразином, с получением сложных эфиров 3-дигалогенметил-пиразол-4-карбоновой кислоты формулы (I).
Особенно предпочтительно использование гидразина, метилгидразина и этил гидразина, из которых еще более предпочтительным является метил-гидразин.
Предпочтительными соединениями общей формулы (I) являются: метиловый эфир 1-метил-3-дифторметил-4-пиразолкарбоновой кислоты, этиловый эфир 1-метил-3-дифторметил-4-пиразолкарбоновой кислоты, метиловый эфир 1-метил-3-хлорфторметил-4-пиразолкарбоновой кислоты, этиловый эфир 1-метил-3-хлорфторметил-4-пиразолкарбоновой кислоты, метиловый эфир 1-метил-3-(трифторметил)фторметил-4-пиразолкарбоновой кислоты, этиловый эфир 1-метил-3-(трифторметил)фторметил-4-пиразолкарбоновой кислоты, из которых особенно предпочтительны этиловый эфир 1-метил-3-дифторметил-4-пиразолкарбоновой кислоты и метиловый эфир 1-метил-3-дифторметил-4-пиразолкарбоновой кислоты.
Реакцию иммониевых солей формулы (IV) с гидразинами формулы (V) предпочтительно осуществляют в присутствии растворителей. Подходящими растворителями являются, например, те же, которые были указаны для предыдущих стадий.
Реакцию с алкилгидразинами можно, например, проводить предпочтительно при температуре от -30 до +80°C, особенно предпочтительно от -20 до +25°C и еще более предпочтительно от -10 до +40°C.
В целях экономичности, предпочтительно проведение реакции при комнатной температуре.
Как особенно благоприятный можно указать тот факт, что образование производных 3-галогеналкил-4-пиразолкарбоновой кислоты при комнатной температуре также протекает с высокой региоселективностью.
Образующиеся в незначительном количестве (<8%) региоизомерные производные 4-галогеналкил-3-пиразолкарбоновой кислоты можно отделить от желаемых продуктов, основываясь на их различных физических свойствах, соответствующими способами, такими как, например, перегонка или кристаллизация, или с помощью простой промывки, например, циклогексаном.
Кроме того, как преимущество можно упомянуть тот факт, что все реакционные стадии способа по настоящему изобретению можно осуществлять одну за другой без промежуточной очистки/выделения промежуточных продуктов.
Производные 3-галогеналкил-4-пиразолкарбоновой кислоты формулы (I) можно, при необходимости, известным образом (Houben-Weyl, Methoden der organischen Chemie, 4. Auflage, Band E5, S 223ff.), например посредством кислотного или щелочного омыления, превратить в 3-галогеналкил-4-пиразолкарбоновые кислоты.
Предпочтительным является щелочное омыление. Его можно осуществлять известными способами, например, путем реакции с основаниями, такими как, например, гидроксиды щелочных металлов, такие как, например, гидроксид лития, натрия или калия, или их водными растворами. Подходящими растворителями являются, например, вода, спирты, такие как, например, метанол, этанол и изопропанол, ароматические углеводороды, такие как, например, толуол, ацетон, пиридин или смеси указанных растворителей.
В контексте настоящего изобретения, предпочтительными 3-галогеналкил-4-пиразолкарбоновыми кислотами являются 1-метил-3-дифторметил-4-пиразолкарбоновая кислота, 1-метил-3-хлорфторметил-4-пиразолкарбоновая кислота, 1-метил-3-(трифторметил)фторметил-4-пиразолкарбоновая кислота, 3-дифторметил-4-пиразолкарбоновая кислота, 3-(трифтор-метил)фторметил-4-пиразолкарбоновая кислота и 3-хлорфторметил-4-пиразолкарбоновая кислота, из которых особенно предпочтительна 1-метил-3-дифторметил-4-пиразолкарбоновая кислота.
Настоящее изобретение объясняется подробнее посредством описанных далее примеров выполнения, однако не ограничивается только ими.
Примеры
Пример 1
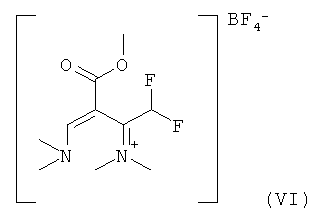
N-[(2E)-1-(дифторметил-3-(диметиламино)-2-(этоксикарбонил)проп2-ен-1-илиден]-N-метилметанаминия тетрафторборат

8,8 г (60 ммоль) N-1,1,2,2-тетрафторэтилдиметиламина растворяли в 50 мл дихлорметана под аргоном и добавляли при комнатной температуре 8,2 г (60 ммоль) комплекса трифторида бора с диэтиловым эфиром. Полученную смесь перемешивали 30 мин и затем добавляли 7,15 г (50 ммоль) этилового эфира диметиламиноакриловой кислоты. После 2 ч перемешивания при комнатной температуре и удаления дихлорметана в вакууме получали 12,4 г продукта (100% выход) в виде желтого масла.
19F-ЯМР (CDCl3) δ=-120,35, (д, 2F, J=51 Гц); -151,2 (c, 4F) м.д.
1H-ЯМР (CDCl3) δ=1,25 (т, 3H); 2,8, (с, 6H), 3,45 (м, 6H); 4,2 (кв, CH2); 6,87 (т, 1Н); 8,16 (с, 1H) м.д.
Пример 2
N-[(2E)-1-(хлорфторметил)-3-(диметиламино)-2-(этоксикарбонил)-проп-2-ен-1-илиден]-N-метилметанаминия тетрафторборат
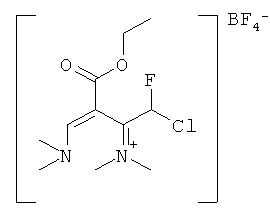
17,6 г (0,1 ммоль) N-1,1,2-трифтор-2-хлорэтилдиметиламина растворяли в 100 мл дихлорметана под аргоном и добавляли при комнатной температуре 13,6 г (0,1 моль) комплекса трифторида бора с диэтиловым эфиром.
После перемешивания в течение 30 мин добавляли 14,3 г (0,1 ммоль) этилового эфира диметиламиноакриловой кислоты и перемешивали 2 ч при комнатной температуре. После удаления дихлорметана в вакууме получают 25,2 г (95%) продукта.
Пример 3
Этил-3-(дифторметил)-1-метил-1Н-пиразол-4-карбоксилат
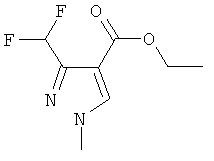
10,0 г (30 ммоль) N-[(2Е)-1-(дифторметил)-3-(диметиламино)-2-(этоксикарбонил)проп-2-ен-1-илиден]-N-метилметанаминия тетрафторбората растворяли в 50 мл ацетонитрила и добавляли 2,3 г метилгидразина. После 2 ч перемешивания при комнатной температуре, ацетонитрил полностью удаляли в вакууме. Перегонкой в вакууме или кристаллизацией из н-гексана получали 5,3 г (86%) продукта с т.пл. 63-65°C.
19F-ЯМР (CDCl3): δ=-117,2 (д) м.д.
1H-ЯМР (CDCl3): δ=1,35 (т, 3H); 3,96 (c, 3H); 4,31 (кв, 2H); 7,10 (т, 1Н), 8,15 (c, 1H) м.д.
Пример 4
Этил 3-(хлорфторметил)-1-метил-1H-пиразол-4-карбоксилат
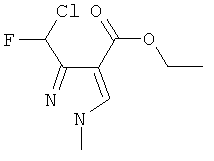
В отличие от Примера 3, в реакцию вводили N-[(2E)-1-(дифторметил)-3-(диметиламино)-2-(этоксикарбонил)проп-2-ен-1-илиден]-N-метилметанаминия тетрафторборат.
19F-ЯМР (CDCl3): δ=-133,8 (д, J=47,5) м.д.
Пример 5
Этил-3-(1,2,2,2-тетрафторэтил)-1-метил-1H-приразол-4-карбоксилат
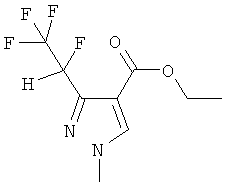
25,3 г (0,1 ммоль) N,N-диэтил-1,1,2,3,3,3-гексафторпропиламина растворяли в 100 мл дихлорметана под аргоном и добавляли при комнатной температуре 13,6 г (0,1 моль) комплекса трифторида бора с диэтиловым эфиром. После перемешивания в течение 30 мин, добавляли 14,3 г (0,1 ммоль) этилового эфира диметиламиноакриловой кислоты и перемешивали 2 ч при комнатной температуре. После удаления дихлорметана в вакууме получают примерно 35 г винамидиниевой соли. 5,6 г метилгидразина растворяли в 40 мл ацетонитрила, и добавляли раствор винамидиниевой соли в 30 мл ацетонитрила при 10°C. После 2 ч перемешивания при комнатной температуре ацетонитрил полностью удаляли в вакууме. Хроматографией на SiO2 выделяют 20 г (82%) продукта в виде масла.
19F-ЯМР (CDCl3): δ=-76,8 (дд, 3F), -191,86 (д.кв, 1F)-м.д.
1H-ЯМР (CDCl3): δ=1,35 (т, 3H); 3,96 (c, 3H); 4,35 (кв, 2H); 6.52 (д.кв., 1H), 8,10 (c, 1H) м.д.
Пример 6
N-[(2E)-1-(дифторметил)-3-метокси-2-(метоксикарбонил)проп-2-ен-1-илиден]-N-метилметанаминия тетрафторборат
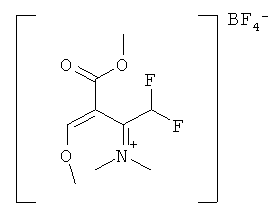
8,7 г (60 ммоль) N-1,1,2,2-тетрафторэтилдиметиламина растворяли в 50 мл дихлорметана под аргоном и при комнатной температуре добавляли 8,2 г (60 ммоль) комплекса трифторида бора с диэтиловым эфиром. Полученную смесь перемешивали в течение 30 мин и затем добавляли 6,38 г (55 ммоль) метилового эфира метоксиакриловой кислоты. После 2 ч перемешивания при комнатной температуре и удаления дихлорметана в вакууме получали 12,4 г продукта (100% выход) в виде желтого масла.
19F-ЯМР (CDCl3) δ=-121,55, (д, 2F, J=51 Гц); -150,2 (c, 4F) м.д.
Пример 7
Метил-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксилат

Реакцию проводят так, как описано в Примере 2, но используют N-[(2E)-1-(дифторметил)-3-метокси-2-(метоксикарбонил)проп2-ен-1-илиден]-N-метилметанаминия тетрафторборат.
Хроматографией на SiO2 выделяют продукт в виде желтого масла.
19F-ЯМР (CDCl3): δ=-117,5 (д) м.д.
Пример 8
Этил-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксилат (однореакторный процесс)

10,8 г N-1,1,2,2-тетрафторэтилдиметиламина растворяли в 50 мл ацетонитрила под аргоном и при комнатной температуре добавляли 26 г трифторида бора в виде 17%-ного раствора в CH3CN. Полученную смесь перемешивали в течение 30 мин и затем добавляли 8,67 г этилового эфира диметиламиноакриловой кислоты. Полученную смесь перемешивали 2 ч при комнатной температуре и затем медленно добавляли к раствору 3,4 г метилгидразина в 10 мл ацетонитрила при 10°C. После 2 ч перемешивания при комнатной температуре, ацетонитрил полностью удаляли в вакууме, к продукту добавляли воду и отфильтровывали. После перегонки в вакууме или промывки циклогексаном получали 10 г продукта, имеющего чистоту 99% и т.пл. 62-63°C.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ РЕГИОСЕЛЕКТИВНОГО СИНТЕЗА ПРОИЗВОДНЫХ 1-АЛКИЛ-3-ГАЛОГЕНАЛКИЛПИРАЗОЛ-4-КАРБОНОВОЙ КИСЛОТЫ | 2009 |
|
RU2498977C9 |
| СПОСОБ РЕГИОСЕЛЕКТИВНОГО СИНТЕЗА ПРОИЗВОДНЫХ 1-АЛКИЛ-3-ГАЛОГЕНАЛКИЛПИРАЗОЛ-4-КАРБОНОВОЙ КИСЛОТЫ | 2013 |
|
RU2661192C2 |
| СПОСОБ РЕГИОСЕЛЕКТИВНОГО СИНТЕЗА ПРОИЗВОДНЫХ 1-АЛКИЛ-3-ГАЛОГЕНАЛКИЛПИРАЗОЛ-4-КАРБОНОВОЙ КИСЛОТЫ | 2009 |
|
RU2498978C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАЗОЛКАРБОНОВОЙ КИСЛОТЫ | 2013 |
|
RU2638112C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ГАЛОГЕНАЛКИЛ-1Н-ПИРАЗОЛОВ | 1996 |
|
RU2169143C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРАЗОЛОВ | 2015 |
|
RU2712192C2 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И АНАЛОГИ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2019 |
|
RU2797316C2 |
| 3(5)-ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПИРАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ P38 | 1998 |
|
RU2249591C2 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И АНАЛОГИ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2019 |
|
RU2799335C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРСОДЕРЖАЩЕГО ПРОИЗВОДНОГО АЦИЛУКСУСНОЙ КИСЛОТЫ, СПОСОБ ПОЛУЧЕНИЯ ФТОРСОДЕРЖАЩЕГО ПРОИЗВОДНОГО СЛОЖНОГО ЭФИРА ПИРАЗОЛКАРБОНОВОЙ КИСЛОТЫ И СПОСОБ ПОЛУЧЕНИЯ ФТОРСОДЕРЖАЩЕГО ПРОИЗВОДНОГО ПИРАЗОЛКАРБОНОВОЙ КИСЛОТЫ | 2009 |
|
RU2456275C2 |
Настоящее изобретение относится к способу получения производных 3-дигалогенметил-пиразол-4-карбоновой кислоты формулы (I) взаимодействием α-фтораминов формулы (III) в присутствии кислот Льюиса с производными акриловой кислоты формулы (II) с получением винамидиниевых солей формулы (IV) и их последующим взаимодействием с гидразинами, и к самим винамидиниевым солям формулы (IV) и промежуточным соединениям формулы (VI). B приведенных ниже структурных формулах радикалы и символы имеют обозначения, указанные в формуле изобретения. Технический результат - разработка более простого и экономичного способа получения 3-дигалогенметил-пиразол-4-карбоновой кислоты. 4 н. и 7 з.п. ф-лы, 8 пр.
1. Способ получения производных 3-дигалогенметил-пиразол-4-карбоновой кислоты формулы (I)

в которой R1 означает C1-12-алкил,
Y означает (C=O)OR6, где R6 означает C1-12-алкил; и
Х представляет собой F, Cl или CF3
взаимодействием α-фтораминов формулы (III)
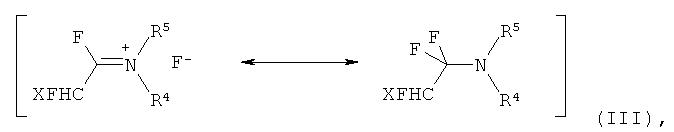
в которой R4 выбран из C1-12-алкилов,
R5 независимо от R4 выбран из С1-12-алкилов, и
Х представляет собой F, Cl или CF3,
в присутствии кислот Льюиса (Z) с производными акриловой кислоты формулы (II)

в которой А выбран из О и NR3,
Y означает (C=O)OR6, где R6 означает С1-12-алкил; и
R2 и R3 независимо друг от друга выбраны из C1-12-алкилов,
с получением винамидиниевых солей формулы (IV)
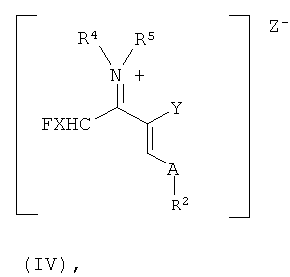
в которой R2, R4, R5, X, Y и А имеют указанные выше значения, и Z- представляет собой анион кислоты Льюиса;
и их последующим взаимодействием с алкилгидразинами формулы (V)
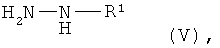
в которой R1 имеет указанное выше значение.
2. Способ по п.1, отличающийся тем, что анион Z- выбран из группы, состоящей из [BF4]-, [AlCl4]-, [AlF4]-, [ZnCl3]-, [PF6]-, [SbF6]-, [SnCl5]-, [BiCl4]-, [GaCl4]-.
3. Способ по п.1 или 2, отличающийся тем, что группа Y представляет собой сложноэфирную группу формулы (C=O)OR6, где R6 означает С1-12-алкил.
4. Способ по п.1 или 2, отличающийся тем, что взаимодействие (IV) с (V) проводят без предварительной очистки/выделения промежуточного продукта (IV).
5. Способ получения производных 3-дигалогенметил-пиразол-4-карбоновой кислоты формулы (I)
в которой R1 означает C1-12-алкил,
Y означает (C=O)OR6, где R6 означает С1-12-алкил; и
Х представляет собой F, Cl или CF3
взаимодействием α-фтораминов формулы (III)
в которой R4 выбран из С1-12-алкилов,
R5 независимо от R4 выбран из С1-12-алкилов, и
Х представляет собой F, Cl или CF3,
с производными акриловой кислоты формулы (II)
в которой А выбран из О и NR3
Y означает (C=O)OR6, где R6 означает С1-12-алкил; и
R2 и R3 независимо друг от друга выбраны из С1-12-алкилов,
и полученная при этом смесь затем подвергается взаимодействию с гидразинами формулы (V)
,
в которой R1 имеет указанное выше значение.
6. Способ по п.5, отличающийся тем, что группа Y представляет собой сложноэфирную группу формулы (C=O)OR6, где R6 означает С1-12-алкил.
7. Винамидиниевая соль формулы (IV)
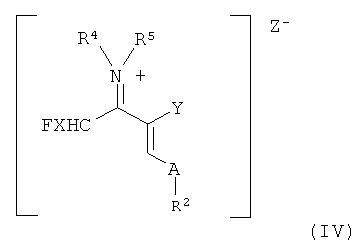
в которой R2, R4, R5, А и Y имеют указанные в п.1 значения и
Z- представляет собой анион кислоты Льюиса.
8. Винамидиниевая соль по п.7, где X=F.
9. Винамидиниевая соль по п.7, отличающаяся тем, что группа Y представляет собой сложноэфирную группу формулы (C=O)OR6, где R6 означает С1-12-алкил.
10. Винамидиниевая соль по одному из пп.7-9, отличающаяся тем, что анион Z- выбран из группы, состоящей из [BF4]-, [AlCl4]-, [AlF4]-, [ZnCl3]-, [PF6]-, [SbF6]-, [SnCl5]-, [BiCl4]-, [GaCl4]-.
11. Соединение формулы (VI)
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| RU 2003125855 A, 10.01.2005. | |||

