Настоящее изобретение относится к способу получения противогрибковых препаратов на основе производных фенилпропаргилового эфира и к способам получения некоторых промежуточных соединений.
Противогрибковые препараты на основе производных фенилпропаргилового эфира, которые можно получать согласно настоящему изобретению, описаны, например, в WO 01/87822. Эти препараты на основе производных фенилпропаргилового эфира соответствуют формуле (А)

включая их оптические изомеры и смеси таких изомеров, в которой
RI представляет собой водород, алкил, циклоалкил или необязательно замещенный арил;
RII и RIII - каждый независимо представляет собой водород или алкил;
RIV является алкилом, алкенилом или алкинилом;
RV, RVI, RVII и RVIII - каждый независимо представляет собой водород или алкил;
RIX представляет собой водород, необязательно замещенный алкил, необязательно замещенный алкенил; или необязательно замещенный алкинил;
Rx представляет собой необязательно замещенный арил, необязательно замещенный гетероарил; и
Z представляет собой галоген, необязательно замещенный арилоксил, необязательно замещенный алкоксил, необязательно замещенный алкенилоксил, необязательно замещенный алкинилоксил, необязательно замещенную арилтиогруппу, необязательно замещенную алкилтиогруппу, необязательно замещенную алкенилтиогруппу, необязательно замещенную алкинилтиогруппу, необязательно замещенный алкилсульфинил, необязательно замещенный алкенилсульфинил, необязательно замещенный алкинилсульфинил, необязательно замещенный алкилсульфонил, необязательно замещенный алкенилсульфонил или необязательно замещенный алкинилсульфонил.
Разные способы получения соединений приведенной выше формулы (А) описаны в WO 01/87822.
Настоящее изобретение относится к другому, альтернативному и предпочтительному пути синтеза фунгицидов на основе производных фенилпропаргилового эфира формулы (I)

в которой:
R представляет собой алкинил;
R1 представляет собой алкил, алкенил, алкинил, циклоалкил, циклоалкил-алкил, фенил и фенилалкил, причем каждая из указанных групп, в свою очередь, содержит один или более одинаковых или разных атомов галогенов; алкоксил; алкенилоксил; алкинилоксил; алкоксиалкил; галогеналкоксил; алкилтиогруппу; галогеналкилтиогруппу; алкилсульфонил; формил; алканоил; гидроксил; галоген; цианогруппу; нитрогруппу; аминогруппу; алкиламиногруппу; диалкиламиногруппу; карбоксил; алкоксикарбонил; алкенилоксикарбонил или алкинилоксикарбонил; и
n является целым числом 0-3.
Термины «алкил», «алкенил» или «алкинил» как в одном заместителе, так и в части другого заместителя представляют собой группы с 1-8 (2-8 в случае алкенила или алкинила) атомами углерода, лучше 1-6 (или 2-6) и предпочтительно 1-4 (или 2-4) атомами углерода.
Конкретные примеры R включают: этинил, проп-1-инил, проп-2-инил, бут-1-инил, бут-2-инил, l-метил-2-бутинил, гекс-1-инил, l-этил-2-бутинил или окт-1-инил. Наиболее предпочтительным является проп-2-инил.
Типичные примеры R1 включают: 4-хлор, 4-бром, 3,4-дихлор, 4- хлор-3-фтор, 3-хлор-4-фтор, 4-метил, 4-этил, 4-пропаргилоксил, 3-метил, 4-фтор, 4-этенил, 4-этинил, 4-пропил, 4-изопропил, 4-трет-бутил, 4-этоксил, 4-этинилоксил, 4-феноксил, 4-метилтиогруппу, 4-метилсульфонил, 4-цианогруппу, 4-нитрогруппу, 4-метоксикарбонил, 3-бром, 3-хлор, 2-хлор, 2,4-дихлор, 3,4,5-трихлор, 3,4-дифтор, 3,4-дибром, 3,4-диметоксил, 3,4-диметил, 3-хлор-4-цианогруппу, 4-хлор-3-цианогруппу, 3-бром-4-метил, 4-метокси-3-метил, 3-фтор-4-метоксил, 4-хлор-3-метил, 4-хлор-3-трифторметил, 4-бром-3-хлор, 4-трифторметил, 4-трифторметоксил, 4-метоксил. Лучше, когда R1 представляет собой 3-галоген, 4-галоген или 3,4-дигалоген; предпочтительно 4-хлор.
Когда n равен 2 или 3, группы R1 могут быть одинаковыми или разными. Лучше n, равный 1 или 2; предпочтительно 1.
Соответственно, первый вариант настоящего изобретения относится к способу получения соединения формулы (I), как описано выше, причем указанный способ включает:
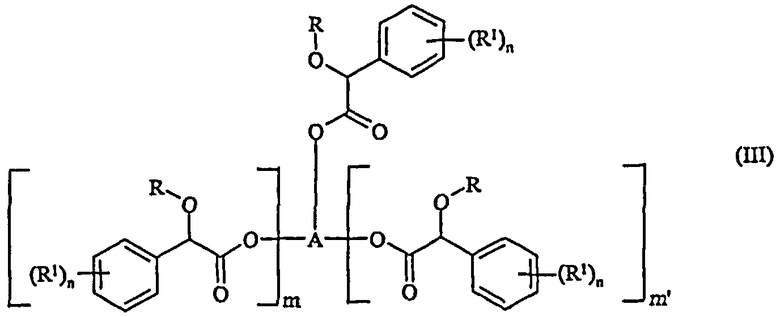
(i) реакцию соединения формулы (III)
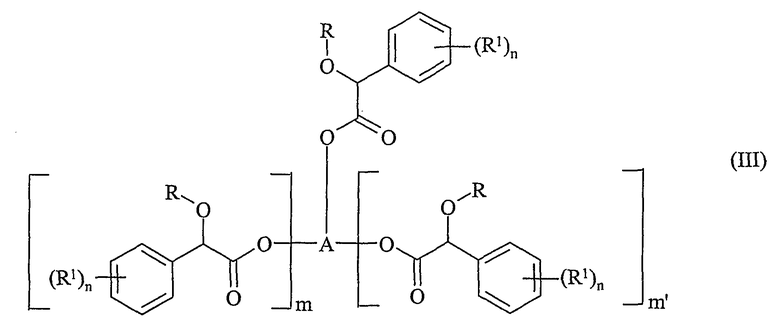
в которой R, R1 и n определены выше;
m и m' независимо равны 0 или 1;
в случае, когда m и m' - оба равны 0, A является алкильной, алкенильной или алкинильной группой (лучше содержащей до восьми атомов углерода), необязательно замещенной одной или более группами, независимо выбранными из галогена, гидроксила, алкоксила, C1-4 диалкиламиногруппы или цианогруппы;
в случае, когда один из m и m' равен 0, а другой равен 1, A представляет собой алкандиильную, алкендиильную или алкиндиильную группу, содержащую по меньшей мере два атома углерода (лучше до восьми атомов углерода), необязательно замещенную одной или более группами, независимо выбранными из галогена, гидроксила, алкоксила, C1-4-диалкиламиногруппы или цианогруппы;
в случае, когда m и m' - оба равны 1, A представляет собой алкантриильную, алкентриильную или алкинтриильную группу, содержащую по меньшей мере три атома углерода (лучше до восьми атомов углерода), необязательно замещенную одной или более группами, независимо выбранными из галогена, гидроксила, алкоксила, C1-4-диалкиламиногруппы или цианогруппы;
и в которой, если группа А содержит три или более атомов углерода, один или более атомов углерода каждый может необязательно быть замещен атомом кислорода при условии, что по меньшей мере один атом углерода находится между двумя атомами кислорода в молекуле,
с соединением формулы (IV)

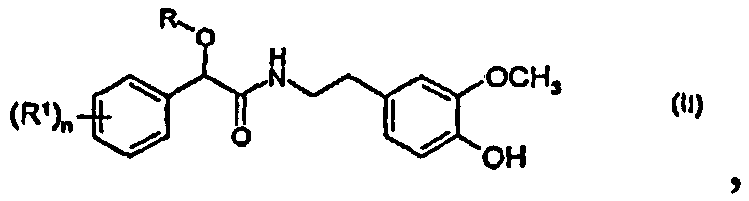
с образованием соединения формулы (II)

в которой R, R1 и n определены выше, и
(ii) реакцию соединения формулы (II), содержащего группировку  , в которой L является уходящей группой, с образованием соединения формулы (I).
, в которой L является уходящей группой, с образованием соединения формулы (I).
Под терминами «алкандиил» и «алкантриил» авторы понимают алкильную группу с двумя или тремя свободными валентностями соответственно (т.е. отсутствуют два или три атома водорода), лучше свободными валентностями при разных атомах углерода.
Под терминами «алкендиил» и «алкентриил» авторы понимают алкенильную группу с двумя или тремя свободными валентностями соответственно, лучше свободными валентностями при разных атомах углерода.
Под терминами «алкиндиил» и «алкинтриил» авторы понимают алкинильную группу с двумя или тремя свободными валентностями соответственно, лучше свободными валентностями при разных атомах углерода.
Подходящие уходящие группы L включают галогены, алкилсульфонаты, галогеналкилсульфонаты и необязательно замещенные арилсульфонаты; и предпочтительно, чтобы L представлял собой хлор или мезилат.
Примеры соединений формулы (III) включают следующие соединения:


Стадию (i) лучше проводить в интервале температур 50-150°C. Реакцию можно проводить в расплаве или в присутствии инертного растворителя, например, толуола, ксилола, хлорбензола и т.п. Температура реакции зависит от реакционной способности сложного эфира. В случае сложных эфиров с низкой реакционной способностью, таких как метиловый эфир, этиловый эфир или бензиловый эфир, можно добавить спирт с высокой реакционной способностью типа диэтиламиноэтанола, этиленгликоль триэтаноламина или пропаргилового спирта или для понижения температуры реакции и во избежание побочных реакций напрямую использовать сложные эфиры указанных спиртов. Обычно температура реакции составляет 70-120°C. При более высоких температурах образуется больше побочных продуктов.
Стадию (ii) лучше проводить в среде наиболее распространенных полярных и неполярных растворителей (например, в углеводородах типа толуола, ксилола или в хлорированных углеводородах, включая хлорбензол, или в простых эфирах, включая ТГФ, диоксан, анизол, или в нитрилах, например ацетонитриле), или в смесях с водой в присутствии оснований, таких как гидроксиды щелочных металлов, гидроксиды щелочноземельных металлов или карбонаты. Лучше, если растворитель или смесь растворителей инертны по отношению к соединению (II) и основанию. Основание можно использовать в широком интервале концентраций, предпочтительно в интервале 1-2 моля на моль соединения (II). Полезно использовать катализатор межфазного переноса типа тройных аммониевых солей в интервале 0,5-10 мол.%. Лучше проводить реакцию в интервале температур 20-150°C, предпочтительно - в интервале 50-100°C.
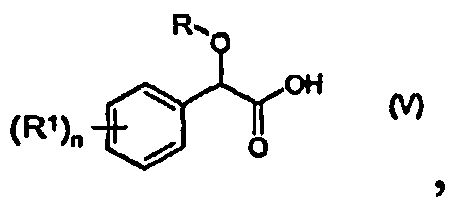
Соединение формулы (III) можно получить из соединения формулы (V)
 ;
;
или из соединения формулы (VI)

или из соединения формулы (VII)
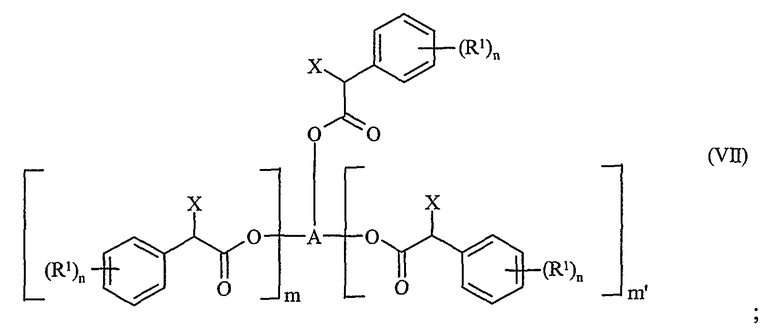
в которой R, R1, m, m', n и A определены выше, и X является уходящей группой. Подходящие уходящие группы включают галоген, такой как фтор, хлор или бром, или алкилсульфонат или арилсульфонат.
Соответственно, второй вариант изобретения предлагает способ получения соединения формулы (I), как описано выше, причем указанный способ включает
(i) (a) этерификацию соединения формулы (V), как описано выше;
или
(b) реакцию соединения формулы (VI), как описано выше, со спиртом формулы  , в которой A, m и m' определены выше; или
, в которой A, m и m' определены выше; или
(c) реакцию указанного выше соединения формулы (VII) со спиртом R-OH, в котором R определен выше, с образованием соединения приведенной выше формулы (III);
(ii) реакцию соединения формулы (III) с соединением указанной выше формулы (IV) с образованием соединения приведенной выше формулы (II); и
(iii) реакцию соединения формулы (II), содержащего группу, в которой L определен выше, с образованием соединения формулы (I).
Стадию (i)(a) лучше проводить в расплаве или в присутствии инертного растворителя, такого как толуол, ксилол, хлорбензол и т.д. Реакция ускоряется при добавлении катализатора, такого как серная кислота, метансульфоновая кислота или п-толуолсульфокислота. Для достижения высокой конверсии образующуюся по реакции воду предпочтительно удалять перегонкой или разлагать химически, например, добавлением, например, триметилового эфира ортомуравьиной кислоты. Реакцию лучше проводить при температуре 0°-150°C, предпочтительно в интервале 50°-100°C.
Стадию (i)(b) лучше проводить в растворителе, таком как углеводород, например в гексане, циклогексане, метилциклогексане или толуоле; в хлоруглеводороде, например дихлорметане или хлорбензоле; в простом эфире, например диэтиловом, трет-бутилметиловом, в диоксане или тетрагидрофуране; или в воде. Можно также использовать в качестве растворителя сам спирт. Можно использовать и смеси таких растворителей. Реакцию проводят в присутствии кислоты, такой как органическая или неорганическая кислота, такой как галогенид водорода, например, хлорид водорода, бромид водорода, или такой как серная кислота или фосфорная кислота. Реакцию лучше проводить при температуре в интервале от -80°C до температуры кипения реакционной смеси, предпочтительно в интервале 0-100°C.
Стадию (i)(c) лучше проводить в присутствии основания, такого как триалкиламин, в отсутствие воды. Реакцию лучше проводить в растворителе, например в углеводороде, включая толуол, ксилол, или в хлорированном углеводороде, например хлорбензоле, или в эфире, таком как ТГФ, диоксан, анизол, или в амиде, таком как ДМФА, в присутствии основания, например карбоната калия, или спирта, например пропаргилового спирта. Температура реакции составляет 0-100°C.
Стадии (ii) и (iii) проводят, как описано выше.
Соединения формулы (V) можно получить из соединения формулы (VIII)

или соединения формулы (IX)

или соединения формулы (X)

в которой R1, n, X и Y определены выше, и каждый может быть одинаковым или разными и представляют собой алкоксигруппу или галоген; лучше C1-4 алкоксил или галоген, предпочтительно метоксил или хлор.
Соответственно, третий вариант изобретения предлагает способ получения соединения формулы (I), как описано выше, причем указанный способ включает
(i) (a) реакцию соединения формулы (VIII), как описано выше, со спиртом R-OH; или
(b) реакцию соединения формулы (IX), как описано выше, со спиртом R-OH в присутствии основания; или
(c) реакцию соединения формулы (X), как описано выше, со спиртом R-OH и тригалогенметаном или тригалогенуксусной кислотой в присутствии основания;
с образованием соединения формулы (V), как описано выше;
(ii) этерификацию соединения формулы (V) с образованием соединения формулы (III), как описано выше;
(iii) реакцию соединения формулы (III) с соединением формулы (IV), как описано выше, с образованием соединения формулы (II), как описано выше; и
(iv) реакцию соединения формулы (II) с соединением  , в котором L определен выше, с образованием соединения формулы (I).
, в котором L определен выше, с образованием соединения формулы (I).
Стадию (i)(a) лучше проводить в присутствии основания, такого как гидроксид щелочного металла или третичный амин. Лучше использовать основание в соотношении 2-10 моль на моль соединения (VIII), предпочтительно 2,5-3,5 моль. Один моль основания расходуется на нейтрализацию карбоксильной кислотной группы соединения (VIII). Реакцию лучше проводить в интервале температур от -50°C до 120°C, предпочтительно в интервале от -10°C до 50°C. В качестве растворителя можно использовать спирт R-OH или дополнительный растворитель, такой как алифатический или ароматический углеводород, галогенированный ароматический углеводород, кетоны, простые эфиры, N-метилпирролидон (NMP) или диметилсульфоксид (ДМСО). Реакцию лучше проводить в отсутствие воды.
Стадию (i)(b) проводят в присутствии основания, такого как гидроксид щелочного или щелочноземельного металла, например, гидроксида натрия или гидроксида калия, алкоголятов натрия или калия, например, метоксида натрия или азотсодержащих соединений, например, l,8-диазабицикло-[5.4.0]-ундец-7-ена (DBU), l,4-диазабицикло-[2.2.2]-октана (DABCO) (известного также как триэтилендиамин). Можно использовать также смеси таких оснований. Реакцию лучше проводить при температуре от -80°C до 150°C, предпочтительно в интервале 30-100°C. Реакцию лучше проводить в растворителе, например в полярном или неполярном органическом растворителе, таком как углеводороды, простые эфиры, амиды, например, ДМЭ, диглим, диоксан, ТГФ, анизол, N-метилпирролидон, ДМСО или спирт; спирт ROH также может быть растворителем.
Стадию (i)(c) лучше проводить при температурах от -80°C до 150°C, предпочтительно в интервале 0-70°C. Тригалогенметаны являются производными метана, в которых три атома водорода замещены одинаковыми или разными галогенами типа фтора, хлора или брома. Примерами таких тригалогенметанов являются хлороформ, бромоформ, хлордибромметан или бромдихлорметан. Подходящие гидроксидные основания включают гидроксиды щелочных или щелочноземельных металлов, такие как гидроксид натрия или гидроксид калия. Реакцию лучше проводить в растворителе, таком как углеводород, например гексан, циклогексан, метилциклогексан или толуол; хлоруглеводород, например дихлорметан или хлорбензол; простой эфир, например диэтиловый эфир, трет-бутилметиловый эфир, диоксан или тетрагидрофуран, или вода. Можно также использовать смеси растворителей. Спирт и/или тригалогенметан можно также использовать в качестве растворителя; в одном варианте спирт R-OH используют как растворитель; еще в одном варианте растворителем служит тригалогенметан.
Стадии (ii)-(iv) проводят, как описано выше.
Соединение определенной выше формулы (VI) можно получить из соединения формулы (XI)
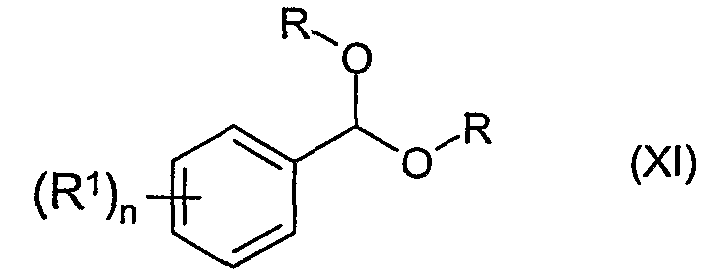
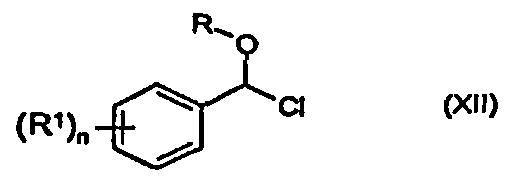
в которой R, R1 и n - такие же, как указано выше, - либо напрямую, либо через соединение формулы (XII)

в которой R, R1 и n - такие же, как указано выше.
Соответственно, четвертый вариант изобретения предлагает способ получения соединения описанной выше формулы (I), причем указанный способ включает
(i) (a) реакцию соединения описанной выше формулы (XI) с цианирующим реагентом; или
(b) (i) реакцию соединения описанной выше формулы (XI) с хлорирующим реагентом с образованием описанного выше соединения формулы (XII) и последующей реакцией соединения формулы (XII) с цианирующим реагентом;
с образованием соединения описанной выше формулы (VI).
(ii) реакцию соединения формулы (VI) со спиртом формулы  , в которой m, m' и A определены выше, с образованием описанного выше соединения формулы (III);
, в которой m, m' и A определены выше, с образованием описанного выше соединения формулы (III);
(iii) реакцию соединения формулы (III) с соединением описанной выше формулы (IV), с образованием соединения описанной выше формулы (II); и
(iv) реакцию соединения формулы (II) с , в котором L определен выше, с образованием соединения формулы (I).
, в котором L определен выше, с образованием соединения формулы (I).
Стадию (i)(a) лучше проводить в присутствии Бренстедовских кислот, таких как сильная минеральная кислота, например, хлорид водорода, бромид водорода или серная кислота, или Льюисовских кислот, таких как соединения элементов III группы, например, трифторид бора, соли металлов, например, соли цинка, такие как хлорид цинка (II), бромид цинка (II), соли железа, такие как хлорид железа (III), соли кобальта, такие как хлорид кобальта (II), соли сурьмы, такие как хлорид сурьмы (V), соли скандия, такие как трифлат скандия (III), соли иттрия, такие как трифлат иттрия (III), соли индия, такие как хлорид индия (III), соли лантана, такие как трифлат лантана (III), или соли висмута, такие как хлорид висмута (III), бромид висмута (III). Предпочтительно использовать кислоту в количествах меньше стехиометрических. Подходящие цианирующие реагенты включают цианид водорода, цианосиланы, такие как триалкилсилилцианид, например, триметилсилилцианид или аналогичные циангидрины. Реакцию предпочтительно проводить в растворителе, таком как углеводород, например гексан, циклогексан, метилциклогексан или толуол; хлоруглеводород, например дихлорметан или хлорбензол; простой эфир, например диэтиловый, трет-бутилметиловый эфиры, диоксан или тетрагидрофуран; амид, например N,N-диметиламид, N,N-диметилацетамид или N-метилпирролидон. Можно также использовать смеси растворителей. Реакцию лучше проводить в интервале температур от -80°C до 150°C, предпочтительно в интервале 0-70°C.
Стадию (i)(b)(i) лучше проводить при температуре от -80°C до 100°C, предпочтительно в интервале 0-25°C. В качестве хлорирующих реагентов лучше использовать органические хлориды, такие как низшие алканоилхлориды, например ацетилхлорид, или хлориды неорганических кислот, например тионилхлориды, сульфурилхлорид или оксихлорид фосфора. Можно также использовать смесь хлорирующих реагентов. Реакцию проводят в подходящем растворителе, таком как углеводород, например гексан, циклогексан, метилциклогексан или толуол; хлоруглеводород, например, дихлорметан или хлорбензол; простой эфир, например диэтиловый, трет-бутилметиловый эфиры, диоксан или тетрагидрофуран. Можно также использовать смеси растворителей.
Стадию (i)(b)(ii) проводят в подходящем растворителе, таком как углеводород, например гексан, циклогексан, метилциклогексан или толуол; хлоруглеводород, например дихлорметан или хлорбензол; простой эфир, например диэтиловый, трет-бутилметиловый эфиры, диоксан или тетрагидрофуран; амиды, например N,N-диметиламид, N,N-диметилацетамид или N-метилпирролидон; или вода. Можно также использовать смеси растворителей. Подходящие цианирующие реагенты включают цианиды металлов, такие как цианиды щелочных или щелочноземельных металлов, например цианиды натрия или калия. Реакцию лучше проводить при температурах от -50°C до 100°C, предпочтительно - 0-40°C.
Стадии (ii)-(iv) проводят, как описано выше.
Описанное выше соединение формулы (VII) можно получить из соединения формулы (XIII)

в которой R1, n и X - такие же, как описано выше, и W является галогеном, предпочтительно хлором.
Соответственно, пятый вариант изобретения предлагает способ получения описанного выше соединения формулы (I), причем указанный способ включает
(i) реакцию соединения описанной выше формулы (XIII) со спиртом формулы
, в которой A, m и m' определены выше, с образованием соединения описанной выше формулы (VII).
(ii) реакцию соединения формулы (VII) со спиртом R-OH, в котором R - такой же, как описано выше, с образованием соединения формулы (III), описанной выше;
(iii) реакцию соединения формулы (III) с соединением приведенной выше формулы (IV) с образованием соединения описанной выше формулы (II); и
(iv) реакцию соединения формулы (II) с соединением  , в котором L определен выше, с образованием соединения формулы (I).
, в котором L определен выше, с образованием соединения формулы (I).
Стадию (i) лучше проводить в присутствии основания, такого как триалкиламин, в обычных условиях реакции хлорангидрида кислоты со спиртом. Например, растворитель может быть спиртом типа пропаргилового спирта, и температура реакции может составлять от -20°C до 150°C, предпочтительно в интервале 0-60°C.
Стадии (ii)-(iv) проводят, как описано выше.
Соединения приведенной выше формулы (VIII) можно получить из соединения формулы (XIV)

в которой R1 и n определены выше.
Соответственно, шестой вариант изобретения предлагает способ получения соединения описанной выше формулы (I), причем указанный способ включает
(i) галогенирование соединения описанной выше формулы (XIV) с образованием соединения описанной выше формулы (VIII);
(ii) реакцию соединения формулы (VIII) со спиртом R-OH, в котором R определен выше, с образованием соединения приведенной выше формулы (V);
(iii) этерификацию соединения формулы (V) с образованием соединения приведенной выше формулы (III);
(iv) реакцию соединения формулы (III) с соединением описанной выше формулы (IV) с образованием соединения определенной выше формулы (III); и
(v) реакцию соединения формулы (II) с , в котором L определен выше, с образованием соединения формулы (I).
Стадию (i) можно проводить в расплаве или в инертном растворителе типа уксусной кислоты или большинства галогенированных ароматических и алифатических растворителей. Для ускорения реакции надо добавить катализатор типа красного фосфора, фосфортрихлорида или бромида, фосфорпентахлорида или бромида, тионилхлорида или тионилбромида, фосгена в количестве 0,01-1,0 моля на моль соединения (XIV), предпочтительно в интервале 0,1-0,5 моль. Галогенирование (XIV) можно осуществить с помощью брома, хлора или соответствующего сукцинимида в интервале температур 50-200°C, предпочтительно 80-150°C.
Стадии (ii)-(v) можно проводить, как описано выше.
Соединения описанной выше формулы (IX) можно получить из соединения описанной выше формулы (X) или соединения формулы (XV)

в которой R1 и n определены выше.
Соответственно, седьмой вариант изобретения предлагает способ получения соединения описанной выше формулы (I), причем указанный способ включает:
(i) (a) присоединение тригалогенметан-аниона к соединению описанной выше формулы (X); или
(b) присоединение тригалогенацетальдегида к соединению описанной выше формулы (XV);
с образованием соединения описанной выше формулы (IX);
(ii) реакцию соединения формулы (IX) со спиртом R-OH и тригалогенметаном в присутствии основания с образованием соединения определенной выше формулы (V);
(iii) этерификацию соединения формулы (V) с образованием соединения приведенной выше формулы (III);
(iv) реакцию соединения формулы (III) с соединением определенной выше формулы (IV) с образованием соединения определенной выше формулы (II); и
(v) реакцию соединения формулы (II) с соединением  , в котором L определен выше, с образованием соединения формулы (I).
, в котором L определен выше, с образованием соединения формулы (I).
Стадию (i)(a) лучше проводить в растворителе, таком как углеводород, например гексан, циклогексан, метилциклогексан или толуол; хлоруглеводород, например дихлорметан или хлорбензол; простой эфир, например диэтиловый, трет-бутилметиловый эфиры, диоксан или тетрагидрофуран; амид, например N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон; или вода. Можно также использовать смеси растворителей. Тригалогенметаны являются производными метана, в котором три атома водорода замещены одинаковыми или разными галогенами типа фтора, хлора или брома. Примерами таких тригалогенметанов являются хлороформ, бромоформ, хлордибромметан или бромдихлорметан. Можно также использовать соли щелочных или щелочноземельных металлов или тригалогенметанкарбоновых кислот в присутствии соответствующих тригалогенметанкарбоновых кислот, таких как натриевая соль трихлоруксусной кислоты или калиевая соль трихлоруксусной кислоты в присутствии трихлоруксусной кислоты. Реакцию лучше проводить в интервале температур от -80°C до 150°C, предпочтительно в интервале 0-70°C.
Стадию (i)(b) проводят в подходящем растворителе, таком как дисульфид углерода; хлоруглеводород, например дихлорметан или хлороформ; ароматическое соединение, например хлорбензол, дихлорбензол, трихлорбензол, нитробензол; простой эфир, например диэтиловый, трет-бутилметиловый эфиры, диоксан или тетрагидрофуран. Можно также использовать смеси растворителей. Тригалогенацетальдегиды являются производными ацетальдегида, в котором три атома водорода замещены одинаковыми или разными атомами галогенов, таких как фтор, хлор или бром. Примерами таких тригалогенацетальдегидов являются трихлорацетальдегид, трибромацетальдегид, хлордибромацетальдегид или бромдихлорацетальдегид. Реакцию лучше проводить при температурах в интервале от -80°C до 150°C, предпочтительно с интервале от -10°C до 70°C.
Стадии (ii)-(v) проводят, как описано выше.
Соединения определенной выше формулы (XI) получают из соединений описанной выше формулы (X).
Соответственно, восьмой вариант настоящего изобретения предлагает способ получения соединения определенной выше формулы (I), причем указанный способ включает:
(i) реакцию соединения приведенной выше формулы (X) со спиртом R-OH в присутствии кислоты с образованием соединения приведенной выше формулы (XI);
(ii) (a) реакцию соединения формулы (XI) с цианирующим реагентом; или
(b) (i) реакцию соединения формулы (XI) с хлорирующим реагентом с образованием соединения приведенной выше формулы (XII) и последующей (ii) реакцией соединения формулы (XII) с цианирующим реагентом
с образованием соединения приведенной выше формулы (VI).
(iii) реакцию соединения формулы (VI) со спиртом формулы
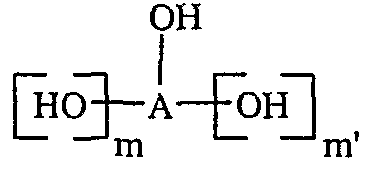 , в которой A, m и m' определены выше, с образованием соединения приведенной выше формулы (III);
, в которой A, m и m' определены выше, с образованием соединения приведенной выше формулы (III);
(iv) реакцию соединения формулы (III) с соединением описанной выше формулы (IV) с образованием соединения указанной выше формулы (II); и
(v) реакцию соединения формулы (II) с соединением  , в котором L определен выше, с образованием соединения формулы (I).
, в котором L определен выше, с образованием соединения формулы (I).
Стадию (i) проводят в присутствии подходящего растворителя, такого как углеводород, например гексан, циклогексан, метилциклогексан или толуол; хлоруглеводород, например дихлорметан или хлорбензол; простой эфир, например диэтиловый, трет-бутилметиловый эфиры, диоксан или тетрагидрофуран. В предпочтительном варианте спирт R-OH используют в качестве растворителя. Можно также использовать смеси растворителей. Реакцию проводят в присутствии кислоты, такой как Бренстедовская кислота, например сильная минеральная кислота, в том числе хлорид водорода, бромид водорода или серная кислота; Льюисовская кислота, такая как соединения элементов III группы, например трифторид бора; соль металла, например соли цинка, такие как хлорид цинка (II), бромид цинка (II), соли железа, такие как хлорид железа (III), соли кобальта, такие как хлорид кобальта (II), соли сурьмы, такие как хлорид сурьмы (V), соли скандия, такие как трифлат скандия (III), соли иттрия, такие как трифлат иттрия (III), соли индия, такие как хлорид индия (III), соли лантана, такие как трифлат лантана (III), или соли висмута, такие как хлорид висмута (III), бромид висмута (III). Предпочтительно использовать кислоту в субстехиометрических количествах. Реакцию можно также проводить в присутствии ортоэфира, такого как ортоэфиры низших алкилкарбоновых кислот и низших алкиловых спиртов, например, триметилортоформиата, триметилортоацетата и триэтилортоформиата или триэтилортоацетата. Предпочтительно использовать ортоэфир, когда продукты реакции (сложный эфир и спирт) можно удалить из реакционной смеси дистилляцией. Реакцию лучше проводить при температурах от -80°C до температуры кипения реакционной смеси, предпочтительно в интервале 0-100°C.
Стадии (ii)-(v) проводят, как описано выше.
Соединения указанной выше формулы (XIII) можно получить из соединений формулы (XVI)
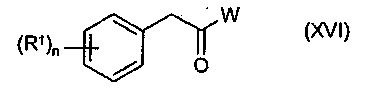
в которой R1, W и n определены выше.
Соответственно, девятый вариант изобретения предлагает способ получения соединения приведенной выше формулы (I), причем указанный способ включает:
(i) галогенирование соединения приведенной выше формулы (XVI) с образованием соединения описанной выше формулы (XIII);
(ii) реакцию соединения формулы (XIII) со спиртом формулы
, в которой A, m и m' определены выше, с образованием соединения определенной выше формулы (VII).
(iii) реакцию соединения формулы (VII) со спиртом R-OH, в котором R определен выше, с образованием соединения описанной выше формулы (III);
(iv) реакцию соединения формулы (III) с соединением описанной выше формулы (IV) с образованием соединения приведенной выше формулы (II); и
(v) реакцию соединения формулы (II) с соединением  , в котором L определен выше, с образованием соединения формулы (I).
, в котором L определен выше, с образованием соединения формулы (I).
Стадию (i) проводят, как описано в патенте Канады 967978, в расплаве или в инертном растворителе, таком как хлорированные углеводороды или хлорированные ароматические соединения, в интервале температур 50-150°C.
Стадии (ii)-(v) проводят, как описано выше.
Соединения формул (IV), (X), (XIV), (XV) и (XVI) известны в данной области, и их получение легко доступно для специалистов.
Альтернативно, соединения формулы (IV) можно получить новым способом согласно следующей схеме реакции:
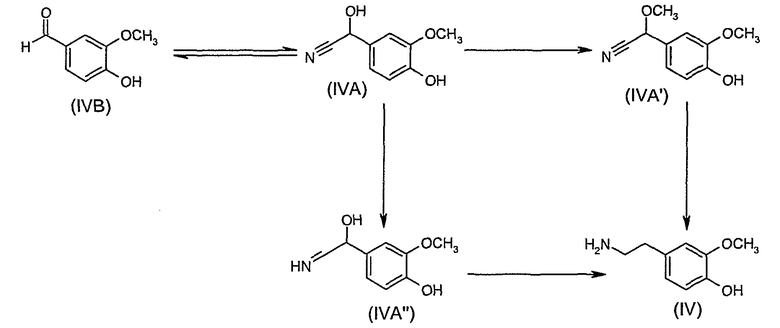
Способы предшествующего уровня техники для получения соединений формулы (IVA) связаны со значительным количеством водных стоков и/или использованием дорогих катализаторов; водные стоки необходимо обрабатывать (путем разложения цианида хлорной известью или пероксидом водорода), что требует больших затрат и приводит к стокам, все еще содержащим токсичные компоненты.
В приведенной схеме соединение (IVB) превращают в соединение (IVA) с помощью одного из следующих способов:
(i) по реакции соединения (IVB) с цианидом, например цианидом натрия или калия (предпочтительно в небольшом избытке), при pH 5-9, предпочтительно 6-7, с последующим снижением рН ниже 3 или
(ii) по реакции соединения (IVB) с HCN в органическом или водном растворителе; или
(iii) по реакции соединения (IVB) с ацетонциангидрином в присутствии каталитического количества цианида или обычного основания.
Соединение (IVA) затем восстанавливают с помощью H2/Pd-C и H2SО4/MeOH. В первом способе H2/Pd-C и H2SО4/MeOH добавляют вместе и способ осуществляют через промежуточное соединение (IVA''); во втором способе первым делом добавляют H2SО4/MeOH с образованием промежуточного соединения (IVA') и последующим восстановлением с помощью H2/Pd-C.
Таким образом, следующий вариант изобретения предлагает способ получения соединения (IV), причем указанный способ включает:
(i) реакцию соединения (IVB)
(a) с цианидом, например цианидом натрия или калия (предпочтительно в небольшом избытке), при рН 5-9, предпочтительно 6-7, с последующим снижением рН ниже 3 или
(b) с HCN в органическом или водном растворителе; или
(c) с ацетонциангидрином в присутствии каталитического количества цианида или обычного основания;
с образованием соединения (IVA), и
(ii) восстановление соединения (IVA) с использованием H2/Pd-C и H2SО4/MeOH через промежуточные соединения (IVA') или (IVA'') и его таутомеров с образованием соединения (IV).
Промежуточные соединения (IVA') и (IVA'') и его «гидроксиенаминный» таутомер также являются новыми веществами и составляют следующий вариант изобретения.
Получение соединения формулы (II) из соединения формулы (III) также является новым предлагаемым способом и соответственно составляет следующий вариант настоящего изобретения.
Многие промежуточные соединения формулы (II), (III), (V), (VI), (VII), (XI) или (XII), особенно когда R1 представляет собой галоген, например 4-хлор, также являются новыми и соответственно предлагают еще один вариант изобретения.
Схема реакций, изображающая все возможные описанные выше реакции, приведена на Фигуре 1.
Далее изобретение будет проиллюстрировано следующими примерами:
Пример 1: 1-(Бис-проп-2-инилоксиметил)-4-хлорбензол-(4-хлорбензальдегид-дипропаргилацеталь) (соединение формулы XI)

4-Хлорбензальдегид (14,3 г) добавляют к пропаргиловому спирту (56,6 г) и концентрированной соляной кислоте (0,1 мл). Реакционную смесь перемешивают и нагревают до 80°C. Затем непрерывно добавляют триметилортоформиат (11,9 г) в течение 1 часа. Реакционную смесь перемешивают при 85°C в течение 5 часов и часть веществ отгоняют. Реакционную смесь охлаждают до комнатной температуры. Добавляют трет-бутилметиловый эфир (200 мл). Органическую фазу промывают 40% раствором кислого сульфита натрия (2 x 200 мл), сушат (сульфат натрия) и упаривают. Получают 1-(бис-проп-2-инилоксиметил)-4-хлорбензол (18,8 г) в виде бесцветного масла.
1H-ЯМР (CDCl3) δ (м.д.): 2,45 (т, 2H); 4,15 (дд, 2H); 4,3 (дд, 2H); 5,85 (с, 1H); 7,35 (д, 2H); 7,45 (д, 2H).
Пример 2: 1-Хлор-4-(хлорпроп-2-инилоксиметил)бензол (соединение формулы XII)

1-(Бис-проп-2-инилоксиметил)-4-хлорбензол (11,7 г) добавляют к ацетилхлориду (19,9 г) и тионилхлориду (0,2 мл) в течение 1 часа. С помощью периодического охлаждения поддерживают температуру 20°C. Реакционную смесь перемешивают при комнатной температуре в течение 20 часов. Реакционную смесь упаривают при 20-30°C в вакууме. Получают 1-хлор-4-(хлор-проп-2-инилоксиметил)бензол (13,4 г) в виде масла.
1H-ЯМР (CDCl3) δ (м.д.): 2,6 (т, 1H); 4,6 (д, 2H); 6,75 (с, 1H); 7,35 (д, 2H); 7,45 (д, 2H).
Пример 3: (4-Хлорфенил)-проп-2-инилоксиацетонитрил (соединение формулы VI)

1-Хлор-4-(хлорпроп-2-инилоксиметил)бензол (13,0 г) добавляют к цианиду натрия (3,1 г) в N,N-диметилформамиде (40 мл) в течение 2 часов при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и затем выливают в воду (200 мл), содержащую гидроксид натрия (4 г). Водную фазу экстрагируют трет-бутилметиловым эфиром (2 x 200 мл). Органические фазы промывают водой (2 x 50 мл), объединяют, сушат (сульфат натрия) и упаривают. Получают (4-хлорфенил)проп-2-инилоксиацетонитрил (9,2 г), очищают флэш-колоночной хроматографией на силикагеле с использованием смеси этилацетат/гексан в качестве элюата.
1H-ЯМР (CDCl3) δ (м.д.): 2,6 (т, 1H); 4,4 (д, 2H); 5,5 (с, 1H); 7,4-7,5 (м, 4H).
Пример 4: (4-Хлорфенил)-проп-2-инилоксиацетонитрил (соединение формулы VI)

В атмосфере азота триметилсилилцианид (3,1 г) добавляют к бромиду висмута (III) (0,22 г) и 1-(бис-проп-2-инилоксиметил)-4-хлорбензолу (6,7 г) в дихлорметане (50 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 48 часов при комнатной температуре и затем выливают в 0,5 M соляную кислоту (50 мл). Органическую фазу отделяют, сушат (сульфат магния) и упаривают. Получают сырой (4-хлорфенил)-проп-2-инилоксиацетонитрил (3,7 г) в виде масла.
1H-ЯМР (CDCl3) δ (м.д.): 2,6 (т, 1H); 4,4 (д, 2H); 5,5 (с, 1H); 7,4-7,5 (м, 4H).
Пример 5: Метиловый эфир (4-хлорфенил)-проп-2-инилоксиуксусной кислоты (соединение формулы III)
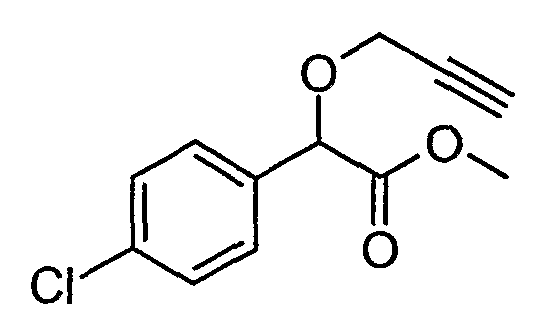
Смесь (4-хлорфенил)-проп-2-инилоксиацетонитрила (6,4 г) и 37% соляной кислоты (12,6 г) в метаноле (40 мл) нагревают с обратным холодильником в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры и добавляют воду (25 мл). Водную фазу экстрагируют этилацетатом (2×25 мл). Органические фазы объединяют, промывают водой (1×25 мл), сушат (сульфат натрия) и упаривают. Получают сырой метиловый эфир (4-хлорфенил)проп-2-инилоксиуксусной кислоты в виде масла.
1H-ЯМР (CDCl3) δ (м.д.): 2,5 (т, 1H);3,7 (с, 3H); 4,15 (дд, 1H); 4,3 (дд, 1H); 5,2 (с, 1H); 7,3-7,5 (м,4H).
Пример 6: (4-Хлорфенил)-проп-2-инилоксиуксусная кислота (соединение формулы V)

a) Смесь гидроксида калия (23,4 г, содержание по анализу 90%) в пропаргиловом спирте (70 мл) добавляют к 4-хлорбензальдегиду (7,2 г) и хлороформу (13,4 г) в пропаргиловом спирте (10 мл) в течение 5 часов при 50°C. Реакционную смесь перемешивают при 50°C еще в течение 3 часов. После охлаждения до комнатной температуры добавляют воду (150 мл). Полученную смесь экстрагируют трет-бутилметиловым эфиром (150 мл). Органическую фазу снова экстрагируют 4M гидроксидом калия (50 мл). Водные щелочные экстракты объединяют и подкисляют (pH<3) концентрированной соляной кислотой. Водную фазу экстрагируют трет-бутилметиловым эфиром (2×150 мл). Органические фазы объединяют, экстрагируют водой (1×100 мл), сушат (сульфат магния) и упаривают. Получают (4-хлорфенил)-проп-2-инилоксиуксусную кислоту (7,7 г) в виде масла, которое затвердевает при стоянии.
b) 4-Хлорбензальдегид (7,2 г) в пропаргиловом спирте (15 мл) нагревают до 50°C. Смесь гидроксида калия (31,2 г, содержание по анализу 90%) в пропаргиловом спирте (150 мл) и смесь бромоформа (13 г) в пропаргиловом спирте (15 мл) добавляют одновременно в течение 1 часа при 50°C. Реакционную смесь перемешивают при 50°C еще 5 часов. После охлаждения до комнатной температуры добавляют воду (150 мл). Полученную смесь экстрагируют трет-бутилметиловым эфиром (150 мл). Органическую фазу снова экстрагируют 4M гидроксидом калия (50 мл). Водные щелочные экстракты объединяют и подкисляют (pH < 3) концентрированной соляной кислотой. Водную фазу экстрагируют трет-бутилметиловым эфиром (2×150 мл). Органические фазы объединяют, экстрагируют водой (1×100 мл), сушат (сульфат магния) и упаривают. Получают (4-хлорфенил)-проп-2-инилоксиуксусную кислоту (10,4 г) в виде масла, которое затвердевает при стоянии.
1H-ЯМР (CDCl3) δ (м.д.): 2,5 (т, 1H); 4,15 (дд, 1H); 4,3 (дд, 1H); 5,2 (с, 1H); 7,3-7,5 (м, 4H); 7,2-9,5 (с, уш., 1H).
Пример 7: 2,2,2-Трихлор-1-(4-хлорфенил)этанол (соединение формулы IX)

Смесь 4-хлорбензальдегида (35,5 г) и трихлоруксусной кислоты (61,5 г) в N,N-диметилформамиде (200 мл) перемешивают при 30-35°C. Натриевую соль трихлоруксусной кислоты (71,5 г) добавляют порциями в течение 20 мин. Периодически необходимо охлаждение. Реакционную смесь перемешивают при 30°C в течение 2 часов. Смесь до конца остается вязкой, и в нее добавляют N,N-диметилформамид (150 мл). Реакционную смесь выливают в воду (700 мл). Водную фазу экстрагируют этилацетатом (600 мл). Органическую фазу отделяют, промывают водой (300 мл), сушат (сульфат магния) и упаривают. Получают 2,2,2-трихлор-1-(4-хлорфенил)этанол в виде масла.
1H-ЯМР (CDCl3) δ (м.д.): 4,1 (с, уш., 1H); 5,2 (с, 1H); 7,3 (д, 2H); 7,55 (д, 2H).
Пример 8: 2,2,2-Трихлор-1-(4-хлорфенил)этанол (соединение формулы IX)

Смесь хлорбензола (1400 г) и трихлорацетальдегида (384 г) перемешивают при 0-2°C. Хлорид алюминия (274 г) добавляют порциями в течение 110 мин при этой же температуре. Смесь периодически охлаждают. Реакционную смесь перемешивают при 0-5°C в течение 5 час. Реакционную смесь выливают в смесь льда с водой (3000 г). Органическую фазу отделяют, промывают три раза водой (по 500 г), сушат (сульфат натрия) и упаривают. Получают 2,2,2-трихлор-1-(4-хлорфенил)этанол в виде масла.
1H-ЯМР (CDCl3) δ (м.д.): 4,1 (с, уш., 1H); 5,2 (с, 1H); 7,3 (д, 2H); 7,55 (д, 2H).
Пример 9: (4-Хлорфенил)-проп-2-инилоксиуксусная кислота (соединение формулы V)

К смеси пропаргилового спирта (300 г) и 2,2,2-трихлор-1-(4-хлорфенил)этанола (501 г) добавляют 15% раствор (1820 г) гидроксида натрия и пропаргилового спирта в течение трех часов при 70-75°C. Смесь периодически охлаждают. Реакционную смесь перемешивают при той же температуре в течение 3 часов. После отгонки большей части растворителя остаток охлаждают до комнатной температуры и добавляют смесь воды с этилацетатом. Органическую фазу снова экстрагируют 2M гидроксидом натрия (50 мл). Объединенный водно-щелочной экстракт подкисляют (pH<3) концентрированной соляной кислотой. Водную фазу дважды экстрагируют этилацетатом. Органические фазы объединяют, экстрагируют водой, сушат (сульфат натрия) и упаривают. Получают (4-хлорфенил)проп-2-инилоксиуксусную кислоту (10,4 г) в виде масла.
1H-ЯМР (CDCl3) δ (м.д.): 2,5 (т, 1H); 4,15 (дд, 1H); 4,3 (дд, 1H); 5,2 (с, 1H); 7,3-7,5 (м, 4H); 7,2-9,5 (с, уш., 1H).
Пример 10: 2-(4-Хлорфенил)-N-[2-(3-метокси-4-проп-2-инилоксифенил)этил]-2-проп-2-инилоксиацетамид (соединение формулы I)
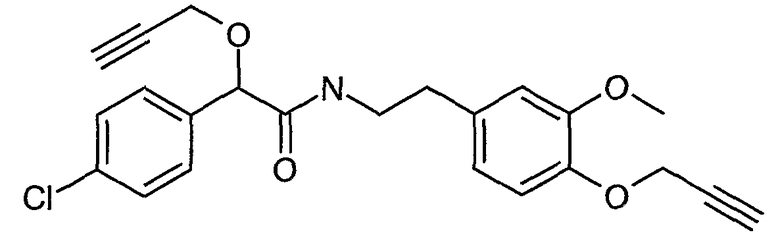
К раствору 1 моль 2-(4-хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамида в 500 мл толуола добавляют 207 г карбоната калия (1,5 моль) и 10 г тетрабутиламмоний бромида. Смесь нагревают до 90°C и добавляют 1,4 моль пропаргилхлорида в виде 35% раствора в толуоле в течение 30 мин. Через 3 часа превращение 2-(4-хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамида заканчивается. Для растворения солей добавляют 500 мл воды и отделяют от фазы продуктов в толуоле. Толуол полностью упаривают при 80°C/20 мбар и заменяют метанолом. Продукт 2-(4-хлорфенил)-N-[2-(3-метокси-4-проп-2-инилоксифенил)этил]-2-проп-2-инилоксиацетамид кристаллизуют из раствора при охлаждении до 0°C, отфильтровывают и промывают 200 мл метанола при 0°C. Продукт сушат при 50°C в вакууме. Получают 315 г 2-(4-хлорфенил)-N-[2-(3-метокси-4-проп-2-инилоксифенил)этил]-2-проп-2-инилоксиацетамида с чистотой 98% по данным ЖХ. Температура плавления =94-96°C.
Пример 11: 2-(4-Хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамид (соединение формулы II)

К раствору 1 моль (4-хлорфенил)-проп-2-инилоксиуксусной кислоты 2-[2-(4-хлорфенил)-2-проп-2-инилоксиацетокси]-этилового эфира («гликоэфира») в 500 г хлорбензола (полученного из 1 моля (4-хлорфенил)проп-2-инилоксиуксусной кислоты) добавляют 1,05 моль 4-(2-аминоэтил)-2-метоксифенола («AE-фенол») и 0,3 моль диэтиламиноэтанола. Реакционную смесь нагревают до 90-100°C и отгоняют хлорбензол в вакууме. После перемешивания в течение 3-4 час при 90-100°C превращение гликоэфира заканчивается. Добавляют 500 г толуола и 250 мл воды. После перемешивания в течение 5 мин при 50-70°C водную фазу отделяют. К толуольной фазе добавляют 250 мл воды и устанавливают pH 0,5-1,0 с помощью 32% водного раствора соляной кислоты для удаления избыточного AE-фенола и диметиламиноэтанола. Водную фазу отделяют и к толуольной фазе 2-(4-хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамида необязательно добавляют 20 г отбеливающего реагента Prolith rapid, перемешивают 30 мин при 50-60°C и затем отфильтровывают. Толуольный фильтрат, содержащий продукт 2-(4-хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамид с выходом 92% (по данным ЖХ), непосредственно используют на следующей стадии. 2-(4-Хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамид можно частично выделить кристаллизацией/фильтрацией из толуолового раствора при -10°C с выходом 224 г (60% в расчете на гликоэфир). Температура плавления = 93-95°C.
Пример 11a: 2-(4-Хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамид (соединение формулы II)

К раствору 1 моль (4-хлорфенил)-проп-2-инилоксиуксусной кислоты 2-[2-(4-хлорфенил)-2-проп-2-инилоксиацетокси]этилового эфира («гликоэфира») в 500 г хлорбензола (полученного из 1 моля (4-хлорфенил)проп-2-инилоксиуксусной кислоты) добавляют 1,05 моль 4-(2-аминоэтил)-2-метоксифенола («AE-фенол») и 0,3 моль диэтиламиноэтанола. Реакционную смесь нагревают до 90-100°C и отгоняют хлорбензол в вакууме. После перемешивания в течение 3-4 час при 90-100°C превращение гликоэфира заканчивается. Добавляют 500 г толуола и 250 мл воды. После перемешивания в течение 5 мин при 50-70°C водную фазу отделяют. К толуольной фазе добавляют 250 мл воды и устанавливают pH 0,5-1,0 с помощью 32% водного раствора соляной кислоты для удаления избыточного AE-фенола и диметиламиноэтанола. Водную фазу отделяют и к толуольной фазе 2-(4-хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамида необязательно добавляют 20 г отбеливающего реагента Prolith rapid, перемешивают 30 мин при 50-60°C и затем отфильтровывают. К толуольному фильтрату добавляют 12% раствор Na2CО3 или 50% раствор K2CO3 и устанавливают рН 8,5-10,5 для удаления побочных продуктов с кислотными фрагментами. Органический слой, содержащий продукт 2-(4-хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамид с выходом 92% (по данным ЖХ), непосредственно используют на следующей стадии. 2-(4-Хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамид можно частично выделить кристаллизацией/фильтрацией из толуольного раствора при -10°C с выходом 224 г (60% от теории в расчете на гликоэфир). Температура плавления = 93-95°C.
Пример 12: 2-Гидроксиэтиловый эфир (4-хлорфенил)проп-2-инилоксиуксусной кислоты или 2-[2-(4-хлорфенил)-2-проп-2-инилоксиацетокси]этиловый эфир (4-Хлорфенил)-проп-2-инилоксиуксусной кислоты (соединение формулы III)

К раствору (4-хлорфенил)проп-2-инилоксиуксусной кислоты (1 моль) в 600 г хлорбензола добавляют 0,75 моль этиленгликоля и 4 г п-толуолсульфокислоты и нагревают в вакууме с обратным холодильником при 90-100°C. Воду реакции отделяют от конденсата и хлорбензол возвращают в реактор. Через 1 час этерификация заканчивается. В конце отгоняют 100 г хлорбензола. Реакционная смесь содержит смесь моно- и диэфиров этиленгликоля, которые непосредственно без выделения превращают в 2-(4-хлорфенил)-N-[2-(4-гидрокси-3-метоксифенил)этил]-2-проп-2-инилоксиацетамид на следующей стадии (пример 11).
Пример 13: Метиловый эфир (4-хлорфенил)проп-2-инилоксиуксусной кислоты (соединение формулы (III))
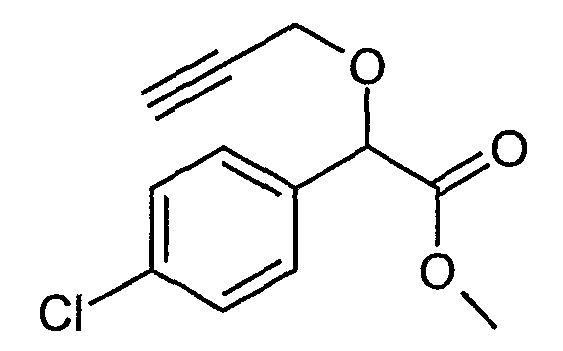
К раствору (4-хлорфенил)проп-2-инилоксиуксусной кислоты (1 моль) в 500 г хлорбензола добавляют 2 моль метанола, 1 моль триметилового эфира ортомуравьиной кислоты и 4 г п-толуолсульфокислоты. Смесь нагревают до 50-60°C и выдерживают в течение 2-3 часов до окончания этерификации (4-хлорфенил)проп-2-инилоксиуксусной кислоты. Низкокипящие вещества типа метанола и метилформиата отгоняют в вакууме при 50-60°C. Раствор «метилового эфира» в хлорбензоле можно непосредственно превратить в II на следующей стадии без выделения. После отгонки растворителя в вакууме получают 245 г масла, содержащего 236 г метилового эфира (4-хлорфенил)проп-2-инилоксиуксусной кислоты (по данным анализа методом ГЖХ).
1H-ЯМР (CDCl3) δ (м.д.): 2,5 (с, HC≡); 3,7 (с, OCH3) (4,2+4,3 (2д, CH2); 5,2 (1с, CH); 7,35(4H, Ar)
Пример 14: (4-Хлорфенил)проп-2-инилоксиуксусная кислота (соединение формулы V)

Загружают в реактор при перемешивании 500 г хлорбензола, 177-187 г гидроксида калия 90-95% (3,0 моль) и 112 г пропаргилового спирта (2 моль). При 15-20°C добавляют раствор 1 моль бром-(4-хлорфенил)уксусной кислоты в 800 г хлорбензола (или реакционную смесь 1 моль бром-(4-хлорфенил)уксусной кислоты/хлорангидрида кислоты, описанную в примере 15a) через капельную воронку в течение 2 часов. Реакционную смесь выдерживают еще в течение 1-2 часов до окончания превращения бром-(4-хлорфенил)уксусной кислоты. Реакционную массу разбавляют 500 мл воды и устанавливают рН 0,5 с помощью соляной кислоты при 35-40°C. Водную фазу отделяют от фазы органического продукта и затем отгоняют 800 г хлорбензола в вакууме при 90-100°C. Оставшийся раствор хлорбензола содержит 218 г (4-хлорфенил)проп-2-инилоксиуксусной кислоты по данным ЖХ-анализа (выход 97% в расчете на бром-(4-хлорфенил)уксусную кислоту). Раствор (4-хлорфенил)проп-2-инилоксиуксусной кислоты в хлорбензоле можно непосредственно использовать на следующей стадии.
«Пропаргиловую кислоту», т.е. (4-хлорфенил)проп-2-инилоксиуксусную кислоту, можно частично выделить концентрированием до 50% раствора и кристаллизацией/фильтрацией при 0°C. Примерно 170 г (4-хлорфенил)проп-2-инилоксиуксусной кислоты можно выделить в кристаллической форме. Температура плавления =69-70°C.
Пример 15: Бром-(4-хлорфенил)уксусная кислота (соединение формулы VIII)

Загружают при перемешивании в реактор с обратным холодильником (связанный со щелочным скруббером) 171 г 4-хлорфенилуксусной кислоты в 750 г хлорбензола и добавляют 41 г фосфортрихлорида (0,3 моль). Смесь нагревают до 100-110°C и через капельную воронку добавляют 280 г брома (1,75 моль) в течение 1 часа. Реакционную смесь перемешивают еще 3-4 часа при 110-115°C до завершения превращения 4-хлорфенилуксусной кислоты (контроль методом ЖХ). Реакционную смесь охлаждают до 50°C и добавляют 100 мл воды. Избыток брома разлагают с помощью раствора NaHSO3. В реакционной смеси устанавливают рН 1 с помощью водного раствора NaOH и затем органический продукт отделяют от водной фазы. Фаза хлорбензола содержит 237 г бром-(4-хлорфенил)уксусной кислоты по данным ЖХ-анализа (выход =95% в расчете на 4-хлорфенилуксусную кислоту). «Бромкислоту», т.е. бром-(4-хлорфенил)уксусную кислоту, можно частично выделить концентрированием до 50% раствора и кристаллизацией/фильтрацией при 0°C. Примерно 200 г бром-(4-хлорфенил)уксусной кислоты можно выделить в кристаллической форме. Температура плавления =92-93°C.
1H-ЯМР (CDCl3) δ (м.д.): 5,3 (с, 1H); 7,4 (4H, Ar); 9,7 (1H, OH).
Пример 15a: Бром-(4-хлорфенил)уксусная кислота (соединение формулы VIII)

В реактор с обратным холодильником (связанный со щелочным скруббером) при перемешивании загружают 171 г 4-хлорфенилуксусной кислоты в 400 г хлорбензола и нагревают до 105°C. Добавляют 42 г тионилхлорида в течение 30 мин при 105-110°C с образованием частично хлорангидрида кислоты. К реакционной смеси добавляют 256 г брома (1,6 моль) в течение 90 мин при 105-108°C. Реакционную смесь перемешивают еще 2-3 часа при 105-108°C, пока не завершится превращение 4-хлорфенилуксусной кислоты (контроль методом ВЭЖХ). Избыток брома отгоняют в виде смеси бром/хлорбензол при 90°C до достижения вакуума 250 мбар и изменения цвета реакционной смеси от коричневого до желтого. Отогнанный бром можно использовать повторно в следующей операции. Реакционную смесь, содержащую смесь бром-(4-хлорфенил)уксусной кислоты и хлорангидрида, разбавляют хлорбензолом до массы 800 г, и ее можно превратить непосредственно в (4-хлорфенил)проп-2-инилоксиуксусную кислоту согласно примеру 14.
Пример 16: Бром-(4-хлорфенил)ацетилхлорид (соединение формулы XIII)
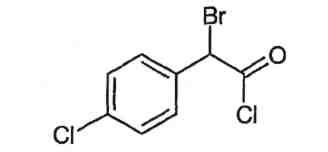
В реактор с обратным холодильником (связанный со щелочным скруббером) при перемешивании загружают 171 г 4-хлорфенилуксусной кислоты (1 моль) в 600 г толуола и 7 г диметилформамида. Смесь нагревают до 50°C и вводят под поверхность 125 г фосгена в течение 2-3 часов. Толуол полностью отгоняют в вакууме и к остатку хлорангидрида 4-хлорфенилуксусной кислоты добавляют 226 г брома при 90°C в течение 1-2 часов. Для полного завершения превращения реакционную смесь перемешивают еще час и затем откачивают для удаления избытка брома. Оранжевый остаток в 290 г содержит примерно 260 г хлорангидрида 4-хлорфенилбромуксусной кислоты (97% в расчете на 4-хлорфенилуксусную кислоту), определенного в виде метилового эфира методом ЖХ.
1H-ЯМР (CDCl3) δ (м.д.): 5,7 (с, 1H); 7,4 (с, 4H, Ar)
Пример 17: Проп-2-иниловый эфир (4-хлорфенил)проп-2-инилоксиуксусной кислоты (соединение формулы III)

К смеси 70 мл пропаргилового спирта и 35 мл N-этилдиизопропиламина добавляют 14 г бром-(4-хлорфенил)ацетилхлорида в течение 15 мин при 0-5°C с образованием соединения формулы VII. Затем реакционную смесь нагревают до 60°C и перемешивают при этой температуре в течение 8 часов с образованием указанного выше соединения формулы (III). Реакционную смесь выливают в 400 мл смеси лед/вода. Устанавливают рН 3 с помощью соляной кислоты и продукт экстрагируют 3 раза диэтиловым эфиром (100 мл). Объединенные экстракты сушат над MgSО4 и растворитель упаривают при 50°C в вакууме. Остается 12 г коричневатого масла.
1H-ЯМР (CDCl3) δ (м.д.): 2,45+2,55 (2с, HC≡); 4,2+4,7 (2кв., CH2); 5,3 (1с, CH); 7,4 (4H, Ar).
Пример 18: 4-(l-Гидроксиацетонитрил)-2-метоксифенол (соединение формулы (IVA))
A1. В 1 л круглодонной колбе суспендировали 80 г (0,52 экв) 4-гидрокси-3-метоксибензальдегида (соединение (IVB), ванилин) в 135 г воды при 5°C. Через 2 часа добавили 90 г (0,64 экв) 35% раствора цианида натрия и 78 г (0,68 экв) 32% соляной кислоты, параллельно регулируя рН на уровне 6,5 и температуру - на уровне 5°C. В конце добавления суспензию перемешивали в течение 6-8 часов при pH 6,5 и 5°C для завершения реакции. Затем установили рН 1-2 с помощью 32% HCl и добавили 160 г метил-трет-бутилового эфира (MTBE) для экстракции циангидрина в органический растворитель. Двухслойную смесь перемешивали при комнатной температуре в течение 1 часа. После этого мешалку остановили, и слои разделились; нижний водный слой отделили и к слою метил-трет-бутилового эфира добавили 0,8 г (0,01 экв) хлоруксусной кислоты для стабилизации циангидрина перед заменой растворителя. Раствор метил-трет-бутилового эфира перегнали при пониженном давлении при 40-60°C (100-500 мбар) и получили 93 г (выход 94%) соединения (IVA) в виде желтого масла или кристаллического вещества.
A2. В 1 л круглодонной колбе суспендируют 100 г (0,64 экв) 4-гидрокси-3-метоксибензальдегида (соединение (IVB), ванилин) в 165 г воды при обычной температуре. Затем полученную суспензию охлаждают при активном перемешивании до 15°C и перемешивают еще 30 мин. Добавляют 130 г (0,8 экв) 30% раствора цианида натрия и 130 г (0,4 экв) 30% серной кислоты при параллельном регулировании в течение 4-6 часов рН на уровне 6,0-6,5 и температуры на уровне 15°С. В конце введения добавки реакционную массу начинают перемешивать до кристаллизации продукта и затем перемешивают суспензию 2 часа при рН 6,5 и 15°C до завершения реакции. Затем устанавливают pH≤1 с помощью примерно 2 г 30% серной кислоты и добавляют 170 г метил-трет-бутилового эфира (MTBE). Продукт экстрагируют в органический слой при перемешивании в течение 1 часа при 25-30°C. После этого мешалку останавливают и слои разделяются, нижний водный слой отделяют и к слою MTBE добавляют 1 г (0,01 экв) хлоруксусной кислоты для стабилизации циангидрина перед заменой растворителя. Раствор в MTBE затем перегоняют при пониженном давлении при 40-60°C (100-500 мбар) и получают 112 г (выход 96%) соединения (IVA) в виде желтого масла или кристаллического остатка.
A3. В 1 л круглодонной колбе суспендируют 160 г (1,03 экв) 4-гидрокси-3-метоксибензальдегида (соединение (IVB), ванилин) в 160 г воды при комнатной температуре. Затем добавляют 4 г метил-трет-бутилового эфира (MTBE) и полученную суспензию охлаждают при интенсивном перемешивании до 15°C. Затем устанавливают рН 7,0-7,5 с помощью примерно 4 г 10% NaOH. Затем к суспензии ванилин/вода при перемешивании добавляют 85 г (1,26 экв) 40% водного раствора HCN в течение 30-60 мин. В конце добавления HCN (если нужно) устанавливают рН 6,5 либо с помощью 20% серной кислоты, либо 10% NaOН. Реакционная масса быстро светлеет, и затем ее перемешивают в течение 3 часов при 15°C и pH 6,0-6,5. Обычно во время перемешивания начинается кристаллизация продукта из прозрачного раствора и сразу по окончании кристаллизации суспензию перемешивают 1-2 часа до завершения реакции.
Затем устанавливают pH≤1,5 добавлением примерно 3 г 20% серной кислоты и затем добавляют 170 г метил-трет-бутилового эфира (MTBE). Продукт экстрагируют в органический слой при перемешивании в течение 1 час при 25-30°C. После этого мешалку останавливают, и слои разделяются, нижний водный слой отделяют и к слою MTBE добавляют 1 г (0,01 экв) хлоруксусной кислоты для стабилизации циангидрина перед заменой растворителя. Затем раствор MTBE перегоняют при пониженном давлении при 40-60°C (100-500 мбар) и получают 180 г (выход 96%) соединения (IVA) в виде желтого масла или кристаллического остатка.
B. В 50 мл круглодонной колбе с механической мешалкой, термометром, обратным холодильником и скруббером для поглощения отходящего газа (1:1 NaOCl:NaOH) создали инертную атмосферу. Раствор HCN в тетрагидрофуране (ТГФ) (17% масс/масс) приготовили до этого опыта по известным литературным методикам. В реактор добавили гидроксид калия (0,026 г, 0,02 экв) и раствор цианида водорода в ТГФ (5,02 мл, 1,5 экв) и затем ТГФ (5 мл). Ванилин (3,07 г) растворили в ТГФ (5 мл) и добавили в реактор при перемешивании в течение нескольких минут. Реакционную массу перемешивали при обычной температуре в течение 3,5 часов (малое количество белого твердого вещества в бледно-желтой жидкости) и затем анализировали количественно методом ВЭЖХ для определения выхода. Конверсия 90%; выход 83%.
C. В колбу, содержащую 8,5 г ацетонциангидрина (1 экв), порциями добавили 15,2 г ванилина (1 экв) в течение 1 часа. После перемешивания в течение 1 часа добавили 0,8 мл 35% водного раствора цианида натрия (0,05 экв). Полученную смесь выдержали при комнатной температуре в течение 5 суток и затем погасили реакцию добавлением 75 г метил-трет-бутилового эфира (MTBE) и 9 г воды. Анализ органического слоя методом ВЭЖХ показал, что ванилинциангидрин образовался с выходом 36%, причем единственным определяемым компонентом был непрореагировавший ванилин.
Пример 19: 4-Аминоэтил-2-метоксифенол (получение соединения формулы IV через промежуточное соединение IVA'')
В 300 мл реактор для работы под давлением поместили 30 мл метанола и 31,1 г 98% серной кислоты (1,41 экв). Добавили суспензию 3,8 г катализатора 5% палладия на древесном угле (0,004 экв) в 10 мл метанола с последующей промывкой 10 мл метанола. В реактор под давлением 5 бар водорода при температуре 20-25°C ввели 100 г 40% раствора ванилинциангидрина в метаноле (1 экв ванилинциангидрина) в течение 4 час и затем промыли 15 мл метанола. Перемешивали 20 мин, затем давление сбросили и добавили 75 мл воды. Эту смесь перемешивали при 45°C для растворения продукта и затем катализатор отфильтровали. Осадок катализатора на фильтре промыли водой (3×25 мл), промывные воды объединили с маточным раствором и получили 315 г раствора продукта, содержащего 10,0% AE-фенола по данным ВЭЖХ (выход 86%).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ФЕНИЛПРОПАРГИЛОВОГО ЭФИРА | 2001 |
|
RU2259353C2 |
| ПРОИЗВОДНЫЕ БЕНЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2248964C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПРОСТОГО ПРОПАРГИЛОВОГО ЭФИРА | 2000 |
|
RU2237058C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРО 3-ГАЛОГЕН-1H-ПИРАЗОЛ-5-КАРБОКСИЛАТЫ, ИХ ПОЛУЧЕНИЕ И ИСПОЛЬЗОВАНИЕ | 2002 |
|
RU2317983C2 |
| ХЛОРМЕТИЛДИАРИЛОКСИРАНЫ | 1991 |
|
RU2125997C1 |
| ПРОИЗВОДНЫЕ ИНДОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1993 |
|
RU2101283C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 4,4-ДИФТОР-3,4-ДИГИДРОИЗОХИНОЛИНА | 2012 |
|
RU2616608C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2170230C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2005 |
|
RU2384562C2 |
| СПОСОБ СТЕРЕОИЗБИРАТЕЛЬНОГО ПОЛУЧЕНИЯ Z-1,2-ДИАРИЛАЛЛИЛХЛОРИДОВ | 1990 |
|
RU2014317C1 |

Заявлен способ получения соединения формулы (I), где n равно 0, а значения заместителей R, R1 приведены в п.1 формулы изобретения, который включает: (i) этерификацию соединения формулы (V) со спиртом формулы 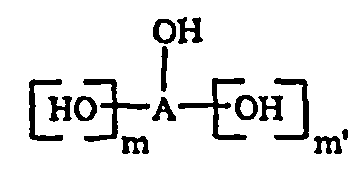 , где m и m' независимо равны 0 или 1; при условии, что оба одновременно не равны 0 и значения А приведены в п.1 формулы изобретения с получением соединения формулы (III); (ii) реакцию соединения формулы (III) и соединения формулы (IV) с образованием соединения формулы (II); и (iii) реакцию соединения формулы (II) с
, где m и m' независимо равны 0 или 1; при условии, что оба одновременно не равны 0 и значения А приведены в п.1 формулы изобретения с получением соединения формулы (III); (ii) реакцию соединения формулы (III) и соединения формулы (IV) с образованием соединения формулы (II); и (iii) реакцию соединения формулы (II) с  , в котором L является уходящей группой, с образованием соединения формулы (I); также заявлены промежуточные соединения формулы (II), формулы (III) и формулы (XII). 4 н. и 11 з.п. ф-лы, 19 пр., 1 ил.
, в котором L является уходящей группой, с образованием соединения формулы (I); также заявлены промежуточные соединения формулы (II), формулы (III) и формулы (XII). 4 н. и 11 з.п. ф-лы, 19 пр., 1 ил.
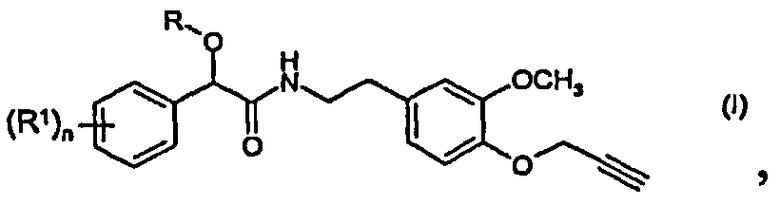


1. Способ получения соединения формулы (I)

в которой:
R представляет собой С2-С8алкинил;
R1 представляет собой С1-С8алкил, С2-С8алкенил, С2-С8алкинил, С3-С8циклоалкил, С3-С8циклоалкил-С1-С8алкил, фенил и фенилС1-С8алкил, причем каждая из указанных групп, в свою очередь, содержит один или более одинаковые или разные атомы галогенов; С1-С8алкоксил; С2-С8алкенилоксил; С2-С8алкинилоксил; С1-С8алкоксиС1-С8алкил; галогенС1-С8алкоксил; С1-С8алкилтиогруппу; галогенС1-С8алкилтиогруппу; C1-С8алкилсульфонил; формил; алканоил; галоген; цианогруппу; нитрогруппу; С1-С4диалкиламиногруппу; карбоксил; C1-С8алкоксикарбонил; С2-С8алкенилоксикарбонил или С2-С8алкинилоксикарбонил; и n равно 1, причем указанный способ включает:
(i) этерификацию соединения формулы (V)

в которой R, R1 и n определены выше со
спиртом формулы

в которой m и m' независимо равны 0 или 1; при условии, что оба одновременно не равны 0;
когда один из m и m' равен 0, а другой равен 1, А представляет собой алкандиильную, алкендиильную или алкиндиильную группу, содержащую по меньшей мере два атома углерода (и лучше содержащую до восьми атомов углерода), необязательно замещенную одной или более группами, независимо выбранными из галогена, гидроксила, С1-8алкоксила, C1-4диалкиламиногруппы или цианогруппы;
когда m и m1 - оба равны 1, А представляет собой алкантриильную, алкентриильную или алкинтриильную группу, содержащую по меньшей мере три атома углерода (лучше содержащую до восьми атомов углерода), необязательно замещенную одной или более группами, независимо выбранными из галогена, гидроксила, С1-8алкоксила, C1-4диалкиламиногруппы или цианогруппы;
и в которой, если группа А содержит три или более атомов углерода, один или более атомов углерода - каждый может необязательно быть замещен атомом кислорода при условии, что по меньшей мере один атом углерода находится между двумя атомами кислорода в молекуле, с получением соединения формулы (III)
в которой R, R1 и n определены выше для соединения формулы (I), и в которой m, m' и А определены выше для соединения формулы
(ii) реакцию соединения формулы (III) с соединением формулы (IV)
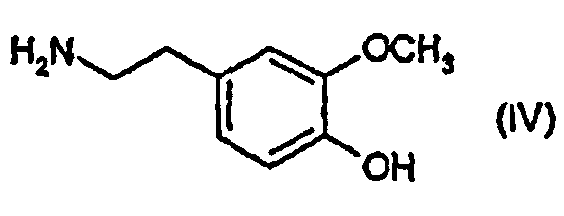
с образованием соединения формулы (II)
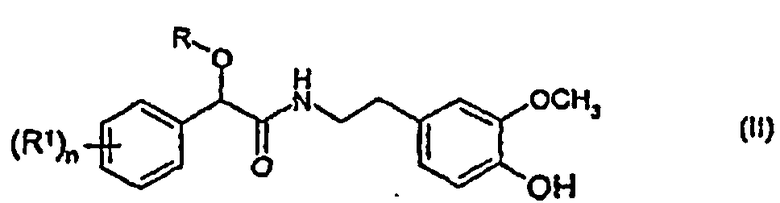
в которой R, R1 и n определены выше, и
(iii) реакцию соединения формулы (II) с  в котором L является уходящей группой, с образованием соединения определенной выше формулы (I).
в котором L является уходящей группой, с образованием соединения определенной выше формулы (I).
2. Способ получения соединения формулы (I) по п.1, в котором соединение формулы (V) получают способом, включающим (а) реакцию соединения формулы (VIII)

в которой R1, n и Х определены выше для соединения формулы (I) по п.1, со спиртом формулы R-OH, в которой R определен выше для соединения формулы (I) по п.1; или (b) реакцию соединения формулы (IX)

в которой R1 и n определены выше для соединения формулы (VIII), и каждый Y может быть одинаковым или разным и представляет собой C1-С8алкоксигруппу или галоген, со спиртом R-OH, в котором R определен выше для соединения формулы (I), в присутствии основания; или с) реакцию соединения формулы (X)

в которой R1 и n определены выше для соединения формулы (I), со спиртом R-OH, в котором R определен выше для соединения формулы (I), и тригалогенметаном в присутствии основания.
3. Способ получения соединения формулы (I) по п.1, в котором соединение формулы (VI) получают способом, включающим (i) (а) реакцию соединения формулы (XI)

в которой R, R1 и n определены выше для соединения формулы (I) по п.1, с цианирующим реагентом; или
(b) (i) реакцию соединения формулы (XI) с хлорирующим реагентом с образованием соединения формулы (XII)

в которой R, R1 и n определены выше,
(ii) с последующей реакцией соединения формулы (XII) с цианирующим реагентом.
4. Способ по п.2, в котором соединение формулы (VIII) получают способом, включающим
(i) галогенирование соединения формулы (XIV)

в которой R1 и n определены выше для соединения формулы (I) по п.1.
5. Способ получения соединения формулы (I) по п.2, в котором соединение формулы (IX) получают способом, включающим (i) (а) присоединение тригалогенметан-аниона к соединению формулы (X)

указанной по п.2, или (b) присоединение тригалогенацетальдегида к соединению формулы (XV)
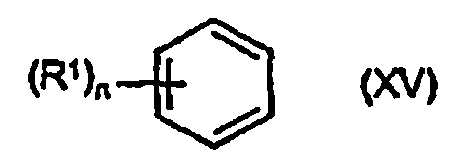
в которой R1 и n определены выше для соединения формулы (I) по п.1.
6. Способ получения соединения формулы (I) по п.3, в котором соединение формулы (XI) получают способом, включающим (i) реакцию соединения формулы (X) по п.2 со спиртом R-OH в присутствии кислоты с образованием соединения формулы (XI) по п.3.
7. Способ по п.1, в котором R представляет собой этинил, проп-1-инил, проп-2-инил, бут-1-инил, бут-2-инил, 1-метил-2-бутинил, гекс-1-инил, 1-этил-2-бутинил или окт-1-инил.
8. Способ по п.7, в котором R представляет собой проп-2-инил.
9. Способ по любому из пп.1-8, в котором R1 представляет собой 4-хлор, 4-бром, 4-метил, 4-этил, 4-пропаргилоксил, 3-метил, 4-фтор, 4-этенил, 4-этинил, 4-пропил, 4-изопропил, 4-трет-бутил, 4-этоксил, 4-этинилоксил, 4-феноксил, 4-метилтиогруппу, 4-метилсульфонил, 4-цианогруппу, 4-нитрогруппу, 4-метоксикарбонил, 3-бром, 3-хлор, 2-хлор, 4-трифторметил, 4-трифторметоксил, 4-метоксил.
10. Способ по п.9, в котором R1 представляет собой 4-хлор.
11. Способ получения соединения формулы (II) по п.1

причем способ включает реакцию соединения формулы (III) по п.1

с соединением формулы (IV)

12. Промежуточное соединение формулы (II)
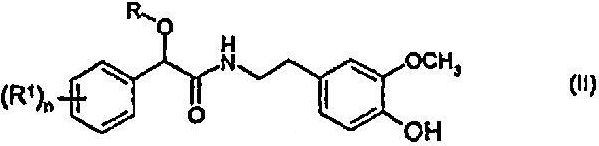
формулы (III)

или формулы (XII)

по пп.1, 11 и 3 соответственно.
13. Промежуточное соединение формулы (XI) по п.3, где R1 представляет собой 4-хлор.
14. Промежуточное соединение по п.12, где R1 представляет собой галоген.
15. Промежуточное соединение по п.14, где R1 представляет собой 4-хлор.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| NAZAROV et al | |||
| "Preparative method of the synthesis of cyanohydrins" J | |||
| GEN | |||
| CHEM. | |||

