Настоящее изобретение относится к производным бензола, содержащим аминную функциональную группу, замещенным алкильной группой и циклоалкильной группой, которые специфически связываются с сигма-рецепторами, в частности с рецепторами периферической нервной системы, к способу получения этих соединений и к их использованию в фармацевтических композициях и, более конкретно, в качестве иммунодепрессантов.
Сигма-рецепторы были выявлены с помощью нескольких лигандов. Прежде всего, можно упомянуть об опиатных соединениях - 6,7-бензоморфанах или SKF-10047 -, в частности о хиральном соединении (+) SKF-10047 (W.R. Martin et al., J. Pharmacol. Exp. Ther. 1976, 197, 517-532; B.R. Martin et al., J. Pharmacol. Exp. Ther. 1984, 231, 539-544). Из этих соединений наиболее часто используют (+)N-аллилнорметазоцин, или (+)NAHM, и (+)пентазоцин. Нейролептический агент галоперидол также является лигандом сигма-рецепторов, как и (+)3-(3-гидроксифенил)-1-пропилпиперидин и (+)3-РРР (В. L. Largent et al., Proc. Nat. Acad. Sci. USA 1984, 81, 4983-4987).
В патенте США 4709094 описаны производные гуанидина, которые являются высокоактивными лигандами, которые специфичны в отношении сигма-рецепторов, и можно упомянуть, в частности, ди-(O-толил)гуанидин, или DTG. Анатомическое распределение сигма-рецепторов в мозге было установлено посредством ауторадиографии после мечения этих рецепторов DTG согласно Е. Weber et al., Proc. Nat. Acad. Sci. USA 1986, 83, 8784-8788, а также лигандами (+)SKF-10047 и (+)3-РРР согласно В. L. Largent et al., J. Pharmacol. Exp. Ther. USA 1986, 238, 739-748. Ауторадиографическое исследование дало возможность четко идентифицировать сигма-рецепторы головного мозга и отличать их от других опиатных рецепторов, а также от фенциклидиновых рецепторов. В частности, сигма-рецепторы распределены в центральной нервной системе и сконцентрированы в стволе головного мозга, лимбической системе и областях, вовлеченных в регуляцию эмоций. Сигма-рецепторы также обнаружены в различных периферических тканях. Различают по меньшей мере два типа сигма-рецепторов: рецепторы сигма-1 и сигма-2. Лиганды типа (+)SKF-10047 избирательно связываются с рецепторами сигма-1, тогда как другие лиганды, такие как DTG, галоперидол или (+)3-РРР демонстрируют значительную аффинность в отношении как сигма-1, так и сигма-2 рецепторов.
В патенте ЕР 461986 описаны соединения формулы

которые избирательно связываются с сигма-рецепторами и которые обладают иммунодепрессивной активностью.
В этом ряду соединений исследован конкретно гидрохлорид (Z)-[3-(3-хлор-4-циклогексилфенил)аллил]циклогексилэтиламина формулы
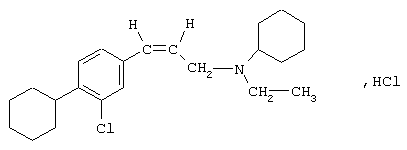
Можно сослаться, например, на Biological Chemistry. 1997, 272 (43), 27107-27115; Immunopharmacology and Immunotoxicology. 1996, 18 (2), 179-191. Однако соединения формулы (А) обладают специфическим свойством, которое можно рассматривать как недостаток. Этим свойством, которое проявляется во время метаболизации, является зависимость от цитохрома, известного как CYP 2D6.
В 1957 году впервые было высказано предположение, что наследственные различия могут быть ответственны за вариации ответной реакции на лекарственные продукты. Окислительный метаболизм демонстрирует значительные вариации между индивидуумами и расами. Исследование, которое проводилось в течение последних 15 лет, показало, что вариации функциональной экспрессии мультигенного семейства цитохрома Р450 (CYP) являются причиной этих различий. Лишь некоторые изоформы цитохрома Р450 из тех, которые уже определены у человека, участвуют в окислительном метаболизме лекарственных продуктов (Xenobiotica, 1986, 16, 367-378). К настоящему времени на основе их клинической важности идентифицированы CYP 1A2, CYP 2A6, CYP 2C9, CYP 2D6, CYP 2C19, CYP 2E1 и CYP 3A4. Установлено, что CYP 3A4, CYP 2D6 и CYP 2C9 сами ответственны (и в различной степени) за 90% окислительного метаболизма лекарственных продуктов. Хотя функциональная экспрессия этих изоформ регулируется и находится под влиянием целого ряда экологических и физиологических факторов, генетические факторы оказывают наиболее явно выраженное влияние, которое подчеркивает важную роль полиморфизма в окислении лекарственных продуктов. Некоторые из этих полиморфизмов были изучены (в частности, полиморфизмы CYP 2C19 и CYP 2D6). Более конкретно, была продемонстрирована клиническая важность полиморфизма CYP 2D6 в 4-гидроксилировании дебризохина (Clin. Pharmacol. Ther., 1991, 50, 233-238). Генетический полиморфизм CYP 2D6 ответственен за проблематичный метаболизм более чем 30 важных лекарственных продуктов и оказывает влияние на вплоть до 10% кавказского населения (медленные метаболисты). В настоящее время показано, что эта изоформа контролирует биотрансформацию лекарственных продуктов, таких как антиаритмические агенты, β-блокаторы, антигипертензивные агенты, агенты против стенокардии, нейролептики и антидепрессанты. За некоторым исключением, эти лекарственные продукты применяют в психиатрии и сердечно-сосудистой терапии для долгосрочного лечения.
Фармакокинетические результаты являются результатами сугубо количественного порядка: медленно метаболизирующие индивидуумы имеют уровень неизмененного продукта, который выше, чем у остальных. Эти количественные различия оказывают существенное клиническое воздействие на молекулы, которые имеют низкий терапевтический индекс.
Таким образом, генетика в значительной степени оказывает влияние на различия в эффективности и побочных эффектах, наблюдаемых среди индивидуумов. Поэтому важно определить, может ли метаболизм лекарственного продукта быть изменен в случае генетического дефицита фермента или нет.
В соответствии с настоящим изобретением обнаружены новые превосходные бензольные производные для сигма-рецепторов, в частности для сигма-рецепторов периферической нервной системы, которые обладают иммунодепрессивной активностью, но имеют низкую скорость метаболизма и/или небольшую степень или отсутствие вовлечения CYP 2D6 в окислительный процесс.
Соединения по данному изобретению также обладают противоопухолевой активностью и, в частности, они ингибируют пролиферацию раковых клеток.
Более того, продемонстрировано, что эти новые соединения обладают активностью в сердечно-сосудистой системе, более конкретно в контролировании частоты сердечных сокращений.
Соединения по данному изобретению также обладают активностью в отношении апоптоза.
Так, в одном из своих аспектов настоящее изобретение относится к соединениям формулы
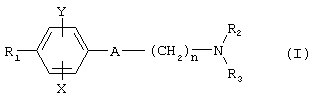
где А представляет собой группу, выбранную из следующих: -С≡С-, -СН=СН-; -СН2-СН2-;
n равно 1 или 2;
Х представляет собой атом водорода, хлора или фтора или метильную или метоксигруппу;
Y представляет собой атом водорода или атом хлора или фтора;
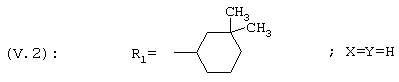
R1 представляет собой циклогексильную группу, монозамещенную, дизамещенную, тризамещенную или тетразамещенную метильной группой; фенильную группу, монозамещенную или дизамещенную атомом фтора или хлора или метоксигруппой; циклогептильную, трет-бутильную, дициклопропилметильную, бицикло[3.2.1]октанильную, 4-тетрагидропиранильную, 4-тетрагидротиопиранильную или 1-или 2-адамантильную или адамантан-2-ольную группу; либо R1 представляет собой фенильную группу, причем понятно, что в этом случае Х и Y иные, чем водород;
R2 представляет собой атом водорода или (С1-С4)алкильную группу, возможно замещенную трифторметильной группой;
R3 представляет собой (С5-С7)циклоалкил;
и солям этих соединений, образованным присоединением фармацевтически приемлемых кислот, а также их сольватам и гидратам.
Термин "алкил" означает линейный или разветвленный насыщенный углеводородный одновалентный радикал.
Термин "(С1-С4)алкил" означает алкильный радикал, включающий в себя от 1 до 4 атомов углерода.
В соответствии с еще одним аспектом данное изобретение относится к соединениям формулы (I), где А представляет собой группу, выбранную из следующих: -С≡С-, -СН=СН-, -СН2-СН2-;
n равно 1 или 2;
Х представляет собой атом водорода, хлора или фтора или метильную или метоксигруппу;
Y представляет собой атом водорода или атом хлора или фтора;
R1 представляет собой циклогексильную группу, монозамещенную, дизамещенную, тризамещенную или тетразамещенную метильной группой; фенильную группу, монозамещенную или дизамещенную атомом фтора или хлора или метоксигруппой; цикпогептильную, трет-бутильную, дициклопропилметильную, бицикло[3.2.1]октанильную, 4-тетрагидропиранильную, 4-тетрагидротиопиранильную или 1-или 2-адамантильную группу; либо R1 представляет собой фенильную группу, причем понятно, что в этом случае Х и Y иные, чем водород;
R2 представляет собой (С1-С4)алкильную группу, возможно замещенную трифторметильной группой;
R3 представляет собой (С5-С7)циклоалкил;
и солям этих соединений, образованным присоединением фармацевтически приемлемых кислот, а также их сольватам и гидратам.
В соответствии с еще одним аспектом данное изобретение относится к соединениям формулы
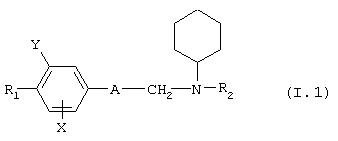
где А представляет собой группу, выбранную из следующих: -С≡С-, -СН=СН-, -СН2-СН2-;
Х представляет собой атом водорода или хлора;
Y представляет собой атом водорода или атом хлора;
R1 представляет собой циклогексил, монозамещенный, дизамещенный, тризамещенный или тетразамещенный метильной группой; фенильную группу, замещенную атомом хлора, метоксигруппой или одним или двумя атомами фтора; трет-бутильную или 1-или 2-адамантильную группу; либо R1 представляет собой фенильную группу, причем понятно, что в этом случае Х и Y оба представляют собой атом хлора;
R2 представляет собой (С2-С3)алкил;
и солям этих соединений, образованным присоединением фармацевтически приемлемых кислот, а также их сольватам и гидратам.
В соответствии с еще одним аспектом данное изобретение относится к соединениям формулы (I) и (I.1), в которых А представляет собой группу -СН=СН-в (Z) конфигурации, и солям этих соединений, образованным присоединением фармацевтически приемлемых кислот, а также их сольватам и гидратам. В соответствии с еще одним аспектом данное изобретение относится к соединениям, как они определены выше, в которых Х представляет собой атом хлора, а Y представляет собой атом водорода или хлора, и солям этих соединений, образованным присоединением фармацевтически приемлемых кислот, а также их сольватам и гидратам.
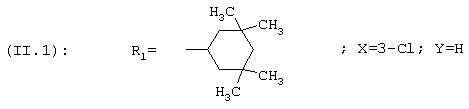
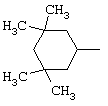
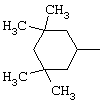
В соответствии с еще одним аспектом данное изобретение относится к соединениям, как они определены выше, в которых R1 представляет собой 3,3,5,5-тетраметилциклогексильную, или 3,3-диметилциклогексильную, или 4,4-диметилциклогексильную группу; фенильную группу, монозамещенную или дизамещенную атомом фтора, или замещенную в положении 4 атомом хлора; или 1-или 2-адамантильную группу; и солям этих соединений, образованным присоединением фармацевтически приемлемых кислот, а также их сольватам и гидратам.
Следующие соединения:
[(Z)-3-(4-адамантан-2-ил-3-хлорфенил)пропен-2-ил]циклогексилэтиламин;
[(Z)-3-(4-адамантан-2-илфенил)пропен-2-ил]циклогексилэтиламин;
{(Z)-3-[4-(4,4-диметилциклогексил)-2-хлорфенил]пропен-2-ил}-циклогексилэтиламин;
[(Z)-3-(4-адамантан-1-ил-3-хлорфенил)пропен-2-ил]циклогексилэтиламин;
[(Z)-3-(4-адамантан-2-ил-3,5-дихлорфенил)пропен-2-ил]циклогексилэтиламин;
[(Z)-3-(4-адамантан-2-ил-3,5-дихлорфенил)пропен-2-ил]циклогексил(2-метилэтил)амин;
а также их соли, образованные присоединением фармацевтически приемлемых кислот, их сольваты и гидраты составляют еще один аспект данного изобретения.
В частности, данное изобретение относится к [(Z)-3-(4-адамантан-2-ил-3,5-дихлорфенил)пропен-2-ил]циклогексилэтиламину, а также его солям, образованным присоединением фармацевтически приемлемых кислот, сольватам и гидратам.
Соли соединений по данному изобретению получают по методикам, хорошо известным специалистам в данной области техники.
Соли соединений формулы (I) по настоящему изобретению включают соли с неорганическими или органическими кислотами, которые дают возможность разделения или подходящей кристаллизации соединений формулы (I), а также фармацевтически приемлемых солей.
Подходящими кислотами, которые могут быть упомянуты, являются пикриновая кислота, щавелевая кислота или оптически активная кислота, например винная кислота, дибензоилвинная кислота, миндальная кислота или камфорсульфоновая кислота и кислоты, которые образуют физиологически приемлемые соли, такие как гидрохлорид, гидробромид, сульфат, гидросульфат, дигидрофосфат, малеат, фумарат, 2-нафталинсульфонат или пара-толуолсульфонат. Гидрохлориды являются наиболее предпочтительными из солей соединений формулы (I). В том случае, когда соединение по данному изобретению содержит один или более чем один асимметрический атом углерода, оптические изомеры этого соединения составляют неотъемлемую часть данного изобретения. Если соединение по данному изобретению проявляет стереоизомерию, например аксиально-экваториального типа или Z-E типа, то данное изобретение включает в себя все стереоизомеры данного соединения.
Настоящее изобретение включает в себя соединения формулы (I) в форме чистых изомеров, а также в форме смеси изомеров в любом соотношении. Соединения (I) выделяют в форме чистых изомеров традиционными способами разделения. Например, можно использовать фракционную перекристаллизацию соли рацемической смеси с оптически активной кислотой или основанием, принцип которой хорошо известен, или стандартные хроматографические способы на хиральной стационарной фазе или нехиральной стационарной фазе; например, можно использовать разделение на силикагеле или C18-привитом силикагеле, элюируя смесями, такими как хлорированные растворители/спирт. Вышеупомянутые соединения формулы (I) включают также соединения, в которых один или более чем атом водорода, углерода или галогена, в частности хлора или фтора, заменены их радиоактивным изотопом, например тритием или углеродом-14. Такие меченые соединения пригодны для исследований метаболизма или фармакокинетики в биохимических тестах в качестве лигандов рецептора.
Функциональные группы, которые могут находиться в молекуле соединений формулы (I) и в промежуточных соединениях, могут быть защищены, перманентно или временно, защитными группами, которые обеспечивают окончательный синтез ожидаемых соединений. Реакции введения и удаления защитных групп осуществляют способами, хорошо известными специалистам в данной области техники. Выражение "временная защитная группа для аминов, спиртов, фенолтиолов или карбоновых кислот" означает защитные группы, такие как группы, описанные в Protective Groups in Organic Synthesis, Greene T. W. and Wuts P. G. M., ed. John Willey and Sons, 1991 и в Protecting Groups, Kocienski P. J., 1994, Georg Thieme Verlag. Специалист в состоянии выбрать подходящие защитные группы. Соединения формулы (I) могут содержать группы-предшественники других функциональных групп, которые последовательно образуются на одной или более чем одной стадии.
Объектом настоящего изобретения также является способ получения соединений формулы (I), отличающийся тем, что:
1) когда А представляет собой группу -C≡C-,
а) либо, если n=1, проводят реакцию Манниха между фенилацетиленовым производным формулы
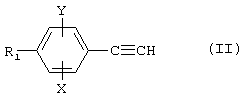
где R1, Х и Y являются такими, как определено для (I), формальдегидом и амином (1) HNR2R3, причем R2 и R3 являются такими, как определено для (I);
б) либо проводят реакцию сочетания Сузуки между соединением формулы

где X, Y, n, R2 и R3 являются такими, как определено для (I), a Z представляет собой бром, йод или трифторметансульфонатную группу (OTf), и производным бора (2) формулы R1-B(OR)2, где R представляет собой атом водорода или алкильную или арильную группу, в присутствии основания и металлического катализатора;
в) либо, когда R1 представляет собой циклогексильную группу, монозамещенную, дизамещенную, тризамещенную или тетразамещенную метильной группой; циклогептильную, 4-тетрагидропиранильную, 4-тетрагидротиопиранильную или адамантильную группу, проводят реакцию сочетания между соединением (1а), в котором Z представляет собой атом йода или брома, и кетоном (3), соответствующим R1, представленным  в присутствии основания с получением промежуточного соединения формулы
в присутствии основания с получением промежуточного соединения формулы
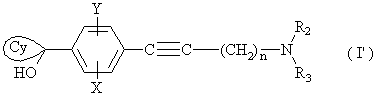
где X, Y, n, R2 и R3 являются такими, как определено для (I); указанное соединение (I’) затем восстанавливают в селективных условиях;
г) либо проводят реакцию сочетания между амином формулы

где n, R2 и R3 являются такими, как определено для (I), и соединением формулы

где R1, Х и Y являются такими, как определено для (I), а Z представляет собой атом брома или йода или трифторметилсульфонатную группу (трифлат, или OTf);
2) когда А представляет собой группу -СН=СН-, проводят гидрирование, водородом в момент выделения или в присутствии циклогексена, соединения формулы (I), где А представляет собой ацетиленовую группу -С≡С-, с получением этиленового соединения (I) в форме смеси Z и Е изомеров, либо это гидрирование проводят в присутствии металлического катализатора на носителе с получением этиленового соединения (I) в Z форме, либо, альтернативно, соединение (I), в котором А представляет собой ацетиленовую группу -С≡С-, подвергают взаимодействию с гидридом металла с получением этиленового соединения (I) в Е форме;
3) когда А представляет собой группу –СН2-СН2-, проводят гидрирование соединения (I), в котором А представляет собой группу -СН=СН-или -C≡C-.
Стадию 1а способа по изобретению проводят при нагревании, предпочтительно при температуре между 80°С и 90°С, в полярном растворителе, таком как 1,2-диметоксиэтан или 1,4-диоксан. Чтобы способствовать реакции конденсации, можно использовать катализатор, например металлическую соль, такую как хлорид меди II или хлорид меди III.
На стадии 1б данного способа реакцию сочетания Сузуки предпочтительно проводят между соединением (Ia), в котором Z представляет собой OTf, и производным бора (2) формулы R1-B(OH)2. Реакцию проводят в присутствии основания, такого как гидроксид, алкоксид, фосфат или карбонат щелочного или щелочно-земельного металла, более конкретно фосфат калия или карбонат натрия. Реакцию проводят в присутствии металлического катализатора, например катализатора на основе меди, олова или, предпочтительно, палладия, такого как тетракис(трифенилфосфин)палладий, возможно с использованием галогенида, такого как хлорид лития, действующего как сокатализатор. Способ осуществляют при нагревании при температуре между 60°С и 80°С в инертном растворителе, таком как толуол или 1,2-диметоксиэтан, или, предпочтительно, в двухфазной среде толуольный/водный раствор, возможно с долей спирта, такого как этанол. Реакция сочетания Сузуки исследована во многих публикациях, таких как, например, Synth. Commun., 1981, 11 (7), 513-519 и J. Org. Chem., 1993, 58 (8), 2201-2208. Бороновые кислоты (2) R1-B(OH)2 имеются в продаже или их синтезируют стандартным способом из соответствующих галогенопроизводных, предпочтительно бромопроизводных R1Br, путем воздействия например триметилборатом в присутствии основания, такого как трет-бутиллитий.
На стадии 1в реакцию сочетания предпочтительно проводят на соединении (Ia), где Z представляет собой атом брома, в присутствии основания, такого как н-бутиллитий, в инертном растворителе, предпочтительно диэтиловом эфире, при низкой температуре, предпочтительно в пределах от -80°С до -70°С. Восстановление (I’) до (I) проводят в селективных условиях, например в соответствии со способом, описанным в Tetrahedron, 1995, 51, 11043-11062, путем воздействия хлортриметилсиланом и йодидом натрия в смеси ацетонитрил/хлорированный растворитель, такой как дихлорметан, с последующей обработкой уксусной кислотой в присутствии цинка, либо, альтернативно, путем воздействия йодоводородной кислоты или путем ионного гидрирования под воздействием тетраборгидрида натрия в трифликовой (трифторметансульфоновой) кислоте.
На стадии 1 г данного способа реакцию сочетания проводят в присутствии палладиевого катализатора, одного или более чем одного третичного амина и возможно хлорида лития. Предпочтительно используют соединение (III), где Z представляет собой трифлат, и данный способ осуществляют в присутствии палладиевого катализатора, такого как тетракис(трифенилфосфин)палладий или дихлорбис(трифенилфосфин)палладий, и возможно сокатализатора, такого как йодид меди. Когда Z представляет собой трифлат, используют также хлорид лития. Эту реакцию сочетания предпочтительно проводят в присутствии триэтиламина и пиридина при температуре дефлегмации реакционной смеси. Для этого типа сочетания, известного как сочетание Соногашира, можно сослаться на J. Org. Chem., 1993, 58, 7368-7376 и 1998, 63, 1109-1118; Syn. Lett., 1995, 1115-1116 и Synthesis, 1987, 981. Для получения соединений (I), где А представляет собой группу -СН=СН-, в Z-форме, гидрирование обычно проводят в присутствии циклогексена и металлического катализатора на носителе, такого как палладий на сульфате бария или карбонате кальция, или никель Ренея, или, предпочтительно, катализатор Линдлара, в растворителе, который является инертным для этой реакционной среды, такого как петролейный эфир или спиртовой растворитель. Для получения соединений (I) в Е-форме предпочтительно используемый гидрид металла представляет собой диизобутилалюминия гидрид (DIBALH) в инертном растворителе, таком как толуол.
Для получения соединений (I), где А представляет собой группу -СН2-СН2-, гидрирование обычно проводят в спирте, например этаноле, в присутствии катализатора, такого как оксид платины или, предпочтительно, палладий-на-угле.
Относительно методов восстановления алкенов и алкинов, используемых выше, можно сослаться на "Catalytic Hydrogenation. Techniques and Applications in Organic Chemistry", Robert L. Augustine, 1965, Narcel Dekker, Inc. New York.
Общий способ получения соединений (I), где А представляет собой ацетиленовую группу -С≡С-, описан на схеме 1, представленной ниже.
СХЕМА 1

На схеме 1 А=-С≡С-, X, Y, n, R1, R2 и R3 являются такими, как определено для (I), R представляет собой атом водорода или алкильную или арильную группу, Z представляет собой атом брома или йода или трифлат, и, когда Z представляет собой бром или йод, Z’ представляет собой трифлат, или же Z’ представляет собой атом брома или йода. Важность природы заместителей Z и Z’ в реакции сочетания, обозначенной Путь Г, конкретизирована ниже.
Соединение (II) получают путем обработки хлоракролеина формулы
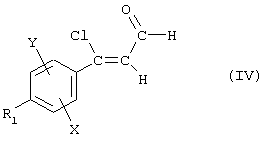
где X, Y и R1 являются такими, как определено для (I), в основной среде, предпочтительно воздействуя гидроксидом натрия в растворителе, таком как тетрагидрофуран или, предпочтительно, 1,4-диоксан, при температуре дефлегмации растворителя.
Хлоракролеин (IV) получают из ацетофенона формулы

где X, Y и R1 являются такими, как определено для (I), воздействуя комплексом Вилсмейера. Используют, например, хлорид (хлорметилен)диметиламмония, представляющий собой коммерческий комплекс Вилсмейера, или комплекс Вилсмейера, полученный из дизамещенного формамида, объединенного с оксалилхлоридом, оксихлоридом фосфора или фосгеном. Этот способ обычно осуществляют в хлорированном растворителе или эфире при температуре между -20°С и 40°С. Более конкретно, используют комплекс Вилсмейера, полученный из диметилформамида и оксалилхлорида в растворителе, таком как дихлорметан или 1,2-диметоксиэтан, при температуре между -10°С и 10°С.
Для этого типа реакции можно сослаться, например, на J. Chem. Soc. (С), 1970, 2484-2488 и Angew. Chem. Internat. Ed., 1963, 2, 98-99.
Ацетофеноны (V) известны или их получают известными способами, такими как способы, описанные в Gazz. Chim. Ital., 1949, 79, 453-457 и J. Am. Chem. Soc., 1947, 69,1651-1652.
Схема 2 иллюстрирует способы, используемые для получения соединений (V).
СХЕМА 2

На схеме 2 X, Y и R1 являются такими, как определено для (I), Су является таким, как определено для (I'), Z представляет собой атом брома или йода или OTf, R представляет собой атом водорода или алкильную или арильную группу, и Р представляет собой защитную группу для кетонной функциональной группы, такую как метил.
Соединения (V) могут быть получены непосредственно из соединений (Va) путем воздействия соединением бора R1-B(OH)2 (2), как описано для превращения из (Ia) в (I). Кетонная функциональная группа соединения (Va) также может быть защищена традиционным образом, например путем воздействия триалкилортоформиатом в соответствующем спирте в присутствии кислоты, такой как пара-толуолсульфоновая кислота.
Полученное таким образом соединение (Vp) подвергают взаимодействию с кетоном  в условиях, описанных для превращения из (Ia) в (I’). С кетонной функциональной группы удаляют защиту путем гидролиза в кислотной среде с получением соединения (V’). Указанное соединение (V’) затем восстанавливают в мягких условиях, описанных для превращения (I’) в (I).
в условиях, описанных для превращения из (Ia) в (I’). С кетонной функциональной группы удаляют защиту путем гидролиза в кислотной среде с получением соединения (V’). Указанное соединение (V’) затем восстанавливают в мягких условиях, описанных для превращения (I’) в (I).
В некоторых случаях, например когда R1 представляет собой 4,4-диметилциклогексильную или 4-тетрагидропиранильную группу, может образовываться промежуточное соединение формулы
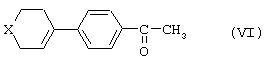
где Х=О или -С(СН3)2, которое после предварительной защиты кетонной функциональной группы и гидрирования, например, в присутствии палладия-на-угле в метаноле и последующего удаления защиты с кетонной функциональной группы кетона дает целевое соединение (V).
Соединение (V), где Х и/или Y иные, чем водород, может быть получено из соединений (V), где Х=Y=Н, способами, хорошо известными специалистам в данной области. Например, когда Х и/или Y представляет собой атом хлора, хлорирование ароматического ядра проводят путем воздействия газообразным хлором в присутствии кислоты Льюиса, предпочтительно трихлорида алюминия, в хлорированном растворителе, таком как дихлорметан, предпочтительно при 0°С.
Соединения (Va) также имеются в продаже или могут быть получены способами, известными специалистам в данной области.
Например, когда Z представляет собой трифлат, соединение (Va) может быть получено, как изображено на схеме 3:
СХЕМА 3

На схеме 3 Х и Y являются такими, как определено для (I). Соединения (VIII) имеются в продаже или их получают стандартными способами.
В соответствии с еще одним из аспектов объектом настоящего изобретения также являются соединения формулы (Iа)

где X, Y, n, R2 и R3 являются такими, как определено для (I), а Z представляет собой атом брома, или атом йода, или OTf. Эти соединения являются новыми и являются ключевыми промежуточными соединениями в синтезе соединений (I).
Настоящее изобретение также относится к способу получения производных (Iа), отличающемуся тем, что:
- либо, когда n=1, проводят реакцию Манниха между фенилацетиленовым производным формулы

где X и Y являются такими, как определено для (I), a Z представляет собой атом брома, или атом йода, или OTf, формальдегидом и амином (1) HNR2R3;
- либо проводят реакцию сочетания между амином формулы

где R2, R3 и n являются такими, как определено для (I), и производным формулы
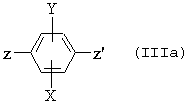
где Х и Y являются такими, как определено для (I), Z представляет собой атом брома, или йода, или трифлат, и Z’ представляет собой атом брома или йода, если Z представляет собой трифлат, в противном случае Z’ представляет собой трифлат, в присутствии палладиевого катализатора, одного или более чем одного третичного амина и возможно хлорида лития.
Реакцию Манниха проводят в таких же условиях, как условия, описанные для превращения из (II) в (I).
Реакцию Соногашира, описанную для сочетания соединений (III) и (4), используют для сочетания соединений (IIIa) и (4). Когда Z представляет собой трифлат, а Z’ представляет собой атом брома или йода, способ осуществляют в отсутствие хлорида лития. С другой стороны, когда Z представляет собой атом брома или йода, а Z’ представляет собой трифлат, способ осуществляют в присутствии хлорида лития. Использование хлорида лития создает возможность управлять реакцией сочетания.
Пропаргиламины (4) (в случае, когда n=1) получают традиционным образом, например согласно Tetrahedron Lett. 1989, 30 (13), 1679-1682, начиная с амина (1) HNR2R3 и 3-бромпропина, путем воздействия карбонатом калия в ацетонитриле при температуре между 50°С и 80°С.
Соединения (III), где Z=OTf, обычно традиционно получают из соответствующих спиртов формулы
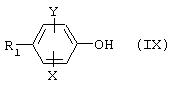
где X, Y и R1 являются такими, как определено для (I), путем воздействия трифторметансульфонового ангидрида (трифликового ангидрида) в пиридине. Сами спирты (IX) получают из соединений формулы

где Z" представляет собой атом брома или йода, способами, описанными ранее для превращения (Iа) в (I) или (Va) в (V). Соединения (IХа) имеются в продаже или их получают способами, которые хорошо известны специалистам в данной области.
Соединение (IIа) получают из хлоракролеина формулы

где Х и Y являются такими, как определено для (I), а Z представляет собой атом брома или йода или OTf, который сам получают из ацетофенона формулы
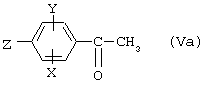
где X, Y и Z являются такими, как определено выше для (IVa), способами, описанными для превращения (IV) в (II) и (V) в (IV).
Соединения по изобретению были подвергнуты биохимическим и фармакологическим исследованиям. Соединения формулы (I) и их фармацевтически приемлемые соли, гидраты и сольваты специфически связываются с сигма-рецепторами, в частности с сигма-рецепторами периферической нервной системы, также известными как рецепторы сигма-2.
Аффинность в отношении рецепторов сигма-1 исследовали in vitro на мембранах головного мозга морских свинок с использованием 3H-(+)-3PPP в качестве лиганда согласно De Haven-Hudkins et al., Life Science, 1993, 53, 41-48. (+)-Пентазоцин специфически связывается с рецепторами сигма-1. Фрагмент мембраны головного мозга морских свинок готовят обычными способами. Препарат мембран (0,3 мг белка/мл) инкубируют в течение 150 минут при 37°С в присутствии 0,5 нМ [3Н]-(+)-пентазоцина. Неспецифическое связывание определяют в присутствии 10 мкМ (+)-пентазоцина. Мембраны затем фильтруют и промывают 3 раза. Отфильтрованный материал анализируют с целью определения специфически связанной фракции [3H]-пентазоцина. В этих условиях соединения по изобретению, примеры которых следуют ниже, имеют значения ИК50 между 0,1 нМ и 100 нМ.
Способность соединений по изобретению взаимодействовать с рецепторами сигма-2 тестировали in vitro на мембранах селезенки крыс, используя в качестве лиганда [3H]-DTG, в соответствии с R. Paul et al. Journal of Neuroimmunology, 1994, 52, 183-192. Препарат мембран (1 мл) инкубируют с 2 нМ [3H]-DTG в течение 90 минут при 20°С. Величину неспецифического связывания оценивают в присутствии 10 мкМ DTG или галоперидола. Мембраны фильтруют и промывают дважды, и отфильтрованный материал анализируют с целью определения количества специфически связанного [3Н]-DTG. Соединения по изобретению обладают активностью в отношении сигма-2 между 1 нМ и 500 нМ.
Соединение примера 44, приведенного ниже, тестировали на связывание на мембранах, извлеченных из клеток млекопитающих и дрожжей, экпрессирующих либо сигма-1, сигма-2, либо стерин-изомеразу человека (Human sterol isomerase (HIS)). Результаты этих испытаний показывают, что соединения по изобретению проявляют очень высокую аффинность к рецепторам сигма-1 и сигма-2 и HIS. В приведенной ниже таблице представлены данные по аффинности соединения примера 44 к указанным рецепторам и HIS по сравнению с данными для известных из предшествующего уровня техники лигандов сигма-рецепторов.
Лиганды/рецепторы
Из таблицы видно, что аффинность типичного представителя заявленных соединений (соединения примера 44) значительно выше аффинности других классических лигандов сигма-рецепторов, которые не связываются с указанными тремя рецепторами одновременно.
1 - Соединения по изобретению также испытывали в тестах на иммунодепрессивную активность.
D-Галактозамин, SEB (энтеротоксин В Staphylococcus) и LPS (липополисахариды) были получены от Sigma Chemical Co (St Louis, МО). SEB содержит менее 0,00029% эндотоксина (тест "лизат амебоцитов лимулус", Bioproduct, Walkersville, MD). Эти молекулы растворяют в фосфатном буферном растворе; соединения по изобретению растворяют в растворе, содержащем 5% этанола, 5% Твин 80 и 90% воды.
Используют самок мышей Balb/C возрастом от 6 до 8 недель, доставленных из питомника Charles River (Франция) и самок мышей C57BL/6 и B6D2F1 возрастом 8 недель, доставленных из питомника IFFA CREDO (Domaine des Oncins, BP 0109, 69592 L'Arbresle Cedex, Франция).
Измерение цитокинов: 5 мышам внутрибрюшинно инъецируют соединения или один растворитель за 30 минут до введения LPS (10 мкг/мышь внутривенно) или перорально за 1 час до LPS. Образцы крови отбирают путем ретро-глазничной или сердечной пункции через 1 час 30 минут после инъекции LPS. Образцы центрифугируют и получают сыворотку крови. Сыворотку крови хранят при -80°С до анализа. Содержание TNF-α и IL-10 определяют с помощью набора ELISA (Genzyme, Cambridge). Тесты проводят согласно инструкциям по использованию.
Токсический шок: Соединения вводят внутрибрюшинно 10 животным. Через 30 минут SEB (Staphylococcus энтеротоксин В, Sigma St. Louis, МО) вводят в количестве 10 мкг/мышь внутривенно, и вводят D-галактозамин (20 мг/мышь, внутрибрюшинно).
Смерть наблюдают через 48 часов.
Болезнь ТПХ (трансплантат-против-хозяина): Тестируемые соединения или только растворитель (в качестве контроля) инъецируют самкам B6D2F1 (H2b × H2d) мышей внутрибрюшинно. Через 4 часа им инъецируют 7,5×107 C57BL/6 (H2b) мононуклеарных клеток селезенки мыши, чтобы инициировать ТПХ. Всех животных умерщвляют через одну неделю после трансплантации и измеряют увеличение массы их селезенки, вызванное ТПХ. Рассчитывают следующий показатель:
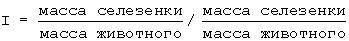
Результаты выражают следующим образом:

где PS - процент спленомегалии.
Измерение Т клеточной пролиферации: Суспензии клеток готовят, используя селезенки мышей Balb/C. Красные кровяные тельца сначала лизируют в ходе краткого гипотонического шока, достигаемого с использованием стерильной дистиллированной воды. Остальные клетки (белые кровяные тельца) дважды промывают культуральной средой (RPMI 1640, содержащей 2% термически инактивированной сыворотки эмбрионов коров, 2 мМ L-глутамина, 1 мМ пирувата натрия, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и 15 мМ PIPES (пиперазин-N,N-бис(2-этансульфоновой кислоты)), заблаговременно доведенной до рН 6,6. Жизнеспособность клеток, определяемая с использованием трипанового синего, всегда превышает 95% при этом способе приготовления. Спленоциты в концентрации 6×106 клеток/мл культивируют с тестируемыми продуктами в плоскодонных 96-луночных планшетах (Falcon, Becton Dickinson, Lincoln Park, NJ) в присутствии 2 мкг/мл SEB. Четыре лунки готовят для каждой концентрации тестируемых продуктов. Инкубацию проводят при 37°С в инкубаторе для клеточных культур (атмосфера: 95% воздух + 5% СO2) в течение 4 дней. Затем в каждую культуральную лунку добавляют 2 мкКи тритиированного тимидина (Amersham, Les Ullis, Франция). Через четыре часа клетки собирают на стекловолокнистом фильтре (Filtermat A, Wallac, Turku, Финляндия), используя скатрон (Pharmacia LKB, Piscataway, NJ). Радиоактивность, включенную и связанную на фильтре, измеряют в подходящем жидкостном сцинтилляционном счетчике (Betaplate, Pharmacia LKB).
Согласно результатам, наблюдаемым во время этих биохимических тестов и поведенческих тестов, соединения по изобретению проявляют иммунодепрессивную активность.
2 - Соединения по изобретению также были подвергнуты испытаниям, демонстрирующим их способность ингибировать пролиферацию опухолевых клеток и раковых клеток.
Измерение пролиферации клеток MDA/MB231 (гормоннезависимого рака груди): Клетки MDA/MB231 сохраняют in vitro путем последовательных переносов в среде Игла, модифицированной по Дульбекко (DMEM) (Gibco Laboratories, Grant Island, NY), содержащей 10% термически инактивированной сыворотки крови эмбрионов коров, 1 мМ пирувата натрия, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина.
Для измерения пролиферации клетки в концентрации 2×105 мл культивируют с тестируемыми продуктами в среде RPMI 1640, содержащей 10 мкг/мл бычьего инсулина (Sigma) и 10 мкг/мл апотрансферрина (Sigma) в плоскодонных 96-луночных планшетах (Falcon, Becton Dickinson, Lincoln Park, NJ). По три лунки готовят для каждой концентрации тестируемых продуктов. Инкубацию проводят при 37°С в инкубаторе для клеточных культур (атмосфера: 95% воздух + 5% СO2) в течение 4 дней. Затем в каждую культуральную лунку добавляют 2 мкКи тритиированного тимидина (Amersham, Les Ullis, Франция). Через двадцать четыре часа клетки отделяют, используя трипсин-EDTA (этилендиаминтетраацетат) (Gibco), и собирают на стекловолокнистом фильтре (Filtermat A, Wallac, Turku, Финляндия), используя скатрон (Pharmacia LKB, Piscataway, NJ). Радиоактивность, включенную и связанную на фильтре, измеряют в подходящем жидкостном сцинтилляционном счетчике (Betaplate, Pharmacia LKB).
3 - Соединения по данному изобретению также были подвергнуты испытаниям, демонстрирующим их ценность в сердечно-сосудистой системе.
Антиаритмические эффекты соединений по изобретению тестировали на реинфузионных аритмиях у анестезированных крыс. Эксперимент проводили на самцах крыс Sprague Dawley с нормальным давлением крови, весящих от 250 до 300 г. Эти животные были доставлены из питомника IFFA CREDO. Животных содержали в стандартных лабораторных условиях и кормили стандартной пищей: АO4 (UAR). Воду давали без ограничений. Метод окклюзии и реперфузии, используемый в этом исследовании, соответствует методам, описанным Manning et al. (Circ. Res., 1984, 55, 545-548) и Kane et al. (Br. J. Pharmacol., 1984, 82, 349-357), модифицированным незначительно.
Животных анестезировали пентобарбиталом натрия в дозе 60 мг/кг внутрибрюшинно, делали им трахеотомию и вентилировали окружающим воздухом (Harward респиратор). Катетер (РЕ10) помещали в яремную вену для внутривенной инъекции тестируемых продуктов. Иглы для подкожных впрыскиваний размещали на четырех лапах животного для записи электрокардиограммы (ЭКГ), обычно DII (Gould ES1000 или на полиграфе Astromed 7400). После выполнения торакотомии на левую переднюю нисходящую коронарную артерию у ее начала накладывали нить для перевязывания артерии. Два конца этой нити пропускали через пластиковую трубку, которую помещали на поверхности сердца непосредственно над коронарной артерией. Коронарную артерию перекрывали, затягивая концы нити в течение 5 минут, и проводили реинфузию, ослабляя затягивание. Температуру животного контролировали и поддерживали при 37°С с помощью гомеотермального покрывала.
Для исследования внутривенного пути продукты растворяли в смеси 75% ПЭГ-400/дистиллированная вода и инъецировали за 5 минут до перевязывания артерии. Продукты инъецировали в объеме 0,1 мл/100 г массы крысы. Контрольная группа получала этот растворитель. Для исследования перорального пути продукты суспендировали в 0,6% метилцеллюлозе и вводили животному, находящемуся в сознании, принудительным кормлением за 120 минут до перевязывания артерии. Продукты вводили в объеме 1 мл/100 г массы крысы. Контрольная группа получала этот растворитель.
Следующие виды аритмий анализировали ЭКГ (электрокардиографией) в ходе реинфузии (исследование, длящееся 10 минут) соласно Ламбетовским соглашениям (Lambeth Conventions) (Cardiovasc., Res., 1988, 22,447-355):
- желудочковые экстрасистолы (ЖЭС),
- желудочковая тахикардию (ЖТ), причем ЖТ представляет собой последовательность по меньшей мере четырех ЖЭС,
- желудочковая фибрилляция (ЖФ),
- и смертность из-за фатальной желудочковой фибрилляции или из-за остановки сердца.
Эти аритмии выражали в виде процента животных, проявляющих это событие (частота).
Животных разделяли на группы из 4-10 животных. Каждое животное получало только одну дозу продукта. Как при внутривенном, так и при пероральном введении эти продукты защищают животное от реинфузионных аритмий, снижая или устраняя смертность и частоту ЖФ. Кроме того, некоторые продукты снижают и/или устраняют частоту ЖФ и ЖЭС, когда их вводят внутривенно.
Вовлечение CYP 2D6 можно продемонстрировать исследованиями метаболизма in vitro на микросомальных фракциях печени человека. Наиболее часто используемая концепция представляет собой ингибирование фермента его специфическим ингибитором хинидином, используемым при 20-кратном значении его Ki, причем Кi представляет собой абсолютное значение константы ингибирования активного начала относительно фермента.
Различные модели позволяют продемонстрировать на конкретных метаболических реакциях вовлечение CYP 2D6.
- Можно использовать микросомальные фракции печени человека, которые содержат все печеночные изоформы человека, инкубируемые в присутствии окислительно-восстановительного кофактора (НАДФН) (никотинамидадениндинуклеотидфосфат) и в отсутствие или в присутствии хинидина при 20-кратном значении его Ki относительно CYP 2D6. Уменьшение метаболизма, наблюдаемое в присутствии хинидина, может быть ассоциировано с ингибированием изоформы CYP 2D6, тем самым обеспечивая его возможное вовлечение в исследуемый метаболический путь(и). Можно также использовать микросомальные фракции, полученные из трансфецированных клеток, которые экспрессируют только одну изоформу цитохрома Р-450 человека (GENTEST Corp.). Можно также использовать гепатоциты человека в первичной культуре, которая способна осуществлять метаболические реакции фазы I и II. В этом случае инкубации осуществляют кинетически в течение 24 часов в присутствии и в отсутствие хинидина, представляющего собой мощный и специфичный ингибитор CYP 2D6. Сослаться можно на J. Pharm. Exp. Ther., 1996, 277. 321-332.
В частности, соединения по изобретению исследовали следующим образом.
- Указанное соединение инкубируют с печеночными микросомальными фракциями человека и НАДФН (окислительно-восстановительного кофактора), а также в присутствии или в отсутствие хинидина. Степень ингибирования метаболизации, наблюдаемая в присутствии хинидина, отражает вовлечение CYP 2D6 в метаболизацию указанного соединения. Этот подход можно использовать, когда метаболизация на печеночных микросомальных фракциях имеет достаточную амплитуду (т.е. больше или равна 10% от количества исходного субстрата).
- Когда метаболизация указанного соединения на микросомах печени слишком незначительна, чтобы служить для точной количественной оценки ингибирования, или когда необходимы дополнительные верификации, в течение 24 часов кинетически осуществляют дополнительные, более глубокие исследования на гепатоцитах человека в первичной культуре. Степень вовлечения CYP 2D6 в общую печеночную метаболизацию затем выявляют по уменьшению характеристического клиренса указанного соединения, возможно наблюдаемого в присутствии хинидина.
- Полученные результаты демонстрируют, что соединения по изобретению обладают низкой степенью метаболизации и/или небольшой степенью вовлечения CYP 2D6 в окислительный процесс.
Было также установлено, что биодоступность соединений по изобретению (в частности, соединения примера 44) у животных и у человека лучше, чем биодоступность близкого по структуре и активности известного (WO 9804251 и пример 5 ЕР 0376850) соединения SR 31747. Экспозицию соединения примера 44 и соединения SR 31747 оценивали в контрольной группе мышей в коллаген-индуцированной модели артрита. Для одной и той же дозы было обнаружено, что экспозиция соединения примера 44 в 10 раз выше, чем экспозиция известного соединения SR 31747. Эти результаты свидетельствуют о лучшем всасывании и/или более низкой степени метаболизации заявленных соединений.
Никаких признаков токсичности не наблюдается при использовании соединений по изобретению в фармакологически активных дозах, и их токсичность, таким образом, совместима с их применением в качестве лекарственных продуктов.
Соединения по настоящему изобретению особенно полезны, и их можно с пользой применять в качестве лекарственных продуктов, в частности, для лечения состояний, при которых желательно снижение иммунологической активности, а также состояний, ассоциированных с воспалительными расстройствами. В качестве не ограничивающих примеров могут быть упомянуты состояния с аутоиммунными компонентами, такие как, например, ревматоидный артрит, системная красная волчанка, состояния, вызванные демиелинизацией, например множественный склероз, болезнь Крона, атопический дерматит, диабет или реакции отторжения трансплантата, реакция "трансплантат-против-хозяина", состояния органного трансплантата, или, альтернативно, аутоиммунный увеит, увеоретинит, болезнь Бехчета, атеросклероз, астма, фиброзные заболевания, идиопатический фиброз легкого, кистозный фиброз, гломерулонефрит, некоторые спондилоартропатии, ревматоидный спондилит, остеоартрит, подагра, резорбция кости и хряща, остеопороз, болезнь Педжета, септический шок, септицемия, эндотоксический шок, респираторный дистресс-синдром взрослых, силикоз, асбестоз, легочный саркоидоз, язвенный колит, амиотрофический боковой склероз, болезнь Альцгеймера, болезнь Паркинсона, диссеминированная красная волчанка, гемодинамический шок, ишемические патологии (инфаркт миокарда, ишемия миокарда, спазм коронарных сосудов, стенокардия, сердечная недостаточность, сердечный приступ), постишемические реинфузионные приступы, малярия, микобактериальные инфекции, менингит, лепра, вирусные инфекции (вирус иммунодефицита человека (ВИЧ), цитомегаловирус, вирус герпеса), связанные со СПИДом оппортунистические инфекции, туберкулез, псориаз, атопический дерматит и контактный дерматит, кахексия и повреждение, связанное с радиацией.
Соединения по изобретению можно также использовать в терапии любого патологического процесса, который вызывает пролиферацию опухолевых клеток. Эта пролиферация клеток может быть либо гормон-чувствительной, либо гормон-нечувствительной. Более конкретно, клинические применения, для которых может быть показано применение этих соединений, включают состояния, являющиеся результатом пролиферации опухолевых клеток, в частности глиобластомы, нейробластомы, лимфомы, миеломы, меланомы, лейкемию, карциномы толстой кишки и колоректальные, эпителиальные, печеночные, легочные карциномы, карциномы молочной железы, яичников, поджелудочной железы, мочевого пузыря или простаты. Таким образом, соединения по изобретению с пользой можно применять в качестве лекарственных продуктов, предназначенных для борьбы с пролиферацией опухолевых клеток, в частности в качестве противоопухолевых агентов или противораковых агентов.
Их можно использовать также в сердечно-сосудистой области, более конкретно для лечения нарушений частоты сердечных сокращений.
Соединения по изобретению можно также использовать в терапии любого патологического процесса, который вызывает пролиферацию опухолевых клеток. Эта пролиферация клеток может быть либо гормон-чувствительной, либо гормон-нечувствительной. Более конкретно, клинические применения, для которых может быть показано применение этих соединений, включают состояния, являющиеся результатом пролиферации опухолевых клеток, в частности глиобластомы, нейробластомы, лимфомы, миеломы, меланомы, лейкемию, карциномы толстой кишки и колоректальные, эпителиальные, печеночные, легочные карциномы, карциномы молочной железы, яичников, поджелудочной железы, мочевого пузыря или простаты. Таким образом, соединения по изобретению с пользой можно применять в качестве лекарственных продуктов, предназначенных для борьбы с пролиферацией опухолевых клеток, в частности в качестве противоопухолевых агентов или противораковых агентов.
Их можно использовать также в сердечно-сосудистой области, более конкретно для лечения нарушений частоты сердечных сокращений.
Соединения по изобретению могут быть также очень полезны, благодаря их нейропротективной активности, а также их активности в отношении апоптоза.
Применение соединений по изобретению для лечения упомянутых выше состояний, а также для приготовления лекарственных продуктов, предназначенных для лечения указанных состояний, составляет неотъемлемую часть данного изобретения.
Объектом настоящего изобретения, таким образом, также являются фармацевтические композиции, содержащие соединение по изобретению или его фармацевтически приемлемую соль, сольват или гидрат и подходящие эксципиенты.
Указанные эксципиенты выбирают в соответствии с фармацевтической формой и желаемым способом введения.
В фармацевтических композициях по настоящему изобретению для перорального, подъязычного, подкожного, внутримышечного, внутривенного, местного, внутритрахеального, интраназального, трансдермального, ректального или внутриглазного введения активные начала формулы (I), приведенные выше, или их возможные соли, сольваты или гидраты можно вводить животным и людям для профилактики или лечения указанных выше расстройств или состояний в виде стандартных лекарственных форм, смешанных с обычными фармацевтическими носителями. Подходящие стандартные формы для введения включают пероральные формы, такие как таблетки, гелевые капсулы, порошки, гранулы и пероральные растворы или суспензии, формы для подъязычного, трансбуккального, внутритрахеального и интраназального введения, формы для подкожного, внутримышечного или внутривенного введения и формы для ректального введения. Для местного применения соединения по изобретению можно использовать в составе кремов, мазей, лосьонов или глазных капель.
Для получения желаемого профилактического или терапевтического эффекта доза активного начала может составлять от 0,2 мг до 15 мг на кг массы тела в сутки.
Каждая стандартная доза может содержать от 10 мг до 300 мг, предпочтительно от 25 мг до 75 мг, активных ингредиентов в комбинации с фармацевтическим носителем. Эту стандартную дозу можно вводить от 1 до 5 раз в сутки, так чтобы введенная суточная доза составляла от 10 мг до 1500 мг, предпочтительно от 25 мг до 375 мг.
При приготовлении твердой композиции в форме таблеток основной активный ингредиент смешивают с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик и тому подобное. Таблетки могут быть покрыты сахарозой, производным целлюлозы или другими подходящими веществами, или, альтернативно, они могут быть обработаны так, чтобы обладать пролонгированной или замедленной активностью и чтобы непрерывно высвобождать предопределенное количество активного начала.
Препарат в виде гелевых капсул получают смешиванием активного ингредиента с носителем и заполнением полученной смесью мягких и твердых гелевых капсул. Препарат в форме сиропа или эликсира или для введения в форме капель может содержать активный ингредиент вместе с подсластителем, предпочтительно некалорийным подсластителем, метилпарабеном и пропилпарбеном в качестве антисептика, а также с усилителем вкуса и аромата и подходящим красителем.
Диспергируемые в воде порошки или гранулы могут содержать активный ингредиент, смешанный с диспергирующими агентами, увлажняющими агентами или суспендирующими агентами, такими как поливинилпирролидон, а также с подсластителями или усилителями вкуса и аромата.
Для ректального введения используют суппозитории, которые изготавливают с использованием связующих, плавящихся при ректальной температуре, например масла какао или полиэтиленгликолей.
Для парентерального введения используют водные суспензии, изотонические солевые растворы или инъецируемые стерильные растворы, которые содержат фармакологически приемлемые диспергирующие агенты и/или увлажняющие агенты, например пропиленгликоль или бутиленгликоль.
Активное начало может быть приготовлено также в форме микрокапсул, возможно с использованием одного или более чем одного носителя или добавок, или, альтернативно, с использованием матриц, таких как полимер или циклодекстрин (пластырь, формы с пролонгированным высвобождением).
Наряду с продуктами формулы (I), приведенной выше, или их фармацевтически приемлемыми солями, сольватами и гидратами, композиции по настоящему изобретению могут содержать другие активные начала, которые можно использовать для лечения осложнений или состояний, указанных выше.
Таким образом, объектом настоящего изобретения также являются фармацевтические композиции, содержащие несколько активных начал в комбинации, одно из которых представляет собой соединение по изобретению.
Подготовительные примеры и примеры, приведенные ниже, иллюстрируют данное изобретение, однако не ограничивают его. Температуры плавления измерены методом 
Спектры ядерного магнитного резонанса получены в диметилсульфоксиде, если это не оговорено особо, при 200 МГц, и химические сдвиги выражены в млн-1.
Использованные ниже сокращения:
s=синглет; m=мультиплет; d=дуплет; t=триплет; q=квартет.
Фенильная группа в соединениях (I) стандартно нумерована следующим образом:

Подготовительный пример 1
1-Бром-4-(1,1-диметоксиэтил)бензол, соединение Vp
(Vp):X=Y=H; Z=Br; P=CH3
Смесь 19,905 г 1-(4-бромфенил)этанона, 101,4 мл метанола, 0,22 г гидрата пара-толуолсульфоновой кислоты и 19,9 мл триметилортоформиата перемешивают в течение 6 часов при комнатной температуре. Раствор нейтрализуют 1% раствором гидроксида калия в метаноле и концентрируют при пониженном давлении. Полученное масло переносят в петролейный эфир, осадок удаляют фильтрацией, и фильтрат упаривают при пониженном давлении. Соединение IVp очищают дистилляцией; выход=96%; точка кипения (т.кип.)=82°С (при давлении 0,03 мбар (3 Па)).
Подготовительный пример 2
4,4-Диметилциклогексанон, соединение 3.1
а) 4,4-Диметилциклогекс-2-енон
1 мл концентрированной серной кислоты добавляют при комнатной температуре к 81 мл бут-3-ен-2-она и 88 мл 2-метилпропиональдегида в 450 мл бензола, после чего реакционную смесь кипятят с обратным холодильником в течение 13 часов для удаления воды путем азеотропного уноса. После охлаждения до комнатной температуры реакционную смесь промывают насыщенным водным раствором бикарбоната натрия, а затем водой. Органическую фазу сушат над сульфатом магния, и растворители выпаривают при пониженном давлении. После дистилляции выделяют 31,1 г ожидаемого соединения; т.кип.=82°С (при давлении 22 мбар (2200 Па)).
б) 31,1 г 4,4-диметилциклогекс-2-енона в 100 мл пентана гидрируют в автоклаве при давлении 5 бар (500 Па) в присутствии 1,6 г 5% палладия-на-угле. Реакционную смесь фильтруют и растворитель выпаривают при пониженном давлении.
Подготовительный пример 3
4-Бром-3,5-дихлорфенол, соединение IXa.1
а) N-(3,5-Дихлорфенил)ацетамид
200 мл пиридина по каплям добавляют к 100 г 3,5-дихлорфениламина в 3000 мл хлороформа, а затем добавляют 90 мл уксусного ангидрида. Реакционную смесь перемешивают в течение 12 часов при комнатной температуре. Растворители выпаривают при пониженном давлении и полученный остаток перекристаллизовывают из 1000 мл этилацетата; точка плавления (т.пл.)=182°С.
б) N-(4-Бром-3,5-дихлорфенил)ацетамид
21,3 мл брома, растворенного в 82 мл уксусной кислоты, добавляют в течение 6 часов к 84,86 г N-(3,5-дихлорфенил)ацетамида и 34 г ацетата натрия в 420 мл уксусной кислоты. После 12 часов при комнатной температуре реакционную смесь нагревают в течение 5 часов при 50°С. Растворители выпаривают при пониженном давлении. Полученный остаток перекристаллизовывают из изопропанола; т.пл.=224°С.
в) 4-Бром-3,5-дихлорфениламин
202 г N-(4-бром-3,5-дихлорфенил)ацетамида и 220 г гидроксида натрия (в виде 50% водного раствора) в 670 мл этиленгликоля перемешивают в течение 5 часов при 120°С, а затем в течение 12 часов при комнатной температуре. Добавляют 3000 мл воды, смесь фильтруют, органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученный остаток перекристаллизовывают из циклогексана; т.пл.=132°С.
г) 100 г 4-Бром-3,5-дихлорфениламина при перемешивании при 5°С добавляют к смеси 125 мл воды и 90 мл концентрированной серной кислоты. В реакционную смесь добавляют 230 г дробленого льда, а затем 29 г нитрита натрия в 70 мл воды и эту реакционную смесь оставляют перемешиваться в течение 15 минут. Реакционную смесь быстро добавляют к смеси, состоящей из 280 мл концентрированной серной кислоты и 200 мл воды, нагретой до 160°С, и эту реакционную смесь оставляют перемешиваться при 160°С в течение 1 часа. Реакционную смесь выливают на воду/дробленый лед и экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью циклргексан/дихлорметан 4/6 (об./об.).
1H ЯМР: 10,5 (s,1H); 7,0 (s,2H).
Подготовительный пример 4
1-[4-(1-Гидрокси-3,3,5,5-тетраметилциклогексил)фенил]этанон, соединение V'.1

27,5 мл 1,6 М раствора н-бутиллития в гексане по каплям добавляют при -78°С к раствору 10 г 1-бром-4-(1,1-диметоксиэтил)бензола (соединение Vp) в 100 мл тетрагидрофурана. Реакционную смесь перемешивают в течение 2 часов при этой температуре. В течение 20 минут добавляют раствор 6,92 мл 3,3,5,5-тетраметилциклогексанона в 20 мл тетрагидрофурана, и эту реакционную смесь перемешивают в течение 1 часа при -78°С. После нагревания до комнатной температуры добавляют 140 мл насыщенного водного раствора хлорида аммония. После расслоения водную фазу экстрагируют диэтиловым эфиром, органические фазы объединяют и сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученное масло очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 95/5 (об./об.); выход=88%; т.пл.=135°С.
Следующие соединения получены таким же способом:
1-[4-(Гидрокси-3,3-диметилциклогексил)фенил]этанон, соединение V'.2

т.пл.=99°С.
1-[4-(Гидроксиадамантан-2-ил)фенил]этанон, соединение V’.3

1H ЯМР: 7,9 (d, 2H); 7,6 (d, 2H); 4,8 (s, 1H); 2,6-1,4 (m, 18H).
1-[4-(Гидрокси-4,4-диметилциклогексил)фенил]этанон, соединение V’.4
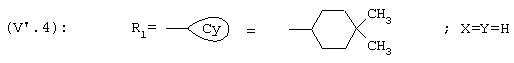
т.пл.=88°С.
Подготовительный пример 5
1-[4-(3,3,5,5-Тетраметилциклогексил)фенил]этанон, соединение V.1

38,1 мл хлортриметилсилана в течение 45 минут добавляют к раствору 40,45 г 1-[4-(гидрокси-3,3,5,5-тетраметилциклогексил)фенил]этанона (соединение V'.1) и 56,21 г йодида натрия в 230 мл безводного ацетонитрила. Во время добавления температуру поддерживают между 35°С и 40°С. После перемешивания в течение 2 часов добавляют 40 мл ацетонитрила и 39,4 мл уксусной кислоты. Затем порциями при перемешивании при комнатной температуре добавляют 29,4 г тонкоизмельченного цинка. Смесь кипятят с обратным холодильником при энергичном перемешивании в течение 4 часов. После охлаждения до комнатной температуры реакционную среду фильтруют через целит, а затем промывают насыщенным водным раствором бикарбоната натрия. Органическую фазу концентрируют при пониженном давлении и полученное масло очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 95/5 (об./об); т.пл.=54°С.
Следующие соединения получены таким же способом:
1-[4-(3,3-Диметилциклогексил)фенил]этанон, соединение V.2
1H ЯМР: 7,8 (d, 2Н); 7,2 (d, 2Н); 2,7 (m, 1H); 2,5 (s, 3Н); 1,8-1,1 (m, 8H); 1,0 (s, 3Н); 0,9 (s, 3Н).
1-(4-Адамантан-2-илфенил)этанон, соединение V.3

т.пл.=75°C.
Подготовительный пример 6
1-[4-(4,4-Диметилциклогекс-1-енил)фенил]этанон, соединение VI.1
а) 1-[4-(1,1-Диметоксиэтил)фенил]-4,4-диметилциклогексанол 328 мл 1,6 М раствора бутиллития в циклогексане добавляют при -78°С к 117 г 1-бром-4-(1,1-диметоксиэтил)бензолу в 1100 мл тетрагидрофурана и эту реакционную смесь перемешивают при -78°С в течение 2 часов. 66 г 4,4-диметилциклогексана, растворенного в 210 мл тетрагидрофурана добавляют при этой же самой температуре и реакционную смесь перемешивают в течение 1 часа при -78°С. Реакционную смесь гидролизуют путем добавления дробленого льда. Органическую фазу отделяют после расслоения фаз, ее сушат над сульфатом натрия и растворители выпаривают при пониженном давлении. Полученное соединение перекристаллизовывают из 500 мл н-гексана; т.пл.=88°С.
б) 99,32 г 1-[4-(1,1-Диметоксиэтил)фенил]-4,4-диметилциклогексанола в 300 мл дихлорметана и 151 г йодида натрия добавляют к 600 мл ацетонитрила в инертной атмосфере и эту реакционную смесь нагревают до 30°С. Добавляют 102 мл хлортриметилсиланхлорида, а затем порциями при 65°С смесь 300 мл ацетонитрила и 47 мл уксусной кислоты и эту реакционную смесь перемешивают в течение 12 часов при комнатной температуре. Реакционную смесь фильтруют и экстрагируют дихлорметаном. Полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 99/1 (об./об.).
Подготовительный пример 7
1-[4-(4,4-Диметилциклогексил)фенил]этанон, соединение V.4
а) 1-(1,1-Диметилэтил)-4-(4,4-диметилциклогекс-1-енил)бензол
36,13 г 1-[4-(4,4-диметилциклогекс-1-енил)фенил]этанона (соединение VI.1) в 250 мл метанола перемешивают в течение 12 часов при комнатной температуре в присутствии 0,5 г пара-толуолсульфоновой кислоты (ПТСА) и 13 мл триметил-орто-формиата. Растворитель частично выпаривают при пониженном давлении. Добавляют 50% раствор гидроксида калия в метаноле и растворители затем выпаривают при пониженном давлении. Полученный остаток переносят в диизопропиловый эфир и растворитель затем выпаривают при пониженном давлении.
б) Полученное в (а) соединение в 250 мл метанола гидрируют в присутствии 3 г 5% палладия-на-угле. Реакционную смесь фильтруют, растворители выпаривают при пониженном давлении и полученный остаток переносят в дихлорметан. Реакционную смесь перемешивают в течение 12 часов в присутствии диоксида кремния и фильтруют, растворители выпаривают при пониженном давлении и полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 99/1 (об./об.); т.пл.=60°С.
Подготовительный пример 8
1-[3-Хлор-4-(3,3,5,5-тетраметилциклогексил)фенил]этанон, соединение V.5
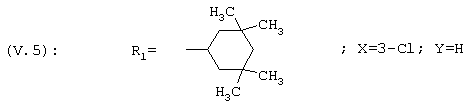
40,25 хлорида алюминия добавляют при 0°С в инертной атмосфере к 350 мл дихлорметана, а затем добавляют 5 г 1-[4-(3,3,5,5-тетраметилциклогексил)фенил]этанона (соединение V.1), растворенного в дихлорметане. После перемешивания в течение 2 часов при 0°С через реакционную смесь барботируют 17,1 мл газообразного газа (d=1,565, измеренная в жидком состоянии при -78°С). После нагревания до комнатной температуры к реакционной смеси добавляют смесь вода/лед. Полученную смесь экстрагируют дихлорметаном, после расслоения фазы разделяют и органическую фазу сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток очищают на силикагелевой колонке, элюируя смесью циклогексан/дихлорметан 7/3 (об./об.); выход=74%; т.пл.=64°С. Следующие дихлорсоединения также выделяют хроматографией:
1-[3,5-Дихлор-4-(3,3,5,5-тетраметилциклогексил)фенил]этанон, соединение V.6

1H ЯМР: 7,9 (1Н, s); 7,8 (s, 1Н); 3,9 (m, 1H); 2,5 (s, 3Н); 2,1 (m, 2H); 1,2 (m, 4H); 1,0 (s, 6H); 0,9 (s, 6H).
1-[3,6-Дихлор-4-(3,3,5,5-тетраметилциклогексил)фенил]этанон, соединение V.7
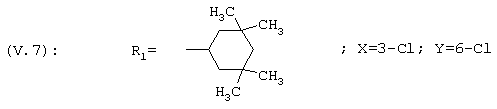
1H ЯМР: 7,6 (s, 1H); 7,2 (s, 1H); 3,3 (m, 1H); 2,6 (s, 3Н); 1,5 (m, 2H); 1,2 (m, 4H); 1,1 (s, 6Н); 0,9 (s, 6Н).
По методике, описанной для соединения V.5, выделены следующие соединения:
1-[3-Хлор-4-(3,3-диметилциклогексил)фенил]этанон, соединение V.8

1H ЯМР: 7,9 (1H, s); 7,8 (d, 1H); 7,4 (d, 1H); 3,1 (m, 1H); 2,5 (s, 3Н); 1,8-1,1 (m, 8H); 0,9 (s, 3Н); 0,8 (s, 3Н).
1-(3-Хлор-4-трет-бутилфенил)этанон, соединение V.9

1H ЯМР: 7,8 (s, 1H); 7,7 (d, 1H); 7,5 (d, 1H); 2,5 (s, 3Н); 1,4 (s, 9H).
1-(3,5-Дихлор-4-циклогексилфенил)этанон, соединение V.10
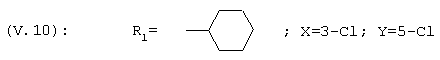
1-[3-Хлор-(4,4-диметилциклогексил)фенил]этанон, соединение V.11
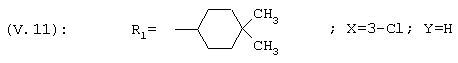
1H ЯМР: 7,9 (s, 1Н); 7,8 (d, 1Н); 7,5 (d, 1Н); 2,8 (m, 1H); 2,5 (s, 3Н); 1,8-1,1 (m, 8H); 0,95 (s, 3Н); 0,9 (s, 3H).
Подготовительный пример 9
1-[(3-Хлор-4-гидрокси)фенил]этанон, соединение VII.1
(VII.1):X=3-Cl; Y=H
167 г трихлорида алюминия добавляют в инертной атмосфере к 63,5 мл 2-хлор-1-метоксибензола в 500 мл 1,2-дихлорэтана, а затем добавляют по каплям 167 г ацетилхлорида, растворенного в 200 мл 1,2-дихлорэтана. Реакционную смесь нагревают при 45°С в течение 48 часов. Реакционную смесь выливают на смесь вода/лед и экстрагируют дихлорметаном, растворители выпаривают при пониженном давлении и полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 90/10 (об./об.). Соединение VII.1 перекристаллизовывают из циклогексана; т.пл.=107°С.
Подготовительный пример 10
Циклогексилэтилпроп-2-иниламин, соединение (4.1)
20 мл 80% 3-бромпропина по каплям добавляют к 30,3 мл циклогексилэтиламина и 29,7 г карбоната калия в 300 мл ацетонитрила. Реакционную смесь нагревают при 50°С в течение 12 часов и при 80°С в течение 6 часов. Полученную смесь фильтруют и растворители выпаривают при пониженном давлении.
Соединение V.1 очищают дистилляцией.
1H ЯМР: 3,3 (s, 2H); 3,0 (s, 1H); 2,5 (q, 2H); 2,4 (m, 1H); 1,8-1,1 (m, 10Н); 1,0 (t, 3Н).
Следующие соединения получают таким же способом:
Циклогексилметилпроп-2-иниламин, соединение 4,2
Циклогексилизопропилпроп-2-иниламин, соединение 4,3
Подготовительный пример 11
Циклогексилэтилбут-3-иниламин, соединение (4,4)
а) Бут-3-ин(4-метилфенил)сульфонат
74,8 г тозилхлорида добавляют к 36 мл пиридина при 80°С. Реакционную смесь охлаждают до 15°С, а затем добавляют 25 г бут-3-ин-1-ола. Реакционную смесь перемешивают при комнатной температуре в течение 12 часов, затем при 15°С добавляют 70 мл воды. Полученную смесь экстрагируют диэтиловым эфиром и органическую фазу затем промывают разбавленным водным раствором серной кислоты, а затем насыщенным водным раствором гидрокарбоната натрия. Органическую фазу сушат над сульфатом натрия и растворители выпаривают при пониженном давлении.
1H ЯМР: 7,8 (d, 2Н); 7,4 (d, 2Н); 4,0 (t, 2H); 3,8 (s, 1Н); 2,5 (t, 2H); 2,4 (s, 3Н)
б) 57,9 г соединения, полученного на стадии (а), 21,7 г гидрокарбоната натрия и 35,7 мл циклогексилэтиламина в 100 мл диметилформамида кипятят с обратным холодильником в течение 12 часов. Реакционную смесь вливают в воду и экстрагируют диэтиловым эфиром. Органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. После дистилляции выделяют ожидаемый амин;
т.кип.=92-94°С (при давлении 13 мбар (1300 Па)).
Подготовительный пример 12
4-Ацетил-2-хлорфенил-трифторметансульфонат, соединение Va.1
(Va.1):X=3-Cl; Y=H; Z=OTf
26,2 мл трифликового ангидрида по каплям добавляют при 0°С к 26,7 г 1-[(3-хлор-4-гидрокси)фенил]этанона (соединение VII.1) в 700 мл пиридина. Реакционную смесь перемешивают при 0°С в течение 36 часов, растворители выпаривают при пониженном давлении и остаток переносят в 0,1 н. раствор соляной кислоты в дихлорметане. После расслаивания фазы разделяют, органические фазы сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 95/5 (об./об.).
1H ЯМР: 8,2 (s, 1H); 8,0 (d, 1H); 7,8 (d, 1H).
Следующие соединения получают таким же способом:
4-Ацетил-2,6-дихлорфенил-трифторметансульфонат, соединение Va.2
(Va.2): X=3-Cl; Y=6-Cl; Z=OTf
1H ЯМР: 8,2 (s, 2H); 2,6 (s, 3H).
4-Бром-2-хлорфенил-трифторметансульфонат, соединение IIIa.1, начиная с 4-бром-2-хлорфенола.
(IIIa.1):Х=3-CL; Y=H
1Н ЯМР: 8,1 (s, 1Н); 7,7 (d, 1H); 7,6 (d, 1H).
Подготовительный пример 13
2-Хлор-4-[3-(циклогексилэтиламино)проп-1-инил]фенил-трифторметансульфонат, соединение Iа.1
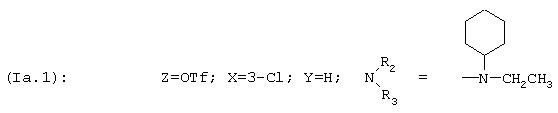
2,14 г циклогексилэтилпроп-2-иниламина (соединение VII.1) добавляют в инертной атмосфере к 4 г 4-бром-3-хлорфенил-трифторметансульфоната (соединение IIIa.1), 0,06 г йодида меди, 10 мл пиридина и 20 мл триэтиламина, после чего добавляют 0,413 г катализатора - дихлорбис(трифенилфосфин)палладияVI. Реакционную смесь кипятят с обратным холодильником в течение 2 часов, а затем выдерживают при комнатной температуре в течение 12 часов. Полученную смесь фильтруют и растворители выпаривают при пониженном давлении. Остаток очищают хроматографией на силикагелевой колонке, элюируя смесью дихлорметан/этанол с градиентом от 100/0 до 99/1 (об./об.). Полученное соединение переносят в дихлорметан и фильтруют и растворители выпаривают при пониженном давлении; выход=76%.
1H ЯМР: 7,8 (s, 1H); 7,6 (d, 1H); 7,5 (d, 1H); 3,6 (s, 2H); 2,6 (q, 2H); 2,4 (m, 1H); 1,9-1,1 (m, 10Н); 0,9 (t,3H).
Подготовительный пример 14
1-[3-Хлор-4-(4-фторфенил)фенил]этанон, соединение V.12
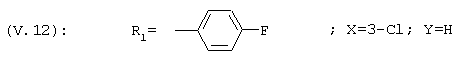
19,7 г 4-ацетил-2-хлорфенил-трифторметансульфоната (соединение Х.1), 10 г 4-фторбензолбороновой кислоты, 2 г тетракис(трифенилфосфин)палладия, 17,9 г карбоната натрия в 84,5 мл воды, 591 мл толуола, 200 мл этанола и 5,51 г хлорида лития перемешивают в инертной атмосфере при 60°С в течение 8 часов. Реакционную смесь затем перемешивают в течение 12 часов при комнатной температуре. Полученную смесь фильтруют и растворители выпаривают из фильтрата при пониженном давлении. Полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 97/3 (об./об.); выход=94%.
1H ЯМР: 8,0 (s, 1Н); 7,9 (d, 1Н); 7,5 (m, 3Н); 7,3 (m, 2H); 2,6 (s, 3Н).
Соединения с V.13 по V.17, приведенные ниже в таблице 1, получены таким же способом.
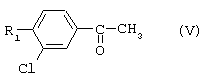
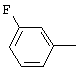

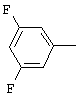
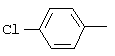
1-(2,6-Дихлорбифенил-4-ил)этанон, соединение V.18

1H ЯМР: 8,0 (s, 2H); 7,4 (m, 3H); 7,2 (m, 2H); 2,6 (s, 3H).
1-(2,6-Дихлор-4’-фторбифенил-4-ил)этанон, соединение V.19

1H ЯМР: 8,0 (s, 2H); 7,3 (m, 4H); 2,6 (s, 3H).
Подготовительный пример 15
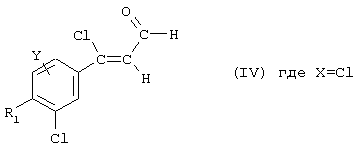
3-Хлор-3-[3-хлор-4-(3,3,5,5-тетраметилциклогексил)фенил]-пропеналь, соединение IV.1

3,51 мл оксалилхлорида по каплям добавляют при температуре между -5°С и 2°С к раствору 3,72 мл диметилформамида и 20 мл безводного дихлорметана и эту реакционную смесь затем перемешивают при комнатной температуре в течение 30 минут. Затем быстро добавляют 3,92 г 1-[3-хлор-4-(3,3,5,5-тетраметилциклогексил)фенил]этанона (соединение V.6), растворенного в 10 мл дихлорметана, после чего реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь вливают в смесь вода/лед, а затем добавляют 20 мл водного 2.84 М раствора этилата натрия. Полученную смесь промывают 50 мл раствора гидрокарбоната натрия и 50 мл воды, после расслаивания фазы разделяют, органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученное масло очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 97/3 (об./об.).
1H ЯМР: 10,2 (d, 1Н); 7,7 (s, 1H); 7,5 (d, 1Н); 7,3 (d, 1H); 6,6 (d, 1H); 3,4 (m, 1Н); 1,5 (m, 2H); 1,3 (m, 4H); 1,1 (s, 6H); 0,9 (s, 6H).
Соединения с IV.2 по IV.17, приведенные ниже в таблицах 2 и 3, получены таким же способом.















Подготовительный пример 16
3-Хлор-4-(3,3,5,5-тетраметилциклогексил)фенилэтин, соединение II.1
5,3 г гидроксида натрия растворяют в 150 мл воды в инертной атмосфере при энергичном перемешивании. Добавляют 80 мл 1.4-диоксана и эту смесь нагревают до температуры дефлегмации. Быстро добавляют 15 г 3-хлор-3-[3-хлор-4-(3,3,5,5-тетраметилциклогексил)фенил]пропеналя (соединение IV.1), растворенного в 130 мл 1,4-диоксана, и реакционную смесь поддерживают при температуре дефлегмации в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь вливают в большой объем дихлорметана. После расслаивания фазы разделяют, органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Остаток очищают хроматографией на силикагелевой колонке, элюируя циклогексаном; выход=80%.
1H ЯМР: 7,5 (s, 1Н); 7,3 (m, 2Н); 4,2 (s, 1Н); 3,2 (m, 1H); 1,4 (m, 2H); 1,2 (m, 4H); 1,0 (s, 6H); 0,9 (s, 6H).
Соединения с II.2 по II.15, приведенные ниже в таблицах 4 и 5, получены таким же способом.











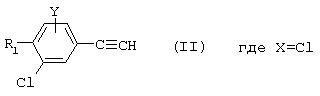

Подготовительный пример 17
3,5-Дифторбензолбороновая кислота, соединение 2.1
91,5 мл трет-бутиллития при -78°С добавляют к 20 г 1-бром-3,5-дифторбензола в 300 мл диэтилового эфира. Реакционную смесь перемешивают в течение 1 часа при -78°С, а затем добавляют 14,2 мл триметилбората. Реакционную смесь перемешивают в течение 1 часа при -78°С и затем в течение 12 часов при комнатной температуре. Добавляют 200 мл 1н. водного раствора соляной кислоты. Полученную смесь экстрагируют диэтиловым эфиром, органическую фазу промывают насыщенным раствором гидрокарбоната натрия и сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Остаток переносят в циклогексан и осадок выделяют фильтрацией.
1H ЯМР: 7,4 (m, 3H); 7,2 (m, 2H).
Подготовительный пример 18
4-Бром-3-хлорацетофенон, соединение Va.3
(Va.3); X=3-Cl; Y=H; Z=Br
Раствор 100 г 4-бромацетофенона в 250 мл дихлорметана по каплям добавляют при 0°С к 133,34 г хлорида алюминия в 600 мл дихлорметана. После перемешивания в течение 2 часов при 0°С в эту среду при 0°С барботируют 28,3 мл предварительно замороженного (-75°С) хлора. Реакционную смесь перемешивают при комнатной температуре в течение 12 часов, а затем гидролизуют. После расслаивания фазы разделяют, водную фазу экстрагируют дихлорметаном, органические фазы сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученный остаток перекристаллизовывают из гексана; выход=57%; т.пл.=80°С.
Подготовительный пример 19
3-Хлор-3-(4-бром-3-хлорфенил)пропеналь, соединение IVa.1
(IVa.1):X=3-Cl; Y=H; Z=Br
15,08 мл оксалилхлорида добавляют при температуре между 3°С и 6°С при энергичном перемешивании к 16 мл диметилформамида в 200 мл дихлорметана. После нагревания до комнатной температуры смесь перемешивают в течение 30 минут, затем добавляют раствор 13,4 г 4-бром-3-хлорацетофенона (соединение Va.3) в 40 мл дихлорметана. Реакционную смесь перемешивают в течение 12 часов при комнатной температуре, а затем гидролизуют добавлением раствора 18,9 г ацетата натрия в 50 мл воды. После перемешивания в течение 30 минут при комнатной температуре, после расслаивания фазы разделяют, водную фазу экстрагируют дихлорметаном, органические фазы сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученный остаток перекристаллизовывают из циклогексана; выход=87%; т.пл.=134°С.
Подготовительный пример 20
[3-(4-Бром-3-хлорфенил)проп-2-инил]циклогексилэтиламин, соединение Ia.2

а) 1-Бром-2-хлор-4-этинилбензол
40 г гидроксида натрия растворяют в инертной атмосфере в 230 мл воды, добавляют 120 мл 1,4-диоксана и эту реакционную смесь нагревают до 80°С. Добавляют 17,5 г 3-хлор-3-(4-бром-3-хлорфенил)пропеналя, растворенного в 400 мл 1,4-диоксана, и эту реакционную смесь перемешивают в течение 30 минут при 80°С. Реакционную смесь оставляют охлаждаться до комнатной температуры, а затем добавляют 2300 мл дихлорметана. После расслаивания фазы разделяют и органическую фазу промывают водой и сушат над сульфатом магния. Соединение, растворенное в смеси дихлорметан/1,4-диоксан, используют в его нынешней форме на следующей стадии.
б)[3-(4-Бром-3-хлорфенил)проп-2-инил]циклогексилэтиламин 36% водный раствор формальдегида добавляют к 10,36 мл этилциклогексиламина в 400 мл 1,2-диметоксиэтана. Этот раствор добавляют к раствору соединения, полученного выше, в присутствии 0,54 г дигидрата хлорида меди II. Реакционную смесь перемешивают в течение 4 часов при кипячении с обратным холодильником, а затем оставляют стоять при комнатной температуре. Полученную смесь фильтруют, растворители выпаривают при пониженном давлении и полученный остаток затем очищают хроматографией на силикагелевой колонке, элюируя смесью дихлорметан/этанол 99/1 (об./об.). Полученное соединение переносят в диэтиловый эфир и через него барботируют хлористый водород. Полученный осадок отфильтровывают и сушат с получением соединения в форме гидрохлорида.
1H ЯМР: 7,7 (d, 1H); 7,6 (s, 1H); 7,2 (d, 1H); 3,5 (s, 2H); 2,6 (q, 2H); 2,4 (m, 1Н); 1,8-1,1 (m, 10Н); 0,9 (t, 3H).
Подготовительный пример 21
2-Хлор-4-(4,4-диметилциклогексил)фенол, соединение IX.1
а) 2-Хлор-4-(1-гидрокси-4,4-диметилциклогексил)фенол
100 мл 1,6 М раствора н-бутиллития в гексане добавляют при -78°С к 15,1 г 4-бром-2-хлорфенола в 150 мл тетрагидрофурана и эту реакционную смесь перемешивают при -78°С в течение 1 часа. Добавляют 10,1 г 4,4-диметилциклогексанона (соединение 3.1) и реакционную смесь перемешивают при -78°С в течение еще 30 минут, а затем при комнатной температуре в течение 12 часов. Реакционную смесь гидролизуют 1н. раствором соляной кислоты и экстрагируют этилацетатом. Органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученное твердое вещество очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат с градиентом от 98/2 до 90/10 (об./об.). Получают 11,8 г твердого вещества.
1H ЯМР: 7,4 (s, 1H); 7,2 (d, 2H); 6,9 (d, 2H); 4,5 (s, 1H); 1,9-1,1 (m, 8H): 0,9 (s, 6H)
б) 50 л 57% водного раствора йодоводородной кислоты добавляют к 11,8 г 2-хлор-4-(1-гидрокси-4,4-диметилциклогексил)фенола в 200 мл уксусной кислоты. Реакционную смесь кипятят с обратным холодильником в течение 3 часов и растворители выпаривают при пониженном давлении. Добавляют 40% водный раствор гидроксида натрия, водный раствор карбоната натрия и затем водный раствор гидросульфата натрия и полученную смесь экстрагируют диэтиловым эфиром. Органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученное соединение очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат 95/5 (об./об.).
1H ЯМР: 9,8 (s, 1H); 7,1 (s, 1H); 7 (d, 1H); 6,9 (d, 1H); 1,9 (m, 1H); 1,6-1,2 (m, 8H); 0,9 (s, 6H)
Соединения с IX.2 по IX.4 получены таким же способом:
4-(Адамантан-2-ил)-3,5-дихлорфенол, соединение IX.2, полученный из соединения IХа.1 и адамантан-2-она
1H ЯМР: 10,1 (s, 1H); 6,8 (s, 2H); 3,4 (s, 1H); 2,4 (s, 2H); 2,3-1,4 (m, 12H)
4-(Адамантан-2-ил)фенол, соединение IX.3
1H ЯМР: 9,1 (s, 1H); 7,1 (d, 2H); 6,7 (d, 2H); 2,8 (s, 1H); 2,4 (s, 2H); 1,9-1,4 (m, 12H)
4-(Адамантан-2-ил)-3-хлорфенол, соединение IX.4
1H ЯМР: 9,8 (s, 1H); 7,1 (s, 1H); 7,0 (d, 1H); 6,9 (d, 1H); 2,8 (s, 1H); 2,3 (m, 2H); 1,9 (m, 5H); 1,7 (m, 5H); 1,5 (m, 2H)
Подготовительный пример 22
4-(Тетрагидропиран-4-ил)фенол, соединение IХ.5
а) 4-(3,6-Дигидропиран-4-ил)фенол
Раствор 100 мл 1,6 м бутиллития в гексане, затем 8,1 г 4-тетрагидропиранона добавляют при -40°С к 12,7 г 4-бромфенола в 150 мл тетрагидрофурана. При этой же температуре к реакционной смеси добавляют 100 мл 1,6 М бутиллития в гексане, после чего добавляют 8,1 г 4-тетрагидропиранона. Реакционную смесь оставляют перемешиваться в течение 18 часов при комнатной температуре, а затем гидролизуют 1н. соляной кислотой. Полученную смесь несколько раз экстрагируют диэтиловым эфиром, органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученное твердое вещество очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат с градиентом от 90/10 до 80/20 (об./об.).
1H ЯМР: 9,4 (s, 1Н); 7,2 (d, 1Н); 6,7 (d, 1Н); 6,0 (t, 1H); 4,1 (d, 2H); 3,7 (t, 2H); 2,4 (t, 2H)
б) 5,5 г 4-(3,6-дигидропиран-4-ил)фенола гидрируют в присутствии 550 мг 10% палладия-на-угле в 100 мл метанола в течение 3 часов. Смесь фильтруют и растворители затем выпаривают при пониженном давлении.
1H ЯМР: 9,1 (s, 1H); 7 (d, 2H); 6,6 (d, 2H); 3,9 (m, 2H); 3,4 (m, 2H); 2,6 (m, 1H); 1,6 (m, 4H)
Следующее соединение получено таким же способом:
4-(4,4-Диметилциклогексил)фенол, соединение IХ.6
1H ЯМР: 9 (s, 1H); 7 (d, 2H); 6,7 (d, 2H); 2,2 (m, 1H); 1,6-1,2 (m, 8H); 0,9 (s, 6H)
Подготовительный пример 23
4-(Адамантан-2-ил)-3,5-дифторфенол. соединение IX.7
а) 2-(2,6-Дифтор-4-метоксифенил)адамантан-2-ол
Получают из 4-бром-3,5-дифторфенилметилового эфира в присутствии одного эквивалента н-бутиллития согласно способу, описанному в подготовительном примере 22 (а).
б) 19 г продукта, полученного на вышеуказанной стадии, 200 мл йодоводородной кислоты и 200 мл уксусной кислоты перемешивают в течение ночи при температуре дефлегмации. После охлаждения до комнатной температуры реакционную смесь вливают в смесь дробленый лед/NaНSО3. После нейтрализации 1н. раствором гидроксида натрия полученную смесь экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом магния и растворители затем выпаривают при пониженном давлении.
Подготовительный пример 24
2-Хлор-4-(4,4-диметилциклогексил)фенил-трифторметансульфонат, соединение III.1
8,2 мл трифликового ангидрида добавляют при 5°С к 9,7 г 2-хлор-4-(4,4-диметилциклогексил)фенола (соединение IX. 1) в 60 мл пиридина и эту реакционную смесь оставляют стоять при 0°С в течение 30 минут, а затем перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь гидролизуют и затем экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученный остаток переносят в толуол и растворители затем выпаривают при пониженном давлении. Полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат с градиентом от 100/0 до 99/1 (об./об.). Получают 15 г соединения.
1H ЯМР: 7,7 (s, 1Н); 7,5 (d, 1Н); 7,4 (d, 1Н); 2,5 (m, 1H); 1,6-1,2 (m, 8H); 0,92 (s, 3H); 0,86 (s, 3Н).
Соединения с III.2 по III.7 получены таким же способом:
4-(Адамантан-2-ил)-3,5-дихлорфенил-трифторметансульфонат, соединение III.2
1H ЯМР: 7,7 (d, 1H); 7,6 (d, 1H); 3,6 (m, 1H); 3,0-1,0 (m, 14H)
4-(Адамантан-2-ил)фенил-трифторметансульфонат, соединение III.3
1H ЯМР: 7,5 (d, 2H); 7,4 (d, 2H); 3,0 (s, 1H); 2,4 (s, 2H); 1,9 (m, 5H); 1,8-1,5 (m, 7H)
4-(Адамантан-2-ил)-3-хлорфенил-трифторметансульфонат, соединение III.4
1H ЯМР: 7,6-7,4 (m, 3Н); 3,0 (s, 1H); 2,4 (m, 2H); 1,9 (m, 5H); 1,8-1,4 (m, 7H)
4-(Адамантан-1-ил)фенил-трифторметансульфонат, соединение III.5
1H ЯМР: 7,5 (d, 2H); 7,3 (d, 2H); 2,1 (m, 3Н); 1,8 (m, 6H); 1,7 (m, 6H)
4-(Тетрагидропиран-4-ил)фенил-трифторметансульфонат, соединение III.6
1H ЯМР: 7,4 (s, 4Н); 3,9 (m, 2Н); 3,4 (m, 2Н); 2,8 (m, 1H); 1,7 (m, 4H)
4-(4,4-Диметилциклогексил)фенил-трифторметансульфонат, соединение III.7
1H ЯМР: 7,4-7,3 (m, 4H); 2,6 (m, 1H); 1,6-1,2 (m, 8Н); 0,93 (s, 3Н); 0,90 (s, 3Н)
Соединения примеров, представленных ниже, за исключением упомянутых особо, представляют собой соединения формулы (I), где:

Пример 1
[3-(4-Адамантан-2-илфенил)проп-2-инил]циклогексилэтиламина гидрохлорид

8,6 мл 36% формальдегида добавляют к 11,2 мл циклогексилэтиламина в 100 мл 1,2-диметоксиэтана и перемешивание продолжают при комнатной температуре в течение 2 часов. Этот раствор добавляют к смеси 16 г 2-(4-этилфенил)адамантана (соединение II.3) и 0,58 г дигидрата хлорида меди II в 400 мл 1,2-диметоксиэтана. Реакционную смесь кипятят с обратным холодильником в течение 2 часов и растворители затем выпаривают при пониженном давлении. Полученное соединение переносят в диэтиловый эфир, через него барботируют хлористый водород, и полученный осадок отфильтровывают и сушат; т.пл.=124°С (HCl·0,5 H2O).
Соединения примеров с 2 по 12, приведенных ниже, получены таким же способом.
Пример 2
{3-[4-(3,3,5,5-Тетраметилциклогексил)фенил]проп-2-инил}-циклогексилэтиламина гидрохлорид

т.пл.=150°С (НСl·0,1 H2O).
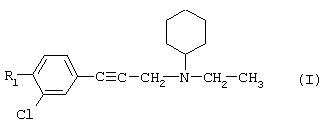
НСl
HCl
HCl
0,1 Н2O
HCl
0,2 Н2O

HCl
0,2 Н2О

HCl
HCl
0,7 Н2O
HCl
(а) 1H ЯМР: 7,4 (m, 2H); 7,3 (d, 1Н); 3.6 (s, 2H); 3,4 (m, 1Н); 2,8 (m, 1H); 2,6 (q, 2H); 1,3-0,9 (m, 27H)
Пример 12
[3-(2,6-Дихлорбифенил-4-ил)проп-2-инил]циклогексилэтиламина гидрохлорид

т.пл.=205°С (HCl).
Пример 13
Соединение, идентичное соединению примера 7, но полученное другим способом.
3-(2-Хлор-3’-фторбифенил-4-ил)проп-2-инил]циклогексилэтиламина гидрохлорид

3,4 г 2-хлор-4-[3-(циклогексилэтиламино)проп-1-инил]фенил-трифторметансульфоната (соединение Iа.1), 1,23 г 3-фторбензолбороновой кислоты, 2,2 г карбоната натрия в 10,4 мл воды, 0,68 г хлорида лития, 75 мл толуола, 25 мл этанола и 0,7 г тетракис(трифенилфосфин)палладия перемешивают в инертной атмосфере при кипячении с обратным холодильником в течение 4 часов. Полученную смесь фильтруют, растворители выпаривают при пониженном давлении и полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью дихлорметан/этанол 99/1 (об./об.). Полученные соединения переносят в диэтиловый эфир и через него барботируют хлористый водород. Полученную смесь фильтруют и растворители выпаривают при пониженном давлении; т.пл.=130°С(НСl·0,2 Н2O).
Соединения примеров 14 и 15, представленные ниже, получены таким же способом:
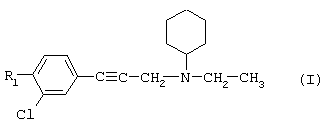

HCl
HCl
0,3 H2O
Пример 16
[3-(4-Адамантан-2-ил-3-хлорфенил)проп-2-инил]циклогексил-этиламина гидрохлорид

а) 2-{2-хлор-4-[3-(цикпогексилэтиламино)проп-1-инил]фенил}-адамантан-2-ол
[3-(4-Бром-3-хлорфенил)пропен-2-инил]циклогексилэтиламина гидрохлорид обрабатывают 1 н. раствором гидроксида натрия в эфире с получением основания. 30,5 мл 15% раствора н-бутиллития в гексане при -75°С добавляют к 17,5 г [3-(4-бром-3-хлорфенил)-пропен-2-инил]циклогексилэтиламина в 200 мл диэтилового эфира и перемешивание продолжают при -75°С в течение 1 часа 30 минут. Далее при -75°С добавляют 7,51 г адамантан-2-она в 100 мл диэтилового эфира и реакционную смесь затем перемешивают в течение 2 часов при -75°С.
Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивание продолжают в течение 1 часа. Реакционную смесь гидролизуют и экстрагируют диэтиловым эфиром, органические фазы сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью дихлорметан/этанол с градиентом от 100/0 до 99/1 (об./об.). Полученное соединение используют непосредственно на следующей стадии.
б) 9,78 г йодида натрия добавляют к 11,12 г полученного выше соединения в 50 мл ацетонитрила и 25 мл дихлорметана, после чего добавляют 6,63 мл хлортриметилсилана. Реакционную смесь перемешивают при 30°С в течение 2 часов с последующим добавлением 25 мл ацетонитрила, 5,12 г цинкового порошка и 2,99 мл уксусной кислоты. Реакционную смесь нагревают при 80°С в течение 3 часов, дают возможность охладиться до комнатной температуры, фильтруют, промывают диэтиловым эфиром и экстрагируют дихлорметаном и растворители затем выпаривают при пониженном давлении. Полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью толуол/этанол 97/3 (об./об.), а затем смесью циклогексан/этилацетат 92,5/7,5 (об./об.). Полученное соединение переносят в диэтиловый эфир. Гидрохлорид получают, барботируя через него хлористый водород, и полученный осадок отфильтровывают и сушат; т.пл.=110°С (НСl·0,3Н2O).
Пример 17
{3-[4-(4,4-Диметилциклогексил)-2-хлорфенил]проп-2-инил}-циклогексилэтиламина гидрохлорид

1,42 г дихлорбис(трифенилфосфин)палладия добавляют в инертной атмосфере к 8,03 г цикпогексилэтилпроп-2-иниламина (соединение 4.1), 15 г [4-(4,4-диметилциклогексил)-2-хлорфенил]трифторметансульфоната (соединение III.1), 0,19 г йодида меди, 3,4 г хлорида лития в 200 мл триэтиламина и 100 мл пиридина. Реакционную смесь кипятят с обратным холодильником в течение 12 часов. Растворители выпаривают при пониженном давлении и полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью циклогексан/этилацетат с градиентом от 95/5 до 90/10 (об./об.). Полученный остаток переносят в диэтиловый эфир. Гидрохлорид отделяют фильтрацией и затем через него барботируют хлористый водород. Полученный остаток перекристаллизовывают из этилацетата.
1H ЯМР: 11 (s, 11Н); 7,6-7,4 (m, 2H); 7,3 (d, 1Н); 4,3 (s, 2H); 3,2 (m, 2H); 1,5 (m, 1H); 2,2-1,1 (m, 22H); 0,9 (d, 6H)
Соединения примеров с 18 по 28, представленные ниже, получают таким же способом:
Пример 18
[4-(4-Адамантан-2-ил-2-хлорфенил)бут-3-инил]циклогексилэтиламина гидрохлорид

1H ЯМР: 7,5 (d, 1H); 7,4 (s, 1H); 7,3 (d, 1H); 3,4-3,2 (m, 4H); 3,1 (m, 2H); 3,0 (s, 1H); 2,4 (s, 2H); 2,0-2,1 (m, 26H).


HCl
0,8 H2O

HCl
HCl
HCl
HCl
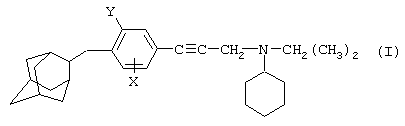
Пример 29
{(Z)-3-[3-Хлор-4-(3,3,5,5-тетраметилциклогексил)фенил]пропен-2-ил}циклогексилэтиламина гидрохлорид

3 г соединения примера 3 в 50 мл петролейного эфира гидрируют в инертной атмосфере при атмосферном давлении в присутствии 3 мл циклогексена и 0,3 г палладия на карбонате кальция, отравленным 3,5% свинца (катализатор Линдлара). Реакционную смесь фильтруют через целит, растворители выпаривают и полученный остаток очищают хроматографией на силикагелевой колонке, элюируя смесью дихлорметан/этанол 95/5 (об./об.). Полученный маслянистый остаток переносят в диэтиловый эфир и через него барботируют хлористый водород. Осадок отфильтровывают и сушат при пониженном давлении. Соединение примера 29 выделяют с выходом 83%; т.пл.=158°С(НСl·0,1 Н2O).
Соединения примеров с 30 по 54, приведенных ниже, получают таким же способом:
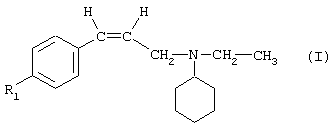

HCl
HCl
HCl
0,3 H2O

HCl
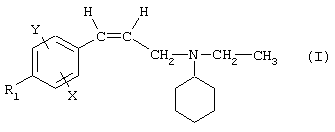
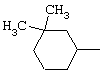
HCl
HCl
0,5 H2O
HCl
0,3 Н2O
HCl
1,1 Н2O
HCl
0,6 Н2O
HCl
HCl
HCl
0,7 H2O
HCl
0,4 Н2O
HCl
HCl
HCl
(а) фумараты получены, начиная с соответствующего основания, следующим образом:
1 г основания растворяют в 50 мл изопропанола. 0,26 г фумаровой кислоты также растворяют при 50°С в 100 мл изопропанола. Раствор, содержащий исходное вещество, вливают в теплый раствор фумаровой кислоты. Реакционную смесь перемешивают в течение 15 минут при комнатной температуре и растворители затем выпаривают при пониженном давлении. Полученные кристаллы промывают этиловым эфиром и затем перекристаллизовывают из ацетонитрила; т.пл.=158°С (фумарат). Малеат получают таким же способом: т.пл.=166°С (малеат).
(б) фумарат получен из соответствующего основания; т.пл.=104°С (фумарат).
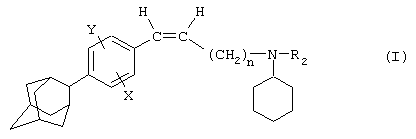
0,75 Н2O
HCl
HCI; H2O
HCl
HCI; 0,2
H2O
(б) получено по той же схеме синтеза, что и соединение примера 44 с использованием соединения 4,2 в качестве исходного вещества.
Пример 53
[(Z)-3-(2,6-Дихлорбифенил-4-ил)пропен-2-ил]циклогексилэтиламина гидрохлорид
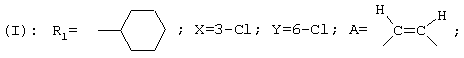
т.пл.=120°С (HCl).
Пример 54
[(Z)-4-(4-Адамантан-2-ил-3-хлорфенил)бут-3-инил]циклогексил-этиламина гидрохлорид
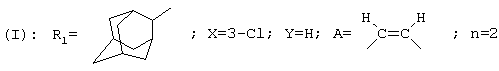
т.пл.=178°С (HCl).
Соединения в таблице 13, представленной ниже, получены по той же схеме синтеза, что и соединение примера 44:
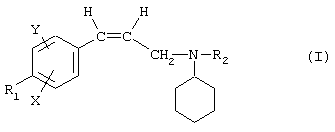

HCl
НОС(O)СF3
HCl
0,2 Н2О
HCl
HCl
HCl
HCl
HCl
HCl
(б) используя 4-бром-2,6-дихлорфенол в качестве исходного вещества (J. Am. Chem. Soc. 1933, 55, 2125-2126).
Пример 64
{(Е)-3-[3-Хлор-4-(3,3,5,5-тетраметилциклогексил)фенил]пропен-2-ил}циклогексилэтиламина гидрохлорид
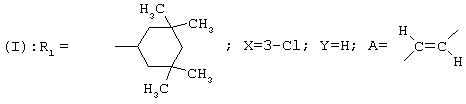
24,3 мл 1М раствора диизобутилалюминия гидрида (DIBALH) в толуоле по каплям добавляют в инертной атмосфере к раствору 4 г соединения примера 4 в 40 мл толуола. Реакционную смесь перемешивают при 40°С в течение 1 часа, а затем вливают в смесь вода/лед и добавляют гидроксид натрия до тех пор, пока рН не станет равным 7. Полученную смесь экстрагируют дихлорметаном, после расслаивания фазы разделяют, органическую фазу сушат над сульфатом магния и растворители выпаривают при пониженном давлении. Остаток переносят в диэтиловый эфир и через него барботируют хлористый водород. Полученный осадок отфильтровывают и сушат: т.пл.=169°С (HCl·0,2 H2O).
Соединения примеров с 65 по 67, представленных ниже, получают способом, описанным для примера 64.
Пример 65
{(Е)-3-[4-(3,3,5,5-тетраметилциклогексил)фенил]пропен-2-ил}циклогексилэтиламина гидрохлорид
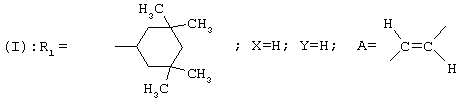
т.пл.=200°С (HCl).
Пример 66
{(Е)-3-[4-(2-Адамантил)фенил]пропен-2-ил}циклогексилэтиламина гидрохлорид

т.пл.=200°С (НСl)
Пример 67
{(Е)-3-[4-(2-Адамантил)-3,5-дихлорфенил]пропен-2-ил}циклогексилэтиламина гидрохлорид
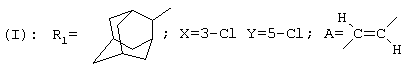
т.пл.=224°С (НСl)
Пример 68
{3-[3-Хлор-4-(3,3,5,5-тетраметилциклогексил)фенил]пропил}-циклогексилэтиламина гидрохлорид

4 г соединения примера 3 гидрируют в присутствии 0.4 г 10% палладия-на-угле и 50 мл этанола. Реакционную смесь фильтруют, фильтрат упаривают при пониженном давлении и полученный остаток очищают на силикагелевой колонке, элюируя смесью толуол/этанол 97/3 (об./об.). Полученный маслянистый остаток переносят в диэтиловый эфир и через него барботируют хлористый водород. Полученный осадок отфильтровывают и сушат; т.пл.=154°С (HCl).
Соединения примеров с 69 по 78, приведенных ниже, получают таким же способом.
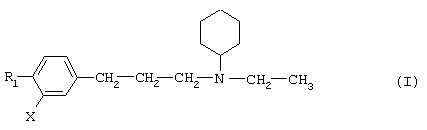

HCl
0,2 Н2O
HCl
0,6 Н2O
HCl
HCl
HCl
1,2 Н2O
HCl
75
HCl
0,7 Н2O
HCl
Пример 77
[3-(2,6-Дихлорфенил-4-ил)пропил]циклогексилэтиламина гидрохлорид
(I): R1= ; Х=3-Cl; Y=6-Сl; А=-СН2-СН2
; Х=3-Cl; Y=6-Сl; А=-СН2-СН2
т.пл.=128°С(НСl)
Пример 78
{3-[4-(2-Адамантил)-3,5-дихлорфенил]пропил}циклогексилэтиламина гидрохлорид
(I): R1=  ; X=3-Cl; Y=5-Cl; А=-СH2-СН2-
; X=3-Cl; Y=5-Cl; А=-СH2-СН2-
т.пл. F=220°С (HCl)
Пример фармацевтической композиции
Ингредиенты тщательно смешивают и таблетки прессуют на таблеточной машине.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ХРОМОНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2010 |
|
RU2545214C2 |
| ПРОИЗВОДНЫЕ БЕНЗОТИАЗИНОВ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВ | 2010 |
|
RU2523791C2 |
| ПРОИЗВОДНЫЕ ПИРИДОИНДОЛОНА, ЗАМЕЩЕННЫЕ В ПОЛОЖЕНИИ 6, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ | 2005 |
|
RU2364595C2 |
| ДИАРИЛОВЫЕ ЭФИРЫ | 2010 |
|
RU2528231C2 |
| НОВЫЕ ИМИДАЗОЛИДИНОВЫЕ СОЕДИНЕНИЯ КАК МОДУЛЯТОРЫ АНДРОГЕННЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2488584C2 |
| ИНДОЛИЛМАЛЕИМИДНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ НАРУШЕНИЙ ИЛИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ Т-ЛИМФОЦИТАМИ И/ИЛИ ПКС | 2003 |
|
RU2340610C2 |
| ЗАМЕЩЕННОЕ ОКСИМОМ АМИДНОЕ СОЕДИНЕНИЕ И СРЕДСТВО ДЛЯ БОРЬБЫ С ВРЕДИТЕЛЯМИ | 2013 |
|
RU2668547C2 |
| СОЕДИНЕНИЯ С МЕДИЦИНСКИМИ ЭФФЕКТАМИ, ОБУСЛОВЛЕННЫМИ ВЗАИМОДЕЙСТВИЕМ С РЕЦЕПТОРОМ ГЛЮКОКОРТИКОИДОВ | 2006 |
|
RU2430086C2 |
| ЗАМЕЩЕННЫЙ 2-АМИНОПИРИДИН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2671212C2 |
| Хинуклидиновые эфиры 1-азагетероциклилуксусной кислоты в качестве антимускариновых средств, способ их получения и их лекарственные композиции | 2012 |
|
RU2628082C2 |
Изобретение относится к новым производным бензола формулы (I),

где А представляет собой группу, выбранную из следующих: -С≡С-, -СН=СН-; -СН2-СН2-; n равно 1 или 2; Х представляет собой атом водорода, хлора или фтора, или метильную, или метоксигруппу; Y представляет собой атом водорода, или атом хлора, или фтора; R1 представляет собой циклогексильную группу, монозамещенную, дизамещенную, тризамещенную или тетразамещенную метильной группой; фенильную группу, монозамещенную или дизамещенную атомом фтора или хлора или метоксигруппой; циклогептильную, трет-бутильную, дициклопропилметильную, 4-тетрагидропиранильную или 1- или 2-адамантильную или адамантан-2-ольную группу; либо R1 представляет собой фенильную группу, причем в этом случае Х и Y оба представляют собой атом хлора; r2 представляет собой атом водорода или (С1-С4)алкильную группу; r3 представляет собой (С5-С7)циклоалкил; и соли этих соединений, образованные присоединением фармацевтически приемлемых кислот, а также их сольваты и гидраты. Изобретение также относится к способам получения соединений формулы (I) и к фармацевтической композиции, способной взаимодействовать с рецепторами сигма-2, на основе этих соединений. Технический результат – получение новых соединений и лекарственных продуктов на их основе в целях лечения аутоиммунных состояний, нарушений частоты сердечных сокращений и борьбы с пролиферацией опухолевых клеток. 8 н. и 10 з.п. ф-лы, 14 табл.

где
А представляет собой группу, выбранную из следующих: -С≡С-, -СН=СН-; -СН2-СН2-;
n равно 1 или 2;
Х представляет собой атом водорода, хлора или фтора или метильную или метоксигруппу;
Y представляет собой атом водорода или атом хлора или фтора;
R1 представляет собой циклогексильную группу, монозамещенную, дизамещенную, тризамещенную или тетразамещенную метильной группой; фенильную группу, монозамещенную или дизамещенную атомом фтора или хлора или метоксигруппой; циклогептильную, трет-бутильную, дициклопропилметильную, 4-тетрагидропиранильную или 1- или 2-адамантильную или адамантан-2-ольную группу; либо R1 представляет собой фенильную группу, причем в этом случае Х и Y оба представляют собой атом хлора;
R2 представляет собой атом водорода или (С1-С4)алкильную группу;
R3 представляет собой (С5-С7)циклоалкил;
и соли этих соединений, образованные присоединением фармацевтически приемлемых кислот, а также их сольваты и гидраты.
А представляет собой группу, выбранную из следующих: -C≡C-, -CH=CH-; -СН2-СН2-;
n равно 1 или 2;
Х представляет собой атом водорода, хлора или фтора или метильную или метоксигруппу;
Y представляет собой атом водорода или атом хлора или фтора;
R1 представляет собой циклогексильную группу, монозамещенную, дизамещенную, тризамещенную или тетразамещенную метильной группой;
фенильную группу, монозамещенную или дизамещенную атомом фтора или хлора или метоксигруппой; циклогептильную, трет-бутильную, дициклопропилметильную, 4-тетрагидропиранильную или 1- или 2-адамантильную группу; либо R1 представляет собой фенильную группу, причем в этом случае Х и Y оба представляют собой атом хлора;
R2 представляет собой (С1-С4)алкил;
R3 представляет собой (С5-С7)циклоалкил;
и соли этих соединений, образованные присоединением фармацевтически приемлемых кислот, а также их сольваты и гидраты.

где
А представляет собой группу, выбранную из следующих: -С≡С-, -СН=СН-; -СН2-СН2-;
Х представляет собой атом водорода или хлора;
Y представляет собой атом водорода или атом хлора;
R1 представляет собой циклогексил, монозамещенный, дизамещенный, тризамещенный или тетразамещенный метильной группой; фенильную группу, замещенную атомом хлора, метоксигруппой или одним или двумя атомами фтора; трет-бутильную или 1- или 2-адамантильную группу; либо R1 представляет собой фенильную группу, причем в этом случае Х и Y оба представляют собой атом хлора;
R2 представляет собой (С2-С3)алкил;
и соли этих соединений, образованные присоединением фармацевтически приемлемых кислот, а также их сольваты и гидраты.
[(Z)-3-(4-адамантан-2-ил-3-хлорфенил)пропен-2-ил]циклогексилэтиламина;
[(Z)-3-(4-адамантан-2-илфенил)пропен-2-ил]циклогексилэтиламина;
{(Z)-3-[4-(4,4-диметилциклогексил)-2-хлорфенил]пропен-2-ил}циклогексилэтиламина;
[(Z)-3-(4-адамантан-2-ил-3,5-дихлорфенил)пропен-2-ил]циклогексилэтиламина;
[(Z)-3-(4-адамантан-2-ил-3,5-дихлорфенил)пропен-2-ил]циклогексил(2-метилэтил)амина;
а также их солей, образованных присоединением фармацевтически приемлемых кислот, и их сольватов и гидратов.
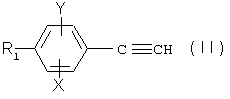
где R1, Х и Y являются такими, как определено для (I), формальдегидом и амином (1) HNR2R3, причем R2 и R3 являются такими, как определено для (I).

где X, Y, n, R2 и R3 являются такими, как определено для (I), a Z представляет собой бром, йод или трифторметансульфонатную группу (OTf), и производным бора (2) формулы R1-B(OR)2, где R представляет собой атом водорода или алкильную или арильную группу, в присутствии основания и металлического катализатора.
 , в присутствии основания с получением промежуточного соединения формулы
, в присутствии основания с получением промежуточного соединения формулы
где X, Y, n, R2 и R3 являются такими, как определено для (I); причем указанное соединение (I') затем восстанавливают в селективных условиях.
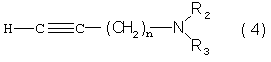
где n, R2 и R3 являются такими, как определено для (I), и соединением формулы

где R1, X и Y являются такими, как определено для (I), a Z представляет собой атом брома или йода или трифторметилсульфонатную группу (трифлат, или OTf).
| (088.а)СЕСО:ОЗНАЯRA-;'?fiTUG-;'?iXlir::""^AS б:-;б;;^;о7екй! ^Л^-ААвторыи И. А. Рабчук | 0 |
|
SU376850A1 |
| WO 9804251 A1, 05.02.1998 | |||
| SILVE S ET AL, Molecular and Cell Biology, 01.06.1996, vol | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| КЕРОСИНОВЫЙ УТЮГ | 1924 |
|
SU2719A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ГЕКСАГИДРОАЗЕПИНОВ ИЛИ ИХ СОЛЕЙ С МИНЕРАЛЬНЫМИ ИЛИ ОРГАНИЧЕСКИМИ КИСЛОТАМИ | 1991 |
|
RU2070194C1 |

