Изобретение относится к созданию библиотеки соединений, содержащей обширное количество соединений, представляющих собой агонисты и антагонисты по отношению к G-белок сопряженным рецепторам.
Представители класса мембранных белков, которые представляют собой G-белок сопряженные рецепторы (G-protein coupled receptors) (также известные как рецепторы, семь раз пронизывающие мембрану, имеющие семь трансмембранных сегментов (7ТМ) или «змеевидные» рецепторы), опосредуют передачу сигналов в клетку в ответ на широкий спектр внеклеточных сигналов, включающих гормоны, нейротрансмиттеры, цитокины и даже вещества из окружающей среды, например, ответственные за запахи и вкусы. В результате взаимодействия лиганда с внеклеточной частью рецептора (обычно, это N-концевая часть рецепторного белка) рецептор временно переходит в активированное состояние (это преобразование обычно обозначают как R+L→R*L, где R - неактивный рецептор, R* - активированный рецептор и L - лиганд).
Активированный (или R*) рецептор затем способен взаимодействовать с представителями семейства G-белков. G-белки представляют собой большое семейство внутриклеточных тримерных белков, связывающих гуаниновые нуклеотиды. После взаимодействия с активированным рецептором (вероятно, по механизму, называемому «ударное сопряжение» (collisional coupling)) G-белок обменивает связанный гуанозиндифосфат (ГДФ) на гуанозинтрифосфат (ГТФ). При этом тример G-белка в ГТФ-связанной форме диссоциирует, высвобождая субъединицу Gα и димер βγ. И Gα и βγ субъединицы затем могут участвовать в дальнейших сигнальных каскадах. Например, субъединица Gα может активировать фермент аденилатциклазу (АЦ), который образует циклический аденозинмонофосфат (цАМФ) из аденозинтрифосфата. Субъединица βγ может активировать ферменты семейства PI-3-киназ. В конечном счете, эти сигналы могут привести к модулированию практически всех аспектов жизнедеятельности клетки, от сокращения до подвижности, от изменения метаболизма до дальнейшей передачи сигналов. Сигнал после активирования затем постепенно «выключается» при помощи различных механизмов. ГТФ, связанный с субъединицей Gα, гидролизуется обратно в ГДФ, что приводит к реассоциации Gα и βγ субъединиц с формированием неактивного тримерного G-белка, связанного с ГДФ. Сам G-белок сопряженный рецептор подвергается фосфорилированию по внутриклеточной C-концевой области, что предотвращает дальнейшее взаимодействие с G-белками. Наконец связанный лиганд также может диссоциировать.
Этот общий сигнальный путь является настолько основополагающим и широко распространенным в физиологии млекопитающих, что до 40% лицензированных фармацевтических препаратов имеют среди своих молекулярных мишеней G-белок сопряженный рецептор. Кроме того, бактерии эволюционировали таким образом, что воздействуют на опосредованную G-белками передачу сигналов с целью нарушения физиологии и иммунитета хозяина: Vibrio cholerae (возбудитель холеры), например, производит белок, известный как холерный токсин, который необратимо ингибирует субъединицу Gα широко распространенных G-белков, называемых Gs. Аналогично, Bordetella pertussis (возбудитель коклюша) производит белок, известный как токсин коклюша, который оказывает аналогичный эффект на другой G-белок, Gi.
Один из подходов для идентификации фармацевтических препаратов, воздействующих на передачу сигналов G-белок сопряженным рецептором, заключался в проведении скрининга библиотек большого числа произвольных соединений на способность препятствовать связыванию лиганда с мембранными препаратами, содержащими рекомбинантные или очищенные G-белок сопряженные рецепторы. Для облегчения обнаружения связывания при проведении такого рода скринингов с высокой пропускной способностью приняты различные способы. Например, при анализе с помощью проксимальной сцинтилляции (scintillation proximity assays) связывание меченного радиоактивным изотопом лиганда с рецептором приводит к сближению радионуклида с молекулой сцинтиллятора, связанной с рецептором, соответственно, при распаде радионуклида испускается излучение, которое можно детектировать и количественно измерять. Кроме того, лиганд может быть помечен флуоресцентно, и связывание выявляют при поляризации флуоресценции (зависящей от уменьшения вращательных степеней свободы флуоресцентной метки при иммоблизации лиганда при его связывании с рецептором).
Хотя применение указанных способов в некоторых случаях имело успех и были выявлены соединения, на основе которых впоследствии были разработаны фармацевтические препараты для человека (например, антагонист рецептора 5НТ3 Ondansetron, применяемый для лечения мигрени), остается множество G-белок сопряженных рецепторов, для которых не были идентифицированы практически никакие подходящие непептидные соединения-агонисты или антагонисты, несмотря на интенсивный скрининг в фармацевтической индустрии. Например, для семейства сопряженных с G-белком хемокиновых рецепторов выявлено незначительное количество специфических непептидных антагонистов и не выявлено агонистов. Поскольку хемокины играют центральную роль в иммунной регуляции, такие молекулы, как ожидают, должны представлять собой чрезвычайно ценные фармацевтические препараты, обладающие иммуномодулирующими свойствами и полезные при лечении широкого спектра заболеваний, включающих воспалительную составляющую.
Два фактора ограничивают вероятный успех программ произвольного скринига: во-первых, существует очень большое количество соединений, которые необходимо проскринировать, и даже при применении наилучшей доступной технологии с высоким выходом и наилучших подходов комбинаторной химии для создания различных библиотек, можно проанализировать лишь небольшую долю всех возможных молекулярных структур. Во-вторых, даже при успешной идентификации исходных соединений коровые фармакофоры часто не пригодны для применения in vivo: исходное соединение или его аналоги могут быть просто слишком токсичными.
Другая основная проблема такого рода подходов «негативного скрининга» (при котором выявляют способность тестируемой библиотеки блокировать связывание меченого лиганда) заключается в том, что большинство из выявленных образцов представляют собой антагонисты рецептора. Лишь немногие из образцов имеют какую-либо агонистическую активность (как предполагают, агонисты должны обладать способностью связываться с рецептором и приводить его в активированное состояние, тогда как антагонистам достаточно только способности связываться с рецептором или лигандом таким образом, предотвращая их взаимодействие), а получение аналогов исходных образцов антагонистов, которые представляли бы собой агонисты, является ненадежным подходом с очень низкой степенью успеха.
Один из способов обойти данную проблему мог бы заключаться в замене библиотеки произвольных соединений на библиотеку молекулярных структур, предварительно отобранных таким образом, чтобы обеспечить значительную долю соединений, связывающихся с G-белок сопряженным рецептором. Такая библиотека в идеальном случае должна содержать как антагонисты, так и агонисты в равном соотношении, и таким образом и те и другие можно было бы легко выявить. Также в идеале основные молекулярные структуры, применяемые в библиотеке, должны быть нетоксичными.
Могут ли быть созданы фактические библиотеки, приближенные к таким идеальным свойствам, или нет, не ясно. Если да, то это потребует существования предполагаемого «идеального» субстрата для G-белок сопряженного рецептора, который бы взаимодействовал с различными G-белок сопряженными рецепторами вне зависимости от и природных лигандных предпочтений. Варьируя заместители у идеализированного субстрата, можно было бы придавать селективность к одному рецептору, принадлежащему к указанному классу в отличие от всех других.
В данной заявке авторы настоящего изобретения описывают «идеальный» субстрат для G-белок сопряженного рецептора. На основании такого «идеального» субстрата авторы настоящего изобретения обеспечивают ряд родственных «остовов», которые могут иметь различные заместители, что приведет к получению агонистов и/или антагонистов ряда различных G-белок сопряженных рецепторов. Изобретение также обеспечивает создание библиотеки вышеуказанных замещенных соединений, а также их применение в ходе скрининга с целью получения лигандов G-белок сопряженных рецепторов с любым заданным набором специфичностей. Таким образом, можно «настраивать» лиганд G-белок сопряженного рецептора, чтобы он имел известный набор свойств (например, лиганд, который обладает агонистической активностью по отношению к рецепторам допамина D2 и в то же самое время антагонистичекой активностью по отношению к 5HT1a (рецепторам) серотонина). В противоположность этому, выявление таких смешанных лигандов из произвольных библиотек является редкой счастливой случайностью.
Необходимые условия для создания библиотек согласно настоящему изобретению включают: (i) идентификацию молекулярного мотива, необходимого для связывания с G-белок сопряженными рецепторами, (ii) остов, включающий указанный молекулярный мотив с сохранением способности связываться с G-белок сопряженным рецептором, но также с обеспечением низкой токсичности и надлежащей стабильности, фармакокинетических и/или фармакодинамических свойств, (iii) легкий способ синтеза с получением различных замещенных форм указанного остова. В основе настоящего изобретения лежит идеальный «субстрат», представленный ниже:
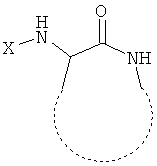
где X представляет собой R-CO- или R-SO2- и R представляет собой различные заместители. Пунктирная линия указывает ограничения для углов связи в пределах молекулярного мотива, например посредством циклизации.
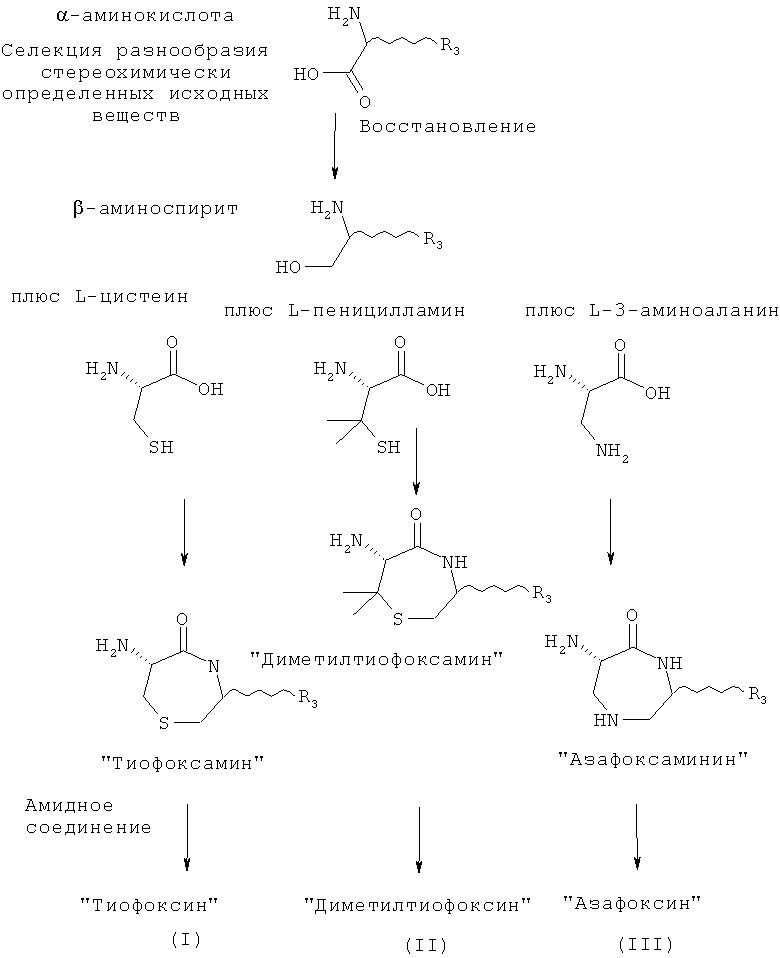
Остовы, предусмотренные данным изобретением, сконструированы с применением широкого спектра доступных α-аминокислот в качестве исходных веществ для создания разнообразных замещенных форм (см., например, Unnatural Amino Acids: Tools for Drug Discovery; Sigma-Aldrich ChemFiles Vol 4 No.6). Каждый из остовов синтезируют на основе комбинации двух α-аминокислот. Первая аминокислота может быть выбрана из цистеина, пеницилламина или 3-аминоаланина. Затем любая аминокислота может быть соединена с первой аминокислотой для создания необходимого разнообразия. Изобретение обеспечивает соединения и их соли общей формулы (I), представляющие собой продукт реакции синтеза с применением цистеина и другой аминокислоты в качестве исходных веществ:
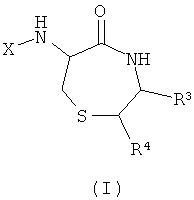
где
X представляет собой -CO-(Y)k-(Z)n или SO2-(Y)k-(Z)n;
k представляет собой 0 или 1;
Y представляет собой циклоалкил или полициклоалкил (например, адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил);
или Y представляет собой циклоалкенил или полициклоалкенил;
каждый Z независимо выбран из водорода или алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила, алкиламино, алкиламиноалкила, алкиламинодиалкила, заряженного алкиламинотриалкила или заряженного алкилкарбоксилатного радикала, содержащего от 1 до 20 атомов углерода;
или каждый Z независимо выбран из фтора, хлора, брома, йода, гидроксигруппы, оксиалкила, амино, аминоалкила, аминодиалкила, заряженного аминотриалкила, или карбоксилатного радикала; и n представляет собой любое целое от 1 до m, где m - максимальное количество замещений, допустимое в циклической группе Y.
Альтернативно, Z может быть выбран из пептидного радикала, например, содержащего от 1 до 4 пептидных звеньев, соединенных пептидными связями (например, пептидного радикала, содержащего от 1 до 4 аминокислотных остатков).
R3 и R4 представляют собой варьируемые заместители, которые наряду с Z различны у отдельных элементов библиотеки.
Этот класс соединений, 6-ациламино-[1,4]тиазепан-5-оны, описывают как «Тиофоксины». Ключевыми структурными характеристиками таких молекул являются наличие лактамамида в кольцевой системе с аминогруппой, присоединенной к атому углерода рядом с лактамкарбонильной группой (6 положение, обозначаемое как α-атом углерода), а также наличие атома серы в положении 1 и R3 и R4 (варьируемых) в положениях 3 и 2 лактамного кольца, соответственно.
Изобретение также обеспечивает соединения и их соли общей формулы (II), представляющие собой продукт реакции синтеза с применением пеницилламина и другой аминокислоты в качестве исходных веществ:
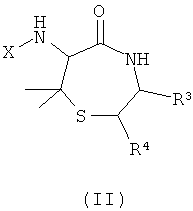
где
X представляет собой -CO-(Y)k-(Z)n или SO2-(Y)k-(Z)n;
k представляет собой 0 или 1;
Y представляет собой циклоалкил или полициклоалкил (например, адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил);
или Y представляет собой циклоалкенил или полициклоалкенил;
каждый Z независимо выбирают из водорода или алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила, алкиламино, алкиламиноалкила, алкиламинодиалкила, заряженного алкиламинотриалкила или заряженного алкилкарбоксилатного радикала, содержащего от 1 до 20 атомов углерода;
или каждый Z независимо выбирают из фтора, хлора, брома, иода, гидроксигруппы, оксиалкила, амино, аминоалкила, аминодиалкила, заряженного аминотриалкила или карбоксилатного радикала; и
n представляет собой любое целое от 1 до m, где m - максимальное количество замещений, допустимое в циклической группе Y.
Альтенативно, Z может быть выбран из пептидного радикала, например, имеющего от 1 до 4 пептидных звеньев, соединенных пептидными связями (например, пептидного радикала, содержащего от 1 до 4 аминокислотных остатков).
R3 и R4 представляют собой различные заместители, которые наряду с Z различны у отдельных элементов библиотеки. R3 и R4 могут быть независимо выбраны как любые заместители, за исключением того, что R3 не может представлять собой -СООН, -COOR', -COSR' или -CONR'R'', где R' и R'' независимо представляют собой любой заместитель и любой или оба из R' и R'' могут представлять собой H. Этот класс соединений, 6-ациламино-7,7-диметил[1,4]тиазепан-5-оны, описывают как «Диметилтиофоксины». Ключевыми структурными характеристиками таких молекул являются наличие лактамамида в кольцевой системе с аминогруппой, присоединенной к атому углерода рядом с лактамкарбонильной группой (6 положение, обозначаемое α-атом углерода), а также наличие атома серы в положении 1 и R3 и R4 (варьируемых) в положениях 3 и 2 лактамного кольца, соответственно. Изобретение также обеспечивает соединения и их соли общей формулы (III), представляющие собой продукт реакции синтеза с применением 3-аминоаланина и другой аминокислоты в качестве исходных веществ:
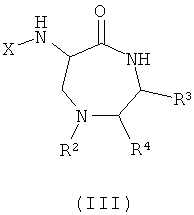
где
X представляет собой -CO-(Y)k-(Z)n или SO2-(Y)k-(Z)n;
k представляет собой 0 или 1;
Y представляет собой циклоалкил или полициклоалкил (например, адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил);
или Y представляет собой циклоалкенил или полициклоалкенил;
каждый Z независимо выбран из водорода или алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила, алкиламино, алкиламиноалкила, алкиламинодиалкила, заряженного алкиламинотриалкила или заряженного алкилкарбоксилатного радикала, содержащего от 1 до 20 атомов углерода;
или каждый Z независимо выбран из фтора, хлора, брома, йода, гидроксигруппы, оксиалкила, амино, аминоалкила, аминодиалкила, заряженного аминотриалкила или карбоксилатного радикала; и n - любое целое от 1 до m, где m - максимальное количество замещений, допустимое в циклической группе Y.
В качестве альтернативы, Z может быть выбран из пептидного радикала, например, имеющего от 1 до 4 пептидных звеньев, соединенных пептидными связями (например, пептидного радикала, содержащего от 1 до 4 аминокислотных остатков).
R2, R3 и R4 представляют собой различные заместители, которые наряду с Z различны у отдельных элементов библиотеки.
Этот класс соединений, 6-ациламино-[1,4]диазепан-5-оны, описывают как «Азафоксины». Ключевыми структурными характеристиками таких молекул являются наличие лактамамида в кольцевой системе с аминогруппой, присоединенной к атому углерода рядом с лактамкарбонильной группой (положение 6, обозначаемое как α-атом углерода), а также наличие азота в 1 положении и R3 и R4 (варьируемых) в 3 и 2 положениях лактамного кольца, соответственно. Дополнительное разнообразие может быть создано при помощи заместителя R2 (варьируемого) в положении N1.
Изобретение также обеспечивает соединения и их соли общей формулы (IV), представляющие собой альтернативный продукт реакции синтеза с применением 3-аминоаланина и другой аминокислоты в качестве исходных веществ:
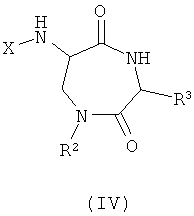
где
X представляет собой -CO-(Y)k-(Z)n или SO2-(Y)k-(Z)n;
k представляет собой 0 или 1;
Y представляет собой циклоалкил или полициклоалкил (например, адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил);
или Y представляет собой циклоалкенил или полициклоалкенил;
каждый Z независимо выбран из водорода или алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила, алкиламино, алкиламиноалкила, алкиламинодиалкила, заряженного алкиламинотриалкила или заряженного алкилкарбоксилатного радикала, содержащего от 1 до 20 атомов углерода;
или каждый Z независимо выбран из фтора, хлора, брома, иода, гидроксигруппы, оксиалкила, амино, аминоалкила, аминодиалкила, заряженного аминотриалкила или карбоксилатного радикала; и n - любое целое от 1 до m, где m - максимальное количество замещений, допустимое в циклической группе Y.
Альтернативно, Z может быть выбран из пептидного радикала, например, имеющего от 1 до 4 пептидных звеньев, соединенных пептидными связями (например, пептидного радикала, содержащего от 1 до 4 аминокислотных остатков).
R2 и R3 представляют собой различные заместители, которые наряду с Z различны для отдельных элементов библиотеки.
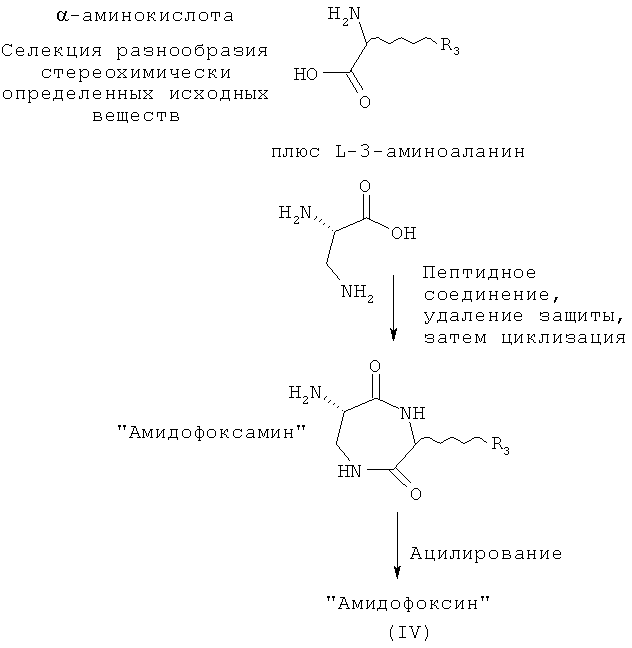
Этот класс соединений, 6-ациламино-[1,4]диазепан-2,5-дионы, описывают как «Амидофоксины». Ключевыми структурными характеристиками молекул являются наличие лактамамида в кольцевой системе с аминогруппой, присоединенной к атому углерода рядом с лактамкарбонильной группой (6 положение, обозначаемое как α-атом углерода), а также наличие азота в положении 1 и карбонильной группы в положении 2 с R3 (варьируемого) в положении 3 лактамного кольца. Дополнительное разнообразие может быть создано при помощи заместителя R2 (варьируемого) в положении N1.
α-атом углерода указанных тиофоксинов, диметилтиофоксинов, азафоксинов и амидофоксинов является асимметричным и, следовательно, соединения согласно настоящему изобретению имеют две возможные энантиомерные формы, то есть R и S конфигурации. Настоящее изобретение охватывает две энантиомерные формы и все комбинации этих форм, в том числе RS рацемические смеси. Для простоты, если в структурных формулах не указана конкретная конфигурация, то следует понимать, что представлены две энантиомерные формы и их смеси.
Все соединения общих формул (I), (II), (III) и (IV) содержат N-заместители в экзоциклической аминогруппе. N-заместитель представляет собой либо карбонамид, либо сульфонамид. Геометрия атома углерода рядом с карбонилом карбонамида или сульфонильной группой сульфонамида («ключевого» углерода) может быть важна для биологической активности молекулы. Природа N-заместителя может быть такова, что кольцо или кольца Y ограничивают углы связи при «ключевом» углероде, делая их по существу тетраэдрическими (т.е. sp3 гибридные связи). Любой заместитель Z может представлять собой заместитель в любом допустимом положении кольца или колец циклической группы Y. В частности, следует отметить, что изобретение охватывает соединения, у которых «ключевой углерод» является и частью циклической группы и сам является замещенным. Определение (Z)n охватывает соединения данного изобретения, не содержащие заместителей (т.е. Z = водород), соединения данного изобретения с одним заместителем (т.е. Z не является водородом и n=1), и также с множественными заместителями (т.е., по меньшей мере, две группы Z не являются водородом и n=2 или более).
Одно из основных преимуществ соединений согласно настоящему изобретению состоит в том, что различные элементы библиотеки могут быть легко синтезированы с применением широкодоступных исходных веществ. Тиофоксины, диметилтиофоксины, азафоксины и амидофоксины представляют собой различные классы соединений (с разнообразными Z, R2 (при наличии), R3 и (при наличии) R4), которые могут быть легко синтезированы из двух различных α-аминокислот. α-аминокислоты представляют собой идеальные исходные вещества для синтеза с целью получения репертуара разнообразных соединений, поскольку известен широкий спектр α-аминокислот (отличающихся только природой заместителя R3), и они коммерчески доступны. Для синтеза тиофоксинов, диметилтиофоксинов и азафоксинов α-аминокислоты можно легко восстановить с получением β-аминоспиртов (с применением защитных групп для сохранения структуры группы R3 при необходимости). Затем различные β-аминоспирты соединяют с цистеином с получением «тиофоксаминов», с пеницилламином с получением «диметилтиофоксаминов» или с 3-аминоаланином с получением «азафоксаминов». Указанные α-аминолактамы можно затем соединить с соответствующим ацилом в качестве боковой цепи при помощи стандартных реакций амидного присоединения (также с применением защитных групп для заместителя R3 при необходимости) с получением тиофоксинов, диметилтиофоксинов или азафоксинов, соответственно.
В отличие от этого, амидофоксины получают при реакции α-аминокислоты (без предварительного ее восстановления до β-аминоспирта) с 3-аминоаланином. Следует отметить, что амидофоксины представляют собой 7-членные кольцевые аналоги хорошо известных дикетопиперазинов, синтезируемых из димеров α-аминокислот, но синтезированные на основе димера β-аминокислоты (3-аминоаланина) и α-аминокислоты.
Образующиеся тиофоксины, диметилтиофоксины, азафоксины и амидофоксины имеют тот же ряд заместителей R3, что и исходная совокупность доступных α-аминокислот. Кроме того, в данной области хорошо известны общие пути синтеза α-аминокислот (например, см. R.M.Williams, Synthesis of Optically Active α-Amino Acids (Pergamon, New York) 1989), делающие возможным создание еще большего разнообразия при необходимости.
В качестве альтернативы или дополнительно, применение непосредственно доступных β-аминоспиртов, таких как эфедрин, с различными заместителями в положении 2 обеспечивает дополнительное разнообразие за счет варьируемой группы R4 в положении 2 кольца тиофоксина, диметилтиофоксина и азафоксина.
В качестве альтернативы или дополнительно, предшествующее восстановительное алкилирование 3-аминоаланина позволяет получение разнообразия по N1 положению азафоксина или амидофоксина (варьирующая R2 группа).
Особенностью настоящего изобретения является легкость стереоконтроля синтеза. В приведенных в качестве примеров путях синтеза применяют дешевые и легко доступные L-цистеин, L-пеницилламин и L-3-аминоаланин для соединения с разнообразными β-аминоспиртами (или непосредственно с α-аминокислотами в случае амидофоксинов). В результате этого получают ряд различных α-аминолактамов, имеющих (S)-конфигурацию. Кроме того, D-цистеин, D-пеницилламин и D-3-аминоаланин также являются легкодоступными и могут быть соединены с таким же рядом β-аминоспиртов (или α-аминокислот) с получением α-аминолактамов (R)-конфигурации. Аналогично, при выборе в качестве исходного вещества соответствующей энантиомерно чистой α-аминокислоты и затем с применением пути синтеза с сохранением стереохимии (например, путей синтеза, приведенных ниже) можно легко синтезировать тиофоксамины, диметилтиофоксамины, азафоксамины и/или амидофоксамины и, следовательно, тиофоксины, диметилтиофоксины, азафоксины и/или амидофоксины с соответствующей конфигурацией атома углерода, несущего R3-заместитель. Например, при синтезе тиофоксина с применением L-аланина и L-цистеин получают (6R)-амино-(3S)-метил-[1,4]тиазапан-5-он. В противоположность этому, при синтезе с применением D-аланина и L-цистеина получают (R,R)-6-амино-3-метил-[1,4]тиазапан-5-он, с применением L-аланина и D-цистеина получают (S,S)-6-амино-3-метил-[1,4]тиазапан-5-он, а с применением D-аланина и D-цистеина получают (6S)-амино-(3R)-метил-[1,4]тиазапан-5-он. В качестве альтернативы, можно выбрать рацемические смеси одного или обоих исходных веществ с получением в качестве продуктов смеси стереоизомеров тиофоксаминов, диметилтиофоксаминов, азафоксаминов и/или амидофоксаминов и, следовательно, производных тиофоксинов, диметилтиофоксинов, азафоксинов и/или амидофоксинов.
Важным является то, что при необходимости возможно осуществление стадий реакций синтеза в различном порядке. При создании библиотеки различных соединений путем комбинаторного синтеза важно получить в конце синтеза как можно большее разнообразие. В идеале последняя стадия синтеза вносит наибольшее разнообразие в библиотеку. Поскольку разнообразие может быть получено на различных стадиях синтеза соединений согласно настоящему изобретению (с варьируемыми Z, R2 (для азафоксинов и амидофоксинов), R3 и R4 (для тиофоксинов, диметилтиофоксинов и азафоксинов)), можно вводить разнообразие в равной степени на каждой стадии (например, создавать библиотеку амидофоксинов с 8 различными группами Z, группами R2 и группами R3, таким образом, получая 512 соединений) или вводить большее разнообразие на одной из стадии синтеза (например, создавать библиотеки с 2 различными группами Z и R2, но с 128 различными группами R3 в библиотеке амидофоксинов, таким образом, получая 512 соединений). В случаях, когда разнообразие вводят по большей части на одной определенной стадии, предпочтительно осуществление данной стадии по возможности в конце пути синтеза. Одно из преимуществ настоящего изобретения, предусмотренное в данной заявке, состоит в том, что можно изменить порядок стадий реакции синтеза. Специфические примеры путей синтеза с получением тиофоксинов, диметилтиофоксинов, азафоксинов и амидофоксинов приведены в примерах ниже. Для каждого класса представлены примеры, в которых стадии реакции выполняют в различном порядке. Однако следует отметить, что существуют другие пути синтеза, которые можно применять в качестве альтернативы, и они входят в область настоящего изобретения.
Предполагается, что синтез компонентов библиотеки можно осуществлять с применением аналогичных способов синтеза, хорошо известных в данной области. Например, синтез можно осуществлять с применением смол или других твердофазных носителей для упрощения введения разнообразия и для облегчения очистки или частичной очистки образующихся компонентов библиотеки. Применение такого рода твердофазного или другого аналогичного способа синтеза, неавтоматизированного, полуавтоматизированного или автоматизированного, для создания библиотеки тиофоксинов, диметилтиофоксинов, азафоксинов или амидофоксины входит в область настоящего изобретения.
Изобретение также обеспечивает фармацевтические композиции, которые в качестве активного компонента содержат соединение общей формулы (I), (II), (III) или (IV), или его фармацевтически приемлемую соль и, по меньшей мере, один фармацевтически приемлемый наполнитель и/или носитель.
Под фармацевтически приемлемой солью понимают, в частности, аддитивные соли неорганических кислот, такие как гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат, бифосфат и нитрат или соли органических кислот, такие как ацетат, малеат, фумарат, тартрат, сукцинат, цитрат, лактат, метансульфонат, р-толуолсульфонат, пальмоат и стереат. Также в область настоящего изобретения, в случаях, когда их можно применять, входят соли, образованные из оснований, например, гидроокиси натрия или калия. Другие примеры фармацевтически приемлемых солей см. в работе "Salt selection for basic drugs", Int. J. Pharm. (1986), 33, 201-217.
Фармацевтическая композиция может быть в твердой форме, например в виде порошков, гранул, таблеток, желатиновых капсул, липосом или суппозиториев. Подходящей твердой основой может служить, например, фосфат кальция, стереат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, карбоксиметилцеллюлоза натрия, поливинилпирролидин и парафин. Другие подходящие фармацевтически приемлемые наполнители и/или носители известны специалистам в данной области.
Фармацевтические композиции согласно настоящему изобретению также могут быть представлены в жидкой форме, например, в виде растворов, эмульсий, суспензий или сиропов. Подходящей жидкой основой может быть, например, вода, органические растворители, такие как глицерол или гликоли, а также их водные смеси в различных пропорциях.
Настоящее изобретение может также обеспечивать применение соединения общей формулы (I), (II), (III) и/или (IV), или его фармацевтически приемлемой соли для получения лекарственного препарата, предназначенного для модулирования активности одного или более членов класса G-белок сопряженных рецепторов.
Настоящее изобретение обеспечивает соединения, композиции и применение соединений общей формулы (I), (II), (III) и (IV) или их фармацевтически приемлемые солей, в которых радикал R1 содержит «ключевой» углерод, двузамещенный одинаковыми или различными группами, выбранными из: алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила и алкиламино радикала.
Настоящее изобретение предусматривает соединения, композиции и применение, где «ключевой» углерод является хиральным.
Настоящее изобретение предусматривает соединения, композиции и применение, где «ключевой» углерод образует sp3 гибридные связи.
Настоящее изобретение предусматривает соединения, композиции и применение, где «ключевой» углерод образует по существу тетраэдрические углы связи.
Соединения общей формулы (I), (II), (III) или (IV), применяемые согласно настоящему изобретению, или их соли, могут быть такими, что кольцо или кольца Y ограничивают углы связи при «ключевом» углероде, делая их по существу тетраэдрическими (т.е. sp3 гибридные связи).
Настоящее изобретение также обеспечивает сульфаниламидные аналоги приведенных в качестве примеров соединений: сульфонил-α-аминолактамные эквиваленты тиофоксинов, диметилтиофоксинов, азафоксинов или амидофоксинов формулы (I), (II), (III) и (IV), соответственно.
Настоящее изобретение охватывает соединения, композиции и их применение, как определено в данной заявке, когда указанные соединения находятся в гидратированной или сольватированной форме.
Амидные и сульфонамидные производные тиофоксинов, диметилтиофоксинов, азафоксиноа и амидофоксинов, описанных в данной заявке, возможно, представляют собой функциональные агонисты и антагонисты G-белок сопряженных рецепторов. Центральная часть, «кор», состоящая из «ключевого» углерода, карбонильной или сульфонильной группы, α-аминогруппы и кольца тиофоксина, диметилтиофоксина, азафоксина или амидофоксина, представляет собой пример лиганда G-белок сопряженного рецептора. Варьируя заместители данного кора, в особенности в положении, содержащем R3 заместитель, можно получить агонисты и антагонисты G-белок сопряженных рецепторов с широким спектром желаемых свойств намного более легким способом, чем при скрининге библиотек произвольных соединений.
В результате, изобретение также обеспечивает библиотеку, которая содержит два или более представителя класса соединений, определяемых общей формулой (I), (II), (III) и/или (IV), такую библиотеку, которую можно скринировать для идентификации молекулы с конкретным требуемым набором свойств в отношении модулирования передачи сигнала от одного (или более) G-белок сопряженного рецептора. Затем можно проводить скрининг вышеупомянутой библиотеки на антагонистическую или агонистическую активность по отношению к вышеупомянутому G-белок сопряженному рецептору (рецепторам) с применением способов, хорошо известных в данной области. Например, можно проводить скрининг указанной библиотеки в отношении способности ее отдельных элементов блокировать связывание меченного радиоактивным изотопом лиганда G-белок сопряженного рецептора с препаратами мембраны, содержащими рекомбинантный или очищенный G-белок сопряженный рецептор. Кроме того, можно проводить скрининг библиотеки на способность отдельных ее элементов стимулировать образование цАМФ в клетках, экспрессирующих рекомбинантный G-белок сопряженный рецептор.
Любое соединение тиофоксина, диметилтиофоксина, азафоксина или амидофоксина согласно настоящему изобретению, обладающее требуемым набором свойств, можно применять в качестве «шаблона» для синтеза аналогичного «карбофоксина» (имеющего углеродную группу, вместо серы, в положении 1 тиофоксина или диметилтиофоксина или азота в положении 1 азафоксина или амидофоксина), например, с применением путей синтеза при помощи реакции обмена с замыканием кольца, которые хорошо известны в данной области (например, Truka, Т.М.; Grubbs, R.Н. Acc. Chem. Res. 2001, 34, 18). Соответствующие пути синтеза с применением реакции обмена с замыканием кольца также охарактеризованы, их можно применять для синтеза «карбофоксиновых» аналогов азафоксинов или амидофоксинов, в которых N в положении 1 является замещенным (т.е., R2≠Н), например, способ Del Valle R.R. & Goodman M.J. Org. Chem. 2004, 69.8946. Предполагается, что такие «карбофоксиновые» соединения являются эффективными агонистами или антагонистами G-белок сопряженных рецепторов.
Изобретение также предусматривает способы лечения, облегчения или профилактики симптомов заболевания или патологического состояния, выбранного из группы, включающей гипертонию, атеросклероз, астму, ожирение, нейродегенеративные заболевания, аутоиммунные заболевания или психопатические заболевания, путем введения пациенту эффективного количества соединения, композиции или лекарственного препарата согласно настоящему изобретению, предназначенного для модулирования активности G-белок сопряженного рецептора.
ОПРЕДЕЛЕНИЯ
Термин «около/примерно» относится к интервалу вокруг рассматриваемого значения. При использовании в данной заявке на патент «около/примерно X» означает интервал от Х минус 10% от Х до Х плюс 10% от X, и предпочтительно интервал от Х минус 5% от Х до Х плюс 5% от X.
Использование числового диапазона в данном описании предназначено для того, чтобы однозначно включить в область изобретения каждое конкретное целое значение данного диапазона и все комбинации значений верхнего и нижнего предела расширенной области данного диапазона. Таким образом, например, диапазон от 1 до 20 атомов углерода, указанный (в числе прочего) в отношении формулы I, охватывает все целые от 1 до 20 и все подобласти каждой комбинации верхних и нижних значений, приведены ли в качестве примеров в явной форме или нет.
При использовании в данном описании термин «включающий» следует понимать и как «включающий/содежащий» и как «состоящий из». Следовательно, если указано, что изобретение относится к «фармацевтической композиции, включающей в качестве активного компонента» соединение, то данные термины относятся и к композициям, в которых могут быть представлены другие активные компоненты, и к композициям, состоящим только из одного определенного активного компонента.
Термин «пептидные звенья», используемый в данной заявке, обозначает следующие 20 природных аминокислотных остатков:
Модифицированные и редкие аминокислотные остатки, как и пептидомиметики, также входят в объем понятия «пептидные звенья». Если не определено иначе, все технические и научные термины, используемые в данной заявке, имеют значение, которое обычно понимается специалистом в области техники, к которой принадлежит данной изобретение. Аналогично, все публикации, заявки на патент, все патенты и другие источники, упомянутые в данной заявке, включены посредством ссылки (если это юридически допустимо).
Следующие примеры иллюстрируют, но никоим образом не ограничивают область изобретения.
ПРИМЕРЫ
В каждом из следующих примеров применяют спектр защитных групп. Функциональные свойства требуемых защитных групп уточнены (т.е. на определенных стадиях требуется применение 2 различных защитных групп, которые удаляют при различных условиях реакции, то есть ортогональных защитных групп), но молекулярный состав не уточнен. Можно применять любую подходящую защитную группу, хорошо известную из уровня техники. Вследствие этого в нижеследующих примерах варьируемые элементы таких защитных групп обозначены как R5, R6, R7 и/или R8. Защитные группы (и, следовательно, заместители R5, R6, R7 и R8) сами не являются частями продукта, входящего в объем охраны настоящего изобретения.
ПРИМЕР 1: Синтез тиофоксинов
Тиофоксины представляют собой продукты реакции сочетания цистеина с β-аминоспиртом (возможно, полученным при реакции восстановления α-аминокислоты). В первой схеме (Схема 1А ниже), вводят группу R1, затем группу R3/R4 и затем соединения циклизуют. Такой путь является оптимальным, если в (значения) R3/R4 требуется внести большее разнообразие, чем в R1.
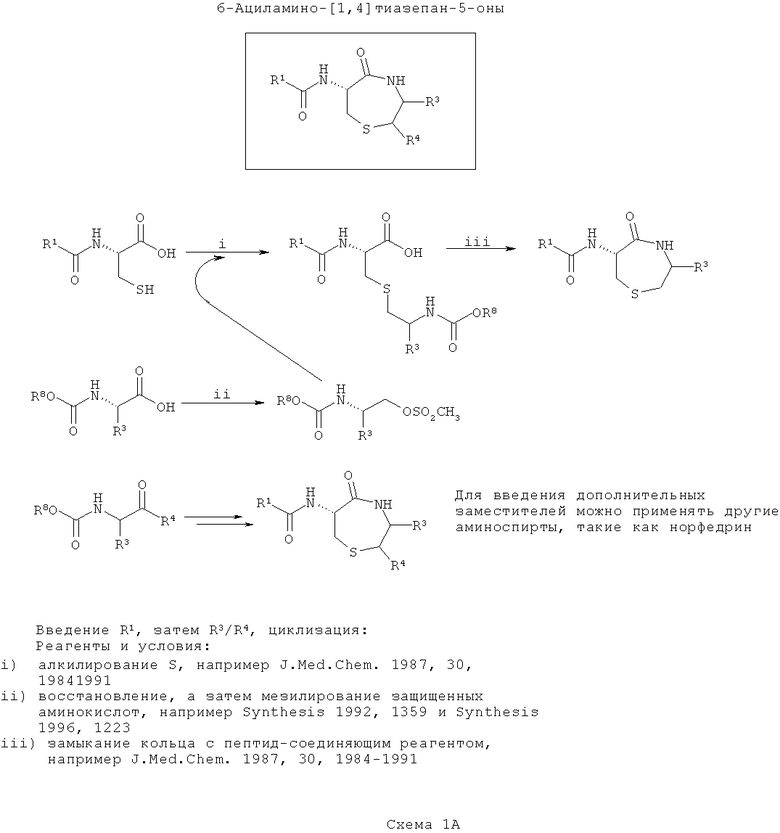
На первой стадии R1-содержащий ацильный (или сульфонамидный) заместитель вводят подходящим способом амидного соединения, несколько из которых хорошо известны в данной области техники, например DCC соединения.
В отдельности, один или более β-аминоспирт (например, эфедрин) получают или синтезируют из α-аминокислот. В каждом случае аминогруппа защищена (например, добавлением группы Boc или получением подходящим образом защищенной α-аминокислоты из коммерчески доступных источников). Вос-защищенные аминокислоты превращают в аминоспирты путем восстановления, а Вос-защищенный β-аминоспирт (купленный или полученный восстановлением аминокислоты) модифицируют так, чтобы обеспечить подходящую уходящую группу для алкилирования гетероатома боковых цепей (в данном случае, атом серы цистеина), например амино-защищенный β-аминоспирт может быть мезилирован. Существует несколько подходящих способов для восстановления и мезилирования, и они хорошо известны в данной области техники (например, Synthesis (1992) 1359 или Synthesis (1996) 1223).
На следующей стадии ацилцистеин алкилируют по центральному атому серы с применением любого из нескольких подходящих способов, которые хорошо известны в данной области (например, J Med Chem. (1987) 30: 1984). Каждый β-аминоспирт реагирует отдельно с подходящим ацилцистеином, при этом на выходе получают библиотеку индивидуальных элементов с различными R1, R2 и R3 в зависимости от выбора доступных ацилцистеинов и β-аминоспиртов. Азот, введенный на этой стадии, обозначен как ω-аминогруппа.
На заключительной стадии S-алкилацилцистеины циклизуют. У ω-амина селективно убирают защиту и его конденсируют с карбоксиэфиром (или после селективного гидролиза соответствующей карбоновой кислоты) с образованием 7-членного кольца с получением библиотеки тиофоксинов. Сходные способы хорошо известны в данной области техники (например, J. Med. Chem (1987) 30: 1984).
На второй схеме (Схема 1Б ниже) вводят группы R3/R4, затем соединения циклизуют и, наконец, вводят группу R1. Такой путь является оптимальным, если в значение R1 требуется ввести большее разнообразие, чем в R3/R4.
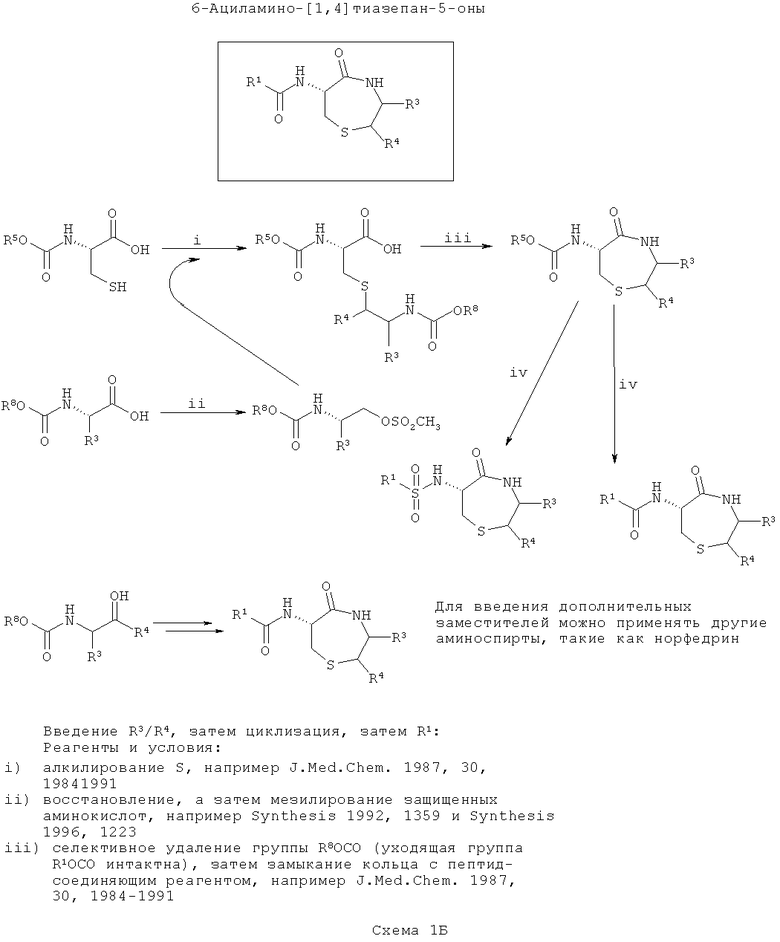
На первой стадии цистеин селективно защищают по α-амину и затем алкилируют по S с N-защищенным β-аминоспиртом, где спирт активируют с образованием уходящей группы, подходящей для нуклеофильного замещения с инверсией стереохимии атома углерода. Введенный азот здесь назван как ω-аминогруппа (аналогичное см. в Tetrahedron, 1999, 55, 10155).
Один или более β-аминоспирт, подходящий для алкилирования (например, эфедрин), получают или синтезируют из α-аминокислот. В каждом случае аминогруппу защищают (например добавлением группы Boc или получением подходящим образом защищенной α-аминокислоты из коммерчески доступных источников). Вос-защищенные аминокислоты превращают в аминоспирты восстановлением, а Вос-защищенный β-аминоспирт (либо купленный, либо полученный восстановлением из аминокислоты) мезилируют. Существует несколько подходящих способов для восстановления и мезилирования, и они хорошо известны в данной области техники (например, Synthesis (1992) 1359 или Synthesis (1996) 1223).
На следующей стадии у ω-амина селективно убирают защиту и его конденсируют с карбоксиэфиром (или после селективного гидролиза соответствующая карбоновая кислота) с образованием 7-членного кольца (согласно J. Med. Chem., 1987, 30, 1984).
На конечной стадии после циклизации у α-амина селективно убирают защиту и ацилируют его, как требуется для введения разнообразия в положение R1 с применением соответствующего реагента для соединения пептидов, некоторые из которых хорошо известны в данной области техники.
На третьей схеме (Схема 1В ниже) вводят группы R3/R4, затем группу R1, и, наконец, соединения циклизуют. Такой путь можно применять, если необходимо ввести большее разнообразие в R1, чем R3/R4.
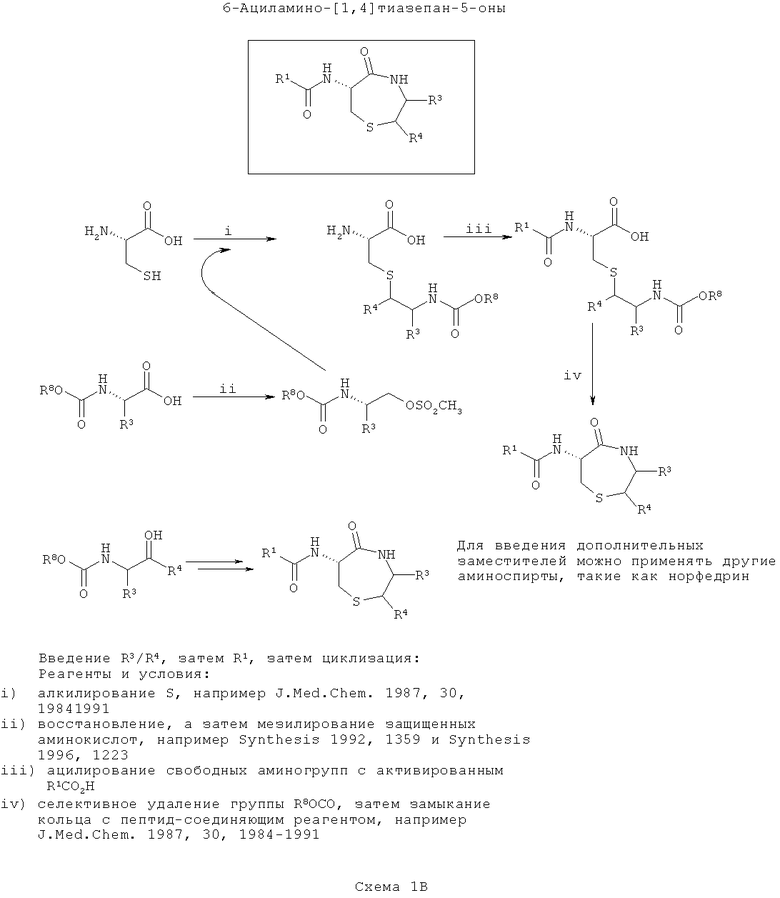
Этот путь сходен со схемой 1Б за исключением того, что ацилирование α-амина цистеина выполняют перед циклизацией (аналогичное см. в Tetrahedron, 1999, 55, 10155).
Эти три схемы иллюстрируют простоту синтеза библиотеки, состоящей из одного или более тиофоксина. В частности, проиллюстрирована возможность выполнения стадий реакции в различном порядке для введения большего разнообразия в различные участки молекулы при сохранении применимости на практике данного способа синтеза.
В литературе уже известно множество родственных структур, в том числе два примера соединений, описанных как лиганды G-белок сопряженного рецептора.
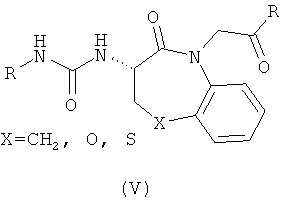

Соединение (V) представляет собой лиганд рецептора гистамина Н2 или гастрина (в зависимости от природы R и X), описанные в Bioorg. Med. Chem (1997) 5: 1411, а соединение (VI) представляет собой лиганд рецептора нейропептида Y, описанный в Bioorg. Med. Chem, (1999) 7: 1703. Хотя они, несомненно, схожи по структуре с тиофоксином, ни одно из этих соединений не является тиофоксином и не входит в объем настоящего изобретения из-за наличия заместителя в положении N4. Однако существование этих соединений подчеркивает высокую плотность лигандов G-белок сопряженных рецепторов, которые, вероятно, должны быть обнаружены в библиотеках, состоящих из или обогащенных тиофоксинами.
ПРИМЕР 2 - Синтез диметилтиофоксинов
Диметилтиофоксины аналогичны тиофоксинам за исключением того, что вместо цистеина применяют пеницилламин. Диметилтиофоксины являются продуктами соединения пеницилламина с β-аминоспиртом (возможно полученным при восстановлении α-аминокислоты). На первой схеме (Схема 2А ниже) вводят группу R1, затем группу R3/R4, а затем соединения циклизуют. Такой путь является оптимальным, если необходимо ввести большее разнообразие в значения заместителей R3/R4, чем R1.
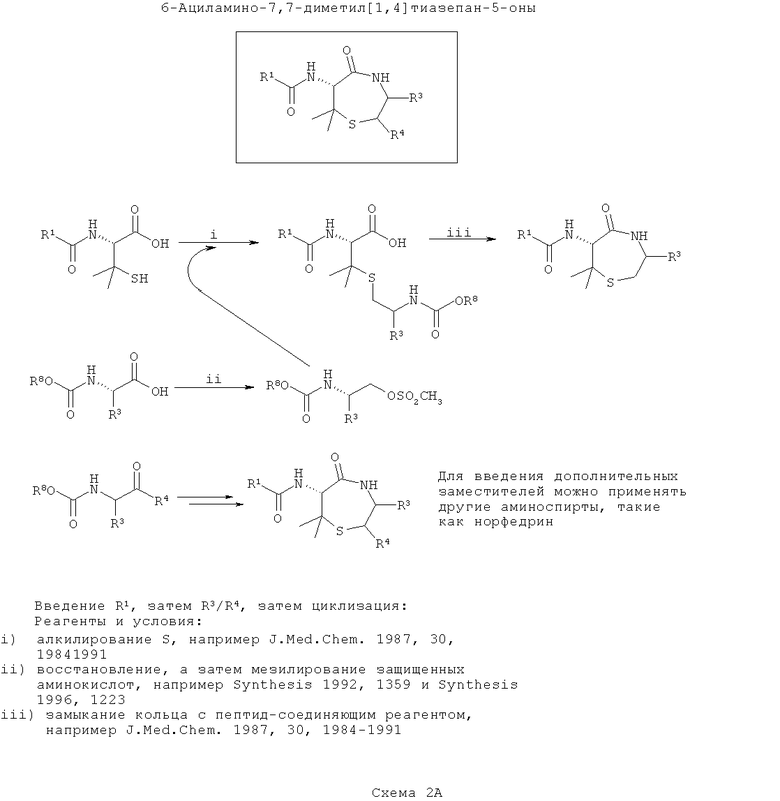
На первой стадии R1-содержащий ацильный (или сульфонамидный) заместитель вводят в пеницилламин подходящим способом амидного сочетания, некоторые из которых хорошо известны в данной области техники, например DCC сочетания.
В отдельности, получают или синтезируют из α-аминокислот один или более β-аминоспирт (например, эфедрин). В каждом случае защищают аминогруппу (например, добавлением группы Boc или получением подходящим образом защищенной α-аминокислоты из коммерчески доступных источников). Вос-защищенные аминокислоты превращают в аминоспирты путем восстановления, а Вос-защищенный β-аминоспирт (купленный или полученный восстановлением аминокислоты) модифицируют так, чтобы обеспечить подходящую уходящую группу для алкилирования гетероатома боковых цепей (в данном случае, атома серы цистеина), например, аминозащищенный β-аминоспирт может быть мезилирован. Существует несколько подходящих способов для восстановления и мезилирования, и они хорошо известны в данной области техники (например, Synthesis (1992) 1359 или Synthesis (1996) 1223).
На следующей стадии ацилпеницилламин алкилируют по центральному атому серы с применением любого из нескольких подходящих способов, которые хорошо известны в данной области техники (например, J. Med Chem. (1987) 30: 1984). Каждый β-аминоспирт реагирует отдельно с подходящим ацилцистеином, образуя на выходе библиотеку индивидуальных элементов с различными R1, R2 и R3 в зависимости от выбора доступных ацилцистеинов и β-аминоспиртов. Азот, введенный на этой стадии, в данном описании обозначают как ω-азот.
На конечной стадии S-алклиацилпеницилламин циклизуют. У ω-амина селективно убирают защиту и его конденсируют с карбоксиэфиром (или после селективного гидролиза соответствующая карбоновая кислота) с образованием 7-членного кольца с получением библиотеки диметилтиофоксинов. Сходные способы хорошо известны в данной области техники (например, J. Med. Chem (1987) 30:1984).
На второй схеме (Схема 2Б ниже) вводят группы R3/R4, затем соединения циклизуют и, наконец, вводят группу R1. Такой путь является оптимальным, если необходимо ввести большее разнообразие в значения R1, чем в R3/R4.
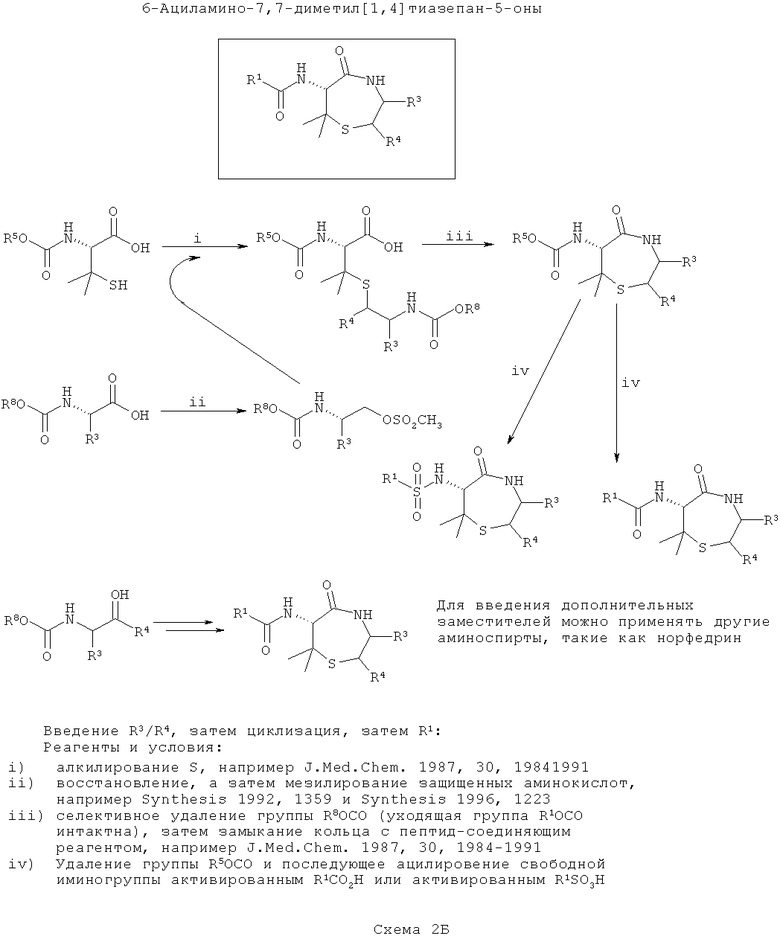
Схема 2Б
На первой стадии пеницилламин селективно защищают по α-амину и затем алкилируют по S с N-защищенным β-аминоспиртом, в котором спирт активируют с образованием уходящей группы, подходящей для нуклеофильного замещения с инверсией стереохимии атома углерода. Введенный азот в данном описании называют ω-аминогруппа (аналогичное см. в Tetrahedron, 1999, 55, 10155).
Получают или синтезируют из α-аминокислот один или более β-аминоспирт, подходящий для алкилирования (например, эфедрин). В каждом случае аминогруппу защищают (например добавлением группы Boc или получением подходящим образом защищенной α-аминокислоты из коммерчески доступных источников). Вос-защищенные аминокислоты превращают в аминоспирты посредством восстановления, а Вос-защищенный β-аминоспирт (либо купленный, либо полученный восстановлением из аминокислоты) мезилируют. Существуют несколько подходящих способов для восстановления и мезилирования, и они хорошо известны в данной области техники (например, Synthesis (1992) 1359 или Synthesis (1996) 1223).
На следующей стадии у ω-амина селективно убирают защиту и конденсируют его с карбоксиэфиром (или после селективного гидролиза соответствующей карбоновой кислоты) с образованием 7-членного кольца (согласно J. Med. Chem., 1987, 30, 1984).
На конечной стадии после циклизации у α-амина селективно убирают защиту и ацилируют, что требуется для введения разнообразия в положение R1, с применением соответствующего реагента для соединения пептидов, некоторые из которых хорошо известны в данной области техники.
На третьей схеме (схема 2В ниже) вводят группы R3/R4, затем группу R1 и, наконец, соединения циклизуют. Такой путь можно применять, если требуется ввести большее разнообразие в значения R1, чем в R3/R4.
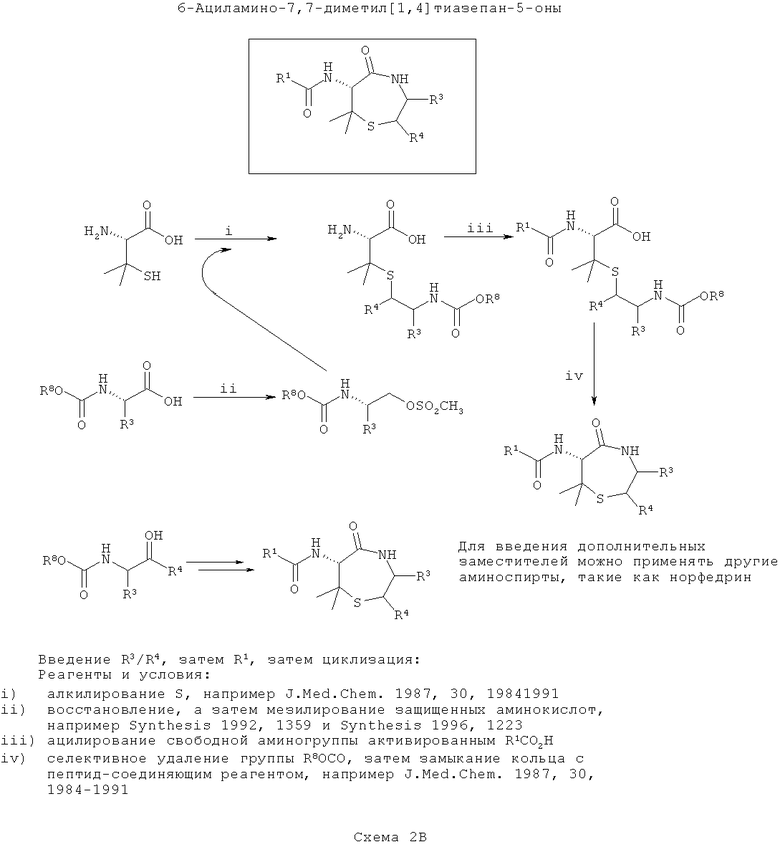
Этот путь сходен со Схемой 2Б за исключением того, что ацилирование α-амина пеницилламина выполняют перед циклизацией (аналогичное см. в Tetrahedron, 1999, 55, 10155).
Эти три схемы иллюстрируют простоту синтеза библиотеки, состоящей из одного или более диметилтиофоксина. В частности, проиллюстрирована возможность выполнения стадий реакции в различном порядке для введения большего разнообразия в различные участки молекулы при сохранении применимости на практике данного способа синтеза.
Известны многие соединения, родственные по структуре диметилтиофоксину (см., например, J. Chem. Soc., Chem. Commum., 1993, 1599 и Liebigs Ann. Reel. 1997, 1711.), поскольку продукты циклизации между пеницилламином и другими α-аминокислотами являются аналогами или промежуточными соединениями в биосинтезе хорошо изученных β-лактамовых антибиотиков, таких как пенициллин. Однако эти аналоги пенициллина содержат карбоксилат или эфирное, тиоэфирное или амидное производное карбоксилата в качестве заместителя в положении 3 кольца.
ПРИМЕР 3 - Синтез азафоксинов
Азафоксины представляют собой продукты реакции сочетания 3-аминоаланина с β-аминоспиртом (возможно, полученном при восстановлении α-аминокислоты). В отличие от серосодержащих лактамов (тиофоксинов и диметилтиофоксинов) введение дополнительного азота в лактамовое кольцо в азафоксинах делает возможным существование дополнительного заместителя (и, следовательно, разнообразия) в гетероатоме кольца. На первой схеме (Схема 3А ниже) вводят группу R1, затем группу R2 (замещение по азоту) и затем группу R3/R4 перед тем, как, наконец, соединения циклизуют. Такой путь является оптимальным, если необходимо ввести большее разнообразие в значения R3/R4, чем в R2 с наименьшим разнообразием по R1.
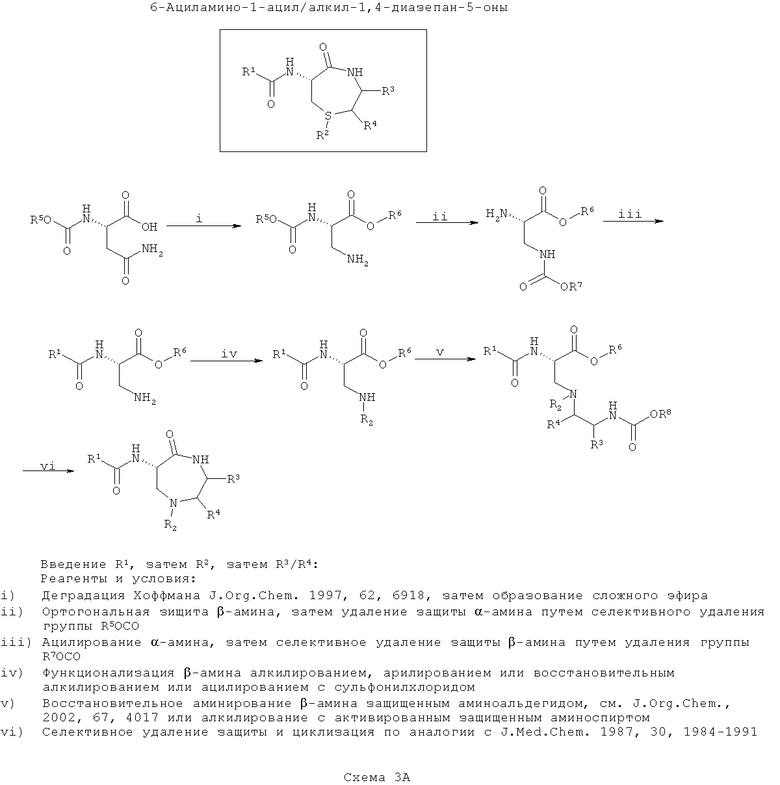
На первой стадии синтезируют защищенный 3-аминоаланин, например, путем деградации по Хоффману защищенного N-α-карбаматэфиром аспарагина (по J. Org. Chem., 1997, 62, 6918), а карбоксильную кислоту этерифицируют (по J. Med. Chem., 1998, 41, 2786).
Для введения заместителя по α-амину свободный β-амин ортогонально защищают и у α-амина селективно удаляют защиту путем удаления карбаматного эфира. Свободный α-амин затем ацилируют, как в предыдущих примерах, для введения функциональности R1.
Затем вводят функциональность R2, например, путем моноалкилирования, моноарилирования или восстановительного алкилирования или ацилирования с сульфонилхлоридом с применением условий реакции, хорошо известных по литературе.
Затем алкилируют β-амин N-защищенным β-аминоспиртом, где спирт активирован для образованиея уходящей группы, подходящей для нуклеофильного замещения с инверсией стереохимии атома углерода (как описано для тиофоксинов и диметилтиофоксинов). Кроме того, β-амин можно конденсировать с N-защищенным α-аминоальдегидом в присутствии восстанавливающего агента с образованием амина (согласно J. Org. Chem., 2002, 67, 4017). Введенный азот здесь указан как ω-аминогруппа. На этой стадии вводят функциональность R3/R4 в зависимости от выбранного β-аминоспирта (возможно полученного из α-аминокислоты).
На конечной стадии (как для тиофоксинов и диметилтиофоксинов) у ω-амина селективно убирают защиту и конденсируют с карбоксиэфиром (или после селективного гидролиза соответствующей карбоксильной кислоты) с образованием 7-членного кольца (согласно J. Med. Chem., 1987, 30, 1984).
На второй схеме (Схема 3Б ниже), вводят группу R1, затем вводят группу (группы) R3/R4 и соединения циклизуют и, наконец, вводят группу R2 (замещение по азоту). Такой путь является оптимальным, если необходимо введение большего разнообразия в значения R2, чем в R3/R4 с наименьшим разнообразием по R1.
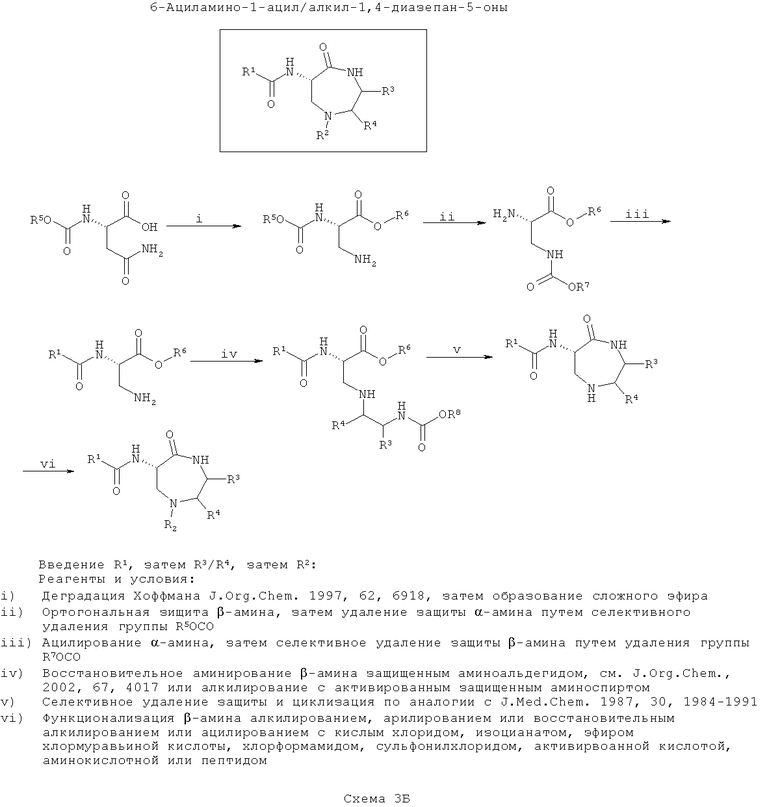
На этой схеме синтезируют защищенный 3-аминоаланин, например, путем деградации по Хоффману N-α-защищенного аспарагина, а β-амин затем ортогонально защищают и у α-амина удаляют защиту и ацилируют и этерифицируют по карбоновой кислоте, как описано для Схемы 3А.
Затем, после селективного удаления защиты у β-амина проводят алкилирование по азоту с N-защищенным β-аминоспиртом, где спирт активирован с образованием уходящей группы, подходящей для нуклеофильного замещения с инверсией стереохимии атома углерода (как описано для тиофоксинов и диметилтиофоксинов). Кроме того, β-амин можно конденсировать с N-защищенным α-аминоальдегидом в присутствии восстанавливающего агента с образованием амина (согласно J. Оrg. Chem., 2002, 67, 4017). Введенный азот здесь обозначен как ω-аминогруппа. Эта стадия вводит функциональность R3/R4 в зависимости от выбранного β-аминоспирта (возможно полученного из α-аминокислоты).
Затем у ω-амина селективно убирают защиту и конденсируют его с карбоксиэфиром (или после селективного гидролиза с соответствующей карбоновой кислотой) с образованием 7-членного кольца (согласно J. Med. Chem., 1987, 30, 1984).
Наконец, вводят функциональность R2, например, посредством моноалкилирования, моноарилирования или восстановительного алкилирования, или ацилирования с сульфонилхлоридом, используя условия реакции, хорошо известные в литературе.
На третьей схеме (Схема 3В ниже), вводят группу R2, затем группу R1 и затем группу (группы) R3/R4 до того, как соединения, наконец, циклизуют. Такой путь является оптимальным, если необходимо введение большего разнообразия в значения R3/R4, чем в R1 с наименьшим разнообразием по R2.
На этой схеме синтезируют защищенный 3-аминоаланин, например, путем деградации по Хоффману N-α-защищенного аспарагина, а карбоновую килоту этерифицируют (как на Схеме 3А, например), затем β-амин моноалкилируют, моноарилируют или сульфонируют как требуется для введения функциональности R2 с применением способов, хорошо известных по литературе. После этого вторичный β-амин защищают ортогонально по отношению к α-амину.
На следующей стадии у α-амина селективно удаляют защиту и ацилируют его, как описано выше, для ввода функциональности R1.
Затем после удаления защиты у β-амина его алкилируют по азоту N-защищенным β-аминоспиртом, где спирт активирован с образованием уходящей группы, подходящей для нуклеофильного замещения с инверсией стереохимии атома углерода (как описано для тиофоксинов и диметилтиофоксинов). Кроме того, β-амин можно конденсировать с N-защищенным α-аминоальдегидом в присутствии восстанавливающего агента с образованием амина (согласно J. Org. Chem., 2002, 67, 4017). Введенный азот называют ω-аминогруппой. Эта стадия вводит функциональность R3/R4 в зависимости от выбранного β-аминоспирта (возможно, полученного из α-аминокислоты).
У ω-амина затем селективно удаляют защиту и конденсируют с карбоксиэфиром (или после селективного гидролиза соответствующей карбоновой кислоты) с образованием 7-членного кольца (согласно J. Med. Chem., 1987, 30, 1984).
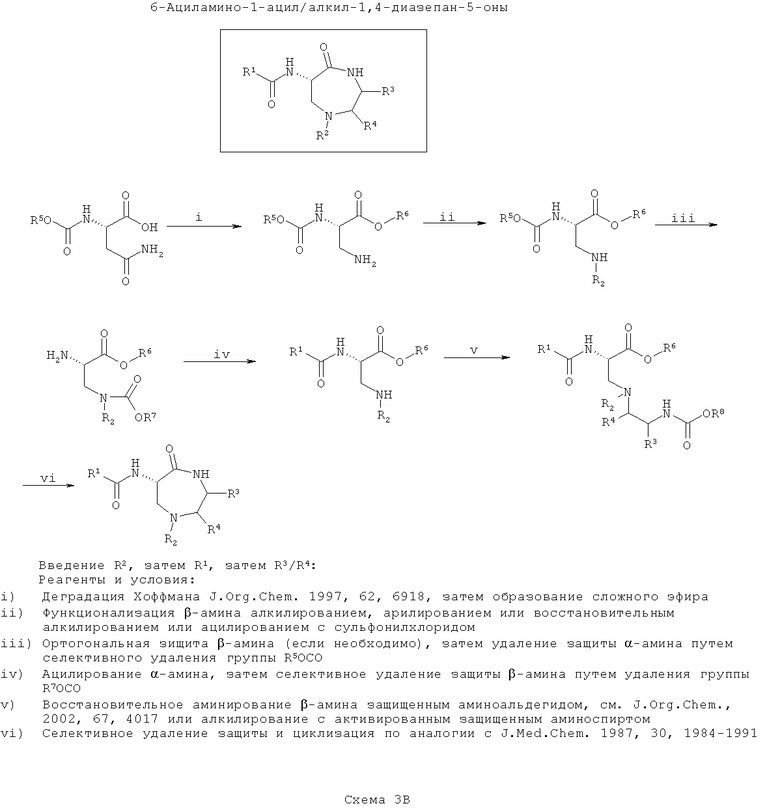
На четвертой схеме (Схема 3Г ниже) вводят группу R2, затем группу (группы) R3/R4. Далее соединения циклизуют и, наконец, вводят группу R1. Такой путь является оптимальным, если необходимо введения большего разнообразия в значения R1, чем в R3/R4 с наименьшим разнообразием по R2.
На этой схеме синтезируют защищенный 3-аминоаланин, например, путем деградации по Хоффману N-α-защищенного аспарагина, а карбоновую кислоту этерифицируют. Затем β-амин моноалкилируют, моноарилируют или сульфонируют как требуется для введения функциональности R2 с применением способов, хорошо известных по литературе, как на Схеме 3В.
На следующей стадии вторичный β-амин алкилируют по азоту N-защищенным β-аминоспиртом, причем спирт активируют с образованием уходящей группы, подходящей для нуклеофильного замещения с инверсией стереохимии атома углерода (как описано для тиофоксинов и диметилтиофоксинов). Кроме того, β-амин можно конденсировать с N-защищенным α-аминоальдегидом в присутствии восстанавливающего агента с образованием амина (согласно J. Org. Chem., 2002, 67, 4017). Введенный азот обозначают как ω-аминогруппу. Эта стадия вводит функциональность R3/R4 в зависимости от выбранного β-аминоспирта (возможно, полученного из α-аминокислоты).
Затем селективно убирают защиту ω-амина и конденсируют его с карбоксиэфиром (или после селективного гидролиза соответствующей карбоновой кислоты) с образованием 7-членного кольца (согласно J. Med. Chem., 1987, 30, 1984).
Наконец, селективно убирают защиту α-амина и ацилируют его, как описано выше для введения функциональности R1.
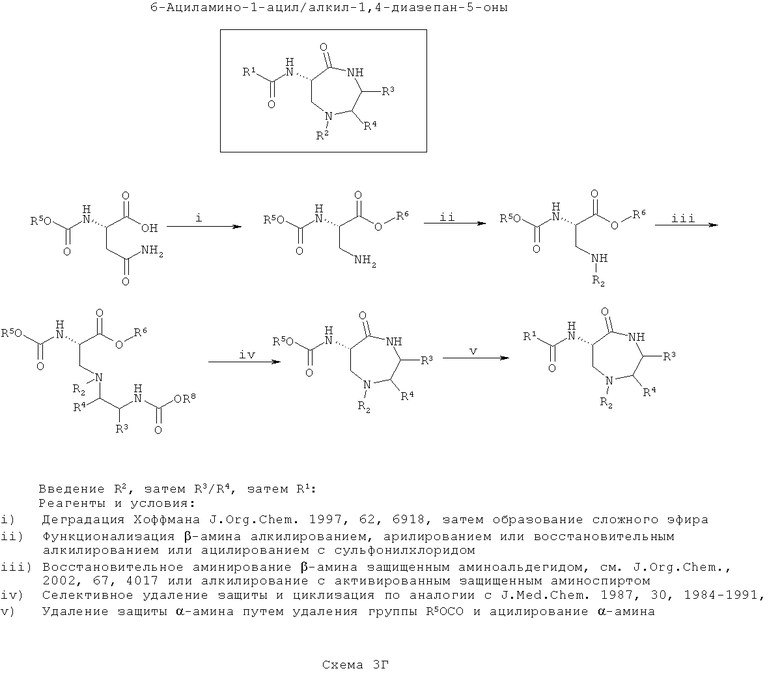
На пятой схеме (Схема 3Д ниже), сначала вводят группу (группы) R3/R4, затем осуществляют циклизацию. Затем вводят группу R1 и затем группу R2. Такой путь является оптимальным, если необходимо введение большего разнообразия в значения R2, чем в значения R1 с наименьшим разнообразием по R3/R4.

На этой схеме синтезируют защищенный 3-аминоаланин, например, путем деградации по Хоффману N-α-защищенного аспарагина, а карбоновую кислоту этерифицируют. Далее, β-амин алкилируют по азоту N-защищенным β-аминоспиртом, где спирт активируют с образованием уходящей группы, подходящей для нуклеофильного замещения с инверсией стереохимии атома углерода (как описано для тиофоксинов и диметилтиофоксинов). Кроме того, β-амин можно конденсировать с N-защищенным α-аминоальдегидом в присутствии восстанавливающего агента с образованием амина (согласно J. Org. Chem., 2002, 67, 4017). Введенный азот обозначают как ω-аминогруппу. На этой стадии вводят функциональность R3/R4 в зависимости от выбранного β-аминоспирта (возможно, полученного из α-аминокислоты).
Затем селективно убирают защиту ω-амина и конденсируют его с карбоксиэфиром (или после селективного гидролиза соответствующей карбоновой кислоты) с образованием 7-членного кольца (согласно J. Med. Chem., 1987, 30, 1984). Вторичный β-амин (в форме кольца) последовательно ортогонально защищают в α-амин.
Далее селективно удаляют защиту α-амина и ацилируют его, как описано выше, для ввода функциональности R1.
Наконец, удаляют защиту β-амина и моноалкилируют, моноарилируют или сульфонируют его, как требуется для вода функциональности R2, с применением способов, хорошо известных по литературе.
На шестой схеме (Схема 3Е ниже), сначала вводят группу (группы) R3/R4, а затем проводят циклизацию. Затем вводят группу R2, а затем группу R1. Такой путь является оптимальным, если требуется введение большего разнообразия в значения R1, чем в значения R2 с наименьшим разнообразием по R3/R4.
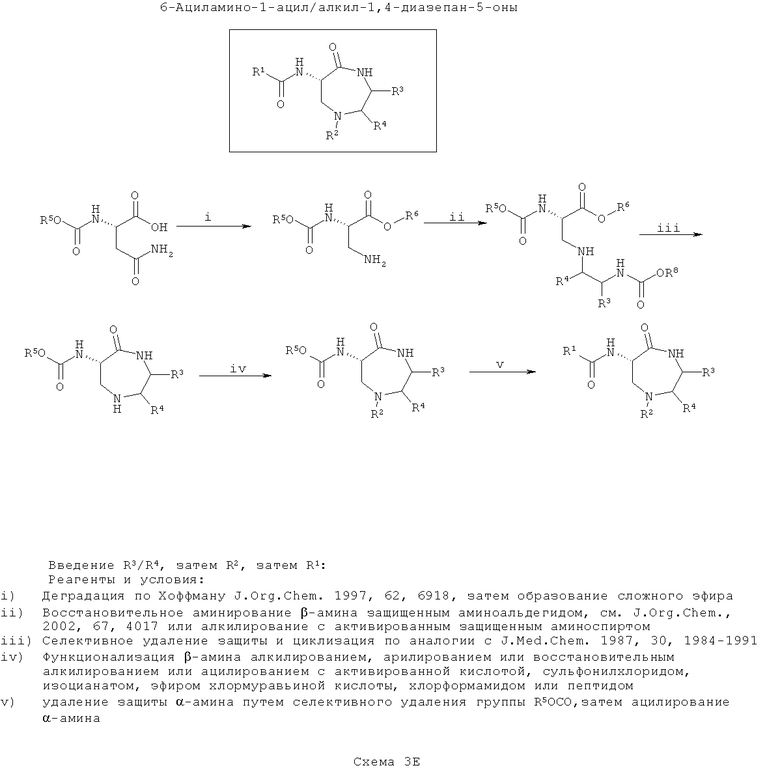
Эта схема сходна со схемой 3Д выше за исключением того, что после стадии циклизации вторичный β-амин (в форме кольца) алкилируют, арилируют или сульфонируют с применением способов, хорошо известных по литературе, а не ортогонально защищают. Это вводит функциональность R2.
На конечной стадии удаляют защиту α-амина и ацилируют его, как описано выше, для ввода функциональности R1.
Приведенные шесть схем иллюстрируют простоту синтеза библиотеки, состоящей из одного или более азафоксинов. В частности, проиллюстрирована возможность выполнения стадий реакции в различном порядке для введения большего разнообразия в различные участки молекулы при сохранении применимости на практике данного способа синтеза.
ПРИМЕР 4 - Синтез амидофоксинов
Амидофоксины представляют собой продукты соединения 3-аминоаланина с α-аминокислотой (в отличие от соединения 3-аминоаланина с β-аминоспиртом, возможно, полученным из α-аминокислоты, с получением азафоксинов, как описано выше). Как и в случае с азафоксинами, но не с тиофоксинами и диметилтиофоксинами, замещение по гетероатому кольца возможно (R2) для введения дополнительного разнообразия. На первой схеме (Схема 4А ниже) вводят группу R1, после чего вводят группу R2 (замещение по азоту), затем группу R3, а затем проводят циклизацию. Такой путь является оптимальным, если необходимо введение большего разнообразия в значения R3, чем в значения R2, с наименьшим разнообразием по R1.
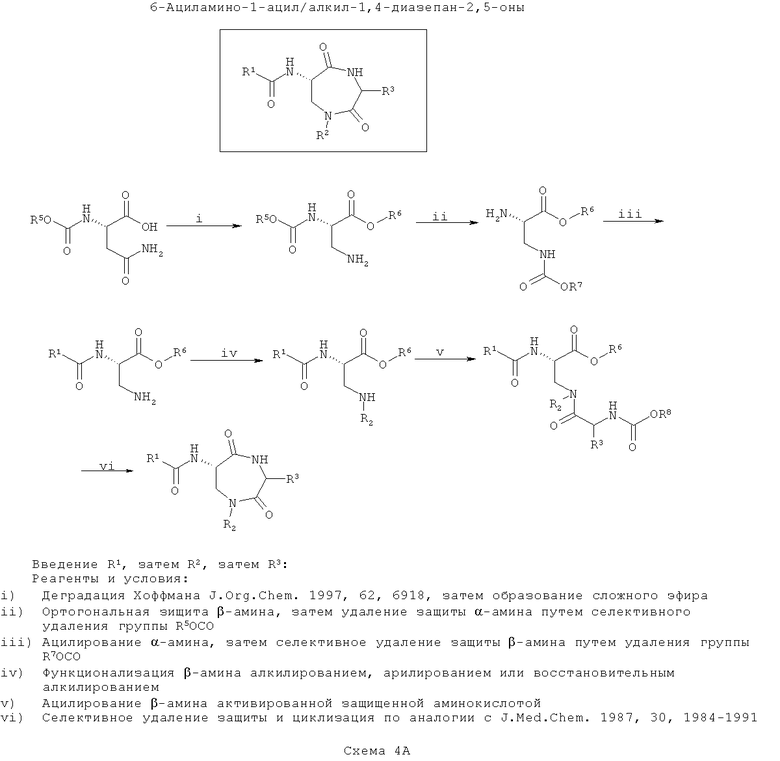
Как и для азафоксинов, на первой стадии идет синтез защищенного 3-аминоаланина, например, путем деградации по Хоффману защищенного N-α-карбаматэфиром аспарагина (согласно J. Org. Chem., 1997, 62, 6918). Затем этерифицируют карбоксигруппу, а группу β-амина защищают ортогонально по отношении к группе α-амина.
На следующей стадии селективно удаляют защиту α-амина и ацилируют его, как описано выше для тиофоксинов (например, на Схемы 1А). На этой стадии вводят функциональность R1.
Далее селективно удаляют защиту β-амина и моноалкилируют или моноарилируют его, как требуется, с применением способов, хорошо известных по литературе, для введения R2.
Вторичный β-амин затем ацилируют N-защищенной α-аминокислотой. Введенный азот называют ω-аминогруппой. Очевидно, что для осуществления этой стадии можно применять любые подходящие способы пептидного сочетания, хорошо известные по литературе. На этой стадии вводят функциональность R3 в зависимости от выбора α-аминокислоты, использованной в реакции.
Наконец, селективно удаляют защиту ω-амина и конденсируют его с карбоксиэфиром (или после селективного гидролиза соответствующей карбоновой кислоты) с образованием 7-членного кольца (например, согласно J. Med. Chem., 1987, 30, 1984).
На второй схеме (Схема 4Б ниже) вводят группу R2, после чего вводят группу R1 (замещение по азоту), затем вводят группу R3, после чего проводят циклизацию. Такой путь является оптимальным, если необходимо введение большего разнообразия в значения R3, чем в значения R1, с наименьшим разнообразием по R2.
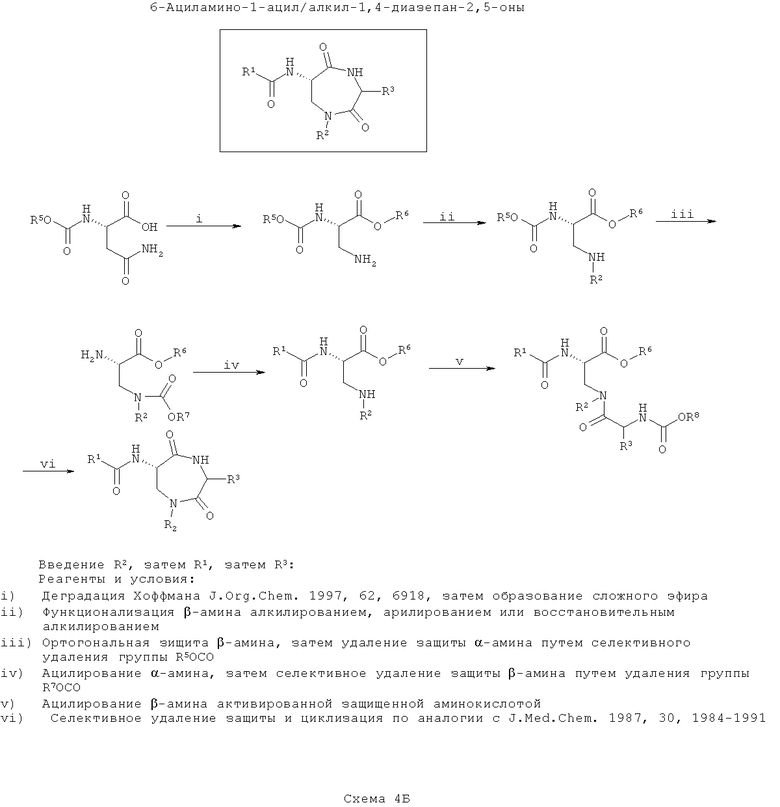
Как на схеме 4А, на первой стадии синтезируют защищенный 3-аминоаланин, например, путем деградации по Хоффману защищенного N-α-карбаматэфира аспарагина (согласно J. Org. Chem., 1997, 62, 6918). Карбоксигруппу этерифицируют, β-амин моноалкилируют или моноарилируют с применением способов, хорошо известных по литературе, перед введением защиты образующейся вторичной β-аминогруппы ортогонально по отношению к α-амину. Это вводит функциональность R2.
На следующей стадии селективно удаляют защиту α-амина и ацилируют его, как описано ниже. На этой стадии вводят функциональность R1.
Затем селективно удаляют защиту вторичного β-амина и ацилируют его N-защищенной α-аминокислотой. Введенный азот назван ω-аминогруппой. Очевидно, что для осуществления этой стадии можно применять любые подходящие способы сочетания пептидов, хорошо известные по литературе. На этой стадии вводят функциональность R3 в зависимости от выбора α-аминокислоты, использованной в реакции.
Наконец, селективно удаляют защиту ω-амина и конденсируют его с карбоксиэфиром (или после селективного гидролиза соответствующей карбоновой кислоты) с образованием 7-членного кольца (например, согласно J. Med. Chem., 1987, 30, 1984).
На третьей схеме (Схема 4В ниже) вводят группу R2, а после группу R3. После циклизации вводят группу R1. Такой путь является оптимальным, если требуется введение большего разнообразия в значения R1, чем в значения R3, с наименьшим разнообразием по R2.
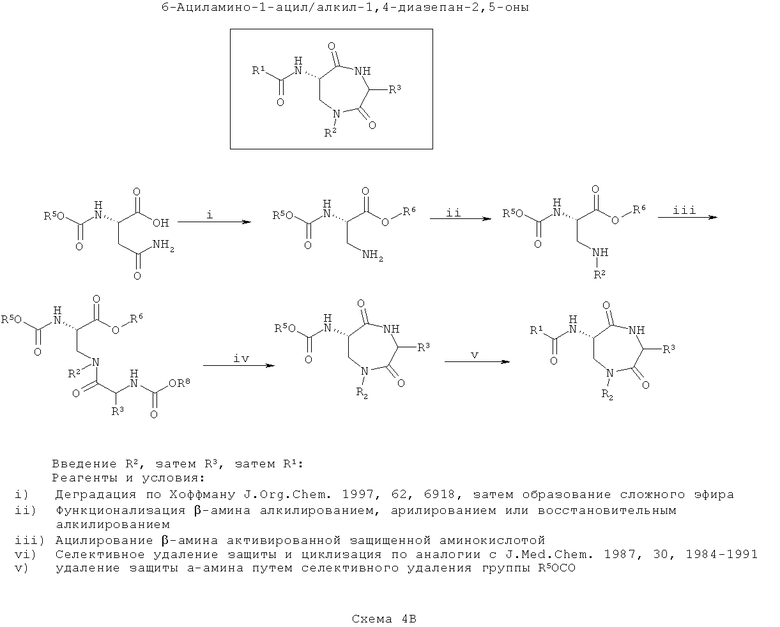
Как на схеме 4Б, на первой стадии синтезируют защищенный 3-аминоаланин, например, путем деградации Хоффмана защищенного N-α-карбаматэфиром аспарагина (согласно J. Org. Chem., 1997, 62, 6918). Карбоксигруппу затем этерифицируют, а β-амин моноалкилируют или моноарилируют с применением способов, хорошо известных по литературе, вводя функциональность R2.
Вторичный β-амин затем ацилируют N-защищенной α-аминокислотой. Введенный азот обозначают как ω-аминогруппу. Очевидно, что для осуществления этой стадии можно применять любые подходящие способы пептидного соединения, хорошо известные по литературе. На этой стадии вводят функциональность R3 в зависимости от выбора α-аминокислоты, использованной в реакции.
Далее селективно удаляют защиту ω-амина и конденсируют его с карбоксиэфиром (или после селективного гидролиза соответствующей карбоновой кислоты) с образованием 7-членного кольца (например, согласно J. Med. Chem., 1987, 30, 1984).
Наконец, селективно удаляют защиту α-амина и ацилируют его, как описано выше. На этом этапе вводят функциональность R1.
Эти три схемы иллюстрируют простоту синтеза библиотеки, состоящей из одного или более азафоксинов. В частности, проиллюстрирована возможность выполнения стадий реакции в различном порядке для введения большего разнообразия в различные участки молекулы при сохранении применимости на практике данного способа синтеза.
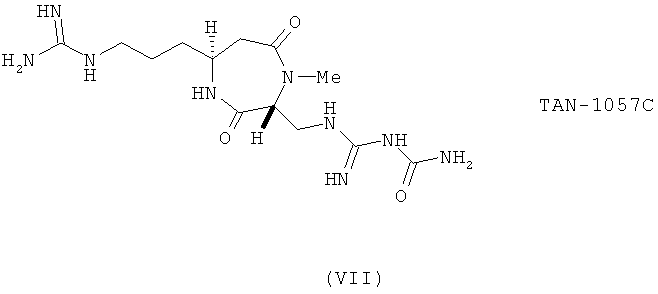
Известно небольшое количество представителей класса амидофоксинов. Например, см. J. Org Chem. (2003) 68: 7893. Однако эти примеры ограничены случаем, когда R3 представляет собой бензил или CH2CH2COR' (где R' варьируют) и их не проверяли на активность в качестве лигандов G-белок сопряженного рецептора. Другие известные схожие структуры, например антибиотик TAN1067C (VII), не являются амидофоксинами и не входят в область настоящего изобретения из-за отсутствия заместителя 3-ациламино или 3-сульфонамид в кольце, а также из-за присутствия заместителя в N1.
Фармакологическое изучение продуктов данного изобретения
Принцип исследований
А: Антагонисты G-белок сопряженных рецепторов
В принципе, соединения согласно настоящему изобретению можно тестировать на антагонистическую активность в отношении некоторого G-белок сопряженного рецептора путем обработки рецептора меченным лигандом при подходящих для связывания условиях в отсутствие и присутствии различных концентраций тестируемого соединения. Затем измеряют количество метки, ассоциированной с рецептором. Если тестируемое соединение способно конкурировать с меченным лигандом за связывание, то количество метки, ассоциированное с рецептором, будет уменьшаться с увеличением концентрации тестируемого соединения. По графику связанного лиганда по отношению к концентрации тестируемого соединения можно оценить аффинность связывания тестируемого соединения с указанным рецептором.
Таким образом, такой анализ требует:
(1) Источник интересующего G-белок сопряженного рецептора. Последовательность каждого члена суперсемейства G-белок сопряженного рецептора человека доступна из последовательности генома человека. Такие последовательности могут быть клонированы в подходящем векторе и экспрессированы в соответствующем типе клеток (например, клетках Jurkat Т, которые, как известно, по существу не имеют эндогенных G-белок сопряженных рецепторов, за исключением рецептора хемокина CXCR4). После выбора подходящего для вектора антибиотика можно отбирать стабильные клеточные линии, экспрессирующие выбранный G-белок сопряженный рецептор на высоком уровне.
Мембранные фракции из линий клеток, экспрессирующих выбранный G-белок сопряженный рецептор, можно получить с применением ряда способов, хорошо известных по литературе. Например, согласно Kuo et al. (Proc. Natl. Acad. Sci. USA (1980) 77: 7039) клетки можно ресуспендировать в 25 мМ буфере HEPES pH 7,5, содержащем 0,25 М сахарозу, 2,5 мМ MgCl2, 2,5 мМ ЭДТА и 50 мМ β-меркаптоэтанола, а также ингибиторы протеазы, такие как PMSF и лейпептин, и вскрыть, с применением гомогенизатора Даунса. Суспензию затем подвергают центрифугированию при 120×g для осаждения не разрушенных клеток и больших клеточных фрагментов, и получают надосадочную жидкость, содержащую малые мембранные фрагменты и компоненты цитозоля. Надосадочную жидкость затем подвергают ультрацентрифугированию при 100,000×g, получая осадок мембранных фрагментов, обогащенных выбранным G-белок сопряженным рецептором. Осадок ресуспендируют в соответствующем буфере для связывания и определяют общую концентрацию белка, используя, например, имеющийся в продаже набор для анализа белка, такой как Coomassie Plus (Pierce). Получение препарата белка можно проводить в объеме с получением стандартизированной общей концентрации белка, например, 1 мг/мл. Стандартизированный препарат можно хранить при -85°С в аликвотах до применения.
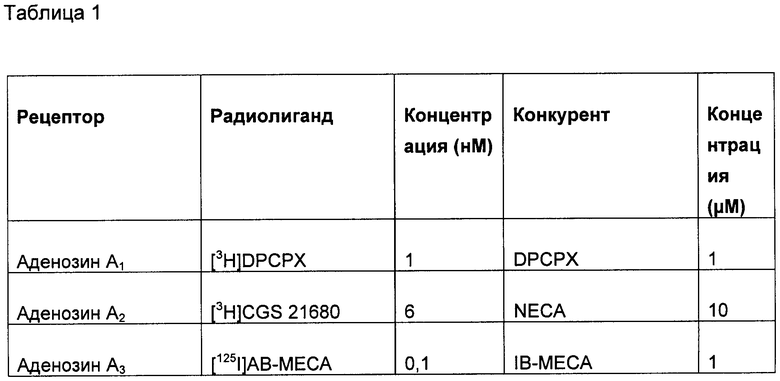
(2) Меченный лиганд с высокой аффинностью к выбранному G-белок сопряженному рецептору. Подходящие лиганды для большинства G-белок сопряженных рецепторов хорошо известны по литературе. Такие лиганды могут быть природными лигандами для рецептора (например, допамин) или могут быть фармакологическим средством (таким как домперидон). Список подходящих лигандов для широкого спектра хорошо исследованных G-белок сопряженных рецепторов приведен в Таблице 1, но специалисту в данной области очевидно, что существуют другие подходящие лиганды для многих из этих рецепторов. Лиганды, наиболее подходящие для этой цели, имеют константу аффинности связывания выбранного рецептора, по меньшей мере, 1 µМ, и предпочтительно меньше, чем 100 нМ, и наиболее предпочтительно меньше, чем 10 нМ.
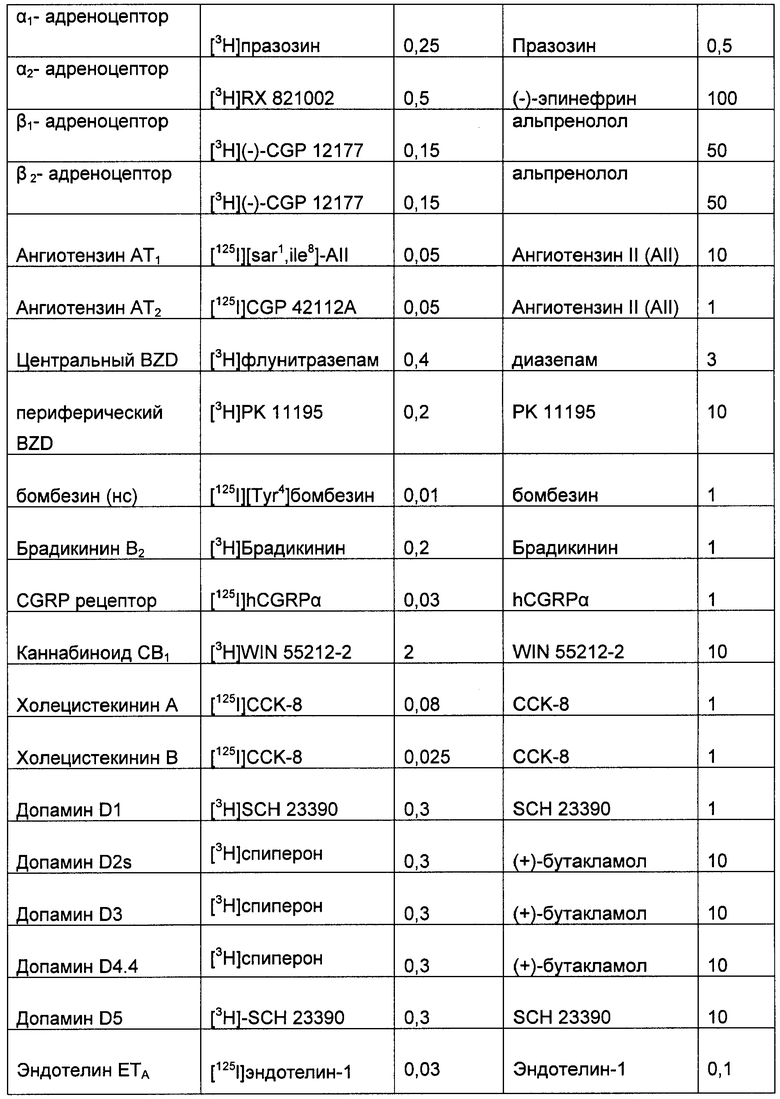
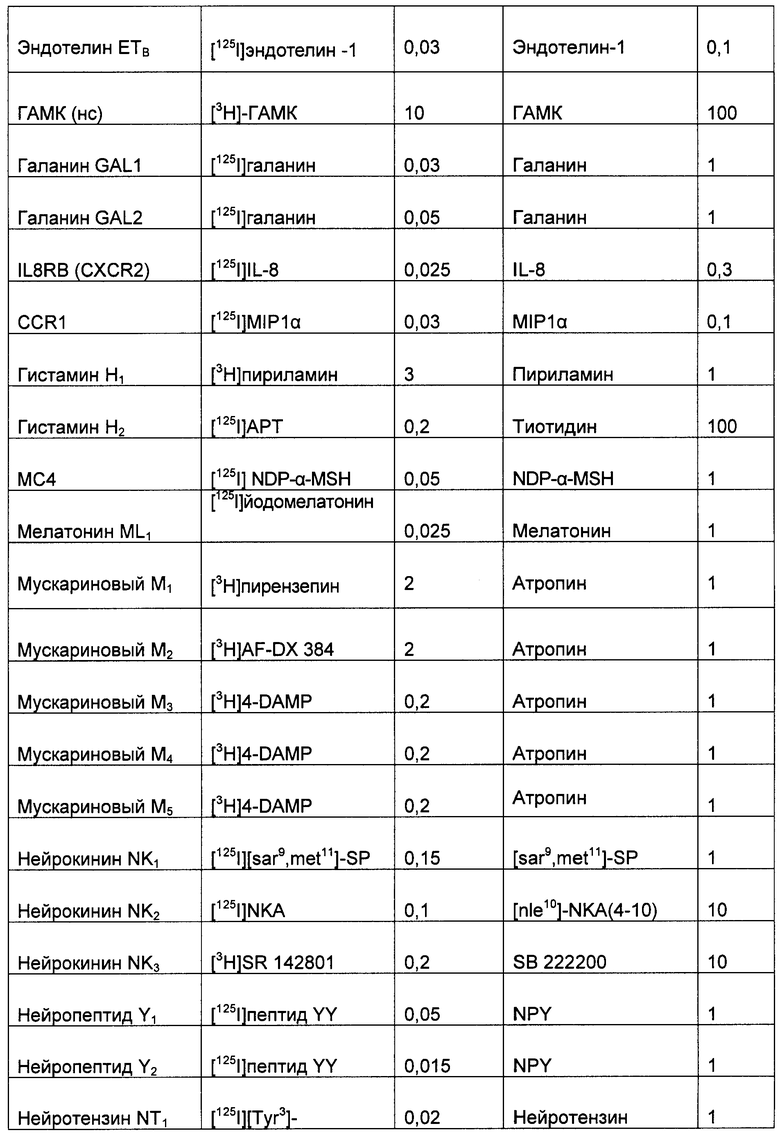
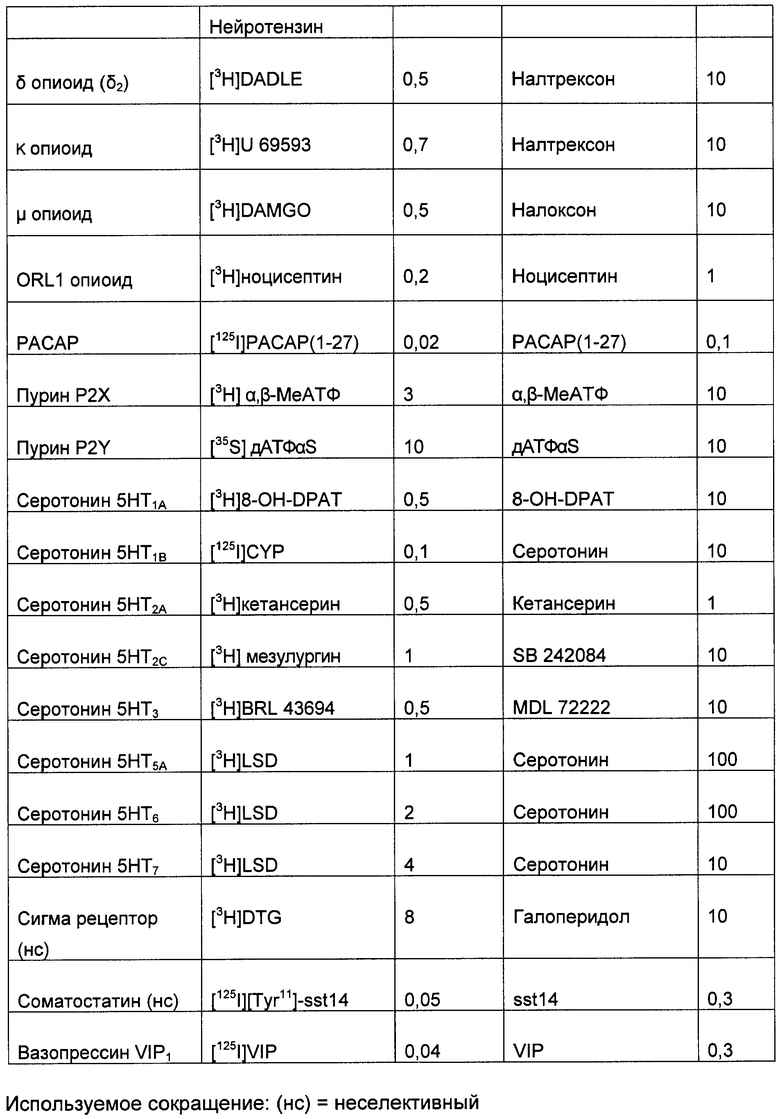
После выбора лиганда, вероятно, необходимо пометить лиганд так, чтобы потом можно было определить количество, связавшееся с выбранным G-белок сопряженным рецептором (хотя может быть возможным выполнить анализ без мечения лиганда, при условии, что доступен чувствительный и точный способ определения количества несвязанного лиганда, например, можно применять твердофазный ИФА (ELISA) для измерения не связавшегося лиганда, таким способом, определяя количество связавшегося лиганда). Подходящие способы мечения лигандов варьируют в зависимости от природы лиганда: Синтетические молекулы можно наиболее легко пометить радионуклидом, таким как 3H, 14С или 35S; пептиды можно наиболее легко пометить косинтетическим биотином (и затем меченным стрептавидином), флуоресцентными метками (такими как флуоресцин изотиоцианат) или радионуклидами (такими как 125I-иодинизация остатков тирозина в пептиде); белки можно наиболее легко пометить флуоресцентными метками (такими как флуоресцинизотиоцианат) или радионуклидами (такими как 125I-йодирование остатков тирозина в белке).
Степень мечения (т.е., доля молекул, несущих метку, в образце) должна быть достаточной, чтобы количество лиганда, связывающегося с рецептором, можно было точно количественно оценить.
С применением этих двух компонентов можно протестировать, модулируют ли соединения согласно настоящему изобретению связывание лиганда с любым данным G-белок сопряженным рецептором, с помощью способов, хорошо известных в данной области техники. Например, в ряде пробирок препарат мембраны смешивают с радиолигандом в концентрации, близкой к константе аффинности для связывания лиганда с выбранным G-белок сопряженным рецептором. В некоторых пробирках соединения согласно настоящему изобретению также добавляют в различных концентрациях. В другие пробирки добавляют в качестве положительного контроля ингибитор (который может быть тем же лигандом в значительном избытке, что и радиоактивно меченный лиганд, но без радиоактивной метки). Как правило, при каждом наборе условий готовят по 3 пробирки. Пробирки затем инкубируют, обычно при 4°С-37°С, более часто при комнатной температуре в течение времени, достаточного для достижения равновесия между свободным и связанным радиолигандом. Обычно, это занимает от 20 минут до 4 часов, а период времени, требуемый для любого данного набора условий реакции, можно определить способами, хорошо известными в данной области техники (например, выполнением эксперимента по времени). Как только равновесие достигнуто, необходимо определить количество связанного радиолиганда. Например, мембраносвязанный рецептор (плюс любой связанный радиолиганд) можно отделить от свободного радиолиганда в растворе путем фильтрации через фильтры (такой как фильтр GF/C, обработанный 1% полиэтиленимином). Фильтры можно затем высушить на воздухе и посчитать количество сцинцилляций для определения добавленной фракции радиолиганда, которая связана с рецептором.
Кроме того, можно осуществлять скрининг соединений согласно настоящему изобретению с применением коммерчески доступных процедур рецепторного скрининга (например, услуги, предлагаемые Cerep, 128 Rue Danton, Paris, France). С помощью таких услуг можно легко идентифицировать члены библиотеки, библиотеки согласно настоящему изобретению, которые модулируют связывание лиганда с одним или более G-белок сопряженных рецепторов.
Соединения, идентифицированные как модулирующие связывание лиганда с одним или более G-белок сопряженным рецептором, используя способы, описанные выше, обычно являются полными антагонистами. Однако для подтверждения антагонистических свойств соединения необходимо провести функциональный анализ. Например, в зависимости от используемого G-белок сопряженного рецептора и/или лиганда стимулируются (или ингибируются) определенные вторичные посредники для передачи сигнала, который представляет собой лиганд. Клетки могут демонстрировать и увеличение (или уменьшение) клеточной концентрации циклического аденозинмонофосфата (цАМФ), различных фосфорилированных соединений, содержащих инозитол (включая I(1,4,5)Р3 и I(1,3,5)Р3), ионов кальция, полиаденозина или других внутриклеточных посредников, известных в данной области, в ответ на присутствие лиганда. Полные антагонисты нивелируют изменение содержания внутриклеточных посредников, которые вызваны природным(и) лигандом(ами), и не оказывают действия в отсутствие природного лиганда. В отличие от этого, полные антагонисты не оказывают действия при добавлении вместе с пригодным(и) лигандом(ами), но мимикрируют изменения во внутриклеточных посредниках, вызванные природным(и) лигандом(ами), при добавлении в отсутствие природного лиганда. Некоторые соединения, включая соединения согласно настоящему изобретению, могут быть частичными антагонистами, частичными агонистами или смешанными агонистами/антагонистами в зависимости от характера действия на внутриклеточные посредники. Несмотря на комплексную фармакологическую характеристику таких соединений, они могут иметь полезные терапевтические свойства при определенных заболеваниях, и известен ряд хорошо проверенных медицинских препаратов, которые являются частичными агонистами, частичными антагонистами или смешанными агонист-антагонистами одного или более G-белок сопряженного рецептора.
Б: Агонисты G-белок сопряженных рецепторов
По существу значительно более трудно тестировать соединения на наличие агонической активности, чем на наличе антагонистической активности, особенно с применением методик скрининга с большой пропускной способностью. Соединения согласно настоящему изобретению, поэтому, вероятно будут особенно полезны для поиска агонистов, по сравнению с общими первичными библиотеками, из-за большего количества агонистов G-белок сопряженного рецептора среди всех элементов библиотек.
Тест для определения агониста G-белок сопряженного рецептора, в принципе, требует системы культур клеток или органов, которая отвечает на природный лиганд выбранного G-белок сопряженного (-ых) рецептора (-ов) желаемой биохимической или физиологической ответной реакцией. Примеры таких ответных реакций включают, но не ограничены ими, изменения во внутриклеточных посредниках (таких как цАМФ, IP(1,4,5)Р3, ионы кальция или полиаденозин), изменения в ферментативной активности (такие как активация протеинкиназ, фосфотаз, метаболических ферментов или транспортных белков), изменения в профиле экспрессии генов, измененный фагоцитоз, измененная секреция белков, измененный уровень пролиферации, сокращение мышечных клеток/ткани, нейротрансмиссию и так далее. Поскольку ответные реакции, такие как эти, являются по существу более сложными для измерения по сравнению со связыванием природного(ых) лиганда(ов) с выбранным(и) G-белок сопряженным(и) рецептором(ами), способы анализа для определения агонистов GPCR являются более трудными, чем для выявления антагонистов.
Общий способ, необходимый для проверки того, является ли соединение согласно настоящему изобретению агонистом одного или более выбранного G-белок сопряженного рецептора, хорошо известен в данной области техники. Клетки подвергают обработке различными концентрациями тестируемого вещества, например, добавлением соединения в подходящий разбавитель (такой как ДМСО, этанол или метанол) в различных концентрациях (например, от примерно 0,1 нМ до примерно 10 мМ) в клеточной культуральной среде на период времени (например, от 1 минут до 48 часов в зависимости от времени ответа, который необходимо измерить) обычно при 37°С. Параллельно, клетки подвергают обработке природным лигандом и оставляют необработанными любыми дополнительными соединениями (в качестве контрольных клеток). В конце инкубационного периода измеряют ответную реакцию, известную в данной области техники, которая имеет место в ответ на связывание природного лиганда с выбранным(и) G-белок сопряженным(и) рецептором(ами). Если соединение согласно настоящему изобретению является агонистом для выбранного(ых) G-белок сопряженных рецепторов, то ответные реакции на тестируемое соединение (в определенных концентрациях) оценивают количественно по аналогии с ответной реакцией на природный лиганд.
Примеры подходящих систем анализа для выявления агонистов для отдельного(ых) G-белок сопряженных рецепторов приведены ниже:
Соматостатин является агонистом для рецепторов sstr2 и sstr5, поэтому ингибирует секрецию гормона роста изолированными клетками гипофиза. Для того, чтобы определить является ли соединение согласно настоящему изобретению агонистом для sstr2 и/или sstr5, клетки гипофиза крысы изолируют и помещают в культуру. Затем клетки инкубируют отдельно или в присутствии соматостатина в концентрации 33 нМ, или в присутствии тестируемого вещества (веществ) в различных концентрациях от примерно 0,1 нМ до примерно 10 мМ при 37°С в течение 24 часов. В конце эксперимента, культуральную среду для клеток удаляют, очищают центрифугированием и анализируют на гормон роста (ГР), например, выполняя имеющиеся в продаже анализы ELISA (твердофазный иммуноферментный анализ) или раидоиммуноанализ. Клетки, обработанные соматостатином, будут продуцировать на 30-90% меньше ГР, чем клетки, инкубированные отдельно. Если соединение согласно настоящему изобретению является агонистом для рецепторов соматостатина, то уровень ГР будет ниже в среде от клеток, обработанных тестируемым соединением (по меньшей мере, при определенных концентрациях), чем в среде от клеток, инкубированных отдельно. Обычно анализ проводят при трех повторениях идентичных по каждому из условий эксперимента для возможности применения подходящего статистического теста (например, дисперсионного анализа ANOVA или непарного t-теста Стьюдента), чтобы показать, что тестируемое вещество приводит к статистически значимому уменьшению секреции ГР и, поэтому, вероятно, обладает активностью агониста для выбранных рецепторов sstr2 и/или sstr5.
Эндотелин-1 является пептидом, передающим сигнал через рецептор ЕТ-А и/или ЕТ-В, вызывая сужение кровеносных сосудов. Для того чтобы определить являются ли соединения согласно настоящему изобретению агонистами для ЕТ-А и/или ЕТ-В, кольца аорты человека (полученные из трансплантируемых донорных сердец) можно поместить в культуру. Кольца затем обрабатывают либо увеличивающимися концентрациями эндотелина-1 (от 0,01 нМ до 100 нМ), или увеличивающимися концентрациями тестируемого вещества (веществ) (от примерно 0,1 нМ до примерно 10 мМ) при 37°С, увеличивая концентрацию соответствующего агента примерно каждые 5 минут. В ходе эксперимента измеряют сокращение кольца аорты с применением тензиометрического датчика, разработанного и коммерчески доступного для таких целей. Кольца, обработанные эндотелином-1, будут сжиматься при увеличении концентрации эндотелина-1, так что ко времени достижения максимальной концентрации сила, действующая на тензиометрический датчик, будет значительно выше, чем до добавления эндотелина-1. Если соединение согласно настоящему изобретению является агонистом для рецепторов эндотелина, то сила, действующая на тензиометрический датчик, будет также выше (по меньшей мере, при определенных концентрациях), чем перед добавлением тестируемого соединения. Обычно анализ проводят при трех повторениях идентичных по каждому из условий эксперимента для возможности применения подходящего статистического теста (например, дисперсионного анализа ANOVA или непарного t-теста Стьюдента), чтобы показать, что тестируемое соединение статистически значимо увеличивает сокращение аорты и, поэтому, вероятно, обладает активностью агониста для выбранных рецепторов ЕТ-А и/или ЕТ-В.
Хемокин SDF-1a представляет собой пептид, передающий сигнал через рецептор CXCR4, вызывая миграцию лейкоцитов. Для того чтобы определить являются ли соединения согласно настоящему изобретению агонистами для CXCR4, культивируемые бессмертные Т-клетки человека (клетки Jurkat Т, например) помещают в верхнюю лунку специализированного коммерчески доступного прибора для анализа миграции из лунки в лунку. Лунки затем помещают в нижние камеры, содержащие только культуральную среду, или в нижние камеру, содержащие SDF-1a в концентрации 75 нМ, или в нижние камеры, содержащие тестируемое соединение (соединения) в различных концентрациях (около 0,1 нМ - 10 мМ), и инкубируют в течение определенного промежутка времени (обычно от 30 минут до 3 часов) при 37°С. В конце число клеток в нижней камере является мерой величины произошедшей миграции. Число клеток в нижней камере можно подсчитать прямым визуальным наблюдением или различными известными способами, например, с помощью инкубации с красителем МТТ, который преобразуется в нерастворимый синий формазан пропорционально количеству имеющихся клеток. В лунках, помещенных в нижнюю камеру, содержащую SDF-1a, число клеток в нижней камере будет в 2-10 раз выше, чем число клеток в нижней камере, содержащих только культуральную среду. Если соединение согласно настоящему изобретению является агонистом CXCR4, то число клеток в нижних камерах, содержащих тестируемое соединение (соединения), также будет выше (по крайней мере, при определенной концентрации), чем в нижних камерах, содержащих только среду. Обычно анализ проводят при трех повторениях идентичных по каждому из условий эксперимента для возможности применения подходящего статистического теста (например, дисперсионного анализа ANOVA или непарного t-теста Стьюдента), чтобы показать, что соединение согласно настоящему изобретению вызывает статистически значимое увеличение миграции лейкоцитов, и следовательно, вероятно, обладает агонистической активностью по отношению к выбранному рецептору CXCR4.
Биологически активный амин адреналин увеличивает концентрацию внутриклеточного цАМФ в сосудистых гладкомышечных клетках. Для определения того, являются ли соединения согласно настоящему изобретению агонистами β-адренорецептора, гладкомышечные клетки сосудов изолируют из грудной аорты крыс и помещают в культуру. Затем клетки инкубируют в отсутствие или в присутствии агониста адреналина сальбутамола при 33 нМ, или в присутствии тестируемого соединения (соединений) в различной концентрации от 0,1 нМ до 10 мМ при 37°С в течение 15 минут. В конце эксперимента, среду клеточной культуры удаляют и промывают клетки 3 раза в ледяном буфере и затем лизируют в соответствующем буфере для лизиса до измерения концентрации внутриклеточного цАМФ, например с применением коммерчески доступного твердофазного ИФА (ELISA) или радиоиммуноанализа. Клетки, на которых воздействовали сальбутамолом, имеют концентрацию внутриклеточного цАМФ выше на 15-150%, по сравнению с клетками без воздействия агониста. Если соединения согласно настоящему изобретению является агонистом β-адренорецептора, тогда концентрация внутриклеточного цАМФ будет более высокой в клетках, на которые воздействовали тестируемым соединением (по крайней мере, в определенной концентрации), чем в клетках, инкубируемых только в среде. Обычно, клеточный лизат готовят в трех повторениях из клеток, обработанных идентично при каждом типе условий эксперимента для возможности применения подходящих статистических тестов (таких как дисперсионный анализ ANOVA или непарный t-тест Стьюдента) для того, чтобы показать, что тестируемое соединение вызывает статистически значимое увеличение концентрации внутриклеточного цАМФ, и следовательно, вероятно, обладает агонистической активностью по отношению к выбранным β-адренорецепторам.
Очевидно, что исследования, такие как приведенные выше в качестве примеров, выявляют агонистов выбранных G-белок сопряженных рецепторов и позволяют провести разделение соединений согласно настоящему изобретению на неактивные соединения и соединения с антагонистической или частичной антагонистичекой активностью по отношению к выбранному G-белок сопряженному рецептору, но не обязательно однозначно идентифицируют выбранный G-белок сопряженный рецептор как молекулярную мишень соединения согласно настоящему изобретению. Например, соединение согласно настоящему изобретению, для которого было показано, что оно увеличивает уровень цАМФ в гладкомышечных клетках сосудов в той же степени, что и агонист β-адренорецептора сальбутамол, может быть агонистом β-адренорецептора (G-белок сопряженного рецептора) или может быть агонистом другого G-белок сопряженного рецептора, который также увеличивает уровень цАМФ (такого как рецептор допамина D2). Кроме того, соединение согласно настоящему изобретению, стимулирующее миграцию лейкоцитов в той же степени, что и SDF-1a может быть агонистом CXCR4 или может являться агонистом другого G-белок сопряженного рецептора, стимулирующего миграцию лейкоцитов (такого как рецептор С5а). Подтверждение, того, что G-белок сопряженный рецептор является молекулярной мишенью, на которую действуют соединения согласно настоящему изобретению как агонисты, требует проведения дополнительных экспериментов с применением специфических антагонистов, уже идентифицированных для выбранного G-белок сопряженного рецептора, или применения рекомбинантных клеточных линий, которые экспрессируют только выбранный G-белок сопряженный рецептор. Например, если миграция лейкоцитов, индуцированная соединением согласно настоящему изобретению, ингибируется добавлением СХСР4-специфичного антагониста AMD3100 в соответствующей концентрации, тогда логично заключить, что CXCR4 является молекулярной мишенью соединения согласно настоящему изобретению. Аналогично, если миграция лейкоцитов, индуцированная соединением согласно настоящему изобретению, наблюдалась при применении клеточной линии, экспрессирующей CXCR4, но отсутствовала в той же клеточной линии, не экспрессирующей CXCR4, тогда логично заключить, что CXCR4 является молекулярной мишенью соединения согласно настоящему изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИГАНДЫ ДЛЯ G-БЕЛОК СОПРЯЖЕННЫХ РЕЦЕПТОРОВ | 2005 |
|
RU2454416C2 |
| МЕЧЕНЫЕ РАДИОАКТИВНЫМ ИЗОТОПОМ ПЕПТИДЫ ДЛЯ ДИАГНОСТИКИ И ТЕРАПИИ | 1996 |
|
RU2171117C2 |
| ПЕПТИДОМИМЕТИЧЕСКИЕ МАКРОЦИКЛЫ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2639523C2 |
| ПРОТИВОВОСПАЛИТЕЛЬНЫЕ АГЕНТЫ | 2005 |
|
RU2410376C2 |
| СТИМУЛЯТОРЫ СЕКРЕЦИИ ГОРМОНА РОСТА | 2006 |
|
RU2416612C9 |
| ИНГИБИТОРЫ IAP | 2013 |
|
RU2728789C2 |
| ИНГИБИТОРЫ IAP | 2013 |
|
RU2593259C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ИНДУКЦИИ АПОПТОЗА РАКОВЫХ КЛЕТОК | 2003 |
|
RU2379056C2 |
| АГОНИСТЫ СОМАТОСТАТИНА | 2002 |
|
RU2259375C2 |
| БЕТА-ЦЕПОЧЕЧНЫЕ МИМЕТИКИ И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2003 |
|
RU2333213C2 |
Изобретение относится к библиотеке для идентификации соединения, способного модулировать передачу сигналов через G-белок сопряженный рецептор, которая включает два или более соединения, которые соответствуют любой из формул (I), (II), (III), (IV) или фармацевтически приемлемые соли указанных соединений, причем указанные соединения имеют разнообразные замещения варьируемыми заместителями. Изобретение также относится к способу получения указанной библиотеки и способу скрининга для идентификации соединений, предназначенных для модулирования активности одного или более представителя класса G-белок сопряженных рецепторов. 16 з.п. ф-лы, 1 табл.
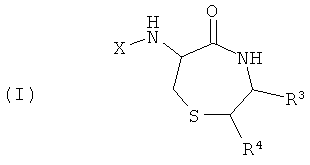
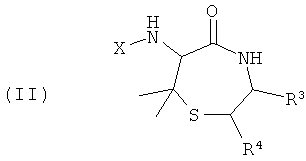

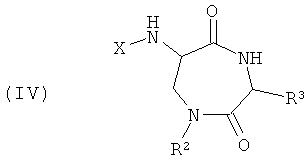
1. Библиотека для идентификации соединения, способного модулировать передачу сигналов через G-белок сопряженный рецептор, которая включает два или более соединений, которые соответствуют любой из формул (I), (II), (III), (IV), или фармацевтически приемлемые соли указанных соединений, причем указанные соединения имеют разнообразные замещения варьируемыми заместителями:
где X представляет собой -CO-R1 или SO2-R1, где R1 представляет собой группу, соседнюю с карбонильной группой карбонамида или сульфонильной группой сульфаниламида;
R1 представляет собой (Y)k-(Z)n;
k представляет собой 0 или 1;
Y представляет собой циклоалкил или полициклоалкил (например адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил);
или Y представляет собой циклоалкенил или полициклоалкенил;
каждый Z независимо выбран из водорода или алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила, алкиламино, алкиламиноалкила, алкиламинодиалкила, заряженного алкиламинотриалкила или заряженного алкилкарбоксилатного радикала, содержащего от 1 до 20 атомов углерода;
или каждый Z независимо выбран из фтора, хлора, брома, йода, гидроксигруппы, оксиалкила, амино, аминоалкила, аминодиалкила, заряженного аминотриалкила или карбоксилатного радикала; и
n представляет собой любое целое от 1 до m, где m - максимальное количество замещений, допустимое в циклической группе Y, или
альтернативно, Z может быть выбран из пептидного радикала, например, содержащего от 1 до 4 пептидных звеньев, соединенных пептидными связями (например пептидного радикала, содержащего от 1 до 4 аминокислотных остатков); и
R3 и R4 независимо представляют собой любые варьируемые заместители;
где X представляет собой -CO-R1 или SO2-R1, при этом R1 представляет собой группу, следующую за карбонильной группой карбонамида или сульфонильной группой сульфаниламида;
R1 представляет собой (Y)k-(Z)n;
k представляет собой 0 или 1;
Y представляет собой циклоалкил или полициклоалкил (например адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил);
или Y представляет собой циклоалкенил или полициклоалкенил;
каждый Z независимо выбирают из водорода или алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила, алкиламино, алкиламиноалкила, алкиламинодиалкила, заряженного алкиламинотриалкила или заряженного алкилкарбоксилатного радикала, содержащего от 1 до 20 атомов углерода;
или каждый Z независимо выбирают из фтора, хлора, брома, йода, гидроксигруппы, оксиалкила, амино, аминоалкила, аминодиалкила, заряженного аминотриалкила или карбоксилатного радикала; и
n представляет собой любое целое от 1 до m, где m - максимальное количество замещений, допустимое в циклической группе Y,
альтернативно, Z может быть выбран из пептидного радикала, например, имеющего от 1 до 4 пептидных звеньев, соединенных пептидными связями (например пептидного радикала, содержащего от 1 до 4 аминокислотных остатков); и
R3 и R4 независимо представляют собой любые варьируемые заместители;
где X представляет собой -CO-R1 или SO2-R1, где R1 представляет собой группу, следующую за карбонильной группой карбонамида или сульфонильной группой сульфаниламида;
R1 представляет собой (Y)k-(Z)n;
k представляет собой 0 или 1;
Y представляет собой циклоалкил или полициклоалкил (например адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил);
или Y представляет собой циклоалкенил или полициклоалкенил;
каждый Z независимо выбран из водорода или алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила, алкиламино, алкиламиноалкила, алкиламинодиалкила, заряженного алкиламинотриалкила или заряженного алкилкарбоксилатного радикала, содержащего от 1 до 20 атомов углерода;
или каждый Z независимо выбран из фтора, хлора, брома, иода, гидрокси, оксиалкила, амино, аминоалкила, аминодиалкила, заряженного аминотриалкила или карбоксилатного радикала; и
n - любое целое от 1 до m, где m - максимальное количество замещений, допустимое в циклической группе Y, или
альтернативно, Z может быть выбран из пептидного радикала, например, имеющего от 1 до 4 пептидных звеньев, соединенных пептидными связями (например пептидного радикала, содержащего от 1 до 4 аминокислотных остатков); и
R2, R3 и R4 независимо представляют собой любые варьируемые заместители;
где X представляет собой -CO-R1 или SO2-R1;
R1 представляет собой (Y)k-(Z)n;
k представляет собой 0 или 1;
Y представляет собой циклоалкил или полициклоалкил (например адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил);
или Y представляет собой циклоалкенил или полициклоалкенил;
каждый Z независимо выбран из водорода или алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила, алкиламино, алкиламиноалкила, алкиламинодиалкила, заряженного алкиламинотриалкила или заряженного алкилкарбоксилатного радикала, содержащего от 1 до 20 атомов углерода;
или каждый Z независимо выбран из фтора, хлора, брома, иода, гидрокси, оксиалкила, амино, аминоалкила, аминодиалкила, заряженного аминотриалкила или карбоксилатного радикала; и
n - любое целое от 1 до m, где m - максимальное количество замещений, допустимое в циклической группе Y, или
альтернативно, Z может быть выбран из пептидного радикала, например, имеющего от 1 до 4 пептидных звеньев, соединенных пептидными связями (например пептидного радикала, содержащего от 1 до 4 аминокислотных остатков), и
R2 и R3 независимо представляют собой любые варьируемые заместители.
2. Библиотека по п.1, отличающаяся тем, что у каждого соединения радикал R1 содержит «ключевой» атом углерода, соседний с карбонильной группой карбонамида или сульфонильной группой сульфаниламида, двузамещенный либо идентичными, либо различными группами, выбранными из: алкила, галогеналкила, алкокси, галогеналкокси, алкенила, алкинила и алкиламино радикала.
3. Библиотека по п.2, отличающаяся тем, что у каждого соединения «ключевой» атом углерода является хиральным.
4. Библиотека по п.2, отличающаяся тем, что у каждого соединения «ключевой» атом углерода образует sp3 гибридные связи.
5. Библиотека по п.2, отличающаяся тем, что у каждого соединения «ключевой» атом углерода образует, по существу, тетраэдральные углы валентных связей.
6. Библиотека по любому из пп.1-5, отличающаяся тем, что у каждого соединения кольцо или кольца Y содержат в основном тетраэдральные углы валентных связей (то есть sp3 гибридные связи) при «ключевом» атоме углерода.
7. Библиотека по любому из пп.1-6, отличающаяся тем, что у каждого соединения формулы (II) R3 не представляет собой карбоксилат, или карбоксилатный эфир (в котором карбоксилат присоединен непосредственно к ациламинолактамному кольцу); или карбоксилатный амид (в котором карбоксилат присоединен непосредственно к ациламинолактамному кольцу); или карбоксилатный тиоэфир (в котором карбоксилат присоединен непосредственно к ациламинолактамному кольцу).
8. Библиотека по любому из пп.1-7, отличающаяся тем, что у каждого соединения формулы (IV) R3 не представляет собой бензил, или R3 не представляет собой CH2CH2COR', где R' может быть любым.
9. Способ скрининга для идентификации соединения, которое модулирует передачу сигнала через G-белок сопряженные рецепторы, включающий:
а) обеспечение библиотеки согласно любому из пп.1-8;
б) проведение скрининга указанной библиотеки; и
в) идентификацию соединения в составе указанной библиотеки, которое модулирует передачу сигнала через G-белок сопряженного рецептора.
10. Способ по п.9, отличающийся тем, что идентифицируемое соединение представляет собой антагонист одного или более G-белок сопряженного рецептора.
11. Способ по п.9, отличающийся тем, что идентифицируемое соединение представляет собой агонист одного или более G-белок сопряженного рецептора.
12. Способ по п.9, отличающийся тем, что G-белок сопряженный рецептор выбран из группы, включающей рецепторы адреналина, эндотелина, хемокина, EDG, VIP/PECAP, допамина, серотонина, пурина, метаботропные рецепторы глутамата, рецепторы ацетилхолина, С5а, fMLP, глюкагона или GLP, NPY, MSH, гормона гликопротеина, активируемые протеазой рецепторы (PARs), рецепторы соматостатина, ангиотензина, холецистокинина или мелатонина.
13. Способ по любому из пп.9-12, включающий следующие стадии:
(а) проведение скрининга библиотеки соединений по любому из пп.1-8;
(б) идентификацию по меньшей мере одного соединения, модулирующего передачу сигнала через G-белок сопряженные рецепторы;
(в) синтез углеродного аналога указанного соединения, в котором гетероатом лактамного кольца, не являющийся частью мотива «идеального» субстрата G-белок сопряженного рецептора (в положении 1), заменен на -СН2- группу; и
(г) обеспечение указанного углеродного аналога (или его фармацевтически приемлемой соли) в выделенной и очищенной форме.
14. Способ получения библиотеки соединений согласно любому из пп.1-8, отличающийся тем, что указанную библиотеку получают путем осуществления реакции разнообразных α-аминокислот и/или β-аминоспиртов с «подходящей» ациламинокислотой, при этом под «подходящей» ациламинокислотой понимают любую ациламинокислоту, которая образует ациламинолактам при реакции с α-аминокислотой или β-аминоспиртом.
15. Способ по п.14, отличающийся тем, что любой радикал Y выбран из группы, состоящей из: адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил.
16. Способ по п.14 или 15, отличающийся тем, что любой радикал Z выбран из пептидного радикала, например, содержащего от 1 до 4 пептидных звеньев, соединенных пептидными связями.
17. Способ по п.16, отличающийся тем, что у любого соединения указанный пептидный радикал содержит от 1 до 4 остатков аминокислот.
| HIROAKI YANAGISAWA et al | |||
| J | |||
| Med | |||
| Chem, vol.30, no.11, 1987, p.p.1984-1991 | |||
| BERNDT SJOBERG et al | |||
| Tetrahedron Letters, no.4, 1965, p.p.281-286 | |||
| EDWARD G | |||
| BREITHOLLE et al | |||
| J | |||
| Org | |||
| Chem., vol.41, no.8, 1976, p.p.1344-1349 | |||
| LAMPARIELLO L R et al | |||
| J | |||
| Org | |||
| Chem., vol.68, no.20, 2003, p.p.7893-7895 | |||
| Способ получения производных пергидротиазепина или их фармацевтически приемлемых кислотно-аддитивных солей | 1987 |
|
SU1787157A3 |

