Уровень техники
Область техники, к которой относится изобретение
Настоящее изобретение относится в целом к бета-цепочечным миметикам, химической библиотеке, относящейся к ним, и их применению.
Описание предшествующего уровня техники
Рандомизированный скрининг молекул на возможную активность в качестве терапевтических агентов проводился в течение многих лет и привел к открытию ряда важных лекарственных средств. Хотя успехи в молекулярной биологии и компьютеризованной химии привели к повышенному интересу в том, что было названо «рациональным конструированием лекарственных средств», такие способы не оказались такими быстрыми или надежными, как первоначально предполагалось. Поэтому в последние годы возобновился интерес и возврат к рандомизированному скринингу лекарственных средств. С этой целью определенные шаги были сделаны в новых технологиях, основанных на разработке комбинаторных библиотек химических соединений и скрининге таких библиотек в поисках на биологически активные члены.
В целом, комбинаторные библиотеки химических соединений являются просто коллекцией молекул. Такие библиотеки различаются химическими разновидностями в библиотеке, а также способами, применяемыми как для образования членов библиотек, так и идентификации того, какие члены взаимодействуют с представляющими интерес биологическими мишенями. Хотя эта область является все еще молодой, способы получения и скрининга библиотек стали уже совсем иными и усовершенствованными. Например, в недавнем обзоре различных комбинаторных химических библиотек соединений идентифицирован ряд таких способов (Dolle, J. Com. Chem., 2(3): 383-433, 2000), включающих применение как «меченых», так и «немеченых» членов библиотек (Janda, Proc. Natl. Acad. Sci. USA 91:10779-10785, 1994).
Первоначально комбинаторные химические библиотеки были, в основном, ограничены членами пептидного или нуклеотидного происхождения. С этой целью способы Houghten et al. иллюстрируют пример метода, названного «двойственно иттерационным» методом, для составления библиотек растворимых комбинаторных пептидов с помощью метода синтеза расщеплением (Nature (London) 354:84-86, 1991; Biotechniques 13:412-421, 1992; Bioorg. Med. Chem. Lett. 3:405-412, 1993). Таким способом получали библиотеки растворимых пептидов, содержащие десятки миллионов членов. Обнаружено, что такие библиотеки являются эффективными в идентификации опиоидных пептидов, таких как метионин- и лейцинэнкефалин (Dooley and Houghten, Life Sci. 52, 1509-1517, 1993), и библиотеку N-ацилированных пептидов применяли для идентификации ацеталинов, которые являются сильнодействующими антагонистами опиоидов (Dooley et al., Proc. Natl. Acad. Sci. USA 90:10811-10815, 1933). Совсем недавно была создана библиотека всех D-аминокислотных опиоидных пептидов и подвергнута скринингу на аналгезирующую активность против мю("μ")-опиоидного рецептора (Dooley et al., Science, 266:2019-2022, 1944).
Хотя комбинаторные библиотеки, содержащие члены пептидного и нуклеотидного происхождения, имеют большую ценность, в данной области все же существует потребность в библиотеках, содержащих члены другого происхождения. Например, традиционные библиотеки пептидов в значительной степени различаются только аминокислотной последовательностью для генерирования членов библиотеки. Хотя общепризнанно, что вторичные структуры пептидов являются важными для биологической активности, такие библиотеки пептидов не придают ограниченную вторичную структуру членам этой библиотеки.
С этой целью некоторые исследователи циклизовали пептиды с дисульфидными мостиками при попытке обеспечить более ограниченную вторичную структуру (Timelty et al., J. Chem. Soc. 1067-68, 1994; Eichler et al., Peptide Res. 7:300-306, 1994). Однако такие циклизованные пептиды обычно являются все еще вполне гибкими и недостаточно биологически доступными, и поэтому были достигнуты только ограниченные успехи.
Совсем недавно были разработаны непептидные соединения, которые более близко имитируют вторичную структуру обращенных витков, обнаруживаемых в биологически активных белках или пептидах. Например, в патенте США № 5440013, выданном Kahn, и опубликованной Международной заявке на патент РСТ WO94/034494 на имя Rahn описаны конформационно затрудненные непептидные соединения, которые имитируют трехмерную структуру обращенных витков.
Хотя значительные успехи были достигнуты в синтезе и идентификации конформационно затрудненных пептидных миметиков, сохраняется потребность в данной области в маленьких молекулах, которые имитируют вторичную структуру пептидов. Имеется также потребность в данной области в библиотеках, содержащих такие члены, а также способах для синтеза и скрининга членов библиотек против представляющих интерес мишеней, в частности биологических мишеней для идентификации биоактивных членов библиотек. Например, в патенте США № 5929237 и являющемся его частичным продолжением патенте США № 6013458, на имя Kahn, описаны также конформационно затрудненные соединения, которые имитируют вторичную структуру областей обращенных витков биологически активных пептидов и белков.
Настоящее соединение также удовлетворяет этим потребностям и обеспечивает дополнительные родственные преимущества предоставлением конформационно затрудненных соединений, которые имитируют вторичную β-цепочечную структуру биологически активных пептидов и белков.
Краткая сущность изобретения
В сущности, настоящее изобретение относится к конформационно затрудненным соединениям, которые имитируют вторичную структуру β-цепочечных структур биологически активных пептидов и белков. Данное изобретение описывает также библиотеки, содержащие такие соединения, а также их синтез и скрининг.
Соединения по настоящему изобретению имеют следующую общую структуру (I):
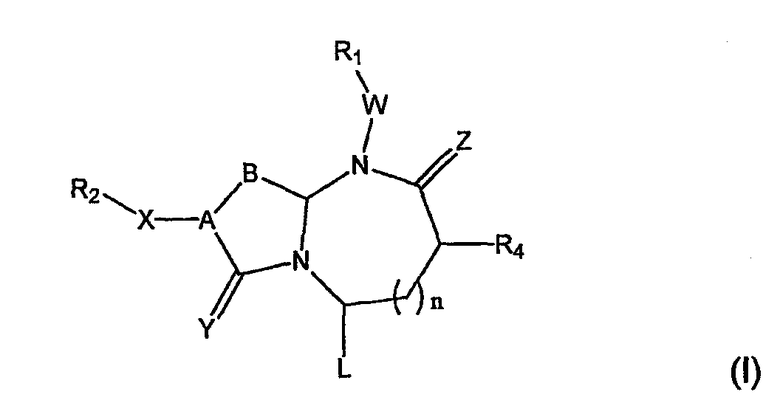
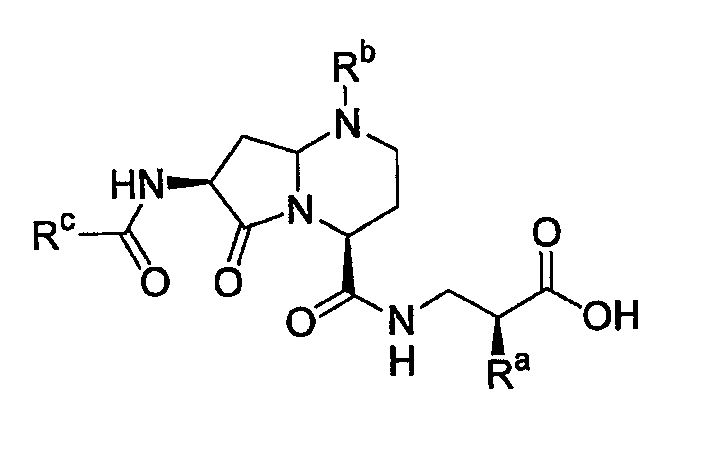
где А представляет собой -(СН)-, -N- или -СН2-N-, В представляет собой -(С=О) или -(СН2)m-, W представляет собой -(С=О)-, -Y(C=O)-, -NH(C=O)- или отсутствует, Х представляет собой -NH-, -NH(C=O)- или отсутствует, Y представляет собой кислород или серу, Z представляет собой кислород или водород, L представляет собой водород, R5, -C(O)NHR3 или его эквиваленты, n равно 0 или 1 и m равно 1 или 2; R1, R2, R3, R4 и R5 являются одинаковыми или различными и независимо выбраны из водорода, остатка боковой цепи аминокислоты или ее производного, остатка молекулы, линкера и твердого носителя, и структуру стереоизомеров указанных соединений.
В одном варианте осуществления Х отсутствует, А представляет собой -N-, B представляет собой -(С=О)-, L представляет собой -С(О)NHR3 и другие группы имеют значения, указанные выше для структуры (I), так что соединения изобретения имеют следующую структуру (I'):
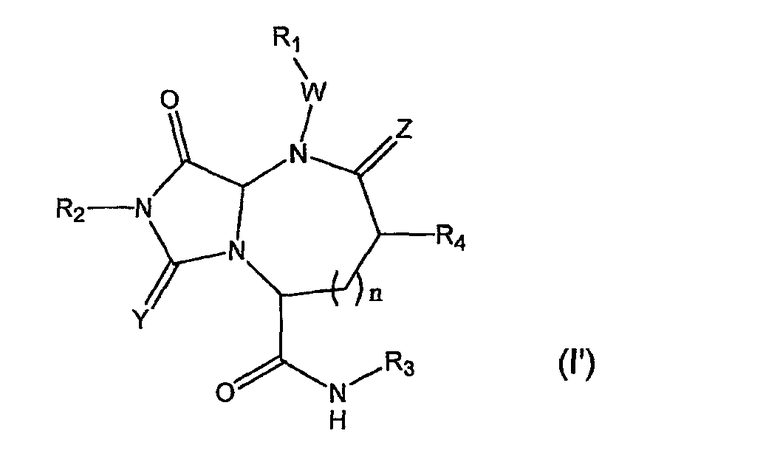
Необязательно, W отсутствует и Z представляет собой кислород.
В одном варианте осуществления Х отсутствует, A представляет собой -N-, B представляет собой -(СН2)m-, L представляет собой -С(О)NHR3- и другие группы имеют значения, указанные выше в связи со структурой (I), так что соединения изобретения имеют следующую структуру (I"):
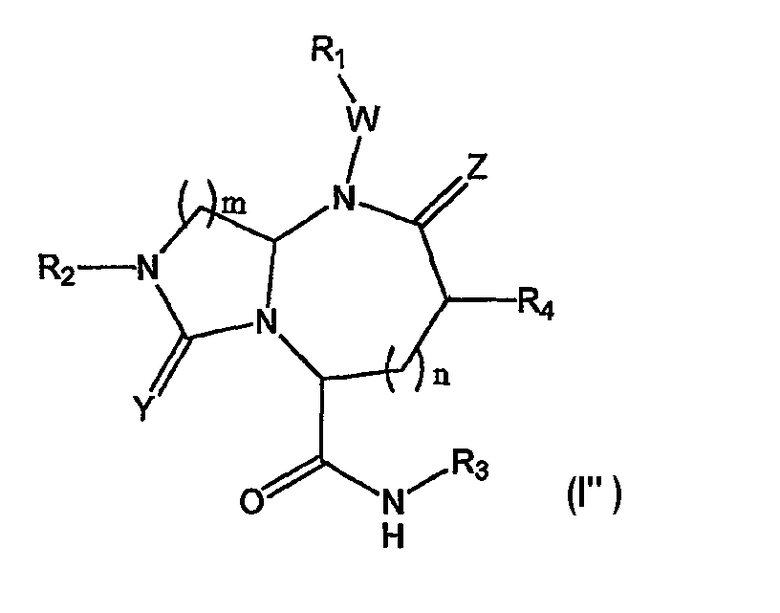
Необязательно, W отсутствует и Z представляет собой кислород.
В одном варианте осуществления Х представляет собой -NH-, A представляет собой -(СН)-, B представляет собой -(СН2)m-, L представляет собой -С(О)NHR3 и другие группы имеют значения, указанные выше в связи со структурой (I), так что соединения изобретения имеют следующую структуру (I"'):
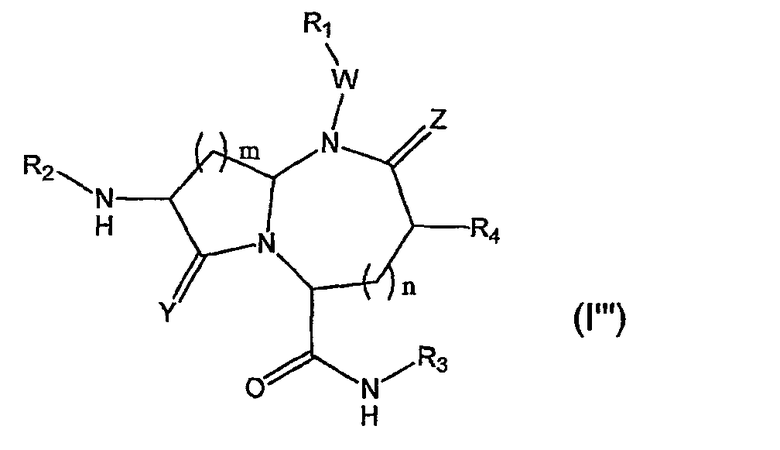
Необязательно, когда Z представляет собой кислород, то W отсутствует.
В одном варианте осуществления А представляет собой -СН2-N-, B представляет собой -(СН2)m-, L представляет собой -С(О)NHR3 и другие группы имеют значения, указанные выше в связи со структурой (I), так что соединения изобретения имеют следующую структуру (I""):
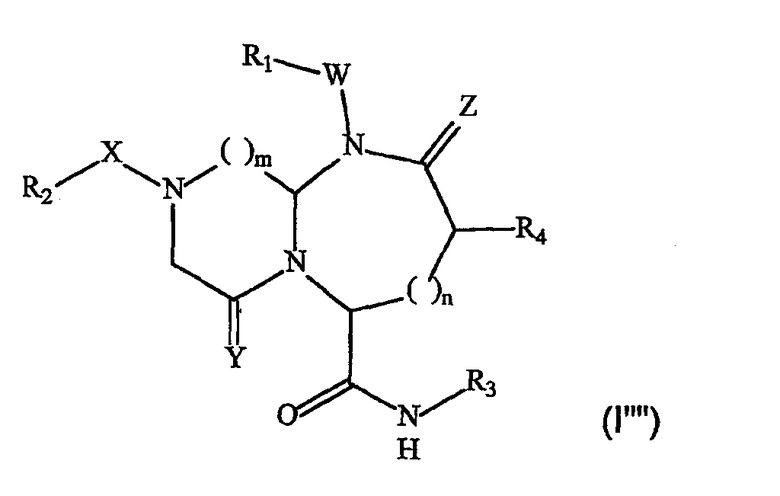
Необязательно, Y представляет собой кислород и/или W отсутствует, и/или Z представляет собой кислород.
Настоящее изобретение относится также к библиотекам, содержащим указанные выше структуры (I), (I'), (I"), (I"') и (I""), а также способам синтеза таких библиотек и способам скрининга их для идентификации биологически активных соединений. Описаны также композиции, содержащие соединение по данному изобретению в комбинации с фармацевтически приемлемым носителем или разбавителем.
Эти и другие аспекты по данному изобретению будут очевидными на основании ссылки на нижеследующее подробное описание и чертежи.
Краткое описание нескольких изображений чертежей
На фиг.1 и 2 показана синтетическая методика получения библиотек по настоящему изобретению и соединений по настоящему изобретению.
На фиг.3 показана синтетическая методика получения библиотеки по настоящему изобретению и соединений по настоящему изобретению, которые более полно описаны в примере 9.
На фиг.4 показана синтетическая методика получения библиотеки по настоящему изобретению и соединений по настоящему изобретению, которые более полно описаны в примере 10.
Подробное описание изобретения
Описаны конформационно затрудненные соединения, которые имитируют вторичную структуру β-цепочечных областей биологически активных пептидов и белков. Такие миметики β-цепочечных структур имеют применение в широком диапазоне областей, включая применение диагностических и терапевтических агентов. Описаны также библиотеки, содержащие миметики β-цепочечных структур по данному изобретению, а также способы скрининга их для идентификации биологически активных членов.
В одном аспекте настоящее изобретение относится к миметикам β-цепочечных структур и химическим библиотекам, содержащим миметики β-цепочечных структур. Миметики β-цепочечной структуры по настоящему изобретению являются применимыми в качестве биоактивных агентов, включающих (но не ограничивающихся перечисленным), применение в качестве диагностических, профилактических и/или терапевтических агентов. Библиотеки миметиков β-цепочечных структур по данному изобретению применяют при идентификации таких биоактивных агентов. На практике по настоящему изобретению библиотеки могут содержать от десятков до сотен тысяч (или больше) индивидуальных миметиков β-цепочечных структур (называемые также здесь «членами»).
В одном аспекте по настоящему изобретению описан миметик β-цепочечной структуры, имеющий следующую структуру (I):
где А представляет собой -(СН)-, -N- или -СН2-N-, В представляет собой -(С=О) или -(СН2)m-, W представляет собой -(С=О)-, -Y(C=O)-, -NH(C=O)- или отсутствует, Х представляет собой -NH-, -NH(C=O)- или отсутствует, Y представляет собой кислород или серу, Z представляет собой кислород или водород (когда Z представляет собой водород, то C=Z представляет собой СН2), L представляет собой водород, R5, -C(O)NHR3 или его эквиваленты, n равно 0 или 1 и m равно 1 или 2; R1, R2, R3, R4 и R5 являются одинаковыми или различными и независимо выбраны из водорода, остатка боковой цепи аминокислоты или ее производного, остатка молекулы, линкера и твердого носителя, и его стереоизомеры.
В одном аспекте изобретения R1, R2, R3, R4 и R5 независимо выбраны из группы, состоящей из амино-С2-5-алкила, гуанидино-С2-5-алкила, С1-4-алкилгуанидино-С2-5-алкила, ди-С1-4-алкилгуанидино-С2-5-алкила, амидино-С2-5-алкила, С1-4-алкиламидино-С2-5-алкила, ди-С1-4-алкиламидино-С2-5-алкила, С1-3-алкокси, фенила, замещенного фенила, (где заместители независимо выбраны из одного или нескольких заместителей из амино, амидино, гуанидина, гидразина, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-4-алкила, С1-3-алкокси, нитро, карбокси, циано, сульфурила или гидроксила), бензила, замещенного бензила (где заместители на бензиле независимо выбраны из одного или нескольких заместителей из амино, амидино, гуанидино, гидразино, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-3-алкокси, нитро, карбокси, циано, сульфурила или гидроксила), нафтила, замещенного нафтила (где заместители независимо выбраны из одного или нескольких заместителей из амино, амидино, гуанидино, гидразина, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-4-алкила, С1-3-алкокси, нитро, карбокси, циано, сульфурила или гидроксила), бисфенилметила, замещенного бисфенилметила (где заместители независимо выбраны из одного или нескольких из заместителей из амино, амидино, гуанидина, гидразина, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-4-алкила, С1-3-алкокси, нитро, карбокси, циано, сульфурила или гидроксила), пиридила, замещенного пиридила (где заместители независимо выбраны из одного или нескольких заместителей из амино, амидино, гуанидино, гидразино, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-4-алкила, С1-3-алкокси, нитро, карбокси, циано, сульфурила или гидрокси), пиридил-С1-4-алкила, замещенного пиридил-С1-4-алкила (где заместители пиридина независимо выбраны из одного или нескольких заместителей из амино, амидино, гуанидино, гидразино, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-4-алкила, С1-3-алкокси, нитро, карбокси, циано, сульфурила или гидроксила), пиримидил-С1-4-алкила, замещенного пиримидил-С1-4-алкила (где заместители пиримидина независимо выбраны из одного или нескольких заместителей из амино, амидино, гуанидина, гидразина, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-4-алкила, С1-3-алкокси или нитро, карбокси, циано, сульфурила или гидроксила), триазин-2-ил-С1-4-алкила, замещенного триазин-2-ил-С1-4-алкила (где заместители триазина независимо выбраны из одного или нескольких заместителей из амино, амидино, гуанидина, гидразина, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-4-алкила, С1-3-алкокси, нитро, карбокси, циано, сульфурила или гидроксила), имидазол-С1-4-алкила, замещенного имидазол-С1-4-алкила (где заместители имидазола независимо выбраны из одного или нескольких заместителей из амино, амидино, гуанидина, гидразина, амидразонила, С1-4-алкиламино, С1-4-диалкиламино, галогена, перфтор-С1-4-алкила, С1-4-алкила, С1-3-алкокси, нитро, карбокси, циано, сульфурила или гидроксила), имидазолинил-С1-4-алкила, N-амидинопиперазинил-N-С0-4-алкила, гидрокси-С2-5-алкила, С1-5-алкиламино-С2-5-алкила, С1-5-диалкиламино-С2-5алкила, N-амидинопиперидинил-С1-4-алкила и 4-аминоциклогексил-С0-2-алкила.
В одном варианте осуществления R1, R2 и R3 являются одинаковыми или различными и представляет собой остаток соединения и R4 выбран из остатка боковой цепи аминокислоты или его производного. В другом варианте осуществления L представляет собой -С(=О)NHR3 и R1, R2 и R3 являются одинаковыми или различными и представляет собой остаток соединения или остаток боковой цепи аминокислоты или его производного и R4 представляет собой водород.
Применяемый здесь термин «остаток боковой цепи аминокислоты» представляет собой любой остаток боковой цепи аминокислоты, присутствующий в существующих в природе белках, включающих в себя (но не ограничивающихся указанным) остатки боковых цепей существующих в природе аминокислот, указанные в таблице 1. Другие остатки боковых цепей существующих в природе аминокислот по данному изобретению включают в себя (но не ограничиваются указанным) остатки боковой цепи 3,5-дибромтирозина, 3,5-дииодтирозина, гидроксилизина, γ-карбоксиглутамата, фосфотирозина и фосфосерина. Кроме того, на практике по данному изобретению можно также применять боковые цепи гликозилированных аминокислот, включающих в себя (но не ограничивающихся перечисленным) гликозилированный треонин, серин и аспарагин.
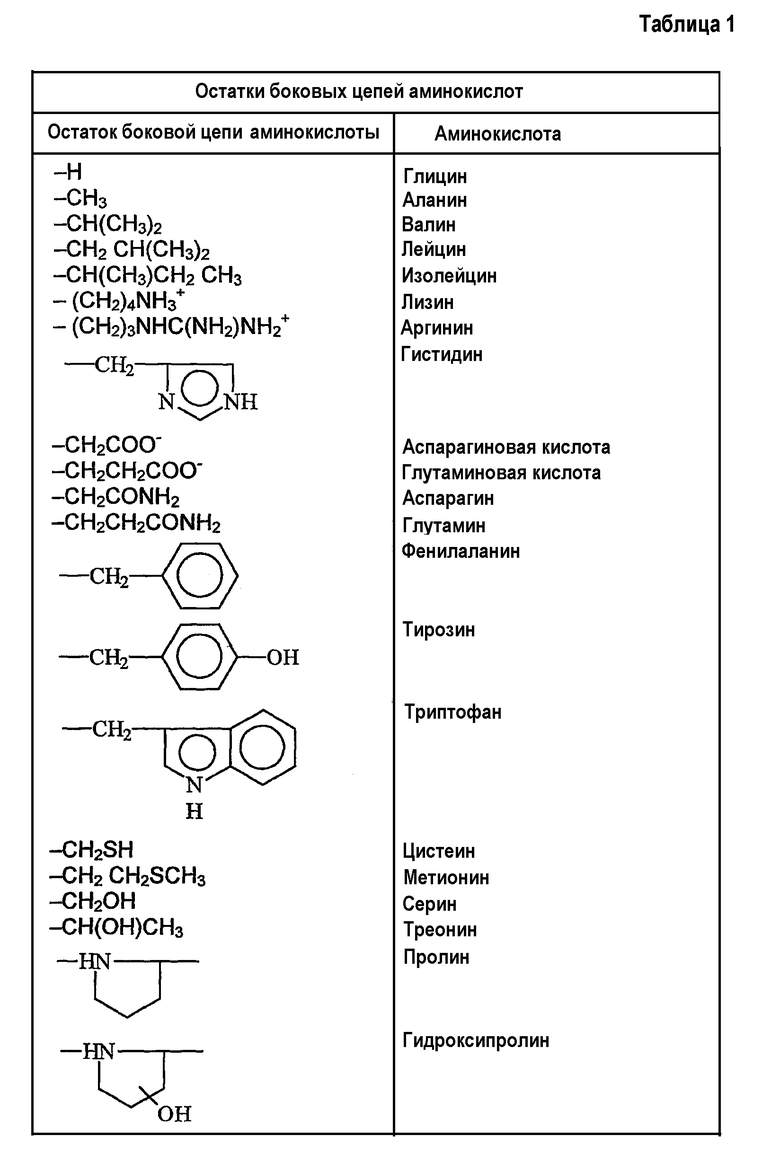
Кроме остатков боковых цепей существующих в природе аминокислот, остатки боковых цепей аминокислот по настоящему изобретению включают в себя различные их производные. Применяемый здесь термин «производное» остатка боковой цепи аминокислоты включает в себя модификации и/или варианты остатков боковых цепей существующих в природе аминокислот. Например, остатки боковых цепей аминокислот аланина, валина, лейцина, изолейцина и фенилаланина могут быть обычно классифицированы как низшие алкильные, арильные или аралкильные остатки. Производные остатков боковых цепей аминокислот включают в себя другие, имеющие неразветвленную или разветвленную цепь, циклические или нециклические, замещенные или незамещенные, насыщенные или ненасыщенные алкильные остатки с низшей цепью, арильные или арилалкильные остатки.
Применяемые здесь термины «остаток соединения» и «остаток молекулы» используют для обозначения любого остатка, агента, соединения, носителя, молекулы, линкера, аминокислоты, пептида или белка, ковалентно присоединенного к миметику β-цепочечной структуры. Присоединение предпочтительно находится в любом из R1-, и/или R2-, и/или R3-положений. Этот термин включают в себя также остатки боковых цепей аминокислот и их производные.
Применяемый здесь термин «алкильные остатки с низшей цепью» содержат 1-12 атомов углерода, «низшие арильные части» содержат 6-12 атомов углерода и «аралкильные части с низшей цепью» содержат 7-12 атомов углерода. Таким образом, в одном варианте осуществления производное боковой цепи аминокислоты выбрано из С1-12-алкила, С6-12-арила и С7-12-арилалкила и в более предпочтительном варианте из С1-7-алкила, С6-10-арила и С7-11-арилалкил.
Производные боковой цепи аминокислоты по данному изобретению дополнительно включает в себя замещенные производные имеющих низшую цепь алкильных остатков, арильных и арилалкильных остатков, где заместитель выбран из (но не ограничивается ими) одного или нескольких химических остатков:
-ОН, -OR, -COOH, -COOR, -CONH2, -NH2, -NHR, -NRR, -SH, -SR, -CO2R, -SO2H, -SOR и галогена (включая F, Cl, Br и I), где в каждом случае R независимо выбран из имеющих неразветвленную или разветвленную цепь, циклических или нециклических, замещенных или незамещенных, насыщенных или ненасыщенных низших алкильных, арильных или аралкильных остатков. В одном аспекте заместитель имеет меньше, чем 18 атомов углерода. Кроме того, циклические, имеющие низшую цепь алкильные, арильные и арилалкильные остатки по данному изобретению включают в себя нафталин, а также гетероциклические соединения, такие как тиофен, пиррол, фуран, имидазол, оксазол, тиазол, пиразол, 3-пирролин, пирролидин, пиридин, пиримидин, пурин, хинолин, изохинолин и карбазол. Производные боковых цепей аминокислот дополнительно включают в себя гетероалкилпроизводные алкильной части имеющих низшую алкильную цепь алкильных и аралкильных остатков, включающих в себя (но не ограничивающихся перечисленным) алкил- и аралкилфосфонаты и силаны.
В одном аспекте изобретения остатки R1, R2 и R3 выбраны из -ОН, -OR, -COR, -COOR, -CONH2, -CONR, -CONRR, -NH2, -NHR, -NRR, -SO2R и -COSR, где в каждом случае R имеет указанные выше значения.
В следующем варианте осуществления и в дополнении к остатку боковой цепи аминокислоты или ее производному (или остатку соединения в случае R1, R2 и R3) R1, R2 или R3 может быть линкером, облегчающим связывание соединения с другим остатком или соединением. Например, соединения по данному изобретению могут быть связаны с одним или несколькими известными соединениями, такими как биотин, для применения в диагностическом анализе или анализе для скрининга. Кроме того, R1, R2 или R3 может быть линкером, связывающим соединение с твердым носителем (таким как носитель, применяемым в синтезе пептида в твердой фазе), или, в альтернативном случае, может быть самим носителем. В этом варианте осуществления связь с другим остатком, или соединением, или твердым носителем находится предпочтительно у R1-, R2- или R3-положения и более предпочтительно у R3-положения.
В варианте осуществления, когда Х отсутствует, А представляет собой N, B представляет собой -С(=О)- и L представляет собой -С(О)NHR3, β-цепочечные соединения по данному изобретению имеют следующую структуру (I'):
где R1, R2, R3, R4, W, Y, Z и n имеют значения, указанные выше. В предпочтительно варианте осуществления R2 и R3 представляют собой остаток соединения, R1 и R4 выбраны из остатков боковых цепей аминокислот.
В варианте осуществления, когда Х отсутствует, А представляет собой N, В представляет собой -(СН2)m- и L представляет собой -С(О)NHR3, миметики β-цепочечных структур по данному изобретению включают в себя следующую структуру (I"):
где R1, R2, R3, R4, W, Y, Z и n имеют значения, указанные выше. В предпочтительно варианте осуществления R2 и R3 представляют собой остаток соединения и R1 и R4 выбраны из остатков боковых цепей аминокислот.
В более конкретном варианте осуществления, когда Х представляет собой -NH-, А представляет собой -(CH)- и В представляет собой -(СН2)m- и L представляет собой -С(О)NHR3, миметики с β-цепочечной структурой имеют следующую структуру (I"'):
где R1, R2, R3, R4, W, Y, Z, m и n имеют значения, указанные выше.
В более конкретном варианте осуществления, когда А представляет собой -CH2-N-, B представляет собой -(СН2)m- и L представляет собой -С(О)NHR3, соединения по данному изобретению имеют следующую структуру (I""):
В формуле необязательно R1, R2, R3, R4, W, Х, Y, Z, m и n имеют значения, указанные выше, W отсутствует, Z представляет собой кислород и Y представляет собой кислород.
Миметики β-цепочечных структур по данному изобретению могут быть получены с применением подходящих молекул исходных компонентов (далее называемых «компонентными частями»). Коротко говоря, в синтезе миметиков β-цепочечных структур, имеющих структуру (I'), первую и вторую компонентные части сочетают с образованием комбинированного первого-второго промежуточного соединения и, если необходимо, сочетают третью и/или четвертую компонентные части с образованием третьего-четвертого промежуточного соединения (или, если он является коммерчески доступным, можно применять одно третье промежуточное соединение), комбинированное первое-второе промежуточное соединение и третье-четвертое промежуточное соединение (или третье промежуточное соединение) затем сочетают, получая при этом первое-второе-третье-четвертое промежуточное соединение (или первое-второе-третье промежуточное соединение), которое циклизуют с образованием миметиков β-цепочечных структур по данному изобретению. В альтернативном варианте миметики β-цепочечных структур структуры (I') могут быть получены последовательным сочетанием индивидуальных компонентных частей либо постадийно в растворе, либо твердофазным синтезом, как обычно практикуется в твердофазном синтезе пептидов.
В контексте по настоящему изобретению «первая компонентная часть» имеет следующую структуру 1:
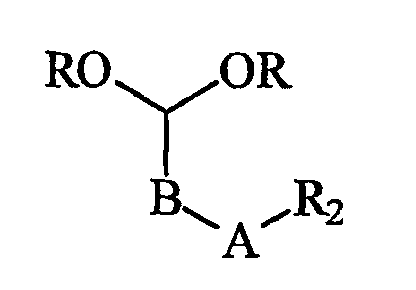
где R2, A и В имеют значения, указанные выше, и R представляет собой защитную группу, подходящую для применения в синтезе пептидов. Подходящие группы R включают в себя алкильные группы и в предпочтительном варианте осуществления R представляет собой метильную группу. Такие первые компонентные части могут быть легко синтезированы восстановительным аминированием посредством сочетания СН(OR)2-(CH2)m-CHO с H2N-R2 или замещением в СН(OR)2-(CH2)m-Br.
Вторая компонентная часть по данному изобретению имеет следующую структуру 2:
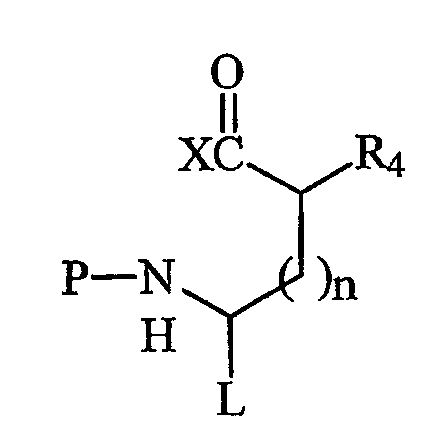
где L и R4 имеют значения, указанные выше, Р представляет собой аминозащитную группу, пригодную для применения в пептидном синтезе, и Х представляет собой уходящую группу активированной группы карбоновой кислоты. Предпочтительные защитные группы включают в себя трет-бутилдиметилсилил (TBDMS), BOC, FMOC и алло(аллилоксикарбонил). Когда L представляет собой -С(О)NHR3, -NHR3 может быть карбоксилзащитной группой. N-защищенные аминокислоты являются коммерчески доступными, например FMOC-аминокислоты являются доступными из различных источников. Превращение указанных N-защищенных аминокислот во вторые компонентные части по данному изобретению может быть легко достигнуто активацией группы карбоновой кислоты N-защищенной аминокислоты. Подходящие активированные группы карбоновых кислот включают в себя галогенангидриды кислот, где Х представляет собой галогенид, такой как хлорид или бромид, ангидриды кислот, где Х представляет собой ацильную группу, такую как ацетил, реакционноспособные эфиры, такие как эфиры N-гидроксисукцинимида и пентафторфениловые эфиры, и другие активированные промежуточные соединения, такие как активное промежуточное соединение, образованное реакцией сочетания с применением карбодиимида, такого как дициклогексилкарбодиимид (DCC).
В случае азидопроизводного аминокислоты, служащего в качестве второй компонентной части, такие соединения могут быть получены из соответствующей аминокислоты реакцией, описанной Zaloom et al. (J. Org. Chem. 46:5173-76, 1981).
«Третья компонентная часть» по данному изобретению имеет следующую структуру 3:
R1-NH2 или R3-NH2
где R1 и R3 имеют значения, указанные выше. Подходящие третьи компонентные части являются коммерчески доступными из различных источников или могут быть легко получены стандартными органическими синтетическими способами, обычно применяемыми для синтеза первичных аминов.
Более конкретно, имеющие структуру (I') миметики β-цепочечных структур по данному изобретению синтезируют взаимодействием первой компонентной части со второй компонентной частью с получением комбинированного первого-второго промежуточного соединения с последующим либо последовательным взаимодействием комбинированного первого-второго промежуточного соединения с третьей компонентной частью, либо с третьей и четвертой компонентными частями с получением комбинированного первого-второго-третьего-четвертого промежуточного соединения и затем циклизацией промежуточного соединения с получением миметика β-цепочечной структуры.
Общий синтез миметика β-цепочечной структуры, имеющего структуру I', можно выполнить следующим способом. Первую компонентную часть 1 сочетают со второй компонентной частью 2 с применением реагента сочетания, такого как фосген, получая при этом, после удаления N-защитной группы, комбинированное первое-второе промежуточное соединение 1-2, как показано ниже:
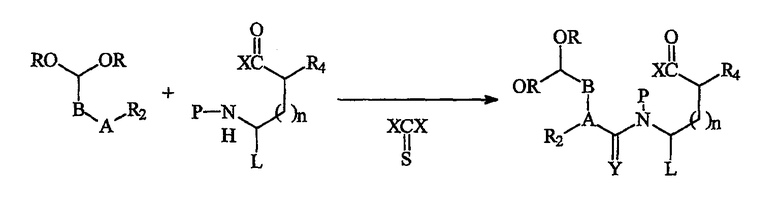
где А, В, L, R, R2, R4, P, X и n имеют значения, указанные выше. Х2С(=S) является примером агента сочетания, можно применять другой тип агентов сочетания. Синтезы репрезентативных компонентных частей по данному изобретению описаны в примерах. Соединения-миметики β-цепочной структуры от (I") до (I"') могут быть получены способами, аналогичными синтезу модульного компонента, описанного выше, но с подходящими модификациями для компонентных частей.
В другом аспекте по данному изобретению описаны библиотеки, содержащие миметики β-цепочечных структур по настоящему изобретению. После составления комплекта библиотеки по настоящему изобретению могут быть подвергнуты скринингу для идентификации индивидуальных членов, обладающих биоактивностью. Такой скрининг библиотек на биоактивные члены может включать в себя, например, оценку связывающей активности членов библиотек или оценку действия членов библиотек в функциональном анализе. Скрининг обычно выполняют контактированием членов библиотеки (или субпопуляции членов библиотеки) с представляющей интерес мишенью, такой как, например, антитело, фермент, рецептор или клеточная линия. Члены библиотек, которые способны взаимодействовать с представляющей интерес мишенью, называют здесь «биоактивными членами библиотек» или «биоактивными миметиками». Например, биоактивным миметиком может быть член библиотеки, который способен связываться с антителом или рецептором, который способен ингибировать фермент или который способен вызывать или антагонизировать функциональную реакцию, ассоциированную, например, с линией клеток. Другими словами, скрининг библиотек по настоящему изобретению определяет, какие члены библиотеки способны взаимодействовать с одной или несколькими специфическими биологическими, представляющими интерес мишенями. Когда взаимодействие имеет место, взаимодействие биоактивного миметика (или миметиков) может быть идентифицировано из числа членов библиотеки. Идентификация одного или (ограниченного числа) биоактивного миметика(ов) из библиотеки дает миметики β-цепочечной структуры, которые сами являются биологически активными и, таким образом, являются применимыми в качестве диагностических, профилактических или терапевтических агентов и могут дополнительно применяться для достижения значительного успеха в идентификации главных соединений в этих областях.
Синтез пептидных миметиков библиотеки по настоящему изобретению можно выполнять с применением известных способов синтеза пептидов в комбинации с первой, второй, третьей и, необязательно, четвертой компонентной частью по данному изобретению. Более конкретно, любая аминокислотная последовательность может быть присоединена к N-концевой и/или С-концевой части конформационно затрудненного соединения. С этой целью миметики могут быть синтезированы на твердом носителе (таком как смола РАМ) известными способами (см., например, John M. Stewart and Janis D. Young, Solid Phase Peptide Synthesis, 1984, Pierce Chemical Comp., Rockford, III) или силилсвязанной смоле присоединением спирта (см. Randolph et al., J. Am. Chem. Soc. 117:5712-14, 1995).
Кроме того, комбинацию способов синтеза как в растворе, так и твердой фазе можно применять для синтеза пептидных миметиков по данному изобретению. Например, твердый носитель можно применять для синтеза линейной пептидной последовательности до точки, в которой конформационно затрудненную β-цепь присоединяют к последовательности. Подходящую структуру миметика конформационно затрудненной β-цепи, которую предварительно синтезировали способами синтеза в растворе, можно затем присоединить в качестве следующей «аминокислоты» для синтеза в твердой фазе (т.е. миметик конформационно затрудненной β-цепи, который имеет как N-концевую часть, так и С-концевую часть, можно применять в качестве следующей аминокислоты, которую нужно присоединить к линейному пептиду). При введении структуры миметика конформационно затрудненной β-цепи в последовательность могут быть затем присоединены дополнительные аминокислоты для завершения образования пептида, связанного с твердым носителем. В альтернативном варианте линейные пептидные последовательности с защищенной N-концевой и С-концевой частями могут быть синтезированы на твердом носителе, отделены от носителя и затем подвергнуты сочетанию с миметиками структур с конформационно ограниченной β-цепью в растворе с применением известных способов сочетания в растворе.
В другом аспекте по данному изобретению описаны способы конструирования библиотек. Традиционные комбинаторные химические способы (см., например, Gallop et al., J. Med. Chem. 37:1233-1251, 1994) делают возможным быстрое получение огромного числа соединений последовательным присоединением реагентов к основному молекулярному каркасу. Комбинаторные способы применяли для конструкции библиотек пептидов, полученных из существующих в природе аминокислот. Например, посредством применения 20 смесей 20 подходящим образом защищенных и разных аминокислот и сочетания каждого с одним из 20 аминокислот создают библиотеку 400 (т.е. 202) дипептидов. Повторение процедуры несколько раз приводит к получению библиотеки дипептидов, содержащей приблизительно 20 биллионов (т.е. 208) октапептидов.
В следующем аспекте по данному изобретению описаны способы скрининга библиотек на биоактивность и выделения биоактивных членов библиотеки. Библиотеки по настоящему изобретению можно подвергнуть скринингу на биоактивность различными способами и методами. Обычно анализ для скрининга проводят (1) контактированием библиотеки с представляющей интерес биологической мишенью, такой как рецептор, и предоставлением возможности достижения связывания между миметиками библиотеки и мишенью и (2) детектированием случая связывания подходящим анализом, таким как калориметрический анализ, описанный Lam et al. (Nature 354: 82-84, 1991) или Griminski et al. (Biotechnology 12:1008-1011, 1994) (обе публикации включены здесь в качестве ссылки). В предпочтительном варианте осуществления члены библиотеки находятся в растворе и мишень иммобилизуют на твердой фазе. В альтернативном варианте библиотека может быть иммобилизована на твердой фазе и может быть зондирована контактированием ее с мишенью в растворе.
Синтез пептидных миметиков библиотеки по настоящему изобретению можно выполнить с применением общей схемы для получения библиотеки β-цепочечных миметиков, как показано на фиг.1. Синтез выбранных пептидных мишеней библиотеки бициклических шаблонов по настоящему изобретению выполняли с применением реакторного блока PlexChem, который имеет 96-луночный планшет. В указанной выше схеме "Pol" представляет собой смолу 2-хлортритилхлорида (Novabiochem), подробная процедура представлена ниже.
Стадия 1. Смолу 2-хлортритилхлорида (1 ммоль/г) и раствор Fmoc-R1-аминокислоты (1,5 экв.) и DIEA (2 экв.) в DCE помещают в 96-луночный блок Robinson (Flexchem). Реакционную смесь встряхивают в течение 12 часов при комнатной температуре. Смолу промывают ДМФА, МеОН и DCM.
Стадия 2. К смоле, набухшей в ДМФА перед реакцией, добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Повторяют стадию удаления защиты и смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор 4-R2-амино-2-Fmoc-аминомасляной кислоты (1,5 экв.), DIC (1,5 экв.) и НОВТ (1,5 экв.) в NMP. После того как реакционную смесь встряхивают в течение 12 часов при комнатной температуре, смолу промывают ДМФА, МеОН и затем DCM.
Стадия 3. К смоле, набухшей в ДМФА перед реакцией, добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Повторяют стадию удаления защиты и смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор 2-(9Н-флуорен-9-илметоксикарбониламино)-5,5-диметоксипентановую кислоту (1,5 экв.), DIC (1,5 экв.) и НОВТ (1,5 экв.) в NMP. Реакционную смесь встряхивают в течение 12 часов при комнатной температуре и затем смолу промывают ДМФА, МеОН и затем DCM.
Стадия 4. К смоле, набухшей в ДМФА перед реакцией, добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Повторяют стадию удаления защиты и смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор коммерчески доступной R3-кислоты (1,5 экв.), DIC (1,5 экв.) и НОВТ (1,5 экв.) в NMP. Реакционную смесь встряхивают в течение 12 часов при комнатной температуре и затем смолу промывают ДМФА, МеОН и затем DCM.
Стадия 5. Смолу обрабатывают муравьиной кислотой (1,2 мл на каждую лунку) в течение 18 часов при комнатной температуре. После этого смолу удаляют фильтрованием, фильтрат концентрируют при пониженном давлении с применением SpeedVac (Servant), получая при этом продукт в виде масла. Эти продукты разбавляют 50% смесью вода/ацетонитрил и затем лиофилизуют после замораживания.
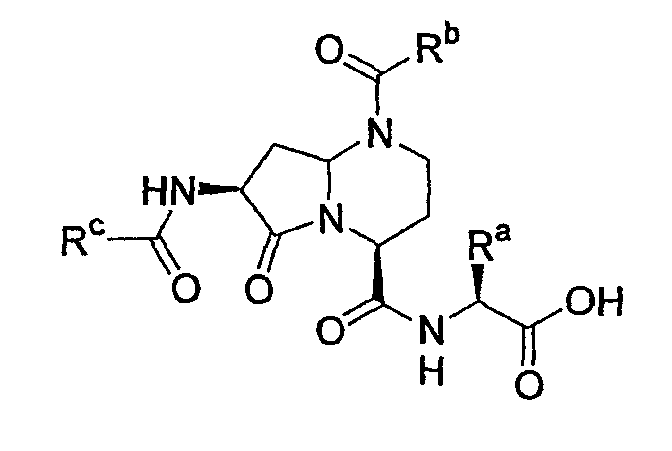
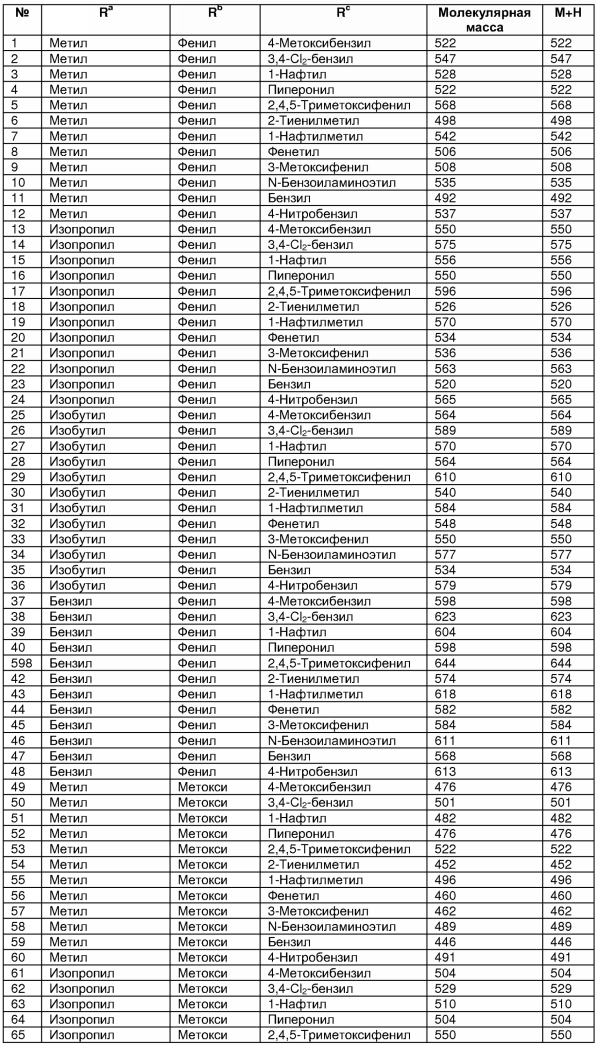
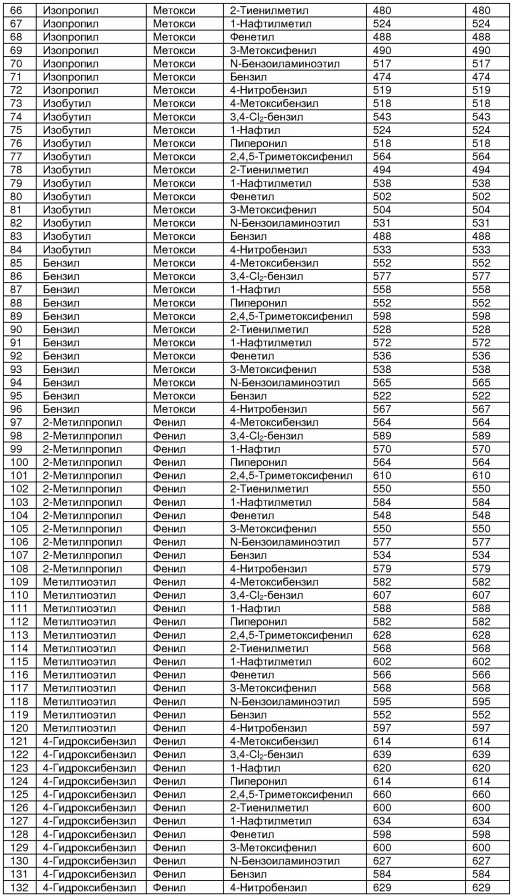
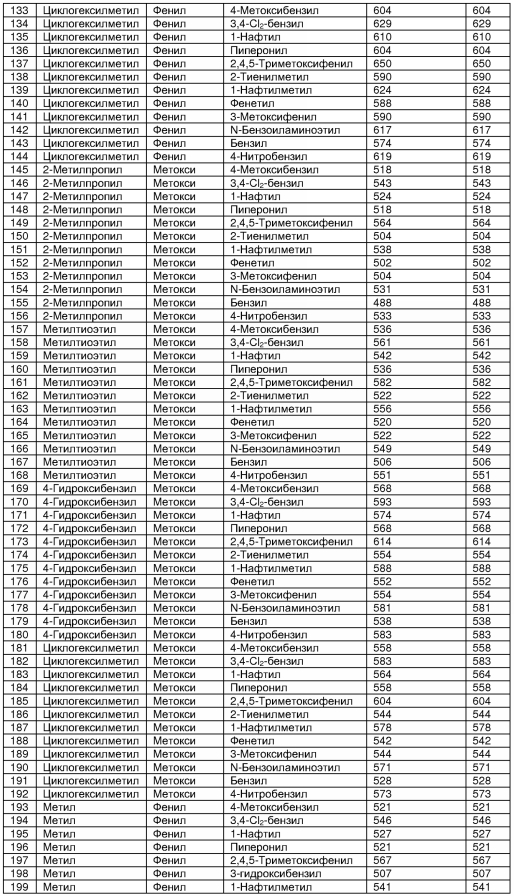
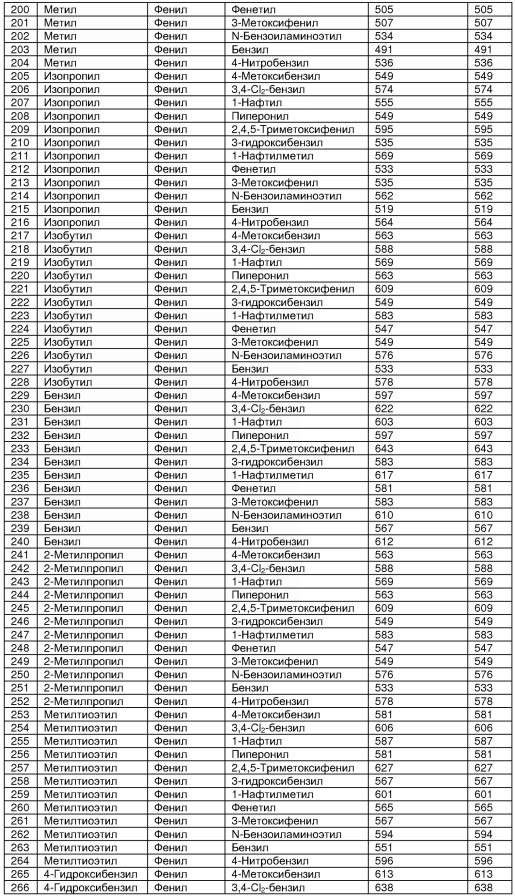
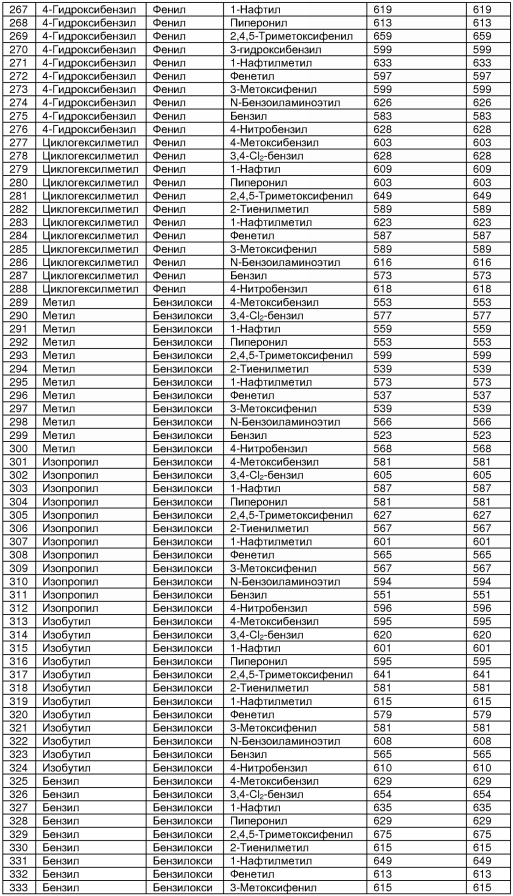
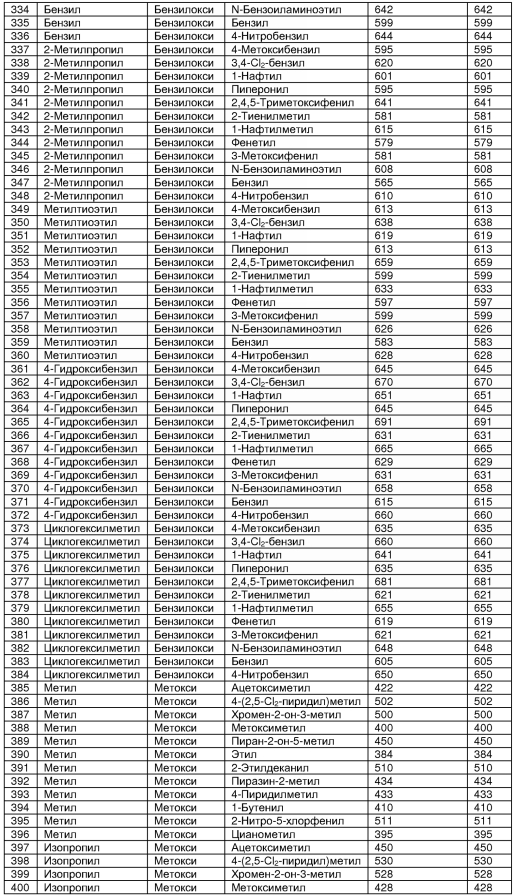
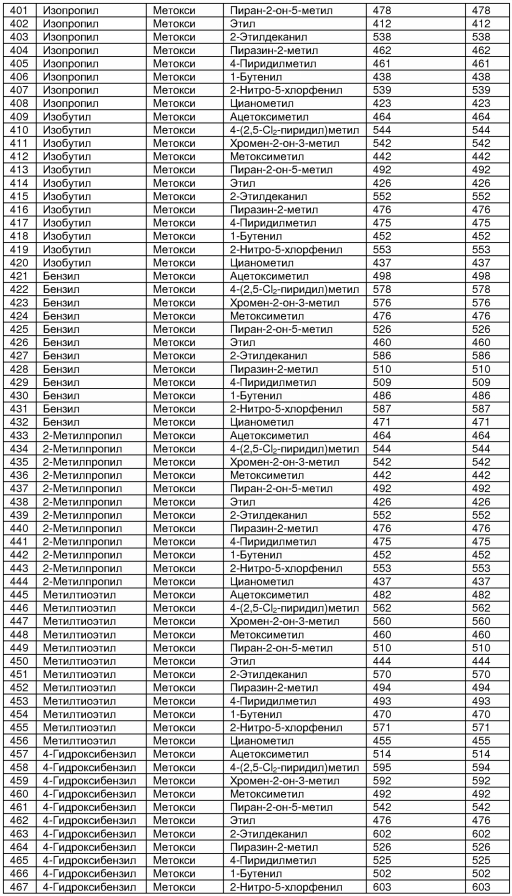
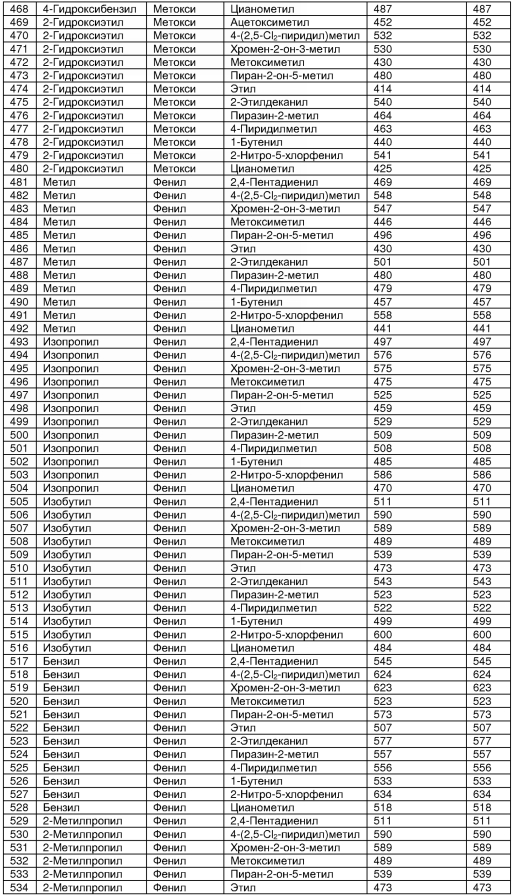
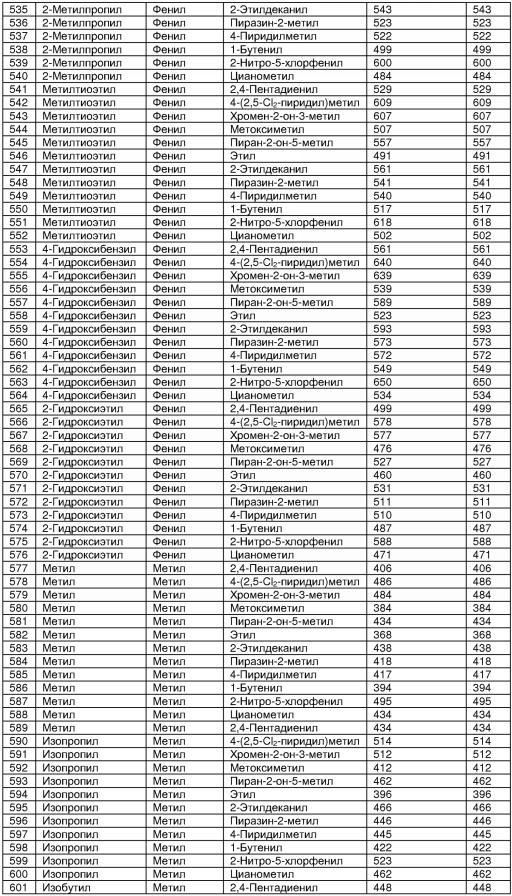
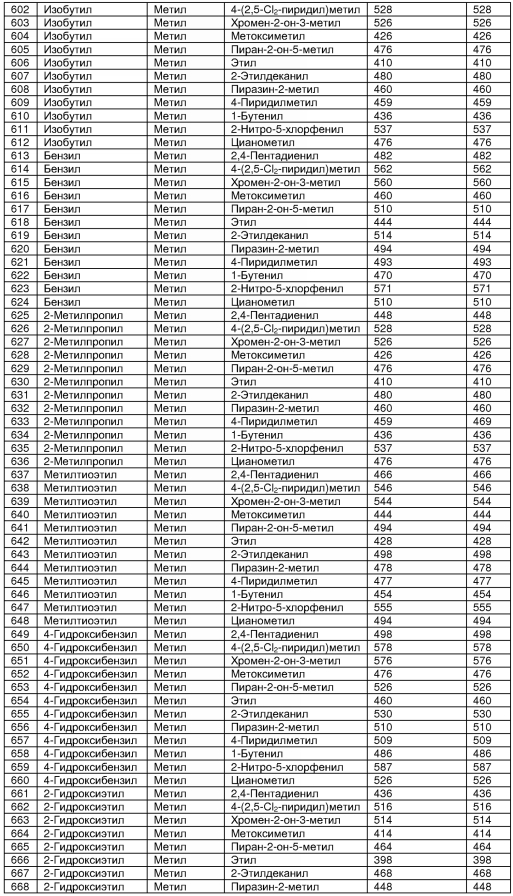
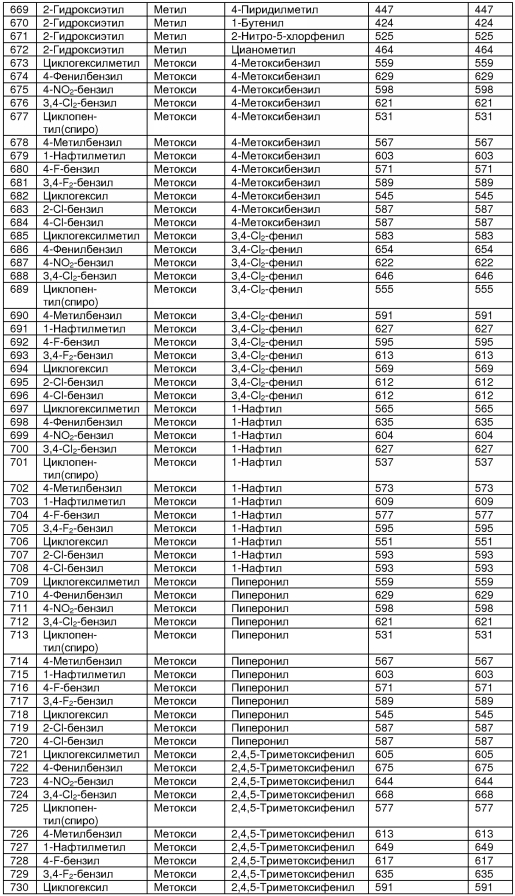
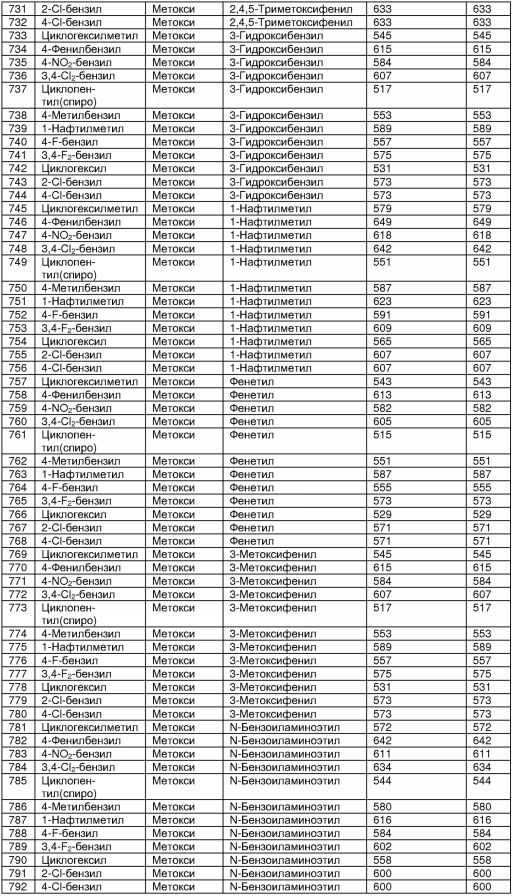
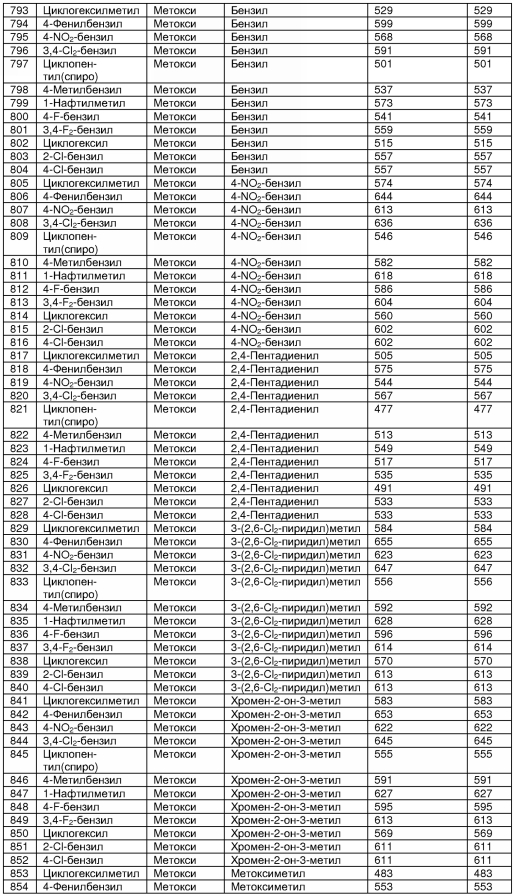
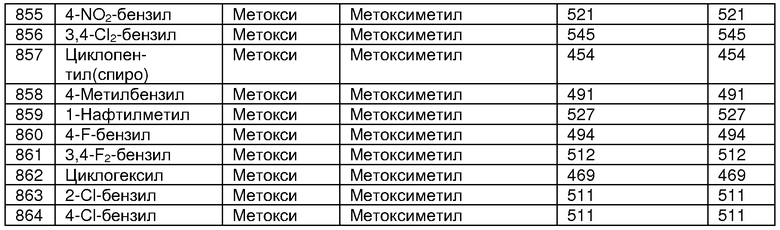
В таблице 2 показана библиотека миметиков β-цепочечной структуры, которая может быть получена по настоящему изобретению, репрезентативное получение ее приводится в примере 9. Соединения таблицы 2 иллюстрируют один аспект изобретения, а именно соединения, у которых А представляет собой -(СН)-, В представляет собой -(СН2)m- с m=1, W представляет собой -(С=О)-, Х представляет собой -NH(C=O)-, Y представляет собой кислород, Z представляет собой водород, так что C=Z представляет собой СН2, L представляет собой -C(=O)NHR3, n=0, R4 представляет собой водород и R1, R2 и R3 являются одинаковыми или различными и независимо выбраны из остатка боковой цепи аминокислоты или его производного, остатка молекулы, линкера и твердого носителя, и их стереоизомеры. В различных вариантах осуществления по данному аспекту изобретения R1, R2 и R3 независимо выбраны из остатков с относительно низкой молекулярной массой, т.е. органических групп, имеющих молекулярные массы между 15 (метил) и 1000 г/моль, и/или, по меньшей мере, один из R1, R2 и R3 представляет собой боковую цепь аминокислоты или ее производное. Например, в соединениях таблицы 2 R3 представляет собой производные аспарагиновой кислоты. В одном аспекте соединения по настоящему изобретению имеют молекулярную массу в диапазоне приблизительно от 440 до 750 г/моль, соединения таблицы 2 обеспечивают многочисленные иллюстрации таких соединений.
ТАБЛИЦА 2
Библиотека бета-цепочечных миметиков
Синтез пептидных миметиков в библиотеке по настоящему изобретению может быть выполнен с применением общей схемы библиотеки миметиков β-цепочечной структуры, как показано на фиг.2. Синтез выбранных пептидных миметиков библиотек и бициклических шаблонов по настоящему изобретению может быть выполнен с применением реакторного блока FlexChem, который имеет 96-луночный планшет. В указанной выше схеме "Pol" представляет собой смолу 2-хлортритилхлорида (Novabiovhem), подробная процедура предложена ниже.
Стадия 1. Смолу 2-хлортритилхлорида (1 ммоль/г) и раствор Fmoc-R1-бета-аминокислоты (1,5 экв.) и DIEA (2 экв.) в DCE помещают в 96-луночный блок Робинзона (Flexchem). Реакционную смесь встряхивают в течение 12 часов при комнатной температуре. Смолу промывают ДМФА, МеОН и затем DCM.
Стадия 2. К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Повторяют стадию удаления защиты и смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор 4-R2-амино-2-Fmoc-аминомасляной кислоты (1,5 экв.), DIC (1,5 экв.), НОВТ (1,5 экв.) в NMP. После встряхивания реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
Стадия 3. К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Повторяют стадию удаления защиты и смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор 2-(9Н-флуорен-9-илметоксикарбониламино)-5,5-диметоксипентановой кислоты (1,5 экв.), DIC (1,5 экв.) и НОВТ (1,5 экв.) в NMP. После встряхивания реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
Стадия 4. К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Повторяют стадию удаления защиты и затем смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор коммерчески доступной R3-кислоты (1,5 экв.), DIC (1,5 экв.) и НОВТ (1,5 экв.) в NMP. После встряхивании реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
Стадия 5. Смолу обрабатывают муравьиной кислотой (1,2 мл на каждую лунку) в течение 18 часов при комнатной температуре. После этого смолу удаляют фильтрованием и фильтрат концентрируют при пониженном давлении с применением SpeedVac (Servant), получая при этом продукт в виде масла. Эти продукты разбавляют 50% смесью вода/ацетонитрил и затем лиофилизуют после замораживания.
В таблице показана библиотека миметиков β-цепочечной структуры, которая может быть получена по настоящему изобретению и репрезентативное получение которой дается в примере 10. Соединения таблицы 3 иллюстрируют один аспект изобретения, а именно соединения, у которых А представляет собой -(СН)-, В представляет собой -(СН2)m- с m=1, W отсутствует, т.е. представляет собой прямую связь между Rb и N гетероциклического кольца, Х представляет собой -NH(C=O)-, Y представляет собой кислород, Z представляет собой водород, так что C=Z представляет собой СН2, L представляет собой -С(=О)NHR3, n=0, R4 представляет собой водород и R1, R2 и R3 являются одинаковыми или различными и независимо выбраны из остатка боковой цепи аминокислоты или его производного, остатка молекулы, линкера и твердого носителя, и их стереоизомеры. В различных вариантах осуществления данного аспекта изобретения R1, R2 и R3 независимо выбраны из остатков с относительно низкой молекулярной массой, т.е. органических групп, имеющих молекулярные массы между 15 (метил) и 1000 г/моль; и/или, по меньшей мере, один из R1, R2 и R3 представляет собой боковую цепь аминокислоты или ее производное. Например, в соединениях таблицы 3 R3 представляет собой производные глутаровой кислоты. В одном аспекте соединения по настоящему изобретению имеют молекулярную массу в диапазоне приблизительно 450-800 г/моль, соединения таблицы 3 обеспечивают многочисленные иллюстрации таких соединений.
ТАБЛИЦА 3
Библиотека миметиков β-цепи
масса
Миметики β-цепочечных структур по настоящему изобретению можно применять в качестве биоактивных агентов, таких как диагностические, профилактические и терапевтические агенты. Соединения предпочтительно изготовляют в фармацевтически приемлемой форме и затем вводят пациенту, нуждающемуся в лечении миметиками β-цепочечных структур по настоящему изобретению.
Таким образом, настоящее изобретение предусматривает фармацевтическую композицию, содержащую соединение со структурами от (I") до (I"'). Для получения фармацевтической композиции, содержащей настоящие соединения, специалист в данной области может применять известные общественности знания и способы, которые известны в соответствующей области техники. Для получения композиции по настоящему изобретению применяют обычно известные разновидности носителей и других добавок. Фармацевтические композиции по данному изобретению могут быть введены стандартным способом при патологическом состоянии, которое нужно лечить, например пероральным, ректальным или парентеральным введением.
Для этих целей соединения по настоящему изобретению могут быть изготовлены способами, известными в данной области, в форме, например, таблеток, капсул, водных или масляных растворов или суспензий, (липидных) эмульсий, диспергируемых порошков, суппозиториев, мазей, кремов, капель и стерильных инъецируемых водных или масляных растворов или суспензий.
Подходящей фармацевтической композицией по настоящему изобретению является композиция, подходящая для перорального введения в стандартной лекарственной форме, такой как, например, таблетка или капсула, которая содержит приблизительно от 1 мг до приблизительно 1 г соединения по данному изобретению.
В другом аспекте фармацевтическая композиция по настоящему изобретению является композицией, подходящей для внутривенной, подкожной или внутримышечной инъекции. Пациент может получать, например, внутривенную, подкожную или внутримышечную дозу приблизительно от 1 мкг/кг до приблизительно 1 г/кг соединения по настоящему изобретению. Внутривенная, подкожная и внутримышечная доза может быть введена посредством болюсной инъекции. В альтернативном варианте внутривенная доза может быть введена непрерывной инфузией на протяжении периода времени.
В альтернативном варианте пациент может получать суточную пероральную дозу, которая приблизительно эквивалентна суточной парентеральной дозе, причем композицию вводят от 1 до приблизительно 4 раз в день.
В следующей таблице иллюстрированы репрезентативные фармацевтические дозированные формы, содержащие соединение или его фармацевтически приемлемую соль, и предназначенные для терапевтического или профилактического применения для людей:
Фармацевтическую композицию, содержащую соединение общей формулы (I), можно применять для индуцирования различных биологических действий, включающих в себя ингибирование протеазы у теплокровного животного, модуляцию относящегося к пептиду фактора транскрипции сигнала клетки у теплокровного животного, и для ингибирования киназы у теплокровного животного. Эти действия можно достичь способом, содержащим введение животному, нуждающемуся в этом, эффективного количества соединения формулы (I).
Кроме того, как описано подробно ниже, миметики β-цепочечных структур по настоящему изобретению могут быть также эффективными для ингибирования МНС-I- и/или МНС-II-презентации пептидов рецепторам Т-клеток у теплокровного животного; для ингибирования связывания пептида с SH2-доменами у теплокровного животного; для ингибирования связывания пептида с SH3-доменами у теплокровного животного; для ингибирования связывания пептида с РТВ-доменами у теплокровного животного; для модуляции связанного с G-белком рецептора (GPCR) и ионного канала у теплокровного животного и для модуляции цитокинов у теплокровного животного.
Ингибирование киназы (включая ингибирование SH2- и SH3-доменов)
В одном аспекте настоящее изобретение предусматривает способ ингибирования киназы у теплокровного животного. Способ содержит введение животному количества соединения по настоящему изобретению, где количество является эффективным для ингибирования киназы. Киназы (известные также как протеинкиназы) являются классом ферментов, которые катализируют реакцию, посредством которой биомолекула (обычно другой фермент) фосфорилируется. Считается, что до 1000 киназ кодируются в геноме млекопитающего (Hunter, Cell 50:823-829, 1987). Большое число киназ делает возможным быструю амплификацию (усиление) сигнала и создание многочисленных точек регуляции.
Фосфорилирование является очень общей ковалентной модификацией, обнаруженной в процессах трансдукции сигналов, и вызывает изменение в активности тех белков, которые становятся фосфорилированными. Киназы, таким образом, являются критическим компонентом путей передачи сигналов. Киназы обычно организуются в несколько модульных функциональных участков или «доменов» (Cohen, G.B., et al. Cell 80:237-248, 1995). Один домен, известный как "SH3", является областью 55-70 аминокислот, которая связывается с обогащенными пролином пептидами, особенно с протяженной цепью. Другим доменом, известным как "SH2", является связывающая фосфотирозин область с длиной приблизительно 100 аминокислот. Считается, что эти два домена принимают участие в узнавании и связывании с белковыми субстратами. Эти, а также другие домены, включая сайты миристоилирования и пальмитоилирования, являются ответственными за сборку полибелковых комплексов, которые направляют каталитический домен к правильным мишеням (Mayer et al. Mol. Cell. Biol. 12:609-618, 1992; and Mayer and Baltimore, Mol. Cell. Biol. 14:2883-2894, 1994). Известно, что, хотя домены SH2 и SH3 присутствуют в некоторых киназах, эти домены присутствуют также в других белках. Соединения по настоящему изобретению можно применять для ингибирования SH2- или SH3-опосредованного связывания в киназе или других белках.
Киназы применяются организмом в очень большом числе разных, но часто взаимосвязанных внутриклеточных механизмах трансдукции сигналов. Например, факторы роста, факторы транскрипции, гормоны, регуляторные белки клеточного цикла и многие другие классы клеточных регуляторов применяют тирозинкиназы в их каскадах передачи сигнала (см., например, Bolen et al. FASEB J. 6:3403-3409, 1992; and Ullrich and Schlessinger, Cell 61:203-212, 1990). Серин/треонинкиназы составляют большинство остальной части семейства киназ.
Одним важным подходом для определения роли и понимания функции ферментов, как in vitro, так и in vivo, является применение специфических ингибиторов ферментов. Если может быть найдено одно или несколько соединений, которые будут ингибировать фермент, ингибитор можно применять для модуляции активности фермента, и могут наблюдаться влияния такого снижения. Такие подходы служат средством в расшифровке многих из путей промежуточного метаболизма и являются также важными в изучении кинетики ферментов и определении каталитических механизмов. Настоящее изобретение предусматривает такие соединения.
Регуляция многих иммунных реакций опосредуется через рецепторы, которые передают сигналы посредством тирозинкиназ, содержащих домен SH2. Активация Т-клеток через антиген-специфический рецептор Т-клеток (TCR) инициирует каскад трансдукции сигнала, приводящего к секреции лимфокина и пролиферации клеток. Одной из самых ранних биохимических реакций после активации TCR является повышение в активности тирозинкиназы. В частности, активация и пролиферация Т-клеток регулируется посредством опосредованной рецептором Т-клеток активации тирозинкиназ р56lck и р59fyn, а также ZAP-70 и Syk (Weiss and Litman, Cell 76:263-274, 1994), которые содержат домены SH2. Дополнительное доказательство указывает на то, что несколько киназ src-семейства (lck, blk, fyn) принимают участие в путях трансдукции сигнала, идущих от рецепторов антигена В-клеток и поэтому могут служить для интеграции стимулов, полученных от нескольких независимых структур рецепторов. Таким образом, ингибиторы, которые блокируют взаимодействие этих киназ, содержащих домен SH2, с их родственными рецепторами, могут служить в качестве иммуносупрессивных агентов, применяемых при аутоиммунных заболеваниях, отторжениях трансплантата или в качестве противовоспалительных агентов, а также в качестве противораковых лекарственных средств в случаях лимфолейкоза.
Кроме того, известны нетрансмембранные РТР-азы, содержащие SH2-домены, причем номенклатура обозначает их SH-PTP1 и SH-PTP2 (Neel, Cell Biology 4:419-432, 1993), SH-PTP1 является идентичной РТР1С, HCP или SHP и SH-PTР2 является известной также как PTP1D или РТР2С. SH-PTP1 экспрессируется при высоких уровнях в гемопоэтических клетках всех линий и всех стадий дифференцировки. Поскольку ген SH-PTP1 идентифицирован как ответственный за материнский (me) фенотип мыши, это обеспечивает основу для предсказания действий ингибиторов, которые могут блокировать его взаимодействие с его клеточными субстратами. Таким образом, можно ожидать, что ингибирование функции SH-PTP1 приведет к ослабленным реакциям Т-клеток на митогенную стимуляцию, пониженной функции NK-клеток и истощению предшественников В-клеток с потенциальными терапевтическими применениями, как описано выше.
Способность соединения по настоящему изобретению связываться с SH2-доменом STAT6 или связываться с SH2-доменом протеинтирозинфосфатазы SH-PTP1 можно демонстрировать процедурами, описанными Payne et al., P.N.A.S. USA 90:4902-4906, 1993). Скрининг библиотеки SH2-связывающих миметиков можно проводить по процедуре Songyang et al., Cell 72:767-778, 1993. См. также процедуру Songyang et al., Current Biology 4:973-982, 1994) для испытания способности соединения действовать в качестве субстрата или ингибитора протеинкиназ.
В соответствии с этим, в одном аспекте настоящее изобретение предусматривает способ ингибирования фосфатазы у теплокровного животного, где способ содержит введение животному количества соединения по настоящему изобретению, которое является эффективным для ингибирования фосфатазы.
При диабете типа 2 (инсулиннезависимом диабете) тирозинфосфатазы (РТР-1b) уравновешивают действие активированных инсулиновым рецептором киназ и могут представлять собой важные мишени для лекарственных средств. Эксперименты in vitro показывают, что инъекция РТРазы блокирует стимулированное инсулином фосфорилирование тирозильных остатков на эндогенных белках. Таким образом, соединения изобретения можно применять для модуляции действия инсулина при диабете.
В другом аспекте настоящее изобретение предусматривает способ ингибирования связывания остатка фосфотирозина в первом белке с SH2-доменом второго белка. Способ содержит контактирование количества соединения по настоящему изобретению с композицией, содержащей первый и второй белок. Количество является эффективным для ослабления связывания между первым и вторым белком, которое имеет место через SH2-домен второго белка и остатки фосфотирозина первого белка.
Ингибирование протеазы
В другом аспекте настоящее изобретение предусматривает способ ингибирования протеаз в организме теплокровного животного. Способ содержит введение животному количества соединения по настоящему изобретению, как здесь описано. Количество является эффективным для ингибирования протеазы в организме животного. В различных вариантах осуществления протеазой является серинпротеаза; протеазой является серинпротеаза, выбранная из тромбина, фактора Х, фактора IX, фактора VII, фактора XI, урокиназы, HCV-протеазы, химазы, триптазы и калликреина; протеазой является тромбин; протеазой является фактор VII и протеаза выбрана из протеазы аспарагиновой кислоты, цистеинпротеазы и металлопротеазы.
Что касается ингибирования протеазы, катепсином В является лизосомная цистеинпротеаза, обычно принимающая участие в проферментном процессинге и обновлении белка. Повышенные уровни активности имеют место при метастазах опухолей (Stoane, B.F. et al., Cancer Metastasis Rev. 9:333-352, 1990), ревматоидном артрите (Werb, Z. Textbook of Rheumatology, Keller, W.N.; Harris W.D.; Ruddy, S.; Siedge, C.S., Eds., 1989, W.B. Saunder Co., Philadelphia, Pa., pp.300-321) и мышечной дистрофии (Katunuma and Kominami, Rev. Physiol. Biochem. Pharmacol. 108:1-20, 1987).
Калпаины представляют собой цитозольные или связанные с мембранами, Са++-активированные протеазы, которые являются ответственными за деградацию цитоскелетных белков в ответ на изменение уровней кальция в клетке. Они способствуют деградации ткани при артрите и мышечной дистрофии (см. Wang and Yuen Trends Pharmacol. Sci. 15:412-419, 1994).
Превращающий интерлейкин фермент (ICE) расщепляет про-IL-1-бета с образованием IL-1-бета, ключевого медиатора воспаления, и, следовательно, можно доказать, что ингибиторы ICE являются применимыми при лечении артрита (см., например, Miller B.E. et al., J. Immunol. 154:1331-1338, 1995). ICE или ICE-подобные протеазы могут также принимать участие в апоптозе (программированная гибель клеток) и, следовательно, играть роль в раковом заболевании, СПИДе, болезни Альцгеймера и других заболеваниях, в которых имеет место разрегулированный апоптоз (см. Barr and Tomei, Biotechnol. 12:487-493, 1994).
ВИЧ-протеаза играет ключевую роль в жизненном цикле ВИЧ, вируса СПИДа. В конечных стадиях вирусного созревания она расщепляет полипротеиновые предшественники до функциональных ферментов и структурных белков ядра вирона. Ингибиторы ВИЧ-протеазы быстро идентифицировали в качестве превосходного терапевтического инструмента (средства) для СПИДА (см. Huff, J.R., J. Med. Chem. 34:2305-2314), и уже доказано, что они являются применимыми при его лечении, что доказано недавним одобрением FDA ритонавира, криксивана и сакуинавира.
Вирус гепатита С (HCV) является основной причиной гепатита не-А и не-В во всем мире на настоящее время. Приблизительно подсчитано, что он инфицирует до 50 миллионов человек. В настоящее время не имеется удовлетворительного лечения, доступного для остановки развития этого ослабляющего здоровье заболевания. Во время жизненного цикла данного вируса продуцируется полипротеин приблизительно из 3000 аминокислот и он протеолитически расщепляется хозяином и вирусными протеазами с продуцированием зрелых продуктов вирусного гена. Серинпротеиназа, расположенная в NS3-белке HCV, расщепляет у четырех специфических сайтах с образованием неструктурных белков, рассматриваемых как неотъемлемые для вирусной репликации. Следовательно, ингибиторы HCV-протеазы являются привлекательными инструментами для разработки лекарственных средств и могут иметь огромную терапевтическую пользу. (Neddermann et al., Biol. Chem. 378:469-476, 1997).
Превращающий ангиотензин фермент (АСЕ) является частью системы ренин-ангиотензин, которая играет центральную роль в регуляции кровяного давления. АСЕ расщепляет ангиотензин I в октапептидный ангилтензин II, сильнодействующий, повышающий кровяное давление агент вследствие его сосудосужающей активности. Доказано, что ингибирование АСЕ является терапевтически применимым при лечении гипертензии (Williams, G.H., N. Engl. J. Med. 319:1517-1525, 1989).
Коллагеназы расщепляют коллаген, основной компонент внеклеточной матрицы (например, соединительной ткани, кожи, кровеносных сосудов). Повышенная активность коллагеназы способствует появлению артрита (Krane et al., Ann. N.Y. Acad. Sci. 580:340-354, 1990), метастаз опухолей (Flug and Korf-Maier, Acta Anat. Basel 152:69-84, 1995) и других заболеваний, заключающихся в разрушении соединительной ткани.
Подобные трипсину серинпротеазы образуют большое и очень селективное семейство ферментов, принимающих участие в гемостазе/коагуляции (Davie and Fujikawa, Ann. Rev. 799-829, 1975) и активации комплемента (Muller-Eberhard, Ann. Rev. Biochem. 44:697-724, 1975). Секвенирование этих протеаз показало присутствие гомологического, подобного трипсину ядра с аминокислотными вставками, которые модифицируют специфичность и которые обычно являются ответственными за взаимодействие с другими макромолекулярными компонентами (Magnusson et al., Miami Winter Symposia 11:203-239, 1976).
Тромбин, подобная трипсину серинпротеаза, действует, обеспечивая ограниченный протеолиз, как при генерации фибрина из фибриногена, так и активации рецептора тромбоцитов, и, таким образом, играет критическую роль в тромбозе и гемостазе (Mann, K.G. Trends Biochem. Sci. 12:229-233, 1987). Тромбин проявляет заметную специфичность в удалении фибринопептидов А и В фибриногена посредством селективного расщепления только двух связей Arg-Gly из ста восьмидесяти одной последовательности Arg- или Lys-Xaa в фибриногене (Biomback, Blood Clotting Enzymology, Seeger, W. H. (ed.), Academic Press, New York, 1967, pp.143-215).
Многие значительные патологические состояния относятся к аномальному гемостазу, включающему острые коронарные синдромы. Аспирин и гепарин широко применяют при лечении пациентов с острыми коронарными синдромами. Однако эти агенты имеют несколько существенных ограничений. Например, тромбоз, осложняющий разрыв атеросклеротической бляшки, имеет тенденцию быть опосредованным тромбином, зависимым от тромбоцитов процессом, который является относительно устойчивым к ингибированию аспирином и гепарином (Fuster et al., N. Engl. J. Med. 326:242-50, 1992).
Ингибиторы тромбина предотвращают образование тромба у мест васкулярного повреждения in vivo. Кроме того, поскольку тромбин является также сильнодействующим фактором роста, который инициирует пролиферацию гладкомышечных клеток в местах механического повреждения в коронарной артерии, ингибиторы блокируют эту пролиферативную реакцию гладкомышечных клеток и снижают рестеноз. Ингибиторы тромбина могут также снижать воспалительную реакцию в васкулярных клеточных оболочках (Harker et al., Am. J. Cardiol. 75:122-16B, 1995).
Кроме того, по меньшей мере, два вполне определенных фактора транскрипции, ядерный фактор (NF) kB и белок-активатор (АР)-1, регулируются внутриклеточным состоянием (системой) восстановление-окисление (редокс). Регуляция экспрессии гена редокс-системой имеет перспективные терапевтические последствия. Например, сайты связывания редокс-регулированных факторов транскрипции NF-kB и АР-1 расположены в области промотора большого числа генов, которые непосредственно вовлекаются в патогенез заболеваний, таких как СПИД, рак, атеросклероз и диабетические осложнения (Sen and Packer, FASEB Journal 10:709-720, 1996). Более конкретно, связывание факторов транскрипции, таких как NF-kB и АР-1, с консенсунсными факторами на ДНК приводится в действие окислительно-антиокислительным гемостазом, особенно равновесием тиол-дисульфид.
В случае NF-kB физиологически подходящий тиол, который играет критическую роль в регуляции функции NF-kB, восстанавливает тиоредоксин или восстанавливает подобный тиоредоксину белок. Тиоредоксин представляет собой важный белок оксидоредуктазу с антиокислительными функциями. Обнаружено, что тиоредоксин позитивно регулирует связывание ДНК активированного NF-kB и, таким образом, повышает экспрессию гена (Schenk et al., Proc. Natl. Acad. Sci. USA 91: 1672-1676, 1994). Тиоредоксин участвовал в восстановлении активированного цитозольного NF-kB (особенно, восстановлении cys-62), который может, таким образом, способствовать его ядерной транслокации и связыванию ДНК (Hayashi et al., J. Biol. Chem. 268:11380-11388, 1993).
Обнаружено также, что активность Fos и Jun в связывании ДНК в комплекс АР-1 регулируется редокс-системой (Abate et al., Science 249:1157-1162, 1990). Каждый белок содержит один консервативный цистеин (фланкированный лизином и аргинином) в его ДНК-связывающем домене. Оказывается, что этот тиол не является частью дисульфидной связи и не может существовать в виде сульфеновой или сульфиновой кислоты в окисленной форме. Ref-1, бифункциональный ядерный белок, также обладающий эндонуклеазной активностью в репарации ДНК, стимулирует связывание ДНК с белком АР-1 посредством восстановления этого регуляторного цистеина. Fos-мутант, у которого критический цистеин заменен серином, вызывал трехкратное повышение в активности связывания ДНК с АР-1 и не был больше объектом редокс-контроля (Okuno et al., Oncogene 8:695-701, 1993). Следовательно, поскольку все из, по меньшей мере, четырех членов семейства fos, 3 членов семейства Jun и, по меньшей мере, 4 членов семейства ATF/CREB факторов транскрипции содержат этот консервативный цистеин, редокс-регулятор факторов транскрипции, по-видимому, широко распространен.
Как указано выше, регуляция факторов транскрипции, таких как NF-kB и АР-1, имеет важное терапевтическое значение. Например, АР-1 является важным медиатором образования опухоли (Yoshioka et al., Proc. Natl. Acad. Sci. USA 92:4972-4976, 1995). Таким образом, соединения, которые подавляют транскрипционную активность АР-1, имеют применение при лечении рака. Кроме того, вследствие его непосредственной роли в регуляции реакций на воспалительные цитокины и эндотоксины, активация NF-kB играет важную роль в развитии хронических заболеваний, таких как ревматоидный артрит, и острых патологических состояний, таких как септический шок. Считается также, что аутоиммунные заболевания, такие как системная красная волчанка (SLE) и болезнь Альцгеймера, заключаются в активации NF-kB. Аналогичного этому, NK-kB играет важную роль в активации экспрессии гена ВИЧ. Считается, что другие состояния, которые связаны с NK-kB, включают в себя грипп, атеросклероз, онкогенез и атаксию-телеангиэктазию (АТ).
Ингибирование оксидоредуктазы
Что касается регуляции факторов транскрипции, соединения по данному изобретению регулируют факторы транскрипции, способность которых связываться с ДНК регулируется восстановлением остатка цистеина клеточной оксидоредуктазой. В одном варианте осуществления фактором транскрипции является NF-kB. В данном варианте осуществления соединения по настоящему изобретению обладают активностью в качестве медиаторов иммунных и/или воспалительных реакций или служат для регуляции роста клеток. В другом варианте осуществления фактором транскрипции является АР-1 и клеточной оксидоредуктазой является Ref-1. В этом варианте осуществления соединения по настоящему изобретению обладают активностью в качестве противовоспалительных и/или противораковых агентов. В следующих вариантах осуществления фактор транскрипции выбран из Myb и глюкокортикоидного рецептора. Другие факторы транскрипции, которые могут быть регулированы в контексте по данному изобретению, включают в себя также: факторы транскрипции семейства NK-kB, такие как Rel-A, c-Rel, Rel-B, p50 и р52; факторы транскрипции семейства АР-1, такие как Fos, FosB, Fra-1, Fra-2, Jun, JunB и JunD; ATF; CREB; STAT-1, -2, -3, -4, -5 и -6; NFAT-1, -2 и -4; MAF; тироидный фактор; IRF; Oct-1 и -2; NF-Y; Erg-1 и USF-43.
В соответствии с этим, в одном аспекте настоящее изобретение предусматривает способ ингибирования оксидоредуктазы у теплокровного животного, содержащий введение животному количества соединения по настоящему изобретению, которое является эффективным для ингибирования оксидоредуктазы. Ингибирование активности оксидоредуктазы можно применять в качестве средства для регуляции транскрипции.
Ингибирование СААХ
В другом аспекте настоящее изобретение предусматривает способ ингибирования СААХ у теплокровного животного. Способ содержит введение животному количества соединения по настоящему изобретению, как здесь описано. Количество является эффективным для обеспечения ингибирования СААХ у животного.
Ras, белковый продукт онкогена ras, является связанным с мембраной белком, принимающим участие в трансдукции сигнала, регулирующей деление и рост клетки. Мутации в гене ras существуют среди наиболее обычных генетических нарушений, связанных с раковыми заболеваниями человека (Barbacid, M. Annu Rev Biochem 56: 779-827, 1987). Эти мутации приводят к сигналу роста, который всегда «включен», что приводит к образованию раковой клетки. Для локализации в клеточной мембране Ras требуется пренилирование цистеина в его С-концевой последовательности СААХ фарнезилтрансферазой (FТаза), где в последовательности СААХ «А» указывается в качестве аминокислоты с гидрофобной боковой цепью и «Х» представляет собой другую аминокислоту. Эта посттрансляционная модификация является критической для ее активности. Обнаружено, что пептидиловые ингибиторы FТазы с последовательностью СААХ блокируют или замедляют рост опухолей в культуре клеток и во всем организме животных (Kohl et al., Science 260: 1934-1937, 1993; Buss and Marsters, Chemistry and Biology 2:787-791, 1995).
Способы для скрининга соединения на его активность ингибирования активности СААХ являются известными в данной области. См., например, патент США № 6391574, в котором описан способ идентификации соединения, которое ингибирует протеолитическое удаление трипептида ААХ у белка СААХ в клетке. См. также патент США № 5990277, в котором описано несколько подходящих анализов, и ссылки Gibbs et al., Cell 77:175, 1994; Gibbs, Cell 65:1, 1991; Maltese, FASEB J. 4:3319, 1990; Moores et al., J. Biol. Chem. 266:14603, 1991; Goldstein et al., J. Biol. Chem. 266:15575, 1991; European Patent 0461869 A2; Casey, J. Lipid Res. 33:1731-1740, 1992; Cox et al., Curr. Opin. Cell Biol. 4:1008-1016. 1992; Garcia et al., J. Biol. Chem. 268:18415-18418, 1993; Vogt et al., J. Biol. Chem. 270:660-664, 1995; Kohl et al., Science, 260:1934-1937, 1993; Garcia et al., J. Biol. Chem., 268:18415-18418, 1993; and Vogt et al., J. Biol. Chem., 270:660-664, 1995).
Молекулы МНС
В другом аспекте настоящее изобретение предусматривает способ ингибирования связывания антигенного пептида с молекулой МНС либо класса один, либо класса два. Способ содержит контактирование соединения по настоящему изобретению с композицией, содержащей антигенный пептид и молекулу МНС либо класса один, либо класса два. Соединение контактирует с антигеном/молекулой в количестве, эффективном для снижения аффинности связывания между двумя видами.
Важным аспектом иммунной системы является ответная реакция Т-клеток. Эта реакция требует, чтобы Т-клетки узнавали и взаимодействовали с комплексами молекул клеточной поверхности, называемыми лейкоцитарными антигенами человека ("HLA") или главными комплексами гистосовместимости человека ("МНС"), и пептидами (см., например, Male et al., Advanced Immunology (J.P. Lipincott Company, 1987). Антигены мобилизуют иммунную реакцию, по меньшей мере, частично, посредством «захватывания» антиген-презентирующей клетки (АРС), которая содержит на своей поверхности гликопротеин класса II, кодируемый геном в основном комплексе гистосовместимости (МНС). Антиген затем презентируется специфической Т-клетке-хелперу в контексте связанного с поверхностью гликопротеина МНС и взаимодействием антиген-специфичного рецептора для Т-клеток с комплексом антиген-МНС, Т-клетка-хелпер стимулируется для опосредования антиген-специфичной иммунной реакции, в том числе индукции функции цитотоксических Т-клеток, индукции функции В-клеток и секреции ряда факторов, помогающих данной реакции и поддерживающих эту реакцию. В одном аспекте изобретения молекула МНС представляет собой HLA-A2.1, HLA-A1 или HLA-A3.1 или любой другой аллель HLA, который присутствует в меланоме пациентов.
Способность соединения по настоящему изобретению связываться с молекулами МНС I может быть демонстрирована, по существу, как описано Elliot et al., Nature 351:402-406, 1991. Аналогично этому, способность соединения по настоящему изобретению связываться с молекулами МНС II может быть демонстрирована процедурой Kwok et al., J. Immunol. 155:2468-2476, 1995.
Белок с доменом 14-3-3
В другом аспекте настоящее изобретение предусматривает способ ингибирования связывания первого пептида со вторым пептидом, который содержит домен 14-3-3, где первый пептид имеет аффинность связывания с доменом 14-3-3 второго домена. Способ содержит контактирование соединения по настоящему изобретению с композицией, содержащей (первый) пептид, который имеет аффинность связывания с доменом 14-3-3 второго белка.
Белки, имеющие домен 14-3-3, и его партнеры связывания описаны в литературе. Эти пептиды можно применять в способе по настоящему изобретению. См., например, Dai and Murakami, J. Neurochem 2003 Jan. 84(1):23-34; Lim et al., J. Biol. Chem. 2002, Oct. 25, 277(43):40997-1008; Parvaresch et al., FEBS Lett 2002 Dec. 18, 532(3):357-62; Eilers et al., Mol Cell Biol 2002 Dec.; 22(24):8514-26; Liu et al., Cancer Res 2002 Nov. 15, 62(22):6475-80; Truong et al., Proteins 2002 Nov. 15, 49(3):321-5; Birkenfeld et al., Biochem J 2003 Jan. 1, 369(Pt 1):45-54; Espejo et al., Biochem J 2002 Nov. 1, 367(Pt 3):697-702; and Benzing et al., J Biol Chem 2002 Sep 6, 277(36):32954-62.
На практике осуществления способов по данному изобретению терапевтически эффективное количество соединения по данному изобретению вводят нуждающемуся в этом теплокровному животному. Например, соединения по данному изобретению могут быть введены теплокровному животному, у которого диагностировано или имеется риск развития состояния, выбранного из любого одного или нескольких следующих заболеваний: болезни Крона, астмы, ревматоидного артрита, ишемии, повреждения при реперфузии, гомологичной болезни (GVHD), бокового амиотрофического склероза (ALS), болезни Альцгеймера, отторжения трансплантата и Т-клеточного лейкоза взрослых пациентов.
Комплекс туберозного склероза
У пациентов, имеющих комплекс туберозного склероза (TSC), обычно развиваются множественные фокальные повреждения в головном мозге, сердце, почках и других тканях (см. например, Gomez, M.R. Brain Dev. 17 (suppl):55-57, 1995). Исследования на клетках млекопитающих обнаружили, что сверхэкспрессия TSC1 (который экспрессирует гамартин) и TSC2 (который экспрессирует туберин) отрицательно регулирует пролиферацию клеток и индуцирует задержку G1/S (см. например, Miloloza, A. et al, Hum. Mol. Genet. 9:1721-1727, 2000). Другие исследования обнаружили, что гамартин и туберин функционируют на уровне комплекса деградации β-катенина и, более конкретно, что указанные белки негативно регулируют стабильность и активность бета-катенина участием в комплексе деградации бета-катенина (см., например, Mak, В.C., et al. J. Biol. Chem. 278(8):5947-5951, 2003). Бета-катенин является белком 95 кДа, который принимает участие в адгезии клеток посредством ассоциации его с членами связанного с мембранами семейства кадгерина и в пролиферации и дифференциации клеток в качестве ключевого компонента пути Wnt/Wingless (см., например, Daniels, D.L., et al., Trends Biochem. Sci. 26:672-678, 2001). Обнаружено, что разрыв этого пути является онкогенным для людей и грызунов. Настоящее изобретение предусматривакт соединения, которые модулируют активность β-катенина, и особенно его взаимодействие с другими белками, и в соответствии с этим их можно применять при лечении TSC.
Для целей иллюстрации, но без ограничения по данному изобретению, предложены следующие примеры.
ПРИМЕРЫ
В препаративных примерах и примерах применяют следующие аббревиатуры:
BMS: Бордиметилсульфид
CbzOSu: Бензилоксикарбонил-N-гидроксисукцинимид
DIC: 1,3-Диизопропилкарбодиимид
DIEA: N,N-Диизопропилэтиламин
DIPEA: N,N-Диизопропилэтиламин
DMAP: N,N-Диметиламинопиридин
ДМФА: Диметилформамид
ДМСО: Диметилсульфоксид
EA: Этилацетат
EDC: Гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида
EDCI: Гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида
FmocOsu: 9-Флуоренилоксикарбонил-N-гидроксисукцинимид
HATU: [Гексафторфосфат 2-(1Н-9-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония]
Hex.: Гексан
НОВТ: N-Гидроксибензотриазол
МС: Метиленхлорид
МеОН: Метанол
-Obn: -О-бензил
PPTS: п-Толуолсульфонат пиридиния
PyBOP: Гексафторфосфат бензотриазол-1-илокситриспирролидинофосфония
p-TsOH: п-Толуолсульфоновая кислота
ТГФ: Тетрагидрофуран
ТСХ: Тонкослойная хроматография
Препаративный пример 1
(1) Получение амида нафталин-2-карбоновой кислоты
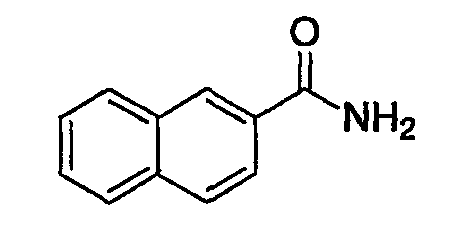
К раствору 2-нафтойной кислоты (25 г, 0,145 моль) в МС (200 мл) добавляют оксалилхлорид (38 мл, 0,4356 моль) и каталитическое количество ДМФА и смесь перемешивают при комнатной температуре в течение 2 час. После выпаривания растворителя сырой ацилхлорид разбавляют МС (200 мл), к которому по каплям при температуре ледяной бани добавляют гидроксид аммония в воде (160 мл). После перемешивания в течение 1 час осажденный продукт собирают фильтрованием с отсасыванием, растирают с гексаном и сушат, получая при этом указанное в заголовке соединение, которое применяют в следующей стадии без дополнительной очистки.
(2) Получение нафталин-2-илметиламина
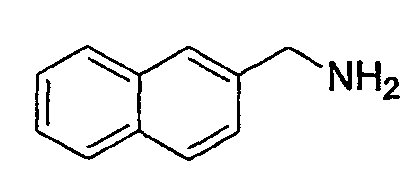
К раствору сырого амида, полученного в указанной выше стадии (1), в ТГФ (100 мл) медленно добавляют BMS (27,5 мл, 0,2904 моль) при 0°С. Образовавшуюся реакционную смесь нагревают до 60°С в течение 3 час, гасят 5% HCl при 0°С, экстрагируют ЕА и промывают 5% HCl. Водные слои объединяют и подщелачивают 1 н. NaOH и снова экстрагируют ЕА. Органические слои объединяют и концентрируют, получая при этом указанное в заголовке соединение (13 г) в виде белого твердого вещества.
Система 1 для ТСХ : МС/МеОН = 90:10 об./об., Rf=0,23.
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 4,07 (с, 2Н), 7,48 (м, 3Н), 7,79 (м, 4Н).
Препаративный пример 2
(1) Получение амида 1Н-индол-2-карбоновой кислоты
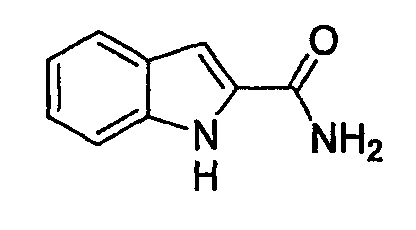
К раствору индол-2-карбоновой кислоты (1 г, 6,21 ммоль) в МС (30 мл) добавляют оксалилхлорид (1,64 мл, 0,1862 ммоль) и каталитического количества ДМФА и смесь перемешивают при комнатной температуре в течение 2 час. После выпаривания растворителя сырой ацилхлорид разбавляют МС (20 мл), к которому по каплям при охлаждении ледяной баней добавляют гидроксид аммония в воде (7 мл). После перемешивания в течение 1 час осажденный продукт собирают фильтрованием с отсасыванием, растирают с гексаном и сушат, получая при этом указанное в заголовке соединение, которое применяют в следующей стадии без дополнительной очистки.
(2) Получение (1Н-индол-2-ил)метиламина
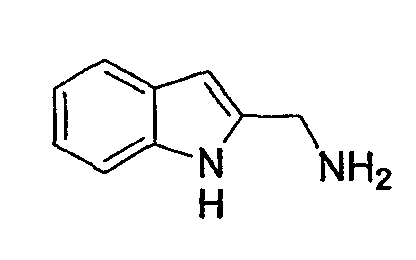
К раствору сырого амида, полученного в указанной выше стадии (1), в ТГФ (30 мл) медленно при 0°С добавляют BMS (1,18 мл, 12,42 моль). Образовавшуюся реакционную смесь нагревают до 60°С в течение 3 час, гасят 5% HCl при 0°С, экстрагируют ЕА и промывают 5% HCl. Водные слои объединяют и подщелачивают 1 н. NaOH и снова экстрагируют ЕА. Органические слои объединяют и концентрируют, получая при этом указанное в заголовке соединение (0,28 г) в виде желтого масла.
Система 1 для ТСХ: МС/МеОН=90:10 об./об., Rf=0,15.
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 3,98 (с, 2Н), 7,08 (м, 3Н), 7,26 (м, 1Н), 7,58 (д, 1Н), 9,10 (ушир.с, 1Н).
Препаративный пример 3
(1) Получение бензилового эфира 2-бензилоксикарбониламино-4-оксомасляной кислоты
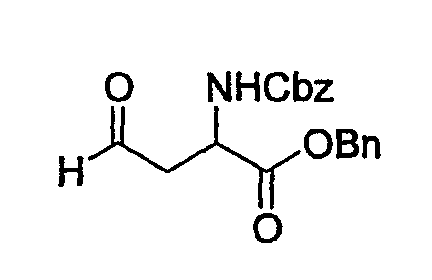
К раствору Z-Asp-OBn (10 г, 0,028 моль) в МС (200 мл) при 0°С добавляют оксалилхлорид (2,93 мл, 0,336 моль) и каталитическое количество ДМФА и смесь перемешивают при комнатной температуре в течение 2 часов. После выпаривания растворителя сырой ацилхлорид растворяют в бензоле (400 мл) и медленно при 0°С добавляют трибутилоловогидрид (15,1 мл, 0,056 моль) и каталитическое количество Pd(0) и смесь перемешивают при комнатной температуре на протяжении ночи. После выпаривания растворителя добавляют смесь эфир (100 мл)/10% KF в воде (100 мл) и смесь перемешивают при комнатной температуре в течение 2 час с последующим фильтрованием, получая при этом двухфазный раствор. Органический слой отделяют и концентрируют с образованием сырого продукта, который очищают колоночной хроматографией, получая при этом указанное в заголовке соединение, Z-Asp-OBn-альдегид (6 г) в виде светло-желтого масла.
Rf: 0,29 в смеси гексан/ЕА (2/1).
(2) Получение бензилового эфира 2-бензилоксикарбониламино-4,4-диметоксимасляной кислоты
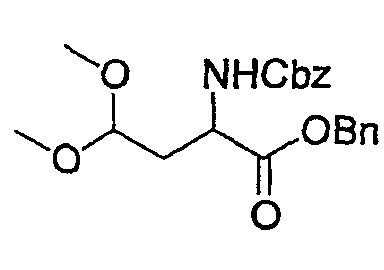
К раствору Z-Asp-OBn-альдегида (6 г, 17,58 ммоль), полученного в указанной выше стадии (1), в МеОН (100 мл) добавляют каталитическое количество p-TsOH и смесь перемешивают при комнатной температуре в течение 5 час. После завершения реакции растворитель выпаривают с получением сырого продукта, который очищают колоночной хроматографией, получая при этом указанное в заголовке соединение, Z-Asp-OBn-ацеталь (5 г) в виде светло-желтого масла.
Rf: 0,32 в смеси Hex./EA (2/1).
(3) Получение 2-бензилоксикарбониламино-4,4-диметоксимасляной кислоты
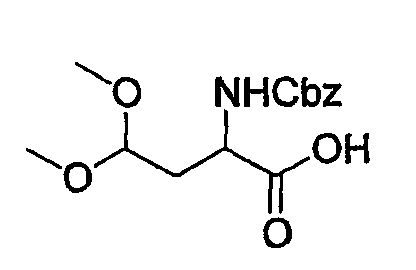
Z-Asp-OBn-ацеталь (0,5 г, 1,29 ммоль), полученный в указанной выше стадии (2), растворяют в смеси ТГФ (20 мл/NaOH (0,11 г, 2,1 ммоль) в воде (20 мл) и перемешивают при комнатной температуре в течение 30 мин. После полного исчезновения исходного материала реакционную смесь концентрируют выпариванием и затем разбавляют смесью вода/ЕА. Водный слой отделяют, подкисляют очень осторожно до рН 4-5 1 н. HCl при 0°С и снова экстрагируют ЕА. Органические слои объединяют и концентрируют, получая при этом указанное в заголовке соединение, Z-Asp-OBn-ацеталь (0,27 г) в виде светло-желтого масла.
Система 1 для ТСХ: гексан/ЕА = 20:10 об./об., Rf=0,10.
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 2,20 (с, 2Н), 3,35 (д, 6Н), 4,52 (м, 2Н), 5,19 (т, 2Н), 5,80 (д, 1Н), 7,37 (ушир.с, 5Н).
(4) Получение 2-амино-4,4-диметоксимасляной кислоты
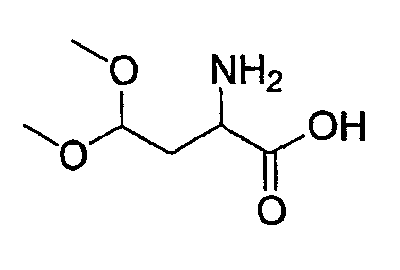
В реакционный сосуд, оснащенный баллоном с газообразным водородом, добавляют раствор Z-Asp-OBn-ацеталя (2,22 г, 5,73 ммоль), полученного в указанной выше стадии (3), в уксусной кислоте (20 мл) и катализатор Pearlman и смесь перемешивают при комнатной температуре на протяжении ночи. Образовавшуюся реакционную смесь фильтруют, концентрируют и лиофилизуют, получая при этом сырой продукт, который применяют в следующей стадии без дополнительной очистки.
(5) Получение 2-(9Н-флуорен-9-илметоксикарбониламино)-4,4-диметоксимасляной кислоты
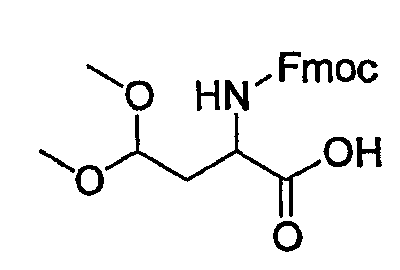
К раствору сырого Asp-OH-ацеталя, полученного в указанной выше стадии (4), в смеси ТГФ (100 мл)/вода (100 мл) добавляют FmocOsu (2,13 г, 6,3 ммоль)/бикарбонат натрия (1,93 г, 22,92 ммоль) и смесь перемешивают при комнатной температуре на протяжении ночи. Образовавшуюся реакционную смесь концентрируют и разбавляют смесью вода/ЕА. Водный слой отделяют, подкисляют очень осторожно до рН 4-5 1 н. HCl при 0°С и снова экстрагируют ЕА. Органические слои объединяют и концентрируют с получением сырого продукта, который очищают колоночной хроматографией, получая при этом указанное в заголовке соединение (1,5 г) в виде пенообразного твердого вещества.
Rf: 0,15 в Hex./ЕА (2/1).
Препаративный пример 4
(1) Получение 2-бензилоксикарбониламинопентадиовой кислоты
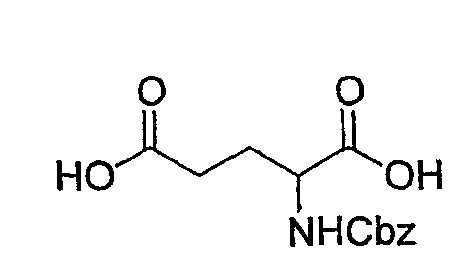
К раствору L-глутаминовой кислоты (20 г, 136 ммоль) в смеси вода/ТГФ (1/1, 400 мл) добавляют бикарбонат натрия (45,7 г, 544 ммоль) и смесь охлаждают до 0°С на ледяной бане. К реакционной смеси добавляют CbzOSu (37,3 г, 150 ммоль) и смесь перемешивают на протяжении ночи при комнатной температуре. После завершения реакции образовавшуюся реакционную смесь экстрагируют ЕА. Водный слой отделяют, подкисляют до рН 2 концентрированной HCl при 0°С и снова экстрагируют ЕА (4 раза). Органические слои концентрируют с получением сырого продукта, который очищают колоночной хроматографией, получая при этом указанное в заголовке соединение (16 г) в виде бесцветного масла.
Rf: 0,2 в МС/МеОН (9/1).
(2) Получение бензилового эфира 4-(2-карбоксиэтил)-5-оксооксазолидин-3-карбоновой кислоты
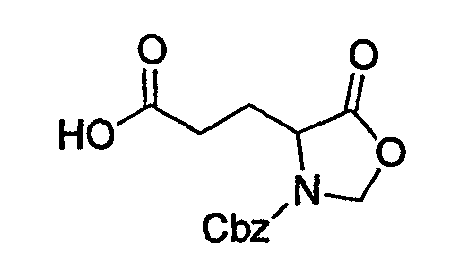
В аппаратуру Дина-Старка помещают N-Cbz-L-глутаминовую кислоту (4 г, 14,22 ммоль), полученную в указанной выше стадии (1), параформальдегид (5 г), каталитическое количество pTsOH, молекулярные сита (5 г) и толуол (100 мл) и смесь кипятят с обратным холодильником до исчезновения исходного материала. Образовавшуюся реакционную смесь охлаждают до комнатной температуры, фильтруют и концентрируют с получением сырого продукта, который очищают колоночной хроматографией, получая при этом указанное в заголовке соединение (2 г) в виде бесцветного масла.
Rf: 0,45 только в ЕА.
(3) Получение бензилового эфира 5-оксо-5-(3-оксопропил)оксазолидин-3-карбоновой кислоты
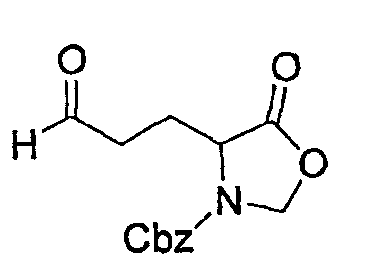
К раствору дизащищенной глутаминовой кислоты (2 г, 6,82 ммоль), полученной в указанной выше стадии (2), в МС (200 мл) при 0°С добавляют оксалилхлорид (0,7 мл, 7,5 ммоль) и каталитическое количество ДМФА и смесь перемешивают при комнатной температуре в течение 2 часов. После выпаривания растворителя образовавшийся сырой ацилхлорид растворяют в ТГФ (400 мл), к которому при 0°С медленно добавляют трибутилоловогидрид (3,86 мл, 14,34 ммоль) и каталитическое количество Pd(0) и смесь перемешивают при комнатной температуре на протяжении ночи. После выпаривания растворителя добавляют смесь эфир (100 мл)/10% KF в воде (100 мл) и смесь перемешивают при комнатной температуре в течение 2 час с последующим фильтрованием с получением двухфазного раствора. Органический слой отделяют и концентрируют, получая при этом сырой продукт, который очищают колоночной хроматографией, получая при этом указанное в заголовке соединение (0,7 г) в виде бесцветного масла.
Rf: 0,23 в смеси гексан/ЕА (4/1)
(4) Получение бензилового эфира 4-(3,3-диметоксипропил)-5-оксооксазолидин-3-карбоновой кислоты
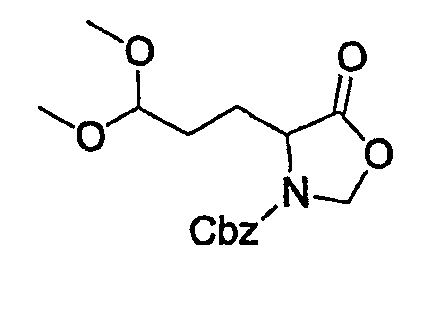
К раствору дизащищенного альдегида (0,7 г, 2,53 ммоль), полученного в указанной выше стадии (3), в МеОН (30 мл) добавляют каталитическое количество pTsOH и смесь перемешивают при комнатной температуре в течение 7 час. После завершения реакции реакционную смесь концентрируют выпариванием растворителя с получением сырого продукта, который очищают колоночной хроматографией, получая при этом указанное в заголовке соединение (0,5 г) в виде бесцветного масла.
Rf: 0,33 в смеси гексан/ЕА (4/1).
(5) Получение 2-бензилоксикарбониламино-5,5-диметоксипентановой кислоты
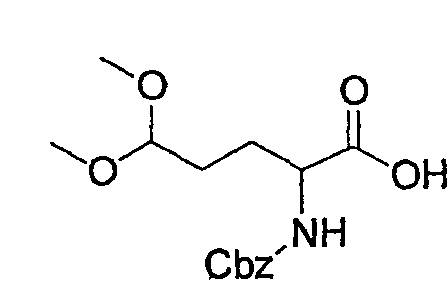
Дизащищенный ацеталь (0,456 г, 1,411 ммоль), полученный в указанной выше стадии (4), растворяют в смеси МеОН (20 мл)/1 н. NaOH (10 мл) и смесь перемешивают при комнатной температуре в течение ночи. После полного исчезновения исходного материала реакционную смесь концентрируют выпариванием растворителя и разбавляют смесью вода/ЕА. Водный слой отделяют, подкисляют очень осторожно до рН 4-5 1 н. HCl при 0°С и снова экстрагируют ЕА. Органические слои объединяют и концентрируют, получая при этом указанное в заголовке соединение (0,35 г) в виде бесцветного масла.
Rf: 0,1 в смеси Hex./ЕА (1/1).
(6) Получение 2-амино-5,5-диметоксипентановой кислоты
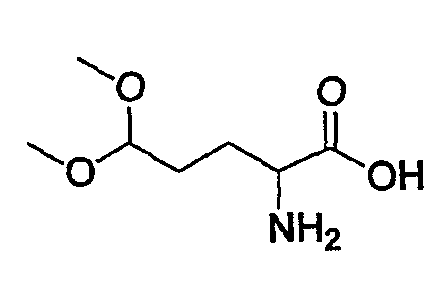
В реакционный сосуд, снабженный баллоном газообразного водорода, добавляют раствор Cbz-ацеталя (0,35 г, 1,13 ммоль), полученного в указанной выше стадии (5), в МеОН (10 мл) и каталитическое количество 10% Pd/C и смесь перемешивают при комнатной температуре на протяжении ночи. Образовавшуюся реакционную смесь фильтруют и концентрируют с получением сырого продукта (0,2 г) в виде бесцветного масла, которое применяют в следующей стадии без дополнительной очистки.
Rf: 0,01 в смеси Hex./ЕА (1/1).
(7) Получение 2-(9Н-флуорен-9-илметоксикарбониламино)-5,5-диметоксипентановой кислоты
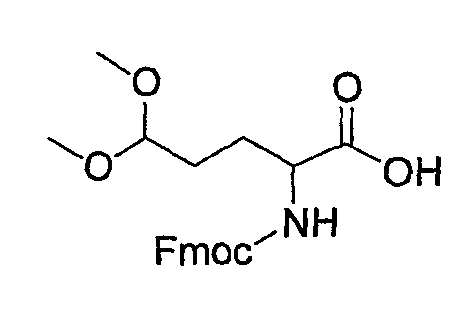
К раствору сырого Glu-OH-ацеталя, полученного в указанной выше стадии (6), в смеси ТГФ (10 мл)/вода (10 мл) добавляют FmocOsu (0,42 г, 1,24 ммоль)/бикарбонат натрия (0,5 г, 5,9 ммоль) и смесь перемешивают при комнатной температуре на протяжении ночи. После выпаривания растворителя образовавшуюся реакционную смесь разбавляют смесью вода/ЕА. Водный слой отделяют и подкисляют очень осторожно до рН 4-5 1 н. HCl при 0°С и снова экстрагируют ЕА. Органические слои объединяют и концентрируют, получая при этом указанное в заголовке соединение (0,19 г) в виде бесцветного масла.
Система 1 для ТСХ: только ЕА, Rf=0,25.
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 1,75 (ушир.м, 4Н), 3,28 (д, 6Н), 3,43 (кв, 1Н), 4,20 (т, 1Н), 4,38 (м, 3Н), 5,62 (д, 1Н), 7,31 (м, 4Н), 7,65 (д, 2Н), 7,75 (д, 2Н).
Препаративный пример 5
(1) Получение 2-трет-бутоксикарбониламино-4-метоксикарбониламиномасляной кислоты
К раствору Вос-Dab-OH (3 г, 13,75 ммоль) в Н2О (50 мл) медленно добавляют NaOH (2,75 г, 68,75 ммоль, 5 экв.) до рН>11, затем добавляют метилхлорформиат (2,6 г, 27,5 ммоль, 2 экв.) в толуоле (50 мл). Образовавшуюся реакционную смесь перемешивают в течение 2 час. Для проведения анализа ТСХ отбирают небольшое количество водной фазы и подкисляют 1 н. HCl. После подтверждения завершения реакции посредством ТСХ органическую фазу отделяют и водную фазу подкисляют 10% раствором HCl и экстрагируют ЕА (5 мл × 2). Органические фазы объединяют, сушат над безводным Na2SO4 и концентрируют в вакууме, получая при этом сырой продукт (3,277 г, 11,86 ммоль, 86%) в виде бесцветного масла.
Система для ТСХ: ЕА, Rf=0,2.
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 1,30˜1,50 (ушир.с, 9Н), 2,00˜2,30 (м, 2Н), 3,10˜3,30 (м, 2Н), 3,70 (ушир.с, 3Н), 4,35 (м, 1Н), 5,40 (м, 1Н), 5,65 (ушир.с, 1Н).
(2) Получение трет-бутилового эфира (1-бензилкарбамоил-3-метоксикарбониламинопропил)карбаминовой кислоты
К раствору 2-трет-бутоксикарбониламино-4-метоксикарбониламиномасляной кислоты (1,1 г, 3,98 ммоль), полученной в указанной выше стадии (1), в ДМФА (20 мл) при 5°С добавляют EDCI (763 мг, 3,98 ммоль, 1 экв.), НОВТ (538 мг, 3,98 ммоль, 1 экв.) и DIEA ((1,4 мл, 7,96 ммоль, 2 экв.) и смесь перемешивают в течение 1 часа. После подтверждения завершения реакции ТСХ реакционный раствор подкисляют 10% HCl при 5°С (до рН˜4) и экстрагируют ЕА (20 мл). Органические фазы объединяют и промывают насыщенным NaHCO3 и насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют в вакууме, получая при этом остаток, который отверждают добавлением ЕА и н-гексана и очищают колоночной хроматографией, получая при этом указанное в заголовке соединение (620 мг, 1,7 ммоль, 43%) в виде белого твердого вещества.
Rf=0,7 (ЕА).
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 1,45 (ушир.с, 9Н), 1,75˜2,10 (м, 2Н), 3,05 (м, 1Н), 3,45 (м, 1Н), 3,65 (с, 3Н), 4,25 (м, 1Н), 4,45 (д, 2Н, J=5,7 Гц), 5,45 (м, 1Н), 7,05 (м, 1Н), 7,20˜7,45 (м, 5Н).
(3) Получение гидрохлорида трет-бутилового эфира (3-амино-3-бензилкарбонилпропил)карбаминовой кислоты
К раствору трет-бутилового эфира (1-бензилкарбамоил-3-метоксикарбониламинопропил)карбаминовой кислоты (1 г, 2,7 ммоль), полученного в указанной выше стадии (2), в 1,4-диоксане (10 мл) добавляют 4 н. HCl в 1,4-диоксане (6,8 мл, 27 ммоль) и смесь перемешивают в течение 2 час. После подтверждения завершения реакции ТСХ реакционный раствор концентрируют и сушат в вакууме, получая при этом указанное в заголовке соединение в виде белого твердого вещества.
Пример 1
1-Бензил-7-метил-6-тиоксогексагидропиримидо[1,6-а]пиримидин-2-он
(А) Получение N-бензил-3-[3-(2-[1,3]диоксолан-2-илэтил)-3-метилтиоуреидо]пропионамида
Суспензию гидрохлорида бензиламидо-β-аланина (1,0 экв.) и N-метилморфолина (2,2 экв.) в дихлорметане обрабатывают тиофосгеном (1,2 экв.) при 0°С в течение 10 мин. Реакционной смеси дают возможность нагреться до комнатной температуры и ее перемешивают в течение дополнительных 2 час. Прозрачный раствор разбавляют этилацетатом и промывают 10% раствором KHSO4, дистиллированной водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток.
Данный продукт растворяют в дихлорметане и обрабатывают 2-(N-метил-2-аминоэтил)-1,3-диоксоланом (0,9 экв.) при 0°С в течение 10 мин. Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение дополнительных 4 час. Реакционную смесь разбавляют этилацетатом и промывают 10% раствором KHSO4, дистиллированной водой, насыщенным раствором NaHCO3, дистиллированной водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют с получением маслянистого остатка. Сырой продукт очищают колоночной хроматографией (силикагель, этилацетат/гексан = 5/2), получая при этом чистый продукт.
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 2,02 (м, 2Н), 2,60 (м, 2Н), 3,18 (с, 3Н), 3,82 (м, 2Н), 3,88 (м, 2Н), 4,03 (м, 4Н), 4,44 (м, 2Н), 4,91 (м, 1Н), 6,84 (ушир.с, 1Н), 7,25-7,38 (м, 5Н);
МС (m/z, ESI), 352 (МН+).
(В) Получение 1-бензил-7-метил-6-тиоксогексагидропиримидо[1,6-a]пиримидин-2-она
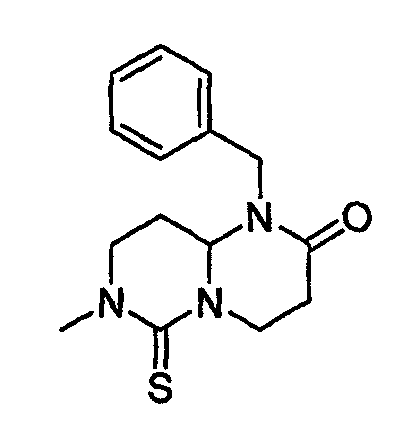
Амид, полученный в указанной выше стадии (А), обрабатывают муравьиной кислотой при 60°С в течение 4 дней. После выпаривания муравьиной кислоты при пониженном давлении остаток очищают препаративной ТСХ (силикагель, этилацетат/метанол = 5/1), получая при этом чистый, указанный в заголовке продукт.
1Н-ЯМР (500 МГц, CDCl3) δ м.д.: 2,05 (м, 1Н), 2,36 (м, 1Н), 2,64 (д, 1Н), 2,96 (м, 1Н), 3,30 (м, 3Н), 3,44 (с, 3Н), 4,42 (д, 1Н), 4,86 (ушир.с, 1Н), 5,08 (д, 1Н), 5,39 (м, 1Н), 7,25-7,38 (м, 5Н);
МС (m/z, ESI), 290 (MH+), 311 (M+Na).
Пример 2
1,7-Дибензил-6-тиоксогексагидропиримидо[1,6-а]пиримидин-2-он
(А) Получение N-бензил-3-[3-бензил-(3,3-диэтоксипропил)тиоуреидо]пропионамида
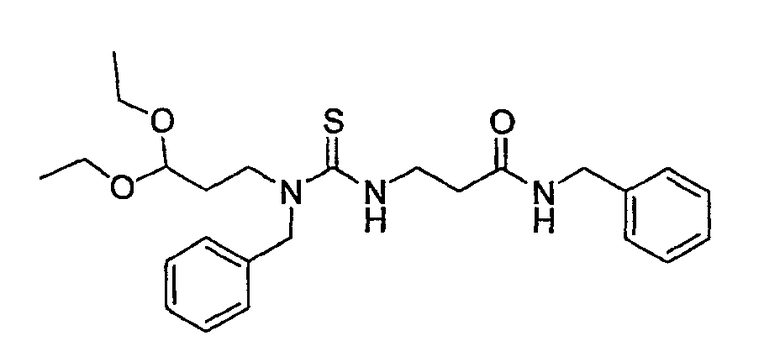
Суспензию гидрохлорида бензиламидо-β-аланина (1,0 экв.) и N-метилморфолина (2,2 экв.) в дихлорметане обрабатывают тиофосгеном (1,2 экв.) при 0°С в течение 10 мин. Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение дополнительных 2 час. Прозрачный раствор разбавляют этилацетатом и промывают 10% раствором KHSO4, дистиллированной водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток. Продукт растворяют в дихлорметане и обрабатывают 2-(N-бензил-1-амино-3,3-диэтоксипропаном (0,9 экв.) при 0°С в течение 10 мин. Реакционной смеси дают возможность нагреться до комнатной температуры и ее перемешивают в течение дополнительных 6 час. Образовавшуюся реакционную смесь разбавляют этилацетатом и промывают 10% раствором KHSO4, дистиллированной водой, насыщенным раствором NaHCO3, дистиллированной водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют с получением маслянистого остатка. Данный сырой продукт очищают колоночной хроматографией (силикагель, этилацетат/гексан = 2/1), получая при этом чистый, указанный в заголовке продукт.
1Н-ЯМР (500 МГц, CDCl3) δ м.д.: 1,22 (т, 6H), 1,95 (м, 2H), 2,60 (м, 2H), 3,46 (м, 2H), 3,60 (ушир.т, 2H), 3,63 (м, 2H), 3,97 (м, 2H), 4,38 (м, 2H), 4,52 (м, 1H), 5,07 (ушир.с, 2H), 6,16 (ушир.с, 1H), 6,98 (ушир.с, 1H), 7,25-7,38 (м, 10H);
MC (m/z, ESI), 458 (MH+).
(В) Получение 1,7-дибензил-6-тиоксогексагидропиримидо[1,6-a]пиримидин-2-она
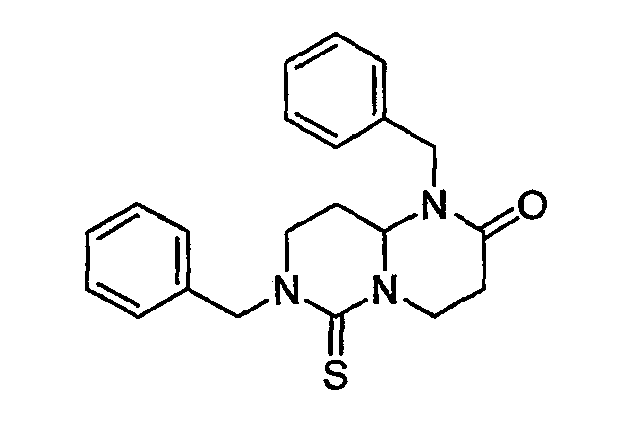
Амид, полученный в указанной выше стадии (А), обрабатывают муравьиной кислотой при 60°С в течение 4 дней. После выпаривания муравьиной кислоты при пониженном давлении остаток очищают препаративной ТСХ (силикагель, этилацетат/метанол = 5/1), получая при этом чистый продукт.
1Н-ЯМР (500 МГц, CDCl3) δ м.д.: 1,94 (м, 1H), 2,24 (м, 1H), 2,62 (м, 1H), 3,01 (м, 1H), 3,18 (м, 1H), 3,43 (м, 1H), 3,62 (м, 1H), 4,39 (д, 1H), 4,51 (м, 1H), 4,91 (м, 1H), 5,02 (д, 1H), 5,26 (д, 1H), 5,53 (м, 1H), 7,25-7,40 (м, 10H);
MC (m/z, APCI), 366 (МН+).
Пример 3
1,7-Дибензилгексагидропиримидо[1,6-а]пиримидин-2,6-дион
(А) Получение N-бензил-3-[3-бензил-(3,3-диэтоксипропил)тиоуреидо]пропионамида
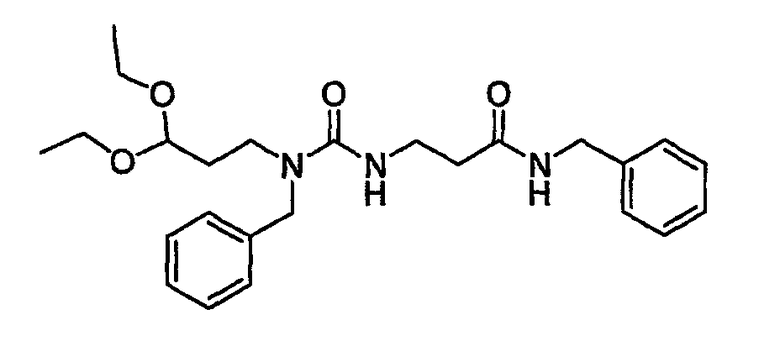
Суспензию гидрохлорида бензиламидо-β-аланина (1,0 экв.) и N-метилморфолина (3,2 экв.) в дихлорметане обрабатывают трифосгеном (0,7 экв.) при 0°С в течение 10 мин. Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение дополнительных 2 час. Прозрачный раствор разбавляют этилацетатом и промывают 10% раствором KHSO4, дистиллированной водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток. Данный продукт растворяют в дихлорметане и обрабатывают 2-(N-бензил-1-амино-3,3-диэтоксипропаном (0,9 экв.) при 0°С в течение 10 мин. Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение дополнительных 4 час. Образовавшуюся реакционную смесь разбавляют этилацетатом и промывают 10% раствором KHSO4, дистиллированной водой, насыщенным раствором NaHCO3, дистиллированной водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток. Данный сырой продукт очищают колоночной хроматографией (силикагель, этилацетат/гексан = 2/1), получая при этом чистый, указанный в заголовке продукт.
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 1,23 (т, 6H), 1,87 (м, 2H), 2,55 (м, 2H), 3,24 (м, 2H), 3,49 (м, 2H), 3,59 (м, 2H), 3,65 (м, 2H), 4,45-4,58 (м, 5H), 5,62 (ушир.с, 1H), 6,57 (ушир.с, 1H), 7,25-7,48 (м, 10H);
MC (m/z, ESI), 442 (MH+).
(В) Получение 1,7-дибензилгексагидропиримидо[1,6-a]пиримидин-2,6-диона
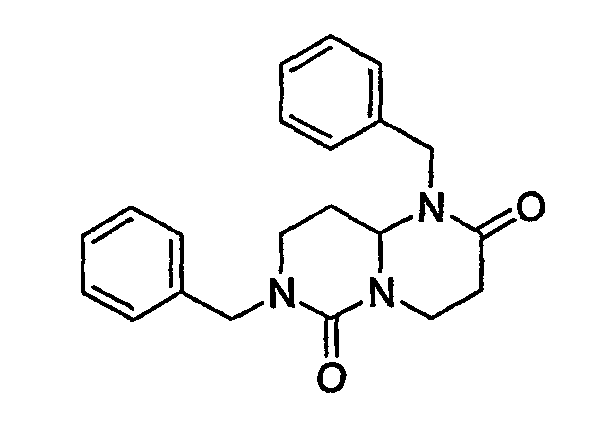
Амид, полученный в указанной выше стадии (А), обрабатывают муравьиной кислотой при 60°С в течение 4 дней. После выпаривания муравьиной кислоты при пониженном давлении остаток очищают препаративной ТСХ (силикагель, этилацетат), получая при этом указанное в заголовке соединение.
1Н-ЯМР (400 МГц, CDCl3) δ м.д.; 1,89 (м, 1Н), 2,19 (м, 1Н), 2,58 (м, 1Н), 2,75 (м, 1Н), 3,02 (м, 3Н), 4,42 (д, j=12,4Hz, 1Н), 4,45 (д, j=2,4Hz, 2Н), 4,65 (м, 1Н), 4,78 (м, 1Н), 4,98 (д, j=12,4Hz, 1Н), 7,25-7,38 (м, 10Н);
MC (m/z, ESI), 350(MH+).
Пример 4
1,7-Дибензил-6-оксооктагидропиримидо[1,6-а]пиримидин-2-он
(А) Получение (3-бром-1-метоксипропан-1-окси)связанной смолы аргогель
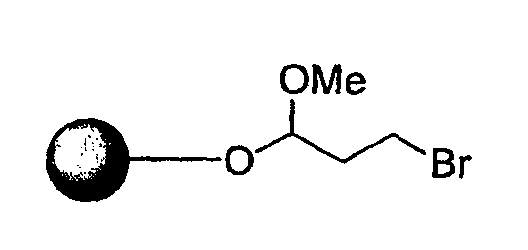
Суспензию сухой смолы аргогель и пара-толуолсульфоната пиридиния (240 мг, 0,96 ммоль) в 1,2-дихлорэтане (15 мл) нагревают для кипячения с обратным холодильником при непрерывном удалении растворителя и следов воды. После удаления приблизительно 5 мл дистиллята добавляют раствор 3-бром-1,1-диметоксипропана (700 мг, 3,84 ммоль) в 1,2-дихлорэтане (5 мл) и смесь выдерживают при кипячении с обратным холодильником в течение 4 час с непрерывным удалением EtOH/EDC, после чего смолу промывают ДМФА и диоксаном с последующей лиофилизацией, получая при этом требуемый продукт.
(В) Получение (3-бензиламино-1-метоксипропан-1-окси)связанной смолы аргогель
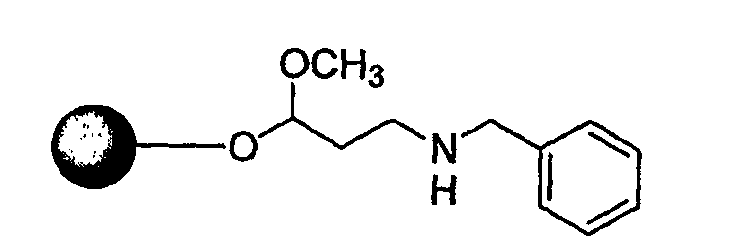
Раствор бензиламина (520 мг, 4,85 ммоль) в ДМСО (4 мл) добавляют к бромацетальной смоле (1 г, 0,48 ммоль) и суспензию встряхивают при 60°С в течение 15 час. Образовавшуюся смолу фильтруют, промывают ДМСО, МеОН и МС и сушат в вакууме на протяжении ночи. Присутствие вторичного амина определяют тестом на хлоранил.
(С) Получение β-аланинбензиламинмочевины
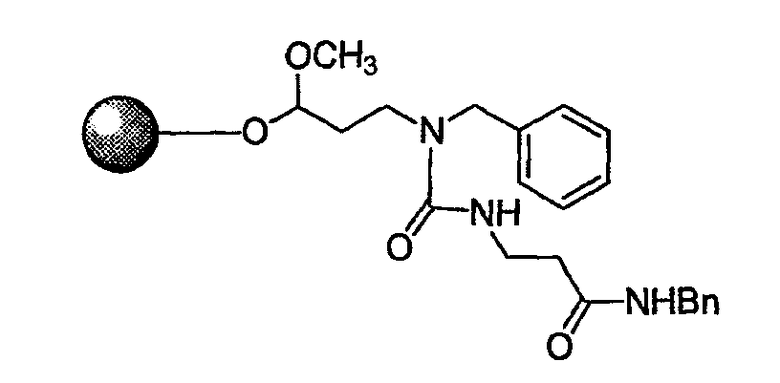
К раствору бензиламида β-аланина в виде соли с HCl (80 мг, 0,36 ммоль) в N-метилморфолине (120 мкл) и МС (2 мл) при комнатной температуре добавляют трифосген (0,72 ммоль). Спустя 10 минут образовавшийся раствор изоцианата добавляют к суспензии содержащей вторичную аминогруппу смолы, полученной в указанной выше стадии (2) (100 мг, 0,048 ммоль), и суспензию выдерживают при встряхивании в течение 3 час при комнатной температуре. Смолу промывают ДМФА, МеОН и МС и завершение реакции проверяют тестом на хлоранил.
(D) Получение 1,7-дибензил-6-оксооктагидропиримидо[1,6-a]пиримидин-2-она
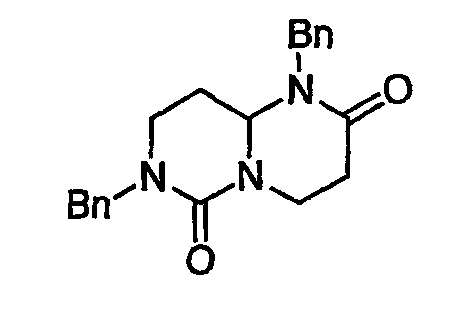
Содержащую группу тиомочевины смолу стадии (С) обрабатывают муравьиной кислотой и выдерживают при встряхивании в течение 15 час. Смолу отфильтровывают и фильтрат концентрируют и очищают хроматографией (силикагель), получая при этом указанное в заголовке соединение.
1Н-ЯМР (500 МГц, CDCl3) δ м.д.: 1,94 (м, 1H), 2,24 (м, 1H), 2,62 (м, 1H), 3,01 (м, 1H), 3,18 (м, 1H), 3,43 (м, 1H), 3,62 (м, 1H), 4,39 (д, 1H), 4,51 (м, 1H), 4,91 (м, 1H), 5,02 (д, 1H), 5,26 (д, 1H), 5,53 (м, 1H), 7,25-7,40 (м, 10H);
MC (m/z, APCI), 366 (МН+).
Пример 5
Бензиловый эфир 1,7-дибензил-2-оксо-6-тиоксооктагидропиримидо[1,6-а]пиримидин-4-карбоновой кислоты
(А) Получение 1-бензилового эфира-4-(9Н-флуорен-9-илметилового) эфира 2-изотиоцианатоянтарной кислоты
К раствору 1-бензилового эфира 2-трет-бутоксикарбониламиноянтарной кислоты (1 г, 3,09 ммоль) в МС добавляют DIC (532 мкл, 1,1 экв.), DMAP (188 мг, 0,5 экв.) и флуоренилметанол (635 мг, 1,05 экв.). После завершения реакции образовавшуюся реакционную смесь промывают 1 н. HCl и насыщенным раствором NaHCO3 и очищают колоночной хроматографией (силикагель), получая при этом сложный флуоренилметиловый эфир (400 мг).
Указанный эфир разбавляют в диоксане (10 мл) и добавляют 4 н. раствор HCl в диоксане и смесь выдерживают при перемешивании в течение 2 час для удаления Вос-защитной группы. После завершения реакции растворитель выпаривают досуха. Соль амина с HCl разбавляют МС и N-метилморфолином и приблизительно при 0°С добавляют тиофосген (1,2 экв.). После завершения реакции смесь промывают 10% раствором KHSO4, дистиллированной водой, насыщенным раствором NaHCO3, дистиллированной водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток. Данный сырой продукт очищают колоночной хроматографией (силикагель, этилацетат/гексан = 1/1), получая при этом чистый, указанный в заголовке продукт.
(С) Бензиловый-флуорениловый эфир тиомочевина аспарагиновой кислоты
Раствор в МС изоцианата (0,5 ммоль), полученного в указанной выше стадии (А), с N-метилморфолином добавляют к суспензии связанной с вторичным амином смолы (200 мг, 0,04 ммоль), получаемой в стадии (В) примера 4, и смесь выдерживают при встряхивании в течение 3 час при комнатной температуре. Образовавшуюся смолу промывают ДМФА, МеОН и МС и завершение реакции контролируют тестом на хлоранил.
(С) Бензиламидтиомочевина аспарагиновой кислоты
Смолу, полученную в указанной выше стадии (С), подвергают набуханию в течение 30 мин в ДМФА (4 мл) и для отщепления флуоренилметильной защитной группы добавляют 25% раствор пиперидина. Образовавшуюся смолу промывают ДМФА, МеОН и МС. Смолу сушат при пониженном давлении и снова подвергают набуханию, добавляют DIC (8 мкл, 0,05 ммоль), HOBt (8 мг, 0,05 ммоль) и DIEA (18 мкл, 0,1 ммоль) для активации кислоты. После встряхивания в течение 30 мин добавляют бензиламин и смесь выдерживают при встряхивании на протяжении ночи, получая при этом требуемую бензиламидозамещенную смолу.
(D) Получение бензилового эфира 1,7-дибензил-2-оксо-6-тиоксооктагидропиримидо[1,6-a]пиримидин-4-карбоновой кислоты
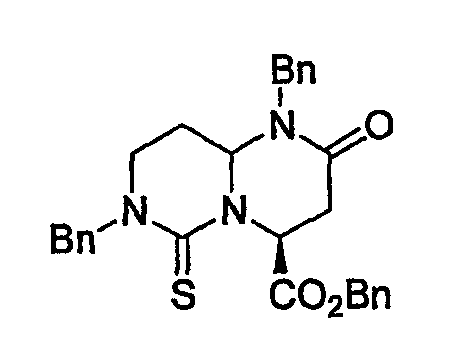
Смолу, полученную в указанной выше стадии (С), подвергают набуханию в МС (4 мл), добавляют PPTS (10 мг) и нагревают в течение 4 час при 60°С, получая при этом указанное в заголовке соединение.
МС (m/z, ESI), 500 (МН+).
Пример 6
Бензиловый эфир 7-бензил-6-тиоксогексагидропиримидо[1,6-а]пиримидин-1-карбоновой кислоты
(А) Получение бензилового эфира {3-[3-бензил-3-(3,3-диметоксипропил)тиоуреидо]пропил}карбаминовой кислоты
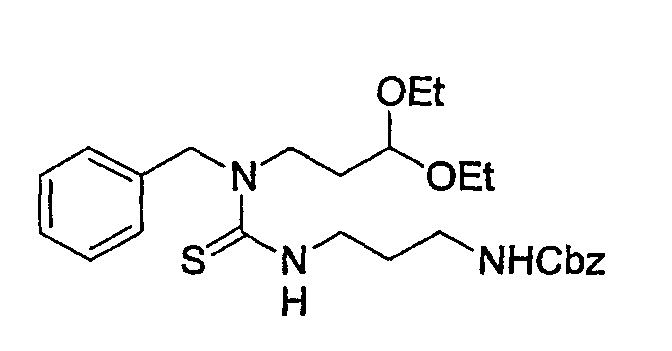
Суспензию Cbz-диаминопропан·HCl (1,0 экв.) и N-метилморфолина (2,2 экв.) в МС обрабатывают тиофосгеном (1,2 экв.) при 0°С в течение 10 мин. Образовавшемуся раствору дают возможность нагреться до комнатной температуры и перемешивают в течение дополнительных 2 час. Образовавшийся прозрачный раствор разбавляют этилацетатом и промывают 10% KHSO4, водой и насыщенным NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток, который растворяют в МС и обрабатывают N-бензил-1-амино-3,3-диэтоксипропаном (0,9 экв.) при 0°С в течение 10 мин и затем дают возможность ему нагреться до комнатной температуры и перемешивают в течение дополнительных 6 час. Образовавшуюся реакционную смесь разбавляют этилацетатом и промывают 10% раствором KHSO4, водой, насыщенным NaHCO3, водой и насыщенным NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток, который затем очищают колоночной хроматографией (силикагель, этилацетат/гексан, 2/1), получая при этом указанное в заголовке соединение.
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 1,17 (т, 6H), 1,5 (ушир.c, 2H), 1,75 (т, 2H), 1,92 (м, 2H), 3,20 (кв, 2H), 3,45 (м, 2H), 3,60 (м, 4H), 3,75 (кв, 2H), 4,51 (т, 1H), 5,06 (c, 4H), 6,75 (ушир.с, 1H), 7,25-7,38 (м, 10H);
MC (m/z, ESI), 442 (М-OEt+).
(В) Получение бензилового эфира 7β-бензил-6-тиоксогексагидропиримидо[1,6-a]пиримидин-1-карбоновой кислоты
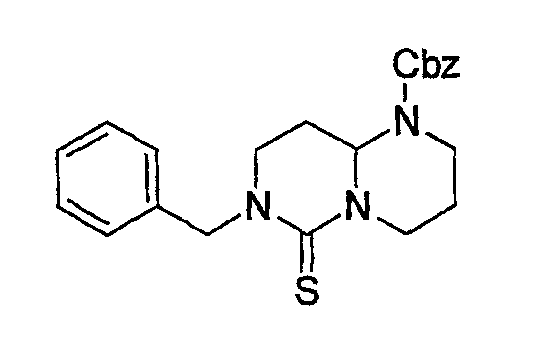
К раствору амида, полученного в указанной выше стадии (А), в МС добавляют PPTS и смесь перемешивают при 70°С на протяжении ночи. Образовавшуюся реакционную смесь концентрируют при пониженном давлении с получением остатка, который очищают препаративной ТСХ (только этилацетат), получая при этом указанное в заголовке соединение.
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 1,89 (м, 2H), 1,95 (м, 1H), 2,63 (м, 1H), 2,80 (м, 1H), 3,10 (м, 1H), 3,45 (м, 1H), 3,89 (м, 1H), 4,01 (м, 1H), 4,39 (д, 1H), 4,51 (м, 1H), 4,92 (м, 2H), 5,10 (м, 2H), 7,16-7,4 (м, 10H);
MC (m/z, ESI): 396 (MH+).
Пример 7
Бензиловый эфир 8-ацетил-6-оксогексагидропиразино[1,2-a]пиримидин-1-карбоновой кислоты
(А) Получение [ацетил-(2,2-диметоксиэтил)амино]уксусной кислоты
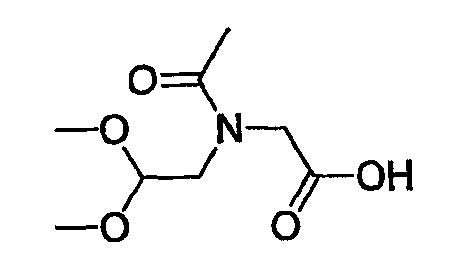
К раствору соли бензилглицина с HCl (1 экв.) в МеОН при комнатной температуре добавляют диметоксиацетальдегид (1,05 экв.) и затем NaCNBH3 (1,2 экв.) и смесь перемешивают в течение 5 час. Образовавшуюся реакционную смесь концентрируют при пониженном давлении, получая при этом маслянистый остаток, который растворяют в МС и промывают насыщенным раствором NaHCO3, водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток, который растворяют в МС и обрабатывают триэтиламином (3 экв.) и ацетилхлоридом (1,1 экв.) при 0°С.
После завершения реакции образовавшуюся реакционную смесь промывают насыщенным раствором NaHCO3, водой и насыщенным раствором NaCl. Органический слой сушат над Na2SO4 и концентрируют, получая при этом маслянистый остаток, который очищают колоночной хроматографией (силикагель, этилацетат), получая при этом чистый продукт. Продукт гидрогенолизируют с применением 10% Pd/C и Н2-содержащего баллона, получая при этом указанное в заголовке соединение, которое применяют в следующей стадии без дополнительной очистки.
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 2,09 (с, 1Н), 2,20 (с, 2Н), 3,40 (д, 6Н), 3,48 (д, 2Н), 4,16 (с, 2Н), 4,44 (м, 1Н).
(В) Получение бензилового эфира (3-{2-[ацетил-(2,2-диметоксиэтил)амино]ацетиламино}пропил)карбаминовой кислоты
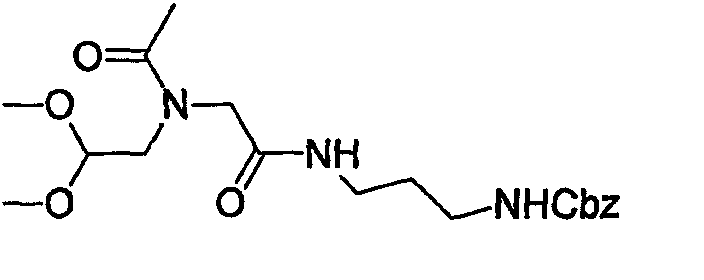
К раствору кислоты (1 экв.), полученной в указанной выше стадии (А), в МС добавляют HATU (1 экв.), DIPEA (3 экв.) и Cbz-диаминопропан·HCl (1,0 экв.) и смесь перемешивают в течение 3 час при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении с получением маслянистого остатка, который очищают препаративной ТСХ, получая при этом указанное в заголовке соединение.
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 1,60 (м, 2H), 2,01 (c, 1H), 2,20 (c, 2H), 3,20 (д, 2H), 3,24 (м, 2H), 3,40 (д, 6H), 3,50 (д, 2H), 4,06 (c, 2H), 4,44 (м, 1H), 5,08 (c, 2H), 5,18 (д, 1H), 6,91 (ушир.д, 1H), 7,16 (ушир.c, 5H);
MC (m/z, ESI): 396 (MH+).
(С) Получение бензилового эфира 8-ацетил-6-оксогексагидропиразино[1,2-a]пиримидин-1-карбоновой кислоты
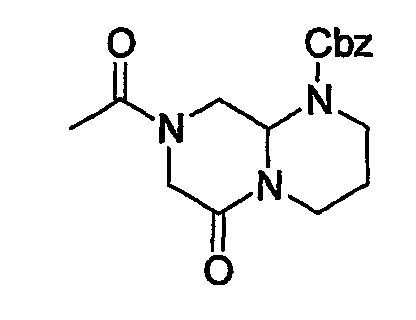
К раствору Cbz-защищенного амидного предшественника, полученного в указанной выше стадии (В), в МС при комнатной температуре добавляют PPTS (1 экв.) и смесь нагревают до 70°С в течение 5 час. Образовавшуюся реакционную смесь концентрируют, получая при этом остаток, который характеризуется следующим образом.
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 1,90 (м, 2H), 2,10 (c, 1H), 2,30 (c, 2H), 2,61 (м, 1H), 2,82 (м, 1H), 3,15 (м, 1H) 3,50 (м, 1H), 3,9 (м, 1H), 4,0 (м, 1H), 4,2 (м, 1H), 4,3 (c, 1H), 4,47 (м, 1H), 5,08-5,18 (м, 2H), 5,28 (ушир.c, 1H), 7,16 (ушир.c, 5H);
MC (m/z, ESI): 332 (MH+).
Пример 8
Метиловый эфир 7-бензоиламино-4-бензилкарбамоил-6-оксогексагидропирроло[1,2-a]пиримидин-1-карбоновой кислоты
(А) Получение бензилового эфира [1-(1-бензилкарбамоил-3-метоксикарбониламинопропилкарбамоил)-3,3-диметоксипропил]карбаминовой кислоты
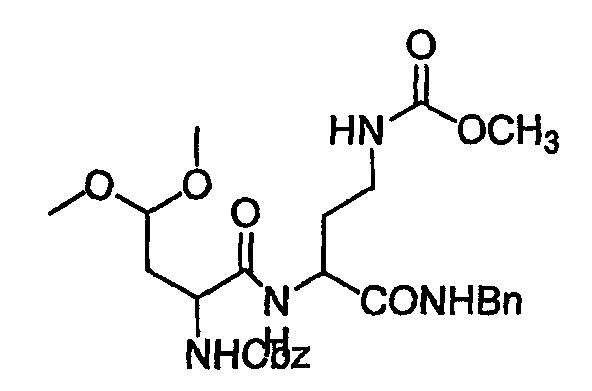
К раствору Cbz-защищенного аминокислотой ацеталя (100 мг, 1,3 экв.), полученного в препаративном примере 3(3), в МС добавляют PyBOP (1 экв. на кислоту), DIPEA (6 экв. на кислоту) и HOBt (1,3 экв.) и смесь перемешивают в течение 30 мин. К реакционной смеси добавляют соль аминобензиламида с HCl (71 мг, 0,27 ммоль) и смесь перемешивают в течение 7 час. Образовавшуюся реакционную смесь промывают насыщенным NaHCO3, водой и насыщенным NaCl. Органический слой сушат над MgSO4 и концентрируют с получением маслянистого остатка, который очищают колоночной хроматографией (силикагель, этилацетат), получая при этом указанное в заголовке соединение (50 мг, выход: 35%).
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 2,1 (т, 2H), 3,05 (м, 1H), 3,50 (cc, 6H), 3,45 (м, 1H), 3,75 (c, 3H), 4,25 (кв, 1H), 4,41 (м, 2H), 4,55 (м, 1H), 5,0 (кв, 2H), 5,3 (м, 1H), 5,95 (м, 1H), 7,2-7,4 (м, 10H).
(В) Получение метилового эфира 4-бензилкарбамоил-7-бензилоксикарбониламино-6-оксогексагидропирроло[1,2-a]пиримидин-1-карбоновой кислоты
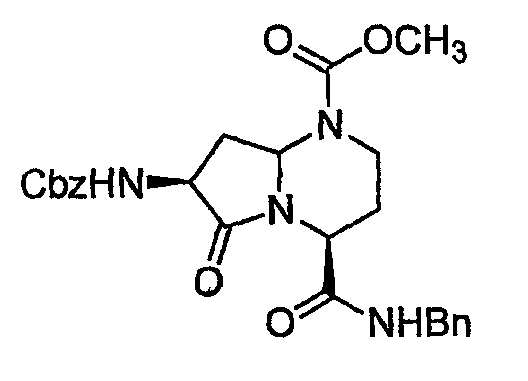
Ацетальамидный предшественник продукта циклизации (5 мг, 0,009 ммоль), полученный в указанной выше стадии (А), растворяют в муравьиной кислоте (1 мл) и перемешивают на протяжении ночи. Образовавшуюся реакционную смесь концентрируют досуха и продукт применяют в следующей стадии без дополнительной очистки.
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 2,25 (м, 2H), 2,61 (т, 2H), 3,24 (м, 1H), 3,50 (c, 3H), 3,55 (м, 1H), 3,95 (м, 1H), 4,45 (м, 2H), 4,65 (д, 1H), 4,8 (м, 2H), 5,3 (м, 1H), 5,7 (д, 1H), 7,15-7,4 (м, 10H), 7,85 (м, 1H).
(С) Получение метилового эфира 7-бензоиламино-4-бензилкарбамоил-6-оксогексагидропирроло[1,2-a]пиримидин-1-карбоновой кислоты
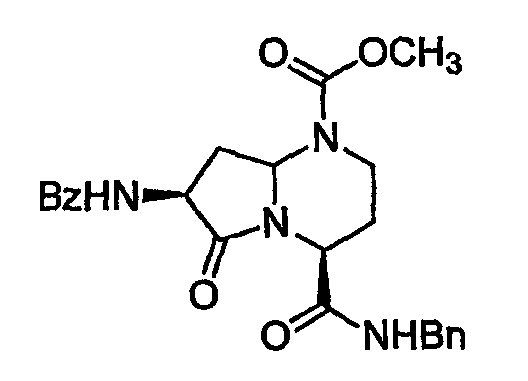
В реакционный сосуд, снабженный баллоном газообразного водорода, при комнатной температуре помещают раствор Cbz-бициклического соединения, полученного в указанной выше стадии (В), в МеОН и Pd/C (1 мг) и смесь перемешивают в течение 2 час. После завершения реакции реакционную смесь фильтруют посредством целитного фильтра для удаления Pd/C и растворитель выпаривают при пониженном давлении. Образовавшийся маслянистый остаток растворяют в МС, к раствору добавляют раствор бензойной кислоты (1,1 экв.) в МС и PyBOP (1,1 экв.), HOBt (1,1 экв.) и DIPEA (3 экв.) и смесь перемешивают в течение 30 мин. К образовавшемуся раствору активированной кислоты добавляют раствор амина и смесь выдерживают при перемешивании в течение 3 часов. Образовавшуюся реакционную смесь концентрируют при пониженном давлении с получением маслянистого остатка, который очищают препаративной ТСХ, получая при этом указанное в заголовке соединение.
1Н-ЯМР (300 МГц, CDCl3) δ м.д.: 2,25 (м, 2H), 2,65 (м, 2H), 3,27 (м, 1H), 3,70 (c, 3H), 3,6 (м, 1H), 4,10 (м, 1H), 4,54 (м, 2H), 4,8 (т, 1H), 5,45 (м, 1H), 7,15-7,42 (м, 10H), 7,9 (д, 1H), 8,31 (т, 1H).
Пример 9
Метиловый эфир 7-бензоиламино-4-(1-карбоксиэтилкарбамоил)-6-оксогексагидропирроло[1,2-a]пиримидин-1-карбоновой кислоты
Синтетическая схема, показывающая методологию примера 9, представлена на фиг.3.
Смолу 2-хлортритилхлорида (200 мг, 1 ммоль/г) и раствор Fmoc-аланина (1,5 экв., коммерчески доступный) и DIEA (2 экв.) в DCE (2 мл) помещают в сосуд с винтовой крышкой. Реакционную смесь встряхивают при комнатной температуре в течение 12 часов. Смолу собирают фильтрованием и промывают ДМФА, МеОН и затем DCM, обеспечивая этим получение первой компонентной части.
К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Стадию снятия защиты повторяют и затем смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор 2-(9Н-флуорен-9-илметоксикарбониламино)-4-метоксикарбониламиномасляной кислоты (1,5 экв. 2-ой компонентной части), DIC (1,5 экв.), НОВТ (1,5 экв.) в NMP. После встряхивания реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Стадию снятия защиты повторяют и смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор 2-(9Н-флуорен-9-илметоксикарбониламино)-5,5-диметоксипентановой кислоты (1,5 экв.), DIC (1,5 экв.), НОВТ (1,5 экв.) в NMP. После встряхивания реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Стадию снятия защиты повторяют и затем смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор коммерчески доступной бензойной кислоты (1,5 экв.), DIC (1,5 экв.), НОВТ (1,5 экв.) в NMP. После встряхивания реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
Смолу обрабатывают муравьиной кислотой (1,2 мл на каждую лунку) в течение 18 часов при комнатной температуре. После этого смолу удаляют фильтрованием и фильтрат концентрируют при пониженном давлении, получая при этом продукт в виде масла.
1Н-ЯМР (300 МГц, MeOH-d4) δ: 1,40 (д, 3Н), 1,90 (м, 1H), 2,20 (м, 1H), 2,30˜2,50 (м, 2H), 3,15 (м, 2H), 3,20 (м, 1Н) 3,45 (c, 3Н), 3,40˜3,60 (м, 1Н), 4,20˜4,40 (м, 2H), 4,70 (т, 1H), 5,40 (т, 1H), 7,25˜7,45 (м, 3Н), 7,75 (д, 2H).
MC (m/z, ESI): 433 (MH+), 455 (MNa+).
Пример 10
Метиловый эфир 7-бензоиламино-4-(2-карбоксипропилкарбамоил)-6-оксогексагидропирроло[1,2-a]пиримидин-1-карбоновой кислоты
Синтетическая схема, показывающая методологию примера 10, представлена на фиг.4.
Смолу 2-хлортритилхлорида (200 мг, 1 ммоль/г) и раствор Fmoc-бета-аланина (1,5 экв.) и DIEA (2 экв.) в DCE (2 мл) помещают в сосуд с винтовой крышкой. Реакционную смесь встряхивают при комнатной температуре в течение 12 часов. Смолу собирают фильтрованием и промывают ДМФА, МеОН и затем DCM, обеспечивая этим получение первой компонентной части.
К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Стадию снятия защиты повторяют и смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор 2-(9Н-флуорен-9-илметоксикарбониламино)-4-метоксикарбониламиномасляной кислоты (1,5 экв. 2-ой компонентной части), DIC (1,5 экв.) и НОВТ (1,5 экв.) в NMP. После встряхивания реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Стадию снятия защиты повторяют и затем смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор 2-(9Н-флуорен-9-илметоксикарбониламино)-5,5-диметоксипентановой кислоты (1,5 экв.), DIC (1,5 экв.), НОВТ (1,5 экв.) в NMP. После встряхивания реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
К смоле, набухшей в ДМФА, перед реакцией добавляют 25% пиперидин в ДМФА. После этого реакционную смесь встряхивают в течение 30 мин при комнатной температуре. Стадию снятия защиты повторяют и затем смесь продуктов промывают ДМФА, МеОН и затем DCM. К смоле добавляют раствор коммерчески доступной бензойной кислоты (1,5 экв.), DIC (1,5 экв.) и НОВТ (1,5 экв.) в NMP. После встряхивания реакционной смеси в течение 12 часов при комнатной температуре смолу промывают ДМФА, МеОН и затем DCM.
Смолу обрабатывают муравьиной кислотой (1,2 мл на каждую лунку) в течение 18 часов при комнатной температуре. После этого смолу удаляют фильтрованием, фильтрат концентрируют при пониженном давлении, получая при этом продукт в виде масла.
1Н-ЯМР (300 МГц, MeOH-d4) δ м.д.: 1,40 (д, 3Н), 1,90 (м, 1H), 2,20 (м, 1H), 2,30˜2,50 (м, 2H), 3,15 (м, 2H), 3,35 (c, 3Н), 3,40˜3,60 (м, 3Н), 4,20˜4,40 (м, 2H), 4,70 (т, 1H), 5,40 (т, 1H), 7,25˜7,45 (м, 3Н), 7,75 (д, 2H).
MC (m/z, ESI): 447 (MH+), 469 (MNa+).
Здесь приводятся различные ссылки, в которых подробно описаны некоторые процедуры, соединения и/или композиции, они включены в качестве ссылки во всей их полноте.
Должно быть понятно, что, хотя конкретные варианты осуществления изобретения были описаны здесь для целей иллюстрации, могут быть сделаны различные модификации, не выходящие за пределы сущности и объема изобретения. В соответствии с этим изобретение не ограничивается ничем, за исключением прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| МИМЕТИКИ С ОБРАТНОЙ КОНФИГУРАЦИЕЙ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2342387C2 |
| НОВЫЕ СОЕДИНЕНИЯ МИМЕТИКИ ОБРАТНОГО ПОВОРОТА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2457210C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ И СПОСОБ ЛЕЧЕНИЯ МЛЕКОПИТАЮЩИХ С ИХ ИСПОЛЬЗОВАНИЕМ | 2002 |
|
RU2310655C2 |
| Производные 2H-[1,4]оксазино- и 2H-[1,4]тиазино[3,2-c]хинолин-3(4H)-онов в качестве ингибиторов ATM и DNA-PK киназ для лечения и/или профилактики онкологических заболеваний | 2024 |
|
RU2835676C1 |
| АЗАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ ОПОСРЕДОВАННЫХ СЕРОТОНИНОМ ЗАБОЛЕВАНИЙ | 2001 |
|
RU2398765C1 |
| КОНДЕНСИРОВАННЫЕ N-ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ CRF | 2004 |
|
RU2382785C2 |
| СУЛЬФОНАМИДНЫЕ ПЕРИ-ЗАМЕЩЕННЫЕ БИЦИКЛЫ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ПОРАЖЕНИЯ АРТЕРИЙ | 2005 |
|
RU2403240C2 |
| ИНГИБИТОРЫ ФЕРМЕНТА, КОНВЕРТИРУЮЩЕГО ИНТЕРЛЕЙКИН-1-β | 1996 |
|
RU2249598C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2005 |
|
RU2348635C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2005 |
|
RU2409582C2 |
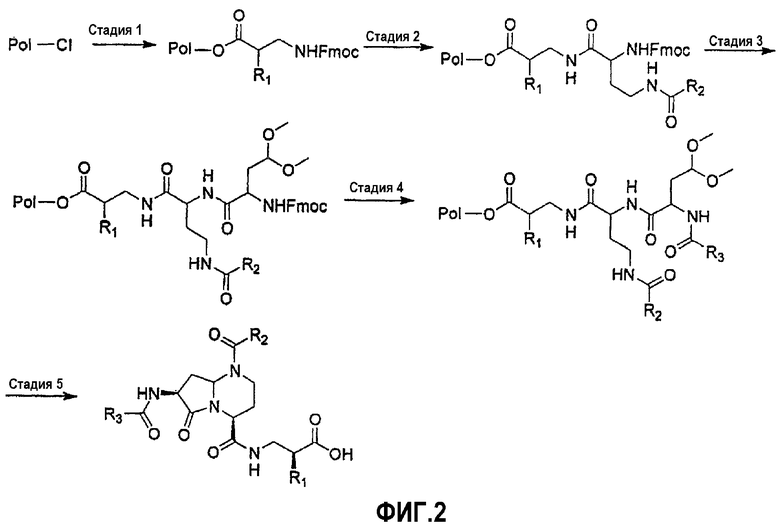
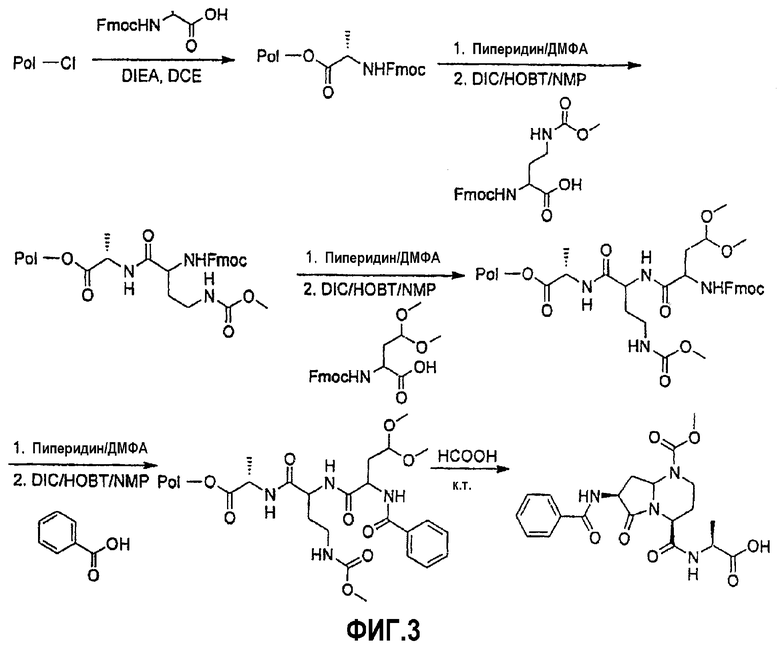
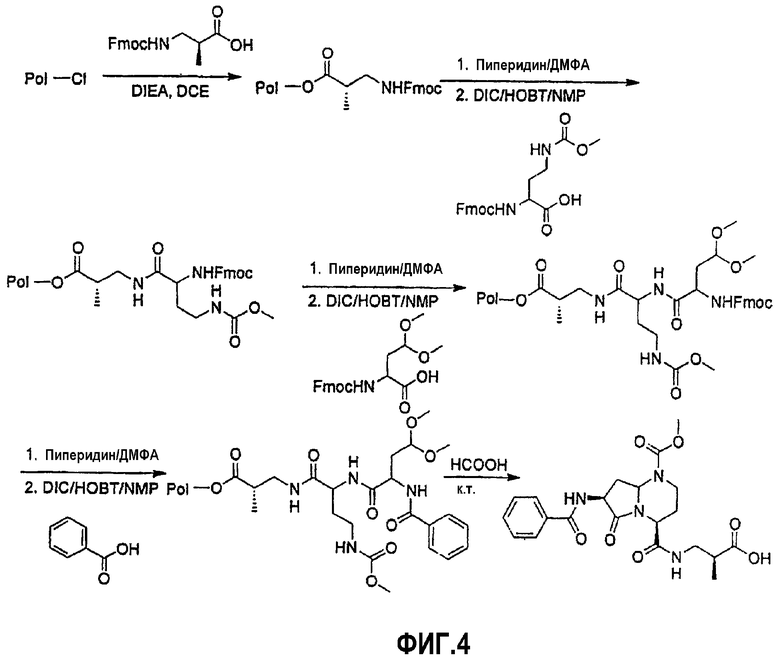
Описываются новые гетероциклические соединения общей формулы I
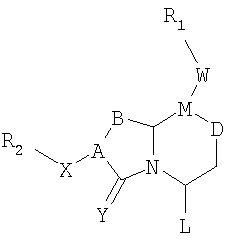
где А представляет собой -(СН)-, -N- или -CH2-N-; В представляет собой -(СН2)-, -(СН2-СН2)-; D представляет собой -(СН2)- или -(С=О)-; W представляет собой -(С=О)- или отсутствует; X представляет собой -NH(C=O)- или отсутствует; Y представляет собой кислород или серу; L представляет собой водород, -C(O)NHR3 или -C(=O)OR4; R1 - С1-12алкил; С6-10арил; С1-6алкокси; С6-10аралкил; С6-10аралкилокси; R2 - насыщенный или ненасыщенный С1-10алкил, возможно замещенный; С1-10алкенил; С6-10аралкилокси; С1-10алкандиенил; С6-10арил, возможно замещенный; С6-10аралкил, возможно замещенный; бензодиоксолил; пиперонил; хроменонил; алкил-С(=О)-; 5-6-членный гетероарил, содержащий один или два атома азота, или атом кислорода, или атом серы и возможно замещенный; R3 - радикал формулы

где Ra - С1-10алкил, возможно замещенный; С1-10алкенил; С3-10циклоалкил; амино; С6-10аралкил:, возможно замещенный; Rd - водород; С6-10аралкил, возможно замещенный; R4 - С6-10аралкил; при условии, что если А означает -(СН)-, тогда В - -(СН2)-, D - -(СН2)-, X - -NH(C=O)- и L - -C(=O)NHR3; если А означает N, тогда В - -(СН2-СН2)- и X отсутствует; если А означает -CH2-N-, тогда В - -(СН2)-, D - -(СН2)-, W - -(С=О)-, X отсутствует и L - водород, и содержащая их фармацевтическая композиция. Новые соединения обладают ингибирующим действием в отношении транскрипции NF-kB и могут быть использованы в медицине. 2 н. и 5 з.п. ф-лы, 3 табл., 4 ил.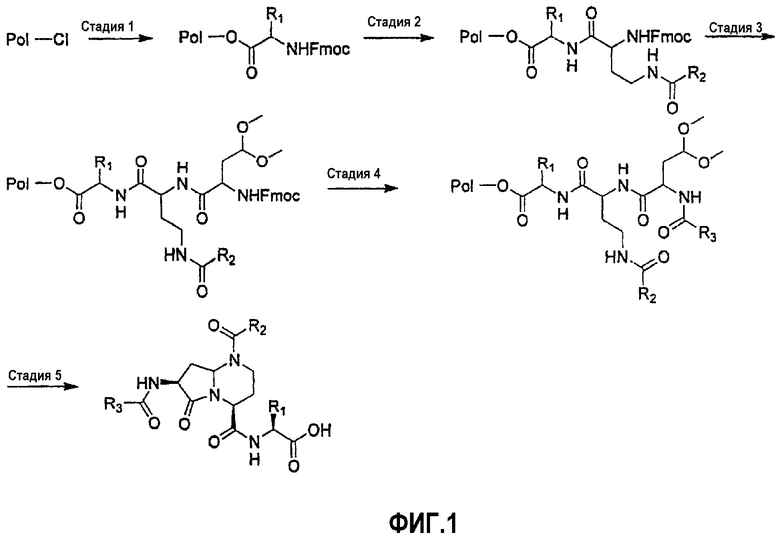
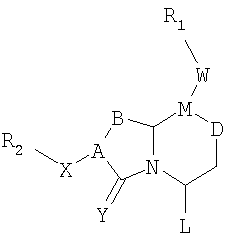
где А представляет собой -(СН)-, -N- или -CH2-N-;
В представляет собой -(СН2)-, -(СН2-СН2)-;
D представляет собой -(СН2)- или -(С=О)-;
W представляет собой -(С=О)- или отсутствует;
X представляет собой -NH(C=O)- или отсутствует;
Y представляет собой кислород или серу;
L представляет собой водород, -C(O)NHR3 или -C(=O)OR4;
R1 выбирают из
С1-12алкила;
С6-10арила;
С1-6алкокси;
С6-10аралкила;
С6-10аралкилокси;
R2 выбирают из
насыщенного или ненасыщенного С1-10алкила, который необязательно замещен одним или более заместителями, выбранными из С1-6алкокси; C1-6алкоксикарбонила; хромен-2-она; CN; бензила; 5-6-членного гетероарила, содержащего один или два атома азота, атом серы или кислорода и который необязательно замещен одним или более заместителями, выбранными из галогена или оксо; формамидила, который необязательно замещен С6-10арилом, необязательно замещенным одним или более заместителями, выбранными из галогена или С1-6алкокси, пиперонилом, С6-10аралкилом, необязательно замещенным одним или более заместителями, выбранными из C1-6алкокси, гидроксила или нитро, С6-10арилкарбамоилалкилом;
С1-10алкенила;
С6-10аралкилокси;
С1-10алкандиенила;
С6-10арила, который необязательно замещен одним или более заместителями, выбранными из С1-6алкокси, галогена, гидроксила или нитро;
С6-10аралкила, который необязательно замещен одним или более заместителями, выбранными из С1-6алкокси, галогена, нитро или гидроксила;
бензодиоксолила;
пиперонила;
хроменонила;
алкил-С(=О)-;
5-6-членного гетероарила, содержащего один или два атома азота, или атом кислорода, или атом серы и который необязательно замещен одним или более заместителями, выбранными из галогена или оксо;
R3 выбирают из
 ,
,  ,
,  ,
,
или бензил;
где Ra выбирают из
С1-10алкила, который необязательно замещен одним или более заместителями, выбранными из гидроксила, С3-10циклоалкила, пиперонила, амино или C1-6алкилтио;
С1-10алкенила;
С3-10диклоалкила;
амино;
С6-10аралкила, который необязательно замещен гидроксилом, галогеном, нитро, С1-6алкилом, С1-6алкенилом или С6-10арилом;
Rd выбирают из
водорода;
С6-10аралкила, который необязательно замещен С1-6алкокси или галогеном;
R4 представляет собой С6-10аралкил;
при условии, что
если А представляет собой -(СН)-, тогда В представляет собой -(СН2)-, D представляет собой -(СН2)-, X представляет собой -NH(С=О)- и L представляет собой -C(=O)NHR3;
если А представляет собой N, тогда В представляет собой -(СН2-СН2)- и X отсутствует;
если А представляет собой -CH2-N-, тогда В представляет собой -(СН2)-, D представляет собой -(СН2)-, W представляет собой -(С=О)-, X отсутствует и L представляет собой водород.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Приспособление для захвата зажигательных авиабомб | 1941 |
|
SU65724A1 |
| US 4448960 А, 15.05.1984 | |||
| Гербицидно-антидотный состав (его варианты) | 1980 |
|
SU984394A3 |

