Настоящее изобретение относится к применению производных 3-аминокапролактама для получения лекарственного препарата, предназначенного для предупреждения или лечения воспалительных заболеваний.
Воспаление является важным компонентом физиологической защиты хозяина. Тем не менее, становится все более понятным, что неправильно протекающие во времени или пространстве воспалительные реакции вносят вклад в широкий спектр различных заболеваний, в том числе таких заболеваний, в которые непосредственно вовлечены лейкоциты (такие как аутоиммунные заболевания, астма или атеросклероз), а также заболеваний, которые традиционно не рассматривали как затрагивающие лейкоциты (такие как остеопороз или болезнь Альцгеймера).
Хемокины представляют собой большое семейство сигнальных молекул, гомологичных интерлейкину-8, которые участвуют в регуляции транспорта лейкоцитов при физиологических и патологических состояниях. Система включает более пятидесяти лигандов и двадцать рецепторов, вовлеченных в сигнализацию хемокином, и имеет необходимую информационную плотность для того, чтобы направлять лейкоциты через сложные иммунорегуляторные процессы из костного мозга на периферию и затем обратно через вторичные лимфоидные органы. Однако такая сложность хемокиновой системы поначалу являлась препятствием для создания фармакологических подходов для модулирования воспалительных ответов через блокаду хемокинового рецептора. Сложность состояла в определении, какой хемокиновый рецептор (рецепторы) необходимо ингибировать для получения терапевтической пользы при данном воспалительном заболевании.
Совсем недавно было описано семейство агентов, блокирующих передачу сигналов (сигналинг) через широкий спектр хемокинов одновременно: Reckless et al., Biochem J. (1999) 340:803-811. Как было показано, первый такой агент, пептид, который назвали «Пептид 3», ингибирует миграцию лейкоцитов, индуцированную пятью различными хемокинами, при этом не оказывая влияние на миграцию в ответ на другие хемоаттрактанты (такие как fMLP или TGF-бета) неизменный. Этот пептид и его аналоги, такие как NR58-3.14.3 (т.е. Sequence ID No.1 c (DCys-DGIn-Dlle-DTrp-DLys-DGIn-DLys-DPro-DAsp-DLeu-DCys)-NH2) в совокупности называют «Ингибиторы Широкого Спектра Хемокинов» (Broad Spectrum Chemokine Inhibitors). Грейнжер с соавторами (Graingerer al., Biochem. Pharm. 65 (2003) 1027-1034) впоследствии показали на ряде животных моделей болезней, что ингибиторы широкого спектра хемокинов имеют потенциально полезную противовоспалительную активность. Интересно, что одновременная блокада множества хемокинов, по-видимому, не связана с острой или хронической токсичностью, что делает данный подход полезной стратегией для разработки новых противовоспалительных медикаментов с такими же преимуществами, как у стероидов, но со сниженными побочными эффектами.
Однако пептиды и пептоидные производные, такие как NR58-3.14.3, не могут являться оптимальным для применения in vivo. Их синтез достаточно дорогостоящий, и они имеют относительно неблагоприятные фармакокинетические и фармакодинамические свойства. Например, NR58-3.14.3 не является биодоступным при пероральном приеме и выводится из плазмы крови, имея период полувыведения менее 30 минут после внутривенной инъекции.
Для идентификации новых препаратов, которые сохраняют противовоспалительные свойства пептида 3 и NR58-3.14.3, но имеют улучшенные характеристики для применения в качестве фармацевтических препаратов, было выбрано две параллельных стратегии. Во-первых, был разработан ряд пептидных аналогов, некоторые из которых имеют более длительный период полужизни в плазме крови по сравнению с NR58-3.14.3, и синтез которых значительно дешевле. Во-вторых, был проведен подробный анализ параметра структура /активность указанных пептидов для выявления ключевых фармакофоров и разработки малых непептидных структур, которые сохраняют полезные свойства исходного пептида.
Указанный второй подход выявил несколько структурно различных серий соединений, которые сохраняли противовоспалительные свойства указанных пептидов, включая 16-амино и 16-аминоалкил производные алкалоида йохимбина, а также ряд N-замещенных 3-аминоглутаримидов. (Ссылки: Fox et al., J Med Chem 45(2002) 360-370: WO 99/12968 и WO 00/42071.) Все эти соединения являются ингибиторами широкого спектра хемокинов, которые сохраняют селективность в отличие от нехемокиновых хемоаттрактантов и было показано, некоторые из них блокируют острое воспаление in vivo.
Наиболее сильнодействующим и селективным из этих соединений был (S)-3-(ундек-10-еноил)-аминоглутаримид (NR58,4), который ингибирует хемокин-индуцированную миграцию in vitro при ЕD50 5 нМ. Однако дальнейшие исследования показали, что аминоглутаримидное кольцо подвержено ферментативной дециклизации в сыворотке крови. Поэтому, в некоторых случаях (например, когда воспаление, подвергаемое лечению, является хроническим, например, при аутоиммунных заболеваниях) эти соединения не могут обладать оптимальными свойствами и лучше применять более стабильные соединения с аналогичными противовоспалительными свойствами.
В качестве одного из подходов для выявлении таких стабильных аналогов были протестированы различные производные (S)-3-(ундек-10-еноил)-аминоглутаримида в отношении их стабильности в сыворотке крови. Одно из таких производных, 6-дезоксо- аналог (S)-3-(ундек-10-еноил)-тетрагидропиридин-2-он, полностью стабилен в сыворотке человека в течение по меньшей мере 7 дней при 37°С, но обладает значительно меньшей активностью по сравнению с исходной молекулой.
Амидные производные 3-аминокапролактама уже были описаны в данной области техники. Например:
- Заявка на патент Японии №09087331 описывает амидные производные 3-аминокарполактама, у которых амидалкильная боковая цепь может содержать от 2 до 30 атомов углерода. Эти соединения были представлены как масложелирующие агенты.
- Патент США №6395282 описывает иммуногенные конъюгаты, включающие молекулу-носитель, связанную с автоиндуктором грамотрицательной бактерии, где упомянутый автоиндуктор может быть амидным производным 3-аминокарполактама, амидалкильная боковая цепь которого может содержать до 34 атомов углерода. Однако терапевтическое применение описано только для конъюгатов, а не для изолированных амидных производных.
- Статья Уэйса и соавторов (Weiss et al. Research Communications in Psychology, Psychiatry and Behavior (1992), 17(3-4), 153-159) описывает серию амидных производных 3-аминокапролактама и среди прочих 3-гексанамидо-DL-ε-капролактам и 3-додеканамидо- DL-ε-капролактам. Эти соединения представлены как обладающие активностью только in vitro, но не оказывающие значительного действия in vivo.
Другими словами, хотя некоторые алкиламидные производные 3-аминокапролактама известны в данной области техники, для амидных производных 3-аминокапролактама не было описано фактического фармацевтического применения.
Изобретение предусматривает применение соединения общей формулы (I) или его фармацевтически приемлемой соли для получения лекарственного препарата, предназначенного для лечения воспалительных заболеваний:
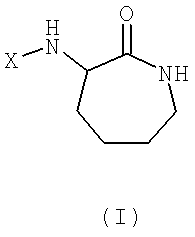
где
Х представляет собой -CO-Y-(R1)n или SO2-Y-(R1)n;
Y представляет собой циклоалкил или полициклоалкил (такой как адамантил, адамантанметил, бициклооктил, циклогексил, циклопропил); или циклоалкенил или полициклоалкенил;
каждый R1 независимо выбран из водорода или линейного или разветвленного алкила, галоалкила, алкокси, галоалкокси, алкенила, алкинила или алкиламино радикала, который содержит от 1 до 20 атомов углерода (например, от 5 до 20 атомов углерода, от 8 до 20 атомов углерода, от 9 до 20 атомов углерода, от 10 до 18 атомов углерода, от 12 до 18 атомов углерода, от 13 до 18 атомов углерода, от 14 до 18 атомов углерода, от 13 до 17 атомов углерода);
или каждый R1 независимо выбран из фтора, хлора, брома, йода, гидрокси, оксиалкил, амино, аминоалкил или аминодиалкил радикала; и
n представляет собой любое целое число от 1 до m, где m представляет собой максимальное количество замещений, допустимых в циклической группе Y.
Атом углерода в 3 позиции карболактамного кольца является асимметричным, и поэтому соединения согласно настоящему изобретению имеют две возможных энантиомерных формы, то есть конфигурации "R" и "S". Настоящее изобретение включает две энантиомерных формы и все комбинации этих форм, включая рацемические смеси "RS". Для упрощения, если в структурных формулах не показана конкретная конфигурация, то следует понимать, что представлены две энантиомерные формы и их смеси.
Предпочтительно соединение общей формулы (I) его фармацевтически приемлемая соль, применяемые согласно данному аспекту изобретения, представляет собой соединение общей формулы (I')
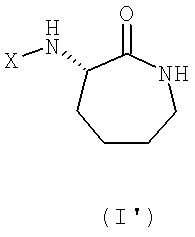
где Х имеет вышеуказанное значение.
Предпочтительно, в соединениях общей формулы (I) или (I') или их фармацевтически приемлемых солях кольцо или кольца Y ограничивают углы связи при альфа-углероде, делая их по существу тетраэдрическими (т.е. sp3 гибридные связи). «Альфа-углерод» представляет собой атом углерода либо в положении 2 (относительно амидкарбонильной группы) или в положении 2 (относительно сульфонамидсульфонильной группы).
Любой заместитель R1 может быть заместителем в любом допустимом положении кольца или колец циклической группы Y. В частности, нужно отметить, что изобретение включает соединения, в которых «альфа-углерод» является и частью циклической группы и сам является замещенным. Определение (R1)n включает соединения согласно настоящему изобретению без заместителей (т.е. R1 = водород), соединения согласно настоящему изобретению однозамещенные (т.е. R1 не является водородом и n=1), и также с несколькими заместителями (т.е., по меньшей мере, две R1 группы не являются водородом и n=2 или больше).
Изобретение также предусматривает фармацевтическую композицию, которая в качестве активного компонента содержит соединение общей формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, один фармацевтически приемлемый наполнитель и/или носитель:
где
Х представляет собой -CO-Y-(R1)n или SO2-Y-(R1)n;
Y представляет собой циклоалкил или полициклоалкил (например, адамантил, адамантанементил, бициклооктил, циклогексил, циклопропил); или - циклоалкенил или полициклоалкенил;
каждый R1 независимо выбран из водорода или алкил, галоалкил, алкокси, галоалкокси, алкенил, алкинил или алкиламино радикала, который содержит от 1 до 20 атомов углерода (например, от 5 до 20 атомов углерода, от 8 до 20 атомов углерода, от 9 до 20 атомов углерода, от 10 до 18 атомов углерода, от 12 до 18 атомов углерода, от 13 до 18 атомов углерода, от 14 до 18 атомов углерода, от 13 до 17 атомов углерода);
или каждый R1 независимо выбран из фтора, хлора, брома, йода, гидрокси, оксиалкил, амино, аминоалкил или аминодиалкил радикала; и n - представляет собой любое целое от 1 до m, где m представляет максимальное количество замещений, допустимое в циклической группе Y. Кроме того, R1 может быть выбран из пептидного радикала, например имеющего от 1 до 4 пептидных звеньев, соединеных пептидной связью (например, пептидный радикал, включающий от 1 до 4 аминокислотных остатков).
Предпочтительно, соединение общей формулы (I) или его фармацевтически приемлемая соль, применяемое согласно данному аспекту изобретения, представляет собой соединение общей формулы (I')
где Х имеет вышеуказанное значение.
Под фармацевтически приемлемой солью понимают, в частности, аддитивные соли неорганических кислот, такие как гидрохлорид, гидробромид, сульфат, фосфат, бифосфат и нитрат или соли органических кислот, такие как ацетат, малеат, фумарат, тартрат, сукцинат, цитрат, лактат, метансульфонат, п-толуолсульфонат, пальмоат и стереат. Также в область настоящего изобретения, в случаях, когда их можно применять, входят соли, образованные из оснований, например, гидроокиси натрия или калия. Другие примеры фармацевтически приемлемых солей см. в работе "Salt selection for basic drugs", Int. J. Pharm. (1986), 33, 201-217.
Фармацевтическая композиция может быть в твердой форме, например, в виде порошка, гранул, таблеток, желатиновых капсул, липосом или суппозиториев. Подходящей твердой основой может служить, например, фосфат кальция, стереат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, поливинилпирролидин и парафин. Другие подходящие фармацевтически приемлемые наполнители и/или носители известны специалистам в данной области техники.
Фармацевтическая композиция согласно настоящему изобретению также может быть представлена в жидкой форме, например, в виде раствора, эмульсии, суспензии или сиропа. Подходящей жидкой основой может быть, например, вода, органические растворители, такие как глицерин или гликоли, а также их водные смеси в различных пропорциях.
Изобретение также предусматривает соединение и его соль общей формулы (I)
где Х представляет собой -CO-Y-(R1)n или SO2-Y-(R1)n;
Y представляет собой циклоалкил или полициклоалкил (например, адамантил, адамантанеметил, бицилооктил, циклогексил, циклопропил);
или циклоалкенил или полициклоалкенил;
каждый R1 независимо выбран из водорода или алкил, галоалкил, алкокси, галоалкокси, алкенил, алкинил или алкиламино-радикала, который содержит от 1 до 20 атомов углерода (например, от 5 до 20 атомов углерода, от 8 до 20 атомов углерода, от 9 до 20 атомов углерода, от 10 до 18 атомов углерода, от 12 до 18 атомов углерода, от 13 до 18 атомов углерода, от 14 до 18 атомов углерода, от 13 до 17 атомов углерода); или каждый R1 независимо выбран из фтора, хлора, брома, йода, гидрокси, оксиалкил, амино, аминоалкил или аминодиалкил-радикала; и
n представляет собой любое целое число от 1 до m, где m представляет собой максимальное количество замещений, допустимых в циклической группе Y.
Кроме того, R1 может быть выбран из пептидного радикала, например, имеющего от 1 до 4 пептидных звеньев, соединенных вместе пептидной связью (например, пептидный радикал, включающий от 1 до 4 аминокислотных остатков).
Предпочтительно, соединение общей формулы (I) или его соль, применяемое согласно данному аспекту изобретения, представляет собой соединение общей формулы (I')
где Х имеет вышеуказанное значение.
Предпочтительно, в соединении общей формулы (I) или (I'), применяемом согласно настоящему изобретению, или в его соли, кольцо или кольца Y ограничивают углы связи при альфа-углероде, делая их по существу тетраэдраэдрическими (т.е. sp3 гибридные связи).
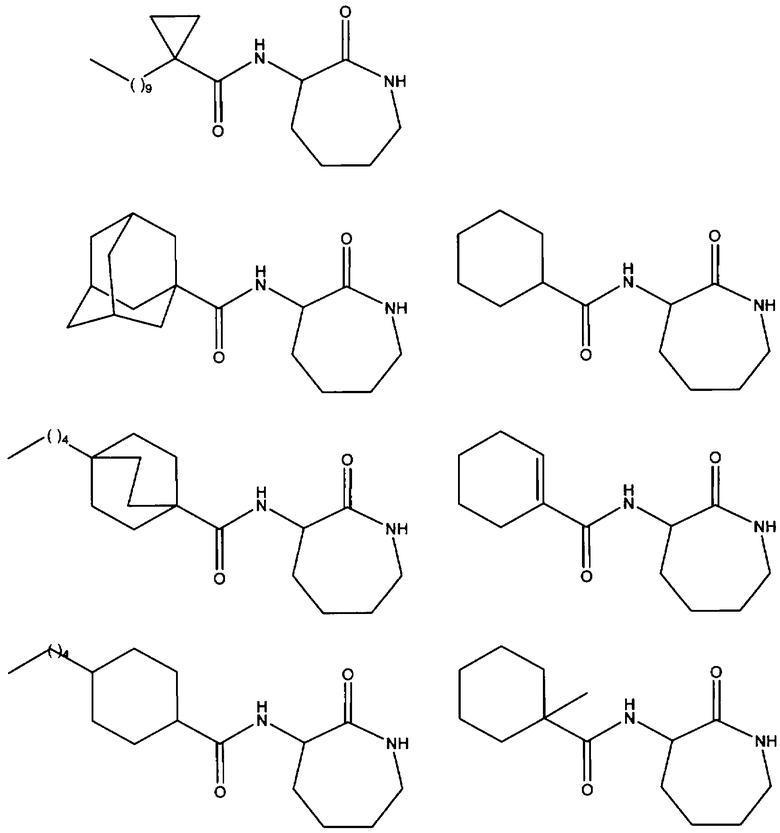
В частности, предпочтительные соединения общей формулы (I) или (I') и их соли в соответствии с любым из аспектов настоящего изобретения выбирают из группы, включающей:
- (S)-3-(циклогексанкарбонил)аминокапролактам;
- (S)-3-(1'-метилциклогексанкарбонил)аминокапролактам;
- (S)-3-(циклогекс-1'-енекарбонил)аминокапролактам;
- (S)-3-(транс-4'-пентилциклогексан-1-карбонил)аминокапролактам;
- (S)-3-(4'-пентил[2,2,2]бицикло-октан-1-карбонил)аминокапролактам;
- (S)-3-(1'-адамантанкарбонил)аминокапролактам;
-(S)-3-(1'-адамантанилметанкарбонил)аминокапролактам;
- (S)-3-(3'-хлор-1'-адамантанкарбонил)аминокапролактам;
- (S)-3-(3',5'-диметил-1'-адамантанкарбонил)аминокапролактам;
- (S)-3-(3',5',7'-триметил-1'-адамантанкарбонил)аминокапролактам;
и их соли.
Наиболее предпочтительное соединение представляет собой (S)-3-(1'-адамантанкарбонил)аминокапролактам и его соли.
Согласно изобретению, также предложены сульфаниламидные аналоги приведенных в качестве примеров соединений, то есть сульфониламинокапролактамные эквиваленты вышеуказанных соединений.
Как упоминалось в рассмотрении предшествующего уровня техники, некоторые алкиламидные производные 3-аминокапролактама известны как таковые (хотя на настоящий момент ни одно из таких соединений не было описано в качестве фармацевтической композиции как таковой или для медицинского применения в отношении противовоспалительных заболеваний).
Изобретение включает соединения, композиции и их применение, как определено, при этом соединения находятся в гидратированной или сольватированной форме.
Амидные производные 3-аминокапролактама, описанные здесь, являются функциональными ингибиторами широкого спектра хемокинов. Синтез их является сравнительно недорогим при применении несложного пути синтеза, представленного в данной заявке; они стабильны в сыворотке крови человека и, следовательно, имеют отличные фармакокинетические свойства; биодоступны при оральном приеме; являются сильнодействующими ингибиторами широкого спектра хемокинов in vitro с высокой селективностью по сравнению с нехемокиновыми хемоаттрактантами; являются сильнодействующими и эффективными противовоспалительными агентами in vivo на модели воспаления у грызунов; их введение не сопровождается какой-либо значительной острой токсичностью при дозах, необходимых для достижения максимального терапевтического эффекта. В целом эти свойства делают амидные производные 3-аминокапролактама противовоспалительными лекарственными препаратами, имеющими преимущества над вышеописанными соединениями.
По сравнению с предшествующим уровнем техники усовершенствование настоящего изобретения заключается в обеспечении 3-аминокапролактамной группы с боковой цепью, которая содержит одно или несколько алкильных/алкенильных колец, которые ограничивают углы связи при альфа-углероде боковой цепи. Соединения в соответствии с настоящим изобретением значительно превосходят соединения с неразветвленными аллеиловыми цепями (алкиламиды или алкилсульфаниламиды).
Пептиды, известные из предшествующего уровня техники (такие как NR58-3.14.3), имеют недостатки, заключающиеся в том, что: (а) они дорогостоящие и требуют твердофазного синтеза (по меньшей мере, для более длинных пептидов) и (b) они очень быстро выводятся через почки и (с) они обычно обладают меньшей активностью.
Аминоглутаримиды, известные из предшествующего уровня техники, являются дешевыми, не выводятся быстро через почки и более активны, однако они не обладают метаболической стабильностью.
Усовершенствование, описанное здесь, состоит в том, что аминокапролактамы, являются дешевыми, не выводятся через почки и даже являются более активными и также метаболически стабильны.
Согласно данному изобретению воспалительные заболевания, для предотвращения или лечения которых можно применять соединения общей формулы (I) или (I') или их фармацевтически приемлемые соли или фармацевтические композиции, или лекарственные препараты, содержащие их в качестве активных компонентов, включают, в частности:
- аутоиммунные заболевания, такие как рассеянный склероз;
- сосудистые заболевания, включающие инсульт, болезни коронарной артерии, инфаркт миокарда, нестабильную стенокардию, атеросклероз или васкулит, например, синдром Бехчета, гигантоклеточный артериит, ревматическую полимиалгию, синдром Вегенера, васкулит синдрома Черджа-Строса, болезнь Шенлейн-Геноха и болезнь Кавасаки;
- вирусную инфекцию или репликацию, например, инфекции или репликацию вирусов, включающих вирус оспы, вирус герпеса (например, Herpesvirus samiri), цитомегаловирус (ЦМВ) или лентивирус;
- астму;
- остеопороз (низкая минеральная плотность костей);
- развитие опухолей
- ревматоидный артрит;
- отторжение органа трансплантанта и/или отсроченное функционирование трансплантанта или органа, например, у пациентов с пересаженной почкой;
- нарушения, характеризующиеся повышенным уровнем TNFα;
- псориаз;
- повреждения кожи;
- заболевания, вызванные внутриклеточными паразитами, такие как малярия или туберкулез;
- аллергию; или
- Болезнь Альцгеймера.
Согласно данному изобретению воспалительные заболевания также включают:
- боковой амиотрофический склероз;
- фиброз (в особенности легочный фиброз, но не ограничивающийся фиброзом в легком);
- формирование адгезий (в особенности в брюшине и тазовой области).
- антиген-индуцированную амнестическую реакцию
- супрессию иммунного ответа.
Эти клинические симптомы попадают под общее определение воспалительных заболеваний или заболеваний, характеризующихся повышенным уровнем TNFα.
В тех случаях, когда это юридически допустимо, изобретение также предусматривает способ лечения, уменьшения выраженности или профилактики симптомов воспалительного заболевания (включая неблагоприятную воспалительную реакцию на любой агент) путем введения пациенту противовоспалительной дозы соединения, композиции или лекарственного препарата, как заявлено в данном описании.
Введение лекарственного препарата согласно изобретению может быть выполнено локально, перорально, парентеральным путем, внутримышечной инъекцией и т.п.
Вводимая доза, предусмотренная для лекарственного препарата, согласно изобретению составляет от 0.1 мг до 10 г в зависимости от типа применяемого активного соединения.
Согласно изобретению соединения общей формулы (I) или (I') могут быть получены с применением способов, описанных ниже.
Получение соединений общей формулы (I) или (I')
Все соединения общей формулы (I') или (I') можно легко получить общими способами, известными специалисту в данной области техники.
Тем не менее, предложен следующий предпочтительный путь синтеза:
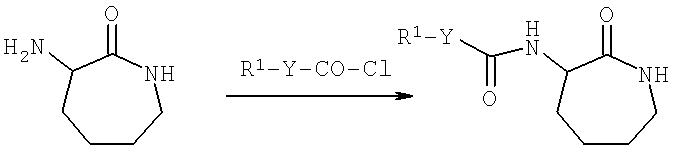
Схема 1
Реакцию, представленную на Схеме 1, можно проводить, например, в хлороформе или дихлорметане. Наиболее предпочтительным растворителем для данной реакции является дихлорметан.
Вышеуказанную реакцию предпочтительно проводят в присутствии основания, например Nа2СО3.
Вышеуказанную реакцию предпочтительно проводят при комнатной температуре (около 25°С) или, главным образом, при температуре между 20 и 50°С.
ОПРЕДЕЛЕНИЯ
Термин «приблизительно» относится к интервалу в области рассматриваемого значения. Как используется в описании данной заявки на патент, «приблизительно X» означает интервал от Х минус 10% от Х до Х плюс 10% от X, и предпочтительно интервал от Х минус 5% от Х до Х плюс 5% от X.
Использование числового диапазона в данном описании предназначено для того, чтобы однозначно включить в область изобретения каждое конкретное целое значение данного диапазона и все комбинации значений верхнего и нижнего предела расширенной области данного диапазона. Таким образом, например, диапазон от 1 до 20 атомов углерода, указанный (в числе прочего) в отношении формулы (I), охватывает все целые числа от 1 до 20 и все подобласти каждой комбинации верхних и нижних значений, приведенных в качестве примеров в явной форме или не приведенных.
В описании настоящей заявки термин «включающий/содержащий» следует понимать и как «включающий/содержащий» и как «состоящий из». Следовательно, если указано, что изобретение относится к «фармацевтической композиции, которая в качестве активного компонента содержит» соединение, то данная терминология относится и к тем композициям, в которых могут быть представлены другие активные компоненты, и к композициям, состоящим только из одного активного компонента, как определено.
Термин «пептидные звенья», используемый здесь, обозначает следующие 20 природных аминокислотных остатков:
Модифицированные и редкие аминокислотные остатки, как и пептидомиметики, также входят в объем понятия «пептидных звеньев»
Если не определено иначе, все технические и научные термины, использованные здесь, имеют значение, которое обычно понимается специалистом в области техники, к которой принадлежит данное изобретение. Аналогично, все публикации, заявки на патенты, все патенты и другие источники, упоминаемые здесь, включены посредством ссылки (если это юридически допустимо).
Следующие примеры иллюстрируют, но никоим образом не ограничивают область изобретения.
На чертеже представлена химическая структура примеров соединений согласно изобретению.
ПРИМЕРЫ
Общая процедура синтеза исходных соединений
Гидрохлориды (R) и (S)-3-аминокапролактама и гидропирролидин-5-карбоксилаты (R,R) и (S,S)-3-аминокапролактама были синтезированы в соответствии с литературными данными (сравнить Boyle et al., J. Org. Chem., (1979), 44, 4841-4847; Rezler et al., J. Med. Chem. (1997), 40, 3508-3515).
Пример 1: (S)-3-(циклогексанкарбонил)амино-капролактам:
(S,S)-3-аминокапролактам гидропирролидин-5-карбоксилата 2 (5 ммоль) и Na2CO3 (15 ммоль) в воде (25 мл) добавили в раствор циклогексанкарбонилхлорида (5 ммоль) в дихлорметане (25 мл) при комнатной температуре и реакционную смесь при перемешивали в течение 12 часов. Затем отделяли органический слой и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушили над Nа2СО3 и концентрировали в в вакууме. Остаток очистили перекристаллизацией из ЕtOАс/гексана с получением лактама (540 мг, 45%); Тпл (ЕtOАс/гексаны) 180-181°С; [α]25 D (с=1, СНСl3)+42.0; vmax/cm-1 3294 (NH), 1668, 1614 (CO), 1537 (NH); δН (500 МГц, CDCl3) 6.89 (1H, д, J 5.5, CHNH), 6.51 (1H, уш. с, CH2NH), 4.48 (1H, дд, J 11, 6, CHNH), 3.30-3.17 (2H, м, СН2NН), 2.11 (1H, тт, J 11.5, 3.5, (СН2)СНСО), 2.01 (1H, уш д, J 13, лактамовое кольцо СН), 1.98-1.92 (1H, м, лактамовое кольцо СН), 1.87-1.70 (6Н, м, лактамовое кольцо СН×2 + циклогексан СН×4), 1.66-1.59 (1H, и, циклогексан СН), 1.47-1.30 (4Н, уш м, лактамовое кольцо СН ×2 + циклогексан СН×2) и 1.23-1.15 (3Н, м, циклогексан СН×3); δС (125 МГц, CDCl3) 175.9, 175.3 (СО), 51.8 (NHCHCO), 45.2 (СН), 42.1, 31.7, 29.6, 29.4, 28.9, 27.9, 25.7 (×2), 25.6 (СН2); m/z (M+ С13Н22N2O2 вычисл. 238.16813) 238.16768.
Пример 2: (S)-3-(1'-метилциклогексанкарбонил)амино-капролактам:
(S,S)-3-аминокапролактам гидропирролидин-5-карбоксилата 2 (5 ммоль) и Nа2СО3 (15 ммоль) в воде (25 мл) добавили в раствор 1-метилциклогексанкарбонилхлорида (5 ммоль) в дихлорметане (25 мл) при комнатной температуре и реакционную смесь перемешивали в течение 12 часов. Затем отделили органический слой и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушили над Nа2СО3 и концентрировали в вакууме. Остаток очищали перекристаллизацией из ЕtOАс/гексана с получением лактама (540 мг, 43%); Тпл (ЕtOАс/гексаны) 168-169°С; [α]25 D (с=1, СНСl3)+33.0; vmax/cm-1 3380, 3241 (NH), 1674, 1638 (CO), 1501 (NH); δН (500 МГц, CDCl3) 7.12 (1H, д, J 5, CHNH), 6.52 (1H, уш с, CH2NH), 4.48 (1Н, ддд, J 11, 5.5, 1.5 CHNH), 3.30-3.16 (2H, м, CH2NH), 2.01 (1H, уш д, J 13, лактамовое кольцо СН), 1.98-1.86 (3Н, м, лактамовое кольцо СН + циклогексан СН×2), 1.85-1.73 (2H, м, лактамовое кольцо СН×2), 1.56-1.47 (2H, м, циклогексан СН×2), 1.47-1.33 (5Н, уш м, лактамовое кольцо СН×2 + циклогексан СН×3) и 1.33-1.25 (3Н, м, циклогексан СН×3); δС (125 МГц, CDCl3) 176.9, 167.0 (СО), 52.0 (NHCHCO), 42.5 (С четв.), 42.1, 35.5 (×2), 31.6, 28.9, 27.9 (СН2), 26.4 (СН3), 25.8, 22.9 (×2) (СН2); m/z (M+ C14H24N2O2 вычисл. 252.18378) 252.18323.
Пример 3: (S)-3-(циклогекс-1'-енкарбонил)амино-капролактам:
(S,S)-3-аминокапролактам гидропирролидин-5-карбоксилата 2 (5 ммоль) и Na2CO3 (15 ммоль) в воде (25 мл) добавили в раствор циклогекс-1-ен-1-карбонилхлорида (5 ммоль) в дихлорметане (25 мл) при комнатной температуре и реакционную смесь перемешивали в течение 12 часов. Затем отделили органический слой и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушивали над Na2CO3 и концентрировали в вакууме. Остаток очистили перекристаллизацией из ЕtOАс/гексана с получением лактама (431 мг, 36%); Тпл (ЕtOАс/гексаны) 151-152°С; [α]25 D (с=1, СНСl3)+57.5; vmax/cm-1 3219 (NH), 1652, 1628 (C=O, C=C), 1515 (NH); δН (500 МГц, CDCl3) 7.12 (1H, д, J 5, CHNH), 6.67 (1H, кн, J 1.5, CH=C), 6.52 (1H, уш с, CH2NH), 4.54 (1Н, ддд, J 11, 5.5, 1.5 CHNH), 3.32-3.18 (2H, м, CH2NH), 2.30-2.17 (2H, м, CH2CH=C), 2.16-2.10 (2H, м, СН=ССН2), 2.07 (1Н, уш д, J 15, лактамовое кольцо СН), 2.00-1.92 (1Н, м, лактамовое кольцо СН), 1.87-1.76 (2H, м, лактамовое кольцо СН×2), 1.68-1.60 (2H, м, циклогексан СН×2), 1.60-1.52 (2H, м, циклогексан СН×2) и 1.50-1.31 (2H, уш м, лактамовое кольцо СН×2); δс (125 МГц, CDCl3) 175.9, 167.4 (СО), 134.0 (СН=С), 132.8 (СН=С), 52.1 (NHCHCO), 42.1, 31.6, 28.9, 27.9, 25.3, 24.0, 22.1, 21.5 (CH2);
m/z (M+ С13Н20N2O2 вычисл. 236.15248) 236.15208.
Пример 4: (S)-3-(транс-4'-пентилциклогексан-1-карбонил)амино-капролактам:
(S,S)-3-аминокапролактам гидропирролидин-5-карбоксилата 2 (7 ммоль) и Nа2СО3 (21 ммоль) в воде (25 мл) добавили в раствор транс-4-пентилциклогексан-1-карбонилхлорида (6 ммоль) в дихлорметане (25 мл) при комнатной температуре и реакционную смесь перемешивали в течение 12 часов. Органический слой затем отделили и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушили над Nа2СО3 и концентрировали в вакууме. Остаток очистили перекристаллизацией из ЕtOАс/гексана с получением лактама (977 мг, 53%); Тпл 182-184°С; [α]25 D (с=1, СНСl3)+32.2; vmax/cm-1 3326 (NH), 1670, 1636 (CO), 1511 (NH); δН (500 МГц, CDCl3) 6.91 (1Н, д, J 5.5, CHNH), 6.87-6.70 (1Н, уш м, CH2NH), 4.44 (1Н, ддд, J 11, 6.0, 1.5, CHNH), 3.28-3.15 (2H, м, CH2NH), 2.08-1.90 (3Н, уш м, кольцо СН×2+(СН2)2СНСО), 1.88-1.72 (6Н, м, кольцо СН + цепь СH2×4), 1.45-1.28 (4Н, уш м, кольцо СН + цепь CH2×2 + цепь СН(СН2)3), 1.27-1.07 (9Н, уш м, кольцо СН + цепь CH2×8) и 0.90-0.79 (5Н, м, цепь CH2+СН3); δС (125 МГц, СDСl3) 176.0, 175.3 (СО), 51.8 (NHCHCO), 45.4 (СН), 41.0 (CH2), 37.1 (CH2), 36.9 (СН), 32.5, 32.4, 32.1, 31.7, 29.6, 29.4, 28.9, 27.9, 26.5, 22.6 (CH2) и 14.0 (СН3); m/z (M+ C18H32N2O2 вычисл. 308.24638) 308.24566.
Пример 5: (S)-3-(4'-пентил[2,2,2]бицикло-октан-1-карбонил)амино-капролактам:
(S,S)-3-аминокапролактам гидропирролидин-5-карбоксилата 2 (5.5 ммоль) и Nа2СО3 (16.5 ммоль) в воде (25 мл) добавили в раствор транс-4-пентилциклогексан-1-карбонилхлорида (4.4 ммоль) в дихлорметане (25 мл) при комнатной температуре и реакционную смесь перемешивали в течение 12 ч. Органический слой затем отделили и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушили над Nа2СО3 и концентрировалив вакууме. Остаток очистили перекристаллизацией из ЕtOАс/гексана с получением лактама (868 мг, 57%); Тпл 195-196°С; [α]25 D (с=1, СНСl3)+28.7; vmax/cm-1 3395, 3254 (NH), 1677, 1626 (CO), 1501 (NH); δН (500 МГц, CDCl3) 6.98 (1H, д, J 5.5, CHNH), 6.77-6.63 (1H, уш м, CH2NH), 4.41 (1H, дд, J 11, 5.5, CHNH), 3.27-3.15 (2H, м, CH2NH), 2.00-1.88 (2Н, уш м, кольцо СН×2), 1.81-1.73 (2Н, уш м, кольцо СН×2), 1.69 (6Н, уш т, J 7.5, цепь ССН2СН2С×6), 1.43-1.30 (8Н, уш м, кольцо СН×2 + цепь CCH2CH2C×6), 1.24 (2Н, сикстет, J 7, СН2СН3), 1.19-1.07 (4Н, м, СН2СH2СН2СН3) 1.05-0.98 (2Н, м, СН2Вu) и 0.82 (3Н, е, J7, СН3); δС (125 МГц, CDCl3) 177.4, 176.1 (СО), 51.9 (NHCHCO), 42.0, 41.2 (CH2), 39.0 (С четв.), 32.7, 31.6, 30.6 (×3) (CH2), 30.4 (С четв.), 28.9, 28.8 (×3), 27.9, 23.3, 22.6 (CH2) и 14.0 (СН3); m/z (M+ С20Н34N2O2 вычисл. 334.26203) 334.26352.
Пример 6: (S)-3-(1'-адамантанкарбонил)аминокапролактам:
(S)-3-аминокапролактам гидрохлорида 2 (1 ммоль) и Na2CO3 (3 ммоль) в воде (15 мл) добавили в раствор 1-адамантанкарбонилхлорида (1 ммоль) в дихлорметане (15 мл) при комнатной температуре и реакционную смесь перемешивали в течение 2 часов. Затем отделили органический слой и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушили над Nа2СО3 и концентрировали в вакууме. Остаток очистили перекристаллизацией из ЕtOАс/гексана с получением (S)-3-(1'-адамантанкарбонил)аминокапролактама (171 мг, 59%); Тпл 256-258°С; [α]25 D (с=1, СНСl3)+29.5; vmax/cm-1 3411, 3259 (NH), 1678, 1626 (CO), 1505 (NH); δН (500 МГц, CDCl3) 7.08 (1H, д, J 5.5, CHNH), 6.67 (1H, уш с, CH2NH), 4.47 (1H, ддд, J 11, 5.5, 1.5, CHNH), 3.32-3.17 (2Н, м, CH2NH), 2.06-1.94 (5Н, м, 2 × кольцо СН+3 × адамантана СН), 1.90-1.75 (8Н, м, 2 × кольцо СН+3 × адамантана CH2), 1.72 (3Н, уш д, J 14.5, 3 × адамантана СНН), 1.68 (3Н, уш д, J 14.5, 3 × адамантана СНН) и 1.47-1.32 (2Н, м, 2 × кольцо СН); δС (125 МГц, CDCl3) 177.2, 175.9 (CO), 51.9 (NHCHCO), 42.2 (CH2N), 40.5 (CCO), 39.0 (3×CH2 адамантана), 36.5 (3×CH2 адамантана), 31.7, 28.9, 28.0 (CH2 лактама), 28.1 (3×СН адамантана); m/z (MH+ C17H27N2O2 вычисл. 291.2073) 291.1994.
Пример 7: (S)-3-(1'-адамантанилметанкарбонил)аминокапролактам:
(S)-3-аминокапролактам гидрохлорида 2 (4 ммоль) и Nа2СО3 (12 ммоль) в воде (50 мл) добавили в раствор 1-адамантанметанкарбонилхлорида (4 ммоль) в дихлорметане (50 мл) при комнатной температуре и реакционную смесь перемешивали в течение 2-х часов. Затем отделили органический слой и экстрагировали водную фазу дополнительным количеством дихлорметана (2×50 мл). Объединенные органические слои высушили над Na2SO4 и концентрировали в вакууме. Остаток перекристаллизовали из ЕtOАс/гексана с получением (S)-3-(1'-адамантилметанкарбонил)аминокапролактама, перекристаллизовали из ЕtOАс с получением белых кристаллов (688 мг, 56%); Тпл 258-260°С; [α]25 D (с=1, СНСl3)+30.7; vmax/см-1 3409, 3255 (NH), 1682, 1611 (CO), 1539 (NH); δН (500 МГц, CDCl3) 6.82 (1H, д, J 5.5, CHNH), 6.77 (1H, уш т, J 5.5, CH2NH), 4.48 (1 Н, ддд, J 11, 6, 1.5, CHNH), 3.28-3.14 (2H, м, CH2NH), 2.04 (1 H, уш д, J 13.5, C-4 H), 1.97-1.86 (6H, м, C-5 H+3 × адамантан СН+СН2СО), 1.84-1.72 (2H, м, С-5 H+С-6 H), 1.63 (3Н, уш д, J 12, адамантан 3×CH2), 1.60-1.54 (9H, м, 9 × адамантан CH2) и 1.47-1.27 (2H, м, C-4 Н+С-6 Н); δС (125 МГц, CDCl3) 175.9 (лактам СО), 170.1 (амид СО), 52.0 (NHCHCO), 51.4 (СН2СО), 42.6 (3 × адамантан СН2), 42.0 (NCH2), 36.7 (3×СН2 адамантан), 32.7 (Cquat адамантан), 31.7 (C-4), 28.8 (С-6), 28.6 (3×СН адамантан), 28.5 (С-5); m/z (M+ C18H28N2O2 вычисл. 304.2151) 304.21430.
Пример 8: (S)-3-(3'-хлор-1'-адамантанкарбонил)аминокапролактам:
(S)-3-аминокапролактам гидрохлорида 2 (3 ммоль) и Nа2СО3 (9 ммоль) в воде (15 мл) добавили в раствор 3-хлор-1-адамантанкарбонилхлорида (3 ммоль) в дихлорметане (15 мл) при комнатной температуре и реакционную смесь перемешивали в течение 12 часов. Затем отделили органический слой и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушили над Nа2СО3 и концентрировали в вакууме. Остаток перекристаллизовали из ЕtOАс/гексана с получением (S)-3-(3'-хлор-1'-адамантанкарбонил)аминокапролактама (621 мг, 64%); Тпл 204-206°С; [α]25 D (с=0.5, СНСl3)+26.2; vmax/см-1 3411, 3267 (NH), 1679, 1630 (CO), 1508 (NH); δН (500 МГц, СDСl3) 7.07 (1H, д, J 5.5, CHNH), 6.65-6.44 (1H, уш м, CH2NH), 4.43 (1H, дд, J 11, 5.5, CHNH), 3.24-3.17 (2H, м, CH2NH), 2.24 (2H, уш с, адамантан СН), 2.20 (2H, уш с, адамантан СН), 2.12-2.03 (4Н, м, адамантан СН), 2.02-1.91 (2H, м, 2 × лактамовое кольцо СН), 1.85-1.72 (6H, м, 2 × кольцо СН+4 × адамантан СН), 1.66-1.55 (2H, м, 2 × адамантан СН) и 1.45-1.31 (2Н, м, 2 × кольцо СН); δС (125 МГц, CDCl3) 175.9, 174.9 (СО), 67.4 (CCl), 51.9 (NHCHCO), 48.6, 46.2 (×2) (3×СН2 адамантан), 44.5 (ССО), 42.1 (СН2N), 37.4, 37.3, 34.5 (3×CH2 адамантан), 31.5 (CH2 лактам), 31.1 (2×СН адамантан), 28.8, 27.9 (СН2 лактам); m/z (MH+ C17H26N2O2Cl вычисл. 325.1683)325.1696.
Пример 9: (S)-3-(3',5'-диметил-1'-адамантанкарбонил)аминокапролактам: (S)-3-аминокапролактам гидрохлорида 2 (1 ммоль) и Nа2СО3 (3 ммоль) в воде (15 мл) добавили к раствору 3,5-диметил-1-адамантанкарбонилхлорида (1 ммоль) в дихлорметане (15 мл) при комнатной температуре, и реакционную смесь перемешивали в течение 12 часов. Затем отделили органический слой и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушили над Nа2СО3 и концентрировали в вакууме. Остаток перекристаллизовали из гексана с получением (S)-3-(3',5'-диметил-1'-адамантанкарбонил)аминокапролактама (200 мг, 63%); Тпл157-158°С; [α]25 D (с=0.5, СНСl3)+26.8; vmax/см-1 3206 (NH), 1647 (CO), 1548 (NH); δН (500 МГц, CDCl3) 7.05 (1H, д, J 5.0, CHNH), 6.49-6.24 (1H, уш м, CH2NH), 4.45 (1Н, ддд, J 11, 5.5, 1.5, CHNH), 3.30-3.16 (2Н, м, CH2NH), 2.12-2.07 (1H, м, адамантан СН), 2.04-1.90 (2Н, м, 2 × лактамовое кольцо СН), 1.86-1.73 (2Н, м, 2 × лактамовое кольцо СН), 1.67 (2Н, уш с, 2 × адамантан СН), 1.51-1.26 (10Н, уш м, 8 × адамантан СН+2 × лактамовое кольцо СН) 1.17-1.09 (2Н, м, адамантан СН) и 0.81 (6Н, с, 2×СН3); δС (125 МГц, CDCl3) 176.9, 176.0 (СО), 51.9 (NHCHCO), 50.6, 45.2 (×2), 42.7 (×2) (СН2 адамантан), 42.4 (ССО), 42.1 (CH2N), 37.7 (СН2 адамантан), 31.6 (СН2 лактам), 31.0 (2×ССН3), 30.4, 29.3 (СН3), 28.9 and 27.9 (СН2 лактам); m/z (MH+ C19H31N2O2 вычисл. 319.2386) 319.2372.
Пример 10: (S)-3-(3',5',7'-триметил-1'-адамантанкарбонил)аминокапролактам: (S)-3-аминокапролактам гидрохлорида 2 (1 ммоль) и Nа2СО3 (3 ммоль) в воде (15 мл) добавили в раствор 3,5,7-триметил-1-адамантанкарбонилхлорида (1 ммоль) в дихлорметане (15 мл) при комнатной температуре и реакционную смесь перемешивали в течение 12 часов. Затем отделили органический слой и экстрагировали водную фазу дополнительным количеством дихлорметана (2×25 мл). Объединенные органические слои высушивали над Nа2СО3 и концентрировали в вакууме. Остаток перекристаллизовали из ЕtOАс/гексана с получением (S)-3-(3',5',7'-триметил-1'-адамантанкарбонил)аминокапролактама (188 мг, 56%); Тпл 177-178°С; [α]25 D (с=0.5, СНСl3)+25.6; vmax/см-1 3377, 3220 (NH), 1677, 1623 (CO), 1514 (NH); δН (500 МГц, CDCl3) 7.06 (1H, д, J 5.0, CHNH), 6.40-6.15 (1H, уш м, CH2NH), 4.46 (1H, ддд, J 11, 5.5, 1.5, CHNH), 3.32-3.17 (2H, м, CH2NH), 2.03-1.92 (2H, м, 2 × лактамовое кольцо СН), 1.86-1.74 (2H, м, 2 × лактамовое кольцо СН) 1.47-1.32 (8Н, м, 2 × кольцо СН+6 × адамантан СН) 1.06 (3Н, уш д, J 12, 3 × адамантан СНН), 1.04 (3Н, уш д, J 12,3 × адамантан СНН) и 0.83 (9Н, с, 3×СН3); δC (125 МГц, CDCl3) 176.8, 176.0 (СО), 51.9 (NHCHCO), 50.0 (3×СН2 адамантан), 44.6 (3×CH2 адамантан), 43.4 (ССО), 42.1 (CH2N), 31.8 (3×ССН3), 31.7 (СН2 лактам), 30.0 (3×СН3), 28.9, 27.9 (СН2 лактам); m/z (MH+ С20Н33N2O2 вычисл. 333.2542) 333.2528.
Фармакологическое изучение продуктов согласно настоящему изобретению
Ингибирование миграции лейкоцитов, индуцированной МСР-1
Принцип анализа
Биологическая активность соединений согласно настоящему изобретению может быть показана путем применения любого из широкого спектра способов функционального анализа миграции лейкоцитов in vitro, без ограничений включающим камеру Бойдена и родственные ей способы анализы миграции из лунки в лунку, анализ миграции в агарозе и камеры прямой визуализации, такие как камера Дунна.
Например, чтобы показать ингибирование миграции лейкоцитов в ответ на хемокины (но не другие хемоаттрактанты), применяли 96-луночную систему микроанализа миграции из лунки в лунку от Neuroprobe (Gaithersburg, MD, USA). В принципе при этом способе анализа используют две камеры, разделенные пористой мембраной. Хемоаттрактант помещают в нижнюю камеру, а клетки - в верхнюю. После инкубации в течение определенного периода времени при 37°С клетки двигаются по направлению к хемоаттрактанту, при этом количество клеток в нижней камере пропорционально активности хемоаттрактанта (относительно серии контролей).
Этот способ анализа можно применять для целого ряда различных популяций лейкоцитов. Например, можно использовать свежевыделенные лейкоциты периферической крови человека. Альтернативно, субпопуляции лейкоцитов можно получить, включая полиморфноядерные клетки или лейкоциты, или моноциты, с применением методов, хорошо известных специалистам в данной области техники, таких как центрифугирование в градиенте плотности, или разделение с помощью магнитных частиц. Альтернативно, можно использовать линии бессмертных клеток, которые широко применяются в качестве моделей лейкоцитов периферической крови человека, включая без ограничений клетки ТНР-1 как модель моноцитов или клетки Jurkat как модель неспециализированных (naive) Т-клеток.
Хотя для того, чтобы показать ингибирование хемокин-индуцированной миграции лейкоцитов можно применять разные условия анализа, здесь приведен конкретный пример.
Материалы
Системы для миграции из лунки в лунку произведены Neuroprobe, Gaithersburg, MD, USA.
Использовали планшеты Chemo Tx (Neuroprobe 101-8) и 30 мкл чистые планшеты (Neuroprobe MP30).
Сбалансированный солевой раствор Джейса приобретен в Sigma (Sigma G-9779).
БСА без жирных кислот приобретен в Sigma (Sigma А-8806).
МТТ, то есть 3-(4,5-диметилтриазол-2-ил)-2,5-дифенилтетразолбромид, приобретен в Sigma (Sigma М-5655).
Среда RPMI-1640 без фенолового красного приобретена в Sigma (Sigma R-8755).
Линию клеток ТНР-1 (Европейская коллекция клеточных культур) использовали в качестве популяции лейкоцитов.
Протокол теста
Следующую методику применяли для тестирования влияния соединений согласно настоящему изобретению на миграцию лейкоцитов, индуцированную МСР-1:
Сначала приготовили клеточную суспензию, которую поместили в верхнюю камеру. Клетки ТНР-1 осадили центрифугированием (770 × g; 4 мин) и промыли сбалансированным солевым раствором Джейса с 1 мг/мл БСА (ССРД+БСА). Затем повторили промывание и осадили клетки перед тем, как ресуспендировать в небольшом количестве ССРД+БСА для подсчета, например, с использованием стандартного гемоцитометра.
Затем доводили объем ССРД+БСА в зависимости от количества клеток таким образом, что окончательная концентрация составляла 4.45 х 106 клеток на мл ССРД+БСА. Это означало, что в каждых 25 мкл раствора, которые помещали в верхнюю камеру планшета, содержалось 100000 клеток ТНР-1.
Для того, чтобы протестировать единственное соединение на его способность ингибировать миграцию, индуцированную МСР-1, необходимо получить 2 порции клеток. Суспензию клеток ТНР-1 с концентрацией 4.45×106 клеток/мл разделили на две части. К одной части добавили тестируемый ингибитор в соответствующей конечной концентрации в соответствующем растворителе (например, 1 мкМ в не более чем 1% ДМСО). Ко второй части, служившей контролем, добавляли равный объем ССРД+БСА с соответствующим растворителем (например, не более чем 1% ДМСО).
Далее готовили раствор хемоаттрактанта, который помещали в нижнюю камеру. МСР-1 разбавляли ССРД+БСА до получения конечной концентрации 25 нг/мл. Этот раствор делили на две части, как и клеточную суспензию. К одной части добавляли тестируемое соединение в той же концентрации, в которой его добавляли в клеточную суспензию, в то время, как ко второй части добавляли равный объем ССРД+БСА с соответствующим растворителем (например, не более чем 1% ДМСО).
Следует отметить, что объем жидкости, который необходимо добавить вместе с анализируемым соединением, нужно учитывать при определении конечной концентрации МСР-1 в растворе для нижней камеры и конечной концентрации клеток в верхней камере.
Как только растворы хемоаттрактанта для нижних лунок и суспензии клеток для верхних камер получены, необходимо собрать камеру для миграции. Поместить 29 мкл соответствующего раствора хемоаттрактанта в нижнюю лунку камеры. Анализы следует выполнять, по меньшей мере, в трех повторениях для определения каждого условия. Как только все нижние камеры наполнены, используют пористую мембрану для камеры согласно инструкциям производителя. Наконец, добавляют 25 мкл соответствующей клеточной суспензии в каждую верхнюю камеру. Для предотвращения испарения аппарат накрывают пластиковой крышкой.
Собранную камеру инкубируют при 37°С, 5% СO2, в течение 2 часов. Суспензию клеток в ССРД+БСА также инкубируют в идентичных условиях в пробирке: эти клетки используют для построения стандартной кривой для определения количества клеток, которые мигрировали в нижнюю камеру при каждом условии.
В конце инкубации жидкую суспензию клеток аккуратно удаляют из верхней камеры и 20 мкл ледяного 20 мМ ЭДТА в ФСБ (фосфатно-солевом буфере) добавляют в верхнюю камеру, и аппарат инкубируют при 4°С в течение 15 мин. С помощью этой процедуры смывают клетки, прикрепившиеся к обратной стороне мембраны, в нижнюю камеру.
После инкубации фильтр аккуратно промывают ССРД+БСА, чтобы смыть ЭДТА, а затем удаляют фильтр.
Количество клеток, мигрировавших в нижнюю камеру при каждом условии, можно затем определить рядом способов, включая прямой подсчет, введение флуоресцентных или радиоактивных меток или с применением витального красителя. Обычно мы используем витальный краситель МТТ. 3 мкл готового раствора МТТ добавляют в каждую лунку, а затем планшет инкубируют при 37°С в течение 1-2 часов, в течение которых ферменты дегидрогеназы в клетках превращают растворимый МТТ в нерастворимый синий формазан, который можно количественно измерить с помощью спектрофотометра.
Одновременно построили стандартную кривую по 8 точкам. Начиная с количества клеток, добавленного в каждую верхнюю камеру (100000), и далее с 2-кратным разбавлением в ССРД+БСА, клетки добавляли в планшет в 25 мкл с добавленными 3 мкл готового раствора МТТ. Планшет для построения стандартной кривой инкубировали рядом с планшетом миграции.
В конце инкубации жидкость осторожно удаляют из нижних камер, таким образом, чтобы не разрушить осадок формазана. После недолгой сушки на воздухе 20 мкл ДМСО добавляют в каждую нижнюю камеру, чтобы растворить синий краситель, а поглощение при 595 нм измерить с применением считывающего устройства для 96-луночного планшета. Поглощение каждой лунки затем интерполировали на стандартную кривую для того, чтобы оценить количество клеток в каждой нижней камере.
Миграцию, стимулированную МСР-1, измеряли путем вычитания среднего числа клеток, которые достигли нижнего отдела в лунках, куда не было добавлено МСР-1, из среднего количества клеток, которые достигли нижнего отдела, где МСР-1 присутствовал в концентрации 25 нг/мл.
Влияние тестируемого вещества высчитывали, сравнивая миграцию, индуцированную МСР-1, в отсутствие и в присутствии различных концентраций тестируемого вещества. Обычно ингибирование миграции выражали как процент от общей МСР-1-индуцированной миграции, которая блокировалась в присутствии соединения. Для большинства соединений график доза-ответ строили путем измерения ингибирования МСР-1 - индуцированной миграции, которое имеет место при диапазоне концентраций соединения (обычно диапазон от 1 нМ до 1 мкМ или выше в случае слабоактивных соединений). Ингибиторную активность каждого соединения затем выражали как концентрацию соединения, необходимую для снижения МСР-1-индуцированной миграции до 50% (концентрация ED50)
Результаты
Протестировали соединения по Примерам 1-8 и 10 и было показано, что в данном тесте ED50 составила 100 нМ или менее.
Энантиоселективность
Можно синтезировать (S)- и (R)-энантиомеры двух разных членов группы аминокапролактама, чтобы установить, является ли биологическая активность энантиоселективной.
Кривые доза-ответ для каждого из соединений, действующих как ингибиторы МСР-1-индуцированной миграции клеток ТНР-1, можно определить, используя анализ миграции из лунки в лунку.
Для применения соединений по изобретению в качестве противовоспалительных агентов in vivo предпочтительно применять чистый (S)-энантиомер соединения, а не рацемическую смесь двух энантиомер или чистый (R)-энантиомер.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВОВОСПАЛИТЕЛЬНЫЕ СРЕДСТВА | 2004 |
|
RU2365585C2 |
| ЛИГАНДЫ ДЛЯ G-БЕЛОК СОПРЯЖЕННЫХ РЕЦЕПТОРОВ | 2005 |
|
RU2454416C2 |
| ПРОТИВОВОСПАЛИТЕЛЬНАЯ КОМПОЗИЦИЯ | 2008 |
|
RU2483730C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-(2-ГЕТЕРОЦИКЛОАЛКИЛ-1-ИЛЭТИЛ)АДАМАНТАН-2-АМИНОВ | 2013 |
|
RU2529201C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ (ВАРИАНТЫ) И СПОСОБ ИНГИБИРОВАНИЯ | 2001 |
|
RU2299201C2 |
| ЦИТОСКЕЛЕТОАКТИВНЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2006 |
|
RU2407745C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ МЕК И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2414455C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2004 |
|
RU2346943C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ ATR | 2012 |
|
RU2677292C2 |
| ИНГИБИТОРЫ ДНК-ПК | 2005 |
|
RU2408596C2 |
Изобретение относится к новым соединениям общей формулы (I) или к их фармацевтически приемлемым солям, или стереоизомерам, обладающим противоспалительными свойствами.
где Х представляет собой -CO-Y-(R1)n;
Y представляет собой (С3-С6)циклоалкил, (С3-С6)циклоалкенил или полициклоалкил, выбранный из группы, включающей адамантил, адамантанметил и бициклооктил; каждый R1 независимо выбран из водорода, (С1-С6)алкила, фтора, хлора, брома или йода; и
n представляет собой целое число от 1 до m, где m представляет собой максимальное число замещений, допустимых для циклической группы Y. 7 з.п. ф-лы, 1 ил.
1. Соединение общей формулы (I)
где X представляет собой -CO-Y-(R1)n;
Y представляет собой (С3-С6)циклоалкил, (С3-С6)циклоалкенил или полициклоалкил, выбранный из группы, включающей адамантил, адамантанметил и бициклооктил;
каждый R1 независимо выбран из водорода, (С1-С6)алкила, фтора, хлора, брома или йода; и
n представляет собой целое число от 1 до m, где m представляет собой максимальное число замещений, допустимых для циклической группы Y;
или фармацевтически приемлемая соль, или стереоизомер указанного соединения.
2. Соединение по п.1, отличающееся тем, что Y независимо выбран из группы, включающей адамантил, адамантанметил, бициклооктил, циклогексил и циклопропил.
3. Соединение по п.1, отличающееся тем, что указанное соединение представляет собой S-энантиомер.
4. Соединение по п.1, в котором кольцо или кольца Y ограничивают углы связей при альфа-атоме углерода, делая их, по существу, тетраэдрическими (sp3-гибридные связи).
5. Соединение по п.1, отличающееся тем, что заместитель R1 представляет собой (С1-С6)алкил с разветвленной цепью.
6. Соединение по п.1, отличающееся тем, что заместитель R1 выбран из водорода, фтора, хлора, брома или йода.
7. Соединение по п.1, отличающееся тем, что указанное соединение выбирают из группы, включающей:
- (S)-3-(циклогексанкарбонил)аминокапролактам;
- (S)-3-(1'-метилциклогексанкарбонил)аминокапролактам;
- (S)-3-(циклогекс-1'-енкарбонил)аминокапролактам;
- (S)-3-(трднс-4'-пентилциклогексан-1-карбонил)аминокапролактам;
- (S)-3-(4'-пентил[2,2,2]бициклооктан-1-карбонил)аминокапролактам;
- (S)-3-(1'-адамантанкарбонил)аминокапролактам;
- (S)-3-(1'-адамантанилметанкарбонил)аминокапролактам;
- (S)-3-(3'-хлор-1'-адамантанкарбонил)аминокапролактам;
- (S)-3-(3',5'-диметил-1'-адамантанкарбонил)аминокапролактам;
- (S)-3-(3',5',7'-триметил-1'-адамантанкарбонил)аминокапролактам;
и фармацевтически приемлемые соли указанных соединений.
8. Соединение по п.1, отличающееся тем, что указанное соединение представляет собой (S)-3-(1'-адамантанкарбонил)аминокапролактам или его фармацевтически приемлемую соль.
| Angelucci L | |||
| et al | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Journal of Medicinal Chemistry, 1993, том 36, №11, стр.1511-1519 | |||
| Nenajdenko V.G | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Russian Chemical Bulletin, International Edition, 2003, том 52, №11, | |||

