ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому способу получения ивабрадина гидрохлорида, применяемого в качестве средства против стенокардии. Кроме того, настоящее изобретение относится к новой аморфной форме ивабрадина гидрохлорида и способу ее получения.
УРОВЕНЬ ТЕХНИКИ
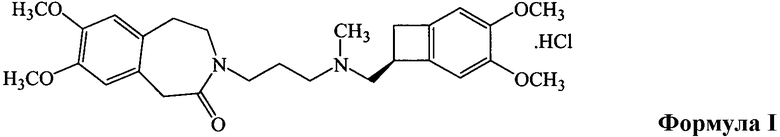
Ивабрадина гидрохлорид формулы I обладает очень ценными фармакологическими и терапевтическими свойствами и применим при многих сердечно-сосудистых заболеваниях, таких как стенокардия, инфаркт миокарда и ассоциированные нарушения ритма, и по химическому названию известен как (S)-7,8-диметокси-3-{3-{N-[(4,5-диметоксибензоциклобут-1-ил)метил]-N-(метил)амино)пропил)-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он
Ивабрадина гидрохлорид впервые описан в патенте США № 5296482. Описанный способ включает реакцию конденсации (S)-N-[(4,5-диметоксибензоциклобут-1-ил)метил]-N-(метил)амина формулы II

с 7,8-диметокси-3-[3-йодпропил]-1,3-дигидро-2H-3-бензазепин-2-оном формулы III

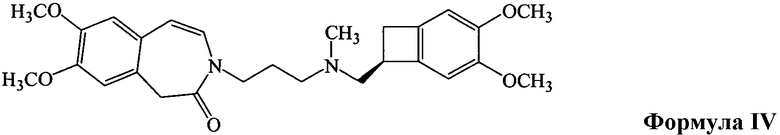
в ацетоне, в присутствии основания, такого как карбонат калия. Полученный бензазепиновое промежуточное соединение формулы IV очищают колоночной хроматографией
и далее восстанавливают гидроксидом палладия в ледяной уксусной кислоте, в атмосфере водорода с получением ивабрадина, который преобразуют в гидрохлорид (соль), воздействуя водной хлористоводородной кислотой
Производное метиламина формулы II получают восстановлением 1-циано-4,5-диметоксибензоциклобутана формулы V

используя боран-тетрагидрофурановый комплекс, который в результате конденсации с этилхлорформиатом с последующим восстановлением алюмогидридом лития в тетрагидрофуране дает рацемат производного метиламина формулы II. С помощью (d)-камфосульфоновой кислоты из рацемата выделяют (+)-изомер производного метиламина формулы II.
Производное бензазепина формулы III получают взаимодействием йодида натрия с 7,8-диметокси-3-[3-хлорпропил]-1,3-дигидро-2H-3-бензазепин-2-оном формулы VI

в ацетоне, и полученное йодсодержащее промежуточное соединение очищают, растворяя его в воде и экстрагируюя дихлорметаном.
Было замечено, что указанный способ страдает от многих недостатков, таких как использование боран-тетрагидрофуранового комплекса, который неустойчив при комнатной температуре, и использование хроматографическиих методов очистки промежуточных соединений и ивабрадина. Хроматографические методы очистки являются утомительными, громоздкими и трудными для использования в промышленном масштабе.
Также не предлагают при получении ивабрадина гидрохлорида использовать водный гидрохлорид, так как удаление хлористоводородной кислоты дистилляцией может привести к разложению и, в результате, к образованию примесей и, следовательно, к необходимости дополнительной очистки. Кроме того, использование при получении бензазепинового промежуточного соединения формулы IV высокоогнеопасного органического растворителя в больших количествах делает этот способ малопривлекательным для крупномасштабного производства.
Указанные выше недостатки требуют альтернативного усовершенствованного способа получения ивабрадина гидрохлорида высокой степени чистоты, который был бы экономически выгодным, воспроизводимым в промышленном масштабе и отвечал требованиям регулирующих органов.
Последующий патент США 7176197 сообщает о кристаллической форме ивабрадина гидрохлорида. «Les Laboratories» также сообщают о нескольких других кристаллических формах, таких как бета, гамма, дельта, бета-d, гамма-d и дельта-d. В известном уровне техники отсутствуют доступные данные, касающиеся существования аморфного ивабрадина гидрохлорида.
Для растворения кристаллических твердых частиц обычно требуется значительное количество энергии вследствие их высокоорганизованных решеткообразных структур. Например, энергия, необходимая для отрыва молекулы лекарственного средства от кристалла, превышает энергию отрыва от аморфной или некристаллической формы. Известно, что аморфные формы ряда лекарственных средств демонстрируют, по сравнению с кристаллической формой, различные параметры растворимости и в некоторых случаях - различную биодоступность. (Econno Т., Chem. Phazm Bull., 1990; 38: 2003-2007). При некоторых терапевтических симптомах один вид биодоступности может оказаться более предпочтительным, чем другой. Аморфная форма цефуроксим аксетила служит примером одного аморфного лекарственного средства, демонстрирующего значительно более высокую биодоступность, чем кристаллические формы. Это приводит к выбору аморфной формы в качестве окончательного вещества для лекарственного средства при разработке фармацевитической лекарственной формы цефуроксим аксетила. Дополнительно, растворимость в воде кристаллического аторвастатина кальция ниже, чем растворимость его аморфной формы, что может приводить к разнице в их биодоступности in vivo. По этой причине, для того чтобы отвечать требованиям регулирующих органов, желательно иметь аморфные формы лекарственных средств, имеющие высокую степень чистоты, а также высокую воспроизводимость способов их получения.
Поэтому, учитывая изложенное выше, желательно предложить эффективный, более экономичный, менее опасный и экологически приемлемый способ получения ивабрадина высокой степени чистоты или его фармацевитически приемлемой соли, при котором снижено образование примесей и, следовательно, можно избежать хроматографической очистки, и который удобен для осуществления в промышленном масштабе. Кроме того, в настоящей заявке на патент, охрану прав на которую испрашивают, была также предложена аморфная форма ивабрадина гидрохлорида.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Согласно одному аспекту настоящего изобретения предлагают способ получения ивабрадина гидрохлорида высокой степени чистоты формулы I, который включает:
a. конденсацию производного метиламина формулы II
с 7,8-диметокси-3-[3-йодпропил]-1,3-дигидро-2H-3-бензазепин-2-оном формулы III
в подходящем полярном растворителе в присутствии основания с получением бензазепинового промежуточного соединения формулы IV
b. восстановление соединения формулы IV с палладием на угле в ледяной уксусной кислоте в атмосфере газообразного водорода с получением ивабрадина; и
с. обрабоботку ивабрадина in situ спиртовым хлористым водородом в подходящем растворителе с получением его гидрохлоридной соли.
Еще одним аспектом настоящего изобретения является получение новой формы ивабрадина гидрохлорида, то есть аморфного ивабрадина гидрохлорида.
В другом аспекте настоящего изобретения предлагают способ получения аморфного ивабрадина гидрохлорида, который включает:
a. гидрирование бензазепинового промежуточного соединения формулы IV палладием на угле в ледяной уксусной кислоте в атмосфере газообразного водорода с получением ивабрадина
b. обработку ивабрадина раствором органической кислоты в подходящем растворителе с получением кислотно-аддитивной соли ивабрадина;
с. необязательную очистку кислотно-аддитивной соли ивабрадина;
d. гидролиз кислотно-аддитивной соли ивабрадина водным основанием в подходящем растворителе; и
e. обработку полученного ивабрадина in situ спиртовым хлористым водородом в органическом растворителе.
В другом аспекте предлагают способ получения ивабрадина гидрохлорида высокой степени чистоты, который включает:
обработку ивабрадина спиртовым хлористым водородом в подходящем растворителе с получением его гидрохлоридной соли.
Еще в одном аспекте настоящего изобретения предлагают способ получения аморфного ивабрадина гидрохлорида, который включает:
обработку ивабрадина раствором органической кислоты в подходящем растворителе с получением кислотно-аддитивной соли ивабрадина;
необязательную очистку кислотно-аддитивной соли ивабрадина;
гидролиз кислотно-аддитивной соли ивабрадина основанием в подходящем растворителе; и
обработку полученного ивабрадина спиртовым хлористым водородом в органическом растворителе.
Согласно еще одному аспекту настоящего изобретения, предлагают способ получения аморфного ивабрадина гидрохлорида, включающего:
a. обработку ивабрадина гидрохлорида основанием в подходящем растворителе; и
b. обработку полученного ивабрадина спиртовым хлористым водородом в органическом растворителе.
Согласно еще одному аспекту настоящего изобретения, предлагают способ получения аморфного ивабрадина гидрохлорида, который включает:
a. при температуре окружающей среды растворение гидрохлорида ивабрадина в смеси низшего алканола и кетона;
b. нагревание раствора до 40-50°C;
с. отгонку растворителя; и
d. отделение аморфного гидрохлорида ивабрадина.
В еще одном аспекте настоящего изобретения предлагают способ получения α-кристаллической формы ивабрадина гидрохлорида, включающий нижеследующие стадии:
a. нагревание раствора ивабрадина гидрохлорида в подходящем растворителе;
b. отгонка некоторой части растворителя;
c. фильтрование реакционной смеси для получения ивабрадина гидрохлорида; и
d. отделение α-кристаллической формы ивабрадина гидрохлорида.
Кислотно-аддитивные соли ивабрадина, получаемые по настоящему изобретению, также образуют патентоспособную часть изобретения. Ивабрадина оксалат выделяют в виде кристаллического твердого вещества, и он представляет собой еще один аспект изобретения, обладающий признаками изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
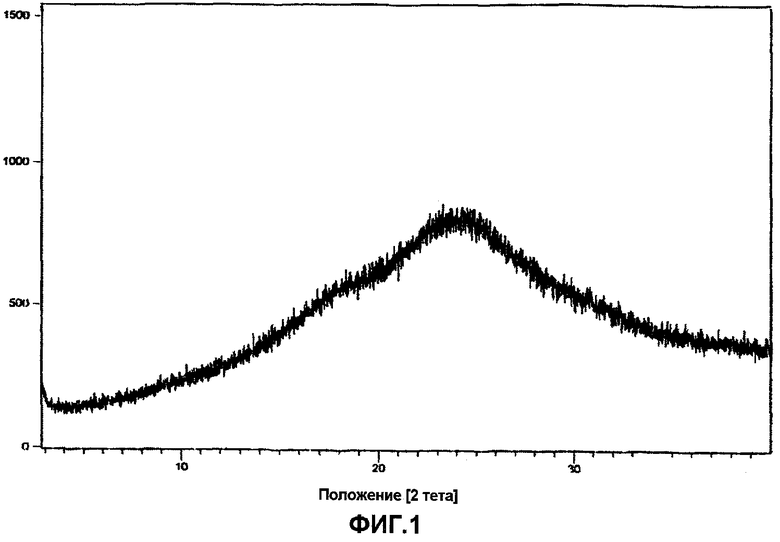
Фиг.1 представляет собой порошковую рентгенограмму аморфного ивабрадина гидрохлорида.
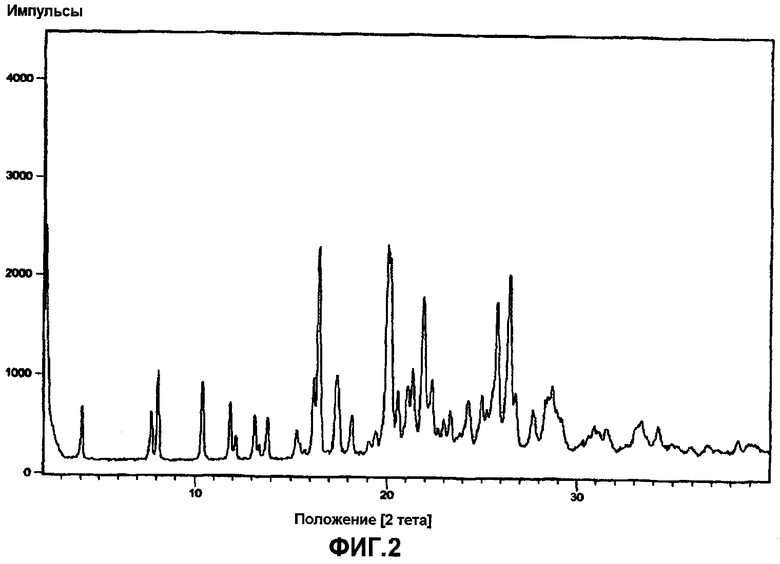
Фиг.2 представляет собой порошковую рентгенограмму α-кристаллической формы ивабрадина гидрохлорида.
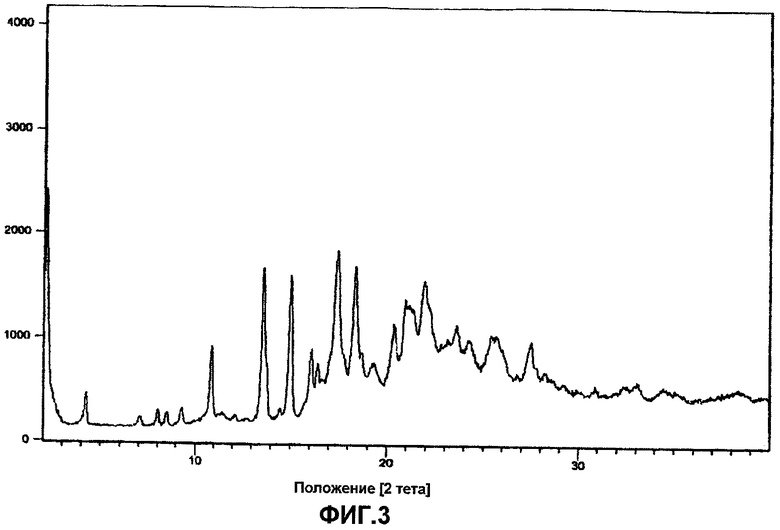
Фиг.3 представляет собой порошковую рентгенограмму ивабрадина оксалата.
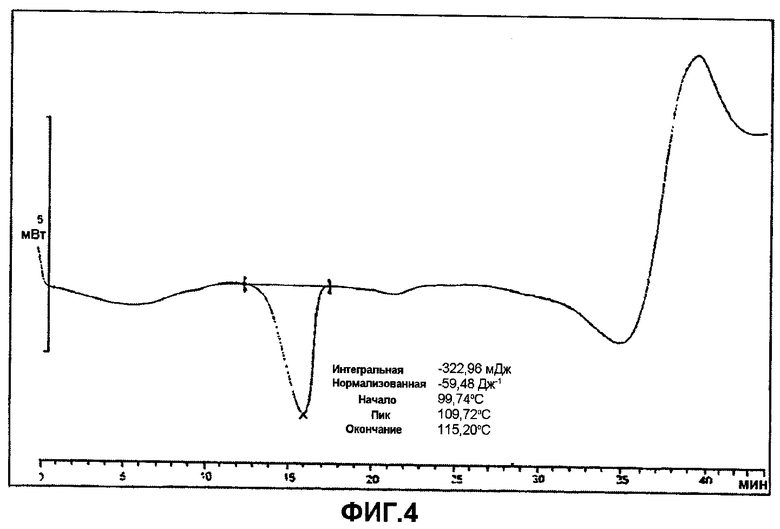
Фиг.4 представляет собой ДСК (DSC) термограмму ивабрадина оксалата (метод дифференциальной сканирующей калориметрии).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к эффективному и предпочтительному для промышленного применения способу получения ивабрадина высокой степени чистоты или его фармацевтически приемлемой соли, в частности, ивабрадина гидрохлорида.
Один аспект настоящего изобретения относится к усовершенствованному способу получения ивабрадина гидрохлорида высокой степени чистоты посредством первоначальной реакции конденсации производного метиламина формулы II c бензазепиновым производным формулы III в присутствии основания в полярном апротонном или протонном растворителе, с получением бензазепинового промежуточного соединения формулы IV. Основание можно выбирать из карбонатов, бикарбонатов и гидроксидов щелочных металлов и, предпочтительно, карбоната калия. Полярный апротонный или протонный растворитель выбирают из тетрагидрофурана, ацетона, ацетонитрила, диметилформамида, диметилсульфоксида, воды, изопропанола, C1-C4 линейных алифатических спиртов, таких как метанол, этанол и так далее, и их смесей. Более предпочтительно растворителем является диметилформамид или диметилсульфоксид и наиболее предпочтительно растворителем является диметилформамид. При реакции конденсации выгодно использовать диметилформамид, так как скорость реакции возрастает, и для завершения реакции требуется всего 2-3 часа, по сравнению с 18 часами в способах известного уровня техники, где используют ацетон. Реакцию проводят при 30-75°C и предпочтительно при 50-60°C. Завершение реакции контролируют с помощью высокоэффективной жидкостной хроматографии. После завершения реакции реакционную массу охлаждают до температуры окружающей среды и разбавляют водой. Продукт экстрагируют из водного слоя в органический растворитель, и некоторые примеси остаются в водном слое. Растворитель можно выбирать из галогенированных углеводородов, таких как метилендихлорид (метиленхлорид), этилендихлорид, четыреххлористый углерод, хлороформ и алифатический сложный эфир, такой как этилацетат, предпочтительно используют метиленхлорид. После этого растворитель отгоняют полностью, и продукт очищают кислотно-основной промывочной обработкой. В частности, остаток обрабатывают хлористоводородной кислотой в воде и промывают таким растворителем, как этилацетат. После этого водный слой нейтрализуют основанием, и требуемое соединение экстрагируют органическим растворителем. Как упоминалось выше, органический растворитель может быть выбран из галогенированных углеводородов и алифатических сложных эфиров, предпочтительно используют этилацетат. Получают продукт высокой степени чистоты, и хроматографической очистки не требуется.
Было также обнаружено, что в процессе конденсации образовывалась также неизвестная примесь, которую нелегко удалять, используя способ выделения, применяемый в известном уровне техники; что, следовательно, снижает чистоту продукта конденсации. Однако при выполнении способа по настоящему изобретению, заметили, что указанная неизвестная примесь может быть легко удалена экстрагированием требуемого продукта из реакционной смеси галогенированными растворителями, такими как метилендихлорид, этилендихлорид, четыреххлористый углерод, хлороформ. Это дополнительно исключает использование трудоемкой хроматографической очистки.
Согласно еще одному аспекту настоящего изобретения, бензазепиновое промежуточное соединения формулы IV наиболее предпочтительно получают конденсацией производного метиламина формулы II с 7,8-диметокси-3-[3-йодпропил]-1,3-дигидро-2H-3-бензазепин-2-оном формулы III в деминерализованной воде и в присутствии основания. Реакцию конденсации осуществляют при 45-60°C, предпочтительно, при 50-55°C, и время до завершения реакции занимает 10-20 часов. Течение реакции контролируют с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Продукт экстрагируют в растворителе, представляющем собой низший алифатический сложный эфир, такой как этилацетат. Затем этилацетатный слой обрабатывают водным раствором кислоты, и полученные слои разделяют. Водный слой обрабатывают основным раствором, и продукт экстрагируют в растворителе, представляющем собой низший алифатический сложный эфир, такой как этилацетат. Этилацетатный слой удаляют известными методами, такими как выпаривание, дистилляция в вакууме или в отсутствие вакуума и т.д. с получением бензазепинового промежуточного соединения формулы IV. Основание, которое используют на стадии конденсации, может быть выбрано из карбонатов, бикарбонатов и гидроксидов щелочных металлов, предпочтительно, карбоната калия.
Промежуточные соединения II и III могут быть получены способами, описанными в известном уровне техники, с минимальными модификациями. В частности, производное метиламина формулы II получают восстановлением 1-циано-4,5-диметоксибензоциклобутана формулы V,
используя такой восстановитель, как борановые комплексы, аналогичные боран-диметилсульфидному комплексу в тетрагидрофуране, который превращают в кислотно-аддитивную соль, обрабатывая кислотой в подходящем растворителе. В частности, гидрохлоридную соль получают обработкой аминопроизводного спиртовым хлористым водородом, эфирным хлористым водородом, такими как этанольный хлористый водород, изопропилацетатный хлористый водород, изопропилэфирный хлористый водород и так далее.
Затем соль хлористоводородной кислоты получаемого аминового промежуточного соединения конденсируют с этилхлорформиатом в присутствии подходящего основания с получением соответствующего амида. Полученный амид восстанавливают алюмогидридом лития в тетрагидрофуране с получением рацемического метиламинового промежуточного соединения формулы II. Рацемическое промежуточное соединение далее разделяют с помощью соответствующего разделяющего агента, такого как (d)-камфосульфокислота с получением требуемого (+)-изомера метиламина формулы II, который необязательно может быть использован в форме масла или может быть отделен.
Указанный способ вполне применим для крупномасштабного производства. Так как боран-диметилсульфидный комплекс устойчив при комнатной температуре, следовательно, не возникает трудностей при обращении с ним и при его хранении на крупномасштабном производстве.
В частности, соединение бензазепина формулы III получают взаимодействием 7,8-диметокси-3-[3-хлорпропил]-1,3-дигидро-2H-3-бензазепин-2-она формулы VI с йодидом натрия в ацетоне, с последующей его очисткой путем промывания взвешенного осадка ацетоном.
Промежуточные соединения формул V и VI получают способами, описанными в литературе (T. Kametani et al, Tetrahedron 1973; vol. 29; pages 73-76 and Reiffer M. et al., J. Med. Chem.; 1990; vol 33 (5): 1496-1504).
Промежуточное соединение формулы V также можно получить взаимодействием 6-бромвератральдегида c цианоуксусной кислотой в присутствии ацетата аммония и в растворителях, необязательно выбираемых из пиридина, толуола, бензола и так далее, и их смесей. Выделенное промежуточное соединение далее восстанавливают боргидридом натрия в присутствии водного основания. Обычно основания выбирают из бикарбоната натрия, карбоната натрия, гидроксида калия, гидроксида натрия или их смесей с получением β-(2-бром-4,5-диметоксифенил)-α-цианопропионовой кислоты. Полученную кислоту после декарбоксилирования в N,N-диметилацетамиде и последующего взаимодействия в жидком аммиаке с амидом натрия или амидом калия в жидком аммиаке преобразуют в 1-циано-4,5-диметоксибензоциклобутан формулы V.
Кроме того, промежуточное соединение формулы VI также можно получить галогенированием 3,4-диметоксифенилуксусной кислоты какими-либо галогенирующими агентами, такими как тионилхлорид, в хлорированных растворителях, таких как метилендихлорид. Полученное хлорсодержащее соединение далее подвергают конденсации с диметилацеталем аминоацетальдегида в присутствии подходящего основания с получением соответствующего амидного производного. Замыкание цикла в полученном амиде с образованием бензазепиновой части осуществляют в присутствии кислоты, например, хлористоводородной кислоты, ледяной уксусной кислоты или их смесей. Полученную бензазепиновую часть далее алкилируют 1-бром-3-хлорпропаном в присутствии подходящего основания, такого как третичный бутилат калия в органическом растворителе с получением 7,8-диметокси-3-[3-хлорпропил]-1,3-дигидро-2H-3-бензазепин-2-она формулы VI. Органические растворители могут быть выбраны из ацетона, диметилсульфоксида, диметилформамида и так далее, или их смесей.
Бензазепиновое промежуточное соединение формулы IV, полученное способами по настоящему изобретению, гидрируют и преобразуют в ивабрадин высокой степени чистоты или его фармацевитически приемлемую соль.
Обычно продукт конденсации, то есть (S)-7,8-диметокси-3-{3-{N-[(4,5-диметоксибензоциклобут-1-ил)метил]-N-(метил)амино)пропил)-1,3-дигидро-2H-3-бензазепин-2-он формулы IV преобразуют в ивабрадина гидрохлорид. Соединение формулы IV каталитически гидрируют, используя в качестве катализатора палладий на угле в уксусной кислоте и при давлении водорода 1-7 кг/см2. Реакцию гидрорования проводят при температуре окружающей среды, и время до завершения реакции составляет 4-10 часов, что контролируют с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Катализатор отфильтровывают, продукт экстрагируют из фильтрата в органический растворитель, или уксусную кислоту удаляют из фильтрата дистилляцией. Органический растворитель состоит из метилендихлорида, этилендихлорида, четыреххлористого углерода, хлороформа. Фильтрат далее обрабатывают разбавленной хлористоводородной кислотой и экстрагируют тем же органическим растворителем.
После этого объединенные экстракты подвергают перегонке, и полученный остаток обрабатывают разбавленной хлористоводородной кислотой. Водный слой для удаления примесей промывают органическим растворителем, таким как этилацетат, и нейтрализуют основанием, таким как водный гидроксид натрия. После этого требуемое соединение экстрагируют органическим растворителем. Органический растворитель может быть выбран из указанных выше растворителей, таких как галогенированный углеводород и алифатические сложные эфиры, предпочтительно используют этилацетат. Ивабрадин необязательно отделяют; в противном случае сам по себе органический слой обрабатывают спиртовым хлористым водородом с получением ивабрадина гидрохлорида высокой степени чистоты. Предпочтительно перед добавлением спиртового хлористого водорода высушивать органический слой, используя такой осушитель, как сульфат натрия или тому подобное.
Предпочтительно, когда ивабрадин формулы I преобразуют в фармацевитически приемлемые кислотно-аддитивные соли, используя смесь спирта и кислоты. В частности, получение гидрохлоридной соли осуществляют, используя спиртовой хлористый водород. Обычно раствор спиртового хлористого водорода получают, пропуская через спирт сухой хлористый водород, следуя описанным способам известного уровня техники. Процентное содержание хлористого водорода в спирте, предпочтительно, выбирают между 10 и 25%. Спирт, используемый в спиртовом растворе хлористого водорода, выбирают из C1-C4 разветвленных или линейных алифатических спиртов и более предпочтительно растворителем является метанол, этанол, н-бутанол или изопропанол, и наиболее предпочтительно растворителем является метанол и изопропанол. Осажденный ивабрадина гидрохлорид выделяют с высоким выходом, а степень чистоты по ВЭЖХ соответствует площади хроматографического пика, превышающей 99,0%. Рентгенограмма показывает, что вещество по природе является, по существу, аморфным, как изображено на фиг.1.
Аморфный ивабрадина гидрохлорид представляет собой новую форму ивабрадина и образует один аспект настоящего изобретения. Аморфный ивабрадина гидрохлорид далее характеризуют термограммой дифференциальной сканирующей калориметрии (DSC), на которой около 194°C показан один эндотермический пик, обусловленный плавлением.
Еще один аспект настоящего изобретения относится к получению аморфного ивабрадина гидрохлорида высокой степени чистоты из ивабрадина через кислотно-аддитивные соли ивабрадина. В частности, ивабрадин обрабатывают раствором органической кислоты в растворителе, выбираемом из низшего алифатического кетона, такого как ацетон; сложного эфира, такого как этилацетат, и нитрила, такого как ацетонитрил, с получением кислотно-аддитивной соли ивабрадина.
Органическая кислота может быть выбрана из уксусной кислоты, пропионовой кислоты, малеиновой кислоты, фумаровой кислоты, винной кислоты, щавелевой кислоты, лимонной кислоты, бензойной кислоты, метансульфоновой кислоты, изетионовой кислоты, бензолсульфоновой кислоты, толуолсульфокислоты и тому подобное. Предпочтительно, кислоту выбирают из щавелевой кислоты, лимонной кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты, толуолсульфокислоты. Наиболее предпочтительной кислотой является щавелевая кислота.
В частности, ивабрадина оксалат получают обработкой ивабрадина щавелевой кислотой в ацетоне, и реакционную массу перемешивают при температуре окружающей среды достаточное время для получения оксалатной соли. Оксалат отделяют фильтрованием и необязательно перекристаллизовывают в ацетонитриле для выделения чистого ивабрадина оксалата. Ивабрадина оксалат выделяют в виде кристаллического твердого вещества, и он может быть охарактеризован c помощью, по меньшей мере, одного из нижеследующих методов анализа: это анализ по Карлу Фишеру или термогравиметрический анализ (ТГА) (TGA), рентгеновская порошковая дифракциия (XRD) или дифференциальная сканирующая калориметрия (DSC).
Кристаллический ивабрадина оксалат характеризуют порошковыми рентгенограммами, имеющими пики при углах дифракции два тета приблизительно 2,04, 2,13, 4,26, 7,06, 8,02, 8,53, 9,32, 10,91, 13,63, 15,07, 16,11, 16,44, 17,48, 18,37, 19,32, 20,38, 20,94, 21,95, 23,61, 24,26, 27,54 и 33,07 градусов, как, по существу, изображено на фиг.3.
Кристаллический ивабрадина оксалат, кроме того, характеризуют термограммой диффернциальной сканирующей калориметрии (DSC), на которой около 110°C показан один эндотермический пик, обусловленный плавлением, как, по существу изображено на фиг.4.
Диффракцию рентгеновских лучей аморфного ивабрадина гидрохлорида и кристаллического ивабрадина оксалата измеряют на дифрактометре модели PANalytical X'Pert Pro, оборудованном медным (Cu) излучателем, и выражают в нижеследующих терминах: «два тета», «d-расстояния» и «относительные интенсивности». Специалист обычного уровня компетентности в данной области понимает, что экспериментальные различия могут возникать вследствие различий в аппаратуре, способах получения образцов или других факторов. DSC анализ осуществляли, используя прибор Mettler Toledo 822 Stare. Перед анализом тигель гофрировали и пробивали отверстие. Масса образцов приблизительно составляла 4-6 мг. Для образцов осуществляли развертку по температуре в интервале 30-250°C со скоростью 5°C/мин. Печь постоянно продували газообразным азотом при скорости потока 80 мл/мин. Использовали типовые 40 мкл алюминиевые тигли, закрытые крышками с одним отверстием.
Для удаления нежелательных примесей предпочтительно очищать кислотно-аддитивную соль ивабрадина известными методами, такими как кристаллизация или суспендирование в подходящем растворителе. Предпочтительно, когда выделенную кислотно-аддитивную соль ивабрадина перекристаллизовывают из подходящего органического растворителя, такого как ацетонитрил.
Чтобы получить ивабрадин, кислотно-аддитивную соль ивабрадина гидролизуют подходящим основанием в деминерализованной воде. Основание может быть выбрано из гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия, бикарбоната натрия и бикарбоната калия, и предпочтительно используют гидроксид натрия. После завершения гидролиза требуемое соединение экстрагируют в органическом растворителе. Органический растворитель может быть выбран из галогенированного углеводорода и алифатических сложных эфиров, и предпочтительно используют этилацетат. Этилацетат отгоняют для выделения ивабрадина в виде масла. Неочищенный ивабрадин далее растворяют в подходящем органическом растворителе, таком как ацетон, этилацетат, предпочтительно, в ацетоне. pH полученного прозрачного раствора устанавливают в интервале 1,0-2,0, используя спиртовой раствор хлористого водорода, и перемешивают в течение 30-60 минут с получением аморфного ивабрадина гидрохлорида высокой степени чистоты. Благоприятно высушивать органический слой перед добавлением спиртового раствора хлористого водорода, используя такой осушитель, как сульфат натрия или тому подобное.
Выделяют аморфный ивабрадина гидрохлорид, удаляя растворитель из реакционной смеси известными способами, такими как выпаривание, дистилляция в вакууме или без вакуума и так далее.
Затем аморфный ивабрадина гидрохлорид необязательно перемешивают в течение 30-60 минут в подходящем органическом растворителе, таком как н-гептан, н-гексан и циклогексан, затем фильтруют и промывают тем же органическим растворителем с получением аморфного ивабрадина гидрохлорида высокой степени чистоты.
В еще одном аспекте настоящего изобретения предлагают способ получения аморфного ивабрадина гидрохлорида, растворяя ивабрадина гидрохлорид в смеси низшего алканола и кетона при температуре окружающей среды; нагревая далее до 40-50°C, отгоняют растворитель в вакууме, и выделяют аморфный ивабрадина гидрохлорид.
Низший алканол может быть выбран из метанола, этанола, пропанола, изопропанола, и предпочтительно используют метанол. Кетон может быть выбран из ацетона, метилэтилкетона, метилизобутилкетона, и предпочтительно используют ацетон. Смесь низшего алканола и кетона используют в соотношении 1:(2-6) (об/об), более предпочтительно 1:3 (об/об) и наиболее предпочтительно 1:2 (об/об). В частности, ивабрадина гидрохлорид растворяют в смеси метанола и ацетона с помощью простого перемешивания при комнатной температуре без предварительного нагревания. Аморфный ивабрадина гидрохлорид отделяют, удаляя растворитель из рекционной смеси известными способами, такими как выпаривание, дистилляция в вакууме или без вакуума, и так далее. Альтернативно, выделение аморфного ивабрадина гидрохлорида высокой степени чистоты осуществляют посредством перемешивания аморфного ивабрадина гидрохлорида в подходящем органическом растворителе, таком как н-гептан, н-гексан и циклогексан в течение приблизительно 30-60 минут, с последующим фильтрованием и промыванием тем же органическим растворителем, с получением аморфного ивабрадина гидрохлорида высокой степени чистоты.
В еще одном аспекте настоящего изобретения аморфный ивабрадина гидрохлорид может быть получен из α-кристаллической формы ивабрадина гидрохлорида. В частности, кристаллический ивабрадина гидрохлорид обрабатывают подходящим основанием в подходящем растворителе, с последующей регенерацией ивабрадина гидрохлорида из раствора изопропиловый спирт/хлористый водород с получением аморфного ивабрадина гидрохлорида. Основание может быть выбрано из гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия, бикарбоната натрия и бикарбоната калия. Растворитель может быть выбран из этилацетата, изорпропилацетата, метилизобутилкетона и ацетона.
В еще одном аспекте настоящего изобретения может быть также получена α-кристаллическая форма ивабрадина гидрохлорида перекристаллизацией ивабрадина гидрохлорида в подходящем органическом растворителе, таком как ацетонитрил, с последующим добавлением низшего алифатического кетона, сложного эфира, неразветвленных, разветвленных или циклических простых эфиров или их смесей, нагреванием смеси, удалением растворителей и регенерированием α-кристаллической формы ивабрадина гидрохлорида.
В еще одном аспекте настоящего изобретения α- кристаллическая форма ивабрадина гидрохлорида может также быть получена из аморфного ивабрадина в подходящем органическом растворителе, таком как низший алифатический кетон, сложный эфир, неразветвленные, разветвленные или циклические простые эфиры или нитрилы, или их смеси. Низшие алифатические кетонные растворители могут быть выбраны из метилизобутилкетона, ацетона или тому подобного. Сложные эфиры могут быть выбраны из этилацетата и изопропилацетата. Простой эфир может быть выбран из изопропилового эфира, тетрагидрофурана и тому подобного. Нитрил может быть ацетонитрилом и тому подобным. Ивабрадина гидрохлорид помещают в подходящий растворитель и нагревают при 60-90°C достаточное время для того, чтобы преобразовать в α-кристаллическую форму ивабрадина гидрохлорида.
После описания изобретения со ссылками на некоторые предпочтительные варианты осуществления изобретения, для специалиста в данной области техники будут очевидны, принимая во внимание описание изобретения, другие варианты осуществления изобретения. Изобретение далее определяют с помощью ссылок на нижеследующие примеры, подробно описывающие получение продукта и способы использования изобретения. Специалистам в данной области техники будут очевидны многие модификации и веществ, и способов, которые могут быть осуществлены на практике без нарушения объема изобретения.
ПРИМЕРЫ
Пример 1: Получение ивабрадина гидрохлорида
Стадия 1: Получение (S)-7,8-диметокси-3-{3-{N-[(4,5-диметоксибензоциклобут-1-ил)метил]-N-(метил)амино)пропил)-1,3-дигидро-2H-3-бензазепин-2-она
При комнатной температуре в смесь (S)-N-[(4,5-диметоксибензоциклобут-1-ил)метил]-N-(метил)амина (42 г) и N,N-диметилформамида (220 мл) загружали 7,8-диметокси-3-[3-йодпропил]-1,3-дигидро-2H-3-бензазепин-2-он (75 г) и карбонат калия (42 г). Реакционную смесь нагревали, и 2 часа перемешивали реакционную массу при 50-55°C. Завершение реакции контролировали с помощью ВЭЖХ/ТСХ. После завершения реакции реакционную массу охлаждали до 25-30°C и разбавляли деминерализованной водой (1000 мл). Рекционную смесь экстрагировали метилендихлоридом (400+200 мл), и слои разделяли. Метиленхлорид отгоняли полностью. К остатку добавляли деминерализованную воду (200 мл) и хлористоводородную кислоту (50 мл), и водный раствор промывали этилацетатом (200 мл ×3). Слои разделяли, и к водному слою при 25-30°C добавляли 50% (м/об) раствор гидроксида натрия (120 мл). Водный слой экстрагировали этилацетатом (400+200 мл), и объединенный этилацетатный слой промывали 5% (м/об) раствором гидроксида натрия (300 мл). Этилацетатный слой сушили над безводным сульфатом натрия, и затем этилацетат полностью отгоняли в вакууме с получением соединения, указанного в названии.
Стадия 2: Получение (S)-7,8-диметокси-3-{3-{N-[(4,5-диметоксибензоциклобут-1-ил)метил]-N-(метил)амино)пропил)-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он гидрохлорида
Соединение бензазепина (85 г), полученное на стадии 1, помещали в уксусную кислоту (700 мл), и гидрировали при комнатной температуре при давлении водорода (1-2 кг), в присутствии Pd/C (10%, 70 г). Далее гидрирование продолжали в течение 6-8 часов при 20°C и давлении газообразного водорода 6-7 кг/см2. После завершения гидрирования (контролируемого по ВЭЖХ), катализатор отфильтровывали, и катализатор промывали водой (800 мл). Затем фильтрат экстрагировали метиленхлоридом (700 мл), и к водному слою добавляли раствор хлористоводородной кислоты (50 мл). Водный слой снова экстрагировали метилендихлоридом (300 мл ×3). Объединенный органический слой отгоняли, и к остатку добавляли деминерализованную воду (400 мл) и раствор хлористоводородной кислоты (90 мл), с последующим добавлением этилацетата (400 мл). Реакционную смесь полчаса перемешивали при 30-35°C, и слои разделяли. Водный слой снова промывали этилацетатом (500 мл). После этого водный слой обрабатывали 50% (м/об) раствором гидроксида натрия (150 мл). Водный слой экстрагировали этилацетатом (400+250 мл); и объединенный этилацетатный слой промывали 5% (м/об), раствором гидроксида натрия (400 мл). Этилацетатный слой сушили над безводным сульфатом натрия, и затем растворитель отгоняли полностью. Остаток помещали в этилацетат (400 мл), и к нему медленно добавляли раствор изопропиловый спирт/хлористый водород (50 мл), и перемешивали 4-5 часов. Полученный продукт фильтровали, промывали этилацетатом (85 мл) и сушили при 55-60°C с получением соединения, указанного в названии, в аморфной форме с чистотой, которая соответствует площади хроматографического пика 98,5% по ВЭЖХ.
Пример 2: Получение бензазепинового промежуточного соединения формулы IV
7,8-диметокси-3-[3-йодпропил]-1,3-дигидро-2H-3-бензазепин-2-он (38 г) и карбонат калия (60 г) при комнатной температуре добавляли к смеси (S)-N-[(4,5-диметоксибензоциклобут-1-ил)метил]-N-(метил)амина (20 г) и деминерализованной воды (100 мл). Реакционную смесь нагревали, и 12-16 часов перемешивали при 50-55°C. После завершения реакции реакционную массу охлаждали до 25-30°C, и продукт экстрагировали в этилацетате (100 мл). Водный слой дополнительно экстрагировали этилацетатом (60 мл). Объединенные слои этилацетата подкисляли водной хлористоводородной кислотой и перемешивали. Слои разделяли, и pH водного слоя устанавливали 10,5-12,5 с помощью водного раствора гидроксида натрия. Водный слой экстрагируют этилацетатом (140+60 мл). Этилацетат полностью отгоняли в вакууме с получением соединения, указанного в названии.
Пример 3: Получение аморфного ивабрадина гидрохлорида
Бензазепиновое промежуточное соединение формулы IV (85 г) помещали в уксусную кислоту (700 мл), и гидрировали при комнстной температуре при давлении водорода (1-2 кг), в присутствии Pd/C (10%, 70 г). Затем, в течение 6-8 часов гидрирование продолжали при 20°C при давлении газообразного водорода 6-7 кг/см2. После завершения гидрирования (контроль осуществляли по ВЭЖХ) катализатор отфильтровывали, и уксусную кислоту удаляли перегонкой. К остатку добавляли деминерализованную воду (360 мл) и хлористоводородную кислоту (40 мл), с последующим добавлением этилацетата (200 мл). Полученную смесь перемешивали, и слои разделяли. Водный слой промывали этилацетатом (100 мл). pH водного слоя устанавливали 10,5-12,5 с помощью водного раствора гидроксида натрия, и продукт экстрагировали в этилацетате (400 мл). Этилацетатный слой сушили над безводным сульфатом натрия, и затем растворитель полностью отгоняли. Остаток помещали в этилацетат (1020 мл) и к нему медленно добавляли щавелевую кислоту (34 г) в ацетоне (68 мл), и 3-4 часа перемешивали при температуре окружающей среды. Полученный ивабрадина оксалат фильтровали и перекристаллизовывали в ацетонитриле. Ивабрадина оксалат помещали в деминерализованную воду (340 мл), и pH рекционной смеси устанавливали 10-12 с помощью водного раствора гидроксида натрия и перемешивали. Затем полученную смесь экстрагируют этилацетатом (340 мл). Этилацетат отгоняли полностью. После чего остаток помещали в ацетон (220 мл) и медленно добавляли метанол/хлористый водород (30-35 мл) и перемешивали 30 минут. Растворитель удаляли перегонкой при пониженном давлении. Затем аморфный продукт, полученный таким образом, перемешивали с н-гептаном в течение 30-40 минут, фильтровали, промывали н-гептаном (50 мл) и сушили при 40-45°C с получением аморфного ивабрадина гидрохлорида со степенью чистоты, которая соответствует площади хроматографического пика 99,9% в методе высокоэффективной жидкостной хроматографии (ВЭЖХ). Из рентгенограммы следует, что продукт имел аморфную природу, как изображено на фиг.1.
Пример 4: Получение аморфного ивабрадина гидрохлорида
Стадия-1: Получение ивабрадина оксалата
Ивабрадин (44 г) растворяли в этилацетате (700 мл) и медленно добавляли щавелевую кислоту (22 г) в ацетоне (50 мл), и 3-4 часа перемешивали при температуре окружающей среды. Ивабрадина оксалат, полученный таким образом, фильтровали, промывали этилацетаом (100 мл) и перекристаллизовывали в ацетонитриле (350 мл). Из рентгенограммы следует, что выделенный ивабрадина оксалат имел кристаллическую природу, что показано на фиг.2.
Стадия-2: Получение аморфного ивабрадина гидрохлорида
Ивабрадина оксалат помещали в деминерализованную воду (200 мл), и добавляли водный раствор гидроксида натрия (100 мл) и перемешивали. Полученную смесь экстрагировали этилацетатом (250 мл). Этилацетатный слой сушили над безводным сульфатом натрия, и затем растворитель полностью отгоняли. Полученный остаток помещали в ацетон (100 мл), медленно добавляли метанол/хлористый водород (7 мл) и 30 минут перемешивали. После этого растворитель удаляли дистилляцией при пониженном давлении. К аморфному продукту добавляли н-гептан (100 мл) и 30 минут перемешивали, фильтровали, промывали н-гептаном (50 мл) и сушили при 40-45°C с получением соединения, указанного в названии.
Пример 5: Получение аморфного ивабрадина гидрохлорида
Ивабрадина гидрохлорид (20 г) растворяли в перемешанной смеси ацетона (100 мл) и метанола (50 мл) при температуре окружающей среды. Раствор нагревали до 50°C, и растворитель отгоняли при пониженном давлении. Аморфный продукт, полученный таким образом, перемешивали с н-гептаном (100 мл). Рекционную смесь фильтровали, промывали с н-гептаном и сушили при 40-45°C в вакууме с получением соединения, указанного в названии.
Пример 6: Получение аморфного ивабрадина гидрохлорида
α-кристаллическую форму ивабрадина гидрохлорида (6 г) помещали в воду (30 мл) и подщелачивали раствором гидроксида натрия (50%). Полученный таким путем ивабрадин экстрагировали этилацетатом (50 мл) и сушили над безводным сульфатом натрия. Растворитель отгоняли. Остаток помещали в этилацетат (30 мл), медленно добавляли раствор изопропиловый спирт/хлористый водород (5 мл) и 2 часа перемешивали. Продукт, полученный таким образом, фильтровали, промывали этилацетатом (6 мл) и сушили при 55-60°C с получением аморфного ивабрадина гидрохлорида, имеющего чистоту, которая по ВЭЖХ соответствует площади хроматографического пика 99,93%.
Пример 7: Получение α-кристаллической формы ивабрадина гидрохлорида
Помещали ивабрадина гидрохлорид (47 г) в ацетонитрил (940 мл) и 30 минут нагревали до 80±2°C до получения прозрачного раствора. После этого половину ацетонитрила отгоняли, и реакционную массу охлаждали, фильтровали и промывали ацетонитрилом с получением влажного осадка продукта на фильтре.
Затем полученный влажный осадок на фильтре растворяли в этилацетате (611 мл) при комнатной температуре, затем смесь нагревали до 75-80°C и сохраняли 60 минут. Рекционную смесь концентрировали, отгоняя приблизительно одну треть этилацетата. Реакционную массу охлаждали, фильтровали, промывали этилацетатом и сушили в вакууме с получением соединения, указанного в названии.
Пример 8: Получение α-кристаллической формы ивабрадина гидрохлорида из аморфной формы
Аморфный ивабрадина гидрохлорид (4 г) помещали в этилацетат (60 мл) и нагревали 30 минут до температуры образования флегмы. После этого одну треть этилацетата отгоняли, и реакционную массу охлаждали до 25-30°C. Полученный продукт фильтровали и сушили с получением соединения, указанного в названии, имеющее чистоту, которая соответствует площади хроматографического пика 99,89% по ВЭЖХ.
Пример 9: Получение α-кристаллической формы ивабрадина гидрохлорида
Ивабрадина гидрохлорид (2 г) помещали в ацетон (200 мл) и нагревали до температуры образования флегмы до получения прозрачного раствора, суспендированное вещество отфильтровывали. После этого отгоняли 60% ацетона, и реакционную массу охлаждали до комнатной температуры. Продукт фильтровали, промывали ацетоном (10 мл) и сушили с получением соединения, указанного в названии, имеющего чистоту, которая соответствует площади хроматографического пика 99,73% по ВЭЖХ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА ИЛИ БЕНЗОТИАЗЕПИНА | 1989 |
|
RU2090562C1 |
| ФОРМА IV ИВАБРАДИНА ГИДРОХЛОРИДА | 2012 |
|
RU2619121C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2026296C1 |
| ФОРМА VI АТОРВАСТАТИНА КАЛЬЦИЯ ИЛИ ЕЕ ГИДРАТЫ | 2002 |
|
RU2294924C2 |
| О-АРИЛЬНЫЕ ЭФИРЫ МОРФИНАНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2123000C1 |
| СИНТЕЗ 2-МЕТИЛ-4-(4-МЕТИЛ-1-ПИПЕРАЗИНИЛ)-10H-ТИЕНО[2,3-b] [1,5]БЕНЗОДИАЗЕПИНА И ЕГО СОЛЕЙ | 2005 |
|
RU2435775C2 |
| Способ получения меркаптозамещенных 2,3,4,5-тетрагидро-1 @ -3-бензазепинов или их солей | 1979 |
|
SU1029827A3 |
| ПОЛУЧЕНИЕ 2-(5-БРОМ-4-(4-ЦИКЛОПРОПИЛНАФТАЛИН-1-ИЛ)-4H-1,2,4-ТРИАЗОЛ-3-ИЛТИО)УКСУСНОЙ КИСЛОТЫ | 2013 |
|
RU2666549C2 |
| (3-ЦИКЛОАЛКИЛ-2,3,4,5-ТЕТРАГИДРО-1Н-БЕНЗО[d]АЗЕПИН-7-ИЛОКСИ)ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ Н3 РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ | 2003 |
|
RU2388752C2 |
| СОЕДИНЕНИЯ | 2003 |
|
RU2327690C2 |
Изобретение относится к кристаллической форме ивабрадина оксалата, характеризующейся дифракционными пиками на рентгенограмме, получаемой порошковой дифракцией рентгеновских лучей при углах 2θ приблизительно 2,04, 2,13, 4,26, 7,06, 8,02, 8,53, 9,32, 10,91, 13,63, 15,07, 16,11, 16,44, 17,48, 18,37, 19,32, 20,38, 20,94, 21,95, 23,61, 24,26, 27,54 и 33,07 градусов; или термограммой, получаемой методом дифференциальной сканирующей калориметрии, демонстрирующей один эндотермический пик около 110°С. Также изобретение относится к способу получения заявленной кристаллической формы. Технический результат - обеспечение кристаллической формы ивабрадина оксалата, применяемой в качестве средства против стенокардии и в качестве промежуточного соединения для получения аморфного ивабрадина гидрохлорида. 2 н. и 4 з.п. ф-лы, 4 ил., 9 пр.
1. Кристаллический ивабрадина оксалат, отличающийся, по меньшей мере, одной из нижеследующих характеристик:
a) дифракционными пиками на рентгенограмме, получаемой порошковой дифракцией рентгеновских лучей при углах 26 приблизительно 2,04, 2,13, 4,26, 7,06, 8,02, 8,53, 9,32, 10,91, 13,63, 15,07, 16,11, 16,44, 17,48, 18,37, 19,32, 20,38, 20,94, 21,95, 23,61, 24,26, 27,54 и 33,07 градусов; или
b) термограммой, получаемой методом дифференциальной сканирующей калориметрии, демонстрирующей один эндотермический пик около 110°С.
2. Кристаллический ивабрадина оксалат по п.1, отличающийся дифракционными пиками на рентгенограмме при углах приблизительно 4,26, 7,06, 8,02, 8,53, 9,32, 10,91, 13,63, 15,07, 16,11, 16,44, 17,48, 18,37, 19,32, 20,38, 20,94, 23,61 и 27,54 градусов 2θ.
3. Способ получения кристаллического ивабрадина оксалата по п.1, включающий стадии:
a) обработку ивабрадина раствором щавелевой кислоты в органическом растворителе с получением ивабрадина оксалата; и
b) необязательно, очистку ивабрадина оксалата с использованием подходящего растворителя.
4. Способ по п.3, в котором на стадии а) органический растворитель выбирают из группы, состоящей из низшего алифатического кетона, сложного эфира и нитрила.
5. Способ по п.3, в котором на стадии а) органический растворитель выбирают из группы, состоящей из низшего алифатического кетона, такого как ацетон; сложного эфира, такого как этилацетат, и нитрила, такого как ацетонитрил.
6. Способ по п.3, в котором на стадии b) растворитель представляет собой ацетонитрил.
| US 5296482 А, 22.03.1994 | |||
| Котел для центрального отопления | 1927 |
|
SU7745A1 |
| Konno Tsutomu: "Physical and chemical changes of medicinals in mixtures with adsorbents in the solid state | |||
| IV | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |

