Область техники, к которой относится изобретение
Настоящее изобретение принадлежит к области органической химии и относится к новому способу очистки 2-метил-4-(4-метил-1-пиперазинил)-10Н-тиено[2,3-b][1,5]бензодиазепина (оланзапина), включающему получение кислотно-аддитивных солей оланзапина и их преобразование в фармацевтически приемлемый чистый и обесцвеченный конечный продукт. Настоящее изобретение относится также к способу получения чистого оланзапина.
Предпосылки создания изобретения
Оланзапин является фармацевтически активным веществом, относящимся к группе нейролептических средств, применяемых для лечения различных психических заболеваний и состояний, таких как, например, расстройства центральной нервной системы, шизофрения, галлюцинации, острый маниакальный синдром, депрессия и им подобные.
С химической точки зрения он принадлежит к группе бензодиазепинов и является 2-метил-4-(4-метил-1-пиперазинил)-10H-тиено[2,3-b][1,5]бензодиазепином (формула 1):
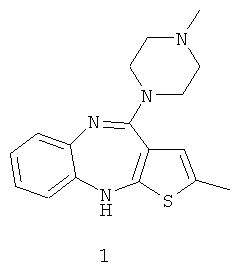
Оланзапин и его аналоги были впервые охвачены общей формулой в патенте Великобритании GB 1,533,235. Но даже если эта ссылка раскрывает общий способ алкилирования для синтеза замещенных по положению 4 производных, метилирование подобного рода не было упомянуто явным образом. Алкилирование осуществляли в этаноле с использованием триэтиламина в качестве основания и R'-Cl в качестве алкилирующего агента, где R' представлял собой различные радикалы.
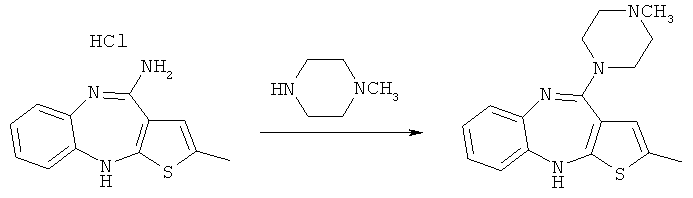
В патенте Великобритании GB 1,533,235 раскрываются два пути синтеза с общими формулами, подробно описанные в основном патенте, европейском патенте ЕР 454436 В1. В европейском патенте ЕР 454436 В1 раскрываются два различных одностадийных процесса получения оланзапина. Первый из описанных процессов представляет собой реакцию гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина с N-метилпиперазином в органическом растворителе, таком как анизол, толуол, диметилформамид или диметилсульфоксид, предпочтительно при температуре от 100 до 150°С, приводящую к оланзапину (схема 1):
Схема 1
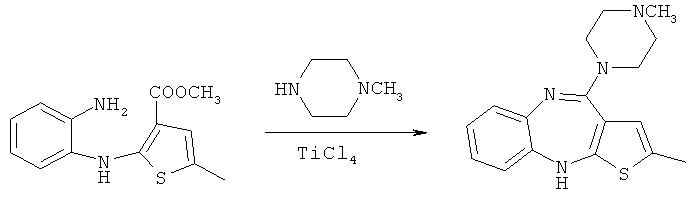
Второй способ, раскрываемый в европейском патенте ЕР 454436 В1, представляет собой реакцию N-метилпиперазина с метил-2-(2-аминоанилино)-5-метилтиофен-3-карбоксилатом в присутствии тетрахлорида титана (схема 2):
Схема 2
В том же патенте упоминается также образование кислотно-аддитивных солей оланзапина и потенциальная возможность их применения в качестве промежуточных соединений в способах очистки оланзапина и в его фармацевтических применениях. Однако ни одна из таковых кислотно-аддитивных солей не была получена или охарактеризована и никакой способ или опыт, использующий какую-либо из таковых кислотно-аддитивных солей, не был раскрыт.
Главной проблемой, связанной с обоими способами синтеза, является окрашивание конечного продукта вследствие использования химии тиофеновых соединений.
Как это раскрывается в европейском патенте ЕР 454436 В1 и его патенте-аналоге, патенте США US 5229382, оланзапин, получаемый согласно первому способу синтеза (схема 1), подвергается после этого очистке посредством перекристаллизации из ацетонитрила, тогда как оланзапин, получаемый согласно второму пути синтеза (схема 2), подвергается после этого очистке с помощью колоночной хроматографии на флорисиле и перекристаллизации из ацетонитрила.
Проблема окрашивания оланзапина хорошо известна и уже сообщалась в европейском патенте ЕР 733635 В1, который ссылается на патент США №5,229,382. В обеих ссылках было показано, что оланзапин может продолжать содержать следы нежелательной окраски даже после очистки с помощью активированного угля. В европейском патенте ЕР 733635 В1 авторы изобретения пробовали решить проблему окрашивания с помощью получения новой кристаллической модификации оланзапина.
Другие способы синтеза для получения оланзапина были описаны в предшествующих работах. Например, международная заявка на изобретение WO 04/000847 раскрывает двухстадийный синтез исходя из гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина через посредство образующегося на промежуточной стадии 2-метил-4-(1-пиперазинил)-10H-тиено[2,3-b][1,5]бензодиазепина (т.е. N-дезметилоланзапина) с последующим восстановительным метилированием по атому азота (с помощью формальдегида и боргидрида металла). Международная заявка на изобретение WO 04/000847 сообщает, что процесс метилирования осуществляют в метаноле. Недостатками способа, раскрываемого в международной заявке на изобретение WO 04/000847, являются низкие выходы и плохое качество конечного продукта.
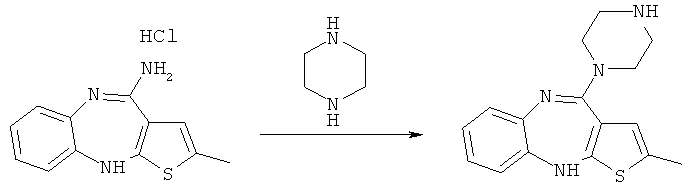
Реакция гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина с пиперазином, приводящая к N-дезметилоланзапину (схема 3), была опубликована в Bioorganic & Medicinal Chemistry Letters, т.7, №1, стр.25-30, 1997. В качестве растворителя использовали смесь диметилсульфоксида и толуола в соотношении 1/4:
Схема 3
Международная заявка на изобретение WO 04/089313 раскрывает кислотно-аддитивные соли, сольваты и сокристаллизованные формы оланзапина и их применение в качестве фармацевтически активного ингредиента в фармацевтическом составе. Получение кислотно-аддитивных солей оланзапина с фумаровой, малеиновой и малоновой кислотами раскрывается в международной заявке на изобретение WO 04/089313. Раскрываемые в данной заявке на изобретение кислотно-аддитивные соли оланзапина демонстрируют удельную растворимость в воде в пределах от 50 мкг/мл до 100 мг/мл.
Специалисту в соответствующей области хорошо известно, что большинство химических реакций протекают не до конца, могут быть обратимы или протекают одновременно с какими-либо другими параллельными реакциями. Исходные вещества или продукты побочных реакций часто обнаруживают в качестве примесей в выделенном основном продукте, который вследствие этого должен быть подвергнут дополнительной очистке. Простейший способ очистки включает различные процедуры перекристаллизации и осаждения, которые, как правило, менее эффективны в тех случаях, когда примеси характеризуются физико-химическими свойствами, очень похожими на свойства основного продукта.
В том случае, когда оланзапин получают согласно одностадийным способам, раскрываемым в европейском патенте ЕР 454436 В1, исходное вещество 4-амино-2-метил-10Н-тиено[2,3-b][1,5]бензодиазепин может быть обнаружено в конечном продукте, оланзапине, в качестве примеси.
Недостатком реакции, опубликованной в упомянутом выше обзоре в журнале Bioorganic & Medicinal Chemistry Letters, т.7, №1, стр.25-30, 1997, является темная окраска продукта.
В случае получения оланзапина с помощью двухстадийного способа, как это также описано в международной заявке на изобретение WO 04/000847, присутствие гидрохлорида 4-амино-2-метил-10Н-тиено[2,3-b][1,5]бензодиазепина не является критическим, но в качестве примесей могут быть обнаружены различные другие сходные продукты, такие как 4-(4-формил-пиперазинил)-2-метил-10Н-тиено[2,3-b][1,5]бензодиазепин и N-дезметилоланзапин. Для всех этих примесей, которые содержат тиенобензодиазепиновую циклическую систему в качестве части каркаса молекулы, таковая циклическая система, представляя существенную часть молекулы, играет определяющую роль в сходстве физико-химических свойств данных примесей и оланзапина.
Как теперь обнаружено, оланзапин не может быть эффективно отделен от своих в большой степени родственных примесей посредством многократной перекристаллизации неочищенного оланзапина.
Вследствие этого было бы желательно разработать альтернативный способ получения фармацевтически приемлемого чистого и обесцвеченного оланзапина.
Краткое изложение сущности изобретения
В настоящее время нами открыт новый и простой способ очистки оланзапина, включающий преобразование оланзапина в его кислотно-аддитивную соль, стадию разделения и последующее выделение оланзапина из его кислотно-аддитивной соли.
В первом варианте осуществления настоящего изобретения его объектом является способ очистки оланзапина, отличающийся тем, что он включает следующие стадии:
а) смешение оланзапина с органической кислотой в органическом растворителе или смеси органических растворителей с целью получения кислотно-аддитивной соли оланзапина,
б) осаждение и выделение кислотно-аддитивной соли оланзапина и
в) преобразование кислотно-аддитивной соли оланзапина в оланзапин.
В другом варианте осуществления настоящего изобретения его объектом является способ получения N-дезметилоланзапина, включающий введение гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина в реакцию с пиперазином в растворителе или в смеси растворителей, включающих по крайней мере один алифатический спирт, имеющий более высокую температуру кипения.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ синтеза 2-метил-4-(4-метил-1-пиперазинил)-10H-тиено[2,3-b][1,5]бензодиазепина (оланзапина) светлой окраски без темно-коричневых или зеленых оттенков, включающий метилирование по атому азота N-дезметилоланзапина с помощью метилирующего агента в необязательном присутствии сильного основания в органическом растворителе или в смеси органических растворителей.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения оланзапина в форме кислотно-аддитивной соли, включающий
а) смешение оланзапина с органической кислотой в растворителе или смеси растворителей и
б) осаждение и выделение кислотно-аддитивной соли оланзапина путем отделения кристаллов.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения оланзапина в форме кислотно-аддитивной соли, включающий следующие стадии: гидрохлорид 4-амино-2-метил-10Н-тиено[2,3-b][1,5]бензодиазепина вводят в реакцию с N-метилпиперазином с целью получения оланзапина и преобразуют получаемый оланзапин в его кислотно-аддитивную соль.
Еще в одном варианте осуществления настоящего изобретения его объектом является оланзапин в форме кислотно-аддитивной соли, причем таковая соль выбрана из группы, включающей соли бензойной кислоты и сульфокислот.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения оланзапина из его кислотно-аддитивной соли путем выделения оланзапина из таковой кислотно-аддитивной соли.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения кристаллической модификации I оланзапина исходя из кислотно-аддитивной соли оланзапина, в котором кристаллы выделяют из органического растворителя.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения кристаллической модификации II оланзапина исходя из кислотно-аддитивной соли оланзапина, в котором кристаллы выделяют из органического растворителя.
Еще в одном варианте осуществления настоящего изобретения его объектом является применение органических кислот в способе получения оланзапина, в котором очистку оланзапина осуществляют через посредство образования его кислотно-аддитивной соли.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения производного N-дезметилоланзапина формулы 2, в которой R означает органический радикал, такой как ацетил, пропионил, хлорацетил и им подобные, включающий введение N-дезметилоланзапина в реакцию с органической кислотой, или замещенной органической кислотой, или производным органической кислоты формулы RX, или с ангидридом органической кислоты. Указанное RX соответствует производному органической кислоты, причем особенно предпочтительным является галогенангидрид органической кислоты, такой как галогенангидрид уксусной кислоты, галогенангидрид пропионовой кислоты, галогенангидрид хлоруксусной кислоты и им подобные, причем Х выбран из группы, включающей Cl, Br или I, особенно же предпочтительным является Cl. В качестве ангидрида органической кислоты может быть применен уксусный ангидрид, пропионовый ангидрид, ангидрид фталевой кислоты и им подобные:
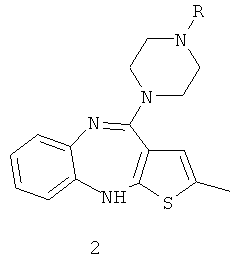
Еще в одном варианте осуществления настоящего изобретения его объектом является оланзапин, полученный исходя из N-дезметилоланзапина с помощью способа метилирования, который приводит к содержанию N-дезметилоланзапина в конечном продукте оланзапине не более 0,1%.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения оланзапина в форме кислотно-аддитивной соли, включающий следующие стадии:
а) введение N-дезметилоланзапина в реакцию с метилирующим агентом с целью получения оланзапина,
б) разбавление получаемой реакционной смеси водой и подкисление ее кислотой,
в) добавление к реакционной смеси органического растворителя и разделение фаз,
г) нейтрализацию получаемой водной фазы и экстракцию оланзапина органическим растворителем с целью получения фазы органического растворителя и
д) добавление к органической фазе с целью получения производного N-дезметилоланзапина формулы 2, замещенного по атому азота, органической кислоты, или замещенной органической кислоты, или производного органической кислоты определенной выше формулы RX, в которой R представляет собой органический радикал, такой как ацетил, пропионил, хлорацетил, а Х выбран из группы, включающей Cl, Br или I, причем особенно предпочтительным является Cl, или ангидрида органической кислоты, как он определен выше,
е) необязательное выпаривание органического растворителя и разбавление остатка вторым органическим растворителем,
ж) добавление органической кислоты либо к получаемому разбавленному раствору либо непосредственно к содержащему оланзапин экстракту, получаемому в результате упомянутой выше экстракции на стадии г), и
з) выделение осажденной кислотно-аддитивной соли оланзапина путем отделения кристаллов.
Еще в одном варианте осуществления настоящего изобретения его объектом является оланзапин, получаемый исходя из N-дезметилоланзапина с помощью способа метилирования по атому азота, который содержит менее 0,05% пиперазин-1,4-бис-4-ил-(2-метил)-10Н-тиено-[2,3-b][1,5]бензодиазепина.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения оланзапина, включающий следующие стадии:
а) преобразование гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]-бензодиазепина в 2-метил-4-(1-пиперазинил)-10H-тиено-[2,3-b][1,5]бензодиазепин,
б) преобразование 2-метил-4-(1-пиперазинил)-10H-тиено[2,3-b][1,5]бензодиазепина в неочищенный оланзапин,
в) преобразование неочищенного оланзапина в его кислотно-аддитивную соль и
г) преобразование кислотно-аддитивной соли оланзапина в оланзапин.
Еще в одном варианте осуществления настоящего изобретения его объектом является фармацевтическая композиция, включающая 2-метил-4-(4-метил-1-пиперазинил)-10Н-тиено[2,3-b][1,5]бензодиазепин (оланзапин), причем оланзапин получен исходя из своей кислотно-аддитивной соли.
Еще в одном варианте осуществления настоящего изобретения его объектом является применение оланзапина, полученного согласно одному из способов, раскрываемых в настоящем изобретении, для изготовления лекарственного средства для лечения различных психических заболеваний и состояний.
Еще в одном варианте осуществления настоящего изобретения его объектом является фармацевтический состав, включающий по крайней мере один фармацевтически приемлемый ингредиент и оланзапин, полученный согласно одному из способов, раскрываемых в настоящем изобретении.
Еще в одном варианте осуществления настоящего изобретения его объектом является применение оланзапина, полученного согласно одному из способов, раскрываемых в настоящем изобретении, для изготовления фармацевтического состава совместно с хотя бы одним фармацевтически приемлемым ингредиентом.
Подробное описание изобретения
Объектом настоящего изобретения является новый способ очистки оланзапина, включающий преобразование оланзапина в его кислотно-аддитивную соль, стадию разделения и выделение оланзапина из таковой кислотно-аддитивной соли.
Специалистам в соответствующей области известно, что образование кислотно-аддитивных солей вещества и их кристаллизация из раствора могут привести к успешной очистке упомянутого вещества от примесей, которые не способны образовывать кислотно-аддитивные соли, а также от примесей, которые могут образовывать подобные соли, но их свойства будут в существенной степени отличаться от таковых для упомянутого вещества. Было обнаружено, что оланзапин, загрязненный родственными ему в высокой степени примесями, может быть эффективно очищен посредством преобразования в свою кислотно-аддитивную соль, предоставляющую отличную возможность очистки, которая может быть осаждена из растворителей. В противоположность этому подобное невозможно в отношении самого оланзапина и некоторых других солей оланзапина, в частности, неорганических солей. Нами обнаружено, что подходящие органические кислоты, которые могут быть применены для получения кислотно-аддитивнных солей оланзапина, предоставляющих возможность очистки, являются карбоновые кислоты с хотя бы одной карбоксильной группой, такие как щавелевая, фумаровая и бензойная кислота, предпочтительно щавелевая кислота. Также могут применяться сульфокислоты.
Способ очистки оланзапина согласно настоящему изобретению включает следующие стадии:
а) смешение оланзапина с органической кислотой в органическом растворителе или смеси органических растворителей с целью получения кислотно-аддитивной соли оланзапина,
б) осаждение и выделение кислотно-аддитивной соли оланзапина и
в) преобразование кислотно-аддитивной соли оланзапина в оланзапин.
Предпочтительная органическая кислота на стадии а) выбрана из группы, включающей сульфокислоты или карбоновые кислоты. Предпочтительные карбоновые кислоты выбраны из группы, включающей щавелевую кислоту, фумаровую кислоту и бензойную кислоту.
Предпочтительный органический растворитель на стадии а) выбран из группы, включающей тетрагидрофуран, ацетон, диметилформамид и ацетонитрил.
Предпочтительная смесь органических растворителей на стадии а) является смесью тетрагидрофурана с по крайней мере одним полярным растворителем. Предпочтительный полярный растворитель выбран из группы, включающей диметилформамид, диметилацетамид, N-метилпирролидон, 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон, 1,3-диметил-2-имидазолидинон, тетраметилмочевину, диметилсульфоксид, сульфолан, ацетон и ацетонитрил.
Предпочтительный способ очистки согласно настоящему изобретению включает следующие подстадии в рамках стадии в):
1) растворение кислотно-аддитивной соли оланзапина в воде,
2) приведение рН получаемого раствора к приблизительно 8-10,
3) экстракцию оланзапина из водной фазы в фазу органического растворителя и
4) выделение кислотно-аддитивной соли оланзапина из фазы органического растворителя путем концентрирования раствора и отделения кристаллов.
В другом варианте осуществления настоящего изобретения его объектом является способ получения оланзапина в форме кислотно-аддитивной соли, отличающийся тем, что он включает следующие стадии:
а) смешение оланзапина с органической кислотой, предпочтительно выбранной из группы, включающей бензойную кислоту и сульфокислоты, в растворителе или смеси растворителей и
б) осаждение и выделение кислотно-аддитивной соли оланзапина путем отделения кристаллов.
Предпочтительно, указанные органический растворитель, смесь органических растворителей и полярный растворитель соответствуют тем же, что были описаны выше.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения оланзапина, предпочтительно в кристаллической форме, отличающийся тем, что последний получают исходя из кислотно-аддитивной соли оланзапина посредством выделения из таковой соли.
Предпочтительно, указанная стадия выделения включает подстадии в рамках стадии в), как они описаны выше.
Подходящие кислотные органические соединения, применяемые на стадии очистки, являются коммерчески доступными. С другой стороны, оланзапин как исходное вещество может быть синтезирован согласно способам синтеза, раскрытым до сего дня в предшествующих работах, например в европейском патенте ЕР 454436 В1, патенте США US 5.229.382, европейском патенте ЕР 733635 В1 или международной заявке на изобретение WO 04/089313.
В настоящее время нами открыты альтернативные способы получения оланзапина, который может быть необязательно подвергнут очистке согласно настоящему изобретению, как это было раскрыто выше.
Соответственно объектом настоящего изобретения также является новый способ синтеза чистого и обесцвеченного оланзапина исходя из гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина, протекающего с образованием N-дезметилоланзапина в качестве промежуточного вещества. Получаемый неочищенный оланзапин может быть впоследствии необязательно подвергнут способу очистки, в котором на первой стадии получают кислотно-аддитивную соль оланзапина, а затем ее выделяют. Примеси, образующиеся в процессе получения неочищенного оланзапина, остаются таким образом в растворе. На дальнейшей стадии очистки кислотно-аддитивную соль оланзапина легко преобразуют в чистый и фармацевтически приемлемый оланзапин без оттенка темного цвета.
Таковой способ очистки, протекающий через посредство преобразования оланзапина в его кислотно-аддитивную соль, может также быть применен к оланзапину, получаемому по одностадийному способу, в котором 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепин реагирует с N-метилпиперазином, что приводит к неочищенному оланзапину, который непосредственно подвергают способу очистки. Очищенный оланзапин, получаемый с помощью способа согласно настоящему изобретению, может быть в конечном итоге получен в различных кристаллических модификациях, таких как модификация I или модификация II.
Помимо вышеизложенного было обнаружено, что в тех случаях, когда кислотно-аддитивную соль оланзапина получают исходя из неочищенного оланзапина, полученного путем двухстадийного синтеза, протекающего через образование N-дезметилоланзапина в качестве промежуточного соединения, N-дезметилоланзапин может осаждаться из растворов в виде кислотно-аддитивной соли и остается в конечном продукте в качестве загрязняющей примеси. Однако, как было весьма неожиданно обнаружено, производные N-дезметилоланзапина, такие как ацетильные производные, не осаждаются из органических растворителей в виде кислотно-аддитивных солей и остаются в смеси после образования кислотно-аддитивной соли из неочищенного оланзапина. Подобным образом N-дезметилоланзапин может быть отделен от оланзапина. Данный способ очистки может быть очень эффективным, обеспечивая ограничение содержания всякой отдельной примеси в оланзапине фармацевтической чистоты на уровне ниже 0,1%, причем данный способ может быть особенно важен для удаления N-дезметилоланзапина, который может быть очень трудно отделить от оланзапина иными способами. Уровень примесей убывает в заметной степени даже в том случае, когда уровень примесей в неочищенном оланзапине высок.
Кроме того, нами обнаружено, что в ходе первой стадии двухстадийного способа синтеза оланзапина (т.е. реакции между 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепином и пиперазином) образуется побочный продукт, идентифицированный как димер исходного соединения, в котором две молекулы гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]-бензодиазепина связаны с атомами азота пиперазина. Химическое название данного побочного продукта - пиперазин-1,4-бис-4-ил-(2-метил)-10Н-тиено-[2,3-b][1,5]бензодиазепин (схема 4):
Схема 4

Анализ с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) показывает, что примесь упомянутого димера образуется в количестве приблизительно 1-4% по сравнению с основным продуктом N-дезметилоланзапином. Нами также обнаружено, что мольный избыток исходного пиперазина по отношению к гидрохлориду 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина в реакционной смеси приводит к меньшим выходам упомянутого димера.
В двухстадийном синтезе оланзапина согласно настоящему изобретению гидрохлорид 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина вводят в реакцию с пиперазином с целью получения N-дезметилоланзапина. Затем получают оланзапин посредством метилирования упомянутого N-дезметилоланзапина (схема 5):
Схема 5
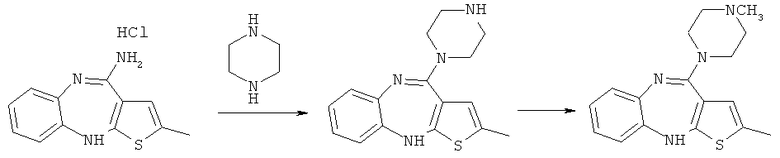
Как было неожиданно обнаружено, оланзапин искомого ярко-желтого цвета может быть получен исходя из исходного продукта темно-коричневой или зеленой окраски (т.е. гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина) в тех случаях, когда оланзапин получают с помощью двухстадийного синтеза согласно настоящему изобретению через посредство выделения N-дезметилоланзапина.
В европейском патенте ЕР 454436 В1 уже был раскрыт синтез оланзапина, включающий введение гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]бензодиазепина в реакцию с избытком N-метилпиперазина в смеси диметилсульфоксида (ДМСО) и толуола состава 1:4, каковая реакция относится к тому же типу (т.е. замещение аминогруппы N-метилпиперазином), что и раскрыт в настоящей заявке на изобретение на первой стадии (т.е. замещение аминогруппы пиперазином) (схема 4).
Нами было неожиданно обнаружено, что в реакции данного типа с точки зрения как скорости реакции, так и ее выхода лучше использовать по крайней мере один разветвленный или неразветвленный алифатический спирт, характеризуемый более высокой температурой кипения. Согласно настоящему изобретению, более высокая температура кипения означает температуру кипения, которая, предпочтительно, превосходит 100°С, более предпочтительно, превосходит 115°С. Предпочтительным алифатическим спиртом с более высокой температурой кипения является н-бутанол. В качестве альтернативы может быть использована смесь растворителей, содержащая хотя бы один разветвленный или неразветвленный алифатический спирт, характеризуемый более высокой температурой кипения, предпочтительно, н-бутанол, и хотя бы один растворитель, не относящийся к спиртам, характеризуемый более высокой температурой кипения. Предпочтительный растворитель, не относящийся к спиртам, характеризуемый более высокой температурой кипения, является ароматическим углеводородным растворителем, в частности ксилолом, толуолом, этилбензолом, анизолом или им подобными. Подходящей смесью является, например, смесь н-бутанола и ксилола или н-бутанола и толуола в соотношении между н-бутанолом и ароматическим углеводородом от 30/70 до 100/0, предпочтительно, в соотношении между 40/60 и 70/30.
Вместо использования избытка пиперазина к реакционной смеси могут быть добавлены дополнительные неорганические или органические основания. Предпочтительными основаниями являются третичные амины, такие как, например, триэтиламин, этилдиизопропиламин или диазабициклооктан.
В тех случаях, когда реакцию осуществляют в системе растворителей, включающей н-бутанол, N-дезметилоланзапин осаждается после добавления к реакционной смеси теплой воды. Получаемый N-дезметилоланзапин уже в существенной степени избавлен от темно-коричневой или зеленой окраски. В дальнейшем возможно избавиться от остающейся окраски путем промывания N-дезметилоланзапина органическим растворителем, таким как сложные эфиры, например этилацетат, изопропилацетат, бутилацетат и им подобные. Теплая вода имеет температуру между, приблизительно, 25°С и, приблизительно, 70°С, предпочтительно, между, приблизительно, 30°С и, приблизительно, 50°С. В результате применения теплой воды липкий комковатый осадок распадается на частицы меньшего размера. Таким образом могут быть достигнуты большее удобство фильтрования и лучшее качество (с точки зрения окраски) продукта.
Вторая стадия синтеза соответствует метилированию по атому азота пиперазиновой группы N-дезметилоланзапина (см. схему 6) с целью получения неочищенного оланзапина. Для метилирования могут быть применены различные метилирующие агенты, например диметилсульфат, или метиловые эфиры сульфокислот, такие как метиловый эфир толуолсульфокислоты, метиловый эфир метансульфокислоты, метиловый эфир трифторметансульфокислоты, или метилгалогениды, предпочтительно, метилиодид. Реакция может быть осуществлена в различных органических растворителях, таких как простые эфиры или циклические простые эфиры, например тетрагидрофуран, кетоны, например ацетон, амиды, например диметилформамид, нитрилы, например ацетонитрил, или спирты или смеси перечисленных растворителей с другими растворителями, предпочтительно, в смеси тетрагидрофурана с полярными растворителями. Подобными полярными растворителями являются амиды, такие как диметилформамид, диметилацетамид и N-метилпирролидон, производные мочевины, такие как 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон, 1,3-диметил-2-имидазолидинон и тетраметилмочевина, и другие растворители, такие как диметилсульфоксид, сульфолан, ацетон, ацетонитрил и им подобные. Подобные смеси тетрагидрофурана и полярных растворителей превосходят другие растворители, обеспечивая более высокое соотношение в продуктах реакции между оланзапином и неметилированными (т.е. N-дезметилоланзапин) и диметилированными продуктами, такими как N,N-диметилоланзапин (схема 6):
Схема 6
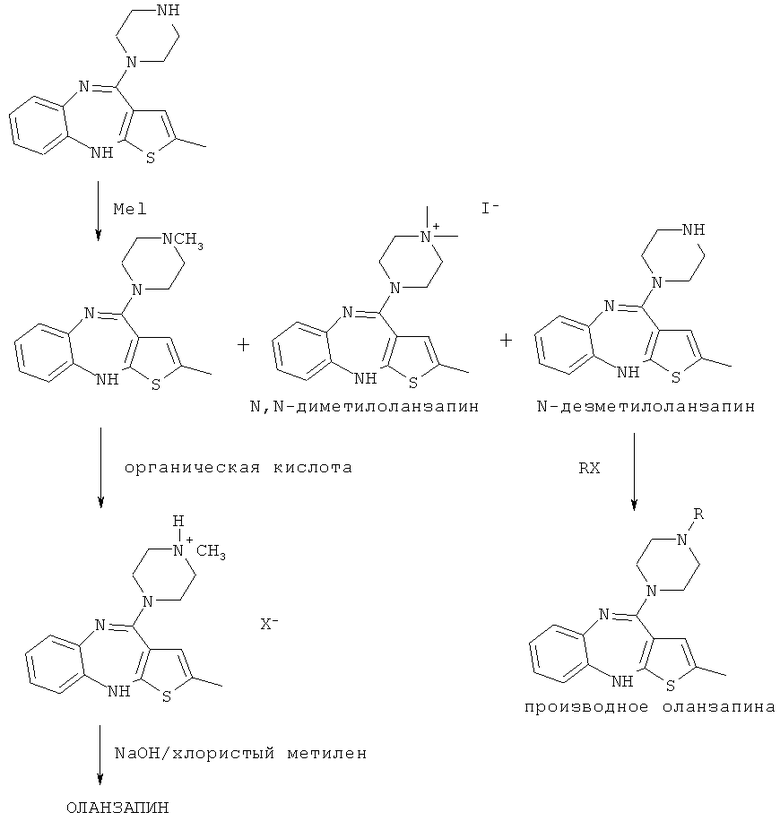
Для данной реакции метилирования предпочтительными являются щелочные условия. Могут быть применены различные амины, такие как триэтиламин, диизопропиламин, дициклогексиламин, этилдиизопропиламин и диазабициклооктан, или сильные основания, включающие щелочные или щелочноземельные металлы, такие как гидроксиды, гидриды или алкоголяты, например гидрид натрия, гидрид кальция, трет-бутилат калия, гидроксид натрия или калия, равно как и иные неорганические основания, такие как карбонат калия или натрия. В результате метилирования N-дезметилоланзапина в указанных условиях получают оланзапин ярко-желтого цвета без какого-либо коричневого или зеленого оттенка, который не нуждается в последующем обесцвечивании.
Как было раскрыто ранее, оланзапин должен быть отделен от N-дезметилоланзапина, остающегося в реакционной смеси после завершения реакции. Сообразно этому, реакционная смесь, получаемая после метилирования N-дезметилоланзапина, может быть сначала экстрагирована органическими растворителями, такими как простые эфиры, например диэтиловый эфир, сложные эфиры, например этилацетат, или, предпочтительно, хлорированными органическими растворителями, такими как хлористый метилен и хлороформ. После разделения фаз органическая фаза может быть промыта водой. После этого к органической фазе добавляют органическую кислоту или замещенную органическую кислоту, или производное органической кислоты, отвечающее формуле RX, как оно определено выше, или ангидрид органической кислоты, как он определен выше, с целью получения замещенного по атому азота производного N-дезметилоланзапина формулы 2:
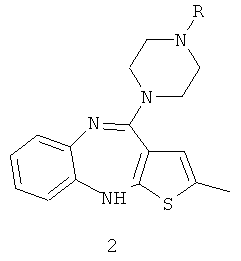
Подходящими реагентами, которые могут быть применены для осуществления этой реакции, являются органические кислоты, замещенные органические кислоты и производные органических кислот, такие как хлоруксусная кислота, хлорэтиламин, бензилбромид, фталевый ангидрид, уксусный ангидрид и им подобные. Для осуществления реакции, в которой образуется вышеупомянутое замещенное по атому азота производное N-дезметилоланзапина формулы 2, могут быть применены различные амины, такие как дициклогексиламин, диизопропиламин, триэтиламин, диизопропилэтиламин, диазабициклооктан, этилендиамин, изопропиламин, бутиламин, диэтиламин, дипропиламин, пропиламин, дибутиламин и им подобные, а также различные неорганические основания, такие как K2CO3, Na2CO3, NaOH, KOH, LiOH, Са(ОН)2, NaH и им подобные.
В качестве заключительной стадии оланзапин может быть необязательно подвергнут очистке путем преобразования в свою кислотно-аддитивную соль согласно изобретению, как это описано выше. В этом случае к реакционной смеси добавляют органическую кислоту, такую как сульфокислоты или карбоновая кислота, предпочтительно, щавелевая, фумаровая или бензойная кислота и им подобные, более предпочтительно, щавелевую кислоту, с целью получения кислотно-аддитивной соли оланзапина, которая может быть осаждена из смеси после охлаждения и может быть отфильтрована. Растворимость в воде получаемой кислотно-аддитивной соли оланзапина со щавелевой кислотой приблизительно достигает около 800 мг/мл.
Кислотно-аддитивная соль оланзапина может быть впоследствии преобразована в чистый оланзапин в кристаллических модификациях I или II путем растворения кислотно-аддитивной соли оланзапина в воде и приведения рН к, приблизительно, 1,0-5,0, предпочтительно, к 2,0, посредством добавления соляной кислоты. К получаемому таким образом раствору добавляют активированный уголь. После перемешивания смеси в течение, приблизительно, 5 минут активированный уголь отфильтровывают и промывают осадок на фильтре водой. Фильтрат и промывную воду объединяют, а затем добавляют низкокипящий органический растворитель, такой как диэтиловый эфир, хлористый метилен, хлороформ, этилацетат и им подобные, предпочтительно, хлористый метилен. Обработка реакционной смеси активированным углем при рН около 2 соответствует окончательной очистке оланзапина и позволяет избавиться от вышеописанного димера (пиперазин-1,4-бис-4-ил-(2-метил)-10Н-тиено-[2,3-b][1,5]бензодиазепина, см. схему 4). На следующей стадии добавляют основание, предпочтительно, неорганическое основание, такое как аммиак, K2CO3, КОН, NaH, Na2CO3, LiOH, Са(ОН)2 и им подобные, более предпочтительно, NaOH, с целью приведения рН к, приблизительно, 7-11, предпочтительно, к, приблизительно, 9-10. После достижения требуемой величины рН смесь может быть экстрагирована низкокипящим органическим растворителем, причем могут быть применены различные растворители, такие как простые эфиры, например диэтиловый эфир, хлорированные углеводороды, например хлористый метилен, хлороформ, сложные эфиры, например этилацетат и им подобные, предпочтительно, хлористый метилен. После экстракции органический растворитель может быть частично отогнан на роторном испарителе, а оставшаяся смесь может быть охлаждена до температуры от, приблизительно, -20°С до, приблизительно, 0°С, предпочтительно, от, приблизительно, -15°С до, приблизительно, -5°С, что приводит к осаждению кристаллической модификации I оланзапина. Получаемый оланзапин содержит крайне низкое количество вышеупомянутого димера (пиперазин-1,4-бис-4-ил-(2-метил)-10Н-тиено-[2,3-b][1,5]бензодиазепина), как то, например, менее 0,05%.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения кристаллической модификации II, в котором участвующий в самой последней стадии органический растворитель полностью отгоняют на роторном испарителе (а не ограничиваются частичной отгонкой, как это указано в предыдущем абзаце), а затем добавляют органический растворитель. Для получения раствора оланзапина, из которого после охлаждения может быть осаждена кристаллическая модификация II оланзапина, могут быть применены различные растворители, такие как простые эфиры, например диэтиловый эфир, нитрилы, например ацетонитрил, сложные эфиры, например этилацетат, и им подобные, предпочтительно, диэтиловый эфир.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения кристаллической модификации I оланзапина через посредство кислотно-аддитивной соли оланзапина. Для получения неочищенного оланзапина применяют реакцию, описанную в европейском патенте ЕР 454436 В1. Реакция включает взаимодействие N-метилпиперазина с гидрохлоридом 4-амино-2-метил-10Н-тиено[2,3-b][1,5]-бензодиазепина. Реакцию осуществляют в присутствии высококипящего органического растворителя, такого как диметилсульфоксид, диметилацетамид, бутанол, диметилформамид, толуол, ксилол, этилбензол, анизол и им подобные, предпочтительно, диметилсульфоксида, при температуре в пределах, приблизительно, 80-150°С, предпочтительно, приблизительно 115-130°С.
Согласно настоящему изобретению, после обработки водой получаемого таким образом раствора, содержащего неочищенный оланзапин, последний экстрагируют из получаемого раствора с помощью органического растворителя, причем могут быть использованы различные растворители, например кетоны, такие как метилизобутилкетон, хлорированные углеводороды, такие как хлористый метилен и хлороформ, предпочтительно, хлористый метилен. После этого добавляют органическую кислоту, такую как карбоновая кислота, например щавелевая, фумаровая, бензойная кислота ли сульфокислоты и им подобные, с целью получения кислотно-аддитивной соли оланзапина. Кислотно-аддитивная соль оланзапина может быть отфильтрована и необязательно растворена в воде и экстрагирована из раствора органическим растворителем, таким как кетоны, например метилизобутилкетон, хлорированные углеводороды, например хлористый метилен и хлороформ. После этого выделяют кислотно-аддитивную соль посредством выпаривания растворителя. Затем получаемую кислотно-аддитивную соль оланзапина преобразуют в оланзапин следующим способом: сначала растворяют ее в воде и приводят рН к, приблизительно, 1,0-5,0, предпочтительно, к 2.0 путем добавления соляной кислоты. К получаемому раствору добавляют активированный уголь. После перемешивания смеси в течение, приблизительно, 5 минут активированный уголь отфильтровывают и промывают осадок на фильтре водой. Фильтрат и промывную воду объединяют, а затем добавляют низкокипящий органический растворитель, такой как диэтиловый эфир, хлористый метилен, хлороформ, этилацетат и им подобные, предпочтительно, хлористый метилен. На следующей стадии добавляют основание, предпочтительно, неорганическое основание, такое как аммиак, K2CO3, КОН, NaH, Na2CO3, LiOH, Са(ОН)2 и им подобные, более предпочтительно, NaOH, с целью приведения рН к, приблизительно, 7-11, предпочтительно, к, приблизительно, 9-10. После достижения требуемой величины рН смесь может быть экстрагирована низкокипящим органическим растворителем, причем могут быть применены различные растворители, такие как простые эфиры, например диэтиловый эфир, хлорированные углеводороды, например хлористый метилен, хлороформ, сложные эфиры, например этилацетат, и им подобные, предпочтительно, хлористый метилен. После экстракции органический растворитель может быть частично отогнан на роторном испарителе, а оставшаяся смесь может быть охлаждена до температуры от, приблизительно, -20°С до, приблизительно, 0°С, предпочтительно, от, приблизительно, -15°С до, приблизительно, -5°С, после чего отфильтровывают осажденную кристаллическую модификацию I оланзапина.
Дальнейший необязательный способ состоит в обработке раствора, остающегося после любой из стадий кристаллизации оланзапина на самой последней стадии синтеза. Раствор, остающийся после того как оланзапин был отфильтрован, может быть непосредственно обработан органической кислотой, такой как карбоновая кислота, например щавелевой, фумаровой, бензойной кислотой, или сульфокислотами и им подобными. Осаждение образующейся таким образом кислотно-аддитивной соли оланзапина либо происходит немедленно либо упомянутый остаточный раствор может быть сначала подвергнут концентрированию посредством выпаривания растворителей, затем же раствор может быть разбавлен другими растворителями или смесью растворителей, лучше подходящей для осаждения кислотно-аддитивной соли, предпочтительно, смесью хлористого метилена и метанола. Получаемая кислотно-аддитивная соль оланзапина может быть далее преобразована в чистый оланзапин с помощью описанной выше методики.
Оланзапин, получаемый с помощью способов согласно настоящему изобретению, и наполнители могут быть объединены в фармацевтический состав согласно известным в соответствующей области способам. Оланзапин, получаемый с помощью способов согласно настоящему изобретению, пригоден для фармацевтического применения в любом фармацевтическом составе.
Оланзапин, получаемый с помощью способов согласно настоящему изобретению и соответствующим образом введенный в фармацевтический состав, может быть затем применен для профилактики и/или лечения различных психических заболеваний и состояний, таких как, например, расстройства центральной нервной системы, шизофрения, галлюцинации, острый маниакальный синдром, депрессия и им подобные.
Объектом настоящего изобретения также является способ лечения психических заболеваний и состояний, таких как, например, расстройства центральной нервной системы, шизофрения, галлюцинации, острый маниакальный синдром, депрессия и им подобные, каковой способ включает введение терапевтически эффективного количества оланзапина совместно с фармацевтически приемлемым разбавителем или носителем.
Примеры
Настоящее изобретение проиллюстрировано, но никоим образом не ограничивается нижеследующими примерами.
Сокращения:
ДМА диметилацетамид
ДМФ диметилформамид
ДМИ 1,3-диметил-2-имидазолидинон
ДМТГП 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон
ДМСО диметилсульфоксид
НМП N-метилпирролидон
Получение N-дезметилоланзапина
Пример 1
Суспендируют 10,7 г гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]-бензодиазепина (0,040 моля) коммерческого качества и темно-коричневого цвета в 70 мл н-бутанола и 30 мл ксилола, затем добавляют пиперазин (31,5 г, 0,37 моля), нагревают смесь до кипения с обратным холодильником и перемешивают при этой температуре еще в течение 4 часов, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции выпаривают растворители и добавляют 200 мл теплой воды. Образующийся осадок отфильтровывают и промывают его 20 мл этилацетата, что приводит к 11,0 г продукта (выход: 92%).
Пример 2
Суспендируют 10,7 г гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]-бензодиазепина (0,040 моля) коммерческого качества и темно-коричневого цвета в 100 мл н-бутанола, затем добавляют пиперазин (31,5 г, 0,37 моля), нагревают смесь до кипения с обратным холодильником и перемешивают при этой температуре еще в течение 8 часов, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции выпаривают растворитель и добавляют 200 мл теплой воды, отфильтровывают образующийся осадок и промывают его 20 мл этилацетата, что приводит к 11,7 г продукта (выход: 97%).
Пример 3
Суспендируют 10,7 г гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]-бензодиазепина (0,040 моля) коммерческого качества и темно-коричневого цвета в 50 мл н-бутанола и 50 мл толуола, затем добавляют пиперазин (31,5 г, 0,37 моля), нагревают смесь до кипения с обратным холодильником и перемешивают при этой температуре еще в течение 7 часов, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции выпаривают растворители и добавляют 200 мл теплой воды, отфильтровывают образующийся осадок и промывают его 20 мл этилацетата, что приводит к 9,7 г продукта (выход: 81%).
Получение неочищенного оланзапина из N-дезметилоланзапина
Пример 4
Растворяют 10 г N-дезметилоланзапина (0,034 моля) в 240 мл тетрагидрофурана и охлаждают при перемешивании до температуры -10°С. Затем добавляют 9,4 мл триэтиламина и 5 мл (0,080 моля) метилиодида. Реакционную смесь перемешивают в течение 4 часов при температуре -10°С, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции добавляют 500 мл опресненной воды и выпаривают тетрагидрофуран, после чего указанное в заглавии соединение кристаллизуется в виде кристаллов желтого цвета. Осадок отфильтровывают и промывают водой, что приводит к 8,6 г неочищенного оланзапина (выход: 82%).
Пример 5
Растворяют 10 г N-дезметилоланзапина (0.034 моля) в 240 мл тетрагидрофурана и охлаждают при перемешивании до температуры -15°С. Затем добавляют 3 г гидроксида натрия и 5 мл (0,080 моля) метилиодида. Реакционную смесь перемешивают в течение 4 часов при температуре -15°С, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции добавляют 500 мл опресненной воды и выпаривают тетрагидрофуран, после чего указанное в заглавии соединение кристаллизуется в виде кристаллов желтого цвета. Осадок отфильтровывают и промывают водой, что приводит к 10.3 г неочищенного оланзапина (выход: 98%).
Пример 6
Растворяют 10 г N-дезметилоланзапина (0,034 моля) в 120 мл тетрагидрофурана и охлаждают при перемешивании до температуры -10°С. Затем добавляют 1,6 г гидрида натрия и 5 мл (0.080 моля) метилиодида. Реакционную смесь перемешивают в течение 3 часов при температуре -10°С, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции добавляют 60 мл опресненной воды и выпаривают тетрагидрофуран, а затем добавляют 200 мл метанола, после чего указанное в заглавии соединение кристаллизуется в виде кристаллов желтого цвета. Осадок отфильтровывают и промывают водой, что приводит к 10,3 г неочищенного оланзапина (выход: 98%).
Пример 7
Растворяют 10 г N-дезметилоланзапина (0,034 моля) в 240 мл тетрагидрофурана и охлаждают при перемешивании до температуры 0°С. Затем добавляют 4,48 г трет-бутилата калия и 5 мл (0,080 моля) метилиодида. Реакционную смесь перемешивают в течение 2 часов при температуре 0°С, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции добавляют 200 мл опресненной воды и выпаривают тетрагидрофуран, после чего указанное в заглавии соединение кристаллизуется в виде кристаллов желтого цвета. Осадок отфильтровывают и промывают водой, что приводит к 10.4 г неочищенного оланзапина (выход: 99%).
Пример 8
Растворяют 10 г N-дезметилоланзапина (0,034 моля) в 200 мл метанола. Затем добавляют 20 г карбоната калия и 4,4 мл (0,046 моля) диметилсульфата. Реакционную смесь перемешивают в течение 2 часов при температуре 25°С, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции добавляют 350 мл опресненной воды и выпаривают метанол, после чего указанное в заглавии соединение кристаллизуется в виде кристаллов желтого цвета. Осадок отфильтровывают и промывают водой, что приводит к 6,3 г неочищенного оланзапина (выход: 60%).
Пример 9
Растворяют 10 г N-дезметилоланзапина (0,034 моля) в 200 мл ацетона и охлаждают при перемешивании до температуры -10°С. Затем добавляют 20 г карбоната калия и 4,4 мл (0,046 моля) диметилсульфата. Реакционную смесь перемешивают в течение 4 часов при температуре -10°С, причем окончание реакции определяют с помощью ВЭЖХ. После окончания реакции добавляют 350 мл опресненной воды и выпаривают ацетон, после чего указанное в заглавии соединение кристаллизуется в виде кристаллов желтого цвета. Осадок отфильтровывают и промывают водой, что приводит к 4,8 г неочищенного оланзапина (выход: 46%).
Получение кислотно-аддитивных солей оланзапина из выделенного неочищенного оланзапина
Получение оксалата оланзапина
Пример 10
К раствору 0,45 г оланзапина в 18 мл ДМИ добавляют раствор 0,26 г щавелевой кислоты в 0,5 мл ДМИ. После перемешивания в течение 10 минут при температуре 25°С начинается кристаллизация. Суспензию перемешивают в течение одного часа при температуре 25°С, после чего продолжают перемешивание в течение одного часа на ледяной бане. Затем продукт выделяют с помощью фильтрования. Продукт промывают 25 мл хлористого метилена и высушивают в течение двух часов при температуре 50°С под вакуумом.
Выход: 0,72 г кристаллического порошка желтого цвета.
tпл.: 235°С
1H ЯМР (300,1 МГц, ДМСО-d6): δ=2,275 (s, 3Н, СН3), 2,625 (s, 4H, NCH 3CO), 2,754 (s, 3Н, СН3), 3,141 (4Н, пиперазинил-Н), 3,193 (s, 4H, СН 2НСН3СО) 3,582 (4H, пиперазинил-Н), 6,436 (s, 1H, тиофенил-Н), 6,579 (s, 4H, HC=CH), 6,651 (s, 6,74 (m, 1H, Ar), 6,909 (m, 3Н, Ar), 7,954 (s, 1H, NH), 9,301 (ушир., 3Н, NH, ОН).
Пример 11
К раствору 0,45 г оланзапина в 18 мл ДМИ добавляют раствор 0,26 г щавелевой кислоты в 0.5 мл ДМА. После перемешивания в течение 10 минут при температуре 25°С начинается кристаллизация. Суспензию перемешивают в течение одного часа при температуре 25°С, после чего продолжают перемешивание в течение одного часа на ледяной бане. Затем продукт выделяют с помощью фильтрования. Продукт промывают 25 мл хлористого метилена и высушивают в течение двух часов при температуре 50°С под вакуумом.
Выход: 0,75 г кристаллического порошка желтого цвета
tпл.: 221°С
1Н ЯМР (300,1 МГц, ДМСО-d6): δ=1,955 (s, 3Н, СН3СОН), (s, 3Н, 2,275 (s, 3Н, СН3), 2,625 (s, 4H, NCH 3CO), 2,707 и 2,754 (2s, 6H, СН3, CONCH3), 2,940 (s, 3Н, СОНСН3), 3,180 (4H, пиперазинил-Н), 3,583 (4H, пиперазинил-Н), 6,416 (s, 1H, тиофенил-Н), 6,416 (m, 1H, Ar), 6,579 (m, 3Н, Ar), 7,846 (s, 1H, NH), 8,787 (ушир., 3Н, NH, ОН).
Пример 12
К суспензии 0,45 г оланзапина в 18 мл ацетонитрила добавляют раствор 0,26 г щавелевой кислоты в 2 мл ацетонитрила. Суспензию перемешивают в течение одного часа при температуре 25°С, после чего продолжают перемешивание в течение одного часа на ледяной бане. Затем продукт выделяют с помощью фильтрования, промывают 25 мл ацетонитрила и высушивают в течение 15 часов при температуре 60°С под вакуумом.
Выход: 0,58 г кристаллического порошка желтого цвета
tпл.: 235°C
Анализ:73,5%
Щавелевая кислота: 24,3%
Ацетонитрил: 3 мол.%
Пример 13
К суспензии 0,45 г оланзапина в 18 мл этанола добавляют раствор 0,26 г щавелевой кислоты в 0,5 мл этанола. После добавления раствора щавелевой кислоты начинается кристаллизация продуктов из раствора. Суспензию перемешивают в течение одного часа при температуре 25°С, после чего продолжают перемешивание в течение одного часа на ледяной бане. Затем продукт выделяют с помощью фильтрования. Продукт промывают 25 мл этанола и высушивают в течение двух часов при температуре 60°С под вакуумом.
Выход: 0,56 г кристаллического порошка желтого цвета
tпл.: 224°С
Анализ: 75,8%
Щавелевая кислота: 24,0%
Пример 14
К раствору 0,45 г оланзапина в 18 мл изопропанола добавляют раствор 0,26 г щавелевой кислоты в 2 мл изопропанола. После добавления раствора щавелевой кислоты начинается кристаллизация. Суспензию перемешивают в течение одного часа при температуре 25°С, после чего продолжают перемешивание в течение одного часа на ледяной бане. Затем продукт выделяют с помощью фильтрования, промывают 25 мл изопропанола и высушивают в течение 15 часов при температуре 60°С под вакуумом.
Выход: 0,60 г кристаллического порошка желтого цвета
tпл.: 230°С
Анализ: 65,18%
Щавелевая кислота: 21,5%
Изопропанол: 94 мол.%
Получение фумарата оланзапина
Пример 15
К раствору 0,45 г оланзапина в 18 мл изопропанола добавляют 0,26 г фумаровой кислоты. Получаемую суспензию перемешивают в течение одного часа при температуре 25°С, после чего продолжают перемешивание в течение одного часа на ледяной бане. Затем продукт выделяют с помощью фильтрования. Продукт промывают 25 мл изопропанола и высушивают в течение 15 часов при температуре 60°С под вакуумом.
Выход: 0,55 г кристаллического порошка желтого цвета
tпл.: 231°С
Анализ: 75,5%
Фумаровая кислота: 17,3%
Изопропанол: 50 мол.%
1Н ЯМР: (300,1 МГц, ДМСО-d6): δ=2,334 (s, 3Н, СН3), 2,443 (s, 3H, СН3), 2,729 (4Н, пиперазинил-Н), 3,434 (4Н, пиперазинил-Н), 5,753 (s, 1,36H, CH2Cl2), 6,346 (s, 1H, тиофенил-Н), 6,579 (s, 4Н, СОНС=СНСО), 6,651 (s, 6,643 (m, 1H, ArH), 6,834 (m, 3H, ArH), 7,634 (s, 1H, NH).
Получение бензоата оланзапина
Пример 16
К раствору 0,45 г оланзапина в 18 мл ацетона добавляют 0,26 г бензойной кислоты. Получаемую суспензию перемешивают в течение одного часа при температуре 25°С, после чего продолжают перемешивание в течение одного часа на ледяной бане. Затем продукт выделяют с помощью фильтрования. Продукт промывают 25 мл ацетона и высушивают в течение 15 часов при температуре 60°С под вакуумом.
Выход: 0,60 г кристаллического порошка желтого цвета.
tпл.: 205°С
Анализ: 70,0%
Бензойная кислота: 26,1%
Ацетон: 5,2 мол.%
1Н ЯМР: (300,1 МГц, ДМСО-d6): δ=2,334 (s, 3Н, СН3), 2,443 (s, 3H, СН3), 2,729 (4Н, пиперазинил-Н), 3,434 (4Н, пиперазинил-Н), 6,365 (s, 1H, тиофенил-Н), 6,579 (s, 4Н, СОНС=СНСО), 6,651 (s, 6,643 (m, 1H, ArH), 6,834 (m, 3H, ArH), 7,686 (s, 1H, NH).
Получение кислотно-аддитивных солей оланзапина непосредственно в процессе синтеза оланзапина, исходя из метилирования N-дезметилоланзапина
Получение оксалата оланзапина
Пример 17
Раствор 12,0 г N-дезметилоланзапина (0,040 моля) в смеси 180 мл ТГФ и 120 мл 1,3-диметил-2-имидазолидинона (ДМИ) охлаждают до температуры приблизительно
-20°С. При температуре -19°С к раствору добавляют сначала 8,19 г диизопропиламина, а затем 13,7 г метилиодида (0,097 моля). После перемешивания реакционной смеси в течение 45 минут при температуре -19°С добавляют 6,4 мл концентрированной соляной кислоты и раствор 6,36 г тиомочевины в 50 мл воды и перемешивают реакционную смесь в течение 15 минут при температуре 20°С.
После добавления 50 мл воды смесь выпаривают на бане, имеющей температуру 35°С, при давлении 50-60 мбар до достижения объема приблизительно 160 мл. Затем добавляют 400 мл воды и 120 мл хлористого метилена и приводят рН к 2,0 с помощью 6 н. HCl. После разделения фаз водную фазу дважды промывают 120 мл хлористого метилена. Добавляют к водной фазе 180 мл хлористого метилена и приводят рН к 9,0 посредством добавления 1 н. NaOH. После перемешивания в течение 5 минут фазы разделяют и дважды экстрагируют водно-щелочную фазу 90 мл хлористого метилена. Органические фазы объединяют, разбавляют смесь 37,5 мл метанола и добавляют в течение 15 минут при перемешивании раствор 7,46 г щавелевой кислоты в 10,5 мл метанола. Получаемую суспензию перемешивают в течение приблизительно 1 часа при температуре приблизительно 20°С, а затем в течение еще 1 часа при температуре приблизительно 0°С.
Продукт выделяют с помощью фильтрования, промывают 100 мл хлористого метилена и высушивают в течение 2 часов при температуре 50°С под вакуумом.
Выход: 15,15 г (69,2%)
tпл.: 228°С
Анализ: 54,1%
Чистота по данным ВЭЖХ: 98,2% площади
N-дезметилоланзапин: 0,95% площади
Щавелевая кислота: 31,6%
ДМИ: 6 мол.%
Хлористый метилен: 0,5 мол.%
Пример 18
Раствор 12,0 г N-дезметилоланзапина (0,040 моля) в 240 мл диметилацетамида (ДМА) охлаждают до температуры приблизительно -20°С. При температуре -20°С к раствору добавляют сначала 8,19 г диизопропиламина, а затем 7,19 г метилиодида (0,050 моля). После перемешивания реакционной смеси в течение 95 минут при температуре -20°С добавляют 6,4 мл концентрированной соляной кислоты и раствор 6,36 г тиомочевины в 50 мл воды и перемешивают реакционную смесь в течение 15 минут при температуре 20°С. Затем добавляют 400 мл воды и 120 мл хлористого метилена и приводят рН к 2,0 с помощью 6 н. HCl. После разделения фаз водную фазу дважды промывают 140 мл хлористого метилена. Затем добавляют к водной фазе 180 мл хлористого метилена и приводят рН к 9.0 посредством добавления 1 н. NaOH. После перемешивания в течение 5 минут фазы разделяют и дважды экстрагируют водно-щелочную фазу 90 мл хлористого метилена. Органические фазы объединяют, добавляют 380 мг уксусного ангидрида и перемешивают смесь в течение 5 минут. Затем смесь разбавляют 37,5 мл метанола и в течение 15 минут добавляют при перемешивании раствор 7,46 г щавелевой кислоты в 10,5 мл метанола. Получаемую суспензию перемешивают в течение приблизительно 1 часа при температуре приблизительно 20°С, а затем в течение еще 1 часа при температуре приблизительно 0°С. Продукт выделяют с помощью фильтрования, промывают 100 мл хлористого метилена и высушивают в течение 15 часов при температуре 25°С под вакуумом.
Выход: 13,76 г (72,0%)
tпл.: 233°С
Анализ: 59,4%
Чистота по данным ВЭЖХ: 98,3% площади
N-дезметилоланзапин: 0,15% площади
Щавелевая кислота: 29,1%
Хлористый метилен: 69,9 мол.%
ДМА: 2,3 мол.%
Пример 19
Раствор 12,0 г N-дезметилоланзапина (0.040 моля) в 240 мл 1,3-диметил-3,4,5,6-тетрагидро-1,3-пиримидинона охлаждают до температуры приблизительно -20°С. При температуре -20°С добавляют к раствору сначала 8,19 г диизопропиламина, а затем 7,57 г метилиодида (0,053 моля). После перемешивания реакционной смеси в течение 60 минут при температуре -20°С добавляют 7,2 мл концентрированной соляной кислоты и раствор 6,36 г тиомочевины в 50 мл воды, нагревают реакционную смесь до температуры 20°С и перемешивают в течение 5 минут при данной температуре. Затем добавляют 400 мл воды и 120 мл хлористого метилена и приводят рН к 2,0 с помощью 6 н. HCl. После разделения слоев дважды промывают водный слой 120 мл хлористого метилена. Добавляют к водной фазе 180 мл хлористого метилена и приводят рН к 9,0 посредством добавления 1 н. NaOH. После перемешивания в течение 5 минут слои разделяют и дважды экстрагируют водно-щелочной слой с помощью 90 мл хлористого метилена. Органические слои объединяют, добавляют 380 мг уксусного ангидрида и перемешивают смесь в течение 5 минут. Затем растворитель выпаривают под вакуумом и растворяют маслянистый остаток в смеси 360 мл хлористого метилена, 37,5 мл метанола и 0,72 мл воды. Добавляют к данному раствору затравку оксалата оланзапина, а затем добавляют в течение 20 минут при перемешивании раствор 7,71 г щавелевой кислоты в 10,5 мл метанола. Получаемую суспензию перемешивают в течение приблизительно 1 часа при температуре приблизительно 25°С, а затем еще в течение 1 часа при температуре приблизительно 0°С. Продукт выделяют с помощью фильтрования, промывают 100 мл хлористого метилена и высушивают в течение 15 часов при температуре 60°С под вакуумом.
Выход: 14,8 г (82,4%) кристаллического порошка желтого цвета
tпл.: 229°С
Анализ: 64,1%
Чистота по данным ВЭЖХ: 99,5% площади
N-дезметилоланзапин: <0,1% площади
Щавелевая кислота: 32,4%
Хлористый метилен: 10,4 мол.% (сушка в течение 24 часов при температуре 50°С)
ДМТГП: 0,5 мол.%
Пример 20
Смесь 30,0 г гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]-бензодиазепина (0,113 моля) и 81 мл N-метилпиперазина (0,729 моля) в 186 мл ДМСО нагревают до температуры 117°С. После перемешивания смеси и пробулькивания через нее азота в течение 17 часов при данной температуре получаемый раствор охлаждают до комнатной температуры (КТ), а затем добавляют 570 мл хлористого метилена и 570 мл воды. После перемешивания смеси в течение 5 минут слои разделяют. Водно-щелочной слой экстрагируют 300 мл хлористого метилена. К объединенным органическим слоям добавляют 250 мл воды и приводят рН к 2.0 посредством добавления 6 М HCl. После разделения слоев дважды экстрагируют органический слой 90 мл воды. Объединенные водно-кислотные слои обрабатывают 4,5 г активированного угля. После перемешивания в течение 5 минут активированный уголь отфильтровывают и промывают осадок на фильтре 100 мл воды. Фильтрат и промывную воду объединяют и после добавления 950 мл хлористого метилена приводят рН к 9,0 посредством добавления 5 М NaOH. После разделения слоев экстрагируют вводно-щелочной слой 125 мл хлористого метилена. Органические слои объединяют и выпаривают под вакуумом. Маслянистый остаток растворяют в смеси 1075 мл хлористого метилена, 140 мл метанола и 3,6 мл воды и нагревают до температуры приблизительно 29-30°С. После добавления к раствору затравки оксалата оланзапина в течение 30 минут добавляют раствор 18,7 г щавелевой кислоты в 27 мл метанола. Получаемую суспензию перемешивают в течение приблизительно 1 часа при температуре приблизительно 25°С, а затем в течение еще 2 часов при температуре приблизительно 0°С. Продукт выделяют с помощью фильтрования, промывают 150 мл хлористого метилена и высушивают в течение 6 часов при температуре 60°С под вакуумом.
Выход: 43,3 г (82,2%) кристаллического порошка желтого цвета
tпл.: 224°С
Анализ: 62,7%
Чистота по данным ВЭЖХ: 99,6% площади
Щавелевая кислота: 26,1%
Хлористый метилен: 22 мол.%
Получение фумарата оланзапина
Пример 21
К раствору оланзапина (получаемого из 12,0 г исходного N-дезметилоланзапина согласно примеру 16) в смеси 360 мл хлористого метилена, 37,5 метанола и 0,72 мг воды добавляют затравку кристаллов фумарата оланзапина и 0,96 г фумаровой кислоты. Получаемую суспензию перемешивают в течение приблизительно 1 часа при температуре 25°С, а затем еще в течение 2 часов при температуре приблизительно 0°С. Продукт выделяют с помощью фильтрования, промывают 150 мл хлористого метилена и высушивают в течение 6 часов при температуре 60°С под вакуумом.
Выход: 11,4 г (65,7%) кристаллического порошка светло-желтого цвета
tпл.: 217°С
Анализ:65,7%
Чистота по данным ВЭЖХ: 97,8% площади
N-дезметилоланзапин: 0,15% площади
Фумаровая кислота: 23,2%
Хлористый метилен: 48 мол.%
Пример 22
Раствор оланзапина, получаемый из 30,0 г гидрохлорида 4-амино-2-метил-10H-тиено[2,3-b][1,5]-бензодиазепина (0,113 моля) и 81 мл N-метилпиперазина (0,729 моля) в 186 мл ДМСО согласно примеру 5 нагревают до температуры 29-30°С. При этой температуре добавляют затравку фумарата оланзапина и 14,4 г фумаровой кислоты. Получаемую суспензию перемешивают в течение приблизительно 1 часа при температуре 29-30°С, а затем еще в течение 2 часов при температуре приблизительно 0°С. Продукт выделяют с помощью фильтрования, промывают 150 мл хлористого метилена и высушивают в течение 6 часов при температуре 60°С под вакуумом.
Выход: 41,9 г (85,2%) кристаллического порошка светло-желтого цвета
tпл.: 217°C
Анализ: 68,5%
Чистота по данным ВЭЖХ: 99,7% площади
Фумаровая кислота: 23,0%
Хлористый метилен: 48 мол.%
Получение кристаллической модификации I чистого оланзапина из оксалата оланзапина
Пример 23
Растворяют 7,40 г оксалата оланзапина в 75 мл воды и приводят рН раствора к 2,0 посредством добавления 6 н. HCl. К получаемому таким образом прозрачному раствору оксалата оланзапина добавляют 0,75 г активированного угля. После перемешивания в течение 5 минут активированный уголь отфильтровывают и промывают осадок на фильтре 50 мл воды. Фильтрат и промывную воду объединяют и после добавления 125 мл хлористого метилена приводят рН объединенной смеси к 9,0 посредством добавления 1 н. NaOH. После перемешивания в течение 5 минут слои разделяют и экстрагируют водную фазу 25 мл хлористого метилена. Органические слои объединяют и после высушивания над карбонатом натрия концентрируют раствор под вакуумом до достижения объема 27 мл. После этого кипятят концентрированный раствор с обратным холодильником при нормальном давлении и после добавления затравки кристаллической модификации I оланзапина раствор немедленно охлаждают на ледяной бане. Добавление указанной затравки продолжают до начала кристаллизации оланзапина. Получаемую суспензию перемешивают в течение 15 минут на ледяной бане, а затем в течение 15 минут при температуре приблизительно -20°С. Затем оланзапин выделяют с помощью фильтрования. Осадок на фильтре промывают 3 мл хлористого метилена, имеющего температуру -20°С. Продукт высушивают в течение двух дней при температуре 25°С под вакуумом.
Выход: 3,63 г (72,6%)
Чистота по данным ВЭЖХ: 99,9%
По данным ИК-спектроскопии вещество идентично эталонному образцу кристаллической модификации I оланзапина.
По рентгенодифракционным данным вещество идентично эталонному образцу кристаллической модификации I оланзапина.
Получение кристаллической модификации II чистого оланзапина из оксалата оланзапина
Пример 24
Растворяют 7,40 г оксалата оланзапина в 75 мл воды и приводят рН раствора к 2,0 посредством добавления 6 н. HCl. К получаемому таким образом прозрачному раствору оксалата оланзапина добавляют 0,75 г активированного угля. После перемешивания в течение 5 минут активированный уголь отфильтровывают и промывают осадок на фильтре 50 мл воды.
Фильтрат и промывную воду объединяют и после добавления 125 мл хлористого метилена приводят рН объединенной смеси к 8-10 посредством добавления 1 н. NaOH. После перемешивания в течение 5 минут слои разделяют и экстрагируют водную фазу 25 мл хлористого метилена. Органические слои объединяют и выпаривают хлористый метилен. Затем добавляют этилацетат, после чего начинается кристаллизация оланзапина. Получаемую суспензию перемешивают в течение 15 минут на ледяной бане. После этого выделяют оланзапин посредством фильтрования. Продукт высушивают в течение двух часов при температуре 60°С под вакуумом.
Выход: 3,4 г
Чистота по данным ВЭЖХ: 99,9%
По данным ИК-спектроскопии вещество идентично эталонному образцу кристаллической модификации II оланзапина.
По рентгенодифракционным данным вещество идентично эталонному образцу кристаллической модификации II оланзапина.
Получение чистого оланзапина из N-дезметилоланзапин через посредство кислотно-аддитивной соли оланзапина
Пример 25
Раствор 20,0 г N-дезметилоланзапина (0,067 моля) в смеси 150 мл ТГФ и 60 мл ДМА охлаждают до температуры приблизительно -15°С. При температуре -15°С сначала добавляют к реакционной смеси 20 мл диизопропиламина, а затем добавляют в течение 30-40 минут 6 мл метилиодида (0,96 моля) в 30 мл ТГФ. После перемешивания реакционной смеси в течение еще 60 минут при температуре от -5 до -10°С добавляют 16 мл концентрированной соляной кислоты в 100 мл воды и раствор 3,3 г тиомочевины в 100 мл воды и перемешивают реакционную смесь в течение 15 минут при температуре 20°С.
Выпаривают ТГФ на бане, имеющей температуру 35°С, при давлении 50-60 мбар до достижения объема приблизительно 200 мл. Затем добавляют 300 мл хлористого метилена и приводят рН к 8,5-9 с помощью 40% NaOH. После разделения фаз дважды промывают водную фазу с помощью 100 мл хлористого метилена. Органические фазы объединяют и пятикратно промывают 100 мл воды. Органические фазы объединяют, добавляют 0,5 мл уксусного ангидрида и перемешивают смесь в течение 5 минут. После этого добавляют в течение 15 минут раствор 10,34 г дигидрата щавелевой кислоты в 40 мл метанола. Получаемую суспензию перемешивают в течение приблизительно 1 часа при температуре приблизительно 20°С, а затем еще в течение 1 часа при температуре приблизительно 0°С. Продукт выделяют с помощью фильтрования, промывают 100 мл хлористого метилена и высушивают в течение 2 часов при температуре 50°С под вакуумом. Выход: 25,1 г.
Растворяют 25 г оксалата оланзапина в 250 мл воды и приводят рН раствора к 2,0 посредством добавления 6 н. HCl. К получаемому таким образом прозрачному раствору оксалата оланзапина добавляют 2,5 г активированного угля. После перемешивания в течение 5 минут активированный уголь отфильтровывают и промывают осадок на фильтре 50 мл воды. Фильтрат и промывную воду объединяют и после добавления 300 мл хлористого метилена приводят рН к 9-10 посредством добавления 10 н. NaOH. После перемешивания в течение 5 минут слои разделяют и экстрагируют водную фазу 50 мл хлористого метилена. Органические слои объединяют и концентрируют раствор под вакуумом до достижения объема 50 мл. Затем концентрированный раствор кипятят с обратным холодильником при нормальном давлении и после добавления затравки кристаллической модификации I оланзапина раствор немедленно охлаждают на ледяной бане. Добавление указанной затравки продолжают до начала кристаллизации оланзапина. Получаемую суспензию перемешивают в течение 15 минут на ледяной бане, а затем в течение 15 минут при температуре -20°С. После этого оланзапин выделяют с помощью фильтрования. Осадок на фильтре промывают 10 мл хлористого метилена, имеющего температуру
-20°С. Продукт высушивают в течение четырех часов при температуре 80°С под вакуумом.
Выход: 11,5 г
В таблице 1 представлены результаты анализа, образующегося в качестве промежуточного вещества оксалата оланзапина и для конечного продукта оланзапина, полученного согласно способу, описанному в примере 16.
Описание анализа методом ВЭЖХ
Анализ методом ВЭЖХ осуществляют с помощью модуля для разделения Waters Alliance 2695, детектора PDA 2996, программного обеспечения Empower 5.0. В качестве буферного раствора используют 15 мМ NaH2PO4, рН=6,2: 2,34 г NaH2PO4×Н2О/1000 мл воды, рН приводят к 6.2 с помощью 5 М NaOH.
Условия проведения хроматографического анализа:
1. Подвижная фаза:
А: Буферный раствор 15 мМ NaH2PO4, pH=6,2/ACN/MeOH 70/20/10 (об./об./об.)
Б: Буферный раствор 15 мМ NaH2PO4, рН=6,2/МеОН 25/75 (об./об.)
2. Колонка: BetaBasic C-8 3 мкм, 100×4.6 мм
3. Температура: 30°С
4. Объемная скорость: 0,55 мл/мин
5. Длина волны: 254 нм
6. Вводимый объем: 5 л
7. Таблица градиентов:
Описание анализа методом 1Н ЯМР
Спектры 1Н-ЯМР регистрируют с помощью спектрометра Bruker Avance 300 с использованием стандартных приборных методик. Образцы растворяют в ДМСО-d6 при концентрации приблизительно 15 мг/мл и регистрируют спектр при температуре внешней среды. Сигнал растворителя используют в качестве внутреннего стандарта: 2,50 м.д. в 1Н ЯМР. Рабочая частота для 1Н составляет 300 МГц.
Изобретение относится к области органической химии, конкретно к способу очистки оланзапина, который включает смешение оланзапина с органической кислотой в органическом растворителе или смеси органических растворителей с получением кислотно-аддитивной соли оланзапина, осаждение и выделение кислотно-аддитивной соли оланзапина и преобразование кислотно-аддитивной соли оланзапина в оланзапин, при этом в качестве органической кислоты используют карбоновую кислоту, которую выбирают из группы, включающей щавелевую, фумаровую и бензойную кислоту. Изобретение также относится к способам получения чистого оланзапина, промежуточных продуктов и кислотно-аддитивных солей оланзапина, которые в свою очередь могут найти применение для получения чистого оланзапина, используемого для изготовления лекарственного средства для лечения психических заболеваний и состояний. 9 н. и 29 з.п. ф-лы, 1 табл.
1. Способ очистки оланзапина, включающий смешение оланзапина с органической кислотой в органическом растворителе или смеси органических растворителей с получением кислотно-аддитивной соли оланзапина, осаждение и выделение кислотно-аддитивной соли оланзапина и преобразование кислотно-аддитивной соли оланзапина в оланзапин, отличающийся тем, что в качестве органической кислоты используют карбоновую кислоту, которую выбирают из группы, включающей щавелевую, фумаровую и бензойную кислоту.
2. Способ по п.1, в котором органический растворитель выбирают из группы, включающей тетрагидрофуран, ацетон, диметилформамид, ацетонитрил и их смесь.
3. Способ по п.1, в котором смесь органических растворителей является смесью тетрагидрофурана с по крайней мере одним полярным растворителем.
4. Способ по п.3, в котором указанный полярный растворитель выбирают из группы, включающей диметилформамид, диметилацетамид, N-метилпирролидон, 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон, 1,3-диметил-2-имидазолидинон, тетраметилмочевину, диметилсульфоксид, сульфолан, ацетон и ацетонитрил.
5. Способ по п.1, отличающийся тем, что преобразование кислотно-аддитивной соли оланзапина в оланзапин включает
1) растворение кислотно-аддитивной соли оланзапина в воде,
2) приведение рН получаемого раствора к приблизительно 8-10,
3) экстракцию оланзапина из водной фазы в фазу органического растворителя и
4) выделение кислотно-аддитивной соли оланзапина из фазы органического растворителя путем концентрирования раствора и отделения кристаллов.
6. Способ синтеза N-дезметилоланзапина путем взаимодействия 4-амино-2-метил-10Н-тиено[2,3-b][1,5]бензодиазепина и пиперазина в растворителе, отличающийся тем, что в качестве растворителя используют по крайней мере один алифатический спирт или его смесь с растворителем, имеющим более высокую температуру кипения.
7. Способ синтеза N-дезметилоланзапина по п.6, в котором упомянутый растворитель включает н-бутанол.
8. Способ синтеза N-дезметилоланзапина по п.6, в котором упомянутая смесь растворителей включает н-бутанол и по крайней мере один ароматический углеводород, имеющий более высокую температуру плавления.
9. Способ синтеза N-дезметилоланзапина по п.6, в котором упомянутый ароматический углеводород, имеющий более высокую температуру плавления, является ксилолом и/или толуолом.
10. Способ синтеза N-дезметилоланзапина по п.6, в котором пиперазин добавляют в избытке по отношению к 4-амино-2-метил-10Н-тиено[2,3-b][1,5]бензодиазепину.
11. Способ синтеза N-дезметилоланзапина по п.6, в котором в реакционную смесь добавляют дополнительное неорганическое или органическое основание.
12. Способ синтеза N-дезметилоланзапина по одному из пп.6-11, отличающийся тем, что после его осуществления N-дезметилоланзапин осаждают с помощью теплой воды и промывают сложными эфирами.
13. Способ синтеза N-дезметилоланзапина по п.12, в котором упомянутые эфиры выбирают из группы, включающей этилацетат, пропилацетат и бутилацетат.
14. Способ получения 2-метил-4-(4-метил-1-пиперазинил)-10Н-тиено[2,3-b][1,5]бензодиазепина (оланзапина), включающий получение N-дезметилоланзапина согласно способу по пп.6-13, метилирование полученного N-дезметилоланзапина по атому азота с помощью метилирующего агента в органическом растворителе или в смеси органических растворителей и очистку оланзапина согласно способу по пп.1-5, с получением оланзапина светлой окраски без темно-коричневых или зеленых оттенков.
15. Способ получения оланзапина в виде кислотно-аддитивной соли, включающий смешение оланзапина с органической кислотой в органическом растворителе или смеси органических растворителей, осаждение и выделение кислотно-аддитивной соли оланзапина путем отделения кристаллов, отличающийся тем, что в качестве органической кислоты используют карбоновую кислоту, которую выбирают из группы, включающей щавелевую, фумаровую и бензойную кислоту.
16. Способ по п.15, в котором упомянутый органический растворитель выбирают из группы, включающей тетрагидрофуран, ацетон, диметилформамид, ацетонитрил и их смесь.
17. Способ по п.15, в котором смесь органических растворителей является смесью тетрагидрофурана с по крайней мере одним полярным растворителем.
18. Способ по п.17, в котором указанный полярный растворитель выбирают из группы, включающей диметилформамид, диметилацетамид, N-метилпирролидон, 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон, 1,3-диметил-2-имидазолидинон, тетраметилмочевину, диметилсульфоксид, сульфолан, ацетон и ацетонитрил.
19. Способ получения оланзапина в виде кислотно-аддитивной соли, включающий реакцию гидрохлорида 4-амино-2-метил-10Н-тиено[2,3-b][1,5]бензодиазепина с N-метилпиперазином с получением оланзапина, отличающийся тем, что осуществляют преобразование полученного оланзапина в кислотно-аддитивную соль, для чего полученную реакционную смесь разбавляют водой, экстрагируют разбавленную реакционную смесь органическим растворителем, выпаривают органическую фазу и разбавляют остаток вторым растворителем, к полученному раствору добавляют карбоновую кислоту, выбранную из группы, включающей щавелевую, фумаровую и бензойную кислоту, для осаждения кислотно-аддитивной соли оланзапина и выделяют осажденную кислотно-аддитивную соль оланзапина посредством отделения кристаллов.
20. Способ получения оланзапина в виде кислотно-аддитивной соли, отличающийся тем, что он включает следующие стадии:
а) введение N-дезметилоланзапина в реакцию с метилирующим агентом с получением оланзапина,
б) разбавление получаемой реакционной смеси водой и подкисление ее кислотой,
в) добавление к реакционной смеси органического растворителя и разделение фаз,
г) нейтрализацию получаемой водной фазы и экстракцию оланзапина органическим растворителем с получением фазы органического растворителя и
д) добавление к органической фазе II органической кислоты или ангидрида органической кислоты,
е) необязательное выпаривание органического растворителя и разбавление остатка вторым органическим растворителем,
ж) добавление карбоновой кислоты, выбранной из группы, включающей щавелевую, фумаровую и бензойную кислоту, либо к получаемому разбавленному раствору, либо непосредственно к содержащему оланзапин экстракту, получаемому в результате упомянутой выше экстракции на стадии г), и
з) выделение осажденной кислотно-аддитивной соли оланзапина путем отделения кристаллов.
21. Способ по п.20, в котором в качестве органического растворителя на стадиях в) и г) используют хлорированный растворитель.
22. Способ по п.21, в котором упомянутый хлорированный растворитель является хлористым метиленом.
23. Способ по п.21, в котором в качестве органического растворителя на стадиях в) и г) используют хлористый метилен, а в качестве упомянутого второго органического растворителя на стадии е) используют метанол.
24. Способ получения кислотно-аддитивной соли оланзапина по одному из пп.15-23, отличающийся тем, что дополнительно включает последующую обработку карбоновой кислотой раствора, оставшегося после выделения конечного оланзапина.
25. Способ по одному из пп.1-5 и 14-24, отличающийся тем, что оланзапин выделяют из полученной кислотно-аддитивной соли в кристаллической форме.
26. Способ по п.25, отличающийся тем, что оланзапин получают из его кислотно-аддитивной соли как описано в пп.1 и 5.
27. Способ по одному из пп.1-5 и 14-26, отличающийся тем, что кристаллы оланзапина выделяют из органического растворителя с получением кристаллической модификации I.
28. Способ по одному из пп.1-5 и 14-26, отличающийся тем, что кристаллы оланзапина выделяют из одного или более органических растворителей с получением кристаллической модификации II.
29. Способ по п.14, отличающийся тем, что способ дополнительно включает стадию получения N-дезметилоланзапина из гидрохлорида 4-амино-2-метил-10Н-тиено[2,3-b][1,5]-бензодиазепина с получением 2-метил-4-(1-пиперазинил)-10Н-тиено[2,3-b][1,5]-бензодиазепина, который преобразуют с помощью метилирующего агента в неочищенный оланзапин, неочищенный оланзапин переводят в его кислотно-аддитивную соль и затем кислотно-аддитивную соль преобразуют в оланзапин.
30. Применение органических кислот, выбранных из группы, включающей щавелевую, фумаровую и бензойную кислоту, в способах получения оланзапина, в которых оланзапин подвергают очистке через посредство образования кислотно-аддитивной соли.
31. Оланзапин в форме кислотно-аддитивной соли с органической кислотой, выбранной из группы, включающей щавелевую, фумаровую и бензойную кислоту.
32. Оланзапин в форме кислотно-аддитивной соли по п.31 для получения очищенного оланзапина (светлой окраски), используемого при изготовлении лекарственного средства для лечения психических заболеваний и состояний.
33. Оланзапин в форме кислотно-аддитивной соли по п.31 для получения очищенного оланзапина (светлой окраски), используемого при изготовлении фармацевтической композиции для лечения психических заболеваний и состояний совместно с по крайней мере одним фармацевтически приемлемым ингредиентом.
34. Оланзапин в форме кислотно-аддитивной соли по п.31 для получения очищенного оланзапина (светлой окраски), используемого для лечения психических заболеваний и состояний, таких как расстройства центральной нервной системы, шизофрения, галлюцинации, острый маниакальный психоз, депрессия и им подобные, при введении в терапевтически эффективном количестве в сочетании с фармацевтически приемлемым разбавителем или носителем.
35. Кислотно-аддитивная соль 2-метил-4-(4-метил-1-пиперазинил)-10Н-тиено[2,3-b][1,5]бензодиазепина и бензойной кислоты.
36. Кислотно-аддитивная соль по п.35 для получения очищенного оланзапина (светлой окраски), используемого при изготовлении лекарственного средства для лечения психических заболеваний и состояний.
37. Кислотно-аддитивная соль по п.35 для получения очищенного оланзапина (светлой окраски), используемого при изготовлении фармацевтической композиции для лечения психических заболеваний и состояний совместно с по крайней мере одним фармацевтически приемлемым ингредиентом.
38. Кислотно-аддитивная соль по п.35 для получения очищенного оланзапина (светлой окраски), используемого для лечения психических заболеваний и состояний, таких как расстройства центральной нервной системы, шизофрения, галлюцинации, острый маниакальный психоз, депрессия и им подобные, при введении в терапевтически эффективном количестве в сочетании с фармацевтически приемлемым разбавителем или носителем.
| Магнитоупругий датчик | 1971 |
|
SU454436A1 |
| WO 03101997 А1, 11.12.2003 | |||
| WO 2004000847 A1, 31.12.2004 | |||
| WO 9618621 A1, 20.06.1996 | |||
| US 20020165225 A1, 07.11.2002. | |||

