Настоящее изобретение относится к фармацевтическим композициям и, более конкретно, к фармацевтической композиции, содержащей соединение формулы (I)
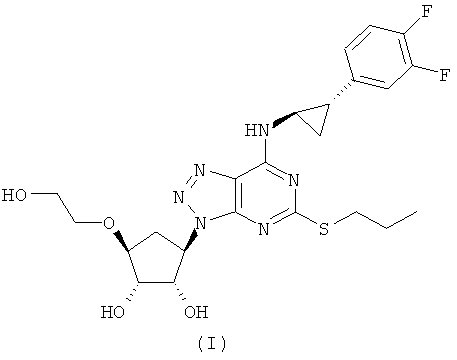
Соединение формулы (I) общепринятым образом называют {1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)-циклопентан-1,2-диол, и здесь ниже оно будет именоваться 'Агент'.
Этот Агент раскрыт в качестве антагониста АДФ-рецептора в международной заявке на патент номер PCT/SE99/02256 (номер публикации WO 00/34283) и в международной заявке на патент номер PCT/SE01/01239 (публикация номер WO 01/92262). Было установлено, что аденозин-5'-дифосфат (АДФ) действует как ключевой медиатор тромбоза. АДФ-индуцированную агрегацию тромбоцитов опосредует рецептор подтипа P2T, локализованный на мембране тромбоцитов. P2T-рецептор (также известный как P2YADP или P2TAC) главным образом участвует в опосредовании агрегации/активации тромбоцитов и представляет собой рецептор, связанный с G-белком, который еще не клонирован. Фармакологические характеристики этого рецептора описаны, например, в Humphries et al., Br. J. Pharmacology (1994), 113, 1057-1063 и Fagura et al., Br. J. Pharmacology (1998) 124, 157-164. Показано, что антагонисты этого рецептора обеспечивают значительные улучшение в сравнении с другими антитромботическими агентами (см. J. Med. Chem. (1999) 42, 213).
Фармацевтические композиции по настоящему изобретению являются пригодными для перорального введения. Одним из качеств, желательных в фармацевтической композиции, пригодной для перорального введения, является биологическая доступность. Биологическая доступность лекарственного средства представляет собой относительное количество введенной дозы, которое достигает большого круга кровообращения в неизмененной форме. Поэтому биологическая доступность важна в определении терапевтически активной концентрации в месте действия. Как высвобождение лекарственного средства из композиции, так и стабильность композиции будут влиять на его биологическую доступность. Поэтому важно, чтобы композиция лекарственного средства высвобождала по существу все количество лекарственного средства (см. Aulton ME, Pharmaceutics - The Science of Dosage Form Design, 2nd Edition, 2002, Churchill Livingstone). Биологическую доступность можно измерять с помощью тестов, известных в данной области, например с помощью стандартного аппарата для растворения согласно Фармакопее США (USP) и стандартной 'био-релевантной' среды для растворения, например FaSSIF (Pharm. Res., 17:439-444, 2000).
Существуют фармацевтические композиции, содержащие Агент, которые удерживают некоторое количество Агента и, следовательно, снижают его биологическую доступность.
Авторы изобретения предложили новую фармацевтическую композицию Агента, которая обладает благоприятными свойствами и решает одну или более проблем, ассоциированных с получением препаратов Агента. В первом аспекте авторы предлагают фармацевтическую композицию, которая является пригодной для перорального введения и которая высвобождает по существу весь Агент. В одном аспекте фармацевтическая композиция высвобождает по меньшей мере 90% Агента. В другом аспекте фармацевтическая композиция высвобождает по меньшей мере 95% Агента. В еще одном аспекте фармацевтическая композиция высвобождает по меньшей мере 97% Агента.
Соответственно, изобретение относится к фармацевтической композиции, содержащей:
Агент;
один или более наполнителей, выбранных из маннита, сорбита, дигидрата двухосновного фосфата кальция, безводного двухосновного фосфата кальция и трехосновного фосфата кальция или их смеси;
один или более связывающих агентов, выбранных из гидроксипропилцеллюлозы, альгиновой кислоты, натриевой
карбоксиметилцеллюлозы, коповидона и метилцеллюлозы или их смеси;
один или более разрыхляющих агентов, выбранных из крахмалгликолята натрия, кроскармеллозы натрия и кросповидона или их смеси;
и один или более смазывающих агентов.
Наполнитель может представлять собой 'растворимый' наполнитель или 'нерастворимый' наполнитель. 'Растворимый' наполнитель представляет собой наполнитель, который является по существу растворимым в воде при температуре окружающей среды. 'Нерастворимый' наполнитель представляет собой наполнитель, который имеет низкую или медленную растворимость в воде при температуре окружающей среды.
В одном аспекте фармацевтическая композиция содержит по меньшей мере один 'растворимый' наполнитель, выбранный из маннита, сорбита, мальтодекстрина, мальтозы и декстрина.
В другом аспекте фармацевтическая композиция содержит один или более 'нерастворимых' наполнителей, выбранных из дигидрата двухосновного фосфата кальция, безводного двухосновного фосфата кальция, частично прежелатинизированного крахмала и трехосновного фосфата кальция.
В одном аспекте фармацевтическая композиция содержит один или более 'растворимых' наполнителей. В другом аспекте фармацевтическая композиция содержит один 'растворимый' наполнитель.
В одном аспекте фармацевтическая композиция содержит один или более 'нерастворимых' наполнителей. В другом аспекте фармацевтическая композиция содержит один 'нерастворимый' наполнитель.
В одном аспекте фармацевтическая композиция содержит один или более связывающих агентов. В другом аспекте фармацевтическая композиция содержит один связывающий агент.
В одном аспекте фармацевтическая композиция содержит один или более разрыхляющих агентов. В другом аспекте фармацевтическая композиция содержит один разрыхляющий агент.
В одном аспект фармацевтическая композиция содержит один или более смазывающий агент. В другом аспекте фармацевтическая композиция содержит один смазывающий агент.
В другом аспекте наполнитель представляет собой смесь маннита и дигидрата двухосновного фосфата кальция.
В одном аспект 'растворимый' наполнитель выбран из маннита и сорбита. В другом аспекте 'растворимый' наполнитель выбран из маннита.
В одном аспекте 'нерастворимый' наполнитель выбран из дигидрата двухосновного фосфата кальция, безводного двухосновного фосфата кальция и трехосновного фосфата кальция. В другом аспекте 'нерастворимый' наполнитель выбран из дигидрата двухосновного фосфата кальция.
В другом аспекте связывающий агент выбран из гидроксипропилцеллюлозы.
В одном аспекте разрыхлитель выбран из крахмалгликолята натрия и кроскармеллозы натрия. В одном аспекте разрыхлитель выбран из крахмалгликолята натрия.
Дополнительные общепринятые эксципиенты, которые могут быть добавлены, включают консерванты, стабилизаторы, антиоксиданты, модификаторы текучести на основе диоксида кремния, агенты против прилипания или скользящие агенты.
Другие подходящие смазывающие агенты и дополнительные эксципиенты, которые могут быть использованы, раскрыты в Handbook of Pharmaceutical Excipients, 2nd Edition, American Pharmaceutical Association; The Theory and Practice of Industrial Pharmacy, 2nd Edition, Lachman, Leon, 1976; Pharmaceutical Dosage Forms: Tablets Volume 1, 2nd Edition, Lieberman, Hebert A., et al. 1989; Modern Pharmaceutics, Banker, Gilbert and Rhodes, Christopher T, 1979; Remington's Pharmaceutical Sciences, 15th Edition, 1975.
Подходящие смазывающие агенты включают, например, стеарат магния, стеариновую кислоту, пальмитиновую кислоту, стеарат кальция, карнаубский воск, гидрогенизированные растительные масла, минеральное масло, полиэтиленгликоли и стеарилфумарат натрия.
В одном аспекте смазывающий агент выбран из стеарата магния и стеарилфумарата натрия. В другом аспекте смазывающий агент представляет собой стеарат магния.
В одном аспекте фармацевтическая композиция содержит от 1% до 50% по массе Агента. В частности, она содержит от 20% до 45% по массе Агента.
В другом аспекте фармацевтическая композиция содержит от 1% до 90% по массе наполнителя. В частности, она содержит от 20% до 70% по массе наполнителя.
В другом аспекте фармацевтическая композиция содержит 1-70% по массе 'растворимого' наполнителя. В частности, она содержит от 20% до 45% по массе 'растворимого' наполнителя.
В другом аспекте фармацевтическая композиция содержит от 1% до 30% по массе 'нерастворимого' наполнителя. В частности, она содержит от 10% до 30% по массе 'нерастворимого' наполнителя.
В другом аспекте фармацевтическая композиция содержит от 2% до 8% по массе связывающего агента. В частности, она содержит от 3% до 6% по массе связывающего агента.
В другом аспекте фармацевтическая композиция содержит от 2% до 6% по массе разрыхляющего агента.
Надо понимать, что один конкретный эксципиент может действовать и как связывающий агент, и как наполнитель, либо как связывающий агент, наполнитель и разрыхлитель. В типичных случаях суммарное количество наполнителя, связывающего агента и разрыхляющего агента составляет, например, от 50% до 90% по массе композиции.
В типичных случаях один или более смазывающий агент присутствует в количестве от 0,5% до 3% и, в частности, от 0,5% до 1% по массе.
В другом аспекте это изобретение относится к фармацевтической композиции, содержащей Агент, маннит, дигидрат двухосновного фосфата кальция, гидроксипропилцеллюлозу, крахмалгликолят натрия и один или более смазывающий агент.
В другом аспекте это изобретение относится к фармацевтической композиции, содержащей:
Агент в количестве от 20% до 45% по массе;
маннит в количестве от 20% до 45% по массе;
дигидрат двухосновного фосфата кальция в количестве от 10% до 30% по массе;
гидроксипропилцеллюлозу в количестве от 3% до 6% по массе;
крахмалгликолят натрия в количестве от 2% до 6% по массе и
один или более смазывающих агентов в количестве от 0,5% до 3% по массе.
Желательно, чтобы физические свойства этих композиций были стабильными при хранении, поскольку изменения, например, в показателях времени распадаемости, скорости растворения или твердости таблетки среди прочего могут влиять на рабочие характеристики продукта. Возможно, что снижение скорости растворения при хранении в условиях тестирования стабильности в соответствии с требованиями Международного совета по гармонизации технических требований к регистрации фармацевтических продуктов, предназначенных для применения человеком (ICH), используемых для определения сроков хранения продукта, может снижать биологическую доступность Агента. Стабильность физических свойств можно измерять способами согласно Фармакопее США (USP) для тестирования времени распадаемости и растворения.
Желательно, чтобы композиции были химически стабильными, поскольку разложение посредством окисления, гидролиза, изомеризации, фотолиза, полимеризации или любого другого способа разложения, либо в результате смешивания с эксципиентами или любого другого способа может приводить к снижению биологической доступности. Химическую стабильность можно измерять подходящим показывающим стабильность хроматографическим методом для определения продуктов разложения (см. Aulton ME, Pharmaceutics - The Science of Dosage Form Design, 2nd Edition, 2002, Churchill Livingstone).
В другом аспекте предложена фармацевтическая композиция, которая пригодна для перорального введения, которая высвобождает по существу весь Агент и имеет желаемый профиль стабильности.
В одном аспекте изобретение относится к фармацевтической композиции, полученной влажным гранулированием.
Гранулирование представляет собой процесс, при котором исходные частицы (порошок) слипаются, образуя большие, состоящие из множества частиц образования, называемые гранулами. Гранулирование в норме начинается после исходного сухого смешивания порошкообразных ингредиентов с достижением практически однородного распределения ингредиентов по всему объему смеси. Методы гранулирования можно подразделить на два типа, а именно методы влажного гранулирования, в которых используют жидкость для образования гранул, и сухие методы, в которых жидкость не используют.
В методах сухого гранулирования исходные частицы порошка агрегируют под давлением (или прессованием). Существуют два основных способа: большую таблетку (также известную как брикет) получают с помощью мощного рабочего таблеточного пресса, либо частицы порошка прессуют между двумя вальцами с получением пластинчатого элемента или 'ленты' вещества (способ, известный как вальцовое прессование). В обоих случаях спрессованный материал размалывают с использованием подходящей техники размола с получением гранулированного материала. Гранулы можно затем прессовать в стандартном таблеточном прессе с получением таблеток.
Влажная грануляция включает получение массы частиц первичного порошка с помощью гранулирующей жидкости. Жидкость содержит растворитель, который можно удалить сушкой и который является нетоксичным. Гранулирующую жидкость можно использовать саму по себе или, что является более типичным, со связывающим агентом (связующим агентом) для обеспечения адгезии частиц в сухом состоянии. Связывающие агенты можно добавлять к системе в виде связывающего раствора (как части гранулирующей жидкости) или в виде сухого вещества, смешанного с частицами первичного порошка. Существует три основных типа влажного гранулятора: сдвиговые грануляторы (такие как планетарные смесители), грануляторы с высоким усилием сдвига (такие как Fielder или Diosna) и грануляторы с псевдоожиженным слоем (такие как Aeromatic или Glatt).
В другом аспекте предложена фармацевтическая композиция, полученная способом влажной грануляции, которая пригодна для перорального введения и высвобождает по существу весь Агент и имеет желаемый профиль стабильности.
В другом аспекте это изобретение относится к фармацевтической композиции, полученной способом влажной грануляции, содержащей Агент, маннит, дигидрат двухосновного фосфата кальция, гидроксипропилцеллюлозу, крахмалгликолят натрия и один или более смазывающих агентов.
В другом аспекте это изобретение относится к фармацевтической композиции, полученной влажной грануляцией с высоким усилием сдвига.
Влажная грануляция с высоким усилием сдвига представляет собой способ, который включает интенсивное сухое смешивание первичных порошков и последующее добавление гранулирующей жидкости, что приводит к образованию гранул. Гранулирующая жидкость содержит летучий растворитель (обычно воду) и может также включать связывающий агент, обеспечивающий адгезию частиц (связывающие агенты также могут быть добавлены сухими в виде порошков к массе препарата, подлежащего грануляции). По сравнению с порошками, из которых они состоят, гранулы обладает большими преимуществами в смысле улучшенных свойств текучести, сниженного риска сегрегации, повышенной гомогенности (информация взята из Aulton ME, Pharmaceutics - The Science of Dosage Form Design, 2nd Edition, 2002, Churchill Livingstone).
В одном аспекте фармацевтическая композиция находится в твердой лекарственной форме, такой как таблетка или капсула. В другом аспекте фармацевтическая композиция находится в форме таблетки.
В другом аспекте это изобретение относится к фармацевтической композиции, полученной способом влажной грануляции с высоким усилием сдвига, содержащей Агент, маннит, дигидрат двухосновного фосфата кальция, гидроксипропилцеллюлозу, крахмалгликолят натрия и один или более смазывающих агентов.
Агент существует в аморфной форме и в четырех существенно различных кристаллических формах (см. международную заявку на патент номер PCT/SE01/01239 (публикация номер WO 01/92262)). В другом аспекте это изобретение относится к фармацевтической композиции, определенной здесь выше, в которой Агент находится в кристаллической форме.
В еще одном аспекте это изобретение относится к фармацевтической композиции, содержащей Агент по существу в виде полиморфа II.
В еще одном аспекте это изобретение относится к фармацевтической композиции, содержащей Агент по существу в виде полиморфа III.
Композиции по этому изобретению, представляющие особый интерес, включают в себя, например, конкретные воплощения, раскрытые здесь ниже в прилагаемом Примере.
Надо понимать, что в соответствии с принципами, хорошо известными в данной области, можно осуществлять модификации способов влажной грануляции, включая порядок добавления компонентов и их просеивание и смешивание перед прессованием в таблетки.
Дополнительный аспект настоящего изобретения составляет способ получения фармацевтической композиции, который включает смешивание Агента с:
одним или более наполнителями, выбранными из маннита, сорбита, мальтодекстрина, мальтозы и декстрина, дигидрата двухосновного фосфата кальция, безводного двухосновного фосфата кальция, частично прежелатинизированного крахмала и трехосновного фосфата кальция или их смеси;
одним или более связывающими агентами, выбранными из гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, альгиновой кислоты, натриевой карбоксиметилцеллюлозы, коповидона и метилцеллюлозы или их смеси;
одним или более разрыхляющими агентами, выбранными из крахмалгликолята натрия, кроскармеллозы натрия и кросповидона или их смеси;
и одним или более смазывающими агентами.
Нижеследующая фармацевтическая композиция предназначена иллюстрировать это изобретение.
Влажный гранулятор с высоким усилием сдвига (Fielder GP1 с емкостью чаши 10 л) использовали в течение 4 минут для сухого смешивания Агента, маннита, дигидрата двухосновного фосфата кальция, гидроксипропилцеллюлозы и крахмалгликолята натрия в количествах, необходимых для получения 2,5 кг суммарного препарата. Воду добавляли через нагнетательный бак при приблизительно 50 г/мин до приблизительно 25% по массе. Суммарное время перемешивания составляло приблизительно 10 минут.
Сушку в псевдоожиженном слое осуществляли с помощью Glatt GPCG1 при 60°C до температуры продукта 42°C. Полученные гранулы размалывали в Quadra Comil 197, размолотые гранулы смешивали со стеаратом магния и из смеси прессовали таблетки.
Настоящее изобретение относится к химико-фармацевтической промышленности и представляет собой фармацевтическую композицию, содержащую: {1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол; наполнитель, представляющий собой смесь маннита и дигидрата двухосновного фосфата кальция; связывающий агент, представляющий собой гидроксипропилцеллюлозу; разрыхляющий агент, представляющий собой крахмалгликолят натрия; и один или более смазывающих агентов. Изобретение обеспечивает получение композиции активного соединения, обладающей высокой стабильностью и высокой биодоступностью активного агента. 11 з.п. ф-лы, 1 пр.
1. Фармацевтическая композиция, содержащая: {1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)циклопентан-1,2-диол;
наполнитель, представляющий собой смесь маннита и дигидрата двухосновного фосфата кальция;
связывающий агент, представляющий собой гидроксипропилцеллюлозу;
разрыхляющий агент, представляющий собой крахмалгликолят натрия;
и один или более смазывающих агентов.
2. Фармацевтическая композиция по п.1, где смазывающий агент выбран из стеарата магния и стеарилфумарата натрия.
3. Фармацевтическая композиция по п.1, где {1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)-циклопентан-1,2-диол присутствует в количестве от 20% до 45% по массе.
4. Фармацевтическая композиция по п.1, где наполнитель присутствует в количестве от 20% до 70% по массе.
5. Фармацевтическая композиция по п.1, где связывающий агент присутствует в количестве от 3% до 6% по массе.
6. Фармацевтическая композиция по п.1, где разрыхляющий агент присутствует в количестве от 2% до 6% по массе.
7. Фармацевтическая композиция по п.1, где смазывающий агент присутствует в количестве от 0,5% до 1% по массе.
8. Фармацевтическая композиция по п.1, где {1S-[1α,2α,3β(1S*,2R*),5β}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)-циклопентан-1,2-диол, по существу, присутствует в форме полиморфа II.
9. Фармацевтическая композиция по п.1, где {1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)-циклопентан-1,2-диол, по существу, присутствует в форме полиморфа III.
10. Фармацевтическая композиция по п.1, содержащая:
{1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]-амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)-циклопентан-1,2-диол в количестве от 20% до 45% по массе;
маннит в количестве от 20% до 45% по массе;
дигидрат двухосновного фосфата кальция в количестве от 10% до 30% по массе;
гидроксипропилцеллюлозу в количестве от 3% до 6% по массе;
крахмалгликолят натрия в количестве от 2% до 6% по массе и
один или более смазывающих агентов в количестве от 0,5% до 3% по массе.
11. Фармацевтическая композиция по любому из пп.1-10, которая получена способом влажного гранулирования.
12. Фармацевтическая композиция по п.11, которая получена способом влажного гранулирования с высоким усилием сдвига.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Handbook of Pharmaceutical Excipients, 5 Edition, London, Chicago, Pharmaceutical Press, 2006 | |||
| Марченко С.И | |||
| КОНСПЕКТ ЛЕКЦИЙ по курсу ТЕХНОЛОГИЯ ЛЕКАРСТВЕННЫХ ФОРМ И ГАЛЕНОВЫХ ПРЕПАРАТОВ для студентов специальности «Технология фармацевтических препаратов» | |||
| Одесский Национальный Политехнический | |||

