Изобретение относится к новым промежуточным соединениям, используемым для получения триазоло(4,5-d)пиримидина, которые пригодны в качестве лекарственных средств.
Адгезия тромбоцитов и их агрегация являются началом возникновения артериального тромбоза. Хотя процесс адгезии тромбоцита к нижней поверхности эндотелия может иметь большое значение в восстановлении поврежденных стенок сосудов, агрегация тромбоцитов, которая запускает этот процесс, может вызвать острую тромботическую окклюзию просвета жизненно важных сосудов, которая приводит к развитию состояний, характеризующихся высокой смертностью, таких как инфаркт миокарда и нестабильная стенокардия. Успех процедур, направленных на предотвращение или на ослабление выраженности указанных состояний, включающих, в частности, тромболизис и ангиопластику, зачастую определяется риском развития окклюзии или повторной окклюзии сосудов, опосредованных участием тромбоцитов.
Агрегация тромбоцитов может происходить в результате различных процессов. Однако независимо от начального стимула, конечным и общим результатом множества таких процессов является объединение тромбоцитов путем прикрепления фибриногена к сайту связывания на мембране - гликопротеину IIb/IIIa (GPIIb/IIIa). Высокая антитромбоцитарная эффективность антител или антагонистов к GPIIb/IIIa объясняется их способностью препятствовать развитию такого конечного общего состояния. Однако указанная эффективность может иметь отношение также к проблемам кровотечения, наблюдаемым при использовании таких средств. Тромбин может приводить к агрегации тромбоцитов по большей части независимо от других путей, однако, скорее всего, значительные количества тромбина не возникают без предварительной активации тромбоцитов через посредство других механизмов. Ингибиторы тромбина, такие как гирудин, представляют собой очень эффективные антитромботические средства, но при этом они могут вызывать обильное кровотечение, поскольку действуют как антитромбоцитарные и противосвертывающие средства (The TIMI 9a Investigators (1994), Circulation 90, pp. 1624-1630; The Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO) IIa Investigators (1994) Circulation 90, pp. 1631-1637; Neuhaus K.L. et al. (1994) Circulation 90, pp. 1638-1642).
Было показано, что аденозин 5'-дифосфат (АДФ) выполняет функцию ключевого медиатора тромбоза. Жизненно важную роль АДФ подтверждает тот факт, что другие средства, такие как адреналин и 5-гидрокситриптамин (5ГТ, серотонин), могут вызывать агрегацию лишь в присутствии АДФ. Ограниченная антитромботическая эффективность аспирина может быть связана с тем, что он блокирует только один источник АДФ, который высвобождается зависимым от тромбоксана образом после адгезии тромбоцитов (см., например, Antiplatelet Trialists' Collaboration (1994), Br. Med. J. 308, pp. 81-106 and Antiplatelet Trialists' Collaboration (1994), Br. Med. J. 308, pp. 159-168). При этом аспирин не оказывает никакого эффекта на агрегацию, возникающую при участии других источников АДФ, таких как поврежденные клетки, или АДФ, высвобождаемый в условиях турбулентного потока крови.
Вызываемая под действием АДФ агрегация тромбоцитов связана с подтипом Р2Т рецептора, расположенным на тромбоцитарной мембране. Р2Т рецептор (известный такие как P2YАДФ или Р2ТАС) участвует прежде всего в промежуточной агрегации/активации тромбоцитов и представляет собой рецептор, связывающийся с G-белком, который еще не был клонирован. Фармакологические характеристики указанного рецептора описаны в литературе (Humphries et al., Br. J. Pharmacology (1994), 113, 1057-1063 and Fagura et al., Br. J. Pharmacology (1998), 124, 157-164). Недавно было обнаружено, что антагонисты к данному рецептору значительно превосходят по своим возможностям другие антитромботические средства (см. J. Med. Chem. (1999) 42, 213). В соответствии с вышесказанным, в настоящее время имеется выраженная потребность найти другие антагонисты Р2Т (P2YАДФ или Р2ТАС), которые могли бы действовать как антитромботические средства.
В заявке на международный патент WO 9905143 раскрывается генерически серия триазоло[4,5-d]пиримидиновых соединений, обладающих активностью в качестве антагонистов Р2Т (P2YАДФ или Р2ТАС). В настоящее время показано, что ряд соединений, рассматриваемых в указанной заявке на международный патент WO 9905143, но специфически в ней не раскрываемые, обладают высокой активностью в рассматриваемом плане в сочетании с удивительно высокой метаболической стабильностью и биодоступностью, так что предполагаемая терапевтическая доза для длительного применения у человека может быть успешной.
Одним из объектов настоящего изобретения является соединение формулы (I):
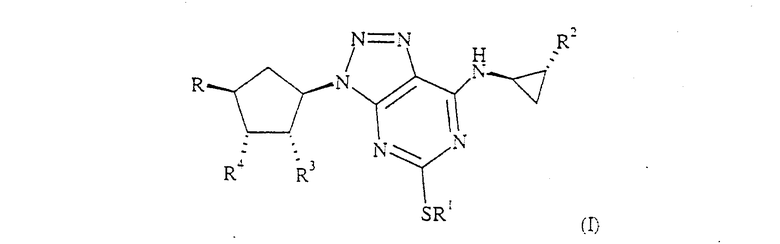
где R1 обозначает C3-5 алкил, необязательно замещенный одним или большим числом атомов галогена;
R2 обозначает фенильную группу, необязательно замещенную одним или большим числом атомов фтора;
R3 и R4 являются, оба, гидроксигруппами;
R обозначает XOH, где X обозначает CH2, OCH2CH2 или связь;
или его фармацевтически приемелемая соль или сольват или сольват такой соли.
При условии, что
когда X обозначает CH2 или связь, R1 не может быть пропилом,
когда X обозначает CH2 и R1 обозначает CH2CH2CF3, бутил или пентил, фенильная группа на R2 должна быть замещена фтором,
когда X обозначает OCH2CH2 и R1 пропил, фенильная группа на R2 должна быть замещена фтором.
Алкильные группы, независимо от того, присутствуют они сами по себе или входят в состав другой группы, являются неразветвленными и полностью насыщенными.
Соответственно R1 обозначает C3-5 алкил, необязательно замещенный одним или большим числом атомов фтора. Предпочтительно, R1 обозначает C3-5 алкил, необязательно замещенный на С-конце тремя атомами фтора. Более предпочтительно, R1 обозначает 3,3,3-трифторпропил, бутил или пропил.
Соответственно R2 обозначает фенил или фенил, замещенный одним или большим числом атомов фтора. Предпочтительно, R2 обозначает фенил, 4-фторфенил или 3,4-дифторфенил.
Соответственно R обозначает XOH, где X обозначает CH2, OCH2CH2 или связь.
Предпочтительно, R обозначает СН2ОН или ОСН2СН2ОН.
Особенно предпочтительные соединения включают:
[1R-[1α,2α,3β(1R*,2S*),5β]]-3-[7-[[2-(4-фторфенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил)-циклопентан-1,2-диол;
[1R-[1α,2α,3β(1R*,2S*),5β]]-3-[7-[[2-(3,4-дифторфенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил)-циклопентан-1,2-диол;
[1S-[1α,2α,3β(1S*,2R*),5β]]-3-[7-[2-(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)-циклопентан-1,2-диол;
[1R-[1α,2α,3β(1R*,2S*),5β]]-3-[5-(бутилтио)-7-[[2-(3,4-дифторфенил)циклопропил]амино]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил)-циклопентан-1,2-диол;
[1S-[1α,2β,3β,4α(1S*,2R*)]]-4-[5-(бутилтио)-7-[[2-(4-фторфенил)циклопропил]амино]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-циклопентан-1,2,3-триол;
[1S-[1α,2α,3β(1S*,2R*),5β]]-3-[7-[[2-(3,4-дифторфенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)-циклопентан-1,2-диол;
[1S-[1α,2α,3β,5β(1S*,2R*)]]-3-(2-гидроксиэтокси)-5-[7-(2-фенилциклопропил)амино]-5-[(3,3,3-трифторпропил)-тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-циклопентан-1,2-диол;
[1S-[1α,2β,3β,4α(1S*,2R*)]]-4-[5-(бутилтио)-7-[[2-(3,4-дифторфенил)циклопропил]амино]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]циклопентан-1,2,3-триол;
[1S-[1α,2α,3β(1S*,2R*),5β]]-3-[5-(бутилтио)-7-[(2-фе-нилциклопропил)амино]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)-циклопентан-1,2-диол;
а также их фармацевтически приемлемые соли или сольваты или сольваты таких солей.
Кроме того, настоящее изобретение относится к способу получения соединения формулы (I), который включает:
(а) взаимодействие соединения формулы (II):
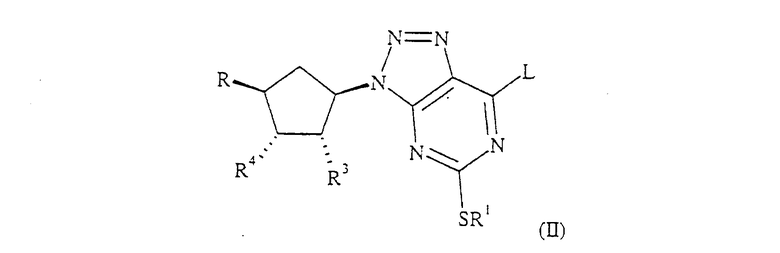
где R, R1, R3 и R4 определены ранее для формулы (I) или представляют собой их защищенные производные, или R3 и R4 вместе образуют связь в 5-членном кольце, или R обозначает CH2CH2OR', где R' обозначает С1-6 алкил или бензил, и L обозначает уходящую группу, такую как галоген или SR, с соединением формулы (III):
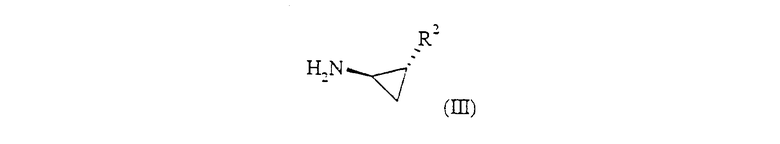
где R2 определен для формулы (I), или представляет собой его защищенное производное,
или где Х обозначает связь;
(б) гидроксилирование соединения формулы (IV):
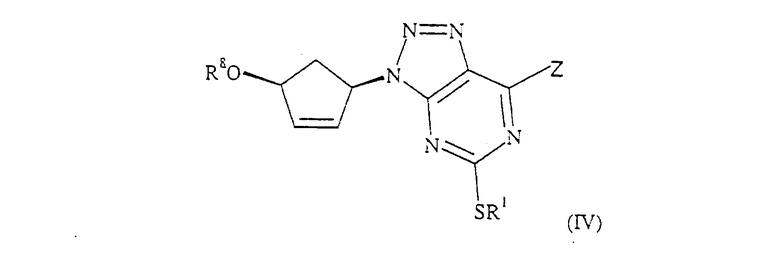
где R1 определен для формулы (I), и R8 обозначает СН2СН2ОР3, где Р3 обозначает Н или защитную группу, или R8 обозначает CH2COOR', где R' обозначает С1-6 алкил или бензил, и Z обозначает NH2 или

где R2 определен для формулы (I),
и для пунктов (а) и (б) необязательно друг за другом и в любом порядке:
преобразование одной или более функциональных групп в другие функциональные группы;
удаление защитных групп;
образование фармацевтически приемлемой соли или сольвата или сольвата такой соли.
Соединения формулы (II) могут взаимодействовать с аминами формулы (III) в присутствии основания, такого как третичный органический амин, в инертном растворителе, таком как дихлорметан, при температуре окружающей среды или при повышенной температуре. Другие приемлемые основания включают неорганические основания, такие как карбонат калия.
Гидроксигруппы R3 и R4 могут быть защищены в виде групп ОР1 и ОР2, где Р1 и Р2 представляют собой защитные группы. Примеры подходящих защитных групп для соединений формулы (II) включают С1-6алкильную (предпочтительно метильную), бензильную, (С1-6алкил)3Si (предпочтительно, трет-бутилдиметилсилильную) и С(О)С1-6-алкильную группу, такую как ацетил. Предпочтительно, две группы Р1 и Р2 вместе с атомами, к которым они присоединены, образуют алкилиденовое кольцо, такое как метилиденовое или изопропилиденовое кольцо. Альтернативно, Р1 и Р2 могут образовывать алкоксиметилиденовое кольцо, такое как этоксиметилиден.
Защитные группы добавляют и удаляют в условиях известных реакций. Использование защитных групп достаточно полно описано в литературе (Protective Groups in Organic Chemistry, edited by J.W.F. McOmie, Plenum Press (1973), and Protective Groups in Organic Synthesis, 2nd edition, T.W.Greene & P.G.M. Wutz, Wiley-Interscience (1991)).
Эфирная защитная группа может быть удалена путем щелочного гидролиза, например, при использовании гидроксида металла, предпочтительно, гидроксида щелочного металла, такого как гидроксид натрия или гидроксид лития, или гидроксида четвертичного аммония в растворителе, таком как водный этанол или водный тетрагидрофуран, при температуре от 10°С до 100°С, предпочтительно при комнатной температуре; или с помощью кислотного гидролиза при использовании минеральной кислоты, такой как HCl, или сильной органической кислоты, такой как трихлоруксусная кислота, в растворителе, таком как водный 1,4-диоксан. Триалкилсилильные защитные группы могут быть удалены при использовании, например, источника иона фтора, в частности, фторида тетра-н-бутиламмония или фтористого водорода. В том случае, когда либо один, либо оба Р1 и Р2 представляют собой С1-6алкил, защитные группы могут быть удалены с помощью трибромида бора. Бензильные группы могут быть удалены гидрогенолизом с использованием катализатора на основе переходного металла, например палладия на угле, в атмосфере водорода, под давлением от 1 до 5 бар, в растворителе, таком как уксусная кислота.
Соединение формулы (II) может быть получено при диазотировании соединения формулы (V):
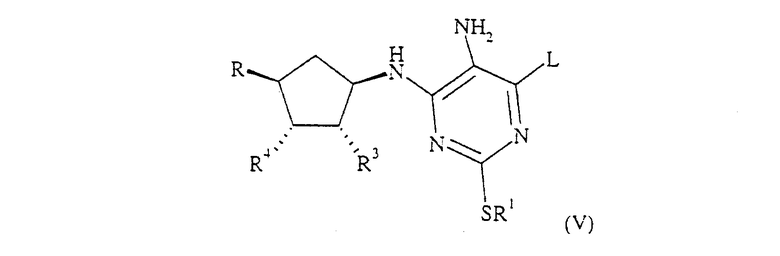
где R1 определен для формулы (I), и R также определен для формулы (I) или представляет собой его защищенное производное, или обозначает OCH2CO2R', где R' обозначает С1-6 алкил или бензил, и L определен выше, тогда как R3 и R4 определены для формулы (I) или представляют собой их защищенные производные, или R3 и R4 вместе образуют связь в 5-членном кольце, с нитритом металла, например нитритом щелочного металла, в особенности с нитритом натрия, в разбавленной водной кислоте, например в 2М HCl, или с нитритом С1-6 алкила, в инертном растворителе, при температуре от примерно -20°С до примерно 100°С. Предпочтительные условия включают использование нитрита изоамила в ацетонитриле при температуре примерно 80°С.
Соединение формулы (V), в котором R обозначает СН2ОН, R3 и R4 обозначают гидроксильные группы или их защищенные производные, и L определен выше, может быть получено при восстановлении соединения формулы (VI):
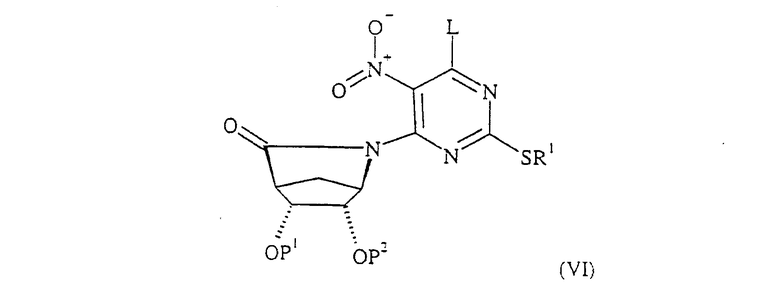
где R1, L, P1 и Р2 определены выше.
Восстановление нитрогруппы может быть осуществлено, например, гидрогенизацией при использовании катализатора на основе переходного металла при комнатной температуре, например, при использовании палладия на угле в атмосфере водорода, предпочтительно под давлением от 1 до 5 атмосфер, в растворителе, например в этаноле, или при использовании железа в кислотном растворителе, таком как уксусная кислота, при температуре примерно 100°.
Восстановление лактама может быть осуществлено с использованием гидридов комплексного металла, такого как алюмогидрид лития, в растворителе, таком как эфир, или предпочтительно, при использовании боргидрида натрия в подходящем растворителе, таком как метанол.
Соединение формулы (VI) может быть получено при взаимодействии соединения формулы (VII):
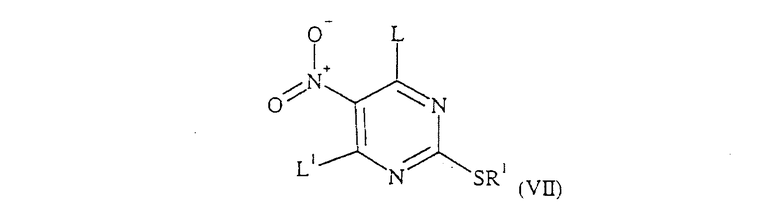
где L и R1 определены выше и L1 обозначает уходящую группу, например атом галогена, при этом предпочтительно L и L1 обозначают одну и ту же группу, с соединением формулы (VIII):
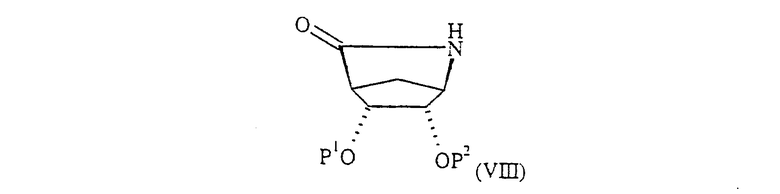
где Р1 и Р2 определены выше, в присутствии основания, такого как С1-6-алкил-М или МН, где М обозначает ион металла, например н-бутиллитий, в инертном растворителе, таком как тетрагидрофуран, при температуре от примерно -10°С до примерно 100°С. Предпочтительно используют гидрид лития в тетрагидрофуране при комнатной температуре.
Одна или несколько функциональных групп могут быть преобразованы в другие функциональные группы с использованием стандартных химических процедур. Соединение, в котором Х обозначает связь, может быть превращено в соединение, в котором Х обозначает О(СН2)2, при обработке основанием и затем LY, где L обозначает уходящую группу, и Y обозначает (СН2)2ОН или его защищенную (CH2)2OH, или Y обозначает CH2COOR', где R' обозначает С1-6 алкил или бензил. Соединение, в котором R обозначает CH2CH2OR' может быть в ходе восстановления преобразовано в соединение, в котором R обозначает О(СН2)2ОН, например, с использованием DIBAL-H®. Группа SR1 может быть подвергнута перегруппировке при окислении серы, с использованием, например, оксонаТМ или mCBPA, с последующей обработкой соединением R1-SM, где R1 обозначает другую R1 группу и М обозначает металл, такой как натрий. Альтернативно, продукт окисления серы может быть обработан с использованием MSH, где М обозначает металл, такой как натрий, с последующей обработкой основанием и R1X, где R1 обозначает другую R1 группу и Х обозначает уходящую группу. Подходящие основания включают N,N-диизо-пропилэтиламин.
Соединение формулы (II), в котором R, R1, R3 и R4 определены выше для формулы (I) или представляют собой их защищенные производные, или R3 и R4 вместе образуют связь в 5-членном кольце, или R обозначает OCH2CO2R', где R' обозначает С1-6 алкил или бензил, и L обозначает уходящую группу, такую как галоген, может быть преобразовано в соединение формулы (II), в котором R, R1, R3 и R4 определены выше и L обозначает NH2, при обработке диазотирующим агентом в присутствии галогенирующего средства, предпочтительно изоамил-нитрита, и тетрабромида углерода.
Соединение формулы (II), в котором R, R1, R3 и R4 определены выше и L обозначает NH2, может быть получено при обработке соединения формулы (II), в котором R, R1, R3 и R4 определены выше и L обозначает уходящую группу, такую как галоген, с аммиаком в растворителе, таком как метанол.
Соединение формулы (V) может быть получено при обработке соединения формулы (XI):
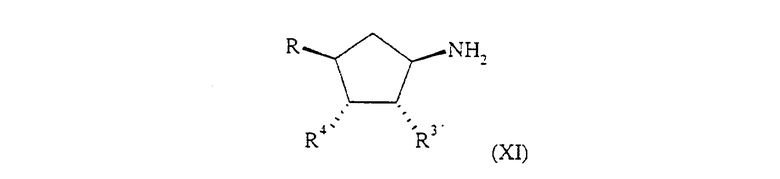
где R, R3 и R4 определены для формулы (I) или представляют собой их защищенные производные, или R обозначает OCH2CO2R', где R' обозначает С1-6 алкил или бензил, или R3 и R4 вместе образуют связь в 5-членном кольце, с соединением формулы (VII), определенной выше, с последующим восстановлением нитрогруппы. Реакцию проводят в инертном растворителе, таком как дихлорметан или 1,4-диоксан, в присутствии ненуклеофильного основания, такого как N,N-диизопропил-амин, при температуре от примерно -20°С до примерно 150°С, предпочтительно при температуре окружающей среды.
Соединения формулы (II), в которых R был определен для формулы (I), R3 и R4 вместе образуют связь в 5-членном кольце и L обозначает SR1 или его защищенное производное, могут быть получены при взаимодействии соединения формулы (XII):
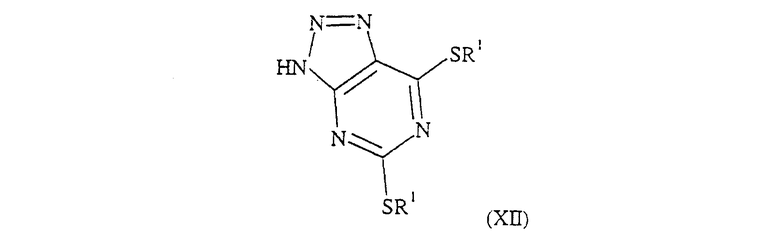
где R1 группа обозначена для формулы (I),
с соединением формулы (XIII):
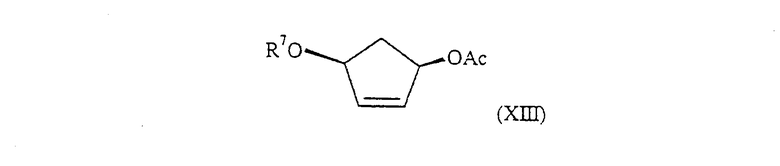
где R7 обозначает Н или его защищенное производное. Реакция может быть проведена в присутствии подходящего комплекса переходного металла, предпочтительно, тетракистрифенилфосфина палладия(0).
Соединения формулы (XII) могут быть получены из соединений формулы (XIV):

*при взаимодействии с соединением R1X, где R1 обозначен для формулы (I), и Х обозначает уходящую группу, такую как галоген, с последующей циклизацией.
Соединения формулы (XI), где R обозначает ОН или его защищенную ОН, и R3 и R4 определены для формулы (I) или представляют собой их защищенные производные, могут быть получены из соединений формулы (XIII), где R7 обозначает Н или защитную группу, путем обработки их бисэфиром или имидодикарбаминовой кислотой с использованием палладиевого катализатора с последующим гидроксилированием двойной связи и необязательно снятием защиты с атома азота. Предпочтительно используют имидодикарбоновую кислоту, бис-(1,1-диметилэтиловый)эфир и тетракистрифенилфосфин палладия(0) с последующим добавлением тетроксида осмия и с удалением защитной группы при помощи соляной кислоты в метаноле.
Соединения формулы (XI), в которой R обозначает OCH2CO2R', где R' обозначает С1-6 алкил и R3 и R4 вместе образуют связь в 5-членном кольце, могут быть получены из соединений формулы (XIII), где R7 обозначает Н или защитную группу, при обработке азидом в присутствии палладиевого катализатора с последующим восстановлением азида и алкилированием спирта, как было описано ранее.
Соединения формулы (XI), в которой R обозначает ОСН2СН2ОН, и R3 и R4 определены для формулы (I) или представляют собой их замещенные производные, могут быть получены из соединений формулы (XI), где R обозначает ОН и R3 и R4 определены для формулы (I) или представляют собой их замещенные производные, при защите атома азота, алкилировании спирта с использованием сложного эфира 2-галоген-уксусной кислоты и с последующим восстановлением указанного эфира и удалением защитной группы от атома азота. Авторы предпочитают осуществлять защиту атома азота в виде карбобензилокси производного с использованием бензилхлорформиата с последующим алкилированием при помощи этилбромацетата и трет-бутоксида калия, далее при восстановлении сложного эфира с помощью боргидрида лития в тетрагидрофуране и удалении защитной группы, находящейся на атоме азота при гидрогенизации в присутствии палладия на угле. Кроме того, авторы рассматривают в качестве предпочтительного тот вариант, в рамках которого спирты R3 и R4 защищаются в виде изопропилиденового кольца.
Амины формулы (III) могут быть получены с помощью описанных в литературе процедур (H.Nishiyama et al., Bull. Chem. Soc. Jpn., 1995, 68, 1247; P. Newman, Optical Resolution Procedures for Chemical Compounds, Vol. 1, Amines and Related Compounds, Optical Resolution and Information Centre; Manhattan College, Riverdale, NY, 1978, p.120; J. Vallgarda et al., J. Chem. Soc. Perkin 1, 1994, 461; International Patent Application WO 9905143).
Все новые соединения представляют собой отдельный объект изобретения.
Соли соединений формулы (I) могут быть образованы при взаимодействии свободной кислоты или ее соли или свободного основания или его соли или производного с одним или более эквивалентами соответствующего основания (например, гидроксида аммония, необязательно замещенного С1-6алкилом или гидроксидом щелочного или щелочноземельного металла) или кислоты (например, галоген-содержащей (в особенности HCl), серной, щавелевой или фосфорной кислоты). Реакция может быть проведена в растворителе или в другой среде, в которой соль нерастворима, или в растворителе, в котором соль растворяется, например в воде, этаноле, тетрагидрофуране или диэтиловом эфире, которые могут быть удалены под вакуумом, например, в процессе лиофильной сушки. Указанная реакция может проводиться как процесс диспропорционирования или может быть осуществлена с использованием ионообменной смолы. Предпочтительны нетоксичные физиологически приемлемые соли, хотя могут использоваться также и другие соли, в частности, в случае выделения или очистки продукта.
Соединения согласно настоящему изобретению действуют как антагонисты Р2Т (P2YАДФ или Р2ТАС) рецептора. Соответственно указанные соединения могут применяться для лечения, включая комбинированную терапию, в особенности, их применение показано в качестве ингибиторов тромбоцитарной активации, агрегации и дегрануляции, в качестве промоторов дезагрегации тромбоцитов, антитромботических средств или в рамках лечения или профилактики нестабильной стенокардии, первичных тромботических осложнений атеросклероза артерий, таких как кровоизлияние тромботической или эмболической природы, преходящие нарушения мозгового кровообращения, заболевания периферических сосудов, инфаркт миокарда при наличии или в отсутствие тромбоза, артериальные осложнения, связанные с проведением инвазивных лечебных процедур при атеросклерозе, таких как ангиопластика, включая коронарную ангиопластику (РТСА), эндартерэктомия, помещение стента и техника коронарной пластики и хирургической пластики других сосудов, тромботические осложнения хирургических или механических повреждений, таких как процедуры по спасению тканей после случайной или хирургической травмы, реконструктивной хирургии, включая пересадку участков кожной и мышечной ткани, состояния, характеризующиеся наличием компонента диффузного тромботического/тромбоцитарного истощения, такие как диссеминированное внутрисосудистое свертывание, тромбоцитопенический акроангиотромбоз, гемолитический уремический синдром, тромботические осложнения септицемии, респираторный дистресс-синдром взрослых, антифосфолипидный синдром, гепарин-индуцированная тромбоцитопения и преэклампсия/эклампсия, или для лечения венозных тромбозов, таких как тромбоз глубоких вен, окклюзия вен, гематологические состояния, такие как миелопролиферативная болезнь, включая тромбоцитемию, серповидно-клеточную анемию; или указанные соединения могут найти применение при профилактике механически индуцированной активации тромбоцитов in vivo, такой как в случае кардио-пульмонарного шунтирования и экстракорпоральной мембранной оксигенации (для профилактики микротромбоэмболии), механически индуцированной активации тромбоцитов in vitro, такой как в случае консервации продуктов крови, например концентрированных препаратов тромбоцитов, или при окклюзионном шунте, например, при почечном диализе и плазмаферезе, в случае вторичного тромбоза после повреждения/воспаления сосудов, такого как васкулит, артериит, гломерулонефрит, воспалительный процесс в кишечнике и реакция отторжения трансплантата органов, в случае состояний, таких как мигрень, феномен Рейнода (Raynaud), а также состояний, при которых тромбоциты могут усугубить течение воспалительного процесса в сосудистой стенке, таких как образование/прогрес-сирование атероматозной бляшки, стеноз/рестеноз, и других воспалительных состояний, таких как астма, при которых тромбоциты и производные от тромбоцитов факторы вовлекаются в течение иммунологического заболевания. Другие показания для применения рассматриваемых соединений включают лечение заболеваний ЦНС и предупреждение роста и распространения опухолей.
Настоящее изобретение относится также к использованию предлагаемых соединений в качестве активного ингредиента при производстве лекарственного средства, предназначенного для лечения или профилактики указанных выше заболеваний. В частности, соединения согласно настоящему изобретению, могут применяться для лечения инфаркта миокарда, кровоизлияния тромботической природы, преходящего нарушения мозгового кровообращения, заболевания периферических сосудов и стабильной и нестабильной стенокардии, в особенности, нестабильной стенокардии. Настоящее изобретение также относится к способу лечения или профилактики указанных выше заболеваний, который включает введение субъекту, страдающему от такого заболевания или чувствительного к нему, терапевтически эффективного количества соединения согласно настоящему изобретению.
Соединение может вводиться местно, например, в легкое или в воздушные пути, в виде растворов, суспензий, аэрозолей HFA и сухих порошковых композиций; или оно может вводиться системно, например, при пероральном употреблении таблеток, пилюль, капсул, сиропов, порошков или гранул или при парентеральном введении их в виде стерильных парентеральных растворов или суспензий, при подкожном введении или при ректальном введении суппозиториев, а также чрескожно.
Соединения согласно настоящему изобретению могут вводиться как сами по себе, так и в составе фармацевтической композиции, включающей соединение согласно изобретению в сочетании с фармацевтически приемлемым разбавителем, адъювантом и/или носителем. Особенно предпочтительными являются композиции, не содержащие материал, способный вызывать неблагоприятные побочные эффекты, например аллергическую реакцию.
Сухие порошковые композиции и HFA аэрозоли, содержащие соединения согласно изобретению, могут быть введены перорально или путем назальной ингаляции. Желательно, чтобы в случае ингаляционного применения указанное соединение было мелко размолото. Соединения согласно настоящему изобретению могут вводиться также с помощью ингалятора для сухого порошка. Указанный ингалятор может представлять собой либо ингалятор с однократной дозой, либо ингалятор, включающий количество, достаточное для многократного применения, а также он может представлять собой сухой порошковый ингалятор, активируемый дыханием.
В одном варианте тонкоизмельченное соединение смешивают с веществом-носителем, например моно-, ди- или полисахаридом, спиртовой формой сахара или другим полиолом. Подходящие носители включают сахара и крахмал. Альтернативно, тонкоизмельченное соединение может быть покрыто другим веществом. Далее порошковая смесь может быть расфасована в твердые желатиновые капсулы, каждая из которых содержит желательную дозу активного соединения.
В другом возможном варианте проводят обработку тонкоизмельченного порошка для получения сферических форм, разбиваемых в ходе ингаляции. Такой сферонизированный порошок может быть введен в качестве лекарственного средства в резервуар многоразового ингалятора, например Turbuhaler®, дозатор которого отмеряет нужную дозу, которую затем вдыхает пациент. При использовании такой системы активное соединение при наличии или в отсутствие вещества-носителя доставляется к пациенту.
Фармацевтическая композиция, содержащая соединение согласно изобретению, может быть представлена в виде таблеток, пилюль, капсул, сиропов, порошков или гранул, предназначенных для перорального введения, а также может иметь вид стерильных парентеральных или подкожных растворов, суспензий для парентерального введения или суппозиториев для ректального введения.
При пероральном введении активное соединение может быть смешано с адъювантом или носителем, например с лактозой, сахарозой, сорбитом, маннитом, крахмалами, такими как картофельный крахмал, кукурузный крахмал или амилопектин, с производными целлюлозы, связующими веществами, такими как желатин или поливинилпирролидон, и замасливателями, такими как стеарат магния, стеарат кальция, полиэтиленгликоль, воски, парафин и др., и затем спрессовано в таблетки. При наличии потребности в покрытых оболочкой таблетках, ядра, полученные в соответствии с описанной процедурой, покрывают концентрированным сахарным раствором, который может включать, например, аравийскую камедь, желатин, тальк, диоксид титана и др. В альтернативном варианте таблетка может быть покрыта подходящим полимером, растворенным либо в легко летучем органическом растворителе, либо в водном растворителе.
Для получения мягких желатиновых капсул указанное соединение может быть смешано, например, с растительным маслом или с полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы соединения при включении в состав такой композиции любого из указанных выше наполнителей для таблеток, например лактозы, сахарозы, сорбита, маннита, крахмалов, производных целлюлозы или желатина. В твердые желатиновые капсулы для их наполнения могут быть введены как жидкие, так и полутвердые композиции.
Жидкие препараты для перорального применения могут иметь вид сиропов или суспензий, например растворов, содержащих нужное соединение, при этом поддерживается баланс между содержанием сахара и смеси этанола, воды, глицерина и пропиленгликоля. Указанные жидкие композиции могут необязательно включать красители, вкусовые добавки, сахарин и карбоксиметилцеллюлозу в качестве загустителя, а также другие, известные в фармацевтической практике наполнители.
Примеры
Ниже приведены неограничивающие примеры, иллюстрирующие настоящее изобретение.
В приведенных примерах спектры ЯМР прописывают на спектрометре Varian Unity Inova 300 или 400 и следующим образом записывают МС спектры: EI спектры получают на спектрометре VG 70-250S или Funnigan Mat Incos-XL, FAB спектры получают на спектрометре VG70-250SEQ, ESI и APCI спектры получают на спектрометре Funnigan Mat SSQ7000 или Micromass Platform. Препаративное разделение методом ВЭЖХ осуществляют в основном с использованием колонок Novapak®, Bondapak® или Hypersil®, наполненных силикагелем BDSC-18 для хроматографирования с обращением фаз. Флэш-хроматографию (указанную в примерах как хроматография на (SiO2)) проводят с использованием силикагеля Fisher Matrix с размером частиц 35-70 мкм. В тех примерах, где отмечено наличие ротамеров в спектре протонного ЯМР, указаны химические смещения лишь для основного ротамера.
Пример 1
[1R-[1α,2α,3β(1R*,2S*),5β]]-3-[7-[[2-(4-фторфенил)-циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил)-циклопентан-1,2-диол
а) [3aS-[1(E),3aα,6α,7aβ]]-1-[3-(4-фторфенил)-1-оксо-2-пропенил]-гексагидро-8,8-диметил-3Н-3а,6-метано-2,1-бензизотиазол-2,2-диоксид
Смесь 3-(4-фторфенил)-2-пропеновой кислоты (3,0 г) и тионилхлорида (5,0 мл) перемешивают при температуре 70°С в течение 1 часа, после чего реакционную смесь концентрируют под пониженным давлением. Осадок дважды делают азеотропным с помощью дихлорметана и затем растворяют в толуоле (10 мл). К суспензии гидрида натрия (60% дисперсия в масле; 0,99 г) в толуоле (40 мл) добавляют раствор [3aS-(3aα,6α,7aβ)]-гексагидро-8,8-диметил-3Н-3а,6-метано-2,1-бензизотиазол-2,2-диоксида (3,89 г) в толуоле (40 мл) и смесь перемешивают в течение 30 минут. Далее к реакционной смеси добавляют указанный выше раствор и полученную суспензию перемешивают в течение 16 часов. После этого добавляют воду (200 мл), собирают органический материал и экстрагируют водную фазу дихлорметаном (3×100 мл). Органический материал собирают, высушивают и концентрируют. Повторная кристаллизация (этанол) дает соединение, указанное в заголовке, в виде бесцветных игольчатых кристаллов (5,92 г).
MS (APCI) 364 (М+Н+, 100%)
б) [3aS-[1(1S*,2S*),3aα,6α,7aβ]]-1-[[2-(4-фторфенил)-циклопропил]карбонил]-гексагидро-8,8-диметил-3Н-3а,6-метано-2,1-бензизотиазол-2,2-диоксид
К раствору продукта на стадии а) (5,90 г) при температуре 0°С добавляют раствор диазометана (2,9 г) в диэтиловом эфире (150 мл) (приготовленный по процедуре, описанной в работе Vogel, Textbook of Practical Organic Chemistry, Fifth Edition, Longman Scientific and Technical, p.432) и ацетата палладия(II) (18 мг) в дихлорметане (350 мл) и полученную реакционную смесь перемешивают при температуре 0°С в течение 5 часов. Затем добавляют уксусную кислоту (5 мл), реакционную смесь промывают насыщенным раствором бикарбоната натрия (200 мл) и органический материал фильтруют через слой силикагеля. После концентрирования под вакуумом осадок повторно кристаллизуют (этанол) с получением соединения, указанного в заголовке, в виде бесцветных игольчатых кристаллов (3,81 г).
MS (APCI) 378 (М+Н+, 100%)
в) (1R-транс)-2-(4-фторфенил)-циклопропанкарбоновая кислота
Суспензию продукта со стадии б) (3,74 г) и моногидрат гидроксида лития (4,11 г) в тетрагидрофуране (100 мл)/воде (3 мл) перемешивают при 50°С в течение 24 часов. Далее реакционную смесь концентрируют под вакуумом, остаток растворяют в воде (100 мл), подкисляют 2 н. HCl и экстрагируют дихлорметаном (3×75 мл). Органический материал высушивают и концентрируют. Очистка (SiO2, при использовании в качестве элюента смеси изогексан:диэтиловый эфир, 2:1) дает соединение, указанное в заголовке, в виде бесцветного твердого вещества (1,78 г).
MS (APCI) 179 (М-Н+, 100%)
г) (1R-транс)-2-(4-фторфенил)циклопропанамин,[R-(R*, R*)]-2,3-дигидроксибутандиоат (1:1)
К раствору продукта со стадии в) (1,78 г) и триэтиламина (2,7 мл) в смеси ацетона/воды (10:1, 23 мл) при температуре 0°С добавляют в течение 5 минут этилхлорформиат (2,0 мл). Раствор выдерживают при температуре 0°С в течение 30 минут, после чего добавляют азид натрия (1,52 г) в воде (6 мл). Еще через 1 час добавляют воду (350 мл) и реакционную смесь экстрагируют толуолом (3×100 мл). Затем органические экстракты объединяют и высушивают, после чего нагревают при температуре кипения с обратным холодильником в течение 2 часов с использованием сита с поддувом. После охлаждения раствора к нему добавляют 6 н. HCl (50 мл) и полученную смесь нагревают при температуре кипения с обратным холодильником в течение 3 часов. Затем добавляют воду (150 мл) и подщелачивают водную фазу 2 н. NaOH (водн.), после чего проводят экстракцию дихлорметаном (3×100 мл). Органическую фазу высушивают и концентрируют. Амин растворяют в этаноле (5 мл) и добавляют раствор L-винной кислоты (1,48 г) в этаноле (20 мл). Через 20 минут собирают твердый материал, что дает соединение, указанное в заголовке, в виде бесцветных игольчатых кристаллов (1,12 г).
ЯМР δН (d6-ДМСО) 1,07-1,39 (1Н, м), 1,22-1,29 (1Н, м), 2,16-2,23 (1Н, м), 2,64-2,70 (1Н, м), 3,95 (2Н, с), 7,06-7,19 (4Н, м)
д) [3aR-[3aα,4α,6α(1R*,2S*),6aα]]-6-[7-[[2-(4-фторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанол
К раствору [3aR-(3aα,4α,6α,6аα)]-6-[7-хлор-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанола (полученного в соответствии с процедурой, раскрытой в заявке на международный патент WO 9703084) и продукта со стадии г) (0,75 г) в дихлорметане (25 мл) добавляют N,N-диизопропил-этиламин (1,29 г). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов, затем промывают водой, высушивают и выпаривают. Остаток очищают (SiO2, с использованием в качестве элюента смеси этилацетат:изогек-сан, 1:1) с получением соединения, указанного в заголовк, (1,25 г).
MS (APCI) 515 (М+Н+, 100%)
е) [3aR-[3aα,4α,6α(1R*,2S*),6aα]]-6-[7-[[2-(4-фторфе-нил)циклопропил]амино]-5-(пропилсульфонил)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанол
К суспензии продукта со стадии д) (1,25 г) в этаноле (25 мл) добавляют 3-хлорпероксибензойную кислоту (70%, 1,8 г) и полученный раствор перемешивают при комнатной температуре в течение 2 часов. Далее реакционную смесь концентрируют, и остаток отбирают этилацетатом (500 мл), промывают 10% водным раствором метабисульфита натрия (2×100 мл) и 10% водным раствором бикарбоната натрия (2×100 мл), после чего высушивают и концентрируют с получением соединения, указанного в заголовке, под е), (1,4 г).
MS (APCI) 547 (М+Н+, 100%)
ж) [[3aR-[3aα,4α,6α(1R*,2S*),6aα]]-6-[7-[[2-(4-фторфенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанол
Гидрат гидросульфида натрия (1,4 г) добавляют к раствору продукта со стадии е) (1,4 г) в диметилсульфоксиде (20 мл) и раствор перемешивают при комнатной температуре в течение 1,5 часа. Затем добавляют солевой раствор (150 мл), смесь подкисляют уксусной кислотой и экстрагируют этилацетатом (3×100 мл). Органическую фазу высушивают и концентрируют и образовавшийся остаток делают азеотропным с помощью толуола (3×100 мл). Далее остаток растворяют в N,N-диметилформамиде (20 мл) и затем добавляют N,N-диизо-пропилэтиламин (0,33 г) и 3,3,3-трифторпропилбромид (0,48 г). После перемешивания в течение 30 минут при температуре 50°С реакционную смесь разбавляют этилацетатом (100 мл), промывают водным солевым раствором (3×100 мл), высушивают и концентрируют, после чего образовавшийся остаток очищают (SiO2, при использовании в качестве элюента смеси изогексан:этилацетат, 1:1) с получением соединения, указанного в заголовке под ж), (1,4 г).
MS (APCI) 569 (М+Н+, 100%)
з) [1R-[1α,2α,3β(1R*,2S*),5β]]-3-[7-[[2-(4-фторфенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1, 2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил)-циклопентан-1,2-диол
Перемешивают в течение 1 часа при комнатной температуре раствор продукта со стадии ж) (1,4 г) в трифторуксусной кислоте (10 мл) и воде (2 мл). Реакционную смесь разбавляют этилацетатом (400 мл) и затем промывают раствором бикарбоната натрия (400 мл), высушивают и выпаривают. Остаток очищают (SiO2 с использованием в качестве элюента смеси метанол:хлороформ, 3:47) с получением целевого соединения (0,44 г).
MS (APCI) 529 (М+Н+, 100%)
ЯМР δН (d6-ДМСО) 9,42 (1Н, д), 7,27-7,22 (2Н, м), 7,14-7,08 (2Н, м), 5,01-4,95 (2Н, м), 4,73-4,70 (2Н, м), 4,44-4,41 (1Н, м), 3,87-3,84 (1Н, м), 3,50-3,45 (2Н, м), 3,26-3,13 (3Н, м), 2,60-2,55 (1Н, м), 2,28-2,20 (2Н, м), 2,10-2,06 (1Н, м), 1,90-1,80 (1Н, м), 1,49-1,46 (1Н, м), 1,33-1,30 (1Н, м)
Пример 2
[1R-[1α,2α,3β(1R*,2S*),5β]]-3-[7-[[2-(3,4-дифторфе-нил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил)-циклопентан-1,2-диол
а) [3aS-[1(E),3aα,6α,7аβ]]-1-[3-(3,4-дифторфенил)-1-оксо-2-пропенил]-гексагидро-8,8-диметил-3Н-3а,6-метано-2,1-бензизотиазол-2,2-диоксид
Промежуточное соединение, указанное в заголовке под а), получают в соответствии с процедурой примера 1, стадия а) с использованием 3-(3,4-дифторфенил)-2-пропеновой кислоты.
MS (APCI) 382 (М+Н+, 100%)
б) [3aS-[1(1S*,2S*),3aα,6α,7aβ]]-1-[[2-(3,4-дифторфе-нил)циклопропил]карбонил]-гексагидро-8,8-диметил-3Н-3а,6-метано-2,1-бензизотиазол-2,2-диоксид
Указанное под б) промежуточное соединение получают в соответствии с процедурой примера 1, стадия б) с использованием продукта, полученного на стадии а).
MS (APCI) 396 (М+Н+, 100%)
в) (1R-транс)-2-(3,4-дифторфенил)-циклопропанкарбоновая кислота
Указанное в заголовке под в) промежуточное соединение получают в соответствии с процедурой примера 1, стадия в) с использованием продукта, полученного на стадии б).
ЯМР δН (CDCl3) 7,06 (1Н, дт, J=10,0 Гц, J=8,5 Гц), 6,93-6,80 (2Н, м), 2,58-2,52 (1Н, м), 1,88-1,82 (1Н, м), 1,66 (1Н, дт, J=9,2 Гц, J=5,2 Гц), 1,34 (1Н, ддд, J=8,5 Гц, J=6,5 Гц, J=4,8 Гц).
г) (1R-транс)-2-(3,4-дифторфенил)циклопропанамин,[R-(R*,R*)]-2,3-дигидроксибутандиоат (1:1)
Указанное в заголовке под г) промежуточное соединение получают в соответствии с процедурой примера 1, стадия г) с использованием продукта, полученного на стадии в).
MS (APCI) 170 (М+Н+, 100%)
д) [3aR-[3aα,4α,6α(1R*,2S*),6aα]]-6-[7-[[2-(3,4-ди-фторфенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанол
К раствору [3aR-(3aα,4α,6α,6aα)]-6-[[5-амино-6-хлор-2-[(3,3,3-трифторпропил)тио]-4-пиримидинил]-амино]-тетра-гидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанола (полученного по методу, раскрытому в заявке на международный патент WO 9703084) (8,1 г) в ацетонитриле (1000 мл) добавляют изоамилнитрит (5,1 мл) и раствор нагревают при температуре 70°С в течение 1 часа. Охлажденную реакционную смесь концентрируют и очищают (SiO2, с использованием в качестве элюента смеси дихлорметан:этилацетат, 4:1) с получением промежуточного продукта, который далее преобразуют в соединение, указанное в заголовке под д), в соответствии со способом примера 1, стадия д) с использованием продукта, полученного на стадии г).
MS (APCI) 587 (М+Н+, 100%)
е) [1R-[1α,2α,3β(1R*,2S*),5β]]-3-[7-[[2-(3,4-дифтор-фенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил)-циклопентан-1,2-диол
Соединение получают в соответствии с процедурой примера 1, стадия з) с использованием продукта, полученного на стадии д).
MS (APCI) 547 (М+Н+, 100%)
ЯМР δН (d6-ДМСО) 9,43 (1Н, д), 7,35-7,28 (2Н, м), 7,14-7,02 (1Н, м), 5,01-4,96 (2Н, м), 4,72-4,69 (2Н, м), 4,42 (1Н, к), 3,87-3,84 (1Н, м), 3,50-3,44 (2Н, м), 3,25-3,12 (3Н, м), 2,58-2,50 (2Н, м), 2,28-2,21 (3Н, м), 1,85-1,80 (1Н, м), 1,52-1,50 (1Н, м), 1,39-1,37 (1Н, м).
Пример 3
[1S-[1α,2α,3β(1S*,2R*),5β]]-3-[7-[2-(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)-циклопентан-1,2-диол
а) (1R-цис)-бис(1,1-диметилэтил)-4-гидрокси-2-циклопентенилимидодикарбонат
К суспензии промытого эфиром гидрида натрия (60% дисперсия в масле; 0,31 г) в тетрагидрофуране (30 мл) добавляют бис-(1,1-диметилэтиловый)эфир имидодикарбоновой кислоты (1,84 г). Смесь перемешивают при 40°С в течение 1 часа. Затем к указанной смеси при температуре окружающей среды добавляют (1S-цис)-4-ацетокси-2-циклопентен-1-ол (0,5 г) и тетракис(трифенилфосфин)палладия(0) (0,18 г). Далее реакционную смесь перемешивают в течение 24 часов, после чего очищают (SiO2, с использованием в качестве элюента смеси этилацетат:гексан, 1:9) с получением продукта, указанного в заголовке, в виде бесцветного твердого вещества (0,90 г).
ЯМР δН (d6-ДМСО) 1,43 (18Н, с), 1,61 (1Н, ддд, J=12,3; 7,7; 6,4 Гц), 2,54 (1Н, дт, J=12,6; 7,4 Гц), 4,51-4,57 (1Н, м), 4,86 (1Н, тк, J=8,0; 1,8 Гц), 4,91 (1Н, д, J=5,4 Гц), 5,71-5,77 (2Н, м).
б) Бис(1-диметилэтиловый) эфир [1R-(1α,2β,3β,4α)]-2,3,4-тригидрокси-циклопентенилимидодикарбоновой кислоты
К раствору продукта, полученного на стадии а) (17,1 г), в тетрагидрофуране (500 мл)/воде (50 мл) добавляют N-метилморфолин-N-оксид (9,4 г) и затем тетроксид осмия (10 мл, 2,5% раствор в трет-бутаноле). Указанную смесь перемешивают при комнатной температуре в течение 4 дней и затем обрабатывают гидросульфитом натрия (6,0 г). Далее суспензию фильтруют через целит и продукт очищают (SiO2, при использовании в качестве элюента смеси этилацетат:гексан, 1:1) с получением соединения, указанного в заголовке под б), (19,1 г).
ЯМР δH (d6-ДМСО) 1,44 (18Н, c), 1,46-1,60 (1Н, м), 1,97-2,05 (1Н, м), 3,55-3,58 (1Н, м), 3,66-3,73 (1Н, м), 4,11-4,21 (2Н, м), 4,54 (1Н, д, J = 4,8 Гц), 4,56 (1Н, д, J = 5,9 Гц), 4,82 (1Н, д, J = 4,6 Гц)
в) Гидрохлорид [3aR-(3aα,4α,6α,6aα)]-6-амино-тетра-гидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ола
Продукт, полученный на стадии б) (17,4 г), в 6 М HCl (100 мл)/метаноле (500 мл) перемешивают в течение 18 часов. Указанную смесь выпаривают и затем делают азеотропной с помощью толуола (4×200 мл) с получением бесцветного порошка (8,7 г). Указанное твердое вещество суспендируют в ацетоне (250 мл), содержащем 2,2-диметоксипропан (25 мл) и конц. HCl (0,2 мл), и затем нагревают при температуре кипения с обратным холодильником в течение 2 часов. Смесь охлаждают, выпаривают и делают азеотропной с помощью толуола (3×200 мл). Далее остаток растворяют в 20% водной уксусной кислоте и перемешивают в течение 2 часов. После этого смесь выпаривают и делают азеотропной с помощью толуола (4×200 мл) с получением соединения, указанного в заголовке под в), (10,1 г).
MS (APCI) 174 (M+H+, 100%).
г) [3aR-(3aα,4α,6α,6aα)]-6-[[6-хлор-5-нитро-2-(пропилтио)-пиримидин-4-ил]-амино]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Раствор продукта, полученного на стадии в), (10,0 г) и N,N-диизопропилэтиламина (35 мл) в тетрагидрофуране (600 мл) перемешивают в течение 1 часа. Далее смесь фильтруют, и полученный раствор добавляют в течение 1 часа к раствору 4,6-дихлор-5-нитро-2-(пропилтио)-пиримидина (полученного по методу, описанному в заявке на международный патент WO 9703084) (25,6 г) в тетрагидрофуране (1000 мл) и перемешивают еще в течение 2 часов. Под вакуумом уменьшают объем растворителя и добавляют этилацетат (1000 мл). Далее смесь промывают водой и органические слои высушивают, выпаривают и очищают (SiO2, при использовании в качестве элюента смеси изогексан:этилацетат) с получением соединения, указанного в заголовке под г), (14,2 г).
MS (APCI) 405 (M+H+, 100%).
д) [3aR-(3aα,4α,6α,6aα)]-6-[[5-амино-6-хлор-2-(пропилтио)-пиримидин-4-ил]амино]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол.
К перемешиваемому раствору продукта, полученного на стадии г), (2,7 г) в уксусной кислоте (100 мл) добавляют порошок железа (3,0 г). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, концентрируют до половинного объема, разбавляют этилацетатом и промывают водой. Органическую фазу высушивают и концентрируют с получением соединения, указанного в заголовке под д), (2,0 г).
MS (APCI) 375 (M+H+, 100%)
е) [3aR-(3aα,4α,6α,6aα)]-6-[7-хлор-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
К раствору продукта, полученного на стадии д), (2,0 г) в ацетонитриле (100 мл) добавляют изоамилнитрит (1,1 мл) и раствор нагревают при температуре 70°С в течение 1 часа. Охлажденную реакционную смесь концентрируют и очищают (SiO2, при использовании в качестве элюента смеси этилацетат: гексан, 1:3) с получением соединения, указанного в заголовке под е), (1,9 г).
MS (APCI) 386 (M+H+, 100%)
ж) [3aR-(3aα,4α,6α,6aα)]-6-[7-амино-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диме-тил-4Н-циклопента-1,3-диоксол-4-ол
Продукт, полученный на стадии е), (13,2 г) в тетрагидрофуране (200 мл), содержащем 0,88 аммиака (5 мл), перемешивают в течение 2 часов, затем концентрируют досуха и остаток распределяют между водой и этилацетатом. Органический материал высушивают и затем концентрируют с получением соединения, указанного в заголовке под ж), (12,5 г).
MS (APCI) 367 (M+H+, 100%)
з) Метиловый эфир [3aR-(3aα,4α,6α,6aα)]-[[6-[7-амино-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол]окси] уксусной кислоты
К раствору продукта, полученного на стадии ж), (0,50 г) в тетрагидрофуране (25 мл) при температуре 0°С добавляют бутиллитий (0,62 мл, 2,5 н. в гексанах). Через 20 минут суспензию обрабатывают раствором метилового эфира трифторметансульфонилоксиуксусной кислоты (0,34 г) (полученного по методу, описанному в Biton, Tetrahedron, 1995, 51, 10513) в тетрагидрофуране (10 мл). Полученный раствор оставляют нагреться до комнатной температуры и затем концентрируют и очищают (SiO2, при использовании в качестве элюента смеси этилацетат:гексан, 4:6) с получением соединения, указанного в заголовке под з), (0,25 г).
MS (APCI) 439 (M+H+, 100%)
и) Метиловый эфир [3aR-(3aα,4α,6α,6aα)]-[[6-[7-бром-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол]окси] уксусной кислоты
Продукт, полученный на стадии з), (1,1 г) и изоамилнитрил (2,4 мл) в бромоформе (30 мл) нагревают при температуре 80°С в течение 30 минут. Охлажденную реакционную смесь очищают (SiO2, при использовании в качестве элюента смеси этилацетат:изогексан, 1:4) с получением соединения, указанного в заголовке под и), (0,44 г).
MS (APCI) 502/4 (M+H+,) 504 (100%)
к) Метиловый эфир [3aR-[3aα,4α,6α(1R*,2S*),6aα]]-[[6-[7-[[2-(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси] уксусной кислоты
К смеси продуктов, полученных на стадии и), (0,80 г) и на стадии г) из примера 2 (0,61 г), в дихлорметане (25 мл) добавляют N,N-диизопропилэтиламин (0,85 мл). Полученный раствор перемешивают при комнатной температуре в течение 16 часов и затем концентрируют под вакуумом. Очистка (SiO2, при использовании в качестве элюента смеси изогексан:этилацетат, 3:1) дает соединение, указанное в заголовке под и), в виде бесцветной пены (0,77 г).
MS (APCI) 591 (M+H+, 100%)
л) [3aR-[3aα,4α,6α(1R*,2S*),6aα]]-2-[6-[[7-[2-(3,4-дифторфенил)циклопропил]амино-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси]-этанол
К охлажденному льдом раствору продукта, полученного на стадии к), (0,76 г) в тетрагидрофуране (1 мл) добавляют DIBAL-H® (1,0М раствор в гексанах, 5,15 мл) и раствор перемешивают при указанной температуре в течение 2 часов. Далее реакционную смесь концентрируют под вакуумом, и остаток растворяют в этилацетате (75 мл). Добавляют насыщенный водный раствор тартрата натрия-калия (75 мл) и смесь энергично перемешивают в течение 16 часов. Собирают органический материал и проводят повторную экстракцию водной фазы этилацетатом (2х50 мл). Объединенный органический материал высушивают, концентрируют и очищают остаток (SiO2, при использовании в качестве элюента смеси изогексан:этилацетат, 1:1) с получением подцелевого соединения, указанного в заголовке под з), (0,63 г).
MS (APCI) 563 (M+H+, 100%)
м) [1S-[1α,2α,3β(1S*,2R*),5β]]-3-[7-(2-(3,4-дифторфенил)циклопропиламино)-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)-циклопентан-1,2-диол
Продукт получают в соответствии с процедурой примера 1, стадия з) с использованием продукта, полученного на стадии л).
MS (APCI) 523 (M+H+, 100%)
ЯМР δH (d6-ДМСО) 8,95 (1Н, д, J = 3,3 Гц), 7,39-7,21 (2Н, м), 7,10-7,00 (1Н, м), 5,12 (1Н, д, J = 6,4 Гц), 5,05 (1Н, д, J = 3,6 Гц), 4,96 (1Н, к, J = 9,0 Гц), 4,62-4,54 (2Н, м), 3,95 (1Н, шс), 3,79-3,73 (1Н, м), 3,55-3,47 (4Н, м), 3,20-3,13 (1Н, м), 2,98-2,81 (2Н, м), 2,63 (1Н, дт, J = 13,6, 8,5 Гц), 2,29-2,21 и 2,16-2,09 (1Н, м), 2,07-2,00 (1Н, м), 1,73-1,33 (4Н, м), 0,99 (3Н, т, J = 7,4 Гц)
Пример 4
[1R-[1α,2α,3β(1R*,2S*),5β]]-3-[5-(бутилтио)-7-[[2-(3,4-дифторфенил)циклопропил]амино]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил) циклопентан-1,2-диол
a) [3aR-(3aα,4α,6α,6aα)]-6-[7-амино-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанол
Продукт получают по методу примера 3, стадия ж) с использованием [3aR-(3aα,4α,6α,6aα)]-6-[7-хлор-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2, 2-диметил-4Н-циклопента-1,3-диоксол-4-метанола (приготовленного в соответствии с процедурой, описанной в заявке на международный патент WO 9703084). Сырой продукт очищают (SiO2, при использовании в качестве элюента смеси метанол:дихлорметан, 1:19) с получением целевого соединения, указанного в заголовке под а).
MS (APCI) 381 (M+H+, 100%).
б) [3aR-(3aα,4α,6α,6aα)]-6-[7-амино-5-(пропилсульфонил)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2, 2-диметил-4Н-циклопента-1,3-диоксол-4-метанол
Продукт получают в соответствии с процедурой примера 1, стадия е) с использованием продукта, полученного на стадии а).
MS (APCI) 413 (M+H+, 100%)
в) [3aR-(3aα,4α,6α,6aα)]-6-[7-амино-5-(бутилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанол
К суспензии гидрида натрия (60%, 1,09 г) в ДМФ (50 мл) добавляют 1-бутантиол (2,38 мл) в ДМФ (25 мл). Через 1 час добавляют по каплям раствор продукта, полученного на стадии б), (3,66 г) в ДМФ (65 мл) и полученную смесь перемешивают в течение ночи. Далее реакционную смесь медленно добавляют к насыщенному водному раствору бикарбоната натрия (1000 мл) и затем экстрагируют этилацетатом (3×200 мл). Органическую фазу высушивают (MgSO4), концентрируют под вакуумом, и остаток очищают (SiO2, при использовании в качестве элюента смеси метанол:дихлорметан, 1:19) с получением соединения, указанного в заголовке под в), (3,32 г).
MS (APCI) 395 (M+H+, 100%)
г) Ацетат [3aR-(3aα,4α,6α,6aα)]-6-[7-амино-5-(бутилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2, 2-диметил-4Н-циклопента-1,3-диоксол-4-метанола
К раствору продукта, полученного на стадии в), (3,3 г) в дихлорметане (50 мл) добавляют пиридин (2,7 мл), 4-диметиламинопиридин (0,4 г) и ангидрид уксусной кислоты (2,0 мл). Смесь перемешивают в течение ночи при комнатной температуре, концентрируют под вакуумом и очищают (SiO2, при использовании в качестве элюента смеси диэтиловый эфир:изогексан, 3:2) с получением соединения, указанного в заголовке под г), (2,7 г).
MS (APCI) 437 (M+H+, 100%)
д) Ацетат [3aR-(3aα,4α,6α,6aα)]-6-[7-бром-5-(бутилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2, 2-диметил-4Н-циклопента-1,3-диоксол-4-метанола
Продукт получают в соответствии с процедурой примера 3, стадия и) с использованием продукта, полученного на стадии г).
MS (APCI) 500/502 (M+H+), 500 (100%).
е) Ацетат [3aR-[3aα,4α,6α(1R*,2S*),6aα]]-6-[5-(бутилтио)-7-[[2-(3,4-дифторфенил)циклопропил]амино]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-метанола
Продукт получают в соответствии с процедурой примера 3, стадия к) с использованием продукта, полученного на стадии г) примера 2, и продукта, полученного на стадии д).
MS (APCI) 589 (M+H+, 100%)
ж) [1R-[1α,2α,3β(1R*,2S*),5β]]-3-[5-(бутилтио)-7-[[2-(3,4-дифторфенил)циклопропил]амино]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(гидроксиметил)-циклопентан-1,2-диол
Продукт, полученный на стадии е), (0,64 г) в 80% водной уксусной кислоте (30 мл) нагревают при температуре 80°С в течение 1 часа. Охлажденную смесь вливают в насыщенный раствор бикарбоната натрия и экстрагируют этилацетатом. Органическую фазу высушивают и концентрируют под вакуумом с получением камеди, которую растворяют в метаноле (50 мл)/10% водном растворе карбоната калия (3 мл). Далее раствор перемешивают в течение 30 минут, нейтрализуют уксусной кислотой и концентрируют под вакуумом. Очистка (SiO2, при использовании в качестве элюента смеси метанол:дихлорметан, 11:9) дает твердое вещество, из которого при повторной кристаллизации получают целевое соединение (0,25 г).
MS (APCI) 507 (M+H+, 100%).
ЯМР δH (d6-ДМСО) 9,34 (1Н, ш), 7,40-7,23 (2Н, м), 7,11-7,00 (1Н, м), 5,06-4,93 (2Н, м), 4,76-4,67 (2Н, м), 4,48-4,38 (1Н, м), 3,91-3,84 (1Н, м), 3,56-3,39 (2Н, м), 3,21-3,08 (1Н, м), 3,03-2,83 (2Н, м), 2,32-2,17 (1Н, м), 2,17-2,03 (2Н, м), 1,91-1,77 (1Н, м), 1,71-1,32 (4Н, м), 1,32-1,17 (2Н, м), 0,81 (3Н, т)
Пример 5
[1S-[1α,2β,3β,4α(1S*,2R*)]]-4-[5-(бутилтио)-7-[[2-(4-фторфенил)циклопропил]амино]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-циклопентан-1,2,3-триол
a) [3aR-[3aα,4α,6α,6aα(1S*,2R*)]]-6-[7-[[(4-фторфенил)циклопропил]амино]-5-(пропилтио)-3H-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Продукт получают в соответствии с процедурой примера 1, стадия д) с использованием продукта примера 1, стадия г) и продукта примера 3, стадия е).
MS (APCI) 501 (M+H+, 100%)
б) [3aR-[3aα,4α,6α,6aα(1S*,2R*)]]-6-[[7-[(4-фторфенил)циклопропил]амино]-5-(пропилсульфонил)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Продукт получают в соответствии с процедурой примера 1, стадия е) с использованием продукта, полученного на стадии а).
MS (APCI) 532 (M+H+, 100%).
в) [3aR-[3aα,4α,6α,6aα(1S*,2R*)]]-6-[7-[[(4-фторфенил)циклопропил]амино]-5-(бутилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1, 3-диоксол-4-ол
Продукт получают в соответствии с процедурой примера 4, стадия в) с использованием продукта, полученного на стадии б).
MS (APCI) 515 (M+H+, 100%)
[1S-[1α,2β,3β,4α(1S*,2R*)]]-4-[5-(бутилтио)-7-[[2-(4-фторфенил)циклопропил]амино]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-циклопентан-1,2,3-триол
Продукт получают в соответствии с процедурой примера 1, стадия з) с использованием продукта, полученного на стадии в).
MS (APCI) 575 (M+H+, 100%)
ЯМР δH (d6-ДМСО) 7,26-7,22 (2Н, м), 7,11 (2Н, т), 4,99-4,90 (1Н, м), 4,67-4,63 (1Н, м), 3,93 (1Н, с), 3,77 (1Н, шс), 3,35-3,13 (1Н, м), 3,00-2,80 (2Н, м), 2,59-2,51 (1Н, м), 2,15-2,11 (1Н, м), 1,91-1,86 (1Н, м), 1,53-1,41 (3Н, м), 1,35-1,30 (1Н, м), 1,22 (2Н, секст.), 0,80 (3Н, т)
Пример 6
[1S-[1α,2α,3β(1S*,2R*),5β)]-3-[7-[[2-(3,4-дифторфенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидрокиэтокси)-циклопентан-1,2-диол
а) [1S-[1α,2α,3β(1S*,2R*),5β)]-3-[7-[[2-(3,4-дифторфенил)циклопропил]амино]-5-(пропилсульфонил)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидрокиэтокси)-циклопентан-1,2-диол
Указанное под а) соединение получают в соответствии с процедурой примера 1, стадия е) с использованием продукта, полученного на стадии м) примера 3.
MS (APCI) 555 (M+H+, 100%)
б)[1S-[1α,2α,3β(1S*,2R*),5β)]-3-[7-[[2-(3,4-дифторфенил)циклопропил]амино]-5-[(3,3,3-трифторпропил)тио]-3H-1, 2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидрокиэтокси)-циклопентан-1,2-диол
Целевое соединение получают в соответствии с процедурой примера 1, стадия ж) с использованием продукта, полученного на стадии а).
MS (APCI) 555 (M+H+, 100%).
ЯМР δH (d6-ДМСО) 9,45 (1Н, д), 7,36-7,05 (3Н, м), 5,05 (1Н, д), 5,02 (1Н, д), 4,95 (1Н, м), 4,60 (2Н, м), 3,95 (1Н, м), 3,86 (1Н, м), 3,47 (4Н, м), 3,30-3,11 (3Н, м), 2,63-2,49 (3Н, м), 2,19 (1Н, м), 2,00 (1Н, м), 1,53 (1Н, м), 1,40 (1Н, м)
Пример 7
[1S-[1α,2α,3β,5β(1S*,2R*)]]-3-(2-гидроксиэтокси)-5-[7-(2-фенилциклопропил)амино]-5-[(3,3,3-трифторпропил)-тио]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-циклопентан-1,2-диол
а) Этиловый эфир (1S-цис)-2-[[4-[[6-хлор-5-нитро-2-[(3,3,3-трифторпропил)тио]-4-пиримидинил]амино]-2-циклопентен-1-ил]окси]-уксусной кислоты
Раствор азида натрия (4,70 г) в дегазированной воде (25 мл) добавляют к раствору (1R,2S)-4-гидрокси-2-циклопентен-1-ил ацетата (9,99 г) в тетрагидрофуране (60 мл) и смесь перемешивают в течение 10 минут. Далее добавляют тетракис(трифенилфосфин)палладия(0) (365 мг) и перемешивают в течение 10 минут. Отделяют водный слой и дважды экстрагируют его этилацетатом. Объединенные органические слои высушивают (MgSO4), концентрируют и очищают хроматографированием на короткой колонке (SiO2, при использовании в качестве элюента смеси этилацетат:изогексан, 1:2) с получением желтого масла. Указанное масло растворяют в тетрагидрофуране (25 мл) и медленно добавляют к суспензии гидрида натрия (2,94 г, 60% дисперсия в масле) в тетрагидрофуране (60 мл) при -78°С. Добавляют раствор этилбромацетата (8,2 мл) в тетрагидрофуране (5 мл) и смеси дают нагреться до 20°С и перемешивают в течение 30 минут. Добавляют водный раствор хлорида аммония и экстрагируют полученную смесь диэтиловым эфиром. Органические слои высушивают (MgSO4), концентрируют и очищают (SiO2, при использовании в качестве элюента смеси диэтиловый эфир:изогексан, 1:5) с получением бесцветного масла. Раствор указанного масла и трифенилфосфина (17,89 г) в тетрагидрофуране (90 мл) перемешивают в течение 10 минут. Далее добавляют воду (15 мл) и раствор перемешивают в течение 18 часов. После этого удаляют под вакуумом растворитель, остаток делают азеотропным с помощью толуола и очищают (SiO2, при использовании в качестве элюентов этилацетата, затем смеси этилацетат:метанол:аммиак, 90:9:1) с получением бледно-желтого масла (7,14 г).
Раствор указанного соединения в тетрагидрофуране (50 мл) добавляют в течение 25 минут к раствору 4,6-дихлор-5-нитро-2-[(3,3,3-трифторпропил)тио]пиримидина (приготовленного по методу, описанному в заявке на международный патент WO 9703084) и N,N-диизопропилэтиламина (77,5 мл) в сухом тетрагидрофуране (100 мл) и затем все перемешивают в течение 30 минут. Далее добавляют воду и смесь экстрагируют диэтиловым эфиром (три раза). Органические слои высушивают (MgSO4), концентрируют и очищают (SiO2, при использовании в качестве элюента смеси этилацетат:изогексан, 1:4) с получением соединения, указанного в заголовке под а), (7,39 г).
MS (APCI) 367/9 (M-(EtO2CCH2O)+), 367 (100%)
б) Этиловый эфир (1S-цис) 2-[[4-[[7-хлор-5-[(3,3,3-трифторпропил)тио]-3H-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-2-циклопентен-1-ил]окси]-уксусной кислоты
Продукт получают в соответствии с процедурой примера 3, стадии д) и е) с использованием продукта, полученного на стадии а).
MS (APCI) 348/50 (M-(EtO2CCH2O)+), 348 (100%).
в) Этиловый эфир (1S-цис) 2-[[4-[[7-амино-5-[(3,3,3-трифторпропил)тио]-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-2-циклопентен-1-ил]окси]-уксусной кислоты
Продукт получают в соответствии с процедурой примера 3, стадия ж) с использованием продукта, полученного на стадии б).
MS (APCI) 433 (M+H+, 100%)
г) [1S-(цис)] 2-[[4-[7-амино-5-[(3,3,3-трифторпропил)-тио]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-2-циклопентен-1-ил]-2-этанол
Продукт получают в соответствии с процедурой примера 3, стадия л) с использованием продукта, полученного на стадии в).
MS (APCI) 391 (M+H+, 100%)
д) [3aR-(3aα,4α,6α,6aα)]-2-[6-[7-амино-5-[(3,3,3-трифторпропил)тио]-3H-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-илокси]этанол
Раствор продукта, полученного на стадии (г), (454 мг), тетроксида осмия (0,17 мл 0,1 М раствора в трет-бутаноле), N-оксид N-метилморфолина (210 мг) и пиридина (0,09 мл) в ацетоне (5 мл) и воде (1 мл) нагревают при температуре 70°С в течение 5 часов. Затем добавляют гидросульфит натрия (330 мг) в воде (1 мл), растворитель удаляют под вакуумом и остаток делают азеотропным с помощью толуола. Раствор указанного остатка, п-толуолсульфоновую кислоту (50 мг) в ацетоне (5 мл) и 2,2-диметоксипропан (2 мл) перемешивают в течение 3 часов. Растворитель удаляют под вакуумом, добавляют водный раствор кислого карбоната натрия и смесь экстрагируют этилацетатом. Органические слои высушивают (MgSO4), концентрируют и очищают (SiO2, при использовании в качестве элюента смеси изогексан:ацетон, 5:2) с получением соединения, указанного в заголовке под д), в виде белого твердого вещества (367 мг).
MS (APCI) 465 (M+H+, 100%)
е) [3aR-(3aα,4α,6α,6aα)]-2-[6-[7-бром-5-[(3,3,3-трифторпропил)тио]-3H-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-илокси]этанол
Продукт получают в соответствии с процедурой примера 3, стадия и) с использованием продукта, полученного на стадии д).
MS (APCI) 528/30 (M+H+), 528 (100%)
ж) [3aR-[3aα,4α,6α(1R*,2S*),6aα)]-2-[6-(7-фенилциклопропил)амино]-5-[(3,3,3-трифторпропил)тио]-3H-1,2,3-триа-золо[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-1,3-диоксол-4-илокси]этанол
Продукт получают в соответствии с процедурой примера 3, стадия к) с использованием продукта, полученного на стадии е), и (1R-транс)-2-фенилциклопропанамин,[R-(R*,R*)]-2,3-дигидроксибутандиоата (1:1) (полученного по методу, описанному в работе L.A. Mitscher et al., J. Med. Chem., 1986, 29, 2044).
MS (APCI) 581 (M+H+, 100%)
з) [1S-[1α,2α,3β,5β(1S*,2R*)]]-3-(2-гидроксиэтокси)-5-[7-(2-фенилциклопропил)амино]-5-[(3,3,3-трифторпропил)-тио]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-циклопентан-1,2-диол
Продукт получают в соответствии с процедурой примера 1, стадия з) с использованием продукта, полученного на стадии ж).
MS (APCI) 540 (M+H+, 100%)
ЯМР δH (d6-ДМСО) 7,35-7,16 (5Н, м), 4,97 (1Н, к), 4,62-4,54 (1Н, м), 3,98-3,92 (1Н, м), 3,78-3,72 (1Н, м), 3,55-3,44 (4Н, м), 3,26-3,19 (2Н, м), 3,16-3,07 (1Н, м), 2,70-2,61 (1Н, м.), 2,58-2,52 (1Н, м.), 2,23-2,18 (1Н, м), 2,05-1,97 (1Н, м), 1,86 (1Н, с), 1,54-1,46 (1Н, м), 1,38-1,30 (1Н, м)
Пример 8
[1S-[1α,2β,3β,4α(1S*,2R*)]]-4-[5-(бутилтио)-7-[[2-(3,4-дифторфенил)циклопропил]амино]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-циклопентан-1,2,3-триол
а) [3aR-[3aα,4α,6α(1S*,2R*),6аα]-6-[[7-[(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3H-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Указанное соединение получают в соответствии с процедурой примера 1, стадия д) с использованием продукта, полученного на стадии е) примера 3 и продукта, полученного на стадии г) примера 2.
MS (APCI) 519 (M+H+, 100%)
б) [3aR-[3aα,4α,6α(1R*,2S*),6аα]]-6-[[7-[(3,4-дифторфенил)циклопропил]амино]-5-(пропилсульфонил)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Указанное соединение получают в соответствии с процедурой примера 1, стадия е) с использованием продукта, полученного на стадии а).
MS (APCI) 551 (M+H+, 100%)
в) [3aR-[3aα,4α,6α(1R*,2S*),6аα]]-6-[5-(бутилтио)-7-[[2-(3,4-дифторфенил)циклопропил]амино]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ол
Указанное соединение получают в соответствии с процедурой примера 4, стадия в) с использованием продукта, полученного на стадии б).
MS (APCI) 533 (M+H+, 100%)
г) [1S-[1α,2β,3β,4α(1S*,2R*)]]-4-[5-(бутилтио)-7-[[2-(3,4-дифторфенил)циклопропил]амино]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]циклопентан-1,2,3-триол
Указанное соединение получают в соответствии с процедурой примера 1, стадия з) с использованием продукта, полученного на стадии в).
ЯМР δH (d6-ДМСО) 7,15-6,98 (3Н, м), 6,67 (1Н, с), 5,11-5,09 (1Н, м), 4,82-4,76 (1Н, м), 4,34-4,21 (3Н, м), 3,7 (1Н, с), 3,2-2,92 (4Н, м), 2,77 (1Н, м), 2,42-2,36 (1Н, м), 2,2-2,18 (1Н, м), 1,42-1,25 (6Н, м), 0,9 (3Н, к)
MS (APCI) 493 (M+H+, 100%)
Пример 9
[1S-[1α,2α,3β(1S*,2R*),5β]]-3-[5-(бутилтио)-7-[(2-фенилциклопропил)амино]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)-циклопентан-1,2-диол
а) Фенилметиловый эфир [3aS-(3aα,4α,6α,6аα)]-[тетрагидро-6-гидрокси-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]-карбаминовой кислоты
К суспензии гидрохлорида [3aR-(3aα,4α,6α,6аα)]-6-аминотетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ола (полученного по методу, описанному в документе WO 9905142) (27,1 г) в 4-метил-2-пентаноне (500 мл) добавляют карбонат калия (39,3 г). После этого добавляют воду (150 мл) и затем по каплям добавляют бензилхлорформиат (23,1 г). Реакционную смесь перемешивают при комнатной температуре в течение 4 часов, после чего отделяют органическую фазу. Водную фазу экстрагируют 4-метил-2-пентаноном (2×50 мл). Объединенный органический материал концентрируют и остаток очищают (SiO2, при использовании в качестве элюента смеси дихлорметан:метанол в соотношениях от 95:5 до 90:10) с получением соединения, указанного под а), (39,23 г).
ЯМР δH (CDCl3) 7,32 (5Н, м), 5,65 (1Н, шс), 5,10 (1Н, шс), 4,59 (1Н, д), 4,48 (1Н, д), 4,27 (1Н, м), 4,19 (1Н, шм), 2,24 (1Н, шс), 1,69 (1Н, д), 1,41 (3Н, с), 1,26 (3Н, с)
б) Фенилметиловый эфир [3aS-(3aα,4α,6α,6аα)]-[2,2-диметил-6-(2-гидроксиэтокси)-тетрагидро-4Н-циклопента-1,3-диоксол-4-ил]-карбаминовой кислоты
К раствору продукта, полученного на стадии а), (39,23 г) в тетрагидрофуране (200 мл) в течение 5 минут добавляют трет-бутоксид калия (3,6 г) в тетрагидрофуране (20 мл). Через 15 минут добавляют по каплям этилбромацетат (3,7 мл) в тетрагидрофуране (10 мл). Смесь перемешивают в течение 10 минут при температуре 0°С и затем добавляют этилбромацетат (3,7 мл × 4). Далее реакционную смесь перемешивают при 0°С в течение еще 2 часов. К полученной суспензии добавляют порциями боргидрид лития (2,79 г), после чего реакционную смесь перемешивают в течение 16 часов при температуре ниже 5°С. К охлажденной смеси добавляют по каплям ледяную уксусную кислоту (23 г). После перемешивания в течение 30 минут добавляют по каплям воду (100 мл) и полученную смесь перемешивают в течение 30 минут. Затем фазы разделяют и проводят экстракцию водной фазы этилацетатом. Объединенный органический материал промывают насыщенным раствором бикарбоната натрия и солевым раствором, затем высушивают и концентрируют. Остаток очищают (SiO2, при использовании в качестве элюента смеси этилацетат: гексан в соотношениях от 25:75 до 50:50) с получением соединения, указанного в заголовке под б), (38,6 г).
MS (APCI) 218 (M+H+, 100%)
в) [3aR-(3aα,4α,6α,6аα)]-2-[[6-амино-2,2-диметилтетрагидро-4Н-циклопента-1,3-диоксол-4-ил]окси]-этанол
Суспензия 5% палладия на угле (4 г) добавляют к раствору продукта, полученного на стадии б) (39,96 г) в этаноле (250 мл), и смесь гидрогенизируют под давлением 1,2 бар в течение 20 часов. Затем отфильтровывают катализатор и концентрируют полученный фильтрат, что дает соединение, указанное в заголовке под в), (23,65 г).
MS (APCI) 160 (M+H+, 100%)
г) 2-(бутилтио)-4,6-дихлорпиримидин-5-амин
Указанное соединение получают в соответствии с процедурой примера 1, стадия д) с использованием 2-(бутилтио)-4,6-дихлор-5-нитро-пиримидина (полученного по методу, описанному в документе DE 2223644).
ЯМР δH (CDCl3) 4,20 (2Н, шс), 3,10 (2Н, т), 1,70 (2Н, м), 1,47 (2Н, м), 0,95 (3Н, т)
д) [3aR-(3aα,4α,6α,6аα)]-2-[[6-[[5-амино-2-(бутилтио)-6-хлор-пиримидин-4-ил]амино]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси]этанол
Соединение получают в соответствии с процедурой примера 3, стадия г) с использованием продуктов, полученных на стадиях в) и г).
MS (APCI) 433 (M+H+, 100%)
е) [3aR-[3aα,4α,6α(1R*,2S*),6аα]]-2-[6-[[5-(бутилтио)-7-хлор-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси]-этанол
Соединение получают в соответствии с процедурой примера 3, стадия е) с использованием продукта, полученного на стадии д).
ЯМР δH (CDCl3) 5,53 (1Н, м), 5,21 (1Н, м), 4,88 (1Н, д), 4,05 (1Н, м), 3,59 (4Н, м), 3,24 (2Н, т), 2,70 (1Н, м), 2,53 (1Н, м), 2,13 (1Н, т), 1,79 (2Н, м), 1,55 (5Н, м), 1,37 (3Н, с), 0,98 (3Н, т)
ж) [3aR-[3aα,4α,6α(1R*,2S*),6аα]]-2-[6-[[5-(бутилтио)-7-[2-фенилциклопропил]амино-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1, 3-диоксол-4-ил]окси]-этанол
Данное соединение получают в соответствии с процедурой примера 3, стадия к) с использованием продукта, полученного на стадии е).
MS (APCI) 541 (M+H+, 100%)
з) [1S-[1α,2α,3β(1S*,2R*),5β]]-3-[5-(бутилтио)-7-[(2-фенилциклопропил)амино]-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)-циклопентан-1,2-диол
Соединение, названное под з), получают в соответствии с процедурой примера 1, стадия з) с использованием продукта, полученного на стадии ж).
MS (APCI) 501 (M+H+, 100%)
ЯМР δH (d6-ДМСО) 9,33 (1Н, д), 7,30 (2Н, м), 7,18 (3Н, м), 5,12 (1Н, д), 5,04 (1Н, д), 4,96 (1Н, к), 4,59 (2Н, м), 3,94 (1Н, с), 3,76 (1Н, м), 3,51 (4Н, м), 3,22 (1Н, м), 2,98(1Н, м), 2,86 (1Н, м), 2,65 (1Н, м), 2,14 (1Н, м), 2,05 (1Н, м), 1,21-1,53 (6Н, м), 0,80 (3Н, т)
Фармакологические данные
Тестирование активности препарата в качестве агониста/антагониста к Р2т (P2YADP или Р2ТАС) проводят следующим образом.
Венозную кровь человека (100 мл) распределяют поровну по 3 пробиркам, каждая из которых содержит 3,2% цитрат натрия (4 мл) в качестве антикоагулянта. Пробирки центрифугируют в течение 15 минут при 240g с получением плазмы, обогащенной тромбоцитами (ПОТ, PRP), к которой добавляют простациклин в количестве 300 нг/мл для стабилизации тромбоцитов в ходе их промывания. Свободную от эритроцитов ПОТ получают при центрифугировании вначале в течение 10 минут при 125g и затем при центрифугировании в течение 15 минут при 640g. Супернатант отбрасывают, и осадок тромбоцитов суспендируют в модифицированном бескальциевом растворе Тирода (Tyrode solution) (10 мл) следующего состава: NaCl - 137 мМ, NaHCO3 - 11,9 мМ, NaH2PO4 - 0,4 мМ, KCl - 2,7 мМ, MgCl2 - 1,1 мМ, декстроза - 5,6 мМ, затем смесь дегазируют пропусканием смеси 95% О2/5% СО2 и поддерживают при 37°С. После добавления еще 300 нг/мл PGI2 объединенную суспензию центрифугируют еще один раз в течение 15 минут при 640g. Супернатант отбрасывают, и тромбоциты суспендируют вначале в 10 мл CFT, после чего добавляют CFT до конечного уровня тромбоцитов 2×105/мл. Указанную конечную суспензию хранят без доступа воздуха в шприцах на 60 мл при температуре 3°С. Для того чтобы прошло восстановление нормального функционирования тромбоцитов из ингибированного под действием PGI2 состояния, в исследованиях по агрегации тромбоциты используют не ранее, чем через 2 часа после окончательного супендирования.
Во всех исследованиях добавляют аликвоты по 3 мл суспензии тромбоцитов в пробирки, содержащие раствор CaCl2 (60 мкл 50 мМ раствора с конечной концентрацией 1 мМ). Добавляют человеческий фибриноген (Сигма, F 4883) и 8-суль-фофенилтеофиллин (8-ФТ, который используют для блокирования любой Р1-агонистской активности соединений) до конечной концентрации 0,2 мг/мл (60 мкл раствора свернувшегося белка с концентрацией 10 мг/мл в солевом растворе) и 300 нМ (10 мкл 15 мМ раствора в 6% глюкозе), соответственно. Затем вносят соответствующее количество тромбоцитов или буфера в объеме 150 мкл в каждую ячейку на 96-гнездном плато. Все измерения для тромбоцитов от каждого донора проводят в тройной повторности.
Активность в качестве агониста/антагониста оценивают следующим образом.
Реакцию агрегации на 96-гнездном плато определяют по изменению поглощения, измеряемому на считывающем устройстве для планшетов при 660 нм. При этом в качестве соответствующего прибора для считывания поглощения используют счетчик Bio-Tec Ceres 900C или Dynatech MRX.
Вначале для определения базового уровня измеряют поглощение каждой ячейки при 660 нм. Затем к каждой ячейке добавляют по 10 мкл солевого раствора или соответствующего раствора тестируемого соединения до достижения конечной концентрации 0; 0,01; 0,1; 1; 10 или 100 мМ. Затем планшеты встряхивают в течение 5 минут на орбитальном шейкере, установленном на режим 10, и измеряют поглощение ячеек при 660 нм. Агрегация в этой точке является показателем агонистской активности исследуемого соединения. Затем в каждую ячейку добавляют солевой раствор или АДФ (30 мМ; 10 мкл 450 мМ раствора) и планшеты встряхивают еще в течение 5 минут, после чего замеряют поглощение при 660 нм.
Антагонистскую активность определяют как % ингибирования относительно контрольного ответа на АДФ, что дает величину ИК50. Взятые в качестве примеров соединения демонстрировали величины ИК50, большие чем 5,0.
Изобретение относится к индивидуальным соединениям: метиловому эфиру [3aR-[3аα,4α,6α(1R*,2S*),6аα]]-[[6-[7-[[2-(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси]уксусной кислоты; [3aR-[3аα,4α,6α(1R*,2S*),6аα]]-6-[[7-[2-(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси]-этанолу; [3aR-[3аα,4α,6α(1R*,2S*),6аα]-6-[[7-[(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-олу. В изобретении также описываются другие индивидуальные соединения. Технический результат: получены новые соединения, которые являются промежуточными соединениями, предназначенными для получения производных триазоло[4,5-d]пиримидина, которые в свою очередь используют для лечения заболеваний, опосредованных Р2Т рецептором. 2 н. и 1 з.п. ф-лы, 9 пр.
1. Соединение, выбранное из:
метилового эфира [3aR-[3аα,4α,6α(1R*,2S*),6аα]]-[[6-[7-[[2-(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]-пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси]уксусной кислоты;
[3aR-[3аα,4α,6α(1R*,2S*),6аα]]-6-[[7-[2-(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси]-этанола;
[3aR-[3аα,4α,6α(1R*,2S*),6аα]-6-[[7-[(3,4-дифторфенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ола.
2. Соединение по п. 1, которое представляет собой
[3aR-[3аα,4α,6α(1R*,2S*),6аα]]-6-[[7-[2-(3,4-дифтор-фенил)циклопропил]амино]-5-(пропилтио)-3Н-1,2,3-триазоло-[4,5-d]пиримидин-3-ил]-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]окси]-этанол формулы (I)
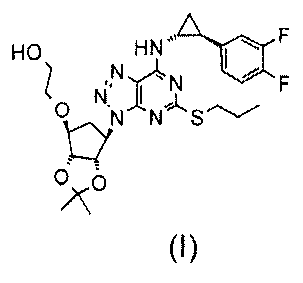
3. Соединение, выбранное из:
фенилметилового эфира [3aS-(3аα,4α,6α,6аα)]-[тетрагидро-6-гидрокси-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]-карбаминовой кислоты;
фенилметилового эфира [3aS-(3аα,4α,6α,6аα)]-[2,2-диметил-6-(2-гидроксиэтокси)-тетрагидро-4Н-циклопента-1,3-диоксол-4-ил]-карбаминовой кислоты;
[3aR-(3аα,4α,6α,6аα)]-2-[[6-амино-2,2-диметил-тетрагидро-4Н-циклопента-1,3-диоксол-4-ил]окси]-этанола.
| ДИДЕЗОКСИДИДЕГИДРОКАРБОЦИКЛИЧЕСКИЕ НУКЛЕОЗИДЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1989 |
|
RU2114846C1 |
| WO 9212718 A1, 06.08.1992 | |||
| Устройство для измерения массового расхода | 1973 |
|
SU521463A1 |

