ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому способу химического синтеза β-(1→6)-связанных глюкозамин-дисахаридов. Такие соединения могут использоваться как производные липида А. Примером производных липида А является ОМ-174-DP ® , впервые выделенный фирмой ОМ Фарма1 из частично разложившихся липополисахаридов Escherichia coli. Данное изобретение включает конструирование и химический синтез новых аналогов липида А, которые лишены обоих сахар-О-ацильных заместителей (по О-3 и О-3') и, следовательно, содержат только N-связанные остатки жирных кислот. Иммунологическая активность таких соединений родственна активности исходного биологического ОМ-174-DP ®.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Липополисахариды (LPS) являются основными соединениями, экспрессируемыми в наружной мембране почти всех грамотрицательных бактерий. Данные амфифильные макромолекулы обладают общей структурой, состоящей из гидрофильного полисахарида (образуемого из скелетного олигосахарида и О-специфичного полисахарида), ковалентно связанного с липофильным фрагментом, называемым липидом А2, который служит в качестве мембранного якоря LPS.
LPS, известные также как эндотоксины, являются сильными стимуляторами систем защиты хозяина как в качестве адъювантов для антигенов3 вакцин, так и в качестве индукторов или возбудителей неспецифичной стойкости к заражению на животных моделях4. Данные амфифильные макромолекулы обладают чрезвычайно сильной иммуностимулирующей активностью5. Биологическая активность LPS является следствием главным образом липидной А составляющей, хотя токсичность липида А зависит строго от его первичной структуры.
В целом, липид А имеет высококонсервативную структуру. Он обычно состоит из β-(1→6)-связанного глюкозамин-дисахаридного скелета, фосфорилированного в положениях О-1 и О-4', и шести или более жирных ацильных групп, связанных в виде сложных эфиров и амидов. Аномерный фосфат (О-1 положение) восстанавливающей части глюкозамина имеет исключительно α конфигурацию. Например, полная химическая структура липида А, выделенного из клеток E. coli (Фиг.1), установленная авторами Имото и др.6, содержит β-(1→6)-связанный глюкозамин-дисахаридный скелет, фосфорилированный в положениях О-1 и О-4' и ацилированный в 2,3 положении (R)-3-гидрокситетрадекановой кислотой, в 2' положении (R)-3-додеканоилокситетрадекановой кислотой, и в 3' положении (R)-3-тетрадеканоилокситетрадекановой кислотой.
Вследствие широкого спектра видов биологической активности был проявлен огромный интерес со стороны как промышленников, так и академических исследовательских лабораторий. Много сил было посвящено химическим модификациям структуры липида А с целью уменьшения природной эндотоксичности исходного соединения и в то же время поддержания или улучшения его благоприятных иммуностимулирующих свойств в восьмидесятых годах Ribi и др. изучили химический процесс с намерением отделить токсические эффекты природного липида А Salmonella Chlamydia RC595 от потенциально полезных иммуномодуляторных эффектов. Данный способ, основанный на селективном гидролизе 1-фосфоногруппы7 и (R)-3-гидрокситетрадеканоильного8 остатка, присоединенного к 3-положению сахара липида А, поставляемого известного иммуностимулятора монофосфорил-липида А (MPL®), который является эффективным адъювантом в профилактических и терапевтических вакцинах9 со значительно пониженной токсичностью по сравнению с его исходным липидом А. Однако MPL®, также как и липид А природного происхождения представляет собой смесь нескольких компонентов вследствие присущей LPS гетерогенности и несовершенных стадий хемоселективного гидролиза или стадий очистки. Вследствие этого иммуностимулятор MPL® включает несколько менее высоко ацилированных соединений в дополнение к основному гексаацильному соединению.
В начале девяностых фирмой ОМ ФАРМА из частично разложившегося LPS1 E. coli было выделено новое производное липида А (ОМ-174-DP ® , Фиг.1). Данное производное лишено обоих сахар-О-ацильных заместителей (в О-3 и O-3') и, следовательно, содержит только N-связанные остатки жирных кислот липида А E. coli, а именно (R)-3-гидрокситетрадеканоильную группу в N-2 и (R)-3-додеканоилокситетрадеканоильную группу в N-2', оставляя таким образом в структуре только три ацильные группы с длинной цепью. Тщательные фармакологические исследования данного нового соединения выявили, что оно обладает сильной противоопухолевой активностью на нескольких in vivo моделях опухоли10 и что оно является эффективным иммуноадъювантом с очень низкой токсичностью.
На протяжении последних двух десятилетий широко изучалась взаимосвязь структура-активность липида А. Shiba с сотрудниками направили серьезные усилия на изучение взаимосвязи структура-активность синтетического липида А E. coli и на разработку химического синтеза таких соединений. Они впервые реализовали химический синтез монофосфорил-липида А E. coli 11 и особенно тщательно подтвердили структуру липида А E. coli с помощью общего химического синтеза, основанного на N-Troc защищенных глюкозаминовых производных.12 Той же группой сообщалось о многих структурных вариациях липида А E. coli в смысле ацильных фрагментов (типов, чисел и положения в сахарном скелете)13 и в смысле гликозилфосфатного фрагмента (фосфоноксиэтильный аналог с α или β конфигурацией в положении 1).14
В 1997 г. они описали наиболее эффективный синтез предшественника липида А.15 К этому времени сообщалось о нескольких неприродных аналогах с модификациями ацильных цепей16 и модификациями гликозилфофатного фрагмента и о синтезе самого липида А.17 Группа опубликовала химический синтез липида А, выделенного из Helicobacter pylori с использованием усовершенствованного способа18. Их публикация включает триацилированный аналог липида А, в котором не хватает обоих сахар-О-ацильных заместителей (в О-3 и O-3'). Однако в дополнение к этому в соединении нет также замещения в 4'-O положении.
Работа Shiba была отправной точкой для более позднего синтеза разнообразных липидов А. В качестве доказательства авторами Kosma с сотрудниками в последнее время синтезированы синтетические тетра- и пентаацильные аналоги липида А Chlamydia, чтобы прояснить роль липида А в связанных с Chlamydia инфекциях.19 Группа Биомира разработала структуру неприродного синтетического липида А, содержащую новые липидные фрагменты, имитирующую встречающиеся в природе происходящие из E. coli и Salmonella производные структуры липида А.20 О химическом синтезе липида А P.gingivalis, триацилированного липида А, содержащего только N-связанные жирнокислотные остатки и с отсутствующей 4'-O-фосфатной группой, также сообщалось авторами Огава с сотрудниками. 21
LPS и родственные им соединения исследовались главным образом как LPS-агонисты. В последние годы родственные липиду А соединения изучались как LPS-антагонисты, которые могут обладать потенциалом в качестве иммуносупрессивных средств, а при аутоиммунных заболеваниях и септицемии путем дезактивирования LPS-индуцируемых агрессивных макрофагов. Например, Qureshi с сотрудниками22 выделили нетоксичный липид А как сильный LPS антагонист из Rhodobacter sphaeroides (Rs-DPLA), а группа Eisai разработала полный синтез предложенной структуры по их собственной методике23 и родственного соединения, а именно Е5564, сильного средства против септицемии.24 Существующие описанные ранее методики синтеза липида А, основанные на окончательном гидрогенолизе11-21, не могли быть применимыми вследствие присутствия в предложенном Rs-DPLA олефиновой функциональности. В последние годы были синтезированы родственные Rs-DPLA и Е5564 соединения.25
Молекулы липополисахаридов и липида А являются иммуностимулирующими агентами, потому что они активируют toll-подобный рецептор 4 (TLR4), однако некоторые LPS могут активировать TLT2, такие как LPS из Porphyromonas gingivalis26. Обычно TLR2 ответные реакции индуцируются только такими агентами, как мурамилпептиды (МРМ), бактериальные липопептиды BLP, пептидогликаны (PGN) и липотейхоевые кислоты (LTA). Очень интересно, изобретатели настоящего изобретения обнаружили в настоящее время, что синтетические соединения изобретения (а не только ОМ-174-DP, происходящий из природных источников, как уже описано в постере27 или в недавнем обзоре28) предпочтительно действуют через TLR2 человека, а не как в случае клеток мышей предпочтительно по ожидаемому TLR4 пути. Данное заметное межвидовое отличие (предпочтительно TLR4 в мышиных клетках и, скорее, TLR2 в клетках человека) ранее не было обнаружено.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Известный уровень техники, обсуждаемый выше, не раскрывает синтетические аналоги липида А, лишенные обоих сахар-О-ацильных заместителей (при О-3 и О-3') и включающие 4'-O-фосфатную группу или альтернативное замещение в 4'-O положении. Такие аналоги липида А обладают благоприятными свойствами и являются полезными в области медицины (человека). Однако такие аналоги липида А могут быть получены из природных источников только с помощью сложных способов, например с помощью процессов специфичного гидролиза. В дополнение к этому получение данных соединений из природных источников с фармацевтически приемлемой чистотой представляет дополнительную проблему, особенно потому, что исходное сырье в основном получают из потенциально патогенных организмов. Ввиду данных проблем целью настоящего изобретения является предоставление таких соединений в синтетической форме. Для этого настоящее изобретение согласно первому аспекту предоставляет новый способ химического синтеза β-(1→6)-связанных глюкозамин-дисахаридов.
Дополнительный аспект изобретения относится к способу, подходящему для обработки продуктов, получаемых с помощью синтетического способа изобретения. Продукты, обрабатываемые с помощью данного способа обработки, обладают измененным физико-химическим строением и в соответствии с предпочтительным воплощением имеют повышенную биологическую активность.
Согласно дополнительным аспектам настоящее соединение относится к соединениям, получаемым по способам изобретения, промежуточным соединениям способа синтеза, композициям, включающим данные соединения, и к применению данных соединений в процессе органического синтеза и/или медицине.
Стоит здесь упомянуть, что соединения изобретения предпочтительно действуют через TLR2 человека.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Важной стадией в способе согласно изобретению является реакция гликозилирования между соединением формулы 10:

в которой
R1 представляет собой группу, выбранную из (C3-C6)алкенила, такого как C3 или C4алкенил, предпочтительно 2-пропенил или 1-пропенил;
X представляет водород, группу, выбранную из бензила или замещенного бензила, такого как 4-метоксибензил или 3,4-диметоксибензил, или 2,5-диметоксибензил, или 2,3,4-триметоксибензил, или 3,4,5-триметоксибензил;
R0 выбран из R5 или R2, где R5 выбран из:
(i) ацильной группы, происходящей из карбоновой кислоты с линейной цепью, имеющей от 2 до 24 атомов углерода, предпочтительно гидроксиацильной группы, такой как 3-гидроксиацильная группа, оксоацильной группы, такой как 3-оксоацильная группа, аминоацильной группы, такой как 3-аминоацильная группа;
(ii) ацилоксиацильной группы, предпочтительно 3-ацилоксиацильной группы, ациламиноацильной группы, предпочтительно 3-ациламиноацильной группы, ацилтиоацильной группы, предпочтительно 3-ацилтиоацильной группы;
(iii) алкилоксиацильной, предпочтительно (C2-C24)алкилоксиацильной группы, алкенилоксиацильной, предпочтительно (C2-C24)алкенилоксиацильной группы, алкинилоксиацильной, предпочтительно (C2-C24)алкинилоксиацильной группы, алкиламиноацильной, предпочтительно (C2-C24)алкиламиноацильной группы, алкениламиноацильной, предпочтительно (C2-C24)алкениламиноацильной группы, алкиниламиноацильной, предпочтительно (C2-C24)алкиниламиноацильной группы; алкилтиоацильной, предпочтительно (C2-C24)алкилтиоацильной группы, алкенилтиоацильной, предпочтительно (C2-C24)алкенилтиоацильной группы, алкинилтиоацильной, предпочтительно (C2-C24)алкинилтиоацильной группы, ацильной группы, происходящей из карбоновой кислоты с разветвленной цепью, имеющей от 2 до 48 атомов углерода, предпочтительно карбоновой кислоты, разветвленной в 3-положении;
где в группах (i), (ii), (iii) углеводородная цепь ацила может быть насыщенной или ненасыщенной, и углеводородная цепь ацила, алкила, алкенила, алкинила может быть разветвленной или линейной и необязательно может быть замещена одной или более группами, независимо выбранными из галогена, такого как фтор, хлор, бром или йод; гидроксила или гидроксильного производного -OY, где Y имеет значения, определенные ниже; амина или производного амина -NHW, где W имеет значения, определенные ниже; группы -OZ, где Z выбран из (f), (g), (h), (i), (k), определенных ниже;
и R2 представляет группу, выбранную из (C1-C6)галогенированного алкоксикарбонила, такого как 2,2,2-трихлорэтоксикарбонил (TROC) или 1,1-диметил-2,2,2-трихлорэтоксикарбонил (TCBOC);
с соединением формулы 7:
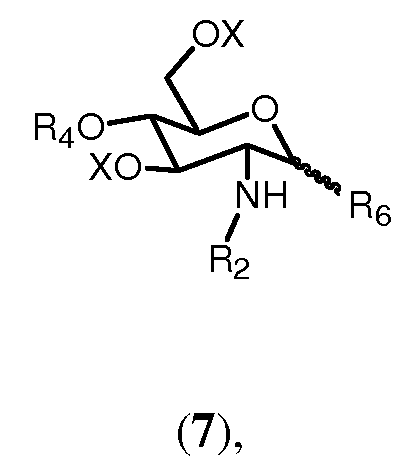
где R4 выбран из
(a) ацильной группы, определенной в (i), (ii) или (iii) для R5;
(b) разветвленной или линейной алкильной, предпочтительно разветвленной или линейной (C1-C24) алкильной группы; разветвленной или линейной алкенильной, предпочтительно разветвленной или линейной (C1-C24) алкенильной группы, разветвленной или линейной алкинильной, предпочтительно разветвленной или линейной (C1-C24) алкинильной группы;
(c) группы -[(C1-C24)алкил]-COOX, -[(C2-C24)алкенил]-COOX или -[(C2-C24)алкинил]-COOX, где X имеет значения, определенные ниже;
(d) группы -[(C1-C24)алкил]-NHW, -[(C2-C24)алкенил]-NHW или -[(C2-C24)алкинил]-NHW, где W имеет значения, определенные ниже;
(e) формилалкильной группы, предпочтительно формил[(C1-C24)алкильной] группы; формилалкенильной группы, предпочтительно формил[(C1-C24)алкенильной] группы; формилалкинильной группы, предпочтительно формил[(C1-C24)алкинильной] группы;
(f) диметоксифосфорильной группы;
(g) группы -P(O)(OY)2, где Y имеет значения, определенные ниже;
(h) группы -P(O)(OH)-O[(C1-C24)алкил]NHW, -P(O)(OH)-O[(C2-C24)алкенил]NHW или -P(O)(OH)-O[(C2-C24)алкинил]NHW, где W имеет значения, определенные ниже;
(i) группы -P(O)(OH)-O[(C1-C24)алкил], -P(O)(OH)-O[(C1-C24)алкенил] или -P(O)(OH)-O[(C1-C24)алкинил];
(j) группы -P(O)(OH)-O[(C1-C24)алкил]-СООХ, -P(O)(OH)-O[(C1-C24)алкенил]-СООХ, -P(O)(OH)-O[(C1-C24)алкинил]-СООХ, где Х имеет значения, определенные выше;
(k) группы -S(O)(OH)2;
(l) защитной группы, выбранной из бензила или замещенного бензила, такого как 4-метоксибензил или 3,4-диметоксибензил, или 2,5-диметоксибензил, или 2,3,4-триметоксибензил, или 3,4,5-триметоксибензил; или из (C3-C6)алкенила, такого как C3 или C4алкенил, предпочтительно 2-пропенил или 1-пропенил;
где алкильная, алкенильная, алкинильная группы могут быть разветвленными или линейными и могут быть незамещенными или необязательно замещены одной или более группами, независимо выбранными из галогена, такого как фтор, хлор, бром или йод; гидроксила или гидроксильного производного -OY, где Y имеет значения, определенные ниже; амина или производного амина -NHW, где W имеет значения, определенные ниже; или группы -OZ, где Z выбран из (f), (g), (h), (i), (j) (k);
и где Y выбран из водорода; (C3-C6)алкенила, такого как C2 или C3алкенил, предпочтительно 2-пропенил или 1-пропенил; группы, выбранной из бензила или замещенного бензила, такого как 4-метоксибензил или 3,4-диметоксибензил, или 2,5-диметоксибензил, или 2,3,4-триметоксибензил, или 3,4,5-триметоксибензил; О-ксилиленовой группы;
и где W выбран из водорода; бензилоксикарбонильной группы или 9-флуоренилметилоксикарбонила;
и где R6 представляет группу, выбранную из трихлорацетимидата, фторида, хлорида, бромида, и Х и R2 имеют значения, определенные выше.
Реакция может осуществляться согласно общему методу гликозилирования, известному в данной области техники, такому как способ, описанный в Angew. Chem., Int. Ed. Engl., (1986), 212. В данном способе используют дихлорметан в качестве растворителя и каталитическое количество кислоты, такой как триметилсилилтрифторметансульфонат. Когда используют данный способ, получают только β-дисахарид согласно формуле 11h.

в которой R1, R2, R4, R0 и Х имеют значения, описанные выше. Связь в виде связи, присоединяющей OR1, указывает, что возможны как α, так и β аномер.
R5 может быть выбран из ацильной группы, определенной в (i), или альтернативно разветвленной ацильной группы, определенной в (ii), (iii). Ацильная группа может быть выбрана из группы, включающей ацилоксиацильную, ациламиноацильную, ацилтиоацильную, (C1-C24)алкилоксиацильную, (C1-C24)алкиламиноацильную и (C1-C24)алкилтиоацильную группу. (Cn-Cn), где n является целым числом, такой как (C1-C24) и (C2-C24), используемый в данном описании, обозначает, что насыщенная или ненасыщенная углеводородная цепь, к которой относится данный символ, может содержать число атомов углерода в интервале, таком как соответственно 1-24 и 2-24 атома углерода, такое как 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22 атома углерода. Ацильная, алкильная, алкенильная и алкинильная углеводородная цепи в ациле и ацильных производных, определенных в (i), (ii) или (iii), может, каждая, индивидуально включать число от 1 до 50 атомов углерода, такое как от 2 до 48 атомов углерода, включая 1-24 атома углерода, такое как 2-24 атома углерода, в частности 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22 атома углерода. В (C2-C24) алкилоксиацильной группе, например, алкильный углеводород может включать от 2 до 24 атомов углерода, и углеводородная цепь ацильного фрагмента может включать от 2 до 24 атомов углерода.
Углеводородная цепь ацильных групп может быть насыщенной или может включать одну или более ненасыщенных углеродных двойных или тройных связей. В дополнение к этому углеводородные цепи ацила, алкила, алкенила и алкинила могут быть разветвленными или линейными и могут быть необязательно замещены одной или более группами, независимо выбранными из галогена, такого как фтор, хлор, бром или йод; гидроксила или гидроксильного производного -OY, где Y имеет значения, определенные выше; амина или производного амина -NHW, где W имеет значения, определенные выше; группы -OZ, где Z выбран из (f), (g), (h), (i), (j),(k), определенных выше.
В случае ацилоксиацильной группы две ацильные группы связаны через атом кислорода, в случае ациламиноацильной группы - через NH группу, и в случае ацилтиоацильной группы - через атом серы. (C1-C24)алкилоксиацильная, (C1-C24)алкиламиноацильная и (C1-C24)алкилтиоацильная группа может быть получена из соответствующей гидрокси жирной кислоты.
Ацильные группы предпочтительно замещены в 3-положении и являются такими как 3-ацилоксиацильная, 3-ациламиноацильная и 3-ацилтиоацильная группа. То же самое применимо к упомянутым выше (C1-C24)алкильным эквивалентам.
Предпочтительно члены группы R5 включают один или два ацильных фрагмента, предпочтительно выбранных из остатков жирной кислоты, остатков гидрокси жирной кислоты и остатков окси жирной кислоты. Когда ацилоксиацильной группой является 3-ацилоксиацильная группа, данные ацильные фрагменты предпочтительно включают остаток 3-гидрокси жирной кислоты или для связанной сложноэфирной связью группы остатка 3-оксо жирной кислоты. Типичными примерами ацилоксиацильных групп являются 3-гидрокси(C4-C24)-жирная кислота-ацилы, которые связаны сложноэфирной связью с 3-гидрокси группой (C1-C24)-карбоновой кислоты. Предпочтительно ацилоксиацильной группой является 3-гидрокси(C8-C18)-жирная кислота-ацил, которая связана сложноэфирной связью с 3-гидрокси группой (C10-C18)-жирной кислоты. Такие ацилоксиацильные группы присутствуют в липидном А компоненте грам-отрицательных бактерий, таких как Escherichia coli, Haemophilus influenzae, Campylobacter jejuni, Rhodocyclus gelatinosus, Chromobacterium violaceum, Neisseria meningitides, Salmonella Minnesota.
В одной группе предпочтительных глюкозамин-дисахаридов согласно изобретению ацилоксиацильной группой, выбранной для R5, является 3-гидрокси-C14-жирная кислота-ацил, связанный сложноэфирной связью с 3-гидрокси группой C12-жирной кислоты, с данной ацилоксиацильной группой в N2'-положении. В еще одном предпочтительном глюкозамин-дисахариде согласно изобретению ацилоксиацильной группой, выбранной для R5, является 3-гидрокси-C14-жирная кислота-ацил, связанный сложноэфирной связью с 3-гидроксигруппой C14-жирной кислоты, и ацилоксиацильная группа находится в N-2' положении.
В еще одном предпочтительном глюкозамин-дисахариде согласно изобретению ацилоксиацильной группой, выбранной для R5, является 3-гидрокси-C14-жирная кислота-ацил, связанный сложноэфирной связью с 3-гидроксигруппой C12-жирной кислоты, с данной ацилоксиацильной группой в N-2 положении. В еще одном предпочтительном глюкозамин-дисахариде согласно изобретению ацилоксиацильной группой, выбранной для R5, является 3-гидрокси-C14-жирная кислота-ацил, связанный сложноэфирной связью с 3-гидроксигруппой C12-жирной кислоты, с ацилоксиацильной группой и в N2-положении, и в N2'-положении.
Когда соединение изобретения включает хиральный центр, изобретение охватывает все R- и S-энантиомеры и любую рацемическую смесь.
Другим выбором для R5 может быть ацильная группа или также ацилоксиацильная группа.
Согласно второй группе дисахаридов согласно изобретению ацильной группой является 3-гидрокси (C4-C24)-жирная кислота, предпочтительно 3-гидрокси (C10-C18)-жирная кислота. 3-Гидроксигруппа такой жирной кислоты может быть защищена группой Х, определенной ранее. В предпочтительных дисахаридах согласно изобретению ацильной группой является 3-гидрокси C14-жирная кислота, в N2-положении или в N2'-положении.
Однако R5 может также быть ацилоксиацильной группой, определенной здесь выше и включающей в число их 3-гидрокси-(C4-C24)жирную кислоту-ацил, который является связанным сложноэфирной связью с 3-гидрокси группой (C1-C20) карбоновой кислоты, предпочтительно 3-гидрокси-(C8-C18)жирную кислоту-ацил, связанный сложноэфирной связью с 3-гидрокси группой (C10-C18) жирной кислоты. Более предпочтительным является дисахарид, в котором R5 в N2-положении представляет собой 3-гидрокси-C14-жирную кислоту-ацил, связанный сложноэфирной связью с 3-гидрокси группой C12-жирной кислоты или C16-жирной кислоты, и в котором R5 в N2'-положении представляет 3-гидрокси-C14-жирную кислоту-ацил, связанный сложноэфирной связью с 3-гидрокси группой C12-жирной кислоты, C12-жирной кислоты или C14-жирной кислоты.
Согласно предпочтительному воплощению первая группа R5 выбрана из подгруппы (i), как она определена, и вторая группа R5 выбрана из подгруппы (ii) или (iii), определенных в п.1 формулы изобретения, где группа R5 в N-2 положении выбрана из (i). В альтернативных воплощениях обе группы R5 выбраны идентично или различно из подгруппы (i) или обе выбраны идентично или различно из подгрупп (ii) или (iii).
Отмечается, что в группе R5 ацильные группы и/или ацильная и алкильная группа могут быть взаимосвязаны.
В данном описании термин “остаток жирной кислоты” означает по существу гидрофобную цепь из C2-C30 атомов, которая может быть линейной, разветвленной, насыщенной, моно- или полиненасыщенной, имеющей один или более гетероатомов, таких как азот, кислород, сера, и которая может быть замещена одним или более заместителями, такими как гидроксил, оксо, ацилокси, алкокси, амино, нитро, циано, галоген, сульфгидрил, при условии, что биологическая активность по существу не испытывает отрицательного воздействия. Пример остатка замещенной жирной кислоты (включая амид-связанный заместитель) описан авторами Onozuka, K. et al. в Int. J. Immunopharmac, Volume 15, страницы 657-664 [1993]).
R4 может быть выбран из (a)-(l), определенных выше. Алкильная, алкенильная, алкинильная цепи в данных заместителях для R4 могут быть разветвленными или линейными и могут быть незамещенными или необязательно замещенными одной или более группами, независимо выбранными из галогена, такого как фтор, хлор, бром или йод; гидроксила или гидроксильного производного -OY, где Y имеет значения, определенные выше; амина или производного амина -NHW, где W имеет значения, определенные выше. Для групп (a), (b), (c), (d), (e) необязательные заместители могут дополнительно включать в их число группу -OZ, где Z выбран из (f), (g), (h), (i), (j), (k). Предпочтительно R4 выбран из (f), (g), (h), (i) или (j), более предпочтительно из (g). Предпочтительно группы (a), (b), (c), (d), (e), (f), (g), (h), (i), (j) включают от 1 до 50 атомов углерода, например от 2 до 24 атомов углерода.
На последующей стадии ряд (С1-С6) галогенированных алкоксикарбонильных защитных групп R2 гидролитически удаляется из соединения формулы 11h. В данном описании ряд обозначает одну или более, если не конкретизировано иным образом. Предпочтительно, чтобы все группы R2 соединения формулы 11h удалялись. Если в качестве R5 выбран R0, тогда соединение формулы 11h будет включать одну группу R2. Если R0 выбран в качестве R2, тогда соединение формулы 11h будет включать две группы R2 и будет предпочтительно удалять обе данные группы. Группы R2 могут быть удалены любыми подходящими средствами, известными специалистам. Специалисту известно, что (С1-С6) галогенированные алкоксикарбонильные защитные группы, такие как Troc, могут удаляться с использованием цинк-медной пары в уксусной кислоте и воде.
Если в качестве R5 выбран R0, тогда получится соединение формулы 12а.
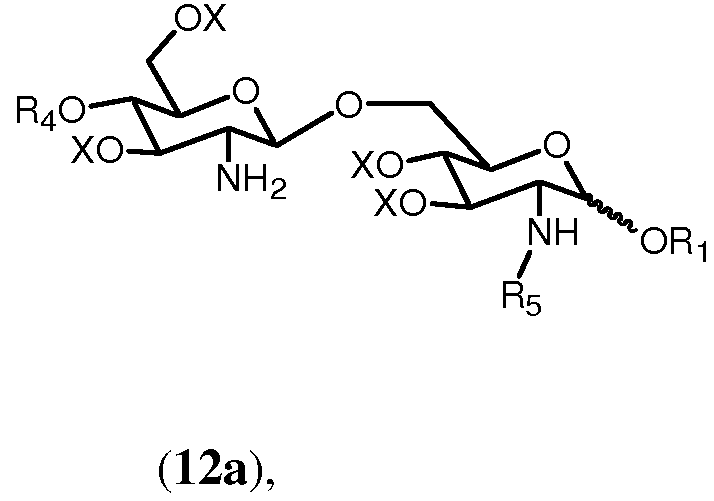
в которой R1, R4, R5 и Х имеют значения, определенные выше. Если R0 выбран в качестве R2 в формуле 11h, тогда получится предпочтительно соединение формулы 12b.
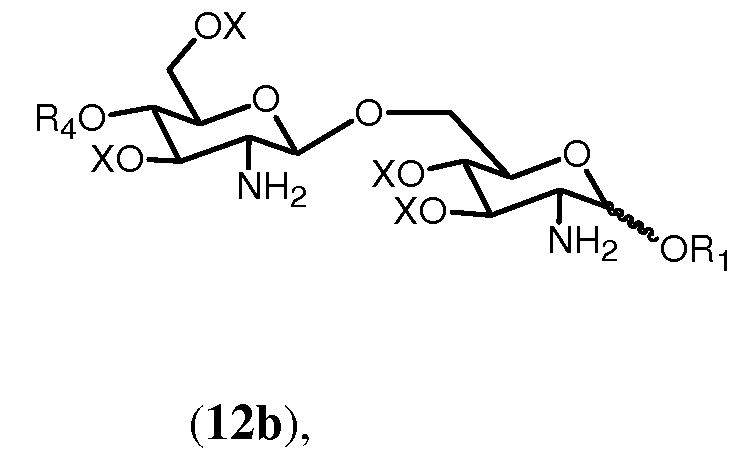
где R1, R4 и Х имеют значения, определенные выше.
К свободной аминогруппе соединения формулы 12a или 12b присоединяют группу R5. Это может достигаться с помощью реакции соединения формулы 12а или 12b с (активированной) карбоновой кислотой, соответствующей указанной группе R5. Реакция может проводиться любым способом, известным специалистам, таким как с использованием конденсирующего агента, такого как изобутилхлорформиат или 1-изобутилокси-2-изобутилоксикарбонил-1,2-дигидрохинолеин или карбодиимид. В реакции соединения 12а (активированная) карбоновая кислота, соответствующая указанной группе R5, может включать группу R5, одинаковую или отличную от группы R5 соединения формулы 12а.
Реакция соединения формулы 12а или 12b с (активированной) карбоновой кислотой, соответствующей указанной группе R5, приводит в результате к образованию соединения формулы 13:
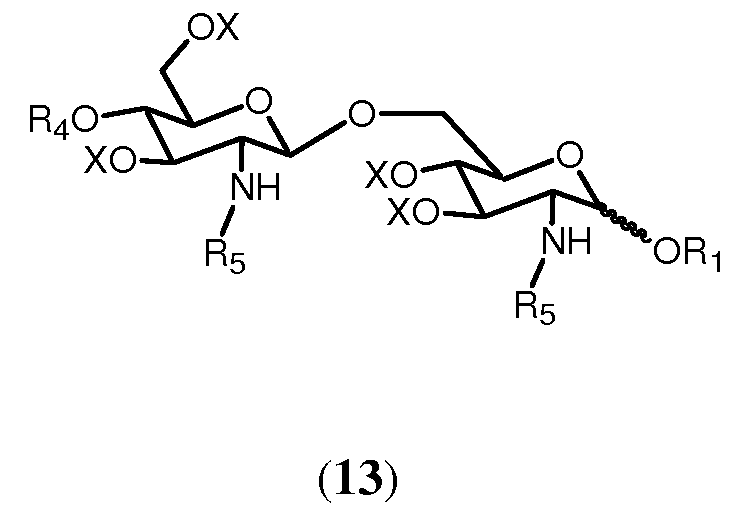
в которой R1, R4, R5 и Х имеют значения, определенные ранее. Группы R5 могут быть одинаковыми или различными. Являются ли группы R5 соединения 13 одинаковыми или различными, может зависеть от того, используется ли в реакции соединение 12а или соединение 12b, и от характера (активированной) карбоновой кислоты, используемой в реакции. Если используется соединение 12b, можно выбрать группу R5 (активированной) карбоновой кислоты, отличную от группы R5 соединения 12b. В данном случае группы R5 соединения 13 будут различаться. Однако группа R5 (активированной) карбоновой кислоты может быть также идентична группе R5 соединения 12b. И ясно очевидно, что в этом случае группы R5 соединения 13 будут идентичными. Если соединение 12а подвергается реакции с одной (активированной) карбоновой кислотой, группы R5 соединения 13 будут идентичными. Однако можно также использовать комбинаторную химию и подвергать реакции соединение 12b с рядом различающихся (активированных) карбоновых кислот. В этом случае будет получаться смесь соединений согласно общей формуле 13, в которых группы R5 являются одинаковыми или различными. Специалист в данной области очевидно понимает, что число различных соединений общей формулы 13 и их соотношения в смеси будут зависеть от числа различных (активированных) карбоновых кислот, используемых в реакции, и от их соотношений. Предпочтительно, чтобы, по меньшей мере, одна из R5 была выбрана из разветвленной ацильной группы, определенной в (ii), (iii). Более предпочтительно в качестве разветвленной ацильной группы выбирается группа R5, присоединенная в N2'-положении.
Полуацеталь формулы 14:
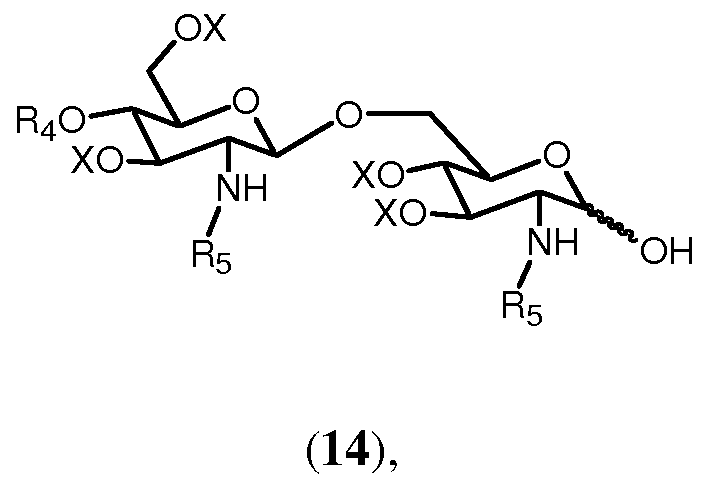
в которой R4, R5 и Х имеют значения, определенные выше, образуется с помощью удаления группы R1 из соединения формулы 13. Снятие защиты (C3-C6)алкенильной группы может достигаться любым способом, известным специалисту. Например, (C3-C6)алкенильная группа может быть удалена двухстадийным превращением. Если (C3-C6)алкенильной группой является, например, 2-пропенил, сначала аллильная группа в 13 может изомеризоваться в 1-пропенил с помощью обработки водород-активируемым иридиевым катализатором, таким как промышленно доступный гексафторфосфат ([бис(метилдифенилфосфин)]-(1,5-циклооктадиен)иридия(I)) в полярном растворителе, таком как тетрагидрофуран (Synthesis, (1981), 305-308). 1-пропенильная группа может затем отщепляться водным источником иода, таким как йод или N-бромсукцинимид. (J. Chem. Soc., Chem. Commun., (1982), 1274). По аналогии могут удаляться различные варианты группы R1.
Соединение 13 и полуацеталь формулы 14 являются важными промежуточными продуктами в способе синтеза согласно изобретению. В зависимости от реакций, проводимых с соединениями 13 и 14, и получающихся в результате их промежуточных соединений может получаться большое число различных защищенных β-(1→6)-связанных глюкозамин-дисахаридов с различными заместителями R8 в О-1 положении. Данные β-(→6)-связанные глюкозамин-дисахариды могут быть представлены общей формулой 15:
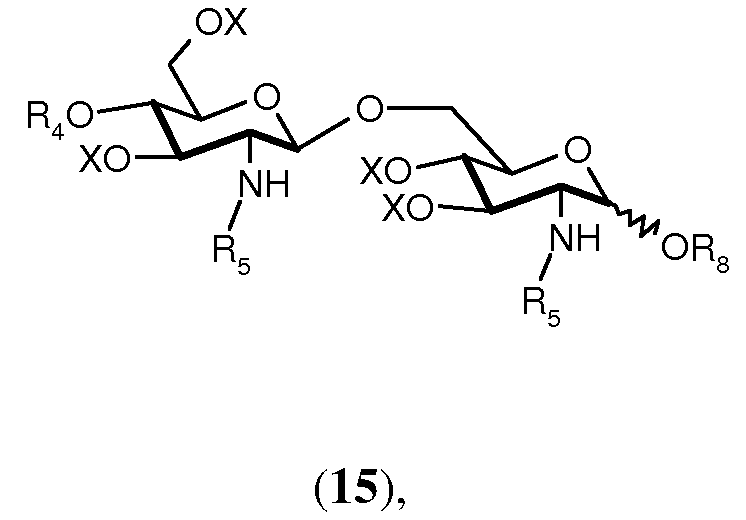
где R4, R5 и Х имеют значения, определенные ранее, и R8 выбран из (a), (b), (c), (d), (e), (f), (g), (h), (i), (j) или (k), определенных ранее для R4.
В одном из воплощений способа синтеза изобретения свободная гидроксильная группа соединения 14 может фосфорилироваться любым известным специалисту способом. Для этого может использоваться промышленно доступный тетрабензилпирофосфат в присутствии подходящего основания в полярном растворителе. Основание может быть выбрано из бис(триметилсилил)амида лития, а растворитель может быть выбран из тетрагидрофурана. Фосфорилирование соединения 14 дает в результате соединение 15а
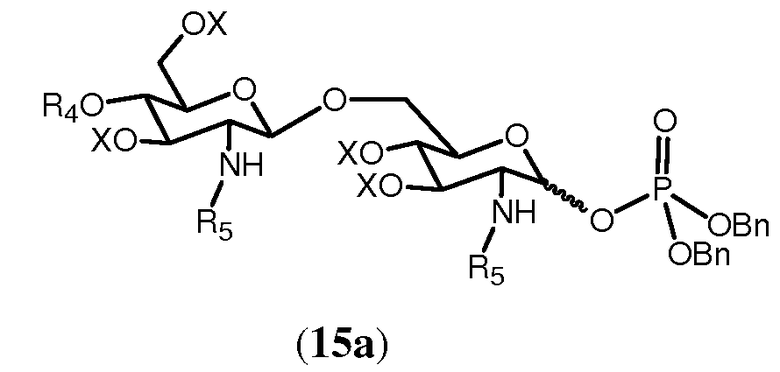
Фосфорилирование может быть полезным для получения соединений, имеющих в О-1 положении заместители, выбранные из (g), (h), (i) или (j), определенных для R4. Если необходимо, фосфатная группа, получаемая в соединении 15а, может далее модифицироваться.
В еще одном воплощении свободная гидроксильная группа соединения 14 может сульфатироваться любым известным специалисту образом. Сульфатирование соединения 14 дает в результате соединение формулы 15b:
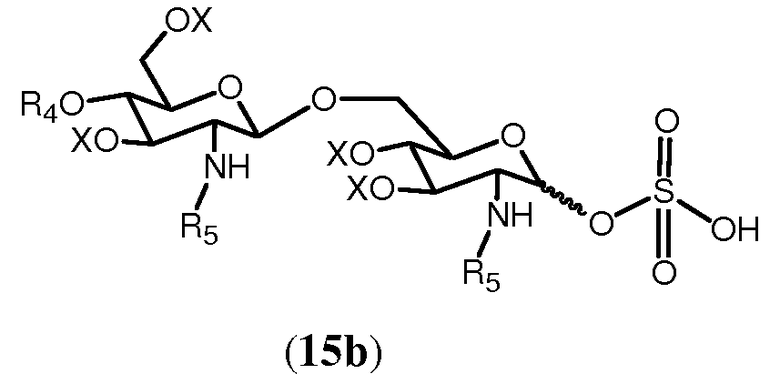
В еще одном воплощении способ согласно изобретению дополнительно предусматривает реакцию свободной гидроксильной группы соединения 14 с (активированной) карбоновой кислотой формулы R8OH, в которой R8 выбран из (а), определенного ранее для R4. Реакция может осуществляться любым известным специалисту образом, таким как в присутствии конденсирующего агента, такого как изобутилхлорформиат или 1-изобутилокси-2-изобутилоксикарбонил-1,2-дигидрохинолеин, или карбодиимид с образованием соединения 15с:

где R4, R5 и Х имеют значения, определенные выше, и R8 выбран из (а), определенного ранее для R4, и R8 может находиться в α- или β-конфигурации, и предпочтительно в β-конфигурации.
В еще одном воплощении группа, которая может функционировать в последующей реакции как уходящая группа, такая как трихлорацетимидатная группа, сочетается со свободной гидроксильной группой соединения 14. Это может осуществляться любым известным специалисту образом, например, с помощью реакции соединения 14 с трихлорацетонитрилом в присутствии неорганического основания, такого как карбонат цезия или карбонат калия, в полярном растворителе, предпочтительно апротонном полярном растворителе, таком как дихлорметан. Данная реакция соединения 14 приводит в результате к соединению формулы 24:
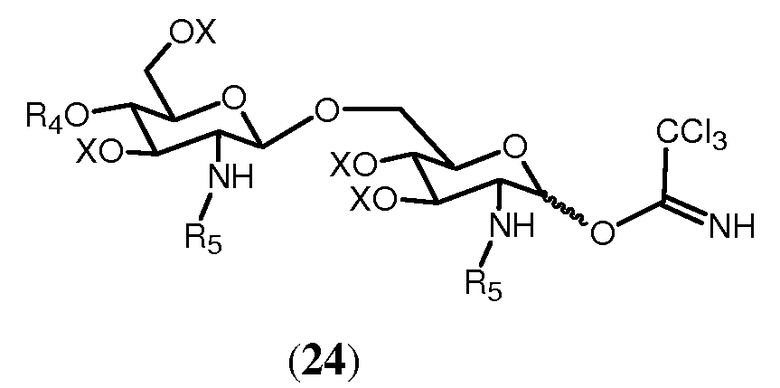
Соединение 24 может подвергаться реакции с органической молекулой R8OH с заменой трихлорацетимидатной группы группой R8. R8 может быть выбран из (b), (c), (d), (e), определенных для R4.
Реакция ацетимидатной группы с органическим спиртом известна специалистам. Она может происходить в полярном растворителе, предпочтительно апротонном полярном растворителе, таком как дихлорметан, в присутствии каталитического количества кислоты, такой как триметилсилилтрифторметансульфонат, и может проводиться по аналогии со способом, описанным в Angew. Chem., Int. Ed. Engl., (1986), 212. Реакция соединения 24 с группой R8 дает в результате соединение формулы 15d:

где R4, R5, R8 и Х имеют значения, определенные выше, и где R8 может находиться в α- или β-конфигурации, и предпочтительно в β-конфигурации.
Соединения 13, 15а, 15b, 15с и 15d могут далее подвергаться реакции так, чтобы удалить любые защитные группы, выбранные из Х, Y, W, отличных от Н. Удаление защитных групп может достигаться в соответствии с известными в технике способами. Например, бензильные защитные группы могут удаляться с помощью гидрогенолиза в присутствии благородного металла, такого как палладий-на-угле. Аллильные группы и аналогичные группы могут быть удалены, как обсуждалось выше для удаления аллильной группы из соединения 13. Удаление 4-метоксибензильной или 3,4-диметоксибензильной, или 2,5-диметоксибензильной, или 2,3,4-триметоксибензильной, или 3,4,5-триметоксибензильной, или фенильной, или 4-метоксифенильной, или 3,4-диметоксифенильной, или 2,5-диметоксифенильной, или 2,3,4-триметоксифенильной, или 3,4,5-триметоксифенильной групп может проводиться с помощью окислительного отщепления, такого как с помощью дихлордицианохинона (DDQ) или аммонийнитрата церия (CAN). О-Ксилиленовая группа и бензилоксикарбонильная группа могут удаляться с помощью гидрогенолиза в присутствии благородного металла, такого как палладий-на-угле. 9-Флуоренилметилоксикарбонил может удаляться основанием, таким как пиперидин, морфолин. Очевидно понятно, что различные защитные группы могут удаляться независимо. Следовательно, перед удалением Х может быть удалена любая защитная группа, присутствующая в R8.
Реакционноспособные группы, первоначально присутствующие в R8 или после удаления защитной группы, могут дальше подвергаться реакции перед удалением (дополнительных) защитных групп. Если R8 включает ряд свободных гидроксильных групп, могут образовываться сложные эфиры, включая фосфатные и сульфатные эфиры, и простые эфиры с помощью известных способов. Свободные гидроксильные группы, кроме того, могут окисляться известными способами с получением карбоновой кислоты или кетона. Если R8 включает ряд групп карбоновой кислоты, по известным способам могут образовываться сложные эфиры или амиды. Если R8 включает ряд свободных аминовых групп, с помощью известных в технике способов могут образовываться амиды. Если R8 включает ряд ненасыщенных углеродных связей, они могут известными способами подвергаться реакции с тетраоксидом осмия с получением α,β-гидроксилированной группы. Свободные гидроксильные группы, такие как α,β-гидроксилированная группа, могут дополнительно подвергаться реакции перед удалением защитных групп.
В дополнение к этому фосфатная группа может метилироваться известными способами, такими как реакция с CH2N2. Следует заметить, что такое метилирование с помощью CH2N2 может происходить до или после удаления защитных групп в β-(1→6)-связанных глюкозамин-дисахаридах, включая защитную группу, выбранную из Х, как определено выше.
В альтернативном воплощении способа изобретения защитные группы соединения 14 удаляют с помощью известных способов, таких как описаны выше.
В дополнительном альтернативном воплощении способа изобретения ненасыщенная связь (C3-C6)алкенильной группы соединения 13, такой как С3 или С2 алкенил, предпочтительно 2-пропенил или 1-пропенил, гидрируется в соответствующий алкил.
В еще одном альтернативном воплощении способа изобретения (С3-С6)алкенильная группа соединения 13 выбирается в виде 2-пропенила, и ненасыщенная связь 2-пропенильной группы подвергается реакции с тетраоксидом осмия по известному способу с получением α,β-гидроксилированной группы. Свободные гидроксильные группы такой α,β-гидроксилированной группы могут далее подвергаться реакции перед удалением защитных групп.
Очевидно ясно, что с помощью способа синтеза согласно изобретению может получаться большое число β-(1→6)-связанных глюкозамин-дисахаридов формулы 1:

где R4', R5' и R8' имеют значения, определенные соответственно ранее для R4, R5 и R8, в которых любой Y или W представляют Н, и где значение R8' дополнительно включает Н.
Соединение 7, которое вовлекается в процесс согласно изобретению, может быть получено с помощью присоединения уходящей группы, выбранной из трихлорацетимидата, фторида, хлорида, бромида, к свободной гидроксильной группе соединения формулы 6:
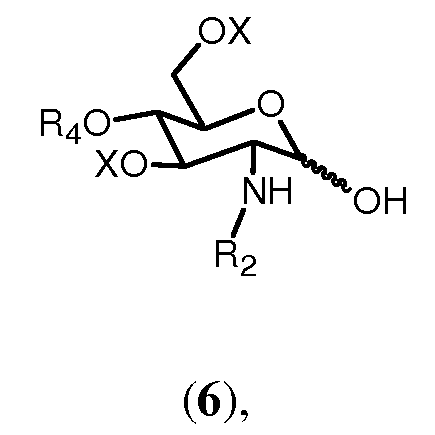
где R2, R4 и Х имеют значения, определенные ранее. Это может проводиться любым подходящим способом, известным в технике. Например, обработка соединения формулы 6 трихлорацетонитрилом, предпочтительно в присутствии основания, более предпочтительно неорганического основания, такого как карбонат цезия или карбонат калия, в полярном растворителе, предпочтительно апротонном полярном растворителе, таком как дихлорметан. Защита хлором и бромом может проводиться по реакции уксусного ангидрида в растворителе, таком как пиридин, и последующей реакции с газообразной соответственно HCl или HBr в уксусной кислоте. Защита фтором может выполняться по реакции с уксусным ангидридом и последующей реакции с трифторидом диациламиносеры (DAST).
Соединение формулы 6 может быть получено путем удаления известными способами группы R1 из соединения формулы 5:
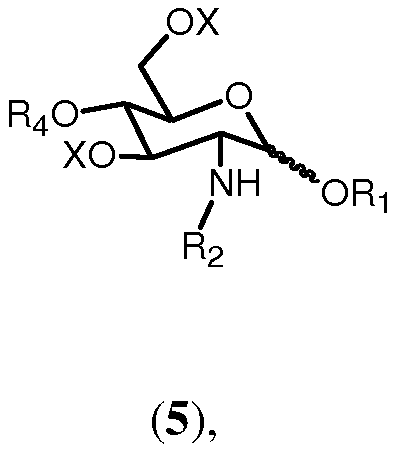
где R1, R2, R4 и Х имеют значения, определенные ранее. Например, снятие защиты аллильной группы может достигаться с помощью двухстадийного превращения. Сначала аллильная группа может изомеризоваться в 1-пропенил с помощью обработки водород-активируемым иридиевым катализатором, таким как промышленно доступный гексафторфосфат ([бис(метилдифенилфосфин)]-(1,5-циклооктадиен)иридия(I) в полярном растворителе, таком как тетрагидрофуран, согласно методу, описанному в Synthesis, (1981), 305-308. Пропенильная группа может затем отщепляться водным источником йода, таким как йод или N-бромсукцинимид. (J. Chem. Soc., Chem. Commun., (1982), 1274).
Соединение формулы 5 может получаться с помощью ряда различных реакций в зависимости от выбора группы R4. Данные реакции могут исходить из соединения формулы 4:
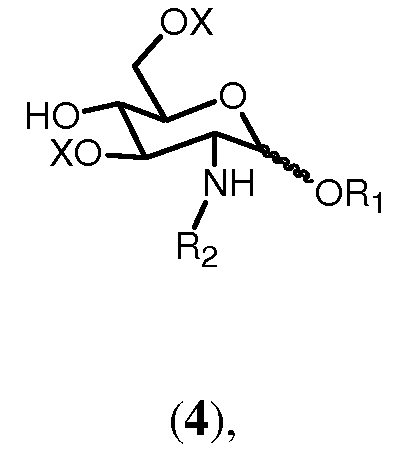
где R1, R2 и Х имеют значения, определенные выше. Исходя из соединения 4, к свободной гидроксильной группе данного соединения в качестве R4 может добавляться ряд различных заместителей. Данные заместители могут присоединяться по общим методам, известным в технике.
Если R4 выбран из (f),(g), (h), (i) или (j), способ согласно изобретению может включать фосфорилирование в подходящих реакционных условиях свободной гидроксильной группы соединения формулы 4:
где R1, R2 и Х имеют значения, определенные ранее. Это может выполняться, например, по реакции с фосфорамидитным реагентом, таким как диарил N,N-диалкилфосфорамидит или диаллил N,N-диалкилфосфорамидит, предпочтительно диаллил N,N-диизопропилфосфорамидит, в присутствии конденсирующего агента, такого как [1H]тетразол, в полярном растворителе, предпочтительно апротонном полярном растворителе. В данной реакции сначала образуется фосфит, который может впоследствии окисляться в фосфат, например, в присутствии ароматической надкарбоновой кислоты, такой как м-хлорпероксибензойная кислота.
Если R4 выбран из (k), способ согласно изобретению может включать сульфатирование в подходящих условиях реакции свободной гидроксильной группы соединения формулы 4
где R1, R2 и Х имеют значения, определенные выше. Это может выполняться, например, по реакции с комплексом триоксида серы, например комплекса триметиламина с триоксидом серы, в полярном растворителе, таком как диметилформамид (ДМФ).
Если R4 выбран из (1), способ согласно изобретению может включать реакцию свободной гидроксильной группы соединения формулы 4:
где R1, R2 и Х имеют значения, определенные выше, с соединением, подходящим для присоединения донорской защитной группы к указанной свободной гидроксильной группе соединения формулы 4. Такая защитная группа соединения донора может быть предпочтительно выбрана из бензил-2,2,2-трихлорацетимидата или замещенного бензил-2,2,2-трихлорацетимидата, такого как 4-метоксибензил-2,2,2-трихлорацетимидат, 3,4-диметоксибензил-2,2,2-трихлорацетимидат, 2,5-диметоксибензил-2,2,2-трихлорацетимидат, 2,3,4-триметоксибензил-2,2,2-трихлорацетимидат или 3,4,5-триметоксибензил-2,2,2-трихлорацетимидат. Альтернативно защитная группа может быть производной (С3-С6)алкенил-2,2,2-трихлорацетимидата, такого как С3 или С4-2,2,2-трихлорацетимидат, предпочтительно 2-пропенил- 2,2,2-трихлорацетимидат или 1-пропенил-2,2,2-трихлорацетимидат. Реакция предпочтительно проводится в полярном растворителе и/или в присутствии кислотного катализатора, такого как трифторметансульфонат олова II или трифторметансульфоновая кислота.
Если R4 выбран из (а), способ согласно изобретению может включать реакцию свободной гидроксильной группы соединения формулы 4:
где R1, R2 и Х имеют значения, определенные выше, с карбоксигруппой (активированной) карбоновой кислоты формулы R4OH, в которой R4 выбран из (а), определенной выше. Реакция предпочтительно проводится в присутствии конденсирующего агента, такого как изобутилхлорформиат или 1-изобутилокси 2-изобутилоксикарбонил-1,2-дигидрохинолеин или карбодиимид.
Если R4 выбран из (b), (c), (d) или (e), способ согласно изобретению может включать реакцию свободной гидроксильной группы соединения формулы 4:
где R1, R2 и Х имеют значения, определенные выше, с производным 2,2,2-трихлорацетимидат-активированного алкилового спирта, соответствующим указанному выбору группы R4 (b), (c), (d) или (e). Реакция предпочтительно проводится в полярном растворителе и/или в присутствии кислотного катализатора, такого как трифторметансульфонат олова II или трифторметансульфоновая кислота. Специалисты очевидно понимают, что 2,2,2-трихлорацетимидат-активируемым спиртовым производным, соответствующим указанному выбору (b), (c), (d) или (e) группы R4, может быть алкил-2,2,2-трихлорацетимидат, такой как, например, пропил-2,2,2-трихлорацетимидат, когда R4 выбран из (b) в качестве алкильной группы. По аналогии с этим можно выбрать другие 2,2,2-трихлоримидат-активируемые спиртовые производные, соответствующие указанному выбору (b), (c), (d) или (e), такие как алкенил-2,2,2-трихлорацетимидат, алкинил-2,2,2-трихлорацетимидат.
Различные заместители группы R4 могут аналогично заместителям группы R8 содержать реакционноспособные группы, такие как гидроксильные группы, аминогруппы, карбоксигруппы или углеродные ненасыщенные связи, такие как двойные связи. Такие реакционноспособные группы в соединении 5 могут далее модифицироваться, например, по реакции, выбранной из образования сложного эфира, амидирования, окисления, гидрирования или α,β-гидроксилирования тетраоксидом осмия.
Соединение 4 может быть получено с помощью восстановительного раскрытия кольца бензилиденовой группы соединения формулы 3:
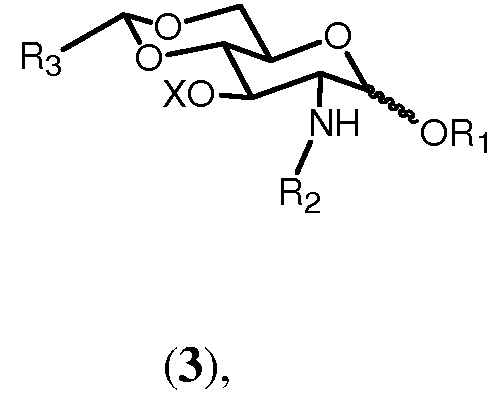
где R1, R2 и Х имеют значения, определенные ранее, и R3 представляет группу, выбранную из ароматического углеводорода, такую как фенильную или 4-метоксифенильную, или 3,4-диметоксифенильную, или 2,5-диметоксифенильную, или 2,3,4-триметоксифенильную, или 3,4,5-триметоксифенильную группу. Реакция может осуществляться по любому методу, известному в данной области, такому как с использованием гидрида, такого как триметиламин-борановый комплекс, и кислоты Льюиса, такой как хлорид алюминия, в полярном растворителе, таком как ТГФ. Данный метод описан в Carbohydrate Research, (2003), 697-703 и в Tetrahedron Lett. (2000), 41, 6843-6847.
Соединение 10, которое в способе изобретения вводится в реакцию вместе с соединением 7 с образованием соединения 11, может быть получено из соединения 9:
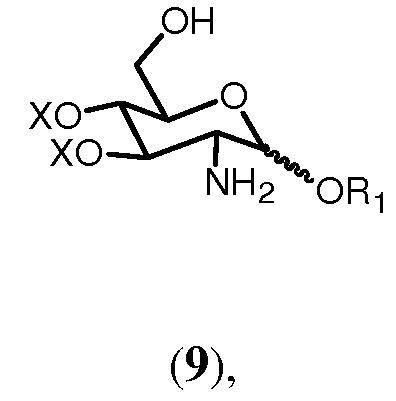
где R1 и Х имеют значения, определенные ранее. Для получения соединения 10 свободную аминогруппу соединения 9 ацилируют по реакции с (активированной) карбоновой кислотой формулы R5OH, где R5 имеет ранее указанные значения. Способ может осуществляться в условиях, известных специалистам, например, со смешанным ангидридом, таким как смешанный ангидрид, получаемый из (R)-3-бензилокситетрадекановой кислоты, описанной в Bull. Chem. Soc. Jpn, (1987), 2197-2204, и алкилхлорформиата, такого как изобутилхлорформиат.
Соединение 9 может образовываться гидролизным расщеплением известными методами группы R2 соединения формулы 8:
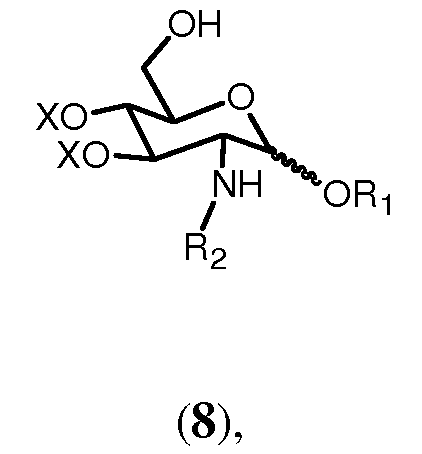
где R1, R2 и Х имеют значения, определенные выше. Например, трихлорэтоксикарбонильная защитная группа (Troc) может удаляться с использованием цинка в уксусной кислоте.
Соединение формулы 8 может получаться восстановительным раскрытием кольца в подходящих условиях реакции бензилиденовой группы соединения формулы 3:
где R1, R2, R3 и Х имеют значения, определенные выше. Для этого может использоваться любой известный метод, такой как с использованием гидрида, такого как диметиламин-борановый комплекс в качестве реагента, и кислоты Льюиса, такой как трифторид бора, в полярном растворителе, таком как дихлорметан.
Соединение формулы 3 может получаться по реакции соединения формулы 2:
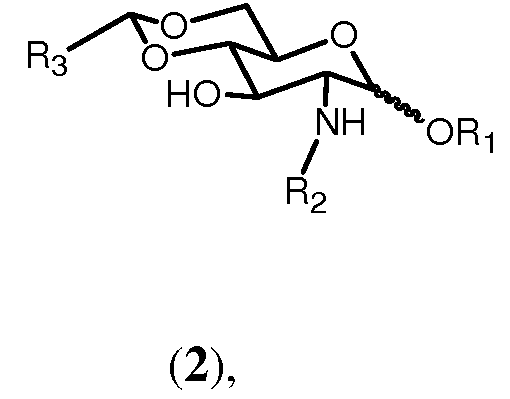
где R1, R2, R3 и Х имеют значения, определенные выше, с соединением, подходящим для введения защитной группы по свободной карбоксильной группе соединения формулы 2. Соединение, являющееся донором защитной группы, предпочтительно выбирается из бензил-2,2,2-трихлорацетимидата, 4-метоксибензил-2,2,2-трихлорацетимидата, 3,4-диметоксибензил-2,2,2-трихлорацетимидата, 2,5-диметоксибензил-2,2,2-трихлорацетимидата, 2,3,4-триметоксибензил-2,2,2-трихлорацетимидата или 3,4,5-триметоксибензил-2,2,2-трихлорацетимидата. Реакция предпочтительно проводится в полярном растворителе и/или в присутствии кислотного катализатора, такого как трифторметансульфонат олова II или трифторметансульфоновая кислота. Подходящие методы описаны в J. Chem. Soc., Chem. Commun., (1981), 1240-1241). Интересно заметить, что не наблюдалось никакой реакции при использовании методики, описанной в Tetrahedron Letters, (2001), 7613-7616 или в Tetrahedron Lett. (2000), 41, 6843-6847 для получения соединения 3, и выделялось только исходное вещество 2. Считают, что данные статьи, как таковые, не раскрывают соединение 3. Соединение 2 получалось, как описано в Liebigs Ann. (1996), 1599-1607.
Согласно дополнительному аспекту изобретение относится к способу обработки глюкозамин-дисахаридов, предпочтительно β-(1→6)-связанных глюкозамин-дисахаридов. Данный способ может использоваться для обработки соединений, получаемых с помощью способа синтеза согласно изобретению. Способ включает
(i) смешивание раствора соединения формулы 1:
где R'4, R'5 и R'8 имеют значения, определенные выше, с твердой смолой с обращенной фазой в условиях, подходящих для связывания, по меньшей мере, части соединения формулы 1 с твердой фазой;
(ii) удаление жидкой фазы и промывку твердой фазы промывочной жидкостью, включающей водную фазу, необязательно буферированную при рН 6-9, предпочтительно 7-8, и наиболее предпочтительно 7,3-7,7, и органическую фазу, которые смешаны в соотношении от 15:1 до 5:1, предпочтительно 9:1 (об./об.);
(iii) удаление промывочной жидкости и элюирование, по меньшей мере, части соединения 1, связанного с твердой фазой, элюирующей жидкостью, включающей водную фазу и органическую фазу, которые смешаны в соотношении между 1:15-1:5, предпочтительно 1:9 (об./об.);
(iv) сбор элюирующей жидкости, включающей некоторое количество соединения 1, и необязательно удаление органической фазы из элюирующей жидкости.
В предпочтительном воплощении способ далее включает доведение рН элюирующей жидкости, включающей некоторое количество соединения формулы 1 до заранее выбранной величины рН, предпочтительно рН 6-9, более предпочтительно 7-8 и наиболее предпочтительно рН 7,3-7,7. При данной величине рН продукты наиболее стабильны.
Неожиданно было найдено, что обработка соединения формулы 1 данным способом дает в результате соединения с повышенной биологической активностью по сравнению с исходным веществом.
Соединения формулы 1 могут связываться со смолой с обращенной фазой в полярном растворителе, таком как С2-С3 органический спирт, необязательно смешанном с водой, таком как смесь воды и 2-пропанола, смешанных в соотношении между 15:1 - 5:1, предпочтительно 9:1 (об./об.). Смолой с обращенной фазой может быть смола VYDAC C18 или любая другая подходящая смола с обращенной фазой.
Органическая фаза промывочной жидкости и/или элюирующей жидкости может включать органический растворитель, такой как полярный органический растворитель, например С2-С3 органический спирт.
Соединение формулы 1 может предоставляться в растворителе, который является подходящим для реакции, в которой защитные группы удаляются с помощью гидрогенолиза. Примером такого растворителя является тетрагидрофуран (ТГФ). Соединения согласно изобретению можно обрабатывать в способе обработки согласно настоящему изобретению непосредственно после их синтеза способом изобретения. Однако предпочтительно сначала очищать соединения изобретения. Очистка может проводиться по известным методам, таким как использование хроматографии с обращенной фазой, предпочтительно ионно-парной хроматографии с обращенной фазой, такой как с использованием фосфата тетрабутиламмония.
Соединения, получаемые с помощью способа синтеза согласно изобретению, представляют собой β-(1→6)-связанные глюкозамин-дисахариды формулы 1:
где R'4, R'5 и R'8 имеют значения, определенные выше. Согласно одному аспекту изобретение относится к данным соединениям. Предпочтительные соединения изобретения представлены в п.47 формулы изобретения и на прилагаемых фигурах. Квалифицированный специалист очевидно понимает, что данные соединения могут существовать в ионизированных формах. Настоящее изобретение относится также к (фармацевтически приемлемым) солям таких ионизированных форм, таким как соли натрия, калия или аммония.
Многие из соединений согласно изобретению являются новыми, что касается их химической структуры. В дополнение к этому соединения согласно изобретению отличаются от соединений с известной химической структурой, но происходящих из природных источников, тем, что они свободны от каких-либо биологических примесей, таких как следы нуклеиновых кислот, и/или пептидов, и/или углеводов. Хотя они и присутствуют в незначительных количествах, присутствие следов данных биологических примесей считается неприемлемым для фармацевтических продуктов. Присутствие биологических примесей может быть определено известными методами, например, выбранными из иммунологических методов или PCR методов. Целью таких методов, в частности, может быть обнаружение клеточных компонентов грам-отрицательных бактерий, таких как E. coli.
В еще одном аспекте изобретение относится к некоторым новым промежуточным соединениям способа согласно изобретению. В частности, согласно данному аспекту изобретение относится к соединениям 3, 7, 8, 10а, 11, 11b, 12b, 12а, 13, 14. Предпочтительные воплощения данного аспекта изобретения относятся к соединениям 3b, 7b, 8b, 10b, 11а, 11с, 12с, 12d, 13b, 14b. Данные соединения могут использоваться в качестве промежуточных соединений, включая исходные вещества, в способе синтеза асимметрично или симметрично замещенных β-(1→6)-связанных глюкозамин-дисахаридов.
Соединения формулы 1 полезны в медицине для лечения теплокровных животных, таких как млекопитающие, включая людей. В частности, соединения изобретения могут использоваться в лечении иммунных расстройств, таких как иммунные нарушения, связанные со сверхпродуцированием воспалительных цитокинов или пониженным продуцированием воспалительных цитокинов. Воспалительные цитокины могут продуцироваться активируемыми Т лимфоцитами, моноцитами, или клетками с присутствующими антигенами и могут принадлежать к группе, состоящей из IL-1β, IL-4, IL-5, IL-6, IL-8, IL-9, IL-13, IFN-γ, TNF-α, или MCP-1. Болезненные состояния, которые можно лечить соединениями согласно изобретению, включают в их число рак, астму, атопический дерматит, аллергический ринит, воспалительную болезнь кишечника, диабет, ревматоидный артрит и другие, при которых благоприятной является регуляция воспалительных цитокинов в сторону увеличения и/или уменьшения. То, что соединения изобретения преимущественно действуют через TLR2 человека, может представлять клинический интерес в лечении рака (Garay et al., 2007). Раковые заболевания, которые можно лечить соединениями изобретения, включают колоректальный рак, рак груди и меланомы.
Соединения изобретения дополнительно могут понижать секрецию гистамина тучными клетками. Они полезны при лечении, включая улучшение или облегчение состояний, при которых вовлечена избыточная секреция гистамина тучными клетками. Такие состояния могут включать аллергические реакции, включая сенную лихорадку (поллиноз), аллергические реакции, вызываемые жалящими или кусающими насекомыми, такими как пчелы и осы, или аллергические реакции на пищевые аллергены.
Благодаря их стимулирующему действию на иммунную систему соединения изобретения далее полезны в качестве компонентов вакцин.
Соединения изобретения могут вводиться субъекту, нуждающемуся в этом, в виде составов, необязательно в сочетании с фармацевтически приемлемым носителем и/или другими эксципиентами оральным, парентеральным, внутривенным, внутриопухолевым, подкожным, ректальным, местным или мукозным (в слизистую оболочку) путем. Возможно введение перитонеальным, подкожным, оральным, интраназальным, подъязычным, внутримышечным или аэрозольным путем. Выбор подходящих интервалов дозировок для соединений изобретения будет зависеть от конкретной активности выбираемого соединения, состояния субъекта и подвергаемого лечению нарушения. Квалифицированный специалист сможет выбрать подходящие интервалы доз с учетом его общих знаний и его опыта в данной области. В случае таких состояний, как астма, атопический дерматит, аллергический ринит, воспалительная болезнь кишечника, диабеты или ревматоидный артрит, подходящими интервалами доз для людей могут быть от 0,01 до 50 мг/кв.м.
Дальнейшие аспекты изобретения относятся к способам, в которых используются и/или синтезируются новые и неочевидные (промежуточные) соединения изобретения. Вследствие полезности и/или получения новых и неочевидных соединений данные способы являются новыми и соответствуют условию изобретательского уровня. Данные процессы могут быть полезны в синтезе асимметрично или симметрично замещенных 1,6-β-дисахаридов, включая соединения изобретения.
Теперь изобретение будет дополнительно проиллюстрировано ссылкой на следующие ниже примеры и сопровождающие фигуры, из которых
Фиг.1 показывает структуру липида А E.coli и OM-174-DP®;
Фиг.2 дает общий обзор воплощения способа синтеза согласно изобретению;
Фиг.3 дает общий обзор предпочтительно воплощения способа синтеза согласно изобретению;
Фиг.4-24 дают общий обзор различных альтернативных путей синтеза для получения соединений формулы 1 и/или их непосредственных предшественников;
Фиг.25 представляет собой график, показывающий NO продуцирование мышиными макрофагами в ответ на соединения изобретения;
Фиг.26 представляет экспериментальные результаты, иллюстрирующие усиление биологической активности β-(1→6)-связанных глюкозамин-дисахаридов при лечении по способу согласно изобретению.
На данных фигурах группы R0, R1, R2, R4, R5, R6, R8, R4', R5', R8', X, Y и W имеют значения, определенные в пунктах формулы изобретения и описании для различных соединений. Bn обозначает бензильную группу, аллил обозначает аллильную группу и Ipr - изопропильную группу.
Молекулярные структуры, представленные на Фиг.1, соответствуют липиду А E. coli и OM-174-DP®, как указывается. На Фиг.1, кроме того, указано обозначение 0-3 и 0-3'.
Фиг.2 дает общий обзор воплощения способа синтеза согласно изобретению. Из приведенного выше описания очевидно ясно, что соединение 7 может вводиться в реакцию с соединением 10 с получением соединения 11h, где R0 представляет R5, или альтернативно с соединением 8 с получением соединения 11h, в котором R0 выбран из R2. В воплощении, показанном на Фиг.2, соединение 7 вводится в реакцию с соединением 10. Это открывает возможность введения различных R5 заместителей в молекулу, которая таким образом может асимметрично замещаться. Симметрично замещенные соединения могут быть получены по реакции соединения 7 с соединением 8 и последующей реакции полученного соединения 11h, в котором R0 выбран из R2 для получения соединения 12b. С соединением 12b последовательность реакций может осуществляться аналогичным образом, чтобы получить соединения, которые являются симметрично замещенными в N-2 и N-2' положении.
Фиг.3 дает общий обзор предпочитаемого воплощения способа синтеза согласно изобретению. В данном воплощении способа согласно изобретению конечным продуктом является асимметрично замещенный OM-174-DP®.
Фиг.4 показывает первую возможную реакцию фосфорилирования свободной гидроксильной группы полуацеталя формулы 14. В данной реакции соединение 14 вводится в реакцию с тетрабензилпирофосфатом в присутствии бис(триметилсилил)амида лития (LiHMDS). Реакция может иметь место в полярном растворителе, таком как ТГФ.
Фиг.5 показывает альтернативную реакцию фосфорилирования свободной гидроксильной группы полуацеталя формулы 14. В данной реакции соединение 14 вводится в реакцию с диалкил N,N-диизопропил-фосфорамидитом в присутствии конденсирующего агента, такого как [1H] тетразол. Реакция может протекать в полярном растворителе, предпочтительно апротонном полярном растворителе. В реакции сначала образуется фосфит. Данный фосфит впоследствии окисляется в защищенный фосфат в присутствии ароматической надкарбоновой кислоты, такой как м-хлорпероксибензойная кислота.
Фиг.6 показывает примерное образование сложного фосфодиэфира по реакции фосфоната с защищенным органическим аминоспиртом формулы HO-(C1-C24)-NHW. После образования фосфодиэфирной связи защитная группа W может быть удалена вместе или отдельно от защитных групп X. Когда группа W удаляется, в то время как группы X остаются в молекуле, свободная аминогруппа может далее модифицироваться, например, путем образования амида с органической кислотой.
Фиг.7 показывает дополнительную альтернативную реакцию модификации фосфатной группы. В данной реакции фосфатная группа метилируется с помощью CH2N2. Реакция, показанная на Фиг.7, проводится на молекуле, в которой ни одна из фосфатных групп не защищена. Очевидно понятно, что когда защищена одна из фосфатных групп, такая как 1-O-фосфатная группа, или 4'-O-фосфатная группа, такая защищенная фосфатная группа не будет метилироваться в реакции. Это открывает возможность выбора модификации любой или обеих фосфатных групп.
Фиг.8 показывает реакцию сульфатирования соединения 14. В реакции соединение 14 вводится в реакцию с комплексом триоксида серы.
Для того, чтобы получить соединения, имеющие углеводородную цепь, присоединенную непосредственно к 1-О-положению, имеется ряд возможностей. Некоторые из них показаны на Фиг.9. Во-первых, можно гидрировать (C3-C6)алкенил, присоединенный к 1-О-положению в соединении формулы 13 в соответствующий алкил. 1-аллильная группа гидрируется в пропильную группу. Во-вторых, можно присоединить углеводородные цепи сначала активированием гидроксильной функции в 1-О-положении соединения 14 и последующей реакцией активированной группы с органическим спиртом. Активирование свободной гидроксильной группы соединения 14 может достигаться по реакции соединения 14 с трихлорацетонитрилом в присутствии неорганического основания, такого как карбонат цезия или карбонат калия. Реакция может происходить в полярном растворителе, предпочтительно апротонном полярном растворителе, таком как дихлорметан. Когда соединение 14 вводится в реакцию с трихлорацетонитрилом в таких условиях, будет образовываться соединение формулы 24. Реакция соединения 24 с органическим спиртом, представленным общей формулой ROH на Фиг.9, будет давать в результате соединение, имеющее углеводородную цепь R, присоединенную в О-1-положении.
Фиг.10 показывает дополнительный пример реакции соединения 24 с органическим спиртом. На Фиг.10 соединение 24 вводится в реакцию с органическим диолом, имеющим 1-24 атома углерода, в котором одна из гидроксильных групп защищена группой Х, предпочтительно PMB. Монозащищенный органический диол представлен общей формулой НО-(C1-C24)-ОХ. На Фиг.10 дополнительно показано, что после присоединения монозащищенного органического диола к О-1-положению защитная группа Х монозащищенного органического диола может селективно удаляться, если она является отличной от группы Х в углеводе. После селективного удаления защитной группы Х монозащищенного органического диола свободная гидроксильная группа может далее модифицироваться, например, фосфорилированием ее по методам, описанным выше. Очевидно понятно, что фосфатная группа может далее модифицироваться, как описано выше.
Фиг.11 показывает реакционную схему, сходную со схемой Фиг.10. Однако на Фиг.11 после удаления защитной группы Х монозащищенного органического диола гидроксильная группа подвергается сульфатированию.
Альтернативно, как показано на Фиг.12, после удаления защитной группы Х монозащищенного органического диола гидроксильная группа может окисляться в карбоксигруппу. Понятно, что карбоксигруппа может далее модифицироваться, например, с помощью образования амида или сложного эфира.
Фиг.13 показывает реакционную последовательность, которая делает возможным введение углеводородной цепи, имеющей α,β-дигидроксизамещение. На данной схеме реакции органический спирт, имеющий ненасыщенную углерод-углеродную двойную связь, вводится в реакцию с соединением 24. Длина углеводородной цепи, соединяющей гидроксильную группу и ненасыщенную связь показанного органического спирта, является переменчивой и включает n углеродных атомов, где n может варьировать между 1 и 24. Хотя показанная ненасыщенная связь органического спирта присутствует на конце органического спирта, очевидно понятно, что она может также присутствовать в каком-либо местоположении в углеводородной цепи. После присоединения органического спирта к 1-О-положению соединения 24 ненасыщенная связь может подвергаться реакции с тетраоксидом осмия для присоединения α,β-дигидрокси к двойной связи. Гидроксильная группа, вводимая данным способом, может далее модифицироваться. Например, с помощью образования фосфата, как показано на Фиг.13, или альтернативно путем образования сульфата, эфиров с органическими кислотами или простых эфиров. На Фиг.13 фосфорилируется только одна гидроксильная группа. Это может достигаться по реакции с незначительным количеством агента фосфорилирования. Очевидно понятно, что в такой реакции будет также образовываться бисфосфат.
Фиг.14 показывает реакционную последовательность, сходную с последовательностью, показанной на Фиг.13. Однако после присоединения α,β-дигидрокси к двойной связи гидроксильные функции сульфатируются.
Фиг.15 показывает реакционную последовательность, сходную с реакционной последовательностью, показанной на Фиг.13. Однако после присоединения α,β-дигидрокси к двойной связи гидроксильные функции подвергаются реакции с окисляющим агентом, таким как NaIO4 с получением карбонильной функции.
На реакционной схеме, показанной на Фиг.16, соединение 24 вводится в реакцию с защищенным органическим аминоспиртом формулы HO-(C1-C24)-NHW. После присоединения защищенного органического аминоспирта защищенная аминофункция может далее подвергаться обработке, как обсуждалось в связи с фиг.6.
Фиг.17 показывает часть реакционной последовательности для получения соединения ОМ-174-МР (соединения 16) из соединения 14b. Подробности реакционной последовательности даются в примерах синтеза.
Фиг.18 показывает часть реакционной последовательности получения соединения ОМ-174-МР-PR (соединения 17) из соединения 14b. Подробности реакционной последовательности даются в примерах синтеза.
Фиг.19 показывает часть реакционной последовательности получения соединения ОМ-174-МР-PD (соединения 19) из соединения 13b через соединение 18. Подробности реакционной последовательности даются в примерах синтеза.
Фиг.20 показывает реакционную последовательность получения соединения ОМ-174-МР-АС (соединения 26) из соединения 14b. Подробности реакционной последовательности представлены в примерах синтеза.
Фиг.21 показывает реакционную последовательность получения соединения ОМ-174-МР-ТЕ (соединения 41с) из соединения 14b. Подробности реакционной последовательности представлены в примерах синтеза.
Фиг.22 показывает реакционную последовательность получения соединения ОМ-174-МР-ЕО (соединения 32) из соединения 18. Подробности реакционной последовательности представлены в примерах синтеза.
Фиг.23 показывает реакционную последовательность получения соединения ОМ-174-МР-ЕР (соединения 33) из соединения 32b. Подробности реакционной последовательности представлены в примерах синтеза.
Фиг.24 показывает реакционную последовательность получения соединения ОМ-174-МР-СМ (соединения 35с) из соединения 32с. Подробности реакционной последовательности представлены в примерах синтеза.
Обсуждаемые выше реакции могут использоваться также для присоединения различных заместителей к О-4'-положению β-(1→6)-связанных глюкозамин-дисахаридов изобретения. Это может достигаться с помощью использования обсуждаемых выше реакций введения заместителей в О-1 положение. Данные реакции могут выполняться сходным образом на свободной гидроксильной группе соединения формулы 4.
Из вышеизложенного ясно, что в О-1- и О-4'-положениях β-(1→6)-связанных глюкозамин-дисахаридов изобретения может присоединяться огромное разнообразие замещений.
Ниже следуют экспериментальные примеры синтеза соединений изобретения и примеры, относящиеся к биологической активности соединений изобретения.
ПРИМЕРЫ СИНТЕЗА
В следующем разделе будет обсуждаться синтез соединений данного изобретения. На Фиг.3 показаны различные синтезированные соединения с их соответствующими номерами обозначения соединений.
Аллил-3-О-бензил-4,6-О-бензилиден-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозид (3b)
К перемешиваемой суспензии аллил-4,6-О-бензилиден-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозида 2b [Liebigs Ann. (1996), 1599-1607](5 г, 10,35 ммол) и коммерчески доступного бензил 2,2,2-трихлорацетимидата (2,9 мл, 15,5 ммол) в простом эфире (200 мл) добавляли трифторметансульфонат олова (863 мг, 2,1 ммол). Смесь перемешивали в течение 17 часов при комнатной температуре, нейтрализовали насыщенным раствором NaHCO3 и концентрировали. Остаток переводили в EtOAc, промывали водой и органическую фазу отделяли и сушили над MgSO4. Растворитель выпаривали и остаток перекристаллизовывали из EtOH, получая соединение 3b (4,43 г, 75%) в виде белого кристаллического твердого вещества. Т.пл. 168,7°С; [α]D + 65 (c 0,24, CHCl3); vmax см-1 3301, 2915, 1709, 1546, 1075, 1013, 693; 1H ЯМР (500 МГц, CDCl3): δ 7,54-7,26 (м, 10H, Ph), 5,90 (м, 1H, CH=СН2), 5,62 (с, 1H, PhCH), 5,32-5,22 (м, 2H, СН=CH 2), 5,10 (д, 1H, J 2,NH 10,0 Гц, NH), 4,95 (AB, 1H, J=11,9 Гц, CH 2Ph), 4,92 (д, 1H, J1,2 3,7 Гц, H-1), 4,81 (д, 1H, J 12,0 Гц, CH 2CCl3), 4,71 (AB, д, 2H, CH 2Ph, CH 2CCl3), 4,30 (дд, 1H, J 6,6 10,1 Гц, J 6,5 4,6 Гц, H-6), 4,19 (м, 1H, OCH 2CH), 4,07-3,97 (м, 2H, H-2, OCH 2CH), 3,88 (м, 1H, H-5), 3,83-3,75 (м, 3H, H-6', H-4, H-3); 13C ЯМР (125,8 МГц, CDCl3): δ 154,3 (C=O), 138,2 (Cq), 137,2 (Cq), 133,2 (CH=CH2), 129,0, 128,7, 128,2, 126,0 (CH аром), 118,3 (CH=CH2), 101,2 (PhCH), 97,3 (C-1), 95,4 (CH2CCl3), 82,7 (C-4), 76,2 (C-3), 74,8-74,4 (CH2CCl3, CH2Ph), 68,9 (C-6), 68,6 (OCH2CH), 62,9 (C-5), 54,9 (C-2); MC-ES 596-594 [M+Na]+; Анализ Вычислено для C26H28Cl3NO7: С, 54,51; H, 4,93; N, 2,44%, Найдено: С, 54,51; H, 4,94; N, 2,34%.
Аллил-3,6-ди-О-бензил-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозид (4b)
К перемешиваемому раствору соединения 3b (1,3 г, 2,27 ммол) и боран триметиламинового комплекса (660 мг, 9,08 ммол) в сухом ТГФ (45 мл) при комнатной температуре добавляли хлорид алюминия (1,81 г, 13,6 ммол). После растворения реагентов по каплям добавляли воду (82 мкл, 4,54 ммол) и продолжали перемешивание при комнатной температуре в течение 30 минут. Реакцию прекращали путем добавления воды (20 мл) с последующим добавлением 1М раствора HCl (20 мл) и разбавлением EtOAc. Органическую фазу отделяли, промывали насыщенным раствором NaCl, сушили над MgSO4 и растворитель удаляли в вакууме. Остаток подвергали флэш-хроматографии на силикагеле (н-гептан/EtOAc, 3:1), получая соединение 4b (1,13 г, 87%) в виде белого твердого вещества. Т.пл. 65°С; [α]D + 64 (c 0,80, CHCl3); vmax см-1 3329, 2915, 1706, 1536, 1044, 730, 694; 1H ЯМР (500 МГц, CDCl3): δ 7,40-7,26 (м, 10H, Ph), 5,90 (м, 1H, CH=СН2), 5,32-5,21 (м, 2H, СН=CH 2), 5,14 (д, 1H, J 2,NH 10,0 Гц, NH), 4,92 (д, 1H, J 1,2 3,7 Гц, H-1), 4,82 (д, 1H, J 12,0 Гц, CH 2CCl3), 4,80 и 4,77 (AB, 2H, J 11,6 Гц, CH 2Ph), 4,67 (д, 1H, J 12,0 Гц, CH 2CCl3), 4,64 и 4,57 (AB, 2H, J 12,0 Гц, CH 2Ph), 4,19 (м, 1H, OCH 2CH), 4,03-3,97 (м, 2H, H-2, OCH 2CH), 3,82-3,68 (м, 4H, H-6, H-6', H-5, H-4), 3,63 (т, 1H, J 2,3 = J 3,4 10,2 Гц, H-3), 2,60 (с, 1H, OH); 13C ЯМР (125,8 МГц, CDCl3): δ 154,2 (C=O), 138,2 (Cq), 137,7 (Cq), 133,4 (CH=CH2), 128,8, 128,5, 128,4, 127,8, 127,7, 127,6 (CH аром), 118,0 (CH=CH2), 96,8 (C-l), 95,4 (CH2CCl3), 80,2 (C-3), 74,6, 74,5, 73,6 (CH2CCl3, 2×CH2Ph), 72,0, 70,2 (C-4, C-5), 69,7 (C-6), 68,3 (OCH2CH), 54,5 (C-2); MC-ES 598-596 [M+Na]+; Анализ Вычислено для C26H30Cl3NO7: С, 54,32; H, 5,26; N, 2,44%, найдено: С, 54,55; H, 5,39; N, 2,39%.
Аллил-3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозид (5b)
К перемешиваемому раствору соединения 4b (1,1 г, 1,91 ммол) и коммерчески доступного раствора 1Н-тетразола в CH3CN (~0,45 M) (8,5 мл, 3,8 ммол) в CH2Cl2 (33 мл) при комнатной температуре добавляли дибензил диметилфосфорамидит (762 мкл; 2,87 ммол). Перемешивание продолжали при комнатной температуре в течение 30 минут и затем раствор охлаждали до -20°С. Затем добавляли раствор mCPBA (57-86%, 1,22 г; 7,00 ммол) в CH2Cl2 (20 мл) и раствор перемешивали в течение 30 минут при -20°С. Добавляли 10% водный раствор тиосульфата натрия (50 мл) и смесь перемешивали в течение 10 минут, затем разбавляли EtOAc и органическую фазу отделяли. Органический слой последовательно промывали 10% водным раствором Na2S2O3 (3×), насыщенным водным раствором NaHCO3 (2×), N раствором HCl (1×) и солевым раствором. Органическую фазу сушили над MgSO4 и растворитель удаляли в вакууме. Остаток подвергали флэш-хроматографии на силикагеле (н-гептан/EtOAc, 4:1), получая соединение 5b (1,25 г, 78%) в виде бесцветного масла. [α]D + 56 (c 1,32, CHCl3); vmax см-1 3301, 2920, 1728, 1542, 1453, 1264, 995, 731, 694; 1H ЯМР (500 МГц, CDCl3): δ 7,40-7,10 (м, 20H, Ph), 5,90 (м, 1H, CH=СН2), 5,33-5,23 (м, 2H, СН=CH 2), 5,13 (д, 1H, J2,NH 10,0 Гц, NH), 4,93 (д, 1H, J 1,2 3,5 Гц, H-1), 4,96-4,82 (м, 4H, 2×CH 2Ph), 4,86 (м, 1H, CH 2CCl3), 4,76 и 4,65 (AB, 2H, J 12,0 Гц, CH 2Ph), 4,73 (м, 1H, CH 2CCl3), 4,60 (т, 1H, J3,4=J 4,5 9,5 Гц, H-4), 4,57 и 4,47 (AB, 2H, J 12,0 Гц, CH 2Ph), 4,21 (м, 1H, OCH 2CH), 4,10 (ддд, 1H, H-2), 4,02 (м, 1H, OCH 2CH), 3,92 (м, 1H, H-5), 3,84 (дд, 1H, J2,3 9,3 Гц, H-3), 3,80 (дд, 1H, J6,5 2,0 Гц, J6,6' 11,0 Гц, H-6), 3,76 (дд, 1H, J6,5 4,7 Гц, H-6'); 13C ЯМР (125,8 МГц, CDCl3): δ 154,0 (C=O), 138,1 (Cq), 137,8 (Cq), 135,8 (Cq), 135,7 (Cq), 133,2 (CH=CH2), 128,6, 128,5, 128,4, 128,3, 128,2, 128,1, 128,0, 127,9, 127,8, 127,7, 127,6, 127,5, 127,4 (CH аром), 118,1 (CH=CH2), 96,4 (C-1), 95,3 (CH2CCl3), 78,4 (C-3), 75,6 (C-4), 74,6, 73,7, 73,3 (CH2CCl3, 2×CH2Ph), 70,2 (C-5), 69,5, 69,4 (2×CH2Ph), 68,4 (C-6, OCH2CH), 54,3 (C-2); MC-ES 858-856 [M+Na]+; Анализ Вычислено для C40H43Cl3NO10P: C, 57,53; H, 5,19; N, 1,68%, Найдено: C, 57,41; H, 5,28; N, 1,74%.
3,6-Ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-D-глюкопираноза (6b)
К перемешиваемому раствору соединения 5b (659 мг; 0,79 ммол) в сухом ТГФ (10 мл) при комнатной температуре добавляли гексафторфосфат [бис(метилдифенилфосфин)]-(1,5-циклооктадиен)иридия(I) (67 мг). После активации иридиевого катализатора водородом в течение 1 мин (слегка красный раствор становится бесцветным) смесь перемешивали в атмосфере азота в течение 1 часа. К реакционной смеси добавляли йод (360 мг, 1,42 ммол) и воду (850 мкл), и перемешивали смесь в течение дополнительных 30 минут. К смеси добавляли 10% водный раствор Na2S2O3, и раствор экстрагировали EtOAc. Органический слой последовательно промывали 10% водным раствором Na2S2O3 (2×) и солевым раствором. Органическую фазу сушили над MgSO4, растворитель удаляли в вакууме и остаток кристаллизовали из смеси н-гептан/EtOAc, получая соединение 6b (419 мг, 67%) в виде бледно-желтого твердого вещества. vmax см-1 3361, 2920, 1716, 1522, 1452, 1216, 1006, 729, 693; 1H ЯМР (500 МГц, CDCl3) для α-аномера: δ 7,40-7,12 (м, 20H, Ph), 5,20 (д, 1H, J 1,2=3,4 Гц, H-1), 5,17 (д, 1H, J 2,NH=9,8 Гц, NH), 4,96-4,40 (м, 10H, 4×CH 2Ph, CH2CC13), 4,45 (т, 1H, J 3,4=J4,5 9,5 Гц, H-4), 4,15 (м, 1H, J 6,5 6,4 Гц, J 4,5 9,5 Гц, H-5), 4,00 (дт, 1H, J 2,NH=J2,3 9,8 Гц, H-2), 3,78 (дд, 1H, H-3), 3,78 (дд, 1H, J 6,5 1,7 Гц, J6,6 ,=11,0 Гц, H-6'), 3,76 (дд, 1H, J 6,5=6,4 Гц, H-6); 13C ЯМР (125,8 МГц, CDCl3) для α-аномера: δ 154,1 (C=O), 137,8 (Cq), 137,7 (Cq), 135,7 (Cq), 135,6 (Cq), 128,6, 128,5, 128,4, 128,3, 128,2, 128,1, 128,0, 127,9, 127,8, 127,7, 127,6, 127,5, 127,4 (CH аром), 95,3 (CH2CCl3), 91,6 (C-1), 77,9 (C-3), 76,1 (C-4), 74,6, 73,7, 73,3 (CH2CCl3, 2×CH2Ph), 70,6 (C-5), 69,5, 69,4 (2×CH2Ph), 68,8 (C-6), 54,7 (C-2); MC-ES 818-816 [M+Na]+; Анализ Вычислено для C37H39Cl3NO10P: C, 55,90; H, 4,94; N, 1,76%, Найдено: C, 55,67; H, 5,18; N, 1,62%.
3,6-Ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-D-глюкопиранозил трихлорацетимидат (7b)
К перемешиваемому раствору соединения 6b (419 мг; 0,53 ммол) в сухом CH2Cl2 (6,5 мл) при комнатной температуре добавляли трихлорацетонитрил (528 мкл; 5,3 ммол) и карбонат цезия (86 мг, 0,26 ммол). После перемешивания в течение 1 часа реакцию гасили насыщенным водным раствором NaHCO3 (5 мл) и раствор экстрагировали. Органический слой промывали солевым раствором, сушили над MgSO4 и растворитель удаляли в вакууме, получая соединение 7b (400 мг) в виде бледно-желтого масла, которое использовали на следующей стадии без дальнейшей очистки.
Аллил-3,4-ди-О-бензил-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозид (8b)
К перемешиваемому раствору соединения 3b (937 мг; 1,63 ммол) и борандиметиламина (482 мг, 8,18 ммол) в CH2Cl2 (18 мл) при 0°С медленно добавляли BF3:Et2O (1 мл, 8,18 ммол). После перемешивания в течение 45 минут реакцию останавливали путем медленного добавления насыщенного водного раствора NaHCO3. Органическую фазу отделяли, промывали насыщенным раствором NaCl и сушили над MgSO4. Растворитель выпаривали и остаток перекристаллизовывали из смеси EtOAc/н-гептан, получая соединение 8b (757 мг, 81%) в виде белого кристаллического твердого вещества. Т.пл. 119,9°С; [α]D + 74 (c 0,59, CHCl3); vmax см-1 3312, 2916, 1702, 1538, 1023, 732, 692; 1H ЯМР (500 МГц, CDCl3): δ 7,40-7,26 (м, 10H, Ph), 5,88 (м, 1H, CH=СН2), 5,30-5,20 (м, 2H, СН=CH 2), 5,07 (д, 1H, J 2,NH 10,0 Гц, NH), 4,89 (д, 1H, J 1,2 3,5 Гц, H-l), 4,87 (д, 1H, J 11,0 Гц, CH 2CCl3), 4,87 и 4,75 (AB, 2H, J 11,0 Гц, CH 2Ph), 4,78 и 4,68 (AB, 2H, J=12,0 Гц, CH 2Ph), 4,68 (д, 1H, J 11,0 Гц, CH 2CCl3), 4,16 (м, 1H, OCH 2CH), 4,02-3,94 (м, 2H, H-2, OCH 2CH), 3,86-3,64 (м, 5H, H-6, H-6', H-5, H-4, H-3), 1,78 (м, 1H, OH); 13C ЯМР (125,8 МГц, CDCl3): δ 154,2 (C=O), 138,0 (Cq), 137,8 (Cq), 133,3 (CH=CH2), 128,5, 128,4, 128,1, 128,0, 127,8, 127,7 (CH аром), 118,0 (CH=CH2), 96,8 (C-1), 95,4 (CH2CCl3), 80,2 (C-3), 78,0 (C-4), 75,2, 75,1, 74,6 (CH2CCl3, 2×CH2Ph), 71,5 (C-5), 68,3 (OCH2CH), 61,6 (C-6), 55,2 (C-2); MC-ES 598-596 [M+Na]+; Анализ Вычислено для C26H30Cl3NO7: C, 54,32; H, 5,26; N, 2,44%, Найдено: C, 54,76; H, 5,53; N, 2,31%.
Аллил-2-амино-3,4-ди-О-бензил-2-дезокси-α-D-глюкопиранозид (9b)
К перемешиваемому раствору соединения 8b (245 мг; 0,43 ммол) в АсОН (6 мл) при комнатной температуре добавляли цинковый порошок (430 мг). После перемешивания в течение ночи суспензию фильтровали через целит, растворитель удаляли в вакууме и остаточный растворитель выпаривали одновременно с толуолом три раза. Остаток переводили в EtOAc, промывали насыщенным водным раствором NaHCO3 и солевым раствором. Органическую фазу отделяли, сушили над MgSO4 и растворитель удаляли в вакууме, получая соединение 9b (157 мг) в виде бесцветного масла, которое использовали на следующей стадии без дальнейшей очистки. Образец соединения очищали с помощью флэш-хроматографии на силикагеле (CH2Cl2/Ацетон, 10:1→1:1), получая соединение 9b в виде белого кристаллического твердого вещества. Т.пл. 85,2°С; [α]D + 98 (c 0,89, CHCl3); vmax см-1 3190, 2899, 1664, 1577, 1496, 1452, 1363, 1024, 737, 695; 1H ЯМР (500 МГц, CDCl3): δ 7,50-7,26 (м, 10H, Ph), 5,92 (м, 1H, CH=СН2), 5,30-5,20 (м, 2H, СН=CH 2), 4,90 (д, 1H, J 1,2 3,5 Гц, H-1), 5,01 и 4,73 (AB, 2H, J 11,0 Гц, CH 2Ph), 4,88 и 4,75 (AB, 2H, J 12,0 Гц, CH 2Ph), 4,16 (дд, 1H, J 5,0 Гц, J 12,9 Гц, OCH 2CH), 4,02 (дд, 1H, J 6,0 Гц, J 12,9 Гц, OCH 2CH), 3,85-3,55 (м, 5H, H-6, H-6', H-5, H-4, H-3), 2,80 (дд, 1H, J2,3 9,4 Гц, H-2); 13C ЯМР (125,8 МГц, CDCl3): δ 138,6 (Cq), 138,1 (Cq), 133,9 (CH=CH2), 128,6, 128,5, 127,9, 127,8, 127,7 (CH аром), 117,3 (CH=CH2), 98,8 (C-l), 83,8, 78,7, 71,9 (C-3, C-4, C-5), 75,6, 74,8 (2×CH2Ph), 68,3 (OCH2CH), 61,4 (C-6), 56,0 (C-2); MC-ES 400 [M+H]+; Анализ Вычислено для C23H29NO5: С, 69,15; H, 7,32; N, 3,51%, Найдено: С, 69,21; H, 7,36; N, 3,25%.
Аллил-2[(R)-3-бензилокситетрадеканоиламино]-3,4-ди-О-бензил-2-дезокси-α-D-глюкопиранозид (10b)
К холодному раствору (-15°С) (R)-3-бензилокситетрадекановой кислоты (145 мг; 0,43 ммол) [Bull. Chem. Soc. Jpn, 60 (1987), 2197-2204] в ТГФ (5 мл) добавляли N-метилморфолин (47 мкл; 0,43 ммол) и изобутилхлорформиат (57 мкл; 0,43 ммол). Реакционную смесь перемешивали при -15°С в течение 30 минут. К реакционной смеси добавляли раствор соединения 9b (157 мг; 0,39 ммол) в ТГФ (5 мл). Перемешивание продолжали в течение ночи при комнатной температуре. Затем добавляли воду и EtOAc, органическую фазу отделяли и водную фазу еще раз экстрагировали EtOAc. Органические слои объединяли, промывали водой и солевым раствором и сушили над MgSO4. Растворитель выпаривали и остаток перекристаллизовывали из MeOH, получая соединение 10b (176 мг, 63% в течение 2 стадий) в виде белого кристаллического твердого вещества Т.пл. 141,3°С; [α]D + 61 (c 0,31, CHCl3); vmax см-1 3297, 2920, 2851, 1637, 1544, 1025, 732, 693; 1H ЯМР (500 МГц, CDCl3): δ 7,40-7,26 (м, 15H, Ph), 6,32 (д, 1H, J2,NH 9,0 ГЦ, NH), 5,76 (м, 1H, CH=СН2), 5,23-5,10 (м, 2H, СН=CH 2), 4,82 и 4,65 (AB, 2H, J 11,0 Гц, CH 2Ph), 4,80 (д, 1H, J 1,2 4,0 Гц, H-1), 4,73 и 4,57 (AB, 2H, J 11,5 Гц, CH 2Ph), 4,54 и 4,49 (AB, 2H, J 11,0 Гц, CH 2Ph), 4,27 (м, 1H, J1,2 4,0 Гц, H-2), 4,00 (м, 1H, OCH 2CH), 3,85-3,62 (м, 7H, OCH 2CH, H-6, H-6', H-5, H-4, H-3, H-3"), 2,40 (дд, 1H, J 3,7, 15,1 Гц, H-2"B), 2,28 (дд, 1H, J=7,6, 15,1 Гц, H-2"A), 1,58 (м, 1H, H-4"A), 1,49 (м, 1H, H-4"B), 1,35-1,12 (м, 18H, 9 CH2), 0,90 (т, 3H, CH3); 13C ЯМР (125,8 МГц, CDCl3): δ 171,2 (C=O), 138,3 (Cq), 138,2 (Cq), 137,9 (Cq), 133,5 (CH=CH2), 128,5, 128,4, 128,3, 128,0, 127,9, 127,8, 127,7, 127,6, 127,5 (CH аром), 117,7 (CH=CH2), 96,9 (C-1), 80,2 (C-3), 78,0 (C-4), 76,6 (C-3"), 74,7 (CH2Ph), 74,6 (CH2Ph), 71,4 (C-5), 71,3 (CH2Ph), 68,3 (OCH2CH), 61,8 (C-6), 52,6 (C-2), 41,5 (C-2"), 33,8 (C-4"), 31,9, 29,6, 29,5, 29,4, 29,3, 25,1, 22,7 (CH2), 14,1 (CH3); MS-ES 739 [M+Na]+; Анализ Вычислено для C44H61NO7: C, 73,81; H, 8,59; N, 1,96%, Найдено: C, 73,62; H, 8,57; N, 1,82%.
Аллил-3,4-ди-О-бензил-6-О-[3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозил]-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-α-D-глюкопиранозид (11a)
К перемешиваемому раствору соединения 10b (231 мг, 0,32 ммол) и имидата соединения 7b (400 мг, 0,42 ммол) в безводном CH2Cl2 (9 мл) при -20°С добавляли 4 Å молекулярные сита. После перемешивания в течение 30 минут добавляли TMSOTf (12 мкл, 64 мкмол) и перемешивание продолжали дополнительно в течение 1 часа. Реакционную смесь фильтровали через целит, разбавляли EtOAc и нейтрализовали насыщенным водным раствором NaHCO3. Органическую фазу отделяли, промывали насыщенным раствором NaCl, сушили над MgSO4. Растворитель выпаривали и остаток перекристаллизовывали из MeOH, получая соединение 11а (335 мг, 70%) в виде белого кристаллического твердого вещества Т.пл. 145,2°С; [α]D + 37 (c 0,12, CHCl3); vmax см-1 3297, 2921, 1713, 1641, 1540, 1453, 1266, 997, 730, 693; 1H ЯМР (500 МГц, CDCl3): δ 7,37-7,14 (м, 35H, Ph), 6,32 (д, 1H, J2,NH 9,0 Гц, NH-2), 5,76 (м, 1H, CH=СН2), 5,23-5,10 (м, 3H, NH-2', СН=CH 2), 4,95-4,42 (м, 15H, CH 2CCl3, 7×CH 2Ph), 4,79 (д, 1H, J 1,2 3,5 Гц, H-1), 4,74 (д, 1H, J 1',2' 10,0 Гц, H-1'), 4,46 (т, 1H, J 3'4'=J4',5', 8,5 Гц, H-4'), 4,32 (м, 1H, J 1,2 4,0 Гц, H-2), 4,25 (AB, 1H, CH 2Ph), 4,13 (м, 1H, H-6A), 4,08 (м, 1H, H-3'), 4,04 (м, 1H, OCH 2CH), 3,90-3,60 (м, 9H, OCH 2CH, H-5', H-6'A, H-6'B, H-6B, H-5, H-4, H-3, H-3"), 3,42 (м, 1H, H-2'), 2,40 (дд, 1H, J 3,7, 15,1 Гц, H-2"B), 2,28 (дд, 1H, J=7,6, 15,1 Гц, H-2"A), 1,58 (м, 1H, H-4"A), 1,49 (м, 1H, H-4"B), 1,35-1,12 (м, 18H, 9 CH2), 0,90 (т, 3Н, СН3); 13C ЯМР (125,8 МГц, CDCl3): δ 171,0 (C=O), 153,7 (C=O), 138,3 (Cq), 138,2 (Cq), 138,1 (Cq), 137,9 (Cq), 137,7 (Cq), 135,7 (Cq), 135,6 (Cq), 133,6 (CH=CH2), 128,5, 128,4, 128,3, 128,2, 128,1, 128,0, 127,9, 127,8, 127,7, 127,6, 127,5, 127,4 (CH аром), 117,7 (CH=CH2), 99,8 (С-1'), 96,9 (C-1), 95,1 (CH2CCl3), 80,6 (C-3), 78,6 (C-3'), 78,0 (C-4), 76,6 (C-3"), 76,2 (C-4'), 74,4 (C-5'), 74,7, 74,2, 73,8, 73,3, 71,3 (CH2CCl3, 5×CH2Ph), 70,2 (C-5), 69,6, 69,4 (2×P-OCH2Ph), 69,0 (C-6 или C-6'), 68,1 (OCH2CH), 67,7 (C-6 или C-6'), 57,3 (C-2'), 52,6 (C-2), 41,6 (C-2"), 33,8 (C-4"), 31,9, 29,6, 29,5, 29,4, 29,3, 25,1, 22,7 (CH2), 14,1 (CH3); MC-ES 1515-1513 [M+Na]+; Анализ Вычислено для C81H98Cl3N2O16P: C, 65,16; H, 6,62; N, 1,88%, Найдено: C, 65,31; H, 6,70; N, 1,77%.
Аллил-6-О-[2-амино-3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-β-D-глюкопиранозил]-2-[(R)-3-бензилокситетрадеканоиламино]-3,4-ди-О-бензил-2-дезокси-α-D-глюкопиранозид (12d)
К перемешиваемому раствору соединения 11а (310 мг, 0,21 ммол) в АсОН/Н2О 9:1 (5 мл) при комнатной температуре добавляли цинк-медную пару (260 мг). После перемешивания в течение 1 часа добавляли цинк-медную пару (260 мг) и операцию повторяли еще раз. Перемешивание продолжалось в течение еще 3 часов и суспензию фильтровали через целит. Растворитель удаляли в вакууме и остаточный растворитель выпаривали одновременно с толуолом три раза. Остаток переводили в EtOAc, промывали насыщенным водным раствором NaHCO3 (2×) и солевым раствором. Органическую фазу отделяли, сушили над MgSO4 и растворитель удаляли в вакууме, получая соединение 12d (273 мг) в виде бесцветного масла, которое использовали на следующей стадии без дальнейшей очистки. MC-ES 1317 [M+H]+, 1339 [M+Na]+.
Аллил-3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-α-D-глюкопиранозид (13b)
К перемешиваемому раствору (R)-3-додеканоилокситетрадекановой кислоты [Bull. Chem. Soc. Jpn, 60 (1987), 2205-2214] (115 мг; 0,27 ммол) в ТГФ (4 мл) при -15°С добавляли последовательно N-метилморфолин (30 мкл; 0,27 ммол) и изобутилхлорформиат (35 мкл; 0,27 ммол). Перемешивание продолжалось в течение 30 минут при -15°С. Затем к реакционной смеси добавляли раствор неочищенного соединения 12b (273 мг; 0,21 ммол) в ТГФ (4 мл). После перемешивания в течение ночи при комнатной температуре растворитель удаляли в вакууме и к остатку добавляли воду. Затем смесь экстрагировали EtOAc, органические фазы последовательно промывали насыщенным водным раствором NaHCO3, солевым раствором и сушили над MgSO4. Растворитель выпаривали и остаток кристаллизовали из MeOH, получая соединение 13b (259 мг, 72% в течение 2 стадий) в виде белого твердого вещества Т.пл. 173°С; [α]D + 30 (c 0,90, CHCl3); vmax см-1 3302, 2919, 2850, 1726, 1636, 1544, 1453, 1357, 1266, 998, 730, 694; 1H ЯМР (500 МГц, CDCl3): δ 7,40-7,10 (м, 35H, Ph), 6,31 (д, 1H, J 2,NH 9,4 Гц, NH-2), 6,01 (д, 1H, J 2',NH' 7,5 ГЦ, NH-2'), 5,76 (м, 1H, CH=СН2), 5,23-5,08 (м, 2H, СН=CH 2), 5,08 (д, 1H, J 1',2' 8,2 Гц, Н-1'), 5,05 (м, 1H, H-3'''), 4,95-4,42 (м, 14H, 7×CH 2Ph), 4,79 (д, 1H, J 1,2 3,4 Гц, H-1), 4,46 (т, 1H, J 3',4'=J 4',5', 8,7 Гц, H-4'), 4,32 (м, 2H, H-2, H-3'), 4,09 (м, 1H, H-6A), 4,03 (м, 1H, OCH 2CH), 3,90-3,60 (м, 9H, OCH 2CH, H-5', H-6'A, H-6'B, H-6B, H-5, H-4, H-3, H-3"), 3,46 (м, 1H, H-2'), 2,40 (дд, 1H, J 3,3, 15,1 Гц, H-2"B), 2,34 (дд, 1H, J 7,2, 15,5 Гц, H-2'''B), 2,28 (дд, 1H, J=7,6, 15,1 Гц, H-2"A), 2,21 (дд, 1H, J 5,0, 15,5 Гц, H-2'''A), 2,11 (т, 2H, J 7,5, H-2””), 1,58 (м, 1H, H-4"A), 1,55-1,36 (м, 3H, H-4"B, 2×H-4'''), 1,35-1,12 (м, 54H, 27 CH2), 0,90 (м, 9H, 3×CH3); 13C ЯМР (125,8 МГц, CDCl3): δ 173,3 (C=O), 171,0 (C=O), 170,0 (C=O), 138,4 (Cq), 138,3 (Cq), 138,2 (Cq), 138,1 (Cq), 137,8 (Cq), 135,7 (Cq), 135,6 (Cq), 133,6 (CH=CH2), 128,5, 128,4, 128,3, 128,2, 128,1, 128,0, 127,9, 127,8, 127,7, 127,6, 127,5, 127,4 (CH аром), 117,6 (CH=CH2), 99,2 (С-1'), 96,8 (C-1), 80,5 (C-3), 78,3 (C-3', C-4), 76,6 (C-3"), 76,0 (C-4'), 74,8, 74,7 (2×CH2Ph), 74,3 (C-5'), 73,2, 73,1 (2×CH2Ph), 71,3 (CH2Ph), 70,5 (C-3'''), 70,3 (C-5), 69,4, 69,3 (2×P-OCH2Ph), 69,1 (C-6 или C-6'), 68,1 (OCH2CH), 67,6 (C-6 или C-6'), 56,7 (C-2'), 52,4 (C-2), 41,6-41,4 (C-2”, C-2'''), 34,4, 34,1, 33,8 (C-4”, C-4''', C-2""), 31,9, 29,9, 29,6, 29,5, 29,4, 29,3, 29,2, 29,0, 25,1, 25,0, 24,9, 22,7 (CH2), 14,1 (CH3); MC-ES 1747 [M+Na]+; Анализ Вычислено для C104H145N2O17P: C, 72,36; H, 8,47; N, 1,62%, Найдено: C, 72,52; H, 8,43; N, 1,51%.
3,4-Ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-D-глюкопираноза (14b)
К перемешиваемому раствору соединения 13b (259 мг; 0,15 ммол) в сухом ТГФ (13 мл) при комнатной температуре добавляли гексафторфосфат [бис(метилдифенилфосфин)]-(1,5-циклооктадиен)иридия(I) (13 мг). После активации иридиевого катализатора водородом в течение 1 мин (слегка красный раствор становится бесцветным) смесь перемешивали в атмосфере азота в течение 1 часа. К реакционной смеси добавляли йод (69 мг, 0,27 ммол) и воду (13 мл) и перемешивали смесь в течение дополнительных 15 минут. К смеси добавляли 10% водный раствор Na2S2O3 и раствор экстрагировали EtOAc. Органический слой последовательно промывали 10% водным раствором Na2S2O3 (2×) и солевым раствором. Органическую фазу сушили над MgSO4, растворитель удаляли в вакууме и остаток кристаллизовали из CH3CN, получая соединение 14b (165 мг, 65%) в виде серого твердого вещества. vmax см-1 3388, 3276, 3062, 2919, 2850, 1726, 1641, 1544, 1453, 1358, 1263, 997, 731, 694; 1H ЯМР (500 МГц, CDC13) для α-аномера: δ 7,40-7,10 (м, 35H, Ph), 6,39 (д, 1H, J 2,NH 9,4 Гц, NH-2), 6,18 (м, 1H, NH-2'), 5,38 (д, 1H, J 1',2' 7,7 Гц, H-1'), 5,12 (м, 1H, J 1,2 3,2 Гц, H-1), 5,05 (м, 1H, H-3'''), 4,95-4,42 (м, 14H, 7×CH 2Ph), 4,50 (м, 1H, H-4'), 4,23 (дт, 1H, J 1,2 3,2 Гц, J 2,NH=J2,3 9,4 Гц, H-2), 4,19 (т, 1H, J 2',3'=J3',4' 8,9 Гц, H-3'), 4,07 (м, 1H, H-5), 3,96 (д, 1H, J 6A,6B 11,4 Гц, H-6A), 3,89-3,80 (м, 2H, H-3", H-6'A), 3,80-3,65 (м, 4H, H-5', H-6'B, H-6B, H-3), 3,35 (м, 1H, H-2'), 3,32 (т, 1H, J 3,4=J 4,5 9,5 Гц, H-4), 2,40-2,20 (м, 4H, 2×H-2", 2×H-2'''), 2,16 (т, 2H, J 7,5, H-2""), 1,58 (м, 1H, H-4"A), 1,55-1,36 (м, 3H, H-4"B, 2×H-4"'), 1,35-1,12 (м, 54H, 27 CH2), 0,90 (м, 9H, 3×CH3); 13C ЯМР (125,8 МГц, CDCl3) для α-аномера: δ 173,9 (C=O), 171,4 (C=O), 170,6 (C=O), 138,4 (Cq), 138,3 (Cq), 138,2 (Cq), 138,1 (Cq), 137,8 (Cq), 135,7 (Cq), 135,6 (Cq), 128,5, 128,4, 128,3, 128,2, 128,1, 128,0, 127,9, 127,8, 127,7, 127,6, 127,5, 127,4 (CH аром), 98,8 (С-1'), 91,7 (C-1), 80,6 (C-3), 78,8 (C-3', C-4), 76,8 (C-3"), 76,2 (C-4'), 74,8 (CH2Ph), 74,2 (C-5'), 73,6, 73,4, 73,3 (3×CH2Ph), 71,8 (C-5), 71,3 (CH2Ph), 70,8 (C-3'''), 69,4, 69,3 (2×P-OCH2Ph), 69,0 (C-6'), 67,6 (C-6), 57,0 (C-2'), 52,9 (C-2), 41,7 (C-2", C-2'''), 34,4, 34,1, 33,8 (C-4", C-4''', C-2""), 31,9, 29,9, 29,6, 29,5, 29,4, 29,3, 29,2, 29,0, 25,1, 25,0, 24,9, 22,7 (CH2), 14,1 (CH3); MC-ES 1708 [M+Na]+; Анализ Вычислено для C101H141N2O17P: C, 71,94; H, 8,43; N, 1,66%, Найдено: C, 71,68; H, 8,35; N, 1,61%.
3,4-Ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-α-D-глюкопиранозилдибензилоксифосфат (15b)
К перемешиваемому раствору соединения 14b (1 г; 0,60 ммол) в ТГФ (90 мл) при -78°С добавляли раствор литий бис(триметилсилил)амида (1М в ТГФ)(1,9 мл, 1,88 ммол). Смесь перемешивали 5 минут, затем добавляли тетрабензилпирофосфат (1,3 г, 2,37 ммол). Перемешивание продолжали при -78°С в течение 2 часов, затем раствор нейтрализовали насыщенным водным раствором NaHCO3 и разбавляли EtOAc. Органическую фазу отделяли, сушили над MgSO4 и растворитель удаляли в вакууме, получая соединение 15b (2 г) в виде бледно-желтого масла, которое использовали на следующей стадии без дальнейшей очистки.
2-дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-α-D-глюкопиранозил дигидрофосфат (1b) (ОМ-174-DP)
Неочищенное соединение 15b (2 г) в ТГФ (100 мл) гидрировали в течение 17 часов в присутствии 5% Pd-C (1,5 г) при комнатной температуре в атмосфере водорода (6 бар). Затем смесь нейтрализовали Et3N (500 мкл) и катализатор удаляли фильтрованием. Фильтрат концентрировали в вакууме и остаток очищали с помощью HPLC согласно изобретению (Метод D), получая соединение 1b (в виде натриевой соли)(342 мг; 48% в течение 2 стадий) в виде белого лиофилизата. [α]D + 14 (c 0,6, H2O); vmax см-1 3305, 2918, 2849, 1713, 1648, 1553, 1465, 1376, 1174, 1039, 915, 719, 654; 1H ЯМР (500 МГц, CDCl3/CD3OD/Пиридин-d5/DCl 37% 5/2/1/1): δ 5,60 (дд, 1H, J 1,2 7,5 Гц, J 1,P 2,0 Гц, H-1), 5,27 (м, 1H, H-3'''), 4,73 (д, 1H, J 1',2' 8,5 Гц, Н-1'), 4,09 (кв, 1H, J 3,4=J 4,5=J 4,P 9,0 Гц, H-4'), 4,05-3,80 (м, 6H, 2×H-6, 2×H-6', H-4, H-3), 4,00 (м, 1H, H-3"), 3,85 (м, 2H, H-2, H-3'), 3,85 (м, 1H, H-2'), 3,50 (м, 2H, H-5, H-5'), 2,67 (м, 2H, J 6,4 Гц, 2×H-2'''), 2,43 (м, 2H, J 6,1 Гц, 2×H-2"), 2,29 (м, 2H, J 7,3, J 15 Гц, 2×H-2""), 1,60-1,36 (м, 6H, 2×H-4", 2×H-4''', 2×H-3""), 1,35-1,12 (м, 52H, 26 CH2), 0,90 (м, 9H, 3×CH3); 13C ЯМР (125,8 МГц, CDCl3/CD3OD/Пиридин-d5/DCl 37% 5/2/1/1): δ 174,9 (C=O), 174,1 (C=O), 172,9 (C=O), 102,4 (С-1'), 94,7 (C-1), 75,3 (C-5'), 73,8 (C-4'), 73,4, 70,4, 70,1, 69,6 (C-3', C-5, C-4, C-3), 71,9 (C-3'''), 69,6 (C-3"), 69,4 (C-6), 60,8 (C-6'), 56,1 (C-2'), 54,8 (C-2), 44,2 (C-2"), 41,7 (C-2'''), 37,7, 35,1, 34,6 (C-4", C-4''', C-2""), 32,3 (C-12", C-12''', C-10""), 30,1-29,6 (C-6"->С-11", C-6'''->С-11''', C-4""->C-9""), 25,7, 25,6, 25,2 (C-5", C-5''', C-3""), 23,1 (C-13", C-13''', С-11””), 14,5 (C-14", C-14''', C-12""); MC-ES 1155 [M+Na-2H]-, 1133 [M-H]-; Анализ Вычислено для C52H97N2O20P2Na3 + H2O: C, 51,23; H, 8,18; N, 2,30%, Найдено: C, 51,11; H, 8,46; N, 2,20%.
2-Дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-α-D-глюкопираноза (16) (ОМ-174-MP)
Соединение 14b (80 мг, 47 мкмол) в ТГФ (50 мл) гидрировали в течение 16 часов в присутствии 5% Pd-C (25 мг) при комнатной температуре в атмосфере водорода (6 бар). Катализатор удаляли фильтрованием и фильтрат концентрировали в вакууме. Остаток очищали с помощью HPLC согласно изобретению (Метод В), получая соединение 16 (в виде натриевой соли)(25 мг; 50%) в виде белого лиофилизата. MS-ES 1053[M-H]-.
Пропил-2-дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-α-D-глюкопиранозид (17) (ОМ-174-MP-PR)
Соединение 13b (100 мг, 58 мкмол) в ТГФ (100 мл) гидрировали в течение 17 часов в присутствии 5% Pd-C (50 мг) при комнатной температуре в атмосфере водорода (6 бар). Затем смесь нейтрализовали Et3N (500 мкл) и катализатор удаляли фильтрованием. Фильтрат концентрировали в вакууме и остаток очищали с помощью HPLC согласно изобретению (Метод D), получая соединение 17 (в виде натриевой соли)(28 мг; 44%) в виде белого лиофилизата. 1H ЯМР (500 МГц, CDCl3/CD3OD/Пиридин-d5/DCl 37% 5/2/1/1): δ 5,25 (м, 1H, H-3'''), 4,72 (д, 1H, J 1,2 3,6 Гц, H-1), 4,70 (д, 1H, J 1',2' 9,9 Гц, Н-1'), 4,16 (кв, 1H, J 3,4=J 4,5=J 4,P 9,2 Гц, H-4'), 4,01 (дд, 1H, J 6A,6B 9,3 Гц, H-6A), 3,95 (м, 1H, H-3"), 3,91 (т, 1H, J 3',4'=J 3',2' 10,0 Гц, H-3'), 3,89 (ддд, 1H, J 1,2 3,6 Гц, J 3,2 10,5 Гц, J NH,2 8,0 Гц, H-2), 3,86-3,81 (м, 4H, 2×H-6', H-6B, H-2'), 3,78 (дд, 1H, J 3,4 9,0 Гц, J 3,2 10,5 Гц, H-3), 3,64 (м, 1H, H-5), 3,56 (т, 1H, J 3,4=J 4,5 8,8 Гц, H-4), 3,54 (м, 1H, OCH 2CH), 3,50 (м, 1H, H-5'), 3,27 (м, 1H, OCH 2CH), 2,63 (м, 2H, J 6,2 Гц, 2×H-2'''), 2,48 (дд, 1H, J 2”,3” 3,4 Гц, J 2”,2” 14,8 Гц, H-2"), 2,39 (дд, 1H, J 2”,3” 8,5 Гц, J 2”,2” 14,8 Гц, H-2"), 2,28 (м, 2H, J 7,3, J 15 Гц, 2×H-2""), 1,70-1,36 (м, 6H, 2×H-4", 2×H-4''', 2×H-3""), 1,55 (м, 2H, OCH2CH2), 1,35-1,12 (м, 52H, 26 CH2), 0,88 (м, 12H, 4×CH3); 13C ЯМР (125,8 МГц, CDCl3/CD3OD/Пиридин-d5/DCl 37% 5/2/1/1): δ 174,7 (C=O), 174,0 (C=O), 172,5 (C=O), 101,9 (С-1'), 97,5 (C-1), 75,3 (C-5'), 75,0 (C-4'), 73,7 (C-3'), 71,7 (C-3'''), 71,6, 71,4, 70,6 (C-5, C-4, C-3), 70,1 (OCH2CH), 69,1 (C-3"), 69,0 (C-6), 60,8 (C-6'), 55,9 (C-2'), 54,4 (C-2), 43,4 (C-2"), 41,5 (C-2'''), 37,4, 35,0, 34,5 (C-4", C-4''', C-2""), 32,3 (C-12", C-12''', C-10""), 30,0-29,6 (C-6"->С-11", C-6'''->С-11''', C-4""->C-9""), 26,1, 25,6, 25,5 (C-5", C-5''', C-3""), 23,0, 22,9 (OCH2CH2CH3, C-13", C-13''', С-11””), 14,5 (C-14", C-14''', C-12""), 10,9 (OCH2CH2CH3); MC-ESI 1141 [M-H+2Na]+, HRMS-ESI вычислено для C55H104N2O17Na2P [M-H+2Na]+ 1141,6868, найдено 1141,6879.
2,3-Дигидроксипропил-3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил}-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-α-D-глюкопиранозид (18)
К перемешиваемому раствору соединения 13b (500 мг; 0,29 ммол) в смеси ТГФ/трет-BuOH/H2O 10:10:1 (15 мл) при комнатной температуре последовательно добавляли 4-метилморфолин N-оксид (NMO)(156 мг; 1,16 ммол) и OsO4 в 2-пропаноле (2,5%; 580 мкл; 58 мкмол). Через 4 часа добавляли насыщенный водный Na2S2O3 и смесь экстрагировали EtOAc. Органическую фазу промывали насыщенным водным раствором Na2S2O3 (2×), солевым раствором и сушили над MgSO4. Растворитель выпаривали и остаток кристаллизовали из EtOH, получая соединение 18 (300 мг, 59%) в виде белого твердого вещества. vmax см-1 3300, 2919, 2850, 1646, 1543, 1453, 1358, 1266, 998, 730, 694; 1H ЯМР (500 МГц, CDCl3): δ 7,40-7,10 (м, 35H, Ph), 6,54 (м, 1H, NH-2'), 6,38 (м, 1H, NH-2), 5,08 (м, 0,5H, H-3'''), 5,03 (м, 0,5H, H-3'''), 4,95-4,42 (м, 14H, 7×CH 2Ph), 4,88 (м, 1H, Н-1'), 4,72 (м, 1H, H-1), 4,52 (м, 1H, H-4'), 4,27 (м, 2H, H-2, H-3'), 4,18-3,30 (м, 15H, OCH 2CH, CH(ОН), CH 2OH, H-2', H-5', 2×H-6', 2×H-6, H-5, H-4, H-3, H-3"), 2,50-2,45 (м, 1H, H-2'''), 2,45-2,35 (м, 1H, H-2"), 2,30-2,20 (м, 2H, H-2", H-2'''), 2,15 (м, 1H, H-2""), 1,65-1,45 (м, 6H, 2×H-4", 2×H-4''', 2×H-3""), 1,35-1,12 (м, 52H, 26 CH2), 0,90 (м, 9H, 3×CH3); 13C ЯМР (125,8 МГц, CDCl3): δ 173,8 (C=O), 173,7 (C=O), 171,1 (C=O), 170,6 (C=O), 170,4 (C=O), 138,2-138,02 (Cq), 137,7 (Cq), 137,6 (Cq), 135,7 (Cq), 135,6 (Cq), 128,4-127,4 (CH аром), 99,4 (С-1'), 99,2 (С-1'), 98,5 (C-1), 98,4 (C-1), 80,6 (C-3), 80,4 (C-3), 78,7, 78,4 (C-3', C-4), 76,8, 76,7 (C-3"), 75,6, 75,5, 75,4, 75,3 (C-4'), 74,8, 74,7 (2×CH2Ph), 74,1 (C-5'), 73,2, 72,6, 72,5, 72,0 (2×CH2Ph), 71,3, 71,2 (CH2Ph), 70,8-70,5 (C-3''', C-5, CH(OH)), 69,4, 69,3 (2×P-OCH2Ph), 68,8, 68,7, 68,6, 68,1 (C-6, (OCH2CH), C-6'), 63,7, 63,6 (CH2OH), 56,2, 56,1 (C-2'), 52,6, 52,5 (C-2), 41,8-41,4 (C-2", C-2'''), 34,4, 34,0, 33,7 (C-4", C-4''', C-2""), 31,9 (C-12", C-12''', C-10""), 29,8-29,1 (C-6"->C-11", C-6'''->С-11”', C-4""->C-9""), 25,2, 25,1, 25,0, 24,9 (C-5", C-5''', C-3""), 22,6 (C-13",C-13''', C-11""), 14,1 (C-14",C-14''', C-12""); MC-ES 1782 [M+Na]+; Анализ Вычислено для C104H147N2O19P: C, 70,96; H, 8,42; N, 1,59%, Найдено: C, 70,44; H, 8,36; N, 1,47%.
2,3-Дигидроксипропил-2-дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-α-D-глюкопиранозид (19) (ОМ-174-MP-PD)
Соединение 18 (113 мг, 64 мкмол) в ТГФ (100 мл) гидрировали в течение 19 часов в присутствии 5% Pd-C (50 мг) при комнатной температуре в атмосфере водорода (6 бар). Затем смесь нейтрализовали Et3N (500 мкл) и катализатор удаляли фильтрованием. Фильтрат концентрировали в вакууме и остаток очищали с помощью HPLC согласно изобретению (Метод D), получая соединение 19 (в виде натриевой соли)(26 мг; 36%) в виде белого лиофилизата. MC-ESI 1173 [M-H+2Na]+, HRMS-ESI рассчитано для C55H104N2O19Na2P [M-H+2Na]+ 1173,6766, найдено 1173,6763.
Трихлорацетимидат 3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил}-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-D-глюкопиранозила (24b)
К перемешиваемому раствору соединения 14b (260 мг; 150 мкмол) в сухом CH2Cl2 (5 мл) при комнатной температуре добавляли трихлорацетонитрил (155 мкл; 1,54 ммол) и карбонат калия (11 мг, 77 мкмол). После перемешивания в течение 1 часа реакционную смесь гасили насыщенным водным раствором NaHCO3 (2 мл) и раствор экстрагировали. Органический слой промывали солевым раствором, сушили над MgSO4 и растворитель удаляли в вакууме, получая соединение 24b (280 мг) в виде бледно-желтого масла, которое использовали на следующей стадии без дальнейшей очистки.
(6-Бензилоксикарбониламиногексил)-3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил}-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-β-D-глюкопиранозид (25)
К перемешиваемому раствору коммерчески доступного 6-бензилоксикарбониламино-1-гексанола (43 мг, 0,17 ммол) и неочищенного имидата соединения 24b (280 мг) в безводном CH2Cl2 (5 мл) при -20°С добавляли 4 Å молекулярные сита. После перемешивания в течение 30 минут добавляли TMSOTf (6 мкл, 31 мкмол) и перемешивание продолжали дополнительно в течение 2 часов. Смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь фильтровали через целит, разбавляли EtOAc и нейтрализовали насыщенным водным раствором NaHCO3. Органическую фазу отделяли, промывали насыщенным раствором NaCl, сушили над MgSO4. Растворитель выпаривали и остаток перекристаллизовывали из MeOH, получая соединение 25 (174 мг, 59% на протяжении 2 стадий) в виде белого кристаллического твердого вещества. MS-ES 1942 [M+Na]+
6-Аминогексил-2-дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-β-D-глюкопиранозид (26) (ОМ-174-MP-АС)
Соединение 25 (102 мг, 53 мкмол) в смеси АсОН/2-пропанол/CH2Cl2 3:3:1 (7 мл) гидрировали в течение 24 часов в присутствии 10% Pd-C (50 мг) при комнатной температуре при атмосферном давлении. Катализатор удаляли фильтрованием и фильтрат концентрировали в вакууме. Остаток очищали с помощью HPLC согласно изобретению (Метод D), получая соединение 26 (в виде натриевой соли)(17 мг; 28%) в виде белого лиофилизата. MS-ES 1153 [M-Н]-.
Тетрадецил-3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил}-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-β-D-глюкопиранозид (41b)
К перемешиваемому раствору коммерчески доступного тетрадеканола (14 мг, 65 мкмол) и неочищенного имидата соединения 24b (98 мг) в безводном CH2Cl2 (2 мл) при -20°С добавляли 4 Å молекулярные сита. После перемешивания в течение 30 минут добавляли TMSOTf (2 мкл, 12 мкмол) и перемешивание продолжали дополнительно в течение 2 часов. Смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь фильтровали через целит, разбавляли EtOAc и нейтрализовали насыщенным водным раствором NaHCO3. Органическую фазу отделяли, промывали насыщенным раствором NaCl, сушили над MgSO4. Растворитель выпаривали и остаток перекристаллизовывали из MeOH, получая соединение 41b (58 мг, 52%) в виде белого кристаллического твердого вещества. 1H ЯМР (500 МГц, CDCl3): δ 7,37-7,11 (м, 35H, Ph), 6,49 (д, 1H, J NH,2 8,0 Гц, NH-2), 6,21 (д, 1H, J NH,2 7,5 Гц, NH-2'), 5,11 (м, 1H, H-3'''), 5,02 (д, 1H, J 1',2' 8,0 Гц, Н-1'), 4,95-4,40 (м, 14H, 7×CH2Ph), 4,61 (д, 1H, H-1), 4,51 (м, 1H, H-4'), 4,31 (т, 1H, J 3',4'=J 3',2' 9,5 Гц, H-3'), 4,09 (м, 1H, H-6), 3,96 (м, 1H, H-3), 3,84 (м, 1H, H-6'), 3,79 (м, 1H, OCH 2CH), 3,79-3,65 (м, 5H, H-6', H-6, H-5', H-5, H-3"), 3,56-3,47 (м, 2H, H-4, H-2'), 3,44 (м, 1H, H-2), 3,36 (м, 1H, OCH 2CH), 2,42-2,23 (м, 4H, 2×H-2", 2×H-2'''), 2,12 (т, 2H, J 7,2 Гц, 2×H-2""), 1,65-1,45 (м, 8H, 2×H-4", 2×H-4''', 2×H-3"", OCH2CH2), 1,35-1,12 (м, 74H, 37 CH2), 0,90 (м, 12H, 4×CH3); 13C ЯМР (125,8 МГц, CDCl3): δ 173,4 (C=O), 171,3 (C=O), 170,1 (C=O), 138,2-138,0 (Cq), 135,7-137,6 (Cq), 128,5-127,4 (CH аром), 99,9 (C-1), 99,2 (С-1'), 80,9 (C-3), 78,5 (C-4), 78,3 (C-3'), 76,3 (C-3"), 75,7 (C-4'), 74,6, 74,9 (2×CH2Ph), 74,2-74,1 (C-5', C-5), 73,2, 73,0 (2×CH2Ph), 71,0 (CH2Ph), 70,6 (C-3'''), 69,6 ((OCH2CH)), 69,4-69,0 (2×P-OCH2Ph, C-6'), 67,3 (C-6), 56,6 (C-2), 56,0 (C-2'), 41,4-41,2 (C-2", C-2'''), 34,4, 34,1, 33,6 (C-4", C-4''', C-2""), 31,9 (C-12", C-12''', C-10"", C-12""'), 29,7-29,1 (C-6"->C-11", С-6'''->С-11''', C-4""->C-9"", C-3””'->C-11””'), 26,1, 25,2, 25,0 (C-5", C-5”', C-3""), 22,7 (C-13", C-13”', C-11"", C-13””'), 14,1 (C-14", C-14”', C-12"", C-14””'); MC-ES 1905 [M+Na]+.
Тетрадецил-2-дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-β-D-глюкопиранозид (41с) (ОМ-174-MP-ТЕ)
Соединение 41b (55 мг, 29 мкмол) в ТГФ (200 мл) гидрировали в течение 17 часов в присутствии 5% Pd-C (20 мг) при комнатной температуре в атмосфере водорода (6 бар). Катализатор удаляли фильтрованием и фильтрат концентрировали в вакууме. Остаток очищали с помощью HPLC согласно изобретению (Метод В), получая соединение 41с (в виде натриевой соли)(10 мг; 27%) в виде белого лиофилизата. MC-ES 1273 [M+Na]+, 1251 [M+Н]+.
Формилметил-3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил}-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-α-D-глюкопиранозид (32с)
К перемешиваемому раствору соединения 18 (300 мг; 0,17 ммол) в ТГФ/Н2О 4:1 (5 мл) при комнатной температуре добавляли перйодат натрия (182 мг; 0,85 ммол). После перемешивания в течение ночи реакционную смесь разбавляли EtOAc и смесь промывали насыщенным водным раствором NaHCO3. Органический слой промывали солевым раствором, сушили над MgSO4 и растворитель удаляли в вакууме. Флэш-хроматография остатка на силикагеле (Петролейный эфир/EtOAc, 2:3) давала соединение 32с (250 мг; 85%) в виде бесцветного масла. [α]D + 26 (c 0,90, CHCl3); 1H ЯМР (500 МГц, CDCl3): δ 9,42 (1H, c, CHO), 7,37-7,10 (м, 35H, Ph), 6,55 (д, 1H, JNH,2 9,2 Гц, NH-2), 6,10 (д, 1H, JNH,2' 7,7 Гц, NH-2'), 5,07 (м, 1H, H-3”'), 4,95 (д, 1H, J1',2' 7,6 Гц, Н-1'), 4,94-4,40 (м, 15H, 7×CH2Ph, H-4'), 4,70 (д, 1H, J1,2 3,6 Гц, H-1), 4,35-4,25 (м, 2H, H-2, H-3'), 4,05 (д, 1H, H-6), 3,95-3,75 (м, 5H, H-5, OCH 2CHO, H-6', H-3”), 3,75-3,65 (м, 3H, H-6', H-5', H-3), 3,60 (м, 1H, H-6), 3,55-3,40 (м, 2H, H-4, H-2'), 2,50-2,15 (м, 4H, 2×H-2", 2×H-2”'), 2,13 (т, 2H, J 7,5 Гц, 2×H-2””), 1,70-1,45 (м, 6H, 2×H-4”, 2×H-4”', 2×H-3””), 1,35-1,12 (м, 52H, 26 CH2), 0,90 (м, 9H, 3×CH3); 13C ЯМР (125,8 МГц, CDCl3): δ 199,26 (CHO), 173,4 (C=O), 171,3 (C=O), 170,2 (C=O), 138,2-138,0 (Cq), 137,6 (Cq), 135,7 (Cq), 135,6 (Cq), 128,7-127,3 (CH аром), 99,5 (С-1'), 98,4 (C-1), 80,2 (C-3), 78,3 (C-4, C-3'), 76,7 (C-3”), 75,9 (C-4'), 74,8, 74,7 (2×CH2Ph), 74,2 (C-5'), 73,2, 73,1, 73,0 (2×CH2Ph, OCH2CHO), 71,2 (CH2Ph, C-5), 70,6 (C-3”'), 69,3, 69,2 (2×P-OCH2Ph), 69,0 (C-6'), 68,1 (C-6), 56,6 (C-2'), 52,3 (C-2), 41,4-41,2 (C-2”, C-2”'), 34,4, 34,0, 33,8 (C-4”, C-4”', C-2””), 31,9 (C-12”, C-12”', C-10””), 29,6-29,1 (C-6”->С-11”, C-6”'->С-11”', C-4””->C-9””), 25,2, 25,1, 24,9 (C-5”, C-5”', C-3””), 22,7 (C-13”, C-13”', С-11””), 14,1 (C-14”, C-14'”, C-12””); MS-ES 1750 [M+Na]+.
2-Гидроксиэтил-3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил}-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-α-D-глюкопиранозид (32b)
К перемешиваемому раствору соединения 32c (119 мг; 69 мкмол) в EtOH/CH2Cl2 4:1 (5 мл) при 0°C добавляли боргидрид натрия (3 мг; 75 мкмол). После перемешивания в течение 10 мин реакционную смесь гасили насыщенным водным раствором NH4Cl и разбавляли CH2Cl2. Органический слой экстрагировали, промывали солевым раствором, сушили над MgSO4. Растворитель выпаривали и остаток перекристаллизовывали из EtOH с получением соединения 32b (100 мг; 84%) в виде белого кристаллического твердого вещества. Т.пл. 175°С (EtOH); [α]D + 27 (c 0,56, CHCl3); vmax см-1 3301, 2920, 2850, 1724, 1649, 1543, 1453, 1358, 1264, 1008, 731, 694; 1H ЯМР (500 МГц, CDC13): δ 7,40-7,10 (м, 35H, Ph), 6,44 (д, 1H, J NH,2' 7,0 Гц, NH-2'), 6,40 (д, 1H, J NH,2 10,1 Гц, NH-2), 5,14 (м, 1H, H-3”'), 5,00-4,42 (м, 14H, 7×CH2Ph), 4,88 (м, 1H, J 1',2' 8,72 Гц, Н-1'), 4,73 (д, 1H, J 1,2 3,6 Гц, H-1), 4,53 (м, 1H, H-4'), 4,47 (м, 1H, H-3'), 4,29 (ддд, 1H, J NH,2=J 2,3 10,1 Гц, J l,2 3,6 Гц, H-2), 4,15 (д, 1H, H-6), 3,99 (м, 1H, H-5), 3,86 (м, 2H, H-6', H-3”), 3,77-3,60 (м, 2H, H-6', H-5'), 3,66 (т, 1H, J 3,4=J 2,3 10,1 Гц, H-3), 3,60-3,33 (м, 7H, H-6, H-4, H-2', OCH 2CH2, OCH2CH2), 2,42 (м, 1H, H-2”'), 2,28 (м, 3H, 2×H-2”, H-2”'), 2,15 (т, 2H, J 7,5 Гц, 2×H-2””), 1,70-1,45 (м, 6H, 2×H-4”, 2×H-4”', 2×H-3””), 1,35-1,12 (м, 52H, 26 CH2), 0,90 (м, 9H, 3×CH3); 13C ЯМР (125,8 МГц, CDCl3): δ 173,3 (C=O), 171,0 (C=O), 170,5 (C=O), 138,2-138,0 (Cq), 137,6 (Cq), 135,7 (Cq), 135,6 (Cq), 128,7-127,3 (CH аром), 98,9 (С-1'), 98,5 (C-1), 80,3 (C-3), 78,6 (C-4), 78,1 (C-3'), 76,7 (C-3”), 75,9 (C-4'), 74,8, 74,7 (2×CH2Ph), 74,0 (C-5'), 73,2, 73,1 (2×CH2Ph), 72,3 (CH2OH), 71,2 (CH2Ph), 70,8 (C-5), 70,6 (C-3”'), 69,3, 69,2 (2×P-OCH2Ph), 68,8 (C-6'), 68,2 (C-6), 61,8 (OCH2CH2), 57,1 (C-2'), 52,5 (C-2), 41,4-41,2 (C-2”, C-2”'), 34,4, 34,0, 33,7 (C-4”, C-4”', C-2””), 31,8 (C-12”, C-12”', C-10””), 29,6-29,0 (C-6”->С-11”, C-6”'->С-11”', C-4””->C-9””), 25,1, 25,0, 24,9 (C-5”, C-5”', C-3””), 22,6 (C-13”, C-13”', C-11””), 14,0 (C-14”, C-14”', C-12””); MC-ES 1753 [M+Na]+.
2-Гидроксиэтил-2-дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-α-D-глюкопиранозид (32) (ОМ-174-MP-ЕО)
Соединение 32b (160 мг, 92 мкмол) в ТГФ (100 мл) гидрировали в течение 17 часов в присутствии 5% Pd-C (50 мг) при комнатной температуре в атмосфере водорода (6 бар). Затем смесь нейтрализовали Et3N (500 мкл) и катализатор удаляли фильтрованием. Фильтрат концентрировали в вакууме и остаток очищали с помощью HPLC согласно изобретению (Метод D), получая соединение 32 (в виде натриевой соли)(50 мг; 49%) в виде белого лиофилизата. MS-ESI 1143 [M-H+2Na]+, HRMS-ESI вычислено для C54H102N2O18Na2P [M-H+2Na]+ 1143,6660, найдено 1143,6659.
2-(Дибензилоксифосфорилокси)этил-3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил}-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-α-D-глюкопиранозид (33b)
К перемешиваемому раствору соединения 32b (125 мг, 72 мкмол) и коммерчески доступного раствора 1Н-тетразола в CH3CN (~0,45 M) (321 мкл, 0,14 ммол) в CH2Cl2 (5 мл) при комнатной температуре добавляли дибензил диметилфосфорамидит (29 мкл; 0,11 ммол). Перемешивание продолжали при комнатной температуре в течение 10 минут и затем раствор охлаждали до -20°С. Затем добавляли раствор mCPBA (57-86%, 46 мг; 0,27 ммол) в CH2Cl2 (3 мл) и раствор перемешивали в течение 30 минут при -20°С. Добавляли 10% водный раствор тиосульфата натрия (5 мл) и смесь перемешивали в течение 10 минут, затем разбавляли EtOAc и органическую фазу отделяли. Органический слой последовательно промывали 10% водным раствором Na2S2O3 (3×), насыщенным водным раствором NaHCO3 (2×), N раствором HCl (1×) и солевым раствором. Органическую фазу сушили над MgSO4 и растворитель удаляли в вакууме. Флэш-хроматография остатка на силикагеле (Петролейный эфир/EtOAc, 1:1-1:2) давала соединение 33b (130 мг; 90%) в виде бесцветного масла. 1H ЯМР (500 МГц, CDCl3): δ 7,40-7,10 (м, 45H, Ph), 7,07 (д, 1H, J NH,2 9,4 Гц, NH-2), 6,56 (д, 1H, J NH,2' 7,4 Гц, NH-2'), 5,09 (м, 1H, H-3”'), 5,05 (м, 1H, J 1',2' 7,8 Гц, Н-1'), 5,00-4,36 (м, 18H, 9×CH2Ph), 4,66 (д, 1H, J1,2 3,2 Гц, H-1), 4,50 (м, 1H, H-4'), 4,43 (м, 1H, H-3'), 4,33 (ддд, 1H, J NH,2=J 2,3 9,4 Гц, J l,2 3,2 Гц, H-2), 4,10 (д, 1H, J6,6 10,0 Гц, H-6), 4,05 (м, 1H, OCH2CH 2OP), 3,93 (м, 1H, OCH2CH 2OP), 3,88 (м, 1H, H-3”), 3,83 (д, 1H, J6',6' 10,0 Гц, H-6'), 3,80 (м, 1H, H-5), 3,73-3,63 (м, 3H, H-5', H-3, OCH 2CH2OP), 3,62 (дд, 1H, H-6), 3,51 (т, 1H, J 3,4=J4,5 9,5 Гц, H-4), 3,38 (ддд, 1H, J NH,2' 7,4 Гц, J 1',2' 7,8 Гц, J 2',3' 8,8 Гц, H-2'), 3,31 (м, 1H, OCH 2CH2OP), 2,44 (дд, 1H, J 2”,3” 7,5 Гц, J 2”,2” 14,7 Гц, H-2”), 2,36 (м, 2H, H-2”', H-2”), 2,23 (дд, 1H, J 2”',3”' 5,3 Гц, J 2”',2”' 15,2 Гц, H-2”'), 2,10 (т, 2H, J 7,5 Гц, 2×H-2””), 1,60-1,40 (м, 6H, 2×H-4”, 2×H-4”', 2×H-3””), 1,35-1,05 (м, 52H, 26 CH2), 0,88 (м, 9H, 3×CH3); 13C ЯМР (125,8 МГц, CDCl3): δ 173,3 (C=O), 171,5 (C=O), 170,3 (C=O), 138,7 (Cq), 138,5 (Cq), 138,3 (Cq), 138,2 (Cq), 138,0 (Cq), 135,8 (Cq), 135,7 (Cq), 135,6 (Cq), 135,5 (Cq), 135,4 (Cq), 128,6-127,8 (CH аром), 99,2 (С-1'), 98,7 (C-1), 80,8 (C-3), 78,1 (C-4, C-3'), 76,8 (C-3”), 76,0 (C-4'), 74,9, 74,8 (2×CH2Ph), 74,2 (C-5'), 73,2, 73,1 (2×CH2Ph), 71,4 (CH2Ph), 70,7-70,6 (C-5, C-3”'), 69,5-69,3 (4×P-OCH2Ph), 69,1 (C-6), 68,0 (C-6'), 67,2 (OCH2CH2O-P), 66,6 (OCH2CH2O-P), 57,0 (C-2'), 52,5 (C-2), 41,6-41,3 (C-2”, C-2”'), 34,4, 34,2, 34,0 (C-4”, C-4”', C-2””), 31,9 (C-12”, C-12”', C-10””), 29,6-29,1 (C-6”->С-11”, C-6”'->С-11”', C-4””->C-9””), 25,2, 25,0, 24,9 (C-5”, C-5”', C-3””), 22,7 (C-13”, C-13”', С-11””), 14,1 (C-14”, C-14”', C-12””).
2-(Фосфонокси)этил-2-дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-α-D-глюкопиранозид (33) (ОМ-174-MP-ЕР)
Соединение 33b (107 мг, 54 мкмол) в ТГФ (80 мл) гидрировали в течение 17 часов в присутствии 5% Pd-C (70 мг) при комнатной температуре в атмосфере водорода (6 бар). Затем смесь нейтрализовали Et3N (500 мкл) и катализатор удаляли фильтрованием. Фильтрат концентрировали в вакууме и остаток очищали с помощью HPLC согласно изобретению (Метод D), получая соединение 33 (в виде натриевой соли)(35 мг; 55%) в виде белого лиофилизата. MS-ESI 1245 [M-2H+3Na]+, HRMS-ESI вычислено для C54H102N2O21Na3P2 [M-2H+3Na]+ 1245,6143, найдено 1245,6136.
2-Карбоксиметил-3,4-ди-О-бензил-6-О-{3,6-ди-О-бензил-4-O-(дибензилоксифосфорил)-2-дезокси-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил}-2-[(R)-3-бензилокситетрадеканоиламино]-2-дезокси-α-D-глюкопиранозид (35d)
К перемешиваемому раствору соединения 32с (100 мг; 58 мкмол), NaH2PO4.H2O (8 мг, 58 мкмол), 2-метил-2-бутена (28 мкл, 260 мкмол) в ТГФ/Н2О 4:1 (5 мл) при комнатной температуре добавляли хлорит натрия (20 мг; 173 мкмол). После перемешивания в течение 6 часов реакционную смесь гасили 1М раствором HCl (1 мл) и разбавляли CH2Cl2. Органический слой экстрагировали, промывали солевым раствором, сушили над MgSO4 и растворитель удаляли в вакууме. Флэш-хроматография остатка на силикагеле (CH2Cl2/Ацетон, 5:1 + 1% АсОН) давала соединение 35d (67 мг; 66%) в виде белого твердого вещества. MC-ES 1766 [M+Na]+.
2-Карбоксиметил-2-дезокси-6-О-[2-дезокси-4-O-(дигидроксифосфорил)-2-[(R)-3-додеканоилокситетрадеканоиламино]-β-D-глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино]-α-D-глюкопиранозид (35c) (ОМ-174-MP-CM)
Соединение 35d (67 мг, 38 мкмол) в ТГФ (20 мл) гидрировали в течение 17 часов в присутствии 5% Pd-C (25 мг) при комнатной температуре в атмосфере водорода (6 бар). Затем смесь нейтрализовали Et3N (500 мкл) и катализатор удаляли фильтрованием. Фильтрат концентрировали в вакууме и остаток очищали с помощью HPLC согласно изобретению (Метод D), получая соединение 35с (в виде натриевой соли)(25 мг; 58%) в виде белого лиофилизата. MS-ESI 1179 [M-2H+3Na]+, HRMS-ESI вычислено для C54H99N2O19Na3P [M-2H+3Na]+ 1179,6273, найдено 1179,6275.
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
1.Метод обработки А
Продукты растворяли в смеси ТГФ/вода (1:1 об./об.). Обработку с помощью препаративной обратной фазы HPLC проводили в следующих условиях:
Колонка: VYDAC C18, 22×250 мм, 10 мкм, 300 Å
Подвижная фаза:
А: Ацетонитрил - вода (1:1 об./об.), 5 мМ тетрабутиламмонийфосфат одноосновный
В: 2-пропанол - вода (9:1 об./об.) 5 мМ тетрабутиламмонийфосфат одноосновный
Скорость потока: 20 мл/мин.
Элюирование:
Детекция: УФ, 210 нм (длина волны)
Фракции, содержащие соединения в виде тетрабутиламмониевой соли, собирали и концентрировали с помощью абсорбции на HPLC, VYDAC C18, 22×250 мм, 10 мкм, 300 Å. Натриевую соль соединения получают путем промывки 200 мМ одноосновным раствором фосфата натрия в воде, рН 4,2 + 2-пропанол (9:1, об./об.)(5 объемов). После удаления избытка одноосновного фосфата натрия путем проведения через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюировали раствором воды + 2-пропанол (1:9 об./об.).
После разбавления водой и удаления растворителя путем лиофилизации получают соединение в виде натриевой соли.
2.Метод обработки В
Продукты растворяли в смеси ТГФ/вода (1:1 об./об.). Обработку с помощью препаративной обратной фазы HPLC проводили в следующих условиях:
Колонка: VYDAC C18, 22×250 мм, 10 мкм, 300 Å
Подвижная фаза:
А: Ацетонитрил - вода (1:1 об./об.), 5 мМ тетрабутиламмонийфосфат одноосновный
В: 2-пропанол - вода (9:1 об./об.) 5 мМ тетрабутиламмонийфосфат одноосновный
Скорость потока: 20 мл/мин.
Элюирование:
Детекция: УФ, 210 нм (длина волны)
Фракции, содержащие соединения в виде тетрабутиламмониевой соли, собирали и концентрировали с помощью абсорбции на HPLC, VYDAC C18, 22×250 мм, 10 мкм, 300 Å. Натриевую соль соединения получают путем промывки 100 мМ двухосновным раствором фосфата натрия - одноосновным раствором фосфата натрия в воде, рН 7,5 + 2-пропанол (9:1, об./об.)(5 объемов) + 2-пропанол (9:1, об./об.)(5 объемов). После удаления избытка одноосновного фосфата натрия - двухосновного фосфата натрия путем проведения через 5 объемов воды + 2-пропанол (1:9 об./об.). После разбавления водой и удаления растворителя путем лиофилизации получают соединение в виде натриевой соли.
3.Метод обработки С
Продукты растворяли в смеси ТГФ/вода (1:1 об./об.). Обработку с помощью препаративной обратной фазы HPLC проводили в следующих условиях:
Колонка: VYDAC C18, 22×250 мм, 10 мкм, 300 Å
Подвижная фаза:
А: Ацетонитрил - вода (1:1 об./об.), 5 мМ тетрабутиламмонийфосфат одноосновный
В: 2-пропанол - вода (9:1 об./об.) 5 мМ тетрабутиламмонийфосфат одноосновный
Скорость потока: 20 мл/мин.
Элюирование:
Детекция: УФ, 210 нм (длина волны)
Фракции, содержащие соединения в виде тетрабутиламмониевой соли, собирали и концентрировали с помощью абсорбции на HPLC, VYDAC C18, 22×250 мм, 10 мкм, 300 Å. Натриевую соль соединения получают путем промывки 200 мМ двухосновным раствором фосфата натрия в воде, рН 9,2 + 2-пропанол (9:1, об./об.)(5 объемов). После удаления избытка двухосновного фосфата натрия путем проведения через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюировали раствором воды + 2-пропанол (1:9 об./об.). После разбавления водой и удаления растворителя путем лиофилизации получают соединение в виде натриевой соли.
4.Метод обработки D
Продукты растворяли в смеси ТГФ/вода (1:1 об./об.). Обработку с помощью препаративной обратной фазы HPLC проводили в следующих условиях:
Колонка: VYDAC C18, 22×250 мм, 10 мкм, 300 Å
Подвижная фаза:
А: Ацетонитрил - вода (1:1 об./об.), 5 мМ тетрабутиламмонийфосфат одноосновный
В: 2-пропанол - вода (9:1 об./об.) 5 мМ тетрабутиламмонийфосфат одноосновный
Скорость потока: 20 мл/мин.
Элюирование:
Детекция: УФ, 210 нм (длина волны)
Фракции, содержащие соединения в виде тетрабутиламмониевой соли, собирали и концентрировали с помощью абсорбции на HPLC, VYDAC C18, 22×250 мм, 10 мкм, 300 Å. Натриевую соль соединения получают путем промывки 10 г/л раствором хлорида натрия в воде, рН 7,0 + 2-пропанол (9:1, об./об.)(5 объемов). После удаления избытка хлорида натрия путем проведения через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюировали раствором воды + 2-пропанол (1:9 об./об.).
После разбавления водой и удаления растворителя путем лиофилизации получают соединение в виде натриевой соли.
5. Мониторинг обработки
После каждой стадии обработки фракции подвергали исследованию с помощью аналитической HPLC хроматографии с обратной фазой в соответствии со следующими условиями:
Колонка: Supelcosil C18, 3 мкм, 4,6×150 мм, 100 Å, Supelco
Подвижная фаза:
А: вода:ацетонитрил (1:1 об./об.), 5 мМ тетрабутиламмонийфосфат одноосновный
В: вода - изопропанол (1:9 об./об.) 5 мМ тетрабутиламмонийфосфат одноосновный
Скорость потока: 1 мл/мин.
Элюирование: А:В градиент (75:25-0:100) в течение 20 минут.
Детекция: УФ, 210 и 254 нм (длина волны)
ПРИМЕР 1:
Секреция IL-6 и TNF-α моноядерными клетками периферической крови человека (PBMC)
Действие на IL-6 и TNF-α синтетических соединений изобретения и сравнение с исходной биологической молекулой
Соединения, испытываемые на секрецию IL-6:
Соединениями изобретения, которые испытывались здесь на секрецию IL-6, были:
Соединение 1b (OM-174-DP), соединение 16 (ОМ-174-МР); соединение 17 (ОМ-174-МР-PR), соединение 19 (ОМ-174-МР-PD) и соединение 26 (ОМ-174-МР-АС).
Кроме того, для сравнения испытывалась также активность биологической исходной молекулы, OM-174-DP (P3).
Затем в еще одной серии экспериментов следующие соединения испытывались на секрецию TNF-α человеческими РВМС:
Соединение 1b (OM-174-DP), соединение 16 (ОМ-174-МР); соединение 17 (ОМ-174-МР-PR), соединение 19 (ОМ-174-МР-PD), соединение 26 (ОМ-174-МР-АС), соединение 41с (ОМ-174-МР-ТЕ), соединение 32 (ОМ-174-МР-ЕО), соединение 33 (ОМ-174-МР-ЕР) и соединение 35с (ОМ-174-МР-СМ).
Введение и объяснение:
Продуцирование IL-6 моноядерными клетками периферической крови человека (РВМС) является важным in vitro испытанием для скрининга новых соединений по способности стимулировать иммунную систему. IL-6 является многофункциональным цитокином, который играет важную роль в защите хозяина, острофазовых реакциях и гематопоэзе.
Фактор некроза опухоли (TNF-α) является плеотропным цитокином, продуцируемым широким множеством типов клеток, главным образом гематопоэтического, но также и не гематопоэтического происхождения. TNF-α необходим для устранения многочисленных инфекционных агентов.
Поэтому активация данных цитокинов соединениями изобретения может представлять важную терапевтическую ценность.
Способы:
Приготовление человеческих РВМС и культуры клеток:
Периферическую кровь от здоровых доноров (Centre de transfusion, Hôpital Universitaire, Женева) центрифугировали для получения желтой оболочки. Желтую оболочку смешивали со сбалансированным физиологическим солевым раствором Хэнкса (HBSS, Sigma, Buchs, Швейцария), наслаивали на Ficoll Paque Plus (Amersham Pharmacia) до 1,077 г/мл и центрифугировали (2800 об./мин, 20°С, 25 мин). Клетки, собранные из промежуточного пространства, промывали дважды в HBSS при 800 об./мин в течение 15 минут при комнатной температуре и таблетированные клетки повторно суспендировали в HBSS. Подсчет клеток производили с использованием клеток Neubauer. Все культуры клеток выполняли в среде RPMI-1640 с добавлением пенициллина (100 ед./мл), стрептомицина (100 мкг/мл), L-глутамина (2 ммол/л) и 10% плодной сыворотки теленка (FCS), все из которых получали от Sigma. Для стимуляции in vitro клетки культивировали в концентрации 1×106 жизнеспособных клеток/лунка.
Стимуляция и измерение IL-6 и TNF-α в супернантах культуры:
РВМС инкубировали при 37°С и в атмосфере 5% СО2 с контролями (см. ниже) и с продуктами изобретения при 1, 5 и 20 мкг/мл для IL-6 и при 0,2, 2 и 20 мкг/мл для секреции TNF-α. Среда: одна RPMI.
Супернатанты культур собирали спустя 24 часа и измеряли концентрацию IL-6 и TNF-α с помощью фермент-связываемого иммуносорбентного анализа (ELISA) (набор человеческого IL-6 и TNF-α ELISA, BD OptEIA, Сан Диего, США) согласно инструкциям производителя. Пределами детекции были соответственно 10 и 8 пг/мл.
Результаты:
Результаты показаны ниже в таблицах:
А: Для IL-6
Объяснение результатов:
LPS индуцировал, как и ожидалось, очень высокие уровни IL-6 из человеческих РВМС даже при наиболее низких испытываемых дозах.
Объяснение результатов:
Обе загрузки ОМ-174 индуцируют секрецию IL-6 человеческими РВМС, а уровни, полученные с синтетической молекулой, были несколько выше, чем уровни, полученные с биологической молекулой Р3.
Объяснение результатов:
В сравнении с биологической исходной молекулой OM-174-DP (загрузка Р3) синтетическая молекула (1b) индуцирует более высокие уровни IL-6 моноцитами человека.
То же самое действительно для ОМ-174-МР (16), поскольку соответствующий биологический продукт не индуцировал продуцирование IL-6 даже при наиболее высокой испытанной дозе (20 мкг/мл, не показано), тогда как родственная синтетическая молекула ОМ-174-МР (соединение 16) индуцирует вплоть до 1348 пг/мл IL-6.
В основном все испытанные синтетические молекулы (1b, 16, 17, 19 и 26) были способны индуцировать секрецию IL-6.
В) Для TNF-α
Объяснение результатов:
LPS индуцировал, как и ожидалось, очень высокие уровни TNF-α человеческими РВМС даже при трех испытанных дозах.
Объяснение результатов:
Обе загрузки ОМ-174 индуцируют секрецию TNF-α человеческими РВМС, а уровни, полученные с синтетической молекулой, были несколько выше, чем уровни, полученные с биологической молекулой Р3.
Объяснение результатов:
За исключением синтезированных молекул (19) и (33) все синтезированные молекулы (1b, 16, 17, 26, 41c и 32) индуцировали высокие уровни TNF-α человеческими моноцитами, что дает основания полагать, что они могли бы действовать как иммуностимулирующие лекарственные средства.
Результаты дают также основания полагать, что могла бы быть стоящей разработка молекул (19, т.е. OM-174-МР-PD и 33, т.е. ОМ-174-МР-ЕР) в качестве “противовоспалительных” лекарственных средств. Очень интересно, что оба соединения (19) и (33) проявляли “противовоспалительные” свойства.
Более того, соединение (19) показало противовоспалительные свойства как “профилактические”, так и “терапевтические” на модели LACK-индуцируемой астмы (см. пример 6), и ингибировали индуцируемую высвобождением соединения 48/80 секрецию гистамина мышиными тучными клетками (см. пример 7). На данной последней модели в примере 7 было активным также соединение (33).
ПРИМЕР 2:
Модификация биологической активности биологического соединения OM-174-DP:
Усиление TNF-α-индицируемой секреции ТНР-1 клетками с помощью оригинального метода очистки исходной биологической молекулы OM-174-DP
Испытуемые соединения:
Соединениями изобретения, испытанными здесь, являются
Исходная биологическая загрузка (GMP004) молекулы изобретения OM-174-DP испытывалась или из исходного раствора, или повторно очищенного, как описано ниже, главным образом путем изменения рН HPLC подвижной фазы.
Введение и объяснение:
Фактор некроза опухоли (TNF-α) является плеотропным цитокином, продуцируемым широким множеством типов клеток, главным образом гематопоэтического, но также и не гематопоэтического происхождения. TNF-α необходим для устранения многочисленных инфекционных агентов (Candida albicans, Listeria monocytogenes, микобактерий …) и проявляет сильные провоспалительные эффекты, например, путем индуцирования экспрессии адгезионных молекул, таких как VCAM-1, межклеточной адгезионной молекулы 1 (ICAM-1), Е-секреции в эндотелиальные клетки и другие типы клеток.
Сверхпродуцирование TNF, однако, вовлечено также в патогенез различных заболеваний, таких как ревматоидный артрит, инсулинзависимый диабет и воспалительная болезнь кишечника, в частности болезнь Крона.
Поэтому TNF-α необходим для запуска иммунологических ответных реакций; однако для того, чтобы избежать воспалительных патологий, следует справляться с данным продуцированием.
Одну партию первоначального биологического продукта OM-174-DP (GMP004) переформулировали в соответствии с двумя методами, описанными ниже:
Методы
1. Метод А
Очистку проводили с помощью препаративной HPLC с обращенной фазой. УФ детекцию проводили при 210 нм. Фракции, содержащие соединения в форме тетрабутиламмониевой соли, собирали и концентрировали с помощью адсорбции на HPLC. Натриевую соль соединения получали с помощью промывки 200 мМ моноосновным раствором фосфата натрия в воде, рН 4,23 + 2-пропанол (9:1, об./об.) (5 объемов). После удаления избытка моноосновного фосфата натрия пропусканием через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюируют раствором воды + 2-пропанол (1:9 об./об.). После разбавления водой и удаления растворителя лиофилизацией получают соединение в виде натриевой соли.
Получаемые соединения затем испытывали с доведением рН (при 7,5) или без него на ТНР-1 клетки для анализа их потенциала индуцировать секрецию TNF-α (см. ниже).
2. Метод В
Очистку проводили с помощью препаративной HPLC с обращенной фазой. УФ детекцию проводили при 210 нм. Фракции, содержащие соединения в форме тетрабутиламмониевой соли, собирали и концентрировали с помощью адсорбции на HPLC. Натриевую соль соединения получали с помощью промывки 100 мМ моноосновным раствором фосфата натрия двухосновного фосфата натрия в воде, рН 7,5 + 2-пропанол (9:1, об./об.) (5 объемов) + 2-пропанол (9:1, об./об.) (5 объемов). После удаления избытка моноосновного фосфата натрия двухосновного фосфата натрия пропусканием через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюируют раствором воды + 2-пропанол (1:9 об./об.). После разбавления водой и удаления растворителя лиофилизацией получают соединение в виде натриевой соли.
Получаемые соединения затем испытывали с доведением рН (при 7,5) или без него на ТНР-1 клетки для анализа их потенциала индуцировать секрецию TNF-α (см. ниже).
Культура ТНР-1 клеток:
ТНР-1, линию лейкемических моноцитных клеток человека, получали от АТСС (Манассас, США)
Клетки ТНР-1 (106 клеток/мл, 200 л/лунку культивировали на 96-луночной плоскодонной планшете с тканевой культурой (Costar) в среде RPMI с добавкой 10% сыворотки человека (HS; Gibco-BRL), содержащего 10 мМ буфер HEPES, 1 мМ пируват, 0,1 М заменимых аминокислот, 2 мМ глутамин, 50 мМ 2-меркаптоэтанол, 100 ед/мл пенициллина и 10 мг/мл стрептомицина (полная среда). Клетки стимулировали различными концентрациями соединений изобретения в течение различных периодов времени при 37°С в увлажняемом инкубаторе с 5% СО2. Супернатанты культуры собирали и хранили при -20°С до определения цитокинов по методу ELISA.
Стимуляция и измерение TNF-α в супернатантах культуры:
Клетки инкубировали при 37°С и в атмосфере 5% СО2 с продуктами изобретения при 0,2, 2 и 20 мкг/мл: среда: одна RPMI.
Супернатанты культур собирали через 24 часа и измеряли концентрацию TNF-α с помощью фермент-связываемого иммуносорбентного анализа (ELISA) (BD OptEIA, Сан Диего, США) согласно инструкциям производителя. Пределом детекции было 8 пг/мл.
Результаты:
Результаты показаны ниже в таблице 2.1:
Объяснение результатов:
Величина TNF-α, полученная с OM-174-DP биологическими продуктами [GMP004] при 20 мкг/мл была 193 пг/мл. Здесь авторы демонстрируют метод очистки В, который усиливает биологическую активность исходного биологического продукта на фактор 3.
В заключение, метод очистки, описанный здесь, мог быть следовательно применен или приспособлен для клиники для усиления терапевтической активности лекарственного средства.
ПРИМЕР 3:
Действие биологической активности 3 синтетических монофосфорильных соединений изобретения: Продуцирование оксида азота мышиными макрофагами, стимулируемыми тремя монофосфорильными синтетическими соединениями изобретения
Испытуемые соединения:
Соединениями изобретения, представленными здесь, являются
Соединение 16 (OM-174-MP); соединение 17 (ОМ-174-МР-PR) и соединение 19 (OM-174-МР-PD).
Введение и объяснение:
Продуцирование оксида азота (NO) макрофагами является важным в in vitro испытании для скрининга новых соединений по способности стимулировать иммунную систему. Он является важной сигнальной молекулой в теле млекопитающих, включая людей, одной из немногочисленных известных газообразных сигнальных молекул.
Молекула оксида азота является свободным радикалом, что делает его очень реакционноспособным и нестабильным. В организме оксид азота синтезируется из аргинина и кислорода с помощью разнообразных ферментов синтазы оксида азота (NOS) и последующего восстановления неорганического нитрата.
Макрофаги продуцируют оксид азота, чтобы убивать вторгающиеся бактерии. В некоторых условиях это может иметь неприятные последствия: молниеносная инфекция (сепсис) вызывает избыточное продуцирование макрофагами оксида азота, что ведет к вазодилатации (расширению кровеносных сосудов), вероятно одной из основных причин гипотензии (низкого кровяного давления) при сепсисе.
Биологические функции оксида азота были открыты в 1980-х, и оксид азота был назван “Молекулой года” журналом Science в 1992 г. Считается, что ежегодно публикуется около 3000 научных статей о биологической роли оксида азота.
Следовательно, его активация соединениями изобретения может представлять важную терапевтическую ценность.
Материалы и методы:
Экспериментальный анализ продуцирования оксида азота мышиными макрофагами: Самцов мышей C57/BL6 шестинедельного возраста (самцы шестинедельного возраста, SPF качество, Charles Rivier, FR) убивали с помощью СО2 ингаляции. Из задней части удаляли бедро, бедренную кость и большеберцовую кость. Костный мозг экстрагировали из просвета инъецированием модифицированной среды Игла Дульбекко (DH) через кость после отрезания обеих концевых частей. После промывки стволовые клетки повторно суспендировали (40000 клеток/мл) в среде DH с добавлением 20% лошадиной сыворотки и 30% супернатанта клеток L929. Клеточную суспензию инкубировали в течение 8 дней в инкубаторе при 37°С в атмосфере 8% СО2 и насыщенной влагой. Макрофаги затем отделяли охлажденным льдом PBS, промывали и повторно суспендировали в среде DH с добавлением 5% фетальной бычьей сыворотки (FCS), аминокислот и антибиотиков (среда DHE). Плотность клеток доводят до 700000 клеток/мл. Непосредственно в микротитровальных планшетах разводят водные растворы продуктов в среде DHE. Продукты испытывают в трех повторениях, и каждая микротитровальная планшета включает отрицательный контроль, составленный из среды. Конечный объем в каждой лунке составляет 100 мкл. 100 мкл клеточной суспензии добавляют к разбавленным продуктам и клетки инкубируют в течение 22 часов в инкубаторе при 37°С в атмосфере 8% СО2, насыщенной влагой. В конце инкубационного периода 100 мкл супернатанта переносят на еще одну микротитровальную планшету и с помощью проведения реакции Griess определяют концентрацию нитрита, продуцируемого в каждом супернатанте. В каждую лунку добавляют 100 мкл реагента Griess (5 мг/мл сульфаниламида + 0,5 мг/мл гидрохлорида N-(1-нафтил)этилендиамина) в 2,5% водной фосфорной кислоте. Микротитровальные планшеты считывают спектрофотометром (SpectraMax Plus, Molecular Devices) при 562 нм против со ссылочной при 690 нм. Концентрация нитрита пропорциональна содержанию образующегося оксида азота. Содержание нитрита определяют на основе стандартной кривой. Результаты даны в виде средней величины ± стандартное отклонение и вычерчиваются графически в виде кривой ответной реакции на дозу.
Результаты:
Результаты показаны на Фиг.25. Три испытанные молекулы способны были индуцировать высокие уровни оксида азота мышиными макрофагами. Соединение 19 (OM-174-МР-PD) было активным при более низких дозах (от 0,01 мкг/мл) в данном испытании, чем соединения 16 (ОМ-174-МР) и 17 (ОМ-174-МР-PR).
ПРИМЕР 4:
Влияние очистки согласно методу В на продуцирование IL-6 моноядерными клетками периферической крови человека ранее неактивных синтетических соединений изобретения, получаемых по методу D:
Испытанные соединения:
Соединениями изобретения, представленными здесь, являются
Партия 14 синтетического продукта OM-174-DP (соединение 1b), обработанного или необработанного по методу D (см. ниже).
Введение и объяснение:
Партия соединения, представленного здесь (синтетического OM-174-DP), была первоначально неактивной, потому что она была получена по методу D, в котором с конечной рН справлялись не должным образом. В действительности партия 14 имела рН 4,88 перед тем, как быть подвергнутой очистке согласно методу В (см. ниже). Очень интересно, что метод очистки В (см. пример 2) значительно увеличивал активность партии 14.
См. также пример 1, что касается описания биологического действия на IL-6.
Метод очистки D
Метод был описан подробно выше.
Синтетический продукт OM-174-DP (1b) (партия 14) растворялся в смеси ТГФ-вода (1:1 об./об.). Очистка проводилась с помощью препаративной HPLC с обращенной фазой. УФ детекция проводилась при 210 нм.
Фракции, содержащие соединения в форме тетрабутиламмониевой соли, собирались и концентрировались с помощью адсорбции на HPLC, VYDAC C18, 22×250 мм, 10 мкм, 300Å. Натриевая соль соединения получается с помощью промывки 10 г/л раствором хлорида натрия в воде, рН 7,0 + 2-пропанол (9:1, об./об.) (5 объемов). После удаления избытка хлорида натрия пропусканием через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюируют раствором воды + 2-пропанол (1:9 об./об.).
После разбавления водой и удаления растворителя лиофилизацией получают соединение в виде натриевой соли. При использовании данного метода (D) конечная рН не регулировалась должным образом. В действительности рН партии 14 была 4,88.
Затем данная партия повторно обрабатывалась по методу В (см. ниже).
Метод очистки В
Метод описан подробно выше. Очистка проводилась с помощью препаративной HPLC с обращенной фазой. УФ детекция проводилась при 210 нм. Фракции, содержащие соединения в форме тетрабутиламмониевой соли, собирались и концентрировались с помощью адсорбции на HPLC. Натриевая соль соединения получается с помощью промывки 100 мМ раствора двухосновного фосфата натрия-моноосновного фосфата натрия в воде, рН 7,5 + 2-пропанол (9:1, об./об.) (5 объемов) + 2-пропанол (9:1, об./об.) (5 объемов). После удаления избытка моноосновного фосфата натрия - двухосновного фосфата натрия пропусканием через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюируют раствором воды + 2-пропанол (1:9 об./об.).
После разбавления водой и удаления растворителя лиофилизацией получают соединение в виде натриевой соли.
Получаемые соединения затем испытывали с доведением рН (при 7,5) или без него на ТНР-1 клетки для анализа их потенциала индуцировать секрецию TNF-α (см. ниже).
Приготовление человеческих РВМС и культуры клеток:
Периферическую кровь от здоровых доноров (Centre de transfusion, Hôpital Universitaire, Женева) центрифугировали для получения желтой оболочки. Желтую оболочку смешивали со сбалансированным физиологическим солевым раствором Хэнкса (HBSS, Sigma, Buchs, Швейцария), наслаивали на Ficoll Paque Plus (Amersham Pharmacia) до 1,077 г/мл и центрифугировали (2800 об./мин, 20°С, 25 мин). Клетки, собранные из промежуточного пространства, промывали дважды в HBSS при 800 об./мин в течение 15 минут при комнатной температуре и таблетированные клетки повторно суспендировали в HBSS. Подсчет клеток производили с использованием клеток Neubauer. Все культуры клеток выполняли в среде RPMI-1640 с добавлением пенициллина (100 ед./мл), стрептомицина (100 мкг/мл), L-глутамина (2 ммол/л) и 10% плодной сыворотки теленка (FCS), все из которых получали от Sigma. Для стимуляции in vitro клетки культивировали в концентрации 1×106 жизнеспособных клеток/лунка.
Стимуляция и измерение IL-6 в супернатантах культуры:
РВМС инкубировали при 37°С и в атмосфере 5% СО2 с продуктами изобретения.
Супернатанты культур собирали спустя 24 часа и измеряли концентрацию IL-6 с помощью фермент-связываемого иммуносорбентного анализа (ELISA) (набор человеческого IL-6 ELISA, BD OptEIA, Сан Диего, США) согласно инструкциям производителя. Предел детекции составлял 10 пг/мл.
Результаты:
Результаты представлены на Фиг.26, которая показывает применение метода В (т.е. использование во время процедуры очистки соответствующей рН) к соединениям изобретения (здесь соединение 1b), который трансформирует неактивное соединение (партия 14) в полностью эффективный активатор человеческих РВМС (партия 39).
ПРИМЕР 5:
Модификация биологической активности соединения 1b (OM-174-DP):
Усиление индуцируемой TNF-α секреции ТНР-1 клетками, дифференцируемыми в макрофаги первоначальным методом очистки различных партий молекулы OM-174-DP
Испытанные соединения:
Соединениями изобретения, представленными здесь, являются
Две биологические партии (Р3 и GMP004) соединения 1b (OM-174-DP) и следующая синтетическая партия (14) обрабатывались по методам А или В (см. ниже). LPS использовали в качестве положительного контроля.
Введение и объяснение:
TNF-α
Фактор некроза опухоли (TNF-α) является плеотропным цитокином, продуцируемым широким множеством типов клеток, главным образом, гематопоэтического, но также и не гематопоэтического происхождения. TNF-α необходим для устранения многочисленных инфекционных агентов (Candida albicans, Listeria monocytogenes, микобактерий …) и проявляет сильные провоспалительные эффекты, например, путем индуцирования экспрессии адгезионных молекул, таких как VCAM-1, межклеточная адгезионная молекула 1 (ICAM-1), или Е-секреции в эндотелиальных клетках и других типах клеток.
Сверхпродуцирование TNF, однако, вовлечено также в патогенез различных заболеваний, таких как ревматоидный артрит, инсулинзависимый диабет и воспалительная болезнь кишечника, в частности болезнь Крона.
Поэтому TNF-α необходим для запуска иммунологических ответных реакций; однако для того, чтобы избежать воспалительных патологий, следует управлять данным продуцированием. Неактивную синтетическую партию (партия “14”, см. пример 4) переформулировали в соответствии с двумя методами, описанными ниже):
Методы
1. Метод А
Очистка проводилась с помощью препаративной HPLC с обращенной фазой. УФ детекция проводилась при 210 нм. Фракции, содержащие соединения в форме тетрабутиламмониевой соли, собирались и концентрировались с помощью адсорбции на HPLC. Натриевая соль соединения получается с помощью промывки 200 мМ моноосновным раствором фосфата натрия в воде, рН 4,23 + 2-пропанол (9:1, об./об.) (5 объемов). После удаления избытка моноосновного фосфата натрия пропусканием через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюируют раствором воды + 2-пропанол (1:9 об./об.). После разбавления водой и удаления растворителя лиофилизацией получают соединение в виде натриевой соли.
Получаемые соединения затем испытывали с доведением рН (при 7,5) или без него на ТНР-1 клетки для анализа их потенциала индуцировать секрецию TNF-α (см. ниже).
2. Метод В
Очистка проводилась с помощью препаративной HPLC с обращенной фазой. УФ детекция проводилась при 210 нм. Фракции, содержащие соединения в форме тетрабутиламмониевой соли, собирались и концентрировались с помощью адсорбции на HPLC. Натриевая соль соединения получается с помощью промывки 100 мМ раствором двухосновного фосфата натрия - моноосновного фосфата натрия в воде, рН 7,5 + 2-пропанол (9:1, об./об.) (5 объемов) + 2-пропанол (9:1, об./об.) (5 объемов). После удаления избытка моноосновного фосфата натрия - двухосновного фосфата натрия пропусканием через 5 объемов воды + 2-пропанол (9:1 об./об.) соединение элюируют раствором воды + 2-пропанол (1:9 об./об.). После разбавления водой и удаления растворителя лиофилизацией получают соединение в виде натриевой соли.
Получаемые соединения затем испытывали с доведением рН (при 7,5) или без него на ТНР-1 клетки для анализа их потенциала индуцировать секрецию TNF-α (см. ниже).
Культура ТНР-1 клеток:
ТНР-1 клетки (см. метод в примере 1) культивируют (5×105 клеток/мл) в RPMI с 10% FCS + 100 нг/мл PMA (Sigma). Через 3 дня прилипшие клетки собирали и доводили до концентрации 3×105 клеток на лунку и инкубировали с продуктами при 37°С с 5% СО2 в течение 6 часов.
Супернатанты культур собирали через 24 часа и измеряли концентрацию TNF-α с помощью фермент-связываемого иммуносорбентного анализа (ELISA) (BD OptEIA, Сан Диего, США) согласно инструкциям производителя. Пределом детекции было 8 пг/мл.
Результаты:
Результаты представлены ниже в 3 различных таблицах:
Объяснение результатов:
LPS индуцирует, как и ожидалось, высокие уровни TNF-α. Продуцирование TNF-α, индуцируемого двумя биологическими партиями (Р3 и GMP004) ОМ-174 было гораздо ниже.
Объяснение результатов:
Результаты ясно показывают, что применение метода В к партии GMP004 сильно увеличивает активность биологической исходной партии GMP004.
Объяснение результатов:
Продукты, закодированные как партия “39”, получали из синтетического соединения, называемого партией “14”, с помощью процессов, соответствующих методу В. Процесс В явно усиливал активность исходной молекулы. В противоположность этому процедура ультразвуковой обработки оказалась без эффекта (ряд или серия “so).
ПРИМЕР 6:
Влияние синтетического OM-174-DP и синтетического OM-174-МР-PD на модели LACK-индуцируемой астмы, “профилактически” и “терапевтически”
Здесь представлен пример in vivo биологической активности двух характерных представителей соединений изобретения. Используя модель аллергической астмы мышей, опубликованной ранее (описано в работе Julia et al. Immunity. 2002 Feb; 16(2):271-83), авторы имели целью исследовать, будет ли i.p. введение синтетических молекул OM-174-DP и OM-174-МР-PD ингибировать воспаление воздушных путей, спровоцированное у LACK-сенсибилизированных мышей. Для данной цели мышей обрабатывали или наряду с индукцией астмы (профилактическая модель) или терапевтически (т.е. три раза после того, как мышей сделали чувствительными к аллергену, протеин LACK). В качестве результатов считывания подсчитывались эозинофилы в бронхоальвеолярных лаважах (BAL), и определялось количество хорошо известных маркеров аллергической астмы, а именно Th2 цитокинов IL-4, IL-5 и IL-13. Кроме того, регистрировался уровень плазматического IgE.
Протокол:
Материал
- В качестве контроля давали физиологический раствор в виде аэрозоля
- Рекомбинантный LACK протеин продуцировался в E.coli и очищался на Ni-NTA колонке для проведения хроматографии по сходству, как описано (Mougneau et al., 1995).
- Гидроксид алюминия (Alum) закупали у фирмы Pierce
- Цитоцентрифугой является Цитоспин 4 (Thermo-Shandon, Cheschire, U.K.), цитослайды закупали у Thermo-Shandon, а красители Wright и Giemsa у Sigma.
- Аэрозоли давали с использованием ультразвукового распылителя Ультрамед (Medicalia, Forenze, Италия)
- Анти-IgE (R35-118) в сочетании с биотином закупали у BD Biosciences (Le Pont de Claix, Франция)
Животные
- Самок мышей BALB/c ByJ 6-недельного возраста закупали в The Centre d'Elevage Janvier, Франция). Мышей содержали в свободных от специфических патогенов условиях и кормили стандартным кормом, поставляемым фирмой Safe (Augy, Франция).
Экспериментальные группы
Испытывали 5 следуюших групп:
- А: ОТРИЦАТЕЛЬНЫЙ контроль:
Необработанные LACK-сенсибилизированные спровоцированные физраствором мыши (3 мыши)
- В: ПОЛОЖИТЕЛЬНЫЙ Контроль:
Необработанные LACK-сенсибилизированные спровоцированные мыши (6 мышей)
- С: 174-DP Профилактическая:
Обработанные OM-174-DP (i.p.) LACK-сенсибилизированные спровоцированные мыши (6 мышей)
- D: 174-МР- PD Профилактическая:
Обработанные OM-174-МР-PD (i.p.) LACK-сенсибилизированные спровоцированные мыши (6 мышей)
- Е: 174-DP Терапевтическая:
Обработанные OM-174-DP (i.p.) LACK-сенсибилизированные спровоцированные мыши (6 мышей)
Лечение и график испытуемой или контрольной групп
Эксперимент начинали в 0 день. В дни 0, 2, 3, 4, 7, 9, 10, 11 и 12 мышам групп С и D вводили i.p. синтетический OM-174-DP (соединение 1b) и OM-174-МР-PD (соединение 19) соответственно в дозе 1 мг/кг (20 мкг на мышь).
Мышей групп Е лечили терапевтически i.p. в дни 15, 17 и 19 вводили лекарство в дозе 1 мг/кг (20 мкг на мышь).
В день 1 и день 8 мышей сенсибилизировали i.p. LACK/Alum. С 16 по 20 день все группы мышей, за исключением группы А, провоцировали аэрозолями раствора LACK (0,15%). Группа А вместо этого получала физраствор (NaCl 0,9%) (группа А) в течение 40 минут.
Метод
А: Бронхоальвеолярные лаважи (BAL) и подсчет эозинофилов.
Всем животным делали бронхоальвеолярные лаважи BAL (санацию обильным проточным орошением).
Через два дня после последней аэрозольной провокации мышам пускали кровь и в их трахею вставляли канюлю. Легкие промывали 3 раза 1 мл подогретого PBS. Клетки промывали PBS и красные кровяные клетки лизировали с использованием лизисного буфера для красных кровяных клеток. Клетки дополнительно промывали в PBS и производили подсчет. Для дифференциального подсчета BAL клеток приготавливали препараты цитоспина и окрашивали с помощью Wright/Giemsa. Для каждого слайда отсчитывали, по меньшей мере, 400 клеток и с помощью микроскопа определяли число лимфоцитов, нейтрофилов, эозинофилов и макрофагов/DC/пневмоцитов (подсчет производился как и других моноядерных клеток). Здесь сообщается только об эозинофилии.
В: Определение легочных цитокинов:
Для анализа числа легочных цитокинов брали легкие, и для приготовления белковых экстрактов использовали левые легкие. Для каждого левого легкого выделяли 400 мкл. Цитокины (IL-4 и IL-13 в первой серии анализов, а затем IL-5 и IFN-γ) измеряли с помощью мультиплексного анализа с использованием FACSArray. Результаты, нормализуемые на содержание белка, представлены в пг/мл.
С: Определение LACK-специфического IgE:
Для анализа специфического IgE группам мышей А, В и G пускали кровь с помощью сердечной пунктуры через два дня после последнего аэрозоля и приготавливали сыворотки. LACK-специфический IgE измеряли с помощью ELISA.
Результаты:
А) Эозинофилия
Характеристика числа эозинофилов в бронхо-альвеолярных лаважах:
Результаты по каждой отдельной мыши и среднее значение каждой испытанной группы показаны ниже в таблице.
По сравнению с положительной астматической группой (В) животные, которых лечили профилактически соединениями 1b и 19, обнаруживают примерно в 8 раз меньшую BAL эозинофилию.
Кроме того, когда животным вводили три раза с терапевтической целью соединение 1b (группа Е), число эозинофилов у них сильно уменьшалось (на фактор 64).
В: Характеристика легочных цитокинов
В первой серии анализов результаты по каждой отдельной мыши и среднее значение каждой группы показаны ниже в таблице:
IL-4 и IL-13 (соединения 1b и 19)
Количества IL-4 и IL-13 сначала анализировали в легких обработанных и необработанных мышей.
Тогда как содержание IL-4 было очень низким до необнаруживаемого у PBS-спровоцированных животных, количество IL-4 у LACK-провоцируемых необработанных (не подвергнутых лечению) контрольных мышей увеличивалось 20-кратно. После профилактического лечения с помощью OM-174-DP (1b) OM-174-МР-PD (19) количество IL-4 в легких уменьшалось в 6 и 4 раза, соответственно (р<0,001 Mann & Whitney) (см. таблицу ниже). Следует заметить, что аналогичное уменьшение количества IL-4 (4-кратное по сравнению с группой В) получалось при терапевтическом лечении синтетической молекулой 1b (группа Е).
PBS
LACK
У PBS-провоцируемых мышей показатель IL-13 был в пределах детекции, но у астматических контрольных мышей определялось количество в виде среднего 50 пг/мл (см. таблицу ниже, группа В). По сравнению с данными астматическими мышами количество IL-13 было в 4 раза ниже у мышей, профилактически обработанных OM-174-DP (соединение 1) (р<0,001 Mann & Whitney), и в 3 раза ниже у мышей, обработанных OM-174-МР-PD (соединение 19) (р<0,001) (см. таблицу ниже).
Следует заметить, что аналогичное уменьшение количества IL-13 (3-кратное по сравнению с группой В) получалось при терапевтическом лечении синтетической молекулой 1b (группа Е).
PBS
LACK
Дополнительные цитокины, измеренные после трех терапевтических введений соединения 1b
Поскольку после терапевтического лечения испытуемыми молекулами эозинофилия воздушных путей резко снижалась, авторы стремились в еще одной серии анализов проверить в содержимом легких дополнительные цитокины (IL-5 и IFN-γ) (только для соединения 1b). Результаты показаны в таблице ниже.
В действительности, при сравнении с легкими не подверженных лечению мышей в легких мышей, подверженных лечению, количества Th2-цитокина: IL-5, сильно снижались. После лечения OM-174-DP (соединением 1b, р<0,01 Mann & Whitney) количества IL-5 снижались в 2,8 раз.
PBS
LACK
Ясно, что по сравнению с группой В, при терапевтическом лечении синтетической молекулой 1b (группа Е) получалось снижение IL-5 цитокина, о котором известно, что он активирует эозинофилы. Данное снижение не было следствием сдвига в сторону TH1 цитокина, поскольку у всех мышей по сравнению с животными группы В уровни IFN-γ не увеличивались (1,5-3 пг/мл) (при лечении мышей соединением 1b уровни IFN-γ даже снижались в два раза, см. таблицу ниже):
PBS
LACK
С: Количественное определение сывороточного аллерген-специфического IgE
Для того, чтобы дополнительно охарактеризовать иммунный статус мышей после терапевтического лечения OM-174-DP (соединением 1b), авторы проанализировали LACK-специфический IgE в сыворотке обработанных мышей и у необработанных мышей по методу ELISA. Результаты по каждой отдельной мыши и средние значения каждой испытанной группы показаны в таблице ниже (терапевтический эксперимент с соединением 1b).
PBS
LACK
В то время как уровни LACK-специфического IgE увеличивались 7-кратно после экспонирования действию LACK аэрозолей, сыворотка мышей, обработанных OM-174-DP, содержала в 2,6 раза меньше LACK-специфического IgE по сравнению с необработанными LACK-провоцируемыми мышами (р<0,05; Mann & Whitney).
Заключение:
В первой части настоящего исследования соединения (1b) и (19) исследовали на их действие при превентивном введении. Известно, что системное введение двух данных синтетических соединений оказывало значительное отрицательное действие на развитие BAL эозинофилии и значительно уменьшали количество IL-4 и IL-13 цитокинов в легких.
Кроме того, здесь показано, что синтетические соединения изобретения обладают также терапевтическим антиастматическим потенциалом, что иллюстрируется результатами, полученными с соединением 1b (сильно и значительно уменьшает аллерген-индуцируемую BAL эозинофилию, легочный IL-4, IL-5 и IL-13, и снижает уровни IgE).
Суммируя вышесказанное, результаты, представленные в примере 6, дают ясный пример того, что соединения изобретения могли бы быть клинически испытаны превентивно или терапевтически против астмы.
ПРИМЕР 7:
Влияние OM-174-DP (соединения 16), OM-174-МР-PD (соединения 19), ОМ-174-МР-ЕР (соединения 33), ОМ-174-МР-СМ (соединения 35с) и ОМ-174-МР-PR (соединения 17) в in vitro анализе на модели, стимулируемой соединением 48/80 секреции гистамина.
С использованием in vitro модели дегрануляции тучных клеток крысы (предлагаемой CRO CEREP, номер по каталогу 2006: 771-с) целью авторов было исследование, будут ли синтетические молекулы OM-174-DP (соединением 1b), OM-174-МР-PD (соединением 19), ОМ-174-МР-ЕР (33), ОМ-174-МР-СМ (35с) и ОМ-174-МР-PR (17) ингибировать секрецию гистамина стимулируемыми соединением 48/80 мастоцитами. Используемый протокол анализа вкратце суммирован ниже:
Протокол
Общая процедура
Экспериментальные условия
Анализ и выражение результатов
Результаты выражены в виде процента специфической активности контроля ((измеренная спец. активность/спец. активность контроля)×100), полученной в присутствии испытуемых соединений.
Величины IC50 (концентрация, вызывающая полумаксимальное ингибирование специфической активности контроля) определялись с помощью анализа нелинейной регрессии кривых ингибирования, получаемых со средними значениями повторных опытов с использованием соответствия кривой уравнения Hill (Y=D+[(A-D)/(1+(C/C50nH)], где Y = специфическая активность, D = минимальная специфическая активность, A = максимальная специфическая активность, C = концентрация соединения, C50 = IC50, и nH = фактор наклона). Данный анализ выполнялся с использованием программного обеспечения, разработанного Cerep (программное обеспечение Hill) и подтверждался сравнением с данными, получаемыми с помощью коммерческой программы SigmaPlot ® 4.0 Windows ® (© 1997 SPSS Inc.).
Испытуемые соединения
Ссылочное соединение
В каждом эксперименте параллельно с испытуемыми соединениями испытывали ссылочные соединения для того, чтобы оценить пригодность оценки. Их испытывали в нескольких концентрациях (для определения ИК50 (IC50)), и данные сравнивались с историческими значениями, определенными CEREP. Анализ считался действительным, если критерии пригодности удовлетворялись согласно соответствующей стандартной процедуре оперирования.
Результаты:
Величины ИК50, определенные для испытуемых соединений и ссылочных (5 различных испытаний) указаны в таблице ниже. Величины ИК50 для ссылочного соединения находятся в принятых пределах исторического среднего значения, полученного на CEREP.
Заключение :
Несомненно, испытанные лекарственные средства являются потенциальными ингибиторами секреции гистамина, индуцируемой стимулируемыми соединением 48/80 тучными клетками. Они являются более активными, чем испытанное ссылочное: SGS.
ПРИМЕР 8:
Влияние OM-174-МР-PD (соединения 19), ОМ-174-МР-ЕР (соединения 33), OM-174-MP (соединения 16) и ОМ-174-МР-PR (соединения 17) на in vitro моделях клеток, экспрессирующих toll-подобные рецепторы человека (TLR)
Химическая структура и происхождение продуктов изобретения (OM-174-DP первоначально было получено из разложившегося LPS из E.coli) могут дать основания полагать, что лекарственные средства могут быть активны посредством TLR рецепторов, более конкретно посредством TLR4 и TLR2. TLR рецепторы экспрессируются главным образом (но не исключительно) иммунными клетками, такими как моноциты, макрофаги, дендритные клетки, Т-клетки и др., и являются ключевыми сенсорами микробных продуктов, которые могут распознаваться хозяином как сигналы опасности. Даже хотя они запускают сначала неспецифичный врожденный иммунитет, активация TLR будет инициировать полный иммунологический каскад, что приведет в результате, в присутствии антигенов, к развитию приобретенного иммунитета.
Клетки, которые существенно экспрессируют обусловленный функциональный TLR ген, являются ценными средствами для многих областей применения, таких как изучение механизма, вовлеченного в TLR распознавание или сигнализацию, и разработка новых потенциальных лекарственных средств. Поэтому целью эксперимента, описанного ниже, является испытание 4 соединений изобретения на данные ключевые адапторы иммунного ответа.
Ответные реакции испытывались на следующих клеточных системах:
a) THP1 синий (лекция об оптической плотности при 625 нм через 48 часов)
b) HEK-TLR2 (IL-8 ELISA через 24 часа)
c) HEK-TLR2-CD14 (IL-8 ELISA через 24 часа)
d) HEK-MD2-TLR4-CD14 (IL-8 ELISA через 24 часа)
а) ТНР-1 Синий
Первая серия эксперимента проводилась на ТНР-1 клетках, которые естественно экспрессируют и TLR2 и TLR4.
ТНР-1 клетки являются моноцитными клетками периферической крови человека. Моноциты играют ключевую роль в природном иммунитете и экспрессируют большинство TLR на различных уровнях. Что касается первичных клеток, ТНР-1 клетки активируют NF-κB и другие факторы транскрипции в ответ на TLR лиганды. В противоположность НЕК293 клеткам, которые были инженерно разработаны для ответа на специфичные TLR агонисты (см. ниже). ТНР-1 клетки естественно экспрессируют TLR гены и все гены, вовлекаемые в сигнальный каскад.
Для облегчения анализа TLR ответа в моноцитах InvivoGen (Тулуза, Франция) предоставила ТНР-1 клоны, устойчиво трансфектируемые NF-κB-индуцируемой репортерной системой, называемой THPI-Blue™. ТНР-1 Синие клетки устойчиво трансфектировались репортерной плазмидой, экспрессирующей секретируемый эмбрионный ген щелочной фосфатазы (SEAP) под контролем промотора, индуцируемого несколькими факторами транскрипции, такими как NF-κB и АР-1. После стимуляции TLR клетки ТНР1- Blue™ активируют факторы транскрипции и впоследствии секреция SEAP, которая легко обнаруживается с использованием QUANTI-Blue\u2122 среды, которая становится пурпурно/синей (голубой) в присутствии SEAP.
Клетки стимулировали контролями и соединениями изобретения согласно инструкциям производителя.
Результаты: увеличенный OD при 625 нм спустя 48 часов:
Результаты по контролям (отрицательный = LPS K12CD25 ультрачистый, TLR4 агонист; и PAM3CSK4 = положительный, TLR2 агонист) даны ниже в таблице. Результаты (выражаемые в виде OD произвольных единиц) показывают средние значения (от двукратных измерений) оптической плотности, считанные при 625 нм, через 48 часов после стимуляции при 37°С контролями (до 1000 нг/мл):
Контроли:
Клеточная линия отвечает четко и на TLR2, и на TLR4 агонисты, PAM3CSK4 и LPS K12CD25 соответственно ультрачистые.
Затем в данном анализе испытывались следующие четыре соединения изобретения OM-174-МР-PD (соединение 19), ОМ-174-МР-ЕР (соединение 33), OM-174-MP (соединение 16) и ОМ-174-МР-PR (соединение 17).
Результаты (среднее от величин OD двух повторов при 625 нм через 48 часов) показаны ниже:
Соединения:
доза (нг/мл)
Соединения (19) и (17) являются хорошими активаторами ТНР-1 клеток, тогда как активность соединений (16) и (33) слабее.
Так как соединения изобретения являются активными на системе, которая экспрессирует и TLR2, и TLR4, авторы даже проверили их активность НЕК клеток, экспрессирующих или только TLR2, или только TLR4, но не одновременно оба TLR.
b) HEK-TLR2
НЕК293 линия клеток была выбрана по ее невыразительной или низкой базальной экспрессии TLR генов. Данные клетки делают возможным эффективный контроль активности TLR с использованием анализа ELISA, такого как IL-8 титрование и системы на основе репортера, которые контролируют индуцируемую TLR NF-κB активацию.
НЕК-TLR2 клетки (Invivogen, Тулуза, Франция) являются инженерно разработанными НЕК293 клетками, устойчиво трансфектируемыми множественными генами из TLR2, которые включают TLR2 и гены, участвующие в распознавании или вовлеченные в сигнальный каскад. Данные клетки секретируют IL-8 после стимуляции TLR2. Эксперименты проводились по инструкциям производителей.
Вкратце, 2×104 клеток/лунка (200 мкл RPMI) инкубируют при 37°С в течение 3 дней (5% СО2). Среду удаляют, и в лунки добавляют 90 мкл RPMI+5%FCS. Затем добавляют агонисты и контроли (10 мкл/лунка). Клетки возвращают в инкубатор на 24 часа. Собирают супернатанты и выполняют IL-8 ELISA по инструкциям производителя.
Результаты: секреция IL-8
Результаты по контролям (отрицательный = LPS K12CD25 ультрачистый, TLR4 агонист; и PAM3CSK4 = положительный, TLR2 агонист) даны ниже в таблице. Результаты (выражаемые в пг/мл IL-8) показывают средние значения (от двукратных измерений) секреции IL-8 через 24 часа после стимуляции контролями (до 1000 нг/мл):
Контроли:
Линия клеток явно отвечает только на TLR2 агонист PAM3CSK4.
Затем в данном анализе испытывали следующие четыре соединения изобретения OM-174-МР-PD (соединение 19), ОМ-174-МР-ЕР (соединение 33), OM-174-MP (соединение 16) и ОМ-174-МР-PR (соединение 17).
Результаты (среднее от величин секретируемого IL-8 двух повторов в пг/мл через 24 часа) показаны ниже:
Соединения:
доза (нг/мл)
Соединения 33 и 17 явно способны активировать секрецию IL-8 посредством TLR2.
с) НЕК-TLR2-CD14
Данные предварительные результаты подтверждались на еще одной клеточной линии экспрессирующими одновременно TLR2 и CD14. Используемая процедура аналогична процедуре, описанной только что выше.
Результаты по контролям и 4 испытанным соединениям показаны в 2 таблицах ниже:
Результаты секреции IL-8
Результаты по контролям (отрицательный = LPS K12CD25 ультрачистый, TLR4 агонист; и PAM3CSK4 = положительный, TLR2 агонист) даны ниже в таблице. Результаты (выражаемые в пг/мл IL-8) показывают средние значения (от двукратных измерений) секреции IL-8 через 24 часа после стимуляции контролями (до 1000 нг/мл):
Контроли:
Как и в случае НЕК-TLR2 линии клеток, НЕК-TLR2-CD14 клетки явно также отвечают только на TLR2 агонист PAM3CSK4.
Соединения:
Затем в данном НЕК-TLR2-CD14 анализе испытывались следующие четыре соединения изобретения:
OM-174-МР-PD (соединение 19), ОМ-174-МР-ЕР (соединение 33), OM-174-MP (соединение 16) и ОМ-174-МР-PR (соединение 17).
Результаты (среднее от величин двух повторов IL-8, секретируемого в пг/мл через 24 часа) показаны ниже:
доза (нг/мл)
Результаты подтверждают результаты, полученные с линией клеток НЕК-TLR2: Соединения 33 и 17 способны активировать секрецию IL-8 посредством TLR2.
d)НЕК-MD2-TLR4-CD14
TLR4 широко изучается, так как он является главным рецептором, вовлекаемым в распознавание липополисахарида (LPS), ответственного за септический шок.
НЕК-MD2-TLR4-CD14 являются высоко чувствительными к LPS. Их получали устойчивой трансфекцией НЕК293 клеток генами TLR4, MD2 и CD14 и NF-κB-индуцируемой репортерной системой. Они секретируют IL-8.
Авторы использовали одну и ту же экспериментальную процедуру, что и для других НЕК клеточных линий, описанную выше.
Результаты по контролям и 4 испытанным соединениям показаны ниже в 2 таблицах:
Результаты: Секреция IL-8
Результаты по контролям (положительный = LPS K12CD25 ультрачистый, TLR4 агонист; и PAM3CSK4 = отрицательный, TLR2 агонист) даны ниже в таблице. Результаты (выражаемые в виде пг/мл IL-8) показывают средние значения (от двукратных измерений) секреции IL-8, через 24 часа после стимуляции контролями (до 1000 нг/мл):
Контроли:
Как и ожидалось, клеточная линия НЕК-MD2-TLR4-CD14 явно отвечает только на TLR4 положительный контрольный ультрачистый LPS-К12, но не на отрицательный TLR2 в отличие от PAM3CSK4.
Соединения:
Затем в данном анализе НЕК-MD2-TLR4-CD14 испытывались следующие четыре соединения изобретения:
OM-174-МР-PD (соединение 19), ОМ-174-МР-ЕР (соединение 33), OM-174-MP (соединение 16) и ОМ-174-МР-PR (соединение 17).
Результаты (среднее от величин секретируемого IL-8 двух повторов в пг/мл через 24 часа) показаны ниже:
Соединения:
доза (нг/мл)
Явно, что в данном TLR4 анализе ни одно из испытанных соединений не является активным.
Общие выводы по данным TLR анализам:
Очень интересно, полученные результаты показывают, что соединения изобретения являются активными на человеческих клетках, экспрессирующих преимущественно TLR2 рецептор человека.
Список литературы


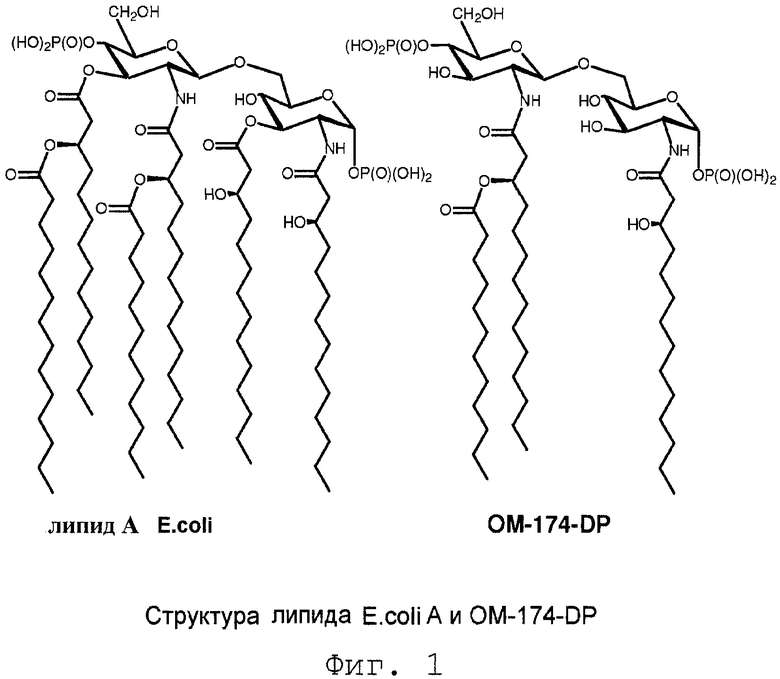
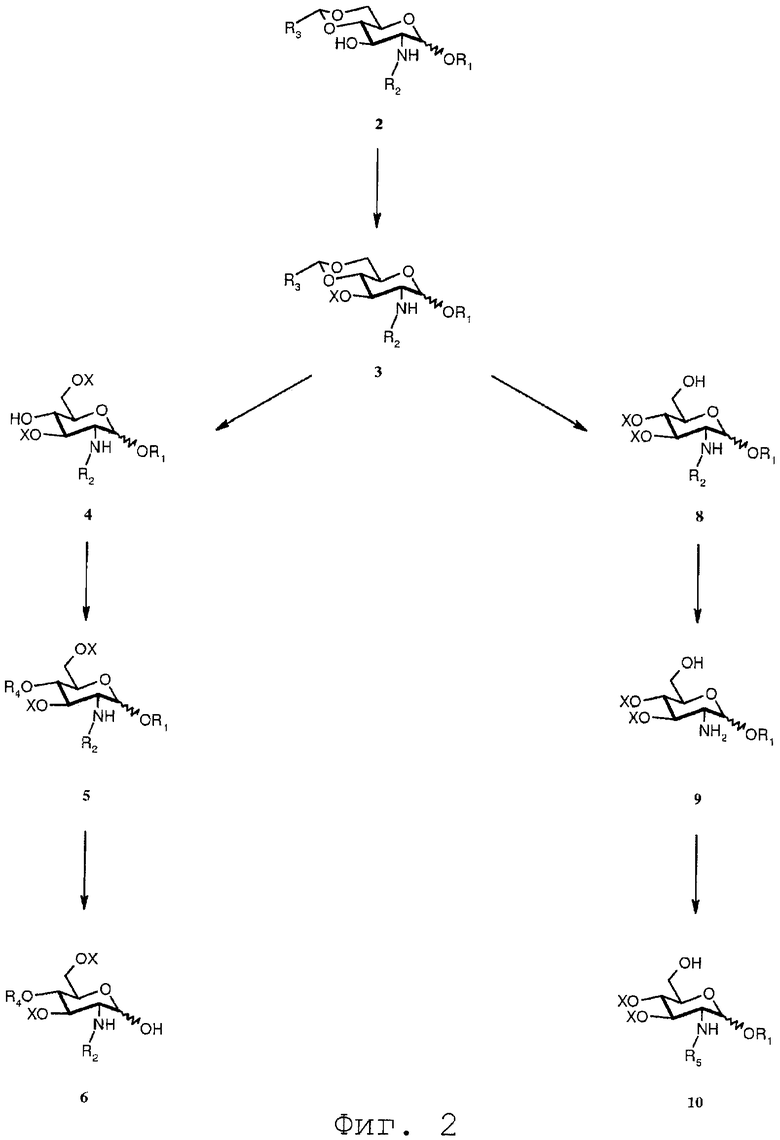
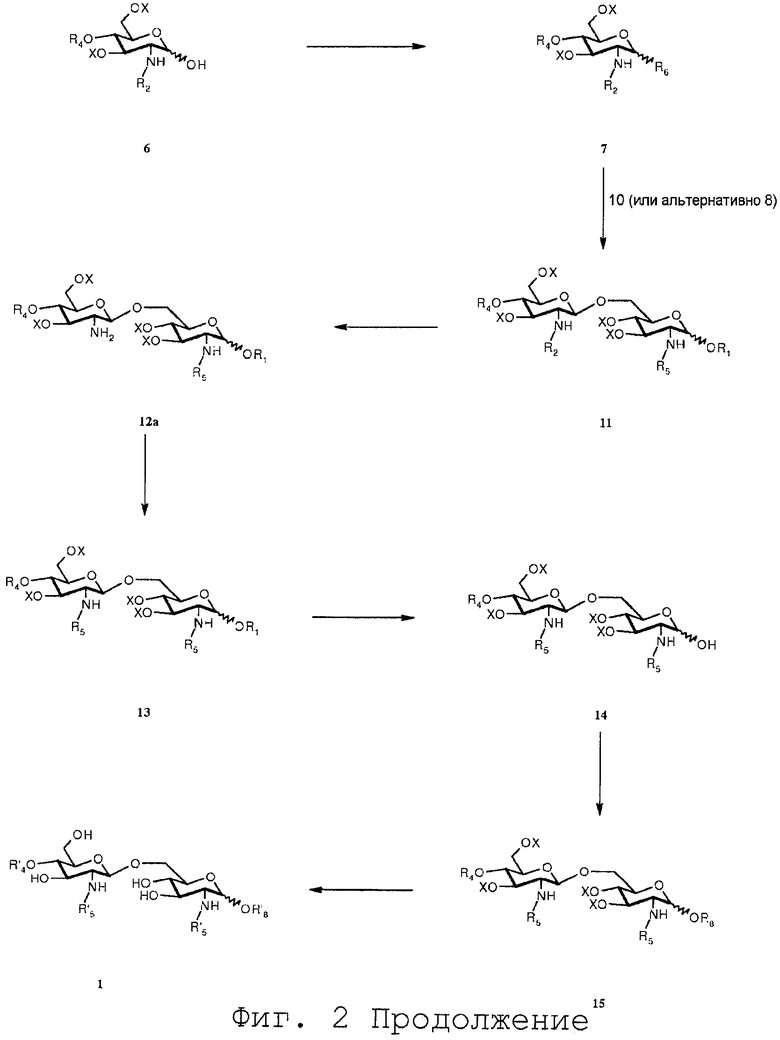
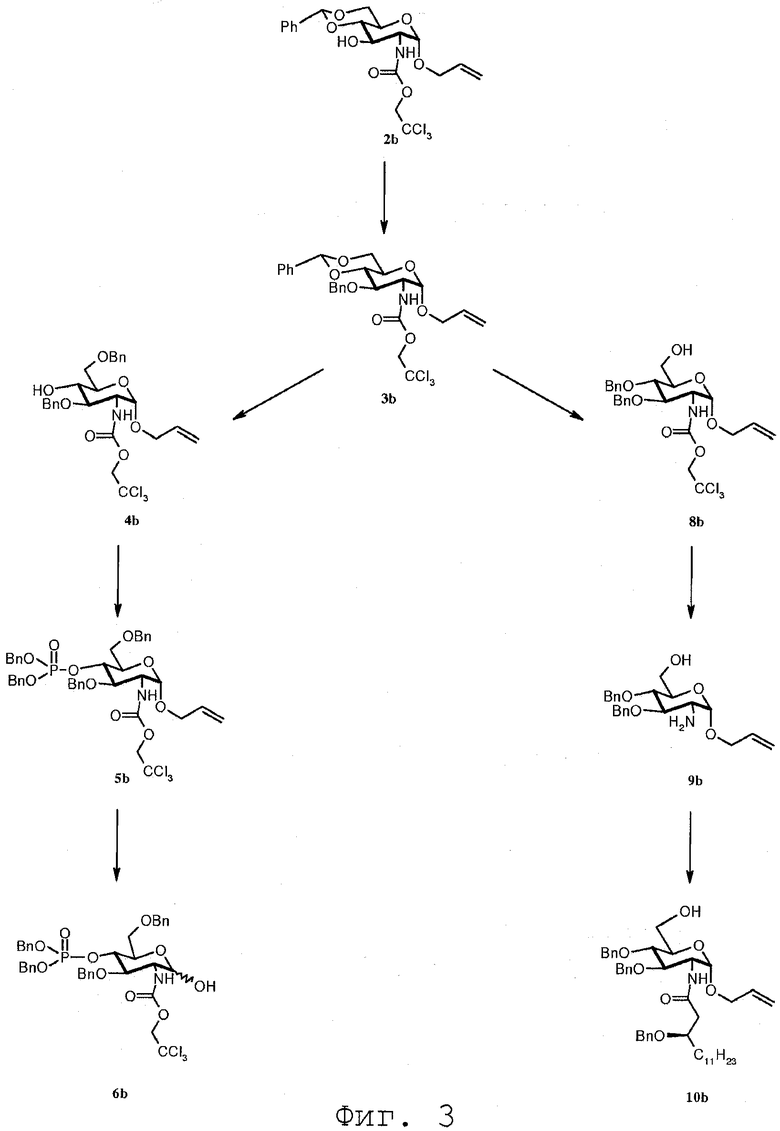
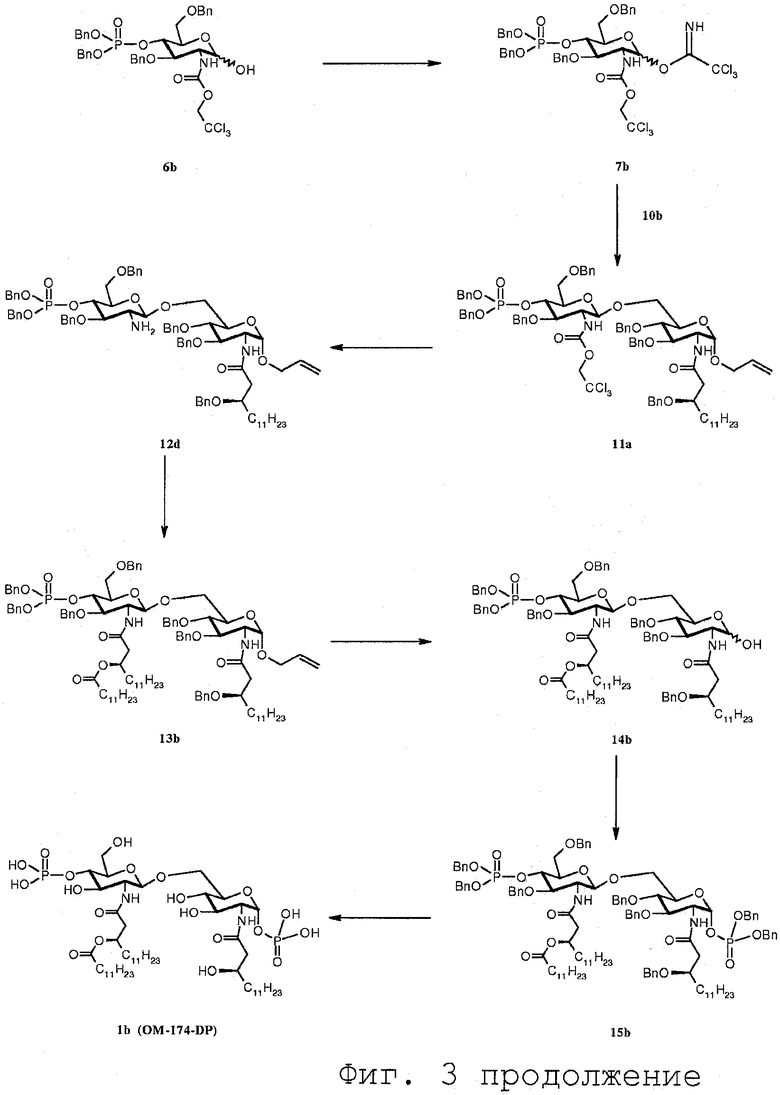
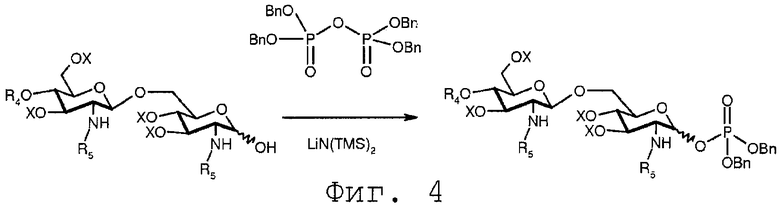
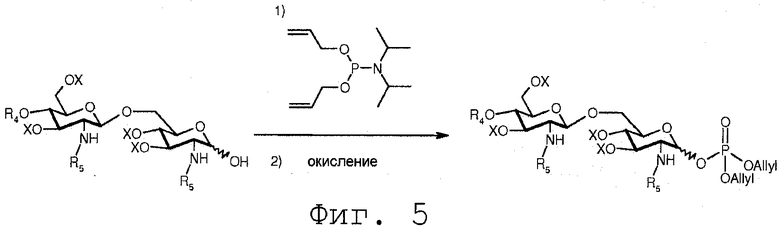
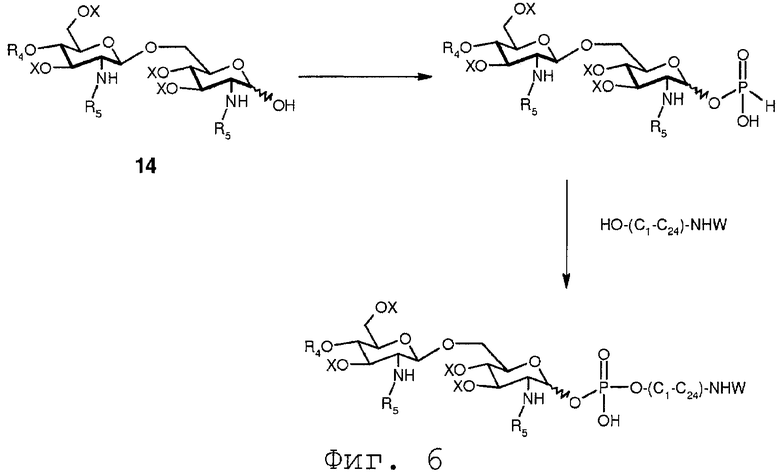
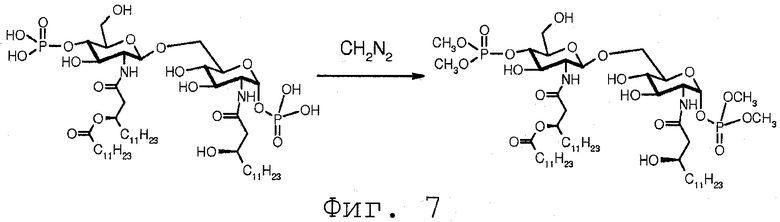
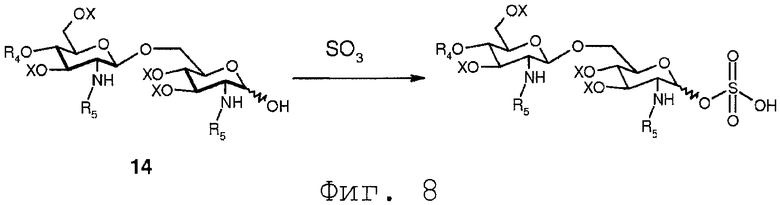
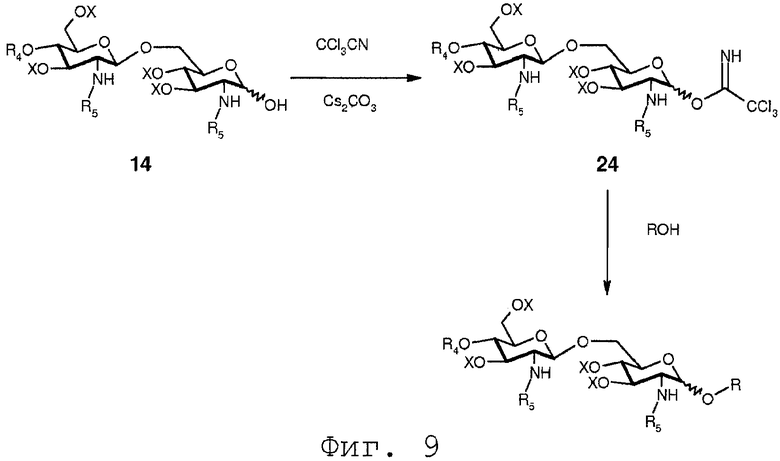
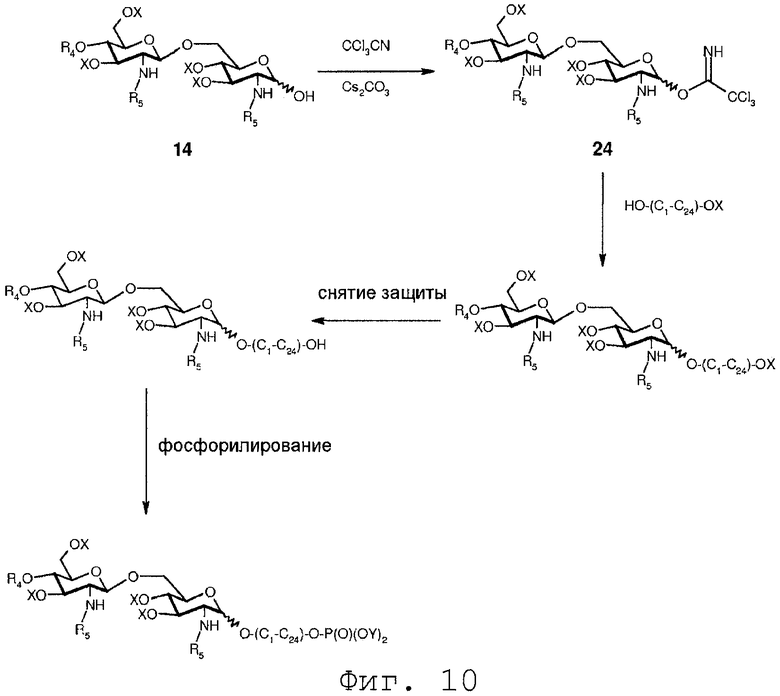
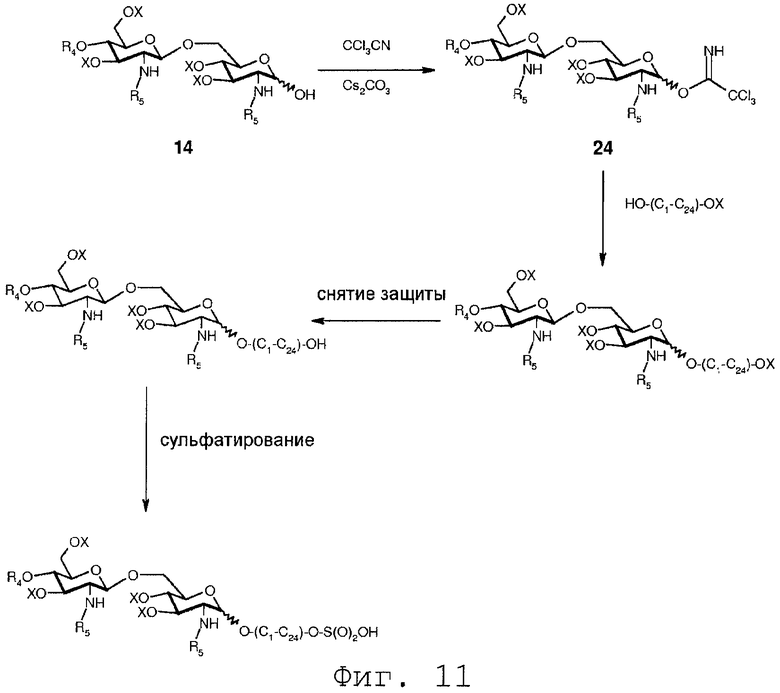

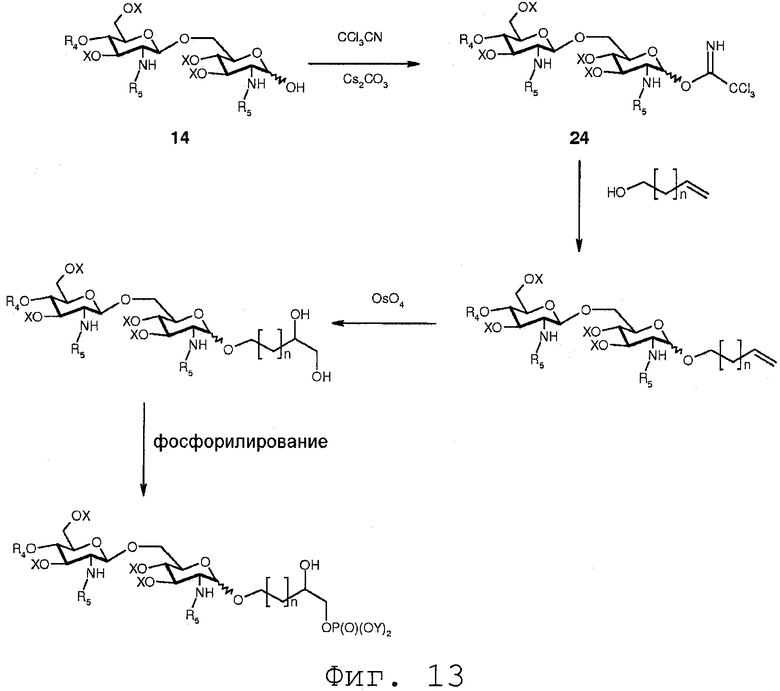
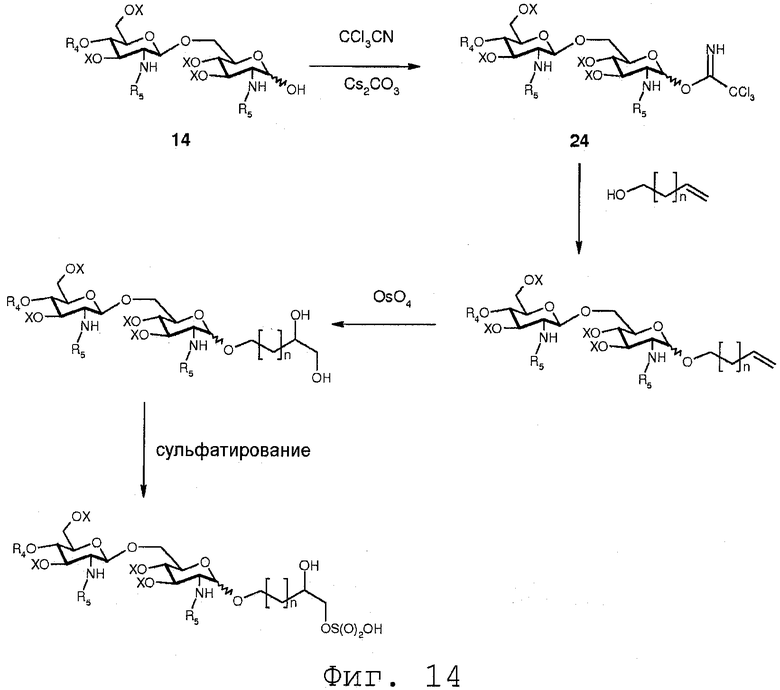
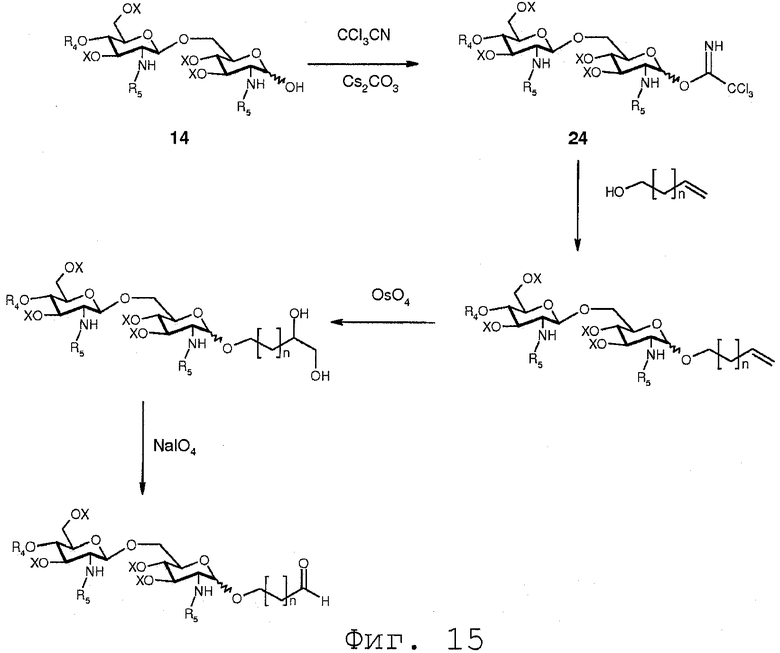
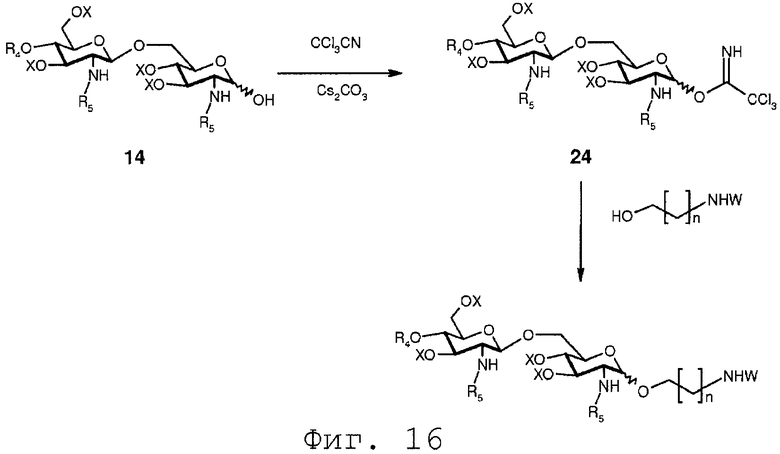
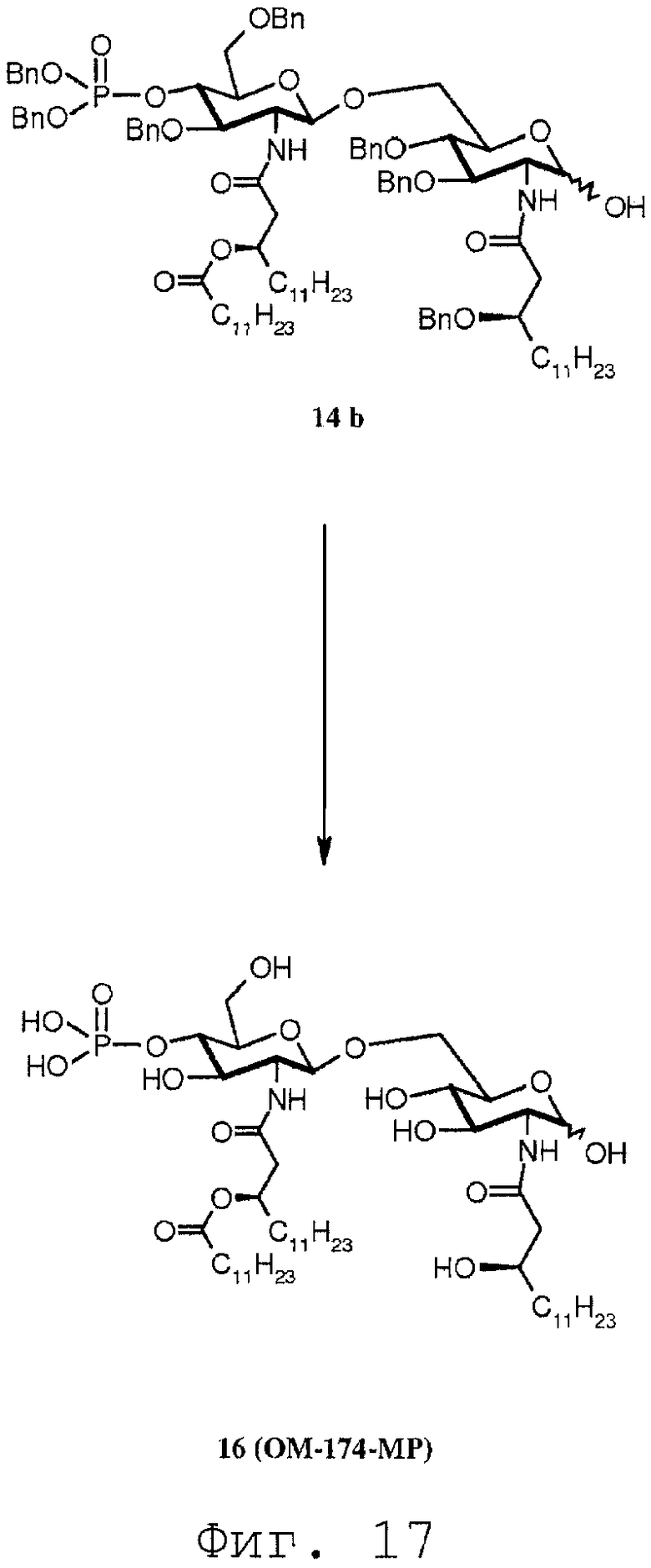
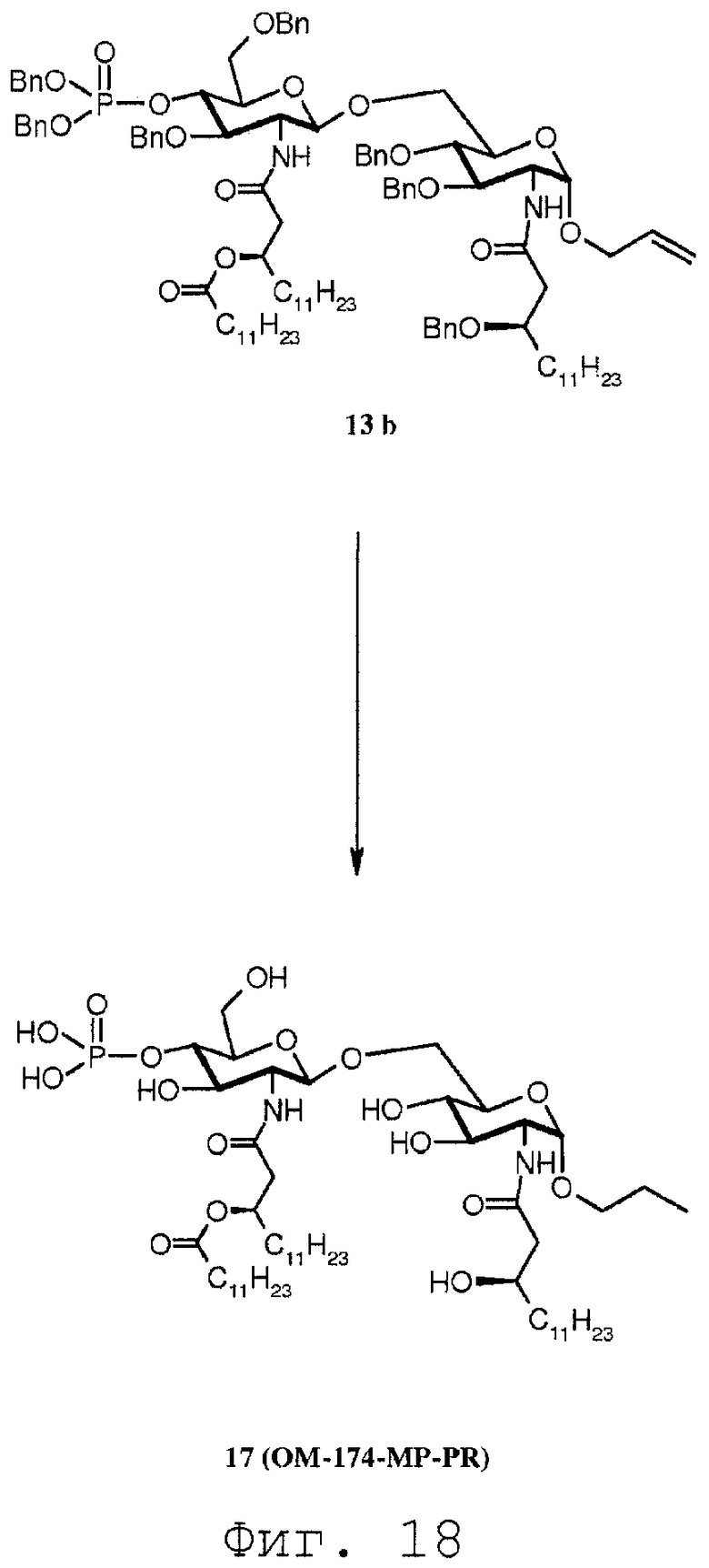
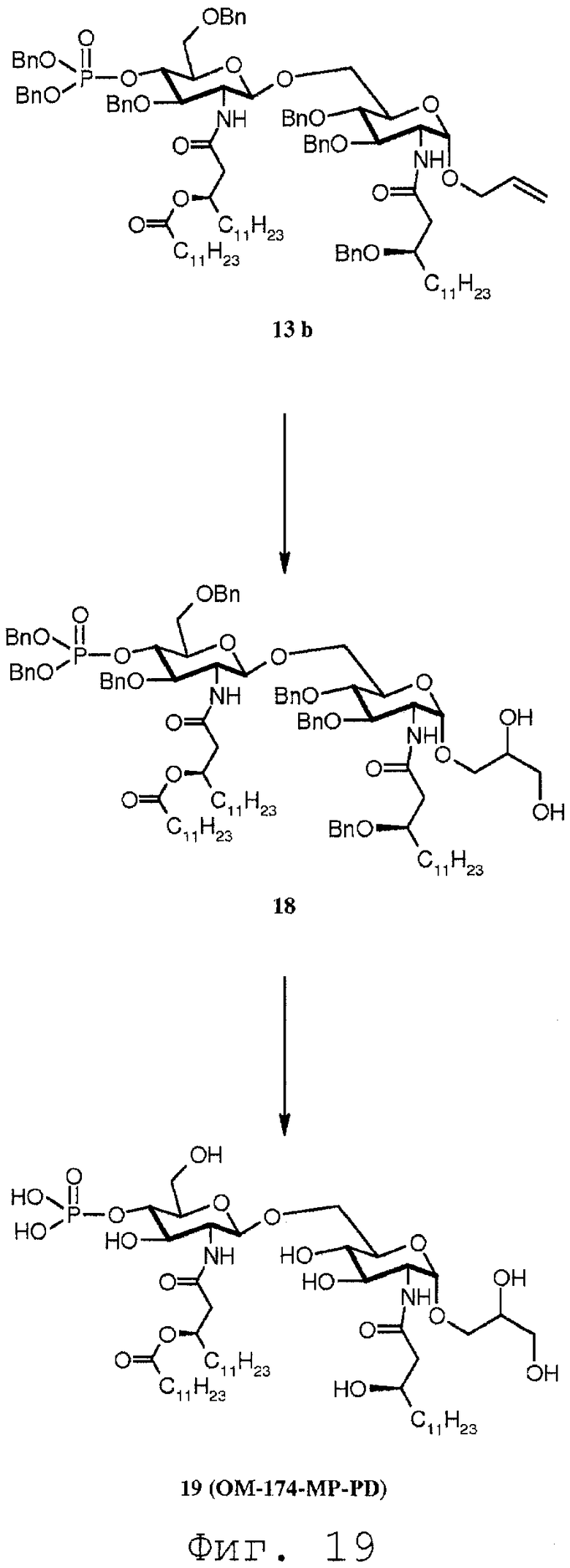
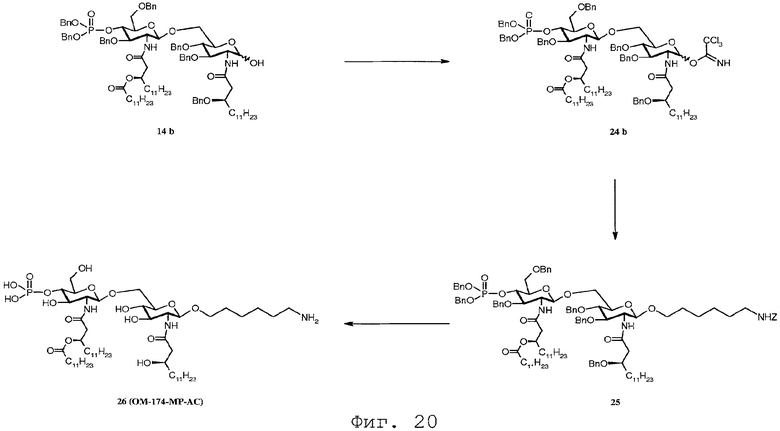
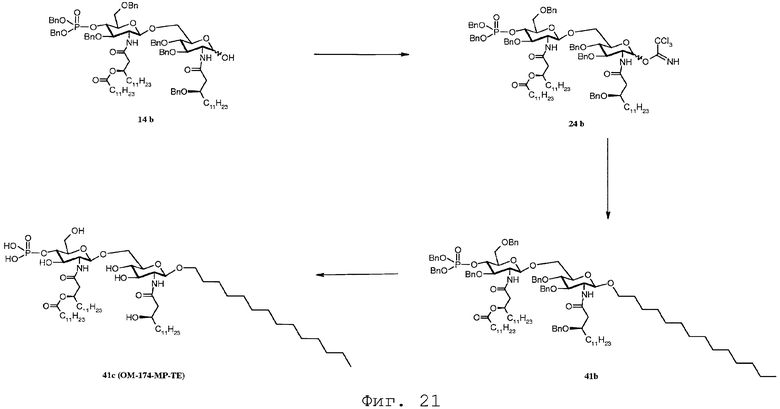
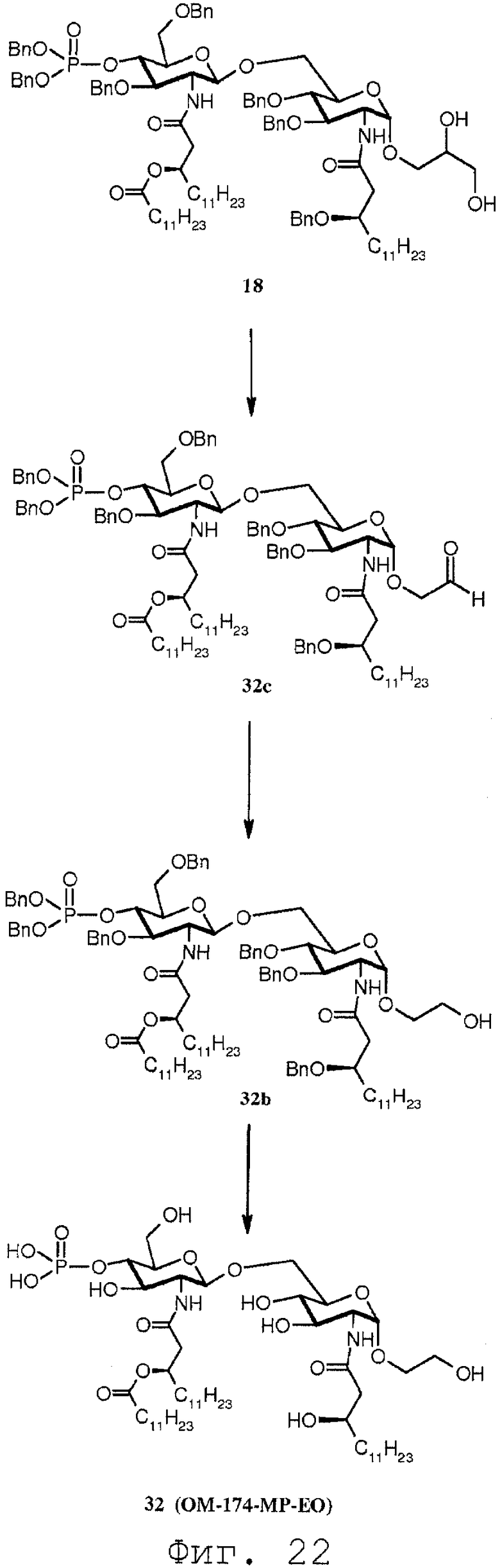
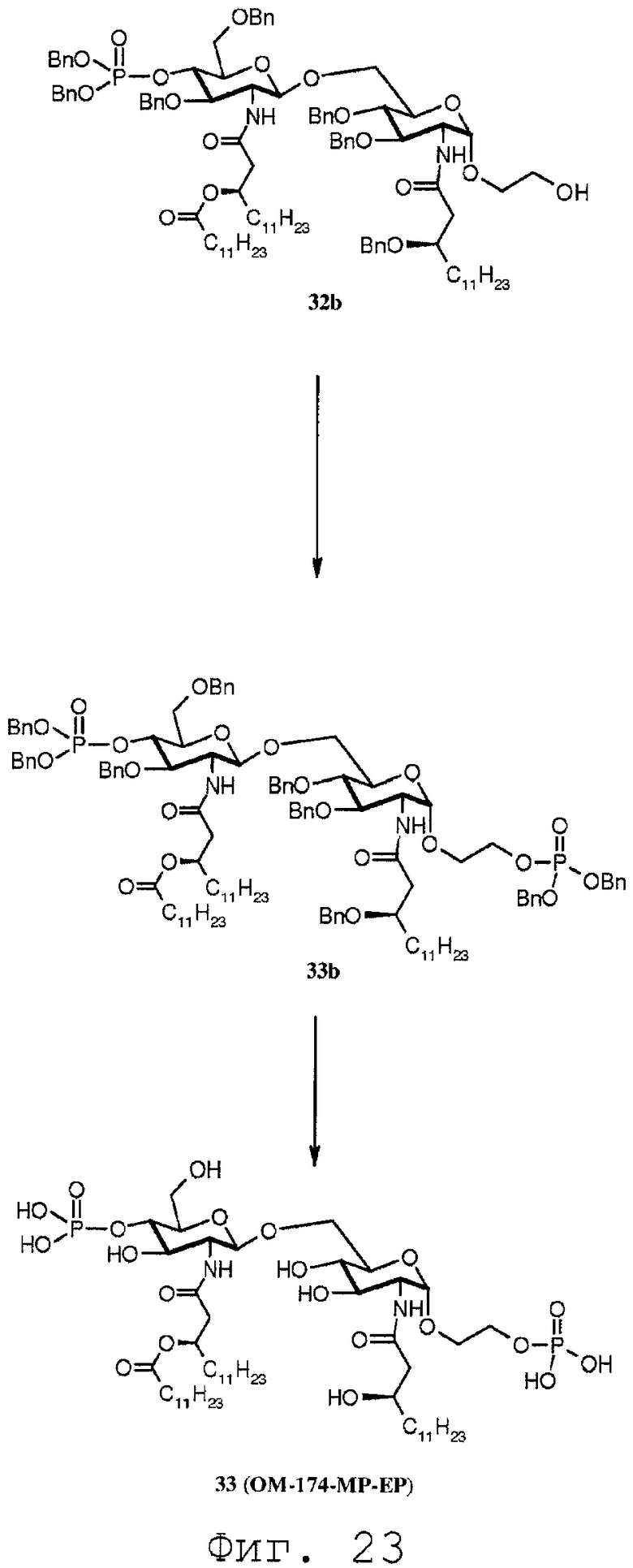
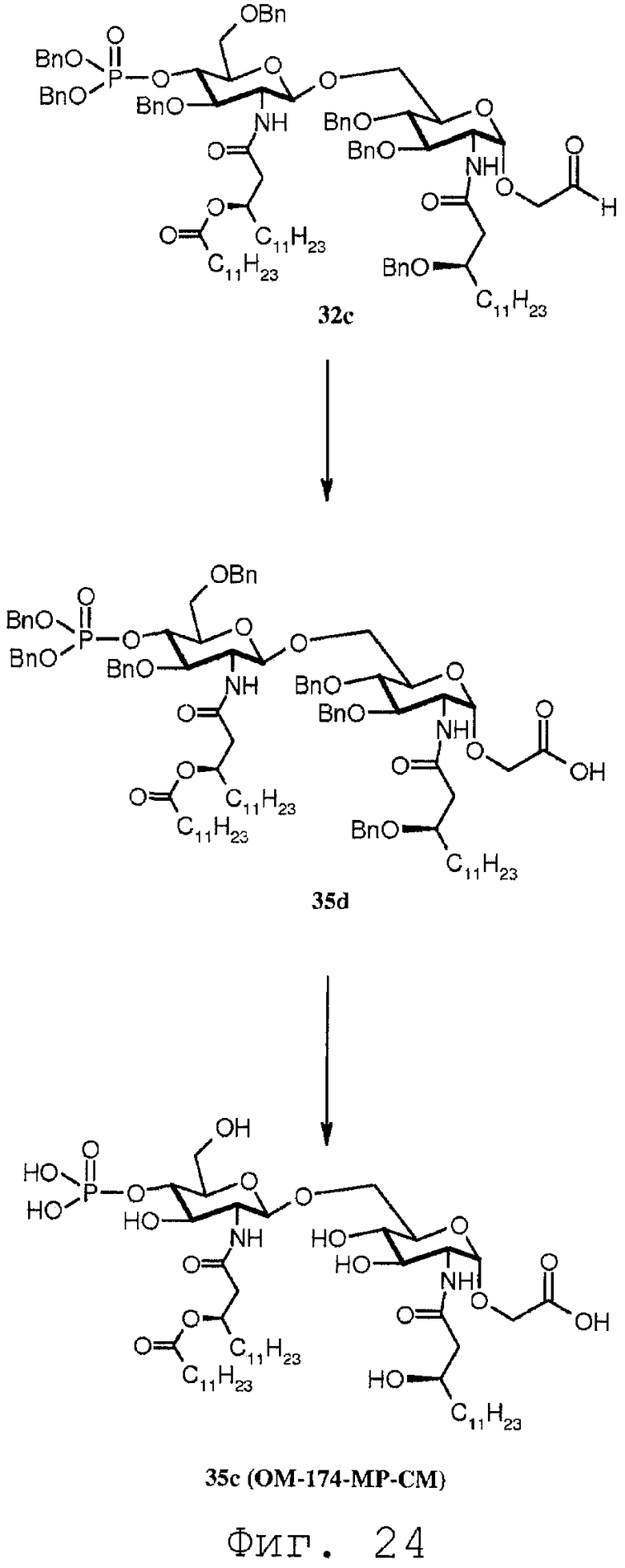
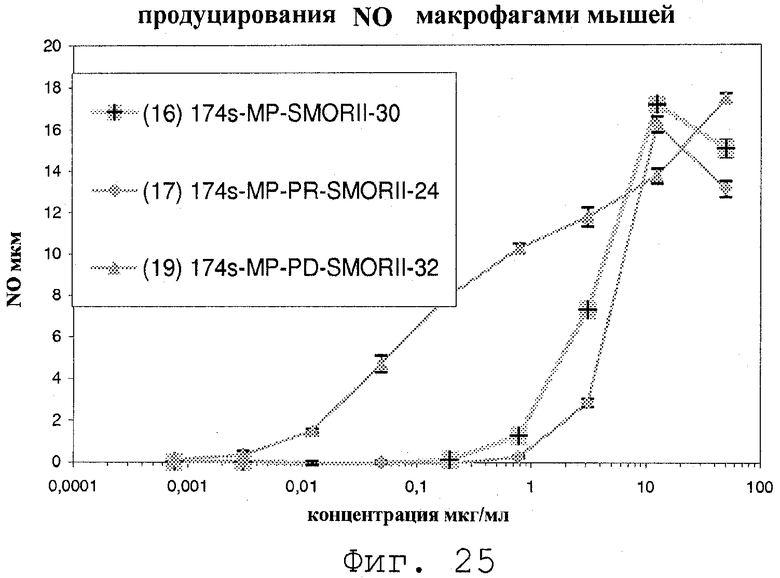

Настоящее изобретение относится к новому способу химического синтеза асимметрично или симметрично замещенного β-(1→6)-связанного глюкозамин-дисахарида формулы (1), а также к способу его очистки. В изобретении заявляются промежуточные соединения, относящиеся к данному способу. Согласно дополнительным аспектам изобретение относится к фармацевтической композиции, включающей указанные соединения, и к применению соединений при лечении расстройства, на которое влияет модулирование активности иммунной системы, включая подавление или активацию иммунной системы, такого как расстройство, выбранное из иммунных расстройств и/или рака. 14 н. и 12 з.п. ф-лы, 8 пр., 26 ил., 32 табл.
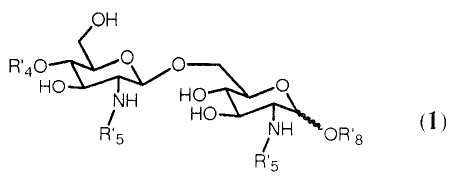
1. Способ получения асимметрично или симметрично замещенного β-(1→6)-связанного глюкозамин-дисахарида, включающий реакцию соединения формулы 10:
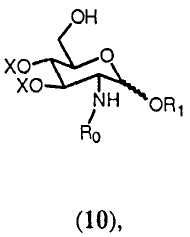
в которой R1 представляет собой группу, выбранную из (С3-С6)алкенила, такого как С3 или С4алкенил;
X представляет водород, группу, выбранную из бензила или замещенного бензила, выбранного из группы, включающей 4-метоксибензил, 3,4-диметоксибензил, 2,5-диметоксибензил, 2,3,4-триметоксибензил или 3,4,5-триметоксибензил;
R0 выбран из R5 или R2, где R5 выбран из:
(i) ацильной группы, происходящей из карбоновой кислоты с линейной цепью, имеющей от 2 до 24 атомов углерода;
(ii) ацилоксиацильной группы, ациламиноацильной группы, ацилтиоацильной группы;
(iii) алкилоксиацильной группы, алкенилоксиацильной группы, алкинилоксиацильной группы, алкиламиноацильной группы, алкениламиноацильной группы, алкиниламиноацильной группы, алкилтиоацильной группы, алкенилтиоацильной группы, алкинилтиоацильной группы, ацильной группы, происходящей из карбоновой кислоты с разветвленной цепью, имеющей от 2 до 48 атомов углерода;
где в группах (i), (ii), (iii) углеводородная цепь ацила может быть насыщенной или ненасыщенной и углеводородная цепь ацила, алкила, алкенила, алкинила может быть разветвленной или линейной и необязательно может быть замещена одной или более группами, независимо выбранными из галогена, такого как фтор, хлор, бром или йод; гидроксила или гидроксильного производного -OY, где Y имеет значения, определенные ниже; амина или производного амина -NHW, где W имеет значения, определенные ниже; группы -OZ, где Z выбран из (f), (g), (h), (i), (j), (k), определенных ниже;
и R2 представляет группу, выбранную из (С1-С6)галогенированного алкоксикарбонила, такого как 2,2,2-трихлорэтоксикарбонил (TROC) или 1,1 -диметил-2,2,2-трихлорэтоксикарбонил (ТСВОС);
с соединением формулы 7
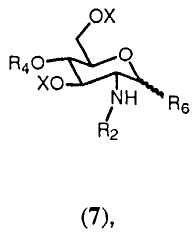
где R4 выбран из
(a) ацильной группы, определенной в (i), (ii) или (iii) для R5;
(b) разветвленной или линейной алкильной группы; разветвленной или линейной алкенильной группы; разветвленной или линейной алкинильной группы;
(c) группы -[(С1-С24)алкил]-СООХ, -[(С2-С24)алкенил] -СООХ или - [(С2-С24)алкинил]-СООХ, где X имеет значения, определенные ниже;
(d) группы -[(С1-С24) алкил]-NHW, -[(С1-С24)алкенил]-NHW или -[(С1-С24)алкинил]-NHW, где W имеет значения, определенные ниже;
(e) формилалкильной группы; формилалкенильной группы; формилалкинильной группы;
(f) диметоксифосфорильной группы;
(g) группы -P(O)(OY)2, где Y имеет значения, определенные ниже;
(h) группы -Р(О)(ОН)-О[(С1-С24)алкил]NHW, -Р(О)(ОН)-О[(С1-С24)алкенил]NHW или -Р(О)(ОН)-О[(С1-С24)алкинил]NHW, где W имеет значения, определенные ниже;
(i) группы -Р(О)(ОН)-О[(С1-С24)алкил], -Р(О)(ОН)-О[(С1-С24)алкенил] или -Р(О)(ОН)-О[(С1-С24)алкинил];
(j) группы -Р(О)(ОН)-О[(С1-С24)алкил]-СООХ, -Р(О)(ОН)-О[(С1-С24)алкенил]-СООХ, -Р(О)(ОН)-О[(С1-С24)алкинил]-СООХ, где X имеет значения, определенные выше;
(k) группы -S(O)(OH)2;
(l) защитной группы, выбранной из бензила или замещенного бензила, такого как 4-метоксибензил или 3,4-диметоксибензил, или 2,5-диметоксибензил, или 2,3,4-триметоксибензил, или 3,4,5-триметоксибензил; или из (С3-С6)алкенила, такого как С3 или С4алкенил; где алкильная, алкенильная, алкинильная группы могут быть разветвленными или линейными и могут быть незамещенными или необязательно замещены одной или более группами, независимо выбранными из галогена, такого как фтор, хлор, бром или йод;
гидроксила или гидроксильного производного -OY, где Y имеет значения, определенные ниже; амина или производного амина -NHW, где W имеет значения, определенные ниже; или группы -OZ, где Z выбран из (f), (g), (h), (i), (j), (k);
и где Y выбран из водорода; (С3-С6)алкенила, такого как С2 или С3 алкенил; группы, выбранной из бензила или замещенного бензила, такого как 4-метоксибензил или 3,4-диметоксибензил, или 2,5-диметоксибензил,
или 2,3,4-триметоксибензил, или 3,4,5-триметоксибензил; О-ксилиленовой группы;
и где W выбран из водорода; бензилоксикарбонильной группы или 9-флуоренилметилоксикарбонила;
и где R6 представляет группу, выбранную из трихлорацетимидата, фторида, хлорида, бромида, и X и R2 имеют значения, определенные выше, в условиях реакции, подходящих для образования соединения формулы 11h:
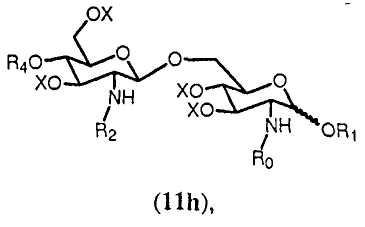
в которой R1, R2, R4, R0 и X имеют значения, определенные выше, причем способ необязательно включает дополнительно одну или более следующих стадий (2)-(11):
(2) реакция соединения формулы 11h в условиях реакции, подходящих для гидролитического удаления ряда групп R2 указанного соединения формулы 11h, с образованием соединения формулы 12а:
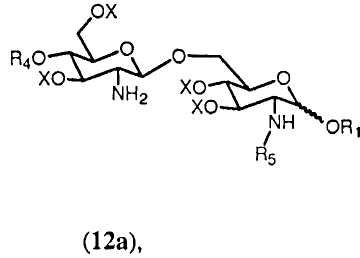
в которой R1, R4, R5 и X имеют значения, определенные выше, когда R0 выбран в качестве R5 в формуле 11h, или соединения формулы 12b:
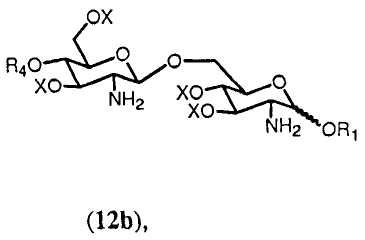
в которой R1, R4 и X имеют значения, определенные выше, когда в формуле 11h в качестве R2 выбран R0.
(3) реакция соединения 12а или 12b в условиях реакции, подходящих для образования амидной связи между свободной аминогруппой указанного соединения формулы 12а или 12b и карбоксигруппой (активированной) карбоновой кислоты формулы R5OH, в которой R5 имеет значения, определенные выше, с образованием соединения формулы 13:
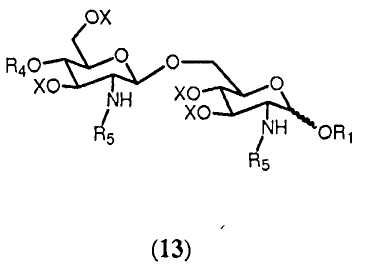
в которой R1, R4, R5 и X имеют значения, определенные выше;
(4) образование полуацеталя формулы 14:
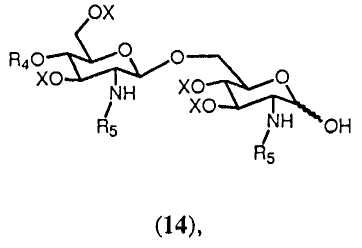
в которой R4, R5 и X имеют значения, определенные выше, с помощью удаления в подходящих реакционных условиях группы R1 из соединения формулы 13, определенной выше;
(5) фосфорилирование свободной гидроксильной группы соединения 14 в условиях реакции, подходящих для образования соединения формулы 15а:
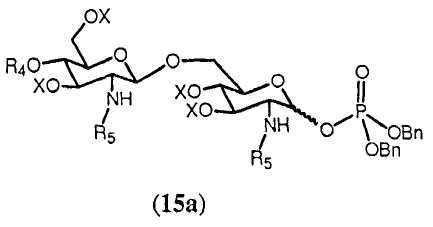
(6) сульфатирование свободной гидроксильной группы соединения 14 в реакционных условиях, подходящих для образования соединения формулы 15b:

(7) реакция свободной гидроксильной группы соединения 14 с (активированной) карбоновой кислотой формулы R8OH, в которой R8 выбран из (а), определенного ранее для R4, с образованием соединения формулы 15с:
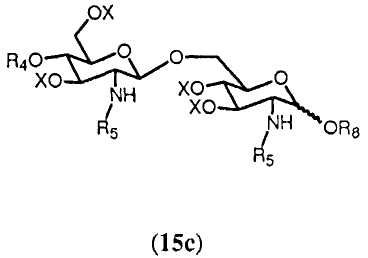
где R4, R5 и X имеют значения, определенные выше, и R8 выбран из (а), определенного ранее для R4;
(8) присоединение уходящей группы, такой как трихлорацетимидатная группа, к свободной гидроксильной группе соединения 14, в условиях реакции, подходящих для образования соединения формулы 24:
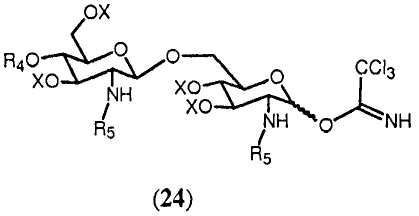
в которой R4, R5 и X имеют значения, определенные ранее;
(9) реакция соединения 24 стадии (8) с органической молекулой R8OH, в которой R8 выбран из (b), (с), (d) или (е), определенных ранее для R4, в условиях реакции, подходящих для образования соединения формулы 15d:
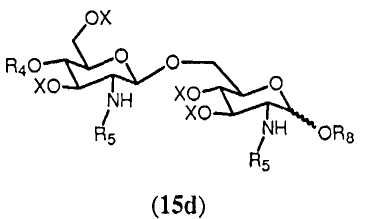
(10) реакция в подходящих реакционных условиях реакционноспособных групп, таких как гидроксильные группы, аминогруппы, карбоксигруппы или углеродные двойные связи, в соединении формулы 15а, 15b, 15c, 15d или 13, например по реакции, выбранной из реакции образования сложного эфира, метилирования, амидирования, окисления, гидрирования или α,β-гидроксилирования тетроксидом осмия, и в котором указанной реакции реакционноспособных групп необязательно предшествует удаление защитных групп, таких как группы Y или W, для высвобождения указанных реакционноспособных групп; и
(11) удаление ряда защитных групп X из соединения 13, 14, 15а, 15b, 15с или 15d, в условиях реакции, подходящих для образования соединения формулы 1:
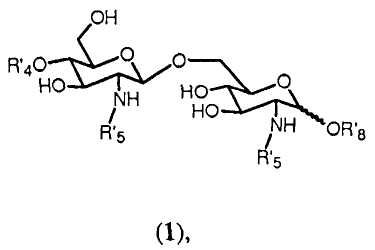
в которой R′4, R′5 и R′7 имеют значения, определенные соответственно ранее для R4, R5 и R7, и R′8 выбран из (а), (b), (с), (d), (e), (f), (g), (h), (i), (j) или (k), определенных ранее для R4, или выбран из Н, и в которой Y и W представляют Н.
2. Способ по п.1, в котором
R1 представляет собой С3 или С4алкенил, выбранный из 2-пропенила или 1-пропенила;
X представляет водород, группу, выбранную из бензила или замещенного бензила, выбранного из группы, включающей 4-метоксибензил, 3,4-диметоксибензил, 2,5-диметоксибензил, 2,3,4-триметоксибензил, или 3,4,5-триметоксибензил;
R0 выбран из R5 или R2, где R5 выбран из
(i) гидроксиацильной группы, такой как 3-гидроксиацильная группа, оксоацильной группы, такой как 3-оксоацильная группа, аминоацильной группы, такой как 3-аминоацильная группа;
(ii) 3-ацилоксиацильной группы, 3-ациламиноацильной группы, 3-ацилтиоацильной группы;
(iii) (С2-С24)алкилоксиацильной группы, (С2-С24)алкенилоксиацильной группы, (С2-С24)алкинилоксиацильной группы, (С2-С24)алкиламиноацильной группы, (С2-С24)алкениламиноацильной группы, (С2-С24)алкиниламиноацильной группы; (С2-С24)алкилтиоацильной группы, (С2-С24)алкенилтиоацильной группы, (С2-С24)алкинилтиоацильной группы, ацильной группы, происходящей из карбоновой кислоты с разветвленной цепью, разветвленной в 3-положении;
где в группах (i), (ii), (iii) углеводородная цепь ацила может быть насыщенной или ненасыщенной и углеводородная цепь ацила, алкила, алкенила, алкинила может быть разветвленной или линейной и необязательно может быть замещена одной или более группами, независимо выбранными из галогена, такого как фтор, хлор, бром или йод; гидроксила или гидроксильного производного -OY, где Y имеет значения, определенные ниже; амина, или производного амина -NHW, где W имеет значения, определенные ниже; группы -OZ, где Z выбран из (f), (g), (h), (i), (j), (k), определенных ниже;
и R2 представляет группу, выбранную из (С1-С6)галогенированного алкоксикарбонила, такого как 2,2,2-трихлорэтоксикарбонил (TROC) или 1,1 -диметил-2,2,2-трихлорэтоксикарбонил (ТСВОС);
R4 выбран из
(a) ацильной группы, определенной в (i), (ii) или (iii) для R5;
(b) разветвленной или линейной (С1-С24)алкильной группы; разветвленной или линейной (С1-С24)алкенильной группы; разветвленной или линейной (С1-С24)алкинильной группы;
(c) группы -[(С1-С24)алкил]-СООХ, -[(С2-С24)алкенил]-СООХ или -[(С2-С24)алкинил]-СООХ, где X имеет значения, определенные ниже;
(d) группы -[(C1-C24)алкил]-NHW, -[(С1-С24) алкенил]-NHW или -[(С1-С24)алкинил]-NHW, где W имеет значения, определенные ниже;
(e) формил[(С1-С24)алкильной]группы; формил [(С1-С24)алкенильной]группы; формил[(С1-С24)алкинильной]группы;
(f) диметоксифосфорильной группы;
(g) группы -P(O)(OY)2, где Y имеет значения, определенные ниже;
(h) группы -Р(О)(ОН)-О[(С1-С24)алкил]NHW, -Р(О)(ОН)-О[(С1-С24)алкенил]NHW или -Р(О)(ОН)-О[(С1-С24)алкинил]NHW, где W имеет значения, определенные ниже;
(i) группы -Р(О)(ОН)-О[(С1-С24)алкил], -Р(О)(ОН)-О[(С1-С24)алкенил] или -Р(О)(ОН)-О[(С1-С24)алкинил];
(j) группы -Р(О)(ОН)-О[(С1-С24)алкил]-СООХ, -Р(О)(ОН)-О[(С1-С24)алкенил]-СООХ, -Р(О)(ОН)-О[(С1-С24)алкинил]-СООХ, где X имеет значения, определенные выше;
(k) группы -S(O)(OH)2;
(l) защитной группы, выбранной из бензила или замещенного бензила, такого как 4-метоксибензил или 3,4-диметоксибензил, или 2,5-диметоксибензил, или 2,3,4-триметоксибензил, или 3,4,5-триметоксибензил; или С3 или С4алкенила, такого как 2-пропенил или 1-пропенил;
где алкильная, алкенильная, алкинильная группы могут быть разветвленными или линейными и могут быть незамещенными или необязательно замещены одной или более группами, независимо выбранными из галогена, такого как фтор, хлор, бром или йод; гидроксила или гидроксильного производного -OY, где Y имеет значения, определенные ниже; амина или производного амина -NHW, где W имеет значения, определенные ниже; или группы -OZ, где Z выбран из (f), (g), (h), (i), (j), (k);
и где Y выбран из водорода; С2 или С3 алкенила, такого как 2-пропенил или 1-пропенил; группы, выбранной из бензила или замещенного бензила, такого как 4-метоксибензил или 3,4-диметоксибензил, или 2,5-диметоксибензил, или 2,3,4-триметоксибензил, или 3,4,5-триметоксибензил; О-ксилиленовой группы;
и где W выбран из водорода; бензилоксикарбонильной группы или 9-флуоренилметилоксикарбонила.
3. Способ по п.1, в котором стадию (3) проводят в присутствии конденсирующего агента, такого как изобутилхлорформиат или 1-изобутилокси-2-изобутилоксикарбонил-1,2-дигидрохинолеин, или карбодиимид.
4. Способ по п.1, в котором на стадии (5) фосфорилирование свободной гидроксильной группы соединения 14, осуществляют при помощи тетрабензилпирофосфата в присутствии подходящего основания, такого как бис(триметилсилил)амид лития, в полярном растворителе, таком как тетрагидрофуран.
5. Способ по п.1, в котором на стадии (6) реакцию сульфатирования осуществляют при помощи комплекса триоксида серы, такого как комплекс триоксида серы с триметиламином, в растворителе, таком как ДМФ.
6. Способ по п.1, в котором стадию (7) проводят в присутствии конденсирующего агента, такого как изобутилхлорформиат или 1-изобутилокси-2-изобутилоксикарбонил-1,2-дигидрохинолеин, или карбодиимид.
7. Способ по п.1, в котором стадию (8) осуществляют с помощью трихлорацетонитрила в присутствии неорганического основания, такого как карбонат цезия или карбонат калия, в полярном растворителе или в апротонном полярном растворителе, или в дихлорметане.
8. Способ по п.1, в котором стадию (11) осуществляют с помощью гидрогенолиза защитных групп в присутствии благородного металла, такого как палладий-на-угле.
9. Синтетический асимметрично или симметрично замещенный β-(1→6)-связанный глюкозамин-дисахарид формулы 1:
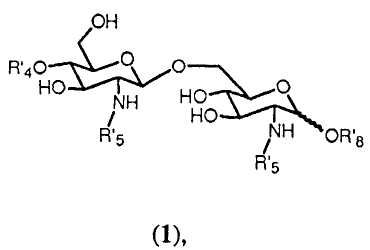
в которой R′4 представляет собой -Р(О)(ОН)2,
R′5 выбран из
(i) ацильной группы, происходящей из карбоновой кислоты с линейной цепью, имеющей от 2 до 24 атомов углерода;
(ii) ацилоксиацильной группы;
где в группах (i) и (ii) углеводородная цепь ацила является насыщенной и необязательно может быть замещена гидроксильной группой;
и R′8 выбран из
(a) разветвленной или линейной алкильной группы, необязательно замещенной гидроксильной группой(группами);
(b) группы -[(С1-С24)алкил]-СООН;
(c) группы -[(С1-С24)алкил]-NH2.
10. Дисахарид по п.9, который выбран из:
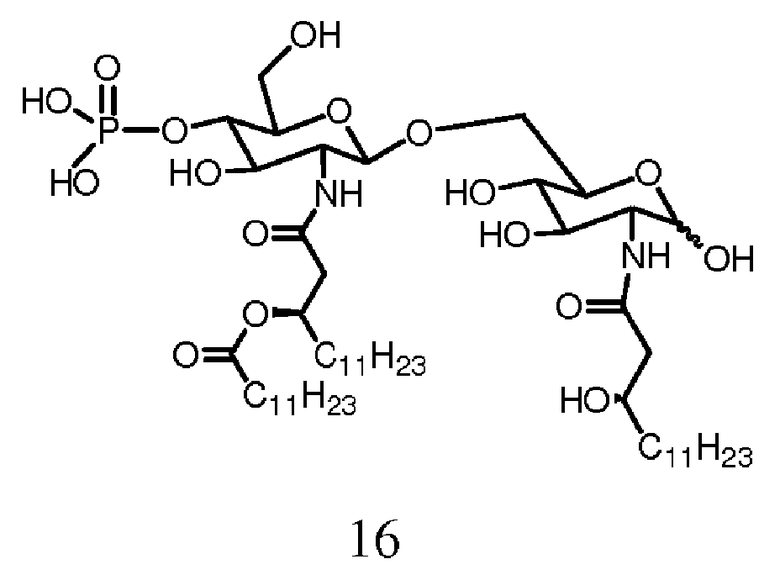
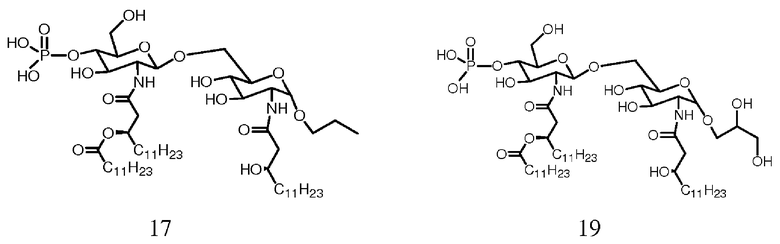
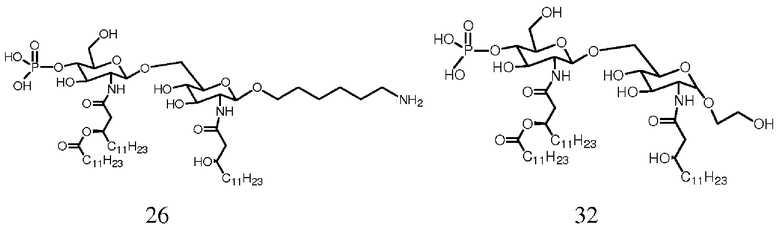

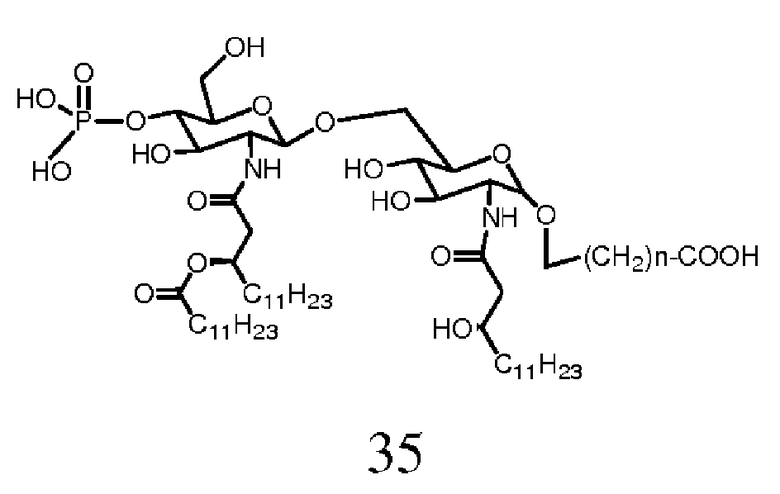
в которой n представляет 0 или целое число от 1 до 21
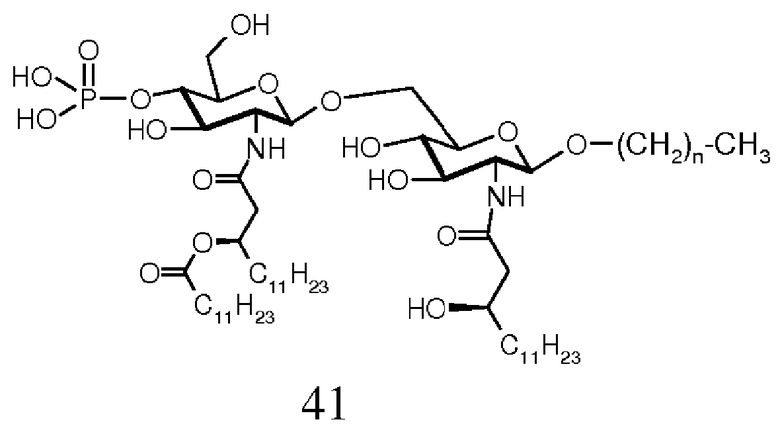
в которой n представляет целое число от 1 до 21, такое как 13.
11. Способ очистки, включающий
(i) смешивание раствора соединения формулы 1, полученного способом по п.1 или 2:

где R4′, R5′ и R8′ имеют значения, определенные ранее, с твердой смолой с обращенной фазой, в условиях, подходящих для связывания, по меньшей мере, части соединения формулы 1 с твердой фазой;
(ii) удаление жидкой фазы и промывку твердой фазы промывочной жидкостью, включающей водную фазу, забуференную при рН 6-9, 7-8 или 7,3-7,7, и органическую фазу, причем водная фаза и органическая фаза смешиваются в соотношении от 15:1 до 5:1 или 9:1 (об./об.);
(iii) удаление промывочной жидкости и элюирование, по меньшей мере, части соединения 1, связанного с твердой фазой, элюирующей жидкостью, включающей водную фазу и органическую фазу, которые смешиваются в соотношении от 1:15 до 1:5 или 1:9 (об./об.);
(iv) сбор элюирующей жидкости, включающей некоторое количество соединения формулы 1;
(v) необязательно удаление органической фазы из элюирующей жидкости, включающей соединение формулы 1.
12. Способ по п.11, дополнительно включающий доведение рН элюирующей жидкости, включающей некоторое количество соединения формулы 1, до заранее выбранной величины рН, или до рН 6-9, или до рН 7-8, или до рН 7,3-7,7.
13. Фармацевтическая композиция, включающая одно или более соединений по п.9 или 10, необязательно в сочетании с подходящим носителем или разбавителем, причем композиция проявляет иммуностимулирующие свойства в отношении toll-подобных рецепторов человека (TLR).
14. Соединение по п.9 или 10 для получения фармацевтической композиции по п.13, для применения при лечении расстройства, на которое влияет модулирование активности иммунной системы, включая подавление или активацию иммунной системы, такого как расстройство, выбранное из иммунных расстройств и/или рака, или для применения в качестве компонента вакцины, при этом расстройство выбрано из
- иммунного расстройства, связанного со сверхпродуцированием воспалительных цитокинов, такого как сверхпродуцирование воспалительных цитокинов, активируемое Т лимфоцитами, моноцитами или антиген-предоставляющими клетками, причем воспалительные цитокины или воспалительные маркеры предпочтительно принадлежат к группе, состоящей из IL-1β, IL-4, IL-5, IL-6, IL-8, IL-9, IL-13, IFN-γ, TNF-α или МСР-1,
- иммунного расстройства, выбранного из группы, состоящей из астмы, атопического дерматита, аллергического ринита, воспалительного заболевания кишечника, диабета и ревматоидного артрита,
- иммунного расстройства, связанного с пониженным продуцированием воспалительных цитокинов,
- расстройства, лечение которого приносит пользу в результате активации иммунной системы человека,
- расстройства, лечение которого приносит пользу в результате снижения секреции гистамина тучными клетками.
15. Соединение формулы 7:

в которой R2, R4, R6 и X имеют значения, определенные ранее в п.1.
16. Соединение по п.15, представляющее собой соединение формулы 7b:
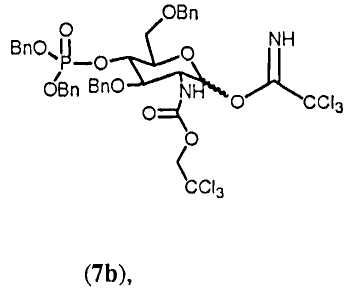
в которой Bn представляет собой бензил.
17. Соединение формулы 10b:
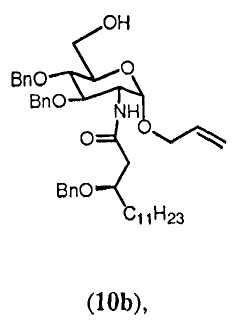
в которой Bn представляет собой бензил.
18. Соединение формулы 11:
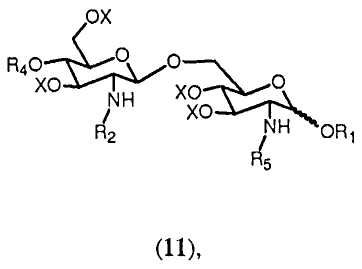
в которой R1, R2, R4, R5 и X имеют значения, определенные ранее в п.1.
19. Соединение по п.18, представляющее собой соединение 11а:
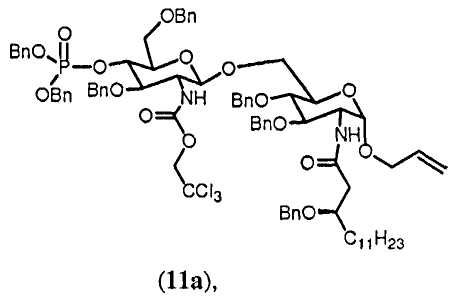
в которой Bn представляет собой бензил.
20. Соединение формулы 11b:
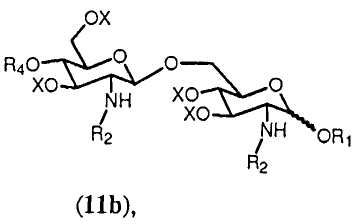
в которой R1, R2, R4 и X имеют значения, определенные ранее в п.1.
21. Соединение формулы 12а:
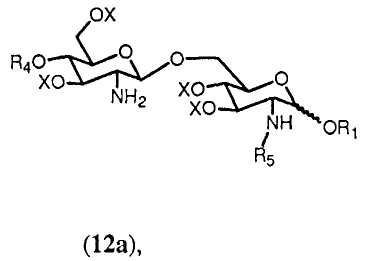
в которой R1, R2, R4, R5 и X имеют значения, определенные ранее в п.1.
22. Соединение формулы 12b:

в которой R1, R4 и X имеют значения, определенные ранее в п.1.
23. Соединение формулы 13:
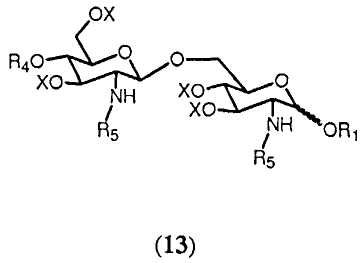
в которой R1, R4, R5 и X имеют значения, определенные ранее в п.1.
24. Соединение формулы 14:
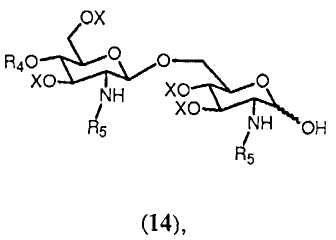
в которой R4, R5 и X имеют значения, определенные ранее в п.1.
25. Соединение, выбранное из группы, включающей
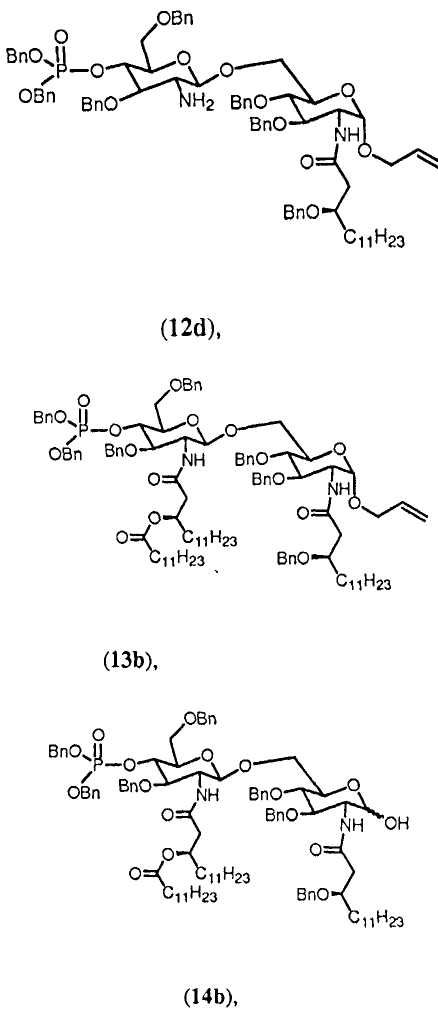
в которых Bn представляет собой бензил.
26. Применение соединения по пп.15-25 в качестве промежуточного соединения, в том числе в качестве исходного вещества, в способе синтеза асимметрично или симметрично замещенного β-(1→6)-связанного глюкозамин-дисахарида.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| Sakai Yasuhiro et al, "Synthesis of Helicobacter pylori lipid A and its analogue using p-(trifluoromethyl)benzyl protecting group", Tetrahedron Letters, 2000, v.41, №35, p.6843-6847 | |||
| Fukase Koichi et al, "Divergent synthesis and biological activities of | |||

