Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям пирролопиридина, их солям, фармацевтическим композициям, на их основе, к терапевтическому применению и способам получения таких соединений. Настоящее изобретение кроме того относится к 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36Cl, 82Br, 76Br, 77Br, 123I, 125I, 124I и 131I изотопно меченным замещенным производным пирролопиридина по настоящему изобретению. В частности, настоящее изобретение относится к 11C, 13C, 14C, 18F, 125I, 15O, 13N, 35S, 2H и 3H изотопам новых пирролопиридиновых соединений и к способам их получения.
Настоящее изобретение также относится к новым соединениям пирролопиридина, которые могут быть пригодны для визуализации агрегатов тау, агрегатов β-листов, агрегатов β-амилоида, агрегатов α-синуклеина или трансактивного ответа ДНК-связывающего белка 43 кДа, и, следовательно, являются полезными в связывании и визуализации агрегатов тау-белка у пациентов с болезнью Альцгеймера. Более конкретно, настоящее изобретение относится к способу применения соединений по настоящему изобретению в качестве индикаторов для визуализации при позитронно-эмиссионной томографии (ПЭТ) для изучения отложений тау-белка в головном мозге in vivo для проведения диагностики болезни Альцгеймера. Кроме того, настоящее изобретение относится к способу измерения клинической эффективности терапевтических средств, направленных на патологию тау.
Уровень техники
Болезнь Альцгеймера представляет собой распространенное нейродегенеративное заболевание, поражающее пожилых людей, что приводит к прогрессирующему ухудшению памяти, потере речи и зрительно-пространственных навыков, и расстройства поведения. Характеристики заболевания включают дегенерацию холинергических нейронов в коре головного мозга, гиппокампе, базальных отделах переднего мозга и других областях головного мозга, нейрофибриллярные клубки и отложение β-амилоидного пептида (Aβ).
При болезни Альцгеймера, два основных белка образуют аномальные полимеры (агрегаты) в головном мозге, которые, как полагают, являются результатом неправильного сворачивания во время агрегации одного из двадцати негомологичных белков человека. Внутриклеточные нейрофибриллярные клубки-NFT из ассоциированного с микротрубочками белка (тау-белка) и внеклеточные ʺамилоидныеʺ бляшки в основном состоят из полимеризованного Aβ-пептида. Оба являются токсичными для нейронов головного мозга и являются результатом волокон, образованных из субъединицы белка путем укладки бета-нитей. См. B. Bulic, E. Mandelkow et al. Angewandte Chemie International Edition, Vol. 48, Issue 10, стр. 1740-1752, 2009 и B. Bulic, et al. J. Med. Chem 2013 June 13; 56(11):4135-55. См. также US2014275040, CN103450152, US2009203903, US2009233945, WO2012106343, US20150031672, WO2010129816, WO2008103615 и US20080027044.
Тубулин-ассоциированная единица, или тау, представляет собой белок, ассоциированный с микротрубочками, который, как полагают, играет решающую роль в этиологии болезни Альцгеймера (БА) на основе нескольких научных доказательств. Во-первых, внутриклеточные агрегаты гиперфосфорилированного тау (NFT) неизменно находятся в головном мозге пациентов с БА и ряда других нейродегенеративных заболеваний. Во-вторых, степень патологии NFT в головном мозге пациентов с болезнью Альцгеймера тесно коррелирует с когнитивной функцией. Наконец, в то время как мутации в тау-белке, как показано не вызывают БА, такие мутации действительно вызывают другую форму деменции, известную как лобно-височная деменция с паркинсонизмом (FTDP). Таким образом, методы, направленные на уменьшение NFT и/или гиперфосфорилированного тау представляют собой болезнь-модифицирующие терапии для БА.
В настоящее время гистологический анализ аутопсийных материалов является основным средством обнаружения агрегатов тау. ПЭТ индикатор тау будет ценным неинвазивным средством для пространственного и временного количественного определения нейрофибриллярных клубков (NFT) в головном мозге человека, поскольку исследования после смерти показали нагрузка NFT лучше коррелирует с когнитивными нарушениями. ПЭТ индикатор тау будет критическим средством, подходящим для заболевания, для количественной оценки стабилизации или уменьшения формирования NFT для болезнь-модифицирующих терапий болезни Альцгеймера. Кроме того, ПЭТ индикатор тау может быть полезен для отбора пациентов для клинических исследований БА. В этом методе ПЭТ индикатор тау может быть разработан в качестве сопутствующей диагностики. В дополнение к БА, существуют и другие нейродегенеративные заболевания, характеризующиеся отложением агрегатов тау (лобно-височная деменция (FTD), прогрессирующий надъядерный паралич (ПНП), кортикобазальная дегенерация (КБД), хроническая травматическая энцефалопатия (ХТЭ), болезнь Пика и тому подобное.).
Таким образом, существует потребность в радиоиндикаторах для нейровизуализации, которые позволили бы визуализацию in vivo патологии тау-белка, тем самым обеспечивая представление об отложении агрегатов тау-белка в головном мозге человека. Эффективный радиоиндикатор для нейровизуализации должен проникать через гематоэнцефалический барьер и обладать высоким сродством и специфичностью к агрегатам тау и, следовательно, должен иметь соответствующую липофильность (logD 1-3) и низкую молекулярную массу (, <450), показывать быстрый клиренс из крови и низкое неспецифическое связывание. Радиоиндикатор нейровизуализации будет играть определенную роль в диагностике путем идентификации пациентов с избыточными агрегатами тау в головном мозге и, следовательно, с риском развития БА, а также дает представление о степени агрегации тау, влиянии на головной мозг с течением времени, корреляции с когнитивной функцией и помощи при анализе эффективности ингибитора тау.
В типичном исследовании ПЭТ, небольшое количество радиоактивного индикатора вводят экспериментальному животному, нормальному человеку или пациенту, который проходит обследование. Радиоактивный индикатор затем циркулирует в крови субъекта и может быть поглощен определенными тканями. Радиоактивный индикатор может преимущественно сохраняться в некоторых из этих тканей в результате специфического ферментативного превращения или посредством специфического связывания с макромолекулярными структурами, такими как белки. Используя сложную визуализирующую аппаратуры для детектирования позитронной эмиссии, количество радиоактивного индикатора затем неинвазивно оценивали в различных тканях организма. Полученные данные анализировали для предоставления количественно пространственной информации биологического процесса in vivo, для которого индикатор был разработан. ПЭТ дает исследователям фармацевтических изысканий возможность оценить биохимические изменения или метаболические действия лекарственного средства-кандидата in vivo в течение длительных периодов времени, и ПЭТ может быть использована для измерения распределения лекарственного средства, таким образом, позволяя оценить фармакокинетики и фармакодинамики конкретного испытуемого лекарственного средства-кандидата. Важно то, что ПЭТ индикаторы могут быть разработаны и использованы для количественного определения присутствия участков связывания в тканях. Следовательно, интерес к ПЭТ-индикаторам для разработки лекарственных средств расширяется на основе развития меченных изотопами биохимических веществ и соответствующих детектирующих устройств для детектирования радиоактивности внешней визуализацией.
В то время как основное использование изотопно-меченых соединений по настоящему изобретению состоит в использовании в позитронно-эмиссионной томографии, которая представляет собой метод анализа in vivo, определенные изотопно-меченые соединения могут быть использованы для других методов анализа, отличных от ПЭТ. В частности, 14C и 3H меченые соединения могут быть использованы в способах in vitro и in vivo для определения связывания, занятости рецепторов и метаболических исследований, включая ковалентное мечение. В частности, различные меченые изотопами соединения находят применение в магнитно-резонансной томографии, авторадиографии и других подобных аналитических методах.
Сущность изобретения
Изобретение относится к классу пирролопиридиновых соединений формулы I, их солям, к фармацевтическим композициям, содержащим их, к диагностическому и терапевтическому применению и к способам получения таких соединений. В частности, настоящее изобретение относится к классу пирролопиридиновых соединений формулы I, которые могут быть полезны для связывания и визуализации агрегатов тау, агрегатов β-листов, агрегатов бета-амилоида или агрегатов альфа-синуклеина, и, следовательно, являются полезными в связывании и визуализации агрегатов тау у пациентов с болезнью Альцгеймера. Настоящее изобретение также относится к применению соединений в качестве средства визуализации для улучшения выявления пациентов, склонных к развитию болезни Альцгеймера, путем определения, присутствия у указанных пациентов избытка агрегатов тау-белка в головном мозге. Настоящее изобретение также относится к поиску средства визуализации, которое будет использоваться для диагностики и наблюдения за развитием БА. Настоящее изобретение также относится к использованию соединения пирролопиридина для мониторинга и оценки лечения, когда лекарственное средство против агрегата тау высвободится. Настоящее изобретение также может быть полезным для визуализации и обнаружения для других нейродегенеративных заболеваний, характеризующихся отложением агрегатов тау, таких как лобно-височная деменция (FTD), прогрессирующий надъядерный паралич (ПНП), кортикобазальная дегенерация (КБД), хроническая травматическая энцефалопатия (ХТЭ), болезнь Пика и тому подобное. Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы I и фармацевтически приемлемый носитель.
Кроме того, включенными являются изотопно-меченые соединения по настоящему изобретению. Другой аспект настоящего изобретения относится к использованию меченых изотопами соединений в качестве радиоиндикаторов нейровизуализации для визуализации in vivo головного мозга в отношении тау-агрегатов при диагностике, мониторинге и/или лечении БА. Другим аспектом настоящего изобретения является использование меченых изотопами соединений в позитронно-эмиссионной томографии, которая представляет собой метод анализа in vivo в диагностике, мониторинге и/или лечения AD. 14C и 3H меченые соединения могут быть использованы в способах in vitro и in vivo для определения связывания, занятости рецепторов и метаболических исследований, включая ковалентное мечение. В частности, различные меченые изотопами соединения находят применение в магнитно-резонансной томографии, авторадиографии и других подобных аналитических методах. Таким образом, другой аспект настоящего изобретения дополнительно относится к меченым изотопом 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36Cl, 82Br, 76Br, 77Br, 123I, 124I, 125I и 131I замещенным производным соединениям пирролопиридина формулы I. В частности, настоящее изобретение относится к 11C, 13C, 14C, 18F, 123I, 15O, 13N, 35S, 2H и 3H изотопам замещенных производных соединений пирролопиридина, композициям и способам их получения и использования в качестве радиоизотопных индикаторов или индикаторов ПЭТ в диагностике и измерении действия соединения при лечении БА. Настоящее изобретение также относится к нетоксичным соединениям, связывающим тау-белок, которые способны быстро пересекать гематоэнцефалический барьер, имеют низкий уровень неспецифического связывания и быстро выводятся из организма. Эти и другие аспекты настоящего изобретения будут реализованы после рассмотрения описания в полном объеме.
Фигуры
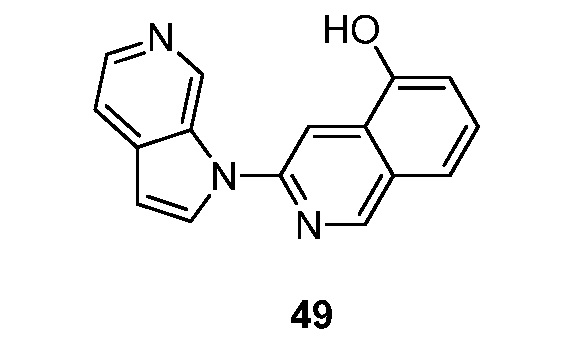
Фиг. 1: показывает насыщение связывания [3H]-6 (Пример 49) в гомогенатах головного мозга БА
Фиг. 2: показывает соединение 6 (немеченое) самовытесняемое [3H]-6 со значением Ki, равным 0,43 нM
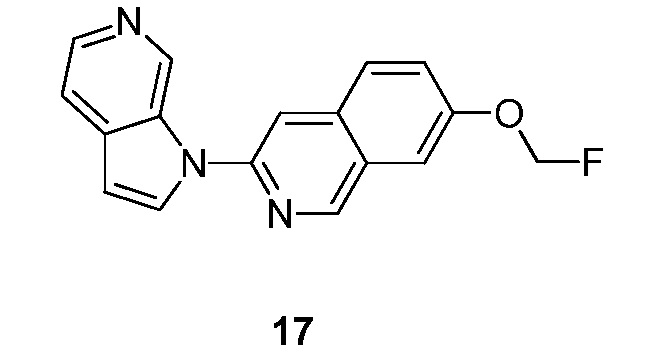
Подробное описание настоящего изобретения
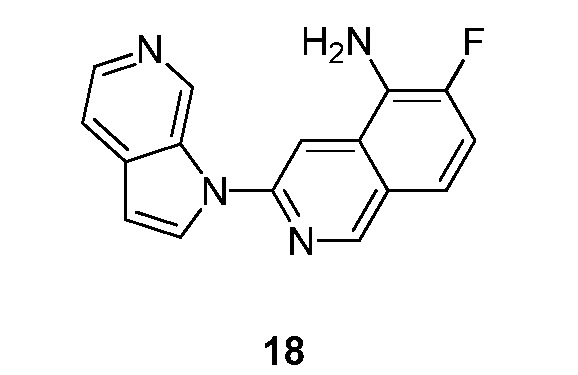
Настоящее изобретение относится к соединениям хинолин амида общей формулы (I)

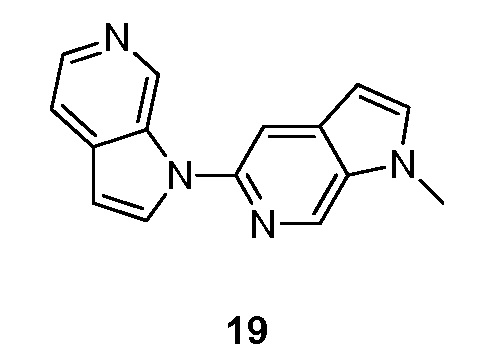
или его фармацевтически приемлемой соли, где;
X представляет собой CH или N;
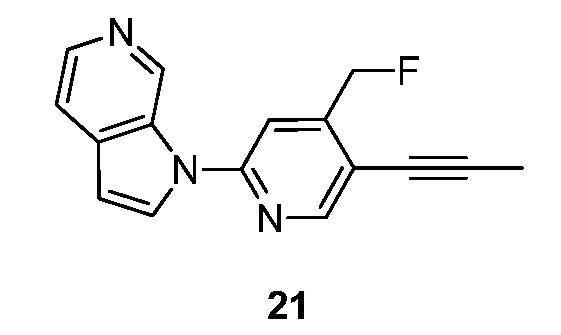
R представляет собой водород или -C1-6алкил, указанный алкил необязательно замещен 1-3 группами Ra;
R1 представляет собой водород, -C1-6алкил, -CN, -(CH2)nNH(CH2)nN(R)2, -C2-6алкенил, -(CH2)nOR или -(CH2)nгалоген;
R2 представляет собой -C1-6алкил, -OC1-6 алкил, -C2-6алкенилR3, -C2-6алкинилR3, -(CH2)nOR, -(CH2)nгалоген, -O(CH2)nгалоген, -C6-10 арил, -C5-10 гетероциклил, -N(R)2, -O(CH2)nRa, -N(CH3)(CH2)nOR, -NRC(O)R, -NH(CH2)nгалоген, -NC(O)C6-10арил, -NC(O)C5-10 гетероциклил, -N(CH3)(CH2)nгалоген, -C(O)NC6-10 арил, указанный алкил, арил и гетероциклил необязательно замещены 1-3 группами Ra;
или соседний R1 может соединяться с R2 с образованием девяти-десяти-членного бициклического кольца вместе с кольцом, к которому R1 и R2 присоединены, необязательно прерываемое N, S и/или O, указанное бициклическое кольцо необязательно замещено 1-3 группами Ra;
R3 представляет собой водород, -C1-6алкил, -(CH2)nгалоген, -(CH2)nN(R)2, -(CH2)nNR(CH2)nN(R)2, -C6-10 арил, -C5-10 гетероарил, указанный алкил, арил и гетероарил, необязательно замещены 1-3 группами Ra;
Ra представляет собой -CN, CF3, -C1-6алкил, -C2-6алкенил, -C2-6алкинил, C6-10 арил, -C5-10 гетероциклил, -CN, NO2, (CH2)nгалоген, -O(CH2)nгалоген, (CH2)nOR, -O(CH2)nC6-10 арил, -(CH2)nN(R)2, -C(O)N(R)2, -N(CH3)(CH2)nOR, -NRCOR, -COR, -NH(CH2)nгалоген, -NC(O)C C6-10 арил, -N(CH3)(CH2)nгалоген, C(O)C6-10 арил или -CO2R, указанный алкил, алкенил, алкинил, арил и гетероциклил необязательно замещены 1-3 группами Rb;
Rb представляет собой водород, -C1-6алкил, -OR, -(CH2)nN(R)2, или галоген;
и
n равен 0-4.
Один из аспектов настоящего изобретения реализуется, когда Rb выбран из группы, состоящей из водорода, метокси, амино, метиламино и гидрокси.
Другой аспект настоящего изобретения реализуется, когда n равно 0. Другой аспект настоящего изобретения реализуется, когда n равно 1. Еще один аспект настоящего изобретения реализуется, когда n равно 2. Еще один аспект настоящего изобретения реализуется, когда n равно 3. Другой аспект настоящего изобретения реализуется, когда n равно 0-2.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда Ra выбран из группы, состоящей из -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда Ra выбран из группы, состоящей из CH3, CH2CH3, OCH3, OH, -(CH2)nNHCH3, -NH2, галогена, -(CH2)nN(CH3)2, NO2, CN, -N(CH3)(CH2)nOH, -N(CH3)(CH2)nF и O(CH2)nF.
Другой аспект настоящего изобретения реализуется, когда X представляет собой CH и все остальные переменные являются такими, как описано ранее.
Другой аспект настоящего изобретения реализуется, когда X представляет собой N и все остальные переменные являются такими, как описано ранее.
Еще один аспект настоящего изобретения реализуется, когда R представляет собой водород.
Еще один аспект настоящего изобретения реализуется, когда R представляет собой необязательно замещенный -C1-6алкил.
Другой аспект настоящего изобретения реализуется, когда R1 представляет собой водород.
Другой аспект настоящего изобретения реализуется, когда R1 -C1-6алкил, указанный алкил необязательно замещен 1-3 группами Ra.
Другой аспект настоящего изобретения реализуется, когда R1 представляет собой CN.
Другой аспект настоящего изобретения реализуется, когда R1 представляет собой -C2-6алкенил, указанный алкенил необязательно замещен 1-3 группами Ra.
Другой аспект настоящего изобретения реализуется, когда R1 представляет собой (CH2)nOR.
Другой аспект настоящего изобретения реализуется, когда R1 представляет собой -(CH2)nN(CH2)nN(R)2.
Еще один аспект настоящего изобретения реализуется, когда R1 представляет собой (CH2)nгалоген.
Понятно, что, когда Х представляет собой N и соседний R1 и R2 не объединяются с образованием бициклического кольца, число R1 заместителей составляет 1-2.
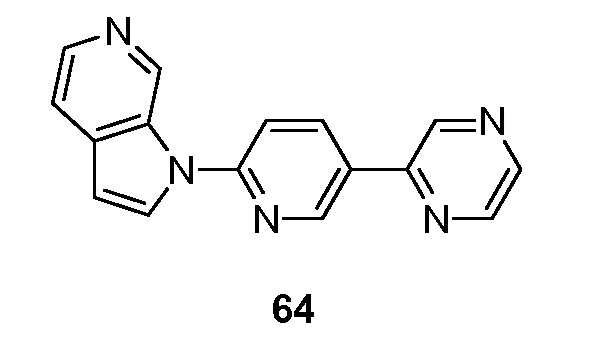
Еще один аспект настоящего изобретения реализуется, когда R2 выбран из группы, состоящей из -C2-6алкенилR3, -C2-6алкинилR3, -NC(O)C6-10 арила, -NC(O)C5-10 гетероциклила, -C6-10 арила и -C5-10 гетероциклила, указанный арил и гетероциклил необязательно замещены 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой -C2-6алкенилR3. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой -C2-6алкинилR3. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой -NC(O) C6-10 арил, указанный арил необязательно замещен. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой -NC(O)C5-10 гетероциклил, указанный гетероциклил необязательно замещен. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой необязательно замещенный -C6-10 арил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда арил из R2 представляет собой необязательно замещенный фенил или нафтил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой необязательно замещенный -C5-10 гетероциклил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероциклил из R2 выбран из группы, состоящей из необязательно замещенного пиридила, тиазолила, пиримидинила, пиперизинила, пиразолила, пиразинила, имидазолила и триазолила.
Другой аспект настоящего изобретения реализуется, когда R3 представляет собой -C1-6алкил, указанный алкил необязательно замещен 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда алкил представляет собой метил, этил или пропил.
Другой аспект настоящего изобретения реализуется, когда R3 представляет собой -(CH2)nгалоген. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда галоген представляет собой фтор или хлор.
Другой аспект настоящего изобретения реализуется, когда R3 представляет собой C6-10 арил, указанный арил необязательно замещен 1-3 группами Ra. Подвариант осуществления настоящего изобретения реализуется, когда арил представляет собой необязательно замещенный фенил или нафтил.
Другой аспект настоящего изобретения реализуется, когда R3 представляет собой -C5-10 гетероарил, указанный гетероарил необязательно замещен 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероарил выбран из группы, состоящей из необязательно замещенного пиридила и тиазолила.
Другой аспект настоящего изобретения реализуется, когда R3 представляет собой -(CH2)nN(R)2 или -(CH2)nNR(CH2)nN(R)2.
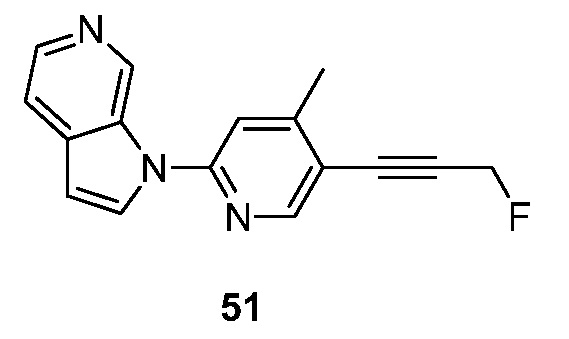
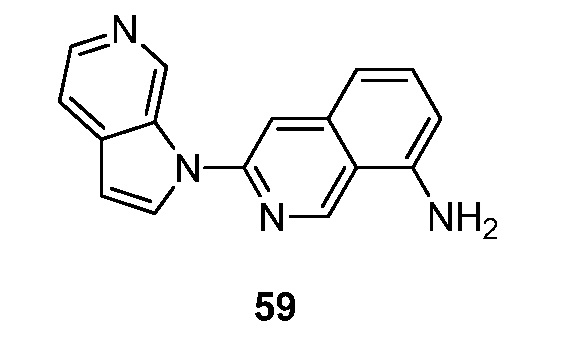
Еще один аспект настоящего изобретения реализуется, когда R2 объединен с соседним R1 и кольцом, к которому R1 и R2 присоединены, с образованием 9-10-членного бициклического кольца, указанное бициклическое кольцо необязательно прервано N, S и/или O, и указанное бициклическое кольцо необязательно замещено 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда бициклическое кольцо необязательно прервано одним из N, S и/или O. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный пирролопиридинил, фуропиридинил, нафтиридинил, тетрагидронафтиридинил, хиназолинил, хинолинил или изохинолинил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный пирролопиридинил. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный нафтиридинил или тетрагидронафтиридинил. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный хиназолинил. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный хинолинил. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный изохинолинил. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный фуропиридинил.
Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой CH и R2 выбран из группы, состоящей из -C2-6алкенилR3, -C2-6алкинилR3, -C6-10 арила и -C5-10 гетероциклила, указанный арил и гетероциклил необязательно замещены 1-3 группами Ra.
Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой CH и R2 представляет собой -C2-6алкенилR3. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R3 выбран из группы, состоящей из метила, этила, пропила, (CH2)nF, -(CH2)nN(R)2, -(CH2)nNR(CH2)nN(R)2, необязательно замещенного фенила, пиридила и тиазолила.
Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой CH и R2 представляет собой -C2-6алкинилR3. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R3 выбран из группы, состоящей из метила, этила, пропила, (CH2)nF, -(CH2)nN(R)2, -(CH2)nNR(CH2)nN(R)2, необязательно замещенного фенила, пиридила и тиазолила.
Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой CH и R2 представляет собой необязательно замещенный -C6-10 арил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой необязательно замещенный фенил или нафтил.
Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой CH и R2 представляет собой необязательно замещенный -C5-10 гетероциклил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой необязательно замещенный O(CH2)nпиридил, NC(O)фенил, C(O)фенил, нафтил, нафтиридинил, пиридил, тиазолил, пиримидинил, тиазолил, пиразинил или имидазолил.
Другой аспект настоящего изобретения реализуется, когда X представляет собой CH и R2 объединяется с соседним R1 с образованием 9-10-членного бициклического кольца, необязательно прерванного N, S и/или O, указанное бициклическое кольцо необязательно замещено 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный пирролопиридинил, фуропиридинил, нафтиридинил, тетрагидронафтиридинил, хинолинил или изохинолинил.
Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой N и R2 выбран из группы, состоящей из -C2-6алкенилR3, -C2-6алкинилR3, -C6-10 арила и -C5-10 гетероциклила, указанный арил и гетероциклил необязательно замещены 1-3 группами Ra.
Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой N и R2 представляет собой -C2-6алкенилR3. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R3 выбран из группы, состоящей из метила, этила, пропила, (CH2)nF, -(CH2)nN(R)2, -(CH2)nNR(CH2)nN(R)2, необязательно замещенного фенила, пиридила и тиазолила.
Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой N и R2 представляет собой -C2-6алкинилR3. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R3 выбран из группы, состоящей из метила, этила, пропила, (CH2)nF, -(CH2)nN(R)2, -(CH2)nNR(CH2)nN(R)2, необязательно замещенного фенила, пиридила и тиазолила.
Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой N и R2 представляет собой необязательно замещенный -C6-10 арил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой необязательно замещенный фенил или нафтил.
Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда X представляет собой N и R2 представляет собой необязательно замещенный -C5-10 гетероциклил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 представляет собой необязательно замещенный O(CH2)nпиридил, NC(O)фенил, C(O)фенил, нафтил, нафтиридинил, пиридил, тиазолил, пиримидинил, тиазолил, пиразинил или имидазолил.
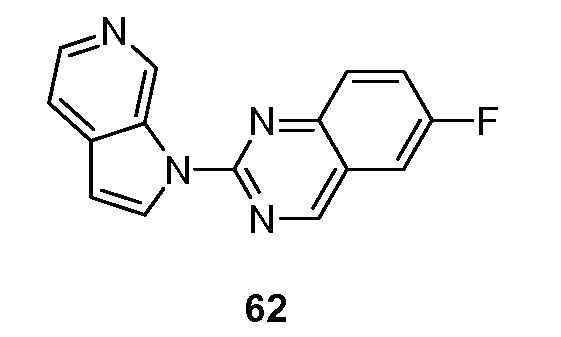
Другой аспект настоящего изобретения реализуется, когда X представляет собой N и R2 объединяется с соседним R1 вместе с кольцом, к которому присоединены R1 и R2, с образованием 9-10-членного бициклического кольца, необязательно прерванного N, S и/или O, указанное бициклическое кольцо необязательно замещено 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный хиназолинил.
Другой аспект настоящего изобретения реализуется, когда соединения формулы I выбраны из меченных изотопами 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36Cl, 82Br, 76Br, 77Br, 123I, 124I, 125I и 131I.
Еще один аспект настоящего изобретения реализуется с соединением структурной формулы Ia:

или его фармацевтически приемлемой солью, где R1 и R2 являются такими, как описано в настоящем документе. Подвариант осуществления формулы Ia реализуется, когда R1 представляет собой водород, галоген, -(CH2)nN(CH2)nN(R)2, необязательно замещенный C1-6 алкил или необязательно замещенный C2-6 алкенил. Подвариант осуществления настоящего изобретения реализуется, когда соединения формулы Ia выбраны из меченных изотопами 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36CL, 82Br, 76Br, 77Br, 123I, 124I, 125I и 131I.
Другой подвариант осуществления формулы Ia реализуется, когда R2 выбран из группы, состоящей из -C2-6алкенилR3, -C2-6алкинилR3, (CH2)nOR, -O(CH2)nгалогена, C6-10 арила, -C5-10 гетероциклила, -N(R)2, -O(CH2)nRa, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена, -NC(O) C6-10 арила, -N(CH3)(CH2)nгалогена, C(O)NC6-10 арила, указанный арил и гетероциклил необязательно замещены 1-3 группами Ra, и R3 выбран из группы, состоящей из водорода, -(CH2)nгалогена, -(CH2)nN(R)2, (CH2)nNR(CH2)nN(R)2, C6-10 арила, -C5-10 гетероарила, указанный арил и гетероарил необязательно замещены 1-3 группами Ra.
Другой подвариант осуществления данного аспекта формулы Iа реализуется, когда R2 выбран из группы, состоящей из C2алкенилR3, C2алкинилR3, O(CH2)nF, N(CH2)nF, N(R)2, и необязательно замещенного O(CH2)nпиридила, NC(O)фенила, C(O)фенила, нафтила, нафтиридинила, пиридила, триазолила, пиримидинила, тиазолила, пиразинила и имидазолила и R3 выбран из группы, состоящей из метила, этила, пропила, (CH2)nF, -(CH2)nN(R)2, -(CH2)nNR(CH2)nN(R)2, необязательно замещенного фенила, пиридила и тиазолила и R1 выбран из группы, состоящей из водорода, фтора или хлора, -(CH2)nN(CH2)nN(R)2 или необязательно замещенного C1-6 алкила и C2-6 алкенила.
Еще один аспект настоящего изобретения формулы I реализуется, когда R2 объединяется с соседним R1 вместе с кольцом, к которому присоединены R1 и R2, с образованием 9-10-членного бициклического кольца, необязательно прерванного N, S, O, указанный бицикл необязательно замещен 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 объединяется с соседним R1 вместе с кольцом, к которому присоединены R1 и R2, с образованием необязательно замещенного нафтиридинила, тетрагидронафтиридинила, фуропиридинила, изохинолинила или пирролопиридинила. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 объединяется с соседним R1 вместе с кольцом, к которому присоединены R1 и R2, с образованием необязательно замещенного нафтиридинила. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 объединяется с соседним R1 вместе с кольцом, к которому присоединены R1 и R2, с образованием необязательно замещенного тетрагидронафтиридинила. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 объединяется с соседним R1 вместе с кольцом, к которому присоединены R1 и R2, с образованием необязательно замещенного фуропиридинила. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 объединяется с соседним R1 вместе с кольцом, к которому присоединены R1 и R2, с образованием необязательно замещенного изохинолинила. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R2 объединяется с соседним R1 вместе с кольцом, к которому присоединены R1 и R2, с образованием необязательно замещенного пирролопиридинила. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда соседний R1 и R2, когда объединены, представлены структурной формулой Ib1, Ib2, Ib3 или Ib4:

где W, W1, W2 и W3 независимо выбраны из -CH- или -N- и R и Ra такие, как описаны первоначально. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда W, W1, W2 и W3 представляют собой все -CH-. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда по меньшей мере, один из W, W1, W2 и W3 представляет собой -N- и другие представляют собой -CH-.
Другой аспект настоящего изобретения формулы I представлен структурной формулой II:
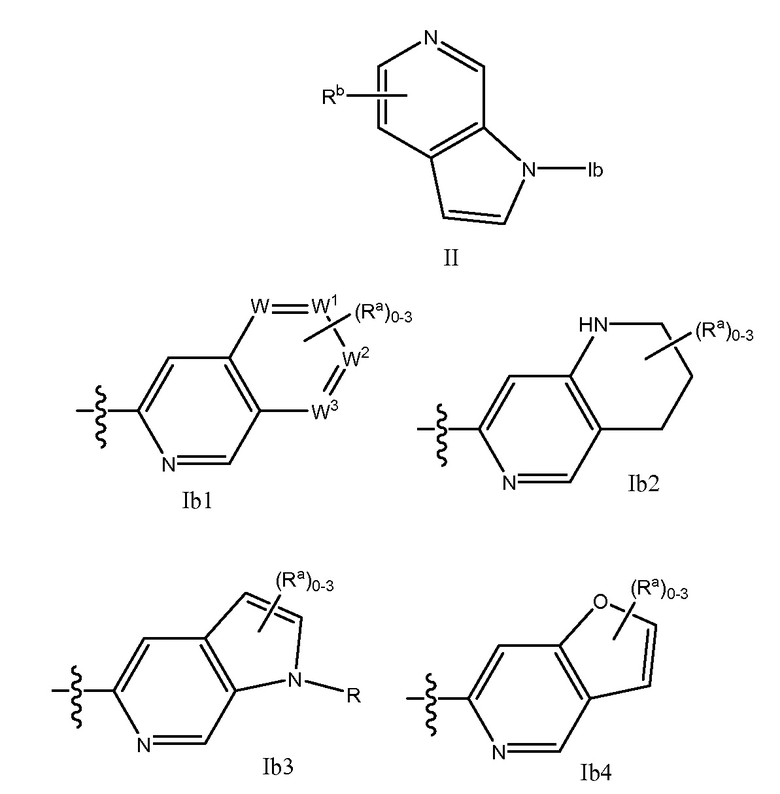
или его фармацевтически приемлемой солью, где Ib= Ib1, Ib2, Ib3 или Ib4, и W, W1, W2, W3, R, Ra и Rb такие, как описано ранее. Подвариант осуществления настоящего изобретения реализуется, когда соединения формулы II выбраны из меченных изотопами 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36CL, 82Br, 76Br, 77Br, 123I, 124I, 125I и 131I. Один из аспектов данного варианта осуществления настоящего изобретения реализуется, когда соединение изотопно меченное 18F. Другой подвариант осуществления настоящего изобретения формулы II реализуется, когда Ra выбран из группы, состоящей из -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда Ra выбран из группы, состоящей из CH3, CH2CH3, OCH3, OH, -(CH2)nNHCH3, -NH2, галогена, -(CH2)nN(CH3)2, NO2, CN, -N(CH3)(CH2)nOH, -N(CH3)(CH2)nF и O(CH2)nF.
Другой вариант осуществления настоящего изобретения формулы II реализуется, когда Ib представляет собой Ib1, Ra выбран из группы, состоящей из -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена, и W, W1, W2 и W3 представляют собой все -CH-.
Другой вариант осуществления настоящего изобретения формулы II реализуется, когда Ib представляет собой Ib1, Ra выбран из группы, состоящей из -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена, и по меньшей мере один из W, W1, W2 и W3 представляет собой -N.
Другой вариант осуществления настоящего изобретения формулы II реализуется, когда Ib представляет собой Ib2 и Ra выбран из группы, состоящей из -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена.
Другой вариант осуществления настоящего изобретения формулы II реализуется, когда Ib представляет собой Ib3 и Ra выбран из группы, состоящей из -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена.
Другой вариант осуществления настоящего изобретения формулы II реализуется, когда Ib представляет собой Ib4 и Ra выбран из группы, состоящей из -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена.
Другой аспект настоящего изобретения формулы I представлен структурной формулой III:

где Ra', Raʺ, Raʺ' и Raiv независимо друг от друга выбраны из водорода и Ra, и Ra и Rb являются такими, как описано ранее. Подвариант осуществления настоящего изобретения реализуется, когда соединения формулы III выбраны из меченных изотопами 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36CL, 82Br, 76Br, 77Br, 123I, 124I, 125I и 131I. Один из аспектов данного варианта осуществления настоящего изобретения реализуется, когда соединение представляет собой изотопно-меченное 18F или 123I. Подвариант осуществления настоящего изобретения формулы III реализуется, когда Ra', Raʺ, Raʺ' и Raiv независимо друг от друга выбраны из группы, состоящей из водорода, -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена и Rb выбран из группы, состоящей из водорода, C1-6алкила, -OR, -(CH2)nN(R)2 или галогена. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда Ra', Raʺ, Raʺ' и Raiv независимо друг от друга выбраны из группы, состоящей из водорода, амино, фтора и иода и Rb выбран из группы, состоящей из водорода, метокси, амино, метиламино, диметиламино и гидрокси. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда Ra', Raʺ, Raʺ' и Raiv независимо друг от друга выбраны из группы, состоящей из водорода, амино, фтора и иода и Rb представляет собой водород. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда один из Ra', Raʺ, Raʺ' и Raiv представляет собой амино, один из Ra', Raʺ, Raʺ' и Raiv представляет собой фтор и другие Ra', Raʺ, Raʺ' и Raiv представляют собой водород и Rb представляет собой водород. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда один из Ra', Raʺ, Raʺ' и Raiv представляет собой амино, один из Ra', Raʺ, Raʺ' и Raiv представляет собой иод и другие Ra', Raʺ, Raʺ' и Raiv представляют собой водород и Rb представляет собой водород. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда один из Ra', Raʺ, Raʺ' и Raiv представляет собой иод и другие Ra', Raʺ, Raʺ' и Raiv представляют собой водород и Rb представляет собой водород. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда один из Ra', Raʺ, Raʺ' и Raiv представляет собой фтор и другие Ra', Raʺ, Raʺ' и Raiv представляют собой водород и Rb представляет собой водород.
Еще один аспект настоящего изобретения формулы I реализуется, когда X представляет собой N и R2 объединяется с соседним R1 с образованием 9-10-членного бициклического кольца, необязательно прерванного N, S и/или O, указанное бициклическое кольцо необязательно замещено 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда образовавшийся бицикл представляет собой необязательно замещенный хиназолинил.
Соединения по настоящему изобретению могут иметь асимметричные центры, хиральные оси и хиральные плоскости, и могут существовать в виде рацематов, рацемических смесей и в виде отдельных диастереомеров, со всеми возможными изомерами, включая оптические изомеры, которые включены в настоящее изобретение. (см. E.L. Eliel and S.H. Wilen Stereochemistry of Carbon Compounds (John Wiley and Sons, New York 1994), в частности, стр. 1119-1190)
Когда любая переменная (например, арил, гетероцикл, R1a, R6 и и тому подобное) более одного раза встречается в любой составляющей, то в каждом случае определение этой переменной не зависит от ее определения в каждом другом случае. Кроме того, комбинации заместителей/или переменных допустимы, если только такие комбинации приводят к стабильным соединениям, являются химически возможными и/или позволяет валентность.
Как используется в настоящем описании термин "алкил" включает как разветвленные, так и линейные насыщенные алифатические углеводородные группы, имеющие определенное число атомов углерода; "алкокси" означает алкильную группу из указанного количества атомов углерода, присоединенную через кислородный мостик.
Термин "галоген" или "гало", используемый в настоящем описании, означает фтор, хлор, бром и иод.
Как используется в настоящем описании, алкенил представляет C2-C6 алкенил.
Как используется в настоящем описании, алкинил представляет C2-C6 алкинил.
Как используется в настоящем описании, "циклоалкил" включает циклические насыщенные алифатические углеводородные группы, имеющие определенное число атомов углерода. Предпочтительно, циклоалкил представляет собой C3-C10 циклоалкил. Примеры таких циклоалкильных групп включают, но ими не ограничиваются, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Как используется в настоящем описании, термин "арил" означает любое стабильное моноциклическое или бициклическое углеродное кольцо, содержащее до 7 членов в каждом кольце, где по меньшей мере одно кольцо является ароматическим. Примеры таких арильных элементов включают фенил, нафтил, тетрагидронафтил, инданил, бифенил, фенантрил, антрил или аценафтил.
Термин гетероциклил, гетероцикл или гетероциклический, как используется в настоящем описании, представляет собой стабильное 5-7-членное моноциклическое или стабильное 8-11-членное бициклическое гетероциклическое кольцо, которое является насыщенным или ненасыщенным и которое состоит из атомов углерода и одного-четырех гетероатомов, выбранных из группы, состоящей из N, O и S, и включает любую бициклическую группу, в которой любое из определенных выше гетероциклических колец конденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено по любому гетероатому или атому углерода, который приводит к созданию стабильной структуры. Термин гетероциклил, гетероцикл или гетероциклический включает гетероарильные фрагменты. Примеры таких гетероциклических фрагментов включают, но ими не ограничиваются, азепинил, бензодиоксолил, бензимидазолил, бензизоксазолил, бензофуразанил, бензопиранил, бензотиопиранил, бензофурил, бензотиазолил, бензотиенил, бензотриазолил, бензоксазолил, хроманил, циннолинил, дигидробензофурил, дигидробензотиенил, дигидробензотиопиранил, сульфон дигидробензотиопиранила, 1,3-диоксоланил, фурил, фуропиридинил, имидазолидинил, имидазолинил, имидазолил, индолинил, индолил, изохроманил, изоиндолинил, изохинолинил, изотиазолидинил, изотиазолил, изотиазолидинил, морфолинил, нафтиридинил, оксадиазолил, 2-оксоазепинил, оксазолил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, пиперидил, пиперазинил, пиридил, пиразинил, пиразолидинил, пиразолил, пиразолопиридинил, пиридазинил, пиримидинил, пирролидинил, пирролил, пирролопиридинил, хиназолинил, хинолинил, хиноксалинил, тетрагидрофурил, тетрагидроизохинолинил, тетрагидрохинолинил, тиаморфолинил, сульфоксид тиаморфолинила, тиазолил, тиазолинил, тиенофурил, тиенотиенил, тиенил и триазолил.
Предпочтительно, гетероциклил выбран из фуропиридинила, имидазолила, индолила, изохинолинилизотиазолила, морфолинила, нафтиридинила, пиперидила, пиперазинила, пиридила, пиразинила, пиразолидинила, пиразолила, пиразолопиридинила, пиридазинила, пиримидинила, пирролидинила, пирролила, пирролопиридинила, хиназолинила, хинолинила, хиноксалинила, тетрагидрофурила, тетрагидроизохинолинила, тетрагидрохинолинила, тиазолила, тиазолинила, тиенофурила, тиенотиенила, тиенила и триазолила.
"Гетероарил" означает любое стабильное моноциклическое или бициклическое углеродное кольцо, содержащее до 7 членов в каждом кольце, где по меньшей мере одно кольцо является ароматическим и где от одного до четырех атомов углерода замещены гетероатомами, выбранными из группы, состоящей из N, О и S. Примеры таких гетероциклических фрагментов включают, но ими не ограничиваются, имидазолил, индолинил, индолил, изохроманил, изоиндолинил, изохинолинил, изотиазолил, нафтиридинил, оксадиазолил, пиридил, пиразинил, пиразолил, пиридазинил, пиримидинил, пирролил, хиназолинил, хинолинил, тетрагидроизохинолинил, тетрагидрохинолинил, тиазолил, тиенофурил, тиенотиенил, тиенил, триазолил и тому подобное.
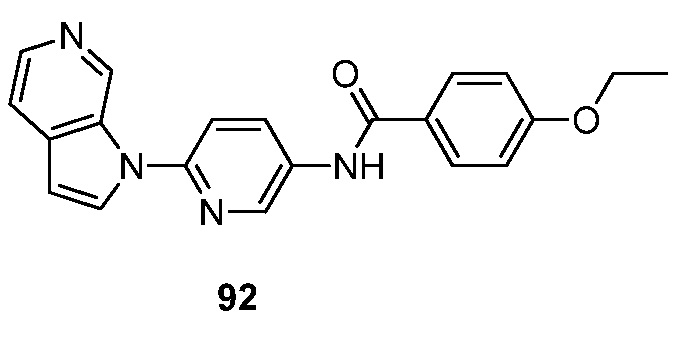
Примеры "эффективного количества" включают количества, делающие возможной визуализацию амилоидного(ых) отложения(й) in vivo, которые обеспечивают приемлемые для фармацевтического применения уровни токсичности и биодоступности и/или предотвращают дегенерацию клеток и токсичность, связанную с образованием фибрилл.
Для использования в медицине соли соединений формулы I должны быть фармацевтически приемлемыми солями. Другие соли могут, однако, быть полезными для получения соединений по настоящему изобретению или их фармацевтически приемлемых солей. Когда соединение по настоящему изобретению является кислотным, термин подходящие "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований, включающих неорганические основания и органические основания. Соли, образованные неорганическими основаниями, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и тому подобного. В особенности предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, образованные фармацевтически приемлемыми органическими нетоксичными основаниями включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин кофеин, холин, N,N 1-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и тому подобное.
Когда соединение по настоящему изобретению является основным, соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изэтиновую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, муциновую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновая кислоту и тому подобные. Особенно предпочтительными являются лимонная, бромистоводородная, хлористоводородная, малеиновая, фосфорная, серная и винная кислоты.
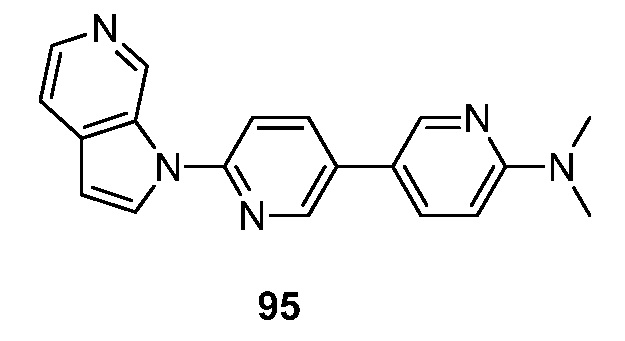
Получение фармацевтически приемлемых солей, описанных выше, и других типичных фармацевтически приемлемых солей более полно описано Berg et al., ʺPharmaceutical Salts,ʺ J. Pharm. Sci., 1977:66:1-19,
Как указано в настоящем документе настоящее изобретение включает меченные изотопами соединения по настоящему изобретению. ʺИзотопно-меченноеʺ, ʺмеченное радиоактивным изотопомʺ соединение, ʺиндикаторʺ, ʺрадиоактивный индикаторʺ, ʺмеченый индикаторʺ или ʺрадиолигандʺ представляет собой соединение, в котором один или несколько атомов замещены или заменены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно обнаруживаемой в природе (то есть, встречающейся в природе). Подходящие радионуклиды (например, ʺобнаруживаемые изотопыʺ), которые могут быть включены в соединения по настоящему изобретению, включают, но ими не ограничиваются, 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36Cl, 82Br, 76Br, 77Br, 123I, 124I и 131I. Меченные изотопами соединения по настоящему изобретению должны быть только обогащены обнаруживаемым изотопом до или выше степени, которая делает возможным обнаружение способом, подходящим для конкретного применения. Радионуклид, который включен в данные меченные радиоактивным изотопом соединения, будет зависеть от конкретного применения этого меченного радиоактивным изотопом соединения. В другом варианте осуществления настоящего изобретения радионуклиды представлены 11C, 13C, 14C, 18F, 15O, 13N, 35S, 2H и 3H и, предпочтительно, 11C и 18F.
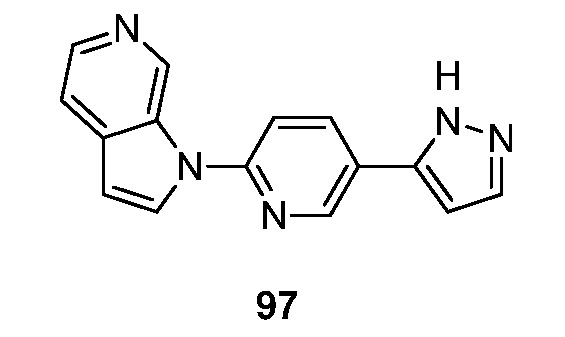
Настоящее изобретение также относится к фармацевтической композиции, содержащей эффективное количество по меньшей мере одного соединения формулы I и фармацевтически приемлемый носитель. Композиция может содержать, но ими не ограничивается, один или несколько буферных средств, смачивающих средств, эмульгаторы, суспендирующие средства, смазывающие вещества, адсорбенты, поверхностно-активные вещества, консерванты и тому подобное. Композиция может быть приготовлена в виде твердого вещества, жидкости, геля или суспензии для перорального введения (например, раствор, болюс, таблетка, порошок, капсула, спрей для рта, эмульсия); парентерального введения (например, подкожной, внутримышечной, внутривенной, эпидуральной инъекцией); местного нанесения (например, крем, мазь, пластырь с регулируемым высвобождением, спрей); интравагинального, ректального, трансдермального, глазного или назального введения.
Настоящее изобретение относится к меченым радиоактивным изотопом производным пирролопиридинила в качестве агентов визуализации тау-белка и соединениям, представляющим собой синтетические предшественники, из которых их получают. Соединения формулы I являются активными в отношении возрастных заболеваний, таких, как болезнь Альцгеймера, а также других тауопатий и нейродегенеративных заболеваний, таких как прогрессирующий супрануклеарный паралич, хроническая травматическая энцефалопатия, лобно-височная деменция, болезнь Пика, кортикобазальная дегенерация и тому подобное. Соединения по настоящему изобретению могут быть также использованы в сочетании с широким спектром средств улучшения когнитивных нарушений. Таким образом, в другом варианте осуществления настоящего изобретения соединение формулы (I) или его фармацевтически приемлемую соль, или фармацевтическую композицию или состав, содержащий соединение формулы (I), вводят одновременно, параллельно, последовательно или раздельно с другим фармацевтически активным соединением или соединениями, используемыми в терапии болезни Альцгеймера, в том числе, например, донепезилом, мемантином, такрином и их эквивалентами и их фармацевтически активными изомером (ами) и метаболитом (ами).
Настоящее изобретение также относится к способу лечения или профилактики связанной с таупатией у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы I. Настоящее изобретение также относится к способу лечения нейродегенеративных заболеваний, таких как деменция, когнитивные нарушения при шизофрении, умеренные когнитивные нарушения, возрастное нарушение памяти, возрастное снижение когнитивных способностей и тому подобное.
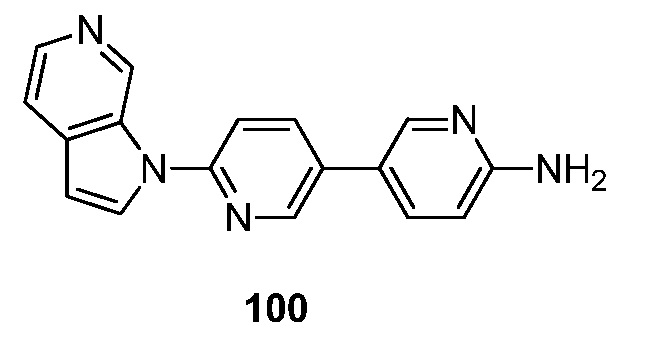
Конечной целью настоящего изобретения является создание радиофармацевтического средства, полезного в визуализации тау-белка, который имеет высокую удельную радиоактивность и высокую селективность ткани-мишени благодаря его высокому сродству к агрегатам тау-белка. Тканеселективность дает возможность дополнительного усиления путем связывания этого высокоселективного радиофармпрепарата с нацеливающими агентами, такими как микрочастицы.
В другом аспекте настоящего изобретения заявляемые соединения обладают неожиданно низким связывающим потенциалом в тау свободном кортикальном сером веществе и прилегающем к нему белом веществе, что обеспечивает улучшенные профили по отношению к связывающему потенциалу в белом веществе.
В соответствии с настоящим изобретением, наиболее предпочтительный способ визуализации отложений тау-белка у пациента, где в качестве визуализирующего агента используют меченное изотопами новое производное пирролопиридина, включает следующие стадии: пациента помещают в положении лежа на спине в ПЭТ-камеру, и вводят достаточное количество (<10 мКи) меченного изотопами производного пирролопиридина в ткани головного мозга пациента. Выполняют эмиссионную томографию головного мозга. Методика выполнения эмиссионной томографии головы хорошо известна специалистам в данной области. Методы ПЭТ описаны Freeman et al., Freeman and Johnson's Clinical Radionuclide Imaging. 3rd. Ed. Vol. 1 (1984); Grune & Stratton, New York; Ennis et Q. Vascular Radionuclide Imaging: A Clinical Atlas, John Wiley & Sons, New York (1983).
Термин "меченый индикатор" относится к любой молекуле, которая может использоваться для отслеживания или обнаружения определенной активности in vivo, например, предпочтительным индикатором является тот, который накапливается в областях, где агрегаты тау можно обнаружить. Предпочтительно, меченый индикатор это тот, который можно видеть в живом экспериментальном животном, здоровом человеке или пациенте (называемый как субъект), например, с помощью позитронно-эмиссионной томографии (ПЭТ). Подходящие индикаторы включают, но ими не ограничиваются, радиоактивные изотопы, флуорохромы, хемилюминесцентные соединения, красители и белки, в том числе ферменты.
Настоящее изобретение также относится к способам определения in vivo активности фермента или другой молекулы. Более конкретно, индикатор, который специфически отслеживает целенаправленную активность, выбирают и метят. В предпочтительном варианте осуществления индикатор отслеживает связывающую активность тау-белка в головном мозге и центральной нервной системе. Индикатор обеспечивает средства для оценки различных нейрональных процессов, включая, регуляцию высвобождения нейротрансмиттеров и долговременную потенциацию. Настоящее изобретение дает исследователям средства для изучения биохимических механизмов боли, тревоги/депрессии, привыкания к лекарственным средствам и абстиненции, нарушения в базальных ганглиях, расстройства пищевого поведения, ожирения, длительной депрессии, обучения и памяти, развития синаптической пластичности, гипоксически-ишемического повреждения и гибели нейронов, эпилептических приступов, обработки визуальной информации, а также патогенеза некоторых нейродегенеративных заболеваний.
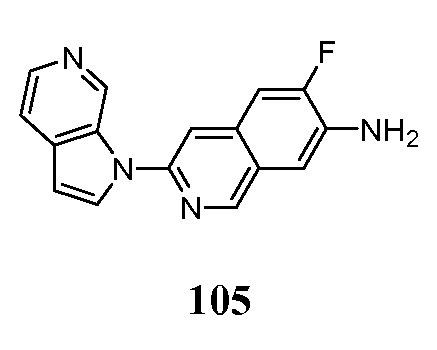
Биомаркеры состояния, прогноза и прогрессирования болезни Альцгеймера, будут полезны для общих диагностических утилит, а также для программ клинической разработки терапевтических средств для лечения болезни Альцгеймера. Настоящее изобретение предоставляет информацию о биомаркере, поскольку пациенты участвуют в клинических испытаниях новых терапий болезни Альцгеймера для помощи в отборе пациентов и отнесение к когортам. Настоящее изобретение будет служить в качестве одного биомаркера состояния заболевания, чтобы найти правильных пациентов в надлежащую когорту исследования PhIIb. Кроме того, настоящее изобретение может служить одним маркером прогноза заболевания в качестве критерия отбора включения для того, чтобы повысить вероятность того, что болезнь будет прогрессировать в группе лечения плацебо, как результат, который преследует недавние клинические испытания AD. Наконец, настоящее изобретение может служить в качестве одного биомаркера прогрессирования заболевания для наблюдения за клиническим течением заболевания пациентов, получающих терапию, и может обеспечить независимую оценку по биомаркеру ответной реакции на лечение терапевтическим средством.
Соединения по настоящему изобретению представляют собой ингибиторы и/или связующие вещества агрегированного тау-белка. Соединения и меченные изотопами их варианты, могут быть полезны для диагностики и/или лечения болезни Альцгеймера, депрессии, шизофрении или болезни Паркинсона. Средства детектирования меток хорошо известны специалистам в данной области техники. Например, изотопные метки могут быть обнаружены с помощью методов визуализации, фотографической пленки или сцинтилляционных счетчиков. В предпочтительном варианте осуществления метку детектируют in vivo в головном мозге субъекта с помощью методов визуализации, например, позитронно-эмиссионной томографией (ПЭТ).
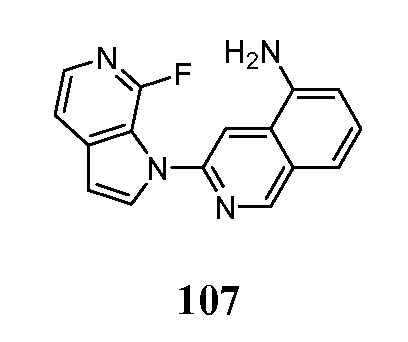
Меченое соединение по настоящему изобретению, предпочтительно, содержит по меньшей мере один радионуклид в качестве метки. Позитрон-излучающие радионуклиды являются все кандидатами для использования. В контексте настоящего изобретения радионуклид, предпочтительно, выбран из 11C, 13C, 14C, 18F, 15O, 13N, 35S, 2H и 3H, более предпочтительно, из 11C и 18F.
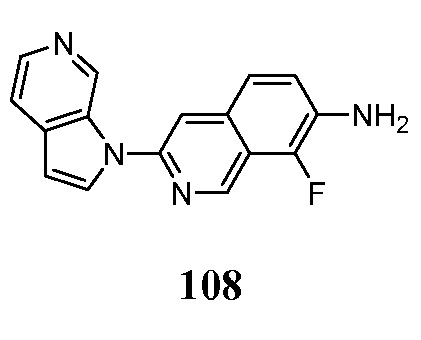
Индикатор может быть выбран в соответствии с выбранным способом детектирования. Перед проведением способа по настоящему изобретению, диагностически эффективное количество меченого или немеченого соединения по настоящему изобретению вводят в живой организм, включая человека.
Диагностически эффективное количество меченого или немеченого соединения по настоящему изобретению, вводимого перед проведением метода in-vivo для настоящего изобретения, находится в пределах диапазона от 0,1 нг до 100 мг на кг массы тела, предпочтительно, в диапазоне от 1 нг до 10 мг на кг массы тела.
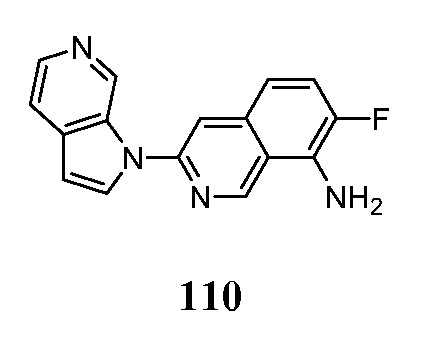
Меченные изотопами соединения по настоящему изобретению получают путем включения изотопа, такого как 11C, 13C, 14C, 18F, 15O, 13N, 35S, 2H и 3H в молекулу подложки. Это достигается за счет использования реагентов, которые имели один или несколько радиоактивных атомов, содержащихся в них, полученных путем помещения их в источник радиоактивности, такой как ядерный реактор, циклотрон и тому подобное. Кроме того многие изотопно меченые реагенты, такие как 2H2O, 3H3CI, 14C6H5Br, ClCH214COCl и тому подобное, являются коммерчески доступными. Изотопно-меченые реагенты затем используются в стандартных синтетических методах органической химии для включения атома или атомов изотопа, в соединение формулы I, как описано ниже. Следующие схемы иллюстрируют, как получить соединения формулы I.
Соединения по настоящему изобретению могут быть пригодны в диагностике, мониторинге и оценки болезни Альцгеймера и других не-БА тауопатий, отличных от болезни Альцгеймера, таких как лобно-височная деменция (FTD), прогрессирующий надъядерный паралич (ПНП), кортикобазальная дегенерация (КБД), хроническая травматическая энцефалопатия (ХТЭ), болезнь Пика, и тому подобное. Другие заболевания, которые могут быть диагностированы с помощью соединений по настоящему изобретению, включают болезнь Паркинсона, легочную гипертензию, хроническое обструктивное заболевание легких (ХОЗЛ), астму, недержание мочи, глаукому, шизофрению, трисомию по 21 хромосоме (синдром Дауна), церебральную амилоидную ангиопатию, дегенеративную деменцию, наследственную церебральную геморрагию с амилоидозом голландского типа (HCHWA-D), болезнь Крейцфельдта-Якоба, прионные болезни, боковой амиотрофический склероз, черепно-мозговую травму, инсульт, панкреатит, миозит с включенными тельцами, другие периферические амилоидозы, диабет, аутизм и атеросклероз.
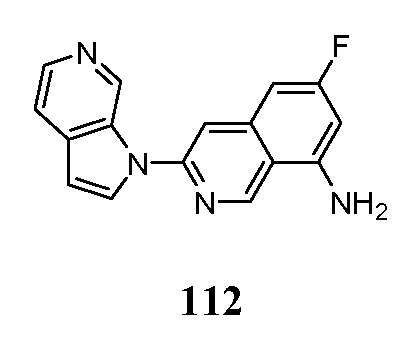
В предпочтительных вариантах осуществления соединения по настоящему изобретению могут быть использованы в диагностике, мониторинге или оценке болезни Альцгеймера, тауопатий, не относящихся к БА, нейродегенеративного заболевания, расстройства познавательной способности, шизофрении, расстройства, связанные с болью и расстройств сна. Например, соединения могут быть полезны для профилактики деменции альцгеймеровского типа, а также для лечения деменции альцгеймеровского типа на ранней стадии, промежуточной стадии или поздней стадии.
Термин "композиция", как используется в настоящем описании, относиться к продукту, содержащему определенные ингредиенты в заранее определенных количествах или пропорциях, а также любому продукту, который образуется непосредственно или опосредованно из сочетания определенных ингредиентов в определенных количествах. Этот термин относительно фармацевтической композиции охватывает продукт, содержащий один или несколько активных ингредиентов и необязательно носитель, содержащий инертные ингредиенты, а также любой продукт, который образуется непосредственно или опосредованно, из комбинации, комплексообразования или агрегации любых двух или нескольких ингредиентов, или из диссоциации одного или нескольких ингредиентов, или из других типов реакций или взаимодействий одного или нескольких ингредиентов.
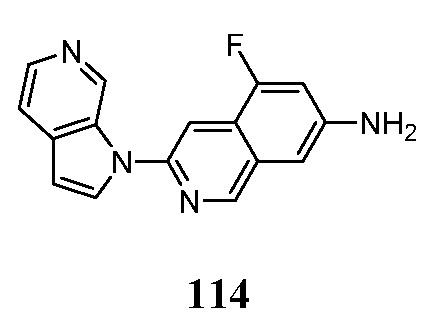
Обычно фармацевтические композиции готовят путем однородного и тщательного перемешивания активного ингредиента с жидким носителем или тонкоизмельченным твердым носителем или с обоими, и затем, если необходимо, формируют продукт в желаемый препарат. В фармацевтической композиции активное соединение, которое представляет собой соединение формул (I)-(VIII), находится в количестве, достаточном для получения желаемого воздействия на протекание или состояние заболевания. Таким образом, фармацевтические композиции по настоящему изобретению включают любую композицию, полученную путем смешивания соединения по настоящему изобретению и фармацевтически приемлемого носителя.
Настоящее изобретение также относится к способу синтеза соединений, используемых в качестве промежуточных соединений при получении соединений по настоящему изобретению.
Соединения по настоящему изобретению могут быть получены согласно способам, представленным нижеследующими схемами и примерами, используя подходящие вещества, и в дальнейшем иллюстрируются нижеследующими конкретными примерами. Однако не следует считать, что соединения, иллюстрируемые в примерах, образуют единственный класс соединений, который предлагается в виде изобретения. Кроме того, примеры иллюстрируют детали получения соединений по настоящему изобретению. Специалистам в данной области, очевидно, что для получения этих соединений могут быть использованы известные вариации условий и способов осуществления нижеприведенных препаративных способов получения. Все температуры выражены в градусах Цельсия, если не указано иное. Масс-спектры (МС) измеряли с помощью масс-спектрометрии с электрораспылительной ионизацией (ESI). 1H-ЯМР-спектры записывали при 400-500 МГц. Соединения, описанные в настоящем документе, синтезировали в виде рацемической смеси, если не указано иное, в экспериментальных методах.
В некоторых случаях конечный продукт может быть дополнительно модифицирован, например, путем манипуляции с заместителями. Такие манипуляции могут включать, но ими не ограничиваются, реакции восстановления, окисления, алкилирования, ацилирования и гидролиза, которые обычно известны специалистам в данной области. В некоторых случаях порядок проведения вышеописанных схем реакций может меняться для облегчения реакции или во избежание нежелательных продуктов реакции. Следующие примеры приведены для того, чтобы изобретение могло быть более понятным. Эти примеры являются только иллюстративными и не должны рассматриваться как ограничивающие изобретение каким-либо образом.
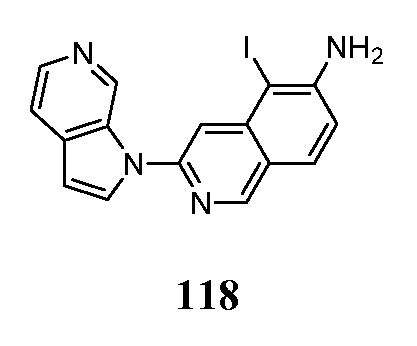
Перечень сокращений
аналит.=аналитический
n-BuLi=н-бутиллитий
ушир.=уширенный
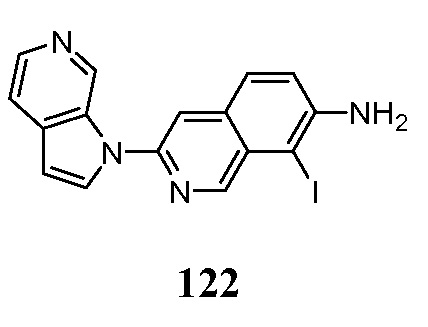
рассч.=рассчитанный
m-CPBA=3-хлорпероксибензойная кислота
д=дублет
DEA=диэтиламин
DIPEA=N,N-диизопропилэтиламин
DMF=диметилформамид
ESI=электрораспылительная ионизация
EtOAc=этилацетат
EtOH=этанол
ВЭЖХ=высокоэффективная жидкофазная хроматография
IPA=изопропиловый спирт
IPAc=изопропилацетат
KF=титрование по методу Карла Фишера (определение количества воды)
KOt-Bu=трет-бутоксид калия
ЖХМС=жидкостная хроматография с масс-спектрометрией
LiHMDS=гексаметил силазан лития
м=мультиплет
MeCN=ацетонитрил
MeOH=метиловый спирт
мПa=миллипаскаль
МС=масс-спектрометрия
MTBE=метил-трет-бутиловый эфир
NHS=нормальная человеческая сыворотка
ЯМР=ядерная магнитно-резонансная спектроскопия
Piv=пивалат, 2,2-диметилпропаноил
Pd/C=палладий на угле
кт=комнатная температура
с=синглет
СФХ=сверхкритическая флюидная хроматография
т=триплет
ТСХ=тонкослойная хроматография
p-TsOH=пара-толуолсульфоновая кислота
THF=тетрагидрофуран
масс.% =процент по массе
Следующие примеры приведены для иллюстрации настоящего изобретения и не должны истолковываться как ограничивающие объем настоящего изобретения каким-либо образом.
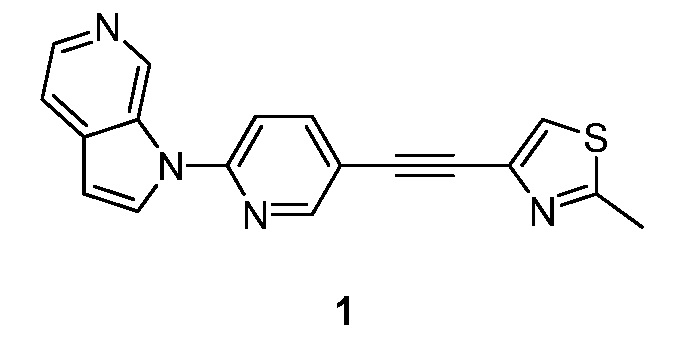
















































































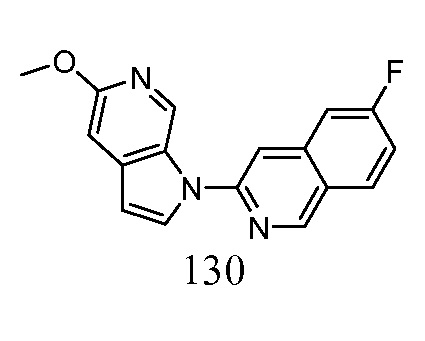
Пример 1
Синтез 1-(5-(2-(2-метилтиазол-4-ил)этинил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (1):
Схема 1
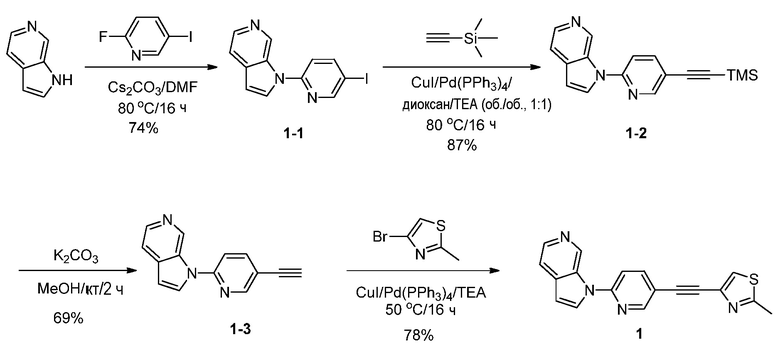
Стадия 1: Синтез 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (1-1). К раствору 1H-пирроло[2,3-с]пиридина (20 г, 0,17 моль) в N,N-диметилформамиде (300 мл) добавляли 2-фтор-5-иодпиридин (45 г, 0,20 моль) и карбонат цезия (110 г, 0,34 моль). Полученную смесь перемешивали в течение 16 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (1 л) и экстрагировали дихлорметаном (3×200 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,5~3% метанола в дихлорметане, с получением 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 322,0 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,61 (с, 1H), 8,77 (д, J=2,0 Гц, 1H), 8,38 (д, J=5,6 Гц, 1H), 8,12-8,09 (м, 1H), 7,81 (д, J=3,6 Гц, 1H), 7,56-7,55 (м, 1H), 7,34-7,28 (м, 1H), 6,72 (д, J=3,6 Гц, 1H).
Стадия 2: Синтез 1-(5-(2-(триметилсилил)этинил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (1-2). К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (4,1 г, 12,5 ммоль) в 1,4-диоксане (50 мл) и триэтиламине (50 мл) добавляли этинилтриметилсилан (3,7 г, 37,4 ммоль), иодид меди (I) (1,4 г, 7,5 ммоль) и тетракис(трифенилфосфин)палладий(0) (1,4 г, 1,2 ммоль). Полученную смесь перемешивали в течение 16 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (100 мл) и органический слой отделяли. Водный слой экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,3~3% метанола в дихлорметане, с получением 1-(5-(2-(триметилсилил)этинил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 292,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,93 (с, 1H), 8,97 (д, J=2,4 Гц, 1H), 8,76 (д, J=1,5 Гц, 1H), 8,51 (д, J=6,3 Гц, 1H), 8,25-8,21 (м, 2H), 8,07 (д, J=8,4 Гц, 1H), 7,24 (д, J=3,3 Гц, 1H), 0,17 (с, 9H).
Стадия 3: Синтез 1-(5-этинилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (1-3). Раствор 1-(5-((триметилсилил)этинил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (4,5 г, 15,4 ммоль) в метаноле (50 мл) обрабатывали карбонатом калия (4,3 г, 30,9 ммоль) в течение 2 часов при температуре окружающей среды. Полученную смесь нейтрализовали уксусной кислотой и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,3~3% метанола в дихлорметане, с получением 1-(5-этинилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 220,0 [M+1]+1; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,95 (с, 1H), 9,00 (д, J=2,4 Гц, 1H), 8,79 (д, J=1,5 Гц, 1H), 8,53 (д, J=6,3 Гц, 1H), 8,28-8,23 (м, 2H), 8,10 (д, J=8,4 Гц, 1H), 7,28 (д, J=3,3 Гц, 1H), 4,58 (с, 1H).
Стадия 4: Синтез 1-(5-(2-(2-метилтиазол-4-ил)этинил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (1). К раствору 1-(5-этинилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (141 мг, 0,64 ммоль) в триэтиламине (10 мл) добавляли 4-бром-2-метилтиазол (178 мг, 1 ммоль), иодид меди (I) (48 мг, 0,26 ммоль) и тетракис(трифенилфосфин)палладий(0) (49 мг, 0,043 ммоль). Полученную смесь перемешивали в течение 16 часов при 50°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~3% метанола в дихлорметане, с получением 1-(5-(2-(2-метилтиазол-4-ил)этинил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 317,0 [M+1]+; 1H ЯМР (400 МГц, CD3OD) δ 9,88 (ушир.с, 1H), 8,79 (д, J=1,6 Гц, 1H), 8,34-8,33 (м, 2H), 8,15-8,12 (м, 1H), 7,85-7,82 (м, 2H), 7,77 (с, 1H), 6,96 (д, J=3,2 Гц, 1H), 2,76 (с, 3H).
Пример 2
Синтез (E)-1-(5-стирилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (2):
Схема 2
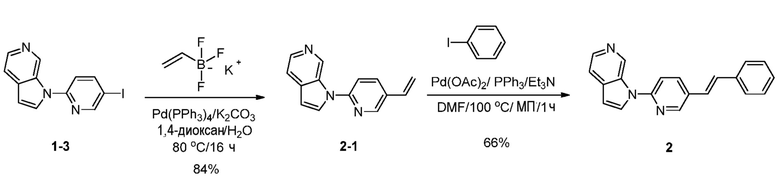
Стадия 1: Синтез 1-(5-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (2-1). К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (2 г, 6,23 ммоль) в 1,4-диоксане (40 мл) и воды (4 мл) добавляли трифтор(винил)борат калия (1,25 г, 9,34 ммоль), карбонат калия (1,72 г, 12,46 ммоль) и тетракис(трифенилфосфин)палладий(0) (0,72 г, 0,62 ммоль). Полученную смесь перемешивали в течение 16 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды, смесь разбавляли водой (100 мл) и органический слой отделяли. Водный слой экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,3~2% метанола в дихлорметане, с получением 1-(5-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 222,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,79 (с, 1H), 8,70 (д, J=2,4 Гц, 1H), 8,35 (д, J=3,6 Гц, 1H), 8,31 (д, J=5,2 Гц, 1H), 8,23 (дд, J=2,0 Гц, 8,4 Гц, 1H), 7,91 (д, J=8,4 Гц, 1H), 7,68 (дд, J=0,8 Гц, 4,4 Гц, 1H), 6,90-6,83 (м, 2H), 6,08 (д, J=17,6 Гц, 1H), 5,46 (д, J=11,2 Гц, 1H).
Стадия 2: Синтез (E)-1-(5-стирилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (2). К раствору 1-(5-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (100 мг, 0,4 ммоль) в N,N-диметилформамиде (2,5 мл) добавляли бис(ацетато)палладий(II), (10 мг, 0,045 ммоль), 1-иодбензол (138 мг, 0,68 ммоль), трифенилфосфин (12 мг, 0,045 ммоль) и триэтиламин (91 мг, 0,9 ммоль). Полученную смесь облучали в микроволновой печи (100 В) в течение 1 часа при 100°C. После охлаждения до температуры окружающей среды, полученную смесь гасили водой (20 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,5~2% метанола в дихлорметане, с получением (E)-1-(5-стирилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 298,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,71 (с, 1H), 8,79 (д, J=2,1 Гц, 1H), 8,56-8,32 (м, 3H), 7,93 (д, J=8,7 Гц, 1H), 7,67-7,64 (м, 3H), 7,48-7,29 (м, 5H), 6,83 (д, J=4,2 Гц, 1H).
Пример 3
Синтез 1-(5-(пиридин-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (3):
Схема 3
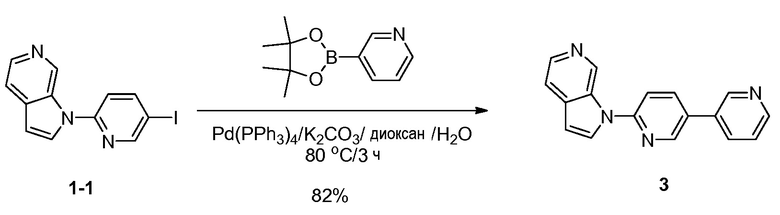
Синтез 1-(5-(пиридин-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (3). К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (100 мг, 0,31 ммоль) в 1,4-диоксане (20 мл) и воде (5 мл) добавляли 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (96 мг, 0,47 ммоль), карбонат калия (129 мг, 0,93 ммоль) и тетракис(трифенилфосфин)палладий(0) (18 мг, 0,016 ммоль). Полученную смесь перемешивали в течение 3 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (100 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~2% метанола в дихлорметане, с получением 1-(5-(пиридин-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 273,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,85 (с, 1H), 9,05-9,01 (м, 2H), 8,66-8,64 (м, 1H), 8,44-8,39 (м, 2H), 8,27-8,23 (м, 2H), 8,04 (д, J=8,7 Гц, 1H), 7,75-7,73 (м, 1H), 7,68-7,54 (м, 1H), 6,89 (д, J=3,3 Гц, 1H).
Пример 4
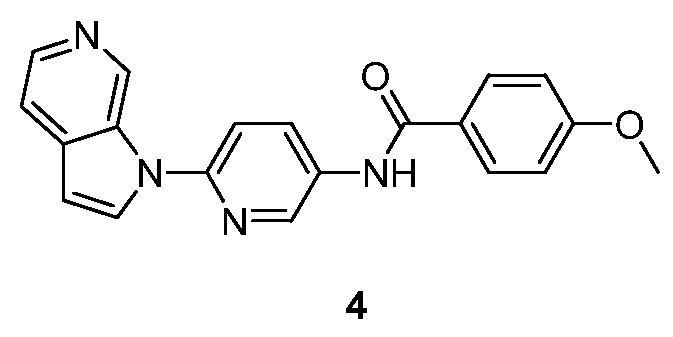
Синтез N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-4-метоксибензамида (4):
Схема 4

Стадия 1: Синтез 1-(5-нитропиридин-2-ил)-1H-пирроло[2,3-с]пиридина (4-1). К раствору 1H-пирроло[2,3-с]пиридина (2 г, 16,9 ммоль) в N,N-диметилформамиде (100 мл) добавляли 2-фтор-5-нитропиридин (2,9 г, 20,3 ммоль) и карбонат цезия (11,0 г, 33,9 ммоль). Полученную смесь перемешивали в течение 16 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (500 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,3~2% метанола в дихлорметане, с получением 1-(5-нитропиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде оранжевого твердого вещества: МС (ESI, m/z): 241,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,95 (с, 1H), 9,46 (д, J=2,4 Гц, 1H), 8,79 (дд, J=2,8 Гц, 6,4 Гц, 1H), 8,50 (д, J=3,6 Гц, 1H), 8,39 (д, J=5,2 Гц, 1H), 8,16 (д, J=9,2 Гц, 1H), 7,72 (д, J=5,2 Гц, 1H), 7,00 (д, J=3,6 Гц, 1H).
Стадия 2: Синтез 6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (4-2). К перемешиваемому раствору 1-(5-нитропиридин-2-ил)-1H-пирроло[2,3-с]пиридина (0,8 г, 3,33 ммоль) в метаноле (50 мл) добавляли палладий на угле (1,0 г, 10% масс./масс.). Полученную смесь выдерживали в атмосфере водорода (1 атм.) в течение 3 часов при температуре окружающей среды. Затем смесь фильтровали через целит и выпаривали фильтрат при пониженном давлении с получением 6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина в виде желтого твердого вещества: МС (ESI, m/z): 211,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,39 (с, 1H), 8,25 (д, J=2,4 Гц, 1H), 8,09 (дд, J=2,8 Гц, 6,4 Гц, 1H), 7,96 (д, J=3,6 Гц, 1H), 7,63 (д, J=5,2 Гц, 1H), 7,48 (д, J=9,2 Гц, 1H), 7,16 (д, J=5,2 Гц, 1H), 6,71 (д, J=3,6 Гц, 1H), 5,49 (ушир.с, 2H).
Стадия 3: Синтез N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-4-метоксибензамида (4). К раствору 6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (100 мг, 0,48 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-метоксибензойную кислоту (109 мг, 0,71 ммоль), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU) (271 мг, 0,71 ммоль) и триэтиламин (144 мг, 1,43 ммоль). Полученный раствор перемешивали в течение 16 часов при температуре окружающей среды в атмосфере азота. Реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (3×60 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,3~2% метанола в дихлорметане, с получением N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-4-метоксибензамида в виде светло-желтого твердого вещества: МС (ESI, m/z): 345,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,4 (с, 1H), 9,66 (с, 1H), 8,96 (д, J=2,4 Гц, 1H), 8,42 (дд, J=2,7 Гц, 3,3 Гц, 1H), 8,27-8,24 (м, 2H), 8,03-8,00 (м, 2H), 7,89 (д, J=9,0 Гц, 1H), 7,66-7,64 (м, 1H), 7,12-7,09 (м, 2H), 6,82 (д, J=3,0 Гц, 1H), 3,85 (с, 3H).
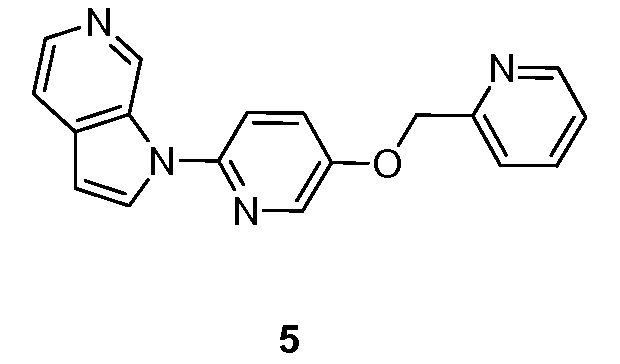
Пример 5
Синтез 1-(5-(пиридин-2-илметокси)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (5):
Схема 5

Стадия 1: Синтез 2-((6-бромпиридин-3-илокси)метил)пиридина (5-1). К раствору 6-бромпиридин-3-ола (0,5 г, 2,87 ммоль) в N,N-диметилформамиде (30 мл) добавляли 2-(бромметил)пиридин гидробромид (0,73 г, 2,87 ммоль) и карбонат калия (1,19 г, 8,62 ммоль). Полученную смесь перемешивали в течение 8 часов при температуре окружающей среды в атмосфере азота. Реакционную смесь гасили водой (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 10~30% этилацетата в петролейном эфире с получением 2-((6-бромпиридин-3-илокси)метил)пиридина в виде твердого вещества красного цвета: МС (ESI, m/z): 265,0, 267,0 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 8,62 (д, J=4,2 Гц, 1H), 8,16 (д, J=3,0 Гц, 1H), 7,77-7,71 (м, 1H), 7,49 (д, J=10,4 Гц, 1H), 7,38 (д, J=11,7 Гц, 1H), 7,29-7,20 (м, 1H), 7,18-7,17 (м, 1H), 5,22 (с, 2H)
Стадия 2: Синтез 1-(5-(пиридин-2-илметокси)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (5). К перемешиваемому раствору 1H-пирроло[2,3-с]пиридина (0,150 г, 1,27 ммоль) в диметилсульфоксиде (50 мл) добавляли 2-бром-5-(пиридин-2-илметокси)пиридин (0,67 г, 2,54 ммоль), диметилглицин (0,13 г, 1,27 ммоль), иодид меди (I) (0,24 г, 1,27 ммоль) и карбонат цезия (1,65 г, 5,08 ммоль). Полученную смесь перемешивали в течение 16 часов при 130°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле, элюировали смесью 0,3~2% метанола в дихлорметане, с получением 1-(5-(пиридин-2-илметокси)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде не совсем белого твердого вещества: МС (ESI, m/z): 303,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,62 (ушир.с, 1H), 8,62-8,60 (м, 1H), 8,43 (д, J=2,7 Гц, 1H), 8,25 (ушир.с, 1H), 8,17 (д, J=5,7 Гц, 1H), 7,90-7,74 (м, 3H), 7,63-7,59 (м, 2H), 7,41-7,36 (м, 1H), 6,80 (д, J=3,3 Гц, 1H), 5,35 (с, 2H).
Пример 6
Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (6):
Схема 6

Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (6). К перемешиваемому раствору 1H-пирроло[2,3-с]пиридина (0,13 г, 1,08 ммоль) в диметилсульфоксиде (15 мл) добавляли иодид меди (I) (0,055 г, 0,29 ммоль), 3-бромизохинолин (0,150 г, 0,72 ммоль), карбонат цезия (0,94 г, 2,88 ммоль) и диметилглицин (0,029 г, 0,29 ммоль). Полученную смесь перемешивали в течение 16 часов при 130°C в атмосфере азота. После охлаждения до температуры окружающей среды реакционную смесь гасили добавлением воды (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,5~2% метанола в дихлорметане, с получением 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин в виде не совсем белого твердого вещества: МС (ESI, m/z): 246,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,70 (с, 1H), 9,43 (с, 1H), 8,36 (д, J=3,3 Гц, 1H), 8,30-8,28 (м, 2 H), 8,21 (д, J=8,1 Гц, 1H), 8,07 (д, J=8,4 Гц, 1H), 7,88-7,82 (м, 1H), 7,71-7,66 (м, 2H), 6,87 (д, J=3,3 Гц, 1H).
Пример 7
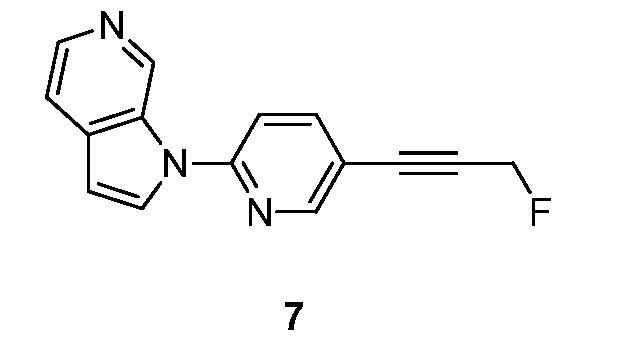
Синтез 1-(5-(3-фторпроп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (7):
Схема 7
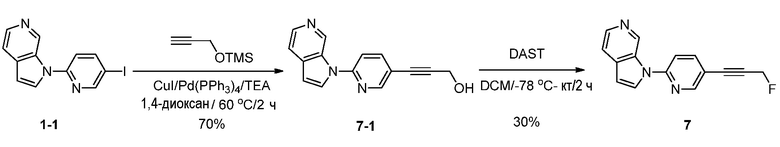
Стадия 1: Синтез 3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)проп-2-ин-1-ола (7-1).
К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (200 мг, 0,62 ммоль) в 1,4-диоксане (20 мл) и триэтиламина (5 мл) добавляли триметил(проп-2-инилокси)силан (96 мг, 0,77 ммоль), иодид меди (I) (71 мг, 0,37 ммоль) и тетракис(трифенилфосфин)палладий(0) (140 мг, 0,12 ммоль). Полученную смесь перемешивали в течение 2 часов при 60°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (100 мл) и органический слой отделяли. Водный слой экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,3~3% метанола в дихлорметане, с получением 3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)проп-2-ин-1-ола в виде бесцветного твердого вещества: МС (ESI, m/z): 250,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,79 (ушир.с, 1H), 8,67 (д, J=1,8 Гц, 1H), 8,34-8,23 (м, 2H), 8,08-8,04 (м, 1H), 7,91 (д, J=8,7 Гц, 1H), 7,67 (д, J=5,1 Гц, 1H), 6,87 (д, J=3,3 Гц, 1H), 5,45 (т, J=6,0 Гц, 1H), 4,37 (д, J=6,0 Гц, 2H).
Стадия 2: Синтез 1-(5-(3-фторпроп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (7).
К раствору 3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)проп-2-ин-1-ола (100 мг, 0,4 ммоль) в дихлорметане (20 мл) добавляли трифторид диэтиламиносеры (DAST) (2 мл) при -78°C. Полученный раствор перемешивали в течение 2 часов при температуре окружающей среды и гасили насыщенным водным раствором бикарбоната натрия (10 мл). Органический слой отделяли и водный слой экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,3~3% метанола в дихлорметане, с получением 1-(5-(3-фторпроп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 252,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,85 (ушир.с, 1H), 8,76 (д, J=2,1 Гц, 1H), 8,37 (д, J=3,3 Гц, 1H), 8,32 (д, J=5,1 Гц, 1H), 8,17-8,14 (м, 1H), 7,96 (д, J=8,7 Гц, 1H), 7,68 (д, J=5,4 Гц, 1H), 6,90 (д, J=3,3 Гц, 1H), 5,49 (с, 1H), 5,33 (с, 1H).
Пример 8
Синтез 1-(1H-пирроло[2,3-с]пиридин-5-ил)-1H-пирроло[2,3-с]пиридина (8):
Схема 8

Стадия 1: Синтез 1-(4-метил-5-нитропиридин-2-ил)-1H-пирроло[2,3-с]пиридина (8-1).
К раствору 1H-пирроло[2,3-с]пиридина (1,0 г, 8,7 ммоль) в N,N-диметилформамиде (30 мл) добавляли карбонат калия (1,6 г, 11,6 ммоль) и 2-хлор-4-метил-5-нитропиридин (1,0 г, 5,8 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 18 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды реакционную смесь разбавляли насыщенным солевым раствором (150 мл) и экстрагировали этилацетатом (2×80 мл). Объединенные органические слои промывали насыщенным солевым раствором (5×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 30~50% этилацетата в петролейном эфире с получением 1-(4-метил-5-нитропиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 255,0 [M+1]+.
Стадия 2: Синтез (E)-N,N-диметил-2-(5-нитро-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)этенамина (8-2). Раствор 1-(4-метил-5-нитропиридин-2-ил)-1H-пирроло[2,3-с]пиридина (140 мг, 0,55 ммоль) в диметилацетале N,N-диметилформамида (20 мл) перемешивали в течение 24 часов при 85°C. Растворитель удаляли при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 50~80% этилацетата в петролейном эфире с получением (E)-N,N-диметил-2-(5-нитро-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)этенамина в виде желтого твердого вещества: МС (ESI, m/z): 310,0 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,59 (с, 1H), 9,05 (с, 1H), 8,39-8,36 (м, 1H), 8,10 (д, J=3,6 Гц, 1H), 7,69 (д, J=7,0 Гц, 1H), 7,54-7,48 (м, 1H), 7,43 (с, 1H), 6,83 (д, J=3,6 Гц, 1H), 6,18 (д, J=12,4 Гц, 1H), 3,12 (с, 6H).
Стадия 3: Синтез 1-(1H-пирроло[2,3-с]пиридин-5-ил)-1H-пирроло[2,3-с]пиридина (8).
К перемешиваемому раствору (E)-N,N-диметил-2-(5-нитро-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)этенамина (40 мг, 0,13 ммоль) в метаноле (10 мл) добавляли палладий на угле (5,0 мг, 10% масс/масс). Полученную смесь выдерживали в атмосфере водорода (1 атм.) в течение 1 часа при температуре окружающей среды. Затем смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении досуха. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~3% метанола в дихлорметане, с получением 1-(1H-пирроло[2,3-с]пиридин-5-ил)-1H-пирроло[2,3-с]пиридина в виде не совсем белого твердого вещества: МС (ESI, m/z): 235,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 11,80 (ушир.с, 1H), 9,48 (с, 1H), 8,78 (с, 1H), 8,22-8,16 (м, 2H), 7,90 (с, 1H), 7,76-7,75 (м, 1H), 7,64-7,63 (м, 1H), 6,76 (д, J=3,2 Гц, 1H), 6,62 (с, 1H).
Пример 9
Синтез 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (9):
Схема 9
Стадия 1: Синтез (E)-5-фтор-2-(гидроксиимино)-2,3-дигидроинден-1-она (9-1). К перемешиваемому раствору 5-фтор-2,3-дигидроинден-1-она (3 г, 20 ммоль) в диэтиловом эфире (50 мл) барботировали сухой гидрохлоридный газ в течение 3 часов при 0°C, затем добавляли изопентилнитрит (4,7 г, 40 ммоль) в течение 5 минут. После перемешивания в течение 3 часов при температуре окружающей среды, твердое вещество собирали фильтрацией и промывали диэтиловым эфиром (3×30 мл) с получением (E)-5-фтор-2-(гидроксиимино)-2,3-дигидроинден-1-она в виде не совсем белого твердого вещества: МС (ESI, m/z): 180,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,67 (с, 1H), 7,85-7,81 (м, 1H), 7,46-7,43 (м, 1H), 7,31-7,28 (м, 1H), 3,79 (с, 2H).
Стадия 2: Синтез 1,3-дихлор-6-фторизохинолина (9-2). К перемешиваемому раствору (E)-5-фтор-2-(гидроксиимино)-2,3-дигидроинден-1-она (3 г, 13,4 ммоль) в фосфор окситрихлориде (50 мл) добавляли пентахлорфосфоран (3,2 г, 15,1 ммоль) при 0°C. Гидрохлоридный газ барботировали в полученный раствор в течение 3 часов при 0°C. Полученную смесь перемешивали в течение 16 часов при 60°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и твердое вещество промывали водой (3×50 мл), сушили в вакуумной печи с получением 1,3-дихлор-6-фторизохинолина в виде темно-серого твердого вещества: МС (ESI, m/z): 216,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,41-8,36 (м, 1H), 8,10 (с, 1H), 7,87-7,83 (м, 1H), 7,78-7,69 (м, 1H).
Стадия 3: Синтез 3-хлор-6-фторизохинолина (9-3). К перемешиваемому раствору 1,3-дихлор-6-фторизохинолина (2,5 г, 11,6 ммоль) в уксусной кислоте (40 мл) и йодистоводородной кислоты (20 мл, 45% водный раствор) добавляли красный фосфор (0,9 г, 28,9 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 4 часов при 100°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении. Остаток растворяли в дихлорметане (100 мл) и промывали насыщенным водным раствором бикарбоната натрия (2×100 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 3-хлор-6-фторизохинолина в виде темно-серого твердого вещества: МС (ESI, m/z): 182,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,24 (с, 1H), 8,32-8,29 (м, 1H), 8,04 (с, 1H), 7,82-7,76 (м, 1H), 7,66-7,61 (м, 1H).
Стадия 4: Синтез 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (9). К перемешиваемому раствору 1H-пирроло[2,3-с]пиридина (100 мг, 0,85 ммоль) и 3-хлор-6-фторизохинолина (231 мг, 1,27 ммоль) в тетрагидрофуране (20 мл) добавляли натрий 2-метилпропан-2-олат (163 мг, 1,69 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-три-изо-пропил-1,1'-бифенил)(2'''-амино-1'',1'''-бифенил-2''-ил)палладий(II) мезилат (33,6 мг, 0,042 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 16 часов при 50°C в атмосфере азота. После охлаждения до температуры окружающей среды реакционную смесь гасили водой (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~5% метанола в дихлорметане, с получением 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 264,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,69 (с, 1H), 9,43 (с, 1H), 8,40-8,25 (м, 4H), 8,01-7,95 (м, 1H), 7,71-7,68 (м, 1H), 7,58-7,51 (м, 1H), 6,89 (с, 1H).
Пример 10
Синтез N,N-диметил-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (10):
Схема 10

Стадия 1: Синтез 6-бром-N,N-диметилпиридин-3-амина (10-1). К раствору 6-бромпиридин-3-амина (300 мг, 1,73 ммоль) в тетрагидрофуране (30 мл) добавляли 1 М раствора бис(триметилсилил)амида (3,46 мл, 3,46 ммоль) в тетрагидрофуране в течение 5 минут при -78°C в атмосфере азота. После перемешивания в течение 30 минут при -78°C, добавляли йодметан (566 мг, 3,99 ммоль). Полученную смесь перемешивали в течение 1 часа при температуре окружающей среды и затем гасили добавлением насыщенного водного раствора хлорида аммония (50 мл). Полученную смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~10% этилацетата в петролейном эфире с получением 6-бром-N,N-диметилпиридин-3-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 201,0, 203,0 [M+1]+.
Стадия 2: Синтез N,N-диметил-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (10).
К перемешиваемому раствору 1H-пирроло[2,3-с]пиридина (100 мг, 0,84 ммоль) в диметилсульфоксиде (30 мл) добавляли 6-бром-N,N-диметилпиридин-3-амин (204 мг, 1,01 ммоль), диметилглицин (52 мг, 0,51 ммоль), иодид меди(I) (97 мг, 0,51 ммоль) и карбонат цезия (1,1 г, 3,39 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 16 часов при 130°C в атмосфере азота. После охлаждения до температуры окружающей среды реакционную смесь гасили водой (80 мл) и экстрагировали этилацетатом (3×80 мл). Объединенные органические слои промывали насыщенным солевым раствором (4×80 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~5% метанола в дихлорметане, с получением N,N-диметил-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 239,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,47 (с, 1H), 8,22 (д, J=5,1 Гц, 1H), 8,11-8,09 (м, 2H), 7,66 (с, 1H), 7,62-7,61 (м, 1H), 7,40-7,35 (м, 1H), 6,75 (д, J=3,3 Гц, 1H), 3,02 (с, 6H).
Пример 11
Синтез 5-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (11):
Схема 11

Стадия 1: Синтез 3-хлор-5-нитроизохинолина (11-1). К раствору 3-хлоризохинолина (2,0 г, 12,2 ммоль) в концентрированной серной кислоте (48 мл) добавляли нитрат калия (1,48 г, 14,6 ммоль) по частям при 0°C. Полученный раствор перемешивали в течение 4 часов при температуре окружающей среды и затем выливали в воду со льдом (300 г). Осадок собирали фильтрацией и сушили с получением 3-хлор-5-нитроизохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 209,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,46 (с, 1H), 8,75-8,72 (м, 1H), 8,66-8,63 (м, 1H), 8,45(с, 1H), 7,94-7,89 (м, 1H).
Стадия 2: Синтез 3-хлоризохинолин-5-амина (11-2). К раствору 3-хлор-5-нитроизохинолина (2,3 г, 11,0 ммоль) в уксусной кислоте (100 мл) добавляли цинковую пыль (3,5 г, 55,1 ммоль) по частям. Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды. Затем полученную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1% метанола в дихлорметане, с получением 3-хлоризохинолин-5-амина в виде не совсем белого твердого вещества: МС (ESI, m/z): 179,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,98 (с, 1H), 8,13 (с, 1H), 7,40-7,35 (м, 1H), 7,28-7,25 (м, 1H), 6,90-6,87 (м, 1H), 6,06 (ушир.с, 2H).
Стадия 3: Синтез трет-бутил 3-хлоризохинолин-5-илкарбамата (11-3). К раствору 3-хлоризохинолин-5-амина (1,6 г, 8,96 ммоль) в дихлорметане (50 мл) добавляли триэтиламин (1,81 г, 17,92 ммоль) и ди-трет-бутилдикарбонат (2,34 г, 10,75 ммоль). Полученную смесь перемешивали в течение 3 часов при температуре окружающей среды и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 2~10% этилацетата в петролейном эфире с получением трет-бутил (3-хлоризохинолин-5-ил)карбамата в виде светло-желтого твердого вещества: МС (ESI, m/z): 279,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,33 (с, 1H), 8,24-8,21 (м, 1H), 7,83-7,77 (м, 2H), 7,57 (с, 1H), 1,32 (с, 9H).
Стадия 4: Синтез трет-бутил 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-илкарбамата (11-4). К раствору 1H-пирроло[2,3-с]пиридина (254 мг, 2,15 ммоль) в тетрагидрофуране (25 мл) добавляли трет-бутил (3-хлоризохинолин-5-ил)карбамат (400 мг, 1,43 ммоль), натрий 2-метилпропан-2-олат (276 мг, 2,87 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-три-изо-пропил-1,1'-бифенил)(2'''-амино-1'',1'''-бифенил-2''-ил)палладий(II) мезилат (114 мг, 0,14 ммоль) в атмосфере азота. Полученный раствор перемешивали в течение 16 часов при 50°C. После охлаждения до температуры окружающей среды, смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~2% метанола в дихлорметане, с получением трет-бутил (3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-ил)карбамата в виде бесцветного твердого вещества: МС (ESI, m/z): 361,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,72 (с, 1H), 9,63 (с, 1H), 9,41 (с, 1H), 8,37-8,31 (м, 1H), 8,29 (с, 2H), 8,07 (д, J=7,6 Гц, 1H), 7,97 (д, J=8,0 Гц, 1H), 7,70 (д, J=4,4 Гц, 1H), 7,68-7,62 (м, 1H), 6,90 (д, J=4,8 Гц, 1H), 1,55 (с, 9 H).
Стадия 5: Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-амина (11-5). К раствору трет-бутил (3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-ил)карбамата (250 мг, 0,69 ммоль) в дихлорметане (20 мл) добавляли 2,2,2-трифторуксусную кислоту (791 мг, 6,94 ммоль). После перемешивания в течение 1 часа при температуре окружающей среды, полученный раствор промывали насыщенным водным раствором бикарбоната натрия (2×50 мл) и органический слой сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,5~1% метанола в дихлорметане, с получением 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-амина в виде не совсем белого твердого вещества: МС (ESI, m/z): 261,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,67 (с, 1H), 9,22 (с, 1H), 8,32-8,26 (м, 3H), 7,68-7,66 (м, 1H), 7,41-7,33 (м, 2H), 6,93-6,84 (м, 2H), 6,12 (ушир.с, 2H).
Стадия 6: Синтез 5-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (11). К раствору 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-амина (100 мг, 0,38 ммоль) в разбавленной серной кислоте (10% водный раствор) добавляли нитрит натрия (53,0 мг, 0,76 ммоль) при 0°C. После перемешивания в течение 20 минут при 0°C, добавляли калий йодид (128 мг, 0,76 ммоль) в виде одной порции. После дополнительных 10 минут, полученную смесь гасили насыщенным раствором сульфита натрия (10 мл) и экстрагировали дихлорметаном (3×25 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1,5% метанола в дихлорметане, с получением 5-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде бесцветного твердого вещества: МС (ESI, m/z): 372,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,61 (с, 1H), 9,39 (с, 1H), 8,46-8,27 (м, 4H), 7,99 (с, 1H), 7,72-7,70 (м, 1H), 7,49-7,45 (м, 1H), 6,91 (д, J=3,3 Гц, 1H).
Пример 12
Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-амина (12):
Схема 12

Стадия 1: Синтез (E)-2-(гидроксиимино)-6-иод-2,3-дигидроинден-1-она (12-1). К раствору 6-иод-2,3-дигидро-1H-инден-1-она (5 г, 19,4 ммоль) в дихлорметане (300 мл) и метаноле (15 мл, насыщенный гидрохлоридом при 0°C) добавляли раствор изопентилнитрита (4,5 г, 38,8 ммоль) в дихлорметане (10 мл) в течение 30 минут при 0°C. Полученный раствор перемешивали в течение 3 часов при температуре окружающей среды. Растворители удаляли частично до приблизительно 50 мл с последующим разбавлением эфиром (100 мл). Твердое вещество собирали фильтрацией с получением (E)-2-(гидроксиимино)-6-иод-2,3-дигидроинден-1-она в виде светло-желтого твердого вещества: МС (ESI, m/z): 288,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 12,75 (с, 1H), 8,06-8,01 (м, 2H), 7,46 (д, J=8,0 Гц, 1H), 3,72 (с, 2H).
Стадия 2: Синтез 1,3-дихлор-7-иодизохинолина (12-2). К раствору (E)-2-(гидроксиимино)-6-иод-2,3-дигидроинден-1-она (5,4 г, 18,81 ммоль) в фосфорилтрихлориде (80 мл) добавляли пентахлорфосфоран (5,9 г, 28,2 ммоль). Затем гидрохлоридный газ барботировали в раствор в течение 3 часов при 0°C. Полученный раствор перемешивали в течение 16 часов при 60°C. После охлаждения до температуры окружающей среды, смесь концентрировали при пониженном давлении и остаток обрабатывали с помощью дихлорметана (100 мл) и насыщенного водного раствора бикарбоната натрия (100 мл). Органический слой отделяли и сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении с получением 1,3-дихлор-7-иодизохинолина в виде коричневого твердого вещества: МС (ESI, m/z): 324,2 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 8,59 (с, 1H), 8,21 (дд, J=1,2 Гц, 8,4 Гц, 1H), 8,17 (с, 1H), 7,85 (д, J=8,8 Гц, 1H).
Стадия 3: Синтез 3-хлор-7-иодизохинолина (12-3). К раствору 1,3-дихлор-7-иодизохинолина (6 г, 18,52 ммоль) в уксусной кислоте (40 мл) добавляли йодистоводородную кислоту (20 мл, 55% масс./масс.) и красный фосфор (1,43 г, 46,3 ммоль). Полученный раствор кипятили с обратным холодильником в течение 16 часов, затем концентрировали при пониженном давлении. Остаток обрабатывали с помощью дихлорметана (100 мл) и насыщенного водного раствора бикарбоната натрия (100 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 2~10% этилацетата в петролейном эфире с получением 3-хлор-7-иодизохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 290,2 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (с, 1H), 8,66 (с, 1H), 8,10 (дд, J=1,2 Гц, 4,8 Гц, 1H), 8,06 (с, 1H), 7,78 (д, J=8,8 Гц, 1H).
Стадия 4: Синтез 7-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (12-4). К раствору 3-хлор-7-иодизохинолина (0,8 г, 2,76 ммоль) в N,N-диметилформамиде (30 мл) добавляли 1H-пирроло[2,3-с]пиридин (0,49 г, 4,15 ммоль) и карбонат цезия (1,80 г, 5,53 ммоль). Полученную смесь перемешивали в течение 24 часов при 120°C. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (5×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 7-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 372,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,70 (с, 1H), 9,39 (с, 1H), 8,70 (с, 1H), 8,36 (д, J=7,2 Гц, 1H), 8,30-8,29 (м, 2H), 8,10 (дд, J=1,6 Гц, 8,4 Гц, 1H), 7,88 (д, J=8,8 Гц, 1H), 7,69 (дд, J=0,8 Гц, 5,2 Гц, 1H), 6,89 (д, J=3,2 Гц, 1H).
Стадия 5: Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-амина (12). К раствору 7-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (300 мг, 0,81 ммоль) в толуоле (30 мл) добавляли дифенилметанимин (220 мг, 1,21 ммоль), трис(дибензилиденацетон)дипалладий(0) (41,8 мг, 0,04 ммоль), 9,9-диметил-4,5-бис(дифенилфосфино)ксантен (46,7 мг, 0,081 ммоль) и карбонат цезия (527 мг, 1,62 ммоль). Полученную смесь кипятили с обратным холодильником в течение 16 часов в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток разбавляли метанолом (30 мл). К полученному раствору добавляли хлористоводородную кислоту (6 мл, 2 н) и полученный раствор перемешивали в течение 3 часов при температуре окружающей среды. Полученный раствор разбавляли раствором бикарбоната натрия (60 мл) и экстрагировали этилацетатом (3×80 мл). Объединенные органические слои промывали насыщенным солевым раствором (30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 261,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,52 (с, 1H), 9,00 (с, 1H), 8,23-8,21 (м, 2H), 7,97 (с, 1H), 7,82 (д, J=10,8 Гц, 1H), 7,66 (д, J=5,1 Гц, 1H), 7,26 (д, J=6,9 Гц, 1H), 7,03 (с, 1H), 6,78 (д, J=3,0 Гц, 1H), 5,79 (ушир.с, 2H).
Пример 13
Синтез 7-метокси-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (13):
Схема 13

Стадия 1: Синтез (E)-2-(гидроксиимино)-6-метокси-2,3-дигидроинден-1-она (13-1).
К перемешиваемому раствору 6-метокси-2,3-дигидроинден-1-она (20 г, 123 ммоль) в диэтиловом эфире (300 мл) барботировали сухой гидрохлоридный газ при 0°C в течение 3 часов, затем добавляли изопентилнитрит (22 г, 185 ммоль). Полученный раствор перемешивали в течение 3 часов при температуре окружающей среды. Твердое вещество собирали фильтрацией и промывали диэтиловым эфиром (3×100 мл) с получением (E)-2-(гидроксиимино)-6-метокси-2,3-дигидроинден-1-она в виде не совсем белого твердого вещества: МС (ESI, m/z): 192,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 12,60 (с, 1H), 7,53 (д, J=8,4 Гц, 1H), 7,32 (д, J=8,4 Гц, 1H), 7,21 (с, 1H), 3,80 (с, 3H), 3,69 (с, 2H).
Стадия 2: Синтез 1,3-дихлор-7-метоксиизохинолина (13-2). К перемешиваемому раствору (E)-2-(гидроксиимино)-6-метокси-2,3-дигидроинден-1-она (5,0 г, 26,2 ммоль) в фосфор окситрихлориде (80 мл) добавляли пентахлорфосфоран (6,0 г, 28,8 ммоль) при 0°C. Гидрохлоридный газ барботировали в полученный раствор в течение 3 часов. Полученную смесь перемешивали в течение 16 часов при 60°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и твердое вещество промывали водой (3×50 мл), сушили в вакуумной печи с получением 1,3-дихлор-7-метоксиизохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 228,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,06 (с, 1H), 8,00 (д, J=9,0 Гц, 1H), 7,60 (дд, J=2,4 Гц, 6,6 Гц, 1H), 7,46 (д, J=2,4 Гц, 1H), 3,97 (с, 3H).
Стадия 3: Синтез 3-хлор-7-метоксиизохинолина (13-3). К перемешиваемому раствору 1,3-дихлор-7-метоксиизохинолина (4,0 г, 17,5 ммоль) в уксусной кислоте (60 мл) и йодистоводородной кислоте (30 мл, 45% водный раствор) добавляли красный фосфор (1,3 г, 43,8 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 4 часов при 100°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении. Остаток растворяли в дихлорметане (100 мл) и промывали насыщенным водным раствором бикарбоната натрия (2×100 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 5~10% этилацетата в петролейном эфире с получением 3-хлор-7-метоксиизохинолина в виде не совсем белого твердого вещества: МС (ESI, m/z): 194,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,00 (с, 1H), 8,00 (с, 1H), 7,89 (д, J=9,0 Гц, 1H), 7,54 (дд, J=2,4 Гц, 6,6 Гц, 1H), 7,46 (д, J=2,4 Гц, 1H), 3,87 (с, 3H).
Стадия 4: Синтез 7-метокси-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (13): К перемешиваемому раствору 1H-пирроло[2,3-с]пиридина (79 мг, 0,52 ммоль) и 3-хлор-7-метоксиизохинолина (100 мг, 1,27 ммоль) в тетрагидрофуране (20 мл) добавляли натрий 2-метилпропан-2-олат (163 мг, 1,69 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-три-изо-пропил-1,1'-бифенил)(2'''-амино-1'',1'''-бифенил-2''-ил)палладий(II) мезилат (33,6 мг, 0,042 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 16 часов при 50°C в атмосфере азота. После охлаждения до температуры окружающей среды, реакционную смесь гасили водой (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~5% метанола в дихлорметане, с получением 7-метокси-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 276,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,64 (с, 1H), 9,30 (с, 1H), 8,31-8,27 (м, 2H), 8,22 (с, 1H), 8,00 (д, J=6,0 Гц, 1H), 7,68 (д, J=5,4 Гц, 1H), 7,61 (д, J=2,4 Гц, 1H), 7,52-7,48 (м, 1H), 6,85 (д, J=3,3 Гц, 1H), 3,95 (с, 3H).
Пример 14
Синтез N-(2-фторэтил)-N-метил-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амин (14):
Схема 14

Стадия 1: Синтез N-метил-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина (14-1). В герметичную трубку добавляли раствор метанамина в этаноле (30 мл, 30% масс./масс.) и 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин (150 мг, 0,57 ммоль, 9). Полученную смесь перемешивали в течение 48 часов при 100°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением N-метил-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 275,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,63 (с, 1H), 8,94 (с, 1H), 8,27-8,24 (м, 2H), 7,83 (с, 1H), 7,80 (д, J=9,0 Гц, 1H), 7,64 (дд, J=0,9 Гц, 4,5 Гц, 1H), 7,04 (дд, J=5,1 Гц, 6,6 Гц, 1H), 6,82 (д, J=3,0 Гц, 1H), 6,76-6,74 (м, 1H), 6,70 (д, J=1,8 Гц, 1H), 2,83 (д, J=4,8 Гц, 3H).
Стадия 2: Синтез N-(2-фторэтил)-N-метил-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина (14). К перемешиваемому раствору N-метил-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина (50 мг, 0,18 ммоль) в N,N-диметилформамиде (10 мл) добавляли гидрид натрия (14 мг, 0,36 ммоль, 60% диспергированный в минеральном масле) при 0°C в атмосфере азота. Полученную смесь перемешивали в течение 30 минут при 0°C с последующим добавлением 1-бром-2-фторэтана (46 мг, 0,36 ммоль). Полученную смесь перемешивали в течение 4 часов при температуре окружающей среды и гасили насыщенным водным раствором хлорида аммония (100 мл). Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением N-(2-фторэтил)-N-метил-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 321,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,68 (с, 1H), 9,04 (с, 1H), 8,38 (д, J=3,3 Гц, 1H), 8,29 (д, J=5,4 Гц, 1H), 7,96 (д, J=9,0 Гц, 1H), 7,92 (с, 1H), 7,75 (д, J=5,4 Гц, 1H), 7,38 (д, J=9,0 Гц, 1H), 7,01 (д, J=1,8 Гц, 1H), 6,90 (д, J=3,0 Гц, 1H), 4,75 (т, J=4,8 Гц, 1H), 4,59 (т, J=4,8 Гц, 1H), 3,93 (т, J=5,1 Гц, 1H), 3,84 (т, J=5,1 Гц, 1H), 3,14 (с, 3H).
Пример 15
Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-ола (15):
Схема 15
Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-ола (15). К перемешиваемому раствору 7-метокси-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (70 мг, 0,25 ммоль, 13) в дихлорметане (15 мл) добавляли трибромборан (64 мг, 0,25 ммоль) при -78°C. Полученную смесь перемешивали в течение 16 часов при температуре окружающей среды, затем гасили добавлением воды (10 мл). Смесь нейтрализовали добавлением карбоната калия и экстрагировали дихлорметаном (5×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~3% метанола в дихлорметане, с получением 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-ола в виде светло-желтого твердого вещества: МС (ESI, m/z): 262,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,26 (с, 1H), 9,58 (с, 1H), 9,21 (с, 1H), 8,28-8,25 (м, 2H), 8,15 (с, 1H), 7,96 (д, J=9,0 Гц, 1H), 7,66 (д, J=5,1 Гц, 1H), 7,44-7,39 (м, 2H), 6,84 (д, J=3,3 Гц, 1H).
Пример 16
Синтез 7-(2-фторэтокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (16):
Схема 16
Синтез 7-(2-фторэтокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (16). К перемешиваемому раствору 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-ола (40 мг, 0,15 ммоль, 15) в N,N-диметилформамиде (5 мл) добавляли карбонат калия (42 мг, 0,31 ммоль) и 1-бром-2-фторэтан (29 мг, 0,23 ммоль). Полученную смесь перемешивали в течение 16 часов при 30°C, затем гасили водой (30 мл) и экстрагировали этилацетатом (3×60 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~3% метанола в дихлорметане, с получением 7-(2-фторэтокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 308,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,90 (с, 1H), 9,38 (с, 1H), 8,94 (д, J=3,3 Гц, 1H), 8,47 (д, J=6,3 Гц, 1H), 8,41 (с, 1H), 8,25 (д, J=6,6 Гц, 1H), 8,05 (д, J=9,0 Гц, 1H), 7,74 (д, J=2,4 Гц, 1H), 7,64-7,60 (м, 1H), 7,26 (д, J=3,3 Гц, 1H), 4,95 (т, J=3,6 Гц, 1H), 4,79 (т, J=3,6 Гц, 1H), 4,51 (т, J=3,6 Гц, 1H), 4,41 (т, J=3,6 Гц, 1H).
Пример 17
Синтез 7-(фторметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (17):
Схема 17

Синтез 7-(фторметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (17). Раствор 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-ола (50 мг, 0,19 ммоль, 15) в N,N-диметилформамиде (10 мл) обрабатывали гидридом натрия (33 мг, 0,83 ммоль, 60% масс./масс., диспергированный в минеральном масле) при 0°C в течение 10 минут с последующим добавлением бромфторметана (108 мг, 0,96 ммоль). После дополнительных 3 часов реакцию гасили насыщенным водным раствором хлорида аммония (30 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 7-(фторметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 294,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,66 (с, 1H), 9,45 (с, 1H), 8,34 (д, J=3,0 Гц, 1H), 8,30-8,27 (м, 2H), 8,12 (д, J=9,0 Гц, 1H), 7,89 (с, 1H), 7,70-7,64 (м, 2H), 6,87 (д, J=2,7 Гц, 1H), 6,15 (с, 1H), 5,97 (с, 1H).
Пример 18
Синтез 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-амина (18):
Схема 18

Стадия 1: Синтез 3-хлор-6-фтор-5-нитроизохинолина (18-1). К раствору 3-хлор-6-фторизохинолина (2 г, 11,01 ммоль, 9-3) в концентрированной серной кислоте (30 мл) добавляли нитрат калия (1,17 г, 11,56 ммоль) при 0°C. Полученную смесь перемешивали в течение 3 часов при температуре окружающей среды затем выливали в смесь лед/вода (200 г). Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 10~20% этилацетата в петролейном эфире с получением 3-хлор-6-фтор-5-нитроизохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 226,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,43 (с, 1H), 8,70-8,65 (м, 1H), 8,07 (с, 1H), 7,70-7,63 (м, 1H).
Стадия 2: Синтез 3-хлор-6-фторизохинолин-5-амина (18-2). К раствору 3-хлор-6-фтор-5-нитроизохинолина (1 г, 4,41 ммоль) в уксусной кислоте (100 мл) добавляли порошок железа (1,27 г, 22,07 ммоль). Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды, затем фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 2~10% этилацетата в петролейном эфире с получением 3-хлор-6-фторизохинолин-5-амина в виде желтого твердого вещества: МС (ESI, m/z): 197,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,06 (с, 1H), 8,26 (с, 1H), 7,51-7,44 (м, 1H), 7,40-7,35 (м, 1H), 6,01 (ушир.с, 2H).
Стадия 3: Синтез трет-бутил 3-хлор-6-фторизохинолин-5-илкарбамата (18-3). К раствору 3-хлор-6-фторизохинолин-5-амина (150 мг, 0,76 ммоль) в дихлорметане (30 мл) добавляли ди-трет-бутилдикарбонат (216 мг, 0,99 ммоль) и триэтиламин (116 мг, 1,14 ммоль). Полученную смесь перемешивали в течение 16 часов при температуре окружающей среды. После этого, смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 2~10% этилацетата в петролейном эфире с получением трет-бутил 3-хлор-6-фторизохинолин-5-илкарбамата в виде светло-желтого твердого вещества: МС (ESI, m/z): 297,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,03 (с, 1H), 7,93-7,88 (м, 1H), 7,83 (с, 1H), 7,41 (т, J=9,3 Гц, 1H), 6,23 (ушир.с, 1H), 1,52 (с, 9H).
Стадия 4: Синтез трет-бутил 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-илкарбамата (18-4). К раствору 1H-пирроло[2,3-с]пиридина (90 мг, 0,76 ммоль) в тетрагидрофуране (15 мл) добавляли трет-бутил (3-хлор-6-фторизохинолин-5-ил)карбамат (150 мг, 0,51 ммоль), натрий 2-метилпропан-2-олат (97 мг, 1,01 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-три-изо-пропил-1,1'-бифенил)(2'''-амино-1'',1'''-бифенил-2''-ил)палладий(II) мезилат (40,2 мг, 0,051 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 16 часов при 50°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1% метанола в дихлорметане, с получением трет-бутил 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-илкарбамата в виде желтого твердого вещества: МС (ESI, m/z): 379,2 [M+1]+; 1H ЯМР (300 МГц, CD3OD) δ 9,62 (с, 1H), 9,34 (с, 1H), 8,25-8,22 (м, 2H), 8,21-8,16 (м, 1H), 8,03 (с, 1H), 7,72 (д, J=5,4 Гц, 1H), 7,57 (т, J=7,8 Гц, 1H), 6,98 (д, J=3,6 Гц, 1H), 1,55 (с, 9H).
Стадия 5: Синтез 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-амина (18). Раствор трет-бутил 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-илкарбамата (65 мг, 0,17 ммоль) в дихлорметане (15 мл) обрабатывали трифторуксусной кислотой (3 мл) в течение 3 часов при температуре окружающей среды. Полученную смесь концентрировали при пониженном давлении и остаток растворяли в дихлорметане (50 мл), промывали насыщенным водным раствором бикарбоната натрия (50 мл). Органический слой сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-амина в виде желтого твердого вещества: МС (ESI, m/z): 279,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,70 (с, 1H), 9,27 (с, 1H), 8,40 (с, 1H), 8,35-8,27 (м, 2H), 7,68-7,66 (м, 1H), 7,45-7,43 (м, 2H), 6,88 (д, J=3,3 Гц, 1H), 6,03 (ушир.с, 2H).
Пример 19
Синтез 1'-метил-1,5'-би(1H-пирроло[2,3-с]пиридина) (19):
Схема 19

Стадия 1: Синтез трет-бутил 6-бром-4-метилпиридин-3-илкарбамата (19-1). К раствору 6-бром-4-метилпиридин-3-амина (2 г, 10,69 ммоль) в дихлорметане (30 мл) добавляли триэтиламин (2,16 г, 21,39 ммоль) и ди-трет-бутилдикарбонат (3,03 г, 13,90 ммоль). Полученный раствор перемешивали в течение 4 часов при температуре окружающей среды, затем концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 5~10% этилацетата в петролейном эфире с получением трет-бутил 6-бром-4-метилпиридин-3-илкарбамата в виде бесцветного твердого вещества: МС (ESI, m/z): 287,1, 289,1 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 8,72 (с, 1H), 7,30 (с, 1H), 6,18 (ушир.с, 1H), 2,26 (с, 3H), 1,55 (с, 9H).
Стадия 2: Синтез трет-бутил 6-бром-4-метилпиридин-3-ил(метил)карбамата (19-2). Раствор трет-бутил 6-бром-4-метилпиридин-3-илкарбамата (3,1 г, 10,8 ммоль) в N,N-диметилформамиде (30 мл) обрабатывали гидридом натрия (0,86 г, 21,6 ммоль, 60% масс./масс., диспергированный в минеральном масле) в течение 10 минут при 0°C с последующим добавлением йодметана (3,1 г, 21,6 ммоль). Полученную смесь перемешивали в течение 1 часа при температуре окружающей среды и гасили добавлением воды (100 мл). Полученную смесь экстрагировали этилацетатом (3×80 мл). Объединенные органические слои промывали насыщенным солевым раствором (4×30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 10~20% этилацетата в петролейном эфире с получением трет-бутил 6-бром-4-метилпиридин-3-ил(метил)карбамата в виде светло-желтого твердого вещества: МС (ESI, m/z): 301,2, 303,2 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 8,11 (с, 1H), 7,48 (с, 1H), 3,14 (с, 3H), 2,21 (с, 3H), 1,31 (с, 9H).
Стадия 3: Синтез 6-бром-N,4-диметилпиридин-3-амина (19-3). Раствор трет-бутил 6-бром-4-метилпиридин-3-ил(метил)карбамата (3,1 г, 10,3 ммоль) в дихлорметане (30 мл) обрабатывали трифторуксусной кислотой (3 мл) в течение 1 часа при температуре окружающей среды. Затем полученный раствор концентрировали при пониженном давлении и остаток растворяли в дихлорметане (100 мл), промывали насыщенным водным раствором бикарбоната натрия (100 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 6-бром-N,4-диметилпиридин-3-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 201,1, 203,1 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 7,66 (с, 1H), 7,11 (с, 1H), 3,48 (ушир.с, 1H), 2,91 (с, 3H), 2,10 (с, 3H).
Стадия 4: Синтез 5-бром-1-метил-1H-пирроло[2,3-с]пиридина (19-4). Раствор 6-бром-N,4-диметилпиридин-3-амина (1 г, 4,97 ммоль) в тетрагидрофуране (30 мл) обрабатывали 2,5 М н-бутиллития (4,97 мл, 12,43 ммоль) в гексане при -78°C в течение 15 минут в атмосфере азота, с последующим добавлением N,N-диметилформамида (0,51 г, 6,96 ммоль). Полученный раствор перемешивали в течение дополнительного 1 часа при температуре окружающей среды и гасили путем добавления насыщенного водного раствора бикарбоната натрия (60 мл). Полученную смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 10~20% этилацетата в петролейном эфире с получением 5-бром-1-метил-1H-пирроло[2,3-с]пиридина в виде масла желтого цвета: МС (ESI, m/z): 211,1, 213,1 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,90 (с, 1H), 7,68 (с, 1H), 7,22 (д, J=2,8 Гц, 1H), 6,44 (д, J=2,4 Гц, 1H), 3,11 (с, 3H).
Стадия 5: Синтез 1'-метил-1,5'-би(1H-пирроло[2,3-с]пиридина) (19). К раствору 5-бром-1-метил-1H-пирроло[2,3-с]пиридина (70 мг, 0,33 ммоль) в тетрагидрофуране (20 мл) добавляли 1H-пирроло[2,3-с]пиридин (59 мг, 0,49 ммоль), натрий 2-метилпропан-2-олат (64 мг, 0,66 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-три-изо-пропил-1,1'-бифенил)(2'''-амино-1'',1'''-бифенил-2''-ил)палладий(II) мезилат (26 мг, 0,033 ммоль). Полученный раствор перемешивали в течение 16 часов при 50°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученный раствор концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1'-метил-1,5'-би(1H-пирроло[2,3-с]пиридина) в виде светло-желтого твердого вещества: МС (ESI, m/z): 249,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 1H), 8,88 (с, 1H), 8,23-8,20 (м, 2H), 7,91 (с, 1H), 7,71 (д, J=3,2 Гц, 1H), 7,66 (д, J=5,6 Гц, 1H), 6,78 (д, J=3,2 Гц, 1H), 6,60 (д, J=2,8 Гц, 1H), 3,97 (с, 3H).
Пример 20
Синтез 6-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (20):
Схема 20

Стадия 1: Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина (20-1). В герметичный сосуд добавляли насыщенный раствор аммиака в метаноле (300 мл) и 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин (2,5 г, 9,5 ммоль, 9). Полученную смесь перемешивали в течение 5 дней при 120°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~10% метанола в дихлорметане, с получением 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 261,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,41 (с, 1H), 8,93 (с, 1H), 8,29-8,23 (м, 2H), 7,82 (д, J=9,0 Гц, 1H), 7,80 (с, 1H), 7,64 (дд, J=0,9 Гц, 4,5 Гц, 1H), 7,04 (дд, J=5,1 Гц, 6,6 Гц, 1H), 6,83-6,78 (м, 2H), 6,19 (с, 2H).
Стадия 2: Синтез 6-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (20). К суспензии 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина (0,15 г, 0,58 ммоль) в воде (20 мл) добавляли концентрированную серную кислоту (2 мл) при температуре окружающей среды. Полученную смесь нагревали до 70-80°C, пока не получали прозрачный раствор. Полученный раствор охлаждали до температуры 0°C и добавляли нитрит натрия (80 мг, 1,15 ммоль) в виде одной порции. После перемешивания в течение 15 минут при 0°C, добавляли калий йодид (0,19 г, 1,15 ммоль). Полученную смесь перемешивали в течение дополнительных 5 минут и гасили насыщенным водным раствором сульфита натрия (5 мл). Полученную смесь экстрагировали дихлорметаном (4×50 мл) и объединенные органические слои сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1% метанола в дихлорметане, с получением 6-иод-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде не совсем белого твердого вещества: МС (ESI, m/z): 372,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,82 (с, 1H), 9,44 (с, 1H), 8,65 (д, J=3,2 Гц, 1H), 8,56 (с, 1H), 8,41 (д, J=5,6 Гц, 1H), 8,29 (с, 1H), 8,05-7,98 (м, 3H), 7,12 (д, J=3,2 Гц, 1H).
Пример 21
Синтез 1-(4-(фторметил)-5-(проп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (21):
Схема 21

Стадия 1: Синтез 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотиновой кислоты (21-1). К перемешиваемому раствору 1H-пирроло[2,3-с]пиридина (1,8 г, 15,2 ммоль) в N,N-диметилформамиде (80 мл) добавляли карбонат цезия (14,9 г, 45,7 ммоль) и метил 5-бром-2-хлоризоникотината (5,0 г, 16,9 ммоль). Полученную смесь перемешивали в течение 3 часов при 100°C. После охлаждения до температуры окружающей среды, полученную смесь фильтровали через целит. Фильтрат разбавляли водой (200 мл) и подкисляли концентрированной водной хлористоводородной кислотой (pH=5~6). Полученную смесь экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×200 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~10% метанола в этилацетате, с получением 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотиновой кислоты в виде светло-желтого твердого вещества: МС (ESI, m/z): 318,0, 320,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 14,3 (ушир.с, 1H), 9,85 (с, 1H), 8,91 (с, 1H), 8,79 (с, 1H), 8,48-8,43 (м, 1H), 8,25 (с, 1H), 8,04 (д, J=6,0 Гц, 1H), 7,13-7,11 (м, 1H).
Стадия 2: Синтез (5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)метанола (21-3). К перемешиваемому раствору 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотиновой кислоты (2,0 г, 6,29 ммоль) в N,N-диметилформамиде (40 мл) добавляли карбонат натрия (2,0 г, 18,86 ммоль) и изопропил карбонохлоридат (1,54 г, 12,57 ммоль) при 0°C. Полученную смесь перемешивали в течение 16 часов при температуре окружающей среды, затем гасили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×100 мл) и сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении с получением сырого 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотиновый изомасляный ангидрид (21-2, 2 г) в виде масла желтого цвета. Полученное сырое желтое масло растворяли в изопропаноле (50 мл) при 0°C и добавляли боргидрид натрия (0,34 г, 9,02 ммоль). Полученную смесь перемешивали в течение 16 часов при температуре окружающей среды и гасили насыщенным водным раствором хлорида аммония (100 мл). Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~10% метанола в дихлорметане, с получением (5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)метанола в виде светло-желтого твердого вещества: МС (ESI, m/z): 304,1, 306,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,65 (с, 1H), 8,68 (с, 1H), 8,29 (д, J=5,4 Гц, 1H), 8,27 (д, J=3,6 Гц, 1H), 7,88 (с, 1H), 7,67 (дд, J=0,9 Гц, 4,5 Гц, 1H), 6,85 (д, J=3,6 Гц, 1H), 5,85 (т, J=5,7 Гц, 1H), 4,61 (д, J=5,4 Гц, 2H).
Стадия 3: Синтез (5-(проп-1-инил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)метанола (21-4). К перемешиваемому раствору (5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)метанола (0,72 г, 2,37 ммоль) и трибутил(проп-1-ин-1-ил)станнана (1,17 г, 3,55 ммоль) в 1,4-диоксане (40 мл) добавляли бис(трифенилфосфин)палладий(II) хлорид (0,17 г, 0,24 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 16 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь гасили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~10% метанола в дихлорметане, с получением (5-(проп-1-ин-1-ил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)метанола в виде светло-желтого твердого вещества: МС (ESI, m/z): 264,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6+D2O) δ 9,94 (с, 1H), 8,93 (д, J=3,3 Гц, 1H), 8,59 (с, 1H), 8,46 (д, J=6,3 Гц, 1H), 8,26 (д, J=6,3 Гц, 1H), 7,96 (с, 1H), 7,26 (д, J=3,3 Гц, 1H), 4,73 (с, 2H), 2,16 (с, 3H).
Стадия 4: Синтез 1-(4-(фторметил)-5-(проп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (21,. К перемешиваемому раствору (5-(проп-1-ин-1-ил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)метанола (50 мг, 0,19 ммоль) в сухом дихлорметане (20 мл) добавляли трифторид диэтиламиносеры (DAST, 92 мг, 0,57 ммоль) при -78°C в атмосфере азота. Полученный раствор перемешивали в течение 4 часов при температуре окружающей среды и гасили насыщенным водным раствором бикарбоната натрия (50 мл). Полученную смесь экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~10% метанола в дихлорметане, с получением 1-(4-(фторметил)-5-(проп-1-ин-1-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 266,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,95 (с, 1H), 8,95 (д, J=3,6 Гц, 1H), 8,69 (с, 1H), 8,48 (д, J=6,0 Гц, 1H), 8,21 (д, J=6,0 Гц, 1H), 8,05 (с, 1H), 7,24 (д, J=3,3 Гц, 1H), 5,78 (с, 1H), 5,62 (с, 1H), 2,17 (с, 3H).
Примеры 22 и 23
Синтез 1-(4-фтор-5-(проп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (22) и 2-метил-6-(1H-пирроло[2,3-с]пиридин-1-ил)фуро[3,2-c]пиридина (23):
Схема 22
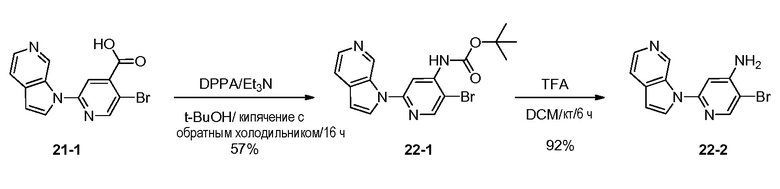
Схема 23
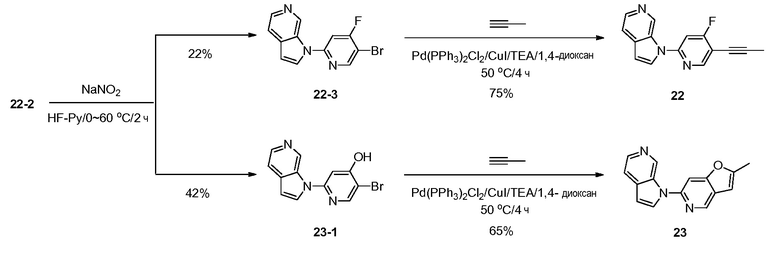
Стадия 1: Синтез трет-бутил (5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-илкарбамата (22-1). К перемешиваемому раствору 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотиновой кислоты (7,80 г, 8,09 ммоль, 21-1) и триэтиламина (2,46 г, 24,27 ммоль) в трет-бутиловом спирте (200 мл) добавляли дифенилфосфоразидат (3,34 г, 12,14 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 16 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток растворяли в дихлорметане (200 мл), промывали водой (200 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением трет-бутил (5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)карбамата в виде желтого твердого вещества: МС (ESI, m/z): 289,2, 291,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,72 (ушир.с, 1H), 8,93 (с, 1H), 8,69 (с, 1H), 8,29 (д, J=5,1 Гц, 1H), 8,20 (с, 1H), 8,17 (д, J=3,3 Гц, 1H), 7,68 (д, J=0,9 Гц, 1H), 6,86 (д, J=3,0 Гц, 1H), 1,48 (с, 9H).
Стадия 2: Синтез 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-амина (22-2). Раствор трет-бутил (5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ил)карбамата (1,71 г, 4,39 ммоль) в дихлорметане (50 мл) обрабатывали трифторуксусной кислотой (5 мл) при температуре окружающей среды в течение 6 часов. Полученный раствор концентрировали при пониженном давлении и остаток растворяли в дихлорметане (100 мл), промывали насыщенным водным раствором бикарбоната натрия (100 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 289,2, 291,2 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,53 (ушир.с, 1H), 8,33 (с, 1H), 8,29-8,27 (м, 1H), 8,07 (д, J=3,3 Гц, 1H), 7,66 (д, J=5,1 Гц, 1H), 7,05 (с, 1H), 6,84 (д, J=3,0 Гц, 1H), 6,61 (ушир.с, 2H).
Стадия 3: Синтез 1-(5-бром-4-фторпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (22-4) и 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ола (23-1). К перемешиваемому раствору 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-амина (0,5 г, 1,73 ммоль) в фтороводороде-пиридине (25 мл, 65-70% масс./масс.) добавляли нитрит натрия (0,72 г, 10,38 ммоль) при 0°C. Полученную смесь перемешивали в течение 2 часов при 60°C. После охлаждения до температуры окружающей среды, полученную смесь гасили водой (100 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-бром-4-фторпиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 292,2, 294,2 [M+1]+; 1H ЯМР (300 МГц, CD3OD) δ 10,08 (ушир.с, 1H), 8,85-8,79 (м, 2H), 8,41 (д, J=3,3 Гц, 1H), 8,23 (д, J=6,3 Гц, 1H), 7,95 (д, J=9,6 Гц, 1H), 7,21 (д, J=3,6 Гц, 1H); и 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ола в виде светло-желтого твердого вещества (210 мг, 42%): МС (ESI, m/z): 290,1, 292,1 [M+1]+; 1H ЯМР (300 МГц, CD3OD) δ 9,95 (ушир.с, 1H), 8,62 (д, J=2,7 Гц, 1H), 8,55 (с, 1H), 8,35 (д, J=6,9 Гц, 1H), 8,20 (д, J=6,0 Гц, 1H), 7,23 (с, 1H), 7,18 (с, 1H).
Стадия 4: Синтез 1-(4-фтор-5-(проп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (22). К перемешиваемому раствору 1-(5-бром-4-фторпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (30 мг, 0,11 ммоль) в 1,4-диоксане (15 мл) барботировали пропин в течение 10 минут при температуре окружающей среды с последующим добавлением триэтиламина (5 мл), иодида меди (I) (2 мг, 10 мкмоль) и бис(трифенилфосфин)палладий(II) дихлорида (3,6 мг, 5,14 мкмоль). Полученную смесь перемешивали в течение 4 часов при 50°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(4-фтор-5-(проп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 252,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,81 (ушир.с, 1H), 8,68 (д, J=10,0 Гц, 1H), 8,40 (с, 1H), 8,32 (д, J=5,2 Гц, 1H), 7,92 (д, J=12,0 Гц, 1H), 7,84 (д, J=5,6 Гц, 1H), 6,98 (с, 1H), 2,13 (с, 3H).
Стадия 5: Синтез 2-метил-6-(1H-пирроло[2,3-с]пиридин-1-ил)фуро[3,2-c]пиридина (23). К перемешиваемому раствору 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ола (30 мг, 0,11 ммоль) в 1,4-диоксане (15 мл) барботировали пропин в течение 10 минут при температуре окружающей среды с последующим добавлением триэтиламина (5 мл), иодида меди (I) (2 мг, 10 мкмоль) и бис(трифенилфосфин)палладий(II) дихлорида (3,6 мг, 5,14 мкмоль). Полученную смесь перемешивали в течение 4 часов при 50°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 2-метил-6-(1H-пирроло[2,3-с]пиридин-1-ил)фуро[3,2-c]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 250,1 [M+1]+; 1H ЯМР (300 МГц, CD3OD) δ 9,62 (ушир.с, 1H), 8,77 (с, 1H), 8,22 (д, J=5,4 Гц, 1H), 8,17 (д, J=3,3 Гц, 1H), 7,83 (с, 1H), 7,73 (д, J=5,4 Гц, 1H), 6,86 (д, J=3,3 Гц, 1H), 6,69 (с, 1H), 2,53 (с, 3H).
Пример 24
Синтез 6-(фторметокси)-2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолина (24):
Схема 24

Стадия 1: Синтез 6-метоксихиназолин-2,4(1H,3H)-диона (24-1). Суспензию 2-амино-4-метоксибензойной кислоты (5 г, 29,9 ммоль) в воде (35 мл) и уксусной кислоте (0,7 мл, 29,7 ммоль) нагревали до 35°C, с последующим добавлением раствора цианата калия (6,1 г, 74,8 ммоль) в воде (10 мл) в течение 5 минут. После дополнительного 1 часа осторожно добавляли гидроксид натрия (12 г, 0,3 моль) и реакционная система становилась прозрачным раствором. Полученный раствор подкисляли добавлением концентрированной хлористо-водородной кислоты (27 мл) и твердое вещество осаждалось. Проводили фильтрование и осадок на фильтре промывали холодной водой (2×30 мл), сушили в вакуумной печи с получением 6-метоксихиназолин-2,4(1H,3H)-диона в виде коричневого твердого вещества: МС (ESI, m/z): 193,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 11,27 (с, 1H), 11,10 (с, 1H), 7,31 (с, 1H), 7,28 (д, J=6,0 Гц, 1H), 7,13 (д, J=5,4 Гц, 1H), 3,76 (с, 3H).
Стадия 2: Синтез 2,4-дихлор-6-метоксихиназолина (24-2). Раствор 6-метоксихиназолин-2,4(1H,3H)-диона (5 г, 26,0 ммоль) в оксихлориде фосфора (30 мл) кипятили с обратным холодильником в течение 16 часов. После охлаждения до температуры окружающей среды, полученный раствор концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 2,4-дихлор-6-метоксихиназолина в виде желтого твердого вещества: МС (ESI, m/z): 229,1 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 7,90 (д, J=9,3 Гц, 1H), 7,61 (дд, J=2,7 Гц, 6,6 Гц, 1H), 7,41 (д, J=2,7 Гц, 1H), 4,00 (с, 3H).
Стадия 3: Синтез 2-хлор-6-метоксихиназолина (24-3). К раствору 2,4-дихлор-6-метоксихиназолина (3 г, 13,1 ммоль) в дихлорметане (50 мл) добавляли цинковую пыль (2,57 г, 39,3 ммоль), водный раствор аммиака (50 мл, 34% масс./масс.) и насыщенный солевой раствор (10 мл). Полученную смесь перемешивали в течение 4 часов при 40°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь фильтровали через целит. Органический слой собирали и водный слой экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 2-хлор-6-метоксихиназолина в виде желтого твердого вещества: МС (ESI, m/z): 195,2 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 9,19 (с, 1H), 7,91 (д, J=9,3 Гц, 1H), 7,61 (дд, J=2,7 Гц, 6,6 Гц, 1H), 7,16 (д, J=2,7 Гц, 1H), 3,96 (с, 3H).
Стадия 4: Синтез 6-метокси-2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолина (24-4). К раствору 2-хлор-6-метоксихиназолина (198 мг, 1,02 ммоль) в N,N-диметилформамиде (20 мл) добавляли 1H-пирроло[2,3-с]пиридин (176 мг, 1,5 ммоль) и карбонат цезия (552 мг, 1,69 ммоль). Полученную смесь перемешивали в течение 2 часов при 80°C. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (60 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 6-метокси-2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолина в виде желтого твердого вещества: МС (ESI, m/z): 277,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,16 (с, 1H), 9,63 (с, 1H), 8,59 (д, J=3,6 Гц, 1H), 8,35 (д, J=5,1 Гц, 1H), 8,05 (д, J=9,3 Гц, 1H), 7,74-7,69 (м, 2H), 7,62 (д, J=2,7 Гц, 1H), 6,88 (д, J=3,6 Гц, 1H), 3,95 (с, 3H).
Стадия 5: Синтез 2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолин-6-ола (24-5). К раствору 6-метокси-2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолина (1 г, 3,62 ммоль) в дихлорметане (50 мл) добавляли трибромборан (4,53 г, 18,10 ммоль) при -78°C. После допонительных 2 часов при температуре окружающей среды, реакционную смесь гасили водой (50 мл) и нейтрализовали гидроксидом натрия (1,45 г, 36,25 ммоль). Органический слой собирали и водный слой экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолин-6-ола в виде желтого твердого вещества: МС (ESI, m/z): 263,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,46 (с, 1H), 10,41 (с, 1H), 9,56 (с, 1H), 8,58 (с, 1H), 8,34 (д, J=5,1 Гц, 1H), 8,00 (д, J=9,3 Гц, 1H), 7,73-7,57 (м, 2H), 7,38 (с, 1H), 6,86 (с, 1H).
Стадия 6: Синтез 6-(фторметокси)-2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолина (24). Раствор 2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолин-6-ола (100 мг, 0,38 ммоль) в N,N-диметилформамиде (15 мл) обрабатывали гидридом натрия (76 мг, 1,91 ммоль, 60% масс./масс., диспергированный в минеральном масле) при 0°C в течение 10 минут с последующим добавлением бромфторметана (65 мг, 0,57 ммоль). После дополнительных 2 часов, полученную смесь гасили водой (50 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 6-(фторметокси)-2-(1H-пирроло[2,3-с]пиридин-1-ил)хиназолина в виде бесцветного твердого вещества: МС (ESI, m/z): 295,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,23 (с, 1H), 9,72 (с, 1H), 8,61 (д, J=3,6 Гц, 1H), 8,37 (д, J=5,1 Гц, 1H), 8,16 (д, J=8,7 Гц, 1H), 7,89-7,84 (м, 2H), 7,71 (д, J=5,1 Гц, 1H), 6,90 (д, J=3,6 Гц, 1H), 6,15 (с, 1H), 5,97 (с, 1H).
Пример 25
Синтез 1-фтор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (25):
Схема 26
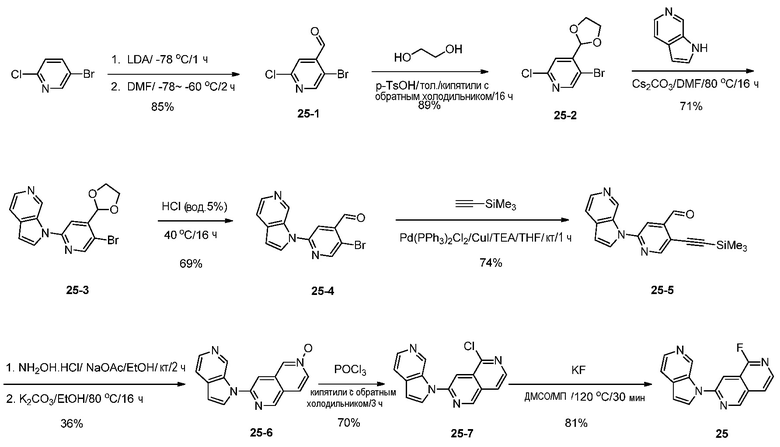
Стадия 1: Синтез 5-бром-2-хлоризоникотинальдегида (25-1). Раствор диизопропиламина (31,3 г, 0,31 моль) в тетрагидрофуране (600 мл) обрабатывали 2,5 М н-бутиллития (109 мл, 0,27 моль) в гексане в течение 30 минут при -78°C, с последующим добавлением раствора 5-бром-2-хлорпиридина (35 г, 0,18 моль) в тетрагидрофуране (200 мл). Полученный раствор перемешивали в течение 30 минут при -78°C, затем добавляли N,N-диметилформамид (40,4 г, 0,55 моль). После дополнительных 30 минут при -60°C, Реакционную смесь гасили насыщенным водным раствором хлорида аммония (1 л) и экстрагировали этилацетатом (3×500 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×500 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~10% этилацетата в петролейном эфире с получением 5-бром-2-хлоризоникотинальдегида в виде светло-желтого твердого вещества: МС (ESI, m/z): 220,2, 222,1 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 10,31 (с, 1H), 8,70 (с, 1H), 7,74 (с, 1H).
Стадия 2: Синтез 5-бром-2-хлор-4-(1,3-диоксолан-2-ил)пиридина (25-2). К раствору 5-бром-2-хлоризоникотинальдегида (15 г, 61,2 ммоль) в толуоле (500 мл) добавляли 4- метилбензолсульфоновую кислоту (11,6 г, 67,4 ммоль) и этан-1,2-диол (7,6 г, 122 ммоль). Полученную смесь кипятили с обратным холодильником в течение 16 часов в атмосфере азота. Воду удаляли с помощью ловушки Дина-Старка. После охлаждения до температуры окружающей среды реакционную смесь гасили водой (300 мл) и нейтрализовали добавлением гидроксида натрия (2,9 г, 74,1 ммоль). Органический слой отделяли и водный слой экстрагировали этилацетатом (3×300 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×300 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении с получением 5-бром-2-хлор-4-(1,3-диоксолан-2-ил)пиридина в виде желтого твердого вещества: МС (ESI, m/z): 264,2, 266,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,69 (с, 1H), 7,59 (с, 1H), 5,93 (с, 1H), 4,13-3,98 (м, 4H).
Стадия 3: Синтез 1-(5-бром-4-(1,3-диоксолан-2-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (25-3). К раствору 5-бром-2-хлор-4-(1,3-диоксолан-2-ил)пиридина (16 г, 54,4 ммоль) в N,N-диметилформамиде (300 мл) добавляли 1H-пирроло[2,3-с]пиридин (6,43 г, 54,4 ммоль) и карбонат цезия (53,2 г, 163 ммоль). Полученную смесь перемешивали в течение 16 часов при 80°C. После охлаждения до температуры окружающей среды, полученную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-бром-4-(1,3-диоксолан-2-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 346,2, 348,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,61 (с, 1H), 8,79 (с, 1H), 8,39 (д, J=3,6 Гц, 1H), 8,30 (д, J=5,4 Гц, 1H), 7,86 (с, 1H), 7,66 (д, J=5,1 Гц, 1H), 6,87 (д, J=3,3 Гц, 1H), 6,04 (с, 1H), 4,21-4,00 (м, 4H).
Стадия 4: Синтез 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида (25-4). К суспензии 1-(5-бром-4-(1,3-диоксолан-2-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (5 г, 13,72 ммоль) в воде (200 мл) добавляли хлористоводородную кислоту (20 мл, 37% масс./масс.). Полученный раствор перемешивали в течение 16 часов при 40°C. После охлаждения до температуры окружающей среды, полученный раствор нейтрализовали добавлением карбоната калия (14 г, 101 ммоль). Твердое вещество собирали фильтрацией и промывали водой (3×50 мл), сушили в вакуумной печи с получением 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида в виде светло-желтого твердого вещества: МС (ESI, m/z): 302,1, 304,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,27 (с, 1H), 9,75 (с, 1H), 8,98 (с, 1H), 8,45 (д, J=3,6 Гц, 1H), 8,38 (д, J=3,3 Гц, 1H), 8,17 (с, 1H), 7,68 (д, J=4,5 Гц, 1H), 6,90 (д, J=3,6 Гц, 1H).
Стадия 5: Синтез 2-(1H-пирроло[2,3-с]пиридин-1-ил)-5-((триметилсилил)этинил)изоникотинальдегида (25-5). К перемешиваемому раствору 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида (4 г, 11,92 ммоль) и триэтиламина (2,41 г, 23,83 ммоль) в тетрагидрофуране (120 мл) добавляли иодид меди (I) (0,23 г, 1,19 ммоль), этинилтриметилсилан (1,76 г, 17,87 ммоль) и бис(трифенилфосфин)палладий(II) дихлорид (0,84 г, 1,19 ммоль) при температуре окружающей среды в атмосфере азота. После дополнительного 1 часа, полученный раствор концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 2-(1H-пирроло[2,3-с]пиридин-1-ил)-5-((триметилсилил)этинил)изоникотинальдегида в виде светло-желтого твердого вещества: МС (ESI, m/z): 320,1 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 10,55 (с, 1H), 8,85 (с, 1H), 7,93 (д, J=3,6 Гц, 1H), 7,83 (д, J=3,3 Гц, 1H), 7,68-7,64 (м, 1H), 7,55-7,45 (м, 2H), 6,77 (д, J=3,0 Гц, 1H), 0,32 (с, 9H).
Стадия 6: Синтез 7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридин 2-оксида (25-6). К перемешиваемому раствору 2-(1H-пирроло[2,3-с]пиридин-1-ил)-5-((триметилсилил)этинил)изоникотинальдегида (3 г, 7,98 ммоль) в этаноле (120 мл) добавляли ацетат натрия (1,31 г, 15,97 ммоль) и гидрохлорид гидроксиламина (0,83 г, 11,97 ммоль) при температуре окружающей среды. Через дополнительные 2 часа, к полученному раствору добавили карбонат калия (2,64 г, 19,16 ммоль) и полученную смесь перемешивали в течение 16 часов при 80°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток растворяли в дихлорметане (100 мл), промывали водой (2×50 мл). Органический слой сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридин 2-оксида в виде светло-желтого твердого вещества: МС (ESI, m/z): 263,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,67 (с, 1H), 9,38 (с, 1H), 8,97 (с, 1H), 8,32-8,30 (м, 2H), 8,24 (т, J=7,2 Гц, 1H), 8,19-8,18 (м, 2H), 7,69 (д, J=5,4 Гц, 1H), 6,91 (д, J=3,3 Гц, 1H).
Стадия 7: Синтез 1-хлор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (25-7). Раствор 7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридин 2-оксида (0,85 г, 3,21 ммоль) в оксихлориде фосфора (30 мл) кипятили с обратным холодильником в течение 3-х часов. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток растворяли в дихлорметане (50 мл), промывали насыщенным водным раствором бикарбоната натрия (20 мл). Органический слой сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-хлор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 281,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,79 (с, 1H), 9,65 (с, 1H), 8,54 (д, J=3,3 Гц, 1H), 8,49 (д, J=2,7 Гц, 1H), 8,34-8,31 (м, 2H), 8,18 (д, J=5,4 Гц, 1H), 7,70 (д, J=5,1 Гц, 1H), 6,93 (д, J=3,3 Гц, 1H).
Стадия 8: Синтез 1-фтор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (25). Смесь 1-хлор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (30 мг, 0,11 ммоль) и фторида калия (60,8 мг, 1,05 ммоль) в диметилсульфоксиде (5 мл) облучали в течение 30 минут при 120°C в микроволной печи (100 Вт). После охлаждения до температуры окружающей среды реакционную смесь гасили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-фтор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 265,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,81 (с, 1H), 9,65 (с, 1H), 8,55 (д, J=3,3 Гц, 1H), 8,39 (с, 1H), 8,32 (д, J=5,4 Гц, 1H), 8,25 (д, J=5,1 Гц, 1H), 8,08 (д, J=5,4 Гц, 1H), 7,69 (д, J=5,7 Гц, 1H), 6,93 (д, J=3,3 Гц, 1H).
Пример 26
Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (26):
Схема 26

Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (26). Раствор 7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридин-2-оксида (100 мг, 0,38 ммоль, 25-6) в N,N-диметилформамиде (10 мл) обрабатывали трихлорфосфином (157 мг, 1,14 ммоль) при 0°C в течение 30 минут. Реакционную смесь гасили насыщенным водным раствором бикарбоната натрия (2 мл) и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 3-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 247,2 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,73 (с, 1H), 9,57 (с, 1H), 9,54 (с, 1H), 8,70 (д, J=5,6 Гц, 1H), 8,49 (с, 1H), 8,40 (д, J=3,2 Гц, 1H), 8,32 (д, J=5,2 Гц, 1H), 8,11 (д, J=5,6 Гц, 1H), 7,70 (д, J=1,2 Гц, 1H), 6,92 (д, J=3,2 Гц, 1H).
Примеры 27 и 28
Синтез 7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридин-1-амина (27) и 1-метокси-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (28):
Схема 27

Синтез 7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридин-1-амина (27) и 1-метокси-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (28). Газообразный аммиак барботировали в раствор 1-хлор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина (20 мг, 0,07 ммоль, 25-7) в метаноле (5 мл) в течение 1 часа при температуре окружающей среды. Полученный раствор выдерживали в течение 16 часов при 80°C в запаянной трубке. После охлаждения до температуры окружающей среды, полученный раствор концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-метокси-7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 277,2 [M+1]+; 1H ЯМР (300 МГц, CD3OD) δ 9,65 (с, 1H), 9,28 (с, 1H), 8,25-8,20 (м, 3H), 8,09 (д, J=6,9 Гц, 1H), 7,67 (д, J=4,8 Гц, 1H), 7,50 (д, J=5,7 Гц, 1H), 6,85 (д, J=3,3 Гц, 1H), 4,16 (с, 3H); и 7-(1H-пирроло[2,3-с]пиридин-1-ил)-2,6-нафтиридин-1-амина в виде светло-желтого твердого вещества (6 мг, 32%): МС (ESI, m/z): 262,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,69 (с, 1H), 9,22 (с, 1H), 8,44 (с, 1H), 8,30-8,27 (м, 2H), 7,95 (д, J=5,7 Гц, 1H), 7,68 (д, J=5,1 Гц, 1H), 7,24 (ушир.с, 2H), 7,13 (д, J=6,0 Гц, 1H), 6,90 (д, J=3,3 Гц, 1H).
Пример 29
Синтез 1-(5-(1-(2-фторэтил)-1H-пиразол-3-ил)пиримидин-2-ил)-1H-пирроло[2,3-с]пиридина (29):
Схема 28
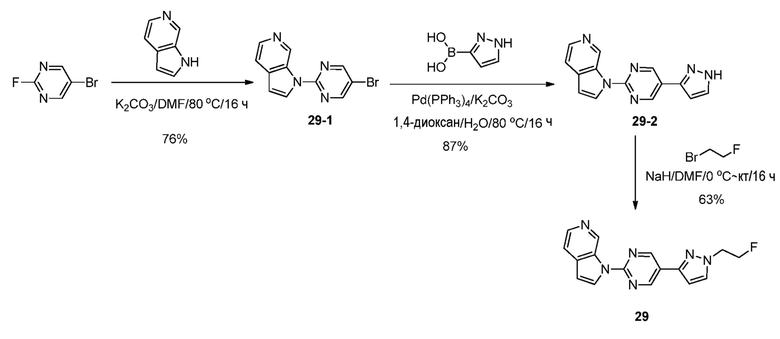
Стадия 1: Синтез 1-(5-бромпиримидин-2-ил)-1H-пирроло[2,3-с]пиридина (29-1). К перемешиваемому раствору 1H-пирроло[2,3-с]пиридина (0,73 г, 6,22 ммоль) и 5-бром-2-фторпиримидина (1 г, 5,65 ммоль) в N,N-диметилформамиде (50 мл) добавляли карбонат калия (1,56 г, 11,30 ммоль). Полученную смесь перемешивали в течение 16 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (4×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~2% метанола в дихлорметане, с получением 1-(5-бромпиримидин-2-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 275,1, 277,1 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,91 (с, 1H), 8,42 (д, J=4,8 Гц, 1H), 8,28 (д, J=5,2 Гц, 1H), 8,01 (с, 2H), 7,52 (т, J=4,4 Гц, 1H), 6,69 (д, J=4,8 Гц, 1H).
Стадия 2: Синтез 1-(5-(1H-пиразол-3-ил)пиримидин-2-ил)-1H-пирроло[2,3-с]пиридина (29-2). Смесь 1-(5-бромпиримидин-2-ил)-1H-пирроло[2,3-с]пиридина (300 мг, 1,09 ммоль), карбоната калия (301 мг, 2,18 ммоль) и тетракис(трифенилфосфин)палладия(0) (126 мг, 0,11 ммоль) в 1,4-диоксане (20 мл) и воды (2 мл) нагревали до 80°C в атмосфере азота, с последующим добавлением (1H-пиразол-3-ил)бороновой кислоты (244 мг, 2,18 ммоль) в виде нескольких порций. После дополнительных 16 часов полученную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-(1H-пиразол-3-ил)пиримидин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 263,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,08 (с, 1H), 9,40 (с, 2H), 9,08 (с, 1H), 8,59 (д, J=6,3 Гц, 1H), 8,33 (д, J=6,3 Гц, 1H), 7,94 (д, J=1,8 Гц, 1H), 7,29 (д, J=3,3 Гц, 1H), 7,03 (д, J=2,4 Гц, 1H).
Стадия 3: Синтез 1-(5-(1-(2-фторэтил)-1H-пиразол-3-ил)пиримидин-2-ил)-1H-пирроло[2,3-с]пиридина (29). Раствор 1-(5-(1H-пиразол-3-ил)пиримидин-2-ил)-1H-пирроло[2,3-с]пиридина (100 мг, 0,38 ммоль) в N,N-диметилформамиде (10 мл) обрабатывали гидридом натрия (76 мг, 1,91 ммоль, 60% масс./масс., диспергированный в минеральном масле) в течение 10 минут при 0°C с последующим добавлением 1-бром-2-фторэтана (242 мг, 1,91 ммоль). После перемешивания в течение 16 часов при температуре окружающей среды, реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-(1-(2-фторэтил)-1H-пиразол-3-ил)пиримидин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 308,9 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,97 (с, 1H), 9,29 (с, 2H), 8,48 (д, J=3,6 Гц, 1H), 8,35 (д, J=5,1 Гц, 1H), 7,95 (с, 1H), 7,70 (д, J=5,1 Гц, 1H), 6,99 (с, 1H), 6,90 (д, J=3,0 Гц, 1H), 4,92 (т, J=4,2 Гц, 1H), 4,75 (т, J=4,2 Гц, 1H), 4,59 (т, J=4,2 Гц, 1H), 4,48 (т, J=4,2 Гц, 1H).
Пример 30
Синтез 1-(5-(3-фторпроп-1-инил)-4-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (30):
Схема 29

Стадия 1: Синтез 5-(3-гидроксипроп-1-инил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида (30-1). К перемешиваемому раствору 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида (3 г, 9,43 ммоль, 25-4) в тетрагидрофуране (100 мл) добавляли триметил(проп-2-ин-1-илокси)силан (1,45 г, 11,32 ммоль), иодид меди (I) (0,18 г, 0,94 ммоль), N-этил-N-изопропилпропан-2-амин (24,3 г, 189 ммоль) и бис(трифенилфосфин)палладий(II) дихлорид (0,33 г, 0,47 ммоль). Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды в атмосфере азота. Полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 5-(3-гидроксипроп-1-ин-1-ил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида в виде желтого твердого вещества: МС (ESI, m/z): 278,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,72 (с, 1H), 8,64 (с, 1H), 8,31-8,30 (м, 2H), 7,88 (с, 1H), 7,86-7,65 (м, 1H), 7,14 (д, J=7,5 Гц, 1H), 6,87 (д, J=3,3 Гц, 1H), 5,45 (т, J=6,0 Гц, 1H), 4,38 (д, J=6,0 Гц, 2H).
Стадия 2: Синтез 5-(3-(трет-бутилдиметилсилилокси)проп-1-инил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида (30-2). К перемешиваемому раствору 5-(3-гидроксипроп-1-ин-1-ил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида (1,8 г, 6,17 ммоль) в N,N-диметилформамиде (50 мл) добавляли триэтиламин (1,25 г, 12,33 ммоль) и трет-бутилхлордиметилсилан (1,39 г, 9,25 ммоль). Полученный раствор перемешивали в течение 2 часов при температуре окружающей среды и гасили водой (150 мл). Полученную смесь экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 5-(3-(трет-бутилдиметилсилилокси)проп-1-инил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида в виде светло-желтого твердого вещества: МС (ESI, m/z): 392,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,37 (с, 1H), 9,82 (с, 1H), 8,90 (с, 1H), 8,52 (д, J=3,6 Гц, 1H), 8,33 (д, J=5,4 Гц, 1H), 8,18 (с, 1H), 7,70 (д, J=5,4 Гц, 1H), 6,92 (д, J=3,3 Гц, 1H), 4,69 (с, 2H), 0,92 (с, 9H), 0,17 (с, 6H).
Стадия 3: Синтез 1-(5-(3-(трет-бутилдиметилсилилокси)проп-1-инил)-4-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (30-3). К смеси 5-(3-(трет-бутилдиметилсилилокси)проп-1-инил)-2-(1H-пирроло[2,3-с]пиридин-1-ил)изоникотинальдегида (0,4 г, 0,92 ммоль) и метилтрифенилфосфонийбромида (0,49 г, 1,38 ммоль) в тетрагидрофуране (20 мл) добавляли 1 М раствора гексаметилдисилазида лития (1 мл, 1 ммоль) в тетрагидрофуране при -20°C в атмосфере азота. Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды и гасили насыщенным водным раствором хлорида аммония (50 мл). Полученную смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-(3-(трет-бутилдиметилсилилокси)проп-1-инил)-4-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 390,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,79 (с, 1H), 8,67 (с, 1H), 8,50 (д, J=3,6 Гц, 1H), 8,29 (д, J=5,4 Гц, 1H), 8,11 (с, 1H), 7,66 (д, J=5,1 Гц, 1H), 7,17-7,07 (м, 1H), 6,88 (д, J=3,6 Гц, 1H), 6,53 (д, J=8,7 Гц, 1H), 5,79 (д, J=11,1 Гц, 1H), 4,43 (с, 2H), 0,92 (с, 9H), 0,17 (с, 6H).
Стадия 4: Синтез 3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)-4-винилпиридин-3-ил)проп-2-ин-1-ола (30-4). Раствор 1-(5-(3-(трет-бутилдиметилсилилокси)проп-1-инил)-4-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (0,2 г, 0,51 ммоль) в дихлорметане (20 мл) обрабатывали триэтиламин тригидрофторидом (66 мг, 0,41 ммоль) в течение 2 часов при температуре окружающей среды. Реакционную смесь гасили насыщенным водным раствором бикарбоната натрия (10 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)-4-винилпиридин-3-ил)проп-2-ин-1-ола в виде светло-желтого твердого вещества: МС (ESI, m/z): 276,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,78 (с, 1H), 8,65 (с, 1H), 8,48 (д, J=3,6 Гц, 1H), 8,29 (д, J=5,4 Гц, 1H), 8,10 (с, 1H), 7,66 (д, J=5,1 Гц, 1H), 7,17-7,07 (м, 1H), 6,88 (д, J=3,6 Гц, 1H), 6,53 (д, J=8,7 Гц, 1H), 5,79 (д, J=11,1 Гц, 1H), 5,46 (т, J=6,0 Гц, 1H), 4,41 (д, J=6,0 Гц, 2H).
Стадия 5: Синтез 1-(5-(3-фторпроп-1-инил)-4-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (30). К перемешиваемому раствору 3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)-4-винилпиридин-3-ил)проп-2-ин-1-ола (140 мг, 0,31 ммоль) в дихлорметане (20 мл) добавляли трифторид диэтиламиносеры (492 мг, 3,05 ммоль) при -78°C. Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды и гасили насыщенным водным раствором бикарбоната натрия (50 мл). Полученную смесь экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-(3-фторпроп-1-инил)-4-винилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 278,1 [M+1]+; 1H ЯМР (300 МГц, CD3OD) δ 9,74 (с, 1H), 8,65 (с, 1H), 8,27 (д, J=3,6 Гц, 1H), 8,21 (д, J=5,4 Гц, 1H), 7,92 (с, 1H), 7,67 (д, J=6,3 Гц, 1H), 7,20-7,11 (м, 1H), 6,84 (д, J=3,9 Гц, 1H), 6,36 (д, J=17,7 Гц, 1H), 5,73 (д, J=11,1 Гц, 1H), 5,39 (с, 1H), 5,36 (с, 1H).
Пример 31
Синтез 1-(5-(4-(2-хлорэтил)пиперидин-1-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (31):
Схема 30
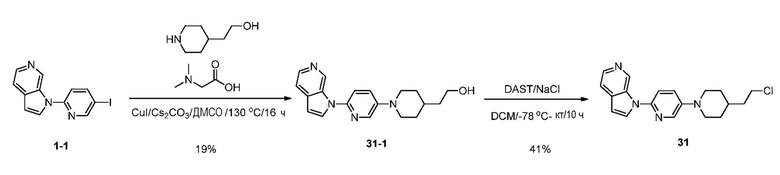
Стадия 1: Синтез 2-(1-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пиперидин-4-ил)этанола (31-1). К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (0,8 г, 2,49 ммоль, 1-1) в диметилсульфоксиде (30 мл) добавляли 2-(пиперидин-4-ил)этанол (0,64 г, 4,98 ммоль), иодид меди (I) (0,28 г, 1,49 ммоль), карбонат цезия (3,24 г, 9,97 ммоль) и 2-(диметиламино)уксусную кислоту (0,1 г, 0,99 ммоль). Полученную смесь перемешивали в течение 16 часов при 130°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 1~2% метанола в дихлорметане, с получением 2-(1-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пиперидин-4-ил)этанола в виде бесцветного твердого вещества: МС (ESI, m/z): 323,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,90 (с, 1H), 8,30-8,27 (м, 2H), 8,06 (д, J=6,0 Гц, 1H), 7,99 (д, J=3,6 Гц, 1H), 7,78 (с, 2H), 6,84 (д, J=3,6 Гц, 1H), 4,35 (т, J=5,7 Гц, 1H), 3,84-3,78 (м, 2H), 3,48-3,43 (м, 2H), 2,78-2,71 (м, 2H), 1,79-1,73 (м, 2H), 1,68-1,54 (м, 1H), 1,42-1,37 (м, 2H), 1,29-1,21 (м, 2H).
Стадия 2: Синтез 1-(5-(4-(2-хлорэтил)пиперидин-1-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (31). К раствору 2-(1-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пиперидин-4-ил)этанола (100 мг, 0,31 ммоль) в дихлорметане (10 мл) добавляли трифторид диэтиламиносеры (DAST, 548 мг, 3,40 ммоль) при -78°C. Полученный раствор перемешивали в течение 10 часов при температуре окружающей среды и гасили насыщенным солевым раствором (30 мл), экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-(4-(2-хлорэтил)пиперидин-1-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 341,1 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,53 (с, 1H), 8,34-8,31 (м, 1H), 8,27 (д, J=2,4 Гц, 1H), 7,88 (д, J=3,2 Гц, 1H), 7,66 (т, J=4,8 Гц, 1H), 7,45-7,43 (м, 2H), 6,76 (д, J=7,2 Гц, 1H), 3,79-3,76 (м, 2H), 3,65 (т, J=7,6 Гц, 2H), 2,89-2,83 (м, 2H), 1,91-1,89 (м, 2H), 1,81-1,77 (м, 3H), 1,49-1,41 (м, 2H).
Пример 32
Синтез 1-(5-(2-фторэтокси)-4-метилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (32):
Схема 31

Стадия 1: Синтез 6-бром-4-метилпиридин-3-ола (32-1). К перемешиваемому раствору 6-бром-4-метилпиридин-3-амина (2,0 г, 10,7 ммоль) в водной 40%-ной серной кислоте (20 мл) добавляли нитрит натрия (1,1 г, 16,1 ммоль) при 0°C. Полученную смесь перемешивали в течение 30 минут при 0°C и 24 часов при температуре окружающей среды. Полученную смесь разбавляли водой (200 мл) и нейтрализовали карбонатом калия. Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1,5% метанола в дихлорметане, с получением 6-бром-4-метилпиридин-3-ола в виде светло-желтого твердого вещества: МС (ESI, m/z): 188,0, 190,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 7,89 (с, 1H), 7,83 (с, 1H), 7,35 (с, 1H), 2,13 (с, 3H).
Стадия 2: Синтез 2-бром-5-(2-фторэтокси)-4-метилпиридина (32-2). К перемешиваемому раствору 6-бром-4-метилпиридин-3-ола (0,50 г, 1,31 ммоль) в N,N-диметилформамиде (20 мл) добавляли карбонат калия (0,55 г, 3,95 ммоль) и 1-бром-2-фторэтан (0,25 мг, 1,99 ммоль). Полученную смесь перемешивали в течение 16 часов при 30°C и гасили водой (150 мл). Полученную смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,5~1% метанола в дихлорметане, с получением 2-бром-5-(2-фторэтокси)-4-метилпиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 234,1, 236,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,07 (с, 1H), 7,48 (с, 1H), 4,85-4,82 (м, 1H), 4,69-4,66 (м, 1H), 4,41-4,38 (м, 1H), 4,31-4,27 (м, 1H), 2,18 (с, 3H).
Стадия 3: Синтез 1-(5-(2-фторэтокси)-4-метилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (32). К перемешиваемой смеси 1H-пирроло[2,3-с]пиридина (70 мг, 0,59 ммоль) и карбоната цезия (770 мг, 2,37 ммоль) в диметилсульфоксиде (20 мл) добавляли 2-бром-5-(2-фторэтокси)-4-метилпиридин (173 мг, 0,59 ммоль), диметилглицин (36,7 мг, 0,36 ммоль) и иодид меди (I) (67,7 мг, 0,36 ммоль). Полученную смесь перемешивали в течение 2 часов при 130°C в атмосфере азота. После охлаждения до температуры окружающей среды реакционную смесь гасили водой (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1% метанола в дихлорметане, с получением 1-(5-(2-фторэтокси)-4-метилпиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 272,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,61 (с, 1H), 8,28 (с, 1H), 8,24 (д, J=5,1 Гц, 1H), 8,19 (д, J=3,3 Гц, 1H), 7,75 (с, 1H), 7,64-7,62 (м, 1H), 6,78 (д, J=3,0 Гц, 1H), 4,89 (т, J=3,6 Гц, 1H), 4,73 (т, J=3,6 Гц, 1H), 4,49 (т, J=3,6 Гц, 1H), 4,39 (т, J=3,6 Гц, 1H), 2,33 (с, 3H).
Пример 33
Синтез 1-(4-метокси-5-(проп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (33):
Схема 32
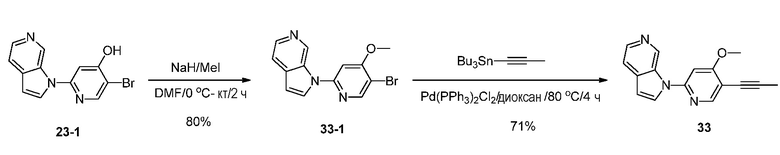
Стадия 1: Синтез 1-(5-бром-4-метоксипиридин-2-ил)-1H-пирроло[2,3-с]пиридина (33-1). Раствор 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-ола (100 мг, 0,35 ммоль, 23-1) в N,N-диметилформамиде (10 мл) обрабатывали гидридом натрия (15 мг, 0,38 ммоль, 60% масс./масс., диспергированный в минеральном масле) в течение 10 минут при 0°C с последующим добавлением йодметана (54 мг, 0,38 ммоль). Полученный раствор перемешивали в течение 2 часов при температуре окружающей среды, затем гасили водой (50 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×20 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-бром-4-метоксипиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 304,1, 306,1 [M+1]+; 1H ЯМР (300 МГц, CD3OD) δ 9,72 (с, 1H), 8,54 (с, 1H), 8,28-8,23 (м, 2H), 7,72 (д, J=5,4 Гц, 1H), 7,37 (с, 1H), 6,87 (д, J=3,3 Гц, 1H), 4,11 (с, 3H).
Стадия 2: Синтез 1-(4-метокси-5-(проп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (33). К перемешиваемому раствору 1-(5-бром-4-метоксипиридин-2-ил)-1H-пирроло[2,3-с]пиридина (60 мг, 0,18 ммоль) в 1,4-диоксане (20 мл) добавляли дибутил(проп-1-ин-1-ил)(пропил)станнан (84 мг, 0,27 ммоль) и бис(трифенилфосфин)палладий(II) хлорид (6 мг, 0,09 ммоль). Полученную смесь перемешивали при 80°С в течение 4 часов в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(4-метокси-5-(проп-1-инил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде светло-желтого твердого вещества: МС (ESI, m/z): 264,1 [M+1]+; 1H ЯМР (300 МГц, CD3OD) δ 9,70 (с, 1H), 8,38 (с, 1H), 8,23-8,21 (м, 2H), 7,68 (д, J=8,1 Гц, 1H), 7,27 (с, 1H), 6,84 (д, J=3,3 Гц, 1H), 4,06 (с, 3H), 2,11 (с, 3H).
Пример 34
Синтез N-(4-метоксифенил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)никотинамида (34):
Схема 33
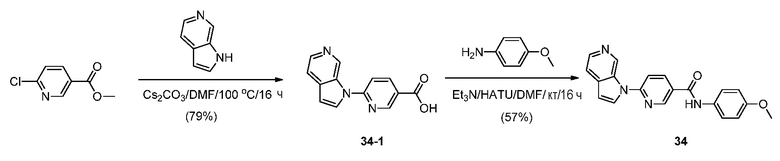
Стадия 1: Синтез 6-(1H-пирроло[2,3-с]пиридин-1-ил)никотиновой кислоты (34-1). К раствору 1H-пирроло[2,3-с]пиридина (1,0 г, 8,5 ммоль) в N,N-диметилформамиде (30 мл) добавляли метил-6-хлорникотинат (1,7 г, 10,2 ммоль) и карбонат цезия (8,3 г, 25,4 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 16 часов при 100°C. После охлаждения до температуры окружающей среды, полученную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~3% метанола (1% уксусная кислота, об./об.) в дихлорметане, с получением 6-(1H-пирроло[2,3-с]пиридин-1-ил)никотиновой кислоты в виде бесцветного твердого вещества: МС (ESI, m/z): 240,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 13,80 (ушир.с, 1H), 10,08 (с, 1H), 9,18 (с, 1H), 9,00 (д, J=3,3 Гц, 1H), 8,55-8,51 (м, 2H), 8,24 (д, J=6,0 Гц, 1H), 8,16 (д, J=8,7 Гц, 1H), 7,28 (д, J=3,6 Гц, 1H).
Стадия 2: Синтез N-(4-метоксифенил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)никотинамида (34). К раствору 6-(1H-пирроло[2,3-с]пиридин-1-ил)никотиновой кислоты (239 мг, 1 ммоль) в N,N-диметилформамиде (30 мл) добавляли 4-метоксибензоламин (246 мг, 2 ммоль), триэтиламин (202 мг, 2 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU, 760 мг, 2 ммоль). Полученный раствор перемешивали в течение 16 часов при температуре окружающей среды, затем гасили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (5×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~2% метанола в дихлорметане, с получением N-(4-метоксифенил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)никотинамида в виде светло-желтого твердого вещества: МС (ESI, m/z): 345,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 10,34 (с, 1H), 9,88 (с, 1H), 9,15 (с, 1H), 8,61-8,52 (м, 1H), 8,43 (д, J=3,3 Гц, 1H), 8,33 (д, J=5,1 Гц, 1H), 8,05 (д, J=8,7 Гц, 1H), 7,72-7,68 (м, 3H), 6,98-6,92 (м, 3H), 3,76 (с, 3H).
Пример 35
Синтез N-(3-фторпропил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (35):
Схема 34

Стадия 1: Синтез трет-бутил 6-бромпиридин-3-илкарбамата (35-1). К раствору 6-бромпиридин-3-амина (5,4 г, 28,9 ммоль) в дихлорметане (200 мл) добавляли триэтиламин (4,4 г, 43,3 ммоль) и ди-трет-бутилдикарбонат (7,6 г, 34,7 ммоль). Полученный раствор перемешивали в течение 4 часов при температуре окружающей среды и гасили водой (300 мл). Органический слой отделяли и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 2~10% этилацетата в петролейном эфире с получением трет-бутил (6-бромпиридин-3-ил)карбамата в виде бесцветного твердого вещества: МС (ESI, m/z): 273,1, 275,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,72 (с, 1H), 8,45 (д, J=1,8 Гц, 1H), 7,83 (д, J=5,4 Гц, 1H), 7,53 (д, J=5,7 Гц, 1H), 1,48 (с, 9H).
Стадия 2: Синтез трет-бутил 6-бромпиридин-3-ил(3-фторпропил)карбамата (35-2). К раствору трет-бутил (6-бромпиридин-3-ил)карбамата (2,0 г, 7,3 ммоль) в диметилформамиде (50 мл) добавляли гидрид натрия (0,6 г, 14,7 ммоль, 60% диспергированный в минеральном масле) при 0°C. После перемешивания в течение 10 минут, 1-фтор-3-иодпропан (2,7 г, 14,7 ммоль) добавляли к полученному раствору. Полученный раствор перемешивали в течение 2 часов при температуре окружающей среды, затем гасили водой (200 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (5×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 5~10% этилацетата в петролейном эфире с получением трет-бутил 6-бромпиридин-3-ил(3-фторпропил)карбамата в виде светло-желтого твердого вещества: МС (ESI, m/z): 333,2, 335,2 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 8,28 (с, 1H), 7,49-7,47 (м, 2H), 4,57 (т, J=5,6 Гц, 1H), 4,45 (т, J=5,6 Гц, 1H), 3,81 (т, J=7,2 Гц, 2H), 2,05-2,00 (м, 1H), 1,99-1,92 (м, 1H), 1,49 (с, 9H).
Стадия 3: Синтез трет-бутил 6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил(3-фторпропил)карбамата (35-3). К раствору 1H-пирроло[2,3-с]пиридина (125 мг, 1,1 ммоль) в диметилсульфоксиде (15 мл) добавляли карбонат цезия (919 мг, 2,8 ммоль), трет-бутил (6-бромпиридин-3-ил)(3-фторпропил)карбамат (235 мг, 0,7 ммоль), диметилглицин (29,1 мг, 0,3 ммоль) и иодид меди (I) (81 мг, 0,4 ммоль) при температуре окружающей среды в атмосфере азота. После перемешивания в течение 16 часов при 130°C, полученную смесь охлаждали до температуры окружающей среды и разбавляли водой (100 мл). Полученную смесь экстрагировали этилацетатом (3×60 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1,5% метанола в дихлорметане, с получением трет-бутил 6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил(3-фторпропил)карбамата в виде светло-желтого твердого вещества: МС (ESI, m/z): 371,1 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,67 (ушир.с, 1H), 8,53 (д, J=2,0 Гц, 1H), 8,40 (ушир.с, 1H), 7,88 (д, J=3,2 Гц, 1H), 7,82 (д, J=7,2 Гц, 1H), 7,61 (д, J=4,4 Гц, 1H), 7,51 (д, J=8,4 Гц, 1H), 6,76 (д, J=3,2 Гц, 1H), 4,75 (т, J=4,8 Гц, 1H), 4,64 (т, J=4,8 Гц, 1H), 4,01 (т, J=4,8 Гц, 1H), 3,95 (т, J=4,8 Гц, 1H), 1,49 (с, 9H), 1,33-1,24 (м, 2H).
Стадия 4: Синтез N-(3-фторпропил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (35). Раствор трет-бутил (6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)(3-фторпропил)карбамата (150 мг, 0,4 ммоль) в дихлорметане (10 мл) обрабатывали трифторуксусной кислотой (3 мл) в течение 2 часов при температуре окружающей среды. Реакционную смесь разбавляли водой (30 мл) и нейтрализовали карбонатом калия. Полученную смесь экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали на колонке с силикагелем, элюировали смесью 0,5~1,5% метанола в дихлорметане, с получением N-(3-фторпропил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина в виде не совсем белого твердого вещества: МС (ESI, m/z): 271,1 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,38 (с, 1H), 8,33 (д, J=5,2 Гц, 1H), 8,02 (с, 1H), 7,80 (с, 1H), 7,60 (д, J=4,4 Гц, 1H), 7,32 (д, J=8,4 Гц, 1H), 7,17-7,11 (м, 1H), 6,71 (д, J=3,2 Гц, 1H), 4,75 (т, J=4,8 Гц, 1H), 4,64 (т, J=4,8 Гц, 1H), 4,05 (ушир.с, 1H), 3,45 (т, J=4,8 Гц, 2H), 2,16-2,12 (м, 1H), 2,10-2,03 (м, 1H).
Пример 36
Синтез N-(2-фторэтил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (36):
Схема 35

Стадия 1: Синтез трет-бутил 6-бромпиридин-3-ил(2-фторэтил)карбамата (36-1). Раствор трет-бутил (6-бромпиридин-3-ил)карбамата (200 мг, 0,73 ммоль, 35-1) в N,N-диметилформамиде (20 мл) обрабатывали гидридом натрия (117 мг, 2,93 ммоль, 60% масс./масс., диспергированный в минеральном масле) при 0°C в течение 40 минут, с последующим добавлением 1-бром-2-фторэтана (186 мг, 1,46 ммоль). Через дополнительных 2 часов при температуре окружающей среды, реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (2×60 мл). Объединенные органические слои промывали насыщенным солевым раствором (5×50 мл) и сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении с получением трет-бутил 6-бромпиридин-3-ил(2-фторэтил)карбамата в виде светло-желтого твердого вещества: МС (ESI, m/z): 319,0, 321,0 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 8,32 (с, 1H), 7,51-7,45 (м, 2H), 4,70 (т, J=4,8 Гц, 1H), 4,58 (т, J=4,8 Гц, 1H), 3,92 (т, J=4,8 Гц, 1H), 3,86 (т, J=4,8 Гц, 1H), 1,46 (с, 9H).
Стадия 2: Синтез трет-бутил 6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил(2-фторэтил)карбамата (36-2). К раствору 1H-пирроло[2,3-с]пиридина (129 мг, 1,09 ммоль) в диметилсульфоксиде (15 мл) добавляли карбонат цезия (951 мг, 2,92 ммоль), трет-бутил (6-бромпиридин-3-ил)(2-фторэтил)карбамат (233 мг, 0,73 ммоль), 2-(диметиламино)уксусную кислоту (30 мг, 0,29 ммоль) и иодид меди(I) (83 мг, 0,44 ммоль) при температуре окружающей среды в атмосфере азота. После перемешивания в течение 16 часов при 120°C, реакционную смесь охлаждали до температуры окружающей среды и разбавляли водой (80 мл). Полученную смесь экстрагировали этилацетатом (2×60 мл) и объединенные органические слои промывали насыщенным солевым раствором (2×50 мл), сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане с получением трет-бутил 6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил(2-фторэтил)карбамата в виде бесцветного твердого вещества: МС (ESI, m/z): 357,2 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,67 (ушир.с, 1H), 8,53 (д, J=2,0 Гц, 1H), 8,40 (ушир.с, 1H), 7,88 (д, J=2,8 Гц, 1H), 7,82 (д, J=7,8 Гц, 1H), 7,62 (д, J=4,4 Гц, 1H), 7,51 (д, J=8,4 Гц, 1H), 6,76 (д, J=3,2 Гц, 1H), 4,76 (т, J=4,8 Гц, 1H), 4,64 (т, J=4,8 Гц, 1H), 4,01 (т, J=4,8 Гц, 1H), 3,94 (т, J=4,8 Гц, 1H), 1,49 (с, 9H).
Стадия 3: Синтез N-(2-фторэтил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (36). Раствор трет-бутил (6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)(2-фторэтил)карбамата (150 мг, 0,41 ммоль) в дихлорметане (10 мл) обрабатывали трифторуксусной кислотой (3 мл) в течение 2 часов при температуре окружающей среды. Полученный раствор концентрировали при пониженном давлении и остаток растворяли в дихлорметане еще раз (50 мл). Полученный раствор промывали насыщенным водным раствором бикарбоната натрия (2×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане с получением N-(2-фторэтил)-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина в виде бесцветного твердого вещества: МС (ESI, m/z): 257,2 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,40 (с, 1H), 8,34 (д, J=5,2 Гц, 1H), 8,06 (д, J=2,8 Гц, 1H), 7,77 (д, J=3,2 Гц, 1H), 7,59 (д, J=5,2 Гц, 1H), 7,39 (т, J=9,6 Гц, 1H), 7,18-7,15 (м, 1H), 6,70 (д, J=3,2 Гц, 1H), 4,77 (т, J=4,8 Гц, 1H), 4,66 (т, J=4,8 Гц, 1H), 4,21 (ушир.с, 1H), 3,56 (т, J=4,8 Гц, 1H), 3,53 (т, J=4,8 Гц, 1H).
Пример 37
Синтез 1-(2-фтор-3,3'-бипиридин-6-ил)-1H-пирроло[2,3-с]пиридина (37):
Схема 36

Стадия 1: Синтез 6-хлор-3,3'-бипиридин-2-амина (37-1). К раствору 3-бром-6-хлорпиридин-2-амина (1 г, 4,82 ммоль) в 1,4-диоксане (40 мл) и воде (4 мл) добавляли пиридин-3-илбороновую кислоту (0,65 г, 5,30 ммоль), карбонат калия (1,33 г, 9,64 ммоль) и тетракис(трифенилфосфин)палладий(0) (0,56 г, 0,48 ммоль). Полученный раствор перемешивали в течение 16 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 6-хлор-3,3'-бипиридин-2-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 206,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 8,66 (с, 1H), 8,57 (дд, J=2,0 Гц, 4,4 Гц, 1H), 7,84-7,82 (м, 1H), 7,48-7,45 (м, 1H), 7,38 (д, J=7,6 Гц, 1H), 6,69 (д, J=11,6 Гц, 1H), 6,28 (ушир.с, 2H).
Стадия 2: Синтез N,N-ди-Boc защищенного 6-хлор-3,3'-бипиридин-2-амина (37-2). К раствору 6-хлор-3,3'-бипиридин-2-амина (0,68 г, 3,31 ммоль) в дихлорметане (30 мл) добавляли ди-трет-бутилдикарбонат (1,51 г, 6,95 ммоль), триэтиламин (0,67 г, 6,61 ммоль) и N,N-диметилпиридин-4-амин (40 мг, 0,33 ммоль). Полученный раствор перемешивали в течение 4 часов при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1% метанола в дихлорметане, с получением N,N-ди-Boc защищенного 6-хлор-3,3'-бипиридин-2-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 406,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,64 (дд, J=1,2 Гц, 4,8 Гц, 1H), 8,59 (д, J=2,1 Гц, 1H), 8,10 (д, J=8,1 Гц, 1H), 7,81-7,75 (м, 2H), 7,58-7,54 (м, 1H), 1,23 (с, 9 H), 1,20 (с, 9H).
Стадия 3: Синтез трет-бутил 6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,3'-бипиридин-2-илкарбамата (3). К раствору трет-бутил N-трет-бутилоксикарбонил-6-хлор-3,3'-бипиридин-2-илкарбамата (0,9 г, 2,22 ммоль) в тетрагидрофуране (40 мл) добавляли 1H-пирроло[2,3-с]пиридин (0,39 г, 3,33 ммоль), натрий 2-метилпропан-2-олат (0,43 г, 4,43 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-три-изо-пропил-1,1'-бифенил)(2'''-амино-1'',1'''-бифенил-2''-ил)палладий(II) мезилат (0,18 г, 0,22 ммоль) в атмосфере азота. Полученный раствор перемешивали в течение 16 часов при 50°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением трет-бутил 6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,3'-бипиридин-2-илкарбамата в виде светло-желтого твердого вещества: МС (ESI, m/z): 388,2 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 10,06 (с, 1H), 9,75 (с, 1H), 8,70 (с, 1H), 8,55 (д, J=4,0 Гц, 1H), 8,40 (д, J=3,2 Гц, 1H), 8,30 (д, J=5,2 Гц, 1H), 8,05 (д, J=8,4 Гц, 1H), 7,92 (д, J=4,4 Гц, 1H), 7,83 (д, J=8,4 Гц, 1H), 7,65 (д, J=5,2 Гц, 1H), 7,48 (дд, J=4,8 Гц, 11,6 Гц, 1H), 6,87 (д, J=3,2 Гц, 1H), 1,24 (с, 9H).
Стадия 4: Синтез 6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,3'-бипиридин-2-амина (37-4). Раствор трет-бутил 6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,3'-бипиридин-2-илкарбамата (0,5 г, 1,29 ммоль) в дихлорметане (30 мл) обрабатывали трифторуксусной кислотой (3 мл) в течение 2 часов при температуре окружающей среды. Полученный раствор концентрировали при пониженном давлении и остаток растворяли в дихлорметане (100 мл), промывали насыщенным водным раствором бикарбоната натрия (100 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,3'-бипиридин-2-амина в виде светло-желтого твердого вещества: МС (ESI, m/z): 288,2 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 10,00 (с, 1H), 8,68 (д, J=5,6 Гц, 1H), 8,58 (т, J=3,6 Гц, 1H), 8,27-8,26 (м, 2H), 7,92 (д, J=8,0 Гц, 1H), 7,62 (д, J=5,2 Гц, 1H), 7,58 (д, J=7,6 Гц, 1H), 7,49 (дд, J=4,8 Гц, 7,6 Гц, 1H), 7,07 (д, J=8,0 Гц, 1H), 6,78 (д, J=3,2 Гц, 1H), 6,33 (ушир.с, 2H).
Стадия 5: Синтез 1-(2-фтор-3,3'-бипиридин-6-ил)-1H-пирроло[2,3-с]пиридина (37). К раствору 6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,3'-бипиридин-2-амина (100 мг, 0,35 ммоль) в фтороводороде-пиридине (3 мл, 64~70% масс./масс.) добавляли нитрит натрия (144 мг, 2,09 ммоль) при -20°C. Полученный раствор перемешивали в течение 2 часов при температуре окружающей среды и разбавляли водой (50 мл). Полученную смесь экстрагировали этилацетатом (3×80 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(2-фтор-3,3'-бипиридин-6-ил)-1H-пирроло[2,3-с]пиридина в виде не совсем белого твердого вещества: МС (ESI, m/z): 291,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,80 (с, 1H), 8,91 (с, 1H), 8,66 (дд, J=1,6 Гц, 4,8 Гц, 1H), 8,48-8,43 (м, 2H), 8,34 (д, J=5,2 Гц, 1H), 8,13 (дд, J=1,2 Гц, 8,0 Гц, 1H), 8,02 (дд, J=1,2 Гц, 8,4 Гц, 1H), 7,70 (д, J=0,8 Гц, 1H), 7,58 (дд, J=4,8 Гц, 8,0 Гц, 1H), 6,93 (д, J=3,2 Гц, 1H).
Пример 38
Синтез 3-(5-фтор-1Н-пирроло[2,3-с]пиридин-1-ил)изохинолина (38):
Схема 37

Стадия 1: Синтез трет-бутил 6-фтор-4-метилпиридин-3-илкарбамата (38-1). К раствору 6-фтор-4-метилпиридин-3-амина (5,0 г, 39,6 ммоль) в дихлорметане (100 мл) добавляли триэтиламин (6,1 г, 59,5 ммоль) и ди-трет-бутилдикарбонат (10,4 г, 47,6 ммоль). Полученную смесь перемешивали в течение 4 часов при температуре окружающей среды, затем концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 5~10% этилацетата в петролейном эфире с получением трет-бутил 6-фтор-4-метилпиридин-3-илкарбамата в виде бесцветного твердого вещества: МС (ESI, m/z): 227,1 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 8,38 (с, 1H), 6,76 (с, 1H), 6,16 (ушир.с, 1H), 2,30 (с, 3H), 1,51 (с, 9H).
Стадия 2: Синтез трет-бутил 5-фтор-1Н-пирроло[2,3-с]пиридин-1-карбоксилата (38-3). Раствор трет-бутил (6-фтор-4-метилпиридин-3-ил)карбамата (3,7 г, 16,3 ммоль) в сухом тетрагидрофуране (50 мл) обрабатывали 2,5 М раствора н-бутиллития (16,3 мл, 40,9 ммоль) в гексане при -78°C в течение 30 минут, с последующим добавлением N,N-диметилформамида (1,4 г, 19,6 ммоль). Через дополнительные 3 часа при температуре окружающей среды, реакционную смесь гасили насыщенным водным раствором хлорида аммония (50 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×100 мл) и сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и остаток растворяли в дихлорметане (25 мл) с последующим добавлением триэтиламина (1,6 г, 16,3 ммоль) и метансульфонилхлорида (1,8 г, 16,3 ммоль). Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 10~20% этилацетата в петролейном эфире с получением трет-бутил 5-фтор-1Н-пирроло[2,3-с]пиридин-1-карбоксилата в виде светло-желтого твердого вещества: МС (ESI, m/z): 237,1 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 9,01 (с, 1H), 7,87 (д, J=3,3 Гц, 1H), 7,14 (с, 1H), 6,64 (д, J=3,6 Гц, 1H), 1,69 (с, 9H).
Стадия 3: Синтез 5-фтор-1Н-пирроло[2,3-с]пиридина (38-4). Раствор трет-бутил 5-фтор-1Н-пирроло[2,3-с]пиридин-1-карбоксилата (1,3 г, 5,5 ммоль) в дихлорметане (20 мл) обрабатывали трифторуксусной кислотой (2 мл) в течение 2 часов при температуре окружающей среды. Полученный раствор концентрировали при пониженном давлении и остаток растворяли в дихлорметане (50 мл), промывали насыщенным водным раствором бикарбоната натрия (100 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 5-фтор-1Н-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 137,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 11,66 (ушир.с, 1H), 8,34 (с, 1H), 7,71 (т, J=2,7 Гц, 1H), 7,17 (с, 1H), 6,51 (т, J=2,1 Гц, 1H).
Стадия 4: Синтез 3-(5-фтор-1Н-пирроло[2,3-с]пиридин-1-ил)изохинолина (38). К раствору 5-фтор-1Н-пирроло[2,3-с]пиридина (50 мг, 0,37 ммоль) в тетрагидрофуране (10 мл) добавляли 3-бромизохинолин (115 мг, 0,55 ммоль), натрий 2-метилпропан-2-олат (70,6 мг, 0,74 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-три-изо-пропил-1,1'-бифенил)(2'''-амино-1'',1'''-бифенил-2''-ил)палладий(II) мезилат (29,2 мг, 0,037 ммоль) в атмосфере азота. Полученную смесь выдерживали в течение 4 часов при 50°C. После охлаждения до температуры окружающей среды, смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 3-(5-фтор-1Н-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде бесцветного твердого вещества: МС (ESI, m/z): 264,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,42 (с, 1H), 9,33 (с, 1H), 8,49 (д, J=3,3 Гц, 1H), 8,29 (с, 1H), 8,21 (д, J=8,1 Гц, 1H), 8,06 (д, J=8,4 Гц, 1H), 7,85 (т, J=8,1 Гц, 1H), 7,68 (т, J=7,8 Гц, 1H), 7,37 (д, J=1,2 Гц, 1H), 6,90 (д, J=3,3 Гц, 1H).
Пример 39
Синтез 7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,2,3,4-тетрагидро-1,6-нафтиридина (39):
Схема 38
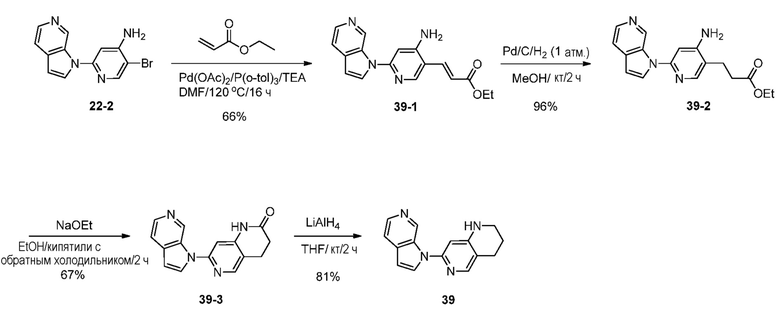
Стадия 1: Синтез (E)-этил 3-(4-амино-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)акрилата (39-1). К раствору 5-бром-2-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-4-амина (1 г, 3,46 ммоль, 22-2) в N,N-диметилформамиде (50 мл) добавляли этилакрилат (1,38 г, 13,83 ммоль), триэтиламин (1,40 г, 13,83 ммоль), ацетат палладия(II) (0,078 г, 0,35 ммоль) и три-o-толилфосфин (0,21 г, 0,69 ммоль) при температуре окружающей среды в атмосфере азота. Полученный раствор перемешивали в течение 16 часов при 120°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~3% метанола в дихлорметане, с получением (E)-этил 3-(4-амино-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)акрилата в виде светло-желтого твердого вещества: МС (ESI, m/z): 309,2 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,65 (с, 1H), 8,56 (с, 1H), 8,36-8,32 (м, 2H), 7,94-7,83 (м, 2H), 6,97 (т, J=4,2 Гц, 2H), 6,90 (ушир.с, 2H), 6,60 (д, J=15,9 Гц, 1H), 4,21 (кв, J=7,8 Гц, 2H), 1,21 (т, J=7,8 Гц, 3H).
Стадия 2: Синтез этил 3-(4-амино-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пропаноата (39-2). К перемешиваемому раствору (E)-этил 3-(4-амино-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)акрилата (0,5 г, 1,62 ммоль) в метаноле (120 мл) добавляли палладий на угле (50 мг, 10% масс./масс.). Полученную смесь выдерживали в атмосфере водорода (1 атм.) в течение 2 часов при температуре окружающей среды. Затем смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении досуха с получением этил 3-(4-амино-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пропаноата в виде светло-желтого твердого вещества: МС (ESI, m/z): 311,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,34 (с, 1H), 8,22 (д, J=5,4 Гц, 1H), 8,02 (д, J=3,3 Гц, 1H), 7,97 (с, 1H), 7,62 (д, J=4,5 Гц, 1H), 6,87 (с, 1H), 6,74 (д, J=7,2 Гц, 1H), 6,26 (ушир.с, 2H), 4,08 (кв, J=7,8 Гц, 2H), 2,75-2,71 (м, 2H), 2,54-2,51 (м, 2H), 1,15 (т, J=7,8 Гц, 3H).
Стадия 3: Синтез 7-(1H-пирроло[2,3-с]пиридин-1-ил)-3,4-дигидро-1,6-нафтиридин-2(1H)-она (39-3). Раствор этил 3-(4-амино-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пропаноата (50 мг, 0,16 ммоль) и этанолат натрия (54,8 мг, 0,81 ммоль) в этаноле (5 мл) кипятили с обратным холодильником в течение 2-х часов. После охлаждения до температуры окружающей среды, полученную смесь нейтрализовали добавлением уксусной кислоты (0,2 мл) и концентрировали при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~3% метанола в дихлорметане, с получением 7-(1H-пирроло[2,3-с]пиридин-1-ил)-3,4-дигидро-1,6-нафтиридин-2(1H)-она в виде бесцветного твердого вещества: МС (ESI, m/z): 265,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,56 (ушир.с, 1H), 9,49 (с, 1H), 8,32 (с, 1H), 8,26 (д, J=5,4 Гц, 1H), 8,07 (д, J=3,3 Гц, 1H), 7,66 (д, J=5,4 Гц, 1H), 7,16 (с, 1H), 6,82 (д, J=2,7 Гц, 1H), 2,96 (т, J=4,8 Гц, 2H), 2,57 (т, J=4,8 Гц, 2H).
Стадия 4: Синтез 7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,2,3,4-тетрагидро-1,6-нафтиридина (39). Раствор 7-(1H-пирроло[2,3-с]пиридин-1-ил)-3,4-дигидро-1,6-нафтиридин-2(1H)-она (28 мг, 0,11 ммоль) в тетрагидрофуране (10 мл) обрабатывали алюмогидридом лития (6 мг, 0,16 ммоль) при температуре окружающей среды в течение 2 часов. Реакционную смесь гасили добавлением декагидрата сульфата натрия (322 мг, 0,1 ммоль) и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~3% метанола в дихлорметане, с получением 7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,2,3,4-тетрагидро-1,6-нафтиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 251,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,43 (с, 1H), 8,21 (д, J=5,4 Гц, 1H), 8,00 (д, J=3,3 Гц, 1H), 7,85 (с, 1H), 7,61 (д, J=5,4 Гц, 1H), 6,91 (с, 1H), 6,79 (д, J=6,3 Гц, 1H), 6,68 (с, 1H), 3,27 (т, J=4,8 Гц, 2H), 2,68 (т, J=6,0 Гц, 2H), 1,83-1,79 (м, 2H).
Пример 40
Синтез 2-фтор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина (40):
Схема 39
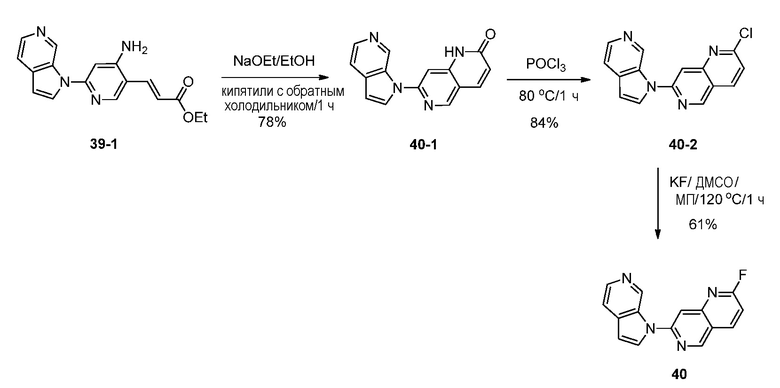
Стадия 1: Синтез 7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридин-2(1H)-она (40-1).
Раствор (E)-этил 3-(4-амино-6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)акрилата (150 мг, 0,49 ммоль, 39-1) в этаноле (5 мл) обрабатывали этанолатом натрия (166 мг, 2,43 ммоль) в течение 1 часа при 78°C. После охлаждения до температуры окружающей среды реакционную смесь гасили водой (2 мл) и нейтрализовали уксусной кислотой (0,2 мл). Полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~5% метанола в дихлорметане, с получением 7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридин-2(1H)-она в виде не совсем белого твердого вещества: МС (ESI, m/z): 263,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,11 (ушир.с, 1H), 9,59 (с, 1H), 8,89 (с, 1H), 8,31 (д, J=5,4 Гц, 1H), 8,18 (д, J=3,6 Гц, 1H), 8,05 (д, J=3,6 Гц, 1H), 7,66-7,64 (м, 1H), 7,49 (с, 1H), 6,88 (д, J=3,3 Гц, 1H), 6,55 (д, J=9,0 Гц, 1H).
Стадия 2: Синтез 2-хлор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина (40-2).
Раствор 7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридин-2(1H)-она (100 мг, 0,38 ммоль) в оксиде трихлорфосфора (5 мл) перемешивали в течение 1 часа при 80°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток поглощали дихлорметаном (50 мл), и промывали насыщенным водным раствором бикарбоната натрия (2×20 мл). Органический слой сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 2-хлор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина в виде не совсем белого твердого вещества: МС (ESI, m/z): 381,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,85 (с, 1H), 9,55 (с, 1H), 8,71 (д, J=8,4 Гц, 1H), 8,50 (д, J=3,3 Гц, 1H), 8,32-8,28 (м, 2H), 7,72-7,61 (м, 2H), 6,93 (д, J=3,3 Гц, 1H).
Стадия 3: Синтез 2-фтор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина (40). Смесь 2-хлор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина (100 мг, 0,37 ммоль) и фторида калия (62 мг, 1,07 ммоль) в диметилсульфоксиде (3 мл) облучали в микроволновой печи (100 Вт) в течение 1 часа при 120°C. После охлаждения до температуры окружающей среды смесь разбавляли водой (20 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенный органический слой промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, элюировали смесью 1~2% метанола в дихлорметане, с получением 2-фтор-7-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина в виде не совсем белого твердого вещества: МС (ESI, m/z): 265,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,08 (с, 1H), 9,59 (с, 1H), 9,16 (д, J=3,3 Гц, 1H), 8,96 (т, J=8,7 Гц, 1H), 8,56-8,51 (м, 2H), 8,13 (т, J=9,0 Гц, 1H), 7,61-7,58 (м, 1H), 7,35-7,58 (д, J=6,6 Гц, 1H).
Пример 41
Синтез 1-(5-(1H-1,2,4-триазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (41):
Схема 40
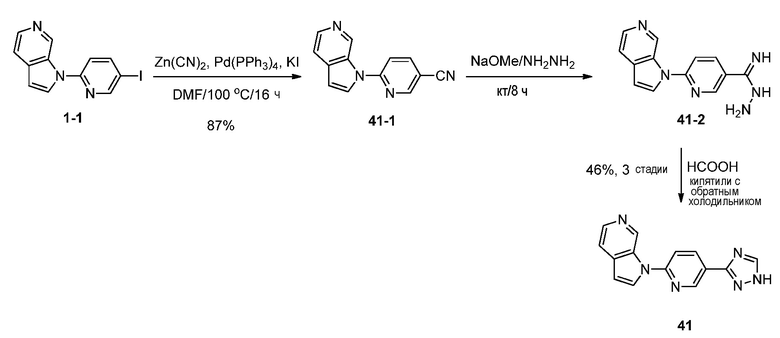
Стадия 1: Синтез 6-(1H-пирроло[2,3-с]пиридин-1-ил)никотинонитрила (41-1). К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (1 г, 3,11 ммоль, 1-1) в N,N-диметилформамиде (50 мл) добавляли дицианоцинк (0,73 г, 6,23 ммоль), тетракис(трифенилфосфин)палладий(0) (0,36 г, 0,31 ммоль) и калий йодид (0,052 г, 0,311 ммоль). Полученную смесь перемешивали в течение 16 часов при 100°C в атмосфере азота. После охлаждения до температуры окружающей среды, реакционную смесь разбавляли водой (150 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (5×80 мл) и сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 20~30% этилацетата в петролейном эфире с получением 6-(1H-пирроло[2,3-с]пиридин-1-ил)никотинонитрила в виде не совсем белого твердого вещества: МС (ESI, m/z): 221,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,85 (с, 1H), 9,07 (д, J=1,2 Гц, 1H), 8,50 (д, J=2,0 Гц, 1H), 8,42 (д, J=3,6 Гц, 1H), 8,34 (д, J=5,2 Гц, 1H), 8,08 (д, J=8,4 Гц, 1H), 7,68 (д, J=5,2 Гц, 1H), 6,94 (д, J=3,2 Гц, 1H).
Стадия 2: Синтез 6-(1H-пирроло[2,3-с]пиридин-1-ил)никотинимидогидразида (41-2). Раствор 6-(1H-пирроло[2,3-с]пиридин-1-ил)никотинонитрила (600 мг, 2,72 ммоль) в метаноле (20 мл) обрабатывали метанолатом натрия (736 мг, 13,63 ммоль) в течение 4 часов при температуре окружающей среды, с последующим добавлением гидразина (190, 5,95 ммоль). Через дополнительные 4 часа при температуре окружающей среды, смесь концентрировали при пониженном давлении и остаток использовали на следующей стадии без дополнительной очистки: (ESI, m/z): 253,2 [M+1]+.
Стадия 3: Синтез 1-(5-(1H-1,2,4-триазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (41). Раствор вышеуказанного сырого 6-(1H-пирроло[2,3-с]пиридин-1-ил)никотинимидогидразида в муравьиной кислоте (15 мл) кипятили с обратным холодильником в течение 16 часов. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~2% метанола в дихлорметане, с получением 1-(5-(1H-1,2,4-триазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 263,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 10,08 (с, 1H), 9,26 (д, J=2,0 Гц, 1H), 9,04 (д, J=3,6 Гц, 1H), 8,68 (ушир.с, 1H), 8,65-8,62 (м, 1H), 8,54 (д, J=6,4 Гц, 1H), 8,30 (д, J=8,4 Гц, 1H), 8,16 (д, J=8,8 Гц, 1H), 7,31 (т, J=3,2 Гц, 1H).
Примеры 42 и 43
Синтез 1-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пиперидин-4-ола (42) и 1-(5-(4-фторпиперидин-1-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (43):
Схема 41

Стадия 1: Синтез 1-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пиперидин-4-ола (42). К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (0,5 г, 1,56 ммоль, 1-1) в диметилсульфоксиде (20 мл) добавляли пиперидин-4-ол (0,3 г, 3,11 ммоль), карбонат цезия (2,1 г, 6,23 ммоль), иодид меди (I) (0,18 г, 0,93 ммоль) и 2-(диметиламино)уксусную кислоту (64,2 мг, 0,62 ммоль). Полученную смесь перемешивали в течение 16 часов при 130°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×60 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пиперидин-4-ола в виде светло-желтого твердого вещества: МС (ESI, m/z): 295,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,91 (с, 1H), 8,30-8,21 (м, 2H), 8,06 (д, J=5,7 Гц, 1H), 8,00 (д, J=3,3 Гц, 1H), 7,62 (с, 2H), 6,84 (д, J=3,3 Гц, 1H), 4,74 (д, J=5,2 Гц, 1H), 3,71-3,57 (м, 3H), 2,98-2,91 (м, 2H), 1,89-1,81 (м, 2H), 1,53-1,42 (м, 2H).
Стадия 2: Синтез 1-(5-(4-фторпиперидин-1-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (43). К раствору 1-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)пиперидин-4-ола (100 мг, 0,34 ммоль) в дихлорметане (10 мл) добавляли трифторид диэтиламиносеры (DAST, 548 мг, 3,40 ммоль) при -78°C. Полученный раствор перемешивали в течение 4 часов при температуре окружающей среды и гасили водой (30 мл), экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-(4-фторпиперидин-1-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 297,1 [M+1]+; 1H ЯМР (400 МГц, CD3OD) δ 8,87 (с, 1H), 8,31(д, J=3,2 Гц, 1H), 8,27 (д, J=6,0 Гц, 1H), 8,06 (д, J=6,0 Гц, 1H), 7,86 (д, J=3,6 Гц, 1H), 6,67 (дд, J=3,2 Гц, 8,8 Гц, 1H), 7,55 (д, J=8,8 Гц, 1H), 6,89 (д, J=3,6 Гц, 1H), 4,93-4,91 (м, 0,5H), 4,84-4,79 (м, 0,5H), 3,53-3,47 (м, 2H), 3,35-3,31 (м, 2H), 2,17-1,94 (м, 4H).
Примеры 44 и 45
Синтез 1-(6'-фтор-3,4'-бипиридин-6-ил)-1H-пирроло[2,3-с]пиридина (44) и Синтез N,N-диметил-6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,4'-бипиридин-2'-амина (45):
Схема 42
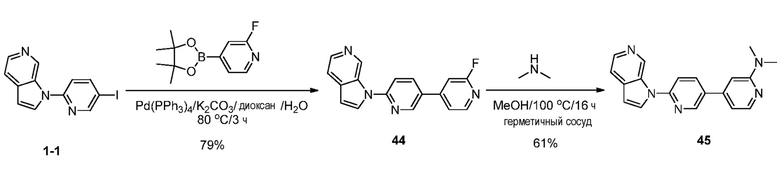
Стадия 1: Синтез 1-(6'-фтор-3,4'-бипиридин-6-ил)-1H-пирроло[2,3-с]пиридина (44). К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (100 мг, 0,31 ммоль, 1-1) в 1,4-диоксане (20 мл) и воде (5 мл) добавляли 2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (105 мг, 0,47 ммоль), карбонат калия (129 мг, 0,93 ммоль) и тетракис(трифенилфосфин)палладий(0) (18 мг, 0,016 ммоль). Полученную смесь перемешивали в течение 3 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (100 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~5% метанола в дихлорметане, с получением 1-(5-(пиридин-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 291,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,87 (с, 1H), 9,16 (д, J=2,1 Гц, 1H), 8,54 (дд, J=2,4 Гц, 8,7 Гц, 1H), 8,43 (д, J=3,3 Гц, 1H), 8,38 (д, J=5,1 Гц, 1H), 8,32 (д, J=5,1 Гц, 1H), 8,07 (д, J=8,7 Гц, 1H), 7,90 (дд, J=1,8 Гц, 3,6 Гц, 1H), 7,76 (с, 1H), 7,69 (дд, J=0,9 Гц, 5,4 Гц, 1H), 6,90 (д, J=3,3 Гц, 1H).
Стадия 2: Синтез N,N-диметил-6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,4'-бипиридин-2'-амина (45). К раствору диметиламина в метаноле (5 мл, 33% масс./масс.) добавляли 1-(6'-фтор-3,4'-бипиридин-6-ил)-1H-пирроло[2,3-с]пиридин (50 мг, 0,17 ммоль). Полученный раствор выдерживали в течение 16 часов при 100°C в герметичном сосуде. После охлаждения до температуры окружающей среды, полученный раствор концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,3~3% метанола в дихлорметане, с получением N,N-диметил-6-(1H-пирроло[2,3-с]пиридин-1-ил)-3,4'-бипиридин-2'-амина в виде бесцветного твердого вещества: МС (ESI, m/z): 316,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,84 (с, 1H), 9,04 (д, J=2,1 Гц, 1H), 8,43-8,39 (м, 2H), 8,30 (д, J=5,4 Гц, 1H), 8,20 (д, J=6,0 Гц, 1H), 8,00 (д, J=8,7 Гц, 1H), 7,67 (дд, J=0,6 Гц, 5,1 Гц, 1H), 7,03-6,89 (м, 2H), 6,89 (д, J=3,3 Гц, 1H).
Пример 46
Синтез 1-(5-(пиримидин-4-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (46):
Схема 43

Стадия 1: Синтез 1-(5-(трибутилстаннил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (46-1). К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (1,51 г, 4,67 ммоль, 1-1) в ацетонитриле (50 мл) добавляли 1,1,1,2,2,2-гексабутилдистаннан (8,13 г, 14,01 ммоль), трифенилфосфин (0,37 г, 1,40 ммоль), триэтиламин (4,73 г, 46,71 ммоль) и ацетат палладия(II) (0,11 г, 0,47 ммоль). Полученный раствор кипятили с обратным холодильником в течение 16 часов в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1-(5-(трибутилстаннил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 486,0 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 9,66 (с, 1H), 9,60 (с, 1H), 8,38 (д, J=5,6 Гц, 1H), 7,95 (д, J=1,6 Гц, 1H), 7,73 (д, J=4,8 Гц, 1H), 7,62 (д, J=6,4 Гц, 1H), 7,48 (д, J=8,4 Гц, 1H), 6,74 (д, J=2,8 Гц, 1H), 1,65-1,61 (м, 6H), 1,42-1,35 (м, 6H), 1,15-1,13 (м, 6H), 0,95-0,91 (м, 9H).
Стадия 2: Синтез 1-(5-(пиримидин-4-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (46). К раствору 1-(5-(трибутилстаннил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (120 мг, 0,25 ммоль) в N,N-диметилформамиде (20 мл) добавляли 4-бромпиримидин (59 мг, 0,37 ммоль), иодид меди (I) (5 мг, 0,026 ммоль) и тетракис(трифенилфосфин)палладий(0) (29 мг, 0,025 ммоль). Полученную смесь перемешивали в течение 4 часов при 40°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (60 мл) и экстрагировали этилацетатом (3×80 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~2% метанола в дихлорметане, с получением 1-(5-(пиримидин-4-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 274,0 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,89 (с, 1H), 9,44 (д, J=2,0 Гц, 1H), 9,32 (д, J=1,2 Гц, 1H), 8,95 (д, J=5,6 Гц, 1H), 8,78 (дд, J=2,4 Гц, 8,8 Гц, 1H), 8,45 (д, J=3,6 Гц, 1H), 8,33 (д, J=5,2 Гц, 1H), 8,28 (дд, J=1,2 Гц, 5,2 Гц, 1H), 8,10 (д, J=8,8 Гц, 1H), 7,68 (д, J=5,2 Гц, 1H), 6,92 (д, J=3,6 Гц, 1H).
Пример 47
Синтез 2-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина (47):
Схема 44
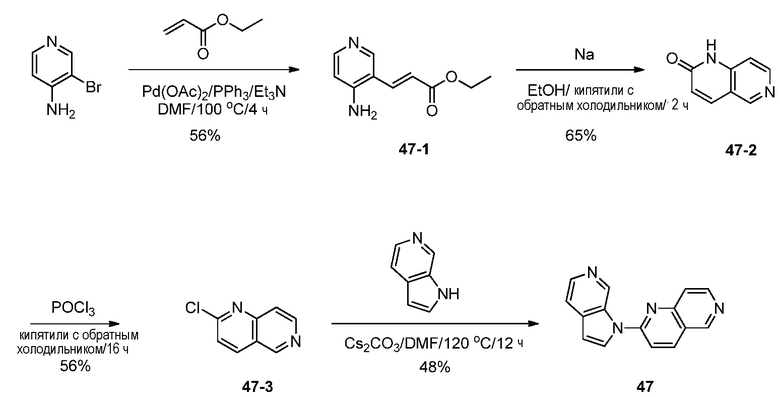
Стадия 1: Синтез (E)-этил 3-(4-аминопиридин-3-ил)акрилата (47-1). К раствору 3-бромпиридин-4-амина (5 г, 28,9 ммоль) в N,N-диметилформамиде (50 мл) добавляли этилакрилат (4,4 г, 43,3 ммоль), трифенилфосфин (1,7 г, 6,4 ммоль), ацетат палладия(II) (0,65 г, 2,9 ммоль) и триэтиламин (2,9 г, 28,9 ммоль). Полученную смесь перемешивали в течение 4 часов при 100°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (200 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (5×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~2% метанола в дихлорметане, с получением (E)-этил 3-(4-аминопиридин-3-ил)акрилата в виде желтого твердого вещества: МС (ESI, m/z): 193,0 [M+1]+; 1H ЯМР (400 МГц, CDCl3) δ 7,86 (с, 1H), 7,65 (д, J=8,0 Гц, 1H), 7,45 (д, J=16,4 Гц, 1H), 7,10 (д, J=8,0 Гц, 1H), 6,65 (д, J=16,4 Гц, 1H), 5,10 (ушир.с, 2H), 4,25 (кв, J=7,2 Гц, 2H), 1,34 (т, J=7,2 Гц, 3H).
Стадия 2: Синтез 1,6-нафтиридин-2(1H)-она (47-2). Натрий (0,96 г, 41,6 ммоль) растворяли в безводном этаноле (60 мл) при 0°C с последующим добавлением (E)-этил 3-(4-аминопиридин-3-ил)акрилата (2,1 г, 10,4 ммоль). Полученный раствор кипятили с обратным холодильником в течение 2-х часов, затем охлаждали до температуры окружающей среды и нейтрализовали уксусной кислотой (2,5 г, 41,6 ммоль). Полученный раствор концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 1,6-нафтиридин-2(1H)-она в виде желтого твердого вещества: МС (ESI, m/z): 147,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,18 (ушир.с, 1H), 8,83 (с, 1H), 8,46 (д, J=5,7 Гц, 1H), 8,00 (д, J=9,3 Гц, 1H), 7,20 (д, J=5,7 Гц, 1H), 6,57 (д, J=9,6 Гц, 1H).
Стадия 3: Синтез 2-хлор-1,6-нафтиридина (47-3). Раствор 1,6-нафтиридин-2(1H)-она (2 г, 13,68 ммоль) в фосфорилтрихлориде (20 мл) кипятили с обратным холодильником в течение 16 часов. После охлаждения до температуры окружающей среды, полученный раствор концентрировали при пониженном давлении и остаток поглощали дихлорметаном (100 мл) и промывали насыщенным водным раствором бикарбоната натрия (100 мл). Органический слой сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 2-хлор-1,6-нафтиридина в виде желтого твердого вещества: МС (ESI, m/z): 165,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,43 (с, 1H), 8,83 (д, J=6,0 Гц, 1H), 8,69 (д, J=11,7 Гц, 1H), 7,88 (д, J=5,7 Гц, 1H), 7,86 (д, J=9,6 Гц, 1H).
Стадия 4: Синтез 2-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина (47). К раствору 1H-пирроло[2,3-с]пиридина (48 мг, 0,41 ммоль) в N,N-диметилформамиде (10 мл) добавляли 2-хлор-1,6-нафтиридин (60 мг, 0,37 ммоль) и карбонат цезия (119 мг, 0,37 ммоль). Полученную смесь перемешивали в течение 12 часов при 120°C в атмосфере азота. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×30 мл) и сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 2-(1H-пирроло[2,3-с]пиридин-1-ил)-1,6-нафтиридина в виде желтого твердого вещества: МС (ESI, m/z): 247,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,24 (ушир.с, 1H), 9,37 (с, 1H), 8,78-8,74 (м, 2H), 8,58 (д, J=3,6 Гц, 1H), 8,38 (д, J=4,8 Гц, 1H), 8,27 (д, J=9,0 Гц, 1H), 8,02 (д, J=6,0 Гц, 1H), 7,71 (д, J=5,1 Гц, 1H), 6,96 (д, J=3,6 Гц, 1H).
Пример 48
Синтез N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-3-(2-фторэтокси)бензамида (48):
Схема 45

Стадия 1: Синтез N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-3-гидроксибензамида (48-1). К раствору 6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-амина (100 мг, 0,48 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-метоксибензойную кислоту (98 мг, 0,71 ммоль), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU) (271 мг, 0,71 ммоль) и триэтиламин (144 мг, 1,43 ммоль). Полученный раствор перемешивали в течение 16 часов при температуре окружающей среды в атмосфере азота. Реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (3×60 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,3~2% метанола в дихлорметане, с получением N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-3-гидроксибензамида в виде светло-желтого твердого вещества: МС (ESI, m/z): 331,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,58 (с, 1H), 9,83 (ушир.с, 1H), 9,67 (с, 1H), 8,97 (д, J=2,4 Гц, 1H), 8,40 (дд, J=2,7 Гц, 9,0 Гц, 1H), 8,28-8,25 (м, 2H), 7,88 (д, J=8,7 Гц, 1H), 7,65 (д, J=5,4 Гц, 1H), 7,45-7,33 (м, 3H), 7,03 (дд, J=2,7 Гц, 9,0 Гц, 1H), 6,83 (д, J=3,3 Гц, 1H).
Стадия 2: Синтез N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-3-(2-фторэтокси)бензамида (48). К раствору N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-3-гидроксибензамида (40 мг, 0,12 ммоль) в N,N-диметилформамиде (10 мл) добавляли 1-бром-2-фторэтан (23 мг, 0,18 ммоль) и карбонат калия (50 мг, 0,36 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 16 часов при 80°C. После охлаждения до температуры окружающей среды, полученную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×50 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением N-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-3-(2-фторэтокси)бензамида в виде бесцветного твердого вещества: МС (ESI, m/z): 377,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 10,58 (с, 1H), 9,68 (ушир.с, 1H), 8,98 (д, J=2,4 Гц, 1H), 8,41 (дд, J=2,7 Гц, 8,7 Гц, 1H), 8,28-8,25 (м, 2H), 7,92 (д, J=8,7 Гц, 1H), 7,72-7,48 (м, 4H), 7,24 (дд, J=2,4 Гц, 7,5 Гц, 1H), 6,84 (д, J=3,6 Гц, 1H), 4,88 (д, J=3,9 Гц, 1H), 4,72 (д, J=3,9 Гц, 1H), 4,41 (д, J=3,9 Гц, 1H), 4,30 (д, J=3,9 Гц, 1H).
Пример 49
Радиохимический синтез [3H]-6
Схема 46

Радиохимический синтез [3H]-6: В сосуд для тритирования добавляли 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин (2,8 мг, 11,4 мкмоль) и черный родий (2,4 мг, 23,3 мкмоль), с последующим добавлением THF (0,3 мл). Сосуд подсоединяли к манифольду для тритирования и выполняли два цикла замораживания-оттаивания (жидкий азот). В то время как реакционная смесь была заморожена, добавляли газообразный тритий (1,02 Ки). Черную суспензию нагревали до комнатной температуры и перемешивали в течение 16 часов. Реакционную смесь замораживали жидким азотом и избыток газообразного трития удаляли, затем сосуд удаляли из реакционного порта. Когда суспензия нагревалась до комнатной температуры, смесь фильтровали через небольшой слой целита с использованием EtOH. Фильтрат концентрировали с получением тонкой пленки, которую помещали в EtOH, концентрировали, помещали в 10 мл EtOH и подсчитывали в результате в общей сложности 494,7 мКи. Анализ с помощью ОФ-ВЭЖХ (Gemini C18, 4,6×150 мм, 254 нм, (55:45) 0,05M pH 9,5 TEAA:CH3CN, 1 мл/мин). Часть партии очищали с помощью ВЭЖХ с обращенной фазой (Gemini C18, 10×250 мм, 254 нм, (60:40) 0,05M pH 9,5 TEAA:CH3CN, 5 мл/мин, с получением партии 135,18 мКи [3H]-6 в 99,4 мл EtOH. Удельная активность была определена как 37,8 Ки/ммоль.
Пример 50
Синтез 6-фтор-3-(5-метокси-1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (L-005587122-000U):

Синтез 6-фтор-3-(5-метокси-1H-пирроло[2,3-с]пиридин-1-ил)изохинолина. К перемешиваемому раствору 1H-пирроло[2,3-с]пиридина (163 мг, 1,10 ммоль) и 3-хлор-6-фторизохинолина 9-3 (200 мг, 1,10 ммоль) в тетрагидрофуране (50 мл) добавляли натрий 2-метилпропан-2-олат (212 мг, 2,20 ммоль) и t-BuXPhos палладий(II) бифенил-2-амин мезилат (351 мг, 0,44 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 3 часов при 55°C в атмосфере азота. После охлаждения до температуры окружающей среды реакционную смесь гасили водой (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~5% метанола в дихлорметане, с получением 6-фтор-3-(5-метокси-1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде светло-желтого твердого вещества (270 мг, 83%): МС (ESI, m/z): 293,9 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,41 (с, 1H), 9,36 (с, 1H), 8,35 (д, J=3,6 Гц, 1H), 8,34-8,30 (дд, J=9,2 Гц, 6,0 Гц, 1H), 8,24 (с, 1H), 7,80 (дд, J=2,0 Гц, 10,0 Гц, 1H), 7,59-7,54 (м, 1H), 7,03 (д, J=0,8 Гц, 1H), 6,77 (д, J=3,6 Гц, 1H), 3,91 (с, 3H).
Радиохимический синтез [18F]-лигандов
Общие способы
[18F]Фторид транспортировали в лабораторию радиохимии анионообменной смоле и элюировали непосредственно перед использованием. Если специально не указано иное, [18F]фторид, содержащий анионообменную смолу, элюировали криптофиксом 222 (Kryptofix222) (7 мг, 19 мкмоль) и K2CO3 (2,1 мг, 15 мкмоль) в ацетонитриле/воде (80/20, 0,7 мл) и переносили в вентилируемый 1 мл V-образный флакон в микроволновом резонаторе. Фторид сушили в токе аргона и СВЧ-нагреве (35 Вт/90°C). Дополнительные аликвоты ацетонитрила (3×0,5 мл) добавляли для азеотропной сушки при 35 Вт/90°C.
Способ синтеза предшественников [18F]-лигандов:
1. Синтез 6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (9a)

Синтез 6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (9a).
Стадия 1: Раствор сульфата меди (1 г, 6,29 ммоль) в воде (5 мл) добавляли к перемешиваемому раствору сульфита натрия (1 г, 7,94 ммоль) в воде (5 мл) при температуре окружающей среды. Через 10 минут, проводили фильтрование и осадок на фильтре промывали водой (3×10 мл) с получением влажного сульфита меди в виде коричневого твердого вещества, который растворяли в насыщенным водном растворе нитрита натрия (50 мл) при температуре окружающей среды.
Стадия 2: 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амин (300 мг, 1,15 ммоль) растворяли в 20% (масс./масс.) водном растворе серной кислоты, (5 мл) с последующим добавлением нитрита натрия (95 мг, 1,38 ммоль) при 0°C. После перемешивания в течение 15 минут, полученный раствор добавляли к вышеуказанному раствору в течение 10 минут при температуре окружающей среды. После перемешивания в течение 10 минут, реакционную смесь гасили 25% (масс./масс.) водным раствором аммиака (5 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1% метанола в дихлорметане, с получением 6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде желтого твердого вещества (31,6 мг, 10%): МС (ESI, m/z): 291,1 [M+1]+; 1H ЯМР (300 МГц, CDCl3) δ 9,81-9,77 (м, 1H), 9,45 (с, 1H), 8,84 (д, J=1,8 Гц, 1H), 8,43-8,41 (м, 1H), 8,35 (д, J=2,1 Гц, 1H), 8,28 (д, J=9,3 Гц, 1H), 8,09 (с, 1H), 8,03 (с, 1H), 7,75-7,68 (м, 1H), 6,88 (т, J=9,0 Гц, 1H).
2. Синтез дитрет-бутил (6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-ил)карбамата (18a):
Стадия 1: Синтез N-(3-хлор-6-нитроизохинолин-5-ил)ацетамида

В 2000-мл 4-горлую круглодонную колбу помещали раствор 3-хлоризохинолин-5-амина (33 г, 184,75 ммоль, 1,00 эквив.) в DCE/THF (550/183 мл), уксусный ангидрид (150,77 г, 1,48 моль, 8,00 эквив.), Bi(NO3)3.5H2O (79,6 г, 184,69 ммоль, 1,00 эквив.). Полученный раствор перемешивали в течение 2 часов при 50°C в масляной бане. Полученную смесь концентрировали в вакууме. Остаток наносили на колонку с силикагелем со смесью этилацетат/петролейный эфир (3:1). Это привело к 19 г смеси (38%) N-(3-хлор-8-нитроизохинолин-5-ил)ацетамида и N-(3-хлор-6-нитроизохинолин-5-ил)ацетамида в виде желтого твердого вещества.
Стадия 2: Синтез N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)ацетамида; N-(8-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)

В 1000-мл 4-горлую круглодонную колбу, продуваемую и поддерживаемую в инертной атмосфере азота, помещали раствор N-(3-хлор-8-нитроизохинолин-5-ил)ацетамида и N-(3-хлор-6-нитроизохинолин-5-ил)ацетамида (19 г смеси, 35,76 ммоль, 1,00 эквив.) в NMP (480 мл), 3st t-BuXPhos предкатализатор (5,7 г, 7,18 ммоль, 0,10 эквив.), Cs2CO3 (70 г, 215,52 ммоль, 3,00 эквив.). Полученный раствор перемешивали в течение 2 часов при 60°C в масляной бане. Затем реакционную смесь гасили путем добавления 3000 мл воды. Полученный раствор экстрагировали 3×500 мл этилацетата и органические слои объединяли. Полученную смесь промывали 2×500 мл воды. Смесь сушили над безводным сульфатом натрия и концентрировали в вакууме. Это привело к 20 г смеси (81%) N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)ацетамида; N-(8-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)ацетамида в виде масла красного цвета.
Стадия 3: Синтез трет-бутил N-ацетил-N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамата

В 1000-мл 4-горлую круглодонную колбу помещали раствор N-(8-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)ацетамид/N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)ацетамида (20 г, 57,58 ммоль, 1,00 эквив.) в DCM/THF (500/50 мл), (Boc)2O (18,8 г, 86,14 ммоль, 1,50 эквив.), TEA (17,46 г, 172,55 ммоль, 3,00 эквив.), DMPA (1,4 г, 11,48 ммоль, 0,20 эквив.). Полученный в результате раствор перемешивали в течение ночи при комнатной температуре. Полученную смесь концентрировали в вакууме. Остаток наносили на колонку с силикагелем со смесью этилацетат/петролейный эфир (3:1). Это привело к 6,8 г (26%) трет-бутил N-ацетил-N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамата в виде желтого твердого вещества.
Стадия 4: Синтез трет-бутил N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамата
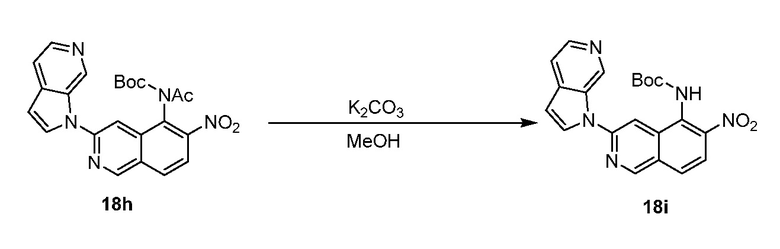
В 250-мл 4-горлую круглодонную колбу помещали раствор трет-бутил N-ацетил-N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамата (6,8 г, 15,20 ммоль, 1,00 эквив.) в метаноле (100 мл), карбонат калия (20,9 г, 151,45 ммоль, 10,00 эквив.). Полученный раствор перемешивали в течение 2 часов при комнатной температуре. Твердые вещества отфильтровывали. Фильтрат концентрировали в вакууме. Остаток наносили на колонку с силикагелем со смесью этилацетата/петролейного эфира (3:1). Это привело к получению 4,8 г (78%) трет-бутил N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамата в виде желтого твердого вещества.
Стадия 5: Синтез трет-бутил N-[(трет-бутокси)карбонил]-N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамата (18a)
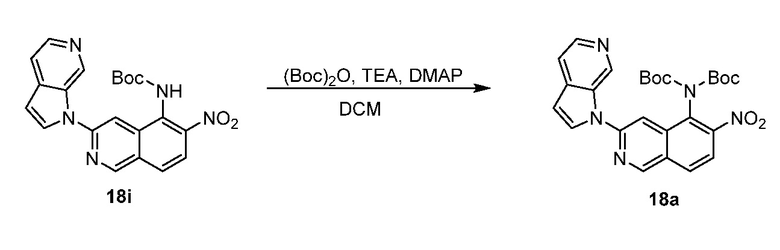
В 100-мл 4-горлую круглодонную колбу помещали трет-бутил N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамат (4,8 г, 11,84 ммоль, 1,00 эквив.), (Boc)2O (3,86 г, 17,69 ммоль, 1,50 эквив.), TEA (3,59 г, 35,48 ммоль, 3,00 эквив.), 4-диметиламинопиридин (290 мг, 2,37 ммоль, 0,20 эквив.), дихлорметан (100 мл). Полученный раствор перемешивали в течение 2 часов при комнатной температуре. Полученную смесь концентрировали в вакууме. Остаток наносили на колонку с силикагелем со смесью этилацетат/петролейный эфир (3:1). Это привело к 3,52 г (59%) трет-бутил N-[(трет-бутокси)карбонил]-N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамата в виде желтого твердого вещества: МС (ESI, m/z): 505 [M+1]+; 1H-ЯМР (300 МГц, CDCl3): δ 1,39 (18H, с), 6,85 (1H, д), 7,65 (1H, д),7,88 (1H, с), 7,95 (1H, д), 8,10 (1H, д), 8,21 (1H, д), 8,43 (1H, д), 9,43 (1H, с), 9,750 (1H, д).
3. Синтез трет-бутил N-[(трет-бутокси)карбонил]-N-(6-нитро-3-{1H-пирроло[2,3-с]пиридин-1-ил}изохинолин-7-ил)карбамата (105a):

Стадия 1: Синтез (4-бром-3-фторфенил)метанола. К раствору 4-бром-3-фторбензойной кислоты (25 г, 114 ммоль) в тетрагидрофуране (150 мл) добавляли 1M раствор борана в тетрагидрофуране (228 мл, 228 ммоль) в течение 1 часа при 0°C. Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили метанолом (200 мл) и концентрировали при пониженном давлении. Остаток растворяли в этилацетате (200 мл), промывали насыщенным солевым раствором (100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении с получением (4-бром-3-фторфенил)метанола в виде бесцветного твердого вещества (23 г, 98%): 1H ЯМР (300 МГц, ДМСО-d6) δ 7,63 (т, J=7,8 Гц, 1H), 7,29-7,25 (м, 1H), 7,12 (т, J=0,9 Гц, 1H), 5,38 (т, J=5,7 Гц, 1H), 4,48 (д, J=5,7 Гц, 2H).
Стадия 2: Синтез 1-бром-4-(хлорметил)-2-фторбензола. К раствору (4-бром-3-фторфенил)метанола (46 г, 0,23 моль) в дихлорметане (500 мл) добавляли сернистый дихлорид (107 г, 0,89 моль) при 0°C с последующим добавлением N,N-диметилформамида (1 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 3 часов. Смесь концентрировали при пониженном давлении и остаток растворяли в этилацетате (150 мл), промывали насыщенным солевым раствором (200 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении с получением 1-бром-4-(хлорметил)-2-фторбензол в виде масла желтого цвета (47,5 г, 95%): 1H ЯМР (300 МГц, ДМСО-d6) δ 7,72 (т, J=7,8 Гц, 1H), 7,49-7,45 (м, 1H), 7,26-7,23 (м, 1H), 4,75 (с, 2H).
Стадия 3: Синтез диэтил 2-(4-бром-3-фторбензил)малоната. Раствор диэтилмалоната (59,8 г, 0,37 моль) в N,N-диметилформамиде (200 мл) обрабатывали гидридом натрия (14,9 г, 0,37 моль) (60% масс./масс., диспергированный в минеральном масле) при температуре окружающей среды в течение 30 минут с последующим добавлением 1-бром-4-(хлорметил)-2-фторбензола (41,5 г, 0,19 моль) в течение 30 минут при 0°C. Полученную смесь перемешивали в течение 16 часов при температуре окружающей среды, а затем гасили насыщенным водным раствором хлорида аммония (1500 мл), экстрагировали этилацетатом (3×500 мл). Объединенные органические слои промывали насыщенным солевым раствором (5×500 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~2% этилацетата в петролейном эфире с получением диэтил 2-(4-бром-3-фторбензил)малоната в виде бесцветного масла (36 г, 34%): МС (ESI, m/z): 346,9, 348,9 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 7,40-7,38 (м, 1H), 7,33 (с, 1H), 7,18-7,16 (м, 1H), 4,13-4,05 (м, 4H), 3,94 (т, J=8,0 Гц, 1H), 3,10 (д, J=8,0 Гц, 2H), 1,15-1,08 (м, 6H).
Стадия 4: Синтез 2-(4-бром-3-фторбензил)малоновой кислоты. Раствор диэтил 2-(4-бром-3-фторбензил)малоната (34 г, 58,8 ммоль) в этаноле (100 мл) обрабатывали 6 н водного раствора гидроксида калия (50 мл, 300 ммоль) при кипении с обратным холодильником в течение 4-х часов. После охлаждения до температуры окружающей среды, полученный раствор концентрировали при пониженном давлении и остаток разбавляли водой (200 мл) и подкисляли концентрированной хлористоводородной кислотой (12 н) до pH=1. Твердое вещество собирали фильтрацией и сушили в вакуумном сушильном шкафу с получением 2-(4-бром-3-фторбензил)малоновой кислоты в виде белого твердого вещества (16 г, 94%): 1H ЯМР (400 МГц, ДМСО-d6) δ 12,89 (ушир.с, 2H), 7,39-7,32 (м, 1H), 7,29 (д, J=13,2 Гц, 1H), 7,17-7,11 (м, 1H), 3,69 (т, J=8,0 Гц, 1H), 3,02 (д, J=8,0 Гц, 2H).
Стадия 5: Синтез 3-(4-бром-3-фторфенил)пропановой кислоты. Суспензию 2-(4-бром-3-фторбензил)малоновой кислоты (16 г, 55 ммоль) в ксилоле (150 мл) кипятили с обратным холодильником в течение 4-х часов. После охлаждения до температуры окружающей среды, ксилол удаляли выпариванием при пониженном давлении с получением 3-(4-бром-3-фторфенил)пропановой кислоты в виде бесцветного твердого вещества (13 г, 94%): 1H ЯМР (400 МГц, ДМСО-d6) δ 12,21 (ушир.с, 1H), 7,32-7,29 (м, 2H), 7,16-7,13 (м, 1H), 2,83 (т, J=7,6 Гц, 2H), 2,56 (д, J=7,6 Гц, 2H).
Стадия 6: Синтез 6-бром-5-фтор-2,3-дигидро-1H-инден-1-она. Смесь 3-(4-бром-3-фторфенил)пропановой кислоты (60 г, 0,24 моль) и полифосфорной кислоты (300 мл) выдерживали при 80°C в течение 160 часов. После охлаждения до температуры окружающей среды, смесь выливали в воду со льдом (1200 г) и экстрагировали этилацетатом (3×500 мл). Объединенные органические слои промывали насыщенным солевым раствором (300 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 5~10% этилацетата в петролейном эфире с получением 6-бром-5-фтор-2,3-дигидро-1H-инден-1-она в виде светло-желтого твердого вещества (26 г, 47%): МС (ESI, m/z): 229,0, 231,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 7,92 (д, J=6,9 Гц, 1H), 7,64 (д, J=8,1 Гц, 1H), 3,08 (т, J=5,4 Гц, 2H), 2,68 (д, J=5,4 Гц, 2H).
Стадия 7: Синтез (E)-6-бром-5-фтор-2-(гидроксиимино)-2,3-дигидро-1H-инден-1-она. К раствору 6-бром-5-фтор-2,3-дигидро-1H-инден-1-она (22 г, 96 ммоль) в диэтиловом эфире (150 мл) и дихлорметана (50 мл) добавляли метанол (25 мл, насыщенный HCl при температуре окружающей среды) и изопентилнитрит (16,9 г, 144 ммоль) при 0°C. Полученную смесь перемешивали при температуре окружающей среды в течение 2 часов. Проводили фильтрование и осадок на фильтре промывали холодным диэтиловым эфиром (2×50 мл) с получением (E)-6-бром-5-фтор-2-(гидроксиимино)-2,3-дигидро-1H-инден-1-она в виде светло-желтого твердого вещества (15 г, 60%): МС (ESI, m/z): 258,0, 260,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 12,78 (с, 1H), 8,06 (д, J=6,9 Гц, 1H), 7,68 (д, J=9,0 Гц, 1H), 3,75 (с, 2H).
Стадия 8: Синтез 1,3,7-трибром-6-фторизохинолина (8). К раствору (E)-6-бром-5-фтор-2-(гидроксиимино)-2,3-дигидро-1H-инден-1-она (13,5 г, 52,3 ммоль) в 1,2-дихлорэтане (200 мл, насыщенный сухой HCl) добавляли трибромид фосфорила (30,0 г, 105 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 2 часов при 80°C. После охлаждения до температуры окружающей среды, смесь концентрировали при пониженном давлении и остаток аккуратно выливали в воду со льдом (500 г). Проводили фильтрование и осадок на фильтре промывали водой (2×100 мл), сушили в вакуумном сушильном шкафу с получением 1,3,7-трибром-6-фторизохинолина в виде светло-желтого твердого вещества (14 г, 63%): МС (ESI, m/z): 384,0, 386,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,38 (д, J=6,6 Гц, 1H), 8,24 (с, 1H), 7,96 (д, J=9,3 Гц, 1H).
Стадия 9: Синтез 3,7-дибром-6-фторизохинолина. К перемешиваемому раствору 1,3,7-трибром-6-фторизохинолина (13,5 г, 35,2 ммоль) в уксусной кислоте (100 мл) добавляли 55%-ный водный раствор HI (50 мл) и красный фосфор (2,7 г, 88,1 ммоль) при температуре окружающей среды. Полученный раствор перемешивали при 110°C в течение 2 часов. После охлаждения до температуры окружающей среды, смесь концентрировали при пониженном давлении. Остаток обрабатывали этилацетатом (300 мл), промывали насыщенным водным раствором бикарбоната натрия (100 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~6% этилацетата в петролейном эфире с получением 3,7-дибром-6-фторизохинолина в виде не совсем белого твердого вещества (7 г, 62%): МС (ESI, m/z): 306,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,15 (с, 1H), 8,68 (д, J=6,9 Гц, 1H), 8,20 (с, 1H), 7,94 (д, J=9,3 Гц, 1H).
Стадия 10: Синтез 3,7-дибромизохинолин-6-амина (10). Раствор 3,7-дибром-6-фторизохинолина (5,0 г, 16,4 ммоль) в 1,4-диоксане (150 мл) барботировали сухим газообразным аммиаком, пока полученный раствор не был насыщен при 0°C. Полученный раствор помещали в автоклав высокого давления и перемешивали при 120°C в течение 24 часов. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1~8% этилацетата в петролейном эфире с получением 3,7-дибромизохинолин-6-амина в виде светло-желтого твердого вещества (2,5 г, 48%): МС (ESI, m/z): 302,7 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 8,76 (с, 1H), 8,27 (с, 1H), 7,73 (с, 1H), 6,93 (с, 1H), 6,34 (ушир.с, 2H).
Стадия 11: Синтез трет-бутил (3,7-дибромизохинолин-6-ил)карбамата (11). К раствору 3,7-дибромизохинолин-6-амина (2,50 г, 8,28 ммоль) в дихлорметане (50 мл) добавляли триэтиламин (2,51 г, 24,84 ммоль), ди-трет-бутилдикарбонат (5,42 г, 24,84 ммоль) и N,N-диметилпиридин-4-амин (51 мг, 0,41 ммоль) при температуре окружающей среды. После дополнительных 4 часов, полученный раствор концентрировали при пониженном давлении и остаток растворяли в метаноле (50 мл) с последующим добавлением карбоната калия (2,5 г, 18,11 ммоль). Полученную смесь перемешивали при 50°C в течение 30 минут. После охлаждения до температуры окружающей среды, проводили фильтрование и фильтрат концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, элюируя 1~5% этилацетата в петролейном эфире с получением трет-бутил (3,7-дибромизохинолин-6-ил)карбамата в виде не совсем белого твердого вещества (1,8 г, 52%): МС (ESI, m/z): 402,8 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,05 (с, 1H), 8,75 (с, 1H), 8,54 (с, 1H), 8,20 (с, 1H), 8,18 (с, 1H), 1,50 (с, 9H).
Стадия 12: Синтез трет-бутил (7-бром-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-ил)карбамата (12). К перемешиваемому раствору трет-бутил (3,7-дибромизохинолин-6-ил)карбамата (1,2 г, 2,98 ммоль) в тетрагидрофуране (10 мл) добавляли 1H-пирроло[2,3-с]пиридин (0,71 г, 5,97 ммоль), t-BuXPhos палладий(II) бифенил-2-амин мезилат (0,71 г, 0,89 ммоль) и натрий 2-метилпропан-2-олат (1,15 г, 11,94 ммоль) при температуре окружающей среды в атмосфере азота. Полученную смесь облучали в микроволновой печи при 70°C в течение 2 часов. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением трет-бутил (7-бром-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-ил)карбамата в виде светло-желтого твердого вещества (0,2 г, 16%): МС (ESI, m/z): 439,0, 441,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,72 (с, 1H), 9,31 (с, 1H), 8,73 (с, 1H), 8,59 (с, 1H), 8,37-8,34 (м, 2H), 8,29-8,28 (м, 2H), 7,67 (д, J=4,8 Гц, 1H), 6,88 (д, J=3,3 Гц, 1H), 1,50 (с, 9H).
Стадия 13: Синтез 7-бром-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина. Раствор трет-бутил (7-бром-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-ил)карбамата (0,47 г, 1,07 ммоль) в дихлорметане (20 мл) обрабатывали 2,2,2-трифторуксусной кислотой (1,14 г, 10 ммоль) в течение 2 часов при температуре окружающей среды. Реакционную смесь гасили водой (50 мл) и нейтрализовали бикарбонатом натрия (0,84 г, 10 ммоль). Полученную смесь экстрагировали дихлорметаном (3×100 мл) и объединенные органические слои сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении с получением 7-бром-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина в виде светло-желтого твердого вещества (0,35 г, 95%): МС (ESI, m/z): 339,0, 341,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,65 (с, 1H), 9,06 (с, 1H), 8,34 (с, 1H), 8,31-8,17 (м, 2H), 7,84 (с, 1H), 7,65 (д, J=5,4 Гц, 1H), 7,10 (с, 1H), 6,82 (д, J=3,0 Гц, 1H), 6,31 (ушир.с, 2H).
Стадия 14: Синтез 7-бром-6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина.
Стадия 1: Раствор сульфата меди (1 г, 6,29 ммоль) в воде (5 мл) добавляли к раствору сульфита натрия (1 г, 7,94 ммоль) в воде (5 мл) при температуре окружающей среды. Через 10 минут, проводили фильтрование и осадок на фильтре промывали водой (3×10 мл) с получением влажного сульфита меди в виде коричневого твердого вещества, который растворяли в насыщенным водном растворе нитрита натрия (50 мл) при температуре окружающей среды.
Стадия 2: К суспензии 7-бром-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-6-амина (0,3 г, 0,88 ммоль) в ацетонитриле (20 мл) и воде (20 мл) добавляли трифторметансульфоновую кислоту (0,65, 4,4 ммоль) с последующим добавлением нитрита натрия (61 мг, 0,88 ммоль) при 0°C. Через 15 минут, полученный раствор добавляли к вышеуказанному раствору в течение 10 минут. Через дополнительные 10 минут добавляли 25% водный раствор аммиака (10 мл) и полученную смесь экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 1% метанола в дихлорметане, с получением 7-бром-6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде светло-желтого твердого вещества (70 мг, 20%): МС (ESI, m/z): 368,8, 370,8 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,75 (с, 1H), 9,55 (с, 1H), 8,86 (с, 1H), 8,77 (с, 1H), 8,47 (с, 1H), 8,37 (с, 1H), 8,33 (д, J=7,2 Гц, 1H), 7,69 (д, J=5,4 Гц, 1H), 6,93 (д, J=3,3 Гц, 1H).
Стадия 15: синтез 6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-амина. К суспензии 7-бром-6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (50 мг, 0,14 ммоль) в толуоле (15 мл) добавляли дифенилметанимин (73,6 мг, 0,41 ммоль), карбонат цезия (88 мг, 0,27 ммоль), XantPhos (15,7 мг, 0,027 ммоль) и трис(дибензилиденацетон)дипалладий (0) (14 мг, 0,014 ммоль) в атмосфере азота. Полученную смесь перемешивали в течение 3 часов при 90°C. После охлаждения до температуры окружающей среды, полученную смесь концентрировали при пониженном давлении и остаток поглощали тетрагидрофураном (20 мл) с последующим добавлением 2 н водного раствора гидрохлорида (3,0 мл). После перемешивания в течение 2 часов при температуре окружающей среды, полученную смесь гасили насыщенным водным раствором бикарбоната натрия (50 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-амина в виде пурпурного твердого вещества (35 мг, 90%): МС (ESI, m/z): 305,9 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,52 (с, 1H), 9,23 (с, 1H), 8,86 (с, 1H), 8,27-8,199 (м, 3H), 7,66 (д, J=5,1 Гц, 1H), 7,57 (с, 1H), 6,97 (ушир.с, 2H), 6,83 (д, J=3,0 Гц, 1H).
Стадия 16: Синтез трет-бутил N-[(трет-бутокси)карбонил]-N-(6-нитро-3-{1H-пирроло[2,3-с]пиридин-1-ил}изохинолин-7-ил)карбамата (105a). К раствору 6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-амина (35 мг, 0,11 ммоль) в N,N-диметилформамиде (10 мл) добавляли триэтиламин (35 мг, 0,34 ммоль), ди-трет-бутилдикарбонат (75 мг, 0,34 ммоль) и N,N-диметилпиридин-4-амин (5 мг, 0,04 ммоль) при температуре окружающей среды. После 1 часа, реакционную смесь гасили водой (50 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×30 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью препаративной тонкослойной хроматографии, элюировали 4% метанола в дихлорметане, с получением трет-бутил N-[(трет-бутокси)карбонил]-N-(6-нитро-3-{1H-пирроло[2,3-с]пиридин-1-ил}изохинолин-7-ил)карбамата в виде желтого твердого вещества (45 мг, 78%): МС (ESI, m/z): 506,0 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,74 (с, 1H), 9,59 (с, 1H), 8,94 (с, 1H), 8,60 (с, 1H), 8,48 (с, 1H), 8,37 (д, J=3,6 Гц, 1H), 8,32 (д, J=5,1 Гц, 1H), 7,71 (д, J=5,4 Гц, 1H), 6,94 (д, J=3,3 Гц, 1H), 1,36 (с, 18H).
Пример 51
Синтез [18F] 1-(5-(1-(2-фторэтил)-1H-пиразол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина ([18F]-104):
Схема 47
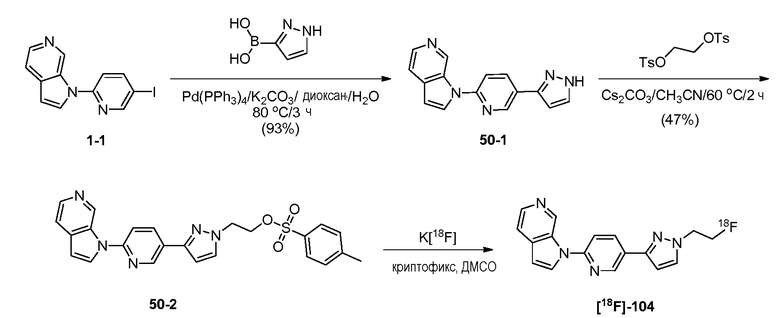
Стадия 1: Синтез 1-(5-(1H-пиразол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (50-1).
К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (100 мг, 0,31 ммоль, 1-1) в 1,4-диоксане (20 мл) и воде (5 мл) добавляли 1H-пиразол-3-илбороновую кислоту (53 мг, 0,47 ммоль), карбонат калия (129 мг, 0,93 ммоль) и тетракис(трифенилфосфин)палладий(0) (18 мг, 0,016 ммоль). Полученную смесь перемешивали в течение 3 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (100 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~5% метанола в дихлорметане, с получением 1-(5-(1H-пиразол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина в виде бесцветного твердого вещества: МС (ESI, m/z): 262,1 [M+1]+; 1H ЯМР (400 МГц, d6-DMSO) δ 13,09 (с, 1H), 9,78 (с, 1H), 9,06 (с, 1H), 8,40 (дд, J=1,6 Гц, 6,8 Гц, 1H), 8,30 (с, 1H), 8,28 (д, J=5,2 Гц, 1H), 7,92 (д, J=8,8 Гц, 1H), 7,89 (с, 1H), 7,67 (д, J=5,6 Гц, 1H), 6,91 (с, 1H), 6,86 (д, J=3,2 Гц, 1H).
Стадия 2: Синтез 2-(3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-1H-пиразол-1-ил)этил 4-метилбензолсульфоната (50-2). К раствору 1-(5-(1H-пиразол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина (0,20 г, 0,77 ммоль) в ацетонитриле (20 мл) добавляли карбонат цезия (0,50 г, 1,53 ммоль) и этан-1,2-диил бис(4-метилбензолсульфонат) (0,42 г, 1,15 ммоль) при температуре окружающей среды. После перемешивания в течение 2 часов при 60°C, реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 0,5~1,5% метанола в дихлорметане, с получением 2-(3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-1H-пиразол-1-ил)этил 4-метилбензолсульфоната в виде не совсем белого твердого вещества: МС (ESI, m/z): 460,2 [M+1]+; 1H ЯМР (300 МГц, d6-DMSO) δ 10,03 (с, 1H), 9,02 (д, J=3,3 Гц, 1H), 8,98 (д, J=2,1 Гц, 1H), 8,52 (д, J=5,7 Гц, 1H), 8,37 (дд, J=2,4 Гц, 6,3 Гц, 1H), 8,30 (д, J=6,3 Гц, 1H), 8,09 (д, J=9,3 Гц, 1H), 7,83 (д, J=2,4 Гц, 1H), 7,60 (д, J=8,4 Гц, 2H), 7,32 (д, J=8,4 Гц, 2H), 7,30 (с, 1H), 6,88 (д, J=1,5 Гц, 1H), 4,45 (ушир.с, 4H), 2,23 (с, 3H).
Стадия 3: Радиохимический синтез [18F] 1-(5-(1-(2-фторэтил)-1H-пиразол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-с]пиридина ([18F]-104): Раствор 2-(3-(6-(1H-пирроло[2,3-с]пиридин-1-ил)пиридин-3-ил)-1H-пиразол-1-ил)этил 4-метилбензолсульфоната (1 мг) в ДМСО или DMF (0,25 мл) добавляли в сосуд для микроволновой печи, содержащий сухой [18F]фторид, выпускную линию удаляли и реакционную смесь нагревали при 110°C (75 Вт) в течение 10 минут. После охлаждения до <50°C, реакционную смесь разбавляли H2O (0,6 мл), смешивали и вводили в полупрепаративную ВЭЖХ колонку. Продукт очищали с использованием колонки Zorbax Eclipse XDB-C-18, 5 мкм, 9,4×250 мм (Agilent), при скорости потока 5 мл/мин. Подвижная фаза представляла собой ацетонитрил/водн. NaH2PO4 (10 мM) от 30 до 70% в 15 мин. Представляющую интерес радиоактивную фракцию собирали, упаривали при отрицательном давлении, разбавляли 0,9% солевым раствором (3 мл) и переносили в стерильный контейнер. Конечный продукт тестировали на химическую и радиохимическую чистоту с помощью аналитической ВЭЖХ системы (Waters) с использованием Gemini, 5 мкм, 4,6×150 мм колонка (Phenomenex) при скорости потока 1 мл/мин. Подвижная фаза представляла собой смесь, состоящую из 50% ацетонитрила и 50% 0,1%-ной трифторуксусной кислоты в воде. Концентрацию [18F]-104 определяли с помощью ультрафиолетового детектора (254 нм). Подтверждение идентичности продукта определяли путем совместной инъекции образца соединения 104, и радиохимическую чистоту определяли с помощью детектора на основе йодида натрия (Bioscan). Время удерживания соединения [18F]-104 составляло 4,2 минуты и радиохимическая чистота была 100%.
Пример 52
Радиохимический синтез [18F] 7-(фторметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина ([18F]-17):
Схема 48

Стадия 1: Синтез 7-(метилтиометокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (51-1). Раствор 3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-ола (0,4 г, 1,53 ммоль, 15) в N,N-диметилформамиде (6 мл) обрабатывали гидридом натрия (80 мг, 2 ммоль, 60% масс./масс., диспергированный в минеральном масле) при 0°C в течение 10 минут, с последующим добавлением (хлорметил)(метил)сульфана (289 мг, 3 ммоль). Полученную смесь перемешивали в течение 2 часов при температуре окружающей среды и гасили насыщенным водным раствором хлорида аммония (30 мл). Полученную смесь экстрагировали этилацетатом (3×60 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×20 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~2% метанола в дихлорметане, с получением 7-(метилтиометокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в виде светло-желтого твердого вещества: МС (ESI, m/z): 322,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,64 (с, 1H), 9,30 (с, 1H), 8,32 (д, J=3,3 Гц, 1H), 8,28 (д, J=5,4 Гц, 1H), 8,24 (с, 1H), 8,04 (д, J=9,0 Гц, 1H), 7,72 (д, J=2,4 Гц, 1H), 7,68 (дд, J=0,9 Гц, 5,1 Гц, 1H), 7,57 (дд, J=2,4 Гц, 9,0 Гц, 1H), 6,86 (д, J=3,3 Гц, 1H), 5,47 (с, 2H), 2,25 (д, J=4,5 Гц, 3H).
Стадия 2: Синтез 3-(1H-пирроло[2,3-с]пиридин-1-ил)-7-((6-(трифторметил)-1H-бензо[d][1,2,3]триазол-1-илокси)метокси)изохинолина (51-3). К раствору 7-(метилтиометокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (0,15 г, 0,46 ммоль) в дихлорметане (10 мл) добавляли сульфурил дихлорид (0,18 г, 1,31 ммоль) при 0°C. Полученный раствор перемешивали в течение 1 часа при температуре окружающей среды и концентрировали при пониженном давлении с получением сырого 7-(хлорметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина. Раствор вышеуказанного сырого 7-(хлорметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина в дихлорметане (10 мл) добавляли к раствору 6-(трифторметил)-1H-бензо[d][1,2,3]триазол-1-ола (0,3 г, 1,48 ммоль) и гидрида натрия (59 мг, 1,48 ммоль) в N,N-диметилформамиде (20 мл) при 0°C. Через дополнительные 3 часа при температуре окружающей среды, реакционную смесь гасили добавлением насыщенного водного раствора хлорида аммония (50 мл) и экстрагировали этилацетатом (3×60 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×20 мл) и сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали смесью 1~3% метанола в дихлорметане, с получением 3-(1H-пирроло[2,3-с]пиридин-1-ил)-7-((6-(трифторметил)-1H-бензо[d][1,2,3]триазол-1-илокси)метокси)изохинолина в виде бесцветного твердого вещества: МС (ESI, m/z): 477,1 [M+1]+; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,68 (с, 1H), 9,42 (с, 1H), 8,36-8,33 (м, 3H), 8,30 (д, J=5,6 Гц, 1H), 8,15 (с, 1H), 8,13-8,11 (м, 1H), 7,91 (с, 1H), 7,77 (д, J=8,8 Гц, 1H), 7,68 (дд, J=0,4 Гц, 5,2 Гц, 1H), 7,64 (дд, J=2,4 Гц, 8,8 Гц, 1H), 6,88 (д, J=3,2 Гц, 1H), 6,48 (с, 2H).
Стадия 3: Радиохимический синтез [18F] 7-(фторметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина ([18F]-17). Раствор 3-(1H-пирроло[2,3-с]пиридин-1-ил)-7-((6-(трифторметил)-1H-бензо[d][1,2,3]триазол-1-илокси)метокси)изохинолина (0,6 мг, 51-3) в ДМСО (0,25 мл) и ацетонитрила (0,15 мл) добавляли в сосуд для микроволновой печи, содержащий сухой [18F]фторид, выпускную линию удаляли и реакционную смесь нагревали при 90°C в течение 5 минут, затем при 110°C в течение 5 минут и 120°C в течение 5 минут (50 Вт). После охлаждения до <50°C, реакционную смесь разбавляли H2O (0,6 мл), смешивали и вводили в полупрепаративную ВЭЖХ колонку. Продукт очищали с использованием колонки Zorbax Eclipse XDB-C-18, 5 мкм, 9,4×250 мм (Agilent), при скорости потока 5 мл/мин. Подвижная фаза представляла собой ацетонитрил/водн. NaH2PO4 (10 мM) от 30 до 70% в 15 мин. Представляющую интерес радиоактивную фракцию собирали, упаривали при отрицательном давлении, разбавляли 0,9% солевым раствором (3 мл) и переносили в стерильный контейнер. Конечный продукт тестировали на химическую и радиохимическую чистоту с помощью аналитической ВЭЖХ системы (Waters) с использованием Xbridge Phenyl, 3,5 мкм, 4,6×150 мм колонка (Waters) при скорости потока 2 мл/мин. Подвижная фаза представляла собой смесь, состоящую из 50% ацетонитрила и 50% 0,1%-ной трифторуксусной кислоты в воде. Концентрацию [18F]-17 определяли с помощью ультрафиолетового детектора (254 нм). Подтверждение идентичности продукта определяли путем совместной инъекции образца соединения 17, и радиохимическую чистоту определяли с помощью детектора на основе йодида натрия (Bioscan). Время удерживания соединения [18F]-17 составляло 5,3 мин.
Пример 53
Радиохимический синтез [18F] 1-(2'-фтор-[3,4'-бипиридин]-6-ил)-1H-пирроло[2,3-с]пиридина ([18F]-44):
Схема 49
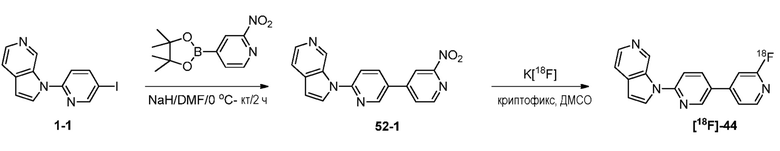
Стадия 1: Синтез 1-(6'-нитро-3,4'-бипиридин-6-ил)-1H-пирроло[2,3-с]пиридина (52-1).
К раствору 1-(5-иодпиридин-2-ил)-1H-пирроло[2,3-с]пиридина (100 мг, 0,31 ммоль) в 1,4-диоксане (30 мл) добавляли бис(пинаколято)диборон (158 мг, 0,62 ммоль), ацетат калия (60 мг, 0,62 ммоль) и [1,1′-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) (23 мг, 0,031 ммоль) при температуре окружающей среды. Полученную смесь перемешивали в течение 3 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды, к смеси добавляли 4-бром-2-нитропиридин (125 мг, 0,62 ммоль) с последующим добавлением карбонат калия (85 мг, 0,62 ммоль), воды (3 мл) и тетракис(трифенилфосфин)палладия(0) (35 мг, 0,031 ммоль). Полученную смесь перемешивали в течение 4 часов при 80°C в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (100 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюировали 0,3~3% метанола в дихлорметане, с получением 1-(6'-нитро-3,4'-бипиридин-6-ил)-1H-пирроло[2,3-с]пиридина в виде желтого твердого вещества: МС (ESI, m/z): 318,1 [M+1]+; 1H ЯМР (300 МГц, ДМСО-d6) δ 9,93 (с, 1H), 9,24 (д, J=2,1 Гц, 1H), 8,81-7,92 (м, 2H), 8,65 (дд, J=2,4 Гц, 8,7 Гц, 1H), 8,47 (д, J=3,3 Гц, 1H), 8,41 (дд, J=1,5 Гц, 5,1 Гц, 1H), 8,33 (д, J=5,1 Гц, 1H), 8,10 (д, J=8,4 Гц, 1H), 7,69 (д, J=5,7 Гц, 1H), 6,93 (д, J=3,6 Гц, 1H).
Стадия 2. Радиохимический синтез [18F] 1-(2'-фтор-[3,4'-бипиридин]-6-ил)-1H-пирроло[2,3-с]пиридина ([18F]-44). Раствор 1-(2'-нитро-[3,4'-бипиридин]-6-ил)-1H-пирроло[2,3-с]пиридина (0,4 мг, 52-1) в ДМСО (0,25 мл) добавляли в сосуд для микроволновой печи, содержащий сухой [18F]фторид, выпускную линию удаляли и реакционную смесь нагревали при 140°C (75 Вт) в течение 3 минут. После охлаждения до <50°C, реакционную смесь разбавляли H2O (0,6 мл), смешивали и вводили в полупрепаративную ВЭЖХ колонку. Продукт очищали с использованием колонки Zorbax Eclipse XDB-C-18, 5 мкм, 9,4×250 мм (Agilent), при скорости потока 5 мл/мин. Подвижная фаза представляла собой ацетонитрил/водн. NaH2PO4 (10 мM) 45/55. Представляющую интерес радиоактивную фракцию собирали, упаривали при отрицательном давлении, разбавляли 0,9% солевым раствором (3 мл) и переносили в стерильный контейнер. Конечный продукт тестировали на химическую и радиохимическую чистоту с помощью аналитической ВЭЖХ системы (Waters) с использованием Gemini, 5 мкм, 4,6×150 мм колонка (Phenomenex) при скорости потока 1 мл/мин. Подвижная фаза представляла собой ацетонитрил/0,1% трифторуксусная кислота в воде (10 мM) от 50 до 85% в течение 10 минут. Концентрацию [18F]-52 определяли с помощью ультрафиолетового детектора (254 нм). Подтверждение идентичности продукта определяли путем совместной инъекции образца соединения 52, и радиохимическую чистоту определяли с помощью детектора на основе йодида натрия (Bioscan). Время удерживания соединения [18F]-52 составляло 6,6 мин.
Пример 54
Радиохимический синтез [18F] d2-7-(фторметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (d2, [18F]-17):
Схема 50
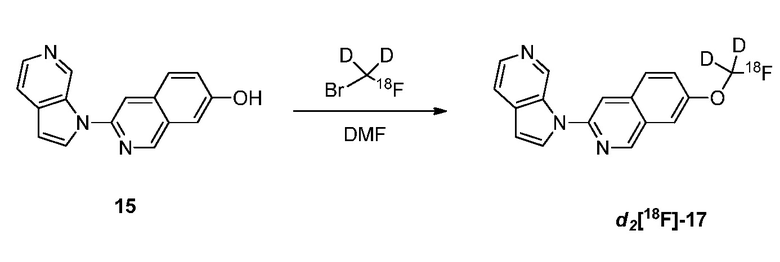
Стадия 1 Радиохимический синтез [d2, 18F] -7-(фторметокси)-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина [d2, 18F]-17): [18F]фторид, содержащий анионообменную смолу, элюировали криптофиксом 222 (Kryptofix222) (7 мг, 19 мкмоль) и K2CO3 (2,1 мг, 15 мкмоль) в ацетонитриле/воде (80/20, 0,7 мл) и переносили в 5-мл V-образный сосуд с отверстием. [18F]Фторид сушили в потоке нагретого азота (95°C). Дополнительные аликвоты ацетонитрила (3×0,5 мл) добавили для азеотропной сушки при 90°C. Дибромметан-d2 (50 мкл) в сухом ацетонитриле добавляли к сухому [18F]фториду и реакционный сосуд герметизировали. Смесь нагревали при 95°C в течение 5 минут. После охлаждения, открывали реакционный сосуд и [d2,18F]бромфторметан переносили через тефлоновую трубку в сосуд 0,9 мл, содержащий 15 (0,36 мг, 1,38 мкмоль) и карбонат цезия (4 мг, 120 мкмоль) в диметилформамиде (300 мкл) при комнатной температуре. Полученную реакционную смесь нагревали в течение 15 минут при 70°C. Раствор переносили в 0,9 мл сосуд, содержащий воду (700 мкл) при комнатной температуре, смешивали и вводили в полупрепаративную ВЭЖХ колонку. Продукт очищали с использованием колонки Zorbax Eclipse XDB-C-18, 5 мкм, 9,4×250 мм (Agilent), при скорости потока 5 мл/мин. Подвижная фаза представляла собой ацетонитрил/водн. NaH2PO4 (10 мM) от 30 до 70% в течение 15 мин. Представляющую интерес радиоактивную фракцию собирали, упаривали при отрицательном давлении, разбавляли 0,9% солевым раствором (3 мл) и переносили в стерильный контейнер. Конечный продукт тестировали на химическую и радиохимическую чистоту с помощью аналитической ВЭЖХ системы (Waters) с использованием Xbridge Phenyl, 3,5 мкм, 4,6×150 мм колонка (Waters) при скорости потока 2 мл/мин. Подвижная фаза представляла собой смесь, состоящую из 50% ацетонитрила и 50% 0,1%-ной трифторуксусной кислоты в воде. [d2, 18F]-17 концентрацию определяли с помощью ультрафиолетового детектора (254 нм). Подтверждение идентичности продукта определяли путем совместной инъекции образца 17, и радиохимическую чистоту определяли с помощью детектора на основе йодида натрия (Bioscan). Время удерживания соединения [d2, 18F]-17 составляло 5,3 мин.
Пример 55
Радиохимический синтез [18F]6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина [18F]-9
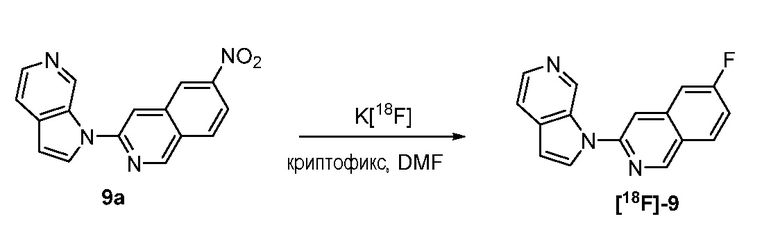
Раствор 6-нитро-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолина (0,3 мг, 9a) в DMF (0,25 мл) добавляли в сосуд для микроволновой печи, содержащий сухой [18F]фторид, выпускную линию удаляли и реакционную смесь нагревали при 140°C (65 Вт) в течение 4 минут. После охлаждения до < 50°C, реакционную смесь разбавляли H2O (0,6 мл), смешивали и вводили в полупрепаративную ВЭЖХ колонку. Продукт очищали с использованием колонки Zorbax Eclipse XDB-C-18, 5 мкм, 9,4×250 мм (Agilent), при скорости потока 5 мл/мин. Подвижная фаза представляла собой ацетонитрил/водн. NaH2PO4 (10 мM) 50-80% в 15 мин. Представляющую интерес радиоактивную фракцию собирали, упаривали при отрицательном давлении, разбавляли 0,9% солевым раствором (3 мл) и переносили в стерильный контейнер. Конечный продукт тестировали на химическую и радиохимическую чистоту с помощью аналитической ВЭЖХ системы (Waters) с использованием Onix Monolithic C-18 100×3,0 мм колонки (Phenomenex) при скорости потока 1 мл/мин. Подвижная фаза представляла собой ацетонитрил/0,1% трифторуксусная кислота в воде: 30/70. Концентрацию [18F]-9 определяли с помощью ультрафиолетового детектора (254 нм). Подтверждение идентичности продукта определяли путем совместной инъекции образца соединения 9, и радиохимическую чистоту определяли с помощью детектора на основе йодида натрия (Bioscan). Время удерживания соединения [18F]-9 составляло 3,2 мин.
Пример 56
Радиохимический синтез [18F] 6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-5-амина. ([18F]-18).
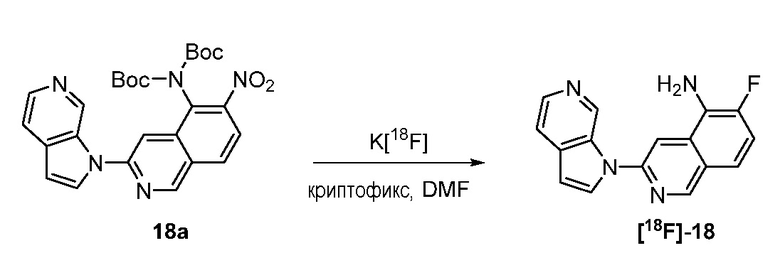
Раствор трет-бутил N-[(трет-бутокси)карбонил]-N-(6-нитро-3-[1H-пирроло[2,3-с]пиридин-1-ил]изохинолин-5-ил)карбамата (1 мг, 18a) в DMF (0,25 мл) добавляли в сосуд для микроволновой печи, содержащий сухой [18F]фторид, выпускную линию удаляли и реакционную смесь нагревали при 90°C (55 Вт) в течение 3 минут, затем 110°C (55 Вт) в течение 3 минут, затем 120°C (55 Вт) в течение 3 минут и 140°C (55 Вт) в течение 3 минут. Добавляли TFA (5% в воде) и смесь нагревали при 110°C (55 Вт) в течение 3 минут. После охлаждения до < 50°C, реакционную смесь вводили в полупрепаративную ВЭЖХ колонку. Продукт очищали с помощью Gemini C6 - Phenyl 110A, 5 мкм, 10×250 мм (Phenomenex), при скорости потока 4 мл/мин. Подвижная фаза представляет собой этанол/ацетат натрия pH 4 (10 мM) 25/75. Представляющую интерес радиоактивную фракцию собирали, упаривали при отрицательном давлении, разбавляли 0,9% солевым раствором (3 мл) и переносили в стерильный контейнер. Конечный продукт тестировали на химическую и радиохимическую чистоту с помощью аналитической ВЭЖХ системы (Agilent) с использованием Xbridge Phenyl 3,5 мкм, 4,6×150 мм колонки (Waters) при скорости потока 2 мл/мин. Подвижная фаза представляла собой ацетонитрил/ацетат натрия pH4 (10 мM): 20/80. Концентрацию [18F]-18 определяли с помощью ультрафиолетового детектора (254 нм). Подтверждение идентичности продукта определяли путем совместной инъекции образца соединения 18, и радиохимическую чистоту определяли с помощью детектора на основе йодида натрия (Bioscan). Время удерживания соединения [18F]-18 составляло 7,1 мин.
Пример 57
Радиохимический синтез [18F]6-фтор-3-(1H-пирроло[2,3-с]пиридин-1-ил)изохинолин-7-амин-([18F]-105).
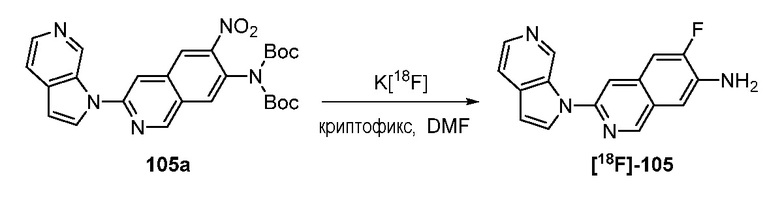
Раствор трет-бутил N-[(трет-бутокси)карбонил]-N-(6-нитро-3-{1H-пирроло[2,3-с]пиридин-1-ил}изохинолин-7-ил)карбамата (1,5 мг, 105a) в DMF (0,25 мл) добавляли в сосуд для микроволновой печи, содержащий сухой [18F]фторид, выпускную линию удаляли и реакционную смесь нагревали при 90°C (55 Вт) в течение 3 минут, затем 110°C (55 Вт) в течение 3 минут, затем 120°C (55 Вт) в течение 3 минут и 140°C (55 Вт) в течение 3 минут. Добавляли TFA (5% в воде) и смесь нагревали при 110°C (55 Вт) в течение 3 минут. После охлаждения до <50°C, реакционную смесь вводили в полупрепаративную ВЭЖХ колонку. Продукт очищали с использованием колонки Zorbax Eclipse XDB-C-18, 5 мкм, 9,4×250 мм (Agilent), при скорости потока 5 мл/мин. Подвижная фаза представляла собой ацетонитрил/водн. NaH2PO4 (10 мM) с градиентом от 30 до 70% в 15 мин. Представляющую интерес радиоактивную фракцию собирали, упаривали при отрицательном давлении, разбавляли 0,9% солевым раствором (3 мл) и переносили в стерильный контейнер. Конечный продукт тестировали на химическую и радиохимическую чистоту с помощью аналитической ВЭЖХ системы (Agilent) с использованием Xbridge Phenyl 3,5 мкм, 4,6×150 мм колонки (Waters) при скорости потока 2 мл/мин. Подвижная фаза представляла собой ацетонитрил/ацетат натрия pH4 (10 мM): 20/80. Концентрацию [18F]-105 определяли с помощью ультрафиолетового детектора (254 нм). Подтверждение идентичности продукта определяли путем совместной инъекции образца соединения 105, и радиохимическую чистоту определяли с помощью детектора на основе йодида натрия (Bioscan). Время удерживания соединения [18F]-105 составляло 5,4 мин.
Способы анализов связывания гомогената ткани
Замороженные образцы головного мозга человека, пораженного болезнью Альцгеймера (БА) закупали у Analytic Biological Services Inc. Они представляли собой посмертные образцы ткани от доноров с клиническим диагнозом БА. Поскольку большую часть белого вещества вырезали из лобной доли с целью обогащения подготовки ткани для серого вещества. Гомогенаты серого вещества мозга, обогащенные лобной корой, получали гомогенизацией ткани в ледяном фосфатно-солевом буфере (PBS), рН 7,4 при 80 мг влажной массы ткани на 1 мл в течение 45 секунд при 4°C на установке 16 Polytron. Гомогенат дополнительно разбавляли ледяным PBS до 30 мг влажной массы ткани на 1 мл и гомогенизировали в течение дополнительной минуты, как описано выше. Гомогенаты делили на аликвоты 5 мл/пробирку и хранили при -70°C до использования.
Для анализа горячего насыщения связывания, различные концентрации радиоактивного лиганда получали в буфере для анализа (PBS плюс 0,1% BSA) плюс 20% ДМСО в диапазоне от 0,02 до 57 нМ [3H]соединения примера 49, 25 мкл радиолиганда добавляли к 225 мкл мембран, разведенных до 0,5 мг/мл в буфере для анализа) для окончательной концентрации радиолиганда в диапазоне от 0,002 до 5,7 нМ и окончательную мембрану из 100 мкг влажной массы/пробирку (инкубация, фильтрация, и определение количества радиоактивного лиганда, используемого в анализе, описаны ниже). Самоблок с немеченым соединением использовали для определения неспецифического связывания. Данные насыщения анализировали, используя программное обеспечение Graphpad/Prism. На фиг.1 показан пример горячего насыщения связывания [3H]-6. Радиолиганд показывает высокое сродство к тау в гомогенатах мозга БА с измеренной константой диссоциации 0,2 нМ.
Для анализа связывания тау методом вытеснения немеченые тестируемые соединения растворяли в ДМСО при 1 мM. Разведения тестируемых соединений по отношению к различным концентрациям делали в 100% ДМСО при 1000x конечной концентрации для анализа и 0,225 мкл аликвот распределяли в планшеты для анализа. Гомогенаты головного мозга разбавляли до 0,5 мг/мл от первоначального объема 30 мг/мл в буфере для анализа, и 200 мкл добавляли в аналитический планшет для конечной концентрации 100 мкг влажной массы/аналитическую пробирку. [3H]-6 получали в 10x конечной концентрации в буфере для анализа плюс 20% ДМСО и 25 мкл добавляли в планшет для анализа для конечной концентрации для анализа 0,25 нМ. Планшет для анализа инкубировали при комнатной температуре (25°C) в течение 90 минут. Несвязанный и связанный лиганд отделяли фильтрованием связанный на фильтр-планшетах GF/C (предварительно обработанных в течение 30 минут 0,2% полиэтиленимином) с помощью харвестера Packard UniFilter и смывали несвязанный 2,5 мл на лунку ледяным 5 мМ Трис при рН 7,4. Фильтр-планшеты сушили в течение 1 часа в вакуумной печи и добавляли 50 мкл/лунку 20-MicroScint. Планшеты считывали 1 мин на лунку с помощью Packard Topcount. Общее количество радиоактивного лиганда, используемого в данном анализе определяли путем подсчета 25 мкл 10х исходного лиганда. Данные анализировали с помощью программного обеспечения Activity Base для получения 4p fit дозозависимого эффекта; и вычисляли значения Ki. Данные Ki тау для типичных соединений по настоящему изобретению приведены в таблице 1. Как показано на фиг.2, соединение 6 (немеченое) самовытесняется [3H]-6 со значением Ki 0,43 нM.
Гомогенаты образцов головного мозга человека с БА и без БА оценивали на их иммунореактивность к анти-Aβ антителу 6E10 и анти-фосфо-тау антителу PHF6 или АТ8. Срезы головного мозга с самыми высокими уровнями PHF6 или AT8 иммунореактивности выбирали для анализа связывания тау и срезы головного мозга с самыми высокими уровнями 6E10 в комбинации с низкими уровнями PHF6 или AT8 иммунореактивности выбирали для анализа связывания амилоида гомогената ткани.
Способ анализа связывания с амилоидом (обратный скрининг) идентичен анализу связывания тау с использованием [3H]-131
в качестве эталонного радиолиганда.
Данные Ki тау и/или Ki амилоида для типичных соединений по настоящему изобретению приведены в таблице 1.
Несмотря на то что настоящее изобретение было описано и проиллюстрировано со ссылкой на некоторые конкретные варианты его осуществления, специалистам в данной области понятно, что допустимы различные адаптации, изменения, модификации, замены, удаления или добавления методик и протоколов без нарушения сущности и объема настоящего изобретения. Поэтому предполагается, что настоящее изобретение определяется объемом прилагаемой формулы изобретения, и что такая формула изобретения будет интерпретироваться настолько широко, насколько это целесообразно.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛО[2,3-С]ПИРИДИНЫ В КАЧЕСТВЕ ВИЗУАЛИЗИРУЮЩИХ АГЕНТОВ ДЛЯ НЕЙРОФИБРИЛЛЯРНЫХ КЛУБКОВ | 2015 |
|
RU2695373C2 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2671571C1 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2647592C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| СУЛЬФОНАМИДНЫЕ ПЕРИ-ЗАМЕЩЕННЫЕ БИЦИКЛЫ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ПОРАЖЕНИЯ АРТЕРИЙ | 2005 |
|
RU2403240C2 |
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2019 |
|
RU2801140C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРОВ 5-НТ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2542103C2 |
| ИНГИБИТОРЫ ОКСИДАЗЫ D-АМИНОКИСЛОТ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2795513C2 |
| ИНГИБИТОРЫ ГЛИКОЛАТОКСИДАЗЫ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2805308C2 |
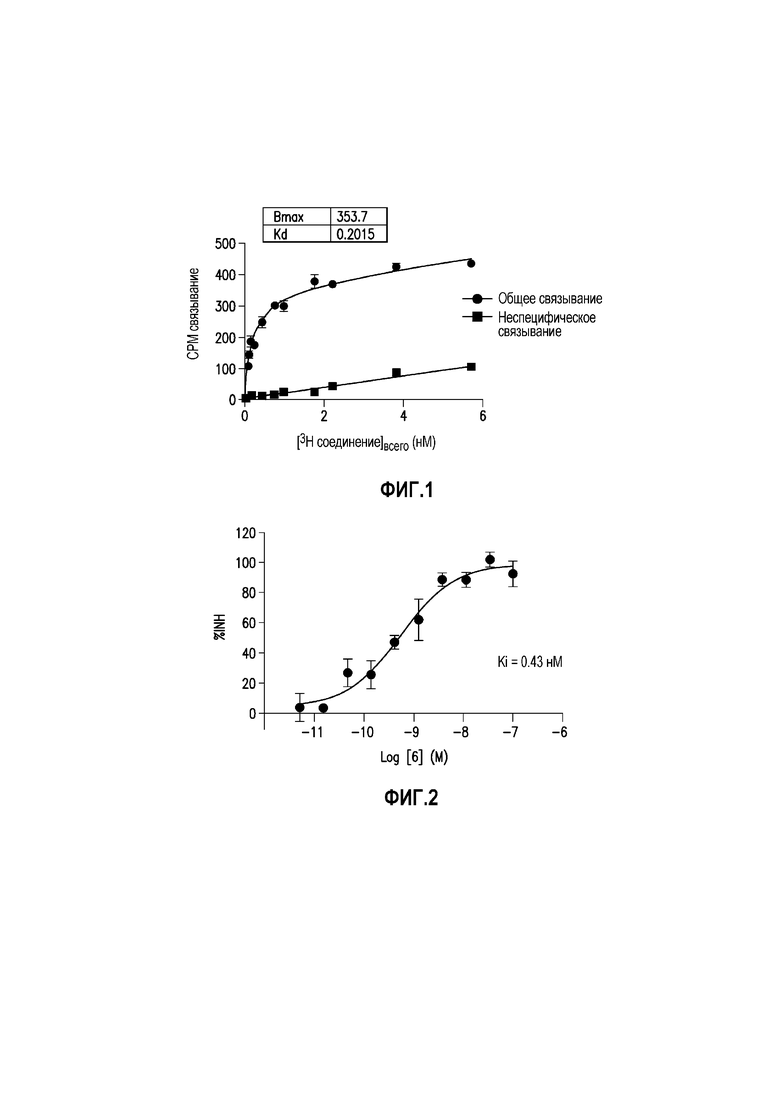
Изобретение относится к соединениям пирролопиридина, а именно к меченному изотопом соединению формулы (I), где соединение представляет собой соединение, меченное изотопом 2H, 3H, 18F. В формуле (I): X представляет собой CH или N; R представляет собой водород или -C1-6алкил, указанный алкил необязательно замещен 1-3 группами Ra; R1 представляет собой водород, -C1-6алкил, -CN, -C2-6алкенил, -(CH2)nOR или -(CH2)nгалоген; R2 представляет собой -C1-6алкил, -OC1-6алкил, -C2-6алкенилR3, -C2-6алкинилR3, пиридин, пиразол, пиперидин, пиримидин, имидазол, триазол, тиазол, пиразин, хинолин, нафтил, -NC(O)C6-10фенил, -C(O)NC6-10арил, указанный алкил, алкенил, алкинил, пиридин, пиразол, пиперидин, пиримидин, имидазол, триазол, тиазол, пиразол, пиразин, хинолин, нафтил, фенил и арил необязательно замещены 1-3 группами Ra; или соседний R1 вместе с R2 могут образовывать фенил, пирол, фуран, пиридин, пиперидин, бициклическое кольцо необязательно замещено 1-3 группами Ra; R3 представляет собой водород, -C1-6алкил, фенил, пиридил, тиазол, которые необязательно замещены 1 группой Ra; Ra представляет собой -C1-6алкил, фенил, пиридин, -CN, NO2, (CH2)nгалоген, -(CH2)nN(R)2, (CH2)nOR, указанный алкил, фенил и пиридин необязательно замещены 1-3 группами Rb; Rb представляет собой водород; n равно 0-4. Также предложены меченное изотопами соединение, выбранное из группы индивидуальных соединений, композиции, содержащие меченное изотопом соединение формулы (I), и способ измерения отложений тау-белка у пациента. Предложенные соединения могут быть использованы в связывании и визуализации агрегатов тау-белка у пациентов с болезнью Альцгеймера и других нейродегенеративных заболеваний, характеризующихся патологией тау-белка, а также для измерения клинической эффективности терапевтических средств для лечения болезни Альцгеймера и других нейродегенеративных заболеваний, характеризующихся патологией тау-белка. 7 н. и 21 з.п. ф-лы, 2 ил., 2 табл., 57 пр.
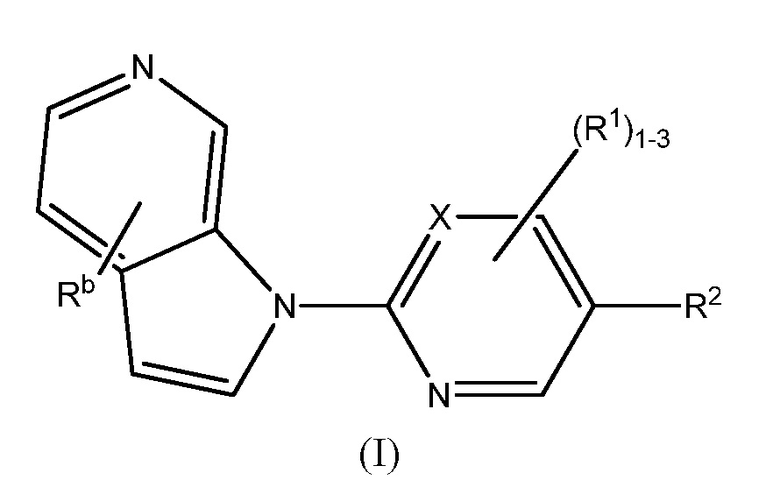
1. Меченное изотопом соединение формулы (I)
 ,
,
где X представляет собой CH или N;
R представляет собой водород или -C1-6алкил, указанный алкил необязательно замещен 1-3 группами Ra;
R1 представляет собой водород, -C1-6алкил, -CN, -C2-6алкенил, -(CH2)nOR или -(CH2)nгалоген;
R2 представляет собой -C1-6алкил, -OC1-6алкил, -C2-6алкенилR3, -C2-6алкинилR3, пиридин, пиразол, пиперидин, пиримидин, имидазол, триазол, тиазол, пиразин, хинолин, нафтил, -NC(O)C6-10фенил, -C(O)NC6-10арил, указанный алкил, алкенил, алкинил, пиридин, пиразол, пиперидин, пиримидин, имидазол, триазол, тиазол, пиразол, пиразин, хинолин, нафтил, фенил и арил необязательно замещены 1-3 группами Ra;
или соседний R1 вместе с R2 могут образовывать фенил, пирол, фуран, пиридин, пиперидин, бициклическое кольцо необязательно замещено 1-3 группами Ra;
R3 представляет собой водород, -C1-6алкил, фенил, пиридил, тиазол, которые необязательно замещены 1 группой Ra;
Ra представляет собой -C1-6алкил, фенил, пиридин, -CN, NO2, (CH2)nгалоген, -(CH2)nN(R)2, (CH2)nOR, указанный алкил, фенил и пиридин необязательно замещены 1-3 группами Rb;
Rb представляет собой водород; и
n равно 0-4, где соединение представляет собой соединение, меченное изотопом 2H, 3H, 18F.
2. Соединение по п.1, где X представляет собой CH.
3. Соединение по п.1, где X представляет собой N.
4. Соединение по любому из пп.1-3, где R2 выбран из группы, состоящей из -C1-6алкила, -C2-6алкенилR3, -C2-6алкинилR3, пиридина, пиримидина, хинолина, нафтила, -NC(O)C6-10фенила, -C(O)NC6-10арила, указанный алкил, алкенил, алкинил, пиридин, пиримидин, хинолин, нафтил, фенил и арил необязательно замещены 1-3 группами Ra.
5. Соединение по любому из пп.1-3, где соседний R1 объединяется с R2 с образованием фенила, пиррола, фурана, пиридина, пиперидина, бициклическое кольцо необязательно замещено 1-3 группами Ra.
6. Соединение по п. 5, где образовавшийся бицикл выбирают из группы, состоящей из необязательно замещенного пирролопиридинила, фуропиридинила, нафтиридинила, тетрагидронафтиридинила, хиназолинила, хинолинила и изохинолинила.
7. Соединение по любому из пп.1, 2 и 4-6, где X представляет собой CH и R2 выбран из группы, состоящей из -C1-6алкила,-C2-6алкенилR3, -C2-6алкинилR3, пиридина, пиримидина, хинолина, нафтила, -NC(O)C6-10фенила, -C(O)NC6-10арила, указанный алкил, алкенил, алкинил, пиридин, пиримидин, хинолин, нафтил, фенил и арил необязательно замещены 1-3 группами Ra и R3 выбран из группы, состоящей из метила, этила, пропила, (CH2)nF, -(CH2)nN(R)2, необязательно замещенного фенила, пиридила и тиазолила.
8. Соединение по любому из пп.1, 2 и 4-7, где X представляет собой CH и R2 объединяется с соседним R1 с образованием фенила, пиррола, фурана, пиридина, пиперидина, указанное бициклическое кольцо необязательно замещено 1-3 группами Ra.
9. Соединение по п.8, где образованное бициклическое кольцо выбирают из группы, состоящей из необязательно замещенного пирролопиридинила, фуропиридинила, нафтиридинила, тетрагидронафтиридинила, хинолинила или изохинолинила.
10. Соединение по любому из пп.1 и 3-6, где X представляет собой N, R2 выбирают из группы, состоящей из –C1-6алкила, -C2-6алкенилR3, -C2-6алкинилR3, пиридина, пиримидина, хинолона, -NC(O)C6-10фенила, -C(O)NC6-10арила, нафтила, где указанные алкил алкенил, алкинил, пиридин, пиримидин, хинолин, нафтил, фенил и арил необязательно замещены 1-3 группами Ra и R3 выбирают из группы, состоящей из метила, этила, пропила, (CH2)nF, -(CH2)nN(R)2, необязательно замещенного фенила, пиридила и тиазолила.
11. Соединение по любому из пп.1 и 3-6, где X представляет собой N и R2 объединяется с соседним R1 с образованием фенила, пиррола, фурана, пиридина, пиперидина, бициклическое кольцо необязательно замещено 1-3 группами Ra.
12. Соединение по п. 1, представленное структурной формулой Ia
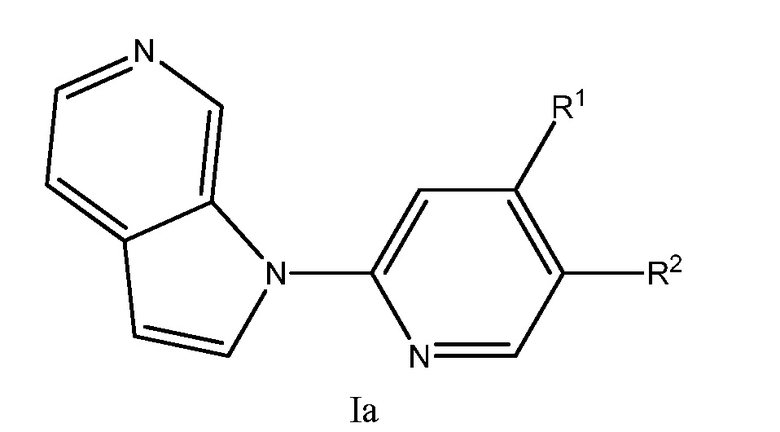 ,
,
или его фармацевтически приемлемая соль.
13. Соединение по п. 12, где R2 выбирают из группы, состоящей из -C1-6алкила, -C2-6алкенилR3, -C2-6алкинилR3, пиридина, пиримидина, хинолина, нафтила, -NC(O)C6-10фенила, C(O)NC6-10 арила, указанный алкил, алкенил, алкинил, пиридин, пиримидин, хинолин, нафтил, фенил и арил необязательно замещены 1-3 группами Ra и R3 выбирают из группы, состоящей из метила, этила, пропила, -(CH2)nF, -(CH2)nN(R)2, необязательно замещенного фенила, пиридила и тиазолила.
14. Соединение по п. 13, где R2 объединяется с соседним R1 с образованием фенила, пиррола, фурана, пиридина, пиперидина, бициклическое кольцо необязательно замещено 1-3 группами Ra.
15. Соединение по п.1, представленное структурной формулой II

или его фармацевтически приемлемая соль, где Ib = Ib1, Ib2, Ib3 или Ib4, и W, W1, W2, W3 независимо друг от друга выбирают из -CH- и -N-, и R, Ra и Rb являются такими, как описано ранее.
16. Соединение по п.1, представленное структурной формулой III
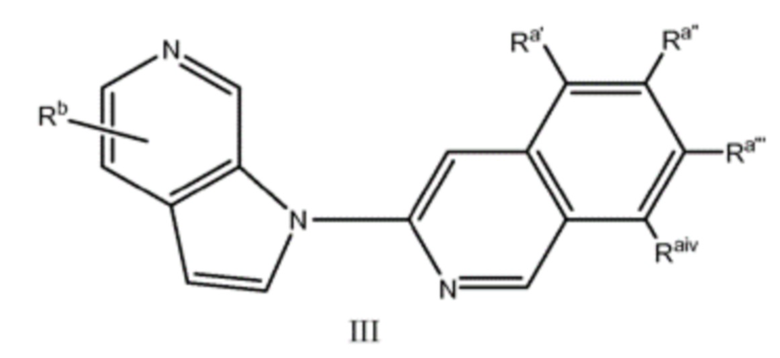 ,
,
где Ra', Raʺ, Raʺ' и Raiv независимо друг от друга выбирают из водорода и Ra и Ra и Rb являются такими, как описано ранее.
17. Соединение по п.16, где Ra', Raʺ, Raʺ' и Raiv независимо друг от друга выбирают из группы, состоящей из водорода, -C1-6алкила, -(CH2)nгалогена, -O(CH2)nгалогена, CN, NO2, (CH2)nOR, -(CH2)nN(R)2, -N(CH3)(CH2)nOR, -NH(CH2)nгалогена и -N(CH3)(CH2)nгалогена.
18. Соединение по любому из пп. 16, 17, где Ra', Raʺ, Raʺ' и Raiv независимо друг от друга выбирают из группы, состоящей из водорода, амино, фтора и иода.
19. Соединение по любому из пп. 17, 18, где один из Ra', Raʺ, Raʺ' и Raiv представляет собой амино, один из Ra', Raʺ, Raʺ' и Raiv представляет собой фтор или йод, остальные Ra', Raʺ, Raʺ' и Raiv представляют собой водород и Rb представляет собой водород.
20. Меченное изотопами соединение, где соединение представляет собой соединение в таблице 1 и 2, меченное изотопами 2H, 3H, 18F, выбранное из группы, состоящей из:
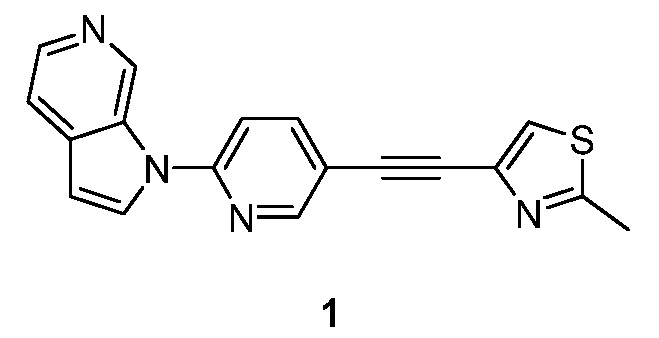



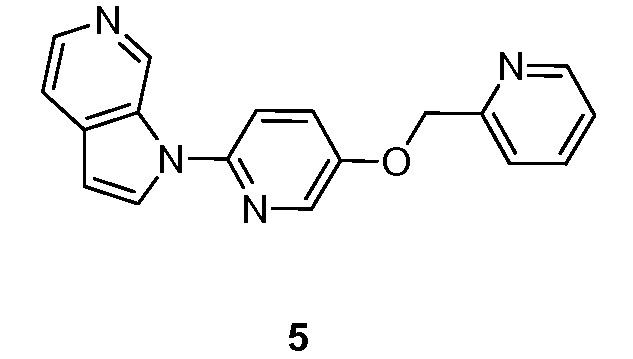
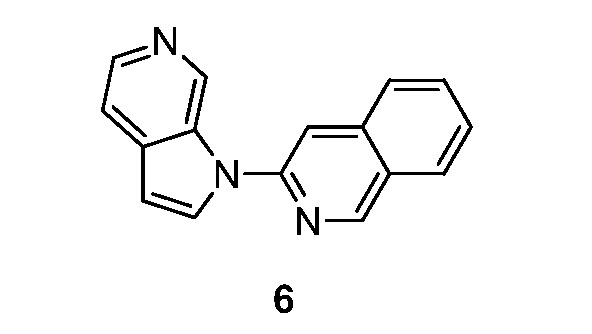
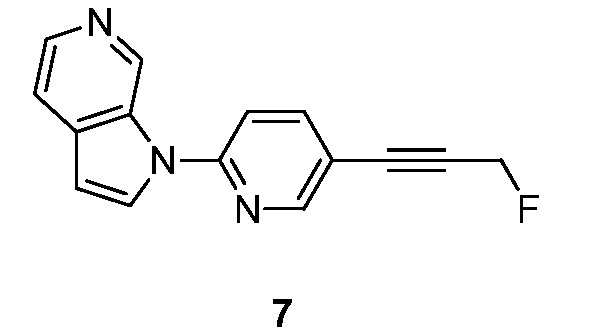
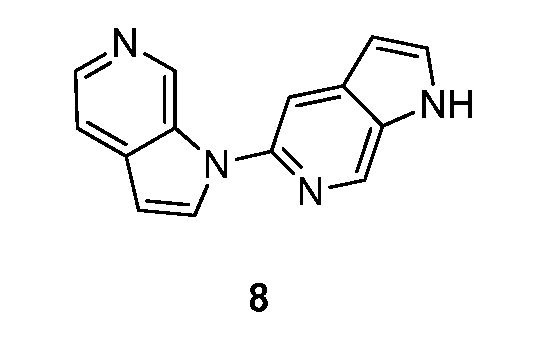


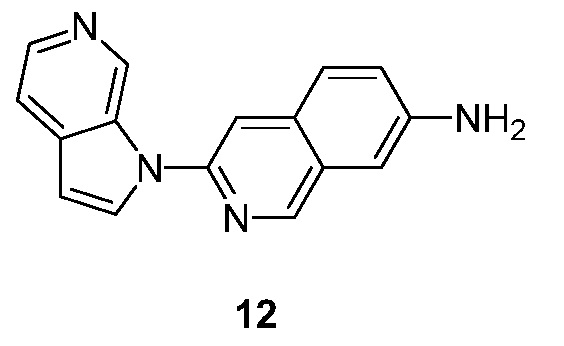
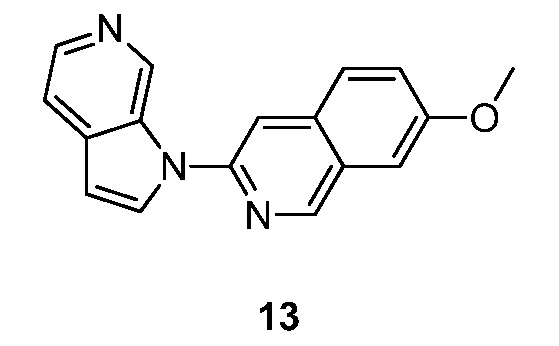
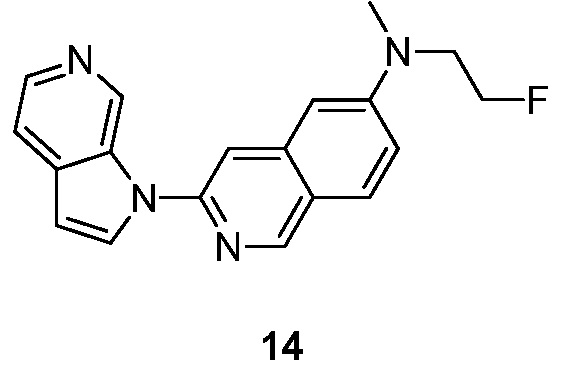
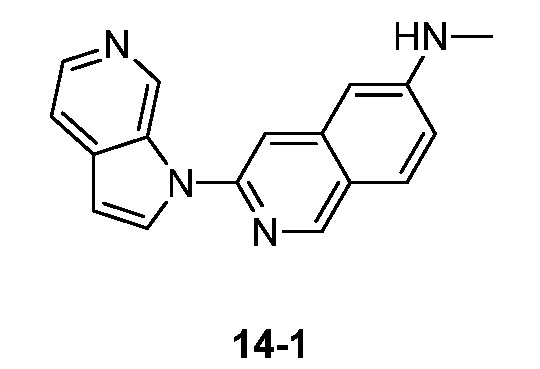


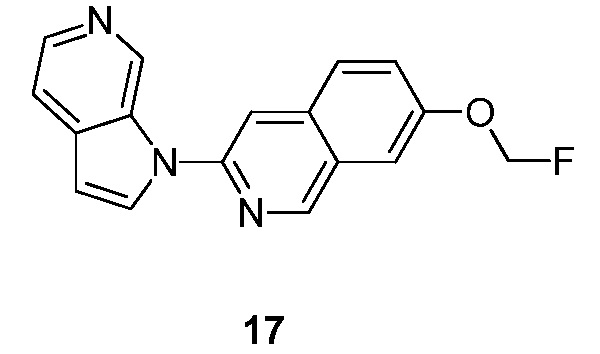





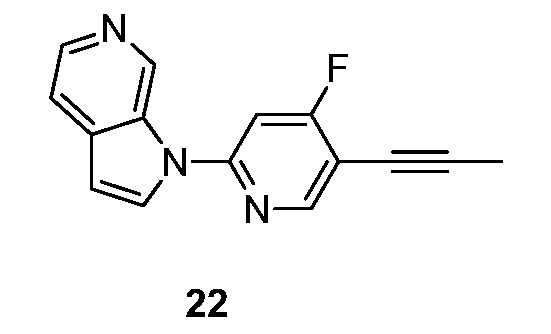

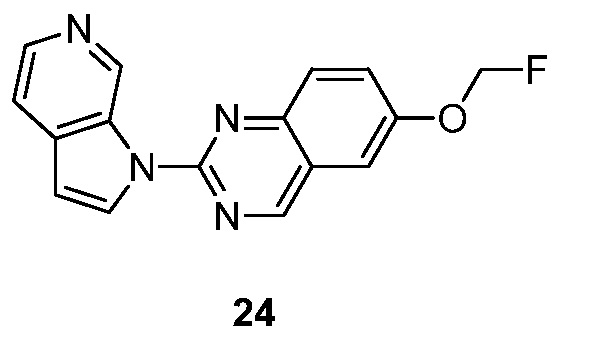



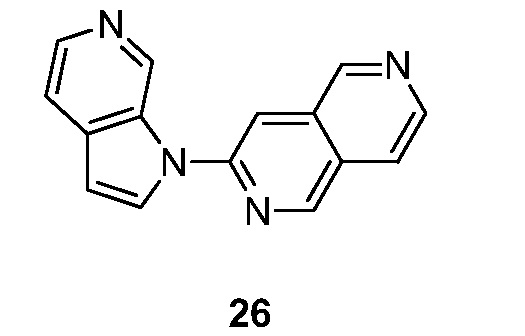
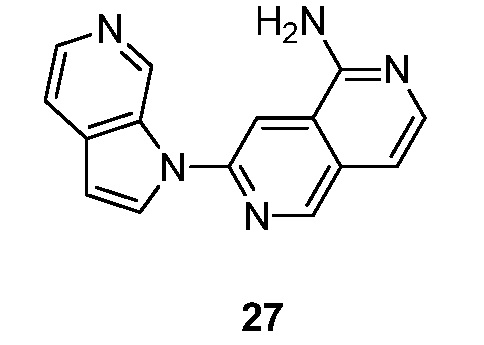
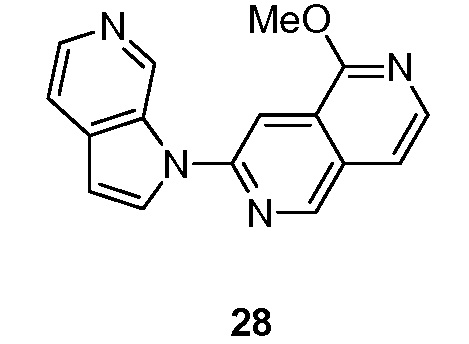


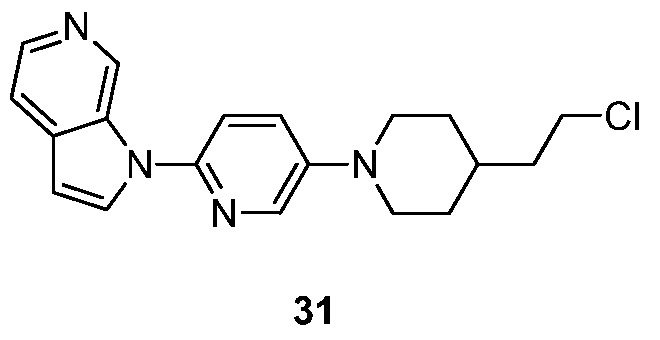


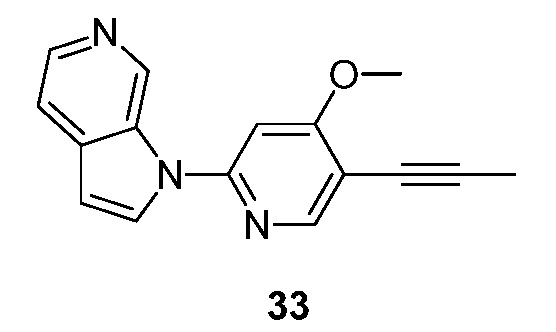



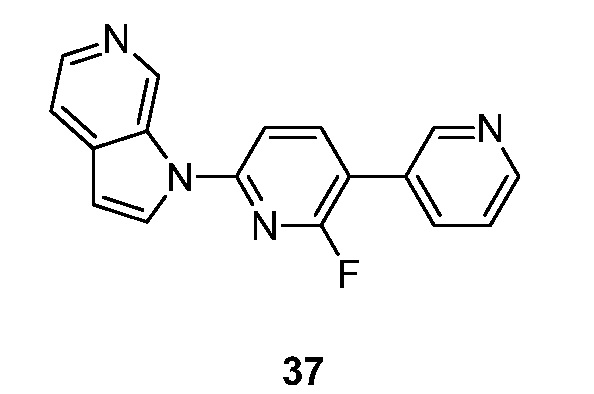
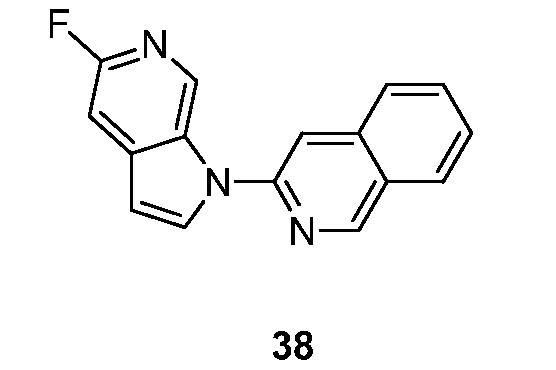
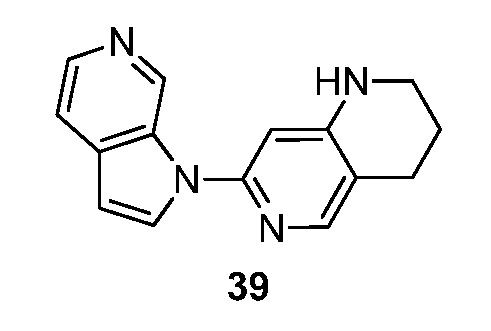

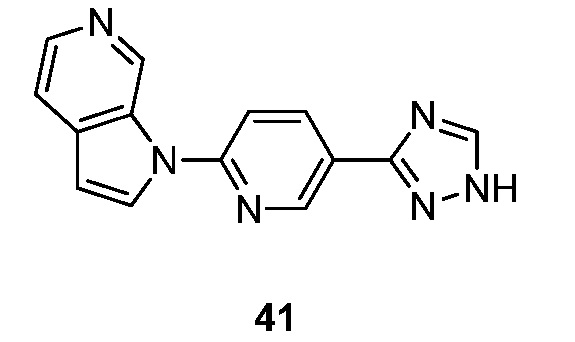
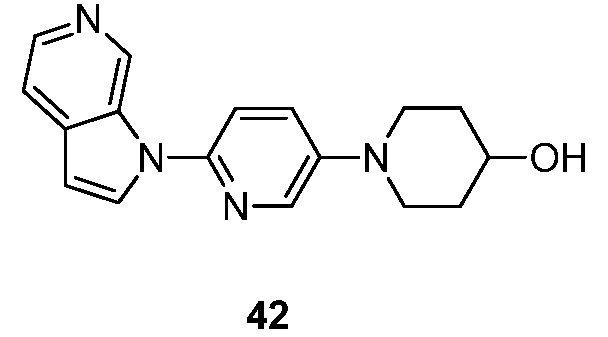

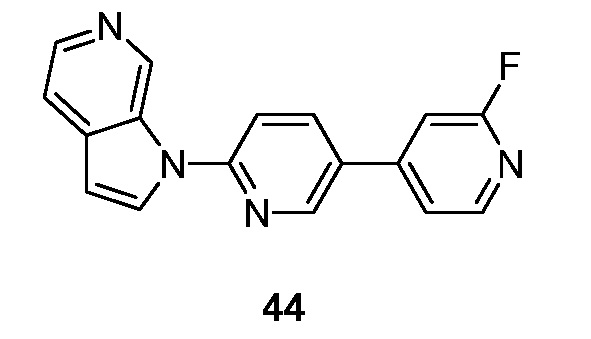

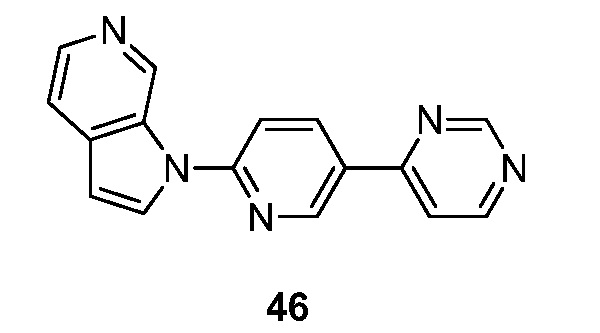


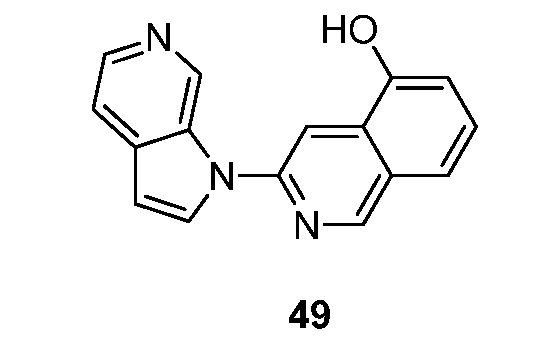



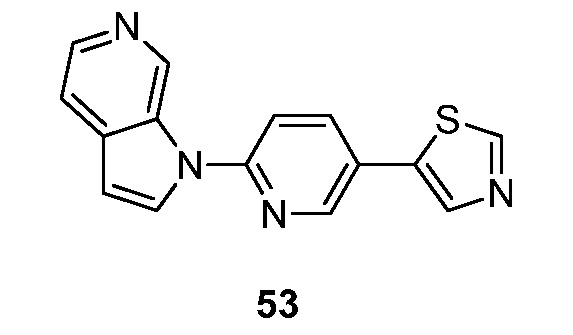

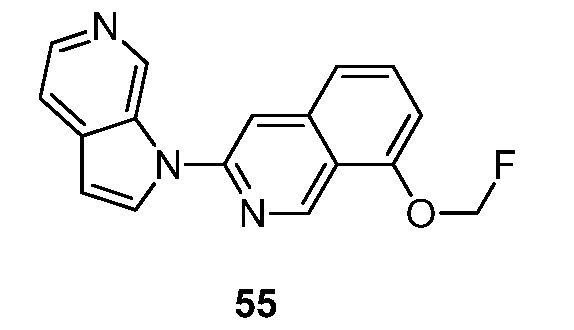
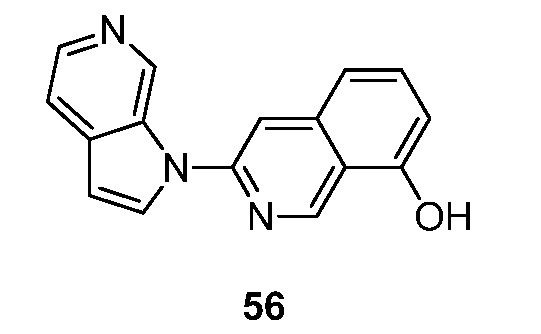
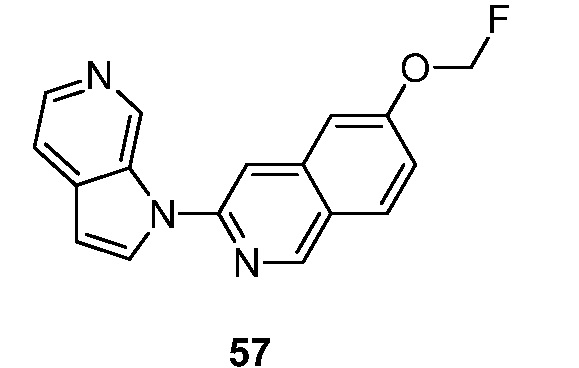

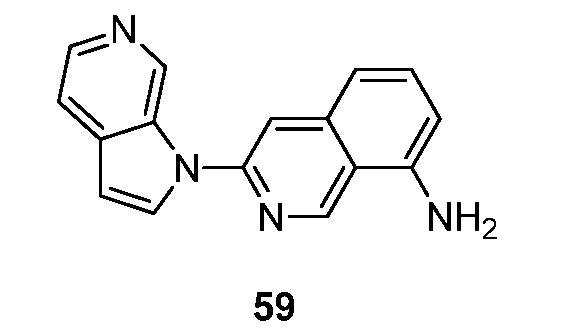

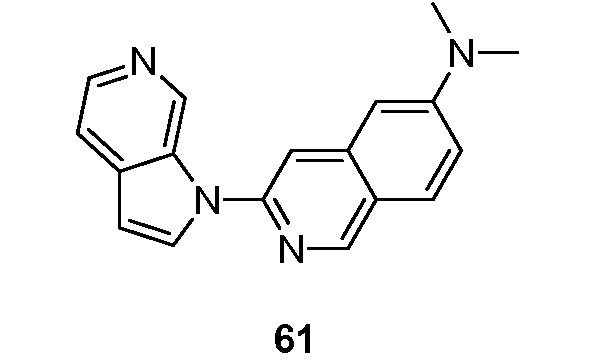

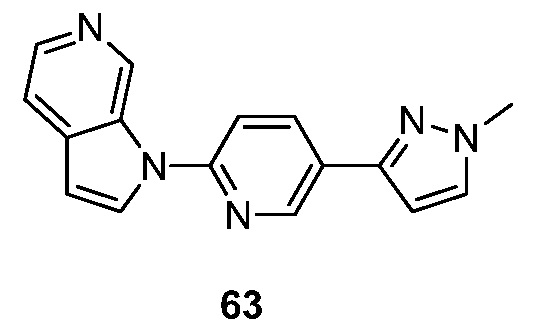
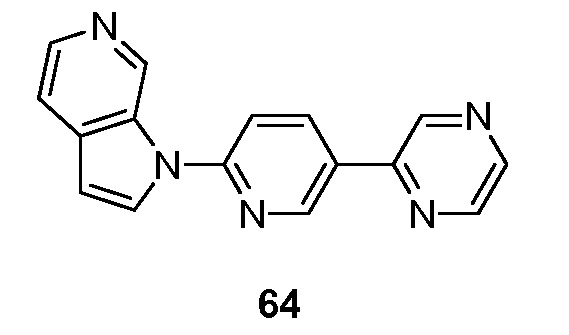
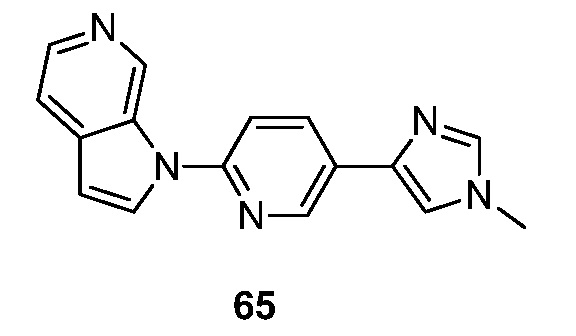




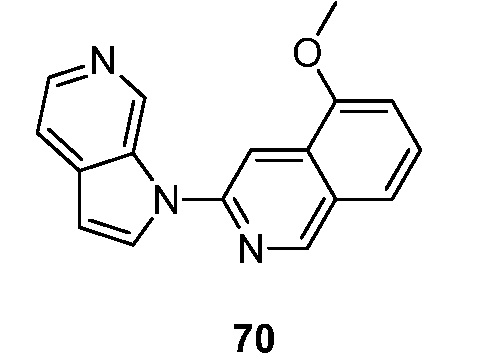
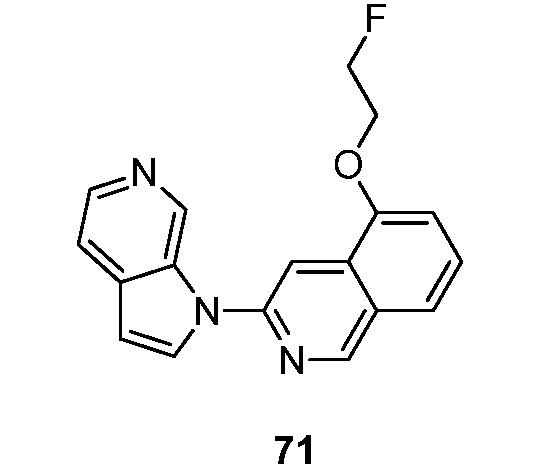
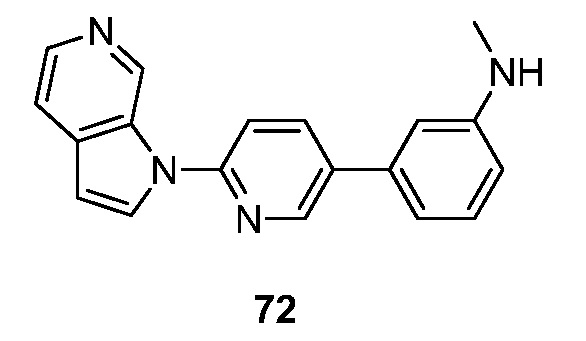

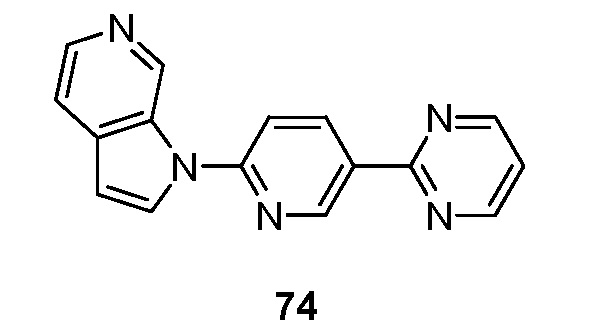


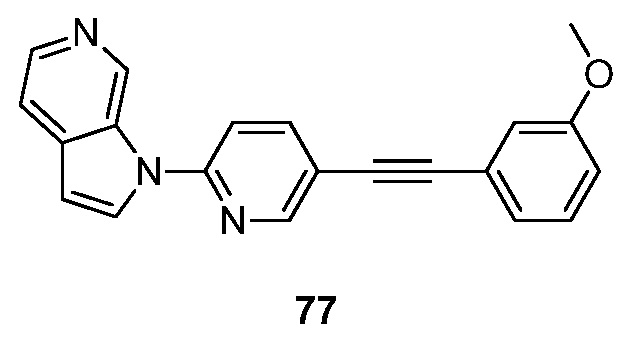
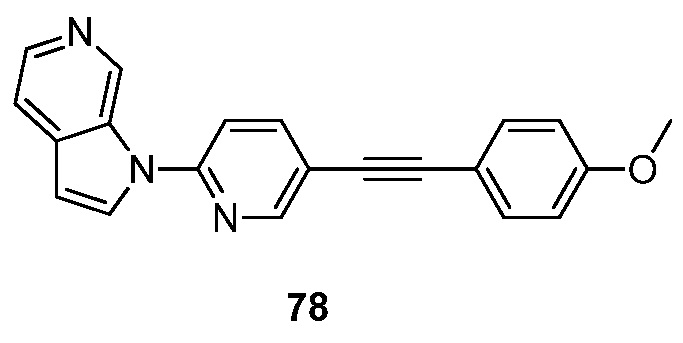


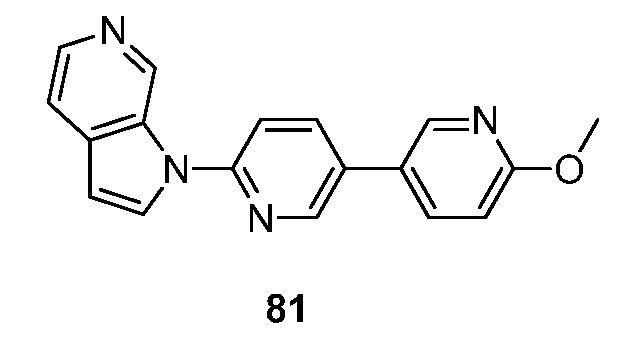
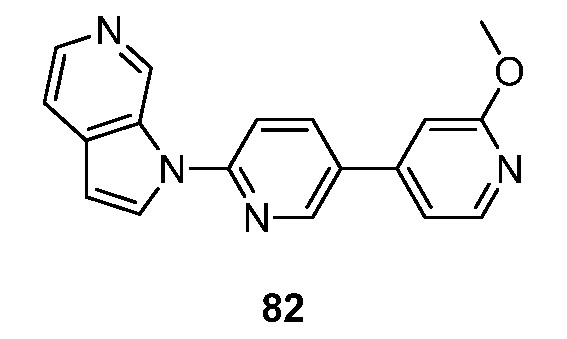
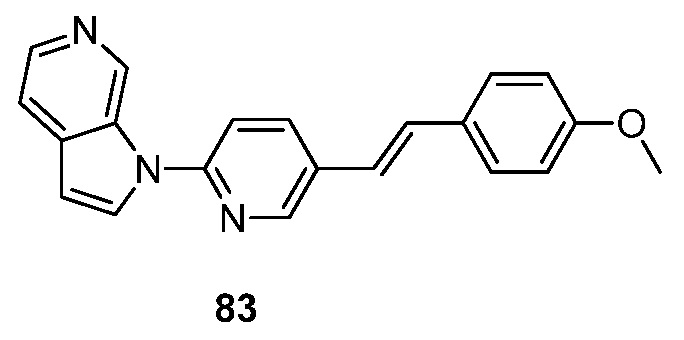




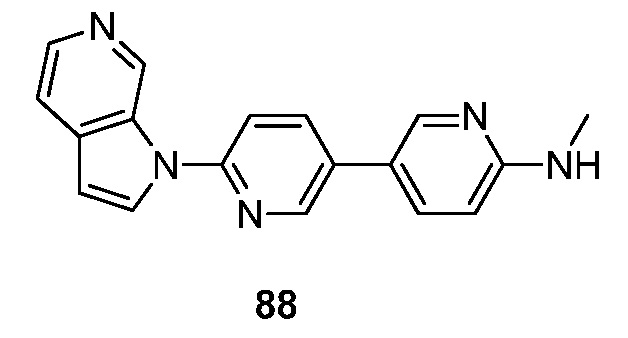


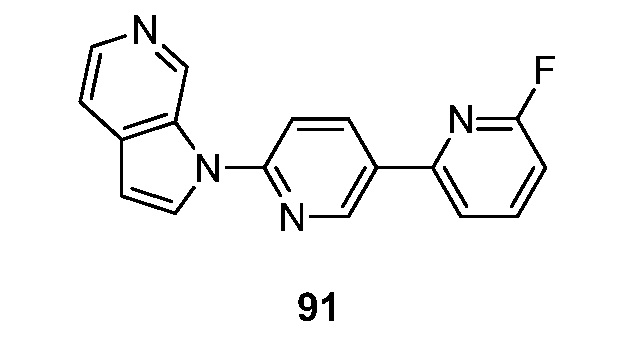



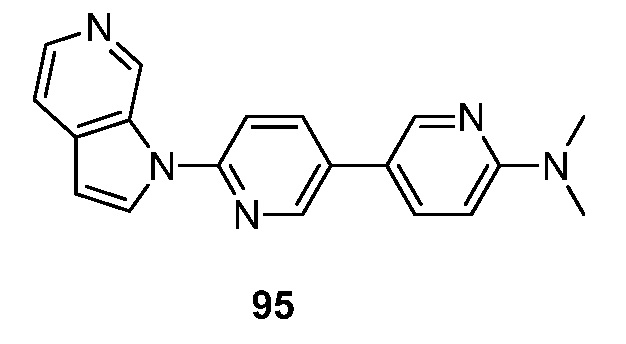
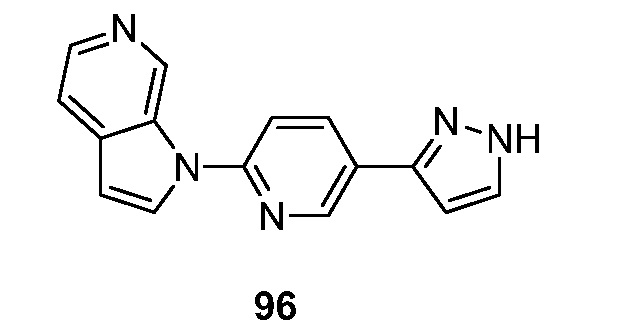






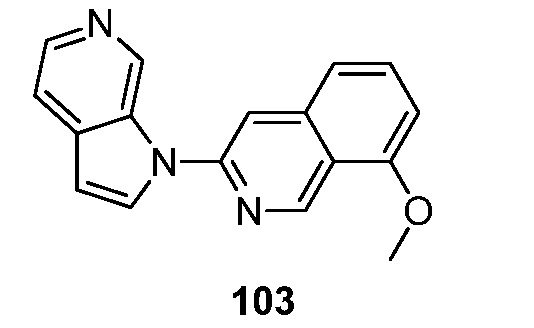

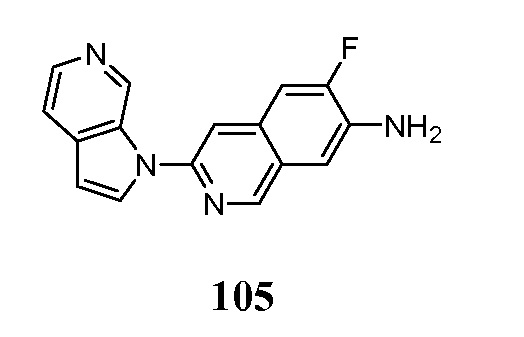
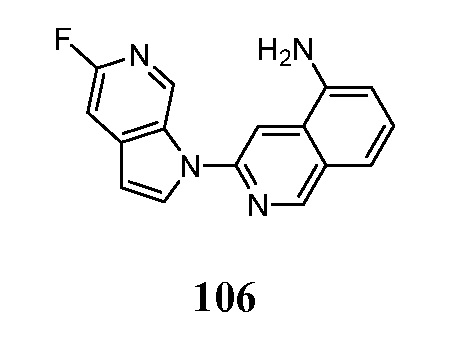
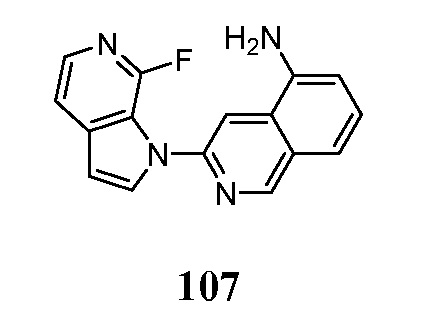
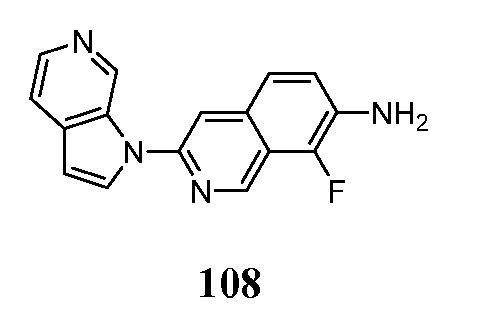
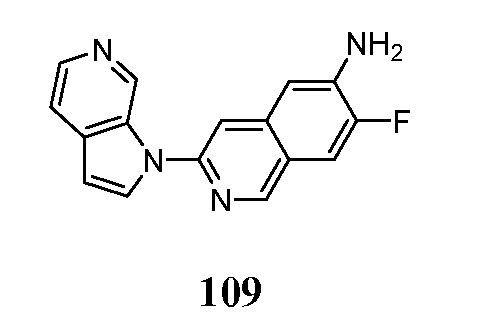
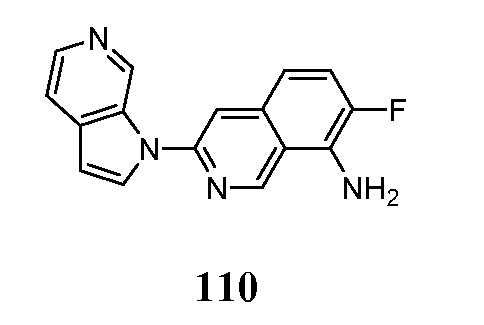


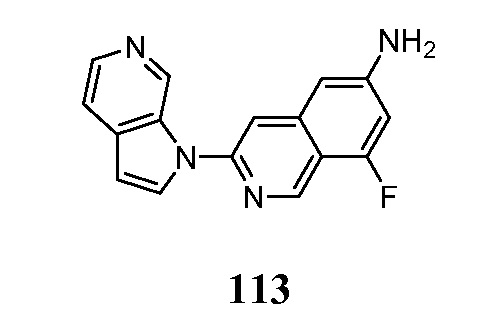
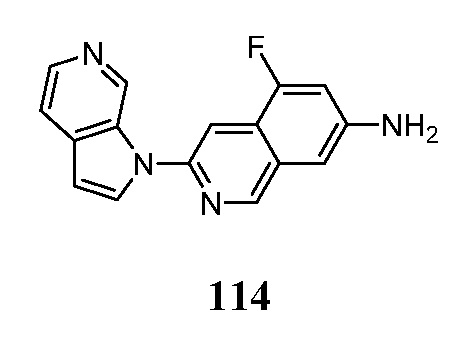
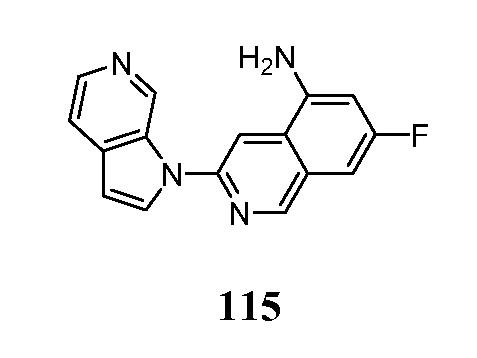

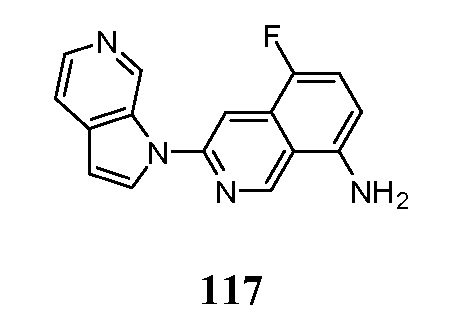


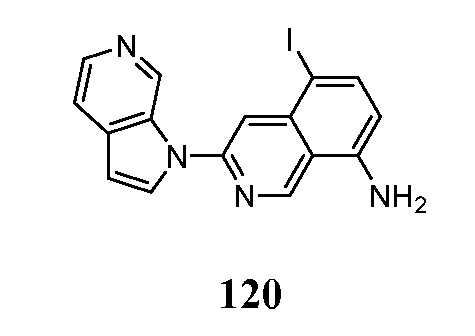
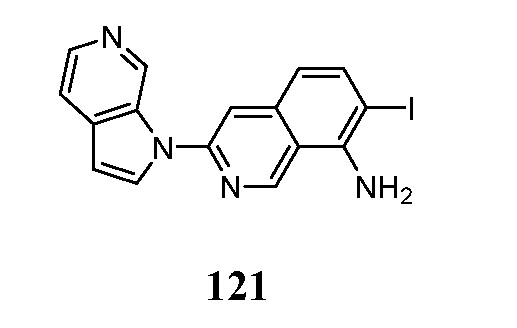
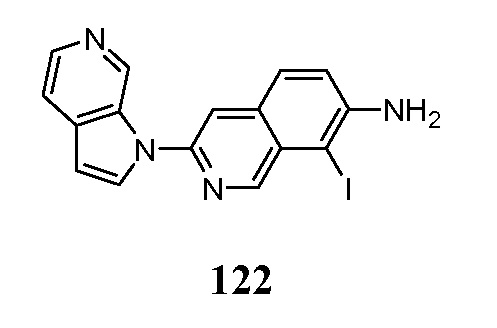
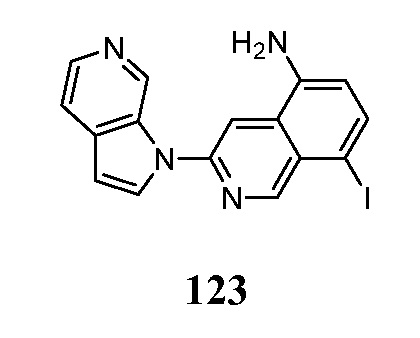
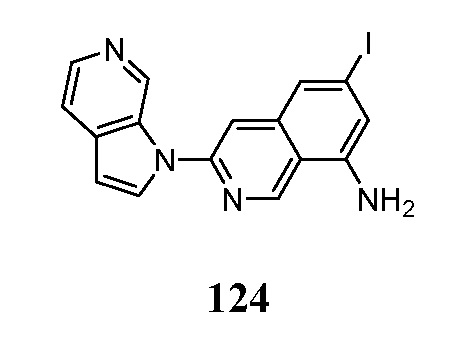

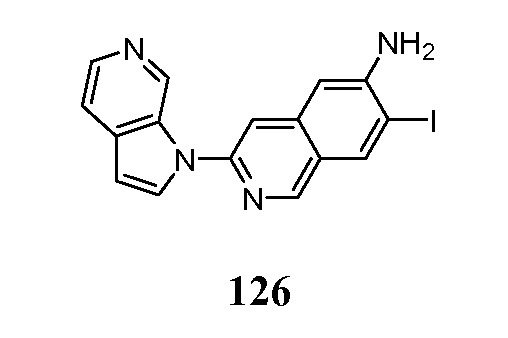


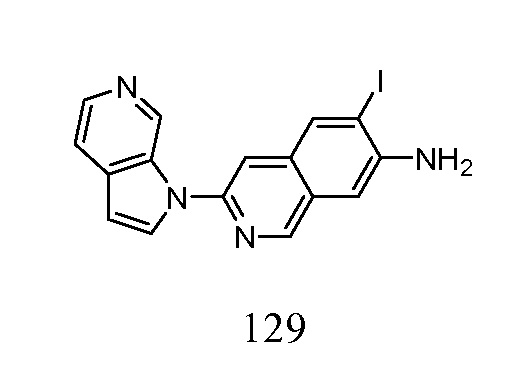

21. Соединение по любому из пп.19, 20, где меченное изотопами соединение представляет собой 18F.
22. Соединение, представленное структурной формулой
 ,
,
которое представляет собой соединение, меченное изотопами 2H, 3H, 18F.
23. Меченное изотопами соединение, представленное структурной формулой [18F]-18
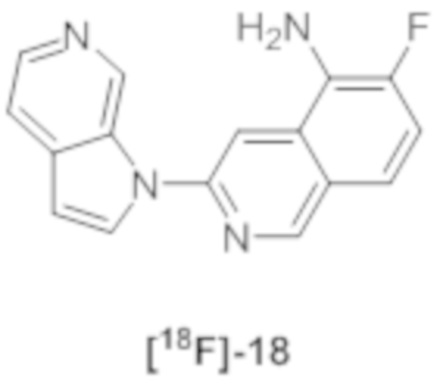 .
.
24. Фармацевтическая композиция, пригодная в качестве радиоизотопного индикатора в диагностике и мониторинге при лечении болезни Альцгеймера, содержащая соединение по п.1 и фармацевтически приемлемый носитель.
25. Композиция для визуализации агрегатов тау-белка, содержащая соединение по п.1 и фармацевтически приемлемый носитель.
26. Способ измерения отложений тау-белка у пациента, включающий стадии введения детектируемого количества соединения формулы I по п.1 и детектирование связывания соединения с отложениями тау-белка в организме пациента.
27. Способ по п.26, в котором обнаружение осуществляют путем проведения позитронно-эмиссионной томографии (ПЭТ), однофотонной эмиссионной компьютерной томографии (ОЭКТ), магнитно-резонансной томографии или авторадиографии.
28. Способ по п.26 для диагностики и мониторинга лечения болезни Альцгеймера или наследственной болезни Альцгеймера.
| WO 2008091195 A1, 31.07.2008 | |||
| US 20120301398 A1, 29.11.2012 | |||
| РАДИОФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2008 |
|
RU2475267C2 |
| US 6696039 B2, 24.02.2004 | |||
| US 20090285756 A1, 19.11.2009 | |||
| МЕЧЕННЫЕ РАДИОАКТИВНОЙ МЕТКОЙ ИНГИБИТОРЫ ПЕРЕНОСЧИКА ГЛИЦИНА 1 | 2009 |
|
RU2512529C2 |

