Изобретение относится к области химико-фармацевтической промышленности и медицины и касается соединений, которые могут быть использованы для создания средств лечения сердечно-сосудистых заболеваний.
Сердечно-сосудистые заболевания остаются ведущей причиной смертности у людей, и одной из наиболее всерьез рассматриваемых причин этого явления - атеросклероз и повышенный уровень липопротеинов низкой плотности в крови. Атеросклероз - это хроническое сердечно-сосудистое заболевание, выражающееся в уплотнении артериальной стенки за счет разрастания соединительной ткани, образовании атеросклеротических бляшек, сужении просвета сосудов, ухудшении кровоснабжения тканей.
Дизрегуляция продукции холестерина приводит к серьезным патологиям. Когда суммарная его продукция (синтезированного и поступившего с пищей) превышает то его количество, которое требуется для построения и ремонта мембран, получения желчных кислот и стероидов, накопление холестерина в сосудах (атероматоз) может вызвать их закупоривание (атеротромбоз). Сердечно-сосудистая патология возникает из-за высокого уровня холестерина и особенно она обусловлена повышенным уровнем липопротеинов низкой плотности (ЛНП).
Современный взгляд на атеросклероз заключается в том, что это - хроническое воспалительное расстройство. И такие противовоспалительные средства как аспирин, обладающий к тому же антиагрегационными свойствами, так же как ингибиторы ангиотензин-конвертирующего фермента и противовоспалительные цитокины, полезны пациентам, пораженным атеросклерозом.
Гипертензию часто ассоциируют с атеросклеротическими повреждениями сосудистой стенки и, следовательно, важными являются исследования, направленные на изучение влияния эффектов антигипертензивных средств на развитие атеросклероза. Согласно некоторым исследованиям, у антагонистов кальция доказаны антиатерогенные свойства, выражающиеся в предотвращении появления атеросклеротических повреждений. Ингибиторы ангиотензин-конвертирующего фермента также проявляют антиатерогенный эффект в опытах на животных. Антагонисты кальция верапамил и амлодипин ингибируют атерогенный эффект, подавляя индуцированную сывороткой клеточную пролиферацию, синтез белка и накопление холестерина в клетках. Оба препарата снижают проявления атеросклероза, показывая антиатеросклеротическую и антиатерогенную активность в культуре клеток. Напротив, периндоприлат не влияет на атеросклеротические параметры, а пропранолол, более того, даже стимулирует и накопление холестерина, пролиферативную активность клеток и протеиновый синтез. По мнению авторов [9], данные, полученные на клеточных культурах, могут отражать и ситуацию in vivo. Исследование пациентов с коронарным атеросклерозом при лечении кальций-антагонистами или пропранололом показало, что последний в этих условиях в определенной степени ингибирует атерогенез. На основе проведенных исследований авторы построили следующий ряд эффективности действия рассмотренных препаратов: амлодипин > верапамил > периндоприл > пропранолол.
К числу лекарственных средств, широко используемых для лечения атеросклероза, несомненно, принадлежат статины, такие как ловастатин, симвастатин, правастатин, аторвастатин, флувастатин.
Важно отметить, что механизм действия статинов связан с ингибированием скорость-лимитирующей стадии биосинтеза холестерина - конверсии β-гидрокси-β-метилглутарил-КоА (HMG-КоА) в мевалонат, которая катализируется ферментом - HMG-KoA редуктазой.
В исследовании [10] отмечено, что несмотря на успехи применения статинов в эффективном снижении уровня холестерина и снижении случаев смертности при сердечно-сосудистых заболеваниях, примерно у двух третей пациентов, которых лечили статинами, все же продолжали выявлять повреждения коронарных сосудов.
В качестве еще одного из перспективных направлений лечения атеросклероза, помимо, например, повышения уровня липидов высокой плотности, рассматривается хроническое торможение развития системных и сосудистых воспалительных процессов [8].
Таким образом, в настоящее время для лечения атеросклероза используется комплекс препаратов, поскольку монотерапия не возможна. Основная причина - отсутствие вещества, обладающего наиболее необходимым спектром фармакологической активности: противовоспалительной, антиагрегационной, антигипертензивной и гипохолестеринемической. Поиск такой молекулы является актуальной задачей фармакологии.
В последние полтора десятилетия в биологии произошли события, повлекшие за собой фундаментальные изменения наших представлений о функционировании самых различных биологических систем. Было обнаружено, что такое низкомолекулярное соединение, как оксид азота (NO), является одним из универсальных и необходимых регуляторов функций клеточного метаболизма. Оксид азота участвует в регуляции тонуса кровеносных сосудов, ингибирует агрегацию тромбоцитов и их адгезию на стенках кровеносных сосудов, функционирует в центральной и вегетативной нервной системе, регулируя деятельность органов дыхания, желудочно-кишечного тракта и мочеполовой системы. Оксид азота играет важную роль в нейротрансмиссии, регуляции иммунитета и защите организма от бактериального поражения.
Оксид азота известен как мощный ингибитор агрегации тромбоцитов. Его образование способствует сохранению текучести крови благодаря торможению тромбогенеза. Факторы, регулирующие возникновение и утилизацию NO в эндогенных условиях, обеспечивают во многом нормальное функционирование сердечно-сосудистой системы.
В связи с изложенным выше важное значение для современной фармакологии приобретает поиск веществ, способствующих появлению NO как агента, угнетающего спонтанную и индуцированную способность кровяных пластинок к склеиванию. Таким образом, поиск новых эффективных соединений, которые могут быть востребованы в медицинской практике, построен в настоящем исследовании на синтезе и биологическом исследовании новых генераторов оксида азота.
В ряду индолинона-3 описаны соединения, обладающие антигипертензивной активностью [2, 4, 5, 6] и способностью генерировать оксид азота [1], но не описаны соединения, способные активировать ферментрастворимую гуанилатциклазу и оказывать антиагрегаторное действие.
Целью изобретения является применение производного индолинона-3 в качестве биологически активного соединения, которое может быть использовано для получения средства для лечения сердечно-сосудистых заболеваний.
Поставленная цель достигается применением соединения, проявляющего свойства экзогенного донора оксида азота, активатора фермента растворимая гуанилатциклаза, ингибитора агрегации тромбоцитов с антигипертензивной активностью.
Настоящее изобретение относится к химии соединений ряда индолинона-3 и конкретно касается производного этого ряда - 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолин-3-он формулы I:
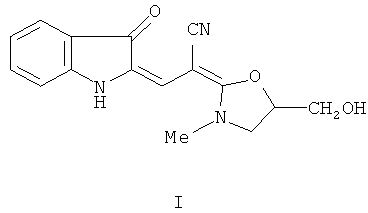
Проводились исследования антиагрегационных свойств, влияния на активность растворимой гуанилатциклазы и генерацию NO соединения I в опытах in vitro, а также на способность оказывать гипотензивное и антигипертензивное действие у животных. Исследовались общетоксические свойства и переносимость соединения формулы I на животных.
Исследования показали, что оно обладает выраженными антиагрегационными свойствами, высвобождает NO и в высокой степени активирует растворимую гуанилатциклазу.
Пример 1
Изучение антиагрегационных свойств проводили по влиянию на агрегацию тромбоцитов плазмы кроликов, индуцированную АДФ (аденозиндифосфат) и арахидоновой кислотой. Агрегацию тромбоцитов оценивали по изменению оптической плотности плазмы. Исследования проводили как при внутривенном введении в сравнении с нитропруссидом натрия, так и при введении перорально в сравнении с ацетилсалициловой кислотой. I применяли в концентрациях 10-3-10-7 (при введении парентерально) и в дозах 4-8 и 12 мг/кг при введении внутрь.
Экспериментально установлено, что соединение формулы I значимо угнетает АДФ-индуцированную агрегацию тромбоцитов.
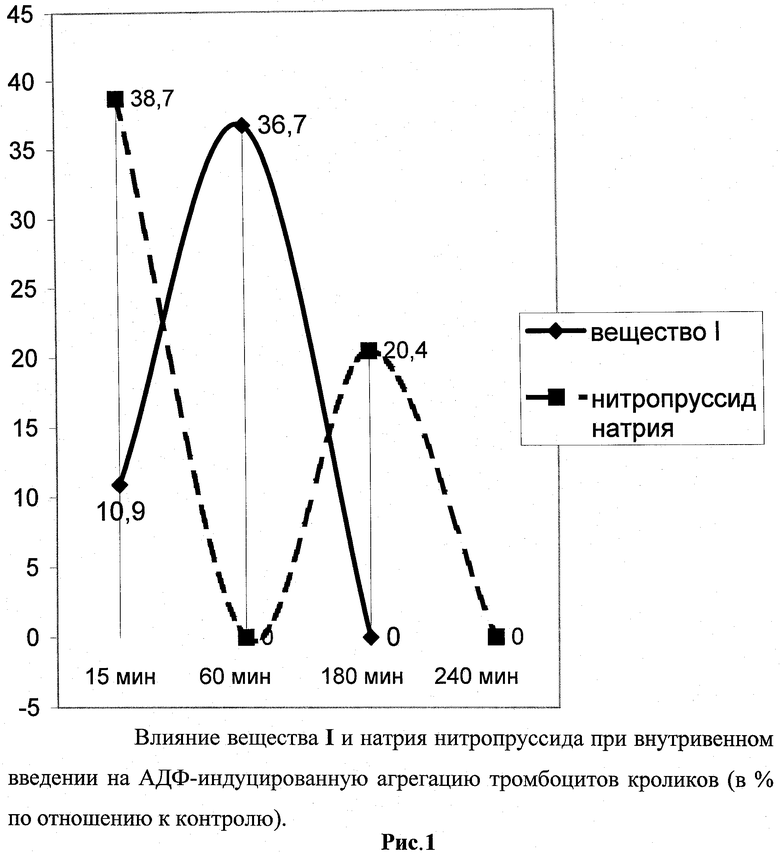
Для окончательного вывода о способности изучаемого соединения угнетать агрегацию тромбоцитов были проведены опыты в условиях живого организма при его внутривенном и пероральном введениях.
Соединение I и натрия нитропруссид (как эталонный лекарственный препарат) вызывали однонаправленное изменение процесса агрегации тромбоцитов - подавляли его. Однако существовали значимые количественные и временные различия в эффекте. Так, натрия нитропруссид активно (достоверно) угнетал агрегацию тромбоцитов уже через 15 мин после введения. Эта временная точка единственная, в которой нам удалось наблюдать активность. Уже через 1 ч эффект исчезал. Торможение агрегации тромбоцитов под действием вещества I наблюдалось тоже через 15 мин после введения (как тенденция), достигало максимальных значений через 60 мин и прекращалось к 3 ч эксперимента (рис.1).
Таким образом, эти серии экспериментов показали способность соединения формулы I угнетать вызываемое АДФ склеивание кровяных пластинок после внутривенного введения.
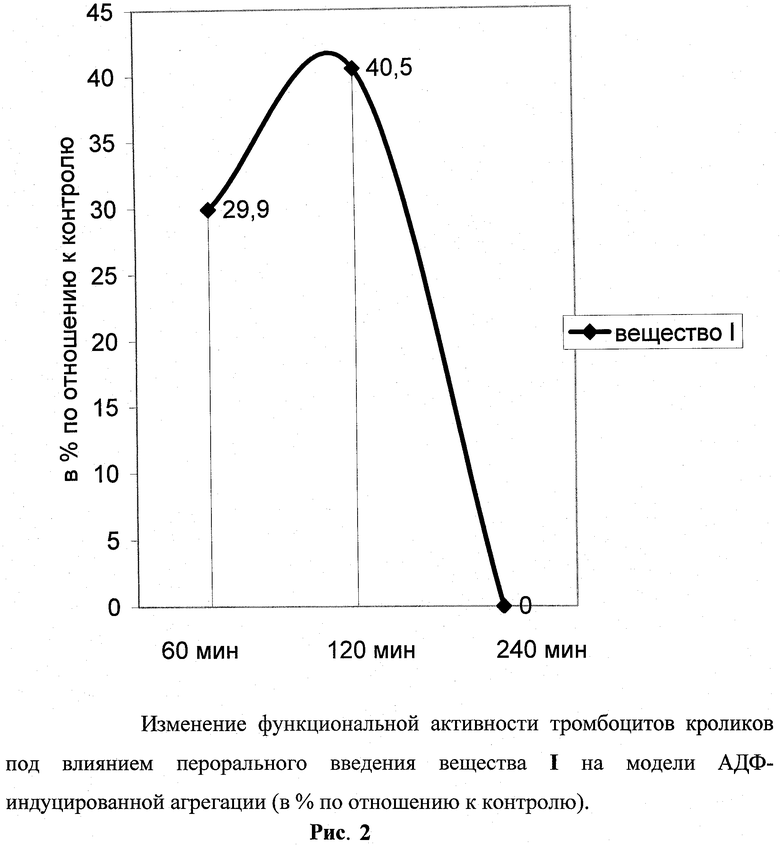
Значительное угнетение процесса агрегации, индуцированного АДФ, выявилось к первому часу (30%), достигало максимума через 2 ч (40%) и, как тенденция (15-25%), сохранялась все 4 ч наблюдения (рис.2).
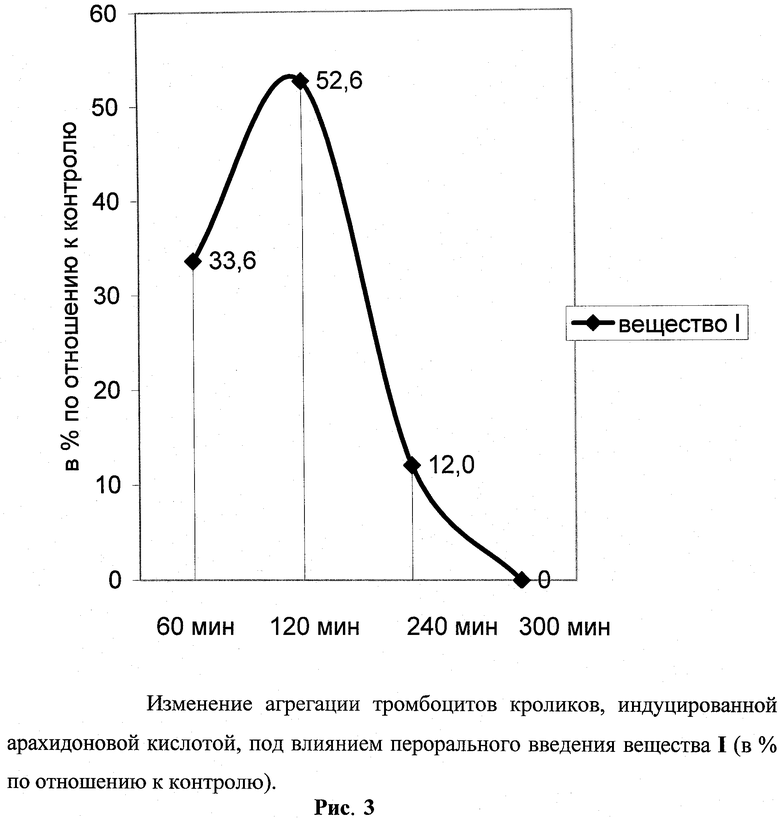
При использовании арахидоновой кислоты (АА) в качестве индуктора агрегации эксперименты выполнялись также в условиях перорального введения. Мы полагали, что такая постановка опытов вполне оправдана возможностью назначения изучаемого вещества (в отличие от нитропруссида натрия) не только внутривенно.
Оказалось, что соединение формулы I начинает действовать уже через 1 ч после аппликации, при этом степень инициации агрегации кровяных пластинок арахидоновой кислотой падала на треть. Еще более значимо действие индуктора (АА) подавлялось ко второму часу - более чем вдвое. К концу периода наблюдения (4 часа) действие АА было сопоставимо с контролем (рис.3).
Эффект соединение формулы I сохранялся свыше 4 ч. Количественно к концу первого часа он выявлялся как тенденция (до 10%), становился статистически значимым через 2 ч (32%) и продолжал расти весь период наблюдения (38% к 4 часу).
Таким образом, агрегация тромбоцитов, индуцированная АА, эффективно подавлялась соединением формулы I, вводимым per os, причем активность проявлялась на протяжении более двух часов.
Мы исследовали также влияние данного соединения на индуцированное формил-метионил-лейцил-фенилаланином (FMLP) образование супероксид анион радикала 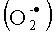 нейтрофилами человека, предварительно ослабленное развитием патологических процессов (гистеоцитоз, гипертоническая болезнь II ст.). Данная постановка опыта была выбрана в связи с тем, что для осуществления завершенного фагоцитоза, сопровождающегося полным уничтожением патогенных микроорганизмов, фагоциты, в том числе и нейтрофилы, образуют так называемые активные формы кислорода (АФК), к которым относятся радикалы кислорода и соединения, легко превращающиеся в такие радикалы. К АФК относятся супероксид анион радикал
нейтрофилами человека, предварительно ослабленное развитием патологических процессов (гистеоцитоз, гипертоническая болезнь II ст.). Данная постановка опыта была выбрана в связи с тем, что для осуществления завершенного фагоцитоза, сопровождающегося полным уничтожением патогенных микроорганизмов, фагоциты, в том числе и нейтрофилы, образуют так называемые активные формы кислорода (АФК), к которым относятся радикалы кислорода и соединения, легко превращающиеся в такие радикалы. К АФК относятся супероксид анион радикал 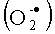 , гидроксильный радикал (ОН•) и гидроксиперекиси липидов (ROO*). Все эти соединения имеют на внешней молекулярной орбитали один неспаренный электрон, что и определяет их высокую реакционную способность, приводящую к взаимодействию с молекулами клетки: белком, РНК, ДНК и др., сопровождающемуся потерей их биологической активности.
, гидроксильный радикал (ОН•) и гидроксиперекиси липидов (ROO*). Все эти соединения имеют на внешней молекулярной орбитали один неспаренный электрон, что и определяет их высокую реакционную способность, приводящую к взаимодействию с молекулами клетки: белком, РНК, ДНК и др., сопровождающемуся потерей их биологической активности.
Следует отметить, что наряду с защитными функциями (инактивация бактериальных токсинов), радикалы кислорода (при длительном действии) часто оказывают пагубное влияние на окружающие клетки, вплоть до их гибели. По этой причине поиск химических соединений, кратковременно усиливающих образование АФК или подавляющих этот процесс, является актуальной задачей современной фармакологии.
После инкубации соединения формулы I с цельной кровью в результате взаимодействия NO с 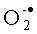 возникает высокореактивное соединение пероксинитрит (ONOO•), который, по-видимому, эффективно влияет на липидные компоненты цитоплазматической мембраны клеток, в том числе остатки АА.
возникает высокореактивное соединение пероксинитрит (ONOO•), который, по-видимому, эффективно влияет на липидные компоненты цитоплазматической мембраны клеток, в том числе остатки АА.
Вследствие этого, возможно, нарушается выделение АА из фосфолипидного слоя плазматических мембран, а следовательно, ее последующий метаболизм с образованием тромбоксана A2 (ТхА2) в тромбоцитах, как известно, являющегося мощным эндогенным вазоконстриктором и проагрегантом, что снижает функциональную активность кровяных пластинок.
Полученные данные позволяют предположить, что одним из возможных механизмов антиагрегационного действия соединения формулы I может оказаться активация процесса образования радикалов кислорода в цельной крови («дыхательный взрыв»), позволяющая снизить уровень тромбогенных факторов.
Модель паренхиматозного кровотечения характеризует гемостаз через стимуляцию тромбообразующего каскада реакций. Как правило, получать на таких моделях существенные сдвиги времени кровотечения с помощью резорбтивно действующих соединений достаточно трудно. Антикоагулянты прямого типа действия (гепарин, гепариноиды) только при высоких дозах способствуют пролонгации кровоточивости на таких моделях. При этом эффект выявляется в разные сроки (до 3 ч после введения). Непрямые антикоагулянты (оксикумарины, индандионы и др.) начинают увеличивать кровоточивость, как правило, после определенного инкубационного периода (от 12 до 48 ч после аппликации). Последнее легко объяснимо, исходя из современного представления их механизма действия: они останавливают биосинтез плазменных факторов свертывания крови II, VII, IX, X, а длительность инкубационного периода определяется количеством этих факторов, наработанных в печени до введения ингибиторов К-витаминредуктазы (оксикумарины, индандионы).
В контрольной группе животных длительность паренхиматозного кровотечения составила 88,9±1,2 с. Опытные группы животных были взяты в эксперимент через 60 минут после перорального введения соединения формулы I. В дозе 8 мг/кг время кровотечения увеличивалось на 71% по сравнению с исходными значениями и еще через 1 час данный эффект усиливался и составил 220,6±3,4 с. К концу опыта (240 мин) этот показатель не отличался от контрольных цифр. В дозе 16 мг/кг время кровотечения увеличивалось двукратно и регистрировалось только в первые 60 мин наблюдения.
Обнаруженный эффект, по-видимому, можно объяснить ранее установленной способностью соединения формулы I угнетать тромбоцитарное звено гемостаза, реализующееся через генерацию оксида азота, регуляцию уровня внутриклеточного кальция в данных форменных элементах крови и др.
Еще одним фактом, свидетельствующим о перспективности данного соединения, является высокое значение LD50 (8900 мг/кг), что соответствует V классу токсичности и позволяет отнести его к практически нетоксичным веществам.
Суммируя все выше изложенное (с учетом проведенных экспериментов, выявивших способность доноров NO угнетать индуцированную агрегацию тромбоцитов), можно констатировать, что заявляемое новое соединение формулы I в организме кроликов при парентеральной (вена) аппликации мощно тормозит склеивание кровяных пластинок.
Можно считать также доказанной способность нового донора NO (соединение I) всасываться из ЖКТ и проявлять свою антиагрегационную активность в условиях энтерального назначения. Разумеется, по сравнению с интравазальным введением она проявляется несколько позже, но сохраняется на значительно большой срок (вещество действовало более 2-4 ч на АА-индуцированную агрегацию и около 3 ч на АДФ-индуцированную).
Результаты испытаний
1. В условиях in vitro антиагрегационная активность вновь синтезированного донора оксида азота - соединения формулы I - не уступает таковой ацетилсалициловой кислоты.
2. Потенциальный антиагрегант диендиамин индольного ряда (соединение формулы I) после внутривенного введения кроликам значительно подавляет индуцированную функциональную активность тромбоцитов. Эффект равен аналогичному у нитропруссида натрия, но превышает его по длительности.
3. При пероральном введении животным соединение формулы I угнетает действие АДФ на тромбоциты.
4. Соединение формулы I нивелирует действие арахидоновой кислоты на активность тромбоцитов.
5. Перспективный антиагрегант - соединение формулы I - на короткий срок усиливает сниженную способность нейтрофилов человека вырабатывать активные формы кислорода.
6. Соединение формулы I статистически существенно тормозит гемостаз у крыс на модели паренхиматозного кровотечения, при этом данный эффект не носит дозозависимый характер.
7. Потенциальный лекарственный препарат (соединение формулы I) относится к V классу практически нетоксичных соединений.
Пример 2
Не менее выраженным и специфическим является влияние соединения формулы I на параметры гемодинамики и, в частности, на систолическое артериальное давление (САД), а также на активность растворимой гуанилатциклазы и на генерацию NO в опытах in vitro.
Эксперименты проводили на спонтанно-гипертензивных (СГК) и нормотензивных (НТ) наркотизированных крысах-самцах при внутривенном введении исследуемых соединений. Оценивали способность соединения снижать артериальное давление у СГК-крыс и предупреждать спазмогенные реакции на норадреналин (НА) и ангиотензин I (AI).
Соединение формулы I применяли в виде раствора в 3% водном ДМСО. В качестве препарата сравнения использовали нитропруссид натрия и нитроглицерин.
Введение соединения формулы I в диапазоне доз 0,01-0,1-0,5 и 1,0 мг/кг вызывало дозозависимый антигипертензивный эффект продолжительностью от 30-40 с до 60 мин и более. Минимальное и кратковременное гипотензивое действие отмечалось в дозе 0,01 мг/кг (снижение АД на 10-12 мм рт.ст. в течение 30 с), а максимальное - в дозе 1,0 мг/кг (снижение АД на 25-30 мм рт.ст. в течение 60 мин).
Препарат сравнения натрия нитропруссид вызывал сходное с максимальным для соединения I снижение АД (снижение на 25 мм рт.ст.) в дозе 0,5 мг/кг продолжительностью 20-30 мин. Эффект нитроглицерина в дозе 0,01 мг/кг был сходен с исследуемым соединением (снижение на 10-15 мм рт.ст в течение 30-40 с), а в более высоких дозах снижение АД не превышало 20-25 мм рт.ст. при продолжительности 5-6 мин (Табл.1).
На нормотензивных наркотизированных крысах изучали влияние соединения формулы I на прессорные эффекты норадреналина (НА, 10-15 мкг/кг), мезатона (М, 5 мкг/кг) и ангиотензина (А1, 5 мкг/кг). Депрессорное действие соединения формулы I оценивали относительно прессорных эффектов контрольных введений указанных анализаторов.
Как показали исследования (Табл.2), соединение формулы I в диапазоне доз 0,01-1,0 мг/кг дозозависимым образом антагонизирует к сосудосуживающему действию НА. Так, при введении соединения в дозах 0,01 и 0,1 мг/кг прессорный эффект НА снижается на 25 и 50% и существенно не изменяется при дальнейшем увеличении доз.
Соединение I в указанных дозах существенно не влияет на прессорное действие М, однако в дозе 0,1 мг/кг на 25% уменьшает прессорный эффект А1.
Таким образом, соединение формулы I эффективно и дозозависимым образом понижает САД у спонтанно гипертензивных животных и превосходит в этом отношении известный препарат нитропруссид. Соединение эффективно также антагонизирует к эффектам прессорных аминов на САД, что свидетельствует о специфическом действии на регуляцию артериального давления (Табл.2).
В опытах на ненаркотизированных СГК-крысах соединение формулы I при внутрижелудочном способе введения также вызывало дозозависимое снижение АД, продолжительность которого увеличивалась пропорционально увеличению дозы (максимальная продолжительность эффекта 5 ч после однократного введения в дозе 50 мг/кг, Табл.3).
Соединение формулы I и аргинин активируют растворимую гуанилатциклазу из тромбоцитов человека. При этом I по активности приближается к L-аргинину. Так, средняя степень активации фермента при применении L-аргинина в концентрации 0,1 mM составляет 2,26 раза, а при использовании I в той же концентрации - 2,12 раза (Табл.4).
Соединение I характеризуется как малотоксичное, хорошо переносимое, практически не вызывающее у животных изменений в общем состоянии, поведении, не нарушающее жизненную цикличность, не вызывающее у них снижения потребления пищи, воды и потерю массы тела. Соединение не вызывает изменений со стороны ЦНС, не проявляет нейротоксичности даже в довольно высоких дозах. Взаимодействие с рядом фармакологических анализаторов не выявило у соединения I специфического ответа, что, однако, не означает отсутствия у него медиаторного механизма действия.
Острая токсичность соединения на мышах составляет при введении внутрь 8,9±0,75 г/кг, кумуляция при введении в течение 28 сут слабая (гибель менее 50%).
Терапевтические дозы для животных находятся в пределах 1/100 от LD50 и составляют 4,0-8,0 и 12,0 мг/кг при внутрижелудочном введении.
Вторым объектом изобретения является способ получения соединения формулы I.
Заявляемое соединение содержит в качестве структурных фрагментов: индолиноновый цикл, цианогруппу в β-положении боковой цепи и замещенный циклический фрагмент, в котором при одном углеродном атоме находятся две сильные электронодонорные группировки - эфирный атом кислорода и замещенная аминогруппа. Включение этих группировок в циклическую систему обеспечивает высокую степень сопряжения их неподеленных электронных пар с двойными связями боковой цепи, что вместе с циклическим индольным атомом азота создает систему, которая по электронным параметрам сходна с гуанидиновой системой. Последнее представляет собой особый интерес, поскольку известно, что именно гуанидиновый фрагмент аргинина является источником оксида азота в живом организме.
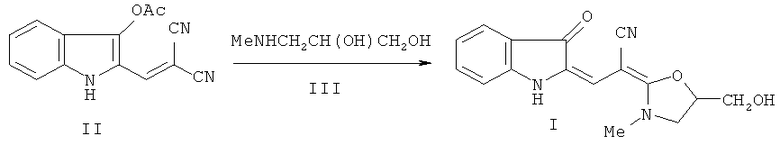
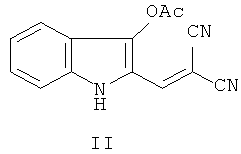
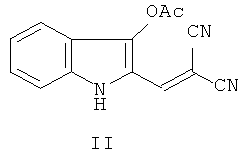
Способ получения соединения, представляющего собой 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолин-3-он формулы I, заключается во взаимодействии нитрила α-циано-β-(3-ацетоксииндолил-2)акриловой кислоты (II) с N-метиламинопропан-2,3-диолом (IIIa) в среде полярного (предпочтительно, изопропилового спирта) растворителя при кипячении по схеме:
Схема заявляемого способа
Недавно нами описан первый случай некаталитического присоединения аминов к цианогруппе [2, 4, 5], заключающийся во взаимодействии соединения II с первичными и вторичными аминами, в том числе и с первичными алканоламинами, например 1-аминопропан-2,3-диолом (IVa) [2, 4] в среде полярного или неполярного растворителя при кипячении с образованием производных 2-[2'-циано-3'-диаминопропенилиден-2']индолинон-3 общей формулы VIII по схеме 1:
Схема 1
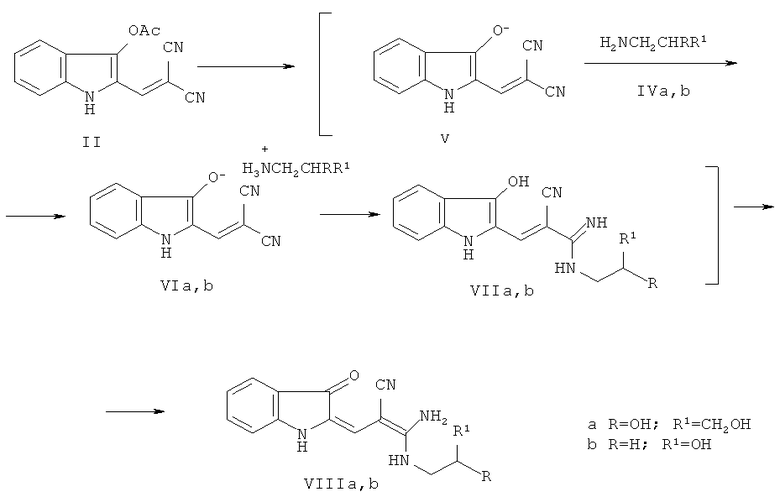
Как видно из схемы 1, сначала происходит дезацетилирование исходного соединения II (на что расходуется 1 моль 1-аминопропан-2,3-диола (IVa)), сопровождающееся солеобразованием (с образованием соли VIa) с участием промежуточного соединения V и амина IVa. При нагревании соль VIa диссоциирует, а высвобождающийся амин присоединяется по связи C≡N с образованием амидина VIIa, который находится в таутомерном равновесии с VIIIa [2, 4]. Как показали наши дальнейшие исследования, аналогично протекает взаимодействие соединения II и с β-этаноламином (IVb) (Пример А).
Синтез и физико-химические характеристики 2-[2'-циано-3'-амино-3'-(2,3-дигидроксипропил)аминопропенилиден-2']индолинон-3 (VIIIa) описаны в [2, 4].1H ЯМР спектр.
Пример А
2-[2'-Циано-3'-амино-3'-(β-гидроксиэтил)аминопропенилиден-2']индолинон-3 (VIIIb).
Смесь 2,5 г (10 ммоль) дициановинильного производного II, 2,5 г (41 ммоль) β-этаноламина IVb и 100 мл изопропилового спирта кипятят при перемешивании 4 ч. Смесь охлаждают, выпавший осадок отфильтровывают, промывают изопропиловым спиртом и эфиром. Получают 1,67 г (62%) 2-[2'-циано-3'-амино-3'-(β-гидроксиэтил)аминопропенилиден-2']индолинона-3 (VIIIb). Т.пл. 209-210°С (разл. из смеси ДМФА-вода, 4:1).
ИК спектр, ν, см-1: 3350, 3240-3060, 2190, 1670, 1605.
Масс-спектр: М+ 270.
Вычислено, %: С 62.21, Н 5.22, N 20.73.
C14H14N4O2.
Найдено, %: С 62.14, Н 5.50, N 20.73.
1Н ЯМР спектр.
УФ спектр, λmax, нм (lg ε): 217 (4.03), 231 (4.18), 257 (3.92),286 (4.01), 310 пл (3.98), 379 (4.33), 490 пл (4.46), 509 (4.64).
Совершенно неожиданные результаты получены при взаимодействии соединения II с N-замещенными алканоламинами, такими как N-метил-β-этаноламин (IIIb) (Пример Б) и N-метилглюкамин (IIIc) (Пример В). Сначала, по-видимому, взаимодействие соединений II и IIIb,с протекает по схеме, аналогичной схеме 1, однако процесс не останавливается на стадии образования соединений диендиаминовой структуры, общей формулы VIII, а происходит дальнейшая оксазолидиновая циклизация с выбросом молекулы аммиака и образованием производных 2-[2'-циано-2'-(3”-метилоксазолидин-2”-илиден)этилиден]-индолинона-3 IX и X. Указанная оксазолидиновая циклизация является неожиданной и непредсказуемой, и необходимым условием ее осуществления оказалось использование N-замещенного алканоламина. Теоретически этот фактор остается пока еще неясным. Ниже приведена схема образования соединений IX-X:
Схема 2
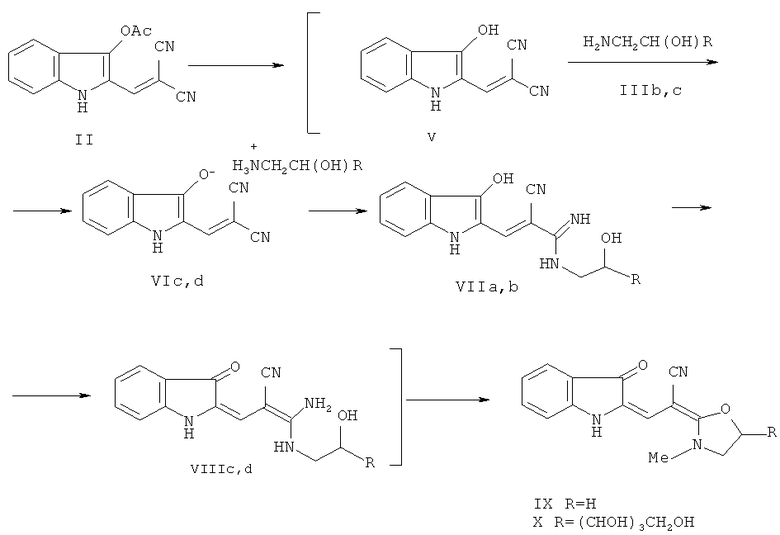
Пример Б
2-[2'-Циано-2'-(3”-метилоксазолидин-2”-илиден)этилиден]индолинон-3 (IX).
Смесь 2.5 г (10 ммоль) дициановинильного производного II, 2.2 г (30 ммоль) N-метиламиноэтанола IIIb и 100 мл изопропилового спирта кипятят при перемешивании 3,5 ч. Смесь охлаждают, выпавший осадок отфильтровывают, промывают изопропиловым спиртом и эфиром. Получают 1,2 г (44%) 2-[2'-циано-(3'-метилоксазолидинилиден-2')этилиден]индолинона-3 (IX). Т.пл. 222-226°С (разл. из смеси ДМФА-метанол, 8:1).
ИК спектр, ν, см-1: 3350, 2200, 1675, 1600.
Масс-спектр: M+ 267.
Вычислено, %: С 67.40, H 4.90, N 15.72 M. 267.
C15H13N3O2.
Найдено, %: С 67.61, Н 4.89, N 15.96.
1Н ЯМР спектр.
УФ спектр, λmax, нм (lg ε): 256 пл. (3.96), 282 (4.09), 310 пл. (4.03), 365 (4.36), 495 (4.41).
Пример В
2-{2'-Циано-2'-[3”-метил-5”-(1,2,3,4-тетрагидроксибутил)оксазолидин-2”-илиден]этилиден}индолинон-3 (X).
Смесь 2,5 г (10 ммоль) дициановинильного производного II, 4 г (20 ммоль) N-метилглюкамина VIIIc и 100 мл изопропилового спирта кипятят при перемешивании 3 ч. Смесь охлаждают, выпавший осадок отфильтровывают, промывают изопропиловым спиртом и эфиром. Получают 1,8 г (45%) 2-{2'-циано-2'-[3”-метил-5”-(1,2,3,4-тетрагидроксибутил)оксазолидин-2”-илиден]-этилиден}-индолинона-3 (X). Т.пл. 237°С (разл. из смеси ДМФА-вода, 4:1).
ИК спектр, ν, см-1: 3430, 3320, 3180, 2190,1650, 1600.
Масс-спектр: М+ не наблюдается, 237 [М - OCH(CHOH)3CH2OH]+, 210 [М-CH3NCH2CH(CHOH)3CH2OH]+.
Вычислено, %: С 58.91, Н 5.46, N 10.85 М 387.
C19H21N3O6.
Найдено, %: С 59.02, Н 5.47, N 10.65.
1Н ЯМР спектр.
УФ спектр, λmax, нм (lg ε): 220 (3.85), 255 пл (3.65), 282 (3.81), 310 пл (3.73), 369 (4.07), 497 (4.13).
Таким образом, оригинальность, необычность и непредсказуемость получения соединения I в указанных выше условиях заключается в том, что до настоящего времени в ряду сопряженных диендиаминов не была известна циклизация подобного типа (с отщеплением аммиака), и возможность такого замыкания неожиданно определяется наличием заместителя при атоме азота применяемого алканоламина.
Нижеприведенный пример 3 иллюстрирует заявляемый способ получения нового соединения I.
Пример 3
2-[2-[5-(Гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолин-3-он (I).
Смесь 2,5 г (10 ммоль) дициановинильного производного II, 2,1 г (20 ммоль) N-метиламинопропандиола IIIa и 100 мл изопропилового спирта кипятят при перемешивании 2 ч. Смесь охлаждают, выпавший осадок отфильтровывают, промывают изопропиловым спиртом и эфиром. Получают 1,55 г (52%) 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолина-3-он (I), Т.пл. 236.5-237.5°C (разл. из смеси ДМФА-вода, 2:1).
ИК спектр, ν, см-1: 3360, 3200, 2180, 1650.
Масс-спектр: М+ 297.
Вычислено, %: С 64.63, Н 5.09, N 14.13 М. 297.
C16H15N3O3.
Найдено, %: С 64.93, Н 5.11, N 14.26.
1Н ЯМР спектр.
1Н ЯМР спектр, (ДМСО-d6), (δ, м.д., J/Гц): 3.94 (2Н, т, J1=J2=9.5, CH 2OH), 3.96 (2Н, к, J1=7, J2=9.5, CH 2OH), 5.33.(1Н, т, J1=J2=6, CH2 OH), 3.54-3.80 (2Н, м, 4”-СН2), 4.91 (1Н, окт, 5”-CH), 3.32 (3Н, с, N-Me), 6.70 (1Н, α-СН), 6.82, 7.22, 7.42, 7.54 (4Н, м, Н(4-7)), 8.44 (1Н, с, N(1)H).
В спектре 13С ЯМР (DMSO-d6) соединения I наблюдаются сигналы ароматических атомов углерода С(4-7) при 113.2, 118.5, 123.2 и 134.2, четвертичных ароматических С-атомов при 121.1 и 151.4, C(1') - 113.2, CN - 120.1, C(2”) - 55.5, C(2”) - 166.2, N(3”)Me - 34.3, C(4”) - 53.3, C(5”) - 80.0 (д, J=156.3 Гц) и CH2OH при 61.0 м.д.
УФ спектр, λmax, нм (lg ε): 222 (4.09), 257 (3.95), 284 (4.07), 310 пл (4.00), 368 (4.34), 497 (4.40).
Синтез исходного соединения II описан в литературе [23, 24] (Схема 3):
Схема 3
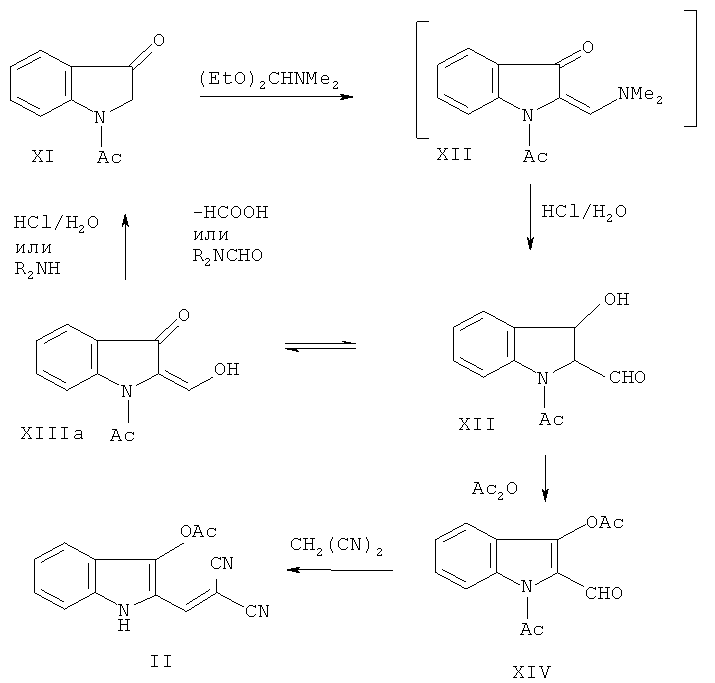
При взаимодействии N-ацетилиндоксила (XI) с диэтиловым ацеталем диметилформамида образуется енаминоиндолинон-3 XII, который без выделения подвергают кислотному гидролизу до 1-ацетил-3-гидрокси-2-формилиндола (XIII) (выход ~60÷70%). Следует отметить, что связь С(1') - С(2) в альдегиде весьма легко расщепляется и при увеличении времени обработки промежуточного соединения XII разбавленной HCl образуется N-ацетилиндоксил XI [1]. 2-Формилиндол XIII - соединение весьма лабильное, не выдерживает длительного хранения, и для проведения реакций этого соединения по формильной группе с СН-кислотами сначала требуется максимально быстрое его превращение в N-ацетил-3-ацетокси-2-формилиндол (XIV). Реакция соединения XIV с малондинитрилом в бензоле в присутствии триэтиламина дает дициановинилиндол II. Нижеследующие примеры иллюстрируют способ синтеза исходного соединения II по схеме 3 [1,3]:
1-Ацетил-2-формил-3-гидроксииндол (XIII) [1]. К суспензии 56 г (0,32 моль) индоксила XI в 640 мл бензола прибавляют 198 мл (~1.24 моль) диэтилацеталя ДМФА и перемешивают 1 ч при 20°С. Образовавшийся раствор кипятят 1 ч. Бензол упаривают на роторном испарителе при температуре в бане не выше 60°С. После удаления бензола остаток растворяют в ~4 л воды и подкисляют (рН 2-3) 60 мл конц. HCl. Через 5 мин1* (1* Через 1 ч осадок отфильтровывают, промывают водой и изопропиловым спиртом. Получают N-ацетилиндоксил с выходом 51%.), выпавший осадок отфильтровывают, промывают водой и изопропиловым спиртом. Выход соединения XIII 45,5 г (70%). Т.пл 122-123°С (из изопропилового спирта).
ИК спектр, ν, см-1: 1700, 1620, 1580.
Масс-спектр: М+ 203.
Вычислено, %: С 65.02, Н 4.47, N 6.89.
C11H9NO3.
Найдено, %: С 64.73, Н 4.50, N 6.90.
Спектр 1Н ЯМР (CDCl3): 10.24 (1Н, уш. с, СНО); 2.79 (3Н, с, NCOCH3); 7.34-7.91 м. д. (4Н, м, аром. протоны).
УФ спектр, λmax (lgε): 230 (4.22), 257 (4.03), 312 (3.95), 356 нм (3.89).
1-Ацетил-2-формил-3-ацетоксииндол (XIV) [1]. Раствор 45,5 г (0,23 моль) альдегида XIII в 130 мл уксусного ангидрида кипятят 10 мин. Охлаждают, образовавшийся осадок отфильтровывают, промывают уксусным ангидридом и эфиром. Выход соединения XIV 31,8 г (58%). Т.пл 111-113°С (из этилацетата).
ИК спектр, ν, см-1: 1785, 1700,1670, 1610.
Масс-спектр: М+ 245.
Вычислено, %: С 63.67, Н 4.52, N 5.71.
C13H11NO4.
Найдено, %: С 63.66, Н 4.52, N 5.64.
Спектр 1H ЯМР (ацетон-d6): 10,15 (1Н, с, СН); 2,45 (3Н, с, ОСОСН3); 2,48 (3Н, с, NCOCH3); 7,3 8-8,12 м. д. (4Н, м, аром. протоны).
Нитрил α-циано-β-(3-ацетоксииндолил-2)акриловой кислоты (II) [3]. К раствору 24,4 г (0,1 моль) 2-формилиндола XIV в 500 мл бензола при 20°С (внешнее охлаждение водой) и перемешивании добавляют 14,4 мл (0,1 моль) триэтиламина и 7,8 г (0,12 моль) малондинитрила. Перемешивают 1,5-2 ч. Выпавший осадок отфильтровывают, промывают бензолом и метанолом. Получают 22,7 г (90%) нитрила II, т.пл 210-212°С (разл., из ацетона).
ИК спектр, ν, см-1: 3340, 2240, 2230, 1770, 1600.
Масс-спектр: М+ 251.
Вычислено, %: С 66.93, Н 3.61, N 16.73.
C14H9N3O2.
Найдено, %: С 67.07, Н 3.42, N 16.63.
Спектр 1H ЯМР (ацетон-d6): 2,47 (3Н, с, ОСОСН3); 8,17 (1Н, с, СН); 10,39 (1Н, с, NH); 7,19-7,68 (4Н, м, аром. протоны).
Спектр 13С ЯМР (DMSO-d6+CD3OD): 168,5 (к, J≈7 Гц, ОСОСН3); 20,7 (к, J≈142 Гц, ОСОСН3); 114,9 (д, J=8,1 Гц, CN); 113,8 (д, J=13,5 Гц, CN); 74,4 (д, J=2 Гц, а-С), 144,3 (д, J=166 Гц, β-CH); 138,8; 138,4 (м, С(2), С(7а)); 119,4 (м, С(3а)); 121,4 (д, J=2 Гц, С(3)); 128,3, 121,4, 119,8, 113,9 (к, J1=160…165 Гц, J≈8 Гц, С(4), С(5), С(6), С(7)).
Выход 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолина-3-он (I), считая на N-ацетилиндоксил по схеме 3, составляет 13,5% (с учетом перекристаллизации конечного продукта).
Узким местом синтеза соединения II по этому способу является синтез 2-формилиндола XIII, а именно операция гидролиза, которая должна осуществляться в строго определенных временных рамках. Понятно, что при масштабировании процесса есть возможность получения загрязненного 2-формилиндола XIII продуктом его дальнейшего гидролиза - N-ацетилиндоксилом или существует опасность выделения чистого N-ацетилиндоксила. Поэтому, с целью избежания технологических трудностей, связанных с выделением промежуточного 2-формилиндола XIII при масштабировании процесса, мы модифицировали схему синтеза соединения II (Схема 4):
Схема 4
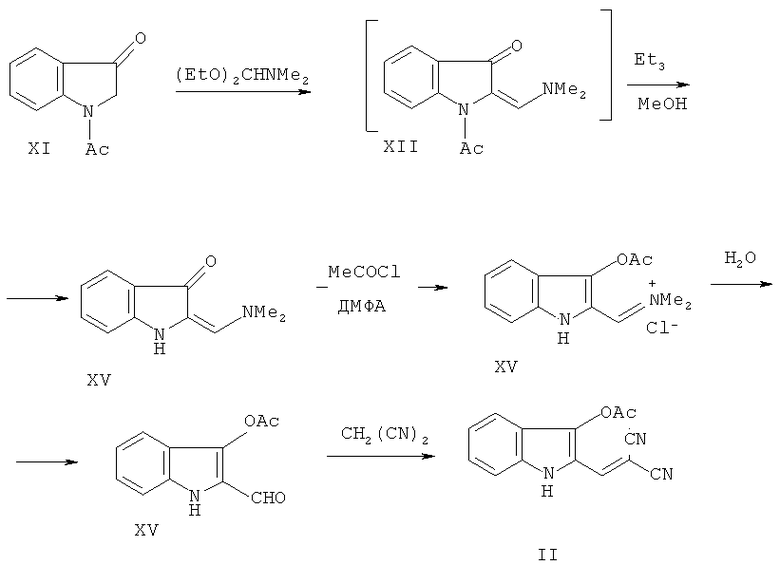
Поставленная цель достигается заявляемым способом получения соединения II, который является ключевым полупродуктом для синтеза биологически активного соединения I.
Синтез исходного соединения II по схеме 4 начинается, так же как и по схеме 3, с взаимодействия N-ацетилиндоксила с ацеталем ДМФА, однако затем в отличие от схемы 3 следует дезацетилирование промежуточного енаминокетона XII триэтиламином в метаноле с образованием енаминокетона XV с выходом 55% [1]. Нами описано ацилирование енаминокетона XV [3] различными ацилгалогенидами. В отличие от метода [3], где описано ацетилирование енаминокетона XV в среде хлористого ацетила, мы проводили процесс ацетилирования в среде ДМФА. В результате этого выделенный хлорид 3-ацетокси-2-диметилиминометилиндола (XVI) после растворения в воде превращен в 3-ацетокси-2-формилиндол (XVII) с выходом 63% против 25% по методу [3]. Реакция 3-ацетокси-2-формилиндола (XXII) с малондинитрилом в бензоле в присутствии триэтиламина дает дициановинилиндол II с высоким выходом. Нижеследующие примеры 4 и 5 иллюстрируют способ синтеза исходного соединения II по схеме 4.
2-Диметиламинометилениндолинон-3 (XV) [1] получают из 7 г (40 ммоль) индоксила XI и 24 мл (120 ммоль) ацеталя ДМФА в 80 мл бензола в условиях синтеза соединения XIII. После удаления бензола остаток растворяют в 60 мл метанола, добавляют 6 мл триэтиламина и кипятят 30 мин. Метанол упаривают. Остаток перемешивают с этилацетатом, осадок отфильтровывают, промывают изопропиловым спиртом и эфиром. Выход енамина XV 4.1 г (55%). Т.пл. 215°С (разл., из изопропилового спирта).
Спектр ИК, ν, см-1: 3250-3100, 1675, 1615.
Масс-спектр: М+ 188.
Найдено, %: С 70.33, Н 6.44, N 14.94.
C11H12N2O.
Вычислено, %: С 70.19, Н 6.43, N 14.88.
Спектр 1H ЯМР (ДМСО-d6), δ, м.д.: 7.01 (1Н, с, СН); 6.78-7.31 (4Н, м, аром. протоны); 3.19, 3.35 (6Н, два с, N(СН3)2); 8.85 (1Н, с, NH).
УФ спектр, λmax (lgε): 254 (4.05), 290 (4.03), 338 (4.32), 437 нм (4.12).
Пример 4
3-Ацетокси-2-формилиндол (XVII). К суспензии 0,38 г (2 ммоля) енаминокетона XV [1] в 2,5 мл ДМФА при охлаждении водой и перемешивании прикапывают 0,31 мл (4,4 ммоля) хлористого ацетила. Смесь перемешивают при комнатной температуре 5 ч. Через 2 ч добавляют еще 0,31 мл хлористого ацетила. Выдерживают ночь в холодильнике (5-7°С). Выпавший осадок отфильтровывают, промывают ДМФА и ацетоном. Получают 0,4 г (75%) хлорида 3-ацетокси-2-диметилиминометилиндола (XVI), который растворяют в 25 мл воды и охлаждают в течение 15 мин льдом. Выпавший осадок отфильтровывают, промывают водой, сушат. Получают 0,26 г (63%) 3-ацетокси-2-формилиндола (XVII). Т.пл. 145-147°С (из смеси вода - изопропиловый спирт, 5:1). Температура плавления смешанной пробы вещества с образцом соединения, полученного по способу [3], не показывает депрессии.
Спектр ИК, ν, см-1: 3280, 1770, 1660, 1620.
Масс-спектр: М+ 203.
Найдено, %: С 65.04, Н 4.64, N 6.76.
C11H9NO3.
Вычислено, %: С 65.02, Н 4.47, N 6.89.
Спектр 1Н ЯМР (CDCl3), δ, м.д.: 9.90 (1Н, с, СНО), 9.17 (1Н, уш. с, NH), 7.12-7.57 (4Н, м, аром. протоны), 2.48 (3Н, с, ОСОСН3).
Пример 5
Нитрил α-циано-β-(3-ацетоксииндолил-2)акриловой кислоты (II). К раствору 0,15 г (0,6 ммоля) 2-формилиндола XVII в 5 мл бензола при 20°С (внешнее охлаждение водой) и перемешивании добавляют 0,01 мл (0,072 ммоля) триэтиламина и 0,05 г (0,72 ммоля) малондинитрила. Перемешивают 0,5 ч. Выпавший осадок отфильтровывают, промывают бензолом и метанолом. Получают 0,15 г (99%) нитрила II. Т.пл 210-212°С (разл., из ацетона). Температура плавления смешанной пробы вещества с образцом соединения, полученного по способу [7], не показывает депрессии.
Выход 2-[2'-циано-2'-(3”-метил-5”-гидроксиметилоксазолидин-2”-илиден)этилиден]индолинона-3 (I), считая на N-ацетилиндоксил по схеме 4, составляет 12,5% (с учетом перекристаллизации конечного продукта).
Разработанные нами способы синтеза заявляемого соединения - 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолина-3-он (I) на основе нитрила II по схемам 3 и 4 позволяют получать это соединение примерно с одинаковыми выходами (13,5÷12,5%). Схема 4 имеет некоторые преимущества технологического характера перед схемой 3, однако, обе эти схемы предусматривают использование дорогого, труднодоступного реагента - диэтилового ацеталя диметилформамида, который применяется в большом избытке. С целью исключения из технологического процесса дорогостоящего реагента - диэтилового ацеталя ДМФА, а также с целью повышения выхода целевого продукта I перед нами стояла задача разработки альтернативного подхода к синтезу соединения I.
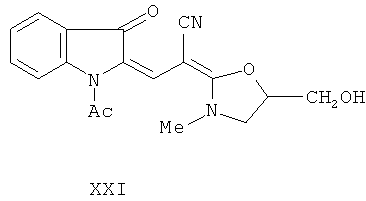
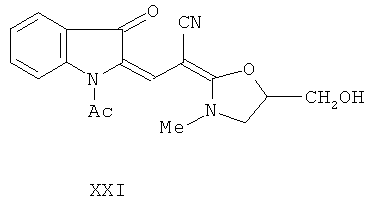
Поставленная цель достигается заявляемым способом получения 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолина-3-он (Схема 5), заключающимся во взаимодействии N-ацетилиндоксила (XI) с этоксиметиленмалондинитрилом (XVIII) с образованием N-ацетил-3-гидрокси-2-дициановинилиндола (XIX), который затем подвергают ацетилированию уксусным ангидридом до N-ацетил-3-ацетокси-2-дициановинилиндола (XX). Последний при взаимодействии с эквимолекулярным количеством N-метиламинопропан-2,3-диола (IIIa) превращается в неизвестный ранее 1-ацетил-2-[2'-циано-2'-(3”-метил-5”-оксиметилоксазолидин-2”-илиден)-этилиден]индолинон-3 (XXI). Снятие ацетильной группы осуществляется действием тритона Б (бензилтриметиламмоний гидроксид, 40% раствор в метаноле) в метаноле с выходом к заявляемому соединению I:
Схема 5
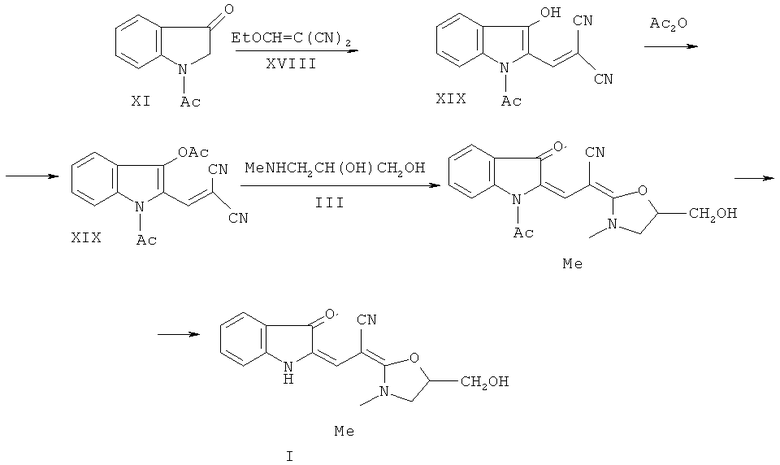
Нижеследующие примеры 6-8 и иллюстрируют способ синтеза заявляемого соединения I по схеме 5.
Пример 6
N-Ацетил-3-ацетокси-2-дициановинилиндол (XX). К суспензии 56 г (0,32 моля) N-ацетилиндоксила в 80 мл пиридина прибавляют 39,04 г (0,32 моля) этоксиметиленмалондинитрила (XVIII). Затем при перемешивании и охлаждении водой прибавляют 44,8 мл (0,32 моля) триэтиламина, температура реакционной массы при этом составляет 25±2°С. Перемешивают при этой температуре 1 ч, высаживают N-ацетил-3-гидрокси-2-дициановинилиндол (XIX) 160 мл уксусного ангидрида при охлаждении льдом. Температура в массе - не выше 30°С. Через 1 ч выдержки при 20°С осадок отфильтровывают, промывают уксусным ангидридом и ацетоном. Сушат на воздухе 1 ч. Получают 79,6 N-ацетил-3-гидрокси-2-дициановинилиндола (XIX), к которому прибавляют 500 мл уксусного ангидрида и при интенсивном перемешивании кипятят 30 мин. Охлаждают, осадок отфильтровывают, промывают уксусным ангидридом и ацетоном. Сушат на воздухе до постоянного веса. Получают 65,34 (69,6%) г N-ацетил-3-ацетокси-2-дициановинилиндола (XX). Т.пл. 250-252°С (с разл. из уксусного ангидрида).
Спектр ИК, ν, см-1: 2225, 1692, 1655, 1607.
Масс-спектр: M+ 293.
Найдено, %: С 65.44, Н 3.42, N 14.12.
C16H11N3O3.
Вычислено, %: С 65.52, Н 3.78, N 14.33.
Спектр 1Н ЯМР (DMSO-d6), δ, м.д.: 2.32 (3Н, с, ОСОСН3), 2.85 (3Н, с, NCOCH3), 7.48 (1Н, т, Н(5)), 7.68 1Н, т, Н(6)), 7.85 (1Н, д, Н(4)), 8.25 (1Н, д, Н(7)), (8.90 (1Н, с, Н(2')).
Пример 7
1-Ацетил-2-[2'-циано-2'-(3”-метил-5”-оксиметилоксазолидин-2”-илиден)-этилиден]индолинон-3 (XXI). Смесь 66,18 г (0,226 моль) N-ацетил-3-ацетокси-2-дициановинилиндола (XX), 47,5 г (0,452 моль) N-метиламинопропан-2,3-диола (VIIIa) и 1400 мл изопропилового спирта кипятят при перемешивании 1 ч. Охлаждают, осадок отфильтровывают, промывают изопропанолом и сушат при 80°С. Получают 66,4 г (86,7%) 1-ацетил-2-[2'-циано-2'-(3”-метил-5”-оксиметилоксазолидин-2”-илиден)-этилиден]индолинона-3 (XXI). Т.пл. 218-220°С (из метанола).
Спектр ИК (в таблетке с KBr), ν, см-1: 2220, 1700, 1680, 1610.
Масс-спектр: М+ 339.
Найдено, %: С 63.71, Н 5.01, N 12.37. C18H17N3O4.
Вычислено, %: С 63.71, Н 5.05, N 12.38.
Спектр 1Н ЯМР (DMSO-d6), (δ, м.д., J Гц): 2.42 (3Н, с, NCOCH3), 3.39 (3Н, с, N(3”)Me), 3.98 (2Н, т, J1=J2=9.5, СН 2ОН), 3.80 (2Н, к, J1=7, J2=9.5, CH 2OH), 5.19 (1H, т, J1=J2=6, CH2 OH), 3.55-3.90 (2Н, м, 4'-СН2), 4.94 (1Н, окт, 5'-СН), 7.20 (1Н, H(1)'), 6.82, 7.23 (1H, д, Н(4)), 7.59-7.67 (2Н, м, Н(5-6)), 8.07 ((1H, д, Н(7)).
Пример 8
2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолин-3-он (I).
К суспензии 66,4 г (0,196 моль) 1-ацетил-2-[2'-циано-2'-(3”-метил-5”-оксиметилоксазолидин-2”-илиден)-этилиден]индолинона-3 (XXI) в 800 мл метанола прибавляют 115,7 мл (0,254 моля) тритона Б и перемешивают при 20°С 30 мин. Осадок отфильтровывают, промывают метанолом, водой и метанолом. Сушат при 80°С, получают 40,6 г (69,7%) 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолин-3-он (I). Перекристаллизовывают из 650 мл смеси ДМФА-вода 2:1. Получают 35,7 г чистого соединения I. Выход соединения I на N-ацетилиндоксил - 37%. Температура плавления смешанной пробы вещества с образцами соединений, полученных по схемам 3 и 4, не показывает депрессии.
Спектры ИК, масс - ЯМР, УФ и полярограммы образцов соединения I, полученных по схемам 3, 4 и 5, идентичны.
Вывод: Разработаны эффективные оригинальные методы синтеза биологически активного соединения - 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолин-3-он (I).
Литература
1. Рябова С.Ю., Трофимкин Ю.И., Алексеева Л.М., Богданова Г.А., Шейнкер Ю.Н., Граник В.Г. // Химия гетероцикл. соедин. 1990. №11. С.1487-1494.
2. Рябова С.Ю., Алексеева Л.М., Граник В.Г., Фаермарк И.Ф., Шварц Г.Я. Патент 2008308 РФ «Производные индолинона-3, обладающие антигипертензивной активностью, и способ их получения».
3. Рябова С.Ю., Трофимкин Ю.И., Алексеева Л.М., Хабарова Л.С., Граник В.Г. // Химия гетероцикл. соедин. 1991. №3. С.343-348.
4. Рябова С.Ю., Трофимкин Ю.И., Граник В.Г., Фаермарк И.Ф., Шварц Г.Я. Патент 2026287 РФ «Производные пирролин-2-она-4 или индолинона-3, обладающие антигипертензивной активностью, и способ их получения».
5. Рябова С.Ю., Трофимкин Ю И., Алексеева Л.М., Кербникова И.Ф., Шварц Г.Я., Граник В.Г. // Хим. - фарм. журн. 1995. Т.29 (9). С.22-29.
6. Хмельницкая Е.Ю., Григорьев Н.Б., Рябова С.Ю., Трофимкин Ю.И., Азимов В.А., Граник В.Г. // Химия гетероцикл. соедин. 2002. №11. С.1540-1546.
7. Feelish М. // J. Cardiovascular. Pharm. 1991. Vol.17 (Suppl 3). P.25.
8. Hansson G.K. // Atheroscler. Thromb. Vasc. Biol. 2001. Vol.21(12). P.1876.
9. Orekhov A.N., Tertov V.V., Pivovarova E.M. // Cardiology. 1998. Vol.89. P.111.
10. Zadelaar S., Kleemann R., Verschuren L., et al. // Atheroscler. Thromb. Vasc. Biol. 2007. Vol.27. P.1706.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛИН-2-ОНА-4 ИЛИ ИНДОЛИНОНА-3, ОБЛАДАЮЩИЕ АНТИГИПЕРТЕНЗИВНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2026287C1 |
| Производные индолинона, обладающих свойствами лигандов мелатонинового рецептора и их применение | 2017 |
|
RU2712036C2 |
| Спироконденсированные производные 2,3-дигидроиндола, их применение в офтальмологии | 2017 |
|
RU2712039C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ПОМОЩЬЮ ЗАМЕЩЕННЫХ ОКСАЗОЛИДИНОНОВ | 2002 |
|
RU2321407C9 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1994 |
|
RU2125560C1 |
| ПРОИЗВОДНЫЕ N-[(1Н-ПИРАЗОЛ-1-ИЛ) АРИЛ]-1Н-ИНДОЛА ИЛИ 1Н-ИНДАЗОЛ-3-КАРБОКСАМИДА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ P2Y12 | 2011 |
|
RU2572593C2 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСАНАЛКЕНОВОЙ КИСЛОТЫ | 1989 |
|
RU2045526C1 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИДИН-2-ОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2163602C2 |
| Способ получения производных бензимидазола | 1977 |
|
SU843751A3 |
| Средство для лечения легочных гипертензий | 2023 |
|
RU2813890C1 |
Изобретение относится к области химико-фармацевтической промышленности и медицины и касается соединений, которые могут быть использованы для создания средств лечения сердечно-сосудистых заболеваний. Предложено применение 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолин-3-она формулы I в качестве биологически активного соединения, проявляющего свойства экзогенного донора оксида азота, активатора фермента растворимая гуанилатциклаза, ингибитора агрегации тромбоцитов с антигипертензивной активностью для получения средства для лечения сердечно-сосудистых заболеваний. Изобретение относится к способу получения 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолина-3-она формулы I, который получают взаимодействием нитрила α-циано-β-(3-ацетоксииндолил-2)акриловой кислоты формулы II с N-метиламинопропан-2,3-диолом (MeHN-CH2CH(OH)СН2ОН (формула IIIa)), в полярном растворителе при кипячении. Также изобретение относится к варианту способа получения 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолина-3-она формулы I, характеризующегося тем, что подвергают взаимодействию N-ацетилиндоксил и этоксиметиленмалондинитрил с образованием N-ацетил-3-гидрокси-2-дициановинилиндола, который затем подвергают ацетилированию уксусным ангидридом до N-ацетил-3-ацетокси-2-дициановинилиндола, с последующим его взаимодействием с эквимолекулярным количеством N-метиламинопропан-2,3-диола (IIIa) с образованием 1-ацетил-2-[2'-циано-2'-(3”-метил-5”-оксиметилоксазолидин-2”-илиден)-этилиден]индолинона-3 формулы XXI, затем в среде метанола удаляют ацетильную группу воздействием тритона Б, представляющего собой 40% раствор бензилтриметиламмоний гидроксида в метаноле, осадок промывают, сушат и очищают путем перекристаллизации из смеси диметилформамид-вода с получением целевого продукта. Технический результат - производное индолинона для создания средств для лечения сердечно-сосудистых заболеваний. 3 н.п. ф-лы, 4 табл., 3 ил., 8 пр.
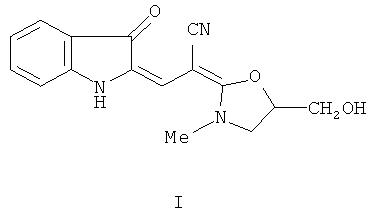
1. Применение 2-[2-[5-(Гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолин-3-он формулы I
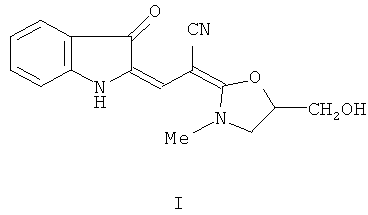
в качестве биологически активного соединения, проявляющего свойства экзогенного донора оксида азота, активатора фермента растворимая гуанилатциклаза, ингибитора агрегации тромбоцитов с антигипертензивной активностью для получения средства для лечения сердечнососудистых заболеваний.
2. Способ получения 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолина-3-она формулы I по п.1, характеризующийся тем, что нитрил α-циано-β-(3-ацетоксииндолил-2)акриловой кислоты формулы II:
подвергают взаимодействию с N-метиламинопропан-2,3-диолом формулы IIIa:
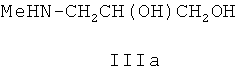
в полярном растворителе при кипячении.
3. Способ получения 2-[2-[5-(гидроксиметил)-3-метил-1,3-оксазолидин-2-илиден]-2-цианоэтилиден]индолина-3-она формулы I по п.1, характеризующийся тем, что подвергают взаимодействию N-ацетилиндоксил и этоксиметиленмалондинитрил с образованием N-ацетил-3-гидрокси-2-дициановинилиндола, который затем подвергают ацетилированию уксусным ангидридом до N-ацетил-3-ацетокси-2-дициановинилиндола, с последующим его взаимодействием с эквимолекулярным количеством N-метиламинопропан-2,3-диола (IIIa) с образованием 1-ацетил-2-[2'-циано-2'-(3”-метил-5”-оксиметилоксазолидин-2”-илиден)-этилиден]индолинона-3 формулы XXI,
затем в среде метанола удаляют ацетильную группу воздействием тритона Б, представляющего собой 40% раствор бензилтриметиламмоний гидроксида в метаноле, осадок промывают, сушат и очищают путем перекристаллизации из смеси диметилформамид-вода с получением целевого продукта.
| СПОСОБ ПОЛУЧЕНИЯ МЕЖМОЛЕКУЛЯРНОГО СОЕДИНЕНИЯ СЯММ-ТРИАЗИНОВ | 1971 |
|
SU425399A3 |
| РЯБОВА С.Ю | |||
| и др | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

