ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к улучшенному способу синтеза для получения гидразидов из гидразина и хлорангидридов. Полученные гидразидные продукты содержат защищенную тиольную группу, которую используют для сшивки калихимицина с моноклональными антителами.
УРОВЕНЬ ТЕХНИКИ
[0002] MYLOTARG® (гемтузумаб озогамицин), также называемый СМА-676 или просто СМА, состоит из моноклонального антитела к антигену CD33, которое связано с калихимицином с помощью гидролизующейся в кислой среде линкерной группы. Когда производное калихимицина связывается с малой бороздкой ДНК, указанное производное нарушает развитие ДНК и в итоге вызывает гибель раковых клеток. Коммерческий продукт был представлен на рынке в качестве первого химиотерапевтического агента с направленным воздействием на антитело под названием MYLOTARG®, а в настоящее время одобрен для лечения острого миелолейкоза (ОМЛ) у пациентов преклонного возраста.
[0003] Агенты, входящие в семейство высокоактивных антибактериальных и противоопухолевых агентов, в совокупности известные как калихимицины или комплекс LL-E33288, описаны в патентах США №№4970198, 4939244 и 5079233. Входящие в указанное семейство агенты можно применять для получения терапевтически полезных иммуноконъюгатов с моноклональными антителами в качестве носителей. Указанные антитела могут представлять собой антитело к антигену CD33 (например, hp67.6), антитело к антигену CD22 (например, G544), антитело к антигену Lewis Y (например, G193), антитело к антигену 5Т4 (например, Н8) или антитело к антигену CD20 (например, ритуксимаб). Агент, принадлежащий к семейству калихимицина, предпочтительно представляет собой N-ацилированный калихимицин, преимущественно N-ацетил-гамма-калихимицин. Агенты, принадлежащие к семейству калихимицинов, содержат метилтрисульфид, который может взаимодействовать с соответствующими тиолами с образованием дисульфидов, в то же время обеспечивая введение гидразидной функциональной группы, подходящей для присоединения производного калихимицина к носителю. Примеры такой реакции с калихимицином приведены в патенте США №5053394. Калихимицингидразидное производное присоединяется к моноклональному антителу через образование гидразона. Например, общий способ присоединения гидразидных производных лекарств к окисленным антителам описан Т.Дж.МакКерном и др. (Т.J.McKearn, et al.) в патенте США №4671958. Патент США №5770701 относится к способу получения форм с направленным действием на основе бисульфидных соединений комплекса LL-E33288. Линкер, представляющий собой 4-(4-ацетил-фенокси)бутановую кислоту, вступает в реакцию конденсации с гидразидным производным калихимицина, предпочтительно N-ацетил-γ-диметилгидразидным производным калихимицина с образованием ацилгидразона, который далее подвергают действию N-гидроксисукцинимида с получением OSu эфира (N-сукцинимидилокси), пригодного для конъюгирования с выбранной биомакромолекулой. Калихимицины содержат ендииновую концевую группу, которая активируется восстановлением -S-S- связи, приводя к разрывам двунитевой ДНК. Таким образом, моноацилированные гидразины, в которых ацильная группа содержит меркаптогруппу, хорошо подходят для связывания калихимицинов с моноклональными антителами. Гидразид 3-метил-3-меркаптобутановой кислоты, также называемый DMH-линкером или CL-332258, является предпочтительным меркапто содержащим N-ацилгидразином для связывания калихимицина с моноклональными антителами и получения, например, гемтузумаб озогамицина (gemtuzumab ozogamicin) или инотузумаб озогамицина (inotuzumab ozogamicin). Производное калихимицина затем активируют для конъюгирования с гуманизированным моноклональным антителом с получением СМА-676. В настоящее время DMH-линкер может быть получен с помощью 5-стадийного процесса через образование гидразида п-метоксибензилтиоэфирной кислоты 5 в качестве промежуточного соединения (Уравнения I-V). В современных способах промышленного получения, осуществляемых в США, присоединению по Михаэлю п-метоксибензилтиола к 3,3-диметилакриловой кислоте способствует добавление пиперидина (Уравнение I).
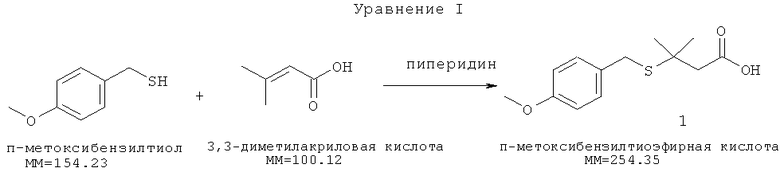
[0004] Полученная тиоэфирная кислота (1) реагирует с оксалилхлоридом в метиленхлориде с образованием хлорангидрида п-метоксибензилтиоэфирной кислоты (2) (Уравнение II)
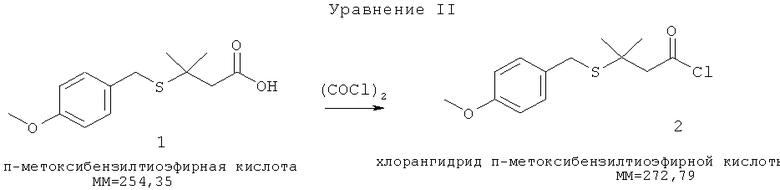
Хлорангидрид (2) медленно добавляют к смеси гидразина/метиленхлорида (в объемном соотношении примерно 28%) при низкой температуре (-70°С). Соответствующий гидразид п-метоксибензилтиоэфира (3) образуется с выходом примерно 74% (Уравнение III).
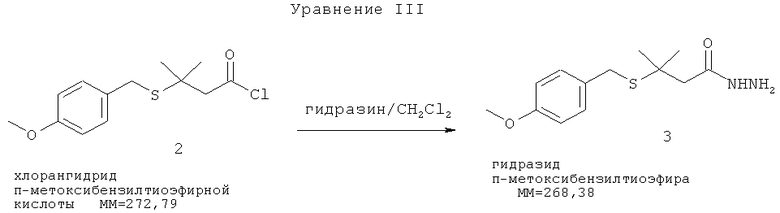
[0005] Тем не менее, целевой продукт, представляющий собой гидразид п-метоксибензилтиоэфира (3), обычно содержит примерно 20% нежелательного побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира (см. Уравнение VI ниже). Удаление защитной бензильной группы в кислой среде (Уравнение IV) с последующей нейтрализацией кислой соли и очисткой (Уравнение V) приводит к получению гидразида 3-метил-3-меркаптобутановой кислоты (5) с выходом 45%.


[0006] Нежелательный побочный продукт, представляющий собой бис-гидразид метоксибензилтиоэфира (6), образуется при взаимодействии продукта, представляющего собой гидразид п-метоксибензилтиоэфир, с исходным веществом, представляющим собой хлорангидрид п-метоксибензилтиоэфирной кислоты (Уравнение VI). Образование этого побочного продукта приводит к уменьшению выхода и качества целевого продукта.

[0007] При использовании первоначального способа получения бис-гидразид метоксибензилтиоэфира (6) образуется в количестве примерно 20%. Очевидно, что присутствие такого или даже большего количества побочного продукта согласно Уравнению III представляется нежелательным. В настоящем изобретении предложен способ, позволяющий преодолеть эту проблему и уменьшить выход нежелательного побочного продукта.
ОПИСАНИЕ ФИГУР
На фигуре 1 представлена технологическая схема получения п-метоксибензилтиоэфирной кислоты (1).
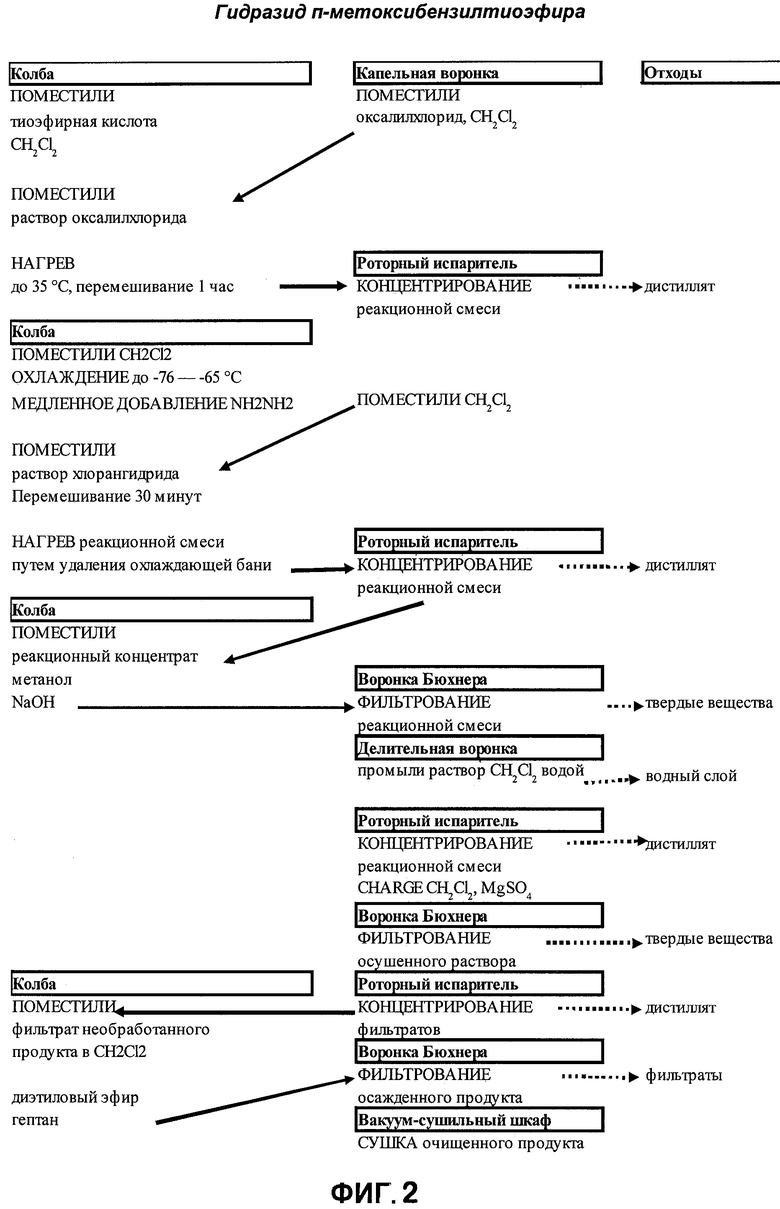
На фигуре 2 представлена технологическая схема получения хлорангидрида п-метоксибензилтиоэфирной кислоты (2) и гидразида п-метоксибензилтиоэфирной кислоты (3).
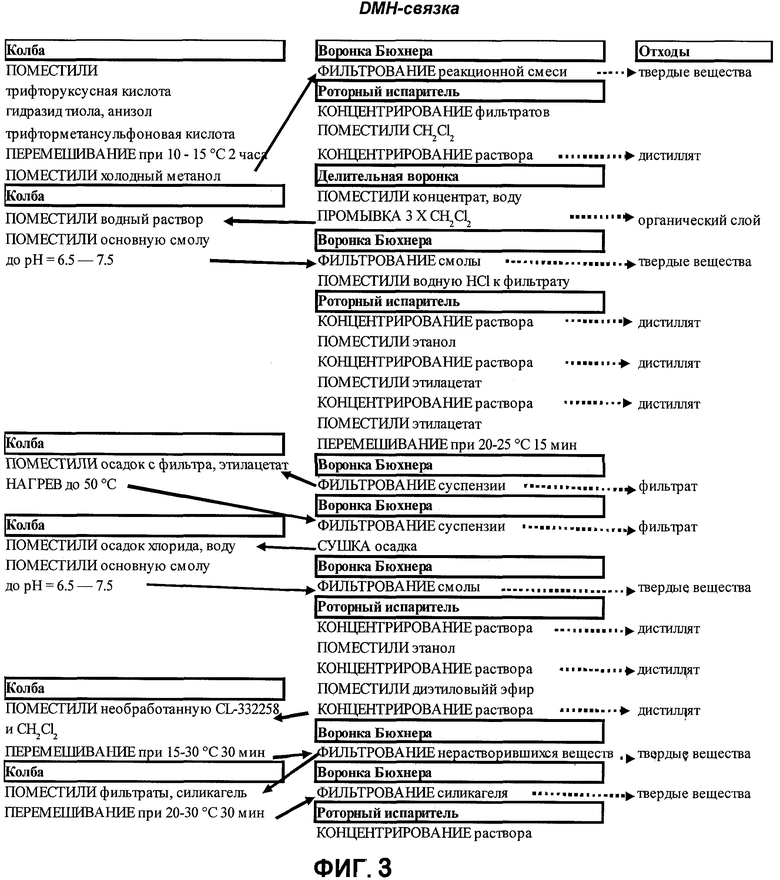
На фигуре 3 представлена технологическая схема получения сшивающей группы DMH (5).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0008] Согласно некоторым вариантам реализации настоящего изобретения предложены способы синтеза гидразидов (например, гидразида 3-метил-3-меркаптобутановой кислоты (4)) при снижении содержания побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира (например, (6)), с примерно 20% до примерно 3-5%. В одном из вариантов реализации изобретения раствор хлорангидрида метоксибензилтиоэфирной кислоты (2) добавляют к перемешиваемой смеси гидразин/метиленхлорид, которая является более разбавленной, чем в первоначальном способе (например, объемное соотношение составляет примерно 14% против 24-32% в первоначальном способе). Предпочтительное объемное соотношение для разбавления составляет примерно 14%. Для получения гидразинсодержащей смеси, к которой добавляют хлорангидрид, в вариантах реализации настоящего изобретения предложено возможное применение, без ограничения, других нереакционноспособных (или инертных) галогенированных растворителей, которые могут использоваться как вместо метиленхлорида, так и вместе с ним. Предпочтительные примеры таких других растворителей включают тетрахлорметан, хлороформ, этилендихлорид и хлорбензол. В некоторых вариантах реализации количество метиленхлорида (или соответствующего инертного растворителя) удвоено, что значительно уменьшает количество нежелательного побочного бис-гидразидного продукта. В некоторых вариантах реализации раствор хлорангидрида метоксибензилтиоэфирной кислоты добавляют к смеси гидразин/метиленхлорид непрерывно с постоянной скоростью, а не по частям. В некоторых вариантах реализации скорость добавления регулируют так, чтобы поддерживать температуру реакции в пределах от -68 до -75°С. В некоторых вариантах реализации применяют скорость перемешивания в пределах 300-400 об./мин в круглодонной колбе или 270 об./мин в колбе Мортона. Некоторые способы согласно настоящему изобретению обеспечивают получение гидразида п-метоксибензилтиоэфира (3) с концентрацией 91.1% и выходом 85%, а также с содержанием бис-гидразида метоксибензилтиоэфира (6) примерно 4-7%.
[0009] В другом варианте реализации изобретения было установлено, что несмотря на улучшения, достигаемые при использовании более разбавленной метиленхлоридной системы, все равно сохранялась необходимость соскабливать замерзший кристаллизовавшийся гидразин со дна и стенок реакционного сосуда. Ранее стандартной практикой было охлаждение раствора метиленхлорида/гидразина до примерно -70°С. Это приводило к значительным количествам гидразина, кристаллизовавшегося и осаждавшегося на стенках сосуда. Для обеспечения участия в реакции всего количества гидразина было необходимо соскабливать вещество со стенок сосуда, чтобы обеспечить возможность перехода указанного вещества в перемешиваемую суспензию. Для предотвращения такой ситуации согласно некоторым вариантам реализации настоящего изобретения была разработана альтернативная методика. Эта альтернативная методика включает охлаждение метиленхлорида до температуры от -68 до -75°С, предпочтительно до -70°С, с последующим медленным добавлением по каплям гидразина к охлажденному метиленхлориду с образованием однородной суспензии. При помощи этой новой методики достигается образование значительно более однородной суспензии гидразина, что минимизирует образование кристаллизовавшегося гидразина на внутренней стороне стенок колбы и уменьшает или устраняет необходимость соскабливать гидразин с колбы. При этом обеспечивается участие в реакции необходимого количества гидразина, что способствует уменьшению образования бис-гидразида метоксибензилтиоэфира (6).
[0010] В некоторых вариантах реализации изобретения предложен способ, включающий непрерывное добавление раствора хлорангидрида метоксибензилтиоэфирной кислоты (2) к сравнительно разбавленной (по сравнению с известным способом) и перемешиваемой охлажденной гетерогенной смеси гидразин/метиленхлорид (предпочтительно с концентрацией гидразина примерно 14%). Раствор хлорангидрида метоксибензилтиоэфирной кислоты добавляют к суспензии гидразин/метиленхлорид непрерывно с примерно постоянной скоростью, а не порциями. Скорость добавления регулируют так, чтобы температура реакции составляла от -68 до -75°С. Предпочтительная скорость перемешивания составляет 300-400 об./мин в круглодонной колбе или 270 об./мин в колбе Мортона. Улучшенные способы согласно некоторым вариантам реализации изобретения обеспечивают уменьшение количества побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира (6), с ранее достигаемого уровня в 20% до примерно 3-5% или ниже. Улучшенный способ синтеза промежуточного соединения 1 обеспечивает повышение эффективности всего способа синтеза гемтузумаб озогамицина.
[0011] В некоторых вариантах реализации изобретения предложен способ получения гидразида из гидразина и хлорангидрида, включающий: а) получение перемешиваемой однородной суспензии гидразина и инертного растворителя; b) непрерывное добавление хлорангидрида к указанной смеси. В другом аспекте этого варианта реализации хлорангидрид добавляют к суспензии по существу по каплям.
[0012] В некоторых вариантах реализации изобретения предложен способ получения гидразидов из галогенангидридов и гидразинов. В данном варианте реализации получение осуществляют путем химической реакции между электрофильным ацилкарбонилом в составе хлорангидрида и нуклеофильным азотом гидразина. Конкретные заместители, присоединенные к ацилкарбонилу, подходящие для целей настоящего изобретения, могут представлять собой любые фрагменты, которые не мешают образованию гидразидной связи, включая фрагменты, содержащие защитные группы, устраняющие помехи для образования гидразидной связи. Применение и удаление защитных групп описано в работе McOmie, Protecting Groups in Organic Chemistry, Plenum Press, NY, 1973, и Greene and Wuts, Protecting Groups in Organic Synthesis, 4nd. Ed., John Wiley & Sons, NY, 2006. Например, в некоторых вариантах реализации изобретения хлорангидрид содержит защищенный тиол. Примеры защищенных тиолов включают, но не ограничиваются ими, бензилтиоэфиры.
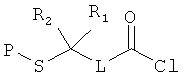
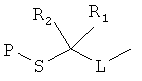
[0013] В другом аспекте изобретения хлорангидриды имеют структуру:
 ,
,
где Р представляет собой тиолзащитную группу, каждый из R1 и R2 выбран из группы, состоящей из C1-С5 алкила, a L представляет собой алкиленовую линкерную группу. Примеры алкиленовых линкерных групп L включают, но не ограничиваются ею, линкерную группу -(СН2)n-, где n представляет собой целое число от 1 до 5. В некоторых вариантах реализации R1 и R2 являются одинаковыми и, например, представляют собой одинаковые C1-С5 алкилы. Примеры C1-С5 алкила включают, но не ограничиваются ими, метил, этил, пропил, бутил, пентил, включая как линейные, так и разветвленные изомеры указанного алкила. Примеры тиолзащитных групп включают, но не ограничиваются ею, бензильную группу, в которой фенильный фрагмент может быть замещенным. Примеры возможных заместителей включают, но не ограничиваются ею, алкоксигруппу, такую как метокси, этокси и т.д. Таким образом, в одном из вариантов реализации изобретения хлорангидрид имеет следующую структуру:
 .
.
[0014] Другим вариантом реализации настоящего изобретения является способ получения гидразида из гидразина и хлорангидрида, включающий, на первой стадии, получение перемешиваемой по существу однородной суспензии, содержащей гидразин и инертный растворитель. В другом аспекте этого варианта реализации инертный растворитель представляет собой метиленхлорид.
[0015] Другим вариантом реализации настоящего изобретения является способ получения гидразида из гидразина и хлорангидрида. В другом аспекте этого варианта реализации гидразидный продукт имеет структуру:
 ,
,
где Р представляет собой тиолзащитную группу, a L, R1 и R2 могут быть такими, как указано выше. В еще одном аспекте этого варианта реализации Р представляет собой бензильную группу, возможно замещенную по фенольному кольцу. В другом аспекте этого варианта реализации Р представляет собой n-метоксибензильную группу, R1 и R2 выбраны из группы, состоящей из C1-С5 алкила, a L представляет собой алкиленовую линкерную группу. Примеры алкиленовых линкерных групп L включают линкерную группу -СН2-, но не ограничиваются этой группой. В одном из вариантов реализации каждый из R1 и R2 представляет собой независимо метил.
[0016] Другим вариантом реализации настоящего изобретения являются гидразидные продукты, полученные в соответствии со способами согласно настоящему изобретению. В одном варианте реализации целевой гидразид имеет структуру:
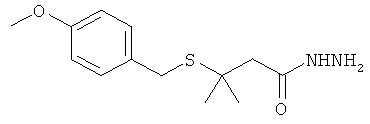
или представляет собой соответствующую соль. В еще одном варианте реализации изобретения гидразид представляет собой гидразид 3-метил-3-меркаптобутановой кислоты.
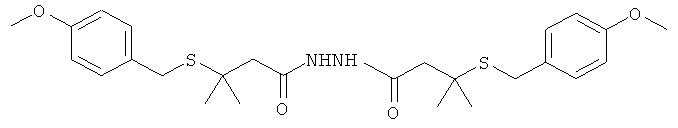
[0017] В еще одном аспекте изобретения целевой гидразид содержит менее 5% бис-гидразидного побочного продукта, имеющего структуру
 ,
,
где R и R′ представляют собой возможно замещенную алкильную, гетероалкильную или гетероалкарильную группы.
[0018] В другом аспекте этого варианта реализации каждый из фрагментов R и R′ в бис-гидразидном побочном продукте представляет собой
 ,
,
где Р представляет собой тиолзащитную группу, каждый из R1 и R2 выбран из группы, состоящей из C1-С5 алкила, a L представляет собой алкиленовую линкерную группу. Примеры алкиленовых линкерных групп L включают, но не ограничиваются ими, -СН2- группы. В другом аспекте этого варианта реализации каждый из R1 и R2 представляет собой независимо метил. В другом аспекте этого варианта реализации Р представляет собой бензильную группу, которая может быть замещена по фенильному кольцу; примеры включают, но не ограничиваются ею, п-метоксибензильную группу.
[0019] Другой вариант реализации изобретения представляет собой способ получения гидразида из гидразина и хлорангидрида, согласно которому гидразидный продукт содержит менее 5% бис-гидразидного побочного продукта, имеющего следующую структуру:
[0020] Другой вариант реализации настоящего изобретения представляет собой способ получения гидразида из гидразина и хлорангидрида, включающий на первой стадии получение перемешиваемой по существу однородной суспензии, содержащей гидразин и инертный растворитель, а на второй стадии - последующее непрерывное добавление хлорангидрида к указанной суспензии. Согласно другому аспекту этого варианта реализации непрерывное добавление раствора хлорангидрида регулируют таким образом, чтобы поддерживать температуру реакции в пределах от примерно -68°С до примерно -75°С. В еще одном аспекте этого варианта реализации суспензия гидразина по существу однородна.
[0021] В другом варианте реализации изобретения гидразидную связь получают в соответствии со способом, включающим стадии: а) охлаждения реакционного сосуда, содержащего перемешиваемый инертный растворитель, до заданной низкой температуры; b) непрерывного добавления гидразина в указанный реакционный сосуд с получением перемешиваемой по существу однородной суспензии, содержащей гидразин и инертный растворитель; с) непрерывного добавления хлорангидрида к указанной суспензии с образованием гидразидной связи. В другом аспекте этого варианта реализации инертный растворитель представляет собой метиленхлорид.
[0022] В другом варианте реализации изобретения суспензию гидразина получают по способу, включающему стадии: а) быстрого охлаждения инертного растворителя до температуры от примерно -68 до примерно -75°С и b) добавления по каплям гидразина, растворенного в инертном растворителе, к указанному охлажденному инертному растворителю. В другом аспекте этого варианта реализации инертный растворитель представляет собой метиленхлорид. В еще одном аспекте этого варианта реализации суспензию гидразина перемешивают со скоростью от примерно 270 до примерно 400 об./мин.
[0023] Другой вариант реализации изобретения представляет собой способ получения иммуноконъюгата агента, принадлежащего к семейству калихимицинов, с моноклональным антителом в качестве носителя, при этом указанный способ включает получение моноацилированного гидразина, в котором ацильная группа содержит S-защищенную меркаптогруппу, согласно предложенному способу; удаление указанной защитной группы и применение полученного гидразида для получения указанного иммуноконъюгата. Другой вариант реализации изобретения представляет собой способ получения гемтузумаб озогамицина или инотузумаб озогамицина, включающий стадии получения линкерного соединения, представляющего собой гидразид 3-метил-3-меркаптобутановой кислоты в соответствии со способом согласно изобретению и применение указанного линкера для получения гемтузумаб озогамицина или инотузумаб озогамицина.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0024] п-Метоксибензилтиол вступает в реакцию Михаэля с 3,3-диметилакриловой кислотой в пиперидине. Количества реагентов влияют на выход реакции. В одном из вариантов реализации количество n-метоксибензилтиола находится в небольшом молярном избытке относительно количества 3,3-диметилакриловой кислоты, например, в диапазоне от 0,354 кг (2,3 моль) и 0,362 кг (2,35 моль). Если количества меньше нижнего предела этого диапазона, то последующие реакции могут протекать не полностью. Если количества больше верхнего предела этого диапазона, то избыток реагентов может усложнять проведение процесса. Реакционную смесь нагревают таким образом, чтобы ее температура не превышала 98°С, в течение по меньшей мере 15 часов для предотвращения повышенного образования примесей. Пиперидин удаляют путем разбавления метиленхлоридом и промывания водной соляной кислотой, а затем водой. Поддержание температуры выше 50°С, но ниже 90°С необходимо до и во время добавления HCl, чтобы предотвратить осаждение продукта реакции. Реакционную смесь далее охлаждают, а затем экстрагируют метиленхлоридом, как указано в экспериментальной части.
[0025] Количества используемых растворителей пропорциональны масштабам реакции для достижения оптимальных результатов и очистки. Получаемый раствор продукта в CH2Cl2 сушат сульфатом магния, очищают, концентрируют под вакуумом, затем разбавляют гептаном для осаждения неочищенного промежуточного соединения, которое отфильтровывают и промывают гептаном. Очистку завершают путем повторного растворения неочищенного вещества в метиленхлориде и переосаждения в гептане. Очищенную п-метоксибензилтиоэфирную кислоту (1) выделяют фильтрованием, промывают гептаном и сушат под вакуумом.
[0026] п-Метоксибензилтиоэфирную кислоту (1) превращают в соответствующий хлорангидрид с использованием оксалилхлорида и метиленхлорида в качестве растворителя. Оксалилхлорид должен присутствовать в молярном избытке по отношению к n-метоксибензилтиоэфирной кислоте для полного протекания реакции. Хлорангидридный продукт выделяют концентрированием под вакуумом для переведения метиленхлорида и избытка оксалилхлорида в маслообразное состояние. Полученное масло разбавляют метиленхлоридом и медленно добавляют в течение примерно 3-5 часов при температуре в пределах от 65 до 75°С к разбавленной смеси гидразина и метиленхлорида.
[0027] Один из аспектов настоящего изобретения представляет собой образование однородной суспензии, содержащей гидразин и инертный растворитель, такой как метиленхлорид. В соответствии с одним вариантом реализации изобретения однородную суспензию получают медленным добавлением по каплям жидкого гидразина к метиленхлориду, предварительно охлажденному до примерно от -68 до -75°С, предпочтительно -70°С, до начала добавления гидразина. Напротив, охлаждение предварительно перемешанного раствора гидразина и метиленхлорида при той же температуре приводит к менее предпочтительному образованию кристаллического гидразина, который собирается на стенках реакционного сосуда. Не желая быть связанными какой-либо конкретной теорией, полагают, что медленное, по каплям, добавление гидразина к предварительно охлажденному метиленхлориду и контролирование максимальной концентрации гидразина в метиленхлориде приводит к образованию меньших по размеру, более однородных кристаллов гидразина, которые остаются суспендированными в перемешиваемой смеси и по существу не замерзают на стенках сосуда. Образование по существу однородной суспензии помогает обеспечивать нахождение гидразина в контакте с перемешиваемым метиленхлоридом и доступность гидразина к реакции с добавляемым раствором хлорангидрида. Образование однородной суспензии устраняет необходимость соскабливания вещества с внутренней поверхности реакционной колбы, которое требовалось в первоначальном способе. К тому же, указанное образование по существу однородной суспензии обеспечивает возможность участия в реакции требуемого количества гидразина, что также приводит к уменьшению количества образующегося бис-гидразида метоксибензилтиоэфира (6).
[0028] Концентрация гидразина в метиленхлориде влияет на количество бис-гидразида метоксибензилтиоэфира (6), который образуется в качестве побочного продукта. В известных способах концентрация гидразина/метиленхлорида составляла примерно от 24 до 32% об. Уменьшение в два раза соотношения гидразин/метиленхлорид (более разбавленный гидразин) до примерно от 12 до 16% об., предпочтительно до примерно 14%, приводит к уменьшению количества образующегося нежелательного бис-гидразида метоксибензилтиоэфира (6) (см. Таблицу 1).
[0029] В некоторых вариантах реализации изобретения раствор хлорангидрида добавляют к суспензии гидразин/метиленхлорид непрерывно с постоянной скоростью, а не порциями. Количество добавляемого хлорангидрида, как и скорость его добавления, влияют на выход целевого гидразида метоксибензилтиоэфира (3). Если добавлено слишком мало хлорангидрида, может образовываться избыточное количество побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира (6). Также, если время добавления хлорангидрида слишком мало, составляя менее 3 часов, может образовываться избыточное количество побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира (6). Скорость добавления регулируют для поддержания температуры реакции в пределах от -68 до -75°С. Если температура реакции возрастает до более высоких значений, также может образовываться избыточное количество бис-гидразида метоксибензилтиоэфира (6). Скорость перемешивания суспензии гидразина предпочтительно составляет от 300 до 400 об./мин в круглодонной колбе или 270 об./мин в колбе Мортона. Оба аспекта улучшенного способа, а именно: использование более разбавленной смеси гидразин/метиленхлорид и образование однородной суспензии, уменьшают количество побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира (6), с примерно 20% до примерно от 3 до 5%. Улучшенные стадии получения гидразина увеличивают общую эффективность синтеза линкера, представляющего собой гидразид 3-метил-3-меркаптобутановой кислоты, вследствие чего также повышается общая эффективность получения MYLOTARG® (гемтузумаб озогамицина).
[0030] После завершения реакции реакционную смесь концентрируют под вакуумом и осадок обрабатывают раствором гидроксида натрия в метаноле (примерно 4-5%). Указанный раствор концентрируют под вакуумом, разбавляют метиленхлоридом, промывают водой, сушат над сульфатом магния, очищают и концентрируют под вакуумом с получением концентрата. Необходимо использовать достаточное количество сульфата магния для полного осушения продукта, чтобы на следующих стадиях не происходило разложения продукта или затруднений при его кристаллизации. Конечный концентрат разбавляют метиленхлоридом в количестве, в 1.33 раза превышающем массу п-метоксибензилтиоэфирной кислоты (1), и этот раствор добавляют к диэтиловому эфиру, взятому в количестве, в 7.6 раза превышающем массу п-метоксибензилтиоэфирной кислоты (1). Алифатический углеводородный растворитель, такой как гептан, гексан, октан или изогексан, предпочтительно гептан, добавляют к образовавшейся суспензии в количестве, в 1.83 раза превышающем массу п-метоксибензилтиоэфирной кислоты (1), для завершения осаждения. Гидразид n-метоксибензилтиоэфира (6) выделяют фильтрованием, промывают гептаном и сушат под вакуумом.
[0031] Гидразид п-метоксибензилтиоэфира (3) обрабатывают трифторметансульфокислотой в присутствии анизола, используя трифторуксусную кислоту в качестве растворителя. Необходимо следить за тем, чтобы во время добавления и последующего протекания реакции температура реакции не превышала примерно 20°С для предотвращения образования нежелательных примесей. По завершении удаления п-метоксибензильной защитной группы реакционную смесь гасят, выливая в метанол, и фильтруют для удаления твердых побочных продуктов. Фильтраты концентрируют под вакуумом, растворяют в воде, промывают метиленхлоридом и обрабатывают анионообменной смолой с получением гидразида 3-метил-3-меркаптобутановой кислоты (5). Смолу удаляют фильтрованием, затем к раствору неочищенного продукта добавляют водный раствор соляной кислоты для образования хлористоводородной соли. Полученное количество продукта концентрируют под вакуумом, растворяют в этаноле, очищают фильтрованием и концентрируют под вакуумом. Этот концентрат разбавляют этилацетатом и концентрируют под вакуумом. Затем вновь осадок разбавляют этилацетатом и выделяют фильтрованием. Влажный осадок нагревают с этилацетатом до примерно от 48 до 55°С, охлаждают, фильтруют и сушат под разрежением. Высушенную хлористоводородную соль превращают в свободное основание путем обработки анионобменной смолой в воде. Смолу удаляют фильтрованием, а фильтраты концентрируют под вакуумом. Концентрат растворяют в этаноле, концентрируют под вакуумом, суспендируют в диэтиловом эфире и концентрируют под вакуумом. В качестве окончательной очистки гидразид 3-метил-3-меркаптобутановой кислоты (5) растворяют в метиленхлориде, очищают фильтрованием и обрабатывают двуокисью кремния, которую затем удаляют фильтрованием. Очищенный продукт выделяют из раствора концентрированием под вакуумом. Согласно предпочтительному способу очистки, показанному в Примере 15, гидразид 3-метил-3-меркаптобутановой кислоты (5) растворяют в 50 частях (объем/масса) метиленхлорида при 20°С±3°С, перемешивают 30 минут и фильтруют. Полученный раствор обрабатывают 0.7-1 частями (масс./масс. относительно массы неочищенного линкерного соединения) силикагеля, перемешивают 30 минут, фильтруют и концентрируют до получения сухого остатка в роторном испарителе. Полученное твердое вещество растирают с гептаном. После выделения и сушки под вакуумом гидразид 3-метил-3-меркаптобутановой кислоты (5) получают в виде сыпучего порошка с примерно 76% выходом.
[0032] Одним из аспектов настоящего изобретения является способ получения гидразида п-метоксибензилтиоэфира с менее чем 5%-ным содержанием нежелательного побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира (6). Этот улучшенный способ включает модифицированный способ проведения реакции сочетания хлорангидрида тиоэфирной кислоты с гидразином с образованием гидразида п-метоксибензилтиоэфира. Стадии указанного способа схематически показаны в Уравнении I. Нежелательный побочный продукт, представляющий собой бис-гидразид метоксибензилтиоэфира (6), образуется при сочетании продукта, представляющего собой гидразид n-метоксибензилтиоэфира (3), с исходным веществом, представляющим собой хлорангидрид n-метоксибензилтиоэфирной кислоты (2). Образование нежелательного бис-гидразида метоксибензилтиоэфира (6) приводит к снижению качества и выхода продукта.
[0033] Согласно другому аспекту настоящего изобретения, предложенный способ в целом может включать более широкое применение. Конкретная последовательность реакций (Уравнение III) может быть обобщена в виде Уравнения VII.
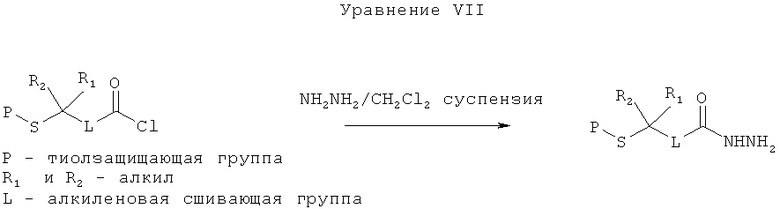
[0034] Когда в настоящем описании указано, что на одной из стадий способа добавление вещества осуществляют непрерывно, это означает, что указанное добавление осуществляют постоянно в течение некоторого времени, а не порциями и не за один раз. Примерами непрерывного добавления являются добавление жидкости по каплям или добавление жидкости непрерывным потоком. В некоторых вариантах реализации непрерывное добавление выполняется путем контролирования скорости добавления вещества, которое реагирует экзотермично, со скоростью, достаточной для поддержания температуры реакции внутри заданного температурного интервала.
[0035] Термин «суспензия» в настоящем описании относится к сочетанию твердой и жидкой фаз, которые тщательно смешивают вместе и, как правило, охлаждают до температуры, при которой присутствуют как твердая, так и жидкая фаза, при этом указанная смесь является исключительно жидкостью при температуре окружающей среды. Иногда суспензией называют смесь жидкой/твердой формы одного и того же вещества, например, смесь лед/вода, в которой лед сравнительно хорошо измельчен и перемешан с жидкой водой. В контексте настоящего изобретения к суспензии можно отнести смесь твердое вещество/ жидкость, образованную комбинацией двух веществ, таких как гидразин и растворитель, например, метиленхлорид. Полагают, что в охлажденной суспензии гидразина/метиленхлорида жидкая фаза содержит смесь метиленхлорида и гидразина, тогда как твердая фаза содержит главным образом гидразин.
[0036] Термин «алкил» включает линейные или разветвленные алкилы, состоящие из 1-10 углеродных атомов, при этом предпочтительными являются низшие алкилы, содержащие от 1 до 5 углеродных атомов. Например, алкил включает метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, трет-пентил, 2-метилбутил, н-гексил, изогексил, гептил, изогептил, октил, изооктил, нонил, децил и т.п. Термин «алкилен» включает линейные и разветвленные бирадикалы алканов, содержащих от 1 до 10 углеродных атомов, такие как метилен (-СН2-), этилен (-СН2СН2-), пропилен, бутилен и пентилен.
[0037] Термин «гетероалкил» относится к описанной выше алкильной группе, в которой один или более углеродных атомов замещены на гетероатом (кислород, серу, азот или фосфор) и которая может содержать дополнительные гетероатомы. Термин «гетероалкарил» относится к гетероалкильному фрагменту, описанному выше, но который дополнительно замещен арильным фрагментом, где указанный арильный фрагмент может быть замещенным. Возможно замещенный арил включает фенил и замещенный фенил. В замещенном фениле один, два или три возможных заместителя могут замещать водород в фенольном кольце и располагаться в орто-, мета- или пара-положении по отношению к метиленовой группе у бензильного атома углерода (или к другому месту присоединения). В Примере 1 метоксигруппа расположена в пара-положении к метиленовой группе. Неограничивающие примеры возможных заместителей в ариле включают C1-С5 алкил, C1-С5 алкокси, C1-С5 галогеналкил, C1-С5 галогеналкокси, в которых атомы водорода, соединенные с углеродными атомами в алкильной или алкоксигруппе, могут быть заменены на атомы галогена, например -CF3 и -OCF3.
Следующие неограничивающие примеры иллюстрируют настоящее изобретение.
ПРИМЕР 1
Первоначальное получение п-метокситиоэфирной кислоты (1)
[0038] В соответствии с Уравнением I в 5-литровую круглодонную колбу, снабженную термопарой, механической мешалкой, обратным холодильником с отверстием для подвода N2 в верхней части и 250 мл капельной воронкой с компенсатором давления, поместили 400 г, 465 мл, 4,70 моль пиперидина. 3,3-диметилакриловую кислоту (215 г, 2,15 моль) добавили порциями в 5-литровую реакционную колбу при перемешивании. Смесь энергично перемешивали в атмосфере N2. Во время добавления температуру реакции поддерживали ниже 35-40°С (Примечание: сильно экзотермично, газовыделение). п-Метоксибензилтиол (386 г, 323 мл, 2,32 моль) добавили с помощью капельной воронки с компенсатором давления в течение 15 минут в (5-литровую) реакционную колбу. Смесь нагрели до 82-88°С при перемешивании в атмосфере N2. Температуру реакции поддерживали в этом интервале в течение 15 минут. Примечание: экзотермично. Прозрачную желтую смесь нагрели до 92-95°С и перемешивали в атмосфере N2 по меньшей мере в течение 15 часов. Для проведения ВЭЖХ взяли 1 мл пробу. Реакцию считали завершенной, когда оставалось менее 3% 3,3-диметилакриловой кислоты, как определяли по площади пика. Реакционную смесь охладили до 70-75°С, убрав колбонагреватель.
[0039] 3М раствор соляной кислоты (1900 мл, 2090 г) добавили через литровую капельную воронку с компенсатором давления к перемешиваемому желтому раствору, поддерживая температуру ниже 90°С. Конечная температура составляла 70-75°С. Смесь охладили до 20-25°С, поместив ее на водяную баню. Метиленхлорид (1600 г, 1210 мл) добавили к гетерогенной смеси. Смесь перемешивали в течение 5 минут. Проверяли рН верхнего водного слоя в колбе и добавляли необходимое количество 3М раствора HCl до достижения значения рН ниже 2. Все содержимое 5-литровой реакционной колбы переносили в 4-литровую делительную воронку и оставляли два слоя разделяться в течение по меньшей мере 10 минут. Органический (нижний) слой переносили из делительной воронки обратно в 5-литровую реакционную колбу. Видный (верхний) слой переносили из делительной воронки в 4-литровую колбу Эрленмейера. 3М раствор соляной кислоты (1050 мл, 1154 г) добавили в 1000 мл капельную воронку с компенсатором давления к раствору метиленхлорида в 5-литровой колбе в течение 10 минут. Проверяли рН верхнего водного слоя в колбе и добавляли необходимое количество 3М HCl до достижения значения рН водного слоя ниже 2. Все содержимое 5-литровой реакционной колбы переносили в 4-литровую делительную воронку. Значение суммарного объема записывали. Два слоя оставляли разделяться по меньшей мере на 10 минут. Органический (нижний) слой переносили из делительной воронки в чистую 4-литровую колбу Эрленмейера. Водный (верхний) слой переносили из делительной воронки в чистую 4-литровую колбу Эрленмейера. Водные слои объединяли в 5-литровой реакционной колбе.
[0040] Метиленхлорид (305 мл, 400 г) добавляли к водному раствору, полученному на предыдущей стадии. Смесь перемешивали по меньшей мере 5 минут. Все содержимое 5-литровой реакционной колбы переносили в 6-литровую делительную воронку, и значение суммарного объема записывали. Смесь оставляли отстаиваться для разделения двух слоев по меньшей мере на 5 минут. Органический (нижний) слой переносили из делительной воронки в 4-литровую колбу Эрленмейера. Раствор метиленхлорида промыли 1000 мл воды. Смесь тщательно перемешивали в течение 1-2 минут и оставляли отстаиваться как минимум на 10 минут. Водный слой отделили. Измерили значение рН водного слоя в колбе. Водные слои объединили и отбросили. К раствору метиленхлорида добавили безводный сульфат магния (110 г) и перемешивали по меньшей мере 15 минут. На 5-литровую колбу нанесли метки уровней 800, 900 и 1000 мл. Используя вакуум-фильтрование, желтую смесь отфильтровали через 15-см воронку Бюхнера с фильтровальной бумагой (Whatman #1) в 5-литровую реакционную колбу. Колбу и осадок на фильтре промыли 300 мл, 400 г метиленхлорида. Раствор метиленхлорида концентрировали, как описано далее: 5-литровую круглодонную колбу снабдили механической мешалкой и дистилляционной насадкой Кляйзена, снабженной термопарой. Дистилляционная насадка была соединена с 30-см холодильником для простой перегонки (simple condenser), который был соединен с алонжем, соединенным с приемной 1-литровой колбой, охлаждавшейся в ледяной бане. Алонж был соединен с охлаждаемой до 0°С ловушкой. Охлаждаемая ловушка была соединена с вакуумным насосом.
[0041] Метиленхлорид из раствора в 5-литровой колбе отгоняли при температуре 15-35°С под вакуумом до достижения объема жидкости в сосуде, равного 900 мл. Дистиллят отбросили. Температуру содержимого 5-литровой колбы установили в пределах 15-20°С. Гептан (2442 мл, 1670 г) добавляли с помощью капельной воронки к перемешиваемому концентрированному раствору по меньшей мере в течение 10 минут. Осадок образовывался после добавления примерно 1000 мл (684 г) гептана. Гетерогенную смесь охлаждали при перемешивании до температуры 0-5°С в течение как минимум 20 минут и выдерживали при такой температуре по меньшей мере 30 минут. Содержимое 5-литровой реакционной колбы отфильтровали через 30-см воронку Бюхнера с фильтровальной бумагой Whatman #1. Фильтрат собирали в 4-литровую колбу для вакуум-фильтрования. 5-литровую реакционную колбу промыли двумя порциями по 310 мл, 212 г, гептана, и раствор добавили к осадку на фильтре. Осадок осушали с помощью вакуум-фильтрования до тех пор, пока сбор фильтрата по существу не прекращался, и по меньшей мере в течение 25 минут. Высота осадка на фильтре составляла примерно 20 мм. Масса осадка составляла примерно 601 г. Фильтрат отбросили. Осадок поместили в 5-литровую круглодонную колбу, снабженную термопарой, механической мешалкой, подводом N2 и 1-литровой капельной воронкой с компенсатором давления. Метиленхлорид (750 мл, 990 г) поместили в 5-литровую реакционную колбу, и перемешивание проводили до полного растворения твердых веществ (примерно 10 минут). Гептан (1060 мл, 725 г) поместили в 5-литровую реакционную колбу. Гетерогенный раствор охладили до 0-5°С, используя ледяную баню, в течение по меньшей мере 15 минут, затем перемешивали по меньшей мере 30 минут. Получили густой гетерогенный раствор. Содержимое 5-литровой реакционной колбы отфильтровали через 30-см воронку Бюхнера с фильтровальной бумагой Whatman #1. Фильтрат собрали в 4-литровую колбу для вакуум-фильтрования. 5-литровую реакционную колбу промыли двумя порциями по 310 мл, 212 г, гептана, и раствор добавили к осадку на фильтре. Осадок осушали с помощью вакуум-фильтрования до тех пор, пока по существу не прекращалось выделение фильтрата (минимум 20 минут). Высота осадка на фильтре составляла примерно 20 мм. Масса осадка составляла примерно 632 г. Фильтрат отбросили. Влажный осадок поместили в эксикатор. Эксикатор с п-метоксибензилтиоэфирной кислотой накрыли чистой фильтровальной бумагой. Продукт сушили в вакуумном сушильном шкафу при 38-40°С и 28-30 дюймах (711,2-762,0 мм рт.ст.) в течение 20 часов.
ПРИМЕР 2
Первоначальное получение хлорангидрида n-метоксибензилтиоэфирной кислоты (2)
[0042] В соответствии с Уравнением II п-метоксибензилтиоэфирную кислоту (400 г, 1,57 моль) поместили в 5-литровую круглодонную колбу, снабженную термопарой, механической мешалкой, обратным холодильником с отверстием для подвода N2 в верхней части и 0,5-литровой капельной воронкой с компенсатором давления. Метиленхлорид (1600 г, 1212 мл) поместили в 5-литровую реакционную колбу. Прозрачный раствор нагрели до 20-25°С. Метиленхлорид (300 г) и оксалилхлорид (110 г, 78 мл) поместили в 0.5-литровую капельную воронку. 350 мл раствора оксалилхлорид/метиленхлорид добавили с помощью капельной воронки, поддерживая температуру реакции 20-30°С. Прозрачный желтый раствор перемешивали при 20-25°С как минимум 30 минут до прекращения выделения пузырьков. Добавление оксалилхлорида повторили. 350 мл раствора оксалилхлорид/метиленхлорид добавили с помощью капельной воронки в реакционную колбу, поддерживая температуру реакции 20-30°С (время добавления примерно 45 минут). Реакционную смесь нагрели до 32-38°С. Перемешиваемый раствор выдерживали в этом температурном интервале как минимум 1 час. Для проведения ВЭЖХ взяли 1 мл пробу. Реакцию считали завершенной, когда оставалось менее 3% исходного вещества, представляющего собой метоксибензилтиоэфирную кислоту. Реакционную смесь охладили до 23-28°С в течение как минимум 5 минут. Раствор перенесли в калиброванную 3-литровую круглодонную колбу. Реакционную колбу промыли 100 мл, 132 г, метиленхлорида, раствор поместили в 3-литровую колбу. Реакционный раствор концентрировали под вакуумом в роторном испарителе с установленной температурой 33-36°С при давлении 25-28 дюймов (635,0-711,2 мм рт.ст.) до удаления летучих веществ. Конечная масса составила 1367 г, а масса нетто хлорангидрида п-метоксибензилтиоэфирной кислоты - 500,3 г. Дистиллят отбросили.
ПРИМЕР 3
Первоначальное получение гидразида п-метоксибензилтиоэфира (3)
[0043] В соответствии с Уравнением III 5-литровую круглодонную колбу Мортона снабдили термопарой, механической мешалкой, обратным холодильником с отверстием для подвода N2 в верхней части и 0,5-литровой капельной воронкой с компенсатором давления. Хлорангидрид п-метоксибензилтиоэфирной кислоты растворили в 500 мл, 660 г, метиленхлорида. Раствор перенесли в 2-литровую колбу Эрленмейера. 500 мл метиленхлорида добавили для получения раствора общим объемом 1300 мл.
[0044] В 5-литровую круглодонную колбу Мортона поместили 2400 г, 1818 мл, метиленхлорида и 256 г, 245 мл, 7,8 моль 98%-го безводного гидразина. Скорость механической мешалки установили на 255-270 об./мин. Мутную суспензию охладили до температуры от -69 до -72°С с использованием смеси сухой лед/ацетон.
[0045] Раствор хлорангидрида добавили по каплям в 5-литровую колбу с помощью 0,5-литровой капельной воронки с компенсатором давления, поддерживая температуру реакции от -68 до -72°С. Было важно поддерживать скорость добавления раствора хлорангидрида п-метоксибензилтиоэфирной кислоты к перемешиваемой суспензии гидразин/метиленхлорид на уровне, обеспечивающем поддержание температуры реакции ниже -67°С. Добавление заканчивали примерно через 3 часа. Перемешиваемую реакционную смесь выдерживали при температуре от -68 до -72°С как минимум 30 минут. Для проведения ВЭЖХ взяли 1 мл пробу. Раствор нагрели до комнатной температуры (20-30°С), убрав ледяную баню.
[0046] Реакционную смесь перенесли в калиброванную 3-литровую круглодонную колбу. Реакционный раствор концентрировали под вакуумом в роторном испарителе при температуре 32-36°С и давлении 25-28 дюймов (635,0-711,2 мм рт.ст.). Все летучие вещества были удалены. Конечная масса составила 1490,7 г, масса нетто неочищенного твердого гидразида п-метоксибензилтиоэфира - 630 г. Дистиллят отбросили.
[0047] Метанол (1250 г, 1580 мл) добавили к неочищенному твердому гидразиду п-метоксибензилтиоэфира, и гетерогенную смесь перемешивали в 5-литровой круглодонной колбе при 33-36°С как минимум 5 минут до образования прозрачного раствора. Раствор неочищенного гидразида п-метоксибензилтиоэфира/метанола перенесли в 5-литровую реакционную колбу.
[0048] 1312 г 4%-го раствора гидроксида натрия в метаноле добавляли в указанную 5-литровую реакционную колбу при 28-34°С в течение 8 минут. Прозрачную смесь перемешивали при 33-36°С в течение 20 минут. Образовался рыхлый осадок.
[0049] Содержимое 5-литровой реакционной колбы отфильтровали через 30-см воронку Бюхнера с фильтровальной бумагой (Whatman #1). (5-литровую) реакционную колбу промыли 200 мл, 158 г, метанола. Фильтрат перенесли в калиброванную 3-литровую круглодонную колбу. Реакционный раствор концентрировали под вакуумом в роторном испарителе при температуре 36-40°С и давлении 25-28 дюймов (635,0-711,2 мм рт.ст.). Все летучие вещества были удалены. Конечная масса составила 1484 г, масса нетто необработанного твердого гидразида п-метоксибензилтиоэфира - 622.7 г. Дистиллят отбросили. Твердое вещество растворили в 700 г, 530 мл, метиленхлорида. Гетерогенную смесь перемешивали в роторном испарителе (без вакуума) при 33-36°С как минимум 10 минут до образования прозрачного раствора.
[0050] Раствор концентрировали под вакуумом в роторном испарителе при температуре 36-40°С и давлении 25-28 дюймов (635,0-711,2 мм рт.ст.). Все летучие вещества были удалены. Конечная масса составила 1494,2 г, масса нетто необработанного твердого гидразида n-метоксибензилтиоэфира - 632.5 г. Дистиллят отбросили. Твердое вещество растворили в 2100 мл, 2772 г, метиленхлорида. Гетерогенную смесь перемешивали при 20-25°С как минимум 5 минут до образования прозрачного раствора. К раствору метиленхлорида добавили 110 г безводного хлорида магния, смесь перемешивали 1 час. Желтую смесь отфильтровали с использованием вакуум-фильтрования через 15-см воронку Бюхнера с фильтровальной бумагой (Whatman #1) в 5-литровую реакционную колбу. Колбу и осадок на фильтре промыли 500 мл, 660 г, метиленхлорида. Фильтрат перенесли в калиброванную 3-литровую круглодонную колбу. Раствор концентрировали под вакуумом в роторном испарителе при температуре 32-35°С и давлении 20-25 дюймов (508,0-635,0 мм рт.ст.). Все летучие вещества были удалены. Конечная масса составила 1090 г, масса нетто необработанного твердого гидразида п-метоксибензилтиоэфира - 643 г. Дистиллят отбросили.
[0051] Полученное твердое вещество растворили в 400 мл, 528 г, метиленхлорида. Гетерогенную смесь перемешивали при 35-40°С как минимум 5 минут до образования прозрачного желтого раствора. 4260 мл, 3040 г, диэтилового эфира поместили в 12-литровую круглодонную колбу, снабженную термопарой, механической мешалкой, подводом N2 и капельной воронкой с компенсатором давления. Эфир в 12-литровой колбе охладили до температуры от 0 до -10°С, используя насыщенный солевой раствор и лед. Желтый раствор гидразида n-метоксибензилтиоэфира в метиленхлориде (приготовленный выше) добавили с помощью капельной воронки к быстро перемешиваемому (300-400 об./мин) диэтиловому эфиру, поддерживая температуру от 0 до -10°С. Добавили 1070 мл, 732 г, гептана. Гетерогенную смесь перемешивали при температуре от 0 до -5°С в течение 20 минут. Содержимое (12-литровой) реакционной колбы отфильтровали через 30-см воронку Бюхнера с фильтровальной бумагой (Whatman #1). Фильтрат собрали в 4-литровую колбу для вакуум-фильтрования. 5-литровую колбу промыли 1070 мл, 732 г, гептана, раствор добавили к осадку. Фильтрат отбросили. Осадок на фильтре сушили с помощью вакуум-фильтрования как минимум 50 минут, до по существу прекращения сбора фильтрата. Высота осадка на фильтре составляла 15 см. Масса осадка составляла 429 г. Влажный осадок перенесли в эксикатор. Эксикатор с n-метоксибензилтиоэфирной кислотой накрыли чистой фильтровальной бумагой. Продукт сушили в вакуумном сушильном шкафу при 38-40°С и 28-30 дюймов (711,2-762,0 мм рт.ст.) в течение по меньшей мере 18 часов.
[0052] Результаты серии типовых реакций, использовавшихся в Примерах 1-3, объединены в Таблице 2.
ПРИМЕР 4
Модифицированное получение гидразида n-метоксибензилтиоэфира (3)
[0053] Для уменьшения содержания побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира (6), были исследованы параметры реакции, влияющие на образование побочного продукта в выделенном продукте, представляющем собой гидразид n-метоксибензилтиоэфира. Повторили методику из Примера 3. При -78°С раствор гидразин/CH2Cl2 является неперемешиваемой замороженной смесью. Когда вязкую массу поместили в реакционную колбу, лопасть мешалки вращалась в воздухе. Раствор хлорангидрида тиоэфира в CH2Cl2 добавлялся по каплям к замороженной смеси гидразин/CH2Cl2 (28% об./об.), поддерживая температуру примерно -72°С. ВЭЖХ в конце добавления (температура составляла -72°С) показала, что реакция прошла в незначительной степени. Это противоречило ожидаемому протеканию быстрой реакции. Указанное несоответствие могло иметь место из-за отсутствия необходимого перемешивания реакционной смеси. По большей части непрореагировавшую реакционную смесь оставили нагреваться. Когда температура достигла примерно -50°С, образовалась перемешиваемая гетерогенная смесь с последующим быстрым разогревом, что привело к мгновенному повышению температуры до -28°С, при этом цвет реакционной смеси сменился с желтого на белый с желтоватым оттенком. На основании вышеуказанного был принят постулат, что неэффективное перемешивание может привести к локализованной реакции, которая способствует образованию бис-гидразида. Нагревание до комнатной температуры и проведение реакции как в Примере 3 привело к образованию бис-гидразида в качестве основного продукта (82%, ВЭЖХ), что намного выше стандартного уровня нежелательного продукта, составляющего 20%. Был сделан вывод о том, что не было обеспечено эффективное смешивание раствора добавляемого хлорангидрида тиоэфирной кислоты (2) в CH2Cl2 с замороженной массой гидразина.
ПРИМЕР 5
Влияние температуры на получение гидразида п-метоксибензилтиоэфира (3)
[0054] В Примере 5 повторили реакцию из Примера 4, но проводили ее при 0°С, а не при примерно -72°С. Раствор хлорангидрида тиоэфирной кислоты в CH2Cl2 добавляли по каплям к перемешиваемому гомогенному раствору гидразина/CH2Cl2 (28% об./об.). В этом случае образовалось 39% (% площади согласно ВЭЖХ) бис-гидразида. Эти условия позволяют сделать вывод о том, что более низкие температуры и перемешивание являются факторами, влияющими на образование нежелательного побочного продукта. Результаты примеров 3.1 и 3.2 представлены в Таблице 3.
ПРИМЕР 6
Влияние концентрации гидразина на получение гидразида п-метоксибензилтиоэфира (3)
[0055] Затем исследовали влияние использования гидразина в более низкой концентрации при низких температурах. Результаты представлены ниже в Таблице 4. Перемешиваемую гетерогенную смесь гидразин/CH2Cl2 при температуре от -65 до -72°С получали разбавлением гидразина в CH2Cl2 до концентраций 5% и 19% против 28% (об./об.). Эксперименты 4.1 и 4.2 проводили при концентрациях гидразин/CH2Cl2 19% и 5%, соответственно, с образованием перемешиваемой гетерогенной смеси, подвергаемой взаимодействию с хлорангидридом тиоэфирной кислоты с образованием целевого продукта, содержавшего 3% и 5% бис-гидразидного побочного продукта, соответственно. При повторении той же реакции с использованием меньшего количества гидразина (в Эксперименте 4.3 в Таблице 4 использовано 5 против 10 моль-эквивалентов) образовалось только 3% бис-гидразида. Стандартное количество гидразина составляет 5 моль-эквивалентов относительно тиоэфирной кислоты. Удвоение количества гидразина до 10 моль-эквивалентов (Таблица 4: Эксперименты 4.1 и 4.2) незначительно повлияло на уровень бис-гидразида в конечном продукте.
[0056] Добавление хлорангидрида к разбавленной гетерогенной смеси гидразин/СН2Cl2 (5% и 19%) с большей скоростью (Эксперименты 4.4 и 4.5 в Таблице 4, в которых скорость 1, а не 0.25 мл/мин) привело к образованию бис-гидразида в количестве 3% и 9%, соответственно. Резкое уменьшение количества бис-гидразида (с 82% до 3%, Эксперимент 3.1 в Таблице 3 и Эксперимент 4.4 в Таблице 4) может быть приписано одному или более следующим факторам: температура, концентрация, количество гидразина, скорость добавления и перемешивание. Образование в Эксперименте 4.5 в Таблице 4 9% бис-гидразида может быть обусловлено первоначальным неэффективным перемешиванием.
ПРИМЕР 7
Влияние температуры на получение гидразида п-метоксибензилтиоэфира (3)
[0057] В Таблице 5 исследовали влияние температуры на содержание бис-гидразида в конечном продукте. В экспериментах, в которых хлорангидрид тиоэфирной кислоты добавляли к смеси гидразин/CH2Cl2 (19 объемных %) при -20°С и -72°С (Таблица 5: Эксперименты 5.1 и 5.2), бис-гидразид образовывался в количестве 28% и 4%, соответственно. Похожие результаты наблюдали в Экспериментах 5.3 и 5.4 (Таблица 5). Данные результаты показывают, что более низкая температура реакции
(~-70°С) необходима для получения более низкого (3-5%) содержания бис-гидразида.
ПРИМЕР 8
Влияние концентрации гидразина на получение гидразида п-метоксибензилтиоэфира (3)
[0058] Исследование экспериментов в Таблице 4 показало, что при температуре добавления примерно -70°С и концентрации гидразина в CH2Cl2, равной как 19%, так и 5%, получены сопоставимые результаты. Это наблюдение было в дальнейшем проверено в Таблице 6. В Экспериментах 6.1, 6.2 и 6.4 в Таблице 6, в которых добавляли хлорангидрид тиоэфирной кислоты к гетерогенной смеси гидразин/CH2Cl2 с концентрацией 19%, 14% и 10%, был получен бис-гидразид в количестве 6%, 13% и 4%, соответственно. Объем реакционной смеси, размеры колбы и скорость перемешивания сохраняли постоянными. Результаты при 19% концентрации были сопоставимы с результатами при 10% концентрации. Эксперимент с 14% концентрацией смеси гидразин/CH2Cl2 был повторен с расчетом на навеску в 30 г (Эксперимент 6.3 в Таблице 6), при этом полученный целевой продукт содержал только 3% бис-гидразида. Наивысшее содержание бис-гидразида в Эксперименте 6.2 обусловлено быстрым добавлением хлорангидрида, что вызвало скачок температуры реакции до -57°С до того, как ее быстро возвратили к необходимому значению.
ПРИМЕР 9
Влияние скорости перемешивания на получение гидразида п-метоксибензилтиоэфира (3)
[0059]
Было проведено исследование влияния перемешивания (Таблица 7). Эксперименты 7.2 и 7.1 показали, что ускоренное перемешивание (400 об./мин против 200 об./мин) приводит к уменьшению количества бис-гидразида (22% против 40%). Более высокое, чем обычно, содержание бис-гидразида в обоих экспериментах могло быть вызвано неэффективным перемешиванием, учитывая начальное содержание (36 мл) смеси гидразин/CH2Cl2 в 50 мл колбе по сравнению с 20 мл жидкости в 100 мл колбе, как показано в Таблице 8. Это значит, что помимо скорости перемешивания необходимо учитывать форму реакционного сосуда и содержание жидкости.
ПРИМЕР 10
Влияние увеличения масштаба процесса на получение гидразида п-метоксибензилтиоэфира (3)
[0060] Получение гидразида п-метоксибензилтиоэфира давало сопоставимые результаты при использовании смеси гидразин/CH2Cl2 как при 19%, так и при 5% концентрации. Хотя смесь гидразин/CH2Cl2 с концентрацией 19% обычно является перемешиваемой смесью при -70°С, существует риск того, что она превратится в замороженную смесь, с трудом поддающуюся перемешиванию. Если проводят процесс в промышленном масштабе (навеска 400 г) со смесью гидразин/CH2Cl2 с концентрацией 5%, может потребоваться применение реакционного сосуда большей вместимости (20-литровая колба Мортона). Однако если процесс проводят с применением смеси гидразин/CH2Cl2 с концентрацией 14%, реакцию можно проводить в стеклянном реакторе: 5-литровой круглодонной колбе Мортона. Когда процесс проводили с применением смеси гидразин/CH2Cl2 с концентрацией 14% и навески 20 г (Эксперимент 8.1 в Таблице 8), выделенный продукт, представляющий собой гидразид п-метоксибензилтиоэфира, содержал 4,4% (% площади, ВЭЖХ) побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира. Применение указанных условий (смесь гидразин/СН2Сl2 с концентрацией 14%) в более крупном масштабе (Эксперимент 8.2 в Таблице 8) привело к получению гидразида п-метоксибензилтиоэфира, загрязненного 4.2% (% площади, ВЭЖХ) побочного продукта, представляющего собой бис-гидразид метоксибензилтиоэфира.
[0061] В первоначальном способе (Таблица 9, Эксперимент 9.1) получали продукт, представляющий собой гидразид п-метоксибензилтиоэфира, с концентрацией 78,4%, при этом указанный продукт содержал 19,3% побочного продукта, представляющего собой бис-гидразид. В модифицированном способе (Таблица 9, Эксперимент 9.2) использовали непрерывное добавление раствора хлорангидрида метоксибензилтиоэфирной кислоты к более разбавленной и перемешиваемой гетерогенной смеси (с концентрацией 14%) гидразин/метиленхлорид. Получили продукт, представляющий собой гидразид п-метоксибензилтиоэфира, с концентрацией 91.1%, содержащий 4.7% побочного бис-гидразидного продукта.
[0062] Приведенные ниже условия обеспечивают низкое содержание образующегося бис-гидразидного побочного продукта и рекомендуются для крупномасштабного производства гидразида п-метоксибензилтиоэфира. Эти условия реакции рассматривают в качестве аспектов некоторых вариантов реализации настоящего изобретения:
1. Скорость добавления раствора хлорангидрида метоксибензилтиокислоты к смеси гидразин/CH2Cl2 регулировали для поддержания температуры в диапазоне от -68 до -75°С.
2. Эффективное перемешивание (30-40% отношение первоначального объема жидкости к размеру реактора) с высокой скоростью перемешивания (300-400 об./мин). Проведение перемешивания при 260-270 об./мин в 5-литровой колбе Мортона.
3. Перемешиваемая однородная смесь гидразин/CH2Cl2 (концентрация от 5 до 19%, об./об.). В одном примере отношение гидразин/CH2Cl2 составляло 14%, об./об.
4. 30-40% отношение первоначального объема жидкости к размеру реактора.
[0063] Приведенные ниже примеры 11-15 иллюстрируют предпочтительный вариант реализации изобретения.
ПРИМЕР 11
Модифицированное получение промежуточного соединения, представляющего собой п-метоксибензилтиоэфирную кислоту (1)
[0064] Собрали установку, содержавшую 5-литровую реакционную колбу, снабженную холодильником, подводом N2, мешалкой и датчиком/контроллером температуры. Пиперидин (0,402 г) поместили в сосуд в атмосфере N2. 3,3-диметилакриловую кислоту (0,215 г) добавляли порциями при перемешивании, затем добавили п-метоксибензилтиол (0,358 г). Реакционную смесь постепенно нагрели до 82-88°С в течение как минимум 15 минут и поддерживали указанную температуру реакции до появления признаков протекания экзотермической реакции. Температура не должна была превышать 95°С. Когда экзотермическая реакция завершилась, нагревание продолжали при 92-98°С и поддерживали указанную температуру в течение как минимум 15 часов.
[0065] Приготовили 3 литра 3М раствора водной HCl. Убрали колбонагреватель и охладили реакционную смесь до 70-75°С. Медленно добавили 1,9 л раствора HCl. Охлаждение продолжали на водяной бане, пока температура содержимого колбы не составила 20-30°С. Добавили 1,64 кг CH2Cl2, и содержимое колбы перемешивали как минимум 5 минут. рН проверяли и поддерживали на уровне <2 с помощью раствора HCl. Реакционную смесь перенесли в делительную воронку, дождались разделения фаз, и нижний органический слой перелили обратно в реакционную колбу. Верхний водный слой перенесли в отдельную колбу. Оставшийся раствор HCl добавили к органической фазе и перемешивали как минимум 5 минут. рН проверяли и поддерживали на уровне <2 с помощью свежеприготовленного раствора HCl.
[0066] Содержимое реакционной колбы поместили обратно в делительную воронку и оставили слои разделяться как минимум на 5 минут. Нижнюю органическую фазу перелили в чистую колбу Эрленмейера, водный слой перелили в реакционную колбу. Водную фазу с предыдущей экстракции добавили в реакционную колбу вместе с CH2Cl2 (0,400 кг). Реакционную смесь перемешивали как минимум 5 минут, затем содержимое реакционной колбы перенесли в делительную воронку и оставили слои разделяться как минимум на 5 минут. Полученную нижнюю органическую фазу объединили с полученной ранее органической фазой и поместили в реакционную колбу. Объединенные водные фазы отбросили. В реакционную колбу добавили воду (1,00 кг) и перемешивали как минимум 5 минут. Смесь поместили в делительную воронку и оставили слои разделяться как минимум на 5 минут. Нижнюю органическую фазу, содержавшую продукт, перелили в колбу Эрленмейера. Водную фазу отбросили. Органический раствор продукта сушили над безводным MgSO4, затем отфильтровали под разрежением в 5-литровую 4-горлую колбу.
[0067] Колбу Эрленмейера и осадок на фильтре промыли CH2Cl2 (0,350 кг) в 5-литровую 4-горлую колбу. CH2Cl2 отогнали до объема 900±50 мл. Температуру концентрата поддерживали в диапазоне 15-20°С, затем концентрат осадили добавлением гептана (1,67 кг). Смесь охладили до 5°С и перемешивали как минимум 30 минут, затем ее отфильтровали под разрежением, осадок промыли гептаном (2×0,272 кг). Была взята проба продукта для анализа потерь при сушке (LOD), фильтрат проверили на содержание растворенных веществ. Если фильтрат содержал >0,110 кг, то его концентрировали и обрабатывали гептаном для осаждения второй порции продукта, как было указано ранее. Влажный продукт взвешивали и вычисляли массу сухого продукта, используя данные LOD. Масса сухого продукта была эквивалентна константе А (кг). Влажный продукт возвращали в 5-литровую колбу и растворяли в CH2Cl2 (минимум 1,75 А × кг, максимум 2,20 × А кг). Гептан (4,56 × А кг) медленно добавляли, вызывая осаждение. Суспензию охладили до 0-5°С, затем выдерживали в течение минимум на 30 минут, после чего ее отфильтровали под разрежением, а осадок на фильтре промыли гептаном (2×0,272 кг). Фильтрование продолжали до тех пор, пока выделение фильтрата по существу не прекратилось. Влажный осадок перенесли в калиброванную посуду, записали массу, затем фильтраты поместили в соответствующие контейнеры для отходов. Осадок сушили в вакуум-сушильном шкафу при температуре не более 38°С до тех пор, пока потери при сушке не стали составлять <1,0%. Образец отдали на анализ.
ПРИМЕР 12А
Получение хлорангидрида п-метоксибензилтиоэфирной кислоты (2)
[0068] Собрали установку, содержавшую 5-литровую реакционную колбу, снабженную холодильником, водяным скруббером, датчиком температуры, 1-литровой капельной воронкой, подводом N2 и мешалкой. CH2Cl2 (1,6 кг) поместили в колбу в атмосфере N2, затем добавили п-метоксибензилтиоэфирную кислоту (0,400 кг) при перемешивании. В капельной воронке приготовили раствор оксалилхлорида (0,220 кг) и CH2Cl2 (0,600 кг). Примерно половину раствора оксалилхлорида добавили, поддерживая температуру в диапазоне 20-30°С (экзотермично!). В течение как минимум 30-минутного перемешивания наблюдалось выделение СО2/СО, затем добавили оставшийся раствор оксалилхлорида, температуру поддерживали в диапазоне 20-30°С. Реакционную смесь перемешивали до окончания выделения газа (примерно 30 мин), затем смесь нагрели до 33-38°С. Эту температуру поддерживали примерно 60 минут до окончания выделения газа. Взяли навеску реакционной смеси, при помощи ВЭЖХ установили количество оставшейся кислоты. Реакцию считали завершенной, когда оставалось не более 5% исходных веществ. Если реакция была не завершена, перемешивание продолжали при 33-38°С в течение часа, затем вновь брали пробу и проводили анализ. Убрали колбонагреватель и охладили реакционную смесь до 20-30°С. Смесь поместили в 3-литровую одногорлую колбу, затем промыли CH2Cl2. Смесь концентрировали в роторном испарителе до удаления большей части летучих веществ.
ПРИМЕР 12Б
Получение промежуточного соединения, представляющего собой гидразид п-метоксибензилтиоэфира (3)
[0069] Собрали установку, содержавшую 5-литровую 4-горлую реакционную колбу Мортона, снабженную холодильником, подводом N2, термопарой, мешалкой и 2-литровой капельной воронкой. CH2Cl2 (1,6 кг) поместили в колбу в атмосфере N2 и охладили до температуры от -75 до -65°С. Безводный гидразин (0,252 кг) добавляли с получением однородной суспензии ледяного гидразина, при этом образования кристаллов гидразина на стенках колбы не происходило. Раствор хлорангидрида тиоэфирной кислоты поместили в капельную воронку и по мере необходимости разбавили CH2Cl2 для получения раствора объемом 1,3 л.
[0070] Раствор хлорангидрида добавляли по каплям с постоянной скоростью в течение как минимум 3 часов, температуру поддерживали в диапазоне от -65 до -75°С (предпочтительно от -70 до -75°С). После завершения добавления реакционную смесь перемешивали при температуре от -65 до 75°С как минимум 30 минут. Смесь нагрели до 20-25°С. Взяли пробу реакционной смеси для ВЭЖХ, при этом реакцию считали завершенной, когда оставалось не более 5% хлорангидрида. Если к этому моменту реакция не завершилась, перемешивание продолжали при 20-25°С как минимум 1 час, затем пробу отбирали вновь. Смесь из 3-литровой колбы концентрировали в роторном испарителе. В соответствии с требованиями смесь в колбе промывали CH2Cl2. Концентрат разбавили метанолом (1,25 кг) и перенесли в 5-литровую реакционную колбу, снабженную мешалкой, термопарой, подводом N2 и, в случае необходимости, промытую метанолом.
[0071] В атмосфере N2 добавили раствор NaOH (0,0640 кг) в метаноле (1,25 кг) и перемешали в течение как минимум 20 минут. Смесь очистили с помощью вакуум-фильтрования и в соответствии с требованиями промыли ее метанолом. Фильтрат перенесли в калиброванную 3-литровую колбу (в соответствии с требованиями промытую метанолом) и концентрировали в роторном испарителе до удаления летучих соединений, затем продолжили концентрировать под вакуумом в течение как минимум 30 минут. После прекращения применения вакуума добавили CH2Cl2 (0,704 кг). Вращение продолжали до растворения концентрата, затем продолжили применять вакуум и концентрировали смесь до образования твердого осадка. Измерили массу осадка и перенесли осадок в делительную воронку, используя CH2Cl2 (2,84 кг). Смесь перемешали до образования раствора.
[0072] Раствор CH2Cl2 промыли двумя порциями воды (1,00 кг каждая). К раствору CH2Cl2 добавили безводный MgSO4 (0,300-0,420 кг), перемешивание проводили примерно 15 минут до образования прозрачного раствора. Смесь отфильтровали под разрежением и промыли CH2Cl2. Фильтрат перенесли в калиброванную 3-литровую колбу и концентрировали в роторном испарителе до образования твердого осадка. Измерили массу осадка и растворили его в CH2Cl2 (не менее 0,532 кг). Раствор перенесли в 1-литровую капельную воронку, присоединенную к 12-литровой четырехгорлой реакционной колбе, снабженной мешалкой, термопарой и подводом N2. Диэтиловый эфир (3,04 кг) поместили в колбу в атмосфере N2. Эфир охладили до температуры от 0 до -10°С.
[0074] Раствор гидразида п-метоксибензилтиоэфира добавили к растворителю, представляющему собой простой эфир, при перемешивании указанного растворителя с высокой скоростью, поддерживая температуру от -10 до 0°С. Добавляемый раствор перенесли в колбу, используя CH2Cl2 (0,0660 кг). Гептан (0,732 кг) поместили в капельную воронку и медленно добавили к жидкой суспензии, вновь поддерживая тот же температурный интервал. Образовавшуюся суспензию перемешивали при этой же температуре минимум 60 минут. Смесь отфильтровали под разрежением через бумагу. Осадок промыли гептаном (2 порции по 0,366 кг) и сушили с помощью вакуум-фильтрования с образованием отфильтрованного осадка. Отфильтрованный осадок поместили в калиброванную посуду и взвесили. Осадок сушили в вакуум-сушильном шкафу (при температуре не выше 38°С) до тех пор, пока потери при сушке не стали ниже 2,0%. Измерили массу сухого гидразида п-метоксибензилтиоэфира и отдали образцы на анализ.
ПРИМЕР 13
Получение тиол-незащищенного промежуточного соединения (4)
[0075] Анионообменную смолу Dowex SRB ОН приготовили путем добавления 2,4 кг смолы в большую воронку Бюхнера и промывания водой (4 порции по 2,4 кг) и последующим промыванием метанолом (4 порции по 1,92 кг). Смолу заполнили водой в химическом стакане так, чтобы вода ее покрывала, и выдерживали ее как минимум 1 час, затем воду отфильтровали. Смолу переместили в контейнер подходящих размеров. Собрали установку, содержавшую 5-литровую колбу, снабженную мешалкой, термопарой, подводом N2 и 250 мл капельной воронкой. Трифторуксусную кислоту (2,8 кг) поместили в колбу в атмосфере N2 и охладили до 5-10°C. Гидразид тиоэфира (0,380 кг) добавляли порциями (экзотермично!), температуру поддерживали в диапазоне 5-15°С. Затем раствор охладили до 0-5°С.
[0076] Трифторметансульфокислоту (0,243 кг) поместили в капельную воронку и добавили к реакционной смеси, поддерживая температуру 0-10°С. После завершения добавления добавили анизол (0,0152 кг). Реакционную смесь перемешивали при 10-15°С как минимум 2 часа до тех пор, пока цвет реакционной смеси не стал темно-красным и больше не менялся. Из смеси отобрали пробу, которую проанализировали методом ТСХ, при этом реакцию считали законченной, если в реакционной смеси содержалось не более 4% исходных веществ. Собрали установку, содержавшую 12-литровую реакционную колбу, снабженную мешалкой, подводом N2 и 2-литровой капельной воронкой. В колбу внесли метанол (3,01 кг). Сосуд охладили до 0-5°С в атмосфере азота. Реакционную смесь перенесли в капельную воронку и затем добавили к охлажденному метанолу с низкой скоростью, поддерживая температуру реакции 0-5°С. Образовался белый осадок. Реакционную колбу промыли дополнительным количеством метанола (0,0790 кг), раствор поместили в капельную воронку. Белую суспензию перемешивали примерно 15 минут при 0-5°С. Смесь отфильтровали под разрежением через бумагу, осадок промыли метанолом (две порции по 0,600 кг). После по существу прекращения выделения фильтрата его поместили в калиброванную 3-литровую колбу (в соответствии с требованиями промытую метанолом) и концентрировали в роторном испарителе до образования полутвердого осадка.
[0077] Осадок перерастворили в метаноле (0,600 кг) и концентрировали снова так же, как и ранее. Осадок перерастворили в CH2Cl2 (0,600 кг) и концентрировали снова так же, как и ранее, измерили массу полученного осадка. Осадок растворили в воде (1,52 кг), и раствор перенесли в 6-литровую делительную воронку. Осадок промыли CH2Cl2 (три порции по 0,927 кг). Органические фазы перенесли в соответствующие контейнеры для отходов. Водную фазу, содержащую продукт, перенесли в химический стакан и довели рН до 6.5-7.5 единиц рН, добавляя набухшую смолу. В случае необходимости рН регулировали трифторуксусной кислотой. После достижения целевого интервала рН суспензию перемешивали примерно 30 минут, рН проверяли снова и, в случае необходимости, доводили до необходимого значения. Смесь отфильтровали под разрежением, смолу промыли водой (2 порции по 0,600 кг). Затем добавили 37% раствор HCl (0,160 кг). Измерили рН и убедились, что значение было <1.5. В случае необходимости добавляли дополнительное количество HCl. Водный раствор перенесли в калиброванную 3-литровую колбу (в соответствии с требованиями промытую водой) и концентрировали в роторном испарителе до образования твердого осадка. Измерили массу осадка. Осадок растворили в абсолютном этаноле (1,20 кг) и концентрировали снова. Осадок перерастворили в абсолютном этаноле (1,65-2,85 кг) и нагрели до 50-65°С. Теплый раствор отфильтровали под разрежением. Фильтрат перенесли в калиброванную 3-литровую колбу, этанольный раствор концентрировали в роторном испарителе до по существу прекращения отгонки. Добавили порциями этилацетат (8,21 кг) и концентрировали так же, как и ранее. Добавили четвертую порцию этилацетата (2,74 кг) и охладили до 20-25°С. Реакционную смесь перемешивали примерно 15 минут. Смесь отфильтровали под разрежением и промыли этилацетатом (2 порции по 0,135 кг). Вакуум-фильтрование продолжали до по существу прекращения выделения фильтрата. Осадок сушили под разрежением примерно 60 минут. Фильтрат отбросили.
[0078] Измерили массу влажного осадка и использовали значение этой массы (в кг) в последующих вычислениях в качестве константы В. Продукт перенесли в 12-литровую реакционную колбу, снабженную холодильником, мешалкой, подводом N2 и термопарой. Добавили этилацетат (45,1 × В кг) в атмосфере азота, суспензию нагрели до 48-53°С при перемешивании. Нагревание прекратили при достижении 50°С. Колбонагреватель убрали, суспензию охладили до 20-25°С. Смесь отфильтровали под разрежением и промыли этилацетатом (2 порции по 0,270 кг). Вакуум-фильтрование продолжали до по существу прекращения выделения фильтрата. Осадок сушили в вакуум-сушильном шкафу при температуре не выше 38°С в течение как минимум 12 часов. Измерили массу осадка и использовали эту массу (в кг) в дальнейших вычислениях.
ПРИМЕР 14
Получение свободного основания гидразида (5)
[0079] Полученный неочищенный продукт, представляющий собой гидрохлорид, смешали с водой (20,0 × С кг) в 12-литровой 4-горлой колбе и перемешивали до образования раствора. рН раствора регулировали при помощи смолы в диапазоне 6,5-7,5 единиц рН. Смесь перемешивали примерно 15 минут, затем рН вновь проверяли и, в случае необходимости, доводили до значения 6,5-7,5 единиц рН. Смесь отфильтровали под разрежением и промыли водой (3,00 × С кг), затем абсолютным этанолом (2 порции по 5,30 × С кг). Вакуум-фильтрование продолжали до по существу окончания выделения фильтрата. Фильтрат переносили в калиброванную 3-литровую колбу и, в случае необходимости, промывали абсолютным этанолом. Смесь концентрировали в роторном испарителе до по существу окончания отгонки. Полученный осадок перерастворили в абсолютном этаноле (1,58 × С кг) и концентрировали так же, как и ранее. Полученный осадок перерастворили в безводном диэтиловом эфире (2,57 × С кг) и концентрировали так же, как и ранее. Как только отгонка прекратилась, осушение продолжали в глубоком вакууме и выпаривание проводили как минимум 2 часа. Массу нетто осадка приняли за константу D. Используя CH2Cl2 (3,98 × D кг), концентрат перенесли в 12-литровую колбу, снабженную перемешивающим устройством, датчиком температуры и подводом азота. Смесь перемешивали при 15-30°С как минимум 30 минут. Смесь отфильтровали, собирая фильтраты в другую 12-литровую колбу. Первую колбу промыли CH2Cl2 (3,98 × D кг), раствор поместили во вторую колбу, пропустив через фильтр. Вторая колба, содержащая смесь, была снабжена перемешивающим устройством, датчиком температуры и подводом азота. При перемешивании добавляли силикагель (0,700 × D кг), перемешивание продолжали примерно 30 минут. Смесь отфильтровали под разрежением, силикагель промывали CH2Cl2 (2 порции по 3,98 × D кг), собирая полученные фильтраты в 10-литровую колбу для вакуум-фильтрования. Смесь концентрировали в роторном испарителе в 1-литровую колбу при температуре примерно 30°С. В соответствии с требованиями 10-литровую колбу промывали CH2Cl2, раствор помещали в испаритель. Отгонку проводили до по существу полного ее прекращения. Роторный испаритель переключили в режим глубокого вакуумирования и продолжили выпаривание в течение 3 часов при 35-40°С.
[0080] Маслообразный продукт кристаллизовали, регулируя температуру бани роторного испарителя в диапазоне 0-5°С и вращая колбу с максимальной скоростью. Выпаривание продолжали примерно 30 минут после затвердевания продукта. Взяли навеску продукта (основания) и исследовали на остаточный метиленхлорид, продукт сушили до приемлемого результата теста на содержание остаточного растворителя. Записали конечную массу, продукт поместили в бутылки из желтого стекла с крышками, содержащими вкладыши из инертного материала, отобрали образцы для анализа.
ВЫХОД
[0081] Конечный выход 5-стадийного способа составлял не менее 33% от теоретического значения (0,105 кг), при этом разница между наибольшим и наименьшим выходами составляла не более 15%. Предельным значением для известного ранее способа было 33-43% от теоретического значения; тем не менее, ожидается, что уменьшение образования побочного продукта, вызванное увеличением количества метиленхлорида в ходе реакции образования гидразида, приведет к увеличению выхода. Фактические выходы проверочных экспериментов будут использоваться для определения диапазона значений выхода для промышленных партий.
ПРИМЕР 15
Конечная очистка гидразида 3-метил-3-меркаптобутановой кислоты, Cl 332258 (DMH-линкера)
[0082] Метиленхлорид (1000 мл, 1325 г) поместили в 2-литровую 4-горлую колбу, снабженную механической мешалкой, подводом N2, обратным холодильником и устройством для контроля температуры. DMH-линкер (20 г) поместили в реакционную колбу. Суспензию перемешивали в атмосфере N2, при 20±3°С как минимум 30 минут. Получившийся мутный раствор DMH-линкера отфильтровали через 350-мл воронку Бюхнера со среднепористым фильтром из пористого стекла. Фильтрат собрали в чистую 2-литровую 4-горлую круглодонную колбу, снабженную механической мешалкой, подводом N2, обратным холодильником и устройством для контроля температуры. Реакционную колбу промыли 20 мл, 26,5 г метиленхлорида, раствор перелили в чистую реакционную колбу. Силикагель (20 г) поместили в раствор в реакционной колбе, поддерживая температуру 15-25°С. Суспензию перемешивали в атмосфере N2 при 20±3°С как минимум 30 минут. Гетерогенную смесь отфильтровали через (350 мл) воронку Бюхнера со среднепористым фильтром из пористого стекла. Фильтрат собрали в чистую 2-литровую одногорлую круглодонную колбу. Реакционную колбу промыли метиленхлоридом (50 мл, 66,3 г), раствор перелили к осадку на фильтре, фильтрат собрали в одногорлую колбу. Фильтрат концентрировали до получения сухого остатка, используя роторный испаритель (температура бани 35±5°С) и водоструйный насос (15-30 мм рт.ст.), а затем глубокий вакуум (7-10 мм рт.ст.). Образовавшееся белое твердое вещество охладили до 0-5°С и сушили в глубоком вакууме при 7 мм рт.ст. в течение 2 часов. н-Гептан (100 мл, 68,4 г) добавили к твердому веществу и перемешивали при комнатной температуре по меньшей мере 10 минут до образования однородной суспензии.
[0083] Продукт выделили с помощью вакуум-фильтрования через фильтровальную бумагу (#1 Whatman) на 15-см воронке Бюхнера. 2-литровую колбу промыли остаточным раствором н-гептана, раствор перенесли к осадку на фильтре, затем колбу промыли н-гептаном (2 порции по 50 мл, 34,2 г). Осадок сушили под разрежением при комнатной температуре в течение как минимум 5 минут. Осадок перенесли в бутыль из желтого стекла, помещенную в вакуумный эксикатор. Влажный осадок сушили с 14,34 г до постоянной массы под вакуумом (<10 мм рт.ст.) при 20-25°С в течение 3 часов. Выход: 14,21 г, 75,9%, теоретический выход: 18,7 г.

Изобретение относится к усовершенствованным способам получения гидразидного продукта из гидразина и хлорангидрида, в частности к способу получения гидразида 3-метил-3-меркаптобутановой кислоты. Способ включает стадии: (а) получения перемешиваемой по существу однородной суспензии, содержащей гидразин и инертный растворитель; и (b) непрерывного добавления хлорангидрида к указанной суспензии, при этом хлорангидрид имеет структуру:
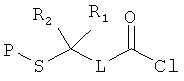
где Р представляет собой тиолзащитную группу; каждый из R1 и R2 выбран из группы, состоящей из C1-С5 алкила; L представляет собой алкиленовый линкер, и при этом непрерывное добавление раствора хлорангидрида регулируют таким образом, чтобы температура реакции поддерживалась в интервале от -65°С до -75°С. Предложенный способ устраняет или ограничивает образование нежелательных бис-гидразидных побочных продуктов. Изобретение также относится к способу получения суспензии гидразина, включающему стадии: (а) охлаждения инертного растворителя до температуры в пределах от -68°С до -75°С; и (b) добавления гидразина по каплям к указанному холодному инертному растворителю; и к способу получения иммуноконъюгата члена семейства калихимицинов с моноклональным антителом в качестве носителя, включающему получение моноацилированного гидразида. 5 н. и 26 з.п. ф-лы, 3 ил., 15 пр., 9 табл.
1. Способ получения гидразидного продукта из гидразина и хлорангидрида, включающий стадии:
(a) получения перемешиваемой по существу однородной суспензии, содержащей гидразин и инертный растворитель; и
(b) непрерывного добавления хлорангидрида к указанной суспензии, при этом хлорангидрид имеет структуру:
где Р представляет собой тиолзащитную группу;
каждый из R1 и R2 выбран из группы, состоящей из C1-С5 алкила;
L представляет собой алкиленовый линкер,
и при этом непрерывное добавление раствора хлорангидрида регулируют таким образом, чтобы температура реакции поддерживалась в интервале от -65°С до -75°С.
2. Способ по п.1, отличающийся тем, что указанный хлорангидрид добавляют к суспензии по каплям.
3. Способ по п.1, отличающийся тем, что указанный хлорангидрид содержит бензилтиоэфирную группу.
4. Способ по п.1, отличающийся тем, что L представляет собой -СН2-.
5. Способ по п.1, отличающийся тем, что каждый из R1 и R2 независимо представляет собой метил.
6. Способ по п.1, отличающийся тем, что Р представляет собой бензильную группу, возможно замещенную по фенильному кольцу.
7. Способ по п.6, отличающийся тем, что Р представляет собой n-метоксибензильную группу.
8. Способ по п.п.1 или 2, отличающийся тем, что указанный хлорангидрид имеет структуру:
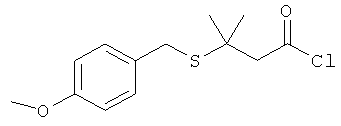
9. Способ по любому из пп.1-7, отличающийся тем, что указанный инертный растворитель представляет собой метиленхлорид.
10. Способ по любому из пп.1-7, отличающийся тем, что указанное гидразидное соединение имеет структуру:
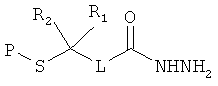
где Р представляет собой тиолзащитную группу;
каждый из R1 и R2 выбран из группы, состоящей из С1-С5алкила;
L представляет собой алкиленовый линкер.
11. Способ по п.10, отличающийся тем, что L представляет собой -CH2-.
12. Способ по п.10, отличающийся тем, что каждый из R1 и R2 независимо представляет собой метил.
13. Способ по п.10, отличающийся тем, что Р представляет собой бензильную группу, возможно замещенную по фенильному кольцу.
14. Способ по п.13, отличающийся тем, что Р представляет собой n-метоксибензильную группу.
15. Способ по п.10, отличающийся тем, что целевой гидразид представляет собой 
или соль указанного соединения.
16. Способ по любому из пп.1-7 или 11-14, отличающийся тем, что указанное гидразидное соединение содержит менее 5% бис-гидразидного побочного продукта, имеющего структуру:
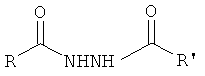
где R и R' представляют собой 
17. Способ по п.16, отличающийся тем, что L представляет собой -СН2-.
18. Способ по п.17, отличающийся тем, что каждый из R1 и R2 представляет собой независимо метил.
19. Способ по п.16, отличающийся тем, что Р представляет собой бензильную группу, возможно замещенную по фенильному кольцу.
20. Способ по п.19, отличающийся тем, что Р представляет собой n-метоксибензильную группу.
21. Способ по п.20, отличающийся тем, что бис-гидразидный побочный продукт имеет структуру:

22. Способ по любому из пп.1-7, 11-15 или 17-21, отличающийся тем, что непрерывное добавление раствора хлорангидрида регулируют для поддержания температуры реакции в диапазоне от примерно -68 до примерно -75°С.
23. Способ получения соединения, содержащего гидразидную линкерую группу, включающий стадии:
(a) охлаждения реакционного сосуда, содержащего перемешиваемый инертный растворитель, до температуры в интервале от -65°С до -75°С;
(b) непрерывного добавления гидразина в указанный реакционный сосуд, приводящее к получению перемешиваемой по существу однородной суспензии, содержащей гидразин и указанный инертный растворитель;
(c) непрерывного добавления хлорангидрида к указанной суспензии, приводящее к образованию гидразидной линкерной группы, при этом хлорангидрид имеет структуру:
где Р представляет собой тиолзащитную группу;
каждый из R1 и R2 выбран из группы, состоящей из C1-C5алкила;
L представляет собой алкиленовый линкер.
24. Способ по п.23, отличающийся тем, что указанный инертный растворитель представляет собой метиленхлорид.
25. Способ получения суспензии гидразина, включающий стадии:
(a) охлаждения инертного растворителя до температуры в пределах от -68°С до -75°С; и
(b) добавления гидразина по каплям к указанному холодному инертному растворителю.
26. Способ по п.25, отличающийся тем, что указанный инертный растворитель представляет собой метиленхлорид.
27. Способ по п.25 или 26, отличающийся тем, что указанную суспензию гидразина перемешивают со скоростью от 270 до 400 об/мин.
28. Способ получения гидразида 3-метил-3-меркаптобутановой кислоты, включающий стадию получения гидразида согласно способу по п.14.
29. Способ получения иммуноконъюгата члена семейства калихимицинов с моноклональным антителом в качестве носителя, включающий получение моноацилированного гидразида, где ацильная группа содержит S-защищенную меркаптогруппу, согласно способу по п.1, удаление указанной защитной группы из S-защищенной меркаптогруппы и применение полученного гидразида для получения указанного иммуноконъюгата.
30. Способ по п.29, отличающийся тем, что указанный полученный гидразид представляет собой гидразид 3-метил-3-меркаптобутановой кислоты.
31. Способ по п.29 или 30, отличающийся тем, что гидразид 3-метил-3-меркаптобутановой кислоты применяют в качестве линкера для получения гемтузумаб озогамицина или инотузумаб озогамицина.
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| US 5635603 A, 03.06.1997 | |||
| HINMAN L M ЕТ AL: "PREPARATION AND CHARACTERIZATION OF MONOCLONAL ANTIBODY CONJUGATES OF THE CALICHEAMICINS: A NOVEL AND POTENT FAMILY OF ANTITUMOR ANTIBIOTICS" CANCER | |||

