Область техники, к которой относится изобретение
Настоящее изобретение относится к способам получения 4-алкокси-3-(ацил или алкил)оксипиколинамидов. Точнее, настоящее изобретение относится к способу получения 4-метокси-3-(ацетил или ацетилоксиметил)оксипиколинамидов из 4-метокси-3-гидроксипиколиновых кислот или 4-метокси-3-ацетилоксипиколиновых кислот.
Уровень техники
В Заявках на Патент США сер. №№ 15/036314 и 15/036316 описываются, среди прочего, некоторые гетероциклические ароматические амиды общей формулы
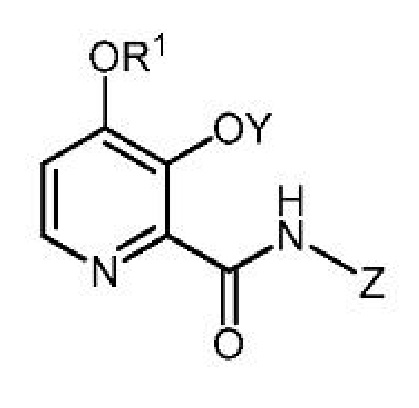
и их применение в качестве фунгицидов. Было бы полезно иметь эффективный и масштабируемый способ получения этих гетероциклических ароматических амидных соединений из недорогого сырья.
Сущность изобретения
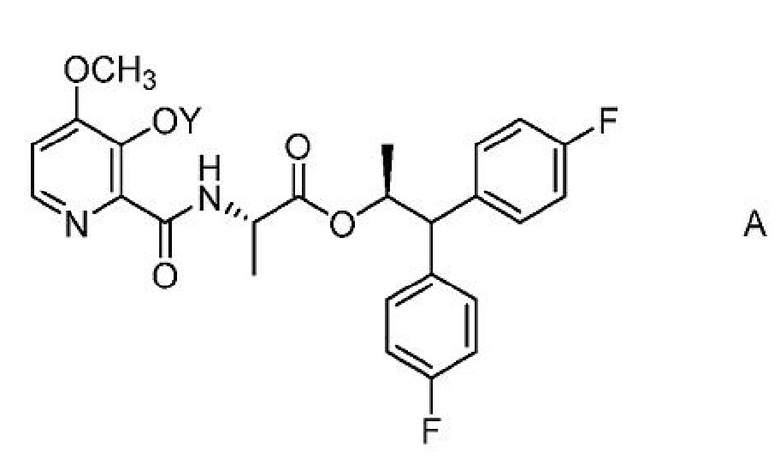
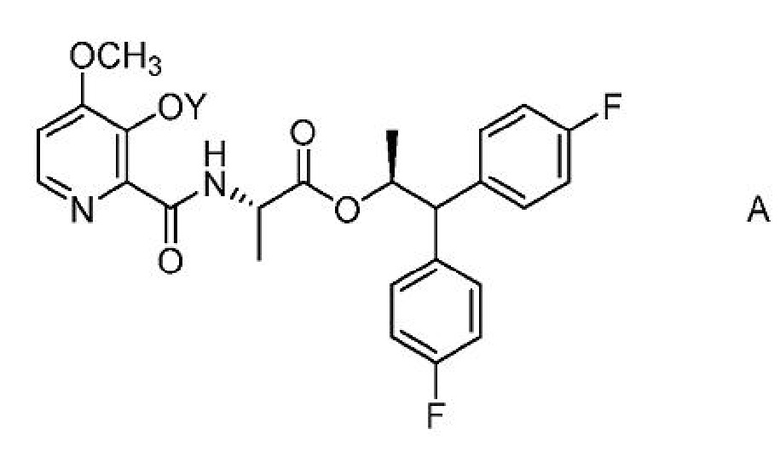
Настоящее изобретение относится к способам получения 4-метокси-3-(ацетил или ацетилоксиметил)оксипиколинамидов формулы A
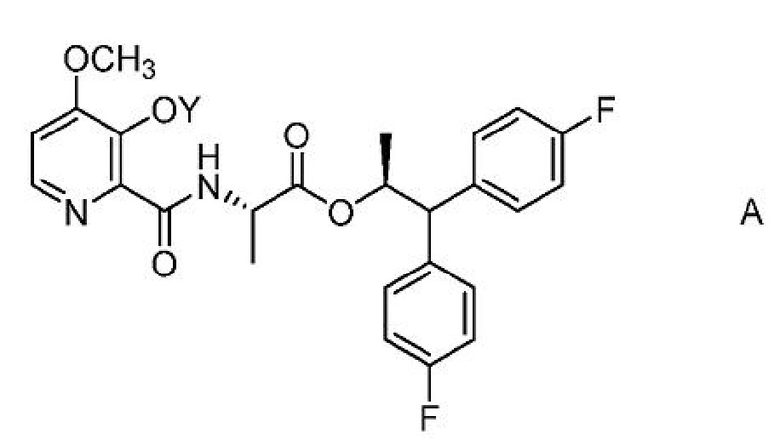
где Y представляет собой СН3СО или CH3COOCH2;
из соединения формулы B или D.
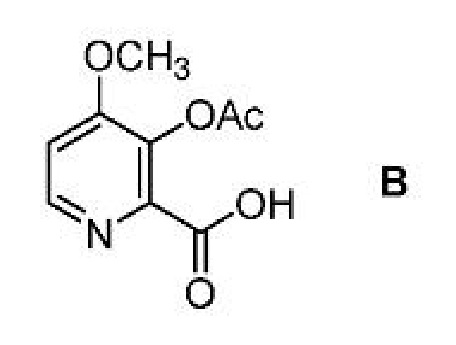
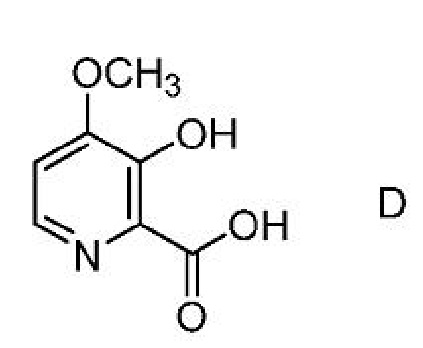
Соединение формулы А, где Y представляет собой СН3СО, может быть получено способом, который включает следующие стадии:
a) получение первой смеси, содержащей соединение формулы B, ацилирующий агент или хлорирующий агент и основание;
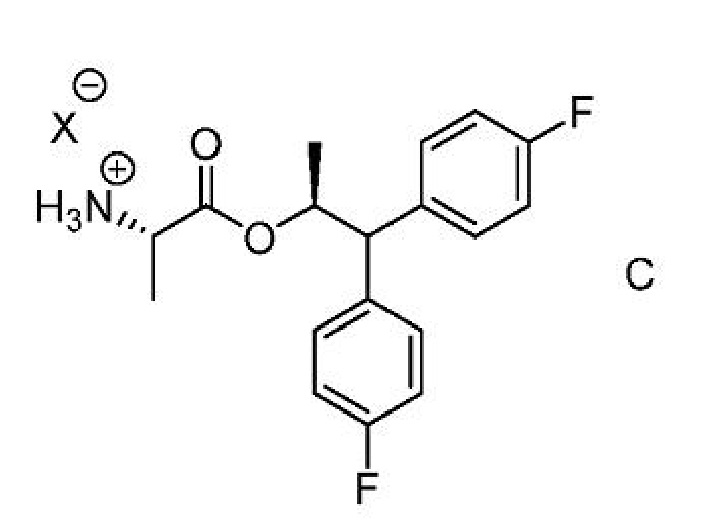
b) добавление соединения формулы C
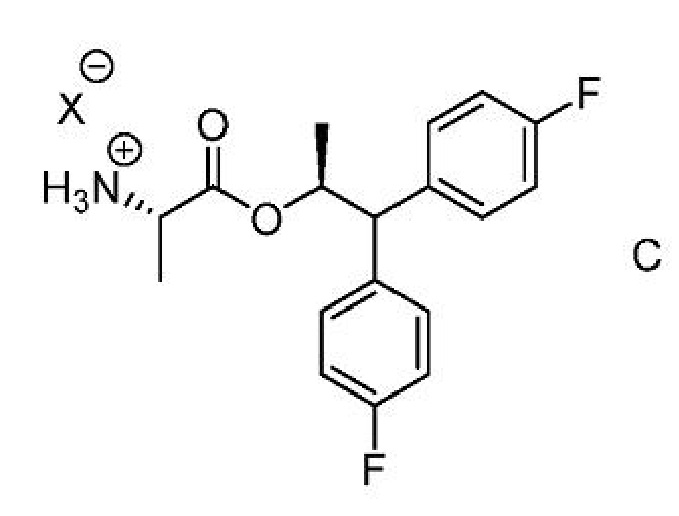
где Х представляет собой Cl, Br, HSO4, Н2PO4 или CH3SO3;
к первой смеси с получением второй смеси; и
с) выделение соединения формулы А из второй смеси, где Y представляет собой ацетил (т.е. СН3СО).
Соединение формулы А, где Y представляет собой СН3СО или CH3COOCH2, может быть получено способом, который включает следующие стадии:
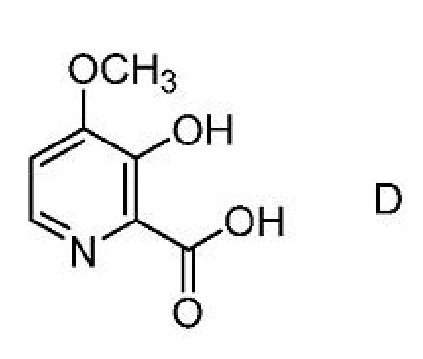
a) получение первой смеси, содержащей соединение формулы D,
ацилирующий агент и основание;
b) добавление соединения формулы C,
где Х представляет собой Cl, Br, HSO4, Н2PO4 или CH3SO3;
к первой смеси с получением второй смеси;
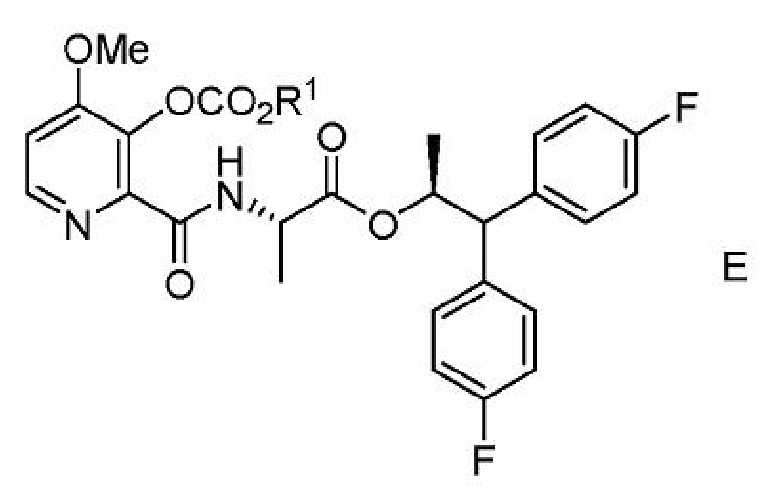
с) выделение соединения формулы Е из второй смеси;
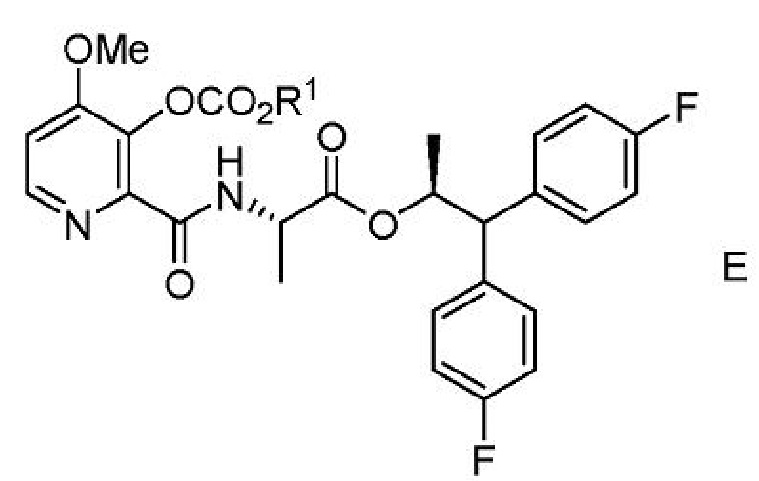
где R1 представляет собой С1-С4 алкил или CH2Ph;
d) получение третьей смеси, содержащей соединение формулы Е, основание щелочного металла и воду;
е) выделение соединения формулы F из третьей смеси;
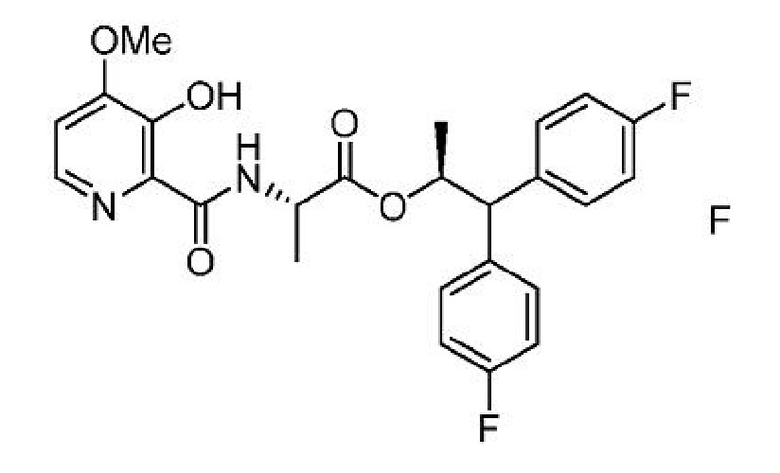
f) получение четвертой смеси, содержащей соединение формулы F, ацетилирующий агент или алкилирующий агент и второе основание; и
g) выделение соединения формулы А из четвертой смеси,
где Y представляет собой СН3СО или CH3COOCH2.
Соединение формулы C,
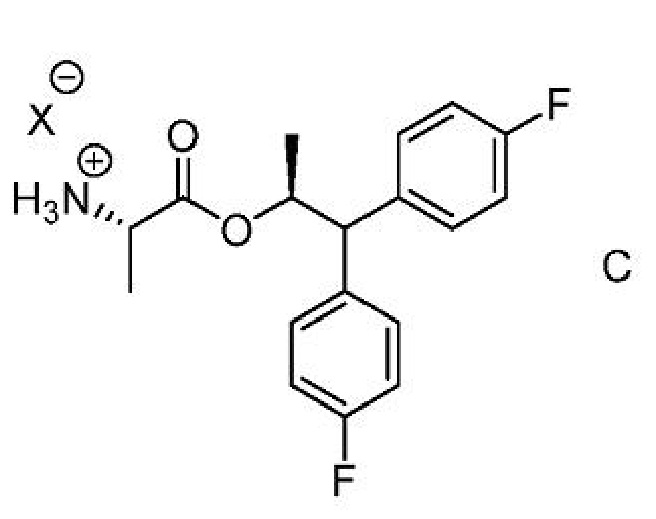
где Х представляет собой Cl, Br, HSO4, Н2PO4 или CH3SO3;
может быть получено способом, который включает следующие стадии:
a) получение первой смеси, содержащей соединение формулы G,
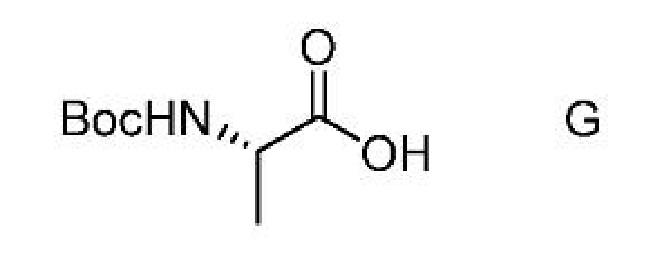
ацилирующий агент и основание;
b) добавление соединения формулы Н
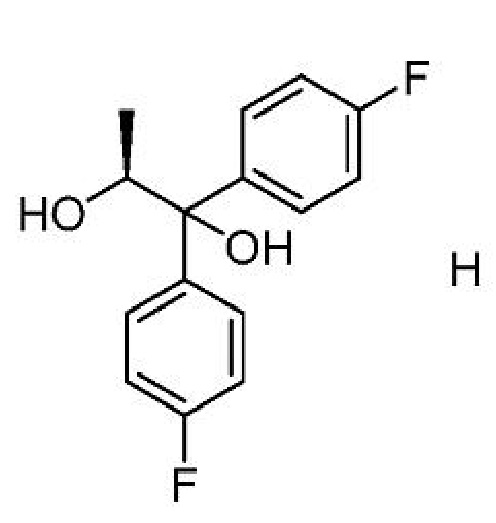
к первой смеси с получением второй смеси;
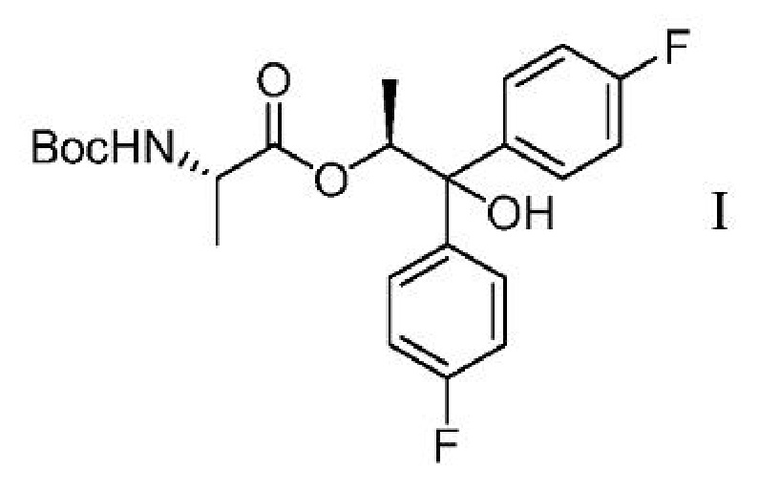
c) выделение соединения формулы I
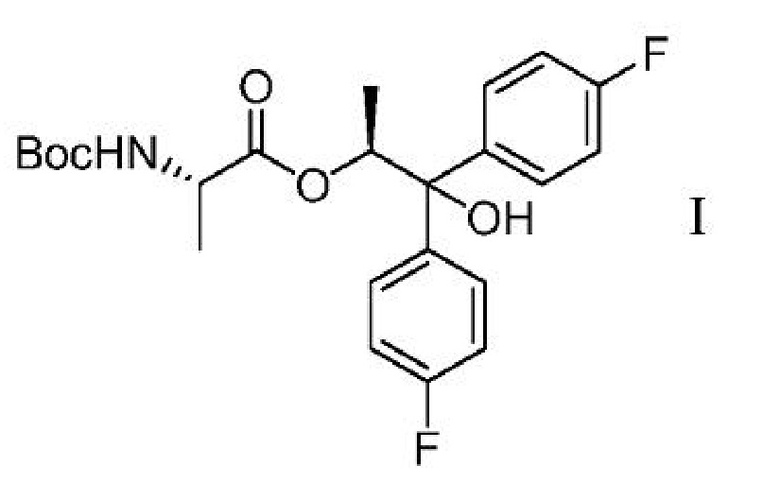
из второй смеси;
d) получение третьей смеси, содержащей соединение формулы I, кислоту и восстановитель;
е) выделение соединения формулы J из третьей смеси;
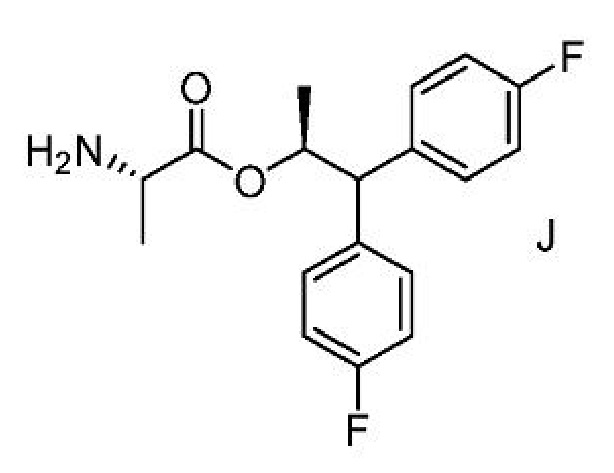
f) получение четвертой смеси, содержащей соединение формулы J и сильную кислоту;
где сильная кислота представляет собой НСl, НВr, Н2SO4, Н3PO4 или CH3SO3H;
и
g) выделение соединения формулы C из четвертой смеси.
Соединение формулы С также может быть получено способом, который включает следующие стадии:
a) получение первой смеси, содержащей соединение формулы H, восстановитель и кислоту;
b) выделение соединения формулы K из первой смеси;
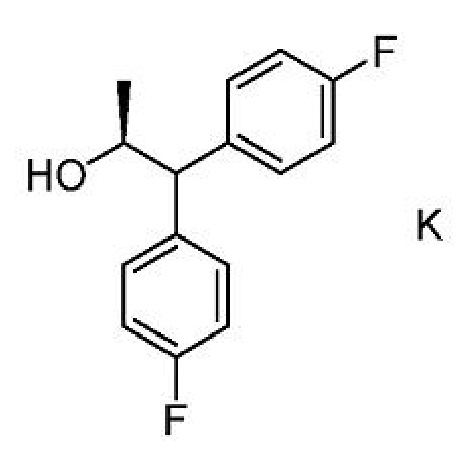
c) получение второй смеси, содержащей соединение формулы G,
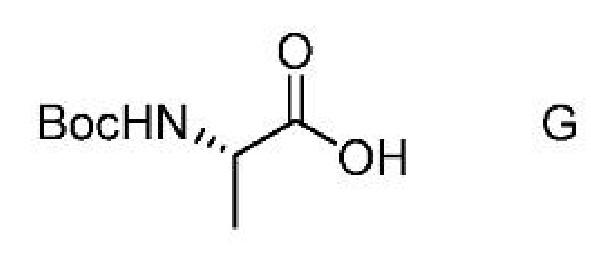
ацилирующий агент и основание;
d) добавление соединения формулы K ко второй смеси с получением третьей смеси;
е) выделение соединения формулы L из третьей смеси;
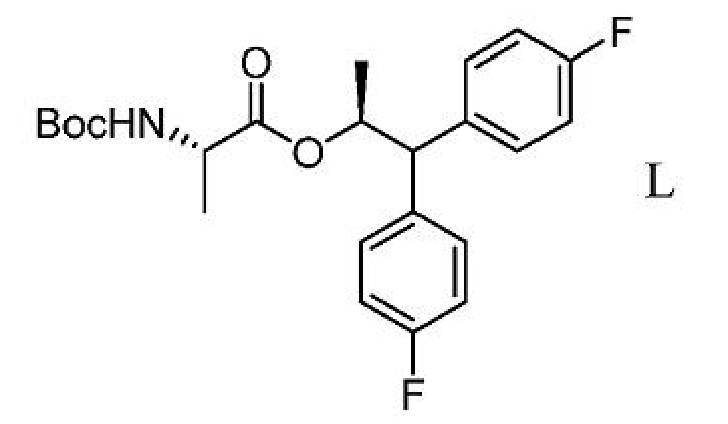
f) получение четвертой смеси, содержащей соединение формулы L и сильную кислоту;
где сильная кислота представляет собой HCl, HBr, H2SO4, H3PO4 или CH3SO3H;
и
g) выделение соединения формулы C из четвертой смеси.
Другой аспект настоящего изобретения относится к новым промежуточным продуктам, полученным в способе по настоящему изобретению, то есть к следующим соединениям:
a)
b)
где R1 представляет собой С1-С4 алкил или PhCH2;
c)
где Х представляет собой Cl, Br, HSO4, Н2PO4 или CH3SO3.
Подробное описание
Термин «алкил» относится к разветвленной, неразветвленной или насыщенной циклической углеродной цепи, включая, но без ограничения, метил, этил, пропил, бутил, изопропил, изобутил, трет-бутил, пентил, гексил, циклопропил, циклобутил, циклопентил, циклогексил и т.п.
Термин «ацил», когда используется в настоящем описании, относится к фрагменту RCO (т.е. RC(O)-), который включен в объем изобретения, где R представляет собой алкил с прямой или разветвленной цепью, содержащий от одного до шести атомов углерода. Конкретные ацильные группы, описанные в настоящем изобретении, включают, например, СН3СО (т.е. ацетильную группу) и CH3COOCH2 (т.е. ацетилоксиметильную группу).
Термины «выделять», «выделяющий» или «выделение», когда используются в настоящем описании, означают частичное или полное удаление или отделение желаемого продукта от других компонентов конечной смеси химического процесса с использованием стандартных методов, таких как, но без ограничения, фильтрование, экстракция, дистилляция, кристаллизация, центрифугирование, растирание, разделение фаз «жидкость-жидкость» или другие способы, известные специалистам в данной области техники. Чистота выделенного продукта может находиться в интервале от <50% до >50%, и выделенный продукт может быть очищен до более высокого уровня чистоты с использованием стандартных методов очистки. Выделенный продукт также может использоваться в последующей стадии способа с очисткой или без нее.
В способах, описанных в настоящем изобретении, пиколинамид формулы A, где Y представляет собой CH3CO, может быть получен связыванием 4-метокси-3-ацетилоксипиколиновой кислоты с 2-аминопропаноатной сложноэфирной частью целевой молекулы. В качестве альтернативы, пиколинамиды формулы A, где Y представляет собой СН3СО или CH3COOCH2, могут быть получены способом, в котором в описанной реакции сочетания используется 4-метокси-3-гидроксипиколиновая кислота с последующим введением группы Y в конечной стадии последовательности способа.
где Y представляет СН3СО или CH3COOCH2.
A. Получение соединения формулы А
Соединение формулы А, где Y представляет СН3СО, может быть получено непосредственно из соединения формулы B способом, показанным на схеме I. Пиколиновая кислота B сначала подвергается активации для реакции сочетания превращением ее в (а) соответствующий смешанный ангидрид с использованием алкил- или бензилхлорформиата или хлорангидрида кислоты и основания или в (b) хлорангидрид кислоты с использованием оксалилхлорида или тионилхлорида и основания. Полученное производное пиколиновой кислоты B в форме смешанного ангидрида или хлорангидрида кислоты может подвергаться обработке аминной солью формулы С, где Х представляет собой Cl, Br, HSO4, H2PO4 или CH3SO3, с получением целевого пиколинамида формулы A (Y представляет собой СН3СО). Соединение формулы А может быть выделено с использованием стандартных методов выделения и очистки. Подходящие растворители для этого способа могут включать дихлорметан (ДХМ), 1,2-дихлорэтан (ДХЭ), изопропилацетат, тетрагидрофуран (ТГФ), 2-MeТГФ и ацетонитрил (ACN).
Схема I
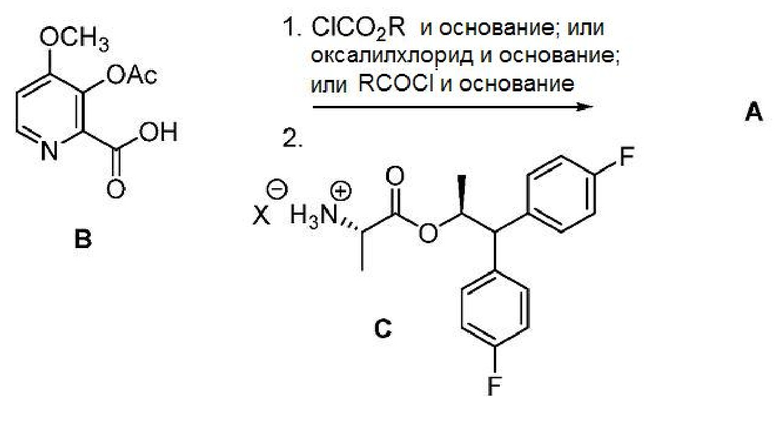
Подходящие хлорформиатные сложные эфиры (т.е. ClCO2R) для использования в данном способе могут включать хлорформиатные сложные эфиры, в которых R представляет собой C1-C4 алкил или бензил. Подходящие хлорангидриды кислот (т.е. RCOCl) для использования в данном способе могут включать хлорангидриды, в которых R представляет собой C1-C4 алкил. Подходящие основания для использования в способе могут включать одно или несколько оснований, выбранных из триэтиламина (TEA), диизопропилэтиламина (DIPEA), пиридина и карбоната калия. В данном способе может использоваться по меньшей мере один, по меньшей мере 2 или по меньшей мере 3 молярных эквивалента основания.
В одном варианте осуществления способ, показанный на схеме I, может осуществляться с использованием сульфирующего соединения вместо алкил- или бензилхлорформата или хлорангидрида кислоты с получением соединения формулы A. В таком способе соединение формулы B подвергается контактированию с сульфирующим соединением формулы B1, где R1 представляет собой C1-C4 алкил, и Z представляет собой Cl или Br, и основанием с получением смешанного ангидрида сульфоновой-карбоновой кислоты формулы В2. После этого ангидрид В2 может подвергаться обработке аминной солью формулы C, где Х представляет собой Cl, Br, HSO4, H2PO4 или CH3SO3, или амином формулы J с получением целевого пиколинамида формулы A (где Y представляет собой СН3СО).
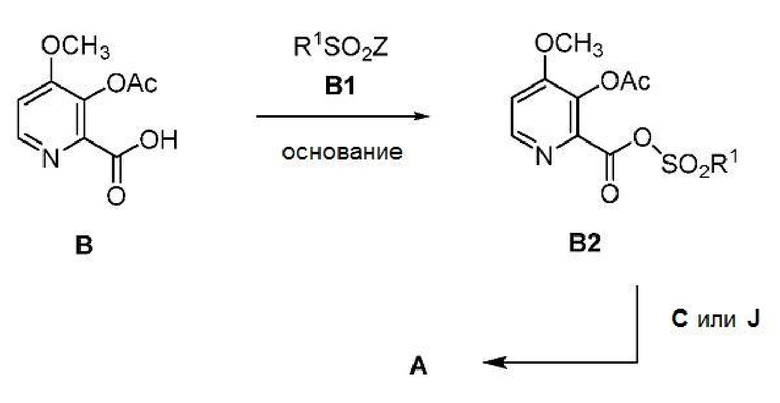
Соединение формулы А также может быть получено из соединения формулы D способом, показанном на схеме II. Пиколиновая кислота D сначала подвергается превращению в соединение формулы D1, которое не выделяют, с использованием по меньшей мере примерно 2 эквивалентов алкил- или бензилхлорформиата формулы ClCO2R, где R представляет собой C1-C4 алкил или бензил, и по меньшей мере примерно 3 эквивалентов основания. Реакционная смесь, содержащая соединение D1, после этого может объединяться с соединением формулы С с получением пиколинамида E. В этих реакциях могут использоваться подходящие основания, такие как, например, TEA, DIPEA или аналогичные триалкиламинные основания. Обработка соединения Е основанием щелочного металла, таким как LiOH, NaOH, KOH или их смеси, в присутствии воды и, необязательно, сорастворителя, такого как, например, тетрагидрофуран (ТГФ), 2- метилтетрагидрофуран (2-MeТГФ), ДМЭ, диоксан, ACN или C1-C4 алкиловый спирт, может приводить к получению соединения формулы F. Ацетилирование соединения F уксусным ангидридом, ацетилхлоридом или другими ацетилирующими агентами, обычно используемыми в данной области техники, и с использованием основания или в условиях реакции Шоттена-Баумана (Schotten-Baumann) может приводить к получению соединения формулы А, где Y представляет СН3СО. Алкилирование соединения F с помощью СН3COOCH2Br и основания может приводить к получению соединения формулы А, где Y представляет собой CH3COOCH2. Основания, используемые в этих реакциях, могут представлять собой по меньшей мере одно основание, выбранное из пиридина, TEA и DIPEA.
Схема II
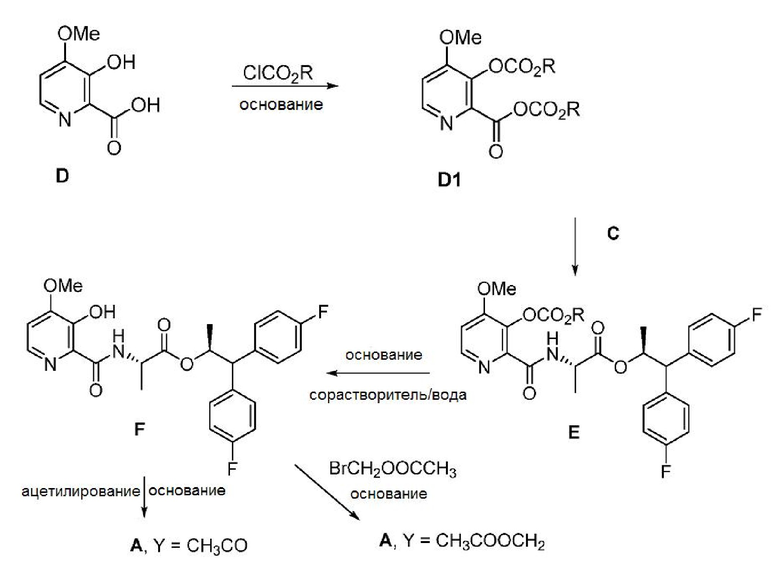
Соединения формул A, E и F могут быть выделены с использованием стандартных методов выделения и очистки.
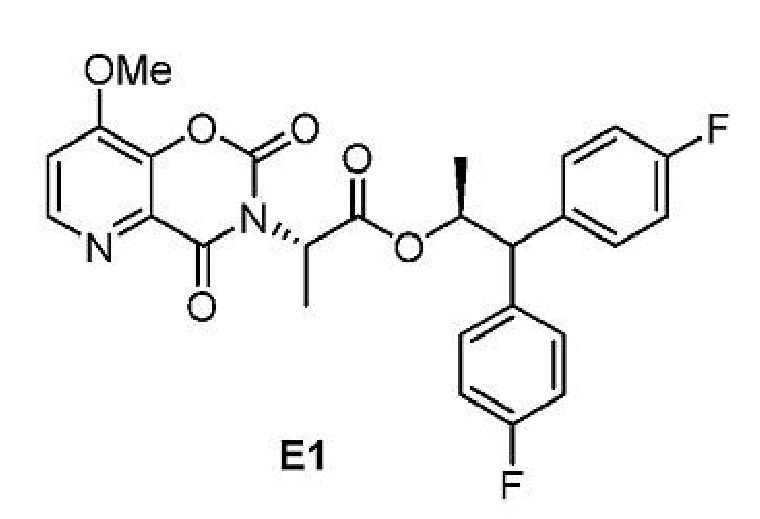
В реакции гидролиза, в результате которой соединение Е подвергается превращению в соединение F, как показано на схеме II, соединение формулы E1 при определенных условиях может быть выделено в качестве промежуточного продукта этой реакции.
В некоторых вариантах осуществления пиколинамид формулы А, где Y представляет собой СН3СО или CH3COOCH2, может быть получен способом с использованием амина формулы J вместо аминной соли формулы С.
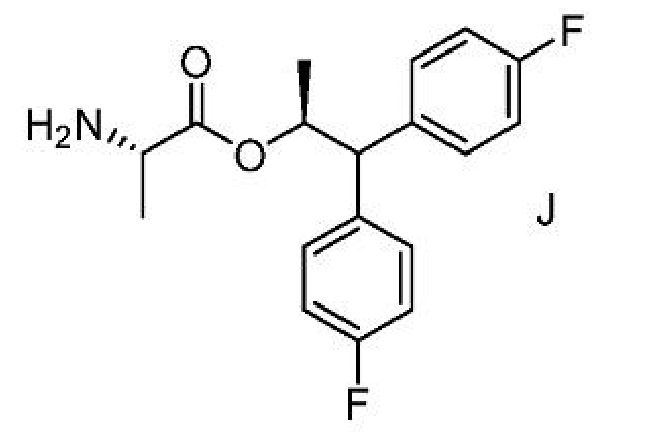
B. Получение соединения формулы C
Соединение формулы С может быть получено двумя различными способами, исходя из диола формулы H. В первом из двух этих способов, показанных на схеме III, диол Н может подвергаться связыванию с BOC-L-аланиновым соединением формулы G с получением соединения формулы I. Реакция сочетания может осуществляться с использованием смешанного ангидридного производного соединения G, которое может быть получено обработкой соединения G хлорангидридом кислоты формулы RCOCl, где R представляет собой C1-C4 алкил, основанием и DMAP (4-(диметиламино)пиридином). Подходящие для этой реакции растворители могут включать один или несколько растворителей, выбранных из ДХМ, ДХЭ, ТГФ, 2-MeТГФ и ACN, и подходящие основания могут включать одно или несколько оснований, выбранных из TEA, DIPEA и пиридина.
Схема III
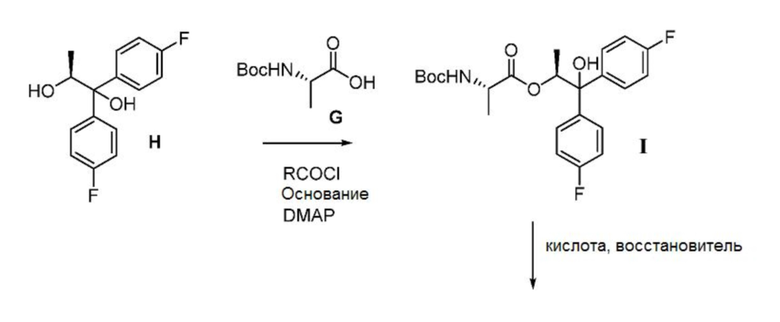
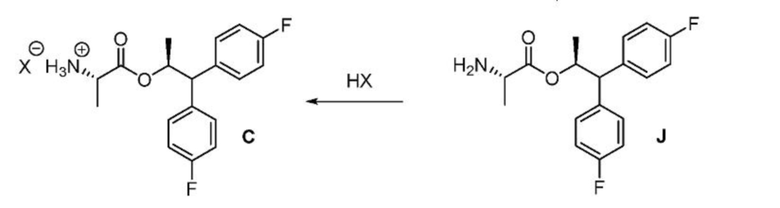
На второй стадии способа, показанного на схеме III, третичная гидроксильная группа и группа ВОС в соединении формулы I удаляются с использованием восстановителя в сочетании с кислотой. Подходящие для этого превращения восстановители могут включать борогидридные реагенты, такие как, но без ограничения, борогидрид натрия и триацетоксиборгидрид натрия, и кремнийорганические гидриды, такие как, например, триэтилсилан, поли(метилгидросилоксан) (ПМГС) и 1,1,3,3-тетраметилдисилоксан (ТМДС). Подходящие кислоты для использования с восстановителями могут включать, но без ограничения, трифторуксусную кислоту и метансульфоновую кислоту. Наконец, соединение J может подвергаться превращению в аминную соль формулы C обработкой сильной кислотой НХ в безводных условиях, где НХ может быть выбрана из НСl, HBr, H2SO4, H3PO4 или CH3SO3H.
Второй способ получения соединения формулы C показан на схеме IV. Диол формулы H может подвергаться обработке кислотой и восстановителем с получением спирта формулы K. Подходящие восстановители для этого превращения включают кремнийорганические гидриды, такие как, например, триэтилсилан, поли(метилгидросилоксан) (ПМГС) и 1,1,3,3-тетраметилдисилоксан (ТМДС), и борогидридные реагенты, такие как, но без ограничения, борогидрид натрия и триацетоксиборогидрид натрия. Подходящие кислоты для использования с восстановителями могут включать, но без ограничения, трифторуксусную кислоту и метансульфоновую кислоту. После этого спирт формулы K может подвергаться реакции сочетания с ВОС-L-аланиновым соединением формулы G для получения соединения формулы L с использованием реагентов и условий, описанных в настоящем описании для получения соединения формулы I в схеме III. Наконец, соединение L может подвергаться превращению в аминную соль формулы C при обработке сильной кислотой HX
Схема IV
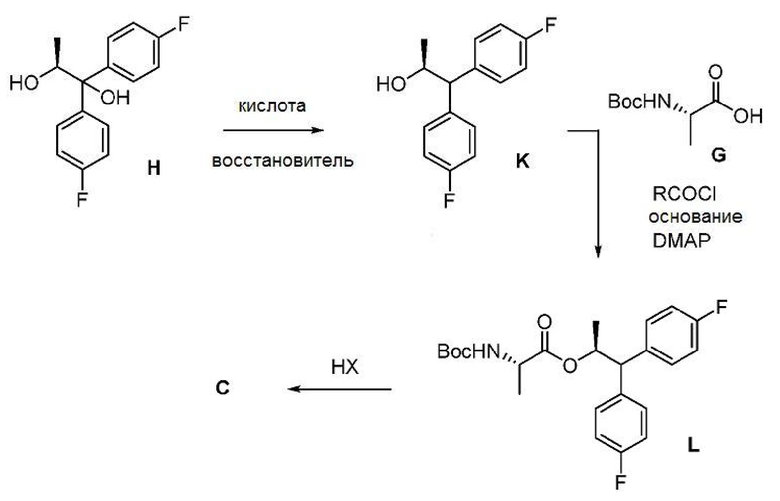
в безводных условиях, где НХ может быть выбрана из HCl, HBr, H2SO4, H3PO4 или CH3SO3H.
C. Получение соединения формулы Н
Диол формулы H может быть получен из (4-фторфенил)магнийбромида и (S)-этиллактата, как описано в настоящем изобретении. Раствор примерно трех молярных эквивалентов (4-фторфенил)магнийбромида в ТГФ может подвергаться обработке (S)-этиллактатом при температуре около 0°С. Диол формулы H может выделяться с использованием стандартных методов выделения и очистки.
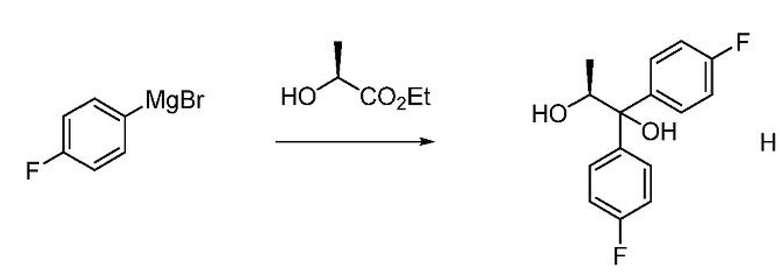
Химическая литература, в которой описывается получение (S)- (1,1-бис-арил)пропан-1,2-диолов, таких как соединение формулы H, включает следующие публикации: (1) Eur. J. Org. Chem. 2005, 1082; (2) Tetrahedron Lett. 1989, 30, 3659; (3) Tetrahedron: Asymmetry, 1990, 1, 199; (4) Патент США 4628120. Для связанных превращений, включающих присоединение арила Гриньяра к (S)-изопропиллактату для синтеза (S)-(1,1-бисарил)пропан-1,2-диолов, см. J. Am. Chem. Soc. 1990, 112, 3949.
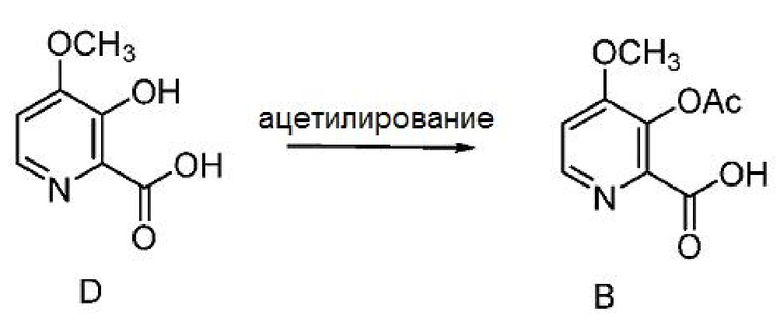
D. Получение соединения формулы B
Превращение 4-метокси-3-гидроксипиколиновой кислоты в 3-ацетокси-соединение формулы B может осуществляться ацетилированием соединения формулы D одним или несколькими ацетилирующими реагентами, выбранными из уксусного ангидрида и ацетилхлорида, основаниями, выбранными из пиридина, алкил-замещенных пиридинов и триалкиламинов, или с использованием условий реакции Шоттена-Баумана.
Продукт, полученный любым из указанных способов, может выделяться стандартными способами, такими как испарение, фильтрация или экстракция, и может быть очищен с помощью стандартных методик, таких как перекристаллизация или хроматография.
Далее представлены примеры, иллюстрирующие описание.
Примеры
Пример 1а. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидo)пропаноат
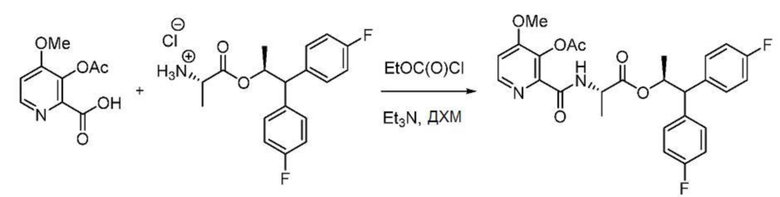
В колбу объемом 100 мл, снабженную магнитной мешалкой, загружают 3-ацетокси-4-метоксипиколиновую кислоту (427 мг, 2 ммоль) и ДХМ (10 мл). Суспензию охлаждают до -5°С. Одной порцией добавляют триэтиламин (445 мг, 4,4 ммоль) с последующим медленным добавлением с помощью шприца этилкарбонхлоридата (0,19 мл, 2 ммоль). Спустя 10 минут добавляют раствор гидрохлорида (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата (712 мг, 2 ммоль) в ДХМ (2 мл). После завершения реакции, в соответствии с ВЭЖХ, к смеси добавляют 20% водный раствор K2CO3 (10 мл) и энергично перемешивают полученную смесь в течение 30 минут при комнатной температуре. Органический слой отделяют. Водный слой экстрагируют ДХМ. Объединенные органические слои промывают водой, 1N HCl и водой. Отделенный органический слой концентрируют с получением пены светло-желтого цвета (1,5 г, 98%): 1Н ЯМР (400 МГц, CDCl3) δ 8,39 (с, 1H), 8,32 (д, J=5,4 Гц, 1H), 7,26-7,16 (м, 4H), 7,03-6,87 (м, 5H), 5,71 (дкв, J=9,6, 6,1 Гц, 1H), 4,55 (дд, J=8,0, 7,1 Гц, 1H), 4,04 (д, J=9,6 Гц, 1H), 3,91 (с, 3H), 2,38 (с, 3H), 1,22 (д, J=6,1 Гц, 3H), 0,99 (д, J=7,2 Гц, 3H); 19F ЯМР (376 МГц, CDCl3) δ -115,61, -115,96; масс-спектрометрия высокого разрешения с электрораспылительной ионизацией (МСВР-ESI) m/z [M+Н]+ вычислено для C27H27F2N2O6 512,1759; найдено 513,1825.
Пример 1b. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидo)пропаноат
Дихлорметан (64,6 л) и 3-ацетокси-4-метоксипиколиновую кислоту (6,82 кг) загружают в реактор из нержавеющей стали с перемешиванием в атмосфере азота при температуре 25-30°С. Раствор охлаждают до -15°С, к раствору по каплям добавляют N,N-диизопропилэтиламин (9,2 кг, 2,2 экв.) и перемешивают полученную смесь в течение 5 минут. К смеси по каплям добавляют этилхлорформиат (3,68 кг, 1,05 экв.) и перемешивают полученную смесь в течение 30 минут. И, наконец, к смеси по каплям добавляют раствор 11,5 кг гидрохлорида (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата в 32,2 л дихлорметана и перемешивают полученную смесь при -15°С в течение 30 минут. Реакционную смесь нагревает до 0-2°С, к смеси добавляют насыщенный водный раствор бикарбоната натрия (57,5 л, 5,75 кг NaHCО3 в 57,5 л воды) и перемешивают смесь в течение 10-15 минут. Водный слой отделяют и экстрагируют дихлорметаном (1 × 57,5 л). Объединенные органические слои промывают водой (1×57,5 л), затем смесью 1N HCl и насыщенного раствора соли (1×64,4 л, 32,2 л в HCl и 32,2 л насыщенного раствора соли). Органический слой сушат над сульфатом натрия (11,5 кг), фильтруют, промывают дихлорметаном (23,0 л) и концентрируют при температуре ниже 40°С в вакууме (500-600 мм рт.ст.) до прекращения отделения дистиллята. К остатку добавляют изопропиловый спирт (23,0 л) и концентрируют смесь при температуре ниже 45°С в вакууме (500-600 мм рт.ст.) с получением густого сиропа. К остатку добавляют изопропиловый спирт (11,5 л) и н-гептан (11,5 л), смесь нагревают до 50-55°С и перемешивают при 50-55°С в течение 30 минут. Раствор охлаждают до 25-30°С, добавляют н-гептан (11,5 л) и перемешивают полученный раствор при 25-30°С в течение 5 часов. Добавляют дополнительную порцию н-гептана (34,5 л) и перемешивают раствор при температуре 25-30°С в течение 6 часов. Образованное твердое вещество собирают фильтрацией, промывают н-гептаном (57,5 л) и сушат при 35-40°С в вакууме (500-600 мм рт.ст.) с получением (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидo)пропаноата в виде не совсем белого порошка (14,74 кг, выход 89,0%). ВЭЖХ (Zorbax SB-Phenyl, (250×4,6) мм, 5,0 мкм; 0,1% муравьиная кислота в смеси вода:ACN 50:50, скорость истечения: 1,0 мл/мин) показывает, что чистота продукта составляет 98,3%.
Пример 1с. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидо)пропаноат
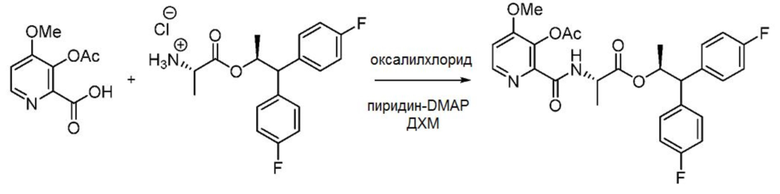
В колбу объемом 500 мл, снабженную магнитной мешалкой и входным отверстием для введения азота, загружают 3-ацетокси-4-метоксипиколиновую кислоту (11,5 г, 54,5 ммоль), ДХМ (140 мл), пиридин (4,84 мл, 59,9 ммоль) и 1 каплю ДМФА. Колбу охлаждают до 0°С и к смеси медленно с помощью шприца добавляют оксалилхлорид (4,77 мл, 54,5 ммоль). Полученный темный раствор перемешивают в течение приблизительно 15 минут. После этого полученный раствор добавляют через канюлю к охлажденной (0°С) суспензии гидрохлорида (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата (19,38 г, 54,5 ммоль) и триэтиламина (15,94 мл, 114 ммоль) в ДХМ (70 мл) в колбе объемом 1 л. После завершения добавления баню удаляют и раствор оставляют нагреваться до комнатной температуры. Завершение реакции определяют с помощью ЖХМС, после этого реакционную смесь выливают в насыщенный водный раствор NH4Cl (200 мл) и переносят смесь в делительную воронку. Органический слой отделяют, водный слой экстрагируют CH2Cl2 (1 × 200 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением пены желтовато-коричневого цвета/черного масла. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: 0-100% этилацетата в гексанах) с получением указанного в заголовке соединения в виде твердой пены розового цвета (14 г, 50,2%, чистота 90%): данные спектроскопии идентичны данным, представленным выше.
Пример 1d. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидo)пропаноат
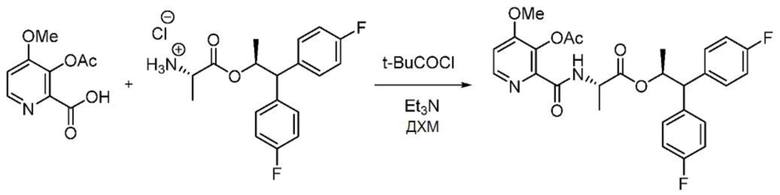
В колбу объемом 100 мл, снабженную магнитной мешалкой и входным отверстием для введения азота, загружают 3-ацетокси-4-метоксипиколиновую кислоту (1,00 г, 4,74 ммоль), ДХМ (23,7 мл) и триэтиламин (0,661 мл, 4,74 ммоль). Колбу охлаждают до 0°С и к реакционной смеси медленно добавляют пивалоилхлорид (0,583 мл, 4,74 ммоль). Реакционную смесь перемешивают в течение 15 минут при 0°С. После этого к смеси одной порцией добавляют гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата (1,685 г, 4,74 ммоль). Реакционную смесь выливают в насыщенный водный раствор NH4Cl и переносят полученную смесь в делительную воронку. Органический слой отделяют, промывают насыщенным водным раствором NaHCО3, насыщенным раствором соли и затем сушат над Na2SO4. Раствор фильтруют и концентрируют с получением не совсем белой пены. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: 0-100% этилацетата в гексанах) с получением указанного в заголовке соединения в виде белой пены (1,7 г, 59,5%, чистота 90%): данные спектроскопии идентичны данным, представленным выше.
Пример 1е. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3- ((этоксикарбонил)окси)-4-метоксипиколинамидo)пропаноат
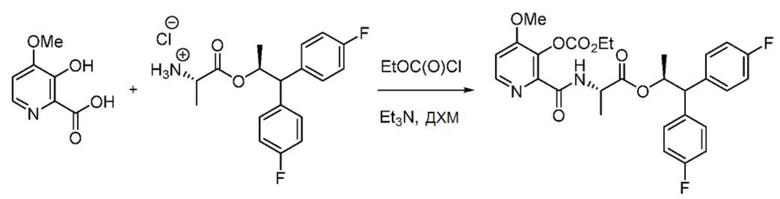
В колбу объемом 250 мл, снабженную магнитной мешалкой, загружают 3-гидрокси-4-метоксипиколиновую кислоту (0,846 г, 5 ммоль) и заполняют азотом. В реакционную колбу добавляют ДХМ (25 мл) и охлаждают полученную белую гетерогенную смесь до 0°С. К смеси добавляют триэтиламин (2,3 мл, 16,5 ммоль), реакционную смесь интенсивно перемешивают в течение 10 минут, и смесь становится гомогенным бесцветным раствором. К реакционной смеси медленно добавляют этилхлорформиат (1,0 мл, 10,5 ммоль), в результате чего начинает образовываться белый осадок. После перемешивания смеси в течение 15 минут при 0°С в колбу одной порцией добавляют гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата (1,78 г, 5,00 ммоль). Реакционную смесь перемешивают при 0°С в течение 3 минут, после чего реакционную смесь гасят 20 мл воды и 5 мл 2N НСl. Двухфазную смесь разбавляют ДХМ и переносят в делительную воронку. Слои разделяют, органический слой сушат с помощью Na2SO4, фильтруют и концентрируют с получением масла бледно-желтого цвета. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: этилацетат/гексан) с получением указанного в заголовке соединения в виде твердого белого вещества (2,3 г, 85%): т.пл. 48-64°С; 1Н ЯМР (400 МГц, CDCl3) δ 8,45-8,25 (м, 2H), 7,38-7,12 (м, 4H), 7,09-6,85 (м, 5H), 5,71 (дкв, J=9,7, 6,2 Гц, 1H), 4,67-4,54 (м, 1H), 4,34 (кв, J=7,1 Гц, 2H), 4,04 (д, J=9,6 Гц, 1H), 3,92 (с, 3H), 1,40 (т, J=7,1 Гц, 3H), 1,22 (д, J=6,2 Гц, 3H), 0,99 (д, J=7,2 Гц, 3H); 13С ЯМР (101 МГц, CDCl3) δ 172,1, 162,2, 161,7 (д, J=246,0 Гц), 161,6 (д, J=245,6 Гц), 159,4, 152,5, 146,8, 141,7, 137,7, 136,9, 136,8, 129,6 (д, J=7,8 Гц), 129,5 (д, J=7,8 Гц), 115,7 (д, 7=21,4 Гц), 115,4 (д, J=21,2 Гц), 110,0, 73,1, 65,4, 56,3, 56,1, 47,8, 19,1, 18,1, 14,1; 19F ЯМР (471 МГц, CDCl3) δ -115,59, -115,95; МСВР-ESI (m/z) [М+Н]+ вычислено для C28H29F2N2O7 543,1937; найдено 543,1932.
Пример 1f. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-((изобутоксикарбонил)окси)-4-метоксипиколинамидo)пропаноат
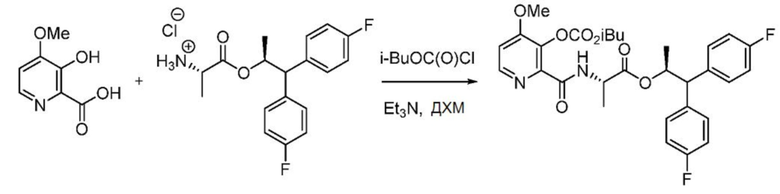
В колбу объемом 250 мл, снабженную магнитной мешалкой, загружают 3-гидрокси-4-метоксипиколиновую кислоту (0,846 г, 5 ммоль) и заполняют колбу азотом. В реакционную колбу добавляют ДХМ (25 мл) и охлаждают полученную белую гетерогенную смесь до 0°С. К смеси добавляют триэтиламин (2,3 мл, 16,5 ммоля), смесь интенсивно перемешивают в течение 10 минут, после чего реакционная смесь становится бесцветным гомогенным раствором. К реакционной смеси медленно добавляют изобутилхлорформиат (1,4 мл, 10,5 ммоль), и в процессе добавления начинает образовываться белый осадок. После перемешивания смеси в течение 15 минут при 0°С в колбу одной порцией добавляют гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата (1,78 г, 5,00 ммоль). Реакционную смесь перемешивают при 0°С в течение 3 минут, после чего гасят добавлением 20 мл воды и 5 мл 2N НСl. Двухфазную смесь разбавляют ДХМ и переносят в делительную воронку. Слои разделяют, органический слой сушат над Na2SO4, фильтруют и концентрируют с получением масла бледно-желтого цвета. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: этилацетат/гексан) с получением указанного в заголовке соединения в виде твердого белого вещества (2,3 г, 81%): т.пл. 47-63°С; 1Н ЯМР (400 МГц, CDCl3) δ 8,38-8,26 (м, 2H), 7,26-7,18 (м, 4H), 7,04-6,88 (м, 5H), 5,71 (дкв, J =9,6, 6,2 Гц, 1H), 4,66-4,51 (м, 1H), 4,07 (д, J=6,7 Гц, 2H), 4,04 (д, J=10,0 Гц, 1H), 3,92 (с, 3H), 2,19-1,98 (м, 1H), 1,22 (д, J=6,1 Гц, 3H), 0,99 (д, J=6,7 Гц, 6H), 0,99 (д, J=7,2 Гц, 3H); 13С ЯМР (101 МГц, CDCl3) δ 172,2, 162,2, 161,73 (д, J=246,0 Гц), 161,65 (д, J=245,6 Гц), 159,4, 152,6, 146,8, 141,7, 137,8, 136,9, 136,9, 129,61 (д, J=7,8 Гц), 129,54 (д, J=8,0 Гц), 115,68 (д, J=21,3 Гц), 115,39 (д, J=21,3 Гц), 109,9, 75,3, 73,1, 56,3, 56,1, 47,8, 27,8, 19,1, 18,9, 18,1; 19F ЯМР (471 МГц, CDCl3) δ -115,59, -115,95; МСВР-ESI (m/z) [М+Н]+ вычислено для C30H33F2N2O7 571,2250; найдено 571,2253.
Пример 1g. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-гидрокси-4-метоксипиколинамидo)пропаноат
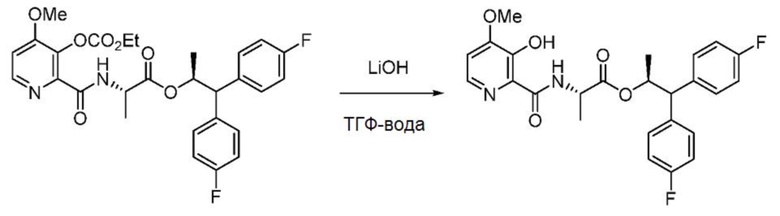
В емкость, снабженную магнитной мешалкой, загружают (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-((этоксикарбонил)окси)-4-метоксипиколинамидo)пропаноат (543 мг, 1 ммоль, используют смесь 8:1 указанного в заголовке исходного материала и продукта: (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-гидрокси-4- метоксипиколинамидo)пропаноата) и ТГФ (5 мл). Гидрат гидроксида лития (71 мг, 1,69 ммоль) помещают в отдельную пробирку, растворяют в воде (2,5 мл) и добавляют в реакционную колбу. Прозрачная бесцветная реакционная смесь сразу же приобретает желтую окраску. Реакционную смесь перемешивают в течение 3 часов при комнатной температуре. Реакционную смесь подкисляют до рН 2 с помощью 2N НСl (0,8 мл) и разбавляют 25 мл этилацетата. Органический слой концентрируют с получением масла желтого цвета. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: этилацетат/гексан) с получением указанного в заголовке соединения в виде белой пены (397 мг, 84%): 1Н ЯМР (400 МГц, CDCl3) δ 12,06 (с, 1H), 8,32 (дд, J=6,7, 4,3 Гц, 1H), 7,98 (д, J =5,2 Гц, 1H), 7,32-7,14 (м, 4H), 7,03-6,89 (м, 4H), 6,87 (д, J=5,2 Гц, 1H), 5,73 (дкв, J=9,8, 6,2 Гц, 1H), 4,61-4,47 (м, 1H), 4,05 (д, J=9,8 Гц, 1H), 3,94 (с, 3H), 1,25 (д, J=6,1 Гц, 3H), 1,07 (д, J=7,2 Гц, 3H); 13С ЯМР (101 МГц, CDCl3) δ 171,6, 168,6, 161,8 (д, J=246,1 Гц), 161,7 (д, J=245,7 Гц), 155,4, 148,8, 140,4, 136,8 (д, J=3,4 Гц), 136,7 (д, J=3,4 Гц), 130,4, 129,5 (д, J=7,8 Гц), 129,5 (д, J=7,8 Гц), 115,7 (д, J=21,3 Гц), 115,4 (д, J=21,3 Гц), 109,5, 73,3, 56,1, 56,1, 47,9, 19,1, 17,7; 19F ЯМР (471 МГц, CDCl3) δ -115,46, -115,80; МСВР-ESI (m/z) [М+Н]+ вычислено для С25H25F2N2O5 471,1726; найдено 471,1724.
Пример 1h. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидo)пропаноат
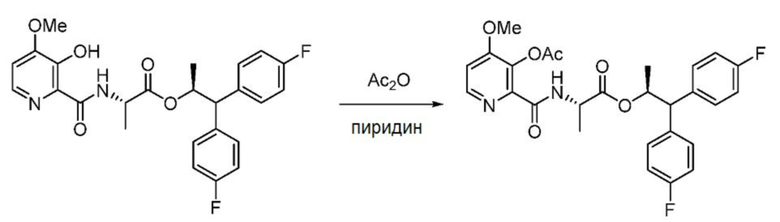
В колбу объемом 2 л, снабженную магнитной мешалкой, загружают (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-гидрокси-4-метоксипиколинамидo)пропаноат (25 г, 51,0 ммоль), пиридин (250 мл) и уксусный ангидрид (250 мл, 2,65 моль). Реакционную смесь перемешивают в течение часа при комнатной температуре и затем растворители удаляют в вакууме. К остатку добавляют гептан и полученную смесь концентрируют. Эту стадию повторяют для обеспечения полного азеотропного удаления любого остаточного растворителя. К остатку добавляют дихлорметан и насыщенный водный раствор хлорида аммония и слои разделяют. Водный слой экстрагируют дихлорметаном (1×), объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением не совсем белой пены. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: этилацетат/гексан) с получением указанного в заголовке соединения в виде белой пены (25,1 г, 95%, чистота 99%): 1Н ЯМР (500 МГц, CDCl3) δ 8,39 (с, уш., 1Н), 8,32 (д, J=5,4 Гц, 1H), 7,26-7,19 (м, 4H), 7,04-6,88 (м, 5H), 5,71 (дкв, J=9,6, 6,1 Гц, 1H), 4,62-4,49 (м, 1H), 4,04 (д, J=9,6 Гц, 1H), 3,90 (с, 3H), 2,38 (с, 3H), 1,22 (д, J=6,2 Гц, 3H), 0,99 (д, J=7,1 Гц, 3H); 13С ЯМР (126 МГц, CDCl3) δ 172,2, 170,3, 162,9, 161,7 (д, J=246,1 Гц), 161,6 (д, J=245,6 Гц), 160,3, 145,7, 144,0, 142,4, 136,9 (д, J=3,3 Гц), 136,8 (д, J=3,4 Гц), 129,6 (д, J=5,9 Гц), 129,5 (д, J=5,8 Гц), 115,7 (д, J=21,3 Гц), 115,4 (д, J=21,1 Гц), 109,6, 73,0, 56,2, 56,1, 48,0, 20,9, 19,2, 17,8; 19F ЯМР (471 МГц, CDCl3) δ -115,60, -115,96; МСВР-ESI (m/z) [M+H]+ вычислено для C27H27F2N2О6 513,1832; найдено 513,1849.
Пример 1i. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетоксиметокси-4-метоксипиколинамидo)пропаноат
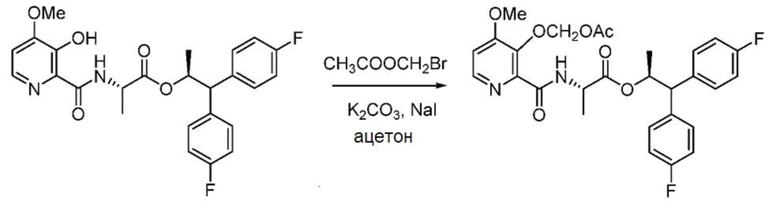
В трехгорлую колбу объемом 500 мл, снабженную магнитной мешалкой, обратным холодильником, термопарой и входным отверстием для введения азота, загружают (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-гидрокси-4-метоксипиколинамидo)пропаноат (4,9 г, 10,42 ммоль)) и ацетон (104 мл). К смеси добавляют твердый карбонат калия (2,88 г, 20,83 ммоля) с последующим добавлением бромметилацетата (1,532 мл, 15,62 ммоль). Добавляют каталитическое количество NaI, смесь нагревают до 50°С и выдерживают при указанной температуре в течение трех часов. Смесь охлаждают, фильтруют и концентрируют. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: этилацетат/гексан) с получением указанного в заголовке соединения в виде белой пены (3,9 г, 69%): 1Н ЯМР (400 МГц, CDCl3) δ 8,26 (д, J=5,4 Гц, 1H), 8,20 (д, J=7,8 Гц, 1H), 7,28-7,18 (м, 4H), 7,02-6,91 (м, 5H), 5,76-5,70 (м, 1H), 5,72 (с, 2H), 4,56 (9, J=7,3 Гц, 1H), 4,05 (д, J=9,7 Гц, 1H), 3,91 (с, 3H), 2,06 (с, 3H), 1,24 (д, J=6,1 Гц, 3H), 1,00 (д, J=7,2 Гц, 3H); 13С ЯМР (101 МГц, CDCl3) δ 172,2, 170,3, 162,9, 161,7 (д, J=246,0 Гц), 161,6 (д, J=245,5 Гц), 160,3, 145,7, 144,0, 142,3, 136,9 (д, J=3,3 Гц), 136,8 (д, J=3,3 Гц), 129,6 (д, J=7,8 Гц), 129,5 (д, J=7,9 Гц), 115,7 (д, J=21,4 Гц), 115,4 (д, J=21,3 Гц), 109,6, 89,5, 73,0, 56,2, 56,1, 48,1, 20,8, 19,1, 17,8; 19F ЯМР (376 МГц, CDCl3) δ -115,59, -115,97; МСВР-ESI (m/z) [М+Н]+ вычислено для C28H29F2N2O7, 543,1937; найдено 543,1948.
Пример 1j. (S,S)-1,1-бис(4-фторфенил)-1-гидроксипропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноат
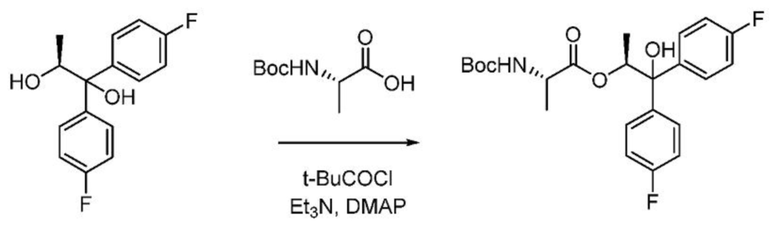
В колбу объемом 250 мл колбу, снабженную магнитной мешалкой, загружают (S)-2-[(трет-бутоксикарбонил)амино]пропановую кислоту (5,68 г, 30,0 ммоль) и ДХМ (125 мл) и охлаждают полученную смесь до 0°С. В реакционную колбу добавляют триэтиламин (8,72 мл, 62,5 ммоль). К реакционной смеси медленно добавляют пивалоилхлорид (3,69 мл, 30,0 ммоль), в процессе чего начинает образовываться белый осадок. Смесь перемешивают в течение 15 минут при 0°С и затем последовательно добавляют (S)-1,1-бис(4-фторфенил)пропан-1,2-диол (6,61 г, 25 ммоль) и N,N-диметилпиридин-4-амин (0,153 г, 1,250 ммоль), что приводит к нагреву смеси до 4,4°С. После указанных добавлений реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Реакционную смесь гасят водой и слои разделяют. Водный слой экстрагируют один раз дихлорметаном. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением масла. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: этилацетат/гексан) с получением указанного в заголовке соединения в виде твердого белого вещества (8,75 г, 80%): т.пл. 50-60°С; 1Н ЯМР (400 МГц, CDCl3) δ 7,50-7,42 (м, 2H), 7,42-7,36 (м, 2H), 7,03-6,94 (м, 4H), 5,91 (кв, J=6,2 Гц, 1H), 4,96 (д, J=7,8 Гц, 1H), 4,20-4,10 (м, 1H), 3,02-2,73 (м, 1H), 1,41 (с, 8Н), 1,18 (д, J=6,3 Гц, 3H), 0,92 (д, J=7,2 Гц, 3H); 13С ЯМР (101 МГц, CDCl3) δ 172,4, 161,9 (д, J=246,7 Гц), 161,9 (д, J=246,7 Гц), 155,0, 140,7 (д, J=3,3 Гц), 138,6 (д, J=2,8 Гц), 127,5 (д, J=8,0 Гц), 127,4 (д, J=8,2 Гц), 115,2 (д, J=21,6 Гц), 80,0, 79,0, 74,9, 49,2, 28,3, 18,0, 14,4 (один пик отсутствует вследствие случайной эквивалентности); 19F ЯМР (376 МГц, CDCl3) δ -115,21, -115,25; МСВР-ESI (m/z) [M+Na]+ вычислено для С23H27F2NNaО5 458,1750; найдено 458,1760.
Пример 1k. (S,S)-1,1-бис(4-фторфенил)-1-гидроксипропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноат
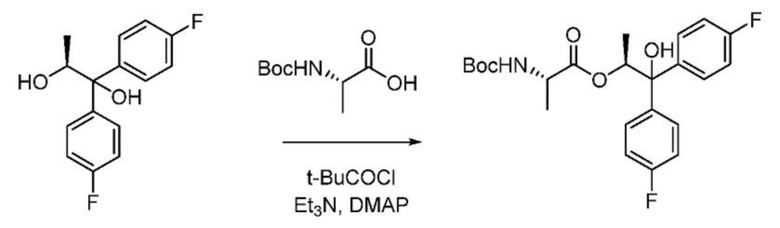
В реактор из нержавеющей стали при температуре 25-30°С загружают с перемешиванием в атмосфере азота безводный ТГФ (49,4 л, 7,6 объема) и Boc-L-аланин (6,3 кг, 1,35 экв.). Реакционную смесь охлаждают до 0-3°С, к смеси по каплям при 0-3°C добавляют триэтиламин (9,7 л, 2,8 экв.) и перемешивают полученную смесь в течение 5 минут. К смеси по каплям при 0-3°C добавляют пивалоилхлорид (4,0 кг, 1,35 экв) и перемешивают смесь при 0-3°С в течение 1 часа. К смеси одной порцией добавляют 4-(диметиламино)пиридин (0,15 кг, 0,05 экв.) и перемешивают полученную смесь в течение 5 минут. И, наконец, к смеси по каплям при 0-3°С добавляют раствор (S)-1,1-бис(4-фторфенил)пропан-1,2-диола в ТГФ (6,5 кг, 1,0 экв. в 19,5 л ТГФ). Реакционную смесь перемешивают при температуре 25-30°С в течение 3 часов. Реакционную смесь концентрируют при температуре ниже 40°С в вакууме (500-600 мм рт.ст.) до прекращения отделения дистиллята. К остатку добавляют этилацетат (49,4 л) и воду (24,7 л) и перемешивают смесь в течение 10 минут. Слои разделяют. Органический слой промывают насыщенным раствором хлорида аммония (1 × 24,7 л), затем насыщенным раствором бикарбоната натрия (1 × 24,7 л) и насыщенным раствором соли (1 × 13,0 л,). Органический слой сушат над сульфатом натрия (3,25 кг), фильтруют, промывают этилацетатом (6,5 л) и концентрируют при температуре ниже 40°С в вакууме (500-600 мм рт.ст.) до прекращения отделения дистиллята. К остатку добавляют гексаны (10,4 л) и концентрируют смесь при температуре ниже 40°С в вакууме (500-600 мм рт.ст.) с получением густого сиропа. Добавляют гексаны (13,0 л) и перемешивают смесь при 25-30°С в течение 10 минут. В раствор вносят затравку аутентичного продукта (13,0 г) и перемешивают раствор при 25-30°С в течение 12 часов. Твердое вещество собирают фильтрацией, промывают гексанами (2 × 6,5 л, 2,0 объема) и сушат при 38-42°С в вакууме (500-600 мм рт.ст.) с получением (S,S)-1,1-бис(4-фторфенил)-1-гидроксипропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноата в виде твердого не совсем белого вещества (8,4 кг, выход 78,4%). ВЭЖХ (Hypersil BDS С18, (250 × 4,6) мм, 5,0 мкм; А: 0,1% ТФУК в воде, В: ACN, скорость истечения: 1,0 мл/мин.) показывает, что чистота продукта составляет 94,0%.
Пример 1l. Гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата
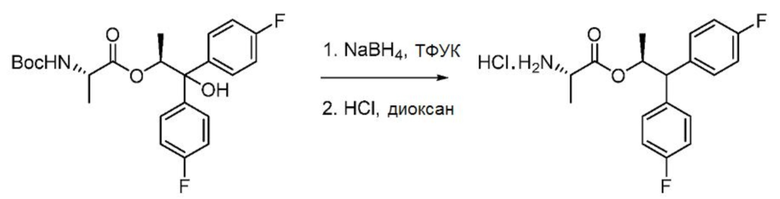
В трехгорлую колбу, снабженную магнитной мешалкой, датчиком температуры и впускным отверстием для введения азота, загружают трифторуксусную кислоту (8,85 мл, 115 ммоль) и охлаждают полученную смесь до 0°С. К смеси медленно добавляют борогидрид натрия (0,434 г, 11,5 ммоль) и затем медленно добавляют (S,S)-1,1-бис(4-фторфенил)-1-гидроксипропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноат (1 г, 2,3 ммоль) в ДХМ (2,3 мл). Реакционную смесь перемешивают при 0°С в течение 1 часа, а затем при комнатной температуре в течение 3 часов. Реакционную смесь гасят с помощью 2 М NaOH до достижения значения рН>12 и разбавляют ДХМ. Органический слой промывают насыщенным раствором соли. Объединенные водные слои экстрагируют один раз ДХМ. Объединенные органические слои концентрируют с получением масла. Сырое свободное основание амина обрабатывают 2 мл 4 М HCl в диоксане и затем концентрируют с получением клейкого масла розового цвета. К остатку добавляют MTBE (2 мл), после чего начинает образовываться белый осадок. Гетерогенную смесь перемешивают в течение 30 минут при 0°С. Вакуумная фильтрация гетерогенной смеси приводит к получению указанного в заголовке соединения в виде твердого белого вещества (355 мг, 40%): 1Н ЯМР (300 МГц, ДМСО-d6) δ 8,38 (с, 3H), 7,56-7,40 (м, 4H), 7,18-7,10 (м, 4H), 5,77 (дкв, J=12,2, 6,2 Гц, 1H), 4,27 (д, J=10,1 Гц, 1H), 3,91 (кв, J=7,1 Гц, 1H), 1,17 (д, J=6,1 Гц, 3H), 0,81 (д, J=7,2 Гц, 3H); 13С ЯМР (101 МГц, ДМСО-d6) δ 169,5, 161,0 (д, J=243,2 Гц), 160,9 (д, J=242,7 Гц), 137,8 (д, J=3,2 Гц), 137,3 (д, J=3,2 Гц), 130,0 (д, J=7,9 Гц), 129,8 (д, J=7,9 Гц), 115,4 (д, J=21,1 Гц), 115,2 (д, J=21,0 Гц), 73,7, 54,7, 47,6, 18,8, 15,0; 19F ЯМР (376 МГц, ДМСО-d6) δ -115,89, -116,29; МС(ESI) m/z 320,1 ([М+Н]+).
Пример 1m. Гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата
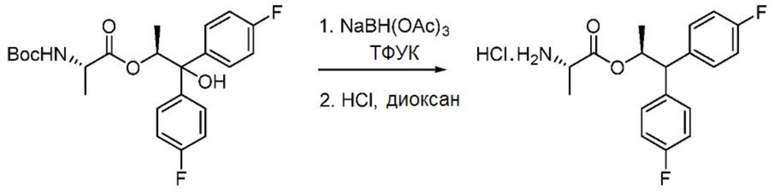
В трехгорлую колбу, снабженную магнитной мешалкой, датчиком температуры и входным отверстием для введения азота, при 0°С загружают триацетоксиборогидрид натрия (4,24 г, 20 ммоль) и трифторуксусную кислоту (15,4 мл, 200 ммоль). Смесь выдерживают в течение 10-15 минут при температуре 0-5°С, после чего добавляют (S,S)-1,1-бис(4-фторфенил)-1-гидроксипропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноат (4,35 г, 10 ммоль) в ДХМ (5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 4-5 часов. Реакционную смесь концентрируют и снова разбавляют ДХМ. Органический слой промывают 20% водным раствором К2СО3. Водный слой экстрагируют дополнительным количеством ДХМ. Объединенные органические слои промывают водой. Органический слой концентрируют с получением масла. Сырое свободное аминное основание разбавляют MTBE, затем обрабатывают 4 М НСl в диоксане (3,0 мл). Начинает образовываться белый осадок. Гетерогенную смесь перемешивают в течение 0,5-1 часа при комнатной температуре. Вакуумная фильтрация приводит к получению указанного в заголовке соединения в виде твердого белого вещества (2,7 г, 75%): данные спектроскопии идентичны данным соединения, выделенного в примере 1l.
Пример 1n. Гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата
В стеклоэмалированный реактор с перемешиванием в атмосфере азота при 25-30°С загружают трифторуксусную кислоту (170,3 л) и охлаждают до 0-2°С. Порциями (4 порции с интервалами 5 мин.) добавляют триацетоксиборогидрид натрия (29,7 кг, 2,7 экв.) при температуре 0-10°С и перемешивают полученную смесь при 13-17°С в течение 30 минут. К смеси по каплям добавляют раствор (S,S)-1,1-бис(4-фторфенил)-1-гидроксипропан-2-ил-2-((трет-бутоксикарбонил)амино)пропионата в дихлорметане (22,7 кг, 1,0 экв, в 22,7 л дихлорметана), поддерживая температуру на уровне 8-10°С, и перемешивают смесь при 13-17°С в течение 2 часов. После завершения реакции реакционную смесь концентрируют при температуре ниже 50°С в вакууме (500-600 мм рт.ст.), затем массу выпаривают совместно с толуолом (2×90,8 л) с получением сиропа бледно-желтого цвета, который растворяют в дихлорметане (227 л). К полученному раствору при температуре 25-30°С медленно добавляют 15% водный раствор хлорида аммония (794,5 л) и перемешивают смесь при 25-30°С в течение 15 минут. Слои разделяют. Водный слой экстрагируют дихлорметаном (2 × 113,5 л), объединенные органические экстракты промывают насыщенным раствором соли (1×113,5 л), сушат над сульфатом натрия (22,7 кг) и фильтруют. Фильтрат концентрируют при температуре ниже 35°С в вакууме (500-600 мм рт.ст.) с получением сиропа бледно-коричневого цвета. К сиропу добавляют MTBE (68,1 л) и н-гептан (22,7 л,) и охлаждают полученную смесь до 8-12°С. К смеси при 8-12°С добавляют 4N НСl в диоксане (20,45 л) и перемешивают смесь в течение 30 минут при 25-30°С. К смеси добавляют н-гептан (113,5 л) и перемешивают смесь при 25-30°С в течение 30 минут. Полученное твердое вещество собирают фильтрацией в атмосфере азота и промывают н-гептаном (68,1 л) с получением первой порции продукта.
Фильтрат концентрируют при температуре ниже 50°С в вакууме (500-600 мм рт.ст.). К остатку добавляют MTBE (45,4 л) и 4N HCl в диоксане (11,4 л) и перемешивают полученную смесь при температуре 25-30°С в течение 1 часа. Раствор концентрируют при температуре ниже 50°С в вакууме (500-600 мм рт.ст.) с получением сиропа коричневого цвета. К сиропу добавляют MTBE (22,7 л) и н-гептан (68,1 л), перемешивают смесь при температуре 25-30°С в течение 5 часов, фильтруют в атмосфере азота и промывают н-гептаном (45,4 л, 2,0 объема) с получением второй порции продукта.
Две порции продукта, 2-пропанол (74,9 л) и н-гептан (74,9 л) загружают в стеклоэмалированный реактор и перемешивают полученную смесь в атмосфере азота при 25-30°С. Полученную смесь нагревают до 75-80°С и выдерживают при 75-80°С в течение 30 минут. Реакционную смесь медленно охлаждают до 25-30°С и выдерживают при 25-30°С в течение 12 часов. Твердое вещество собирают фильтрацией, промывают 50% раствором 2-пропанола в н-гептане (68,1 л) и сушат при 40-45°С в вакууме (500-600 мм рт.ст.) с получением чистого гидрохлорида (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата в виде не совсем белого порошка (11,54 кг, выход 62,2%). ВЭЖХ (Zorbax 300 SCX, (250 × 4,6) мм, 5,0 мкм; 55:45 [200 мм фосфатный буфер (рН 3):ACN], скорость истечения: 2,0 мл/мин.) показывает, что чистота продукта составляет 94,0%.
Пример 1о. Гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата
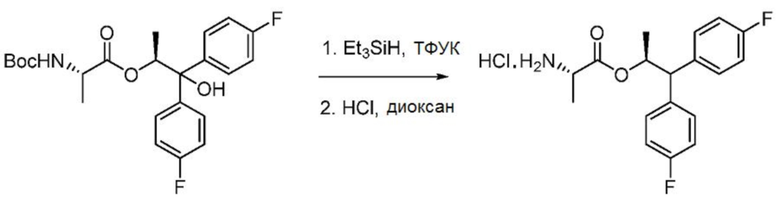
В колбу объемом 25 мл, снабженную магнитной мешалкой, датчиком температуры и входным отверстием для введения азота, загружают (S,S)-1,1-бис(4-фторфенил)-1-гидроксипропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноат (3,0 г, 6,89 ммоль) с последующим добавлением CH2Cl2 (10 мл) и триэтилсилана (4,4 мл, 27,56 ммоль, 4 экв.). Внутреннюю температуру колбы поддерживают на уровне 4°С с помощью ледяной бани. К смеси в течение 15 минут добавляют трифторуксусную кислоту (10 мл, 130 ммоль, 19 экв.). Во время добавления внутренняя температура не поднимается выше 8°С. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 4 часов. ЖХ-МС показывает полное превращение исходных веществ в продукт. Растворитель удаляют при пониженном давлении и совместным испарением с CH2Cl2 (3 × 20 мл). Полученное масло растворяют в CH2Cl2 (50 мл) и небольшими порциями в течение 10 минут добавляют к насыщенному раствору NaHCО3 (100 мл). Водный слой экстрагируют CH2Cl2 (25 мл), объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют при пониженном давлении. Масло растворяют в MTBE (15 мл) и добавляют 4N HCl в диоксане (1,8 мл), в процессе чего образуется белый осадок. Собранное твердое вещество перекристаллизовывают из смеси 2-пропанол/гептан, собирают фильтрацией, промывают гептаном и сушат в вакуумной печи при 50°С с получением 1,85 г конечного продукта с выходом 75%. Данные спектроскопии идентичны данным соединения, выделенного в примере 1h.
Пример 1p. Гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата
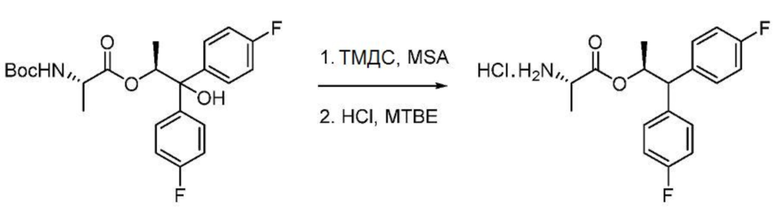
В трехгорлой круглодонной колбе объемом 250 мл, снабженной магнитной мешалкой, термопарой и входным отверстием для введения азота, смесь MsOH (15,0 мл, 230,0 ммоль) и CH2Cl2 (15 мл) охлаждают до 1°С. К смеси добавляют 1,1,3,3-тетраметилдисилоксан (ТМДС) (4,1 мл, 23,0 ммоль). К смеси медленно, в течение часа, по каплям добавляют раствор (S,S)-1,1-бис(4-фторфенил)-1-гидроксипропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноата (10,1 г, 23,0 ммоль) в CH2Cl2 (15 мл), поддерживая внутреннюю температуру ниже 3°С. После завершения добавления реакционную смесь перемешивают в течение 45 минут, после чего ВЭЖХ анализ показывает, что реакция завершена. К смеси медленно добавляют водный раствор карбоната натрия (насыщенный, 200 мл), поддерживая внутреннюю температуру ниже 20°C. Смесь переносят в делительную воронку. Слои разделяют. Водный слой экстрагируют CH2Cl2 (20 мл × 1). Объединенные органические слои являются мутными, их промывают насыщенным раствором соли (20 мл × 1), сушат над сульфатом натрия и фильтруют с получением не совсем белой слизи. Сырой продукт растворяют в MTBE (125 мл) и при перемешивании добавляют HCl (3 М в CPME, 11,5 мл). Белое твердое вещество собирают и промывают гептаном (50 мл). Продукту дают возможность высохнуть в течение ночи в вытяжном шкафу. Получают 8,03 г твердого вещества (выход 98%, чистота 89% в соответствии с ВЭЖХ с внутренним стандартом). 1Н ЯМР (400 МГц, CDCl3) δ 8,64 (уш. с, 3Н), 7,21 (тдд, J=7,2, 5,1, 2,0 Гц, 4H), 6,98 (тд, J=8,6, 6,2 Гц, 4H), 5,77-5,59 (м, 1H), 4,05 (д, J=10,0 Гц, 1H), 3,96 (кв, J=7,2 Гц, 1H), 1,23 (д, J=6,1 Гц, 3H), 1,14 (д, J=7,2 Гц, 3H). 19F ЯМР (376 МГц, CDCl3) δ -115,16, -115,52. МС (ESI) m/z 320,1 ([М+Н]+).
Пример 1q. (S)-1,1-бис(4-фторфенил)пропан-2-ол
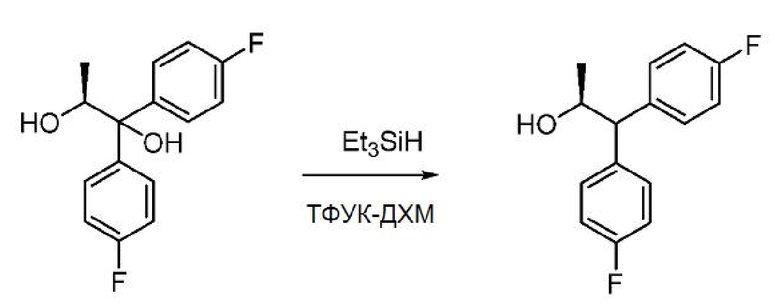
В колбу объемом 5 л загружают (S)-1,1-бис(4-фторфенил)пропан-1,2-диол (120 г, 431 ммоль) и ДХМ (1200 мл). Смесь охлаждают до 0°С и в колбу последовательно добавляют триэтилсилан (689 мл, 4314 ммоль) и ТФУК (332 мл, 4314 ммоль). Добавление осуществляют в течение 12 минут, в процессе добавления температура поднимается от -3°С до -2°С. Смесь перемешивают при 0°С в течение 1 часа. После этого реакционную смесь нейтрализуют 4 N NaOH (~1,2 л), оставляя ее на ледяной бане, до достижения значения рН ~10. Слои разделяют и водную фазу экстрагируют дихлорметаном (1×). Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением 159 г масла бледно-желтого цвета. Сырой продукт загружают в хроматографической колонку с диоксидом кремния ISCO (1,5 кг) и элюируют продукт с градиентом: EtOAc/гексан, получая 90,3 г продукта в виде твердого белого вещества (83%). 1Н ЯМР (400 МГц, хлороформ-d) δ 7,35-7,28 (м, 2H), 7,25-7,18 (м, 2H), 7,05-6,93 (м, 4H), 4,45 (м, 1H), 3,79 (д, J=8,3 Гц, 1H), 1,63 (д, J=3,7 Гц, 1H), 1,17 (д, J=6,1 Гц, 3H). 13С ЯМР (101 МГц, CDCl3) δ 161,8 (д, J=245,7 Гц), 161,6 (д, J=245,4 Гц), 138,2 (д, J=3,3 Гц), 137,0 (д, J=3,3 Гц), 130,2 (д, J=7,8 Гц), 129,6 (д, J=7,9 Гц), 115,7 (д, J=21,2 Гц), 115,5 (д, J=21,0 Гц), 70,1, 58,6, 21,6. 19F ЯМР (376 МГц, CDCl3) δ -115,84, -116,16. МС (ESI) m/z 231,3 ([М-OH]+). Анализ хиральной ВЭЖХ проводят с использованием колонки Chiralpak IA (250×4,6 мм, P/N: 80325), используя в качестве подвижной фазы смесь 85% гексанов (0,1% трифторуксусной кислоты) и 15% изопропанола (0,1% трифторуксусной кислоты) (впрыск 10 мкл). С использованием УФ-детектора, установленного на 265 нм, энантиомер #1 (основной) элюируют на 6,2 мин., энантиомер #2 (минорный) элюируют на 6,8 мин. Определение стереочистоты показывает энантиомерный избыток 98%.
Пример 1r. (S)-1,1-бис(4-фторфенил)пропан-2-ол
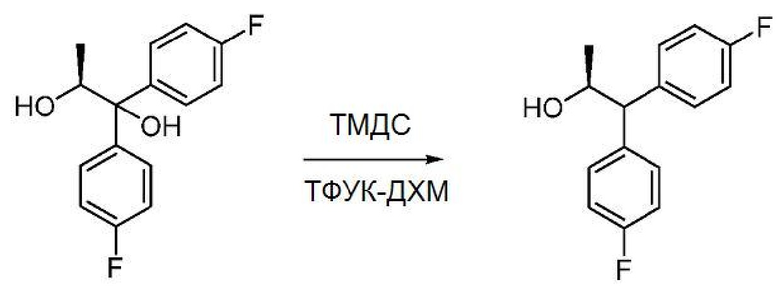
В трехгорлую круглодонную колбу объемом 50 мл, снабженную магнитной мешалкой, термопарой и входным отверстием для введения азота, загружают (S)-1,1-бис(4-фторфенил)пропан-1,2-диол (1,0 г, 3,8 ммоль), СН2Сl2 (3 мл) и ТДМС (2,0 мл, 1,1 ммоль). По каплям добавляют ТФУК (5,8 мл, 7,6 ммоль). Спустя 30 минут реакция завершается, как показывает ВЭЖХ анализ. Реакционную смесь промывают насыщенным водным раствором карбоната натрия (20 мл × 2). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают с помощью колоночной хроматографии (SiO2, 0-40% EtOAc в гексанах) с получением бесцветного масла (0,68 г, выход 75%). 1Н ЯМР (400 МГц, CDCl3) δ 7,38-7,29 (м, 2H), 7,25-7,16 (м, 2H), 7,07-6,89 (м, 4H), 4,51-4,43 (м, 1H), 3,80 (д, J=8,2 Гц, 1H), 1,53 (уш. с, 1H), 1,19 (д, J=6,1 Гц, 3H). 19F ЯМР (376 МГц, CDCl3) δ -115,86, -116,20.
Пример 1s. (S)-1,1-бис(4-фторфенил)пропан-2-ол
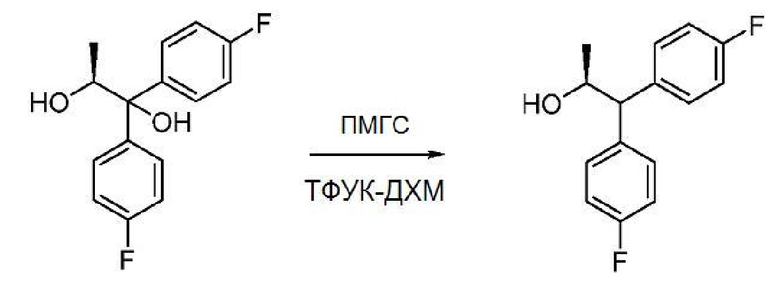
В трехгорлую круглодонную колбу объемом 100 мл, снабженную магнитной мешалкой, термопарой и входным отверстием для введения азота, загружают (S)-1,1-бис(4-фторфенил)пропан-1,2-диол (1,23 г, 4,67 ммоль) и CH2Cl2 (53 мл) и охлаждают полученный раствор до 0°С. Добавляют чистый ПМГС (МN=1700-3200, 2,9 г) с последующим добавлением по каплям чистой ТФУК (5,4 г, 46,7 ммоль). Спустя 80 минут реакционную смесь гасят добавлением 50 мл 1 М NaOH. Добавляют CH2Cl2 (30 мл). Водный слой отделяют и экстрагируют дополнительным количеством CH2Cl2 (2 × 35 мл). Объединенные органические слои промывают насыщенным раствором соли, сушат (Na2SО4) и упаривают на роторном испарителе. Сырой продукт очищают колоночной хроматографией (SiO2, элюирование с градиентом: 0-45% EtOAc в гексанах) с получением бесцветного масла (0,613 г, выход 53%). 1Н ЯМР (400 МГц, CDCl3) δ 7,38-7,29 (м, 2H), 7,25-7,16 (м, 2H), 7,07-6,89 (м, 4H), 4,51-4,43 (м, 1H), 3,80 (д, J=8,2 Гц, 1H), 1,53 (уш. с, 1H), 1,19 (д, J=6,1 Гц, 3H). 19F ЯМР (376 МГц, CDCl3) δ -115,86, -116,20. Анализ хиральной ВЭЖХ показывает наличие единственного энантиомера.
Пример 1t. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноат
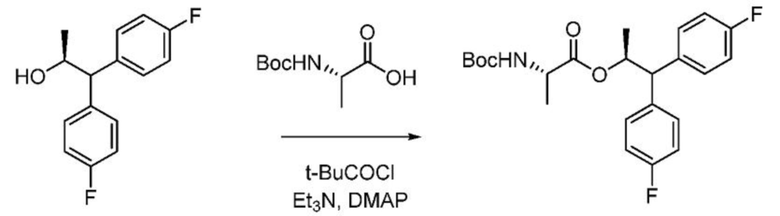
В колбу объемом 250 мл, снабженную магнитной мешалкой, загружают (S)-2-((трет-бутоксикарбонил)амино)пропановую кислоту (0,91 г, 4,8 ммоль) и ДХМ (20 мл) и охлаждают полученную смесь до 0°С. К смеси добавляют триэтиламин (1,4 мл, 10 ммоль). К смеси медленно добавляют пивалоилхлорид (0,59 мл, 4,8 ммоль), в процессе чего начинает образовываться белый осадок. После перемешивания в течение 15 минут при 0°С к смеси добавляют (S)-1,1-бис(4-фторфенил)пропан-2-ол (993 мг, 4,0 ммоль) с последующим добавлением N,N-диметилпиридин-4-амина (49 мг, 0,4 ммоль) и перемешивают полученную реакционную смесь в течение ночи при комнатной температуре. Реакционную смесь гасят водой и слои разделяют. Водный слой экстрагируют один раз дихлорметаном. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением бесцветного масла. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: этилацетат/гексан) с получением указанного в заголовке соединения в виде белой пены (1,4 г, 83%): 1Н ЯМР (300 МГц, CDCl3) δ 7,29-7,17 (м, 4H), 7,03-6,92 (м, 4H), 5,71 (дкв, J=9,8, 6,2 Гц, 1H), 4,94 (д, J=8,0 Гц, 1H), 4,12 (кв, J=7,1 Гц, 1H), 4,02 (д, J=9,9 Гц, 1H), 1,42 (с, 9H), 1,22 (д, J=6,2 Гц, 3H), 0,84 (д, J=7,2 Гц, 3H); 13С ЯМР (126 МГц, CDCl3) δ 172,8, 161,7 (д, J=246,1 Гц), 161,7 (д, J=245,6 Гц), 154,9, 137,0 (д, J=3,3 Гц), 136,8 (д, J=3,4 Гц), 129,5 (д, J=7,9 Гц), 129,5 (д, J=7,8 Гц), 115,7 (д, J=21,3 Гц), 115,4 (д, J=21,3 Гц), 79,8, 72,9, 56,2, 49,2, 28,3, 19,2, 18,1; 19F ЯМР (376 МГц, CDCl3) δ -115,56, -115,97; МC (ESI) m/z 442,1 ([M+Na]+).
Пример 1u. Гидрохлорид (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-аминопропаноата
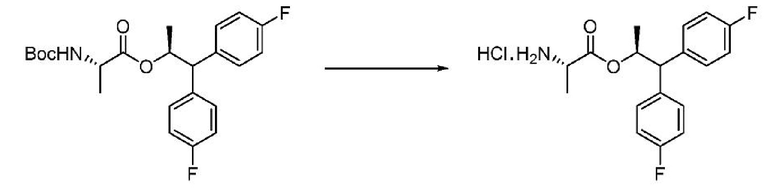
В одногорлую колбу объемом 3 л, снабженную магнитной мешалкой, загружают (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-((трет-бутоксикарбонил)амино)пропаноат (130 г, 294 ммоль) и диоксан (100 мл). К смеси при перемешивании при комнатной температуре (20°С) добавляют HCl в диоксане (750 мл, 3 моль, 4 М раствор). Реакционную смесь перемешивают в течение ночи и затем концентрируют в вакууме с получением липкой пены желтовато-коричневого цвета. К остатку добавляют диэтиловый эфир (1,75 л) и энергично перемешивают гетерогенную смесь в течение 30 минут. Смесь фильтруют, промывают диэтиловым эфиром, затем гексаном и сушат в вакууме с получением продукта в виде твердого белого вещества (104,7 г, 100%): 1Н ЯМР (300 МГц, ДМСО-d6) δ 8,38 (с, 3H), 7,56-7,40 (м, 4H), 7,18-7,10 (м, 4H), 5,77 (дкв, J=12,2, 6,2 Гц, 1H), 4,27 (д, J=10,1 Гц, 1H), 3,91 (кв, J=7,1 Гц, 1H), 1,17 (д, J=6,1 Гц, 3H), 0,81 (д, J=7,2 Гц, 3H); 13С ЯМР (101 МГц, ДМСО-d6) δ 169,5, 161,0 (д, J=243,2 Гц), 160,9 (д, J=242,7 Гц), 137,8 (д, J=3,2 Гц), 137,3 (д, J=3,2 Гц), 130,0 (д, J=7,9 Гц), 129,8 (д, J=7,9 Гц), 115,4 (д, J=21,1 Гц), 115,2 (д, J=21,0 Гц), 73,7, 54,7, 47,6, 18,8, 15,0; 19F ЯМР (376 МГц, ДМСО-d6) δ -115,89, -116,29; МС(ESI) m/z 320,1 ([М+Н]+).
Пример 1v. (S)-1,1-бис(4-фторфенил)пропан-1,2-диол
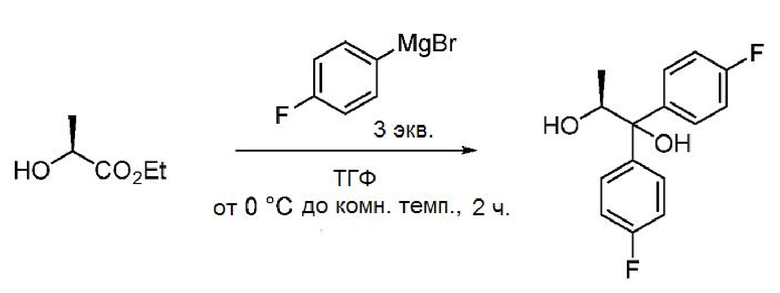
В трехгорлую колбу объемом 5 л, снабженную мешалкой с верхним приводом, датчиком внутренней температуры и капельной воронкой, в атмосфере азота загружают (4-фторфенил)магнийбромид (1600 мл, 1600 ммоль, 1 М в ТГФ). Смесь охлаждают до 0°С, к смеси с помощью капельной воронки медленно (40 минут) добавляют раствор (S)-этиллактата (60 г, 483 ммоль) в ТГФ (500 мл), поддерживая температуру не выше 0°С. Все еще холодную (4°С) реакционную смесь гасят насыщенным водным раствором NH4Cl (250 мл) и перемешивают до тех пор, пока температура реакционной смеси не достигнет температуры окружающей среды. Жидкий слой декантируют от твердого белого вещества. Белое твердое вещество суспендируют в EtOAc, суспензию фильтруют и промывают EtOAc. Объединенные органические фазы концентрируют в вакууме. Остаток переносят в EtOAc, загружают смесь в делительную воронку и промывают водой. Органическую фазу сушат над Na2SО4, фильтруют и концентрируют в вакууме с получением масла желтого цвета. Сырой продукт растворяют в ацетонитриле (500 мл) и экстрагируют гексаном (2 × 300 мл). Ацетонитрильный слой сушат над Na2SО4, фильтруют и концентрируют с получением 117 г масла желтого цвета. Сырой продукт очищают хроматографией (1,5 кг ISCO картридж с силикагелем; элюирование с градиентом: EtOAc/гексан) с получением 88,3 г твердого белого вещества (66%). 1Н ЯМР (400 МГц, хлороформ-d) δ 7,59-7,49 (м, 2H), 7,41-7,32 (м, 2H), 7,07-6,92 (м, 4H), 4,74 (квд, J=6,2, 3,8 Гц, 1H), 3,00 (с, 1H), 1,81 (д, J=3,8 Гц, 1H), 1,08 (д, J=6,3 Гц, 3H). 13С ЯМР (101 МГц, CDCl3) δ 161,9 (д, J=246,8 Гц), 161,7 (д, J=246,0 Гц), 141,2 (д, J=3,3 Гц), 139,6 (д, J=3,2 Гц), 128,1 (д, J=7,9 Гц), 127,4 (д, J=8,0 Гц), 115,4 (д, J=21,3 Гц), 115,0 (д, J=21,3 Гц), 79,3, 71,5, 16,9. 19F ЯМР (376 МГц, CDCl3) δ -115,3, -115,9. МС (ESI) m/z 263,1 ([МН]-).
Пример 1w. (S)-1,1-бис(4-фторфенил)пропан-1,2-диол
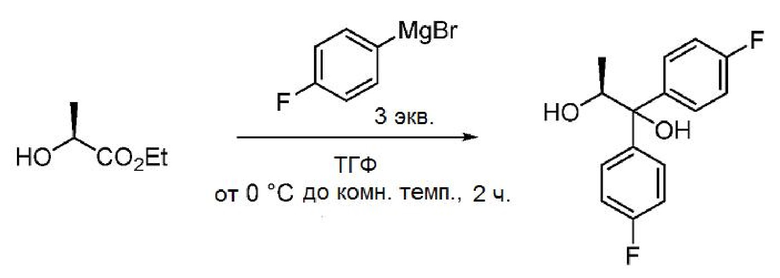
Магниевую стружку (12,6 кг, 3,5 экв.) и безводный тетрагидрофуран (115,6 л) загружают при перемешивании в атмосфере азота в реактор из нержавеющей стали при 25-30°С. Реакционную смесь нагревают до 40-45°С. К реакционной смеси по каплям при 50-55°С добавляют раствор 4-бромфторбензола в тетрагидрофуране (81,35 кг, 3,25 экв. 4-бромфторбензола в 115,6 л ТГФ) и выдерживают полученную смесь при указанной температуре в течение 30 минут. Реакционную смесь охлаждают до 0-3°С, а затем по каплям в течение 2,0 часов добавляют раствор этил-L-лактата в тетрагидрофуране (17,0 кг, 1,0 экв. этил-L-лактата в 84,2 л ТГФ) при 0-3°С и выдерживают в указанных условиях в течение 30 минут. К смеси по каплям при 0-10°С в течение 2,0 часов добавляют насыщенный раствор хлорида аммония (119,0 л, 41,65 кг хлорида аммония в 119,0 л воды). Реакционную смесь фильтруют и твердое вещество промывают этилацетатом (3 × 125,8 л). Фильтрат загружают обратно в реактор и промывают насыщенным раствором соли (1 × 85,0 л, 5,0 об.). Водный слой повторно экстрагируют этилацетатом (1 × 125,8 л, 7,4 об.), объединенные органические слои промывают насыщенным раствором соли (1 × 85,0 л, 5,0 об.), сушат над сульфатом натрия (8,5 кг, 0,5 об.), фильтруют и упаривают при температуре 40-45°С в вакууме (500-600 мм рт.ст.) с получением масла бледно-желтого цвета. Добавляют гексаны (85,0 л, 5,0 об.) и концентрируют смесь при температуре ниже 45°С в вакууме (500-600 мм рт.ст.) до прекращения образования дистиллята. К остатку добавляют гексаны (119,0 л), смесь перемешивают в течение 15 минут, охлаждают до 8-12°С и выдерживают в течение 1 часа. Твердый продукт собирают фильтрацией и промывают гексанами (1 × 17,0 л). Полученное влажное твердое вещество загружают обратно в реактор, добавляют 2% MTBE в гексанах (119,0 мкл, 7,0 об.) и перемешивают при температуре 25-30°С в течение 30 минут. Образованный осадок собирают фильтрацией, промывают гексанами (51,0 л) и сушат при 35-40°С в вакууме (500-600 мм рт.ст.) с получением (S)-1,1-бис(4-фторфенил)пропан-1,2-диола в виде порошка бледно-желтого цвета (26,0 кг, выход 68,3%). ВЭЖХ (Hypersil BDS-С18, (250 × 4,6) мм, 5,0 мкм; А: 0,1% ТФУК в воде, В: ACN, скорость истечения: 1,0 мл/мин.) показывает, что чистота продукта составляет 95,1%.
Пример 1x. 3-(Ацетилокси)-4-метоксипиколиновая кислота
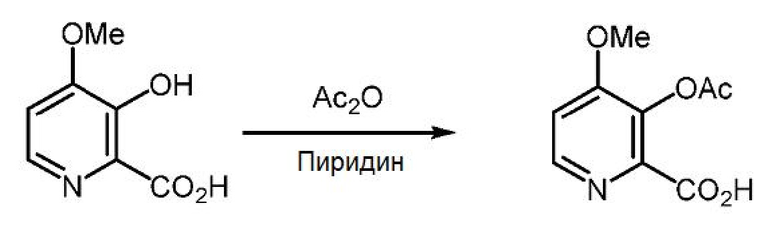
3-Гидрокси-4-метоксипиколиновую кислоту (5,0 г, 29,6 ммоль) суспендируют в 50 мл пиридина и 50 мл уксусного ангидрида при температуре окружающей среды. Спустя 1 час раствор приобретает желтую окраску, и раствор перемешивают в течение ночи. Раствор упаривают при 45°С (2 мм рт.ст.) с получением 6,28 г твердого вещества рыжевато-коричневого цвета (выход 99%, т.пл.=132-134°С). 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,32 (с, 1H), 8,43 (д, J=5,5 Гц, 1H), 7,40 (д, J=5,5 Гц, 1H), 3,91 (с, 3H), 2,27 (с, 3H). 13С ЯМР (101 МГц, ДМСО-d6) δ 167,95, 164,81, 158,34, 147,87, 142,77, 136,18, 110,87, 56,59, 20,27. МСВР (m/z) вычислено для C9H9NO5 211,0478, найдено 211,0481 ([М]+).
Пример 1y. 3-(Ацетилокси)-4-метоксипиколиновая кислота
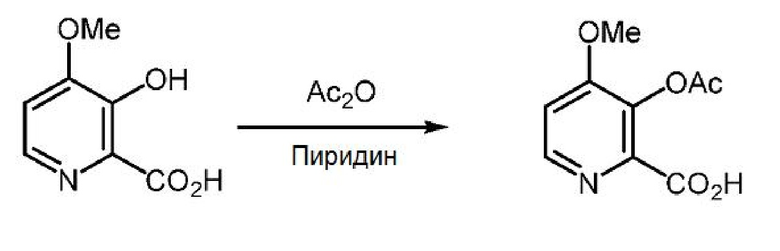
Пиридин (5,7 л, 1,0 об.), 3-гидрокси-4-метоксипиколиновую кислоту (5,7 кг, 1,0 экв.) и уксусный ангидрид (15,73 л, 5,0 экв.) загружают в стеклоэмалированный реактор с перемешиванием в атмосфере азота при 25-30°С. Реакционную смесь перемешивают при 25-30°С в течение 18 часов. После завершения реакции добавляют 30% раствор MTBE в гексанах (28,5 л, 5,0 об., 8,55 л MTBE в 19,95 л гексанов) и перемешивают смесь при 25-30°С в течение 2 часов. Твердое вещество собирают фильтрацией, промывают 20% MTBE в гексанах (34,2 л, 6,0 объема, 6,8 л МТБЭ в 27,4 л гексана) и оставляют сушить. Твердый продукт сушат при 25-30°С в вакууме (500-600 мм рт.ст.) с получением 3-(ацетилокси)-4-метоксипиколиновой кислоты в виде порошка бледно-желтого цвета (6,85 кг, выход 96,3%). ВЭЖХ (Zorbax SB-Aq, (250 × 4,6) мм, 5,0 мкм; А: 0,1% ТФУК в воде, В: ацетонитрил, скорость истечения: 1,0 мл/мин.) показывает, что чистота продукта составляет 98,5%.
Пример 1z. (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидо)пропаноат
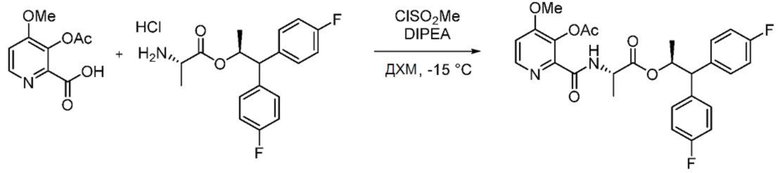
В колбу, снабженную магнитной мешалкой и входным отверстием для введения азота, загружают 3-ацетокси-4-метоксипиколиновую кислоту (2,97 г, 14,05 ммоль) и ДХМ (28,1 мл). Добавляют DIPEA (9,82 мл, 56,2 ммоль) и охлаждают реакционную смесь до -15°С. К смеси медленно добавляют метансульфонилхлорид (1,095 мл, 14,05 ммоль). После перемешивания в течение 15 минут к смеси добавляют гидрохлорид (S)-1,1-бис(4-фторфенил)пропан-2-ил-L-аланината (5 г, 14,05 ммоль). Смесь перемешивают в течение 90 минут, после чего оставляют ее нагреваться. Раствор выливают в насыщенный раствор NH4Cl (водный раствор) в делительную воронку. Слои разделяют и органический слой промывают смесью 1:1 насыщенный раствор соли/1N HCl, затем водой. ДХМ слой сушат над Na2SO4, фильтруют и концентрируют с получением масла. Сырой продукт очищают с помощью хроматографии на силикагеле (элюирование с градиентом: этилацетат/гексан) с получением указанного в заголовке соединения в виде белой пены (2,9 г, выход 38% выход): 1Н ЯМР (400 МГц, CDCl3) δ 8,39 (с, 1H), 8,32 (д, J=5,4 Гц, 1H), 7,26-7,16 (м, 4H), 7,03-6,87 (м, 5H), 5,71 (дкв, J=9,6, 6,1 Гц, 1H), 4,55 (дд, J=8,0, 7,1 Гц, 1H), 4,04 (д, J=9,6 Гц, 1H), 3,91 (с, 3H), 2,38 (с, 3H), 1,22 (д, J=6,1 Гц, 3H), 0,99 (д, J=7,2 Гц, 3H); 19F ЯМР (376 МГц, CDCl3) δ -115,61, -115,96; МСВР-ESI (m/z) [М+Н]+ вычислено для C27H27F2N2O6 512,1759; найдено 513,1825.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ 1,2,3,4-ТЕТРАГИДРОХИНОКСАЛИНА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2804127C2 |
| СПИРОЦИКЛИЧЕСКИЕ АМИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ S1P | 2011 |
|
RU2602800C2 |
| ПИРИМИДИНАМИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2006 |
|
RU2420519C2 |
| ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2761626C2 |
| БОРСОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE4 | 2019 |
|
RU2793936C2 |
| ПИРИМИДИНГИДРАЗИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2008 |
|
RU2464262C2 |
| СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ ОКСАДИАЗОЛ, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2789456C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2013 |
|
RU2667788C2 |
| ПРОЛЕКАРСТВА ЛЕВОДОПА, КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2365580C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
Изобретение относится к области химической технологии. Описана группа изобретений, включающая способ получения (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидo)пропаноата (варианты), а так же способ получения сложного 2-аминопропаноатного эфира из 1,1-бис(4-фторфенил)пропан-1,2-диола (варианты). В одном из вариантов реализации способ получения соединения (S,S)-1,1-бис(4-фторфенил)пропан-2-ил-2-(3-ацетокси-4-метоксипиколинамидo)пропаноата включает связывание 4-метокси-3-ацетилоксипиколиновой кислоты или 4-метокси-3-гидроксипиколиновой кислоты со сложным 2-аминопропаноатным эфиром полученным из 1,1-бис(4-фторфенил)пропан-1,2-диола. 4 н. и 29 з.п. ф-лы, 26 пр.
1. Способ получения соединения формулы А,
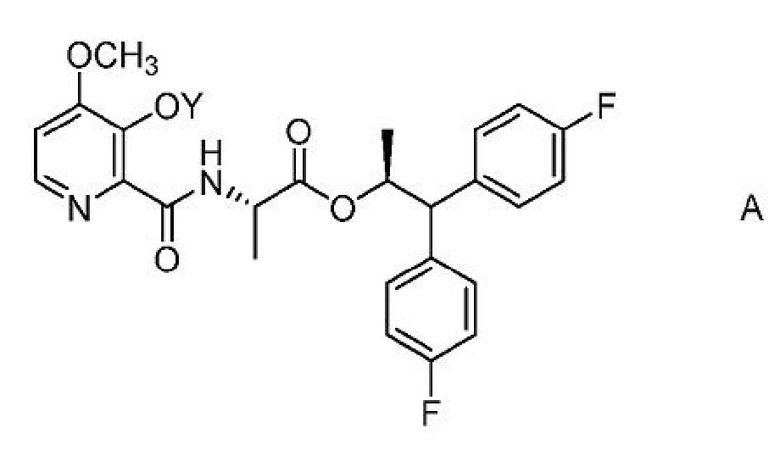
где Y представляет собой СН3СО;
из соединения формулы В,
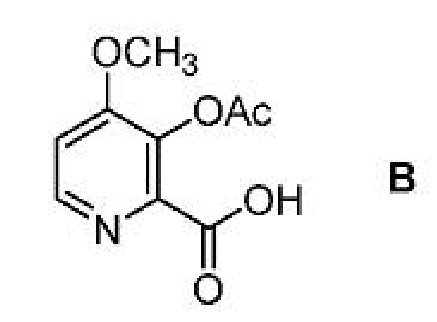
который включает следующие стадии:
a) получение первой смеси, содержащей соединение формулы В, ацилирующий агент или хлорирующий агент и основание;
b) добавление по меньшей мере одного соединения, выбранного из соединений формулы С
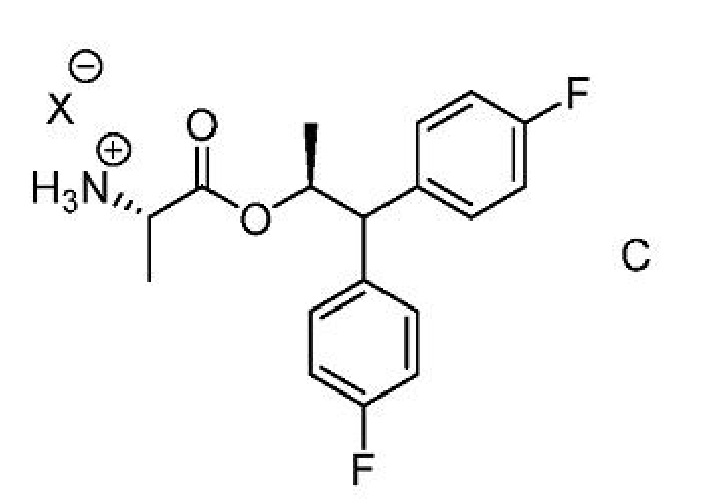
где Х представляет собой Cl, Br, HSO4, Н2PO4 или CH3SO3;
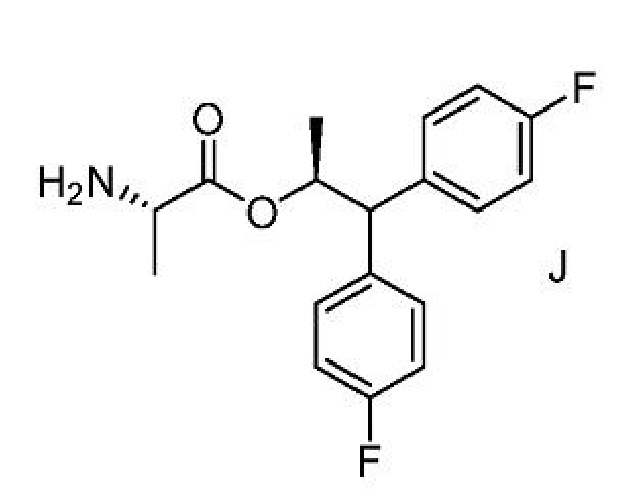
и формулы J
к первой смеси с получением второй смеси; и
с) выделение соединения формулы А из второй смеси.
2. Способ по п. 1, в котором ацилирующий агент представляет собой алкилхлорформиат формулы ClCO2R, где R представляет собой С1-С4 алкил или бензил, или хлорангидрид формулы RCOCl, где R представляет собой С1-С4 алкил.
3. Способ по п.1, в котором хлорирующий агент представляет собой оксалилхлорид или тионилхлорид.
4. Способ по п.1, в котором основание может быть выбрано из группы, в которую входят триэтиламин (ТEA), диизопропилэтиламин (DIPEA), пиридин, карбонат кальция и их смеси.
5. Способ по п.1, в котором первая смесь дополнительно содержит растворитель, выбранный из группы, включающей дихлорметан (ДХМ), 1,2-дихлорэтан (ДХЭ), изопропилацетат, тетрагидрофуран (ТГФ), 2-МeТГФ, ацетонитрил (ACN) и их смеси.
6. Способ получения соединения формулы А,
где Y представляет собой СН3СО или СН3СООСН2;
который включает следующие стадии:
a)получение первой смеси, содержащей соединение формулы D,
ацилирующий агент и первое основание;
b) добавление по меньшей мере одного соединения из соединений формулы С,
где Х представляет собой Cl, Br, HSO4, Н2PO4 или CH3SO3;
и формулы J
к первой смеси с получением второй смеси; и
с) выделение соединения формулы Е из второй смеси;
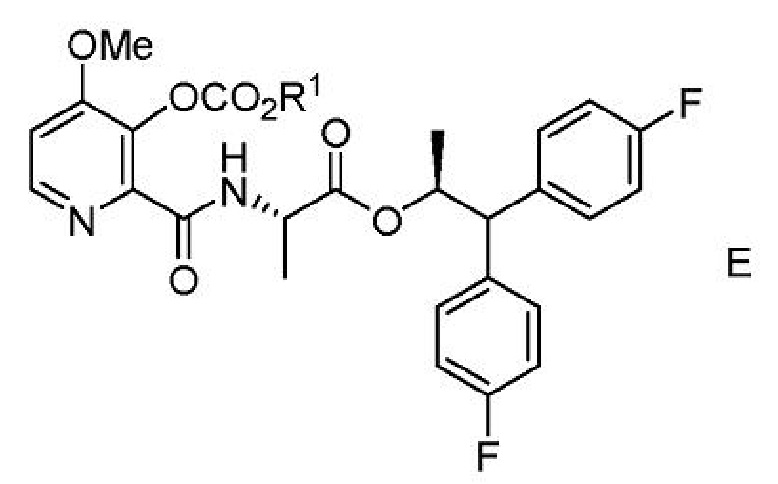
где R1 представляет собой С1-С4 алкил или бензил;
d) получение третьей смеси, содержащей соединение формулы Е, основание щелочного металла и воду;
e) выделение соединения формулы F из третьей смеси;
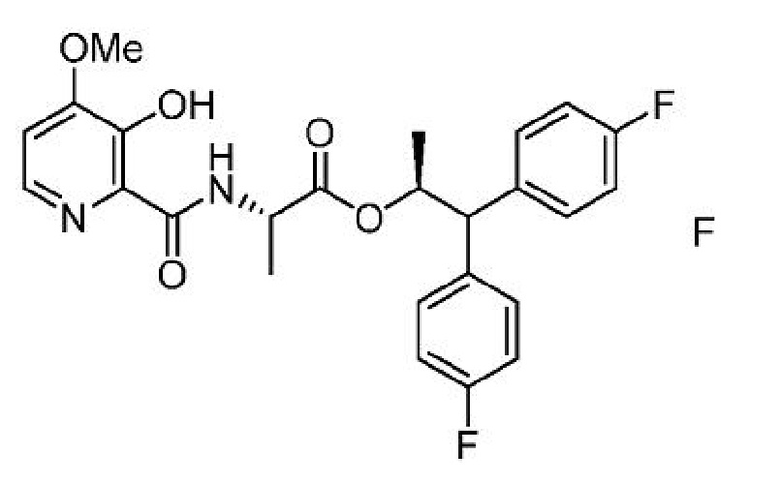
f) получение четвертой смеси, содержащей соединение формулы F, ацетилирующий агент или алкилирующий агент и второе основание; и
g) выделение соединения формулы А из четвертой смеси.
7. Способ по п.6, в котором ацилирующий агент представляет собой алкилхлорформиат формулы ClCO2R, где R представляет собой С1-С4 алкил или бензил.
8. Способ по п.6, в котором первая смесь дополнительно содержит растворитель, выбранный из группы, включающей дихлорметан (ДХМ), 1,2-дихлорэтан (ДХЭ), ацетонитрил (ACN), изопропилацетат, ТГФ, 2-МeТГФ и их смеси.
9. Способ по п.6, в котором первое основание может быть выбрано из группы, в которую входят триэтиламин (ТЕА), диизопропилэтиламин (DIPEA), пиридин и карбонат калия.
10. Способ по п.6, в котором используется примерно 3 эквивалента первого основания и примерно 2 эквивалента ацилирующего агента.
11. Способ по п.6, в котором основание щелочного металла может быть выбрано из группы, включающей LiOH, NaOH, KOH и их смеси.
12. Способ по п.6, в котором третья смесь дополнительно содержит сорастворитель, выбранный из группы, включающей ТГФ, 2-МeТГФ, ДМЭ, диоксан, ACN и С1-С4 спирт.
13. Способ по п.6, в котором ацетилирующий агент может быть выбран из уксусного ангидрида и ацетилхлорида.
14. Способ по п.6, в котором алкилирующий агент представляет собой CH3COOCH2Br.
15. Способ по п.6, в котором второе основание может быть выбрано из группы, в которую входят пиридин, ТEА, DIPEA и их смеси.
16. Способ получения соединения формулы С,
где Х представляет собой Cl, Br, HSO4, Н2PO4 или CH3SO3;
который включает следующие стадии:
a) получение первой смеси, содержащей соединение формулы G,
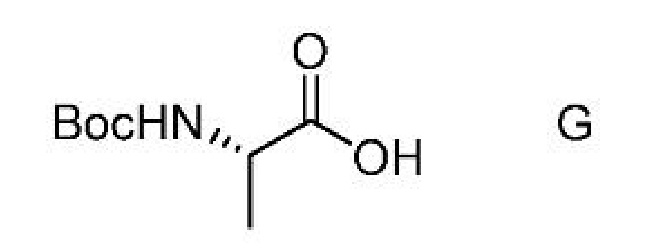
ацилирующий агент и основание;
b) добавление соединения формулы Н
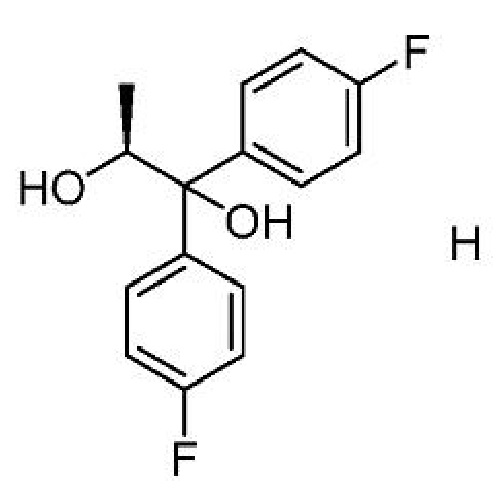
к первой смеси с получением второй смеси;
с) выделение соединения формулы I
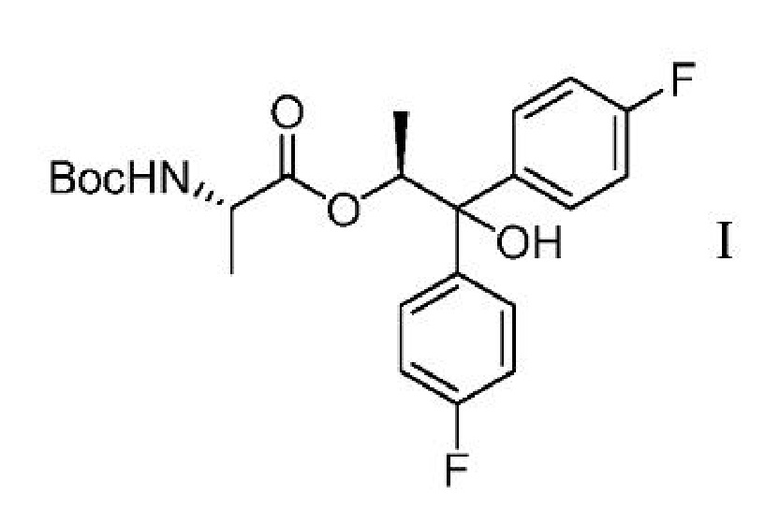
из второй смеси;
d) получение третьей смеси, содержащей соединение формулы I, кислоту и восстановитель;
e) выделение соединения формулы J из третьей смеси;
f) получение четвертой смеси, содержащей соединение формулы J и сильную кислоту;
где сильная кислота представляет собой НСl, НВr, Н2SO4, Н3PO4 или CH3SO3H; и
g) выделение соединения формулы С из четвертой смеси.
17. Способ по п.16, в котором ацилирующий агент представляет собой хлорангидрид кислоты формулы RCOCl, где R представляет собой С1-С4 алкил.
18. Способ по п.16, в котором первая смесь дополнительно включает растворитель, выбранный из группы, в которую входят ДХМ, ДХЭ, ACN, изопропилацетат, ТГФ, 2-МеТГФ и их смеси.
19. Способ по п.16, в котором основание может быть выбрано из группы, включающей ТЕА, DIPEA, пиридин и их смеси.
20. Способ по п.16, дополнительно включающий добавление DMAP (4-(диметиламино)пиридина) к первой смеси.
21. Способ по п.16, в котором кислота в третьей смеси представляет собой трифторуксусную кислоту или метансульфоновую кислоту.
22. Способ по п.16, в котором восстановитель выбирают из группы, включающей борогидрид натрия, триацетоксиборогидрид натрия и кремнийорганический гидрид.
23. Способ по п.22, в котором кремнийорганический гидрид выбирают из группы, включающей триэтилсилан, поли(метилгидросилоксан) и 1,1,3,3-тетраметилдисилоксан.
24. Способ по п.16, в котором четвертую смесь выдерживают в безводных условиях.
25. Способ получения соединения формулы С,
где Х представляет собой Cl, Br, HSO4, Н2PO4 или CH3SO3;
который включает следующие стадии:
a) получение первой смеси, содержащей соединение формулы Н,
восстановитель и кислоту;
b) выделение соединения формулы K из первой смеси;
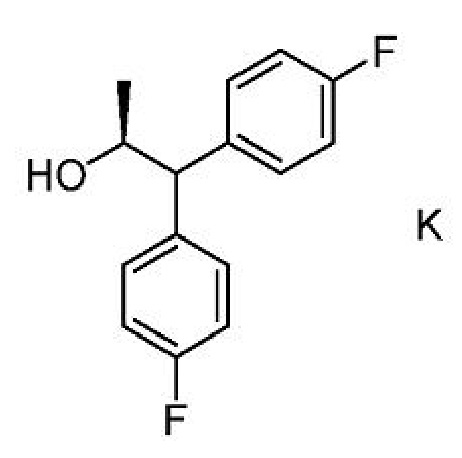
с) получение второй смеси, содержащей соединение формулы G,
ацилирующий агент и основание;
f) добавление соединения формулы K ко второй смеси с получением третьей смеси;
g) выделение соединения формулы L из третьей смеси;
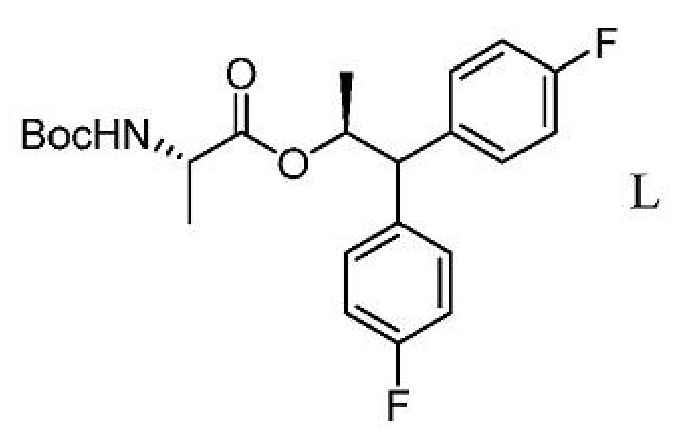
h) получение четвертой смеси, содержащей соединение формулы L и сильную кислоту;
где сильная кислота представляет собой НСl, НВr, Н2SO4, Н3PO4 или CH3SO3H; и
i) выделение соединения формулы С из четвертой смеси.
26. Способ по п.25, в котором кислота в первой смеси представляет собой трифторуксусную кислоту или метансульфоновую кислоту.
27. Способ по п.25, в котором восстановитель выбирают из группы, включающей борогидрид натрия, триацетоксиборогидрид натрия и кремнийорганический гидрид.
28. Способ по п.27, в котором кремнийорганический гидрид выбирают из группы, включающей триэтилсилан, поли(метилгидросилоксан) и 1,1,3,3-тетраметилдисилоксан.
29. Способ по п.25, в котором ацилирующий агент представляет собой хлорангидрид кислоты формулы RCOCl, где R представляет собой С1-С4 алкил.
30. Способ по п.25, в котором вторая смесь дополнительно содержит растворитель, выбранный из группы, включающей ДХМ, ДХЭ, ACN, изопропилацетат, ТГФ, 2-MeТГФ и их смеси.
31. Способ по п.24, в котором основание во второй смеси может быть выбрано из группы, включающей ТЕА, DIPEA, пиридин и их смеси.
32. Способ по п.25, дополнительно включающий добавление DMAP (4-(диметиламино)пиридина) ко второй смеси.
33. Способ по п.25, в котором четвертую смесь выдерживают в безводных условиях.
| US 20140187588 A1 03.07.2014 | |||
| US 20060040995 A1 23.02.2006 | |||
| US 9212141 B2 15.12.2015 | |||
| US 20150181867 A1 02.07.2015 | |||
| RU 2010118481 A 20.11.2011. |

